Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - ARATANA THERAPEUTICS, INC. | petx-20151231xex322.htm |
| EX-31.2 - EX-31.2 - ARATANA THERAPEUTICS, INC. | petx-20151231xex312.htm |
| EX-21.1 - EX-21.1 - ARATANA THERAPEUTICS, INC. | petx-20151231xex211.htm |
| EX-31.1 - EX-31.1 - ARATANA THERAPEUTICS, INC. | petx-20151231xex311.htm |
| EX-32.1 - EX-32.1 - ARATANA THERAPEUTICS, INC. | petx-20151231xex321.htm |
| EX-23.1 - EX-23.1 - ARATANA THERAPEUTICS, INC. | petx-20151231xex231.htm |
| EX-10.11 - EX-10.11 - ARATANA THERAPEUTICS, INC. | petx-20151231ex10116a054.htm |
| XML - IDEA: XBRL DOCUMENT - ARATANA THERAPEUTICS, INC. | R9999.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
|
|
FORM 10-K
|
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2015
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 12 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number: 001-35952
|
|
ARATANA THERAPEUTICS, INC.
|
|
|
Delaware |
38-3826477 |
|
|
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification Number) |
11400 Tomahawk Creek Parkway, Suite 340
Leawood, KS 66211
(913) 353-1000
(Address of principal executive offices, zip code and telephone number, including area code)
Securities Registered Pursuant to Section 12(b) of the Act:
|
Title of Each Class |
Name of Exchange on Which Registered |
|
|
Common Stock, par value $0.001 per share |
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
|
|
Indicate by check mark whether the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act of 1933 Yes: ☐ No: ☒
Indicate by check if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes: ☐ No: ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 and 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes: ☒ No: ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes: ☒ No: ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer |
☐ |
Accelerated filer |
☒ |
|||
|
|
|
|
|
|||
|
Non-accelerated filer |
☐ |
Smaller reporting company |
☐ |
|||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes: ☐ No: ☒
The approximate aggregate market value of the common stock held by non-affiliates of the registrant based upon the closing price of the registrant’s common stock on The NASDAQ Global Market on June 30, 2015 was $405,168,742.
As of March 10, 2016, there were 35,355,266 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant’s definitive proxy statement to be filed in connection with the registrant’s 2016 annual meeting of stockholders are incorporated by reference into Part III of this Form 10-K.
ARATANA THERAPEUTICS, INC.
FORM 10-K
For the Fiscal Year Ended December 31, 2015
|
Page |
||||
| 1 | ||||
|
Item 1. |
1 | |||
|
Item 1A. |
24 |
|||
|
Item 1B. |
43 | |||
|
Item 2. |
43 |
|||
|
Item 3. |
43 |
|||
|
Item 4. |
43 |
|||
| 43 | ||||
|
Item 5. |
43 |
|||
|
Item 6. |
46 |
|||
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
48 |
||
|
Item 7A. |
69 |
|||
|
Item 8. |
69 |
|||
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
69 |
||
|
Item 9A. |
69 |
|||
|
Item 9B. |
70 |
|||
|
71 |
||||
|
Item 10. |
71 |
|||
|
Item 11. |
71 |
|||
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
71 |
||
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
71 |
||
|
Item 14. |
71 |
|||
|
72 |
||||
|
Item 15. |
72 |
Aratana Therapeutics and our logo are two of our trademarks that are used in this filing. This filing also includes trademarks, tradenames and service marks that are the property of other organizations. Solely for convenience, trademarks and tradenames referred to in this filing appear without the ® and ™ symbols, but those references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or that the applicable owner will not assert its rights, to these trademarks and tradenames.
Cautionary Note Regarding Forward-Looking Statements
Except for historical information, the matters discussed in this Annual Report on Form 10-K for the fiscal year ended December 31, 2015 (“Annual Report”) are forward looking statements that involve risks, uncertainties and assumptions that, if they never materialize or if they prove incorrect, could cause our consolidated results to differ materially from those expressed or implied by such forward-looking statements. The Company makes such forward-looking statements under the “Safe Harbor” section of the Private Securities Litigation Reform Act of 1995. Actual future results may vary materially from those projected, anticipated, or indicated in any forward-looking statements as a result of various important factors, including those set forth in Item 1A of this Annual Report under the heading “Risk Factors.” Readers should also carefully review the risk factors described in the other documents that we file from time to time with the SEC. In this Annual Report, the words “anticipates,” “believes,” “expects,” “intends,” “future,” “could,” “estimates,” “plans,” “would,” “should,” “potential,” “continues” and similar words or expressions (as well as other words or expressions referencing future events, conditions or circumstances) identify forward-looking statements. Forward-looking statements also include the assumptions underlying or relating to any of the foregoing statements. The forward-looking statements contained in this Annual Report include, but are not limited to, statements related to: industry trends; market conditions; management’s plans and objectives for product development and commercialization; anticipated timing of regulatory submissions and approvals; investments in research and development; business prospects and collaborations; anticipated financial performance, including future revenues; expected liquidity and capitalization; our ability to protect our intellectual property from third-party claims; changes in accounting principles; changes in actual or assumed tax liabilities; expectations regarding tax exposures; anticipated reinvestment of future earnings; ability to repay our indebtedness; and our intentions regarding the use of cash. All forward-looking statements included in this document are based on information available to us on the date hereof. We will not undertake and specifically decline any obligation to update any forward looking statements.
Our Company
We are a pet therapeutics company focused on licensing, developing and commercializing innovative biopharmaceutical products for companion animals. We operate in one business segment, which sits at the intersection of the more than $60 billion annual U.S. pet market and the more than $23 billion annual worldwide animal health market.
Cats and dogs are the most popular pet species in the United States and Europe: there are approximately 86 million cats and 78 million dogs in the United States and 99 million cats and 81 million dogs in Europe. An estimated 68% of households in the United States have at least one pet. We believe that the role of pets in the family has significantly evolved over the last two decades. Many pet owners consider pets important members of their families, and they have been increasingly willing to spend money to maintain the health of their pets. Consequently, pets are living longer and, as they do, are exhibiting many of the same signs and symptoms of disease as aging humans, such as arthritis, cancer, obesity, diabetes and heart disease. Today veterinarians have comparatively few drugs at their disposal that have been specifically approved for use in pets. As a result, veterinarians often must resort to using products approved for use in humans, but not approved, or even formally studied, in pets, relying on key opinion leaders and literature, rather than regulatory review and approval.
We believe that pets deserve therapeutics that have been specifically studied and approved by regulatory authorities for each species, and that veterinarians and pet owners will increasingly demand that therapeutics be demonstrated safe and effective in pets before using them. We also believe there is an opportunity to leverage the investment in the human biopharmaceutical industry to bring therapeutics to pets in a capital and time-efficient manner.
Aratana Therapeutics, Inc. was incorporated on December 1, 2010 under the laws of the State of Delaware. In October 2013, we acquired Vet Therapeutics, Inc. (“Vet Therapeutics”), and in January 2014, we acquired Okapi Sciences NV (“Okapi Sciences”, which was renamed Aratana Therapeutics NV and is referred to as “Aratana NV” for all post-acquisition references). In addition to these acquisitions, we have completed several licensing transactions to further build our pipeline.
Our current portfolio includes therapeutic candidates in development consisting of small molecule pharmaceuticals and large molecule biologics that target what we believe to be large opportunities in serious medical conditions in pets. Our lead product candidates in development include small molecules directed at treating osteoarthritis pain and inflammation (AT-001 for dogs, also known as grapiprant for dogs or GALLIPRANT®), appetite stimulation (AT-002 for dogs, also known as capromorelin for dogs or ENTYCE®) and post-operative pain (AT-003 for dogs, also known as bupivacaine liposome injectable suspension for dogs or NOCITA®). On January 25, 2016, we submitted our administrative new animal drug application (“NADA”) for GALLIPRANT and anticipate approval from the U.S. Food and Drug Administration’s (“FDA”) Center for Veterinary Medicine (“CVM”) on or before our Animal
1
Drug User Fee Act (“ADUFA”) date of March 25, 2016. We plan to submit our administrative NADA for ENTYCE in March 2016, and we anticipate approval from the CVM on or before our ADUFA date which will be approximately 60 days thereafter. We anticipate submitting an NADA for NOCITA in the summer of 2016. We are also working to have AT-001, AT-002, and AT-003 approved in Europe, and on February 17, 2016 we announced that we had filed a marketing authorization application for AT-001 for dogs with the European Medicines Agency (“EMA”). We are also working to advance AT-001, AT-002 and AT-003 for cats. We believe that the potential commercial opportunity for AT-001, AT-002 and AT-003 is attractive. We also have two United States Veterinary Biological Product Licenses from the United States Department of Agriculture (“USDA”), AT-004 (also known as BLONTRESS®) and AT-005 (also known as TACTRESSTM ), to aid in the treatment of lymphoma for dogs. We believe that the commercial opportunity for the first generation monoclonal antibodies, BLONTRESS and TACTRESS, is very modest. We have also filed for a United States Veterinary Biological Product License from the USDA for AT-014, a novel her2/neu-directed cancer immunotherapy for the treatment of canine osteosarcoma, and we continue to anticipate conditional licensure for AT-014 by the end of 2016.
Our Strategy
Our strategy is to in-license proprietary technology from human biopharmaceutical companies and academia or leverage existing insights in human biology applicable to pets and to develop therapeutics specifically for use in pets. We seek to identify human therapeutics that have demonstrated safety and effectiveness in at least two species and are in, or have completed, Phase I or Phase II clinical trials in humans, with well-developed active pharmaceutical ingredients (“API”), advanced process chemistry and manufacturing. We also seek to identify products already in development for pets and to license or acquire these products. To date we have in-licensed and are further developing pharmaceutical compounds from companies such as Pacira Pharmaceuticals, Inc. (“Pacira”), RaQualia Pharma Inc. (“RaQualia”), Advaxis, Inc. (“Advaxis”), VetStem BioPharma, Inc. (“VetStem”) and Atopix Therapeutics Ltd. (“Atopix”).
Our goal is to become a leading provider of therapeutics developed and approved specifically for the treatment of unmet medical needs in pets. We are a pet-focused company and we intend to help shape and define the pet therapeutics market. We plan to accomplish this by:
|
|
• |
|
Leveraging our management team’s established experience in the human biopharmaceutical and animal health industries. In order to successfully execute our plan, we have assembled an experienced management team consisting of veterinarians, physicians, scientists and other professionals that apply the core principles of drug development to the medical needs of pets. The members of our senior management team have over 100 years of combined experience in the animal health and human biopharmaceutical industries, as well as a strong track record of successfully developing and commercializing therapeutics for pets. |
|
|
• |
|
Advancing our existing compounds to regulatory approval or licensure. We have assembled a pipeline of product candidates, including small molecule pharmaceuticals and large molecule biologics. These product candidates are in various stages of development for the treatment of cats or dogs, or both. Our target indications include pain and inflammation associated with osteoarthritis, appetite stimulation, post-operative pain, lymphoma, osteosarcoma, atopic dermatitis and ocular herpes. We plan to submit NADAs for the small molecule pharmaceuticals to the CVM for several of these product candidates starting in 2016 and to make similar regulatory filings to the EMA starting in 2016. For certain of our large molecule biologics, we plan to submit applications to the USDA for product licensure. |
|
|
• |
|
Using a direct sales organization and distributors to commercialize our products in the United States. We intend to employ a direct sales organization for fully licensed products, complemented by strategic distributor relationships intended to extend our commercial reach, to market our products in the United States. Our direct sales organization and distributors will sell products directly to veterinarians, who in turn typically sell pet therapeutics products to pet owners at a mark-up. In light of the veterinarian’s goal of improving the health of pets and the ability to generate revenue from the sale of therapeutic products, we believe veterinarians are motivated to prescribe innovative therapeutics that are safe, effective and supported by reliable clinical data and regulatory approval. We expect to continue a limited direct commercial effort with veterinary oncologists in 2016 and following the receipt of approvals for GALLIPRANT, ENTYCE and NOCITA, which we anticipate in 2016, we expect to begin commercializing product candidates GALLIPRANT in 2016, ENTYCE in late-2016 or shortly thereafter and NOCITA in 2016. |
|
|
• |
|
Engaging active collaborators to build a commercial presence. We have also in-licensed the rights in other geographies outside the U.S. for the use of our compounds in animal health, and we intend to seek regulatory approval for our pet therapeutics in Europe and potentially other countries. Based on the stage of our lead programs, we are currently exploring collaborations with companies that have an established commercial presence in other geographies that are looking for additional product candidates. We believe there are several animal health companies that desire innovative pet therapeutics. We also believe these companies may be interested in collaborating with us to provide EMA-approved best-in-class or first-in-class therapeutic products. |
2
|
|
• |
|
Continuing to expand our product pipeline by in-licensing additional compounds. We believe the pet therapeutics market is significantly underserved and have identified for further pursuit more than 20 therapeutic areas that overlap with areas of human biopharmaceutical development. Pursuant to our corporate strategy, we seek to identify compounds that have demonstrated safety and effectiveness in at least two species and are in, or have completed, Phase I or Phase II clinical trials in humans. We are looking for compounds with well-developed API process chemistry. Once identified, we seek to obtain exclusive, worldwide rights to these compounds in the animal health field. Each of our current compounds is covered by patents and other intellectual property that provide for a multi-year period of market exclusivity. Additionally, we intend to seek opportunities to collaborate with companies where we can provide commercialization for their approved products, or close-to-approved, pet therapeutic product candidates. |
Research and Development
Our pharmaceutical drug development programs focus on the development of novel compounds that target what we believe to be large opportunities in serious medical conditions in pets. We are building a proprietary research and development pipeline both through the application of our proprietary technologies and through strategic agreements that provide access to promising product development opportunities within our therapeutic focus. Our current product candidates are animal pharmaceuticals and certain biologics regulated by the CVM and immune-mediated biologics including monoclonal antibodies and therapeutic cancer vaccines regulated by the USDA. Our pipeline consists of three product candidates in final stages of submission to the CVM, two products with USDA licensure, one product candidate in the submission stage to the USDA, several product candidates in pilot studies, and additional early research programs.
We have incurred and will continue to incur research and development expense as we develop our business. Our research and development expenses were $24.9 million, $20.0 million and $10.9 million for the years ended December 31, 2015, 2014 and 2013, respectively.
Development Programs at the FDA
To begin the development process for our product candidates in the United States, we establish an Investigational New Animal Drug (“INAD”) file with the CVM. We then hold a pre-development meeting with the CVM to reach a general agreement on the plans for providing the data necessary to fulfill requirements for an NADA. During development, we submit pivotal protocols to the CVM for review and concurrence prior to conducting the required studies. We gather and submit data on manufacturing, safety and effectiveness to the CVM for review, and this review is conducted according to timelines specified in the ADUFA. Once all data have been submitted and reviewed for each technical section – safety, effectiveness and Chemistry, Manufacturing and Controls (“CMC”) – the CVM issues us a technical section complete letter as each section review is completed, and when the three letters have been issued, we compile a draft of the Freedom of Information Summary, the proposed labeling, and all other relevant information, and submit these as an administrative NADA for CVM review. Generally, if there are no deficiencies in the submission, the NADA is issued within 60 days after submission of the administrative NADA.
A separate approval either as an original or supplemental NADA is required for each species. In addition, where appropriate, we seek additional indications and additional formulations to extend the lifecycle of our product candidates. By exploring new uses and methods, we may potentially be able to extend the patent life of our product candidates and further achieve differentiation on in the marketplace.
3
The following tables identify the most advanced product candidates being developed under the FDA CVM regulations and their current regulatory status:
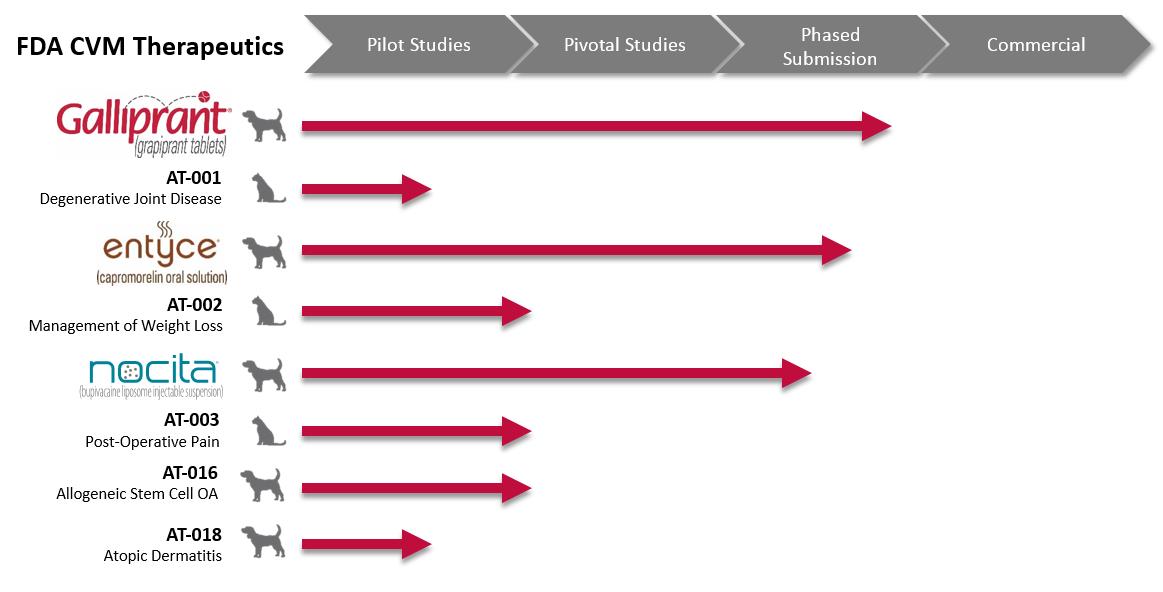

4
The product candidates that we believe are closest to commercialization and are a high priority for us include GALLIPRANT, ENTYCE and NOCITA, each of which we anticipate being approved for use in dogs in the United States. Product candidates being developed from the same active ingredient for other species are generally considered separate product candidates, and therefore we refer to these molecules in species other than dogs with AT-XXX and species designation or the generic name and species designation (for instance, AT-002 for cats or capromorelin for cats). Similarly, when referring to additional potential indications, we also revert to the AT-XXX nomenclature with the new indication or generic name with the new indication (for instance, the designation would be ENTYCE for appetite stimulation in dogs but AT-002 for weight gain in dogs or capromorelin for weight gain in dogs). In addition, while GALLIPRANT is used in the U.S. reflecting the fact that the FDA has reviewed the trade name, we refer to the product candidate outside of the U.S. as AT-001. We believe the naming conventions in pet therapeutics, while potentially cumbersome, are important from a regulatory perspective so that veterinarians know specifically what products are approved in what species, for what indications and in what geographies.
AT-001 (grapiprant)
AT-001 is a selective antagonist of the prostaglandin E2 (PGE 2) EP4 receptor (EP4 prostaglandin receptor antagonist, or EP4 PRA) that we in-licensed from RaQualia. EP4 is one of four G-protein coupled PGE 2 receptors (EP1, EP2, EP3 and EP4) located on the membrane of various cells in the mammalian body. The EP4 receptor predominantly mediates PGE 2-elicited pain. The specific effects of the binding of PGE 2 to the EP4 receptor include vasodilation, increased permeability, angiogenesis and production of pro-inflammatory mediators. The EP4 receptor mediates PGE 2-elicited sensitization of sensory neurons, and studies published in the Journal of Pharmacology and Experimental Therapeutics and in the British Journal of Pharmacology have demonstrated that EP4 is a major receptor in mediating pain associated with both rheumatoid arthritis and osteoarthritis and inflammation. AT-001 binds selectively to the EP4 receptor with high affinity thus blocking it from PGE 2-mediated pain and inflammation.
GALLIPRANT
During 2015, we received the target animal safety technical section complete letter, the technical section complete letter for CMC, and the technical section complete letter for effectiveness from the CVM for GALLIPRANT (grapiprant for dogs), an EP4 PRA to treat osteoarthritis pain and inflammation in dogs. The technical section complete letter for efficacy is based on results from a randomized, prospective, blinded, placebo-controlled multi-site pivotal study of 285 client-owned dogs evaluating the efficacy of Galliprant for dogs with osteoarthritis. The primary endpoint of the study was an owner assessment of success rate based on pre-determined improvements in the Pain Severity Score (PSS) and the Pain Interference Score (PIS) of the Canine Brief Pain Inventory on day 28. The study showed Galliprant met statistically significant (p<0.05) efficacy success criteria using the validated CBPI owner assessment tool as the primary endpoint. On Day 28, The CBPI success rate in dogs receiving Galliprant was 48 percent compared to 31 percent in dogs receiving placebo. Dogs receiving Galliprant showed statistically significant improvement in both pain severity scores and pain interference scores when compared to placebo over the 28-day period. The most common adverse reactions were emesis, diarrhea/soft stool and decreased appetite. There were no clinically relevant changes in blood chemistry. We finalized label negotiations, completed the other minor sections, and submitted an administrative NADA on January 25, 2016. Approval is anticipated on or before our ADUFA date on March 25, 2016, which if approved, is expected to enable us to commence commercialization of GALLIPRANT in the United States in the fall of 2016. Also, on February 17, 2016, we announced that we had filed a marketing authorization application for grapiprant for dogs with the EMA, and we anticipate regulatory approval in Europe as early as 2017.
AT-002 (capromorelin)
AT-002, which we also in-license from RaQualia, is currently being investigated for two separate potential indications in dogs, appetite stimulation and weight gain, and for one indication, management of weight loss, in cats. AT-002 is a potent and selective ghrelin agonist. Ghrelin is a 28-amino acid peptide hormone, also referred to as the hunger hormone, produced predominantly in the stomach. It is the endogenous ligand of the ghrelin receptor, also known as growth hormone secretagogue receptor (“GHS-R”). By activation of the ghrelin receptor, ghrelin stimulates appetite and growth hormone secretion, and also exhibits a role in regulation of gastrointestinal motility and energy balance. Ghrelin binds to specific receptors and affects signaling in the hypothalamus, interacting with other hormones to cause the feeling of hunger and stimulate food intake. In addition to its effects on appetite, ghrelin stimulates growth hormone secretion by activation of GHS-Rs in the pituitary. This effect acts to build lean body mass, which has been shown to result in increased strength in frail, elderly people. The appetite stimulation and GH-releasing activity of AT-002 has been demonstrated in laboratory cats and dogs where AT-002 treatment results in increased food intake and weight gain. Similarly, chronic oral dosing of AT-002 in a dog GLP toxicology study stimulated appetite, weight gain and caused increased plasma growth hormone levels.
ENTYCE
During 2015, we received the target animal safety technical section complete letter and the CMC technical section complete letter, and on February 23, 2016, we announced the receipt of the technical section complete letter for effectiveness from the CVM for ENTYCE for appetite stimulation in dogs. The technical section complete letter for effectiveness was based on results from a double-masked, randomized, placebo-controlled pivotal study of 244 dogs evaluating the efficacy of ENTYCE for dogs with various medical conditions that demonstrated reduced or no appetite for a minimum of two days. The primary endpoint of the study was an owner assessment of appetite using an “increased,” “no change” or “decreased” scoring system. The statistically
5
significantly results (p=0.0078) of the owner assessment showed 68.6 percent of dogs had an increased appetite compared to 44.6 percent of the dogs receiving placebo. The secondary endpoints of the study showed ENTYCE successfully increased the owner appetite assessment questionnaire score (56.2 percent compared to 26.8 percent in placebo; p=0.0071) and increased body weight in dogs enrolled in the study (76.0 percent compared to 44.6 percent in placebo; p=0.0012). Mild adverse reactions were observed in the treatment and placebo groups including diarrhea and vomiting; hypersalivation, abdominal discomfort, flatulence and nausea was observed in a small number of dogs in the treatment group.
We anticipate submitting an administrative NADA by the end of March 2016, which if approved, is expected to enable us to commence commercialization of ENTYCE in the United States in late-2016 or shortly thereafter. We are continuing our interactions with European national agencies, and believe that our discussions will lead to the successful development of capromorelin outside the U.S. We believe that the first claim in dogs for Europe will be either acute appetite stimulation or a chronic use weight gain claim. We also intend to pursue capromorelin for weight gain in dogs in the U.S. as a label extension after approval of the appetite stimulation claim.
AT-003 (bupivacaine liposome injectable suspension)
AT-003, which we in-licensed from Pacira, is being investigated for post-operative pain in dogs and cats. The product was approved for use in humans as a local, post-operative analgesic by the FDA in October 2011 and is marketed by Pacira for use in controlling post-operative pain following various types of surgical procedures. AT-003 is a 1.3% bupivacaine liposome injectable suspension consisting of microscopic, spherical multivesicular liposomes, which is Pacira’s proprietary drug delivery system. Bupivacaine is released from the particles by mechanisms involving reorganization of the barrier lipid membranes and subsequent diffusion of the drug occurs over an extended period of time. The formulation has been shown to extend the duration of human post-operative analgesia from approximately six to eight hours, to as long as 72 hours in some instances, which can eliminate the need for follow-on post-operative administration of other pain drugs. Additionally, the slower uptake of the bupivacaine into the systemic circulation helps avoid high plasma concentration and presumably lowers the risk of systemic toxicity.
Bupivacaine is a local anesthetic that prevents the generation and conduction of nerve impulses, apparently by increasing the threshold for electrical excitation in the nerve, by slowing the propagation of the nerve impulse, and by reducing the rate of rise in the action potential. Bupivacaine has a history of use in the United States of more than 30 years and its pharmacology, pharmacodynamics and toxicology in laboratory animals and humans are well understood. Bupivacaine is widely used by veterinary surgeons.
NOCITA
During 2015, we received the target animal safety technical section complete letter and on February 22, 2016, we announced the receipt of the technical section complete letter for effectiveness from the CVM for NOCITA for post-operative pain management in dogs undergoing knee surgery. The technical section complete letter for effectiveness was based on data from a multi-center, placebo-controlled, randomized and masked field study of 182 client-owned dogs undergoing cranial cruciate ligament stabilization surgery. Results from the study showed NOCITA met efficacy success criteria of no rescue analgesia required as assessed by trained observers using the Glasgow Composite Measure Pain Scale-Short Form. The primary endpoint for effectiveness was evaluated in the first 24 hours and showed NOCITA provided statistically significant (p=0.0322) success rates (68.8 percent) compared to placebo (36.5 percent). The secondary endpoint confirmed NOCITA demonstrated statistically significant success rates for dogs compared to placebo for the 24-48-hour interval (64.3 percent to 34.6 percent respectively; p=0.0402) and for the 48-72-hour interval (61.6 percent to 32.7 percent respectively; p=0.0432), supporting effective use of NOCITA for up to 72 hours of analgesia. Failures were carried forward to each subsequent interval. Adverse reactions in the study were infrequent and included discharge from the incision, incisional inflammation and vomiting.
During 2015, we received a response to our first CMC technical section submission and we re-submitted the CMC technical section in December 2015. We have an ADUFA date of June 7, 2016 for a response from the CVM on the CMC technical section. Accordingly, we anticipate submitting an administrative NADA in the summer of 2016, which if approved, is expected to enable us to commence commercialization of NOCITA in the United States in late-2016. We are continuing our interactions with European national agencies, and believe that our discussions will lead to the successful development of AT-003 for dogs outside the U.S.
AT-001, AT-002 and AT-003 for cats
We are also investigating the use of our lead molecules in cats, which would require separate development studies and separate regulatory approvals.
AT-001 for cats
We have completed three proof of concept studies in client-owned cats, including a recently completed placebo controlled pilot field study of 64 cats with degenerative joint disease to develop a study design, establish a dose regimen, and test various endpoints. Earlier studies and the most recent pilot field study indicate that while there is an efficacy signal for AT-001 for cats, further work is needed to establish an acceptable tolerability profile. We continue to conduct pilot studies to better understand the therapeutic window before deciding if and how we can proceed to a pivotal field effectiveness study.
6
AT-002 for cats
During the third quarter of 2015, we announced the results of a multi-site field pilot study of AT-002 in approximately 40 cats diagnosed with chronic kidney disease and documented weight loss. AT-002-treated cats had statistically significant increases in body weight compared to placebo after 90 days with differences beginning on day 30 and AT-002 was well tolerated. We believe that the results of this pilot study sufficiently inform us in designing and proceeding with the pivotal field effectiveness study which we anticipate commencing in 2016.
AT-003 for cats
During the third quarter of 2015, we announced the results of a placebo-controlled pilot field study of AT-003 for cats with postsurgical pain, which demonstrated improvements in pain evaluations compared to placebo that approached statistical significance. We believe that the results of this pilot study sufficiently inform us in designing and proceeding with the pivotal field effectiveness study which we anticipate commencing in 2016.
We also have additional programs that continue to advance through the CVM regulatory process, including AT-016 and AT-018.
AT-016 for dogs
AT-016 is an adipose-derived allogeneic stem cell product candidate for the potential treatment of osteoarthritis pain in dogs. We have an exclusive license agreement with VetStem for the rights to commercialize AT-016. AT-016 may provide long-term relief of pain and disability caused by osteoarthritis through regeneration of joint damage. AT-016 could provide immediately available point-of-care and does not require daily administration. If approved, we believe that AT-016 would be the first FDA-regulated “off the shelf” regenerative cell therapy for the treatment of osteoarthritis in dogs. In 2015, VetStem completed enrollment of a dose confirmation study for AT-016 of approximately 90 dogs with osteoarthritis and announced positive results, including that AT-016 demonstrated a statistically significant improvement in treatment versus placebo (p<0.05). VetStem received FDA feedback on the manufacturing methods section of CMC and first qualifying batch of drug product for use in clinical trials. If approved by the CVM, AT-016 could be commercially available in the United States in 2018.
AT-018 for dogs
We are developing AT-018 as an oral CRTH2 antagonist for the potential treatment of atopic dermatitis, focusing initially on developing the product candidate for dogs. This product candidate is in-licensed from Atopix following an option period. CRTH2 is a G protein-coupled receptor expressed selectively by key cells that mediate allergic responses, and it has been shown to play an important role in both allergic sensitization and effector responses to allergen. CRTH2 antagonists block mast cell-dependent activation of Th2 lymphocytes, basophils and eosinophils, interrupting an important immune pathway. We are currently conducting a multi-center, masked, placebo-controlled, randomized pilot field study in client-owned dogs and intend to close enrollment by the end of the first quarter 2016. We intend to initiate an additional pilot field study in 2016 in preparation to plan for the pivotal program.
Development Programs at the USDA
There are many parallels between the requirements to receive approvals for a veterinary pharmaceutical product candidate and certain veterinary biologics product candidates. The terminology differs but the three main components are the same: efficacy, manufacturing, and safety. USDA regulations are designed to ensure that veterinary biologics are pure, safe, potent and effective. The differences compared to pharmaceutical product regulations are based on the immunological nature of the mode of action of the product and the manufacturing process involving living organisms. In cases of emergencies, which means there is no approved product available, the USDA will issue a time-limited conditional license after the manufacturing and safety requirements have been substantially fulfilled and a reasonable expectation of efficacy has been established. The applicant has to continue the pivotal efficacy program and product testing validation. The conditional license can be extended if reasonable progress towards full licensure can be demonstrated.
7
A unique requirement for veterinary biologics in the United States is that manufacturers must hold a U.S. Veterinary Biologics Establishment License to produce licensed veterinary biologicals. An establishment license will only be issued if at least one biological product qualifies for a license. Applications for veterinary biologics establishments include articles of incorporation for the applicant, qualifications of veterinary biologics personnel for key employees, water quality statement, facility blueprints, plot plans, and legends. Products in our pipeline regulated by the USDA are:
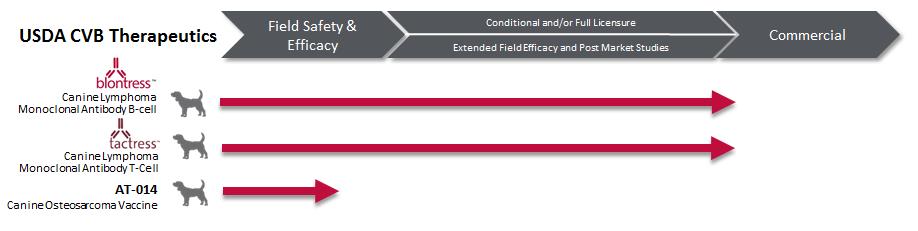
BLONTRESS and TACTRESS
BLONTRESS is a caninized monoclonal antibody approved by the USDA Center for Veterinary Biologics (“CVB”), in January 2015 under a full license as an aid for the treatment of B-cell lymphoma in dogs.
TACTRESS is a caninized monoclonal antibody approved by the CVB in January 2016 under a full license as an aid for the treatment of T-cell lymphoma in dogs.
We believe the full licensures of BLONTRESS and TACTRESS represent the only two full licensures for a species-specific monoclonal antibody within the animal health industry. However, scientific studies suggest that BLONTRESS and TACTRESS are not as specific to the target as expected. In addition, we have been performing various clinical activities to gain further knowledge around the efficacy of BLONTRESS and TACTRESS and to better elucidate how these products work in combination with chemotherapy-based protocols, which is the current standard of care. With respect to TACTRESS, our analysis of the results from two randomized, placebo-controlled post-marketing studies (T-CHOMP and T-LAB) indicate that TACTRESS did not seem to be adding significant progression free survival. With respect to BLONTRESS, prior to March 2016, it had only been available in regulatory studies, investigator-initiated studies, the on-going Mini B-CHOMP study, and for limited compassionate use. Three small investigator-initiated studies have been reported that support use of BLONTRESS in combination with chemotherapy. The larger Mini B-CHOMP study will compare the effectiveness of two cycles of CHOP plus BLONTRESS against historical benchmarks for a standard of care CHOP protocol. We expect to report the results of Mini B-CHOMP in mid-2016.
We expect that some oncologists will continue to use TACTRESS in certain, limited settings. Feedback from oncology advisors and oncologists is that while median progression-free survival would have been important for broad use, there is interest to explore the product in individual dogs especially given the limited effective treatment options in canine T-cell lymphoma. With respect to BLONTRESS, our market research and interactions with veterinary oncologists indicate that high specificity, including binding and depletion, will likely be necessary to drive wide adoption of monoclonal antibody therapy given that canine B-cell lymphoma is generally chemotherapy sensitive. Given that there are not alternative monoclonal antibodies available to veterinarian oncologists, we intend to maintain product availability and build awareness of monoclonal antibody therapy with BLONTRESS and TACTRESS while we pursue second generation monoclonal antibodies and other product concepts in lymphoma.
AT-014 for dogs
In March 2014, we entered into an exclusive license agreement with Advaxis for a novel her2/neu-directed cancer immunotherapy for the treatment of canine osteosarcoma and other cancers. In July 2014, we submitted for a product license with the USDA for AT-014 and we are in ongoing communication with the USDA to agree on the data requirements in support of U.S. Veterinary Biological Licensure. In 2015, we received data from a clinical study in 18 client-owned dogs with osteosarcoma treated with AT-014. The median survival time (“MST”) of a historic control group was 316 days. The MST of the treatment group had not been reached with 80% of dogs (15 dogs) surpassing the MST of the control group (p<0.001) and 60% still alive. In March 2015, we received notice that the USDA accepted the data from this study to provide reasonable expectation of efficacy to support conditional licensure for AT-014. We have started the process to transfer manufacturing from Advaxis to a USDA-licensed contract manufacturer of our choice. We are planning on further safety studies for USDA licensure. We anticipate receipt of a conditional USDA license by the end of 2016.
8
Early Stage Pipeline, Exclusive Option Programs and De Novo Product Generation
We also have additional programs targeting indications such as lymphoma and other cancers in multiple species, canine allergy, feline calicivirus, feline herpesvirus, feline immunodeficiency virus and periodontal disease. We can provide no assurance, however, that regulatory approval of our product candidates will be obtained.
As part of our product selection and development effort, we enter into option agreements with human biopharmaceutical companies to access certain product candidates. These agreements are for a predetermined period of time and enable us to perform additional due diligence and further evaluate the product candidate prior to entering into a license. We negotiate the terms of the license at the time of the option agreement and those terms become effective only if we exercise the option. Using this strategy, we have the ability to perform due diligence on multiple molecules in the same therapeutic class. In addition to exploring external product candidates in our exclusive option program, we occasionally secure rights to additional product candidates in conjunction with licensing deals. We typically designate such external and internal product candidate opportunities with a letter from the Greek alphabet. We currently have three programs secured: AT-Epsilon for melanoma, AT-Zeta for hemangiosarcoma, and AT-Iota for periodontal disease in dogs.
We operate two development sites, one in San Diego, California and the other in Leuven, Belgium, where we are engaged in the identification of new product candidates. At our facility in San Diego, we are developing patent protected, species-specific, monoclonal antibodies against biological targets of known activity. From our internal efforts, we have two programs, AT-Eta and AT-Theta as monoclonal antibodies against undisclosed targets. At our Leuven facility, historically Okapi Sciences (and then Aratana NV) had also been engaged in pre-clinical discovery efforts and certain production animal research and development. However, during the third quarter of 2015, we reviewed certain operations of our wholly owned subsidiary, Aratana NV and as a result, we made the strategic decision to wind down pre-clinical discovery efforts being performed at Aratana NV and focus future efforts of Aratana NV on clinical assets, the development of core legacy programs, i.e. AT-001, AT-002 and AT-003 for EU approval, business development and monetization of production animal assets and know-how obtained in the acquisition of Okapi Sciences. To facilitate this reorganization, ViroVet BVBA (“ViroVet”), a variable interest entity, was formed and during the third quarter of 2015, we began to transition employees from Aratana NV to ViroVet. We plan to transition additional selected Aratana NV employees, assets and liabilities over the next six months to ViroVet to further pursue the research and development of production animal products. These employees are expected to focus on the advancement of production animal assets/know-how and the securing of additional funding for future operations.
Manufacturing and Supply Chain
We have no internal manufacturing capabilities for the active pharmaceutical ingredients (or “APIs”) and drug products regulated by the CVM. To ensure dependable and high quality supply of API and formulated products for our clinical studies, we have chosen to rely on Current Good Manufacturing Practices (“cGMP”) compliant contract manufacturers rather than devote capital and manpower toward developing or acquiring internal manufacturing facilities. We believe we have or will have sufficient supply of formulated drugs to conduct each of our currently contemplated studies.
We have identified contract manufacturers to provide commercial supplies of the APIs and formulated drugs for our lead pharmaceutical product candidates, other than NOCITA, for which we expect to obtain marketing approval. These contract manufacturers have established track records of quality product supply and significant experience with regulatory requirements of both CVM and EMA. We are currently transferring the manufacturing technology process for GALLIPRANT and ENTYCE and scale-up required for commercialization. We believe we have or will have sufficient supply of formulated drugs to meet our commercial forecast.
For NOCITA, Pacira is our exclusive supplier of AT-003 and, under our December 2012 supply agreement with Pacira, is responsible for supplying us with finished drug product in vials. We are responsible for the labeling, packaging and shipping of the product. We must submit rolling forecasts to Pacira, with a portion of each forecast constituting a binding commitment. The term of the supply agreement extends for as long as the license agreement with Pacira continues in force. The license agreement has a term of fifteen years, until December 5, 2027, after which we have the option to renew the term for an additional five years. Pacira may terminate the supply agreement if we fail to make an undisputed payment, if we breach a material provision of the agreement, or if Pacira ceases manufacture of the product. Pacira also has the unilateral right to change its manufacturing process for the product. In this case, if we cannot reach agreement on the terms of continued supply of AT-003 meeting current specifications and Pacira decides that it is no longer commercially reasonable to supply us with product meeting such specifications, then Pacira may terminate the supply agreement.
We manufacture our monoclonal antibody products at our USDA-licensed facility in San Diego, California. We perform all steps for production including cell line development, assay development, production in batch mode, fill and finish, release of products, and packaging. We believe our production capacity will provide modest but sufficient product quantities to meet the commercial supply requirements of BLONTRESS and TACTRESS and development effort for second generation products. As noted above, for AT-014, we have started the process to transfer manufacturing from Advaxis to a third party USDA-licensed contract manufacturer. We have the ability to expand the relationship with the contract manufacturer, subject to our needs and growth of current and/or future biological products.
9
Sales and Marketing
We intend to commercialize any approved products in the United States through a direct sales organization and to augment that sales organization with select national and regional distributors. Outside the United States, we intend to engage in strategic collaborations to commercialize any approved products. Additionally, we seek to collaborate with companies where we can commercialize their approved pet therapeutic products in the United States.
Our sales organization is designed to sell any approved products to our customers, the companion animal veterinarians, who will in turn sell our products to pet owners at a mark-up. We believe that our products will be used by both primary care veterinarians and specialty veterinarians. According to industry sources, approximately one-third of companion animal practice revenue comes from prescription drug sales, vaccinations and non-prescription medicines. In light of the veterinarian’s goal of improving the health of pets and the ability to generate revenue from the sale of therapeutic products, we believe veterinarians are motivated to prescribe innovative therapeutics that are safe, effective and supported by reliable clinical data and regulatory approval. Veterinarians diagnose the medical problem, prescribe the medication, and directly sell or assist the pet owner in acquiring the medication. In pet health the pet owner is typically the payer as third-party insurance covers less than 5% of United States pet owners. Because the payments are the responsibility of the pet owner, the pet owner tends to be more involved in the purchase decision and more price conscious than what is seen in human health.
Based on a recent survey that we conducted, key attributes of companies that veterinarians find important include: standing behind their products, providing products that improve the standard of care, providing products exclusively to veterinarians, developing game changing products that are approved by FDA, having a knowledgeable sales force and offering continuing education. We believe that if customers view us favorably when considering these attributes, we can benefit from the loyalty and trust of our customers.
There are roughly 66,000 companion animal veterinarians today and they practice primarily in urban and suburban areas. Approximately 45,000 veterinarians are found in private primary care practices. Approximately 10,000 veterinarians are in corporate primary care practices with another approximately 11,000 veterinarians in specialty hospitals. Specialty veterinarians have become boarded in one of 22 specialties such as internal medicine, oncology or dermatology. Our commercial approach involves identifying our likely customers and developing strategies to efficiently reach and service them.
To prepare for the expected commercial introduction of our products, we have begun pre-launch marketing activities. Our marketing team is working closely with our development team to identify the key differentiating features and benefits of our products. We have developed labeling, pet-friendly formulations and user-friendly packaging to meet the needs of veterinarians and pet owners. We have filed or registered trademarks for our lead products in the key pet health markets and we have conducted primary market research with key opinion leaders, veterinarians and pet owners to assist in the establishment of the optimal product positioning and pricing. We continue to prepare peer-reviewed journal articles and scientific presentations that are being delivered at veterinary conferences and that can potentially be used as key elements of our promotional materials upon commercial introduction.
Currently, we have approximately 15 employees in our commercial team consisting of a medical director, medical science liaisons, a sales director, a sales operations director, a marketing director, and marketing support, pharmacovigilance, customer service and other operations personnel. We expect to add approximately 24 sales representatives, additional operations, marketing and medical personnel in the second half of 2016 to coincide with the anticipated launch of our first primary care products. These sales representatives will generate awareness, gain trial, educate veterinary staff, merchandise the hospitals and support our distributor partners. We plan to assist veterinarians with communications they can use to educate their clients, and we also plan to develop on-demand communications on the various diseases our products and product candidates are designed to treat, and their associated conditions, that pet owners can access. For large corporate accounts, we intend to sell directly to those organizations and we intend to service these accounts with key account teams.
In addition to a direct sales organization in the United States, we believe that we can use distributors to expand our commercial reach in an efficient manner. National veterinary distributors cover approximately 90% of the companion animal veterinary hospitals. There are also a number of buying groups or group purchasing organizations in the animal health industry that have formed to gain volume-based pricing advantages. These organizations often work through a preferred distributor and these agreements are also set up on an annual contractual basis. We intend to strategically balance our direct sales organization with national and regional distributors in a manner that optimizes our commercial efforts and allows us to provide coverage to a more expansive group of veterinary practices while incrementally growing our direct sales organization.
The pet supplies market in the United States is approximately one-third of the global market as measured by Euromonitor International and Vetnosis data suggest that the US represents one-third of the global animal health market. The pet market in Europe is similar in size to North America, and Latin America, Asia and the rest-of-the world constitutes the final one-third. We are working to complete the clinical and regulatory work required to get several of our products approved in Europe, including AT-001, AT-002 and AT-003. However, rather than build a direct commercial organization outside of the United States, we are seeking to form a commercial collaboration arrangement with one of the global animal health companies who have commercial capabilities in Europe and other areas. We believe we will be able to identify a licensee that will pay us attractive upfront payments, milestone payments and royalties for commercial rights to our products. Our objective is to work with a licensee to create global brands for our products even if the primary marketing responsibility is split geographically. Also, once we have secured a collaboration, we plan to explore ways to potentially work together in the United States to ensure successful commercial launches since the potential collaborators that we are
10
considering also have significant commercial presence in the United States. We hope to secure an outside-the-United States collaboration arrangement by mid-2016.
GALLIPRANT (grapiprant tablets)
GALLIPRANT is a small molecule EP4 prostaglandin receptor antagonist being developed to treat osteoarthritis pain and inflammation in dogs. See “Development Programs at the FDA-GALLIPRANT” for additional information.
Medical need
Osteoarthritis is the most common inflammatory joint disease in pets. The prevalence of osteoarthritis increases with age, usually occurring in dogs aged nine years or older, but it can occur even in young animals. According to industry sources, the number of pets diagnosed with arthritis has significantly increased over the past five years and an estimated 13% of all geriatric dogs, and 22% of geriatric large and giant breed dogs, are diagnosed with arthritis. We believe many dogs with arthritis remain undiagnosed and the incidence is approximately 20% in dogs. Osteoarthritis is a progressive disease that can first manifest itself with periodic signs of stiffness or lameness and can progress to where the pet is experiencing constant joint pain and stiffness. Osteoarthritis is diagnosed in animals by the veterinarian using clinical signs and radiographs. The disease is incurable, but treatment can improve the dog’s quality of life. Treatment includes a combination of rest, avoidance of overexertion, reduction in weight, proper exercise and a regimen of pain and anti-inflammatory drugs. In some cases, surgery to relieve the pain or correct deformities or instability might also be employed.
Currently available treatments and their limitations
Analgesic and anti-inflammatory drugs are often necessary to control pain in dogs with osteoarthritis. The currently approved products for control of the pain and inflammation associated with osteoarthritis in dogs are NSAIDs from the class of cyclooxygenase (“COX”) inhibitors. The arachidonic acid pathway constitutes the main mechanism for the production of pain and inflammation in osteoarthritis. This pathway also controls other important body functions such as kidney regulation, gastrointestinal mucosal protection, thrombosis and blood flow through the enzymatic synthesis of mediators in multiple steps along the pathway. Three COX isoenzymes have been identified—COX-1, COX-2 and COX-3. COX-2 initiates the biosynthesis of prostaglandin-I 2 or PGI 2 and prostaglandin-E 2 or PGE 2. PGI 2 affects gastrointestinal mucosa, kidney function and blood flow. PGE 2 also affects gastrointestinal mucosa and is a key mediator of pain and inflammation. The inhibition of COX enzymes to provide relief from pain and inflammation is the mode of action of NSAIDs. The first product approved for the control of pain and inflammation associated with dog osteoarthritis was Rimadyl (carprofen) in 1996. The introduction of this product created a product category around a previously unmet medical need and fundamentally changed the management of chronic pain in dogs. Rimadyl is a moderately selective inhibitor of COX-2 (Coxib) and has demonstrated selective inhibition of COX-2 versus COX-1 in dogs. While side effects in most dogs are generally mild and typical of the Coxib NSAID class, some dogs have an idiosyncratic sensitivity that results in hepatic and/or gastrointestinal toxicity and, in extreme cases, death. As a result, Coxib NSAID label language contains bolded warnings and specifies that baseline blood tests should be conducted, and pets should be periodically monitored using blood tests to check for any toxic effects.
Market opportunity
We believe there is a significant market opportunity for treatment of osteoarthritis in dogs. According to market research, 14.7 million dogs are diagnosed with osteoarthritis each year. Of those dogs, 9.7 million are being treated for the condition, and 2.4 million are treated with NSAIDs for more than 20 days, representing a $180 million market. According to market research, the total NSAID ex-manufacturer market in the United States is $357 million per year, $177 million of which represents treatment for acute pain and $180 million of which represents treatment for chronic pain.
We believe that GALLIPRANT will be used by veterinarians looking for alternatives to Coxib NSAIDs especially in dogs who do not tolerate Coxib NSAIDs or might otherwise be treated with nutritional supplements or alternative therapies. According to our market research, 94% of surveyed veterinarians stated that they were extremely likely or very likely to use GALLIPRANT assuming competitive pricing. In addition, veterinarians indicated a willingness to use GALLIPRANT in a variety of osteoarthritis disease states: severe (77%), moderate (65%) and mild (38%). A large majority of veterinarians surveyed indicated that they would adopt this product within the first year of it becoming available, and more than half of veterinarians said they would use GALLIPRANT earlier in the disease process versus the current marketed Coxib NSAIDs.
ENTYCE (capromorelin oral solution)
ENTYCE is a small molecule ghrelin agonist being developed for appetite stimulation in dogs. See “Development Programs at the FDA-ENTYCE” for additional information.
Medical need
The control of hunger and satiety involves a complex system in mammals. In many acute and chronic disease states, as well as with aging, lack of appetite is a problem and can fuel a downward spiral. Malnutrition and decreased muscle mass can result from inadequate food intake regardless of the underlying condition. In humans, doctors can rationalize with the patients the importance of maintaining nutrition despite the lack of natural appetite and there are medical therapeutics approved in humans to treat inappetence. Veterinarians and pet owners cannot successfully rationalize with pets about the importance of maintaining nutrition and there are no
11
approved medical therapeutics to treat inappetence in pets. This can be a frustrating clinical situation for the veterinarian and pet owner and often contributes to the decision to euthanize a pet. In a recent survey of veterinarians 81% believed that stimulation of appetite and weight gain in dogs represented a significant unmet need. Fear, pain, stress, trauma, organic disease, dental disease, oral fractures and cancer are all possible causes of inappetence in pets. For example, in pets undergoing cancer treatment, the cancer therapy is commonly stopped when the pet loses appetite and body weight.
According to our market research, inappetence is seen in approximately 23% of dogs who receive chemotherapy, although in clinical studies we observe inappetence rates to be lower but still clinically meaningful. We believe that, if approved for such indications, ENTYCE could be an important potential medicine in managing inappetence and weight in dogs with cancer. As a second example, inappetence and weight loss commonly occurs in conjunction with chronic renal failure (“CRF”). Dietary therapy with a diet that is designed for dogs with renal insufficiency is recommended regardless of the severity of disease. Unfortunately, many of the therapeutic diets that are prescribed may be less palatable to pets than normal diets. We believe that, if approved, ENTYCE could be an important medicine in managing inappetence and weight loss that occurs in connection with CRF. Other possible uses include inappetence associated with liver disease, cardiac disease, pancreatitis and gastrointestinal disorders.
Currently available treatments and their limitations
The first goal of therapy for inappetence is to correct the underlying cause. Often veterinarians will begin treatment of inappetence by recommending a change to a highly palatable diet such as tuna for cats and chicken or beef for dogs. Depending on the severity of the condition, the animal may be supported with fluids and electrolytes until the diagnosis of the underlying condition is made and effective treatment is initiated where possible. Prolonged or severe inappetence may require hospitalization and feeding tube placement. There are no drugs approved for the treatment of inappetence in cats and dogs. Drug therapy to address inappetence has focused primarily on human drugs affecting the central nervous system control of feeding such as benzodiazepines, cyproheptadine and mirtazapine. However, these drugs are not approved for veterinary use, have limited effectiveness and are contraindicated for cats with hepatic lipidosis. We have seen veterinarians using Cerenia (maropitant), which is indicated for the control of vomiting to determine whether the dog is inappetent due to nausea. We believe a significant number of veterinarians are not prescribing these therapies due to their limited safety and efficacy.
Market opportunity
We believe there is a significant market opportunity for a therapeutic product that is safe and can effectively stimulate appetite in pets. According to market research, 9.8 million dogs in the United States are inappetent each year, and 4.1 million of such dogs are treated for the condition (2.3 million dogs are being treated for acute inappetence and 1.8 million dogs are being treated for chronic inappetence). The large majority of veterinarians (87%) surveyed stated that they are extremely likely or very likely to use ENTYCE in chronic conditions, which is 40 days of therapy per year. By contrast 63% of veterinarians said they would use the product in acute conditions, which is 4 days of therapy per year.
NOCITA (bupivacaine liposome injectable suspension)
NOCITA is being developed to potentially provide local post-operative analgesia for cranial cruciate ligament surgery in dogs. NOCITA is a long-acting local anesthetic designed to provide up to 72 hours of post-operative pain control. See “Development Programs at the FDA-NOCITA” for additional information.
Medical need
Veterinarians perform approximately 20 million dog surgeries each year. Approximately 50% of dog surgeries are spays and neuters, while other common surgeries include cruciate repairs, fracture repairs, and cancer surgery. There is no established protocol for the use of pain medications in these surgeries and pain management practices have traditionally been based on the veterinarian’s views on the level of pain associated with a specific surgical procedure and the perceived pain tolerance of the pets. Recently, as pet owners have begun requesting analgesia for their pets’ painful conditions, veterinarians have made advances in treating pain in pets. Furthermore, animal research demonstrates that pain can have a detrimental effect on healing, and pain experts in academia and specialty practices are advocating more use of local anesthesia for pain control.
Currently available treatments and their limitations
The most widely used drugs approved for treatment of post-operative pain are Coxib NSAIDs and opioids in dogs and Coxib NSAIDs and buprenorphine and butorphanol in cats. In surgeries associated with the most severe post-operative pain, opioids including fentanyl are commonly used. Fentanyl is a controlled narcotic drug, and pets are often kept in the hospital while receiving fentanyl. In our experience, the majority of fentanyl is dispensed as fentanyl patches, although such use in pets has not been approved. In 2012, Nexcyon received FDA approval for a transdermal fentanyl solution, and Elanco launched the product in 2013 under the tradename Recuvyra. Use has been limited due to a number of label requirements relating to human safety and application. We believe that there are unmet needs in pets undergoing these more painful surgeries, especially if effective and extended pain relief could be achieved with a non-narcotic medicine. The same group of NSAIDs approved to treat the pain and inflammation associated with osteoarthritis in dogs are used for post-operative pain. Some of these drugs can be given in the veterinary hospital as an injection, and then oral formulations are dispensed to the owner for a few days of treatment at home. Among the drugs used for post-operative pain, some have been approved by the CVM, while others are used off label. The most commonly used post-operative pain medication in dogs is Rimadyl, which has been approved by the CVM for this use. Coxib NSAIDs have demonstrated significant side effects that result in
12
prescribed monitoring of dog health during their use. Consequently, we believe veterinarians would appreciate a drug for post-operative use that was effective, but also safe on the liver, gastrointestinal system and kidneys.
Market opportunity
We believe that there is a significant market opportunity for the treatment of post-operative pain in dogs. According to market research, approximately 20 million dogs in the United States undergo surgery per year and of such dogs, 5.8 million have very painful expensive surgeries for which we believe veterinarians may consider using NOCITA. In a survey of veterinarians, 76% responded that they believe that NOCITA offers a unique solution to post-operative pain and 39% said they would use the product for cranial cruciate ligament and fracture repairs. A significant number said they are also likely to use the product in amputations and other orthopedic surgeries. Due to the high price of treatment with NOCITA relative to generic opioids and Coxib NSAIDs, we anticipate that veterinarians will choose to use NOCITA in these more expensive and painful surgical procedures.
BLONTRESS (canine lymphoma monoclonal antibody, B-cell) and TACTRESS (canine lymphoma monoclonal antibody, T-cell)
TACTRESS is currently available to all veterinary oncologists. Feedback from our oncology advisors and oncologists is that while median progression-free survival would have been important for broad use, there is interest to explore the product in individual dogs especially given the limited effective treatment options in canine T-cell lymphoma. Hence, we expect that some oncologists will continue to use TACTRESS in certain, limited settings. We made BLONTRESS available in March 2016.
We believe the revenue and gross margin opportunity for the first generation monoclonal antibodies are very modest. However, given that there are not alternative monoclonal antibodies available to veterinary oncologists, we intend to maintain product availability and build awareness of lymphoma and monoclonal antibody therapy with BLONTRESS and TACTRESS while we pursue second generation monoclonal antibodies and other product concepts in lymphoma.
Competition
The development and commercialization of new animal health medicines is highly competitive, and we expect considerable competition from major pharmaceutical, biotechnology and specialty animal health medicines companies. As a result, there are, and likely will continue to be, extensive research and substantial financial resources invested in the discovery and development of new animal health medicines. Our potential competitors include large animal health companies, such as Zoetis; Merck Animal Health, the animal health division of Merck & Co., Inc.; Merial, the animal health division of Sanofi S.A.; Elanco, the animal health division of Eli Lilly and Company; Bayer Animal Health, the animal health division of Bayer AG; Boehringer Ingelheim Animal Health, the animal health division of Boehringer Ingelheim GmbH; Virbac Group; Ceva Animal Health; Vetoquinol and Dechra Pharmaceuticals PLC. We are also aware of several smaller early stage animal health companies, such as Nexvet, Jaguar Animal Health, Parnell Pharmaceuticals, VetDC and Kindred Bio that are developing products for use in the pet therapeutics market.
If approved, we expect GALLIPRANT will face competition from Rimadyl, marketed by Zoetis, and generic Carprofen, Deramaxx, marketed by Elanco, Previcox, marketed by Merial, Metacam, marketed by Boehringer Ingelheim, and generic Meloxicam. Nexvet is developing a monoclonal antibody for osteoarthritis pain. Parnell is developing Zydax, an injectable product for osteoarthritis, a direct competitor for GALLIPRANT. ENTYCE is entering a new market where there are not products approved for veterinary use to stimulate appetite. However, we are aware that some veterinarians utilize mirtazapine, a human generic antidepressant with known side effects and limited effectiveness, to treat inappetence. We expect NOCITA will compete primarily with the Coxibs and injectable buprenorphine and anesthetics, such as bupivacaine, which is not approved for non-human use but is widely used by veterinarians. Recuvyra fentanyl transdermal solution received approval in the United States and Europe for control of postoperative pain from surgical procedures in dogs. We are also unaware of any approved products for the treatment of lymphoma in dogs, although we are aware of a number of biotechnology companies such as Nexvet, Karyopharm and VetDC that are developing products for the treatment of lymphoma in dogs, including some that have received Minor-Use-Minor-Species (“MUMS”) designation. We expect that BLONTRESS and TACTRESS will face competition from human generic chemotherapies, although we are unaware of any companies that are actively promoting this use. Among the larger animal health companies, Merial recently received USDA conditional licensure for a lymphoma vaccine based on technology similar to their melanoma vaccine. We know of no direct competitor for AT-014 in osteosarcoma.
We are an emerging company with a limited history of operations and many of our competitors have substantially more resources than we do, including both financial and technical resources. In addition, many of our competitors have more experience than we have in the development, manufacture, regulation and worldwide commercialization of animal health medicines. We are also competing with academic institutions, governmental agencies and private organizations that are conducting research in the field of animal health medicines.
Our competition will be determined in part by the potential indications for which our products are developed and ultimately approved by regulatory authorities. Additionally, the timing of market introduction of some of our potential products or of competitors’ products may be an important competitive factor. Accordingly, the speed with which we can develop our compounds, complete target animal studies and approval processes, and supply commercial quantities to market are expected to be important competitive factors. We expect that competition among products approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price and patent position.
13
Intellectual Property and License Agreements
We seek to protect our products and technologies through a combination of patents, regulatory exclusivity, and proprietary know-how. Our goal is to obtain, maintain and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our current compounds and any future compounds for development, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the United States and abroad. However, even patent protection may not always afford us with complete protection against competitors who seek to circumvent our patents.
We depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as that of our advisors, consultants and other contractors, none of which is patentable. To help protect our proprietary know-how, which is not patentable, and inventions for which patents may be difficult to obtain or enforce, we rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we generally require all of our employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
Exclusive License Agreements with RaQualia
In December 2010, we entered into two agreements with RaQualia pursuant to which we exclusively licensed intellectual property rights relating to AT-001 and AT-002 in the animal health field. Pursuant to these agreements we obtained the rights to certain patents in the United States and other jurisdictions. The patents relating to AT-001 include composition of matter claims as well as claims to methods of use of AT-001. The patent rights relating to the use of AT-001 further include methods of preparing the compounds of interest and salts, polymorphs and intermediates thereof, as well as certain combination therapies. Additionally, we licensed from RaQualia additional patent rights relating to AT-002 that include composition of matter claims as well as claims to methods of use of AT-002. Under these agreements, we were granted exclusive, worldwide licenses to develop, manufacture and commercialize AT-001 and AT-002 in the field of animal health, except that we cannot develop, manufacture or commercialize injectable AT-001 products in Japan, South Korea, China or Taiwan. We have the right to grant sublicenses to third parties under these agreements. Under our agreement with RaQualia, we are responsible for using commercially reasonable efforts to develop and commercialize AT-001 and AT-002. The patent that we believe covers the crystalline form of the AT-001 compound to be marketed expires on February 21, 2027 and is expected to be eligible for a patent term extension of 2.5 to 3 years to 2029-2030. There is also a patent on methods of producing the AT-002 compound which expires on February 1, 2020 with potential term extension of about 2.5 years to 2022 or early 2023. Each of these potential patent term extensions are dependent on certain FDA approval dates of commercial use of the corresponding product. The patents and applications licensed under these agreements are expected to expire between 2016 and 2034.
We are responsible for contingent milestone payments upon achievement of development and regulatory milestones and royalties on net sales of licensed products, subject to certain potential offsets and deductions, under each of the AT-001 and AT-002 agreements, and the royalty percentage is in the mid-single digits. We must also pay to RaQualia a portion of royalties we receive from any sublicensees, subject to a minimum royalty on net sales by such sublicensees. Our royalty obligations apply on a country-by-country and licensed product-by-licensed product basis, and end upon the expiration or abandonment of all patents with valid claims covering a licensed product in a given country.
Each of the AT-001 and AT-002 agreements continues until terminated. RaQualia may terminate the AT-001 agreement or the AT-002 agreement if we fail to pay any undisputed fee under the relevant agreement and do not cure such failure within 60 days after RaQualia notifies us of such failure. We may terminate the AT-001 agreement or the AT-002 agreement, or any license granted under either agreement, on a patent-by-patent and country-by-country basis at will, upon 30 days’ prior written notice to RaQualia. Once all of the patents licensed under the AT-001 agreement or the AT-002 agreement have expired or been abandoned, the licenses granted under the relevant agreement become fully-paid and irrevocable.
Exclusive License Agreement with Pacira
In December 2012, we entered into an exclusive license agreement and related exclusive supply agreement with Pacira Pharmaceuticals, Inc., or Pacira. Under the license agreement, we were granted an exclusive, worldwide license to develop and commercialize, but not to manufacture, AT-003 in the veterinary field. Pursuant to this agreement we obtained the rights to certain patent rights relating to AT-003 including composition of matter claims and methods of use thereof. The patents and applications relating to AT-003 are expected to expire between 2015 and 2031.
We have the right to grant sublicenses to third parties outside the United States upon Pacira’s approval. Any sublicenses we wish to grant to third parties within the United States must be discussed with Pacira and approved by Pacira in its sole discretion and good faith reasonable business judgment. We are responsible for using commercially reasonable efforts to develop and commercialize AT-003, and for launching AT-003 within a specified time period following regulatory approval in certain countries.
We paid Pacira an upfront fee and are responsible for contingent milestone payments upon the achievement of certain development and commercial milestones and for royalties on net sales of AT-003 by us and our affiliates, with a tiered royalty percentage in the low- to mid-20s. We must pay Pacira a royalty on net sales of AT-003 by us and our affiliates, subject to certain reductions. We must also pay to Pacira a percentage of all payments we receive from any sublicensee, subject to certain offsets, and under certain
14
circumstances, share a portion of Pacira’s royalty payment obligations to its third-party licensors. We are responsible for meeting minimum annual revenue requirements for AT-003 beginning the fifth year after the first commercial sale of AT-003. If we fail to meet these requirements, either we or Pacira may terminate the license agreement.
The term of the license agreement extends for 15 years, until December 5, 2027, after which we have the option to renew the term for an additional five years. Pacira may terminate the agreement in its entirety if we fail to pay any amount due within a specified time period, or on a country-by-country basis if we fail to achieve certain regulatory, clinical and commercial milestones within certain timeframes. We may terminate the agreement on a country-by-country basis either upon the entry of a generic competitor, or at will outside the United States or the European Union. Either we or Pacira may terminate the agreement if the other party materially breaches or files for bankruptcy and fails to cure such breach within a specified time period, or if we do not pay the minimum annual revenue requirements referenced above. The agreement automatically terminates if Pacira terminates the related supply agreement and if certain circumstances involving a U.S. sublicensee occur and we do not meet certain financial obligations to Pacira.
Vet Therapeutics
As part of our Vet Therapeutics acquisition, we acquired a patent family related to the speciesization of antibodies that cover all Vet Therapeutics products with an issued patent expiring in 2029. We also acquired a patent family related to antibody constant domain regions and uses thereof, which also covers all Vet Therapeutics products and has an issued U.S. patent expiring in 2032. Finally, we acquired patent filings that cover specific canine monoclonal antibodies directed to various targets, including an issued U.S. patent directed to the canine CD 52 development antibody, which will expire in 2029.
Aratana NV
As part of our January 2014 acquisition of Okapi Sciences, we acquired certain patent rights that cover formulations of AT-006 and methods of making the active ingredient of AT-006. These applications, if granted into patents, would expire in 2032 and 2031, respectively. We also have a license to certain patent rights that covers the composition and methods of use of AT-007, AT-008 and AT-011. These patent rights, if the patent applications included therein issue, will expire in 2031.
Regulatory
The development, approval and sale of animal health products are governed by the laws and regulations of each country in which we intend to sell our products. To comply with these regulatory requirements, we have established processes and resources to provide oversight of the development and launch of our products and their maintenance in the market.
United States
Three federal regulatory agencies regulate the health aspects of animal health products in the United States: the FDA; the USDA; and the Environmental Protection Agency (“EPA”).
The CVM at the FDA regulates animal pharmaceuticals under the Food, Drug and Cosmetics Act. The CVB at the USDA regulates veterinary vaccines and some biologics pursuant to the Virus, Serum, Toxin Act. The EPA regulates veterinary pesticides under the Federal Insecticide, Fungicide and Rodenticide Act. Many topical products used for treatment of flea and tick infestations are regulated by the EPA.
Our current product candidates are animal pharmaceuticals regulated by the CVM and animal biologics regulated by the USDA. Manufacturers of animal health pharmaceuticals, including us, must show their products to be safe, effective and produced by a consistent method of manufacture. The CVM’s basis for approving a drug application is documented in a Freedom of Information Summary. We will be required to conduct post-approval monitoring of FDA- and EMA-approved pharmaceutical products and to submit reports of product quality defects, adverse events or unexpected results to the CVM’s Surveillance and Compliance group.
European Union
The EMA regulates the scientific evaluation of applications for marketing authorizations via the centralized procedure for medicines developed by pharmaceutical companies for use in the EU. Its veterinary review section is distinct from the review section for human drugs. The Committee for Medicinal Products for Veterinary Use (“CVMP”), is responsible for scientific review of the submissions for animal pharmaceuticals and vaccines. However, the European Commission is responsible for the grant of EU marketing authorizations. Once a centralized marketing authorization is granted by the European Commission, it is valid throughout the European Economic Area (“EEA”) (meaning the 28 member states of the EU plus, by extension pursuant to the EEA Agreement, Norway, Iceland and Liechtenstein). The centralized procedure is mandatory for approval of certain veterinary medicines, including those derived from biotechnology processes and veterinary medicines for use as growth or yield enhancers. Other veterinary medicines may be approved centrally if the product contains a new active substance or if the applicant can demonstrate to the CVMP that the product is sufficiently innovative. We believe our current product candidates contain new active substances or are sufficiently innovative and thus will be subject to central approval.
For all other products, the competent authorities of the EU Member States are responsible for granting marketing authorizations for products that are sold in their markets. Applicants who intend to market such products in more than one Member State may seek marketing authorizations under the mutual recognition procedure or the decentralized procedure, which are procedures designed to streamline and harmonize approval in multiple EU Member States. If the product has already been authorized in one Member State,
15
the mutual recognition procedure facilitates mutual recognition of the existing authorization, so called reference Member State approval, in another Member State. The decentralized procedure, on the other hand, may be used in cases where the product has not received a marketing authorization in any Member State. Under this procedure, the applicant submits an identical dossier to each relevant Member State, and one, known as the reference Member State, takes the lead in reviewing the application. Under both procedures, other member states are required to accept the reference Member State’s view on the approvability of the product unless they can identify significant public health reasons not to do so.
In general, the requirements for regulatory approval of an animal health product in the EU are similar to those in the United States, requiring demonstrated evidence of purity, safety, efficacy and consistency of manufacturing processes.
Rest of World
Each other country has its own regulatory requirements for approving and marketing veterinary pharmaceuticals. For example, in Brazil, the Ministry of Agriculture, Livestock Products and Supply (“MAPA”), is responsible for the regulation and control of pharmaceuticals, biologicals and feed additives for animal use. MAPA’s regulatory activities are conducted through the Secretary of Agricultural Defense and its Livestock Products Inspection Department. In addition, regulatory activities are conducted at a local level through the Federal Agriculture Superintendence. These activities include the inspection and licensing of both manufacturing and commercial establishments for veterinary products, as well as the submission, review and approval of pharmaceuticals, biological and feed additives.
In Australia, the Australian Pesticides and Veterinary Medicines Authority (“APVMA”), is the Australian government statutory authority for the registration of all agricultural and veterinary products. The APVMA assesses applications from manufacturers of veterinary pharmaceuticals and related products.
Many country specific regulatory laws contain provisions that include requirements for labeling, safety, efficacy and manufacturers’ quality control procedures to assure the consistency of the products, as well as company records and reports. With the exception of the EU, the regulatory agencies of most other countries generally refer to the FDA, USDA, EMA, and other international animal health entities, including the World Organization for Animal Health and the Codex Alimentarius Commission, in establishing standards and regulations for veterinary pharmaceuticals and vaccines.
Other Regulatory Considerations
Regulatory rules relating to human food safety, food additives, or drug residues in food will not apply to the products we currently are developing because our products are not intended for use in food production animals.
Advertising and promotion of animal health products is controlled by regulations in many countries. These rules generally restrict advertising and promotion to those claims and uses that have been reviewed and endorsed by the applicable agency. We will conduct a review of advertising and promotional material for compliance with the local and regional requirements in the markets where we sell pet therapeutics.
Requirements for Approval of Veterinary Pharmaceuticals for Pets
As a condition to regulatory approval for sale of animal products, regulatory agencies worldwide require that a product to be used for pets be demonstrated to:
|
|
• |
|
be safe for the intended use in the intended species; |
|
|
• |
|
have substantial evidence of effectiveness for the intended use; |
|
|
• |
|
have a defined manufacturing process that ensures that the product can be made with high quality consistency; and |
|
|
• |
|
be safe for humans handling the product and for the environment. |
Safety. To determine that a new veterinary drug is safe for use, regulatory bodies will require us to provide data from a safety study generated in laboratory cats and dogs tested at doses higher than the intended label dose, over a period of time determined by the intended length of dosing of the product. In the case of the CVM, the design and review of the safety study and the study protocol are completed prior to initiation of the study to help assure that the data generated will meet FDA requirements. These studies are conducted under rigorous quality control, including GLP, to assure integrity of the data. They are designed to clearly define a safety margin, identify any potential safety concerns, and establish a safe dose for the product. This dose and effectiveness is evaluated in the pivotal field effectiveness study where the product is studied in the animal patient population in which the product is intended to be used. Field safety data, obtained in a variety of breeds and animals kept under various conditions, are evaluated to assure that the product will be safe in the target population. Safety studies are governed by regulations and regulatory pronouncements that provide the parameters of required safety studies and are utilized by regulatory bodies in the United States, the European Union, Japan and other countries.
Effectiveness. Early pilot studies may be done in laboratory cats or dogs to establish effectiveness and the dose range for each product. Data on how well the drug is absorbed when dosed by different routes and the relationship of the dose to the effectiveness are studied. When an effective dose is established, a study protocol to test the product in real world conditions is developed prior to beginning the
16
study. In the case of the CVM, the pivotal effectiveness field study protocol is submitted for review and concurrence prior to study initiation, to help assure that the data generated will meet requirements. The pivotal field effectiveness study must be conducted with the formulation of the product that is intended to be commercialized, and is a multi-site, randomized, controlled study, generally with a placebo control. To reduce bias in the study, individuals doing the assessment are not told whether the subject is in the group receiving the treatment being tested or the placebo group. For pharmaceuticals, in both the United States and the European Union, the number of patients enrolled in the pivotal field effectiveness studies is required to be approximately 100 to 150 animal subjects treated with the test product and a comparable number of subjects in the control group that receive the placebo. In many cases, a pivotal field study may be designed with clinical sites in both the European Union and the United States, and this single study may satisfy regulatory requirements in both the European Union and the United States.
Chemistry, Manufacturing and Controls. To assure that the product can be manufactured consistently, regulatory agencies will require us to provide documentation of the process by which the API is made and the controls applicable to that process that assure the API and the formulation of the final commercial product meet certain criteria, including purity and stability. After a product is approved, we will be required to communicate with the regulatory bodies any changes in the procedures or manufacturing site. For FDA and EMA approvals, both pharmaceutical API and commercial formulations are required to be manufactured at facilities that practice cGMP.
Environmental and Human Safety. We will not be required under United States law to provide an environment impact statement for products currently in development if the products are given at the home of the pet’s owner or in a veterinary hospital. If products might result in some type of environmental exposure or release, the environmental impact must be assessed. For approval in the EU, a risk assessment for potential human exposure will be required.
Labeling, All Other Information, and Freedom of Information Summary. We also will be required to submit the intended label for the product, and also any information regarding additional research that has been conducted with the drug, to the CVM and other regulatory bodies for review. We will draft, and submit for regulatory review, the Freedom of Information Summary for use in the United States. This summary outlines the studies and provides substantial information that CVM uses to assess the drug’s safety and effectiveness and then publishes on its website.
Regulatory Process at the FDA
To begin the development process for our products in the United States, we establish an INAD, file with the CVM. We then hold a pre-development meeting with the CVM to reach a general agreement on the plans for providing the data necessary to fulfill requirements for an NADA. During development, we submit pivotal protocols to the CVM for review and concurrence prior to conducting the required studies. We gather and submit data on manufacturing, safety and effectiveness to the CVM for review, and this review is conducted according to timelines specified in the ADUFA. Once all data have been submitted and reviewed for each technical section – safety, effectiveness and CMC – the CVM issues us a technical section complete letter as each section review is completed, and when the three letters have been issued, we compile a draft of the Freedom of Information Summary, the proposed labeling, and all other relevant information, and submit these as an administrative NADA for CVM review. Generally, if there are no deficiencies in the submission, the NADA is issued within 60 days after submission of the administrative NADA. After approval, we will be required to collect reports of adverse events and submit them on a regular basis to the CVM.
The CVM has an alternative approval process for drugs used in minor species, or for drugs that are used for a ‘minor use’ in a major species. This process is called MUMS which stands for minor use, minor species. For example, if it can be documented that the population of cats or dogs that contract a specific condition is below a specified number, a company can apply to the CVM for MUMS designation. Once designation has been granted, then we must submit the same safety and CMC data as required for a full NADA, and also submit some evidence of effectiveness. After a review period, the CVM can then grant a conditional approval. This approval allows for the commercialization of the product, while completing the pivotal effectiveness study required for a full NADA. Because in many cases the CMC section of the submission takes the longest, MUMS conditional approval may not shorten the time to commercialization. Following submission, review and approval of the pivotal field effectiveness study, the CVM may grant a full NADA.
Requirements for Approval of Certain Veterinary Biologics for Pets
There are many parallels between the requirements to receive approvals for a veterinary pharmaceutical product candidates and certain veterinary biologics product candidates. The terminology differs but the three main components are the same: efficacy, manufacturing, and safety. USDA regulations are designed to ensure that veterinary biologics are pure, safe, potent and effective. The differences compared to pharmaceutical product regulations are based on the immunological nature of the mode of action of the product and the manufacturing process involving living organisms.
Efficacy. Documentation requirements depend significantly on product type and typically include data from preliminary dose determination studies and master seed immunogenicity/efficacy studies.
Safety. Typical safety documentation includes safety data from laboratory animal studies, typically rodents, studies in host animals, typically laboratory dogs or cats, in biocontainment, and field safety studies conducted in client-owned animals.
Manufacturing. The required documentation must include an Outline of Production, Master Seed Reports, and Summary Information Formats, or SIFs, for novel live biological products and products based on recombinant DNA technology. SIFs contain additional
17
safety and identity data to establish proper biocontainment requirements and to conduct confirmatory testing. Other supportive documentation is product-type specific and includes in-process procedures and corresponding validation reports, potency test development report, stability reports, and veterinary biologics production and test for satisfactory three consecutive prelicensing serials (numbered lots) of product.
Other information. This includes labels or label sketches.
A unique requirement for veterinary biologics in the United States is that manufacturers must hold a U.S. Veterinary Biologics Establishment License to produce licensed veterinary biologicals. An establishment license will only be issued if at least one biological product qualifies for a license. Applications for veterinary biologics establishments include articles of incorporation for the applicant, qualifications of veterinary biologics personnel for key employees, water quality statement, facility blueprints, plot plans, and legends.
Regulatory Process at the USDA
Applicants are encouraged to contact the CVB early in the product development process. A licensing reviewer will be assigned to help with the regulatory process. Initially, the CVB will confirm that the proposed product meets the definition of a veterinary biologic and is subject to regulation by the CVB. The CVB then recommends that applicants submit a licensing plan, including pivotal study protocols, to the CVB for review and comment prior to initiating work that will be used to support product licensure. The USDA provides a complete list of guidance documents named “Veterinary Services Memorandums” that lay out the data requirements and regulatory process. Applicants that do not hold a U.S. Veterinary Biologics Establishment License need to submit the required documentation for the establishment and the product concurrently.
Study protocols and reports can be submitted any time after the initial applications have been made. The administrative process is facilitated by forms (APHIS Forms) that accompany the submissions and capture regulatory actions. There is no requirement to submit parts of dossiers or entire dossiers. The CVB provides official responses to submissions in hard copy mail indicating if more data are needed or that the submission was satisfactory to support licensure. When master seed and master cell reports have been found to be satisfactory, samples have to be submitted to the CVB laboratory for confirmatory testing. Once all requirements have been satisfactorily met, the CVB will issue a veterinary biological product license.
In cases of emergencies, which means there is no approved product available, the USDA will issue a time-limited conditional license after the manufacturing and safety requirements have been substantially fulfilled and a reasonable expectation of efficacy has been established. The applicant has to continue the pivotal efficacy program and product testing validation. The conditional license can be extended if reasonable progress towards full licensure can be demonstrated.
There are no statutory review times. Submissions enter the review queue in chronological order. Hence predictions of development timelines and time to approval are difficult to make. However, we believe the typical time to achieve conditional licensure is approximately three years and the typical time to achieve full licensure is approximately five years.
Furthermore, while the CVB regulates certain biologics (for instance, based on the immunological nature of the mode of action) the CVM regulates other biologics in a manner described under “Regulatory Process at the FDA.”
European Regulatory Process
The EMA is responsible for coordinating scientific evaluation of applications for marketing approval via the centralized procedure for pet therapeutics in the EU. To perform these evaluations the EMA established a specific scientific committee, the CVMP. The CVMP considers applications submitted by companies for the marketing approval of individual pet therapeutics and evaluates whether or not the medicines meet the necessary quality, safety and efficacy requirements. Assessments conducted by the CVMP are based on scientific criteria and are intended to ensure that pet therapeutics reaching the marketplace have a positive benefit-risk balance in favor of the pet population they are intended for. Based on the CVMP’s recommendation, a centralized marketing authorization is granted by the European Commission, which allows the product to be marketed throughout the EEA. The CVMP is also responsible for various post-authorization and maintenance activities, including the assessment of modifications or extensions to an existing marketing authorization.
To obtain a centralized marketing authorization from the European Commission, we must submit a marketing authorization application called a dossier. The dossier is the EMA’s equivalent of the FDA’s NADA and includes data from studies showing the quality, safety and efficacy of the product. The CVMP reviews and evaluates the dossier. For any dossier, a rapporteur and co-rapporteur are appointed from the members of the CVMP. Their role is to lead the scientific evaluation and prepare the assessment report. The rapporteur can utilize experts to assist it in performing its assessment. The report is critiqued by the co-rapporteur and other members of the CVMP before the CVMP makes its determination. The final opinion of the CVMP is generally given within 210 days of the submission of a dossier.
For products that are not eligible for centralized approval, the competent authorities of the EU Member States are responsible for granting marketing authorizations for products that are sold in their markets. Such products may be approved nationally in one Member State, or in multiple Member States via the mutual recognition procedure or the decentralized procedure.
In the EU, products for MUMS are eligible for regulatory incentives such as free scientific advice and fee reductions. These incentives may apply, for example, if it can be documented that the population of cats or dogs that contract a specific condition is below a
18
specified number in Europe. However, the EMA recently announced that fee reductions are only applicable to products indicated for food-producing species. An applicant may apply to the EMA for MUMS classification for any product irrespective of the intended route of approval (i.e. centralized, decentralized or national approval) and incentives may be requested for all routes of authorization. The CVMP has established guidelines specific to MUMS for data requirements, which apply to all sections of the application, i.e. quality, safety and efficacy. Consequently, there may be scope for a reduced quality data package. Similarly, the safety and efficacy sections might be abridged to a certain extent (more flexibility for the combination of dose-determination, dose-confirmation and field studies) provided reasonable evidence of safety and effectiveness are submitted. However, the CVMP and national veterinary medicines regulators have significant discretion in this respect. Overall, data requirements for demonstrating quality, efficacy and safety in the target species for minor use indications of a new medicine will be determined on a case-by-case basis and any potential applicant should seek scientific advice on specific data requirements to guide its research and development activities.
There are three different procedures to receive a marketing authorization (regulatory approval) in Europe, the decentralized procedure (“DCP”), the mutual recognition procedure (“MRP”), and the centralized procedure (“CP”). The centralized procedure is mandatory for certain products and technologies, for example biopharmaceuticals, gene therapy products, somatic cell therapeutic products or certain therapeutic areas, for example oncology or neurodegenerative disorders. Otherwise the sponsor can opt between CP and DCP.
An application for CP is submitted to the European Medicines Agency (“EMA”) which coordinates the scientific evaluation. To perform these evaluations the EMA established a specific scientific committee, the CVMP. The CVMP evaluates whether or not the medicines meet the necessary quality, safety and efficacy requirements. Assessments conducted by the CVMP are based on scientific criteria and are intended to ensure that pet therapeutics reaching the marketplace have a positive benefit-risk balance in favor of the pet population they are intended for. Based on the CVMP’s recommendation, a centralized marketing authorization is granted by the European Commission, which allows the product to be marketed in any of the EU states. The CVMP is also responsible for various post-authorization and maintenance activities, including the assessment of modifications or extensions to an existing marketing authorization.
To obtain authorization from the EMA through CP, we must submit a marketing authorization application called a dossier, which consists of four parts. Part 1 includes administrative information, part 2 quality documentation, part 3 safety documentation, and part 4 efficacy documentation. For any dossier, a rapporteur and co-rapporteur are appointed from the members of the CVMP. Their role is to lead the scientific evaluation and prepare the assessment report. The rapporteur can utilize experts to assist it in performing its assessment. The entire regulatory assessment period is limited to 210 days, which is divided in three periods. After an initial review period of 120 days the sponsor receives a list of questions and the clock is stopped. With the submission on the response the clock starts again and after a 60-days review period the CVMP discusses a draft opinion and the clock is stopped. If felt necessary, an oral explanation is offered. In the last 30 days of the review period the CVMP finalizes the opinion and the assessment report.
A DCP can be used for products that have not been approved in any of the EU member states and do not fall under mandatory CP. The sponsor selects one Reference Member State (“RMS”) and one or more Concerned Member States (“CMS”). The RMS leads the scientific evaluation and with the input from CMS issues the initial and final assessment report. The regulatory assessment period is similar to the CP and divided into two periods of 120 and 90 days, respectively. The procedure ends with a consensus decision and leads to products approval in the RMS and CMS.
The MRP must be used for products that have been approved in at least one EU member state either by national procedure or DCP. The MRP uses an existing and if needed updated assessment report to extend marketing authorizations to more EU member states
Segment and Geographic Information
We operate in one business segment and have operations in the United States and Belgium. See our consolidated financial statements for further information regarding our segment, including revenues and loss from operations. See Note 2 “Summary of Significant Accounting Policies” to our consolidated financial statements for total assets, and geographic information including revenues and long-lived assets.
Employees
As of December 31, 2015, we had a total of 57 employees, including 55 full-time employees.
As of March 10, 2016, we have a total of 58 employees, including 56 full-time employees. We have a total of 20 employees with D.V.M., V.M.D., M.D. or Ph.D. degrees. Within our workforce, 23 employees are engaged in research and development and 35 in business development, marketing and sales, finance, legal, human resources, facilities, information technology and general management and administration.
19
Available Information
We maintain a website at www.aratana.com. We make available free of charge on our website our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to these reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to the Securities and Exchange Commission.
Executive Officers of the Registrant
The executive officers of Aratana Therapeutics, Inc. as of March 10, 2016, are as follows:
Steven St. Peter, M.D., age 49, is one of our founders and has served as our President and Chief Executive Officer since September 2012. He has been a member of our Board of Directors since December 2010 and served as the Chairman of our Board of Directors from December 2010 to September 2012. Dr. St. Peter was a Managing Director of MPM Asset Management LLC from January 2004 to May 2012, where he focused his investments on both venture and buyout transactions across the pharmaceuticals and medical technology industries. He has previous investment experience from Apax Partners and The Carlyle Group, two private equity firms. Dr. St. Peter was previously an Assistant Clinical Professor of medicine at Columbia University. He received his M.D. from Washington University and completed his residency and fellowship at the Hospital of the University of Pennsylvania. Prior to his medical training, he was an investment banker at Merrill Lynch. Dr. St. Peter also holds an M.B.A. from the Wharton School of Business at the University of Pennsylvania and a B.A. in Chemistry from the University of Kansas. Dr. St. Peter has served as a director of PharmAthene, Inc., a publicly-traded biodefense company, since August 2007 and is currently a member of its governance and nominating committee. Dr. St. Peter has also served as a member of the Board of Directors of the Kansas City Area Life Sciences Institute since March 2014 and as a member of the Board of Directors of the Greater Kansas City Foundation since November 2015. Dr. St. Peter’s previous board experience includes MPM Acquisition Corp., Proteon Therapeutics, Inc., Rhythm Pharmaceuticals, Inc. and Syndax Pharmaceuticals, Inc. As noted above, Dr. St. Peter is a member of our Board of Directors and as such, we believe Dr. St. Peter is qualified to serve on our Board because of his diverse background as a venture capital investor, investment banker, physician and director of several healthcare companies, which brings a unique perspective to our Board.
Ernst Heinen, D.V.M., Ph.D., age 53, has served as our Chief Development Officer since March 5, 2014. In addition, he served as our Head of Drug Evaluation and Development from June 2012 until March 5, 2014. From 1990 to 2012, Dr. Heinen held positions of increasing responsibility at Bayer Animal Health, the animal health division of Bayer AG, where he ultimately served as Vice President of Research & Development and Veterinary Technical Services, Pets. Dr. Heinen currently serves on the Kansas State University Olathe Advisory Board and previously served on the boards of the Kansas City Area Development Council and the Center for Animal Health Innovation, and he is the author of dozens of scientific articles and presentations focused on the animal health industry. Dr. Heinen received a veterinary degree and a D.V.M. in veterinary microbiology from the Justus-Liebig-University of Giessen Veterinary School in Giessen, Germany, and is a certified specialist in veterinary microbiology.
Linda Rhodes, V.M.D., Ph.D., age 66, has served as our Chief Scientific Officer since September 2012. She served as a member of our Board of Directors from February 2011 to March 5, 2014. In addition, she served as our Chief Executive Officer from February 2011 to September 2012. In 2001, Dr. Rhodes was a founding partner of AlcheraBio LLC, an animal health consulting and contract research firm, which was acquired in October 2008 by Argenta, a New Zealand animal health formulations and contract manufacturing organization, and she served as its Vice President of Clinical Development from February 2008 to February 2011. She is an Adjunct Professor for the Graduate School of Animal Science at Rutgers University and is a member of the Board of Directors of the Alliance for Contraception in Cats and Dogs, a non-profit organization. She also serves as a member of the Scientific Advisory Board of the Found Animals Foundation. She has been a member of the Board of Directors of ImmuCell Corporation, a publicly-traded animal health company, since 2005, and is currently the chair of its compensation committee. From 1998 to 2001, she was a director of production animal development projects and new technology assessment at Merial Ltd., an animal health company. Prior to that role, she held various research positions at Merck Research Laboratories and Sterling Winthrop Drug Company. She has held several teaching positions and worked as a bovine veterinarian in private practice. She earned her Ph.D. in Physiology/Immunology from Cornell University and her V.M.D. from the University of Pennsylvania School of Veterinary Medicine, graduating summa cum laude. She also holds a Bachelor of Arts degree from Sarah Lawrence College.
Julia A. Stephanus, age 57, has served as our Chief Commercial Officer since January 2013. From September 2010 through December 2012, Ms. Stephanus was director of the global pet franchise for Ceva Animal Health, where she oversaw the commercial development of new products as well as global marketing for strategic pet products. In 2006, Ms. Stephanus founded Summit VetPharm, the developer of Vectra, a pet parasiticide product line, and served as its President and Chief Executive Officer until it was acquired by Ceva Animal Health in August 2010. Prior to founding Summit VetPharm, Ms. Stephanus worked in various sales and marketing positions for Pfizer Inc. and its legacy companies, where she had the commercial responsibility for, among other things, the development and global launch of two highly-profitable pet products: Rimadyl, the first NSAID approved for osteoarthritis in dogs, and Revolution, the first topical endectocide for heartworm and fleas in cats and dogs. Ms. Stephanus has been a board member of BioKansas, a not-for-profit bioscience advocacy organization, since November 2015. She received a B.A. from Indiana University and has attended executive education programs at Harvard, Columbia and the Wharton School of Business at the University of Pennsylvania.
20
Craig A. Tooman, age 50, has served as our Chief Financial Officer since November 2013 and our Treasurer since January 2014. He was a member of our Board of Directors from April 2012 to November 2013, before accepting the CFO role. Mr. Tooman previously served as the Chief Executive Officer of Avanzar Medical, Inc., a privately-held company focused on commercial oncology opportunities, from February 2012 until November 2014. Mr. Tooman was also the founder and principal of Stockbourne LLC, a firm that provides strategic business and financial advisory services, a position he held from January 2011 to November 2013. From July 2010 to January 2011, Mr. Tooman was the Senior Vice President of Finance and Chief Financial Officer of Ikaria Inc., a biotherapeutics company. From January 2005 to July 2010, Mr. Tooman was the Executive Vice President of Finance and Chief Financial Officer at Enzon Pharmaceuticals, a biopharmaceutical company. Prior to that, Mr. Tooman was the Senior Vice President of Strategic Planning and Corporate Communications at ILEX Oncology, Inc. and the Vice President of Investor Relations at Pharmacia Corporation. Mr. Tooman previously served on the Board of Directors of Insite Vision Incorporated, a publicly-traded ophthalmological company, from September 2011 to November 2015. He has a B.A. in Economics from Kalamazoo College and M.B.A. in Finance from the University of Chicago.
Non-Employee Directors
The non-employee directors of Aratana Therapeutics, Inc. as of March 10, 2016 are as follows:
Wendy L. Yarno, age 61, has been a member of our Board of Directors since October 2013 and currently serves as the Chairperson of the Board. Ms. Yarno retired in September 2008 from Merck & Co., Inc. following a 26-year career there in commercial and human resource positions of increasing seniority, most recently Chief Marketing Officer before she retired. In that role, Ms. Yarno led a global organization charged with all aspects of supporting pre-and post-launch commercialization of pharmaceuticals in more than 20 therapeutic areas. Prior to this role, she served as General Manager, Cardiovascular/Metabolic U.S. Business Unit, where she had P&L responsibility for Merck’s largest therapeutic area, and as Senior Vice President, Human Resources. From September 2010 through September 2011, Ms. Yarno was the Chief Marketing Officer of HemoShear LLC, a biotechnology research company and leading developer of human cell-based surrogate systems for discovery and assessment of new drug compounds. Ms. Yarno has served as a Director and member of the governance and nominating committee and compensation committee of St. Jude Medical, Inc., a Fortune 500 medical device company, since 2002 and has served as a Director of Medivation, Inc., a publicly-traded biopharmaceutical company, since 2013 and is currently a member of its governance and nominating committee and audit committee and chair of its compensation committee. Ms. Yarno also served as a Director and member of the compensation committee of Durata Therapeutics, Inc., a publicly-traded pharmaceutical company, from August 2014 until November 2014 when Durata was acquired by Actavis plc. Ms. Yarno received a B.S. in Business Administration from Portland State University and an M.B.A from Temple University. We believe Ms. Yarno is qualified to serve on our Board based on her extensive experience in commercialization of pharmaceutical products and in human resource management in the pharmaceutical industry.
Laura A. Brege, age 58, has been a member of our Board of Directors since February 2014. In September 2015, Ms. Brege became managing director of Cervantes Life Sciences Partners, LLC, a healthcare advisory and consulting company. She also served as President and Chief Executive Officer of Nodality, Inc., a privately-held life sciences company, from September 2012 to July 2015. Prior to joining Nodality, from January 2011 to January 2012, Ms. Brege was the Executive Vice President, Corporate Affairs of Onyx Pharmaceuticals, Inc. From October 2007 to January 2011, she was the Chief Operating Officer, and from June 2006 to October 2007, she was the Executive Vice President and Chief Business Officer of Onyx Pharmaceuticals. From 1999 to 2006, Ms. Brege was a General Partner at Red Rock Capital Management, a venture capital firm. Previously, Ms. Brege served as Chief Financial Officer at companies such as COR Therapeutics, Inc., a biotechnology company, and Flextronics, Inc., a supply-chain solutions company. Ms. Brege currently also serves on the Board of Directors of publicly-traded Acadia Pharmaceuticals, Inc., Dynavax Technologies Corporation, Pacira Pharmaceuticals, Inc. and Portola Pharmaceuticals, Inc. Ms. Brege has served as a Director of Acadia since May 2008 and is currently a member of its audit committee and has served as a Director and chair of the audit committee of Dynavax since February 2015. Ms. Brege has served as a Director of Pacira since June 2011 and is currently the chair of its audit committee and a member of its nominating and governance committee and has served as a Director and member of the audit committee of Portola since January 2015. Ms. Brege previously served as a member of the Board of Directors of publicly-traded Angiotech Pharmaceuticals, Inc. from 2007 to 2011 and Delcath Systems, Inc. from 2012 to December 2014. Ms. Brege earned her undergraduate degrees from Ohio University and has an M.B.A. from the University of Chicago. We believe Ms. Brege is qualified to serve on our Board based on her strong background in finance and her extensive executive leadership experience in the life sciences and biotechnology industries, including her service as a public company director and in various executive officer roles.
David L. Brinkley, age 58, has been a member of our Board of Directors since March 2014. Mr. Brinkley worked for Theravance, Inc., a publicly-traded biopharmaceutical company, from 2000 to 2013, most recently as the Head of Business Development from November 2008 to July 2013. Mr. Brinkley had previously served as Senior Vice President, Commercial Development at Theravance from September 2000 through December 2007, when he left to start a consulting practice. From 1996 to 2000 he served as Worldwide Team Leader for Viagra at Pfizer Inc., leading the team that had full responsibility for the global launch and marketing of Viagra. Mr. Brinkley joined Pfizer in 1995 through its acquisition of SmithKline Beecham’s Animal Health operations and was Director of New Product Planning before leading the Viagra launch team. Mr. Brinkley held various management positions with SmithKline Animal Health from 1983 to 1995. Mr. Brinkley previously served on the Board of Directors of Ziarco Pharma Ltd., a privately-held pharmaceutical company. Mr. Brinkley holds an M.A. with honors in International Economics from the School of Advanced International Studies of the Johns Hopkins University and a B.A. in International Relations from Kent State University, where he
21
graduated with University Honors. We believe Mr. Brinkley is qualified to serve on our Board due to his extensive leadership experience in the biopharmaceutical industry, including his roles at Theravance and Pfizer.
Robert “Rip” Gerber, age 53, has been a member of our Board of Directors since October 2012. Since January 2015, Mr. Gerber has served as an executive at Vlocity, Inc., a software company, including as Chief Marketing Officer and Head of Alliances since June 2015. From July 2009 to January 2015, he served as the President and Chief Executive Officer of Locaid Technologies, Inc., a telecommunications software company. From June 2006 to June 2009, Mr. Gerber served as a member of the advisory board of SignalDemand Inc., a private firm focused on producing margin optimization software. From May 2004 to May 2006, Mr. Gerber served as Chief Marketing Officer and Senior Vice President of Intellisync Corporation, a public company and provider of data synchronization software to consumer mobile devices. Prior to that role, he served as Senior Vice President at Carlson Companies, Inc., one of the largest family-held corporations in the United States. Mr. Gerber was also on the founding executive team of Commtouch Software, Inc., where, as Chief Marketing Officer, he was a lead executive in taking the Company public in 1999. Earlier in his career, Mr. Gerber was a consultant for Deloitte & Touche LLP, a public accounting firm. Mr. Gerber serves on the Board of Directors of LocationSmart, a privately-held location software company and on the board of trustees of Drew School, a private high school located in San Francisco. He holds an M.B.A. from Harvard Business School and a B.S. in Chemical Engineering from the University of Virginia. We believe Mr. Gerber is qualified to serve on our Board because of his experience as an entrepreneur and his extensive background in operational, marketing and strategic planning.
Irvine “Irv” O. Hockaday, Esq., age 79, has been a member of our Board of Directors since August 2014. Mr. Hockaday is the retired President and Chief Executive Officer of Hallmark Cards, Inc. Prior to joining Hallmark in 1983, Mr. Hockaday served as President and Chief Executive Officer of Kansas City Southern Industries, Inc. He was a member of the Hallmark Board of Directors from 1978 through 2001. Mr. Hockaday has been on the Board of Directors of the Estee Lauder Companies, Inc. since 2001 and is currently lead Director and chair of its audit committee. Mr. Hockaday is a former Director or Lead Director of Crown Media Holdings, Inc., Dow Jones & Company, Inc., Ford Motor Company and Sprint Nextel Corporation. He currently holds various civic positions including trustee of the Hall Family Foundation and board member of Kansas City Area Life Sciences Institute and has previously served as chairman of the board of the Tenth District Federal Reserve Bank. He graduated with an A.B. in English from Princeton University in 1958 and from the University of Michigan Law School with a J.D. in 1961. We believe Mr. Hockaday is qualified to serve on our Board due to his extensive experience as a Chief Executive Officer and board member of public companies.
Merilee Raines, age 60, has been a member of our Board of Directors since February 2014. Ms. Raines served as Chief Financial Officer of IDEXX Laboratories, Inc., a publicly-traded company providing diagnostic and IT products and services primarily to the companion animal health market, from October 2003 until her retirement in May 2013. Ms. Raines also served as Executive Vice President of IDEXX Laboratories from July 2012 to May 2013, and as Corporate Vice President, Finance of IDEXX Laboratories from May 1995 to July 2012. Ms. Raines has served as a Director of Watts Water Technologies, Inc., a publicly-traded manufacturer of products and systems focused on control, conservation and quality of water, since 2011, and is currently a member of its nominating and corporate governance committee and chair of its audit committee. Ms. Raines has also served as a Director of Affymetrix, Inc., a publicly-traded provider of life sciences products and molecular diagnostic products, since January 2015, and is currently a member of its audit committee. Ms. Raines earned a bachelor’s degree in mathematics from Bowdoin College and an M.B.A. from the University of Chicago. We believe Ms. Raines is qualified to serve on our Board based on her experience as an executive of a public company in the animal health industry and her extensive financial expertise, including her role as Chief Financial Officer of IDEXX Laboratories and her service on the audit committees of Watts Water Technologies and Affymetrix.
Robert P. Roche, age 60, has been a member of our Board of Directors since June 2014. Mr. Roche is the founding member of Robert Roche Associates, LLC, a consulting firm providing guidance to the pharmaceutical and healthcare industries. Mr. Roche created this firm upon his retirement from Cephalon, Inc., a biopharmaceutical company, in February 2010. Mr. Roche joined Cephalon in January 1995 as the Vice President of Sales and Marketing and was named Executive Vice President, Worldwide Pharmaceutical Operations of Cephalon in 2005. Before joining Cephalon, Mr. Roche served as Director and Vice President, Worldwide Strategic Product Development, for SmithKline Beecham’s central nervous system and gastrointestinal products business. Mr. Roche also was Managing Director of SmithKline’s pharmaceutical operations in the Philippines. Prior to that, he held senior marketing positions in Canada and Spain and had product planning responsibilities for SmithKline in Latin America. Mr. Roche began his pharmaceutical career in 1982 with SmithKline as a U.S. pharmaceutical sales representative. Mr. Roche has served as a Director of Antares Pharma, Inc., a publicly-traded specialty pharmaceutical company, since July 2013 and is currently a member of its governance and nominating committee and audit committee. Mr. Roche is also a director of Paragon Bioservices, Inc., a privately-held contract development and manufacturing organization. He formerly served as a Director of LifeCell Corp. until its acquisition in 2008, EKR Therapeutics until its acquisition in 2012, NuPathe Inc. until its acquisition in February 2014 and Civitas Therapeutics until its acquisition in November 2014. He also serves on the boards of Bryn Mawr Hospital and Westtown School. Mr. Roche earned his B.A. from Colgate University and his M.B.A. from The Wharton School at the University of Pennsylvania. We believe Mr. Roche is qualified to serve on our Board due to his executive and board leadership experience in the global pharmaceutical industry and his extensive commercial operations and product launch background.
John Vander Vort, Esq., age 51, has been a member of our Board of Directors since September 2012. Mr. Vander Vort is currently a Managing Director at Pilot House Associates, LLC, a family investment office based in Boston which he joined in September 2014. Prior to this role, Mr. Vander Vort was a Managing Director and the Chief Operating Officer of Charlesbank Capital Partners, a
22
private equity firm. Mr. Vander Vort joined Charlesbank in September 2013 from MPM Asset Management LLC, a venture capital firm, where he served as a Managing Director, the Chief Operating Officer and the Chief Compliance Officer since May 2005, and he served on the Board of Directors of MPM Acquisition Corp., a public shell company, from February 2008 to November 2010. Prior to joining MPM Asset Management, from May 2003 until May 2005, he worked as Portfolio Manager for DuPont Capital Management. Prior to that, he was a General Partner and co-founder of BlueStream Ventures, a venture capital firm. Previously, he was a Managing Director at Dain Rauscher Wessels (now the Royal Bank of Canada), where he was the head of the West Coast networking and communications investment banking group and served as an advisor to leading venture-backed technology companies. Mr. Vander Vort began his career as a corporate transaction attorney in the San Francisco office of Cooley Godward, where he represented venture capital firms and venture-backed companies. Mr. Vander Vort earned his B.A. from Amherst College and his J.D. from The University of Chicago Law School. We believe Mr. Vander Vort is qualified to serve on our Board because of his background in venture capital, significant legal experience and business acumen.
23
Investing in our common stock involves a high degree of risk. You should carefully consider the important risks described below, as well as the other information contained in or incorporated by reference into our public filings with the Securities and Exchange Commission, before deciding whether to invest in our common stock. The occurrence of any of the events or developments described below could harm our business, financial condition, results of operations and growth prospects. In such an event, the market price of our common stock could decline, and you may lose all or part of your investment.
Risks Related to Our Business
We have a limited operating history and have incurred significant losses since our inception and we anticipate that we will continue to incur losses for the foreseeable future, and our limited operating history makes it difficult to assess our future viability.
We are a development-stage biopharmaceutical company in the pet therapeutics industry transitioning into a commercial enterprise, with a limited operating history. Biopharmaceutical product development in the pet therapeutics industry is a highly speculative undertaking and involves a substantial degree of risk. We currently have a product pipeline with multiple products under development including two biologics, BLONTRESS and TACTRESS, for which we received full licenses from the USDA, but for which we do not expect to receive significant revenues. We expect to receive FDA approval for, product candidates GALLIPRANT, ENTYCE and NOCITA in 2016 and subsequently to commence commercialization.
We are not profitable and have incurred losses in each year since our inception in December 2010. In addition, as an early stage company, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical industry. We continue to incur significant research and development and other expenses related to our ongoing operations. Our net loss for the year ended December 31, 2015 was $84.1 million, for the year ended December 31, 2014 was $38.8 million and for the year ended December 31, 2013 was $6.9 million. As of December 31, 2015, we had an accumulated deficit of $152.0 million and we had $86.2 million in cash, cash equivalents and short-term investments. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for, our product candidates and begin to commercialize them if they are approved by the FDA, or for our biologic products, the USDA. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital.
We may require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product portfolio expansion, product development, other operations or commercialization efforts.
Since our inception, a majority of our resources have been dedicated to the in-licensing, acquisition and research and development of our current product candidates. Completing the development and obtaining regulatory approval of our product candidates will require substantial funds. We also have an active in-licensing effort focused on identifying human therapeutics for development and commercialization as pet therapeutics. We believe that we will continue to expend substantial resources for the foreseeable future for the development and commercialization of our current product candidates and any future product candidates we may choose to pursue. These expenditures will include costs associated with identifying potential product candidates, licensing or acquisition payments, conducting target animal studies, completing other research and development, obtaining regulatory approvals and manufacturing and supply, as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise. Because the outcome of any target animal study is uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of any of our current or future product candidates. As of the date of the filing of this Annual Report on Form 10-K, we believe that our existing cash, cash equivalents and short-term investments will allow us to fund our operations and our debt obligations through December 31, 2016. However, our operating plan may change as a result of many factors currently unknown to us, and we may seek additional funds sooner than planned through public or private equity or debt financings or other sources, such as strategic collaborations. Such financing may result in dilution to stockholders, imposition of debt covenants and repayment obligations, or other restrictions that may affect our business. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Our future capital requirements depend on many factors, including, but not limited to:
|
|
• |
|
the scope, progress, results and costs of researching and developing any of our current or future product candidates and conducting target animal studies; |
|
|
• |
|
the amount and timing of any milestone payments or royalties we must pay pursuant to our current or future license agreements or collaboration agreements; |
|
|
• |
|
the timing of, and the costs involved in, obtaining regulatory approvals for any of our current or future product candidates; |
|
|
• |
|
the upfront and other payments, and associated costs, related to identifying, acquiring and in-licensing new product candidates; |
24
|
• |
the number and characteristics of the product candidates we pursue; |
||
|
• |
whether we acquire any other companies, assets, intellectual property or technologies in the future; |
||
|
|
• |
|
our ability to partner with companies with an established commercial presence in Europe and/or other countries to provide our products in that market; |
|
|
• |
|
the cost of commercialization activities for any of our current or future product candidates, including marketing, sales and distribution costs; |
|
|
• |
|
the cost of manufacturing our current and future product candidates and any products we successfully commercialize; |
|
|
• |
|
our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements and the potential costs and financial terms to amend or terminate such relationships or other arrangements, including litigation costs and the outcome of such litigation; |
|
|
• |
|
whether we are required to repay grant amounts that we received from foreign, U.S. and/or state governments; |
|
|
• |
|
the expenses needed to attract and retain skilled personnel; |
|
|
• |
|
the costs associated with being a public company; and |
|
|
• |
|
the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent rights, including litigation costs. |
Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, reduce or terminate:
|
|
• |
|
our target animal studies or other development activities for our current or future product candidates; |
|||||
|
|
• |
|
our establishment of sales and marketing capabilities or other activities that may be necessary to launch and/or commercialize any of our current or future product candidates; or |
|||||
|
|
• |
|
|
our in-licensing and acquisition efforts and expansion of our product portfolio. |
||||
We recognized substantial intangible asset impairment losses in the third quarter of 2015 and may be required to recognize additional non-cash impairment losses in the future.
During the third quarter of 2015, we recorded a non-cash impairment charge of $43.4 million related to our intangible assets BLONTRESS, TACTRESS, AT-007 and AT-011. At December 31, 2015, we had $15.1 million of remaining intangible assets on our balance sheet, compared to $62.3 million at December 31, 2014. We could experience material impairment losses in the future. Certain factors, including negative pre-clinical study results and reduced market potential, might have a negative impact on the carrying value of our intangible assets. For example, with respect to BLONTRESS and TACTRESS, which as of December 31, 2015 have carrying values of $5.5 million and $0.6 million, respectively, these products are expected to continue to be available or become available to oncologists, but we believe the revenue and gross margin opportunity for these first generation monoclonal antibodies will be very modest. Therefore, we are pursuing second generation monoclonal antibodies and other product concepts in lymphoma which are intended to deliver break-through benefits. We intend to build awareness of monoclonal antibody therapy with BLONTRESS and TACTRESS and effectively bridge to second generation product candidates which are several years away. We also intend to improve the gross margins for any second generation products. If we determine that we are likely to be unsuccessful in developing product candidates with break-through benefits at an economic gross margin, we would consider phasing out BLONTRESS and TACTRESS depending on product volumes, and fully impairing the value of these intangible assets. The process of testing intangible assets for impairment involves numerous judgments, assumptions and estimates made by management including expected future profitability, cash flows and the fair values of assets and liabilities, which inherently reflect a high degree of uncertainty and may be affected by significant variability. If the business climate deteriorates, then actual results may not be consistent with these judgments, assumptions and estimates, and our intangible assets may become further impaired in future periods. This would in turn have an adverse impact on our business, financial condition and results of operations.
Unstable market and economic conditions may have serious adverse consequences on our business.
Our business may be adversely affected by the recent unpredictable and unstable market conditions. If the current equity and credit markets deteriorate further, or do not improve, it may make any necessary debt or equity financing more difficult to obtain and more costly. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our development programs, commercialization efforts, financial performance and stock price and could require us to delay or abandon plans for our target animal studies and/or the commercialization of any approved products. In addition, difficult economic conditions may limit pet owners’ discretionary funds, which could in turn limit their ability to purchase pet therapeutics. A tight spending climate for pet owners could negatively affect our ability to generate revenues from any approved products. Further, we rely on third-parties for several aspects of our business, including contract manufacturers for the manufacture of our products and licensors of
25
pharmaceutical compounds. During challenging and uncertain economic times and in difficult credit markets, there may be a disruption or delay in the performance of our third party contractors and other collaborators. If such third parties are unable to satisfy their commitments to us, or if they become bankrupt or insolvent, our agreements with such parties may terminate, and our business and results of operations would likely be adversely affected.
The terms of our credit facility place restrictions on our operating and financial flexibility.
Effective as of October 16, 2015, we and Vet Therapeutics, Inc., and together with us, the borrowers, entered into a Loan and Security Agreement, with Pacific Western Bank, or Pacific Western, as collateral agent and the lenders party thereto from time to time, or the lenders, including Pacific Western and Oxford Finance, LLC, that is secured by substantially all of the borrowers’ personal property other than intellectual property. The outstanding principal balance under the loan agreement was $35.0 million under the term loan facility and $5.0 million under the revolving facility at December 31, 2015.
The loan agreement contains customary representations and warranties and customary affirmative and negative covenants, including, among others, limits or restrictions on the borrowers’ ability to incur liens, incur indebtedness, make certain restricted payments, make certain investments, merge, consolidate, make an acquisition, enter into certain licensing arrangements and dispose of certain assets. The loan agreement also contains customary events of default that entitle the lenders to cause the borrowers’ indebtedness under the loan agreement to become immediately due and payable. The events of default, some of which are subject to cure periods, include, among others, a non-payment default, a covenant default, the occurrence of a material adverse change, the occurrence of an insolvency, a material judgment default, defaults regarding other indebtedness and certain actions by governmental authorities. Upon the occurrence and for the duration of an event of default, an additional default interest rate equal to 4% per annum will apply to all obligations owed under the loan agreement and would provide Pacific Western, as collateral agent, with the right to exercise remedies against us and the collateral securing the loan agreement, including foreclosure against our properties securing the loan agreement, including our cash.
The loan agreement requires that the borrowers receive unrestricted net cash proceeds of at least $45.0 million from partnering transactions and/or the issuance of equity securities from October 16, 2015 to October 16, 2016. The loan agreement also requires that the borrowers have at least three products fully USDA- or FDA-approved for commercialization by December 31, 2016. Additionally, the loan agreement requires that the borrowers maintain certain minimum liquidity (approximately $20.0 million) at all times. At December 31, 2015, the borrowers were in compliance with the minimum liquidity covenant. Our ability to make scheduled payments on or to refinance our indebtedness depends on our future performance and ability to raise additional sources of cash, which is subject to economic, financial, competitive and other factors beyond our control. If we are unable to generate sufficient cash to service our debt, we may be required to adopt one or more alternatives, such as selling assets, restructuring our debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. If we desire to refinance our indebtedness, our ability to do so will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
Although we have two fully approved products, we are substantially dependent on the success of our current product candidates.
We currently have two products approved for commercial distribution, BLONTRESS and TACTRESS, however we do not expect to receive significant revenues from sales of these products. To date, we have invested substantial efforts and financial resources in the in-licensing, research and development of GALLIPRANT, ENTYCE and NOCITA, which, prior to our acquisition of Vet Therapeutics and Okapi Sciences, were our only product candidates and are still in development. We expect to receive FDA approval for GALLIPRANT, ENTYCE and NOCITA in 2016, and begin commercializing product candidates GALLIPRANT in 2016 and ENTYCE in late-2016 or shortly thereafter and NOCITA in 2016.
Our near-term prospects, including our ability to finance our company and to enter into strategic collaborations and generate revenue, will depend heavily on the successful development and commercialization of our current product candidates. The development and commercial success of our current product candidates will depend on a number of factors, including the following:
|
|
• |
|
our ability to demonstrate to the satisfaction of the CVM, the USDA and the European Medicines Agency, or EMA, or the applicable EU Member State national competent authorities, the safety and efficacy of our product candidates and for biologics, the potency and purity of our product candidates and to obtain regulatory approval in the United States and Europe; |
||||
|
|
• |
|
our ability to establish and maintain strategic collaborations, licensing or other arrangements, the financial terms of such agreements and the potential costs and financial terms to amend or terminate such relationships or other arrangements, including litigation costs; |
||||
|
|
• |
|
the prevalence and severity of adverse side effects, including a continued acceptable safety profile of the product following approval; |
||||
|
|
• |
|
achieving and maintaining compliance with all regulatory requirements applicable to our product candidates; |
||||
26
|
|
• |
|
the availability, perceived advantages, relative cost, relative safety and relative efficacy of alternative and competing treatments; |
||||
|
|
• |
|
the effectiveness of our commercialization efforts, including the effectiveness of marketing, sales and distribution strategy and operations, whether performed solely by us or in collaboration with others; |
||||
|
|
• |
|
the ability of us or our third-party manufacturers to manufacture supplies of any of our current or future product candidates and to develop, validate and maintain commercially viable manufacturing processes that are compliant with cGMP; |
||||
|
|
• |
|
our ability to successfully launch commercial sales of our current product candidates, assuming CVM, USDA or EMA approval is obtained, whether alone or in collaboration with others; |
||||
|
|
• |
|
our ability to enforce our intellectual property rights in and to our product candidates and avoid third-party patent interference, third-party initiated and U.S. Patent and Trademark Office (“PTO”)-initiated administrative patent proceedings or patent infringement claims; |
||||
|
|
• |
|
our success in educating veterinarians and pet owners about the benefits, administration and use of our product candidates; and |
||||
|
|
• |
|
acceptance of our product candidates as safe and effective by veterinarians, pet owners and the animal health community. |
Many of these factors are beyond our control. Accordingly, we cannot assure you that we will ever be able to generate revenue through the sale of our product candidates. If we are not successful in commercializing one or more of our product candidates, or are significantly delayed in doing so, our business will be materially harmed and the value of your investment could substantially decline.
The development of our biologic product candidates is dependent upon relatively novel technologies and compliance with complex regulatory requirements.
As a result of our acquisition of Vet Therapeutics, we are developing biologics, including animal antibodies, for pets. Identification, optimization and manufacturing of therapeutic animal biologics is a relatively new field in which unanticipated difficulties or challenges could arise. While many biologics have been approved for use in humans, very few have been approved for use in animals, except for vaccines. There are unique risks and uncertainties with biologics, the development, manufacturing and sale of which are subject to regulations that are often as complex and extensive as the regulations applicable to other small molecule products. We anticipate that our animal biologics will continue to be regulated by the USDA, rather than CVM, and the regulatory standards that the USDA may require for novel biologics may be more difficult to satisfy than we anticipate.
We may be unable to obtain regulatory approval for our existing or future product candidates under applicable regulatory requirements. The denial or delay of any such approval would delay commercialization efforts and adversely impact our potential to generate revenue, our business and our results of operations.
Our product candidates are in various stages of development, and our business currently depends entirely on their successful development, regulatory approval and commercialization. With the exception of BLONTRESS and TACTRESS, we currently have no products approved for sale, and we may never obtain regulatory approval to commercialize any of our other current or future product candidates. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of pet therapeutics products are subject to extensive regulation by the CVM, the USDA, the EMA and other regulatory authorities in the United States and other countries, whose regulations differ from country to country. We are not permitted to market our products in the United States until we receive approval of a New Animal Drug Application, or NADA, from the CVM or a full product license from the USDA with respect to our biologic products, or in Europe until we receive approval from the European Commission or applicable EU State national competent authorities.
Even if we receive approval of an NADA, USDA product license or foreign regulatory filing for our product candidates, the CVM, the USDA or the applicable foreign regulatory body may approve our product candidates for a more limited indication than we originally requested, and the CVM or the USDA may not approve the labeling that we believe is necessary or desirable for the successful commercialization of our product candidates. Any delay in obtaining, or inability to obtain, applicable regulatory approval would delay or prevent commercialization of our product candidates and would materially adversely impact our business and prospects.
Even if our current or future product candidates obtain regulatory approval, they may never achieve market acceptance or commercial success.
Even if we obtain CVM, USDA, EMA or other regulatory approvals, our current or future products may not achieve market acceptance among veterinarians and pet owners, and may not be commercially successful. For example, BLONTRESS and TACTRESS both received a full license from the USDA, however, we impaired the value of these assets during the third quarter of 2015, and do not anticipate that these products will generate significant revenues, as recent studies indicate that these products are not as specific to the targets as expected and have uncertain clinical benefits. Market acceptance of any of our current or future products will depend on a number of factors, including:
|
|
• |
|
the safety of our products as demonstrated in our target animal studies; |
27
|
|
• |
|
the indications for which our products are approved; |
|
• |
the acceptance by veterinarians and pet owners of the products as safe and effective treatments; |
||
|
|
• |
|
the consistent and reliable supply and manufacture of the products; |
|
|
• |
|
the proper training and administration of our products by veterinarians; |
|
|
• |
|
the potential and perceived advantages of our products over alternative treatments, including generic medicines and products approved for use by humans that are used off label; |
|
|
• |
|
the cost of treatment in relation to alternative treatments and willingness to pay for our products, if approved, on the part of veterinarians and pet owners; |
|
|
• |
|
the willingness of pet owners to pay for our treatments, relative to other discretionary items, especially during economically challenging times; |
|
|
• |
|
the relative convenience and ease of administration; |
|
• |
the prevalence and severity of adverse side effects; and |
||
|
|
• |
|
the effectiveness of our sales and marketing efforts and those of our collaborators. |
Because we expect sales of GALLIPRANT, ENTYCE and NOCITA, if approved, to generate substantially all of our product revenues for the foreseeable future, the failure of these product candidates to gain market acceptance or achieve commercial success would adversely affect our financial results and require us to seek additional financing.
We may not realize all of the anticipated benefits of acquisitions, or those benefits may take longer to realize than expected. We may also encounter significant unexpected difficulties in integrating acquired businesses.
We have made, and may continue to make, acquisitions. The overall integration of any acquired businesses may result in material unanticipated problems, expenses, liabilities, competitive responses, and diversion of management’s attention. The difficulties of combining the operations of acquired companies include, among others:
|
|
• |
|
the diversion of management’s attention to integration matters; |
|
|
• |
|
difficulties in achieving anticipated cost savings, synergies, business opportunities and growth prospects from combining any acquired businesses with our company; |
|
|
• |
|
difficulties in the integration of operations and systems; |
|
|
• |
|
difficulties in the assimilation of employees; |
|
|
• |
|
challenges in attracting and retaining key personnel; and |
|
|
• |
|
challenges in maintaining previously-established relationships with licensors and licensees. |
Many of these factors will be outside of our control and any one of them could result in increased costs and diversion of management’s time and energy, which could materially impact our business, financial condition and results of operations. In addition, even if the operations of any acquired businesses are integrated successfully, we may not realize the full benefits of the transaction, including the synergies or growth opportunities that we expect. For example, we acquired BLONTRESS and TACTRESS in connection with our acquisition of Vet Therapeutics, Inc. and, in the third quarter of 2015, we recorded an impairment charge of $20.2 million and $8.6 million, respectively, in relation to these intangible assets. It is possible that we will fully impair the value of these assets in the future. Other expected benefits of our acquisitions may not be achieved within the anticipated time frame, or at all.
In addition, through acquisitions, we may assume liabilities, losses or costs for which we are not indemnified or insured or for which our indemnity or insurance is inadequate. Any such liabilities may have a material adverse effect on our financial position or results of operations.
Development of pet therapeutics is an expensive and lengthy process with an uncertain outcome, and results of earlier studies may not be predictive of future study results.
Development of pet therapeutics is expensive and can take many years to complete, and its outcome is inherently uncertain. To gain approval to market a pet therapeutic for a particular species of pet, we must provide the CVM, the USDA or foreign regulatory authorities, as applicable, with data from animal safety and effectiveness studies that adequately demonstrate the safety and efficacy of that product in the target animal for the intended indication applied for in the NADA, product license or other regulatory filing. We rely on contract research organizations, or CROs, and other third parties to ensure the proper and timely conduct of most of our studies and development efforts and, while we have agreements governing their committed activities, we have limited influence over their actual performance. Failure can occur at any time during the development process. Success in prior target animal studies or in the treatment of human beings with a product candidate does not ensure that our target animal studies will be successful and the results of
28
development efforts by other parties may not be indicative of the results of our target animal studies and other development efforts. Product candidates in our studies may fail to show the desired safety and efficacy despite showing such results in initial data or previous human or animal studies conducted by other parties. Even if our studies and other development efforts are completed, the results may not be sufficient to obtain regulatory approval for our product candidates.
Once our target animal studies commence, we may experience delays in such studies and other development efforts and we do not know whether planned studies will begin on time, need to be redesigned or be completed on schedule, if at all. Pet therapeutics studies can be delayed or discontinued for a variety of reasons, including delay or failure to:
|
|
• |
|
reach agreement on acceptable terms with prospective CROs and study sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
|
|
• |
|
complete target animal studies due to deviations from study protocol; |
|
|
• |
|
address any safety concerns that arise during the course of testing; |
|
|
• |
|
address any conflicts with new or existing laws or regulations; |
|
|
• |
|
add new study sites; or |
|
|
• |
|
manufacture sufficient quantities of formulated drug for use in studies. |
If we experience delays in the completion of, or terminate any development efforts for our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our development efforts will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of our development efforts may also ultimately lead to the denial of regulatory approval of our product candidates.
Our product candidates, if approved, will face significant competition and our failure to effectively compete may prevent us from achieving significant market penetration.
The development and commercialization of new animal health medicines is highly competitive, and we expect considerable competition from major pharmaceutical, biotechnology and specialty animal health medicines companies. As a result, there are, and will likely continue to be, extensive research and substantial financial resources invested in the discovery and development of new animal health medicines. Our potential competitors include large animal health companies, such as Zoetis, Inc.; Merck Animal Health, the animal health division of Merck & Co., Inc.; Merial, the animal health division of Sanofi S.A.; Elanco, the animal health division of Eli Lilly and Company; Bayer Animal Health, the animal health division of Bayer AG; Boehringer Ingelheim Animal Health, the animal health division of Boehringer Ingelheim GmbH; Virbac Group; Ceva Animal Health; Vetoquinol and Dechra Pharmaceuticals PLC. We are also aware of several smaller early stage animal health companies such as Nexvet, Jaguar Animal Health, Parnell Pharmaceuticals, VetDC and Kindred Bio that are developing products for use in the pet therapeutics market.
If approved, we expect that GALLIPRANT will face competition from Rimadyl, marketed by Zoetis; generic Carprofen and Deramaxx, marketed by Elanco; Previcox, marketed by Merial; Metacam, marketed by Boehringer Ingelheim; and generic Meloxicam. Nexvet is developing a monoclonal antibody for osteoarthritis pain. Parnell is developing Zydax, an injectable product for osteoarthritis, a direct competitor for GALLIPRANT. We expect ENTYCE to enter a new market where there are no veterinary approved products for appetite stimulation. However, we are aware that some veterinarians utilize mirtazapine, a human generic antidepressant to attempt to treat inappetence. We expect NOCITA will compete primarily with the Coxibs and injectable buprenorphine and anesthetics, such as bupivacaine, which is not approved for non-human use but is widely used by veterinarians. Recuvyra fentanyl transdermal solution received approval in the United States and Europe for control of postoperative pain from surgical procedures in dogs. We are also unaware of any approved products for the treatment of lymphoma in dogs, although we are aware of a number of biotechnology companies such as Nexvet, Karyopharm and VetDC that are developing products for the treatment of lymphoma in dogs, including some that have received MUMS designation. We expect that BLONTRESS and TACTRESS will face competition from human generic chemotherapies, though we are unaware of any companies that are actively promoting this use. Among the larger animal health companies, Merial is developing a lymphoma vaccine based on technology similar to their melanoma vaccine. We know of no direct competitor for AT-014 in osteosarcoma.
Many of our competitors have substantially more resources than we do, including both financial and technical resources. In addition, many of our competitors have more experience than we have in the development, manufacture, regulation and worldwide commercialization of animal health medicines. We are also competing with academic institutions, governmental agencies and private organizations that are conducting research in the field of animal health medicines.
Our competition will be determined in part by the potential indications for which our products are developed and ultimately approved by regulatory authorities. Additionally, the timing of market introduction of some of our potential products or of competitors’ products may be an important competitive factor. Accordingly, the speed with which we can develop our compounds, complete target animal studies and approval processes, and supply commercial quantities to market are expected to be important competitive factors. We
29
expect that competition among products approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price and patent position.
If we are not successful in identifying, licensing or acquiring, developing and commercializing additional product candidates, our ability to expand our business and achieve our strategic objectives would be impaired.
Although we will continue to focus on the continued development and potential approval of our current product candidates, a key element of our strategy is to identify, license or acquire, develop and commercialize a portfolio of products to serve the pet therapeutics market. We derive potential pet therapeutic product candidates from molecules and compounds discovered or developed as part of human biopharmaceutical research. We expect to enter into license arrangements with third parties to provide us with rights to human health compounds for purposes of our business. Such agreements are typically complex and require time to negotiate and implement. If we enter into these arrangements, we may not be able to maintain these relationships or establish new ones in the future on acceptable terms or at all. If we are unable to access human health-generated molecules and compounds to conduct research and development on cost-effective terms, our ability to develop new products could be limited. In some instances, human biopharmaceutical companies may be unwilling to license us their products or compounds for development as pet therapeutics because of perceived regulatory and commercial risks, including the risk that the FDA could delay or halt an ongoing human development trial if the same compound, when studied in animals, produces an unexplained adverse event or death, and the risk that, if the same compound is developed for humans and pets, and the human version is priced significantly higher than the pet version, which is usually the case, human patients would attempt to use the cheaper animal version of the drug. Even if we successfully identify and license potential product candidates, we may still fail to yield product candidates for development and commercialization for many reasons, including the following:
|
|
• |
|
competitors may develop alternatives that render our product candidates obsolete; |
|
|
|
• |
|
product candidates we develop may nevertheless be covered by third parties’ patents or other exclusive rights; |
|
|
|
• |
|
a product candidate may on further study be shown to have harmful side effects in pets or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
|
|
|
• |
|
a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
|
|
|
• |
|
a product candidate may not be accepted as safe and effective by veterinarians, pet owners and the pet therapeutics community. |
|
If we fail to develop and successfully commercialize other product candidates, our business and future prospects may be harmed and our business will be more vulnerable to any problems that we encounter in developing and commercializing our current and future product candidates.
If we fail to attract and keep senior management and key scientific and commercial personnel, we may be unable to successfully develop any of our current or future product candidates, conduct our in-licensing and development efforts and commercialize any of our current or future product candidates.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We are highly dependent upon our senior management team as well as our senior scientists and sales and marketing team. The loss of services of any of these individuals could delay or prevent the successful development of our current or future product pipeline, completion of our planned development efforts or the commercialization of our product candidates.
In addition, we could experience difficulties attracting and retaining qualified employees in the future. For example, competition for qualified personnel in the animal health fields is intense due to the limited number of individuals who possess the skills and experience required by our industry. We will need to hire additional personnel as we expand our development and commercial activities. We may not be able to attract and retain quality personnel on acceptable terms, or at all. In addition, to the extent we hire personnel from competitors, we may be subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output. Failure to attract and retain highly qualified personnel could have an adverse effect on our business and our ability to develop and commercialize our product candidates.
We rely completely on third-party manufacturers to manufacture the supplies for the development of our small molecule and antiviral product candidates and we intend to rely on third-party manufacturers to produce commercial quantities of any of these approved drug candidates.
With respect to our small molecules and antiviral programs, we do not currently have, nor do we currently plan to acquire, the internal infrastructure or capability to manufacture the formulated drug for use in the conduct of our target animal studies. We also lack the resources and the capability to manufacture any of our product candidates on a scale necessary for commercialization. We will need to rely on contract manufacturers to provide commercial supplies of the formulated drugs for all small molecule and antiviral products. For example, for AT-003, we have entered into a commercial supply agreement with Pacira to supply the formulated drug. If this supply agreement terminates for any reason, or Pacira does not produce the necessary quantities, we may be unable to arrange for
30
alternative supply of AT-003 in a timely manner, on commercially reasonable terms, or at all. Our agreement with Pacira may terminate due to factors outside of our control, including if Pacira ceases to manufacture, for any reason, the formulated drug. With respect to AT-003 and our other product candidates, any delay in our ability to identify and contract with a replacement or an initial third-party contract manufacturer, as applicable, on commercially reasonable terms, or at all, would have an adverse impact upon our business.
The facilities used by our contract manufacturers to manufacture the active pharmaceutical ingredients and formulated drugs may be subject to inspections by one or more regulatory bodies. We do not control the manufacturing processes used by, and we are completely dependent on, our contract manufacturers to comply with cGMP for the manufacture of both active pharmaceutical ingredients and finished drug products. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control and quality assurance practices and to engage qualified personnel. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and complies with regulatory requirements, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. If the CVM, the USDA or the EMA does not approve our contract manufacturers’ facilities used for the manufacture of our product candidates, or if any such agency withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would adversely impact our ability to develop and obtain regulatory approval for or market our product candidates, if approved.
We and our third-party contractors are continuing to refine and improve the manufacturing process for our product candidates, certain aspects of which are complex and unique. We may encounter difficulties with new or existing manufacturing processes. In addition, to manufacture our product candidates in the quantities that we believe would be required to meet anticipated market demand, our third-party manufacturers may need to increase manufacturing capacity, which could involve significant challenges and may require additional regulatory approvals. Neither we nor our third-party manufacturers may successfully complete any manufacturing scale-up activities required to increase existing manufacturing capabilities in a timely manner, or at all.
Furthermore, we rely on our contract manufacturers to obtain any raw materials necessary to manufacture our products, and we do not have any control over the process or timing of the acquisition of these materials. If there is a disruption to our or our third-party manufacturers’ relevant operations, we will have no other means of producing our product candidates until they restore the affected facilities or we or they procure alternative manufacturing facilities or raw materials. Additionally, any damage to or destruction of our third-party manufacturers’ facilities or equipment may significantly impair our ability to manufacture product candidates on a timely basis.
Biologics manufacturing is difficult and costly, and may not be commercially viable.
We acquired a USDA-licensed manufacturing facility for our biologic products as part of our acquisition of Vet Therapeutics. Manufacturing of our pet biologics is a relatively new field in which unanticipated difficulties or challenges could arise. For example, we announced in February 2015 that there was a potential quality issue in the manufacturing of one serial lot of BLONTRESS. We completed an investigation into the serial lot and identified an environmental contaminant in certain of the vials in the lot, and in agreement with the USDA, that serial lot was destroyed. Manufacturing biologics, especially in large quantities, is complex and may require the use of technologies that we may need to develop. Such manufacturing requires facilities specifically designed and validated for this purpose as well as sophisticated quality assurance and quality control procedures. Biologics can also be costly to manufacture. Manufacturing biologics may be more technically challenging, time-consuming and expensive than we anticipate.
During 2016, we plan to manufacture BLONTRESS and TACTRESS at our USDA-licensed facility in San Diego. We perform all steps for production including cell line development, assay development, production in batch mode, fill and finish, release of products and packaging. There is no assurance that we will be able to manufacture these or other biologics at full commercial scale and at an economical cost. We may experience supply constraints or other supply challenges as we attempt to increase manufacturing capacity. We may encounter difficulties with new or existing manufacturing processes and we may experience delays, may be required to seek additional regulatory approvals and/or may incur significant expense in scaling up the capacity and operations to meet anticipated demand for future biologics products. If there is a disruption to our relevant operations in San Diego, we will have no other means of producing our products until operations are restored or we transition to alternative manufacturing facilities. Additionally, any damage to or destruction of our facilities or equipment may significantly impair our ability to manufacture products on a timely basis.
We currently rely on third parties to conduct our target animal studies and certain other development efforts. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize our current or future product candidates.
We currently do not conduct our target animal studies, and we rely on CROs and/or academic institutions to conduct these studies. The third parties with whom we contract for the execution of our studies play a significant role in the conduct of these studies and the subsequent collection and analysis of data. However, these third parties are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount or timing of resources that they devote to our programs. Although we rely on these third parties to conduct our studies, we remain responsible for ensuring that each of our studies is conducted in accordance with the development plan and protocol. Moreover, the CVM, the USDA and EMA require us to comply with regulations and standards, commonly referred to as current good clinical practices, or cGCPs, or good laboratory practices, or GLPs, for conducting, monitoring, recording and reporting the results of our studies to ensure that the data and results are scientifically credible and accurate.
31
In addition, the execution of target animal studies and the subsequent compilation and analysis of the data produced requires coordination among various parties. If the third parties conducting our target animal studies do not perform their contractual duties or obligations, experience work stoppages, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our development protocols or cGCPs, or for any other reason, we may need to enter into new arrangements with alternative third parties, which could be difficult and costly, and our target animal studies may be extended, delayed or terminated or may need to be repeated. If any of the foregoing were to occur, the regulatory approval for and commercialization of the product candidate being tested in any such study may be delayed or require us to utilize additional resources.
Our ability to market our product candidates, if approved, will be limited to use for the treatment of the indications for which they are approved, and if we want to expand the indications for which we may market our product candidates, we will need to obtain additional CVM, USDA or EMA approvals, which may not be granted.
We are seeking CVM approval in the United States for AT-001 directed at treating osteoarthritis pain and inflammation in dogs and for the treatment of pain and inflammation associated with degenerative joint disease in cats, AT-002 for the appetite stimulation of for dogs and management of weight loss in chronically diseased cats, and AT-003 for the treatment of post-operative pain in dogs and cats. In addition, we have received a full license from the USDA for BLONTRESS as an aid for the treatment of B-cell lymphoma in dogs and for TACTRESS as an aid for the treatment of T-cell lymphoma in dogs. If our product candidates are approved, we may market or advertise them only for the treatment of indications for which they are approved, which could limit their adoption by veterinarians and pet owners. We may attempt to develop, promote and commercialize new treatment indications and protocols for our product candidates in the future, but we cannot predict when or if we will receive the approvals required to do so. In addition, we would be required to conduct additional target animal studies to support our applications, which would utilize additional resources and may produce results that do not result in CVM, USDA or EMA approvals. If we do not obtain additional CVM, USDA or EMA approvals, our ability to expand our business will be limited.
We currently have a small commercial organization. If we are unable to expand sales capabilities on our own or through third parties, we may not be able to market and sell significant amounts of our current or future product candidates, if approved, or generate product revenue.
We currently have a small commercial organization. In order to commercialize any of our current or future product candidates in the United States and any jurisdictions outside the United States, including GALLIPRANT, ENTYCE and NOCITA, we must build our marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. We expect to expand our direct sales organization in the United States, complemented by distributors, to commercialize our product candidates, which will be expensive and time-consuming. Because we have limited prior experience in the marketing, sale and distribution of pet therapeutics, there are significant risks involved in building and managing a sales organization, including our ability to hire, retain and motivate qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, and effectively oversee a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of our products. Outside of the United States we intend to collaborate with companies with an established commercial presence to market our products in those locations. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our current product candidates or any future product candidates that receive regulatory approval. If we are not successful in commercializing any of our current or future product candidates, either on our own or through collaborations with one or more distributors, our future product revenue will suffer and we would incur significant additional losses.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
Since our initial public offering in June 2013, we have grown from approximately 15 full-time employees to approximately 60 full-time employees as of March 10, 2016. We will need to continue to expand our managerial, operational, financial and other resources in order to manage our operations and target animal studies, continue our development activities and commercialize any of our current or future product candidates. Our management and personnel, systems and facilities currently in place may not be adequate to support this future growth. Our need to effectively execute our growth strategy requires that we:
|
|
• |
|
manage our target animal studies and other development efforts effectively; |
|
|
• |
|
identify, recruit, maintain, motivate and integrate additional employees; |
|
|
• |
|
manage our internal development efforts effectively while carrying out our contractual obligations to third parties; and |
|
|
• |
|
continue to improve our operational, financial and management controls, reporting systems and procedures. |
Any failure to successfully manage our growth could have a material adverse effect on our ability to effectively carry out our target animal studies, continue our development activities and commercialize our product candidates.
We are incurring significant costs as a result of operating as a public company, and our management is expected to devote substantial time to new compliance initiatives.
32
As a publicly-traded company, we have incurred and will continue to incur significant legal, accounting and other expenses that we did not incur when we were a private company, particularly after we are no longer an “emerging growth company” as defined under the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. In addition, new and changing laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd-Frank Wall Street Reform and Consumer Protection Act and the rules and regulations promulgated and to be promulgated thereunder, as well as under the Sarbanes-Oxley Act, the JOBS Act, and the rules and regulations of the U.S. Securities and Exchange Commission, or SEC, and The NASDAQ Global Market, have created uncertainty for public companies and increased our costs and time that our Board of Directors and management must devote to complying with these rules and regulations. We expect these rules and regulations to continue to increase our legal and financial compliance costs and lead to a diversion of management time and attention from revenue-generating activities.
Furthermore, the need to establish the corporate infrastructure demanded of a public company may divert management’s attention from implementing our growth strategy, which could prevent us from improving our business, results of operations and financial condition.
The restatement of our consolidated financial statements for the year ended December 31, 2013 has subjected us to a number of additional risks and uncertainties, including increased possibility of legal proceedings.
Our management determined that our consolidated financial statements for the year ended December 31, 2013 should be restated due to errors relating to the valuation of the intangible asset BLONTRESS in connection with our October 2013 acquisition of Vet Therapeutics, Inc. As a result of the restatement, we have become subject to a number of risks and uncertainties, including possible shareholder or other litigation, in connection with the restatement.
Management previously identified a material weakness in our internal control over financial reporting for the year ended 2013, which could have a significant adverse effect on our business and the price of our common stock.
Our management is required to report annually on the effectiveness of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act, or Section 404. The rules governing the standards that must be met for our management to assess our internal control over financial reporting are complex and require significant documentation, testing and possible remediation.
We identified a material weakness in our internal controls relating to the valuation of the intangible asset BLONTRESS in connection with our October 2013 acquisition of Vet Therapeutics, Inc., and concluded that our internal control over financial reporting as of December 31, 2014 was ineffective. We remediated this material weakness in the second quarter of 2015.
In the future, we may identify additional material weaknesses or significant deficiencies, and we may not be able to remediate them in time to meet the deadline imposed by the Sarbanes-Oxley Act for compliance with the requirements of Section 404. In addition, we may encounter problems or delays in completing the implementation of any requested improvements and receiving a favorable attestation report from our independent registered public accounting firm, if such a report is required. We will be unable to issue securities in the public markets through the use of a shelf registration statement if we are not in compliance with Section 404. Furthermore, failure to achieve and maintain an effective internal control environment could materially adversely affect our business, reduce the market’s confidence in our common stock, adversely affect the price of our common stock and limit our ability to report our financial results accurately and timely.
Changes in distribution channels for pet therapeutics could negatively impact our market share, margins and distribution of our products.
In most markets, pet owners typically purchase their pet therapeutics directly from veterinarians. Pet owners increasingly could purchase pet therapeutics from sources other than veterinarians, such as Internet-based retailers, “big-box” retail stores or other over-the-counter distribution channels. This trend has been demonstrated by the significant shift away from the veterinarian distribution channel in the sale of parasiticides and vaccines in recent years. Pet owners also could decrease their reliance on, and visits to, veterinarians as they rely more on Internet-based animal health information. Because we expect to market our pet prescription products through the veterinarian distribution channel, any decrease in visits to veterinarians by pet owners could reduce our market share for such products and materially adversely affect our operating results and financial condition. In addition, pet owners may substitute human health products for pet therapeutics if human health products are deemed to be lower-cost alternatives.
Legislation has also been proposed in the United States, and may be proposed in the United States or abroad in the future, that could impact the distribution channels for our pet products. For example, such legislation may require veterinarians to provide pet owners with written prescriptions and disclosure that the pet owner may fill prescriptions through a third party, which may further reduce the number of pet owners who purchase their pet therapeutics directly from veterinarians. Such requirements may lead to increased use of generic alternatives to our products or the increased substitution of our products with other pet therapeutics or human health products if such other products are deemed to be lower-cost alternatives. Many states already have regulations requiring veterinarians to provide prescriptions to pet owners upon request and the American Veterinary Medical Association has long-standing policies in place to encourage this practice.
Over time, these and other competitive conditions may increase our reliance on Internet-based retailers, “big-box” retail stores or other over-the-counter distribution channels to sell our pet products. Any of these events could materially adversely affect our operating results and financial condition.
33
Consolidation of our customers could negatively affect the pricing of our products.
Veterinarians are our primary customers. In recent years, there has been a trend towards the concentration of veterinarians in large clinics and hospitals. If this trend towards consolidation continues, these customers could attempt to improve their profitability by leveraging their buying power to obtain favorable pricing. The resulting decrease in our prices could have a material adverse effect on our operating results and financial condition.
Our ability to use our net operating loss carryforwards to offset future taxable income may be subject to certain limitations.
As of December 31, 2015, we had net operating loss carryforwards, or NOLs, for federal and state income tax purposes of $44.2 million and $42.4 million, respectively, which may be available to offset our future taxable income, if any. Our federal NOLs begin to expire in 2031, and our state NOLs begin to expire in 2020. In general, under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, a corporation that undergoes an “ownership change” is subject to limitations on its ability to use its pre-change net operating loss carryforwards to offset future taxable income. If the Internal Revenue Service challenges our analysis that our existing NOLs will not expire before utilization due to previous ownership changes, or if we undergo an ownership change in the future, our ability to use our NOLs could be limited by Section 382 of the Code. Future changes in our stock ownership, some of which are outside of our control, could result in an ownership change under Section 382 of the Code. Furthermore, our ability to use NOLs of companies that we may acquire in the future may be subject to limitations. For these reasons, we may not be able to use a material portion of the NOLs reflected on our consolidated balance sheet, even if we attain profitability. We have not completed a study to assess whether an ownership change has occurred, or whether there have been multiple ownership changes since its formation, due to significant complexity and related costs associated with such a study.
Generic products may be viewed as more cost-effective than our products.
We may face competition from products produced by other companies, including generic (or non-patented) alternatives to any of our products. We will depend on patents to provide us with exclusive marketing rights for some of our products. As of December 31, 2015 we had licensed an extensive portfolio of issued patents or pending patent applications relating to our AT-001, AT-002 and AT-003 products covering various composition of matter claims as well as methods of treatment and methods of manufacturing our products. In addition, as part of our Vet Therapeutics acquisition, we acquired a patent family related to the speciesization of antibodies that covers all Vet Therapeutics products. We also acquired a patent family related to antibody constant domain regions and uses thereof, which also covers all Vet Therapeutics products. We also acquired pending patent applications that cover specific canine monoclonal antibodies directed to various targets, including an allowed U.S. patent directed to the canine CD-52 development antibody. Further, as part of our acquisition of Okapi Sciences, we acquired two patent applications that cover formulations of AT-006 and commercially-viable methods of making the active ingredient of AT-006. We also have a license to an issued U.S. patent that covers the active ingredient of AT-007. We also have patent applications in the United States, Europe and other countries that cover therapeutic uses of AT-007. Finally, we have in-licensed a patent portfolio for AT-008 that covers the composition and use of AT-008 through 2024 and 2027, respectively.
The protection afforded to our patents, which varies from country to country, is limited by the scope and applicable terms of our patents and the availability of legal remedies in the applicable country. As a result, we may face competition from lower-priced generic alternatives to many of our products. Generic competitors are becoming more aggressive in terms of pricing, and generic products are an increasing percentage of overall animal health sales in certain regions. In addition, private label products may compete with our products. If pet therapeutics customers increase their use of new or existing generic or private label products, our operating results and financial condition could be materially adversely affected.
Our pet therapeutics are subject to unanticipated safety or efficacy concerns, which may harm our reputation.
Unanticipated safety or efficacy concerns can arise with respect to pet therapeutics, whether or not scientifically or clinically supported, leading to product recalls, withdrawals or suspended or declining sales, as well as product liability, and other claims. For example, although BLONTRESS and TACTRESS both received a full license from the USDA, we impaired the value of these assets during the third quarter of 2015 and do not anticipate that these products will generate significant revenues, as recent studies indicate that these products are not as specific to the targets as expected and have uncertain clinical benefits. In addition, we depend on positive perceptions of the safety and quality of our products, and pet therapeutics generally, by our customers, veterinarians and end-users, and such concerns may harm our reputation. These concerns and the related harm to our reputation could materially adversely affect our operating results and financial condition, regardless of whether such reports are accurate.
Our business and operations would suffer in the event of system failures or security breaches.
Our internal computer systems and those of our current and future contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Any system failure or security breach by employees or others may pose a risk that sensitive data, including data from our target animal studies, intellectual property, trade secrets or personal information belonging to us may be exposed to unauthorized persons or to the public. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of data from completed or future studies could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely on third parties to manufacture our product candidates, and similar events relating to their computer systems could also have a material adverse effect on
34
our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability, the further development and commercialization of our product candidates could be delayed, and the trading price of our common stock could be adversely affected.
Risks Related to Intellectual Property
We currently own two issued patents and several patent applications, license the issued patents covering our small molecule product candidates and have limited rights to prosecute and enforce those licensed patents.
We currently own one issued patent related to the speciesization of antibodies that covers all Vet Therapeutics products, an issued patent related to antibody constant domain regions and uses thereof, which also covers all Vet Therapeutics products, a patent related to canine monoclonal antibodies directed to canine CD-52 and a filed patent application that specifically covers our canine monoclonal antibodies directed to canine CD-20, as well as pending related applications and foreign counterpart patents and applications.
We also own a patent family relating to our AT-002 product candidate, covering a method of treating inappetence using AT-002. Applications in this patent family are pending in Australia, Argentina, Canada, China, European Patent Office, Japan and Taiwan. We cannot assure you that a patent based on any of these patent applications will ever be issued. We do not own any patents or patent applications relating to AT-001 or AT-003. We have exclusive license agreements in the field of animal health with RaQualia, pursuant to which we license key intellectual property relating to AT-001 and AT-002, and with Pacira pursuant to which we license key intellectual property relating to AT-003. Under each of the license agreements, RaQualia and Pacira retain ownership over the licensed patents and patent applications and retain control over the maintenance and prosecution of the licensed patents and patent applications. In the case of AT-003, we have no control over the manner in which Pacira chooses to maintain or prosecute its patent and patent applications and have no right to continue to prosecute any patents or patent applications that Pacira elects to abandon. We do not have the right to enforce patents licensed from Pacira against any third-party infringement, although we have certain limited rights to request our licensor to enforce such patents against infringement.
If we cannot obtain ownership of or adequate license rights to issued patents covering our product candidates or we cannot prosecute or enforce licensed patents, our business, results of operations, financial condition and prospects would be adversely affected.
If we fail to comply with our obligations under our intellectual property licenses with third parties, we could lose license rights that are essential to our business.
We are party to license agreements for our product candidates that are essential to our business. These license agreements impose various payment and performance obligations on us. If we fail to comply with these obligations, RaQualia or Pacira, as applicable, may have the right to terminate the relevant license agreement, in which event we would not be able to develop or commercialize AT-001, AT-002 and/or AT-003, as the case may be.
If we lose such license rights, our business, results of operations, financial condition and prospects would be adversely affected. We may enter into additional licenses in the future and if we fail to comply with obligations under those agreements, we could suffer adverse consequences.
We may not own any intellectual property rights we develop with respect to AT-003 or be able to share our licensed patent rights to AT-003 with future collaborators.
Our license agreement with Pacira contains certain obligations and restrictions on our ability to develop and commercialize AT-003. All of the intellectual property rights that we develop with respect to AT-003 will be owned by Pacira upon termination of this license agreement. If we wish to enter into any collaboration agreements relating to AT-003, Pacira has the right to approve all of our sublicenses. Furthermore, Pacira has a right of first negotiation for shared commercialization rights to AT-003 in the United States. These restrictions may impair or delay our ability to engage third parties to commercialize AT-003.
We may become subject to third parties’ claims alleging infringement of patents and proprietary rights or seeking to invalidate our patents or proprietary rights, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development and commercialization of our current or future product candidates.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the field of pet therapeutics, as well as patent challenge proceedings, including interference and administrative law proceedings before the U.S. PTO and oppositions and other comparable proceedings in foreign jurisdictions. Recently, under U.S. patent reform laws, new procedures including inter party review and post grant review have been implemented. As stated below, the novel implementation of such reform laws presents uncertainty regarding the outcome of challenges to our patents in the future. We cannot assure you that any of our current or future product candidates will not infringe existing or future patents. Because we have not conducted a formal freedom to operate analysis for patents related to our products, we may not be aware of patents that have already been issued that a third party might assert are infringed by one of our current or future product candidates. Because patent applications can take many years to issue and may be confidential for eighteen months or more after filing, there also may be applications now pending of which we are unaware and which may later result in issued patents that we may infringe by commercializing any of our current or future product candidates.
35
We may be subject to third-party claims in the future against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing a third party’s patents. If a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product candidate that is the subject of the suit. As a result of patent infringement claims, or in order to avoid potential claims, we or our collaborators may choose to seek, or be required to seek, a license from the third party and would most likely be required to pay license fees, milestone payments, royalties or other payments. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or forced to redesign it, or to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. Even if we are successful in defending such claims, infringement and other intellectual property litigation can be expensive and time-consuming to litigate and divert management’s attention from our core business. Any of these events could harm our business significantly.
In addition to infringement claims against us, if third parties have prepared and filed patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference proceedings in the U.S. PTO to determine the priority of invention. Third parties may also attempt to initiate reexamination, post grant review or inter party review of our patents in the U.S. PTO. We may also become involved in similar opposition proceedings in the European Patent Office or similar offices in other jurisdictions regarding our intellectual property rights with respect to our products and technology. Moreover, we may face claims from non-practicing entities, which have no relevant product revenue and against whom our own patent portfolio may thus have no deterrent effect.
If our efforts to protect the proprietary nature of the intellectual property related to any of our current or future product candidates are not adequate, we may not be able to compete effectively in our market.
We rely upon a combination of patents, trade secret protection, confidentiality and license agreements to protect the intellectual property related to our current product candidates and our development programs.
Composition-of-matter patents on the active pharmaceutical ingredient are generally considered to be the strongest form of intellectual property protection for pharmaceutical products, including pet therapeutics, as such patents provide protection without regard to any particular method of use or manufacture. Method-of-use patents protect the use of a product for the specified method. This type of patent does not prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside the scope of the patented method. Moreover, even if competitors do not actively promote their product for our targeted indications, veterinarians may recommend that pet owners use these products off label, or pet owners may do so themselves. Although off-label use may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute. Method of manufacturing patents protect a specific way to make a product and do not prevent a third party from making the product by a different method and then using the product for our uses. We cannot be certain that the claims in our patent applications will be considered patentable by the U.S. PTO and courts in the United States, or by the patent offices and courts in foreign countries.
The strength of patents in the field of pet therapeutics involves complex legal and scientific questions and can be uncertain. The patent applications that we own or license may fail to result in issued patents in the United States or in other foreign countries. Even if the patents do successfully issue, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our products or our intellectual property or prevent others from designing around our claims. If the breadth or strength of protection provided by the patents and patent applications we own, in-license or pursue with respect to any of our current or future product candidates is threatened, it could threaten our ability to commercialize any of our current or future product candidates. Further, if we encounter delays in our development efforts, the period of time during which we could market any of our current or future product candidates under patent protection would be reduced. Since patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to file any patent application related to our product candidates. Furthermore, for patent applications in which claims are entitled to a priority date before March 16, 2013, an interference proceeding can be provoked by a third party or instituted by the U.S. PTO to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. For patent applications containing a claim not entitled to a priority date before March 16, 2013, there is a greater level of uncertainty in the patent law due to the passage of the America Invents Act, which brings into effect significant changes to the U.S. patent laws that have yet to be well defined, and which introduces new procedures for challenging pending patent applications and issued patents. A primary change under this reform is creating a “first to file” system in the United States, which requires us to minimize the time from invention to filing of a patent application.
We also rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. We cannot be certain that we have executed such agreements with all parties, including our collaborators and contract manufacturers, who may have helped to develop our intellectual property or had access to our proprietary information, nor that our agreements will not be breached. We cannot guarantee that our
36
trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, results of operations and financial condition.
Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe our patents, or patents that may be issued to us in the future, or the patents of our licensors that are licensed to us. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, if we or one of our future collaborators were to initiate legal proceedings against a third party to enforce a patent covering our current product candidates, or one of our future products, the defendant could counterclaim that our patent is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the U.S. PTO, or made a materially misleading statement, during prosecution. Third parties may also raise similar claims before the U.S. PTO, even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our current or future product candidates. Such a loss of patent protection could have a material adverse impact on our business.
Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be unsuccessful, it could have an adverse effect on the price of our common stock.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation. The Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. PTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing owned or licensed patents and patents that we might obtain in the future.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The U.S. PTO, the European Patent Office and various other foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case, which would have an adverse effect on our business.
We have pending trademark applications for our company name in the United States and certain other countries, and we have pending trademark applications in the United States for certain product candidates, and we have pending trademark applications for these product candidates in certain other countries; however, registration is not yet complete for certain of these filings, and failure to finally secure these registrations could adversely affect our business.
We have two pending trademark applications and have obtained four trademark registrations for our company name and design marks in the United States, and we have eleven pending foreign trademark applications for our company name and design marks and we have obtained eleven foreign registrations for these marks, although we cannot make assurances that the trademarks will become registered. We have nine pending trademark applications in the United States for commercial trade names for our current product
37
candidates, and we have obtained seven U.S. registrations for these candidates, and we have seventeen pending foreign applications and twenty-nine foreign registrations for these candidates, although we cannot make assurances that the trademarks applications will be registered. During trademark registration proceedings, we have in the past and may in the future receive rejections. Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the U.S. PTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings have in the past and may in the future be filed against our trademarks, and our trademarks may not survive such proceedings. Additionally, we may need to enforce our trademark rights against third parties and expend significant additional resources to enforce such rights against infringements. Moreover, any name we propose to use with our product candidates in the United States must be approved by the CVM, the USDA or the EMA, regardless of whether we have registered it, or applied to register it, as a trademark. The CVM typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the CVM, the USDA or the EMA object to any of our proposed proprietary product names (which they have done in the past done and may do in the future), we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the CVM, the USDA or the EMA.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on product candidates throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued or licensed patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions, including in Europe where our Aratana NV facilities are located. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology, pharmaceutical or animal health companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise improperly used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial cost and be a distraction to our management and employees.
Risks Related to Government Regulation
The regulatory approval process is uncertain, requires us to utilize significant resources, and may prevent us from obtaining approvals for the commercialization of some or all of our product candidates.
The research, testing, manufacturing, labeling, approval, selling, import, export, marketing and distribution of pet therapeutics are subject to extensive regulation by the CVM, the USDA or the EMA and other regulatory authorities in the United States and other countries, which regulations differ from country to country. We are not permitted to market any of our current or future product candidates in the United States until we receive approval of an NADA from the CVM or a product license from the USDA. Obtaining approval of an NADA from CVM or a product license from the USDA can be an uncertain process that requires us to utilize significant resources. The CVM, the USDA or any foreign regulatory bodies can delay, limit or deny approval of any of our product candidates for many reasons, including:
|
|
• |
|
we are unable to demonstrate to the satisfaction of the CVM, the USDA, the EMA or the applicable foreign regulatory body that the product candidate is safe and effective for the requested indication; |
|
|
• |
|
the CVM, the USDA or the applicable foreign regulatory body may disagree with our interpretation of data from our target animal studies and other development efforts; |
|
|
• |
|
we may be unable to demonstrate that the product candidate’s benefits outweigh any safety or other actual or perceived risks; |
|
|
• |
|
the CVM, the USDA or the applicable foreign regulatory body may require additional studies; |
38
|
|
• |
|
the CVM, the USDA or the applicable foreign regulatory body may not approve of the formulation, labeling and/or the specifications of our current and future product candidates; |
|
|
• |
|
the CVM, the USDA or the applicable foreign regulatory body may fail to approve our manufacturing processes or facilities, or the manufacturing processes or facilities of third-party manufacturers with which we contract; and |
|
|
• |
|
the approval policies or regulations of the CVM, USDA or the applicable foreign regulatory body may significantly change in a manner rendering the data from our studies insufficient for approval. |
Failure to comply with CVM and other applicable United States and foreign regulatory requirements may subject us to administrative or judicially imposed sanctions, including: warning letters, civil and criminal penalties, injunctions, withdrawal of approved products from the market, product seizure or detention, product recalls, total or partial suspension of production, and refusal to approve pending NADAs or product licenses or supplements to approved NADAs or product licenses.
Regulatory approval of an NADA or supplement NADA, or of a product license, is not guaranteed, and the approval process requires us to utilize significant resources, may take several years, and is subject to the substantial discretion of the CVM, the USDA or the EMA. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon or repeat studies, or perform additional studies. If any of our current or future product candidates fails to demonstrate safety and efficacy in our studies, or for any other reason does not gain regulatory approval, our business and results of operations will be materially and adversely harmed.
Even if we receive regulatory approval for any of our current or future product candidates, we will be subject to ongoing CVM, USDA or EMA obligations and continued regulatory review, which may result in significant additional expense. Additionally, any product candidates, if approved, will be subject to labeling and manufacturing requirements and could be subject to other restrictions. Failure to comply with these regulatory requirements or the occurrence of unanticipated problems with our products could result in significant penalties.
Any regulatory approvals that we or any of our collaborators receive for any of our current or future product candidates may be subject to conditions of approval or limitations on the approved indicated uses for which the product may be marketed, or may contain requirements for potentially costly surveillance to monitor the safety and efficacy of the product candidate. In addition, if the CVM, the USDA or the EMA approves any of our current or future product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP, GLP and GCP, for any studies that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
|
• |
restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
||||||
|
|
• |
|
fines, warning letters or holds on target animal studies; |
||||
|
|
• |
|
refusal by the CVM, the USDA or the EMA to approve pending applications or supplements to approved applications filed by us or our strategic collaborators, or suspension or revocation of product license approvals; |
||||
|
|
• |
|
product seizure or detention, or refusal to permit the import or export of products; and |
||||
|
|
• |
|
injunctions or the imposition of civil or criminal penalties. |
||||
The CVM’s, USDA’s or the EMA’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business.
Failure to obtain regulatory approvals in foreign jurisdictions for our product candidates would prevent us from marketing our products internationally.
In order to market any product outside of the United States, including in the EEA (which is comprised of the 28 member states of the European Union plus Norway, Iceland and Liechtenstein) and many other foreign jurisdictions, separate regulatory approvals are required. More concretely, in the EEA, pet therapeutics can only be commercialized after obtaining a Marketing Authorization (“MA”). Before granting the MA, the EMA or the competent national authorities of the member states of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
39
The approval procedures vary among countries and can involve additional studies and testing, and the time required to obtain approval may differ from that required to obtain CVM or USDA approval. Animal studies conducted in one country may not be accepted by regulatory authorities in other countries. Approval by the CVM or USDA does not ensure approval by regulatory authorities in other countries, and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other foreign countries or by the CVM or the USDA. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. The foreign regulatory approval process may include all of the risks associated with obtaining CVM or USDA approval. We may not be able to file for regulatory approvals or to do so on a timely basis and, even if we do file them, we may not receive necessary approvals to commercialize our products in any market.
If approved, any of our current or future products may cause or contribute to adverse medical events that we are required to report to the CVM, USDA and regulatory authorities in other countries and, if we fail to do so, we could be subject to sanctions that would materially harm our business.
If we are successful in commercializing any of our current or future products, regulations of the CVM, the USDA and of the regulatory authorities in other countries require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the CVM, USDA and regulatory authorities in other countries could take action including criminal prosecution, the imposition of civil monetary penalties, seizure of our products, or delay in approval or clearance of future products.
Legislative or regulatory reforms with respect to pet therapeutics may make it more difficult and costly for us to obtain regulatory clearance or approval of any of our current or future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, CVM and USDA regulations and guidance are often revised or reinterpreted by the CVM and USDA in ways that may significantly affect our business and our products. Similar changes in laws or regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the United States or in other countries may impose additional costs or lengthen review times of any of our current or future product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
|
|
• |
|
changes to manufacturing methods; |
|
|
• |
|
recall, replacement, or discontinuance of certain products; and |
|
|
• |
|
additional record keeping. |
Each of these would likely entail substantial time and cost and could materially harm our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm our business, financial condition, and results of operations.
Our research and development relies on evaluations in animals, which may become subject to bans or additional regulations.
As a biopharmaceutical company with a focus on pet therapeutics, the evaluation of our existing and new products in animals is required to register our products. Animal testing in certain industries has been the subject of controversy and adverse publicity. Some organizations and individuals have attempted to ban animal testing or encourage the adoption of additional regulations applicable to animal testing. To the extent that the activities of such organizations and individuals are successful, our research and development, and by extension our operating results and financial condition, could be materially adversely affected. In addition, negative publicity about us or our industry could harm our reputation.
Risks Related to Our Common Stock
Our stock price may be volatile and you may not be able to resell shares of our common stock at or above the price you paid.
The trading price of our common stock is volatile with trading prices ranging from $2.56 per share to $29.32 per share since our initial public offering in June 2013. The price of our common stock has been and could continue to be subject to wide fluctuations, including due to results from, and any delays in, our current and future target animal studies. For example, on September 25, 2015, following our announcement that we do not believe that BLONTRESS or TACTRESS will capture the desired lymphoma market opportunity, the price of our common stock fell from $17.49 on September 24, 2015 to $10.67 on September 25, 2015, a 39% reduction. The price of our common stock could be volatile in the future in response to various factors, some of which are beyond our control. These factors include those discussed in this “Risk Factors” section and others, such as:
40
|
|
• |
|
announcements of regulatory approval or disapproval of any of our current or future product candidates; |
|
|
• |
|
failure or discontinuation of any of our research programs; |
|
|
• |
|
the termination of any of our existing license agreements; |
|
|
• |
|
announcements relating to future licensing or development agreements; |
|
|
• |
|
delays in the commercialization of our current or future product candidates; |
|
|
• |
|
acquisitions and sales of new product candidates, technologies or businesses; |
|
|
• |
|
manufacturing and supply issues related to our current or future product candidates for our development programs and commercialization; |
|
|
• |
|
quarterly variations in our results of operations or those of our future competitors; |
|
|
• |
|
changes in earnings estimates or recommendations by securities analysts; |
|
|
• |
|
announcements by us or our competitors of new product candidates, significant contracts, commercial relationships, acquisitions or capital commitments; |
|
|
• |
|
developments with respect to intellectual property rights; |
|
|
• |
|
our commencement of, or involvement in, litigation; |
|
|
• |
|
any major changes in our Board of Directors or management; |
|
|
• |
|
new legislation in the United States or other countries relating to the sale or pricing of pet therapeutics; |
|
|
• |
|
CVM or USDA or other U.S. or foreign regulatory actions affecting us or our industry; |
|
|
• |
|
product liability claims, other litigation or public concern about the safety of our product candidates or future products; |
|
|
• |
|
market conditions in the animal health sector and in the pet therapeutics market; |
|
|
• |
|
low daily trading volumes in our stock; and |
|
|
• |
|
general economic conditions in the United States and abroad. |
In addition, the stock market in general, or the market for stocks in our industry or industries related to our industry, may experience extreme volatility unrelated to the operating performance of the issuer. These broad market fluctuations may adversely affect the trading price or liquidity of our common stock. In the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action or other litigation against the issuer. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our management would be diverted from the operation of our business.
We are an “emerging growth company,” as defined in the JOBS Act, and as a result of the reduced disclosure and governance requirements applicable to emerging growth companies, our common stock may be less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) of 2018, (b) in which we have total annual gross revenue of at least $1.0 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million as of the prior June 30, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
If we sell shares of our common stock in future financings, stockholders may experience immediate dilution and, as a result, our stock price may decline.
We may from time to time issue additional shares of common stock at a discount from the current trading price of our common stock. As a result, our stockholders would experience immediate dilution upon the sale of any shares of our common stock at such discount. In addition, as opportunities present themselves, we may enter into financing or similar arrangements in the future, including the
41
issuance of debt securities, preferred stock or common stock. If we issue common stock or securities convertible into common stock, our common stockholders would experience additional dilution and, as a result, our stock price may decline.
Our principal stockholders and management own a significant percentage of our stock and will be able to influence matters subject to stockholder approval.
As of March 10, 2016, our executive officers, directors, holders of 5% or more of our capital stock and their respective affiliates beneficially owned approximately 50% of our voting stock. These stockholders will have the ability to influence us through this ownership position. For example, these stockholders may be able to influence elections of directors, amendments of our organizational documents, or approvals of any merger, sale of assets or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that stockholders may believe to be in their best interest.
Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management.
Our restated certificate of incorporation and amended and restated bylaws contain provisions that could delay or prevent changes in control or changes in our management without the consent of our Board of Directors. These provisions include the following:
|
|
• |
|
a classified Board of Directors with three-year staggered terms, which may delay the ability of stockholders to change the membership of a majority of our Board of Directors; |
|
|
• |
|
no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; |
|
|
• |
|
the exclusive right of our Board of Directors to elect a director to fill a vacancy created by the expansion of the Board of Directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our Board of Directors; |
|
|
• |
|
the ability of our Board of Directors to authorize the issuance of shares of preferred stock and to determine the terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquirer; |
|
|
• |
|
the ability of our Board of Directors to alter our bylaws without obtaining stockholder approval; |
|
|
• |
|
the required approval of the holders of at least two-thirds of the shares entitled to vote at an election of directors to adopt, amend or repeal our bylaws or repeal the provisions of our restated certificate of incorporation regarding the election and removal of directors; |
|
|
• |
|
a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders; |
|
|
• |
|
the requirement that a special meeting of stockholders may be called only by the chairman of the Board of Directors, the chief executive officer, the president or the Board of Directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; and |
|
|
• |
|
advance notice procedures that stockholders must comply with in order to nominate candidates to our Board of Directors or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of us. |
In addition, these provisions would apply even if we were to receive an offer that some stockholders may consider beneficial.
We are also subject to the anti-takeover provisions contained in Section 203 of the Delaware General Corporation Law. Under Section 203, a corporation may not, in general, engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other exceptions, the Board of Directors has approved the transaction.
We do not currently intend to pay dividends on our common stock, and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our common stock.
We do not currently intend to pay any cash dividends on our common stock for the foreseeable future. We currently intend to invest our future earnings, if any, to fund our growth. Additionally, the terms of our Loan and Security Agreement restrict our ability to pay dividends. Therefore, you are not likely to receive any dividends on your common stock for the foreseeable future. Since we do not intend to pay dividends, your ability to receive a return on your investment will depend on any future appreciation in the market value of our common stock. There is no guarantee that our common stock will appreciate or even maintain the price at which our holders have purchased it.
42
Item 1B. Unresolved Staff Comments
Not applicable.
Our corporate headquarters is located in Leawood, Kansas, where we lease and occupy approximately 17,600 square feet of office space pursuant to a lease that expires on February 28, 2021. We also maintain additional corporate office space of approximately 2,300 square feet in Boston, Massachusetts pursuant to a lease that expires on August 31, 2016.
Additionally we lease approximately 3,300 square feet of office, laboratory and manufacturing space in San Diego, California that expires on May 31, 2016, and we lease approximately 8,000 square feet of office and research laboratory space in Leuven, Belgium that expires on October 30, 2018.
We are not currently a party to any material legal proceedings.
Item 4. Mine Safety Disclosures
Not applicable.
Item 5. Market For Registrant’s Common Equity, Related Stockholder Matters, and Issuer Purchases of Equity Securities
Market Information
Our common stock has been publicly traded on the NASDAQ Global Select Market under the symbol “PETX” since our initial public offering on June 26, 2013. The following table sets forth, for the periods indicated, the high and low intraday sale prices of our common stock as reported by The NASDAQ Global Select Market.
|
High |
Low |
||||
|
2015 |
|||||
|
Fourth quarter |
$ |
9.48 |
$ |
5.20 | |
|
Third quarter |
$ |
19.99 |
$ |
7.71 | |
|
Second quarter |
$ |
16.92 |
$ |
11.69 | |
|
First quarter |
$ |
20.63 |
$ |
15.14 | |
|
High |
Low |
||||
|
2014 |
|||||
|
Fourth quarter |
$ |
18.16 |
$ |
9.20 | |
|
Third quarter |
$ |
17.94 |
$ |
9.60 | |
|
Second quarter |
$ |
18.94 |
$ |
11.12 | |
|
First quarter |
$ |
25.50 |
$ |
16.63 | |
As of March 10, 2016, there were approximately 66 holders of record and 35,355,266 shares of our common stock outstanding.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock. We intend to retain future earnings, if any, to finance the operation and expansion of our business and do not anticipate paying any cash dividends in the foreseeable future. In addition, unless waived, the terms of our loan agreement with Pacific Western Bank and Oxford Finance LLC limit our ability to pay cash dividends. Any future determination related to dividend policy will be made at the discretion of our Board of Directors after considering our financial condition, results of operations, capital requirements, business prospects and other factors the Board of Directors deems relevant, and subject to the restrictions contained in our current or future financing instruments.
43
Unregistered Sales of Equity Securities
None.
Repurchases of Common Stock
The repurchase activity for the three months ended December 31, 2015, was as follows:
|
Total number of |
Maximum number of |
|||||||||
|
Total number |
Average |
shares purchased |
shares that may yet be |
|||||||
|
of shares |
price paid |
as part of publicly |
purchased under the |
|||||||
|
purchased |
per share |
announced plan or program |
plan or program |
|||||||
|
October 1 - October 31 |
859 |
(1) |
$ |
8.37 |
— |
N/A |
||||
|
November 1 - November 30 |
— |
— |
— |
N/A |
||||||
|
December 1 - December 31 |
— |
— |
— |
N/A |
||||||
|
Total |
859 |
$ |
8.37 |
— |
N/A |
|||||
|
(1) |
For the three months ended December 31, 2015, 859 shares of restricted stock were withheld to satisfy employee tax withholding obligations arising in conjunction with the vesting of restricted stock pursuant to our 2013 Incentive Award Plan. |
44
Stock Performance Graph
This performance graph shall not be deemed “soliciting material” or to be “filed” with the SEC for purposes of Section 18 of the Securities Exchange Act of 1934, as amended the (“Exchange Act”), or otherwise subject to the liabilities under that Section, and shall not be deemed to be incorporated by reference into any of our filings under the Securities Act of 1933, as amended, or the Exchange Act.
The following graph shows a comparison from June 27, 2013 (the date our common stock commenced trading on the NASDAQ Global Select Market) through December 31, 2015 of the cumulative total return for our common stock, the NASDAQ Composite Index (the “NASDAQ Composite”), the Standard & Poor’s 500 Stock Index (the “S&P 500”), and the NASDAQ Biotechnology Index (the “NBI”). The graph assumes that $100 was invested at the market close on June 27, 2013 in the common stock of Aratana Therapeutics, Inc., the NASDAQ Composite, the S&P 500 and the NBI and data for the NASDAQ Composite, the S&P 500, and the NBI assumes reinvestments of dividends. The stock price performance of the following graph is not necessarily indicative of future stock price performance.
45
Item 6. Selected Financial Data
The following tables set forth selected consolidated financial data of our company as of and for each of the years in the five-year period ended December 31, 2015, and should be read in conjunction with Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and notes thereto included elsewhere in this Annual Report on Form 10-K.
We have derived the consolidated statements of operations for the years ended December 31, 2015, 2014 and 2013 and the consolidated balance sheet data as of December 31, 2015 and December 31, 2014 from our audited consolidated financial statements included in this Annual Report on Form 10-K in Item 8. “Financial Statements and Supplementary Data.” The selected historical balance sheet data as of December 31, 2013, December 31, 2012 and December 31, 2011, presented below has been derived from our audited financial statements not included in this 2015 Annual Report. The revenue data for the years ended December 31, 2012 and 2011 is derived from our audited combined financial statements not included in this 2015 Annual Report.
For a discussion of certain factors that materially affect the comparability of the selected consolidated financial data or cause the data reflected herein not to be indicative of our future results of operations or financial condition, see Item 1A. “Risk Factors”, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and notes of our consolidated financial statements included elsewhere in this report.
|
YEAR ENDED DECEMBER 31, |
|||||||||||||||
|
2015 |
2014 |
2013 |
2012 |
2011 |
|||||||||||
|
( in thousands) |
|||||||||||||||
|
Revenues: |
|||||||||||||||
|
Licensing and collaboration revenue |
$ |
— |
$ |
500 |
$ |
15 |
$ |
— |
$ |
— |
|||||
|
Product sales |
678 | 267 | 108 |
— |
— |
||||||||||
|
Total revenues |
678 | 767 | 123 |
— |
— |
||||||||||
|
Cost and expenses |
|||||||||||||||
|
Cost of product sales |
365 | 333 | 108 |
— |
— |
||||||||||
|
Royalty expense |
84 | 72 | 1 |
— |
— |
||||||||||
|
Research and development |
24,964 | 19,985 | 10,925 | 7,291 | 2,196 | ||||||||||
|
Selling, general and administrative |
19,819 | 17,938 | 8,572 | 2,987 | 1,274 | ||||||||||
|
In-process research and development |
— |
2,157 |
— |
1,500 |
— |
||||||||||
|
Amortization of acquired intangible assets |
1,544 | 1,891 | 298 |
— |
— |
||||||||||
|
Impairment of acquired intangible assets |
43,398 |
— |
— |
— |
— |
||||||||||
|
Total costs and expenses |
90,174 | 42,376 | 19,904 | 11,778 | 3,470 | ||||||||||
|
Loss from operations |
(89,496) | (41,609) | (19,781) | (11,778) | (3,470) | ||||||||||
|
Other income (expense) |
|||||||||||||||
|
Interest income |
189 | 123 | 75 | 21 | 6 | ||||||||||
|
Interest expense |
(1,585) | (1,060) | (432) |
— |
— |
||||||||||
|
Other income (expense), net |
5,140 | 2,287 | 478 | 121 |
— |
||||||||||
|
Total other income (expense) |
3,744 | 1,350 | 121 | 142 | 6 | ||||||||||
|
Loss before income taxes |
(85,752) | (40,259) | (19,660) | (11,636) | (3,464) | ||||||||||
|
Income tax benefit |
1,698 | 1,443 | 12,722 | ||||||||||||
|
Net loss |
$ |
(84,054) |
$ |
(38,816) |
$ |
(6,938) |
$ |
(11,636) |
$ |
(3,464) | |||||
|
Modification of Series A convertible preferred stock |
— |
— |
— |
— |
(276) | ||||||||||
|
Unaccreted dividends on convertible preferred stock |
— |
— |
— |
(2,035) | (902) | ||||||||||
|
Net loss attributable to common stockholders(1) |
$ |
(84,054) |
$ |
(38,816) |
$ |
(6,938) |
$ |
(13,671) |
$ |
(4,642) | |||||
|
Net loss per share attributable to common stockholders, basic and diluted |
$ |
(2.45) |
$ |
(1.30) |
$ |
(0.63) |
$ |
(34.53) |
$ |
(15.43) | |||||
|
Weighted average shares outstanding, basic and diluted |
34,355,525 | 29,767,429 | 11,059,382 | 395,918 | 300,841 | ||||||||||
__________________
46
|
(1) |
Net loss available to common stockholders and basic and diluted net loss per common share are computed consistent with annual per share calculations described in Notes 2 and 15 of our consolidated financial statements included elsewhere in this Annual Report on Form 10-K. |
|
AS OF DECEMBER 31, |
|||||||||||||||
|
2015 |
2014 |
2013 |
2012 |
2011 |
|||||||||||
|
(in thousands) |
|||||||||||||||
|
Consolidated Balance Sheet Data: |
|||||||||||||||
|
Cash, cash equivalents and short-term investments |
$ |
86,202 |
$ |
98,072 |
$ |
45,754 |
$ |
20,355 |
$ |
12,384 | |||||
|
Working capital(1) |
83,335 | 90,441 | 31,307 | 17,546 | 11,720 | ||||||||||
|
Total assets |
147,066 | 207,903 | 112,343 | 21,222 | 12,573 | ||||||||||
|
Total long-term debt, net of discount |
39,710 | 14,963 | 9,310 |
— |
— |
||||||||||
|
Total convertible preferred stock(2) |
— |
— |
— |
39,197 | 22,155 | ||||||||||
|
Total stockholders’ equity (deficit) |
$ |
101,550 |
$ |
181,832 |
$ |
83,390 |
$ |
(21,555) |
$ |
(10,271) | |||||
__________________
|
(1) |
We define working capital as current assets less current liabilities. |
|
(2) |
Consists of our Series A, A-1, B and C preferred stock. |
47
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and the related notes and other financial information included elsewhere in this Annual Report on Form 10-K. Some of the information contained in this discussion and analysis or set forth elsewhere in this annual report, including information with respect to our plans and strategy for our business, and expectations regarding product development and licensing, includes forward-looking statements that involve risks and uncertainties. You should review the “Risk Factors” section of this annual report for a discussion of important factors that could cause our actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis or elsewhere in this annual report.
Overview
We are a pet therapeutics company focused on licensing, developing and commercializing innovative biopharmaceutical products for companion animals. We operate in one business segment: pet therapeutics. Our current product portfolio includes multiple therapeutic candidates in development consisting of small molecule pharmaceuticals and large molecule biologics that target large opportunities in serious medical conditions in pets.
Our lead products in development include small molecules directed at treating osteoarthritis pain and inflammation (GALLIPRANT), appetite stimulation (ENTYCE) and post-operative pain (NOCITA).
We have incurred significant net losses since our inception. We incurred net losses of $84.1 million, $38.8 million and $6.9 million for the years ended December 31, 2015, 2014, and 2013, respectively. These losses have resulted principally from costs incurred in connection with in-licensing our product candidates, research and development activities and general and administrative costs associated with our operations. As of December 31, 2015, we had a deficit accumulated since inception of $152.0 million and cash, cash equivalents and short-term investments of $86.2 million.
We expect to continue to incur operating losses for the next several years as we work to develop and commercialize our product candidates. As a result, we will seek to fund our operations through corporate collaborations and licensing arrangements, as well as public or private equity offerings or further debt financings. We cannot assure you that such funds will be available on terms favorable to us, if at all. Arrangements with collaborators or others may require us to relinquish rights to certain of our technologies or product candidates. In addition, there is a risk that we may never successfully complete development of our product candidates, obtain adequate patent protection for our technology, obtain necessary regulatory approval for our product candidates or achieve commercial viability for any approved product candidates. If we are not able to raise additional capital on terms acceptable to us, or at all, as and when needed, we may be required to curtail our operations which could include delaying the commercial launch of our products, discontinuing product development programs, or granting rights to develop and market products or product candidates that we would otherwise prefer to develop and market ourselves. As disclosed in Note 10 to the consolidated financial statements, we have a term loan and revolving credit facility with a principal balance of $40.0 million as of December 31, 2015. The terms of this agreement require us to receive unrestricted net cash proceeds of at least $45.0 million from partnering transactions and/or the issuance of equity securities from October 16, 2015 to October 16, 2016. The loan agreements also require that we have at least three products fully USDA-or FDA-approved for commercialization by December 31, 2016. If these conditions are not met, the Company may be required to repay the loan prior to December 31, 2016. As of the date of the filing of this Annual Report on Form 10-K, we believe that our existing cash, cash equivalents and short-term investments of $86.2 million, will allow us to fund our operations, including, if necessary, the repayment of our long-term debt, at least through December 31, 2016.
For more information regarding our business and the animal health industry, see Item 1. “Business”.
Recent Developments
GALLIPRANT Files in U.S.
On January 25, 2016, we filed an administrative NADA with the CVM for GALLIPRANT. The ADUFA date for approval has been set for March 25, 2016. If approved, we anticipate commercial availability of GALLIPRANT to veterinarians in the fall of 2016.
GALLIPRANT Files in Europe
On February 17, 2016, we filed Marketing Authorization Application with the EMA for GALLIPRANT. The EMA has started reviewing the submission and if approved, we anticipate marketing authorization in 2017.
Technical Section Complete Letters for Effectiveness
In February 2016 we received technical section complete letters for effectiveness from the CVM for both ENTYCE and NOCITA. The technical section complete letter for effectiveness is one of three major technical section complete letters required to file an administrative NADA for approval.
For more information regarding research and development, manufacturing and sales and marketing refer to applicable sections in Item 1. “Business”.
48
Financial Overview
Revenues
Licensing and collaboration revenue consist solely as a result of research and development services performed with AT-006.
Product sales consist primarily of our caninized monoclonal antibody, TACTRESS, which was offered to veterinary oncology practices during 2015.
Costs and expenses
Cost of product sales consists primarily of the cost of direct materials, direct labor, and overhead costs associated with the manufacturing of our products.
Royalty expense consists of royalty expenses associated with third-party intellectual property. In 2014 and 2015, royalty expense includes third party royalties for licensed technologies pertaining to BLONTRESS and TACTRESS. Royalties are either a minimum amount per year or a percentage of net product sales.
Research and development expenses consist primarily of costs associated with our product development efforts (new product R&D and product lifecycle development), overhead costs associated with R&D and expenses related to regulatory approval of our products. Product development efforts costs consist primarily of contracted development costs, manufacturing costs, wages, stock-based compensation, and employee benefits for all employees engaged in scientific research and development functions. Overhead costs associated with R&D consists of other operational costs related to our research and development activities, including facility-related expenses, license payments made under our licensing agreements, regulatory, professional and consulting fees, travel costs, and allocated corporate costs.
We have been developing our lead programs in parallel and typically use our employee and infrastructure resources across multiple development programs. We track contracted development costs by development compound but do not allocate personnel or other internal costs related to development to specific programs or development compounds. These expenses are included in personnel costs and other internal costs, respectively.
Selling, general and administrative expenses consist primarily of personnel costs, including salaries, related benefits and stock-based compensation for employees in commercial, administration, finance, information technology, human resources, legal, and business development. Selling, general and administrative expenses also includes allocated rent and other facilities costs; conference and sponsorship activities, information technology services, professional and consulting fees for general and commercial business purposes, for accounting and tax services, business development activities, general legal services; and travel and other costs.
In-process research and development (“IPR&D”) expense consists of costs associated with acquired in-licensed technology that has not reached technological feasibility in animal health indications and has no alternative future use in the field of animal health, including upfront payments.
Amortization of acquired intangible assets consists primarily of the amortization expense for identifiable finite-lived intangible assets that have been acquired through business combinations. These assets consist of, but are not limited to intellectual property rights for currently marketed products.
Impairment of acquired intangible assets consists primarily of impairment charges for intangible assets that have been acquired through business combinations whose carrying amounts exceeded their fair value.
Other Income (Expense)
Interest income consists of interest earned on our cash, cash equivalents and short-term investments.
Interest expense consists of interest incurred on our borrowings.
A more detailed description of our Loan and Security Agreement is available under the caption “Liquidity and Capital Resources.”
Other income (expense), net consist primarily of various items including net gains (losses) on asset disposals, grant income from various government programs, and net gains (losses) on derivative financial instruments and marketable securities.
Income tax benefit is a result of the increased losses in 2015, limited to the amount of future taxable income from the reversal of taxable temporary differences that arose from the Okapi Sciences NV acquisition during 2014.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of our consolidated financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, revenues, costs and expenses and related disclosures during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those described below. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for
49
making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our consolidated financial statements appearing elsewhere in this filing, we believe that the estimates and assumptions involved in the following accounting policies may have the greatest potential impact on our consolidated financial statements.
Revenue Recognition
We recognize revenue when all of the following conditions are met:
|
· |
there is persuasive evidence of an agreement or arrangement; |
|
· |
delivery of products has occurred or services have been rendered; |
|
· |
the seller’s price to the buyer is fixed or determinable; and |
|
· |
collectibility is reasonably assured. |
Our principal revenue streams and their respective accounting treatments are discussed below:
(i) Product sales - Revenue for the sale of products is recognized when delivery has occurred and substantially all the risks and rewards of ownership have been transferred to the customer. Revenue for the sale of products are recorded net of sales returns, allowances and discounts.
(ii) Licensing and collaboration revenue - Revenues derived from product out-licensing arrangements typically consist of an initial up-front payment on inception of the license and subsequent milestone payments contingent on the achievement of certain clinical and sales milestones. Product out licensing arrangements may require us to provide multiple deliverables to the licensee which would require the total selling price to be allocated between each of the separable elements in the arrangement based on their relative selling prices. An element is considered separable if it has value to the customer on a stand-alone basis. The selling price used for each separable element will be based on vendor specific objective evidence (“VSOE”) if available, third party evidence if VSOE is not available, or estimated selling price if neither VSOE nor third party evidence is available. Revenue is then recognized as each of the separable elements to which the revenue has been allocated is delivered.
Milestone payments which are non-refundable, non-creditable and contingent on achieving certain development, regulatory, or commercial milestones are recognized as revenues either on achievement of such milestones or over the period we have continuing substantive performance obligations.
Research and Development
As part of the process of preparing our consolidated financial statements, we are required to estimate accrued research and development expenses. Examples of estimated accrued expenses include fees paid to clinical research organizations (“CROs”), in connection with target animal studies, to investigative sites in connection with target animal studies, to contract manufacturers in connection with the production of active pharmaceutical ingredient, and formulated drug, and to other parties for outsourced chemistry services.
We review new and open contracts and communicate with applicable internal and vendor personnel to identify services that have been performed on our behalf and estimate the level of service performed and the associated costs incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost for accrued expenses. The majority of our service providers invoice us monthly in arrears for services performed or as milestones are achieved in relation to our contract manufacturers. We make estimates of our accrued expenses as of each consolidated balance sheet date.
We base our accrued expenses related to target animal studies on our estimates of the services received and efforts expended pursuant to contracts with CROs that conduct and manage target animal studies on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of animals and the completion of development milestones. We estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the related expense accrual accordingly on a prospective basis. If we do not identify costs that have been incurred or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates. To date, we have not made any material adjustments to our estimates of accrued research and development expenses or the level of services performed in any reporting period presented in this document.
Impairment of Intangible Assets and Goodwill
Indefinite-lived IPR&D intangible assets are assessed for impairment at least annually. In addition, all intangible assets are reviewed for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. Factors that we consider in deciding when to perform an impairment review include significant underperformance of the business in relation to expectations, significant negative industry or economic trends, and significant changes or planned changes in the use of the assets. If an impairment review is performed to evaluate a long-lived asset for recoverability, we compare forecasts of
50
undiscounted cash flows for definite-lived intangible assets and discounted cash flows for indefinite-lived IPR&D intangible assets expected to result from the use and eventual disposition of the long-lived asset to its carrying value. An impairment loss would be recognized when estimated undiscounted (definite-lived) or discounted (indefinite-lived) future cash flows expected to result from the use of an asset are less than its carrying amount. The impairment loss would be based on the excess of the carrying value of the impaired asset over its fair value, determined based on discounted cash flows. In the quarter ended September 30, 2015 and to date, the Company has recorded $43.4 million of impairment losses on intangible assets (see Note 8 to our consolidated financial statements).
We completed our annual indefinite-lived IPR&D intangible assets impairment testing during the fourth quarter of 2015. We elected to bypass the qualitative assessment. For purposes of impairment testing, the fair value of the indefinite-lived IPR&D intangible assets was determined by using the framework of ASC 820, Fair Value Measurement. When determining the fair value of the indefinite-lived IPR&D intangible assets, we revisited all assumptions used in measuring the indefinite-lived IPR&D intangible assets at the time of acquisition, and evaluated and considered new and updated data and information available. We noted the fair values for all indefinite-lived IPR&D intangible assets were greater than the carrying value. As such, no impairment was recognized during the fourth quarter of 2015.
We completed our annual goodwill impairment testing during the third quarter of 2015. We elected to bypass the qualitative assessment. We determined as of the testing date that we consisted of one operating segment which is comprised of one reporting unit. In performing step one of the assessment, we determined that our fair value, determined to be our market capitalization, was greater than our carrying value, determined to be stockholder’s equity. Based on this result, step two of the assessment was not required to be performed, and we determined there was no impairment of goodwill during the third quarter of 2015.
During the fourth quarter of 2015, we completed an interim goodwill impairment assessment due to a decline in our market capitalization. In performing step one of the assessment, we determined that fair value exceeded our carrying value by 93%. Based on this result, step two of the assessment was not required to be performed, and we determined there was no impairment of goodwill during the fourth quarter of 2015.
Subsequent to December 31, 2015, we experienced further declines and fluctuations in our market capitalization. The extent and duration of any decline in our market capitalization after December31, 2015, will be assessed in future periods against our carrying value to assess if there is an indication of impairment of our goodwill which was $39.8 million as of December 31, 2015.
Stock-Based Compensation
We measure stock-based awards granted to employees and directors at fair value on the date of grant and recognize the corresponding compensation expense of those awards, net of estimated forfeitures, over the requisite service period, which is generally the vesting period of the respective award. Stock-based compensation related to restricted stock awards is based on the market value of our common stock on the date of grant and is recognized as expense, net of forfeitures, ratably over the requisite service period. Generally, we issue stock-based awards with only service-based vesting conditions and record compensation expense for these awards using the straight-line method. Our intention is to grant stock-based awards with exercise prices equivalent to the fair value of our common share as of the date of grant.
We account for all stock-based awards issued to non-employees based on the fair value of the award on each measurement date. Stock-based awards granted to non-employees are subject to revaluation at each reporting date over their vesting terms. As a result, the charge to operations for non-employee awards with vesting conditions is affected each reporting period by changes in the fair value of our common stock.
The fair value of each stock-based award is estimated using the Black-Scholes option-pricing model. Prior to 2014, due to the lack of company-specific historical and implied volatility information, we estimated our expected volatility based on the historical volatility of our publicly-traded peer companies. Beginning in the first quarter of 2014 we began to base expected volatility on the historical volatility of our common stock as adequate historical data regarding the volatility of our common stock price had become available. The expected term of our awards has been determined utilizing the “simplified” method as we do not have sufficient historical experience for option grants overall, rendering existing historical experience irrelevant to expectations for current grants. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is based on the fact that we have never paid cash
51
dividends and do not expect to pay any cash dividends in the foreseeable future. The assumptions we used to determine the fair value of stock-based compensation granted in each period were as follows, presented on a weighted average basis:
|
YEAR ENDED DECEMBER 31, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Risk-free interest rate |
1.38 |
% |
1.88 |
% |
1.59 |
% |
||||||
|
Expected term (in years) |
6.1 | 6.1 | 6.1 | |||||||||
|
Expected volatility |
70 |
% |
84 |
% |
66 |
% |
||||||
|
Expected dividend yield |
— |
% |
— |
% |
— |
% |
||||||
These assumptions represent our best estimates, but the estimates involve inherent uncertainties and the application of our judgment. As a result, if factors change and we use significantly different assumptions or estimates, our stock-based compensation expense could be materially different. We recognize compensation expense for only the portion of awards that are expected to vest. In developing a forfeiture rate estimate, we have considered our historical experience to estimate pre-vesting forfeitures. If our actual forfeiture rate is materially different from the estimate, our stock-based compensation expense could be different from what we have recorded in the current period. We had an aggregate of $10.4 million and $4.6 million of unrecognized stock-based compensation expense for options outstanding and restricted stock awards, respectively, as of December 31, 2015, which is expected to be recognized over a weighted-average of 2.37 years for stock options and 1.68 years for restricted stock.
Business Combinations
Our business acquisitions were made at a price above the fair value of the assets acquired and liabilities assumed, resulting in goodwill, based on our expectations of synergies and other benefits of combining the businesses. These synergies and benefits include elimination of redundant facilities, functions and staffing; use of our existing commercial infrastructure to expand sales of the products of the acquired businesses; and use of the commercial infrastructure of the acquired businesses to expand product sales in a cost-efficient manner.
Significant judgment is required in estimating the fair value of intangible assets and in assigning their respective useful lives. The fair value estimates are based on available historical information and on future expectations and assumptions deemed reasonable by management, but which are inherently uncertain.
We generally employ the income method to estimate the fair value of intangible assets, which is based on forecasts of the expected future cash flows attributable to the respective assets. Significant estimates and assumptions inherent in the valuations reflect a consideration of other marketplace participants, and include the amount and timing of future cash flows (including expected growth rates and profitability), the underlying product life cycles, economic barriers to entry, a brand’s relative market position and the discount rate applied to the cash flows. Unanticipated market or macroeconomic events and circumstances may occur, which could affect the accuracy or validity of the estimates and assumptions.
Net assets acquired are recorded at their fair value and are subject to adjustment upon finalization of the fair value analysis. We are not aware of any information that indicates the final fair value analysis will differ materially from the preliminary estimates.
Contingent consideration is recorded as a liability and measured at fair value using a discounted cash flow model utilizing significant unobservable inputs, including the probability of achieving each of the potential milestones and an estimated discount rate commensurate with the risks of the expected cash flows attributable to the milestones. Significant increases or decreases in any of the probabilities of success would result in a significantly higher or lower fair value, respectively, and commensurate changes to this liability. At each reporting date, we revalue the contingent consideration obligations to the reporting date fair values and record increases and decreases in the fair values as income or expense in the consolidated statements of operations until actual settlement occurs.
Increases or decreases in the fair values of the contingent consideration obligations may result from changes in discount periods and rates, changes in the timing and amount of earn-out criteria and changes in probability assumptions with respect to the likelihood of achieving the various earn-out criteria.
On January 6, 2014, we acquired Okapi Sciences, a Leuven, Belgium based company with a proprietary antiviral platform and three clinical/development stage product candidates. The aggregate purchase price was approximately $44,439, which consisted of $14,139 in cash, a promissory note in the principal amount of $15,134 with a maturity date of December 31, 2014, and a contingent consideration of up to $16,308 with an acquisition fair value of $15,166. The promissory note bore interest at a rate of 7% per annum, payable quarterly in arrears, and was subject to mandatory prepayment in the event of a specified equity financing by us. On February 4, 2014, the promissory note and accrued interest was paid in cash in the amount of $15,158. On March 17, 2014, the contingent consideration was settled in cash in the amount of $15,235.
52
Income Taxes
We account for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the consolidated financial statements or in our tax returns. Deferred taxes are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect in the years in which the differences are expected to reverse. Changes in deferred tax assets and liabilities, other than those arising from business combinations, are recorded in the provision for income taxes. We assess the likelihood that our deferred tax assets will be recovered from future sources of taxable income and, to the extent we believe, based upon the weight of available evidence, that it is more likely than not that all or a portion of deferred tax assets will not be realized, a valuation allowance is established through a charge to income tax expense. Potential for recovery of deferred tax assets is evaluated by estimating the future taxable profits expected and considering prudent and feasible tax planning strategies.
We account for uncertainty in income taxes recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the consolidated financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includes the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate as well as the related net interest and penalties.
JOBS Act
On April 5, 2012, the Jumpstart Our Business Startups Act (“JOBS Act”), was signed into law. The JOBS Act contains provisions that, among other things, reduce certain reporting requirements for an “emerging growth company.” As an “emerging growth company” we are electing not to take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision not to take advantage of the extended transition period is irrevocable.
In addition, we are in the process of evaluating the benefits of relying on the other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act, if as an “emerging growth company” we choose to rely on such exemptions, we may not be required to, among other things, (i) provide an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404, and (ii) comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the consolidated financial statements (auditor discussion and analysis). These exemptions will apply for a period of five years following the completion of our initial public offering, which such fifth anniversary will occur in 2018, or until we no longer meet the requirements of being an “emerging growth company,” whichever is earlier.
53
Results of Operations
Comparison of the Years Ended December 31, 2015 and 2014
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Revenues: |
|||||||||
|
Licensing and collaboration revenue |
$ |
— |
$ |
500 | (100.0) |
% |
|||
|
Product sales |
678 | 267 | 153.9 |
% |
|||||
|
Total revenues |
678 | 767 | (11.6) |
% |
|||||
|
Costs and expenses: |
|||||||||
|
Cost of product sales |
365 | 333 | 9.6 |
% |
|||||
|
Royalty expense |
84 | 72 | 16.7 |
% |
|||||
|
Research and development |
24,964 | 19,985 | 24.9 |
% |
|||||
|
Selling, general and administrative |
19,819 | 17,938 | 10.5 |
% |
|||||
|
In-process research and development |
— |
2,157 | (100.0) |
% |
|||||
|
Amortization of acquired intangible assets |
1,544 | 1,891 | (18.4) |
% |
|||||
|
Impairment of acquired intangible assets |
43,398 |
— |
NA |
||||||
|
Other income (expense): |
|||||||||
|
Interest income |
189 | 123 | 53.7 |
% |
|||||
|
Interest expense |
(1,585) | (1,060) | 49.5 |
% |
|||||
|
Other income/(expense), net |
5,140 | 2,287 | 124.7 |
% |
|||||
|
Income tax benefit |
1,698 | 1,443 | 17.7 |
% |
|||||
Revenues
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Revenues: |
|||||||||
|
Licensing and collaboration revenue |
$ |
— |
$ |
500 | (100.0) |
% |
|||
|
Product sales |
678 | 267 | 153.9 |
% |
|||||
|
Total |
$ |
678 |
$ |
767 | (11.6) |
% |
|||
Total revenue decreased $0.1 million, or 12%, in 2015 compared to 2014. This decrease was primarily due to a decrease of $0.5 million in collaboration revenue related to AT-006 as a result of research collaboration completed under our license agreement with Elanco in 2014, while no research collaboration was completed in 2015, partially offset by an increase of $0.4 million in product sales of TACTRESS, driven by TACTRESS being made available to more customers during 2015 and being available for the entire year in 2015, unlike in 2014 when it was available for only three months. Given the clinical results from T-CHOMP, T-LAB, T-CEP and the scientific studies in September 2015, we believe that TACTRESS revenues will be modest. In the first quarter of 2015, we received a $3.0 million payment from Elanco Animal Health, Inc. (“Elanco”) relating to the milestone achieved when BLONTRESS
54
received a full license from the USDA. This was offset by $3.0 million in consideration we paid in the first quarter of 2015 for the return of the commercial and manufacturing license of BLONTRESS previously granted to Elanco. These amounts were netted resulting in zero licensing and collaboration revenue in the year ended December 31, 2015.
Cost of Product Sales
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Cost of product sales |
$ |
365 |
$ |
333 | 9.6 |
% |
|||
Cost of product sales increased $0.1 million, or 10%, in 2015 compared with 2014, primarily as result of increased sales volume of TACTRESS. In 2014, the cost of product sales included costs related to the sales of AT-004 which was sold to our former commercial licensee on a cost plus margin basis. No such sales occurred in 2015.
Research and development expense
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Contracted development costs |
$ |
17,589 |
$ |
11,758 | 49.6 |
% |
|||
|
Personnel costs |
5,726 | 5,624 | 1.8 |
% |
|||||
|
Other costs |
1,649 | 2,603 | (36.7) |
% |
|||||
|
Total research and development |
$ |
24,964 |
$ |
19,985 | 24.9 |
% |
|||
Research and development expense increased by $5.0 million in 2015 as compared to 2014. This increase was primarily due to a $5.8 million increase in contracted development costs as a result of the advancement of lead programs, as well as an increased number of programs and related studies, and a $0.1 million increase in personnel costs primarily related to increased staff, partially offset by a $1.0 million decrease in other costs related to supporting multiple facilities. During the year ended December 31, 2015, we made license payments associated with licensing additional technology, success milestone payments of $0.1 million for the AT-016 program, $0.5 million in milestone payments for the AT-018 program and $0.3 million for entering into the AT-Iota option program for periodontal disease in dogs.
We expect in 2016, research and development expenses to be related to technology transfer to contract manufacturing suppliers for commercial supply for our lead programs and regulatory expenses related to product filings. We also expect to make significant payments related to achievement of development and regulatory milestones. See Note 12 for further information on potential milestone payments.
55
Royalty expense
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Royalty expense |
$ |
84 |
$ |
72 | 16.7 |
% |
|||
Royalty expense increased $12,000 in 2015 as compared to 2014, primarily as a result in the increased sales of TACTRESS.
Selling, general and administrative expense
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Selling, general and administrative |
$ |
19,819 |
$ |
17,938 | 10.5 |
% |
|||
Selling, general and administrative expense increased by $1.9 million in 2015 as compared to 2014.
The increase is primarily due to activities in preparation for the commercialization of additional product candidates for which we anticipate regulatory approval in 2016, which included increased sales and marketing headcount and corporate infrastructure. We also incurred sponsorship and other expenses in conjunction with participation in several key veterinary conferences to build awareness of Aratana and our product candidates. The increase in selling, general and administrative expenses also includes $1.5 million as a result of increased stock-based compensation. This increase was partially offset by a credit of $1.2 million to reduce the fair value of the contingent consideration to zero as a result of the agreement with the Vet Therapeutics’ shareholders.
We expect selling, general and administrative expense to continue to increase as we build out our sales organization and corporate infrastructure in the support of the expected commercialization of GALLIPRANT, ENTYCE and NOCITA.
In-process research and development expense
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
In-process research and development expenses |
$ |
— |
$ |
2,157 | (100.0) |
% |
|||
In-process research and development expense decreased by $2.2 million in 2015 as compared to 2014. We did not acquire any in-process research and development programs during 2015. In 2014, we entered into exclusive license agreements with Advaxis, VetStem Biopharma and Atopix for $0.7 million, $0.5 million and $1.0 million, respectively.
56
Amortization of acquired intangible assets
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Amortization of acquired intangible assets |
$ |
1,544 |
$ |
1,891 | (18.4) |
% |
|||
Amortization of acquired intangible assets decreased by $0.3 million in 2015 as compared to 2014. The decrease reflects the impact of the impairment of BLONTREES and TACTRESS in 2015. The new carrying value resulting from the impairment charge resulted in the lower expense being recognized. Lower carrying values due to the 2015 impairment charges will result in lower amortization of acquired intangible assets for the impaired intangibles in future periods.
Impairment of acquired intangible assets
Impairment of acquired intangible assets expense was $43.4 million in 2015. No impairment charges were recognized in 2014. The impairment of acquired intangible expense is related to impairment charges of BLONTRESS ($20.2 million), TACTRESS ($8.6 million), AT-007 ($8.7 million), and AT-011 ($5.9 million) incurred in the third quarter of 2015. For more information regarding the impairment charges, see Note 8 to the consolidated financial statements.
Other income (expense)
Changes in the components of other income (expense) were as follows:
Interest income
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Interest income |
$ |
189 |
$ |
123 | 53.7 |
% |
|||
Interest income increased by $66,000 in 2015 as compared to 2014. The increase primarily relates to interest earned related to deposits held at Square 1 Bank and short-term investments in reverse repurchase agreements.
Interest expense
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Interest expense |
$ |
(1,585) |
$ |
(1,060) | 49.5 |
% |
|||
Interest expense increased by $0.5 million in 2015 as compared to 2014. This increase was due to interest expense related to our Loan and Security Agreement, as discussed below in, “Financial Condition, Liquidity and Capital Resources – Indebtedness”, which was entered into during October 2015. Accretion of the debt discount and deferred financing costs totaled $0.3 million, which is non-cash interest included in our interest expense above.
57
Other income
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Other income |
$ |
5,140 |
$ |
2,287 | 124.7 |
% |
|||
Other income increased by $2.9 million in 2015 as compared to 2014. The increase in 2015 is primarily related to the following non-recurring transactions: $3.5 million gain on the sale of Advaxis stock, $1.3 million gain related to the increase in fair value of the Advaxis warrant and a $0.3 million gain on the sale of shares received from the exercise of the Advaxis warrant.
The 2014 activity is related primarily to the successful development and delivery of the API to RaQualia. During the fourth quarter of 2014, we recognized $0.8 million of deferred income related to the payment received from RaQualia at execution and received another $0.8 million of income from the successful delivery. We also recorded other income of $63,000 associated with a research and development voucher program grant agreement with the Kansas Bioscience Authority (“KBA”).
Income tax benefit
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Income tax benefit |
$ |
1,698 |
$ |
1,443 | 17.7 |
% |
|||
Income tax benefit increased by $0.3 million in 2015 as compared to 2014. The income tax benefit recognized during 2015 and 2014 is due to losses incurred in Aratana NV. The increase in income tax benefit in 2015 is due to the increased losses in 2015, limited to
58
the amount of future taxable income from the reversal of taxable temporary differences that arose from the Okapi Sciences NV acquisition during 2014.
Comparison of the Years Ended December 31, 2014 and 2013
|
YEAR ENDED |
||||||||
|
DECEMBER 31, |
||||||||
|
2014 |
2013 |
% CHANGE |
||||||
|
(Dollars in thousands) |
||||||||
|
Revenues: |
||||||||
|
Licensing and collaboration revenue |
$ |
500 |
$ |
15 |
3,233% |
|||
|
Product sales |
267 | 108 |
147% |
|||||
|
Total |
767 | 123 |
524% |
|||||
|
Costs and expenses: |
||||||||
|
Cost of product sales |
333 | 108 |
208% |
|||||
|
Royalty expense |
72 | 1 |
7,100% |
|||||
|
Research and development |
19,985 | 10,925 |
83% |
|||||
|
Selling, general and administrative |
17,938 | 8,572 |
109% |
|||||
|
In-process research and development |
2,157 |
— |
NM |
|||||
|
Amortization of acquired intangible assets |
1,891 | 298 |
535% |
|||||
|
Other income |
||||||||
|
Interest income |
123 | 75 |
64% |
|||||
|
Interest expense |
(1,060) | (432) |
145% |
|||||
|
Other income |
2,287 | 478 |
378% |
|||||
|
Income tax benefit |
1,443 | 12,722 |
(89)% |
|||||
Revenues
|
YEAR ENDED |
||||||||
|
DECEMBER 31, |
||||||||
|
2014 |
2013 |
% CHANGE |
||||||
|
(Dollars in thousands) |
||||||||
|
Revenues: |
||||||||
|
Licensing and collaboration revenue |
$ |
500 |
$ |
15 |
3,233% |
|||
|
Product sales |
267 | 108 |
147% |
|||||
|
Total |
$ |
767 |
$ |
123 |
524% |
|||
Total revenue for the year ended December 31, 2014 was $0.8 million compared to total revenue of $0.1 million in 2013. This increase was primarily due to an increase of $0.5 million in collaboration revenue related to AT-006 as a result of research and development completed under our license agreement with Elanco and an increase of $0.1 million for BLONTRESS product sold at a cost plus margin basis to our former commercial licensee. In 2014, we also recorded $40,000 in product sales of TACTRESS, which
59
was granted conditional approval in early 2014. In 2013, we reported $0.1 million in revenue as a result of BLONTRESS product sold to Elanco.
During 2014, BLONTRESS for the treatment of B-cell lymphoma in dogs, was licensed in the United States and Canada to Elanco. Under the terms of the agreement, we received a royalty for commercial product sales and were responsible for the manufacturing of BLONTRESS. During 2014, we recorded $0.2 million in product sales for BLONTRESS. On February 24, 2015, we and our commercial licensee Elanco terminated by mutual consent an exclusive commercial license agreement for BLONTRESS in the U.S and Canada, reverting control of all commercial, development and manufacturing activities of BLONTRESS back to us. TACTRESS, our product for the treatment of T-cell lymphoma in dogs, was granted conditional approval in early 2014 and we commenced sales during the fourth quarter of 2014 recording product sales of $40,000.
Cost of product sales
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2014 |
2013 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Cost of product sales |
$ |
333 |
$ |
108 | 208 |
% |
|||
|
Total cost of product sales |
$ |
333 |
$ |
108 | 208 |
% |
|||
The cost of product sales includes direct material, direct labor, stock-based compensation and overhead costs associated with manufacturing products. During 2014, we manufactured and provided product to Elanco on a cost plus margin basis per the license agreement. During the fourth quarter of 2014, we began reporting the cost of the TACTRESS product for the commercial sales we generated during the quarter. Cost of product sales increased $0.2 million due to the increased volume of BLONTRESS sold to Elanco and we began selling TACTRESS during 2014.
Research and development expense
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2014 |
2013 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Contracted development costs |
$ |
11,758 |
$ |
6,527 | 80.1 |
% |
|||
|
Personnel costs |
5,624 | 2,676 | 110.2 |
% |
|||||
|
Other costs |
2,603 | 1,722 | 51.2 |
% |
|||||
|
Total research and development |
$ |
19,985 |
$ |
10,925 | 82.9 |
% |
|||
Research and development expense increased by $9.1 million for the year ended December 31, 2014 as compared to 2013. This increase was primarily due to a $5.2 million increase in contracted development costs as a result of the advancement of lead programs, as well as an increased number of programs and related studies, a $2.9 million increase in personnel costs primarily related to increased staff as a results of the Vet Therapeutics and Okapi Sciences acquisitions and a $0.9 million increase in other costs related to supporting multiple facilities and an increased number of programs under development. We expect research and development expense will increase for the foreseeable future as we continue to advance our portfolio of therapeutic candidates. At this time, due to the inherently unpredictable nature of our development, we cannot reasonably estimate or predict the nature, specific timing or estimated costs of the efforts that will be necessary to complete the development of our product candidates.
60
Royalty expense
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2014 |
2013 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Royalty expense |
$ |
72 |
$ |
1 | 7,100 |
% |
|||
|
Total royalty expense |
$ |
72 |
$ |
1 | 7,100 |
% |
|||
We are required to pay third party royalties for licensed technology associated with those products that are sold commercially. During 2013 and 2014, we recognized royalty expense for net product sales allocable to our monoclonal antibody targets and recombinant antibodies. In 2014, we recognized $72,000 in royalty expenses which includes an accrued minimum royalty of $70,000 related to the Crucell Commercial Agreement. During 2013, this minimum royalty expense was borne by Vet Therapeutics before the acquisition.
Selling, general and administrative expense
|
YEAR ENDED |
|||||||
|
DECEMBER 31, |
|||||||
|
2014 |
2013 |
% CHANGE |
|||||
|
(Dollars in thousands) |
|||||||
|
Selling, general and administrative |
$ |
17,938 |
$ |
8,572 | 109% | ||
Selling, general and administrative expense increased by $9.4 million for the year ended December 31, 2014 compared to the year ended December 31, 2013. This increase was primarily due to a $4.6 million increase in stock-based compensation expense due to increased stock prices and an increase in the number of grants provided to new employees, $1.6 million in costs associated with our commercial infrastructure and business development initiatives; and a $1.6 million increase in personnel-related costs, which was the result of increased staffing, employee benefits, travel and supplies.
In-process research and development expense
|
YEAR ENDED |
|||||||
|
DECEMBER 31, |
|||||||
|
2014 |
2013 |
% CHANGE |
|||||
|
(Dollars in thousands) |
|||||||
|
In-process research and development expenses |
$ |
2,157 |
$ |
— |
NM |
||
In-process research and development expense increased by $2.2 million for the year ended December 31, 2014. In 2014, we incurred $0.7 million, $0.5 million and $1.0 million of in-process research and development expense under our agreements with Advaxis, VetStem and Atopix, respectively. We did not acquire any in-process research and development programs during 2013, other than those acquired in the acquisition of Vet Therapeutics, which were capitalized.
61
Amortization of acquired intangible assets
|
YEAR ENDED |
|||||||
|
DECEMBER 31, |
|||||||
|
2014 |
2013 |
% CHANGE |
|||||
|
(Dollars in thousands) |
|||||||
|
Amortization of acquired intangible assets |
$ |
1,891 |
$ |
298 | 535% | ||
Amortization expense increased by $1.6 million for the year ended December 31, 2014. This increase is due to a full year of amortization of BLONTRESS and 11 months of amortization of TACTRESS. We began amortizing TACTRESS intangible over its 20 year estimated useful life during the period as conditional approval was received.
Other income (expense)
Changes in the components of other income (expense) were as follows:
Interest income
|
YEAR ENDED |
|||||||
|
DECEMBER 31, |
|||||||
|
2014 |
2013 |
% CHANGE |
|||||
|
(Dollars in thousands) |
|||||||
|
Interest income |
$ |
123 |
$ |
75 | 64% | ||
Interest income increased by $48,000 for the year ended December 31, 2014 compared to the year ended December 31, 2013. The increase primarily relates to interest earned related to deposits held at Square 1 Bank and short-term investments in reverse repurchase agreements. We also had increased deposits and investments in 2014 due to the additional proceeds acquired in public offerings in January 2014 and September 2014.
Interest expense
|
YEAR ENDED |
|||||||
|
DECEMBER 31, |
|||||||
|
2014 |
2013 |
% CHANGE |
|||||
|
(Dollars in thousands) |
|||||||
|
Interest expense |
$ |
(1,060) |
$ |
(432) | 145% | ||
Interest expense increased by $0.6 million for the year ended December 31, 2014 compared to the year ended December 31, 2013. This increase was due to interest expense related to our Credit Facility, which was entered into during March 2013 and notes payable associated with the acquisitions of Vet Therapeutics and Okapi Sciences. Accretion of the debt discount and deferred financing costs totaled $41,000, which is non-cash interest included in our interest expense above.
62
Other income
|
YEAR ENDED |
|||||||
|
DECEMBER 31, |
|||||||
|
2014 |
2013 |
% CHANGE |
|||||
|
(Dollars in thousands) |
|||||||
|
Other income |
$ |
2,287 |
$ |
478 | 378% | ||
Other income increased by $1.8 million during the year ended December 31, 2014 compared to the year ended December 31, 2013. This activity is related primarily to the successful development and delivery of the API to RaQualia. During the fourth quarter of 2014, we recognized $0.8 million of deferred income related to the payment received from RaQualia at execution and received another $0.8 million of income from the successful delivery. We also recorded other income associated with research and development voucher program grant agreement with the KBA, which was executed in March 2012. We were eligible to receive up to $1.3 million over an estimated two-year period, in the form of a quarterly reimbursement of 33% of costs incurred during that period for pre-formulation, formulation, manufacture and pivotal studies associated with the AT-001 and AT-002 programs, to the extent that such costs are incurred with specifically named Kansas companies. From inception through December 31, 2014, we have received $0.6 million under this agreement.
Income tax benefit
|
YEAR ENDED |
|||||||
|
DECEMBER 31, |
|||||||
|
2014 |
2013 |
% CHANGE |
|||||
|
(Dollars in thousands) |
|||||||
|
Income tax benefit |
$ |
1,443 |
$ |
12,722 |
(89)% |
||
Income tax benefit decreased by $11.3 million for the year ended December 31, 2014. This income tax benefit recognized in 2013 is due to the change in judgment on our existing valuation allowance against our deferred tax assets, following the Vet Therapeutics acquisition during 2013. The income tax benefit recognized during 2014 is due to losses incurred in Aratana NV.
Financial Condition, Liquidity and Capital Resources
Our financial condition is summarized as follows:
|
DECEMBER 31, 2015 |
DECEMBER 31, 2014 |
% CHANGE |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Financial assets: |
|||||||||
|
Cash and cash equivalents |
$ |
26,755 |
$ |
9,823 | 172.4 |
% |
|||
|
Marketable securities - short-term |
747 | 249 | 200.0 |
% |
|||||
|
Reverse repurchase agreements |
58,700 | 88,000 | (33.3) |
% |
|||||
|
Marketable securities - long-term |
— |
2,452 | (100.0) |
% |
|||||
|
Derivative financial instruments |
— |
1,108 | (100.0) |
% |
|||||
|
Total cash, cash equivalents, marketable securities and short-term investments |
$ |
86,202 |
$ |
101,632 | (15.2) |
% |
|||
|
Borrowings: |
|||||||||
63
|
Loan payable |
$ |
39,710 |
$ |
14,963 | 165.4 |
% |
|||
|
Working capital: |
|||||||||
|
Current assets |
$ |
89,019 |
$ |
99,909 | (10.9) |
% |
|||
|
Current liabilities |
5,684 | 9,468 | (40.0) |
% |
|||||
|
Total working capital |
$ |
83,335 |
$ |
90,441 | (7.9) |
% |
We have incurred significant net losses since our inception. We incurred net losses of $84.1 million, $38.8 million and $6.9 million for the years ended December 31, 2015, 2014, and 2013, respectively. These losses have resulted principally from costs incurred in connection with in-licensing our product candidates, research and development activities and general and administrative costs associated with our operations. As of December 31, 2015, we had an accumulated deficit of $152.0 million and cash, cash equivalents and short-term investments of $86.2 million.
We expect to continue to incur operating losses for the next several years as we work to develop and commercialize our product candidates. As a result, we will seek to fund our operations through corporate collaborations and licensing arrangements, as well as public or private equity offerings or further debt financings. If we are not able to raise additional capital on terms acceptable to us, or at all, as and when needed, we may be required to curtail our operations which could include delaying the commercial launch of our products, discontinuing product development programs, or granting rights to develop and market products or product candidates that we would otherwise prefer to develop and market ourselves. Our failure to raise capital as and when needed would have a negative impact on our financial condition and our ability to pursue our business strategies as this capital is necessary for us to perform the research and development and commercial activities required to generate future revenue streams. Arrangements with collaborators or others may require us to relinquish rights to certain of our technologies or product candidates. In addition, there is a risk that we may never successfully complete development of our product candidates, obtain adequate patent protection for our technology, obtain necessary regulatory approval for our product candidates or achieve commercial viability for any approved product candidates. As disclosed in Note 10 to the consolidated financial statements, we have a term loan and revolving credit facility with a principal balance of $40.0 million as of December 31, 2015. The terms of this agreement require us to receive unrestricted net cash proceeds of at least $45.0 million from partnering transactions and/or the issuance of equity securities from October 16, 2015 to October 16, 2016. The loan agreements also require that we have at least three products fully USDA-or FDA-approved for commercialization by December 31, 2016. If these conditions are not met, the Company may be required to repay the loan prior to December 31, 2016. As of the date of the filing of this Annual Report on Form 10-K, we believe that our existing cash and cash equivalents, short-term investments of $86.2 million, will allow us to fund our operations, including, if necessary, the repayment of our long-term debt, at least through December 31, 2016.
Cash, Cash Equivalents and Investments
Until required for another use in our business, we typically invest our cash reserves in bank deposits, certificates of deposit, and other interest bearing debt instruments in accordance with our investment policy. It is our policy to mitigate credit risk in our cash reserves and investments by maintaining a well-diversified portfolio that limits the amount of exposure as to institution, maturity, and investment type. The value of our investments, however, may be adversely affected by increases in interest rates, instability in the global financial markets that reduces the liquidity of securities included in our portfolio, and by other factors which may result in declines in the value of the investments. Each of these events may cause us to record charges to reduce the carrying value of our investment portfolio if the declines are other-than-temporary or sell investments for less than our acquisition cost which could adversely impact our financial position and our overall liquidity.
Sales Agreement
On October 16, 2015, we entered into a Sales Agreement with Barclays Capital Inc. (“Barclays”) pursuant to which we may sell from time to time, at our option, up to an aggregate of $52.0 million of shares of our common stock (the “Shares”) through Barclays, as sales agent. Sales of the Shares, if any, will be made under our previously filed and currently effective Registration Statement on Form S-3 (Reg. No. 333-197414), by means of ordinary brokers’ transactions on The NASDAQ Global Market or otherwise. Additionally, under the terms of the Sales Agreement, the Shares may be sold at market prices, at negotiated prices or at prices related to the prevailing market price. We will pay Barclays a commission of 2.75% of the gross proceeds from the sale of the Shares, if any. As of the date of this filing, we have not sold any shares pursuant to the Sales Agreement.
64
Indebtedness
On October 16, 2015, we and Vet Therapeutics (together the “Borrowers”) entered into a Loan and Security Agreement with Pacific Western Bank (“Pacific Western Bank”) as collateral agent (“Collateral Agent”) and a lender and Oxford Finance LLC as a lender (“Oxford” and together with Pacific Western Bank, the “Lenders”), pursuant to which the Lenders agreed to make available to the Borrowers, term loans in an aggregate principal amount up to $35.0 million (the “Term Loan”), and a revolving credit facility in an aggregate principal amount up to $5.0 million (the “Revolving Line” and together with the Term Loan, the “Credit Extensions”), subject to certain conditions to funding. A term loan was made on October 16, 2015 in an aggregate principal amount equal to $35.0 million, and an advance under the Revolving Line was made on October 16, 2015 in an aggregate principal amount equal to $5.0 million. The Borrowers are required to make interest-only payments on the Term Loan for 18 months, and beginning on May 1, 2017, are required to make payments of principal and accrued interest on the Term Loan in equal monthly installments over a term of 30 months. The interest-only period can be extended by one year to May 1, 2018 if the Borrowers have at least four products fully USDA- or FDA-approved, plus another product conditionally- or fully-approved, in each case for commercialization by December 31, 2016, and agree to certain other financial covenants with the Lenders. The Credit Extensions bear interest per annum at the greater of (i) 6.91% or (ii) 3.66% plus the prime rate, which is customarily defined. All principal and accrued interest on the Term Loan are due on October 16, 2019 (the “Term Loan Maturity Date”), and all principal and accrued interest on the Revolving Line are due on October 16, 2017 (the “Revolving Maturity Date”).
The Borrowers used approximately $15.0 million of the proceeds from the Credit Extensions to repay all the amounts owed under their Loan and Security Agreement, dated as of March 4, 2013, as amended, with Pacific Western Bank (as successor in interest to Square 1 Bank) and the lenders party thereto.
As security for their obligations under the Loan Agreement, the Borrowers granted a security interest in substantially all of their existing and after-acquired assets except for their intellectual property and certain other customary exclusions. Subject to customary exceptions, the Borrowers are not permitted to encumber their intellectual property.
Upon execution of the Loan Agreement, the Borrowers were obligated to pay a facility fee to the Lenders of $0.2 million, and an agency fee to the Collateral Agent of $0.1 million. In addition, the Borrowers are or will be obligated to pay a final payment fee equal to 3.30% of such Term Loan being prepaid or repaid with respect to the Term Loans upon the earliest to occur of the Term Loan Maturity Date, the acceleration of any Term Loan or the prepayment of a Term Loan. The Borrowers will also be obligated to pay a termination fee equal to 3.30% of the highest outstanding amount of the Revolving Line with respect to the Revolving Line upon the earliest to occur of the Revolving Maturity Date, the acceleration of the Revolving Line or the termination of the Revolving Line. The Borrowers will also be obligated to pay an unused-line fee equal to 0.25% per annum of the average unused portion of the Revolving Line.
The Loan Agreement contains customary representations and warranties and customary affirmative and negative covenants, including, among others, limits or restrictions on the Borrowers’ ability to incur liens, incur indebtedness, make certain restricted payments, make certain investments, merge, consolidate, make an acquisition, enter into certain licensing arrangements and dispose of certain assets. In addition, the Loan Agreement contains customary events of default that entitle the Lenders to cause the Borrowers’ indebtedness under the Loan Agreement to become immediately due and payable. The events of default, some of which are subject to cure periods, include, among others, a non-payment default, a covenant default, the occurrence of a material adverse change, the occurrence of an insolvency, a material judgment default, defaults regarding other indebtedness and certain actions by governmental authorities. Upon the occurrence and for the duration of an event of default, an additional default interest rate equal to 4% per annum will apply to all obligations owed under the Loan Agreement.
The Loan Agreement requires that the Borrowers receive unrestricted net cash proceeds of at least $45.0 million from partnering transactions and/or the issuance of equity securities from October 16, 2015 to October 16, 2016. The Loan Agreement also requires that the Borrowers have at least three products fully USDA- or FDA approved for commercialization by December 31, 2016. Additionally, the Loan Agreement requires that the Borrowers maintain certain minimum liquidity (approximately $20.0 million) at all times. At December 31, 2015, the Borrowers were in compliance with all financial covenants.
Working Capital
We define working capital as current assets less current liabilities. The decrease in working capital from December 31, 2014, reflects a decrease in total current assets of $10.9 million and a decrease in current liabilities of $3.8 million. The decrease in total current assets was primarily driven by a decrease in cash and cash equivalents and short-term investments. The decrease in total current liabilities is primarily a result of the reclassification of the current portion-loan payable to loan payable (long-term) and the payment of contingent consideration during 2015.
65
Cash Flows
The following table shows a summary of our cash flows for the periods set forth below:
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
(Dollars in thousands) |
|||||||||
|
Net cash used in operating activities |
$ |
(38,495) |
$ |
(32,188) |
$ |
(16,151) | |||
|
Net cash provided by (used in) investing activities |
$ |
33,663 |
$ |
(100,125) |
$ |
(29,235) | |||
|
Net cash provided by financing activities |
$ |
21,895 |
$ |
100,978 |
$ |
72,497 | |||
Net cash used in operating activities
During the year ended December 31, 2015, net cash used in operating activities was $38.5 million. We had a pretax loss of $84.1 million which includes a gain on sale of marketable securities of $3.9 million, an adjustment of a non-cash expense for stock-based compensation of $8.6 million, a non-cash depreciation and amortization expense of $1.8 million, and an impairment of intangible assets loss of $43.4 million, partially offset by a non-cash change in fair value of contingent consideration of $1.2 million, a non-cash change in fair value of derivative instruments of $1.3 million, and a non-cash deferred income tax benefit of $1.7 million. Our net losses were primarily attributed to the impairment of intangible assets, research and development activities related to our programs and our selling, general and administrative expenses. Net cash used in changes in our working capital consisted primarily of a decrease in accounts payable of $1.0 million, partially offset by an increase in accrued expenses and other liabilities of $0.7 million, and an increase in inventories of $0.5 million. The decrease in accounts payable primarily related to the timing of payments made for our outsourced research and development activities.
During the year ended December 31, 2014, net cash used in operating activities was $32.2 million. We had a pretax loss of $40.3 million. Net cash used in operating activities primarily resulted from the adjustments of a non-cash expense for stock-based compensation of $7.1 million, IPR&D expense of $2.2 million, non-cash depreciation and amortization expense of $2.0 million, non-cash deferred income tax benefit of $1.4 million and working capital changes of $2.7 million. Our net losses were primarily attributable to research and development activities related to our research and development programs and our selling, general and administrative expenses, offset by $0.8 million revenue in the period. Net cash used by changes in our working capital consisted primarily of a decrease of $1.1 million in accounts payable, a decrease of $0.9 million in deferred income, and increase $0.5 million in accrued expenses, offset by uses of cash related to increase of $0.6 million in prepaid expenses. The increase in accrued expenses primarily related to the timing of payments made for our contracted research and development activities. The increase in prepaid expenses relates primarily to research and development agreements.
During the year ended December 31, 2013, net cash used in operating activities was $16.2 million. We had a pretax loss of $19.7 million. Net cash used in operating activities primarily resulted from the non-cash deferred income tax benefit of $12.7 million and from the net loss of $6.9 million, which includes adjustments of a non-cash expense for stock-based compensation of $1.0 million and working capital changes of $1.8 million. Our net losses were primarily attributed to research and development activities related to our AT-001, AT-002, AT-003, BLONTRESS and TACTRESS programs and our general and administrative expenses, as we had no significant revenue in the period. Net cash provided by changes in our working capital consisted primarily of an increase of $1.0 million in accounts payable, an increase of $0.2 million in accrued expenses, offset by uses of cash related to increase of $0.2 million in prepaid expenses. The increase in accrued expenses primarily related to the timing of payments made for our contracted research and development activities. The increase in prepaid expenses relates primarily to research and development agreements.
Net cash provided by (used in) investing activities
During the year ended December 31, 2015, net cash provided by investing activities was $33.7 million, which related to $2,079 million from the proceeds of maturities of investments and $7.4 million from the sales of marketable securities, partially offset by $2,051 million for the purchase of investments and $2.2 million for purchases of property and equipment. The increase in cash as compared to 2014 was primarily due to increased investments in reverse repurchase agreements and the exercise of Advaxis warrant and subsequent sale of stock.
During the year ended December 31, 2014, net cash used in investing activities was $100.1 million, which related to the purchase of short-term investments for $371.2 million, as well as cash paid for the Okapi Sciences acquisition of $12.1 million, partially offset by proceeds from the maturities of investments of $286.7 million.
During the year ended December 31, 2013, net cash used in investing activities was $29.2 million, which related to the acquisition and related expenses for Vet Therapeutics of $31.0 million. In addition, $6.9 million of short-term marketable securities matured, partially offset by the purchase of $5.2 million of marketable securities.
66
Net cash provided by financing activities
During the year ended December 31, 2015, net cash provided by financing activities was $21.9 million. Net cash provided by financing activities primarily resulted from net cash received from our Loan and Security Agreement of $24.8 million, partially offset by cash paid for contingent consideration of $3.0 million to the former shareholders of Vet Therapeutics.
During the year ended December 31, 2014, net cash provided by financing activities was $101.0 million. Net cash provided by financing activities primarily resulted from net proceeds of $137.2 million, net of commissions, raised in conjunction with public offerings, and proceeds of $0.2 million received from the exercise of stock options. This was partially offset by payments of $18.1 million related to payments of promissory notes, $15.2 million paid for contingent consideration, $2.2 million paid for public offering costs and $1.1 million paid for repurchase of common stock.
During the year ended December 31, 2013, net cash provided by financing activities was $72.5 million. Net cash provided by financing activities primarily resulted from net proceeds of $36.9 million, net of commissions, raised in conjunction with our initial public offering, net proceeds of $19.7 million from private investment in a public entity, net proceeds of $14.9 million from our Credit Facility, $3.4 million raised from the private placement of our series C convertible preferred stock prior to our initial public offering of common stock, and proceeds of $0.2 million received from the exercise of stock options. This was partially offset by payments of $2.6 million related to our initial public offering.
Future Funding Requirements
We anticipate that we will continue to incur net losses for the next several years due to expenses for our development programs, including continuing studies in both cats and dogs for our programs in the United States and Europe and the in-licensing or acquisition of additional compounds for development as pet therapeutics.
In addition, we intend to hire additional personnel to build out a commercial sales force in the United States in anticipation of USDA and CVM approval of our products.
As of the date of the filing of this Annual Report on Form 10-K, we believe that our cash, cash equivalents and short-term investments, will fund our operations including, if necessary, the repayment of our long-term debt through December 31, 2016. However, our operating plan may change as a result of many factors currently unknown to us, and we may seek additional funds sooner than planned, through public or private equity or debt financings or other sources, such as strategic collaborations. Such financing may result in dilution to stockholders, imposition of debt covenants and repayment obligations, or other restrictions that may affect our business. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed in the section of this filing entitled “Risk Factors.”
Our future capital requirements depend on many factors, including, but not limited to:
|
· |
the results of our target animal studies for our current and future product candidates; |
|
· |
the amount and timing of any milestone payments or royalties we must pay pursuant to our current or future license agreements or collaboration agreements; |
|
· |
the timing of, and the costs involved in, obtaining regulatory approvals for any of our current or future product candidates; |
|
· |
the upfront and other payments, and associated costs, related to our identifying, acquiring and in-licensing new product candidates; |
|
· |
the number and characteristics of the product candidates we pursue; |
|
· |
the scope, progress, results and costs of researching and developing any of our current or future product candidates and conducting target animal studies; |
|
· |
whether we acquire any other companies, assets, intellectual property or technologies in the future; |
|
· |
our ability to partner with companies with an established commercial presence in Europe to provide our products in that market; |
|
· |
the cost of commercialization activities, if any of our current or future product candidates are approved for sale, including marketing, sales and distribution costs; |
|
· |
cost of manufacturing our current and future product candidates and any products we successfully commercialize; |
|
· |
our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements; |
|
· |
whether we are required to repay amounts that we received from the KBA, repurchase the shares of our capital stock owned by the KBA or repay Kansas income tax credits allocated to some of our investors; |
67
|
· |
whether we are able to service our debt and satisfy debt covenants; |
|
· |
the expenses needed to attract and retain skilled personnel; |
|
· |
the costs associated with being a public company; and |
|
· |
the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation. |
Contractual Obligations and Commitments
The following table summarizes our contractual obligations as of December 31, 2015 and the effect such obligations are expected to have on our liquidity and cash flows in future periods:
|
PAYMENTS DUE BY FISCAL YEAR |
|||||||||||||||
|
LESS THAN |
MORE THAN |
||||||||||||||
|
TOTAL |
1 YEAR |
1-3 YEARS |
3-5 YEARS |
5 YEARS |
|||||||||||
|
(In Thousands) |
|||||||||||||||
|
Loan payable (1) |
$ |
50,017 |
$ |
3,377 |
$ |
34,373 |
$ |
12,267 |
$ |
— |
|||||
|
Early exercise of stock options (2) |
30 | 29 | 1 |
— |
— |
||||||||||
|
Manufacturing (3) |
2,250 | 100 | 2,150 |
— |
— |
||||||||||
|
Minimum royalty (4) |
942 | 73 | 149 | 153 | 567 | ||||||||||
|
Operating leases (5) |
2,785 | 598 | 1,195 | 917 | 75 | ||||||||||
|
Total (6) |
$ |
56,024 |
$ |
4,177 |
$ |
37,868 |
$ |
13,337 |
$ |
642 | |||||
_________________
|
(1) |
Represents the contractually required principal and interest payments on our Loan and Security Agreement in accordance with the required payment schedule. Amounts associated with future interest payments to be made were calculated using the interest rate in effect as of December 31, 2015, which was 7.16%. |
|
(2) |
Reflects the amount recorded as a liability for the early exercise of a stock-based awards. The amount will be reclassified to equity on a ratable basis as the award vests. |
|
(3) |
The table includes minimum order commitments based upon an agreed upon demand forecast established each year. |
|
(4) |
The table includes the minimum royalty payment due each year based upon a Commercial License Agreement with Crucell Holland B.V. (“Crucell”) under which we received a commercial license to prepare recombinant antibodies. We are required to pay single digit royalties on net product sales by us allocable to Crucell’s producer cells and/or producer cell know-how, if any. We are required to pay Crucell a minimum royalty of $70 that is subject to a yearly inflation index adjustment. |
|
(5) |
The table above includes payments for office equipment and rent for the lease of the corporate headquarters in Leawood, Kansas through February 2020, for offices in Boston, Massachusetts through August 2016, for Vet Therapeutics in San Diego, California through May 2016 and for offices and laboratory space in Leuven, Belgium through 2020. |
|
(6) |
The table above excludes flat rate royalty payments and/or milestone payments of up to $119.6 million that will become due in connection with various agreements. The milestones payments will become due as we achieve development, regulatory and commercial milestones and the royalty payments will be paid upon product sales. |
Other Funding Commitments
As of December 31, 2015, we have several on-going development programs in various stages in the regulatory process. Our most significant expenditures are payments to clinical research and contract manufacturing organizations. The contracts are generally cancellable, with notice, at our option.
Contingent Development, Regulatory and Commercial Milestone Payments
Based on our development plans as of December 31, 2015, we have committed to make potential future milestone payments to third parties of up to approximately $119.6 million, of which $80.4 million are for commercial milestones, as part of our various collaborations, including licensing and development programs. Approximately $68.9 million of the commercial milestones relate to the achievement of various sales thresholds. Payments under these agreements generally become due and payable only upon achievement of certain development, regulatory or commercial milestones. Because the achievement of these milestones had not
68
occurred or was not considered probable as of December 31, 2015, such contingencies have not been recorded in our financial statements, except for $0.5 million due to our former commercial licensee of BLONTRESS.
We paid $0.2 million in the fourth quarter of 2015 and as of December 31, 2015, we anticipated paying an additional $14.7 million of milestone payments during the next 12 months, provided that various development, regulatory or commercial milestones are achieved. As of the date of this filing $3.0 million of the $14.7 million of milestones have been achieved. Amounts related to contingent milestone payments are not considered contractual obligations as they are contingent on the successful achievement of certain development, regulatory approval and commercial milestones. These milestones may not be achieved.
Contingent Consideration Related to Business Combinations
During the three months ended March 31, 2015, we settled the remaining contingent consideration related to our October 2013, acquisition of Vet Therapeutics, Inc. in the amount of $3.0 million. With the settlement of this contingent consideration we no longer have any contingent consideration related to business combinations.
Off-Balance Sheet Arrangements
We have not engaged in the use of any off-balance sheet arrangements, such as structured finance entities or special purpose entities.
New Accounting Standards
For discussion of our new accounting standards, see noted to consolidated financial statements-Note 2. “Summary of Significant Accounting Policies-New Accounting Standards”.
69
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Interest Rate Fluctuation Risk
Our cash, cash equivalents and short-term investments as of December 31, 2015 consisted primarily of cash, certificates of deposit and reverse repurchase agreements. Our primary exposure to market risk for our cash, cash equivalents and short-term investments is interest income sensitivity, which is affected by changes in the general level of U.S. interest rates. However, because of the short-term nature of the instruments in our portfolio, a sudden change in market interest rates would not be expected to have a material impact on our financial condition or results of operations.
We have borrowed $40.0 million under our Loan and Security Agreement, consisting of a $35.0 million term loan and a $5.0 million revolving credit facility. We are obligated to make interest-only payments on the term loan until May 1, 2017, and thereafter are required to make payments of principal and accrued interest on the term loan in equal monthly installments over a term of 30 months. We are obligated to make interest-only payments on the revolving loan until October 16, 2017. The term loan and revolving credit facility bear interest per annum at the greater of (i) 6.91% or (ii) 3.66% plus the prime rate. Given the amounts outstanding and available under the Loan and Security Agreement, and the interest rate paid to date, we do not believe a 1.0% increase in the interest rate would have a material effect on our financial condition or results of operations.
Foreign Exchange Risk
We are exposed to market risk associated with foreign currency exchange rate fluctuations, and this market risk was further enhanced as a result of our acquisition of Okapi Sciences during January 2014. We face exposure to movements in foreign currency exchange rates whenever we enter into transactions with third parties that are denominated in currencies other than our functional currency. Intercompany transactions between entities that use different functional currencies also expose us to foreign currency risk.
Item 8. Financial Statements and Supplementary Data
The financial statements and supplementary data are listed under Item 15(a) and have been filed as part of this Annual Report on Form 10-K on the pages indicated.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Limitations on Effectiveness of Controls and Procedures
In designing and evaluating our disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated, as of the end of the period covered by this Annual Report on Form 10-K, the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended). Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of December 31, 2015.
Management’s Annual Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) under the Securities Exchange Act of 1934, as amended.
Our management conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2015 based on the criteria set forth in “Internal Control-Integrated Framework (2013 Framework)” issued by the Committee of Sponsoring Organizations of the Treadway Commission.
Based on this assessment, management concluded that as of December 31, 2015, our internal control over financial reporting was effective.
As we are an emerging growth company, our independent registered public accounting firm is not required to attest to the effectiveness of our internal control over financial reporting.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended) identified in connection with the evaluation of our internal control performed during the fiscal
70
quarter ended December 31, 2015 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
None.
71
Item 10. Directors, Executive Officers and Corporate Governance
The information concerning the Company’s executive officers and directors is contained in Part I of this Annual Report on Form 10-K. The rest of the information required to be disclosed by this item will be contained under the headings “Section 16(a) Beneficial Ownership Reporting Compliance,” “Corporate Governance – Code of Ethics,” and “Committees of the Board” in the Proxy Statement for the Company’s 2016 Annual Meeting of Stockholders and is incorporated herein by reference.
Item 11. Executive Compensation
The information required to be disclosed by this item will be contained under the headings “Executive and Director Compensation” and “Compensation Committee Interlocks and Insider Participation” in the Proxy Statement for the Company’s 2016 Annual Meeting of Stockholders and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required to be disclosed by this item will be contained under the headings “Executive and Director Compensation – Equity Compensation Plan Information” and “Security Ownership of Certain Beneficial Owners and Management” in the Proxy Statement for the Company’s 2016 Annual Meeting of Stockholders and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required to be disclosed by this item will be contained under the headings “Certain Relationships” and “Corporate Governance – Director Independence” in the Proxy Statement for the Company’s 2016 Annual Meeting of Stockholders and is incorporated herein by reference.
Item 14. Principal Accounting Fees and Services
The information required to be disclosed by this item will be contained under the heading “Independent Registered Public Accounting Firm Fees and Other Matters” in the Proxy Statement for the Company’s 2016 Annual Meeting of Stockholders and is incorporated herein by reference.
72
Item 15. Exhibits, Financial Statement Schedules
(a)(1), (a)(2) and (c). The response to this portion of Item 15 is submitted as a separate section of this report commencing on page F-1.
(a)(3) and (b). Exhibits (numbered in accordance with Item 601 of Regulation S-K).
73
|
|
|
Incorporated by Reference _______________________________________________ |
|
|||
|
Exhibit Number |
Exhibit Description |
Form |
File No. |
Exhibit |
Filing Date |
Filed/ Furnished Herewith |
|
2.1† |
Agreement and Plan of Merger, dated October 13, 2013, by and among Aratana Therapeutics, Inc., Vet Therapeutics, Inc., Jayhawk Acquisition Corporation and Jeffrey Miles, as stockholders’ representative |
8-K |
001-35952 |
10.1 |
10/16/13 |
|
|
2.2 |
Stock Purchase Agreement, dated January 6, 2014, by and among Aratana Therapeutics, Inc., Wildcat Acquisition BVBA, the Sellers of Okapi Sciences NV listed on Annex A thereto and Thuja Capital Healthcare Fund BV, as the Sellers’ representative |
8-K |
001-35952 |
10.1 |
1/7/14 |
|
|
3.1 |
Restated Certificate of Incorporation |
8-K |
001-35952 |
3.1 |
7/3/13 |
|
|
3.2 |
Amended and Restated Bylaws |
8-K |
001-35952 |
3.2 |
7/3/13 |
|
|
4.1 |
Specimen stock certificate evidencing the shares of common stock |
S-1/A |
333-187372 |
4.1 |
6/6/13 |
|
|
4.2 |
Second Amended and Restated Investors’ Rights Agreement, dated as of December 28, 2012, as amended May 22, 2013 |
S-1/A |
333-187372 |
10.1 |
5/23/13 |
|
|
10.1†† |
Form of Indemnification Agreement for Directors and Officers |
S-1 |
333-187372 |
10.3 |
3/20/13 |
|
|
10.2†† |
Employment Agreement, dated September 6, 2012, by and between Steven St. Peter and Aratana Therapeutics, Inc., as amended April 26, 2013 |
S-1/A |
333-187372 |
10.4 |
5/23/13 |
|
|
10.3†† |
Employment Agreement, dated March 5, 2014, by and between Linda Rhodes and Aratana Therapeutics, Inc. |
8-K |
001-35952 |
10.1 |
3/10/14 |
|
|
10.4†† |
Employment Agreement, dated December 18, 2012, by and between Julia Stephanus and Aratana Therapeutics, Inc., as amended April 29, 2013 |
S-1/A |
333-187372 |
10.7 |
5/23/13 |
|
|
10.5†† |
Employment Agreement, dated March 12, 2013, by and between Ernst Heinen and Aratana Therapeutics, Inc., as amended April 29, 2013 |
S-1/A |
333-187372 |
10.8 |
5/23/13 |
|
|
10.6†† |
Employment Agreement, dated November 8, 2013, by and between Craig Tooman and Aratana Therapeutics, Inc. |
8-K |
001-35952 |
10.1 |
11/14/13 |
|
|
10.7(a)†† |
Aratana Therapeutics, Inc. 2010 Equity Incentive Plan |
S-1/A |
333-193324 |
10.8(a) |
1/28/14 |
|
|
10.7(b)†† |
Amendment No. 1 to 2010 Equity Incentive Plan |
S-1 |
333-187372 |
10.9(b) |
3/20/13 |
|
|
10.7(c)†† |
Amendment No. 2 to 2010 Equity Incentive Plan |
S-1 |
333-187372 |
10.9(c) |
3/20/13 |
|
|
10.7(d)†† |
Form of Stock Option Grant Notice and Stock Option Agreement under 2010 Equity Incentive Plan |
S-1 |
333-187372 |
10.9(d) |
3/20/13 |
|
|
10.8(a)†† |
Aratana Therapeutics, Inc. 2013 Incentive Award Plan |
S-8 |
333-187372 |
99.1 |
1/21/14 |
|
|
10.8(b)†† |
Form of Stock Option Grant Notice and Stock Option Agreement under 2013 Incentive Award Plan |
S-1/A |
333-187372 |
10.10(b) |
4/30/13 |
|
|
10.8(c)†† |
Form of Restricted Stock Grant Notice and Restricted Stock Agreement under 2013 Incentive Award Plan |
S-1/A |
333-187372 |
10.10(c) |
4/30/13 |
|
|
10.9†† |
Non-Employee Director Compensation Program, as amended |
10-Q |
001-35952 |
10.5 |
11/6/15 |
|
74
|
10.10 |
Office Building Lease, dated as of October 8, 2015, by and between Academy 1740, Inc. and Aratana Therapeutics, Inc. |
8-K |
001-35952 |
10.1 |
10/13/15 |
|
|
10.11 |
Subordination, Non-Disturbance and Attornment Agreement, dated as of January 29, 2016, by and between Aratana Therapeutics, Inc., Academy 1740, Inc., and Commerce Bank |
|
|
|
|
* |
|
10.12 |
Loan and Security Agreement, dated as of October 16, 2015, by and between Pacific Western Bank, as collateral agent, the lenders listed on Schedule 1.1 thereto, Oxford Finance LLC, and Aratana Therapeutics, Inc. and Vet Therapeutics, Inc. |
8-K |
001-35952 |
10.2 |
10/16/15 |
|
|
10.13 |
Sales Agreement, dated as of October 16, 2015, by and between Barclays Capital Inc. and Aratana Therapeutics, Inc. |
8-K |
001-35952 |
10.1 |
10/16/15 |
|
|
10.14† |
Exclusive IP License Agreement for RQ-00000005, dated December 27, 2010, by and between Aratana Therapeutics, Inc. and RaQualia Pharma Inc. |
S-1/A |
333-187372 |
10.18 |
6/6/13 |
|
|
10.15 |
First Amendment to the Exclusive IP License Agreement for RQ-00000005, dated July 12, 2012, by and between Aratana Therapeutics, Inc. and RaQualia Pharma Inc. |
S-1/A |
333-187372 |
10.19 |
4/11/13 |
|
|
10.16† |
Exclusive IP License Agreement for RQ-00000007, dated December 27, 2010, by and between Aratana Therapeutics, Inc. and RaQualia Pharma Inc. |
S-1/A |
333-187372 |
10.20 |
6/6/13 |
|
|
10.17 |
First Amendment to the Exclusive IP License Agreement for RQ-00000007, dated July 12, 2012, by and between Aratana Therapeutics, Inc. and RaQualia Pharma Inc. |
S-1/A |
333-187372 |
10.21 |
4/11/13 |
|
|
10.18 |
Letter Agreement regarding RQ-00000008 Technology, dated July 12, 2012, by and between RaQualia Pharma Inc. and Aratana Therapeutics, Inc. |
S-1/A |
333-187372 |
10.23 |
4/11/13 |
|
|
10.19† |
Exclusive License, Development and Commercialization Agreement, effective as of December 5, 2012, by and between Pacira Pharmaceuticals, Inc. and Aratana Therapeutics, Inc. |
S-1/A |
333-187372 |
10.24 |
4/11/13 |
|
|
10.20† |
Supply Agreement, dated December 5, 2012, by and between Pacira Pharmaceuticals, Inc. and Aratana Therapeutics, Inc. |
S-1/A |
333-187372 |
10.25 |
4/11/13 |
|
|
10.21 |
Promissory Note of Wildcat Acquisition BVBA, dated January 6, 2014 |
8-K |
001-35952 |
10.2 |
1/7/14 |
|
|
21.1 |
Subsidiaries of Aratana Therapeutics, Inc. |
|
|
|
|
* |
|
23.1 |
Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm |
|
|
|
|
* |
|
31.1 |
Rule 13a-14(a) / 15d-14(a) Certification of Chief Executive Officer |
|
|
|
|
* |
|
31.2 |
Rule 13a-14(a) / 15d-14(a) Certification of Chief Financial Officer |
|
|
|
|
* |
|
32.1 |
Section 1350 Certification of Chief Executive Officer** |
|
|
|
|
** |
|
32.2 |
Section 1350 Certification of Chief Financial Officer** |
|
|
|
|
** |
75
|
101.INS |
XBRL Instance Document |
|
|
|
|
* |
|
101.SCH |
XBRL Taxonomy Extension Schema Document |
|
|
|
|
* |
|
101.CAL |
XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
* |
|
101.DEF |
XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
* |
|
101.LAB |
XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
* |
|
101.PRE |
XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
|
* |
†Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment pursuant to Rule 24b-2 under the Securities Exchange Act of 1934.
††Management contract or compensatory plan or arrangement.
* Filed herewith.
** Furnished herewith.
76
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
ARATANA THERAPEUTICS, INC. |
||
|
BY: |
/s/ Steven St. Peter |
|
|
Steven St. Peter, M.D. President and Chief Executive Officer |
||
Date: March 15, 2016
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
SIGNATURE |
TITLE |
DATE |
||
|
/s/ Steven St. Peter Steven St. Peter, M.D. |
President, Chief Executive Officer and Director (principal executive officer) |
March 15, 2016 |
||
|
/s/ Craig Tooman Craig Tooman |
Chief Financial Officer and Treasurer (principal financial and accounting officer) |
March 15, 2016 |
||
|
/s/ Wendy L. Yarno Wendy L. Yarno |
Chairperson of the Board of Directors |
March 15, 2016 |
||
|
/s/ Laura A. Brege Laura A. Brege |
Director |
March 15, 2016 |
||
|
/s/ David L. Brinkley David L. Brinkley |
Director |
March 15, 2016 |
||
|
/s/ Robert Gerber Robert “Rip” Gerber |
Director |
March 15, 2016 |
||
|
/s/ Irvine O. Hockaday Irvine “Irv” O. Hockaday, Esq. |
Director |
March 15, 2016 |
||
|
/s/ Merilee Raines Merilee Raines |
Director |
March 15, 2016 |
||
|
/s/ Robert P. Roche Robert P. Roche |
Director |
March 15, 2016 |
||
|
/s/ John Vander Vort John Vander Vort, Esq. |
Director |
March 15, 2016 |
||
77
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Aratana Therapeutics, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, of comprehensive loss, of changes in convertible preferred stock and stockholders’ equity (deficit) and of cash flows present fairly, in all material respects, the financial position of Aratana Therapeutics, Inc. and its subsidiaries at December 31, 2015 and December 31, 2014, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2015 in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We conducted our audits of these statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
As discussed in Note 2 to the consolidated financial statements, the Company changed the manner in which it classifies deferred taxes in 2015 due to the adoption of Accounting Standards Update 2015-17, Balance Sheet Classification of Deferred Taxes.
As discussed in Note 1 to the consolidated financial statements, the Company has long term debt which may require repayment during 2016. Also as discussed in Note 1, the Company will require additional financing to fund future operations. Management’s plans in regard to this matter are described in Note 1.
/s/ PricewaterhouseCoopers LLP
Boston, Massachusetts
March 15, 2016
F-2
CONSOLIDATED BALANCE SHEETS
(Amounts in thousands, except share and per share data)
|
DECEMBER 31, 2015 |
DECEMBER 31, 2014 |
|||||
|
Assets |
||||||
|
Current assets: |
||||||
|
Cash and cash equivalents |
$ |
26,755 |
$ |
9,823 | ||
|
Short-term investments |
59,447 | 88,249 | ||||
|
Accounts receivable, net |
60 | 352 | ||||
|
Inventories |
1,306 | 427 | ||||
|
Prepaid expenses and other current assets |
1,451 | 900 | ||||
|
Deferred tax asset |
— |
158 | ||||
|
Total current assets |
89,019 | 99,909 | ||||
|
Property and equipment, net |
2,555 | 620 | ||||
|
Long-term marketable securities |
— |
2,452 | ||||
|
Goodwill |
39,781 | 41,398 | ||||
|
Intangible assets, net |
15,067 | 62,323 | ||||
|
Restricted cash |
350 |
— |
||||
|
Other long-term assets |
294 | 1,201 | ||||
|
Total assets |
$ |
147,066 |
$ |
207,903 | ||
|
Liabilities and Stockholders’ Equity |
||||||
|
Current liabilities: |
||||||
|
Accounts payable |
$ |
1,400 |
$ |
1,532 | ||
|
Accrued expenses |
4,247 | 3,229 | ||||
|
Current portion – contingent consideration |
— |
4,248 | ||||
|
Deferred tax liability |
— |
413 | ||||
|
Other current liabilities |
37 | 46 | ||||
|
Total current liabilities |
5,684 | 9,468 | ||||
|
Loans payable, net |
39,710 | 14,963 | ||||
|
Deferred tax liability |
— |
1,610 | ||||
|
Other long-term liabilities |
122 | 30 | ||||
|
Total liabilities |
45,516 | 26,071 | ||||
|
Commitments and contingencies (Notes 12 and 16) |
||||||
|
Stockholders’ equity: |
||||||
|
Common stock, $0.001 par value; 100,000,000 shares authorized at December 31, 2015 and December 31, 2014, 34,563,816 and 34,147,861 issued and outstanding at December 31, 2015 and December 31, 2014, respectively |
35 | 34 | ||||
|
Treasury stock |
(1,088) | (1,081) | ||||
|
Additional paid-in capital |
263,941 | 254,993 | ||||
|
Accumulated deficit |
(152,018) | (67,964) | ||||
|
Accumulated other comprehensive loss |
(9,320) | (4,150) | ||||
|
Total stockholders’ equity |
101,550 | 181,832 | ||||
|
Total liabilities and stockholders’ equity |
$ |
147,066 |
$ |
207,903 | ||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
CONSOLIDATED STATEMENTS OF OPERATIONS
(Amounts in thousands, except share and per share data)
|
2015 |
2014 |
2013 |
|||||||
|
Revenues |
|||||||||
|
Licensing and collaboration revenue |
$ |
— |
$ |
500 |
$ |
15 | |||
|
Product sales |
678 | 267 | 108 | ||||||
|
Total revenues |
678 | 767 | 123 | ||||||
|
Costs and expenses |
|||||||||
|
Cost of product sales |
365 | 333 | 108 | ||||||
|
Royalty expense |
84 | 72 | 1 | ||||||
|
Research and development |
24,964 | 19,985 | 10,925 | ||||||
|
Selling, general and administrative |
19,819 | 17,938 | 8,572 | ||||||
|
In-process research and development |
— |
2,157 |
— |
||||||
|
Amortization of acquired intangible assets |
1,544 | 1,891 | 298 | ||||||
|
Impairment of acquired intangible assets |
43,398 |
— |
— |
||||||
|
Total costs and expenses |
90,174 | 42,376 | 19,904 | ||||||
|
Loss from operations |
(89,496) | (41,609) | (19,781) | ||||||
|
Other income (expense) |
|||||||||
|
Interest income |
189 | 123 | 75 | ||||||
|
Interest expense |
(1,585) | (1,060) | (432) | ||||||
|
Other income (expense), net |
5,140 | 2,287 | 478 | ||||||
|
Total other income (expense) |
3,744 | 1,350 | 121 | ||||||
|
Loss before income taxes |
$ |
(85,752) |
$ |
(40,259) |
$ |
(19,660) | |||
|
Income tax benefit |
1,698 | 1,443 | 12,722 | ||||||
|
Net loss |
$ |
(84,054) |
$ |
(38,816) |
$ |
(6,938) | |||
|
Net loss per share, basic and diluted |
$ |
(2.45) |
$ |
(1.30) |
$ |
(0.63) | |||
|
Weighted average shares outstanding, basic and diluted |
34,355,525 | 29,767,429 | 11,059,382 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(Amounts in thousands, except share and per share data)
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
Net loss |
$ |
(84,054) |
$ |
(38,816) |
$ |
(6,938) | |||
|
Other comprehensive loss: |
|||||||||
|
Foreign currency translation adjustment |
(3,918) | (5,402) |
— |
||||||
|
Unrealized gain on available-for-sale securities |
2,622 | 1,252 |
— |
||||||
|
Net gain reclassified into income on sale of available-for-sale securities |
(3,874) |
— |
— |
||||||
|
Other comprehensive loss |
(5,170) | (4,150) |
— |
||||||
|
Comprehensive loss |
$ |
(89,224) |
$ |
(42,966) |
$ |
(6,938) | |||
The accompanying notes are an integral part of these consolidated financial statements
F-5
CONSOLIDATED STATEMENTS OF CHANGES IN CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY/(DEFICIT)
(Amounts in thousands, except share data)
|
Accumulated |
Treasury |
Total |
||||||||||||||||||||||||
|
Series A-C |
Additional |
Other |
Stock |
Stockholders' |
||||||||||||||||||||||
|
Convertible |
Common Stock |
Paid-In |
Accumulated |
Comprehensive |
at |
Equity |
||||||||||||||||||||
|
Preferred Stock |
Shares |
Par Value |
Capital |
Deficit |
Loss |
at cost |
(Deficit) |
|||||||||||||||||||
|
Balance at December 31, 2012 |
20,241,207 | 39,197 | 830,823 | 1 | 654 | (22,210) |
— |
— |
(21,555) | |||||||||||||||||
|
Issuance of Series C convertible preferred stock, net of issuance cost of $19 |
693,571 | 2,756 |
— |
— |
— |
— |
— |
— |
— |
|||||||||||||||||
|
Impact of initial public offering on stockholders' equity (deficit): |
— |
— |
||||||||||||||||||||||||
|
Effect of a 1 for 1.662 reverse split on Preferred Stock |
(8,338,699) |
— |
— |
— |
— |
— |
— |
— |
— |
|||||||||||||||||
|
Conversion of shares of preferred stock to common stock |
(12,596,079) | (41,953) | 12,596,079 | 13 | 41,940 |
— |
— |
— |
41,953 | |||||||||||||||||
|
Dividends for preferred shares issued upon initial public offering |
— |
— |
755,823 | 1 | (1) |
— |
— |
— |
— |
|||||||||||||||||
|
Initial public offering of common stock, net of $5,394 of offering costs |
— |
— |
6,612,500 | 6 | 34,274 |
— |
— |
— |
34,280 | |||||||||||||||||
|
Issuance of common stock related to Vet Therapeutics, Inc. merger |
— |
— |
624,997 | 1 | 14,699 |
— |
— |
— |
14,700 | |||||||||||||||||
|
Issuance of common stock related to private investment in public entity |
— |
— |
1,234,375 | 1 | 19,749 |
— |
— |
— |
19,750 | |||||||||||||||||
|
Compensation expense related to stock options and restricted awards |
— |
— |
— |
— |
1,025 |
— |
— |
— |
1,025 | |||||||||||||||||
|
Vesting of restricted stock awards |
— |
— |
230,298 |
— |
— |
— |
— |
— |
— |
|||||||||||||||||
|
Vesting of stock awards early exercised |
— |
— |
270,419 |
— |
118 |
— |
— |
— |
118 | |||||||||||||||||
|
Issuance of common stock related to option exercises |
— |
— |
270,173 |
— |
57 |
— |
— |
— |
57 | |||||||||||||||||
|
Net loss |
— |
— |
— |
— |
— |
(6,938) |
— |
— |
(6,938) | |||||||||||||||||
|
Balance at December 31, 2013 |
— |
— |
23,425,487 | 23 | 112,515 | (29,148) |
— |
— |
83,390 | |||||||||||||||||
|
Public offering of common stock, net of $10,627 of offering costs |
— |
— |
10,325,000 | 11 | 135,067 |
— |
— |
— |
135,078 | |||||||||||||||||
|
Compensation expense related to stock options and restricted awards |
— |
— |
— |
— |
7,130 |
— |
— |
— |
7,130 | |||||||||||||||||
|
Vesting of restricted stock awards |
— |
— |
240,528 |
— |
— |
— |
— |
— |
— |
|||||||||||||||||
|
Repurchase of common stock |
— |
— |
(77,367) |
— |
— |
— |
— |
(1,081) | (1,081) | |||||||||||||||||
|
Vesting of stock awards early exercised |
— |
— |
152,272 |
— |
56 |
— |
— |
— |
56 | |||||||||||||||||
|
Issuance of common stock related to option exercises |
— |
— |
81,941 |
— |
225 |
— |
— |
— |
225 | |||||||||||||||||
|
Other comprehensive loss |
— |
— |
— |
— |
— |
— |
(4,150) |
— |
(4,150) | |||||||||||||||||
|
Net loss |
— |
— |
— |
— |
— |
(38,816) |
— |
— |
(38,816) | |||||||||||||||||
|
Balance at December 31, 2014 |
— |
$ |
— |
34,147,861 |
$ |
34 |
$ |
254,993 |
$ |
(67,964) |
$ |
(4,150) |
$ |
(1,081) |
$ |
181,832 | ||||||||||
|
Compensation expense related to stock options and restricted awards |
— |
— |
— |
— |
8,592 |
— |
— |
— |
8,592 | |||||||||||||||||
|
Vesting of restricted stock awards |
— |
— |
205,387 |
— |
— |
— |
— |
— |
— |
|||||||||||||||||
|
Repurchase of common stock |
— |
— |
(859) |
— |
— |
— |
— |
(7) | (7) | |||||||||||||||||
|
Vesting of stock awards early exercised |
— |
— |
121,014 | 1 | 44 |
— |
— |
— |
45 | |||||||||||||||||
F-6
|
Issuance of common stock related to option exercises |
— |
— |
90,413 |
— |
312 |
— |
— |
— |
312 | |||||||||||||||||
|
Other comprehensive loss |
— |
— |
— |
— |
— |
— |
(5,170) |
— |
(5,170) | |||||||||||||||||
|
Net loss |
— |
— |
— |
— |
— |
(84,054) |
— |
— |
(84,054) | |||||||||||||||||
|
Balance at December 31, 2015 |
— |
$ |
— |
34,563,816 |
$ |
35 |
$ |
263,941 |
$ |
(152,018) |
$ |
(9,320) |
$ |
(1,088) |
$ |
101,550 |
The accompanying notes are an integral part of these consolidated financial statements
F-7
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Amounts in thousands)
|
YEAR ENDED |
||||||||
|
DECEMBER 31, |
||||||||
|
2015 |
2014 |
2013 |
||||||
|
Cash flows from operating activities |
||||||||
|
Net loss |
$ |
(84,054) |
$ |
(38,816) |
$ |
(6,938) | ||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
|
Acquired in-process research and development |
— |
2,157 |
— |
|||||
|
Stock-based compensation expense |
8,592 | 7,130 | 1,025 | |||||
|
Depreciation and amortization expense |
1,840 | 2,043 | 313 | |||||
|
Impairment of acquired intangible assets |
43,398 |
— |
— |
|||||
|
Gain on sale of marketable securities |
(3,874) |
— |
— |
|||||
|
Non-cash interest expense |
129 | 41 | 23 | |||||
|
Creditor fees |
(150) |
— |
— |
|||||
|
Change in fair value of contingent consideration |
(1,248) | (133) | 305 | |||||
|
Change in fair value of derivative instruments |
(1,274) | (465) |
— |
|||||
|
Deferred tax benefit |
(1,698) | (1,443) | (12,722) | |||||
|
Changes in operating assets and liabilities: |
||||||||
|
Accounts receivable, net |
281 | (102) |
— |
|||||
|
Inventories |
(879) | (372) |
— |
|||||
|
Prepaid expenses |
(595) | (642) | (249) | |||||
|
Other assets |
(31) | (45) | (14) | |||||
|
Accounts payable |
(117) | (1,143) | 1,527 | |||||
|
Accrued expenses and other liabilities |
1,185 | 482 | 579 | |||||
|
Deferred income |
— |
(880) |
— |
|||||
|
Net cash used in operating activities |
(38,495) | (32,188) | (16,151) | |||||
|
Cash flows from investing activities |
||||||||
|
Purchases of property and equipment, net |
(2,245) | (471) | (94) | |||||
|
Cash paid for acquisitions, net of cash received |
— |
(12,075) | (30,994) | |||||
|
Proceeds from sales of marketable securities |
7,456 |
— |
— |
|||||
|
Purchase of investments |
(2,050,594) | (371,449) | (5,169) | |||||
|
Proceeds from maturities of investments |
2,079,396 | 286,670 | 6,881 | |||||
|
Purchase of derivative instruments |
— |
(643) |
— |
|||||
|
Purchase of in-process research and development |
— |
(2,157) |
— |
|||||
|
Change in restricted cash |
(350) |
— |
141 | |||||
|
Net cash provided by (used in) investing activities |
33,663 | (100,125) | (29,235) | |||||
|
Cash flows from financing activities |
||||||||
|
Proceeds from the issuance of Series C convertible preferred stock, net of issuance costs |
— |
— |
3,406 | |||||
|
Proceeds from the issuance of debt, net of discount |
24,779 |
— |
14,914 | |||||
|
Repurchase of common stock |
(7) | (1,081) |
— |
|||||
|
Proceeds from stock option exercises |
312 | 225 | 153 | |||||
|
Repurchase, early exercised stock |
— |
— |
(6) | |||||
F-8
|
Proceeds from public offering, net of commission |
— |
137,220 | 36,897 | |||||
|
Payments for public offering costs |
(189) | (2,153) | (2,617) | |||||
|
Issuance of common stock private investment in public entity |
— |
— |
19,750 | |||||
|
Cash paid for promissory notes |
— |
(18,067) |
— |
|||||
|
Cash paid for contingent consideration |
(3,000) | (15,166) |
— |
|||||
|
Net cash provided by financing activities |
21,895 | 100,978 | 72,497 | |||||
|
Effect of exchange rate on cash |
(131) | 74 |
— |
|||||
|
Net increase in cash and cash equivalents |
16,932 | (31,261) | 27,111 | |||||
|
Cash and cash equivalents, beginning of year |
9,823 | 41,084 | 13,973 | |||||
|
Cash and cash equivalents, end of year |
$ |
26,755 |
$ |
9,823 | 41,084 | |||
|
Supplemental disclosure of cash flow information |
||||||||
|
Cash paid for interest, net of amounts capitalized |
$ |
1,057 |
$ |
942 |
$ |
357 | ||
|
Supplemental disclosure of noncash investing and financing activities: |
||||||||
|
Non-cash exercise of warrant |
$ |
750 |
$ |
— |
$ |
— |
||
|
Conversion of preferred stock into common stock |
$ |
— |
$ |
— |
$ |
41,953 | ||
|
Payment of dividends on preferred stock |
$ |
— |
$ |
— |
$ |
4,535 | ||
|
Issuance of common stock relating to Vet Therapeutics, Inc. acquisition |
$ |
— |
$ |
— |
$ |
14,700 | ||
|
Contingent consideration relating to Vet Therapeutics, Inc. acquisition |
$ |
— |
$ |
— |
$ |
4,115 | ||
|
Note payable related to Vet Therapeutics, Inc. acquisition |
$ |
— |
$ |
— |
$ |
3,000 | ||
|
Contingent consideration relating to Okapi Sciences NV acquisition |
$ |
— |
$ |
15,166 |
$ |
— |
||
|
Note payable related to Okapi Sciences NV acquisition |
$ |
— |
$ |
15,134 |
$ |
— |
The accompanying notes are an integral part of these consolidated financial statements.
F-9
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
1. The Company and Basis of Presentation
The Company
Aratana Therapeutics, Inc., including its subsidiaries (the “Company,” or “Aratana”) was incorporated on December 1, 2010 under the laws of the State of Delaware. The Company is a pet therapeutics company focused on licensing, developing and commercializing of innovative biopharmaceutical products for companion animals. The Company has one operating segment: pet therapeutics.
Since its inception, the Company has devoted substantially all of its efforts to research and development, recruiting management and technical staff, building a commercial infrastructure, acquiring operating assets and raising capital.
The Company is subject to risks common to companies in the biotechnology and pharmaceutical industries. There can be no assurance that the Company’s licensing efforts will identify viable product candidates, that the Company’s research and development will be successfully completed, that adequate protection for the Company’s technology will be obtained, that any products developed will obtain necessary government regulatory approval or that any approved products will be commercially viable. The Company operates in an environment of substantial competition from other animal health companies. In addition, the Company is dependent upon the services of its employees and consultants, as well as third-party contract research organizations and manufacturers and collaborators.
Basis of Presentation
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“GAAP”) and on a basis which assumes that the Company will continue as a going concern and which contemplates the realization of assets and satisfaction of liabilities and commitments in the normal course of business.
The Company has incurred recurring losses and negative cash flows from operations and has an accumulated deficit of $152,018 as of December 31, 2015. The Company expects to continue to generate operating losses for the foreseeable future. The Company believes that its cash, cash equivalents and short-term investments on hand, will be sufficient to fund operations including, if necessary, the repayment of its long term debt, at least through December 31, 2016. As disclosed in Note 10 to the consolidated financial statements, the Company has a term loan and revolving credit facility with a principal balance of $40,000 as of December 31, 2015. The terms of this agreement require the Company to receive unrestricted net cash proceeds of at least $45,000 from partnering transactions and/or the issuance of equity securities from October 16, 2015 to October 16, 2016. The loan agreements also require that the Company have at least three products fully USDA-or FDA-approved for commercialization by December 31, 2016. If these conditions are not met, the Company may be required to repay the loan prior to December 31, 2016. The Company expects an increase in investment related to its commercial activities, including expanding commercial operations, milestones related to approval and commencement of commercial sales, and manufacturing activities in preparation for product launches. As a result, the Company may need additional capital to fund its operations beyond December 31, 2016, which the Company may obtain from corporate collaborations and licensing arrangements, or other sources, such as public or private equity and debt financings. The future viability of the Company beyond December 31, 2016 is dependent on its ability to raise additional capital to finance its operations, to fund on-going research and development costs, commercialization of its product candidates and satisfy debt covenants. If the Company is not able to raise additional capital on terms acceptable to it, or at all, as and when needed, it may be required to curtail its operations which could include delaying the commercial launch of its products, discontinuing product development programs, or granting rights to develop and market products or product candidates that it would otherwise prefer to develop and market itself. The Company’s failure to raise capital as and when needed would have a negative impact on its financial condition and its ability to pursue its business strategies as this capital is necessary for it to perform the research and development and commercial activities required to generate future revenue streams.
2. Summary of Significant Accounting Policies
Consolidation
The Company’s consolidated financial statements include its financial statements, and those of its wholly-owned subsidiaries and a consolidated variable interest entity. Intercompany balances and transactions are eliminated in consolidation.
To determine if the Company holds a controlling financial interest in an entity, the Company first evaluates if it is required to apply the variable interest entity (“VIE”) model to the entity. Where the Company holds current or potential rights that give it the power to direct the activities of a VIE that most significantly impact the VIE’s economic performance combined with a variable interest that gives it the right to receive potentially significant benefits or the obligation to absorb potentially significant losses, the Company is the primary beneficiary of that VIE. When changes occur to the design of an entity, the Company reconsiders whether it is subject to the VIE model. The Company continuously evaluates whether it is the primary beneficiary of a consolidated VIE.
F-10
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates, judgments and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Estimates are periodically reviewed in light of changes in circumstances, facts and experience. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company classifies all highly liquid investments with stated maturities of three months or less from the date of purchase as cash equivalents. Cash equivalents consisted of certificates of deposit (“CDs”) at December 31, 2015, and CDs and overnight reverse repurchase agreements at December 31, 2014.
Restricted Cash
Pursuant to the terms of the Loan and Security Agreement, the Company has posted collateral to Square 1 Bank N.A., a division of Pacific Western Bank, to collateralize corporate credit card services. The Company classifies the collateral as restricted cash.
Short-term Investments
The Company classifies reverse repurchase agreements other than overnight reverse repurchase agreements as short-term investments and as available-for-sale. Short-term investments for both 2015 and 2014 include reverse repurchase agreements and CDs with original maturities greater than three months.
Marketable Securities
The Company classifies all highly liquid investments with stated maturities of greater than three months from the date of purchase as marketable securities. The Company determines the appropriate classification of investments in marketable securities at the time of purchase and re-evaluates such designation at each consolidated balance sheet date. The Company classifies and accounts for marketable securities as available-for-sale. The Company may or may not hold securities with stated maturities greater than 12 months until maturity. After consideration of the risk versus reward objectives, as well as the Company’s liquidity requirements, the Company may sell these securities prior to their stated maturities. These securities are viewed as being available to support current operations. As a result, the Company classifies securities with maturities beyond 12 months as long-term assets as long-term marketable securities in the consolidated balance sheet. The Company reports available-for-sale investments at fair value as of each consolidated balance sheet date and records any unrealized gains and losses as a component of stockholders’ equity (deficit). At December 31, 2014, investments in equity securities were classified as long-term marketable securities and were carried at fair value, as determined by quoted market prices, and gains were recorded as a component of other comprehensive loss. No such investments were held as of December 31, 2015. The cost of securities sold is determined on a specific identification basis, and realized gains and losses are included in other income (expense) in the consolidated statements of operations. If any adjustment to fair value reflects a decline in the value of the investment, the Company considers available evidence to evaluate the extent to which the decline is “other than temporary” and recognizes the impairment by releasing other comprehensive income to the consolidated statement of operations. There were no such adjustments necessary during the years ended December 31, 2015 and 2014.
Accounts Receivable, net
Accounts receivable are uncollateralized customer obligations due under normal trade terms generally requiring payment within 30 days of the invoice date.
The Company provides an allowance for doubtful accounts equal to the estimated losses that will be incurred in collection of accounts receivable. This estimate is based on the current review of existing receivables and historical experience in the industry. The allowance and associated accounts receivable are reduced when the receivables are determined to be uncollectible.
Inventories
The Company states inventories at the lower of cost or market and consist of raw materials, work-in-process and finished goods. Cost is determined by the average cost method for raw materials and standard cost for work-in-process and finished goods, which approximates actual cost. Market is considered the lower of prevailing replacement cost or net realizable value. Inventories acquired in business combinations are recorded at fair value as of acquisition date.
F-11
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Property and Equipment, net
The Company records property and equipment at historical cost or, in the case of a business combination, at fair value on the date of the business combination, less accumulated depreciation and amortization. Depreciation amortization expense is recognized using the straight-line method over the following estimated useful lives:
|
Laboratory and office equipment |
3–10 years |
|
|
Computer software and equipment |
3–5 years |
|
|
Furniture |
3–7 years |
|
|
Vehicles |
3–5 years |
|
|
Leasehold improvements |
3–10 years |
Leasehold improvements are amortized over the shorter of the life of the related asset or the term of the lease.
Expenditures for repairs and maintenance of assets are charged to expense as incurred. Costs of major additions and betterments are capitalized and depreciated on a straight-line basis over their useful lives. When property and equipment are disposed of, the cost and respective accumulated depreciation and amortization are removed from the books. Any gain or loss on disposal is recorded in the consolidated statements of operations.
Goodwill
Goodwill relates to amounts that arose in connection with the Company’s business combinations (Note 17) and represents the difference between the purchase price and the estimated fair value of the identifiable tangible and intangible net assets when accounted for using the acquisition method of accounting. Goodwill is not amortized, but is subject to periodic review for impairment.
The Company tests goodwill at the reporting unit level for impairment on an annual basis and between annual tests, if events and circumstances indicate impairment may exist. Events that would indicate impairment and trigger an interim impairment assessment include, but are not limited to, current economic and market conditions, including a decline in market capitalization, a significant adverse change in legal factors, business climate or operational performance of the business and an adverse action or assessment by a regulator.
The Company completed its annual goodwill impairment testing during the third quarter of 2015. The Company elected to bypass the qualitative assessment. The Company determined as of the testing date that it consisted of one operating segment which is comprised of one reporting unit. In performing step one of the assessment, the Company determined that its fair value, determined to be its market capitalization, was greater than its carrying value, determined to be stockholders’ equity. Based on this result, step two of the assessment was not required to be performed, and the Company determined there was no impairment of goodwill during the third quarter of 2015.
During the fourth quarter of 2015, the Company completed an interim impairment assessment due to a decline in its market capitalization. In performing step one of the assessment, the Company determined that its fair value exceeded its carrying value, by 93%. Based on this result, step two of the assessment was not required to be performed, and the Company determined there was no impairment of goodwill during the fourth quarter of 2015.
Subsequent to December 31, 2015, the Company experienced further declines and fluctuations in its market capitalization. The extent and duration of any decline in the Company’s market capitalization will be assessed in future periods against the Company’s carrying value to assess if there is an indication of impairment of the Company’s goodwill which was $39,781 as of December 31, 2015.
Intangible Assets, net
The Company’s intangible assets consist of intellectual property rights acquired for currently marketed products and intellectual property rights acquired for in-process research and development (“IPR&D”). All of the Company’s intangible assets were recorded in connection with the Company’s business combinations (Note 17). The Company’s intangible assets are recorded at fair value at the time of their acquisition. The Company amortizes intangible assets over their estimated useful lives once the acquired technology is developed into a commercially viable product.
The estimated useful lives of the individual categories of intangible assets are based on the nature of the applicable intangible asset and the expected future cash flows to be derived from the intangible asset. Amortization of intangible assets with finite lives
F-12
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
is recognized over the time the intangible assets are estimated to contribute to future cash flows. The Company amortizes finite-lived intangible assets using the straight-line method.
Indefinite-lived IPR&D intangible assets are assessed for impairment at least annually. In addition, all intangible assets are reviewed for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. Factors the Company considers in deciding when to perform an impairment review include significant underperformance of the business in relation to expectations, significant negative industry or economic trends, and significant changes or planned changes in the use of the assets. If an impairment review is performed to evaluate a long-lived asset for recoverability, the Company compares forecasts of undiscounted cash flows for definite-lived intangible assets and discounted cash flows for indefinite-lived IPR&D intangible assets expected to result from the use and eventual disposition of the long-lived asset to its carrying value. An impairment loss would be recognized when estimated undiscounted (definite-lived) or discounted (indefinite-lived) future cash flows expected to result from the use of an asset are less than its carrying amount. The impairment loss would be based on the excess of the carrying value of the impaired asset over its fair value, determined based on discounted cash flows.
The Company completed its annual indefinite-lived IPR&D intangible assets impairment testing during the fourth quarter of 2015. The Company elected to bypass the qualitative assessment. For purposes of impairment testing, the fair value of the indefinite-lived IPR&D intangible assets was determined by using the framework of ASC 820, Fair Value Measurement. When determining the fair value of the indefinite-lived IPR&D intangible assets, the Company revisited all assumptions used in measuring the indefinite-lived IPR&D intangible assets at the time of acquisition, and evaluated and considered new and updated data and information available. The Company noted the fair values for all indefinite-lived IPR&D intangible assets were greater than the carrying value. As such, no impairment was recognized as of the annual testing date.
In the quarter ended September 30, 2015 and to date, the Company recorded $43,398 of impairment losses on intangible assets (Note 8).
Derivative Financial Instruments
The Company accounts for its derivative instruments as either assets or liabilities and carries them at fair value. The Company’s sole derivative (Note 9) was a warrant to purchase common stock and was adjusted to fair value through current income as it was not designated as a hedging instrument. In 2015, the Company exercised the warrant and subsequently sold the shares of common stock received upon exercise.
Foreign Currency
With the acquisition of Okapi Sciences (Note 17) in 2014, the Company is exposed to effects of foreign currency from translation. Transactions in foreign currencies are translated into the relevant functional currency at the rate of exchange at the date of the transaction. Transaction gains and losses are recognized in other income (expense) in the consolidated statements of operations. The results of operations for subsidiaries, whose functional currency is not the U.S. Dollar, are translated into the U.S. Dollar at the average rates of exchange during the period, with the subsidiaries’ balance sheets translated at the rates accumulated at the balance sheet date. The cumulative effect of these exchange rate adjustments is included in a separate component of other comprehensive income (loss) in the consolidated balance sheets. Gains and losses arising from intercompany foreign currency transactions are included in loss from operations unless the gains and losses arise from long-term investments in subsidiaries. Gains and losses from long-term investments in subsidiaries are included in a separate component of other comprehensive income (loss).
Business Combinations
The Company’s business acquisitions were made at a price above the fair value of the assets acquired and liabilities assumed, resulting in goodwill, based on the Company’s expectations of synergies and other benefits of combining the businesses. These synergies and benefits include elimination of redundant facilities, functions and staffing; use of the Company’s existing commercial infrastructure to expand sales of the products of the acquired businesses; and use of the commercial infrastructure of the acquired businesses to expand product sales in a cost-efficient manner.
Significant judgment is required in estimating the fair value of intangible assets and in assigning their respective useful lives. The fair value estimates are based on available historical information and on future expectations and assumptions deemed reasonable by management, but which are inherently uncertain.
The Company generally employs the income method to estimate the fair value of intangible assets, which is based on forecasts of the expected future cash flows attributable to the respective assets. Significant estimates and assumptions inherent in the valuations reflect a consideration of other marketplace participants, and include the amount and timing of future cash flows (including expected growth rates and profitability), the underlying product life cycles, economic barriers to entry, a brand’s relative market position and the discount rate applied to the cash flows. Unanticipated market or macroeconomic events and circumstances may occur, which could affect the accuracy or validity of the estimates and assumptions.
F-13
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Net assets acquired are recorded at their fair value and are subject to adjustment upon finalization of the fair value analysis. The Company is not aware of any information that indicates the final fair value analysis will differ materially from the preliminary estimates.
Contingent consideration is recorded as a liability and measured at fair value using a discounted cash flow model utilizing significant unobservable inputs, including the probability of achieving each of the potential milestones and an estimated discount rate commensurate with the risks of the expected cash flows attributable to the milestones. Significant increases or decreases in any of the probabilities of success would result in a significantly higher or lower fair value, respectively, and commensurate changes to this liability. At each reporting date, we revalue the contingent consideration obligations to the reporting date fair values and record increases and decreases in the fair values as income or expense in the consolidated statements of operations until actual settlement occurs.
Increases or decreases in the fair values of the contingent consideration obligations may result from changes in discount periods and rates, changes in the timing and amount of earn-out criteria and changes in probability assumptions with respect to the likelihood of achieving the various earn-out criteria.
On January 6, 2014, the Company acquired Okapi Sciences, a Leuven, Belgium based company with a proprietary antiviral platform and three clinical/development stage product candidates. The aggregate purchase price was approximately $44,439, which consisted of $14,139 in cash, a promissory note in the principal amount of $15,134 with a maturity date of December 31, 2014, and a contingent consideration of up to $16,308 with an acquisition fair value of $15,166. The promissory note bore interest at a rate of 7% per annum, payable quarterly in arrears, and was subject to mandatory prepayment in the event of a specified equity financing by the Company. On February 4, 2014, the promissory note and accrued interest was paid in cash in the amount of $15,158. On March 17, 2014, the contingent consideration was settled in cash in the amount of $15,235.
Deferred Public Offering Costs
The Company capitalizes certain legal, accounting and other third-party fees that are directly associated with in-process equity financings as other assets until such financings are consummated. After consummation of the equity financing, these costs are recorded in stockholders’ equity (deficit) as a reduction of additional paid-in capital generated as a result of the offering. Should it no longer be considered probable that the equity financing will be consummated, the deferred offering costs would be expensed immediately as a charge to operating expenses in the consolidated statements of operations. On October 16, 2015, the Company entered into a Sales Agreement with Barclays Capital Inc. (“Barclays”) pursuant to which the Company may sell from time to time, at its option, up to an aggregate of $52,000 of shares of its common stock (the “Shares”) through Barclays, as sales agent. As of the date of this filing, the Company has not sold any shares under the Sales Agreement. The Company recorded $189 and $0 of deferred public offering costs as of December 31, 2015 and 2014, respectively.
Income Taxes
The Company accounts for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the consolidated financial statements or in the Company’s tax returns. Deferred taxes are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect in the years in which the differences are expected to reverse. Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. The Company assesses the likelihood that its deferred tax assets will be recovered from future taxable income and, to the extent it believes, based upon the weight of available evidence, that it is more likely than not that all or a portion of deferred tax assets will not be realized, a valuation allowance is established through a charge to income tax expense. Potential for recovery of deferred tax assets is evaluated by estimating the future taxable profits expected and considering prudent and feasible tax planning strategies.
The Company accounts for uncertainty in income taxes recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the consolidated financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includes the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate as well as the related net interest and penalties.
Debt Issuance Costs, net
Debt issuance costs, net represent legal and other direct costs related to the Company’s Loan and Security Agreement (Note 10). These costs are recorded as an offset to the carrying value of loans payable on the consolidated balance sheet at the time they are incurred and are amortized to interest expense through the scheduled final principal payment date. During the year ended December 31, 2015, the Company capitalized $360 of debt issuance costs and recognized $129 of amortization expense related to debt
F-14
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
issuance costs. During the year ended December 31, 2014, the Company recorded $11 of debt issuance costs and recognized $41 of amortization expense related to debt issuance costs.
Revenue Recognition
The Company recognizes revenue when all of the following conditions are met:
|
· |
there is persuasive evidence of an agreement or arrangement; |
|
· |
delivery of products has occurred or services have been rendered; |
|
· |
the seller’s price to the buyer is fixed or determinable; and |
|
· |
collectibility is reasonably assured. |
The Company’s principal revenue streams and their respective accounting treatments are discussed below:
(i) Product sales - Revenue for the sale of products is recognized when delivery has occurred and substantially all the risks and rewards of ownership have been transferred to the customer. Revenue for the sale of products are recorded net of sales returns, allowances and discounts.
(ii) Licensing and collaboration revenues - Revenues derived from product out-licensing arrangements typically consist of an initial up-front payment on inception of the license and subsequent milestone payments contingent on the achievement of certain clinical and sales milestones. Product out licensing arrangements may require the Company to provide multiple deliverables to the licensee which would require the total selling price to be allocated between each of the separable elements in the arrangement based on their relative selling prices. An element is considered separable if it has value to the customer on a stand-alone basis. The selling price used for each separable element will be based on vendor specific objective evidence (“VSOE”) if available, third party evidence if VSOE is not available, or estimated selling price if neither VSOE nor third party evidence is available. Revenue is then recognized as each of the separable elements to which the revenue has been allocated is delivered.
Milestone payments which are non-refundable, non-creditable and contingent on achieving certain development, regulatory, or commercial milestones are recognized as revenues either on achievement of such milestones or over the period the Company has continuing substantive performance obligations.
Research and Development Costs
Research and development costs are expensed as incurred. Included in research and development costs are wages, stock-based compensation and employee benefits, and other operational costs related to the Company’s research and development activities, including facility-related expenses, external costs of outside contractors engaged to conduct both preclinical and clinical studies and allocation of corporate costs. Payments received from external parties to fund the Company’s research and development activities are used to reduce the Company’s research and development expenses. If IPR&D is acquired in an asset purchase then the acquired IPR&D is expensed on its acquisition date. Future costs to develop these assets are recorded to research and development expense as they are incurred.
Patent Costs
All patent-related costs incurred in connection with filing and prosecuting patent applications are recorded as selling, general and administrative expenses as incurred, as recoverability of such expenditures is uncertain.
Shipping
Shipping costs are included in cost of product sales.
Sales Tax
The Company collects and remits taxes assessed by various governmental authorities. These taxes may include sales, use and value added taxes. These taxes are recorded on a net basis and are excluded from sales.
Accounting for Stock-Based Compensation
The Company’s stock-based compensation program grants awards that may consist of stock options and restricted stock awards. The fair values of stock option grants are determined as of the date of grant using the Black-Scholes option pricing method. This method incorporates the fair value of the Company’s common stock at the date of each grant and various assumptions such as the risk-free interest rate, expected volatility based on the volatility of the Company’s common stock price, expected dividend yield, and expected term of the options. The fair values of restricted stock awards are determined based on the fair value of the Company’s common stock. Prior to the Company’s initial public offering of its common stock in June 2013, the fair value of the common stock was determined by management and the Board of Directors, on the date of grant. Beginning in the first quarter of 2014, the Company began to base
F-15
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
expected volatility on the historical volatility of its common stock, as adequate historical data regarding the volatility of its common stock price had become available.
The fair values of the stock-based awards, including the effect of estimated forfeitures, are then expensed over the requisite service period, which is generally the award’s vesting period. The Company classifies stock-based compensation expense in the consolidated statements of operations in the same manner in which the respective award recipient’s payroll costs are classified.
For stock-based awards granted to consultants and nonemployees, compensation expense is recognized over the period during which services are rendered by such consultants and nonemployees until completed. At the end of each financial reporting period prior to completion of the service, the value of these awards is re-measured using the then-current fair value of the Company’s common stock and updated assumption inputs in the Black-Scholes option pricing model.
For stock-based awards granted to employees under the 2010 Equity Incentive Plan (the “2010 Plan”), the Company allows employees to exercise certain awards prior to vesting. However, the employee may not sell or transfer these awards prior to vesting. For most of these awards, the Company has the right, but not the obligation, to repurchase any unvested (but issued) shares of common stock upon termination of employment or service at the lesser of (1) the original purchase price per share or (2) the fair value of the common share on the date of termination. If a stock option is early exercised in this circumstance, the consideration received for an exercise of an option is considered a deposit of the exercise price, and the related dollar amount is recorded as a liability. The unvested shares and liability are reclassified to equity as the award vests. The Company has 90 days from the effective termination of employment or service to repurchase unvested shares that are issued upon the exercise of a stock option prior to its vesting. If, after 90 days, the Company has elected not to repurchase the unvested shares, the shares would become vested in full. The Company would then apply modification accounting and any resulting compensation expense would be immediately recognized related to the award. Upon vesting, these shares would be considered issued and outstanding shares of common stock for accounting purposes.
In addition, the Company has granted restricted stock awards subject to repurchase to one employee, under which the Company has the right, but not the obligation, upon termination of the holder’s employment or service, to repurchase unvested shares at the greater of (1) the original purchase price per share or (2) the fair value of the common share on the date of termination (Note 14).
Comprehensive Loss
In addition to the Company’s net loss, comprehensive loss during the years ended December 31, 2015 and 2014 includes foreign currency translation adjustments related to the translation of foreign subsidiaries’ balance sheets and unrealized holding gains and losses on available-for-sale securities. For the year ended December 31, 2013, there was no difference between net loss and comprehensive loss.
Net Loss Per Share
The Company follows the two-class method when computing net loss per share, as the Company has issued shares that meet the definition of participating securities. The two-class method determines net loss per share for each class of common and participating securities according to dividends declared or accumulated and participation rights in undistributed earnings. The two-class method requires income available to common stockholders for the period to be allocated between common and participating securities based upon their respective rights to receive dividends as if all income for the period had been distributed.
Restricted stock awards granted by the Company entitle the holder of such awards to dividends declared or paid by the Board of Directors, regardless of whether such awards are unvested, as if such shares were outstanding common shares at the time of the dividend. However, the unvested restricted stock awards are not entitled to share in the residual net assets (deficit) of the Company. Accordingly, in periods in which the Company reports a net loss or a net loss attributable to common stockholders resulting from preferred stock dividends, accretion or modifications, net losses are not allocated to participating securities. The Company reported a net loss in each of the years ended December 31, 2015, 2014 and 2013.
Basic net loss per share is computed by dividing the net loss by the weighted average number of shares of common stock outstanding for the period. Diluted net loss is computed by adjusting net loss to reallocate undistributed earnings based on the potential impact of dilutive securities, including outstanding stock options. Diluted net loss per share is computed by dividing the diluted net loss by the weighted average number of shares of common stock, including potential dilutive shares of common stock assuming the dilutive effect of potentially dilutive securities. For periods in which the Company has reported net losses, diluted net loss per share is the same as basic net loss per share, since their impact would be anti-dilutive to the calculation of net loss per share. Diluted net loss per share is the same as basic net loss per share for each of the years ended December 31, 2015, 2014 and 2013.
Concentration of Credit Risk and of Significant Suppliers and Customers
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash, cash equivalents, short-term investments and accounts receivable. At December 31, 2015, the Company’s short-term investments included reverse repurchase agreements that are tri-party, have maturities of three months or less at the time of investment and the underlying collateral is U.S. government securities including U.S. treasuries, agency debt and agency mortgage securities. At December 31, 2015
F-16
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
and 2014, all of the Company’s fixed income marketable securities were invested in CDs insured by the Federal Deposit Insurance Corporation (“FDIC”). The Company also generally maintains balances in various operating accounts in excess of federally insured limits at two accredited financial institutions. The Company does not believe that it is subject to unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
The Company is dependent on a small number of third-party manufacturers to supply active pharmaceutical ingredients (“API”), and formulated drugs for research and development activities in its programs and commercial supply, which would be adversely affected by a significant interruption in supply.
Fair Value Measurements
Certain assets and liabilities are carried at fair value under GAAP. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. A fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last is considered unobservable, is used to measure fair value:
|
· |
Level 1—Quoted prices in active markets for identical assets or liabilities. |
|
· |
Level 2—Observable inputs (other than Level 1 quoted prices) such as quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities, or other inputs that are observable or can be corroborated by observable market data. |
|
· |
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to determining the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques. |
Segment and Geographic Information
Segment Assets
The Company manages its operations as a single segment for the purposes of assessing performance and making operating decisions. The Company is a pet therapeutics company developing compounds to address unmet and under-served medical needs in companion animals. All assets were held in the United States and Belgium as of December 31, 2015 and December 31, 2014. Total assets were $147,066 and $207,903 at December 31, 2015, and 2014, respectively.
Revenue by Geographic Region
|
YEAR ENDED DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
(dollars in thousands) |
|||||||||
|
Revenues |
|||||||||
|
United States |
$ |
678 |
$ |
267 |
$ |
123 | |||
|
Belgium |
— |
500 |
NA |
||||||
|
Total revenues |
$ |
678 |
$ |
767 |
$ |
123 | |||
Long Lived Assets by Geographic Region
|
DECEMBER 31, |
||||||
|
2015 |
2014 |
|||||
|
(dollars in thousands) |
||||||
|
Long-lived assets |
||||||
|
United States |
$ |
2,460 |
$ |
459 | ||
|
Belgium |
95 | 161 | ||||
|
Total long-lived assets |
$ |
2,555 |
$ |
620 | ||
F-17
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
New Accounting Standards
Revenue from Contracts with Customers
In May 2014, the Financial Accounting Standards Board (“FASB”) issued guidance on recognizing revenue in contracts with customers. The guidance affects any entity that either enters into contracts with customers to transfer goods or services or enters into contracts for the transfer of nonfinancial assets unless those contracts are within the scope of other standards (e.g., insurance contracts or lease contracts). This guidance will supersede the revenue recognition requirements in topic, Revenue Recognition, and most industry-specific guidance. This guidance also supersedes some cost guidance included in subtopic, Revenue Recognition – Construction-Type and Production-Type Contracts. In addition, the existing requirements for the recognition of a gain or loss on the transfer of nonfinancial assets that are not in a contract with a customer (e.g., assets within the scope of topic, Property, Plant, and Equipment, and tangible assets within the scope of topic, Intangibles – Goodwill and Other) are amended to be consistent with the guidance on recognition and measurement (including the constraint on revenue) in this guidance.
The core principle of the guidance is that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services.
These changes become effective for the Company on January 1, 2017 and early adoption is not permitted. The standard permits the use of either the retrospective or cumulative effect transition method. The Company is currently assessing the impact, if any, this new guidance will have on its consolidated financial statements.
In July 2015, the FASB approved a one-year delay in the effective date of the new revenue standard. These changes become effective for the Company on January 1, 2018, and early adoption is permitted but not before the original effective date of January 1, 2017. The Company is currently assessing the impact, if any, this new guidance will have on its consolidated financial statements.
Interest—Imputation of Interest: Simplifying the Presentation of Debt Issuance Costs
In April 2015, the FASB issued guidance which requires debt issuance costs to be presented in the balance sheet as a direct deduction from the associated debt liability. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2015, and interim periods within those fiscal years. Early adoption is permitted and is to be applied on a retrospective basis. The Company elected to early adopt this guidance retrospectively starting in the fourth quarter of 2015. The Company determined that the retrospective application did not have had a material impact on its consolidated financial statements.
Inventory – Simplifying the Measurement of Inventory
In July 2015, the FASB issued guidance which requires entities to measure most inventory “at lower of cost and net realizable value” thereby simplifying the current guidance under which an entity must measure inventory at the lower of cost or market. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2016, and interim periods within those fiscal years. Early adoption is permitted and is to be applied on a prospective basis. The Company does not expect that this new guidance will have a material impact on its consolidated financial statements.
Income Taxes – Balance Sheet Classification of Deferred Taxes
In November 2015, the FASB issued guidance which requires entities to present deferred tax assets and deferred tax liabilities as noncurrent in a classified balance sheet. This guidance simplifies the current guidance in ASC Topic 740, Income Taxes, which requires entities to separately present deferred tax assets and liabilities as current and noncurrent in a classified balance sheet. This guidance is effective for fiscal years beginning after December 15, 2016, and interim periods within those annual periods, with early adoption permitted. During the fourth quarter of 2015, the Company early adopted the guidance as of December 31, 2015 on a prospective basis; prior periods were not retrospectively adjusted. Due to a full valuation allowance recorded against its deferred tax assets as of December 31, 2015, the Company’s consolidated balance sheet contains no deferred tax balances.
Leases
In February 2016, the FASB issued guidance which requires, for operating leases, a lessee to recognize a right-of-use asset and a lease liability, initially measured at the present value of the lease payments, in its balance sheet. The standard also requires a lessee to recognize a single lease cost, calculated so that the cost of the lease is allocated over the lease term, on a generally straight-line basis. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. Early adoption is permitted and is to be applied on a modified retrospective transition. The Company is currently assessing the effect that adoption of this guidance will have on its consolidated financial statements.
F-18
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
3. Fair Value of Financial Assets and Liabilities
Financial Assets and Liabilities Measured at Fair Value on a Recurring Basis
The following financial assets and liabilities are measured at fair value on a recurring basis using quoted prices in active markets for identical assets (Level 1); significant other observable inputs (Level 2); and significant unobservable inputs (Level 3).
|
FAIR VALUE MEASUREMENTS AS OF |
|||||||||||||||
|
CARRYING |
DECEMBER 31, 2015 USING: |
||||||||||||||
|
VALUE |
LEVEL 1 |
LEVEL 2 |
LEVEL 3 |
TOTAL |
|||||||||||
|
Assets: |
|||||||||||||||
|
Cash equivalents: |
|||||||||||||||
|
Certificates of deposit |
$ |
6,972 |
$ |
— |
$ |
6,972 |
$ |
— |
$ |
6,972 | |||||
|
Money market fund |
35 | 35 |
— |
— |
35 | ||||||||||
|
Short-term investments: |
|||||||||||||||
|
Short-term marketable securities – certificates of deposit |
747 |
— |
747 |
— |
747 | ||||||||||
|
Reverse repurchase agreements |
58,700 |
— |
58,700 |
— |
58,700 | ||||||||||
|
$ |
66,454 |
$ |
35 |
$ |
66,419 |
$ |
— |
$ |
66,454 | ||||||
|
FAIR VALUE MEASUREMENTS AS OF |
|||||||||||||||
|
CARRYING |
DECEMBER 31, 2014 USING: |
||||||||||||||
|
VALUE |
LEVEL 1 |
LEVEL 2 |
LEVEL 3 |
TOTAL |
|||||||||||
|
Assets: |
|||||||||||||||
|
Cash equivalents: |
|||||||||||||||
|
Certificates of deposit |
$ |
6,972 |
$ |
— |
$ |
6,972 |
$ |
— |
$ |
6,972 | |||||
|
Money market fund |
45 |
— |
45 |
— |
45 | ||||||||||
|
Short-term investments: |
|||||||||||||||
|
Short-term marketable securities – certificate of deposit |
249 |
— |
249 |
— |
249 | ||||||||||
|
Reverse repurchase agreements |
88,000 |
— |
88,000 |
— |
88,000 | ||||||||||
|
Long-term marketable securities: |
|||||||||||||||
|
Common stock |
2,452 | 2,452 |
— |
— |
2,452 | ||||||||||
|
Derivative financial instruments |
1,108 |
— |
1,108 |
— |
1,108 | ||||||||||
|
$ |
98,826 |
$ |
2,452 |
$ |
96,374 |
$ |
— |
$ |
98,826 | ||||||
|
Liabilities: |
|||||||||||||||
|
Contingent consideration |
$ |
4,248 |
$ |
— |
$ |
— |
$ |
4,248 |
$ |
4,248 | |||||
|
$ |
4,248 |
$ |
— |
$ |
— |
$ |
4,248 |
$ |
4,248 | ||||||
Certain estimates and judgments are required to develop the fair value amounts shown above. The fair value amounts shown above are not necessarily indicative of the amounts that the Company would realize upon disposition, nor do they indicate the Company’s intent or ability to dispose of the financial instrument.
The following methods and assumptions were used to estimate the fair value of each material class of financial instrument:
|
· |
Cash equivalents – the fair value of the cash equivalents has been determined to be amortized cost or has been based on the quoted prices in active markets or exchanges for identical assets. |
F-19
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
|
· |
Reverse repurchase agreements – the fair value of reverse repurchase agreements has been determined to be amortized cost given the short duration of the agreements. |
|
· |
Marketable securities (long-term) – the fair value of marketable securities has been based on quoted prices in active markets or exchanges for identical assets. |
|
· |
Marketable securities (short-term) – the fair value of marketable securities has been determined to be amortized cost given the short duration of the agreements. |
|
· |
Derivative financial instruments – the fair value of the derivative instruments has been estimated using a modified Black-Scholes model. Inputs into the Black-Scholes model include interest rates, stock volatilities and dividend data. |
|
· |
Contingent consideration – the fair value of the contingent consideration payable has been estimated using the income approach using a probability weighted discounted cash flow method. Inputs into the discounted cash flow method include the probability of and period in which the relevant milestone event is expected to be achieved and the discount rate to be applied in calculating the present values of the relevant milestones. |
Transfers Between Levels of the Fair Value Hierarchy
Transfers between levels of the fair value hierarchy are reported at the beginning of the reporting period in which they occur. During the year ended December 31, 2014, transfer of investments between Level 1 and Level 2 were $2,452. In the third quarter of 2014, the Company re-classified its common stock investment in Advaxis (Note 12) to a Level 1 investment from Level 2, because the Company was no longer precluded from selling shares due to restrictions imposed by Rule 144 of the Securities Act of 1933. Previously, the Company calculated a lack of marketability discount on the fair value of the Advaxis common stock because of trading restrictions on the shares. The Company considered the inputs used to calculate the lack of marketability discount Level 2 inputs and, as a result, the Company classified this investment as Level 2. The Company determined the lack of marketability discount by using a Black-Scholes model to value a hypothetical put option to approximate the cost of hedging the stock until the restriction ended.
During the years ended December 31, 2015 and 2013, there were no transfers between levels of the fair value hierarchy.
Financial Assets and Liabilities Measured at Fair Value on a Recurring Basis Using Significant Unobservable Inputs (Level 3)
The change in the fair value of the Company’s contingent consideration payable as of December 31, 2015, which is measured at fair value on a recurring basis using significant unobservable inputs (Level 3), was as follows:
Contingent consideration
|
2015 |
2014 |
|||||
|
As of January 1, |
$ |
4,248 |
$ |
4,115 | ||
|
Initial recognition of contingent consideration payable |
— |
15,166 | ||||
|
Cash settlement of contingent consideration earned |
(3,000) | (15,235) | ||||
|
Derecognition of remaining contingent consideration recorded in the consolidated statement of operations (within selling, general and administrative) |
(1,248) | 202 | ||||
|
As of December 31, |
$ |
— |
$ |
4,248 | ||
On January 2, 2015, the Company was granted a full product license for BLONTRESS® (also known as AT-004). The approval resulted in $3,000 of the contingent consideration being earned and due to the former Vet Therapeutics, Inc. (“Vet Therapeutics”) shareholders per the terms of Vet Therapeutics merger agreement. Further, on February 24, 2015, in connection with the mutual termination of the Elanco Animal Health, Inc. (“Elanco”) Agreement for BLONTRESS (Note 12), the Company obtained consent from the shareholder representative of the former Vet Therapeutics shareholders that the $3,000 payment shall cause the Company to have no further obligation or liability under the merger agreement. The Company paid the $3,000 contingent consideration in March 2015. During the year ended December 31, 2015, the Company recorded a credit of $1,248 to selling, general and administrative expense to reduce the fair value of the contingent consideration to zero as a result of the agreement with the Vet Therapeutics shareholders.
F-20
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Financial Assets and Liabilities that are not Measured at Fair Value on a Recurring Basis
The carrying amounts and estimated fair value of the Company’s financial assets and liabilities which are not measured at fair value on a recurring basis was as follows:
|
DECEMBER 31, 2015 |
||||||
|
CARRYING VALUE |
FAIR VALUE |
|||||
|
Financial liabilities: |
||||||
|
Loan payable (Level 2) |
$ |
39,710 |
$ |
40,569 | ||
|
DECEMBER 31, 2014 |
||||||
|
CARRYING VALUE |
FAIR VALUE |
|||||
|
Financial liabilities: |
||||||
|
Loan payable (Level 2) |
$ |
14,963 |
$ |
14,933 | ||
Certain estimates and judgments were required to develop the fair value amounts. The fair value amount shown above is not necessarily indicative of the amounts that the Company would realize upon disposition, nor do they indicate the Company’s intent or ability to dispose of the financial instrument.
The fair value of loan payable was estimated using discounted cash flow analysis discounted at current rates.
Fair value information about the intangible assets that were impaired during the year ended December 31, 2015 (Note 8) was as follows:
|
FAIR VALUE |
|||||||||||||||
|
CARRYING |
DECEMBER 31, 2015 |
||||||||||||||
|
VALUE |
LEVEL 1 |
LEVEL 2 |
LEVEL 3 |
IMPAIRMENT |
|||||||||||
|
Intellectual property rights for currently marketed products |
$ |
6,057 |
$ |
— |
$ |
— |
$ |
6,057 |
$ |
28,862 | |||||
|
Intellectual property rights acquired for in-process research and development |
9,010 |
— |
— |
9,010 | 14,536 | ||||||||||
|
$ |
15,067 |
$ |
— |
$ |
— |
$ |
15,067 |
$ |
43,398 | ||||||
The fair value amount is presented as of the date of impairment, as these assets are not measured at fair value on a recurring basis. (Note 8).
The fair value reflects intangible assets written down to fair value during the year ended December 31, 2015. Fair-value was determined using the income approach, specifically the multi-period excess earnings method, a form of a discounted cash flow method. The Company started with a forecast of all the expected net cash flows associated with the asset and then it applied an asset-specific discount rate to arrive at a net present value amount. Some of the more significant estimates and assumptions inherent in this approach include: the amount and timing of the projected net cash flows, which includes the expected impact of competitive legal and/or regulatory forces on the product and the impact of technological risk associated with IPR&D assets; the discount rate, which seeks to reflect the various risks inherent in the projected cash flows; and the tax rate, which seeks to incorporate the geographic diversity of the projected cash flows.
F-21
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
4. Investments
Marketable Securities
Marketable securities consisted of the following:
|
DECEMBER 31, 2015 |
||||||||||||
|
GROSS |
GROSS |
|||||||||||
|
AMORTIZED |
UNREALIZED |
UNREALIZED |
FAIR |
|||||||||
|
COST |
GAINS |
LOSSES |
VALUE |
|||||||||
|
Short-term marketable securities: |
||||||||||||
|
Certificates of deposit |
$ |
747 |
$ |
— |
$ |
— |
$ |
747 | ||||
|
Total |
$ |
747 |
$ |
— |
$ |
— |
$ |
747 | ||||
|
DECEMBER 31, 2014 |
||||||||||||
|
GROSS |
GROSS |
|||||||||||
|
AMORTIZED |
UNREALIZED |
UNREALIZED |
FAIR |
|||||||||
|
COST |
GAINS |
LOSSES |
VALUE |
|||||||||
|
Short-term marketable securities: |
||||||||||||
|
Certificate of deposit |
$ |
249 |
$ |
— |
$ |
— |
$ |
249 | ||||
|
Long-term marketable securities: |
||||||||||||
|
Common stock |
1,200 | 1,252 |
— |
2,452 | ||||||||
|
Total |
$ |
1,449 |
$ |
1,252 |
$ |
— |
$ |
2,701 | ||||
At December 31, 2015 and 2014, short-term marketable securities consisted of investments that mature within one year. Short-term marketable securities are recorded as short-term-investments in the consolidated balance sheets.
At December 31, 2014, unrealized gains in the amount of $1,252 were recorded as a component of other comprehensive loss.
Reverse Repurchase Agreements
The Company, as part of its cash management strategy, may invest excess cash in reverse repurchase agreements. All reverse repurchase agreements are tri-party and have maturities of three months or less at the time of investment. The underlying collateral is U.S. government securities including U.S. treasuries, agency debt and agency mortgage securities. The underlying collateral posted by each counterparty is required to cover 102% of the principal amount and accrued interest after the application of a discount to fair value.
5. Inventories
Inventories are stated at the lower of cost or market and comprised of the following:
|
DECEMBER 31, |
DECEMBER 31, |
|||||
|
2015 |
2014 |
|||||
|
Raw materials |
$ |
120 | 115 | |||
|
Work-in-process |
441 |
$ |
206 | |||
|
Finished goods |
745 | 106 | ||||
|
$ |
1,306 |
$ |
427 | |||
F-22
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
6. Property and Equipment, Net
Property and equipment, net consisted of the following:
|
DECEMBER 31, |
DECEMBER 31, |
|||||
|
2015 |
2014 |
|||||
|
Laboratory and office equipment |
$ |
527 |
$ |
478 | ||
|
Computer equipment and software |
2,039 | 68 | ||||
|
Furniture |
132 | 71 | ||||
|
Vehicles |
11 | 34 | ||||
|
Leasehold improvements |
91 | 102 | ||||
|
Construction in process |
185 | 49 | ||||
|
Total property and equipment |
2,985 | 802 | ||||
|
Less: Accumulated depreciation and amortization |
(430) | (182) | ||||
|
Property and equipment, net |
$ |
2,555 |
$ |
620 | ||
Depreciation expense was $296, $152 and $16 for the years ended December 31, 2015, 2014 and 2013, respectively. No significant gains/losses were recognized during the years ended December 31, 2015, 2014 and 2013.
7. Goodwill
Goodwill is recorded as an indefinite-lived asset and is not amortized for financial reporting purposes but is tested for impairment on an annual basis or when indications of impairment exist. No goodwill impairment losses have been recognized. Goodwill is not expected to be deductible for income tax purposes. The Company performs its annual impairment test of the carrying value of the Company’s goodwill during the third quarter of each year.
The Company completed its annual goodwill impairment testing during the third quarter of 2015. The Company elected to bypass the qualitative assessment. The Company determined as of the testing date that it consisted of one operating segment which is comprised of one reporting unit. In performing step one of the assessment, the Company determined that its fair value, determined to be its market capitalization, was greater than its carrying value, determined to be stockholders’ equity. Based on this result, step two of the assessment was not required to be performed, and the Company determined there was no impairment of goodwill during the third quarter of 2015.
The Company during the fourth quarter of 2015, completed an interim impairment assessment due to a decline in market capitalization. In performing step one of the assessment, the Company determined that its fair value exceeded its carrying value by 93%. Based on this result, step two of the assessment was not required to be performed, and the Company determined there was no impairment of goodwill during the fourth quarter of 2015.
Subsequent to December 31, 2015, the Company experienced further declines and fluctuations in its market capitalization. The extent and duration of any decline in the Company’s market capitalization will be assessed in future periods against the Company’s carrying value to assess if there is an indication of impairment of the Company’s goodwill which was $39,781 as of December 31, 2015.
F-23
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Goodwill as of December 31, 2015, was as follows:
|
GROSS |
IMPAIRMENT |
NET |
|||||||
|
CARRYING AMOUNT |
LOSSES |
CARRYING VALUE |
|||||||
|
Goodwill |
$ |
39,781 |
$ |
— |
$ |
39,781 | |||
The change in the net book value of goodwill for the years ended December 31, 2015 and 2014, was as follows:
|
2015 |
2014 |
|||||
|
As of January 1, |
$ |
41,398 |
$ |
25,646 | ||
|
Acquisition |
— |
17,909 | ||||
|
Effect of foreign currency exchange |
(1,617) | (2,157) | ||||
|
As of December 31, |
$ |
39,781 |
$ |
41,398 | ||
8. Intangible Assets, net
The change in the net book value of intangible assets for the years ended December 31, 2015 and 2014, was as follows:
|
2015 |
2014 |
|||||
|
As of January 1, |
$ |
62,323 |
$ |
38,354 | ||
|
Acquisitions |
— |
29,400 | ||||
|
Amortization expense |
(1,544) | (1,891) | ||||
|
Effect of foreign currency exchange |
(2,314) | (3,540) | ||||
|
Impairment |
(43,398) |
— |
||||
|
As of December 31, |
$ |
15,067 |
$ |
62,323 | ||
The estimated useful lives of the individual categories of intangible assets were based on the nature of the applicable intangible asset and the expected future cash flows to be derived from the intangible asset. Amortization of intangible assets with finite lives is recognized over the shorter of the respective lives of the agreement or the period of time the intangible assets are expected to contribute to future cash flows. The Company amortizes finite-lived intangible assets using the straight-line method. Amortization of intangible assets for the years ended December 31, 2015, 2014 and 2013 amounted to $1,544, $1,891 and $298, respectively. Indefinite-lived IPR&D intangible assets are not amortized until a product reaches its first conditional license or approval, then they are amortized over their estimated useful life.
Summary of estimated aggregate amortization expense of intangible assets for each of the five succeeding years as of December 31, 2015, was as follows:
|
YEAR ENDING DECEMBER 31, |
|||
|
2016 |
$ |
381 | |
|
2017 |
381 | ||
|
2018 |
381 | ||
|
2019 |
381 | ||
|
2020 |
$ |
381 |
F-24
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Unamortized Intangible Assets
In January 2014, the Company completed its acquisition of Okapi Sciences (Note 17). The Company acquired certain identifiable intangible assets which relate to IPR&D.
Unamortized intangible assets as of December 31, 2015 and 2014, were as follows:
|
NET |
||||||
|
CARRYING |
||||||
|
VALUE |
||||||
|
AS OF DECEMBER 31, |
||||||
|
Unamortized intangible assets: |
2015 |
2014 |
||||
|
Intellectual property rights acquired for in-process research and development |
$ |
9,010 |
$ |
25,860 | ||
Impairment of Unamortized Intangible Assets
AT-007
Based on the completed pilot study results for AT-007 received late in the first quarter of 2015, the Company determined additional pilot studies were needed before advancing to a pivotal program. During the third quarter of 2015, the Company met with key opinion leaders to review the pilot study results, determine the relevant treatment population and develop acceptable study protocols. The Company then determined that the best course forward would be to study the product in a more limited clinical setting. The market forecast in the more limited clinical setting required the Company to reassess the target market size and change the treatment regimen assumed at the time of acquisition. Furthermore, the Company concluded that the likely customers for this product would be shelters and non-profits, thereby putting pressure on pricing. Also, while a Minor-Use-Minor-Species (“MUMS”) designation would potentially be available for the more limited indication in the United States, the MUMS pathway would not be available in Europe since other products are available there for this disease. The above factors caused the Company to record an impairment charge of $8,717, resulting in the net carrying value of $2,202 for AT-007. The Company intends to explore out-licensing AT-007, and it may consider advancing the program internally at a later time. The Company determined the fair value using a multi-period excess earnings method, a form of the income approach, which incorporates the estimated future cash flows to be generated from this technology. Excess earnings are the earnings remaining after deducting the market rates of return on the estimated values of contributory assets, including debt-free net working capital, tangible, and intangible assets. The excess earnings are thereby calculated for each year of a multi-year projection period and discounted to present value. Accordingly, the primary components of this method consist of the determination of excess earnings and an appropriate rate of return.
AT-011
For AT-011, the Company has been conducting early pre-development studies, including lead selection and proof of concept on several molecules. During the third quarter of 2015, the Company completed its evaluation of AT-011. Based on this evaluation, the Company determined that none of the molecules being evaluated were suitable for advancement in development. As such, the
F-25
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Company decided to abandon the development of AT-011, resulting in an impairment charge of $5,819, the full carrying value of AT-011.
Unfavorable outcomes of the Company’s development activities or the Company’s estimates of the market opportunities for the product candidates could result in impairment charges in future periods.
Amortized Intangible Assets
Amortized intangible assets as of December 31, 2015, were as follows:
|
GROSS |
NET |
|||||||||||
|
CARRYING |
ACCUMULATED |
CARRYING |
AVERAGE |
|||||||||
|
Amortized intangible assets: |
VALUE |
AMORTIZATION |
VALUE |
USEFUL LIFE |
||||||||
|
Intellectual property rights for currently marketed products: |
||||||||||||
|
BLONTRESS |
$ |
28,572 |
$ |
23,103 |
$ |
5,469 | 20 |
Years |
||||
|
TACTRESSTM(1) |
$ |
10,080 |
$ |
9,492 |
$ |
588 | 8.25 |
Years |
||||
(1) TACTRESS received a conditional license in early 2014 which initiated amortization for this intangible.
Accumulated amortization includes both amortization expense and asset impairment charges. Asset impairment charges to date are $20,228 and $8,634 for BLONTRESS and TACTRESSTM (also known as AT-005), respectively.
Impairment of Amortized Intangible Assets
BLONTRESS and TACTRESS
Since the Vet Therapeutics acquisition (October 2013) the Company has been performing various scientific and clinical activities to gain further knowledge around the science and efficacy of BLONTRESS and TACTRESS. The Company’s analysis of the clinical results indicated that TACTRESS did not seem to be adding significant progression free survival in canine T-cell lymphoma. In addition, scientific studies suggested that TACTRESS is not as specific to the target as expected. Given these clinical and scientific results, the Company no longer believes that TACTRESS will capture the desired T-cell lymphoma market opportunity.
TACTRESS is commercially available to all oncologists. The Company expects that some oncologists will continue to use TACTRESS in certain, limited settings. Feedback from its oncology advisors and oncologists is that while median progression-free survival would have been important for broad use, there is interest to explore the product in individual dogs especially given the limited effective treatment options in canine T-cell lymphoma.
With respect to BLONTRESS, scientific studies suggest that BLONTRESS is not as specific to the target as previously expected. The Company’s market research and interactions with veterinary oncologists indicate that high specificity, including binding and depletion, will likely be necessary to drive wide adoption of monoclonal antibody therapy given that canine B-cell is generally chemotherapy sensitive. Furthermore, the Company is aware of other emerging therapies that will compete in the B-cell lymphoma market, and believes that products with break-through benefit will dominate the market.
The first generation products, BLONTRESS and TACTRESS, are expected to continue to be available or become available to oncologists insofar as they are USDA licensed and the Company is manufacturing these products today. The Company believes the revenue and gross margin opportunity for the first generation monoclonal antibodies will be very modest and given that there are not alternative monoclonal antibodies available to veterinarian oncologists, the Company intends to maintain product availability. The Company intends to build awareness of monoclonal antibody therapy with BLONTRESS and TACTRESS and pursue second generation monoclonal antibodies and other product concepts in lymphoma. These second generation products are likely several years away. The Company also intends to improve the gross margins for the second generation products. If the Company determines that it is unlikely to be successful in developing products with sufficient benefits at an economic gross margin, it would consider phasing out BLONTRESS and TACTRESS depending on product volumes.
The Company deemed the events and market projections described above to be indicators of potential impairment of its finite-lived intangible assets of BLONTRESS and TACTRESS during the third quarter of 2015. The Company performed impairment testing for intangible assets BLONTRESS and TACTRESS as of September 30, 2015 and recorded an impairment expense of $20,228 and $8,634, resulting in a net carrying value of $5,546 and $606 for BLONTRESS and TACTRESS, respectively.
F-26
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
In conjunction with the Company’s impairment assessment, the Company re-evaluated the useful lives of BLONTRESS and TACTRESS and concluded that the useful life of BLONTRESS will remain at 20 years and the useful life of TACTRESS will be adjusted to 8.25 years. The conclusion of no change in the useful life for BLONTRESS was determined due to the fact that if and when competing therapies come, BLONTRESS for canine B-cell lymphoma is likely to have some demand that can be fulfilled at lower but economically viable production volumes. The new lower useful life for TACTRESS was concluded due to the Company determining that if and when competing therapies come, fulfilling the residual demand for TACTRESS for canine T-cell lymphoma at the lower production volumes would no longer be economically viable. The lower volume assumptions for TACTRESS, reflect the fact that the incidence of canine B-cell lymphoma is three-to-four times the incidence of canine T-cell lymphoma.
The Company determined the fair value for both BLONTRESS and TACTRESS using a multi-period excess earnings method, a form of the income approach, which incorporates the estimated future cash flows to be generated from these technologies. Excess earnings are the earnings remaining after deducting the market rates of return on the estimated values of contributory assets, including debt-free net working capital, tangible and intangible assets. The excess earnings are thereby calculated for each year of a multi-year projection period and discounted to present value. Accordingly, the primary components of this method consist of the determination of excess earnings and an appropriate rate of return.
Prior to the impairment assessment in the third quarter of 2015 as discussed above, the Company had conducted an impairment assessment in the first quarter of 2015 following the Company and Elanco mutually terminating the Elanco Agreement associated with BLONTRESS (Note 8) and concluded that the BLONTRESS intangible asset was not impaired at that time of the termination. As part of the assessment, the Company considered that the product would be made available in the second half of 2015, as well as manufacturing expenses, technology royalties, post-approval studies, marketing, and selling expenses to commercialize the product.
Unfavorable outcomes of the Company’s development activities or the Company’s estimates of the market opportunities for the product candidates could result in impairment charges in future periods.
9. Derivative Financial Instruments
The Company records all derivatives in the consolidated balance sheets at fair value in other long-term assets. In 2015, the Company’s derivative financial instrument, the Advaxis warrant, was not designated as a hedging instrument and was adjusted to fair value through earnings in other income (expense). During the year ended December 31, 2015, the Company exercised the Advaxis warrant (Note 12) and subsequently sold the shares of common stock received upon exercise.
The Company’s derivative instrument at gross fair value:
|
FAIR VALUE OF DERIVATIVES NOT |
||||||
|
DESIGNATED AS HEDGING INSTRUMENT |
||||||
|
DECEMBER 31, 2015 |
DECEMBER 31, 2014 |
|||||
|
Derivative assets: |
||||||
|
Warrant (Notes 3 and 12) |
$ |
— |
$ |
1,108 | ||
The gain recognized in other income (expense) for the year ended:
|
GAIN RECOGNIZED IN |
|||||||||
|
OTHER INCOME (EXPENSE) |
|||||||||
|
YEAR ENDED |
|||||||||
|
DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
Derivative assets: |
|||||||||
|
Warrant |
$ |
1,274 |
$ |
465 |
$ |
NA |
|||
During the year ended December 31, 2015, the Company exercised the warrant and recognized a gain of $1,274 in other income (expense), and subsequently sold the shares of common stock received upon exercise and recognized a gain of $341 in other income (expense).
F-27
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
10. Debt
Loan and Security Agreement
On October 16, 2015, the Company entered into a Loan and Security Agreement with Pacific Western Bank (“Pacific Western Bank”) as collateral agent and a lender and Oxford Finance LLC (“Oxford” and together with Pacific Western Bank, the “Lenders”), pursuant to which the Lenders agreed to make available to the Company, term loans in an aggregate principal amount up to $35,000 (the “Term Loan”) and a revolving credit facility in an aggregate principal amount up to $5,000 (the “Revolving Line” and together with the Term Loan, the “Credit Extensions”) subject to certain conditions to funding. A term loan was made on October 16, 2015 in an aggregate principal amount equal to $35,000, and an advance under the Revolving Line was made on October 16, 2015 in an aggregate principal amount equal to $5,000. The Company is required to make interest-only payments on the Term Loan for 18 months, and beginning on May 1, 2017, is required to make payments of principal and accrued interest on the Term Loan in equal monthly installments over a term of 30 months. The interest-only period can be extended by one year to May 1, 2018 if the Company has at least four products fully USDA- or FDA-approved, plus another product conditionally or fully approved, in each case for commercialization by December 31, 2016, and agree to certain other financial covenants with the Lenders. The Credit Extensions bear interest per annum at the greater of (i) 6.91% or (ii) 3.66% plus the prime rate, which is customarily defined. All principal and accrued interest on the Term Loan are due on October 16, 2019 (the “Term Loan Maturity Date”), and all principal and accrued interest on the Revolving Line are due on October 16, 2017 (the “Revolving Maturity Date”).
The Company used approximately $15,000 of the proceeds from the Credit Extensions to repay all the amounts owed under the Credit Facility, dated as of March 4, 2013, as amended, with Pacific Western Bank (as successor in interest to Square 1 Bank) and the lenders party thereto. The Company’s outstanding debt consisted of term loans under the Credit Facility of $15,000 at a fixed annual rate of 5.50%. The Company was obligated to make interest-only payments on any loans funded under the Credit Facility until June 13, 2016,
As security for the obligations under the Loan Agreement, the Company granted a security interest in substantially all of its existing and after-acquired assets except for its intellectual property and certain other customary exclusions. Subject to customary exceptions, the Company is not permitted to encumber its intellectual property.
Upon execution of the Loan Agreement, the Company was obligated to pay a facility fee to the Lenders of $150, and an agency fee to the Collateral Agent of $100. In addition, the Company is or will be obligated to pay a final payment fee equal to 3.30% of such Term Loan being prepaid or repaid with respect to the Term Loans upon the earliest to occur of the Term Loan Maturity Date, the acceleration of any Term Loan or the prepayment of a Term Loan. The Company will also be obligated to pay a termination fee equal to 3.30% of the highest outstanding amount of the Revolving Line with respect to the Revolving Line upon the earliest to occur of the Revolving Maturity Date, the acceleration of the Revolving Line or the termination of the Revolving Line. The Company will also be obligated to pay an unused-line fee equal to 0.25% per annum of the average unused portion of the Revolving Line.
The Loan Agreement contains customary representations and warranties and customary affirmative and negative covenants, including, among others, limits or restrictions on the Company’s ability to incur liens, incur indebtedness, make certain restricted payments, make certain investments, merge, consolidate, make an acquisition, enter into certain licensing arrangements and dispose of certain assets. In addition, the Loan Agreement contains customary events of default that entitle the Lenders to cause the Company’s indebtedness under the Loan Agreement to become immediately due and payable. The events of default, some of which are subject to cure periods, include, among others, a non-payment default, a covenant default, the occurrence of a material adverse change, the occurrence of an insolvency, a material judgment default, defaults regarding other indebtedness and certain actions by governmental authorities. Upon the occurrence and for the duration of an event of default, an additional default interest rate equal to 4% per annum will apply to all obligations owed under the Loan Agreement.
The Loan Agreement requires that the Company receive unrestricted net cash proceeds of at least $45,000 from partnering transactions and/or the issuance of equity securities from October 16, 2015 to October 16, 2016. The Loan Agreement also requires that the Company has at least three products fully USDA- or FDA approved for commercialization by December 31, 2016. Additionally, the Loan Agreement requires that the Company maintain certain minimum liquidity (approximately $20,000) at all times. At December 31, 2015, the Company was in compliance with all financial covenants. If these covenants are not met, the Company may be required to repay the Credit Extensions prior to December 31, 2016.
Impairment charges related to goodwill and intangible assets have no impact on the Company’s compliance with financial covenants contained in the Company’s Loan and Security Agreement.
On the issuance date of the Credit Extensions, the Company accounted for a portion of the transaction as a debt modification of the Credit Facility dated March 4, 2013 with Pacific Western Bank and a portion as a new financing for the Credit Extensions from Oxford. In conjunction with the refinancing, the Company incurred $556 in lender and legal fees, of which $360 were recorded in the consolidated balance sheet as a reduction in note payable and $196 were expensed as interest expense. Debt issuance fees are amortized throughout the life of Credit Extensions using the straight line method which materially approximates effective interest rate method. Final payment and termination fees related to the Credit Extensions are being accreted to loan payable over the life of the
F-28
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Credit Extensions using the straight line method which materially approximates effective interest rate method. Amortization of debt issuance costs was $129 and $41 for the years ended December 31, 2015 and 2014, respectively.
The Company’s loan payable balance as of December 31, 2015 was as follows:
|
Principal amounts |
|||
|
Term Loan, due October 16, 2019 |
$ |
35,000 | |
|
Revolving Line, due October 16, 2017 |
5,000 | ||
|
Add: accretion of final payment and termination fees |
77 | ||
|
Less: unamortized debt issuance costs |
(367) | ||
|
Loan payable |
$ |
39,710 |
Estimated future principal payments under the Loan and Security Agreement are as follows:
|
YEARS ENDING DECEMBER 31, |
|||
|
2016 |
$ |
— |
|
|
2017 |
15,500 | ||
|
2018 |
14,000 | ||
|
2019 |
10,500 | ||
|
2020 |
— |
||
|
Thereafter |
— |
||
|
Total |
$ |
40,000 |
During the years ended December 31, 2015 and 2014, the Company recognized $1,579 and $875 of interest expense, respectively.
F-29
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
11. Accrued Expenses, Other Current Liabilities and Other Long-Term Liabilities
Accrued expenses, other current liabilities and other long-term liabilities consisted of the following:
|
DECEMBER 31, |
DECEMBER 31, |
|||||
|
2015 |
2014 |
|||||
|
Accrued expenses: |
||||||
|
Accrued payroll and related expenses |
$ |
1,922 |
$ |
2,017 | ||
|
Accrued professional fees |
388 | 429 | ||||
|
Accrued royalty expense |
1 | 72 | ||||
|
Accrued interest expense |
238 | 41 | ||||
|
Accrued research and development costs |
1,111 | 663 | ||||
|
Accrued milestone |
500 |
— |
||||
|
Accrued other |
87 | 7 | ||||
|
Total accrued expenses |
$ |
4,247 |
$ |
3,229 | ||
|
Other current liabilities: |
||||||
|
Early exercise of stock-based awards |
$ |
29 | 46 | |||
|
Other |
8 |
$ |
— |
|||
|
$ |
37 |
$ |
46 | |||
|
Other long-term liabilities: |
||||||
|
Deferred income |
$ |
122 |
$ |
30 | ||
|
$ |
122 |
$ |
30 | |||
12. Agreements
RaQualia Pharma Inc. (“RaQualia”)
On December 27, 2010, the Company entered into two Exclusive License Agreements with RaQualia (the “RaQualia Agreements”) that granted the Company global rights, subject to certain exceptions for injectables in Japan, Korea, China and Taiwan for development and commercialization of licensed animal health products for compounds RQ-00000005 (ENTYCE®, also known as AT-002) and RQ-00000007 (GALLIPRANT®, also known as AT-001). The Company will be required to pay RaQualia milestone payments associated with GALLIPRANT and ENTYCE of up to $10,000 and $8,500, respectively, upon the Company’s achievement of certain development, regulatory and commercial milestones, as well as mid-single digit royalties on the Company’s product sales, if any. As of December 31, 2015, the Company had not accrued or paid any milestone or royalty payments since execution of the RaQualia Agreements.
It is possible that multiple milestones related to the RaQualia Agreements are achieved within the next twelve months totaling $11,500. As of the date of this filing, the Company had achieved milestones totaling $3,000.
On July 12, 2012, the Company entered into an API Development Agreement with RaQualia (the “RaQualia API Agreement”) to develop the active pharmaceutical ingredient in relation to compound RQ-00000007 (AT-001). Under the terms of the RaQualia API Agreement, RaQualia was required to pay $800 to the Company upon execution of the agreement. The Company was also eligible to receive another $800 payment for the successful development and delivery of the API to RaQualia. The Company delivered the API to RaQualia during the fourth quarter of 2014. In October 2014, the Company received notice that API delivered to RaQualia was accepted. Per the terms of the RaQualia API Agreement, the Company was entitled to a payment of $800 for the successful development and delivery of the API to RaQualia. The Company recognized $1,600 in other income, consisting of the $800 deferred income related to the payment received at execution and the $800 payment from RaQualia based on the successful delivery, during the fourth quarter of 2014. The RaQualia API Agreement expired by its own terms upon the Company receiving a technical section complete letter for CMC in 2015.
F-30
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Pacira Pharmaceuticals, Inc. (“Pacira”)
On December 5, 2012, the Company entered into an Exclusive License, Development, and Commercialization Agreement with Pacira (the “Pacira Agreement”) that granted the Company global rights for development and commercialization of licensed animal health products for NOCITA® (also known as AT-003). The Company will be required to pay Pacira milestone payments of up to $42,000 upon the Company’s achievement of certain development, regulatory $2,000 and commercial $40,000 milestones, as well as tiered royalties on the Company’s product sales, if any. To date the Company had paid $500 in milestone payments and no royalties, and no amounts were accrued at December 31, 2015.
It is possible that multiple milestones related to the Pacira Agreement are achieved within the next twelve months totaling $2,000.
Other
BLONTRESS and TACTRESS Agreements
The Company has entered into agreements with counterparties to utilize technology in the production of BLONTRESS and TACTRESS. These agreements require the Company to pay low to mid-single digit royalties on net product sales of BLONTRESS and TACTRESS, and one of the agreements requires a minimum royalty of $70 that is subject to a yearly inflation index adjustment. The Company may also be required to pay up to $405 in sales milestones, based on future sales of certain products. These agreements would also apply to any other products that would utilize the technology. During the years ended December 31, 2015, 2014 and 2013, the Company had recognized $84, $72 and $70 in royalty expense, respectively, related to these agreements.
Elanco Animal Health, Inc.
On December 6, 2012, Vet Therapeutics entered into an Exclusive Commercial License Agreement with Elanco (the “Elanco Agreement”), under which Vet Therapeutics granted a commercial license to Elanco for BLONTRESS for the United States and Canada.
On January 2, 2015, the Company was granted a full product license from the USDA for BLONTRESS. The approval resulted in a $3,000 milestone payment being earned and due to the Company per the terms of the Elanco Agreement. During the first quarter of 2015, the Company recognized $3,000 of licensing revenue related to the milestone payment.
On February 24, 2015, the Company and Elanco agreed to terminate the Elanco Agreement. In consideration for the return of the commercial license granted to Elanco, the Company paid Elanco $2,500 in March 2015, and will be required to pay an additional $500 upon the first commercial sale by the Company. At that time the Company determined that it is probable that the $500 payment will be paid, and recorded the $500 as a current liability in the first quarter of 2015. The Company currently believes the first commercial sale will occur in March 2016. The Company recorded the $3,000 paid to Elanco as a reduction in revenues received from Elanco as the payment was to re-acquire rights that the Company had previously licensed to Elanco.
On February 25, 2016, the Company and Elanco agreed to amend the terms related to the $500 payment due upon the first commercial sale by the Company. Under the amended terms, upon the first commercial sale, the Company will be required to pay quarterly, a small royalty per vial sold until $500 in royalties are paid or the end of two years. After two years, the Company will be required to pay Elanco $500 plus 10% interest, compounded annually against the unpaid balance, less any royalties paid during the two years. If during the two years following the first commercial sale, the Company withdraws BLONTRESS from the market and ceases all commercialization the remaining royalty and related interest is no longer payable.
Kansas Bioscience Authority (“KBA”) Programs
On March 6, 2012, the Company was awarded a research and development grant from KBA, a non-principal owner independent entity of the State of Kansas. The Company received $641 during the life of the agreement which concluded in May 2014. During the years ended December 31, 2015, 2014 and 2013, the Company recognized income of $0, $62 and $478, respectively.
Pursuant to Kansas law, the Company may be required to repay any financial assistance received from the KBA, which may include an obligation to repurchase the shares of its capital stock purchased by the KBA, subject to the discretion of the KBA, if the Company relocates the operations in which the KBA invested outside of the State of Kansas within ten years after receiving such financial assistance. Further, pursuant to the agreement accompanying the voucher award, the KBA may terminate the agreement and require the Company to repay the grant if it initiates procedures to dissolve and wind up or if it ceases operations within the State of Kansas within 10 years following the final grant payment. The Company has determined these contingencies to be within its control and will only account for the repayment of the equity and grant if it becomes probable that the Company is going to relocate the operations in which the KBA invested outside of the State of Kansas within the ten-year period or for the repayment of only the grant if it becomes probable that the Company is going to initiate procedures to dissolve and wind up or cease operations within the State of Kansas within the ten-year period.
F-31
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Advaxis Inc. (“Advaxis”)
On March 19, 2014, the Company entered into an Exclusive License Agreement with Advaxis (the “Advaxis Agreement”) that granted the Company global rights for development and commercialization of licensed animal health products for Advaxis’ ADXS-cHER2 for the treatment of osteosarcoma in dogs (“AT-014”) and three additional cancer immunotherapy products for the treatment of three other types of cancer. Under the terms of the Advaxis Agreement, the Company paid $2,500 in exchange for the license, 306,122 shares of common stock, and a warrant to purchase 153,061 shares of common stock. The consideration was allocated to the common stock and warrant based on their fair values on the date of issuance of $1,200 and $643, respectively. The remaining consideration of $657 was allocated to the licensed technology. On the date of acquisition, the licensed technology had not reached technological feasibility in animal health indications and had no alternative future use in the field of animal health. Accordingly, in-process research and development of $657 was expensed upon acquisition. The Company will be required to pay Advaxis milestone payments of up to an additional $6,000 in clinical and regulatory milestones for each of the four products, assuming approvals in both cats and dogs, in both the United States and the European Union. In addition, the Company agreed to pay up to $28,500 in commercial milestones, as well as tiered royalties ranging from mid-single digit to 10% on the Company’s product sales, if any. As of December 31, 2015, the Company had not accrued or paid any milestone or royalty payments since execution of the Advaxis Agreement.
The Company does not expect to achieve milestones related to the Advaxis Agreement within the next twelve months.
Under the terms of the subscription agreement, the Company acquired 306,122 shares of common stock and a warrant to purchase another 153,061 shares of common stock for $1,843. The warrant is exercisable through March 19, 2024, at an exercise price of $4.90 per share of common stock and is to be settled through physical share issuance or net share settlement where the total number of issued shares is based on the amount the market price of common stock exceeds the exercise price of $4.90 on date of exercise. Neither the common stock nor warrant have registration rights. The Company allocated the consideration of $1,843 to Advaxis common stock ($1,200) and the Advaxis warrant ($643) based on their respective fair values and recorded the purchase in marketable securities and other long-term assets, respectively. See Note 3 for subsequent fair value matters related to the Advaxis common stock and warrant.
In January 2015, Aratana sold 124,971 shares of Advaxis common stock for proceeds of $1,500 and recognized a gain of $1,010 in other income (expense). Further in April 2015, the Company sold the remaining 181,151 shares of Advaxis common stock for proceeds of $3,233, recognizing a gain of $2,523 in other income (expense) during the second quarter of 2015.
In May 2015, the Company, through net share settlement, exercised the Advaxis warrant for a total exercise price equivalent to $750 and received 116,411 net shares of Advaxis common stock. Subsequently, the Company sold this Advaxis common stock for proceeds of $2,724, a gain of $341, recorded in other income (expense) during the second quarter of 2015.
VetStem BioPharma, Inc. (“VetStem”)
On June 12, 2014, the Company entered into an Exclusive License Agreement with VetStem (the “VetStem Agreement”) that granted the Company the exclusive United States rights for commercialization and development of VetStem’s allogeneic stem cells being developed for the treatment of pain and inflammation of canine osteoarthritis (“AT-016”). VetStem is responsible for the development and obtaining regulatory approval of AT-016 and the Company is responsible for the commercialization of licensed products. Under the terms of the VetStem Agreement, the Company paid an initial license fee of $500. On the date of acquisition, the licensed technology had not reached technological feasibility in animal health indications and had no alternative future use in the field of animal health.
Accordingly, in-process research and development of $500 was expensed upon acquisition. The Company will be required to pay VetStem milestone payments of up to $4,200 upon VetStem’s achievement of certain development and regulatory milestones, as well as tiered royalties in the low double digit percentages on the Company’s product sales, if any. The Company could be required to pay to VetStem up to $3,600 in reimbursement for development expenses, unless additional monies are approved by the joint steering committee. In 2015, the Company paid milestones of $300. No royalties or milestone payments were accrued as of December 31, 2015.
The Company believes it is possible, but not probable that VetStem might achieve multiple milestones totaling $700 related to the VetStem Agreement within the next twelve months.
Atopix Therapeutics Ltd.
On October 10, 2014, the Company entered into an Exclusive License Agreement with Atopix (the “Atopix Agreement”) that granted the Company an exclusive global license for development and commercialization of animal health products containing the active pharmaceutical ingredient included in Atopix’s CRTH2 antagonist product for the treatment of atopic dermatitis (“AT-018”). Under the terms of the Atopix Agreement, the Company paid an initial license fee of $1,000. On the date of acquisition, the licensed technology had not reached technological feasibility in animal health indications and had no alternative future use in the field of animal health. Accordingly, in-process research and development of $1,000 was expensed upon acquisition. The Company will be
F-32
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
required to pay Atopix milestone payments of up to an additional $4,000 in clinical and regulatory milestones, assuming approvals in both cats and dogs, in both the United States and the European Union, as well as tiered royalties in the mid-single digits on the Company’s product sales, if any. As of December 31, 2015 and December 31, 2014, the Company had paid $500 and $0 in milestone payments respectively, no royalties, and no milestones or royalty payments had been accrued.
The Company does not expect to achieve milestones related to the Atopix Agreement within the next twelve months.
Exclusive Option Programs
The Company’s business model is to identify innovative proprietary compounds from human biopharmaceutical companies and to develop these product candidates into regulatory-approved therapeutics specifically for use in pets. To this end, the Company has developed a process in which, in exchange for a fee, it enters into a time-limited option agreement (the “Exclusive Option Program”) with a biopharmaceutical company (the “Potential Licensor”). During the option period the Company obtains from the Potential Licensor the data and information necessary to perform studies to evaluate the compound itself or through a cooperative research and development agreement. Once the Company has evaluated the compound, it can choose to either terminate the Exclusive Option Program with no further obligation, or exercise the option to enter into an exclusive, worldwide license agreement (the “License Agreement”) to develop and commercialize products for non-human animal health applications. The fee associated with the Exclusive Option Program is generally non-refundable and non-creditable.
The principal terms of any License Agreement entered into by the Company will generally consist of an exclusive, world-wide license to all non-human animal health applications in exchange for an upfront license fee, milestone payments upon the achievement of certain regulatory milestones, as well as royalties on sales.
During the years ended December 31, 2015, 2014 and 2013, the Company recognized research and development expenses of $270, $307 and $915, respectively, due to Exclusive Option Programs.
13. Common Stock
As of December 31, 2015, there were 34,563,816 shares of the Company’s common stock outstanding, net of 441,800 shares of unvested restricted common stock. As of December 31, 2014, there were 34,147,861 shares of the Company’s common stock outstanding, net of 561,651 shares of unvested restricted common stock.
Authorized Common Stock
In February 2013, the Board of Directors of the Company approved an amendment of the Company’s Certificate of Incorporation and increased the number of authorized shares of common stock to 25,041,667. On July 2, 2013, the Company increased the number of authorized shares of its common stock from 25,041,667 to 100,000,000, par value $0.001 per share.
Voting Rights
Each share of common stock entitles the holder to one vote on all matters submitted to a vote of the Company’s stockholders. Common stockholders are entitled to receive dividends, as may be declared by the Board of Directors, if any. As of December 31, 2015 and 2014, the Board of Directors had not declared any dividends in any period.
Stock-Based Awards
During the year ended December 31, 2013, the Company issued common stock pursuant to the 2010 Equity Incentive Plan (Note 14) and the 2013 Incentive Award Plan (Note 14). During the years ended December 31, 2015 and 2014, the Company did not reacquire any unvested shares of common stock from its terminated employees that had been issued upon the exercise of a stock option prior to its vesting. During the years ended December 31, 2015 and 2014, the Company issued common stock pursuant to the 2013 Incentive Award Plan (Note 14).
Reverse Stock Split
On May 22, 2013, the Company effected a 1-for-1.662 reverse stock split of its issued and outstanding shares of common stock. No fractional shares were issued in connection with the reverse stock split. Accordingly, all share and per share amounts for all periods presented in these consolidated financial statements and notes thereto have been adjusted retroactively, where applicable, to reflect the reverse stock split.
Initial Public Offering
In July 2013, the Company completed the initial public offering of its common stock in which the Company issued and sold 6,612,500 shares of common stock at a public offering price of $6.00 per share. The Company received net proceeds of approximately $34,274 after deducting underwriting discounts and commissions of approximately $2,777 and other offering expenses of approximately $2,617.
F-33
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Preferred Convertible Stock Conversion
On June 26, 2013, the holders of at least 75% of the then-outstanding shares of Series A Preferred Stock elected and consented to the automatic conversion of each outstanding share of Preferred Stock into shares of common stock immediately prior to the consummation of the public offering contemplated by the Company’s Registration Statement on Form S-1 (No. 333-187372).
Immediately prior to the consummation of the initial public offering, all shares of the Company’s then-outstanding convertible preferred stock and accumulated dividends automatically converted into an aggregate of 13,351,902 shares of common stock.
Vet Therapeutics Merger
On October 15, 2013, the Company completed the acquisition of Vet Therapeutics, in which the Company issued 624,997 shares of common stock to the former equity and stock option holders of Vet Therapeutics. The fair value of the common stock at time of the acquisition was determined to be $14,700.
Private Placement
On October 13, 2013, the Company completed a private placement in which the Company issued and sold 1,234,375 shares of its common stock for $16.00 an aggregate purchase price of $19,750, or $16.00 per share.
Public Offerings
On February 3, 2014, the Company completed a public offering of its common stock in which the Company issued and sold 5,150,000 shares of common stock at a public offering price of $19.00 per share. The Company received net proceeds of approximately $90,507 after deducting underwriting discounts and commissions of approximately $5,871 and other offering expenses of approximately $1,472.
On September 22, 2014, the Company completed a public offering of its common stock in which the Company issued and sold 5,175,000 shares of common stock at a public offering price of $9.25 per share. The Company received net proceeds of approximately $44,827 after deducting underwriting discounts and commissions of approximately $2,872 and other offering expenses of approximately $412.
Sales Agreement
On October 16, 2015, the Company entered into a Sales Agreement with Barclays Capital Inc. (“Barclays”) pursuant to which the Company may sell from time to time, at its option, up to an aggregate of $52,000 of shares of its common stock (the “Shares”) through Barclays, as sales agent. Sales of the Shares, if any, will be made under the Company’s previously filed and currently effective Registration Statement on Form S-3 (Reg. No. 333-197414), by means of ordinary brokers’ transactions on The NASDAQ Global Market or otherwise. Additionally, under the terms of the Sales Agreement, the Shares may be sold at market prices, at negotiated prices or at prices related to the prevailing market price. The Company will pay Barclays a commission of 2.75% of the gross proceeds from the sale of the Shares, if any. As of the date of this filing the Company has not sold any shares pursuant to the Sales Agreement.
14. Stock-Based Awards
2010 Equity Incentive Plan
In 2010, the Company’s Board of Directors adopted the 2010 Equity Incentive Plan (the “2010 Plan”). The 2010 Plan provided for the Company to sell or issue common stock or restricted common stock and to grant incentive stock options or nonqualified stock options for the purchase of common stock with a maximum term of ten years to employees, members of the Board of Directors and consultants of the Company. With the adoption and approval of the 2013 Incentive Award Plan (the “2013 Plan”), no further awards will be granted from the 2010 plan.
The 2010 Plan permits the exercise of stock options granted under the plan before the options are fully vested. If a stock option is early exercised in this circumstance, the issued common stock is subject to restrictions on the sale or transfer by the holder that lapse according to the vesting terms of the early-exercised stock option. Unvested shares may not be sold or transferred by the holder. In the event of termination of the holder’s employment, any unvested shares received upon early exercise are subject to repurchase by the Company, typically at the lesser of (1) the original purchase price per share or (2) the fair value of the common share on the date of termination. During the year ended December 31, 2012, the Company granted three restricted stock awards under the 2010 Plan that were subject to repurchase at the greater of (1) the original purchase price per share or (2) the fair value of the common share on the date of termination. Only one of these three restricted stock awards remain outstanding as of December 31, 2015.
Under the 2010 Plan, the Company has 90 days from the effective termination of the holder’s employment or service to repurchase unvested shares that are issued upon the exercise of a stock award prior to its vesting. If, after 90 days, the Company elects not to repurchase these unvested shares, the shares become vested in full. The Company would then apply modification accounting and any
F-34
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
resulting compensation expense would be immediately recognized related to the award. Upon vesting, these shares would be considered issued and outstanding shares of common stock for accounting purposes.
Stock Options
At the year ended December 31, 2015, 71,459 shares of restricted common shares issued due to early exercises were unvested and subject to repurchase. Early exercise is not considered an exercise for accounting purposes and, therefore, any payment for unvested shares is recognized as a liability at the original exercise price. As of December 31, 2015 and 2014, the liability related to the early exercise of awards was $30 and $76, respectively, and was recorded in other current liabilities and other long-term liabilities. During the year ended December 31, 2013, the Company repurchased 33,447 unvested shares from terminated employees. No early exercised stock option shares were repurchased by the Company during the years ended December 31, 2015 and 2014.
Activity related to stock options for the year ended December 31, 2015, was as follows:
|
WEIGHTED |
||||||||||
|
SHARES |
WEIGHTED |
AVERAGE |
||||||||
|
ISSUABLE |
AVERAGE |
REMAINING |
AGGREGATE |
|||||||
|
UNDER |
EXERCISE |
CONTRACTUAL |
INTRINSIC |
|||||||
|
OPTIONS |
PRICE |
TERM |
VALUE |
|||||||
|
(IN YEARS) |
||||||||||
|
Outstanding as of December 31, 2014 |
170,466 |
$ |
1.71 | 8.05 |
$ |
2,746 | ||||
|
Granted |
— |
— |
||||||||
|
Exercised |
(57,652) | 0.43 | ||||||||
|
Forfeited |
(26,324) | 0.45 | ||||||||
|
Expired |
— |
— |
||||||||
|
Outstanding as of December 31, 2015 |
86,490 |
$ |
2.95 | 7.09 |
$ |
228 | ||||
|
Options vested and expected to vest as of December 31, 2015 |
85,385 |
$ |
2.92 | 7.09 |
$ |
227 | ||||
|
Options exercisable as of December 31, 2015 |
86,213 |
$ |
2.94 | 7.09 |
$ |
228 | ||||
No stock options were granted in 2015 and 2014, for the year ended December 31, 2013, the weighted average grant date fair value of stock options granted was $ 2.48. For the years ended December 31, 2015, 2014, and 2013, the total intrinsic value of options exercised was $786, $871 and $4,840, respectively. For the years ended December 31, 2015, 2014 and 2013 the total fair value of awards vested during the period was $140, $765 and $98, respectively. The Company received cash proceeds of $25, $19 and $153 from the exercise of stock options for the years ended December 31, 2015, 2014 and 2013, respectively, of which $0, $0 and $97 were from the early exercise of stock options, respectively.
During 2014, there were three awards subject to modification accounting under ASC 718-20-35-3 through 35-4. Per terms of separation with a former employee, all unvested shares of restricted stock held by the employee became fully vested upon the employee’s employment termination. In addition, six months of accelerated vesting was granted for the former employee’s two stock option awards. As the employee would have forfeited the unvested awards upon termination according to the awards’ original terms, the awards would not be expected to vest under the original vesting conditions. The accelerated/full vesting of the unvested awards resulted in a Type III modification. The incremental fair value was equal to the fair value of the awards on the modification date. This amount was recognized immediately as the awards did not require further service. The incremental expense for the stock option awards and restricted stock award was $327 and $649, respectively.
During August 2013, the Company modified two stock option awards granted to the Company’s former President, for the purchase of 269,817 shares of common stock in the aggregate. The modifications included the expiration of options to purchase 9,228 shares of common stock, and extended the expiration date of options from August 9, 2013 to January 31, 2014. No additional stock-based compensation expense was recognized as a result of this modification.
Restricted Common Stock
The Company’s 2010 Plan provided for the award of restricted common stock. The Company has granted restricted common stock with time-based vesting conditions. Unvested shares of restricted common stock may not be sold or transferred by the holder. These restrictions lapse according to the time-based vesting.
F-35
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
During the year ended December 31, 2013, the Company issued 76,496 shares of restricted common stock for no proceeds. The vesting of these awards is time-based, with terms between two and four years.
Activity related to restricted stock for the year ended December 31, 2015, was as follows:
|
WEIGHTED |
|||||
|
AVERAGE GRANT |
|||||
|
SHARES |
DATE FAIR VALUE |
||||
|
Unvested restricted common stock as of December 31, 2014 |
91,334 |
$ |
0.94 | ||
|
Restricted common stock issued |
— |
— |
|||
|
Restricted common stock vested |
(54,256) | 0.37 | |||
|
Restricted common stock forfeited |
— |
— |
|||
|
Unvested restricted common stock as of December 31, 2015 |
37,078 |
$ |
0.36 | ||
No restricted stock was granted in 2015 and 2014. For the year ended December 31, 2013, the weighted average grant date fair value of restricted stock granted was $2.59. For the years ended December 31, 2015, 2014 and 2013, the total fair value of restricted shares vested was $731, $2,615 and $2,257, respectively. As of December 31, 2015, 2014 and 2013, 37,078, 91,334 and 237,740 shares of common stock related to restricted stock awards were unvested and subject to repurchase, respectively.
2013 Incentive Award Plan
In 2013, the Company’s Board of Directors adopted and stockholders approved the 2013 Plan which became effective upon the day prior to the effective date of the Company’s initial public offering. The 2013 Plan currently allows for the issuance of up to 3,222,298 shares of common stock, plus any additional shares represented by the 2010 Plan that are forfeited or lapse unexercised. The number of shares of common stock that may be issued under the plan is also subject to an annual increase on January 1st of each calendar year beginning in 2014 and ending in 2023, equal to the lesser of (i) 1,203,369 shares, (ii) 4% of the shares of common stock outstanding on the final day of the immediately preceding calendar year and (iii) and amount determined by the Board of Directors. As of December 31, 2015, there were 847,103 shares available for future grant under the 2013 Plan. On January 1, 2016, the annual increase was determined to be 1,203,369.
The 2013 Plan is administered by the Compensation Committee of the Board of Directors, which selects the individuals eligible to receive awards, determines or modifies the terms and condition of the awards granted, accelerates the vesting schedule of any award and generally administers and interprets the 2013 Plan. The 2013 Plan permits the granting of incentive and nonqualified stock options, with terms of up to ten years and the granting of restricted stock, restricted stock units, performance stock awards, dividend equivalent rights, stock payments (i.e. unrestricted stock), cash bonuses and stock appreciation rights to employees, consultants, and non-employee directors.
Stock Options
During the year ended December 31, 2015, the Company granted under the 2013 Plan stock options for the purchase of 523,350 shares of common stock to certain employees and non-employee directors. The vesting conditions for most of these awards are time-based, and the awards typically vest 25% after one year and monthly thereafter for the next 36 months. Awards typically expire after 10 years.
F-36
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Activity related to stock options for the year ended December 31, 2015, was as follows:
|
WEIGHTED |
||||||||||
|
SHARES |
WEIGHTED |
AVERAGE |
||||||||
|
ISSUABLE |
AVERAGE |
REMAINING |
AGGREGATE |
|||||||
|
UNDER |
EXERCISE |
CONTRACTUAL |
INTRINSIC |
|||||||
|
OPTIONS |
PRICE |
TERM |
VALUE |
|||||||
|
(IN YEARS) |
||||||||||
|
Outstanding as of December 31, 2014 |
1,481,866 |
$ |
17.13 | 8.96 |
$ |
3,835 | ||||
|
Granted |
523,350 | 16.14 | ||||||||
|
Exercised |
(32,761) | 8.76 | ||||||||
|
Forfeited |
(182,945) | 18.96 | ||||||||
|
Expired |
(61,311) | 23.55 | ||||||||
|
Outstanding as of December 31, 2015 |
1,728,199 |
$ |
16.57 | 8.31 |
$ |
— |
||||
|
Options vested and expected to vest as of December 31, 2015 |
1,642,972 |
$ |
16.59 | 8.31 |
$ |
— |
||||
|
Options exercisable as of December 31, 2015 |
610,406 |
$ |
16.54 | 7.88 |
$ |
— |
||||
For the years ended December 31, 2015, 2014 and 2013, the weighted average grant date fair value of stock options granted was $10.09, $12.84 and $9.16, respectively. For the years ended December 31, 2015, 2014 and 2013, the total intrinsic value of options exercised was $267, $214 and $0 (no exercises occurred), respectively. For the years ended December 31, 2015, 2014 and 2013, the total fair value of awards vested during the period was $5,660, $1,888 and $0 (no awards vested), respectively. The Company received cash proceeds of $287, $206 and $0 (no exercises occurred) from the exercise of stock options for the years ended December 31, 2015, 2014 and 2013, respectively.
Restricted Common Stock
The Company’s 2013 Plan provides for the award of restricted common stock. The Company has granted restricted common stock typically with time-based vesting conditions, having terms of between several months and four years. The awards granted in 2015 to executives and non-executives typically vest in three annual installments of 33.3% each year for three years. In 2016, the vesting conditions for executive awards changed so that the awards vest in 12 quarterly installments of 8.33% per quarter for three years, similar to the vesting conditions for executive awards in 2014. Awards granted to consultants typically vest in accordance with the expected term length of the consulting arrangement. Unvested shares of restricted common stock may not be sold or transferred by the holder. These restrictions lapse according to the time-based vesting.
Activity related to restricted stock for the year ended December 31, 2015, was as follows:
|
WEIGHTED |
|||||
|
AVERAGE GRANT |
|||||
|
SHARES |
DATE FAIR VALUE |
||||
|
Unvested restricted common stock as of December 31, 2014 |
277,844 |
$ |
16.92 | ||
|
Restricted common stock issued |
230,400 | 17.14 | |||
|
Restricted common stock vested |
(151,131) | 15.85 | |||
|
Restricted common stock forfeited |
(23,850) | 14.00 | |||
|
Unvested restricted common stock as of December 31, 2015 |
333,263 |
$ |
17.77 | ||
For the years ended December 31, 2015, 2014 and 2013, the weighted average grant date fair value of restricted common stock granted was $17.14, $16.73 and $18.91, respectively. For the years ended December 31, 2015, 2014 and 2013, the total fair value of
F-37
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
restricted common stock vested was $1,893, $94 and $25, respectively. The Company received no proceeds for any of the restricted common stock granted during the years ended December 31, 2015, 2014 and 2013.
Stock-Based Compensation
The fair value of each stock option award is estimated using the Black-Scholes option-pricing model. Prior to 2014, due to the lack of company- specific historical and implied volatility information the Company estimated its expected volatility based on the historical volatility of the Company’s publicly-traded peer companies. Beginning in the first quarter of 2014, the Company began to base expected volatility on historical volatility of the Company’s common stock, as adequate historical data regarding the volatility of the Company’s common stock price had become available. The expected term of the Company’s stock options has been determined utilizing the “simplified” method as the Company has insufficient historical experience for option grants overall, rendering existing historical experience irrelevant to expectations for current grants. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is based on the fact that the Company has never paid cash dividends and does not expect to pay any cash dividends in the foreseeable future.
The relevant data used to determine the value of the stock option grants, presented on a weighted average basis, was as follows:
|
YEAR ENDED DECEMBER 31, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Risk-free interest rate |
1.38 |
% |
1.88 |
% |
1.59 |
% |
||||||
|
Expected term (in years) |
6.1 | 6.1 | 6.1 | |||||||||
|
Expected volatility |
70 |
% |
84 |
% |
66 |
% |
||||||
|
Expected dividend yield |
— |
% |
— |
% |
— |
% |
||||||
Compensation expense related to restricted stock granted to employees and non-employee directors is equal to the excess, if any, of the fair value of the Company’s common stock on date of grant over the original purchase price per share, multiplied by the number of shares of restricted common stock issued for employees. Compensation expense related to restricted stock granted to non-employees is equal to the excess, if any, of the fair value of the Company’s common stock on date of vesting over the original purchase price per share, multiplied by the number of shares of restricted common stock vesting.
The Company recognizes compensation expense for only the portion of awards that are expected to vest. In developing a forfeiture rate estimate, the Company has considered its historical experience to estimate pre-vesting forfeitures for service-based awards. The impact of a forfeiture rate adjustment will be recognized in full in the period of adjustment, and if the actual forfeiture rate is materially different from the Company’s estimate, the Company may be required to record adjustments to stock-based compensation expense in future periods.
The Company recorded stock-based compensation expense related to stock options and restricted stock as follows:
|
YEAR ENDED DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
Research and development |
$ |
1,646 |
$ |
1,611 |
$ |
419 | |||
|
Cost of product sales |
118 | 48 |
— |
||||||
|
Selling, general and administrative |
6,828 | 5,471 | 606 | ||||||
|
$ |
8,592 |
$ |
7,130 |
$ |
1,025 | ||||
As of December 31, 2015, the Company had an aggregate of $10,372 and $4,570 of unrecognized stock-based compensation expense for options outstanding and restricted stock awards, respectively, which is expected to be recognized over 2.37 years and 1.68 years, respectively.
F-38
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
15. Net Loss Per Share
Basic and diluted net loss per share attributable to common stockholders was calculated as follows:
|
YEAR ENDED DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
Numerator: |
|||||||||
|
Net loss attributable to common stockholders |
$ |
(84,054) |
$ |
(38,816) |
$ |
(6,938) | |||
|
Denominator: |
|||||||||
|
Weighted average shares outstanding – basic and diluted |
34,355,525 | 29,767,429 | 11,059,382 | ||||||
|
Net loss per share attributable to common stockholders – basic and diluted (1) |
$ |
(2.45) |
$ |
(1.30) |
$ |
(0.63) | |||
__________________
|
(1) |
All per share amounts and shares outstanding for all periods reflect the 1-for-1.662 reverse stock split, which was effective May 22, 2013. |
Stock options for the purchase of 1,814,689, 1,789,305, and 1,294,146 shares of common stock were excluded from the computation of diluted net loss per share attributable to common stockholders for the years ended December 31, 2015, 2014 and 2013, respectively, because those options had an anti-dilutive impact due to the net loss attributable to common stockholders incurred for the period.
16. Commitments and Contingencies
Operating Leases
Future minimum lease payments for operating leases as of December 31, 2015, were as follows:
|
YEAR ENDING DECEMBER 31, |
|||
|
2016 |
598 | ||
|
2017 |
605 | ||
|
2018 |
590 | ||
|
2019 |
466 | ||
|
2020 and thereafter |
526 | ||
|
Total |
$ |
2,785 |
The Company leases facilities and certain operating equipment under operating leases expiring through 2021. The Company incurred rent expense of $678, $565 and $177 for the years ended December 31, 2015, 2014 and 2013, respectively.
Contingent Consideration Obligations
The Company determines the acquisition date fair value of the contingent consideration obligation based on a probability-weighted approach derived from the overall likelihood of achieving certain specified future milestones, such as certain regulatory and manufacturing milestones. The fair value measurement is based on significant inputs not observable in the market and thus represents a Level 3 measurement (Note 3), as defined in fair value measurement accounting. The resultant probability-weighted earn-out payments are discounted using a discount rate based upon the Company’s current borrowing rate. At each reporting date, the Company revalues the contingent consideration obligations to the reporting date fair values and record increases and decreases in the fair values as income or expense in the consolidated statements of operations.
Increases or decreases in the fair values of the contingent consideration obligations may result from changes in discount periods and rates, changes in the timing and amount of earn-out criteria and changes in probability assumptions with respect to the likelihood of achieving the various earn-out criteria.
F-39
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
The Company had a contractual contingent purchase price consideration obligation related to acquisitions, as follows (in thousands):
|
Maximum |
|||||||||||||||||
|
Remaining |
Remaining |
||||||||||||||||
|
Earn-out |
Earn-out |
Estimated |
|||||||||||||||
|
Potential |
Period |
Fair Value |
Payments |
||||||||||||||
|
Acquisition |
as of |
as of |
as of |
made |
|||||||||||||
|
Date Fair |
December 31, |
December 31, |
December 31, |
during |
|||||||||||||
|
Acquisition |
Acquisition Date |
Value |
2015 |
2015 |
2015 |
2015 |
|||||||||||
|
Vet Therapeutics |
October 15, 2013 |
$ |
3,810 |
$ |
— |
— |
$ |
— |
$ |
3,000 | |||||||
|
$ |
— |
— |
$ |
— |
$ |
3,000 | |||||||||||
On January 2, 2015 the Company was granted a full product license for BLONTRESS. The approval resulted in $3,000 of the contingent consideration being earned and due to the former Vet Therapeutics shareholders per the terms of Vet Therapeutics merger. Further, on February 24, 2015 in connection with the mutual termination of the NAH Agreement for BLONTRESS, the Company’s obligation to pay additional consideration to the Vet Therapeutics shareholders upon achievement of certain manufacturing milestones ended. The Company paid the $3,000 contingent consideration in March 2015. During the first quarter of 2015, the Company recorded a credit of $1,248 to reduce the fair value of the contingent consideration to zero as a result of the agreement the Vet Therapeutics shareholders.
Litigation and Contingencies Related to Use of Intellectual Property
From time to time, the Company may become subject to legal proceedings, claims and litigation arising in the ordinary course of business. The Company currently is not a party to any threatened or pending litigation. However, third parties might allege that the Company or its licensors are infringing their patent rights or that the Company is otherwise violating their intellectual property rights. Such third parties may resort to litigation against the Company or its licensors, which the Company has agreed to indemnify. With respect to some of these patents, the Company expects that it will be required to obtain licenses and could be required to pay license fees or royalties, or both. These licenses may not be available on acceptable terms, or at all. A costly license, or inability to obtain a necessary license, could have a material adverse effect on the Company’s financial condition, results of operations or cash flows. The Company accrues contingent liabilities when it is probable that future expenditures will be made and such expenditures can be reasonably estimated.
Indemnification Agreements
In the ordinary course of business, the Company may provide indemnifications of varying scope and terms to customers, vendors, lessors, business partners, and other parties with respect to certain matters including, but not limited to, losses arising out of breach of such agreements, from services to be provided by the Company, or from intellectual property infringement claims made by third parties. In addition, the Company has entered into indemnification agreements with certain of its officers and members of its Board of Directors that will require the Company, among other things, to indemnify them against certain liabilities that may arise by reason of their status or service as directors or officers. The maximum potential amount of future payments the Company could be required to make under these indemnification agreements is, in many cases, not readily quantifiable. To date, the Company has not incurred any material costs as a result of such indemnifications. The Company does not believe that the outcome of any claims under indemnification arrangements will have a material effect on its financial position, results of operations or cash flows, and it has not accrued any liabilities related to such obligations in its consolidated financial statements as of December 31, 2015 or 2014.
17. Business Combinations
Acquisition of Okapi Sciences
On January 6, 2014, the Company acquired Okapi Sciences, a Leuven, Belgium based company with a proprietary antiviral platform and three clinical/development stage product candidates. This acquisition further expanded the existing Company pipeline. The aggregate purchase price was approximately $44,439, which consisted of $14,139 in cash, a promissory note in the principal amount of $15,134 with a maturity date of December 31, 2014, and a contingent consideration of up to $16,308 with an acquisition fair value of $15,166. The promissory note bore interest at a rate of 7% per annum, payable quarterly in arrears, and was subject to mandatory prepayment in the event of a specified equity financing by the Company. On February 4, 2014, the promissory note and accrued
F-40
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
interest was paid in cash in the amount of $15,158. On March 17, 2014, the contingent consideration was settled in cash in the amount of $15,235.
Included in the Company’s consolidated statements of operations for the year ended December 31, 2014 is revenue totaling approximately $452 related to Okapi Sciences.
The acquisition-date fair value of the consideration transferred to the sellers of Okapi Sciences, less cash acquired, was $43,376, which consisted of the following:
|
Cash consideration |
$ |
14,139 | |
|
Fair value of promissory note |
15,134 | ||
|
Fair value of contingent consideration |
15,166 | ||
|
Fair value of total consideration |
44,439 | ||
|
Less cash acquired |
(1,063) | ||
|
Total consideration transferred, net of cash acquired |
$ |
43,376 |
Fair Value of Contingent Consideration: The Company agreed to pay up to $16,308 on or prior to April 7, 2014, subject to mandatory prepayment in cash in the event of a specified future equity financing, provided that if not paid in cash by April 7, 2014, payment was to be made in the form of shares of the Company’s common stock-based on the average closing price of the Company’s common stock during the 10-trading day period ending April 4, 2014, subject to a maximum of 1,060,740 shares and a minimum of 707,160 shares. This contingent consideration was recorded as a liability and measured at fair value using a probability-weighted model utilizing significant observable and unobservable inputs, including the volatility in the market price of the Company’s common stock, the expected probability of settling the contingent consideration in either cash or shares and an estimated discount rate commensurate with the risks of these outcomes. The analysis resulted in an estimated fair value of contingent consideration of $15,166. The contingent consideration was settled March 17, 2014 for $15,235 and the difference between the initial fair value amount and settlement amount was $69 which is reflected as a charge to selling, general and administrative expenses in the consolidated statement of operations.
The acquisition of Okapi Sciences was accounted for as a business combination under the acquisition method of accounting. Accordingly, the assets acquired and liabilities assumed were recorded at fair value with the remaining purchase price recorded as goodwill. The assets acquired and the liabilities assumed from Okapi Sciences have been recorded at their fair values at the date of acquisition, being January 6, 2014. The Company’s consolidated financial statements and results of operations include the results of Okapi Sciences from January 6, 2014.
F-41
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
The Company’s allocation of the purchase price to the assets acquired and liabilities assumed was as follows:
|
Cash |
$ |
1,063 | |
|
Accounts receivable |
149 | ||
|
Other receivables |
60 | ||
|
Prepaid expenses and other current assets |
82 | ||
|
Property and equipment |
217 | ||
|
Other long-term assets |
18 | ||
|
Identifiable intangible assets |
29,400 | ||
|
Accounts payable and accrued expenses |
(586) | ||
|
Deferred revenue |
(83) | ||
|
Deferred tax liabilities, net |
(3,786) | ||
|
Long-term debt |
(4) | ||
|
Total identifiable net assets |
26,530 | ||
|
Goodwill |
17,909 | ||
|
Total net assets acquired |
44,439 | ||
|
Less: |
|||
|
Promissory note |
15,134 | ||
|
Contingent consideration |
15,166 | ||
|
Cash paid |
$ |
14,139 |
The following are the intangible assets acquired by drug program and their estimated useful lives as of the date of the acquisition:
|
FAIR VALUE |
USEFUL LIFE |
||||
|
AT-006 |
$ |
3,400 |
13 years |
||
|
AT-007 |
13,500 |
15 years |
|||
|
AT-008 |
5,300 |
13 years |
|||
|
AT-011 |
7,200 |
14 years |
|||
|
Total intangible assets |
$ |
29,400 | |||
The identifiable intangible assets recognized by the Company as a result of the Okapi Sciences acquisition relate to Okapi Sciences technology, and consist primarily of its intellectual property related to Okapi Sciences AT-006, AT-007, AT-008 and AT-011 programs, and the estimated net present value of future cash flows from commercial agreements related to the AT-006 program.
All Okapi Sciences programs, which were considered IPR&D at the acquisition date, were valued using a multi-period excess earnings method, a form of the income approach, which incorporates the estimated future cash flows to be generated from this technology. Excess earnings are the earnings remaining after deducting the market rates of return on the estimated values of contributory assets, including debt-free net working capital, tangible, and intangible assets. The excess earnings are thereby calculated for each year of a multi-year projection period and discounted to present value. Accordingly, the primary components of this method consist of the determination of excess earnings and an appropriate rate of return.
The Company will not amortize the assets related to the Okapi Sciences programs until commercialization has been achieved.
The valuation analysis conducted by the Company determined that the aggregate fair value of identifiable assets acquired less the aggregate fair value of identifiable liabilities assumed by the Company is less than the purchase price. As the purchase price exceeds the fair value of assets and liabilities acquired or assumed, goodwill will be recognized. Goodwill is calculated as the difference between the Okapi Sciences acquisition date fair value of the consideration transferred and the fair values of the assets acquired and
F-42
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
liabilities assumed. The goodwill is not expected to be deductible for income tax purposes. Goodwill is recorded as an indefinite-lived asset and is not amortized but tested for impairment on an annual basis or when indications of impairment exist.
The difference between the total consideration and the fair value of the net assets acquired of $17,909 was recorded as Goodwill in the consolidated balance sheet. This goodwill represents the excess of the purchase price over the fair value of the tangible and identifiable intangible assets acquired and liabilities assumed, principally representing the tax attributes of the acquisition and certain operational and strategic synergies such as advancement toward becoming a commercial company and acquiring a proprietary antiviral platform.
Acquisition of Vet Therapeutics, Inc.
On October 15, 2013, the Company acquired Vet Therapeutics, a San Diego, California based company with a proprietary antibody-based biologics platform. This acquisition further expanded the existing Company pipeline. The aggregate purchase price was approximately $51,515, which consisted of $30,005 in cash, 624,997 shares of Aratana’s common stock with an acquisition date fair value of $14,700, a promissory note in the principal amount of $3,000 with a maturity date of December 31, 2014, and contingent consideration of up to $5,000 with an acquisition-date fair value of $3,810. The promissory note bore interest at a rate of 7% per annum, payable quarterly in arrears, and was subject to prepayment in the event of a specified equity financing by the Company. The Company could have paid up to $5,000 in contingent cash consideration in connection with the achievement of certain regulatory and manufacturing milestones for BLONTRESS. On February 4, 2014 the promissory note and accrued interest was paid in cash in the amount of $3,020. On March 5, 2015 the contingent consideration was settled in the amount of $3,000.
Included in the Company’s consolidated statements of operations for the years ended December 31, 2014 and 2013, is revenue totaling approximately $273 and $123, respectively, related to Vet Therapeutics. The amount of goodwill from this acquisition is not deductible for tax purposes.
The acquisition-date fair value of the consideration transferred to the sellers of Vet Therapeutics, less cash acquired, was $51,503, which consisted of the following:
|
Cash consideration |
$ |
30,005 | |
|
Fair value of promissory note |
3,000 | ||
|
Fair value of merger shares |
14,700 | ||
|
Fair value of contingent consideration |
3,810 | ||
|
Fair value of total consideration |
51,515 | ||
|
Less cash acquired |
(12) | ||
|
Total consideration transferred, net of cash acquired |
$ |
51,503 |
Fair Value of Merger Shares: The Company agreed to transfer 624,997 unregistered shares of its common stock without registration rights to former Vet Therapeutics stock and option holders. On October 15, 2013, the closing date of the Vet Merger, the fair market value of Aratana’s publicly traded common stock was $27.67 per share. In order to determine the fair value of consideration transferred to Vet Therapeutics stock and option holders related to the Merger Shares, the Company applied a discount for the lack of marketability of 15% to the Company’s closing stock price on the closing date of the Vet Merger to account for the lack of access to an active public market for these shares. This resulted in aggregate purchase consideration related to the Merger Shares of $14,700.
Fair Value of Contingent Consideration: The Company agreed to pay up to $5,000 in contingent cash consideration in connection with the achievement of certain regulatory and manufacturing milestones for BLONTRESS. Contingent consideration is recorded as a liability and measured at fair value using a discounted cash flow model utilizing significant unobservable inputs, including the probability of achieving each of the potential milestones and an estimated discount rate commensurate with the risks of the expected cash flows attributable to the milestones. This resulted in aggregate contingent purchase consideration of $3,810. Significant increases or decreases in any of the probabilities of success would result in a significantly higher or lower fair value, respectively, and commensurate changes to this liability. The fair value of contingent consideration and the associated liability was adjusted to fair value at each reporting date until actual settlement occurred, with the changes in fair value reflected in earnings.
The acquisition of Vet Therapeutics was accounted for as a business combination under the acquisition method of accounting. Accordingly, the assets acquired and liabilities assumed were recorded at fair value with the remaining purchase price recorded as goodwill. The assets acquired and the liabilities assumed from Vet Therapeutics have been recorded at their fair values at the date of
F-43
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
acquisition, being October 15, 2013. The Company’s consolidated financial statements and results of operations include the results of Vet Therapeutics from October 16, 2013.
In the year ended December 31, 2013, the Company incurred expenses totaling $1,369 relating to the Vet Therapeutics acquisition, which were recorded within selling, general and administrative expenses in the Company’s consolidated statement of operations.
The Company’s allocation of the purchase price to the assets acquired and liabilities assumed was as follows:
|
Cash |
$ |
12 | ||
|
Inventories |
173 | |||
|
Other current assets |
5 | |||
|
Property and equipment |
73 | |||
|
Other long-term assets |
3 | |||
|
Identifiable intangible assets |
38,652 | |||
|
Accounts payable and accrued expenses |
(273) | |||
|
Deferred revenue |
(55) | |||
|
Deferred tax liabilities, net |
(12,722) | |||
|
Total identifiable net assets |
$ |
25,868 | ||
|
Goodwill |
25,646 | |||
|
Total net assets acquired |
$ |
51,514 | ||
|
Less: |
||||
|
Merger shares |
14,700 | |||
|
Promissory note |
3,000 | |||
|
Contingent consideration |
3,810 | |||
|
Cash paid |
$ |
30,005 |
The following are the intangible assets acquired by drug program and their estimated useful lives as of the date of the acquisition:
|
FAIR VALUE |
USEFUL LIFE |
||||
|
AT-004 (Antibody for B-cell lymphoma) |
$ |
28,572 |
20 years |
||
|
AT-005 (Antibody for T-cell lymphoma) |
10,080 |
20 years |
|||
|
Total intangible assets subject to amortization |
$ |
38,652 | |||
The identifiable intangible assets recognized by the Company as a result of the Vet Therapeutics acquisition relate to Vet Therapeutics’ technology, and consist primarily of its intellectual property related to Vet Therapeutics’ B-cell and T-cell antibodies, and the estimated net present value of future cash flows from commercial agreements related to the B-cell technology.
The Vet Therapeutics B-cell technology, which is now referred to as BLONTRESS, was valued using the discounted cash flow method, a form of the income approach, which incorporates the estimated royalty income and milestone payments to be generated from this technology. The estimated cash flows are then discounted to present value. Accordingly, the primary components of this method consist of the determination of cash flows, the probability of achieving and the anticipated timing of the milestone payments, and an appropriate rate of return.
The Vet Therapeutics T-cell technology, which was considered in-process research and development at the acquisition date and which is now referred to as TACTRESS, was valued using a multi-period excess earnings method, a form of the income approach, which incorporates the estimated future cash flows to be generated from this technology. Excess earnings are the earnings remaining after deducting the market rates of return on the estimated values of contributory assets, including debt-free net working capital, tangible, and intangible assets. The excess earnings are thereby calculated for each year of a multi-year projection period and discounted to
F-44
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
present value. Accordingly, the primary components of this method consist of the determination of excess earnings and an appropriate rate of return.
For the B-cell technology, the Company will recognize straight-line amortization expense over the estimated useful life of the asset. The Company began amortizing the asset related to the T-cell technology when the conditional license was issued during January of 2014.
The difference between the total consideration and the fair value of the net assets acquired of $25,646 was recorded to Goodwill in the consolidated balance sheet. This goodwill represents the excess of the purchase price over the fair value of the tangible and identifiable intangible assets acquired and liabilities assumed, principally representing the tax attributes of the acquisition and certain operational and strategic synergies such as advancement toward becoming a commercial company and acquiring a proprietary antibody-based biologics platform focused on the treatment of lymphoma.
Pro Forma Financial Information
The following pro forma financial information summarizes the combined results of operations for the Company as though the acquisition of Okapi Sciences occurred on January 1, 2013 and acquisition of Vet Therapeutics occurred on January 1, 2012. The unaudited pro forma financial information was as follows:
|
YEAR ENDED DECEMBER 31, |
||||||
|
2014 |
2013 |
|||||
|
(Unaudited) |
(Unaudited) |
|||||
|
Revenue |
$ |
767 |
$ |
2,570 | ||
|
Loss from operations |
(41,314) | (22,977) | ||||
|
Loss before income taxes |
$ |
(39,979) |
$ |
(24,448) | ||
|
Net loss per share before income taxes – basic and diluted |
$ |
(1.34) |
$ |
(1.95) | ||
Pro forma results include non-recurring pro forma adjustments that were directly attributable to the business combinations. The following material non-recurring pro forma adjustments relating to charges recorded in 2014 have been assumed to have occurred in 2013 for pro forma purposes:
|
· |
Pre-tax increase in income of $440 in 2014, relating to acquisition-related transaction costs incurred by the Company and Okapi Sciences. |
Additionally, the following material non-recurring pro forma adjustments relating to charges recorded in 2013 have been assumed to have occurred in 2012 for pro forma purposes:
|
· |
Pre-tax increase in income of $1,639 for 2013, relating to acquisition-related transaction costs incurred by the Company and Vet Therapeutics. |
The pro forma financial information for all periods presented has been calculated after adjusting the results of the Company and Okapi Sciences and Vet Therapeutics to reflect the business combination accounting effects resulting from these acquisitions including the amortization expenses from acquired intangible assets, the depreciation expenses from acquired tangible assets, the stock-based compensation expense for unvested stock options and restricted stock units assumed and the related tax effects as though the acquisition occurred as of January 1, 2013 for Okapi Sciences and January 1, 2012 for Vet Therapeutics. The pro forma financial information is for informational purposes only and is not indicative of the results of operations that would have been achieved if the acquisition had taken place at the beginning of the Company’s 2013 fiscal year.
F-45
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
18. Income Taxes
The components of income from continuing operations before income taxes benefit were as follows:
|
YEAR ENDED DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
U.S. |
$ |
(65,481) |
$ |
(34,256) |
$ |
(19,660) | |||
|
Non-U.S. |
(20,271) | (6,003) |
— |
||||||
|
Income from continuing operations |
$ |
(85,752) |
$ |
(40,259) |
$ |
(19,660) | |||
The components of the income tax benefit were as follows:
|
YEAR ENDED DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
Current: |
|||||||||
|
Federal |
$ |
— |
$ |
— |
$ |
||||
|
State |
— |
— |
|||||||
|
Deferred: |
|||||||||
|
Federal |
— |
— |
10,782 | ||||||
|
State |
— |
— |
1,940 | ||||||
|
Foreign |
1,698 | 1,443 |
— |
||||||
|
Total |
$ |
1,698 |
$ |
1,443 |
$ |
12,722 | |||
A reconciliation of the U.S. federal statutory income tax rate to the Company’s effective income tax rate was as follows:
|
YEAR ENDED DECEMBER 31, |
||||||||||
|
2015 |
2014 |
2013 |
||||||||
|
Federal statutory income tax rate |
34.0 |
% |
34.0 |
% |
34.0 |
% |
||||
|
State income taxes, net of federal tax benefit |
2.5 | 1.1 | 7.2 | |||||||
|
Non-deductible expenses |
(1.1) | (3.0) | (2.5) | |||||||
|
Research credits |
0.4 | 0.8 | 0.9 | |||||||
|
Losses benefitted/(not benefitted) |
(33.8) | (29.3) | 25.3 | |||||||
|
Total |
2.0 |
% |
3.6 |
% |
64.9 |
% |
||||
F-46
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Net deferred tax assets consisted of the following:
|
YEAR ENDED DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
Net operating loss carry forwards |
$ |
26,670 |
$ |
12,196 |
$ |
3,180 | |||
|
Capitalized start-up costs |
6,645 | 7,151 | 3,751 | ||||||
|
Tax credit carry forwards |
1,308 | 1,062 | 784 | ||||||
|
Other temporary differences |
3,451 | 1,469 | 1,421 | ||||||
|
Capitalized research and development, net |
11,911 | 10,378 | 5,788 | ||||||
|
Total deferred tax assets |
49,985 | 32,256 | 14,924 | ||||||
|
Valuation allowance |
(46,885) | (14,747) | (3,118) | ||||||
|
Net deferred tax assets |
3,100 | 17,509 | 11,806 | ||||||
|
Intangibles, net |
(3,041) | (19,356) | (11,790) | ||||||
|
Depreciation |
(59) | (18) | (16) | ||||||
|
Total deferred tax liabilities |
(3,100) | (19,374) | (11,806) | ||||||
|
Net deferred tax liability |
$ |
— |
$ |
(1,865) |
$ |
— |
|||
As of December 31, 2015, the Company had net operating loss carryforwards for federal and state income tax purposes of $44,247 and $42,381, respectively, which begin to expire in fiscal year 2031 and 2020, respectively. Approximately $1,209 of the federal and state net operating loss carryforwards relate to excess tax deductions from share-based payments, the tax benefits of which would be credited to additional paid-in capital when the deductions reduce cash taxes payable.
As of December 31, 2015, the Company had federal and state research and development tax credit carryforwards of $935 and $567, respectively, which begin to expire in fiscal year 2031 and until utilized, respectively. Additionally, $4,038 of excess tax deductions from share-based payments were capitalized in 2014 and are being amortized over 15 years for tax purposes. The tax benefits of these items would be credited to additional paid-in capital when the deductions reduce cash taxes payable. The Company has approximately $31,546 of foreign net operating loss carryforward, which may be carried forward indefinitely.
Management of the Company has evaluated the positive and negative evidence bearing upon the realizability of its deferred tax assets, which are comprised principally of net operating loss carryforwards and research and development credits. Under the applicable accounting standards, management has considered the Company’s history of losses and concluded that it is more likely than not that the Company will not recognize the benefits of net federal and state deferred tax assets. Accordingly, a full valuation allowance of the net U.S. deferred tax asset had been established at December 31, 2015 and 2014.
The Company recognized a deferred tax benefit of approximately $1,698 during 2015 for losses incurred in Belgium. On January 6, 2014, we acquired Okapi Sciences. As a result of the acquisition, we recorded approximately $3,786 of net deferred tax liability primarily related to the step-up of intangible assets for book purposes. The taxable temporary difference from the acquisition is considered a source of future taxable income in determining the realizability of our deferred tax assets. During 2014, we recognized approximately $1,443 of income tax benefit for losses incurred in Aratana NV. During 2013, as a result of the acquisition of Vet Therapeutics, the Company recorded approximately $12,722 of net deferred tax liability, primarily related to the step-up of intangible assets for book purposes. The taxable temporary difference from the acquisition is considered as a source of future taxable income in determining the realizability of the Company’s deferred tax assets. The Company recognized a deferred tax benefit of approximately $12,722 due to a release of the valuation allowance against the Company’s federal deferred tax assets of $10,782 and a change in the valuation allowance against our state deferred tax assets of $1,940.
Utilization of the net operating loss and research and development credit carryforwards may be subject to a substantial annual limitation under Section 382 of the Internal Revenue Code of 1986 due to ownership change limitations that have occurred previously or that could occur in the future. These ownership changes may limit the amount of net operating loss and research and development credit carryforwards that can be utilized annually to offset future taxable income and tax, respectively. The Company has not completed a study to assess whether an ownership change has occurred, or whether there have been multiple ownership changes since its formation, due to significant complexity and related costs associated with such a study.
F-47
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Changes in the valuation allowance for deferred tax assets during the years ended December 31, 2015, 2014 and 2013, were as follows:
|
YEAR ENDED DECEMBER 31, |
|||||||||
|
2015 |
2014 |
2013 |
|||||||
|
Valuation allowance as of beginning of year |
$ |
14,747 |
$ |
3,118 |
$ |
8,065 | |||
|
Decreases recorded as income tax benefit |
— |
— |
(4,947) | ||||||
|
Increases due to acquisitions |
— |
271 |
— |
||||||
|
Increases due to operations |
32,138 | 11,358 |
— |
||||||
|
Valuation allowance as of end of year |
$ |
46,885 |
$ |
14,747 |
$ |
3,118 | |||
The Company has not recorded any amounts for unrecognized tax benefits as of December 31, 2015 and 2014. The Company will recognize interest and penalties related to uncertain tax positions in income tax expense. As of December 31, 2015 and 2014, the Company had no accrued interest or penalties related to uncertain tax positions and no amounts have been recognized in the Company’s consolidated statements of operations.
The Company files tax returns as prescribed by the tax laws of the jurisdictions in which it operates. The Company’s major taxing jurisdictions include the U.S. federal and states, and Belgium. In the normal course of business, the Company is subject to examination by federal and state jurisdictions, where applicable. The Company’s federal income tax return for 2013 is currently under examination by the Internal Revenue Service. The Company does not expect that the examination would result in any significant adjustments to the taxable loss previously reported. The Company’s tax years are still open under statute from 2012 to the present. The Company’s policy is to record interest and penalties related to income taxes as part of its income tax expense in the consolidated statements of operations.
On December 18, 2015, the Consolidated Appropriations Act, 2016 (Pub. L. 114-113)("the 2015 Act") was signed into law, retroactively reinstated research credit for qualified research expenses ("QREs") paid or incurred in 2015, and made the credit permanent. Under the accounting guidance on this topic, the effects are recognized as a component of income tax expense or benefit from continuing operations in the consolidated financial statements for the interim or annual period that includes the enactment date.
19. Variable Interest Entity
ViroVet BVBA
During the third quarter of 2015, the Company reviewed certain operations of its wholly owned subsidiary, Aratana NV. As a result, the Company made the strategic decision to wind down pre-clinical discovery efforts being performed at Aratana NV and focus future efforts of Aratana NV on clinical assets, the development of core legacy programs, i.e. AT-001, AT-002 and AT-003 for EU approval, business development and monetization of production animal assets and know-how obtained in the acquisition of Okapi Sciences. To facilitate this reorganization, the Company via Aratana NV, along with the former General Manager of Aratana NV, the current General Manager of Aratana NV and a consultant to the Company formed ViroVet BVBA (“ViroVet”) during the third quarter. During the third quarter of 2015, the Company began to transition employees from Aratana NV to ViroVet. The Company plans to transition selected Aratana NV employees, assets and liabilities over the next six months to ViroVet to further pursue the research and development of production animal products. These employees will be focused on the advancement of production animal assets/know-how and the securing of additional funding for future operations.
Except for the financing matters described below, the Company will have little to no involvement in the operations of ViroVet.
Equity Investment
In July 2015, the Company paid $2 and committed another $4 for 28% ownership interest in ViroVet. The Company has no further obligation to provide any further capital.
Convertible Loan Agreement
On September 11, 2015, Aratana NV and ViroVet executed a convertible loan agreement in which Aratana NV agreed to loan ViroVet $335 (€300) on September 15, 2015. The proceeds from the loan require ViroVet to use the monies towards the development and operations of ViroVet in accordance with the budget prepared by ViroVet. The loan bears interest at 7% and is unsecured.
F-48
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
Primary Beneficiary
The Company determined ViroVet is a VIE and it had a controlling financial interest in ViroVet due to the Company having the power to direct the activities of ViroVet that most significantly impact ViroVet’s economic performance and having the obligation to absorb losses or receive benefits. The Company will continue to consolidate ViroVet unless a reconsideration event occurs, for example, an equity financing.
Total assets and liabilities of the Company’s consolidated VIE were not material as of December 31, 2015.
For the year ended December 31, 2015, ViroVet’s net loss and non-controlling interest was not material and is included in the Company’s consolidated statement of operations. Creditors in ViroVet only have recourse to the assets owned by the VIE and not to the Company’s general credit. The Company currently does not have implicit support arrangements with ViroVet.
20. Related Party Transactions
MPM Asset Management, LLC
The Company subleased office space (Heartland House in Kansas City, Kansas) and received office related services from MPM Asset Management, LLC, formerly an affiliate of two of the Company’s principal stockholders. Rent paid in each of the years ended December 31, 2015, 2014 and 2013, was $50, $67 and $67, respectively. This sublease ended December 31, 2015.
The Company leased office space (Boston) and received certain office-related services. The term of the agreement was from February 9, 2013 through December 31, 2013. The Company then leased this space month-to-month through June 2014. Rent and services paid in the years ended December 31, 2014 and 2013, was $60 and $52, respectively.
MPM Heartland House, LLC
The Company leased its former corporate headquarters office space in Kansas City, Kansas from MPM Heartland House, LLC, a company in which the current Chief Executive Officer and President of the Company, also a director of the Company, is the principal owner. Rent paid for the years ended December 31, 2015, 2014 and 2013, were $131, $113 and $60, respectively. The most recent lease period was from May 1, 2013 to December 31, 2015. The Company believes the terms of the lease agreement with MPM Heartland House were no less favorable than those that the Company could have obtained from an unaffiliated third party. Also, the Company had a services agreement with MPM Heartland House, LLC which included the lease of the furniture, janitorial and other services to care for the property. Service charges for the years ended December 31, 2015, 2014 and 2013, were $33, $33 and $5, respectively.
Indemnification Agreements
The Company has entered into indemnification agreements with each of its directors and executive officers. These agreements, among other things, require the Company or will require the Company to indemnify each director (and in certain cases their related venture capital funds) and executive officer to the fullest extent permitted by Delaware law, including indemnification of expenses such as attorneys’ fees, judgments, fines and settlement amounts incurred by the director or executive officer in any action or proceeding, including any action or proceeding by or in right of the Company, arising out of the person’s services as a director or executive officer.
F-49
ARATANA THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share data)
21. Selected Quarterly Financial Data (unaudited)
Selected unaudited quarterly financial data for each of the quarters in the years ended December 31, 2015 and 2014 (in thousands, except per share data), was as follows:
|
2015 |
|||||||||||||
|
First |
Second |
Third |
Fourth |
||||||||||
|
Quarter |
Quarter |
Quarter |
Quarter |
||||||||||
|
Net revenue |
$ |
156 |
$ |
230 |
$ |
229 |
$ |
63 | |||||
|
Gross profit |
46 | 121 | 91 | 55 | |||||||||
|
Net loss |
(8,774) | (7,983) | (54,442) | (12,855) | |||||||||
|
Basic and diluted– loss per common share: |
|||||||||||||
|
Net loss per common share (1) |
$ |
(0.26) |
$ |
(0.23) |
$ |
(1.58) |
$ |
(0.37) | |||||
|
Weighted average number of common shares outstanding, basic and diluted |
34,193,994 | 34,278,105 | 34,405,646 | 34,540,001 | |||||||||
|
2014 |
|||||||||||||
|
First |
Second |
Third |
Fourth |
||||||||||
|
Quarter |
Quarter |
Quarter |
Quarter |
||||||||||
|
Net revenue |
$ |
176 |
$ |
300 |
$ |
43 |
$ |
248 | |||||
|
Gross profit |
176 | 300 | 43 | (85) | |||||||||
|
Net loss |
(9,152) | (9,278) | (10,130) | (10,256) | |||||||||
|
Basic and diluted– loss per common share: |
|||||||||||||
|
Net loss per common share (1) |
$ |
(0.34) |
$ |
(0.32) |
$ |
(0.35) |
$ |
(0.30) | |||||
|
Weighted average number of common shares outstanding, basic and diluted |
26,765,565 | 28,761,326 | 29,348,375 | 34,118,255 | |||||||||
__________________
|
(1) |
Net loss available to common stockholders and basic and diluted net loss per common share are computed consistent with annual per share calculations described in Notes 2 and 15 (Net Loss Per Share) of its consolidated financial statements included elsewhere in this Annual Report on Form 10-K. |
F-50