Attached files
| file | filename |
|---|---|
| EX-21 - EX-21 - NovoCure Ltd | nvcr-ex21_305.htm |
| EX-32.1 - EX-32.1 - NovoCure Ltd | nvcr-ex321_306.htm |
| EX-31.2 - EX-31.2 - NovoCure Ltd | nvcr-ex312_308.htm |
| EX-32.2 - EX-32.2 - NovoCure Ltd | nvcr-ex322_307.htm |
| EX-10.25 - EX-10.25 - NovoCure Ltd | nvcr-ex1025_883.htm |
| EX-10.24 - EX-10.24 - NovoCure Ltd | nvcr-ex1024_673.htm |
| EX-31.1 - EX-31.1 - NovoCure Ltd | nvcr-ex311_309.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
x |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2015
or
|
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 001-37565
NovoCure Limited
(Exact Name of Registrant as Specified in Its Charter)
|
Jersey (State or Other Jurisdiction of Incorporation or Organization) |
98-1057807 (I.R.S. Employer Identification No.) |
|
|
|
|
Le Masurier House La Rue Le Masurier St. Helier, Jersey JE2 4YE (Address of Principal Executive Offices)
|
|
Registrant’s telephone number, including area code: +44 (0) 15 3475 6700
Securities registered pursuant to Section 12(b) of the Act:
|
|
Title of each class |
|
|
Name of each exchange on which registered |
|
|
Ordinary shares, no par value per share |
NASDAQ Global Select Market |
||||
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.:
|
Large accelerated filer |
o |
|
Accelerated filer |
o |
|
Non-accelerated filer |
x |
(Do not check if a smaller reporting company) |
Smaller reporting company |
o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The registrant was not a public company as of the last business day of its most recently completed second fiscal quarter and, therefore, cannot calculate the aggregate market value of its voting and non-voting common equity held by non-affiliates as of such date.
The number of shares of the registrant’s ordinary shares outstanding as of February 26, 2016 was 84,426,720.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement for its 2016 annual meeting of stockholders are incorporated by reference into Items 10, 11, 12, 13, and 14 of Part III of this Form 10-K. Such definitive proxy statement will be filed with the Securities and Exchange Commission within 120 days after the end of the registrant’s fiscal year ended December 31, 2015.
|
|
|
Page |
|
ii |
||
|
Item 1. |
1 |
|
|
Item 1A. |
31 |
|
|
Item 1B. |
59 |
|
|
Item 2. |
59 |
|
|
Item 3. |
59 |
|
|
Item 4. |
59 |
|
|
Item 5. |
61 |
|
|
Item 6. |
64 |
|
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
65 |
|
Item 7A. |
76 |
|
|
Item 8. |
77 |
|
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
107 |
|
Item 9A. |
107 |
|
|
Item 9B. |
107 |
|
|
Item 10. |
108 |
|
|
Item 11. |
108 |
|
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
108 |
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
108 |
|
Item 14. |
108 |
|
|
Item 15. |
109 |
|
|
112 |
||
i
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
In addition to historical facts or statements of current condition, this report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements contained in this report are based on our current plans, expectations, hopes, beliefs, intentions or strategies concerning future developments and their impact on us. Forward-looking statements contained in this report constitute our expectations or forecasts of future events as of the date this report was filed with the Securities and Exchange Commission and are not statements of historical fact. You can identify these statements by the fact that they do not relate strictly to historical or current facts. Such statements may include words such as “anticipate,” “will,” “estimate,” “expect,” “project,” “intend,” “should,” “plan,” “believe,” “hope,” and other words and terms of similar meaning in connection with any discussion of, among other things, future operating or financial performance, strategic initiatives and business strategies, regulatory or competitive environments, our intellectual property and delivery system research and development. In particular, these forward-looking statements include, among others, statements about:
|
|
· |
our research and development, clinical trial and commercialization activities and projected expenditures; |
|
|
· |
the further commercialization of Optune and our delivery system candidates; |
|
|
· |
our business strategies and the expansion of our sales and marketing efforts in the United States and in other countries; |
|
|
· |
the market acceptance of Optune and our other delivery systems by patients, physicians, third-party payers and others in the healthcare and scientific community; |
|
|
· |
our plans to pursue the use of TTFields delivery systems for the treatment of other solid tumor cancers; |
|
|
· |
our estimates regarding revenues, expenses, capital requirements and needs for additional financing; |
|
|
· |
our ability to obtain regulatory approvals for additional indications and any future delivery systems; |
|
|
· |
our ability to acquire the supplies needed to manufacture our delivery systems from third-party suppliers; |
|
|
· |
our ability to manufacture adequate supply; |
|
|
· |
our ability to secure adequate coverage from third-party payers to reimburse us for our delivery systems; |
|
|
· |
our ability to maintain and develop our intellectual property position; |
|
|
· |
our cash needs; and |
|
|
· |
our prospects, financial condition and results of operations. |
These forward-looking statements involve a number of risks and uncertainties (some of which are beyond our control) or other assumptions that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. Should one or more of these risks or uncertainties materialize, or should any of our assumptions prove incorrect, actual results may vary in material respects from those projected in these forward-looking statements. Some of these factors are described in Part I, Item IA, Risk Factors, of this Annual Report on Form 10-K. We do not intend to update publicly any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
ii
Below is a description of our business. Please refer to the “Glossary of terms” set forth at the end of this Item 1 as you review this section.
Overview
We are a commercial-stage oncology company developing a novel, proprietary therapy called TTFields for the treatment of solid tumor cancers. TTFields is a low-toxicity anti-mitotic treatment that uses low-intensity, intermediate frequency, alternating electric fields to exert physical forces on key molecules inside cancer cells, disrupting the basic machinery necessary for normal cell division, leading to cancer cell death. Physicians have typically treated patients with solid tumors using one or a combination of three principal treatment modalities—surgery, radiation and pharmacological therapies. Despite meaningful advancements in each of these modalities, a significant unmet need to improve survival and quality of life remains. We believe we will establish TTFields as a new treatment modality for a variety of solid tumors that increases survival without significantly increasing side effects when used in combination with other cancer treatment modalities.
We were founded in 2000 and operated as a development stage company through December 31, 2011. We initially received FDA approval for Optune, our first TTFields delivery system, in 2011 for use as a monotherapy treatment for adult patients with glioblastoma brain cancer, or GBM, following confirmed recurrence after chemotherapy. We have built a commercial organization and launched Optune in the United States, Germany, Switzerland and Japan, which we refer to as our currently active markets. In November 2014, our phase 3 pivotal trial of Optune in combination with chemotherapy for patients with newly diagnosed GBM met its endpoints after a protocol pre-specified interim analysis showed significant improvements in both progression free and overall survival.
In October 2015, we received U.S. Food and Drug Administration (FDA) approval to market and sell Optune for the treatment of adult patients with newly diagnosed glioblastoma in combination with temozolomide. We are actively marketing for that indication in the United States, Germany and Switzerland. In the same month, we also received CE mark for our newly designed second generation Optune system and have since made it available to all new patients in Europe. In December 2015, we submitted the registration dossier for premarketing approval of Optune to treat newly diagnosed GBM to the Japanese Pharmaceutical and Medical Device Agency (JPMDA). In the same month, we also filed a 180-day PMA supplement application with the FDA seeking approval to market the second generation of Optune in the United States for its approved indications. We believe that TTFields will transform the standard of care for patients with GBM.
The fourth quarter of 2015 was marked by substantial growth versus the prior quarter driven primarily by the October 2015 FDA approval for Optune in newly diagnosed glioblastoma. This growth was largely achieved prior to the peer-reviewed publication of the successful EF-14 newly diagnosed glioblastoma phase 3 pivotal trial in the Journal of the American Medical Association (JAMA) on December 15, 2015. 557 prescriptions were received in the fourth quarter of 2015, an increase of 109% versus the prior year quarter. 499 of the prescriptions were received in the U.S. and 58 were received outside of the United States, primarily in Europe. For the full year, 1,777 prescriptions were received in 2015, an increase of 151% versus 2014. 1,607 of the prescriptions were received in the U.S. and 170 were received outside of the U.S., primarily in Europe.
Prescriptions are a leading indicator of demand. The conversion of prescriptions to new patients is driven by the prescription fill rate and the time to fill. Our prescription fill rate for the year ended December 31, 2015 was 73%. The relationship between filled prescriptions and active patients is primarily driven by treatment duration. There were 605 active patients on Optune therapy at December 31, 2015, an increase of 169% versus December 31, 2014. 529 of the active patients were in the United States and 76 of the active patients were outside of the U.S., primarily in Europe.
We have researched the biological effects of TTFields extensively. Because TTFields are delivered regionally, act only on mitotic cells and are tuned to target cancer cells of a specific size, there is minimal damage to healthy cells. We believe our pre-clinical and clinical research demonstrates that TTFields’ mechanism of action affects fundamental aspects of cell division and can have broad applicability across a variety of solid tumors. We have demonstrated in pre-clinical studies that TTFields can offer additive or synergistic benefits in combination with radiation and chemotherapy, which may lead to greater efficacy than either modality alone, without appearing to potentiate the systemic toxicities of either radiation or chemotherapy. In addition to our clinical and commercial progress in GBM, we are currently planning or conducting clinical trials evaluating the use of TTFields in brain metastases, non-small-cell lung cancer (NSCLC), pancreatic cancer, ovarian cancer and mesothelioma.
1
We own all commercialization rights to TTFields in oncology, and have a patent and intellectual property portfolio that, as of December 31, 2015, consists of a total of 54 issued patents, including 36 issued in the United States, as well as 45 additional patent applications on file. We believe we will maintain exclusive rights to market TTFields for all solid tumor indications in our key markets through the life of our patents.
GBM—our first approved and commercialized indication
GBM is the most common and aggressive form of primary brain cancer. We estimate approximately 27,500 patients are diagnosed with GBM annually in the United States, the top five European Union markets and Japan. GBM has few effective treatment options at present and provides our first opportunity to transform the standard of care for a solid tumor cancer to include TTFields.
We launched Optune in the United States for the treatment of recurrent GBM in 2011 and more recently in our other currently active markets. The majority of recurrent GBM patients are treated at large academic cancer centers and our commercial organization historically focused on these centers. Following FDA approval of Optune for newly diagnosed GBM, we are expanding our commercial organization to cover community oncology practices, where we believe a majority of newly diagnosed GBM patients are being treated. As of December 31, 2015, we have trained physicians at 244 clinical centers in the United States, at 62 clinical centers in Europe, including 45 in Germany, and at 61 clinical centers in Japan. As of December 31, 2015, our global sales force consisted of 33 full-time employees, 31 in the United States and 2 in Europe.
We market Optune consistent with our FDA-approved labeling, which presents the data from the EF-14 phase 3 pivotal trial. The EF-14 trial randomized 695 patients to receive either temozolomide, the established standard of care chemotherapy for newly diagnosed GBM, or TTFields in combination with temozolomide. In November 2014, a protocol pre-specified analysis of the first 315 patients demonstrated the trial met its powered endpoints of significant extension of both progression free survival, or PFS, and overall survival, or OS, in patients treated with TTFields in combination with temozolomide versus temozolomide alone. The analysis results demonstrated that:
|
· |
patients treated with TTFields, in combination with temozolomide, in the intent-to-treat population, demonstrated a statistically significant increase in PFS compared to temozolomide alone (median PFS of 7.2 months compared to 4.0 months, hazard ratio=0.62, p=0.001); |
|
· |
patients treated with TTFields, in combination with temozolomide, in the as-treated population, demonstrated a statistically significant increase in OS compared to temozolomide alone (median OS of 20.5 months compared to 15.6 months, hazard ratio=0.66, p=0.004); and |
|
· |
the two-year survival rate among patients treated with TTFields in combination with temozolomide, in the as-treated population, was 48% compared to 32% among patients treated with temozolomide alone (p=0.0058). |
The trial’s independent data monitoring committee recommended that patients receiving temozolomide alone be allowed to cross over immediately to receive TTFields. Following FDA approval of this recommendation in December 2014, we allowed patients receiving temozolomide alone to cross over. In October 2015, we received FDA approval to market and sell Optune for the treatment of adult patients with newly diagnosed glioblastoma in combination with temozolomide. We are actively marketing for that indication in the United States, Germany and Switzerland and for the treatment of recurrent GBM in Japan.
The peer-reviewed results of the EF-14 trial were published in the JAMA in December 2015. The JAMA article concluded that adding TTFields to maintenance temozolomide chemotherapy significantly prolonged progression-free and overall survival in newly diagnosed GBM. We believe that TTFields will transform the standard of care for patients with GBM.
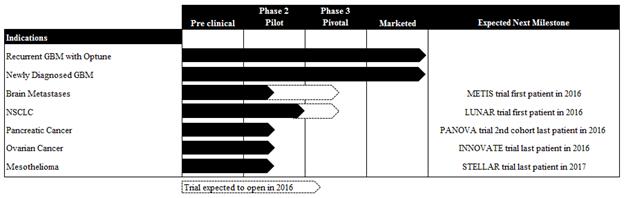
Our clinical pipeline
We have performed extensive pre-clinical research on TTFields and their effects in multiple solid tumor cancers. We believe we have gained a deep understanding of the underlying mechanism of action and the multiple pathways through which TTFields exert their effects within the dividing cancer cells. Our research shows that TTFields have an anti-mitotic effect in over 15 different solid tumor types in culture and in eight in vivo tumor models. In vitro and in vivo studies combining TTFields with radiation or chemotherapy, in multiple tumor types, have demonstrated at least additive efficacy, or stronger efficacy than the effect of either treatment alone, and in some cases synergistic efficacy, or stronger efficacy than the sum of the effects of both treatments. An increase in cancer cell sensitivity to chemotherapy when used in combination with TTFields in the range of one to two orders of magnitude suggests additivity, while an increase in the range of three to four orders of magnitude suggests synergism. Certain in vitro experiments using TTFields have suggested both additivity and synergism when used in combination with chemotherapy, as the presence of TTFields was shown to increase cancer cell sensitivity to chemotherapy from approximately 275 times to over 1,250 times depending on the
2
mechanism of action of the particular chemotherapy. The upper end of this range was observed in testing with taxane-based chemotherapies.
In recent preclinical research in an in vivo model of NSCLC in mice (called Lewis Lung Carcinoma), our researchers found that the use of TTFields in combination with a type of immunotherapy referred to as PD-1 inhibitors led to additive efficacy in the treatment of tumors in the lungs. PD-1 (programmed death receptor 1) functions primarily as a “stop signal” that limits immune cell effectiveness within a tumor by binding to its ligand, PDL1, expressed on many tumor cells. A PD-1 inhibitor seeks to interfere with that “stop signal” so that immune cells will continue to attack the tumor cells potentially leading to immune cell mediated destruction of tumor cells. Our researchers found that:
|
|
· |
TTFields did not lower immune cell infiltration into the tumor, potentially allowing for a full immune response to be mounted against the tumor cells once the PD1 pathway is blocked by PD1 inhibitors. |
|
|
· |
After one week of treating mice with NSCLC using TTFields directed to the lungs and/or a PD1 inhibitor, tumor volumes were lower for mice treated with the combination treatment versus either treatment alone at a statistically significant level (p<0.05); and |
|
|
· |
PDL1, the ligand which binds PD-1 on immune cells was maximized on the immune cells themselves in the combination treatment. This may indicate that the immune cells are protecting themselves from autoimmunity (attack of the immune system against normal cells in the body – in this case, the immune cells themselves) in the presence of TTFields. Increased PDL1 expression on immune cells is also a known indicator of increased immune activation. |
In separate pre-clinical research published in an abstract in November 2015, researchers at Tel-Aviv Medical Center found that activated immune cells treated with TTFields showed little or no decrease in several major immune activity parameters, including secretion of certain anti-tumoral molecules (known as cytokines), increased PD-1 expression on the immune cells seen when these cells are activated and degranulation (a process by which cells extrude internal substances into their surroundings, which is necessary for normal immune function of immune cells).
While we work to verify and expand on these results, this research to date indicates that TTFields in combination with PD-1 immunotherapies may be another possible treatment paradigm for solid tumors that should be investigated clinically, particularly for those solid tumors whose standard of care currently includes (or is evolving to include) PD-1 immunotherapies.
In addition to our clinical trials in GBM, we completed a phase 2 pilot trial in advanced NSCLC in 2010 and presented data on the first cohort of our phase 2 pilot trial in advanced pancreatic cancer in 2016. Both trials suggest an increase in PFS and OS for patients receiving TTFields in combination with chemotherapy compared to historical control data.
We believe our success in delaying disease progression and extending survival in GBM patients, our pre-clinical data and our early clinical data in additional indications validate the potential of TTFields to become a new therapeutic modality for a variety of solid tumors. We have developed a pipeline strategy to advance TTFields through phase 2 pilot and phase 3 pivotal trials across multiple solid tumor types, and anticipate expanding our clinical pipeline over time to study the safety and efficacy of TTFields for additional solid tumor indications.
Our competitive advantages
We believe our key competitive advantages are:
3
|
solid tumors with an annual incidence of approximately 1.1 million people in the United States alone. Currently, we have ongoing and completed clinical trials for indications with an incidence of approximately 350,000 people annually in the United States. We believe that the global incident population of target solid tumors provides us with significant additional commercial opportunities. |
|
· |
Immediate commercial opportunity for Optune in GBM. We are currently marketing Optune for the treatment of both newly diagnosed and recurrent GBM in the United States, Germany and Switzerland and for the treatment of recurrent GBM in Japan. We expect that TTFields will transform the standard of care for patients with GBM. |
|
· |
Pipeline of phase 2 pilot trials in five additional indications. In addition to our GBM clinical programs, we have invested in a variety of clinical programs in other solid tumors. We have completed a phase 2 trial in NSCLC, and are currently enrolling patients in phase 2 pilot trials for brain metastases, pancreatic cancer, ovarian cancer and mesothelioma. We expect to continue investing in our pipeline over time to broaden our commercial opportunity. |
|
· |
Established commercial organization and supply chain. We have established our commercial organization and believe we have the experience, expertise and infrastructure to scale our sales and marketing efforts in our key markets. In addition to our commercial organization, we believe we have established a scalable supply chain. |
|
· |
Significant barriers to entry. We own all commercialization rights to TTFields in oncology and have a patent and intellectual property portfolio that, as of December 31, 2015, includes 54 issued patents, 36 of which are issued in the United States, as well as 45 additional patent applications on file. We have patent protection through 2031 in the United States and through 2026 in other key markets. We believe we will maintain exclusive rights to TTFields for all solid tumor indications in our key markets for the life of our patents. In addition, even after the expiration of our U.S. patents, potential market entrants applying low-intensity, alternating electric fields to solid tumors in the United States will have to undertake their own clinical trials and PMA submissions to the FDA to demonstrate equivalence to TTFields to market a competing product. |
Our strategies for growth
Our objective is to establish TTFields as a new modality for the treatment of a variety of solid tumors. Our key strategies include the following:
|
· |
Drive adoption of Optune in GBM. We plan to use the data from our phase 3 pivotal EF-14 trial and our commercial organization to transform the standard of care for patients with GBM and to drive adoption of Optune by physicians and patients. We are expanding our direct sales force to call on physicians, including medical and radiation oncologists, who treat newly diagnosed GBM patients. We expect to further expand our commercial organization following regulatory approvals for additional indications. |
|
· |
Advance clinical development of TTFields. We plan to advance our clinical pipeline and evaluate other solid tumor indications that we believe can be targeted with TTFields. |
|
· |
Evaluate the use of TTFields in combination with other solid tumor therapies. We are conducting research and we are supporting independent research into the optimal combinations of TTFields with radiation or pharmacological therapies to expand the population of patients who may benefit from TTFields. For example, we believe that TTFields may be combined with radiation or chemotherapy to allow for dose reductions, leading to reduced toxicity while achieving the same or better treatment outcomes. |
|
· |
Continue to improve our TTFields delivery systems. We plan to continue to develop and enhance our TTFields delivery systems, which include our proprietary NovoTAL software, to improve performance and to provide the optimal patient experience across a variety of approved and potential clinical indications. |
Cancer and solid tumors
Cancer is a disease characterized by unregulated growth of abnormal cells. Normal cells are preprogrammed with genetic information informing them of their function throughout the body. Cells reproduce using a process called mitosis, creating genetic copies of themselves. When normal cells become damaged, the body uses several repair processes to restore function. When normal cells cannot be repaired, they undergo a process of preprogrammed cell death, or apoptosis. Cancer cells avoid the body’s repair and apoptosis pathways and undergo uncontrolled rapid replication, which can lead to the formation of a tumor.
Today, solid tumors are typically treated using one or a combination of three principal treatment modalities—surgery, radiation and pharmacological therapies.
4
|
More often, surgery is used to reduce the size of a tumor prior to the initiation of additional treatment modalities, such as radiation and pharmacological therapies. |
|
· |
Radiation—Radiation is a non-invasive solid tumor therapy that transfers energy to tissues, causing damage to biologically important molecules such as DNA. Radiation kills tumor cells or slows their growth when delivered at high doses. Radiation may be given before, during or after surgery and may also be given before, during or after other tumor treatments to shrink the tumor or to kill tumor cells that might remain. While effective in killing most solid tumors, radiation injures healthy tissues, leading to numerous potential toxic side effects, including bone marrow suppression and inflammation of the esophagus and the mucosal lining of the gastrointestinal tract. These side effects typically result in significant weakening of the patient, discomfort, nausea, vomiting and immune compromise. In the brain, the cumulative dose of radiation is limited due to long-term side effects on normal brain function, including cognitive decline and memory impairment. Advances in radiation have focused largely on limiting the exposure of healthy tissues to toxic radiation. |
|
· |
Pharmacological therapies—Chemotherapy, one of the earliest pharmacological tumor therapies, kills rapidly proliferating tumor cells by interacting with specific molecular pathways critical to DNA replication or cell reproduction, including mitosis. Chemotherapy acts in an indiscriminate manner, killing all dividing cells, including healthy as well as tumor cells, leading to a variety of side effects. In addition, many solid tumors reside in areas of the body that have poor accessibility to systemically delivered chemotherapies. For example, in GBM, the blood-brain barrier reduces the access of chemotherapies to the tumor. |
Newer agents, such as targeted cancer therapies, are intended to block the growth and spread of cancer by interfering with specific molecules, or molecular targets, in cancer cells. Targeted therapies are designed to kill cancer cells that express the target while attempting to spare normal cells. Targeted therapies have added some benefit to the treatment of solid tumors. However, they have a significant limitation because normal cells often possess variations of the target, leading to damage to healthy tissues. Targeted therapies are often used in combination with one or more traditional chemotherapies to increase efficacy, which can lead to expanded side effects. In addition, tumor cells often develop resistance to both chemotherapies and targeted therapies through mutations, which renders these therapies less effective or ineffective over time.
More recently, immuno-oncology has emerged as a promising therapy for solid tumors. This approach aims to harness a patient’s immune system to fight solid tumors and has shown extension of survival in certain solid tumors, such as previously treated metastatic malignant melanoma and NSCLC patients, but can also lead to serious side effects in some patients. Similar to the advancements in the development of targeted therapies for cancer, it is widely anticipated that immuno-oncologic approaches to treat solid tumors will be used in combination with existing and newer treatments to increase their overall effectiveness and overcome tumor resistance pathways.
Surgery, radiation and pharmacological therapies have been used as monotherapies or in combination to treat solid tumors for over 100 years. While significant advancements have been made to each of these treatment modalities, they still represent an imperfect solution to cancer care in terms of efficacy and side effects.
Tumor treating fields (TTFields)
TTFields consist of low-intensity, alternating electric fields that operate at intermediate frequencies, changing polarity hundreds of thousands of times a second. TTFields’ anti-mitotic mechanism of action is based on disruption of key electrically charged molecules essential to the mitotic process by which all cells divide. Interference with these key molecules leads to cell death through multiple pathways.
Cell reproduction begins with the replication of the cell’s genetic content. The cell’s DNA is copied to produce two identical copies. Following DNA replication, the cell enters mitosis, a well-orchestrated series of events that lead to the formation of two identical progeny of the original reproducing cell, called daughter cells. Each of the newly formed daughter cells has all the necessary molecular and genetic content to reproduce itself.
At the early stages of mitosis, a geometrically organized set of molecular strands or ropes is formed at two opposing ends of the cell, by self-assembly of many thousands of molecules called tubulin dimers. This mitotic spindle acts as a molecular motor to move pairs of exact copies of DNA to the equator of the cell. After the DNA is organized in one plane in the middle of the cell, the mitotic spindle begins to shorten, pulling one copy of the DNA to each side of the reproducing cell. In parallel to the formation of the mitotic spindle, a circular band, known as a cytokinetic band, forms on the membrane exactly surrounding the DNA plane at the equator of the cell. The location of this band is determined by a group of molecules called septins that are guided by the cell to specific locations on the cell membrane during mitosis. Once the DNA has been pulled to the two opposing ends of the cell by the mitotic spindle, the cytokinetic band begins to contract, physically pinching the cell into an hour glass shape with a narrow bridge between the two forming cells, called the mitotic furrow. At the end of mitosis, the furrow narrows until the membrane between the two forming daughter cells is pinched apart and disconnects in a process called cytokinesis.
5
There are two well established physical processes that TTFields use to exert their anti-mitotic effect: alignment of large molecules with the direction of the applied field and physical displacement of molecules and organelles.
|
· |
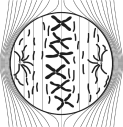
At early stages of mitosis, after the cell has made an exact copy of its genetic content, referred to as metaphase, the mitotic spindle is formed. The mitotic spindle acts as a group of molecular ropes that grab the two copies of the genetic content and pull each copy to an opposite side of the dividing cell. These ropes are formed by self-assembly of many thousands of identical molecules called tubulin dimers. Electrically, tubulin dimers have one highly positive end and one highly negative end. When TTFields are activated, the intracellular environment experiences a uniform electric field and tubulin dimers align with the field instead of with the mitotic spindle. This in turn does not allow the spindle to form properly, and the cells often cannot complete the division process. Cells that cannot complete division and remain arrested in mitosis will ultimately undergo programed cell death, known as apoptosis. In addition, the fact that the mitotic spindle does not form properly leads to improper separation of the two copies of the genetic content into two groups. If these cells do complete mitosis and split into two daughter cells, the genetic content may no longer be evenly divided between the daughter cells. The resulting daughter cells with incomplete genetic content can no longer replicate and will eventually die. |
|
The application of TTFields to the cell during metaphase aligns the tubulin dimers in the direction of the fields, disrupting the formation of the mitotic spindle, and leads to arrested mitosis and subsequent apoptosis
|
|
|
|
|
|
|
· |
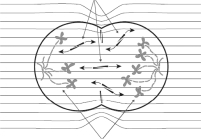
In order for the cell to split physically into two cells at the later stages of mitosis, referred to as anaphase, a cytokinetic band forms on the cell surface or membrane. This cytokinetic band must be placed exactly at the equator of the cell, so that when it contracts it will pinch the membrane into two identical daughter cells, each containing one exact copy of the same genetic material. The precise localization of this band depends on septins, which signal its location on the cell surface. Septins, like tubulin dimers, have one negative and one positive end, making them a target for TTFields. By rotating septins and aligning them in the direction of the field, TTFields lead to improper localization of the cytokinetic band, so that when the cell enters cytokinesis and the band receives a signal to contract, the cell is now torn into multiple small bubbles, or blebbing, instead of two equally sized daughter cells, leading to cell death. |
|
The application of TTFields to the cell during anaphase aligns septins in the direction of the fields, leading to improper formation of the cytokinetic band and subsequent membrane blebbing |
|
|
|
|
|
|
· |
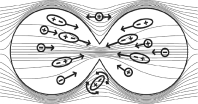
In cells that successfully proceed through the above-described stages of mitosis, the hourglass shape that forms when the cytokinetic band contracts at the cell equator causes the cell to experience a non-uniform electric field. In a non-uniform electric field many macromolecules and intracellular organelles experience electric forces pushing them toward the area of higher field intensity. During cytokinesis, the field intensity is highest at the center of the cell where the membrane is pinching off into two daughter cells, an area called the mitotic furrow. TTFields concentrate macromolecules and organelles toward the mitotic furrow, leading to structural disruption and cell death. |
|
The application of TTFields to the cell during anaphase and telophase, or cytokinesis, pushes charged and polar subcellular structures towards the mitotic furrow between the dividing cells, leading to cell destruction |
The intensity of the electric field within the cell depends on the frequency of the applied TTFields. Optimal tuning of field frequency to the specific cell type increases electric field intensity and non-uniformity within the cell, maximizing TTFields’ anti-mitotic effect. Pre-clinical data has shown that TTFields’ effects on different tumor cells are specific to the frequency of the electrical field. The optimal frequency for each cell type is dictated by physical properties of the cell, including cell size (which is inversely related to the optimal frequency), membrane thickness, resistance and capacitance. Tumor cells are typically a different size than the normal surrounding cells and may also exhibit other differences in membrane properties. We believe that the ability to frequency tune TTFields is a significant factor in our ability to deliver treatment to the solid tumor site without harming surrounding tissue and normal cell growth.
6
A TTFields delivery system includes a portable electric field generator, transducer arrays, rechargeable batteries and accessories. The electric fields are delivered through the non-invasive, insulated transducer arrays that are placed directly on the skin in the region surrounding the tumor. The therapy is designed to be delivered continuously throughout the day and night.
The portable field generator is designed to allow patients to go about their daily activities while receiving continuous cancer treatment. Transducer arrays are connected to the electric field generator to deliver therapy. Transducer arrays are made of ceramic discs with a very high dielectric constant that are capable of efficient delivery of TTFields into the body and incorporate precision temperature sensors designed to ensure safety. The self-adhesive transducer arrays are placed on the skin after shaving any hair in the treatment area. The sterile, single-use transducer arrays are changed when hair growth or hydrogel dissipation reduces array adhesion to the skin, which is typically two to three times per week for our GBM patients. Each battery provides two to three hours of therapy per charge. The field generator can be run from a standard power outlet for use when the patient is sleeping or stationary. We provide the patient with a specially designed bag to carry the electric field generator and a battery.
We plan to use the same field generator technology across all solid tumor indications for which TTFields are approved. We will specifically target individual solid tumor types by tuning TTFields to the appropriate frequency based on tumor cell size and adjusting the output power to treat the required tissue volume. Our transducer arrays have been developed and are in use, either commercially or clinically, for application on the head, chest and abdomen.
In October 2015, we received CE Mark for our newly designed second generation of Optune, which is less than half the weight and size of the current version, and have since made it available to all new patients in Europe. In December 2015, we filed a 180-day PMA supplement application with the FDA seeking approval of the second generation of Optune for the approved indications. Assuming that we do not receive comments or requests for additional information from the FDA, we hope to begin marketing the second generation Optune system in the United States in the third quarter of 2016. We plan to continue to improve all aspects of TTFields delivery to improve ease of use for patients.
TTFields penetrate the entire volume of tissue between the arrays. Unlike other forms of energy, such as radiation, the strength of the fields does not attenuate over distance. The distribution of the field within a certain part of the body depends on the exact layout of the transducer arrays and the passive electrical properties, mainly resistance, of the different tissues between them. Physicians or company personnel optimize the placement for each patient using a proprietary software package called NovoTAL, based on morphometric measurements of the patient’s anatomy according to a recent MRI scan and the location of the tumor.
Benefits of TTFields
We believe TTFields offer a number of distinct benefits that will lead to its establishment as a principal solid tumor treatment modality alongside surgery, radiation and pharmacological therapies, including:
|
· |
Targeted effect on solid tumors. We believe TTFields have a targeted effect on dividing solid tumor cells and limited effect on healthy tissues due to their mechanism of action and regional delivery. |
|
|
· |
Acts only on mitotic cells. Based on our research, TTFields do not appear to damage non-mitotic cells since the highly charged tubulin and septin proteins, which the TTFields target, are not assembled when a cell is not in mitosis. In addition, the hourglass shape of cells undergoing cytokinesis is not seen in non-dividing cells. We believe this lack of impact on non-mitotic cells is a significant factor in TTFields’ mild side effect profile as a monotherapy and the limited incremental side effects when used in combination with other cancer treatment modalities. In contrast, radiation and chemotherapy do not differentiate well between healthy cells and rapidly dividing tumor cells, causing damage to healthy tissues. |
|
|
· |
Specific to a certain size. TTFields are tuned to target cells of a certain size and with specific membrane properties. Healthy cells in the tissues surrounding or adjacent to a tumor often have different sizes and/or membrane properties than the tumor cells themselves, which leads us to believe that these healthy dividing cells are only minimally affected by TTFields. In contrast, while many chemotherapies also target only dividing cells, they often do not differentiate between dividing cells in healthy surrounding tissues and tumor cells, leading to side effects like hair loss, mucositis and bone marrow suppression. |
|
|
· |
Regional delivery. TTFields are regionally delivered to the tumor site rather than systemically delivered throughout the body. As a result, the parts of the body not covered by TTFields are generally not affected, and no systemic toxicities have been observed to date. In contrast, chemotherapy generally is systemic. As it circulates throughout the body, it does not discriminate between healthy tissues and tumors, causing systemic side effects. |
7
|
· |
Access to sanctuary sites. Certain organs in the body are considered sanctuary sites, since chemotherapy does not enter these organs at sufficiently efficacious doses. For example, chemotherapy doses are limited in the brain due to the blood brain barrier and in the pancreas due to stromal effects. TTFields are not delivered through the bloodstream and can be applied to both the brain and the pancreas, overcoming this known limitation of chemotherapy. |
|
· |
Complementary to other treatment modalities. We believe TTFields may be combined with existing and future treatments for many solid tumors, offering the potential for more effective, safer treatment. In our pre-clinical and clinical experience to date, TTFields do not appear to potentiate the systemic toxicities of radiation or chemotherapy when administered in combination with either treatment. Also, pre-clinical evidence has shown that the combination of TTFields with radiation, immune therapy or chemotherapy may lead to additive or synergistic efficacy. For example, certain in vitro experiments using TTFields have suggested both additivity and synergism when used in combination with chemotherapy, as the presence of TTFields was shown to increase cancer cell sensitivity to chemotherapy from approximately 275 times to over 1,250 times depending on the mechanism of action of the particular chemotherapy. The upper end of this range was observed in testing with taxane-based chemotherapies. |
GBM—our first approved and commercialized indication
The first indication for TTFields that we pursued was GBM, the most common form of primary brain cancer. GBM is an aggressive disease for which there are few effective treatment options. Prior to the approval of Optune for the treatment of newly diagnosed GBM,,the median overall survival in that patient population was approximately 15 months with standard therapies. TTFields represents the first improvement in clinical outcomes in the treatment of patients with GBM in over 10 years. We received FDA approval for Optune in 2011 for use as a monotherapy treatment for adult patients with recurrent GBM. In November 2014, we presented results from the protocol pre-specified analysis of our phase 3 pivotal trial of TTFields for patients with newly diagnosed GBM. This analysis demonstrated a significant improvement in progression free survival and overall survival. In October 2015, we received FDA approval to market and sell Optune for the treatment of adult patients with newly diagnosed glioblastoma in combination with temozolomide. We have commenced our commercial launch for that indication in the United States, Germany and Switzerland and for the treatment of recurrent GBM in Japan.
We estimate that approximately 12,500 people are diagnosed with GBM or tumors that typically progress to GBM in the United States each year. Of this population, we estimate that approximately 9,300 patients are candidates for treatment with Optune based on the rate of disease progression and medical eligibility..
We estimate that approximately 13,500 patients in the top five EU markets (France, Germany, Italy, Spain and the United Kingdom) are diagnosed with GBM or tumors that typically progress to GBM each year. Of this population, we estimate that approximately 10,000 patients are candidates for treatment with Optune based on the rate of disease progression and medical eligibility.
We estimate that approximately 1,500 patients in Japan are diagnosed with GBM or tumors that typically progress to GBM each year. Of this population, we estimate that approximately 1,100 patients will be candidates for treatment with Optune for newly diagnosed GBM once approved by the JPMDA. Of that population, we estimate that approximately 650 patients are candidates for treatment with Optune for recurrent GBM, Optune’s currently-approved indication in Japan.
The last clinical trial to show a statistically significant survival benefit in GBM was published in 2005 when concomitant and adjuvant temozolomide was added to the prior standard of care of surgical resection, followed by adjuvant radiotherapy. The median overall survival for radiation was 12.1 months versus 14.6 months with radiation plus temozolomide. Since 2005, temozolomide has become the standard of care chemotherapy for newly diagnosed GBM and all subsequent Phase 3 GBM trials have included temozolomide in the control arm. Standard of care temozolomide in these Phase 3 trials has consistently shown a median overall survival of approximately 15 months and a median two-year survival of less than 30%. No significant advances in GBM patient survival have been made since 2005, and a significant unmet need to improve survival and quality of life remains.
8
GBM was an optimal initial target for TTFields because the tumor rarely metastasizes outside the brain, allowing us to evaluate the effect of TTFields on the entire scope of the disease. We began to evaluate the use of TTFields for the treatment of GBM in 2004. We initially ran a phase 2 pilot clinical trial, EF-07, which included 10 recurrent GBM patients who were treated with TTFields alone as salvage therapy and 10 newly diagnosed GBM patients who were treated with a combination of TTFields and maintenance temozolomide after having undergone surgery and radiation with adjuvant temozolomide. Median time to disease progression for the recurrent GBM patients was 26.1 weeks and median overall survival was 62.2 weeks, more than double the reported medians of historical control patients. Median overall survival for the newly diagnosed GBM patients was greater than 39 months in the TTFields in combination with temozolomide arm versus 14.6 months for matched historical control patients who received maintenance temozolomide alone. As of December 1, 2014, four of the 20 patients were alive more than eight years after receiving TTFields in the EF-07 trial. Based on the promising results of the EF-07 trial, we conducted two randomized phase 3 pivotal trials for TTFields in GBM:
|
· |
EF-11 to evaluate TTFields as a monotherapy for the treatment of patients with recurrent GBM, which we believe established clinical validation and enabled commercial proof of concept; and |
|
· |
EF-14 to evaluate TTFields in combination with temozolomide for the treatment of patients with newly diagnosed GBM, which we believe will transform the standard of care for patients with newly diagnosed and recurrent GBM. |
Our phase 3 pivotal trial in recurrent GBM (EF-11)
We received FDA approval in 2011 to market Optune for use as a monotherapy treatment for adult patients with recurrent GBM. The FDA approved Optune based on the EF-11 trial, which was a randomized, active standard of care controlled phase 3 pivotal clinical trial. While the trial did not achieve its primary endpoint of superiority, the trial results indicate that monotherapy treatment with Optune provides patients with clinically comparable extension of survival compared to chemotherapy and that patients treated with Optune alone had significantly fewer side effects and an overall better quality of life than patients treated with chemotherapy alone.
|
· |
Overview—The EF-11 trial was a multicenter, randomized (1:1), active controlled clinical trial of 237 adults with recurrent GBM. Participants received either TTFields as a monotherapy (n=120) or the physician’s choice of chemotherapy (n=117). Chemotherapies chosen for the active control arm included mainly bevacizumab, nitrosureas and temozolomide. More than 80% of patients had failed two or more prior lines of chemotherapy and 20% of the patients had failed bevacizumab prior to enrollment. The primary endpoint for the trial was OS. The secondary endpoints included progression free survival at six months, or PFS6, radiological response rate, one-year survival rate, adverse event severity and frequency and quality of life. |
|
· |
Efficacy—Overall survival times for patients treated with TTFields alone and active chemotherapy were 6.6 months and 6.0 months, respectively (hazard ratio = 0.86; p=0.27). PFS was not significantly different between the groups and PFS6 was numerically higher in the TTFields arm (21.4% vs. 15.2%). The FDA determined that these results represented clinically comparable efficacy outcomes. The overall radiographic response rate was higher for the group treated with TTFields compared with the group treated with chemotherapy (14.0% vs. 9.6%, respectively; p=0.19). Three patients treated with TTFields had complete responses compared to no patients treated with active chemotherapy. A group of principal investigators from the trial published long-term follow-up data on the trial indicating that 8% of the TTFields-treated patients had a long-term survival at 48 months compared to no long-term survivors in the chemotherapy treated group. |
The EF-11 trial demonstrated that patient compliance is important for successful outcomes. Patients who used TTFields more than 75% of the time had a significant survival advantage compared to those who used it less than 75% of the time (median survival was 7.8 months compared to 4.5 months, respectively; p<0.05).
|
· |
Safety and quality of life—Patients treated with TTFields experienced significantly fewer treatment-related adverse events than those treated with active chemotherapy. Specifically, there were significantly fewer hematological, infectious and gastrointestinal adverse events in the TTFields-treated patients than in those treated with chemotherapy. The most commonly reported side effect from the delivery of TTFields was a mild-to-moderate rash on the skin beneath the transducer arrays, which affected 16% of patients. Patients receiving TTFields reported better quality-of-life scores compared to patients treated with active chemotherapy. Importantly, patients reported better quality-of-life outcomes specifically related to cognitive and emotional functioning. |
9
Our commercial registry (PRiDe)
At the time of our initial commercial launch of Optune for recurrent GBM in 2011, we established a patient registry aimed at capturing information related to the use of TTFields in the real-world commercial setting, which we refer to as PRiDe. We collected Optune treatment data and OS data from all 457 recurrent GBM patients who commenced treatment with Optune in the United States between October 2011 and November 2013. Key findings from this peer-reviewed published data include:
|
· |
Compelling overall efficacy—Median OS was significantly greater with TTFields in PRiDe than in the EF-11 trial (9.6 months vs. 6.6 months; p=0.0003). OS rates were more than double for TTFields patients in PRiDe than in the EF-11 trial (one-year: 44% vs. 20%; two-year: 30% vs. 9%); |
|
· |
Efficacy correlated to compliance—Patients for whom compliance data was available (n=287) who used Optune more than 75% of the time (the recommended minimum is 18 hours per day) had a significant survival advantage compared to those who used it less than 75% of the time (median survival was 13.5 months compared to 4.0 months, respectively; p<0.0001); and |
|
· |
Consistent safety profile—No unexpected adverse events were detected in PRiDe. As in the EF-11 trial, the most frequent side effects were mild to moderate skin reactions associated with application of the transducer arrays. |
Our phase 3 pivotal trial in newly diagnosed GBM (EF-14)
We began enrolling patients at 80 centers in the EF-14 phase 3 pivotal trial in 2009 to study the efficacy and safety of TTFields in combination with temozolomide for the treatment of newly diagnosed GBM in comparison with temozolomide alone. The primary endpoint of the trial was PFS and a powered secondary endpoint was OS. A protocol pre-specified analysis of the EF-14 phase 3 pivotal trial was presented in November 2014. The analysis demonstrated that TTFields met both of the trial’s powered endpoints with significant extension of both PFS and OS. The analysis was conducted by the trial’s independent data monitoring committee on the first 315 patients with a minimum of 18 months follow-up. In October 2015, we received FDA approval to market and sell Optune for the treatment of adult patients with newly diagnosed glioblastoma in combination with temozolomide. In December 2015, the EF-14 results were published in JAMA. The JAMA article concluded that adding TTFields to maintenance temozolomide chemotherapy significantly prolonged progression-free and overall survival in newly diagnosed GBM.
Clinical trial design
The EF-14 phase 3 pivotal trial enrolled newly diagnosed GBM patients following completion of concomitant radiation with temozolomide. Patients were randomized 2:1 to receive either continuous TTFields in combination with monthly maintenance temozolomide or maintenance temozolomide alone. Randomization was stratified by extent of resection (biopsy, partial resection or gross total resection) and O(6)-methylguanine-DNA methyltransferase, or MGMT, methylation status, which are both known prognostic factors in newly diagnosed GBM. In prior clinical studies, positive MGMT status was correlated with better survival outcomes for newly diagnosed GBM patients treated with radiation and temozolomide. Patients on both arms of the study had follow up MRIs performed once every two months until treatment termination. MRIs were assessed for progression by a blinded central radiology group. Upon disease progression or temozolomide toxicity, patients were allowed to change to a second line chemotherapy. Patients on TTFields continued to receive TTFields until the earlier of the second disease progression or 24 months. Analysis of PFS was performed in the intent-to-treat population, based on a blinded review of patient MRIs. Analysis of OS was analyzed in a pre-specified as-treated population, which excluded 11 patients who crossed over at progression to receive TTFields in the commercial setting against protocol.
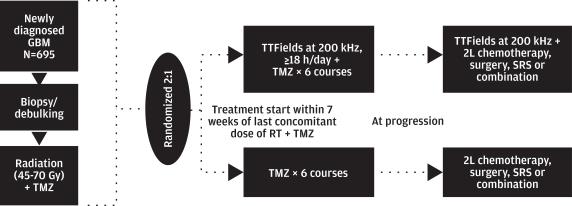
Definitions: TMZ= temozolomide; SRS= stereotactic radiosurgery; 2L= second line; RT= radiation therapy
10
Baseline characteristics were balanced between the two groups. The median age was 58 years old for patients receiving temozolomide alone and 57 years old for patients treated with TTFields in combination with temozolomide. Sixty-six percent of the participants were male. The median Karnofsky performance score, a standard way of measuring the ability of cancer patients to perform ordinary tasks independently, was 90. The percentage of patients that had either a gross total resection or partial resection was 90% for patients receiving temolozomide alone and 89% for patients treated with TTFields in combination with temolozomide. Tumor tissue for MGMT testing was available for 72% of the patients; the MGMT methylation frequency was 38% and 41% for TTFields in combination with temozolomide and temozolomide alone arm, respectively. Ninety-five percent of the patients were Caucasian, and over 60% of the patients were treated in the United States. Tumor location in the brain was also comparable. The median time from diagnosis to randomization was 3.8 months in both arms. Median time from end of radiation to randomization was 36 and 38 days, respectively. Median time from randomization to initiation of TTFields was five days. Both PFS and OS were measured from the time of randomization, not the time of diagnosis.
Clinical trial results
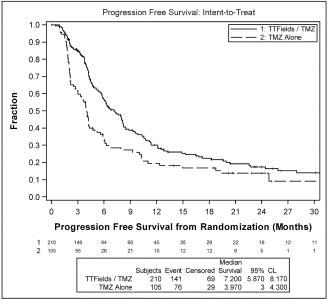
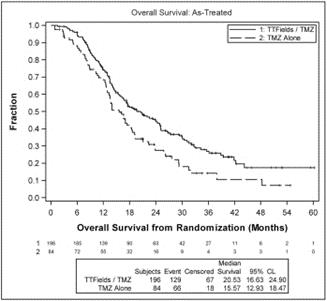
The analysis demonstrated that patients treated with TTFields in combination with temozolomide experienced a significant extension in PFS based on blinded central radiology review and lived significantly longer than patients treated with temozolomide alone:
|
· |
patients treated with TTFields, in combination with temozolomide, in the intent-to-treat population, demonstrated a statistically significant increase in PFS compared to temozolomide alone (median PFS of 7.2 months compared to 4.0 months, hazard ratio=0.62, p=0.001); |
|
· |
patients treated with TTFields, in combination with temozolomide, in the as-treated population, demonstrated a statistically significant increase in OS compared to temozolomide alone (median OS of 20.5 months compared to 15.6 months, hazard ratio=0.66, p=0.004); and |
|
· |
the two-year survival rate among patients treated with TTFields in combination with temozolomide, in the as-treated population, was 48% compared to 32% among patients treated with temozolomide alone (p=0.0058). |
The following graph presents PFS data in the intent-to-treat population from our analysis:
11
The following graph presents OS data in the as-treated population from our analysis:
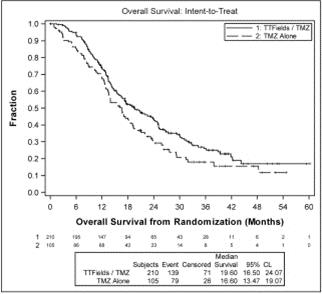
In order to test the effect on OS of crossover of patients from the temozolomide arm to receive TTFields, OS was also analyzed in the intent-to-treat population including all patients randomized according to their original randomization group. This sensitivity analysis showed that even when crossover patients were included, patients treated with TTFields in combination with temozolomide lived significantly longer than patients treated with temozolomide alone (median OS of 19.6 months compared to 16.6 months, hazard ratio=0.74, p=0.034).
The following graph presents OS data in the intent-to-treat population from our analysis:
A post-hoc analysis of all 695 patients enrolled in the EF-14 trial was performed after an average follow up of approximately 12 months for all patients. This analysis, which was presented at the American Society of Clinical Oncology annual meeting in 2015, confirmed that TTFields in combination with temozolomide extends both PFS and OS significantly compared to temozolomide alone.
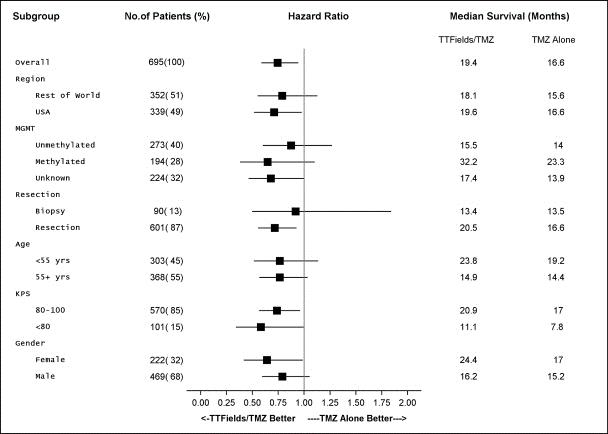
The significant extension of PFS and OS in patients receiving TTFields in combination with temozolomide in the EF-14 trial was seen in all patient subgroups and was not specific to any prognostic subgroup or tumor genetic marker. PFS and OS were extended in patients with either MGMT methylated or unmethylated tumors.
12
The following table presents details of the subgroup analyses for OS in the EF-14 trial:
13
The following table presents details of the subgroup analyses for PFS in the EF-14 trial:
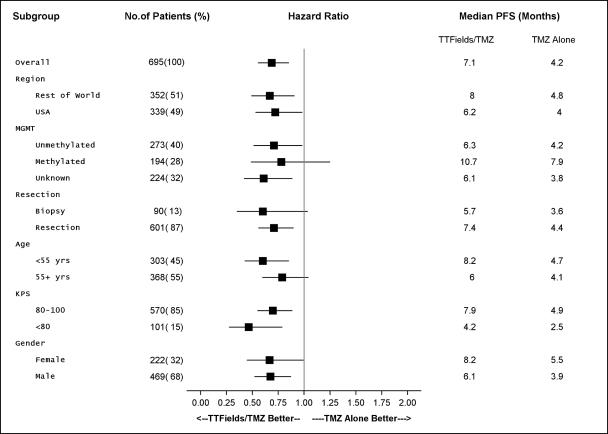
Definitions: TMZ = temozolomide; PFS = progression free survival; MGMT = O-6-methylguanine-DNA methyltransferase; KPS = Karnofsky performance score
The EF-14 trial was designed such that patients continued TTFields through first disease progression. A post-hoc analysis presented in November 2015 at the 20th Annual Society of Neuro-Oncology Meeting compared 144 patients who received Tumor Treating Fields in combination with second line therapy at first disease recurrence with 60 patients who received second line therapy alone at first disease recurrence.
14
The analysis showed that patients treated with TTFields in combination with second line therapy lived significantly longer than patients treated with second line therapy alone. The median overall survival from first progression increased from 9.2 months in the second line therapy alone arm to 11.8 months in the TTFields in combination with second line therapy arm. The overall survival benefit was statistically significant with a hazard ratio of 0.695 and a log rank p value of 0.0489.
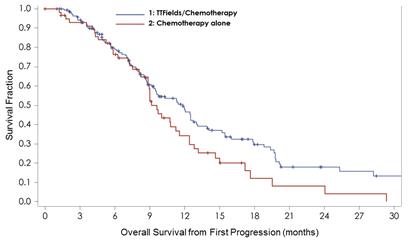
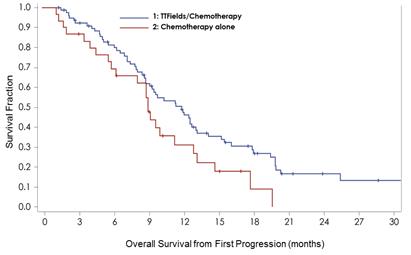
A further analysis of the first recurrence survival data was performed to isolate the patient population who received bevacizumab as the backbone of their second line therapy. Fifty percent of the patients treated with second line therapy alone received bevacizumab and 55% of the patients treated TTFields in combination with second line therapy received bevacizumab.
The resulting analysis showed that patients treated with TT Fields in combination with bevacizumab live significantly longer than patients treated with bevacizumab alone. The overall survival benefit was statistically significant with a hazard ratio of 0.606 and a log rank p value of 0.0428.
Safety and quality-of-life results
The TTFields in combination with temozolomide arm was not associated with any significant increase in systemic toxicities compared with temozolomide alone. The overall incidence, distribution and severity of adverse events were similar in patients treated with TTFields in combination with temozolomide compared to those treated with temozolomide alone. The only notable exception was, as expected, a higher incidence of localized skin toxicity related to the skin beneath the transducer arrays in patients treated with TTFields. Some mild to moderate skin irritation was observed in 45% of patients, severe skin reaction (grade 3) was observed in 1% of patients. In addition, TTFields had no negative impact on patient quality of life, performance status or cognitive function.
A prospective analysis of the EF-14 trial population, presented in November 2015 at the 20th Annual Society of Neuro Oncology Meeting, collected data until disease progression on the secondary endpoints of quality of live, cognitive function and ability to perform activities of daily life as measured by the EORTC quality of life questionnaire, the mini-mental status exam and Karnofsky
15
performance scores. Newly diagnosed GBM patients receiving TTFields therapy together with temozolomide treatment had stable cognitive and functional status throughout treatment. No difference in Karnofsky performance score or mini-mental status exam was seen between the two treatment groups over time. These two analyses indicate that patients’ ability to perform regular daily activities did not decline over time and that cognitive function was maintained throughout treatment in both treatment arms.
We believe based on the reported EF-14 trial data that newly diagnosed patients treated with Optune in combination with temozolomide will have a longer treatment duration than what we have observed in the recurrent GBM population. Median treatment duration was 2.3 months for recurrent GBM patients in the EF-11 trial and 4.1 months in the PRiDe dataset. Median treatment duration increased to 9.0 months in the EF-14 trial.
Our commercial capabilities
We established commercial operations in the United States following FDA approval of Optune for the treatment of recurrent GBM in 2011. We believe that the majority of recurrent GBM patients in the United States are treated by physicians in approximately 200 large clinical centers, most of which are major academic teaching hospitals. This allowed us to focus our initial commercial efforts and optimize our distribution and support services to a well-defined customer base.
We have expanded our U.S. commercial team to cover community oncology practices as well as the large cancer centers in light of the FDA’s approval of Optune for newly diagnosed GBM. Our U.S. sales force expanded to 31 full-time employees as of December 31, 2015 compared to 14 full-time employees as of December 31, 2014. We have also expanded our target physician specialties to include medical and radiation oncologists, as these specialists play a role in the treatment decisions for newly diagnosed GBM. We believe this will allow us to significantly grow the number of patients treated with Optune and the number of physicians and centers that we target. We believe that time spent in the initial physician outreach to share clinical data and the supporting education process is critical to establishing durable market penetration and anticipate we will be able to grow and maintain Optune adoption within this expanded customer base with effective targeting and a scalable sales force driven by demand. As of December 31, 2015, we have trained physicians at 244 clinical centers in the United States.
We established commercial operations in Europe in 2014, focusing initially on Germany. Our European sales force consisted of 2 full-time employees as of December 31, 2015. As of December 31, 2015, we have trained physicians at 62 clinical centers in Europe, including 45 in Germany. As in the United States, we believe we will be able to grow and maintain Optune adoption with effective targeting and a scalable sales force driven by demand.
We recently initiated commercial operations in Japan in March 2015 upon our approval in recurrent GBM. In December 2015, we submitted the registration dossier for premarketing approval of Optune to treat newly diagnosed GBM to the JPMDA. Treatment for GBM patients in Japan occurs mainly at large academic hospitals. As of December 31, 2015, we have trained physicians at over 61 clinical centers in Japan. In addition to our commercial functions, we provide health care professionals with educational support and Optune training through a geographically distributed team of clinical science liaisons. We also have a dedicated team of device support specialists who are available locally throughout our commercial markets to assist patients starting Optune and to resolve any technical difficulties patients may encounter. Patients also have access to 24/7 technical support by phone or e-mail as needed. We also work to secure reimbursement for Optune on behalf of patients. We believe our patient-centric approach will enable us to maximize our commercial opportunity by driving patient adoption and therapy compliance.
Our existing commercial and supply chain capabilities allow us to satisfy the initial market demand for Optune that followed FDA approval for newly diagnosed GBM. We plan to scale our commercial organization over time to maximize clinical adoption of Optune therapy.
We believe that we can leverage our commercial capabilities for the GBM market in the United States, Europe and Japan to address additional future indications. We believe that cancer patients are increasingly seeking treatment at large oncology practices, which will allow our sales representatives to market for multiple indications within one practice.
Our clinical pipeline
Based on the results of our pre-clinical research, we have developed a pipeline strategy to advance TTFields through phase 2 pilot and phase 3 pivotal trials across multiple solid tumor types, as described in greater detail below. In addition, we anticipate expanding our clinical pipeline over time to apply TTFields to additional solid tumor indications.
16
We believe brain metastases will be our next label expansion beyond GBM. We have an ongoing European phase 2 pilot trial, or the COMET trial, in brain metastases originating from NSCLC, and we plan to open a phase 3 pivotal trial, or the METIS trial, in 2016 in the United States subject to final protocol approval by the FDA. Metastatic cancer is cancer that has spread from the place where it first started to another place in the body. In metastasis, cancer cells break away from where they first formed (the primary cancer), travel through the blood or lymph system, and form new tumors (the metastatic tumors) in other parts of the body.
The exact incidence of brain metastases is unknown because no national cancer registry documents brain metastases, but it has been estimated that 98,000 to 170,000 new cases are diagnosed in the United States each year. We believe approximately 40% of brain metastases are seeded from NSCLC. Brain metastases cause an estimated 20% of all cancer deaths in the United States annually.
As with GBM, brain metastases are commonly treated with a combination of surgery and radiation. Chemotherapy is often given for the primary tumor; however, many chemotherapy agents do not cross the blood brain barrier and are thus ineffective in the treatment of brain metastases. When brain metastases appear, they are either surgically resected or irradiated using stereotactic radiosurgery (SRS) when possible. Whole brain radiation therapy, or WBRT, although effective in delaying progression or recurrence of brain metastases when given either before or after SRS, is associated with neurotoxicity leading to the development of dementia with a significant decline in cognitive and emotional functioning. Thus, WBRT is often delayed until later in the disease course and is often used as a last resort. This practice results in a window of unmet need after localized surgery and SRS are used and before WBRT is administered to delay or prevent additional seeding of brain metastases.
We believe TTFields could be an effective treatment for patients with brain metastases. We have published pre-clinical data showing TTFields can prevent metastatic seeding in vivo. Based on these pre-clinical results and the established safety and efficacy of TTFields in GBM, we commenced the COMET trial examining TTFields as a monotherapy compared to supportive care alone after SRS in patients with brain metastases originating from NSCLC. The primary endpoint of the trial will be local disease control in the brain. In December 2015, we submitted a pre-submission package to the FDA with the protocol for our METIS trial for discussion and feedback. The open-label randomized study will test the effectiveness of TTFields following SRS compared with watchful waiting after SRS alone in patients with brain metastases stemming from non-small cell lung cancer. We plan to open the trial to 240 patients in mid-2016 subject to FDA approval of an investigational device exemption (IDE). With an expected 18 months of follow-up, we anticipate phase 3 pivotal data will be available for presentation in 2019 or 2020.
Non-small cell lung cancer (NSCLC)
We have completed a phase 2 pilot trial in advanced NSCLC and are planning a randomized phase 3pivotal trial in NSCLC, or the LUNAR trial. Lung cancer is the leading cause of cancer-related death in the United States. NSCLC accounts for approximately 85% of all lung cancers. The incidence of NSCLC in the United States is approximately 185,000 new cases annually. We have received approval to market our NovoTTF-100L system, our TTFields delivery system designed for the treatment of NSCLC, in combination with standard of care chemotherapy in Europe, based on the efficacy results from our clinical trials completed to date, as described further below.
Physicians use different combinations of surgery, radiation and pharmacological therapies to treat NSCLC, depending on the stage of the disease. Surgery, which may be curative in a subset of patients, is usually used in early stages of the disease. Unfortunately, most patients are likely to relapse sometime after initial surgery. For patients with locally advanced NSCLC, the standard of care is chemotherapy and radiation with or without surgery. Most frequently, however, NSCLC patients are diagnosed at an advanced stage when the cancer has spread outside of the lungs, leaving chemotherapy as the only treatment option. Despite the many advances in chemotherapy made in recent decades, treatment outcomes remain inadequate, especially for patients with the squamous histology, which clinical results suggest may be resistant to or cannot be treated with therapies such as bevacizumab and pemetrexed.
Phase 2 pilot trial design
Based on our pre-clinical findings, we conducted a phase 2 pilot trial to evaluate the safety and efficacy of TTFields in the treatment of advanced NSCLC (both squamous and adenocarcinoma histologies). Results of this study were published in 2013. To treat NSCLC, we developed the NovoTTF-100L system to deliver TTFields regionally to the lungs, mediastinum and liver. The delivery system output settings differ in frequency and intensity from Optune based on the findings of our pre-clinical studies in NSCLC. The pilot study was a single-arm, open-label, historically-controlled, multi-center trial. The primary endpoints of the trial were safety and PFS. The secondary endpoints were OS and response rates. Results of the pemetrexed Phase 3 FDA registration trial were used as historical controls in this trial.
17
A total of 42 patients were recruited to the study at four centers in Switzerland with a minimum follow-up of six months. Before entering the trial, each patient’s disease had progressed after chemotherapy treatment. Patients were 18 years of age or older (median 63 years old) with an Eastern Cooperative Oncology Group, or ECOG, performance of 0-2. ECOG is a standard scoring system of a patient’s general health and well-being running from zero (full health) to five (death). Patients with significant co-morbidities, pregnancy or pacemakers were excluded. All patients were treated with the NovoTTF-100L system in combination with standard of care pemetrexed chemotherapy.
Phase 2 pilot trial results
Efficacy results based on 41 evaluable patients (n=34 non-squamous histology, including 32 adenocarcinoma histology; n=7 squamous histology) showed both PFS and OS for patients receiving TTFields in combination with pemetrexed increased compared to historical control data for pemetrexed alone. Median PFS in the TTFields-treated group was 6.5 months (compared to 2.9 months in pemetrexed historical controls) and median OS was 13.8 months (compared to 8.3 months in historical controls). OS was essentially the same for patients with adenocarcinoma and squamous NSCLC when treated with TTFields and pemetrexed concomitantly (12.8 months and 13.8 months, respectively). As pemetrexed is now known to be inactive in squamous NSCLC, we believe that the OS effect seen in patients in this study with this subtype of lung cancer is due to TTFields alone. Based on these data, the investigators concluded that TTFields in combination with pemetrexed was well tolerated and did not lead to an increase in toxicity. Finally, the data suggests potentially higher efficacy of TTFields in combination with pemetrexed compared to pemetrexed alone.
Adverse events reported in this combination study were comparable to those reported with pemetrexed alone, suggesting minimal added toxicities due to TTFields. TTFields applied to the chest did not cause arrhythmias or other cardiac or pulmonary toxicity for patients in this study. The only delivery system-related toxicities were mild-to-moderate local rash beneath the transducer arrays, which was seen in most of the patients. This condition was managed by topical corticosteroids and transducer array relocation. In persistent cases, either oral corticosteroids were used or transient treatment breaks were made for several days.
Planned phase 3 pivotal trial
Our planned LUNAR trial will examine TTFields in combination with a platinum-based chemotherapy and a taxane versus a platinum-based chemotherapy and a taxane alone as a first-line therapy for the squamous histology of NSCLC. Paclitaxel is a taxane that interferes with microtubule function during mitosis. We expect the LUNAR trial to commence in 2016, subject to final protocol approval by the FDA. With an expected 18 months of follow-up, we anticipate Phase 3 data will be available for presentation in 2019. Given recent clinical results and FDA approval of certain immunotherapies for this indication, we are currently evaluating the study protocol for the LUNAR trial.
Pancreatic cancer
We have an ongoing phase 2 pilot trial in pancreatic cancer, or the PANOVA trial, with two cohorts of 20 patients each, and have accelerated planning of a phase 3 pivotal trial based upon the positive results shown in the first cohort. Pancreatic cancer is one of the most lethal cancers, as most cases are diagnosed once the cancer is at a late stage and/or has metastasized to other parts of the body. It is the fourth most frequent cause of death from cancer in the United States and is responsible for 7% of all U.S. cancer-related deaths. In contrast to the decrease in mortality from other cancers over the past decade, pancreatic cancer death rates have been slowly increasing in the United States. Pancreatic cancer prognosis remains very poor, with a five-year survival of less than 6%. The National Cancer Institute estimated that, in 2015, there were approximately 49,000 new cases of pancreatic cancer diagnosed and approximately 40,000 deaths in the United States from pancreatic cancer.
Phase 2 pilot trial design
Based on our pre-clinical findings, we initiated PANOVA, a phase 2 pilot trial in advanced pancreatic adenocarcinoma examining TTFields in combination with chemotherapy, in the first quarter of 2014.
The first cohort was designed to test the feasibility, safety and preliminary efficacy of TTFields in combination with gemcitabine and included 20 patients with advanced pancreatic cancer whose tumors could not be removed surgically and who had not received chemotherapy or radiation therapy prior to the clinical trial. Results of the first cohort were presented at the American Society of Clinical Oncology Gastrointestinal Cancers Symposium in January 2016. To treat pancreatic cancer, we use the NovoTTF-100L(P) system, a device identical to the NovoTTF-100L system previously developed to treat lung cancer, to deliver TTFields regionally to the pancreas, liver and entire upper abdomen. The first cohort of the phase 2 pilot trial was a single-arm, open-label, historically-controlled, multi-center trial. The primary endpoint of the trial was safety and feasibility. The secondary endpoints were PFS, OS,
18
response rates, six month progression free survival rate and one year survival rate. Results of the nab-paclitaxel phase 3 FDA registration trial were used as historical controls in this trial.
In the first cohort, a total of 20 patients were recruited to the study at five centers in Spain and one center in Switzerland with a minimum follow-up of six months. Patients were 18 years of age or older (median 73 years old) with a ECOG performance of 0-1. Eighty percent of patients had an ECOG score of 1. Patients with significant co-morbidities, pregnancy or pacemakers were excluded. Twelve patients (60%) had distant metastases, while the others had locally advanced disease. Median compliance with TTFields was 78%, with median treatment duration of five months. In the first cohort, all patients were treated with the NovoTTF-100L(P) system in combination with gemcitabine chemotherapy.
Following the approval of nab-paclitaxel, a taxane-based chemotherapy, for the treatment of advanced pancreatic cancer, we expanded this study to include 20 additional patients that will be treated with TTFields in combination with nab-paclitaxel and gemcitabine. We expect to finish enrollment of the second patient cohort study in 2016.
Phase 2 pilot trial first cohort results
Efficacy results based on the 20 patients in the first cohort showed that PFS and OS of patients treated with TTFields combined with gemcitabine were more than double those of gemcitabine-treated historical controls. Median PFS in the TTFields-treated group was 8.3 months (compared to 3.7 months in gemcitabine historical controls) and median OS was 14.9 months (compared to 6.7 months in gemcitabine historical controls). Median one-year survival was 55% (compared to 22% in gemcitabine historical controls). Thirty percent of the evaluable tumors (n=19) had partial responses (compared to 7% with gemcitabine alone) and another 30% had stable disease.
Adverse events reported in this combination study were comparable to those reported with gemcitabine alone, suggesting minimal added toxicities due to TTFields. The only delivery system-related toxicities were mild-to-moderate local rash beneath the transducer arrays, which was seen in 10 patients. This condition was managed by topical corticosteroids and transducer array relocation. In persistent cases, either oral corticosteroids were used or transient treatment breaks were made for several days. Based on the results from this first cohort, the investigators concluded that TTFields in combination with gemcitabine was well tolerated and did not lead to an increase in toxicity for advanced pancreatic cancer patients. Finally, the data suggests potentially higher efficacy of TTFields in combination with gemcitabine compared to gemcitabine alone.
Ovarian cancer
We have an ongoing phase 2 pilot trial in ovarian cancer, or the INNOVATE trial, and plan to open a hpase 3 pivotal trial if the results from our INNOVATE trial are promising. Ovarian cancer is the fifth most common cause of cancer death in women in the United States. The National Cancer Institute estimated that in 2015, there were approximately 21,000 new cases of ovarian cancer diagnosed and approximately 14,000 deaths in the United States. Ovarian cancer incidence increases with age, and the median age at time of diagnosis is 63 years old. The five-year survival rate is 44%, and the majority of patients present at advanced stage with 60% having metastatic disease.
In October 2014, we commenced the INNOVATE trial examining TTFields in combination with weekly paclitaxel for recurrent ovarian cancer. The INNOVATE trial is designed to test the feasibility, safety and preliminary efficacy of TTFields in combination with paclitaxel and is expected to enroll 30 patients. We expect to complete enrollment in 2016 and we anticipate phase 2 pilot trial data will be available for presentation in 2017.
Mesothelioma
We have an ongoing phase 2 pilot trial in mesothelioma, or the STELLAR trial. Malignant mesothelioma is a rare thoracic solid tumor cancer that occurs in approximately 3,000 patients in the United States annually. Asbestos exposure has been strongly associated with the development of mesothelioma, which may occur many years following the exposure. The prognosis of mesothelioma patients is very poor, with a median OS of approximately 11 months in most reported studies. Mesothelioma is often limited to the thoracic cavity and progresses regionally making it an attractive target for TTFields.
We commenced the STELLAR trial in mesothelioma in 2015. The STELLAR trial is an open-label trial designed to test the efficacy and safety of TTFields in combination with pemetrexed combined with cisplatin or carboplatin in patients with unresectable, previously untreated malignant mesothelioma compared to a historical, published control. The STELLAR trial is anticipated to enroll 80 patients from Germany, Italy and Poland. We expect to finish enrollment of the trial in 2017. We anticipate phase 2 pilot trial data will be available for presentation in 2018.
19
We developed Optune, formerly known as NovoTTF-100A, for cranial indications, and NovoTTF-100L for torso indications, and we are developing additional delivery systems for other indications in our pipeline. We have also developed our propriety NovoTAL transducer array layout planning software. Given the opportunity for TTFields in treating a wide variety of solid tumors, including GBM and other solid tumor cancers we are investigating, we continue to develop and enhance our TTFields delivery systems.
We have recently completed development of improved field generator circuitry, which minimizes power loss and requires a smaller battery. We have incorporated these advancements into a re-designed second generation Optune delivery system, which is less than half of the size and weight of the current system, with improved functionality for physicians and patients. In October 2015, we received CE Mark for our newly designed second generation of Optune, which is less than half the weight and size of the current version, and have since made it available to all new patients in Europe. In December 2015, we filed a 180-day PMA supplement application with the FDA seeking approval of the second generation of Optune for the approved indications. Assuming that we do not receive comments or requests for additional information from the FDA, we hope to begin marketing the second generation Optune system in the United States in the third quarter of 2016.
We plan to continue to improve all aspects of TTFields delivery to improve ease of use for patients. We also plan to improve the NovoTAL transducer array layout planning software to allow more flexibility and ease of use by prescribing physicians.
Manufacturing
We have established flexible capacity in our principal manufacturing supply chain while driving costs down. We outsource production of all of our system components to qualified partners. Disposable transducer array manufacturing, the dominant activity in our manufacturing supply chain, includes several specialized processes. Production of the durable system components follows standard electronic medical device methodologies.
We have formal supply agreements with our third-party manufacturing partners. We hold safety stocks of single source components to protect our production capacity.
We currently source the ceramic discs used in the transducer arrays for Optune from Harris Corporation (formerly known as Exelis Inc.), which is our single-source supplier for these components. We do not currently have alternate suppliers for the ceramic discs, but we are in the process of identifying a second source supplier.
Our current agreement with Harris Corporation continues through July 21, 2017, following which time the agreement will automatically renew for up to three successive two-year periods unless either we provide timely written notice of non-renewal (for any reason) or Harris Corporation provides timely written notice of non-renewal (if we fail to satisfy certain minimum purchase requirements). We currently expect that this agreement will be renewed. In addition to certain other customary termination rights, Harris Corporation can terminate this agreement with 90 days’ written notice if we breach any of our material obligations under the agreement. Agreements with our other suppliers range from terms of four years to ten years and are terminable by either party, generally between 180 days’ and 12 months’ written notice. See “Risk factors—We depend on single-source suppliers for some of our components. The loss of these suppliers could prevent or delay shipments of Optune, delay our clinical trials or otherwise adversely affect our business.”
Transducer array production represents the main area of focus for the supply chain team. Over the last eighteen months, we and our third-party manufacturer have automated the assembly process for the main array components. We utilize standardized printed circuit board continuous flow production techniques including automatic handling and gluing of the ultra-high dielectric constant ceramic transducer elements. Automation has substantially increased capacity while lowering costs. We believe the automated line can be easily duplicated to add capacity as needed.
Billing and reimbursement
We provide Optune directly to patients in the United States, Germany and Switzerland following receipt of a prescription order. We bear the financial risk of securing payment from patients and third-party payers in these markets. We distribute our product through hospitals in Japan and bill a monthly fee to the hospital for its use. The monthly charge for Optune is $21,000 in the United States and €21,000 in the European Union.
20
In the United States, total cash payments of $31.0 million, net of indirect taxes paid, received during the twelve months ended December 31, 2015 were recorded as revenues for Optune therapy provided to patients in the current period and prior periods. These cash payments represent an average of approximately $14,000 per charged month in the United States. The difference between billed and paid amounts consists of indirect taxes, disputed underpayments, patient financial assistance, charitable care and discounts. Our average time to collect on billed charges in 2015 was between four and five months. The “payment amount” and “average time to collect” metrics do not include our experience with patients covered by the Medicare fee-for-service program, as we have not received material payments from that program and the invoices remain open as we appeal the coverage denials. We currently employ reimbursement experts devoted to securing coverage and payment for Optune from third-party payers. Physicians in the United States may separately bill patients or third-party payers for medical services related to initiating and managing treatment with Optune, including generating personalized transducer array layouts with NovoTAL.
We are aware of eight commercial payers in the United States that have issued positive coverage policies that deem Optune as medically necessary for the treatment of newly diagnosed or recurrent glioblastoma. In total, we estimate that approximately 97 million people in the United States have access to Optune for one or both indications under these coverage policies. For our U.S. patients covered by commercial third-party payers and privately administered government-sponsored plans, specifically Medicare Advantage, Tricare (U.S. military) and federal employee plans, we have been successful in securing coverage for approximately 98% of these patients as of December 31, 2015. Approximately 20% of our active U.S. patients were beneficiaries of the Medicare fee-for-service program as of December 31, 2015. The Medicare fee-for-service program has denied coverage for our claims to date, and we are actively appealing these coverage denials.
The Centers for Medicare and Medicaid Services (CMS) have issued Healthcare Common Procedure Coding System (HCPCS) Level II codes that are specific to our delivery systems. The codes are E0766 “Electrical stimulation device used for cancer treatment, includes all accessories, any type” and A4555 “Electrode/transducer for use with electrical stimulation device used for cancer treatment, replacement only.” We bill payers for a single monthly fee at the start of each month of therapy using the E0766 code and the A4555 code is used as a non-billable tracking code by certain payers.
We started commercial activity in Germany in 2014. We have hired personnel with expertise in reimbursement to support the launch of Optune. We are able to bill healthcare payers in Germany for individual cases currently, and each case is evaluated individually on its merits and under the payer’s specific rules for such cases. We are also pursuing defined coverage and pricing terms in Germany. The German healthcare reimbursement system is a mix of public and private payers all operating under government regulatory oversight. Medical device-based therapies are eligible for reimbursement under multiple pathways with varying clinical and effectiveness evidence requirements for each pathway. In Switzerland we are preparing our application to the Federal Office of Public Health to secure a defined reimbursement rate for Optune.
In Japan we are preparing our application to the Japanese Ministry of Health, Labour and Welfare (“MHLW”) to secure a defined reimbursement rate for Optune. We have initiated a small commercial launch in Japan, and until we secure MHLW reimbursement approval, our launch efforts will focus on the privately insured patient population.
Intellectual property
As of December 31, 2015, our patent portfolio consists of a total of 54 issued patents, including 36 issued U.S. patents, three issued EU patents, one issued Canadian patent, eight issued Japanese patents and five issued Chinese patents. The U.S. patents have expected expiration dates between 2021 and 2031. The EU, Canadian, Japanese and Chinese patents have expected expiration dates between 2021 and 2026. We have also filed over 45 additional patent applications that, if issued, may protect aspects of our platform beyond 2034 in the United States, Europe, Canada, Japan and China.
In addition to our patent portfolio, we further protect our intellectual property by maintaining the confidentiality of our trade secrets, know-how and other confidential information. Given the length of time and expense associated with bringing delivery systems candidates through development and regulatory approval to the market place, the healthcare industry has traditionally placed considerable importance on obtaining patent protection and maintaining trade secrets, know-how and other confidential information for significant new technologies, products and processes.
Our policy is to require each of our employees, consultants and advisors to execute a confidentiality agreement before beginning their employment, consulting or advisory relationship with us. These agreements generally provide that the individual must keep confidential and not disclose to other parties any confidential information developed or learned by the individual during the course of their relationship with us except in limited circumstances. These agreements also generally provide that we own, or the individual is required to assign to us, all inventions conceived by the individual in the course of rendering services to us.
21
On February 10, 2015, we entered into a settlement agreement, or the Settlement Agreement, with the Technion, whereby we agreed to resolve certain potential disputes among us, the Technion and Professor Yoram Palti, our Chief Technology Officer and a member of our board of directors, arising out of certain intellectual property that Professor Palti developed while affiliated with the Technion and that Professor Palti has assigned to us. In settlement of these potential disputes, we agreed to pay the Technion an aggregate of $7.5 million, including $1.0 million that was paid on the date of the agreement, an additional $1.0 million that was paid upon the completion of the IPO and an additional $5.5 million that will be payable within five business days (1) if we achieve $250.0 million of cumulative net sales since inception at the end of any given quarter or (2) upon consummation of an M&A transaction, which includes any merger to the extent it involves a change of control, the sale of all or substantially all of our assets or shares, the sale of or exclusive license to our intellectual property or a similar transaction.
In addition, pursuant to the terms of the Settlement Agreement, we issued 1,005,210 ordinary shares to the Technion, and further granted the Technion an option to acquire an additional 1,005,210 ordinary shares, which is exercisable at any time until the first to occur of (1) 12 months following the IPO and (2) immediately prior to the sale of the company for cash or publicly traded stock. There is no exercise price on this option, and to date this option has not been exercised; however, the Technion will have until 12 months after the completion of the IPO to exercise its option. No royalties are owed to the Technion or Professor Palti.
In 2005, we granted an exclusive license to a third party, NovoBiotic LLC, to certain of our key intellectual property for use outside the field of oncology. We are not entitled to any future revenues from this license.
We may be unable to obtain, maintain and protect the intellectual property rights necessary to conduct our business and may be subject to claims that we infringe, misappropriate or otherwise violate the intellectual property rights of others. For more information see “Risk factors—Risks relating to intellectual property.”
Competition
The market for cancer treatments is intensely competitive, subject to rapid change and significantly affected by new product and treatment introductions and other activities of industry participants. The general bases of competition are overall effectiveness, side effect profile, availability of reimbursement and general market acceptance of a product as a suitable cancer treatment.
TTFields is our proprietary method for treating cancer. As such, there is no direct competition from other TTFields delivery systems. We believe our intellectual property rights would provide an obstacle to the introduction of TTFields delivery systems by a competitor, and we intend to protect and enforce our intellectual property. In addition, even after the expiration of our U.S. patents, potential market entrants applying low-intensity, alternating electric fields to solid tumors in the United States will have to undertake their own clinical trials and regulatory submissions to prove equivalence to TTFields, a necessary step in receiving regulatory approvals for a competing product.
When used as a monotherapy for solid tumor cancers, TTFields competes with a number of existing cancer treatment alternatives, including surgery, radiation and pharmacological therapies. These treatments have been developed, commercialized and established in the market by large, well-capitalized companies with significantly more market share and resources than we have. We believe that our direct competition is limited because we intend to use TTFields in combination with cancer treatments offered by other oncology companies.
We also intend to study the use of TTFields in combination with new entrants in the market, including new pharmacological agents such as PD-1 inhibitors and others in the field of immuno-oncology. In this regard, we expect to be less susceptible to competition from such emerging new pharmacological agents.
Government regulation
Our delivery systems and operations are subject to extensive regulation by the FDA and by agencies and notified bodies of the countries or regions in which we develop and market our delivery systems. In addition, our delivery systems must meet the requirements of a large and growing body of international standards that govern the pre-clinical and clinical testing, manufacturing, labeling, certification, storage, recordkeeping, advertising, promotion, export and marketing and distribution, among other things, of TTFields and our delivery systems.
Advertising and promotion of medical devices, in addition to being regulated by the FDA, are also regulated by the Federal Trade Commission and by state regulatory and enforcement authorities. Recently, promotional activities for FDA-regulated products of other companies have been the subject of enforcement action brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the federal Lanham Act and similar state laws, competitors and others can initiate litigation relating to advertising claims. In addition, we are required to meet regulatory requirements in countries outside the United States, which can change rapidly with relatively short notice.
22
Our research, development and clinical programs, as well as our manufacturing and marketing operations, are subject to extensive regulation in the United States and other countries. Most notably, all of our delivery systems sold in the United States are subject to the Federal Food, Drug, and Cosmetic Act, or the FDCA, as implemented and enforced by the FDA. Marketing our delivery systems sold in the United States requires FDA approval. Foreign countries may require similar or more onerous approvals to manufacture or market these delivery systems.
Failure by us or by our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities, which may result in any number of regulatory enforcement actions.
Food and Drug Administration
The FDA regulates the development, testing, manufacturing, labeling, storage, recordkeeping, promotion, marketing, distribution and service of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. In addition, the FDA regulates the export of medical devices manufactured in the United States to international markets and the importation of medical devices manufactured abroad.
The FDA governs the following activities that we perform or that are performed on our behalf:
|
· |
product design, development and manufacture; |
|
· |
product safety, testing, labeling and storage; |
|
· |
record keeping procedures; |
|
· |
product marketing, sales and distribution; and |
|
· |
post-marketing surveillance, complaint handling, medical device reporting, reporting of deaths, serious injuries or device malfunctions and repair or recall of products. |
We have registered three of our facilities with the FDA. The FDA has broad post-market and regulatory enforcement powers. We are subject to announced and unannounced inspections by the FDA to determine our compliance with the Quality System Regulation, or QSR, and other regulations and these inspections include the manufacturing facilities of our suppliers.
FDA’s premarket clearance and approval requirements
Unless an exemption applies, before we can commercially distribute medical devices in the United States, we must obtain, depending on the type of device, either prior 510(k) clearance or PMA from the FDA. The FDA classifies medical devices into one of three classes. Devices deemed to pose lower risks are placed in either class I or II, which typically requires the manufacturer to submit to the FDA a premarket notification requesting permission to commercially distribute the device. This process is generally known as 510(k) clearance. Some low-risk devices are exempted from this requirement. Devices deemed by the FDA to pose the greatest risks, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in class III, requiring PMA approval.
Premarket approval (PMA) pathway
Optune, which is the only delivery system we have marketed in the United States, is classified as a Class III device as it is deemed a life-sustaining device. Accordingly, we were required to receive PMA approval for Optune, which the FDA granted in April 2011 and October 2015 for the treatment of recurrent and newly diagnosed GBM, respectively, in adult patients. We expect that we will be required to receive PMA approval for future indications (and the applicable delivery systems for such indications) using TTFields.
A PMA must be supported by extensive data, including from technical tests, pre-clinical studies and clinical trials, manufacturing information and intended labeling to demonstrate, to the FDA’s satisfaction, the safety and effectiveness of a medical device for its intended use. During the PMA review period, the FDA will typically request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The FDA may or may not accept the panel’s recommendation. In addition, the FDA will generally conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with QSRs. Prior to approval of the Optune PMA for the treatment of recurrent GBM, we and our critical component suppliers were each inspected by the FDA.
23
New PMAs or PMA supplements are required for modifications that affect the safety or effectiveness of our delivery systems, including, for example, certain types of modifications to a delivery system’s indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require any or as extensive clinical data as the original PMA required, or the convening of an advisory panel. The FDA requires a company to make the determination as to whether a new PMA or PMA supplement application is to be filed. If a company determines that neither a new PMA or PMA supplement application is required for modifications, it must nevertheless notify the FDA of these modifications in its PMA Annual Report. The FDA may review a company’s decision and require the filing of an application.
We have received approval for a number of PMA supplements since approval of the PMA for recurrent GBM, including for modifications to Optune’s electric field generator, transducer arrays, software, manufacturing processes and labeling. Most recently, in October 2015, we received FDA approval to expand our label for Optune to include the treatment of newly diagnosed GBM. In December 2015, we also filed a PMA supplement application for our next-generation delivery system for Optune, as described under “—Our engineering developments.” Future modifications may be considered by us as the need arises, some of which we may deem to require a PMA supplement application and others to require reporting in our PMA Annual Report.
Clinical trials
Clinical trials are generally required to support a PMA. Such trials generally require an investigational device exemption application, or IDE, approved in advance by the FDA for a specified number of patients and study sites, unless the product is deemed a nonsignificant risk device eligible for more abbreviated IDE requirements. Clinical trials are subject to extensive monitoring, recordkeeping and reporting requirements. Clinical trials must be conducted under the oversight of an institutional review board, or IRB, for the relevant clinical trial sites and must comply with FDA regulations, including those relating to good clinical practices. To conduct a clinical trial, we also are required to obtain the patients’ informed consent in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations. We, the FDA or the respective IRB could suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA approval to market the product in the United States.
Post-approval studies are also typically required as a condition of PMA approval to demonstrate reasonable assurance of safety and effectiveness. Such studies are conducted in the post-market setting with the approved device, often to address the long-term use of the device or other discrete questions that may have been raised based on the clinical data from the IDE clinical study. The FDA required a post-approval study as a condition of approval for Optune for recurrent GBM. We have obtained approval of the protocol for this study and are currently enrolling patients.
Foreign approvals and CE mark
Sales and marketing of medical devices outside of the United States are subject to foreign regulatory requirements that vary widely from country to country. These include the requirement to affix a CE mark to our medical devices in the European Union. Whether or not we have obtained FDA approval, our delivery systems must be subject to conformity assessment procedure in which a notified body can be involved. Apart from low risk medical devices (Class I with no measuring function and which are not sterile), where the manufacturer can issue a declaration of conformity based on a self-assessment of the conformity of its products with the Essential Requirements laid down in the Medical Devices Directive, a conformity assessment procedure requires the intervention of a notified body. The notified body typically audits and examines products’ technical file and the quality system for the manufacture, design and final inspection of our devices before issuing a CE Certificate of Conformity demonstrating compliance with the relevant Essential Requirements or the quality system requirements laid down in the relevant Annexes to the Medical Devices Directive. Following the issuance of this CE Certificate of Conformity, we can draw up a declaration of conformity and affix the CE mark to the delivery systems covered by this CE Certificate of Conformity and the declaration of conformity. The time required to CE mark our delivery systems or to obtain approval from other foreign authorities may be longer or shorter than that required for FDA approval. Pursuant to a mutual recognition agreement, our products bearing a CE mark may be exported to Switzerland. In the European Union, a clinical study must receive a positive opinion from a local ethics committee and approval from the competent authority in the applicable EU member states in which the clinical study is conducted. When a clinical study relates to a CE marked medical device that will be used as part of the study according to its CE mark intended purpose, the approval of the competent authorities is not required. In Japan, we must obtain approvals from the Japanese Ministry of Health, Labour and Welfare to market our delivery systems. The foreign regulatory approval process includes all the risks associated with FDA regulation, as well as country-specific regulations.
24
Pervasive and continuing regulation
After a device is placed on the market, numerous regulatory requirements apply. These include:
|
· |
product listing and establishment registration, which helps facilitate FDA inspections and other regulatory action; |
|
· |
QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process; |
|
· |
labeling regulations and FDA and equivalent foreign competent authority prohibitions against the promotion of products for uncleared, unapproved or off-label use or indication; |
|
· |
approval of product modifications that affect the safety or effectiveness of one of our delivery systems that has been approved or is the subject of a CE Certificate of Conformity; |
|
· |
medical device reporting regulations, which require that manufacturers comply with FDA or equivalent foreign competent authority requirements to report if their device may have caused or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the device or a similar device were to recur; |
|
· |
post-approval restrictions or conditions, including post-approval study commitments; |
|
· |
post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device; |
|
· |
the FDA’s and equivalent foreign competent authority’s recall authority, whereby they can ask, or under certain conditions order, device manufacturers to recall from the market a product that is in violation of governing laws and regulations; |
|
· |
the Sunshine Act and similar state and foreign laws, which require reporting of payments and other transfers of value to healthcare practitioners periodically; |
|
· |
regulations pertaining to voluntary recalls; and |
|
· |
notices of corrections or removals. |
Our delivery systems could be subject to voluntary recall if we, the FDA or an equivalent foreign competent authority determine, for any reason, that our delivery systems pose a risk of injury or are otherwise defective. Moreover, the FDA can order a mandatory recall if there is a reasonable probability that our delivery system would cause serious adverse health consequences or death.
The FDA has broad post-market and regulatory enforcement powers. We are subject to unannounced inspections by the FDA to determine our compliance with the QSR and other regulations, and these inspections include the manufacturing facilities of our subcontractors. Failure by us or by our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other equivalent foreign authorities, which may result in sanctions, including, but not limited to:
|
· |
untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
|
· |
unanticipated expenditures to address or defend such actions; |
|
· |
customer notifications for repair, replacement and/or refunds; |
|
· |
recall, detention or seizure of our delivery systems; |
|
· |
operating restrictions or partial suspension or total shutdown of production; |
|
· |
refusing or delaying our requests for approval of delivery system candidates or a modified version of Optune; |
|
· |
withdrawal of PMA approvals or suspension, variation or withdrawal of CE Certificates of Conformity that have already been granted; |
|
· |
refusal to grant export approval for our delivery systems; or |
|
· |
criminal prosecution. |
To date, our facility and those of our critical suppliers have been inspected by the FDA in order to obtain FDA approval of Optune. We and one of our critical component suppliers also were inspected by the FDA in 2012 and 2015. Another one of our suppliers was inspected in the fall of 2013. No inspectional observations were identified and no FDA Form 483s were issued following these inspections.
25
DME accreditation and licensing
We are subject to accreditation and licensing requirements as a DME supplier in most states. Certain of these states require that DME providers maintain an in-state location. Although we believe we are in compliance with all applicable state regulations regarding licensure requirements, if were found to be noncompliant, we could lose our accreditation or licensure in that state, which could prohibit us from selling our current or future delivery systems to patients in that state.
Healthcare regulatory matters
In addition to FDA restrictions on the marketing of medical devices, in recent years several other types of U.S. federal and state laws have been applied to restrict certain business practices in the healthcare industry and penalize unlawful conduct. These laws include anti-kickback, self-referral and false claims statutes.
The U.S. federal anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between device manufacturers on one hand and prescribers and purchasers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly and practices that involve remuneration intended to induce ordering, purchasing or recommending of a medical device may be subject to scrutiny if they do not qualify for an exemption or safe harbor. In some cases, our practices may not meet all of the criteria for safe harbor protection from anti-kickback liability.
As a DME supplier, we also are subject to a U.S. federal self-referral law, commonly known as the Stark law, which prohibits Medicare payments for DME ordered by physicians who, personally or through an immediate family member, have ownership interests in or compensation arrangements with the furnishing supplier. The Stark law contains a number of specific exceptions that, if met, permit physicians who have certain financial relationships with a DME supplier to make referrals to that entity.
The False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. In recent years, the government has pursued a number of cases under the False Claims Act in connection with the off-label promotion of medical products. In addition, violation of the federal anti-kickback statute and of the Stark law may be actionable under the False Claims Act.
The majority of states also have statutes or regulations similar to the federal anti-kickback, self-referral and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, that apply regardless of the payer.
The Health Insurance Portability and Accountability Act of 1996, or HIPAA, mandated the adoption of standards for the exchange of electronic health information in an effort to encourage overall administrative simplification and enhance the effectiveness and efficiency of the healthcare industry. Ensuring privacy and security of patient information is one of the key factors driving the legislation. We believe we are in substantial compliance with the applicable HIPAA regulation.
HIPAA also included a number of federal criminal provisions, including for healthcare fraud and for false statements relating to healthcare matters. The healthcare fraud provision prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payers. The false statements provision prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Many states have similar healthcare fraud laws or insurance fraud laws that apply to claims for healthcare reimbursement.
Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, criminal fines and imprisonment.
Legislation similar to U.S. anti-kickback, self-referral and false claims statutes have been adopted in foreign countries, including a number of EU member states.
26
The Sunshine Act requires manufacturers of drugs, medical devices, biologicals or medical supplies that participate in U.S. federal health care programs to track and then report certain payments and items of value given to U.S. physicians and U.S. teaching hospitals, which are defined as Covered Recipients. The Sunshine Act requires that manufacturers collect this information on a yearly basis and then report it to Centers for Medicare & Medicaid Services by the 90th day of each subsequent year. We have adopted policies and codes of conduct regarding our interactions with Covered Recipients and believe we are in substantial compliance with the Sunshine Act. However, our failure to adhere to these requirements could materially adversely impact our business and financial results.
In addition, the U.S. Foreign Corrupt Practices Act prohibits corporations and individuals from engaging in certain activities to obtain or retain business outside the United States or to influence a person working in an official capacity in a foreign country. It is illegal to pay, offer to pay or authorize the payment of anything of value to any official of another country, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in that capacity. Legislation similar to the U.S. Foreign Corrupt Practices Act has been adopted in foreign countries, including a number of EU member states.
Employees
As of December 31, 2015, we had 318 full-time employees, of which 58 were in sales and marketing, 44 were in research and development, 5 were in medical affairs, 49 were device support specialists and 162 were in operations support, finance, legal and administration. As of December 31, 2015, we had 207 employees located in the United States, 57 employees in Israel, 45 employees located in Europe and 9 employees located in Japan. We believe relations with our employees are good.
27
Adenocarcinoma histology is a cancer of epithelial tissue that has glandular origin, glandular characteristics or both. Adenocarcinoma of the lung is a subtype of lung cancer that responds to certain chemotherapies such as pemetrexed.
Advanced non-small cell lung cancer (NSCLC) is any type of epithelial lung cancer other than small cell lung carcinoma (SCLC). As a class, NSCLCs are relatively insensitive to chemotherapy, compared to small cell lung carcinoma. Advanced NSCLC refers to NSCLC that cannot be treated surgically due to spread of the cancer.
Anaphase is the stage of mitosis when chromosomes are split and the sister chromatids move to opposite poles of the cell.
Anti-mitotic is the description of a substance or treatment that interferes with the replication process of proliferating cells, such as cancer cells.
Apoptosis is the process of programmed cell death characterized by specific biochemical events that lead to characteristic cell changes and death. These changes include blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, chromosomal DNA fragmentation and global mRNA decay.
Bevacizumab, trade name Avastin (Genentech/Roche), is an angiogenesis inhibitor, a drug that slows the growth of new blood vessels. Bevacizumab is a recombinant humanized monoclonal antibody that blocks angiogenesis by inhibiting vascular endothelial growth factor A (VEGF-A). VEGF-A is a chemical signal that stimulates angiogenesis in a variety of diseases, especially in cancer.
Blebbing is the formation of bubble-like structures, or blebs, out of the cell membrane. Blebbing is a sign of different types of damage to cells including apoptosis.
Blinded central radiology review is when a group of radiologists assesses whether a cancer tumor has grown or not without knowing which treatment each patient received.
Blood-brain barrier is a highly selective barrier that separates the circulating blood from the brain. The blood brain barrier restricts many types of chemotherapy and other drugs from entering the brain.
Brain metastases are a form of cancer that has metastasized (spread) to the brain from another location in the body, such as the lung.
Cytokinetic band is a ring formed out of contractile molecules (mainly actin and myosin) that is located at the equator between two daughter cells of a replicating parental cell. The contraction of this ring pinches the cell membrane in two, leading to the formation of two independent daughter cells.
Cytokinesis is the process by which a single cell is physically divided to form two daughter cells.
Daughter cells refer to the two identical cells formed during cell division when a dividing cell, known as the mother cell, grows and divides to produce two identical cells.
Dielectric constant is a physical property of a material that determines what part of the electric field magnitude will be reduced within the volume of the material. Materials with higher dielectric constants decrease the field amplitude less.
Gemcitabine, brand name Gemzar, is a chemotherapy drug manufactured and marketed by Eli Lilly and Company. Its indications are the treatment of advanced ovarian cancer, metastatic breast cancer, NSCLC and pancreatic cancer. Gemcitabine is a chemotherapy of the nucleoside analog family, which interfere with the formation of DNA.
Histology is the microscopic anatomy of cells and tissues which is classically used to differentiate between the cellular origins of different cancer types.
Karnofsky performance score (KPS) is a quantification of ability to perform activities of daily life independently. The score is used in 10-point intervals from 0 (dead) to 100 (perfect independence in activities of daily life).
Mediastinum is the central compartment of the chest cavity surrounded by loose connective tissue, and is a region that contains a group of structures within the chest. The mediastinum contains mainly the heart and its vessels, the esophagus, trachea, nerves, thymus and lymph nodes of the central chest.
28
Mesothelioma is a form of cancer that develops from cells of the mesothelium, the protective lining that covers many of the internal organs of the body. Mesothelioma is most commonly caused by exposure to asbestos. The most common anatomical site for mesothelioma is the pleura (the outer lining of the lungs and internal chest wall).
Metaphase is a stage of mitosis in the cell cycle in which chromosomes are at their most condensed and coiled stage. These chromosomes, carrying genetic information, align in the equator of the cell before being separated into each of the two daughter cells.
Methylation is a biochemical process used by cells to silence certain genes, i.e. to turn off their expression as proteins within a cell.
MGMT is a gene that encodes the enzyme O-6-methylguanine-DNA methyltransferase. When this gene undergoes a chemical reaction called methylation, it is inactivated. Tumor cells that express more of the enzyme MGMT are resistant to certain types of chemotherapy, such as temozolomide. Therefore methylated (inactive) MGMT gene is a marker for tumor cells that are more sensitive to these types of chemotherapy.
Mini Mental State Exam (MMSE) is a quantification of a patient’s cognitive capabilities. The score is used in one point intervals from one to 30, with a higher score indicating better cognitive capabilities.
Mitosis is a part of the cell replication process by which chromosomes in a cell nucleus are separated into two identical sets of chromosomes, each in its own nucleus. In general, mitosis (division of the nucleus) is followed by cytokinesis, which divides the cytoplasm, organelles and cell membrane into two new cells containing roughly equal shares of these cellular components.
Mitotic furrow, or the cleavage furrow, is the indentation of the cell’s surface that begins the progression of cleavage, by which cells undergo cytokinesis, the final splitting of the membrane, in the process of cell replication. The same proteins responsible for muscle contraction, actin and myosin, begin the process of forming the cleavage furrow.
Mitotic spindle, or the spindle apparatus, refers to the subcellular structure of cells that separates chromosomes between daughter cells during cell division. Besides chromosomes, the spindle apparatus is composed of hundreds of proteins. Microtubules comprise the most abundant components of the machinery.
Mucositis is a painful inflammation and ulceration of the mucous membranes lining the digestive tract, usually as an adverse effect of chemotherapy and radiotherapy treatment for cancer.
Nitrosureas compounds are DNA alkylating agents and are often used in chemotherapy. They can cross the blood–brain barrier, making them candidates in the treatment of brain tumors.
Overall survival (OS) is a measure of how long a person lives from a certain starting time point such as diagnosis of a disease or randomization into a clinical trial. Doctors often use median overall survival time to estimate the patient’s prognosis or the effectiveness of a treatment. The FDA considers extension of OS the gold standard in effectiveness of cancer treatments.
Pemetrexed, brand name Alimta, is a chemotherapy drug manufactured and marketed by Eli Lilly and Company. Its indications are the treatment of pleural mesothelioma and non-small cell lung cancer. Pemetrexed is chemically similar to folic acid and is in the class of chemotherapy drugs called folate antimetabolites.
Phase 2 pilot trial is a trial intended to provide initial evidence of efficacy of a treatment in a specific disease state. In medical device trials, the term pilot trial also indicates that the trial will assess safety and tolerability of a device prior to definitive efficacy testing in a pivotal trial.
Progression free survival (PFS) is a measure of how long a patient remains alive without a growing cancer tumor from a certain time point such as diagnosis of a disease or randomization into a clinical trial. Doctors often use median PFS to estimate the patient’s prognosis or the effectiveness of a treatment.
PFS6 is the percentage of patients in a group who are alive and did not have tumor growth six months after entering a clinical trial. PFS6 is correlated with OS in recurrent glioblastoma and is sometimes used as a surrogate endpoint for OS in clinical trials.
Platinum-based chemotherapy drugs, informally called platins, are chemotherapeutic agents to treat cancer. The main dose-limiting side effect of cancer treatment with platinum compounds is neurotoxicity, which causes peripheral neuropathies. Platinum-based chemotherapy inhibits DNA repair and/or DNA synthesis in cancer cells.
29
Salvage therapy is a form of treatment given after a disease does not respond to standard treatment. The term is usually used to describe a final attempt to treat a deadly disease, often with little hope of extending the patient’s life.
Septins are a group of proteins found primarily in cells of animals. Different septins form protein complexes with each other. These complexes can further assemble into filaments. Assembled as such, septins function in cells by localizing other proteins, either by providing a scaffold to which proteins can attach, or by preventing diffusion of molecules from one compartment of the cell to another.
Squamous histology refers to a cancer of a kind of epithelial cell, the squamous cell. These cells are the main part of the epidermis of the skin, and this cancer is one of the major forms of skin cancer. However, squamous cells also occur in the lining of the digestive tract, lungs and other areas of the body. Squamous histology non-small cell lung cancer is a subtype of NSCLC that is usually resistant to many types of chemotherapy, such as pemetrexed.
Stereotactic radiosurgery (SRS) is a distinct neurosurgical discipline that utilizes externally generated ionizing radiation focused to damage very precisely defined targets in the head or spine without the need to make an incision. SRS is often used to treat brain metastases either alone or in combination with standard surgery.
Stromal effects refer to the role of tumor cell microenvironment (stroma) in tumor development and therapy resistance. It is known that the surrounding stroma contributes to pancreatic cancer development and progression, and may contribute to its high resistance to chemotherapy.
Taxane is a type of chemotherapy whose principal mechanism of action is the disruption of microtubule function. Microtubules are essential to cell division, and taxanes stabilize tubulin to tubulin connections in the microtubule, thereby inhibiting the process of cell division, leading to inhibition of mitosis.
Temozolomide, brand names Temodar and Temodal and Temcad, is an oral chemotherapy drug. It is an alkylating agent used as a treatment of some brain cancers, e.g., as a second-line treatment for astrocytoma and a first-line treatment for glioblastoma.
Tubulin dimers are the paired combination of a-tubulin and ß-tubulin, the proteins that make up microtubules in dividing cells. These structural proteins have a very high electric dipole, i.e., there is a large difference between the electric charge on either end of the dimer.
Tumor genetic marker is a gene that is found in a tumor and has either implications regarding prognosis or selection of therapies for a certain patient.
Unmethylated – methylation is a biochemical process used by cells to silence certain genes, i.e. to turn off their expression as proteins within a cell. When a gene is not methylated (or unmethylated) it is expressed as a protein within the cell.
Unresectable means cannot be safely removed surgically without causing unacceptable damage to normal organ function.
Whole brain radiation therapy (WBRT) is a type of external radiation therapy used to treat patients who have cancer in the brain. It is often used to treat patients whose cancer has spread to the brain, or who have more than one tumor or tumors that cannot be removed by surgery or treated with stereotactic radiotherapy. Radiation is given to the whole brain over a period of many weeks.
30
An investment in our ordinary shares involves a high degree of risk. You should carefully consider all of the information in this Annual Report on Form 10-K, including the risks and uncertainties described below, before you decide to buy our ordinary shares. Any of the following risks could have a material adverse effect on our business, prospects, financial condition and results of operations. In any such case, the trading price of our ordinary shares could decline, and you could lose all or part of your investment. In assessing these risks, you should also refer to the other information contained in this Annual Report on Form 10-K, including our consolidated financial statements and the related notes.
Risks relating to our business, TTFields and our delivery systems
Our business and prospects depend heavily on Optune, which is currently FDA-approved only for GBM. If we are unable to increase sales of Optune, obtain regulatory approvals for and commercialize Optune or our other delivery system candidates for the treatment of additional indications or are significantly delayed or limited in doing so, our business and prospects will be materially harmed.
Although we have received the FDA regulatory approvals for Optune for treatment of adult patients with newly diagnosed GBM in combination with temozolomide and recurrent GBM and the Japanese Ministry of Health, Labour and Welfare regulatory approvals for Optune for the treatment of recurrent GBM, and have affixed a CE mark to our TTFields delivery systems for certain indications in the European Union, such approvals and the CE mark affixed to our delivery systems do not guarantee future revenues for these indications, and until we receive FDA approval for the use of TTFields delivery systems for other indications, almost all of our revenues in the United States will derive from sales of Optune for newly diagnosed and recurrent GBM. The commercial success of Optune and any other delivery systems and our ability to generate and maintain revenues from the use of these delivery systems will depend on a number of factors, including:
|
· |
our ability to develop, obtain regulatory approval for and commercialize Optune and our other TTFields delivery system candidates for additional indications; |
|
· |
our ability to successfully commercialize Optune and our other delivery system candidates for approved indications in our key markets; |
|
· |
the acceptance of TTFields by patients and the healthcare community, including physicians and third-party payers (both private and public), as therapeutically effective and safe relative to the cost and safety of alternative therapies; |
|
· |
the ability to obtain and maintain sufficient coverage or reimbursement by private and public third-party payers; |
|
· |
the ability of our third-party manufacturers to manufacture Optune and other delivery systems in sufficient quantities with acceptable quality; |
|
· |
our ability to provide marketing and distribution support for Optune and our other delivery system candidates; |
|
· |
results of future clinical studies relating to TTFields or our competitors’ products; |
|
· |
the label and promotional claims allowed by the FDA and by the applicable rules on the promotion of medical devices in other foreign jurisdictions, such as in the EU member states; |
|
· |
the maintenance of our existing regulatory approvals in the United States, the European Union, Switzerland and Japan; and |
|
· |
the consequences of any reportable adverse events occurring in the United States, the European Union or other foreign jurisdictions. |
In addition, sales of Optune are limited to approved indications, which vary by geography, and the FDA label for Optune is limited in certain respects (for example, it is not approved for use in the brain stem, and is limited for use by adults ages 22 and older). Optune is also less efficacious in the cerebellum, which may reduce the number of GBM patients to whom it may be prescribed.
Our ability to generate future revenues will depend on achieving regulatory approval of, and eventual commercialization of, our most advanced delivery system candidates. However, obtaining regulatory approval of our delivery systems is not guaranteed. Our near-term prospects are substantially dependent on our ability to obtain regulatory approvals on the timetable we have anticipated, and thereafter to further successfully commercialize our delivery systems. If we are not able to receive such approvals or to further commercialize our delivery systems, or are significantly delayed or limited in doing so, our business and prospects will be materially harmed and we may need to delay our initiatives or even significantly curtail operations.
31
To date, we have incurred substantial operating losses.
We were founded in 2000, operated as a development stage company through December 31, 2011 and have incurred substantial operating losses to date. In assessing our prospects, you must consider the risks and difficulties frequently encountered by companies in new and rapidly evolving markets, particularly companies engaged in the development and sales of oncology products. These risks include our ability to:
|
· |
continue to develop and enhance Optune and our delivery system candidates; |
|
· |
obtain regulatory clearance to commercialize new delivery systems and enhance or modify our existing delivery systems; |
|
· |
increase our sales, marketing and distribution organization to commercialize our delivery systems; |
|
· |
perform clinical research and trials on TTFields; |
|
· |
establish and increase awareness and acceptance of our delivery systems; |
|
· |
implement and successfully execute our business and marketing strategy; |
|
· |
respond effectively to competitive pressures and developments; |
|
· |
maintain, protect and expand our intellectual property portfolio; |
|
· |
expand our presence and commence operations in our key markets; |
|
· |
attract, retain and motivate qualified personnel; and |
|
· |
grow our organization to support our operations and our clinical pipeline and planned commercialization efforts. |
We anticipate incurring significant costs associated with commercializing our delivery systems for approved indications. Our expenses could increase beyond expectations if we are required by the FDA, or other regulatory agencies, domestic or foreign, to change manufacturing processes for our delivery systems, or to perform clinical, nonclinical or other types of studies in addition to those that we currently anticipate. Our revenues are dependent, in part, upon the size of the markets in the jurisdictions in which we receive regulatory approval, the accepted price for our delivery systems and the ability to obtain reimbursement at such price. If the number of our addressable patients is not as significant as we estimate, the indications approved by regulatory authorities is narrower than we expect or the population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenues. If we are not able to generate significant revenues, we may never become profitable.
We can also be negatively affected by general economic conditions. We may not have insight into trends that could emerge and negatively affect our business. As a result of these or other risks, our business strategy might not be successful.
If we do not achieve our projected research and development and commercialization goals in the timeframes we announce or expect, our business would be harmed and we may need to raise additional capital to fund our operations.
For planning purposes, we estimate the timing of the accomplishment of various scientific, clinical, regulatory and other goals, which we sometimes refer to as milestones. These milestones may include the commencement or completion of scientific studies and clinical trials, the submission of regulatory filings in the United States and other foreign jurisdictions and the receipt of regulatory approvals in such jurisdictions. From time to time, we may publicly announce the expected timing of some of these milestones. All of these milestones are based on a variety of assumptions. The actual timing of the achievement of these milestones can vary dramatically from our estimates, in many cases for reasons beyond our control, depending on numerous factors, including:
|
· |
the rate of progress, costs and results of our research and development activities and clinical trials; |
|
· |
our ability to identify and enroll patients who meet clinical trial eligibility criteria; |
|
· |
the extent of scheduling conflicts with participating clinicians and clinical institutions; |
|
· |
the occurrence of unanticipated adverse events during clinical trials; |
|
· |
the receipt of approvals by our competitors and by us of our delivery systems and our competitors’ products; |
|
· |
our ability to achieve coverage and reimbursement milestones with private and governmental third-party payers; |
|
· |
our ability to access sufficient, reliable and cost-effective supplies of components used in the manufacture of our delivery systems and delivery system candidates, including the transducer arrays and other materials; |
|
· |
our ability to develop a sales and marketing organization and/or enter into sales and marketing collaborations for Optune and, if approved, our delivery system candidates; and |
|
· |
other actions by regulators. |
32
For example, our key milestones include FDA approval of our second-generation Optune delivery system for GBM; and other clinical development milestones for other indications. We can provide no assurance that we will achieve these milestones on our expected timetable, or at all.
If we do not achieve these milestones in the timeframes we expect, and/or if we are unable to obtain sufficient additional funds through financings, the proceeds from long-term loans, strategic collaborations or the license or sale of certain of our assets on a timely basis when necessary, we may be required to reduce expenses by delaying, reducing or curtailing the development of our delivery systems and we may need to raise additional capital to fund our operations, which we may not be able to obtain on favorable terms, if at all. If we fail to commence or complete, or experience delays in or are forced to curtail, our proposed clinical programs or otherwise fail to adhere to our projected development goals in the timeframes we announce or expect (or within the timeframes expected by analysts or investors), or we fail to raise any required additional capital, any of such events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
We may not be successful in our efforts to create a pipeline of delivery system candidates for future indications for TTFields and successfully commercialize them, or we may expend our resources on indications that do not yield a successful approval and fail to capitalize on other indications that may be more profitable or for which there is a greater likelihood of success.
We are pursuing clinical development of TTFields to treat a variety of solid tumors. For these future indications, we are at an early stage of development and we do not have approvals. Further, we do not intend to pursue indications involving solid tumors of the throat or extremities, and TTFields would not be efficacious for non-solid tumor cancers like lymphoma or other blood cancers.
Even if we are successful in continuing to build our pipeline, obtaining regulatory approvals and commercializing our delivery system candidates for additional indications are prone to risks of failure, including the significant risk that the development of our delivery system candidates for any potential indications will fail to demonstrate adequate efficacy or an acceptable safety profile, gain regulatory approval and become commercially viable. We cannot provide you any assurance that we will be able to advance any of these additional indications through the development and commercialization process. Our research programs may initially show promise in addressing additional indications, yet fail to yield approvals or commercialization for many reasons, including the following:
|
· |
we may not be able to assemble sufficient resources to pursue clinical trials for additional indications; |
|
· |
our delivery system candidates may not succeed in pre-clinical or clinical testing; |
|
· |
our delivery systems may on further study be shown to have harmful side effects for other indications or other characteristics that indicate they are unlikely to be effective or otherwise do not meet applicable regulatory criteria for such indications; |
|
· |
competitors may develop alternative treatments that render our delivery systems obsolete or less attractive; |
|
· |
the market for TTFields may change so that the continued development of our pipeline as currently contemplated is no longer appropriate; |
|
· |
our delivery systems may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
|
· |
our delivery systems may not be accepted as safe, effective, convenient or otherwise desirable by patients, the medical community or third-party payers. |
If any of these events occur, we may be forced to delay or abandon our development efforts for our anticipated pipeline, which would have a material adverse effect on our business and prospects and could potentially cause us to cease operations. Moreover, any such events in respect of any particular indication and/or delivery system candidate may have a negative effect on the approval process for other indications and/or result in losing approval of approved delivery systems for other indications, which may exacerbate the harm to our business and prospects.
33
We have limited experience in commercializing Optune and, to the extent we do not successfully develop this ability or contract with a third party to assist us, we may not be able to successfully commercialize our delivery systems that may be approved for commercial sale.
We currently have a small sales and marketing organization, and we may not be able to successfully develop adequate sales and marketing capabilities to achieve our growth objectives. The growth of our sales and marketing organization will require us to commit significant additional management and other resources. We will have to compete with other pharmaceutical and life sciences companies to recruit, hire, train and retain the sales and marketing personnel that we anticipate we will need. If we are unable to establish adequate sales and marketing capabilities, we will need to enter into sales and distribution agreements to market some or all of our delivery systems that may be approved for commercial sale. In addition, because Optune and future delivery systems require physician training and education, our sales and marketing organization must grow substantially as we expand our approved indications and markets. As a consequence, our expenses associated with building up and maintaining our sales force and marketing capabilities may be disproportionate to the revenues we may be able to generate on sales of Optune and other delivery systems.
If we are unable to establish adequate sales and marketing capabilities or successful distribution relationships, we may fail to realize the full sales potential of some or all of our delivery system candidates, and we may not be able to achieve the necessary growth in a cost-effective manner or realize a positive return on our investment. If we establish distribution agreements with other companies, we generally would not have control over the resources or degree of effort that any of these third parties may devote to our delivery systems, and if they fail to devote sufficient time and resources to the marketing of such delivery systems, or if their performance is substandard, it will adversely affect our revenues.
We may not be successful in achieving market acceptance of TTFields by healthcare professionals, patients and/or third-party payers in the timeframes we anticipate, or at all, which could have a material adverse effect on our business, prospects, financial condition and results of operations.
Our business model is predicated on achieving market acceptance of TTFields as a monotherapy or in combination with well-established cancer treatment modalities like surgery, radiation and chemotherapy. We may not achieve market acceptance of Optune and other TTFields delivery systems we develop in the amount of time that we have anticipated, or at all, for a number of different reasons. As a general matter, we may not achieve market acceptance of TTFields because of the following factors, among others:
|
· |
it may be difficult to gain broad acceptance of TTFields because it is a new technology and involves a novel delivery system, and as such physicians may be reluctant to prescribe TTFields delivery systems without prior experience or additional data or training; |
|
· |
it may be difficult to gain broad acceptance at community hospitals where the number of patients seeking cancer treatment may be more limited than at larger medical centers, and such community hospitals may not be willing to invest in the resources necessary for their physicians to become trained to use TTFields, which could lead to reluctance to prescribe our TTFields delivery systems; |
|
· |
patients may be reluctant to elect to use our TTFields delivery systems, including Optune, for various reasons, including a perception that the treatment is untested; |
|
· |
the delivery systems may have some side effects (for example, dermatitis where the transducer arrays are placed) and the delivery system cannot be worn in all circumstances (for example, it cannot get wet and is difficult to wear in high temperatures); and |
|
· |
the price of the TTFields delivery systems includes a monthly fee for use of the delivery system (including the transducer arrays), so as the duration of the treatment course increases, the price will increase correspondingly, and, when used in combination with other treatments, the overall cost of treatment will be greater than using a single type of treatment. |
In particular, Optune may not achieve market acceptance because of the following additional factors (which may apply to our future delivery systems, to varying degrees):
|
· |
achieving patient acceptance is difficult because GBM is a devastating disease with a poor prognosis, and not all patients with short lifespans are willing to comply with Optune therapy requirements, such as extended use of Optune, carrying around a battery pack and shaving their heads (which may be of particular concern to women), and other patients may forego Optune treatment for cosmetic or mobility reasons; |
|
· |
achieving patient compliance is difficult because the recommended average daily use of Optune is at least 18 hours a day, requiring patients to wear the delivery system nearly continuously, which to some extent restricts physical mobility because the battery must be frequently recharged, and the patient or a caregiver must ensure that it remains continuously operable; |
34
|
· |
the need to wear Optune nearly continuously in order to achieve efficacy of TTFields may also impact the pool of patients to whom physicians may be willing to prescribe treatment, as physicians may be reluctant to treat patients who are physically frail or lack caregiver support with Optune, and efficacy may also be limited in instances where patients take a break from the delivery system when experiencing skin rashes, while bathing or swimming because Optune cannot get wet, or while traveling because Optune batteries cannot be taken on airplanes, and although we ship batteries to patients, there is inevitably a disruption in continuous use; and |
|
· |
side effects reported by GBM patients treated with a combination of TTFields and temozolomide, including dermatitis where the transducer arrays are placed, headaches, weakness, falls, fatigue, muscle twitching and skin ulcers (and there may be additional side effects not yet observed). |
In addition, even if we are successful in achieving market acceptance of Optune for GBM, we may be unsuccessful in achieving market acceptance of TTFields as a treatment for other solid tumor cancers, such as brain metastases, NSCLC, pancreatic cancer, ovarian cancer, mesothelioma and other solid tumor cancers, because certain radiation or chemotherapies may remain the preferred standard of care for these indications.
There may be other factors that are presently unknown to us that also may negatively impact our ability to achieve market acceptance of TTFields delivery systems. If we do not achieve market acceptance of our delivery systems in the timeframes we anticipate, or are unable to achieve market acceptance at all, our business, prospects, financial condition and results of operations could be materially adversely affected, and our stock price could decline.
Failure to secure and maintain adequate coverage and reimbursement from third-party payers could adversely affect acceptance of our delivery systems and reduce our revenues.
We expect that the vast majority of our revenues will come from third-party payers either directly to us in markets where we provide our delivery systems to patients or indirectly via payments made to hospitals or other entities providing our delivery systems to patients. Private payers in the United States cover the largest segment of the population, with the remainder either uninsured or covered by governmental payers. We anticipate that the majority of the third-party payers outside the United States will be government agencies, government sponsored entities or other payers operating under significant regulatory requirements from national or regional governments.
Medical treatments may not be reimbursed by third-party payers based on a number of factors, such as a determination that it is experimental, not medically necessary or not appropriate for a particular patient. Currently, we are aware that in the United States several payers have issued policies that deny coverage for Optune on one or more of these bases. Additionally, private commercial and government payers may be permitted to consider the cost of a treatment in approving coverage or in setting payment for the treatment.
Private and government payers in the United States and around the world are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of U.S. federal and state governments and governments around the world. Adoption of additional price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our revenues and operating results. If third-party payers do not consider our delivery system or the combination of our delivery system with additional treatments to be cost-justified under a required cost-testing model, they may not cover our delivery systems for their populations or, if they do, the level of payment may not be sufficient to allow us to sell our delivery systems on a profitable basis.
Reimbursement for the treatment of patients with medical devices in the EU member states, Switzerland and Japan is governed by complex mechanisms established on a national level in each country. In the European Union, these mechanisms vary widely among the EU member states and evolve constantly, reflecting the efforts of these countries to reduce public spending on healthcare. As a result, obtaining reimbursement for the treatment of patients with medical devices has become more challenging. Outside the United States, the European Union and Japan, reimbursement systems vary significantly by country. We cannot, therefore, guarantee that the treatment of patients with Optune or any of our future delivery systems would be reimbursed in any of the EU member states, Switzerland, Japan or any other country.
35
We provide financial assistance to patients to defray their out-of-pocket costs for Optune, and therefore, absorb any unreimbursed costs of patients who begin treatment and are unable to pay for the costs of their treatment not covered by insurance. Our costs associated with this program could increase if payers increase the cost-sharing burden of patients.
Our failure to secure or maintain adequate coverage or reimbursement for Optune or any of our future delivery systems by third-party payers in the United States or in the other jurisdictions in which we market Optune or any of our future delivery systems, could have a material adverse effect on our business, financial condition and results of operations and cause our stock price to decline.
We may not be successful securing and maintaining reimbursement codes necessary to facilitate accurate and timely billing for Optune, future delivery systems and physician services attendant to TTFields therapy.
Third-party payers, healthcare systems, government agencies or other groups often issue reimbursement codes to facilitate billing for products and physician services used in the delivery of medicine. Within the United States, the billing codes most directly related to Optune and future delivery systems are contained in the Healthcare Common Procedure Coding System, or HCPCS code set. The HCPCS code set contains Level I codes that describe physician services, also known as Common Procedural Terminology codes, or CPT codes, and Level II codes that primarily describe products. The Centers for Medicare and Medicaid Services, or CMS, is responsible for issuing the HCPCS Level II codes. The American Medical Association issues HCPCS Level I codes.
We have secured unique HCPCS Level II codes that describe Optune and we are able to use these codes in the United States to bill third-party payers. Loss of these codes or any alteration in the payment attached to these codes would materially impact our operating results.
No CPT codes exist to describe physician services related to the delivery of TTFields therapy. We may not be able to secure CPT codes for physician services related to Optune based on the relatively low incidence of GBM. Our future revenues and results may be affected by the absence of CPT codes, as physicians may be less likely to adopt the therapy when not adequately reimbursed for the time, effort, skill, practice expense and malpractice costs required to provide the therapy to patients.
We have not secured codes to describe our delivery systems or to document physician services related to the delivery of TTFields therapy in markets outside the United States. Absence of these codes may affect the future growth of our business.
There is no assurance that Medicare or the Medicare Administrative Contractors will provide coverage or adequate payment rates for Optune or our future delivery systems.
Approximately 20% of our patients are beneficiaries under the Medicare fee-for-service program as of December 31, 2015. Failure to secure coverage and adequate payment from Medicare would reduce our revenues and may also affect the coverage and payment decisions of other third-party payers in the United States.
Medicare has the authority to issue national coverage determinations or to defer coverage decisions to its regional Medicare Administrative Contractors, or MACs. Medicare has not issued a national coverage determination for Optune. The four MACs that currently administer the durable medical equipment benefit for Medicare, or DME MACs, have each issued local coverage determination policies stating that Optune is not reasonable and necessary for the treatment of recurrent GBM. Medicare is in the process of consolidating the administration of the four DME MAC jurisdictions under just two contractors, which may negatively affect our ability to petition individual medical policy decision-makers at the MACs for coverage. The continuing absence of a positive coverage determination from Medicare or the DME MACs would materially affect our future revenues.
Additionally, Medicare has the authority to publish the price of durable medical equipment products. Medicare may publish prices for Optune or future delivery systems that do not reflect then current prices for Optune or future delivery systems. Medicare price schedules are frequently referenced by private payers in the United States and around the world. Medicare would materially reduce our revenues and operating results by publishing a price for Optune or future delivery systems that is not based on the actual price of Optune or future delivery systems within the private payer market.
CMS implemented a demonstration project in 2012 to require prior authorization for certain Durable Medical Equipment, Prosthetics, Orthotics and Supplies items. Claims for services that did not receive prior authorization before they were rendered will be automatically denied. In the event Medicare provides coverage for Optune in the future and Optune is added to the list of items requiring prior authorization that may reduce our ability to bill and secure payment for patients who would otherwise be covered to use Optune under the Medicare fee-for-service program.
We are unable to bill our existing Medicare fee-for-service patients for amounts not paid by Medicare. Therefore, we will absorb the costs of treatment for amounts not paid by Medicare.
36
Further, as of July 2013, no new Medicare coverage denial appeals are being assigned to Administrative Law Judges and no new Medicare coverage cases are being scheduled as of December 31, 2015. Thus, we anticipate that we will experience a significant delay in securing payment for Medicare patients when Medicare’s DME MACs deny coverage for patients who start therapy.
We depend on single-source suppliers for some of our components. The loss of these suppliers could prevent or delay shipments of Optune, delay our clinical trials or otherwise adversely affect our business.
We source some of the key components of Optune from only a single vendor. If any one of these single-source suppliers were to fail to continue to provide components to us on a timely basis, or at all, our business and reputation could be harmed. For example, we currently have a single source for the ceramic discs used in the transducer arrays for Optune, which we source from Harris Corporation. We currently do not have alternate suppliers for these ceramic discs, and our existing supplier would be difficult for us to replace because the ceramic discs are manufactured with specialized electrical properties designed specifically for Optune, and as such there are few vendors available to produce these components, and those that can supply them may not be able to do so on terms that are commercially favorable to us. We are in the process of identifying a second source for the ceramic discs, but we can provide no assurance that we will secure an alternate supplier on favorable terms or in time to support our commercialization efforts, or at all. Our current agreement with Harris Corporation continues through July 21, 2017, following which time the agreement will automatically renew for up to three successive two-year periods unless either we provide timely written notice of non-renewal (for any reason) or Harris Corporation provides timely written notice of non-renewal (if we fail to satisfy certain minimum purchase requirements). We currently expect that this agreement will be renewed. In addition to certain other customary termination rights, Harris Corporation can terminate this agreement with 90 days’ written notice if we breach any of our material obligations under the agreement.
Agreements with our other suppliers range from terms of four years to ten years and are terminable by either party, generally between 180 days’ and 12 months’ written notice. Establishing additional or replacement suppliers for any components of our delivery systems, and obtaining any additional regulatory approvals required to add or replace suppliers, will take a substantial amount of time and could result in increased costs and impair our ability to produce Optune, which would have a material adverse effect on our business, prospects, financial condition and results of operations. We may have difficulty obtaining similar components from other suppliers that are acceptable to the FDA or foreign regulatory authorities, or to comply with the Essential Requirements laid down in Annex I to the Directive 93/42/EEC concerning medical devices, commonly known as the Medical Devices Directive, which are the minimum requirements governing design and manufacturing in the European Union. The risks associated with the failure of our suppliers to comply with strictly enforced regulatory requirements as described below are exacerbated by our dependence on single-source suppliers. Furthermore, since some of these suppliers are located outside of the United States, we are subject to foreign export laws and United States import and customs regulations, which complicate and could delay shipments of components to us.
We are currently seeking second-source suppliers, which we expect to have under contract over the next few years, but we can provide no assurance we will achieve this on this timeframe or at all. Various steps must be taken before signing up these suppliers, including qualifying these suppliers in accordance with regulatory requirements.
If we experience any delay or deficiency in the quality of components supplied to us by third-party suppliers, or if we have to switch to replacement suppliers, we may face additional regulatory delays and the manufacture and delivery of Optune would be interrupted for an extended period of time, which could materially adversely affect our business, prospects, financial condition and results of operations. In addition, we may be required to obtain prior regulatory approval if we use different suppliers or components. Such changes could affect our FDA regulatory approvals and the compliance of our delivery systems with the Essential Requirements laid down in Annex I to the Medical Devices Directive and the validity of our current CE Certificates of Conformity. If we are required to obtain prior regulatory approval from the FDA or foreign regulatory authorities or to conduct a new conformity assessment procedure and obtain new CE Certificates of Conformity in the EU to use different suppliers or components for our delivery systems, regulatory approval or the CE Certificates of Conformity for our delivery systems may not be received on a timely basis, or at all, which would have a material adverse effect on our business, prospects, financial condition and results of operations.
Quality control problems with respect to delivery systems and components supplied by third-party vendors could have a material adverse effect on our reputation, our clinical trials or the commercialization of Optune and our future delivery systems and, as a result, a material adverse effect on our business, prospects, financial condition and results of operations.
Our delivery systems, which are manufactured by third parties, are highly technical and are required to meet exacting specifications. Any quality control problems that we experience with respect to the delivery systems and components supplied by third-party vendors could have a material adverse effect on our reputation, our attempts to complete our clinical trials or the commercialization of Optune and our future delivery systems. The failure of our suppliers to comply with strictly enforced regulatory requirements could expose us to regulatory action, including warning letters, product recalls, suspension or termination of distribution, product seizures or civil penalties. If we experience any delay or deficiency in the quality of products supplied to us by third-party suppliers, or if we have to switch to replacement suppliers, we may face additional regulatory delays and the manufacture and delivery of our delivery systems
37
would be interrupted for an extended period of time, which would materially adversely affect our business, prospects, financial condition and results of operations.
If the third parties on which we rely to conduct our clinical trials and to assist us with pre-clinical research and development do not perform as contractually required or expected, we may not be able to obtain regulatory approvals for our future delivery systems or commercialize our future delivery systems.
We do not have the ability to independently conduct our pre-clinical and clinical trials for our delivery systems and we must rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories, to conduct such trials. We and these third parties are required to comply with current good clinical practices, or cGCPs, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for TTFields in clinical development. Regulatory authorities enforce these cGCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of these third parties fail to comply with applicable cGCP regulations, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or foreign regulatory authorities may require us to perform additional nonclinical or clinical trials before approving our approved applications. We cannot be certain that, upon inspection or review of our files, such regulatory authorities will determine that any of our clinical trials comply with the cGCP regulations.
Any third parties conducting our clinical trials are not and will not be our employees and, except for remedies available to us under our agreements with such third parties, we cannot control whether or not they devote sufficient time and resources to our ongoing pre-clinical, clinical and nonclinical programs. These third parties may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical studies or other cancer treatment development activities, which could affect their performance on our behalf. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for our delivery systems or successfully commercialize our delivery systems on a timely basis, if at all, and our business, prospects and results of operations may be adversely affected.
If we encounter difficulties enrolling patients in our clinical trials, our clinical trials could be delayed or otherwise adversely affected.
The timely completion of clinical trials depends, among other things, on our ability to enroll a sufficient number of patients who remain in the trial until its conclusion. We may experience difficulties in patient enrollment in our clinical trials for a variety of reasons, including:
|
· |
the severity of the disease under investigation; |
|
· |
the size and nature of the patient population; |
|
· |
the patient eligibility criteria defined in the protocol; |
|
· |
the nature of the trial protocol, including the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects; |
|
· |
clinicians’ and patients’ perceptions as to the potential advantages and side effects of TTFields in relation to other available therapies, including any new drugs or treatments that may be approved for the indications we are pursuing; |
|
· |
availability of other clinical trials; |
|
· |
patient referral practices of physicians; |
|
· |
the ability to monitor patients adequately during and after treatment; |
|
· |
the availability of appropriate clinical trial investigators, support staff and proximity of patients to clinical sites; |
|
· |
our ability to obtain and maintain patient consents; and |
|
· |
the risk that patients enrolled in clinical trials will choose to withdraw from or otherwise not be able to complete a clinical trial. |
Patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive follow-up to assess the safety and effectiveness of TTFields or if they determine that the treatments received under the trial protocols are not attractive or involve unacceptable risks or discomforts. Patients may also not participate in our clinical trials if they choose to participate in contemporaneous clinical trials of competing products. In addition, the inclusion of critically ill patients in our clinical trials may result in deaths or other adverse medical events for reasons that may not be related to TTFields, or, in those trials where TTFields is being tested in combination with one or more other therapies, for reasons that may be attributable to the other therapies,
38
but which can nevertheless negatively affect clinical trial results. If we have difficulty enrolling a sufficient number or diversity of patients to conduct our clinical trials as planned, we may need to delay or terminate ongoing or planned clinical trials, either of which would have an adverse effect on our business.
Continued testing of Optune or our other delivery system candidates may not yield successful results and could reveal currently unknown safety hazards associated with TTFields.
Our research and development programs are designed to test the safety and efficacy of TTFields through extensive pre-clinical and clinical testing. Even if our ongoing and future clinical trials are completed as planned, we cannot be certain that their results will support our claims or that the FDA and other regulatory authorities will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our delivery system candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a delivery system candidate and may delay development of others. It is also possible that patients enrolled in clinical trials will experience adverse side effects that have not been previously observed. In addition, our pre-clinical studies and clinical trials for our delivery system candidates involve a relatively small patient population, and as a result, these studies and trials may not be indicative of future results.
We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent further commercialization of Optune and any of our delivery system candidates, including the following:
|
· |
safety and efficacy results for Optune and any of our delivery system candidates obtained in our pre-clinical and clinical testing may be inconclusive or may not be predictive of results obtained in future clinical trials, following long-term use or in much larger populations; |
|
· |
unanticipated adverse events may occur during TTFields’ clinical trials; |
|
· |
the data collected from clinical trials of our delivery system candidates may not reach statistical significance due to limited sample size or otherwise be sufficient to support FDA or other regulatory approval; and |
|
· |
our delivery system candidates may not produce the desired effects or may result in adverse health effects or other characteristics that are not currently known that preclude additional regulatory approval or limit their commercial use if approved. |
To date, patients treated with Optune in our EF-11 and EF-14 clinical trials have experienced treatment-related side effects, including dermatitis where the transducer arrays are placed, headaches, weakness, falls, fatigue, muscle twitching and skin ulcers. There may be additional side effects observed in future clinical trials and/or through real-world experience with patients using Optune or our other TTFields delivery system candidates. Undesirable side effects caused by our delivery systems could cause us or regulatory authorities to interrupt, delay or terminate clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other regulatory authorities. Results of our clinical trials could reveal a high and unacceptable severity and prevalence of side effects.
If unacceptable side effects arise in the development of our delivery system candidates, we could suspend or terminate our clinical trials or the FDA or other regulatory authorities could order us to cease clinical trials or deny approval of our delivery system candidates for any or all targeted indications, narrow the approved indications for use or otherwise require restrictive product labeling or marketing, or require further clinical trials, which may be time-consuming and expensive and may not produce results supporting FDA or other regulatory approval of our delivery system candidates in a specific indication. Treatment-related side effects could also affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We expect to have to train medical personnel using our delivery system candidates to understand the side effect profiles for our clinical trials and upon any commercialization of any of our delivery system candidates. Inadequate training in recognizing or managing the potential side effects of our delivery system candidates could result in patient injury or death. Any of these occurrences may harm our business, prospects and financial condition significantly.
Any delay or termination of our clinical trials will delay the filing of our delivery systems submissions for regulatory approvals and ultimately our ability to commercialize our delivery systems and generate revenues. Furthermore, we may abandon delivery system candidates that we previously believed to be promising. Any of these events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
39
We face competition from numerous competitors, most of whom have far greater resources than we have, which may make it more difficult for us to achieve significant market penetration and which may allow them to develop additional oncology treatments to compete with TTFields.
The oncology market is intensely competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. As a monotherapy, TTFields primarily competes with radiation and pharmacological therapies. We may face additional competition as advancements are made in the field of immuno-oncology and to date, we have not conducted any clinical trials where TTFields is used in combination with an immuno-oncological therapy. Many of our competitors are large, well-capitalized companies with significantly more market share and resources than we have. As a consequence, they are able to spend more aggressively on product development, marketing, sales and other initiatives than we can. Many of these competitors have:
|
· |
significantly greater name recognition and experience; |
|
· |
established relations with healthcare professionals, patients and third-party payers; |
|
· |
established distribution networks; |
|
· |
additional product lines, and the ability to offer rebates or bundle products to offer higher discounts or more competitive pricing or other incentives to gain a competitive advantage; and/or |
|
· |
greater financial and human resources for research and development, sales and marketing, patent litigation and/or acquisitions. |
Although we believe TTFields represents a treatment modality that can be used in combination with other cancer treatment modalities, our current competitors or other companies may at any time develop additional drugs and devices for the treatment of GBM and other solid tumors that could reduce the benefits of using our TTFields delivery systems. If an existing or future competitor develops a product that proves to be superior or comparable to Optune or any of our future delivery systems, our revenues may decline. In addition, some of our competitors may compete by changing the price of their cancer treatments. If these competitors’ products were to gain acceptance by healthcare professionals, patients or third-party payers, a downward pressure on prices could result. If prices were to fall, we may not be able to improve our gross margins or sales growth sufficiently to achieve profitability.
As we expand, we may experience difficulties managing our growth.
Our anticipated growth will place a significant strain on our management and on our operational and financial resources and systems. Failure to manage our growth effectively could materially adversely affect us. Additionally, our anticipated growth will increase the demands placed on our suppliers, resulting in an increased need for us to carefully monitor the available supply of components and quality assurance. Any failure by us to manage our growth effectively could have an adverse effect on our ability to achieve our development and commercialization goals.
Because of the specialized nature of our business, the termination of relationships with our key management and scientific personnel may prevent us from developing TTFields, conducting clinical trials and obtaining any necessary financing. Further, the inability to recruit and retain additional personnel may have an adverse effect on our ability to successfully operate our business.
For the majority of our history, Asaf Danziger and Dr. Eilon Kirson have played a significant role in our research efforts. Mr. Danziger is our Chief Executive Officer and a director of our company and Dr. Kirson is our Chief Science Officer and Head of Research and Development. We are highly dependent on these individuals, and they have played a critical role in our research and development programs, clinical trials and financing. Additionally, we have several scientific personnel with significant expertise in TTFields, some of whom are critical to our research and development efforts. The loss of the services of either of these key members of our company or any of our scientific personnel may prevent us from achieving our business objectives.
In addition to Mr. Danziger and Mr. Kirson, William F. Doyle, our chairman of the board of directors, has also played a significant role providing strategic oversight, leading our financing efforts and acting as a public ambassador for the company. Mr. Doyle is the managing member of WFD Ventures LLC, a manager of venture capital partnerships. WFD Ventures LLC is the manager of WFD Ventures Fund A and the sole member of WFD-GP II, LLC, which is the general partner of WFD Ventures Fund II. Collectively, Mr. Doyle manages venture capital partnerships that beneficially own approximately 26% of our ordinary shares at December 31, 2015. We currently have a consulting agreement with Mr. Doyle pursuant to which we pay Mr. Doyle quarterly cash payments of $75,000. We are in discussions with Mr. Doyle regarding possible employment with us. Due to certain existing obligations as managing member of WFD Ventures LLC, Mr. Doyle is restricted from receiving equity incentive compensation from us. At such time when Mr. Doyle is not subject to these restrictions (either through termination or waiver), we desire to negotiate an employment agreement with Mr. Doyle that, subject to successful conclusion and board of directors approval, is likely to include an increase in
40
cash compensation and a significant equity award under our 2015 omnibus incentive plan. The loss of Mr. Doyle’s services to our company may also prevent us from achieving our business objectives.
The competition for qualified personnel in the oncology field is intense, and we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. Our future success depends upon our ability to attract, retain and motivate highly skilled employees. In order to commercialize TTFields successfully, we will be required to expand our workforce, particularly in the areas of research and development and clinical trials, sales and marketing and supply chain management. These activities will require the addition of new personnel and the development of additional expertise by existing management personnel. We face intense competition for qualified individuals from numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well as academic and other research institutions. We may not be able to attract and retain these individuals on acceptable terms or at all. Failure to do so would materially harm our business.
Changes in tax or other laws, regulations or treaties, changes in our status under U.S. or non-U.S. laws or adverse determinations by taxing or other governmental authorities could increase our tax burden or otherwise affect our financial condition or results of operations, as well as subject our shareholders to additional taxes.
The amount of taxes we pay is subject to a variety of tax laws in the various jurisdictions in which we and our subsidiaries are organized and operate. Our domestic and international tax liabilities are dependent on the location of earnings among these various jurisdictions. Such tax liabilities could be affected by changes in tax or other laws, treaties and regulations as well as the interpretation or enforcement thereof by tax or other governmental entities in any relevant jurisdiction. The amount we pay in tax to any particular jurisdiction depends, in part, on the correct interpretation of the tax laws in such jurisdiction, and we have made a number of determinations as to the effect of such tax laws in our particular circumstances. For example, while our U.S. operations are subject to U.S. federal income tax, we believe that a significant portion of our non-U.S. operations are generally not subject to U.S. tax other than withholding taxes in certain circumstances. In some cases, the determinations we have made as to the effect of the tax laws in a particular jurisdiction depend on the continuing effectiveness of administrative rulings we have received from the tax authorities in that jurisdiction, while in other cases, our determinations are based on the reasoned judgment of our tax advisors. Although we believe that we are in compliance with the administrative rulings we have received, that the assumptions made by our tax advisors in rendering their advice remain correct, and that as a result we are in compliance with applicable tax laws in the jurisdictions where we and our subsidiaries are organized and operate, a taxing authority in any such jurisdiction may challenge our interpretation of those laws and assess us or any of our subsidiaries with additional taxes.
Additionally, from time to time, proposals have been made and legislation has been introduced (for example, the Swiss Corporate Tax Reform III, or CTR III) to change the tax laws, regulations or interpretations thereof (possibly with retroactive effect) of various jurisdictions or limit tax treaty benefits that, if enacted, could materially increase our tax burden, increase our effective tax rate or otherwise have a material adverse impact on our financial condition and results of operations. As an example, recent U.S. legislative proposals would broaden the circumstances under which a foreign corporation like us would be considered a U.S. resident for U.S. federal income tax purposes, in addition to other U.S. legislative proposals that could have a material adverse impact on us by overriding certain tax treaties and limiting the treaty benefits on certain payments, which could increase our tax liability. We cannot predict whether or when any of these potential changes in law might become effective in any jurisdiction.
While we monitor proposals and other developments that would materially impact our tax burden and effective tax rate and investigate our options accordingly, we could still be subject to increased taxation on a going forward and retroactive basis no matter what action we undertake if certain legislative proposals or regulatory changes are enacted, certain tax treaties are amended and/or our interpretation of applicable tax or other laws is challenged and determined to be incorrect. In particular, any alternative interpretations of applicable tax laws asserted by a tax authority or changes in tax laws, regulations or accounting principles that limit our ability to take advantage of tax treaties between jurisdictions, modify or eliminate the deductibility of various currently deductible payments, increase the tax burden of operating or being resident in a particular country, result in transfer pricing adjustments or otherwise require the payment of additional taxes, may have a material adverse effect on our cash flows, financial condition and results of operations.
We believe our ordinary shares should not be treated as stock of a passive foreign investment company, or PFIC, for U.S. federal income tax purposes in the current taxable year or in a future taxable year, but this conclusion is a factual determination that is made annually and thus may be subject to change. If we were to be treated as a PFIC, this could result in adverse U.S. federal income tax consequences to U.S. persons that hold our ordinary shares.
Based on the composition of our assets and the nature of our income, we believe that our shares should not be treated as stock of a PFIC for U.S. federal income tax purposes, but this conclusion is a factual determination that is made annually and thus may be subject to change.
41
A non-U.S. corporation will be treated as a PFIC for U.S. federal income tax purposes in any taxable year in which a specified percentage of its gross income is “passive income” or a specified percentage of its assets produce or are held for the production of passive income (“passive assets”), including cash. If we are treated as a PFIC, and a U.S. person that holds our ordinary shares, either directly or indirectly, did not make one of the applicable available elections, such U.S. person would be subject to adverse U.S. federal income tax consequences on distributions with respect to the ordinary shares to the extent the distributions are “excess distributions,” which are generally distributions in excess of a normal rate of distribution as calculated for PFIC purposes. Gain realized on the sale or other disposition of the ordinary shares would generally not be treated as capital gain, but rather would be treated as if such U.S. person had realized such gain and certain “excess distributions” ratably over the holding period for the ordinary shares and would be taxed at the highest tax rate in effect for each such year to which the gain was allocated, together with an interest charge in respect of the tax attributable to each such year. Partial redemptions would also be treated as excess distributions. We will, upon request from any shareholder, prepare and provide information as necessary for “qualified electing fund” elections but we make no representation as to the availability of “mark to market” elections that may mitigate the consequences of our being a PFIC to any U.S. investor. Prospective U.S. investors should consult their own U.S. tax advisors regarding the potential application of the PFIC rules.
Product liability suits, whether or not meritorious, could be brought against us due to alleged defective delivery systems or for the misuse of our delivery systems. These suits could result in expensive and time-consuming litigation, payment of substantial damages and an increase in our insurance rates.
If our current or future delivery systems are defectively designed or manufactured, contain defective components or are misused, or if someone claims any of the foregoing, whether or not meritorious, we may become subject to substantial and costly litigation. For example, we may be sued if our delivery systems cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. This may occur if Optune is misused or damaged, has a sudden failure or malfunction (including with respect to safety features) or is otherwise impaired due to wear and tear. Even absent a product liability suit, malfunctions of the device or misuse by the physician or patient would need to be remedied swiftly in order to maintain continuous use and ensure efficacy of TTFields.
Any product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the delivery system, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of Optune and our delivery system candidates. Even successful defense may require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
|
· |
decreased demand for our TTFields delivery systems; |
|
· |
injury to our reputation; |
|
· |
withdrawal of clinical trial participants and inability to continue clinical trials; |
|
· |
initiation of investigations by regulators; |
|
· |
costs to defend the related litigation; |
|
· |
a diversion of management’s time and our resources; |
|
· |
substantial monetary awards to trial participants or patients; |
|
· |
product recalls, withdrawals or labeling, marketing or promotional restrictions; |
|
· |
loss of revenues; |
|
· |
exhaustion of any available insurance and our capital resources; |
|
· |
the inability to commercialize any delivery system candidate; and |
|
· |
a decline in our share price. |
Product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizable damage awards against us. We may not have sufficient insurance coverage for all claims. Any product liability claims brought against us, with or without merit, could increase our product liability insurance rates or prevent us from securing continuing coverage, could harm our reputation in the industry and could reduce revenues. Product liability claims in excess of our insurance coverage would be paid out of cash reserves, if any, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Even if our agreements with our manufacturers and suppliers entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
42
Regional instability in Israel may adversely affect business conditions and may disrupt our business and negatively affect our revenues and results of operations.
We have research facilities located in Israel, and one of our key suppliers, which is both a component supplier and finished good manufacturer, manufactures its goods in one physical location in Israel. Accordingly, political, economic and military conditions in Israel may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel and its Arab neighbors, as well as incidents of civil unrest. In recent years, these have included hostilities between Israel and Hezbollah in Lebanon and Hamas in the Gaza strip, both of which resulted in rockets being fired into Israel, causing casualties and disruption of economic activities. While we did not sustain damages from the conflicts with Hezbollah or Hamas, our Israeli operations, which are located in Haifa, in northern Israel, are within range of Hezbollah missiles and we or our immediate surroundings may sustain damages in a missile attack, which could adversely affect our operations. Recent political events, including political uprisings, social unrest and regime change, in various countries in the Middle East and North Africa have weakened the stability of those countries, which could result in extremists coming to power. Any future armed conflicts or political instability in the region could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Parties with whom we do business have sometimes declined to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in the agreements.
Additionally, several countries, principally in the Middle East, restrict doing business with Israeli companies, and additional countries and groups may impose similar restrictions if hostilities in Israel or political instability in the region continue or increase. If recent regime changes and civil wars in neighboring states result in the establishment of fundamentalist Islamic regimes or governments more hostile to Israel, or if Egypt or Jordan abrogates its respective peace treaty with Israel, Israel could be subject to additional political, economic and military confines, which could result in a material adverse effect on our operations.
In addition, our business insurance only covers certain specified events associated with war or terrorism in the Middle East, and may not cover all such events. Additionally, although the Israeli government currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, this government coverage may not be maintained, or may be insufficient to cover all losses we incur, even if available. Any losses or damages incurred by us could have a material adverse effect on our business.
If any of our facilities are damaged or our clinical, research and development or other business processes interrupted, our business could be seriously harmed.
We conduct our business in a limited number of facilities in the United States, Switzerland, Israel and Japan. Additionally, one of our key suppliers, which is both a component supplier and finished goods manufacturer, manufactures its goods in one physical location in Israel. Damage or extended periods of interruption to our or our suppliers’ or manufacturers’ corporate, development or research facilities due to fire, natural disaster, power loss, communications failure, unauthorized entry, terrorist attacks or other events could cause us to cease or delay development of some or all of our delivery systems. Our internal computer systems may fail or suffer security breaches, which could result in a material disruption of our business. Our business may be seriously harmed by such delays and interruption.
We have significant debt service obligations and may incur additional indebtedness in the future, which could adversely affect our financial condition and results of operations and our ability to react to changes in our business.
As of December 31, 2015, we had $25.2 million of indebtedness outstanding under our Loan and Security Agreement dated as of January 7, 2015, between us, as borrower, and Biopharma Secured Investments III Holdings Cayman LP, as lender (“Term Loan Credit Facility”), and availability to borrow an additional $75.0 million thereunder. We may incur additional indebtedness in the future, including draws under our Term Loan Credit Facility. Our existing indebtedness and any additional indebtedness we may incur could require us to divert funds identified for other purposes for debt service and impair our liquidity position.
The fact that a substantial portion of our cash flow from operations could be needed to make payments on our indebtedness could have important consequences, including the following:
|
· |
increasing our vulnerability to general adverse economic and industry conditions or increased interests rates; |
|
· |
reducing the availability of our cash flow for other purposes; |
|
· |
limiting our flexibility in planning for or reacting to changes in our business and the markets in which we operate, which would place us at a competitive disadvantage compared to our competitors that may have less debt; |
|
· |
limiting our ability to borrow additional funds for working capital, capital expenditures and other investments; and |
|
· |
failing to comply with the covenants in our debt agreements could result in all of our indebtedness becoming immediately due and payable. |
43
Our ability to obtain necessary funds through borrowing, as well as our ability to service our indebtedness, will depend on our ability to generate cash flow from operations. Our ability to generate cash is subject to general economic, financial, competitive, legislative, regulatory and other factors that are beyond our control. If our business does not generate sufficient cash flow from operations or if future borrowings are not available to us under our Term Loan Credit Facility or otherwise in amounts sufficient to enable us to fund our liquidity needs, our financial condition and results of operations may be adversely affected. Our inability to make scheduled payments on our debt obligations in the future would require us to refinance all or a portion of our indebtedness on or before maturity, sell assets or seek additional equity investment. We may not be able to take any of such actions on a timely basis, on terms satisfactory to us or at all.
Covenants in our debt agreements restrict our operational flexibility.
Our Term Loan Credit Facility contains usual and customary restrictive covenants relating to the operation of our business, including restrictions on our ability:
|
· |
to incur or guarantee additional indebtedness; |
|
· |
to incur or permit to exist certain liens; |
|
· |
to enter into certain sale and lease-back transactions; |
|
· |
to make certain investments, loans and advances; |
|
· |
to effect certain mergers, consolidations, asset sales and acquisitions; |
|
· |
to pay dividends on, or redeem or repurchase, capital stock, enter into transactions with affiliates or materially change our business; and |
|
· |
to repay or modify certain other agreements with respect to other material indebtedness or modify our organizational documents. |
In addition, our Term Loan Credit Facility has a minimum liquidity covenant, which is tested quarterly. We must also meet certain annual pro forma net sales requirements.
Risks relating to regulation
Our delivery system candidates must undergo rigorous pre-clinical and clinical testing and we must obtain regulatory approvals, which could be costly and time-consuming and subject us to unanticipated delays or prevent us from marketing any delivery systems.
Our research and development activities, as well as the manufacturing and marketing of Optune and our delivery system candidates, are subject to regulation, including regulation for safety, efficacy and quality, by the FDA in the United States and comparable authorities in other countries. FDA regulations and the regulations of comparable foreign regulatory authorities are wide-ranging and govern, among other things:
|
· |
the conduct of pre-clinical and clinical studies; |
|
· |
product design, development, manufacturing and testing; |
|
· |
product labeling; |
|
· |
product storage and shipping; |
|
· |
premarket clearance, approval and conformity assessment procedures; |
|
· |
premarket clearance, approval and conformity assessment procedures for modifications introduced in marketed products; |
|
· |
post-approval market surveillance and monitoring; |
|
· |
reporting of adverse events or incidents and implementation of corrective actions, including product recalls; |
|
· |
pricing and reimbursement; |
|
· |
interactions with healthcare professionals; |
|
· |
advertising and promotion; and |
|
· |
product sales and distribution. |
44
Clinical testing can be costly and take many years, and the outcome is uncertain and susceptible to varying interpretations. Moreover, success in pre-clinical and early clinical trials does not ensure that large-scale trials will be successful or predict final results. Acceptable results in early trials may not be replicable in later trials. A number of companies in the oncology industry have suffered significant setbacks in advanced clinical trials, even after promising results in earlier trials. Negative or inconclusive results or adverse medical events during a clinical trial could cause it to be redone or terminated. In addition, failure to construct appropriate clinical trial protocols could result in the test or control group experiencing a disproportionate number of adverse events and could cause a clinical trial to be suspended, redone or terminated. We cannot be certain if or when the FDA, a foreign regulatory agency or our notified body (a private organization designated in an EU member state to conduct conformity assessment procedures under the Medical Devices Directive) might request additional studies, under what conditions such studies might be requested, or what the size or length of any such studies might be. The clinical trials of our delivery system candidates may not be completed on schedule, the FDA, foreign regulatory agencies or our notified body may order us to stop or modify our research, or these agencies or our notified body may not ultimately approve or issue a CE Certificate of Conformity for any of our delivery system candidates for commercial sale. While we have received regulatory approval for Optune for treatment of adult patients with recurrent GBM in the United States, the FDA required us to initiate a post-approval study and we have met this requirement. The data collected from our clinical trials may not be sufficient to support regulatory approval in the United States, Japan and other countries or to obtain CE Certificate of Conformity in the European Union for our various future delivery system candidates. Even if we believe the data collected from our clinical trials are sufficient, the FDA, equivalent foreign regulatory bodies and notified bodies have substantial discretion in the assessment and approval or conformity assessment processes and may disagree with our interpretation of the data. Our failure to adequately demonstrate the safety and efficacy of any of our delivery system candidates would delay or prevent regulatory approval in the United States, Japan and other countries or the CE marking in the European Union of our delivery system candidates, which could prevent us from achieving profitability.
We currently market Optune in the United States, as well as certain EU member states, Switzerland and Japan. We intend to market our TTFields delivery systems in a number of additional international markets. Although certain of our delivery systems have been approved for commercialization in Australia, Switzerland and Israel and are CE marked in the European Union, in order to market our delivery systems in other foreign jurisdictions and for other indications, we must obtain separate regulatory approvals and CE Certificates of Conformity. The requirements governing the conduct of clinical trials and manufacturing and marketing of our delivery system candidates outside the United States vary widely from country to country. Foreign regulatory approvals and CE Certificates of Conformity may take longer to obtain than FDA approvals and can require, among other things, additional testing and different clinical trial designs. Foreign regulatory approval and CE marking processes include essentially all of the risks associated with the FDA approval processes. Some foreign agencies must also approve prices of the delivery systems. Approval of a product by the FDA does not ensure approval of the same product by the health authorities of other countries or CE marking of Optune in the European Union and vice versa. In addition, changes in regulatory policy in the United States or in foreign countries for the approval or CE marking of a medical device during the period of product development and regulatory agency review or notified body review of each submitted new application may cause delays or rejections. Upcoming changes in the EU rules governing the CE marking of medical devices may also have a potential impact on the CE marking of our delivery systems in the European Union. On September 26, 2012, the European Commission adopted a package of legislative proposals designed to replace the existing regulatory framework for medical devices in the European Union. On October 22, 2013, the European Parliament voted on an amended draft of the Regulation. The proposed text is currently being discussed by the Council of the European Union, or the Council. If and when adopted, the proposed new legislation may prevent or delay the CE marking of our delivery systems under development or impact our ability to modify our currently CE marked delivery systems on a timely basis. On June 19, 2015, the Council came to a common position concerning the Council’s proposed amendments to the two draft regulations intended to replace the current Medical Devices Directive, the Active Implantable Medical Devices Directive and the In Vitro Diagnostic Medical Devices Directive. Negotiations among the Council, the European Parliament and the European Commission are ongoing. Depending on the outcome of the negotiations, the regulation on medical devices and the regulation on in vitro diagnostic medical devices could be definitively adopted in mid-2016.
We may choose to, or may be required to, suspend, repeat or terminate our clinical trials if they are not conducted in accordance with regulatory requirements, the results are negative or inconclusive or the trials are not well designed.
Clinical trials must be conducted in accordance with the FDA’s cGCPs and the equivalent laws and regulations applicable in other jurisdictions in which the clinical trials are conducted. The clinical trials are subject to oversight by the FDA, foreign regulatory agencies, ethics committees and institutional review boards at the medical institutions where the clinical trials are conducted. In addition, clinical trials must be conducted with delivery system candidates produced under the FDA’s Good Manufacturing Practices, or GMP, and in accordance with the applicable regulatory requirements in the other jurisdictions in which the clinical trials are conducted. The conduct of clinical trials may require large numbers of test patients. Patient enrollment is a function of many factors, including the size of the patient population for the target indication, the proximity of patients to clinical sites, the eligibility criteria for the trial, the existence of competing clinical trials and the availability of alternative or new treatments. Clinical trials may be suspended by the FDA or by a foreign regulatory agency at any time if the FDA or the foreign regulatory agency finds deficiencies in the conduct of these trials or it is believed that these trials expose patients to unacceptable health risks.
45
We, the FDA or foreign regulatory agencies might delay or terminate our clinical trials of a delivery system candidate for various reasons, including:
|
· |
the delivery system candidate may have unforeseen adverse side effects; |
|
· |
the time required to determine whether the delivery system candidate is effective may be longer than expected; |
|
· |
we may not agree with the FDA, a foreign regulatory authority or an ethics committee regarding the protocol for the conduct of a clinical trial; |
|
· |
fatalities may occur during a clinical trial due to medical problems that may or may not be related to clinical trial treatments; |
|
· |
the delivery system candidate may not appear to be more effective than current therapies; |
|
· |
there may be insufficient patient enrollment in the clinical trials; or |
|
· |
we may not be able to produce sufficient quantities of the delivery system candidate to complete the trials. |
Furthermore, the process of obtaining and maintaining regulatory approvals in the United States and other foreign jurisdictions and CE Certificates of Conformity in the European Union for new therapeutic products is lengthy, expensive and uncertain. It can vary substantially, based on the type, complexity and novelty of the product involved. Accordingly, any of our delivery system candidates could take a significantly longer time than we expect to, or may never, gain regulatory approval or obtain CE Certificates of Conformity in the European Union, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Healthcare reform and other legislative and regulatory changes in the United States and in other countries may adversely affect our business and financial results.
In response to perceived increases in healthcare costs in recent years, there have been and continue to be proposals by the U.S. federal government, state governments, regulators and third-party payers to control these costs and, more generally, to reform the United States healthcare system. In the United States, the Patient Protection and Affordable Care Act, or the PPACA, was enacted in 2010 with a goal of reducing the cost of healthcare and substantially changing the way healthcare is financed by both government and private insurers.
In addition, other legislative changes have been proposed and adopted in the United States since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011 created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This included aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013. On January 2, 2013, the American Taxpayer Relief Act, or the ATRA, was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals.
The U.S. Congress could pass additional healthcare laws in the future, including those that affect coverage and reimbursement for healthcare items and services, including our delivery systems. The Centers for Medicare and Medicaid Services, or CMS, also could implement regulatory changes that could affect coverage and reimbursement for our delivery systems as well. In addition, various healthcare reform proposals have also emerged at the state level. We cannot predict to what extent future healthcare initiatives will be implemented at the federal or state level or the effect any future legislation or regulation will have on us.
We believe that substantial uncertainty remains regarding the net effect of the PPACA on our business, including uncertainty over how benefit plans purchased on exchanges will cover our products, how the expansion of the Medicaid program will affect access to our products, the effect of risk-sharing payment models such as Accountable Care Organizations and other value-based purchasing programs on coverage for our product, and the effect of the general increase in Federal oversight of healthcare payers. The taxes imposed by the new federal legislation and the expansion in government’s role in the U.S. healthcare industry may result in decreased revenues, lower reimbursements by payers for our delivery systems and reduced medical procedure volumes, all of which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Specifically, beginning in January 2013, PPACA imposed a 2.3% excise tax on the constructive sale price in the United States of certain medical devices by a manufacturer, producer or importer of such devices. This tax was suspended for two years beginning January 1, 2016 and ending December 31, 2017.
46
The competent authorities in the EU member states are increasingly active in their goal of reducing public spending on healthcare. We cannot, therefore, guarantee that the treatment of patients with Optune would be reimbursed in any of the EU member states or, if successfully included on reimbursement lists will remain thereon. If adopted, the recent proposals of the European Commission for new rules governing medical devices in the European Union could impose additional requirements on manufacturers of medical devices placed on the market in the European Union. Failure to comply with these new requirements may affect our ability to market our delivery systems in the European Union.
We are subject to extensive regulation by the FDA and equivalent foreign authorities, which could restrict the sales and marketing of Optune and could cause us to incur significant costs. In addition, we may become subject to additional foreign regulation as we increase our efforts to sell Optune outside of the United States.
We sell Optune, and expect to sell our delivery system candidates, subject to extensive regulation by the FDA and numerous other federal, state and foreign governmental authorities. These regulations are broad and relate to, among other things, the conduct of pre-clinical and clinical studies, product design, development, manufacturing, labeling, testing, product storage and shipping, premarket clearance and approval, conformity assessment procedures, premarket clearance and approval of modifications introduced in marketed products, post-market surveillance and monitoring, reporting of adverse events and incidents, pricing and reimbursement, interactions with healthcare professionals, advertising and promotion and product sales and distribution. Although we have received FDA approval to market Optune in the United States for the treatment of adult patients with newly diagnosed GBM (in combination with temozolomide) and recurrent GBM, we will require additional FDA approval to market Optune for other indications. We may be required to obtain approval of a new PMA or PMA supplement application for modifications made to Optune. This approval process is costly and uncertain, and it could take one to three years, or longer, from the time the application is filed with the FDA. We may make modifications in the future that we believe do not or will not require additional approvals. If the FDA disagrees, and requires new PMAs or PMA supplements for the modifications, we may be required to recall and to stop marketing the modified versions of Optune.
In addition, before our delivery systems can be marketed in the European Union, they must obtain a CE Certificate of Conformity from a notified body. New therapeutic uses of CE marked medical devices falling outside the scope of the current CE Certificate of Conformity require a completely new conformity assessment before the device can be CE marked and marketed in the European Union for the new intended purpose.
These processes can be expensive and lengthy and entail significant fees. The process preceding CE marking of a medical device in the European Union could also be expensive and lengthy and its outcome would be uncertain. We may make modifications in the future that we believe do not or will not require additional approvals or the notification of our notified body and potentially additional conformity assessment to permit maintenance of current CE Certificate of Conformity. If the competent authorities of the EU member states or our notified body disagree and require the conduct of a new conformity assessment procedure and the modification of the existing CE Certificate of Conformity or the issuance of a new certificate, we may be required to recall or suspend the marketing of the modified versions of Optune.
In the United States and other jurisdictions, we also are subject to numerous post-marketing regulatory requirements, which include quality system regulations related to the manufacturing of our delivery systems, labeling regulations and medical device reporting regulations, which require us to report to the FDA or other foreign regulatory authorities and notified bodies if our delivery systems cause or contribute to a death or serious injury, or malfunction in a way that would likely cause or contribute to a death or serious injury. In addition, these regulatory requirements may change in the future in a way that adversely affects us. If we fail to comply with present or future regulatory requirements that are applicable to us, we may be subject to enforcement action by the FDA or other foreign regulatory authorities and notified bodies, which may include any of the following sanctions:
|
· |
untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
|
· |
unanticipated expenditures to address or defend such actions; |
|
· |
patient notification, or orders for repair, replacement or refunds; |
|
· |
voluntary or mandatory recall or seizure of our current or future delivery systems; |
|
· |
administrative detention by the FDA of medical devices believed to be adulterated or misbranded; |
|
· |
operating restrictions, suspension or shutdown of production; |
|
· |
refusal or delay of our requests for PMA approval for new intended uses or modifications to Optune; |
|
· |
refusal or delay of our requests for PMA approval of new delivery systems; |
|
· |
refusal or delay in obtaining CE Certificates of Conformity for new intended uses or modifications to Optune; |
47
|
· |
suspension, variation or withdrawal of the CE Certificates of Conformity granted by our notified body in the EU member states; |
|
· |
operating restrictions; |
|
· |
suspension or withdrawal of PMA approvals that have already been granted; |
|
· |
refusal to grant export approval for Optune or any delivery system candidates; or |
|
· |
criminal prosecution. |
The occurrence of any of these events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Modifications to Optune or any of our future approved delivery systems may require approvals of new PMA or PMA supplement applications, modified or new CE Certificates of Conformity or even require us to cease promoting or to recall the modified versions of Optune until such clearances, approvals or modified or new CE Certificates of Conformity are obtained, and the FDA, foreign regulatory authorities or our notified body may not agree with our conclusions regarding whether new approvals are required.
Any modification to a device approved through the PMA pathway that impacts the safety or effectiveness of the device requires submission to the FDA and FDA approval of a PMA supplement application or even a new PMA application, as the case may be. The FDA requires a company to make the determination as to whether a new PMA or PMA supplement application is to be filed, but the FDA may review the company’s decision. For example, in the past, we have made initial determinations that certain modifications did not require the filing of a new PMA or PMA supplement application and have notified the FDA of these changes in our PMA Annual Report, but after its review of our PMA Annual Report, the FDA requested that we submit these modifications to the FDA as a PMA supplement application. We submitted a PMA supplement application for our second generation Optune delivery system in December 2015. From time to time, we may make other changes to the software and packaging and may submit additional PMA supplement applications for these changes. FDA may conduct a facility inspection as part of its review and approval process. In addition, it is possible that the FDA will require a human factors study (user interface). It is also possible that the FDA may require clinical data, although we do not expect that clinical data will be required, as the indications for use, patient contacting element (transducer arrays) and the treatment itself have not changed. We can provide no assurance that we will receive FDA approval for these changes on a timely basis, or at all. We also may make additional changes in the future that we may determine do not require the filing of a new PMA or PMA supplement application. The FDA may not agree with our decisions regarding whether the filing of new PMAs or PMA supplement applications are required.
In addition, any substantial change introduced to a medical device CE marked in the European Union or to the quality system review by our notified body requires a new conformity assessment of the device and can lead to changes to the CE Certificates of Conformity or the preparation of a new CE Certificate of Conformity. Substantial changes include, among others, the introduction of a new intended purpose of the device, a change in its design or a change in the company’s suppliers. Responsibility for determination that a modification constitutes a substantial change lies with the manufacturer of the medical device. We must inform the notified body that conducted the conformity assessment of the delivery systems we market or sell in the European Union of any planned substantial changes to our quality system or changes to our devices which could affect compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive or the devices’ intended purpose. The notified body will then assess the changes and verify whether they affect the delivery system’s conformity with the Essential Requirements laid down in Annex I to the Medical Devices Directive or the conditions for the use of the device. If the assessment is favorable, the notified body will issue a new CE Certificate of Conformity or an addendum to the existing CE Certificate of Conformity attesting compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive. There is a risk that the competent authorities of the EU member states or our notified body may disagree with our assessment of the changes introduced to our delivery systems. The competent authorities of the EU member states or our notified body also may come to a different conclusion than the FDA on any given product modification.
If the FDA disagrees with us and requires us to submit a new PMA or PMA supplement application for then-existing modifications and/or the competent authorities of the EU member states or our notified body disagree with our assessment of the change introduced in a product, we may be required to cease promoting or to recall the modified product until we obtain approval and/or until a new conformity assessment has been conducted in relation to the product, as applicable. In addition, we could be subject to significant regulatory fines or other penalties. Furthermore, our delivery systems could be subject to recall if the FDA or the competent authorities of the EU member states or our notified body determine, for any reason, that our delivery systems are not safe or effective or that appropriate regulatory submissions were not made. Any recall or requirement that we seek additional approvals or clearances could result in significant delays, fines, increased costs associated with modification of a product, loss of revenues and potential operating restrictions imposed by the FDA or the competent authorities of the EU member states or our notified body. Delays in receipt or failure to receive approvals, the loss of previously received approvals, the failure to conduct appropriate conformity assessments prior to CE marking of our delivery systems, or the failure to comply with any other existing or future regulatory requirements, could reduce our sales, profitability and future growth prospects.
48
We will spend considerable time and money complying with federal, state and foreign regulations in addition to FDA regulations, and, if we are unable to fully comply with such regulations, we could face substantial penalties.
We are subject to extensive regulation by the U.S. federal government and the states and foreign countries in which we conduct our business. U.S. federal government healthcare laws apply when we submit a claim on behalf of a U.S. federal healthcare program beneficiary, or when a customer submits a claim for an item or service that is reimbursed under a U.S. federal government-funded healthcare program, such as Medicare or Medicaid. The laws that affect our ability to operate our business in addition to the Federal Food, Drug, and Cosmetic Act and FDA regulations include, but are not limited to, the following:
|
· |
the federal anti-kickback statute, which prohibits offering or providing remuneration of any kind for the purpose of inducing or rewarding referrals for items or services reimbursable by a federal healthcare program; |
|
· |
the U.S. federal False Claims Act, or the False Claims Act, which prohibits submitting false claims or causing the submission of false claims to the federal government; |
|
· |
Medicare laws and regulations that prescribe requirements for coverage and payment, including the conditions of participation for DME suppliers, and laws prohibiting false claims for reimbursement under Medicare and Medicaid; |
|
· |
healthcare fraud statutes that prohibit false statements and improper claims to any third-party payer; |
|
· |
the federal physician self-referral prohibition, commonly known as the Stark law, which prohibits physicians from referring Medicare patients to an entity for the provision of certain designated health services (including DME) if the physician (or a member of the physician’s immediate family) has an impermissible financial relationship with that entity; |
|
· |
similar state anti-kickback, false claims, insurance fraud and self-referral laws, which may not be limited to government-reimbursed items, as well as state laws that require us to maintain permits or licenses to distribute durable medical equipment; |
|
· |
state accreditation and licensing requirements applicable to DME providers; |
|
· |
the U.S. Foreign Corrupt Practices Act, which can be used to prosecute companies in the United States for arrangements with physicians or other parties outside the United States if the physician or party is a government official of another country and the arrangement violates the law of that country; |
|
· |
the Federal Trade Commission Act, the Lanham Act and similar federal and state laws regulating advertising and consumer protection; |
|
· |
the Physician Payments Sunshine Act, or the Sunshine Act, and similar state and foreign laws, which require reporting of payments and other transfers of value to health care practitioners periodically; and |
|
· |
the laws and codes of practices applicable in the EU member states, Switzerland and Japan concerning the marketing and promotion of medical devices, interactions with healthcare professionals, consumer protection, comparative advertising and unfair commercial practices, data protection, anti-corruption, bribery and reimbursement of medical devices. |
The laws and codes of practices applicable to us are subject to evolving interpretations. Moreover, certain federal and state laws regarding healthcare fraud and abuse and certain foreign laws regarding interactions with healthcare professionals are broad and we may be required to restrict certain of our practices to be in compliance with these laws. Similar law exists in the European Union, individual EU member states and other foreign countries. These laws are complemented by EU or national profession codes of practices. Healthcare fraud and abuse laws also are complex and even minor, inadvertent irregularities can potentially give rise to claims that a statute has been violated. If a governmental authority were to conclude that we are not in compliance with applicable laws and regulations, we and our officers and employees could be subject to severe criminal and civil penalties, including, for example, exclusion from participation as a supplier of delivery systems to beneficiaries covered by federal healthcare programs. For example, most states require us to maintain a license as a DME provider. The Medicare program requires that we maintain accreditation with an independent quality body. Loss of this accreditation would result in loss of our billing privileges to Medicare.
49
Any violation of these laws or equivalent foreign laws and codes of practices regarding interactions with healthcare professionals and bribery could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Similarly, if there is a change in law, regulation or administrative or judicial interpretations, we may have to change our business practices or our existing business practices could be challenged as unlawful, which likewise could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. In addition, although we believe that we have the required licenses, permits and accreditation to dispense our delivery systems, a regulator could find that we need to obtain additional licenses or permits. We also may be subject to audits, mandatory reaccreditation and other requirements in order to maintain our billing privileges. Failure to satisfy those requirements or successfully address any issues identified in an audit could cause us to lose our privileges to bill public and private payers. If we are required to obtain permits or licenses that we do not already possess, we also may become subject to substantial additional regulation or incur significant expense. Any penalties, damages, fines, curtailment or restructuring of our operations would adversely affect our ability to operate our business, our prospects and our financial results. In addition, any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
If we, our contract manufacturers or our component suppliers fail to comply with the FDA’s quality system regulations or equivalent regulations established in foreign countries, the manufacturing and distribution of our delivery systems could be interrupted, and our delivery system sales and results of operations could suffer.
We, our contract manufacturers and our component suppliers are required to comply with the FDA’s quality system regulations and the equivalent quality system requirements imposed by the laws and regulations in other jurisdictions, which are a complex regulatory framework that covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our delivery systems. All aspects of our supply chain are subject to periodic inspections and audits by the FDA, notified bodies and other regulatory authorities to ensure continuing compliance. We and the two critical finished goods manufacturers listed in our PMA were inspected by the FDA in the first half of 2012 and again in the fall of 2013. No material inspectional observations were identified and no FDA Form 483s were issued following these inspections. The December 2015 submission of the 180-day PMA supplement for the second generation Optune system may result in the need for another inspection in order to gain approval, We cannot assure you that our facilities or our contract manufacturers’ or component suppliers’ facilities would pass any future quality system inspection. If our or any of our contract manufacturers’ or component suppliers’ facilities fails a quality system inspection, the manufacturing or distribution of our delivery systems could be interrupted and our operations disrupted. Failure to take adequate and timely corrective action in response to an adverse quality system inspection could force a suspension or shutdown of our packaging and labeling operations or the manufacturing operations of our contract manufacturers, and lead to suspension, variation or withdrawal of our regulatory approvals or a recall of our delivery systems. If any of these events occurs, we may not be able to provide our customers with TTFields delivery systems that they require on a timely basis, our reputation could be harmed and we could lose customers, any or all of which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Our delivery systems may in the future be subject to recalls that could harm our reputation, business and financial results.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is a reasonable probability that the device would cause serious injury or death. In addition, foreign governmental bodies have the authority to require the recall of our delivery systems in the event of material deficiencies or defects in design or manufacture. Distributors and manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our manufacturers could occur as a result of component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. The FDA requires that certain classifications of recalls be reported to the FDA within ten working days after the recall is initiated. Requirements for the reporting of product recalls to the competent authorities are imposed in other jurisdictions in which our delivery systems are or would be marketed in the future. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA or to the competent authorities of other countries. In the future, we may initiate voluntary recalls involving our delivery systems that we determine do not require notification of the FDA or to other equivalent non-U.S. authorities. If the FDA or the equivalent non-U.S. authorities disagree with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA and the equivalent non-U.S. authorities could take enforcement action if we fail to report the recalls when they were conducted. Recalls of any of our delivery systems would divert managerial and financial resources and could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
50
If our delivery systems cause or contribute to a death or a serious injury, or malfunction in certain ways, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting regulations and the equivalent regulations applicable in foreign jurisdictions in which our delivery systems are or may be marketed in the future, medical device manufacturers are required to report to the FDA and to the equivalent non-U.S. authorities information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. If we fail to report these events to the FDA or to the equivalent foreign authorities within the required timeframes, or at all, the FDA or the equivalent foreign authorities could take enforcement action against us. Any such adverse event involving our delivery systems also could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
We may be subject to fines, penalties or injunctions if we are determined to be promoting the use of our delivery systems for unapproved or off-label uses.
Medical devices may be marketed only for the indications for which they are approved. Our promotional materials and training materials must comply with FDA regulations and other applicable laws and regulations governing the promotion of our delivery systems in the United States and foreign jurisdictions. Currently, Optune is approved for treatment of adult patients with newly diagnosed GBM (in combination with temozolomide) and recurrent GBM in the United States. In the European Union and Switzerland, we have CE marked the Optune delivery system for the treatment of newly diagnosed GBM (in combination with temozolomide), recurrent GBM, and advanced NSCLC (in combination with standard-of-care chemotherapy). Optune is also approved in Israel and Japan for the treatment of recurrent GBM and in Australia for the treatment of recurrent GBM and newly diagnosed GBM (in combination with temozolomide).
If the FDA or the competent authorities in other jurisdictions, including the EU member states, determine that our promotional materials or training constitutes promotion of an unapproved use, they could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, an injunction, seizure, civil fines and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and commercialization of Optune and future delivery systems would be impaired.
U.S. legislative or FDA regulatory reforms may make it more difficult and costly for us to obtain regulatory approval of our delivery system candidates and to manufacture, market and distribute our delivery systems after approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our delivery systems. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of future delivery system candidates. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our delivery systems. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be. Any change in the laws or regulations that govern the clearance and approval processes relating to our current and future delivery systems could make it more difficult and costly to obtain clearance or approval for new delivery systems, or to produce, market and distribute Optune. Significant delays in receiving clearance or approval, or the failure to receive clearance or approval for our new delivery systems would have an adverse effect on our ability to expand our business in the United States.
As a DME supplier, if we are found to have violated laws protecting the confidentiality of patient health information, we could be subject to civil or criminal penalties, which could increase our liabilities and harm our reputation or our business.
There are a number of federal and state laws protecting the confidentiality of certain patient health information, including patient records, and restricting the use and disclosure of that protected information, as well as data protection laws applicable in other jurisdictions, such as the EU member states. In particular, the U.S. Department of Health and Human Services promulgated patient privacy rules under HIPAA. These privacy rules protect medical records and other personal health information by limiting their use and disclosure, giving individuals the right to access, amend and seek accounting of their own health information and limiting most use and disclosures of health information to the minimum amount reasonably necessary to accomplish the intended purpose.
51
The collection and use of personal health data in the European Union is governed by the provisions of Directive 95/46/EC of the European Parliament and of the Council of October 24, 1995, on the protection of individuals with regard to the processing of personal data and on the free movement of such data, commonly known as the Data Protection Directive. The EU member states have adopted national laws and regulations transposing the EU Data Protection Directive into their national laws. The Data Protection Directive and related national laws impose a number of requirements, including an obligation to seek the consent of individuals to whom the personal data relates, the information that must be provided to the individuals, notification of data processing obligations to the competent national data protection authorities of EU member states and the security and confidentiality of the personal data. The Data Protection Directive also imposes strict rules on the transfer of personal data out of the European Union to the United States. We are currently assessing the impact of the recent EU decision overturning the EU-United States data protection safe harbor. Failure to comply with the requirements of the Data Protection Directive and the related national data protection laws of EU member states may result in fines and other administrative penalties.
We are affected by and subject to environmental laws and regulations that could be costly to comply with or that may result in costly liabilities.
We are affected by federal, state, foreign and local laws and regulations, including those that impose various environmental controls on the manufacturing, transportation, storage, use and disposal of batteries and hazardous chemicals and other materials used in, and hazardous waste produced by, the manufacturing of our delivery systems. We incur and expect to continue to incur costs to comply with these environmental laws and regulations. Additional or modified environmental laws and regulations, including those relating to the manufacture, transportation, storage, use and disposal of materials used to manufacture our delivery systems or restricting disposal or transportation of batteries, may be imposed that may result in higher costs.
In addition, we cannot predict the effect that additional or modified environmental laws and regulations may have on us, our third-party suppliers of equipment, batteries and our delivery systems or our customers. For example, we and our suppliers rely on an exemption from the European Directive 2011/65/EU relating to the restriction of the use of certain hazardous substances in electrical and electronic equipment relating to lead content in our transducer arrays. To the extent this exemption is revoked, it may have a material impact on our business and results of operations.
Regulations on the transportation of lithium ion batteries may affect our business.
The Air Line Pilots Association International has called on the U.S. government to prohibit shipments of lithium-ion batteries on cargo and passenger planes pending new regulations, in light of recent incidents involving a battery pack for an electric bicycle and more recently lithium ion batteries in a shipment of electronic cigarettes that may have been a contributing factor in a fire on a FedEx cargo plane. Rechargeable lithium-ion batteries are not as flammable and can be put out with fire extinguishers, but the National Transportation Safety Board has issued a series of recommendations calling for tighter regulation and testing of the batteries. In March 2014, the U.S. Department of Transportation and the Pipeline and Hazardous Materials Safety Administration issued new standards to strengthen safety conditions for the shipment of lithium-ion batteries and cells. The new rules enhance packaging and hazard communication requirements for lithium-ion batteries transported by air, adopt separate shipping descriptions for lithium-ion batteries, revise provisions for the transport of small and medium lithium-ion batteries packed with, or contained in, equipment, and harmonize the provisions for the transport of low production and prototype lithium cells and batteries with the International Civil Aviation Organization’s Technical Instructions and the International Maritime Dangerous Goods Code. In February 2015, the U.S. Postal Service revised its policies so that shipping carriers are not permitted to ship packages solely containing lithium-ion batteries internationally. Consequently, we use vendors other than the U.S. Postal Service to ship our lithium-ion batteries.
Additionally, lithium ion batteries are classified as Class 9—Miscellaneous Dangerous Goods by the International Air Transport Association, or IATA. Our batteries have passed the UN 3480 test for transport as cargo called out in the IATA guidelines and, as such, when they are properly packaged and labeled (with a class 9 sticker) they can be shipped by air transport as cargo. However, our batteries are not allowed on passenger aircraft according to the IATA standards. Consequently, we offer to ship batteries for patients who are traveling by air. If additional restrictions are put in place that limit our ability to ship our delivery systems by air freight or on water borne cargo, it could have an adverse effect on our supply chain, our inventory management procedures and processes and our ability to fill prescriptions and service patients in a timely manner, which could have a material adverse effect on our business, prospects, financial condition and results of operations.
52
Risks relating to intellectual property
If we are unable to protect our proprietary technology, trade secrets or know-how, we may not be able to operate our business profitably. Similarly, if we fail to sustain, further build and enforce our intellectual property rights, competitors may be able to develop competing therapies.
Our success depends, in part, on our ability to obtain and maintain protection for our delivery systems and technologies under the patent laws or other intellectual property laws of the United States and other countries. The standards that the U.S. Patent and Trademark Office, or USPTO, and its foreign counterparts use to grant patents are not always applied predictably or uniformly and can change. Consequently, we cannot be certain as to whether pending patent applications will result in issued patents, and we cannot be certain as to the type and extent of patent claims that may be issued to us in the future. Any issued patents may not contain claims that will permit us to stop competitors from using similar technology.
Our existing and future patent portfolio also may be vulnerable to legal challenges. The standards that courts use to interpret patents are not always applied predictably or uniformly and can change, particularly as new technologies develop. On September 16, 2011, President Obama signed into law the Leahy-Smith America Invents Act, or AIA, a significant patent law reform. The AIA implements a first-inventor-to-file standard for patent approval, changes the legal standards for patentability and creates a post-grant review system. As a result of the uncertainties of patent law in general, and surrounding the interpretation of the AIA in particular, we cannot predict with certainty how much protection, if any, will be given to our patents if we attempt to enforce them and they are challenged in court. Any attempt to enforce our intellectual property rights may also be time-consuming and costly, may divert the attention of management from our business, may ultimately be unsuccessful or may result in a remedy that is not commercially valuable. Such attempts may also provoke third parties to assert claims against us or result in our intellectual property being narrowed in scope or declared to be invalid or unenforceable.
In addition, we rely on certain proprietary trade secrets, know-how and other confidential information. We have taken measures to protect our unpatented trade secrets, know-how and other confidential information, including the use of confidentiality and assignment of inventions agreements with our employees, consultants and certain contractors. It is possible, however, that these persons may breach or challenge the agreements, that our trade secrets may otherwise be misappropriated or that competitors may independently develop or otherwise discover our trade secrets. There is therefore no guarantee that we will be able to obtain, maintain and enforce the intellectual property rights that may be necessary to protect and grow our business and to provide us with a meaningful competitive advantage, and our failure to do so could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
The oncology industry is characterized by patent and other intellectual property litigation and disputes, and any litigation, dispute or claim against us may cause us to incur substantial costs, could place a significant strain on our financial resources, divert the attention of management from our business, harm our reputation and require us to remove certain delivery systems from the market.
Whether a product infringes a patent or violates other intellectual property rights involves complex legal and factual issues, the determination of which is often uncertain. Any intellectual property dispute, even a meritless or unsuccessful one, would be time consuming and expensive to defend and could result in the diversion of our management’s attention from our business and result in adverse publicity, the disruption of research and development and marketing efforts, injury to our reputation and loss of revenues. Any of these events could negatively affect our business, prospects, financial condition and results of operations.
Third parties may assert that TTFields, Optune, our other delivery system candidates, the methods employed in the use of our delivery systems or other activities infringe on U.S. or foreign patents. Such claims may be made by competitors seeking to obtain a competitive advantage or by other parties, many of whom have significantly larger intellectual property portfolios than we have. Additionally, in recent years, individuals and groups have begun purchasing intellectual property assets for the purpose of making claims of infringement and attempting to extract settlements from companies like ours. The risk of infringement claims is exacerbated by the fact that there are numerous issued and pending patents relating to the treatment of cancer. Because patent applications can take many years to issue, and in many cases remain unpublished for many months after filing, there may be applications now pending of which we are unaware that may later result in issued patents that our delivery systems may infringe. There could also be existing patents that one or more components of our delivery systems may inadvertently infringe. As the number of competitors in the market for the treatment of cancer grows, the possibility of inadvertent patent infringement by us or a patent infringement claim against us increases. To the extent we gain greater market visibility, our risk of being subject to such claims is also likely to increase.
53
If a third party’s patent was upheld as valid and enforceable and we were found to be infringing, we could be prevented from making, using, selling, offering to sell or importing Optune or other delivery system candidates, unless we were able to obtain a license under that patent or to redesign our systems to avoid infringement. A license may not be available at all or on terms acceptable to us, and we may not be able to redesign our delivery systems to avoid any infringement. Modification of our delivery systems or development of delivery system candidates to avoid infringement could require us to conduct additional clinical trials and to revise our filings with the FDA and other regulatory bodies, which would be time-consuming and expensive. If we are not successful in obtaining a license or redesigning our delivery systems, we may be unable to make, use, sell, offer to sell or import our delivery systems and our business could suffer. We may also be required to pay substantial damages and undertake remedial activities, which could cause our business to suffer.
We may also be subject to claims alleging that we infringe or violate other intellectual property rights, such as copyrights or trademarks, may have to defend against allegations that we misappropriated trade secrets, and may face claims based on competing claims of ownership of our intellectual property. The confidentiality and assignment of inventions agreements that our employees, consultants and other third parties sign may not in all cases be enforceable or sufficient to protect our intellectual property rights. In addition, we may face claims from third parties based on competing claims to ownership of our intellectual property.
We also employ individuals who were previously employed at other medical device companies, and as such we may be subject to claims that such employees have inadvertently or otherwise used or disclosed the alleged trade secrets or other proprietary information of their former employers.
Any such litigation, dispute or claim could be costly to defend and could subject us to substantial damages, injunctions or other remedies, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
We entered into a settlement agreement in February 2015 with the Technion, whereby we agreed to resolve certain potential disputes among us, the Technion and Professor Yoram Palti arising out of certain intellectual property that Professor Palti developed while affiliated with the Technion and which Professor Palti has assigned to us. As part of the settlement, we have a contingent obligation to pay the Technion $5.5 million upon the earlier of our achieving $250.0 million of cumulative net sales since our inception, as defined in the Settlement Agreement, and any merger, consolidation, reorganization or sale or other disposition of all or substantially all of our assets.
The patent rights on which we rely to protect the intellectual property underlying TTFields delivery systems may not be adequate, which could enable third parties to use our technology or market competing products, which would harm our continued ability to compete in the market.
Our success will depend in part on our continued ability to develop or acquire commercially valuable patent rights and to protect these rights adequately. The scope of some of our patents are limited to certain ranges. For example, some of our patents protect low-intensity (1-3 V/cm), intermediate frequency (100-300 kHz) alternating electric fields, but do not cover intensities and frequencies for electric fields that are outside of these ranges. While intensities and frequencies of electric fields outside of these ranges have not yet proven to be effective treatment modalities, that may not be the case in the future. Our patent position is generally uncertain and involves complex legal and factual questions. The risks and uncertainties that we face with respect to our patents and other related rights include the following:
|
· |
the pending patent applications we have filed may not result in issued patents or may take longer than we expect to result in issued patents; |
|
· |
the pending patent applications and patents we own may be subject to interference proceedings or similar disputes over the priority of the inventions claimed; |
|
· |
the claims of any patents that are issued may not provide meaningful protection; |
|
· |
we may not be able to develop additional proprietary technologies that are patentable; |
|
· |
changes in patent laws or their interpretation in the United States and other countries (including the recently enacted AIA) could diminish the value of our patents, narrow the scope of our patent protection or adversely affect our ability to obtain new patents; |
|
· |
obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by government patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements; |
|
· |
other parties may challenge patents, patent claims or patent applications licensed or issued to us, and such patents, patent claims or patent applications may be narrowed or found to be invalid or unenforceable; and |
|
· |
other companies may design around technologies we have patented or developed. |
54
We also may fail to apply for or be unable to obtain patent rights in some foreign countries. In addition, the legal systems of certain countries may not protect our rights to the same extent as the laws of the United States, which could affect our ability to enforce patent rights effectively in such foreign countries. For a variety of reasons, we may decide not to file for patent protection for certain of our intellectual property. Our patent rights underlying TTFields, Optune and our other delivery systems may not be adequate, and our competitors or customers may design around our proprietary technologies or independently develop similar or alternative technologies or products that are equal or superior to ours without infringing on any of our patent rights. In addition, the patents licensed or issued to us may not provide a competitive advantage, and may be insufficient to prevent others from commercializing products similar or identical to ours. The occurrence of any of these events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
We have limited foreign intellectual property rights outside of our key markets and may not be able to protect our intellectual property rights throughout the world.
We have limited intellectual property rights outside of our key markets. In some countries outside the United States, we do not have any intellectual property rights, and our intellectual property rights in other countries outside the United States have a different scope and strength compared to our intellectual property rights in the United States. Consequently, we will not be able to prevent third parties from practicing our inventions in all countries outside the United States. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but where enforcement rights are not as strong as those in the United States. These products may compete with our delivery systems, and our patents or other intellectual property rights may not be effective or adequate to prevent such competition.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our delivery systems.
As is the case with other medical device companies, our success is heavily dependent on our intellectual property rights, and particularly on our patent rights. Obtaining and enforcing patents in the medical device industry involves both technological and legal complexity, and is therefore costly, time consuming and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents once obtained. Depending on decisions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could further negatively impact the value of our patents, narrow the scope of available patent protection or weaken the rights of patent owners.
Risks relating to our ordinary shares
We are an emerging growth company, and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our ordinary shares less attractive to investors.
We are an emerging growth company, as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including the ability to include only two years of audited financial statements and only two years of related management’s discussion and analysis of financial condition and results of operations disclosure, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act or any Public Company Accounting Oversight Board requirements regarding mandatory audit firm rotation or supplemental disclosures regarding the audit, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the market value of our ordinary shares held by non-affiliates exceeds $700.0 million as of any June 30 before that time, in which case, we would become a large accelerated filer under SEC rules and would no longer be an emerging growth company as of the following December 31. We cannot predict if investors will find our ordinary shares less attractive because we may rely on these exemptions. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market for our ordinary shares and our share price may be more volatile.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
55
There was no public market for our ordinary shares prior to our initial public offering, and an active trading market for our ordinary shares may not develop. As a result, you may not be able to resell your ordinary shares at or above the price you paid, or at all.
Prior to our initial public offering in October 2015 (“IPO”), there was no public market for our ordinary shares and we cannot predict the extent of investor interest in us following our IPO. An active trading market for our ordinary shares may not develop or be maintained after our IPO and the market price of our ordinary shares may decline, from time to time. The lack of an active market for our ordinary shares may impact our shareholders’ ability to sell their shares at the time they wish to sell or at a price that they consider reasonable.
The market price for our ordinary shares may be volatile, which could result in substantial losses to you.
The market price for our ordinary shares may be volatile and subject to wide fluctuations in response to factors such as publication of clinical studies relating to Optune, our other delivery system candidates or a competitor’s product, actual or anticipated fluctuations in our quarterly results of operations, changes in financial estimates by securities research analysts, negative publicity, studies or reports, changes in the economic performance or market valuations of other companies that operate in our industry, changes in the availability of third-party reimbursement in the United States or other countries, changes in governmental regulations or in the status of our regulatory approvals or applications, announcements by us or our competitors of material acquisitions, strategic partnerships, joint ventures or capital commitments, intellectual property litigation, release of lock-up or other transfer restrictions on our outstanding ordinary shares, and economic or political conditions in the United States, Israel or elsewhere. In addition, the performance, and fluctuation in market prices, of other foreign companies that have listed their securities in the United States may affect the volatility in the price of and trading volumes of our ordinary shares. Volatility in global capital markets, as was experienced during the global financial crisis beginning in 2008 and during the recent European sovereign debt crisis, as well as volatility resulting from the recent economic slowdown in Asia, could also have an adverse effect on the market price of our ordinary shares. Furthermore, the securities market has from time to time experienced significant price and volume fluctuations that are not related to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price and liquidity of our ordinary shares.
Our ordinary shares are issued under the laws of Jersey, which may not provide the level of legal certainty and transparency afforded by incorporation in a U.S. state.
We are incorporated under the laws of the Bailiwick of Jersey, Channel Islands. Jersey legislation regarding companies is largely based on English corporate law principles. However, there can be no assurance that Jersey law will not change in the future or that it will serve to protect investors in a similar fashion afforded under corporate law principles in the United States, which could adversely affect the rights of investors.
U.S. shareholders may not be able to enforce civil liabilities against us.
We are a Jersey entity with most of our assets located outside of the United States. Although we have appointed an agent for service of process in the United States for purposes of U.S. federal securities laws, a number of our directors and executive officers and a number of directors of each of our subsidiaries are not residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce against them judgments obtained in U.S. courts predicated upon the civil liability provisions of the federal securities laws of the United States.
We have been advised by our Jersey lawyers that the courts of Jersey would recognize any final and conclusive judgment under which a sum of money is payable (not being a sum payable in respect of taxes or other charges of a like nature or in respect of a fine or other penalty) obtained against us in the courts of any other territory in respect of certain enforceable obligations in accordance with the principles of private international law as applied by Jersey law (which are broadly similar to the principles accepted under English common law) and such judgment would be sufficient to form the basis of proceedings in the Jersey courts for a claim for liquidated damages in the amount of such judgment. In such proceedings, the Jersey courts would not re-hear the case on its merits save in accordance with such principles of private international law. Obligations may not necessarily be enforceable in Jersey in all circumstances or in accordance with their terms; and in particular, but without limitation: (a) any agreement purporting to provide for a payment to be made in the event of a breach of such agreement would not be enforceable to the extent that the Jersey courts were to construe such payment to be a penalty that was excessive, in that it unreasonably exceeds the maximum damages that an obligee could have suffered as a result of the breach of an obligation; (b) the Jersey courts may refuse to give effect to any provision in an agreement that would involve the enforcement of any foreign revenue or penal laws; and (c) the Jersey courts may refuse to allow unjust enrichment or to give effect to any provisions of an agreement (including provisions relating to contractual interest on a judgment debt) that it considers usurious.
56
Our annual and quarterly results may fluctuate due to a number of factors and, as a result, could fall below investor expectations or estimates by securities research analysts, which may cause the trading price of our ordinary shares to decline.
Our revenues and results of operations are difficult to predict, and potentially may vary significantly from period to period. As a result of a number of factors, many of which are beyond our control, it is possible that results of operations for future periods may be below the expectations of public market analysts and investors, which could cause our stock price to decline. Factors that may affect our quarterly results include, but are not limited to:
|
· |
failure to obtain regulatory approval for our delivery systems; |
|
· |
failure to effectively commercialize our delivery systems; |
|
· |
competition; and |
|
· |
changes in the laws and regulations that affect our operations. |
As a result, investors should not rely on year-to-year or quarter-to-quarter comparisons of results of operations as an indication of future performance.
Substantial future sales of our ordinary shares in the public market, or the perception that such sales may occur, could cause the price of our ordinary shares to decline.
Sales of our ordinary shares in the public market, or the perception that these sales may occur, could cause the market price of our ordinary shares to decline. All ordinary shares sold in our IPO (other than any shares acquired by our affiliates) are freely transferable without restriction or additional registration under the Securities Act of 1933, as amended, or the Securities Act. Substantially all of the remaining ordinary shares outstanding after our IPO in October 2015 will be available for sale upon the expiration of the 180-day lock-up period, subject to volume, notice and manner of sale restrictions as applicable to our affiliates in the United States under Rule 144 and Rule 701 under the Securities Act. Any or all of these ordinary shares may be released prior to expiration of the lock-up period at the discretion of J.P. Morgan Securities LLC. To the extent ordinary shares are released before the expiration of the lock-up period and these ordinary shares are sold into the market, the market price of our ordinary shares could decline.
Our executive officers, directors and principal shareholders may exert control over us and may be able to exercise influence over matters subject to shareholder approval.
Our executive officers and directors, together with their respective affiliates, beneficially own approximately 33.6% of our outstanding ordinary shares as of December 31, 2015. Accordingly, these shareholders, if they act together, will be able to exercise substantial influence over all matters requiring shareholder approval, including the election of directors and approval of corporate transactions, such as a merger. This concentration of ownership could have the effect of delaying or preventing a change in our control or otherwise discouraging a potential acquirer from attempting to obtain control of us, which in turn could have a material adverse effect on the market value of our ordinary shares.
Our memorandum and articles of association contain anti-takeover provisions that could adversely affect the rights of holders of our ordinary shares.
Our amended and restated memorandum and articles of association, referred to as the memorandum and articles of association, contain certain provisions that could limit the ability of third parties to acquire control of our company, including a provision for a classified board of directors and a provision that grants authority to our board of directors to issue from time to time one or more classes of preferred shares without action by our shareholders and to determine, with respect to any class of preferred shares, the terms and rights of that class. The provisions could have the effect of depriving our shareholders of the opportunity to sell their ordinary shares at a premium over the prevailing market price by discouraging third parties from seeking to obtain control of our company in a tender offer or similar transactions.
Our management has broad discretion over the use and investment of the net proceeds we received in our initial public offering and may not apply the proceeds in ways that increase the value of your investment.
Our management has broad discretion over the use and investment of the net proceeds we received from our IPO, and you will be relying on, and may not agree with, the judgment of our management regarding the application of these net proceeds. Our management intends to use the net proceeds for working capital and general corporate purposes, including clinical trials, research and development and continued commercialization of Optune and future delivery systems. The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business and cause the price of our ordinary shares to decline. Pending these uses, we may invest the net proceeds we receive from our IPO in a manner that does not produce income or loses value.
57
If securities or industry analysts do not publish research or publish unfavorable or inaccurate research about our business, our share price and trading volume could decline.
The trading market for our ordinary shares will continue to depend, in part, on the research and reports that securities or industry analysts publish about us or our business. We may be unable to sustain coverage by well-regarded securities and industry analysts. If either none or only a limited number of securities or industry analysts maintain coverage of our company, or if these securities or industry analysts are not widely respected within the general investment community, the trading price for our ordinary shares would be negatively impacted. In the event we obtain securities or industry analyst coverage, if one or more of the analysts who cover us downgrade our ordinary shares or publish inaccurate or unfavorable research about our business, the price of our ordinary shares would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our ordinary shares could decrease, which might cause the price of our ordinary shares and trading volume to decline.
We incur significant costs on an ongoing basis as a result of operating as a company whose ordinary shares are publicly traded in the United States, and our management will be required to devote substantial time to new compliance initiatives, including after we are no longer an emerging growth company.
As a company whose shares are publicly traded in the United States, we incur significant legal, accounting and other expenses on an ongoing basis. In addition, the Sarbanes-Oxley Act of 2002, or Sarbanes-Oxley Act, and the rules of the SEC and The NASDAQ Stock Market LLC, or NASDAQ, have imposed various requirements on public companies, including requirements for the establishment and maintenance of effective disclosure controls and internal control over financial reporting. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
When the emerging growth company exemptions under the JOBS Act cease to apply, we expect to incur additional expenses and devote increased management effort toward ensuring compliance with reporting requirements. We cannot predict or estimate the amount of additional costs we may incur as a result of losing our emerging growth company status or the timing of such costs.
If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired and investors’ views of us could be harmed.
As a public company, we are required to maintain internal control over financial reporting and to report any material weaknesses in such internal controls. In addition, beginning with our annual report on Form 10-K for our fiscal year ending December 31, 2016 to be filed in 2017, we will be required to furnish a report by management on the effectiveness of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act. We are in the process of designing, implementing and testing the internal control over financial reporting required to comply with this obligation, which process is time-consuming, costly and complicated. In addition, our independent registered public accounting firm will be required to attest to the effectiveness of our internal control over financial reporting beginning with our annual report on Form 10-K following the date on which we are no longer an emerging growth company, which may be up to five full years following the date of our IPO in October 2015. Our compliance with Section 404 of the Sarbanes-Oxley Act will require that we incur substantial accounting expense and expend significant management efforts. We currently do not have an internal audit function, and we will need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge. If we are not able to comply with the requirements of Section 404 in a timely manner, or if we or our independent registered public accounting firm identify deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our ordinary shares could decline and we could be subject to sanctions or investigations by NASDAQ, the SEC or other regulatory authorities, which would require additional financial and management resources.
Our ability to implement our business plan successfully and comply with Section 404 requires us to be able to prepare timely and accurate financial statements. We expect that we will need to continue to improve existing, and implement new, operational and financial systems, procedures and controls to manage our business effectively. Any delay in the implementation of, or disruption in the transition to, new or enhanced systems, procedures or controls, may cause our operations to suffer and we may be unable to conclude that our internal control over financial reporting is effective and to obtain an unqualified report on internal controls from our auditors when required under Section 404 of the Sarbanes-Oxley Act. Moreover, we cannot be certain that these measures would ensure that we implement and maintain adequate controls over our financial processes and reporting in the future. Even if we were to conclude, and, when required, our auditors were to concur, that our internal control over financial reporting provided reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, because of its inherent limitations, internal control over financial reporting may not prevent or detect fraud or misstatements or omissions.
58
We have never declared or paid cash dividends on our ordinary shares and we do not anticipate paying dividends in the foreseeable future and, as a result, you must rely on price appreciation of our ordinary shares for a return on your investment.
We have never declared or paid cash dividends on our ordinary shares. We currently intend to retain our funds and any future earnings to support the operation, growth and development of our business. Any future determination to declare cash dividends will be made at the discretion of our board of directors and subject to compliance with applicable laws and covenants under our current or any future credit facilities, which may restrict or limit our ability to pay dividends, and the form, frequency and amount of dividends will depend upon our future operations and earnings, capital requirements and surplus, general financial condition, contractual restrictions and other factors that our board of directors may deem relevant. We do not anticipate paying any cash dividends on our ordinary shares in the foreseeable future. As a result, a return on your investment will only occur if our share price appreciates. There is no guarantee that our ordinary shares will appreciate in value or even maintain the price at which you purchased your ordinary shares. You may not realize a return on your investment in our ordinary shares and you may even lose your entire investment in our ordinary shares.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We currently lease space in several countries, including the following properties with square footage in the approximate amounts noted below:
|
|
· |
35,000 square feet of office and warehouse space in Portsmouth, New Hampshire which houses our U.S. operations center; |
|
|
· |
11,600 square feet of office space in Malvern, Pennsylvania which houses our finance, legal and information technology operations; |
|
|
· |
11,400 square feet of office and warehouse space in Lucerne, Switzerland which houses our global supply chain and European commercial operations; |
|
|
· |
10,900 square feet of office and laboratory space in Haifa, Israel which houses our research and development operations; |
|
|
· |
8,000 square feet of office space in New York, New York which houses our U.S. commercial operations; and |
|
|
· |
4,500 square feet of office space in Tokyo, Japan which houses our Japanese operations. |
We believe that our current facilities are adequate for our present purposes, but we may need additional space as we continue to grow and expand our operations. We believe that suitable additional or alternative office, laboratory, and manufacturing space would be available as required in the future on commercially reasonable terms.
We currently are not subject to any material pending legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURES
None.
Executive Officers of the Registrant
Our executive officers are elected by and serve at the discretion of our board of directors. The following table lists information about our executive officers:
|
Name |
|
Age |
|
Position |
|
Asaf Danziger |
|
49 |
|
Chief Executive Officer and Director |
|
|
|
|
|
|
|
Michael Ambrogi |
|
52 |
|
Chief Operating Officer |
|
|
|
|
|
|
|
Wilhelmus Groenhuysen |
|
58 |
|
Chief Financial Officer |
|
|
|
|
|
|
|
Eilon Kirson, M.D., Ph.D. |
|
47 |
|
Chief Science Officer and Head of Research and Development |
|
|
|
|
|
|
|
Peter Melnyk |
|
53 |
|
Chief Commercial Officer |
59
Asaf Danziger has served as our Chief Executive Officer since 2002 and has been a director of NovoCure since 2012. From 1998 to 2002, Mr. Danziger was CEO of Cybro Medical, a subsidiary of Imagyn Medical Technologies, Inc. Mr. Danziger holds a B.Sc. in material engineering from Ben Gurion University of the Negev, Israel. We believe that Mr. Danziger is qualified to serve on our board of directors due to his service as our Chief Executive Officer and his extensive knowledge of our company and industry.
Michael Ambrogi has been our Chief Operating Officer since 2010 and previously served as our U.S. General Manager from 2006 to 2010. Mr. Ambrogi has overall responsibility for our ongoing operations, engineering, manufacturing and service and human resources activities worldwide. From 1991 to 2006, Mr. Ambrogi worked for Deka Research and Development Corporation, inventor Dean Kamen’s research and development firm, last serving a general manager. Mr. Ambrogi led Deka’s teams on many products including the Baxter HomeChoice peritoneal dialysis machine, the Davol Hydroflex surgical irrigation device, the Cordis Crowne Stent and the J&J IBOT 3000 mobility system. Earlier in his career, from 1988 to 1990, Mr. Ambrogi was a consultant with McKinsey & Company. Mr. Ambrogi holds a S.B. in mechanical engineering from MIT.
Wilhelmus Groenhuysen has been our Chief Financial Officer since 2012. From 2007 to 2011, Mr. Groenhuysen worked for Cephalon, Inc., last serving as executive vice president and chief financial officer, where he had responsibility for worldwide finance, commercial operations and risk management. From 1987 to 2007, Mr. Groenhuysen worked for Philips Group in various assignments in Europe, Asia and the United States, the latest of which started in 2002 when he was promoted to chief financial officer and senior vice president of Philips Electronics North America Corporation. Mr. Groenhuysen holds a Master’s Degree in Business Economics from VU University Amsterdam and graduated as a Registered Public Controller at VU University Amsterdam.
Eilon Kirson, M.D., Ph.D. has been our Chief Science Officer and Head of Research and Development since 2012 and previously served as our Chief Medical Officer from 2006 to 2012. Dr. Kirson has led the development of TTFields from pre-clinical testing to large, multi-center phase 3 studies and through multiple regulatory approvals. Dr. Kirson previously served as head of electrophysiology at Carmel Biosensors Ltd. Dr. Kirson received his B.Med.Sc., M.D. and Ph.D. in Physiology-Biophysics from the Hebrew University of Jerusalem and served his residency in neurology at Sheba Medical Center, Tel Ha-Shomer Hospital, Israel.
Peter Melnyk has been our Chief Commercial Officer since 2011. Mr. Melnyk has overall responsibility for directing our global marketing and sales efforts. Mr. Melnyk was previously senior vice president for sales and marketing at OSI Pharmaceuticals, Inc. where he led the global commercialization efforts for Tarceva from 2003 to 2011. Prior to OSI, Mr. Melnyk was executive director of oncology at Pfizer Inc. and a director of oncology at Bristol-Myers Squibb Company. Mr. Melnyk holds a B.Sc in animal science and M.Sc in reproductive endocrinology from McGill University.
60
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our ordinary shares are quoted on the NASDAQ Global Select Market under the symbol “NVCR.” Our ordinary shares began to be quoted on the NASDAQ Global Select Market on October 2, 2015. The following table sets forth the range of high and low sale prices for our ordinary shares as reported on the NASDAQ Global Select Market for the period indicated below.
|
|
|
High |
|
|
Low |
|
||
|
2015 |
|
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
30.89 |
|
|
$ |
15.01 |
|
Holders of Ordinary Shares
As of February 26, 2016, there were 148 holders of record of our ordinary shares. On February 26, 2016, the last reported sale price of our ordinary shares as reported on the NASDAQ Global Select Market was $11.17 per share.
Dividend Policy
We have not paid any dividends on our ordinary shares since our inception and do not anticipate paying any dividends on our ordinary shares in the foreseeable future.
Performance Graph
This graph is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference into any of our filings under the Securities Act of 1933, as amended (the “Securities Act”), or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
61
The following graph shows the total stockholder return of an investment of $100 in cash at market close on October 2, 2015 (the first day of trading of our ordinary shares) through December 31, 2015 for (1) our ordinary shares, (2) the Russell 2000 Index, and (3) the Nasdaq Biotechnology Index. Pursuant to applicable SEC rules, all values assume reinvestment of the full amount of all dividends; however, no dividends have been declared on our ordinary shares to date. The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
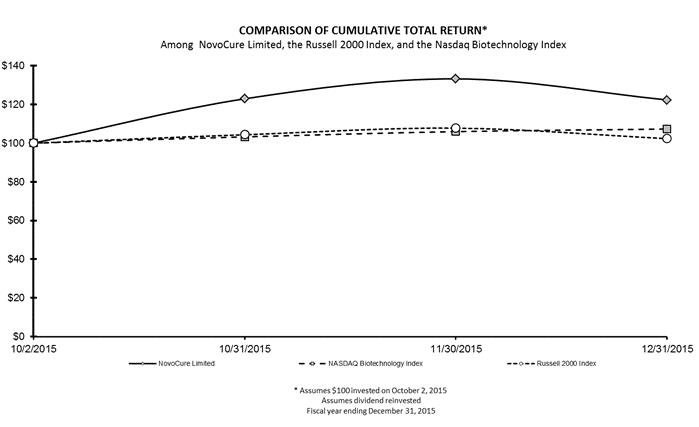
TOTAL RETURN ANNUAL COMPARISON
CUMULATIVE TOTAL RETURN SUMMARY
|
|
|
|
|
10/2/2015 |
|
|
10/31/2015 |
|
11/30/2015 |
|
12/31/2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NovoCure Limited |
|
Return % |
|
|
|
|
|
22.92 |
|
8.37 |
|
-8.17 |
|
|
|
Cum $ |
|
|
100.00 |
|
|
122.92 |
|
133.21 |
|
122.32 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NASDAQ Biotechnology Index |
|
Return % |
|
|
|
|
|
3.25 |
|
2.62 |
|
1.25 |
|
|
|
Cum $ |
|
100 |
|
|
103.25 |
|
105.96 |
|
107.29 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Russell 2000 Index |
|
Return % |
|
|
|
|
|
4.35 |
|
3.25 |
|
-5.02 |
|
|
|
Cum $ |
|
|
100.00 |
|
|
104.35 |
|
107.74 |
|
102.33 |
62
Recent Sales of Unregistered Securities
(a) From January 1, 2015 to December 31, 2015, we have issued the following securities, which include convertible preferred shares and options to acquire our ordinary shares. We believe that each of the following instances was exempt from registration under the Securities Act in reliance on Regulation S under the Securities Act, under Section 4(a)(2) of the Securities Act regarding transactions not involving a public offering and under Rule 701 promulgated under the Securities Act:
|
Purchaser |
|
Date of sale or issuance |
|
Number of securities |
|
|
Consideration |
|
||
|
Ordinary shares (1) |
|
March 2015 |
|
1,005,210 |
|
|
|
- |
|
|
|
Series J Preferred Share Purchasers |
|
June 2015 |
|
4,068,500 |
|
|
$94.6 million |
|
||
|
Options to Purchase Ordinary Shares (2) |
|
January 1, 2015 to December 31, 2015 |
|
|
4,113,603 |
|
|
|
- |
|
|
Issuance of Ordinary shares upon IPO and exercise of over-allotment, net |
|
October2015 |
|
|
7,876,195 |
|
|
$157.5 million |
|
|
|
Ordinary Share Option and Warrants Exercisers |
|
January 1, 2015 to December 31, 2015 |
|
|
731,665 |
|
|
$2.04 million |
|
|
|
(1) |
Ordinary shares issued to the Technion. See Note 14c to our consolidated financial statements included in Part II, Item 8 of this Annual Report on Form 10-K. |
|
(2) |
Number of securities includes an option to purchase 1,005,210 ordinary shares issued to the Technion in March 2015. See Note 14c to our consolidated financial statements included in Part II, Item 8 of this Annual Report on Form 10-K. |
(b) On October 7, 2015, we closed our IPO, in which we sold an aggregate of 7,500,000 ordinary shares at a price to the public of $22.00 per share. On October 19, 2015, the underwriters to the IPO partially exercised their over-allotment option, in which we sold an aggregate of 376,195 ordinary shares at a price to the public of $22.00 per share. We received net proceeds from the IPO and the over-allotment option of $157.5 million, after deducting the underwriting discounts, commissions and offering expenses payable by us. The offer and sale of all of the ordinary shares in the IPO and the over-allotment option were registered under the Securities Act pursuant to a registration statement on Form S-1 (File No. 333-206681), which was declared effective by the SEC on October 1, 2015 (the “Registration Statement”).
There has been no material change in the planned use of proceeds from our IPO as described in the Registration Statement. No amount of proceeds has been used to date, as we had sufficient cash on hand prior to the IPO to fund our near-term activities. We primarily invested the proceeds received in short-term, interest-bearing investment-grade securities and government securities. None of the offering proceeds were paid directly or indirectly to any of our directors or officers (or their associates) or persons owning 10.0% or more of any class of our equity securities or to any other affiliates.
Issuer Purchases of Equity Securities
Not applicable.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table gives information about our ordinary shares that may be issued upon the exercise of stock options under all of our existing equity compensation plans as of December 31, 2015, including the 2003 Share Option Plan (the “2003 Plan”), the 2013 Share Option Plan (the “2013 Plan”) and the 2015 Omnibus Incentive Plan (the “2015 Plan”).
Equity Compensation Plan Information
|
Plan Category |
|
(a) Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights(1) |
|
|
(b) Weighted Average Exercise Price of Outstanding Options, Warrants and Rights |
|
|
(c) Number of Securities Remaining Available for Future Issuance (Excludes Securities Reflected in Column (a)) |
|
|||
|
Equity compensation plans approved by shareholders |
|
|
10,134,829 |
|
|
|
8.20 |
|
|
|
14,947,667 |
|
|
Equity compensation plans not approved by shareholders (4) |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Total |
|
|
10,134,829 |
|
|
|
8.20 |
|
|
|
14,947,667 |
|
63
ITEM 6. SELECTED FINANCIAL DATA
(In thousands, except per share data)
The following tables set forth our consolidated statements of operations data:
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands, except share and per share data |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Net revenues |
|
$ |
33,087 |
|
|
$ |
15,490 |
|
|
$ |
10,359 |
|
|
Cost of revenues |
|
|
20,610 |
|
|
|
10,036 |
|
|
|
7,013 |
|
|
Gross profit |
|
|
12,477 |
|
|
|
5,454 |
|
|
|
3,346 |
|
|
Operating costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research, development and clinical trials |
|
|
43,748 |
|
|
|
40,381 |
|
|
|
34,797 |
|
|
Sales and marketing |
|
|
38,861 |
|
|
|
21,177 |
|
|
|
16,406 |
|
|
General and administrative |
|
|
33,864 |
|
|
|
24,052 |
|
|
|
16,602 |
|
|
Total operating costs and expenses |
|
|
116,473 |
|
|
|
85,610 |
|
|
|
67,805 |
|
|
Operating loss |
|
|
(103,996 |
) |
|
|
(80,156 |
) |
|
|
(64,459 |
) |
|
Financial expenses, net |
|
|
(3,151 |
) |
|
|
(144 |
) |
|
|
(12,558 |
) |
|
Loss before income taxes |
|
|
(107,147 |
) |
|
|
(80,300 |
) |
|
|
(77,017 |
) |
|
Income taxes |
|
|
4,434 |
|
|
|
382 |
|
|
|
353 |
|
|
Net loss |
|
$ |
(111,581 |
) |
|
$ |
(80,682 |
) |
|
$ |
(77,370 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per ordinary share |
|
$ |
(3.67 |
) |
|
$ |
(6.46 |
) |
|
$ |
(6.73 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of ordinary shares used in computing basic and diluted net loss per share |
|
|
30,401,603 |
|
|
|
12,490,017 |
|
|
|
11,498,392 |
|
Non-cash share-based compensation expense included in costs and expenses:
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Cost of revenues |
|
$ |
174 |
|
|
$ |
32 |
|
|
$ |
52 |
|
|
Research, development and clinical trials |
|
|
2,529 |
|
|
|
820 |
|
|
|
1,137 |
|
|
Sales and marketing |
|
|
2,496 |
|
|
|
1,104 |
|
|
|
791 |
|
|
General and administrative |
|
|
6,661 |
|
|
|
2,668 |
|
|
|
3,140 |
|
|
Total share-based compensation expense |
|
$ |
11,860 |
|
|
$ |
4,624 |
|
|
$ |
5,120 |
|
Consolidated balance sheet data:
|
|
|
December 31, |
|
|||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
||
|
Cash and cash equivalents |
|
$ |
119,423 |
|
|
$ |
57,613 |
|
|
Short-term investments |
|
|
150,001 |
|
|
|
44,999 |
|
|
Total assets |
|
|
307,336 |
|
|
|
117,876 |
|
|
Working capital |
|
|
265,277 |
|
|
|
94,161 |
|
|
Current liabilities |
|
|
28,627 |
|
|
|
17,669 |
|
|
Long term liabilities |
|
|
27,889 |
|
|
|
2,332 |
|
|
Total shareholders' equity |
|
$ |
250,820 |
|
|
$ |
97,875 |
|
64
|
|
|
Year Ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Net cash used in operating activities |
|
$ |
(99,884 |
) |
|
$ |
(74,244 |
) |
|
$ |
(52,717 |
) |
|
Net cash provided by (used in) investing activities |
|
|
(115,295 |
) |
|
|
(46,182 |
) |
|
|
10,402 |
|
|
Net cash provided by financing activities |
|
|
276,989 |
|
|
|
2,145 |
|
|
|
183,318 |
|
|
Net increase (decrease) in cash and cash equivalents |
|
$ |
61,810 |
|
|
$ |
(118,281 |
) |
|
$ |
141,003 |
|
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) is intended to provide information to assist you in better understanding and evaluating our financial condition and results of operations. We encourage you to read this MD&A in conjunction with our consolidated financial statements included in Part II, Item 8 of this Annual Report on Form 10-K and the “Risk Factors” contained in Part I, Item 1A of this Annual Report on Form 10-K.
Overview
We are a commercial-stage oncology company developing a novel, proprietary therapy called TTFields for the treatment of solid tumor cancers. TTFields is a low-toxicity anti-mitotic treatment that uses low-intensity, intermediate frequency, alternating electric fields to exert physical forces on key molecules inside cancer cells, disrupting the basic machinery necessary for normal cell division, leading to cancer cell death. Physicians have typically treated patients with solid tumors using one or a combination of three principal treatment modalities — surgery, radiation and pharmacological therapies. Despite meaningful advancements in each of these modalities, a significant unmet need to improve survival and quality of life remains. We believe we will establish TTFields as a new treatment modality for a variety of solid tumors that increases survival without significantly increasing side effects when used in combination with other cancer treatment modalities.
We view our operations and manage our business in one operating segment. We have incurred significant losses and cumulative negative cash flows from operations since our founding in 2000. Our net losses were $111.6 million for the year ended December 31, 2015, $80.7 million for the year ended December 31, 2014 and $77.4 million for the year ended December 31, 2013. As of December 31, 2015, 2014 and 2013 we had an accumulated deficit of $388.1 million, $276.5 million and $195.8 million, respectively. Our net losses primarily resulted from costs incurred in connection with our pre-clinical and clinical trial programs, costs incurred in our commercial launch efforts and general and administrative costs necessary to operate as a multi-national oncology business. To date, we have financed our operations primarily through the issuance and sale of our convertible preferred shares, ordinary shares and the proceeds from long-term loans. As of December 31, 2015, we had received a total of $614.0 million from the sale of our convertible preferred shares, including the sale of our Series J convertible preferred stock in June 2015 for net proceeds of $94.6 million, all of which were converted into our ordinary shares after the consummation of our IPO, and including $157.5 million proceeds from our IPO including the partial exercise of the underwriters' overallotment option and the exercise of ordinary share warrants and options.
In October 2015, we received FDA approval to market and sell Optune for the treatment of adult patients with newly diagnosed glioblastoma in combination with temozolomide. We are actively marketing for that indication in the United States, Germany and Switzerland. In the same month, we also received CE mark for our newly designed second generation Optune system and have since made it available to all new patients in Europe. In December 2015, we submitted the registration dossier for premarketing approval of Optune to treat newly diagnosed GBM to the JPMDA. The same month, we also filed a 180-day PMA supplement application with the FDA, seeking approval to market the second generation of Optune in the United States for its approved indications. We believe that TTFields will transform the standard of care for patients with newly diagnosed and recurrent GBM.
We expect to continue to incur significant expenses and operating losses for at least the next several years. We expect our research, development and clinical trials expenses to increase in connection with our ongoing activities, and as additional indications enter late-stage clinical development. In addition, we expect to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. We may need additional funding to support the continuation of our operating activities. Until we can generate substantial revenues (which may not occur), we expect to finance our cash needs through the proceeds of our IPO and availability under our Term Loan Credit Facility, and possibly also from collaborations, strategic alliances, licensing arrangements and other marketing and distribution arrangements. We will need to generate significant revenues to achieve profitability, and we may never do so.
65
Net revenues
Substantially all of our revenues are derived from patients using our TTFields delivery system, marketed as Optune in our currently active markets. We charge patients or their third-party healthcare payers on a monthly basis. Our potential revenues per patient are determined by the monthly fee we collect and the number of months that the patient remains on therapy. The monthly charge is $21,000 in the United States and €21,000 in the European Union.
We account for revenues when cash is collected and all other revenue recognition criteria have been met. When we have sufficient history of collections from the third party payers or third-party payer groups and can demonstrate that we can make a reasonable estimate of amounts that will ultimately be collected, we will recognize revenues ratably over the term of patients’ use of Optune and approved future delivery systems. Revenues are presented net of indirect taxes, including the U.S. medical device excise.
The difference between billed and paid amounts consists of disputed underpayments, patient financial assistance and discounts. Patient assistance programs include assistance to reduce cost-share burdens imposed on patients by the payer (for example, co-insurance and co-payments), and the terms and conditions of our assistance program vary by market.
Cost of revenues
We contract with third-party manufacturers that manufacture our TTFields delivery systems. Our cost of revenues is primarily comprised of the following:
|
|
· |
disposable transducer arrays; |
|
|
· |
depreciation expense for the field equipment, including the electric field generator used by patients; and |
|
|
· |
personnel, warranty and overhead costs such as facilities, freight and depreciation of property, plant and equipment associated with managing our inventory, warehousing and order fulfillment functions. |
The cost of revenues reported for the years ended December 31, 2015, 2014 and 2013 reflects costs incurred for patients receiving TTFields treatment in the period. Revenue recognized in any period includes collections from amounts billed primarily in prior periods and, to a lesser extent, in the current period. Gross margin as a percentage of revenues is also affected by the timing of revenue recognition based on cash collections, which often result in costs being incurred in one period that relate to revenues recognized in a later period.
Operating expenses
Our operating expenses consist of research, development and clinical trials, sales and marketing and general and administrative expenses. Personnel costs are a significant component for each category of operating expenses and consist of wages, benefits and bonuses. Personnel costs also include share-based compensation. We expect personnel costs to continue to increase as we hire new employees to continue to grow our business.
Research, development and clinical trials
Our research, development and clinical trials activity is focused on advancing TTFields through clinical trials across multiple solid tumor types and improving our delivery systems. Research, development and clinical trials costs, including direct and allocated expenses, are expensed as incurred and consist primarily of the following:
|
|
· |
personnel costs (including share-based compensation) for those employees involved in our research, development, clinical trial, regulatory and medical affairs activities; |
|
|
· |
costs to conduct research, development and clinical trial activity through agreements with contract research organizations and other third parties; |
|
|
· |
facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities, depreciation of leasehold improvements and equipment and laboratory and other supplies; |
|
|
· |
manufacturing expense associated with TTFields delivery systems, including durable components and disposable arrays, utilized in clinical trials and other research; and |
|
|
· |
professional fees related to regulatory approvals and conformity assessment procedures. |
66
We have incurred significant expenditures related to conducting clinical studies to develop TTFields in multiple solid tumor indications. The following table summarizes our principal clinical programs for TTFields for the years ended December 31, 2015, 2014 and 2013:
|
|
|
Year Ended December 31, |
|
|||||||||
|
U.S. dollars in thousands, except share and per share data |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Personnel costs |
|
$ |
15,194 |
|
|
$ |
13,443 |
|
|
$ |
12,176 |
|
|
General research and development |
|
|
13,198 |
|
|
|
12,199 |
|
|
|
8,384 |
|
|
Materials |
|
|
5,267 |
|
|
|
5,397 |
|
|
|
6,224 |
|
|
Newly diagnosed GBM |
|
|
8,726 |
|
|
|
8,415 |
|
|
|
7,677 |
|
|
Brian metastases |
|
|
253 |
|
|
|
157 |
|
|
|
181 |
|
|
Pancreatic Cancer |
|
|
370 |
|
|
|
292 |
|
|
|
155 |
|
|
Ovarian Cancer |
|
|
254 |
|
|
|
223 |
|
|
|
- |
|
|
Mesothelioma |
|
|
486 |
|
|
|
255 |
|
|
|
|
|
|
Research, development and clinical trials |
|
$ |
43,748 |
|
|
$ |
40,381 |
|
|
$ |
34,797 |
|
Personnel costs include all pre-clinical, clinical and other research and development company personnel. General research and development costs include costs related to pre-clinical, engineering, regulatory, intellectual property, advisors and subcontractors, travel and other. Materials include the costs of all equipment, arrays and other disposables for use in the clinical trials. Clinical trial costs in these periods include contract research organization services, data managing services, clinic and lab costs, as well as clinical sites costs.
We expect our research and development expenses to increase in absolute dollars as we continue to advance TTFields and develop new delivery systems to address current and possible future indications. We are in different stages in clinical programs evaluating TTFields as a treatment for brain metastases, NSCLC, pancreatic cancer, ovarian cancer and mesothelioma. We also expect to continue or begin a number of significant clinical programs in the future for other solid tumor indications.
Sales and marketing
Sales and marketing expenses consist primarily of personnel costs (including share-based compensation), travel expenses, marketing and promotional activities, and facilities costs. Over the next few years, we expect to continue to make significant expenditures associated with selling and marketing our delivery systems, primarily in connection with the continued commercialization of Optune in the United States, Europe and Japan for the treatment of our approved indications.
General and administrative
General and administrative expenses consist primarily of personnel costs (including share-based compensation), professional fees and facilities costs. General and administrative personnel costs include our executive, finance, human resources and legal functions. These costs also include our contributions to support industry and patient groups. Our professional fees consist primarily of accounting, legal and other consulting costs. We expect that general and administrative expenses will increase in absolute dollars to support our growth. In addition, we expect to incur significant legal and accounting costs related to compliance with SEC rules and regulations, including the costs of achieving and maintaining compliance with Section 404 of the Sarbanes-Oxley Act and compliance with NASDAQ rules, as well as insurance, investor relations and other costs associated with being a public company.
Financial expenses, net
Financial expenses, net primarily consists of credit facility interest expense and related debt issuance costs, interest income from cash balances and short-term investments and gains (losses) from foreign currency transactions. Our functional currency is the U.S. dollar. We have historically held substantially all of our cash balances in U.S. dollar denominated accounts to minimize the risk of translational currency exposure.
Income taxes
Our income taxes for the years ended December 31, 2015, 2014 and 2013 are due to statutory tax liabilities incurred by certain subsidiaries.
67
Critical accounting policies and estimates
In accordance with U.S. GAAP, in preparing our financial statements, we must make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amounts of net revenues and expenses during the reporting period. We develop and periodically change these estimates and assumptions based on historical experience and on various other factors that we believe are reasonable under the circumstances. Actual results may differ from these estimates.
The critical accounting policies requiring estimates, assumptions and judgments that we believe have the most significant impact on our consolidated financial statements are described below.
Revenue recognition
The TTFields delivery system, currently marketed for newly diagnosed and recurrent GBM as Optune, is comprised of two main components: (1) an electric field generator and (2) transducer arrays and related accessories that are disposable supplies to the device, or the transducer arrays. We retain title to the electric field generator, and the patient is provided replacement transducer arrays and technical support for the device during the term of treatment. The electric field generator and transducer arrays are always supplied and function together and are not sold on a standalone basis.
Revenues are recognized when persuasive evidence of an arrangement exists, delivery of the electric field generator and transducer arrays has occurred, the price is fixed or determinable and collectability is reasonably assured. The evidence of an arrangement generally consists of a prescription, a patient service agreement and the verification of eligibility and insurance with the patient’s third-party insurance company. We generally bill third-party payers a monthly rental fee for use of Optune by patients. As such, we take assignment of the benefit and risk of collection from the third-party payer. Patients often have out-of-pocket costs for the amount not covered by their third-party payer and we bill the patient directly for the amounts of their co-pays and deductibles subject to our patient assistance programs.
For the reported periods, all revenues were recognized when cash was collected and all other revenue recognition criteria have been met as the price is not fixed or determinable and the collectability cannot be reasonably assured. The price is not fixed or determinable since we do not have sufficient history with third-party payers to reliably estimate their individual payment patterns and as such cannot reliably estimate the amount that would be ultimately collected. Once sufficient history is established and we can demonstrate that we can reliably estimate the amounts that would be ultimately collected per third-party payer or payer group and the above criteria are met, we will recognize revenues ratably over the term of the patient’s use of Optune.
Revenues are presented net of indirect taxes incurred in the reported period, including the U.S. medical device excise tax, regardless of whether the revenues associated with those taxes are reported on a cash basis.
Share-based compensation
Under the Financial Accounting Standards Board’s (FASB) Accounting Standards Codification (ASC) 718, Compensation—Stock Compensation, we measure and recognize compensation expense for share options granted to our employees and directors based on the fair value of the awards on the date of grant. The fair value of share options is estimated at the date of grant using the Black-Scholes option pricing model that requires management to apply judgment and make estimates, including:
|
|
· |
the fair value of our ordinary shares on the date of grant until our IPO determined as discussed below; |
|
|
· |
the expected term of the stock option award, which we calculate using the simplified method, in accordance with ASC No.718-10-S99-1 (SAB No. 110) as we have insufficient historical information regarding our stock options to provide a basis for an estimate; |
|
|
· |
the expected share price volatility of our underlying ordinary shares, which we estimate based on the historical volatility of a representative group of publicly traded biopharmaceutical and medical technology companies with similar characteristics to us for a period matching the expected term assumption; |
|
|
· |
the risk-free interest rate, which we base on the yield curve of U.S. Treasury securities with periods commensurate with the expected term of the options being valued; and |
|
|
· |
the expected dividend yield, which we estimate to be zero based on the fact that we have never paid cash dividends and have no present intention to pay cash dividends. |
68
We recognize share-based compensation costs only for those shares expected to vest over the requisite vesting period of the award, which is generally the option vesting term of four years, using the accelerated method. ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Our forfeiture rate is based on an analysis of our actual forfeitures since the adoption of our 2003 Share Option Plan.
The table below summarizes the assumptions that were used to estimate the fair value of the options granted to employees during the periods presented:
|
|
|
Year ended December 31, |
|
|||||||
|
|
|
2015 |
|
2014 |
|
|
2013 |
|
||
|
Expected term (years) |
|
6.25 |
|
|
6.25 |
|
|
|
6.25 |
|
|
Expected volatility |
|
59%-65.8% |
|
73.1%-75.3% |
|
|
70.4%-75.9% |
|
||
|
Risk-free interest rate |
|
1.7%-2.1% |
|
1.9%-2.3% |
|
|
1.4%-2.0% |
|
||
|
Dividend yield |
|
0% |
|
0% |
|
|
0% |
|
||
We incurred share-based compensation expense of $11.9 million, $4.6 million, and $5.1 million during the years ended December 31, 2015, 2014 and 2013, respectively. As of December 31, 2015, we have unrecognized compensation expense of $25.0 million, which is expected to be recognized over a weighted average period of approximately 3.39 years. We expect to continue to grant share options in the future, and to the extent that we do, our recognized share-based compensation expense will likely increase.
If any of the assumptions used in the Black-Scholes option pricing model change significantly, share-based compensation for future awards may differ materially from the awards granted previously.
Our management and board of directors determined the fair value of our ordinary shares based on a number of objective and subjective factors consistent with the methodologies, approaches and assumptions consistent with the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation, or the AICPA Guide. These factors included the hiring of key personnel, contemporaneous third-party valuations of our ordinary shares, our financial condition and prospects as of such date, the status of our research and development efforts, the public trading price of comparable companies for the March 5, 2015 grant, the lack of marketability of our ordinary shares as a private company, risk factors relevant to our business, capital markets conditions generally and the prices of our preferred shares sold to investors in arm’s-length transactions, and the rights, preferences and privileges of our preferred shares relative to our ordinary shares.
The per share estimated fair value of ordinary shares in the table below represents the determination of the fair value of our ordinary shares as of the date of grant, taking into consideration the various objective and subjective factors described above. The following table presents the grant dates and related exercise prices of stock options granted to employees and consultants from January 1, 2013 through December 31, 2015:
|
Grant date |
|
Options granted |
|
|
Exercise price ($) |
|
|
Fair value per Ordinary share ($) |
|
|||
|
February 20, 2013 |
|
|
669,050 |
|
|
|
7.03 |
|
|
|
7.03 |
|
|
July 24, 2013 |
|
|
180,923 |
|
|
|
7.04 |
|
|
|
7.04 |
|
|
October 23, 2013 |
|
|
193,935 |
|
|
|
7.28 |
|
|
|
7.28 |
|
|
February 26, 2014 |
|
|
511,460 |
|
|
|
7.48 |
|
|
|
7.48 |
|
|
April 23, 2014 |
|
|
136,281 |
|
|
|
7.52 |
|
|
|
7.52 |
|
|
August 3, 2014 |
|
|
73,908 |
|
|
|
7.58 |
|
|
|
7.58 |
|
|
October 22, 2014 |
|
|
438,148 |
|
|
|
7.73 |
|
|
|
7.73 |
|
|
March 5, 2015 |
|
|
1,637,887 |
|
|
|
14.37 |
|
|
|
14.37 |
|
|
April 22, 2015 |
|
|
158,752 |
|
|
|
15.60 |
|
|
|
15.60 |
|
|
October 1, 2015 |
|
|
921,488 |
|
|
|
22.00 |
|
|
|
22.00 |
|
|
October 21, 2015 |
|
|
140,466 |
|
|
|
20.20 |
|
|
|
20.20 |
|
|
December 17, 2015 |
|
|
249,800 |
|
|
|
27.50 |
|
|
|
27.50 |
|
So long as our ordinary shares are publicly traded in a liquid market, we will rely on the daily trading price of our ordinary shares when we estimate the fair value of options granted.
69
Property and equipment and field equipment are stated at cost, net of accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful life of the relevant asset. We make estimates of the useful life of our property and equipment and field equipment, based on similar assets purchased in the past and our historical experience with such similar assets, in order to determine the depreciation expense to be recorded for each reporting period.
Our field equipment consists of equipment being utilized under rental agreements accounted for on a monthly basis as an operating lease, as well as service pool equipment. Service pool equipment is equipment owned and maintained by us that is swapped for equipment that needs repair or maintenance by us while being used by a patient. We record a provision for any excess, lost or damaged equipment when warranted based on an assessment of the equipment.
We assess impairment whenever events or changes in circumstances indicate that the carrying amount of the asset is impaired or the estimated useful life is no longer appropriate. Circumstances such as changes in technology or in the way an asset is being used may trigger an impairment review. For the reported periods, no impairment losses have been identified.
Inventories
Inventories are stated at the lower of cost or market. We regularly evaluate the ability to realize the value of inventory. If actual demand for our delivery systems declines or market conditions are less favorable than those projected, inventory write-offs may be required. For the reported periods, no write-offs have been made.
Income taxes
As part of the process of preparing our consolidated financial statements, we are required to calculate our income taxes based on taxable income by jurisdiction. We make certain estimates and judgments in determining our income taxes, including assessment of our uncertain tax positions, for financial statement purposes. Significant changes to these estimates may result in an increase or decrease to our tax provision in the subsequent period when such a change in estimate occurs.
Uncertain tax positions are based on estimates and assumptions that have been deemed reasonable by management. Our estimates of unrecognized tax benefits and potential tax benefits may not be representative of actual outcomes.
Results of operations
The following tables set forth our consolidated statements of operations data:
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands, except share and per share data |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Net revenues |
|
$ |
33,087 |
|
|
$ |
15,490 |
|
|
$ |
10,359 |
|
|
Cost of revenues |
|
|
20,610 |
|
|
|
10,036 |
|
|
|
7,013 |
|
|
Gross profit |
|
|
12,477 |
|
|
|
5,454 |
|
|
|
3,346 |
|
|
Operating costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research, development and clinical trials |
|
|
43,748 |
|
|
|
40,381 |
|
|
|
34,797 |
|
|
Sales and marketing |
|
|
38,861 |
|
|
|
21,177 |
|
|
|
16,406 |
|
|
General and administrative |
|
|
33,864 |
|
|
|
24,052 |
|
|
|
16,602 |
|
|
Total operating costs and expenses |
|
|
116,473 |
|
|
|
85,610 |
|
|
|
67,805 |
|
|
Operating loss |
|
|
(103,996 |
) |
|
|
(80,156 |
) |
|
|
(64,459 |
) |
|
Financial expenses, net |
|
|
(3,151 |
) |
|
|
(144 |
) |
|
|
(12,558 |
) |
|
Loss before income taxes |
|
|
(107,147 |
) |
|
|
(80,300 |
) |
|
|
(77,017 |
) |
|
Income taxes |
|
|
4,434 |
|
|
|
382 |
|
|
|
353 |
|
|
Net loss |
|
$ |
(111,581 |
) |
|
$ |
(80,682 |
) |
|
$ |
(77,370 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per ordinary share |
|
$ |
(3.67 |
) |
|
$ |
(6.46 |
) |
|
$ |
(6.73 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of ordinary shares used in computing basic and diluted net loss per share (1) |
|
|
30,401,603 |
|
|
|
12,490,017 |
|
|
|
11,498,392 |
|
70
Share-based compensation expense included in costs and expenses:
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Cost of revenues |
|
$ |
174 |
|
|
$ |
32 |
|
|
$ |
52 |
|
|
Research, development and clinical trials |
|
|
2,529 |
|
|
|
820 |
|
|
|
1,137 |
|
|
Sales and marketing |
|
|
2,496 |
|
|
|
1,104 |
|
|
|
791 |
|
|
General and administrative |
|
|
6,661 |
|
|
|
2,668 |
|
|
|
3,140 |
|
|
Total share-based compensation expense |
|
$ |
11,860 |
|
|
$ |
4,624 |
|
|
$ |
5,120 |
|
The following table includes certain commercial patient operating statistics for and as of the end of the periods presented.
|
|
|
Year ended December 31, |
|
|||||||||
|
Operating statistics |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Prescriptions received in period (1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
|
1,607 |
|
|
|
669 |
|
|
|
506 |
|
|
Europe and Middle East |
|
|
167 |
|
|
|
38 |
|
|
|
4 |
|
|
Japan |
|
|
3 |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
1,777 |
|
|
|
707 |
|
|
|
510 |
|
|
Active patients at period end (2) |
|
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
|
529 |
|
|
|
211 |
|
|
|
180 |
|
|
Europe and Middle East |
|
|
74 |
|
|
|
14 |
|
|
|
4 |
|
|
Japan |
|
|
2 |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
605 |
|
|
|
225 |
|
|
|
184 |
|
|
(1) |
A “prescription received” is a commercial order for Optune that is received from a physician certified to treat patients with TTFields therapy for a patient not previously on TTFields therapy. Orders to renew or extend treatment are not included in this total. In the future, we may have regulatory approvals and commercial programs for multiple clinical indications, at which time we will recognize a commercial order as a prescription for the same patient for each clinical indication treated. For example, in the future, a patient may have a prescription for the treatment of lung cancer and a prescription for the treatment of brain metastases from the lung cancer. |
|
(2) |
An “active patient” is a patient who is on TTFields therapy under a commercial prescription order as of the measurement date, including patients who may be on a temporary break from treatment and who plan to resume treatment in less than 60 days. |
Year ended December 31, 2015 compared to year ended December 31, 2014
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2015 |
|
|
2014 |
|
|
Change |
|
|
% Change |
|
||||
|
Net revenues |
|
$ |
33,087 |
|
|
$ |
15,490 |
|
|
$ |
17,597 |
|
|
|
114 |
% |
Net revenues. Substantially all of our revenues are derived from patients using our TTFields delivery system, marketed as Optune in our currently active markets. Total cash payments received during the year ended December 31, 2015 were recorded as revenues for Optune provided to patients in the current and prior periods. Net revenues increased by $17.6 million, or 114%, to $33.1 million for the year ended December 31, 2015 from $15.5 million for the year ended December 31, 2014. The increase was primarily due to an increase of $16.0 million in U.S. commercial sales of Optune, primarily driven by an increase in the number of U.S. certified centers at December 31, 2014 versus at December 31, 2015, from 154 to 244, respectively, and the increased demand for Optune therapy after the initial presentation of the EF-14 phase 3 pivotal trial data in November 2014 and FDA approval for Optune in newly diagnosed GBM in October 2015 and an increase of $1.6 million in commercial sales of Optune in our currently active markets in Europe.
Cost of revenues. Our cost of revenues is comprised primarily of (i) cost of the disposable transducer arrays purchased from third-party manufacturers, (ii) depreciation expense for the field equipment, including the electric field generator used by patients and (iii) personnel, warranty and overhead costs such as facilities, freight and depreciation of property, plant and equipment associated with managing our inventory, warehousing and order fulfillment functions. Our cost of revenues increased by $10.6 million, or 105%, to $20.6 million for the year ended December 31, 2015 from $10.0 million for the year ended December 31, 2014.
71
The increase was primarily due to an increase of $6.4 million in transducer arrays shipped to commercial patients, , $0.5 million for field equipment depreciation and personnel costs of $2.0 million.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2015 |
|
|
2014 |
|
|
Change |
|
|
% Change |
|
||||
|
Research, development and clinical trials |
|
$ |
43,748 |
|
|
$ |
40,381 |
|
|
$ |
3,367 |
|
|
|
8 |
% |
|
Sales and marketing |
|
|
38,861 |
|
|
|
21,177 |
|
|
$ |
17,684 |
|
|
|
84 |
% |
|
General and administrative |
|
|
33,864 |
|
|
|
24,052 |
|
|
$ |
9,812 |
|
|
|
41 |
% |
|
|
|
$ |
116,473 |
|
|
$ |
85,610 |
|
|
$ |
30,863 |
|
|
|
36 |
% |
Research, development and clinical trials expenses. Research, development and clinical trials expenses increased by $3.4 million, or 8%, to $43.7 million for the year ended December 31, 2015 from $40.4 million for the year ended December 31, 2014. The change is primarily due to the overall increase in clinical and research activities led to an increase of $1.6 million in personnel costs (including share-based compensation), third party research (including investigator sponsored trials) expenses increased by $1.0 million and an increase by $0.8 million in expenses related to our clinical trials including phase 2 pilot trials and our phase 3 pivotal EF – 14 trial which is in phasing out stage. Total non-cash share-based compensation for research, development and clinical trials personnel for the year ended December 31, 2015 and 2014 was $2.5 million and $0.8 million, respectively.
Sales and marketing expenses. Sales and marketing expenses increased by $17.7 million, or 84%, to $38.9 million for the year ended December 31, 2015 from $21.2 million for the year ended December 31, 2014. The increase was driven by an increase of $4.7 million of personnel costs (including share-based compensation), an increase of $13.0 million in advertising, marketing and other related expenses to support the launch of Optune for the treatment of newly diagnosed GBM. Total non-cash share-based compensation for sales and marketing personnel for the year ended December 31, 2015 and 2014 was $2.5 million and $1.1 million, respectively.
General and administrative expenses. General and administrative expenses increased by $9.8 million, or 41%, to $33.9 million for the year ended December 31, 2015 from $24.1 million for the year ended December 31, 2014. The increase was primarily related to an increase in personnel costs of $7.3 million (including share-based compensation) to support the growth and operation of our business, an increase of $2.5 million relating to legal services, facilities and costs associated with preparing the Company for the IPO and professional services related to our new SAP ERP system. Total non-cash share-based compensation for general and administrative personnel for the year ended December 31, 2015 and 2014 was $6.7 million and $2.7 million, respectively.
Financial expenses, net. Financial expenses, net increased by $3.0 million, or 2,088%, to $3.1 million for the year ended December 31, 2015 from $0.1 million for the year ended December 31, 2014. The change is primarily due to interest expense, including amortization expense of the discount and deferred issuance costs, related to our Term Loan Credit Facility entered into in January 2015.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2015 |
|
|
2014 |
|
|
Change |
|
|
% Change |
|
||||
|
Income taxes |
|
$ |
4,434 |
|
|
$ |
382 |
|
|
$ |
4,052 |
|
|
|
1061 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income taxes. Income taxes increase by $4.0 million, or 1,061 %, to $4.4 million for the year ended December 31, 2015 from $0.4 million for the year ended December 31, 2014. The change was primarily attributable to an increase in the statutory tax provision related to Switzerland, the United States and Japan.
Year ended December 31, 2014 compared to year ended December 31, 2013
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2014 |
|
|
2013 |
|
|
Change |
|
|
% Change |
|
||||
|
Net revenues |
|
$ |
15,490 |
|
|
$ |
10,359 |
|
|
$ |
5,131 |
|
|
|
50 |
% |
Net revenues. Net revenues increased by $5.1 million, or 50%, to $15.5 million for the year ended December 31, 2014 from $10.4 million for the year ended December 31, 2013. The increase was primarily due to an increase of $4.7 million in U.S. commercial sales of Optune, driven in part by the increased marketing efforts in the United States for recurrent GBM and in part by the announcement of the EF-14 results in November 2014, which we believe led to more sales of Optune for recurrent GBM, and to an increase of $0.4 million in commercial sales of Optune in our currently active markets in Europe.
Cost of revenues. Our cost of revenues increased by $3.0 million, or 43%, to $10 million for the year ended December 31, 2014 from $7 million for the year ended December 31, 2013. The increase was primarily driven by an increase in active patients resulting in
72
increased transducer array shipments and associated warehousing and order fulfillment personnel costs.an increase of $1.8 million in transducer arrays shipped to U.S. commercial patients, an increase of $0.3 million in transducer arrays shipped to patients in Europe, an increase of $0.4 million for field equipment depreciation and increased personnel costs of $0.5 million associated with new employees.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2014 |
|
|
2013 |
|
|
Change |
|
|
% Change |
|
||||
|
Research, development and clinical trials |
|
$ |
40,381 |
|
|
$ |
34,797 |
|
|
$ |
5,584 |
|
|
|
16 |
% |
|
Sales and marketing |
|
|
21,177 |
|
|
|
16,406 |
|
|
$ |
4,771 |
|
|
|
29 |
% |
|
General and administrative |
|
|
24,052 |
|
|
|
16,602 |
|
|
$ |
7,450 |
|
|
|
45 |
% |
|
|
|
$ |
85,610 |
|
|
$ |
67,805 |
|
|
$ |
17,805 |
|
|
|
26 |
% |
Research, development and clinical trials expenses. Research, development and clinical trials expenses increased by $5.6 million, or 16%, to $40.4 million for the year ended December 31, 2014 from $34.8 million for the year ended December 31, 2013. The change is primarily due to an increase in clinical trials expenses of $3.5 million mainly related to EF-14, an increase of $1.3 million of personnel costs (including share-based compensation), and an increase of $0.4 million for regulatory expenses related to the preparation of approval applications to the Japanese Ministry of Health, Labor and Welfare for the treatment of recurrent GBM and the FDA in the United States for the treatment of newly diagnosed GBM. In addition, there was $0.4 million increase in facility expenses.
Sales and marketing expenses. Sales and marketing expenses increased by $4.8 million, or 29%, to $21.2 million for the year ended December 31, 2014 from $16.4 million for the year ended December 31, 2013. The increase was driven by an increase of $3.2 million of personnel costs (including share-based compensation), an increase of $0.7 million in advertising and other marketing expenses and a $0.9 million increase related to facility, freight and travel expenses.
General and administrative expenses. General and administrative expenses increased by $7.5 million, or 45%, to $24.1 million for the year ended December 31, 2014 from $16.6 million for the year ended December 31, 2013. The increase was primarily related to an increase in personnel costs of $3.0 million due to increased headcount to support the growth and operation of our business, an increase of $1.3 million for costs of professional services relating to the implementation of our new SAP ERP system and legal services, and an increase of $1.0 million related to facility and travel costs. In addition, there was an expense related to a provision for settlement with the Technion of $1.9 million.
Financial expenses, net. Financial expenses, net decreased by $12.4 million, or 98%, to $0.1 million for the year ended December 31, 2014 from $12.6 million for the year ended December 31, 2013. The change was primarily due to repayment of an outstanding $52.0 million principal amount loan at the end of 2013, which contributed $12.5 million to the 2013 financial expenses.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2014 |
|
|
2013 |
|
|
Change |
|
|
% Change |
|
||||
|
Income taxes |
|
$ |
382 |
|
|
$ |
353 |
|
|
$ |
29 |
|
|
|
8 |
% |
Income taxes. Income taxes did not significantly change from 2014 as compared to 2013. The minor change was primarily attributable to an increase in our provision for current taxes in Switzerland offset by a decrease in provision for unrecognized tax benefits.
Liquidity and capital resources
We have incurred significant losses and cumulative negative cash flows from operations since our founding in 2000. As of December 31, 2015, we had an accumulated deficit of $388.1 million. We expect to continue to incur losses until our delivery systems achieve market acceptance. To date, we have primarily financed our operations through the issuance and sale of our convertible preferred shares and the proceeds from long-term loans. As of December 31, 2015, we had received a total of $614.0 million from the sale of our convertible preferred shares, including the sale of our Series J convertible preferred stock in June 2015 for net proceeds of $94.6 million, all of which were converted into our ordinary shares after the consummation of our IPO, and including $157.5 million proceeds from our IPO including the exercise of the underwriters' overallotment option and the exercise of ordinary share warrants and options.
73
As of December 31, 2015 and December 31, 2014, we had $269.4 million and $102.6 million, respectively, of cash, cash equivalents, and short-term investments. In 2013, we raised $194.2 million through the issuance of our Series I convertible preferred shares. In January 2015, we entered into the Loan and Security Agreement dated as of January 7, 2015, between us, as borrower, and Biopharma Secured Investments III Holdings Cayman LP, as lender, or the Term Loan Credit Facility, for up to $100.0 million, of which we drew $25.0 million on entering into the facility. In June 2015, we raised net proceeds of $94.6 million through the issuance of our Series J convertible preferred shares. Our IPO closed on October 7, 2015 and we issued and sold 7,876,195 ordinary shares. We received cash proceeds of $157.5 million from the IPO and the partial exercise of the underwriters’ overallotment option, net of underwriting discounts and commissions and offering expenses. We believe our cash, cash equivalents and short-term investments as of December 31, 2015 are sufficient for our operations for at least the next 12 months based on our existing business plan and our ability to control the timing of significant expense commitments. We expect that our research, development and clinical trials expenses, sales and marketing expenses and general and administrative expenses will continue to increase over the next several years. As a result, we may need to raise additional capital to fund our operations.
The following summary of our cash flows for the periods indicated has been derived from our consolidated financial statements, which are included elsewhere in this Annual Report:
|
|
|
Year Ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Net cash used in operating activities |
|
$ |
(99,884 |
) |
|
$ |
(74,244 |
) |
|
$ |
(52,717 |
) |
|
Net cash provided by (used in) investing activities |
|
|
(115,295 |
) |
|
|
(46,182 |
) |
|
|
10,402 |
|
|
Net cash provided by financing activities |
|
|
276,989 |
|
|
|
2,145 |
|
|
|
183,318 |
|
|
Net increase (decrease) in cash and cash equivalents |
|
$ |
61,810 |
|
|
$ |
(118,281 |
) |
|
$ |
141,003 |
|
Operating activities
Net cash used in operating activities primarily represents our net loss for the periods presented. Adjustments to net loss for non-cash items include depreciation, share-based compensation and accrued interest. Operating cash flows are also impacted by changes in operating assets and liabilities, principally inventories, prepaid expenses, trade payables and accrued expenses.
Net cash used in operating activities was $99.8 million for the year ended December 31, 2015, as compared to $74.2 million for the year ended December 31, 2014, reflecting a net loss of $111.6 million, and a change of $4.3 million in our net operating assets and liabilities offset by non-cash charges of $16.1 million. The change in our net operating assets and liabilities was primarily the result of an increase in our inventories of $10.1 million necessary to meet anticipated demand, an increase in other receivables of $5.1 million, offset by an increase in trade payables of $7.0 million and other payables of $3.6 million. Non-cash charges included $11.9 million of share-based compensation, $3.2 million of depreciation and $1.0 million of accrued interest and amortization related to our Term Loan Credit Facility.
Net cash used in operating activities was $74.2 million in 2014, reflecting a net loss of $80.7 million, offset by non-cash charges of $6.6 million and a change of $0.1 million in our net operating assets and liabilities. The change in our net operating assets and liabilities was the result of an increase in receivables and prepaid expenses of $1.2 million, a decrease of $0.5 million in trade payables and an increase in inventory of $1.6 million primarily due to commercial sales in the United States, offset by an increase in other payables of $2.3 million and an increase in other long-term liabilities of $0.8 million. Non-cash charges included $4.6 million of share-based compensation and $2.0 million of depreciation.
Net cash used in operating activities was $52.7 million in 2013, reflecting a net loss of $77.4 million, offset by non-cash charges of $17.7 million and a change of $7.0 million in our net operating assets and liabilities. The change in our net operating assets and liabilities was primarily the result of an increase in trade payables of $5.7 million due to increased activity, an increase in other payables of $3.0 million, a decrease in inventory of $1.5 million and an increase in other long-term liabilities of $0.3 million, offset by an increase in our receivables and prepaid expenses of $3.6 million. Non-cash charges included $5.1 million of share-based compensation, $1.2 million of depreciation, accrued interest of $6.0 million related to a long-term loan and $5.4 million in related amortization expenses.
Investing activities
Our investing activities consist primarily of capital expenditures to purchase property and equipment and field equipment, as well as investments in and redemptions of our short-term investments.
74
Net cash provided by investing activities was $115.3 million in the year ended December 31, 2015 attributable to our receipt of $104.0 million from the maturity of short-term investments, offset by the purchase of new short-term investments of $209.0 million, purchases of $4.7 million of property and equipment and purchases of $5.6 million of field equipment. Net cash used in investing activities for the same period in 2014 was $46.2 million, attributable to the purchase of $93.0 million of short-term investments, purchases of property and equipment of $0.8 million and purchases of field equipment of $1.5 million was offset by receipt of $138.0 million from the maturity of short-term investments and a decrease in restricted cash of $1.1 million.
Net cash provided by investing activities in 2013 was $10.4 million attributable to proceeds from redemption of bank deposits of $15.0 million, offset by the purchase of property and equipment of $2.2 million, the purchase of field equipment of $1.4 million and an increase in restricted cash of $1.0 million.
Financing activities
To date, our primary financing activities have been the sale of our convertible preferred shares, our IPO and the proceeds from long-term loans.
Net cash provided by financing activities was $277.0 million for the year ended December 31, 2015, attributable to the net proceeds from the issuance of Series J preferred shares of $94.6 million and borrowings under our Term Loan Credit Facility of $22.9 million and $157.5 from our IPO and the partial exercise of the overallotment option by the underwriters and in addition $2.0 million from the exercise of options and warrants. Net cash provided by financing activities was $2.1 million in 2014, attributable to net proceeds received from the exercise of options. Net cash provided by financing activities was $183.3 million in 2013 attributable mainly to proceeds from the issuance of our Series I preferred shares of $191.7 million and proceeds from a long-term loan of $49.6 million, offset by the repayment of the long-term loan (including interest) of $58.0 million.
Term Loan Credit Facility
Our material outstanding indebtedness consists of our Term Loan Credit Facility, which provides for up to $100.0 million of borrowings in up to four draws, the first of which was made on January 30, 2015 in the amount of $25.0 million. Interest on the outstanding loan is 10% annually, payable quarterly in arrears. As of December 31, 2015, the aggregate principal balance of amounts outstanding under the Term Loan Credit Facility was $25.2 million. The commitments made by the lender to make additional term loans terminate on June 30, 2016. We may prepay the term loans, in whole, at any time, and must prepay in the event of a change of control, in each case, subject to a pay-down fee, prepayment premium and/or make-whole payment. The funding fee payable on the amount drawn on the funding date is 1.5%, the pay-down fee on all principal payments to be paid on the date such payments are made is 0.75% and the pre-payment fee if we prepay outstanding loan amounts prior to the first, second or third year from the initial funding date is 3.0%, 2.0% or 1.0%, respectively.
All obligations under the Term Loan Credit Facility are guaranteed by certain of our current and future domestic direct and indirect subsidiaries. In addition, the obligations under the Term Loan Credit Facility are secured by a first-priority security interest in substantially all of the property and assets of, as well as the equity interests owned by, us and the other guarantors.
The Term Loan Credit Facility has a minimum liquidity covenant, which is tested quarterly. In addition, we must meet certain pro forma net sales requirements. The Term Loan Credit Facility contains other customary covenants. As of December 31, 2015, we were in compliance with the Term Loan Credit Facility covenants.
Contractual obligations and commitments
The following summarizes our significant contractual obligations as of December 31, 2015:
|
|
|
December 31, |
|
|||||||||||||||||||||||||
|
Contractual Obligations: |
|
2016 |
|
|
2017 |
|
|
2018 |
|
|
2019 |
|
|
2020 |
|
|
After |
|
|
Total |
|
|||||||
|
|
|
(in thousands) |
|
|||||||||||||||||||||||||
|
Operating leases |
|
$ |
2,320 |
|
|
$ |
2,171 |
|
|
$ |
1,152 |
|
|
$ |
987 |
|
|
$ |
591 |
|
|
$ |
1,261 |
|
|
$ |
8,482 |
|
|
Term Loan Credit Facility (1) |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
25,188 |
|
|
|
|
|
|
|
25,188 |
|
|
Leasehold improvement financing and other loans |
|
$ |
63 |
|
|
$ |
69 |
|
|
$ |
73 |
|
|
$ |
23 |
|
|
$ |
24 |
|
|
$ |
65 |
|
|
$ |
317 |
|
|
(1) |
The Term Loan Credit Facility has a fixed per annum interest rate of 10.0%. Interest due is excluded from the table. |
75
There was no material changes in our commitments under contractual obligations in the year ended December 31, 2015.
The total amount of unrecognized tax expenses (benefits) for uncertain tax positions was $1.6 million and $0.3 million at December 31, 2015 and December 31, 2014, respectively. Payment of these obligations would result from settlements with taxing authorities. Due to the difficulty in determining the timing of settlements, these obligations are not included in the above table. We do not expect a significant tax payment related to these obligations within the next year.
We also have employment agreements with certain employees that require the funding of a specific level of payments if certain events, such as a change in control or termination without cause, occur. In the course of normal business operations, we also have agreements with contract service providers to assist in the performance of our research and development (including clinical trials) and manufacturing activities. We could also enter into additional collaborative research, contract research, manufacturing and supplier agreements in the future, which may require up-front payments and even long-term commitments of cash.
We also have a contingent obligation to pay the Technion $5.5 million within five business days (1) if we achieve $250.0 million of cumulative net sales since inception at the end of any given quarter or (2) upon consummation of a merger or acquisition transaction, which includes any merger to the extent it involves a change of control, the sale of all or substantially all of our assets or shares, the sale of or exclusive license to our intellectual property or a similar transaction.
Off-balance sheet arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements as defined under SEC rules.
JOBS Act election
The JOBS Act permits an “emerging growth company” such as us to take advantage of an extended transition period to comply with new or revised accounting standards applicable to public companies. We have irrevocably elected to “opt out” of the exemption for the delayed adoption of certain accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to market risks in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates. Our market risk exposure is primarily a result of fluctuations in interest rates and foreign currency exchange rates. We do not hold or issue financial instruments for trading purposes.
Interest rate sensitivity
Our exposure to market risk for changes in interest rates relates primarily to our investment portfolio. Our cash, cash equivalents and short-term investment accounts as of December 31, 2015 totaled $269.4 million and consist of cash, cash equivalents and short-term investments with maturities of less than one year from the date of purchase. Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of the interest rates in the United States. However, because of the short-term nature of the instruments in our portfolio, a 10% change in market interest rates would not be expected to have a material impact on our financial condition or our results of operations.
Foreign currency exchange risk
Our consolidated results of operations and cash flow are subject to fluctuations due to changes in foreign currency exchange rates. All of our revenues are generated in the local currency for commercial markets. Our expenses are generally denominated in the currencies in which our operations are located, which is primarily in Germany, Israel, Japan, Switzerland, and the United States. Our consolidated results of operations and cash flows are, therefore, subject to fluctuations due to changes in foreign currency exchange rates and may be adversely affected in the future due to changes in foreign exchange rates. The effect of a hypothetical 10% change in foreign currency exchange rates applicable to our business would not have a material impact on our historical consolidated financial statements. We do not hedge our foreign currency exchange risk.
76
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
NovoCure Limited
Index to consolidated financial statements
77
Report of independent registered public accounting firm
To the board of directors and shareholders of
NovoCure Limited
We have audited the accompanying consolidated balance sheets of NovoCure Limited and subsidiaries (the “Company”) as of December 31, 2014 and 2015, and the related consolidated statements of operations, comprehensive loss, changes in shareholders’ equity and cash flows for each of the three years in the period ended December 31, 2015. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of the Company and subsidiaries at December 31, 2014 and 2015 and the consolidated results of their operations and their cash flows for each of the three years in the period ended December 31, 2015, in conformity with U.S. generally accepted accounting principles.
|
Tel-Aviv, Israel |
|
/s/ KOST FORER GABBAY & KASIERER |
|
March 1, 2016 |
|
A Member of Ernst & Young Global |
78
NovoCure Limited and subsidiaries
Consolidated balance sheets
|
|
|
December 31, |
|
|||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Current Assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
119,423 |
|
|
$ |
57,613 |
|
|
Short-term investments |
|
|
150,001 |
|
|
|
44,999 |
|
|
Restricted cash |
|
|
87 |
|
|
|
61 |
|
|
Receivables and prepaid expenses |
|
|
10,799 |
|
|
|
5,711 |
|
|
Inventories |
|
|
13,594 |
|
|
|
3,446 |
|
|
Total current assets |
|
|
293,904 |
|
|
|
111,830 |
|
|
Long-term Assets: |
|
|
|
|
|
|
|
|
|
Property and equipment, net |
|
|
6,552 |
|
|
|
3,732 |
|
|
Field equipment, net |
|
|
6,029 |
|
|
|
2,017 |
|
|
Severance pay fund |
|
|
79 |
|
|
|
70 |
|
|
Other long-term assets |
|
|
772 |
|
|
|
227 |
|
|
Total long-term assets |
|
|
13,432 |
|
|
|
6,046 |
|
|
Total Assets |
|
$ |
307,336 |
|
|
$ |
117,876 |
|
The accompanying notes are an integral part of the consolidated financial statements.
79
NovoCure Limited and subsidiaries
|
|
|
December 31, |
|
|||||
|
U.S. dollars in thousands, except share and per share data |
|
2015 |
|
|
2014 |
|
||
|
Liabilities And Shareholders’ Equity |
|
|
|
|
|
|
|
|
|
Current Liabilities: |
|
|
|
|
|
|
|
|
|
Trade payables |
|
$ |
16,755 |
|
|
$ |
10,033 |
|
|
Other payables and accrued expenses |
|
|
11,872 |
|
|
|
7,636 |
|
|
Total current liabilities |
|
|
28,627 |
|
|
|
17,669 |
|
|
Long-term Liabilities: |
|
|
|
|
|
|
|
|
|
Long-term loan, net of discount and issuance costs |
|
|
23,097 |
|
|
|
— |
|
|
Employee benefit liabilities |
|
|
2,057 |
|
|
|
246 |
|
|
Other long-term liabilities |
|
|
2,735 |
|
|
|
2,086 |
|
|
Total long-term liabilities |
|
|
27,889 |
|
|
|
2,332 |
|
|
Total Liabilities |
|
|
56,516 |
|
|
|
20,001 |
|
|
Commitments and Contingencies |
|
|
|
|
|
|
|
|
|
Shareholders’ Equity: |
|
|
|
|
|
|
|
|
|
Share capital - |
|
|
|
|
|
|
|
|
|
Ordinary shares - No par value, Unlimited shares authorized; Issued and outstanding: 83,778,581 shares and 13,431,414 shares at December 31, 2015 and December 31, 2014 respectively; |
|
|
— |
|
|
|
— |
|
|
Preferred shares - No par value, Unlimited shares authorized; Issued and outstanding: Zero shares and 58,676,017 shares at December 31, 2015 and December 31, 2014 respectively; |
|
|
— |
|
|
|
— |
|
|
Additional paid-in capital |
|
|
640,406 |
|
|
|
374,375 |
|
|
Accumulated other comprehensive loss |
|
|
(1,505 |
) |
|
|
— |
|
|
Accumulated deficit |
|
|
(388,081 |
) |
|
|
(276,500 |
) |
|
Total shareholders’ equity |
|
|
250,820 |
|
|
|
97,875 |
|
|
Total Liabilities and Shareholders’ Equity |
|
$ |
307,336 |
|
|
$ |
117,876 |
|
The accompanying notes are an integral part of the consolidated financial statements.
80
NovoCure Limited and subsidiaries
Consolidated statements of operations
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands, except share and per share data |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Net revenues |
|
$ |
33,087 |
|
|
$ |
15,490 |
|
|
$ |
10,359 |
|
|
Cost of revenues |
|
|
20,610 |
|
|
|
10,036 |
|
|
|
7,013 |
|
|
Gross profit |
|
|
12,477 |
|
|
|
5,454 |
|
|
|
3,346 |
|
|
Operating costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research, development and clinical trials |
|
|
43,748 |
|
|
|
40,381 |
|
|
|
34,797 |
|
|
Sales and marketing |
|
|
38,861 |
|
|
|
21,177 |
|
|
|
16,406 |
|
|
General and administrative |
|
|
33,864 |
|
|
|
24,052 |
|
|
|
16,602 |
|
|
Total operating costs and expenses |
|
|
116,473 |
|
|
|
85,610 |
|
|
|
67,805 |
|
|
Operating loss |
|
|
(103,996 |
) |
|
|
(80,156 |
) |
|
|
(64,459 |
) |
|
Financial expenses, net |
|
|
(3,151 |
) |
|
|
(144 |
) |
|
|
(12,558 |
) |
|
Loss before income taxes |
|
|
(107,147 |
) |
|
|
(80,300 |
) |
|
|
(77,017 |
) |
|
Income taxes |
|
|
4,434 |
|
|
|
382 |
|
|
|
353 |
|
|
Net loss |
|
$ |
(111,581 |
) |
|
$ |
(80,682 |
) |
|
$ |
(77,370 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per ordinary share |
|
$ |
(3.67 |
) |
|
$ |
(6.46 |
) |
|
$ |
(6.73 |
) |
|
Weighted average number of ordinary shares used in computing basic and diluted net loss per share |
|
|
30,401,603 |
|
|
|
12,490,017 |
|
|
|
11,498,392 |
|
The accompanying notes are an integral part of the consolidated financial statements.
81
NovoCure Limited and subsidiaries
Consolidated statements of comprehensive loss
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Net loss |
|
|
(111,581 |
) |
|
|
(80,682 |
) |
|
|
(77,370 |
) |
|
Other comprehensive loss, net of tax : |
|
|
|
|
|
|
|
|
|
|
|
|
|
Pension benefit plan, net of tax |
|
|
(1,505 |
) |
|
|
- |
|
|
|
- |
|
|
Total comprehensive loss |
|
|
(113,086 |
) |
|
|
(80,682 |
) |
|
|
(77,370 |
) |
The accompanying notes are an integral part of the consolidated financial statements.
82
NovoCure Limited and subsidiaries
Statements of changes in shareholders’ equity
|
|
|
Ordinary shares |
|
|
Preferred shares |
|
|
Additional paid-in capital |
|
|
Accumulated other comprehensive loss |
|
|
Accumulated deficit |
|
|
Total shareholders’ equity |
|
||||||
|
U.S. dollars in thousands, except share data |
|
Shares |
|
|
Shares |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
Balance as of January 1, 2013 |
|
|
11,419,786 |
|
|
|
45,315,765 |
|
|
|
167,873 |
|
|
|
- |
|
|
|
(118,448 |
) |
|
|
49,425 |
|
|
Share-based compensation to employees |
|
|
- |
|
|
|
|
|
|
|
5,120 |
|
|
|
- |
|
|
|
- |
|
|
|
5,120 |
|
|
Issuance of Series I preferred shares, net (a) |
|
|
- |
|
|
|
13,360,252 |
|
|
|
191,738 |
|
|
|
- |
|
|
|
- |
|
|
|
191,738 |
|
|
Issuance of warrants, net (b) |
|
|
- |
|
|
|
- |
|
|
|
2,864 |
|
|
|
- |
|
|
|
- |
|
|
|
2,864 |
|
|
Exercise of options |
|
|
471,635 |
|
|
|
- |
|
|
|
2 |
|
|
|
- |
|
|
|
- |
|
|
|
2 |
|
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(77,370 |
) |
|
|
(77,370 |
) |
|
Balance as of December 31, 2013 |
|
|
11,891,421 |
|
|
|
58,676,017 |
|
|
|
367,597 |
|
|
|
- |
|
|
|
(195,818 |
) |
|
|
171,779 |
|
|
Share-based compensation to employees |
|
|
- |
|
|
|
- |
|
|
|
4,624 |
|
|
|
- |
|
|
|
- |
|
|
|
4,624 |
|
|
Exercise of options and warrants |
|
|
1,539,993 |
|
|
|
- |
|
|
|
2,154 |
|
|
|
- |
|
|
|
- |
|
|
|
2,154 |
|
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(80,682 |
) |
|
|
(80,682 |
) |
|
Balance as of December 31, 2014 |
|
|
13,431,414 |
|
|
|
58,676,017 |
|
|
|
374,375 |
|
|
|
- |
|
|
|
(276,500 |
) |
|
|
97,875 |
|
|
Share-based compensation to employees |
|
|
- |
|
|
|
- |
|
|
|
11,860 |
|
|
|
- |
|
|
|
- |
|
|
|
11,860 |
|
|
Exercise of options and warrants |
|
|
731,665 |
|
|
|
- |
|
|
|
2,038 |
|
|
|
- |
|
|
|
- |
|
|
|
2,038 |
|
|
Issuance of Series J preferred shares, net (c) |
|
|
- |
|
|
|
4,068,500 |
|
|
|
94,599 |
|
|
|
- |
|
|
|
- |
|
|
|
94,599 |
|
|
Issuance of shares and options in respect of settlement, net of fair value of shares provided as indemnification (Note 14c) |
|
|
(1,005,210 |
) |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Issuance of ordinary shares upon IPO and exercise of over-allotment, net (d) |
|
|
7,876,195 |
|
|
|
- |
|
|
|
157,534 |
|
|
|
- |
|
|
|
- |
|
|
|
157,534 |
|
|
Conversion of preferred shares to ordinary shares |
|
|
62,744,517 |
|
|
|
(62,744,517 |
) |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Other comprehensive loss, net of tax benefit of $165 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(1,505 |
) |
|
|
- |
|
|
|
(1,505 |
) |
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(111,581 |
) |
|
|
(111,581 |
) |
|
Balance as of December 31, 2015 |
|
|
83,778,581 |
|
|
|
- |
|
|
|
640,406 |
|
|
|
(1,505 |
) |
|
|
(388,081 |
) |
|
|
250,820 |
|
|
(a) |
Net of issuance expenses of $2,440 |
|
(b) |
Net of issuance expenses of $115 |
|
(c) |
Net of issuance expenses of $319 |
|
(d) |
Net of issuance expenses (including underwriter fees) of $15,742 |
The accompanying notes are an integral part of the consolidated financial statements.
83
NovoCure Limited and subsidiaries
Consolidated statements of cash flows
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(111,581 |
) |
|
$ |
(80,682 |
) |
|
$ |
(77,370 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
3,153 |
|
|
|
1,962 |
|
|
|
1,216 |
|
|
Asset write-downs and impairment |
|
|
46 |
|
|
|
23 |
|
|
|
19 |
|
|
Accrued interest expense |
|
|
672 |
|
|
|
- |
|
|
|
6,070 |
|
|
Share-based compensation to employees |
|
|
11,860 |
|
|
|
4,624 |
|
|
|
5,120 |
|
|
Amortization of discount (premium) |
|
|
329 |
|
|
|
(19 |
) |
|
|
5,432 |
|
|
Increase in receivables and prepaid expenses |
|
|
(5,088 |
) |
|
|
(1,192 |
) |
|
|
(3,611 |
) |
|
Decrease (increase) in inventories |
|
|
(10,148 |
) |
|
|
(1,554 |
) |
|
|
1,479 |
|
|
Increase in other long-term assets |
|
|
(381 |
) |
|
|
(44 |
) |
|
|
(44 |
) |
|
Increase (decrease) in trade payables |
|
|
6,961 |
|
|
|
(492 |
) |
|
|
5,695 |
|
|
Increase in other payables and accrued expenses |
|
|
3,579 |
|
|
|
2,324 |
|
|
|
2,955 |
|
|
Increase in employee benefit liabilities, net |
|
|
133 |
|
|
|
42 |
|
|
|
20 |
|
|
Increase in other long-term liabilities |
|
|
581 |
|
|
|
764 |
|
|
|
302 |
|
|
Net cash used in operating activities |
|
$ |
(99,884 |
) |
|
$ |
(74,244 |
) |
|
$ |
(52,717 |
) |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of property and equipment |
|
|
(4,667 |
) |
|
|
(849 |
) |
|
|
(2,201 |
) |
|
Purchase of field equipment |
|
|
(5,604 |
) |
|
|
(1,470 |
) |
|
|
(1,426 |
) |
|
Decrease (increase) in restricted cash |
|
|
(26 |
) |
|
|
1,117 |
|
|
|
(1,019 |
) |
|
Proceeds from maturity of short-term investments |
|
|
104,000 |
|
|
|
93,000 |
|
|
|
15,048 |
|
|
Purchase of short-term investments |
|
|
(208,998 |
) |
|
|
(137,980 |
) |
|
|
- |
|
|
Net cash provided by (used in) investing activities |
|
$ |
(115,295 |
) |
|
$ |
(46,182 |
) |
|
$ |
10,402 |
|
The accompanying notes are an integral part of the consolidated financial statements.
84
NovoCure Limited and subsidiaries
Consolidated statements of cash flows
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of shares, net |
|
$ |
252,133 |
|
|
$ |
- |
|
|
$ |
191,738 |
|
|
Proceeds from long-term loan, net |
|
|
22,886 |
|
|
|
- |
|
|
|
49,432 |
|
|
Repayment of long-term loans |
|
|
- |
|
|
|
- |
|
|
|
(58,047 |
) |
|
Proceeds from issuance of other long-term loans |
|
|
- |
|
|
|
54 |
|
|
|
193 |
|
|
Repayment of other long-term loans |
|
|
(63 |
) |
|
|
(63 |
) |
|
|
- |
|
|
Exercise of options and warrants |
|
|
2,038 |
|
|
|
2,154 |
|
|
|
2 |
|
|
Purchase of shares in respect of settlement |
|
|
(5 |
) |
|
|
- |
|
|
|
- |
|
|
Net cash provided by financing activities |
|
$ |
276,989 |
|
|
$ |
2,145 |
|
|
$ |
183,318 |
|
|
Increase (decrease) in cash and cash equivalents |
|
|
61,810 |
|
|
|
(118,281 |
) |
|
|
141,003 |
|
|
Cash and cash equivalents at the beginning of the year |
|
|
57,613 |
|
|
|
175,894 |
|
|
|
34,891 |
|
|
Cash and cash equivalents at the end of the year |
|
$ |
119,423 |
|
|
$ |
57,613 |
|
|
$ |
175,894 |
|
|
Supplemental cash flow activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid during the year for: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Income taxes |
|
$ |
1,489 |
|
|
$ |
282 |
|
|
$ |
689 |
|
|
Interest |
|
$ |
1,688 |
|
|
$ |
25 |
|
|
$ |
7,131 |
|
|
Non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of property and equipment |
|
$ |
- |
|
|
$ |
239 |
|
|
$ |
585 |
|
The accompanying notes are an integral part of the consolidated financial statements.
85
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 1: Organization and Basis of Presentation
a. NovoCure Limited (including its consolidated subsidiaries, the “Company”) was incorporated in Jersey and is principally engaged in the development, manufacture and commercialization of tumor treating fields (“TTFields”) for the treatment of solid tumors. Since inception, the Company has devoted substantially all of its efforts to developing a family of products to deliver TTFields for a variety of solid tumor indications, raising capital and recruiting personnel. The Company commenced selling and marketing activities in the United States at the end of 2011, in certain countries in Europe in 2014 and in Japan in 2015.
b. On October 7, 2015, the Company completed its initial public offering (the “IPO”) by issuing 7,876,195 ordinary shares (including exercise of overallotments) and raising net proceeds of $157,534. In September 2015, the Company’s shareholders approved the restructuring of the Company’s share capital by converting the Company’s ordinary and preferred shares to no par value shares and by effecting a sub division of the issued and outstanding share capital of the Company based on a proportion of 1: 5.913 (“Share Split Ratio”), such that each ordinary and preferred share nominal value of £0.01 of the Company, was divided into 5.913 shares of such applicable class of shares of the Company each with no par value. It was also resolved to apply the Split Ratio to the Company’s outstanding options and warrants, in accordance with their terms. All share and per share information included in these consolidated financial statements has been retroactively adjusted to reflect the conversion to no par value shares and the Share Split Ratio.
Note 2: Significant accounting policies
The consolidated financial statements are prepared according to United States generally accepted accounting principles (“U.S. GAAP”).
a. Use of estimates:
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. The Company evaluates on an ongoing basis its assumptions, including those related to contingencies, deferred taxes, tax liabilities, useful-life of field equipment and share-based compensation costs. The Company’s management believes that the estimates, judgment and assumptions used are reasonable based upon information available at the time they are made. These estimates, judgments and assumptions can affect the reported amounts of assets and liabilities at the dates of the consolidated financial statements, and the reported amounts of net revenue and expenses during the reporting period. Actual results could differ from those estimates.
b. Financial statements in U.S. dollars:
The accompanying financial statements have been prepared in U.S. dollars in thousands.
The Company finances its operations in U.S. dollars and a substantial portion of its costs and revenues from its primary markets is incurred in U.S. dollars. As such, the Company’s management believes that the dollar is the currency of the primary economic environment in which the Company operates. Thus, the functional and reporting currency of the Company is the U.S. dollar.
Transactions and balances denominated in U.S. dollars are presented at their original amounts. Monetary accounts maintained in currencies other than the dollar are re-measured into dollars in accordance with Accounting Standards Codification No. 830-10, “Foreign Currency Matters.” All transaction gains and losses of the re-measurement of monetary balance sheet items are reflected in the consolidated statements of operations as financial income or expenses, as appropriate.
c. Principles of consolidation:
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. Intercompany transactions and balances, including profits from intercompany sales not yet realized outside the Company, have been eliminated upon consolidation.
d. Cash equivalents:
Cash equivalents are short-term, highly liquid investments that are readily convertible into cash with an original maturity of three months or less at the date acquired.
86
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
e. Short-term investments and restricted cash:
1. Short-term investments:
The Company accounts for investments in debt securities in accordance with ASC 320, “Investments-Debt and Equity Securities.” Management determines the appropriate classification of its investments in marketable debt securities at the time of purchase and reevaluates such determinations at each balance sheet date. For the years ended December 31, 2015 and 2014, all securities are classified as held-to-maturity since the Company has the intent and ability to hold the securities to maturity and, accordingly, debt securities are stated at amortized cost.
The amortized cost of held-to-maturity securities is adjusted for amortization of premiums and accretion of discounts to maturity and any other than temporary impairment losses. Such amortization and interest are included in the consolidated statement of operations as financial income or expenses, as appropriate.
For the three years ended December 31, 2015, no impairment losses other than temporary impairment losses have been identified.
2. Restricted cash:
The Company has restricted cash used as security for bank guarantees related to the use of Company credit cards.
f. Inventories:
Inventories are stated at the lower of cost or market. Cost is determined using the weighted average method. The Company regularly evaluates the ability to realize the value of inventory. If actual demand for the Company’s delivery systems deteriorates, or market conditions are less favorable than those projected, inventory write-offs may be required.
There were no inventory write-offs for the years ended December 31, 2015, 2014 and 2013.
g. Property and equipment:
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets at the following rates:
|
|
|
% |
|
Computers and laboratory equipment |
|
15 - 33 |
|
Office furniture |
|
6 - 33 |
|
Production equipment |
|
20 |
|
Leasehold improvements |
|
Over the shorter of the term of the lease or its useful life |
h. Field equipment under operating leases:
Field equipment is stated at cost, net of accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful life of the field equipment which was determined to be two years. Field equipment consists of equipment being utilized under rental agreements accounted for in accordance with ASC 840 on a monthly basis as an operating lease, as well as “service pool” equipment. Service pool equipment is equipment owned and maintained by the Company that is swapped for equipment that needs repairs or maintenance by the Company while being rented by a patient. The Company records a provision for any excess, lost or damaged equipment when warranted based on an assessment of the equipment. Write-downs for equipment are included in cost of revenues. During the years ended December 31, 2015, 2014 and 2013, write downs for $36, $12 and $19, respectively, had been identified.
87
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
i. Impairment of long-lived assets:
The Company’s long-lived assets are reviewed for impairment in accordance with ASC 360-10, “Property, Plant and Equipment,” whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. Recoverability of an asset to be held and used is measured by a comparison of the carrying amount of an asset to the future undiscounted cash flows expected to be generated by the asset. If such asset is considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the asset exceeds its fair value. During the three years ended December 31, 2015, no impairment losses have been identified.
j. Other long-term assets:
Long term lease deposits in respect of office rent and vehicles under operating leases and restricted deposits are presented in other long-term assets.
k. Revenue recognition:
The TTFields delivery system (“System”) for GBM, Optune, is comprised of two main components: (1) an Electric Field Generator (the “device”) and (2) Transducer Arrays and related accessories that are disposable supplies to the device (“disposables”). Title is retained by the Company for the device and the patient is provided replacement disposables and technical support for the device during the rental period. The device and disposables are always supplied and functioning together and are not sold on a standalone basis.
Revenues are recognized when persuasive evidence of an arrangement exists, delivery of the system has occurred, the price is fixed or determinable and collectability is reasonably assured. The evidence of an arrangement generally consists of a prescription, a patient service agreement and the verification of eligibility and insurance with the patient’s third-party insurance company (“payer”). The Company generally bills third-party payers a monthly fee for use of the System by patients. As such, the Company takes assignment of benefits and risk of collection from the third-party payer. Patients have out-of-pocket costs for the amount not covered by their payer and the Company bills the patient directly for the amounts of their co-pays and deductible, subject to the Company’s patient assistance programs.
For the reported periods, all revenues are recognized when cash is collected assuming that all other revenue recognition criteria have been met, as the price is not fixed or determinable and the collectability cannot be reasonably assured. The price is not fixed or determinable since the Company does not have sufficient history with payers to reliably estimate their individual payment patterns and as such cannot reliably estimate the amount that would be ultimately collected. Once sufficient history is established and the Company can reliably estimate the amounts that would be ultimately collected per payer/payer group and the above criteria are met, the Company will recognize revenues from the use of the System on an accrual basis ratably over the lease term.
Revenues are presented net of indirect taxes, which include excise tax of $1,457 , $1,010, and $584 for the years ended December 31, 2015, 2014 and 2013, respectively, and other indirect tax of $818, $266 and $300 for the years ended December 31, 2015, 2014 and 2013, respectively.
l. Charitable care:
The Company provides Optune to patients who meet certain criteria under its charitable care policy without charge. Because the Company does not pursue collection of amounts determined to qualify as charity, they are not reported as revenue. The Company's costs of care provided under charitable care were: $1,376, $836 and $254 for the years ended December 31, 2015, 2014 and 2013, respectively. These estimates were determined by applying a ratio of costs to gross charges multiplied by the Company's gross charitable care charges.
m. Research, development and clinical trials:
Research, development and clinical trials, including direct and allocated expenses are expensed as incurred.
88
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
n. Shipping and handling costs:
The Company does not bill its customers for shipping and handling costs associated with shipping its delivery systems to its customers. These direct shipping and handling costs of $1,385, $553 and $431 for the years ended December 31, 2015, 2014 and 2013, respectively are included in selling and marketing costs.
o. Accounting for share-based payments:
The Company accounts for share-based compensation in accordance with ASC 718, “Compensation—Stock Compensation.” ASC 718 requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as an expense over the requisite service periods in the Company’s consolidated statements of operations.
The Company recognizes compensation costs net of a forfeiture rate only for those shares expected to vest using the accelerated method over the requisite service period of the award, which is generally the option vesting term of four years. ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
The Company selected the Black-Scholes option-pricing model as the most appropriate fair value method for its option awards. The option-pricing model requires a number of assumptions, of which the most significant are the share price expected, expected volatility and the expected option term.
Prior to the IPO, the fair value of ordinary shares underlying the options was historically determined by management and the board of directors. Because there was no public market for the Company’s ordinary shares, the board of directors determined fair value of an ordinary share at the time of grant of the option by considering a number of objective and subjective factors including operating and financial performance, the lack of liquidity of share capital, general and industry specific economic outlook and valuations performed amongst other factors. For the period from January 1, 2015 through the IPO and for the two years ended December 31, 2014, the Company’s board of directors determined the fair value of ordinary shares for the reported periods, among other factors, based on valuations performed using the hybrid method, which is the hybrid between the probability weighted expected return method (PWERM) and the option pricing method, as the Company began to consider IPO activities commencing in January 2012.
The computation of expected volatility is based on actual historical share price volatility of comparable companies. Expected term of options granted is calculated using the average between the vesting period and the contractual term to the expected term of the options in effect at the time of grant. The Company has historically not paid dividends and has no foreseeable plans to pay dividends and, therefore, uses an expected dividend yield of zero in the option pricing model. The risk-free interest rate is based on the yield of U.S. treasury bonds with equivalent terms.
p. Fair value of financial instruments:
The carrying amounts of cash and cash equivalents, short-term investments, restricted cash, receivables and prepaid expenses, trade payables and other accounts payable and accrued expenses approximate their fair value due to the short-term maturity of such instruments. Based upon the borrowing terms and conditions currently available to the Company, the carrying values of the long-term loans approximate fair value.
The Company accounts for certain assets and liabilities at fair value under ASC 820, “Fair Value Measurements and Disclosures.” Fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or a liability.
The hierarchy below lists three levels of fair value based on the extent to which inputs used in measuring fair value are observable in the market. The Company categorizes each of its fair value measurements in one of these three levels based on the lowest level input that is significant to the fair value measurement in its entirety.
The three levels of inputs that may be used to measure fair value are as follows:
Level 1 - Observable inputs that reflect quoted prices (unadjusted) for identical assets or liabilities in active markets;
89
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Level 2 - Includes other inputs that are directly or indirectly observable in the marketplace, other than quoted prices included in Level 1, such as quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets with insufficient volume or infrequent transactions, or other inputs that are observable (model-derived valuations in which significant inputs are observable), or can be derived principally from or corroborated by observable market data; and
Level 3 - Unobservable inputs which are supported by little or no market activity.
The availability of observable inputs can vary from instrument to instrument and is affected by a wide variety of factors, including, for example, the type of instrument, the liquidity of markets and other characteristics particular to the transaction. To the extent that valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment and the instrument are categorized as Level 3.
q. Basic and diluted net loss per share:
The Company applies the two class method as required by ASC 260-10, “Earnings Per Share.” ASC 260-10 requires the income or loss per share for each class of shares (ordinary and preferred shares) to be calculated assuming 100% of the Company’s earnings are distributed as dividends to each class of shares based on their contractual rights. No dividends were declared or paid during the reported periods.
According to the provisions of ASC 260-10, the Company’s preferred shares are not participating securities in losses and, therefore, are not included in the computation of net loss per share.
Basic and diluted net loss per share is computed based on the weighted average number of ordinary shares outstanding during each year. Diluted loss per share is computed based on the weighted average number of ordinary shares outstanding during the period, plus dilutive potential shares considered outstanding during the period, in accordance with ASC 260-10. Basic and diluted net loss per ordinary share was the same for each period presented as the inclusion of all potential ordinary shares (all preferred shares, options and warrants) outstanding was anti-dilutive.
r. Income taxes:
The Company accounts for income taxes in accordance with ASC 740-10, “Income Taxes.” ASC 740-10 prescribes the use of the liability method whereby deferred tax asset and liability account balances are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company provides a valuation allowance, to reduce deferred tax assets to their estimated realizable value, if needed.
The Company established reserves for uncertain tax positions based on the evaluation of whether or not the Company’s uncertain tax position is “more likely than not” to be sustained upon examination. The Company records interest and penalties pertaining to its uncertain tax positions in the financial statements as income tax expense.
s. Concentration of risks:
Financial instruments that potentially subject the Company to concentration of credit risk consist principally of cash and cash equivalents, restricted cash and short-term investments.
Cash and cash equivalents and restricted cash are invested in major banks or financial institutions in Jersey, the United States, Israel, Luxemburg, Switzerland, Japan and Germany. Such investments may be in excess of insured limits and are not insured in other jurisdictions. Generally, these investments may be redeemed upon demand and, therefore, bear minimal risk.
The Company has no off-balance sheet concentrations of credit risk such as foreign exchange contracts, option contracts or other foreign hedging arrangements.
In 2015, two payers represented $5,595 and $2,512 or 17% and 8% of net revenues, respectively. In 2014, the same two payers represented $2,372 and $2,014 or 15% and 12% of net revenues, respectively and in 2013, the same two payers represented $2,056 and $1,160 or 18% and 10% of net revenues, respectively.
90
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The majority of the Company’s employees in Israel have subscribed to Section 14 of Israel’s Severance Pay Law, 5723-1963 (“Section 14”). Pursuant to Section 14, the Company’s employees covered by this section are entitled to monthly deposits at a rate of 8.33% of their monthly salary, made on their behalf by the Company. Payments in accordance with Section 14 release the Company from any future severance liabilities in respect of those employees. Neither severance pay liability nor severance pay fund under Section 14 for such employees is recorded on the Company’s consolidated balance sheet.
With regard to employees in Israel that are not subject to Section 14, the Company’s liability for severance pay is calculated pursuant to Israeli Severance Pay Law, based on the most recent salary of the relevant employees multiplied by the number of years of employment as of the balance sheet date. These employees are entitled to one month’s salary for each year of employment or a portion thereof. The Company’s liability for these employees is fully provided for through monthly deposits to the employees’ pension and management insurance policies and an accrual. The value of these deposits is recorded as an asset on the Company’s consolidated balance sheet.
The carrying value of the deposited funds is based on the cash surrender value and includes profits accumulated up to the balance sheet date. The deposited funds may be withdrawn only upon the fulfillment of the obligation pursuant to the Israeli Severance Pay Law. Severance pay expense for the years ended December 31, 2015, 2014 and 2013 amounted to $356, $307 and $288, respectively.
u. Retirement and pension plans:
The Company has a 401(k) retirement savings plan for its U.S. employees. Each eligible employee may elect to contribute a portion of the employee’s compensation to the plan. The Company does not make any matching contributions to the plan.
The Company has a defined benefit plan with a pension fund for its Swiss employees, whereby the employee and the Company contribute to the pension fund. The Company accounts for its obligation, in accordance with ASC 715, "Compensation – Retirement Benefits" (see note 9).
The pension expense for the years ended December 31, 2015, 2014 and, 2013 was $404, $205 and $70, respectively.
v. Contingent liabilities:
The Company accounts for its contingent liabilities in accordance with ASC 450, “Contingencies.” A provision is recorded when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated.
With respect to legal matters, provisions are reviewed and adjusted to reflect the impact of negotiations, estimated settlements, legal rulings, advice of legal counsel and other information and events pertaining to a particular matter. As of December 31, 2015 and 2014, the Company was not a party to any ligation that could have a material adverse effect on the Company’s business, financial position, results of operations or cash flows.
w. Other comprehensive loss:
The Company accounts for comprehensive loss in accordance with ASC 220, "Comprehensive Income". This statement establishes standards for the reporting and display of comprehensive loss and its components. Comprehensive loss generally represents all changes in shareholders' equity during the period except those resulting from investments by, or distributions to, shareholders. The accumulated other comprehensive loss, net of taxes, at December 31, 2015 relates to a pension liability.
x. Recently issued accounting pronouncements:
In November 2015, the Financial Accounting Standards Board ("FASB") issued an accounting standards update for income taxes, which requires deferred tax assets and liabilities to be classified as noncurrent on the consolidated balance sheets. The new accounting guidance is effective for annual reporting periods beginning after December 15, 2016 and interim periods therein. Early adoption is permitted for all entities as of the beginning of interim or annual reporting periods. The Company early adopted the new guidance for the year ended December 31, 2015 and has applied the amendment on a prospective basis.
91
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
In April 2015, the FASB amended the existing accounting standards for imputation of interest. The amendments require that debt issuance costs related to a recognized debt liability be presented on the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. The recognition and measurement guidance for debt issuance costs are not affected by these amendments. The Company is required to adopt the guidance in the first quarter of fiscal 2017. Early adoption is permitted. The amendments should be applied retrospectively with the adjusted balance sheet of each individual period presented, in order to reflect the period-specific effects of applying the new guidance. The Company early adopted the new guidance for the year ended December 31, 2015.The adoption of this standard resulted in a reclassification of the debt issuance costs of $1.4 million for the year ended December 31, 2015, from Other long-term assets to Long-term loan, net of discount and issuance costs on the consolidated balance sheet. There was no impact to the consolidated statements of operations, comprehensive loss or cash flows.
In May 2014, the FASB amended the existing accounting standards for revenue recognition. The amendments are based on the principle that revenue should be recognized to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. In August 2015, the FASB issued an accounting standard update for a one-year deferral of the effective date, with an option of applying the standard on the original effective date, which for the Company is the first quarter of fiscal 2017. In accordance with this deferral, the Company is required to adopt these amendments no later than the first quarter of fiscal 2018. The amendments may be applied retrospectively to each prior period presented or retrospectively with the cumulative effect recognized as of the date of initial application. The Company is continuing to evaluate the impact of these amendments and the transition alternatives on its consolidated financial statements.
Note 3: Cash and Cash equivalents and Short-term investments
|
|
a. |
Cash and cash equivalents: |
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Cash |
|
$ |
75,421 |
|
|
$ |
47,614 |
|
|
Money market funds |
|
|
44,002 |
|
|
|
- |
|
|
U.S. treasury bills |
|
|
- |
|
|
|
9,999 |
|
|
Total cash and cash equivalents |
|
$ |
119,423 |
|
|
$ |
57,613 |
|
|
|
b. |
Short-term investments |
The Company invests in marketable U.S. Treasury Bills (“T-bills”) that are classified as held-to-maturity securities. The amortized cost and recorded basis of the T-bills are presented as short-term investments in the amount of $150,001 and $44,999, as of December 31, 2015 and 2014, respectively and their estimated fair value as of December 31, 2015 and 2014 was $149,978 and $44,999, respectively.
Note 4: Receivables and prepaid expenses
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Advances and receivables from suppliers |
|
$ |
7,323 |
|
|
$ |
4,262 |
|
|
Government authorities |
|
|
1,955 |
|
|
|
501 |
|
|
Prepaid expenses |
|
|
1,290 |
|
|
|
853 |
|
|
Others |
|
|
231 |
|
|
|
95 |
|
|
|
|
$ |
10,799 |
|
|
$ |
5,711 |
|
92
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Raw materials |
|
$ |
3,518 |
|
|
$ |
526 |
|
|
Work in process |
|
|
4,618 |
|
|
|
1,280 |
|
|
Finished goods |
|
|
5,458 |
|
|
|
1,640 |
|
|
|
|
$ |
13,594 |
|
|
$ |
3,446 |
|
Note 6: Property and equipment, net
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Cost: |
|
|
|
|
|
|
|
|
|
Computers and laboratory equipment |
|
$ |
6,734 |
|
|
$ |
3,745 |
|
|
Office furniture |
|
|
1,245 |
|
|
|
995 |
|
|
Production equipment |
|
|
857 |
|
|
|
- |
|
|
Leasehold improvements |
|
|
1,653 |
|
|
|
1,343 |
|
|
Total cost |
|
$ |
10,489 |
|
|
$ |
6,083 |
|
|
Accumulated depreciation and amortization |
|
|
(3,937 |
) |
|
|
(2,351 |
) |
|
Depreciated cost |
|
$ |
6,552 |
|
|
$ |
3,732 |
|
Depreciation expense was $1,348, $886 and $520 for the years ended December 31, 2015, 2014 and 2013, respectively.
In 2015, the Company implemented a new ERP system and capitalized costs incurred related to the system according to FASB ASC 350-40, "Accounting for the costs of Computer Software Developed or Obtained for Internal Use". As of December 31, 2015, the Company capitalized an accumulated amount of $2,803. Amortization for the year ended December 31, 2015 was $250.
Note 7: Field equipment, net
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Field equipment |
|
$ |
9,226 |
|
|
$ |
3,942 |
|
|
Less: accumulated depreciation |
|
|
(3,197 |
) |
|
|
(1,925 |
) |
|
Field equipment, net |
|
$ |
6,029 |
|
|
$ |
2,017 |
|
Depreciation expense was $1,555, $1,076 and 696 for the years ended December 31, 2015, 2014 and 2013, respectively.
Note 8: Other payables and accrued expenses
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Employees and payroll accruals |
|
$ |
8,258 |
|
|
$ |
5,846 |
|
|
Taxes payable and others |
|
|
2,850 |
|
|
|
693 |
|
|
Provision for settlement |
|
|
- |
|
|
|
1,000 |
|
|
Other |
|
|
764 |
|
|
|
97 |
|
|
|
|
$ |
11,872 |
|
|
$ |
7,636 |
|
93
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 9: Employee benefit obligations
The Company sponsors a defined benefit plan (the “Swiss Plan”) for all its employees in Switzerland for retirement benefits, as well as benefits on death or long-term disability. The liability in respect of the Swiss Plan is the projected benefit obligation calculated using the projected unit credit method. The projected benefit obligation as of December 31, 2015 represents the actuarial present value of the estimated future payments required to settle the obligation that is attributable to employee service rendered before that date. Swiss Plan assets are recorded at fair value. Pension expense is presented in the payroll expenses in the various functions in which the employees are engaged. Actuarial gains and losses arising from differences between the actual and the expected return on the Swiss Plan assets are recognized in accumulated other comprehensive income (loss) and amortized over the requisite service period. The plan is part of a collective pension foundation run by an insurance company. The Company and the employees pay retirement contributions, which are defined as a percentage of the employees’ covered salaries. The foundation, in turn, has all its risks (disability, death, longevity) and future benefits managed and guaranteed by the insurance company. Interest is credited to the employees’ account at the minimum rate provided in the Swiss Plan, payment which is guaranteed by the insurance contract, which represents the Swiss Plan’s primary asset. The targeted allocation for these funds is as follows:
|
Asset Allocation by Category as of December 31, 2015: |
|
|
|
|
|
Asset Category: |
|
Asset allocation (%) |
|
|
|
Debt Securities |
|
|
75 |
|
|
Real Estate |
|
|
16 |
|
|
Equity Securities |
|
|
3 |
|
|
Others |
|
|
6 |
|
|
Total |
|
|
100 |
|
94
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The following table sets forth the Swiss Plan’s funded status and amounts recognized in the consolidated financial statements for the year ended December 31, 2015:
|
|
|
December 31, |
|
|
|
|
|
2015 |
|
|
|
Change in Benefit Obligation |
|
|
|
|
|
Projected benefit obligation at beginning of year |
|
$ |
- |
|
|
Interest cost |
|
|
47 |
|
|
Company service cost |
|
|
312 |
|
|
Employee contributions |
|
|
189 |
|
|
Prior service cost |
|
|
158 |
|
|
Benefits paid |
|
|
4,023 |
|
|
Actuarial loss |
|
|
1,494 |
|
|
Projected benefit obligation at end of year |
|
$ |
6,223 |
|
|
Change in Plan Assets |
|
|
|
|
|
Fair value of plan assets at beginning of year |
|
$ |
- |
|
|
Actual return on plan assets |
|
|
(63 |
) |
|
Employer contributions |
|
|
284 |
|
|
Employee contributions |
|
|
189 |
|
|
Benefits paid |
|
|
4,023 |
|
|
Fair value of plan assets at end of year |
|
$ |
4,433 |
|
|
|
|
|
|
|
|
Funded Status at End of year |
|
|
|
|
|
Excess of obligation over assets |
|
$ |
(1,790 |
) |
|
|
|
|
|
|
|
Change in Accrued Benefit Liability |
|
|
|
|
|
Accrued benefit asset/(liability) at beginning of year |
|
$ |
- |
|
|
Company contributions made during year |
|
|
284 |
|
|
Net periodic benefit cost for year |
|
|
(404 |
) |
|
Net decrease in accumulated other comprehensive loss |
|
|
(1,670 |
) |
|
Accrued benefit liability at end of year |
|
$ |
(1,790 |
) |
|
|
|
|
|
|
|
|
|
December 31, |
|
|
|
|
|
2015 |
|
|
|
Non - current plan assets |
|
$ |
4,433 |
|
|
Non - current liability |
|
|
6,223 |
|
|
Accrued benefit liability at end of year |
|
$ |
(1,790 |
) |
|
Projected Benefit Payments |
|
|
|
|
|
2016 |
|
$ |
8 |
|
|
2017 |
|
|
13 |
|
|
2018 |
|
|
19 |
|
|
2019 |
|
|
25 |
|
|
2020 |
|
|
32 |
|
|
2021 - 2024 |
|
$ |
264 |
|
95
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The fair value of the plan assets is the estimated cash surrender value of the insurance contract at December 31, 2015. The level of inputs used to measure fair value was Level 2.
|
|
|
Year ended December 31, |
|
|
|
|
|
2015 |
|
|
|
Net Periodic Benefit Cost |
|
|
|
|
|
Service cost |
|
$ |
312 |
|
|
Interest cost |
|
|
47 |
|
|
Expected return on plan assets |
|
|
(38 |
) |
|
Amortization of prior service costs |
|
|
14 |
|
|
Amortization of transition obligation |
|
|
69 |
|
|
Total net periodic benefit cost |
|
$ |
404 |
|
|
|
|
|
|
|
|
Weighted average assumptions: |
|
|
|
|
|
Discount rate as of December 31 |
|
|
1.00% |
|
|
Expected long-term rate of return on assets |
|
|
1.00% |
|
|
Rate of compensation increase |
|
|
1.00% |
|
|
Mortality and disability assumptions (*) |
|
BVG 2010 GT |
|
|
(*) Mortality data used for actuarial calculation.
Note 10: Long-term loan, net of discount and issuance costs
In January 2013, the Company entered into a Credit Agreement (the “Credit Agreement”) with the lenders named therein (the “Lenders”) for a three-year term (the “Credit Facility”). Upon the closing of the Credit Agreement in January 2013, the Company withdrew the full $52,000 and issued the Lenders 975,644 warrants to purchase preferred H shares (converted to Ordinary shares as described in note 14a) at an exercise price of $18.77 per share. The Credit Facility accrued an 11% per year payment-in-kind interest charge, capitalized as additional principal quarterly, and a quarterly cash coupon of 3-month US$ LIBOR, subject to a LIBOR floor of 0.5% plus 1.5% per annum. The Company recorded the relative fair value of the warrants of $2,864 as shareholder’s equity and as a discount to the related loan outstanding. The total discount of $4,927 (including an original issue discount of $2,000) and additional deferred issuance costs of $500 in respect of the loan are amortized to interest expense over the three-year term using the effective interest method.
In December 2013, the Company repaid the entire outstanding Credit Facility of $58,000 of principal and accrued interest. Upon repayment of the Credit Facility, the discount and the deferred issuance costs were recorded immediately as interest expense. Financial expenses related to the Credit Facility for the year ended December 31, 2013 were $12,577.
In January 2015, the Company entered into a five-year term loan agreement (the “Loan Agreement”) with a lender to draw up to $100,000. In January 2015, the Company drew $25,000 from the lender. The Company may draw the remaining $75,000 at its option at any time through June 30, 2016. Interest on the outstanding loan is 10% annually, payable quarterly in arrears. In addition, there is a 1.5% funding fee payable on the amount drawn on the funding date, a 0.75% pay-down fee on all principal amount repayments to be paid on the date such payments of principal are made and a pre-payment fee of 3.0%, 2.0% or 1.0% if the Company prepays outstanding loan amounts prior to the first, second or third year, respectively, from the initial funding date. The entire outstanding principal loan is due on January 2020. The loan is secured by a first priority security interest in substantially all assets of the Company. The Loan Agreement sets forth certain affirmative and negative covenants with which the Company must comply on a quarterly basis commencing March 31, 2015 through the term of loan. As of December 31, 2015, the Company complies with its debt covenants.
The total discount of $491 and additional issuance costs of $1,739 are presented net of the loan and are amortized to interest expense over the five year term of the loan using the effective interest method.
96
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 11: Other long-term liabilities
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Deferred rent liability |
|
$ |
785 |
|
|
$ |
590 |
|
|
Leasehold improvements financing and other (see a and b below) |
|
|
254 |
|
|
|
321 |
|
|
Unrecognized tax benefits (Note 13e) |
|
|
1,565 |
|
|
|
308 |
|
|
Provision for settlement (Note 14c) |
|
|
- |
|
|
|
867 |
|
|
Other |
|
|
131 |
|
|
|
- |
|
|
|
|
$ |
2,735 |
|
|
$ |
2,086 |
|
a. In July 2013, the Company entered into a loan agreement with the landlord of its facility in Switzerland whereby the landlord will offer a loan of up to CHF 400 for the purpose of financing leasehold improvements in the facility. As of December 31, 2015 and 2014, the Company received CHF 220 ($232) of this financing. The principal and interest is due in monthly payments from January 1, 2014 through December 31, 2018 and bears an annual interest of 5%.
b. In May 2013 and January 2014, the Company entered into an agreement with the landlord of one of its facilities in the U.S. and an agreement with a leasing company of $226 for the purpose of financing leasehold improvements in the facility and a lease of machinery, respectively. The loan and interest is due in monthly payments from June 1, 2013 through May 1, 2023 and bears an annual interest of 7%.
The above principal leasehold improvement financing repayments as of December 31, 2015 are as follows:
|
2016 |
|
$ |
63 |
|
|
2017 |
|
|
69 |
|
|
2018 |
|
|
73 |
|
|
2019 |
|
|
23 |
|
|
2020 |
|
|
24 |
|
|
Thereafter |
|
|
65 |
|
|
|
|
|
317 |
|
|
Less: current portion of long-term loans |
|
|
(63 |
) |
|
Long-term loans, net of current portion |
|
$ |
254 |
|
Note 12: Commitments and contingent liabilities
a. The facilities of the Company are leased under various operating lease agreements for periods ending no later than 2023. The Company also leases motor vehicles under various operating leases, which expire on various dates, the latest of which is in 2018.
Future minimum lease payments under non-cancelable operating leases as of December 31, 2015, are as follows:
|
2016 |
$ |
2,320 |
|
|
2017 |
|
2,171 |
|
|
2018 |
|
1,152 |
|
|
2019 |
|
987 |
|
|
2020 |
|
591 |
|
|
Thereafter |
|
1,261 |
|
|
|
$ |
8,482 |
|
Lease and rental expense for the years ended December 31, 2015, 2014 and 2013 was $2,194, $1,794, and $1,356, respectively.
97
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
As of December 31, 2015 and 2014 the Company pledged bank deposits of $133 and $130, respectively, to cover bank guarantees in respect of its leases of operating facilities and obtained guarantees by the bank for the fulfillment of the Company’s lease commitments of $283 and $281, respectively.
b. For an additional commitment, see note 14c.
Note 13: Income taxes
a. The provision (benefit) for income taxes is comprised of:
Loss before income taxes:
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
United States (U.S.) |
|
$ |
(55,087 |
) |
|
$ |
(22,015 |
) |
|
$ |
(15,698 |
) |
|
Non-U.S. |
|
|
(52,060 |
) |
|
|
(58,285 |
) |
|
|
(61,319 |
) |
|
|
|
$ |
(107,147 |
) |
|
$ |
(80,300 |
) |
|
$ |
(77,017 |
) |
Income tax expense (benefit):
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Current: |
|
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
891 |
|
|
$ |
65 |
|
|
$ |
(18 |
) |
|
Non-U.S. |
|
|
3,678 |
|
|
|
324 |
|
|
|
308 |
|
|
Total current |
|
|
4,569 |
|
|
|
389 |
|
|
|
290 |
|
|
Deferred: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-U.S. |
|
|
(135 |
) |
|
|
(7 |
) |
|
|
63 |
|
|
Total deferred |
|
$ |
(135 |
) |
|
$ |
(7 |
) |
|
$ |
63 |
|
|
|
|
$ |
4,434 |
|
|
$ |
382 |
|
|
$ |
353 |
|
b. The Company is a resident taxpayer in Jersey and as such is not generally subject to Jersey tax on remitted foreign earnings. Therefore, the Company has chosen to present the note below using the notional U.S. federal income tax rate of 35% when presenting the Company's reconciliation of the income tax provision. A reconciliation of the provision for income taxes compared with the amounts at the notional federal statutory rate was:
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Loss before income taxes |
|
$ |
(107,147 |
) |
|
$ |
(80,300 |
) |
|
$ |
(77,017 |
) |
|
Statutory tax rate |
|
|
35 |
% |
|
|
35 |
% |
|
|
35 |
% |
|
Notional U.S. federal income taxes at statutory rate |
|
$ |
(37,501 |
) |
|
$ |
(28,105 |
) |
|
$ |
(26,956 |
) |
|
Non-deductible expenses |
|
|
2,621 |
|
|
|
1,242 |
|
|
|
1,411 |
|
|
Foreign tax rate differential |
|
|
18,573 |
|
|
|
20,261 |
|
|
|
20,965 |
|
|
Change in valuation allowance |
|
|
19,550 |
|
|
|
7,226 |
|
|
|
4,727 |
|
|
Unrecognized tax expense (benefit) |
|
|
1,257 |
|
|
|
(242 |
) |
|
|
206 |
|
|
Other |
|
|
(66 |
) |
|
|
- |
|
|
|
- |
|
|
Income tax expenses |
|
$ |
4,434 |
|
|
$ |
382 |
|
|
$ |
353 |
|
|
Effective tax rate |
|
|
-4.14 |
% |
|
|
-0.48 |
% |
|
|
-0.46 |
% |
The Company's tax rate is affected by the tax rates in the jurisdictions in which the Company operates, the relative amount of income earned by jurisdiction, jurisdictions with a statutory tax rate less than the U.S. tax rate of 35% and the relative amount of losses or income for which no tax benefit or expense was recognized due to a valuation allowance.
98
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
c. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and liabilities are as follows:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Allowance for doubtful accounts |
|
$ |
11,504 |
|
|
$ |
4,001 |
|
|
Revenue recognition (timing differences) |
|
|
21,972 |
|
|
|
9,042 |
|
|
Net operating loss carryforwards |
|
347 |
|
|
850 |
|
||
|
Other temporary differences |
|
952 |
|
|
482 |
|
||
|
Total gross deferred tax assets |
|
$ |
34,775 |
|
|
$ |
14,375 |
|
|
Less: valuation allowance |
|
|
(33,476 |
) |
|
|
(13,926 |
) |
|
Total deferred tax assets |
|
$ |
1,299 |
|
|
$ |
449 |
|
|
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
Fixed assets |
|
|
1,008 |
|
|
|
442 |
|
|
Total gross deferred tax liabilities |
|
$ |
1,008 |
|
|
$ |
442 |
|
|
Net deferred tax assets |
|
$ |
291 |
|
|
$ |
7 |
|
d. Carryforward loss:
As of December 31, 2015, the Company's Luxembourg subsidiary has $1.2 million of net operating loss carry forwards (NOLs) available for utilization in future years.
e. Accounting for uncertainty in income taxes (“ASC 740”):
A reconciliation of the beginning and ending balances of uncertain tax benefits is as follows:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Balance at beginning of the year |
|
$ |
308 |
|
|
$ |
549 |
|
|
Additions for tax positions related current year |
|
|
848 |
|
|
|
79 |
|
|
Additions for tax positions related to prior years |
|
|
409 |
|
|
|
- |
|
|
Reduction related to lapse of applicable statute of limitations |
|
|
- |
|
|
|
(320 |
) |
|
Balance at the end of the year |
|
$ |
1,565 |
|
|
$ |
308 |
|
The Company recognizes interest and penalties related to unrecognized tax benefits in tax expense. During the years ended December 31, 2015, 2014 and 2013, the Company accrued $26, $2 and $20, respectively, for interest and penalties expenses related to uncertain tax positions.
f. The Company's Israeli subsidiary is currently under an income tax audit for the tax years 2011 through 2013. There are no other ongoing income tax audits.
99
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Share capital is composed as follows:
|
|
|
Issued and outstanding |
|
|||||
|
|
|
Number of shares |
|
|||||
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Ordinary shares no par value |
|
|
83,778,581 |
|
|
|
13,431,414 |
|
|
Series A Convertible Preferred shares no par value |
|
|
- |
|
|
|
4,177,235 |
|
|
Series B Convertible Preferred shares no par value |
|
|
- |
|
|
|
4,590,439 |
|
|
Series C Convertible Preferred shares no par value |
|
|
- |
|
|
|
2,261,666 |
|
|
Series D Convertible Preferred shares no par value |
|
|
- |
|
|
|
4,451,913 |
|
|
Series E Convertible Preferred shares no par value |
|
|
- |
|
|
|
7,790,861 |
|
|
Series F Convertible Preferred shares no par value |
|
|
- |
|
|
|
12,864,362 |
|
|
Series G Convertible Preferred shares no par value |
|
|
- |
|
|
|
5,106,269 |
|
|
Series H Convertible Preferred shares no par value |
|
|
- |
|
|
|
4,073,020 |
|
|
Series I Convertible Preferred shares no par value |
|
|
- |
|
|
|
13,360,252 |
|
|
Total |
|
|
- |
|
|
|
58,676,017 |
|
a. Investment rounds:
1) Series I Convertible Preferred shares:
In 2013, the Company sold to investors 13,360,252 Series I Convertible Preferred shares at a purchase price of $14.53 per share, for total consideration of $191,738, net of issuance expenses. Prior to conversion of the Series I Convertible Preferred shares into ordinary shares as a result of the IPO, Series I Convertible Preferred shares were senior to the other series of preferred shares on payment of the liquidation preference ($14.53 per share), but otherwise had similar participating preferred rights, dividend rights and voting rights to the other series of preferred shares.
2) Series J Preferred shares:
In June 2015, the Company sold to investors 4,068,500 Series J Convertible Preferred shares at a price per share of $23.33, for a total consideration of $94,599 (net of issuance expenses of $319). Prior to conversion of the Series J Convertible Preferred shares into ordinary shares as a result of the IPO, such shares
were senior to the other series of preferred shares on payment of the liquidation preference (equal to $23.33 per share), but otherwise had similar participating preferred rights, dividend rights and voting rights of the other series of preferred shares.
b. Rights, preferences and restrictions:
On October 7, 2015, the Company completed the IPO of its ordinary shares by issuing 7,876,195 ordinary shares (including exercise of overallotments) and raising net proceeds of $157,534, at which time the Series A through J Convertible Preferred shares converted into ordinary shares and ceased to exist. The rights, preferences and restrictions of the Series A through J Convertible Preferred shares were, and the rights, preferences and restrictions of ordinary shares are, as follows:
Conversion — Each Convertible Preferred share was convertible into ordinary shares (i) at any time at the holder’s option or (ii) automatically upon a qualified initial public offering, which is defined in the Company’s articles of association in effect prior to the IPO as an offering in which the aggregate gross proceeds received by the Company are (1) at least $50,000 if the equity securities of the Company that are sold in such offering are listed or traded on the New York Stock Exchange (NYSE) or the National Association of Securities Dealers Automated Quotations (NASDAQ) or (2) at least $100,000 if the equity securities of the Company that are sold in such offering are listed or traded on any other public stock exchange. The IPO constituted a qualified initial public offering and, therefore, triggered conversion of the Series A through J Convertible Preferred shares.
100
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Each of the Series A, B, C, D, E, F, G, H, I and J Convertible Preferred shares converted to ordinary shares on a 1-for-1 ratio, following adjustment for any recapitalizations, splits, ordinary share dividends and similar standard anti-dilution events.
Dividend rights — The holders of Series A through J Convertible Preferred shares were entitled to receive on a pari passu basis, prior and in preference to the declaration or payment of any dividend or distribution to the holders of ordinary shares on an as-converted basis when, as and if any dividend or distribution is declared by the Company’s board of directors in respect of ordinary shares, up to an amount equal to the applicable original issue price for such preferred shares.
Liquidation preference — The holders of Series A through J Convertible Preferred shares have a liquidation preference equal to $1.52, $1.09, $1.77, $1.80, $1.80, $2.23, $8.64, $13.5, $14.53, and $23.33 per share, respectively, plus any declared but unpaid dividends.
Preferred shareholders were entitled to their liquidation preferences based on their seniority. Series J Convertible Preferred shareholders would have been the first to receive their liquidation preferences and they would have been followed by the holders of Series I, H, G, F, E, D, C, B and A Convertible Preferred shares in turn.
Voting rights — Each holder of ordinary shares is and, prior to conversion into ordinary shares as a result of the IPO, each holder of Series A, B, C, D, E, F, G, H, I or J Convertible Preferred shares was, entitled to one vote.
c. Settlement agreement:
In February 2015, the Company entered into a settlement agreement (the “Settlement Agreement”) with a third party to resolve certain potential disputes regarding intellectual property developed by the Company’s founder and previously assigned to the Company. In exchange for a release of potential disputes from the third party, the Company paid $1,000 on execution of the Settlement Agreement and agreed to pay an additional $1,000 (the “Additional Payment”) at the earliest of (i) 18 months after signing of the Agreement, (ii) an IPO or (iii) the earlier of consummation of a merger/acquisition (“M&A”) or achievement of a Cumulative Net Sales milestone of $250,000 (as defined pursuant to the Agreement). The Company also agreed to pay an additional $5,500 on the earlier of (i) achievement of the Cumulative Net Sales milestone per above or (ii) consummation of a merger or acquisition transaction. In addition, the Company agreed to issue 1,005,210 ordinary shares (the “Settlement Shares”) to the third party and to grant to the third party options to purchase 1,005,210 ordinary shares (the “Settlement Options”) that are fully vested and at no cost. The Settlement Options terminate at the earlier of (i) 12 months subsequent to an IPO or (ii) immediately prior to a merger or acquisition transaction.
In February 2015, the Company contemporaneously entered into a Letter of Agreement (“Letter of Agreement”) with a Company founder and a related party of the founder (together, the “Founder”) pursuant to which the Founder indemnified the Company for compensation incurred to the third party by providing 2,010,420 ordinary shares which were redeemed and cancelled (the “Redeemed Shares”) in March 2015 to the Company at par value. The Founder also may be obligated to pay an additional $2,000 in cash to the Company upon its request out of the net proceeds from the sale of any ordinary shares by the Founder in a private transaction or following the consummation of a qualified initial public offering (as defined above in Note 14a—Conversion) in an open market transaction if the closing price of the ordinary shares is at least 80% of the price per share for which the ordinary shares were sold in the IPO (after deducting underwriting discounts and commissions and offering expenses). In March 2015, the Company provided the Settlement Shares and Settlement Options to the third party. On October 7, 2015, the Company completed the IPO of its ordinary shares and the Additional Payment was paid in October 2015.
Accordingly, for the year ended December 31, 2014, in accordance with ASC 450, the Company recorded a provision for a net settlement expense of $1,867 in general and administrative expense, reflecting the present value of the cash obligation of $2,000 and the fair value of the Settlement Shares issued and Settlement Options granted to the third party, net of the fair value of the Redeemed Shares provided by the Founder as consideration, which amounted to nil as presented in the statement of shareholder’s equity, in connection with the indemnification provided and the Letter of Agreement.
d. Warrants:
As part of the Series B, C, D and E Convertible Preferred share investment agreements, the investors received warrants to purchase ordinary shares. The Company accounted for these warrants as equity instruments based on the guidance of ASC 815, “Derivatives and Hedging”, ASC 480-10, “Distinguishing Liabilities from Equity”, its related FASB staff positions, ASC 815-40 “Contracts in Entity’s Own Stock” and the AICPA Technical Practice Aid for accounting for preferred shares and warrants, including the roadmap for accounting for freestanding financial instruments indexed to, and potentially settled in, a company’s own stock.
101
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Significant terms of the warrants to purchase ordinary shares that were issued to purchasers of the Series B, C, D and E Convertible Preferred shares are as follows as of December 31, 2015 and 2014:
|
|
|
Warrants for ordinary shares |
|
|
Exercise price per share |
|
||||||
|
Expiration date |
|
2015 |
|
|
2014 |
|
|
|
|
|
||
|
October 11, 2015 (1) |
|
|
- |
|
|
|
565,411 |
|
|
|
3.54 |
|
|
May 8, 2016 (1) |
|
|
1,108,050 |
|
|
|
1,112,983 |
|
|
|
3.59 |
|
|
July 31, 2017 |
|
|
556,678 |
|
|
|
556,678 |
|
|
|
3.59 |
|
|
January 22, 2018 |
|
|
556,678 |
|
|
|
556,678 |
|
|
|
3.59 |
|
|
July 21, 2018 |
|
|
834,355 |
|
|
|
834,355 |
|
|
|
3.59 |
|
|
|
|
|
3,055,761 |
|
|
|
3,626,105 |
|
|
|
|
|
|
(1) |
Warrants to purchase 570,344 ordinary shares were exercised in 2015, resulting in the issuance of 570,344 ordinary shares. |
Pursuant to the Credit Agreement (see Note 10), the Company issued the Lenders 975,644 warrants to purchase Series H Convertible Preferred shares at an exercise price of $18.77 per share. The warrants were exercised on a cashless basis in January 2016.
e. Share option plans:
In 2003, the Company and its shareholders approved and adopted the 2003 Share Option Plan (the “2003 Plan”), which provided for the grant of options to the Company’s officers, directors, employees and advisors. The options granted generally have a four-year vesting period and expire ten years after the date of grant. Since March 2013, when the 2003 Plan expired, the Company has made grants pursuant to the 2013 Share Option Plan (as described below) and, following completion of the IPO in October 2015, all future equity grants will be made under the 2015 Omnibus Incentive Plan (as described below); however, any awards granted under the 2003 Plan that were outstanding as of the IPO continue to be subject to the terms and conditions of the 2003 Plan and the applicable option award agreement.
In 2013, the Company and its shareholders approved and adopted the 2013 Equity Incentive Share Option Plan (the “2013 Plan”), which provided for the grant of options to the Company’s officers, directors, advisors, management and other key employees. The options granted generally have a four-year vesting period and expire ten years after the date of grant. Options granted under the 2013 Plan that are cancelled or forfeited before expiration become available for future grant.
In February and March 2015, the Company’s board of directors and its shareholders approved an increase in the number of ordinary shares reserved for grant of options pursuant to the 2013 Plan by 2,956,500 ordinary shares to 13,198,224 ordinary shares. Following completion of the IPO in October 2015, all future equity grants will be made under the 2015 Omnibus Incentive Plan (as described below); accordingly, as of December 31, 2015, there are no options available for future grants under the 2013 Plan. Any awards granted under the 2013 Plan that were outstanding as of the IPO continue to be subject to the terms and conditions of the 2013 Plan and the applicable option award agreement.
In August 2015, the Company’s board of directors adopted and established the 2015 Omnibus Incentive Plan (the “2015 Plan”). The Company’s shareholders approved the 2015 Plan in September 2015. Under the 2015 Plan, the Company can issue various types of equity compensation awards such as restricted shares, performance shares, restricted stock units, performance units, long-term cash award and other share-based awards. The options granted generally have a four-year vesting period and expire ten years after the date of grant. Options granted under the 2015 Plan that are cancelled or forfeited before expiration become available for future grant.
On December 31, 2015, in accordance with the terms of the 2015 Plan, the number of shares available for issuance under the 2015 Plan automatically increased by 4% of the Company’s outstanding ordinary shares as of December 30, 2015. As a result, the number of shares available for issuance under the 2015 Plan increased from 12,900,000 shares to 16,251,143 shares. As of December 31, 2015, 14,947,667 ordinary shares are available for grant under the 2015 Plan.
102
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The fair value of share-based awards was estimated using the Black-Scholes option-pricing model for all grants, with the following underlying assumptions:
|
|
|
Year ended December 31, |
|
|||||||
|
|
|
2015 |
|
2014 |
|
|
2013 |
|
||
|
Expected term (years) |
|
6.25 |
|
|
6.25 |
|
|
|
6.25 |
|
|
Expected volatility |
|
59%-65.8% |
|
73.1%-75.3% |
|
|
70.4%-75.9% |
|
||
|
Risk-free interest rate |
|
1.74%-2.05% |
|
1.9%-2.3% |
|
|
1.4%-2.0% |
|
||
|
Dividend yield |
|
0% |
|
0% |
|
|
0% |
|
||
A summary of the status of the Company’s options to purchase ordinary shares as of December 31, 2015 and changes during the year ended on that date is presented below:
|
|
|
Year ended December 31, 2015 |
|
|||||||||
|
|
|
Number of options |
|
|
Weighted average exercise price |
|
|
Aggregate intrinsic value |
|
|||
|
Outstanding at beginning of year |
|
|
7,426,159 |
|
|
|
3.98 |
|
|
|
|
|
|
Granted |
|
|
3,108,393 |
|
|
|
18.01 |
|
|
|
|
|
|
Exercised |
|
|
(183,911 |
) |
|
|
3.08 |
|
|
|
|
|
|
Forfeited and cancelled |
|
|
(215,812 |
) |
|
|
8.81 |
|
|
|
|
|
|
Outstanding at end of year |
|
|
10,134,829 |
|
|
|
8.20 |
|
|
|
144,776 |
|
|
Exercisable options |
|
|
5,521,406 |
|
|
|
3.04 |
|
|
|
106,684 |
|
|
Vested and expected to vest |
|
|
10,007,130 |
|
|
|
8.11 |
|
|
|
143,835 |
|
The total equity-based compensation expense related to all of the Company’s equity-based awards recognized for the years ended December 31, 2015, 2014 and 2013, was comprised as follows:
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Cost of revenues |
|
$ |
174 |
|
|
$ |
32 |
|
|
$ |
52 |
|
|
Research, development and clinical trials |
|
|
2,529 |
|
|
|
820 |
|
|
|
1,137 |
|
|
Sales and marketing |
|
|
2,496 |
|
|
|
1,104 |
|
|
|
791 |
|
|
General and administrative |
|
|
6,661 |
|
|
|
2,668 |
|
|
|
3,140 |
|
|
Total share-based compensation expense |
|
$ |
11,860 |
|
|
$ |
4,624 |
|
|
$ |
5,120 |
|
As of December 31, 2015, there were unrecognized compensation costs of $25,008, which are expected to be recognized over a weighted average period of approximately 3.39 years.
The weighted average grant date fair values of the Company’s options granted during the years ended December 31, 2015, 2014 and 2013 were $10.64, $5.08 and $4.59 per share, respectively. The weighted average grant date fair values of the Company’s unvested options for the years ended December 31, 2015,2014 and 2013 were $8.66, $4.26 and $3.42 per share, respectively, and the unvested options for the years ended December 31, 2015,2014 and 2013 were 4,613,423, 2,934,974 and 3,125,543, respectively.
The weighted average grant date fair values of the Company’s vested options during the years ended December 31, 2015, 2014 and 2013 were $3.66, $2.89 and $2.26, respectively, and the vested options for the years ended December 31, 2015,2014 and 2013 were 1,235,880, 1,166,974 and 1,030,933, respectively. The weighted average grant date fair values of the Company’s options forfeited and cancelled during the years ended December 31, 2015, 2014 and 2013 were $ 5.73, $3.59 and $1.84, respectively.
The aggregate intrinsic values for the options exercised during the years ended December 31, 2015, 2014 and 2013 were $3,546, $3,339 and $3,431, respectively. The aggregate intrinsic value is calculated as the difference between the per share exercise price and the deemed fair value of the Company’s ordinary shares for each share subject to an option multiplied by the number of shares subject to options at the date of exercise. The Company deemed the fair value of the Company’s ordinary shares to be $22.36, $7.73 and $7.28 per share as of December 31, 2015, 2014, and 2013, respectively.
103
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The options outstanding as of December 31, 2015 are as follows:
|
Exercise price |
|
|
Number of options outstanding as of December 31, 2015 |
|
|
Weighted average remaining contractual term |
|
|
Number of options exercisable as of December 31, 2015 |
|
|
Weighted average remaining contractual term |
|
|||||
|
$ |
|
|
|
|
|
|
(years) |
|
|
|
|
|
|
(years) |
|
|||
|
|
0.17 |
|
|
|
1,402,757 |
|
|
|
0.71 |
|
|
|
1,402,757 |
|
|
|
0.71 |
|
|
|
0.23 |
|
|
|
440,531 |
|
|
|
3.42 |
|
|
|
440,531 |
|
|
|
3.42 |
|
|
|
0.38 |
|
|
|
488,331 |
|
|
|
4.78 |
|
|
|
488,331 |
|
|
|
4.78 |
|
|
|
3.44 |
|
|
|
1,779,072 |
|
|
|
5.88 |
|
|
|
1,728,710 |
|
|
|
5.89 |
|
|
|
6.72 |
|
|
|
850,401 |
|
|
|
6.67 |
|
|
|
642,321 |
|
|
|
6.66 |
|
|
|
6.83 |
|
|
|
92,227 |
|
|
|
6.95 |
|
|
|
69,308 |
|
|
|
6.95 |
|
|
|
7.03 |
|
|
|
613,174 |
|
|
|
7.14 |
|
|
|
307,309 |
|
|
|
7.14 |
|
|
|
7.04 |
|
|
|
135,992 |
|
|
|
7.46 |
|
|
|
67,982 |
|
|
|
7.46 |
|
|
|
7.28 |
|
|
|
167,921 |
|
|
|
7.65 |
|
|
|
83,946 |
|
|
|
7.65 |
|
|
|
7.48 |
|
|
|
504,365 |
|
|
|
8.14 |
|
|
|
130,509 |
|
|
|
8.13 |
|
|
|
7.52 |
|
|
|
110,560 |
|
|
|
8.24 |
|
|
|
29,925 |
|
|
|
8.23 |
|
|
|
7.58 |
|
|
|
52,032 |
|
|
|
8.49 |
|
|
|
13,006 |
|
|
|
8.49 |
|
|
|
7.73 |
|
|
|
431,053 |
|
|
|
8.77 |
|
|
|
107,755 |
|
|
|
8.77 |
|
|
|
14.37 |
|
|
|
1,616,011 |
|
|
|
9.16 |
|
|
|
9,016 |
|
|
|
8.84 |
|
|
|
15.60 |
|
|
|
146,926 |
|
|
|
9.32 |
|
|
|
- |
|
|
|
- |
|
|
|
20.20 |
|
|
|
140,466 |
|
|
|
9.81 |
|
|
|
- |
|
|
|
- |
|
|
|
22.00 |
|
|
|
913,210 |
|
|
|
9.76 |
|
|
|
- |
|
|
|
- |
|
|
|
27.50 |
|
|
|
249,800 |
|
|
|
9.97 |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
|
10,134,829 |
|
|
|
6.56 |
|
|
|
5,521,406 |
|
|
|
4.63 |
|
In August, 2015, the Company’s board of directors adopted an employee share purchase plan (“ESPP”) subject to shareholder approval, which was obtained in September, 2015. The Company adopted the ESPP to encourage and enable eligible employees to acquire ownership of the Company’s ordinary shares purchased through accumulated payroll deductions on an after-tax basis. The ESPP is intended to be an “employee stock purchase plan” within the meaning of Section 423 of the Code and the provisions of the ESPP will be construed in a manner consistent with the requirements of such section. The Company intends to begin offerings under the ESPP at a future date determined by the compensation committee, in its sole discretion. As of December 31, 2015 no shares have been offered under the ESPP.
Under the ESPP, initially an aggregate of 830,000 ordinary shares could be purchased by eligible employees who become participants in the ESPP; which amount shall be automatically increased on December 31 of each year during the term of the ESPP to an amount equal to 1% of the total number of ordinary shares outstanding on December 30 of such year unless otherwise determined by the board of directors. On December 31, 2015, the number of shares available for issuance under the ESPP increased from 837,785 shares to 1,667,785 shares. As of December 31, 2015, 1,667,785 ordinary shares are available for grant under the ESPP.
104
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 15: Financial expenses, net
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Financial expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest expense |
|
$ |
(2,373 |
) |
|
$ |
(41 |
) |
|
$ |
(7,158 |
) |
|
Amortization of credit facility costs |
|
|
(329 |
) |
|
|
- |
|
|
|
(5,432 |
) |
|
Foreign currency transaction losses |
|
|
(356 |
) |
|
|
(104 |
) |
|
|
(157 |
) |
|
Others |
|
|
(177 |
) |
|
|
(142 |
) |
|
|
(78 |
) |
|
|
|
$ |
(3,235 |
) |
|
$ |
(287 |
) |
|
$ |
(12,825 |
) |
|
Financial income: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
$ |
84 |
|
|
$ |
143 |
|
|
$ |
267 |
|
|
Total financial expenses, net |
|
$ |
(3,151 |
) |
|
$ |
(144 |
) |
|
$ |
(12,558 |
) |
Note 16: Basic and diluted net loss per share
The following table sets forth the computation of the Company’s basic and diluted net loss per ordinary share:
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
Net loss attributable to ordinary shares as reported |
|
$ |
(111,581 |
) |
|
$ |
(80,682 |
) |
|
$ |
(77,370 |
) |
|
Shares used in computing net loss per ordinary share, basic and diluted |
|
|
30,401,603 |
|
|
|
12,490,017 |
|
|
|
11,498,392 |
|
|
Net loss per ordinary share, basic and diluted |
|
$ |
(3.67 |
) |
|
$ |
(6.46 |
) |
|
$ |
(6.73 |
) |
For the years ended December 31, 2015, 2014 and 2013, all outstanding preferred shares, options and warrants have been excluded from the calculation of the diluted net loss per share since their effect was anti-dilutive.
Note 17: Subcontractor
The Company is dependent upon sole source suppliers for certain key components used in its delivery systems. The Company’s management believes that in most cases other suppliers could provide similar components at comparable terms. A change of suppliers which requires FDA or other regulatory approval, however, could cause a material delay in manufacturing and a possible loss of sales, which could adversely affect the Company’s operating results and financial position.
Note 18: Supplemental information
The following table presents long-lived assets by location:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
United States |
|
|
6,600 |
|
|
|
3,468 |
|
|
Switzerland |
|
|
4,204 |
|
|
|
1,259 |
|
|
Israel |
|
$ |
1,376 |
|
|
$ |
1,009 |
|
|
Others |
|
|
401 |
|
|
|
13 |
|
|
|
|
$ |
12,581 |
|
|
$ |
5,749 |
|
105
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The Company’s net revenues by geographic region, based on the patient’s location are summarized as follows:
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||
|
United States |
|
|
30,961 |
|
|
|
14,951 |
|
|
|
10,330 |
|
|
Europe, Israel and Japan |
|
|
2,126 |
|
|
$ |
539 |
|
|
$ |
29 |
|
|
|
|
$ |
33,087 |
|
|
$ |
15,490 |
|
|
$ |
10,359 |
|
106
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
(a) Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on that evaluation, the Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K have been designed and are functioning effectively to provide reasonable assurance that the information required to be disclosed by us in reports filed under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. We believe that a controls system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
(b) Management’s Annual Report on Internal Control over Financial Reporting
This report does not include a report of management’s assessment regarding internal control over financial reporting due to a transition period established by rules of the SEC for newly public companies.
(c) Attestation Report of the Registered Public Accounting Firm
This report does not include an attestation report of our registered public accounting firm due to an exemption established by the JOBS Act for “emerging growth companies.”
(d) Changes in Internal Control over Financial Reporting
There have been no changes in our internal control over financial reporting during our most recent fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
On February 24, 2016, the Compensation Committee of our Board of Directors ratified and adopted a form of stock option award agreement for use in Germany (the “Award Agreement”). The Award Agreement was adopted under the Company’s 2015 Omnibus Incentive Plan (the “Plan”) in order to meet specific legal and tax requirements in Germany. A summary of the material terms of the Plan is set forth in our Amendment No. 2 to Form S-1 Registration Statement filed with the SEC on October 1, 2015 under the section entitled “2015 omnibus incentive plan”, beginning on page 137, and is incorporated herein by reference. That summary and the foregoing description are qualified in their entirety by reference to the text of the Plan and the Award Agreement and the other forms of grant notice and award agreements under the Plan, which are filed as Exhibits 10.5, 10.17 through 10.23 and 10.25 to this Annual Report on Form 10-K and incorporated herein by reference.
On February 24, 2016, we entered into Amendment No. 2 to the Consulting Services Agreement with William F. Doyle dated as of June 24, 2014. Amendment No. 2 extended the term of the Consulting Services Agreement for one year commencing as of January 1, 2016 and continuing through December 31, 2016. All other terms of the Consulting Services Agreement remain the same.
107
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Directors
The information required by Item 10 is incorporated herein by reference to the information contained under the caption “Proposal 1—Election of Directors” in our definitive proxy statement related to the 2016 annual meeting of stockholders.
Executive Officers
The information concerning our executive officers required by this Item 10 is provided under the caption “Executive Officers of the Registrant” in Part I hereof.
Section 16(a) Beneficial Ownership Reporting Compliance
The information concerning Section 16(a) Beneficial Ownership Reporting Compliance by our directors and executive officers is incorporated by reference to the information contained under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in our definitive proxy statement related to the 2016 annual meeting of stockholders.
Audit Committee
The information required by this Item 10 is incorporated by reference to the information contained in our definitive proxy statement related to the 2016 annual meeting of stockholders.
Code of Ethics
The information concerning our Code of Ethics is incorporated by reference to the information contained under the caption “Governance of the Company—Does the Company have a “Code of Ethics’?” in our definitive proxy statement related to the 2016 annual meeting of stockholders.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item 11 is incorporated by reference to the information contained in our definitive proxy statement related to the 2016 annual meeting of stockholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by Item 12 is incorporated by reference to the information contained in our definitive proxy statement related to the 2016 annual meeting of stockholders.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by Item 13 is incorporated by reference to the information contained in our definitive proxy statement related to the 2016 annual meeting of stockholders.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by Item 14 is incorporated by reference to the information contained in our definitive proxy statement related to the 2016 annual meeting of stockholders.
108
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) DOCUMENTS FILED AS PART OF THIS REPORT
The following is a list of our consolidated financial statements and our subsidiaries and supplementary data included in this Annual Report on Form 10-K under Item 8 of Part II hereof:
|
1. FINANCIAL STATEMENTS AND SUPPLEMENTAL DATA |
|
|
|
|
|
|
Report of Independent Registered Public Accounting Firm. |
|
|
|
|
|
Consolidated Balance Sheets as of December 31, 2015 and 2014. |
|
|
|
|
|
Consolidated Statements of Operations for the years ended December 31, 2015, 2014 and 2013. |
|
|
|
|
|
Consolidated Statement of Comprehensive Loss for the years ended December 31, 2015, 2014 and 2013. |
|
|
|
|
|
Consolidated Statements of Shareholders’ Equity for the years ended December 31, 2015, 2014 and 2013. |
|
|
|
|
|
Consolidated Statements of Cash Flows for the years ended December 31, 2015, 2014 and 2013. |
|
|
|
|
|
Notes to Consolidated Financial Statements. |
|
|
|
|
2. FINANCIAL STATEMENT SCHEDULES |
|
|
|
|
Schedules are omitted because they are not applicable or are not required, or because the required information is included in the consolidated financial statements or notes thereto.
109
The following is a list of exhibits filed as part of this Annual Report on Form 10-K. Where so indicated by footnote, exhibits that were previously filed are incorporated by reference. For exhibits incorporated by reference, the location of the exhibit in the previous filing is indicated.
EXHIBIT INDEX
|
Exhibit Number |
|
|
|
Incorporated by Reference |
|
Filed Herewith |
|||||
|
|
Exhibit Description |
|
Form |
|
Date |
|
Number |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
|
3.1 |
|
Memorandum and Articles of Association |
|
S-1/A |
|
9/21/15 |
|
3.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
Form of Ordinary Shares Certificate |
|
S-1/A |
|
9/21/15 |
|
4.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.2 |
|
Eleventh Amended and Restated Investors Rights Agreement, dated June 1, 2015 |
|
DRS |
|
6/24/15 |
|
4.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.3 |
|
Tenth Amended and Restated Registration Rights Agreement, dated June 1, 2015 |
|
DRS |
|
6/24/15 |
|
4.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1 |
|
Loan and Security Agreement between the Company and Biopharma Secured Investments III Holdings Cayman LP, dated January 7, 2015 |
|
DRS |
|
6/24/15 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2 |
|
Strategic Supplier Agreement between the Company and ITT Corporation (assigned to Exelis Inc.), dated July 21, 2011, as amended on April 29, 2014† |
|
DRS |
|
6/24/15 |
|
10.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3 |
|
2003 Share Option Plan# |
|
DRS |
|
6/24/15 |
|
10.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4 |
|
2013 Share Option Plan# |
|
DRS |
|
6/24/15 |
|
10.4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5 |
|
2015 Omnibus Incentive Plan# |
|
S-1/A |
|
9/21/15 |
|
10.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.6 |
|
Employment Agreement with Asaf Danziger and the Company, dated October 1, 2002# |
|
DRS |
|
6/24/15 |
|
10.6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7 |
|
Employment Agreement with Wilhelmus C.M. Groenhuysen and the Company, dated December 23, 2011†# |
|
DRS |
|
6/24/15 |
|
10.7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.8 |
|
Employment Agreement with Kinyip Gabriel Leung and the Company, dated August 24, 2011†# |
|
DRS |
|
6/24/15 |
|
10.8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.9 |
|
Employment Agreement with Eilon Kirson and the Company, dated June 2, 2002†# |
|
DRS |
|
6/24/15 |
|
10.9 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.10 |
|
Consulting Agreement with Palti Consultants Ltd. and the Company, dated May 1, 2002†# |
|
DRS |
|
6/24/15 |
|
10.10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.11 |
|
Consulting Agreement with William F. Doyle, dated June 24, 2014†# |
|
DRS |
|
6/24/15 |
|
10.11 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.12 |
|
Form of Indemnification Agreement |
|
S-1/A |
|
9/21/15 |
|
10.12 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.13 |
|
Settlement Agreement with the Technion, dated February 10, 2015 |
|
DRS/A |
|
8/11/2015 |
|
10.13 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.14 |
|
Director Compensation Plan# |
|
S-1/A |
|
9/21/15 |
|
10.14 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.15 |
|
Employee Share Purchase Plan# |
|
S-1/A |
|
9/21/15 |
|
10.15 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.16 |
|
Amendment to Consulting Agreement with William F. Doyle, dated September 16, 2015# |
|
S-1/A |
|
9/21/15 |
|
10.16 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.17 |
|
Form of Non-Qualified Stock Option Agreement pursuant to the 2015 Omnibus Incentive Plan (U.S. individuals)# |
|
S-1/A |
|
9/21/15 |
|
10.17 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
110
|
Exhibit Number |
|
|
|
Incorporated by Reference |
|
Filed Herewith |
|||||
|
|
Exhibit Description |
|
Form |
|
Date |
|
Number |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
|
|
Form of Incentive Stock Option Agreement pursuant to the 2015 Omnibus Incentive Plan (U.S. individuals)# |
|
S-1/A |
|
9/21/15 |
|
10.18 |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.19 |
|
2015 Omnibus Incentive Plan, including 2015 Omnibus Incentive Plan Sub-Plan for Grantees Subject to Israeli Taxation and 2015 Omnibus Incentive Plan Sub-Plan for Switzerland# |
|
8-K |
|
12/22/15 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.20 |
|
Form of Incentive Stock Option Agreement pursuant to the 2015 Omnibus Incentive Plan for use in connection with the 2015 Omnibus Incentive Plan Sub-Plan for Grantees Subject to Israeli Taxation (non-102(b) grants)# |
|
8-K |
|
12/22/15 |
|
10.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.21 |
|
Form of Incentive Stock Option Agreement pursuant to the 2015 Omnibus Incentive Plan for use in connection with the 2015 Omnibus Incentive Plan Sub-Plan for Grantees Subject to Israeli Taxation (102(b) grants)# |
|
8-K |
|
12/22/15 |
|
10.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.22 |
|
Form of Stock Option Award Agreement based on the 2015 Omnibus Incentive Plan for use in connection with the 2015 Omnibus Incentive Plan Sub-Plan for Switzerland# |
|
8-K |
|
12/22/15 |
|
10.4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.23 |
|
Form of Incentive Stock Option Agreement pursuant to the 2015 Omnibus Incentive Plan for use in Japan# |
|
8-K |
|
12/22/15 |
|
10.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.24 |
|
Amendment No. 2 to Consulting Services Agreement with William F. Doyle, dated February 24, 2016# |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.25 |
|
Form of Stock Option Award Agreement based on the 2015 Omnibus Incentive Plan for use in Germany# |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
21 |
|
Subsidiaries |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
Certification of Principal Executive Officer Required Under Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
Certification of Principal Financial Officer Required Under Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.1* |
|
Certification of Principal Executive Officer Required Under Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. §1350 |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.2* |
|
Certification of Principal Financial Officer Required Under Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. §1350 |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
XBRL Instance Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.SCH |
|
XBRL Taxonomy Extension Schema Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.CAL |
|
XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.DEF |
|
XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.LAB |
|
XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.PRE |
|
XBRL Extension Presentation Linkbase Document |
|
|
|
|
|
|
|
X |
|
|
* |
The certifications attached as Exhibits 32.1 and 32.2 that accompany this Annual Report on Form 10-K are not deemed filed with the Securities and Exchange Commission and are not to be incorporated by reference into any filing of NovoCure Limited under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Form 10-K, irrespective of any general incorporation language contained in such filing. |
|
† |
Confidential treatment has been granted for certain information set forth in this exhibit. Such information has been omitted and filed separately with the Securities and Exchange Commission. |
|
# |
Compensation plans and arrangements for executive officers and others. |
This Annual Report on Form 10-K includes trademarks of NovoCure Limited and other persons. All trademarks or trade names referred to herein are the property of their respective owners.
111
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: March 1, 2016
|
|
|
Novocure limited |
|
|
|
|
|
|
|
|
|
By: |
/s/ Asaf Danziger |
|
|
|
|
Asaf Danziger |
|
|
|
|
Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
|
Date: |
|
Signature |
|
Title |
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Asaf Danziger |
|
Chief Executive Officer and Director (Principal Executive Officer) |
|
|
|
Asaf Danziger |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Wilhelmus Groenhuysen |
|
Chief Financial Officer (Principal Financial and Accounting Officer) |
|
|
|
Wilhelmus Groenhuysen |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ William F. Doyle |
|
Chairman and Director |
|
|
|
William F. Doyle |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Kinyip Gabriel Leung |
|
Vice Chairman and Director |
|
|
|
Kinyip Gabriel Leung |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Yoram Palti, M.D., Ph.D. |
|
Director |
|
|
|
Yoram Palti, M.D., Ph.D. |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ William Burkoth |
|
Director |
|
|
|
William Burkoth |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Timothy Langloss |
|
Director |
|
|
|
Timothy Langloss |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Louis J. Lavigne, Jr. |
|
Director |
|
|
|
Louis J. Lavigne, Jr. |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Robert J. Mylod, Jr. |
|
Director |
|
|
|
Robert J. Mylod, Jr. |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Gert Lennart Perlhagen |
|
Director |
|
|
|
Gert Lennart Perlhagen |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ Charles G. Phillips III |
|
Director |
|
|
|
Charles G. Phillips III |
|
|
|
|
|
|
|
|
|
March 1, 2016 |
|
/s/ William A. Vernon |
|
Director |
|
|
|
William A. Vernon |
|
112