Attached files
| file | filename |
|---|---|
| EX-12 - STATEMENT RE: COMPUTATION OF RATIO OF EARNINGS TO FIXED CHARGES - ELI LILLY & Co | lly-20151231x10kexhibit12.htm |
| EX-32 - SECTION 1350 CERTIFICATION - ELI LILLY & Co | lly-20151231x10kexhibit32.htm |
| EX-21 - LIST OF SUBSIDIARIES AND AFFILIATES - ELI LILLY & Co | lly-20151231x10kexhibit21.htm |
| EX-23 - CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM - ELI LILLY & Co | lly-20151231x10kexhibit23.htm |
| EX-31.1 - RULE 13A-14(A) CERTIFICATION OF JOHN C. LECHLEITER, PH.D., CHAIRMAN OF THE BOARD - ELI LILLY & Co | lly-20151231x10kexhibit311.htm |
| EX-31.2 - RULE 13A-14(A) CERTIFICATION OF DERICA W. RICE, EXECUTIVE VICE PRESIDENT, GLOBAL - ELI LILLY & Co | lly-20151231x10kexhibit312.htm |
| EX-10.3 - ELI LILLY AND COMPANY SHAREHOLDER VALUE AWARD (FOR EXECUTIVE OFFICERS) - ELI LILLY & Co | lly-20151231x10kexhibit103.htm |
United States
Securities and Exchange Commission
Washington, D.C. 20549
Form 10-K
Annual report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
for the fiscal year ended December 31, 2015
Commission file number 001-06351
Eli Lilly and Company
An Indiana corporation | I.R.S. employer identification no. 35-0470950 | |
Lilly Corporate Center, Indianapolis, Indiana 46285 | ||
(317) 276-2000 | ||
Securities registered pursuant to Section 12(b) of the Exchange Act:
Title of Each Class | Name of Each Exchange On Which Registered | |
Common Stock (no par value) | New York Stock Exchange | |
7 1/8% Notes Due June 1, 2025 | New York Stock Exchange | |
6.77% Notes Due January 1, 2036 | New York Stock Exchange | |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 under the Securities Act. Yes þ No o
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act. Yes o No þ
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months, and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark whether the Registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the Registrant was required to submit and post such files). Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in the definitive proxy statement incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 under the Exchange Act. (Check one):
Large accelerated filer þ | Accelerated filer o | Non-accelerated filer o | Smaller reporting company o | |||
Indicate by check mark whether the Registrant is a shell company as defined in Rule 12b-2 under the Exchange Act: Yes o No þ
Aggregate market value of the common equity held by non-affiliates computed by reference to the price at which the common equity was last sold as of the last business day of the Registrant’s most recently completed second fiscal quarter (Common Stock): approximately $81,473,000,000
Number of shares of common stock outstanding as of February 12, 2016: 1,106,093,485
Portions of the Registrant’s Proxy Statement to be filed on or about March 21, 2016 have been incorporated by reference into Part III of this report.
1
Eli Lilly and Company
Form 10-K
For the Year Ended December 31, 2015
Table of Contents
Page | ||||
2
Forward-Looking Statements
This Annual Report on Form 10-K includes forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934 (Exchange Act). Forward-looking statements include all statements that do not relate solely to historical or current facts, and can generally be identified by the use of words such as “may,” “believe,” “will,” “expect,” “project,” “estimate,” “intend,” “anticipate,” “plan,” “continue,” or similar expressions.
In particular, information appearing under “Business,” “Risk Factors” and “Management's Discussion and Analysis of Financial Condition and Results of Operations” includes forward-looking statements. Forward-looking statements inherently involve many risks and uncertainties that could cause actual results to differ materially from those projected in these statements. Where, in any forward-looking statement, we express an expectation or belief as to future results or events, it is based on management's current plans and expectations, expressed in good faith and believed to have a reasonable basis. However, we can give no assurance that any such expectation or belief will result or will be achieved or accomplished. The following include some but not all of the factors that could cause actual results or events to differ materially from those anticipated:
• | the timing of anticipated regulatory approvals and launches of new products; |
• | market uptake of recently launched products; |
• | competitive developments affecting current products; |
• | the expiration of intellectual property protection for certain of our products; |
• | our ability to protect and enforce patents and other intellectual property; |
• | the impact of actions of governmental and private payers affecting pricing of, reimbursement for, and access to pharmaceuticals; |
• | regulatory compliance problems or government investigations; |
• | regulatory actions regarding currently marketed products; |
• | unexpected safety or efficacy concerns associated with our products; |
• | issues with product supply stemming from manufacturing difficulties or disruptions; |
• | regulatory changes or other developments; |
• | changes in patent law or regulations related to data-package exclusivity; |
• | litigation involving past, current or future products as we are largely self-insured; |
• | unauthorized disclosure or misappropriation of trade secrets or other confidential data stored in our information systems, networks, and facilities, or those of third parties with whom we share our data; |
• | changes in tax law; |
• | changes in foreign currency exchange rates, interest rates, and inflation; |
• | asset impairments and restructuring charges; |
• | changes in accounting standards promulgated by the Financial Accounting Standards Board and the Securities and Exchange Commission; |
• | acquisitions and business development transactions and related integration costs; |
• | information technology system inadequacies or operating failures; |
• | reliance on third-party relationships and outsourcing arrangements; and |
• | the impact of global macroeconomic conditions. |
Investors should not place undue reliance on forward-looking statements. You should carefully read the factors described in the “Risk Factors” section of this Annual Report on Form 10-K for a description of certain risks that could, among other things, cause our actual results to differ from these forward-looking statements.
All forward-looking statements speak only as of the date of this report and are expressly qualified in their entirety by the cautionary statements included in this report. Except as is required by law, we expressly disclaim any obligation to publicly release any revisions to forward-looking statements to reflect events after the date of this report.
3
Part I
Item 1. | Business |
Eli Lilly and Company (the “company” or “registrant” or "Lilly") was incorporated in 1901 in Indiana to succeed to the drug manufacturing business founded in Indianapolis, Indiana, in 1876 by Colonel Eli Lilly. We discover, develop, manufacture, and market products in two business segments—human pharmaceutical products and animal health products.
The mission of our human pharmaceutical business is to make medicines that help people live longer, healthier, more active lives. Our vision is to make a significant contribution to humanity by improving global health in the 21st century. Most of the products we sell today were discovered or developed by our own scientists, and our success depends to a great extent on our ability to continue to discover, develop, and bring to market innovative new medicines.
Our animal health business, operating through our Elanco division, develops, manufactures, and markets products for both food animals and companion animals. Elanco food animal products help the food industry produce an abundant supply of safe, nutritious and affordable food. Elanco companion animal products help pets live longer, healthier, happier lives.
We manufacture and distribute our products through facilities in the United States (U.S.), Puerto Rico, and 14 other countries. Our products are sold in approximately 125 countries.
Human Pharmaceutical Products
Our human pharmaceutical products include:
Endocrinology products, including:
• | Humalog®, Humalog Mix 75/25™, and Humalog Mix 50/50™, insulin analogs for the treatment of diabetes |
• | Humulin®, human insulin of recombinant DNA origin for the treatment of diabetes |
• | Trajenta®, for the treatment of type 2 diabetes |
• | Jentadueto®, a combination tablet of linagliptin (Trajenta) and metformin hydrochloride for use in the treatment of type 2 diabetes |
• | Jardiance®, for the treatment of type 2 diabetes (approved in the U.S., Europe, and Japan in 2014) |
• | Trulicity®, for the treatment of type 2 diabetes (approved in the U.S. and Europe in 2014 and Japan in 2015) |
• | Glyxambi®, a combination tablet of linagliptin and empagliflozin (Jardiance) for the treatment of type 2 diabetes (approved in the U.S. in 2015) |
• | Synjardy®, a combination tablet of empagliflozin and metformin hydrochloride for the treatment of type 2 diabetes (approved in the U.S. and Europe in 2015) |
• | Basaglar® (insulin glargine injection), a long-acting human insulin analog for the treatment of diabetes (launched in Japan in 2015 and in Europe in 2015 under the trade name Abasaglar®). Basaglar was also approved in the U.S. in 2015; under an agreement settling patent litigation with Sanofi-Aventis U.S. LLC (Sanofi) regarding Sanofi's insulin glargine product, we will have the ability to launch Basaglar in the U.S. on December 15, 2016. Under the terms of the agreement, Sanofi has granted us a royalty-bearing license so we can manufacture and sell Basaglar in the Kwikpen™ device globally. |
• | Forteo®, for the treatment of osteoporosis in postmenopausal women and men at high risk for fracture and for glucocorticoid-induced osteoporosis in men and postmenopausal women |
4
• | Evista®, for the prevention and treatment of osteoporosis in postmenopausal women and for the reduction of the risk of invasive breast cancer in postmenopausal women with osteoporosis and postmenopausal women at high risk for invasive breast cancer |
• | Humatrope®, for the treatment of human growth hormone deficiency and certain pediatric growth conditions |
• | Axiron®, a topical solution of testosterone, applied by underarm applicator, for replacement therapy in men for certain conditions associated with a deficiency or absence of testosterone |
Neuroscience products, including:
• | Cymbalta®, for the treatment of major depressive disorder, diabetic peripheral neuropathic pain, generalized anxiety disorder, fibromyalgia, and chronic musculoskeletal pain due to chronic low back pain or chronic pain due to osteoarthritis |
• | Zyprexa®, for the treatment of schizophrenia, acute mixed or manic episodes associated with bipolar I disorder, and bipolar maintenance |
• | Strattera®, for the treatment of attention-deficit hyperactivity disorder |
• | Prozac®, for the treatment of major depressive disorder, obsessive-compulsive disorder, bulimia nervosa, and panic disorder |
• | Amyvid®, a radioactive diagnostic agent for positron emission tomography imaging of beta-amyloid neuritic plaques in the brains of adult patients with cognitive impairment who are being evaluated for Alzheimer's disease and other causes of cognitive decline |
Oncology products, including:
• | Alimta®, for the first-line treatment, in combination with another agent, of advanced non-small cell lung cancer (NSCLC) for patients with non-squamous cell histology; for the second-line treatment of advanced non-squamous NSCLC; as monotherapy for the maintenance treatment of advanced non-squamous NSCLC in patients whose disease has not progressed immediately following chemotherapy treatment; and in combination with another agent, for the treatment of malignant pleural mesothelioma |
• | Erbitux®, indicated both as a single agent and with another chemotherapy agent for the treatment of certain types of colorectal cancers; and as a single agent, in combination with chemotherapy, or in combination with radiation therapy for the treatment of certain types of head and neck cancers |
• | Cyramza®, for the treatment of various cancers, with approvals as follows: |
◦ | approved in 2014 in the U.S. and the European Union (EU), and in Japan in 2015, both as a single agent and in combination with another agent as a second-line treatment of advanced or metastatic gastric cancer |
◦ | approved in 2014 in the U.S., and in the EU in 2016, in combination with another agent as a second-line treatment of metastatic NSCLC |
◦ | approved in 2015 in the U.S., and in the EU in 2016, as a second-line treatment of metastatic colorectal cancer |
• | Gemzar®, for the treatment of pancreatic cancer; in combination with other agents, for the treatment of metastatic breast cancer, NSCLC, and advanced or recurrent ovarian cancer; and in the EU for the treatment of bladder cancer |
• | Portrazza™, approved in 2015 in the U.S. for use in combination with other agents as a first-line treatment of metastatic squamous NSCLC, and approved in 2016 in the EU for use in combination with other agents as a first-line treatment for epidermal growth factor receptor expressing squamous NSCLC |
Cardiovascular products, including:
• | Cialis®, for the treatment of erectile dysfunction and benign prostatic hyperplasia |
5
• | Effient®, for the reduction of thrombotic cardiovascular events (including stent thrombosis) in patients with acute coronary syndrome who are managed with an artery-opening procedure known as percutaneous coronary intervention (PCI), including patients undergoing angioplasty, atherectomy, or stent placement |
• | ReoPro®, for use as an adjunct to PCI for the prevention of cardiac ischemic complications |
Animal Health Products
Our products for food animals include:
• | Rumensin®, a cattle feed additive that improves feed efficiency and growth and also controls and prevents coccidiosis |
• | Posilac®, a protein supplement to improve milk productivity in dairy cows |
• | Paylean® and Optaflexx®, leanness and performance enhancers for swine and cattle, respectively |
• | Tylan®, an antibiotic used to control certain diseases in cattle, swine, and poultry |
• | Micotil®, Pulmotil®, and Pulmotil AC®, antibiotics used to treat respiratory disease in cattle, swine, and poultry, respectively |
• | Coban®, Monteban®, and Maxiban®, anticoccidial agents for use in poultry |
• | Surmax® (sold as Maxus® in some countries), a performance enhancer for swine and poultry |
• | Imrestor™, a biopharmaceutical that restores neutrophil function in peri-parturient dairy cows |
Our products for companion animals include:
• | Trifexis®, a monthly chewable tablet for dogs that kills fleas, prevents flea infestations, prevents heartworm disease, and controls intestinal parasite infections |
• | Comfortis®, a chewable tablet that kills fleas and prevents flea infestations on dogs |
• | Onsior®, a non-steroidal short-term pain reliever for cats administered orally or by injection |
• | Interceptor Plus®, a canine heartworm drug that fights tapeworms in addition to hookworms, roundworms, and whipworms |
• | Osurnia®, a gel formulation treatment for canine ear canal infection or inflammation |
6
On January 1, 2015, we completed our acquisition of Novartis Animal Health (Novartis AH) in an all-cash transaction for $5.28 billion. Novartis AH operates in approximately 40 countries. The combined organization has added several hundred products to our animal health product portfolio, expanded our global commercial presence, and augmented our animal health manufacturing and research and development. In particular, it has provided Elanco with a greater commercial presence in the companion animal and swine markets, expanded Elanco’s presence in equine and vaccines areas, and created an entry into the aquaculture market. Acquired Novartis AH products include:
• | Denagard®, an antibiotic for the control and treatment of respiratory and enteric diseases in swine and poultry |
• | Milbemax™, a broad-spectrum intestinal wormer which, if given monthly, also offers prevention against heartworm |
• | Sentinel® (outside the U.S.), a monthly tablet for the prevention of flea populations, the concurrent prevention of heartworm disease and the treatment of roundworms, hookworms, and whipworms in dogs |
• | Atopica®, for the treatment of chronic manifestations of atopic dermatitis in dogs and for the symptomatic treatment of chronic allergic dermatitis in cats |
• | Fortekor®, for the treatment of congestive heart failure in dogs and reduction of proteinurea associated with chronic kidney disease in cats |
Marketing
We sell most of our products worldwide. We adapt our marketing methods and product emphasis in various countries to meet local customer needs.
Human Pharmaceuticals—United States
In the U.S., we distribute human pharmaceutical products principally through independent wholesale distributors, with some sales directly to pharmacies. In 2015, 2014, and 2013, three wholesale distributors in the U.S.—AmerisourceBergen Corporation, McKesson Corporation, and Cardinal Health, Inc.—each accounted for between 8 percent and 19 percent of our consolidated total revenue. No other distributor accounted for more than 10 percent of consolidated total revenue in any of those years.
We promote our major human pharmaceutical products in the U.S. through sales representatives who call upon physicians and other health care professionals. We advertise in medical journals, distribute literature and samples of certain products to physicians, and exhibit at medical meetings. In addition, we advertise certain products directly to consumers in the U.S., and we maintain websites with information about our major products. We supplement our employee sales force with contract sales organizations as appropriate to leverage our own resources and the strengths of our partners in various markets.
We maintain special business groups to service wholesalers, pharmacy benefit managers, managed care organizations, government and long-term care institutions, hospitals, and certain retail pharmacies. We enter into arrangements with these organizations providing for discounts or rebates on our products.
Human Pharmaceuticals—Outside the United States
Outside the U.S, we promote our human pharmaceutical products primarily through sales representatives. While the products marketed vary from country to country, endocrinology products constitute the largest single group in total revenue. Distribution patterns vary from country to country. In most countries in which we operate, we maintain our own sales organizations, but in some smaller countries we market our products through independent distributors.
Human Pharmaceutical Marketing Collaborations
Certain of our human pharmaceutical products are marketed in arrangements with other pharmaceutical companies, including the following:
• | We and Boehringer Ingelheim have a diabetes alliance under which we jointly develop and commercialize Trajenta, Jentadueto, Jardiance, Glyxambi, Synjardy, and Basaglar in major markets. |
7
• | We co-promote Cymbalta in Japan with Shionogi & Co. Ltd. |
• | Through September 30, 2015, Erbitux was marketed in the U.S. and Canada by Bristol-Myers Squibb (BMS). Effective October 1, 2015, BMS transferred to us all commercialization rights for Erbitux in those two countries. Outside the U.S. and Canada, Erbitux is commercialized by Merck KGaA, and we receive royalties from Merck KGaA. |
• | Effient is co-promoted with us by Daiichi Sankyo Co., Ltd. (Daiichi Sankyo) in the U.S., Brazil, Mexico, and certain other countries. Through the end of 2015, we also co-promoted Effient with Daiichi Sankyo in major European markets. Effective January 2016, Daiichi Sankyo is exclusively promoting Effient in major European markets; however, the economic results for these countries will continue to be shared in the same proportion as under the previous arrangement. We retain sole marketing rights in Canada, Australia, Russia, and certain other countries. Daiichi Sankyo retains sole marketing rights in Japan and certain other countries. |
For additional information, see Item 8, "Financial Statements and Supplementary Data—Note 4, Collaborations and Other Arrangements."
Animal Health Products
Our Elanco animal health business unit employs field salespeople throughout the U.S. and has an extensive sales force outside the U.S. Elanco sells its products primarily to wholesale distributors. Elanco promotes its products primarily to producers and veterinarians for food animal products and to veterinarians for companion animal products. Elanco also advertises certain companion animal products directly to pet owners in markets where it is consistent with allowable promotional practices.
Competition
Our human pharmaceutical products compete globally with products of many other companies in highly competitive markets. Our animal health products compete globally with products of animal health care companies as well as pharmaceutical, chemical, and other companies that operate animal health businesses.
Important competitive factors for both human pharmaceutical and animal health products include effectiveness, safety, and ease of use; price and demonstrated cost-effectiveness; marketing effectiveness; and research and development of new products, processes, and uses. Most new products that we introduce must compete with other branded or generic products already on the market or products that are later developed by competitors. If competitors introduce new products or delivery systems with therapeutic or cost advantages, our products can be subject to decreased sales, progressive price reductions, or both.
We believe our long-term competitive success depends upon discovering and developing (either alone or in collaboration with others) or acquiring innovative, cost-effective human pharmaceutical and animal health products that provide improved outcomes and deliver value to payers, and continuously improving the productivity of our operations in a highly competitive environment. There can be no assurance that our research and development efforts will result in commercially successful products, and it is possible that our products will become uncompetitive from time to time as a result of products developed by our competitors.
Generic Pharmaceuticals
One of the biggest competitive challenges we face is from generic pharmaceuticals. In the U.S. and the EU, the regulatory approval process for human pharmaceuticals (other than biological products (biologics)) exempts generics from costly and time-consuming clinical trials to demonstrate their safety and efficacy, allowing generic manufacturers to rely on the safety and efficacy of the innovator product. Therefore, generic manufacturers generally invest far less than we do in research and development and can price their products much lower than our branded products. Accordingly, when a branded non-biologic human pharmaceutical loses its market exclusivity, it normally faces intense price competition from generic forms of the product. Public and private payers typically encourage the use of generics as alternatives to brand-name drugs in their healthcare programs. Laws in the U.S. generally allow, and in many cases require, pharmacists to substitute generic drugs that have been rated under government procedures to be essentially equivalent to a brand-name drug. Where substitution is mandatory, it must be made unless the prescribing physician expressly forbids it. In many countries outside the U.S., intellectual property protection is weak, and we must compete
8
with generic or counterfeit versions of our products. Many of our animal health products also compete with generics.
Biosimilars
Several of our current products, including Cyramza, Erbitux, Trulicity, and Portrazza, and many of the new molecular entities (NMEs) in our research pipeline are biologics. Competition for Lilly’s biologics may be affected by the approval of follow-on biologics, also known as biosimilars. A biosimilar is a subsequent version of an an approved innovator biologic that, due to its physical/structural similarity to the original product, is approved based on an abbreviated data package that relies in part on the full testing required of the originator product. Globally, governments have or are developing regulatory pathways to approve biosimilars as alternatives to innovator-developed biologics, but the patent for the existing, branded product must expire in a given market before biosimilars may enter that market. The extent to which a biosimilar, once approved, will be substituted for the innovator biologic in a way that is similar to traditional generic substitution for non-biologic products, is not yet entirely clear, and will depend on a number of regulatory and marketplace factors that are still developing.
Biosimilars may present both competitive challenges and opportunities. For example, with our partner Boehringer Ingelheim we have developed Basaglar, a new insulin glargine product which has the same amino acid sequence as the product currently marketed by a competitor. Our product has launched in the EU and Japan, and can be launched in the U.S. on December 15, 2016.
U.S. Private Sector Payer Consolidation
In the U.S. private sector, consolidation and integration among healthcare providers is also a major factor in the competitive marketplace for human pharmaceuticals. Health plans and pharmaceutical benefit managers have been consolidating into fewer, larger entities, thus enhancing their purchasing strength and importance.
Payers typically maintain formularies which specify coverage (the conditions under which drugs are included on a plan's formulary) and reimbursement (the associated out-of-pocket cost to the consumer). Formulary placement can lead to reduced usage of a drug for the relevant patient population due to coverage restrictions, such as prior authorizations and formulary exclusions, or due to reimbursement limitations which result in higher consumer out-of-pocket cost, such as non-preferred co-pay tiers, increased co-insurance levels, and higher deductibles. Consequently, pharmaceutical companies compete for formulary placement not only on the basis of product attributes such as efficacy, safety profile, or patient ease of use, but also by providing rebates. Price is an increasingly important factor in formulary decisions, particularly in treatment areas in which the payer has taken the position that multiple branded products are therapeutically comparable. These downward pricing pressures could negatively affect our future consolidated results of operations.
Patents, Trademarks, and Other Intellectual Property Rights
Overview
Intellectual property protection is critical to our ability to successfully commercialize our life sciences innovations and invest in the search for new medicines. We own, have applied for, or are licensed under, a large number of patents in the U.S. and many other countries relating to products, product uses, formulations, and manufacturing processes. In addition, as discussed below, for some products we have additional effective intellectual property protection in the form of data protection under pharmaceutical regulatory laws.
The patent protection anticipated to be of most relevance to human pharmaceuticals is provided by national patents claiming the active ingredient (the compound patent), particularly those in major markets such as the U.S., various European countries, and Japan. These patents may be issued based upon the filing of international patent applications, usually filed under the Patent Cooperation Treaty (PCT). Patent applications covering the compounds are generally filed during the Discovery Research Phase of the drug discovery process, which is described in the “Research and Development” section below. In general, national patents in each relevant country are available for a period of 20 years from the filing date of the PCT application, which is often years prior to the launch of a commercial product. Further patent term adjustments and restorations may extend the original patent term:
9
• | Patent term adjustment is a statutory right available to all U.S. patent applicants to provide relief in the event that a patent is delayed during examination by the United States Patent and Trademark Office (USPTO). |
• | Patent term restoration is a statutory right provided to U.S. patents that claim inventions subject to review by the U.S. Food and Drug Administration (FDA). A single patent for a human pharmaceutical product may be eligible for patent term restoration to make up for a portion of the time invested in clinical trials and the FDA review process. Patent term restoration is limited by a formula and cannot be calculated until product approval due to uncertainty about the duration of clinical trials and the time it takes the FDA to review an application. There is a five-year cap on any restoration, and no patent may be extended for more than 14 years beyond FDA approval. Some countries outside the U.S. also offer forms of patent term restoration. For example, Supplementary Protection Certificates are sometimes available to extend the life of a European patent up to an additional five years. Similarly, in Japan, Korea, and Australia, patent terms can be extended up to five years, depending on the length of regulatory review and other factors. |
Loss of effective patent protection for human pharmaceuticals typically results in the loss of effective market exclusivity for the product, which can result in severe and rapid decline in sales of the product. However, in some cases the innovator company may be protected from approval of generic or other follow-on versions of a new medicine beyond the expiration of the compound patent through manufacturing trade secrets, later-expiring patents on methods of use or formulations, or data protection that may be available under pharmaceutical regulatory laws. The primary forms of data protection are as follows:
• | Regulatory authorities in major markets generally grant data package protection for a period of years following new drug approvals in recognition of the substantial investment required to complete clinical trials. Data package protection prohibits other manufacturers from submitting regulatory applications for marketing approval based on the innovator company’s regulatory submission data for the drug. The base period of data package protection depends on the country. For example, the period is five years in the U.S. (12 years for new biologics as described below), 10 years in the EU, and eight years in Japan. The period begins on the date of product approval and runs concurrently with the patent term for any relevant patent. |
• | Under the Biologics Price Competition and Innovation Act of 2010, the FDA has the authority to approve biosimilars. A competitor seeking approval of a biosimilar must file an application to show its molecule is highly similar to an approved innovator biologic and include a certain amount of safety and efficacy data which the FDA will determine on a case-by-case basis. Under the data protection provisions of this law, the FDA cannot approve a biosimilar application until 12 years after initial marketing approval of the innovator biologic, subject to certain conditions. |
• | In the U.S., the FDA has the authority to grant additional data protection for approved drugs where the sponsor conducts specified testing in pediatric or adolescent populations. If granted, this “pediatric exclusivity” provides an additional six months, which are added to the term of data protection as well as to the term of any relevant patents, to the extent these protections have not already expired. |
• | Under the U.S. orphan drug law, a specific use of a drug or biological product can receive "orphan" designation if it is intended to treat a disease or condition affecting fewer than 200,000 people in the U.S., or affecting more than 200,000 people but not reasonably expected to recover its development and marketing costs through U.S. sales. Among other benefits, orphan designation entitles the particular use of the drug to seven years of market exclusivity, meaning that the FDA cannot (with limited exceptions) approve another marketing application for the same drug for the same indication until expiration of the seven-year period. Unlike pediatric exclusivity, the orphan exclusivity period is independent of and runs in parallel with any applicable patents. |
Outside the major markets, the adequacy and effectiveness of intellectual property protection for human pharmaceuticals varies widely. Under the Trade-Related Aspects of Intellectual Property Agreement (TRIPs) administered by the World Trade Organization, more than 140 countries have agreed to provide non-discriminatory protection for most pharmaceutical inventions and to assure that adequate and effective rights are available to patent owners. Implementation of this agreement differs between developed and developing
10
countries, with many developing countries limiting protection for biopharmaceutical products under their interpretation of “flexibilities” allowed under the agreement. Thus, certain types of patents, such as those on new uses of compounds or new forms of molecules, are not available in many developing countries. Further, many developing countries do not provide effective data package protection even though it is specified in TRIPs.
Certain of our Elanco animal health products are covered by patents or other forms of intellectual property protection. Historically, upon loss of effective market exclusivity for our animal health products, we have not generally experienced the rapid and severe declines in revenues that are common in the human pharmaceutical segment.
There is no assurance that the patents we are seeking will be granted or that the patents we hold will be found valid and enforceable if challenged. Moreover, patents relating to particular products, uses, formulations, or processes do not preclude other manufacturers from employing alternative processes or marketing alternative products or formulations that compete with our patented products. In addition, competitors or other third parties may assert claims that our activities infringe patents or other intellectual property rights held by them, or allege a third-party right of ownership in our existing intellectual property.
Our Intellectual Property Portfolio
We consider intellectual property protection for certain products, processes, uses, and formulations—particularly with respect to those products discussed below—to be important to our operations. For many of our products, in addition to the compound patent, we hold other patents on manufacturing processes, formulations, or uses that may extend exclusivity beyond the expiration of the compound patent.
The most relevant U.S. patent protection or data protection for our top-selling or recently launched patent-protected marketed products is as follows:
• | Alimta is protected by a compound patent (July 2016) plus pediatric exclusivity (January 2017), and a vitamin regimen patent (2021) plus pediatric exclusivity (2022). |
• | Cialis is protected by compound and use patents (November 2017). |
• | Cyramza is protected by biologics data package protection (2026). |
• | Effient is protected by a compound patent (April 2017) and patents covering methods of using Effient with aspirin (2023). |
• | Forteo is protected by patents primarily covering its formulation and related processes (2018) and use patents (2019). |
• | Jardiance, and the related combination products Glyxambi and Synjardy, are protected by a compound patent (2025 not including possible patent extension). |
• | Portrazza is protected by a compound patent (2025 not including possible patent extension), and by biologics data package protection (2027). |
• | Strattera is protected by a patent covering its use in treating attention deficit-hyperactivity disorder (2016) plus pediatric exclusivity (May 2017). |
• | Trajenta and Jentadueto are protected by a compound patent (2023), and Boehringer Ingelheim has applied for a patent extension to 2025 under the patent restoration laws. |
• | Trulicity is protected by a compound patent (2024 not including possible patent extension) and by biologics data package protection (2026). |
11
Outside the U.S., important patent protection or data protection includes:
• | Alimta in major European countries (compound patent December 2015, vitamin regimen patent 2021) and Japan (compound patent December 2015, patents covering use to treat cancer concomitantly with vitamins 2021) |
• | Cialis in major European countries (compound patent November 2017) |
• | Cymbalta in Japan (data package protection 2018). In major European countries, our Cymbalta data package protection expired in 2014, and we experienced the entry of generic competitors in 2015 in these markets. |
• | Forteo in Japan (data package protection 2018; patent covering its formulation and related process 2019). |
• | Zyprexa in Japan (patent for schizophrenia expired December 2015; patent for bipolar mania will expire April 2016). |
U.S. patent protection or data protection for our new molecular entities that have been submitted for regulatory review is as follows (additional information about these molecules is provided in Item 7, "Management’s Discussion and Analysis—Late-Stage Pipeline”):
• | Ixekizumab - compound patent 2026 (not including possible patent extension); biologics data package protection for 12 years after approval |
• | Baricitinib - compound patent 2030 (not including possible patent extension) |
Worldwide, we sell all of our major products under trademarks that we consider in the aggregate to be important to our operations. Trademark protection varies throughout the world, with protection continuing in some countries as long as the mark is used, and in other countries as long as it is registered. Registrations are normally for fixed but renewable terms.
Patent Licenses
Most of our major products were discovered in our own laboratories and are not subject to significant license agreements. Two of our largest products, Cialis and Alimta, are subject to patent assignments or licenses granted to us by others.
• | The compound patent for Cialis is the subject of a license agreement with GlaxoSmithKline (Glaxo), which assigns to us exclusively all rights in the compound. The agreement calls for royalties of a single-digit percentage of net sales. The agreement is not subject to termination by Glaxo for any reason other than a material breach by Lilly of the royalty obligation, after a substantial cure period. |
• | The compound patent for Alimta is the subject of a license agreement with Princeton University, granting us an irrevocable exclusive worldwide license to the compound patents for the lives of the patents in the respective territories. The agreement calls for royalties of a single-digit percentage of net sales. The agreement is not subject to termination by Princeton for any reason other than a material breach by Lilly of the royalty obligation, after a substantial cure period. Alimta is also the subject of a worldwide, nonexclusive license to certain patents owned by Takeda Pharmaceutical Company Limited. The agreement calls for royalties of a single-digit percentage of net sales in countries covered by a relevant patent. The agreement is subject to termination for material default and failure to cure by Lilly and in the event that Lilly becomes bankrupt or insolvent. |
Patent Challenges
In the U.S., the Drug Price Competition and Patent Term Restoration Act of 1984, commonly known as the Hatch-Waxman Act, made a complex set of changes to both patent and new-drug-approval laws for human pharmaceuticals. Before the Hatch-Waxman Act, no drug could be approved without providing the FDA complete safety and efficacy studies, i.e., a complete New Drug Application (NDA). The Hatch-Waxman Act authorizes the FDA to approve generic versions of innovative human pharmaceuticals (other than biologics) without such information by filing an Abbreviated New Drug Application (ANDA). In an ANDA, the generic manufacturer must demonstrate only “bioequivalence” between the generic version and the NDA-approved drug—not safety and efficacy.
12
Absent a patent challenge, the FDA cannot approve an ANDA until after the innovator’s patents expire. However, after the innovator has marketed its product for four years, a generic manufacturer may file an ANDA alleging that one or more of the patents listed in the innovator’s NDA are invalid or not infringed. This allegation is commonly known as a “Paragraph IV certification.” The innovator must then file suit against the generic manufacturer to protect its patents. The FDA is then prohibited from approving the generic company’s application for a 30- to 42-month period (which can be shortened or extended by the trial court judge hearing the patent challenge). If one or more of the NDA-listed patents are challenged, the first filer(s) of a Paragraph IV certification may be entitled to a 180-day period of market exclusivity over all other generic manufacturers.
Generic manufacturers use Paragraph IV certifications extensively to challenge patents on innovative human pharmaceuticals. In addition, generic companies have shown an increasing willingness to launch “at risk,” i.e., after receiving ANDA approval but before final resolution of their patent challenge. We are currently in litigation with numerous generic manufacturers in Hatch-Waxman litigation involving Alimta and Effient, among other products. For more information on Hatch-Waxman litigation involving the company, see Item 8, “Financial Statements and Supplementary Data—Note 15, Contingencies” and Item 3, "Legal Proceedings."
The passage of the America Invents Act in 2011 added a new procedure to U.S. patent law. This procedure, inter partes review (IPR), allows any member of the public to file a petition with the USPTO seeking the review of any issued U.S. patent. IPRs are conducted before Administrative Patent Judges in the USPTO using a lower standard of proof than used in Federal District Court. In addition, the challenged patents are not accorded the presumption of validity as they are in Federal District Court. We are now seeing instances where generic drug companies and some investment funds are attempting to invalidate our patents by filing IPR challenges in the USPTO. For more information, see Item 8, “Financial Statements and Supplementary Data—Note 15, Contingencies.”
Outside the U.S., the legal doctrines and processes by which pharmaceutical patents can be challenged vary widely. In recent years, we have experienced an increase in patent challenges from generic manufacturers in many countries outside the U.S., and we expect this trend to continue. For more information on administrative challenges and litigation involving our Alimta patents in Europe and Japan, see Item 8, “Financial Statements and Supplementary Data—Note 15, Contingencies.”
Government Regulation
Regulation of Our Operations
Our operations are regulated extensively by numerous national, state, and local agencies. The lengthy process of laboratory and clinical testing, data analysis, manufacturing development, and regulatory review necessary for governmental approvals is extremely costly and can significantly delay product introductions. Promotion, marketing, manufacturing, and distribution of human pharmaceutical and animal health products are extensively regulated in all major world markets. We conduct extensive post-marketing surveillance of the safety of the products we sell. In addition, our operations are subject to complex federal, state, local, and foreign laws and regulations concerning the environment, occupational health and safety, and privacy. Animal health product regulations address the administration of the product in or on the animal, and in the case of food animal products, the impact on humans who consume the food as well as the impact on the environment at the production site. The laws and regulations affecting the manufacture and sale of current products and the discovery, development, and introduction of new products will continue to require substantial effort, expense, and capital investment.
Of particular importance is the FDA in the U.S. Pursuant to the Federal Food, Drug, and Cosmetic Act, the FDA has jurisdiction over all of our human pharmaceutical products and certain animal health products in the U.S. and administers requirements covering the testing, safety, effectiveness, manufacturing, quality control, distribution, labeling, marketing, advertising, dissemination of information, and post-marketing surveillance of those products. The U.S. Department of Agriculture and the U.S. Environmental Protection Agency also regulate some animal health products.
The FDA extensively regulates all aspects of manufacturing quality for human pharmaceuticals under its current Good Manufacturing Practices (cGMP) regulations. Outside the U.S., our products and operations are subject to similar regulatory requirements, notably by the European Medicines Agency in the EU and the Ministry of Health, Labor and Welfare in Japan. Specific regulatory requirements vary from country to country. We make substantial investments of capital and operating expenses to implement comprehensive, company-
13
wide quality systems in our manufacturing, product development, and process development operations to ensure sustained compliance with cGMP and similar regulations. However, in the event we fail to adhere to these requirements in the future, we could be subject to interruptions in production, fines and penalties, and delays in new product approvals. Certain of our products are manufactured by third parties, and their failure to comply with these regulations could adversely affect us through failure to supply product to us or delays in new product approvals.
The marketing, promotional, and pricing practices of human pharmaceutical manufacturers, as well as the manner in which manufacturers interact with purchasers and prescribers, are subject to various other U.S. federal and state laws, including the federal anti-kickback statute and the False Claims Act and state laws governing kickbacks, false claims, unfair trade practices, and consumer protection. These laws are administered by, among others, the Department of Justice (DOJ), the Office of Inspector General of the Department of Health and Human Services, the Federal Trade Commission, the Office of Personnel Management, and state attorneys general. Over the past several years, the FDA, the DOJ, and many of these other agencies have increased their enforcement activities with respect to pharmaceutical companies and increased the inter-agency coordination of enforcement activities. Several claims brought by these agencies against Lilly and other companies under these and other laws have resulted in corporate criminal sanctions and very substantial civil settlements.
The U.S. Foreign Corrupt Practices Act of 1977 (FCPA) prohibits certain individuals and entities, including U.S. publicly traded companies, from promising, offering, or giving anything of value to foreign officials with the corrupt intent of influencing the foreign official for the purpose of helping the company obtain or retain business or gain any improper advantage. The FCPA also imposes specific recordkeeping and internal controls requirements on U.S. publicly traded companies. As noted above, outside the U.S., our business is heavily regulated and therefore involves significant interaction with foreign officials. Additionally, in many countries outside the U.S., the health care providers who prescribe human pharmaceuticals are employed by the government and the purchasers of human pharmaceuticals are government entities; therefore, our interactions with these prescribers and purchasers are subject to regulation under the FCPA.
In addition to the U.S. application and enforcement of the FCPA, the various jurisdictions in which we operate and supply our products have laws and regulations aimed at preventing and penalizing corrupt and anticompetitive behavior. In recent years, several jurisdictions, including China, Brazil, and the United Kingdom (U.K.), have enhanced their laws and regulations in this area, increased their enforcement activities, and/or increased the level of cross-border coordination and information sharing.
It is possible that we could become subject to additional administrative and legal proceedings and actions, which could include claims for civil penalties (including treble damages under the False Claims Act), criminal sanctions, and administrative remedies, including exclusion from U.S. federal and other health care programs. It is possible that an adverse outcome in future actions could have a material adverse impact on our consolidated results of operations, liquidity, and financial position.
Regulations Affecting Human Pharmaceutical Pricing, Reimbursement, and Access
In the U.S., we are required to provide rebates to the federal government and respective state governments on their purchases of our human pharmaceuticals under state Medicaid and Medicaid Managed Care programs (minimum of 23.1 percent plus adjustments for price increases over time) and rebates to private payers who cover patients in certain types of health care facilities that serve low-income and uninsured patients (known as 340B facilities). No rebates are required at this time in the Medicare Part B (physician and hospital outpatient) program where reimbursement is set on an "average selling price plus 4.3 percent" formula. Drug manufacturers are required to provide a discount of 50 percent of the cost of branded prescription drugs for Medicare Part D participants who are in the “doughnut hole” (the coverage gap in Medicare prescription drug coverage). Additionally, an annual fee is imposed on pharmaceutical manufacturers and importers that sell branded prescription drugs to specified government programs.
Rebates are also negotiated in the private sector. We give rebates to private payers who provide prescription drug benefits to seniors covered by Medicare and to private payers who provide prescription drug benefits to their customers. These rebates are affected by the introduction of competitive products and generics in the same class.
14
In most international markets, we operate in an environment of government-mandated cost-containment programs, which may include price controls, international reference pricing (to other countries’ prices), discounts and rebates, therapeutic reference pricing (to other, often generic, pharmaceutical choices), restrictions on physician prescription levels, and mandatory generic substitution.
Globally, public and private payers are increasingly restricting access to human pharmaceuticals based on assessments of comparative effectiveness and value, including through the establishment of formal health technology assessment processes. In addition, third party organizations, including professional associations, academic institutions, and non-profit entities associated with payers, are conducting and publishing comparative effectiveness and cost/benefit analyses on medicines, the impact of which are uncertain at this time.
We cannot predict the extent to which our business may be affected by these or other potential future legislative or regulatory developments. However, in general we expect that state, federal, and international legislative and regulatory developments could have further negative effects on pricing and reimbursement for our human pharmaceutical products.
Research and Development
Our commitment to research and development dates back more than 100 years. We invest heavily in research and development because we believe it is critical to our long-term competitiveness. At the end of 2015, we employed approximately 8,730 people in human pharmaceutical and animal health research and development activities, including a substantial number of physicians, scientists holding graduate or postgraduate degrees, and highly skilled technical personnel. Our research and development expenses were $4.80 billion in 2015, $4.73 billion in 2014, and $5.53 billion in 2013.
Our internal human pharmaceutical research focuses primarily on the areas of cancer, diabetes, neurodegeneration, immunology, and pain. We have a strong biotechnology research program, with more than half of our clinical-stage pipeline currently consisting of biologics. In addition to discovering and developing NMEs, we seek to expand the value of existing products through new uses, formulations, and therapeutic approaches that provide additional value to patients. Across all our therapeutic areas, we are increasingly focusing our efforts on tailored therapeutics, seeking to identify and use advanced diagnostic tools and other information to identify specific subgroups of patients for whom our medicines—or potentially those of other companies—will be the best treatment option.
To supplement our internal efforts, we collaborate with others, including academic institutions and research-based pharmaceutical and biotechnology companies. We use the services of physicians, hospitals, medical schools, and other research organizations worldwide to conduct clinical trials to establish the safety and effectiveness of our human pharmaceutical products. We actively seek out external investments in research and technologies that hold the promise to complement and strengthen our own efforts. These investments can take many forms, including licensing arrangements, co-development and co-marketing agreements, co-promotion arrangements, joint ventures, and acquisitions.
Our Elanco animal health innovation strategy is focused on identifying and developing promising technologies and potential products from internal and external sources to meet unmet veterinary needs. Our animal health scientists also leverage discoveries from our human health laboratories to develop products to enhance the health and wellbeing of farm animals and pets.
15
Human pharmaceutical development is time-consuming, expensive, and risky. On average, only one out of many thousands of molecules discovered by researchers ultimately becomes an approved medicine. The process from discovery to regulatory approval can take over a decade. Drug candidates can fail at any stage of the process, and even late-stage drug candidates sometimes fail to receive regulatory approval or achieve commercial success. After approval and launch of a product, we expend considerable resources on post-marketing surveillance and additional clinical studies to collect data and understand the benefits and potential risks of medicines as they are used as therapeutics. The following describes in more detail the research and development process for human pharmaceutical products:
Phases of New Drug Development
• | Discovery Research Phase |
The earliest phase of new drug research and development, the discovery phase, can take many years. Scientists identify, design, and synthesize promising molecules, screening tens of thousands of molecules for their effect on biological “targets” that appear to play an important role in one or more diseases. Targets can be part of the body, such as a protein, receptor, or gene; or foreign, such as a virus or bacteria. Some targets have been proven to affect disease processes, but often the target is unproven and may later prove to be irrelevant to the disease or to yield insufficient clinical benefit. Molecules that have the desired effect on the target and meet other design criteria become “candidate” molecules and move to the next phase of development. The probability of any one candidate molecule becoming a commercial product is extremely low.
• | Early Development Phase |
The early development phase involves refining candidate molecules, understanding how to manufacture them efficiently, and completing initial testing for safety and efficacy. Safety testing is done first in laboratory tests and animals as necessary, to identify toxicity and other potential safety issues that would preclude use in humans. In general, the first human tests (often referred to as Phase I) are conducted in small groups of healthy volunteers to assess safety and find the potential dosing range. After a safe dose has been established, the drug is typically administered to small populations of patients (Phase II) to look for initial signs of efficacy in treating the targeted disease, or biomarkers of the disease, and to continue to assess safety. In parallel, scientists work to identify safe, effective, and economical manufacturing processes. Long-term animal studies continue to test for potential safety issues. Of the molecules that enter the early development phase, approximately 10 percent move on to the product phase. The early development phase can take several years to complete.
• | Product Phase |
Product phase (Phase III) molecules have already demonstrated safety and, typically, shown initial evidence of efficacy. As a result, these molecules generally have a higher likelihood of success. The molecules are tested in much larger patient populations to demonstrate efficacy to a predetermined level of statistical significance and to continue to develop the safety profile. These trials are generally global in nature and are designed to generate the data necessary to submit the molecule to regulatory agencies for marketing approval. The potential new drug is generally compared with existing competitive therapies, placebo, or both. The resulting data is compiled and may be submitted to regulatory agencies around the world. Phase III testing varies by disease state, but can often last from three to four years.
• | Submission Phase |
Once a molecule is submitted to regulatory agencies, the time to final marketing approval can vary from several months to several years, depending on variables such as the disease state, the strength and complexity of the data presented, the novelty of the target or compound, and the time required for the agency(ies) to evaluate the submission. There is no guarantee that a potential medicine will receive marketing approval, or that decisions on marketing approvals or indications will be consistent across geographic areas.
16
We believe our investments in research, both internally and in collaboration with others, have been rewarded by the large number of new molecules and new indications for existing molecules that we have in all stages of development. We currently have approximately 50 drug candidates across all stages of human testing and a larger number of projects in preclinical development. Among our new investigational molecules currently in the product phase of development or awaiting regulatory approval or launch are potential therapies for various cancers, Alzheimer’s disease, pain, migraines, rheumatoid arthritis, psoriasis, psoriatic arthritis, and severe hypoglycemia. We are studying many other drug candidates in the earlier stages of development in our chosen priority areas. We are also developing new uses, formulations, or delivery methods for many of these molecules as well as several currently marketed products. See Item 7, "Management's Discussion and Analysis—Late-Stage Pipeline," for more information on certain of our product candidates.
Raw Materials and Product Supply
Most of the principal materials we use in our manufacturing operations are available from more than one source. However, we obtain certain raw materials principally from only one source. In the event one of these suppliers was unable to provide the materials or product, we generally seek to maintain sufficient inventory to supply the market until an alternative source of supply can be implemented. However, in the event of an extended failure of a supplier, it is possible that we could experience an interruption in supply until we established new sources or, in some cases, implemented alternative processes.
The majority of our revenue comes from products produced in our own facilities. Our principal active ingredient manufacturing occurs at four owned sites in the U.S. as well as owned sites in Ireland, Puerto Rico, and the U.K. Finishing operations, including formulation, filling, assembling, delivery device manufacturing, and packaging, take place at a number of sites throughout the world. We utilize third parties for certain active ingredient manufacturing and finishing operations.
We manage our supply chain (including our own facilities, contracted arrangements, and inventory) in a way that should allow us to meet all expected product demand while maintaining flexibility to reallocate manufacturing capacity to improve efficiency and respond to changes in supply and demand. To maintain a stable supply of our products, we use a variety of techniques including comprehensive quality systems, inventory management, and back-up sites.
However, human pharmaceutical and animal health production processes are complex, highly regulated, and vary widely from product to product. Shifting or adding manufacturing capacity can be a very lengthy process requiring significant capital expenditures, process modifications, and regulatory approvals. Accordingly, if we were to experience extended plant shutdowns at one of our own facilities, extended failure of a contract supplier, or extraordinary unplanned increases in demand, we could experience an interruption in supply of certain products or product shortages until production could be resumed or expanded.
Quality Assurance
Our success depends in great measure upon customer confidence in the quality of our products and in the integrity of the data that support their safety and effectiveness. Product quality arises from a total commitment to quality in all parts of our operations, including research and development, purchasing, facilities planning, manufacturing, distribution, and dissemination of information about our medicines.
Quality of production processes involves strict control of ingredients, equipment, facilities, manufacturing methods, packaging materials, and labeling. We perform tests at various stages of production processes and on the final product to assure that the product meets all regulatory requirements and Lilly internal standards. These tests may involve chemical and physical chemical analyses, microbiological testing, testing in animals, or a combination. Additional assurance of quality is provided by corporate quality-assurance groups that audit and monitor all aspects of quality related to human pharmaceutical and animal health manufacturing procedures and systems in company operations and at third-party suppliers.
Executive Officers of the Company
The following table sets forth certain information regarding our executive officers. Except as otherwise noted, all executive officers have been employed by the company in management or executive positions during the last five years.
17
The term of office for each executive officer expires on the date of the annual meeting of the Board of Directors, to be held on May 2, 2016, or on the date his or her successor is chosen and qualified. No director or executive officer has a “family relationship” with any other director or executive officer of the company, as that term is defined for purposes of this disclosure requirement. There is no understanding between any executive officer and any other person pursuant to which the executive officer was selected.
Name | Age | Offices and Business Experience |
John C. Lechleiter, Ph.D. | 62 | Chairman (since January 2009), President (since October 2005), Chief Executive Officer (since April 2008), and a Director (since October 2005) |
Melissa S. Barnes | 47 | Senior Vice President, Enterprise Risk Management and Chief Ethics and Compliance Officer (since January 2013) |
Enrique A. Conterno | 49 | Senior Vice President and President, Lilly Diabetes (since November 2009) |
Maria A. Crowe | 56 | President, Manufacturing Operations (since January 2012) |
Stephen F. Fry | 50 | Senior Vice President, Human Resources and Diversity (since February 2011) |
Michael J. Harrington | 53 | Senior Vice President and General Counsel (since January 2013) |
Jan M. Lundberg, Ph.D. | 62 | Executive Vice President, Science and Technology, and President, Lilly Research Laboratories (since January 2010) |
Susan Mahony, Ph.D. | 51 | Senior Vice President and President, Lilly Oncology (since February 2011) |
Barton R. Peterson | 57 | Senior Vice President, Corporate Affairs and Communications (since June 2009) |
Derica W. Rice | 51 | Executive Vice President, Global Services (since January 2010) and Chief Financial Officer (since May 2006) |
David A. Ricks | 48 | Senior Vice President and President, Lilly Bio-Medicines (since January 2012) |
Jeffrey N. Simmons | 48 | Senior Vice President and President, Elanco Animal Health (since January 2008) |
Fionnuala M. Walsh | 56 | Senior Vice President, Global Quality (since July 2007) |
Alfonso Zulueta | 53 | Senior Vice President and President, Emerging Markets (since January 2014) |
Employees
At the end of 2015, we employed approximately 41,275 people, including approximately 23,425 employees outside the U.S. A substantial number of our employees have long records of continuous service.
Financial Information Relating to Business Segments and Classes of Products
You can find financial information relating to our business segments and classes of products in Item 8, "Financial Statements and Supplementary Data—Note 18, Segment Information." That information is incorporated here by reference.
The relative contribution of any particular product to our consolidated revenue changes from year to year. This is due to several factors, including the introduction of new products by us and by other manufacturers and the introduction of generic pharmaceuticals upon patent expirations. Our product revenues are generally not seasonal.
Financial Information Relating to Foreign and Domestic Operations
You can find financial information relating to foreign and domestic operations in Item 8, “Financial Statements and Supplementary Data—Note 18, Segment Information.” That information is incorporated here by reference. To date, our overall operations abroad have not been significantly deterred by local restrictions on the transfer of funds from branches and subsidiaries located abroad, including the availability of U.S. dollar exchange. We cannot predict what effect these restrictions or the other risks inherent in foreign operations, including possible nationalization, might have on our future operations or what other restrictions may be imposed in the future. In addition, changing currency values can either favorably or unfavorably affect our financial position, liquidity, and results of operations. We mitigate certain foreign exchange risks through various hedging techniques including the use of foreign currency contracts.
18
Available Information on Our Website
Our company website is http://www.lilly.com. None of the information accessible on or through our website is incorporated into this Form 10-K. We make available through the website, free of charge, our company filings with the Securities and Exchange Commission (SEC) as soon as reasonably practicable after we electronically file them with, or furnish them to, the SEC. These include our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy statements, registration statements, and any amendments to those documents. The company website link to our SEC filings is
http://investor.lilly.com/sec.cfm.
In addition, the Corporate Governance portion of our website includes our corporate governance guidelines, board and committee information (including committee charters), and our articles of incorporation and by-laws. The link to our corporate governance information is http://www.lilly.com/about/corporate-governance/Pages/corporate-governance.aspx.
We will provide paper copies of our SEC filings free of charge upon request to the company’s secretary at the address listed on the front of this Form 10-K.
Item 1A. | Risk Factors |
In addition to the other information contained in this Form 10-K, the following risk factors should be considered carefully in evaluating our company. It is possible that our business, financial condition, liquidity, or results of operations could be materially adversely affected by any of these risks. Certain of these risks could also adversely affect the company's reputation.
• | Pharmaceutical research and development is very costly and highly uncertain; we may not succeed in developing or acquiring commercially successful products sufficient in number or value to replace revenues of products losing intellectual property protection. |
There are many difficulties and uncertainties inherent in human pharmaceutical research and development and the introduction of new products. There is a high rate of failure inherent in new drug discovery and development. To bring a drug from the discovery phase to market can take over a decade and often costs in excess of $1 billion. Failure can occur at any point in the process, including in later stages after substantial investment. As a result, most funds invested in research programs will not generate financial returns. New product candidates that appear promising in development may fail to reach the market or may have only limited commercial success because of efficacy or safety concerns, inability to obtain necessary regulatory approvals or payer reimbursement or coverage, limited scope of approved uses, difficulty or excessive costs to manufacture, or infringement of the patents or intellectual property rights of others. Regulatory agencies are establishing increasingly high hurdles for the efficacy and safety of new products; delays and uncertainties in drug approval processes can result in delays in product launches and lost market opportunity. In addition, it can be very difficult to predict sales growth rates of new products.
We cannot state with certainty when or whether our products now under development will be approved or launched; whether we will be able to develop, license, or otherwise acquire additional product candidates or products; or whether our products, once launched, will be commercially successful. We must maintain a continuous flow of successful new products and successful new indications or brand extensions for existing products sufficient both to cover our substantial research and development costs and to replace sales that are lost as profitable products lose intellectual property exclusivity or are displaced by competing products or therapies. Failure to do so in the short-term or long-term would have a material adverse effect on our business, results of operations, cash flows, financial position, and prospects.
19
• | We depend on products with intellectual property protection for most of our revenues, cash flows, and earnings; we have lost or will lose effective intellectual property protection for many of those products in the next several years, which may result in rapid and severe declines in revenues. |
A number of our top-selling human pharmaceutical products have recently lost, or will lose in the next several years, significant patent protection and/or data protection in the U.S. as well as key countries outside the U.S., as illustrated in the tables below:
Product | U.S. Revenues (2015) ($ in millions) | Percent of Worldwide Revenues (2015) | Patent / Data Protection - U.S. | ||
Cialis | $ | 1,256.8 | 6% | Compound and use patents November 2017 | |
Alimta | 1,162.4 | 6% | Compound patent plus pediatric exclusivity January 2017; vitamin regimen patent plus pediatric exclusivity 2022 | ||
Forteo | 612.4 | 3% | Formulation and related process patents 2018; use patents 2019 | ||
Strattera | 502.1 | 3% | Use patent plus pediatric exclusivity May 2017 | ||
Effient | 417.6 | 2% | Compound patent April 2017; use patents 2023 | ||
Product | Revenues Outside U.S. (2015) ($ in millions) | Percent of Worldwide Revenues (2015) | Patent / Data Protection - Major Europe / Japan | ||
Alimta | $ | 1,330.7 | 7% | Major European countries: compound patent December 2015, vitamin regimen patent 2021 Japan: compound patent December 2015, use patents to treat cancer concomitantly with vitamins 2021 | |
Cialis | 1,053.9 | 5% | Major European countries: compound patent November 2017 | ||
Cymbalta | 883.0 | 4% | Major European countries: data package protection 2014 Japan: data package protection 2018 | ||
Zyprexa | 783.6 | 4% | Japan: Patent for schizophrenia December 2015; for bipolar mania April 2016 | ||
Forteo | 735.9 | 4% | Japan: Data package protection 2018; formulation and related process patent 2019 | ||
Certain other significant products no longer have effective exclusivity through patent protection or data protection. For non-biological products, loss of exclusivity (whether by expiration or as a consequence of litigation) typically results in the entry of one or more generic competitors, leading to a rapid and severe decline in revenues, especially in the U.S. Historically, outside the U.S. the market penetration of generics following loss of exclusivity has not been as rapid or pervasive as in the U.S.; however, generic market penetration is increasing in many markets outside the U.S., including Japan, Europe, and many countries in the emerging markets. For biological products (such as Humalog, Humulin, Erbitux, and Cyramza), loss of exclusivity may or may not result in the near-term entry of competitor versions (i.e., biosimilars) due to development timelines, manufacturing challenges, and/or uncertainties in the regulatory pathways for approval of the competitor versions. See Item 7, “Management’s Discussion and Analysis—Executive Overview—Other Matters,” and Item 1, "Business—Patents, Trademarks, and Other Intellectual Property Rights," for more details.
• | Our long-term success depends on intellectual property protection; if our intellectual property rights are invalidated, circumvented, or weakened, our business will be adversely affected. |
Our long-term success depends on our ability to continually discover, develop, and commercialize innovative new pharmaceutical products. Without strong intellectual property protection, we would be unable to generate the returns necessary to support the enormous investments in research and development and capital as well as other expenditures required to bring new drugs to the market.
20
Intellectual property protection varies throughout the world and is subject to change over time. In the U.S., the Hatch-Waxman Act provides generic companies powerful incentives to seek to invalidate our human pharmaceutical patents; as a result, we expect that our U.S. patents on major pharmaceutical products will be routinely challenged in litigation and administrative proceedings, and may not be upheld. We face many generic manufacturer challenges to our patents outside the U.S. as well. The entry of generic competitors typically results in rapid and severe declines in sales. In addition, competitors or other third parties may claim that our activities infringe patents or other intellectual property rights held by them. If successful, such claims could result in our being unable to market a product in a particular territory or being required to pay damages for past infringement or royalties on future sales. See Item 1, “Business—Patents, Trademarks, and Other Intellectual Property Rights,” Item 3, "Legal Proceedings," and Item 8, "Financial Statements and Supplementary Data—Note 15, Contingencies," for more details.
• | Our human pharmaceutical business is subject to increasing government price controls and other public and private restrictions on pricing, reimbursement, and access for our drugs, which could have a material adverse effect on our business. |
Public and private payers are taking increasingly aggressive steps to control their expenditures for human pharmaceuticals by placing restrictions on pricing and reimbursement for, and patient access to, our medications. These pressures could negatively affect our future revenues and income.
In the U.S., public concern over prices for specialty and brand name pharmaceuticals continues to drive the legislative debate. These policy and political issues increase the risk that taxes, fees, rebates or other federal and state measures may be enacted. Key health policy proposals affecting biopharmaceuticals include a reduction in biologic data exclusivity, modifications to Medicare Parts B and D, new language that would allow the Department of Health and Human Services to negotiate prices for biologics and drugs on the specialty tier in Part D, and state-level proposals to reduce the cost of pharmaceuticals purchased by government health care programs. Savings projected under these proposals are targeted as a means to fund both health care expenditures and non-health care initiatives, or to manage federal and state budgets.
In the U.S. private sector, health plans and pharmaceutical benefit managers have been consolidating into fewer, larger entities, thus enhancing their purchasing strength and importance. Payers typically maintain formularies specifying which drugs are covered and the cost to the consumer. Non-preferred formulary placement, including the exclusion of a drug from a formulary, typically leads to its reduced usage in the patient population. Consequently, pharmaceutical companies compete to have their branded products included by, among other things, providing offsetting rebates. Price is an increasingly important factor in formulary decisions, particularly in treatment areas in which payers take the position that multiple branded products are therapeutically comparable.
The main coverage expansion provisions of the Affordable Care Act (ACA) are now in effect through both the launch of state-based exchanges and the expansion of Medicaid. An emerging trend has been the prevalence of benefit designs containing high patient out-of-pocket costs for pharmaceuticals. In addition to the coverage expansions, many employers in the commercial market, driven in part by ACA changes such as the 2020 implementation of the excise tax on employer-sponsored health care coverage for which there is an excess benefit (the so-called "Cadillac tax"), continue to evaluate strategies such as private exchanges and wider use of consumer-driven health plans to reduce their healthcare liabilities over time. At the same time, the broader paradigm shift towards quality-based reimbursement and the launch of several value-based purchasing initiatives are placing demands on the pharmaceutical industry to offer products with proven real-world outcomes data and a favorable economic profile.
International operations also are generally subject to extensive price and market regulations. Cost-containment measures exist in a number of countries, including additional price controls and mechanisms to limit reimbursement for our products. Such policies are expected to increase in impact and reach, given the pressures on health care budgets that come from a growing aging population and ongoing economic challenges. In addition, governments in many emerging markets are becoming increasingly active in expanding health care system offerings. Given the budget challenges of increasing health care coverage for citizens, policies may be proposed that promote generics only and reduce current and future access to human pharmaceutical products.
We expect pricing, reimbursement, and access pressures from both governments and private payers inside and outside the U.S. to become more severe. For more details, see Item 1, “Business—Regulations Affecting Human Pharmaceutical Pricing, Reimbursement, and Access,” and Item 7, “Management’s Discussion and Analysis—Executive Overview—Other Matters.”
21
• | We face intense competition from multinational pharmaceutical companies, biotechnology companies, and lower-cost generic and biosimilar manufacturers, and such competition could have a material adverse effect on our business. |
We compete with a large number of multinational pharmaceutical companies, biotechnology companies, and generic pharmaceutical companies. To compete successfully, we must continue to deliver to the market innovative, cost-effective products that meet important medical needs. Our product revenues can be adversely affected by the introduction by competitors of branded products that are perceived as superior by the marketplace, by generic or biosimilar versions of our branded products, and by generic or biosimilar versions of other products in the same therapeutic class as our branded products. Our revenues can also be adversely affected by treatment innovations that eliminate or minimize the need for treatment with drugs. See Item 1, “Business—Competition,” for more details.
• | Changes in foreign currency rates can materially affect our revenue, cost of sales, and operating expenses. |
As a global company with substantial operations outside the U.S., we face foreign currency risk exposure from fluctuating currency exchange rates, primarily the U.S. dollar against the euro, Japanese yen, and British pound, and the British pound against the euro. While we manage a portion of these exposures through hedging and other risk management techniques, significant fluctuations in currency rates can have a material impact, either positive or negative, on our revenue, cost of sales, and operating expenses.
• | Unanticipated changes in our tax rates or exposure to additional tax liabilities could increase our income taxes and decrease our net income. |
We are subject to income taxes in the U.S. and numerous foreign jurisdictions. Changes in the relevant tax laws, regulations, administrative practices, principles, and interpretations could adversely affect our future effective tax rates. The U.S. and a number of other countries are actively considering or enacting changes in this regard. For example, the Obama administration proposed changes to the manner in which the U.S. would tax the international income of U.S.-based companies, including unremitted earnings of foreign subsidiaries. Other tax proposals under discussion or introduced in the U.S. Congress could change the tax rate and manner in which U.S. companies would be taxed. Additionally, the Organisation for Economic Co-operation and Development issued its final recommendations of international tax reform proposals to influence international tax policy in major countries in which we operate. While outcomes of these initiatives continue to develop and remain uncertain, changes to key elements of the U.S. or international tax framework could have a material adverse effect on our consolidated operating results and cash flows.
See Item 8, "Financial Statements and Supplementary Data—Note 13, Income Taxes," for more details.
• | Regulatory compliance problems could be damaging to the company. |
The marketing, promotional, and pricing practices of human pharmaceutical manufacturers, as well as the manner in which manufacturers interact with purchasers, prescribers, and patients, are subject to extensive regulation. Many companies, including us, have been subject to claims related to these practices asserted by federal, state, and foreign governmental authorities, private payers, and consumers. These claims have resulted in substantial expense and other significant consequences to us. It is possible that we could become subject to such investigations and that the outcome could include criminal charges and fines, penalties, or other monetary or non-monetary remedies, including exclusion from U.S. federal and other health care programs. In addition, regulatory issues concerning compliance with cGMP regulations (and comparable foreign regulations) for pharmaceutical products can lead to product recalls and seizures, fines and penalties, interruption of production leading to product shortages, and delays in the approvals of new products pending resolution of the issues. See Item 1, “Business—Regulation of our Operations,” for more details.
• | The loss, theft, or inadvertent disclosure of our confidential data could impair our valuable intellectual property, harm our competitive position, and/or expose us to regulatory penalties and other costs. |
A great deal of confidential information owned by both us and our alliances is stored in our information systems, networks, and facilities or those of third parties. This includes valuable trade secrets and intellectual property, corporate strategic plans, marketing plans, customer information, and personally identifiable information (such as employee and patient information). Some of this information is created, accessed, and/or maintained by third parties. The confidentiality of this information may be breached in a variety of ways, including but not limited to negligent or wrongful conduct by employees or others with
22
permitted access to our systems and data, or wrongful conduct by certain governments, hackers, unethical competitors, or former workforce members. The rapid growth of factors such as mobile computing capacity, high-speed Internet access, and social media exacerbates the risk of information security breaches.
The theft or unauthorized disclosure of confidential information could impair our ability to secure and maintain intellectual property rights, cause damage to company operations and reputation, and cause us to lose trade secrets or other competitive advantages. Unauthorized disclosure of personally identifiable information could expose us to sanctions for violations of data privacy laws and regulations and could damage the public trust in our company. Information security breaches may be very difficult to detect, and once detected, their impact may be very difficult to assess. To date, the information security breaches of which we have become aware have been infrequent in occurrence and, to the extent we have been able to measure their financial impact on our consolidated results of operations, such impact has not been material. We have invested and continue to invest to prevent, monitor, detect, and respond to information security breaches by strengthening our employee awareness and training, information technology systems, and business processes, and strengthening data protection requirements for third parties that handle our confidential information. However, despite these efforts, we expect information security breaches to continue, and there can be no assurance that these efforts will prevent information security breaches that would have a material adverse effect on our business.
• | Worsening economic conditions could adversely affect our business and operating results. |
While human pharmaceuticals have not generally been sensitive to overall economic cycles, prolonged economic slowdowns could lead to decreased utilization of drugs, affecting our sales volume. Declining tax revenues attributable to economic downturns increase the pressure on governments to reduce health care spending, leading to increasing government efforts to control drug prices and utilization. Additionally, some customers, including governments or other entities reliant upon government funding, may be unable to pay in a timely manner for our products. Also, if our customers, suppliers, or collaboration partners experience financial difficulties, we could experience slower customer collections, greater bad debt expense, and performance defaults by suppliers or collaboration partners.
• | Pharmaceutical products can develop unexpected safety or efficacy concerns, which could have a material adverse effect on revenues and income. |
Human pharmaceutical products receive regulatory approval based on data obtained in controlled clinical trials of limited duration. After approval, the products are used for longer periods of time by much larger numbers of patients; we and others (including regulatory agencies and private payers) collect extensive information on the efficacy and safety of our marketed products by continuously monitoring the use of our products in the marketplace. In addition, we or others may conduct post-marketing clinical studies on efficacy and safety of our marketed products. New safety or efficacy data from both market surveillance and post-marketing clinical studies may result in product label changes that could reduce the product's market acceptance and result in declining sales. Serious safety or efficacy issues that arise after product approval could result in voluntary or mandatory product recalls or withdrawals from the market. Safety issues could also result in costly product liability claims.
• | We face many product liability claims and are self-insured; we could face large numbers of claims in the future, which could adversely affect our business. |
We are subject to a substantial number of product liability claims involving Actos®, Byetta®, Cymbalta, and Prozac among other products. See Item 8, “Financial Statements and Supplementary Data—Note 15, Contingencies,” and Item 3, “Legal Proceedings,” for more information on our current product liability litigation. Because of the nature of pharmaceutical products, we could become subject to large numbers of product liability claims for these or other products in the future, which could require substantial expenditures to resolve and, if involving marketed products, could adversely affect sales of the product. Due to a very restrictive market for product liability insurance, we are self-insured for product liability losses for all our currently marketed products.
• | Manufacturing difficulties or disruptions could lead to product supply problems. |
Pharmaceutical and animal health manufacturing is complex and highly regulated. Manufacturing difficulties at our facilities or contracted facilities, or the failure or refusal of a contract manufacturer to supply contracted quantities, could result in product shortages, leading to lost revenue. Such difficulties or disruptions could result from quality or regulatory compliance problems, natural disasters, mechanical or information technology system failures, or inability to obtain sole-source raw or intermediate materials. In addition, given
23
the difficulties in predicting sales of new products and the very long lead times necessary for the expansion and regulatory qualification of pharmaceutical manufacturing capacity, it is possible that we could have difficulty meeting demand for new products. See Item 1, “Business—Raw Materials and Product Supply,” for more details.
• | We depend on information technology systems and infrastructure to operate our business; system inadequacies or operating failures could harm our business. |
We rely to a large extent on the efficient and uninterrupted operation of complex information technology systems and networks, some of which are within the company and some of which are outsourced. These systems and networks are potentially vulnerable to corruption, damage, or interruption from a variety of sources, including energy or telecommunications failures, breakdowns, natural disasters, terrorism, war, computer malware or other malicious intrusions, and random attacks. To date, system interruptions have been infrequent and have not had a material impact on our consolidated results of operations. We have implemented measures to prevent, respond to, and minimize the impact of system interruptions. However, there can be no assurance that these efforts will prevent future interruptions that would have a material adverse effect on our business.
• | Reliance on third-party relationships and outsourcing arrangements could adversely affect our business. |
We utilize third parties, including suppliers, alliances with other pharmaceutical and biotechnology companies, and third-party service providers, for selected aspects of product development, the manufacture and commercialization of certain products, support for information technology systems, and certain financial transactional processes. For example, we outsource the day-to-day management and oversight of our clinical trials to contract research organizations. Outsourcing these functions involves the risk that the third parties may not perform to our standards or legal requirements, may not produce reliable results, may not perform in a timely manner, may not maintain the confidentiality of our proprietary information, or may fail to perform at all. Failure of these third parties to meet their contractual, regulatory, confidentiality, or other obligations to us could have a material adverse effect on our business.
• | Our animal health segment faces risks related to increased generic competition, food and animal safety concerns, factors affecting global agricultural markets, and other risks. |
The animal health operating segment may be impacted by, among other things, increased generic competition; increased sales of companion animal products by non-veterinarian retail outlets; emerging restrictions and bans on the use of antibacterials in food-producing animals; perceived adverse effects on human health linked to the consumption of food derived from animals that utilize our products; increased regulation or decreased governmental support relating to the raising, processing, or consumption of food-producing animals; an outbreak of infectious disease carried by animals; adverse weather conditions and the availability of natural resources; adverse global economic conditions affecting agricultural markets; and failure of our research and development, acquisition, and licensing efforts to generate new products. The failure to manage these risks could have a material adverse effect on our revenues and income.
• | Integration of the Novartis Animal Health business could lead to additional unplanned expenses and be disruptive to operations. |
We are continuing to integrate into our operations the Novartis AH business which we purchased in January 2015. This complex global integration is a multi-year process and could still be disruptive to the ongoing operations of the Elanco business or to certain corporate support functions. Unexpected delays and difficulties in the integration could lead to additional expenses and disruption to ongoing operating results.
Item 1B. | Unresolved Staff Comments |
None.
24
Item 2. | Properties |
Our principal domestic and international executive offices are located in Indianapolis. At December 31, 2015, we owned 13 production and distribution sites in the U.S. and Puerto Rico. Together with the corporate administrative offices, these facilities contain an aggregate of approximately 10.7 million square feet of floor area dedicated to production, distribution, and administration. Major production sites include Indianapolis and Clinton, Indiana; Carolina, Puerto Rico; and Branchburg, New Jersey.
We own production and distribution sites in 14 countries outside the U.S. and Puerto Rico, containing an aggregate of approximately 5.4 million square feet of floor area. Major production sites include facilities in France, Ireland, China, the U.K., Spain, and Italy.
In the U.S., our research and development facilities contain an aggregate of approximately 3.9 million square feet of floor area, primarily consisting of owned facilities located in Indianapolis. We also lease smaller sites in San Diego and New York City. Outside the U.S., we own smaller research and development facilities in the U.K., Spain, Australia, and lease smaller sites in China.
The 2015 acquisition of Novartis AH added 11 owned sites and 26 leased sites totaling approximately 1.4 million square feet and approximately 500 thousand square feet of floor area, respectively. These locations include a mix of office, research and development, and production and distribution space.
We believe that none of our properties is subject to any encumbrance, easement, or other restriction that would detract materially from its value or impair its use in the operation of the business. The buildings we own are of varying ages and in good condition.
Item 3. | Legal Proceedings |
We are a party to various currently pending legal actions, government investigations, and environmental proceedings, and we anticipate that such actions could be brought against us in the future. The most significant of these matters are described below or, as noted, in Item 8, "Financial Statements and Supplementary Data—Note 15, Contingencies." While it is not possible to determine the outcome of the legal actions, investigations, and proceedings brought against us, we believe that, except as otherwise specifically noted in Item 8, "Financial Statements and Supplementary Data—Note 15, Contingencies," the resolution of all such matters will not have a material adverse effect on our consolidated financial position or liquidity, but could be material to our consolidated results of operations in any one accounting period.
Legal Proceedings Described in Note 15 to the Consolidated Financial Statements
See Item 8, "Financial Statements and Supplementary Data—Note 15, Contingencies," for information on various legal proceedings, including but not limited to:
• | The patent litigation and administrative proceedings involving Alimta and Effient |
• | The product liability litigation involving Actos, Byetta, Cymbalta, and Prozac |
• | The employee litigation in Brazil. |
That information is incorporated into this Item by reference.
Other Product Liability Litigation
We are named as a defendant in approximately 410 Axiron product liability lawsuits in the U.S. involving approximately 560 plaintiffs. In more than one-third of the cases, other manufacturers of testosterone are named as co-defendants. Nearly all of these lawsuits have been consolidated in a federal multi-district litigation in the U.S. District Court for the Northern District of Illinois. A small number of lawsuits has been filed in state courts. The cases generally allege cardiovascular and related injuries. We believe these claims are without merit and are prepared to defend against them vigorously.
25
Other Patent Litigation
Boehringer Ingelheim, our partner in marketing and development of Trajenta, is engaged in various U.S. patent litigation matters involving Trajenta/Jentadueto in accordance with the procedures set out in the Drug Price Competition and Patent Term Restoration Act of 1984. Eleven groups of companies submitted Abbreviated New Drug Applications seeking approval to market generic versions of Trajenta prior to the expiration of Trajenta/Jentadueto patents, alleging certain patents, including in some allegations the compound patent, are invalid or would not be infringed.
In Canada, several generic companies challenged the validity of our Zyprexa patent. In September 2012, the Canadian Court of Appeals affirmed the lower court's decision that the patent was invalid for lack of utility. In 2013, our petition for leave to appeal the decision to the Supreme Court of Canada was denied. Two of the generic companies, Apotex Inc. (Apotex) and Teva Canada Limited (Teva), pursued claims for damages arising from our enforcement of the patent under Canadian regulations. In April 2014, the Supreme Court of Canada dismissed Apotex's damages suit. Teva's claim for damages remains, and the total amount of damages that may be awarded to Teva will be determined through a separate trial, which is scheduled for May 2016.
Other Matters
In September 2015, we were advised that the U.S. Attorney’s office for the Eastern District of Pennsylvania and the Civil Division of the DOJ are conducting an inquiry concerning the treatment by various pharmaceutical companies, including us, of certain distribution service agreements with wholesalers when calculating and reporting Average Manufacturer Prices in connection with the Medicaid drug rebate program. We are voluntarily responding to this request.
Under the Comprehensive Environmental Response, Compensation, and Liability Act, commonly known as "Superfund," we have been designated as one of several potentially responsible parties with respect to the cleanup of fewer than 10 sites. Under Superfund, each responsible party may be jointly and severally liable for the entire amount of the cleanup.
We are also a defendant in other litigation and investigations, including product liability, patent, employment, and premises liability litigation, of a character we regard as normal to our business.
Item 4. | Mine Safety Disclosures |
Not applicable.
26
Part II
Item 5. | Market for the Registrant’s Common Equity, Related Stockholder Matters, and Issuer Purchases of Equity Securities |
You can find information relating to the principal market for our common stock and related stockholder matters at Item 6, "Selected Financial Data (unaudited)" and Item 8, "Financial Statements and Supplementary Data—Note 19, Selected Quarterly Data (unaudited).” That information is incorporated here by reference.
The following table summarizes the activity related to repurchases of our equity securities during the fourth quarter ended December 31, 2015:
Period | Total Number of Shares Purchased (in thousands) | Average Price Paid per Share | Total Number of Shares Purchased as Part of Publicly Announced Plans or Programs (in thousands) | Approximate Dollar Value of Shares that May Yet Be Purchased Under the Plans or Programs (dollars in millions) | ||||||
October 2015 | 1,207.5 | $ | 82.81 | 1,207.5 | $ | 3,103.4 | ||||
November 2015 | 957.0 | 81.40 | 957.0 | 3,025.5 | ||||||
December 2015 | 881.1 | 85.12 | 881.1 | 2,950.5 | ||||||
Total | 3,045.6 | 83.04 | 3,045.6 | |||||||
During the fourth quarter of 2015, we repurchased $252.9 million of shares associated with our $5.00 billion share repurchase program announced in October 2013.
27
PERFORMANCE GRAPH
This graph compares the return on Lilly stock with that of the Standard & Poor’s 500 Stock Index and our peer group for the years 2011 through 2015. The graph assumes that, on December 31, 2010, a person invested $100 each in Lilly stock, the S&P 500 Stock Index, and the peer groups' common stock. The graph measures total shareholder return, which takes into account both stock price and dividends. It assumes that dividends paid by a company are reinvested in that company’s stock.
Value of $100 Invested on Last Business Day of 2010
Comparison of Five-Year Cumulative Total Return Among Lilly, S&P 500 Stock Index, and Peer Group(1)
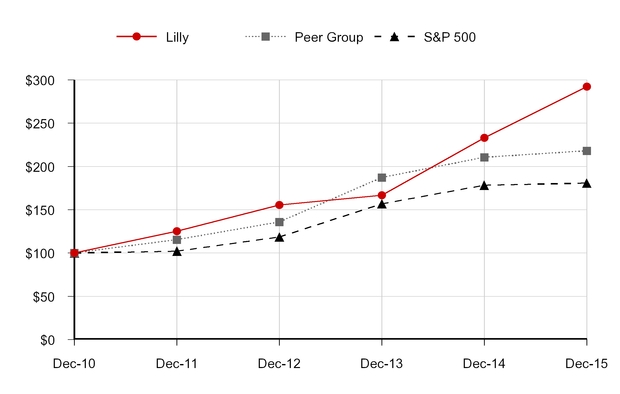
Lilly | Peer Group | S&P 500 | ||||||||||
Dec-10 | $ | 100.00 | $ | 100.00 | $ | 100.00 | ||||||
Dec-11 | $ | 125.15 | $ | 115.41 | $ | 102.11 | ||||||
Dec-12 | $ | 155.52 | $ | 135.93 | $ | 118.45 | ||||||
Dec-13 | $ | 166.77 | $ | 187.14 | $ | 156.82 | ||||||
Dec-14 | $ | 233.07 | $ | 210.73 | $ | 178.28 | ||||||
Dec-15 | $ | 292.20 | $ | 218.03 | $ | 180.75 | ||||||
(1) | We constructed the peer group as the industry index for this graph. It comprises the public companies in the pharmaceutical and biotech industries that we used to benchmark the compensation of executive officers for 2015 (other than Allergan Inc.): Abbott Laboratories; AbbVie Inc.; Amgen Inc.; AstraZeneca PLC; Baxter International Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Celgene Corporation; Gilead Sciences Inc.; GlaxoSmithKline plc; Johnson & Johnson; Medtronic plc; Merck & Co., Inc.; Novartis AG.; Pfizer Inc.; and Sanofi. The peer group total shareholder return reflected above excludes Allergan Inc. as it was acquired in 2015. |
28
Item 6. Selected Financial Data (unaudited)
ELI LILLY AND COMPANY AND SUBSIDIARIES (Dollars in millions, except revenue per employee and per-share data) | 2015 | 2014 | 2013 | 2012 | 2011 | ||||||||||||||
Operations | |||||||||||||||||||
Revenue | $ | 19,958.7 | $ | 19,615.6 | $ | 23,113.1 | $ | 22,603.4 | $ | 24,286.5 | |||||||||
Cost of sales | 5,037.2 | 4,932.5 | 4,908.1 | 4,796.5 | 5,067.9 | ||||||||||||||
Research and development | 4,796.4 | 4,733.6 | 5,531.3 | 5,278.1 | 5,020.8 | ||||||||||||||
Marketing, selling, and administrative | 6,533.0 | 6,620.8 | 7,125.6 | 7,513.5 | 7,879.9 | ||||||||||||||
Other | 802.1 | 328.4 | (341.2 | ) | (392.9 | ) | 968.4 | ||||||||||||
Income before income taxes | 2,790.0 | 3,000.3 | 5,889.3 | 5,408.2 | 5,349.5 | ||||||||||||||
Income taxes | 381.6 | 609.8 | 1,204.5 | 1,319.6 | 1,001.8 | ||||||||||||||
Net income | 2,408.4 | 2,390.5 | 4,684.8 | 4,088.6 | 4,347.7 | ||||||||||||||
Net income as a percent of revenue | 12.1 | % | 12.2 | % | 20.3 | % | 18.1 | % | 17.9 | % | |||||||||
Net income per share—diluted | $ | 2.26 | $ | 2.23 | $ | 4.32 | $ | 3.66 | $ | 3.90 | |||||||||
Dividends declared per share | 2.01 | 1.97 | 1.96 | 1.96 | 1.96 | ||||||||||||||
Weighted-average number of shares outstanding—diluted (thousands) | 1,065,720 | 1,074,286 | 1,084,766 | 1,117,294 | 1,113,967 | ||||||||||||||
Financial Position | |||||||||||||||||||
Current assets(1) | $ | 12,573.6 | $ | 11,928.3 | $ | 12,820.4 | $ | 12,790.3 | $ | 13,884.6 | |||||||||
Current liabilities(1) | 8,229.6 | 9,741.0 | 8,123.8 | 7,341.5 | 8,508.6 | ||||||||||||||
Property and equipment—net | 8,053.5 | 7,963.9 | 7,975.5 | 7,760.2 | 7,760.3 | ||||||||||||||
Total assets(1) | 35,568.9 | 36,307.6 | 35,210.8 | 33,316.1 | 33,216.5 | ||||||||||||||
Long-term debt | 7,972.4 | 5,332.8 | 4,200.3 | 5,519.4 | 5,464.7 | ||||||||||||||
Total equity | 14,590.3 | 15,388.1 | 17,640.7 | 14,773.9 | 13,535.6 | ||||||||||||||
Supplementary Data | |||||||||||||||||||
Return on total equity | 16.1 | % | 13.7 | % | 29.5 | % | 27.8 | % | 31.4 | % | |||||||||
Return on assets(1) | 6.8 | % | 6.8 | % | 14.1 | % | 12.5 | % | 13.5 | % | |||||||||
Capital expenditures | $ | 1,066.2 | $ | 1,162.6 | $ | 1,012.1 | $ | 905.4 | $ | 672.0 | |||||||||
Depreciation and amortization | 1,427.7 | 1,379.0 | 1,445.6 | 1,462.2 | 1,373.6 | ||||||||||||||
Effective tax rate | 13.7 | % | 20.3 | % | 20.5 | % | 24.4 | % | 18.7 | % | |||||||||
Revenue per employee | $ | 484,000 | $ | 501,000 | $ | 609,000 | $ | 590,000 | $ | 638,000 | |||||||||
Number of employees | 41,275 | 39,135 | 37,925 | 38,350 | 38,080 | ||||||||||||||
Number of shareholders of record | 28,000 | 29,300 | 31,900 | 33,600 | 35,200 | ||||||||||||||
(1) Amounts have been adjusted to reflect the retrospective application of Accounting Standards Update 2015-17 Income Taxes: Balance Sheet Classification of Deferred Taxes. See Note 2 to consolidated financial statements.
29
Item 7. | Management’s Discussion and Analysis of Results of Operations and Financial Condition |
RESULTS OF OPERATIONS
(Tables present dollars in millions, except per-share data)
Executive Overview
This section provides an overview of our financial results, recent product and late-stage pipeline developments, and other matters affecting our company and the pharmaceutical industry. Earnings per share (EPS) data is presented on a diluted basis.
Financial Results
The following table summarizes our key operating results:
Year Ended, December 31, | Percent Change from | |||||||||
2015 | 2014 | 2014 | ||||||||
Revenue | $ | 19,958.7 | $ | 19,615.6 | 2 | % | ||||
Gross margin | 14,921.5 | 14,683.1 | 2 | % | ||||||
Gross margin as a percent of revenue | 74.8 | % | 74.9 | % | ||||||
Operating expense (1) | $ | 11,329.4 | $ | 11,354.4 | — | % | ||||
Acquired in-process research and development | 535.0 | 200.2 | NM | |||||||
Asset impairment, restructuring, and other special charges | 367.7 | 468.7 | (22 | )% | ||||||
Net income | 2,408.4 | 2,390.5 | 1 | % | ||||||
Earnings per share | 2.26 | 2.23 | 1 | % | ||||||
(1) Operating expense consists of research and development and marketing, selling, and administrative expenses.
NM - not meaningful
Revenue and gross margin increased slightly in 2015. Operating expense in 2015 remained essentially flat as a decrease in marketing, selling, and administrative expense was largely offset by increased research and development expense. Net income and EPS increased slightly in 2015 as a higher gross margin, lower income taxes, and decreased asset impairment, restructuring, and other special charges were largely offset by increased acquired in-process research and development (IPR&D) charges and lower other income.
The following highlighted items affect comparisons of our 2015 and 2014 financial results:
2015
Acquisitions (Note 3 to the consolidated financial statements)
• | We recognized expense of $153.0 million (pretax), or $0.10 per share, related to the fair value adjustments to Novartis Animal Health (Novartis AH) acquisition date inventory that has been sold. |
Acquired IPR&D (Notes 3 and 4 to the consolidated financial statements)
• | We recognized acquired IPR&D charges of $535.0 million (pretax), or $0.33 per share, related to upfront fees paid in connection with various collaboration agreements primarily with Pfizer, Inc. (Pfizer), as well as the consideration paid to acquire the worldwide rights to Locemia Solutions' (Locemia) intranasal glucagon. |
30
Asset Impairment, Restructuring, and Other Special Charges (Note 5 to the consolidated financial statements)
• | We recognized charges of $367.7 million (pretax), or $0.25 per share, related to severance costs, integration costs, and intangible asset impairments. |
Debt Repurchase (Notes 7 and 10 to the consolidated financial statements)
• | We recognized net charges of $152.7 million (pretax), or $0.09 per share, attributable to the debt extinguishment loss of $166.7 million from the purchase and redemption of certain fixed-rate notes, partially offset by net gains from non-hedging interest rate swaps and foreign currency transactions associated with the related issuance of lower interest rate euro-denominated notes. |
2014
Acquired IPR&D (Notes 3 and 4 to the consolidated financial statements)
• | We recognized acquired IPR&D charges of $200.2 million (pretax), or $0.12 per share, related to acquired IPR&D from various collaboration agreements. |
Collaborations (Note 4 to the consolidated financial statements)
• | We recognized income of $92.0 million (pretax), or $0.06 per share, related to the transfer of our linagliptin and empagliflozin commercial rights in certain countries to Boehringer Ingelheim. |
Asset Impairment, Restructuring, and Other Special Charges (Note 5 to the consolidated financial statements)
• | We recognized charges of $468.7 million (pretax), or $0.38 per share, related to severance costs associated with our ongoing cost containment efforts to reduce our cost structure and global workforce, and asset impairments primarily associated with the closure of a manufacturing site in Puerto Rico. |
Other
• | We recognized a marketing, selling, and administrative expense of $119.0 million (non-tax deductible), or $0.11 per share, for an extra year of the United States Branded Prescription Drug Fee (U.S. Drug Fee) due to final regulations issued by the Internal Revenue Service (IRS) which required us to accelerate into 2014 the recording of an expense for the 2015 fee. |
Late-Stage Pipeline
Our long-term success depends to a great extent on our ability to continue to discover and develop innovative pharmaceutical products and acquire or collaborate on molecules currently in development by other biotechnology or pharmaceutical companies. We currently have approximately 50 potential new drugs in human testing or under regulatory review, and a larger number of projects in preclinical research.
The following new molecular entity (NME) was approved by regulatory authorities in at least one of the major geographies for use in the disease described. The quarter in which the NME initially was approved in any major geography is shown in parentheses:
Necitumumab* (Portrazza™) (Q4 2015)—an anti-epidermal growth factor receptor monoclonal antibody for the treatment of metastatic squamous non-small cell lung cancer (NSCLC).
The following NMEs have been submitted for regulatory review in at least one of the major geographies for potential use in the diseases described. The quarter in which each NME initially was submitted for any indication is shown in parentheses:
Ixekizumab* (Q1 2015)—a neutralizing monoclonal antibody to interleukin-17A for the treatment of psoriasis and psoriatic arthritis.
Baricitinib (Q1 2016)—a Janus tyrosine kinase inhibitor for the treatment of moderately-to-severely active rheumatoid arthritis (in collaboration with Incyte Corporation).
31
The following NMEs and diagnostic agent are currently in Phase III clinical trial testing for potential use in the diseases described. The quarter in which each NME and diagnostic agent initially entered Phase III for any indication is shown in parentheses:
Abemaciclib (Q3 2014)—a small molecule cell-cycle inhibitor, selective for cyclin-dependent kinases 4 and 6 for the treatment of metastatic breast cancer and NSCLC.
CGRP monoclonal antibody* (Q2 2015)—a once-monthly subcutaneously injected calcitonin gene-related peptide (CGRP) antibody for the treatment of cluster headache and migraine prevention.
Intranasal glucagon* (Q3 2013)—a glucagon nasal powder formulation for the treatment of severe hypoglycemia in patients with diabetes treated with insulin.
Olaratumab* (Q3 2015)—a human lgG1 monoclonal antibody for the treatment of advanced soft tissue sarcoma.
Solanezumab* (Q2 2009)—an anti-amyloid beta monoclonal antibody for the treatment of preclinical and mild Alzheimer’s disease.
Tanezumab* (Q3 2008)—an anti-nerve growth factor monoclonal antibody for the treatment of osteoarthritis pain, chronic low back pain, and cancer pain (in collaboration with Pfizer).
Tau Imaging Agent** (Q3 2015)—a positron emission tomography (PET) tracer intended to image tau (or neurofibrillary) tangles in the brain, which are an indicator of Alzheimer's disease.
* | Biologic molecule subject to the United States (U.S.) Biologics Price Competition and Innovation Act |
** | Diagnostic agent |
The following table reflects the status of each NME and diagnostic agent within our late-stage pipeline including developments since January 1, 2015:
Compound | Indication | U.S. | Europe | Japan | Developments |
Cardiovascular | |||||
Evacetrapib | High-risk vascular disease | Terminated | Announced decision to discontinue further development in October 2015. | ||
32
Compound | Indication | U.S. | Europe | Japan | Developments |
Endocrinology | |||||
Basal insulin peglispro | Type 1 diabetes | Terminated | Announced decision to discontinue further development in December 2015. | ||
Type 2 diabetes | Terminated | ||||
Intranasal glucagon | Severe hypoglycemia | Phase III | Acquired worldwide rights to intranasal glucagon in October 2015. See Note 3 to the consolidated financial statements for information on the acquisition. | ||
Jardiance® | Type 1 diabetes | Phase III | Initiated Phase III study in July 2015. | ||
Type 2 diabetes | Launched | Launched in first quarter of 2015 in Japan. In first quarter of 2016, the U.S. Food and Drug Administration (FDA) accepted data from long-term clinical trial investigating cardiovascular (CV) outcomes in adults with type 2 diabetes at high risk for CV events. Also submitted CV data to European regulatory authorities in fourth quarter of 2015. Glyxambi®, combination tablet of empagliflozin and linagliptin, approved in the U.S. and launched in first quarter of 2015. Submitted to European regulatory authorities in fourth quarter of 2015. Synjardy®, combination tablet of empagliflozin and metformin hydrochloride, approved and launched in Europe in second and third quarters of 2015, respectively. Approved and launched in the U.S. in third and fourth quarters of 2015, respectively. | |||
Basaglar® (new insulin glargine product) | Type 1 diabetes | Approved | Launched | First launch in Europe and Japan in second and third quarter of 2015, respectively. Approved in the U.S. in fourth quarter of 2015. See Note 4 to the consolidated financial statements for information on the U.S. approval. | |
Type 2 diabetes | Approved | Launched | |||
Trulicity® | Type 2 diabetes | Launched | Launched in certain European countries in first quarter of 2015. In Japan, approved and launched in third quarter of 2015. | ||
33
Compound | Indication | U.S. | Europe | Japan | Developments |
Immunology | |||||
Baricitinib | Rheumatoid arthritis | Submitted | Phase III | Announced in February, September, and October 2015 top-line results of three Phase III trials which all met primary endpoints. Submitted to regulatory authorities in the U.S. and Europe in first quarter of 2016. | |
Ixekizumab | Psoriasis | Submitted | Submitted to regulatory authorities in the U.S., Europe, and Japan in first, second, and third quarter of 2015, respectively. | ||
Psoriatic arthritis | Phase III | Submitted | Announced in April 2015 top-line results of Phase III trial which met primary endpoints. Submitted to regulatory authorities in Japan in third quarter of 2015. | ||
Neuroscience | |||||
CGRP monoclonal antibody | Cluster headache | Phase III | Initiated first Phase III study in June 2015. Granted Fast Track Designation(1) from the FDA in June 2015. | ||
Migraine prevention | Phase III | Initiated Phase III study in January 2016. | |||
Solanezumab | Preclinical Alzheimer's disease | Phase III | Phase III study in asymptomatic Alzheimer's disease is ongoing. | ||
Mild Alzheimer's disease | Phase III | Enrollment in the ongoing Phase III study completed. In July 2015, announced clinical trial results from previous Phase III studies indicating the treatment effect was preserved in patients with mild Alzheimer's disease who received solanezumab earlier in disease, compared to patients beginning treatment at later point. | |||
Tanezumab | Osteoarthritis pain | Phase III | FDA clinical hold lifted in March 2015. Certain Phase III studies resumed in July 2015. | ||
Chronic low back pain | Phase III | ||||
Cancer pain | Phase III | ||||
Tau imaging agent | Alzheimer's disease | Phase III | Initiated Phase III study in September 2015. | ||
34
Compound | Indication | U.S. | Europe | Japan | Developments |
Oncology | |||||
Abemaciclib | Metastatic breast cancer | Phase III | Phase III studies are ongoing. Announced that abemaciclib was granted Breakthrough Therapy Designation(2) by the FDA. | ||
NSCLC | Phase III | Phase III study is ongoing. | |||
Cyramza® | Gastric cancer (first-line) | Phase III | Initiated Phase III study in January 2015. | ||
Gastric cancer (second-line) | Launched | Launched in certain European countries in first quarter of 2015. In Japan, approved in first quarter of 2015 and launched in second quarter of 2015. | |||
NSCLC (first-line) | Phase III | Initiated Phase III study in May 2015. | |||
NSCLC (second-line) | Launched | Submitted | Launched in the U.S. in first quarter of 2015. Submitted in Japan in third quarter of 2015. Approved in Europe and launched in certain European countries in first quarter of 2016. | ||
Liver cancer (second-line) | Phase III | Initiated Phase III study in July 2015. | |||
Metastatic colorectal cancer (second-line) | Launched | Submitted | Approved and launched in the U.S. in second quarter of 2015. Submitted in Japan in second quarter of 2015. Approved in Europe and launched in certain European countries in first quarter of 2016. | ||
Urothelial (bladder) cancer (second-line) | Phase III | Initiated Phase III study in July 2015. | |||
Olaratumab | Soft tissue sarcoma | Phase III | Announced that olaratumab was granted Breakthrough Therapy Designation(2) by the FDA. In third quarter of 2015, announced intention to submit U.S. and European regulatory applications based on Phase II clinical trial data. Initiated a rolling submission to FDA in fourth quarter of 2015. Submission to European regulatory authorities expected in 2016. Initiated Phase III study of olaratumab in soft tissue sarcoma in September 2015. | ||
Portrazza | Metastatic squamous NSCLC (first-line) | Launched | Approved | Phase Ib/II | Approved and launched in the U.S. in fourth quarter of 2015. Approved in Europe in first quarter of 2016. |
(1) The FDA Fast Track designation is designed to facilitate the development, and expedite the review, of drugs which treat a serious or life-threatening condition and fill an unmet medical need.
(2) The Breakthrough Therapy Designation is designed to expedite the development and review of potential medicines that are intended to treat a serious condition where preliminary clinical evidence indicates that the treatment may demonstrate substantial improvement over available therapy on a clinically significant endpoint.
35
There are many difficulties and uncertainties inherent in pharmaceutical research and development and the introduction of new products. A high rate of failure is inherent in new drug discovery and development. The process to bring a drug from the discovery phase to regulatory approval can take over a decade and cost more than $1 billion. Failure can occur at any point in the process, including late in the process after substantial investment. As a result, most research programs will not generate financial returns. New product candidates that appear promising in development may fail to reach the market or may have only limited commercial success. Delays and uncertainties in the regulatory approval processes in the U.S. and in other countries can result in delays in product launches and lost market opportunities. Consequently, it is very difficult to predict which products will ultimately be approved.
We manage research and development spending across our portfolio of molecules, and a delay in, or termination of, any one project will not necessarily cause a significant change in our total research and development spending. Due to the risks and uncertainties involved in the research and development process, we cannot reliably estimate the nature, timing, completion dates, and costs of the efforts necessary to complete the development of our research and development projects, nor can we reliably estimate the future potential revenue that will be generated from a successful research and development project. Each project represents only a portion of the overall pipeline, and none is individually material to our consolidated research and development expense. While we do accumulate certain research and development costs on a project level for internal reporting purposes, we must make significant cost estimations and allocations, some of which rely on data that are neither reproducible nor validated through accepted control mechanisms. Therefore, we do not have sufficiently reliable data to report on total research and development costs by project, by preclinical versus clinical spend, or by therapeutic category.
Other Matters
Novartis Animal Health Acquisition
On January 1, 2015, we completed our acquisition of Novartis AH in an all-cash transaction for $5.28 billion. Novartis AH operates in approximately 40 countries. We acquired Novartis AH’s nine manufacturing sites, six dedicated research and development facilities, a global commercial infrastructure with a portfolio of approximately 600 products, a pipeline with more than 40 projects in development, and more than 3,000 employees. The combined organization has increased our animal health product portfolio, expanded our global commercial presence, and augmented our animal health manufacturing and research and development. In particular, it has provided Elanco with a greater commercial presence in the companion animal and swine markets, expanded Elanco’s presence in equine and vaccines areas, and created an entry into the aquaculture market. As a condition to the clearance of the transaction under the Hart-Scott-Rodino Antitrust Improvement Act, following the closing of the acquisition of Novartis AH, we divested certain companion animal assets in the U.S. related to the Sentinel® canine parasiticide franchise to Virbac Corporation for approximately $410 million. The Novartis AH business we retained generated revenue of approximately $1.1 billion in 2014.
36
Patent Matters
We depend on patents or other forms of intellectual-property protection for most of our revenues, cash flows, and earnings. The loss of U.S. patent exclusivity for Cymbalta® in December 2013 and Evista® in March 2014, resulted in the immediate entry of generic competitors and a rapid and severe decline in revenue from the affected products, having, in the aggregate, a material adverse effect on our consolidated results of operations and cash flows.
We lost our data package protection for Cymbalta in major European countries in 2014. In 2015, we saw the entry of generic competition in all major European markets. The loss of exclusivity for Cymbalta in the European markets has caused a rapid and severe decline in revenue for the product, which over time will, in the aggregate, have a material adverse effect on our consolidated results of operations and cash flows. We also lost patent exclusivity for the schizophrenia indication in December 2015 for Zyprexa® in Japan. We will lose our patent protection for the bipolar mania indication in April 2016 for Zyprexa in Japan. Generic versions of Zyprexa were approved in Japan in February 2016. We cannot speculate whether the generic company will apply for pricing and proceed to launch.
Additionally, as described in Note 15 to the consolidated financial statements, the Alimta® vitamin regimen patent, which provides us with patent protection for Alimta through June 2021 in Japan and major European countries, and through May 2022 in the U.S., has been challenged in each of these jurisdictions. Our compound patent for Alimta will expire in the U.S. in January 2017, and expired in major European countries and Japan in December 2015. We expect that the entry of generic competition for Alimta into these markets following the loss of effective patent protection will cause a rapid and severe decline in revenue for the product, which will, in the aggregate, have a material adverse effect on our consolidated results of operations and cash flows. We are aware that a generic competitor has received approval to market a generic version of Alimta in a major European country, although we are not aware of whether this competitor's product has entered the market. Notwithstanding our patents, generic versions of Alimta were approved in Japan in February 2016. We filed preliminary injunctions against four generic competitors. We do not anticipate generic competitors to proceed to launch prior to the completion of the Sawai invalidation trial, as described in Note 15 to the consolidated financial statements.
The U.S. compound patent for Humalog® expired in 2013. Thus far, the loss of compound patent protection for Humalog has not resulted in a rapid and severe decline in revenue. Global regulators have different legal pathways to approve similar versions of Humalog and to date none have been approved in the U.S. or Europe. We are aware that other manufacturers have efforts underway to develop a similar version of Humalog, and it is difficult to predict the likelihood, timing, and impact of these products entering the market.
Foreign Currency Exchange Rates
As a global company with substantial operations outside the U.S., we face foreign currency risk exposure from fluctuating currency exchange rates, primarily the U.S. dollar against the euro, Japanese yen, and British pound, and the British pound against the euro. While we manage a portion of these exposures through hedging and other risk management techniques, significant fluctuations in currency rates can have a substantial impact, either positive or negative, on our revenue, cost of sales, and operating expenses. Over the past two years, we have seen significant foreign currency rate fluctuations as the U.S. dollar strengthened compared to several other foreign currencies, including the euro, British pound, and Japanese yen. While there is uncertainty in the future movements in foreign exchange rates, these fluctuations could negatively impact our future consolidated results of operations.
37
Trends Affecting Pharmaceutical Pricing, Reimbursement, and Access
United States
In the U.S., public concern over prices for specialty and brand name pharmaceuticals continues to drive the legislative debate. These policy and political issues increase the risk that taxes, fees, rebates or other federal and state measures may be enacted. Key health policy proposals affecting biopharmaceuticals include a reduction in biologic data exclusivity, modifications to Medicare Parts B and D, new language that would allow the Department of Health and Human Services to negotiate prices for biologics and drugs on the specialty tier in Part D, and state-level proposals to reduce the cost of pharmaceuticals purchased by government health care programs. Savings projected under these proposals are targeted as a means to fund both health care expenditures and non-health care initiatives, or to manage federal and state budgets.
In the U.S. private sector, consolidation and integration among U.S. healthcare providers is also a major factor in the competitive marketplace for human pharmaceuticals. Health plans and pharmaceutical benefit managers have been consolidating into fewer, larger entities, thus enhancing their purchasing strength and importance. Payers typically maintain formularies which specify coverage (the conditions under which drugs are included on a plan's formulary) and reimbursement (the associated out-of-pocket cost to the consumer). Formulary placement can lead to reduced usage of a drug for the relevant patient population due to coverage restrictions, such as prior authorizations and formulary exclusions, or due to reimbursement limitations which result in higher consumer out-of-pocket cost, such as non-preferred co-pay tiers, increased co-insurance levels and higher deductibles. Consequently, pharmaceutical companies compete for formula placement not only on the basis of product attributes such as greater efficacy, fewer side effects, or greater patient ease of use, but also by providing rebates. Price is an increasingly important factor in formulary decisions, particularly in treatment areas in which the payer has taken the position that multiple branded products are therapeutically comparable. These downward pricing pressures could negatively affect future consolidated results of operations.
The main coverage expansion provisions of the Affordable Care Act (ACA) are now in effect through both the launch of state-based exchanges and the expansion of Medicaid. An emerging trend has been the prevalence of benefit designs containing high out-of-pocket costs for patients, particularly for pharmaceuticals. In addition to the coverage expansions, many employers in the commercial market, driven in part by ACA changes such as the 2020 implementation of the excise tax on employer-sponsored health care coverage for which there is an excess benefit (the so-called "Cadillac tax"), continue to evaluate strategies such as private exchanges and wider use of consumer-driven health plans to reduce their healthcare liabilities over time. At the same time, the broader paradigm shift towards quality-based reimbursement and the launch of several value-based purchasing initiatives have placed demands on the pharmaceutical industry to offer products with proven real-world outcomes data and a favorable economic profile.
International
International operations also are generally subject to extensive price and market regulations. Cost-containment measures exist in a number of countries, including additional price controls and mechanisms to limit reimbursement for our products. Such policies are expected to increase in impact and reach, given the pressures on national and regional health care budgets that come from a growing aging population and ongoing economic challenges. In addition, governments in many emerging markets are becoming increasingly active in expanding health care system offerings. Given the budget challenges of increasing health care coverage for citizens, policies may be proposed that promote generics only and reduce current and future access to human pharmaceutical products.
38
Tax Matters
We are subject to income taxes in the U.S. and numerous foreign jurisdictions. Changes in the relevant tax laws, regulations, administrative practices, principles, and interpretations could adversely affect our future effective tax rates. The U.S. and a number of other countries are actively considering or enacting changes in this regard. For example, the Obama administration proposed changes to the manner in which the U.S. would tax the international income of U.S.-based companies, including unremitted earnings of foreign subsidiaries. Other tax proposals under discussion or introduced in the U.S. Congress could change the tax rate and manner in which U.S. companies would be taxed. Additionally, the Organisation for Economic Co-operation and Development issued its final recommendations of international tax reform proposals to influence international tax policy in major countries in which we operate. While outcomes of these initiatives continue to develop and remain uncertain, changes to key elements of the U.S. or international tax framework could have a material adverse effect on our consolidated operating results and cash flows.
Operating Results—2015
Revenue
The following table summarizes our revenue activity by jurisdiction:
Year Ended, December 31, | Change in | |||||||||||||
2015 | 2014 | Dollars | Percent | |||||||||||
U.S. (1) | $ | 10,097.4 | $ | 9,134.1 | $ | 963.3 | 11 | % | ||||||
Outside U.S. | 9,861.3 | 10,481.5 | (620.2 | ) | (6 | )% | ||||||||
Revenue | $ | 19,958.7 | $ | 19,615.6 | $ | 343.1 | 2 | % | ||||||
Numbers may not add due to rounding.
(1) U.S. revenue includes revenue in Puerto Rico.
The following are components of the change in revenue compared to the prior year:
2015 vs. 2014 | ||||||
U.S. | Outside U.S. | Consolidated | ||||
Volume | 6 | % | 9 | % | 8 | % |
Price | 5 | % | (2 | )% | 1 | % |
Foreign exchange rates | — | % | (13 | )% | (7 | )% |
Percent change | 11 | % | (6 | )% | 2 | % |
Numbers may not add due to rounding.
In the U.S., the volume increase in 2015 was driven by the inclusion of revenue from Novartis AH and increased volumes for several pharmaceutical products, partially offset by the residual impact of the loss of exclusivity for Cymbalta and Evista.
Outside the U.S., the volume increase in 2015 was driven by the inclusion of revenue from Novartis AH and increased volumes for several pharmaceutical products.
On a pro forma basis, which reflects the 2014 revenues of Novartis AH as described in Note 3 to the consolidated financial statements, our consolidated volume in 2015 would have increased by 2 percent compared with 2014.
39
The following table summarizes our revenue activity in 2015 compared with 2014:
Year Ended | Year Ended | |||||||||||||||||
December 31, 2015 | December 31, 2014 | Percent Change from | ||||||||||||||||
Product | U.S.(1) | Outside U.S. | Total | Total | 2014 | |||||||||||||
Humalog | $ | 1,772.3 | $ | 1,069.6 | $ | 2,841.9 | $ | 2,785.2 | 2 | |||||||||
Alimta | 1,162.4 | 1,330.7 | 2,493.1 | 2,792.0 | (11 | ) | ||||||||||||
Cialis® | 1,256.8 | 1,053.9 | 2,310.7 | 2,291.0 | 1 | |||||||||||||
Forteo® | 612.4 | 735.9 | 1,348.3 | 1,322.0 | 2 | |||||||||||||
Humulin® | 764.4 | 543.0 | 1,307.4 | 1,400.1 | (7 | ) | ||||||||||||
Cymbalta | 144.6 | 883.0 | 1,027.6 | 1,614.7 | (36 | ) | ||||||||||||
Zyprexa | 156.7 | 783.6 | 940.3 | 1,037.3 | (9 | ) | ||||||||||||
Strattera® | 502.1 | 281.9 | 784.0 | 738.5 | 6 | |||||||||||||
Effient® | 417.6 | 105.4 | 523.0 | 522.2 | — | |||||||||||||
Cyramza | 277.7 | 106.1 | 383.8 | 75.6 | NM | |||||||||||||
Trulicity | 207.7 | 41.0 | 248.7 | 10.2 | NM | |||||||||||||
Evista | 61.7 | 175.6 | 237.3 | 419.8 | (43 | ) | ||||||||||||
Other pharmaceutical products(2) | 738.4 | 785.1 | 1,523.5 | 1,472.0 | 3 | |||||||||||||
Animal health products | 1,541.2 | 1,639.8 | 3,181.0 | 2,346.6 | 36 | |||||||||||||
Total net product revenue | 9,616.0 | 9,534.6 | 19,150.6 | 18,827.2 | 2 | |||||||||||||
Collaboration and other revenue(3) | 481.4 | 326.7 | 808.1 | 788.4 | 2 | |||||||||||||
Revenue | $ | 10,097.4 | $ | 9,861.3 | $ | 19,958.7 | $ | 19,615.6 | 2 | |||||||||
(1) | U.S. revenue includes revenue in Puerto Rico. |
(2) Other pharmaceutical products includes revenue of $175.6 million and $46.1 million in 2015 and 2014, respectively, for Erbitux®. The 2015 revenue is primarily associated with net product revenue from third parties subsequent to the transfer of commercialization rights in the U.S. and Canada (collectively, North America) from Bristol-Myers Squibb Company and E.R. Squibb (collectively, BMS) to us in the fourth quarter. See Note 4 to the consolidated financial statements.
(3) | Collaboration and other revenue consists primarily of revenue associated with Trajenta® (which includes Jentadueto®) of $356.8 million and $328.8 million in 2015 and 2014, respectively, as well as royalties for Erbitux prior to the transfer of commercialization of rights in North America from BMS to us of $309.4 million and $327.2 million in 2015 and 2014, respectively. See Note 4 to the consolidated financial statements. |
NM - not meaningful
Revenues of Humalog, our injectable human insulin analog for the treatment of diabetes, increased 9 percent in the U.S., driven by higher realized prices and, to a lesser extent, increased volume. Revenues outside the U.S. decreased 8 percent, driven by the unfavorable impact of foreign exchange rates, partially offset by higher volume.
Revenues of Alimta, a treatment for various cancers, decreased 5 percent in the U.S., driven by decreased demand and, to a lesser extent, lower realized prices. Revenues outside the U.S. decreased 15 percent, driven by the unfavorable impact of foreign exchange rates and, to a lesser extent, lower realized prices, partially offset by increased volume.
Revenues of Cialis, a treatment for erectile dysfunction and benign prostatic hyperplasia, increased 21 percent in the U.S., driven by higher realized prices. Revenues outside the U.S. decreased 16 percent, driven by the unfavorable impact of foreign exchange rates.
Revenues of Forteo, an injectable treatment for osteoporosis in postmenopausal women and men at high risk for fracture and for glucocorticoid-induced osteoporosis in men and postmenopausal women, increased 14 percent in the U.S., driven by higher realized prices, partially offset by decreased volume. Revenues outside the U.S. decreased 6 percent, driven by the unfavorable impact of foreign exchange rates, partially offset by increased volume.
40
Revenues of Humulin, an injectable human insulin for the treatment of diabetes, increased 7 percent in the U.S., driven by higher realized prices and, to a lesser extent, wholesaler buying patterns, partially offset by decreased demand. Revenues outside the U.S. decreased 21 percent, driven by decreased volume, primarily due to the loss of a government contract in Brazil, and the unfavorable impact of foreign exchange rates.
Revenues of Cymbalta, a product for the treatment of major depressive disorder, diabetic peripheral neuropathic pain, generalized anxiety disorder, chronic musculoskeletal pain, and the management of fibromyalgia, decreased 66 percent in the U.S. due to the loss of U.S. patent exclusivity in December 2013. Revenues outside the U.S. decreased 26 percent, driven by the unfavorable impact of foreign exchange rates and the loss of exclusivity in Europe in 2014.
Revenues of Zyprexa, a treatment for schizophrenia, acute mixed or manic episodes associated with bipolar I disorder, and bipolar maintenance, increased 31 percent in the U.S., driven by adjustments to the return reserve resulting from the expiration of the period to return expired product for credit. Revenues outside the U.S. decreased 15 percent, driven primarily by the unfavorable impact of foreign exchange rates. We lost patent exclusivity for Zyprexa in Japan in December 2015. Zyprexa revenues in Japan were $415.9 million in 2015, compared with $466.2 million in 2014. The revenue decrease in Japan was due to the unfavorable impact of foreign exchange rates.
Revenues of Strattera, a treatment for attention-deficit hyperactivity disorder, increased 11 percent in the U.S., driven by higher realized prices and, to a lesser extent, increased demand. Revenues outside the U.S. decreased 1 percent, driven by the unfavorable impact of foreign exchange rates, largely offset by increased volume.
Revenues of Effient, a product for the reduction of thrombotic cardiovascular events (including stent thrombosis) in patients with acute coronary syndrome who are managed with an artery-opening procedure known as percutaneous coronary intervention, including patients undergoing angioplasty, atherectomy, or stent placement, increased 6 percent in the U.S., driven by higher realized prices, partially offset by decreased demand. Revenues outside the U.S. decreased 17 percent, driven primarily by the unfavorable impact of foreign exchange rates.
Revenues of Evista, a product for the prevention and treatment of osteoporosis in postmenopausal women and for reduction of risk of invasive breast cancer in postmenopausal women with osteoporosis and postmenopausal women at high risk for invasive breast cancer, decreased 70 percent in the U.S., due to the loss of patent exclusivity in March 2014. Revenues outside the U.S. decreased 17 percent, driven primarily by the unfavorable impact of foreign exchange rates.
Revenues of animal health products in the U.S. increased 21 percent and animal health product revenues outside the U.S. increased 53 percent. The increases were driven by the inclusion of revenue from Novartis AH.
On a pro forma basis, which reflects the 2014 revenues of Novartis AH as described in Note 3 to the consolidated financial statements, revenues of animal health products in the U.S. would have decreased 1 percent, driven primarily by decreased volume in food animal products. Revenues outside the U.S. would have decreased 13 percent, driven by the unfavorable impact of foreign exchange rates and decreased volume in companion animal products, partially offset by higher realized prices and volume for food animal products.
Gross Margin, Costs, and Expenses
Gross margin as a percent of total revenue was 74.8 percent in 2015, essentially flat compared with 2014 as the unfavorable impacts of the inclusion of Novartis AH and inventory step-up and amortization costs were offset by the favorable impact of foreign exchange rates on international inventories sold.
Research and development expenses increased 1 percent to $4.80 billion in 2015, driven primarily by higher late-stage clinical development costs, the inclusion of Novartis AH, and an increase in charges associated with the termination of late-stage molecules, primarily evacetrapib and basal insulin peglispro, of approximately $135 million, partially offset by the favorable impact of foreign exchange rates.
41
Marketing, selling, and administrative expenses decreased 1 percent to $6.53 billion in 2015, due to the favorable impact of foreign exchange rates and a 2014 charge associated with the U.S. Drug Fee, partially offset by the inclusion of Novartis AH and expenses related to new product launches.
We recognized acquired IPR&D charges of $535.0 million in 2015 resulting from various collaboration agreements, primarily with Pfizer, as well as the consideration paid to acquire the worldwide rights to Locemia's intranasal glucagon. There were $200.2 million of acquired IPR&D charges in 2014 related to various collaboration agreements, including charges associated with the transfer of commercial rights to us, from Boehringer Ingelheim, of the new insulin glargine product in certain countries where it was not yet approved. See Notes 3 and 4 to the consolidated financial statements for additional information.
We recognized asset impairment, restructuring, and other special charges of $367.7 million in 2015. The charges relate to severance costs, integration costs for Novartis AH, and asset impairments. In 2014, we recognized charges of $468.7 million for asset impairment, restructuring, and other special charges. The charges included severance costs, asset impairments primarily associated with the closure of a manufacturing site in Puerto Rico, and integration costs for the then-pending acquisition of Novartis AH. See Note 5 to the consolidated financial statements for additional information.
Other—net, (income) expense was income of $100.6 million in 2015, compared with income of $340.5 million in 2014. Other income in 2015 included net gains of $236.7 million on investments, partially offset by a net charge of $152.7 million related to the repurchase of $1.65 billion of debt. Other income in 2014 included net gains of $216.4 million on investments and $92.0 million of income associated with the transfer of commercial rights to linagliptin and empagliflozin in certain countries from us to Boehringer Ingelheim. See Notes 4 and 17 to the consolidated financial statements for additional information.
Our effective tax rate was 13.7 percent in 2015, compared with 20.3 percent in 2014. The effective tax rate for 2014 reflects the impact of a $119.0 million nondeductible charge associated with the U.S. Drug Fee. The decrease in the tax rate for 2015 compared with 2014 is primarily due to a favorable tax impact of the net charges related to the repurchase of debt, acquired IPR&D, and asset impairment, restructuring, and other special charges. See Note 13 to the consolidated financial statements for additional information.
Operating Results—2014
Financial Results
The following table summarizes our key operating results:
Year Ended, December 31, | Percent Change from | |||||||||
2014 | 2013 | 2013 | ||||||||
Revenue | $ | 19,615.6 | $ | 23,113.1 | (15 | )% | ||||
Gross margin | 14,683.1 | 18,205.0 | (19 | )% | ||||||
Gross margin as percent of revenue | 74.9 | % | 78.8 | % | ||||||
Operating expense (1) | $ | 11,354.4 | $ | 12,656.9 | (10 | )% | ||||
Acquired in-process research and development | 200.2 | 57.1 | NM | |||||||
Asset impairment, restructuring, and other special charges | 468.7 | 120.6 | NM | |||||||
Net income | 2,390.5 | 4,684.8 | (49 | )% | ||||||
Earnings per share | 2.23 | 4.32 | (48 | )% | ||||||
(1) Operating expense consists of research and development and marketing, selling, and administrative expenses.
NM - not meaningful
Revenue and gross margin decreased in 2014. The decrease in operating expense in 2014 was due to decreases in both research and development and marketing, selling, and administrative expenses. The decreases in net income and EPS for 2015 were due to lower gross margin, higher asset impairment, restructuring, and other special charges, and decreased other income, partially offset by lower operating expenses, and income tax expense.
42
Certain items affect the comparisons of our 2014 and 2013 results. The 2014 highlighted items are summarized in the "Results of Operations—Executive Overview" section. The 2013 highlighted items are summarized as follows:
Acquired IPR&D (Note 3 to the consolidated financial statements)
• | We recognized acquired IPR&D charges of $57.1 million (pretax), or $0.03 per share, resulting from our acquisition of rights for a CGRP monoclonal antibody (see "Results of Operations—Executive Overview—Late-Stage Pipeline" section). |
Collaborations (Note 4 to the consolidated financial statements)
• | We recognized income of $495.4 million (pretax), or $0.29 per share, related to the transfer to Amylin Pharmaceuticals, Inc. (Amylin) of exenatide commercial rights in all markets outside the U.S. |
Asset Impairment, Restructuring, and Other Special Charges (Note 5 to the consolidated financial statements)
• | We recognized charges of $120.6 million (pretax), or $0.08 per share, primarily related to severance costs, as well as asset impairment costs associated with the closure of a packaging and distribution facility in Germany. |
Revenue
The following table summarizes our revenue activity by jurisdiction:
Year Ended, December 31, | Change in | |||||||||||||
2014 | 2013 | Dollars | Percent | |||||||||||
U.S. (1) | $ | 9,134.1 | $ | 12,889.7 | $ | (3,755.6 | ) | (29 | )% | |||||
Outside U.S. | 10,481.5 | 10,223.4 | 258.1 | 3 | % | |||||||||
Revenue | $ | 19,615.6 | $ | 23,113.1 | $ | (3,497.5 | ) | (15 | )% | |||||
Numbers may not add due to rounding.
(1) U.S. revenue includes revenue in Puerto Rico.
The following are components of the change in revenue compared to the prior year:
2014 vs. 2013 | ||||||
U.S. | Outside U.S. | Consolidated | ||||
Volume | (28 | )% | 7 | % | (13 | )% |
Price | (1 | )% | (1 | )% | (1 | )% |
Foreign exchange rates | — | % | (3 | )% | (2 | )% |
Percent change | (29 | )% | 3 | % | (15 | )% |
Numbers may not add due to rounding.
In the U.S., the volume decrease in 2014 was due to lower demand for Cymbalta and Evista following patent expirations, and to a lesser extent, to wholesaler buying patterns.
43
The following table summarizes our revenue activity in 2014 compared with 2013:
Year Ended | Year Ended | |||||||||||||||||
December 31, 2014 | December 31, 2013 | Percent Change from | ||||||||||||||||
Product | U.S.(1) | Outside U.S. | Total | Total | 2013 | |||||||||||||
Alimta | $ | 1,229.5 | $ | 1,562.5 | $ | 2,792.0 | $ | 2,703.0 | 3 | |||||||||
Humalog | 1,627.6 | 1,157.6 | 2,785.2 | 2,611.2 | 7 | |||||||||||||
Cialis | 1,039.9 | 1,251.1 | 2,291.0 | 2,159.4 | 6 | |||||||||||||
Cymbalta | 420.5 | 1,194.2 | 1,614.7 | 5,084.4 | (68 | ) | ||||||||||||
Humulin | 713.1 | 687.0 | 1,400.1 | 1,315.8 | 6 | |||||||||||||
Forteo | 539.0 | 783.0 | 1,322.0 | 1,244.9 | 6 | |||||||||||||
Zyprexa | 119.8 | 917.5 | 1,037.3 | 1,194.8 | (13 | ) | ||||||||||||
Strattera | 452.5 | 286.0 | 738.5 | 709.2 | 4 | |||||||||||||
Effient | 394.5 | 127.7 | 522.2 | 508.7 | 3 | |||||||||||||
Evista | 207.2 | 212.6 | 419.8 | 1,050.4 | (60 | ) | ||||||||||||
Other pharmaceutical products | 647.5 | 910.3 | 1,557.8 | 1,672.3 | (7 | ) | ||||||||||||
Animal health products | 1,274.4 | 1,072.2 | 2,346.6 | 2,151.5 | 9 | |||||||||||||
Total net product revenue | 8,665.5 | 10,161.7 | 18,827.2 | 22,405.6 | (16 | ) | ||||||||||||
Collaboration and other revenue(2) | 468.6 | 319.8 | 788.4 | 707.5 | 11 | |||||||||||||
Revenue | $ | 9,134.1 | $ | 10,481.5 | $ | 19,615.6 | $ | 23,113.1 | (15 | ) | ||||||||
(1) U.S. revenue includes revenue in Puerto Rico.
(2) Collaboration and other revenue consists primarily of royalties for Erbitux and revenue associated with Trajenta.
Revenues of Alimta increased 2 percent in the U.S., driven by increased volume. Revenues outside the U.S. increased 5 percent, driven by increased volume, partially offset by the unfavorable impact of foreign exchange rates and lower realized prices.
Revenues of Humalog increased 7 percent in the U.S., driven by increased demand, partially offset by lower realized prices as a result of payer contracts and greater Medicaid and Medicare utilization, as well as wholesaler buying patterns. Revenues outside the U.S. increased 6 percent, driven by increased volume and, to a lesser extent, higher realized prices, partially offset by the unfavorable impact of foreign exchange rates.
Revenues of Cialis increased 10 percent in the U.S., driven by higher realized prices, partially offset by wholesaler buying patterns. Revenues outside the U.S. increased 3 percent, driven by higher realized prices and increased volume, partially offset by the unfavorable impact of foreign exchange rates.
Revenues of Cymbalta decreased 89 percent in the U.S. due to the loss of U.S. patent exclusivity in December 2013. Revenues outside the U.S. increased 6 percent, driven by increased volume, partially offset by the unfavorable impact of foreign exchange rates.
Revenues of Humulin increased 5 percent in the U.S., primarily driven by increased demand, partially offset by wholesaler buying patterns. Revenues outside the U.S. increased 8 percent, driven by increased volume, partially offset by the unfavorable impact of foreign exchange rates.
Revenues of Forteo increased 5 percent in the U.S., driven by higher realized prices, partially offset by decreased volume. Revenues outside the U.S. increased 7 percent, driven by increased volume, primarily in Japan, partially offset by the unfavorable impact of foreign exchange rates, primarily the Japanese yen.
Revenues of Zyprexa decreased 3 percent in the U.S. Revenues outside the U.S. decreased 14 percent, driven by decreased volume, the unfavorable impact of foreign exchange rates, primarily the Japanese yen, and lower realized prices.
Revenues of Strattera increased 1 percent in the U.S., driven by higher realized prices, partially offset by decreased volume. Revenues outside the U.S. increased 9 percent, driven by increased volume, primarily in Japan, partially offset by the unfavorable impact of foreign exchange rates, primarily the Japanese yen.
44
Revenues of Effient increased 5 percent in the U.S., driven by higher realized prices, partially offset by wholesaler buying patterns. Revenues outside the U.S. decreased 3 percent, driven by lower volume.
Revenues of Evista decreased 73 percent in the U.S., due to the loss of U.S. patent exclusivity in March 2014. Revenues outside the U.S. decreased 24 percent, driven primarily by the expiration of a supply agreement in 2013, and to a lesser extent the unfavorable impact of foreign exchange rates.
Animal health product revenues in the U.S. increased 4 percent, driven by increased volume in food animal products and higher realized prices, partially offset by decreased volume in companion animal products due to competitive pressure. Revenues outside the U.S. increased 16 percent, driven by increased volume in food animal products, due in part to the acquisition of Lohmann SE and, to a lesser extent, higher realized prices, partially offset by the unfavorable impact of foreign exchange rates.
Gross Margin, Costs, and Expenses
Gross margin as a percent of total revenue was 74.9 percent in 2014, a decrease of 3.9 percentage points compared with 2013, driven primarily by lower sales of Cymbalta and Evista following U.S. patent expirations.
Research and development expenses decreased 14 percent to $4.73 billion in 2014, driven primarily by lower late-stage clinical development costs. Research and development expenses in 2013 included $97.2 million of milestone payments made to Boehringer Ingelheim following regulatory submissions for empagliflozin.
Marketing, selling, and administrative expenses decreased 7 percent to $6.62 billion in 2014, driven primarily by the reduction in U.S. sales and marketing activities for Cymbalta and Evista, as well as ongoing cost containment efforts, partially offset by an additional $119.0 million charge in 2014 associated with the U.S. Drug Fee, an annual non-tax deductible fee enacted by the Patient Protection and Affordable Care Act that is imposed on us and others engaged in the business of manufacturing or importing branded prescription drugs. The final regulations issued by the IRS in 2014, accelerated the expense recognition criteria for the fee obligation by one year, from the year in which the fee is paid to the year in which the sales used to calculate the fee occur. This change resulted in the need to expense two years of the U.S. Drug Fee in 2014 to account for the fee imposed and paid in 2014 and the fee that would be imposed and paid in 2015.
We recognized acquired IPR&D charges of $200.2 million in 2014 resulting from our collaboration agreements with Adocia, AstraZeneca UK Limited, and Immunocore Limited in addition to charges associated with the transfer of commercial rights to us, from Boehringer Ingelheim, of the new insulin glargine product in certain countries where it was not yet approved. There were $57.1 million of acquired IPR&D charges in 2013 related to the acquisition of rights for the CGRP antibody. See Notes 3 and 4 to the consolidated financial statements for additional information.
We recognized asset impairment, restructuring, and other special charges of $468.7 million in 2014. These charges included $225.5 million of severance costs and $243.2 million of asset impairment and other special charges consisting primarily of a $180.8 million asset impairment charge related to our decision to close and sell a manufacturing plant located in Puerto Rico. In 2013, we recognized asset impairment, restructuring, and other special charges of $120.6 million. These charges included $30.0 million of asset impairments primarily associated with the closure of a packaging and distribution facility in Germany and $90.6 million of severance costs. See Note 5 to the consolidated financial statements for additional information.
Other—net, (income) expense was income of $340.5 million in 2014, compared with income of $518.9 million in 2013. Other income in 2014 included net gains of $216.4 million on investments and $92.0 million of income related to the transfer of commercial rights to linagliptin and empagliflozin in certain countries from us to Boehringer Ingelheim. Other income in 2013 was primarily comprised of $495.4 million related to the termination of the exenatide collaboration with Amylin. See Notes 4 and 17 to the consolidated financial statements for additional information.
Our effective tax rate was 20.3 percent in 2014, compared with 20.5 percent in 2013. See Note 13 to the consolidated financial statements for additional information.
45
FINANCIAL CONDITION
As of December 31, 2015, cash and cash equivalents was $3.67 billion, a decrease of $205.2 million, compared with $3.87 billion at December 31, 2014. Refer to the Consolidated Statements of Cash Flows for additional details on the significant sources and uses of cash for the years ended December 31, 2015 and December 31, 2014.
In addition to our cash and cash equivalents, we held total investments of $4.43 billion and $5.52 billion as of December 31, 2015 and December 31, 2014, respectively. See Note 7 to the consolidated financial statements for additional details.
As of December 31, 2015, total debt was $7.98 billion, a slight decrease of $43.0 million compared with $8.02 billion at December 31, 2014. This decrease is due primarily to $2.68 billion of net repayments of short-term commercial paper borrowings, the repayment of $1.78 billion of fixed-rate notes in connection with the purchase and redemption of certain U.S. dollar-denominated notes in June 2015, and, to a lesser extent, the decrease in fair value of our hedged debt. These decreases were largely offset by the issuance of $4.45 billion of fixed-rate notes during 2015. At December 31, 2015, we had a total of $1.30 billion of unused committed bank credit facilities, $1.20 billion of which is available to support our commercial paper program. See Note 10 to the consolidated financial statements for additional details. We believe that amounts accessible through existing commercial paper markets should be adequate to fund short-term borrowing needs.
For the 130th consecutive year, we distributed dividends to our shareholders. Dividends of $2.00 per share and $1.96 per share were paid in 2015 and 2014, respectively. In the fourth quarter of 2015, effective for the dividend to be paid in the first quarter of 2016, the quarterly dividend was increased to $0.51 per share, resulting in an indicated annual rate for 2016 of $2.04 per share.
Capital expenditures of $1.07 billion during 2015 were $96.4 million less than in 2014. We expect 2016 capital expenditures to be approximately $1.1 billion.
In 2015, we repurchased $749.5 million of shares under the $5.00 billion share repurchase program previously announced in October 2013.
See "Results of Operations—Executive Overview—Other Matters" section for information regarding the actual or anticipated effect of losses of exclusivity for Cymbalta (U.S. and Europe), Evista (U.S.), Alimta (U.S., Europe, and Japan), and Zyprexa (Japan).
At December 31, 2015, we had an aggregate of $7.12 billion of cash and investments at our foreign subsidiaries. A significant portion of this amount would be subject to tax payments if such cash and investments were repatriated to the U.S. We record U.S. deferred tax liabilities for certain unremitted earnings, but when foreign earnings are expected to be indefinitely reinvested outside the U.S., no accrual for U.S. income taxes is provided. We believe cash provided by operating activities in the U.S. and planned repatriations of foreign earnings for which tax has been provided should be sufficient to fund our domestic operating needs, dividends paid to shareholders, share repurchases, and capital expenditures. Various risks and uncertainties, including those discussed in "Forward-Looking Statements" and Item 1A, “Risk Factors,” may, however, affect our operating results and cash generated from operations.
Both domestically and abroad, we continue to monitor the potential impacts of the economic environment; the creditworthiness of our wholesalers and other customers, including foreign government-backed agencies and suppliers; the uncertain impact of health care legislation; and various international government funding levels.
In the normal course of business, our operations are exposed to fluctuations in interest rates and currency values. These fluctuations can vary the costs of financing, investing, and operating. We address a portion of these risks through a controlled program of risk management that includes the use of derivative financial instruments. The objective of controlling these risks is to limit the impact on earnings of fluctuations in interest and currency exchange rates. All derivative activities are for purposes other than trading.
Our primary interest rate risk exposure results from changes in short-term U.S. dollar interest rates. In an effort to manage interest rate exposures, we strive to achieve an acceptable balance between fixed and floating rate debt positions and may enter into interest rate derivatives to help maintain that balance. Based on our overall interest rate exposure at December 31, 2015 and 2014, including derivatives and other interest rate risk-sensitive instruments, a hypothetical 10 percent change in interest rates applied to the fair value of the instruments as of December 31, 2015 and 2014, respectively, would not have a material impact on earnings, cash flows, or fair values of interest rate risk-sensitive instruments over a one-year period.
46
Our foreign currency risk exposure results from fluctuating currency exchange rates, primarily the U.S. dollar against the euro, Japanese yen, and British pound, and the British pound against the euro. We face foreign currency exchange exposures when we enter into transactions arising from subsidiary trade and loan payables and receivables denominated in foreign currencies. We also face currency exposure that arises from translating the results of our global operations to the U.S. dollar at exchange rates that have fluctuated from the beginning of the period. We may enter into foreign currency forward or option derivative contracts to reduce the effect of fluctuating currency exchange rates (principally the euro, the British pound, and the Japanese yen). Our corporate risk-management policy outlines the minimum and maximum hedge coverage of such exposures. Gains and losses on these derivative contracts offset, in part, the impact of currency fluctuations on the existing assets and liabilities. We periodically analyze the fair values of the outstanding foreign currency derivative contracts to determine their sensitivity to changes in foreign exchange rates. A hypothetical 10 percent change in exchange rates (primarily against the U.S. dollar) applied to the fair values of our outstanding foreign currency derivative contracts as of December 31, 2015 and 2014, would not have a material impact on earnings, cash flows, or financial position over a one-year period. This sensitivity analysis does not consider the impact that hypothetical changes in exchange rates would have on the underlying foreign currency denominated transactions.
Off-Balance Sheet Arrangements and Contractual Obligations
We have no off-balance sheet arrangements that have a material current effect or that are reasonably likely to have a material future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures, or capital resources. We acquire and collaborate on potential products still in development and enter into research and development arrangements with third parties that often require milestone and royalty payments to the third party contingent upon the occurrence of certain future events linked to the success of the asset in development. Milestone payments may be required contingent upon the successful achievement of an important point in the development life cycle of the pharmaceutical product (e.g., approval for marketing by the appropriate regulatory agency or upon the achievement of certain sales levels). If required by the arrangement, we may make royalty payments based upon a percentage of the sales of the pharmaceutical product in the event that regulatory approval for marketing is obtained. Because of the contingent nature of these payments, they are not included in the table of contractual obligations below.
Individually, these arrangements are not material in any one annual reporting period. However, if milestones for multiple products covered by these arrangements were reached in the same reporting period, the aggregate charge to expense could be material to the results of operations or cash flows in that period. See Note 4 to the consolidated financial statements for additional details. These arrangements often give us the discretion to unilaterally terminate development of the product, which would allow us to avoid making the contingent payments; however, we are unlikely to cease development if the compound successfully achieves milestone objectives. We also note that, from a business perspective, we view these payments as positive because they signify that the product is successfully moving through development and is now generating or is more likely to generate cash flows from sales of products.
47
Our current noncancelable contractual obligations that will require future cash payments are as follows:
Payments Due by Period | |||||||||||||||||||
(Dollars in millions) | Total | Less Than 1 Year | 1-3 Years | 3-5 Years | More Than 5 Years | ||||||||||||||
Long-term debt, including interest payments(1) | $ | 10,880.2 | $ | 192.0 | $ | 1,792.4 | $ | 939.2 | $ | 7,956.6 | |||||||||
Capital lease obligations | 15.8 | 4.9 | 8.5 | 2.4 | — | ||||||||||||||
Operating leases | 934.4 | 132.5 | 244.9 | 181.1 | 375.9 | ||||||||||||||
Purchase obligations(2) | 13,786.7 | 12,982.2 | 678.9 | 122.7 | 2.9 | ||||||||||||||
Other long-term liabilities reflected on our balance sheet(3) | 2,054.8 | — | 381.2 | 189.5 | 1,484.1 | ||||||||||||||
Total | $ | 27,671.9 | $ | 13,311.6 | $ | 3,105.9 | $ | 1,434.9 | $ | 9,819.5 | |||||||||
(1) Our long-term debt obligations include both our expected principal and interest obligations and our interest rate swaps. We used the interest rate forward curve at December 31, 2015, to compute the amount of the contractual obligation for interest on the variable rate debt instruments and swaps.
(2) We have included the following:
• | Purchase obligations consisting primarily of all open purchase orders as of December 31, 2015. Some of these purchase orders may be cancelable; however, for purposes of this disclosure, we have not distinguished between cancelable and noncancelable purchase obligations. |
• | Contractual payment obligations with each of our significant vendors, which are noncancelable and are not contingent. |
(3) We have included long-term liabilities consisting primarily of our nonqualified supplemental pension funding requirements and deferred compensation liabilities. We excluded long-term income taxes payable of $868.9 million, because we cannot reasonably estimate the timing of future cash outflows associated with those liabilities.
The contractual obligations table is current as of December 31, 2015. We expect the amount of these obligations to change materially over time as new contracts are initiated and existing contracts are completed, terminated, or modified.
APPLICATION OF CRITICAL ACCOUNTING ESTIMATES
In preparing our financial statements in accordance with accounting principles generally accepted in the U.S., we must often make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses, and related disclosures. Some of those judgments can be subjective and complex, and consequently actual results could differ from those estimates. For any given individual estimate or assumption we make, it is possible that other people applying reasonable judgment to the same facts and circumstances could develop different estimates. We believe that, given current facts and circumstances, it is unlikely that applying any such other reasonable judgment would cause a material adverse effect on our consolidated results of operations, financial position, or liquidity for the periods presented in this report. Our most critical accounting estimates have been discussed with our audit committee and are described below.
Revenue Recognition and Sales Return, Rebate, and Discount Accruals
We recognize revenue from sales of products at the time title of goods passes to the buyer and the buyer assumes the risks and rewards of ownership. Provisions for returns, rebates, and discounts are established in the same period the related sales are recorded.
48
Sales Returns - Background and Uncertainties
We regularly review the supply levels of our significant products sold to major wholesalers in the U.S. and in major markets outside the U.S., primarily by reviewing periodic inventory reports supplied by our major wholesalers and available prescription volume information for our products, or alternative approaches. We attempt to maintain U.S. wholesaler inventory levels at an average of approximately one month or less on a consistent basis across our product portfolio. Causes of unusual wholesaler buying patterns include actual or anticipated product-supply issues, weather patterns, anticipated changes in the transportation network, redundant holiday stocking, and changes in wholesaler business operations. In the U.S., the current structure of our arrangements does not provide an incentive for speculative wholesaler buying and provides us with data on inventory levels at our wholesalers. When we believe wholesaler purchasing patterns have caused an unusual increase or decrease in the sales of a major product compared with underlying demand, we disclose this in our product sales discussion if we believe the amount is material to the product sales trend; however, we are not always able to accurately quantify the amount of stocking or destocking in the retail channel. Wholesaler stocking and destocking activity historically has not caused any material changes in the rate of actual product returns.
When sales occur, we estimate a reserve for future product returns related to those sales. This estimate is based on several factors, including: historical return rates, expiration date by product (generally, 24 to 36 months after the initial sale of a product to our customer), and estimated levels of inventory in the wholesale and retail channels, among others, as well as any other specifically-identified anticipated returns due to known factors such as the loss of patent exclusivity, product recalls and discontinuances, or a changing competitive environment. We maintain a returns policy that allows U.S. pharmaceutical customers to return product for dating issues within a specified period prior to and subsequent to the product's expiration date. Following the loss of exclusivity for a patent-dependent product, we expect to experience an elevated level of product returns as product inventory remaining in the wholesale and retail channels expires. Adjustments to the returns reserve may be required in the future based on revised estimates to our assumptions, which would have an impact on our consolidated results of operations. We record the return amounts as a deduction to arrive at our net product sales. Once the product is returned, it is destroyed. Actual product returns have been less than 2 percent of our net sales over the past three years and have not fluctuated significantly as a percentage of sales. We expect the ratio of actual product returns as a percentage of net sales to increase in future periods as we begin to experience elevated return levels for Cymbalta following the loss of patent exclusivity in the U.S. market.
Sales Rebates and Discounts - Background and Uncertainties
We establish sales rebate and discount accruals in the same period as the related sales. The rebate and discount amounts are recorded as a deduction to arrive at our net product sales. Sales rebates and discounts that require the use of judgment in the establishment of the accrual include Medicaid, managed care, Medicare, chargebacks, long-term care, hospital, patient assistance programs, and various other programs. We base these accruals primarily upon our historical rebate and discount payments made to our customer segment groups and the provisions of current rebate and discount contracts.
The largest of our sales rebate and discount amounts are rebates associated with sales covered by Medicaid and managed care contracts. In determining the appropriate accrual amount, we consider our historical Medicaid and managed care rebate payments by product as a percentage of our historical sales as well as any significant changes in sales trends (e.g., patent expiries), an evaluation of the current Medicaid and managed care contracts, the percentage of our products that are sold via Medicaid and managed care contracts, and our product pricing. Although we accrue a liability for Medicaid and managed care rebates at the time we record the sale (when the product is shipped), the Medicaid and managed care rebate related to that sale is typically paid up to six months later. Because of this time lag, in any particular period our rebate adjustments may incorporate revisions of accruals for several periods.
Most of our rebates outside the U.S. are contractual or legislatively mandated and are estimated and recognized in the same period as the related sales. In some large European countries, government rebates are based on the anticipated budget for pharmaceutical payments in the country. A best estimate of these rebates, updated as governmental authorities revise budgeted deficits, is recognized in the same period as the related sale. If our estimates are not reflective of the actual pharmaceutical costs incurred by the government, we adjust our rebate reserves.
49
Financial Statement Impact
We believe that our accruals for sales returns, rebates, and discounts are reasonable and appropriate based on current facts and circumstances. Our global rebate and discount liabilities are included in sales rebates and discounts on our consolidated balance sheet. Our global sales return liability is included in other current liabilities and other noncurrent liabilities on our consolidated balance sheet. As of December 31, 2015, a 5 percent change in our global sales return, rebate, and discount liability would have led to an approximate $158 million effect on our income before income taxes.
The portion of our global sales return, rebate, and discount liability resulting from sales of our products in the U.S. was 87 percent and 88 percent as of December 31, 2015 and 2014, respectively.
The following represents a roll-forward of our most significant U.S. pharmaceutical sales return, rebate, and discount liability balances, including Medicaid and managed care:
(Dollars in millions) | 2015 | 2014 | |||||
Sales return, rebate, and discount liabilities, beginning of year | $ | 2,241.4 | $ | 2,215.5 | |||
Reduction of net sales due to sales returns, discounts, and rebates(1) | 6,245.1 | 4,707.8 | |||||
Cash payments of discounts and rebates | (5,927.9 | ) | (4,681.9 | ) | |||
Sales return, rebate, and discount liabilities, end of year | $ | 2,558.6 | $ | 2,241.4 | |||
(1) Adjustments of the estimates for these returns, rebates, and discounts to actual results were less than 1.5 percent of consolidated net sales for each of the years presented.
Product Litigation Liabilities and Other Contingencies
Background and Uncertainties
Product litigation liabilities and other contingencies are, by their nature, uncertain and are based upon complex judgments and probabilities. The factors we consider in developing our product litigation liability reserves and other contingent liability amounts include the merits and jurisdiction of the litigation, the nature and the number of other similar current and past litigation cases, the nature of the product and the current assessment of the science subject to the litigation, and the likelihood of settlement and current state of settlement discussions, if any. In addition, we accrue for certain product liability claims incurred, but not filed, to the extent we can formulate a reasonable estimate of their costs based primarily on historical claims experience and data regarding product usage. We accrue legal defense costs expected to be incurred in connection with significant product liability contingencies when both probable and reasonably estimable.
We also consider the insurance coverage we have to diminish the exposure for periods covered by insurance. In assessing our insurance coverage, we consider the policy coverage limits and exclusions, the potential for denial of coverage by the insurance company, the financial condition of the insurers, and the possibility of and length of time for collection. Due to a very restrictive market for product liability insurance, we are self-insured for product liability losses for all our currently marketed products. In addition to insurance coverage, we also consider any third-party indemnification to which we are entitled, including the nature of the indemnification, the financial condition of the indemnifying party, and the possibility of and length of time for collection.
Financial Statement Impact
The litigation accruals and environmental liabilities and the related estimated insurance recoverables have been reflected on a gross basis as liabilities and assets, respectively, on our consolidated balance sheets.
Retirement Benefits Assumptions
Background and Uncertainties
Defined benefit pension plan and retiree health benefit plan costs include assumptions for the discount rate, retirement age, and expected return on plan assets. These assumptions have a significant effect on the amounts reported. In addition to the analysis below, see Note 14 to the consolidated financial statements for additional information regarding our retirement benefits.
50
Annually, we evaluate the discount rate and the expected return on plan assets in our defined benefit pension and retiree health benefit plans. We use an actuarially determined, plan-specific yield curve of high quality, fixed income debt instruments to determine the discount rates. In evaluating the expected return on plan assets, we consider many factors, with a primary analysis of current and projected market conditions, asset returns and asset allocations (approximately 80 percent of which are growth investments); and the views of leading financial advisers and economists. We may also review our historical assumptions compared with actual results, as well as the discount rates and expected return on plan assets of other companies, where applicable. In evaluating our expected retirement age assumption, we consider the retirement ages of our past employees eligible for pension and medical benefits together with our expectations of future retirement ages.
Financial Statement Impact
If the 2015 discount rate for the U.S. defined benefit pension and retiree health benefit plans (U.S. plans) were to change by a quarter percentage point, income before income taxes would change by $42.9 million. As of January 1, 2016, we changed the method used to estimate the service and interest cost components of the net periodic pension and retiree health benefit plan costs. Prior to this change, the service and interest costs were determined using a single weighted-average discount rate based on yield curves of high quality, fixed income debt instruments used to measure the benefit obligation at the beginning of the period. This new method uses the spot yield curve approach to estimate the service and interest costs by applying the specific spot rates along the yield curve to the projected cash outflows of our obligations. The new method provides a more precise measure of interest and service costs by improving the correlation between the projected benefit cash flows and the specific spot yield curve rates. The change does not affect the measurement of the total benefit obligations as the change in service and interest costs is recorded in the actuarial gains and losses recorded in accumulated other comprehensive loss. We will account for this as a change in estimate prospectively beginning in the first quarter of 2016. The decrease in the 2016 service and interest costs is expected to be approximately $110 million compared to the previous method.
If the 2015 expected return on plan assets for U.S. plans were to change by a quarter percentage point, income before income taxes would change by $22.7 million. If our assumption regarding the 2015 expected age of future retirees for U.S. plans were adjusted by one year, our income before income taxes would be affected by $50.4 million. The U.S. plans, including Puerto Rico, represent approximately 75 percent of both the total projected benefit obligation and total plan assets at December 31, 2015.
Impairment of Indefinite-Lived and Long-Lived Assets
Background and Uncertainties
We review the carrying value of long-lived assets (both intangible and tangible) for potential impairment on a periodic basis and whenever events or changes in circumstances indicate the carrying value of an asset may not be recoverable. We determine impairment by comparing the projected undiscounted cash flows to be generated by the asset to its carrying value. If an impairment is identified, a loss is recorded equal to the excess of the asset’s net book value over its fair value, and the cost basis is adjusted.
Goodwill and indefinite-lived intangible assets are reviewed for impairment at least annually and when certain impairment indicators are present. When required, a comparison of fair value to the carrying amount of assets is performed to determine the amount of any impairment.
Several methods may be used to determine the estimated fair value of acquired IPR&D, all of which require multiple assumptions. We utilize the “income method,” as described in Note 8 to the consolidated financial statements.
For acquired IPR&D assets, the risk of failure has been factored into the fair value measure and there can be no certainty that these assets ultimately will yield a successful product, as discussed previously in the “Late-Stage Pipeline” section. The nature of the pharmaceutical business is high-risk and requires that we invest in a large number of projects to build a successful portfolio of approved products. As such, it is likely that some acquired IPR&D assets will become impaired in the future.
Estimates of future cash flows, based on what we believe to be reasonable and supportable assumptions and projections, require management’s judgment. Actual results could vary materially from these estimates.
51
Income Taxes
Background and Uncertainties
We prepare and file tax returns based on our interpretation of tax laws and regulations and record estimates based on these judgments and interpretations. In the normal course of business, our tax returns are subject to examination by various taxing authorities, which may result in future tax, interest, and penalty assessments by these authorities. Inherent uncertainties exist in estimates of many tax positions due to changes in tax law resulting from legislation, regulation, and/or as concluded through the various jurisdictions’ tax court systems. We recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such a position are measured based on the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate resolution. The amount of unrecognized tax benefits is adjusted for changes in facts and circumstances. For example, adjustments could result from significant amendments to existing tax law, the issuance of regulations or interpretations by the taxing authorities, new information obtained during a tax examination, or resolution of an examination. We believe our estimates for uncertain tax positions are appropriate and sufficient to pay assessments that may result from examinations of our tax returns. We recognize both accrued interest and penalties related to unrecognized tax benefits in income tax expense.
We have recorded valuation allowances against certain of our deferred tax assets, primarily those that have been generated from net operating losses and tax credit carryforwards in certain taxing jurisdictions. In evaluating whether we would more likely than not recover these deferred tax assets, we have not assumed any future taxable income or tax planning strategies in the jurisdictions associated with these carryforwards where history does not support such an assumption. Implementation of tax planning strategies to recover these deferred tax assets or future income generation in these jurisdictions could lead to the reversal of these valuation allowances and a reduction of income tax expense.
Financial Statement Impact
As of December 31, 2015, a 5 percent change in the amount of the uncertain tax positions and the valuation allowance would result in a change in net income of $20.2 million and $29.5 million, respectively.
Acquisitions
Background and Uncertainties
To determine whether acquisitions or licensing transactions qualify as a business combination or as an asset acquisition, we make certain judgments, which include assessing whether the acquired set of activities would meet the definition of a business under the relevant accounting rules. This involves determining the inputs, processes, and outputs associated with the acquired set of activities.
If the acquired set of activities meets the definition of a business, assets acquired and liabilities assumed are required to be recorded at their respective fair values as of the acquisition date. The excess of the purchase price over the fair value of the acquired net assets, where applicable, is recorded as goodwill. If the acquired set of activities does not meet the definition of a business, the transaction is recorded as an acquisition of assets and, therefore, any acquired IPR&D that does not have an alternative future use is charged to expense at the acquisition date, and goodwill is not recorded. Refer to Note 3 to the consolidated financial statements for additional information.
The judgments made in determining estimated fair values assigned to assets acquired and liabilities assumed in a business combination, as well as estimated asset lives, can materially affect our consolidated results of operations. The fair values of intangible assets, including acquired IPR&D, are determined using information available near the acquisition date based on expectations and assumptions that are deemed reasonable by management. Depending on the facts and circumstances, we may deem it necessary to engage an independent valuation expert to assist in valuing significant assets and liabilities.
The fair values of identifiable intangible assets are primarily determined using an "income method," as described in Note 8 to the consolidated financial statements.
52
The fair value of any contingent consideration liability that results from a business combination is determined using a market approach based on quoted market values, significant other observable inputs for identical or comparable assets or liabilities, or a discounted cash flow analysis. Estimating the fair value of contingent consideration requires the use of significant estimates and judgments, including, but not limited to, revenue and the discount rate.
Financial Statement Impact
As of December 31, 2015, a 5 percent change in the contingent consideration liability would result in a change in income before income taxes of $33.5 million.
LEGAL AND REGULATORY MATTERS
Information relating to certain legal proceedings can be found in Note 15 to the consolidated financial statements and is incorporated here by reference.
FINANCIAL EXPECTATIONS FOR 2016
For the full year of 2016, we expect EPS to be in the range of $2.83 to $2.93. We anticipate that total revenue will be between $20.2 billion and $20.7 billion. Excluding the unfavorable impact of foreign exchange rates, we expect revenue growth from a number of established products including Humalog, Trajenta, Cialis, Forteo, Strattera, Erbitux, and animal health products, as well as higher revenues from new products including Cyramza, Trulicity, Jardiance, Portrazza, and Basaglar. We expect this revenue growth to be partially offset by lower revenue from Alimta as a result of increased competitive pressures.
We anticipate that gross margin as a percent of revenue will be approximately 74 percent in 2016. Research and development expenses are expected to be in the range of $4.8 billion to $5.0 billion. Other—net, (income) expense is expected to be income of up to $75 million. Marketing, selling, and administrative expenses are expected to be in the range of $6.0 billion to $6.2 billion.
The 2016 tax rate is expected to be approximately 21 percent.
Capital expenditures are expected to be approximately $1.1 billion.
Amortization associated with the transfer of Erbitux commercialization rights included in our 2016 financial guidance is subject to final acquisition accounting adjustments.
Various risks and uncertainties, including those discussed in "Forward-Looking Statements" and Item 1A, “Risk Factors,” may cause our actual results to differ materially from these forward-looking statements.
Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
You can find quantitative and qualitative disclosures about market risk (e.g., interest rate risk) in Item 7 at “Management’s Discussion and Analysis—Financial Condition.” That information is incorporated in this report by reference.
53
Item 8. | Financial Statements and Supplementary Data |
Consolidated Statements of Operations
ELI LILLY AND COMPANY AND SUBSIDIARIES (Dollars in millions and shares in thousands, except per-share data) | Year Ended December 31 | 2015 | 2014 | 2013 | ||||||||||
Revenue | $ | 19,958.7 | $ | 19,615.6 | $ | 23,113.1 | ||||||||
Costs, expenses, and other: | ||||||||||||||
Cost of sales | 5,037.2 | 4,932.5 | 4,908.1 | |||||||||||
Research and development | 4,796.4 | 4,733.6 | 5,531.3 | |||||||||||
Marketing, selling, and administrative | 6,533.0 | 6,620.8 | 7,125.6 | |||||||||||
Acquired in-process research and development (Notes 3 and 4) | 535.0 | 200.2 | 57.1 | |||||||||||
Asset impairment, restructuring, and other special charges (Note 5) | 367.7 | 468.7 | 120.6 | |||||||||||
Other—net, (income) expense (Note 17) | (100.6 | ) | (340.5 | ) | (518.9 | ) | ||||||||
17,168.7 | 16,615.3 | 17,223.8 | ||||||||||||
Income before income taxes | 2,790.0 | 3,000.3 | 5,889.3 | |||||||||||
Income taxes (Note 13) | 381.6 | 609.8 | 1,204.5 | |||||||||||
Net income | $ | 2,408.4 | $ | 2,390.5 | $ | 4,684.8 | ||||||||
Earnings per share: | ||||||||||||||
Basic | $ | 2.27 | $ | 2.23 | $ | 4.33 | ||||||||
Diluted | 2.26 | 2.23 | 4.32 | |||||||||||
Shares used in calculation of earnings per share: | ||||||||||||||
Basic | 1,061,913 | 1,069,932 | 1,080,874 | |||||||||||
Diluted | 1,065,720 | 1,074,286 | 1,084,766 | |||||||||||
See notes to consolidated financial statements.
54
Consolidated Statements of Comprehensive Income
ELI LILLY AND COMPANY AND SUBSIDIARIES (Dollars in millions) | Year Ended December 31 | 2015 | 2014 | 2013 | ||||||||||
Net income | $ | 2,408.4 | $ | 2,390.5 | $ | 4,684.8 | ||||||||
Other comprehensive income (loss): | ||||||||||||||
Change in foreign currency translation gains (losses) | (859.8 | ) | (961.4 | ) | 36.2 | |||||||||
Change in net unrealized gains and losses on securities | (138.1 | ) | (162.2 | ) | 204.3 | |||||||||
Change in defined benefit pension and retiree health benefit plans (Note 14) | 572.9 | (1,327.6 | ) | 2,592.2 | ||||||||||
Change in effective portion of cash flow hedges | (42.0 | ) | (14.5 | ) | (123.8 | ) | ||||||||
Other comprehensive income (loss) before income taxes | (467.0 | ) | (2,465.7 | ) | 2,708.9 | |||||||||
Provision for income taxes related to other comprehensive income (loss) items | (121.9 | ) | 476.6 | (914.5 | ) | |||||||||
Other comprehensive income (loss) (Note 16) | (588.9 | ) | (1,989.1 | ) | 1,794.4 | |||||||||
Comprehensive income | $ | 1,819.5 | $ | 401.4 | $ | 6,479.2 | ||||||||
See notes to consolidated financial statements.
55
Consolidated Balance Sheets
ELI LILLY AND COMPANY AND SUBSIDIARIES (Dollars in millions, shares in thousands) | December 31 | 2015 | 2014 | |||||||
Assets | ||||||||||
Current Assets | ||||||||||
Cash and cash equivalents (Note 7) | $ | 3,666.4 | $ | 3,871.6 | ||||||
Short-term investments (Note 7) | 785.4 | 955.4 | ||||||||
Accounts receivable, net of allowances of $44.3 (2015) and $55.0 (2014) | 3,513.0 | 3,234.6 | ||||||||
Other receivables | 558.6 | 566.7 | ||||||||
Inventories (Note 6) | 3,445.8 | 2,740.0 | ||||||||
Prepaid expenses and other | 604.4 | 560.0 | ||||||||
Total current assets | 12,573.6 | 11,928.3 | ||||||||
Other Assets | ||||||||||
Restricted cash (Note 3) | — | 5,405.6 | ||||||||
Investments (Note 7) | 3,646.6 | 4,568.9 | ||||||||
Goodwill (Note 8) | 4,039.9 | 1,758.1 | ||||||||
Other intangibles, net (Note 8) | 5,034.8 | 2,884.2 | ||||||||
Sundry | 2,220.5 | 1,798.6 | ||||||||
Total other assets | 14,941.8 | 16,415.4 | ||||||||
Property and equipment, net (Note 9) | 8,053.5 | 7,963.9 | ||||||||
Total assets | $ | 35,568.9 | $ | 36,307.6 | ||||||
Liabilities and Equity | ||||||||||
Current Liabilities | ||||||||||
Short-term borrowings and current maturities of long-term debt (Note 10) | $ | 6.1 | $ | 2,688.7 | ||||||
Accounts payable | 1,338.2 | 1,128.1 | ||||||||
Employee compensation | 967.0 | 759.0 | ||||||||
Sales rebates and discounts | 2,560.1 | 2,068.8 | ||||||||
Dividends payable | 539.0 | 530.3 | ||||||||
Income taxes payable (Note 13) | 358.9 | 93.5 | ||||||||
Other current liabilities | 2,460.3 | 2,472.6 | ||||||||
Total current liabilities | 8,229.6 | 9,741.0 | ||||||||
Other Liabilities | ||||||||||
Long-term debt (Note 10) | 7,972.4 | 5,332.8 | ||||||||
Accrued retirement benefits (Note 14) | 2,160.3 | 2,562.9 | ||||||||
Long-term income taxes payable (Note 13) | 868.9 | 998.5 | ||||||||
Other noncurrent liabilities | 1,747.4 | 2,284.3 | ||||||||
Total other liabilities | 12,749.0 | 11,178.5 | ||||||||
Commitments and Contingencies (Note 15) | ||||||||||
Eli Lilly and Company Shareholders' Equity (Notes 11 and 12) | ||||||||||
Common stock—no par value Authorized shares: 3,200,000 Issued shares: 1,106,063 (2015) and 1,111,437 (2014) | 691.3 | 694.6 | ||||||||
Additional paid-in capital | 5,552.1 | 5,292.3 | ||||||||
Retained earnings | 16,011.8 | 16,482.7 | ||||||||
Employee benefit trust | (3,013.2 | ) | (3,013.2 | ) | ||||||
Accumulated other comprehensive loss (Note 16) | (4,580.7 | ) | (3,991.8 | ) | ||||||
Cost of common stock in treasury | (90.0 | ) | (91.4 | ) | ||||||
Total Eli Lilly and Company shareholders' equity | 14,571.3 | 15,373.2 | ||||||||
Noncontrolling interests | 19.0 | 14.9 | ||||||||
Total equity | 14,590.3 | 15,388.1 | ||||||||
Total liabilities and equity | $ | 35,568.9 | $ | 36,307.6 | ||||||
See notes to consolidated financial statements.
56
Consolidated Statements of Shareholders' Equity
ELI LILLY AND COMPANY AND SUBSIDIARIES (Dollars in millions, shares in thousands) | Common Stock | Additional Paid-in Capital | Retained Earnings | Accumulated Other Comprehensive Loss | Common Stock in Treasury | Employee Benefit Trust | Shareholders' Equity | ||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||
Balance at January 1, 2013 | 1,146,493 | $ | 716.6 | $ | 4,963.1 | $ | 16,088.2 | $ | (3,797.1 | ) | 2,850 | $ | (192.4 | ) | $ | (3,013.2 | ) | $ | 14,765.2 | ||||||||||||||
Net income | 4,684.8 | 4,684.8 | |||||||||||||||||||||||||||||||
Other comprehensive income (loss), net of tax | 1,794.4 | 1,794.4 | |||||||||||||||||||||||||||||||
Cash dividends declared per share: $1.96 | (2,102.8 | ) | (2,102.8 | ) | |||||||||||||||||||||||||||||
Retirement of treasury shares | (32,406) | (20.3 | ) | (1,677.8 | ) | (32,406) | 1,698.1 | — | |||||||||||||||||||||||||
Purchase for treasury | 30,400 | (1,600.0 | ) | (1,600.0 | ) | ||||||||||||||||||||||||||||
Issuance of stock under employee stock plans, net | 3,541 | 2.2 | (58.0 | ) | (11) | 0.7 | (55.1 | ) | |||||||||||||||||||||||||
Stock-based compensation | 144.9 | 144.9 | |||||||||||||||||||||||||||||||
Balance at December 31, 2013 | 1,117,628 | 698.5 | 5,050.0 | 16,992.4 | (2,002.7 | ) | 833 | (93.6 | ) | (3,013.2 | ) | 17,631.4 | |||||||||||||||||||||
Net income | 2,390.5 | 2,390.5 | |||||||||||||||||||||||||||||||
Other comprehensive income (loss), net of tax | (1,989.1 | ) | (1,989.1 | ) | |||||||||||||||||||||||||||||
Cash dividends declared per share: $1.97 | (2,108.1 | ) | (2,108.1 | ) | |||||||||||||||||||||||||||||
Retirement of treasury shares | (12,579) | (7.9 | ) | (792.1 | ) | (12,579) | 800.0 | — | |||||||||||||||||||||||||
Purchase for treasury | 12,579 | (800.0 | ) | (800.0 | ) | ||||||||||||||||||||||||||||
Issuance of stock under employee stock plans, net | 6,388 | 4.0 | 86.3 | (23) | 2.2 | 92.5 | |||||||||||||||||||||||||||
Stock-based compensation | 156.0 | 156.0 | |||||||||||||||||||||||||||||||
Balance at December 31, 2014 | 1,111,437 | 694.6 | 5,292.3 | 16,482.7 | (3,991.8 | ) | 810 | (91.4 | ) | (3,013.2 | ) | 15,373.2 | |||||||||||||||||||||
Net income | 2,408.4 | 2,408.4 | |||||||||||||||||||||||||||||||
Other comprehensive income (loss), net of tax | (588.9 | ) | (588.9 | ) | |||||||||||||||||||||||||||||
Cash dividends declared per share: $2.01 | (2,136.0 | ) | (2,136.0 | ) | |||||||||||||||||||||||||||||
Retirement of treasury shares | (9,877 | ) | (6.2 | ) | (743.3 | ) | (9,877 | ) | 749.5 | — | |||||||||||||||||||||||
Purchase for treasury | 9,877 | (749.5 | ) | (749.5 | ) | ||||||||||||||||||||||||||||
Issuance of stock under employee stock plans, net | 4,503 | 2.9 | 42.0 | (14 | ) | 1.4 | 46.3 | ||||||||||||||||||||||||||
Stock-based compensation | 217.8 | 217.8 | |||||||||||||||||||||||||||||||
Balance at December 31, 2015 | 1,106,063 | $ | 691.3 | $ | 5,552.1 | $ | 16,011.8 | $ | (4,580.7 | ) | 796 | $ | (90.0 | ) | $ | (3,013.2 | ) | $ | 14,571.3 | ||||||||||||||
See notes to consolidated financial statements.
57
Consolidated Statements of Cash Flows
ELI LILLY AND COMPANY AND SUBSIDIARIES (Dollars in millions) | Year Ended December 31 | 2015 | 2014 | 2013 | ||||||||||
Cash Flows from Operating Activities | ||||||||||||||
Net income | $ | 2,408.4 | $ | 2,390.5 | $ | 4,684.8 | ||||||||
Adjustments to Reconcile Net Income to Cash Flows from Operating Activities: | ||||||||||||||
Depreciation and amortization | 1,427.7 | 1,379.0 | 1,445.6 | |||||||||||
Change in deferred income taxes | (748.4 | ) | 36.8 | 265.9 | ||||||||||
Stock-based compensation expense | 217.8 | 156.0 | 144.9 | |||||||||||
Acquired in-process research and development | 535.0 | 200.2 | 57.1 | |||||||||||
Income related to termination of the exenatide collaboration with Amylin Pharmaceuticals, Inc. (Note 4) | — | — | (495.4 | ) | ||||||||||
Net proceeds from (payments for) terminations of interest rate swaps | (186.1 | ) | 340.7 | — | ||||||||||
Other non-cash operating activities, net | 36.4 | 13.8 | 25.1 | |||||||||||
Other changes in operating assets and liabilities, net of acquisitions and divestitures: | ||||||||||||||
Receivables—(increase) decrease | (304.5 | ) | 117.4 | (152.7 | ) | |||||||||
Inventories—(increase) decrease | (736.3 | ) | (307.1 | ) | (286.5 | ) | ||||||||
Other assets—(increase) decrease | (338.8 | ) | 411.5 | 116.5 | ||||||||||
Accounts payable and other liabilities—increase (decrease) | 461.6 | (371.7 | ) | (70.3 | ) | |||||||||
Net Cash Provided by Operating Activities | 2,772.8 | 4,367.1 | 5,735.0 | |||||||||||
Cash Flows from Investing Activities | ||||||||||||||
Purchases of property and equipment | (1,066.2 | ) | (1,162.6 | ) | (1,012.1 | ) | ||||||||
Disposals of property and equipment | 92.6 | 15.3 | 179.4 | |||||||||||
Cash released (restricted) for pending acquisition (Note 3) | 5,405.6 | (5,405.6 | ) | — | ||||||||||
Proceeds from sales and maturities of short-term investments | 2,161.8 | 4,054.1 | 3,320.1 | |||||||||||
Purchases of short-term investments | (842.2 | ) | (1,637.8 | ) | (1,531.0 | ) | ||||||||
Proceeds from sales of noncurrent investments | 3,068.4 | 11,009.4 | 11,235.0 | |||||||||||
Purchases of noncurrent investments | (3,226.5 | ) | (9,802.7 | ) | (14,041.9 | ) | ||||||||
Proceeds from sale of product rights | 410.0 | — | — | |||||||||||
Purchase of product rights | — | (308.3 | ) | (24.1 | ) | |||||||||
Purchases of in-process research and development | (560.0 | ) | (95.0 | ) | (57.1 | ) | ||||||||
Cash paid for acquisitions, net of cash acquired (Note 3) | (5,283.1 | ) | (551.4 | ) | (43.7 | ) | ||||||||
Other investing activities, net | (133.6 | ) | (24.5 | ) | (97.4 | ) | ||||||||
Net Cash Provided by (Used for) Investing Activities | 26.8 | (3,909.1 | ) | (2,072.8 | ) | |||||||||
Cash Flows from Financing Activities | ||||||||||||||
Dividends paid | (2,127.3 | ) | (2,101.2 | ) | (2,120.7 | ) | ||||||||
Net change in short-term borrowings | (2,680.6 | ) | 2,680.6 | — | ||||||||||
Proceeds from issuance of long-term debt | 4,454.7 | 992.9 | — | |||||||||||
Repayments of long-term debt | (1,955.7 | ) | (1,034.8 | ) | (10.5 | ) | ||||||||
Purchases of common stock | (749.5 | ) | (800.0 | ) | (1,698.1 | ) | ||||||||
Other financing activities, net | 139.2 | 187.4 | — | |||||||||||
Net Cash Used for Financing Activities | (2,919.2 | ) | (75.1 | ) | (3,829.3 | ) | ||||||||
Effect of exchange rate changes on cash and cash equivalents | (85.6 | ) | (341.5 | ) | (21.5 | ) | ||||||||
Net increase (decrease) in cash and cash equivalents | (205.2 | ) | 41.4 | (188.6 | ) | |||||||||
Cash and cash equivalents at beginning of year | 3,871.6 | 3,830.2 | 4,018.8 | |||||||||||
Cash and Cash Equivalents at End of Year | $ | 3,666.4 | $ | 3,871.6 | $ | 3,830.2 | ||||||||
See notes to consolidated financial statements.
58
Notes to Consolidated Financial Statements
ELI LILLY AND COMPANY AND SUBSIDIARIES
(Tables present dollars in millions, except per-share data)
Note 1: Summary of Significant Accounting Policies
Basis of presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (GAAP). The accounts of all wholly-owned and majority-owned subsidiaries are included in the consolidated financial statements. Where our ownership of consolidated subsidiaries is less than 100 percent, the noncontrolling shareholders’ interests are reflected as a separate component of equity. All intercompany balances and transactions have been eliminated.
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses, and related disclosures at the date of the financial statements and during the reporting period. Actual results could differ from those estimates. We issued our financial statements by filing with the Securities and Exchange Commission and have evaluated subsequent events up to the time of the filing.
Certain reclassifications have been made to prior periods in the consolidated financial statements and accompanying notes to conform with the current presentation.
All per-share amounts, unless otherwise noted in the footnotes, are presented on a diluted basis, that is, based on the weighted-average number of outstanding common shares plus the effect of incremental shares from our stock-based compensation programs.
Revenue recognition
We recognize revenue from sales of products at the time title of goods passes to the buyer and the buyer assumes the risks and rewards of ownership. Provisions for returns, discounts, and rebates are established in the same period the related sales are recognized.
In arrangements involving the delivery of more than one element (e.g., research and development), marketing and selling, manufacturing, and distribution), each required deliverable is evaluated to determine whether it qualifies as a separate unit of accounting. Our determination is based on whether the deliverable has "standalone value" to the customer. If a deliverable does not qualify as a separate unit of accounting, it is combined with the other applicable undelivered item(s) within the arrangement and these combined deliverables are treated as a single unit of accounting. The arrangement's consideration that is fixed or determinable is then allocated to each separate unit of accounting based on the relative selling price of each deliverable.
Initial fees we receive in collaborative and other similar arrangements from the partnering of our compounds under development are generally deferred and amortized into income through the expected product approval date. Initial fees may also be received for out-licensing agreements that include both an out-license of our marketing rights to commercialized products and a related commitment to supply the products. When we have determined that the marketing rights do not have standalone value, the initial fees received are generally deferred and amortized to income as net product sales over the term of the supply agreement.
Royalty revenue from licensees, which is based on third-party sales of licensed products and technology, is recorded as earned in accordance with the contract terms when third-party sales can be reasonably measured and collection of the funds is reasonably assured. This royalty revenue is included in collaboration and other revenue.
Profit-sharing due from our collaboration partners, which is based upon gross margins reported to us by our partners, is recognized as collaboration and other revenue as earned.
Developmental milestone payments earned by us are generally recorded in other–net, (income) expense. We immediately recognize the full amount of developmental milestone payments due to us upon the achievement of the milestone event if the event is objectively determinable and the milestone is substantive in its entirety. A milestone is considered substantive if the consideration earned 1) relates solely to past performance, 2) is
59
commensurate with the enhancement in the pharmaceutical or animal health product's value associated with the achievement of the important event in its development life cycle, and 3) is reasonable relative to all of the deliverables and payment terms within the arrangement. If a milestone payment to us is part of a multiple-element commercialization arrangement and is triggered by the initiation of the commercialization period (e.g., regulatory approval for marketing or launch of the product) or the achievement of a sales-based threshold, we amortize the payment to income as we perform under the terms of the arrangement. See Note 4 for specific agreement details.
Research and development expenses and acquired in-process research and development
Research and development expenses include the following:
• | Research and development costs, which are expensed as incurred. |
• | Milestone payment obligations incurred prior to regulatory approval of the product, which are accrued when the event requiring payment of the milestone occurs. |
Acquired in-process research and development (IPR&D) expense includes the initial costs of IPR&D projects, acquired directly in a transaction other than a business combination, that do not have an alternative future use.
Earnings per share
We calculate basic earnings per share (EPS) based on the weighted-average number of common shares outstanding and incremental shares from potential participating securities. We calculate diluted EPS based on the weighted-average number of common shares outstanding, including incremental shares from our stock-based compensation programs.
Foreign Currency Translation
Operations in our subsidiaries outside the United States (U.S.) are recorded in the functional currency of each subsidiary which is determined by a review of the environment where each subsidiary primarily generates and expends cash. The results of operations for our subsidiaries outside the U.S. are translated from functional currencies into U.S. dollars using the weighted average currency rate for the period. Assets and liabilities are translated using the period end exchange rates. The U.S. dollar effects that arise from translating the net assets of these subsidiaries are recorded in other comprehensive income (loss).
Other significant accounting policies
Our other significant accounting policies are described in the remaining appropriate notes to the consolidated financial statements.
60
Note 2: Implementation of New Financial Accounting Pronouncements
The following table provides a brief description of accounting standards that have not yet been adopted that could have a material effect on our financial statements:
Standard | Description | Effective Date | Effect on the financial statements or other significant matters | |||
Accounting Standards Update 2014-09, Revenue from Contracts with Customers | This standard will replace existing revenue recognition standards and will require entities to recognize revenues to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. An entity can apply the new revenue standard retrospectively to each prior reporting period presented or with the cumulative effect of initially applying the standard recognized at the date of initial application in retained earnings. | This standard is effective January 1, 2018, but we are permitted to adopt this standard one year earlier if we choose. We are evaluating our anticipated date of adoption. | There are areas within the standard that are currently under review and reconsideration by the Financial Accounting Standards Board, including accounting for licensing transactions, which could lead to updates to the standard. As the outcomes of this review and reconsideration could lead to significant changes to the standard, we are still in the process of determining our approach to the adoption of the standard, as well as the anticipated impact to our consolidated financial statements. | |||
Accounting Standards Update 2016-01, Financial Instruments - Overall: Recognition and Measurement of Financial Assets and Financial Liabilities | This standard will require entities to recognize changes in the fair value of equity investments with readily determinable fair values in net income (except for investments accounted for under the equity method of accounting or those that result in consolidation of the investee). An entity should apply the new standard through a cumulative effect adjustment to retained earnings as of the beginning of the fiscal year of adoption. | This standard is effective January 1, 2018. Early adoption of the majority of the amendments in this standard is not permitted, however, early application of certain amendments is permitted. We intend to fully adopt this standard on January 1, 2018. | We are currently unable to estimate the impact of adopting this standard as the significance of the impact will depend upon our equity investments on hand as of the date of adoption. | |||
61
The following table provides a brief description of an accounting standard that has been adopted:
Standard | Description | Effective Date | Effect on the financial statements or other significant matters | |||
Accounting Standards Update 2015-17, Income Taxes: Balance Sheet Classification of Deferred Taxes | This standard simplifies the presentation of deferred income taxes and requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. An entity can apply this new standard either prospectively to all deferred tax liabilities and assets or retrospectively to all periods presented. | We have adopted this standard for the annual period beginning on January 1, 2015. | We applied this standard retrospectively. As a result of adopting this standard, all deferred tax liabilities and assets have been classified as noncurrent. The balance sheet as of December 31, 2014 was retrospectively adjusted which resulted in reductions to prepaid expenses and other of $251.5 million, sundry of $584.2 million, and deferred income tax liabilities of $1.47 billion. Other noncurrent liabilities as of December 31, 2014 were increased by $630.8 million as a result of the adoption of this standard. | |||
Note 3: Acquisitions
During 2015 and 2014, we completed the acquisitions of Novartis Animal Health (Novartis AH) and Lohmann SE (Lohmann AH), respectively. Additionally, on October 1, 2015, Bristol-Myers Squibb Company and E.R. Squibb (collectively, BMS) transferred to us their commercialization rights with respect to Erbitux® in the U.S. and Canada (collectively, North America) through a modification of our existing arrangement. These transactions were accounted for as business combinations under the acquisition method of accounting. See Note 4 for additional information related to the Erbitux arrangement. The assets acquired and liabilities assumed were recorded at their respective fair values as of the acquisition date in our consolidated financial statements. The determination of estimated fair value required management to make significant estimates and assumptions. The excess of the purchase price over the fair value of the acquired net assets, where applicable, has been recorded as goodwill. The results of operations of these acquisitions are included in our consolidated financial statements from the date of acquisition.
In addition to the acquisitions of businesses, we also acquired assets in development in 2015, 2014, and 2013 which are further discussed below in Product and Other Acquisitions and in Note 4. Upon acquisition, the acquired IPR&D related to these products was immediately written off as an expense because the products had no alternative future use. For the years ended December 31, 2015, 2014, and 2013, we recorded acquired IPR&D charges of $535.0 million, $200.2 million, and $57.1 million, respectively. The charges were associated with the transactions discussed below in Product and Other Acquisitions, the 2015 upfront fee of $200.0 million related to tanezumab, and the 2014 charge of $55.2 million related to the transfer to us of Boehringer Ingelheim's rights to co-promote our new insulin glargine product in countries where it was not yet approved. See Note 4 for additional information related to the tanezumab and Boehringer Ingelheim arrangements.
62
Acquisitions of Businesses
Novartis AH Acquisition
Overview of Transaction
On January 1, 2015, we acquired from Novartis AG all of the shares of certain Novartis subsidiaries and all of the assets and liabilities of other Novartis subsidiaries that are exclusively related to the Novartis AH business in an all-cash transaction for a total purchase price of $5.28 billion. As of December 31, 2014, there was $5.41 billion of cash held in escrow for the pending acquisition of Novartis AH. This cash was classified as restricted cash, a noncurrent asset, on our consolidated balance sheet.
As a condition to the clearance of the transaction under the Hart-Scott-Rodino Antitrust Improvements Act, following the closing of the acquisition of Novartis AH, we divested certain animal health assets in the U.S. related to the Sentinel® canine parasiticide franchise to Virbac Corporation for approximately $410 million.
The acquired Novartis AH business consists of the research and development, manufacture, marketing, sale and distribution of veterinary products to prevent and treat diseases in pets, farm animals, and farmed fish. Under the terms of the agreement, we acquired manufacturing sites, research and development facilities, a global commercial infrastructure and portfolio of products, a pipeline of projects in development, and employees.
Assets Acquired and Liabilities Assumed
The following table summarizes the amounts recognized for assets acquired and liabilities assumed as of the acquisition date:
Estimated Fair Value at January 1, 2015 | |||
Inventories | $ | 380.2 | |
Acquired in-process research and development | 298.0 | ||
Marketed products(1) | 1,953.0 | ||
Property and equipment | 199.9 | ||
Assets held for sale (primarily the U.S. Sentinel rights) | 422.7 | ||
Accrued retirement benefits | (108.7 | ) | |
Deferred income taxes | (60.1 | ) | |
Other assets and liabilities - net | (73.0 | ) | |
Total identifiable net assets | 3,012.0 | ||
Goodwill(2) | 2,271.1 | ||
Total consideration transferred - net of cash acquired | $ | 5,283.1 | |
(1) These intangible assets, which will be amortized to cost of sales on a straight-line basis over their estimated useful lives, are expected to have a weighted average useful life of 19 years.
(2) The goodwill recognized from this acquisition is attributable primarily to expected synergies that we believe will result from combining the operations of Novartis AH with our legacy animal health business, future unidentified projects and products, and the assembled workforce of Novartis AH. Approximately $950 million of the goodwill associated with this acquisition is estimated to be deductible for tax purposes.
Actual and Supplemental Pro Forma Information
Our consolidated statement of operations for the year ended December 31, 2015 includes Novartis AH revenue of $1.02 billion. Novartis AH has been partially integrated into our animal health segment and as a result of these integration efforts, certain parts of the animal health business are operating on a combined basis, and we cannot distinguish the operations between Novartis AH and our legacy animal health business.
63
The following unaudited pro forma financial information presents the combined consolidated results of our operations with Novartis AH as if the portion of Novartis AH that we retained after the sale to Virbac had been acquired as of January 1, 2014. We have adjusted the historical consolidated financial information to give effect to pro forma events that are directly attributable to the acquisition. The unaudited pro forma financial information is not necessarily indicative of what our consolidated results of operations would have been had we completed the acquisition at the beginning of 2014. In addition, the unaudited pro forma financial information does not attempt to project the future results of operations of our combined company.
Unaudited Pro Forma Consolidated Results | |||||||
2015 | 2014 | ||||||
Revenue | $ | 19,958.7 | $ | 20,696.7 | |||
Net income | 2,518.1 | 2,127.9 | |||||
Diluted earnings per share | 2.36 | 1.98 | |||||
The unaudited pro forma financial information above reflects primarily the following pro forma pretax adjustments:
• | Additional amortization expense of approximately $104 million for the year ended December 31, 2014, related to the fair value of identifiable intangible assets acquired. |
• | Additional cost of sales in 2014, and a corresponding reduction in cost of sales in 2015, of approximately $153 million related to the fair value adjustments to acquisition date inventory that has been sold in the year ended December 31, 2015. |
• | A decrease to pro forma net income of approximately $112 million in the year ended December 31, 2014, associated with an increase to interest expense related to the incremental debt that we issued to partially finance the acquisition and a reduction of interest income associated with investments which would have been used to partially fund the acquisition. |
In addition, all of the above adjustments were adjusted for the applicable tax impact. The taxes associated with the adjustments above reflect the statutory tax rates in the various jurisdictions where the fair value adjustments occurred.
Lohmann AH Acquisition
On April 30, 2014, we acquired Lohmann AH, a privately-held company headquartered in Cuxhaven, Germany, through a stock purchase for a total purchase price of $591.2 million, comprised of $551.4 million of net cash plus $39.8 million of assumed debt. Lohmann AH was a global leader in poultry vaccines. As part of this transaction, we acquired the rights to a range of vaccines, commercial capabilities, and manufacturing sites in Germany and the U.S. The acquisition was not material to our consolidated financial statements.
The following table summarizes the amounts recognized for assets acquired and liabilities assumed as of the acquisition date:
Estimated Fair Value at April 30, 2014 | |||
Marketed products | $ | 275.4 | |
Other intangible assets | 23.9 | ||
Property and equipment | 81.9 | ||
Deferred income taxes | (92.7 | ) | |
Other assets and liabilities - net | 51.1 | ||
Total identifiable net assets | 339.6 | ||
Goodwill(1) | 251.6 | ||
Total consideration transferred - net of cash acquired | $ | 591.2 | |
(1) Goodwill associated with this acquisition is not deductible for tax purposes.
64
Product and Other Acquisitions
The following table summarizes our product and other acquisitions which are discussed in detail below:
Counterparty | Compound(s) or Therapy | Acquisition Month | Phase of Development(1) | Acquired IPR&D Expense | ||||
Innovent Biologics, Inc. (Innovent) | Monoclonal antibody targeting protein CD-20 Immuno-oncology molecule cMet monoclonal antibody | March 2015 | Pre-clinical(2) | $ | 56.0 | |||
Hanmi Pharmaceutical Co., Ltd. (Hanmi) | BTK Inhibitor - HM71224 | April 2015 | Phase I | 50.0 | ||||
BioNTech AG (BioNTech) | Cancer immunotherapies | May 2015 | Pre-clinical | 30.0 | ||||
Locemia Solutions | Intranasal glucagon | October 2015 | Phase III | 149.0 | ||||
Undisclosed | Technology collaboration | December 2015 | N/A | 25.0 | ||||
Halozyme Therapeutics, Inc. (Halozyme) | Recombinant human hyaluronidase enzyme - rHuPH20 | December 2015 | N/A | 25.0 | ||||
Immunocore Limited (Immunocore) | T cell-based cancer therapies | July 2014 | Pre-clinical | 45.0 | ||||
AstraZeneca UK Limited (AstraZeneca) | Oral beta-secretase cleaving enzyme inhibitor - AZD3293 | September 2014 | Phase I | 50.0 | ||||
Adocia | BioChaperone Lispro | December 2014 | Phase I | 50.0 | ||||
Arteaus Therapeutics | Calcitonin gene-related peptide (CGRP) monoclonal antibody | December 2013 | Phase II | 57.1 | ||||
(1) The phase of development presented is as of the date of the arrangement.
(2) Prior to acquisition, Innovent's monoclonal antibody targeting protein CD-20 had received investigational new drug approval in China to begin Phase I development.
In connection with the arrangements described herein, our partners may be entitled to future royalties based on sales should these products be approved for commercialization and/or milestones based on the successful progress of the drug candidate through the development process.
Our collaboration agreement with Innovent is to develop and commercialize a portfolio of cancer treatments. In China, we will be responsible for the commercialization efforts, while Innovent will lead the development and manufacturing efforts. Innovent also has co-promotion rights in China. We will be responsible for development, manufacturing, and commercialization efforts of Innovent's pre-clinical immuno-oncology molecules outside of China. Separate from the collaboration, we will continue the development of our cMet monoclonal antibody gene outside of China.
Our collaboration agreement with Hanmi is to develop and commercialize Hanmi's compound being investigated for the treatment of autoimmune and other diseases. We received rights to the molecule for all indications on a worldwide basis excluding China, Hong Kong, Taiwan, and Korea. We will be responsible for leading development, regulatory, manufacturing, and commercial efforts in our territories.
Our research collaboration with BioNTech is to discover novel cancer immunotherapies.
Our global collaboration and license agreement with Halozyme is to develop and commercialize products combining our proprietary compounds with Halozyme's ENHANZE™ platform to aid in the dispersion and absorption of other injected therapeutic drugs.
65
Our co-discovery and co-development collaboration with Immunocore is to research and potentially develop pre-clinical novel T cell-based cancer therapies.
Our collaboration agreement with AstraZeneca is for the worldwide co-development and co-commercialization of AstraZeneca’s molecule being investigated for the potential treatment of Alzheimer’s disease. We are responsible for leading development efforts, while AstraZeneca will be responsible for manufacturing efforts. If successful, both parties will take joint responsibility for commercialization of AZD3293. Under the agreement, both parties will share equally in the ongoing development costs, gross margins, and certain other costs associated with commercialization of the molecule.
Our collaboration agreement with Adocia is for the worldwide development and commercialization of Adocia's ultra-rapid insulin, a molecule being developed for the treatment of patients with type 1 and type 2 diabetes. We will be responsible for leading development, manufacturing, and commercialization efforts.
We acquired all development and commercial rights from Arteaus Therapeutics for a CGRP antibody being studied as a potential treatment for the treatment of cluster headache and migraine prevention.
Note 4: Collaborations and Other Arrangements
We often enter into collaborative and other similar arrangements to develop and commercialize drug candidates. Collaborative activities may include research and development, marketing and selling (including promotional activities and physician detailing), manufacturing, and distribution. These arrangements often require milestone and royalty or profit-share payments, contingent upon the occurrence of certain future events linked to the success of the asset in development, as well as expense reimbursements or payments to the collaboration partner. Elements within a collaboration are separated into individual units of accounting if they have standalone value from other elements within the arrangement. In these situations, the arrangement consideration is allocated to the elements on a relative selling price basis. Revenues related to products we sell pursuant to these arrangements are included in net product revenues, while other sources of revenue (e.g., royalties and profit sharing due from our partner) are included in collaboration and other revenue.
The following table summarizes our collaboration and other revenue recognized:
2015 | 2014 | 2013 | |||||||||
Collaboration and other revenue | $ | 808.1 | $ | 788.4 | $ | 707.5 | |||||
Operating expenses for costs incurred pursuant to these arrangements are reported in their respective expense line items, net of any payments due to or reimbursements due from our collaboration partners, with such reimbursements being recognized at the time the party becomes obligated to pay. Each collaboration is unique in nature, and our more significant arrangements are discussed below.
66
Boehringer Ingelheim Diabetes Collaboration
We and Boehringer Ingelheim have a global agreement to jointly develop and commercialize a portfolio of diabetes compounds. Currently, included in the collaboration are Boehringer Ingelheim’s oral diabetes products: Trajenta®, Jentadueto®, Jardiance®, Glyxambi®, and Synjardy®, as well as our Basaglar®.
The table below summarizes significant regulatory and commercialization events and milestones (received) paid for the compounds included in this collaboration:
Product Family | Product Status | Milestones (Earned) Expensed(1) | Milestones (Deferred) Capitalized(2) | |||||||||||||
U.S. | Europe | Japan | Year | Amount | Year | Amount | ||||||||||
Trajenta(3) | Launched 2011 | Launched 2011 | Launched 2011 | 2015 | $ | — | 2015 | $ | — | |||||||
2014 | — | 2014 | — | |||||||||||||
2013 | — | 2013 | — | |||||||||||||
Cumulative(6) | 446.4 | |||||||||||||||
Jardiance(4) | Launched 2014 | Launched 2014 | Launched 2015 | 2015 | — | 2015 | — | |||||||||
2014 | — | 2014 | 299.5 | |||||||||||||
2013 | 97.2 | 2013 | — | |||||||||||||
Cumulative(6) | 299.5 | |||||||||||||||
Basaglar | Approved(5) | Launched 2015 | Launched 2015 | 2015 | — | 2015 | — | |||||||||
2014 | — | 2014 | (62.5 | ) | ||||||||||||
2013 | (50.0 | ) | 2013 | — | ||||||||||||
Cumulative(6) | (62.5 | ) | ||||||||||||||
(1) Milestones earned for Basaglar as a result of regulatory submissions were recorded as income in other-net, (income) expense. Milestones expensed for Jardiance as a result of regulatory submissions were recorded as research and development expenses.
(2) In connection with the regulatory approvals of Basaglar in Europe and Japan, milestone payments received were recorded as deferred revenue and are being amortized through the term of the collaboration (2029) to collaboration and other revenue. In connection with the regulatory approvals of Trajenta and Jardiance, milestone payments made were capitalized as intangible assets and are being amortized through the term of the collaboration to cost of sales.
(3) Jentadueto is included in the Trajenta family of product results.
(4) Glyxambi and Synjardy are included in the Jardiance family of product results.
(5) In September 2015, we entered into a settlement agreement to resolve patent infringement litigation filed by Sanofi-Aventis U.S. LLC (Sanofi), which markets Lantus® (insulin glargine). As part of the settlement agreement, the parties agreed that Basaglar can be launched in the U.S. beginning on December 15, 2016. Basaglar received U.S. Food and Drug Administration (FDA) approval in December 2015. As a result of receiving FDA approval, we received $187.5 million in 2016, which was recorded as deferred revenue and will be amortized through the term of the collaboration to collaboration and other revenue upon product launch.
(6) The cumulative amount represents the total amounts as of the end of the reporting period that have been (deferred) or capitalized since the start of this collaboration.
In October 2014, we and Boehringer Ingelheim agreed upon certain changes to the operational and financial structure of our diabetes collaboration. Under the revised agreement the companies have continued their co-promotion work in 17 countries, representing over 90 percent of the collaboration’s anticipated market opportunity. In the other countries, the companies exclusively commercialize the respective molecules they brought to the collaboration. The modifications became effective at the end of 2014 and changed the financial terms related to the modified countries; however, the financial impact resulting from the revised terms of the agreement in these countries has not been and is not anticipated to be material. As a result of these changes, we recorded a gain of $92.0 million in 2014 related to the transfer to Boehringer Ingelheim of our license rights to co-promote linagliptin and empagliflozin in these countries, which was recorded as income in other–net, (income) expense. We also incurred a charge of $55.2 million related to the transfer to us of Boehringer Ingelheim's rights to co-promote Basaglar in countries where it was not yet approved, which was recorded as acquired IPR&D expense.
67
With the exception of the countries affected by the amendment to the collaboration agreement, the companies share equally the ongoing development costs, commercialization costs and gross margin for any product resulting from the collaboration. We record our portion of the gross margin associated with Boehringer Ingelheim's compounds as collaboration and other revenue. We record our sales of Basaglar to third parties as net product revenues with the payments made to Boehringer Ingelheim for their portion of the gross margin recorded as cost of sales. For all compounds under this collaboration, we record our portion of the commercialization costs as marketing, selling, and administrative expense. Each company will also be entitled to potential performance payments on sales of the molecules they contribute to the collaboration.
The following table summarizes our revenue recognized with respect to the Trajenta family of products:
2015 | 2014 | 2013 | |||||||||
Collaboration and other revenue | $ | 356.8 | $ | 328.8 | $ | 249.2 | |||||
Our revenues related to the Jardiance family of products were not significant for the years ended December 31, 2015 and 2014. Our revenues related to Basaglar were not significant for the year ended December 31, 2015.
Erbitux
We have several collaborations with respect to Erbitux. The most significant collaborations are in Japan, and prior to the transfer of commercialization rights in the fourth quarter of 2015, the U.S. and Canada (Bristol-Myers Squibb Company); and worldwide except North America (Merck KGaA). Certain rights to Erbitux outside North America will remain with Merck KGaA (Merck) upon expiration of that agreement.
The following table summarizes our revenue recognized with respect to Erbitux:
2015 | 2014 | 2013 | |||||||||
Net product revenues - BMS | $ | 23.3 | $ | 46.1 | $ | 58.5 | |||||
Net product revenues - third party | 152.3 | — | — | ||||||||
Collaboration and other revenue | 309.4 | 327.2 | 315.2 | ||||||||
Revenue | $ | 485.0 | $ | 373.3 | $ | 373.7 | |||||
Bristol-Myers Squibb Company
Pursuant to commercial agreements with BMS, we had been co-developing Erbitux in North America with BMS exclusively. A separate agreement grants co-exclusive rights among Merck, BMS, and us in Japan and expires in 2032. On October 1, 2015, BMS transferred their commercialization rights to us with respect to Erbitux in North America pursuant to a modification of our existing arrangement, and we began selling Erbitux at that time. This modification did not affect our rights with respect to Erbitux in other jurisdictions. In connection with the modification of terms, we will provide consideration to BMS based upon a tiered percentage of net sales of Erbitux in North America estimated to average 38 percent through September 2018. The transfer of the commercialization rights was accounted for as an acquisition of a business.
68
The following table summarizes the preliminary amounts recognized for assets acquired and liabilities assumed as of the acquisition date:
Estimated Fair Value at October 1, 2015 | |||
Marketed products(1) | $ | 602.1 | |
Deferred tax asset | 232.2 | ||
Deferred tax liability | (228.2 | ) | |
Other assets and liabilities - net | 57.2 | ||
Total identifiable net assets | $ | 663.3 | |
Total consideration - contingent consideration liability(2) | $ | (663.3 | ) |
(1) These intangible assets will be amortized to cost of sales using the straight-line method through the co-development period in North America as set forth in the original agreement, which was scheduled to expire in September 2018.
(2) See Note 7 for discussion on the estimation of the contingent consideration liability.
The final determination of these amounts will be completed as soon as possible but no later than one year from the acquisition date and may result in asset and liability fair values that differ from preliminary estimates, but it is not expected that these differences will be material to our consolidated financial statements.
Including the Erbitux business as if we had acquired it on January 1, 2015, our combined consolidated unaudited pro forma revenue would have been approximately $20.2 billion for the year ended December 31, 2015. This unaudited pro forma financial information adjusts the historical consolidated revenue to give effect to pro forma events that are directly attributable to the acquisition. There would have been no material change to our historical consolidated net income. The unaudited pro forma financial information is not necessarily indicative of what our consolidated revenues would have been had we completed the acquisition at the beginning of 2015. In addition, the unaudited pro forma financial information does not attempt to project the future results of operations of our combined company.
Until the effective date of the transfer of the business, the arrangements between us and BMS were as set forth in this paragraph. Erbitux research and development and other costs were shared by both companies according to a predetermined ratio. Responsibilities associated with clinical and other ongoing studies were apportioned between the parties under the agreements. Collaborative reimbursements due to us for supply of clinical trial materials, for research and development, and for a portion of marketing, selling, and administrative expenses were recorded as a reduction to the respective expense line items on the consolidated statement of operations. We received a distribution fee in the form of a royalty from BMS, based on a percentage of net sales in North America, which was recorded in collaboration and other revenue. Royalties due to third parties were recorded as a reduction of collaboration and other revenue, net of any royalty reimbursements due from third parties. We were responsible for the manufacture and supply of all requirements of Erbitux in bulk-form active pharmaceutical ingredient (API) for clinical and commercial use in North America, and BMS purchased all of its requirements of API from us, subject to certain stipulations per the agreement. Sales of Erbitux API to BMS were reported in net product revenues.
Merck KGaA
A development and license agreement grants Merck exclusive rights to market Erbitux outside of North America until December 2018. A separate agreement grants co-exclusive rights among Merck, BMS, and us in Japan and expires in 2032. This agreement was amended in 2015 to grant Merck exclusive commercialization rights in Japan but did not result in any changes to our rights.
Merck manufactures Erbitux for supply in its territory as well as for Japan. We receive a royalty on the sales of Erbitux outside of North America, which is included in collaboration and other revenue as earned. Royalties due to third parties are recorded as a reduction of collaboration and other revenue, net of any royalty reimbursements due from third parties.
Effient®
We are in a collaborative arrangement with Daiichi Sankyo Co., Ltd. (Daiichi Sankyo) to develop, market, and promote Effient. Marketing rights for major territories are shown below. We and Daiichi Sankyo each have exclusive marketing rights in certain other territories.
69
Territory | Marketing Rights | Selling Party | ||
U.S. | Co-promotion | Lilly | ||
Major European markets | Co-promotion | Pre-January 1, 2016, Lilly Post-January 1, 2016, Daiichi Sankyo | ||
Japan | Exclusive | Daiichi Sankyo | ||
Beginning January 1, 2016, while major European markets continue to be a co-promotion territory under the terms of our arrangement, Daiichi Sankyo exclusively promotes Effient in these markets. The economic results for the major European markets continue to be shared in the same proportion as they were previously.
The parties share approximately 50/50 in the profits, as well as in the costs of development and marketing in the co-promotion territories. A third party manufactures bulk product, and we continue to produce the finished product for our exclusive and co-promotion territories, as well as the major European markets.
We record net product revenue in our exclusive and co-promotion territories where we are the selling party. Profit-share payments due to Daiichi Sankyo for co-promotion countries where we are the selling party are recorded as marketing, selling, and administrative expenses. Beginning January 1, 2016, any profit-share payments due to us from Daiichi Sankyo for the major European markets will be recorded as collaboration and other revenue. We also record our share of the expenses in these co-promotion territories as marketing, selling, and administrative expenses. In our exclusive territories, we pay Daiichi Sankyo a royalty specific to these territories. All royalties due to Daiichi Sankyo and the third-party manufacturer are recorded in cost of sales.
The following table summarizes our revenue recognized with respect to Effient:
2015 | 2014 | 2013 | |||||||||
Revenue | $ | 523.0 | $ | 522.2 | $ | 508.7 | |||||
Baricitinib
We have a worldwide license and collaboration agreement with Incyte Corporation (Incyte) which provides us the development and commercialization rights to its Janus tyrosine kinase inhibitor compound, now known as baricitinib, and certain follow-on compounds, for the treatment of inflammatory and autoimmune diseases. Incyte has the right to receive tiered, double-digit royalty payments on future global sales with rates ranging up to 20 percent if the product is successfully commercialized. The agreement provides Incyte with options to co-develop these compounds on an indication-by-indication basis by funding 30 percent of the associated development costs from the initiation of a Phase IIb trial through regulatory approval in exchange for increased tiered royalties ranging up to percentages in the high twenties. In 2010, Incyte exercised its option to co-develop baricitinib in rheumatoid arthritis. The agreement also provides Incyte with an option to co-promote in the U.S. and calls for us to make payments to Incyte associated with certain development, success-based regulatory, and sales-based milestones. In 2016, we incurred milestone-related expenses of $55.0 million in connection with regulatory submissions in the U.S. and Europe. These regulatory submission milestones will be recorded as research and development expenses in 2016. In the future, Incyte will be eligible to receive up to $360.0 million of additional payments from us contingent upon certain development and success-based regulatory milestones as well as an additional $150.0 million of potential sales-based milestones.
Solanezumab
We have an agreement with an affiliate of TPG-Axon Capital (TPG) whereby TPG funded a portion of the Phase III development of solanezumab. Under the agreement, TPG’s obligation to fund solanezumab costs ended in 2011. In exchange for its funding, TPG may receive success-based sales milestones totaling approximately $70 million and mid-single digit royalties contingent upon the successful development of solanezumab. The royalties would be paid for approximately 10 years after launch of a product.
70
Tanezumab
In October 2013, we entered into a collaboration agreement with Pfizer Inc. (Pfizer) to jointly develop and globally commercialize tanezumab for the treatment of osteoarthritis pain, chronic low back pain and cancer pain. Under the agreement, the companies share equally the ongoing development costs and, if successful, in gross margins and certain commercialization expenses. Following the FDA's decision in March 2015 to lift the partial clinical hold on tanezumab, certain Phase III trials resumed in July 2015. Upon the FDA's lifting of the partial clinical hold and the decision to continue the collaboration with Pfizer, we paid an upfront fee of $200.0 million which was expensed as acquired IPR&D. In addition to this fee, we may pay up to $350.0 million in success-based regulatory milestones and up to $1.23 billion in a series of sales-based milestones, contingent upon the commercial success of tanezumab. Both parties have the right to terminate the agreement under certain circumstances.
Exenatide
In November 2011, we agreed with Amylin Pharmaceuticals, Inc. (Amylin) to terminate our collaborative arrangement for the joint development, marketing, and selling of Byetta® (exenatide injection) and other forms of exenatide such as Bydureon® (exenatide extended-release for injectable suspension). Under the terms of the termination agreement, Amylin made a one-time, upfront payment to us of $250.0 million and agreed to make other payments. Upon completion of the acquisition of Amylin by Bristol-Myers Squibb Company in August 2012, Amylin's obligation of $1.26 billion was paid in full. We would also receive a $150.0 million milestone payment contingent upon FDA approval of a once-monthly suspension version of exenatide.
Commercial operations were transferred to Amylin in the U.S. in late 2011. Outside the U.S., we transferred to Amylin exenatide commercial rights and control in all markets during the first quarter of 2013. All income allocated to the business outside the U.S. that was transferred during the first quarter of 2013 was recognized as a gain on the disposition of a business in other–net, (income) expense, net of the goodwill allocated to the business transferred.
Under the terms of our prior arrangement, we reported as net product revenues 100 percent of sales outside the U.S. We paid Amylin a percentage of the gross margin of exenatide sales outside of the U.S., and these costs were recorded in cost of sales. This arrangement for the commercial operations outside the U.S. continued until those rights were transferred to Amylin during the first quarter of 2013.
The following table summarizes the revenue and other income recognized with respect to exenatide for the year ended December 31, 2013:
2013 | |||
Revenue | $ | 133.1 | |
Income related to termination of the exenatide collaboration with Amylin(1) | 495.4 | ||
(1) Presented in other-net, (income) expense.
Our revenue from exenatide was not significant in 2014. We have not recorded any additional revenue from exenatide in 2015 and will not do so in future periods.
Summary of Commission and Profit-Share Payments
The following table summarizes our aggregate amount of marketing, selling, and administrative expense associated with our commission and profit-sharing obligations for the collaborations and other arrangements described above:
2015 | 2014 | 2013 | |||||||||
Marketing, selling, and administrative | $ | 213.2 | $ | 211.2 | $ | 203.7 | |||||
71
Note 5: Asset Impairment, Restructuring, and Other Special Charges
The components of the charges included in asset impairment, restructuring, and other special charges in our consolidated statements of operations are described below.
2015 | 2014 | 2013 | |||||||||
Severance: | |||||||||||
Human pharmaceutical products | $ | 81.5 | $ | 225.5 | $ | 90.6 | |||||
Animal health | 59.5 | — | — | ||||||||
Total severance | 141.0 | 225.5 | 90.6 | ||||||||
Asset impairment and other special charges: | |||||||||||
Human pharmaceutical products | 24.6 | 204.4 | 30.0 | ||||||||
Animal health | 202.1 | 38.8 | — | ||||||||
Total asset impairment and other special charges | 226.7 | 243.2 | 30.0 | ||||||||
Asset impairment, restructuring, and other special charges | $ | 367.7 | $ | 468.7 | $ | 120.6 | |||||
Severance costs recognized during the year ended December 31, 2015 resulted primarily from actions taken to reduce our cost structure and the integration of Novartis AH. Severance costs recognized during the years ended December 31, 2014 and 2013 related to ongoing cost containment efforts as we continued our initiatives to reduce our cost structure and global workforce. Substantially all of the severance costs incurred during the year ended December 31, 2015 are expected to be paid by the end of 2016, and substantially all of the severance costs incurred during the years ended December 31, 2014 and 2013 have been paid.
Asset impairment and other special charges recognized during year ended December 31, 2015 resulted primarily from integration costs and asset impairments due to product rationalization and site closures resulting from our acquisition and integration of Novartis AH.
Asset impairment and other special charges recognized during the year ended December 31, 2014 resulted primarily from a $180.8 million asset impairment charge related to our decision to close and sell a manufacturing plant located in Puerto Rico. The manufacturing plant was written down to its estimated fair value, which was based primarily on recent sales of similar assets.
Asset impairment and other special charges recognized during the year ended December 31, 2013 resulted from costs associated with the closure of a packaging and distribution facility in Germany.
In January 2016, we approved a plan to close an animal health manufacturing plant located in Ireland. As a result of this action, we expect to record charges of approximately $100 million in our animal health business segment during the first quarter of 2016.
Note 6: Inventories
We state all inventories at the lower of cost or market. We use the last-in, first-out (LIFO) method for the majority of our inventories located in the continental U.S. Other inventories are valued by the first-in, first-out (FIFO) method. FIFO cost approximates current replacement cost.
Inventories at December 31 consisted of the following:
2015 | 2014 | ||||||
Finished products | $ | 1,053.4 | $ | 838.0 | |||
Work in process | 2,058.1 | 1,715.4 | |||||
Raw materials and supplies | 403.0 | 315.0 | |||||
Total (approximates replacement cost) | 3,514.5 | 2,868.4 | |||||
Reduction to LIFO cost | (68.7 | ) | (128.4 | ) | |||
Inventories | $ | 3,445.8 | $ | 2,740.0 | |||
Inventories valued under the LIFO method comprised $1.30 billion and $1.09 billion of total inventories at December 31, 2015 and 2014, respectively.
72
Note 7: Financial Instruments
Financial instruments that potentially subject us to credit risk consist principally of trade receivables and interest-bearing investments. Wholesale distributors of life-science products account for a substantial portion of trade receivables; collateral is generally not required. The risk associated with this concentration is mitigated by our ongoing credit-review procedures and insurance. A large portion of our cash is held by a few major financial institutions. We monitor our exposures with these institutions and do not expect any of these institutions to fail to meet their obligations. Major financial institutions represent the largest component of our investments in corporate debt securities. In accordance with documented corporate policies, we monitor the amount of credit exposure to any one financial institution or corporate issuer. We are exposed to credit-related losses in the event of nonperformance by counterparties to risk-management instruments but do not expect any counterparties to fail to meet their obligations given their high credit ratings.
We consider all highly liquid investments with a maturity of three months or less from the date of purchase to be cash equivalents. The cost of these investments approximates fair value.
Substantially all of our investments in debt and marketable equity securities are classified as available-for-sale. Investment securities with maturity dates of less than one year from the date of the balance sheet are classified as short-term. Available-for-sale securities are carried at fair value with the unrealized gains and losses, net of tax, reported in other comprehensive income (loss). The credit portion of unrealized losses on our debt securities considered to be other-than-temporary is recognized in earnings. The remaining portion of the other-than-temporary impairment on our debt securities is then recorded, net of tax, in other comprehensive income (loss). The entire amount of other-than-temporary impairment on our equity securities is recognized in earnings. We do not evaluate cost-method investments for impairment unless there is an indicator of impairment. We review these investments for indicators of impairment on a regular basis. Investments in companies over which we have significant influence but not a controlling interest are accounted for using the equity method with our share of earnings or losses reported in other–net, (income) expense. We own no investments that are considered to be trading securities.
Our derivative activities are initiated within the guidelines of documented corporate risk-management policies and offset losses and gains on the assets, liabilities, and transactions being hedged. Management reviews the correlation and effectiveness of our derivatives on a quarterly basis.
For derivative contracts that are designated and qualify as fair value hedges, the derivative instrument is marked to market with gains and losses recognized currently in income to offset the respective losses and gains recognized on the underlying exposure. For derivative contracts that are designated and qualify as cash flow hedges, the effective portion of gains and losses on these contracts is reported as a component of accumulated other comprehensive loss and reclassified into earnings in the same period the hedged transaction affects earnings. Hedge ineffectiveness is immediately recognized in earnings. Derivative contracts that are not designated as hedging instruments are recorded at fair value with the gain or loss recognized in current earnings during the period of change.
We may enter into foreign currency forward or option contracts to reduce the effect of fluctuating currency exchange rates (principally the euro, the British pound, and the Japanese yen). Foreign currency derivatives used for hedging are put in place using the same or like currencies and duration as the underlying exposures. Forward and option contracts are principally used to manage exposures arising from subsidiary trade and loan payables and receivables denominated in foreign currencies. These contracts are recorded at fair value with the gain or loss recognized in other–net, (income) expense. We may enter into foreign currency forward and option contracts and currency swaps as fair value hedges of firm commitments. Forward contracts generally have maturities not exceeding 12 months. At December 31, 2015, we had outstanding foreign currency forward commitments to purchase 1.17 billion U.S. dollars and sell 1.06 billion euro; commitments to purchase 1.85 billion euro and sell 2.04 billion U.S. dollars; commitments to purchase 187.7 million British pounds and sell 258.4 million euro; commitments to purchase 288.3 million U.S. dollars and sell 190.6 million British pounds, and commitments to purchase 561.7 million U.S. dollars and sell 67.78 billion Japanese yen, which will all settle within 30 days.
73
Foreign currency exchange risk is also managed through the use of foreign currency debt. Our euro-denominated notes issued in June 2015 and discussed in Note 10, which had a carrying amount of $2.27 billion as of December 31, 2015, have been designated as, and are effective as, economic hedges of net investments in certain of our euro-denominated foreign operations. Accordingly, foreign currency translation gains or losses due to spot rate fluctuations on the euro-denominated notes are included as a component of other comprehensive income (loss). During the year ended December 31, 2015, we recorded a pretax foreign currency translation gain of $5.6 million from the euro-denominated notes.
In the normal course of business, our operations are exposed to fluctuations in interest rates which can vary the costs of financing, investing, and operating. We address a portion of these risks through a controlled program of risk management that includes the use of derivative financial instruments. The objective of controlling these risks is to limit the impact of fluctuations in interest rates on earnings. Our primary interest-rate risk exposure results from changes in short-term U.S. dollar interest rates. In an effort to manage interest-rate exposures, we strive to achieve an acceptable balance between fixed- and floating-rate debt and investment positions and may enter into interest rate swaps or collars to help maintain that balance.
Interest rate swaps or collars that convert our fixed-rate debt to a floating rate are designated as fair value hedges of the underlying instruments. Interest rate swaps or collars that convert floating-rate debt to a fixed rate are designated as cash flow hedges. Interest expense on the debt is adjusted to include the payments made or received under the swap agreements. Cash proceeds from or payments to counterparties resulting from the termination of interest rate swaps are classified as operating activities in our consolidated statement of cash flows. At December 31, 2015, substantially all of our total long-term debt is at a fixed rate. We have converted approximately 40 percent of our long-term fixed-rate notes to floating rates through the use of interest rate swaps.
We may enter into forward contracts and designate them as cash flow hedges to limit the potential volatility of earnings and cash flow associated with forecasted sales of available-for-sale securities.
We also may enter into forward-starting interest rate swaps, which we designate as cash flow hedges, as part of any anticipated future debt issuances in order to reduce the risk of cash flow volatility from future changes in interest rates. Upon completion of a debt issuance and termination of the swap, the change in fair value of these instruments is recorded as part of other comprehensive income (loss) and is amortized to interest expense over the life of the underlying debt.
The Effect of Risk Management Instruments on the Consolidated Statement of Operations
The following effects of risk-management instruments were recognized in other–net, (income) expense:
2015 | 2014 | 2013 | |||||||||
Fair value hedges: | |||||||||||
Effect from hedged fixed-rate debt | $ | (11.9 | ) | $ | 156.9 | $ | (308.2 | ) | |||
Effect from interest rate contracts | 11.9 | (156.9 | ) | 308.2 | |||||||
Cash flow hedges: | |||||||||||
Effective portion of losses on equity contracts reclassified from accumulated other comprehensive loss(1) | — | 129.0 | — | ||||||||
Effective portion of losses on interest rate contracts reclassified from accumulated other comprehensive loss | 13.7 | 9.0 | 9.0 | ||||||||
Net (gains) losses on foreign currency exchange contracts not designated as hedging instruments | (28.2 | ) | (20.4 | ) | 15.4 | ||||||
(1) Realized gains on the sale of underlying equity securities recognized in other-net, (income) expense were $260.8 million during the year ended December 31, 2014. There were no realized gains on the sale of underlying equity securities during the years ended December 31, 2015 and 2013.
During the years ended December 31, 2015, 2014, and 2013, net losses related to ineffectiveness, as well as net losses related to the portion of our risk-management hedging instruments, fair value hedges, and cash flow hedges that were excluded from the assessment of effectiveness, were not material.
Fair Value Hedges
During the years ended December 31, 2015 and 2014, we terminated certain interest rate swaps designated as fair value hedges with an aggregate notional amount of $876.0 million and $1.30 billion, respectively. The termination of certain interest rate swaps in 2015 was in connection with the note purchase and redemption discussed at Note 10. As a result of the termination, we received cash of $20.2 million and $340.7 million in
74
2015 and 2014, respectively, which represented the fair value of the interest rate swaps at the time of termination. In 2015, the related fair value adjustment was recorded as an increase to the carrying value of the underlying notes and was included as a component of the debt extinguishment loss. In 2014, the related fair value was recorded as an increase to the carrying value of the underlying notes and is being amortized into earnings as a reduction of interest expense over the remaining life of the underlying debt.
Cash Flow Hedges
The effective portion of equity contracts and forward-starting interest rate swaps in designated cash flow hedging relationships recorded in other comprehensive income (loss) was as follows:
2015 | 2014 | 2013 | |||||||||
Equity contracts | $ | — | $ | 149.6 | $ | (149.6 | ) | ||||
Forward-starting interest rate swaps | (56.7 | ) | (164.7 | ) | 16.7 | ||||||
Upon issuance of the underlying fixed-rate notes in March 2015, which are discussed in Note 10, we terminated forward-starting interest rate contracts in designated cash flow hedging instruments with an aggregate notional amount of $1.35 billion and paid $206.3 million in cash to the counterparties for settlement. The settlement amount represented the fair value of the forward-starting interest rate contracts at the time of termination and was recorded in other comprehensive income (loss).
During the year ended December 31, 2014, we sold all of the underlying equity securities that had been in designated cash flow hedging relationships. At the time of the sales, we reclassified to earnings the accumulated other comprehensive loss related to the cash flow hedges and the previously unrealized gains on the underlying equity securities.
During the next 12 months, we expect to reclassify from accumulated other comprehensive loss to earnings $14.8 million of pretax net losses on cash flow hedges of the variability in expected future interest payments on our floating rate debt.
75
Fair Value of Financial Instruments
The following tables summarize certain fair value information at December 31 for assets and liabilities measured at fair value on a recurring basis, as well as the carrying amount and amortized cost of certain other investments:
Fair Value Measurements Using | |||||||||||||||||||||||
Description | Carrying Amount | Cost (1) | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | Fair Value | |||||||||||||||||
December 31, 2015 | |||||||||||||||||||||||
Cash equivalents | $ | 1,644.4 | $ | 1,644.4 | $ | 1,637.0 | $ | 7.4 | $ | — | $ | 1,644.4 | |||||||||||
Short-term investments: | |||||||||||||||||||||||
U.S. government and agency securities | $ | 153.2 | $ | 153.4 | $ | 153.2 | $ | — | $ | — | $ | 153.2 | |||||||||||
Corporate debt securities | 625.8 | 626.9 | — | 625.8 | — | 625.8 | |||||||||||||||||
Asset-backed securities | 3.3 | 3.3 | — | 3.3 | — | 3.3 | |||||||||||||||||
Other securities | 3.1 | 3.1 | — | 3.1 | — | 3.1 | |||||||||||||||||
Short-term investments | $ | 785.4 | $ | 786.7 | |||||||||||||||||||
Noncurrent investments: | |||||||||||||||||||||||
U.S. government and agency securities | $ | 284.5 | $ | 286.0 | $ | 283.5 | $ | 1.0 | $ | — | $ | 284.5 | |||||||||||
Corporate debt securities | 1,962.6 | 1,995.8 | — | 1,962.6 | — | 1,962.6 | |||||||||||||||||
Mortgage-backed securities | 153.3 | 154.7 | — | 153.3 | — | 153.3 | |||||||||||||||||
Asset-backed securities | 441.9 | 443.1 | — | 441.9 | — | 441.9 | |||||||||||||||||
Other securities | 4.1 | 4.1 | — | 4.1 | — | 4.1 | |||||||||||||||||
Marketable equity securities | 128.9 | 74.8 | 128.9 | — | — | 128.9 | |||||||||||||||||
Other investments(2) | 671.3 | 671.3 | |||||||||||||||||||||
Noncurrent investments | $ | 3,646.6 | $ | 3,629.8 | |||||||||||||||||||
December 31, 2014 | |||||||||||||||||||||||
Cash equivalents | $ | 2,443.5 | $ | 2,443.5 | $ | 2,415.5 | $ | 28.0 | $ | — | $ | 2,443.5 | |||||||||||
Short-term investments: | |||||||||||||||||||||||
U.S. government and agency securities | $ | 185.5 | $ | 185.6 | $ | 156.5 | $ | 29.0 | $ | — | $ | 185.5 | |||||||||||
Corporate debt securities | 767.4 | 766.7 | — | 767.4 | — | 767.4 | |||||||||||||||||
Other securities | 2.5 | 2.5 | — | 2.5 | — | 2.5 | |||||||||||||||||
Short-term investments | $ | 955.4 | $ | 954.8 | |||||||||||||||||||
Noncurrent investments: | |||||||||||||||||||||||
U.S. government and agency securities | $ | 756.7 | $ | 757.5 | $ | 747.5 | $ | 9.2 | $ | — | $ | 756.7 | |||||||||||
Corporate debt securities | 2,462.7 | 2,468.9 | — | 2,462.7 | — | 2,462.7 | |||||||||||||||||
Mortgage-backed securities | 217.0 | 217.6 | — | 217.0 | — | 217.0 | |||||||||||||||||
Asset-backed securities | 477.8 | 478.0 | — | 477.8 | — | 477.8 | |||||||||||||||||
Other securities | 3.2 | 3.2 | — | 3.2 | — | 3.2 | |||||||||||||||||
Marketable equity securities | 204.8 | 44.0 | 204.8 | — | — | 204.8 | |||||||||||||||||
Other investments(2) | 446.7 | 446.7 | |||||||||||||||||||||
Noncurrent investments | $ | 4,568.9 | $ | 4,415.9 | |||||||||||||||||||
(1) For available-for-sale debt securities, amounts disclosed represent the securities' amortized cost.
(2) Primarily includes investments accounted for under the cost method and equity method for which fair value disclosures are not applicable.
76
Fair Value Measurements Using | |||||||||||||||||||
Description | Carrying Amount | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | Fair Value | ||||||||||||||
Short-term commercial paper borrowings | |||||||||||||||||||
December 31, 2015 | $ | — | $ | — | $ | — | $ | — | $ | — | |||||||||
December 31, 2014 | (2,680.6 | ) | — | (2,680.6 | ) | — | (2,680.6 | ) | |||||||||||
Long-term debt, including current portion | |||||||||||||||||||
December 31, 2015 | $ | (7,978.5 | ) | $ | — | $ | (8,172.0 | ) | $ | — | $ | (8,172.0 | ) | ||||||
December 31, 2014 | (5,340.9 | ) | — | (5,722.1 | ) | — | (5,722.1 | ) | |||||||||||
Fair Value Measurements Using | |||||||||||||||||||
Description | Carrying Amount | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | Fair Value | ||||||||||||||
December 31, 2015 | |||||||||||||||||||
Risk-management instruments | |||||||||||||||||||
Interest rate contracts designated as hedging instruments: | |||||||||||||||||||
Sundry | $ | 70.1 | $ | — | 70.1 | $ | — | $ | 70.1 | ||||||||||
Other noncurrent liabilities | (0.4 | ) | — | (0.4 | ) | — | (0.4 | ) | |||||||||||
Foreign exchange contracts not designated as hedging instruments: | |||||||||||||||||||
Other receivables | 13.1 | — | 13.1 | — | 13.1 | ||||||||||||||
Other current liabilities | (17.3 | ) | — | (17.3 | ) | — | (17.3 | ) | |||||||||||
Contingent consideration liability(1): | |||||||||||||||||||
Other current liabilities | (243.7 | ) | — | — | (243.7 | ) | (243.7 | ) | |||||||||||
Other noncurrent liabilities | (427.2 | ) | — | — | (427.2 | ) | (427.2 | ) | |||||||||||
December 31, 2014 | |||||||||||||||||||
Risk-management instruments | |||||||||||||||||||
Interest rate contracts designated as hedging instruments: | |||||||||||||||||||
Sundry | $ | 102.5 | $ | — | $ | 102.5 | $ | — | $ | 102.5 | |||||||||
Other current liabilities | (149.5 | ) | — | (149.5 | ) | — | (149.5 | ) | |||||||||||
Other noncurrent liabilities | (0.7 | ) | — | (0.7 | ) | — | (0.7 | ) | |||||||||||
Foreign exchange contracts not designated as hedging instruments: | |||||||||||||||||||
Other receivables | 9.1 | — | 9.1 | — | 9.1 | ||||||||||||||
Other current liabilities | (14.0 | ) | — | (14.0 | ) | — | (14.0 | ) | |||||||||||
(1) The contingent consideration liability relates to the Erbitux arrangement with BMS discussed in Note 4.
Risk-management instruments above are disclosed on a gross basis. There are various rights of setoff associated with certain of the risk-management instruments above that are subject to an enforceable master netting arrangement or similar agreements. Although various rights of setoff and master netting arrangements or similar agreements may exist with the individual counterparties to the risk-management instruments above, individually, these financial rights are not material.
We determine our Level 1 and Level 2 fair value measurements based on a market approach using quoted market values, significant other observable inputs for identical or comparable assets or liabilities, or discounted cash flow analyses. The fair value of equity method investments and other investments is not readily available.
77
The fair value of the Erbitux contingent consideration liability was estimated using a discounted cash flow analysis and Level 3 inputs, including projections representative of a market participant view for net sales in North America over the three-year period ending in September 2018 and an estimated discount rate. The amount to be paid is calculated as a tiered percentage of net sales (see Note 4) and will, therefore, vary directly with increases and decreases in net sales of Erbitux in North America. There is no cap on the amount that may be paid pursuant to this arrangement.
The table below summarizes the contractual maturities of our investments in debt securities measured at fair value as of December 31, 2015:
Maturities by Period | |||||||||||||||||||
Total | Within 1 Year | After 1 Year Through 5 Years | After 5 Years Through 10 Years | After 10 Years | |||||||||||||||
Fair value of debt securities | $ | 3,631.8 | $ | 785.4 | $ | 2,516.8 | $ | 164.0 | $ | 165.6 | |||||||||
A summary of the fair value of available-for-sale securities in an unrealized gain or loss position and the amount of unrealized gains and losses (pretax) in accumulated other comprehensive loss follows:
2015 | 2014 | ||||||
Unrealized gross gains | $ | 68.0 | $ | 171.9 | |||
Unrealized gross losses | 52.5 | 18.3 | |||||
Fair value of securities in an unrealized gain position | 764.5 | 1,778.8 | |||||
Fair value of securities in an unrealized loss position | 2,933.4 | 3,129.2 | |||||
We periodically assess our investment securities for other-than-temporary impairment losses. Other-than-temporary impairment losses recognized during the year ended December 31, 2015 totaled $42.6 million and related primarily to our equity method and other investments. Other-than-temporary impairment losses recognized during the years ended December 31, 2014, and 2013 totaled $12.5 million and $11.3 million, respectively.
For fixed-income securities, the amount of credit losses are determined by comparing the difference between the present value of future cash flows expected to be collected on these securities and the amortized cost. Factors considered in assessing the credit losses include the position in the capital structure, vintage and amount of collateral, delinquency rates, current credit support, and geographic concentration.
For equity securities, factors considered in assessing other-than-temporary impairment losses include the length of time and the extent to which the fair value has been less than cost, the financial condition and near term prospects of the issuer, our intent and ability to retain the securities for a period of time sufficient to allow for recovery in fair value, and general market conditions and industry specific factors.
As of December 31, 2015, the securities in an unrealized loss position include primarily fixed-rate debt securities of varying maturities. The value of fixed-income securities is sensitive to changes in the yield curve and other market conditions. Approximately 85 percent of the securities in a loss position are investment-grade debt securities. As of December 31, 2015, we do not intend to sell, and it is not more likely than not that we will be required to sell, the securities in a loss position before the market values recover or the underlying cash flows have been received, and there is no indication of default on interest rate or principal payments for any of our debt securities.
Activity related to our investment portfolio, substantially all of which related to available-for-sale securities, was as follows:
2015 | 2014 | 2013 | |||||||||
Proceeds from sales | $ | 4,733.3 | $ | 14,609.5 | $ | 13,753.5 | |||||
Realized gross gains on sales | 255.1 | 353.5 | 49.5 | ||||||||
Realized gross losses on sales | 10.3 | 29.4 | 15.4 | ||||||||
Realized gains and losses on sales of investments are computed based upon specific identification of the initial cost adjusted for any other-than-temporary declines in fair value that were recorded in earnings.
78
Note 8: Goodwill and Other Intangibles
Goodwill
Goodwill by segment at December 31 was as follows:
2015 | 2014 | ||||||
Human pharmaceutical products | $ | 1,366.5 | $ | 1,359.4 | |||
Animal health | 2,673.4 | 398.7 | |||||
Total goodwill | $ | 4,039.9 | $ | 1,758.1 | |||
Goodwill results from excess consideration in a business combination over the fair value of identifiable net assets acquired. Goodwill is not amortized but is reviewed for impairment at least annually and when impairment indicators are present. When required, a comparison of implied fair value to the carrying amount of goodwill is performed to determine the amount of any impairment. The increase in goodwill for the animal health segment in 2015 is a result of the acquisition of Novartis AH (Note 3).
No impairments occurred with respect to the carrying value of goodwill for the years ended December 31, 2015, 2014, and 2013.
Other Intangibles
The components of intangible assets other than goodwill at December 31 were as follows:
2015 | 2014 | ||||||||||||||||||||||
Description | Carrying Amount— Gross | Accumulated Amortization | Carrying Amount— Net | Carrying Amount— Gross | Accumulated Amortization | Carrying Amount— Net | |||||||||||||||||
Finite-lived intangible assets: | |||||||||||||||||||||||
Marketed products | $ | 7,528.0 | $ | (2,706.4 | ) | $ | 4,821.6 | $ | 5,684.3 | $ | (2,915.6 | ) | $ | 2,768.7 | |||||||||
Other | 151.1 | (115.5 | ) | 35.6 | 149.3 | (45.2 | ) | 104.1 | |||||||||||||||
Total finite-lived intangible assets | 7,679.1 | (2,821.9 | ) | 4,857.2 | 5,833.6 | (2,960.8 | ) | 2,872.8 | |||||||||||||||
Indefinite-lived intangible assets: | |||||||||||||||||||||||
Acquired in-process research and development | 177.6 | — | 177.6 | 11.4 | — | 11.4 | |||||||||||||||||
Other intangibles | $ | 7,856.7 | $ | (2,821.9 | ) | $ | 5,034.8 | $ | 5,845.0 | $ | (2,960.8 | ) | $ | 2,884.2 | |||||||||
Marketed products consist of the amortized cost of the rights to assets acquired in business combinations and approved for marketing in a significant global jurisdiction (U.S., Europe, and Japan) and capitalized milestone payments. For transactions other than a business combination, we capitalize milestone payments incurred at or after the product has obtained regulatory approval for marketing.
Other finite-lived intangibles consist primarily of the amortized cost of licensed platform technologies that have alternative future uses in research and development, manufacturing technologies, and customer relationships from business combinations.
Acquired IPR&D consists of the related costs capitalized, adjusted for subsequent impairments, if any. The costs of acquired IPR&D projects acquired directly in a transaction other than a business combination are capitalized if the projects have an alternative future use; otherwise, they are expensed immediately. The fair values of acquired IPR&D projects acquired in business combinations are capitalized as other intangible assets.
79
Several methods may be used to determine the estimated fair value of other intangibles acquired in a business combination. We utilize the “income method,” which is a Level 3 fair value measurement and applies a probability weighting that considers the risk of development and commercialization to the estimated future net cash flows that are derived from projected revenues and estimated costs. These projections are based on factors such as relevant market size, patent protection, historical pricing of similar products, and expected industry trends. The estimated future net cash flows are then discounted to the present value using an appropriate discount rate. This analysis is performed for each asset independently. The acquired IPR&D assets are treated as indefinite-lived intangible assets until completion or abandonment of the projects, at which time the assets are tested for impairment and amortized over the remaining useful life or written off, as appropriate.
See Note 3 for further discussion of intangible assets acquired in recent business combinations and Note 4 for additional discussion of recent capitalized milestone payments. The increases in marketed products and acquired IPR&D assets in 2015 are primarily due to the acquisition of Novartis AH and the transfer of the Erbitux commercialization rights discussed in Notes 3 and 4, respectively.
Other indefinite-lived intangible assets are reviewed for impairment at least annually and when impairment indicators are present. When required, a comparison of fair value to the carrying amount of assets is performed to determine the amount of any impairment. When determining the fair value of indefinite-lived acquired IPR&D assets for impairment testing purposes, we utilize the "income method" discussed above. Finite-lived intangible assets are reviewed for impairment when an indicator of impairment is present. No material impairments occurred with respect to the carrying value of other intangible assets for the years ended December 31, 2015, 2014 and 2013.
Intangible assets with finite lives are capitalized and are amortized over their estimated useful lives, ranging from 3 to 20 years. As of December 31, 2015, the remaining weighted-average amortization period for finite-lived intangible assets is approximately 12 years.
Amortization expense related to finite-lived intangible assets was as follows:
2015 | 2014 | 2013 | |||||||||
Amortization expense | $ | 631.8 | $ | 535.9 | $ | 555.0 | |||||
The estimated amortization expense associated with our current finite-lived intangible assets for each of the next five years is as follows:
2016 | 2017 | 2018 | 2019 | 2020 | |||||||||||||||
Estimated amortization expense | $ | 674.7 | $ | 641.8 | $ | 480.4 | $ | 302.7 | $ | 301.4 | |||||||||
Amortization expense is included in either cost of sales, marketing, selling, and administrative or research and development depending on the nature of the intangible asset being amortized.
80
Note 9: Property and Equipment
Property and equipment is stated on the basis of cost. Provisions for depreciation of buildings and equipment are computed generally by the straight-line method at rates based on their estimated useful lives (12 to 50 years for buildings and 3 to 25 years for equipment). We review the carrying value of long-lived assets for potential impairment on a periodic basis and whenever events or changes in circumstances indicate the carrying value of an asset may not be recoverable. Impairment is determined by comparing projected undiscounted cash flows to be generated by the asset to its carrying value. If an impairment is identified, a loss is recorded equal to the excess of the asset’s net book value over its fair value, and the cost basis is adjusted.
At December 31, property and equipment consisted of the following:
2015 | 2014 | ||||||
Land | $ | 220.6 | $ | 205.2 | |||
Buildings | 6,786.5 | 6,516.2 | |||||
Equipment | 7,988.5 | 7,609.7 | |||||
Construction in progress | 1,665.3 | 1,698.2 | |||||
16,660.9 | 16,029.3 | ||||||
Less accumulated depreciation | (8,607.4 | ) | (8,065.4 | ) | |||
Property and equipment, net | $ | 8,053.5 | $ | 7,963.9 | |||
Depreciation expense related to property and equipment and rental expense for all leases, including contingent rentals (not material), was as follows:
2015 | 2014 | 2013 | |||||||||
Depreciation expense | $ | 717.6 | $ | 759.1 | $ | 774.8 | |||||
Rental expense | 225.7 | 227.3 | 227.2 | ||||||||
Capitalized interest costs were not material for the years ended December 31, 2015, 2014, and 2013.
Assets under capital leases included in property and equipment, net on the consolidated balance sheets, capital lease obligations entered into, and future minimum rental commitments are not material.
Note 10: Borrowings
Debt at December 31 consisted of the following:
2015 | 2014 | ||||||
Short-term commercial paper borrowings | $ | — | $ | 2,680.6 | |||
1.00 to 7.13 percent long-term notes (due 2017-2045) | 7,700.1 | 4,872.8 | |||||
Other long-term debt, including capitalized leases | 23.1 | 33.1 | |||||
Unamortized debt issuance costs | (37.1 | ) | (20.4 | ) | |||
Fair value adjustment on hedged long-term notes | 292.4 | 455.4 | |||||
Total debt | 7,978.5 | 8,021.5 | |||||
Less current portion | (6.1 | ) | (2,688.7 | ) | |||
Long-term debt | $ | 7,972.4 | $ | 5,332.8 | |||
There were no outstanding borrowings under our commercial paper program at December 31, 2015. We had $2.68 billion in outstanding borrowings under our commercial paper program at December 31, 2014, with a weighted-average effective borrowing rate of 0.18 percent.
At December 31, 2015, we had a total of $1.30 billion of unused committed bank credit facilities, which consisted primarily of a $1.20 billion credit facility that expires in August 2020 and is available to support our commercial paper program. During the year ended December 31, 2015, our $2.00 billion 364-day credit facility expired unused and was not renewed. There were no amounts outstanding under the revolving credit facilities during the years ended December 31, 2015 and 2014. Compensating balances and commitment fees are not material, and there are no conditions that are probable of occurring under which the lines may be withdrawn.
In March 2015, we issued $600.0 million of 1.25 percent fixed-rate notes due March 1, 2018, $800.0 million of 2.75 percent fixed-rate notes due June 1, 2025, and $800.0 million of 3.70 percent fixed-rate notes
81
due March 1, 2045 with interest to be paid semi-annually. The proceeds from the issuance of the notes were used primarily to repay outstanding commercial paper issued in connection with our January 2015 acquisition of Novartis AH.
In June 2015, we issued euro-denominated notes consisting of €600.0 million of 1.00 percent fixed-rate notes due June 2, 2022, €750.0 million of 1.63 percent fixed-rate notes due June 2, 2026, and €750.0 million of 2.13 percent fixed-rate notes due June 3, 2030 with interest to be paid annually. The net cash proceeds of the offering of $2.27 billion were used primarily to purchase and redeem certain higher interest rate U.S. dollar-denominated notes and to repay outstanding commercial paper. We paid $1.95 billion to purchase and redeem notes with an aggregate principal amount of $1.65 billion and a net carrying value of $1.78 billion in June 2015, resulting in a pretax debt extinguishment loss of $166.7 million, which was included in other–net, (income) expense in our consolidated statement of operations during the year ended December 31, 2015.
In February 2014, we issued $600.0 million of 1.95 percent and $400.0 million of 4.65 percent fixed-rate notes with interest to be paid semi-annually and maturity dates of March 15, 2019, and June 15, 2044, respectively. Current maturities of long-term notes of $1.00 billion were repaid in March 2014.
The aggregate amounts of maturities on long-term debt for the next five years are as follows:
2016 | 2017 | 2018 | 2019 | 2020 | |||||||||||||||
Maturities on long-term debt | $ | 6.1 | $ | 635.2 | $ | 803.2 | $ | 601.8 | $ | 0.6 | |||||||||
We have converted approximately 40 percent of our long-term fixed-rate notes to floating rates through the use of interest rate swaps. The weighted-average effective borrowing rates based on long-term debt obligations and interest rates at December 31, 2015 and 2014, including the effects of interest rate swaps for hedged debt obligations, were 2.67 percent and 3.69 percent, respectively.
The aggregate amount of cash payments for interest on borrowings, net of capitalized interest, are as follows:
2015 | 2014 | 2013 | |||||||||
Cash payments for interest on borrowings | $ | 129.6 | $ | 140.4 | $ | 139.7 | |||||
In accordance with the requirements of derivatives and hedging guidance, the portion of our fixed-rate debt obligations that is hedged as a fair value hedge, is reflected in the consolidated balance sheets as an amount equal to the sum of the debt’s carrying value plus the fair value adjustment representing changes in fair value of the hedged debt attributable to movements in market interest rates subsequent to the inception of the hedge.
Note 11: Stock-Based Compensation
Our stock-based compensation expense consists of performance awards (PAs), shareholder value awards (SVAs), and restricted stock units (RSUs). We recognize the fair value of stock-based compensation as expense over the requisite service period of the individual grantees, which generally equals the vesting period. We provide newly issued shares of our common stock and treasury stock to satisfy the issuance of PA, SVA, and RSU shares. We classify tax benefits resulting from tax deductions in excess of the compensation cost recognized for stock-based compensation as a financing cash flow in the consolidated statements of cash flows.
Stock-based compensation expense and the related tax benefits were as follows:
2015 | 2014 | 2013 | |||||||||
Stock-based compensation expense | $ | 217.8 | $ | 156.0 | $ | 144.9 | |||||
Tax benefit | 76.2 | 54.6 | 50.7 | ||||||||
At December 31, 2015, additional stock-based compensation awards may be granted under the 2002 Lilly Stock Plan for not more than 100.6 million shares.
82
Performance Award Program
PAs are granted to officers and management and are payable in shares of our common stock. The number of PA shares actually issued, if any, varies depending on the achievement of certain pre-established earnings-per-share targets over a two-year period. PA shares are accounted for at fair value based upon the closing stock price on the date of grant and fully vest at the end of the measurement period. The fair values of PAs granted for the years ended December 31, 2015, 2014, and 2013 were $70.34, $48.81, and $50.19, respectively. The number of shares ultimately issued for the PA program is dependent upon the earnings achieved during the vesting period. Pursuant to this program, approximately 0.5 million shares, 0.7 million shares, and 0.7 million shares were issued during the years ended December 31, 2015, 2014, and 2013, respectively. Approximately 0.5 million shares are expected to be issued in 2016. As of December 31, 2015, the total remaining unrecognized compensation cost related to nonvested PAs was $69.5 million, which will be amortized over the weighted-average remaining requisite service period of 12 months.
Shareholder Value Award Program
SVAs are granted to officers and management and are payable in shares of our common stock. The number of shares actually issued, if any, varies depending on our stock price at the end of the three-year vesting period compared to pre-established target stock prices. We measure the fair value of the SVA unit on the grant date using a Monte Carlo simulation model. The model utilizes multiple input variables that determine the probability of satisfying the market condition stipulated in the award grant and calculates the fair value of the award. Expected volatilities utilized in the model are based on implied volatilities from traded options on our stock, historical volatility of our stock price, and other factors. Similarly, the dividend yield is based on historical experience and our estimate of future dividend yields. The risk-free interest rate is derived from the U.S. Treasury yield curve in effect at the time of grant. The weighted-average fair values of the SVA units granted during the years ended December 31, 2015, 2014, and 2013 were $54.81, $41.97, and $45.17, respectively, determined using the following assumptions:
(Percents) | 2015 | 2014 | 2013 | |||||
Expected dividend yield | 2.50 | % | 3.50 | % | 3.50 | % | ||
Risk-free interest rate | 0.79 | .08-.71 | .08-.43 | |||||
Volatility | 20.37 | 18.87-21.56 | 18.95-22.37 | |||||
Pursuant to this program, approximately 1.4 million shares, 1.4 million shares, and 1.5 million shares were issued during the years ended December 31, 2015, 2014, and 2013, respectively. Approximately 1.0 million shares are expected to be issued in 2016. As of December 31, 2015, the total remaining unrecognized compensation cost related to nonvested SVAs was $59.9 million, which will be amortized over the weighted-average remaining requisite service period of 20 months.
Restricted Stock Units
RSUs are granted to certain employees and are payable in shares of our common stock. RSU shares are accounted for at fair value based upon the closing stock price on the date of grant. The corresponding expense is amortized over the vesting period, typically three years. The fair values of RSU awards granted during the years ended December 31, 2015, 2014, and 2013 were $71.69, $52.72, and $54.10, respectively. The number of shares ultimately issued for the RSU program remains constant with the exception of forfeitures. Pursuant to this program, 0.9 million, 1.2 million, and 1.1 million shares were granted and approximately 0.9 million, 0.9 million, and 0.8 million shares were issued during the years ended December 31, 2015, 2014, and 2013, respectively. Approximately 0.6 million shares are expected to be issued in 2016. As of December 31, 2015, the total remaining unrecognized compensation cost related to nonvested RSUs was $103.0 million, which will be amortized over the weighted-average remaining requisite service period of 24 months.
Note 12: Shareholders' Equity
During 2015, 2014, and 2013, we repurchased $749.5 million, $800.0 million and $500.0 million, respectively, of shares associated with our $5.00 billion share repurchase program announced in 2013. As of December 31, 2015, there were $2.95 billion of shares remaining in that program. During 2013, we repurchased $1.10 billion of shares, completing our $1.50 billion share repurchase program announced in 2012.
83
We have 5.0 million authorized shares of preferred stock. As of December 31, 2015 and 2014, no preferred stock has been issued.
We have an employee benefit trust that held 50.0 million shares of our common stock at both December 31, 2015 and 2014, to provide a source of funds to assist us in meeting our obligations under various employee benefit plans. The cost basis of the shares held in the trust was $3.01 billion at both December 31, 2015 and 2014, and is shown as a reduction in shareholders’ equity. Any dividend transactions between us and the trust are eliminated. Stock held by the trust is not considered outstanding in the computation of EPS. The assets of the trust were not used to fund any of our obligations under these employee benefit plans during the years ended December 31, 2015, 2014, and 2013.
Note 13: Income Taxes
Deferred taxes are recognized for the future tax effects of temporary differences between financial and income tax reporting based on enacted tax laws and rates. Federal income taxes are provided on the portion of the income of foreign subsidiaries that is expected to be remitted to the U.S. and be taxable. When foreign earnings are expected to be indefinitely reinvested outside the U.S., no accrual for U.S. income taxes is provided.
We recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such a position are measured based on the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate resolution.
Following is the composition of income tax expense:
2015 | 2014 | 2013 | |||||||||
Current: | |||||||||||
Federal | $ | 660.5 | $ | 168.9 | $ | 259.1 | |||||
Foreign | 422.0 | 406.2 | 553.2 | ||||||||
State | 47.5 | (2.1 | ) | 126.3 | |||||||
Total current tax expense | 1,130.0 | 573.0 | 938.6 | ||||||||
Deferred: | |||||||||||
Federal | (689.6 | ) | (83.3 | ) | 297.0 | ||||||
Foreign | (66.0 | ) | 120.2 | (28.2 | ) | ||||||
State | 7.2 | (0.1 | ) | (2.9 | ) | ||||||
Total deferred tax (benefit) expense | (748.4 | ) | 36.8 | 265.9 | |||||||
Income taxes | $ | 381.6 | $ | 609.8 | $ | 1,204.5 | |||||
84
Significant components of our deferred tax assets and liabilities as of December 31 are as follows:
2015 | 2014 | ||||||
Deferred tax assets: | |||||||
Compensation and benefits | $ | 1,034.6 | $ | 976.3 | |||
Purchases of intangible assets | 637.2 | 473.3 | |||||
Tax credit carryforwards and carrybacks | 294.2 | 279.4 | |||||
Tax loss carryforwards and carrybacks | 247.8 | 265.5 | |||||
Contingent consideration | 214.6 | — | |||||
Product return reserves | 212.1 | 241.8 | |||||
Other comprehensive loss on hedging transactions | 129.7 | 115.3 | |||||
Debt | 111.3 | 176.0 | |||||
Other | 628.6 | 623.2 | |||||
Total gross deferred tax assets | 3,510.1 | 3,150.8 | |||||
Valuation allowances | (590.3 | ) | (601.1 | ) | |||
Total deferred tax assets | 2,919.8 | 2,549.7 | |||||
Deferred tax liabilities: | |||||||
Inventories | (771.3 | ) | (684.6 | ) | |||
Intangibles | (756.3 | ) | (582.6 | ) | |||
Property and equipment | (411.6 | ) | (424.7 | ) | |||
Prepaid employee benefits | (317.8 | ) | (275.8 | ) | |||
Unremitted earnings | (218.8 | ) | (737.1 | ) | |||
Financial instruments | (152.6 | ) | (276.8 | ) | |||
Total deferred tax liabilities | (2,628.4 | ) | (2,981.6 | ) | |||
Deferred tax assets (liabilities) - net | $ | 291.4 | $ | (431.9 | ) | ||
The deferred tax asset and related valuation allowance amounts for U.S. federal and state net operating losses and tax credits shown above have been reduced for differences between financial reporting and tax return filings.
Based on filed tax returns, we have tax credit carryforwards and carrybacks of $668.5 million available to reduce future income taxes; $180.5 million, if unused, will expire by 2021. The remaining portion of the tax credit carryforwards is related to federal tax credits of $93.4 million, international tax credits of $100.6 million, and state tax credits of $294.0 million, all of which are substantially reserved.
At December 31, 2015, based on filed tax returns we had net operating losses and other carryforwards for international and U.S. federal income tax purposes of $462.0 million: $38.0 million will expire by 2020; $354.0 million will expire between 2020 and 2035; and $70.0 million of the carryforwards will never expire. Net operating losses and other carryforwards for international and U.S. federal income tax purposes are partially reserved. Deferred tax assets related to state net operating losses of $93.2 million and other state carryforwards of $8.8 million are fully reserved.
Domestic and Puerto Rican companies contributed approximately 35 percent, 20 percent, and 60 percent for the years ended December 31, 2015, 2014, and 2013, respectively, to consolidated income before income taxes. We have a subsidiary operating in Puerto Rico under a tax incentive grant effective through the end of 2016. A similar, new tax incentive grant will begin in 2017 and will be in effect for 15 years.
At December 31, 2015, U.S. income taxes have not been provided on approximately $26.5 billion of unremitted earnings of foreign subsidiaries as we consider these unremitted earnings to be indefinitely invested for continued use in our foreign operations. Additional tax provisions will be required if these earnings are repatriated in the future to the U.S. Due to complexities in the tax laws and assumptions that we would have to make, it is not practicable to determine the amount of the related unrecognized deferred income tax liability.
85
Cash payments of income taxes were as follows:
2015 | 2014 | 2013 | |||||||||
Cash payments of income taxes | $ | 969.0 | $ | 729.7 | $ | 1,255.6 | |||||
Following is a reconciliation of the income tax expense applying the U.S. federal statutory rate to income before income taxes to reported income tax expense:
2015 | 2014 | 2013 | |||||||||
Income tax at the U.S. federal statutory tax rate | $ | 976.5 | $ | 1,050.1 | $ | 2,061.3 | |||||
Add (deduct): | |||||||||||
International operations, including Puerto Rico | (565.2 | ) | (344.8 | ) | (778.3 | ) | |||||
General business credits | (69.2 | ) | (44.3 | ) | (175.6 | ) | |||||
Other | 39.5 | (51.2 | ) | 97.1 | |||||||
Income taxes | $ | 381.6 | $ | 609.8 | $ | 1,204.5 | |||||
The American Taxpayer Relief Act of 2012, which included the reinstatement of the research tax credit for the year 2012, was enacted in early 2013. Therefore, the research tax credits for the years 2012 and 2013 are both included in 2013 with general business credits.
A reconciliation of the beginning and ending amount of gross unrecognized tax benefits is as follows:
2015 | 2014 | 2013 | |||||||||
Beginning balance at January 1 | $ | 1,338.8 | $ | 1,136.4 | $ | 1,534.3 | |||||
Additions based on tax positions related to the current year | 131.3 | 126.4 | 142.5 | ||||||||
Additions for tax positions of prior years | 116.6 | 132.6 | 251.5 | ||||||||
Reductions for tax positions of prior years | (45.2 | ) | (32.1 | ) | (358.2 | ) | |||||
Settlements | (446.2 | ) | (4.2 | ) | (404.9 | ) | |||||
Lapses of statutes of limitation | (4.0 | ) | (3.5 | ) | (24.9 | ) | |||||
Changes related to the impact of foreign currency translation | (24.7 | ) | (16.8 | ) | (3.9 | ) | |||||
Ending balance at December 31 | $ | 1,066.6 | $ | 1,338.8 | $ | 1,136.4 | |||||
The total amount of unrecognized tax benefits that, if recognized, would affect our effective tax rate was $404.1 million and $638.8 million at December 31, 2015 and 2014, respectively.
We file income tax returns in the U.S. federal jurisdiction and various state, local, and non-U.S. jurisdictions. We are no longer subject to U.S. federal, state and local, or non-U.S. income tax examinations in most major taxing jurisdictions for years before 2007.
During 2013, we reached resolution on the remaining matters related to tax years 2008–2009 that were not settled as part of a previous U.S. examination. Considering the impact of this resolution on periods that have not yet been examined, as well as its impact on tax asset carryforwards, there was an immaterial benefit to our consolidated results of operations. We made cash payments of approximately $135 million related to tax years 2008–2009 after application of available tax credit carryforwards and carrybacks.
The U.S. examination of tax years 2010-2012 commenced during the fourth quarter of 2013 and is expected to conclude in the first quarter of 2016. In December 2015, we executed a closing agreement with the Internal Revenue Service which effectively settled certain matters for tax years 2010-2012. Accordingly, we have reduced our gross uncertain tax positions by approximately $320 million in 2015. There was an immaterial impact to our consolidated results of operations during the fourth quarter related to issues settled in the closing agreement. In the first quarter of 2016, we anticipate reaching resolution on the remaining issues under examination and estimate that our gross uncertain tax positions will further be reduced by approximately $100 million to $150 million. Additionally, we do not anticipate that resolution of the U.S. examination will result in a material change to our consolidated financial position, and we expect that up to $250 million of cash payments will be due upon resolution of these tax years. We expect the U.S. examination of tax years 2013-2014, and possibly 2015, to begin in 2016.
We recognize both accrued interest and penalties related to unrecognized tax benefits in income tax expense. We recognized income tax (benefit) expense related to interest and penalties as follows:
2015 | 2014 | 2013 | |||||||||
Income tax (benefit) expense | $ | 13.2 | $ | 35.9 | $ | (10.9 | ) | ||||
86
At December 31, 2015 and 2014, our accruals for the payment of interest and penalties totaled $216.3 million and $207.2 million, respectively.
Note 14: Retirement Benefits
We use a measurement date of December 31 to develop the change in benefit obligation, change in plan assets, funded status, and amounts recognized in the consolidated balance sheets at December 31 for our defined benefit pension and retiree health benefit plans, which were as follows:
Defined Benefit Pension Plans | Retiree Health Benefit Plans | ||||||||||||||
2015 | 2014 | 2015 | 2014 | ||||||||||||
Change in benefit obligation: | |||||||||||||||
Benefit obligation at beginning of year | $ | 12,012.4 | $ | 9,976.4 | $ | 1,553.5 | $ | 1,757.2 | |||||||
Benefit obligation assumed in Novartis AH acquisition | 334.7 | — | 9.9 | — | |||||||||||
Service cost | 315.7 | 240.9 | 45.1 | 33.0 | |||||||||||
Interest cost | 476.8 | 472.6 | 62.6 | 85.6 | |||||||||||
Actuarial (gain) loss | (812.4 | ) | 1,996.3 | (113.5 | ) | 293.5 | |||||||||
Benefits paid | (437.8 | ) | (421.2 | ) | (77.5 | ) | (76.1 | ) | |||||||
Plan amendments | (0.4 | ) | (2.4 | ) | — | (533.6 | ) | ||||||||
Foreign currency exchange rate changes and other adjustments | (169.8 | ) | (250.2 | ) | (12.7 | ) | (6.1 | ) | |||||||
Benefit obligation at end of year | 11,719.2 | 12,012.4 | 1,467.4 | 1,553.5 | |||||||||||
Change in plan assets: | |||||||||||
Fair value of plan assets at beginning of year | 9,835.7 | 9,481.7 | 1,918.7 | 1,879.6 | |||||||
Fair value of plan assets assumed in Novartis AH acquisition | 235.9 | — | — | — | |||||||
Actual return on plan assets | 90.4 | 813.6 | 85.1 | 157.4 | |||||||
Employer contribution | 404.1 | 127.2 | 17.4 | (42.2 | ) | ||||||
Benefits paid | (437.8 | ) | (421.2 | ) | (77.5 | ) | (76.1 | ) | |||
Foreign currency exchange rate changes and other adjustments | (132.7 | ) | (165.6 | ) | — | — | |||||
Fair value of plan assets at end of year | 9,995.6 | 9,835.7 | 1,943.7 | 1,918.7 | |||||||
Funded status | (1,723.6 | ) | (2,176.7 | ) | 476.3 | 365.2 | |||||||||
Unrecognized net actuarial loss | 4,552.7 | 5,114.9 | 347.9 | 439.5 | |||||||||||
Unrecognized prior service (benefit) cost | 32.5 | 43.5 | (574.8 | ) | (666.7 | ) | |||||||||
Net amount recognized | $ | 2,861.6 | $ | 2,981.7 | $ | 249.4 | $ | 138.0 | |||||||
Amounts recognized in the consolidated balance sheet consisted of: | |||||||||||||||
Sundry | $ | 261.6 | $ | 211.2 | $ | 722.1 | $ | 609.4 | |||||||
Other current liabilities | (63.8 | ) | (62.3 | ) | (6.9 | ) | (6.9 | ) | |||||||
Accrued retirement benefits | (1,921.4 | ) | (2,325.6 | ) | (238.9 | ) | (237.3 | ) | |||||||
Accumulated other comprehensive (income) loss before income taxes | 4,585.2 | 5,158.4 | (226.9 | ) | (227.2 | ) | |||||||||
Net amount recognized | $ | 2,861.6 | $ | 2,981.7 | $ | 249.4 | $ | 138.0 | |||||||
The unrecognized net actuarial loss and unrecognized prior service cost (benefit) have not yet been recognized in net periodic pension costs and are included in accumulated other comprehensive loss at December 31, 2015.
A change to our U.S. retiree health benefit plan was approved in 2014 and communicated to retirees in January 2015. Beginning in 2016, Medicare-eligible retirees and Medicare-eligible dependents will choose health care coverage from insurance providers through a private Medicare supplement marketplace, while still receiving financial support from us. This change decreased our retiree health benefit obligation and increased our unrecognized prior service benefit as of December 31, 2014 by $520.8 million.
87
During 2016, we expect the following components of accumulated other comprehensive loss to be recognized as components of net periodic benefit cost:
Defined Benefit Pension Plans | Retiree Health Benefit Plans | ||||||
Unrecognized net actuarial loss | $ | 285.9 | $ | 19.2 | |||
Unrecognized prior service (benefit) cost | 11.9 | (85.8 | ) | ||||
Total | $ | 297.8 | $ | (66.6 | ) | ||
We do not expect any plan assets to be returned to us in 2016.
The following represents our weighted-average assumptions as of December 31:
Defined Benefit Pension Plans | Retiree Health Benefit Plans | ||||||||||
(Percents) | 2015 | 2014 | 2013 | 2015 | 2014 | 2013 | |||||
Discount rate for benefit obligation | 4.3 | 4.0 | 4.9 | 4.5 | 4.1 | 5.0 | |||||
Discount rate for net benefit costs | 4.0 | 4.9 | 4.3 | 4.1 | 5.0 | 4.3 | |||||
Rate of compensation increase for benefit obligation | 3.4 | 3.4 | 3.4 | ||||||||
Rate of compensation increase for net benefit costs | 3.4 | 3.4 | 3.4 | ||||||||
Expected return on plan assets for net benefit costs | 7.4 | 8.1 | 8.4 | 8.0 | 8.5 | 8.8 | |||||
We annually evaluate the expected return on plan assets in our defined benefit pension and retiree health benefit plans. In evaluating the expected rate of return, we consider many factors, with a primary analysis of current and projected market conditions; asset returns and asset allocations; and the views of leading financial advisers and economists. We may also review our historical assumptions compared with actual results, as well as the assumptions and trend rates utilized by similar plans, where applicable.
Given the design of our retiree health benefit plans, heathcare-cost trend rates do not have a material impact on our financial condition or results of operations.
The following benefit payments, which reflect expected future service, as appropriate, are expected to be paid as follows:
2016 | 2017 | 2018 | 2019 | 2020 | 2021-2025 | ||||||||||||||||||
Defined benefit pension plans | $ | 458.4 | $ | 465.2 | $ | 480.3 | $ | 498.8 | $ | 518.5 | $ | 2,987.5 | |||||||||||
Retiree health benefit plans | 70.7 | 75.3 | 78.3 | 81.1 | 84.0 | 464.2 | |||||||||||||||||
Amounts relating to defined benefit pension plans with projected benefit obligations in excess of plan assets were as follows at December 31:
2015 | 2014 | ||||||
Projected benefit obligation | $ | 10,054.1 | $ | 10,537.2 | |||
Fair value of plan assets | 8,069.7 | 8,149.2 | |||||
Amounts relating to defined benefit pension plans and retiree health benefit plans with accumulated benefit obligations in excess of plan assets were as follows at December 31:
Defined Benefit Pension Plans | Retiree Health Benefit Plans | ||||||||||||||
2015 | 2014 | 2015 | 2014 | ||||||||||||
Accumulated benefit obligation | $ | 2,028.1 | $ | 2,179.8 | $ | 245.8 | $ | 244.2 | |||||||
Fair value of plan assets | 844.9 | 700.9 | — | — | |||||||||||
The total accumulated benefit obligation for our defined benefit pension plans was $10.75 billion and $10.88 billion at December 31, 2015 and 2014, respectively.
88
Net pension and retiree health benefit expense included the following components:
Defined Benefit Pension Plans | Retiree Health Benefit Plans | ||||||||||||||||||||||
2015 | 2014 | 2013 | 2015 | 2014 | 2013 | ||||||||||||||||||
Components of net periodic (benefit) cost: | |||||||||||||||||||||||
Service cost | $ | 315.7 | $ | 240.9 | $ | 287.1 | $ | 45.1 | $ | 33.0 | $ | 49.9 | |||||||||||
Interest cost | 476.8 | 472.6 | 437.2 | 62.6 | 85.6 | 98.1 | |||||||||||||||||
Expected return on plan assets | (782.3 | ) | (756.6 | ) | (701.9 | ) | (150.0 | ) | (146.4 | ) | (130.7 | ) | |||||||||||
Amortization of prior service (benefit) cost | 10.4 | 3.6 | 3.7 | (91.1 | ) | (37.6 | ) | (35.6 | ) | ||||||||||||||
Recognized actuarial loss | 383.2 | 282.3 | 414.7 | 38.0 | 20.7 | 100.5 | |||||||||||||||||
Net periodic (benefit) cost | $ | 403.8 | $ | 242.8 | $ | 440.8 | $ | (95.4 | ) | $ | (44.7 | ) | $ | 82.2 | |||||||||
As of January 1, 2016, we changed the method used to estimate the service and interest cost components of the net periodic pension and retiree health benefit plan costs. This new method uses the spot yield curve approach to estimate the service and interest costs by applying the specific spot rates along the yield curve to the projected cash outflows of our obligations. Previously, those costs were determined using a single weighted-average discount rate. The new method provides a more precise measure of interest and service costs by improving the correlation between the projected benefit cash flows and the specific spot yield curve rates. The change does not affect the measurement of the total benefit obligations as the change in service and interest costs is recorded in the actuarial gains and losses recorded in accumulated other comprehensive loss. We will account for this change as a change in estimate prospectively beginning in the first quarter of 2016.
The following represents the amounts recognized in other comprehensive income (loss) for the year ended December 31, 2015:
Defined Benefit Pension Plans | Retiree Health Benefit Plans | ||||||
Actuarial gain arising during period | $ | 120.4 | $ | 48.6 | |||
Plan amendments during period | 0.4 | — | |||||
Amortization of prior service (benefit) cost included in net income | 10.4 | (91.1 | ) | ||||
Amortization of net actuarial loss included in net income | 383.2 | 38.0 | |||||
Foreign currency exchange rate changes and other | 58.8 | 4.2 | |||||
Total other comprehensive income (loss) during period | $ | 573.2 | $ | (0.3 | ) | ||
We have defined contribution savings plans that cover our eligible employees worldwide. The purpose of these plans is generally to provide additional financial security during retirement by providing employees with an incentive to save. Our contributions to the plans are based on employee contributions and the level of our match. Expenses under the plans totaled $162.4 million, $153.3 million, and $147.7 million for the years ended December 31, 2015, 2014, and 2013, respectively.
We provide certain other postemployment benefits primarily related to disability benefits and accrue for the related cost over the service lives of employees. Expenses associated with these benefit plans for the years ended December 31, 2015, 2014, and 2013 were not material.
Benefit Plan Investments
Our benefit plan investment policies are set with specific consideration of return and risk requirements in relationship to the respective liabilities. U.S. and Puerto Rico plans represent approximately 75 percent of our global investments. Given the long-term nature of our liabilities, these plans have the flexibility to manage an above-average degree of risk in the asset portfolios. At the investment-policy level, there are no specifically prohibited investments. However, within individual investment manager mandates, restrictions and limitations are contractually set to align with our investment objectives, ensure risk control, and limit concentrations.
We manage our portfolio to minimize concentration of risk by allocating funds within asset categories. In addition, within a category we use different managers with various management objectives to eliminate any significant concentration of risk.
Our global benefit plans may enter into contractual arrangements (derivatives) to implement the local investment policy or manage particular portfolio risks. Derivatives are principally used to increase or decrease
89
exposure to a particular public equity, fixed income, commodity, or currency market more rapidly or less expensively than could be accomplished through the use of the cash markets. The plans utilize both exchange-traded and over-the-counter instruments. The maximum exposure to either a market or counterparty credit loss is limited to the carrying value of the receivable, and is managed within contractual limits. We expect all of our counterparties to meet their obligations. The gross values of these derivative receivables and payables are not material to the global asset portfolio, and their values are reflected within the tables below.
The defined benefit pension and retiree health benefit plan allocation for the U.S. and Puerto Rico currently comprises approximately 80 percent growth investments and 20 percent fixed-income investments. The growth investment allocation encompasses U.S. and international public equity securities, hedge funds, private equity-like investments, and real estate. These portfolio allocations are intended to reduce overall risk by providing diversification, while seeking moderate to high returns over the long term.
Public equity securities are well diversified and invested in U.S. and international small-to-large companies across various asset managers and styles. The remaining portion of the growth portfolio is invested in private alternative investments.
Fixed-income investments primarily consist of fixed-income securities in U.S. treasuries and agencies, emerging market debt obligations, corporate bonds, mortgage-backed securities, and commercial mortgage-backed obligations.
Hedge funds are privately owned institutional investment funds that generally have moderate liquidity. Hedge funds seek specified levels of absolute return regardless of overall market conditions, and generally have low correlations to public equity and debt markets. Hedge funds often invest substantially in financial market instruments (stocks, bonds, commodities, currencies, derivatives, etc.) using a very broad range of trading activities to manage portfolio risks. Hedge fund strategies focus primarily on security selection and seek to be neutral with respect to market moves. Common groupings of hedge fund strategies include relative value, tactical, and event driven. Relative value strategies include arbitrage, when the same asset can simultaneously be bought and sold at different prices, achieving an immediate profit. Tactical strategies often take long and short positions to reduce or eliminate overall market risks while seeking a particular investment opportunity. Event strategy opportunities can evolve from specific company announcements such as mergers and acquisitions, and typically have little correlation to overall market directional movements. Our hedge fund investments are made through limited partnership interests primarily in fund-of-funds structures to ensure diversification across many strategies and many individual managers. Plan holdings in hedge funds are valued based on net asset values (NAVs) calculated by each fund or general partner, as applicable, and we have the ability to redeem these investments at NAV.
Private equity-like investment funds typically have low liquidity and are made through long-term partnerships or joint ventures that invest in pools of capital invested in primarily non-publicly traded entities. Underlying investments include venture capital (early stage investing), buyout, and special situation investing. Private equity management firms typically acquire and then reorganize private companies to create increased long term value. Private equity-like funds usually have a limited life of approximately 10-15 years, and require a minimum investment commitment from their limited partners. Our private investments are made both directly into funds and through fund-of-funds structures to ensure broad diversification of management styles and assets across the portfolio. Plan holdings in private equity-like investments are valued using the value reported by the partnership, adjusted for known cash flows and significant events through our reporting date. Values provided by the partnerships are primarily based on analysis of and judgments about the underlying investments. Inputs to these valuations include underlying NAVs, discounted cash flow valuations, comparable market valuations, and may also include adjustments for currency, credit, liquidity and other risks as applicable. The vast majority of these private partnerships provide us with annual audited financial statements including their compliance with fair valuation procedures consistent with applicable accounting standards.
Real estate is composed of both public and private holdings. Real estate investments in registered investment companies that trade on an exchange are classified as Level 1 on the fair value hierarchy. Real estate investments in funds measured at fair value on the basis of NAV provided by the fund manager are classified as Level 3. These NAVs are developed with inputs including discounted cash flow, independent appraisal, and market comparable analyses.
90
Other assets include cash and cash equivalents and mark-to-market value of derivatives.
The cash value of the trust-owned insurance contract is invested in investment-grade publicly traded equity and fixed-income securities.
Other than hedge funds, private equity-like investments, and real estate, which are discussed above, we determine fair values based on a market approach using quoted market values, significant other observable inputs for identical or comparable assets or liabilities, or discounted cash flow analyses.
The fair values of our defined benefit pension plan and retiree health plan assets as of December 31, 2015 by asset category are as follows:
Fair Value Measurements Using | |||||||||||||||
Asset Class | Total | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | |||||||||||
Defined Benefit Pension Plans | |||||||||||||||
Public equity securities: | |||||||||||||||
U.S. | $ | 414.8 | $ | 187.1 | $ | 227.7 | $ | — | |||||||
International | 2,340.6 | 956.3 | 1,384.3 | — | |||||||||||
Fixed income: | |||||||||||||||
Developed markets | 1,316.6 | 175.7 | 1,140.9 | — | |||||||||||
Emerging markets | 385.7 | 10.1 | 375.3 | 0.3 | |||||||||||
Private alternative investments: | |||||||||||||||
Hedge funds | 3,073.3 | 14.5 | 1,537.9 | 1,520.9 | |||||||||||
Equity-like funds | 1,221.3 | — | 67.4 | 1,153.9 | |||||||||||
Real estate | 516.3 | 329.5 | 10.8 | 176.0 | |||||||||||
Other | 727.0 | 431.3 | 295.7 | — | |||||||||||
Total | $ | 9,995.6 | $ | 2,104.5 | $ | 5,040.0 | $ | 2,851.1 | |||||||
Retiree Health Benefit Plans | |||||||||||||||
Public equity securities: | |||||||||||||||
U.S. | $ | 40.0 | $ | 18.2 | $ | 21.8 | $ | — | |||||||
International | 144.7 | 51.5 | 93.2 | — | |||||||||||
Fixed income: | |||||||||||||||
Developed markets | 61.3 | — | 61.3 | — | |||||||||||
Emerging markets | 36.9 | — | 36.9 | — | |||||||||||
Private alternative investments: | |||||||||||||||
Hedge funds | 272.3 | — | 148.7 | 123.6 | |||||||||||
Equity-like funds | 104.5 | — | — | 104.5 | |||||||||||
Cash value of trust owned insurance contract | 1,209.7 | — | 1,209.7 | — | |||||||||||
Real estate | 33.2 | 33.2 | — | — | |||||||||||
Other | 41.1 | 25.0 | 16.1 | — | |||||||||||
Total | $ | 1,943.7 | $ | 127.9 | $ | 1,587.7 | $ | 228.1 | |||||||
No material transfers between Level 1, Level 2, or Level 3 occurred during the year ended December 31, 2015.
91
The activity in the Level 3 investments during the year ended December 31, 2015 was as follows:
Fixed Income: Emerging Markets | Hedge Funds | Equity-like Funds | Real Estate | Total | |||||||||||||||
Defined Benefit Pension Plans | |||||||||||||||||||
Beginning balance at January 1, 2015 | $ | 1.8 | $ | 1,583.1 | $ | 1,071.4 | $ | 165.9 | $ | 2,822.2 | |||||||||
Actual return on plan assets, including changes in foreign exchange rates: | |||||||||||||||||||
Relating to assets still held at the reporting date | (0.1 | ) | 89.4 | 78.3 | (6.1 | ) | 161.5 | ||||||||||||
Relating to assets sold during the period | — | (111.5 | ) | — | — | (111.5 | ) | ||||||||||||
Purchases, sales, and settlements, net | (0.3 | ) | (40.1 | ) | 4.2 | 16.2 | (20.0 | ) | |||||||||||
Transfers into (out of) Level 3 | (1.1 | ) | — | — | — | (1.1 | ) | ||||||||||||
Ending balance at December 31, 2015 | $ | 0.3 | $ | 1,520.9 | $ | 1,153.9 | $ | 176.0 | $ | 2,851.1 | |||||||||
Retiree Health Benefit Plans | |||||||||||||||||||
Beginning balance at January 1, 2015 | $ | 0.2 | $ | 124.0 | $ | 92.3 | $ | — | $ | 216.5 | |||||||||
Actual return on plan assets, including changes in foreign exchange rates: | |||||||||||||||||||
Relating to assets still held at the reporting date | — | 14.7 | 10.3 | — | 25.0 | ||||||||||||||
Relating to assets sold during the period | — | (11.2 | ) | — | — | (11.2 | ) | ||||||||||||
Purchases, sales, and settlements, net | — | (3.9 | ) | 1.9 | — | (2.0 | ) | ||||||||||||
Transfers into (out of) Level 3 | (0.2 | ) | — | — | — | (0.2 | ) | ||||||||||||
Ending balance at December 31, 2015 | $ | — | $ | 123.6 | $ | 104.5 | $ | — | $ | 228.1 | |||||||||
92
The fair values of our defined benefit pension plan and retiree health plan assets as of December 31, 2014 by asset category are as follows:
Fair Value Measurements Using | |||||||||||||||
Asset Class | Total | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | |||||||||||
Defined Benefit Pension Plans | |||||||||||||||
Public equity securities: | |||||||||||||||
U.S. | $ | 411.4 | $ | 183.8 | $ | 227.6 | $ | — | |||||||
International | 2,337.8 | 999.7 | 1,338.1 | — | |||||||||||
Fixed income: | |||||||||||||||
Developed markets | 1,230.7 | 112.2 | 1,118.5 | — | |||||||||||
Emerging markets | 374.7 | 8.7 | 364.2 | 1.8 | |||||||||||
Private alternative investments: | |||||||||||||||
Hedge funds | 3,277.6 | — | 1,694.5 | 1,583.1 | |||||||||||
Equity-like funds | 1,146.6 | — | 75.2 | 1,071.4 | |||||||||||
Real estate | 569.0 | 403.1 | — | 165.9 | |||||||||||
Other | 487.9 | 229.8 | 258.1 | — | |||||||||||
Total | $ | 9,835.7 | $ | 1,937.3 | $ | 5,076.2 | $ | 2,822.2 | |||||||
Retiree Health Benefit Plans | |||||||||||||||
Public equity securities: | |||||||||||||||
U.S. | $ | 39.2 | $ | 17.2 | $ | 22.0 | $ | — | |||||||
International | 158.9 | 58.8 | 100.1 | — | |||||||||||
Fixed income: | |||||||||||||||
Developed markets | 61.8 | — | 61.8 | — | |||||||||||
Emerging markets | 35.5 | — | 35.3 | 0.2 | |||||||||||
Private alternative investments: | |||||||||||||||
Hedge funds | 282.7 | — | 158.7 | 124.0 | |||||||||||
Equity-like funds | 92.3 | — | — | 92.3 | |||||||||||
Cash value of trust owned insurance contract | 1,189.2 | — | 1,189.2 | — | |||||||||||
Real estate | 39.0 | 39.0 | — | — | |||||||||||
Other | 20.1 | 7.6 | 12.5 | — | |||||||||||
Total | $ | 1,918.7 | $ | 122.6 | $ | 1,579.6 | $ | 216.5 | |||||||
No material transfers between Level 1, Level 2, or Level 3 occurred during the year ended December 31, 2014.
93
The activity in the Level 3 investments during the year ended December 31, 2014 was as follows:
Fixed Income: Developed Markets | Fixed Income: Emerging Markets | Hedge Funds | Equity-like Funds | Real Estate | Total | ||||||||||||||||||
Defined Benefit Pension Plans | |||||||||||||||||||||||
Beginning balance at January 1, 2014 | $ | 15.9 | $ | — | $ | 1,440.4 | $ | 993.5 | $ | 153.4 | $ | 2,603.2 | |||||||||||
Actual return on plan assets, including changes in foreign exchange rates: | |||||||||||||||||||||||
Relating to assets still held at the reporting date | (0.4 | ) | 0.1 | 44.6 | 108.2 | 0.2 | 152.7 | ||||||||||||||||
Relating to assets sold during the period | (0.8 | ) | — | — | — | — | (0.8 | ) | |||||||||||||||
Purchases, sales, and settlements, net | (3.3 | ) | 1.7 | 98.1 | (30.3 | ) | 12.3 | 78.5 | |||||||||||||||
Transfers into (out of) Level 3 | (11.4 | ) | — | — | — | — | (11.4 | ) | |||||||||||||||
Ending balance at December 31, 2014 | $ | — | $ | 1.8 | $ | 1,583.1 | $ | 1,071.4 | $ | 165.9 | $ | 2,822.2 | |||||||||||
Retiree Health Benefit Plans | |||||||||||||||||||||||
Beginning balance at January 1, 2014 | $ | 1.6 | $ | — | $ | 120.6 | $ | 88.9 | $ | — | $ | 211.1 | |||||||||||
Actual return on plan assets, including changes in foreign exchange rates: | |||||||||||||||||||||||
Relating to assets still held at the reporting date | (0.1 | ) | — | 1.2 | 6.0 | — | 7.1 | ||||||||||||||||
Relating to assets sold during the period | (0.1 | ) | — | — | — | — | (0.1 | ) | |||||||||||||||
Purchases, sales, and settlements, net | (0.3 | ) | 0.2 | 2.2 | (2.6 | ) | — | (0.5 | ) | ||||||||||||||
Transfers into (out of) Level 3 | (1.1 | ) | — | — | — | — | (1.1 | ) | |||||||||||||||
Ending balance at December 31, 2014 | $ | — | $ | 0.2 | $ | 124.0 | $ | 92.3 | $ | — | $ | 216.5 | |||||||||||
In 2016, we expect to contribute approximately $40 million to our defined benefit pension plans to satisfy minimum funding requirements for the year, along with approximately $5 million of additional discretionary contributions.
Note 15: Contingencies
We are a party to various legal actions and government investigations. The most significant of these are described below. It is not possible to determine the outcome of these matters, and we cannot reasonably estimate the maximum potential exposure or the range of possible loss in excess of amounts accrued for any of these matters; however, we believe that, except as noted below with respect to the Alimta® patent litigation and administrative proceedings, the resolution of all such matters will not have a material adverse effect on our consolidated financial position or liquidity, but could possibly be material to our consolidated results of operations in any one accounting period.
Litigation accruals, environmental liabilities, and the related estimated insurance recoverables are reflected on a gross basis as liabilities and assets, respectively, on our consolidated balance sheets. With respect to the product liability claims currently asserted against us, we have accrued for our estimated exposures to the extent they are both probable and reasonably estimable based on the information available to us. We accrue for certain product liability claims incurred but not filed to the extent we can formulate a reasonable estimate of their costs. We estimate these expenses based primarily on historical claims experience and data regarding product usage. Legal defense costs expected to be incurred in connection with significant product liability loss contingencies are accrued when both probable and reasonably estimable.
Alimta Patent Litigation and Administrative Proceedings
A number of generic manufacturers are seeking approvals in various countries to market generic forms of Alimta prior to the expiration of our vitamin regimen patents, alleging that those patents are invalid, not infringed, or both. We believe our Alimta vitamin regimen patents are valid and enforceable against these
94
generic manufacturers. However, it is not possible to determine the ultimate outcome of the proceedings, and accordingly, we can provide no assurance that we will prevail. An unfavorable outcome could have a material adverse impact on our future consolidated results of operations, liquidity, and financial position. We expect that a loss of exclusivity for Alimta would result in a rapid and severe decline in future revenues for the product in the relevant market.
U.S. Patent Litigation and Administrative Proceedings
We are engaged in various U.S. patent litigation matters involving Alimta brought pursuant to procedures set out in the Drug Price Competition and Patent Term Restoration Act of 1984 (the Hatch-Waxman Act). More than ten Abbreviated New Drug Applications (ANDAs) seeking approval to market generic versions of Alimta prior to the expiration of our vitamin regimen patent (expiring in 2021 plus pediatric exclusivity expiring in 2022) have been filed by a number of companies, including Teva Parenteral Medicines, Inc. (Teva) and APP Pharmaceuticals, LLC (APP). These companies have also alleged the patent is invalid. In February 2016, we filed a lawsuit alleging infringement against Dr. Reddy's Laboratories in response to their recently filed abbreviated application.
In October 2010, we filed a lawsuit in the U.S. District Court for the Southern District of Indiana against Teva, APP and two other defendants seeking rulings that the U.S. vitamin regimen patent is valid and infringed (the Teva/APP litigation). Teva and APP stipulated to infringement of our vitamin regimen patent, with the contingency that Teva and APP would be permitted to litigate the issue of infringement if the U.S. Supreme Court vacated an en banc decision of the U.S. Court of Appeals for the Federal Circuit that dealt with issues of liability related to infringement (Akamai v. Limelight Networks). Thus, the sole issue before the district court was to determine patent validity.
Trial occurred in August 2013. In March 2014, the court ruled that the asserted claims of the vitamin regimen patent are valid. In June 2014, the U.S. Supreme Court vacated the Akamai decision, and the U.S. District Court for the Southern District of Indiana held a hearing on the issue of infringement in May 2015. In September 2015, the district court ruled that the vitamin regimen patent would be infringed by the generic challengers' proposed products. Teva and APP filed a notice of appeal with respect to all of the district court’s substantive decisions. A decision from the U.S. Court of Appeals for the Federal Circuit is expected in late 2016.
Throughout the course of 2012 through 2015, we filed similar lawsuits against other ANDA defendants seeking a ruling that our patents are valid and infringed. Some of these cases have been stayed pending the outcome of the Teva/APP litigation, and these parties have agreed to be bound by the outcome of the Teva/APP litigation.
In 2015, Neptune Generics LLC and Sandoz International GmbH each submitted petitions to the United States Patent and Trademark Office (USPTO), seeking inter partes review (IPR) of our vitamin regimen patent by the USPTO. The USPTO is expected to decide whether to institute an IPR by mid-2016. If instituted, then the final written decision on the merits will be issued by the USPTO by mid-2017.
European Patent Litigation and Administrative Proceedings
Generic manufacturers filed an opposition to the European Patent Office's (EPO) decision to grant us a vitamin regimen patent. The Opposition Division of the EPO upheld the patent and the generic manufacturers lodged an appeal. The EPO appeal hearing was scheduled for November 2015. In October 2015 the generic manufacturers withdrew the appeal, and the hearing was canceled. As a result, the original EPO decision upholding the patent is now final.
In addition, in the United Kingdom (U.K.), Actavis Group ehf and other Actavis companies (collectively, Actavis) filed litigation asking for a declaratory judgment that commercialization of certain salt forms of pemetrexed (the active ingredient in Alimta) would not infringe the vitamin regimen patents in the U.K., Italy, France, and Spain. In May 2014, the trial court ruled that the vitamin regimen patents for Alimta would not be infringed by commercialization of alternative salt forms of pemetrexed, after expiration of the compound patents in December 2015. We appealed, and in June 2015, the U.K. Court of Appeal reversed the trial court, ruling that the Alimta vitamin regimen patent in the U.K. would be indirectly infringed by commercialization of Actavis' products as proposed prior to the patent's expiration in June 2021. The Court of Appeal also held there was no difference between the law in the U.K. and that in France, Italy, and Spain as it relates to indirect infringement, and so reversed the trial court's decision granting declarations of noninfringement over the Alimta vitamin regimen patents in those countries. In February 2016, the U.K. Supreme Court granted our and Actavis' requests for permission to appeal different aspects of the judgment.
95
In February 2016, the trial court ruled that Actavis’ commercialization of a different proposed product would not infringe the patent in the U.K., Italy, France, and Spain. We will seek permission to appeal this ruling.
We commenced separate infringement proceedings against certain Actavis companies in Germany. Following a trial, in April 2014, the German trial court ruled in our favor. The defendants appealed, and after a hearing in March 2015, the appellate court overturned the trial court and ruled that our vitamin regimen patent in Germany would not be infringed by a dipotassium salt form of pemetrexed. In January 2016, the German Federal Supreme court granted our request to appeal this matter. A hearing is scheduled for mid-2016.
In separate proceedings, in December 2015 we applied for and obtained a preliminary injunction against Hexal AG (Hexal), which had stated their intention to launch a generic disodium salt product in Germany. Hexal has appealed.
We are aware that a generic competitor has received approval to market a generic version of Alimta in a major European market, although we are not aware of whether the competitor's product has entered the market.
Japanese Administrative Proceedings
Three companies have filed separate demands for invalidation of our two vitamin regimen patents with the Japanese Patent Office (JPO). In November 2015, the JPO issued a written decision in the invalidation trial initiated by Sawai Pharmaceutical Co., Ltd. (Sawai), and joined by four other companies, upholding both vitamin regimen patents. These patents provide intellectual property protection for Alimta until June 2021. Sawai and Teva, one of the other companies in the Sawai invalidation trial, have filed appeals in both cases. The remaining invalidation trials initiated by the other parties are currently suspended.
Notwithstanding our patents, generic versions of Alimta were approved in Japan in February 2016. We filed preliminary injunctions against four generic competitors. We do not anticipate generic competitors to proceed to launch prior to the completion of the Sawai invalidation trial.
Effient Patent Litigation and Administrative Proceedings
We, along with Daiichi Sankyo, Daiichi Sankyo, Inc., and Ube Industries (Ube) are engaged in U.S. patent litigation involving Effient brought pursuant to procedures set out in the Hatch-Waxman Act. More than ten different companies have submitted ANDAs seeking approval to market generic versions of Effient prior to the expiration of Daiichi Sankyo’s and Ube’s patents (expiring in 2023) covering methods of using Effient with aspirin, and alleging the patents are invalid. One of these ANDAs also alleges that the compound patent for Effient (expiring in April 2017) is invalid.
Beginning in March 2014, we filed lawsuits in the U.S. District Court for the Southern District of Indiana against these companies, seeking a ruling that the patents are valid and infringed. These cases have been consolidated. Four generic companies have agreed to be bound by the outcome of the consolidated case.
In 2015, several generic pharmaceutical companies filed petitions with the USPTO, requesting IPR of the method patents. In September 2015, the USPTO granted the generic pharmaceutical companies' request and scheduled review in mid-2016. In light of these petitions, the district court in the consolidated lawsuit stayed the case with respect to all parties.
We believe the Effient patents are valid and enforceable against these generic manufacturers. However, it is not possible to determine the outcome of the proceedings, and accordingly, we can provide no assurance that we will prevail. We expect a loss of exclusivity for Effient would result in a rapid and severe decline in future revenues for the product in the relevant market.
Actos® Product Liability Litigation
We have been named along with Takeda Chemical Industries, Ltd., and Takeda affiliates (collectively, Takeda) as a defendant in approximately 6,500 product liability cases in the U.S. related to the diabetes medication Actos, which we co-promoted with Takeda in the U.S. from 1999 until 2006. In general, plaintiffs in these actions allege that Actos caused or contributed to their bladder cancer. Almost all of the active cases have been consolidated in federal multi-district litigation (MDL) in the Western District of Louisiana or are pending in a coordinated state court proceeding in California or a coordinated state court proceeding in Illinois.
In April 2015, Takeda announced they will pay approximately $2.4 billion to resolve the vast majority of the U.S. product liability lawsuits involving Actos, including the case of Allen, et al. v. Takeda Pharmaceuticals, et al., no. 6:12-md-00064, in which a judgment of approximately $28 million was entered against Takeda and a
96
judgment of approximately $9 million was entered against us. In September 2015, Takeda announced that more than 96 percent of eligible claimants have opted into the resolution program that was announced in April 2015. Takeda is now evaluating the submissions to determine whether they satisfy various criteria specified under the terms of the resolution program. Takeda expects the resolution program to become effective upon completion of that review.
Although the vast majority of U.S. product liability lawsuits involving Actos are included in the resolution program announced in April 2015, there may be additional cases pending against Takeda and us following completion of the resolution program. Our agreement with Takeda calls for Takeda to defend and indemnify us against our losses and expenses with respect to the U.S. litigation arising out of the manufacture, use, or sale of Actos and other related expenses in accordance with the terms of the agreement. We believe we are entitled to full indemnification of our losses and expenses in the U.S. cases; however, there can be no guarantee we will ultimately be successful in obtaining full indemnification.
We are also named along with Takeda as a defendant in four purported product liability class actions in Canada related to Actos, including two in Ontario (Casseres et al. v. Takeda Pharmaceutical North America, Inc., et al. and Carrier et al. v. Eli Lilly et al.), one in Quebec (Whyte et al. v. Eli Lilly et al.), and one in Alberta (Epp v. Takeda Canada et al.). We promoted Actos in Canada until 2009.
We believe these lawsuits are without merit, and we and Takeda are prepared to defend against them vigorously.
Byetta Product Liability Litigation
We are named as a defendant in approximately 510 Byetta product liability lawsuits in the U.S. involving approximately 1,035 plaintiffs. Approximately 110 of these lawsuits, covering about 630 plaintiffs, are filed in California state court and coordinated in a Los Angeles Superior Court. Approximately 395 lawsuits, covering about 400 plaintiffs, are filed in federal court, the majority of which are coordinated in a MDL in the U.S. District Court for the Southern District of California. The remaining approximately five lawsuits, representing about five plaintiffs, are in various state courts. Approximately 450 of the lawsuits, involving approximately 690 plaintiffs, contain allegations that Byetta caused or contributed to the plaintiffs' cancer (primarily pancreatic cancer or thyroid cancer). The federal and state trial courts granted summary judgment in favor of us and co-defendants on the claims alleging pancreatic cancer; those rulings are being appealed by the plaintiffs. We are aware of approximately 10 additional claimants who have not yet filed suit. These additional claims allege damages for pancreatic cancer or thyroid cancer. We believe these lawsuits and claims are without merit and are prepared to defend against them vigorously.
Cymbalta® Product Liability Litigation
In October 2012, we were named as a defendant in a purported class-action lawsuit in the U.S. District Court for the Central District of California (Saavedra et al v. Eli Lilly and Company) involving Cymbalta. The plaintiffs, purporting to represent a class of all persons within the U.S. who purchased and/or paid for Cymbalta, asserted claims under the consumer protection statutes of four states, California, Massachusetts, Missouri, and New York, and sought declaratory, injunctive, and monetary relief for various alleged economic injuries arising from discontinuing treatment with Cymbalta. In December 2014, the district court denied the plaintiffs' motion for class certification. Plaintiffs filed a petition with the U.S. Court of Appeals for the Ninth Circuit requesting permission to file an interlocutory appeal of the denial of class certification, which was denied. Plaintiffs filed a second motion for certification under the consumer protection acts of New York and Massachusetts. The district court denied that motion for class certification in July 2015. The district court dismissed the suit and plaintiffs are appealing to the U.S. Court of Appeals for the Ninth Circuit.
Additionally, we are named in approximately 140 lawsuits involving approximately 1,300 plaintiffs filed in various federal and state courts alleging injuries arising from discontinuation of treatment with Cymbalta. Counsel for plaintiffs in the federal court proceedings filed a petition seeking to have then-filed cases and an unspecified number of future cases coordinated into a federal MDL in the U.S. District Court for the Central District of California. In December 2014, the Judicial Panel on Multidistrict Litigation (JPML) denied the plaintiffs' petition for creation of an MDL. Plaintiffs’ counsel subsequently filed a second petition seeking MDL consolidation, which petition was denied by the JPML in October 2015. There have been approximately 35 individual and multi-plaintiff cases filed in California state court. Most of those cases have been centralized in a California Judicial Counsel Coordination Proceeding pending in Los Angeles. The first individual product liability cases were tried in August 2015 and resulted in defense verdicts against four plaintiffs. Two of those plaintiffs are appealing the verdicts against them.
97
We believe all these Cymbalta lawsuits and claims are without merit and are prepared to defend against them vigorously.
Prozac® Product Liability Litigation
We are named as a defendant in approximately 10 U.S. lawsuits primarily related to allegations that the antidepressant Prozac caused or contributed to birth defects in the children of women who ingested the drug during pregnancy. We are aware of approximately 515 additional claims related to birth defects, which have not yet been filed. We believe these lawsuits and claims are without merit and are prepared to defend against them vigorously.
Brazil–Employee Litigation
Our subsidiary in Brazil, Eli Lilly do Brasil Limitada (Lilly Brasil), is named in a lawsuit brought by the Labor Attorney for 15th Region in the Labor Court of Paulinia, State of Sao Paulo, Brazil, alleging possible harm to employees and former employees caused by exposure to heavy metals at a former Lilly manufacturing facility in Cosmopolis, Brazil, operated by the company between 1977 and 2003. The plaintiffs allege that some employees at the facility were exposed to benzene and heavy metals; however, Lilly Brasil maintains that these alleged contaminants were never used in the facility. In May 2014, the labor court judge ruled against Lilly Brasil. The judge's ruling orders Lilly Brasil to undertake several actions of unspecified financial impact, including paying lifetime medical insurance for the employees and contractors who worked at the Cosmopolis facility more than six months during the affected years and their children born during and after this period. While we cannot currently estimate the range of reasonably possible financial losses that could arise in the event we do not ultimately prevail in the litigation, the judge has estimated the total financial impact of the ruling to be approximately 1.0 billion Brazilian real (approximately $255 million as of December 31, 2015) plus interest. We strongly disagree with the decision and filed an appeal in May 2014.
We are also named in approximately 25 lawsuits filed in the same court by individual former employees making similar claims. We believe these lawsuits are without merit and are prepared to defend against them vigorously.
Product Liability Insurance
Because of the nature of pharmaceutical products, it is possible that we could become subject to large numbers of product liability and related claims in the future. Due to a very restrictive market for product liability insurance, we are self-insured for product liability losses for all our currently marketed products.
98
Note 16: Other Comprehensive Income (Loss)
The following table summarizes the activity related to each component of other comprehensive income (loss):
(Amounts presented net of taxes) | Foreign Currency Translation Gains (Losses) | Unrealized Net Gains (Losses) on Securities | Defined Benefit Pension and Retiree Health Benefit Plans | Effective Portion of Cash Flow Hedges | Accumulated Other Comprehensive Loss | ||||||||||||||
Beginning balance at January 1, 2013 | $ | 426.8 | $ | 72.5 | $ | (4,195.2 | ) | $ | (101.2 | ) | $ | (3,797.1 | ) | ||||||
Other comprehensive income (loss) before reclassifications | 36.2 | 138.9 | 1,387.1 | (86.5 | ) | 1,475.7 | |||||||||||||
Net amount reclassified from accumulated other comprehensive loss | — | (6.2 | ) | 319.0 | 5.9 | 318.7 | |||||||||||||
Net other comprehensive income (loss) | 36.2 | 132.7 | 1,706.1 | (80.6 | ) | 1,794.4 | |||||||||||||
Balance at December 31, 2013 | 463.0 | 205.2 | (2,489.1 | ) | (181.8 | ) | (2,002.7 | ) | |||||||||||
Other comprehensive income (loss) before reclassifications | (961.4 | ) | 105.2 | (1,098.5 | ) | (15.2 | ) | (1,969.9 | ) | ||||||||||
Net amount reclassified from accumulated other comprehensive loss | — | (210.7 | ) | 185.6 | 5.9 | (19.2 | ) | ||||||||||||
Net other comprehensive income (loss) | (961.4 | ) | (105.5 | ) | (912.9 | ) | (9.3 | ) | (1,989.1 | ) | |||||||||
Balance at December 31, 2014 | (498.4 | ) | 99.7 | (3,402.0 | ) | (191.1 | ) | (3,991.8 | ) | ||||||||||
Other comprehensive income (loss) before reclassifications | (861.8 | ) | 38.6 | 155.0 | (36.9 | ) | (705.1 | ) | |||||||||||
Net amount reclassified from accumulated other comprehensive loss | — | (128.2 | ) | 234.9 | 9.5 | 116.2 | |||||||||||||
Net other comprehensive income (loss) | (861.8 | ) | (89.6 | ) | 389.9 | (27.4 | ) | (588.9 | ) | ||||||||||
Ending Balance at December 31, 2015 | $ | (1,360.2 | ) | $ | 10.1 | $ | (3,012.1 | ) | $ | (218.5 | ) | $ | (4,580.7 | ) | |||||
The tax effects on the net activity related to each component of other comprehensive income (loss) for the years ended December 31, were as follows:
Tax (expense) benefit | 2015 | 2014 | 2013 | ||||||||
Foreign currency translation gains (losses) | $ | (2.0 | ) | $ | — | $ | — | ||||
Unrealized net gains (losses) on securities | 48.5 | 56.7 | (71.6 | ) | |||||||
Defined benefit pension and retiree health benefit plans | (183.0 | ) | 414.7 | (886.1 | ) | ||||||
Effective portion of cash flow hedges | 14.6 | 5.2 | 43.2 | ||||||||
Provision for income taxes related to other comprehensive income (loss) items | $ | (121.9 | ) | $ | 476.6 | $ | (914.5 | ) | |||
Except for the tax effects of foreign currency translation gains (losses) related to our euro-denominated notes (see Note 7), income taxes were not provided for foreign currency translation.
99
Reclassifications Out of Accumulated Other Comprehensive Loss | ||||||||||||
Details about Accumulated Other Comprehensive Loss Components | Year Ended December 31, | Affected Line Item in the Consolidated Statements of Operations | ||||||||||
2015 | 2014 | 2013 | ||||||||||
Amortization of defined benefit items: | ||||||||||||
Prior service benefits, net | $ | (80.7 | ) | $ | (34.0 | ) | $ | (31.9 | ) | (1) | ||
Actuarial losses | 421.2 | 303.0 | 515.2 | (1) | ||||||||
Total before tax | 340.5 | 269.0 | 483.3 | |||||||||
Tax benefit | (105.6 | ) | (83.4 | ) | (164.3 | ) | Income taxes | |||||
Net of tax | 234.9 | 185.6 | 319.0 | |||||||||
Unrealized gains/losses on available-for-sale securities: | ||||||||||||
Realized gains, net | (209.3 | ) | (324.1 | ) | (12.0 | ) | Other—net, (income) expense | |||||
Impairment losses | 12.0 | — | 2.4 | Other—net, (income) expense | ||||||||
Total before tax | (197.3 | ) | (324.1 | ) | (9.6 | ) | ||||||
Tax expense | 69.1 | 113.4 | 3.4 | Income taxes | ||||||||
Net of tax | (128.2 | ) | (210.7 | ) | (6.2 | ) | ||||||
Other, net of tax | 9.5 | 5.9 | 5.9 | Other—net, (income) expense | ||||||||
Total reclassifications for the period (net of tax) | $ | 116.2 | $ | (19.2 | ) | $ | 318.7 | |||||
(1) These accumulated other comprehensive loss components are included in the computation of net periodic pension cost (see Note 14).
Note 17: Other–Net, (Income) Expense
Other–net, (income) expense consisted of the following:
2015 | 2014 | 2013 | |||||||||
Income related to termination of the exenatide collaboration with Amylin (Note 4) | $ | — | $ | — | $ | (495.4 | ) | ||||
Interest expense | 161.2 | 148.8 | 160.1 | ||||||||
Interest income | (87.0 | ) | (121.0 | ) | (119.7 | ) | |||||
Debt extinguishment loss (Note 10) | 166.7 | — | — | ||||||||
Other income | (341.5 | ) | (368.3 | ) | (63.9 | ) | |||||
Other–net, (income) expense | $ | (100.6 | ) | $ | (340.5 | ) | $ | (518.9 | ) | ||
For the year ended December 31, 2015, other income is primarily related to net gains on investments (Note 7). For the year ended December 31, 2014, other income is primarily related to net gains on investments (Note 7) and income related to the transfer to Boehringer Ingelheim of our license rights to co-promote linagliptin and empagliflozin in certain countries (Note 4).
100
Note 18: Segment Information
We operate in two business segments—human pharmaceutical products and animal health. Our business segments are distinguished by the ultimate end user of the product—humans or animals. Performance is evaluated based on profit or loss from operations before income taxes. The accounting policies of the individual segments are the same as those described throughout the notes to the consolidated financial statements.
Our human pharmaceutical products segment includes the discovery, development, manufacturing, marketing, and sales of human pharmaceutical products worldwide in the following therapeutic areas: endocrinology, neuroscience, oncology, cardiovascular, and other. We lost U.S. patent exclusivity for Cymbalta in December 2013 and Evista® in March 2014, which resulted in the immediate entry of generic competitors and a rapid and severe decline in revenue.
Our animal health segment, operating through our Elanco animal health division, includes the development, manufacturing, marketing, and sales of animal health products worldwide for both food and companion animals. Animal health products include Rumensin®, Posilac®, Tylan®, Denagard®, Maxiban®, Optaflexx®, and other products for livestock and poultry, as well as Trifexis®, Comfortis®, and other products for companion animals. The animal health segment amounts for the year ended December 31, 2015 include the results of operations from Novartis AH, which was acquired on January 1, 2015 (Note 3).
Most of our pharmaceutical products are distributed through wholesalers that serve pharmacies, physicians and other health care professionals, and hospitals. For the years ended December 31, 2015, 2014, and 2013, our three largest wholesalers each accounted for between 8 percent and 19 percent of consolidated total revenue. Further, they each accounted for between 9 percent and 16 percent of accounts receivable as of December 31, 2015 and 2014. Animal health products are sold primarily to wholesale distributors.
We manage our assets on a total company basis, not by operating segment, as the assets of the animal health business are intermixed with those of the pharmaceutical products business. Therefore, our chief operating decision maker does not review any asset information by operating segment and, accordingly, we do not report asset information by operating segment.
We are exposed to the risk of changes in social, political, and economic conditions inherent in foreign operations, and our results of operations and the value of our foreign assets are affected by fluctuations in foreign currency exchange rates.
The following table summarizes our revenue activity:
2015 | 2014 | 2013 | ||||||||||||
Segment revenue—to unaffiliated customers: | ||||||||||||||
Human pharmaceutical products: | ||||||||||||||
Endocrinology: | ||||||||||||||
Humalog® | $ | 2,841.9 | $ | 2,785.2 | $ | 2,611.2 | ||||||||
Forteo® | 1,348.3 | 1,322.0 | 1,244.9 | |||||||||||
Humulin® | 1,307.4 | 1,400.1 | 1,315.8 | |||||||||||
Trajenta | 356.8 | 328.8 | 249.2 | |||||||||||
Trulicity® | 248.7 | 10.2 | — | |||||||||||
Evista | 237.3 | 419.8 | 1,050.4 | |||||||||||
Other Endocrinology | 696.4 | 672.9 | 832.9 | |||||||||||
Total Endocrinology | 7,036.8 | 6,939.0 | 7,304.4 | |||||||||||
101
2015 | 2014 | 2013 | ||||||||||||
Neuroscience: | ||||||||||||||
Cymbalta | 1,027.6 | 1,614.7 | 5,084.4 | |||||||||||
Zyprexa® | 940.3 | 1,037.3 | 1,194.8 | |||||||||||
Strattera® | 784.0 | 738.5 | 709.2 | |||||||||||
Other Neuroscience | 183.5 | 206.0 | 227.8 | |||||||||||
Total Neuroscience | 2,935.4 | 3,596.5 | 7,216.2 | |||||||||||
Oncology: | ||||||||||||||
Alimta | 2,493.1 | 2,792.0 | 2,703.0 | |||||||||||
Erbitux | 485.0 | 373.3 | 373.7 | |||||||||||
Cyramza® | 383.8 | 75.6 | — | |||||||||||
Other Oncology | 147.9 | 152.1 | 191.8 | |||||||||||
Total Oncology | 3,509.8 | 3,393.0 | 3,268.5 | |||||||||||
Cardiovascular: | ||||||||||||||
Cialis® | 2,310.7 | 2,291.0 | 2,159.4 | |||||||||||
Effient | 523.0 | 522.2 | 508.7 | |||||||||||
Other Cardiovascular | 234.3 | 240.3 | 255.1 | |||||||||||
Total Cardiovascular | 3,068.0 | 3,053.5 | 2,923.2 | |||||||||||
Other pharmaceuticals | 227.7 | 287.0 | 249.3 | |||||||||||
Total human pharmaceutical products | 16,777.7 | 17,269.0 | 20,961.6 | |||||||||||
Animal health | 3,181.0 | 2,346.6 | 2,151.5 | |||||||||||
Revenue | $ | 19,958.7 | $ | 19,615.6 | $ | 23,113.1 | ||||||||
Segment profits: | ||||||||||||||
Human pharmaceutical products | $ | 4,026.7 | $ | 3,604.6 | $ | 5,521.0 | ||||||||
Animal health | 597.9 | 621.8 | 605.6 | |||||||||||
Total segment profits | $ | 4,624.6 | $ | 4,226.4 | $ | 6,126.6 | ||||||||
Reconciliation of total segment profits to consolidated income before taxes: | ||||||||||||||
Segment profits | $ | 4,624.6 | $ | 4,226.4 | $ | 6,126.6 | ||||||||
Other profits (losses): | ||||||||||||||
Inventory fair value adjustment related to Novartis AH (Note 3) | (153.0 | ) | — | — | ||||||||||
Acquired in-process research and development (Notes 3 and 4) | (535.0 | ) | (200.2 | ) | (57.1 | ) | ||||||||
Asset impairment, restructuring, and other special charges (Note 5) | (367.7 | ) | (468.7 | ) | (120.6 | ) | ||||||||
Debt repurchase charges, net(1) (Note 10) | (152.7 | ) | — | — | ||||||||||
Amortization of intangible assets(2) (Note 8) | (626.2 | ) | (530.2 | ) | (555.0 | ) | ||||||||
Income related to transfer of linagliptin and empagliflozin rights in certain countries to Boehringer Ingelheim (Note 4) | — | 92.0 | — | |||||||||||
U.S. Branded Prescription Drug Fee | — | (119.0 | ) | — | ||||||||||
Income related to termination of the exenatide collaboration with Amylin Pharmaceuticals, Inc. (Note 4) | — | — | 495.4 | |||||||||||
Consolidated income before taxes | $ | 2,790.0 | $ | 3,000.3 | $ | 5,889.3 | ||||||||
(1) We recognized pretax net charges of $152.7 million for the year ended December 31, 2015, attributable to the debt extinguishment loss of $166.7 million from the purchase and redemption of certain fixed-rate notes, partially offset by net gains from non-hedging interest rate swaps and foreign currency transactions associated with the related issuance of euro-denominated notes.
(2) In 2015, the measurement of segment profitability was changed to exclude certain amortization of intangible assets. The prior periods have been adjusted to conform with the 2015 presentation.
102
Depreciation and software amortization expense included in our segment profits was as follows:
2015 | 2014 | 2013 | |||||||||
Human pharmaceutical products | $ | 720.7 | $ | 790.0 | $ | 838.8 | |||||
Animal health | 80.8 | 58.8 | 51.8 | ||||||||
Total depreciation expense included in segment profits | $ | 801.5 | $ | 848.8 | $ | 890.6 | |||||
For internal management reporting presented to the chief operating decision maker, certain costs are fully allocated to our human pharmaceutical products segment and therefore are not reflected in the animal health segment's profit. Such items include costs associated with treasury-related financing, global administrative services, certain acquisition-related transaction costs, and certain manufacturing costs.
2015 | 2014 | 2013 | ||||||||||||
Geographic Information | ||||||||||||||
Revenue—to unaffiliated customers(1): | ||||||||||||||
United States | $ | 10,097.4 | $ | 9,134.1 | $ | 12,889.7 | ||||||||
Europe | 3,943.6 | 4,506.7 | 4,338.4 | |||||||||||
Japan | 2,033.1 | 2,027.1 | 2,063.8 | |||||||||||
Other foreign countries | 3,884.6 | 3,947.7 | 3,821.2 | |||||||||||
Revenue | $ | 19,958.7 | $ | 19,615.6 | $ | 23,113.1 | ||||||||
Long-lived assets(2): | ||||||||||||||
United States | $ | 4,576.8 | $ | 4,566.2 | $ | 4,649.6 | ||||||||
Europe | 2,306.4 | 2,401.5 | 2,469.7 | |||||||||||
Japan | 89.2 | 80.4 | 81.1 | |||||||||||
Other foreign countries | 1,724.2 | 1,499.1 | 1,540.9 | |||||||||||
Long-lived assets | $ | 8,696.6 | $ | 8,547.2 | $ | 8,741.3 | ||||||||
(1) Revenue is attributed to the countries based on the location of the customer.
(2) Long-lived assets consist of property and equipment, net, and certain sundry assets.
103
Note 19: Selected Quarterly Data (unaudited)
2015 | Fourth | Third | Second | First | ||||||||||||
Revenue | $ | 5,375.6 | $ | 4,959.7 | $ | 4,978.7 | $ | 4,644.7 | ||||||||
Cost of sales | 1,389.2 | 1,236.9 | 1,218.4 | 1,192.7 | ||||||||||||
Operating expenses(1) | 3,242.6 | 2,719.1 | 2,804.9 | 2,562.8 | ||||||||||||
Acquired in-process research and development | 199.0 | — | 80.0 | 256.0 | ||||||||||||
Asset impairment, restructuring, and other special charges | 144.9 | 42.4 | 72.4 | 108.0 | ||||||||||||
Other—net, (income) expense | (44.7 | ) | (86.5 | ) | 123.3 | (92.7 | ) | |||||||||
Income before income taxes | 444.6 | 1,047.8 | 679.7 | 617.9 | ||||||||||||
Net income | 478.4 | 799.7 | 600.8 | 529.5 | ||||||||||||
Earnings per share—basic | 0.45 | 0.75 | 0.57 | 0.50 | ||||||||||||
Earnings per share—diluted | 0.45 | 0.75 | 0.56 | 0.50 | ||||||||||||
Dividends paid per share | 0.50 | 0.50 | 0.50 | 0.50 | ||||||||||||
Common stock closing prices: | ||||||||||||||||
High | 87.52 | 89.98 | 86.59 | 76.36 | ||||||||||||
Low | 76.98 | 78.26 | 70.89 | 68.41 | ||||||||||||
2014 | Fourth | Third | Second | First | ||||||||||||
Revenue | $ | 5,121.3 | $ | 4,875.6 | $ | 4,935.6 | $ | 4,683.1 | ||||||||
Cost of sales | 1,253.1 | 1,267.0 | 1,189.7 | 1,222.7 | ||||||||||||
Operating expenses(1) | 2,985.6 | 2,915.3 | 2,859.3 | 2,594.2 | ||||||||||||
Acquired in-process research and development | 105.2 | 95.0 | — | — | ||||||||||||
Asset impairment, restructuring, and other special charges | 401.0 | 36.3 | — | 31.4 | ||||||||||||
Other—net, (income) expense | (137.2 | ) | (93.5 | ) | (53.8 | ) | (56.0 | ) | ||||||||
Income before income taxes | 513.6 | 655.5 | 940.4 | 890.8 | ||||||||||||
Net income | 428.5 | 500.6 | 733.5 | 727.9 | ||||||||||||
Earnings per share—basic | 0.40 | 0.47 | 0.68 | 0.68 | ||||||||||||
Earnings per share—diluted | 0.40 | 0.47 | 0.68 | 0.68 | ||||||||||||
Dividends paid per share | 0.49 | 0.49 | 0.49 | 0.49 | ||||||||||||
Common stock closing prices: | ||||||||||||||||
High | 72.83 | 66.59 | 63.10 | 59.85 | ||||||||||||
Low | 61.90 | 60.35 | 58.21 | 50.73 | ||||||||||||
(1) Includes research and development and marketing, selling, and administrative expenses.
Our common stock is listed on the New York Stock Exchange (NYSE), NYSE Euronext, and SIX Swiss Exchange.
104
Management’s Reports
Management’s Report for Financial Statements—Eli Lilly and Company and Subsidiaries
Management of Eli Lilly and Company and subsidiaries is responsible for the accuracy, integrity, and fair presentation of the financial statements. The statements have been prepared in accordance with generally accepted accounting principles in the United States and include amounts based on judgments and estimates by management. In management’s opinion, the consolidated financial statements present fairly our financial position, results of operations, and cash flows.
In addition to the system of internal accounting controls, we maintain a code of conduct (known as "The Red Book") that applies to all employees worldwide, requiring proper overall business conduct, avoidance of conflicts of interest, compliance with laws, and confidentiality of proprietary information. All employees must take training annually on The Red Book and are required to report suspected violations. A hotline number is published in The Red Book to enable employees to report suspected violations anonymously. Employees who report suspected violations are protected from discrimination or retaliation by the company. In addition to The Red Book, the CEO and all financial management must sign a financial code of ethics, which further reinforces their fiduciary responsibilities.
The consolidated financial statements have been audited by Ernst & Young LLP, an independent registered public accounting firm. Their responsibility is to examine our consolidated financial statements in accordance with generally accepted auditing standards of the Public Company Accounting Oversight Board (United States). Ernst & Young’s opinion with respect to the fairness of the presentation of the statements is included in Item 8 of our annual report on Form 10-K. Ernst & Young reports directly to the audit committee of the board of directors.
Our audit committee includes four nonemployee members of the board of directors, all of whom are independent from our company. The committee charter, which is available on our website, outlines the members’ roles and responsibilities and is consistent with enacted corporate reform laws and regulations. It is the audit committee’s responsibility to appoint an independent registered public accounting firm subject to shareholder ratification, approve both audit and non-audit services performed by the independent registered public accounting firm, and review the reports submitted by the firm. The audit committee meets several times during the year with management, the internal auditors, and the independent public accounting firm to discuss audit activities, internal controls, and financial reporting matters, including reviews of our externally published financial results. The internal auditors and the independent registered public accounting firm have full and free access to the committee.
We are dedicated to ensuring that we maintain the high standards of financial accounting and reporting that we have established. We are committed to providing financial information that is transparent, timely, complete, relevant, and accurate. Our culture demands integrity and an unyielding commitment to strong internal practices and policies. Finally, we have the highest confidence in our financial reporting, our underlying system of internal controls, and our people, who are objective in their responsibilities and operate under a code of conduct and the highest level of ethical standards.
Management’s Report on Internal Control Over Financial Reporting—Eli Lilly and Company and Subsidiaries
Management of Eli Lilly and Company and subsidiaries is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934. We have global financial policies that govern critical areas, including internal controls, financial accounting and reporting, fiduciary accountability, and safeguarding of corporate assets. Our internal accounting control systems are designed to provide reasonable assurance that assets are safeguarded, that transactions are executed in accordance with management’s authorization and are properly recorded, and that accounting records are adequate for preparation of financial statements and other financial information. A staff of internal auditors regularly monitors, on a worldwide basis, the adequacy and effectiveness of internal accounting controls. The general auditor reports directly to the audit committee of the board of directors.
105
We conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in "2013 Internal Control—Integrated Framework" issued by the Committee of Sponsoring Organizations of the Treadway Commission. Our evaluation excluded the current year acquisition of Novartis Animal Health. The operations acquired from Novartis AG represented approximately 3% of our consolidated total assets and 5% of our consolidated net sales as of and for the year ended December 31, 2015.
Based on our evaluation under this framework, we concluded that our internal control over financial reporting was effective as of December 31, 2015. However, because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The internal control over financial reporting has been assessed by Ernst & Young LLP as of December 31, 2015. Their responsibility is to evaluate whether internal control over financial reporting was designed and operating effectively.
John C. Lechleiter, Ph.D. | Derica W. Rice | |
Chairman, President, and Chief Executive Officer | Executive Vice President, Global Services and Chief Financial Officer | |
February 19, 2016
106
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Eli Lilly and Company
We have audited the accompanying consolidated balance sheets of Eli Lilly and Company and subsidiaries as of December 31, 2015 and 2014, and the related consolidated statements of operations, comprehensive income, shareholders' equity, and cash flows for each of the three years in the period ended December 31, 2015. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Eli Lilly and Company and subsidiaries at December 31, 2015 and 2014, and the consolidated results of their operations and their cash flows for each of the three years in the period ended December 31, 2015, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 2 to the consolidated financial statements, the Company changed its method for classifying deferred tax liabilities and assets as a result of the adoption of the amendments to the FASB Accounting Standards Codification resulting from Accounting Standards Update No. 2015-17, Balance Sheet Classification of Deferred Taxes.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Eli Lilly and Company and subsidiaries’ internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated February 19, 2016, expressed an unqualified opinion thereon.
 | |
Indianapolis, Indiana
February 19, 2016
107
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Eli Lilly and Company
We have audited Eli Lilly and Company and subsidiaries’ internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). Eli Lilly and Company and subsidiaries’ management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
As indicated in the accompanying Management’s Report on Internal Control Over Financial Reporting, management’s assessment of and conclusion on the effectiveness of internal control over financial reporting did not include the internal controls of Novartis Animal Health, which is included in the 2015 consolidated financial statements of Eli Lilly and Company and subsidiaries and constituted 3% of total assets as of December 31, 2015 and 5% of revenues for the year then ended. Our audit of internal control over financial reporting of Eli Lilly and Company and subsidiaries also did not include an evaluation of the internal control over financial reporting of Novartis Animal Health.
108
In our opinion, Eli Lilly and Company and subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the 2015 consolidated financial statements of Eli Lilly and Company and subsidiaries and our report dated February 19, 2016 expressed an unqualified opinion thereon.
| |
Indianapolis, Indiana
February 19, 2016
109
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
Item 9A. | Controls and Procedures |
Disclosure Controls and Procedures
Under applicable Securities and Exchange Commission (SEC) regulations, management of a reporting company, with the participation of the principal executive officer and principal financial officer, must periodically evaluate the company’s “disclosure controls and procedures,” which are defined generally as controls and other procedures designed to ensure that information required to be disclosed by the reporting company in its periodic reports filed with the SEC (such as this Form 10-K) is recorded, processed, summarized, and reported on a timely basis.
Our management, with the participation of John C. Lechleiter, Ph.D., chairman, president, and chief executive officer, and Derica W. Rice, executive vice president, global services and chief financial officer, evaluated our disclosure controls and procedures as of December 31, 2015, and concluded that they are effective.
Internal Control over Financial Reporting
Dr. Lechleiter and Mr. Rice provided a report on behalf of management on our internal control over financial reporting, in which management concluded that the company’s internal control over financial reporting is effective at December 31, 2015. Our evaluation excluded the 2015 acquisition of Novartis Animal Health (Novartis AH). In addition, Ernst & Young LLP, the company’s independent registered public accounting firm, provided an attestation report on the company’s internal control over financial reporting as of December 31, 2015. You can find the full text of management’s report and Ernst & Young’s attestation report in Item 8, and both reports are incorporated by reference in this Item.
Changes in Internal Controls
During the fourth quarter of 2015, there were no changes in our internal control over financial reporting that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. This evaluation excluded the operations of Novartis AH, which we acquired on January 1, 2015. As part of the ongoing integration activities, we will complete an assessment of Novartis AH's existing controls and incorporate our controls and procedures into the acquired operations, as appropriate.
110
Item 9B. | Other Information |
Disclosure Pursuant to Section 219 of the Iran Threat Reduction and Syria Human Rights Act of 2012
Section 219 of the Iran Threat Reduction and Syria Human Rights Act of 2012 (ITRSHRA), which added Section 13(r) of the Exchange Act, requires disclosure by public companies of, among other things, certain transactions involving the Government of Iran, as well as entities and individuals designated under Executive Order 13382. ITRSHRA requires companies to disclose these types of transactions even if they were permissible under U.S. law or were conducted by a non-U.S. subsidiary in accordance with the local law under which such entity operates.
As a global pharmaceutical company, we conduct business in multiple jurisdictions throughout the world. Our activities have included supplying medicines for patient use in Iran. We ship our products to Iran and conduct related activities in accordance with our corporate policies and licenses issued by the U.S. Department of the Treasury's Office of Foreign Assets Control.
Pursuant to U.S. government authorizations, we, through a non-U.S. subsidiary, shipped our products to authorized customers in Iran. We recently completed a review in which we determined that between the last quarter of 2013 and the first half of 2015, some of these shipments, which were arranged by a third-party logistics company, were sent to Iran on aircraft owned or operated by Iran Air, which is a designated air carrier under Executive Order 13382. Neither we nor our affiliate entered into agreements with nor made any direct payments to Iran Air. We generated no gross revenues or net profits during the relevant periods attributable to the use of this carrier. Our affiliate paid air freight expenses associated with these shipments to the third-party logistics company in the total amount of approximately $51,700 using exchange rates in effect at the time of each shipment. We have voluntarily self-disclosed this matter to the U.S. government. We also have instructed the third-party logistics company that arranged these shipments from our non-U.S. affiliate not to use Iran Air to ship our products in the future, and we have implemented additional controls to address this issue.
111
Part III
Item 10. | Directors, Executive Officers, and Corporate Governance |
Directors and Executive Officers
Information relating to our Board of Directors is found in our Proxy Statement to be dated on or about March 21, 2016 (the Proxy Statement) under “Board of Directors” and is incorporated in this report by reference.
Information relating to our executive officers is found at Item 1, "Business—Executive Officers of the Company.”
Code of Ethics
We have adopted a code of ethics that complies with the applicable SEC and New York Stock Exchange requirements. The code is set forth in:
• | The Red Book, a comprehensive code of ethical and legal business conduct applicable to all employees worldwide and to our Board of Directors; and |
• | Code of Ethical Conduct for Lilly Financial Management, a supplemental code for our chief executive officer and all members of financial management that focuses on accounting, financial reporting, internal controls, and financial stewardship. |
Both documents are online on our website at http://www.lilly.com/about/business-practices/ethics-compliance/Pages/ethics-compliance.aspx. In the event of any amendments to, or waivers from, a provision of the code affecting the chief executive officer, chief financial officer, chief accounting officer, controller, or persons performing similar functions, we intend to post on the above website within four business days after the event a description of the amendment or waiver as required under applicable SEC rules. We will maintain that information on our website for at least 12 months. Paper copies of these documents are available free of charge upon request to the company’s secretary at the address on the front of this Form 10-K.
Corporate Governance
In our proxy statements, we describe the procedures by which shareholders can recommend nominees to our board of directors. There have been no changes in those procedures since they were last published in our proxy statement of March 23, 2015.
The board has appointed an audit committee consisting entirely of independent directors in accordance with applicable SEC and New York Stock Exchange rules for audit committees. The members of the committee are Michael L. Eskew (chair), Katherine Baicker, Kathi P. Seifert, and Jackson P. Tai. The board has determined that Messrs. Eskew and Tai are audit committee financial experts as defined in the SEC rules.
Item 11. | Executive Compensation |
Information on director compensation, executive compensation, and compensation committee matters can be found in the Proxy Statement under “Director Compensation,” "Committees of the Board of Directors -- Compensation Committee," "Compensation Discussion and Analysis," and “Executive Compensation.” That information is incorporated in this report by reference.
112
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Security Ownership of Certain Beneficial Owners and Management
Information relating to ownership of the company’s common stock by management and by persons known by the company to be the beneficial owners of more than five percent of the outstanding shares of common stock is found in the Proxy Statement under “Ownership of Company Stock.” That information is incorporated in this report by reference.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table presents information as of December 31, 2015, regarding our compensation plans under which shares of Lilly common stock have been authorized for issuance.
Plan category | (a) Number of securities to be issued upon exercise of outstanding options, warrants, and rights | (b) Weighted-average exercise price of outstanding options, warrants, and rights | (c) Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | ||||
Equity compensation plans approved by security holders | 368,914 | $ | 56.20 | 100,636,160 | |||
Equity compensation plan not approved by security holders | — | — | — | ||||
Total | 368,914 | 56.20 | 100,636,160 | ||||
Item 13. | Certain Relationships and Related Transactions, and Director Independence |
Related Person Transactions
Information relating to three related person transactions and the board’s policies and procedures for approval of related person transactions can be found in the Proxy Statement under “Highlights of the Company’s Corporate Governance—Conflicts of Interest and Transactions with Related Persons.” That information is incorporated in this report by reference.
Director Independence
Information relating to director independence can be found in the Proxy Statement under “Director Independence” and is incorporated in this report by reference.
Item 14. | Principal Accountant Fees and Services |
Information related to the fees and services of our principal independent accountants, Ernst & Young LLP, can be found in the Proxy Statement under “Item 3. Proposal to Ratify the Appointment of Principal Independent Auditor—Services Performed by the Independent Auditor” and “Independent Auditor Fees.” That information is incorporated in this report by reference.
Item 15. | Exhibits and Financial Statement Schedules |
(a)1. Financial Statements
The following consolidated financial statements of the company and its subsidiaries are found at Item 8:
• | Consolidated Statements of Operations—Years Ended December 31, 2015, 2014, and 2013 |
• | Consolidated Statements of Comprehensive Income—Years Ended December 31, 2015, 2014, and 2013 |
• | Consolidated Balance Sheets—December 31, 2015 and 2014 |
• | Consolidated Statements of Shareholders' Equity—Years Ended December 31, 2015, 2014, and 2013 |
• | Consolidated Statements of Cash Flows—Years Ended December 31, 2015, 2014, and 2013 |
• | Notes to Consolidated Financial Statements |
(a)2. Financial Statement Schedules
The consolidated financial statement schedules of the company and its subsidiaries have been omitted because they are not required, are inapplicable, or are adequately explained in the financial statements.
Financial statements of interests of 50 percent or less, which are accounted for by the equity method, have been omitted because they do not, considered in the aggregate as a single subsidiary, constitute a significant subsidiary.
113
(a)3. Exhibits
2.1 | Stock and Asset Purchase Agreement between Novartis AG and Eli Lilly and Company dated as of April 22, 2014 | |
2.2 | First Amendment to Stock and Asset Purchase Agreement between Novartis AG and Eli Lilly and Company dated as of December 17, 2014 | |
3.1 | Amended Articles of Incorporation | |
3.2 | Bylaws, as amended | |
4.1 | Indenture with respect to Debt Securities dated as of February 1, 1991, between Eli Lilly and Company and Deutsche Bank Trust Company Americas, as successor trustee to Citibank, N.A., Trustee | |
4.2 | Agreement dated September 13, 2007 appointing Deutsche Bank Trust Company Americas as Successor Trustee under the Indenture listed above | |
10.1 | 2002 Lilly Stock Plan, as amended(1) | |
10.2 | Form of Performance Award under the 2002 Lilly Stock Plan(1) | |
10.3 | Form of Shareholder Value Award under the 2002 Lilly Stock Plan(1) | |
10.4 | The Lilly Deferred Compensation Plan, as amended(1) | |
10.5 | The Lilly Directors’ Deferral Plan, as amended(1) | |
10.6 | The Eli Lilly and Company Bonus Plan, as amended(1) | |
10.7 | The Eli Lilly and Company Executive Officer Incentive Plan(1) | |
10.8 | 2007 Change in Control Severance Pay Plan for Select Employees, as amended(1) | |
12 | Statement re: Computation of Ratio of Earnings to Fixed Charges | |
21 | List of Subsidiaries | |
23 | Consent of Independent Registered Public Accounting Firm | |
31.1 | Rule 13a-14(a) Certification of John C. Lechleiter, Ph.D., Chairman of the Board, President, and Chief Executive Officer | |
31.2 | Rule 13a-14(a) Certification of Derica W. Rice, Executive Vice President, Global Services and Chief Financial Officer | |
32 | Section 1350 Certification | |
101 | Interactive Data File | |
(1) Indicates management contract or compensatory plan.
Signatures
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
Eli Lilly and Company
By | /s/ John C. Lechleiter | |
John C. Lechleiter, Ph.D. | ||
Chairman of the Board, President, and Chief Executive Officer | ||
February 19, 2016
114
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below on February 19, 2016 by the following persons on behalf of the Registrant and in the capacities indicated.
Signature | Title | |
/s/ John C. Lechleiter, Ph.D. | Chairman of the Board, President, and Chief Executive Officer, and a Director (principal executive officer) | |
JOHN C. LECHLEITER, Ph.D. | ||
/s/ Derica W. Rice | Executive Vice President, Global Services and Chief Financial Officer (principal financial officer) | |
DERICA W. RICE | ||
/s/ Donald A. Zakrowski | Vice President, Finance and Chief Accounting Officer (principal accounting officer) | |
DONALD A. ZAKROWSKI | ||
/s/ Ralph Alvarez | Director | |
RALPH ALVAREZ | ||
/s/ Katherine Baicker, Ph.D. | Director | |
KATHERINE BAICKER, Ph.D. | ||
/s/ Michael L. Eskew | Director | |
MICHAEL L. ESKEW | ||
/s/ J. Erik Fyrwald | Director | |
J. ERIK FYRWALD | ||
/s/ R. David Hoover | Director | |
R. DAVID HOOVER | ||
/s/ Karen N. Horn, Ph.D. | Director | |
KAREN N. HORN, Ph.D. | ||
/s/ William G. Kaelin, Jr., M.D. | Director | |
WILLIAM G. KAELIN, JR., M.D. | ||
/s/ Juan R. Luciano | Director | |
JUAN R. LUCIANO | ||
/s/ Ellen R. Marram | Director | |
ELLEN R. MARRAM | ||
/s/ Franklyn G. Prendergast, M.D., Ph.D. | Director | |
FRANKLYN G. PRENDERGAST, M.D., Ph.D. | ||
/s/ Marschall S. Runge, M.D., Ph.D. | Director | |
MARSCHALL S. RUNGE, M.D., Ph.D. | ||
/s/ Kathi P. Seifert | Director | |
KATHI P. SEIFERT | ||
/s/ Jackson P. Tai | Director | |
JACKSON P. TAI | ||
115
Trademarks Used In This Report
Trademarks or service marks owned by Eli Lilly and Company or its subsidiaries or affiliates, when first used in this report, appear with an initial capital and are followed by the symbol ® or ™, as applicable. In subsequent uses of the marks in the report, the symbols may be omitted.
Actos® is a trademark of Takeda Pharmaceutical Company Limited.
ENHANZE™ is a trademark of Halozyme Therapeutics, Inc.
Bydureon® and Byetta® are trademarks of Amylin Pharmaceuticals, Inc.
Glyxambi®, Jardiance®, Jentadueto®, Synjardy® and Trajenta® are trademarks of Boehringer Ingelheim GmbH.
Lantus® is a trademark of Sanofi-Aventis Deutschland GmbH.
Sentinel® is a trademark of Virbac Corporation.
116
Index to Exhibits
The following documents are filed as part of this report:
Exhibit | Location | |||
2.1 | Stock and Asset Purchase Agreement between Novartis AG and Eli Lilly and Company dated as of April 22, 2014 | Incorporated by reference to Exhibit 2 to the Company's Report on Form 10-Q for the quarter ended June 30, 2014 | ||
2.2 | First Amendment to Stock and Asset Purchase Agreement between Novartis AG and Eli Lilly and Company dated as of December 17, 2014 (confidential treatment requested for certain information in this Amendment) | Incorporated by reference to Exhibit 2.2 to the Company's Report on Form 10-K for the year ended December 31, 2014 | ||
3.1 | Amended Articles of Incorporation | Incorporated by reference to Exhibit 3.1 to the Company's Report on Form 10-K for the year ended December 31, 2013 | ||
3.2 | Bylaws, as amended | Incorporated by reference to Exhibit 99 to the Company’s Report on Form 8-K filed February 27, 2012 | ||
4.1 | Indenture with respect to Debt Securities dated as of February 1, 1991, between Eli Lilly and Company and Deutsche Bank Trust Company Americas, as successor trustee to Citibank, N.A., Trustee | Incorporated by reference to Exhibit 4.1 to the Company’s Registration Statement on Form S-3, Registration No. 333-186979 | ||
4.2 | Agreement dated September 13, 2007 appointing Deutsche Bank Trust Company Americas as Successor Trustee under the Indenture listed above | Incorporated by reference to Exhibit 4.2 to the Company’s Report on Form 10-K for the year ended December 31, 2008 (SEC File No. 001-06351, Film No. 09640420) | ||
10.1 | 2002 Lilly Stock Plan, as amended | Incorporated by reference to Exhibit 10 to the Company’s Report on Form 10-Q for the quarter ended September 30, 2012 | ||
10.2 | Form of Performance Award under the 2002 Lilly Stock Plan | Incorporated by reference to Exhibit 10.2 to the Company's Report on Form 10-K for the year ended December 31, 2014 | ||
10.3 | Form of Shareholder Value Award under the 2002 Lilly Stock Plan | Attached | ||
10.4 | The Lilly Deferred Compensation Plan, as amended | Incorporated by reference to Exhibit 10.5 to the Company's Report on Form 10-K for the year ended December 31, 2013 | ||
10.5 | The Lilly Directors’ Deferral Plan, as amended | Incorporated by reference to Exhibit 10 to the Company’s Report on Form 10-Q for the quarter ended September 30, 2009 (SEC File No. 001-06351, Film No. 091147352) | ||
10.6 | The Eli Lilly and Company Bonus Plan, as amended | Incorporated by reference to Exhibit 10.7 to the Company's Report on Form 10-K for the year ended December 31, 2013 | ||
10.7 | The Eli Lilly and Company Executive Officer Incentive Plan | Incorporated by reference to Appendix B to the Company’s proxy statement on Schedule 14A filed March 7, 2011 (SEC File No. 001-06351, Film No. 11666753) | ||
117
Exhibit | Location | |||
10.8 | 2007 Change in Control Severance Pay Plan for Select Employees, as amended | Incorporated by reference to Exhibit 10 to the Company’s Report on Form 10-Q for the quarter ended September 30, 2010 (SEC File No. 001-06351, Film No. 101149876) | ||
12 | Statement re: Computation of Ratio of Earnings to Fixed Charges | Attached | ||
21 | List of Subsidiaries | Attached | ||
23 | Consent of Registered Independent Public Accounting Firm | Attached | ||
31.1 | Rule 13a-14(a) Certification of John C. Lechleiter, Ph.D., Chairman of the Board, President, and Chief Executive Officer | Attached | ||
31.2 | Rule 13a-14(a) Certification of Derica W. Rice, Executive Vice President, Global Services and Chief Financial Officer | Attached | ||
32 | Section 1350 Certification | Attached | ||
101 | Interactive Data File | Attached | ||
118