Attached files
| file | filename |
|---|---|
| EX-23.2 - EX-23.2 - INVIVO THERAPEUTICS HOLDINGS CORP. | a2227601zex-23_2.htm |
| EX-31.1 - EX-31.1 - INVIVO THERAPEUTICS HOLDINGS CORP. | a2227601zex-31_1.htm |
| EX-32.2 - EX-32.2 - INVIVO THERAPEUTICS HOLDINGS CORP. | a2227601zex-32_2.htm |
| EX-31.2 - EX-31.2 - INVIVO THERAPEUTICS HOLDINGS CORP. | a2227601zex-31_2.htm |
| EX-23.1 - EX-23.1 - INVIVO THERAPEUTICS HOLDINGS CORP. | a2227601zex-23_1.htm |
| EX-32.1 - EX-32.1 - INVIVO THERAPEUTICS HOLDINGS CORP. | a2227601zex-32_1.htm |
Use these links to rapidly review the document
TABLE OF CONTENTS
Item 8. CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2015 |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
FOR THE TRANSITION PERIOD FROM TO |
||
COMMISSION FILE NUMBER 000-52089
INVIVO THERAPEUTICS HOLDINGS CORP.
(Exact name of registrant as specified in its charter)
| Nevada (State or other jurisdiction of incorporation or organization) |
36-4528166 (I.R.S. Employer Identification No.) |
|
One Kendall Square, Suite B14402, Cambridge, Massachusetts (Address of principal executive offices) |
02139 (Zip Code) |
(617) 863-5500
Registrant's telephone number, including area code
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class to be so registered | Name of exchange on which registered | |
|---|---|---|
| Common Stock, $0.00001 par value | The Nasdaq Stock Market, LLC |
Securities registered pursuant to Section 12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer ý | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant as of June 30, 2015, the last business day of the registrant's most recently completed second fiscal quarter, was $433,011,482, based on a per share price of $16.15, which was the closing price of the registrant's common stock on the Nasdaq Capital Market on such date.
As of February 26, 2016, the number of shares outstanding of the registrant's common stock, $0.00001 par value per share, was 27,597,896.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant's definitive proxy statement for its 2016 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K to the extent stated herein. Such proxy statement will be filed with the Securities and Exchange Commission within 120 days of the registrant's fiscal year ended December 31, 2015.
INVIVO THERAPEUTICS HOLDINGS CORP.
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2015
2
PART I
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains "forward-looking statements" within the meaning of Section 27A of the Securities Act of 1933, as amended (the "Securities Act"), and Section 21E of the Securities Exchange Act of 1934, as amended (the "Exchange Act"). Statements, other than statements of historical facts, contained in this Annual Report on Form 10-K regarding future events, our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management, are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as "may," "might," "will," "should," "intends," "expects," "plans," "goals," "projects," "anticipates," "believes," "estimates," "predicts," "potential," or "continue" or the negative of these terms or other comparable terminology, and include statements about the market potential for treatment of acute and chronic spinal cord injury, the sufficiency of our existing capital resources for continuing operations in 2016, the safety, feasibility, and clinical effectiveness of our Neuro-Spinal Scaffold implant, the expected completion of our pivotal probable benefit study of the Neuro-Spinal Scaffold and its related clinical development, and our ability to develop collaborations and partnerships to support our business plan. These forward-looking statements are only predictions, are uncertain and involve substantial known and unknown risks, uncertainties and other factors which may cause our actual results, levels of activity or performance to be materially different from any future results, levels of activity or performance expressed or implied by these forward-looking statements. Such factors include, among others, the following:
- •
- our limited operating history and history of net losses;
- •
- our ability to raise substantial additional capital to finance our planned operations and to continue as a going concern;
- •
- our ability to successfully commercialize our current and future product candidates, including our Neuro-Spinal Scaffold;
- •
- our ability to successfully complete clinical trials and obtain and maintain regulatory approval of our product candidates;
- •
- our ability to protect and maintain our intellectual property and licensing arrangements;
- •
- our reliance on third parties to conduct testing and clinical trials;
- •
- market acceptance of our technology and products;
- •
- our ability to promote, manufacture and sell our products, either directly or through collaborative and other arrangements with third
parties;
- •
- our ability to attract and retain key personnel; and
- •
- other factors set forth in the "Risk Factors" section of this Annual Report on Form 10-K and in subsequent filings we make with the Securities and Exchange Commission.
We cannot guarantee future results, levels of activity or performance. You should not place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K. These cautionary statements should be considered with any written or oral forward-looking statements that we may issue in the future. Except as required by applicable law, including the securities laws of the United States, we do not intend to update any of the forward-looking statements to conform these statements to reflect actual results, later events or circumstances or to reflect the occurrence of unanticipated events.
As used herein, "we," "us," "our" or the "Company" means InVivo Therapeutics Holdings Corp., together with its consolidated subsidiaries, unless otherwise noted.
3
Overview
We are a research and clinical-stage biomaterials and biotechnology company with a focus on treatment of spinal cord injuries (SCI). Our mission is to redefine the life of the SCI patient, and we are developing treatment options intended to provide meaningful improvement in patient outcomes following SCI. Our approach to treating acute SCIs is based on our investigational Neuro Spinal Scaffold implant, an investigational bioresorbable polymer scaffold that is designed for implantation at the site of injury within a spinal cord contusion and is intended to treat acute spinal cord injury. We believe the Neuro Spinal Scaffold implant is the only SCI therapy in development focused solely on treating acute SCI directly at the epicenter of the injury, and incorporates intellectual property licensed under an exclusive, world-wide license from Boston Children's Hospital ("BCH") and the Massachusetts Institute of Technology ("MIT"). We are continually evaluating other technologies and therapeutics that may be complementary to our development of the Neuro-Spinal Scaffold implant or offer the potential to bring us closer to our goal of redefining the life of the SCI patient. Recently we entered into exclusive license/assignment agreements with the University of California, San Diego and James Guest, M.D., Ph.D. covering delivery methods and devices for our pre-clinical Bioengineered Neural Trails™ injection program.
Market Opportunity
Our clinical program is intended to address the lack of successful treatments for SCIs. The current management of acute SCI is a surgical approach consisting of spine stabilization and an external decompression procedure of uncertain value. We believe the market opportunity for our Neuro-Spinal Scaffold implant is significant. It is estimated that approximately 276,000 people are currently living in the United States with paralysis due to spinal cord injury (chronic SCI), and approximately 12,500 individuals in the United States will become fully or partially paralyzed each year (acute SCI). The regulatory approval pathway for a Humanitarian Device Exemption (HDE) we are initially pursuing would, if U.S. Food and Drug Administration (FDA) approval is granted, cover a potential population of up to 4,000 acute SCI patients each year. This population includes patients afflicted with complete spinal cord injury, i.e., paraplegia or tetraplegia, and excludes gunshot or other penetrating wounds). SCI can lead to permanent paralysis, sensory impairment, and autonomic, bowel, bladder, and sexual dysfunction. Future products, which may include use of stem cells or drug ingredients may enable the treatment of a broader population such as patients with incomplete and/or chronic paralysis and would require separate regulatory approval.
Since 1973, the National Spinal Cord Injury Statistical Center ("NSCISC") at the University of Alabama has been commissioned by the U.S. government to maintain a national database of spinal cord injury statistics. The financial impact of spinal cord injuries, as reported by the NSCISC, is substantial. Direct costs, which include hospital and medical expenses, modification of the home, and personal assistance, are highest in the first year after injury. According to the fact sheet published by NSCISC titled "Spinal Cord Injury—Facts and Figures at a Glance" in conjunction with its 2015 Annual Report, (i) during the first year, average "cost of care" ranges from $347,484 to $1,064,716, depending on the severity of the injury, (ii) the net present value ("NPV") to maintain a quadriplegic injured at age 25 for life is $4,724,181, and (iii) the NPV to maintain a paraplegic injured at age 25 for life is $2,310,104. These costs place a tremendous financial burden on families, insurance providers, and government agencies. Moreover, despite such a significant financial investment, the patient often remains disabled for life because current medical interventions address only the symptoms of SCI rather than the underlying neurological cause. We believe our approach could represent an important advance in the treatment of SCIs.
4
The ASIA Impairment Scale: The American Spinal Injury Association (ASIA) in collaboration with the International Spinal Cord Society (ISCOS) has developed a neurologic examination tool for assessing spinal cord injury known as the International Standards for Neurological Classification of Spinal Cord Injury (ISNCSCI). Results of the ISNCSCI examination are used to determine the ASIA Impairment Scale (AIS) classification.
Patients with complete spinal cord injury are classified as AIS A. Patients with incomplete spinal cord injury have partial sensory and/or motor function below the level of injury and are classified as AIS B (partial sensory function), AIS C (partial sensory and motor function) or AIS D (partial sensory and increased motor function, i.e. can move at least half of the muscles against gravity). Patients who have a complete return of sensory and motor function are classified as AIS E.
These classifications are based upon the ISNCSCI examination in which the examiner performs a neurologic examination to assess sensory function of the entire body and motor function of the upper and lower extremities.
Our Clinical and Pre-Clinical Programs
We currently have a clinical development program for acute SCI and a pre-clinical development program for chronic SCI.
Neuro-Spinal Scaffold™ implant for acute SCI
Our leading product under development is our Neuro-Spinal Scaffold implant, an investigational bioresorbable polymer scaffold that is designed for implantation at the site of injury within a spinal cord contusion. The Neuro-Spinal Scaffold implant is intended to provide support to the surrounding tissue after injury, minimizing expansion areas of necrosis, and supporting endogenous healing/repair processes following injury. This form of appositional healing harbors the promise of sparing white matter, increasing neural sprouting, and diminishing post-traumatic cyst formation.
The Neuro-Spinal Scaffold implant is composed of two biocompatible and bioresorbable polymers that are cast to form a highly porous investigational product:
- •
- Poly lactic-co-glycolic acid (PLGA), a polymer that is widely used in resorbable
sutures and provides the biocompatible support for Neuro-Spinal Scaffold implant; and
- •
- Poly-L-Lysine (PLL), a positively charged polymer commonly used to coat surfaces in order to promote cellular attachment.
Because of the complexity of spinal cord injuries, it is likely that multi- modal therapies will be required in order to maximize positive outcomes in SCI patients. In the future, we may attempt to further enhance the performance of our Neuro-Spinal Scaffold by multiple combination strategies involving electrostimulation devices, additional biomaterials, drugs approved by the U.S. Food & Drug Administration ("FDA"), or growth factors.
We expect the Neuro-Spinal Scaffold will be regulated by the FDA as a Class III medical device, please see below "Government Regulation" for additional information on the regulatory pathway for the Neuro-Spinal Scaffold.
Pre-Clinical and Non-Clinical Studies relating to the Neuro-Spinal Scaffold
SCI can result in permanent paralysis, sensory impairment, and autonomic, bowel, bladder, and sexual dysfunction. These functional deficits result from damage to or loss of cells (neurons and glia) in the affected region of the spinal cord, either from the initial mechanical trauma or through secondary mechanisms that persists for several weeks. The ability of potential treatments for SCI to mitigate loss of function or promote recovery can be evaluated with non- clinical models using different species and
5
different methods of inducing SCI. In our pre-clinical studies, we utilized rat, non-human primate, and pig models because each exhibits a pattern of neuropathology following SCI that is similar to human SCI. Hemisection injury models, in which sections of spinal cord are surgically removed, are useful in the evaluation of treatment strategies that involve device implantation. Unilateral hemisection models preserve function on one side of the cord, resulting in improved recovery of bladder and bowel function. We, therefore, evaluated the bioresorbable polymer scaffold device in both rats and non-human primates with unilateral hemisection injury. Because most human SCIs are non-penetrating contusion injuries resulting from rapid compression of spinal tissue by intrusion of bone or disc material following mechanical disruption of the vertebral column, we also evaluated the bioresorbable polymer scaffold device in rat and pig models of spinal contusion injury.
The first non-clinical study was conducted by founding scientists of our wholly-owned subsidiary in rats with surgically induced unilateral spinal cord hemisection injury. This study (see Teng, Y. D., Lavik, E. B., Qu, X., Park, K. I., Ourednik, J., Zurakowski, D., Langer, R., and Snyder, E. Y., Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells, Proceedings of the National Academy of Sciences 99, pg. 3024-3029, 2002) demonstrated the baseline safety and efficacy of porous, biodegradable scaffolds fabricated from PLGA-PLL polymer. Subsequently, the safety and efficacy of implantation of the bioresorbable polymer scaffold device was evaluated in rats with spinal cord contusion injury. Initial studies indicated that 24 hours after contusion injury was an appropriate time for device implantation based on both histological evaluation and ex vivo MRI techniques. Based on these results, larger rat contusion studies were performed in our laboratory. Functional recovery was evaluated with the 21-point Basso, Beattie, and Bresnahan (BBB) locomotor rating scale to assess open field locomotion. In this model, the BBB score was not improved by the scaffold device. However, implantation of the bioresorbable polymer scaffold device into the necrotic zone of the injured spinal cord resulted in appositional healing and tissue remodeling that preserved spinal cord architecture. Morphometric analysis of spinal sections stained with hematoxylin & eosin revealed that non-implanted rats with contusion injury developed large cavities surrounded by a thin rim of spared white matter. In contrast, rats treated with the implanted bioresorbable polymer scaffold device demonstrated decreased cavity volume along with increased amounts of spared and remodeled tissue at the lesion epicenter. Cavitation following spinal contusion injury, particularly if progressive, can impair recovery and result in serious clinical symptoms. These results indicate that implantation of the bioresorbable polymer scaffold device in the acutely injured rat spinal cord can provide the benefit of preserving spinal cord architecture through reduced cavitation, and promotion of white matter sparing and tissue remodeling.
The spinal cord anatomy of non-human primates is very similar to that of humans. We performed a series of studies in African green monkeys in order to evaluate the bioresorbable polymer scaffold device in a non-human primate. Our first study in African green monkeys established that unilateral thoracic hemisection SCI (a new model in this species) produced a consistent functional deficit, and we observed a consistently positive response to scaffold implantation (see Pritchard, C. D., Slotkin, J. R., Yu, D., Dai, H., Lawrence, M. S., Bronson, R. T., Reynolds, F. M., Teng, Y. D., Woodard, E. J., and Langer, R. S. Establishing a model spinal cord injury in the African green monkey for the preclinical evaluation of biodegradable polymer scaffolds seeded with human neural stem cells, Journal of Neuroscience Methods 188, pg. 258- 269, 2010). We then conducted two larger studies evaluating the safety and efficacy of the bioresorbable polymer scaffold device in the African green monkey. The extent and time course of functional recovery in biopolymer implant treated primates was assessed with video capture and KinemaTracer evaluation of locomotor behavior with synchronous EMG recording along with locomotor observation rating. When the results of these two studies were combined and analyzed together, we found that implantation of the bioresorbable polymer scaffold device resulted in an increase in remodeled tissue in the region of the hemisection compared to non-implant controls, and improved recovery of locomotion in subjects with full unilateral hemisection lesions.
6
The pig has been used as a large animal model of spinal cord contusion injury due to similarities in size and structure to the human spinal cord. We evaluated the surgical feasibility of implanting the bioresorbable polymer scaffold device in a spinal cord after a contusion injury in the pig model. Severe contusion injuries were created in Gottingen pigs with a weight drop apparatus. At approximately 4, 6, and 24 hours after contusion injury, pigs underwent the bioresorbable polymer scaffold device surgical implantation procedure. At each time point, a large volume of necro-hemorrhagic fluid and debris rapidly effluxed from the injury site, releasing built-up pressure and resulting in a substantial cavity in the center of the spinal cord. Increased spinal tissue pressure after contusion injury results in reduced blood perfusion and ischemia in damaged spinal tissue, and is an important contributor to the pathophysiology of spinal cord injury. As part of our study, we placed bioresorbable polymer scaffold devices into the resulting contusion-induced spinal cord cavity. We measured intraspinal pressure (using catheter pressure probes) at the contusion epicenter in the pigs before, during, and after the surgical procedure. As expected, contusion injury elevated intraspinal tissue pressure compared to normal values. Surgical implantation of the bioresorbable polymer scaffold device resulted in a return of intraspinal tissue pressure to physiologically normal levels.
Taken together, the results from these non-clinical studies in two rat spinal cord injury models, in the African green monkey unilateral hemisection injury model, and the pig contusion injury model, demonstrate that the bioresorbable polymer scaffold device, surgically implanted at the epicenter of the wound after an acute spinal cord injury, acts by appositional healing to spare spinal cord tissue, decrease post-traumatic cyst formation, and decrease spinal cord tissue pressure in preclinical models of spinal cord contusion injury.
Completed Pilot Study
We conducted an early feasibility human pilot study of our Neuro-Spinal Scaffold under our approved Investigational Device Exemption application (IDE) for the treatment of complete, traumatic acute spinal cord injury. The FDA approved the study, which was intended to capture the safety and feasibility of the Neuro-Spinal Scaffold for the treatment of complete functional spinal cord injury, as well as to gather preliminary evidence of the clinical effectiveness of the Neuro-Spinal Scaffold.
The pilot study was initially approved for five subjects in up to six clinical sites across the United States, and was later modified to increase the number of allowable clinical sites to up to 20 and to permit enrollment of up to 10 subjects. The pilot study was initially staggered such that each patient that met the eligibility criteria would be followed for three months prior to enrolling the next patient in the study. In December 2014, barring significant safety issues, the FDA approved an expedited enrollment plan. We enrolled five subjects in the pilot study between October 2014 and September 2015. As discussed below, the FDA has approved a pivotal probable benefit study, the INSPIRE study, that includes data from the patients enrolled in the pilot study.
The INSPIRE Study
Our Neuro-Spinal Scaffold implant is currently being studied in a pivotal probable benefit study formally known as The INSPIRE Study: InVivo Study of Probable Benefit of the Neuro-Spinal Scaffold for Safety and Neurologic Recovery in Subjects with Complete Thoracic AIS A Spinal Cord Injury. The FDA approved converting the pilot study into the INSPIRE study in January 2016. The purpose of the study is to evaluate whether the Neuro-Spinal Scaffold implant is safe and demonstrates probable benefit for the treatment of complete T2-T12/L1 spinal cord injury. The primary endpoint is defined as the proportion of patients achieving an improvement of at least one AIS grade by 6 months post-implantation.
The INSPIRE study is currently approved to enroll up to 12 patients, but we expect that the FDA will approve the full 20 patients, inclusive of the five pilot patients, following the review of the
7
complete 6-month data package for the first five patients. The FDA has requested this data package to include complete and objective comparisons of post-operative and pre-implants magnetic (MRI) findings for each patient to assess the possibility of cyst formation in certain patients. We plan to submit this five-patient, 6-month data package in the second quarter of 2016. We anticipate this will be the only study required for marketing approval under the HDE regulatory pathway. We are targeting completion of the study, which includes completion of enrollment, follow-up, and submission of the HDE application, in 2017.
We have seen promising neurologic outcomes and a favorable profile in the five enrolled pilot study subjects.
Patient
|
Date of Implantation |
Neurologic Level of Injury |
Neurologic Outcome to Date | |||
|---|---|---|---|---|---|---|
1 |
Oct. 2014 | T11 | Converted from AIS A to AIS C at Month 1 with substantial ongoing lower limb motor and sensory improvement through Month 12 | |||
2 |
Jan. 2015 | T7 | Remains AIS A but with marked bowel and bladder improvement through Month 12 | |||
3 |
June 2015 | T4 | Converted from AIS A to AIS B at Month 1 with additional sensory improvement (from mid-chest to mid-abdomen) through Month 6 | |||
4 |
Aug. 2015 | T3 | Remains AIS A at Month 6 | |||
5 |
Sept. 2015 | T8 | Remains AIS A at Month 3 | |||
6 |
Feb. 2016 | T10 | In follow up |
In February 2016, we received approval of a protocol amendment for The INSPIRE Study. The amended protocol establishes the Objective Performance Criterion (OPC), which is a measure of study success used in clinical studies designed to demonstrate safety and probable benefit in support of a Humanitarian Device Exemption approval. The OPC for The INSPIRE Study is defined as 25% or more of the patients in the study demonstrating an improvement of at least one AIS grade by six months post-implantation. Since The INSPIRE Study is designed to enroll 20 patients with complete (AIS A) spinal cord injuries (inclusive of the 5 patients enrolled in the company's pilot trial), the OPC equates to having five patients convert to any other AIS grade by six months post-implantation.
Bioengineered Neural Trails™ injection program for chronic SCI
In December 2015, we announced our preclinical Bioengineered Neural Trails injection program for the treatment of chronic spinal cord injury. Bioengineered Neural Trails are injectable combinations of biomaterials and neural stem cells (NSCs) delivered using minimally-invasive surgical instrumentation and techniques to create trails across the chronic injury site. To support this program, we recently entered into an exclusive license agreement with University of California, San Diego and an assignment agreement with James Guest, M.D., Ph.D., for issued patents covering technology related to the Bioengineered Neural Trails program, and we also have filed a provisional application in support of the Bioengineered Neural Trails injection program. We expect that our Bioengineered Neural Trails injection investigational product will be regulated by the FDA as a combination product, and we are targeting a pre-Investigational New Drug meeting with the FDA by the end of 2016. For further information on the regulatory pathway for the Bioengineered Neural Trails injection product, please see "Government Regulation" below.
Intellectual Property
We rely on a combination of patents, licenses, trade secrets and non-disclosure agreements to develop, protect and maintain our intellectual property. Our patent portfolio includes patents and
8
patent applications. We seek to develop or obtain intellectual property that we believe might be useful or complementary with our products and technologies, including by way of licenses or acquisitions of other companies or intellectual property from third parties.
We hold an exclusive worldwide license to a broad suite of patents co-owned by BCH and MIT covering the use of a wide range of polymers to treat SCI, and to promote the survival and proliferation of human stem cells in the spinal cord (the "BCH License"). Issued patents and pending patent applications licensed under the BCH License cover the technology underlying our Neuro-Spinal Scaffold implant and the use of a wide range of biomaterial scaffolding for treating SCI by itself or in combination with drugs, growth factors or human stem cells. The BCH License covers seven issued United States patents and four issued international patents expiring between 2018 and 2027, and one pending United States patent and 10 pending international patents.
The BCH License has a 15-year term, or as long as the life of the last expiring patent right under the license, whichever is longer, unless terminated earlier by BCH. In connection with our acquisition of the BCH License, we submitted to a 5-year development plan to BCH and MIT that includes certain targets and projections related to the timing of product development and regulatory approvals. We are required to either meet the stated targets and projections in the plan, or notify BCH and revise the plan. BCH has the right to terminate the BCH License for failure by us to either meet the targets and projections in the plan or our failure submit an acceptable revision to the plan within a 60-day cure period after notification by BCH that we are not in compliance with the plan. We are currently in compliance with our plan.
We have the right to sublicense the patents covered by the BCH License, and have full control and authority over the development and commercialization of any products that use the licensed technology, including clinical trial design, manufacturing, marketing, and regulatory filings,. We also own the rights to the data generated pursuant to the BCH License, whether generated by us or a sublicensee. We have the first right of negotiation with BCH and MIT for a 30-day period to any improvements to the intellectual property covered by the BCH License.
We are required to pay certain fees and royalties under the BCH License. We paid an initial fee upon execution of the BCH License and are required to pay an amendment fee if we expand the field of use under the BCH License. We are also required to make milestone payments upon completing various phases of product development, including upon (i) filing with the FDA of the first investigational new drug application and IDE application for a product that uses the licensed technology; (ii) enrollment of the first patient in Phase II testing for a product that uses the licensed technology; (iii) enrollment of the first patient in Phase III testing for a product that uses the licensed technology; (iv) FDA approval of first new drug application or related application for a product that uses the licensed technology, and (v) first market approval in any country outside the United States for a product that uses the licensed technology. Each year prior to the release of a licensed product, we are also required to pay a maintenance fee for the BCH License. Further, we are required to make ongoing payments based on any sublicenses we grant to manufacturers and distributors. Following commercialization, we are required to make ongoing royalty payments equal to a percentage of net sales of any product that uses the licensed technology.
In addition to the rights it licensed under the BCH license, InVivo has additional rights relating to the Neuro-Spinal Scaffold. InVivo and MIT co-own patent application No. U.S. 14/232,525 ("Poly((lactic-co-glycolic acid)-b-lysine) and process for synthesizing a block copolymer of PLGA and PLL- (poly-e-cbz-l-lysine)"). InVivo also owns patent application No. U.S. 13/793,231 ("Protective packaging with product preparation features incorporated") and US patent application No. U.S. 13/930,829 ("cupped forceps").
To support our Bioengineered Neural Trails injection program, we recently entered into agreements with the University of California, San Diego (UC San Diego) and
9
James Guest, M.D., Ph.D., to expand our intellectual property portfolio. We entered into an exclusive license agreement with UC San Diego for an issued patent and into an assignment agreement with Dr. Guest for an issued patent. We also have filed a provisional application in support of the Bioengineered Neural Trails injection program with the USPTO.
Government Regulation
The testing, manufacturing, and potential labeling, advertising, promotion, distribution, import and marketing of our products are subject to extensive regulation by governmental authorities in the U.S. and in other countries. In the U.S., the FDA, under the Public Health Service Act, the Federal Food, Drug and Cosmetic Act (FDCA), and their implementing regulations, regulates biologics and medical device products. In addition, our products under development are subject to extensive regulation by other U.S. federal and state regulatory bodies and comparable authorities in other countries. To ensure that medical products distributed domestically are safe and effective for their intended use, the FDA and comparable authorities in other countries have imposed regulations that govern, among other things, the following activities that we or our partners perform or will perform:
- •
- product design and development;
- •
- product testing;
- •
- product manufacturing;
- •
- product labeling;
- •
- product storage;
- •
- premarket clearance, approval or CE marking of products;
- •
- advertising and promotion;
- •
- product marketing, sales and distribution; and
- •
- post-market surveillance reporting, including reporting of death or serious injuries.
The labeling, advertising, promotion, marketing and distribution of biopharmaceuticals, or biologics and medical devices also must be in compliance with the FDA requirements which include, among others, standards and regulations for off-label promotion, industry sponsored scientific and educational activities, promotional activities involving the internet, and direct-to-consumer advertising. In addition, the Federal Trade Commission, or FTC, also regulates the advertising of many medical devices. The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the issuance of a warning letter directing us to correct deviations from regulatory standards and enforcement actions that can include seizures, injunctions and criminal prosecution. . In addition, under the federal Lanham Act and similar state laws, competitors and others can initiate litigation relating to advertising claims.
The FDA has broad post-market and regulatory enforcement powers. As with medical devices, manufacturers of biologics and combination products are subject to unannounced inspections by the FDA to determine compliance with applicable regulations, and these inspections may include the manufacturing facilities of some of our subcontractors. Failure by manufacturers or their suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities. Potential FDA enforcement actions include:
- •
- warning letters, fines, injunctions, consent decrees and civil penalties;
- •
- unanticipated expenditures to address or defend such actions
- •
- customer notifications for repair, replacement, refunds;
10
- •
- recall, detention or seizure of our products;
- •
- operating restrictions or partial suspension or total shutdown of production;
- •
- refusing or delaying our requests for 510(k) clearance or premarket approval of new products or modified products;
- •
- operating restrictions;
- •
- withdrawing 510(k) clearances on PMA approvals that have already been granted;
- •
- refusal to grant export approval for our products; or
- •
- criminal prosecution.
FDA Regulation—Medical Device Products
FDA's Premarket Clearance and Approval Requirements
Unless an exemption applies, each medical device we wish to commercially distribute in the U.S. will require either prior 510(k) clearance or prior premarket approval from the FDA. The FDA classifies medical devices into one of three classes. Devices deemed to pose lower risk are placed in either Class I or II, which requires the manufacturer to submit to the FDA a premarket notification which must be cleared by the FDA before the medical device may be distributed commercially. This process is known as 510(k) clearance. Most Class I devices are exempt from this requirement. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in Class III, requiring premarket approval or approval of a humanitarian device exemption. We expect the Neuro-Spinal Scaffold implant will be regulated by the FDA as a Class III medical device.
Premarket Approval Pathway
A premarket approval application must be submitted if the device cannot be cleared through the 510(k) process. A premarket approval application, or PMA, must be supported by extensive data including, but not limited to, technical, preclinical and other nonclinical, clinical, and manufacturing and labeling information to demonstrate to the FDA's satisfaction the safety and effectiveness of the device for its intended use.
If the FDA determines that a PMA submission is sufficiently complete, the FDA will accept the application for filing and begin an in-depth review of the submitted information. By statute, the FDA has 180 days to review the "accepted application," although, generally, review of the application can take between one and three years, and it may take significantly longer. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance with quality system regulations. New PMAs or PMA supplements are required for modifications that affect the safety or effectiveness of the device, including, for example, certain types of modifications to the device's indication for use, manufacturing process, labeling and design. Premarket approval supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA, and may not require as extensive clinical data or the convening of an advisory panel.
11
Humanitarian Device Exemption
Alternatively, a Class III device may qualify for FDA approval to be distributed under a Humanitarian Device Exemption (HDE) rather than a PMA. For a device to be eligible for an HDE, it must be first designated by the FDA as a Humanitarian Use Device (HUD) intended to benefit patients in the treatment or diagnosis of a disease or condition that affects fewer than 4,000 individuals in the United States per year. The HDE also requires that there must be no other comparable device available to provide therapy for this condition. An HDE application is similar in form and content to a PMA application and, although exempt from the effectiveness requirements of a PMA, an HDE does require sufficient information for FDA to determine that the device does not pose an unreasonable or significant risk of illness or injury, and that the probable benefit to health outweighs the risk of injury or illness from its use. In addition, a HUD may only be used in facilities that have established a local institutional review board, or IRB, to supervise clinical testing of devices, and after an IRB has approved the use of the device to treat or diagnose the specific disease.
In addition, except in certain circumstances, products approved under an HDE cannot be sold for an amount that exceeds the costs of research and development, fabrication, and distribution of the device (i.e., for profit). Currently, a product is only eligible to be sold for profit after receiving HDE approval if the device (1) is intended for the treatment or diagnosis of a disease or condition that occurs in pediatric patients or in a pediatric subpopulation, and such device is labeled for use in pediatric patients or in a pediatric subpopulation in which the disease or condition occurs; or (2) is intended for the treatment or diagnosis of a disease or condition that does not occur in pediatric patients or that occurs in pediatric patients in such numbers that the development of the device for such patients is impossible, highly impracticable, or unsafe. If an HDE-approved device does not meet either of the eligibility criteria, the device cannot be sold for profit. We expect our Neuro-Spinal Scaffold will meet the eligibility criteria to be sold for a profit.
Clinical Trials
Clinical trials are almost always required to support a PMA or HDE application. If the device presents a "significant risk" to human health as defined by the FDA, the FDA requires the device sponsor to submit an investigational device exemption application, or IDE, to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a "non-significant risk" device in which case, and IDE will not be required, although the clinical trial must meet other requirements including IRB approval. Clinical trials for a significant risk device may begin once the IDE is approved by the FDA and the appropriate IRB at each clinical trial sites. Future clinical trials may require that we obtain an IDE from the FDA prior to commencing clinical trials and that the trial be conducted with the oversight of an IRB at the clinical trial site. Our clinical trials must be conducted in accordance with FDA regulations and federal and state regulations concerning human subject protection, including informed consent and healthcare privacy. A clinical trial may be suspended by FDA or at a specific site by the relevant IRB at any time for various reasons, including a belief that the risks to the trial participants outweigh the benefits of participation in the clinical trial. Even if a clinical trial is completed, the results of our clinical testing may not demonstrate the safety and efficacy of the device, or may be equivocal or otherwise not be sufficient to obtain approval of our product.
12
Pervasive and Continuing FDA Regulation
After a device is placed on the market, numerous regulatory requirements continue to apply. These include:
- •
- product listing and establishment registration, which helps facilitate FDA inspections and other regulatory action;
- •
- Quality System Regulation, or QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design,
testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process;
- •
- labeling regulations and FDA prohibitions against the promotion of products for uncleared or unapproved indications or other off-label
uses;
- •
- clearance of product modifications that could significantly affect safety or efficacy or that would constitute a major change in
intended use of one of our cleared devices;
- •
- approval of product modifications that affect the safety or effectiveness of one of our approved devices;
- •
- medical device reporting regulations, which require that manufacturers comply with FDA requirements to report if their device may have
caused or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the device or a similar
device were to recur;
- •
- post-approval restrictions or conditions, including post-approval study commitments;
- •
- post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and
effectiveness data for the device;
- •
- the FDA's recall authority, whereby it can ask, or under certain conditions order, device manufacturers to recall from the market a
product that is in violation of governing laws and regulations;
- •
- regulations pertaining to voluntary recalls; and
- •
- notices of corrections or removals.
We and any third-party manufacturers that we use must register with the FDA as medical device manufacturers and must obtain all necessary state permits or licenses to operate our business. As manufacturers, we and any third-party manufacturers that we use are subject to announced and unannounced inspections by the FDA to determine our compliance with quality system regulation and other regulations. We have not yet been inspected by the FDA. We believe that we are in substantial compliance with quality system regulation and other regulations.
Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
- •
- warning letters, fines, injunctions, consent decrees and civil penalties;
- •
- unanticipated expenditures to address or defend such actions
- •
- customer notifications for repair, replacement, refunds;
- •
- recall, detention or seizure of our products;
- •
- operating restrictions or partial suspension or total shutdown of production;
- •
- refusing or delaying our requests for 510(k) clearance or premarket approval of new products or modified products;
13
- •
- operating restrictions;
- •
- withdrawing 510(k) clearances on PMA approvals that have already been granted;
- •
- refusal to grant export approval for our products; or
- •
- criminal prosecution.
Regulatory Pathway for Neuro-Spinal Scaffold implant
We expect the Neuro-Spinal Scaffold will be regulated by the FDA as a Class III medical device. A Class III medical device typically will require FDA approval of a Pre-Market Approval (PMA) Application before we can begin selling the product in the United States. A PMA application must be supported by extensive data including, but not limited to, technical information regarding device design and development, preclinical and clinical trials, data and manufacturing and labeling to support the FDA's determination that there is reasonable assurance that the device is safe and effective for its intended use.
Alternatively, a Class III device may qualify for FDA approval to be distributed under a Humanitarian Device Exemption (HDE) rather than a PMA. For a device to be eligible for an HDE, it must be first designated by the FDA as a Humanitarian Use Device (HUD) intended to benefit patients in the treatment or diagnosis of a disease or condition that affects fewer than 4,000 individuals in the United States per year. The FDA granted HUD designation for our Neuro-Spinal Scaffold implant in 2013. In 2015, we received conditional approval from the FDA to convert our ongoing pilot study into a pivotal probable benefit study, which condition to approval was lifted, and for which full approval subsequently granted, in January 2016.
In the future, if our Neuro-Spinal Scaffold is approved via either the PMA or HDE pathway, modifications or enhancements that could significantly affect the safety or effectiveness of the device or that constitute a major change to the intended use of the device will require new PMA or HDE application and approval. Other changes may require a supplement or other change notification that must be reviewed and approved by the FDA. Modified devices for which a new PMA or HDE application, supplement or notification is required cannot be distributed until the application is approved by the FDA. An adverse determination or a request for additional information could delay the market introduction of new products, which could have a material adverse effect on our business, financial condition and results of operations. We may not be able to obtain PMA or HDE approval in a timely manner, if at all, for the Neuro-Spinal Scaffold implant or any future devices or modifications to Neuro-Spinal Scaffold implant or such devices for which we may submit a PMA or HDE application.
European Economic Area (EEA)
Sales of medical devices are subject to foreign government regulations, which vary substantially from country to country. In order to market our products outside the United States, we must obtain regulatory approvals or CE Certificates of Conformity and comply with extensive safety and quality regulations. The time required to obtain approval by a foreign country or to obtain a CE Certificate of Conformity may be longer or shorter than that required for FDA clearance or approval, and the requirements may differ. In the EEA, we are required to obtain Certificates of Conformity before drawing up an EC Declaration of Conformity and affixing the CE mark to our medical devices. Many other countries, such as Australia, India, New Zealand, Pakistan and Sri Lanka, accept CE Certificates of Conformity or FDA clearance or approval although others, such as Brazil, Canada and Japan require separate regulatory filings.
In the EEA, our devices are required to comply with the Essential Requirements laid down in Annex I to the Council Directive 93/42/EEC of 14 June 1993 concerning medical devices, known as the Medical Devices Directive. Compliance with these requirements entitles us to affix the CE mark to our
14
medical devices, without which they cannot be commercialized in the EEA. To demonstrate compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive and obtain the right to affix the CE mark to our medical devices, we must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. Except for low risk medical devices (Class I with no measuring function and which are not sterile), where the manufacturer can issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the Essential Requirements laid down in the Medical Devices Directive, a conformity assessment procedure requires the intervention of a Notified Body. This is an organization designated by the competent authorities of a EEA country to conduct conformity assessments. The Notified Body would typically audit and examine products' Technical File and the quality system for the manufacture, design and final inspection of our devices before issuing a CE Certificate of Conformity demonstrating compliance with the relevant Essential Requirements laid down in Annex I to the Medical Devices Directive. Following the issuance of this CE Certificate of Conformity, we can draw up an EC Declaration of Conformity and affix the CE mark to the products covered by this CE Certificate of Conformity and the EC Declaration of Conformity. We have not applied for CE Mark for the Neuro-Spinal Scaffold.
After the product has been CE marked and placed on the market in the EEA, we must comply with a number of regulatory requirements relating to:
- •
- registration/notification of medical devices in individual EEA countries;
- •
- pricing and reimbursement of medical devices;
- •
- establishment of post-marketing surveillance and adverse event reporting procedures;
- •
- Field Safety Corrective Actions, including product recalls and withdrawals;
- •
- marketing and promotion of medical devices; and
- •
- interactions with physicians.
Failure to comply with these requirements may result in enforcement measures being taken against us by the competent authorities of the EEA countries. These can include fines, administrative penalties, compulsory product withdraws, injunctions and criminal prosecution. Such enforcement measures would have an adverse effect on our capacity to market our products in the EEA and, consequently, on our business and financial position. Such failures may also lead to cancelation, suspension, or variation of our CE Certificates of Conformity by our Notified Body.
Further, the advertising and promotion of our products in the EEA is subject to the provisions of the Medical Devices Directive, Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other national legislation in the individual EEA countries governing the advertising and promotion of medical devices. These laws may limit or restrict the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals.
FDA Regulation—Combination Products/Biologics
We believe that our Bioengineered Neural Trails under development may be defined as combination products consisting of two or more regulated components, that is, a biologic and a medical device. In the U.S., a combination product is assigned by the FDA to one of the agency's centers, such as the Center for Biologics Evaluation and Research (CBER) or Center for Devices and Radiological Health (CDRH) with the chosen center to take the lead in pre-marketing review and approval of the combination product. Other FDA centers also may review the product in regard to matters that are within their expertise. The FDA selects the lead center based on an assessment of the combination
15
product's "primary mode of action." Some products also may require approval or clearance from more than one FDA center.
To determine which FDA center or centers will review a combination product submission, companies may submit a request for assignment to the FDA. Those requests may be handled formally or informally. In some cases, jurisdiction may be determined informally based on FDA experience with similar products. However, informal jurisdictional determinations are not binding on the FDA. Companies also may submit a formal Request for Designation to the FDA Office of Combination Products. The Office of Combination Products will review the request and make its jurisdictional determination within 60 days of receiving a Request for Designation. Stem cell-based therapies are typically regulated under the jurisdiction of CBER typically requiring an Investigational New Drug (IND) application and a biologic license application, or BLA, for marketing approval.
The IND and BLA Approval Process
Biological products must satisfy the requirements of the Public Health Services Act and its implementing regulations. In order for a biologic product to be legally marketed in the U.S., the product must have a BLA approved by the FDA.
The steps for obtaining FDA approval of a BLA to market a biopharmaceutical, or biologic product in the U.S. include:
- •
- completion of preclinical laboratory tests, animal studies and formulation studies under the FDA's GLP regulations;
- •
- submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin and
which must include IRB approval at each clinical site before the trials may be initiated;
- •
- performance of adequate and well-controlled clinical trials in accordance with good clinical practices, or GCPs, to establish the
safety, purity, and potency of the product for each indication;
- •
- submission to the FDA of a BLA, which contains detailed information about the chemistry, manufacturing and controls for the product,
reports of the outcomes of the clinical trials, and proposed labeling and packaging for the product;
- •
- the FDA's acceptance of the BLA for filing;
- •
- for any biological product containing an active ingredient not previously approved, automatic referral to an appropriate advisory
committee for review prior to approval, unless the FDA decides otherwise;
- •
- satisfactory review of the contents of the BLA by the FDA, including the satisfactory resolution of any questions raised during the
review or by the advisory committee, if applicable;
- •
- satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess
compliance with cGMP requirements, to assure that the facilities, methods and controls are adequate to ensure the product's identity, strength, quality and purity; and
- •
- FDA approval of the BLA.
Preclinical or nonclinical studies include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies, and may be conducted before or after an IND is submitted.
An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about issues such as the conduct of the trials as outlined in
16
the IND. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed.
Clinical trials are subject to extensive monitoring, recordkeeping and reporting requirements. Clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial sites and must comply with FDA requirements, including but not limited to those relating to GCP. Clinical trials involving drugs and biologics are typically conducted in three sequential phases. The phases may overlap or be combined. A fourth, or post-approval, phase may include additional clinical trials. These phases are described generally below.
- •
- Phase I. Phase I clinical trials involve
the initial introduction of the drug into healthy human subjects to test for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or
life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
- •
- Phase II. Phase II clinical trials usually
involve studies in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific, targeted indications, to
determine dosage tolerance and optimal dosage, and to identify possible adverse effects and safety risks.
- •
- Phase III. Phase III involves studies
undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical sites. These studies are intended to establish the overall
risk-benefit ratio of the product and provide an adequate basis for product labeling.
- •
- Post-Approval (Phase IV). Post-approval clinical trials are required of or agreed to by a sponsor as a condition of, or subsequent to marketing approval. Further, if the FDA becomes aware of new safety information about an approved product, it is authorized to require post-approval trials of the biological product. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of biologics approved under accelerated approval regulations. Failure to promptly conduct Phase IV clinical trials could result in withdrawal of approval for products approved under accelerated approval regulations.
Clinical testing may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted under an IND and may, at its discretion, reevaluate, alter, suspend, or terminate the testing based upon the data accumulated to that point and the FDA's assessment of the risk/benefit ratio to the patient. The FDA or the sponsor may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request that additional preclinical studies or clinical trials be conducted as a condition to product approval. Additionally, new government requirements may be established that could delay or prevent regulatory approval of our products under development. Furthermore, IRBs have the authority to suspend clinical trials in their respective institutions at any time for a variety of reasons, including safety issues.
Assuming successful completion of the required clinical testing, the results of the preclinical studies and of the clinical trials, together with other detailed information, including information on the chemistry, manufacture and composition of the product, are submitted to the FDA in the form of a BLA requesting approval to market the product for one or more indications. In most cases, the BLA must be accompanied by a substantial user fee. The FDA will initially review the BLA for completeness before it accepts the BLA for filing. There can be no assurance that the submission will be accepted for filing or that the FDA may not issue a refusal-to-file, or RTF. If a RTF is issued, there is opportunity
17
for dialogue between the sponsor and the FDA in an effort to resolve all concerns. If the BLA submission is accepted for filing, the FDA will begin an in-depth review of the BLA to determine, among other things, whether a product is safe and effective for its intended use and whether the product is being manufactured in accordance with cGMP to assure and preserve the product's identity, strength, quality and purity. If the biological product contains a new active ingredient not previously approved, the BLA automatically will be referred to an appropriate advisory committee for review prior to approval of the biological product, unless the FDA decides otherwise and specifies such reasons in a complete response letter to the sponsor. The FDA, however, is not bound by the opinion of the advisory committee.
Companies also may seek fast track designation for their products. Fast track products are those that are intended for the treatment of a serious or life-threatening condition and that demonstrate the potential to address unmet medical needs for such a condition. If awarded, the fast track designation applies to the product only for the indication for which the designation was received. Fast track products are eligible for two means of potentially expediting product development and FDA review of BLAs. First, a fast track product may be approved on the basis of either a clinical endpoint or a surrogate endpoint that is reasonably likely to predict clinical benefit. Approvals of this kind may be subject to requirements for appropriate post-approval studies to validate the surrogate endpoint or otherwise confirm the effect on the clinical endpoint, and to certain other conditions. Second, if the FDA determines after review of preliminary clinical data submitted by the sponsor that a fast track product may be effective, it may begin review of portions of a BLA before the sponsor submits the complete BLA, thereby accelerating the date on which review of a portion of the BLA can begin. There can be no assurance that any of our other products will receive designation as fast track products. Even if they are designated as fast track products, we cannot assure you that our products will be reviewed or approved more expeditiously for their fast track indications than would otherwise have been the case, or that such products will be approved promptly, or at all. Furthermore, the FDA can revoke previously-granted fast track status at any time.
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the therapeutic product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-approval clinical trials to verify and further define the drug's clinical benefit and safety profile. There can be no assurance that any of our products will receive accelerated approval. Even if accelerated approval is granted, the FDA may withdraw such approval if the sponsor fails to conduct the required post-approval clinical trials, or if the post-approval clinical trials fail to confirm the early benefits seen during the accelerated approval process.
Fast-Track designation and accelerated approval should be distinguished from priority review although products awarded fast track status may also be eligible for priority review. Products regulated by the CBER may receive priority review if they provide significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious or life-threatening disease. Products awarded priority review are given abbreviated review goals by the agency. The FDA has agreed to a performance goal of reviewing products awarded priority review within six months, whereas products under standard review receive a ten-month target. The review process, however, is often significantly extended by FDA requests for additional information or clarification regarding information already provided in the submission. Priority review is requested at the time the BLA is submitted, and the FDA makes a decision as part of the agency's review of the application for filing.
18
If granted, Fast-Track designation, accelerated approval, and priority review may expedite the approval process, but they do not change the standards for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. Data obtained from clinical activities are not always conclusive, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions, such as post approval studies, on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and further FDA review and approval.
Post-Approval Requirements
After regulatory approval of a product is obtained, companies are required to comply with a number of post-approval requirements relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping. For example, as a condition of approval of a BLA, the FDA may require post-approval testing and surveillance to monitor the product's safety or efficacy. In addition, holders of an approved BLA are required to keep extensive records, to report certain adverse reactions and production deviations and problems to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for their products. If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities, or previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
Regulatory Pathway for Bioengineered Neural Trails
Our Bioengineered Neural Trails injection investigational product is expected to be regulated as a combination product. Combination products are therapeutic and diagnostic products that combine drugs, devices, and/or biological products. As described above, a combination product is assigned to an FDA center based on a determination of the "primary mode of action" of the combination product. Stem cell-based therapies are regulated under the jurisdiction of Center for Biologics Evaluation and Research, or CBER, typically requiring an IND and a BLA for marketing approval. The formal jurisdiction assignment process is achieved through the request for designation process. We are targeting a pre-IND meeting with the FDA by the end of 2016.
Research and Development Expenditures
Our research and development expenditures, which include research and development related to our product candidates, were $10,058,000, $10,273,000 and $10,553,000 in 2015, 2014, and 2013, respectively.
Competition
We have many potential competitors, including major drug companies, specialized biotechnology firms, academic institutions, government agencies and private and public research institutions. Many of these competitors have significantly greater financial and technical resources than us, and superior experience and expertise in research and development, preclinical testing, design and implementation of clinical trials, regulatory processes and obtaining regulatory approval for products, production and
19
manufacturing, and sales and marketing of approved products. Smaller or early-stage companies and research institutions may also prove to be significant competitors, particularly if they have collaborative arrangements with larger and more established biotechnology companies. We will also face competition from these parties in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and registering subjects for clinical trials.
In order to compete effectively, we will have to make substantial investments in development, clinical testing, manufacturing and sales and marketing, or partner with one or more established companies. There is no assurance that we will be successful in having any of our products approved or gaining significant market share for any of our products. Our technologies and products also may be rendered obsolete or noncompetitive as a result of products introduced by our competitors.
Manufacturing
We have developed a proprietary manufacturing process to build our Neuro-Spinal Scaffold implant. We manufacture our implants following FDA regulations for design controls using two fully operational manufacturing cleanrooms located at our facility in Cambridge, Massachusetts. These two cleanrooms are validated to ISO 14644-1 Class ISO-7 (Class 10k) and Class ISO-8 (Class 100k) cleanroom standards, respectively. In addition, the manufacturing process contains numerous quality control steps including in- process and final inspection. Currently, we are working with two preferred vendors for our critical raw materials; however, these materials are also available from other vendors. We are currently manufacturing our Neuro-Spinal Scaffold implant to support our pivotal probable benefit clinical study.
Sales and Marketing
If we obtain approval from the FDA, or another foreign regulatory body, to commercialize our products, we plan to establish a direct sales force to sell our products to major markets in the United States and to sell through distributors in foreign markets. We anticipate the direct sales force, once and if established, would focus its efforts on maximizing revenue through product training, placement and support. We would also seek to establish strong relationships with orthopedic spine surgeons and neurosurgeons and would expect to provide a high level of service for the products including providing on-site assistance and service during procedures. In addition, we expect to implement medical education programs intended for outreach to practitioners in physical medicine and rehabilitation centers and patient advocacy groups. We may also seek corporate partners with expertise in commercialization.
Compliance with Environmental, Health and Safety Laws
In addition to FDA regulations discussed above, we are also subject to evolving federal, state and local environmental, health and safety laws and regulations. In the past, compliance with environmental, health and safety laws and regulations has not had a material effect on our capital expenditures. We believe that we comply in all material respects with existing environmental, health and safety laws and regulations applicable to us.
Employees
As of December 31, 2015, we had 30 employees. None of our employees is represented by a labor union, and we consider our employee relations to be good. We also utilize a number of consultants to assist with research and development and regulatory activities. We believe that our future success will depend in part on our continued ability to attract, hire and retain qualified personnel.
20
Corporate Information
We incorporated under the laws of the state of Nevada on April 2, 2003 as Design Source, Inc. On October 26, 2010, we acquired the business of InVivo Therapeutics Corporation, which was founded in 2005, and are continuing the existing business operations of InVivo Therapeutics Corporation as our wholly-owned subsidiary. We changed our name to InVivo Therapeutics Holdings Corp. in connection with the acquisition.
Our offices are located at One Kendall Square, Suite B14402, Cambridge, Massachusetts 02139, and our telephone number is 617-863-5500. Our website is www.invivotherapeutics.com. Information contained on, or accessible through, our website is not a part of, and is not incorporated by reference into, this Annual Report.
Available Information
We make available free of charge on or through the Investor Relations link on our website, www.invivotherapeutics.com, all materials that we file electronically with the Securities and Exchange Commission ("SEC"), including our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports. Information appearing on our website is not a part of, and is not incorporated in, this Annual Report.
You may also read and copy any materials filed by us with the SEC at the SEC's Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549, and you may obtain information on the operation of the Public Reference Room by calling the SEC in the United States at 1-800-SEC-0330. In addition, the SEC maintains an Internet website at www.sec.gov that contains reports, proxy and information statements and other information that we file electronically with the SEC.
21
Certain factors may have a material adverse effect on our business, financial condition, and results of operations. You should consider carefully the risks and uncertainties described below, in addition to other information contained in this Annual Report on Form 10-K, including our consolidated financial statements and related notes. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that adversely affect our business. If any of the following risks actually occurs, our business, financial condition, results of operations, and future prospects could be materially and adversely affected.
Risks Related to Our Business
We have a limited operating history and have incurred significant losses since our inception.
We have incurred net losses each year since our inception, including net losses of $33.3 million for the year ended December 31, 2015, and $18.3 million for the year ended December 31, 2014. As of December 31, 2015, we had an accumulated deficit of $133.6 million. To date, we have not commercialized any products or generated any revenues from the sale of products, and we do not expect to generate any product revenues in the foreseeable future. We do not know whether or when we will generate revenue or become profitable.
We have devoted most of our financial resources to research and development, including our clinical and preclinical development activities related to our Neuro-Spinal Scaffold implant. Overall, we expect our research and development expenses to be substantial and to increase for the foreseeable future as we continue the development and clinical investigation of our current and future products. Our lead product candidate, Neuro-Spinal Scaffold implant, is currently being studied in a pivotal probable benefit study and, as a result, we expect that it could be several years, if ever, before we have a product candidate ready for commercialization. Even if we obtain regulatory approval to market our Neuro-Spinal Scaffold implant or other products, our future revenues will depend upon the size of any markets in which our products have received approval, our ability to achieve sufficient market acceptance, reimbursement from third-party payors and other factors.
We anticipate that we will continue to incur substantial losses for the foreseeable future and may never achieve or maintain profitability.
We expect to continue to incur significant expenses and increasing net losses for at least the next several years. We expect our expenses will increase substantially in connection with our ongoing activities, as we:
- •
- continue our pivotal probable benefit study of Neuro-Spinal Scaffold implant;
- •
- continue the research and development of our other product candidates;
- •
- have our product candidates manufactured for clinical trials and for commercial sale;
- •
- establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval;
- •
- maintain, protect and expand our intellectual property portfolio; and
- •
- continue our research and development efforts for new product opportunities.
To become and remain profitable, we must succeed in developing and commercializing our product candidates with significant market potential. This will require us to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of our product candidates, developing additional product candidates, obtaining regulatory approval for these product
22
candidates and manufacturing, marketing and selling any products for which we may obtain regulatory approval. We are only in the initial stages of most of these activities. We may never succeed in these activities and, even if we do, may never generate revenues that are significant enough to achieve profitability.
Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable could depress the value of our Company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or even continue our operations. A decline in the value of our Company could cause you to lose all or part of your investment.
There is substantial doubt about our ability to continue as a going concern, which will affect our ability to obtain future financing and may require us to curtail our operations.
Our financial statements as of December 31, 2015 were prepared under the assumption that we will continue as a going concern. The independent registered public accounting firm that audited our 2015 financial statements, in their report, included an explanatory paragraph referring to our recurring losses since inception and expressing substantial doubt in our ability to continue as a going concern. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty. At December 31, 2015, we had cash and cash equivalents of $20.2 million. Our ability to continue as a going concern depends on our ability to obtain additional equity or debt financing, attain further operating efficiencies, reduce expenditures, and, ultimately, to generate revenue.
We will need additional funding in the future. If we are unable to raise capital when needed, we could be forced to delay, reduce or eliminate our product development programs or commercialization efforts.
We expect our expenses to increase in connection with our ongoing activities, particularly as we conduct our pivotal probable benefit study of, and seek regulatory approval for, our Neuro-Spinal Scaffold implant. In addition, if we obtain regulatory approval for any of our product candidates, we expect to incur significant commercialization expenses related to manufacturing, marketing, sales and distribution. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts.
As of December 31, 2015, our consolidated cash balance was approximately $20.2 million. We believe our current cash and cash equivalents are adequate to fund our operations into the fourth quarter of 2016. As a result, our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern in their report on our financial statements. Our future funding requirements, both near- and long-term, will depend on many factors, including, but not limited to:
- •
- the scope, progress, results and costs of preclinical development, laboratory testing and clinical studies for our Neuro-Spinal Scaffold implant and
any other product candidates that we may develop or acquire;
- •
- future clinical trial results of our Neuro-Spinal Scaffold implant;
- •
- the timing of, and the costs involved in, obtaining regulatory approvals for the Neuro-Spinal
Scaffold implant if our pivotal probable benefit study is successful, and the outcome of regulatory review of the Neuro-Spinal
Scaffold implant;
- •
- the cost and timing of future commercialization activities for our products, if any of our product candidates are approved for marketing, including product manufacturing, marketing, sales and distribution costs;
23
- •
- the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval;
- •
- the cost of having our product candidates manufactured for clinical trials in preparation for regulatory approval and in preparation
for commercialization;
- •
- the cost and delays in product development as a result of any changes in regulatory oversight applicable to our product candidates;
- •
- our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such
agreements;
- •
- the cost and timing of establishing sales, marketing and distribution capabilities;
- •
- the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing our intellectual property portfolio;
- •
- the efforts and activities of competitors and potential competitors;
- •
- the effect of competing technological and market developments; and
- •
- the extent to which we acquire or invest in businesses, products and technologies.
Identifying potential product candidates and conducting preclinical testing and clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain regulatory approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. Our commercial revenues, if any, will be derived from sales of products that we do not expect to be commercially available for several years, if at all. Accordingly, we will need to continue to rely on additional financing to achieve our business objectives. Adequate additional financing may not be available to us on acceptable terms, or at all, and if we are not successful in raising additional capital, we may not be able to continue as a going concern
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our product candidates on unfavorable terms to us.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings and other third-party funding alternatives including license and collaboration agreements. To raise additional capital or pursue strategic transactions, we may in the future sell additional shares of our common stock or other securities convertible into or exchangeable for our common stock which will dilute the ownership interest of our current stockholders, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our current stockholders. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our product candidates, future revenue streams or research programs, or grant licenses on terms that may not be favorable to us or that may reduce the value of our common stock. If we are unable to raise additional funds when needed, we may be required to delay, limit, reduce or terminate our product development or commercialization efforts for our Neuro-Spinal Scaffold implant or any other product candidates that we develop or acquire.
24
We license certain technology underlying the development of our Neuro-Spinal Scaffold from BCH and MIT, and the loss of the license would result in a material adverse effect on our business, financial position and operating results and cause the market value of our common stock to decline.
We license technology from BCH and MIT that is integrated into our Neuro-Spinal Scaffold implant under an exclusive license. Under the license agreement, we have agreed to milestone payments and to meet certain reporting obligations. In the event that we were to breach any of the obligations under the agreement and fail to timely cure, BCH and MIT would have the right to terminate the agreement upon notice. In addition, BCH and MIT have the right to terminate our license upon the bankruptcy or receivership of the Company. If we are unable to continue to use or license this technology on reasonable terms, or if this technology fails to operate properly, we may not be able to secure alternatives in a timely manner and our ability to develop our products could be harmed.
We depend heavily on the success of one product candidate, Neuro-Spinal Scaffold implant, which is currently being studied in a pivotal probable benefit study. Even if we obtain favorable clinical results, we may not be able to obtain regulatory approval for, or successfully commercialize, our Neuro-Spinal Scaffold implant.
We currently have only one product candidate, Neuro-Spinal Scaffold implant, in clinical development, and our business depends almost entirely on the successful clinical development, regulatory approval and commercialization of that product candidate, which may never occur. We currently have no products available for sale, generate no revenues from sales of any products, and we may never be able to develop marketable products. Our Neuro-Spinal Scaffold implant, which is currently being studied in an ongoing pivotal probable benefit study, will require substantial additional clinical development, testing, manufacturing process development, and regulatory approval before we are permitted to commence its commercialization. Our other product candidate, Bioengineered Neural Trails™, is in preclinical development. The clinical trials of our product candidates are, and the manufacturing and marketing of our product candidates will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries where we intend to test and, if approved, market any product candidates. Before obtaining regulatory approval via the HDE pathway for the commercial sale of any product candidate, we must demonstrate through extensive preclinical testing and clinical trials that the product candidate does not pose an unreasonable or significant risk of illness or injury, and that the probable benefit to health outweighs the risk of injury or illness from its use, taking into account the probable risks and benefits of currently available devices or alternative forms of treatment. Alternatively, if we were to seek PMA approval for our product candidates, that would require demonstration that the product is safe and effective for use in each target indication. This process can take many years. Of the large number of medical devices in development in the United States, only a small percentage successfully complete the FDA regulatory approval process and are commercialized. Accordingly, even if we are able to obtain the requisite capital to continue to fund our development and clinical programs, we may be unable to successfully develop or commercialize our Neuro-Spinal Scaffold implant or any other product candidate..
We may experience delays in our ongoing pivotal probable benefit study for our Neuro-Spinal Scaffold implant, and we do not know whether future clinical trials of our Neuro-Spinal Scaffold implant, or other future product candidates, will begin on time, need to be redesigned, enroll patients on time or be completed on schedule, if at all.
Before we can obtain regulatory approval for the sale of our Neuro-Spinal Scaffold implant, we must complete the pivotal probable benefit study. Our Neuro-Spinal Scaffold implant is currently being studied in a 20 subject pivotal study under our approved IDE application for the treatment of complete traumatic acute spinal cord injury. Our preclinical testing to date has been limited in nature and we cannot predict whether more extensive clinical testing will obtain similar results. Even though the initial
25
results of our clinical studies in humans are promising, our results may subsequently fail to meet the safety and efficacy standards required to obtain regulatory approvals. Our pivotal probable benefit study may not be successfully completed or may take longer than anticipated because of any number of factors, including potential delays in the enrollment of subjects in the study, the availability of scaffolds to supply to our clinical sites, failure to demonstrate safety and efficacy of our Neuro-Spinal Scaffold implant, lack of adequate funding to continue the clinical trial, or unforeseen safety issues.
In additional, clinical trials can be delayed or aborted for a variety of reasons, including delay or failure to:
- •
- obtain regulatory approval to commence future clinical trials;
- •
- reach agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of
which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
- •
- obtain institutional review board, or IRB, approval at each site;
- •
- recruit, enroll and retain patients through the completion of clinical trials;
- •
- maintain clinical sites in compliance with trial protocols through the completion of clinical trials;
- •
- address any patient safety concerns that arise during the course of the trial;
- •
- initiate or add a sufficient number of clinical trial sites; or
- •
- manufacture sufficient quantities of our product candidate for use in clinical trials.
We could encounter delays if a clinical trial is suspended or terminated by us, by the relevant IRBs at the sites at which such trials are being conducted, by the Data Safety Monitoring Board, or DSMB, for such trial or by the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, or changes in laws or regulations. Any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly.
We may find it difficult to enroll patients in our clinical studies, which could delay or prevent clinical studies of our product candidates.
Identifying and qualifying patients to participate in clinical studies of our product candidates is critical to our success. The timing of our clinical studies depends on the speed at which we can enroll patients to participate in testing our product candidates. If we have difficulty enrolling a sufficient number of patients to conduct our clinical studies as planned, we may need to delay, limit or terminate ongoing or planned clinical studies, any of which would have an adverse effect on our business.
Patient enrollment is affected by a number of factors including:
- •
- severity of the disease or condition under investigation;
- •
- design of the study protocol;
- •
- size and nature of the patient population;
- •
- eligibility criteria for and design of the study in question;
26
- •
- perceived risks and benefits of the product candidate under study;
- •
- proximity and availability of clinical study sites for prospective patients;
- •
- availability of competing therapies and clinical studies;
- •
- efforts to facilitate timely enrollment in clinical studies;
- •
- patient referral practices of physicians; and
- •
- ability to monitor patients adequately during and after treatment.
We may not be able to initiate or continue clinical studies if we cannot enroll a sufficient number of eligible patients to participate in the clinical studies required by regulatory agencies. If we have difficulty enrolling a sufficient number of patients to conduct our clinical studies as planned, we may need to delay, limit or terminate ongoing or planned clinical studies, any of which would have an adverse effect on our business.
Clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
The results of preclinical studies and early clinical trials of new medical devices do not necessarily predict the results of later-stage clinical trials. The design of our clinical trials is based on many assumptions about the expected effects of our product candidates, and if those assumptions are incorrect, the trials may not sufficiently produce results to support regulatory applications. We are currently pursuing marketing approval via our HDE which requires us to show the device does not pose an unreasonable or significant risk of illness or injury, and that the probable benefit of health outweighs the risk of injury or illness from its use. Preliminary results may not be confirmed upon full analysis of the detailed results of an early clinical trial. Product candidates in later stages of clinical trials may fail to show safety and probable benefit sufficient to support intended use claims despite having progressed through initial clinical testing. The data collected from clinical trials of our product candidates may not be sufficient to obtain regulatory approval in the United States or elsewhere. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate's profile. Because of the uncertainties associated with clinical development and regulatory approval, we cannot determine if or when we will have an approved product for commercialization or achieve sales or profits.
Our products and operations are subject to extensive governmental regulation both in the United States and abroad, and our failure to comply with applicable requirements could cause our business to suffer.
Our medical device and biologic products and operations are subject to extensive regulation by the FDA and various other federal, state and foreign governmental authorities. Government regulation of medical devices and biologic products is meant to assure their safety and effectiveness, and includes regulation of, among other things:
- •
- design, development and manufacturing;
- •
- testing, labeling, content and language of instructions for use and storage;
- •
- clinical trials;
- •
- product safety;
- •
- marketing, sales and distribution;
- •
- regulatory clearances and approvals including premarket clearance and approval;
- •
- conformity assessment procedures;
27
- •
- product traceability and record keeping procedures;
- •
- advertising and promotion;
- •
- product complaints, complaint reporting, recalls and field safety corrective actions;
- •
- post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead
to death or serious injury;
- •
- post-market studies; and
- •
- product import and export.
The regulations to which we are subject are complex and have tended to become more stringent over time. Regulatory changes could result in restrictions on our ability to carry on or expand our operations, higher than anticipated costs or lower than anticipated sales.
Before we can market or sell a new regulated medical device product in the United States, we must obtain clearance under Section 510(k) of the Federal Food, Drug and Cosmetic Act (FDCA), approval of a PMA application, or approval of a HDE, unless the device is specifically exempt from premarket review. Our Neuro-Spinal Scaffold implant is expected to be regulated by the FDA as a Class III medical device, requiring either PMA or HDE approval. A HUD designation was granted for the Neuro-Spinal Scaffold implant in 2013, opening the HDE pathway.
In the PMA approval process, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data. Modifications to products that are approved through a PMA application generally need FDA approval. The process of obtaining a PMA is costly and generally takes from one to three years, or even longer, from the time the application is submitted to the FDA until an approval is obtained.
An HDE application is similar in form and content to a PMA application and, although exempt from the effectiveness requirements of a PMA, an HDE does require sufficient information for FDA to determine that the device does not pose an unreasonable or significant risk of illness or injury, and that the probable benefit to health outweighs the risk of injury or illness from its use. Like a PMA, changes to HDE devices generally need FDA approval.
Biological products must satisfy the requirements of the Public Health Services Act and its implementing regulations. In order for a biologic product to be legally marketed in the U.S., the product must have a BLA approved by the FDA. The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete.
The FDA can delay, limit or deny clearance or approval of a product for many reasons, including:
- •
- we may not be able to demonstrate to the FDA's satisfaction that our products are safe and effective for their intended uses;
- •
- the data from our preclinical studies and clinical trials may be insufficient to support clearance or approval, where required; and
- •
- the manufacturing process or facilities we use may not meet applicable requirements.
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions that may prevent or delay approval or clearance of our products under development or impact our ability to modify our currently approved or cleared products on a timely basis.
In addition, even after we have obtained the proper regulatory clearance or approval to market a product, the FDA has the power to require us to conduct postmarketing studies. Failure to conduct
28
required studies in a timely manner could result in the revocation of approval for the product that is subject to such a requirement and could also result in the recall or withdrawal of the product, which would prevent us from generating sales from that product in the United States.
Failure to comply with applicable laws and regulations could jeopardize our ability to sell our products and result in enforcement actions such as:
- •
- warning letters;
- •
- fines;
- •
- injunctions;
- •
- civil penalties;
- •
- termination of distribution;
- •
- recalls or seizures of products;
- •
- delays in the introduction of products into the market;
- •
- total or partial suspension of production;
- •
- refusal of the FDA or other regulator to grant future clearances or approvals;
- •
- withdrawals or suspensions of current clearances or approvals, resulting in prohibitions on sales of our products; and/or
- •
- in the most serious cases, criminal penalties.
Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales and have a material adverse effect on our reputation, business, results of operations and financial condition.
We must obtain FDA approval before we can sell any of our products in the United States and approval of similar regulatory authorities in countries outside the United States before we can sell our products in such countries. We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our products if such approval is denied or delayed.
The development, manufacture and marketing of our products are subject to government regulation in the United States and other countries. In the United States and most foreign countries, we must complete rigorous preclinical testing and extensive human clinical trials that demonstrate the safety and efficacy of a product in order to apply for regulatory approval to market the product. If the FDA grants regulatory approval of a product, the approval may be limited to specific indications or limited with respect to its distribution. Expanded or additional indications for approved devices may not be approved, which could limit our potential revenues. Foreign regulatory authorities may apply similar limitations or may refuse to grant any approval. Consequently, even if we believe that preclinical and clinical data are sufficient to support regulatory approval for our products, the FDA and foreign regulatory authorities may not ultimately grant approval for commercial sale in any jurisdiction. If our products are not approved, our ability to generate revenues will be limited and our business will be adversely affected.
There are risks associated with pursuing FDA approval via an HDE pathway, including the possibility that the approval could be withdrawn in the future if FDA subsequently approves another device for the same intended use, as well as limitations on the ability to profit from sales of the product.
Except in certain circumstances, products approved under an HDE cannot be sold for an amount that exceeds the costs of research and development, fabrication, and distribution of the device (i.e., for
29
profit). Currently, a product is only eligible to be sold for profit after receiving HDE approval if the device (1) is intended for the treatment or diagnosis of a disease or condition that occurs in pediatric patients or in a pediatric subpopulation, and such device is labeled for use in pediatric patients or in a pediatric subpopulation in which the disease or condition occurs; or (2) is intended for the treatment or diagnosis of a disease or condition that does not occur in pediatric patients or that occurs in pediatric patients in such numbers that the development of the device for such patients is impossible, highly impracticable or unsafe. If an HDE-approved device does not meet either of the eligibility criteria, the device cannot be sold for profit.
If we or our suppliers fail to comply with ongoing FDA regulatory requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain regulatory clearance or approval, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections by the FDA. In particular, we and our third-party suppliers will be required to comply with the FDA's Quality System Regulation or QSRs. These FDA regulations cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of products. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA. If we, or our manufacturers, fail to adhere to QSR requirements, this could delay production of our product candidates and lead to fines, difficulties in obtaining regulatory clearances, recalls, enforcement actions, including injunctive relief or consent decrees, or other consequences, which could, in turn, have a material adverse effect on our financial condition or results of operations.
In addition, we and our suppliers are required to comply with Good Manufacturing Practices, or GMPs, and Good Tissue Practices, or GTPs, with respect to any human cells and biologic products we may develop, and International Standards Organization, or ISO, regulations for the manufacture of our products and other regulations which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of any product for which we obtain clearance or approval. Manufacturing may also be subject to controls by the FDA for parts of the combination products that FDA may find are controlled by the biologics regulations.
The FDA audits compliance with the QSR and other similar regulatory requirements through periodic announced and unannounced inspections of manufacturing and other facilities. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in any of the following enforcement actions:
- •
- untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties;
- •
- unanticipated expenditures to address or defend such actions;
- •
- customer notifications or repair, replacement, refunds, recall, detention or seizure of our products;
- •
- operating restrictions or partial suspension or total shutdown of production;
- •
- refusing or delaying our requests for premarket approval of new products or modified products;
- •
- withdrawing PMA approvals that have already been granted;
- •
- refusal to grant export approval for our products; or
- •
- criminal prosecution.
30
Any of these sanctions could have a material adverse effect on our reputation, business, results of operations and financial condition.
If our medical device products, or malfunction of our medical device products, cause or contribute to a death or a serious injury before or after approval, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting regulations, medical device manufacturers with approved products are required to report to the FDA information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. Any such serious adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. In the context of our ongoing clinical trial, we report adverse events in accordance with IDE regulations. No serious adverse events involving our products have been reported in the past, however we cannot guarantee that such events will not occur in the future. Any corrective action, whether voluntary or involuntary, and either pre- or post-market, needed to address any serious adverse events will require the dedication of our time and capital, distract management from operating our business and may harm our reputation and financial results.
In addition, if our products are approved for commercialization, the FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is reasonable probability that the device would cause serious injury or death. A government-mandated or voluntary recall by us or one of our partners could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our commercialized products would divert managerial and financial resources and have an adverse effect on our reputation, results of operations and financial condition, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers' demands. We may also be subject to liability claims, be required to bear other costs, or take other actions that may have a negative impact on our future sales and our ability to generate profits.
Further, under the FDA's medical device reporting, or MDR, regulations, we are required to report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. Repeated product malfunctions may result in a voluntary or involuntary product recall, which could divert managerial and financial resources, impair our ability to manufacture our products in a cost-effective and timely manner, and have an adverse effect on our reputation, results of operations and financial condition.
If we obtain approval for our products, we may be subject to enforcement action if we engage in improper marketing or promotion of our products.
We are not permitted to promote or market our investigational products. After approval, our promotional materials and training methods must comply with FDA and other applicable laws and regulations, including the prohibition of the promotion of unapproved, or off-label, use. Surgeons may use our products off-label, as the FDA does not restrict or regulate a surgeon's choice of treatment within the practice of medicine. However, if the FDA determines that our promotional materials or training constitutes promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our
31
promotional or training materials to constitute promotion of an off-label use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and adoption of the products could be impaired. In addition, the off-label use of our products may increase the risk of product liability claims. Product liability claims are expensive to defend and could divert our management's attention, result in substantial damage awards against us, and harm our reputation.
Some of our future products will be viewed by the FDA as combination products comprised of a biologic and medical device component, and the review of combination products is often more complex and more time consuming than the review of other types of products.
It is possible that some of our products, including our Bioengineered Neural Trails injection, may be regulated by the FDA as combination products. For a combination product, the FDA must determine which center or centers within the FDA will review the product candidate and under what legal authority the product candidate will be reviewed. We are currently developing our regulatory strategies with respect to which regulatory pathway will be necessary to obtain clearance or approval, if medical device clearance or approval is required at all. We believe that the biologic component, as well as the associated biomaterial component, of the Bioengineered Neural Trails injection will be reviewed by the Center for Biologics Evaluation and Research, or CBER. The delivery tools associated with that product may be reviewed by the Center for Devices and Radiological Health, or CDRH either in consultation with CBER as part of the Biologics License Application, or BLA, or separately as a medical device. The process of obtaining FDA marketing clearance or approval is lengthy, expensive, and uncertain, and we cannot be sure that our biologic-device combination products, or any other products, will be cleared or approved in a timely fashion, or at all. In addition, the review of combination products is often more complex and more time consuming than the review of a product candidate under the jurisdiction of only one center within the FDA. We cannot be sure that the FDA will not select to have our combination products reviewed and regulated by only one FDA center and/or different legal authority, in which case the path to regulatory approval would be different and could be more lengthy and costly. If the FDA does not approve or clear our products in a timely fashion, or at all, our business and financial condition will be adversely affected.
If we cannot protect, maintain and, if necessary, enforce our intellectual property rights, our ability to develop and commercialize products will be adversely impacted.
Our success, in large part, depends on our ability to protect and maintain the proprietary nature of our technology. We and our licensors must prosecute and maintain our existing patents and obtain new patents. Some of our proprietary information may not be patentable, and there can be no assurance that others will not utilize similar or superior solutions to compete with us. We cannot guarantee that we will develop proprietary products that are patentable, and that if issued, any patent will give a competitive advantage or that such patent will not be challenged by third parties. The process of obtaining patents can be time consuming with no certainty of success, as a patent may not issue or may not have sufficient scope or strength to protect the intellectual property it was intended to protect. We cannot assure you that our means of protecting our proprietary rights will suffice or that our others will not independently develop competitive technology or design around patents or other intellectual property rights issued to us. Even if a patent is issued, it does not guarantee that it is valid or enforceable. Any patents that we or our licensors have obtained or obtain in the future may be challenged, invalidated or unenforceable. If necessary, we may initiate actions to protect our intellectual property, which can be costly and time consuming.
32
If third parties successfully claim that we infringe their intellectual property rights, our ability to continue to develop and commercialize products could be delayed or prevented.
Third parties may claim that we or our licensors are infringing on or misappropriating their proprietary information. Other organizations are engaged in research and product development efforts that may overlap with our products. Such third parties may currently have, or may obtain in the future, legally blocking proprietary rights, including patent rights, in one or more products or methods under development or consideration by us. These rights may prevent us from commercializing products, or may require us to obtain a license from the organizations to use the technology. We may not be able to obtain any such licenses that may be required on reasonable financial terms, if at all, and cannot be sure that the patents underlying any such licenses will be valid or enforceable. There may be rights that we are not aware of, including applications that have been filed but not published that, when issued, could be asserted against us. These third parties could bring claims against us that would cause us to incur substantial expenses and, if successful, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us, we could be forced to stop or delay research and development of the product that is the subject of the suit. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our trade secrets or other confidential information could be compromised by disclosure during this type of litigation.
We will face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
In general, the biotechnology industry is subject to intense competition and rapid and significant technological change. We have many potential competitors, including major drug companies, specialized biotechnology firms, academic institutions, government agencies and private and public research institutions. Many of these competitors have significantly greater financial and technical resources than us, and superior experience and expertise in research and development, preclinical testing, design and implementation of clinical trials, regulatory processes and approval for products, production and manufacturing, and sales and marketing of approved products. Large and established companies compete in the biotechnology market. In particular, these companies have greater experience and expertise in securing government contracts and grants to support their research and development efforts, conducting testing and clinical trials, obtaining regulatory approvals to market products, manufacturing such products on a broad scale and marketing approved products. Smaller or early-stage companies and research institutions may also prove to be significant competitors, particularly if they have collaborative arrangements with larger and more established biotechnology companies. We will also face competition from these parties in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and registering subjects for clinical trials.
In order to effectively compete, we will have to make substantial investments in development, clinical testing, manufacturing and sales and marketing, or partner with one or more established companies. There is no assurance that we will be successful in having our products approved or gaining significant market share for any of our products. Our technologies and products also may be rendered obsolete or noncompetitive as a result of products introduced by our competitors.
We will depend upon strategic relationships to develop, exploit and manufacture our products. If these relationships are not successful, we may not be able to capitalize on the market potential of these products.
The near and long-term viability of our products will depend, in part, on our ability to successfully establish new strategic collaborations with biotechnology companies, hospitals, insurance companies and government agencies. Establishing strategic collaborations is difficult and time-consuming. Potential collaborators may reject collaborations based upon their assessment of our financial, regulatory or intellectual property position. If we fail to establish a sufficient number of collaborations on acceptable
33
terms, we may not be able to commercialize our products or generate sufficient revenue to fund further research and development efforts.
Even if we establish new collaborations, these relationships may never result in the successful development or commercialization of any of our product candidates for reasons both within and outside of our control.
We have limited experience manufacturing our Neuro-Spinal Scaffold implant for clinical-study scale and no experience for commercial scale.
To date, we have manufactured our Neuro-Spinal Scaffold implant on a small scale, including sufficient supply that is needed for our clinical studies. We may encounter unanticipated problems in the scale-up process that will result in delays in the manufacturing of the Neuro-Spinal Scaffold implant, and therefore, delay our clinical studies. During our clinical trials, we are subject to FDA regulations requiring manufacturing of our scaffolds with the FDA requirements for design controls and subject to inspections by regulatory agencies. Our failure to comply with applicable regulations may result in delays and interruptions to our product supply while we seek to secure another supplier that meets all regulatory requirements. If we are unable to scale up our manufacturing to meet requirements for our clinical studies, we may be required to rely on contract manufacturers. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured the product ourselves, including the possible breach of the manufacturing agreements by the third parties because of factors beyond our control, and the possibility of termination or nonrenewal of the agreements by the third parties because of our breach of the manufacturing agreement or based on their own business priorities.
There are a limited number of suppliers that can provide materials to us. Any problems encountered by such suppliers may detrimentally impact us.
We rely on third-party suppliers and vendors for certain of the materials used in the manufacture of our products or other of our product candidates. Any significant problem experienced by one of our suppliers could result in a delay or interruption in the supply of materials to us until such supplier resolves the problem or an alternative source of supply is located. Any delay or interruption could negatively affect our operations.
If the third parties on which we rely to conduct our laboratory testing, animal and human clinical trials do not perform as contractually required or expected, we may not be able to obtain regulatory approval for or commercialize our products.
We have been, and will continue to be, dependent on third-party CROs, medical institutions, investigators, and contract laboratories to conduct certain of our laboratory testing, animal and human clinical studies. We are responsible for confirming that each of our clinical trials is conducted in accordance with our approved plan and protocol. Moreover, the FDA and foreign regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. Our reliance on these third parties does not relieve us of these responsibilities and requirements. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval or successfully commercialize our products on a timely basis, if at all, and our business, operating results and prospects may be adversely affected..
34
The results of our clinical trials may not support our product candidate claims or may result in the discovery of adverse side effects.
Our ongoing research and development, preclinical testing and clinical trial activities are subject to extensive regulation and review by numerous governmental authorities both in the United States and abroad. We are currently conducting a pivotal study of our Neuro-Spinal Scaffold implant to gather information about the product's safety and probable benefit. In the future we may conduct clinical trials to support approval of new products. Clinical studies must be conducted in compliance with FDA regulations or the FDA may take enforcement action. The data collected from these clinical studies may ultimately be used to support market clearance for these products. Even if our clinical trials are completed as planned, we cannot be certain that their results will support our product candidate claims or that the FDA will agree with our conclusions regarding them. Success in preclinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and preclinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a product candidate and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our product submissions and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate's profile.
We may encounter difficulties in managing our growth and expanding our operations successfully.
As we seek to advance our product candidates through clinical trials and commercialization, we will need to expand our development, regulatory, manufacturing, marketing and sales capabilities or contract with third parties to provide these capabilities for us. As our operations expand, we expect that we will need to manage additional relationships with various strategic collaborators, suppliers and other third parties. Future growth will impose significant added responsibilities on members of our management. Our future financial performance and our ability to commercialize our Neuro-Spinal Scaffold implant, if approved, and any other product candidates, and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to manage our development efforts and clinical trials effectively and hire, train and integrate additional management, administrative and, if necessary, sales and marketing personnel. We may not be able to accomplish these tasks, and our failure to accomplish any of them could prevent us from successfully growing our Company.
If approved, our products will require market acceptance to be successful. Failure to gain market acceptance would impact our revenues and may materially impair our ability to continue our business.
Even if we receive regulatory approvals for the commercial sale of our products, the commercial success of our products will depend on, among other things, their acceptance by physicians, patients, third-party payors such as health insurance companies and other members of the medical community as a therapeutic and cost-effective alternative to competing products and treatments. Physicians and hospitals will need to establish training and procedures to utilize and implement our Neuro-Spinal Scaffold implant, and there can be no assurance that these parties will adopt the use of our device or develop sufficient training and procedures to properly utilize it. Market acceptance of, and demand for, any product that we may develop and commercialize will depend on many factors, both within and outside of our control. If our product candidates fail to gain market acceptance, we may be unable to earn sufficient revenue to continue our business.
35
If we obtain approval for our products, their commercial success will depend in part upon the level of reimbursement we receive from third parties for the cost of our products to users.
The commercial success of any product will depend, in part, on the extent to which reimbursement for the costs of our products and related treatments will be available from third-party payors such as government health administration authorities, private health insurers, managed care programs, and other organizations. Adequate third-party insurance coverage may not be available for us to establish and maintain price levels that are sufficient for us to continue our business or for realization of an appropriate return on investment in product development.
Acquisitions of companies, businesses or technologies may substantially dilute our stockholders and increase our operating losses.
We may make acquisitions of businesses, technologies or intellectual property rights that we believe would to be necessary, useful or complementary to our current business. Any such acquisition may require assimilation of the operations, products or product candidates and personnel of the acquired business and the training and integration of its employees, and could substantially increase our operating costs, without any offsetting increase in revenue. Acquisitions may not provide the intended technological, scientific or business benefits and could disrupt our operations and divert our limited resources and management's attention from our current operations, which could harm our existing product development efforts. While we may use cash or equity to finance a future acquisition, it is likely we would issue equity securities as a portion or all of the consideration in any acquisition. The issuance of equity securities for an acquisition could be substantially dilutive to our stockholders. Any investment made in, or funds advanced to, a potential acquisition target could also significantly adversely affect our results of operation and could further reduce our limited capital resources. Any acquisition or action taken in anticipation of a potential acquisition or other change in business activities could substantially depress the price of our stock. In addition, our results of operations may suffer because of acquisition related costs or the post-acquisition costs of funding the development of an acquired technology or product candidates or operation of the acquired business, or due to amortization or impairment costs for acquired goodwill and other intangible assets.
Our success depends on our ability to retain our management and other key personnel.
We depend on our senior management as well as key scientific personnel. The loss of any of these individuals could harm our business and significantly delay or prevent the achievement of research, development or business objectives. Competition for qualified employees is intense among biotechnology companies, and the loss of qualified employees, or an inability to attract, retain and motivate additional highly skilled employees could hinder our ability to successfully develop marketable products.
Our future success also depends on our ability to identify, attract, hire, train, retain and motivate other highly skilled scientific, technical, marketing, managerial and financial personnel. Although we will seek to hire and retain qualified personnel with experience and abilities commensurate with our needs, there is no assurance that we will succeed despite our collective efforts. The loss of the services of any of our senior management or other key personnel could hinder our ability to fulfill our business plan and further develop and commercialize our products and services. Competition for personnel is intense, and any failure to attract and retain the necessary technical, marketing, managerial and financial personnel would have a material adverse effect on our business, prospects, financial condition and results of operations.
36
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from collaborators, prospective licensees and other third parties. In addition, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of these third parties or our employees' former employers. We may also be subject to claims that former employees, collaborators or other third parties have an ownership interest in our patents or other intellectual property. We may be subject to ownership disputes in the future arising, for example, from conflicting obligations of consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against these claims, litigation could result in substantial cost and be a distraction to our management and employees.
We are subject to a pending securities class action and derivative lawsuit, which could divert management's attention and harm our business.
We are the subject of a securities class action lawsuit. The lawsuit, filed in July 2014, alleges violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements related to the timing and completion of the clinical study of our Neuro-Spinal Scaffold implant. That lawsuit was dismissed with prejudice in April 2015. Plaintiffs filed an appeal of that dismissal, which will be heard by the United States Court of Appeals for the First Circuit on April 6, 2016. No assurance can be provided that we will be successful in defending against this appeal or, if the dismissal is overturned, in defending the underlying lawsuit. Nor can we be certain that insurance proceeds will be sufficient to cover any liability under such claims.
We are also the subject of a securities derivative lawsuit. The lawsuit, filed in August 2015, alleges breaches of fiduciary duties related to the claimed false and misleading statements that are the subject of the securities class action. That lawsuit was dismissed with prejudice in February 2016. The time for filing an appeal of that dismissal has not yet expired. If an appeal of the dismissal is filed, we cannot provide any assurance that we will be successful in defending against the appeal or, if the dismissal is overturned, in defending the underlying lawsuit. Nor can we be certain that insurance proceeds will be sufficient to cover any liability under such claims.
Further, the amount of time that will be required to resolve these lawsuits is unpredictable and these actions may divert management's attention from the day-to-day operations of our business, which could adversely affect our business, results of operations and cash flows.
We face potential product liability claims, and, if successful claims are brought against us, we may incur substantial liability and costs.
We will have exposure to claims for product liability. Product liability coverage for the healthcare industry is expensive and sometimes difficult to obtain. We may not be able to maintain such insurance on acceptable terms or be able to secure increased coverage if the commercialization of our products progresses, nor can we be sure that existing or future claims against us will be covered by our product liability insurance. Moreover, the existing coverage of our insurance policy or any rights of indemnification and contribution that we may have may not be sufficient to offset existing or future claims. A successful claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable terms, if at all. Even if a claim is not successful, defending such a
37
claim would be time-consuming and expensive, may damage our reputation in the marketplace, and would likely divert our management's attention.
We are subject to environmental, health and safety laws. Failure to comply with such environmental, health and safety laws could cause us to become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to various environmental, health and safety laws and regulations, including those relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and humans, emissions and wastewater discharges, and the use and disposal of hazardous or potentially hazardous substances used in connection with our research. Any of these laws or regulations could cause us to incur additional expense or restrict our operations. Compliance with environmental laws and regulations may be expensive, and current or future environmental regulations may impair our research and development efforts.
Risks Related to Investment in Our Securities
The price of our common stock may become volatile, which could lead to losses by investors and costly securities litigation.
The trading price of our common stock is likely to be highly volatile and could fluctuate in response to factors such as:
- •
- the status, completion and/or results of our clinical trials;
- •
- actual or anticipated variations in our operating results;
- •
- announcements of developments by us or our competitors;
- •
- regulatory actions regarding our products;
- •
- announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments;
- •
- adoption of new accounting standards affecting our industry;
- •
- additions or departures of key personnel;
- •
- sales of our common stock or other securities in the open market; and
- •
- other events or factors, many of which are beyond our control.
The stock market is subject to significant price and volume fluctuations. In the past, following periods of volatility in the market price of a company's securities, securities class action litigation has often been initiated against such company. Litigation initiated against us, whether or not successful, could result in substantial costs and diversion of our management's attention and resources, which could harm our business and financial condition.
Investors may experience dilution of their ownership interests because of the future issuance of additional shares of our common stock.
As of December 31, 2015, there were outstanding warrants to purchase 1,156,979 shares of our common stock, and outstanding options to purchase 3,253,310 shares of our common stock. We expect to issue additional equity awards to compensate employees, consultants and directors, and may issue additional shares to raise capital, to acquire other companies or technologies, to pay for services, or for other corporate purposes. Any such issuances will have the effect of diluting the interest of current stockholders. The future issuance of any such additional shares of common stock may create downward pressure on the trading price of the common stock. There can be no assurance that we will not be
38
required to issue additional shares, warrants or other convertible securities in the future in conjunction with any capital raising efforts, including at a price (or exercise prices) below the price at which shares of our common stock are currently quoted on the Nasdaq Global Market.
Anti-takeover effects of certain provisions of our articles of incorporation and Nevada state law may discourage or prevent a takeover.
Our articles of incorporation divide our Board of Directors into three classes, with three-year staggered terms. The classified board provision could increase the likelihood that, in the event an outside party acquired a controlling block of our stock, incumbent directors nevertheless would retain their positions for a substantial period, which may have the effect of discouraging, delaying or preventing a change in control. In addition, Nevada has a business combination law, which prohibits certain business combinations between Nevada corporations and "interested stockholders" for three years after the interested stockholder first becomes an interested stockholder, unless the corporation's board of directors approves the combination in advance. In addition, we may become subject to Nevada's control share laws. A corporation is subject to Nevada's control share law if it has more than 200 stockholders, at least 100 of whom are stockholders of record and residents of Nevada, and if the corporation does business in Nevada, including through an affiliated corporation. This control share law may have the effect of discouraging corporate takeovers. Currently, we believe that we have less than 100 stockholders of record who are residents of Nevada, and are therefore not subject to the control share laws.
The provisions of our articles of incorporation and Nevada's business combination and control share laws make it more difficult for a third party to acquire us and make a takeover more difficult to complete, even if such a transaction were in our stockholders' interest or might result in a premium over the market price for our common stock.
Item 1B. UNRESOLVED STAFF COMMENTS
None.
We lease approximately 26,150 square feet of office, laboratory and manufacturing space in Cambridge, Massachusetts. The lease commenced in November 2011, and is for an initial term of six years and three months, with one five-year extension. We believe the facility is adequate to meet our current needs and that additional space will be available on commercially reasonable terms as needed.
Lawsuit with Former Employee
In November 2013, we filed a lawsuit against Francis Reynolds, our former Chairman, Chief Executive Officer and Chief Financial Officer, in Middlesex Superior Court, Middlesex County, Massachusetts (InVivo Therapeutics Holdings Corp. v. Reynolds, Civil Action No. 13-5004). The complaint alleges breaches of fiduciary duties, breach of contract, conversion, misappropriation of corporate assets, unjust enrichment, corporate waste, and seeks monetary damages and an accounting. The lawsuit involves approximately $500,000 worth of personal and/or exorbitant expenses that we allege Mr. Reynolds inappropriately caused us to pay while he was serving as our Chief Executive Officer, Chief Financial Officer, President and Chairman of our Board of Directors. On December 6, 2013, Mr. Reynolds answered the complaint, and filed counterclaims against us and the members of our Board of Directors at that time. The counterclaims allege two counts of breach of contract, two counts of breach of the covenant of good faith and fair-dealing, and tortious interference with a contract, and seek monetary damages and a declaratory judgment. The counterclaims involve Mr. Reynolds's
39
allegations that we and the Board interfered with the performance of his duties under the terms of his employment agreement, and that Mr. Reynolds was entitled to additional shares upon the exercise of certain stock options. On January 9, 2014, we, along with the directors named in the counterclaims, filed our answer. The parties are currently conducting pre-trial discovery. No judgments or rulings are pending at this stage.
Shareholder Matters and Investigations
On July 31, 2014, a putative securities class action lawsuit was filed in the United States District Court for the District of Massachusetts, naming the Company and Mr. Reynolds, as defendants (the "Securities Class Action"). The lawsuit alleges violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements related to the timing and completion of the clinical study of our Neuro Spinal Scaffold. The plaintiff seeks class certification for purchasers of our common stock during the period from April 5, 2013 through August 26, 2013 and unspecified damages. On April 3, 2015, the United States District Court for the District of Massachusetts dismissed the plaintiff's claim with prejudice. Plaintiff filed a notice of appeal of this decision on May 4, 2015. A mandatory mediation conference was held on September 10, 2015. Following that conference, on October 5, 2015, plaintiff/appellant filed his opening brief with the United States Court of Appeals for the First Circuit. The Company and the individual defendants/appellees filed their answering brief on November 5, 2015, and plaintiff/appellant filed his reply brief on December 10, 2015. The Court of Appeals has scheduled oral arguments for April 6, 2016.
On January 23, 2015, Shawn Luger, a purported shareholder of the Company, sent us a letter demanding that the Board of Directors take action to remedy purported breaches of fiduciary duties allegedly related to the claimed false and misleading statements that are the subject of the Securities Class Action (the "Shareholder Demand"). Our Board of Directors completed its investigation of the matters raised in the Shareholder Demand and voted unanimously not to pursue any litigation against any current or former director, officer or employee of the Company with respect to the matters set forth in the Shareholder Demand.
On August 14, 2015, Shawn Luger filed a shareholder derivative lawsuit in the Superior Court of Suffolk County for the Commonwealth of Massachusetts on behalf of the Company against certain present and former board members and company executives alleging the same breaches of fiduciary duties purportedly set forth in the Shareholder Demand. On February 5, 2016, the Superior Court of Suffolk County dismissed the plaintiff's claims with prejudice. The plaintiff's time to appeal the dismissal has not expired.
In addition to the derivative lawsuit and the appeal of the Securities Class Action, we received investigation subpoenas from the Boston Regional Office of the Securities and Exchange Commission ("SEC") and the Massachusetts Securities Division of the Secretary of the Commonwealth of Massachusetts ("MSD") requesting corporate documents also concerning, among other topics, the allegations raised in the Securities Class Action and the Shareholder Demand. We responded to the MSD's subpoena on September 22, 2014 and October 8, 2014. On February 18, 2015, we received a second subpoena from the MSD requesting additional documents and information related to the same topics. We responded to this second subpoena on March 24, 2015. On October 21, 2015, we received a letter from the SEC notifying the Company that it has concluded its investigation of the Company and that it does not intend to recommend an enforcement action against the Company.
Item 4. MINE SAFETY DISCLOSURES
Not applicable.
40
Item 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market Information
Our Common Stock is currently listed for trading on the Nasdaq Global Market under the symbol "NVIV." From October 29, 2010 through April 16, 2015, our Common Stock was quoted on the OTCQB under the same symbol. The following table shows the high and low bid prices for our Common Stock for our two most recent fiscal years:
Fiscal Quarter Ended
|
High Bid | Low Bid | |||||
|---|---|---|---|---|---|---|---|
December 31, 2014 |
$ | 6.40 | $ | 1.96 | |||
September 30, 2014 |
$ | 4.84 | $ | 1.86 | |||
June 30, 2014 |
$ | 9.00 | $ | 3.72 | |||
March 31, 2014 |
$ | 10.68 | $ | 5.96 | |||
Fiscal Quarter Ended
|
High Bid | Low Bid | |||||
|---|---|---|---|---|---|---|---|
December 31, 2015 |
$ | 11.80 | $ | 6.55 | |||
September 30, 2015 |
$ | 17.65 | $ | 7.33 | |||
June 30, 2015 |
$ | 19.68 | $ | 11.20 | |||
March 31, 2015 |
$ | 12.48 | $ | 5.04 | |||
These market quotations reflect inter-dealer prices, without retail mark-up, markdown or commissions and may not necessarily represent actual transactions. The prices give effect to the 1-for-4 reverse stock split of our outstanding shares of Common Stock that occurred on April 8, 2015. The high and low bid prices listed have been rounded up to the next two decimal places.
Dividends
We have never declared or paid cash dividends. We do not intend to pay cash dividends on our Common Stock for the foreseeable future, but currently intend to retain any future earnings to fund the development and growth of our business. The payment of cash dividends if any, on the Common Stock will rest solely within the discretion of our Board of Directors and will depend, among other things, upon our earnings, capital requirements, financial condition, and other relevant factors.
Holders
As of February 26, 2016, we had approximately 250 stockholders of record. This figure does not reflect persons or entities that hold their stock in nominee or "street" name through various brokerage firms.
Equity Compensation Plans
The information required to be disclosed by Item 201(d) of Regulation S-K, "Securities Authorized for Issuance Under Equity Compensation Plans," is incorporated herein by reference. Refer to Item 12 of Part III of this Annual Report on Form 10-K for additional information.
Recent Sales of Unregistered Securities
None.
41
Issuer Repurchases of Equity Securities
None.
Performance Graph
The following performance graph and related information shall not be deemed to be "soliciting material" or to be "filed" with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act, each as amended, except to the extent that we specifically incorporate it by reference into such filing.
The graph below compares the cumulative total returns of our Common Stock to the NASDAQ Composite index and the NASDAQ Biotechnology index based on the period from December 31, 2010 through December 31, 2015. The graph assumes $100 was invested on December 31, 2010 in our Common Stock and in each of the comparative indices and assumes reinvestment of dividends, if any.
The comparisons shown in the graph below are based on historical data. We caution that the stock price performance showing in the graph below is not necessarily indicative of, nor is it intended to forecast, the potential future performance of our Common Stock.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among InVivo Therapeutics Holdings Corp, the NASDAQ Composite Index,
and the NASDAQ Biotechnology Index
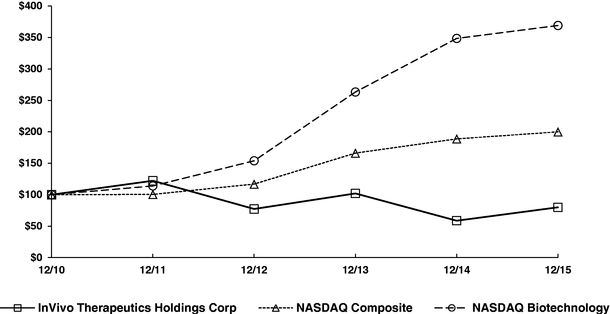
- *
- $100 invested on 12/31/10 in stock or index, including reinvestment of dividends. Fiscal year ending December 31.
| |
December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
12/10 | 12/11 | 12/12 | 12/13 | 12/14 | 12/15 | |||||||||||||
InVivo Therapeutics Holdings Corp |
$ | 100.00 | $ | 122.22 | $ | 77.33 | $ | 102.04 | $ | 58.67 | $ | 80.00 | |||||||
NASDAQ Composite |
$ | 100.00 | $ | 100.53 | $ | 116.92 | $ | 166.19 | $ | 188.78 | $ | 199.95 | |||||||
NASDAQ Biotechnology |
$ | 100.00 | $ | 113.92 | $ | 153.97 | $ | 263.29 | $ | 348.49 | $ | 369.06 | |||||||
42
The selected financial data presented below is derived from our audited consolidated financial statements. You should read the selected historical combined financial data set forth below in conjunction with "Management's Discussion and Analysis of Financial Condition and Results of Operations" in Item 7 of Part II to this Annual Report. Unless otherwise indicated, all amounts in this Item 6 are presented in thousands, except share and per- share data. All share amounts give effect to the 1-for-4 reverse stock split of our outstanding shares of Common Stock that occurred on April 8, 2015.
InVivo Therapeutics Holdings Corp.
Consolidated Balance Sheets
| |
December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | 2012 | 2011 | |||||||||||
ASSETS : |
||||||||||||||||
Current assets: |
||||||||||||||||
Cash and cash equivalents |
$ | 20,194 | $ | 13,459 | $ | 13,980 | $ | 12,825 | $ | 4,364 | ||||||
Restricted cash |
361 | 422 | 602 | 601 | 548 | |||||||||||
Prepaid expenses and other current assets |
184 | 1,072 | 20 | 144 | 104 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total current assets |
20,739 | 14,953 | 14,602 | 13,570 | 5,016 | |||||||||||
Property and equipment, net |
938 | 1,605 | 2,337 | 2,312 | 520 | |||||||||||
Other assets |
115 | 135 | 157 | 180 | 166 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total assets |
$ | 21,792 | $ | 16,693 | $ | 17,096 | $ | 16,062 | $ | 5,702 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
LIABILITIES AND STOCKHOLDERS' EQUITY (DEFICIT): |
||||||||||||||||
Current liabilities: |
||||||||||||||||
Accounts payable |
$ | 521 | $ | 569 | $ | 899 | $ | 1,152 | $ | 567 | ||||||
Loan payable-current portion |
395 | 320 | — | — | 51 | |||||||||||
Note payable-current portion |
— | 18 | 74 | — | — | |||||||||||
Capital lease payable-current portion |
— | — | 3 | 33 | 31 | |||||||||||
Derivative warrant liability |
1,907 | 7,224 | — | 14,585 | 35,473 | |||||||||||
Accrued expenses |
765 | 1,044 | 1,292 | 1,021 | 618 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total current liabilities |
3,588 | 9,175 | 2,268 | 16,791 | 36,740 | |||||||||||
Loan payable-less current portion |
1,275 | 1,600 | 1,920 | 1,578 | 84 | |||||||||||
Note payable-less current portion |
— | — | 18 | — | — | |||||||||||
Capital lease payable-less current portion |
— | — | — | 3 | 38 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total liabilities |
4,863 | 10,775 | 4,206 | 18,372 | 36,862 | |||||||||||
| | | | | | | | | | | | | | | | | |
Commitments and contingencies |
||||||||||||||||
Stockholders' equity (deficit): |
||||||||||||||||
Common stock, $0.00001 par value(1) |
1 | 1 | 1 | 1 | 1 | |||||||||||
Additional paid-in capital |
150,497 | 106,172 | 94,798 | 40,842 | 16,656 | |||||||||||
Accumulated deficit |
(133,569 | ) | (100,255 | ) | (81,909 | ) | (43,153 | ) | (47,817 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Total stockholders' equity (deficit) |
16,929 | 5,918 | 12,890 | (2,310 | ) | (31,160 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Total liabilities and stockholders' equity (deficit) |
$ | 21,792 | $ | 16,693 | $ | 17,096 | $ | 16,062 | $ | 5,702 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
- (1)
- Authorized—50,000,000 shares; issued and outstanding—27,555,948, 23,453,000, and 19,693,434 at December 31, 2015, 2014 and 2013, respectively. Authorized—25,000,000 shares; issued and outstanding—16,470,280 and 13,440,118 shares at December 31, 2012 and 2011, respectively.
43
InVivo Therapeutics Holdings Corp.
Consolidated Statement of Operations
| |
Years Ended December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | 2012 | 2011 | |||||||||||
Operating expenses: |
||||||||||||||||
Research and development |
$ | 10,058 | $ | 10,273 | $ | 10,533 | $ | 6,376 | $ | 4,103 | ||||||
General and administrative |
12,340 | 7,566 | 8,472 | 6,403 | 4,556 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total operating expenses |
22,398 | 17,839 | 19,005 | 12,779 | 8,659 | |||||||||||
| | | | | | | | | | | | | | | | | |
Operating loss |
(22,398 | ) | (17,839 | ) | (19,005 | ) | (12,779 | ) | (8,659 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Other income (expense): |
||||||||||||||||
Interest income |
60 | 5 | 15 | 35 | 9 | |||||||||||
Interest expense |
(172 | ) | (136 | ) | (130 | ) | (72 | ) | (13 | ) | ||||||
Modification of warrants |
— | — | (765 | ) | — | — | ||||||||||
Derivatives gain (loss) |
(10,804 | ) | (376 | ) | (18,871 | ) | 17,480 | (26,065 | ) | |||||||
| | | | | | | | | | | | | | | | | |
Other income (expense), net |
(10,916 | ) | (507 | ) | (19,751 | ) | 17,443 | (26,069 | ) | |||||||
| | | | | | | | | | | | | | | | | |
Net income (loss) |
$ | (33,314 | ) | $ | (18,346 | ) | $ | (38,756 | ) | $ | 4,664 | $ | (34,728 | ) | ||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Net income (loss) per share, basic |
$ | (1.26 | ) | $ | (0.83 | ) | $ | (2.10 | ) | $ | 0.30 | $ | (2.68 | ) | ||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Net income (loss) per share, diluted |
$ | (1.26 | ) | $ | (0.83 | ) | $ | (2.10 | ) | $ | 0.26 | $ | (2.68 | ) | ||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Weighted average number of common shares outstanding, basic |
26,461,374 | 22,080,761 | 18,497,922 | 15,806,725 | 12,973,718 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Weighted average number of common shares outstanding, diluted |
26,461,374 | 22,080,761 | 18,497,922 | 17,979,855 | 12,973,718 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
We have derived our statements of operations data for the years ended December 31, 2011 and 2012 and our balance sheet data as of December 31, 2013, 2012 and 2011 from our audited financial statements which are not included in this Annual Report. We have derived our statements of operations data for the years ended December 31, 2015, 2014 and 2013 and our balance sheet data as of December 31, 2015 and 2014 from our audited financial statements appearing elsewhere in this Annual Report on Form 10-K. Our audited financial information is prepared and presented in accordance with generally accepted accounting principles in the U.S. (U.S. GAAP).
44
Supplementary Quarterly Financial Data (Unaudited—In thousands)
| |
Quarter Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
December 31, 2015 |
September 30, 2015 |
June 30, 2015 |
March 31, 2015 |
|||||||||
Operating expenses: |
|||||||||||||
Research and development |
$ | 2,777 | $ | 2,432 | $ | 2,546 | $ | 2,303 | |||||
General and administrative |
2,481 | 3,437 | 3,214 | 3,208 | |||||||||
| | | | | | | | | | | | | | |
Total operating expenses |
5,258 | 5,869 | 5,760 | 5,511 | |||||||||
| | | | | | | | | | | | | | |
Operating loss |
(5,258 | ) | (5,869 | ) | (5,760 | ) | (5,511 | ) | |||||
| | | | | | | | | | | | | | |
Other income (expense): |
|||||||||||||
Interest income |
48 | 9 | 2 | 1 | |||||||||
Interest expense |
(67 | ) | (39 | ) | (32 | ) | (34 | ) | |||||
Derivatives gain (loss) |
544 | 3,591 | (4,653 | ) | (10,286 | ) | |||||||
| | | | | | | | | | | | | | |
Other income (expense), net |
(525 | ) | 3,561 | (4,683 | ) | (10,319 | ) | ||||||
| | | | | | | | | | | | | | |
Net loss |
$ | (4,733 | ) | $ | (2,308 | ) | $ | (10,443 | ) | $ | (15,830 | ) | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| |
Quarter Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
December 31, 2014 |
September 30, 2014 |
June 30, 2014 |
March 31, 2014 |
|||||||||
Operating expenses: |
|||||||||||||
Research and development |
$ | 1,595 | $ | 2,385 | $ | 3,051 | $ | 3,242 | |||||
General and administrative |
2,249 | 1,800 | 1,688 | 1,829 | |||||||||
| | | | | | | | | | | | | | |
Total operating expenses |
3,844 | 4,185 | 4,739 | 5,071 | |||||||||
| | | | | | | | | | | | | | |
Operating loss |
(3,844 | ) | (4,185 | ) | (4,739 | ) | (5,071 | ) | |||||
| | | | | | | | | | | | | | |
Other income (expense): |
|||||||||||||
Interest income |
1 | 2 | 1 | 1 | |||||||||
Interest expense |
(33 | ) | (35 | ) | (35 | ) | (33 | ) | |||||
Derivatives gain (loss) |
(4,508 | ) | 3,005 | 1,127 | — | ||||||||
| | | | | | | | | | | | | | |
Other expense, net |
(4,540 | ) | 2,972 | 1,093 | (32 | ) | |||||||
| | | | | | | | | | | | | | |
Net loss |
$ | (8,384 | ) | $ | (1,213 | ) | $ | (3,646 | ) | $ | (5,103 | ) | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
45
Item 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion should be read in conjunction with our consolidated financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K. The following discussion contains forward- looking statements that involve risks and uncertainties that could cause actual results or events to differ materially from those expressed or implied by such forward-looking statements as a result of many important factors, including those set forth in Part I of this Annual Report on Form 10-K under the caption "Risk Factors." Please see "Special Note Regarding Forward-Looking Statements" in Part I above. We do not undertake any obligation to update forward-looking statements to reflect events or circumstances occurring after the date of this Annual Report on Form 10-K.
All share amounts give effect to the 1-for-4 reverse stock split of our outstanding shares of Common Stock that occurred on April 8, 2015.
Introduction
This Management's Discussion and Analysis of our financial condition and results of operations are based on our financial statements, which management has prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate such estimates and judgments, including those described in greater detail below. We base estimates on historical experience and on various other factors that management believes are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Business Overview
We are a research and clinical-stage biomaterials and biotechnology company with a focus on treatment of spinal cord injuries (SCI). Our mission is to redefine the life of the SCI patient, and we are developing treatment options intended to provide meaningful improvement in patient outcomes following SCI. Our approach to treating acute SCIs is based on our investigational Neuro Spinal Scaffold implant, an investigational bioresorbable polymer scaffold that is designed for implantation at the site of injury within a spinal cord contusion and is intended to treat acute spinal cord injury. We believe the Neuro Spinal Scaffold implant is the only SCI therapy in development focused solely on treating acute SCI directly at the epicenter of the injury, and incorporates intellectual property licensed under an exclusive, world wide license from Boston Children's Hospital ("BCH") and the Massachusetts Institute of Technology ("MIT"). We are continually evaluating other technologies and therapeutics that may be complementary to our development of the Neuro-Spinal Scaffold implant or offer the potential to bring us closer to our goal of redefining the life of the SCI patient. Recently we entered into exclusive license/assignment agreements with the University of California, San Diego and James Guest, M.D., Ph.D. covering delivery methods and devices for our preclinical Bioengineered Neural Trails™ injection program.
Overall, we expect our research and development expenses to be substantial and to increase for the foreseeable future as we continue the development and clinical investigation of our current and future products. However, expenditures on research and development programs are subject to many uncertainties, including whether we develop our products with a partner or independently or acquire products. At this time, due to the uncertainties and inherent risks involved in our business, we cannot estimate in a meaningful way the duration of, or the costs to complete, our research and development
46
programs or whether, when or to what extent we will generate revenues or cash inflows from the commercialization and sale of any of our products. While we are currently focused on advancing our Neuro-Spinal Scaffold implant, our future research and development expenses will depend on the determinations we make as to the scientific and clinical prospects of each product candidate, as well as our ongoing assessment of the regulatory requirements and each product's commercial potential. In addition, we may make acquisitions of businesses, technologies or intellectual property rights that we believe would be necessary, useful or complementary to our current business. Any investment made in a potential acquisition could affect our results of operations and reduce our limited capital resources, and any issuance of equity securities in connection with a potential acquisition could be substantially dilutive to our stockholders.
There can be no assurance that we will be able to successfully develop or acquire any product, or that we will be able to recover our development or acquisition costs, whether upon commercialization of a developed product or otherwise. We cannot provide assurance that any of our programs under development or any acquired technologies or products will result in products that can be marketed or marketed profitably. If our development-stage programs or any acquired products or technologies do not result in commercially viable products, our results of operations could be materially adversely affected.
We were incorporated on April 2, 2003, under the name of Design Source, Inc. On October 26, 2010, we acquired the business of InVivo Therapeutics Corporation, which was founded in 2005, and continued the existing business operations of InVivo Therapeutics Corporation as our wholly-owned subsidiary.
Critical Accounting Policies and Estimates
Our consolidated financial statements, which appear in Item 8 of this Annual Report on Form 10-K, have been prepared in accordance with accounting principles generally accepted in the United States, which require that the management make certain assumptions and estimates and, in connection therewith, adopt certain accounting policies. Our significant accounting policies are set forth in Note 2 in the Notes to Consolidated Financial Statements. Of those policies, we believe that the policies discussed below may involve a higher degree of judgment and may be more critical to an accurate reflection of our financial condition and results of operations.
Stock-Based Compensation
Stock options are granted with an exercise price at fair market value at the date of the grant. The stock options generally expire ten years from the date of grant. Stock option awards vest upon terms determined by our Board of Directors.
We recognize compensation costs resulting from the issuance of stock-based awards to employees, non-employees and directors as an expense in the statement of operations over the service period based on a measurement of fair value for each stock-based award. The fair value of each option grant was estimated as of the date of grant using the Black-Scholes option-pricing model. The fair value is amortized as compensation cost on a straight-line basis over the requisite service period of the awards, which is generally the vesting period. We use historical data, as well as subsequent events occurring prior to the issuance of the consolidated financial statements, to estimate option exercise and employee departure within the valuation model. The expected term of options granted under our stock plans is based on the average of the contractual term (generally, 10 years) and the vesting period (generally, 48 months). The risk-free rate is based on the yield of a U.S. Treasury security with a term consistent with the option. See Note 12, "Stock Options," in the Notes to Consolidated Financial Statements in Item 8 of this Annual Report on Form 10-K for more information about the assumptions underlying these estimates.
47
Derivative Instruments
Certain of our issued and outstanding warrants to purchase Common Stock contain anti-dilution provisions. These warrants do not meet the requirements for classification as equity and are recorded as derivative warrant liabilities. We used valuation methods and assumptions that consider among other factors the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates consistent with those discussed in Note 11, "Derivative Instruments" in the Notes to Consolidated Financial Statements in Item 8 of this Annual Report on Form 10-K in estimating the fair value for these warrants. Such derivative warrant liabilities are initially recorded at fair value with subsequent changes in fair value charged (credited) to operations in each reporting period. The fair value of the derivative warrant liability is most sensitive to changes in the fair value of the underlying Common Stock and the estimated volatility of our Common Stock.
Research and Development and General and Administrative Expenses
Research and development expenses consist primarily of costs incurred for the development of our product candidates, which include:
- •
- employee-related expenses, including salaries, benefits, travel and stock-based compensation expense;
- •
- expenses incurred under agreements with CROs and clinical sites that conduct our clinical studies;
- •
- facilities, depreciation, and other expenses, which include direct and allocated expenses for rent and maintenance of facilities,
insurance, and other supplies;
- •
- costs associated with our research platform and preclinical activities;
- •
- costs associated with our regulatory, quality assurance and quality control operations; and
- •
- amortization of intangible assets.
Research and development costs are expensed as incurred. We cannot determine with certainty the duration and completion costs of the current or future clinical studies of our product candidates or if, when, or to what extent we will generate revenues from the commercialization and sale of any of our product candidates that obtain regulatory approval. We may never succeed in achieving regulatory approval for any of our product candidates. The duration, costs, and timing of clinical studies and development of our product candidates will depend on a variety of factors, including:
- •
- the scope, rate of progress, and expense of our ongoing as well as any additional clinical studies and other research and development
activities we undertake;
- •
- future clinical study results;
- •
- uncertainties in clinical study enrollment rates;
- •
- changing standards for regulatory approval; and
- •
- the timing and receipt of any regulatory approvals.
A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA, or another regulatory authority were to require us to conduct clinical studies beyond those that we currently anticipate will be required for the completion of clinical development of a product candidate or if we experience significant delays in enrollment in any of our clinical studies, we could be required to expend significant additional financial resources and time on the completion of clinical development for our product candidates.
48
We accrue costs associated with third parties related to our research and development expenses based on our estimate of site management, monitoring, and project management costs. We maintain regular communication with third parties to develop these estimates.
General and administrative expenses consist primarily of salaries and related costs for personnel, including stock-based compensation and travel expenses for our employees in executive, operational, finance, legal, business development, commercial and human resource functions. Other general and administrative expenses include facility-related costs, professional fees for accounting, tax and legal and consulting services, directors' fees and expenses associated with obtaining and maintaining patents.
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our continued research and development and potential commercialization of our product candidates. Additionally, if and when we believe a regulatory approval of the first product candidate appears likely, we anticipate an increase in payroll and related expenses as a result of our preparation for commercial operations, especially as it relates to the sales and marketing of our product candidates.
Recent Accounting Pronouncements
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements—Going Concern, on disclosure of uncertainties about an entity's ability to continue as a going concern. This guidance addresses management's responsibility in evaluating whether there is substantial doubt about a company's ability to continue as a going concern and to provide related footnote disclosures. The guidance is effective for fiscal years ending after December 15, 2016 and for annual and interim periods thereafter, with early adoption permitted. The Company is currently in the process of evaluating the impact of the adoption of this ASU on the financial statements.
In April 2015, the Financial Accounting Standards Board (the "FASB") issued Accounting Standards Update ("ASU") 2015-03, "Interest—Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs". ASU 2015-03 is intended to simplify the presentation of debt issuance costs by requiring that debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. The recognition and measurement guidance for debt issuance costs are not affected by the amendments in this ASU. This new guidance is effective for fiscal years beginning after December 15, 2015 and interim periods within those fiscal years. Early adoption is permitted. The Company is currently in the process of evaluating the impact of the adoption of this ASU on the financial statements.
Results of Operations
Comparison of the Years Ended December 31, 2015 and 2014 (in thousands, except share and per share amounts)
Research and Development Expenses
Research and development expenses decreased by $215 to $10,058 for the year ended December 31, 2015 from $10,273 for the year ended December 31, 2014. After adjusting for the insurance settlement related to business interruption, ($621 for 2014), the research and development expenses were $10,894 for 2014. The decrease in adjusted research and development expenses for 2015 of $836 is primarily attributable to lower consulting costs ($612), testing costs ($375), Packaging and Lab Supplies ($359) and lower compensation related expenses related to the 2014 reduction in force ($564) and other various expenses ($338). These reductions were partly offset by higher clinical trial costs ($729), stock comp expense ($147) and bonuses ($536). There was a higher bonus expense in
49
2015 compared to 2014 due to the fact that in 2014 the accrual which related to the 2013 bonus accrual was reversed because of the Company's decision not paying out the 2013 bonuses.
General and Administrative Expenses
General and administrative expenses increased by $4,774 to $12,340 for the year ended December 31, 2015 from $7,566 for the year ended December 31, 2014. The increase in general and administrative expenses for 2015 is primarily attributable to higher legal costs ($1,361), related to the SEC and MSD inquiries as well as the Class Action law suit, higher stock compensation expense ($1,789), higher investor relation expense and NASDAQ listing fees ($425), an increase in Board and audit fees ($251), consulting costs ($387) and other various expenses ($561).
Interest Income
Interest income increased by $55 to $60 for the year ended December 31, 2015 from $5 for the year ended December 31, 2014. The increase is related to interest earned on our short-term investments.
Interest Expense
Interest expense increased by $36 to $172 for the year ended December 31, 2015 from $136 for the year ended December 31, 2014. The increase in interest expense is due the amortization of the premium or discount values of our short-term investments compared to the maturity value.
Derivatives Gain (Loss)
Derivative losses increased by $10,428 to a loss of $10,804 for the year ended December 31, 2015 from a loss of $376 for the year ended December 31, 2014. The 2015 loss of $10,804 reflects the increase in the fair value of derivative warrant liability which was due primarily to the increase in the fair value of the underlying Common Stock, the decreasing term to expiration of the warrants as well as the exercise of approximately 78% of the outstanding warrants during 2015.
Comparison of the Years Ended December 31, 2014 and 2013 (in thousands, except share and per share amounts)
Research and Development Expenses
Research and development expenses, as reported, decreased by $260 to $10,273 for the year ended December 31, 2014 from $10,533 for the year ended December 31, 2013. After adjusting for the insurance settlements related to business interruption, ($621 for 2014 and $1,100 for 2013), the research and development expenses were $10,894 and $11,633 for 2014 and 2013 respectively. The decrease in adjusted research and development expenses for 2014 of $739 is primarily attributable to lower compensation and stock compensation expenses of $777 related to the reduction in force during the second quarter of 2014, lower consulting costs of $190, reduction in lab supplies of $142, and other various expenses of $23 which were partly offset by increases in our clinical trial costs of $347.
General and Administrative Expenses
General and administrative expenses decreased by $906 to $7,566 for the year ended December 31, 2014 from $8,472 for the year ended December 31, 2013. The decrease in general and administrative expenses for 2014 was primarily attributable to lower travel and entertainment expenses of $341, lower recruiting fees of $329, lower facilities costs of $216, lower consulting expenses of $159 and lower donation expenses of $100 and a decrease in other various expenses of $140. These cost reductions
50
were partly offset by higher stock compensation expense of $252 and higher insurance premiums of $127.
Interest Expense
Interest expense increased by $6 to $136 for the year ended December 31, 2014 from $130 for the year ended December 31, 2013. The increase in interest expense is due to an increase in borrowing under the loans payable.
Derivatives Gain (Loss)
Derivative losses decreased by $18,495 to a loss of $376 for the year ended December 31, 2014 from a loss of $18,871 for the year ended December 31, 2013. The 2014 loss of $376 reflects the increase in the fair value of derivative warrant liability which is due primarily to the increase in the fair value of the underlying Common Stock. The 2013 loss of $18,871 was related to the redemption of the investor warrants from offerings prior to 2013.
Loss from Modification of Warrants
The loss from modification of warrants was $765 for the year ended December 31, 2013. No such modification occurred in the year ended December 31, 2014.
Liquidity and Capital Resources
Since inception, we have devoted substantially all of our efforts to business planning, research and development, recruiting management and technical staff, acquiring operating assets and raising capital. At December 31, 2015, our accumulated deficit was $133,569.
At December 31, 2015, we had total assets of $21,792 and total liabilities of $4,863, resulting in stockholders' equity of $16,929, and had a net loss of $33,314 for the year ended December 31, 2015. We have not achieved profitability and may not be able to realize sufficient revenue to achieve or sustain profitability in the future. We do not expect to be profitable in the next several years, but rather expect to incur additional operating losses. We have limited liquidity and capital resources and must obtain significant additional capital resources in order to fund our operations and sustain our product development efforts, for acquisition of technologies and intellectual property rights, for preclinical and clinical testing of our anticipated products, pursuit of regulatory approvals, acquisition of capital equipment, laboratory and office facilities, establishment of production capabilities, for selling, general and administrative expenses and other working capital requirements. We also expect that we will need to raise additional capital through a combination of equity offerings, debt financings, other third party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements
Since our inception, we have historically financed our operations primarily through the sale of equity-related securities. At December 31, 2015, our consolidated cash and cash equivalents balance was $20,194. We believe our current cash and cash equivalents are adequate to fund our operations into the fourth quarter of 2016. In January 2015, we closed a registered direct offering of an aggregate of 2,000,000 shares of our common stock, resulting in net proceeds of approximately $11,038. In July 2015, we entered into a Sales Agreement with Cowen and Company, LLC ("Cowen") allowing us to issue and sell from time to time up to $50 million in shares of our Common Stock through an "at the market" equity offering program (the "ATM"). To date, we have raised approximately $3,442, through the ATM, net of a 3% commission of the gross proceeds from the sale of shares under the ATM due to Cowen, as our sales agent in the ATM, and other transaction-related expenses.
51
We intend to pursue opportunities to obtain additional financing in the future through equity and/or debt financings. We have filed with the SEC, and the SEC declared effective, a universal shelf registration statement which permits us to issue up to $100 million worth of registered equity securities, of which we utilized $12 million in our January 2015 offering and approximately $3.5 million in our ATM. We may additionally raise use up to approximately $46.5 million under the ATM. Under this effective shelf registration, we also have the flexibility to issue registered securities, from time to time, in one or more separate offerings or other transactions with the size, price and terms to be determined at the time of issuance. Registered securities issued using this shelf may be used to raise additional capital to fund our working capital and other corporate needs, for future acquisitions of assets, programs or businesses, and for other corporate purposes.
We may pursue various other dilutive and non-dilutive funding alternatives depending upon the results of our ongoing pivotal probable benefit study and the extent to which we require additional capital to proceed with development of some or all of our product candidates on expected timelines. The source, timing and availability of any future financing will depend principally upon market conditions and the status of our clinical development programs. Funding may not be available when needed, at all, or on terms acceptable to us. Lack of necessary funds may require us, among other things, to delay, scale back or eliminate some or all of our research and product development programs, planned clinical trials, and our capital expenditures or to license our potential products or technologies to third parties.
We may alternatively engage in cost-cutting efficiencies in an attempt to extend the Company's cash resources as long as possible. If we are unable to raise capital or achieve cost-cutting measures, substantial doubt about our ability to continue as a going concern exists.
Net cash used in operating activities for the year ended December 31, 2015 was $16,329 and the most significant drivers of which were our net loss of $33,314 and offsetting non-cash derivative warrant liability expense of $10,804 and stock share based compensation of $4,666.
Net cash used in investing activities for the year ended December 31, 2015 totaled $5 for purchase of capital equipment.
Net cash provided by financing activities was approximately $23,069 for the year ended December 31, 2015 consisting of the proceeds from our January 2015 offering ($11,038) the exercise of warrants and stock options ($8,857) and the funds raised by the ATM ($3,442). This cash was partly offset by the repayment of loan principal and note principal ($268).
Off Balance Sheet Arrangements
We do not have any off balance sheet arrangements that have or are reasonably likely to have a current or future material effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures, or capital resources.
Contractual Obligations
The following summarizes our significant contractual obligations at December 31, 2015, and the effects such obligations are expected to have on our liquidity and cash flows in future periods:
| |
Payments Due | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Contractual Obligations
|
Total | Less than 1 year |
1 - 3 years | |||||||
Long-term debt |
$ | 1,670 | $ | 395 | $ | 1,275 | ||||
Operating lease payments |
3,644 | 1,263 | 2,381 | |||||||
| | | | | | | | | | | |
Total |
$ | 5,314 | $ | 1,658 | $ | 3,656 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
52
Commitments
See Note 16, "Commitments and Contingencies," in the Notes to Consolidated Financial Statements in Item 8 of this Annual Report on Form 10-K for information.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to market risk related to changes in interest rates. We do not use derivative financial instruments for speculative or trading purposes. Our interest-earning assets consist of cash and cash equivalents of $20,149, or 93% of our total assets at December 31, 2015, and $13,459, or 81% of our total assets at December 31, 2014. Interest income earned on these assets was $60 in 2015 and $5 in 2014. Our interest income is sensitive to changes in the general level of interest rates, primarily U.S. interest rates. At December 31, 2015, our cash equivalents were primarily composed of money market accounts comprised of U.S. Treasury debt securities and repurchase agreements.
53
Item 8. CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Index to Consolidated Financial Statements
All share number and share prices presented in this Item 8 have been adjusted to reflect the 1-for-4 reverse stock split of the Company's Common Stock affected on April 8, 2015.
54
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To
the Board of Directors and Stockholders of InVivo Therapeutics Holdings Corp.:
Cambridge, Massachusetts
We have audited the accompanying consolidated balance sheets of InVivo Therapeutics Holdings Corp. and Subsidiary as of December 31, 2015, and the related consolidated statements of operations, changes in stockholders' equity, and cash flows for the year ended December 31, 2015. We also have audited InVivo Therapeutics Holdings Corp.'s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission in 2013. InVivo Therapeutics Holdings Corp.'s management is responsible for these financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on these financial statements and an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audit of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinions.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (a) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (b) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (c) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of InVivo Therapeutics Holdings Corp. and Subsidiary as of December 31, 2015, and the results of its operations and its cash flows for each of the years in the year period December 31, 2015, in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, InVivo Therapeutics Holdings Corp. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on criteria
55
established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission in 2013.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the annual financial statements, the Company has incurred recurring losses from operations which raises substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1 to the financial statements. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ RSM US LLP
Boston,
MA
March 4, 2016
56
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of InVivo Therapeutics Holdings Corp.:
We have audited the accompanying consolidated balance sheet of InVivo Therapeutics Holdings Corp. as of December 31, 2014, and the related consolidated statements of operations, changes stockholders' equity (deficit) and cash flows for each of the two years in the period ended December 31, 2014. These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of InVivo Therapeutics Holdings Corp. as of December 31, 2014, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
/s/ Wolf & Company, P.C.
Boston,
Massachusetts
March 11, 2015
57
InVivo Therapeutics Holdings Corp.
Consolidated Balance Sheets
(In thousands, except share and per-share data)
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
ASSETS: |
|||||||
Current assets: |
|||||||
Cash and cash equivalents |
$ | 20,194 | $ | 13,459 | |||
Restricted cash |
361 | 422 | |||||
Prepaid expenses and other current assets |
184 | 1,072 | |||||
| | | | | | | | |
Total current assets |
20,739 | 14,953 | |||||
Property and equipment, net |
938 | 1,605 | |||||
Other assets |
115 | 135 | |||||
| | | | | | | | |
Total assets |
$ | 21,792 | $ | 16,693 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
LIABILITIES AND STOCKHOLDERS' EQUITY: |
|||||||
Current liabilities: |
|||||||
Accounts payable |
$ | 521 | $ | 569 | |||
Loan payable, current portion |
395 | 320 | |||||
Note payable, current portion |
— | 18 | |||||
Derivative warrant liability |
1,907 | 7,224 | |||||
Deferred rent payable, current portion |
115 | 114 | |||||
Accrued expenses |
374 | 539 | |||||
| | | | | | | | |
Total current liabilities |
3,312 | 8,784 | |||||
Loan payable, net of current portion |
1,275 | 1,600 | |||||
Deferred rent payable, net current portion |
276 | 391 | |||||
| | | | | | | | |
Total liabilities |
4,863 | 10,775 | |||||
| | | | | | | | |
Commitments and contingencies |
|||||||
Stockholders' equity: |
|||||||
Common stock, $0.00001 par value, authorized—50,000,000 shares; issued and outstanding—27,555,948 and 23,453,000 shares at December 31, 2015 and 2014, respectively |
1 | 1 | |||||
Additional paid-in capital |
150,497 | 106,172 | |||||
Accumulated deficit |
(133,569 | ) | (100,255 | ) | |||
| | | | | | | | |
Total stockholders' equity |
16,929 | 5,918 | |||||
| | | | | | | | |
Total liabilities and stockholders' equity |
$ | 21,792 | $ | 16,693 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
See notes to the consolidated financial statements.
58
InVivo Therapeutics Holdings Corp.
Consolidated Statements of Operations
(In thousands, except share and per-share data)
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Operating expenses: |
||||||||||
Research and development |
$ | 10,058 | $ | 10,273 | $ | 10,533 | ||||
General and administrative |
12,340 | 7,566 | 8,472 | |||||||
| | | | | | | | | | | |
Total operating expenses |
22,398 | 17,839 | 19,005 | |||||||
| | | | | | | | | | | |
Operating loss |
(22,398 | ) | (17,839 | ) | (19,005 | ) | ||||
| | | | | | | | | | | |
Other income (expense): |
||||||||||
Interest income |
60 | 5 | 15 | |||||||
Interest expense |
(172 | ) | (136 | ) | (130 | ) | ||||
Modification of warrants |
— | — | (765 | ) | ||||||
Derivative loss |
(10,804 | ) | (376 | ) | (18,871 | ) | ||||
| | | | | | | | | | | |
Other expense, net |
(10,916 | ) | (507 | ) | (19,751 | ) | ||||
| | | | | | | | | | | |
Net loss |
$ | (33,314 | ) | $ | (18,346 | ) | $ | (38,756 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per share, basic and diluted |
$ | (1.26 | ) | $ | (0.83 | ) | $ | (2.10 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted average number of common shares outstanding, basic and diluted |
26,461,374 | 22,080,761 | 18,497,922 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
See notes to the consolidated financial statements.
59
InVivo Therapeutics Holdings Corp.
Consolidated Statements of Changes in Stockholders' Equity (Deficit)
| |
Common Stock | |
|
Total Stockholders' Equity (Deficit) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Additional Paid-in Capital |
Accumulated Deficit |
||||||||||||||
| |
Shares | Amount | ||||||||||||||
Balance as of December 31, 2012 |
16,470,281 | 1 | 40,842 | (43,153 | ) | (2,310 | ) | |||||||||
Share-based compensation expense |
— | — | 3,136 | — | 3,136 | |||||||||||
Issuance of common stock upon exercise of warrants |
3,056,211 | — | 15,952 | — | 15,952 | |||||||||||
Issuance of common stock upon exercise of stock options |
147,221 | — | 455 | — | 455 | |||||||||||
Issuance of common stock to 401(k) plan |
19,721 | — | 192 | — | 192 | |||||||||||
Fair value of derivative warrant liability reclassified to additional paid-in capital |
— | — | 33,456 | — | 33,456 | |||||||||||
Incremental fair value from warrant modification |
— | — | 765 | — | 765 | |||||||||||
Net income |
— | — | — | (38,756 | ) | (38,756 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balance as of December 31, 2013 |
19,693,434 | 1 | 94,798 | (81,909 | ) | 12,890 | ||||||||||
Share-based compensation expense |
— | — | 2,730 | — | 2,730 | |||||||||||
Issuance of common stock in public offering |
3,500,312 | — | 7,770 | — | 7,770 | |||||||||||
Issuance of common stock for services |
74,626 | — | 477 | — | 477 | |||||||||||
Issuance of common stock upon exercise of warrants |
9,975 | — | 12 | — | 12 | |||||||||||
Issuance of common stock upon exercise of stock options |
132,900 | — | 212 | — | 212 | |||||||||||
Issuance of common stock to 401(k) plan |
41,753 | — | 173 | — | 173 | |||||||||||
Net loss |
— | — | — | (18,346 | ) | (18,346 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balance as of December 31, 2014 |
23,453,000 | 1 | 106,172 | (100,255 | ) | 5,918 | ||||||||||
Share-based compensation expense |
— | — | 4,666 | — | 4,666 | |||||||||||
Issuance of common stock in public offerings |
2,388,245 | — | 14,480 | — | 14,480 | |||||||||||
Issuance of common stock upon exercise of warrants |
1,379,575 | — | 7,789 | — | 7,789 | |||||||||||
Issuance of common stock upon exercise of stock options |
316,177 | — | 1,068 | — | 1,068 | |||||||||||
Fair value of derivative warrant liability reclassified to additional paid-in capital |
— | — | 16,121 | — | 16,121 | |||||||||||
Fractional shares issued due to reverse stock split |
1,514 | — | — | — | — | |||||||||||
Issuance of common stock to 401(k) plan |
17,437 | — | 201 | — | 201 | |||||||||||
Net loss |
— | — | — | (33,314 | ) | (33,314 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balance as of December 31, 2015 |
27,555,948 | 1 | 150,497 | (133,569 | ) | 16,929 | ||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
See notes to the consolidated financial statements.
60
InVivo Therapeutics Holdings Corp.
Consolidated Statements of Cash Flows
(In thousands)
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Cash flows from operating activities: |
||||||||||
Net loss |
$ | (33,314 | ) | $ | (18,346 | ) | $ | (38,756 | ) | |
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||
Depreciation and amortization expense |
689 | 752 | 740 | |||||||
Non-cash derivative losses |
10,804 | 376 | 18,871 | |||||||
Non-cash loss from modification of warrants |
— | — | 765 | |||||||
Common stock issued to 401(k) plan |
201 | 173 | 192 | |||||||
Common stock issued for services |
— | 477 | — | |||||||
Share-based compensation expense |
4,666 | 2,730 | 3,136 | |||||||
Changes in operating assets and liabilities: |
||||||||||
Restricted cash |
61 | 180 | — | |||||||
Prepaid expenses |
888 | (363 | ) | 124 | ||||||
Insurance receivable |
— | (689 | ) | — | ||||||
Other assets |
3 | 4 | 5 | |||||||
Accounts payable |
(48 | ) | (330 | ) | (254 | ) | ||||
Accrued expenses |
(279 | ) | (248 | ) | 271 | |||||
| | | | | | | | | | | |
Net cash used in operating activities |
(16,329 | ) | (15,284 | ) | (14,906 | ) | ||||
| | | | | | | | | | | |
Cash flows from investing activities: |
||||||||||
Non-cash disposals of property and equipment |
— | 45 | — | |||||||
Purchases of property and equipment |
(5 | ) | (47 | ) | (749 | ) | ||||
| | | | | | | | | | | |
Net cash used in investing activities |
(5 | ) | (2 | ) | (749 | ) | ||||
| | | | | | | | | | | |
Cash flows from financing activities: |
||||||||||
Proceeds from exercise of stock options |
1,068 | 212 | 456 | |||||||
Proceeds from exercise of warrants |
7,789 | 12 | 15,952 | |||||||
Proceeds from issuance of note payable |
— | — | 150 | |||||||
Repayment of note payable |
(18 | ) | (56 | ) | (57 | ) | ||||
Principal payments on capital lease obligation |
— | (21 | ) | (33 | ) | |||||
Proceeds from loans payable |
— | — | 342 | |||||||
Repayment of loans payable |
(250 | ) | — | — | ||||||
Proceeds from issuance of common stock and warrants |
14,480 | 14,618 | — | |||||||
| | | | | | | | | | | |
Net cash provided by financing activities |
23,069 | 14,765 | 16,810 | |||||||
| | | | | | | | | | | |
Increase (decrease) in cash and cash equivalents |
6,735 | (521 | ) | 1,155 | ||||||
Cash and cash equivalents at beginning of period |
13,459 | 13,980 | 12,825 | |||||||
| | | | | | | | | | | |
Cash and cash equivalents at end of period |
$ | 20,194 | $ | 13,459 | $ | 13,980 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Supplemental disclosure of cash flow information and non-cash investing and financing activities: |
||||||||||
Cash paid for interest |
$ | 121 | $ | 132 | $ | 125 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Cash paid for taxes |
$ | — | $ | — | $ | — | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Fair value of warrants issued in connection with underwriting agreement |
$ | — | $ | 6,848 | $ | — | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Reclassification of derivative warrant liability to additional paid-in capital |
$ | 16,121 | $ | — | $ | 33,456 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
See notes to the consolidated financial statements.
61
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements
(In thousands, except share and per-share data)
1. NATURE OF OPERATIONS AND GOING CONCERN
Business
InVivo Therapeutics Holdings Corp. ("InVivo" or the "Company") is a pioneering biomaterials and biotechnology company with a focus on the treatment of spinal cord injuries. Its proprietary technologies incorporate intellectual property that is licensed under an exclusive, world-wide license from Boston Children's Hospital and the Massachusetts Institute of Technology, as well as intellectual property that has been developed internally in collaboration with its advisors and partners.
Since its inception, InVivo has devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, acquiring operating assets and raising capital. InVivo historically financed its operations primarily through the sale of equity-related securities. At December 31, 2015, the consolidated cash balance was $20,194. InVivo believes its current cash and cash equivalents are adequate to fund its operations into the fourth quarter of 2016. Invivo has not achieved profitability and may not be able to realize sufficient revenue to achieve or sustain profitability in the future. InVivo does not expect to be profitable in the next several years, but rather expects to incur additional operating losses. InVivo has limited liquidity and capital resources and must obtain significant additional capital resources in order to sustain its product development efforts, for acquisition of technologies and intellectual property rights, for preclinical and clinical testing of its anticipated products, pursuit of regulatory approvals, acquisition of capital equipment, laboratory and office facilities, establishment of production capabilities, for selling, general and administrative expenses and other working capital requirements. InVivo expects that it will need additional capital to fund its operations, which it may raise through a combination of equity offerings, debt financings, other third party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements.
Going Concern
The accompanying consolidated financial statements have been prepared on a basis that assumes that the Company will continue as a going concern and that contemplates the continuity of operations, realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since inception in devoting substantially all of its efforts toward research and development and has an accumulated loss of $133,569 at December 31, 2015. During the year ended December 31, 2015, the Company generated a net loss of $33,314 used cash in operations of $16,329 and the Company expects that it will continue to generate operating losses for the foreseeable future. The Company believes that its cash balance at December 31, 2015 of $20,194 is adequate to fund operations at budgeted levels into the fourth quarter 2016. The Company's ability to execute its operating plan beyond that date depends on its ability to obtain additional funding via the sale of equity and/or debt securities, a strategic transaction or otherwise. The Company plans to continue to actively pursue financing alternatives, but there can be no assurance that it will obtain the necessary funding. The accompanying consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
62
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
2. SIGNIFICANT ACCOUNTING POLICIES
A summary of the significant accounting policies followed by the Company in the preparation of the financial statements is as follows:
Use of estimates
The process of preparing financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of assets and liabilities at the date of financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates and changes in estimates may occur.
Basis of presentation and principles of consolidation
The consolidated financial statements include the accounts of InVivo Therapeutics Holdings Corp. and its wholly-owned subsidiary, InVivo Therapeutics Corporation. All significant intercompany balances and transactions have been eliminated in consolidation.
Cash and cash equivalents
The Company considers only those investments that are highly liquid, readily convertible to cash, and that mature within three months from date of purchase to be cash equivalents. Marketable investments are those with original maturities in excess of three months.
At December 31, 2015 and 2014, cash equivalents were comprised of money market funds and other short-term investments.
Cash and cash equivalents consist of the following:
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Cash on deposit |
$ | 116 | $ | 269 | |||
Money market funds and other short-term investments |
20,078 | 13,190 | |||||
| | | | | | | | |
Total cash and cash equivalents |
$ | 20,194 | $ | 13,459 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Restricted cash
At December 31, 2015 and 2014, the restricted cash of $361 and $422, respectively, represents a $50 and $111 respectively, security deposit related to the Company's credit card account, and, for each year, a $311 standby letter of credit in favor of a landlord (see Note 16).
Financial instruments
The carrying amounts reported in the Company's consolidated balance sheets for cash and cash equivalents and accounts payable approximate fair value based on the short-term nature of these instruments. The carrying value of note and loans payable approximates their fair value due to the market terms.
63
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
2. SIGNIFICANT ACCOUNTING POLICIES (Continued)
Property and equipment
Property and equipment are carried at cost. Depreciation and amortization expense is provided over the estimated useful lives of the assets using the straight-line method. A summary of the estimated useful lives is as follows:
Classification
|
Estimated Useful Life | |
|---|---|---|
Computer hardware |
5 years | |
Software |
3 years | |
Office furniture and equipment |
5 years | |
Research and lab equipment |
5 years | |
Leasehold improvements |
Remaining life of lease |
Research and development expenses
Costs incurred for research and development are expensed as incurred.
Concentrations of credit risk
Financial instruments which potentially subject the Company to concentrations of credit risk consist principally of cash and cash equivalents. The Company maintains cash in commercial banks, which may at times exceed Federally Insured limits. The Company has not experienced any loss in such accounts. The Company believes it is not exposed to any significant credit risk on cash and cash equivalents.
Segment information
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision making group, in making decisions regarding resource allocation and assessing performance. To date, the Company has viewed its operations and manages its business as principally one operating segment, which is developing and commercializing biopolymer scaffolding devices for the treatment of spinal cord injuries. As of December 31, 2015 and 2014, all of the Company's assets were located in one location in the United States.
Income taxes
For federal and state income taxes, deferred tax assets and liabilities are recognized based upon temporary differences between the financial statement and the tax basis of assets and liabilities. Deferred income taxes are based upon prescribed rates and enacted laws applicable to periods in which differences are expected to reverse. A valuation allowance is recorded when it is more likely than not that some portion or all of the deferred tax assets will not be realized. Accordingly, the Company provides a valuation allowance, if necessary, to reduce deferred tax assets to amounts that are realizable. Tax positions taken or expected to be taken in the course of preparing the Company's tax returns are required to be evaluated to determine whether the tax positions are "more-likely-than-not" of being sustained by the applicable tax authority.
64
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
2. SIGNIFICANT ACCOUNTING POLICIES (Continued)
Tax positions not deemed to meet a more-likely-than-not threshold would be recorded as a tax expense in the current year. There were no material uncertain tax positions that require accrual or disclosure to the financial statements as of December 31, 2015 or 2014. Tax years subsequent to 2011 remain open to examination by U.S. federal and state tax authorities.
Impairment of long-lived assets
The Company continually monitors events and changes in circumstances that could indicate that carrying amounts of long-lived assets may not be recoverable. An impairment loss is recognized when expected cash flows are less than an asset's carrying value. Accordingly, when indicators of impairment are present, the Company evaluates the carrying value of such assets in relation to the operating performance and future undiscounted cash flows of the underlying assets. The Company's policy is to record an impairment loss when it is determined that the carrying value of the asset may not be recoverable. No impairment charges were recorded for the years ended December 31, 2015, 2014 and 2013.
Share-based payments
The Company recognizes compensation costs resulting from the issuance of stock-based awards to employees, non-employees and directors as an expense in the Company's statement of operations over the service period based on a measurement of fair value for each stock-based award. The fair value of each option grant is estimated as of the date of grant using the Black-Scholes option-pricing model. The fair value is amortized as compensation cost on a straight-line basis over the requisite service period of the awards, which is generally the vesting period.
Derivative instruments
The Company generally does not use derivative instruments to hedge exposures to cash-flow or market risks; however, certain warrants to purchase Common Stock that do not meet the requirements for classification as equity are classified as liabilities. In such instances, net-cash settlement is assumed for financial reporting purposes, even when the terms of the underlying contracts do not provide for a net-cash settlement. Such financial instruments are initially recorded at fair value with subsequent changes in fair value charged (credited) to operations in each reporting period. If these instruments subsequently meet the requirements for classification as equity, the Company reclassifies the fair value to equity.
Net income (loss) per common share
Basic net income (loss) per share of Common Stock has been computed by dividing net income (loss) by the weighted average number of shares outstanding during the period. Diluted net income per share of Common Stock has been computed by dividing net income by the weighted average number of shares outstanding plus the dilutive effect, if any, of outstanding stock options, warrants and convertible securities. Diluted net loss per share of Common Stock has been computed by dividing the net loss for the period by the weighted average number of shares of Common Stock outstanding during such period. In a net loss period, options, warrants related to the May 2014 capital raise, which include
65
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
2. SIGNIFICANT ACCOUNTING POLICIES (Continued)
anti-dilution provisions, and convertible securities are anti-dilutive and therefore excluded from diluted loss per share calculations.
Recent accounting pronouncements
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements—Going Concern, on disclosure of uncertainties about an entity's ability to continue as a going concern. This guidance addresses management's responsibility in evaluating whether there is substantial doubt about a company's ability to continue as a going concern and to provide related footnote disclosures. The guidance is effective for fiscal years ending after December 15, 2016 and for annual and interim periods thereafter, with early adoption permitted. The Company is currently in the process of evaluating the impact of the adoption of this ASU on the financial statements.
In April 2015, the Financial Accounting Standards Board (the "FASB") issued Accounting Standards Update ("ASU") 2015-03, "Interest—Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs". ASU 2015-03 is intended to simplify the presentation of debt issuance costs by requiring that debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. The recognition and measurement guidance for debt issuance costs are not affected by the amendments in this ASU. This new guidance is effective for fiscal years beginning after December 15, 2015 and interim periods within those fiscal years. Early adoption is permitted. The Company is currently in the process of evaluating the impact of the adoption of this ASU on the financial statements.
3. PROPERTY AND EQUIPMENT
Property and equipment, net consisted of the following:
| |
2015 | 2014 | |||||
|---|---|---|---|---|---|---|---|
Computer software and hardware |
$ | 562 | $ | 562 | |||
Research and lab equipment |
1,874 | 1,873 | |||||
Leasehold improvements |
392 | 390 | |||||
Office Equipment |
792 | 790 | |||||
Less accumulated depreciation and amortization |
(2,682 | ) | (2,010 | ) | |||
| | | | | | | | |
Property and equipment, net |
$ | 938 | $ | 1,605 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Depreciation and amortization expense for the years ended December 31, 2015, 2014, and 2013 was $672, $735 and $723, respectively. Maintenance and repairs are charged to expense as incurred, while any additions or improvements are capitalized. During 2015, the Company had no disposals.
66
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
4. INTANGIBLE ASSETS
Intangible assets, included in "other assets," consisted of patent licensing fees paid to license intellectual property (see Note 15). The Company is amortizing the license fee as a research and development expense over the 15-year term of the license.
| |
2015 | 2014 | |||||
|---|---|---|---|---|---|---|---|
Patent licensing fee |
$ | 200 | $ | 200 | |||
Accumulated amortization |
(104 | ) | (86 | ) | |||
| | | | | | | | |
|
$ | 96 | $ | 114 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
For each of the years ended December 31, 2015, 2014, and 2013, the amortization expense was $17. Amortization expense in each of the next five years is also expected to be $17 per year.
5. ACCRUED EXPENSES
Accrued expenses consisted of the following:
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Accrued payroll |
$ | 85 | $ | 49 | |||
Accrued vacation |
81 | 72 | |||||
Accrued legal |
— | 360 | |||||
Other accrued expenses |
208 | 58 | |||||
| | | | | | | | |
|
$ | 374 | $ | 539 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
6. FAIR VALUES OF ASSETS AND LIABILITIES
The Company groups its assets and liabilities generally measured at fair value in three levels, based on the markets in which the assets and liabilities are traded and the reliability of the assumptions used to determine fair value.
Level 1—Valuation is based on quoted prices in active markets for identical assets or liabilities. Level 1 assets and liabilities generally include debt and equity securities that are traded in an active exchange market. Valuations are obtained from readily available pricing sources for market transactions involving identical assets or liabilities.
Level 2—Valuation is based on observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3—Valuation is based on unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. Level 3 assets and liabilities include financial instruments whose value is determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value requires significant management judgment or estimation.
67
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
6. FAIR VALUES OF ASSETS AND LIABILITIES (Continued)
The Company uses valuation methods and assumptions that consider among other factors the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments.
Assets and liabilities measured at fair value on a recurring basis are summarized below:
| |
At December 31, 2015 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Level 1 | Level 2 | Level 3 | Fair Value | |||||||||
Cash equivalents |
$ | 20,078 | $ | — | $ | — | $ | 20,078 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Derivative warrant liability |
$ | — | $ | 1,907 | $ | — | $ | 1,907 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| |
At December 31, 2014 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Level 1 | Level 2 | Level 3 | Fair Value | |||||||||
Cash equivalents |
$ | 13,190 | $ | — | $ | — | $ | 13,190 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Derivative warrant liability |
$ | — | $ | 7,224 | $ | — | $ | 7,224 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
7. NOTE PAYABLE
In May 2013, the Company entered into a contract for the purchase of an enterprise resource planning ("ERP") system for $150. The total cost for the ERP system, including interest, is $159, with an implicit interest rate of approximately 6%. Pursuant to the terms of this non-cancelable purchase agreement in effect at December 31, 2015, there are no future minimum principal payments due to the fact that it has been paid in full. In the third quarter of 2013, the Company decided to abandon the implementation of the ERP system. As such, the ERP system cost of $150 was fully expensed in 2013. The Company reserves the right to implement the ERP system at a future date.
8. LOAN PAYABLE
In October 2012, the Company entered into a loan agreement with the Massachusetts Development Finance Agency ("MassDev"). The loan agreement provided the Company with a $2,000 line of credit from the Commonwealth of Massachusetts's Emerging Technology fund, with $200 to be used for working capital purposes and the remainder to be used for the purchase of capital equipment. The annual interest rate is fixed at 6.5% with interest-only payments for the first thirty months, commencing on November 1, 2012, and then equal interest and principal payments over the next fifty-four months, with the final maturity of the loan on October 5, 2019. Commencing on May 1, 2015, equal monthly principal payments of $41 will be due until loan maturity on October 5, 2019. Therefore, for the years ending December 31, 2016, 2017, 2018, and 2019, principal payments of $395, $423, $451, and $400, respectively, will be due. In October 2012, as part of the agreement, the Company issued MassDev a warrant for the purchase of 9,037 shares of its Common Stock. The warrant has a seven-year term and is exercisable at $6.64 per share. The fair value of the warrant was determined to be $32 and was recorded as a deferred financing cost, and is being amortized to interest expense over a seven-year period commencing in October 2012. Amortization of the deferred financing cost for the years ended December 31, 2015, 2014, and 2013 was $5, and was included in interest expense in the
68
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
8. LOAN PAYABLE (Continued)
Company's consolidated statements of operations. The equipment line of credit is secured by substantially all the assets of the Company, excluding intellectual property. During 2013, the Company drew on the line and received proceeds of $342, for capital equipment. Interest expense related to this loan was $126, $127, and $120 for the year ended December 31, 2015, 2014, and 2013, respectively.
At December 31, loans payable consisted of the following:
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
MassDev Loan |
$ | 1,670 | $ | 1,920 | |||
Less: current portion |
395 | 320 | |||||
| | | | | | | | |
|
$ | 1,275 | $ | 1,600 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
9. INCOME TAXES
No provision or benefit for federal or state income taxes has been recorded, as the Company has incurred a net loss for all of the periods presented, and the Company has provided a full valuation allowance against its deferred tax assets.
At December 31, 2015, the Company had U.S. federal and Massachusetts net operating loss carryforwards of $77,456 and $69,682, respectively, of which federal carryforwards will expire in varying amounts beginning in 2026. Massachusetts net operating losses began to expire in 2011. Utilization of net operating losses may be subject to substantial annual limitations due to the "change in ownership" provisions of the Internal Revenue Code, and similar state provisions. The annual limitations may result in the expiration of net operating losses before utilization. The Company also had research and development tax credit carryforwards at December 31, 2015 of $1,010 which will begin to expire in 2021 unless previously utilized.
Significant components of the Company's net deferred tax asset are as follows:
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Net operating loss carryforward |
$ | 30,014 | $ | 22,490 | |||
Research and development credit carryforward |
942 | 801 | |||||
Stock-based compensation |
3,307 | 2,265 | |||||
Depreciation and amortization |
(48 | ) | (124 | ) | |||
Accrued expenses |
186 | 226 | |||||
Charitable contributions |
96 | 112 | |||||
| | | | | | | | |
Subtotal |
34,497 | 25,770 | |||||
Valuation allowance |
(34,497 | ) | (25,770 | ) | |||
| | | | | | | | |
Net deferred taxes |
$ | — | $ | — | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The Company has maintained a full valuation allowance against its deferred tax assets in all periods presented. A valuation allowance is required to be recorded when it is more likely than not
69
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
9. INCOME TAXES (Continued)
that some portion or all of the net deferred tax assets will not be realized. Since the Company cannot be assured of generating taxable income and thereby realizing the net deferred tax assets, a full valuation allowance has been provided. In the years ended December 31, 2015 and 2014, the valuation allowance increased by $8,727 and $6,290, respectively.
The Company has no uncertain tax positions at December 31, 2015 and 2014 that would affect its effective tax rate. The Company does not anticipate a significant change in the amount of uncertain tax positions over the next twelve months. Since the Company is in a loss carryforward position, the Company is generally subject to U.S. federal and state income tax examinations by tax authorities for all years for which a loss carryforward is available.
Income tax benefits computed using the federal statutory income tax rate differs from the Company's effective tax rate primarily due to the following:
| |
December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Statutory rate |
(34.0 | )% | (34.0 | )% | (34.0 | )% | ||||
State taxes, net of benefit |
(3.5 | )% | (4.8 | )% | (2.6 | )% | ||||
Permanent differences: |
||||||||||
Derivative losses |
11.0 | % | 0.7 | % | 17.1 | % | ||||
Other |
0.3 | % | 2.6 | % | 0.0 | % | ||||
R&D tax credit |
(0.4 | )% | (1.0 | )% | (0.4 | )% | ||||
Other |
0.4 | % | 2.2 | % | 0.0 | % | ||||
Increase in valuation reserve |
26.2 | % | 34.3 | % | 19.9 | % | ||||
| | | | | | | | | | | |
Effective tax rate |
0.0 | % | 0.0 | % | 0.0 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
10. COMMON STOCK
The Company has authorized 50,000,000 shares of Common Stock, $0.00001 par value per share, of which 27,555,948, shares were issued and outstanding as of December 31, 2015 and 23,453,000 shares were issued and outstanding as of December 31, 2014.
During the year ended December 31, 2015, the Company issued an aggregate of 316,177 shares of common stock upon the exercise of stock options, including stock options to purchase 52,224 shares of common stock exercised through cashless exercise provisions resulting in the issuance of 14,961 shares of common stock and stock options to purchase 301,216 shares of common stock exercised for cash, providing cash proceeds of $1,068.
During the year ended December 31, 2015, the Company issued an aggregate of 1,379,575 shares of common stock upon the exercise of warrants, including warrants to purchase 40,955 shares of common stock exercised through cashless exercise provisions resulting in the issuance of 25,052 shares of common stock and warrants to purchase 1,354,523 shares of common stock exercised for cash, providing net cash proceeds of $7,789.
70
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
10. COMMON STOCK (Continued)
During the year ended December 31, 2015, the Company issued an aggregate of 17,437 shares of Common Stock with a fair value of $201 to the Company's 401(k) plan as a matching contribution.
In January 2015, the Company closed a registered direct offering of an aggregate of 2,000,000 shares of common stock, resulting in net proceeds of $11,038.
As part of the adjustment to reflect the 1-for-4 reverse stock split on April 8, 2015, 1,514 shares were issued to account for the fractional roundup of shareholders.
In July 2015, the Company entered into a Sales Agreement (the "Sales Agreement") with Cowen and Company, LLC ("Cowen") pursuant to which the Company may issue and sell from time to time shares of Common Stock having aggregate sales proceeds of up to $50 million through an "at the market" equity offering program under which Cowen acts as the Company's sales agent. The Company is required to pay Cowen a commission of 3% of the gross proceeds from the sale of shares of Common Stock under the Sales Agreement. The Company issued 388,245 shares of common stock under the Sales Agreement during the year ended December 31, 2015, providing cash proceeds of $3,442, net through this facility.
During the year ended December 31, 2014, the Company issued an aggregate of 132,900 shares of Common Stock upon the exercise of stock options and received cash proceeds of $212.
During the year ended December 31, 2014, the Company issued an aggregate of 9,975 shares of Common Stock upon the exercise of warrants, including warrants to purchase 15,655 shares of Common Stock exercised through cashless exercise provisions resulting in the issuance of 6,903 shares of common stock and warrants to purchase 3,072 shares of Common Stock exercised for cash, providing cash proceeds of $12.
During the year ended December 31, 2014, the Company issued an aggregate of 41,753 shares of Common Stock with a fair value of $173 to the Company's 401(k) plan as a matching contribution.
In January 2014, the Company issued 27,212 and 5,594 shares of Common Stock to Michael J. Astrue, the Company's then-Interim Chief Executive Officer, and Gregory D. Perry, the Company's then-Interim Chief Financial Officer, respectively, in lieu of executive cash bonuses. Such shares had an aggregate fair value of approximately $282.
In December 2014, the Company issued 41,821 shares of Common Stock to certain employees of the Company in lieu of cash bonuses. Such shares had an aggregate fair value of approximately $195.
During the year ended December 31, 2014, the Company closed an underwritten public offering of an aggregate of 3,500,312 shares of common stock and warrants to purchase up to an aggregate of 1,750,156 shares of common stock, at a price to the public of $4.60 per share of common stock and $0.00001 per warrant. The net proceeds to the Company, after deducting underwriting discounts and offering expenses, were approximately $14,600. The warrants have a per share price of $5.75, or 125% of the public offering of the common stock, and expire on May 9, 2019.
During the year ended December 31, 2013, the Company issued an aggregate of 147,221 shares of Common Stock upon the exercise of stock options and received cash proceeds of $456.
71
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
10. COMMON STOCK (Continued)
During the year ended December 31, 2013, the Company issued an aggregate of 3,056,211 shares of Common Stock upon the exercise of warrants, including warrants to purchase 156,759 shares of Common Stock exercised through cashless exercise provisions and warrants to purchase 2,953,334 shares of Common Stock exercised for cash, providing cash proceeds of $15,952.
Common Stock Reserves
As of December 31, 2015, the Company had the following reserves established for the future issuance of Common Stock as follows:
Reserves for the exercise of warrants |
1,156,779 | |||
Reserves for the exercise of stock options |
6,736,923 | |||
| | | | | |
Total Reserves |
7,893,702 | |||
| | | | | |
| | | | | |
| | | | | |
11. DERIVATIVE INSTRUMENTS
Certain warrants issued to investors and the placement agent warrants in the fourth quarter of 2010 had provisions that included anti-dilution protection and, under certain conditions, granted the right to the holder to require the Company to repurchase the warrant. Accordingly through March 2013, these warrants were accounted for as derivative liabilities. In the quarter ended March 31, 2013, $476 was reclassified from Derivative warrant liability to Additional paid-in capital related to warrants exercised. In May 2013 outstanding investor warrants totaling 11,726,343 warrants, were exercised and the fair value of $25,241was reclassified from Derivative warrant liability to Additional paid-in capital.
On May 17, 2013, the Company completed its offer to exchange certain of its outstanding warrants to purchase shares of the Company's common stock (the "Eligible Warrants") for new warrants (the "New Warrants") with the same terms except (i) the expiration date of the New Warrants was extended two years and (ii) weighted average anti-dilution provisions were removed from the New Warrants (the "Offer"). The Eligible Warrants consisted of (i) warrants to purchase common stock dated October 26, 2010, issued in connection with the closing of a merger (the "Merger Warrants") and (ii) warrants to purchase common stock issued to the placement agent as compensation for services in connection with each closing of a private placement which occurred on October 26, 2010, November 10, 2010 and December 3, 2010 (the "Placement Agent Warrants"). In connection with the Offer, Merger Warrants to purchase 255,000 shares of the Company's common stock and Placement Agent Warrants to purchase 3,064,091 shares of the Company's common stock were tendered and accepted for exchange for New Warrants to purchase an aggregate of 3,319,091 shares of the Company's common stock. Due to the modification of the terms, the Eligible Warrants were revalued prior to modification and immediately after modification as of May 17, 2013. This resulted in a non-cash charge of $765 which was recorded in Other expense as Loss from modification of warrants. Since the New Warrants are not accounted for as derivative liabilities, the fair value of these warrants after modification of $7,738 was reclassified from Derivative warrant liability to Additional paid-in capital.
The warrants issued in connection with the May 2014 public offering to purchase 1,750,156 shares of the Common Stock (see Note 10) have anti- dilution protection provisions and, under certain conditions, grant the right to the holder to require the Company to re-price the warrant. Accordingly
72
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
11. DERIVATIVE INSTRUMENTS (Continued)
these warrants are accounted for as derivative warrant liabilities. The Company used a Binomial Lattice option pricing model and assumptions that consider, among other factors, the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments. Changes in fair value of the derivative financial instruments are recognized currently in the Company's consolidated statement of operations as a derivative gain or loss. The warrant derivative losses are non-cash expenses and for the 12 months ended December 31, 2015, 2014 and 2013, a loss of $10,804, $376 and $18,871, respectively, were included in other income (expenses) in the Company's consolidated statement of operations.
The Company uses the Binomial Lattice option pricing model and assumptions that consider among other factors the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments. The fair value of these derivative instruments at December 31, 2015 and 2014 was $1,907 and $7,224, respectively, and was included as a derivative warrant liability, a current liability. Changes in fair value of the derivative financial instruments are recognized currently in the consolidated statement of operations as a derivative gain or loss.
The assumptions used principally in determining the fair value of warrants were as follows:
| |
Year Ended December 31, | |||||
|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||
Risk free interest rate |
0.65% | 1.47% | NA | |||
Expected dividend yield |
0% | 0% | NA | |||
Contractual term |
3.4 years | 4.4 years | NA | |||
Expected volatility |
100% | 119% | NA | |||
The primary underlying risk exposure pertaining to the warrants is the change in fair value of the underlying Common Stock for each reporting period.
The table below presents the changes in derivative warrant liability during the years ended December 31, 2015, 2014, and 2013:
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Balance at beginning of year |
$ | 7,224 | $ | — | $ | 14,585 | ||||
Issuance of warrants |
— | 6,848 | — | |||||||
Increase (decrease) in the fair value of the warrants |
10,804 | 376 | 18,871 | |||||||
Fair value of derivative warrant liability reclassified to additional paid in capital |
(16,121 | ) | — | (33,456 | ) | |||||
| | | | | | | | | | | |
Balance at end of year |
$ | 1,907 | $ | 7,224 | $ | — | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
12. STOCK OPTIONS
In 2007, the Company adopted the 2007 Employee, Director and Consultant Stock Plan (the "2007 Plan"). Pursuant to the 2007 Plan, the Company's Board of Directors (or committees and/or executive
73
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
12. STOCK OPTIONS (Continued)
officers delegated by the Board of Directors) may grant incentive and nonqualified stock options to the Company's employees, officers, directors, consultants and advisors. As of December 31, 2015, there were options to purchase an aggregate of 240,863 shares of common stock outstanding under the 2007 Plan and no shares available for future grants under the 2007 Plan.
On October 26, 2010, the Company's Board of Directors adopted, and the Company's shareholders subsequently approved, the 2010 Equity Incentive Plan (as subsequently amended, the "2010 Plan"). The 2010 Plan provides for grants of incentive stock options to employees, and nonqualified stock options and restricted Common Stock to employees, consultants and non-employee directors of the Company.
In April 2015, the Company's Board of Directors adopted, and the Company' shareholders subsequently approved, the 2015 Equity Incentive Plan (the "2015 Plan"). The 2015 Plan provides for grants of incentive stock options to employees, and nonqualified stock, restricted Common Stock, restricted stock units and stock appreciation rights to employees, consultants and directors of the Company. As of December 31, 2015, the total number of shares authorized for issuance under the 2015 Plan is 4,322,355 shares, consisting of 4,000,000 shares plus 322,355 shares that remained available for grant under the 2010 Plan. Upon approval of the 2015 Plan by the Company's shareholders on June 16, 2016, the 2010 Plan was terminated and no additional shares or share awards have been subsequently granted under the 2010 Plan.
As of December 31, 2015, there were outstanding options to purchase an aggregate of 946,760 and 2,065,687 shares of common stock under the 2015 Plan and 2010 Plan, respectively. Options issued under the Plans are exercisable for up to 10 years from the date of issuance.
Options issued under the 2007, 2010, and 2015 Plan (collectively, the "Plans") are exercisable for up to 10 years from the date of issuance.
In March 2015, the Company's Board of Directors adopted, and the Company's shareholders subsequently approved, the Employee Stock Purchase Plan (the "ESPP"). The ESPP allows employees to buy company stock twice a year through after-tax payroll deductions at a discount from market. The board of directors initially authorized 187,500 shares for issuance under the ESPP. Commencing on the first day of fiscal 2016 and on the first day of each fiscal year thereafter during the term of the ESPP, the number of shares of common stock reserved for issuance shall be increased by the lesser of (i) 1% of our outstanding shares of Common Stock on such date, (ii) 50,000 shares or (iii) a lesser amount determined by the Board. In no event shall the aggregate number of shares reserved for issuance during the term of the ESPP exceed 1,250,000 shares.
The 2015 ESPP is considered a compensatory plan with the related compensation cost recognized over the six month offering period. As of December 31, 2015, approximately $49 of employee payroll deductions which have been withheld since July 1, 2015, the commencement of the offering period and are included in accrued expenses in the accompanying balance sheet. The compensation expense related to the 2015 ESPP for the year ended December 31, 2015 was $41. In January 2016, 6,948 shares that were purchased as of December 31, 2015 were issued under the 2015 ESPP.
74
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
12. STOCK OPTIONS (Continued)
Share-based compensation
For stock options issued and outstanding for the years ended December 31, 2015, 2014 and 2013, the Company recorded non-cash, stock-based compensation expense of $4,666, $2,730 and $3,136, respectively, net of forfeitures.
The fair value of each option award is estimated on the date of grant using the Black-Scholes option pricing model that uses the assumptions noted in the following table. Due to its limited operating history and limited number of sales of its Common Stock, the Company estimated its volatility in consideration of a number of factors including the volatility of comparable public companies. The Company uses historical data, as well as subsequent events occurring prior to the issuance of the financial statements, to estimate option exercises and employee terminations within the valuation model. The expected term of options granted under the Plans, all of which qualify as "plain vanilla," is based on the average of the contractual term (10 years) and the vesting period (generally, 48 months). For non-employee options, the expected term is the contractual term. The risk-free rate is based on the yield of a U.S. Treasury security with a term consistent with the option.
The assumptions used principally in determining the fair value of options granted were as follows:
| |
December 31, | |||||
|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||
Risk-free interest rate |
1.53 - 1.89% | 1.62 - 2.06% | 0.77 - 2.52% | |||
Expected dividend yield |
0% | 0% | 0% | |||
Expected term (employee grants) |
6.00 years | 6.03 years | 6.25 years | |||
Expected volatility |
116% | 124% | 102% | |||
A summary of option activity as of December 31, 2015 and changes for the year then ended are presented below:
Options
|
Shares | Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term in Years |
Aggregate Intrinsic Value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Outstanding at December 31, 2014 |
2,606,737 | $ | 6.58 | ||||||||||
Granted |
1,093,009 | $ | 8.67 | ||||||||||
Forfeited |
(92,997 | ) | $ | 8.66 | |||||||||
Exercised |
(353,439 | ) | $ | 4.28 | |||||||||
| | | | | | | | | | | | | | |
Outstanding at December 31, 2015 |
3,253,310 | $ | 7.47 | 8.08 | $ | 3,416 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Vested at December 31, 2015 |
1,349,752 | $ | 6.81 | 6.65 | $ | 2,459 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Vested and expected to vest at December 31, 2015 |
2,625,248 | $ | 7.37 | 7.82 | $ | 3,073 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The weighted average grant-date fair value of options granted during years ended December 31, 2015, 2014 and 2013 was $7.37, $6.15, and $7.76 per share, respectively. The total fair value of options that vested in years ended December 31, 2015, 2014, and 2013 was $5,144, $2,329 and $1,985, respectively. As of December 31, 2015, there was $8,193 of total unrecognized compensation expense,
75
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
12. STOCK OPTIONS (Continued)
related to non-vested share-based option compensation arrangements. The unrecognized compensation expense is estimated to be recognized over a period of 2.88 years at December 31, 2015.
13. WARRANTS
The following table presents information about warrants to purchase Common Stock issued and outstanding at December 31, 2015:
Year Issued
|
Classification | Number of Warrants |
Exercise Price |
Date of Expiration | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
2010 |
Equity | 354,342 | $ | 5.60 | 10/26/2017 - 12/3/2017 | |||||
2010 |
Equity | 314,882 | $ | 4.00 | 9/26/2015 - 12/3/2015 | |||||
2011 |
Equity | 85,785 | $ | 12.04 | 12/21/2016 | |||||
2012 |
Equity | 6,054 | $ | 6.64 | 10/5/2019 | |||||
2014 |
Liability | 395,716 | $ | 5.75 | 5/9/2019 | |||||
| | | | | | | | | | | |
Total |
1,156,779 | |||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted average exercise price |
$ | 5.71 | ||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted average life in years |
2.29 | |||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
14. EMPLOYEE BENEFIT PLAN
In November 2006, the Company adopted a 401(k) plan (the "Plan") covering all employees. Employees must be 21 years of age in order to participate in the Plan. Under the Plan, the Company has the option to make matching contributions. For the years ended December 31, 2015, 2014 and 2013, the Company made matching contributions in the form of shares of Common Stock. For the years ended December 31, 2015, 2014, and 2013, the Company issued 17,437, 41,753, and 19,721 shares of Common Stock, respectively, with related fair values of $201, $173, and $192, respectively, which were recorded as expense in the statement of operations.
15. INTELLECTUAL PROPERTY LICENSE
In July 2007, the Company entered into a world-wide exclusive license (the "BCH License") for patents co-owned by BCH and MIT initially covering the use of biopolymers to treat spinal cord injuries, and to promote the survival and proliferation of human stem cells in the spinal cord. During 2011, the BCH License was amended, and the Company obtained additional rights for use in the field of peripheral nerve injuries. The BCH License, as amended, has a 15- year term, or as long as the life of the last expiring patent right, whichever is longer, unless terminated earlier by the licensor, under certain conditions as defined in the related license agreement. In connection with the BCH License, the Company paid an initial $75 licensing fee and is required to pay certain annual maintenance fees, milestone payments and royalties. During 2011, the Company paid $75 to expand the license and, at December 31, 2011, accrued $50 for a milestone payment. License fees and milestone payments are capitalized and total $200 at December 31, 2015 (see Note 4). Maintenance and royalty costs are expensed as incurred.
76
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
16. COMMITMENTS AND CONTINGENCIES
Leases
On November 30, 2011 and as amended on September 17, 2012, the Company entered into a commercial lease for 26,150 square feet of office, laboratory and manufacturing space in Cambridge, Massachusetts (as amended, the "Cambridge Lease"). The term of the Cambridge Lease is six years and three months, with one five-year extension option. The terms of the Cambridge Lease require a standby letter of credit in the amount of $311 (see Note 2).
The Cambridge Lease contains rent holidays and rent escalation clauses. The Company recognizes rent expense on a straight-line basis over the term of the Cambridge Lease and records the difference between the amount charged to expense and the rent paid as a deferred rent liability. As of December 31, 2015, the amount of deferred rent liability is $391 and is included in accrued expenses.
It is the Company's policy to assess whether improvements made to the space rented under operating leases should be accounted for as "lessor" or "lessee" assets. Such costs are recorded as leasehold improvements, which are amortized to rent expense over the term of the Cambridge Lease. As of December 31, 2015, such leasehold improvements totaled $392 and are $185, net of accumulated depreciation.
Pursuant to the terms of the non-cancelable lease agreements in effect at December 31, 2015, the future minimum rent commitments are as follows:
Year Ended December 31,
|
|
|||
|---|---|---|---|---|
2016 |
$ | 1,263 | ||
2017 |
1,289 | |||
2018 |
1,092 | |||
| | | | | |
Total |
$ | 3,644 | ||
| | | | | |
| | | | | |
| | | | | |
Total rent expense for the years ended December 31, 2015, 2014, and 2013, including month-to-month leases, was $1,123, $1,148 and $1,125, respectively.
On September 4, 2013, the Company entered into a legal settlement agreement for $286 in connection with the Cambridge Lease. The settlement has been included in the deferred rent liability and the benefit will be amortized over the remainder of the term of the Cambridge Lease.
Litigation
Lawsuit with Former Employee
In November 2013, we filed a lawsuit against Francis Reynolds, our former Chairman, Chief Executive Officer and Chief Financial Officer, in Middlesex Superior Court, Middlesex County, Massachusetts (InVivo Therapeutics Holdings Corp. v. Reynolds, Civil Action No. 13-5004). The complaint alleges breaches of fiduciary duties, breach of contract, conversion, misappropriation of corporate assets, unjust enrichment, corporate waste, and seeks monetary damages and an accounting. The lawsuit involves approximately $500 worth of personal and/or exorbitant expenses that we allege Mr. Reynolds inappropriately caused us to pay while he was serving as our Chief Executive Officer, Chief Financial Officer, President and Chairman of our Board of Directors. On December 6, 2013,
77
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
16. COMMITMENTS AND CONTINGENCIES (Continued)
Mr. Reynolds answered the complaint, and filed counterclaims against us and the former members of our Board of Directors. The counterclaims allege two counts of breach of contract, two counts of breach of the covenant of good faith and fair-dealing, and tortious interference with a contract, and seek monetary damages and a declaratory judgment. The counterclaims involve Mr. Reynolds's allegations that we and the Board interfered with the performance of his duties under the terms of his employment agreement, and that Mr. Reynolds was entitled to additional shares upon the exercise of certain stock options. On January 9, 2014, we, along with the directors named in the counterclaims, filed our answer. The parties are currently conducting pre-trial discovery. No judgments or rulings are pending at this stage.
Shareholder Matters and Investigations
On July 31, 2014, a putative securities class action lawsuit was filed in the United States District Court for the District of Massachusetts, naming the Company and Mr. Reynolds, as defendants (the "Securities Class Action"). The lawsuit alleges violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements related to the timing and completion of the clinical study of the Company's Neuro-Spinal Scaffold implant. The plaintiff seeks class certification for purchasers of the Company's common stock during the period from April 5, 2013 through August 26, 2013 and unspecified damages. On April 3, 2015, the United States District Court for the District of Massachusetts dismissed the plaintiff's claim with prejudice.
On May 4, 2015, plaintiff filed a notice of appeal of this decision. A mandatory mediation conference was held on September 10, 2015. Following that conference, on October 5, 2015, plaintiff/appellant filed his opening brief with the United States Court of Appeals for the First Circuit. The Company and the individual defendants/appellees filed their answering brief on November 5, 2015, and plaintiff/appellant filed his reply brief on December 10, 2015. The Court of Appeals has scheduled oral argument for April 6, 2016.
On January 23, 2015, Shawn Luger, a purported shareholder of the Company, sent the Company a letter demanding that the Board of Directors take action to remedy purported breaches of fiduciary duties allegedly related to the claimed false and misleading statements that are the subject of the Securities Class Action (the "Shareholder Demand"). The Board of Directors completed its investigation of the matters raised in the Shareholder Demand and voted unanimously not to pursue any litigation against any current or former director, officer or employee of the Company with respect to the matters set forth in the Shareholder Demand.
On August 14, 2015, Shawn Luger filed a shareholder derivative lawsuit in the Superior Court of Suffolk County for the Commonwealth of Massachusetts on behalf of InVivo Therapeutics against certain present and former board members and company executives alleging the same breaches of fiduciary duties purportedly set forth in the Shareholder Demand. On February 5, 2016, the Superior Court of Suffolk County dismissed the plaintiff's claims with prejudice. The plaintiff's time to appeal the dismissal has not expired.
In addition to the derivative lawsuit and the appeal of the Securities Class Action, the Company received investigation subpoenas from the Boston Regional Office of the Securities and Exchange Commission ("SEC") and the Massachusetts Securities Division of the Secretary of the Commonwealth
78
InVivo Therapeutics Holdings Corp.
Notes to Consolidated Financial Statements (Continued)
(In thousands, except share and per-share data)
16. COMMITMENTS AND CONTINGENCIES (Continued)
of Massachusetts ("MSD") requesting corporate documents also concerning, among other topics, the allegations raised by the Securities Class Action and the Shareholder Demand. The Company responded to the MSD's subpoena on September 22, 2014 and October 8, 2014. On February 18, 2015, the Company received a second subpoena from the MSD requesting additional documents and information related to the same topics. The Company responded to this second subpoena on March 24, 2015. On October 21, 2015, the Company received a letter from the SEC notifying the Company that it has concluded its investigation of the Company and that it does not intend to recommend an enforcement action against the Company.
17. INSURANCE CLAIM
During the year ended December 31, 2014, the Company settled an insurance claim of $621 for business interruption that covered the disruption of the Company's operations at its facility in Cambridge, Massachusetts caused by water damage that occurred in September 2014. The insurance settlement reimburses the Company for costs incurred as a result of the disruption and is included as reduction of research and development expense in the consolidated statement of operations for the year ended December 31, 2014. The settlement receivable is included in other current assets in the consolidated balance sheet for the year ended December 31, 2014.
During the year ended December 31, 2013, the Company received insurance proceeds of approximately $1,100 from the settlement of a business interruption claim that covered the disruption of the Company's operations at its facility in Cambridge, Massachusetts caused by water damage that occurred in November 2012. The insurance settlement reimbursed the Company for costs incurred as a result of the disruption and is included as reduction of research and development expense in the consolidated statement of operations for the year ended December 31, 2013.
18. RELATED PARTY
The Company has entered into a consulting agreement with Dr. Robert Langer, a member of the Company's Scientific Advisory Board and a holder of over 5% of InVivo's common stock, for certain consulting services. Dr. Langer was one of the original co-founders of InVivo. Pursuant to the terms of the agreement, the Company has agreed to pay Dr. Langer $250 per year in consulting fees.
19. SUBSEQUENT EVENTS
The Company has evaluated all events or transactions that occurred after December 31, 2015. In the judgement of management, there were no material events that impacted the consolidated financial statements or disclosures.
79
Item 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
Item 9A. CONTROLS AND PROCEDURES
Evaluation of Our Disclosure Controls
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is (i) recorded, processed, summarized, and reported within the time periods specified in the SEC's rules and forms, and (ii) such information is accumulated and communicated to our management, our chief executive officer and our chief financial officer, to allow timely decisions regarding required disclosure. As of the end of the period covered by this Annual Report on Form 10-K, we carried out an evaluation, under the supervision and with the participation of our management, including our principal executive and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rule 13a 15(e) under the Exchange Act. Based on this evaluation, our principal executive officer and principal financial officer concluded that, as of December 31, 2015, our disclosure controls and procedures were effective.
Management's Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control over financial reporting is designed to provide reasonable assurances regarding the reliability of financial reporting and the preparation of our consolidated financial statements in accordance with U.S. generally accepted accounting principles, or GAAP. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree or compliance with the policies or procedures may deteriorate.
With the participation of our chief executive officer and our chief financial officer, our management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2015 based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework) ("COSO"). Based upon our assessment and the COSO criteria, management concluded that our internal control over financial reporting was effective as of December 31, 2015.
Limitations on Effectiveness of Controls and Procedures
Our management, including our chief executive officer and our chief financial officer, does not expect that our disclosure controls and procedures or our internal controls will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include, but are not limited to, the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. The design of any system of controls is also based, in part. upon certain assumptions about the likelihood of future events and there can be no assurance that any design will succeed in achieving its stated goals under all
80
potential future conditions. Over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
Changes in Internal Controls over Financial Reporting
During the fiscal quarter ended December 31, 2015, there have been no changes in our internal control over financial reporting that have materially affected or are reasonably likely to materially affect our internal controls over financial reporting.
81
None.
Item 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required under this Item is incorporated herein by reference to the information regarding directors, executive officers and corporate governance included in our proxy statement for our 2016 Annual Meeting of Stockholders.
Code of Ethics
We previously adopted a Code of Business Conduct and Ethics that applies to all employees, officers and directors of our Company, including our principal executive officer, principal financial officer and principal accounting officer or controller, or persons performing similar functions. Our Code of Business Conduct and Ethics is available in the "Investor Relations" section of our website at www.invivotherapeutics.com. A copy of our Code of Business Conduct and Ethics can also be obtained free of charge by contacting our Secretary, c/o InVivo Therapeutics Holdings Corp., One Kendall Square, Suite B14402, Cambridge, Massachusetts 02139. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding any amendment to, or waiver from, a provision of our Code of Business Conduct and Ethics by posting such information on our website.
Item 11. EXECUTIVE COMPENSATION
The information required under this Item is incorporated herein by reference to the information regarding executive compensation included in our proxy statement for our 2016 Annual Meeting of Stockholders.
Item 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required under this Item is incorporated herein by reference to the information regarding security ownership of certain beneficial owners and management and related stockholder matters included in our proxy statement for our 2016 Annual Meeting of Stockholders.
Item 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required under this Item is incorporated herein by reference to the information regarding certain relationships and related transactions and director independence included in our proxy statement for our 2016 Annual Meeting of Stockholders.
Item 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required under this Item is incorporated herein by reference to the information regarding principal accounting fees and services included in our proxy statement for our 2016 Annual Meeting of Stockholders.
82
Item 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
Financial Statements.
The financial statements listed in the Index to Consolidated Financial Statements appearing in Item 8 are filed as part of this report.
Financial Statement Schedules.
All financial statement schedules have been omitted as they are either not required, not applicable, or the information is otherwise included.
Exhibits.
The exhibits listed in the Exhibit Index immediately preceding the exhibits are filed as part of this report.
83
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| INVIVO THERAPEUTICS HOLDINGS CORP. | ||||||
Date: March 4, 2016 |
By: |
/s/ STEVEN F. MCALLISTER |
||||
| Name: | Steven F. McAllister | |||||
| Title: | Chief Financial Officer | |||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ MARK D. PERRIN Mark D. Perrin |
Chief Executive Officer and Chairman of the Board (Principal Executive Officer) | March 4, 2016 | ||
/s/ STEVEN F. MCALLISTER Steven F. McAllister |
Chief Financial Officer (Principal Financial and Accounting Officer) |
March 4, 2016 |
||
/s/ JOHN A. MCCARTHY, JR. John A. McCarthy, Jr. |
Director |
March 4, 2016 |
||
/s/ KENNETH DIPIETRO Kenneth DiPietro |
Director |
March 4, 2016 |
||
/s/ DANIEL R. MARSHAK Daniel R. Marshak |
Director |
March 4, 2016 |
||
/s/ C. ANN MERRIFIELD C. Ann Merrifield |
Director |
March 4, 2016 |
||
/s/ RICHARD J. ROBERTS Richard J. Roberts |
Director |
March 4, 2016 |
84
| 2.1 | Agreement and Plan of Merger, dated October 4, 2010, by and between Design Source, Inc. and InVivo Therapeutics Holdings Corp. (incorporated by reference from Exhibit 2.2 to the Company's Current Report on Form 8-K, as filed with the SEC on October 6, 2010). | ||
| 2.2 | Agreement and Plan of Merger and Reorganization, dated as of October 26, 2010, by and among InVivo Therapeutics Holdings Corp. (f/k/a Design Source, Inc.), a Nevada corporation, InVivo Therapeutics Acquisition Corp., a Delaware corporation and InVivo Therapeutics Corporation, a Delaware corporation (incorporated by reference from Exhibit 2.1 to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | ||
| 3.1 | Articles of Incorporation of InVivo Therapeutics Holdings Corp., as amended (incorporated by reference from Exhibit 3.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2011, as filed with the SEC on November 14, 2011). | ||
| 3.2 | Amended and Restated Bylaws of InVivo Therapeutics Holdings Corp. (incorporated by reference from Exhibit 3.1 to the Company's Current Report on Form 8-K, as filed with the SEC on April 24, 2012). | ||
| 3.3 | Certificate of Change Pursuant to NRS 78.209 filed with the Nevada Secretary of State, dated March 23, 2015 (incorporated by reference from Exhibit 3.1 to the Company's Current Report on Form 8-K, as filed with the SEC on March 24, 2015). | ||
| 4.1 | Form of Bridge Warrant of InVivo Therapeutics Corporation (incorporated by reference from Exhibit 4.1 to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | ||
| 4.2 | Form of Investor Warrant of InVivo Therapeutics Holdings Corp. (incorporated by reference from Exhibit 4.3 to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | ||
| 4.3(i) | Form of Warrant of InVivo Therapeutics Holdings Corp. ($1.00 exercise price) issued to Placement Agent (incorporated by reference from Exhibit 4.2 to the Company's Current Report on Form 8-K, as filed with the SEC on December 9, 2010). | ||
| 4.3(ii) | Form of Warrant of InVivo Therapeutics Holdings Corp. ($1.40 exercise price) issued to Placement Agent (incorporated by reference from Exhibit 4.3 to the Company's Current Report on Form 8-K, as filed with the SEC on December 9, 2010). | ||
| 4.4 | Form of Warrant of InVivo Therapeutics Holdings Corp. issued to Bridge Lenders (incorporated by reference from Exhibit 4.5 to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | ||
| 4.5 | Warrant dated June 17, 2011 issued to Square 1 Bank (incorporated by reference from Exhibit 4.7 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2011, as filed with the SEC on March 15, 2012). | ||
| 4.6 | Specimen Common Stock Certificate (incorporated by reference from Exhibit 4.8 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2011, as filed with the SEC on March 15, 2012). | ||
85
| 4.7 | Warrant dated October 5, 2012 issued to Massachusetts Development Finance Agency (incorporated by reference from Exhibit 4.1 to the Company's Current Report on Form 8-K, as filed with the SEC on October 9, 2012). | ||
| 4.8 | Form of New Warrant issued on May 17, 2013 in exchange for Merger Warrants (incorporated by reference from Exhibit (a)(1)(D)(1) to the Company's Tender Offer Statement on Schedule TO (File No. 005-85686), as filed with the SEC on April 8, 2013). | ||
| 4.9 | Form of New Warrant issued on May 17, 2013 in exchange for Placement Agent Warrants (incorporated by reference from Exhibit (a)(1)(D)(3) to the Company's Tender Offer Statement on Schedule TO (File No. 005-85686), as filed with the SEC on April 8, 2013) | ||
| 10.1 | * | InVivo Therapeutics Corp. 2007 Employee, Director and Consultant Stock Plan (incorporated by reference from Exhibit 10.9 to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | |
| 10.2(i) | * | Form of Incentive Stock Option Agreement by and between InVivo Therapeutics Corp. and participants under the 2007 Employee, Director and Consultant Stock Plan (incorporated by reference from Exhibit 10.11(i) to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | |
| 10.2(ii) | * | Form of Non-Qualified Stock Option Agreement by and between InVivo Therapeutics Corp. and participants under the 2007 Employee, Director and Consultant Stock Plan (incorporated by reference from Exhibit 10.11(ii) to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | |
| 10.3 | * | InVivo Therapeutics Holdings Corp. 2010 Equity Incentive Plan, as amended (incorporated by reference to Appendix A to the Company's Schedule 14A Proxy Statement, as filed with the SEC on April 19, 2013). | |
| 10.4(i) | * | Form of Incentive Stock Option Agreement by and between InVivo Therapeutics Holdings Corp. and participants under the 2010 Equity Incentive Plan (incorporated by reference from Exhibit 10.12(i) to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2010, as filed with the SEC on March 24, 2011). | |
| 10.4(ii) | * | Form of Non-Qualified Stock Option Agreement by and between InVivo Therapeutics Holdings Corp. and participants under the 2010 Equity Incentive Plan (incorporated by reference from Exhibit 10.12(ii) to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2010, as filed with the SEC on March 24, 2011). | |
| 10.5 | Form of Scientific Advisory Board Agreement entered into by InVivo Therapeutics Corp. (incorporated by reference from Exhibit 10.13 to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | ||
| 10.6 | Exclusive License Agreement dated July 2007 between InVivo Therapeutics Corporation and Children's Medical Center Corporation (incorporated by reference from Exhibit 10.1 to Amendment No. 2 to the Company's Quarterly Report on Form 10-Q/A for the quarter ended March 31, 2011, as filed with the SEC on July 18, 2011). | ||
| 10.7 | Amendment One to the Exclusive License, dated May 12, 2011, by and between Children's Medical Center Corporation and InVivo Therapeutics Corporation (incorporated by reference from Exhibit 10.22 to the Amendment No. 4 to the Company's Registration Statement on Form S-1/A (File No. 333-171998), as filed with the SEC on July 19, 2011). | ||
| 10.8 | Form of Indemnification Agreement (for directors and officers) (incorporated by reference from Exhibit 10.19 to the Company's Registration Statement on Form S-1 (File No. 333-171998), as filed with the SEC on February 1, 2011). |
86
| 10.9 | Lease Agreement, dated November 30, 2011, between InVivo Therapeutics Corporation and RB Kendall Fee, LLC (incorporated by reference from Exhibit 10.25 to the Company's Registration Statement on Form S-1 (File No. 333-178584), as filed with the SEC on December 16, 2011). | ||
| 10.10 | Lease Guaranty, dated November 30, 2011, by InVivo Therapeutics Holdings Corp. (incorporated by reference from Exhibit 10.26 to the Company's Registration Statement on Form S-1 (File No. 333-178584), as filed with the SEC on December 16, 2011). | ||
| 10.11 | First Amendment of Lease between InVivo Therapeutics Corporation and RB Kendall Fee, LLC, dated September 17, 2012 (incorporated by reference from Exhibit 10.31 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2012, as filed with the SEC on March 12, 2013). | ||
| 10.12 | Common Stock Purchase Warrant dated December 21, 2011 and issued by the Company to Ingenieria E Inversiones Ltda. (incorporated by reference from Exhibit 10.2 to the Company's Current Report on Form 8-K, as filed with the SEC on December 22, 2011). | ||
| 10.13 | * | InVivo Therapeutics Holdings Corp. Annual Cash Bonus Plan for Executive Officers (incorporated by reference from Exhibit 10.2 to the Company's Current Report on Form 8-K, as filed with the SEC on March 8, 2012). | |
| 10.14 | Promissory Note dated October 5, 2012 in favor of Massachusetts Development Finance Agency (incorporated by reference from Exhibit 10.1 to the Company's Current Report on Form 8-K, as filed with the SEC on October 9, 2012). | ||
| 10.15 | * | Employment Agreement, dated as of August 22, 2013, between the Company and Michael J. Astrue (incorporated by reference from Exhibit 10.26 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2013, as filed with the SEC on March 17, 2014). | |
| 10.16 | * | Employment Agreement, dated as of September 16, 2013, between the Company and Gregory D. Perry (incorporated by reference from Exhibit 10.27 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2013, as filed with the SEC on March 17, 2014). | |
| 10.17 | * | Employment Agreement, dated as of December 23, 2013, between the Company and Mark D. Perrin (incorporated by reference from Exhibit 10.28 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2013, as filed with the SEC on March 17, 2014). | |
| 10.18 | * | Employment Agreement, dated as of December 31, 2013, between the Company and Steven F. McAllister (incorporated by reference from Exhibit 10.29 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2013, as filed with the SEC on March 17, 2014). | |
| 10.19 | * | Amendment to the December 31, 2013 Employment Agreement, dated as of April 29, 2014, between the Company and Steven F. McAllister. | |
| 10.20 | * | Amended and Restated Employment Agreement, dated as of May 30, 2014, between the Company and Steven F. McAllister (incorporated by reference from Exhibit 10.1 to the Company's Current Report on Form 8-K, as filed with the SEC on May 30, 2014). | |
87
| 10.21 | * | Second Amended and Restated Employment Agreement, dated as of June 17, 2014, between the Company and Steven F. McAllister (incorporated by reference from Exhibit 10.1 to the Company's Current Report on Form 8-K, as filed with the SEC on June 23, 2014). | |
| 10.22 | Letter Agreement, dated as of December 10, 2014, between the Company and H.C. Wainwright & Co., LLC (incorporated by reference from Exhibit 10.1 to the Company's Current Report on Form 8-K, as filed with the SEC on January 29, 2015). | ||
| 10.23 | Securities Purchase Agreement, dated as of January 28, 2015, between the Company and the purchasers signatory thereto (incorporated by reference from Exhibit 10.1 to the Company's Current Report on Form 8-K/A, as filed with the SEC on January 29, 2015). | ||
| 10.24 | * | InVivo Therapeutics Holdings Corp. Employee Stock Purchase Plan (incorporated by reference from Exhibit 10.1 to the Company's Current Report on Form 8-K, as filed with the SEC on June 16, 2015). | |
| 10.25 | * | InVivo Therapeutics Holdings Corp. 2015 Equity Incentive Plan (incorporated by reference from Exhibit 10.2 to the Company's Current Report on Form 8-K, as filed with the SEC on June 16, 2015). | |
| 10.26 | * | Letter Agreement regarding Amendments to Employment Agreement, dated as of July 21, 2015, by and between Mark D. Perrin and InVivo Therapeutics Holding Corp. (incorporated by reference from Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2015, as filed with the SEC on November 4, 2015). | |
| 10.27 | * | Letter Agreement regarding Amendments to Employment Agreement, dated as of July 21, 2015, by and between Steven F. McAllister and InVivo Therapeutics Holdings Corp. (incorporated by reference from Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2015, as filed with the SEC on November 4, 2015). | |
| 10.28 | * | Employment Agreement, dated July 21, 2015, by and between Thomas R. Ulich, M.D and InVivo Therapeutics Holdings Corp. (incorporated by reference from Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2015, as filed with the SEC on November 4, 2015). | |
| 10.29 | * | Employment Agreement, dated August 3, 2015, by and between Tamara L. Joseph and InVivo Therapeutics Holdings Corp. (incorporated by reference from Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2015, as filed with the SEC on November 4, 2015). | |
| 21 | Subsidiaries of InVivo Therapeutics Holdings Corp. (incorporated by reference from Exhibit 21.1 to the Company's Current Report on Form 8-K, as filed with the SEC on November 1, 2010). | ||
| 23.1 | Consent of RSM US LLP | ||
| 23.2 | Consent of Wolf & Company, P.C. | ||
| 31.1 | Certification by the Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | ||
| 31.2 | Certification by the Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | ||
88
| 32.1 | Certification of Principal Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | ||
| 32.2 | Certification of Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | ||
| 101.INS | XBRL Instance Document. | ||
| 101.SCH | XBRL Taxonomy Extension Schema Document. | ||
| 101.CAL | XBRL Taxonomy Calculation Linkbase Document. | ||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document. | ||
| 101.LAB | XBRL Taxonomy Label Linkbase Document. | ||
| 101.PRE | XBRL Taxonomy Presentation Linkbase Document. |
- *
- Management contract or compensatory plan or arrangement filed in response to Item 15(a)(3) of Form 10-K.
89