Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 - Moleculin Biotech, Inc. | v431913_ex23-1.htm |
| EX-99.1 - EXHIBIT 99.1 - Moleculin Biotech, Inc. | v431913_ex99-1.htm |
| EX-99.2 - EXHIBIT 99.2 - Moleculin Biotech, Inc. | v431913_ex99-2.htm |
| EX-99.3 - EXHIBIT 99.3 - Moleculin Biotech, Inc. | v431913_ex99-3.htm |
| EX-10.12 - EXHIBIT 10.12 - Moleculin Biotech, Inc. | v431913_ex10-12.htm |
As filed with the Securities and Exchange Commission on February 16, 2016.
Registration No. 333-209323
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 1 TO
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Moleculin Biotech, Inc.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 2834 | 47-4671997 |
| (State or Other Jurisdiction of | (Primary Standard Industrial | (I.R.S. Employer |
| Incorporation or Organization) | Classification Code Number) | Identification Number) |
2575 West Bellfort, Suite 333
Houston, Texas 77054
(713) 300-5160
(Address, Including Zip Code, and Telephone Number, Including Area Code, of Registrant’s Principal Executive Offices)
Mr. Walter Klemp, Chief Executive Officer
2575 West Bellfort, Suite 333
Houston, Texas 77054
(713) 300-5160
(Name, Address, Including Zip Code, and Telephone Number, Including Area Code, of Agent For Service)
Copies to:
| Cavas S. Pavri | Jack I. Kantrowitz, Esq. |
| Schiff Hardin LLP | DLA Piper LLP (US) |
| 100 N. 18th, Suite 300 | 1251 Avenue of the Americas |
| Philadelphia, PA 19103 | New York, New York 10020 |
| Telephone: (202) 724-6847 | Telephone: (212) 335-4500 |
| Fax: (202) 778-6460 | Fax: (212) 335-4501 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this Registration Statement.
If any of the securities being registered on this form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Securities Exchange Act of 1934. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ |
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | x |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement is filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell, nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion Dated February 16, 2016
Up to 2,000,000 Shares
Moleculin Biotech, Inc.
Common Stock
This is an initial public offering of Moleculin Biotech, Inc.
We are offering a minimum of 1,400,000 and a maximum of 2,000,000 shares of common stock in this offering.
Prior to this offering, there has been no public market for our common stock. It is currently estimated that the initial public offering price per share will be between $5.00 and $6.00. We have applied to list the common stock on the Nasdaq Capital Market under the symbol “MBRX”. If the application is approved, trading of our common stock on the Nasdaq Capital Market is expected to begin within five days after the initial issuance of the common stock. If our common stock is not approved for listing on the Nasdaq Capital Market, we will not consummate this offering.
We are an “emerging growth company” as defined in Section 2(a) of the Securities Act of 1933, as amended, and we have elected to comply with certain reduced public company reporting requirements.
An investment in our common stock involves significant risks. You should carefully consider the risk factors beginning on page 11 of this prospectus before you make your decision to invest in our shares of common stock.
Neither the Securities and Exchange Commission nor any other regulatory body has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
| Underwriting | Proceeds to Us, Before | ||
| Public Offering Price | Commissions (1) | Expenses (2) | |
| Per share | $ | $ | $ |
| Total minimum offering | $ | $ | $ |
| Total maximum offering. | $ | $ | $ |
| (1) | For the purpose of estimating the underwriting commissions, we have assumed that the underwriters will receive their maximum commission on all sales made in this offering, plus an advisory fee of $50,000. The underwriters will also be entitled to reimbursement of their out-of-pocket expenses incurred in connection with this offering, which shall not exceed $25,000, and fees and expenses of their counsel, not to exceed $100,000. |
| (2) | We estimate the total expenses of this offering, excluding the underwriting commissions, will be approximately $500,000 if all 2,000,000 shares are sold in this offering. Because this is a best efforts offering, the actual public offering amount, underwriting commissions and proceeds to us are not presently determinable and may be substantially less than the total maximum offering set forth above. See “Underwriting” beginning on page 73 of this prospectus for more information on this offering and the underwriter arrangements. |
Bonwick Capital Partners LLC is acting as the sole representative of the underwriters and, together with Network 1 Financial Securities, Inc., is acting as co-manager for this offering. The underwriters are selling shares of our common stock in this offering on a best efforts basis. We do not intend to close this offering unless we sell at least a minimum number of 1,400,000 shares of common stock, at the price per share set forth in the table above, and otherwise satisfy the listing conditions to trade our common stock on the NASDAQ Capital Market. This offering will terminate on [*], 2016 (60 days after the date of this prospectus) or [*], 2016, if extended, unless we sell the maximum number of shares of common stock set forth above before that date or we decide to terminate this offering prior to that date. The gross proceeds of this offering will be deposited at Signature Bank in an escrow account established by us, until we have sold a minimum of 1,400,000 shares of common stock and otherwise satisfy the listing conditions to trade our common stock on the Nasdaq Capital Market. Once we satisfy the minimum stock sale and Nasdaq listing conditions, the funds will be released to us. In the event we do not sell a minimum of 1,400,000 shares of common stock and raise minimum gross proceeds of $7,000,000 by [*], 2016 or [*], 2016, if extended, all funds received will be promptly returned to investors without interest or offset. See “Prospectus Summary - The Offering” on page 7.
Delivery of the shares of our common stock is expected to be made on or about [*], 2016.
 |
N E T W O R K 1 F I N A N C I A L S E C U R I T I E S, I N C. |
| Bonwick Capital Partners LLC | Network 1 Financial Securities, Inc. |
The date of this prospectus is , 2016
Table of Contents
Through and including , 2016 (the 25th day after the date of this prospectus), all dealers effecting transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to unsold allotments or subscriptions.
No dealer, salesperson or other person is authorized to give any information or to represent anything not contained in this prospectus. You must not rely on any unauthorized information or representations. This prospectus is an offer to sell only the shares offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus is current only as of its date.
Market data and certain industry data and forecasts used throughout this prospectus were obtained from internal company surveys, market research, consultant surveys, publicly available information, reports of governmental agencies and industry publications and surveys. Industry surveys, publications, consultant surveys and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable, but that the accuracy and completeness of such information is not guaranteed. While we are not aware of any misstatements regarding the industry data presented in this prospectus, our estimates involve risks and uncertainties and are subject to change based on various factors, including those discussed under the heading “Risk Factors” in this prospectus.
| i |
This summary highlights information contained elsewhere in this prospectus. This summary does not contain all of the information that you should consider before deciding to invest in our common stock. You should read this entire prospectus carefully, including the “Risk Factors” section, our historical financial statements and the notes thereto, and unaudited pro forma financial information, each included elsewhere in this prospectus. The terms “MBI”, “the Company”, “our”, or “we” refer to Moleculin Biotech, Inc. and, unless the context otherwise requires, its predecessors.
Overview
We are a preclinical and clinical-stage pharmaceutical company focused on the development of anti-cancer drug candidates, many of which are based on license agreements with The University of Texas System on behalf of the M.D. Anderson Cancer Center, which we refer to as MD Anderson. Our lead drug candidate is liposomal Annamycin, which we refer to as Annamycin, an anthracycline intended for the treatment of relapsed or refractory acute myeloid leukemia, or AML. Annamycin has been in clinical trials pursuant to an Investigational New Drug application, or IND, that had been filed with the U.S. Food and Drug Administration, or FDA. Due to a lack of development activity by a prior drug developer, this IND was terminated. However, we intend to apply for a new IND based on the same data that supported the original IND, updated for subsequent clinical data, and to commence a Phase II clinical trial for Annamycin funded with the proceeds from this offering. We have two other drug development projects in progress, one involving a portfolio of small molecules, which we refer to as the WP1066 Portfolio, focused on the modulation of key regulatory transcription factors involved in the progression of cancer, and the WP1122 Portfolio, a suite of molecules targeting the metabolic processes involved in cancer in general, and glioblastoma (the most common form of brain tumor) in particular.
We have been granted royalty-bearing, worldwide, exclusive licenses for the patent and technology rights related to our WP1066 Portfolio and WP1122 Portfolio drug technologies, as these patent rights are owned by MD Anderson. The Annamycin drug substance is no longer covered by any existing patent protection. We intend to submit patent applications for formulation, synthetic process and reconstitution related to our Annamycin drug product candidate, although there is no assurance that we will be successful in obtaining such patent protection. Independently from potential patent protection, we believe Annamycin will qualify for Orphan Drug status, which could entitle us to market exclusivity of up to 7 and 10 years from the date of approval of a New Drug Application (NDA) and Marketing Authorization (MA), in the US and the European Union (EU), respectively. However, there can be no assurance that such status will be granted. Separately, the FDA may also grant market exclusivity of up to five years for newly approved new chemical entities (of which Annamycin would be one), but there can be no assurance that such exclusivity will be granted or, if granted, for how long.
Our Drug Candidates
Annamycin
Our lead product candidate is Annamycin, an anthracycline intended to target the treatment of relapsed or refractory AML. Anthracyclines are a class of chemotherapy drugs designed to disrupt the DNA of, and eventually destroy, targeted cancer cells. They are considered to be among the most effective anticancer drugs developed and are used to treat a range of cancers, including leukemias, lymphomas, and breast, stomach, uterine, ovarian, bladder, and lung cancers. The effectiveness of currently approved anthracyclines is limited, however, by their propensity to cause heart damage (cardiotoxicity) and for cancer cells to become resistant by natural cell defense (multidrug resistance) mechanisms.
Leukemia is a cancer of the white blood cells and acute forms of leukemia can manifest quickly and leave patients with limited treatment options. AML is the most common type of acute leukemia in adults. It occurs when a clone of leukemic progenitor white blood cells proliferates in the bone marrow, suppressing the production of normal blood cells. Currently, the only viable option for acute leukemia patients is a bone marrow transplant, which is successful in a significant number of patients. However, in order to qualify for a bone marrow transplant, the patient’s leukemia cells must be decreased to a sufficiently low level. This begins with a therapy of combining two chemotherapeutic drugs, which always includes an anthracycline, in inducing remission (a complete response, or CR), which therapy has not improved since it was first used in the 1970s and we estimate that this induction therapy has the same cure rate of about 20% as at that time. Unfortunately, the current clinically approved anthracyclines are cardiotoxic, which can result in damage to the heart and limit the dosage amount that may be administered to patients. Additionally, the tumor cells often present de novo or developed resistance to the first line anthracycline, often through what is called “multidrug resistance”, enabling them to purge themselves of the available anthracyclines. Consequently, there remains no effective therapy for these patients and most will succumb quickly to their leukemia. If a patient’s leukemia reappears before they can be prepared for a bone marrow transplant, they are considered to have “relapsed”. If a patient fails to achieve a sufficient response from the induction therapy to qualify for a bone marrow transplant, they are considered to be “refractory” (resistant to therapy). Together, this group of relapsed and refractory AML patients constitutes our primary focus for treatment with Annamycin and our intent is to pursue FDA approval for Annamycin as a second-line induction therapy for adult relapsed and refractory AML patients.
1
We believe that pursuing approval as a second line induction therapy for adult relapsed or refractory AML patients is the shortest path to regulatory approval, but we also believe that one of the most important potential uses of Annamycin is in the treatment of children with either AML or ALL (acute lymphoblastic leukemia, which is more common in children). Accordingly, we also intend to pursue approval for pediatric use when practicable.
Annamycin is a unique liposome formulated anthracycline (also referred to in literature as “L-Annamycin”). It has been tested in 6 clinical trials and 114 patients without any reporting of cardiotoxicity and, in the two of those clinical trials focused on leukemia, with fewer dose-limiting toxicities than are normally experienced with doxorubicin (one of the leading first-line anthracyclines used for induction therapy). Each of these trials was conducted by previous holders of the intellectual property surrounding Annamycin and not by our company. Annamycin demonstrated efficacy in 8 of 16 patients in a Phase I study in adult relapsed or refractory AML patients, with 6 of 14 patients completely clearing leukemic blasts. A 30 patient dose-ranging Phase I/II study in ALL demonstrated a similar efficacy profile, with 3 of 10 patients treated with the maximum tolerable dose clearing their leukemic blasts to a level sufficient to qualify for a bone marrow transplant. One of these patients went on to receive a successful curative bone marrow transplant. The other two of these three patients died of tumor lysis syndrome, a condition resulting from the overloading of their system with the debris from leukemic blast cells destroyed by the induction therapy. Armed with the knowledge of this potential, prophylactic pretreatment known to protect patients from the effects of tumor lysis syndrome will be deployed in future trials. Based on the results of the above clinical trials, we believe Annamycin is different from currently approved induction therapy drugs in four key ways: (i) it has demonstrated clinical activity in a patient population for whom there are currently no effective therapies, (ii) it appears to be capable of avoiding the “multi-drug resistance” mechanisms that often limit the effectiveness of currently approved anthracyclines; (iii) it has been shown to be non-cardiotoxic in animal models, when compared with doxorubicin and no events of cardiotoxicity have been reported from the use of Annamycin in 114 patients; and (iv) in AML cell lines, it has been shown to be more potent than one of the leading approved drugs.
Based on initial conversations with the FDA, because of the serious unmet medical need, we believe Annamycin may qualify for a “Special Protocol Assessment” providing for accelerated approval based on our planned Phase II clinical trial. In order to negotiate the Special Protocol Assessment with the FDA, the dose-ranging Phase I/II clinical trial discussed above, which was conducted by a previous holder of the intellectual property surrounding Annamycin, must be independently audited, which is an expensive and time consuming process. In addition, since the original IND was terminated for lack of development activity by the prior drug developer, we must apply for a new IND, based on the data that supported the original IND, updated for subsequent clinical activity. The ultimate negotiation with the FDA will determine the size and efficacy endpoints for the registration trial. We can provide no assurance that the audit of the most recent clinical trial will confirm the results reported by the prior drug developer or that we will be successful in obtaining a new IND or in negotiating a Special Protocol Assessment with the FDA. If we are successful, we estimate such process, including auditing the prior clinical trial results, will take approximately six months or more from the closing of this offering.
We also intend to pursue Orphan Drug status for Annamycin. The prevalence ceiling for qualifying rare diseases under the US Orphan Drug Act is 200,000 patients and proportionally similar guidelines exist in the EU. The most recently published prevalence statistics from the National Cancer Institute reported that an estimated 37,726 patients had acute myeloid leukemia in the United States as of January 1, 2011, and trend data since that publication would indicate that the prevalence today should still be well below the 200,000 patient limitation for Orphan Drugs, which would permit Annamycin for the treatment of acute myeloid leukemia to qualify for Orphan Drug status. Annamycin already qualified for Orphan Drug status with its prior developer and we intend to repeat that process. However, we can provide no assurance that we will be successful in obtaining Orphan Drug status for Annamycin.
The WP1066 Portfolio
We have a license agreement with MD Anderson pursuant to which we have been granted a royalty-bearing, worldwide, exclusive license for the patent and technology rights related to WP1066 and its close analogs, molecules targeting the modulation of key oncogenic transcription factors.
2
In vitro testing has shown a high level of activity for WP1066 against a wide range of solid tumors, and in vivo testing has shown significant activity against head and neck, pancreatic, stomach, and ovarian cancers, as well as metastatic melanoma and glioblastoma, among others. In vivo testing in mouse tumor models has shown that WP1066 inhibits tumor growth, blocks angiogenesis (a process that provides necessary blood supply to tumors) and increases survival.
With respect to our WP1066 Portfolio, we must complete pre-clinical toxicology testing, along with additional chemistry, manufacturing and control work to fully characterize the drug, establish the desired formulation and develop reference standards for future drug release, among others, prior to submitting an application for an IND. We currently expect clinicians at MD Anderson to submit an IND for this drug candidate by early 2016.
An analog of WP1066, referred to as WP1220, was previously the subject of an IND (WP1220 was referred to as MOL4239 for purposes of this IND) related to use of the molecule in the topical treatment of psoriasis. Clinical trials were commenced on WP1220 in the US, but were terminated early due to limited efficacy in the topical treatment of psoriatic plaques. Notwithstanding its limitations in treating psoriasis, our pre-clinical research has shown WP1220 to be effective in inhibiting cutaneous T-cell lymphoma (CTCL) in multiple CTCL cell lines. Based on this encouraging data, we are collaborating with a Polish drug development company, Dermin Sp. Zo. O., or Dermin, which has received a Polish government grant to begin a clinical trial in Poland for the topical treatment of early stage CTCL patients.
We also conducted a Phase II clinical trial for WP1066 for the topical treatment of psoriasis, however this trial was terminated early as a significant number of patients experienced a non-permanent worsening of their psoriatic plaques after extended use of the drug, suggesting that its use as a topical agent for non-life threatening diseases such as psoriasis will require further study to optimize dosing and scheduling regimens.
The WP1122 Portfolio
We have a license agreement with MD Anderson pursuant to which we have been granted a royalty-bearing, worldwide, exclusive license for the patent and technology rights related to our WP1122 Portfolio and similar molecules targeting the treatment of glioblastoma multiform, or GBM, and related central nervous system, or CNS, malignancies.
We believe this technology has the potential to target a wide variety of solid tumors, which eventually become resistant to all treatments, and thereby provide a large and important opportunity for novel drugs. Notwithstanding this potential, we are focused on the treatment of central nervous system malignancies and especially GBM. Although less prevalent than some larger categories of solid tumors, cancers of the central nervous system are particularly aggressive and resistant to treatment. The prognosis for such patients can be particularly grim and the treatment options available to their physicians are among the most limited of any cancer.
The American Cancer Society has estimated 23,770 new cases of brain and other nervous system cancers will occur in the United States in 2016, resulting in 16,050 deaths. Despite the severity and poor prognosis of these tumors, there are few FDA-approved drugs on the market.
We have proof of concept data for our WP1122 Portfolio, including data on survival of animals subjected to xenografts of human brain tumors, as well as biodistribution and pharmacokinetics. In non-optimal doses and treatment regimes, our WP1122 Portfolio performed equal to or better than the current market leader, temozolomide and provided for superior survival for animals treated in combination with temozolomide.
Risks Relating to Our Business
As a preclinical and clinical-stage, pharmaceutical company, our business and ability to execute our business strategy are subject to a number of risks of which you should be aware before you decide to buy our securities. In particular, you should consider the following risks, which are discussed more fully in the section entitled “Risk Factors”:
| · | we currently do not have regulatory approval for our lead drug candidate, Annamycin, or any other product candidates, in the United States or elsewhere, although we plan to conduct clinical trials in the United States for Annamycin and other drug candidates in the future, there is no assurance that we will be successful in our clinical trials or receive regulatory approval in a timely manner, or at all; |
3
| · | a portion of our business is dependent upon the intellectual property rights that we license from MD Anderson, and in the past we have been in breach of these license agreements. Although we are currently in compliance with our obligations under our license agreements, there is no assurance that in the future we will not again be in breach of these agreements. Additionally, the intellectual property that we have licensed from MD Anderson may have been developed under a funding agreement with the United States government. To the extent that is the case, our license agreements with and the intellectual property rights we have licensed from MD Anderson are subject to such a funding agreement and any superior rights that the U.S. government may have with respect to the licensed intellectual property; |
| · | our lead drug candidate, Annamycin, is not the subject of any patent protection, and, although we intend to apply for formulation and method-of-use patents for Annamycin, there is no assurance that we will be successful in obtaining such patents and, even if we are successful, such patents generally offer less protection than original composition of matter patents; |
| · | unforeseen side effects from any of our product candidates could arise either during clinical development. For example, in the most recent Phase I/II dose-ranging clinical trial of Annamycin, two patients succumbed to tumor lysis syndrome (TLS) resulting from the debris created by Annamycin killing the targeted leukemic blasts more rapidly than anticipated; |
| · | we do not currently carry product liability insurance covering any of our drug candidates and, although we intend to obtain product liability insurance for future clinical trial liability that we may incur, there can be no assurance that we will secure adequate coverage or that, even if we do so, any such coverage will be sufficient to prevent the exposure of our operations to significant potential liability in the future; |
| · | the patents we have licensed from MD Anderson may not be valid or enforceable and may not protect us against competitors who challenge those licensed patents, obtain their own patents that may have an adverse effect on our ability to conduct business, or are able to otherwise circumvent our patents. Additionally, our products and technologies are complex and one patent may not be sufficient to protect our products where a series of patents may be needed. Further, we may not have the necessary financial resources to enforce or defend our patents or patent applications. In addition, any patent applications we may have made or may make relating to inventions for our actual or potential products and technologies may not result in patents being issued or may result in patents that provide insufficient or incomplete coverage for our inventions; |
| · | third parties may claim that the manufacture, use or sale of our technologies infringe their intellectual property rights. As with any litigation where such claims may be asserted, we may have to seek licenses, defend infringement actions or challenge the validity of those patents in the patent office or the courts. If these are not resolved favorably, we may not be able to continue to develop and commercialize our product candidates. Even if we were able to obtain rights to a third party’s intellectual property, these rights may be non-exclusive, thereby giving our competitors potential access to the same intellectual property. If we are found liable for infringement or are not able to have these patents declared invalid or unenforceable, we may be liable for significant monetary damages, encounter significant delays in bringing products to market or be precluded from participating in the manufacture, use or sale of products or technologies by patents of others. We may not have identified, or be able to identify in the future, U.S. or foreign patents that pose a risk of potential infringement claims; |
| · | we have completed and will in the future complete related party transactions that were not and will not be conducted on an arm’s length basis. We acquired the rights to the license agreement with MD Anderson covering our WP1122 Portfolio held by IntertechBio Corporation, a company affiliated with certain members of our management and board of directors. We acquired the rights to all data related to the development of Annamycin held by AnnaMed, Inc., a company affiliated with certain members of our management and board of directors. Prior to the effective date of the registration statement of which this prospectus is a part, Moleculin LLC will be merged with and into our company. Moleculin LLC is affiliated with certain members of our management and board of directors. In addition, prior to the effective date of the registration statement of which this prospectus is a part, we will enter into an agreement with Houston Pharmaceuticals, Inc., or HPI, whereby HPI will agree to terminate its option to sublicense certain rights to the WP1066 Portfolio and to enter into a co-development agreement with us. Waldemar Priebe and Don Picker are shareholders of HPI. Since these transactions were not conducted on an arm’s length basis, it is possible that the terms were less favorable to us than in an arm’s length transaction. |
| · | we have never been profitable, have not generated significant revenue to date and we expect to incur significant additional losses to fund our clinical trials; |
| · | we will require substantial additional funding beyond the proceeds of the offering to which this prospectus relates to complete the development and commercialization of our drug candidates, and such funding may not be available on acceptable terms or at all; |
| · | our development work is dependent in part on collaborations with other drug development companies and funding from government and philanthropic funding sources and there can be no assurance that such collaborations and funding will continue in the future; |
| · | our short-to-medium term prospects depend largely on our ability to develop and commercialize one drug candidate, Annamycin, and our ability to generate revenues in the future will depend heavily on the successful development and commercialization of Annamycin; |
| · | we may be subject to delays in our clinical trials, which could result in increased costs and delays or limit our ability to obtain regulatory approval for Annamycin and/or any other potential drug candidates; |
| · | we have never commercialized any of our drug candidates, including Annamycin, and, even if approved, our drug candidates may not be accepted by healthcare providers or healthcare payors; and |
| · | we may be unable to maintain and protect our intellectual property assets, which could impair the advancement of our pipeline and commercial opportunities. |
4
Company Information and Moleculin Merger
We were organized as a Delaware corporation in July 2015. In August 2015, in exchange for the issuance of 630,000 shares of common stock, we acquired the rights to the license agreement with MD Anderson covering our WP1122 Portfolio held by IntertechBio Corporation, a company affiliated with certain members of our management and board of directors. In August 2015, in exchange for the issuance of 1,431,000 shares of common stock, we acquired the rights to all data related to the development of Annamycin held by AnnaMed, Inc., a company affiliated with certain members of our management and board of directors.
Prior to the effective date of the registration statement of which this prospectus is a part, Moleculin LLC, a Texas limited liability company, will be merged with and into our company. Moleculin LLC is the holder of the license agreement with MD Anderson covering the WP1066 Portfolio. As a result of the merger, we will issue the holders of Moleculin LLC equity interests an aggregate of 1,000,000 shares of our common stock. As part of the acquisition process of Moleculin LLC by MBI, we will provide operating loans for Moleculin in the estimated range of $300,000 to $310,000 for Moleculin to meet its license obligations to MD Anderson, and maintain its current level of operations. These loans will be in the form of promissory notes payable with an interest rate of 8% from Moleculin to us that will be forgiven at the completion of the IPO. As of the date hereof, MBI has secured a total of $615,000 of convertible indebtedness and estimates that it will need approximately an additional $535,000 to $635,000 to complete the IPO process.
Prior to the effective date of the registration statement of which this prospectus is a part, in exchange for 629,000 shares of our common stock, we will enter into an agreement with Houston Pharmaceuticals, Inc., or HPI, whereby HPI will agree to terminate its option to sublicense certain rights to the WP1066 Portfolio and to enter into a co-development agreement with us, allowing us to benefit from HPI’s grant-funded development efforts currently under way.
Except as otherwise noted, the narrative discussion and other information set forth in this prospectus assumes the completion of the merger described above and reflects, as appropriate, the effects of the merger. Unless the context indicates otherwise, the terms “our,” “we,” and “us,” refer to Moleculin Biotech, Inc. after the merger.
Conversion of our Outstanding Notes
Unless otherwise indicated, this prospectus gives effect to the automatic conversion of $615,000 in aggregate principal amount of our 8% unsecured promissory notes that we have issued as of the date hereof into an aggregate of 3,749,557 shares of our common stock contemporaneously with the closing of this offering. The foregoing does not include any shares of common stock issuable upon conversion of the accrued interest on such convertible notes. However, we expect to issue additional shares of our common stock as payment of the aggregate amount of interest at the same conversion rate as principal conversion. The financial statements of the Company in this prospectus do not reflect the conversion of any of these unsecured promissory notes. The conversion of the unsecured promissory notes on our financial statements will reduce our debt by $615,000 plus accrued interest as a result of the extinguishment thereof, and to increase our stockholders’ equity by the same amount as a result of the issuance of the additional shares of common stock. No holder of these notes will be permitted to convert such notes to the extent that the holder or any of its affiliates would beneficially own in excess of 4.99% of our common stock after such conversion. The number of shares set forth above assumes no such limitation on the conversion of the notes.
Implications of Being an Emerging Growth Company
We qualify as an “emerging growth company” as the term is used in The Jumpstart Our Business Startups Act of 2012 (JOBS Act), and therefore, we may take advantage of certain exemptions from various public company reporting requirements, including:
| · | a requirement to only have two years of audited financial statements and only two years of related selected financial data and management’s discussion and analysis; |
| · | exemption from the auditor attestation requirement on the effectiveness of our internal controls over financial reporting; |
| · | reduced disclosure obligations regarding executive compensation; and |
| · | exemptions from the requirements of holding a nonbinding advisory stockholder vote on executive compensation and any golden parachute payments. |
5
We may take advantage of these provisions for up to five years or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we have more than $1.0 billion in annual revenues, have more than $700 million in market value of our capital stock held by non-affiliates or issue more than $1.0 billion of non-convertible debt over a three-year period. We may choose to take advantage of some, but not all, of the available benefits of the JOBS Act. We have taken advantage of some of the reduced reporting requirements in this prospectus. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold stock. In addition, the JOBS Act provides that an emerging growth company can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Our principal executive offices are located at 2575 West Bellfort, Suite 333, Houston, Texas 77054. Our telephone number is (713) 300-5160. Our website address is www.moleculin.com. The information on or accessible through our website is not part of this prospectus.
6
The Offering
| Common stock we are offering | 1,400,000 shares (minimum) to 2,000,000 shares (maximum) | |
| Common stock outstanding immediately before this offering | 6,661,000 shares | |
| Common stock outstanding immediately after this offering (1) | [*] shares (minimum) to [*] shares (maximum) | |
| Offering price | $[*] per share | |
| Best efforts offering | The underwriters are selling the shares of our common stock offered in this prospectus on a “best efforts” basis and are not required to sell any specific number or dollar amount of the shares offered by this prospectus, but will use their best efforts to sell such shares. However, one of the conditions to our obligation to sell any of the shares through the underwriters is that, upon the closing of the offering, our common stock would qualify for listing on the Nasdaq Capital Market. In order to list, the Nasdaq Capital Market requires that, among other criteria, at least 1,000,000 publicly-held shares of our common stock be outstanding, the shares be held in the aggregate by at least 300 round lot holders, the market value of the publicly-held shares of our common stock be at least $15.0 million, our stockholders’ equity after giving effect to the sale of our shares in this offering be at least $4.0 million, the bid price per share of our common stock be $4.00 or more, and there be at least three registered and active market makers for our common stock. We do not intend to close this offering unless we sell a minimum of 1,000,000 shares of common stock and otherwise satisfy the listing conditions to trade our common stock on the Nasdaq Capital Market. | |
| Use of proceeds |
Based on an estimated initial public offering price of $5.50 per share, which is the midpoint of the offering price range, we estimate that the net proceeds to us from this offering, assuming we sell a minimum of 1,400,000 shares, will be approximately $6.7 million and, assuming we sell all 2,000,000 shares, will be approximately $9.7 million, after payment of underwriting commissions and our estimated offering expenses. However, this is a best efforts offering, and there is no assurance that we will sell any shares or receive any proceeds. We intend to use the proceeds from this offering to: apply for a new IND for Annamycin, audit the Phase I/II clinical trial conducted by a prior developer, negotiate a Special Protocol Assessment with the FDA, commence a Phase II clinical trial for Annamycin, facilitate the maintenance of our licenses and ongoing development work relating to the WP1066 and WP1122 Portfolios and for working capital. See “Use of Proceeds” for more information. |
|
| Risk Factors | See “Risk Factors” and other information appearing elsewhere in this prospectus for a discussion of factors you should carefully consider before deciding whether to invest in our common stock. | |
| Lock-up | We, our directors, executive officers, and certain shareholders have agreed not to offer, issue, sell, contract to sell, encumber, grant any option for the sale of or otherwise dispose of any of our securities for a period of 12 months following the closing of the offering of the shares, and thereafter such individuals have agreed to a leak-out of their shares for a period ending on the third anniversary of this offering. See “Shares Eligible for Future Sale” for more information. |
7
| Escrow | The gross proceeds of this offering will be deposited at Signature Bank, New York, New York, in an escrow account established by us. Purchasers are to make payment for the shares they purchase by (i) delivering to the escrow agent, Signature Bank, at 261 Madison Avenue, New York, New York 10016, checks made payable to the order of “Signature Bank, as Escrow Agent for Moleculin Biotech,” or (ii) wire transfer to Signature Bank, ABA No. [*], 261 Madison Avenue, New York, New York 10016, for credit to Signature Bank, as Escrow Agent for Moleculin Biotech, Account No. [*]. All checks received by the underwriter will be delivered to Signature Bank for deposit into the escrow account not later than 12:00 p.m. on the business day immediately following receipt. The funds will be held in escrow until we receive a minimum of $7.0 million (assuming the offering price is at the low end of the price range) and otherwise satisfy the listing conditions to trade our common stock on the Nasdaq Capital Market, at which time the funds will be released to us. Any funds received in excess of the foregoing minimum amount and following the satisfaction of the Nasdaq listing requirements will immediately be available to us. If we do not receive the minimum amount by [*], 2016 (60 days after the date of this prospectus) or [*], 2016, if extended, all funds will be returned to purchasers in this offering on the next business day after the offering’s termination, without charge, deduction or interest. Prior to [*], 2016, in no event will funds be returned to you. You will only be entitled to receive a refund of your subscription if we do not raise the minimum amount set forth above and satisfy the Nasdaq listing conditions by [*], 2016 (10 days after the date of this prospectus). | |
| Proposed listing symbol | “MBRX“ |
(1) The number of shares of common stock to be outstanding after this offering includes (i) 1,000,000 shares of common stock to be issued to the members of Moleculin LLC, (ii) the issuance of 629,000 shares of common stock in connection with our agreement HPI; and (iii) the issuance of 3,749,558 shares of common stock upon the conversion of $450,000 in principal amount of our outstanding bridge notes. The number of shares does not give effect to:
| · | 2,500,000 shares of common stock available for issuance under the Moleculin Biotech, Inc. 2015 Stock Plan; and |
| · | between 98,000 shares (assuming the minimum offering is completed) and 140,000 shares (assuming the maximum offering is completed) of common stock issuable upon the exercise of the warrants issued to the representatives of the underwriters. |
Unless otherwise indicated, all information in this prospectus reflects and assumes the filing and effectiveness of our amended and restated certificate of incorporation and the adoption of our amended and restated bylaws, each of which will occur immediately prior to the completion of this offering.
8
Summary Financial Data
We have derived the following summary statement of operations data for period from July 28, 2015 (Inception) to September 30, 2015 and the summary balance sheet data as of September 30, 2015 from MBI’s audited financial statements included elsewhere in this prospectus. We have derived the following summary statements of operations data for the years ended December 31, 2014 and 2013 and the summary statements of financial position data as of December 31, 2014 and 2013 from Moleculin’s audited financial statements included elsewhere in this prospectus. The summary statements of operations data for the nine months ended September 30, 2014 and 2015 and the summary statement of financial position data as of September 30, 2015 for Moleculin have been derived from the unaudited interim financial statements included elsewhere in this prospectus. Moleculin’s unaudited interim financial statements have been prepared on the same basis as its annual financial statements and, in the opinion of Moleculin’s management, reflect all adjustments, which include only normal recurring adjustments necessary for a fair presentation of its financial position and results of operations for these periods. The historical results of MBI and Moleculin are not necessarily indicative of the results that may be expected in the future, and the interim results should not necessarily be considered indicative of results that may be expected for the full year or any other period. You should read the following summary financial data together with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and financial statements of MBI and Moleculin and related notes included elsewhere in this prospectus.
| From July 28, 2015 (Inception) to September 30, 2015 | ||||
| Statement of Operations Data -MBI | ||||
| Revenue | $ | - | ||
| Research and development expense | 38,409 | |||
| General and administrative expense | 184,344 | |||
| Other expense | 1,562 | |||
| Net loss | $ | (224,315 | ) | |
| Net loss per common share | $ | (0.05 | ) | |
| September 30, 2015 | ||||
| Balance Sheet Data - MBI | ||||
| Cash and cash equivalents | $ | 87,340 | ||
| Working capital deficit | (222,254 | ) | ||
| Total assets | 110,340 | |||
| Accumulated deficit | (224,315 | ) | ||
| Total stockholders’ deficit | (222,254 | ) | ||
9
| Nine Months Ended September 30, | Year Ended December 31, | |||||||||||||||
| 2015 | 2014 | 2014 | 2013 | |||||||||||||
| (unaudited) | ||||||||||||||||
| Statements of Operations Data - Moleculin | ||||||||||||||||
| Revenue | $ | - | $ | - | $ | - | $ | - | ||||||||
| Research and development expense | 270,991 | 238,467 | 798,785 | 2,341,676 | ||||||||||||
| General and administrative expense | 267,423 | 649,344 | 839,556 | 1,161,282 | ||||||||||||
| Depreciation expense | 8,469 | 8,254 | 11,005 | 11,005 | ||||||||||||
| Other (income) expense | 334,338 | 55,011 | 159,740 | (849 | ) | |||||||||||
| Net loss | $ | (881,221 | ) | $ | (951,076 | ) | $ | (1,809,086 | ) | $ | (3,513,114 | ) | ||||
| September 30, 2015 | December 31, 2014 | December 31, 2013 | ||||||||||
| (unaudited) | ||||||||||||
| Statements of Financial Position Data - Moleculin | ||||||||||||
| Cash and cash equivalents | $ | 298,350 | $ | 524,477 | $ | 353,366 | ||||||
| Working capital (deficit) | (2,948,701 | ) | (1,095,276 | ) | 1,336,585 | |||||||
| Total assets | 317,185 | 781,448 | 627,728 | |||||||||
| Accumulated deficit | (14,718,926 | ) | (13,837,705 | ) | (12,028,619 | ) | ||||||
| Total members’ deficit | (3,348,740 | ) | (2,467,519 | ) | (1,070,685 | ) | ||||||
10
Risk Factors
Investing in our common stock involves a high degree of risk. You should carefully consider each of the following risks, together with all other information set forth in this prospectus, including the financial statements and the related notes, before making a decision to buy our common stock. If any of the following risks actually occurs, our business could be harmed. In that case, the trading price of our common stock could decline, and you may lose all or part of your investment.
Risks Relating to Our Business
We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.
We intend to use the proceeds from this offering to advance Annamycin through clinical development. Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. We will require substantial additional future capital in order to complete clinical development and commercialize Annamycin. If the FDA requires that we perform additional nonclinical studies or clinical trials, our expenses would further increase beyond what we currently expect and the anticipated timing of any potential approval of Annamycin would likely be delayed. Further, there can be no assurance that the costs we will need to incur to obtain regulatory approval of Annamycin will not increase.
We will continue to require substantial additional capital to continue our clinical development and commercialization activities. Because successful development of our product candidates is uncertain, we are unable to estimate the actual amount of funding we will require to complete research and development and commercialize our products under development.
The amount and timing of our future funding requirements will depend on many factors, including but not limited to:
| · | whether the results of our audit of the most recent Phase I/II clinical trial conducted by a prior developer comport with our understanding of the informal summary of the trial results; |
| · | whether we are successful in obtaining a Special Protocol Assessment, or SPA, with the FDA related to Annamycin; |
| · | the progress, costs, results of and timing of our clinical trials for Annamycin; |
| · | the outcome, costs and timing of seeking and obtaining FDA and any other regulatory approvals; |
| · | the progress of our collaborative drug development partners, which is dependent upon their continued access to grant funding; |
| · | the costs associated with securing and establishing commercialization and manufacturing capabilities; |
| · | market acceptance of our product candidates; |
| · | the costs of acquiring, licensing or investing in businesses, products, product candidates and technologies; |
| · | our ability to maintain, expand and enforce the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
| · | our need and ability to rely on data to be generated by our sublicensee partner, Dermin; |
| · | our need and ability to hire additional management and scientific and medical personnel; |
| · | the effect of competing drug candidates and new product approvals; |
| · | our need to implement additional internal systems and infrastructure, including financial and reporting systems; and |
11
| · | the economic and other terms, timing of and success of our existing licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future. |
Some of these factors are outside of our control. Based upon our currently expected level of operating expenditures, we believe that we will be able to fund our operations through mid-2017. This period could be shortened if there are any significant increases in planned spending on development programs or more rapid progress of development programs than anticipated. We do not believe that our existing capital resources are sufficient to enable us to complete the development and commercialization of Annamycin, if approved, or to initiate any clinical trials or additional development work needed for any other drug candidates, other than as described above. Accordingly, we expect that we will need to raise additional funds in the future.
We may seek additional funding through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. Additional funding may not be available to us on acceptable terms or at all. In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. In addition, the issuance of additional shares by us, or the possibility of such issuance, may cause the market price of our shares to decline.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail one or more of our research or development programs. We also could be required to seek funds through arrangements with collaborative partners or otherwise that may require us to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us.
We have completed and will in the future complete related party transactions that were not and will not be conducted on an arm’s length basis.
We acquired the rights to the license agreement with MD Anderson covering our WP1122 Portfolio held by IntertechBio Corporation, a company affiliated with certain members of our management and board of directors. We acquired the rights to all data related to the development of Annamycin held by AnnaMed, Inc., a company affiliated with certain members of our management and board of directors. Prior to the effective date of the registration statement of which this prospectus is a part, Moleculin LLC will be merged with and into our company. Moleculin LLC is affiliated with certain members of our management and board of directors. Prior to the effective date of the registration statement of which this prospectus is a part, we will enter into an agreement with HPI whereby HPI will agree to terminate its option to sublicense certain rights to the WP1066 Portfolio and to enter into a co-development agreement with us. Waldemar Priebe and Don Picker are shareholders of HPI.
None of the foregoing transactions were conducted on an arm’s length basis. As such, it is possible that the terms were less favorable to us than in an arm’s length transaction.
Our ability to retain the development rights to the WP1066 Portfolio will require us to make up to $1.75 million in future payments to HPI, in addition to payments of shares of our common stock and cash made prior to the completion of this offering, pursuant to the development agreement we intend to enter into with HPI prior to this offering.
Our acquisition of Moleculin LLC prior to this offering will provide us with the rights to the license agreement Moleculin LLC has with MD Anderson covering the WP1066 Portfolio. However, Moleculin LLC previously granted HPI an option to obtain an exclusive sub-license to develop the WP1066 Portfolio in all non-dermatological fields. Prior to this offering, we will enter into two agreements with HPI. The first agreement will terminate HPI’s option to obtain the aforementioned exclusive sublicense in exchange for a payment of $100,000 and the issuance of 629,000 shares of our common stock. The second agreement, the HPI Out-Licensing Agreement will be a technology rights and development license agreement that will provide HPI with a non-exclusive sublicense to develop the WP1066 Portfolio. Pursuant to this HPI Out-Licensing Agreement, we will agree to make payments to HPI of $750,000 over a three-year period commencing after this offering in exchange for HPI allowing us to access any data, information or know-how resulting from the research and development conducted by HPI. Notwithstanding our obligation to make the foregoing payments, the HPI Out-Licensing Agreement does not obligate HPI to conduct any specific research or to meet any milestones. Pursuant to the HPI Out-Licensing Agreement, we have the right within three years of the date we enter into the agreement to buy-out from HPI all rights granted to HPI under the agreement for a payment of $1.0 million. If we do not exercise the foregoing buy-out right within three years, the license granted to HPI shall convert into an exclusive license even as to our company. As such, if we do not exercise the buy-out right for any reason, we will no longer have access to the non-dermatology uses of the WP1066 Portfolio and all amounts paid to HPI prior to such date will have value only to the extent that the data, information and know-how may be applicable to dermatology applications of the WP1066 Portfolio. We do not expect to maintain a reserve of $1.0 million to make the buy-out payment and, as such, we will need to raise additional funds to make the buy-out payment. We cannot assure you that such additional funding will be available on satisfactory terms, or at all.
We have never been profitable, we have no products approved for commercial sale, and to date we have not generated any revenue from product sales. As a result, our ability to reduce our losses and reach profitability is unproven, and we may never achieve or sustain profitability.
We have never been profitable and do not expect to be profitable in the foreseeable future. We have not yet submitted any drug candidates for approval by regulatory authorities in the United States or elsewhere. From inception, July 28, 2015 to September 30, 2015,we incurred a net loss of $224,315 and Moleculin incurred a net loss of $881,221 for the nine-month period ended September 30, 2015. Moleculin incurred net losses of $1,809,086 and $3,513,114 for the years ended December 31, 2014 and 2013, respectively. We had an accumulated deficit of $224,315 and Moleculin had an accumulated deficit of $14,718,926 as of September 30, 2015.
To date, we have devoted most of our financial resources to our corporate overhead and research and development, including our drug discovery research, preclinical development activities and clinical trials. We have not generated any revenues from product sales. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for Annamycin, prepare for and begin the commercialization of any approved products, and add infrastructure and personnel to support our continuing product development efforts. We anticipate that any such losses could be significant for the next several years. If Annamycin or any of our other drug candidates fails in clinical trials or does not gain regulatory approval, or if our drug candidates do not achieve market acceptance, we may never become profitable. As a result of the foregoing, we expect to continue to experience net losses and negative cash flows for the foreseeable future. These net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders' equity and working capital.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. In addition, our expenses could increase if we are required by the FDA to perform studies or trials in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of any of our drug candidates. The amount of future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues.
We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on an annual basis, which may make it difficult to predict our future performance.
We are a preclinical and clinical-stage pharmaceutical company with a limited operating history. Our operations to date have been limited to acquiring our technology portfolio. We have not yet commenced any clinical trials or obtained any regulatory approvals for any of our drug candidates. Consequently, any predictions made about our future success or viability may not be as accurate as they could be if we had a longer operating history or approved products on the market. Our operating results are expected to significantly fluctuate from quarter-to-quarter or year-to-year due to a variety of factors, many of which are beyond our control. Factors relating to our business that may contribute to these fluctuations include:
12
| · | any delays in regulatory review and approval of our product candidates in clinical development, including our ability to receive approval from the FDA for Annamycin; |
| · | delays in the commencement, enrollment and timing of clinical trials; |
| · | difficulties in identifying patients suffering from our target indications; |
| · | the success of our clinical trials through all phases of clinical development; |
| · | potential side effects of our product candidate that could delay or prevent approval or cause an approved drug to be taken off the market; |
| · | our ability to obtain additional funding to develop drug candidates; |
| · | our ability to identify and develop additional drug candidates beyond Annamycin and our WP1066 and WP1122 Portfolios; |
| · | competition from existing products or new products that continue to emerge; |
| · | the ability of patients or healthcare providers to obtain coverage or sufficient reimbursement for our products; |
| · | our ability to adhere to clinical trial requirements directly or with third parties such as contract research organizations (CROs); |
| · | our dependency on third-party manufacturers to manufacture our products and key ingredients; |
| · | our ability to establish or maintain collaborations, licensing or other arrangements, particularly with MD Anderson; |
| · | our ability to defend against any challenges to our intellectual property including, claims of patent infringement; |
| · | our ability to enforce our intellectual property rights against potential competitors; |
| · | our ability to secure additional intellectual property protection for our developing drug candidates and associated technologies; |
| · | our ability to attract and retain key personnel to manage our business effectively; and |
| · | potential product liability claims. |
Accordingly, the results of any historical quarterly or annual periods should not be relied upon as indications of future operating performance.
We cannot be certain that Annamycin will receive regulatory approval, and without regulatory approval we will not be able to market Annamycin.
Our business currently depends largely on the successful development and commercialization of Annamycin. Our ability to generate revenue related to product sales, if ever, will depend on the successful development and regulatory approval of Annamycin for the treatment of relapsed or refractory acute myeloid leukemia, or AML.
We currently have no products approved for sale and we cannot guarantee that we will ever have marketable products. The development of a product candidate and issues relating to its approval and marketing are subject to extensive regulation by the FDA in the United States and regulatory authorities in other countries, with regulations differing from country to country. We are not permitted to market our product candidates in the United States until we receive approval of a NDA from the FDA. We have not submitted any marketing applications for any of our product candidates.
13
NDAs must include extensive preclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each desired indication. NDAs must also include significant information regarding the chemistry, manufacturing and controls for the product. Obtaining approval of a NDA is a lengthy, expensive and uncertain process, and we may not be successful in obtaining approval. The FDA review processes can take years to complete and approval is never guaranteed. If we submit a NDA to the FDA, the FDA must decide whether to accept or reject the submission for filing. We cannot be certain that any submissions will be accepted for filing and review by the FDA. Regulators in other jurisdictions have their own procedures for approval of product candidates. Even if a product is approved, the FDA may limit the indications for which the product may be marketed, require extensive warnings on the product labeling or require expensive and time-consuming clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the United States and Europe also have requirements for approval of drug candidates with which we must comply with prior to marketing in those countries. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure that we will be able to obtain regulatory approval in any other country. In addition, delays in approvals or rejections of marketing applications in the United States, Europe or other countries may be based upon many factors, including regulatory requests for additional analyses, reports, data, preclinical studies and clinical trials, regulatory questions regarding different interpretations of data and results, changes in regulatory policy during the period of product development and the emergence of new information regarding our product candidates or other products. Also, regulatory approval for any of our product candidates may be withdrawn.
If we are unable to obtain approval from the FDA, or other regulatory agencies, for Annamycin and our other product candidates, or if, subsequent to approval, we are unable to successfully commercialize Annamycin or our other product candidates, we will not be able to generate sufficient revenue to become profitable or to continue our operations.
Any statements in this prospectus indicating that Annamycin has demonstrated preliminary evidence of efficacy are our own and are not based on the FDA’s or any other comparable governmental agency’s assessment of Annamycin and do not indicate that Annamycin will achieve favorable efficacy results in any later stage trials or that the FDA or any comparable agency will ultimately determine that Annamycin is effective for purposes of granting marketing approval.
Delays in the commencement, enrollment and completion of clinical trials could result in increased costs to us and delay or limit our ability to obtain regulatory approval for Annamycin and our other product candidates.
Delays in the commencement, enrollment and completion of clinical trials could increase our product development costs or limit the regulatory approval of our product candidates. We do not know whether any future trials or studies of our other product candidates will begin on time or will be completed on schedule, if at all. The start or end of a clinical study is often delayed or halted due to changing regulatory requirements, manufacturing challenges, including delays or shortages in available drug product, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparative drug or required prior therapy, clinical outcomes or financial constraints. For instance, delays or difficulties in patient enrollment or difficulties in retaining trial participants can result in increased costs, longer development times or termination of a clinical trial. Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the eligibility criteria for the clinical trial, that include the age and condition of the patients and the stage and severity of disease, the nature of the protocol, the proximity of patients to clinical sites and the availability of effective treatments and/or availability of investigational treatment options for the relevant disease.
A product candidate can unexpectedly fail at any stage of preclinical and clinical development. The historical failure rate for product candidates is high due to scientific feasibility, safety, efficacy, changing standards of medical care and other variables. The results from preclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in later phase clinical trials of the product candidate. We, the FDA or other applicable regulatory authorities may suspend clinical trials of a product candidate at any time for various reasons, including, but not limited to, a belief that subjects participating in such trials are being exposed to unacceptable health risks or adverse side effects, or other adverse initial experiences or findings. We may not have the financial resources to continue development of, or to enter into collaborations for, a product candidate if we experience any problems or other unforeseen events that delay or prevent regulatory approval of, or our ability to commercialize, product candidates, including:
| · | inability to obtain sufficient funds required for a clinical trial; |
14
| · | inability to reach agreements on acceptable terms with prospective CROs and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| · | negative or inconclusive results from our clinical trials or the clinical trials of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program; |
| · | serious and unexpected drug-related side effects experienced by subjects in our clinical trials or by individuals using drugs similar to our product candidates; |
| · | conditions imposed by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
| · | delays in enrolling research subjects in clinical trials; |
| · | high drop-out rates and high fail rates of research subjects; |
| · | inadequate supply or quality of product candidate components or materials or other supplies necessary for the conduct of our clinical trials; |
| · | greater than anticipated clinical trial costs; |
| · | poor effectiveness of our product candidates during clinical trials; or |
| · | unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or vendor. |
We have never conducted a clinical trial or submitted an NDA before, and any product candidate we advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval.
Clinical failure can occur at any stage of our clinical development. Clinical trials may produce negative or inconclusive results, and our collaborators or we may decide, or regulators may require us, to conduct additional clinical trials or nonclinical studies. In addition, data obtained from trials and studies are susceptible to varying interpretations, and regulators may not interpret our data as favorably as we do, which may delay, limit or prevent regulatory approval. Success in preclinical studies and early clinical trials does not ensure that subsequent clinical trials will generate the same or similar results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. A number of companies in the pharmaceutical industry, including those with greater resources and experience than us, have suffered significant setbacks in clinical trials, even after seeing promising results in earlier clinical trials.
In addition, the design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. We may be unable to design and execute a clinical trial to support regulatory approval. Further, clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts.
If Annamycin is found to be unsafe or lack efficacy, we will not be able to obtain regulatory approval for it and our business would be harmed.
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes in trial protocols, differences in composition of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants. We do not know whether any clinical trials we or any of our potential future collaborators may conduct will demonstrate the consistent or adequate efficacy and safety that would be required to obtain regulatory approval and market any products. If we are unable to bring Annamycin to market, or to acquire other products that are on the market or can be developed, our ability to create long-term stockholder value will be limited.
Our product candidates may have undesirable side effects that may delay or prevent marketing approval, or, if approval is received, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
15
Unforeseen side effects from any of our product candidates could arise either during clinical development or, if Annamycin is approved, after the approved product has been marketed. For example, in the most recent Phase I/II dose-ranging clinical trial of Annamycin, two patients succumbed to tumor lysis syndrome (TLS) resulting from the debris created by Annamycin killing the targeted leukemic blasts more rapidly than anticipated. Now that this potential has been identified, prophylactic measures known to protect patients from TLS will be deployed in future clinical trials, but there can be no assurance that such measures will be effective or that other adverse events may not emerge related to our drug. Additional or unforeseen side effects from Annamycin or any of our other product candidates could arise either during clinical development or, if approved, after the approved product has been marketed.
The range and potential severity of possible side effects from therapies such as Annamycin are significant. A showing that Annamycin causes undesirable or unacceptable side effects could interrupt, delay or halt clinical trials and result in the failure to obtain or suspension or termination of marketing approval from the FDA and other regulatory authorities, or result in marketing approval from the FDA and other regulatory authorities only with restrictive label warnings.
If any of our product candidates receives marketing approval and we or others later identify undesirable or unacceptable side effects caused by such products:
| · | regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies; |
| · | we may be required to change instructions regarding the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| · | we may be subject to limitations on how we may promote the product; |
| · | sales of the product may decrease significantly; |
| · | regulatory authorities may require us to take our approved product off the market; |
| · | we may be subject to litigation or product liability claims; and |
| · | our reputation may suffer. |
Any of these events could prevent us or our potential future collaborators from achieving or maintaining market acceptance of the affected product or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from the sale of our products.
If the FDA does not find the manufacturing facilities of our future contract manufacturers acceptable for commercial production, we may not be able to commercialize any of our product candidates.
We do not intend to manufacture the pharmaceutical products that we plan to sell. We are currently utilizing contract manufacturers for the production of the active pharmaceutical ingredients and the formulation of drug product for our trials of Annamycin that we will need to conduct prior to seeking regulatory approval. However, we do not have agreements for supplies of Annamycin or any of our other product candidates and we may not be able to reach agreements with these or other contract manufacturers for sufficient supplies to commercialize Annamycin if it is approved. Additionally, the facilities used by any contract manufacturer to manufacture Annamycin or any of our other product candidates must be the subject of a satisfactory inspection before the FDA approves the product candidate manufactured at that facility. We are completely dependent on these third-party manufacturers for compliance with the requirements of U.S. and non-U.S. regulators for the manufacture of our finished products. If our manufacturers cannot successfully manufacture material that conform to our specifications and the FDA’s current good manufacturing practice standards, or cGMP, and other requirements of any governmental agency whose jurisdiction to which we are subject, our product candidates will not be approved or, if already approved, may be subject to recalls. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured our product candidates, including:
| · | the possibility that we are unable to enter into a manufacturing agreement with a third party to manufacture our product candidates; |
| · | the possible breach of the manufacturing agreements by the third parties because of factors beyond our control; and |
16
| · | the possibility of termination or nonrenewal of the agreements by the third parties before we are able to arrange for a qualified replacement third-party manufacturer. |
Any of these factors could cause the delay of approval or commercialization of our product candidates, cause us to incur higher costs or prevent us from commercializing our product candidates successfully. Furthermore, if any of our product candidates are approved and contract manufacturers fail to deliver the required commercial quantities of finished product on a timely basis at commercially reasonable prices and we are unable to find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volumes and quality and on a timely basis, we would likely be unable to meet demand for our products and could lose potential revenue. It may take several years to establish an alternative source of supply for our product candidates and to have any such new source approved by the government agencies that regulate our products.
We have no sales, marketing or distribution experience and we will have to invest significant resources to develop those capabilities or enter into acceptable third-party sales and marketing arrangements.
We have no sales, marketing or distribution experience. To develop sales, distribution and marketing capabilities, we will have to invest significant amounts of financial and management resources, some of which will need to be committed prior to any confirmation that Annamycin or any of our other product candidates will be approved by the FDA. For product candidates where we decide to perform sales, marketing and distribution functions ourselves or through third parties, we could face a number of additional risks, including that we or our third-party sales collaborators may not be able to build and maintain an effective marketing or sales force. If we use third parties to market and sell our products, we may have limited or no control over their sales, marketing and distribution activities on which our future revenues may depend.
We may not be successful in establishing and maintaining development and commercialization collaborations, which could adversely affect our ability to develop certain of our product candidates and our financial condition and operating results.
Because developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities and marketing approved products are expensive, we may seek to enter into collaborations with companies that have more experience. Additionally, if any of our product candidates receives marketing approval, we may enter into sales and marketing arrangements with third parties with respect to our unlicensed territories. If we are unable to enter into arrangements on acceptable terms, if at all, we may be unable to effectively market and sell our products in our target markets. We expect to face competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement and they may require substantial resources to maintain. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements for the development of our product candidates.
When we collaborate with a third party for development and commercialization of a product candidate, we can expect to relinquish some or all of the control over the future success of that product candidate to the third party. For example, have formed a collaboration with a Polish drug development company called Dermin, where we have provided them with sub-license rights to our technologies for use in limited territories in exchange for their use of Polish government grant funding to pay for development costs we would otherwise have to fund ourselves. With the exception of Annamycin, Dermin’s territories are primarily Poland and lesser surrounding countries, but not including any of the major European markets (UK, Germany, France, Spain and Italy). In the case of Annamycin, Dermin’s territories also include Germany, but we retain the right to repurchase that territory for $500,000 at any time in the future.
One or more of our collaboration partners may not devote sufficient resources to the commercialization of our product candidates or may otherwise fail in their commercialization. The terms of any collaboration or other arrangement that we establish may contain provisions that are not favorable to us. In addition, any collaboration that we enter into may be unsuccessful in the development and commercialization of our product candidates. In some cases, we may be responsible for continuing preclinical and initial clinical development of a product candidate or research program under a collaboration arrangement, and the payment we receive from our collaboration partner may be insufficient to cover the cost of this development. If we are unable to reach agreements with suitable collaborators for our product candidates, we would face increased costs, we may be forced to limit the number of our product candidates we can commercially develop or the territories in which we commercialize them. As a result, we might fail to commercialize products or programs for which a suitable collaborator cannot be found. If we fail to achieve successful collaborations, our operating results and financial condition could be materially and adversely affected.
17
Our success depends greatly on the success of Annamycin’s development for the treatment of relapsed or refractory AML, and our pipeline of product candidates beyond this lead indication is extremely early stage and limited.
Other than Annamycin, our other two drug candidates, our WP1066 Portfolio and our WP1122 Portfolio, are each in the early stages of development. As such, we are dependent on the success of Annamycin in the near term. We cannot provide you any assurance that we will be able to successfully advance any of these indications through the development process.
We face competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. We have competitors in the United States, Europe and other jurisdictions, including major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical and generic drug companies and universities and other research institutions. Many of our competitors have greater financial and other resources, such as larger research and development staff and more experienced marketing and manufacturing organizations than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing pharmaceutical products. These companies also have significantly greater research, sales and marketing capabilities and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or FDA approval or discovering, developing and commercializing drugs for the diseases that we are targeting before we do or may develop drugs that are deemed to be more effective or gain greater market acceptance than ours. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. In addition, many universities and private and public research institutes may become active in our target disease areas. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis, technologies and drug products that are more effective or less costly than any of our product candidates that we are currently developing or that we may develop, which could render our products obsolete or noncompetitive.
A number of attempts have been made or are under way to provide an improved treatment for AML. Drugs attempting to target a subset of AML patients who present with particular anomalies involving a gene referred to as FLT3 are currently in clinical trials. Other approaches to improve the effectiveness of induction therapy are in early stage clinical trials and, although they do not appear to address the underlying problems with anthracyclines, we can provide no assurance that such improvements, if achieved, would not adversely impact the need for improved anthracyclines. A modified version of doxorubicin designed to reduce cardiotoxicity is in clinical trials for the treatment of sarcoma and, although this drug does not appear to address multidrug resistance and is not currently intended for the treatment of acute leukemia, we can provide no assurance that it will not become a competitive alternative to Annamycin. Although we are not aware of any other single agent therapies in clinical trials that would directly compete against Annamycin in the treatment of relapsed and refractory AML, we can provide no assurance that such therapies are not in development, will not receive regulatory approval and will reach market before our drug candidate Annamycin. In addition, any such competing therapy may be more effective and / or cost-effective than ours.
If our competitors market products that are more effective, safer or less expensive or that reach the market sooner than our future products, if any, we may not achieve commercial success. In addition, because of our limited resources, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our technologies or product candidates obsolete, less competitive or not economical.
Annamycin does not have any patent protections, including composition of matter patent protection.
We intend to pursue patents with claims directed to Annamycin drug product formulations and the methods of use of Annamycin to treat relapsed or refractory AML and other conditions, and methods for its synthesis, as the composition of matter patent protection for Annamycin has expired. As a result, competitors may be able to offer and sell products so long as these competitors do not infringe any other patents that third parties or we hold, including formulation, synthesis and method of use patents. However, method of use patents, in particular, are more difficult to enforce than composition of matter patents because of the risk of off-label sale or use of the subject compounds. Physicians are permitted to prescribe an approved product for uses that are not described in the product's labeling. Although off-label prescriptions may infringe our method of use patents, the practice is common across medical specialties and such infringement is difficult to prevent or prosecute. Off-label sales would limit our ability to generate revenue from the sale of Annamycin, if approved for commercial sale.
The intellectual property rights we have licensed from MD Anderson are subject to the rights of the U.S. government.
We have obtained a royalty-bearing, worldwide, exclusive license to intellectual property rights, including patent rights related to our WP1066 Portfolio and WP1122 Portfolio drug product candidates from MD Anderson. However, the rights we have obtained pursuant to our license agreement with MD Anderson are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. Additionally, to the extent there is any conflict between our license agreement with MD Anderson, applicable laws or regulations, and MD Anderson’s funding agreement with the U.S. government, applicable laws or regulations and the terms of the funding agreement will prevail. Therefore, there is a risk that the intellectual property rights we have licensed from MD Anderson may be non-exclusive or void if a funding agreement related to the licensed technology between MD Anderson and the U.S. government does exist and depending on the terms of such an agreement.
18
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
We may from time to time seek to enforce our intellectual property rights against infringers when we determine that a successful outcome is probable and may lead to an increase in the value of the intellectual property. If we choose to enforce our patent rights against a party, then that individual or company has the right to ask the court to rule that such patents are invalid or should not be enforced. Additionally, the validity of our patents and the patents we have licensed may be challenged if a petition for post grant proceedings such as inter-partes review and post grant review is filed within the statutorily applicable time with the U.S. Patent and Trademark Office (USPTO). These lawsuits and proceedings are expensive and would consume time and resources and divert the attention of managerial and scientific personnel even if we were successful in stopping the infringement of such patents. In addition, there is a risk that the court will decide that such patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of such patents is upheld, the court will refuse to stop the other party on the ground that such other party's activities do not infringe our intellectual property rights. In addition, in recent years the U.S. Supreme Court modified some tests used by the USPTO in granting patents over the past 20 years, which may decrease the likelihood that we will be able to obtain patents and increase the likelihood of a challenge of any patents we obtain or license.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industries, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees, or we, have used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
We will need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth.
As of January 27, 2016, we had one full-time and two part-time employees. As we advance our product candidates through preclinical studies and clinical trials, we will need to increase our product development, scientific and administrative headcount to manage these programs. In addition, to meet our obligations as a public company, we may need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel and consultants.
We may not be able to attract or retain qualified management, finance, scientific and clinical personnel and consultants due to the intense competition for qualified personnel and consultants among biotechnology, pharmaceutical and other businesses. If we are not able to attract and retain necessary personnel and consultants to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
19
We are highly dependent on the development, regulatory, commercialization and business development expertise of our management team, key employees and consultants. We currently do not have any employment agreements with any of our management team or key employees. If we lose one or more of our executive officers or key employees or consultants, our ability to implement our business strategy successfully could be seriously harmed. Any of our executive officers or key employees or consultants may terminate their employment at any time. Replacing executive officers, key employees and consultants may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition to hire and retain employees and consultants from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel and consultants. Our failure to retain key personnel or consultants could materially harm our business.
In addition, we have scientific and clinical advisors and consultants who assist us in formulating our research, development and clinical strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us and typically they will not enter into non-compete agreements with us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services. In addition, our advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours.
Our acting chief executive officer and our chief operating officer are currently working for us on a part-time basis.
Two of our key employees are currently part-time and provide services for other biotechnology development efforts. Specifically, Walter Klemp, our chairman and acting chief executive officer is also the chief executive officer of Soliton, Inc., a medical device development company whose business operations we do not believe conflict with those of our company, and Donald Picker, our president and chief operating officer, is currently also serving as chief operating officer of two other biotechnology companies, whose business operations we do not believe conflict with those of our company. Both Messrs. Klemp and Picker currently provide services as needed by us, which we estimate does not exceed 10 hours per week. Upon closing of this offering, Mr. Picker will become a full time officer of our company and Mr. Klemp will continue to provide part time services to us as needed. As we progress, we will need to identify a suitable CEO who can dedicate substantially full time to our company. We can provide no assurance that we will be able to successfully identify and retain a qualified candidate for this position.
Upon the closing of this offering, we do not expect that our insurance policies will cover all of our business exposures thus leaving us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. In particular, we do not carry product liability insurance covering any clinical trials liability that we may incur. Although we intend to obtain such insurance before we commence any clinical trials, there can be no assurance that we will secure adequate insurance coverage or that any such insurance coverage will be sufficient to protect our operations to significant potential liability in the future. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our financial position and results of operations.
Risks Relating to this Offering of Our Common Stock
There has been no public market for our common stock and an active market may not develop or be sustained, which could limit your ability to sell shares of our common stock.
There currently is no public market for our common stock, and our common stock will not be traded in the open market prior to this offering. Although we intend to list the common stock on the Nasdaq Capital Market in connection with this offering, an adequate trading market for the common stock may not develop or be sustained after this offering. The initial public offering price will be determined by negotiations between the underwriters and our board of directors and may not be representative of the market price at which our shares of common stock will trade after this offering. In particular, we cannot assure you that you will be able to resell your shares at or above the initial public offering price.
The best efforts structure of this offering may yield insufficient gross proceeds to fully execute on our business plan.
The underwriters are offering shares of our common stock in this offering on a best efforts basis. The underwriters are not required to sell any specific number or dollar amount of common stock, but will use their best efforts to sell the shares offered by us. It is a condition to this offering that, upon the closing of the offering, our common stock would qualify for listing on the Nasdaq Capital Market. In order to list, the Nasdaq Capital Market requires that, among other criteria, at least 1,000,000 publicly-held shares of our common stock be outstanding, the shares be held in the aggregate by at least 300 round lot holders, the market value of the publicly-held shares of our common stock be at least $15.0 million, our stockholders’ equity after giving effect to the sale of our shares in this offering be at least $4.0 million, the bid price per share of our common stock be $4.00 or more, and there be at least three registered and active market makers for our common stock. As a “best efforts” offering, there can be no assurance that we will successfully raise this minimum amount, that the offering will satisfy the listing conditions required to trade our common stock on the Nasdaq Capital Market or that the offering contemplated by this prospectus will ultimately be completed or will result in any proceeds being made available to us.
20
The success of this offering will impact, in large part, our ability to cover expenses and finance operations over the next 13 to 19 months. If no shares are sold in this offering, or if we sell only the minimum number of shares yielding insufficient gross proceeds, we may be unable to cover our expenses, sufficiently fund operations or fully execute on our business plan. This could potentially result in a material adverse effect on our business, prospects, financial condition and results of operations.
If securities or industry analysts do not publish research or reports about us, or if they adversely change their recommendations regarding our common stock, then our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us, our industry and our market. If no analyst elects to cover us and publish research or reports about us, the market for our common stock could be severely limited and our stock price could be adversely affected. In addition, if one or more analysts ceases coverage of us or fails to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline. If one or more analysts who elect to cover us issue negative reports or adversely change their recommendations regarding our common stock, our stock price could decline.
Purchasers in this offering will experience immediate and substantial dilution in net tangible book value.
The initial public offering price is substantially higher than the net tangible book value of each outstanding share of our common stock. Purchasers of common stock in this offering will experience immediate and substantial dilution on a book value basis. The dilution per share in the net tangible book value per share of common stock will be $5.07 per share if the minimum number of shares are sold and $4.87 per share if the maximum number of shares are sold, based on an estimated $5.50 initial public offering price, which is the midpoint of the offering price range, and assuming, for purposes of the dilution calculations contained in this prospectus and the conversion of all of our outstanding 8% unsecured promissory notes into an aggregate of 3,749,557 shares of our common stock contemporaneously with the closing of this offering (exclusive of shares issuable for accrued interest under such notes). No holder of these notes will be permitted to convert such notes to the extent that the holder or any of its affiliates would beneficially own in excess of 4.99% of our common stock after such conversion. The number of shares set forth above assumes no such limitation on the conversion of the notes. If stock options and warrants to purchase shares of common stock are exercised, there would be further dilution. See “Dilution.”
Your ownership may be diluted if additional capital stock is issued to raise capital, to finance acquisitions or in connection with strategic transactions.
We intend to seek to raise additional funds, finance acquisitions or develop strategic relationships by issuing equity or convertible debt securities in addition to the shares issued in this offering, which would reduce the percentage ownership of our existing stockholders. Our board of directors has the authority, without action or vote of the stockholders, to issue all or any part of our authorized but unissued shares of common or preferred stock. Prior to the effective date of the registration statement of which this prospectus is a part, our certificate of incorporation will be amended to authorize us to issue up to 75,000,000 shares of common stock and 5,000,000 shares of preferred stock. Future issuances of common or preferred stock would reduce your influence over matters on which stockholders vote and would be dilutive to earnings per share. In addition, any newly issued preferred stock could have rights, preferences and privileges senior to those of the common stock. Those rights, preferences and privileges could include, among other things, the establishment of dividends that must be paid prior to declaring or paying dividends or other distributions to holders of our common stock or providing for preferential liquidation rights. These rights, preferences and privileges could negatively affect the rights of holders of our common stock, and the right to convert such preferred stock into shares of our common stock at a rate or price that would have a dilutive effect on the outstanding shares of our common stock.
The concentration of our common stock ownership by our current management will limit your ability to influence corporate matters.
Upon completion of this offering, and assuming the conversion of all our outstanding 8% unsecured promissory notes contemporaneously with the closing of this offering, our directors and executive officers will beneficially own and will be able to vote in the aggregate approximately [*]% of our outstanding common stock if the minimum number of shares are sold and approximately [*]% of our outstanding common stock if the maximum number of shares offered are sold. As such, our directors and executive officers, as stockholders, will continue to have the ability to exert significant influence over all corporate activities, including the election or removal of directors and the outcome of tender offers, mergers, proxy contests or other purchases of common stock that could give our stockholders the opportunity to realize a premium over the then-prevailing market price for their shares of common stock. This concentrated control will limit your ability to influence corporate matters and, as a result, we may take actions that purchasers in this offering do not view as beneficial. In addition, such concentrated control could discourage others from initiating changes of control. In such cases, the perception of our prospects in the market may be adversely affected and the market price of our common stock may decline.
21
Certain provisions in our organizational documents could enable our board of directors to prevent or delay a change of control.
Our organizational documents contain provisions that may have the effect of discouraging, delaying or preventing a change of control of, or unsolicited acquisition proposals, that a stockholder might consider favorable. These include provisions:
| • | prohibiting the stockholders from acting by written by consent; |
| • | requiring advance notice of director nominations and of business to be brought before a meeting of stockholders; |
| • | requiring a majority vote of the outstanding shares of common stock to amend the bylaws; and |
| • | limiting the persons who may call special stockholders’ meetings. |
Furthermore, our board of directors has the authority to issue shares of preferred stock in one or more series and to fix the rights and preferences of these shares without stockholder approval. Any series of preferred stock is likely to be senior to our common stock with respect to dividends, liquidation rights and, possibly, voting rights. The ability of our board of directors to issue preferred stock also could have the effect of discouraging unsolicited acquisition proposals, thus adversely affecting the market price of our common stock.
In addition, Delaware law makes it difficult for stockholders that recently have acquired a large interest in a corporation to cause the merger or acquisition of the corporation against the directors’ wishes. Under Section 203 of the Delaware General Corporation Law, a Delaware corporation may not engage in any merger or other business combination with an interested stockholder for a period of three years following the date that the stockholder became an interested stockholder except in limited circumstances, including by approval of the corporation’s board of directors.
We have no intention of declaring dividends in the foreseeable future.
The decision to pay cash dividends on our common stock rests with our board of directors and will depend on our earnings, unencumbered cash, capital requirements and financial condition. We do not anticipate declaring any dividends in the foreseeable future, as we intend to use any excess cash to fund our operations. Investors in our common stock should not expect to receive dividend income on their investment, and investors will be dependent on the appreciation of our common stock to earn a return on their investment.
We will incur increased costs as a result of being a publicly-traded company.
As a company with publicly-traded securities, we will incur additional legal, accounting and other expenses not presently incurred. In addition, the Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, as well as rules promulgated by the SEC and the national securities exchange on which we list, requires us to adopt corporate governance practices applicable to U.S. public companies. These rules and regulations will increase our legal and financial compliance costs.
We may be exposed to risks relating to evaluations of controls required by Sarbanes-Oxley Act of 2002.
Pursuant to Sarbanes-Oxley Act of 2002, our management will be required to report on, and our independent registered public accounting firm may in the future be required to attest to, the effectiveness of our internal control over financial reporting. Although we prepare our financial statements in accordance with accounting principles generally accepted in the United States of America, our internal accounting controls may not meet all standards applicable to companies with publicly traded securities. If we fail to implement any required improvements to our disclosure controls and procedures, we may be obligated to report control deficiencies and our independent registered public accounting firm may not be able to certify the effectiveness of our internal controls over financial reporting. In either case, we could become subject to regulatory sanction or investigation. Further, these outcomes could damage investor confidence in the accuracy and reliability of our financial statements.
22
Failure to continue improving our accounting systems and controls could impair our ability to comply with the financial reporting and internal controls requirements for publicly traded companies.
As a public company, we operate in an increasingly demanding regulatory environment, which requires us to comply with the Sarbanes-Oxley Act of 2002, and the related rules and regulations of the SEC. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud.
We have implemented a system of internal controls over financial reporting and are preparing the documentation necessary to perform the evaluation needed to comply with Section 404(a) of the Sarbanes-Oxley Act. However, we may need to retain additional finance capabilities and build our financial infrastructure as a public company. Section 404(a) of the Sarbanes-Oxley Act requires annual management assessments of the effectiveness of our internal control over financial reporting, starting with the second annual report that we would expect to file with the SEC. However, for as long as we remain an “emerging growth company” as defined in the JOBS Act, we have and intend to continue to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act. We may continue to take advantage of these reporting exemptions until we are no longer an “emerging growth company.” If we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed and investors could lose confidence in our reported financial information.
The protection provided by the federal securities laws relating to forward-looking statements does not apply to us. The lack of this protection could harm us in the event of an adverse outcome in a legal proceeding relating to forward-looking statements made by us.
Although federal securities laws provide a safe harbor for forward-looking statements made by a public company that files reports under the federal securities laws, this safe harbor is not available to certain issuers, including issuers that do not have their equity traded on a recognized national securities exchange. Our common stock does not trade on any recognized national securities exchange. As a result, we will not have the benefit of this safe harbor protection in the event of any legal action based upon a claim that the material provided by us contained a material misstatement of fact or was misleading in any material respect because of our failure to include any statements necessary to make the statements not misleading. The lack of this protection in a contested proceeding could harm our financial condition.
As an “emerging growth company” under the Jumpstart Our Business Startups Act, or JOBS Act, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements.
As an “emerging growth company” under the JOBS Act, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements. We are an emerging growth company until the earliest of:
| • | the last day of the fiscal year during which we have total annual gross revenues of $1 billion or more; |
| • | the last day of the fiscal year following the fifth anniversary of this offering; |
| • | the date on which we have, during the previous 3-year period, issued more than $1 billion in non-convertible debt; or |
| • | the date on which we are deemed a “large accelerated issuer” as defined under the federal securities laws. |
For so long as we remain an emerging growth company, we will not be required to:
| • | have an auditor report on our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002; |
| • | comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (auditor discussion and analysis); |
| • | submit certain executive compensation matters to shareholders advisory votes pursuant to the “say on frequency” and “say on pay” provisions (requiring a non-binding shareholder vote to approve compensation of certain executive officers) and the “say on golden parachute” provisions (requiring a non-binding shareholder vote to approve golden parachute arrangements for certain executive officers in connection with mergers and certain other business combinations) of the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010; |
23
| • | include detailed compensation discussion and analysis in our filings under the Securities Exchange Act of 1934, as amended, and instead may provide a reduced level of disclosure concerning executive compensation; |
| • | may present only two years of audited financial statements and only two years of related Management’s Discussion and Analysis of Financial Condition and Results of Operations, or MD&A; and |
| • | are eligible to claim longer phase-in periods for the adoption of new or revised financial accounting standards under §107 of the JOBS Act. |
We intend to take advantage of all of these reduced reporting requirements and exemptions, including the longer phase-in periods for the adoption of new or revised financial accounting standards under §107 of the JOBS Act. Our election to use the phase-in periods may make it difficult to compare our financial statements to those of non-emerging growth companies and other emerging growth companies that have opted out of the phase-in periods under §107 of the JOBS Act.
Certain of these reduced reporting requirements and exemptions were already available to us due to the fact that we also qualify as a “smaller reporting company” under SEC rules. For instance, smaller reporting companies are not required to obtain an auditor attestation and report regarding management’s assessment of internal control over financial reporting; are not required to provide a compensation discussion and analysis; are not required to provide a pay-for-performance graph or CEO pay ratio disclosure; and may present only two years of audited financial statements and related MD&A disclosure.
Under the JOBS Act, we may take advantage of the above-described reduced reporting requirements and exemptions for up to five years after our initial sale of common equity pursuant to a registration statement declared effective under the Securities Act of 1933, or such earlier time that we no longer meet the definition of an emerging growth company. In this regard, the JOBS Act provides that we would cease to be an “emerging growth company” if we have more than $1.0 billion in annual revenues, have more than $700 million in market value of our common stock held by non-affiliates, or issue more than $1.0 billion in principal amount of non-convertible debt over a three-year period. Further, under current SEC rules, we will continue to qualify as a “smaller reporting company” for so long as we have a public float (i.e., the market value of common equity held by non-affiliates) of less than $75 million as of the last business day of our most recently completed second fiscal quarter.
We cannot predict if investors will find our securities less attractive due to our reliance on these exemptions. If investors were to find our common stock less attractive as a result of our election, we may have difficulty raising all of the proceeds we seek in this offering.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
We make forward-looking statements under the “Summary,” “Risk Factors,” “Business,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in other sections of this prospectus. In some cases, you can identify these statements by forward-looking words such as “may,” “might,” “should,” “would,” “could,” “expect,” “plan,” “anticipate,” “intend,” “believe,” “estimate,” “predict,” “potential” or “continue,” and the negative of these terms and other comparable terminology. These forward-looking statements, which are subject to known and unknown risks, uncertainties and assumptions about us, may include projections of our future financial performance based on our growth strategies and anticipated trends in our business. These statements are only predictions based on our current expectations and projections about future events. There are important factors that could cause our actual results, level of activity, performance or achievements to differ materially from the results, level of activity, performance or achievements expressed or implied by the forward-looking statements. In particular, you should consider the numerous risks and uncertainties described under “Risk Factors.”
While we believe we have identified material risks, these risks and uncertainties are not exhaustive. Other sections of this prospectus describe additional factors that could adversely impact our business and financial performance. Moreover, we operate in a very competitive and rapidly changing environment. New risks and uncertainties emerge from time to time, and it is not possible to predict all risks and uncertainties, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
Although we believe the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, level of activity, performance or achievements. Moreover, neither we nor any other person assumes responsibility for the accuracy or completeness of any of these forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. We are under no duty to update any of these forward-looking statements after the date of this prospectus to conform our prior statements to actual results or revised expectations, and we do not intend to do so.
24
Forward-looking statements include, but are not limited to, statements about:
| · | our ability to obtain additional funding to develop our product candidates; |
| · | the need to obtain regulatory approval of our product candidates; |
| · | the success of our clinical trials through all phases of clinical development; |
| · | compliance with obligations under intellectual property licenses with third parties; |
| · | any delays in regulatory review and approval of product candidates in clinical development; |
| · | our ability to commercialize our product candidates; |
| · | market acceptance of our product candidates; |
| · | competition from existing products or new products that may emerge; |
| · | potential product liability claims; |
| · | our dependency on third-party manufacturers to supply or manufacture our products; |
| · | our ability to establish or maintain collaborations, licensing or other arrangements; |
| · | our ability and third parties’ abilities to protect intellectual property rights; |
| · | our ability to adequately support future growth; and |
| · | our ability to attract and retain key personnel to manage our business effectively. |
We caution you not to place undue reliance on the forward-looking statements, which speak only as of the date of this prospectus in the case of forward-looking statements contained in this prospectus.
Based on an estimated initial public offering price of $5.50 per share, which is the midpoint of the offering price range, we estimate that the net proceeds from this offering, after deducting underwriting commissions and expenses payable by us and other offering expenses payable by us, will be approximately $6.7 million if we sell a minimum of 1,400,000 shares and approximately $9.7 million if we sell all 2,000,000 shares of our common stock in this offering. However, this is a best efforts offering and there is no assurance that we will sell any shares or receive any proceeds.
We intend to use the proceeds from this offering as follows:
| Assuming Minimum Offering | Assuming Maximum Offering | |||||||
| Costs to prepare for filings with FDA in advance of our Annamycin clinical trial | $ | 380,000 | (6%) | $ | 380,000 | (4%) | ||
| Commence a Phase II clinical trial for Annamycin (including manufacturing of product candidate for use) (1) | $ | 2,730,000 | (44%) | $ | 3,600,000 | (42%) | ||
| License maintenance and IP prosecution costs | $ | 800,000 | (11%) | $ | 1,100,000 | (10%) | ||
| Research | $ | 100,000 | (2%) | $ | 150,000 | (2%) | ||
| Working capital | $ | 2,700,000 | (37%) | $ | 4,550,000 | (42%) | ||
(1) If we complete the maximum offering, we estimate that we will have sufficient funds to complete the majority of the Phase II clinical trials for Annamycin. If we complete the minimum offering, we estimate that we will require additional financing of at least $1.1 million to complete the trial plus such additional working capital to fund our operations during the pendency of the trial. The timing and costs of clinical trials are difficult to predict and as such the foregoing estimates may prove to be inaccurate.
We believe the net proceeds of this offering, together with our and Moleculin’s cash and cash equivalents, including the remaining proceeds from our recent private placement of our 8% unsecured promissory notes, will be sufficient to meet our cash, operational and liquidity requirements for at least 12 months if we sell a minimum of 1,400,000 shares and for at least 16 months if we sell all 2,000,000 shares of our common stock in this offering.
As of the date of this prospectus, we cannot specify with certainty all of the particular uses for the net proceeds to us from this offering. Accordingly, our management will have broad discretion in the application of these proceeds. Net offering proceeds not immediately applied to the uses summarized above will be invested in short-term investments such as money market funds, commercial paper, U.S. treasury bills and similar securities investments pending their use.
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain earnings, if any, to finance the growth and development of our business. We do not expect to pay any cash dividends on our common stock in the foreseeable future. Payment of future dividends, if any, will be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements, restrictions contained in any financing instruments, provisions of applicable law and other factors the board deems relevant.
25
The following table sets forth our cash and cash equivalents and capitalization as of September 30, 2015 on:
| · | an actual basis; and |
| · | a pro forma as adjusted basis after giving effect to: (1) (a) the sale of a minimum of 1,400,000 shares of our common stock in this offering at an estimated initial public offering price of $5.50 per share, which is the midpoint of the offering price range, and our receipt of the estimated $6.7 million in net proceeds from this offering, after deducting underwriting commissions and estimated offering expenses payable by us, and (b) the sale of all 2,000,000 shares of our common stock in this offering at an estimated initial public offering price of $5.50 per share, which is the midpoint of the offering price range, and our receipt of the estimated $9.7 million in net proceeds from this offering, after deducting underwriting commissions and estimated offering expenses payable by us; (2) the conversion of all of our outstanding 8% unsecured promissory notes in principal amount of $615,000 into 3,749,557 shares of our common stock contemporaneously with the closing of this offering (exclusive of shares issuable for accrued interest under such notes); (3) our planned acquisition of Moleculin; and (4) the issuance of 629,000 shares of common stock in connection with our agreement with HPI. |
You should read this capitalization table together with “Use of Proceeds,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and the related notes appearing elsewhere in this prospectus.
| At September 30, 2015 | ||||||||||||
| Actual | Pro Forma – As adjusted Minimum | Pro Forma – As adjusted Maximum | ||||||||||
| Cash and cash equivalents | $ | 87,340 | $ | 7,355,916 | $ | 10,424,916 | ||||||
| Notes payable | 250,000 | - | - | |||||||||
| Stockholders’ equity (deficit): | ||||||||||||
| Preferred stock, $0.001 par value: 5,000,000 shares authorized, no shares issued and outstanding | - | - | - | |||||||||
| Common stock, $0.001 par value: 75,000,000 shares authorized; 6,661,000 shares issued and outstanding, actual; 13,439,557 and 14,039,557 shares issued and outstanding, pro forma as adjusted – minimum and pro forma as adjusted – maximum, respectively (1) | 6,661 | 13,440 | 14,040 | |||||||||
| Additional paid-in capital | - | 16,228,721 | 19,297,121 | |||||||||
| Subscription receivable | (4,600 | ) | (4,600 | ) | (4,600 | ) | ||||||
| Accumulated deficit | (224,315 | ) | (3,683,815 | ) | (3,683,815 | ) | ||||||
| Total stockholders’ equity (deficit) | (222,254 | ) | 12,553,746 | 15,622,746 | ||||||||
| Total capitalization | $ | 27,746 | $ | 12,553,746 | $ | 15,622,746 | ||||||
| (1) | The number of shares of common stock to be outstanding after this offering includes (i) 1,000,000 shares of common stock to be issued to the members of Moleculin LLC, (ii) the issuance of 629,000 shares of common stock in connection with our agreement HPI; and (iii) the issuance of 3,749,558 shares of common stock upon the conversion of $615,000 in principal amount of our outstanding bridge notes. |
26
The number of shares does not give effect to:
| · | 2,500,000 shares of common stock available for issuance under the Moleculin Biotech, Inc. 2015 Stock Plan; and |
| · | between 98,000 shares (assuming the minimum offering is completed) and 140,000 shares (assuming the maximum offering is completed) of common stock issuable upon the exercise of the warrants issued to the representatives of the underwriters. |
27
Purchasers of our common stock in this offering will experience an immediate dilution of net tangible book value per share from the initial public offering price. Dilution in net tangible book value per share represents the difference between the amount per share paid by the purchasers of shares of common stock and the net tangible book value per share immediately after this offering.
As of September 30, 2015, our net tangible book value was $(825,060), or $(0.07) per share of common stock. Net tangible book value per share represents our total tangible assets, less our total liabilities, divided by the number of outstanding shares of our common stock.
Dilution represents the difference between the amount per share paid by purchasers in this offering and the pro forma net tangible book value per share of common stock after the offering. After giving effect to the sale of 1,400,000 shares of common stock (minimum) and 2,000,000 shares of common stock (maximum) in this offering at an estimated offering price of $5.50 per share, which is the midpoint of the offering price range, and after deducting underwriting commissions and estimated offering expenses payable by us, our pro forma net tangible book value would have been $0.43 (minimum) and $0.63 (maximum) per share. This represents an immediate increase in pro forma net tangible book value of $0.50 (minimum) and $0.70 (maximum) per share to our existing stockholders and immediate dilution of $5.07 (minimum) and $4.87 (maximum) per share to new investors purchasing shares at the proposed public offering price. The following table illustrates the dilution in pro forma net tangible book value per share to new investors as of September 30, 2015:
| Minimum | Maximum | |||||||
| Assumed initial public offering price per share | $ | 5.50 | $ | 5.50 | ||||
| Net tangible book value per share at September 30, 2015* | (0.07 | ) | (0.07 | ) | ||||
| Increase in net tangible book value per share to the existing stockholders attributable to this offering | 0.50 | 0.70 | ||||||
| Adjusted net tangible book value per share after this offering | 0.43 | 0.63 | ||||||
| Dilution in net tangible book value per share to new investors | 5.07 | 4.87 | ||||||
*Including adjustments for the acquisition of Moleculin, the conversion of our 8% convertible notes, and the issuance of 629,000 shares to HPI.
The following tables set forth, as of September 30, 2015, the number of shares of common stock purchased from us, the total consideration paid to us and the average price per share paid by the existing holders of our common stock and the price to be paid by new investors at an estimated public offering price of $5.50 per share, which is the midpoint of the offering price range.
Minimum Offering
| Shares Purchased | Total Consideration | Average Price Per Share |
||||||||||||||||||
| Number | Percent | Amount | Percent | |||||||||||||||||
| Existing investors before this offering | $ | $ | ||||||||||||||||||
| Investors purchasing shares in this offering | 1,400,000 | $ | 7,700,000 | $ | 5.50 | |||||||||||||||
| Total | $ | |||||||||||||||||||
Maximum Offering
| Shares Purchased | Total Consideration | Average Price Per Share |
||||||||||||||||||
| Number | Percent | Amount | Percent | |||||||||||||||||
| Existing investors before this offering | $ | $ | ||||||||||||||||||
| Investors purchasing shares in this offering | 2,000,000 | $ | 11,000,000 | $ | 5.50 | |||||||||||||||
| Total | ||||||||||||||||||||
28
The number of shares of common stock set forth in the tables above under “Existing investors before this offering” includes (i) 1,000,000 shares of common stock to be issued to the members of Moleculin LLC, (ii) the issuance of 629,000 shares of common stock in connection with our agreement HPI; and (iii) the issuance of 3,749,558 shares of common stock upon the conversion of $615,000 in principal amount of our outstanding bridge notes.
29
UNAUDITED PRO FORMA COMBINED FINANCIAL INFORMATION
The following unaudited pro forma combined financial information is based on the historical financial statements of Moleculin Biotech, Inc. after giving effect to our planned acquisition of Moleculin, LLC (“Moleculin”), our initial public offering and the assumptions, reclassifications and adjustments described in the accompanying notes to the unaudited pro forma combined financial information.
The unaudited pro forma combined statements of operations for the nine months ended September 30, 2015 is presented as if our acquisition of Moleculin occurred on January 1, 2014 and were carried forward through to September 30, 2015. The unaudited pro forma combined balance sheet as of September 30, 2015 is presented as if the acquisition of Moleculin had occurred on September 30, 2015.
These pro forma combined financial statements include adjustments for our planned acquisition because we believe the acquisition is probable under the standards of Rule 3-05 of Regulations S-X. We have based the pro forma adjustments upon available information and certain assumptions that we believe are reasonable under the circumstances. We describe in greater detail the assumptions underlying the pro forma combined financial statements in the notes to the unaudited pro forma combined financial statements. In many cases, we based these assumptions on preliminary information and estimates. The actual adjustments to our pro forma combined financial statements will depend upon a number of factors and additional information that will be available on or after the closing date of this offering. Accordingly, the actual adjustments that will appear in our financial statements will differ from these pro forma adjustments, and those differences may be material.
We account for our proposed acquisitions of Moleculin using the acquisition method of accounting for business combinations under accounting principles generally accepted in the United States of America, with MBI being considered the acquiring entity. Under the acquisition method of accounting, the total consideration paid is allocated to an acquired company’s tangible and intangible assets, net of liabilities, based on their estimated fair values as of the acquisition date. We have not completed the acquisition of Moleculin, and therefore, the estimated purchase price and fair value of Moleculin’s assets to be acquired and liabilities assumed is preliminary. Once we complete our final valuation process for our planned acquisitions, we may report changes to the value of the assets acquired and liabilities assumed, as well as the amount of goodwill, and those changes could differ materially from what we present here.
The unaudited pro forma combined financial information is not intended to represent or be indicative of our consolidated results of operations or financial position that we would have reported had the Moleculin acquisition been completed as of the date presented, and should not be taken as a representation of our future consolidated results of operations or financial position. The unaudited pro forma combined financial information does not reflect any operating efficiencies and/or cost savings that we may achieve with respect to combining the companies.
The following unaudited financial information should be read with the accompanying notes, the financial statements of Moleculin and the notes thereto included elsewhere in this prospectus and the financial statements of MBI and the notes thereto included elsewhere in this prospectus.
30
Moleculin Biotech, Inc.
Unaudited Pro Forma Combined Statement of Operations
For the nine months ended September 30, 2015
| Moleculin | MBI | Pro Forma Adjustments | Note | Pro Forma Combined | ||||||||||||||
| Revenue | $ | - | $ | - | $ | - | $ | - | ||||||||||
| Operating expenses: | ||||||||||||||||||
| Research and development | 270,991 | 38,409 | - | 309,400 | ||||||||||||||
| General and administrative | 267,423 | 184,344 | - | 451,767 | ||||||||||||||
| Depreciation and amortization | 8,469 | - | - | (a) | 8,469 | |||||||||||||
| Total operating expenses | 546,883 | 222,753 | - | 769,636 | ||||||||||||||
| Net loss from operations | (546,883 | ) | (222,753 | ) | - | (769,636 | ) | |||||||||||
| Other income (expense): | ||||||||||||||||||
| Other income | 549 | - | - | 549 | ||||||||||||||
| Interest expense | (334,887 | ) | (1,562 | ) | 317,543 | (b) | (18,906 | ) | ||||||||||
| Total other income (expense) | (334,338 | ) | (1,562 | ) | 317,543 | (18,357 | ) | |||||||||||
| Net loss | $ | (881,221 | ) | $ | (224,315 | ) | $ | 317,543 | (c) | $ | (787,993 | ) | ||||||
| Loss per common share - basic and diluted | $ | (0.05 | ) | $ | (0.08 | ) | ||||||||||||
| Weighted average number of common shares outstanding - basic and diluted | 4,320,015 | 5,378,557 | (f)(g)(h)(i)(k)(l) | 9,698,572 | ||||||||||||||
31
Moleculin Biotech, Inc.
Unaudited Pro Forma Combined Statement of Operations
For the year ended December 31, 2014
| Moleculin | MBI | Pro Forma Adjustments | Note | Pro Forma Combined | ||||||||||||||
| Revenue | $ | - | $ | - | $ | - | $ | - | ||||||||||
| Operating expenses: | ||||||||||||||||||
| Research and development | 798,785 | - | - | 798,785 | ||||||||||||||
| General and administrative | 839,556 | - | - | 839,556 | ||||||||||||||
| Depreciation and amortization | 11,005 | - | - | (a) | 11,005 | |||||||||||||
| Total operating expenses | 1,649,346 | - | - | 1,649,346 | ||||||||||||||
| Net loss from operations | (1,649,346 | ) | - | - | (1,649,346 | ) | ||||||||||||
| Other income (expense): | ||||||||||||||||||
| Other income | 800 | - | - | 800 | ||||||||||||||
| Interest expense | (160,540 | ) | - | 156,789 | (b) | (3,751 | ) | |||||||||||
| Total other income (expense) | (159,740 | ) | - | 156,789 | (2,951 | ) | ||||||||||||
| Net loss | $ | (1,809,086 | ) | $ | - | $ | 156,789 | (c) | $ | (1,652,297 | ) | |||||||
| Loss per common share - basic and diluted | $ | (0.31 | ) | |||||||||||||||
| Weighted average number of common shares outstanding - basic and diluted | 5,378,557 | (f)(g)(h)(i)(k)(l) | 5,378,557 | |||||||||||||||
32
Moleculin Biotech, Inc.
Unaudited Pro Forma Combined Balance Sheet
As of September 30, 2015
| Moleculin | MBI | Pro Forma Adjustments | Pro Forma Combined | Adjustments for IPO | Pro Forma Combined As Adjusted | |||||||||||||||||||
| ASSETS | ||||||||||||||||||||||||
| Current Assets: | ||||||||||||||||||||||||
| Cash and cash equivalents | $ | 298,350 | $ | 87,340 | $ | - | $ | 385,690 | 309,226 | (g) | $ | 7,355,916 | ||||||||||||
| 6,661,000 | (k) | |||||||||||||||||||||||
| Prepaid expenses and other current assets | 4,709 | 23,000 | - | 27,709 | - | 27,709 | ||||||||||||||||||
| Total Current Assets | 303,059 | 110,340 | - | 413,399 | 6,970,226 | 7,383,625 | ||||||||||||||||||
| Property and equipment, net of accumulated depreciation | 14,126 | - | - | 14,126 | - | 14,126 | ||||||||||||||||||
| Intangibles | - | - | - | (d) | - | - | - | |||||||||||||||||
| Goodwill | - | - | 6,717,806 | (e) | 6,717,806 | - | 6,717,806 | |||||||||||||||||
| Total Assets | $ | 317,185 | $ | 110,340 | $ | 6,717,806 | 7,145,331 | 6,970,226 | $ | 14,115,557 | ||||||||||||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||||||||||||||||||
| Current Liabilities: | ||||||||||||||||||||||||
| Accounts payable and accrued liabilities | $ | 1,160,571 | $ | 63,010 | $ | (95,519 | )(f) | 1,128,062 | - | $ | 1,128,062 | |||||||||||||
| Accounts payable and accrued liabilities- related party | 56,194 | 19,584 | (56,194 | )(f) | 19,584 | - | 19,584 | |||||||||||||||||
| Common unit payable | 548,630 | - | (548,630 | )(i) | - | - | - | |||||||||||||||||
| Notes payable | 55,774 | - | - | 55,774 | (55,774 | )(g) | - | |||||||||||||||||
| Convertible notes payable | 962,000 | 250,000 | (962,000 | )(f) | 250,000 | (250,000 | )(j) | - | ||||||||||||||||
| Convertible notes payable- related parties | 468,591 | - | (468,591 | )(f) | - | - | - | |||||||||||||||||
| Total Current Liabilities | 3,251,760 | 332,594 | (2,130,934 | ) | 1,453,420 | (305,774 | ) | 1,147,646 | ||||||||||||||||
| Long-term note payable | 414,165 | - | - | 414,165 | - | 414,165 | ||||||||||||||||||
| Total Liabilities | 3,665,925 | 332,594 | (2,130,934 | ) | 1,867,585 | (305,774 | ) | 1,561,811 | ||||||||||||||||
| Stockholders’ Equity (Deficit): | ||||||||||||||||||||||||
| Preferred units | 10,620,186 | - | (10,620,186 | )(h) | - | - | - | |||||||||||||||||
| Common units | 750,000 | - | (750,000 | )(h) | - | - | - | |||||||||||||||||
| Common stock | - | 6,661 | 293 | (f) | 7,661 | 1,825 | (g) | 13,440 | ||||||||||||||||
| 707 | (h) | 1,925 | (j) | |||||||||||||||||||||
| 1,400 | (k) | |||||||||||||||||||||||
| 629 | (l) | |||||||||||||||||||||||
| Additional paid-in capital | - | - | 1,613,368 | (f) | 5,499,000 | 363,175 | (g) | 16,228,721 | ||||||||||||||||
| 3,885,632 | (h) | 248,075 | (j) | |||||||||||||||||||||
| | 6,659,600 | (k) | ||||||||||||||||||||||
| 3,458,871 | (l) | |||||||||||||||||||||||
| Subscription receivable | - | (4,600 | ) | - | (4,600 | ) | - | (4,600 | ) | |||||||||||||||
| Accumulated deficit | (14,718,926 | ) | (224,315 | ) | 14,718,926 | (h) | (224,315 | ) | (3,459,500 | )(l) | (3,683,815 | ) | ||||||||||||
| Total Stockholders’ Equity (Deficit) | (3,348,740 | ) | (222,254 | ) | 8,848,740 | 5,277,746 | 7,276,000 | 12,553,746 | ||||||||||||||||
| Total Liabilities and Stockholders’ Equity (Deficit) | $ | 317,185 | $ | 110,340 | $ | 6,717,806 | $ | 7,145,331 | $ | 6,970,226 | $ | 14,115,557 | ||||||||||||
33
Moleculin Biotech, Inc.
Notes to Unaudited Pro Forma Combined Financial Information
Note 1 – Basis of presentation
The unaudited pro forma combined balance sheet as of September 30, 2015, and the unaudited pro forma combined statements of operations for the nine months ended September 30, 2015 and the year ended December 31, 2014 are based on the historical financial statements of Moleculin Biotech, Inc. and Moleculin, LLC after giving effect to our planned acquisition of Moleculin and the assumptions, reclassifications and adjustments described in the accompanying notes to the unaudited pro forma combined financial information. The actual acquisition of Moleculin is to occur just prior to the date of effectiveness of the registration statement of which this prospectus forms a part.
We account for business combinations pursuant to Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (ASC) 805, Business Combinations. In accordance with ASC 805, MBI uses its best estimates and assumptions to assign fair value to the tangible and intangible assets acquired and liabilities assumed at the acquisition date. Goodwill as of the acquisition date is measured as the excess of purchase consideration over the fair value of net tangible and identifiable intangible assets acquired.
The fair values assigned to Moleculin’s tangible assets acquired and liabilities assumed are based on management’s estimates and assumptions. The estimated fair values of these assets acquired and liabilities assumed are considered preliminary and are based on the information and the account balances that were available as of September 30, 2015. We believe that the information provides a reasonable basis for estimating the fair values of assets acquired and liabilities assumed. We expect to finalize the valuation of the net tangible and intangible assets as soon as practicable, but not later than one year from the acquisition date.
The unaudited pro forma combined financial information is not intended to represent or be indicative of our results of operations or financial position that would have been reported had the acquisition been completed as of the date presented, and should not be taken as a representation of our future consolidated results of operations or financial position. The unaudited pro forma combined financial information does not reflect any operating efficiencies and/or cost savings that we may achieve with respect to the combined companies.
For purposes of these unaudited combined pro forma statements of operations, the acquisition of Moleculin and the conversion of outstanding Moleculin convertible debt and equity units outstanding are assumed to have occurred on January 1, 2014. The pro forma statement of operations for the nine months ended September 30, 2015 combined the results of Moleculin for the nine months ended September 30, 2015 and our results for the period from inception (July 28, 2015) through September 30, 2015.
The unaudited pro forma condensed combined balance sheet as of September 30, 2015 is presented as if the Moleculin acquisition had occurred on September 30, 2015.
Note 2 - Preliminary purchase price allocation
The Company plans to acquire Moleculin immediately prior to the date of effectiveness of the registration statement of which this prospectus forms a part. Moleculin is the holder of the license agreement with MD Anderson covering the WP1066 Portfolio. Upon the acquisition, the Company will issue the following common shares to Moleculin’s security holders:
| Shares | Consideration paid | |||||||
| To convertible note holders (1) | 293,393 | $ | 1,613,661 | |||||
| To Moleculin Preferred Unit holders | 635,946 | 3,497,703 | ||||||
| To Moleculin Common Unit holders | 70,661 | 388,636 | ||||||
| Total | 1,000,000 | $ | 5,500,000 | |||||
| (1) | On an “as converted” basis. Holders of Moleculin’s convertible debt will convert their debt into Moleculin units prior to MBI’s acquisition of Moleculin. |
The consideration paid was calculated based on an estimated offering price of $5.50 per share.
34
The following table shows the preliminary allocation of the purchase price for Moleculin to the identifiable assets, liabilities assumed and pro forma goodwill:
| Amount | ||||
| Purchase price | $ | 5,500,000 | ||
| Cash and cash equivalents | 298,350 | |||
| Prepaid insurance expense | 4,709 | |||
| Property and equipment | 14,126 | |||
| Intangibles | [ ] | |||
| Total identifiable assets | 317,185 | |||
| Accounts payable and accrued liabilities | 1,065,052 | |||
| Note payable | 55,774 | |||
| Long-term note payable | 414,165 | |||
| Total liabilities assumed | 1,534,991 | |||
| Net liabilities assumed | 1,217,806 | |||
| Total pro forma goodwill | $ | 6,717,806 | ||
The unaudited pro forma combined financial information includes various assumptions, including those related to the preliminary purchase price allocation of the assets acquired and liabilities assumed of Moleculin based on management’s best estimates of fair value. The final purchase price allocation may vary based on final appraisals, valuations and analyses of the fair value of the acquired assets and assumed liabilities. Accordingly, the pro forma adjustments are preliminary and have been made solely for illustrative purposes.
We plan to engage a third-party valuation specialist to assist us in valuing the assets acquired and liabilities assumed for acquisition. The assumptions will be updated on the consummation of the initial public offering.
The following table sets forth the preliminary components of identifiable intangible assets acquired and their estimated useful lives:
| Preliminary Fair Value | Useful Life (years) | |||||
| Agreements with MD Anderson | $ | [ ] | ||||
| In-process research and development | [ ] | Indefinite | ||||
| Total intangible assets | $ | [ ] | ||||
Agreements with MD Anderson represents the preliminary estimated fair value of Moleculin’s prior and current agreements Moleculin has regarding research and development efforts through and with MD Anderson.
In-process research and development represents the preliminary estimated fair value derived from the results of Moleculin’s research and development activities regarding WP1066 Portfolio conducted before the acquisition. The in-process research and development intangible has indefinite life and is not amortized.
The fair value of the intangible assets acquired will be estimated mainly using the income approach based on the discounted future net cash flows related to those intangible assets.
The purchase price and resulting goodwill will vary based on the initial offering price of MBI’s common shares upon consummation of this offering. In preparing the pro forma financial information, the Company estimates the expected purchase price based on an estimated initial offering price of $5.50 per share. The following schedule shows the impact to the goodwill value based on a range of our initial offering price:
| Purchase price | Goodwill | |||||||
| As presented in the pro forma combined result | 5,500,000 | $ | 6,717,806 | |||||
| 10% increase in initial offering price | 6,050,000 | 7,267,806 | ||||||
| 10% decrease in initial offering price | 4,950,000 | 6,167,806 | ||||||
35
Note 3 — Pro forma adjustments
The pro forma adjustments are based on our preliminary estimates and assumptions that are subject to change. The following adjustments have been reflected in the unaudited pro forma condensed combined financial information:
| (a) | Reflects the estimated amortization expense related to the acquired intangible assets discussed at Note 3(d). We based the estimated useful lives of acquired intangible assets on the amount and timing in which we expect to receive an economic benefit. We assigned these intangible assets a useful life of [ ] years based upon a number of factors, including contractual agreements and economic factors (including known technological advances, size and duration of the market for the product, time and costs to commercialize and market, and potential customers associated with the intangibles) pertaining to the combined companies. |
The estimate of fair value and useful life could be impacted by a variety of factors including legal, regulatory, contractual, competitive, economic or other factors. Increased knowledge about these factors could result in a change to the estimated fair value of the intangible asset and/or the useful life from what we have assumed in these unaudited pro forma combined financial statements. In addition, the combined effect of any such changes could result in a significant increase or decrease to the related amortization expense estimates.
The amortization of intangible assets of our planned acquisition assumes that the asset was acquired on January 1, 2014 and amortized over the period associated with the statement of operations. For the nine months ended September 30, 2015, the pro forma adjustment for the amortization expenses related to the intangible assets acquired was $[ ]. For the year ended December 31, 2014, the pro forma adjustment for the amortization expenses related to the intangible assets acquired was $[ ].
| (b) | Reflects the removal of interest expense accrued and the amortization of deferred financing cost and debt discount related to the convertible notes payable to third parties and related parties. For the nine months ended September 30, 2015 and the year ended December 31, 2014, interest expense related to convertible notes payable to third parties and related parties was $317,543 and $156,789, respectively. |
| (c) | We have not reflected any pro forma adjustments to reflect the tax impact of the pro forma adjustments, as we believe that the tax impact would not be material. |
| (d) | Reflects the preliminary fair value adjustment of $[ ] for intangible assets. |
| (e) | Reflects the preliminary estimate of goodwill, which represents the excess of the purchase price over the fair value of Moleculin’s identifiable assets acquired and liabilities assumed as shown in Note 2. The stock offering price is assumed to be $5.50 per share. A difference in our offering price would change the amount of goodwill created under the proposed transaction. If the offering price goes up, goodwill will increase and if the offering price goes down, goodwill will decrease. There are no other impacts to the pro forma financial statements. |
| (f) | Reflects the settlement of Moleculin’s convertible notes outstanding as of September 30, 2015 and associated accrued interest by issuing 293,393 shares of the Company’s common stock, which include 270,617 shares for the conversion of note principal and 22,776 shares for the accrued interest associated with the convertible notes. |
| (g) | Reflects the payment of a note with $55,774 cash upon the Company’s initial public offering and the receipt of $365,000 cash for notes issued subsequent to September 30, 2015. The $365,000 notes are automatically converted into the 1,825,000 common shares upon the IPO. |
| (h) | Reflects the issuance of 706,607 common shares of MBI at the price of our common stock to be sold in this offering (currently assumed at $5.50 per share) to the members of Moleculin. The adjustment also reflects the elimination of Moleculin’s members’ equity accounts. |
| (i) | Common unit payable represents common units of Moleculin issuable to MD Anderson pursuant to an agreement entered on June 21, 2010. On October 8, 2015, the Company entered into a letter agreement with MD Anderson where MD Anderson forfeited its right to the common units. Moleculin recorded a gain related to the forfeiture in October 2015. |
| (j) | Reflects issuance of 1,924,557 common shares for the conversion of $250,000 convertible notes and associated accrued interest upon the IPO. The adjustment does not include shares issuable for accrued interest owed to the noteholders on the date of our IPO. |
| (k) | Reflects net proceeds from the minimum offering. |
| (l) | Reflects the issuance of 629,000 common shares of MBI at the price of our common stock to be sold in this offering (currently assumed at $5.50 per share) to HPI according to a co-development agreement. |
36
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
This Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) contains certain forward-looking statements. Historical results may not indicate future performance. Our forward-looking statements reflect our current views about future events, are based on assumptions and are subject to known and unknown risks and uncertainties that could cause actual results to differ materially from those contemplated by these statements. Factors that may cause differences between actual results and those contemplated by forward-looking statements include, but are not limited to, those discussed in “Risk Factors.” We undertake no obligation to publicly update or revise any forward-looking statements, including any changes that might result from any facts, events, or circumstances after the date hereof that may bear upon forward-looking statements. Furthermore, we cannot guarantee future results, events, levels of activity, performance, or achievements.
Highlights
We are a preclinical and clinical-stage pharmaceutical development company organized as a Delaware corporation in July 2015 to focus on the development of anti-cancer drug candidates. We have three active drug development projects. Our lead drug candidate is liposomal Annamycin, which is referred to as Annamycin, an anthracyline intended for the treatment of relapsed or refractory acute myeloid leukemia, or AML. Annamycin has been in clinical trials pursuant to an investigational new drug application, or IND, that had been filed with the U.S. Food and Drug Administration, or FDA. Due to a lack of development activity by a prior drug developer, this IND was terminated, however we intend to apply for a new IND based on the same data that supported the original IND, updated for subsequent clinical data, and to commence a Phase II clinical trial for Annamycin.
We have two other active drug development projects in progress. One of them involves a collection of small molecules we refer to as the WP1066 Portfolio that will be obtained via our merger with Moleculin, LLC (“Moleculin”) and is focused on the modulation of key regulatory transcription factors involved in the progression of cancer. The other, which we call the WP1122 Portfolio, is a suite of molecules targeting the metabolic processes involved in cancer in general, and glioblastoma in particular that we acquired from IntertechBio Corporation. Both of these technologies are licensed on a worldwide exclusive basis from The University of Texas M.D. Anderson Cancer Center, or MD Anderson.
Overview
MBI was founded in 2015 in order to combine and consolidate the development efforts involving several MD Anderson anti-cancer technologies. This effort began with the acquisition of the Annamycin development project from AnnaMed, Inc., or AnnaMed, followed by the acquisition of the license rights to the WP1122 Portfolio from IntertechBio Corporation, or IntertechBio. Further, we began negotiations to create a co-development agreement with Houston Pharmaceuticals, Inc., or HPI, which will culminate with the merger of Moleculin and MBI coincident with this offering allowing us to gain control of the WP1066 Portfolio.
AnnaMed was formed in 2012 to take over the development of Annamycin from a prior drug development company, Callisto Pharmaceuticals, Inc., or Callisto. Callisto ceased development work on Annamycin leading to the termination of its IND by the FDA. In order to satisfy unmet license obligations, Callisto agreed to transfer all available Annamycin data to AnnaMed, which data we will now use to apply for a new IND.
IntertechBio was formed in 2009 to license and begin development on the WP1122 Portfolio. In August 2015, IntertechBio agreed to assign all license rights to us in exchange for our common stock.
Moleculin was formed in 2006 and has been working to develop the WP1066 Portfolio it licensed from MD Anderson. Prior to the effective date of the registration statement of which this prospectus is a part, Moleculin will be merged with and into MBI. As a result of the merger, we will issue the holders of Moleculin LLC equity interests an aggregate of 1,000,000 shares of our common stock.
Since Moleculin commenced operations in 2006, substantially all of its efforts have been focused on research, development and the advancement of our WP1066 Portfolio. Moleculin has not generated any revenue from product sales and, as a result, has incurred significant losses. Moleculin incurred a net loss of $1.8 million and $3.5 million for the years ended December 31, 2014 and 2013, respectively, and net losses of $0.9 million and $1.0 million for the nine months ended September 30, 2015 and 2014, respectively. We anticipate these losses will increase as we continue our development of, seek regulatory approval for, and, if approved, begin to commercialize Annamycin, and as we develop other product candidates.
37
Neither Moleculin nor MBI has manufacturing facilities and all manufacturing activities are contracted out to third parties. Additionally, Moleculin currently utilizes third-party clinical research organizations to carry out clinical trials. Neither Moleculin nor MBI have a sales organization.
JOBS Act and Recent Accounting Pronouncements
The recently enacted JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies.
We have implemented all new accounting pronouncements that are in effect and may impact our financial statements and we do not believe that there are any other new accounting pronouncements that have been issued that might have a material impact on our financial position or results of operations.
Critical Accounting Policies and Significant Judgments and Estimates
The financial statements have been prepared in accordance with generally accepted accounting principles in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe that the following accounting policies are the most critical to aid in fully understanding and evaluating our reported financial results, and they require our most difficult, subjective or complex judgments, resulting from the need to make estimates about the effect of matters that are inherently uncertain.
Beneficial Conversion Feature
From time to time, we may issue convertible notes that have conversion prices that create an embedded beneficial conversion feature on the issuance date. A beneficial conversion feature exists on the date a convertible note is issued when the fair value of the underlying common stock to which the note is convertible into is in excess of the remaining unallocated proceeds of the note after first considering the allocation of a portion of the note proceeds to the fair value of any attached equity instruments, if any related equity instruments were granted with the debt. We estimate the fair value of our common stock using the most recent selling price available. The intrinsic value of the beneficial conversion feature is recorded as a debt discount with a corresponding amount to additional paid-in capital. The debt discount is amortized to interest expense over the life of the note using the effective interest method.
Research and Development Costs
We record accrued expenses for estimated costs of our research and development activities conducted by third-party service providers, which include the conduct of pre-clinical studies and clinical trials and contract manufacturing activities. We record the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced, and we include these costs in accrued liabilities in the balance sheets and within research and development expense in the statement of operations. These costs are a significant component of our research and development expenses. We record accrued expenses for these costs based on the estimated amount of work completed and in accordance with agreements established with these third parties.
We estimate the amount of work completed through discussions with internal personnel and external service providers as to the progress or stage of completion of the services and the agreed-upon fee to be paid for such services. We make significant judgments and estimates in determining the accrued balance in each reporting period. As actual costs become known, we adjust our accrued estimates. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed, the number of patients enrolled and the rate of patient enrollment may vary from our estimates and could result in us reporting amounts that are too high or too low in any particular period. Our accrued expenses are dependent, in part, upon the receipt of timely and accurate reporting from clinical research organizations and other third-party service providers. To date, there have been no material differences from our accrued expenses to actual expenses.
Income Taxes
Moleculin elected to be taxed as a partnership under the Internal Revenue Code. Moleculin pays no U.S. taxes on its earnings. Moleculin’s net earnings/losses are passed through to its members and, as such, it reports no income tax expense or liability.
38
Impairment of Long-Lived Assets
Management reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount may not be realizable or at a minimum annually during the fourth quarter of the year. If an evaluation is required, the estimated future undiscounted cash flows associated with the asset are compared to the asset’s carrying value to determine if an impairment of such asset is necessary. The effect of any impairment would be to expense the difference between the fair value of such asset and its carrying value.
Components of our Results of Operations and Financial Condition
Operating expenses
We classify our operating expenses into three categories: research and development, general and administrative and depreciation.
Research and development. Research and development expenses consist primarily of:
| • | costs incurred to conduct research, such as the discovery and development of our product candidates; |
| • | costs related to production of clinical supplies, including fees paid to contract manufacturers; |
| • | fees paid to clinical consultants, clinical trial sites and vendors, including clinical research organizations in conjunction with implementing and monitoring our clinical trials and acquiring and evaluating clinical trial data, including all related fees, such as patient screening fees, laboratory work and statistical compilation and analysis; and |
| • | costs related to compliance with drug development regulatory requirements. |
We recognize all research and development costs as they are incurred. Clinical trial costs, contract manufacturing and other development costs incurred by third parties are expensed as the contracted work is performed.
We expect our research and development expenses to increase in the future as we advance our product candidates into and through clinical trials and pursue regulatory approval of our product candidates in the United States and Europe. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming. The actual probability of success for our product candidates may be affected by a variety of factors including: the quality of our product candidates, early clinical data, investment in our clinical program, competition, manufacturing capability and commercial viability. We may never succeed in achieving regulatory approval for any of our product candidates. As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenue from the commercialization and sale of our product candidates.
General and administrative. General and administrative expense consists of personnel related costs, which include salaries, as well as the costs of professional services, such as accounting and legal, facilities, information technology and other administrative expenses. We expect our general and administrative expense to increase following the completion of this offering due to the anticipated growth of our business and related infrastructure as well as accounting, insurance, investor relations and other costs associated with becoming a public company.
Depreciation. Depreciation expense consists of depreciation on our property and equipment. We depreciate our assets over their estimated useful life. We estimate machinery and equipment to have a 5-year life and furniture and fixtures to have a 7-year life.
Other income (expense), net
Other income (expense), net consists of interest expense associated with our notes payable and interest income earned on our cash and investment balances.
Statements of Operations - MBI
The following table sets forth the components of MBI’s statements of operations for the period from July 28, 2015 (Inception) to September 30, 2015.
| From July 28, 2015 (Inception) to September 30, 2015 | ||||
| Operating expenses: | ||||
| Research and development | $ | 38,409 | ||
| General and administrative | 184,344 | |||
| Total operating expenses | 222,753 | |||
| Interest expense | (1,562 | ) | ||
| Net loss | $ | (224,315 | ) | |
Research and Development Expense. Research and development expense was $38,409 for the period from July 28, 2015 (Inception) to September 30, 2015. The expense is mainly related to patent prosecution fees and travel expense of our research and development personnel.
General and Administrative Expense. General and administrative expense was $184,344 for the period from July 28, 2015 (Inception) to September 30, 2015. The expense mainly included professional fees to our consultants, attorney, accountants and our previous investment banker and financial advisor for services related to our initial public offering.
Interest expense. Interest expense included expense accrued on our convertible promissory notes issued in August and September 2015 bearing interest at the rate of 8% per annum.
Statements of Operations - Moleculin
The following table sets forth the components of Moleculin’s unaudited statements of operations for the nine-month periods ended September 30, 2015 and 2014.
39
| Nine Months Ended September 30, | ||||||||||||
| 2015 | 2014 | Change | ||||||||||
| Operating expenses: | ||||||||||||
| Research and development | $ | 270,991 | $ | 238,467 | $ | 32,524 | ||||||
| General and administrative | 267,423 | 649,344 | (381,921 | ) | ||||||||
| Depreciation expense | 8,469 | 8,254 | 215 | |||||||||
| Total operating expenses | 546,883 | 896,065 | (349,182 | ) | ||||||||
| Other expense | (334,338 | ) | (55,011 | ) | (279,327 | ) | ||||||
| Net loss | $ | (881,221 | ) | $ | (951,076 | ) | $ | 69,855 | ||||
Research and Development Expense. Research and development expense was approximately $271,000 for the nine months ended September 30, 2015 as compared to $238,000 for the same period in the prior year. This increase of $33,000 was driven primarily by clinical trial costs incurred during the first nine months of 2015 as compared to the same period in 2014.
General and Administrative Expense. General and administrative expense for the nine months ended September 30, 2015 was $267,000 as compared to $649,000 for the same period in the prior year. This $382,000 decrease in general and administrative expense related primarily to a reduction in payroll and related personnel costs of approximately $245,000 and a reduction in professional services fees of $109,000.
Depreciation Expense. Depreciation expense was $8,000 for each of the nine months ended September 30, 2015 and 2014. Depreciation is generated from our fixed assets.
Other expense. Other expense for the nine months ended September 30, 2015 was approximately $334,000 as compared to other expense of $55,000 for the same period in the prior year. This increase of $279,000 in expense related primarily to an increase of $279,000 in interest expense accrued on our convertible promissory notes issued late in 2014 and amortization of debt discount and deferred financing cost associated with those notes.
The following table sets forth the components of Moleculin’s audited statements of operations for the years ended December 31, 2014 and 2013.
| 2014 | 2013 | Change | ||||||||||
| Operating expenses: | ||||||||||||
| Research and development | $ | 798,785 | $ | 2,341,676 | $ | (1,542,891 | ) | |||||
| General and administrative | 839,556 | 1,161,282 | (321,726 | ) | ||||||||
| Depreciation expense | 11,005 | 11,005 | - | |||||||||
| Total operating expenses | 1,649,346 | 3,513,963 | (1,864,617 | ) | ||||||||
| Other income (expense) | (159,740 | ) | 849 | (160,589 | ) | |||||||
| Net loss | $ | (1,809,086 | ) | $ | (3,513,114 | ) | $ | 1,704,028 | ||||
Research and Development Expense. Research and development expense consists primarily of the discovery and development of our product candidates; costs related to production of clinical supplies, including fees paid to contract manufacturers, fees paid to clinical consultants, clinical trial sites and vendors, including clinical research organizations in conjunction with implementing and monitoring our clinical trials and acquiring and evaluating clinical trial data; and costs related to compliance with drug development regulatory requirements.
40
Research and development expense was approximately $799,000 for the year ended December 31, 2014 as compared to $2.3 million for the same period in the prior year. This decrease of over $1.5 million was driven primarily by a reduction in clinical trial costs in 2014 of approximately $368,000; a decrease in consulting costs to comply with regulatory requirements of $163,000; a decrease of approximately $121,000 in manufacturing costs for clinical supplies; and a decrease in research costs of approximately $864,000. This decrease in research costs is due primarily to the expense related to a stock issuance to MD Anderson in 2003 in accordance with Moleculin’s license agreement entered in 2010, which was not repeated in 2014, as well as the cost of a sponsored research agreement with MD Anderson recognized in 2013.
General and Administrative Expense. General and administrative expense primarily consists of personnel costs, travel, information technology, facilities, and professional service fees. Professional services fees primarily consist of legal, accounting and consulting costs.
General and administrative expense for the year ended December 31, 2014 was approximately $840,000 as compared to approximately $1.2 million for the same period in the prior year. The $322,000 decrease in general and administrative expense is related primarily to a reduction in payroll and related personnel costs of $78,000, a reduction in travel expenses of $92,000, and a reduction in professional services fees of $87,000.
We expect general and administrative expense to increase in future periods due to additional legal, accounting, insurance, investor relations and other costs associated with being a public company.
Depreciation Expense. Depreciation expense was approximately $11,000 for each of the years ended December 31, 2014 and 2013.
Other income (expense). Other expense for the year ended December 31, 2014 was $160,000 as compared to other income of $1,000 for the same period in the prior year. This increase of $161,000 in expense relates primarily to interest accrued on our convertible promissory notes issued in 2014.
Liquidity and Capital Resources
Since Moleculin’s inception and through September 30, 2015, Moleculin has funded its operations primarily through the sale and issuance of convertible preferred units and convertible and non-convertible promissory notes. During 2013, Moleculin sold 2,019,116 units of Preferred Series A-4 at $1 per unit to various members. From May to November 2014, Moleculin issued various convertible notes to its creditors. The note proceeds were $1.5 million. These notes bear interest at 8% per annum and are due on the earlier of June 30, 2016 or the consummation of a liquidation event (which event will include the merger between MBI and Moleculin to be completed prior to the effective date of the registration statement of which this prospectus forms a part), unless these notes are converted.
As of September 30, 2015, Moleculin had approximately $298,000 in cash and MBI had approximately $87,000 in cash.
In August and September 2015, we issued two 8% convertible notes in an aggregate of $250,000 in principal amount of convertible notes to one investor, which principal and accrued interest will automatically convert into shares of common stock upon the closing of this offering at a conversion rate of $0.1299 per share. In October 2015, we issued two 8% convertible notes in an aggregate of $200,000 in principal amount of convertible notes to two investors, which principal and accrued interest will automatically convert into shares of common stock upon the closing of this offering at a conversion rate of $0.20 per share. At the time of such purchase in October 2015, the convertible note investors and we agreed that the investors would fund us up to the filing of our initial non-confidential registration statement an additional $165,000 on the same terms as in the October financing. Pursuant to such agreement, such amount was received in January 2016.
None of the foregoing convertible notes will be convertible by the holder of such notes to the extent (and only to the extent) that the holder or any of its affiliates would beneficially own in excess of 4.99% of our common stock. For purposes of the limitation described in this paragraph, beneficial ownership and all determinations and calculations are determined in accordance with Section 13(d) of the Exchange Act and the rules and regulations promulgated thereunder.
As part of the acquisition process of Moleculin LLC by MBI, we will provide operating loans for Moleculin in the estimated range of $300,000 to $310,000 for Moleculin to meet its license obligations with MD Anderson, and maintain its current level of operations. These loans will be in the form of promissory notes payable with an interest rate of 8% from Moleculin to us that will be forgiven at the completion of the IPO. As of the date hereof, MBI has secured a total of $615,000 of convertible notes and estimates that the company will need approximately an additional $535,000 to $635,000 to complete the IPO process.
In January and February 2016, we sold 53,000 shares of our common stock at a purchase price per share of $3.00 in a private placement.
We believe that our and Moleculin’s existing cash and cash equivalents, our remaining proceeds from our recent private placement of 8% unsecured promissory notes and shares of common stock, together with the net proceeds from this offering, will be sufficient to fund our planned operations for approximately 16 months, assuming completion of the maximum offering, and 12 months, assuming completion of the minimum offering, including through preliminary data readout of our planned Phase II registration trial for Annamycin.
41
We do not expect to generate revenue from product sales unless and until we successfully complete development of, obtain regulatory approval for and begin to commercialize one or more of our product candidates, which we expect will take a number of years and is subject to significant uncertainty. Accordingly, we anticipate that we will need to raise additional capital to fund our future operations. Until such time that we can generate substantial revenue from product sales, if ever, we expect to finance our operating activities through a combination of equity offerings and debt financings and we may seek to raise additional capital through strategic collaborations. However, we may be unable to raise additional funds or enter into such arrangements when needed on favorable terms, or at all, which would have a negative impact on our financial condition and could force us to delay, limit, reduce or terminate our development programs or commercialization efforts or grant to others rights to develop or market product candidates that we would otherwise prefer to develop and market ourselves. Failure to receive additional funding could cause us to cease operations, in part or in full. Furthermore, even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital due to favorable market conditions or strategic considerations.
The following table sets forth the primary sources and uses of cash for the periods indicated for Moleculin:
| Nine months ended September 30, | Year ended December 31, | |||||||||||||||||||||||
| 2015 | 2014 | Change | 2014 | 2013 | Change | |||||||||||||||||||
| (Restated) | ||||||||||||||||||||||||
| Net Cash Used in Operating Activities | $ | (458,427 | ) | $ | (878,722 | ) | $ | 420,295 | $ | (1,354,145 | ) | $ | (2,617,402 | ) | $ | 1,263,257 | ||||||||
| Net Cash Provided by Investing Activities | 232,300 | - | 232,300 | - | - | - | ||||||||||||||||||
| Net Cash Provided by Financing Activities | - | 782,756 | (782,756 | ) | 1,525,256 | 2,019,116 | (493,860 | ) | ||||||||||||||||
| Net increase (decrease) in cash and cash equivalents | $ | (226,127 | ) | $ | (95,966 | ) | $ | (130,161 | ) | $ | 171,111 | $ | (598,286 | ) | $ | 769,397 | ||||||||
Cash used in operating activities
Net cash used in operating activities was approximately $458,000 and $879,000 for the nine months ended September 30, 2015 and 2014, respectively. The $421,000 decrease in net cash used in operating activities was primarily due to a decrease in spending in research and development, specifically in sponsored research performed by MD Anderson, as well as decrease in payments made to professionals and employees.
Net cash used in operating activities was approximately $1,354,000 and $2,617,000 for the years ended December 31, 2014 and 2013, respectively. The $1,263,000 decrease in net cash used in operating activities was primarily due to decreases in spending in research and development of $1.0 million, specifically in research performed by MD Anderson, and in general and administrative of $0.3 million attributable to reductions in payroll and personnel related costs.
Cash provided by investing activities
Net cash provided by investing activities was approximately $232,000 and $0 for the nine months ended September 30, 2015 and 2014, respectively. The increase was due to a collection of long-term receivable from Dermin, Sp. Zo. O. to whom the Company had provided funding assistance for Dermin’s efforts in developing and commercializing certain pharmaceutical products.
Cash provided by financing activities
Net cash provided by financing activities was approximately $783,000 for the nine months ended September 30, 2014. Cash was provided by issuance of convertible notes payable in 2014 of approximately $783,000. There were no financing activities during the nine months ended September 30, 2015.
Net cash provided by financing activities was approximately $1,525,000 and $2,019,000 for the years ended December 31, 2014 and 2013, respectively. The $493,000 decrease in net cash provided by financing activities was primarily due to cash provided by issuance of preferred units in 2013 of $2.0 million, offset by cash provided in issuance of convertible notes payable in 2014 of $1.5 million.
42
Overview
We are a preclinical and clinical-stage, pharmaceutical company focused on the development of anti-cancer drug candidates, many of which are based on license agreements with The University of Texas System on behalf of the M.D. Anderson Cancer Center, which we refer to as MD Anderson. Our lead drug candidate is liposomal Annamycin, which we refer to as Annamycin, an anthracycline intended for the treatment of relapsed or refractory acute myeloid leukemia, or AML. Annamycin has been in clinical trials pursuant to an Investigational New Drug application, or IND, that had been filed with the FDA. Due to a lack of development activity by a prior drug developer, this IND was terminated. However we intend to apply for a new IND based on the same data that supported the original IND, updated for subsequent clinical data and to commence a Phase II clinical trial for Annamycin with the proceeds from this offering. We have two other drug development projects in process, one involving a collection of small molecules, which we refer to as the WP1066 Portfolio, focused on the modulation of key regulatory transcription factors involved in the progression of cancer, and the WP1222 Portfolio, a suite of molecules targeting the metabolic processes involved in cancer in general and glioblastoma (the most common form of brain tumor) in particular. We also intend to sponsor ongoing research at MD Anderson in order to improve and expand our drug development pipeline.
We have been granted royalty-bearing, worldwide, exclusive licenses for the patent and technology rights related to our WP1066 Portfolio and WP1122 Portfolio drug technologies, as these patent rights are owned by MD Anderson. The Annamycin drug substance is no longer covered by any existing patent protection. We intend to submit patent applications for formulation, synthetic process and reconstitution related to our Annamycin drug product candidate, although there is no assurance that we will be successful in obtaining such patent protection. Independently from potential patent protection, we believe Annamycin will qualify for Orphan Drug status, which would entitle us to market exclusivity of 7 and 10 years from the date of approval of a New Drug Application (NDA) and Marketing Authorization (MA), in the United States and the European Union (EU), respectively. Separately, the FDA may also grant market exclusivity of up to 5 years for newly approved new chemical entities (of which Annamycin would be one), but there can be no assurance that such exclusivity will be granted or, if granted, for how long.
Corporate History
We were founded in 2015 by Walter Klemp (our chairman and acting CEO), Dr. Don Picker (our President and Chief Operating Officer) and Dr. Waldemar Priebe of MD Anderson (Chairman of our Scientific Advisory Board) in order to combine and consolidate development efforts involving several MD Anderson anti-cancer technologies. This effort began with the acquisition of the Annamycin development project from AnnaMed, Inc., or AnnaMed, followed by the acquisition of the license rights to the WP1122 Portfolio from IntertechBio Corporation, or IntertechBio. Further, we began negotiations to create a co-development agreement with Houston Pharmaceuticals, Inc., or HPI, which will culminate with the merger of Moleculin and MBI coincident with this offering allowing us to gain control of the WP1066 Portfolio.
AnnaMed was formed in 2012 to take over the development of Annamycin from a prior drug development company, Callisto Pharmaceuticals, Inc., or Callisto. Callisto ceased development work on Annamycin, leading to the termination of its IND by the FDA. In order to satisfy unmet license obligations, Callisto agreed to transfer all available Annamycin data to AnnaMed, which data we will use to apply for a new IND. In 2012, AnnaMed out-licensed development rights in a limited territory to a Polish special purpose drug development company called Dermin in exchange for Dermin’s development work based on its successful effort to obtain Polish government grant funding to assist in the development of Annamycin. Since that time, such grant funding has been used to produce Annamycin in preparation for future clinical trials. In August 2015, we entered into a rights transfer agreement with AnnaMed pursuant to which in exchange for 1,431,000 shares of our common stock AnnaMed agreed to transfer any and all data it had regarding the development of Annamycin and the Annamycin IND, including all trade secrets, know-how, confidential information and other intellectual property rights held by AnnaMed.
IntertechBio was formed in 2009 to license and begin development on the WP1122 Portfolio. The WP1122 Portfolio was also out-licensed to Dermin, which was awarded a Polish government grant to assist in drug development efforts. In August 2015, IntertechBio agreed to assign all license rights to us in exchange for our common stock.
Moleculin was formed in 2006 and has been working to develop the WP1066 Portfolio it licensed from MD Anderson. As a part of the formation of Moleculin, an agreement was reached with HPI to limit Moleculin’s development efforts to uses in dermatology only, leaving non-dermatology indications to HPI. From 2006 to 2014, Moleculin raised a total of approximately $11 million through the sale of equity and debt securities and an additional $2 million through an out-licensing agreement with a Japanese dermatology drug development company, Maruho, Ltd., or Maruho. Since 2012, Moleculin has conducted a series of clinical trials focused on the topical treatment of psoriasis. Although those trials have demonstrated drug activity, the results to date are not conclusive enough to warrant full commercialization as a topical dermatology drug. Additional study is required to determine optimal dosing and scheduling regimens for the topical treatment of psoriatic plaques. As a result of this additional complexity with regard to the potential treatment of psoriasis, Maruho did not elect to continue its funding of the psoriasis drug development effort and, therefore, forfeited their license rights to the WP1066 Portfolio that had been granted to it by Moleculin. Dermin is planning to clinically evaluate a candidate from the WP1066 Portfolio for the topical treatment of cutaneous T-cell lymphoma, or CTCL. Moleculin does not have control of the clinical plan or timeline for Dermin’s development effort.
43
Prior to the effective date of the registration statement of which this prospectus is a part, Moleculin will be merged with and into our company. As a result of the merger, we will issue the equity interests holders of Moleculin an aggregate of 1,000,000 shares of our common stock. The merger agreement contains mutual representations and warranties between the parties. Pursuant to the merger agreement, we will agree for a period of six years to indemnify and hold harmless each present and former director and/or officer of Moleculin whom Moleculin would have had the power to indemnify under Delaware that is made a party or threatened to be made a party to any threatened, pending or completed proceeding or claim by reason of the fact that he or she was a director or officer of the Moleculin prior to the effective time of the merger and arising out of actions or omissions of the indemnified party in any such capacity occurring at or prior to the effective time of the merger against any losses or damages reasonably incurred in connection with any claim. To our knowledge, no such proceeding or claim exists or has been threatened on the date hereof and Moleculin has made representations to this effect in the merger agreement. As additional consideration payable to the Moleculin unit holders, we will agree pursuant to the merger agreement that if drugs for dermatology indications are successfully developed by us (or our successors) using any of the Existing IP Assets, then the Moleculin unit holders, in the aggregate, will be entitled to receive a 2.5% royalty on the net revenues generated by such drugs. Any such net revenues would include a deduction for license fees or royalty obligations payable to MD Anderson for such Existing IP Assets. The merger agreement will define “Existing IP Assets” to mean all intellectual property, licensed by us and Moleculin as of the time of the merger, including, without limitation, the intellectual property licensed from MD Anderson under the Patent and Technology License Agreement entered into by and between IntertechBio Corporation and MD Anderson dated April 2, 2012, as amended, and the Patent and Technology License Agreement dated June 21, 2010, as amended, between MD Anderson and Moleculin, but excluding any intellectual property relating to Annamycin. The right to receive the contingent royalty payments described herein are for drugs developed only for dermatology indications, and do not include drugs developed for any other indications. We have no obligation of any nature to pursue the development of any drugs for dermatology indications.
From 2006 through 2015, HPI has been engaged in research related to the use of the WP1066 Portfolio for the treatment of non-dermatology cancers and has received grant awards totaling approximately $4.9 million toward this effort. Prior to the effective date of the registration statement of which this prospectus is a part, we will enter into a co-development agreement with HPI whereby HPI will continue its grant-funded research and make all resulting data available for our use in exchange for a development fee. We may buy HPI out of its co-development rights in the WP1066 Portfolio at our option. Please see the section “Business – License Agreements” for a description of our proposed agreement with HPI.
Neither our founders nor MBI have or will have any ownership stake in Dermin, our Polish grant-funded licensee. No Dermin-related grant money is expected to be paid directly to us, but rather the sublicense agreements require Dermin to share resulting data, which we believe will reduce or eliminate costs we might otherwise have to incur directly. There can be no assurance, however, that Dermin will continue to receive the funds they have been awarded or that such funds will be spent by Dermin in a manner that will benefit MBI. The sublicensed territories granted to Dermin for the WP1066 Portfolio are Poland, Ukraine, Czech Republic, Hungary, Romania, Slovakia, Belarus, Lithuania, Latvia and Estonia. For the WP1122 Portfolio, the territory expanded to include Russia and Kazakhstan. For Annamycin, the territory is further expanded to include Netherlands, Turkey, Belgium, Switzerland, Austria, Sweden, Greece, Portugal, Norway, Denmark, Ireland, Finland, Luxembourg, Iceland, Uzbekistan, Georgia, Armenia, Azerbaijan and Germany. However, in the case of Germany, this territory may be reclaimed by us for a payment of $500,000 to Dermin.
The following summarizes the transactional history and common relationships among HPI, IntertechBio, Moleculin, MBI, Callisto, AnnaMed, MD Anderson, and our officers and major shareholders:
| · | Houston Pharmaceuticals, Inc.: Prior to the effective date of the registration statement of which this prospectus is a part, we will enter into a co-development agreement with HPI whereby HPI will continue its grant-funded research and make all resulting data available for our use in exchange for a development fee. Waldemar Priebe, chairman of our Scientific Advisory Board, and Don Picker, our president and chief operating officer, are shareholders of HPI, and Dr. Priebe has the voting and dispositive power over the shares held by HPI. |
| · | IntertechBio Corporation: In August 2015, in exchange for the issuance of 630,000 shares of common stock, we acquired the rights to the license agreement with MD Anderson covering our WP1122 Portfolio held by IntertechBio Corporation. Waldemar Priebe, chairman of our Scientific Advisory Board, and Don Picker, our president and chief operating officer, are shareholders of IntertechBio and control the voting and dispositive power over the shares held by IntertechBio. |
| · | Moleculin LLC: Prior to the effective date of the registration statement of which this prospectus is a part, Moleculin will be merged with and into our company. Moleculin is the holder of the license agreement with MD Anderson covering our WP 1066 Portfolio. As a result of the merger, we will issue the equity interests holders of Moleculin an aggregate of 1,000,000 shares of our common stock. Waldemar Priebe, chairman of our Scientific Advisory Board, Walter Klemp, our chairman and acting chief executive officer, and Don Picker, our president and chief operating officer, are members of Moleculin and will receive 6,046 shares, 22,795 shares and 6,046 shares, respectively, of our common stock as a result of the merger. In addition, Walter Klemp and Don Picker are members of the board of Moleculin. |
| · | AnnaMed, Inc.: In August 2015, in exchange for 1,431,000 shares of our common stock, we acquired the rights to the Annamycin data related to the original Annamycin IND and the development of Annamycin held by AnnaMed, Inc., a company controlled by Walter Klemp, our chairman and acting chief executive officer. |
| · | Callisto Pharmaceuticals, Inc.: Our president and chief operating officer, Don Picker and our chief medical officer, Robert Shepard, were chief operating officer and chief medical officer, respectively, of Callisto. Since 2009, neither individual has had any relationship or ownership with Callisto. |
| · | MD Anderson: Both Moleculin and IntertechBio have entered into license agreements with MD Anderson. See “Business – Our Licenses Agreements” below. Waldemar Priebe, chairman of our Scientific Advisory Board, is a Professor of Medicinal Chemistry, Department of Experimental Therapeutics, Division of Cancer Medicine, at the University of Texas MD Anderson Cancer Center. |
44
Our Drug Candidates
Annamycin
Our lead product candidate is Annamycin. We intend to use the proceeds from this offering to conduct a Phase II clinical trial for Annamycin as a monotherapy for the treatment of relapsed or refractory acute myeloid leukemia, or AML.
Market for Annamycin
Leukemia is a cancer of the white blood cells and acute forms of leukemia can manifest quickly and leave patients with limited treatment options. AML is the most common type of acute leukemia in adults. It occurs when a clone of leukemic progenitor white blood cells proliferates in the bone marrow, suppressing the production of normal blood cells. Currently, the only viable option for acute leukemia patients is a bone marrow transplant, which is successful in a significant number of patients. However, in order to qualify for a bone marrow transplant, the patient’s leukemia cells must be decreased to a sufficiently low level. This begins with a therapy of combining two chemotherapeutic drugs, which always includes an anthracycline, in inducing remission (a complete response, or CR), which therapy has not improved since it was first used in the 1970s and we estimate that this induction therapy has the same cure rate of about 20% as at that time. Unfortunately, the current clinically approved anthracyclines are cardiotoxic, which can result in damage to the heart and limit the dosage amount that may be administered to patients. Additionally, the tumor cells often present de novo or developed resistance to the first line anthracycline, often through what is called “multidrug resistance”, enabling them to purge themselves of the available anthracyclines. Consequently, there remains no effective therapy for these patients and unfortunately most will succumb quickly to their leukemia. If a patient’s leukemia reappears before they can be prepared for a bone marrow transplant, they are considered to have “relapsed”. If a patient fails to achieve a sufficient response from the induction therapy to qualify for a bone marrow transplant, they are considered to be “refractory” (resistant to therapy). Together, this group of relapsed and refractory AML patients constitutes our primary focus for treatment with Annamycin and our intent is to pursue FDA approval for Annamycin as a second-line induction therapy for adult relapsed and refractory AML patients.
45
We believe that pursuing approval as a second line induction therapy for adult relapsed or refractory AML patients is the shortest path to regulatory approval, but we also believe that one of the most important potential uses of Annamycin is in the treatment of children with either AML or ALL (acute lymphoblastic leukemia, which is more common in children). Accordingly, we also intend to pursue approval for pediatric use when practicable.
Of the estimated 18,860 U.S. cases of acute myeloid leukemia diagnosed in 2014, an estimated 97% were adult and although exact numbers are not available, we estimate that 70% to 80% (approximately 13,000 to 14,000 patients) were expected to relapse or be resistant to first-line therapy.
Prior Clinical Trials for Annamycin
Annamycin is a unique liposome formulated anthracycline (also referred to in literature as “L-Annamycin”). It has been tested in 6 clinical trials and 114 patients without any reported cardiotoxicity and, in the two clinical trials focused on leukemia, with fewer dose-limiting toxicities than are normally expected with doxorubicin (one of the leading first-line anthracyclines used for induction therapy). Each of these trials was conducted by Callisto Pharmaceuticals, Inc., the previous holder of the intellectual property surrounding Annamycin, and not by our company. Annamycin demonstrated efficacy in 8 of 16 patients in a Phase I study in adult relapsed or refractory AML patients, with 6 of 14 patients completely clearing leukemic blasts. A 30 patient dose-ranging Phase I/II study in ALL demonstrated a similar efficacy profile, with 3 of 10 patients treated with the maximum tolerable dose clearing their leukemic blasts to a level sufficient to qualify for a bone marrow transplant. One of these patients went on to receive a successful curative bone marrow transplant. The other two of these three patients died of tumor lysis syndrome, a condition resulting from the overloading of their system with the debris from leukemic blast cells destroyed by the induction therapy. Armed with the knowledge of this potential, prophylactic pretreatment known to protect patients from the effects of tumor lysis syndrome will be deployed in future trials. Based on the results of the above clinical trials, we believe Annamycin is different from currently approved induction therapy drugs in four key ways: (i) it has demonstrated clinical activity in a patient population for whom there are currently no effective therapies, (ii) it appears to be capable of avoiding the “multi-drug resistance” mechanisms that often limit the effectiveness of currently approved anthracyclines; (iii) it has been shown to be non-cardiotoxic in animal models when compared with doxorubicin and no events of cardiotoxicity have been reported from the use of Annamycin in 114 patients; and (iv) in AML cell lines, it has been shown to be more potent than one of the leading approved drugs.
Lack of Cardiotoxicity
One of the key dose-limiting toxicities associated with currently available anthracyclines like doxorubicin is their propensity to induce life-threatening heart damage. This is especially problematic for pediatric leukemia patients whose life spans can be severely shortened by the very induction therapy designed to cure them of acute leukemia. In the animal model relied upon by the FDA as an indicator of human cardiotoxicity, the non-liposomal (free) form of Annamycin has been shown to be significantly less likely than doxorubicin to create heart lesions in mice and the liposomal formulation (L-Annamycin) has been shown in animal models to have reduced cardiotoxicity to the point where it is unlikely to cause harm to human patients. This lack of cardiotoxicity means L-Annamycin may be able to be used more aggressively in helping patients achieve remission without the potentially deadly long-term side effects of cardiotoxicity. This would be especially valuable in the case of pediatric acute leukemia (both AML and ALL) where long-term survival can be greatly impacted by cardiotoxicity.
Circumventing Multidrug Resistance
In addition to cardiotoxicity, the effectiveness of currently approved anthracyclines is limited by their propensity for succumbing to “multidrug resistance”, whereby transporters (one type of which is referred to as a “P-glycoprotein pump”) develop on the outer surface of cells to expel drugs like doxorubicin as a natural defense mechanism. The dosing of current therapies cannot be increased in an attempt to overcome multidrug resistance because of the likelihood of cardiotoxicity and other serious side effects. This limitation prevents adequate dosing of current anthracyclines to produce lasting remission. Annamycin appears to resist being expelled by P-glycoprotein pumps and other similar tested multidrug resistance transporters and thereby would seem to circumvent multidrug resistance. This characteristic has been shown in pre-clinical testing to allow for higher drug uptake in diseased cells, which we believe should allow for more effective induction therapy with less risk to the patient.
46
In order for anthracyclines to provide effective induction therapy, they must be allowed to accumulate in leukemic cells sufficiently to enter the cell’s nucleus where they damage the cell’s DNA and induce apoptosis (cell death). As induction therapy progresses, however, the targeted cells can develop a natural defense mechanism to prevent the anthracycline activity. The graphic below provides a simplified depiction of the formation of a P-glycoprotein pump on the outer surface (membrane) of a leukemic cell. As typical anthracyclines enter the cell, they are attracted to such pumps and are expelled (referred to as “efflux”) before they can accumulate sufficiently to serve their purpose. In contrast, Annamycin avoids such pumps and is allowed to accumulate sufficiently to destroy the leukemia cell despite the presence of the multidrug resistance mechanisms.
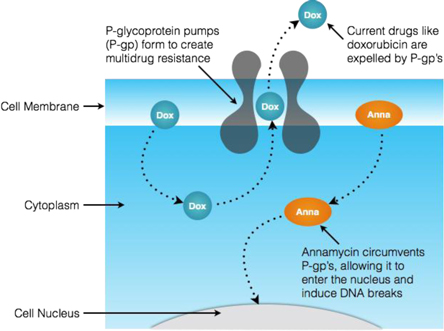
Based on initial conversations with the FDA, because of the serious unmet medical need, we believe Annamycin may qualify for a “Special Protocol Assessment” providing for accelerated approval based on our planned Phase II clinical trial. In order to negotiate the Special Protocol Assessment with the FDA, the dose-ranging Phase I/II clinical trial discussed above, which was conducted by a previous holder of the intellectual property surrounding Annamycin, must be independently audited, which is an expensive and time consuming process. In addition, since the original IND was terminated for lack of development activity by the prior drug developer, we must apply for a new IND, based on the data that supported the original IND updated for subsequent clinical activity. The ultimate negotiation with the FDA will determine the size and efficacy endpoints for the registration trial. We can provide no assurance that the audit of the most recent clinical trial will confirm the results reported by the prior drug developer or that we will be successful in obtaining a new IND or in negotiating a Special Protocol Assessment with the FDA, and if we are successful, we estimate such process, including auditing the prior clinical trial results, will take approximately six months or more from the closing of this offering.
We also intend to pursue Orphan Drug status for Annamycin. The prevalence ceiling for qualifying rare diseases under the US Orphan Drug Act is 200,000 patients and proportionally similar guidelines exist in the EU. The most recently published prevalence statistics from the National Cancer Institute reported that an estimated 37,726 patients had acute myeloid leukemia in the United States as of January 1, 2011, and trend data since that publication would indicate that the prevalence today should still be well below the 200,000 patient limitation for Orphan Drugs, qualifying Annamycin for the treatment of acute myeloid leukemia to qualify for Orphan Drug status. Annamycin already qualified for Orphan Drug status with its prior developer and we intend to repeat that process. However, we can provide no assurance that we will be successful in obtaining Orphan Drug status for Annamycin.
47
The WP1066 Portfolio
We have a license agreement with MD Anderson pursuant to which we have been granted a royalty-bearing, worldwide, exclusive license for the patent and technology rights related to our WP1066 Portfolio and its close analogs, molecules targeting the modulation of key oncogenic transcription factors.
Clinical Testing of WP1066 Portfolio
In vitro testing has shown a high level of activity for WP1066 against a wide range of solid tumors, and in vivo testing has shown significant activity against head and neck, pancreatic, stomach, and ovarian cancers, as well as metastatic melanoma and glioblastoma, among others. In vivo testing in mouse tumor models has shown that WP1066 inhibits tumor growth, blocks angiogenesis (a process that provides necessary blood supply to tumors) and increases survival.
With respect to our WP1066 Portfolio, we must complete pre-clinical toxicology testing, along with additional chemistry, manufacturing and control work to fully characterize the drug, establish the desired formulation and develop reference standards for future drug release, among others prior to submitting and application for IND. We currently expect clinicians at MD Anderson to submit an IND for this drug candidate in the first quarter of 2016.
An analog of WP1066, referred to as WP1220, was previously the subject of an IND (WP1220 was referred to as MOL4239 for purposes of this IND) related to use of the molecule in the topical treatment of psoriasis. Clinical trials were commenced on WP1220 in the US, but were terminated early due to limited efficacy in the topical treatment of psoriatic plaques. Notwithstanding its limitations in treating psoriasis, our pre-clinical research has shown WP1220 to be effective in inhibiting cutaneous T-cell lymphoma (CTCL) in multiple CTCL cell lines. Based on this encouraging data, we are collaborating with a Polish drug development company, Dermin, which has received Polish government grant money to begin a clinical trial in Poland for the topical treatment of early stage CTCL patients. CTCL is a potentially deadly form of skin cancer for which there are limited treatment options.
We also conducted a Phase II clinical trial for WP1066 for the topical treatment of psoriasis, however this trial was terminated early as a significant number of patients experienced a non-permanent worsening of their psoriatic plaques after extended use of the drug, suggesting that its use as a topical agent for non-life threatening diseases such as psoriasis will require further study to optimize dosing and scheduling regimens.
Scientific Rationale for WP1066 Portfolio
Cellular biology depends upon signaling mechanisms to regulate functions such as cell growth, death and adaptation. Signal “transduction” is such a mechanism that converts an upstream stimulus to a cell into a specific cellular response. Signal transduction starts with a signal to a receptor or via a compound capable of passing through the cell membrane and ends with a change in cell function. The end result of this signal is often the activation of “transcription”, whereby genetic information is expressed and, in the case of oncogenic transcription, disease processes are initiated or maintained.
Receptors span the cell membrane, with part of the receptor outside and part inside the cell. When a chemical signal represented by a specific protein binds to the outer portion of the receptor, it conveys another signal inside the cell. Often there is a cascade of signals within the cell, wherein an upstream inducer starts a chain of events that resembles a domino effect. Collectively, this sequence is referred to as a “signaling pathway.” Eventually, the signal creates a change in the cell function by changing the expression of specific genes and production of specific proteins within the cell, and again, in the case of tumor development, such expression results in unwanted oncogenic processes.
Importantly, while normal healthy cell function relies on signaling mechanisms, diseases are capable of co-opting these mechanisms with negative consequences. Oncogenic processes (including inflammation and proliferation) depend upon signaling pathways that are responsible for coordinating functions such as cell growth, survival and cell differentiation. A particular class of proteins referred to as Signal Transducers and Activators of Transcription (such proteins are “STATs”) regulates the process of disease cell survival and proliferation, angiogenesis and immune system function and is persistently activated in a large number of human inflammatory processes and in hyper-proliferating diseases. Because certain of these proteins are known to be co-opted by tumor cells, we refer to them as “oncogenic transcription factors,” of which certain STATs are a subset.
48
Some STATs, such as STAT3, can be activated by any one of many different upstream inducers, making them very difficult to target by blocking just one or more of these upstream inducers. We believe that blocking a targeted STAT directly rather than via its multiple upstream inducers should result in greater efficacy with lower toxicity.
In the diagram shown below, any one of many different pathways (categorized as Growth Factor Receptors, Cytokine Receptors and Non-Receptor Tyrosine Kinases) triggers the activation of STAT3 proteins in a process called “phosphorylation”. In this process, phosphates attach to corresponding receptors on STAT3 and, eventually, two phosphorylated STAT3 proteins (“p-STAT3”) bind together in a pair referred to as a “dimer”. Once the dimer is formed, it enters the cell nucleus and triggers gene transcription. Conversely, if we reduce the presence of p-STAT3 before dimers can be formed, we can prevent the triggering of gene transcription and effectively inhibit the disease process.
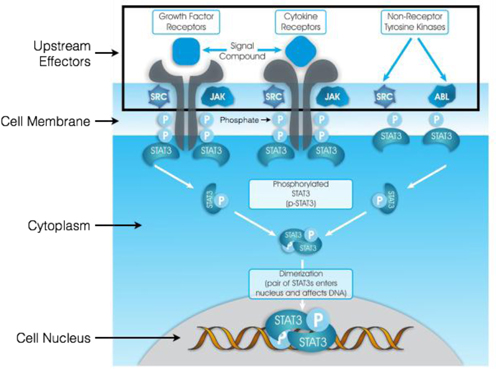
The upstream effectors shown in the above diagram (SRC, JAK and ABL) are just some of those capable of activating STAT3 once they themselves are activated by a variety of signal compounds. The complexity and diversity of pathways capable of activating STAT3 make it very difficult to develop effective drugs that attempt to target the upstream effectors. Furthermore, many of these upstream pathways are necessary for normal healthy cell function, so blocking them indiscriminately can lead to unwanted toxicities.
Published research has identified STAT3 as a master regulator of a wide range of tumors and clearly links STAT3 activation with the progression of these tumors. For this reason, it is believed that direct inhibition of p-STAT3 should be an effective way to reduce or eliminate the progression of these diseases.
Many research efforts have been directed toward development of specific methods to control activation of STAT3, but most have focused on targeting the upstream effectors of these pathways like growth factors, cytokines, and specific kinases including Janus kinases (JAKs). However, we believe that the multifactorial nature of the activation of STAT3 limits the effectiveness of such upstream approaches. Since the activity of p-STAT3 is a final and determinative step in triggering unwanted transcription, we believe it is preferable to inhibit p-STAT3 more directly and independently from upstream effectors.
49
We believe the WP1066 Portfolio represents a novel class of agents capable of hitting multiple targets, including p-STAT3, regardless of their upstream method of activation. By inhibiting the presence of p-STAT3, WP1066 directly attacks tumor cells, as has been demonstrated in numerous preclinical tests involving a wide range of tumor cells. We believe the effectiveness of WP1066 is not only the result of attacking tumors directly, but also indirectly by stimulating the immune system, increasing the patient’s natural ability to fight off tumor development. STAT1 is believed to stimulate T-cell activity and thereby the immune system responsible for fighting tumors. WP1066 has been shown to increase the activity of STAT1 at the same time it inhibits the activity of p-STAT3. We believe this dual activity makes WP1066 a uniquely promising anticancer drug candidate.
We believe the combination of the direct and indirect effects of WP1066 are to be credited with significant tumor suppression and increased survival in a number of in vivo cancer models. Below is one example showing a dramatic increase in survival by treating mice with metastatic melanoma with WP1066.
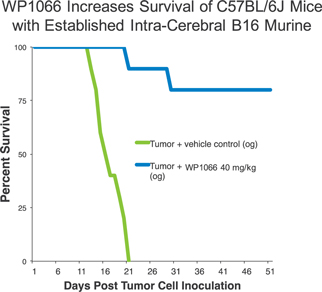
The WP1122 Portfolio
We have a license agreement with MD Anderson pursuant to which we have been granted a royalty-bearing, worldwide, exclusive license for the patent and technology rights related to our WP1122 Portfolio and similar molecules targeting the treatment of GBM and related CNS malignancies.
We believe this technology has the potential to target a wide variety of solid tumors, which eventually become resistant to all treatments, and thereby provide a large and important opportunity for novel drugs. Notwithstanding this potential, we are focused on the treatment of central nervous system malignancies and especially GBM. Although less prevalent than some larger categories of solid tumors, cancers of the central nervous system are particularly aggressive and resistant to treatment. The prognosis for such patients can be particularly grim and the treatment options available to their physicians are among the most limited of any cancer.
The American Cancer Society has estimated 23,770 new cases of brain and other nervous system cancers will occur in the United States in 2016, resulting in 16,050 deaths. Despite the severity and poor prognosis of these tumors, there are few FDA-approved drugs on the market.
We have proof of concept data for WP1122, including in vitro activity against cancer cell lines, as well as data on survival of animals subjected to xenografts of human brain tumors, including data regarding biodistribution and pharmacokinetics. In non-optimal doses and treatment regimes, WP1122 performed equal to or better than the current market leader, temozolomide and provided for superior survival for animals treated in combination with temozolomide.
50
Scientific Rationale for WP1122
Science has recognized that many cancer cells have a unique metabolism, distinct from that of normal cells. Dubbed the “Warburg Effect” by its discoverer, tumors rely preferentially on glycolysis for the metabolism of glucose, even in the presence of abundant oxygen, for energy (adenosine triphosphate (ATP)) production. This alternative form of energy production makes cancer cells as much as 17 times more dependent on glucose than normal cells.
The fundamental mechanism for imaging actively growing tumors using positron emission tomography (PET scans) is the Warburg Effect. A radiolabeled glucose decoy called F18DG accumulates disproportionately in tumors because of their dramatically increased rate of glucose uptake and accumulation.
Researchers have theorized that if a tumor’s access to glucose could be blocked the tumor could be starved out of existence. Previous attempts at targeting the metabolism of tumor cells have failed due to the rapid metabolism and short half-life (minutes) of the drugs being investigated. Efforts to target tumor metabolism in the brain were further thwarted by the inability to get glycolytic inhibitors into the brain in sufficient/therapeutic amounts due to the presence of what is called the “blood brain barrier”.
We believe WP1122 has the potential for developing a technology platform for enabling increased cellular uptake, increased drug half-life and, importantly, an increased ability to cross the blood brain barrier, enabling greater uptake in the brain. Our approach was inspired by the same principle that distinguishes morphine from heroin. Heroin is chemically the diacetyl ester of morphine. While morphine has a limited ability to cross the blood brain barrier (making it a good candidate for pain killing without impairing mental function), its diacetyl form, heroin, has the ability to accumulate in the brain by 10 to 100 times more than morphine. Once across the blood brain barrier, the acetyl groups are cleaved off by natural enzyme esterases, leaving pure morphine to accumulate in the brain. Similarly, we believe, based on pre-clinical testing, that WP1122, the diacetyl form of a glucose analog and decoy known as “2-DG”, crosses the blood brain barrier where its acetyl groups are cleaved off, allowing the resulting 2-DG to accumulate in the brain at a much higher rate than free 2-DG can do by itself.
Adding to the difficulty in getting free 2-DG across the blood brain barrier in therapeutic quantities is the relatively short half-life of 2-DG. The free form of 2-DG is rapidly metabolized and rendered ineffective within minutes of entering the body. In contrast, WP1122 has a half-life of approximately 6 hours, making it much more feasible to deliver quantities adequate for a therapeutic effect. In addition, we believe WP1122 may represent an improvement to current PET scan diagnostics techniques because of its unique ability to cross the blood brain barrier.
Overview of the market for our anti-cancer drugs
Cancer is the second leading cause of death in the United States behind heart disease. In 2014, an estimated 14.5 million people in the United States were living with a past or current diagnosis of cancer and, in 2016, the National Institutes of Health estimates that nearly 1.7 million new cases will be diagnosed and almost 600,000 Americans will die from cancer.
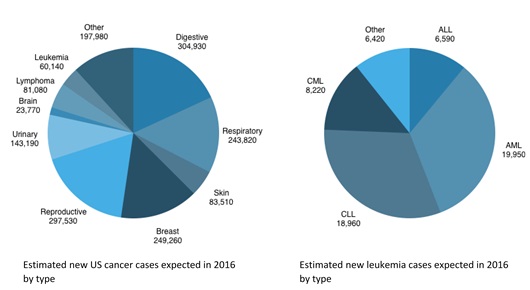
Source: American Cancer Society - Cancer Facts & Figures 2016
Digestive, reproductive, breast and respiratory cancers comprise 65% of expected cancer diagnoses in 2016, while cancers like leukemia and brain tumors are considered “rare diseases”. Leukemia in particular, can be divided into acute, chronic and other, with acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) comprising 26,540 of the estimated 60,140 new cases expected in the United States this year.
The worldwide cancer drug business has been estimated to represent approximately $100 billion in annual sales. Our lead drug, Annamycin, is in a class of drugs referred to as anthracyclines, which are chemotherapy drugs designed to destroy the DNA of targeted cancer cells. The most common approved anthracyclines are daunorubicin and doxorubicin and, prior to the expansion of their generic equivalents, annual revenues generated from anthracyclines have been estimated in the range of $600 million. Acute leukemia is one of a number of cancers that are treated with anthracyclines. One industry report estimates that annual drug revenues generated from the demand for AML-related thereapies in the United States, United Kingdom, France, Germany, Italy and Spain were in the range of $151 million in 2012, and we believe that this number may increase if and when improved AML treatments are available.
Our other two active development projects have applications (among others) in the treatment of brain tumors, another rare disease for which there are few available treatments. The leading brain tumor drug is temozolomide, a drug introduced under the brand name Temodar. In 2012, one industry source reported annual revenues of approximately $882 million for Temodar before the expiration of its patent protection, at which point generic versions of the drug began to enter the market and reduce prices.
The Orphan Drug Act of 1983 and other legislative initiatives provide incentives and in some cases, accelerated approval pathways for companies that pursue the development of treatments for rare diseases and diseases for which there are few or no acceptable available treatment alternatives. Over the last 10 years, an increasing number of companies have begun using these designations to obtain new drug approvals for drugs where patent coverage has expired and/or where accelerated approval appears possible. An IMS Health report estimated that, in 2013, the sale of drugs with full or partial Orphan Drug status represented approximately $29 billion in revenue. We consider obtaining Orphan Drug status and accelerated approval pathways to be an important part of our development strategy for our drug candidates. Notwithstanding these potential opportunities, we can provide no assurance that our drugs will receive Orphan Drug status or any other special designation that could, among other things, provide for accelerated approval.
51
Our License Agreements
We acquired the rights and obligations under the Patent and Technology License Agreement entered into by and between IntertechBio and MD Anderson dated April 2, 2012 (the “IntertechBio Agreement”). Pursuant to that license agreement, IntertechBio obtained a royalty-bearing, worldwide, exclusive license to intellectual property including patent rights related to our WP1122 Portfolio and to our drug product candidate, WP1122. In consideration, IntertechBio agreed to make payments to MD Anderson, including an up-front payment, license documentation fee, annual maintenance fee, milestone payments and minimum annual royalty payments for sales of products developed under the license agreement. Specifically, under the IntertechBio Agreement, IntertechBio agreed to pay a nonrefundable upfront documentation fee in the amount of $80,000; annual maintenance fee in the amount of $10,000 on the first anniversary of the effective date of the IntertechBio Agreement, $20,000 on the second anniversary of the effective date of the agreement, $40,000 on the third anniversary of the effective date of the agreement, $60,000 on the fourth anniversary of the effective date of the agreement, $80,000 on the fifth anniversary of the effective date of the agreement and $100,000 on the sixth anniversary of the effective date of the agreement, except that such payments will no longer be due upon the first sale of a licensed product. Under the IntertechBio Agreement, IntertechBio also agreed to make a minimum annual royalty in the amount of $200,000 for the first anniversary following the first sale of a licensed product, $400,000 for the second anniversary following the first sale of a licensed product, and $600,000 for the third year following the first sale of a licensed product. IntertechBio also agreed to make one-time milestone payments in the amount of $200,000 upon commencement of a Phase II study for a licensed product, $250,000 upon commencement of a Phase III study for a licensed product, $400,000 upon filing of a New Drug Application (“NDA”) for a licensed product and $500,000 upon receipt of market approval for sale of a licensed product. MD Anderson has the right to terminate the agreement upon advanced notice in the event of a default by IntertechBio. The agreement will also be terminated immediately upon IntertechBio’s insolvency. Additionally, per an October 2015 amendment to the IntertechBio Agreement, MD Anderson has the right to terminate the license agreement if (i) a preclinical toxicology program for a licensed product is not initiated within one year of the of effective date of the amendment, (ii) an investigational new drug application (IND) is not filed with the Food and Drug Administration (FDA) for a Phase I study for a licensed product within three years of the effective date of the amendment, or (iii) a Phase I study for a licensed product is not commenced within five years of the effective date of the amendment. The IntertechBio Agreement will expire upon the expiration of the licensed intellectual property. In November 2015, the IntertechBio Agreement and amendments thereto were assigned to MBI. Under the assignment, we have assumed the rights and obligations of IntertechBio including, without limitation, the right to manufacture, have manufactured, use, import, offer to sell and/or sell products worldwide for any indication under the licensed intellectual property with the right to sublicense. However, the rights obtained pursuant to the assignment of the IntertechBio Agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by us. We are not required to issue MD Anderson any equity in our company upon the completion of any milestones.
Upon completion of our acquisition of Moleculin LLC, we will obtain a royalty-bearing, worldwide, exclusive license to intellectual property rights, including patent rights related to our WP1066 drug product candidate from MD Anderson. Moleculin entered into a June 2010 Patent and Technology License Agreement with MD Anderson (the “Moleculin Agreement”). Under the Moleculin Agreement, Moleculin obtained the right to manufacture, have manufactured, use, import, offer to sell and/or sell products worldwide for any indication under the licensed intellectual property with the right to sublicense. In consideration, Moleculin agreed to make payments to MD Anderson including an up-front payment, milestone payments and minimum annual royalty payments for sales of products developed under the license agreement. Specifically, under the Moleculin Agreement, Moleculin agreed to pay a nonrefundable upfront documentation fee in the amount of $223,585; annual maintenance fee in the amount of $20,000 on the first anniversary of the effective date of the Moleculin Agreement, which shall increase in $10,000 increments on an annual basis thereafter up to a maximum of $100,000, except that such payments will no longer be due upon marketing approval in any country of a licensed product. Under the Moleculin Agreement, Moleculin also agreed to make a minimum annual royalty payment to MD Anderson in the amount of $200,000 after the first sale of a licensed product. Moleculin also agreed to make one-time milestone payments in the amount of $150,000 upon commencement of the first Phase III study for a licensed product within the United States, Europe, China or Japan; $500,000 upon submission of the first NDA for a licensed product in the United States; and $600,000 upon receipt of the first marketing approval for sale of a licensed product in the United States. MD Anderson has the right to terminate the Moleculin Agreement upon advanced notice in the event of a default by Moleculin. Moleculin has the right to terminate the Moleculin Agreement for any reason upon advanced written notice to MD Anderson. The Moleculin Agreement will also be terminated immediately upon Moleculin’s insolvency. Per an October 2015 amendment to the Moleculin Agreement, MD Anderson relinquished any rights to any equity previously due MD Anderson in Moleculin. Upon completion of our acquisition of Moleculin LLC, will assume the rights and obligations of Moleculin LLC including, without limitation, the right to manufacture, have manufactured, use, import, offer to sell and/or sell products worldwide for any indication under the licensed intellectual property with the right to sublicense. However, the rights we have obtained pursuant to the assignment of the Moleculin Agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by us. We are not required to issue MD Anderson any equity in our company upon the completion of any milestones.
52
Moleculin LLC out-licensed certain intellectual property rights including rights covering the WP1066 drug product candidate to Dermin (“Moleculin Out-License Agreement”). The licensed intellectual property includes rights obtained by Moleculin pursuant to a license agreement with MD Anderson (“Moleculin-MD Anderson Agreement”). Under the Moleculin Out-License Agreement, Moleculin granted Dermin a royalty-bearing, exclusive license to manufacture, have manufactured, use, import, offer to sell and/or sell products in the field of human therapeutics under the licensed intellectual property in the countries of Belarus, Czech Republic, Estonia, Hungary, Latvia, Lithuania, Poland, Romani, Slovakia and Ukraine (“licensed territories”). Additionally, Moleculin agreed to develop and provide a dossier containing data related to the licensed subject matter to Dermin. In consideration, Dermin agreed to make payments to Moleculin, including upfront development fees, annual royalty payments, sublicense fee, and milestone payments. Specifically, under the Moleculin Out-License Agreement, Dermin agreed to make a nonrefundable upfront dossier development payment in the amount of $100,000; a service fee in the amount of $100,000 for assistance provided to Dermin in securing additional funding; and royalty payments on sales of any licensed product at a rate of no less than the royalty rate due to MD Anderson under the Moleculin-MD Anderson Agreement plus 1%. Dermin also agreed to provide a percentage of certain consideration Dermin receives pursuant to sublicense agreements in the amount of 25% prior to completion of a Phase IIb clinical study in a licensed territory and 10% on or after completion of a Phase IIb clinical study in a licensed territory, provided, however, if the sublicense fee is less than the sublicense fee due to MD Anderson under the Moleculin-MD Anderson Agreement, then Dermin shall be obligated to pay no less than the amount due to MD Anderson plus 5%. Also under the Moleculin Out-License Agreement, Dermin agreed to pay all out-of-pocket expenses incurred by Moleculin in filing, prosecuting and maintaining the licensed patents for which the license has been granted. The parties to the Moleculin Out-License Agreement each have the right to terminate the agreement upon advanced notice in the event of a default by the other party. Dermin has the right to terminate the agreement if (i) Moleculin fails to timely provide the dossier to Dermin after Moleculin’s filing of an NDA for a licensed product in the United States; or (ii) Moleculin does not cooperate in assisting Dermin to secure funds to develop the licensed subject matter. Upon completion of our acquisition of Moleculin, we will assume the rights and obligations of Moleculin under the Moleculin Out-License Agreement.
We acquired the rights and obligations to the Patent and Technology License Agreement entered into by and between IntertechBio and Dermin dated April 15, 2011 (the “IntertechBio Out-License Agreement”). Pursuant to that license agreement, IntertechBio out-licensed intellectual property rights, including rights covering the WP1122 drug product candidate obtained from MD Anderson pursuant to the IntertechBio Agreement to Dermin. Under the IntertechBio Out-License Agreement, IntertechBio granted Dermin a royalty-bearing, exclusive license to manufacture, have manufactured, use, import, offer to sell and/or sell products in the field of human therapeutics under the licensed intellectual property in the countries of Belarus, Russia, Kazakhstan, Uzbekistan, Turkmenistan, Czech Republic, Estonia, Hungary, Latvia, Lithuania, Poland, Romania, Slovakia and Ukraine (“licensed territories”). Additionally, IntertechBio agreed to develop and provide a dossier containing data related to the licensed subject matter to Dermin. In consideration, Dermin agreed to make payments to IntertechBio, including upfront development fees, annual royalty payments, sublicense fees, and milestone payments. Specifically, under the IntertechBio Out-licensing Agreement, Dermin agreed to make a nonrefundable upfront dossier development fee in the amount of $35,000; a service fee in the amount of $40,000 for assistance provided to Dermin in securing additional funding; and royalty payments on sales of any licensed product at a rate of no less than the royalty rate due to MD Anderson under the IntertechBio Agreement plus 2%. Dermin also agreed to provide a percentage of certain consideration Dermin receives pursuant to sublicense agreements in the amount of 25% prior to completion of a Phase IIb clinical study in a licensed territory and 10% on or after completion of a Phase IIb clinical study in the licensed territories, provided, if the sublicense fee is less than the sublicense fee due to MD Anderson under the IntertechBio Agreement, then Dermin shall be obligated to pay not less than the amount due to MD Anderson plus 5%. Also under the IntertechBio Out-Licensing Agreement, Dermin agreed to pay all out-of-pocket expenses incurred by IntertechBio in filing, prosecution and maintaining the licensed patents for which the license has been granted. The parties to the IntertechBio Out-licensing Agreement each have the right to terminate the agreement upon advanced notice in the event of a default by the other party. Dermin has the right to terminate the agreement if (i) IntertechBio fails to timely provide the dossier to Dermin after IntertechBio’s filing of an NDA for a licensed product in the United States; or (ii) IntertechBio does not cooperate in assisting Dermin to secure funds to develop the licensed subject matter.
We will enter into an out-licensing agreement with HPI, pursuant to which we will grant HPI certain intellectual property rights, including rights covering the potential drug candidate, WP1066 (“HPI Out-Licensing Agreement”). Under the HPI Out-Licensing Agreement we will be required to make quarterly payments in the amount of $37,500 for the first four quarters following the effective date of the HPI Out-Licensing Agreement and $75,000 per quarter for the following eight quarters thereafter in consideration for the right to development data related to the development of licensed products. Notwithstanding our obligation to make the foregoing payments, the HPI Out-Licensing Agreement does not obligate HPI to conduct any specific research or to meet any milestones. Upon payment in the amount of $1,000,000 to HPI within three years of the effective date of the HPI Out-Licensing Agreement we will regain all rights to the licensed subject matter and rights to any and all development data and any regulatory submissions including any IND, NDA or ANDA related to the licensed subject matter. In the event that we do not exercise our right to regain our rights to the licensed subject matter within three years of the effective date of the HPI Out-Licensing Agreement the license granted to HPI shall convert to an exclusive license upon HPI’s written notice and we shall be obligated to transfer all existing data relating to licensed subject matter including any development data and any IND to HPI. The HPI Out-Licensing Agreement will be rendered void in its entirety if we are unable to raise gross proceeds of at least $7,000,000 in this offering no later than June 30, 2016.
53
Competition
We operate in a highly competitive segment of the pharmaceutical market, which market is highly competitive as a whole. We face competition from numerous sources including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies, and private and public research institutions. Many of our competitors may have significantly greater financial, product development, manufacturing and marketing resources. Additionally, many universities and private and public research institutes are active in cancer research, and some may be in direct competition with us. We may also compete with these organizations to recruit scientists and clinical development personnel. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
The unmet medical need for more effective cancer therapies is such that anticancer drugs are, by far, the leading class of drugs in development. These include a wide array of products against cancer targeting many of the same indications as our drug candidates. While the introduction of newer targeted agents may result in extended overall survival, induction therapy regimens are likely to remain a cornerstone of cancer treatment in the foreseeable future.
There are a number of established therapies that may be considered competitive for the cancer indications for which we intend to develop our lead product, Annamycin. A key consideration when treating AML patients is whether the patient is suitable for intensive therapy. The standard of care for the treatment of newly diagnosed AML patients who can tolerate intensive therapy is cytarabine in combination with an anthracycline (i.e., doxorubicin or daunorubicin) – for some patients, primarily those less than 60 years of age, a stem cell transplant could also be considered if the induction regimen is effective in attaining a CR (Complete Response). The regimen of cytarabine in combination with an anthracycline has been the standard of care for decades. A patient not suitable for intensive therapy may be offered the option for low-intensity therapy such as low-dose cytarabine, azacitidine or decitabine. It should be noted that, in the U.S., these are not approved by the FDA for the treatment of AML patients and there remains no effective therapy for these patients or for relapsed or refractory AML. The initial focus for Annamycin development is in patients for whom the standard induction regimen has failed. Also, several major pharmaceutical companies and biotechnology companies are aggressively pursuing new cancer development programs for the treatment of AML.
A number of attempts have been made or are under way to provide an improved treatment for AML. Drugs attempting to target a subset of AML patients who present with particular anomalies involving a gene referred to as FLT3 are currently in clinical trials. Other approaches to improve the effectiveness of induction therapy are in early stage clinical trials and, although they do not appear to address the underlying problems with anthracyclines, we can provide no assurance that such improvements, if achieved, would not adversely impact the need for improved anthracyclines. A modified version of doxorubicin designed to reduce cardiotoxicity is in clinical trials for the treatment of sarcoma and, although this drug does not appear to address multidrug resistance and is not currently intended for the treatment of acute leukemia, we can provide no assurance that it will not become a competitive alternative to Annamycin. Although we are not aware of any other single agent therapies in clinical trials that would directly compete against Annamycin in the treatment of relapsed and refractory AML, we can provide no assurance that such therapies are not in development, will not receive regulatory approval and will reach market before our drug candidate Annamycin. In addition, any such competing therapy may be more effective and / or cost-effective than ours.
Government Regulation
Government authorities in the U.S., at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing. The pharmaceutical drug product candidates that we develop must be approved by the FDA before they may be legally marketed.
In the United States, the FDA regulates pharmaceutical products under the Federal Food, Drug and Cosmetic Act, and implementing regulations. Pharmaceutical products are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a non-biological pharmaceutical product may be marketed in the U.S. generally involves the following:
54
| · | Completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices or other applicable regulations; |
| · | Submission to the FDA of an Investigational New Drug, or IND, which must become effective before human clinical studies may begin; |
| · | Performance of adequate and well-controlled human clinical studies according to the FDA’s current good clinical practices (“GCP”), to establish the safety and efficacy of the proposed pharmaceutical product for its intended use; |
| · | Submission to the FDA of an NDA for a new pharmaceutical product; |
| · | Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the pharmaceutical product is produced to assess compliance with the cGMP, to assure that the facilities, methods and controls are adequate to preserve the pharmaceutical product’s identity, strength, quality and purity; |
| · | Potential FDA audit of the preclinical and clinical study sites that generated the data in support of the NDA; and |
| · | FDA review and approval of the NDA. |
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources and approvals are inherently uncertain.
Before testing any compounds with potential therapeutic value in humans, the pharmaceutical product candidate enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the pharmaceutical product candidate. These early proof-of-principle studies are done using sound scientific procedures and thorough documentation. The conduct of the single and repeat dose toxicology and toxicokinetic studies in animals must comply with federal regulations and requirements including good laboratory practices. The sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA has concerns and notifies the sponsor. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical study can begin. If resolution cannot be reached within the 30-day review period, either the FDA places the IND on clinical hold or the sponsor withdraws the application. The FDA may also impose clinical holds on a pharmaceutical product candidate at any time before or during clinical studies due to safety concerns or non-compliance. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical studies to begin, or that, once begun, issues will not arise that suspend or terminate such clinical study.
Clinical studies involve the administration of the pharmaceutical product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the clinical study sponsor’s control. Clinical studies are conducted under protocols detailing, among other things, the objectives of the clinical study, dosing procedures, subject selection and exclusion criteria, how the results will be analyzed and presented and the parameters to be used to monitor subject safety. Each protocol must be submitted to the FDA as part of the IND. Clinical studies must be conducted in accordance with GCP. Further, each clinical study must be reviewed and approved by an independent institutional review board (“IRB”) at, or servicing, each institution at which the clinical study will be conducted. An IRB is charged with protecting the welfare and rights of study participants and considers such items as whether the risks to individuals participating in the clinical studies are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical study subject or his or her legal representative and must monitor the clinical study until completed.
Human clinical studies are typically conducted in three sequential phases that may overlap or be combined:
| · | Phase 1: The pharmaceutical product is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases such as cancer, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients, with a goal of characterizing the safety profile of the drug and establishing a maximum tolerable dose (“MTD”). Our pharmaceutical products fall into this latter category because its products are intended to treat cancer and contain cytotoxic agents. Hence, our Phase 1 studies are conducted in late-stage cancer patients whose disease has progressed after treatment with other agents. |
55
| · | Phase 2: With the maximum tolerable dose established in a Phase 1 trial, the pharmaceutical product is evaluated in a limited patient population at the MTD to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases, to determine dosage tolerance, optimal dosage and dosing schedule and to identify patient populations with specific characteristics where the pharmaceutical product may be more effective. |
| · | Phase 3: Clinical studies are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These clinical studies are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. The studies must be well controlled and usually include a control arm for comparison. One or two Phase 3 studies are usually required by the FDA for an NDA approval, depending on the disease severity and other available treatment options. In some instances, where a Special Protocol Assessment is granted by the FDA, an NDA approval may be obtained based on Phase 2 clinical data with the understanding that the approved drug can be sold subject to a confirmatory Phase 3 trial to be conducted post-approval. |
Post-approval studies, or Phase 4 clinical studies, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication. The FDA also may require post-marketing testing, known as Phase 4 testing, risk minimization action plans and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical studies may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical study at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical study at its institution if the clinical study is not being conducted in accordance with the IRB’s requirements or if the pharmaceutical product has been associated with unexpected serious harm to patients.
Concurrent with clinical studies, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the pharmaceutical product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the pharmaceutical product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final pharmaceutical product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the pharmaceutical product candidate does not undergo unacceptable deterioration over its shelf life.
The results of product development, preclinical studies and clinical studies, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the pharmaceutical product, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of substantial user fees. A waiver of such fees may be obtained under certain limited circumstances.
The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act (“PDUFA”), the FDA has 10 months after the 60-day filing date in which to complete its initial review of a standard NDA and respond to the applicant, and six months after the 60-day filing date for a priority NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs.
56
After the NDA submission is accepted for filing, the FDA reviews the NDA application to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel pharmaceutical products or pharmaceutical products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the pharmaceutical product approval process, the FDA also will determine whether a risk evaluation and mitigation strategy (“REMS”) is necessary to assure the safe use of the pharmaceutical product. If the FDA concludes that a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without a REMS, if required.
Before approving an NDA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites as well as the site where the pharmaceutical product is manufactured to assure compliance with GCP and cGMP. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. In addition, the FDA will require the review and approval of product labeling.
The NDA review and approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical studies are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA. The complete response letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical studies. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing which involves clinical studies designed to further assess pharmaceutical product safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Expedited Development and Review Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new pharmaceutical products that meet certain criteria. Specifically, new pharmaceutical products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider for review sections of the NDA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, if the FDA determines that the schedule is acceptable and if the sponsor pays any required user fees upon submission of the first section of the NDA.
Any product submitted to the FDA for market, including a Fast Track program, may also be eligible for other FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or if it offers a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new pharmaceutical product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Pharmaceutical products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that the products may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a pharmaceutical product receiving accelerated approval perform adequate and well-controlled post-marketing clinical studies. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
57
Post-Approval Requirements
Any pharmaceutical products for which the Company receives FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, prohibitions on promoting pharmaceutical products for uses or in patient populations that are not described in the pharmaceutical product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities and promotional activities involving the internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, actions by the U.S. Department of Justice and/or U.S. Department of Health and Human Services’ Office of Inspector General, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Although physicians may prescribe legally available pharmaceutical products for off-label uses, manufacturers may not directly or indirectly market or promote such off-label uses.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products. Manufacturers of our products are required to comply with applicable FDA manufacturing requirements contained in the FDA’s cGMP regulations. cGMP regulations require, among other things, quality control and quality assurance, as well as the corresponding maintenance of records and documentation. Pharmaceutical product manufacturers and other entities involved in the manufacture and distribution of approved pharmaceutical products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of the FDA approval of the use of our pharmaceutical product candidates, some of our products covered by U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved pharmaceutical product is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent unless an extension is obtained. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and renders a decision on the application for any patent term extension or restoration. In the future, we may be able to apply for extension of patent term for one of our currently licensed patents or any future owned patents to add patent life beyond its current expiration date, depending upon the expected length of the clinical studies and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the U.S. Food, Drug, and Cosmetic Act can also delay the submission or the approval of certain applications of other companies seeking to reference another company’s NDA. Currently seven years of reference product exclusivity are available to pharmaceutical products designated as Orphan Drugs, during which the FDA may not approve generic products relying upon the reference product’s data. Pediatric exclusivity is another type of regulatory market exclusivity in the U.S. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of pediatric clinical studies in accordance with an FDA-issued “Written Request” for such a clinical study.
58
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical product candidates for which we obtain regulatory approval. In the United States and in markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part upon the availability of reimbursement from third-party payors. Third-party payors include government payors such as Medicare and Medicaid, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a pharmaceutical product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the pharmaceutical product. Third-party payors may limit coverage to specific pharmaceutical products on an approved list, or formulary, which might not, and frequently does not, include all of the FDA-approved pharmaceutical products for a particular indication. Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. A payor’s decision to provide coverage for a pharmaceutical product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. In addition, in the United States there is a growing emphasis on comparative effectiveness research, both by private payors and by government agencies. We may need to conduct expensive pharmaco-economic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Our pharmaceutical product candidates may not be considered medically necessary or cost-effective. To the extent other drugs or therapies are found to be more effective than our products, payors may elect to cover such therapies in lieu of our products and/or reimburse our products at a lower rate.
Different pricing and reimbursement schemes exist in other countries. In the European Community, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed upon. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical studies that compare the cost-effectiveness of a particular pharmaceutical product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any pharmaceutical product candidates for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased and we expect this will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
International Regulation
In addition to regulations in the United States, there are a variety of foreign regulations governing clinical studies and commercial sales and distribution of our current and future product candidates. Whether or not FDA approval is obtained for a product, approval of a product must be obtained by the comparable regulatory authorities of foreign countries, or jurisdictions such as the EU, before clinical studies or marketing of the product can commence in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical studies, product licensing, pricing and reimbursement vary greatly from country to country. In addition, certain regulatory authorities may require us to repeat previously conducted preclinical and/or clinical studies under specific criteria for approval in their respective country or jurisdictions, which may delay and/or increase the cost of approval in certain markets targeted for approval by us.
59
Employees
As of January 27, 2016, we had one full-time employee and two part-time employees, and accordingly, a high percentage of the work performed for our development projects is outsourced to qualified independent contractors.
Legal Proceedings
We are not subject to any litigation.
Properties
Our corporate and executive offices are in located in a leased facility in Houston, Texas. The current lease is month-to-month but is expected to be renegotiated in the next six months. We believe our facilities are sufficient to meet our current needs and that suitable space will be available as and when needed. We do not own any real property.
60
The following table sets forth certain information regarding our executive officers and directors as of January 27, 2016.
| Name | Age | Position | ||
| Walter V. Klemp | 56 | Chairman and Acting Chief Executive Officer | ||
| Louis Ploth, Jr. | 61 | Chief Financial Officer | ||
| Donald Picker | 70 | President and Chief Operating Officer | ||
| Robert E. George | 65 | Director Nominee | ||
Michael D. Cannon |
70 | Director Nominee | ||
| Jacqueline Northcut | 54 | Director Nominee |
Set forth below is biographical information about each of the individuals named in the tables above:
Walter V. Klemp - Chairman of the Board and Acting Chief Executive Officer. Mr. Klemp is a co-founder of our company, and has served as our chairman and acting chief executive officer since July 2015. Since 2006, Mr. Klemp has served as the chairman, co-founder and part-time chief executive officer of Moleculin LLC. Since November 2011, Mr. Klemp has also served as chief executive officer of Soliton, Inc., a medical device company focused on developing new technology for use in aesthetics. Mr. Klemp served as president and chief executive officer of Zeno Corporation from 2004 to April 2011, where he developed and marketed dermatology devices and drugs from concept through FDA approval and market launch. From 1987 to 2000, Mr. Klemp served as chief executive officer and chairman of Drypers Corporation, a publicly traded multinational consumer products company that was listed as #1 on the INC 500 List of America’s Fastest Growing Companies. We believe that Mr. Klemp’s history with our company and background, coupled with his extensive experience in the medical field, provide him with the qualifications to serve as a director. Mr. Klemp currently provides services as needed by us, which we estimate does not exceed 10 hours per week.
Louis Ploth, Jr. - Chief Financial Officer. Mr. Ploth has served as our chief financial officer since August 2015 and as our president from August 2015 until January 2016. From August 2011 until August 2015, Mr. Ploth worked as a financial and business development consultant at his own firm, Corporate Administrative Solutions, Inc. (“CAS”). Prior to starting CAS, Mr. Ploth worked as a consultant from February 2010 to August 2011. Mr. Ploth was previously employed by Repros Therapeutics Inc. from October 1993 through February 2010 where he served as chief financial officer, and secretary since January 2001 through August 2009 and vice president, business development from January 2001 through April 2009. Mr. Ploth has a B.S. degree from Montclair State University.
Donald Picker, PhD - President and Chief Operating Officer. Dr. Picker joined Moleculin in 2007 as its chief executive officer and has served as our chief operating officer since July 2015 and as our president since January 2016. In 2007, Dr. Picker became the chief executive officer of IntertechBio. From 2006 through 2007, Dr. Picker was the President of Tapestry Pharmaceuticals. From 1998 to 2003, Dr. Picker was CEO of Synergy Pharmaceuticals. Synergy was merged into Callisto Pharmaceuticals where he was vice present of research and development until 2006. Dr. Picker led the development of carboplatin and cisplatin from concept to FDA approval. These drugs were among the first metals based chemotherapy drugs every approved by the FDA. Dr. Picker received his BS degree from Brooklyn Polytechnic University and his PhD from SUNY Albany in 1975. Dr. Picker is currently devoting only part of his work time to us. Dr. Picker currently provide services as needed by us, which we estimate does not exceed 10 hours per week. Upon closing of this offering, Dr. Picker will become a full time officer of our company.
Robert E. George - Director Nominee. Mr. George has agreed to join our board of directors upon the date we are listed on the Nasdaq Capital Market. He was a partner with the international accounting firm of PricewaterhouseCoopers (PWC) for 27 years until 2010, where his client service sectors included healthcare, among others. Mr. George currently serves as Chairman of the Audit Committee for The University of Texas Health Science Center at Houston and, since June 2011, has been a member of The University of Texas at Austin, McCombs Graduate School of Business accounting faculty. Since January 2014, he also has been the Director of Energy Programs and Conferences for the Professional Development Institute. His past experience includes being an Editorial Advisor to the American Institute of Certified Public Accountants Journal of Accountancy and a contributing author for two accounting books. Mr. George graduated with accounting honors from the University of North Texas.
Michael D. Cannon - Director Nominee. Mr. Cannon has agreed to join our board of directors upon the date we are listed on the Nasdaq Capital Market. Between 1997 and 2004, Mr. Cannon was the Chief Science Officer, EVP and a Director of SICOR, Inc., a U.S. public pharmaceutical company, until its acquisition by Teva Pharmaceutical Industries, Inc. SICOR focused on generic finished dosage injectable pharmaceuticals, active pharmaceutical ingredients and generic biopharmaceuticals. While at SICOR, he oversaw the acquisition and development of the biological business, including initiation and management of international partnerships, as well as on the design, construction, and licensure of protein manufacturing facilities. From July 2005 to December 2009, Mr. Cannon was a member of the scientific advisory board of Trevi Health Ventures LP, a New York investment fund specializing in health care investments. From May 2005 until December 2011, Mr. Cannon was a partner in a private partnership formed to evaluate and perform preliminary development of intellectual property in the healthcare sector. Since 2005, Mr. Cannon has served as a board member for several private companies. Mr. Cannon currently serves on the board of directors of 4 privately held biotech companies, including 2 focused on cancer-related treatments, and has been a partner and advisor in several biotech venture firms. Mr. Cannon has a degree in chemistry from Fordham College.
Jacqueline Northcut - Director Nominee. Ms. Northcut has agreed to join our board of directors upon completion of our listing on the Nasdaq Capital Market. Ms. Northcut served as President and CEO of BioHouston from 2002 until 2015. BioHouston, Inc. is a non-profit organization founded by Houston, Texas area academic/research institutions to assist in the commercialization efforts of development stage life science and biotechnology companies. During this same period, Ms. Northcut became the founder and CEO of the Texas BioAlliance, an organization created to further assist with development efforts around the state. In these capacities she organized the Texas Life Science Forum and the Texas Life Science CEO Summit, helping connect Biotech CEOs with funding, technical, regulatory and political resources. She also created the Women in Science with Excellence program, honoring women in the Houston scientific community. Ms. Northcut was previously a Partner in the international accounting firm of Arthur Andersen & Co., where she worked from 1984 to 2002. Ms. Northcut received her BBS from Harding University in 1984.
61
Director Independence
The rules of the Nasdaq Stock Market, or the Nasdaq Rules, require a majority of a listed company’s board of directors to be composed of independent directors within one year of listing. In addition, the Nasdaq Rules require that, subject to specified exceptions, each member of a listed company’s audit, compensation and nominating and governance committees be independent. Under the Nasdaq Rules, a director will only qualify as an independent director if, in the opinion of our board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. The Nasdaq Rules also require that audit committee members satisfy independence criteria set forth in Rule 10A-3 under the Securities Exchange Act of 1934, as amended, or the Exchange Act. In order to be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee, accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or otherwise be an affiliated person of the listed company or any of its subsidiaries. In considering the independence of compensation committee members, the Nasdaq Rules require that our board of directors must consider additional factors relevant to the duties of a compensation committee member, including the source of any compensation we pay to the director and any affiliations with the company.
Our board of directors undertook a review of the composition of our board of directors and its committees and the independence of each director. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our board of directors has determined that each of our directors, with the exception of Messrs. Klemp and [*], are independent as defined under the Nasdaq Rules. Our board of directors has appointed [*] to the audit committee, and [*] to our compensation committee, and such directors satisfy the independence standards for these committees established by the Securities and Exchange Commission, or SEC, and the Nasdaq Rules, as applicable. In addition, our board of directors appointed [*] to our nominating and governance committee.
62
Committees of the Board of Directors
Our board of directors has established an audit committee, a compensation committee and a nominating and governance committee. Each of these committees will operate under a charter that will be approved by our board of directors prior to this offering.
Audit Committee. Our audit committee consists of three independent directors. The members of the audit committee are [*], who will chair the committee, [*] and [*]. The audit committee consists exclusively of directors who are financially literate. In addition, [*] will be considered an “audit committee financial expert” as defined by the SEC’s rules and regulations.
The audit committee responsibilities include:
| • | overseeing the compensation and work of and performance by our independent auditor and any other registered public accounting firm performing audit, review or attestation services for us; |
| • | engaging, retaining and terminating our independent auditor and determining the terms thereof; |
| • | assessing the qualifications, performance and independence of the independent auditor; |
| • | evaluating whether the provision of permitted non-audit services is compatible with maintaining the auditor’s independence; |
| • | reviewing and discussing the audit results, including any comments and recommendations of the independent auditor and the responses of management to such recommendations; |
| • | reviewing and discussing the annual and quarterly financial statements with management and the independent auditor; |
| • | producing a committee report for inclusion in applicable SEC filings; |
| • | reviewing the adequacy and effectiveness of internal controls and procedures; |
| • | establishing procedures regarding the receipt, retention and treatment of complaints received regarding the accounting, internal accounting controls, or auditing matters and conducting or authorizing investigations into any matters within the scope of the responsibility of the audit committee; and |
| • | reviewing transactions with related persons for potential conflict of interest situations. |
Compensation Committee. Our compensation committee consists of three independent directors. The members of the Compensation Committee are [*], who will chair the committee, [*] and [*]. The committee has primary responsibility for:
| • | reviewing and recommending all elements and amounts of compensation for each executive officer, including any performance goals applicable to those executive officers; |
| • | reviewing and recommending for approval the adoption, any amendment and termination of all cash and equity-based incentive compensation plans; |
| • | once required by applicable law, causing to be prepared a committee report for inclusion in applicable SEC filings; |
| • | approving any employment agreements, severance agreements or change of control agreements that are entered into with the CEO and certain executive officers; and |
| • | reviewing and recommending the level and form of non-employee director compensation and benefits. |
Nominating and Governance Committee. The Nominating and Governance Committee consists of three independent directors. The members of the Nominating and Governance Committee are [*], who will chair the committee, [*] and [*]. The Nominating and Governance Committee’s responsibilities include:
| • | recommending persons for election as directors by the stockholders; |
| • | recommending persons for appointment as directors to the extent necessary to fill any vacancies or newly created directorships; |
| • | reviewing annually the skills and characteristics required of directors and each incumbent director’s continued service on the board; |
63
| • | reviewing any stockholder proposals and nominations for directors; |
| • | advising the board of directors on the appropriate structure and operations of the board and its committees; |
| • | reviewing and recommending standing board committee assignments; |
| • | developing and recommending to the board Corporate Governance Guidelines, a Code of Business Conduct and Ethics and other corporate governance policies and programs and reviewing such guidelines, code and any other policies and programs at least annually; |
| • | making recommendations to the board as to determinations of director independence; and |
| • | making recommendations to the board regarding corporate governance based upon developments, trends, and best practices. |
The Nominating and Governance Committee will consider stockholder recommendations for candidates for the board of directors.
Our bylaws provide that, in order for a stockholder’s nomination of a candidate for the board to be properly brought before an annual meeting of the stockholders, the stockholder’s nomination must be delivered to the Secretary of the company no later than 120 days prior to the one year anniversary date of the prior year’s annual meeting.
Code of Business Conduct and Ethics
Prior to the effectiveness of the registration statement of which this prospectus forms a part, we will adopt a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. Following this offering, a copy of the code will be made available on the Corporate Governance section of our website, which is located at www. moleculin.com. If we make any substantive amendments to, or grant any waivers from, the code of business conduct and ethics for any officer or director, we will disclose the nature of such amendment or waiver on our website or in a current report on Form 8-K filed with the SEC.
Compensation of Executive Officers
Summary Compensation Table
MBI was formed in July 2015. To provide you with a complete picture of the compensation paid to our executive officers for the fiscal year ended 2014 when they acted in executive capacities for Moleculin and MBI, the following table and the related notes set forth information relating to the compensation paid to each of the named executive officers by Moleculin and us.
| Name and Principal Position | Year | Salary($) | Total ($) | |||||||||
| Moleculin: | ||||||||||||
| Walter V. Klemp, Chairman and Acting Chief Executive Officer | 2014 | 162,600 | 162,600 | |||||||||
| Don Picker, President and Chief Operating Officer MBI: | 2014 | 214,600 | 214,600 | |||||||||
| Louis Ploth, Jr., Chief Financial Officer (1) | 2014 | — | — | |||||||||
| (1) | Mr. Ploth was not employed by Moleculin LLC. |
Employment Agreements
None of our named executive officers are currently party to any employment agreements.
Upon the closing of this offering, we intend to enter into an employment agreement with Mr. Klemp to continue to serve as our acting chief executive officer for a three-year term with annual extensions by mutual agreement. The employment agreement will provide for an annual salary of $300,000 per year. To the extent we retain a full-time chief executive officer during the term of the employment agreement, the agreement will convert to a consulting agreement.
Equity Awards
We had no equity awards outstanding as of September 30, 2015.
64
Director Compensation
We do not currently have any independent board members. Mr. Klemp does not receive any additional compensation for serving as a director.
Scientific Advisory Board
Our executive team is supported by our scientific advisory board, the members of which include scientists experienced in the fields in which we pursue. The members of our Scientific Advisory Board currently serve without compensation, but we intend to compensate the members in the future based on our utilization of their time.
Waldemar Priebe, PHD, Chair of Scientific Advisory Board (SAB). Waldemar Priebe is a founder of and one of the founding scientists at Moleculin and serves as head of our Scientific Advisory Board. Dr. Priebe is also a Professor of Medicinal Chemistry at the Department of Experimental Therapeutics at The University of Texas MD Anderson Cancer Center. Dr. Priebe led the discovery of the molecules that form the basis for our lead drug candidates. As a founder or founding scientist at a number of successful biotechnology firms such as Aronex Pharmaceuticals, Houston Pharmaceuticals, Reata Pharmaceuticals, and IntertechBio, Dr. Priebe has been integral in advancing several drugs through the pipeline, three of which are currently in clinical development. He has also developed several new small molecule compounds that have been licensed as potential drugs.
Charles Conrad, MD, SAB Member. Charles Conrad is one of the founding scientists at Moleculin and has more than 15 years of industry experience and has served as a Principal Investigator (PI) or co-PI in numerous clinical trials. Dr. Conrad worked as a Professor of Neuro-Oncology and also held a joint appointment in the Department of Experimental Therapeutics at the University of Texas MD Anderson Cancer Center. He has been a founding scientist at a number of biotechnology firms, including Zeno Corporation and DNAtrix. Dr. Conrad recently joined Texas Oncology where he is the Director of New Drug Development for The Austin Brain Tumor Center.
2015 Stock Plan
We have adopted a 2015 Stock Plan (the “Plan”). The Plan is a stock-based compensation plan that provides for discretionary grants of stock options, stock awards and stock unit awards to key employees and non-employee directors. The purpose of the Plan is to recognize contributions made to our company and its subsidiaries by key employees and non-employee directors and to provide them with additional incentive to achieve the objectives of our company. The following is a summary of the Plan.
Administration. The Plan will be administered by our board of directors, or, once constituted, the Compensation Committee of the board of directors (we refer to body administering the Plan as the “Committee”). The Committee will have full authority to select the individuals who will receive awards under the Plan, determine the form and amount of each of the awards to be granted and establish the terms and conditions of awards.
Number of Shares of Common Stock. The number of shares of the common stock that may be issued under the Plan is 2,500,000. Shares issuable under the Plan may be authorized but unissued shares or treasury shares. If there is a lapse, forfeiture, expiration, termination or cancellation of any award made under the Plan for any reason, the shares subject to the award will again be available for issuance. Any shares subject to an award that are delivered to us by a participant, or withheld by us on behalf of a participant, as payment for an award or payment of withholding taxes due in connection with an award will not again be available for issuance, and all such shares will count toward the number of shares issued under the Plan. The number of shares of common stock issuable under the Plan is subject to adjustment, in the event of any reorganization, recapitalization, stock split, stock distribution, merger, consolidation, split-up, spin-off, combination, subdivision, consolidation or exchange of shares, any change in the capital structure of the company or any similar corporate transaction. In each case, the Committee has the discretion to make adjustments it deems necessary to preserve the intended benefits under the Plan. No award granted under the Plan may be transferred, except by will, the laws of descent and distribution.
Eligibility. All employees designated as key employees for purposes of the Plan and all non-employee directors are eligible to receive awards under the Plan. On November 1, 2015, three key employees and all non-employee directors were eligible to participate in the Plan.
Awards to Participants. The Plan provides for discretionary awards of stock options, stock awards and stock unit awards to participants. Each award made under the Plan will be evidenced by a written award agreement specifying the terms and conditions of the award as determined by the Committee in its sole discretion, consistent with the terms of the Plan.
Stock Options. The Committee has the discretion to grant non-qualified stock options or incentive stock options to participants and to set the terms and conditions applicable to the options, including the type of option, the number of shares subject to the option and the vesting schedule; provided that the exercise price of each stock option will be the closing price of the common stock on the date on which the option is granted (“fair market value”), each option will expire ten years from the date of grant and no dividend equivalents may be paid with respect to stock options. It is intended that stock options qualify as “performance-based compensation” under Section 162(m) of the Code and thus be fully deductible by us for federal income tax purposes, to the extent permitted by law.
In addition, an incentive stock option granted to a key employee is subject to the following rules: (i) the aggregate fair market value (determined at the time the option is granted) of the shares of common stock with respect to which incentive stock options are exercisable for the first time by a key employee during any calendar year (under all incentive stock option plans of the company and its subsidiaries) cannot exceed $100,000, and if this limitation is exceeded, that portion of the incentive stock option that does not exceed the applicable dollar limit will be an incentive stock option and the remainder will be a non-qualified stock option; (ii) if an incentive stock option is granted to a key employee who owns stock possessing more than 10% of the total combined voting power of all class of stock of the company, the exercise price of the incentive stock option will be 110% of the closing price of the common stock on the date of grant and the incentive stock option will expire no later than five years from the date of grant; and (iii) no incentive stock option can be granted after ten years from the date the Plan was adopted.
Stock Awards. The Committee has the discretion to grant stock awards to participants. Stock awards will consist of shares of common stock granted without any consideration from the participant or shares sold to the participant for appropriate consideration as determined by the Board. The number of shares awarded to each participant, and the restrictions, terms and conditions of the award, will be at the discretion of the Committee. Subject to the restrictions, a participant will be a shareholder with respect to the shares awarded to him or her and will have the rights of a shareholder with respect to the shares, including the right to vote the shares and receive dividends on the shares; provided that dividends otherwise payable on any performance-based stock award will be held by us and will be paid to the holder of the stock award only to the extent the restrictions on such stock award lapse, and the Committee in its discretion can accumulate and hold such amounts payable on any other stock awards until the restrictions on the stock award lapse.
65
Stock Units. The Committee has the discretion to grant stock unit awards to participants. Each stock unit entitles the participant to receive, on a specified date or event set forth in the award agreement, one share of common stock or cash equal to the fair market value of one share on such date or event, as provided in the award agreement. The number of stock units awarded to each participant, and the terms and conditions of the award, will be at the discretion of the Committee. Unless otherwise specified in the award agreement, a participant will not be a shareholder with respect to the stock units awarded to him prior to the date they are settled in shares of common stock. The award agreement may provide that until the restrictions on the stock units lapse, the participant will be paid an amount equal to the dividends that would have been paid had the stock units been actual shares; provided that dividend equivalents otherwise payable on any performance-based stock units will be held by us and paid only to the extent the restrictions lapse, and the Committee in its discretion can accumulate and hold such amounts payable on any other stock units until the restrictions on the stock units lapse.
Performance-Based Awards. The Committee has the discretion to establish restrictions on the stock awards that qualify the awards as “performance-based compensation” under Section 162(m) of the Code so that they are fully deductible by us for federal income tax purposes. In such case, the Committee may establish performance goals for certain performance periods and targets for achievement of the performance goals, and the restrictions on the stock subject to the award will lapse if the performance goals and targets are achieved for the designated performance period. The performance goals will be based on one or more of the following criteria: (i) net earnings or net income (before or after taxes); (ii) earnings per share; (iii) net sales or revenue growth; (iv) net operating profit or income (including as a percentage of sales); (v) return measures (including, but not limited to, return on assets, capital, invested capital, equity, sales, or revenue); (vi) cash flow (including, but not limited to, operating cash flow, free cash flow, cash flow return on equity, and cash flow return on investment); (vii) earnings before or after taxes, interest, depreciation, and/or amortization; (viii) gross or operating margins; and (ix) share price (including, but not limited to, growth measures and total shareholder return). Performance goals may be absolute in their terms or measured against or in relationship to the performance of other companies or indices selected by the Committee. The performance goals may be particular to one or more lines of business or subsidiaries or may be based on our performance as a whole. The performance goals may be identical for all participants for a given performance period or, at the discretion of the Committee, may differ among participants. In addition, performance goals may be adjusted for any events or occurrences (including acquisition expenses, extraordinary charges, losses from discontinued operations, restatements and accounting charges and restructuring expenses), as may be determined by the Committee.
Payment for Stock Options and Withholding Taxes. The Committee may make one or more of the following methods available for payment of any award, including the exercise price of a stock option, and for payment of the minimum required tax obligation associated with an award: (i) cash; (ii) cash received from a broker-dealer to whom the holder has submitted an exercise notice together with irrevocable instructions to deliver promptly to us the amount of sales proceeds from the sale of the shares subject to the award to pay the exercise price or withholding tax; (iii) by directing us to withhold shares of common stock otherwise issuable in connection with the award having a fair market value equal to the amount required to be withheld; and (iv) by delivery of previously acquired shares of common stock that are acceptable to the Committee and that have an aggregate fair market value on the date of exercise equal to the exercise price or withholding tax, or certification of ownership by attestation of such previously acquired shares.
Provisions Relating to a “Change in Control” of the Company. Notwithstanding any other provision of the Plan or any award agreement, in the event of a “Change in Control” of the company, the Committee has the discretion to provide that all outstanding awards will become fully exercisable, all restrictions applicable to all awards will terminate or lapse, and performance goals applicable to any stock awards will be deemed satisfied at the highest target level. In addition, upon such Change in Control, the Committee has sole discretion to provide for the purchase of any outstanding stock option for cash equal to the difference between the exercise price and the then fair market value of the common stock subject to the option had the option been currently exercisable, make such adjustment to any award then outstanding as the Committee deems appropriate to reflect such Change in Control and cause any such award then outstanding to be assumed by the acquiring or surviving corporation after such Change in Control.
Amendment of Award Agreements; Amendment and Termination of the Plan; Term of the Plan. The Committee may amend any award agreement at any time, provided that no amendment may adversely affect the right of any participant under any agreement in any material way without the written consent of the participant, unless such amendment is required by applicable law, regulation or stock exchange rule.
66
The Board may terminate, suspend or amend the Plan, in whole or in part, from time to time, without the approval of the shareholders, unless such approval is required by applicable law, regulation or stock exchange rule, and provided that no amendment may adversely affect the right of any participant under any outstanding award in any material way without the written consent of the participant, unless such amendment is required by applicable law, regulation or rule of any stock exchange on which the shares are listed.
Notwithstanding the foregoing, neither the Plan nor any outstanding award agreement can be amended in a way that results in the repricing of a stock option. Repricing is broadly defined to include reducing the exercise price of a stock option or cancelling a stock option in exchange for cash, other stock options with a lower exercise price or other stock awards. (This prohibition on repricing without shareholder approval does not apply in case of an equitable adjustment to the awards to reflect changes in the capital structure of the company or similar events.)
No awards may be granted under the Plan on or after the tenth anniversary of the effective date of the Plan.
RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
We were incorporated in July 2015. In August 2015, in exchange for the issuance of 630,000 shares of common stock, we acquired the rights to the license agreement with MD Anderson covering our WP1122 Portfolio held by IntertechBio Corporation. Waldemar Priebe, chairman of our Scientific Advisory Board, and Don Picker, our president and chief operating officer, are shareholders of IntertechBio and control the voting and dispositive power over the shares held by IntertechBio.
In August 2015, in exchange for the issuance of 1,431,000 shares of common stock, we acquired the rights to the Annamycin data related to the original Annamycin IND and the development of Annamycin held by AnnaMed, Inc., a company controlled by Walter Klemp.
Prior to the effective date of the registration statement of which this prospectus is a part, in exchange for the issuance of 629,000 shares of common stock, we will enter into an agreement with HPI whereby HPI will agree to terminate its option to sublicense certain rights to the WP1066 Portfolio and to enter into a co-development agreement with us, allowing us to benefit from HPI’s grant-funded development efforts currently under way. Waldemar Priebe and Don Picker are shareholders of HPI, and Dr. Priebe will have the voting and dispositive power over the shares held by HPI. For a detailed discussion of the HPI co-development agreement, see “Business – Our License Agreements.”
Prior to the effective date of the registration statement of which this prospectus is a part, Moleculin LLC, a Texas limited liability company, will be merged with and into our company. Moleculin is the holder of the license agreement with MD Anderson covering our WP 1066 Portfolio. As a result of the merger, we will issue the equity interests holders of Moleculin an aggregate of 1,000,000 shares of our common stock. Waldemar Priebe, Walter Klemp and Don Picker are members of Moleculin and will receive 6,046 shares, 22,795 shares and 6,046 shares, respectively, of our common stock as a result of the merger. In addition, Walter Klemp and Don Picker are members of the board of Moleculin.
As part of the acquisition process of Moleculin LLC by MBI, we will provide operating loans for Moleculin in the estimated range of $200,000 to $225,000 for Moleculin to meet its license obligations with MD Anderson, (of which $188,000 are due on January 31, 2016) and maintain its minimal operations. These loans will be in the form of promissory notes payable with an interest rate of 8% from Moleculin to us that will be forgiven at the completion of the IPO.
In August 2015, in Messrs. Klemp, Picker and Priebe purchased 1,000,000 shares, 500,000 shares and 3,000,000 shares of our common stock, respectively, at a purchase price of $0.001 per share.
On July 1, 2013, Moleculin agreed to reimburse HPI for laboratory space used by Moleculin from January 2010 through December 2013 in the amount of $31,764. Moleculin stopped using the space in 2014. On September 1, 2013, Moleculin agreed to reimburse HPI for laboratory equipment used by Moleculin for the period from January 2009 through December 2013 in the amount of $35,000. Moleculin stopped using the equipment in 2014. During the year ended December 31, 2013, Moleculin paid $100,000 for laboratory services provided by HPI for the five years prior to August 1, 2013. We do not intend to utilize HPI for such services in the future.
Policies and Procedures for Related Party Transactions
Our audit committee charter will provide that our audit committee will be responsible for reviewing and approving in advance any related party transaction. This will cover, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships in which we were or are to be a participant, where the amount involved exceeds $120,000 and a related person had or will have a direct or indirect material interest, including, without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. All of the transactions described in this section occurred prior to the creation of our audit committee and the adoption of this policy.
67
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth information, as of January 27, 2016, regarding beneficial ownership of our common stock by:
| • | each of our directors; | |
| • | each of our executive officers; | |
| • | all directors and executive officers as a group; and | |
| • | each person, or group of affiliated persons, known by us to beneficially own more than five percent of our shares of common stock. |
Beneficial ownership is determined according to the rules of the SEC, and generally means that person has beneficial ownership of a security if he or she possesses sole or shared voting or investment power of that security, and includes options that are currently exercisable or exercisable within 60 days. Each director or officer, as the case may be, has furnished us with information with respect to beneficial ownership. Except as otherwise indicated, we believe that the beneficial owners of common stock listed below, based on the information each of them has given to us, have sole investment and voting power with respect to their shares, except where community property laws may apply. Except as otherwise noted below, the address for each person or entity listed in the table is c/o Moleculin Biotech, Inc., 2575 West Bellfort, Suite 333, Houston, Texas 77054.
| Shares beneficially owned prior to Offering (1) | Percentage owned prior to Offering (1) | Percentage beneficially owned after Offering | ||||||||||||||
| Minimum | Maximum | |||||||||||||||
| Name and Address of Beneficial Owner | ||||||||||||||||
| Directors and Executive Officers | ||||||||||||||||
| Walter V. Klemp | 2,453,795 (2) | |||||||||||||||
| Donald Picker | 1,136,046 (3)(4) | |||||||||||||||
| Louis Ploth | - | |||||||||||||||
| Directors and Executive Officers as a Group (3 persons) | 3,670,841 | |||||||||||||||
| 5% or greater shareholders | ||||||||||||||||
| Waldemar Priebe | 4,257,834 (3)(5)(6) | |||||||||||||||
| AnnaMed, Inc. | 1,431,000 (2) | |||||||||||||||
| IntertechBio Corp. | 630,000 (3) | |||||||||||||||
| Houston Pharmaceuticals, Inc. | 629,000 (6) | |||||||||||||||
| * | Less than 1%. |
(1) Assumes and gives effect to (i) the completion of the merger with Moleculin and the issuance of 1,000,000 shares in the aggregate to Moleculin’s equity holders, and (ii) the completion of the issuance of 629,000 shares of common stock to HPI in exchange for HPI’s agreement to terminate its option to sublicense certain rights to the WP1066 Portfolio and to enter into a co-development agreement with us.
(2) The 1,431,000 shares held by AnnaMed, Inc. have been included in the amount for Mr. Klemp. Mr. Klemp has voting and dispositive power over the shares held by AnnaMed, Inc. Includes 22,795 shares issuable to Mr. Klemp upon the completion of the merger with Moleculin.
(3) The 630,000 shares held by IntertechBio Corp. have been included in the amounts for Drs. Picker and Priebe. Drs. Picker and Priebe have voting and dispositive power over the shares held by IntertechBio Corp.
(4) Includes 6,046 shares issuable to Dr. Picker upon the completion of the merger with Moleculin.
(5) Includes 17,834 shares issuable to Dr. Priebe upon the completion of the merger with Moleculin.
(6) The 629,000 shares held by HPI have been included in the amount for Dr. Priebe. Dr. Priebe has voting and dispositive power over the shares held by HPI.
68
The following summary is a description of the material terms of our capital stock and is not complete. You should also refer to the Moleculin Biotech, Inc. certificate of incorporation and bylaws, which are included as exhibits to the registration statement of which this prospectus forms a part, and the applicable provisions of the Delaware General Corporation Law.
Our amended and restated certificate of incorporation to be in effect prior to the completion of this offering will authorized us to issue up to 75,000,000 shares of common stock, par value $0.001 per share, and 5,000,000 shares of preferred stock, $0.001 par value per share. Our 8% unsecured promissory notes will be automatically converted into 3,749,557 shares of common stock contemporaneously with the closing of this offering (exclusive of shares issuable for accrued interest under such notes). No holder of these notes will be permitted to convert such notes to the extent that the holder or any of its affiliates would beneficially own in excess of 4.99% of our common stock after such conversion. The number of shares set forth above assumes no such limitation on the conversion of the notes. After giving effect to the conversion of our notes contemporaneously with the closing of this offering, we will have [*] shares of common stock outstanding (if the minimum number of shares are sold) and [*] shares of common stock outstanding (if the maximum number of shares are sold) immediately after the closing of this offering.
Common Stock
Shares of our common stock have the following rights, preferences and privileges:
Voting
Each holder of common stock is entitled to one vote for each share of common stock held on all matters submitted to a vote of stockholders. Any action at a meeting at which a quorum is present will be decided by a majority of the voting power present in person or represented by proxy, except in the case of any election of directors, which will be decided by a plurality of votes cast. There is no cumulative voting.
Dividends
Holders of our common stock are entitled to receive dividends when, as and if declared by the our board of directors out of funds legally available for payment, subject to the rights of holders, if any, of any class of stock having preference over the common stock. Any decision to pay dividends on our common stock will be at the discretion of our board of directors. Our board of directors may or may not determine to declare dividends in the future. See “Dividend Policy.” The board’s determination to issue dividends will depend upon our profitability and financial condition any contractual restrictions, restrictions imposed by applicable law and the SEC, and other factors that our board of directors deems relevant.
Liquidation Rights
In the event of a voluntary or involuntary liquidation, dissolution or winding up of the company, the holders of our common stock will be entitled to share ratably on the basis of the number of shares held in any of the assets available for distribution after we have paid in full, or provided for payment of, all of our debts and after the holders of all outstanding series of any class of stock have preference over the common stock, if any, have received their liquidation preferences in full.
Other
Our issued and outstanding shares of common stock are fully paid and nonassessable. Holders of shares of our common stock are not entitled to preemptive rights. Shares of our common stock are not convertible into shares of any other class of capital stock, nor are they subject to any redemption or sinking fund provisions.
Preferred Stock
We are authorized to issue up to 5,000,000 shares of preferred stock. Our certificate of incorporation authorizes the board to issue these shares in one or more series, to determine the designations and the powers, preferences and relative, participating, optional or other special rights and the qualifications, limitations and restrictions thereof, including the dividend rights, conversion or exchange rights, voting rights (including the number of votes per share), redemption rights and terms, liquidation preferences, sinking fund provisions and the number of shares constituting the series. Our board of directors could, without stockholder approval, issue preferred stock with voting and other rights that could adversely affect the voting power and other rights of the holders of common stock and which could have the effect of making it more difficult for a third party to acquire, or of discouraging a third party from attempting to acquire, a majority of our outstanding voting stock.
69
Convertible Notes
In August and September 2015, we issued two 8% convertible notes in an aggregate of $250,000 in principal amount of convertible notes to one investor, which principal and accrued interest will automatically convert into shares of common stock upon the closing of this offering at a conversion rate of $0.1299 per share. In October 2015, we issued two 8% convertible notes in an aggregate of $200,000 in principal amount of convertible notes to two investors, which principal and accrued interest will automatically convert into shares of common stock upon the closing of this offering at a conversion rate of $0.20 per share. None of the foregoing convertible notes will be convertible by the holder of such notes to the extent (and only to the extent) that the holder or any of its affiliates would beneficially own in excess of 4.99% of our common stock. For purposes of the limitation described in this paragraph, beneficial ownership and all determinations and calculations are determined in accordance with Section 13(d) of the Exchange Act and the rules and regulations promulgated thereunder.
The holders of our convertible notes have agreed to the following lock-up of the common stock issuable upon conversion of the notes:
| · | begin 90 days after the initial closing of this offering and until the one-year anniversary of the initial closing of this offering, (a) a holder of our convertible notes will be able to sell 1% of the number of shares of common stock into which such note is converted at the closing of the offering on a monthly basis, subject to a maximum sale on any trading day of 4% of the daily volume; (b) if our common stock price is over $7.00 per share for five consecutive trading days then the holder can sell up to 3% of the number of shares of common stock underlying the note on a monthly basis, subject to a maximum sale on any trading day of 4% of the daily volume; (c) if our common stock price is over $10.00 per share for five consecutive trading days then the holder can sell up to an additional 5% of the number of shares of common stock underlying the note on a monthly basis, subject to a maximum sale on any trading day of 7% of the daily volume; and (d) if our common stock price is over $14.00 per share then the holder is not restricted from making any sales until such time as our common stock price falls back below $14.00 per share; and |
| · | thereafter, until the two-year anniversary of the initial closing of this offering, the holder can sell on any trading day 10% of the daily volume; provided that if our common stock price is over $10.00 per share then the holder is not restricted from making any sales until such time as the common stock falls back below $10.00 per share. |
Certificate of Incorporation and Bylaw Provisions
Our certificate of incorporation and bylaws include a number of anti-takeover provisions that may have the effect of encouraging persons considering unsolicited tender offers or other unilateral takeover proposals to negotiate with our board of directors rather than pursue non-negotiated takeover attempts. These provisions include:
Advance Notice Requirements. Our bylaws establish advance notice procedures with regard to stockholder proposals relating to the nomination of candidates for election as directors or new business to be brought before meetings of stockholders. These procedures provide that notice of stockholder proposals must be timely and given in writing to our corporate Secretary. Generally, to be timely, notice must be received at our principal executive offices not fewer than 120 calendar days prior to the first anniversary date on which our notice of meeting and related proxy statement were mailed to stockholders in connection with the previous year’s annual meeting of stockholders. The notice must contain the information required by the bylaws, including information regarding the proposal and the proponent.
Special Meetings of Stockholders. Our bylaws provides that special meetings of stockholders may be called at any time by only the Chairman of the Board, the Chief Executive Officer, the President or the board of directors, or in their absence or disability, by any vice president.
No Written Consent of Stockholders. Our certificate of incorporation and bylaws provide that any action required or permitted to be taken by stockholders must be effected at a duly called annual or special meeting of stockholders and may not be effected by any consent in writing by such stockholders.
Exclusive Forum Provision. Our certificate of incorporation provides that the Court of Chancery of the State of Delaware shall be the sole and exclusive forum for (i) any derivative action or proceeding brought our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors or officers to us or our stockholders, (iii) any action asserting a claim against us arising pursuant to any provision of the Delaware General Corporation Law (the “DGCL”), or our certificate of incorporation or the bylaws, and (iv) any action asserting a claim against us governed by the internal affairs doctrine. This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and employees. Alternatively, a court could find these provisions of our certificate of incorporation to be inapplicable or unenforceable in respect of one or more of the specified types of actions or proceedings, which may require us to incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business and financial condition.
70
Amendment of Bylaws. Our stockholders may amend any provisions of our bylaws by obtaining the affirmative vote of the holders of a majority of each class of issued and outstanding shares of our voting securities, at a meeting called for the purpose of amending and/or restating our bylaws.
Preferred Stock. Our certificate of incorporation authorizes our board of directors to create and issue rights entitling our stockholders to purchase shares of our stock or other securities. The ability of our board to establish the rights and issue substantial amounts of preferred stock without the need for stockholder approval may delay or deter a change in control of us. See “Preferred Stock” above.
Delaware Takeover Statute
We are subject to Section 203 of the DGCL which, subject to certain exceptions, prohibits a Delaware corporation from engaging in any “business combination” (as defined below) with any interested stockholder for a period of three years following the date that such stockholder became an interested stockholder, unless: (1) prior to such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; (2) on consummation of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding those shares owned (x) by persons who are directors and also officers and (y) by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to this plan will be tendered in a tender or exchange offer; or (3) on or subsequent to such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2⁄3% of the outstanding voting stock that is not owned by the interested stockholder.
Section 203 of the DGCL defines generally “business combination” to include: (1) any merger or consolidation involving the corporation and the interested stockholder; (2) any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; (3) subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; (4) any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; or (5) the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. In general, Section 203 defines an “interested stockholder” as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by such entity or person.
Limitations on Liability and Indemnification of Officers and Directors
Our certificate of incorporation and bylaws limit the liability of our officers and directors and provide that we will indemnify our officers and directors, in each case, to the fullest extent permitted by the Delaware General Corporation Law. We expect to obtain additional directors’ and officers’ liability insurance coverage prior to the completion of this offering.
Listing
We intend to apply to list our common stock on the Nasdaq Capital Market under the symbol “MBRX”.
Transfer Agent
The transfer agent for our common stock is [*].
71
SHARES ELIGIBLE FOR FUTURE SALE
Future sales of substantial amounts of common stock in the public market after this offering could adversely affect market prices prevailing from time to time and could impair our ability to raise capital through the sale of our equity securities. We are unable to estimate the number of shares of common stock that may be sold in the future.
After giving effect to the conversion of our notes contemporaneously with the closing of this offering, we will have [*] shares of common stock outstanding (if the minimum number of shares are sold) and [*] shares of common stock outstanding (if the maximum number of shares are sold) immediately after the closing of this offering. All of the shares sold in this offering will be freely tradable without restriction under the Securities Act unless purchased by one of our affiliates as that term is defined in Rule 144 under the Securities Act, which generally includes directors, officers or 10% stockholders. None of the holders of shares of our common stock or securities exercisable for or convertible into shares of our common stock have any registration rights.
Lock-Up
Management Lock-Up
Our executive officers, directors, and major stockholders, have agreed with the underwriters not to offer, sell, dispose of or hedge any shares of our common stock, subject to specified limited exceptions and extensions described elsewhere in this prospectus, during the period continuing through the date that is twelve months (subject to extension) after the date of this prospectus. After such twelve month period and until 24 months from the closing of this offering, such individuals and entities may sell their shares pursuant to the following criteria:
| · | if our common stock price is over $7.00 per share for five consecutive trading days then the holder can sell up to 3% of their holdings on a monthly basis, subject to a maximum sale on any trading day of 4% of the daily volume; |
| · | if our common stock price is over $10.00 per share for five consecutive trading days then the holder can sell up to an additional 5% of their holdings on a monthly basis, subject to a maximum sale on any trading day of 7% of the daily volume; and |
| · | if our common stock price is over $14.00 per share then the holder is not restricted from making any sales until such time as our common stock price falls back below $14.00 per share. |
From the end of the preceding 24 month period until the three-year anniversary of the initial closing of this offering, the holders can sell on any trading day 10% of the daily volume; provided that if our common stock price is over $10.00 per share then the holder is not restricted from making any sales until such time as the common stock falls back below $10.00 per share.
Moleculin LLC Members Lock-Up
As a result of our planned merger with Moleculin LLC, we will issue the holders of Moleculin LLC equity interests an aggregate of 1,000,000 shares of our common stock. Subject to and except as described above under “-Management Lock-Up,” such holders will agree to the following lock-up on the shares of our common stock that they receive:
| · | no sales may be made for six months from the closing date of this offering; |
| · | starting on the first day after the initial six-month period, the holders will be able to sell collectively 27,000 shares; |
| · | starting on the first day after seven months from the closing date of this offering, the holders will be able to sell collectively an additional 68,000 shares; |
| · | starting on the first day after eight months from the closing date of this offering, the holders will be able to sell collectively an additional 49,000 shares; |
| · | starting on the first day after nine months from the closing date of this offering, the holders will be able to sell collectively an additional 87,000 shares; |
| · | starting on the first day after ten months from the closing date of this offering, through the last day of the fifteenth month from the closing date of this offering, each holder may sell the lesser of 5,000 shares or 10% of their original holdings per month, subject to a cap of 3% of the previous day’s value; and |
| · | starting on the first day after the sixteenth month from the closing date of this offering, each holder can sell on any trading day 10% of the daily volume; provided that if our common stock price is over $10.00 per share then the holder will not be restricted from making any sales until such time as the common stock falls back below $10.00 per share. |
See Section “Description of Capital Stock - Convertible Notes” above for a discussion of the lock-up of the shares underlying such notes.
Rule 144
Shares of common stock held by any of our affiliates, as that term is defined in Rule 144 of the Securities Act, as well as shares held by our current stockholders, may be resold only pursuant to further registration under the Securities Act or in transactions that are exempt from registration under the Securities Act. In general, under Rule 144 as currently in effect, beginning 90 days after our Form S-1 Registration Statement becomes effective, any of our affiliates would be entitled to sell, without further registration, within any three-month period a number of shares that does not exceed the greater of:
| • | 1% of the number of shares of common stock then outstanding, which will equal approximately [*] shares immediately after this offering; or |
| • | the average weekly trading volume of the common stock during the four calendar weeks preceding the filing of a Form 144 with respect to the sale. |
Sales under Rule 144 by our affiliates will also be subject to manner of sale provisions and notice requirements and to the availability of current public information about us.
Stock Plan
We intend to file a registration statement on Form S-8 under the Securities Act of 1933, as amended, which will register 2,500,000 shares of common stock underlying stock options or restricted stock awards for issuance under our 2015 Stock Plan. Subject to any vesting requirements, these shares registered on Form S-8 will be eligible for resale in the public markets without restriction, subject to Rule 144 limitations applicable to affiliates.
72
We have entered into an underwriting agreement with Bonwick Capital Partners LLC, for itself and as sole representative (the “Representative”) of the underwriters named therein, with respect to the shares of our common stock in this offering. Under the terms and subject to the conditions contained in the underwriting agreement, we have agreed to issue and sell to the public through the underwriters, and the underwriters have agreed to offer and sell, up to 2,000,000 shares of our common stock, on a best efforts basis.
The underwriting agreement provides that the obligation of the underwriters to arrange for the offer and sale of the shares of our common stock, on a best efforts basis, is subject to certain conditions precedent, including but not limited to (i) receipt of a listing approval letter from the Nasdaq Capital Market, (ii) delivery of legal opinions and (iii) delivery of auditor comfort letters. The underwriters are under no obligation to purchase any shares of our common stock for their own account. As a “best efforts” offering, there can be no assurance that the offering contemplated hereby will ultimately be consummated, or even if consummated that we will in fact obtain a listing on the Nasdaq Capital Market. The underwriters may, but are not obligated to, retain other selected dealers that are qualified to offer and sell the shares and that are members of the Financial Industry Regulatory Authority, Inc. The underwriters propose to offer the shares to investors at the public offering price, and will receive the underwriting commissions, set forth on the cover of this prospectus. The gross proceeds of this offering will be deposited at Signature Bank, New York, New York, in an escrow account established by us, until we have sold a minimum of 1,400,000 shares of common stock and otherwise satisfy the listing conditions to trade our common stock on the Nasdaq Capital Market unless sooner withdrawn or canceled by us or the Underwriters, the offering will continue until (i) not less than 1,400,000 shares of common stock have been sold, or (ii) close of business on [*], 2016 (60 days after the date of this Prospectus), unless extended by us and the Underwriters to not later than [*], 2016 (90 days after the date of this Prospectus). Once we satisfy the minimum stock sale and Nasdaq listing conditions, the funds will be released to us.
We anticipate the shares of our common stock will be listed on the Nasdaq Capital Market under the symbol “MBRX.” In order to list, the Nasdaq Capital Market requires that, among other criteria, at least 1,000,000 publicly-held shares of our common stock be outstanding, the shares be held in the aggregate by at least 300 round lot holders, the market value of the publicly-held shares of our common stock be at least $15.0 million, our stockholders’ equity after giving effect to the sale of our shares in this offering be at least $4.0 million, the bid price per share of our common stock be $4.00 or more, and there be at least three registered and active market makers for our common stock. If the application is approved, trading of our shares on the Nasdaq Capital Market is expected to begin within five days after the date of initial issuance of the common stock.
The following table and the two succeeding paragraphs summarize the underwriting compensation and estimated expenses we will pay:
| Public Offering Price | Underwriting Commissions | Proceeds to Us, Before Expenses | ||||||||||
| Per share | $ | $ | $ | |||||||||
| Total minimum offering | $ | $ | $ | |||||||||
| Total maximum offering | $ | $ | $ | |||||||||
We have agreed to reimburse the underwriters for expenses incurred relating to the offering, including all actual fees and expenses incurred by the underwriters in connection with, among other things, due diligence costs, the underwriters’ “road show” expenses, which shall not exceed $25,000, and the fees and expenses of the underwriters’ counsel. The fees and expenses of underwriters’ counsel shall not exceed $100,000. We estimate that the total expenses of this offering, excluding underwriting commissions described above, will be approximately $500,000. We have also agreed to pay the representative a financial advisory fee of $50,000 at the closing of this offering.
As additional compensation to the underwriters, upon consummation of this offering, we will issue to the underwriters or their designees warrants to purchase an aggregate number of shares of our common stock equal to 7% of the number of shares of common stock issued in this offering, at an exercise price per share equal to 125.0% of the initial public offering price (the “Underwriter Warrants”). The Underwriter Warrants and the underlying shares of common stock will not be exercised, sold, transferred, assigned, or hypothecated or be the subject of any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the Underwriter Warrants by any person for a period of 180 days from the effective date of the registration statement for this offering in accordance with FINRA Rule 5110. The Underwriter Warrants will expire on the fifth anniversary of the effective date of the registration statement for this offering.
The underwriters have informed us that they may provide an allowance not in excess of $[*] per share to other dealers out of the underwriters’ commission of $[*] per share. No underwriters or selling group members will receive any fees or warrants in connection with the purchase by any of our officers or directors or their respective affiliates of shares of common stock in this offering.
73
A prospectus in electronic format may be made available on the websites maintained by the underwriters, or selling group members, if any, participating in the offering. The underwriters may agree to allocate a number of shares to selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriters and selling group members that may make Internet distributions on the same basis as other allocations.
We have agreed that we will not: (i) offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend, or otherwise transfer or dispose of, directly or indirectly, any shares of capital stock of our company or any securities convertible into or exercisable or exchangeable for shares of capital stock of our company; (ii) file or cause to be filed any registration statement with the SEC relating to the offering of any shares of capital stock of our company or any securities convertible into or exercisable or exchangeable for shares of capital stock of our company; or (iii) enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of capital stock of our company, whether any such transaction described in clause (i), (ii) or (iii) above is to be settled by delivery of shares of capital stock of our company or such other securities, in cash or otherwise, in each case without the prior consent of the Representative for a period of twelve months after the date of this prospectus, other than (A) the shares of our common stock to be sold hereunder, (B) the issuance by us of shares of our common stock upon the exercise of a stock option or warrant or the conversion of a security outstanding on the date of this offering, hereafter issued pursuant to our currently existing or hereafter adopted equity compensation plans or employment or consulting agreements or arrangements of which the representative has been advised in writing or which have been filed with the Commission or (C) the issuance by us of stock options or shares of capital stock of our company under any currently existing or hereafter adopted equity compensation plan or employment/consulting agreements or arrangements of our company.
Our directors, executive officers and holders of 5% or more of our outstanding shares of common stock will enter into lock-up agreements with the representative prior to the commencement of this offering pursuant to which each of these persons or entities, will not, without the prior written consent of the Representative, (i) offer, sell, dispose of or hedge any shares of our common stock, during the period continuing through the date that is twelve months (subject to extension) after the date of this prospectus; (ii) after such twelve month period and until 24 months from the closing of this offering, such individuals and entities may sell their shares pursuant to the following criteria: (A) if our common stock price is over $7.00 per share for five consecutive trading days then the holder can sell up to 3% of such holder’s holdings on a monthly basis, subject to a maximum sale on any trading day of 4% of the daily volume; (B) if our common stock price is over $10.00 per share for five consecutive trading days then the holder can sell up to an additional 5% of such holder’s holdings on a monthly basis, subject to a maximum sale on any trading day of 7% of the daily volume; and (C) if our common stock price is over $14.00 per share then the holder is not restricted from making any sales until such time as our common stock price falls back below $14.00 per share; (iii) from the end of the 24 month period referenced in clause (ii) above until the three-year anniversary of the initial closing of this offering, the holders can sell on any trading day 10% of the daily volume; provided that if our common stock price is over $10.00 per share then the holder is not restricted from making any sales until such time as the common stock falls back below $10.00 per share. Subject to and except as described in this paragraph, the holders of the Moleculin LLC equity interests will agree to the lock-up of the shares of our common stock they receive as set forth under the caption “Shares Eligible for Future Sale – Moleculin LLC Members Lock-up.” In addition, the holders of our convertible notes have agreed to the lock-up of the shares of our common stock issuable upon conversion of these notes as set forth under the caption “Description of Capital Stock – Convertible Notes.”
The underwriting agreement provides that we will indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, or contribute to payments the underwriters may be required to make in respect thereof.
We have applied to have our common stock approved for listing on the Nasdaq Capital Market under the symbol “MBRX.” If the application is approved, trading of our common stock on the Nasdaq Capital Market is expected to begin within five days after the date of initial issuance of the common stock. We will not consummate and close this offering without a listing approval letter from the Nasdaq Capital Market. Our receipt of a listing approval letter is not the same as an actual listing on the Nasdaq Capital Market. The listing approval letter will serve only to confirm that, if we sell a number of shares in this best efforts offering sufficient to satisfy applicable listing criteria, our common stock will in fact be listed.
Prior to this offering, there has been no public market for our common stock. The initial public offering price has been determined by negotiations between us and the underwriters. In determining the initial public offering price, we and the underwriters have considered a number of factors including:
| · | the information set forth in this prospectus and otherwise available to the underwriters; |
| · | our prospects and the history and prospects for the industry in which we compete; |
| · | an assessment of our management; |
| · | our prospects for future earnings; |
| · | the general condition of the securities markets at the time of this offering; |
| · | the recent market prices of, and demand for, publicly traded common stock of generally comparable companies; and |
| · | other factors deemed relevant by the underwriters and us. |
74
Neither we nor the underwriters can assure investors that an active trading market will develop for shares of our common stock, or that the shares will trade in the public market at or above the initial public offering price.
The validity of the securities offered hereby will be passed upon for us by Schiff Hardin LLP, Washington, DC. DLA Piper LLP (US), New York, New York is acting as counsel to the underwriters in connection with this offering.
The financial statements of Moleculin Biotech, Inc. as of September 30, 2015 and for the period from July 28, 2015 (inception) to September 30, 2015 and of Moleculin LLC as of and for the years ended December 31, 2014 and 2013 included in this prospectus have been audited by GBH CPAs, PC, an independent registered public accounting firm, as stated in their reports appearing herein. Such financial statements have been so included in reliance upon the report of such firm given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act for the shares of common stock being offered by this prospectus. This prospectus, which is part of the registration statement, does not contain all of the information included in the registration statement and the exhibits. For further information about us and the common stock offered by this prospectus, you should refer to the registration statement and its exhibits. References in this prospectus to any of our contracts or other documents are not necessarily complete, and you should refer to the exhibits attached to the registration statement for copies of the actual contract or document. You may read and copy any document that we file at the SEC’s public reference room located at 100 F Street, NE, Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the public reference rooms. SEC filings are also available to the public at the SEC’s website at www.sec.gov.
We will be subject to the reporting and information requirements of the Exchange Act and, as a result, will file periodic and current reports, proxy statements and other information with the SEC. We expect to make our periodic reports and other information filed with or furnished to the SEC, available, free of charge, through our website as soon as reasonably practicable after those reports and other information are filed with or furnished to the SEC. Additionally, these periodic reports, proxy statements and other information will be available for inspection and copying at the public reference room and website of the SEC referred to above.
75
INDEX TO FINANCIAL STATEMENTS
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
of
Moleculin Biotech, Inc.
Houston, Texas
We have audited the accompanying balance sheet of Moleculin Biotech, Inc. as of September 30, 2015 and the related statements of operations, changes in stockholders’ deficit, and cash flows for the period from July 28, 2015 (Inception) to September 30, 2015. Moleculin Biotech, Inc.’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Moleculin Biotech, Inc. as of September 30, 2015 and the results of its operations and cash flows for the period from July 28, 2015 (Inception) to September 30, 2015 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that Moleculin Biotech, Inc. will continue as a going concern. As discussed in Note 1 to the financial statements, Moleculin Biotech, Inc. incurred an accumulated loss and has not yet generated any revenue from operations that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ GBH CPAs, PC
GBH CPAs, PC
www.gbhcpas.com
Houston, Texas
December 3, 2015
| F-2 |
Balance Sheet
| September 30, 2015 | ||||
| ASSETS | ||||
| Current Assets: | ||||
| Cash and cash equivalents | $ | 87,340 | ||
| Prepaid expenses | 23,000 | |||
| Total Assets | $ | 110,340 | ||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||
| Current Liabilities: | ||||
| Accounts payable and accrued liabilities | $ | 63,010 | ||
| Accounts payable and accrued liabilities – related parties | 19,584 | |||
| Notes payable | 250,000 | |||
| Total Liabilities | 332,594 | |||
| Commitments and contingencies | ||||
| Stockholders’ Deficit: | ||||
| Common stock, $0.001 par value; 20,000,000 shares authorized, 6,661,000 shares issued and outstanding | 6,661 | |||
| Subscription receivable | (4,600 | ) | ||
| Accumulated deficit | (224,315 | ) | ||
| Total Stockholders’ Deficit | (222,254 | ) | ||
| Total Liabilities and Stockholders’ Deficit | $ | 110,340 | ||
See accompanying notes to financial statements.
| F-3 |
Statement of Operations
| From July 28, 2015 (Inception) to September 30, 2015 | ||||
| Revenue | $ | - | ||
| Operating expenses: | ||||
| Research and development | 38,409 | |||
| General and administrative | 184,344 | |||
| Total operating expenses | 222,753 | |||
| Loss from operations | (222,753 | ) | ||
| Other expense: | ||||
| Interest expense | (1,562 | ) | ||
| Total other expense | (1,562 | ) | ||
| Net loss | $ | (224,315 | ) | |
| Loss per common share - basic and diluted | $ | (0.05 | ) | |
| Weighted average number of common shares outstanding - basic and diluted | 4,320,015 | |||
See accompanying notes to financial statements.
| F-4 |
Statement of Changes in Stockholders’ Deficit
From July 28, 2015 (Inception) to September 30, 2015
| Common Stock | Subscription | Accumulated | ||||||||||||||||||
| Shares | Amount | Receivable | Deficit | Total | ||||||||||||||||
| Balance at July 28, 2015 (Inception) | - | $ | - | $ | - | $ | - | $ | - | |||||||||||
| Issuance of shares for cash | 4,600,000 | 4,600 | (4,600 | ) | - | - | ||||||||||||||
| Shares issued for licenses used for research and development | 2,061,000 | 2,061 | - | - | 2,061 | |||||||||||||||
| Net loss | - | - | - | (224,315 | ) | (224,315 | ) | |||||||||||||
| Balance at September 30, 2015 | 6,661,000 | $ | 6,661 | $ | (4,600 | ) | $ | (224,315 | ) | $ | (222,254 | ) | ||||||||
See accompanying notes to financial statements.
| F-5 |
Statement of Cash Flows
| From July 28, 2015 (Inception) to September 30, 2015 | ||||
| Cash Flows From Operating Activities: | ||||
| Net loss | $ | (224,315 | ) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||
| Shares issued for licenses used for research and development | 2,061 | |||
| Changes in operating assets and liabilities: | ||||
| Prepaid expenses | (23,000 | ) | ||
| Accounts payable and accrued liabilities | 63,010 | |||
| Accounts payable and accrued liabilities – related parties | 19,584 | |||
| Net Cash Used In Operating Activities | (162,660 | ) | ||
| Cash Flows From Financing Activities: | ||||
| Proceeds from notes payable | 250,000 | |||
| Net Cash Provided By Financing Activities | 250,000 | |||
| Net change in cash and cash equivalents | 87,340 | |||
| Cash at beginning of period | - | |||
| Cash at end of period | $ | 87,340 | ||
| Supplemental cash flow disclosures: | ||||
| Cash paid for interest | $ | - | ||
| Cash paid for income taxes | $ | - | ||
| Non-cash investing and financing activities: | ||||
| Shares subscribed | $ | 4,600 | ||
See accompanying notes to financial statements.
| F-6 |
Notes to Financial Statements
Note 1 - Description of Business and Summary of Significant Accounting Policies
Nature of Business - The Company is a preclinical and clinical-stage pharmaceutical company organized as a Delaware corporation in July 2015 to focus on the development of anti-cancer drug candidates. The Company has two in-licensed drug development technologies.The lead drug candidate is Lipsosomal Annamycin, which is referred to as Annamycin, an anthracyline that targets the treatment of relapsed or refractory acute myeloid leukemia, or AML. In August 2015, the Company entered into a rights transfer agreement with AnnaMed, Inc. (“AnnaMed”), a company affiliated with certain members of our management and board of directors, pursuant to which, in exchange for 1,431,000 shares of our common stock, AnnaMed agreed to transfer any and all data it had regarding the development of Annamycin and the Annamycin IND, including all trade secrets, know-how, confidential information and other intellectual property rights held by AnnaMed. Annamycin has been in clinical trials pursuant to an investigational new drug application, or IND, that had been filed with the U.S. Food and Drug Administration, or FDA. This IND was terminated due to a lack of activity by a prior drug developer who was developing the drug for a different indication. We intend to apply for a new IND based on the same data that supported the original IND, updated for subsequent clinical data, and to commence a Phase II clinical trial for Annamycin.
The Company also has a second drug development technology, WP1122, a molecule targeting the treatment of glioblastoma through metabolic inhibition, which was developed at The University of Texas M.D. Anderson Cancer Center (“MD Anderson”).
On August 11, 2015, the Company entered into a rights transfer agreement with IntertechBio Corporation (“IntertechBio”), a company affiliated with certain members of our management and board of directors, whereby IntertechBio agreed to assign its license or sublicense its license to certain metabolic inhibitor technology owned by MD Anderson. In consideration, the Company issued 630,000 common shares to Intertech Bio. IntertechBio agreed to make payments to MD Anderson including an up-front payment, license documentation fee, annual maintenance fee, milestone payments and minimum annual royalty payments for sales of products developed under the license agreement. The Company has assumed the rights and obligations of IntertechBio under the license agreement with MD Anderson. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by the Company.
The Company intends to file a registration statement on Form S-1 with respect to the Company’s initial public offering of shares of its common stock (“IPO”) to fund the development of its technologies and also intends to acquire Moleculin, LLC, a Texas limited liability company. Moleculin, LLC is the holder of a license agreement with MD Anderson covering technology referred to as WP1066 Portfolio, which is focused on the modulation of key oncogenic transcription factors. Immediately prior to the declaration of effectiveness of the registration statement on Form S-1, Moleculin, LLC will be merged with and into MBI, which will survive the merger.
Use of Estimates in Financial Statement Presentation - The preparation of these financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Going Concern - These financial statements have been prepared on a going concern basis, which assumes the Company will continue to realize its assets and discharge its liabilities in the normal course of business. The continuation of the Company as a going concern is dependent upon the continued financial support from its stockholders, the ability of the Company to obtain necessary equity financing to continue operations, and the attainment of profitable operations. As of September 30, 2015, the Company has incurred an accumulated loss of $224,315 since inception, had a working capital deficit of $222,254, and had not yet generated any revenue from operations. These factors raise substantial doubt regarding the Company’s ability to continue as a going concern. These financial statements do not include any adjustments to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
Cash and Cash Equivalents - The Company considers all highly liquid accounts with original maturities of three months or less to be cash equivalents. At September 30, 2015, all of the Company’s cash was deposited in one bank.
Beneficial Conversion Feature - From time to time, the Company may issue convertible notes that have conversion prices that create an embedded beneficial conversion feature on the issuance date. A beneficial conversion feature exists on the date a convertible note is issued when the fair value of the underlying common stock to which the note is convertible into is in excess of the remaining unallocated proceeds of the note after first considering the allocation of a portion of the note proceeds to the fair value of any attached equity instruments, if any related equity instruments were granted with the debt. The Company estimates the fair value of its common stock using the most recent selling price available. The intrinsic value of the beneficial conversion feature is recorded as a debt discount with a corresponding amount to additional paid-in capital. The debt discount is amortized to interest expense over the life of the note using the effective interest method.
Income Taxes - The Company uses the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and the tax bases of reported assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company must then assess the likelihood that the resulting deferred tax assets will be realized. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized.
| F-7 |
Research and Development Costs - Research and development costs are expensed as incurred. Research and development reimbursements and grants are recorded by the Company as a reduction of research and development costs.
Subsequent Events - The Company’s management reviewed all material events through the date these financial statements were issued for subsequent event disclosure consideration.
Recent Accounting Pronouncements - The Company does not believe that any recently issued effective pronouncements, or pronouncements issued but not yet effective, if adopted, would have a material effect on the accompanying financial statements.
Note 2 - Related Party Transactions
As of September 30, 2015, the Company owed $10,281 to its Chief Financial Officer and $9,303 to its Chief Operating Officer for due and unpaid compensation and reimbursable expenses.
Note 3 - Notes Payable
On August 31, 2015, the Company entered into an unsecured convertible promissory note with a third party, in the amount of $125,000. The note bears interest at 8.0% per annum and matures on the earlier of April 1, 2016 or the completion of an IPO of the Company’s securities. However, if the completion of the IPO occurs prior to April 1, 2016, the note shall be automatically converted according to its terms into shares of the Company’s common stock at a conversion price of $0.1299 per share upon the Company’s IPO.
On September 3, 2015, the Company entered into a second unsecured convertible promissory note with a third party in the amount of $125,000. The note bears interest at 8.0% per annum and matures on the earlier of April 1, 2016 or the completion of an IPO of the Company’s securities. However, if the completion of the IPO occurs prior to April 1, 2016, the note shall be automatically converted according to its terms into shares of Company common stock at a conversion price equal to $0.1299 per share upon the Company’s IPO.
The convertible notes were analyzed for a beneficial conversion feature at which time it was concluded that a beneficial conversion feature did not exist.
Note 4 - Equity
In August 2015, the Company entered into an agreement to issue 4,600,000 shares of common stock to its director and officers for subscriptions of $4,600 cash to be received. As of September 30, 2015, the Company had not collected the proceeds for the subscriptions.
On August 11, 2015, the Company was granted an assignment of a license agreement between MD Anderson and IntertechBio Corporation, a Company affiliated with certain members of our management and board of directors, in exchange for 630,000 shares of the Company’s common stock. The sublicense gives the Company rights to access certain metabolic inhibitor technology owned by MD Anderson that had been licensed to IntertechBio Corporation. The shares were valued at a total of $630 and the related expense included in research and development costs.
On August 21, 2015, the Company acquired the right to the intellectual property of AnnaMed, Inc., a company affiliated with certain members of our management and board of directors, in exchange for 1,431,000 shares of the Company’s common stock. The license gives the Company full ownership rights to the data package supporting the FDA IND Number 46869 (the “Annamycin IND”), allowing the Company to resubmit a request for IND to the FDA to begin development work on Annamycin. The shares were valued at a total of $1,431 and the related expense included in research and development costs.
| F-8 |
Note 5 - Income Taxes
The Company is subject to United States federal and state income taxes at an approximate rate of 35%. The reconciliation of the provision for income taxes at the United States federal and state statutory rate compared to the Company’s income tax expense as reported is as follows:
| From July 28, 2015 (Inception) to September 30, 2015 | ||||
| Income tax benefit computed at the statutory rate | $ | 78,510 | ||
| Change in valuation allowance | (78,510 | ) | ||
| Provision for income taxes | $ | - | ||
Significant components of the Company’s deferred tax assets after applying enacted corporate income tax rates are as follows:
| As of September 30, 2015 | ||||
| Deferred income tax assets | ||||
| Net operating losses | $ | 78,510 | ||
| Valuation allowance | (78,510 | ) | ||
| Net deferred income tax assets | $ | - | ||
The Company has an operating loss carry forward of approximately $224,000, which expires commencing in 2035.
Note 6 – Commitments and contingencies
On August 5, 2015, the Company entered into a letter agreement with Burnham Securities Inc. (“Burnham”) to engage Burnham as an exclusive financial advisor of the Company for 12 months. Pursuant to the agreement, the Company agreed to: a) pay success fees equal to 7% of the gross proceeds from any form of financing; b) issue warrants to purchase 7% of the Company’s equity securities sold with a cashless exercise provision, exercisable at a price per share of the equity securities paid by investors in the transaction. In addition, the Company agreed to reimburse Burnham for all of its reasonable out-of-pocket expenses. Upon execution of this agreement, the Company also paid Burnham a $50,000 advisory fee and agreed to pay an additional advisory fee of $50,000 upon completion of the Company’s IPO. As of September 30, 2015, the Company has accrued $17,500 success fees related to the notes issued to in August and September 2015. See Note 3.
Note 7 - Subsequent Events
On October 6, 2015, the Company entered into an unsecured promissory note with a third party in the amount of $150,000. The note bears interest at 8.0% per annum and matures on the earlier of April 1, 2016 or the completion of an IPO of the Company’s securities. However, if the completion of the IPO occurs prior to April 1, 2016, the note shall be automatically converted according to its terms into shares of the Company common stock at a conversion price equal to $0.20 per share upon the Company’s IPO.
On October 8, 2015, IntertechBio Corporation entered into a letter agreement with MD Anderson where MD Anderson agreed to receive past due maintenance fees and patent expenses of $98,108 owed by IntertechBio Corporation in four installments. The past due amount is related to certain metabolic inhibitor technology license that was assigned to the Company by IntertechBio Corporation and was owed by IntertechBio Corporation prior to the Company’s acquisition of the license. In order to have the license in good standing, the Company agreed to pay MD Anderson the $98,108 on behalf of IntertechBio Corporation. The Company made a prepayment of $23,000 in September 2015 before this letter agreement was reached.
| F-9 |
On October 19, 2015, the Company, through IntertechBio Corporation, amended the Patent and Technology License Agreement with MD Anderson. Pursuant to the amendment, the Company will pay milestone payments as follows:
| Phase | Amount | |||
| Commencement of Phase II Study for a licensed product | $ | 200,000 | ||
| Commencement of Phase III Study for a licensed product | $ | 250,000 | ||
| Filing of a New Drug Application for a licensed product | $ | 400,000 | ||
| Receipt of market approval for a licensed product | $ | 500,000 | ||
On October 15, 2015, the Company entered into an exclusive option agreement with MD Anderson granting the Company a period of time to evaluate the potential of acquiring and to negotiate the purchase of an exclusive license in the field of cancer therapeutics. The option period extends from October 15, 2015 through May 11, 2016. MD Anderson shall not offer the license to any other third party during the option period. In consideration of the option granted, the Company paid $5,000 to MD Anderson.
On October 28, 2015, the Company entered into an unsecured convertible promissory note with a third party in the amount of $50,000. The note bears interest at 8.0% per annum and matures on the earlier of April 1, 2016 or the completion of an IPO of the Company’s securities. However, if the completion of the IPO occurs prior to April 1, 2016, the note shall be converted according to its terms into shares of the Company common stock at a conversion price equal to $0.20 per share upon the Company’s IPO.
Note 8 - Additional Subsequent Event (UNAUDITED)
The Company issued two 8% convertible notes in an aggregate of $200,000 in principal amount of convertible notes to two investors as described in Note 7. At the time of such purchase in October 2015, the convertible note investors and the Company agreed that the investors would fund the Company up to the filing of the Company’s initial non-confidential registration statement an additional $165,000 on the same terms as in the October financing. Pursuant to such agreement, such amounts were received in January 2016. The new notes bear interest at 8.0% per annum and matures on the earlier of April 1, 2016 or the completion of an IPO of the Company’s securities. However, if the completion of the IPO occurs prior to April 1, 2016, the note shall be converted according to its terms into shares of the Company common stock at a conversion price equal to $0.20 per share upon the Company’s IPO.
In January and February 2016, the Company sold 53,000 common shares for $159,000 cash.
| F-10 |
Statements of Financial Position
As of September 30, 2015 and December 31, 2014
(Unaudited)
| September 30, | December 31, | |||||||
| 2015 | 2014 | |||||||
| ASSETS | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 298,350 | $ | 524,477 | ||||
| Prepaid expenses and other current assets | 4,709 | 2,076 | ||||||
| Total Current Assets | 303,059 | 526,553 | ||||||
| Other receivables | - | 232,300 | ||||||
| Property and equipment, net of accumulated depreciation | 14,126 | 22,595 | ||||||
| Total Assets | $ | 317,185 | $ | 781,448 | ||||
| LIABILITIES AND MEMBERS’ DEFICIT | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable and accrued liabilities | $ | 1,160,571 | $ | 1,047,455 | ||||
| Accounts payable and accrued liabilities - related party | 56,194 | 25,744 | ||||||
| Common units payable | 548,630 | 548,630 | ||||||
| Note payable | 55,774 | - | ||||||
| Convertible notes payable, net of $119,049 discount and $53,877 financing costs | 962,000 | - | ||||||
| Convertible notes payable- related parties, net of $45,489 discount | 468,591 | - | ||||||
| Total Current Liabilities | 3,251,760 | 1,621,829 | ||||||
| Long-term note payable | 414,165 | 414,165 | ||||||
| Convertible notes payable, net of $237,664 discount and $107,558 financing costs | - | 789,705 | ||||||
| Convertible notes payable - related parties, net of $90,811 discount | - | 423,268 | ||||||
| Total Liabilities | 3,665,925 | 3,248,967 | ||||||
| Commitments and contingencies | ||||||||
| Members’ Deficit: | ||||||||
| Preferred units, 16,321,558 units authorized, 13,515,820 units issued and outstanding | 10,620,186 | 10,620,186 | ||||||
| Common units, 4,727,074 authorized, 4,178,444 units issued and outstanding | 750,000 | 750,000 | ||||||
| Accumulated deficit | (14,718,926 | ) | (13,837,705 | ) | ||||
| Total Members’ Deficit | (3,348,740 | ) | (2,467,519 | ) | ||||
| Total Liabilities and Members’ Deficit | $ | 317,185 | $ | 781,448 | ||||
See accompanying notes to the financial statements.
| F-11 |
Statements of Operations
For the Nine Months Ended September 30, 2015 and 2014
(Unaudited)
| 2015 | 2014 | |||||||
| Revenue | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development | 270,991 | 238,467 | ||||||
| General and administrative | 267,423 | 649,344 | ||||||
| Depreciation expense | 8,469 | 8,254 | ||||||
| Total operating expenses | 546,883 | 896,065 | ||||||
| Loss from operations | (546,883 | ) | (896,065 | ) | ||||
| Other income (expense): | ||||||||
| Other income | 549 | 396 | ||||||
| Interest expense | (334,887 | ) | (55,407 | ) | ||||
| Total other income (expense) | (334,338 | ) | (55,011 | ) | ||||
| Net loss | $ | (881,221 | ) | $ | (951,076 | ) | ||
See accompanying notes to the financial statements.
| F-12 |
Statements of Cash Flows
For the Nine Months Ended September 30, 2015 and 2014
(Unaudited)
| 2015 | 2014 | |||||||
| (Restated) | ||||||||
| Cash Flows From Operating Activities: | ||||||||
| Net loss | $ | (881,221 | ) | $ | (951,076 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation expense | 8,469 | 8,254 | ||||||
| Amortization of debt discount | 163,937 | 32,476 | ||||||
| Amortization of deferred costs | 53,681 | - | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (2,633 | ) | 4,867 | |||||
| Accounts payable and accrued liabilities | 168,890 | 11,508 | ||||||
| Accounts payable and accrued liabilities - related parties | 30,450 | 15,249 | ||||||
| Net Cash Used In Operating Activities | (458,427 | ) | (878,722 | ) | ||||
| Cash Flows From Investing Activities: | ||||||||
| Collection from other receivable | 232,300 | - | ||||||
Net Cash Provided By Investing Activities | 232,300 | - | ||||||
| Cash Flows From Financing Activities: | ||||||||
| Proceeds from issuance of convertible notes payable | - | 268,677 | ||||||
| Proceeds from issuance of convertible notes payable - related parties | - | 514,079 | ||||||
| Net Cash Provided By Financing Activities | - | 782,756 | ||||||
| Net change in cash and cash equivalents | (226,127 | ) | (95,966 | ) | ||||
| Cash and cash equivalents at beginning of period | 524,477 | 353,366 | ||||||
| Cash and cash equivalents at end of period | $ | 298,350 | $ | 257,400 | ||||
| Supplemental cash flow disclosures: | ||||||||
| Cash paid for interest | $ | - | $ | - | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| Non-cash investing and financing activities: | ||||||||
| Long-term note payable issued to settle accounts payable | $ | - | $ | 414,165 | ||||
| Note payable issued to settle accounts payable | $ | 55,774 | $ | - | ||||
| Beneficial conversion feature of convertible notes | $ | - | $ | 195,689 | ||||
See accompanying notes to the financial statements.
| F-13 |
Notes to Financial Statements
(Unaudited)
Note 1 - Description of Business and Summary of Significant Accounting Policies
Nature of Business - Moleculin, LLC, a Texas limited liability company, (the “Company” or “Moleculin”) is an early-stage Houston-based pharmaceutical company formed in December 2006 to create novel therapies for skin disorders and the development of cancer related technology. Although the Company previously focused on the development of treatments for skin disorders, the Company’s current focus is on the development of drugs for cancer treatment.
Moleculin intends to be acquired by Moleculin Biotech, Inc. (“MBI”) by merging into MBI. The intended transaction is scheduled to occur immediately prior to the declaration of effectiveness of MBI’s registration statement on Form S-1 with respect to MBI’s initial public offering of its common stock.
Basis of Presentation - Unaudited Interim Financial Information - The accompanying unaudited interim financial statements and related notes have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for interim financial information, and in accordance with the rules and regulations of the United States Securities and Exchange Commission with respect to Form 10-Q and Article 8 of Regulation S-X. Accordingly, they do not include all of the information and footnotes required by U.S. GAAP for complete financial statements. The unaudited interim financial statements furnished reflect all adjustments (consisting of normal recurring adjustments) which are, in the opinion of management, necessary for a fair statement of the results for the interim periods presented. Interim results are not necessarily indicative of the results for the full year. These unaudited interim financial statements should be read in conjunction with the audited financial statements of the Company for the year ended December 31, 2014 and notes thereto contained in this prospectus.
Use of Estimates in Financial Statement Presentation - The preparation of these financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of these financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Going Concern - These financial statements have been prepared on a going concern basis, which assumes the Company will continue to realize its assets and discharge its liabilities in the normal course of business. The continuation of the Company as a going concern is dependent upon the continued financial support from its members, the ability of the Company to obtain necessary equity financing to continue operations, and the attainment of profitable operations. The Company has suffered recurring losses from operating and has insufficient working capital. As of September 30, 2015, the Company has incurred accumulated deficit of $14,718,926 since inception, had a working capital deficit of $2,948,701, and has not yet generated any significant revenue from operations. These factors raise substantial doubt regarding the Company’s ability to continue as a going concern. These financial statements do not include any adjustments to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
Cash and Cash Equivalents - The Company considers all highly liquid accounts with original maturities of three months or less to be cash equivalents. Balances held by the Company are typically in excess of Federal Deposit Insurance Corporation’s insured limits ($250,000 per depositor per bank). At September 30, 2015 and December 31, 2014, all of the Company’s cash was deposited in one bank.
Property and Equipment - Property and equipment are recorded at cost and depreciated on a straight-line basis over estimated useful lives. Leasehold improvements are recorded at cost and depreciated on a straight-line basis over the shorter of the estimated useful life or the lease term. When assets are retired or sold, the cost and related accumulated depreciation are removed from the accounts, and any related gain or loss is reflected in income (loss) for the period. Expenditures for major additions which extend the useful lives of assets are capitalized. Minor replacements, maintenance and repairs, which do not improve or extend the life of such assets are charged to expense as incurred.
| F-14 |
Impairment of Long-Lived Assets - Management reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount may not be realizable or at a minimum annually during the fourth quarter of the year. If an evaluation is required, the estimated future undiscounted cash flows associated with the asset are compared to the asset’s carrying value to determine if an impairment of such asset is necessary. The effect of any impairment would be to expense the difference between the fair value of such asset and its carrying value. For the nine months ended September 30, 2015, there were no impairments recorded.
Fair Value of Financial Instruments - Fair value is defined as the price that would be received to sell an asset, or paid to transfer a liability, in an orderly transaction between market participants. A fair value hierarchy has been established for valuation inputs that give the highest priority to quoted prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs. The fair value hierarchy is as follows:
Level 1 Inputs - Unadjusted quoted prices in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date.
Level 2 Inputs - Inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly. These might include quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (such as interest rates, volatilities, prepayment speeds, credit risks, etc.) or inputs that are derived principally from or corroborated by market data by correlation or other means.
Level 3 Inputs - Unobservable inputs for determining the fair values of assets or liabilities that reflect an entity’s own assumptions about the assumptions that market participants would use in pricing the assets or liabilities.
Income Taxes - The Company elected to be taxed as a partnership under the Internal Revenue Code. The Company pays no U.S. taxes on its earnings. The Company’s net earnings/losses are passed through to the Company’s members and, as such, reports no income tax expense or liability.
Revenue Recognition - The Company recognizes revenues when persuasive evidence of an arrangement exists, delivery has occurred or services have been provided, the purchase price is fixed or determinable and collectability is reasonably assured. The Company may receive cost reimbursement-based grants. For these grants, revenues are based on costs incurred that are specifically covered under reimbursement arrangements. These revenues are recognized as grant-related expenses are incurred by the Company or its subcontractors.
Research and Development Costs - Research and development costs are expensed as incurred.
Subsequent Events - The Company’s management reviewed all material events through the date these financial statements were issued for subsequent event disclosure consideration.
Recent Accounting Pronouncements - In June 2014, the FASB issued ASU 2014-10, Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements. ASU 2014-10 eliminates the distinction of a development stage entity and certain related disclosure requirements, including the elimination of inception-to-date information on the statements of operations, cash flows and stockholders’ equity. The amendments in ASU 2014-10 will be effective prospectively for annual reporting periods beginning after December 15, 2014, and interim periods within those annual periods, however early adoption is permitted. The Company evaluated and adopted ASU 2014-10 as of December 31, 2014.
In April 2015, the FASB issued ASU 2015-03, Interest – Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs. The amended guidance requires that debt issuance costs be presented in the balance sheet as a direct reduction from the carrying amount of the recognized debt liability, consistent with the treatment of debt discounts. Amortization of debt issuance costs is to be reported as interest expense. The updated guidance is effective for reporting periods beginning after December 15, 2015, and interim periods within those annual periods, however early adoption is permitted. The Company evaluated and adopted ASU 2015-13 as of December 31, 2014.
| F-15 |
The Company does not believe that any other recently issued effective pronouncements, or pronouncements issued but not yet effective, if adopted, would have a material effect on the accompanying financial statements.
Note 2 – Other Receivables
On October 27, 2010, the Company entered into a Patent and Technology Development and License Agreement with Dermin, Sp. Zo. O. (“Dermin”), a Polish limited liability company, to assist in the development and commercialization of certain pharmaceutical products within a described licensed territory in exchange for royalty payments. On December 19, 2011, the Company advanced $200,000 to Dermin providing funding assistance to Dermin’s efforts in developing and commercializing these pharmaceutical products in and around the Poland European region. Further, throughout the year 2012, the Company paid $32,300 of expenses on behalf of Dermin. As of December 31, 2014, the Company had $232,300 long-term note receivable from Dermin which was collected in August 2015.
Note 3 - Property and Equipment
Property and equipment consisted of the following at September 30, 2015 and December 31, 2014:
| Lives | September 30, 2015 | December 31, 2014 | ||||||||||
| Leasehold improvements | 1 year | $ | 81,602 | $ | 81,602 | |||||||
| Machinery and equipment | 5 years | 57,327 | 57,327 | |||||||||
| Furniture and fixtures | 7 years | 7,955 | 7,955 | |||||||||
| Less: accumulated depreciation | (132,758 | ) | (124,289 | ) | ||||||||
| Property and equipment, net | $ | 14,126 | $ | 22,595 | ||||||||
Depreciation expense for the nine months ended September 30, 2015 and 2014 was $8,469 and $8,254, respectively.
Note 4 – Note Payable
On September 8, 2015, the Company issued an unsecured promissory note to Hilltop Research in the amount of $55,774 to settle an outstanding payable. The note matures on June 30, 2016 and accrues interest at 6.0% per annum. In the event that the Company or a significant portion of the Company’s assets are acquired by MBI prior to the maturity date, the loan becomes due and payable upon closing of MBI’s initial public offering.
Note 5 – Long-Term Notes Payable
On September 16, 2014, the Company issued a promissory note in the amount of $414,165 to Davos Chemical Corporation to settle an outstanding accounts payable balance. The note bears interest at an annual rate of 6.0% and matures on the earlier of December 31, 2016 or the completion of one of the following prior to December 31, 2016:
| a) | The Company (i) is awarded a grant from the Cancer Prevention and Research Institute of Texas (“CPRIT”) of at least $5,000,000, and (ii) is deemed by CPRIT to have received the 50% matching funds required by the CPRIT grant; or |
| b) | The Company concludes a privately placed sale of the Company’s Series A Preferred Units of at least $3,000,000. |
| F-16 |
At September 30, 2015 and December 31, 2014, accrued interest related this note was $25,802 and $7,217, respectively.
Note 6 – Convertible Notes Payable and Convertible Notes Payable – Related Parties
From May to November 2014, the Company issued various convertible notes to its creditors. Note proceeds from third and related parties were $1,011,177 and $514,079, respectively. These notes bear interest at 8% per annum and are due on the earlier of June 30, 2016 or the consummation of a liquidation event, unless these notes are converted, pursuant to the following:
| a) | Automatic conversion upon CPRIT qualified financing- When the Company is awarded a grant from the CPRIT of at least $3,500,000 and is deemed by CPRIT to have received the 50% matching funds required by the CPRIT grant. The conversion price is $0.80 per unit in the event of a CPRIT qualified financing. |
| b) | First tier automatic conversion - When the Company concludes a privately placed sale of the Company’s Series A Preferred Units of at least $3,000,000 and is at a pre-money valuation of not less than $20,000,000. The conversion price is 80% of the lowest per unit purchase price in the privately placed sale in the event of a first tier automatic conversion. |
| c) | Second tier automatic conversion - When the Company concludes a privately placed sale of the Company’s Series A Preferred Units of at least $2,000,000. The conversion price is 70% of the lowest per unit purchase price in the privately placed sale in the event of a second tier automatic conversion. |
| d) | Voluntary conversion - At any time after the earlier of June 30, 2016 or a Liquidation Event, the note holders can elect to convert the notes into the Company’s Preferred Units at $0.80 per unit. |
A liquidation event means a) the acquisition of the Company by another entity; b) a sale, exclusive license, lease or other conveyance of all or substantially all of the Company’s assets; c) any liquidation, dissolution of the Company; d) the acquisition of an aggregate of 50% or more of the Company’s voting power by a party; e) in a bankruptcy proceeding. Upon a liquidation event, the note holders is entitle to 150% of the then outstanding principal and unpaid interest.
These convertible notes were analyzed for a beneficial conversion feature at which time it was concluded that a beneficial conversion feature existed. The beneficial conversion feature was measured using the commitment-date stock price and was determined to be $283,732 for third party notes and $128,520 for related party notes. This amount was recorded as a debt discount and is amortized to interest expense over the term of these notes. For the nine months ended September 30, 2015 and 2014, amortization of debt discount for third and related parties were $163,937 and $32,476, respectively.
As of September 30, 2015, accrued interest for third party and related party notes were $95,519 and $56,194, respectively. As of December 31, 2014, accrued interest for third party and related party notes were $27,605 and $25,744, respectively.
On September 30, 2014, the Company entered into an agreement with Brompton Group NA, LLC (“Brompton”) for a non-exclusive arrangement to identify investors or lenders interested in providing financing for the Company. In exchange for their services, the Company agreed to pay Brompton 15% of the proceeds received from any financing provided by the investors introduced by Brompton. Brompton agreed to receive two-thirds of the fee, at their option, in either cash or in the form of the same securities offered in the financing being offered to the identified investors or lenders. The remaining fee should be paid in the form of the same securities offered in the financing. Of the $1,254,874 debt raised from third party investors during the fourth quarter of 2014, $825,000 was raised through investors introduced by Brompton. As such, $123,750 of finder’s fee was recorded as debt discount and amortized over the life of the notes. For the nine months ended September 30, 2015 and 2014, interest expense associated with the amortization of the finder’s fee was $53,681 and $0, respectively.
| F-17 |
Note 7 - Commitments and Contingencies
MD Anderson Agreements
On June 21, 2010, the Company entered into a patent and technology license agreement with the University of Texas M.D. Anderson Cancer Center (“MD Anderson”) under which the Company is obligated to make upfront and periodic progress payments. The agreement allows the Company to obtain from MD Anderson an exclusive, worldwide royalty-bearing license to research, develop, manufacture, have manufactured, use, import, offer to sell or sell products comprising or made through the use of certain patent and technology rights owned by MD Anderson. In consideration, the Company agreed to pay for all out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining patent rights. Pursuant to the agreement, the Company also agreed to pay: a) an annual maintenance fee starting at $20,000 per year and increase by $10,000 each year up to a maximum of $100,000; b) royalties equal to 2.5% of net sales for licensed products approved for dermatological use; and c) royalties equal to 4.0% of net sales for licensed products approved for all other uses. The 2.5% and 4.0% royalties could be further reduced if certain criteria are met. Annual minimum royalties are $200,000 following the first sale after marketing approval is obtained from MD Anderson by the Company for any licensed products. In addition, the Company will be required to issue 3.5% of the total issued and outstanding common units of the Company on a fully diluted basis to MD Anderson at the time the Company commences its first phase II trial of a drug covered by the agreement. The Company is also required to pay certain milestone payments.
On October 8, 2015, the Company entered into a letter agreement with MD Anderson for the Company’s past due fees to MD Anderson in the amount of $691,186 of which $300,000 has been paid prior to the letter agreement. Pursuant to the letter agreement, MD Anderson agreed to receive the remaining past due fee in three installments: a) $125,000 due on October 31, 2015; b) $175,000 due on January 31, 2016; and c) $91,186 due on April 30, 2016. The Company paid $125,000 to MD Anderson on November 2, 2015.
On October 19, 2015, the agreement was amended for the milestone payments. The amended milestone payments are as follows:
| Phase | Amount | |||
| Commencement of Phase III Study for first licensed drug/product | $ | 150,000 | ||
| Submission of the first NDA | $ | 500,000 | ||
| Receipt of first marketing approval | $ | 600,000 | ||
The amendment also removed the term regarding the issuance of 3.5% of the Company outstanding units.
For the nine months ended September 30, 2015, the Company recorded liabilities to and made payments to MD Anderson of $132,844 and $6,586, respectively.
Dermin Agreement
On October 27, 2010, the Company entered into a Patent and Technology Development and License Agreement with Dermin. The agreement provided Dermin with sub-license rights to the Company’s technologies for use in limited territories in exchange for Dermin’s use of Polish government grant funding to pay for development costs the Company would otherwise have been required to fund. Dermin’s territories are primarily Poland and surrounding countries, but not including any of the major European markets (UK, Germany, France, Spain and Italy). Pursuant to the agreement, Dermin has to pay the Company a running royalty on sales of license products. As of September 30, 2015, no sales have occurred and no royalty paid to the Company by Dermin.
Lease Agreements
On July 31, 2007, the Company entered into a rental agreement for office space at 2575 West Belfort, Houston, Texas. The agreement was renewed on July 26, 2010 for one year. After July 2011, the lease continues on a month-to-month basis. Therefore, there are no future minimum obligations on the lease. Rent expense for the nine months ended September 30, 2015 and 2014 was $22,026 and $21,800, respectively, and is included in general and administrative expenses on the statements of operations.
| F-18 |
Note 8 - Equity
In March 2011, the Company amended its articles of incorporation to create two classes of interests referred to as ‘Series A Preferred Membership Interests’ and ‘Common Membership Interests’, with each interest issued being measured as Units (Series A Preferred Units and Common Units, respectively). The Series A Preferred Units are further broken down into 4 series including Series A-1, Series A-2, Series A-3 and Series A-4.
Pursuant to Moleculin LLC's Company Agreement, the Board shall distribute net cash, as defined in the Company Agreement, to the members at such times and in such amounts as the Board may determine in the following order and priority:
| a) | First, to the Series A Preferred members in proportion to each such members’ original capital contribution until such members receive net cash equal to 150% of their original capital contribution. |
| b) | Second, to the members in proportion to their percentage interests until Series A Preferred members have received net cash equal to 300% of their original capital contribution. |
| c) | Third, to the common members in proportion to each common member’s common units until the common members have receive aggregate distributions of net cash equal to the then total amount of distributions to all members for all company fiscal years multiplied by the common members’ aggregate percentage interests; and |
| d) | Fourth, to the members in accordance with their percentage interests. |
As of September 30, 2015 and December 31, 2014, the Preferred Units are summarized as follows:
| Units Outstanding | ||||||||||||
| Series | September 30, 2015 | December 31, 2014 | Contribution | |||||||||
| Series A-1 | 283,604 | 283,604 | $ | 150,000 | ||||||||
| Series A-2 | 1,595,552 | 1,595,552 | 600,000 | |||||||||
| Series A-3 | 1,942,400 | 1,942,400 | 513,672 | |||||||||
| Series A-4 | 9,694,264 | 9,694,264 | 9,356,514 | |||||||||
| Total | 13,515,820 | 13,515,820 | $ | 10,620,186 | ||||||||
During the first quarter of 2013, the Company commenced the first Phase II Study of a licensed product in the patent and technology license agreement between the Company and MD Anderson. Pursuant to the agreement, the Company was obligated to issue MD Anderson 548,630 common units, representing 3.5% of its issued and outstanding equity units. These common units were valued at their estimated fair value of $548,630, which was recorded as a research and development expense. In October 2015, MD Anderson agreed to forfeit its right to these units. The Company recorded a gain on liability extinguishment of $548,630 upon the forfeiture.
Note 9 - Restatement
The Company has previously reported the collection of $232,000 long-term note receivable from Dermin as cash flow from operating activities while it should have been reported as cash flow from investing activities. The statement of cash flow for the nine months ended September 30, 2015 has been revised to correct the error.
Note 10 - Subsequent Event
On October 15, 2015, the Company entered into a $5,000 exclusive option agreement with MD Anderson allowing the Company a period of time to evaluate the potential of acquiring and to negotiate the purchase of an exclusive license in the field of cancer therapeutics. The option period extends from October 15, 2015 through August 31, 2016. MD Anderson shall not offer a license to any third party during the option period.
On January 2, 2016, the Company purchased 189,070 common units from one of the Company's unit holders for $100 cash.
| F-19 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Members of
Moleculin, LLC
Houston, TX
We have audited the accompanying statements of financial position of Moleculin, LLC as of December 31, 2014 and 2013 and the related statements of operations, changes in members’ deficit, and cash flows for the years then ended. Moleculin, LLC’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Moleculin, LLC as of December 31, 2014 and 2013 and the results of its operations and its cash flows for each of the years then ended in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that Moleculin, LLC will continue as a going concern. As discussed in Note 1 to the financial statements, Moleculin, LLC has suffered recurring losses from operations and has insufficient working capital that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ GBH CPAs, PC
GBH CPAs, PC
www.gbhcpas.com
Houston, Texas
December 3, 2015
| F-20 |
Statements of Financial Position
As of December 31, 2014 and 2013
| 2014 | 2013 | |||||||
| ASSETS | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 524,477 | $ | 353,366 | ||||
| Prepaid expenses and other current assets | 2,076 | 8,462 | ||||||
| Total Current Assets | 526,553 | 361,828 | ||||||
| Other receivables | 232,300 | 232,300 | ||||||
| Property and equipment, net of accumulated depreciation | 22,595 | 33,600 | ||||||
| Total Assets | $ | 781,448 | $ | 627,728 | ||||
| LIABILITIES AND MEMBERS’ DEFICIT | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable and accrued liabilities | $ | 1,047,455 | $ | 1,149,783 | ||||
| Accounts payable and accrued liabilities - related party | 25,744 | - | ||||||
| Common units payable | 548,630 | 548,630 | ||||||
| Total Current Liabilities | 1,621,829 | 1,698,413 | ||||||
| Long-term note payable | 414,165 | - | ||||||
| Convertible notes payable, net of $237,664 discount and $107,558 financing costs | 789,705 | - | ||||||
| Convertible notes payable - related parties, net of $90,811 discount | 423,268 | - | ||||||
| Total Liabilities | 3,248,967 | 1,698,413 | ||||||
| Commitments and contingencies | ||||||||
| Members’ Deficit: | ||||||||
| Preferred units, 16,321,558 units authorized, 13,515,820 units issued and outstanding | 10,620,186 | 10,207,934 | ||||||
| Common units, 4,727,074 authorized, 4,178,444 units issued and outstanding | 750,000 | 750,000 | ||||||
| Accumulated deficit | (13,837,705 | ) | (12,028,619 | ) | ||||
| Total Members’ Deficit | (2,467,519 | ) | (1,070,685 | ) | ||||
| Total Liabilities and Members’ Deficit | $ | 781,448 | $ | 627,728 | ||||
See accompanying notes to the financial statements.
| F-21 |
Statements of Operations
For the Years Ended December 31, 2014 and 2013
| 2014 | 2013 | |||||||
| Revenue | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development | 798,785 | 2,341,676 | ||||||
| General and administrative | 839,556 | 1,161,282 | ||||||
| Depreciation expense | 11,005 | 11,005 | ||||||
| Total operating expenses | 1,649,346 | 3,513,963 | ||||||
| Loss from operations | (1,649,346 | ) | (3,513,963 | ) | ||||
| Other income (expense): | ||||||||
| Other income | 800 | 849 | ||||||
| Interest expense | (160,540 | ) | - | |||||
| Total other income (expense) | (159,740 | ) | 849 | |||||
| Net loss | $ | (1,809,086 | ) | $ | (3,513,114 | ) | ||
See accompanying notes to the financial statements.
| F-22 |
Statements of Changes in Members’ Deficit
For the Years Ended December 31, 2014 and 2013
| Preferred Units | Common Units | Accumulated | ||||||||||||||||||||||
| Units | Amount | Units | Amount | Deficit | Total | |||||||||||||||||||
| Balance at December 31, 2012 | 11,496,704 | $ | 8,188,818 | 4,178,444 | $ | 750,000 | $ | (8,515,505 | ) | $ | 423,313 | |||||||||||||
| Issuance of preferred units for cash | 2,019,116 | 2,019,116 | - | - | - | 2,019,116 | ||||||||||||||||||
| Net loss | - | - | - | - | (3,513,114 | ) | (3,513,114 | ) | ||||||||||||||||
| Balance at December 31, 2013 | 13,515,820 | 10,207,934 | 4,178,444 | 750,000 | (12,028,619 | ) | (1,070,685 | ) | ||||||||||||||||
| Beneficial conversion feature of convertible notes | - | 412,252 | - | - | - | 412,252 | ||||||||||||||||||
| Net loss | - | - | - | - | (1,809,086 | ) | (1,809,086 | ) | ||||||||||||||||
| Balance at December 31, 2014 | 13,515,820 | $ | 10,620,186 | 4,178,444 | $ | 750,000 | $ | (13,837,705 | ) | $ | (2,467,519 | ) | ||||||||||||
See accompanying notes to the financial statements.
| F-23 |
Statements of Cash Flows
For the Years Ended December 31, 2014 and 2013
| 2014 | 2013 | |||||||
| Cash Flows From Operating Activities: | ||||||||
| Net loss | $ | (1,809,086 | ) | $ | (3,513,114 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation expense | 11,005 | 11,005 | ||||||
| Amortization of debt discount | 83,777 | - | ||||||
| Amortization of deferred costs | 16,192 | - | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | 6,386 | (3,462 | ) | |||||
| Accounts payable and accrued liabilities | 317,673 | 339,539 | ||||||
| Accounts payable and accrued liabilities - related parties | 19,908 | - | ||||||
| Common units payable | - | 548,630 | ||||||
| Net Cash Used In Operating Activities | (1,354,145 | ) | (2,617,402 | ) | ||||
| Cash Flows From Financing Activities: | ||||||||
| Proceeds from issuance of preferred units | - | 2,019,116 | ||||||
| Proceeds from issuance of convertible notes payable | 1,011,177 | - | ||||||
| Proceeds from issuance of convertible notes payable - related parties | 514,079 | - | ||||||
| Net Cash Provided By Financing Activities | 1,525,256 | 2,019,116 | ||||||
| Net change in cash and cash equivalents | 171,111 | (598,286 | ) | |||||
| Cash and cash equivalents at beginning of year | 353,366 | 951,652 | ||||||
| Cash and cash equivalents at end of year | $ | 524,477 | $ | 353,366 | ||||
| Supplemental cash flow disclosures: | ||||||||
| Cash paid for interest | $ | - | $ | - | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| Non-cash investing and financing activities: | ||||||||
| Long-term note payable issued to settle accounts payable | $ | 414,165 | $ | - | ||||
| Beneficial conversion feature of convertible notes | $ | 412,252 | $ | - | ||||
See accompanying notes to the financial statements.
| F-24 |
Notes to Financial Statements
Note 1 - Description of Business and Summary of Significant Accounting Policies
Nature of Business - Moleculin, LLC, a Texas limited liability company, (the “Company” or “Moleculin”) is an early-stage Houston-based pharmaceutical company formed in December 2006 to create novel therapies for skin disorders and the development of cancer related technology. Although the Company previously focused on the development of treatments for skin disorders, the Company’s current focus is on the development of drugs for cancer treatment.
Moleculin intends to be acquired by Moleculin Biotech, Inc. (“MBI”) through merging with MBI Acquisition Corp., a Delaware corporation wholly owned by MBI, which will survive the merger. The intended transaction is scheduled to occur immediately prior to the declaration of effectiveness of MBI’s registration statement on Form S-1 with respect to MBI’s initial public offering.
Use of Estimates in Financial Statement Presentation - The preparation of these financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of these financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Going Concern - These financial statements have been prepared on a going concern basis, which assumes the Company will continue to realize its assets and discharge its liabilities in the normal course of business. The continuation of the Company as a going concern is dependent upon the continued financial support from its members, the ability of the Company to obtain necessary equity financing to continue operations, and the attainment of profitable operations. The Company has suffered recurring losses from operating and has insufficient working capital. As of December 31, 2014, the Company has incurred an accumulated deficit of $13,837,705 since inception, had a working capital deficit of $1,095,276, and has not yet generated any significant revenue from operations. These factors raise substantial doubt regarding the Company’s ability to continue as a going concern. These financial statements do not include any adjustments to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
Cash and Cash Equivalents - The Company considers all highly liquid accounts with original maturities of three months or less to be cash equivalents. Balances held by the Company are typically in excess of Federal Deposit Insurance Corporation’s insured limits ($250,000 per depositor per bank). At December 31, 2014 and 2013, all of the Company’s cash was deposited in one bank.
Property and Equipment – Property and equipment are recorded at cost and depreciated on a straight-line basis over estimated useful lives. Leasehold improvements are recorded at cost and depreciated on a straight-line basis over the shorter of the estimated useful life or the lease term. When assets are retired or sold, the cost and related accumulated depreciation are removed from the accounts, and any related gain or loss is reflected in income (loss) for the period. Expenditures for major additions which extend the useful lives of assets are capitalized. Minor replacements, maintenance and repairs which do not improve or extend the life of such assets are charged to expense as incurred.
Impairment of Long-Lived Assets - Management reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount may not be realizable or at a minimum annually during the fourth quarter of the year. If an evaluation is required, the estimated future undiscounted cash flows associated with the asset are compared to the asset’s carrying value to determine if an impairment of such asset is necessary. The effect of any impairment would be to expense the difference between the fair value of such asset and its carrying value. For the years ended December 31, 2013 and 2014, there were no impairments recorded.
| F-25 |
Fair Value of Financial Instruments - Fair value is defined as the price that would be received to sell an asset, or paid to transfer a liability, in an orderly transaction between market participants. A fair value hierarchy has been established for valuation inputs that give the highest priority to quoted prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs. The fair value hierarchy is as follows:
Level 1 Inputs - Unadjusted quoted prices in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date.
Level 2 Inputs - Inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly. These might include quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (such as interest rates, volatilities, prepayment speeds, credit risks, etc.) or inputs that are derived principally from or corroborated by market data by correlation or other means.
Level 3 Inputs - Unobservable inputs for determining the fair values of assets or liabilities that reflect an entity’s own assumptions about the assumptions that market participants would use in pricing the assets or liabilities.
Income Taxes - The Company elected to be taxed as a partnership under the Internal Revenue Code. The Company pays no U.S. taxes on its earnings. The Company’s net earnings/losses are passed through to the Company members and, as such, reports no income tax expense or liability.
Revenue Recognition - The Company recognizes revenues when persuasive evidence of an arrangement exists, delivery has occurred or services have been provided, the purchase price is fixed or determinable and collectability is reasonably assured. The Company may receive cost reimbursement-based grants. For these grants, revenues are based on costs incurred that are specifically covered under reimbursement arrangements. These revenues are recognized as grant-related expenses are incurred by the Company or its subcontractors.
Research and Development Costs - Research and development costs are expensed as incurred.
Subsequent Events - The Company’s management reviewed all material events through the date these financial statements were issued for subsequent event disclosure consideration.
Recent Accounting Pronouncements - In June 2014, the FASB issued ASU 2014-10, Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements. ASU 2014-10 eliminates the distinction of a development stage entity and certain related disclosure requirements, including the elimination of inception-to-date information on the statements of operations, cash flows and stockholders’ equity. The amendments in ASU 2014-10 will be effective prospectively for annual reporting periods beginning after December 15, 2014, and interim periods within those annual periods, however early adoption is permitted. The Company evaluated and adopted ASU 2014-10 as of December 31, 2014.
In April 2015, the FASB issued ASU 2015-03, Interest – Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs. The amended guidance requires that debt issuance costs be presented in the balance sheet as a direct reduction from the carrying amount of the recognized debt liability, consistent with the treatment of debt discounts. Amortization of debt issuance costs is to be reported as interest expense. The updated guidance is effective for reporting periods beginning after December 15, 2015, and interim periods within those annual periods, however early adoption is permitted. The Company evaluated and adopted ASU 2015-03 as of December 31, 2014.
The Company does not believe that any other recently issued effective pronouncements, or pronouncements issued but not yet effective, if adopted, would have a material effect on the accompanying financial statements.
Note 2 – Other Receivables
On October 27, 2010, the Company entered into a Patent and Technology Development and License Agreement with Dermin, Sp. Zo. O. (“Dermin”), a Polish limited liability company, to assist in the development and commercialization of certain pharmaceutical products within a described licensed territory in exchange for royalty payments. On December 19, 2011, the Company advanced $200,000 to Dermin providing funding assistance to Dermin’s efforts in developing and commercializing these pharmaceutical products in and around the Poland European region. Further, throughout the year 2012, the Company paid $32,300 of expenses on behalf of Dermin. As of December 31, 2014 and 2013, the Company had $232,300 long-term note receivable from Dermin which was collected in August 2015.
| F-26 |
Note 3 - Property and Equipment
Property and equipment consisted of the following at December 31, 2014 and 2013:
| Lives | 2014 | 2013 | ||||||||||
| Leasehold improvements | 1 year | $ | 81,602 | $ | 81,602 | |||||||
| Machinery and equipment | 5 years | 57,327 | 57,327 | |||||||||
| Furniture and fixtures | 7 years | 7,955 | 7,955 | |||||||||
| Less: accumulated depreciation | (124,289 | ) | (113,284 | ) | ||||||||
| Property and equipment, net | $ | 22,595 | $ | 33,600 | ||||||||
Depreciation expense for each of the years ended December 31, 2014 and 2013 was $11,005.
Note 4 – Long-Term Notes Payable
On September 16, 2014, the Company issued a promissory note in the amount of $414,165 to Davos Chemical Corporation to settle an outstanding accounts payable balance. The note bears interest at an annual rate of 6.0% and matures on the earlier of December 31, 2016 or the completion of one of the following prior to December 31, 2016:
| a) | The Company (i) is awarded a grant from the Cancer Prevention and Research Institute of Texas (“CPRIT”) of at least $5,000,000, and (ii) is deemed by CPRIT to have received the 50% matching funds required by the CPRIT grant; or |
| b) | The Company concludes a privately placed sale of the Company’s Series A Preferred Units of at least $3,000,000. |
At December 31, 2014, accrued interest related this note was $7,217.
Note 5 – Convertible Notes Payable and Convertible Notes Payable – Related Parties
From May to November 2014, the Company issued various convertible notes to its creditors. Note proceeds from third and related parties were $1,011,177 and $514,079, respectively. These notes bear interest at 8% per annum and are due on the earlier of June 30, 2016 or the consummation of a liquidation event, unless these notes are converted, pursuant to the following:
| a) | Automatic conversion upon CPRIT qualified financing- When the Company is awarded a grant from the CPRIT of at least $3,500,000 and is deemed by CPRIT to have received the 50% matching funds required by the CPRIT grant. The conversion price is $0.80 per unit in the event of a CPRIT qualified financing. |
| b) | First tier automatic conversion - When the Company concludes a privately placed sale of the Company’s Series A Preferred Units of at least $3,000,000 and is at a pre-money valuation of not less than $20,000,000. The conversion price is 80% of the lowest per unit purchase price in the privately placed sale in the event of a first tier automatic conversion. |
| c) | Second tier automatic conversion - When the Company concludes a privately placed sale of the Company’s Series A Preferred Units of at least $2,000,000. The conversion price is 70% of the lowest per unit purchase price in the privately placed sale in the event of a second tier automatic conversion. |
| d) | Voluntary conversion - At any time after the earlier of June 30, 2016 or a Liquidation Event, the note holders can elect to convert the notes into the Company’s Preferred Units at $0.80 per unit. |
| F-27 |
A liquidation event means a) the acquisition of the Company by another entity; b) a sale, exclusive license, lease or other conveyance of all or substantially all of the Company’s assets; c) any liquidation, dissolution of the Company; d) the acquisition of an aggregate of 50% or more of the Company’s voting power by a party; e) in a bankruptcy proceeding. Upon a liquidation event, the note holders is entitle to 150% of the then outstanding principal and unpaid interest.
These convertible notes were analyzed for a beneficial conversion feature at which time it was concluded that a beneficial conversion feature existed. The beneficial conversion feature was measured using the commitment-date stock price and was determined to be $283,732 for third party notes and $128,520 for related party notes. This amount was recorded as a debt discount and is amortized to interest expense over the term of these notes. For the year ended December 31, 2014, amortization of debt discount for third and related parties were $46,068 and $37,709, respectively.
As of December 31, 2014, accrued interest for third party and related party notes were $27,605 and $25,744, respectively.
On September 30, 2014, the Company entered into an agreement with Brompton Group NA, LLC (“Brompton”) for a non-exclusive arrangement to identify investors or lenders interested in providing financing for the Company. In exchange for their services, the Company agreed to pay Brompton 15% of the proceeds received from any financing provided by the investors introduced by Brompton. Brompton agreed to receive two-thirds of the fee, at their option, in either cash or in the form of the same securities offered in the financing being offered to the identified investors or lenders. The remaining fee should be paid in the form of the same securities offered in the financing. Of the $1,254,874 debt raised from third party investors, $825,000 was raised through investors introduced by Brompton. As such, $123,750 of finder’s fee was recorded as debt discount and amortized over the life of the notes. For the year ended December 31, 2014, interest expense associated with the amortization of the finder’s fee was $16,192.
Note 6 - Commitments and Contingencies
MD Anderson Agreements
On June 21, 2010, the Company entered into a patent and technology license agreement with the University of Texas M.D. Anderson Cancer Center (“MD Anderson”) under which the Company is obligated to make upfront and periodic progress payments. The agreement allows the Company to obtain from MD Anderson an exclusive, worldwide royalty-bearing license to research, develop, manufacture, have manufactured, use, import, offer to sell or sell products comprising or made through the use of certain patent and technology rights owned by MD Anderson. In consideration, the Company agreed to pay for all out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining patent rights. Pursuant to the agreement, the Company also agreed to pay: a) an annual maintenance fee starting at $20,000 per year and increase by $10,000 each year up to a maximum of $100,000; b) royalties equal to 2.5% of net sales for licensed products approved for dermatological use; and c) royalties equal to 4.0% of net sales for licensed products approved for all other uses. The 2.5% and 4.0% royalties could be further reduced if certain criteria are met. Annual minimum royalties are $200,000 following the first sale after marketing approval is obtained from MD Anderson by the Company for any licensed products. In addition, the Company will be required to issue 3.5% of the total issued and outstanding common units of the Company on a fully diluted basis to MD Anderson at the time the Company commences its first phase II trial of a drug covered under the agreement. The Company is also required to pay certain milestone payments.
On October 8, 2015, the Company entered into a letter agreement with MD Anderson for the Company’s past due fees to MD Anderson in the amount of $691,186 of which $300,000 had been paid prior to the letter agreement. Pursuant to the letter agreement, MD Anderson agreed to receive the remaining past due fee in three installments: a) $125,000 due on October 31, 2015; b) $175,000 due on January 31, 2016; and c) $91,186 due on April 30, 2016. The Company paid $125,000 to MD Anderson on November 2, 2015. The Company and MD Anderson have agreed to defer the $175,000 payment due January 31, 2016 until the earlier of April 30, 2016 or the completion of this offering.
| F-28 |
On October 19, 2015, the agreement was amended for the milestone payments. The amended milestone payments are as follows:
| Phase | Amount | |||
| Commencement of Phase III Study for first licensed drug/product | $ | 150,000 | ||
| Submission of the first NDA | $ | 500,000 | ||
| Receipt of first marketing approval | $ | 600,000 | ||
The amendment also removed the term regarding the issuance of 3.5% of the Company outstanding units.
For the year ended December 31, 2013, the Company recorded a liability to and paid MD Anderson $1,214,651 and $260,000, respectively. For the year ended December 31, 2014, the Company recorded a liability to and paid MD Anderson $323,912 and $50,000, respectively.
Dermin Agreement
On October 27, 2010, the Company entered into a Patent and Technology Development and License Agreement with Dermin. The agreement provided Dermin with sub-license rights to the Company’s technologies for use in limited territories in exchange for Dermin’s use of Polish government grant funding to pay for development costs the Company would otherwise have been required to fund. Dermin’s territories are primarily Poland and surrounding countries, but not including any of the major European markets (UK, Germany, France, Spain and Italy). Pursuant to the agreement, Dermin is required to pay the Company a running royalty on sales of license products. As of December 31, 2014, no sales have occurred and no royalty paid to the Company by Dermin.
Lease Agreements
On July 31, 2007, the Company entered into a rental agreement for office space at 2575 West Belfort, Houston, Texas. The agreement was renewed on July 26, 2010 for one year. After July 2011, the lease continues on a month-to-month basis. Therefore, there are no future minimum obligations on the lease. Rent expense for the years ended December 31, 2013 and 2014 was $28,600 and $26,600, respectively, and is included in general and administrative expenses on the statements of operations.
Note 7 - Equity
In March 2011, the Company amended its articles of incorporation to create two classes of interests referred to as ‘Series A Preferred Membership Interests’ and ‘Common Membership Interests’, with each interest issued being measured as Units (Series A Preferred Units and Common Units, respectively). The Series A Preferred Units are further broken down into 4 series including Series A-1, Series A-2, Series A-3 and Series A-4.
Pursuant to Moleculin LLC's Company Agreement, the Board shall distribute net cash, as defined in the Company Agreement, to the members at such times and in such amounts as the Board may determine in the following order and priority:
| e) | First, to the Series A Preferred members in proportion to each such members’ original capital contribution until such members receive net cash equal to 150% of their original capital contribution. |
| f) | Second, to the members in proportion to their percentage interests until Series A Preferred members have received net cash equal to 300% of their original capital contribution. |
| g) | Third, to the common members in proportion to each common member’s common units until the common members have receive aggregate distributions of net cash equal to the then total amount of distributions to all members for all company fiscal years multiplied by the common members’ aggregate percentage interests; and |
| h) | Fourth, to the members in accordance with their percentage interests. |
| F-29 |
During 2013, the Company sold 2,019,116 units of Preferred Series A-4 at $1 per units to various members. As of December 31, 2014 and 2013, the Preferred Units are summarized as follows:
| Units Outstanding | ||||||||||||
| Series | December 31, 2014 | December 31, 2013 | Contribution | |||||||||
| Series A-1 | 283,604 | 283,604 | $ | 150,000 | ||||||||
| Series A-2 | 1,595,552 | 1,595,552 | 600,000 | |||||||||
| Series A-3 | 1,942,400 | 1,942,400 | 513,672 | |||||||||
| Series A-4 | 9,694,264 | 9,694,264 | 9,356,514 | |||||||||
| Total | 13,515,820 | 13,515,820 | $ | 10,620,186 | ||||||||
During the first quarter of 2013, the Company commenced the first Phase II Study of a licensed product in the patent and technology license agreement between the Company and MD Anderson. Pursuant to the agreement, the Company was obligated to issue MD Anderson 548,630 common units representing 3.5% of its issued and outstanding equity units. These common units were valued at their estimated fair value of $548,630, which was recorded as a research and development expense. In October 2015, MD Anderson agreed to forfeit its right to these units.
Note 8 – Related Party Transactions
On July 1, 2013, the Company agreed to reimburse Houston Pharmaceuticals, Inc. (“HPI”), an entity controlled by a member of the Company’s advisory board, for laboratory space used by the Company from January 2010 through December 2013 in the amount of $31,764. Rent expense was allocated back to those periods, with rent expense of $7,941 recorded in the year ended December 31, 2013. The Company stopped using the space in 2014.
On September 1, 2013, the Company agreed to reimburse HPI for laboratory equipment used by the Company for the period from January 2009 through December 2013 in the amount of $35,000. Rent expense was allocated back to those periods, with rent expense of $7,000 recorded in the year ended December 31, 2013. The Company stopped using the equipment in 2014.
During the year ended December 31, 2013, the Company also paid $100,000 for laboratory services provided by HPI for the five years prior to August 1, 2013.
Note 9 - Subsequent Events
On September 8, 2015, the Company issued an unsecured promissory note to Hilltop Research in the amount of $55,774 to settle an outstanding payable. The note matures on June 30, 2016 and accrues interest at 6.0% annum. In the event the Company or a significant portion of the Company’s assets are acquired by MBI prior to the maturity date, the loan will be paid by MBI on closing of MBI’s initial public offering.
On October 15, 2015, the Company entered into a $5,000 exclusive option agreement with MD Anderson allowing the Company a period of time to evaluate the potential of acquiring and to negotiate the purchase of an exclusive license in the field of cancer therapeutics. The option period extends from October 15, 2015 through August 31, 2016. MD Anderson shall not offer a license to any third party during the option period.
| F-30 |
Up to 2,000,000 Shares
Moleculin Biotech, Inc.
Common Stock
Bonwick Capital Partners LLC
Network 1 Financial Securities, Inc.
Through and including , 2016 (the 25th day after the date of this prospectus), all dealers effecting transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth the estimated costs and expenses to be incurred in connection with the issuance and distribution of the securities of Moleculin Biotech, Inc. (the “Registrant”) which are registered under this Registration Statement on Form S-1 (this “Registration Statement”), other than underwriting discounts and commissions. All amounts are estimates except the Securities and Exchange Commission registration fee and the Financial Industry Regulatory Authority, Inc. filing fee.
The following expenses will be borne solely by the Registrant.
| Amount to be | ||||
| Paid | ||||
| SEC Registration fee | $ | 1,292.99 | ||
| Financial Industry Regulatory Authority, Inc. filing fee | $ | 2,500 | ||
| Nasdaq Listing fees | $ | 75,000 | ||
| Printing and engraving expenses | $ | * | ||
| Legal fees and expenses | $ | * | ||
| Accounting fees and expenses | $ | * | ||
| Transfer Agent’s fees | $ | * | ||
| Miscellaneous fees and expenses | $ | * | ||
| Total | * | |||
| * | To be provided by amendment. |
Item 14. Indemnification of Directors and Officers.
Pursuant to Section 145 of the Delaware General Corporation Law (the “DGCL”), a corporation shall have the power to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than a derivative action by or in the right of such corporation) by reason of the fact that such person is or was a director, officer, employee or agent of such corporation, or serving at the request of such corporation in such capacity for another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred in connection with such action, suit or proceeding, if such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interests of such corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
The DGCL also permits indemnification by a corporation under similar circumstances for expenses (including attorneys’ fees) actually and reasonably incurred by such persons in connection with the defense or settlement of a derivative action or suit, except that no indemnification shall be made in respect of any claim, issue or matter as to which such person shall have been adjudged to be liable to such corporation unless the Delaware Court of Chancery or the court in which such action or suit was brought shall determine upon application that such person is fairly and reasonably entitled to indemnity for such expenses which such court shall deem proper.
To the extent a present or former director or officer is successful in the defense of such an action, suit or proceeding referenced above, or in defense of any claim, issue or matter therein, a corporation is required by the DGCL to indemnify such person for actual and reasonable expenses incurred in connection therewith. Expenses (including attorneys’ fees) incurred by such persons in defending any action, suit or proceeding may be paid in advance of the final disposition of such action, suit or proceeding upon in the case of a current officer or director, receipt of an undertaking by or on behalf of such person to repay such amount if it is ultimately determined that such person is not entitled to be so indemnified.
The DGCL provides that the indemnification described above shall not be deemed exclusive of other indemnification that may be granted by a corporation pursuant to its bylaws, disinterested directors’ vote, stockholders’ vote and agreement or otherwise.
| II-1 |
Section 102(b)(7) of the DGCL enables a corporation, in its certificate of incorporation or an amendment thereto, to eliminate or limit the personal liability of a director to the corporation or its stockholders for monetary damages for violations of the directors’ fiduciary duty, except (i) for any breach of the director’s duty of loyalty to the corporation or its stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (iii) pursuant to Section 174 of the DGCL (providing for liability of directors for unlawful payment of dividends or unlawful stock purchases or redemptions) or (iv) for any transaction from which a director derived an improper personal benefit. The Registrant’s certificate of incorporation provides for such limitations on liability for its directors.
The DGCL also provides corporations with the power to purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation in a similar capacity for another corporation, partnership, joint venture, trust or other enterprise, against any liability asserted against him or her in any such capacity or arising out of his or her status as such, whether or not the corporation would have the power to indemnify him or her against such liability as described above. In connection with this offering, the Registrant will obtain liability insurance for its directors and officers. Such insurance would be available to its directors and officers in accordance with its terms.
The Registrant’s certificate of incorporation in requires the Registrant to indemnify and hold harmless, to the fullest extent permitted by applicable law as it presently exists or may hereafter be amended, any person (a “covered person”) who was or is made or is threatened to be made a party or is otherwise involved in any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (a “proceeding”) by reason of the fact that he or she is or was a director, officer or member of a committee of the Registrant, or, while a director or officer of the Registrant, is or was serving at the request of the Registrant as a director or officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise or non-profit entity, including service with respect to employee benefit plans, against all liability and loss suffered and expenses (including attorneys’ fees), judgment, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with a proceeding.
In addition, under the Registrant’s certificate of incorporation, in certain circumstances, the Registrant shall pay the expenses (including attorneys’ fees) incurred by a covered person in defending a proceeding in advance of the final disposition of such proceeding; provided, however, that the Registrant shall not be required to advance any expenses to a person against whom the Registrant directly brings an action, suit or proceeding alleging that such person (1) committed an act or omission not in good faith or (2) committed an act of intentional misconduct or a knowing violation of law. Additionally, an advancement of expenses incurred by a covered person shall be made only upon delivery to the Registrant of an undertaking, by or on behalf of such covered person, to repay all amounts so advanced if it shall ultimately be determined by final judicial decision from which there is no further right to appeal or otherwise in accordance with Delaware law that such covered person is not entitled to be indemnified for such expenses.
The foregoing statements are subject to the detailed provisions of Section 145 of the DGCL and the full text of the Registrant’s certificate of incorporation, which is filed as Exhibit 3.1 hereto. Reference is made to the form of underwriting agreement to be filed as Exhibit 1.1 hereto for provisions providing that the underwriters are obligated under certain circumstances, to indemnify our directors, officers and controlling persons against certain liabilities under the Securities Act of 1933, as amended.
Item 15. Recent Sales of Unregistered Securities.
Except as set forth below, in the three years preceding the filing of this Registration Statement, the Registrant has not issued any securities that were not registered under the Securities Act:
In August 2015, Messrs. Klemp, Picker and Priebe purchased 1,000,000 shares, 500,000 shares and 3,000,000 shares of our common stock, respectively, at a purchase price of $0.001 per share.
In August 2015, in exchange for the issuance of 630,000 shares of common stock, we acquired the rights to the license agreement with MD Anderson covering our WP1122 Portfolio held by IntertechBio Corporation, a company affiliated with Messrs. Priebe and Picker. In August 2015, in exchange for the issuance of 1,431,000 shares of common stock, we acquired the rights to the Annamycin IND and all data related to the Annamycin IND or the development of Annamycin held by AnnaMed, Inc., a company affiliated with Mr. Klemp.
Prior to the effective date of the registration statement of which this prospectus is a part, Moleculin LLC, a Texas limited liability company, will be merged with and into MBI Acquisition Corp., a Delaware corporation and our wholly owned subsidiary, which will survive the merger. Moleculin LLC is the holder of the license agreement with MD Anderson covering our WP 1066 Portfolio. As a result of the merger, we will issue the equity interests holders of Moleculin LLC an aggregate of 1,000,000 shares of our common stock. Messrs. Klemp, Picker and Priebe are members of Moleculin LLC and will receive shares of our common stock as a result of the merger.
| II-2 |
In August and September 2015, the Registrant issued two 8% convertible notes in an aggregate of $250,000 in principal amount of convertible notes to one investor, which principal and accrued interest will automatically convert into shares of common stock upon the closing of this offering at a conversion rate of $0.1299 per share. In October 2015, the Registrant issued two 8% convertible notes in an aggregate of $200,000 in principal amount of convertible notes to two investors, which principal and accrued interest will automatically convert into shares of common stock upon the closing of this offering at a conversion rate of $0.20 per share. At the time of such purchase in October 2015, the convertible note investors and the Registrant agreed that the investors would fund the Registrant up to the filing of its initial non-confidential registration statement an additional $165,000 on the same terms as in the October financing. Pursuant to such agreement, such amounts were received in January 2016.
In January and February 2016, the Registrant sold 53,000 shares of common stock for a total purchase price of $159,000 to 7 investors.
None of these transactions involved any underwriters’ underwriting discounts or commissions, or any public offering. We believe that each of the following issuances was exempt from registration under the Securities Act in reliance on Section 4(a)(2) of the Securities Act regarding transactions not involving a public offering.
Item 16. Exhibits and Financial Statement Schedules.
(a) Exhibits: Reference is made to the Exhibit Index following the signature pages hereto, which Exhibit Index is hereby incorporated into this Item.
(b) Financial Statement Schedules: All schedules are omitted because the required information is inapplicable or the information is presented in the financial statements and the related notes.
Item 17. Undertakings
The undersigned hereby undertakes:
(a) The undersigned Registrant hereby undertakes to provide to the underwriter at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.
(b) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the provisions referenced in Item 14 of this Registration Statement, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered hereunder, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question of whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
(c) The undersigned Registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this Registration Statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this Registration Statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new Registration Statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
| II-3 |
SIGNATURES
Pursuant to the requirements of the Securities Act, the registrant has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the city of Houston, Texas, on February 16, 2016.
| MOLECULIN BIOTECH, INC. | ||
| (Registrant) | ||
| By: | /s/ Walter V. Klemp | |
| Walter V. Klemp | ||
| Director and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed below by the following persons in the capacities and on the dates indicated:
| SIGNATURE | TITLE | DATE | ||
| /s/ Walter V. Klemp | ||||
| Walter V. Klemp | Chief Executive Officer and Sole Director | February 16, 2016 | ||
| (Principal Executive Officer) | ||||
| /s/ Louis Ploth, Jr. | ||||
| Louis Ploth, Jr. | Chief Financial Officer | February 16, 2016 | ||
| (Principal Financial Officer and Principal Accounting Officer) | ||||
| * | ||||
| Donald Picker | President and Chief Operating Officer | February 16, 2016 |
| *By: | /s/ Walter V. Klemp | ||
| Walter V. Klemp, | |||
| Attorney-in-fact |
EXHIBIT INDEX
| Exhibit Number |
Description |
| 1.1 | Form of Underwriting Agreement* |
| 3.1 | Amended and Restated Certificate of Incorporation to be filed prior to offering* |
| 3.2 | Amended and Restated Bylaws of to be filed prior to offering * |
| 5 | Opinion of Schiff Hardin LLP as to legality of the securities being registered* |
| 10.1 | Moleculin Biotech, Inc. 2015 Incentive Plan * |
| 10.2 | Rights Transfer Agreement between Moleculin Biotech, Inc. and AnnaMed, Inc.* |
| 10.3 | Patent and Technology License Agreement dated June 21, 2010 by and between The Board of Regents of the University of Texas System and Moleculin, LLC* |
| 10.4 | Amendment No. 1 to the Patent and Technology License Agreement dated June 21, 2010 by and between The Board of Regents of the University of Texas System and Moleculin, LLC* |
| 10.5 | Patent and Technology License Agreement dated April 2, 2012 by and between The Board of Regents of the University of Texas System and Intertech Bio Corporation* |
| 10.6 | Amendment No. 1 to the Patent and Technology License Agreement dated June 21, 2010 by and between The Board of Regents of the University of Texas System and Intertech Bio Corporation* |
| 10.7 | Amended and Restated Patent and Technology Development and License Agreement by and between Annamed, Inc. and Dermin Sp. z.o.o* |
| 10.8 | Patent and Technology Development and License Agreement dated April 15, 2011 by and between Intertech Bio Corporation and Dermin Sp. z.o.o* |
| 10.9 | Patent and Technology Development and License Agreement dated October 27, 2010 by and between Moleculin, LLC and Dermin Sp. z.o.o* |
| 10.10 | Rights Transfer Agreement dated between Moleculin Biotech, Inc. and Intertech Bio Corporation dated August 11, 2015* |
| 10.11 | Rights Transfer Agreement dated between Moleculin Biotech, Inc. and Annamed dated August 11, 2015* |
| 10.12 | Form of Escrow Deposit Agreement by and among Moleculin Biotech, Inc., Signature Bank and the Underwriters |
| 21 | Subsidiaries of the Registrant* |
| 23.1 | Consent of Independent Registered Public Accounting Firm |
| 23.2 | Consent of Schiff Hardin LLP (included in Exhibit 5)* |
| 99.1 | Consent of Director Nominee Robert E. George |
| 99.2 | Consent of Director Nominee Michael D. Cannon |
| 99.3 | Consent of Director Nominee Jacqueline Northcut |
| * | To be filed by amendment. |