Attached files
| file | filename |
|---|---|
| EX-5.1 - EXHIBIT 5.1 - AEOLUS PHARMACEUTICALS, INC. | exh5_1.htm |
| EX-23.1 - EXHIBIT 23.1 - AEOLUS PHARMACEUTICALS, INC. | exh23_1.htm |
As filed with the Securities and Exchange Commission on February 12, 2016
Registration No. 333-209119
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
______________
AMENDMENT NO. 1 TO
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
AEOLUS PHARMACEUTICALS, INC.
(Exact Name of Issuer in Its Charter)
|
Delaware
|
2834
|
56-1953785
|
|
(State or Other Jurisdiction of
Incorporation or Organization)
|
(Primary Standard Industrial
Classification Number)
|
(I.R.S. Employer
Identification No.)
|
Aeolus Pharmaceuticals, Inc.
26361 Crown Valley Parkway, Suite 150
Mission Viejo, California 92691
(949) 481-9825
(Address, including zip code, and telephone number,
including area code, of registrant's principal executive offices)
______________
John McManus
President and Chief Executive Officer
Aeolus Pharmaceuticals, Inc.
26361 Crown Valley Parkway, Suite 150
Mission Viejo, California 92691
(949) 481-9825
(Name, address, including zip code, and telephone number,
including area code, of agent for service)
______________
Copies of all communications to:
|
Brian J. Lynch
Drinker Biddle & Reath LLP
One Logan Square, Suite 2000
Philadelphia, Pennsylvania 19103-6996
(215) 988-1119
|
______________
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement becomes effective
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer
|
☐
|
Accelerated filer
|
☐
|
|
Non-accelerated filer
|
☐ (Do not check if a smaller
reporting company)
|
Smaller reporting company
|
☒
|
______________
CALCULATION OF REGISTRATION FEE
|
Title of Each
Class of Securities
to be Registered
|
Amount to be
Registered(1)
|
Proposed
Maximum
Offering Price
Per Security
|
Proposed
Maximum
Aggregate
Offering Price
|
Amount of
Registration Fee
|
||||||||||||
|
Common stock, par value $0.01 per share
|
15,629,676
|
$
|
0.22
|
(2)
|
$
|
3,438,528.72
|
$
|
346.26
|
||||||||
|
Common stock, $0.01 par value per share, issuable upon the exercise of warrants
|
37,298,249
|
$
|
0.22
|
(3)
|
$
|
8,205,614.78
|
$
|
826.31
|
||||||||
|
Common stock, $0.01 par value per share, issuable upon the conversion of Series C Convertible Preferred Stock
|
20,454,546
|
$
|
[ ]
|
(4)
|
$
|
4,500,000
|
$
|
453.15
|
||||||||
|
Common stock, $0.01 par value per share, issuable upon the exercise of warrants
|
50,000
|
$
|
0.26
|
(3)
|
$
|
13,000
|
$
|
1.31
|
||||||||
|
Total:
|
73,432,471
|
$
|
16,157,143.50
|
$
|
1,627.03
|
(5) | ||||||||||
(1) In accordance with Rule 416 under the Securities Act, the registrant is also registering hereunder an indeterminate number of shares that may be issued and resold resulting from stock splits, stock dividends or similar transactions.
(2) Estimated solely for purposes of calculating the registration fee in accordance with Rule 457(c) under the Securities Act, using the average of the high and low prices as reported on the OTC Bulletin Board on February 2, 2016.
(3) Estimated solely for purposes of calculating the registration fee in accordance with Rule 457(g) under the Securities Act, the proposed maximum offering price per share is based on the exercise price of the warrants.
(4) Estimated solely for purposes of calculating the registration fee in accordance with Rule 457(i) under the Securities Act, the proposed maximum offering price per share is based on the equivalent purchase price per share of common stock underlying the Series C Convertible Preferred Stock.
(5) Previously paid.
______________________
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell the securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to completion, dated February 12, 2016
PRELIMINARY PROSPECTUS
73,432,471
Common Stock
This prospectus relates to the offer and sale from time to time by the selling stockholders identified in this prospectus of up to 73,432,471 shares of our common stock, par value $0.01 per share. These shares include (i) 15,629,676 shares of common stock issued and outstanding, (ii) 20,454,546 shares of common stock issuable upon conversion of Series C Convertible Preferred Stock and (iii) 37,298,249 shares of common stock issuable upon exercise of warrants, all of which were issued in connection with a private placement which closed in December of 2015. This prospectus also relates to 50,000 shares of our common stock issuable upon the exercise of warrants that we issued in 2014. The shares of common stock and warrants were issued in transactions made in reliance on Section 4(a)(2) of the Securities Act of 1933, as amended, which we refer to as the Securities Act, and Rule 506 promulgated thereunder. For additional information regarding the private placements, please see "Description of the Shares included in this Prospectus" beginning on page 8 of this prospectus.
We are not selling any common stock under this prospectus and will not receive any of the proceeds from the sale of shares by the selling stockholders; however, we will receive the proceeds of any cash exercise of the warrants.
The selling stockholders may sell the shares from time to time at the market price quoted on the OTC Bulletin Board (or any stock exchange on which our common stock may be listed in the future) at the time of offer and sale, or at prices related to such prevailing market prices, in negotiated transactions or in a combination of such methods of sale directly or through brokers. See "Plan of Distribution" beginning on page 109 for additional information on how the selling stockholders may conduct sales of their shares of common stock.
In addition to the shares that may be offered under this prospectus, we also have registered shares for sale by certain other selling stockholders, who may offer and sell up to 117,512,184 shares under a separate prospectus. See "Risk Factors – Risks Related to Owning Our Stock; Substantial blocks of our total outstanding common stock may be sold into the market through existing registration statements already on file with the SEC. If there are substantial sales of shares of our common stock, the price of our common stock could decline"
Other than underwriting discounts and commissions, and transfer taxes, if any, we have agreed to bear certain expenses incurred in connection with the registration and sale of the common stock offered by the selling stockholders.
Our common stock is quoted on the OTC Bulletin Board under the symbol "AOLS." On February 2, 2016, the closing price of our common stock was $0.20 per share.
Investing in our common stock involves a high degree of risk. See "Risk Factors" beginning on page 8 for certain risks you should consider before purchasing any shares.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved these securities, or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
______________
The date of this prospectus is February 12, 2016
TABLE OF CONTENTS
|
Page
|
|
|
Prospectus Summary
|
1
|
|
Risk Factors
|
8
|
|
Special Note Regarding Forward-Looking Statements
|
27
|
|
Description of the Shares Included in this Prospectus
|
28
|
|
Use of Proceeds
|
30
|
|
Determination of Offering Price
|
31
|
|
Market Information / Price Range Of Common Stock / Dividends
|
32
|
|
Management's Discussion and Analysis of Financial Condition and Results of Operations
|
34
|
|
Business
|
42
|
|
Management
|
80
|
|
Compensation of Directors
|
84
|
|
Executive Compensation
|
86
|
|
Certain Relationships and Related Party Transactions
|
90
|
|
Security Ownership of Certain Beneficial Owners and Management
|
92
|
|
Selling Stockholders
|
95
|
|
Description of Capital Stock
|
105
|
|
Plan of Distribution
|
110
|
|
Legal Matters
|
112
|
|
Experts
|
112
|
|
Additional Information
|
112
|
|
Incorporation of Certain Information by Reference
|
113
|
|
Index to Financial Statements
|
F-1
|
You should only rely on the information contained in this prospectus. We have not, and the selling stockholders have not, authorized anyone to provide you with additional information or information different from that contained in this prospectus. We are not making an offer to sell these securities in any jurisdiction where an offer or sale is not permitted. You should assume that the information appearing in this prospectus is accurate as of the date on the front cover of this prospectus only, regardless of the time of delivery of this prospectus or of any sale of our securities. Our business, prospects, financial condition and results of operations may have changed since that date.
This document may only be used where it is legal to sell these securities. Certain jurisdictions may restrict the distribution of these documents and the offering of these securities. We require persons receiving these documents to inform themselves about, and to observe any, such restrictions. We have not taken any action that would permit an offering of these securities or the distribution of these documents in any jurisdiction that requires such action.
_________________________________
We own or have rights to trademarks or trade names that we use in conjunction with the operation of our business. Each trademark, trade name or service mark of any other company appearing in this prospectus belongs to its holder. Use or display by us of other parties' trademarks, trade names or service marks is not intended to and does not imply a relationship with, or endorsement or sponsorship by us of, the trademark, trade name or service mark owner
_________________________________
Industry and Market Data
Unless otherwise indicated, the market data and certain other statistical information used throughout this prospectus are based on independent industry publications, government publications, reports by market research firms or other published independent sources. Although we believe these third-party sources are reliable, we have not independently verified the information. Except as otherwise noted, none of the sources cited in this prospectus has consented to the inclusion of any data from its reports, nor have we sought their consent. In addition, some data are based on our good faith estimates. Such estimates are derived from publicly available information released by independent industry analysts and third-party sources, as well as our own management's experience in the industry, and are based on assumptions made by us based on such data and our knowledge of such industry and markets, which we believe to be reasonable. However, except as otherwise noted, none of our estimates have been verified by any independent source. Our estimates and assumptions involve risks and uncertainties and are subject to change based on various factors, including those discussed in the "Risk Factors" section of this prospectus and the other information contained herein. These and other factors could cause our actual results to differ materially from those expressed in the estimates and assumptions.
PROSPECTUS SUMMARY
This summary highlights certain information contained elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our common stock. You should carefully read the entire prospectus, including ''Risk Factors'' and our financial statements and related notes before you decide whether to invest in our common stock. Investing in our common stock involves risks. See ''Risk Factors'' beginning on page 3. All dollar amounts referred to in this prospectus are in U.S. dollars unless otherwise indicated. Any discrepancies in the tables included herein between the amounts listed and the totals thereof are due to rounding.
Unless otherwise indicated or unless the context otherwise requires, all references in this document to "we," "us," "our," the "Company" and similar expressions are references to Aeolus Pharmaceuticals, Inc.
Our Company and Business
We are a Southern California-based biopharmaceutical company leveraging significant U.S. Government funding to develop a platform of novel compounds for use in biodefense, fibrosis, oncology, infectious disease and diseases of the central nervous system. The platform consists of approximately 180 compounds licensed from the University of Colorado ("UC") Duke University ("Duke") and National Jewish Health ("NJH").
Our lead compound, AEOL 10150 ("10150"), is being developed under contract with the Biomedical Advanced Research and Development Authority ("BARDA" and the "BARDA Contract"), a division of the U.S. Department of Health and Human Services ("HHS"), as a medical countermeasure ("MCM") against the pulmonary sub-syndrome of acute radiation syndrome ("Pulmonary Acute Radiation Syndrome" or "Lung-ARS") and the delayed effects of acute radiation exposure ("DEARE"). Lung-ARS is caused by acute exposure to high levels of radiation due to a nuclear detonation or radiological event.
We are also developing 10150 for the treatment of lung fibrosis, including idiopathic pulmonary fibrosis ("IPF") and other fibrotic diseases. This new development program was created based upon the data generated from animal studies in Lung-ARS and chemical gas exposure under the BARDA Contract and National Institutes of Health ("NIH") grants. On March 17, 2015, we announced that 10150 was granted Orphan Drug Designation for IPF by the U.S. Food and Drug Administration ("FDA"). The Company plans to initiate a Phase 1 safety study in patients with IPF in 2016. After we have completed safety studies, we plan to initiate efficacy studies in patients with IPF. AEOL 10150 has previously been tested in two Phase I human clinical trials with no drug-related serious adverse events reported.
We are also developing 10150 for use in combination with radiation therapy for cancer as a treatment to reduce side effects caused by radiation toxicity and improve local tumor control. Pre-clinical studies at Duke, the University of Maryland and Loma Linda University have demonstrated that 10150 protects healthy, normal tissue, while not interfering with the benefit of radiation therapy or chemotherapy in prostate and lung cancer. Additional studies have demonstrated that 10150 enhances the anti-tumor activity of chemotherapy and radiation. A significant portion of the development work funded by the BARDA contract is applicable to the development program for radiation oncology, including safety, toxicology, pharmacokinetics and Chemistry, Manufacturing and Controls ("CMC"). After we have completed safety studies, we plan to initiate studies to demonstrate efficacy in protecting against the toxic side effects related to radiation therapy.
We are also developing 10150 as a MCM for exposure to chemical vesicants (e.g., chlorine gas, sulfur mustard gas and phosgene gas) and nerve agents (e.g., sarin gas and soman gas) with grant money from the NIH Countermeasures Against Chemical Threats ("NIH-CounterACT") program. 10150 has consistently demonstrated safety and efficacy in animal studies of chemical gas exposure and nerve gas exposure.
The Company is developing a second compound, AEOL 11114B ("11114"), as a treatment for Parkinson's disease. Research funded by the Michael J Fox Foundation for Parkinson's disease ("MJFF") demonstrated the neuro-protective activity of 11114 in mouse and rat models of Parkinson's disease. The compounds were invented by Aeolus in collaboration with Brian J. Day, PhD at National Jewish Health and Manisha Patel, PhD at the University of Colorado, Anschutz Medical Campus, Department of Pharmaceutical Sciences in collaboration with the Company. We have obtained worldwide, exclusive licenses to develop the compounds from NJH and the UC. We plan to complete the remaining work to file an Investigational New Drug ("IND") application with the FDA by 2017.
1
In April 2015, we announced the discovery of a new compound, AEOL 20415 ("20415"), which has demonstrated anti-inflammatory and anti-infective properties, and could be effective in treating cystic fibrosis and combatting anti-biotic resistant bacteria. The compound was developed under collaboration between Brian J. Day, PhD at National Jewish Health in Denver, Colorado and Aeolus Pharmaceuticals. We have obtained a worldwide, exclusive license to develop the rights to the compound from NJH. We plan to complete the remaining work to file an Investigational New Drug ("IND") application with the FDA by 2017.
Finally, we have a pipeline of approximately 180 additional compounds. We expect that the development of additional compounds in our portfolio is dependent on our finding non-dilutive capital sources to fund such pipeline opportunities.
Recent Developments
On December 10, 2015, we entered into securities purchase agreements with certain accredited investors to sell and issue (i) an aggregate of 10,215,275 common units issued at a purchase price of $0.22 per unit, and (ii) 4,500 preferred stock units issued to Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P., for an aggregate purchase price of $4.5 million, resulting in aggregate gross proceeds to the Company of approximately $6.75 million (the "2015 Securities Placement"). Each common unit consisted of one share of the Company's common stock and a five year warrant to purchase one share of the Company's common stock, subject to adjustment. The preferred stock units collectively consisted of (i) 4,500 shares of our Series C Convertible Preferred Stock (the "Series C Preferred Stock") that are collectively convertible into an aggregate of 20,454,546 shares of the Company's common stock and (ii) warrants to purchase an aggregate of 20,454,546 shares of our common stock, in each case subject to adjustment. Each of the foregoing warrants has an initial exercise price of $0.22 per share.
On September 29, 2015, the Company received funding in the form of convertible promissory notes (the "BVF Notes") from Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P. The BVF Notes (i) had an aggregate principal balance of $1,000,000, (ii) accrued interest at a rate of 6% per annum, (iii) had a scheduled maturity date of September 28, 2016 and (iv) were subject to automatic conversion into Company equity securities, provided a qualified financing of not less than $4 million occurred. Following the completion of the 2015 Securities Placement, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of our common stock and warrants to purchase an additional 5,414,402 shares of our common stock at an exercise price per share of $0.22 subject to adjustment (the "BVF Convert"). As a result, the BVF Notes are no longer outstanding as of the date of this prospectus.
Strategy
Our strategy is pursue the development of our promising platform of anti-fibrotic, anti-inflammatory, anti-infective and anti-oxidant compounds that address important unmet medical indications of clinical and national strategic importance. Our objective is to use non-dilutive capital whenever possible.
To date, we, and/or our research collaborators, have been awarded more than $149 million in non-dilutive U.S. government funding in the form of grants and contracts from federal agencies, such as BARDA, NIH-NIAID and NIH-CounterACT. Additional research has been conducted on our compounds with funding from private foundations, such as the MJFF and Citizens United for Research in Epilepsy ("CURE").
The expected benefit of this strategy is threefold. First, safety, toxicology, pharmacokinetic and CMC work funded by the government and foundations is applicable to our traditional commercial development programs. As an example, significant work funded under the BARDA contract for Lung-ARS has generated data that can be used to support our New Drug Applications ("NDA") for pulmonary fibrosis and/or radiation therapy for cancer.
2
Second, cost-plus development contracts, like our contract with BARDA, include funds for overhead and profit. These overhead and profit streams have significantly reduced our cash burn rate, which reduces our need to raise capital and incur dilution.
Third, some government contracts, such as the Lung ARS contract with BARDA are designed to lead to the acquisition of the product under development by the US Government for use as a MCM in the Strategic National Stockpile ("SNS" or the "Stockpile"). Government procurement could result in significant revenue to the Company, which could be used to further the development of the product in other indications or for the development of other promising products. Procurements may be made if either the drug meets the requirements for approval by the U.S. Food and Drug Administration (the "FDA") under the "Animal Rule" or prior to Animal Rule approval following the filing of a pre-Emergency Use Authorization ("EUA") application. Most of BARDA's procurements to date have been following the filing of a pre-EUA application.
The amount of any potential procurement is undisclosed by BARDA at this time and is unknown to us. Based on publicly available information, as well as other procurements made by the agency after pre-EUA applications, we believe the agency may purchase sufficient courses of therapy to treat a minimum of one hundred thousand people, with options to purchase an additional two hundred thousand courses of treatment. If purchases of such volumes occurred, the revenue to the Company could provide funding to advance numerous clinical studies, including potentially large Phase III programs in lung fibrosis and radiation therapy for cancer. This funding could allow us to fund studies with less dependence on collaborative partnering arrangements and future equity offerings, which is consistent with our strategy to deploy non-dilutive capital wherever possible to develop our compounds for unmet medical indications and thereby generate value for our stockholders. In addition, purchases of such volumes of drug could make the Company profitable.
Business Overview
We are developing a platform of compounds with anti-fibrotic, anti-inflammatory, anti-infective and anti-oxidant activity based on technology discovered and researched at Duke University, the University of Colorado and National Jewish Health, developed by Drs. Irwin Fridovich, Brian Day and others. Dr. Day is our Chief Scientific Officer.
Our lead compound, 10150, is a metalloporphyrin specifically designed to neutralize reactive oxygen and nitrogen species. The neutralization of these species reduces oxidative stress, inflammation, and subsequent tissue damage-signaling cascades resulting from radiation or chemical exposure. We are developing 10150 as a MCM for national defense and for use in oncology and treating lung fibrosis.
Our most extensive development program to date is the advanced development of 10150 for Lung-ARS and DEARE. On February 11, 2011, we signed a cost-plus contract with BARDA for the development of 10150 as a MCM against Lung-ARS. BARDA is the government agency responsible for the advanced development and purchase of medical countermeasures for chemical, biological, radiological and nuclear threats. The contract contemplates the advanced development of 10150 through approval by the FDA under 21 CFR Part 314 Subpart I and Part 601 Subpart H (the "Animal Rule.") The Animal Rule allows for approval of drugs using only animal studies when human clinical trials cannot be conducted ethically. The ultimate goal of the BARDA Contract is to complete all of the work necessary to obtain FDA approval for 10150 as a MCM for Lung-ARS. In addition, the BARDA Contract is designed to generate the data that would allow for acquisition of the drug by BARDA prior to FDA approval under a pre-Emergency Use Authorization.
Pursuant to the BARDA Contract we were awarded approximately $10.4 million for the base period of the contract. On April 16, 2012, we announced that BARDA had exercised two options under the BARDA Contract worth approximately $9.1 million. On September 17, 2013, we announced that BARDA had exercised $6.0 million in additional contract options. On May 07, 2014, we announced that BARDA had exercised a Contract Modification worth approximately $1.8 million. The Contract Modification allowed Aeolus to reconcile actual costs incurred with billings under the original provisional indirect billing rate. It established a new provisional indirect billing rate and placed a cap on the current and future provisional indirect billing rates. On June 26, 2015, we announced that BARDA had exercised $3.0 million in additional contract options under its advanced research and development contract for AEOL 10150. The June option exercise brings the total contract value exercised as of September 30, 2015 to approximately $30.3 million. We may receive up to an additional $88.1 million in options exercisable over the remainder of the BARDA Contract. Options are exercised based on the progress of the development program, including the completion of clinical trials or manufacturing tasks under previously exercised options.
3
The final goal of the contract is to achieve FDA approval for 10150 and the development of commercial manufacturing capability. In order to achieve these goals, we believe it will be necessary to exercise the majority of the options in the contract. We also believe that BARDA is likely to continue to exercise options as long as 10150 continues to demonstrate efficacy and safety in animal testing for Lung-ARS. In the event we begin sales to the U.S. government following the filing of a pre-EUA application, we believe that BARDA is likely to exercise the majority of the remaining options under the contract. One of the requirements of an EUA is that the development program continue towards the goal of FDA approval. If all of the options are exercised by BARDA, the total value of the contract would be approximately $118.4 million.
There are no existing treatments for Lung-ARS or DEARE and we are not aware of any compounds in development that have shown efficacy in increasing survival when administered after exposure to radiation. 10150 has demonstrated efficacy in two animal models (mouse and non-human primate) when administered after exposure to radiation. The U.S. government's planning scenario for a radiation incident is a 10 kiloton detonation of a nuclear device in a major American city. It is estimated that several hundred thousand civilians would be exposed to high doses of radiation in this scenario.
The BARDA contract is also designed to complete the work necessary for 10150 to be purchased for the US Strategic National Stockpile (the "SNS"). BARDA currently acquires drugs for the SNS through a Special Reserve Fund (the "SRF") created under Project BioShield and reauthorized under the Pandemic All-Hazards Preparedness Reauthorization Act of 2013. Although the final goal of the contract is full FDA approval under the Animal Rule, BARDA may purchase product prior to FDA approval following the filing of a pre-EUA application. BARDA has made numerous acquisitions of compounds that were not approved by the FDA, but were the subject of a pre-EUA filing. Procurements from BARDA following a pre-EUA application could result in a significant increase in revenues for Aeolus and potential profitability.
In August 2014, we filed an Investigational New Drug ("IND") application with the Division of Medical Imaging Products of the U.S. Food & Drug Administration ("FDA") for 10150 as a treatment for Lung-ARS. On September 4, 2014, the Company announced positive results from a study in non-human primates exposed to lethal radiation and treated with 10150. The study demonstrated that administration of 10150 24 hours after exposure to lethal radiation impacted survival at six months post-exposure as follows: survival in the 60 day treatment group was 50%, compared to 25% survival in the radiation only untreated control group. The data from this study, combined with development work completed in manufacturing and human safety data, will form the basis for a pre-EUA application. On September 22, 2014, we received a letter from the FDA placing our proposed Phase I study in healthy normal volunteers for 10150 as a treatment for Lung-ARS on clinical hold. (See - "Future Development Plans" and "Risk Factors – Risks Related to Our Business; Our commercialization efforts may be adversely impacted by an FDA clinical hold on our Phase I clinical trial in Lung-ARS.") The FDA provided the Company with specific concerns that need to be addressed in order to allow for the initiation of studies in healthy normal volunteers. In consultation with BARDA, the Company has submitted a mitigation strategy to the FDA for their review and feedback. The Company filed a complete response to the FDA's question in January 2016 and expects the FDA to review and respond to that submission within 30 days.
We also benefit from research funded by grants from the NIH CounterACT program for the development of 10150 as a MCM for the effects of nerve gas (e.g., sarin and soman) and chemical vesicant gasses (e.g., mustard gas, phosgene gas and chlorine gas) exposure. Funding for this indication is provided directly to the research institution and does not flow through our financial statements. Continued funding is generally dependent on continuing evidence of efficacy in animal trials. In October 2011, we announced that National Jewish Health was awarded a $12.5 million grant from NIH CounterACT to continue the development of 10150 as a MCM against chlorine gas exposure. Also included in the grant is support for research in looking at tissue plasminogen activator ("TPA") and Silabilin, which are not Aeolus assets, as MCMs against sulfur mustard gas exposure. The ultimate objective of the sulfur mustard and chlorine gas work at National Jewish Health will be to complete all work necessary to initiate pivotal efficacy studies in animals for both indications. This would include: running efficacy studies in the rat model for higher doses of sulfur mustard and chlorine gas; establishing endpoints, optimal dosing and duration of treatment for pivotal efficacy studies; and characterizing the natural history from sulfur mustard and chlorine gas damage.
4
We are also funded by grant money from the NIH CounterACT program and the National Institute of Neurological Disorders and Stroke ("NINDS") for the development of 10150 as a MCM for the effects of nerve gas (e.g., sarin and soman) exposure. NIH-CounterACT awarded a contract on September 24, 2011 worth approximately $735,000, to the University of Colorado to develop 10150 as a MCM against nerve agents. Work performed with this initial funding has demonstrated that 10150 significantly improved survival when administered with current treatment in a pilocarpine model for nerve gas exposure. In September 2013, we announced that Dr. Manisha Patel at the University of Colorado had been awarded a $4.3 million grant from NINDS to further develop as a MCM for exposure to sarin gas and other nerve agents.
Substantially all of the past costs for the Lung-ARS program have been funded by the BARDA Contract. To date, the chlorine, phosgene, mustard gas and nerve agent programs have been funded by NIH-CounterACT and NINDS through programs at National Jewish Health, the University of Colorado, and the United States Army Medical Research Institute for Chemical Defense ("USAMRICD").
We are also developing 10150 for the treatment of lung fibrosis, including idiopathic pulmonary fibrosis ("IPF") and other fibrotic diseases. This new development program was created based upon the data generated from animal studies in Lung-ARS and chemical gas exposure under the BARDA Contract and NIH grants. On March 17, 2015, we announced that 10150 was granted Orphan Drug Designation for IPF by the U.S Food and Drug Administration ("FDA"). The Company plans to initiate a Phase 1 safety study in patients with IPF in 2016. After we have completed safety studies, we plan to initiate efficacy studies in patients with IPF. We expect a portion of the net proceeds from the 2015 Securities Placement will be used to fund a portion of the future costs of work associated with IPF and other fibrotic diseases.
We are also developing 10150 for use in combination with radiation therapy for cancer as a treatment to reduce side effects caused by radiation toxicity and to improve local tumor control. Pre-clinical studies at Duke University and Loma Linda University have demonstrated that 10150 does not interfere with the benefit of radiation therapy or chemotherapy in prostate and lung cancer. Additional studies have shown that 10150 enhances the anti-tumor activity of radiation and chemotherapy. A significant portion of the development work funded by the BARDA contract is applicable to the development program for radiation oncology, including safety, toxicology, pharmacokinetics and Chemistry, Manufacturing and Controls ("CMC"). After we have completed safety studies, we plan to initiate studies to demonstrate efficacy in ameliorating the toxic side effects related to radiation therapy and, potentially, enhancing tumor control. We expect a portion of the net proceeds from the 2015 Securities Placement will be used to fund a portion of the future costs of work related to radiation therapy opportunities.
10150 has been tested in two human Phase I safety studies where it was well-tolerated and no adverse events were observed. Efficacy has been demonstrated in animal models for Lung-ARS, chlorine gas exposure, phosgene gas exposure, sulfur mustard gas exposure (lungs and skin) and nerve gas exposure. In both mouse and non-human primate ("NHP") studies for Lung-ARS, 10150 treated groups showed significantly reduced weight loss, inflammation, oxidative stress, lung damage, and most importantly, mortality. Therapeutic efficacy has been demonstrated when 10150 is administered 24 hours after exposure to radiation, a requirement for consideration as a radiation MCM for the SNS.
Following the events at the Fukushima nuclear plant in Japan in 2011, we performed radiation exposure studies in mice at the request of Japanese researchers to determine how the administration of AEOL 10150 would impact the use of leukocyte growth factors ("LGF") used to treat the hematopoietic or bone marrow syndrome of ARS ("H-ARS"). Data showed that 10150 does not interfere with the efficacy of LGF (in this case Amgen's Neupogen®). Additionally, the study demonstrated that administration of Neupogen®, the current standard of care for H-ARS, increased damage to the lungs. When 10150 was administered with Neupogen® this damage was significantly reduced. We believe that this finding may have important implications for the potential procurement of 10150 for the SNS. In September 2013, BARDA announced that it had entered into a procurement and inventory management agreement with Amgen to provide Neupogen® for the SNS. On March 30, 2015, the FDA approved Neupogen® for the treatment of H-ARS.
5
We have an active Investigational New Drug Application ("IND") on file with the FDA for AEOL 10150 as a potential treatment for amyotrophic lateral sclerosis ("ALS"). At this time, we do not have any plans to continue development of 10150 for ALS.
We have already completed two Phase I safety studies in 50 humans (39 receiving drug and 13 control) demonstrating that 10150 is safe and well tolerated. CMC work has been completed, pilot lots have been prepared and production is being scaled up under the BARDA Contract.
The Company is developing a second compound, AEOL 11114 ("11114"), as a treatment for Parkinson's disease. Research funded by the Michael J Fox Foundation for Parkinson's disease ("MJFF") demonstrated the neuro-protective activity of 11114 in mouse and rat models of Parkinson's disease. The compounds were invented by Aeolus in collaboration with Brian J. Day, PhD at National Jewish Health and Manisha Patel, PhD at the University of Colorado, Anschutz Medical Campus, Department of Pharmaceutical Sciences in collaboration with the Company. We have obtained worldwide, exclusive licenses to develop the compounds from NJH and the UC. We plan to complete the remaining work to file an Investigational New Drug ("IND") application with the FDA by 2017.
In April 2015, we announced the discovery of a new compound, AEOL 20415 ("20415"), that has demonstrated anti-inflammatory and anti-infective properties, and could be effective in treating cystic fibrosis and combatting anti-biotic resistant bacteria. The compound was developed under a collaboration between Brian J. Day, PhD at National Jewish Health in Denver, Colorado and Aeolus Pharmaceuticals. We have obtained a worldwide, exclusive license to develop the rights to the compound from NJH. We plan to complete the remaining work to file an Investigational New Drug ("IND") application with the FDA by 2017.
Risks Associated with Our Business
Our business is subject to numerous risks. Please see the "Risk Factors" section beginning on page 8 of this prospectus.
Corporate Information
We were incorporated in the State of Delaware in 1994. Our common stock trades on the OTC Bulletin Board under the symbol "AOLS." Our principal executive offices are located at 26361 Crown Valley Parkway, Suite 150, Mission Viejo, California 92691, and our phone number at that address is (949) 481-9825. Our website address is www.aeoluspharma.com. However, the information in, or that can be accessed through, our web site is not part of the registration statement of which this prospectus forms a part. We also make available free of charge through our website our most recent annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and any amendments to those reports, as soon as reasonably practicable after such material is electronically filed with or furnished to the Securities and Exchange Commission, which we refer to as the SEC.
6
THE OFFERING
|
Common stock offered by us
|
None
|
|
Common stock offered by selling stockholders
|
73,432,471, including (i) 15,629,676 shares of common stock issued and outstanding as of the date of this prospectus, (ii) 20,454,456 shares of common stock issuable upon conversion of Series C Convertible Preferred Stock (the "Conversion Shares") and (iii) 37,298,249 shares of common stock issuable upon the exercise of warrants (the "Warrant Shares").
|
|
Charter Amendment
|
In order to facilitate the potential future issuance of Conversion Shares and Warrant Shares, the Company expects to complete a Charter Amendment (defined below) on or about March 10, 2016. As used in this prospectus, "Charter Amendment" means an amendment to the Company's Certificate of Incorporation to increase the authorized number of shares of common stock available for issuance by the Company or to effect a reverse common stock split without altering the number of shares of common stock previously authorized.
|
|
Factors affecting the issuance of Conversion Shares and Warrant Shares
|
The Company is not obligated to issue Conversion Shares or Warrant Shares (i) if the holder, including its affiliates, would thereby beneficially own in excess of 9.99% of the Company's common stock and (ii) in no case prior to March 10, 2016.
|
|
OTC Bulletin Board Symbol
|
"AOLS"
|
|
Proceeds to us
|
We will not receive any proceeds from the sale of the shares of common stock covered by this prospectus. However, we will receive the proceeds of any cash exercise of the warrants.
|
|
Risk factors
|
Investing in our common stock involves certain risks. You should read "Risk Factors" beginning on page 8 for a discussion of factors that you should consider carefully before deciding whether to purchase shares of our common stock.
|
7
RISK FACTORS
You should carefully consider the following information about risks described below, together with the other information contained in this prospectus and in our other filings with the SEC, before you decide to buy or maintain an investment in our common stock. We believe the risks described below are the risks that are material to us as of the date of this prospectus. If any of the following risks actually occur, our business, financial condition, results of operations and future growth prospects would likely be materially and adversely affected. In these circumstances, the market price of our common stock could decline, and you may lose all or part of your investment.
Risks Related to Our Business
We have operated at a loss and will likely continue to operate at a loss for the foreseeable future.
We have incurred significant historical losses and had an accumulated deficit of approximately $186,630,000 as of September 30, 2015. While the vast majority of this accumulated deficit relates to development programs that were abandoned before 2014 (our "Discontinued Development Programs"), our current development programs (primarily 10150, and to a lesser extent 11114 and 20415) continue to generate losses. During the years ended September 30, 2015 and 2014, we incurred losses of $2,628,000 and $80,000, respectively. Our operating losses have been due primarily to our expenditures for research and development on our drug candidates and for general and administrative expenses and our lack of significant, or sufficient, revenues to offset all of the expenditures. We are likely to continue to incur operating losses until such time, if ever, that we generate significant recurring revenues from product sales, whether to the U.S. government for the Strategic National Stockpile or to the general healthcare community for commercial indications, like oncology, epilepsy or Parkinson's Disease. We anticipate it will take a minimum of two years (and possibly longer) for us to generate recurring revenues. We expect that it will take at least that long before the development of any of our licensed, or other current potential, products is completed, marketing approvals are obtained from the FDA and commercial sales of any of these products can begin, or that we might receive a procurement from the U.S. Government following a pre-Emergency Use Authorization application or Animal Rule Approval.
We may need substantial additional funding to continue our operations and may be unable to raise capital when needed, or at all, which would force us to delay, curtail or eliminate our clinical programs and our product development programs.
We may need to raise substantial additional capital to fund human clinical trials and continue our research and development, unless and until we receive a procurement of sufficient size from the U.S. Government for the Strategic National Stockpile. In addition, we may need to raise substantial additional capital to enforce our proprietary rights, defend, in litigation or otherwise, any claims that we infringe third party patents or other intellectual property rights; and commercialize, for non-government related indications, any of our products that may be approved by the FDA or any international regulatory authority.
As of December 31, 2015, we had cash of approximately $5,444,000. As of December 31, 2015, our monthly cash requirements to operate our business that are not reimbursed under the BARDA Contract are approximately $204,000. To the extent we do not have sufficient cash to fund our working capital requirements; we may not be able to pay our payables timely, which may cause vendors to cease providing services to us.
In order to fund on-going operating cash requirements, or to accelerate or expand our oncology and other programs we will need to raise significant additional funds. We are continuously considering strategic and financial options available to us, including public or private equity offerings, debt financings or collaboration arrangements. If we raise additional funds by issuing securities, our stockholders will experience dilution of their ownership interest. Debt financings, if available, may involve restrictive covenants and require significant interest payments. If we do not receive additional financing to fund our operations not reimbursed under the BARDA Contract, or if BARDA does not exercise any additional options under the BARDA Contract, and we are unable to raise sufficient capital for operations, we would have to discontinue some or all of our activities, merge with or sell, lease or license some or all of our assets to another company, or cease operations entirely, and our stockholders might lose all or part of their investments.
8
In addition, if our non-biodefense, commercial development program shows scientific progress, we will need significant additional funds to move therapies through the preclinical stages of development and clinical trials. If we are unable to raise the amount of capital necessary, or do not receive a sufficient procurement from the U.S. Government for the Strategic National Stockpile, to complete development and reach commercialization of any of our catalytic antioxidant products, we will need to delay or cease development of one or more of these products or partner with another company for the development and commercialization of these products.
We have a history of operating losses and expect to continue to incur substantial losses and may never become profitable.
We have no products approved for commercialization in the United States or abroad. Our drug candidates are still being developed, and all but 10150 are still in early stages of development. Our drug candidates will require significant additional development, clinical trials, regulatory clearances or approvals by the FDA and additional investment before they can be commercialized in the United States.
Our likelihood of achieving profitability will depend on numerous factors, including success in:
• developing our existing drug candidates and developing and testing new drug candidates;
• protecting our intellectual property;
• establishing our competitive position;
• achieving third-party collaborations;
• receiving regulatory approvals;
• manufacturing and marketing products; and
• receiving government funding and identifying new government funding opportunities.
Many of these factors will depend on circumstances beyond our control. We may not achieve sufficient revenues for profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis in the future. If revenues grow more slowly than we anticipate, or if operating expenses exceed our expectations or cannot be adjusted accordingly, then our business, results of operations, financial condition and cash flows will be materially and adversely affected.
As of September 30, 2015, we had an accumulated deficit of $186,630,000 from our research, development and other activities, the vast majority of which is related to Discontinued Development Programs. We have not generated material revenues from product sales and do not expect to generate product revenues sufficient to support us for at least several more years. As a result, we may not be successful in obtaining sufficient financing on commercially reasonable terms, or at all. Our requirements for additional capital may be substantial and will be dependent on many factors, including the success of our research and development efforts, our ability to commercialize and market products, our ability to successfully pursue our licensing and collaboration strategy, the receipt of government funding, competing technological developments, costs associated with the protection of our intellectual property and any future change in our business strategy.
Our research and development ("R&D") activities are at an early stage and therefore might never result in viable products.
Our catalytic antioxidant program is in the early stages of development, involves unproven technology, requires significant further R&D and regulatory approvals and is subject to the risks of failure inherent in the development of products or therapeutic procedures based on innovative technologies. These risks include the possibilities that:
| • | any or all of these proposed products or procedures are found to be unsafe or ineffective or otherwise fail to receive necessary regulatory approvals; |
| • | the proposed products or procedures are not economical to market or do not achieve broad market acceptance; |
| • | third parties hold proprietary rights that preclude us from marketing the proposed products or procedures; and |
| • | third parties market a superior or equivalent product. |
9
Further, the timeframe for commercialization of any product is long and uncertain because of the extended testing and regulatory review process required before marketing approval can be obtained. We may not be able to successfully develop or market any of our proposed products or procedures. If we are not able to successfully market any product, our business will suffer.
Our commercialization efforts may be adversely impacted by an FDA clinical hold on our Phase I clinical trial in Lung-ARS.
In September of 2014, the Company received notice from the FDA that our IND for 10150 for Lung ARS has been placed on a clinical hold following their review of our data. A clinical hold is an order that the FDA issues to a trial sponsor to suspend all ongoing clinical trials and delay all proposed trials. With this clinical hold, any patients in an ongoing clinical trial cannot receive treatment with 10150. Therefore, initiation of this Phase I clinical trial will be delayed indefinitely and may not occur at all. In particular, until the FDA lifts the clinical hold, or partially lifts the full clinical hold, we will be unable to submit any new clinical trial protocols to the FDA under our IND for 10150 for Lung ARS. If the FDA does not lift the full clinical hold, we may be unable to pursue the development of 10150 for Lung ARS.
The FDA provided the Company with specific concerns that need to be addressed in order to allow for the initiation of studies in healthy normal volunteers. In consultation with BARDA, the Company submitted a mitigation strategy to the FDA for their review and requested a meeting with the FDA to discuss and receive feedback. On March 12, 2015, the Company met with the FDA and received feedback and further guidance on the requirements for release of the clinical hold. In its written and verbal comments to the Company, the FDA requested that the Company provide an explanation for positive results seen in the Ames Assay testing of AEOL 10150, and indicated that if the Company could produce data to explain why results in four in-vivo gentox studies have been negative, but that results in the Ames were positive, the FDA would release the clinical hold. A complete response to the FDA's question was filed in January 2016 and we expect a response within 30 days. There can be no assurance, however, that the FDA will release the clinical hold.
Our ability to advance our commercialization objectives may be limited by limitation on our access to capital, the absence of any current ability to manufacture our drug candidates on a commercial scale and the absence of any internal marketing capabilities and external marketing arrangements.
In light of the limited number of employees we currently have, our limited resources, and the early stage of our commercial development efforts, our future success may be limited. In addition, there are significant uncertainties as to our ability to access potential sources of capital, and we may not be able to enter into industry partner collaborations that would provide us with liquidity sources on terms acceptable to us, or at all. These uncertainties may be due to a variety of factors, including evolving or adverse conditions in the pharmaceutical industry or in the economy in general or based on the prospects of our commercial programs. Even if we are successful in obtaining collaboration for our commercial programs, we may have to relinquish rights to technologies, product candidates or markets that we might otherwise develop ourselves. These same risks apply to any attempt to out-license our compounds. Moreover, even if our commercialization efforts advance, we currently do not have the capability to manufacture any of our drug candidates on a commercial scale and our ability to develop this capability will be limited since it will be resource intensive and costly. Finally, our product candidates are being developed for large therapeutic markets. We believe these markets are best approached by partnering with established biotechnology or pharmaceutical companies that have broad sales and marketing capabilities. We are pursuing marketing collaborations of this type as part of our search for development partners. However, we may not be able to enter into any marketing arrangements for any of our products on satisfactory terms or at all.
If our products are not successfully developed and eventually approved by the FDA, we may be forced to reduce or terminate our operations.
All of our drug candidates are at various stages of development and must be approved by the FDA or similar foreign governmental agencies before they can be marketed. The process for obtaining FDA and foreign regulatory approval is both time-consuming and costly, with no certainty of a successful outcome. This process typically requires extensive preclinical and clinical testing, which may take longer or cost more than we anticipate, and may prove unsuccessful due to numerous factors. Drug candidates that may appear to be promising at early stages of development may not successfully reach the market for a number of reasons. The results of preclinical and initial clinical testing of these drug candidates may not necessarily indicate the results that will be obtained from later or more extensive testing. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier trials.
10
Numerous factors could affect the timing, cost or outcome of our drug development efforts, including the following:
• difficulty in securing research laboratories to conduct research activities;
• difficulty in securing centers to conduct trials;
• difficulty in enrolling patients in conformity with required protocols or projected timelines;
• unexpected adverse reactions by patients in trials;
• difficulty in obtaining clinical supplies of the product;
| • | changes in the FDA's or other regulatory body's requirements for our testing during the course of that testing; |
• inability to generate statistically significant data confirming the efficacy of the product being tested;
• modification of the drug during testing; and
• reallocation of our limited financial and other resources to other clinical programs.
It is possible that none of the products we develop will obtain the regulatory approvals necessary for us to begin commercializing them. The time required to obtain FDA and other approvals is unpredictable but often can take years following the commencement of clinical trials, depending upon the nature of the drug candidate. Any analysis we perform on data from clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenues from the particular drug candidate and we may not have the financial resources to continue to develop our drug candidates and, as a result, may have to terminate our operations.
If we do not reach the market with our products before our competitors offer products for the same or similar uses, or if we are not effective in marketing our products, our revenues from product sales, if any, will be reduced.
We face intense competition in our development activities. Many of our competitors are fully integrated pharmaceutical companies, biotechnology companies or more established bioterrorism product companies, which have substantially greater financial, technical, sales and marketing and human resources than we do. These companies might succeed in obtaining regulatory approval for competitive products more rapidly than we can for our products. In addition, competitors might develop technologies and products that are less expensive and perceived to be safer or more effective than those being developed by us, which could impair our product development and render our technology obsolete.
We are and expect to remain dependent upon collaborations with third parties for the development of new products, and adverse events involving these collaborations could prevent us from developing and commercializing our drug candidates and achieving profitability.
We currently license from third parties, and do not own, rights under patents and certain related intellectual property for the development of our drug candidates. In addition, we expect to enter into agreements with third parties to license rights to our drug candidates. We might not be able to enter into or maintain these agreements on terms favorable to us, if at all. Further, if any of our current licenses were to expire or terminate, our business, prospects, financial condition and results of operations could be materially and adversely affected.
Our research and development activities rely on technology licensed from third parties, and termination of any of those licenses would result in loss of significant rights to develop and market our products, which would impair our business, prospects, financial condition and results of operations.
We have exclusive worldwide rights to our antioxidant small molecule technology through license agreements with Duke and the NJH. Each license generally may be terminated by the licensor if we fail to perform our obligations under the agreement, including obligations to develop the compounds and technologies under license. If terminated, we would lose the right to develop the products, which could adversely affect our business, prospects, financial condition and results of operations. The license agreements also generally require us to meet specified milestones or show reasonable diligence in development of the technology. If disputes arise over the definition of these requirements or whether we have satisfied the requirements in a timely manner, or if any other obligations in the license agreements are disputed by the other party, the other party could terminate the agreement, and we could lose our rights to develop the licensed technology.
11
If new technology is developed from these licenses, we may be required to negotiate certain key financial and other terms, such as royalty payments, for the licensing of this future technology with these research institutions, and it might not be possible to obtain any such license on terms that are satisfactory to us, or at all.
We now rely, and will continue to rely, heavily on third parties for product and clinical development, manufacturing, marketing and distribution of our products.
We currently depend heavily and will depend heavily in the future on third parties for support in product development, clinical development, manufacturing, marketing and distribution of our products. The termination of some or all of our existing collaborative arrangements, or our inability to establish and maintain collaborative arrangements, could have a material adverse effect on our ability to continue or complete clinical development of our products.
We rely on contract clinical research organizations ("CROs") for various aspects of our clinical development activities including clinical trial monitoring, data collection and data management. As a result, we have had and continue to have less control over the conduct of clinical trials, the timing and completion of the trials, the required reporting of adverse events and the management of data developed through the trial than would be the case if we were relying entirely upon our own staff. Although we rely on CROs to conduct our clinical trials, we are responsible for confirming that each of our clinical trials is conducted in accordance with the investigational plan and protocol. Moreover, the FDA and foreign regulatory agencies require us to comply with GCPs for conducting, recording and reporting the results of clinical trials to assure that the data and results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements.
The third parties on which we rely may have staffing difficulties, may undergo changes in priorities or may become financially distressed, adversely affecting their willingness or ability to conduct our trials. We may experience unexpected cost increases that are beyond our control. Any failure of such CROs to successfully accomplish clinical trial monitoring, data collection and data management and the other services they provide for us in a timely manner and in compliance with regulatory requirements could have a material adverse effect on our ability to complete clinical development of our products and obtain regulatory approval. Problems with the timeliness or quality of the work of a CRO may lead us to seek to terminate the relationship and use an alternate service provider. However, making such changes may be costly and would likely delay our trials, and contractual restrictions may make such a change difficult or impossible. Additionally, it may be difficult to find a replacement organization that can conduct our trials in an acceptable manner and at an acceptable cost.
If BARDA opts not to exercise its options under the BARDA Contract, we would be dependent upon grants from NIH for continued development of 10150 for Lung-ARS, or we would need to curtail our development program in this area significantly and we may be placed at a competitive disadvantage in addressing this market opportunity.
During the fiscal years ended September 30, 2015 and 2014, we received 100% of our revenues from our agreement with BARDA, for the development of 10150 as a MCM against Lung-ARS. These revenues have funded some of our personnel and other R&D costs and expenses. Pursuant to the BARDA Contract, we received approximately $10.4 million under the base period of the contract from February 2011 to April 2012. On April 9, 2012, we announced that BARDA had issued a Notice of Intent to Exercise two options valued at $9.1 million. On April 16, 2012, BARDA exercised the two options. On September 17, 2013, we announced that BARDA had exercised $6.0 million in additional contract options. On May 5, 2014, we announced that BARDA had exercised $1.8 million in additional contract options. On June 26, 2015, we announced that BARDA had exercised $3.0 million in additional contract options, bringing the total value to date to approximately $30.3 million. We may receive up to an additional $88.1 million in options exercisable over the remaining years of the contract. If all of the options are exercised by BARDA, the total value of the contract would be approximately $118.4 million.
12
Under the terms of the BARDA Contract, BARDA may elect not to exercise some or all of the additional options. Because a significant portion of our current revenues are generated from the BARDA Contract, if BARDA does not exercise its options under the BARDA Contract, our ability to develop 10150 as an MCM for Lung-ARS could be negatively impacted, which could harm our competitive position and materially and adversely affect our business, financial condition and results of operations. In general, we believe that future exercise of options under the contract will depend on successful completion of tasks under exercised options and continued demonstration of efficacy.
Necessary reliance on the "Animal Rule" in conducting trials is time-consuming and expensive.
To obtain FDA approval for our drug candidate for a bioterrorism indication under current FDA regulations, we are required to utilize animal model studies for efficacy and provide animal and human safety data under the "Animal Rule." For many of the biological and chemical threats, animal models are not yet available, and as such we are developing, or will have to develop, appropriate animal models, which is a time-consuming and expensive research effort. Further, we may not be able to sufficiently demonstrate the animal correlation to the satisfaction of the FDA, as these corollaries are difficult to establish and are often unclear. The FDA may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies, refuse to approve our products, or place restrictions on our ability to commercialize those products. Further, other countries do not, at this time, have established criteria for review and approval of these types of products outside their normal review process; i.e., there is no "Animal Rule" equivalent, and consequently we may not be able to make a submission for marketing approval in foreign countries based on such animal data.
Additionally, few facilities in the U.S. and internationally have the capability to test animals with radiation, nerve agents, or other lethal biotoxins or chemical agents or otherwise assist us in qualifying the requisite animal models. We have to compete with other biodefense companies for access to this limited pool of highly specialized resources. We therefore may not be able to secure contracts to conduct the testing in a predictable timeframe, cost-effectively or at all.
Even if we succeed in commercializing our drug candidates, we may not become profitable and manufacturing problems or side effects discovered at later stages can further increase costs of commercialization.
Any drugs resulting from our research and development efforts may not become commercially available. Even if we succeed in developing and commercializing our drug candidates, we may never generate sufficient or sustainable revenues to enable us to be profitable. Even if effective, a product that reaches the market may be subject to additional clinical trials, changes to or re-approvals of our manufacturing facilities or a change in labeling if we or others identify side effects or manufacturing problems after a product is on the market. This could harm sales of the affected products and could increase the cost and expenses of commercializing and marketing them. It could also lead to the suspension or revocation of regulatory approval for the products.
We and our contract manufacturing organizations ("CMOs") will also be required to comply with the applicable FDA current good manufacturing practice ("cGMP") regulations. These regulations include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Manufacturing facilities are subject to inspection by the FDA. These facilities must be approved to supply licensed products to the commercial marketplace. We and our contract manufacturers may not be able to comply with the applicable cGMP requirements and other FDA regulatory requirements. Should we or our contract manufacturers fail to comply, we could be subject to fines or other sanctions or could be prohibited from marketing any products we develop.
Political or social factors may delay or impair our ability to market our products and our business may be materially adversely affected.
13
Products developed to treat diseases caused by, or to combat the threat of, bioterrorism will be subject to changing political and social environments. The political and social responses to bioterrorism have been unpredictable. Political or social pressures may delay or cause resistance to bringing our products to market or limit pricing of our products, which would harm our business. Changes to favorable laws, such as Project BioShield, could have a material adverse effect on our business, prospects, financial condition and results of operations.
Legislation limiting or restricting liability for medical products used to fight bioterrorism is new, and we cannot be certain that any such protection will apply to our products or if applied what the scope of any such coverage will be.
The U.S. Public Readiness Act was signed into law in December 2005 (the "Public Readiness Act") and creates general immunity for manufacturers of countermeasures, including security countermeasures (as defined in Section 319F-2(c)(1)(B) of the Public Readiness Act), when the U.S. Secretary of Health and Human Services issues a declaration for their manufacture, administration or use. The declaration is meant to provide general immunity from all claims under state or federal law for loss arising out of the administration or use of a covered countermeasure. Manufacturers are excluded from this protection in cases of willful misconduct. The Secretary of Health and Human Services may not make declarations that would cover any of our other drug candidates or the U.S. Congress may not act in the future to reduce coverage under the Public Readiness Act or it may repeal it altogether.
Upon a declaration by the Secretary of Health and Human Services, a compensation fund would be created to provide "timely, uniform and adequate compensation to eligible individuals for covered injuries directly caused by the administration or use of a covered countermeasure." The "covered injuries" to which the program applies are defined as serious physical injuries or death. Individuals are permitted to bring a willful misconduct action against a manufacturer only after they have exhausted their remedies under the compensation program. A willful misconduct action could be brought against us if an individual(s) has exhausted his or her remedies under the compensation program, which could thereby expose us to liability. Furthermore, the Secretary of Health and Human Services may not issue a declaration under the Public Readiness Act to establish a compensation fund. We may also become subject to standard product liability suits and other third party claims if products we develop which fall outside of the Public Readiness Act cause injury or if treated individuals subsequently become infected or otherwise suffer adverse effects from such products.
Healthcare reform measures and other statutory or regulatory changes could adversely affect our business.
The pharmaceutical and biotechnology industries are subject to extensive regulation, and from time to time legislative bodies and governmental agencies consider changes to such regulations that could have significant impact on industry participants. For example, in light of certain highly-publicized safety issues regarding certain drugs that had received marketing approval, the U.S. Congress is considering various proposals regarding drug safety, including some which would require additional safety studies and monitoring and could make drug development more costly. We are unable to predict what additional legislation or regulation, if any, relating to safety or other aspects of drug development may be enacted in the future or what effect such legislation or regulation would have on our business.
The business and financial condition of pharmaceutical and biotechnology companies are also affected by the efforts of governments, third-party payors and others to contain or reduce the costs of healthcare to consumers. In the United States and various foreign jurisdictions there have been, and we expect that there will continue to be, a number of legislative and regulatory proposals aimed at changing the healthcare system, such as the Affordable Care Act and proposals relating to the reimportation of drugs into the U.S. from other countries (where they are then sold at a lower price) and government control of prescription drug pricing. The roll-out, pendency or approval of such proposals could affect our commercialization efforts and result in a decrease in our share price or limit our ability to raise capital or to obtain strategic collaborations or licenses.
We will need to enter into collaborative arrangements for the manufacturing and marketing of our drug candidates, or we will have to develop the expertise, obtain the additional capital and invest the resources to perform those functions internally.
14
We do not have the staff or facilities to manufacture or market any of the drug candidates being developed in our catalytic antioxidant program. As a result, we will need to enter into collaborative arrangements to commercialize, manufacture and market products that we expect to emerge from our catalytic antioxidant program, or develop the expertise within Aeolus. We might not be successful in entering into such third party arrangements on terms acceptable to us, if at all. If we are unable to obtain or retain third-party manufacturing or marketing on acceptable terms, we may be delayed in our ability to commercialize products, which could have a material adverse effect on our business, prospects, financial condition and results of operations. Substantial additional funds and personnel would be required if we needed to establish our own manufacturing or marketing operations. We may not be able to obtain adequate funding or establish these capabilities in a cost-effective or timely manner, which could have a material adverse effect on our business, prospects, financial condition and results of operations.
A failure to obtain or maintain patent and other intellectual property rights would allow others to develop and sell products similar to ours, which could impair our business, prospects, financial condition and results of operations.
The success of our business depends, in part, on our ability to establish and maintain adequate protection for our intellectual property, whether owned by us or licensed from third parties. We rely primarily on patents in the United States and in other key markets to protect our intellectual property. If we do not have adequate patent protection, other companies could develop and sell products that compete directly with ours, without incurring any liability to us. Patent prosecution, maintenance and enforcement on a global basis are time-consuming and expensive, and many of these costs must be incurred before we know whether a product covered by the claims can be successfully developed or marketed.
Even if we expend considerable time and money on patent prosecution, a patent application may never issue as a patent. We can never be certain that we were the first to invent the particular technology or that we were the first to file a patent application for the technology because patent applications in the United States and elsewhere are not typically published for public inspection for at least 18 months from the date when they are filed. It is always possible that a competitor is pursuing a patent for the same invention in the United States as we are and has an earlier invention date. In some jurisdictions outside of the United States, priority of invention is determined by the earliest effective filing date, not the date of invention. Consequently, if a third party pursues the same invention and has an earlier filing date, patent protection outside the United States would be unavailable to us. Also, outside the United States, an earlier date of invention cannot overcome a date of publication that precedes the earliest effective filing date. Accordingly, the patenting of our proposed products would be precluded outside the United States if a prior publication anticipates the claims of a pending application, even if the date of publication is within a year of the filing of the pending application.
Even if patents issue, the patent claims allowed might not be sufficiently broad to offer adequate protection for our technology against competitive products. Patent protection differs from country to country, giving rise to increased competition from other products in countries where patent coverage is either unavailable, weak or not adequately enforced, if enforced at all. Once a patent issues, we still face the risk that others will try to design around our patent or will try to challenge the validity of the patent. The cost of defending against a challenge to one or more of our patents could be substantial and even if we prevailed, there could be no assurance that we would recover damages.
If a third party were to bring an infringement claim against us, we would incur significant costs in our defense; if the claim were successful, we would need to develop non-infringing technology or obtain a license from the successful patent holder, if available.
Our business also depends on our ability to develop and market products without infringing on the proprietary rights of others or being in breach of our license agreements. The pharmaceutical industry is characterized by a large number of patents, patent filings and frequent and protracted litigation regarding patent and other intellectual property rights. Many companies have numerous patents that protect their intellectual property rights. Third parties might assert infringement claims against us with respect to our drug candidates and future products. If litigation were required to determine the validity of a third party's claims, we could be required to spend significant time and financial resources, which could distract our management and prevent us from furthering our core business activities, regardless of the outcome. If we did not prevail in the litigation, we could be required to pay damages, license a third party's technology, which may not be possible on terms acceptable to us, or at all, or discontinue our own activities and develop non-infringing technology, any of which could prevent or significantly delay pursuit of our development activities.
15
Protection of trade secret and confidential information is difficult, and loss of confidentiality could eliminate our competitive advantage.
In addition to patent protection, we rely on trade secrets, proprietary know-how and confidential information to protect our technology. We use confidentiality agreements with our employees, consultants and collaborators to maintain the proprietary nature of this technology. However, confidentiality agreements can be breached by the other party, which would make our trade secrets and proprietary know-how legally available for use by others. There is generally no adequate remedy for breach of confidentiality obligations. In addition, the competitive advantage afforded by trade secrets is limited because a third party can independently discover or develop something identical to our own trade secrets or know-how, without incurring any liability to us.
In addition, if our current or former employees, consultants or collaborators were to use information improperly obtained from others (even if unintentional), we may be subject to claims as to ownership and rights in any resulting know-how or inventions.
We are highly dependent upon an extremely limited number of executive officers and key employees. If we cannot retain or hire additional qualified personnel or maintain our collaborations, our programs could be delayed and may be discontinued.
As of December 31, 2015 we had four full-time employees. We utilize consultants to assist with our operations and are highly dependent on the services of our executive officers. We do not maintain "key person" life insurance on any of our personnel. We also are dependent on our collaborators for our research and development activities. The loss of key executive officers or collaborators could delay progress in our research and development activities or result in their termination entirely.
We believe that our future success will depend in large part upon our ability to attract and retain highly skilled scientific and managerial personnel. We face intense competition for these kinds of personnel from other companies, research and academic institutions, government entities and other organizations. If we fail to identify, attract and retain personnel, we may be unable to continue the development of our drug candidates, which would have a material adverse effect on our business, prospects, financial condition and results of operations.
We face the risk of product liability claims that could exceed our insurance coverage and deplete our cash resources.
The pharmaceutical and biotechnology industries expose us to the risk of product liability claims alleging that use of our drug candidates caused an injury or harm. These claims can arise at any point in the development, testing, manufacture, marketing or sale of pharmaceutical products and may be made directly by patients involved in clinical trials of our products, by consumers or healthcare providers or by organizations selling our products. Product liability claims can be expensive to defend, even if the product did not actually cause the alleged injury or harm.
Insurance covering product liability claims becomes increasingly expensive as a product candidate moves through the development pipeline to commercialization. We have limited product liability insurance coverage (currently capped at $5 million) for our clinical trials and this coverage may not be sufficient to cover us against some or all potential losses due to liability, if any, or to the expenses associated with defending against liability claims. A product liability claim successfully asserted against us could exceed our insurance coverage, require us to use our own cash resources and have a material adverse effect on our business, financial condition and results of operations.
In addition, some of our licensing and other agreements with third parties require or might require us to maintain product liability insurance. If we cannot maintain acceptable amounts of coverage on commercially reasonable terms in accordance with the terms set forth in these agreements, the corresponding agreements would be subject to termination.
16
The costs of compliance with environmental, safety and similar laws could increase our cost of doing business or subject us to liability in the event of noncompliance.
Our business is subject to regulation under state and federal laws regarding occupational safety, laboratory practices, environmental protection and the use, generation, manufacture, storage and disposal of hazardous substances. We may be required to incur significant costs in the future to comply with existing or future environmental and health and safety regulations. Our research activities involve the use of hazardous materials, chemicals and radioactive compounds. Although we believe that our procedures for handling such materials comply with applicable state and federal regulations, we cannot eliminate the risk of contamination or injury from these materials. In the event of contamination, we could be liable for any resulting damages, which could have a material adverse effect on our business, financial condition and results of operations.
We are subject to intense competition that could materially impact our operating results.
We may be unable to compete successfully against our current or future competitors. The pharmaceutical, biopharmaceutical, biotechnology and bioterrorism industries are characterized by intense competition and rapid and significant technological advancements. Many companies, research institutions and universities are working in a number of areas similar to our primary fields of interest to develop new products. There also is intense competition among companies seeking to acquire products that already are being marketed. Many of the companies with which we compete have or are likely to have substantially greater research and product development capabilities and financial, technical, scientific, manufacturing, marketing, distribution and other resources than at least some of our present or future strategic partners or licensees.
As a result, these competitors may:
• succeed in developing competitive products sooner than us or our strategic partners or licensees;
| • | obtain FDA and other regulatory approvals (including EUA approvals) for their products before approval of any of our products; |
| • | obtain patents that block or otherwise inhibit the development and commercialization of our drug candidates; |
• develop products that are safer or more effective than our products;
• devote greater resources to marketing or selling their products;
• introduce or adapt more quickly to new technologies or scientific advances;
• introduce products that render our products obsolete;
• withstand price competition more successfully than us or our strategic partners or licensees;
• negotiate third-party strategic alliances or licensing arrangements more effectively; or
• take advantage of other opportunities more readily.
Currently, as discussed under "Business; Competition" there are no drugs approved for Lung-ARS. There is one drug, Amifostine, approved for use in reducing side effects from radiation therapy. However, there are also many companies working to develop pharmaceuticals that act as radiation protection agents.
Acceptance of our products in the marketplace is uncertain, and failure to achieve market acceptance will harm our business.
Even if approved for marketing, our products may not achieve market acceptance. The degree of market acceptance will depend upon a number of factors, including:
• the receipt of regulatory approvals for the indications that we are studying;
| • | the establishment and demonstration in the medical community of the safety, clinical efficacy and cost-effectiveness of our products and their potential advantages over existing therapeutic products; |
• marketing and distribution support;
• the introduction, market penetration and pricing strategies of competing and future products; and
| • | coverage and reimbursement policies of governmental and other third-party payors such as insurance companies, health maintenance organizations and other plan administrators. |
17
Physicians, patients, payors or the medical community in general may be unwilling to accept, purchase, utilize or recommend any of our products.
We may be required to make milestone payments and other payments relating to the commercialization of our products.
Our agreements by which we acquired rights to our drug candidates provide for milestone payments by us upon the occurrence of certain regulatory filings and approvals related to the acquired products. In the event that we successfully develop our drug candidates, these milestone payments could be significant. In addition, our agreements require us to pay a royalty interest on worldwide sales. Also, any future license, collaborative or other agreements we may enter into in connection with our development and commercialization activities may require us to pay significant milestone, license and other payments in the future.
We are continually evaluating our business strategy, and may modify this strategy in light of developments in our business and other factors.
We continue to evaluate our business strategy and, as a result, may modify this strategy in the future. In this regard, we may, from time to time, focus our development efforts on different drug candidates or may delay or halt the development of our drug candidates. In addition, as a result of changes in our strategy, we may also change or refocus our existing drug discovery, development, commercialization and manufacturing activities.
Our short-term investments, marketable securities and restricted investments, if any, are subject to certain risks which could materially adversely affect our overall financial position.
We invest our cash in accordance with an established internal policy and customarily in instruments which historically have been highly liquid and carried relatively low risk. However, the capital and credit markets have been experiencing extreme volatility and disruption. Over the past few years, the volatility and disruption have reached unprecedented levels. We maintain a portfolio of investments in short-term investments, marketable debt securities and restricted investments, which are recorded at fair value. Certain of these transactions expose us to credit risk in the event of default of the issuer. To minimize our exposure to credit risk, we invest in securities with strong credit ratings. Should any of our short-term investments, marketable securities or restricted investments lose value or have their liquidity impaired, it could materially and adversely affect our overall financial position by imperiling our ability to fund our operations and forcing us to seek additional financing sooner than we would otherwise. Such financing may not be available on commercially attractive terms or at all.
Our insurance policies are expensive and protect us only from some business risks, which could leave us exposed to significant, uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. We currently maintain general liability, property, auto, workers' compensation, products liability, fiduciary and directors' and officers' insurance policies. We do not know, however, if we will be able to maintain existing insurance with adequate levels of coverage. For example, the premiums for our directors' and officers' insurance policy have increased in the past and may increase in the future, and this type of insurance may not be available on acceptable terms or at all in the future. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our cash position and results of operations.
We may have a limitation on the use of net operating loss carryforwards and tax credits.
Our ability to utilize our net operating loss carryforwards, or NOLs, and tax credits may be limited if we undergo or have undergone an ownership change, as defined in Section 382 of the Internal Revenue Code, as a result of changes in the ownership of outstanding stock. An ownership change generally occurs if the percentage of stock owned by one or more stockholders who own, directly or indirectly, 5% or more of the value of our outstanding stock (or are otherwise treated as 5% stockholders under Section 382 and the regulations promulgated thereunder) has increased by more than 50 percentage points over the lowest percentage of our outstanding stock owned by these stockholders at any time during the testing period, which is generally the three-year period preceding the potential ownership change. In the event of an ownership change, Section 382 imposes an annual limitation on the amount of post-ownership change taxable income a corporation may offset with pre-ownership change NOLs.
18
We are exposed to risks if we are unable to comply with changes to laws affecting public companies, including the Sarbanes-Oxley Act of 2002 and the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, and also to increased costs associated with complying with such laws.
Laws and regulations affecting public companies in the U.S., including the provisions of the Sarbanes-Oxley Act of 2002 and the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, will cause us to incur increased costs as we evaluate the implications of new rules and respond to new requirements. Delays or a failure to comply with the new laws, rules and regulations could result in enforcement actions, the assessment of other penalties and civil suits. These laws and regulations make it more expensive for us under indemnities provided by us to our officers and directors and may make it more difficult for us to obtain certain types of insurance, including liability insurance for directors and officers; as such, we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, or as executive officers. We may be required to hire additional personnel and utilize additional outside legal, accounting and advisory services - all of which could cause our general and administrative costs to increase beyond what we currently have planned.
Our corporate compliance program cannot guarantee that we are in compliance with all potentially applicable regulations.
The development, manufacturing, pricing, sales, coverage and reimbursement of our products, together with our general operations, are subject to extensive regulation by federal, state and other authorities within the United States and numerous entities outside of the United States. While we have developed and instituted a corporate compliance program based on what we believe are the current best practices, governmental authorities may not find that our business practices comply with current or future administrative or judicial interpretations of potentially applicable laws and regulations. If we fail to comply with any of these laws and regulations, we could be subject to a range of regulatory actions, including suspension or termination of clinical trials, the failure to approve a product candidate, restrictions on our products or manufacturing processes, withdrawal of products from the market, significant fines, or other sanctions or litigation.
Risks Related to Our Dependence on U.S. Government Grants and Contracts
Even with the BARDA Contract, we may not be able to fully fund our research and development of 10150.
The BARDA Contract is a cost-plus-fixed-fee reimbursement contract that only reimburses certain specified activities that have been previously authorized by BARDA. Additional activities may be needed and, if so, BARDA may not reimburse us for these activities. Performance under the BARDA Contract requires that we comply with appropriate regulations and operational mandates. Our ability to be regularly and fully reimbursed for our activities will depend on our ability to comply and demonstrate compliance with such requirement.
The BARDA Contract award does not guarantee that we will be successful in future clinical trials or that 10150 will be approved by the FDA.
The BARDA Contract provides a cost-plus-fixed-fee reimbursement opportunity for certain specified clinical and development activities, but we remain fully responsible for conducting these activities. The award of BARDA Contract does not guarantee that any of these activities will be successful. Our inability to be successful with certain key clinical or development activities could jeopardize our ability to obtain FDA approval for 10150.
Most of our immediately foreseeable future revenues are contingent upon grants, contracts or purchases from the U.S. government and we may not achieve sufficient, if any, revenues from these agreements to attain profitability.
For the foreseeable future, we believe our main customer, if any, will be national governments, primarily the U.S. government. However, we may not ultimately be successful in receiving any future revenues or grants from such governments. The process of obtaining government contracts is lengthy and uncertain and we may have to compete with other companies for each contract. We may not be awarded any contracts to supply the U.S. or other governments with our drug candidates or products as such awards may be made, in whole or in part, to our competitors. If the U.S. government makes significant future contract awards for the supply to the Strategic National Stockpile of a competing product, our business will be harmed and it is unlikely that we will ultimately be able to supply that particular treatment or product to foreign governments or other third parties. Further, changes in government budgets and agendas, or advances by our competitors, may result in a decreased and de-prioritized emphasis on procuring the biodefense products we are developing.
19
The U.S. government's determination to award any contracts may be challenged by an interested party, such as another bidder, at the Government Accountability Office ("GAO") or in federal court. If such a challenge is successful, a contract may be terminated.
The laws and regulations governing procurements of goods and services by the U.S. government provide procedures by which other bidders and other interested parties may challenge the award of a government contract. If we are awarded a government contract, such challenges or protests could be filed even if there are not any valid legal grounds on which to base the protest. If any such protests are filed, the government agency may decide to suspend our performance under the contract while such protests are being considered by the GAO or the applicable federal court, thus potentially delaying delivery of goods and services and payment. In addition, we could be forced to expend considerable funds to defend any potential award. If a protest is successful, the government may be ordered to terminate the contract and reselect bids. The government could even be directed to award a potential contract to one of the other bidders.
Our business may become subject to audit by the U.S. government and a negative audit could adversely affect our business.
U.S. government agencies such as the Defense Contract Audit Agency (the "DCAA"), routinely audit and investigate government contractors. These agencies review a contractor's performance under its contracts, cost structure and compliance with applicable laws, regulations and standards.
The DCAA also reviews the adequacy of, and a contractor's compliance with, its internal control systems and policies, including the contractor's purchasing, property, estimating, compensation and management information systems. Any costs found to be improperly allocated to a specific contract will not be reimbursed, while such costs already reimbursed must be refunded. If an audit uncovers improper or illegal activities, we may be subject to civil and criminal penalties and administrative sanctions, including:
• termination of contracts;
• forfeiture of profits;
• suspension of payments;
• fines; and
• suspension or prohibition from conducting business with the U.S. government.
In addition, we could suffer serious reputational harm if allegations of impropriety were made against us.
Laws and regulations affecting government contracts make it more costly and difficult for us to successfully conduct our business.
We must comply with numerous laws and regulations relating to the formation, administration and performance of government contracts, which can make it more difficult for us to retain our rights under these contracts. These laws and regulations affect how we conduct business with government agencies. Among the most significant government contracting regulations that affect our business are:
| • | the Federal Acquisition Regulations, and agency-specific regulations supplemental to the Federal Acquisition Regulations, which comprehensively regulate the procurement, formation, administration and performance of government contracts; |
20
|
•
|
business ethics and public integrity obligations, which govern conflicts of interest and the hiring of former government employees, restrict the granting of gratuities and funding of lobbying activities and incorporate other requirements such as the Anti-Kickback Act and Foreign Corrupt Practices Act;
|
| • | export and import control laws and regulations; and |
| • | laws, regulations and executive orders restricting the use and dissemination of information classified for national security purposes and the exportation of certain products and technical data. |
Foreign governments typically also have laws and regulations governing contracts with their respective agencies. These foreign laws and regulations could affect how we conduct business and, in some instances, impose added costs on our business. Any changes in applicable laws and regulations could restrict our ability to obtain contracts, which could limit our ability to conduct our business and materially adversely affect our revenues and results of operations.
Because we depend on clinical research centers and other contractors for clinical and non-clinical testing, including testing under the "Animal Rule", and for certain research and development activities, the results of our clinical trial, non-clinical animal efficacy studies, and research and development activities are largely beyond our control.
The nature of studies, clinical trials and our business strategy of outsourcing substantially all of our research and development and manufacturing work require that we rely on clinical research centers and other contractors to assist us with research and development, clinical and non-clinical testing (including animal efficacy studies under the "Animal Rule"), patient enrollment and other activities. As a result, our success depends largely on the success of these third parties in performing their responsibilities. Although we prequalify our contractors and believe that they are fully capable of performing their contractual obligations, we cannot directly control the adequacy and timeliness of the resources and expertise that they apply to these activities. Furthermore, we have to compete with other biodefense companies for access to this limited pool of highly specialized resources. If our contractors do not perform their obligations in an adequate and timely manner or we are unable to enter into contracts with them because of prior commitments to our competitors, the pace of clinical or non-clinical development, regulatory approval and commercialization of our drug candidates could be significantly delayed and our prospects could be adversely affected.
Data obtained from clinical trials is susceptible to varying interpretations, which could delay, limit or prevent regulatory clearances.
Data already obtained, or obtained in the future, from pre-clinical studies, non-clinical studies and clinical trials does not necessarily predict the results that will be obtained from later pre-clinical studies and clinical trials. Moreover, pre-clinical and clinical data are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after promising results in earlier trials. The failure to adequately demonstrate the safety and effectiveness of an intended product under development could delay or prevent regulatory clearance of the drug candidate, which would result in delays to commercialization and could materially harm our business. Our studies and clinical trials may not demonstrate sufficient levels of safety and efficacy necessary to obtain the requisite regulatory approvals for our drugs, and our proposed drugs may not be approved for marketing.
We may encounter delays or rejections based on additional government regulation from future legislation or administrative action or changes in FDA policy during the period of development, clinical trials and FDA regulatory review. We may encounter similar delays in foreign countries. If any of our products are approved for commercialization, sales of the products outside the U.S. would be subject to foreign regulatory approvals that vary from country to country. The time required to obtain approvals from foreign countries may be shorter or longer than that required for FDA approval, and requirements for foreign licensing may differ from FDA requirements. We may be unable to obtain requisite approvals from the FDA or foreign regulatory authorities, and even if obtained, such approvals may not be on a timely basis, or they may not cover the uses that we request.
Even if we do ultimately receive FDA approval for any of our drug candidates, these drug candidates will be subject to extensive ongoing regulation, including regulations governing manufacturing, labeling, packaging, testing, dispensing, prescription and procurement quotas, record keeping, reporting, handling, shipment and disposal of any such drug. Failure to obtain and maintain required registrations or to comply with any applicable regulations could further delay or preclude development and commercialization of our drugs and subject us to enforcement action.
21
Unfavorable provisions in government contracts, some of which may be customary, may harm our business, financial condition and operating results.
Government contracts customarily contain provisions that give the government substantial rights and remedies, many of which are not typically found in commercial contracts, including provisions that allow the government to:
• terminate existing contracts, in whole or in part, for any reason or no reason;
• unilaterally reduce or modify contracts or subcontracts, including equitable price adjustments;
| • | cancel multi-year contracts and related orders if funds for contract performance for any subsequent year become unavailable; |
• decline to exercise an option to renew a contract;
• exercise an option to purchase only the minimum amount specified in a contract;
• decline to exercise an option to purchase the maximum amount specified in a contract;
• claim rights to products, including intellectual property, developed under the contract;
• take actions that result in a longer development timeline than expected;
• audit and object to the contractor's contract-related costs and fees, including allocated indirect costs;
• direct the course of a development program in a manner not chosen by the government contractor;
• suspend or debar the contractor from doing business with the government or a specific government agency;
• pursue criminal or civil remedies under the False Claims Act and False Statements Act; and
• control or prohibit the export of products.
Generally, government contracts contain provisions permitting unilateral termination or modification, in whole or in part, at the government's convenience. Under general principles of government contracting law, if the government terminates a contract for convenience, the terminated company may recover only its incurred or committed costs, settlement expenses and profit on work completed prior to the termination.
If the government terminates a contract for default, the defaulting company is entitled to recover costs incurred and associated profits on accepted items only and may be liable for excess costs incurred by the government in procuring undelivered items from another source. Some government contracts grant the government the right to use, for or on behalf of the U.S. government, any technologies developed by the contractor under the government contract. If we were to develop technology under a contract with such a provision, we might not be able to prohibit third parties, including our competitors, from using that technology in providing products and services to the government.
Risks Related to Owning Our Stock
Our principal stockholders own a significant percentage of our outstanding common stock and are, and will continue to be, able to exercise significant influence over our affairs.
As of December 31, 2015, Xmark Opportunity Partners, LLC ("Xmark") possessed voting power over 96,931,944 shares, or 64%, of our outstanding common stock as of such date, through its management of Goodnow Capital, L.L.C. ("Goodnow"), Xmark Opportunity Fund, L.P., Xmark Opportunity Fund, Ltd. and Xmark JV Investment Partners, LLC (collectively, the "Xmark Funds"). As a result, Xmark is able to determine a significant part of the composition of our board of directors, holds significant voting power with respect to matters requiring stockholder approval and is able to exercise significant influence over our operations. The interests of Xmark may be different than the interests of other stockholders on these and other matters. This concentration of ownership also could have the effect of delaying or preventing a change in our control or otherwise discouraging a potential acquirer from attempting to obtain control of us, which could reduce the price of our common stock.
David Cavalier, an employee and our Chairman of the board of directors and Mitchell D. Kaye, a Director, are affiliated with Xmark, collectively possessed voting power of 64% of our outstanding common stock as of December 31, 2015. Accordingly, Mr. Cavalier and Mr. Kaye currently have, and will continue to have, a significant influence over the outcome of all corporate actions requiring stockholder approval.
22
Our executive officers and directors and holders of greater than five percent of our outstanding common stock, together with entities that may be deemed affiliates of, or related to, such persons or entities, beneficially owned greater than 66% of our outstanding common stock as of December 31, 2015. As a result, these stockholders, acting together, may be able to control our management and affairs and matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions, such as mergers, consolidations or the sale of substantially all of our assets. The interests of our current major stockholders may not always coincide with the interests of other stockholders and they may take actions to advance their respective interests to the detriment of other stockholders.
We may need to sell additional shares of our common stock, preferred stock or other securities to meet our capital requirements and these future sales could cause dilution and adversely affect our stock price.
Sales of substantial amounts of capital stock, or the perception that such sales could occur, could adversely affect the prevailing market price of the common stock and our ability to raise capital. We may issue additional common stock in future financing transactions or as incentive compensation for our executive management and other key personnel, consultants and advisors. Issuing any equity securities would be dilutive to the equity interests represented by our then-outstanding shares of common stock. The market price for our common stock could decrease as the market takes into account the dilutive effect of any of these issuances.
In the event of the conversion of our preferred stock and exercises of currently outstanding options and warrants, the ownership interests of our current stockholders could be substantially diluted, which would reduce the market price of our common stock and could make it more difficult for us to raise funds in the future.
As of December 31, 2015, we had 151,559,745 shares of common stock outstanding. We may grant to our employees, directors and consultants, options to purchase shares of our common stock under our 2004 Stock Incentive Plan. In addition, as of December 31, 2015, options to purchase 12,572,591 shares were outstanding, with a weighted average exercise price of $0.41 per share, and 25,000,000 shares were reserved for issuance under the 2004 Stock Incentive Plan. In addition, as of December 31, 2015, warrants to purchase 53,247,877 shares of common stock were outstanding at exercise prices ranging from $0.22 to $2.50 per share, with a weighted exercise price of $0.24 per share.
In connection with prior collaborations and financing transactions, we also issued 526,080 shares of Series B preferred stock and warrants to purchase 896,037 shares of Series B preferred stock at an exercise price of $0.01 per share to affiliates of Elan Corporation, plc ("Elan"). These securities generally are exercisable and convertible at the option of the Elan affiliates. Each of the Series B preferred shares is convertible into one share of common stock. The warrant has a term of five years, a cashless exercise provision and customary anti-dilution adjustments in the event of stock splits, stock combination, reorganizations and similar events.
In connection with the 2015 Securities Placement, we issued 4,500 shares of Series C Preferred Stock Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P. Each share of Series C Preferred Stock are collectively convertible into an aggregate of shares of our common stock and (ii) warrants to purchase shares of our common stock, in each case subject to adjustment, provided that no conversion may be effected that would result in the holders of Series C Convertible Preferred Stock owning more than 9.99% of our common stock on a fully converted to common stock basis. The conversion of all or a portion of these securities would dilute the ownership interests of our stockholders.
Substantial blocks of our total outstanding common stock may be sold into the market through existing registration statements already on file with the SEC. If there are substantial sales of shares of our common stock, the price of our common stock could decline.
The price of our common stock could decline if there are substantial sales of our common stock, particularly sales by our directors, executive officers and significant stockholders. As of the date of this prospectus, substantially all of our outstanding common stock and substantially all common stock underlying outstanding warrants (excluding all shares covered by this prospectus) are covered by existing registration statements already on file with the SEC and/or otherwise freely tradeable without restrictions or further registration under the Securities Act. The market price of our common stock could decline as a result of the sale of a substantial number of our common stock or due to the perception in the market that the holders of a large number of shares intend to sell their common stock.
23
Our common stock is not listed on a national securities exchange, is illiquid and is characterized by low and/or erratic trading volume, and the intraday per share price of our common stock has fluctuated from $0.20 to $0.51 during the last two completed fiscal years.
Our common stock is quoted on the OTCQB under the symbol "AOLS." An active public market for our common stock is unlikely to develop as long as we are not listed on a national securities exchange. We plan to use commercially reasonable efforts in the future to make application to list our common stock for trading upon notice of listing with NASDAQ, or such other exchange upon which the Company is eligible to meet such exchange's listing criteria. However, we can provide no assurances that we will be able to successfully list our common stock after application or satisfy listing criteria after any such approval or initial listing. Even if listed, the market for our stock may be impaired because of the limited number of investors, the significant ownership stake of Xmark, and our small market capitalization, which is less than that authorized for investment by many institutional investors.
Historically, the public market for our common stock has been characterized by low and/or erratic trading volume, often resulting in price volatility. For the fiscal year ended September 30, 2015, the average daily trading volume for our common stock was approximately 32,400 shares. In addition, the price of our common stock has been volatile. Our common stock had a closing price of $0.24 on October 1, 2014 and ended fiscal year 2015 at a closing price of $0.24. During the twelve month period ended September 30, 2015, our common stock had a low closing price of $0.20, which occurred on October 7, 2014, and had a high closing price of $0.41, which occurred on May 1, 2015.
The market price of our common stock is subject to wide fluctuations due to factors that we cannot control, including the results of preclinical and clinical testing of our products under development, decisions by collaborators regarding product development, regulatory developments, market conditions in the pharmaceutical and biotechnology industries, future announcements concerning our competitors, adverse developments concerning proprietary rights, public concern as to the safety or commercial value of any products and general economic conditions.
Furthermore, the stock market has experienced significant price and volume fluctuation unrelated to the operating performance of particular companies. These market fluctuations can adversely affect the market price and volatility of our common stock.
If registration rights that we have previously granted are exercised, or if we grant additional registration rights in the future, the price of our common stock may be adversely affected.
Upon receiving notice from Elan, we are obligated to register with the SEC shares of common stock underlying the Series B Convertible Preferred Stock and warrants to purchase Series B Convertible Preferred Stock held by the Elan affiliates. If these securities are registered with the SEC, they may be sold in the open market. We expect that we also will be required to register any securities sold in future private financings. The sale of a significant amount of shares in the open market, or the perception that these sales may occur, could cause the trading price of our common stock to decline or become highly volatile.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and bylaws may delay or prevent an acquisition of us or a change in our management. These provisions include a prohibition on certain actions by written consent of our stockholders and the ability of our board of directors to issue up to 7,145,000 shares of "blank check" preferred stock without stockholder approval. As a result, our board of directors has the power to issue shares without stockholder approval, and such shares can be issued with such rights, preferences, and limitations as may be determined by our board of directors. The rights of the holders of common stock will be subject to, and may be adversely affected by, the rights of any holders of preferred stock that may be issued in the future. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the General Corporation Law of the State of Delaware, which prohibits stockholders owning in excess of 15% of our outstanding voting stock from merging or combining with us. These provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
24
We do not expect to pay cash dividends on our common stock for the foreseeable future.
We have never paid cash dividends on our common stock and do not anticipate that any cash dividends will be paid on the common stock for the foreseeable future. The payment of any cash dividend by us will be at the discretion of our board of directors and will depend on, among other things, our earnings, capital, regulatory requirements and financial condition. Furthermore, the terms of some of our financing arrangements directly limit our ability to pay cash dividends on our common stock.
We have previously identified a material weakness in our internal control over financial reporting and disclosure control procedures, and any inability to maintain effective internal control over financial reporting or disclosure controls could have a material adverse effect on our business.
As of September 30, 2015, we carried out an evaluation, under the supervision and with the participation of our management, including our President and Chief Executive Officer (our Principal Executive Officer) and Chief Financial Officer (our Principal Financial and Accounting Officer), of the effectiveness of our disclosure controls and procedures required by Rule 13a-15 under the Exchange Act, as amended. Based upon that evaluation, our Principal Executive Officer and Principal Financial and Accounting Officer have concluded that our disclosure controls and procedures were not effective as of September 30, 2015 to provide reasonable assurance that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms.
In connection with the preparation of the Annual Report on Form 10-K for the fiscal year 2015, we determined that we applied the incorrect accounting treatment for the redemption feature and redemption feature liability associated with the promissory notes issued in the fourth quarter of fiscal year 2015. The promissory notes contain a redemption feature that require the issuer to bifurcate and fair value the derivative redemption feature as a separate financial instrument at each reporting period. As a result of the determination that we used an incorrect accounting treatment for the redemption feature in the promissory notes, our initial balance sheet liabilities were understated for the quarter ended September 30, 2015 and management determined a material weakness existed as of September 30, 2015.
A material weakness is a significant deficiency, or combination of deficiencies, in internal control over financial reporting that results in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected. As a result of the determination that our balance sheet liabilities did not include the fair value treatment of the derivative features in the promissory notes issued for the quarter ended September 30, 2015 management has determined that a material weakness existed as of September 30, 2015.
Management believes the material weakness is due to a deficiency in technical resources over financial reporting. As a result of the material weakness, management is evaluating mitigating controls to minimize errors related to complex transactions.
25
A failure to maintain adequate internal controls over our financial and management systems could cause errors in our financial reporting, which could cause a loss of investor confidence and result in a decline in the price of our Common Shares.
Our public company reporting obligations and our anticipated commercial development objectives will likely strain our financial and management systems, internal controls and employees. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. In order to maintain and, if required, improve our disclosure controls and procedures and internal control over financial reporting to meet this standard, significant resources and management oversight may be required. If we have a future material weakness or deficiency in our internal control over financial reporting, we may not detect errors on a timely basis and our financial statements may be materially misstated. Effective internal controls are necessary for us to produce reliable financial reports and are important to prevent fraud. As a result, our failure in this regard to satisfy these requirements on a timely basis could result in us being subject to regulatory action and a loss of investor confidence in the reliability of our financial statements, both of which in turn could cause the market value of our Common Shares to decline and affect our ability to raise capital and advance commercialization objectives.
26
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the "Exchange Act"), that relate to future events or our future financial performance. You can identify forward-looking statements by terminology such as "may," "might," "will," "could," "should," "would," "expect," "plan," "anticipate," "believe," "estimate," "predict," "intend," "potential" or "continue" or the negative of these terms or other comparable terminology. Our actual results might differ materially from any forward-looking statement due to various risks, uncertainties and contingencies, including but not limited to those identified herein under "Risk Factors" beginning on page 8 of this prospectus, as well as those discussed in our other filings with the Securities and Exchange Commission and the following:
| • | our need for, and our ability to obtain, additional funds; |
| • | our ability to obtain grants to develop our drug candidates; |
| • | our ability to demonstrate efficacy and safety in animal testing for Lung-Acute Radiation Syndrome (or Lung ARS) |
| • | uncertainties regarding the effectiveness of proceeds deployment and our actual future use of proceeds from our 2015 Securities Placement; |
| • | uncertainties relating to non-clinical studies, clinical trials and regulatory reviews and approvals; |
| • | uncertainties relating to our pre-clinical studies and trials and regulatory reviews and approvals; |
| • | uncertainties regarding our ability to successfully advance our Phase I study for Lung ARS and other commercialization studies and projects involving AEOL 10150, in light of the clinical hold placed on our Lung ARS study by the Food & Drug Administration on September 22, 2014; |
| • | uncertainties regarding whether our compounds could inhibit formation of fibrosis in the lungs. |
| • | uncertainties concerning whether we can position our compounds for a pre-Emergency Use Authorization application or we can obtain procurements from the Biomedical Advanced Research and Development Authority following any such application; |
| • | our dependence on a limited number of therapeutic compounds; |
| • | the early stage of the drug candidates we are developing; |
| • | the acceptance of any future products by physicians and patients; |
| • | competition with and dependence on collaborative partners; |
| • | loss of key consultants, management or scientific personnel; |
| • | our ability to obtain adequate intellectual property protection and to enforce these rights; and |
| • | our ability to avoid infringement of the intellectual property rights of others. |
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. We disclaim any intention or obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
27
DESCRIPTION OF THE SHARES INCLUDED IN THIS PROSPECTUS
2015 Securities Placement Terms; Limitations on Conversion and Exercise
On December 10, 2015, we entered into securities purchase agreements with certain accredited investors to sell and issue (i) an aggregate of 10,215,275 common units issued at a purchase price of $0.22 per unit, and (ii) 4,500 preferred stock units issued to Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P., for an aggregate purchase price of $4.5 million, resulting in aggregate gross proceeds to the Company of approximately $6.75 million (the "2015 Securities Placement"). Each common unit consisted of one share of the Company's common stock and a five year warrant to purchase one share of the Company's common stock, subject to adjustment. The preferred stock units collectively consist of (i) 4,500 shares of our Series C Preferred Stock that are collectively convertible into an aggregate of 20,454,546 shares of the Company's common stock and (ii) warrants to purchase an aggregate of 20,454,546 shares of our common stock, in each case subject to adjustment. Each of the foregoing warrants has an initial exercise price of $0.22 per share. Neither the warrants nor the Series C Preferred Stock may be exercised or converted until after 90 days following the date of issuance.
The Series C Preferred Stock and warrants issued in the 2015 Securities Placement contain provisions restricting the conversion or exercise of such securities in circumstances where such event would result in the holder and its affiliates to beneficially own in excess of 9.99% of the Company's common stock.
On September 29, 2015, the Company received funding in the form of convertible promissory notes (the "BVF Notes") from Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P. The BVF Notes (i) had an aggregate principal balance of $1,000,000, (ii) accrued interest at a rate of 6% per annum, (iii) had a scheduled maturity date of September 28, 2016 and (iv) were subject to automatic conversion into Company equity securities, provided a qualified financing of not less than $4 million occurred. Following the completion of the 2015 Securities Placement, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of our common stock and warrants to purchase an additional 5,414,402 shares of our common stock at an exercise price per share of $0.22 subject to adjustment. As a result, the BVF Notes are no longer outstanding as of the date of this prospectus.
In connection with the 2015 Securities Placement, we entered into a Registration Rights Agreement with the investors (the "RRA"). Pursuant to the RRA, we agreed to file a registration statement with the SEC, within 45 days from closing to register the resale of the shares of our common stock and shares of common stock issuable upon exercise of the warrants issued in the 2015 Securities Placement, and shares of our common stock issuable upon conversion of the Series C Preferred Stock (collectively, the "Registrable Securities"). We also agreed to use our best efforts to have the registration statement declared effective as promptly as possible after the filing thereof, but in any event within 120 days (150 days if the we receive comments from the SEC) from the filing date.
In the event (i) the registration statement has not been filed by the agreed upon filing date, (ii) an acceleration request has not been filed within five trading days of the date which we are notified that the registration statement will not be reviewed by the SEC staff or is not subject to further review and comment by the SEC staff, (iii) the registration statement has not been declared effective by the required effectiveness date, or (iv) sales cannot be made pursuant to such registration statement for any reason (other than by reason of a permissible delay under the terms of the RRA) after the registration statement has been declared effective by the SEC (each such event, a "Registration Default"), then we have agreed to pay each of the investors as liquidated damages an amount equal to 0.5% of the purchase price paid by each such investor with respect to any Registrable Securities then held and not registered pursuant to an effective registration statement, per each 30-day period or portion thereof during which the Registration Default remains uncured thereafter. However, liquidated damages, if any, payable as a result of any Registration Default shall cease to accrue, in any event, after the date that is six (6) months after the closing.
We granted the investors in the 2015 Securities Placement customary indemnification rights in connection with the registration statement. These investors have also granted us customary indemnification rights in connection with the registration statement.
28
The foregoing description of the 2015 Securities Placement does not purport to be complete and is qualified in its entirety by reference to the Form of RRA, attached as Exhibit 10.2 to the Form 8-K we filed with the SEC on December 15, 2015.
Placement Agent Warrants in connection with the 2015 Securities Placement
In connection with the 2015 Securities Placement we issued an additional 1,214,027 warrants to purchase common stock to placement agents and their affiliates and employees for services rendered in connection with the 2015 Securities Placement (the "2015 Placement Agent Warrants").
Common Stock Subject to Warrants Issued Outside of the 2015 Securities Placement
This prospectus also relates to 50,000 shares of our common stock issuable upon the exercise of warrants that we issued in 2014.
29
USE OF PROCEEDS
All proceeds from the sale of our common stock covered by this prospectus will belong to the selling stockholders who offer and sell their shares. We will not receive any proceeds from the sale of the common stock by the selling stockholders. A portion of the shares covered by this prospectus are issuable upon exercise of warrants to purchase our common stock. Upon any exercise of the warrants for cash, the selling stockholders would pay us the exercise price of the warrants. Under certain limited conditions set forth in the warrants, the warrants are exercisable on a cashless basis. If any warrants are exercised on a cashless basis, we would not receive any cash payment from the selling stockholders upon any exercise of such warrants.
30
DETERMINATION OF OFFERING PRICE
This offering is being made solely to allow the selling stockholders to offer and sell shares of common stock to the public. The selling stockholders may offer for resale some or all of their shares at the time and price that they choose. On any given day, the price per share is likely to be based on the quoted price for the common stock on the OTC Bulletin Board on the date of sale, unless shares are sold in private transactions. Consequently, we cannot currently make a determination of the price at which shares offered for resale pursuant to this prospectus may be sold.
31
MARKET INFORMATION / PRICE RANGE OF COMMON STOCK / DIVIDENDS
Price Range of Common Stock
Our common stock is quoted on the OTC Bulletin Board under the symbol "AOLS." The following sets forth the quarterly high and low trading prices as reported by the OTC Bulletin Board for the periods indicated. These prices are based on quotations between dealers, which do not reflect retail mark-up, markdown or commissions, and do not necessarily represent actual transactions.
|
High
|
Low
|
|||||||
|
Fiscal Year Ending September 30, 2013
|
||||||||
|
October 1, 2012 through December 31, 2012
|
$
|
0.39
|
$
|
0.23
|
||||
|
January 1, 2013 through March 31, 2013
|
$
|
0.42
|
$
|
0.28
|
||||
|
April 1, 2013 through June 30, 2013
|
$
|
0.46
|
$
|
0.32
|
||||
|
July 1, 2013 through September 30, 2013
|
$
|
0.34
|
$
|
0.22
|
||||
|
Fiscal Year Ending September 30, 2014
|
||||||||
|
October 1, 2013 through December 31, 2013
|
$
|
0.29
|
$
|
0.23
|
||||
|
January 1, 2014 through March 31, 2014
|
$
|
0.33
|
$
|
0.23
|
||||
|
April 1, 2014 through June 30, 2014
|
$
|
0.36
|
$
|
0.24
|
||||
|
July 1, 2014 through September 30, 2014
|
$
|
0.51
|
$
|
0.23
|
||||
|
Fiscal Year Ending September 30, 2015
|
||||||||
|
October 1, 2014 through December 31, 2014
|
$
|
0.29
|
$
|
0.20
|
||||
|
January 1, 2015 through March 31, 2015
|
$
|
0.35
|
$
|
0.21
|
||||
|
April 1, 2015 through June 30, 2015
|
$
|
0.41
|
$
|
0.28
|
||||
|
July 1, 2015 through September 30, 2015
|
$
|
0.32
|
$
|
0.22
|
||||
|
Fiscal Year Ending September 30, 2016
|
||||||||
|
October 1, 2015 through December 31, 2015
|
$
|
0.30
|
$
|
0.18
|
||||
|
January 1, 2016 through February 2, 2016
|
$
|
0.25
|
$
|
0.18
|
||||
On February 2, 2016, the last reported sales price of our common stock on the OTC Bulletin Board was $0.20 per share.
Approximate Number of Equity Security Holders
As of December 31, 2015 the number of record holders of our common stock was 89, and we estimate that the number of beneficial owners was approximately 2,504.
Dividend Policy
We have never paid a cash dividend on our common stock and we do not anticipate paying cash dividends on our common stock in the foreseeable future. A future cash dividend, if paid, would enable the Xmark Entities to dispose of up to 59.1 million shares of common stock that were previously subject to warrants. Such parties are bound by an agreement which provides that the Xmark Entities will not transfer the shares issued upon exercise of the Xmark Warrants (the "Xmark Warrant Shares") until the Company either (i) declares a cash dividend on its common stock or otherwise makes a cash distribution or (ii) effects a Change of Control, subject in each case to the terms of the Xmark Warrant Agreement. See "Certain Relationships and Related Party Transactions – 2013 Warrant Repricing, Exercise and Lockup Agreement."
32
Moreover, any additional preferred stock to be issued and any future credit facilities might contain restrictions on our ability to declare and pay dividends on our common stock. We plan to retain all earnings, if any, for the foreseeable future for use in the operation of our business and to fund future growth.
33
MANAGEMENT'S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Introduction
You should read the following discussion in conjunction with our consolidated financial statements and the notes appearing elsewhere in this Annual Report on Form 10-K. The following discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in the forward-looking statements as a result of various factors, including those discussed in Item 1A – "Risk Factors" and elsewhere in this Annual Report on Form 10-K.
Overview
We are developing a new class of catalytic antioxidant compounds as medical countermeasures ("MCM") against nuclear, radiological and chemical weapons as well as for diseases and disorders of the respiratory system, central nervous system and oncology.
Our lead compound, AEOL 10150 ("10150"), is being developed:
| · | as a MCM for the pulmonary and delayed effects of acute radiation exposure under contract with the U.S Government |
| · | as a MCM for exposure to chemical gas and nerve gas exposure with grant funding from the U.S. Government |
| · | as a treatment for idiopathic pulmonary fibrosis |
| · | as a treatment to reduce the side effects of radiation therapy for and/or potentially improve the control of solid tumors in oncology |
Our other development programs include:
| · | development AEOL 1114B ("11114") as a treatment for Parkinson's disease |
| · | development of AEOL 20415 ("20415") as a treatment for infectious disease |
We have reported positive safety results from two Phase I clinical trials of 10150 with no serious adverse events noted.
We had a net loss of $2,628,000 and $80,000 for the fiscal years ended September 30, 2015 and 2014, respectively. We had an accumulated deficit of approximately $186,630,000 at September 30, 2015. We have not yet generated any revenue from product sales and we cannot provide assurances that we will receive any product revenue from non-government sales in the foreseeable future, if at all.
We have not had any recurring revenue from product sales. Therefore, we have relied on public or private equity offerings, debt financings, collaboration arrangements and grants to finance our operations.
Corporate Matters
On February 11, 2011, we signed a five-year, cost-plus contract with BARDA for the development of 10150 as a MCM against Lung-ARS (the "BARDA Contract"). BARDA is the government agency responsible for the advanced development and purchase of medical countermeasures for chemical, biological, radiological and nuclear threats. The contract fully funds the advanced development of 10150 through approval by the FDA under 21 CFR Part 314 Subpart I and Part 601 Subpart H (the "Animal Rule.") The Animal Rule allows for approval of drugs using only animal studies when human clinical trials cannot be conducted ethically.
34
Pursuant to the BARDA Contract we were awarded approximately $10.4 million for the base period of the contract (from February 2011 to April 2012). On April 16, 2012, we announced that BARDA had exercised two options under the BARDA Contract worth approximately $9.1 million. On September 17, 2013, we announced that BARDA had exercised $6.0 million in additional contract options. On May 07, 2014, we announced that BARDA had exercised a Contract Modification worth approximately $1.8 million. On June 26, 2015, we announced that BARDA had exercised $3.0 million in additional contract options under its advanced research and development contract for AEOL 10150. The June option exercise brings the total exercised contract value to date to approximately $30.3 million. We may receive up to an additional $88.1 million in options exercisable over the remaining years of the contract. Options are exercised based on the progress of the development program, including the completion of clinical trials or manufacturing tasks under previously exercised options. The final goal of the contract is to achieve FDA approval for 10150 and the development of commercial manufacturing capability. In order to achieve these goals, we believe it will be necessary to exercise the majority of the options in the contract. We also believe that BARDA is likely to continue to exercise options as long as 10150 continues to perform in testing for Lung-ARS. In the event we begin sales to the U.S. government following a pre-EUA application, we believe that BARDA is highly likely to exercise the majority of the remaining options under the contract. One of the requirements of an EUA is that the development program continue towards the goal of FDA approval. If all of the options are exercised by BARDA, the total value of the contract would be approximately $118.4 million.
Research and development activities under the contract to date include animal efficacy studies, animal model development with radiation survival curve studies, dosing studies, bulk drug manufacturing, bulk drug and final drug product manufacturing, validation testing, compliance studies, stability studies and the filing of an orphan drug status application and a fast track designation application with the FDA.
Among the key deliverables accomplished in the program, we delivered a final report on the survival data from our efficacy study in non-human primates, prepared a Fast Track filing for AEOL 10150 in Lung-ARS, completed murine extended natural history, duration of treatment and delayed treatment studies, completed studies to identify biomarkers for use in Lung-ARS studies, completed additional pharmacokinetic pharmacodynamics work and completed a absorption, distribution, metabolism and excretion ("ADME") study in the new formulation for AEOL 10150, manufactured pilot scale batches of bulk drug substance and final drug product, and generated stability data on the new bulk drug substance and new final drug product.
In the event BARDA exercises the remaining options under the contract, we expect to conduct, among other things, bulk drug and final drug product manufacturing, stability studies, animal pivotal efficacy studies, human clinical safety studies and Phase I, Phase II and pre-new drug application ("NDA") meetings and applications with the FDA.
On September 4, 2014, Aeolus presented the results and deliverables that had been produced during the first 28 months of the contract at an "In-Progress Review" meeting with BARDA, and requested the exercise of additional contract options. At the meeting, Aeolus presented results from a large study in non-human primates (NHPs) demonstrating that treatment with 10150 resulted in survival in the 60 day treatment group at 50%, compared to 25% survival in the radiation only untreated control group at 180 days after radiation exposure to the whole thorax.
On September 22, 2014, Aeolus received notification from the FDA that its proposed Phase I safety study in healthy normal volunteers had been placed on clinical hold pending additional information from the company. Although the Phase I safety study is part of the development plan with BARDA, the clinical hold does not prevent progress on the other elements of the Contract related to animal efficacy studies, manufacturing or other areas.
For additional developments under the BARDA Agreement preceding our 2015 fiscal year, please see Item 1 "Business – BARDA Contract; Background and Recent Developments."
Results of Operations
Fiscal Year Ended September 30, 2015 Compared to Fiscal Year Ended September 30, 2014
We had a net loss of $2,628,000 for the fiscal year ended September 30, 2015, versus a net loss of $80,000 for the fiscal year ended September 30, 2014.
35
Revenue for the fiscal year ended September 30, 2015 was approximately $3,111,000, compared to $9,631,000 revenue for the fiscal year ended September 30, 2014. The revenue is from the collaboration with BARDA announced on February 11, 2011. Since being awarded the BARDA Contract, we generate contract revenue from a cost-plus fee arrangement. Revenues on reimbursable contracts are recognized as costs are incurred, based on allowable costs incurred during the period, plus any recognizable earned fee. We consider fixed fees under cost-plus fee contracts to be earned in proportion to the allowable costs incurred in performance of the contract. Revenue was higher in the prior year primarily due to the timing of work related to the BARDA contract and the $1,778,000 Contract Modification.
Research and Development
Research and development expenses decreased by $3,457,000, or 50%, to approximately $3,509,000 for the fiscal year ended September 30, 2015 from approximately $6,966,000 for the fiscal year ended September 30, 2014. R&D expenses were lower during the fiscal year ended September 30, 2015 versus September 30, 2014 due to the timing of work related to the BARDA Contract. For the fiscal year ended September 30, 2015, Preclinical fees decreased by $1,960,000 due to costs associated with the BARDA Contract. Consultant expenses decreased about $672,000 in 2015 over the comparable period in 2014 due to work related to the BARDA contract. Manufacturing expenses decreased about $665,000. We currently have 8 development programs in progress: studies of 10150 as a medical countermeasure against the effects of radiation on the lungs, against the effects of sulfur mustard gas and chlorine gas on the lungs, against the effects of radiation on the lungs, against the effects of nerve gas exposure, as a prophylaxis against normal tissue injury during radiotherapy treatment for cancer, as a treatment for IPF and studies of 1114B, 11203 and 11207 as potential treatments for Parkinson's disease and epilepsy.
R&D expenses for our programs have totaled approximately $62,558,000 from inception through September 30, 2015. Because of the uncertainty of our research and development and clinical studies, we are unable to predict the total level of spending on the program or the program completion date. Future R&D expenses will be determined primarily by the exercise of options under the BARDA contract and the possible initiation of human clinical studies in oncology. We anticipate that much of the R&D expense, except for the possible oncology studies, will be reimbursed by the BARDA Contract.
General and Administrative
General and administrative ("G&A") expenses include corporate costs required to support Aeolus, our employees and consultants and our stockholders. These costs include personnel and outside costs in the areas of legal, human resources, investor relations and finance. Additionally, we include in general and administrative expenses such costs as rent, repair and maintenance of equipment, utilities, information technology and procurement costs that we need to support the corporate functions listed above.
G&A expenses decreased approximately $517,000, or 19%, to approximately $2,228,000 for the fiscal year ended September 30, 2015 from about $2,745,000 for the fiscal year ended September 30, 2014. Consulting stock expense decreased by about $444,000 as a result of decreased awards for the period. Legal expense decreased by about $34,000 as a result of lower SEC filing and financing costs.
Other Income or Expense
Other Expense increased approximately $2,000 for the fiscal year ended September 30, 2015, from zero for the fiscal year ended September 30, 2014 as result of issuance of convertible promissory notes during the fiscal year ended September 30, 2015.
Fiscal Year Ended September 30, 2014 Compared to Fiscal Year Ended September 30, 2013
We had a net loss of $80,000 for the fiscal year ended September 30, 2014, versus a net loss of $3,208,000 (including a non-cash loss for increases in valuation of warrants of $510,000) for the fiscal year ended September 30, 2013.
36
Revenue for the fiscal year ended September 30, 2014 was approximately $9,631,000, compared to $3,928,000 revenue for the fiscal year ended September 30, 2013. The revenue is from the collaboration with BARDA announced on February 11, 2011. Since being awarded the BARDA Contract, we generate contract revenue from a cost-plus fee arrangement. Revenues on reimbursable contracts are recognized as costs are incurred, based on allowable costs incurred during the period, plus any recognizable earned fee. We consider fixed fees under cost-plus fee contracts to be earned in proportion to the allowable costs incurred in performance of the contract. Revenue was higher primarily due to the timing of work related to the BARDA contract and the $1,778,000 Contract Modification.
Research and Development
Research and development expenses increased by $3,606,000, or 107%, to approximately $6,966,000 for the fiscal year ended September 30, 2014 from approximately $3,360,000 for the fiscal year ended September 30, 2013. R&D expenses were higher during the fiscal year ended September 30, 2014 versus September 30, 2013 due to the timing of work related to the BARDA Contract. For the fiscal year ended September 30, 2014, consultant expenses increased by $323,000 due to costs associated with the BARDA Contract. Preclinical fees increased about $3,334,000 in 2014 over the comparable period in 2013 due to work related to the BARDA contract. Manufacturing expenses decreased about $143,000. We currently have 8 development programs in progress: studies of 10150 as a medical countermeasure against the effects of radiation on the lungs, against the effects of sulfur mustard gas and chlorine gas on the lungs, against the effects of radiation on the lungs, against the effects of nerve gas exposure, as a treatment for cancer, as a treatment for IPF and studies of 1114B, 11203 and 11207 as potential treatments for Parkinson's disease and epilepsy.
R&D expenses for our antioxidant program have totaled approximately $59,049,000 from inception through September 30, 2014. Because of the uncertainty of our research and development and clinical studies, we are unable to predict the total level of spending on the program or the program completion date. Future R&D expenses will be determined primarily by the exercise of options under the BARDA contract and the possible initiation of human clinical studies in oncology. We anticipate that much of the R&D expense, except for the possible oncology studies, will be reimbursed by the BARDA Contract.
General and Administrative
General and administrative ("G&A") expenses include corporate costs required to support Aeolus, our employees and consultants and our stockholders. These costs include personnel and outside costs in the areas of legal, human resources, investor relations and finance. Additionally, we include in general and administrative expenses such costs as rent, repair and maintenance of equipment, utilities, information technology and procurement costs that we need to support the corporate functions listed above.
G&A expenses decreased approximately $521,000, or 16%, to approximately $2,745,000 for the fiscal year ended September 30, 2014 from about $3,266,000 for the fiscal year ended September 30, 2013. Consulting stock expense decreased by about $218,000 as a result of decreased awards for the period. Legal expense decreased by about $168,000 as a result of lower SEC filing and financing costs.
Other Income or Expense
Other Expense decreased approximately $510,000, or 100%, to zero for the fiscal year ended September 30, 2014, from approximately $510,000 for the fiscal year ended September 30, 2013. Warrant liability expense decreased $510,000 as result of all warrants subject to warrant liability accounting treatment being exercised during the fiscal year ended September 30, 2013.
Liquidity and Capital Resources
As of September 30, 2015, we had approximately $94,000 of cash and cash equivalents, a decrease of $1,422,000 from September 30, 2014. The decrease in cash was primarily due to cash used in operations and the 2014 BARDA Contract Modification described below, which had the effect of increasing our cash as of September 30, 2014. In order to fund on-going operating cash requirements, or to accelerate or expand our oncology and other programs, we may need to raise significant additional funds.
37
On May 5, 2014, the Company received a BARDA Contract Modification for $1,778,000 which reimbursed the Company for additional overhead expenses under the contract that were previously unbilled. The Contract Modification also updated our provisional indirect billing rate and placed a cap on the current and future provisional indirect billing rates. The Company estimates and accrued $304,000 in additional revenue for the fiscal year ended September 30, 2015 as a result of the new overhead rate.
We had a net loss of $2,628,000 for the fiscal year ended September 30, 2015, compared to a net loss of $80,000 for the fiscal year ended September 30, 2014. For the same periods, we had cash outflows from operations of approximately $2,443,000 and cash inflows from operations of approximately $323,000, respectively, with the outflows increasing in 2015 due to lower revenue from BARDA.
Our ongoing future cash requirements will depend on numerous factors, particularly the progress of our development programs, clinical trials and our ability to negotiate and complete collaborative agreements or out-licensing arrangements. In order to help fund our on-going operating cash requirements, we intend to seek new collaborations for our antioxidant research program that include initial cash payments and on-going research support. In addition, we might sell additional shares of our stock and/or convertible debentures and explore other strategic and financial alternatives, including a merger with another company, the sale of stock and/or debt, the establishment of new collaborations for current research programs that include initial cash payments and ongoing research support and the out-licensing of our compounds for development by a third party. We expect to incur additional losses and negative cash flow from operations for several more years.
Under the BARDA Contract, substantially all of the costs of the development of 10150 as a medical countermeasure for pulmonary injuries resulting from an acute exposure to radiation from a radiological/nuclear accident or attack, particularly injuries associated with ARS or DEARE would be paid for by the U.S. government through BARDA funding. We recognized approximately $3,111,000 in revenue during the fiscal year ended September 30, 2015 related to the BARDA Contract.
We do not have any revenues from product sales and, therefore, we rely on investors, grants and collaborations to finance our operations. We generate limited revenue from reimbursable, cost-plus fee R&D contracts and grants. Revenues on reimbursable contracts are recognized as costs are incurred, generally based on allowable costs incurred during the period, plus any recognizable earned fee. We consider fixed fees under cost-plus fee contracts to be earned in proportion to the allowable costs incurred in performance of the contract.
Since the terms of the BARDA Contract include provisions to cover some general corporate overhead as well as a small provision for profit, the result on our liquidity is that our cash burn is related to the level of non-BARDA contract activities that we pursue. In order to fund on-going operating cash requirements, or to further accelerate or expand our programs, we expect to need to raise significant additional funds in order to pursue non-BARDA contract programs, including our oncology program.
There are significant uncertainties as to our ability to access potential sources of capital. We may not be able to enter into any collaboration on terms acceptable to us, or at all, due to conditions in the pharmaceutical industry or in the economy in general or based on the prospects of our catalytic antioxidant program. Even if we are successful in obtaining collaboration for our antioxidant program, we may have to relinquish rights to technologies, product candidates or markets that we might otherwise develop ourselves. These same risks apply to any attempt to out-license our compounds.
Similarly, due to market conditions, the illiquid nature of our stock and other possible limitations on equity offerings, we may not be able to sell additional securities or raise other funds on terms acceptable to us, if at all. Any additional equity financing, if available, could result in substantial dilution to existing stockholders.
Our forecast of the period of time through which our financial resources will be adequate to support our operations is forward-looking information, and actual results could vary.
38
Recent Developments
On December 10, 2015, we completed the 2015 Securities Placement by entering into securities purchase agreements with certain accredited investors to sell and issue (i) an aggregate of 10,215,275 common units issued at a purchase price of $0.22 per unit, and (ii) 4,500 preferred stock units issued to Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P., for an aggregate purchase price of $4.5 million, resulting in aggregate gross proceeds to the Company of approximately $6.75 million. Each common unit consists of one share of the Company's common stock and a five year warrant to purchase one share of the Company's common stock, subject to adjustment. The preferred units collectively consist of (i) 4,500 shares of Series C Preferred Stock that are collectively convertible into an aggregate of 20,454,546 common shares and (ii) warrants to purchase an aggregate of 20,454,546 Common Shares, in each case subject to adjustment. The foregoing warrants have an initial exercise price of $0.22 per share. On September 29, 2015, the Company received funding in the form of the BVF Notes. Following the completion of the 2015 Securities Placement, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of the Company's common stock and warrants to purchase an additional 5,414,402 shares of the Company's common stock at an exercise price per share of $0.22 subject to adjustment. As a result, the BVF Notes were no longer outstanding as of the date of this prospectus. Although the foregoing transactions provide us with additional liquidity, there can be no assurance that such funds will be sufficient to enable us to advance our development objectives. See "Risk Factors -- We may need substantial additional funding to continue our operations and may be unable to raise capital when needed, or at all, which would force us to delay, curtail or eliminate our clinical programs and our product development programs."
Contractual Obligations
Our contractual obligations (in thousands) as of September 30, 2015 were as follows:
|
Payments due by period
|
||||||||||||||||||||
|
Contractual Obligations
|
Total
|
Less
than 1
Year
|
1-3
Years
|
3-5
Years
|
More
than 5
Years
|
|||||||||||||||
|
Short and long-term debt
|
$
|
1,000
|
$
|
1,000
|
$
|
—
|
$
|
—
|
$
|
—
|
||||||||||
|
Capital lease obligations
|
—
|
—
|
—
|
—
|
—
|
|||||||||||||||
|
Operating leases
|
9
|
9
|
—
|
—
|
—
|
|||||||||||||||
|
Purchase obligations
|
—
|
—
|
—
|
—
|
—
|
|||||||||||||||
|
Total
|
$
|
1,009
|
$
|
1,009
|
$
|
—
|
$
|
—
|
$
|
—
|
||||||||||
Off Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources as defined under the rules of SEC Release No. FR-67. We do not have any capital leases.
Relationship with Goodnow Capital, LLC and Xmark Opportunity Partners, LLC
In July 2003, we initiated a series of transactions that led to our corporate reorganization and recapitalization. We obtained an aggregate of $8,000,000 in secured bridge financing in the form of convertible promissory notes we issued to Goodnow Capital, LLC ("Goodnow"). A portion of this financing allowed us to pay our past due payables and become current. We used the remainder for our operations, including a toxicology study for our catalytic antioxidant compounds under development as a treatment for ALS.
We completed our corporate reorganization on November 20, 2003. The reorganization involved the merger of our former parent company into one of our wholly owned subsidiaries. Subsequent to our 2003 reorganization, we completed a number of equity and debt financings, the majority of which included Xmark as investors. As of September 30, 2015, Xmark Opportunity Partners, LLC, through its management of Goodnow and the Xmark Funds and options held by David Cavalier, an affiliate of Xmark, an employee of the Company and the Chairperson of our Board of Directors, had voting power of 64% of our outstanding common stock and had beneficial ownership, calculated based on SEC requirements, of approximately 64% of our common stock. As a result of this significant ownership, Xmark Opportunity Partners, LLC and its affiliates is able to control future actions voted on by our stockholders.
39
In addition, under the terms of the warrants to purchase up to 61,822,749 shares of our common stock issued to Xmark on October 6, 2009 as well as subsequent warrant issuances on July 30, 2010, August 11, 2010 and December 28, 2010 (collectively, the "Xmark Warrants"), if we were to pay a dividend on our common stock the exercise price of these warrants would be reset from $0.28 per share or $0.50 per share, as applicable, to $0.01 per share and the warrant holders would also receive any such dividend paid. The Xmark Warrants also contain a provision that provides for the reduction of the exercise price to $0.01 upon a change of control and anti-dilution provisions in the event of a stock dividend or split, dividend payment or other issuance, reorganization, recapitalization or similar event. In addition, the Xmark Warrants, among other restrictions, prohibit the sale of Aeolus to an entity other than one that is publicly traded.
Effective February 19, 2013, the Company and each of Xmark JV Investment Partners, LLC, Xmark Opportunity Fund, Ltd. and Xmark Opportunity Fund, L.P. (collectively, the "Xmark Entities") entered into a Warrant Repricing, Exercise and Lockup Agreement (the "Xmark Warrant Agreement") pursuant to which the Company agreed to reduce the exercise price of outstanding warrants to purchase an aggregate of up to 59,149,999 shares of Common Stock held by the Xmark Entities (the "Xmark Warrants") to $0.01 per share. In consideration for the reduction of the exercise price of the Xmark Warrants, each of the Xmark Entities agreed to immediately exercise all of the Xmark Warrants by cashless exercise. The Xmark Warrant Agreement also provides that the Xmark Entities will not transfer the shares issuable upon exercise of the Xmark Warrants (the "Xmark Warrant Shares") until the Company either (i) declares a cash dividend on its common stock or otherwise makes a cash distribution or (ii) effects a Change of Control, subject in each case to the terms of the Xmark Warrant Agreement.
Critical Accounting Policies and Estimates
Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America, which require us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues, expenses and related disclosure of contingent assets and liabilities. We evaluate our estimates, judgments and the policies underlying these estimates on a periodic basis as the situation changes, and regularly discuss financial events, policies, and issues with our independent registered public accounting firm and members of our audit committee. We routinely evaluate our estimates and policies regarding revenue recognition; clinical trial, preclinical, manufacturing and patent related liabilities; license obligations; inventory; intangible assets; share-based payments; and deferred tax assets.
We generally enter into contractual agreements with third-party vendors to provide clinical, preclinical and manufacturing services in the ordinary course of business. Many of these contracts are subject to milestone-based invoicing and the contract could extend over several years. We record liabilities under these contractual commitments when we determine an obligation has been incurred, regardless of the timing of the invoice. Patent-related liabilities are recorded based upon various assumptions or events that we believe are the most reasonable to each individual circumstance, as well as based upon historical experience. License milestone liabilities and the related expense are recorded when the milestone criterion achievement is probable. We have not recognized any assets for inventory, intangible items or deferred taxes as we have yet to receive regulatory approval for any of our compounds. Any potential asset that could be recorded in regards to any of these items is fully reserved. In all cases, actual results may differ from our estimates under different assumptions or conditions.
Revenue Recognition
We do not currently generate revenue from product sales, but do generate revenue from the BARDA Contract. We recognize revenue from the BARDA Contract in accordance with the authoritative guidance for revenue recognition. Revenue is recognized when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller's price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured. We also comply with the authoritative guidance for revenue recognition regarding arrangements with multiple deliverables.
40
The BARDA Contract is classified as a "cost-plus-fixed-fee" contract. We recognize government contract revenue in accordance with the authoritative guidance for revenue recognition including the authoritative guidance specific to federal government contracts. Reimbursable costs under the contract primarily include direct labor, subcontract costs, materials, equipment, travel, and indirect costs. In addition, we receive a fixed fee under the BARDA Contract, which is unconditionally earned as allowable costs are incurred and is not contingent on success factors. Reimbursable costs under this BARDA Contract, including the fixed fee, are generally recognized as revenue in the period the reimbursable costs are incurred and become billable. We receive regular updates from our subcontractors regarding estimated completion of individual projects. Management evaluates the status of each project with respect to budgeted work completed, actual work completed, and cost of actual work completed. We are required to provide BARDA with monthly reports in addition to our bi-weekly conference calls with BARDA regarding the progress of each project.
41
BUSINESS
General
Overview
Aeolus Pharmaceuticals, Inc. ("we," "us" or "Aeolus") is a Southern California-based biopharmaceutical company leveraging significant U.S. Government funding to develop a platform of novel compounds for use in biodefense, fibrosis, oncology, infectious disease and diseases of the central nervous system. The platform consists of approximately 180 compounds licensed from the University of Colorado ("UC") Duke University ("Duke") and National Jewish Health ("NJH").
Our lead compound, AEOL 10150 ("10150"), is being developed under contract with the Biomedical Advanced Research and Development Authority ("BARDA" and the "BARDA Contract"), a division of the U.S. Department of Health and Human Services ("HHS"), as a medical countermeasure ("MCM") against the pulmonary sub-syndrome of acute radiation syndrome ("Pulmonary Acute Radiation Syndrome" or "Lung-ARS") and the delayed effects of acute radiation exposure ("DEARE"). Lung-ARS is caused by acute exposure to high levels of radiation due to a nuclear detonation or radiological event.
We are also developing 10150 for the treatment of lung fibrosis, including idiopathic pulmonary fibrosis ("IPF") and other fibrotic diseases. This new development program was created based upon the data generated from animal studies in Lung-ARS and chemical gas exposure under the BARDA Contract and National Institutes of Health ("NIH") grants. On March 17, 2015, we announced that 10150 was granted Orphan Drug Designation by the U.S. Food and Drug Administration ("FDA"). The Company plans to initiate a Phase 1 safety study in patients with IPF in 2016. After we have completed safety studies, we plan to initiate efficacy studies in patients with IPF. AEOL 10150 has previously been tested in two Phase I human clinical trials with no drug-related serious adverse events reported.
We are also developing 10150 for use in combination with radiation therapy for cancer as a treatment to reduce side effects caused by radiation toxicity and improve local tumor control. Pre-clinical studies at Duke, the University of Maryland and Loma Linda University have demonstrated that 10150 protects healthy, normal tissue, while not interfering with the benefit of radiation therapy or chemotherapy in prostate and lung cancer. Additional studies have demonstrated that 10150 enhances the anti-tumor activity of chemotherapy and radiation. A significant portion of the development work funded by the BARDA contract is applicable to the development program for radiation oncology, including safety, toxicology, pharmacokinetics and Chemistry, Manufacturing and Controls ("CMC"). After we have completed safety studies, we plan to initiate studies to demonstrate efficacy in protecting against the toxic side effects related to radiation therapy and/or the improvement of local tumor control.
We are also developing 10150 as a MCM for exposure to chemical vesicants (e.g., chlorine gas, sulfur mustard gas and phosgene gas) and nerve agents (e.g., sarin gas and soman gas) with grant money from the NIH Countermeasures Against Chemical Threats ("NIH-CounterACT") program. 10150 has consistently demonstrated safety and efficacy in animal studies of chemical gas exposure and nerve gas exposure.
The Company is developing a second compound, AEOL 11114B ("11114"), as a treatment for Parkinson's disease. Research funded by the Michael J Fox Foundation for Parkinson's disease ("MJFF") demonstrated the neuro-protective activity of 11114 in mouse and rat models of Parkinson's disease. The compounds were invented by Aeolus in collaboration with Brian J. Day, PhD at National Jewish Health and Manisha Patel, PhD at the University of Colorado, Anschutz Medical Campus, Department of Pharmaceutical Sciences in collaboration with the Company. We have obtained worldwide, exclusive licenses to develop the compounds from NJH and the UC. We plan to complete the remaining work to file an Investigational New Drug ("IND") application with the FDA by 2017.
In April 2015, we announced the discovery of a new compound, AEOL 20415 ("20415"), which has demonstrated anti-inflammatory and anti-infective properties, and could be effective in treating cystic fibrosis and combatting anti-biotic resistant bacteria. The compound was developed under collaboration between Brian J. Day, PhD at National Jewish Health in Denver, Colorado and Aeolus Pharmaceuticals. We have obtained a worldwide, exclusive license to develop the rights to the compound from NJH. We plan to complete the remaining work to file an Investigational New Drug ("IND") application with the FDA by 2017.
42
Finally, we have a pipeline of approximately 180 additional compounds. We expect that the development of additional compounds in our portfolio is dependent on our finding non-dilutive capital sources to fund such pipeline opportunities.
Recent Developments
On December 10, 2015, we entered into securities purchase agreements with certain accredited investors to sell and issue (i) an aggregate of 10,215,275 common units issued at a purchase price of $0.22 per unit, and (ii) 4,500 preferred stock units issued to Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P., for an aggregate purchase price of $4.5 million, resulting in aggregate gross proceeds to the Company of approximately $6.75 million (the "2015 Securities Placement"). Each common unit consists of one share of the Company's common stock and a five year warrant to purchase one share of the Company's common stock, subject to adjustment. The preferred units collectively consist of (i) 4,500 shares of Series C Preferred Stock that are collectively convertible into an aggregate of 20,454,546 common shares and (ii) warrants to purchase an aggregate of 20,454,546 Common Shares, in each case subject to adjustment. The warrants have an initial exercise price of $0.22 per share.
On September 29, 2015, the Company received funding in the form of convertible promissory notes (the "BVF Notes") from Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P. The BVF Notes (i) has an aggregate principal balance of $1,000,000, (ii) accrued interest at a rate of 6% per annum, (iii) had a scheduled maturity date of September 28, 2016 and (iv) were subject to automatic conversion into Company equity securities, provided a qualified financing of not less than $4 million occurred. On December 11, 2015, following the completion of the 2015 Securities Placement, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of the Company's common stock and warrants to purchase an additional 5,414,402 shares of the Company's common stock at an exercise price per share of $0.22 subject to adjustment. As a result, the BVF Notes are no longer outstanding as of the date of this prospectus.
Strategy
Our strategy is pursue the development of our promising platform of anti-fibrotic, anti-inflammatory, anti-infective and anti-oxidant compounds that address important unmet medical indications of clinical and national strategic importance. Our objective is to use non-dilutive capital whenever possible.
To date, we, and/or our research collaborators, have been awarded more than $149 million in non-dilutive U.S. government funding in the form of grants and contracts from federal agencies, such as BARDA, NIH-NIAID and NIH-CounterACT. Additional research has been conducted on our compounds with funding from private foundations, such as the MJFF and Citizens United for Research in Epilepsy ("CURE").
The expected benefit of this strategy is threefold. First, safety, toxicology, pharmacokinetic and CMC work funded by the government and foundations is applicable to our traditional commercial development programs. As an example, significant work funded under the BARDA contract for Lung-ARS has generated data that can be used to support our New Drug Applications ("NDA") for pulmonary fibrosis and/or radiation therapy for cancer.
Second, cost-plus development contracts, like our contract with BARDA, include funds for overhead and profit. These overhead and profit streams have significantly reduced our cash burn rate, which reduces our need to raise capital and incur dilution.
Third, some government contracts, such as the Lung ARS contract with BARDA are designed to lead to the acquisition of the product under development by the US Government for use as a MCM in the Strategic National Stockpile ("SNS" or the "Stockpile"). Government procurement could result in significant revenue to the Company, which could be used to further the development of the product in other indications or for the development of other promising products. Procurements may be made if either the drug meets the requirements for approval by the U.S. Food and Drug Administration (the "FDA") under the "Animal Rule" or prior to Animal Rule approval following the filing of a pre-Emergency Use Authorization ("EUA") application. Most of BARDA's procurements to date have been following the filing of a pre-EUA application.
43
The amount of any potential procurement is undisclosed by BARDA at this time and is unknown to us. Based on publicly available information, as well as other procurements made by the agency after pre-EUA applications, we believe the agency may purchase sufficient courses of therapy to treat a minimum of one hundred thousand people, with options to purchase an additional two hundred thousand courses of treatment. If purchases of such volumes occurred, the revenue to the Company could provide funding to advance numerous clinical studies, including potentially large Phase III programs in lung fibrosis and radiation therapy for cancer. This funding could allow us to fund studies with less dependence on collaborative partnering arrangements and future equity offerings, which is consistent with our strategy to deploy non-dilutive capital wherever possible to develop our compounds for unmet medical indications and thereby generate value for our stockholders. In addition, purchases of such volumes of drug could make the Company profitable.
Business Overview
We are developing a platform of compounds with anti-fibrotic, anti-inflammatory, anti-infective and anti-oxidant activity based on technology discovered and researched at Duke University, the University of Colorado and National Jewish Health, developed by Drs. Irwin Fridovich, Brian Day and others. Dr. Day is our Chief Scientific Officer.
Our lead compound, 10150, is a metalloporphyrin specifically designed to neutralize reactive oxygen and nitrogen species. The neutralization of these species reduces oxidative stress, inflammation, and subsequent tissue damage-signaling cascades resulting from radiation or chemical exposure. We are developing 10150 as a MCM for national defense and for use in oncology and treating lung fibrosis.
Our most extensive development program to date is the advanced development of 10150 for Lung-ARS and DEARE. On February 11, 2011, we signed a cost-plus contract with BARDA for the development of 10150 as a MCM against Lung-ARS. BARDA is the government agency responsible for the advanced development and purchase of medical countermeasures for chemical, biological, radiological and nuclear threats. The contract contemplates the advanced development of 10150 through approval by the FDA under 21 CFR Part 314 Subpart I and Part 601 Subpart H (the "Animal Rule.") The Animal Rule allows for approval of drugs using only animal studies when human clinical trials cannot be conducted ethically. The ultimate goal of the BARDA Contract is to complete all of the work necessary to obtain FDA approval for 10150 as a MCM for Lung-ARS. In addition, the BARDA Contract is designed to generate the data that would allow for acquisition of the drug by BARDA prior to FDA approval under a pre-Emergency Use Authorization.
Pursuant to the BARDA Contract we were awarded approximately $10.4 million for the base period of the contract (from February 2011 to April 2012). On April 16, 2012, we announced that BARDA had exercised two options under the BARDA Contract worth approximately $9.1 million. On September 17, 2013, we announced that BARDA had exercised $6.0 million in additional contract options. On May 07, 2014, we announced that BARDA had exercised a Contract Modification worth approximately $1.8 million. The Contract Modification allowed Aeolus to reconcile actual costs incurred with billings under the original provisional indirect billing rate. It established a new provisional indirect billing rate and placed a cap on the current and future provisional indirect billing rates. On June 26, 2015, we announced that BARDA had exercised $3.0 million in additional contract options under its advanced research and development contract for AEOL 10150. The June option exercise brings the total contract value exercised as of September 30, 2015 to approximately $30.3 million. We may receive up to an additional $88.1 million in options exercisable over the remainder of the BARDA Contract. Options are exercised based on the progress of the development program, including the completion of clinical trials or manufacturing tasks under previously exercised options.
The final goal of the contract is to achieve FDA approval for 10150 and the development of commercial manufacturing capability. In order to achieve these goals, we believe it will be necessary to exercise the majority of the options in the contract. We also believe that BARDA is likely to continue to exercise options as long as 10150 continues to demonstrate efficacy and safety in animal testing for Lung-ARS. In the event we begin sales to the U.S. government following the filing of a pre-EUA application, we believe that BARDA is likely to exercise the majority of the remaining options under the contract. One of the requirements of an EUA is that the development program continue towards the goal of FDA approval. If all of the options are exercised by BARDA, the total value of the contract would be approximately $118.4 million.
44
There are no existing treatments for Lung-ARS or DEARE and we are not aware of any compounds in development that have shown efficacy in increasing survival when administered after exposure to radiation. 10150 has demonstrated efficacy in two animal models (mouse and non-human primate) when administered after exposure to radiation. The U.S. government's planning scenario for a radiation incident is a 10 kiloton detonation of a nuclear device in a major American city. It is estimated that several hundred thousand civilians would be exposed to high doses of radiation in this scenario.
The BARDA contract is also designed to complete the work necessary for 10150 to be purchased for the US Strategic National Stockpile (the "SNS"). BARDA currently acquires drugs for the SNS through a Special Reserve Fund (the "SRF") created under Project BioShield and reauthorized under the Pandemic All-Hazards Preparedness Reauthorization Act of 2013. Although the final goal of the contract is full FDA approval under the Animal Rule, BARDA may purchase product prior to FDA approval following the filing of a pre-EUA application. BARDA has made numerous acquisitions of compounds that were not approved by the FDA, but were the subject of a pre-EUA filing. Procurements from BARDA following a pre-EUA application could result in a significant increase in revenues for Aeolus and potential profitability.
In August 2014, we filed an Investigational New Drug ("IND") application with the Division of Medical Imaging Products of the U.S. Food & Drug Administration ("FDA") for 10150 as a treatment for Lung-ARS. On September 4, 2014, the Company announced positive results from a study in non-human primates exposed to lethal radiation and treated with 10150. The study demonstrated that administration of 10150 24 hours after exposure to lethal radiation impacted survival at six months post-exposure as follows: survival in the 60 day treatment group was 50%, compared to 25% survival in the radiation only untreated control group. The data from this study, combined with development work completed in manufacturing and human safety data, will form the basis for a pre-EUA application. On September 22, 2014, we received a letter from the FDA placing our proposed Phase I study in healthy normal volunteers for 10150 as a treatment for Lung-ARS on clinical hold. (See - "Future Development Plans" and "Risk Factors – Risks Related to Our Business; Our commercialization efforts may be adversely impacted by an FDA clinical hold on our Phase I clinical trial in Lung-ARS.") The FDA provided the Company with specific concerns that need to be addressed in order to allow for the initiation of studies in healthy normal volunteers. In consultation with BARDA, the Company has submitted a mitigation strategy to the FDA for their review and feedback. The Company filed a complete response to the FDA's question in January 2016 and expects the FDA to review and respond to that submission within 30 days.
We also benefit from research funded by grants from the NIH CounterACT program for the development of 10150 as a MCM for the effects of nerve gas (e.g., sarin and soman) and chemical vesicant gasses (e.g., mustard gas, phosgene gas and chlorine gas) exposure. Funding for this indication is provided directly to the research institution and does not flow through our financial statements. Continued funding is generally dependent on continuing evidence of efficacy in animal trials. In October 2011, we announced that National Jewish Health was awarded a $12.5 million grant from NIH CounterACT to continue the development of 10150 as a MCM against chlorine gas exposure. Also included in the grant is support for research in looking at tissue plasminogen activator ("TPA") and Silabilin, which are not Aeolus assets, as MCMs against sulfur mustard gas exposure. The ultimate objective of the sulfur mustard and chlorine gas work at National Jewish Health will be to complete all work necessary to initiate pivotal efficacy studies in animals for both indications. This would include: running efficacy studies in the rat model for higher doses of sulfur mustard and chlorine gas; establishing endpoints, optimal dosing and duration of treatment for pivotal efficacy studies; and characterizing the natural history from sulfur mustard and chlorine gas damage.
We are also funded by grant money from the NIH CounterACT program and the National Institute of Neurological Disorders and Stroke ("NINDS") for the development of 10150 as a MCM for the effects of nerve gas (e.g., sarin and soman) exposure. NIH-CounterACT awarded a contract on September 24, 2011 worth approximately $735,000, to the University of Colorado to develop 10150 as a MCM against nerve agents. Work performed with this initial funding has demonstrated that 10150 significantly improved survival when administered with current treatment in a pilocarpine model for nerve gas exposure. In September 2013, we announced that Dr. Manisha Patel at the University of Colorado had been awarded a $4.3 million grant from NINDS to further develop as a MCM for exposure to sarin gas and other nerve agents.
45
Substantially all of the past costs for the Lung-ARS program have been funded by the BARDA Contract. To date, the chlorine, phosgene, mustard gas and nerve agent programs have been funded by NIH-CounterACT and NINDS through programs at National Jewish Health, the University of Colorado, and the United States Army Medical Research Institute for Chemical Defense ("USAMRICD").
We are also developing 10150 for the treatment of lung fibrosis, including idiopathic pulmonary fibrosis ("IPF") and other fibrotic diseases. This new development program was created based upon the data generated from animal studies in Lung-ARS and chemical gas exposure under the BARDA Contract and NIH grants. On March 17, 2015, we announced that 10150 was granted Orphan Drug Designation for IPF by the U.S Food and Drug Administration ("FDA"). The Company plans to initiate a Phase 1 safety study in patients with IPF in 2016. After we have completed safety studies, we plan to initiate efficacy studies in patients with IPF. We expect a portion of the net proceeds from the 2015 Securities Placement will be used to fund a portion of the future costs of work associated with IPF and other fibrotic diseases.
We are also developing 10150 for use in combination with radiation therapy for cancer as a treatment to reduce side effects caused by radiation toxicity and to improve local tumor control. Pre-clinical studies at Duke University and Loma Linda University have demonstrated that 10150 does not interfere with the benefit of radiation therapy or chemotherapy in prostate and lung cancer. Additional studies have shown that 10150 enhances the anti-tumor activity of radiation and chemotherapy. A significant portion of the development work funded by the BARDA contract is applicable to the development program for radiation oncology, including safety, toxicology, pharmacokinetics and Chemistry, Manufacturing and Controls ("CMC"). After we have completed safety studies, we plan to initiate studies to demonstrate efficacy in toxic side effects related to radiation therapy and/or improvement of local tumor control. We expect a portion of the net proceeds from the 2015 Securities Placement will be used to fund a portion of the future costs of work related to radiation therapy opportunities.
10150 has been tested in two human Phase I safety studies where it was well-tolerated and no adverse events were observed. Efficacy has been demonstrated in animal models for Lung-ARS, chlorine gas exposure, phosgene gas exposure, sulfur mustard gas exposure (lungs and skin) and nerve gas exposure. In both mouse and non-human primate ("NHP") studies for Lung-ARS, 10150 treated groups showed significantly reduced weight loss, inflammation, oxidative stress, lung damage, and most importantly, mortality. Therapeutic efficacy has been demonstrated when 10150 is administered 24 hours after exposure to radiation, a requirement for consideration as a radiation MCM for the SNS.
Following the events at the Fukushima nuclear plant in Japan in 2011, we performed radiation exposure studies in mice at the request of Japanese researchers to determine how the administration of AEOL 10150 would impact the use of leukocyte growth factors ("LGF") used to treat the hematopoietic or bone marrow syndrome of ARS ("H-ARS"). Data showed that 10150 does not interfere with the efficacy of LGF (in this case Amgen's Neupogen®). Additionally, the study demonstrated that administration of Neupogen®, the current standard of care for H-ARS, increased damage to the lungs. When 10150 was administered with Neupogen® this damage was significantly reduced. We believe that this finding may have important implications for the potential procurement of 10150 for the SNS. In September 2013, BARDA announced that it had entered into a procurement and inventory management agreement with Amgen to provide Neupogen® for the SNS. On March 30, 2015, the FDA approved Neupogen® for the treatment of H-ARS.
We have an active Investigational New Drug Application ("IND") on file with the FDA for AEOL 10150 as a potential treatment for amyotrophic lateral sclerosis ("ALS"). At this time, we do not have any plans to continue development of 10150 for ALS.
We have already completed two Phase I safety studies in 50 humans (39 receiving drug and 13 control) demonstrating that 10150 is safe and well tolerated. CMC work has been completed, pilot lots have been prepared and production is being scaled up under the BARDA Contract.
46
The Company is developing a second compound, AEOL 11114 ("11114"), as a treatment for Parkinson's disease. Research funded by the Michael J Fox Foundation for Parkinson's disease ("MJFF") demonstrated the neuro-protective activity of 11114 in mouse and rat models of Parkinson's disease. The compounds were invented by Aeolus in collaboration with Brian J. Day, PhD at National Jewish Health and Manisha Patel, PhD at the University of Colorado, Anschutz Medical Campus, Department of Pharmaceutical Sciences in collaboration with the Company. We have obtained worldwide, exclusive licenses to develop the compounds from NJH and the UC. We plan to complete the remaining work to file an Investigational New Drug ("IND") application with the FDA by 2017.
In April 2015, we announced the discovery of a new compound, AEOL 20415 ("20415"), that has demonstrated anti-inflammatory and anti-infective properties, and could be effective in treating cystic fibrosis and combatting anti-biotic resistant bacteria. The compound was developed under a collaboration between Brian J. Day, PhD at National Jewish Health in Denver, Colorado and Aeolus Pharmaceuticals. We have obtained a worldwide, exclusive license to develop the rights to the compound from NJH. We plan to complete the remaining work to file an Investigational New Drug ("IND") application with the FDA by 2017.
Aeolus' Catalytic Antioxidant Program
We established our research and development program to explore and exploit the therapeutic potential of small molecule catalytic antioxidants. We have achieved our initial research objectives and begun to extend our preclinical accomplishments into non-clinical studies, clinical trials and drug development programs, as well as into the exploration of novel approaches to address inflammation and infection.
Our catalytic antioxidant program is designed to:
| · | Retain the catalytic mechanism and high antioxidant efficiency of the natural enzymes, and |
| · | Create and develop stable and small molecule antioxidants without the limitations of SOD so that they: |
| o | Have broader antioxidant activity, |
| o | Have better tissue penetration, |
| o | Have a longer life in the body, and |
| o | Are not proteins, which are more difficult and expensive to manufacture. |
We created a class of small molecules that consume reactive oxygen and nitrogen species catalytically; that is, these molecules are not themselves consumed in the reaction. Our class of compounds is a group of manganoporphyrins (an anti-oxidant containing manganese) that retain the benefits of antioxidant enzymes, are active in animal models of disease and, unlike the body's own enzymes, have properties that make them suitable drug development candidates.
Our most advanced compound, 10150 (Figure 1), is a small molecule, broad-based, catalytic antioxidant that has shown the ability to scavenge a broad range of reactive oxygen species, or free radicals. As a catalytic antioxidant, 10150 mimics, and thereby amplifies, the body's natural enzymatic systems for eliminating these damaging compounds. Because oxygen- and nitrogen-derived reactive species are believed to have an important role in the pathogenesis of many diseases, we believe that our catalytic antioxidants and 10150 may have a broad range of potential therapeutic uses. 10150 has been tested extensively as a subcutaneous injection. Recently, we have broadened the potential utility of 10150 by developing an oral formulation that would allow for more chronic dosing and could potentially reduce the cost of goods given the lower cost of pills and capsules compared to sterile injections.
47
Figure 1
|
AEOL 10150 Overview
|
|
|
Product Type
|
√ Catalytic antioxidants
|
|
(manganoporphyrin)
|
|
|
Administration Route
|
√ Subcutaneous administration; self-injection possible
√ Oral formulation developed
|
|
Indications in Development
|
√ Radiation Oncology
√ Pulmonary Fibrosis
|
|
√ Pulmonary ARS/DEARE
|
|
|
√ Sulfur Mustard; Chlorine Gas; Nerve Gas
|
|
|
Technical Readiness Level (TRL)
|
√ TRL 7/8 for Pulmonary Effects
|
|
of ARS/DEARE
|
|
|
Regulatory Status
|
√ Active IND (IND-67741)
|
|
Phase I (3 studies, 50 patients)
|
|
|
√ IND on Clinical Hold (IND-112103)
|
|
AEOL 10150 has shown efficacy in a variety of animal models as a protectant against pulmonary damage and diseases including: lung fibrosis, radiation injury, sulfur mustard gas exposure, chlorine gas exposure and phosgene gas exposure, as well as against neurological damage and diseases including: nerve gas exposure, epilepsy, ALS, and stroke and against diabetes. We filed an IND for AEOL 10150 in April 2004, under which human safety trials in ALS patients were conducted as more fully described below under the heading "10150 Clinical Program to Date." For a more detailed description of antioxidants see the section below under the heading "Background on Antioxidants." 10150 has been granted orphan drug designation for ALS, Lung ARS and Idiopathic Pulmonary Fibrosis by the Office of Orphan Products at the FDA.
AEOL 10150 Medical Countermeasure Development Program
We and our research partners have been awarded in excess of $149 million for the development of 10150 as a dual-use, broad spectrum medical countermeasure. The table below details the indications currently under development and the sources of funding from the US Government.
|
Indication
|
Funding Source
|
Amount of Grant/Contract
|
Research Partners
|
|
Lung-ARS
|
BARDA
|
Up to $118.4 million
|
University of Maryland
|
|
Chlorine Gas
|
NIH CounterACT
|
$20.3 million
|
National Jewish Health
|
|
Mustard Gas
|
NIH CounterACT
|
Part of the NIH-CounterACT grant above
|
National Jewish Health
University of Colorado
|
|
Nerve Agents
|
NIH CounterACT
|
$5 million
|
University of Colorado
|
48
10150 as a potential medical countermeasure against the effects of Pulmonary Acute Radiation Syndrome (Lung ARS)
Overview
The U.S Government's current planning scenario for a nuclear attack is a 10 kiloton detonation in a major American city. For purposes of comparison, the yield of the bomb dropped on Hiroshima in World War II was approximately 16 kilotons. Such an attack would potentially expose hundreds of thousands of citizens to acute, high dose, ionizing radiation and the lethal effects of Acute Radiation Syndrome ("ARS").
ARS is not a single disease, but a series of sub-syndromes that follow exposure to ionizing radiation. BARDA is pursuing separate development plans for each sub-syndrome. 10150 is the only compound in advanced development with BARDA for Lung-ARS.
Immediately after exposure, the first syndromes of ARS typically experienced by those exposed are the acute hematopoietic (bone marrow) syndrome ("H-ARS") and early-onset GI-ARS, both of which can be lethal. However, depending on the level and location of radiation exposure, the lethal effects of both H-ARS and early-onset GI-ARS may be reduced with proper treatment, including supportive care (fluids and antibiotics) and LGFs like Amgen's Neupogen®.
In September 2013, BARDA announced that it had entered into a vendor-managed supply agreement with Amgen to supply its LGF, Neupogen®, to the SNS as a treatment for H-ARS. Although Neupogen® is an FDA-approved drug for neutropenia, it is not approved for H-ARS and would be used under a pre-EUA application. The procurement of Neupogen® for the SNS is significant for 10150 and its potential role in the treatment of ARS. A 2011 murine study conducted at Indiana University at the request of Japanese researchers confirmed that 10150 does not interfere with the positive effects of Neupogen® in H-ARS and the two products in combination were safe and well-tolerated. More importantly, this study also demonstrated that treatment of H-ARS with Neupogen® exacerbates radiation damage to the lung, even at sub-lethal doses of radiation. Treatment with Neupogen® in combination with 10150 significantly reduced the lung damage. We believe that the use of Neupogen® in treating H-ARS makes the use of 10150 crucial in managing the lung effects of acute radiation exposure. On March 30, 2015, the FDA approved Neupogen® for the treatment of H-ARS.
When patients survive H-ARS and GI-ARS, respiratory failure becomes the major cause of death. Research has shown that damage associated with the exposure to upper half body irradiation or total body irradiation is an acute, but delayed, onset of radiation pneumonitis (inflammation of lung tissue) followed by lung fibrosis (scarring caused by inflammation).The incidence of radiation pneumonitis rises very steeply at relatively low radiation doses. This is Lung-ARS, the syndrome that 10150 is being developed to treat.
We believe it is in the government's interest in to provide care not only for survival from the short-term effects of radiation exposure following an event (e.g., H-ARS and GI-ARS), but also to provide care for the delayed effects of radiation exposure, such as Lung-ARS. There are no current FDA-approved or EUA-approved therapies for Lung-ARS. We believe 10150 is the only drug in advanced development with BARDA for Lung-ARS.
Pre-clinical studies
In a 2013 study run under the BARDA contract at the University of Maryland at Baltimore, a total of 120, CBA/J mice were exposed to 14.6 Gray of whole thorax irradiation ("WTLI"). Four cohorts of animals were treated with daily doses of 5mg, 10 mg, 25 mg or 40 mg/kg of 10150 beginning 24 hours after exposure for a total for 28 days. The results are shown in the table below. Survival at six months post-exposure in the optimal treatment group of 25mg/kg of 10150 improved to 40 percent, compared to 10 percent survival in the radiation only group. In addition, animals receiving 10150 showed significant protection of the lungs as measured by differences in wet lung weights and breathing frequency. This study confirms previous studies in animals that demonstrate 10150's protection of the lungs from radiation exposure.
49
|
Treatment
|
Survival
|
Number of Animals
|
|
Radiation Only
|
10%
|
20
|
|
Radiation + 5 mg/kg AEOL 10150
|
16%
|
19
|
|
Radiation + 10 mg/kg AEOL 10150
|
16%
|
19
|
|
Radiation + 25 mg/kg AEOL 10150
|
40%
|
20
|
|
Radiation + 40 mg/kg AEOL 10150
|
30%
|
20
|
A number of other preclinical studies by researchers at the University of Indiana, the University of Maryland and Duke University have demonstrated the efficacy of 10150 in the protection of healthy, normal tissue from damage due to blast and fractionated radiation exposure.
In 2011, we announced positive results from study of 10150 and Neupogen® as combination therapy for treatment of ARS. The study was conducted by Christie Orschell, PhD of Indiana University. The primary endpoint of the study was to determine drug-drug interactions between Neupogen® and 10150, as well as to monitor safety and tolerability of the two treatments given simultaneously. Results of the study confirmed that 10150 does not interfere with the positive effects of Neupogen® on the hematopoietic, or bone marrow, syndrome of Acute Radiation Syndrome ("ARS"), and the two products in combination were safe and well tolerated. In 2012, we announced further data from this study, which demonstrated that treatment of the hematopoetic sub-syndrome of acute radiation syndrome ("Heme-ARS") with Neupogen® exacerbates radiation damage to the lung. The study also confirmed that treatment with 10150 in combination with Neupogen® significantly reduced the lung damage.
The study entitled "Pilot Study to Test the Effects of Aeolus 10150 on Neupogen®-Induced ANC Recovery in Sub-Lethally Irradiated C57Bl/6 Mice" was initiated at the request of Shigetaka Asano, MD of Waseda University and Arinobu Tojo, MD, PhD and Tokiko Nagamura, MD at the Institute of Medical Science at the University of Tokyo to determine whether there would be any interference with the demonstrated efficacy of Neupogen® as a medical countermeasure against the hematopoietic complications of radiation exposure. In previous treatment of radiation accident victims at Tokai-mura, Dr. Asano and others were able to use Granulocyte Colony Stimulating Factor ("G-CSF") and supportive care to enable victims of 8 to 12 Gy exposure to survive the hematopoietic ("heme") syndrome. Unfortunately, these patients later died due to lung and multi-organ complications. As 10150 has shown separate efficacy against lung and GI complications in mice and in Lung-ARS in non-human primates, this study was undertaken to evaluate whether the Neupogen® and Aeolus 10150 compounds would be beneficial if used in tandem.
The use of Neupogen® or other G-CSFs or Neulasta® or other Granulocyte-Macrophage Colony Stimulating Factor ("GM-CSF") products is recommended by the Radiation Emergency Assistance Center/Training Site (REAC/TS) at radiation exposures greater than 2 to 3 Gy to mitigate damage to the hematopoietic system. REAC/TS is a response asset of the U.S. Department of Energy and provides treatment capabilities and consultation assistance nationally and internationally. In animal studies G-CSF's have been shown to be effective in increasing survival at levels up to 7.5 Gy due to their positive effects on the hematopoietic damage created by radiation exposure. BARDA began procuring Neupogen® and Sanofi-Aventis' drug Leukine® for the Strategic National Stockpile in September 2013.
LGFs as a class have not demonstrated an effect on the two other major sub-syndromes -- GI and Lung. 10150 has demonstrated efficacy in treating the GI sub-syndrome in pilot studies conducted by NIH-NIAID, by protecting crypt cells and reducing diarrhea. More extensive studies of the drug in treating the pulmonary effects of radiation at Duke University and the University of Maryland have shown improved survival and enhanced lung function and improved histology at exposures up to 15 Gy in mice and 11.5 Gy in non-human primates. These exposure levels caused death in 100 percent of animals that were not treated with 10150. Studies at Duke University have also shown a significant survival advantage for animals treated with 10150 after 15 Gy upper half body irradiation, which causes lethal damage to both the GI tract and the lungs.
In summary, 10150 has consistently shown a survival advantage and protective effect against the lung and delayed effects of acute radiation exposure when administered 24 hours or more after exposure. Additionally, the current standard of care for the acute ARS syndromes, LGF administration, exacerbates damage to the lungs and 10150 has demonstrated efficacy in reducing that damage.
50
Non-clinical studies
In a 2014 study run under the BARDA contract at the University of Maryland at Baltimore, a total of 80 rhesus macaque monkeys were exposed to 10.74 Gray of WTLI. Three cohorts of animals were treated with 25 mg/kg of 10150 beginning 24 hours after exposure. The three cohorts then received daily doses of 10150 for 28 days, 60 days or 28 days followed by a pause of 32 days and then an additional 28 days of treatment. As we announced in September 2014, survival at six months post-exposure in the 60 day treatment group was 50%, compared to 25% survival in the radiation-only untreated control group. This study confirms previous studies in non-human primate and mouse models that demonstrate 10150's protection of the lungs from radiation exposure. We plan to publish the detailed results of the study with our research collaborators as soon as possible.
In May 2015, we announced secondary endpoints from the study. The data from these endpoints demonstrated that administration of 10150 for 60 days beginning 24 hours after exposure to 10.74 Gy of radiation:
| · | Increased mean and median overall survival time |
| · | Increased mean and median survival time in subjects that did not survive to 180 days |
| · | Increased time to onset of increased respiratory rate, a clinical measure of lung injury |
| · | Decreased mortality in subjects with elevated respiratory rate |
| · | Decreased wet lung weight in all animals, suggesting less parenchymal damage and edema |
| · | Increased Sp02, a measure of compensated lung function |
| · | Diminished radiographic evidence of pneumonitis and fibrosis during the later stages of the study (days 90 -180) |
In addition, a new approach to investigating lipids, metabolites and proteins in pathophysiological process, matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) was employed in the study to measure potential biomarkers for lung injury in tissue samples from study subject. Analysis using MALDI-MSI showed that the molecular profile for the naïve (un-irradiated) lung is statistically distinct from irradiated lung and treatment with AEOL 10150 shifts the molecular profile back towards the naïve lung. Two prospective biomarkers found in irradiated, damaged lung tissue were not detectable in either naïve or 10150-treated samples.
In 2010, we initiated a study to test the efficacy of 10150 as an MCM to nuclear and radiological exposure in non-human primates ("NHPs"). The study was designed to test the efficacy of 10150 as a treatment for Lung-ARS and to begin establishing an animal model that can be validated and could be utilized by the FDA for approval of an MCM for Pulmonary Acute Radiation Syndrome under the "Animal Rule". The FDA "Animal Rule" enumerates criteria whereby the FDA can rely on animal efficacy data when "evidence is needed to demonstrate efficacy of new drugs against lethal or permanently disabling toxic substances when efficacy studies in humans, ethically cannot be conducted." The criteria are discussed below.
Results from the study were published in the journal Health Physics, Volume 106, Number 1 (January 2014) under the title "A Pilot Study in Rhesus Macaques to Assess the Treatment Efficacy of A Small Molecular Weight Catalytic Metalloporphyrin Antioxidant (AEOL 10150) in Mitigating Radiation-induced Lung Damage." The primary objective of the study was to determine if 10150 could mitigate radiation-induced lung injury and enhance survival in rhesus macaques exposed to whole thorax lung irradiation ("WTLI") and administered supportive care. Two cohorts of NHPs were exposed to 11.5Gy LINAC-derived photon radiation in the WTLI protocol. The control cohort had n=6 and 10150-treated cohort was n=7.This model showed 100% incidence of severe radiation-induced lung damage. 10150 was administered subcutaneously at 5mg/kg beginning at day 1 post WTLI and continued as a single, daily injection for 28 consecutive days. The final results were presented at the 14th International Congress of Radiation Research in Warsaw, Poland in September 2011. Key findings in the study include:
51
| 1. | Exposure of the whole thorax to 11.5 Gy resulted in radiation-induced lung injury in all NHPs in the study and proved 100% fatal in the control animals, despite supportive care including dexamethasone. 11.5 Gy is, therefore, equal to or greater than the LD100/180dose for the WTLI model. |
| 2. | 10150, as administered in this pilot study (daily for 28 days at a dose of 5mg/kg subcutaneously), demonstrated potential efficacy in mitigating against fatal radiation-induced lung injury. Treatment with the drug resulted in 28.6% survival following exposure to a radiation dose that proved to be 100% fatal in the untreated control group. |
| 3. | Serial CT scans demonstrated less quantitative radiographic injury (pneumonitis, fibrosis, effusions) in the 10150-treated cohort, suggesting that the drug reduces the severity of the radiographically detectable lung injury. |
| 4. | Dexamethasone administration yielded a transient benefit on both clinical and radiographic evidence of pneumonitis. The 10150 treated cohort required 1/3 less dexamethasone support due to reduced pulmonary injury in the 10150 treated group, resulting in less frequent clinical "triggers" (respiratory rate≥80) to treat with dexamethasone. |
| 5. | The results of this pilot study are encouraging and suggest that treatment with 10150 results in reduced clinical, radiographic and anatomic evidence of radiation-induced lung injury, which also results in improved survival. 10150 merits further study as a post-exposure MCM against radiation-induced lung injury. |
In rodents, non-human primates and humans, radiation of the lungs can cause reduced breathing capacity, pneumonitis, fibrosis, weight loss and death and is characterized by oxidative stress, inflammation and elevated macrophage counts. 10150 has proven to be an effective countermeasure to radiation exposure of the lungs in mice and rats in published studies such as Rabbani et al Int J Rad Oncol Biol Phys 67:573-80, 2007, Rabbani et al Free Rad Res 41:1273-82, 2007 and Gridley et al Anticancer Res 27:3101-9, 2007.
Future Development Plans
Our objective is to develop 10150 as an MCM against Lung-ARS via the FDA's "Animal Rule". This development pathway requires demonstration of the key study efficacy parameter of 10150 treatment in two animal models relevant to the human radiation response and its treatment, demonstration of safety in humans, demonstration of relevant dosing and administration in humans, and clear identification of the mechanism of radiation-induced damage to the lung and its amelioration by the drug candidate.
10150 has several distinct advantages as an MCM, including the following:
| · | Demonstrated survival increase in animal studies of lung ARS when administered 24 hours after exposure, |
| · | Demonstrated reduction in lung fibrosis in animal studies when administered up to 24 hours post exposure, |
| · | Demonstrated histological improvement in lung tissue post-radiation exposure, |
| · | Addresses an unmet medical need as an MCM to Lung-ARS, |
| · | Established safety profile in both clinical and pre-clinical studies, |
| · | Subcutaneous self-administration possible by exposed individuals during emergency, |
| · | Rapid administration, allowing large numbers of patients to be treated quickly, |
| · | Original formulation stable for up to 4½ years at 0–8°C and 1 year at room temperature, |
| · | New formulation stability tested in bulk drug for 2 years at room temperature (25°C) and refrigerated conditions (2-8°C); stability testing in bulk drug will continue to three years, |
| · | New formulation stability tested in final drug product to 18 months under room temperature (25°C), accelerated conditions (40°C) and refrigerated conditions (2-8°C); stability testing in final drug product will continue to 5 years, |
52
| · | Requires no non-standard storage conditions (i.e., not photosensitive), |
| · | Currently in development as an adjunct to radiation therapy and lung fibrosis; if approved will provide a pre-existing distribution and stockpile resource at oncology centers in the event of a radiological emergency, |
| · | Demonstrated advantage when used in combination with Neupogen®, |
| · | Demonstrated potential as both a therapeutic and prophylactic, |
| · | Demonstrated efficacy against sulfur mustard gas, phosgene gas, chlorine gas and nerve agent exposures, |
| · | Potential dual use as an adjunct treatment for cancer patients receiving radiation therapy and treatment of idiopathic pulmonary fibrosis, subject to separate FDA approvals for these indications. |
We believe that in order to receive approval from the FDA for Lung-ARS with the FDA, we will need to demonstrate efficacy in animal models and demonstrate product safety. We also plan to request Fast Track status for this indication. If the FDA accepts our Fast Track request, a rolling NDA submission process is enabled, a key step in achieving Priority Review. The FDA determines within 45 days of a company's request, made once the complete NDA is submitted, whether a Priority or Standard Review designation will be assigned.
In August 2014, the Company filed an IND (IND#112103) for 10150 for Lung-ARS with the Division of Medical Imaging Products ("DMIP") at FDA. On September 22, 2014, the Company received a letter from FDA placing the proposed Phase I study in healthy normal volunteers on clinical hold. In January 2015, the Company provided additional data to the DMIP including a mitigation plan to address the DMIP's concerns and requested a "Type A" meeting to discuss the plan. On March 12, 2015, the Company met with the DMIP and received updated guidance for the removal of the clinical hold. The DMIP indicated that it would remove the clinical hold if the Company conducts a mini-pig skin study, and demonstrates that 10150 is not mutagenic by (i) performing studies to support its hypothesized non-mutagenic mechanism of action ("MOA") as responsible for positive findings in prior in-vitro genotoxicity studies (the "Genotox MOA Work") or (ii) performing a transgenic mouse carcinogenicity study ("Mouse Carc Study") to definitively determine the mutagenic potential of 10150.
In the first quarter of 2015, the Company initiated a mini-pig skin study in accordance with the DMIP's request. The study has been completed and results were submitted to the FDA in January 2016. In June 2015, the Company received an option exercise under its Lung ARS development contract with BARDA that provides the funding for the Genotox MOA Work requested by the DMIP. The Genotox MOA Work was completed in December 2015 and the Company believes that preliminary results provide the data to support the hypothesized non-mutagenic mechanism of action. A complete response incorporating this data was submitted to the FDA in January 2016 with a request for removal of the clinical hold. The Company expects the FDA to respond within 30 days.
In August 2015, the Company also initiated the initial portion of the Mouse Carc Study. This study is expected to be complete in the fourth quarter of calendar 2016, and will be used in the event that the Company is unable to meet the FDA's request through the Genotox MOA Work. If the Genotox MOA Work is acceptable to the FDA, the Company expects to initiate a phase 1 study in healthy normal volunteers in 2016.
In addition to the phase 1 study in healthy, normal volunteers, we plan to initiate studies in patients with cancer and/or lung fibrosis, and will use this data together with the existing data we have in patients with Amyotrophic Lateral Sclerosis ("ALS") to respond to the DMIP's concerns. As discussed above, we also plan to file separate 10150 IND's with the Respiratory division of the FDA and the Oncology division of the FDA in the first half of 2016 and initiate Phase I studies in patients with pulmonary fibrosis and in patients receiving radiation therapy for cancer upon FDA approval of those INDs. We also believe that this is the most efficient way to generate the safety data required for a pre-EUA application for Lung-ARS.
The FDA's "Animal Rule" enumerates criteria whereby the FDA can rely on animal efficacy data when evidence is needed to demonstrate efficacy of new drugs against lethal or permanently disabling toxic substances when efficacy studies in humans cannot be ethically conducted. The criteria are as follows:
| · | Knowledge of the mechanism of radiation-induced damage to the lung and its amelioration by the candidate drug. |
53
| · | Pharmacokinetic and pharmacodynamic analysis to provide information on relevant dose and administration schedule. |
| · | Direct correlation of key study parameters (e.g., survival or major morbidity) with the desired clinical benefit in humans. |
| · | Collection of efficacy data in two species relevant to the human radiation response and its treatment unless otherwise justified under GLP-compliant conditions. |
| · | A Phase I safety trial using the same product and formulation as used in the pivotal trial(s) is required. |
Demonstrate Efficacy in Animal Models
Under the BARDA contract, we have developed and validated mouse and NHP models for Lung-ARS. We have also presented these models to the FDA and they have concurred with our design and our development plan for demonstrating efficacy. We believe that the efficacy data produced in pivotal studies using validated models will provide the data required to demonstrate efficacy of 10150 at the dose and schedule proposed for licensure
Demonstrate Product Safety
For product approval under the "Animal Rule", we will also demonstrate product safety using the same product and formulation used in the animal efficacy trials and proposed for use in humans. Demonstration of safety includes preclinical demonstration of safety via the standard pre-clinical studies and analyses methods and Phase I safety trials sufficient to demonstrate product safety in the target patient population. We believe our proposed safety studies in healthy normal volunteers, planned studies in pulmonary fibrosis and radiation therapy, as well as completed phase 1 studies in ALS may be utilized to demonstrate safety for this indication. We expect that all studies in healthy normal volunteers will be funded by BARDA, and believe that BARDA may fund or co-fund studies in other indications to provide the human safety data required for approval under the animal rule.
Competition
Currently there are no FDA-approved drugs for the treatment of Lung-ARS. We are also not aware of any other drug candidates that have demonstrated the ability to protect the lungs from radiation when administered after exposure, which we believe is a critical aspect of the development of an MCM against the effects of acute radiation syndrome. We are also not aware of any drugs with advanced development funding from BARDA for Lung-ARS.
However, in general, we face significant competition for U.S. government funding for both development and procurements of an MCM for biological, chemical and nuclear threats, diagnostic testing systems and other emergency preparedness countermeasures. The U.S. federal government has currently allocated a significant amount of research funding to the development of countermeasures against the effects of radiation exposure. As a result, there are many drug candidates under development as a possible countermeasure against the various effects and sub-syndromes of radiation exposure.
Funding and Funding Options
On February 11, 2011, we signed a five-year, cost-plus contract with BARDA for the development of 10150 as a MCM against Lung-ARS (the "BARDA Contract"). BARDA is the government agency responsible for the advanced development and purchase of medical countermeasures for chemical, biological, radiological and nuclear threats. The contract funds the advanced development of 10150 through approval by the United States Food & Drug Administration ("FDA") under the "Animal Rule." The Animal Rule allows for approval of drugs using only animal studies when human clinical trials cannot be conducted ethically.
Pursuant to the BARDA Contract we were awarded approximately $10.4 million for the base period of the contract (from February 2011 to April 2012). On April 16, 2012, we announced that BARDA had exercised two options under the BARDA Contract worth approximately $9.1 million. On September 17, 2013, we announced that BARDA had exercised $6.0 million in additional contract options. On May 07, 2014, we announced that BARDA had exercised a Contract Modification worth approximately $1.8 million. On June 26, 2015, we announced that BARDA had exercised $3.0 million in additional contract options under its advanced research and development contract for AEOL 10150. The June option exercise brings the total contract value exercised as of September 30, 2015 to approximately $30.3 million. We may receive up to an additional $88.1 million in options exercisable over the remaining years of the contract. Options are exercised based on the progress of the development program, including the completion of clinical trials or manufacturing tasks under previously exercised options. The final goal of the contract is to achieve FDA approval for 10150 and the development of commercial manufacturing capability. In order to achieve these goals, we believe it will be necessary to exercise the majority of the options in the contract. We also believe that BARDA is likely to continue to exercise options as long as 10150 continues to demonstrate efficacy and safety in testing for Lung-ARS. In the event we begin sales to the U.S. government under an EUA, we believe that BARDA is likely to exercise the majority of the remaining options under the contract. One of the requirements of an EUA is that the development program continue towards the goal of FDA approval. If all of the options are exercised by BARDA, the total value of the contract would be approximately $118.4 million.
54
Since we have been awarded the BARDA Contract, substantially all of the past costs associated with the research and development of 10150 as a MCM for Lung-ARS have been covered by the BARDA Contract and we expect substantially all future costs would be covered by the BARDA Contract; however, our expectation in this regards is primarily dependent on continued positive results with 10150 in animal studies and the general risks of doing business with the government. See Risk Factors – "Risks Related to Our Dependence on U.S. Government Grants and Contracts." We expect that a portion of the net proceeds from the 2015 Securities Placement will also be used to fund part of the future costs of the Lung ARS program that are not funded by the BARDA Contract.
10150 as a potential medical countermeasure against the effects of mustard gas
Overview
Sulfur mustards, of which sulfur mustard gas ("SM") is a member, are a class of related cytotoxic, vesicant chemical warfare agents with the ability to form large blisters on exposed skin and cause pneumonitis and fibrosis in the lungs. In their pure form most sulfur mustards are colorless, odorless, viscous liquids at room temperature. When used as warfare agents they are usually yellow-brown in color and have an odor resembling mustard plants, garlic or horseradish. Mustard agents, including sulfur mustard, are regulated under the 1993 Chemical Weapons Convention. Three classes of chemicals are monitored under this Convention, with sulfur and nitrogen mustard grouped in the highest risk class, "schedule 1." However, concerns about its use in a terrorist attack have led to resurgence in research to develop a protectant against exposure.
Mustard gas is a strong vesicant (blister-causing agent). Due to its alkylating properties, it is also strongly mutagenic (causing damage to the DNA of exposed cells) and carcinogenic (cancer causing). Those exposed usually suffer no immediate symptoms. Within 4 to 24 hours the exposure develops into deep, itching or burning blisters wherever the mustard contacted the skin; the eyes (if exposed) become sore and the eyelids swollen, possibly leading to conjunctivitis and blindness. At very high concentrations, if inhaled, it causes bleeding and blistering within the respiratory system, damaging the mucous membrane and causing pulmonary edema. Blister agent exposure over more than 50% body surface area is usually fatal.
The NIH awarded a five-year, $7.8 million Center of Excellence grant to National Jewish Health and the University of Colorado Health Sciences Center, both in Denver, Colorado. This Center of Excellence was developed to focus on sulfur mustard toxicity in the lung and skin with the long-term goal to develop an effective treatment for mustard gas induced injury in lung and skin.10150 has been identified by the National Jewish Health Center of Excellence as a lead compound for its center, and research work there has been focused on further testing and studies of 10150.
Research in the area of mustard gas-mediated lung injury has provided experimental evidence that the mechanisms of these injuries are directly linked to the formation of reactive oxygen and nitrogen species and that superoxide dismutase and catalase can improve injury responses. This theory has led to the hypothesis that the administration of catalytic antioxidant therapy can protect against mustard gas-induced acute lung and dermal injury.
Non-clinical studies
55
In July 2013, we announced that four separate studies conducted at USAMRICD using 85 rats and comparing 2 different 10150 dosing regimens conclusively demonstrated that 10150 improves survival against an LD60-70 sulfur mustard gas exposure. 10150 improved sulfur mustard gas survival up to 82% over 48 hours. The improvement in survival seen with 10150 treated animals after sulfur mustard gas exposure correlated with improvements in clinical scores, blood oxygenation and airway obstruction. The best improvements in survival and lung function occurred with the 10150 dosing regimen of 5 mg/kg body weight given every 4 hours by subcutaneous injection (p < 0.0004).
The primary endpoints in these studies were survival and blood oxygen saturation. Secondary endpoints included clinical scores, blood gas and histopathology for cast formations. 10150 or placebo was given to rats one hour after sulfur mustard vapor exposure and repeated every 4 or 8 hours. Forty-eight hours after exposure, lung edema was assessed by changes in the Broncho alveolar lavage (BAL) protein levels. At euthanasia, 48 hours after exposure, 10150 significantly improved (p < 0.0001) pulse oximetry, and 10150 treated rats had improved blood oxygenation throughout the study period. Treatment with 10150 also restored blood gas parameters to near normal levels, including: pO2 (p < 0.001), pCO2 (p < 0.0016) and pH (p < 0.0006). Finally, 10150 treatment reduced airway cast formation by 50% at 24 hours (p < 0.017).
In prior studies 10150 reduced lung edema (p < 0.05), decreased SM-induced increases in macrophages (p < 0.05) and epithelial cells in BAL fluid (p < 0.05). In all three measurements 10150 provided approximately 100 percent protection -- with levels approximating that of the control animals in the study. These results indicate that 10150 significantly improves survival and attenuates lung injury from mustard gas exposure and may provide an effective countermeasure against mustard gas-induced lung injury.
The first whole mustard gas study was completed in October 2009. In June 2010, National Jewish Health and Lovelace Respiratory Research Institute reported results from a second whole mustard study confirming that 10150 protects lungs from whole mustard gas exposure in rats. The two studies demonstrated that10150 protects lungs from whole mustard gas exposure in rats. The primary objective of the study was to determine whether administration of 10150, after exposure, reduces the severity of acute lung injury induced by mustard gas. 10150 was given to rats one hour after sulfur mustard exposure and repeated every 6 hours. Twenty-four hours after exposure, lung edema was assessed by changes in the BAL protein levels. 10150 significantly reduced (p<0.05) mustard gas-induced lung edema as measured by BAL protein levels. In addition, 10150 decreased SM-induced increase in the numbers of BAL neutrophils. These results indicate that 10150 can attenuate lung injury from mustard gas exposure and may provide an effective countermeasure against mustard gas-induced lung injury.
Future Development Plans
We plan to meet with the FDA in 2016 to discuss a pivotal rat study and a second animal model for this indication. We expect to file an IND for the sulfur mustard indication in the second half of 2016. We will also seek to launch the two pivotal efficacy studies required for approval by the FDA under the "Animal Rule" as well as complete the necessary safety studies as further described under the heading "10150 as a potential medical countermeasure against the effects of Pulmonary Acute Radiation Syndrome – Future Development Plans – Demonstrate Product Safety."
Competition
There are currently no effective treatments for mustard gas exposure.
However, in general, we face significant competition for U.S. government funding for both development and procurements of medical countermeasures for biological, chemical and nuclear threats, diagnostic testing systems and other emergency preparedness countermeasures. The U.S. federal government has currently allocated a significant amount of research funding to the development of countermeasures against bioterrorism. As a result, there are many drug candidates under development as a possible countermeasure against chemical threat agents.
56
Funding Options
This development program to date has been funded under the NIH-CounterACT Program and we expect that future efficacy studies necessary for approval by the FDA, including the pivotal rat study and second animal model described above, will be funded by either the NIH-CounterACT program, the Department of Defense or BARDA.
10150 as a potential medical countermeasure against the effects of chlorine gas.
Overview
Chlorine gas is a toxic gas that inflicts airway injury through primary oxidative stress and secondary inflammation. Chlorine inhalation was recently used in terrorist/insurgent attacks on military and civilian populations and has caused numerous industrial, transportation, swimming pool, and household accidents, as well as deaths to members of the U.S. military in the past. Chlorine gas, also known as bertholite, was first used as a weapon in World War I. Chlorine gas was also used against the local population and coalition forces in the first Iraq War in the form of chlorine bombs.
One of the primary goals of the NIH-CounterACT program is to assist in the development of safe and effective medical countermeasures designed to prevent, diagnose, and treat the conditions caused by potential and existing chemical agents of terrorism and which can be added to the SNS. The SNS is maintained by the Centers for Disease Control and Prevention ("CDC"). The SNS now includes CHEMPACKS that are forward-deployed in secure, environmentally controlled areas throughout the United States available for rapid distribution in case of emergency. CHEMPACKS were developed for threats like chemical and nerve gas where treatment must be administered in less than 24 hours and shipment of MCMs from SNS warehouse locations is not practicable. The CDC has established a diagnostic response network for the detection of nerve agents, mustard, cyanide and toxic metals.
Worldwide, independent of warfare and chemical terrorism, chlorine is the greatest single cause of major toxic release incidents (16.Davis DS, Dewolf GB, Ferland KA, et al. Accidental Release of Air Toxins. Park Ridge, New Jersey: NDC; 1989:6-9.). In the U.S., there are about 5-6,000 exposures per year resulting in, on average, about one death, 10 major, 400-500 moderate, and 3-4,000 minor adverse outcomes. Chlorine gas causes damage to upper and lower respiratory tracts. While chlorine is an irritant, its intermediate water solubility may delay emergence of upper airway symptoms for several minutes. No specific beneficial therapies are available. 10150 has demonstrated a decrease in airway injury, inflammation, oxidative damage, hyperreactivity and cell proliferation after acute chlorine gas inhalation in mice, and therefore could be a possible beneficial therapy for chlorine gas inhalation injury to the airways.
Pre-clinical studies
Under a grant from NIH CounterACT, researchers from National Jewish Health and McGill University have completed a series of preliminary studies demonstrating that 10150 protects lungs from chlorine gas exposure in mice and rats. The primary objective of these studies was to determine whether administration of 10150, after exposure, reduces the severity of acute lung injury and asthma-like symptoms induced by chlorine gas. In 2010, 10150 was given to mice at a 5 mg/kg subcutaneous dose one hour after chlorine gas exposure (100 ppm for 5 minutes) and repeated every 6 hours. Twenty-four hours after exposure, lung inflammation was assessed by changes in Broncho alveolar lavage ("BAL") cellularity and neutrophil influx. 10150 significantly reduced (p<0.05, n=6/group) chlorine gas-induced lung inflammation as measured by BAL fluid cellularity levels by 40% that appeared to be due to limiting neutrophil influx. 10150 also significantly attenuated (p<0.05, n=6) the degree of asthma-like airway reactivity induced by chlorine gas exposure by 40%. These results indicate that 10150 can attenuate lung injury and asthma-like symptoms from chlorine gas exposure and may provide an effective countermeasure against chlorine gas-induced lung injury. The data from this study was published in December 2010 as AEOL10150: A novel therapeutic for rescue treatment after toxic gas lung injury by Toby McGovern, Brian J. Day, Carl W. White, William S. Powell and James G. Martin in the journal Free Radical Biology and Medicine.
National Jewish Health replicated the mice studies previously conducted by McGill University in rats to determine whether 10150 mitigates lung damage due to chlorine gas exposure in multiple studies between 2011 and 2013. In these studies, 10150 significantly reduced protein, white blood cell, red blood cell, macrophage and neutrophil counts in Broncho alveolar lavage fluid. Additional studies are ongoing at National Jewish Health.
57
Future Development Plans
We plan to develop a second animal model and to launch the two pivotal efficacy studies required for approval by the FDA under the "Animal Rule." We believe that the safety and CMC work being done under the BARDA Lung-ARS further described under the heading "10150 as a potential medical countermeasure against the effects of Pulmonary Acute Radiation Syndrome – Future Development Plans" will be sufficient to satisfy the safety and CMC requirements for an NDA filing.
Competition
There are currently no effective treatments for chlorine gas exposure.
However, in general, we face significant competition for U.S. government funding for both development and procurements of MCMs for biological, chemical and nuclear threats, diagnostic testing systems and other emergency preparedness countermeasures. The U.S. federal government has currently allocated a significant amount of research funding to the development of countermeasures against bioterrorism. As a result, there are many drug candidates under development as a possible countermeasure against chemical threat agents.
Funding Options
This development program to date has been funded under the NIH-CounterACT Program and we expect that future efficacy studies necessary for approval by the FDA will be funded by the NIH-CounterACT program or BARDA.
AEOL 10150 as a potential medical countermeasure against the effects of nerve gas
Overview
Nerve agents, such as sarin gas, have been used in Syria, Iraq and Japan and pose a threat to civilian and military personnel. Sarin gas exposure can cause pain, weakness, vomiting, diarrhea and changes in heart rate within minutes to 18 hours after exposure. High levels of exposure can cause convulsions, paralysis, breathing problems and death. BARDA classifies nerve agents as a priority threat.
10150 is the focus of a sponsored research grant awarded by the National Institutes of Health (NIH), National Institute of Neurological Disorders and Stroke (NINDS) and Office of the Director (OD) to the University of Colorado to test its potential efficacy as a MCM against nerve agents.
Pre-clinical studies
In September 2013, we announced that researchers, led by Dr. Manisha Patel at the University of Colorado, completed a pilot study demonstrating that 10150 provides neuroprotection, decreases oxidative stress, and significantly improves survival in rats exposed to pilocarpine – a surrogate for the nerve agents soman and sarin gas. These data were presented at the 6th Annual CounterACT Countermeasures Against Chemical Threats Network Research Symposium in Washington, D.C.
The current standard of care for nerve agent exposure is Atropine® and benzodiazepines. In this study 10150 significantly improved the survival of animals exposed to pilocarpine. Injection of 10150 sixty minutes after pilocarpine in the presence of standard therapy resulted in 100% survival and near complete inhibition of oxidative stress indices in the hippocampus.
Future Development Plans
58
On September 10th 2013, NIH-NINDS notified Dr. Patel that a total of $4.3 million had been awarded for her project titled "Neuroprotective Effects of AEOL 10150 against organophosphate toxicity." The research work is being conducted in Dr. Patel's lab at the University of Colorado, National Jewish Health as well as at the USAMRICD. Funding for this research has been awarded under a grant supported by the CounterACT Program, National Institutes of Health Office of the Director (NIH OD), and the NINDS
Competition
The current standard of care for nerve agent exposure is Atropine® and benzodiazepines. 10150 would be added to current standard of care to improve outcomes. We believe that other companies are developing potential treatments for nerve gas, although it is difficult to evaluate their success unless data is announced publicly
However, in general, we face significant competition for U.S. government funding for both development and procurements of medical countermeasures for biological, chemical and nuclear threats, diagnostic testing systems and other emergency preparedness countermeasures. The U.S. federal government has currently allocated a significant amount of research funding to the development of countermeasures against bioterrorism. As a result, there are many drug candidates under development as a possible countermeasure against chemical threat agents.
Funding Options
This development program to date has been funded under the NIH-CounterACT Program and NINDS and we expect that future efficacy studies necessary for approval by the FDA will also be funded by the $4.3 million grant to Dr. Patel.
AEOL 10150 in Radiation Therapy
Overview
According to the American Cancer Society, cancer is the second leading cause of death by disease, representing one out of every four deaths in the United States. Approximately 572,000 Americans were expected to die of cancer in 2011. In 2012, about 1.6 million new cancer cases were expected to be diagnosed in the United States. According to the Radiological Society of North America, about 50 to 60 percent of cancer patients are treated with radiation at some time during their disease.
Combinations of surgery, chemotherapy and radiation treatments are the mainstay of modern cancer therapy. Success is often determined by the ability of patients to tolerate the most aggressive, and most effective, treatment regimens. Radiation therapy-induced toxicity remains a major factor limiting radiation doses. The ability to deliver maximal radiation doses for treatment of tumors without injury to surrounding normal tissue has important implications in oncology therapeutic outcomes because higher doses of radiation therapy may improve both local tumor control and patient survival.
Advances in the tools of molecular and cellular biology have enabled researchers to develop a better understanding of the underlying mechanisms responsible for radiation therapy-induced normal tissue injury. For decades ionizing radiation has been known to increase production of free radicals, which is reflected by the accumulation of oxidative damaged cellular macromolecules.
As one example of radiation-induced damage to adjacent normal tissue, radiation therapy may injure pulmonary tissue either directly via generation of ROS or indirectly via the action on parenchymal and inflammatory cells through biological mediators such as TGF-β and pro-inflammatory cytokines. Since the discovery of SOD, it is apparent that these enzymes provide an essential line of defense against ROS. SODs and SOD mimics, such as 10150, act by catalyzing the degradation of superoxide radicals into oxygen and hydrogen peroxide. SODs are localized intra/extracellularly, are widely expressed throughout the body, and are important in maintenance of redox status (the balance between oxidation and reduction). Previous studies have demonstrated that treating irradiated animal models with SOD delivered by injection of the enzyme through liposome/viral-mediated gene therapy or insertion of human SOD gene can ameliorate radiation therapy-induced damage. For an illustrative example of the radiation therapy reaction see Figure 2.
59
Figure 2
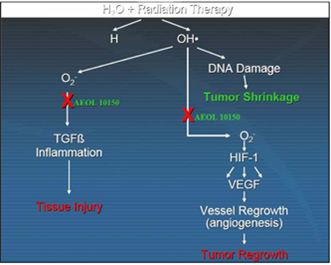
Figure 2 above shows the dual mechanism of action of radiation therapy and the application of 10150 to the process.
In vitro studies have demonstrated that 10150 reduces the formation of lipid peroxides, forms of ROS, and that it inactivates biologically important ROS molecules such as superoxide, hydrogen peroxide and peroxynitrite. 10150 inactivates these ROS by one or two electron oxidation or reduction reactions in which the oxidation state of the manganese moiety in 10150 changes. 10150 is not consumed in the reaction and it continues to inactivate such ROS molecules as long as it is present at the target site. Preclinical models and human safety studies suggest 10150 is not metabolized in the body and is excreted in feces and urine.
Pre-clinical studies
Figure 3
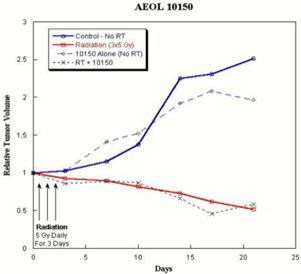
60
Figure 3: Relative tumor volumes of human prostate tumor implants in nude mice: Implants of well-vascularized PC3 tumors were grown to substantial size prior to receiving fractionated radiation (5 Gy daily for three days).10150 (7.5 mg/kg/bid) was administered subcutaneously commencing on the first day of irradiation and continued for 20 days. Other groups of mice received either no irradiation, irradiation only or 10150 without irradiation.
Due to the similar mechanisms of actions between radiation therapy (in oncology) and radiation exposure (from nuclear events), we believe that the pre-clinical studies performed for the development of 10150 as a potential medical countermeasure against the effects of Lung-ARS, as described herein, also provide support for the development of 10150 in oncology, to be used in combination with radiation therapy.
We have performed several additional studies specifically for this indication to ensure the use of an antioxidant in radioprotection of normal adjacent tissue does not interfere with the efficacy of tumor radiotherapy. A number of preclinical, in vivo studies have addressed this issue and have demonstrated that 10150 does not negatively impact tumor radiotherapy.
In one study (Vujaskovic, et al. of Duke University), human prostate tumors (PC3) grown in nude mice to substantial size were irradiated with 5 Gy per day for 3 days for a total of 15 Gy. 10150 at 7.5 mg/kg/bid was administered subcutaneously on the first day of radiation and continued for either of two time courses: when tumor volume reached 5 times the initial volume or for twenty days. The receding tumor volume curves for irradiation only and for irradiation plus 10150 demonstrated no difference. Therefore 10150 did not interfere with the radiation effect on prostate tumors.
In another study of prostate cancer tumors (Gridley, et al of Loma Linda University), mouse prostate cancer cell line RM-9 was injected subcutaneously into C57/Bl6 mice, followed by up to 16 days of 10150 delivered intraperitoneally at 6 mg/kg/day. On day seven, a single dose of radiation (10 Gy) was delivered. Therefore, the mice received compound for seven days prior to radiation. The results of this study demonstrated that 10150 does not protect the prostate tumor against radiation, however, 10150 showed a trend towards increasing the effectiveness of the radiation treatment. The primary effect appears to be in down-regulation of radiation induced HIF-1 expression and VEGF and up-regulation of IL-4. Thus, 10150, through its down-regulation of VEGF, may inhibit formation of blood vessels (i.e., angiogenesis) required for tumor re-growth and protects normal tissues from damage induced by radiation and chemotherapy.
In another study (Vujaskovic, et al. of Duke University), mice were implanted with human NSCLC tumors and treated with all potential combinations of paclitaxel, radiation and 10150 to determine the impact on tumor growth. The results showed that 10150 did not impact the effects of either radiation therapy or paclitaxel. Further, the study indicated that the greatest impact in inhibiting tumor growth was with the regimen that included all three courses of treatment, radiation, paclitaxel and10150.
61
Figure 4
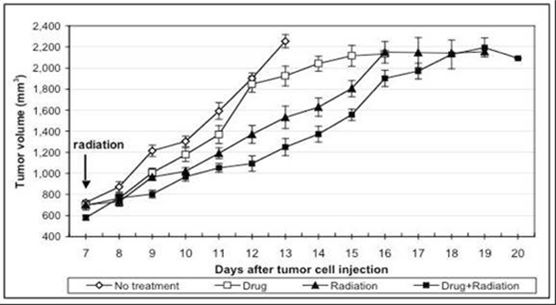
Figure 4 above measures tumor volume against time after implantation of RM-9 tumor cells and shows that 10150 treatment resulted in inhibition of tumor re-growth in a study performed by Dr. Gridley of Loma Linda University. Daily intraperitoneal injections of 10150 were initiated on day 1. At 12 days, approximately one half of each tumor-bearing group and control mice with no tumor were euthanized for in vitro analyses; remaining mice/group were followed for tumor growth and euthanized individually when maximum allowed tumor volume was attained. Each point represents the mean +/- standard error of the mean. Two-way analysis of the variance for days 8 to 14 revealed that group and time had highly significant main effects (Ps<0.001) and a group x time interaction was noted (P<0.001).
Figure 5
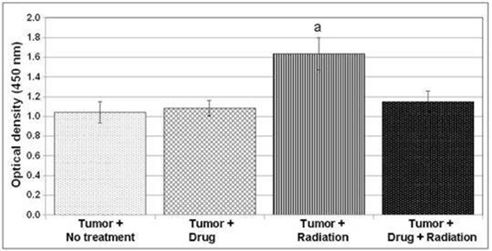
Figure 5 above shows the HIF-1 Expression in prostate tumors and the impact of the treatment of 10150 in a study by Dr. Gridley of Loma Linda University.
62
Figure 6
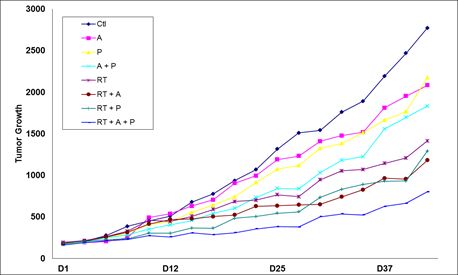
Figure 6 above shows impact on tumor growth in mice that were implanted with human NSCLC tumors and treated with all potential combinations of paclitaxel (P), Radiation (RT) and 10150 (A).
In summary, the data obtained in these preclinical studies suggest that the post-irradiation, long-term delivery of 10150 may be protective against radiation-induced lung injury, as assessed by histopathology and immunohistochemistry. Oxidative stress, inflammation and hypoxia, which play important roles in the pathogenesis of radiation mediated fibrosis, were shown to be reduced in animals treated with higher doses of 10150. Studies have also shown that 10150 does not adversely impact tumor response to radiation therapy. Thus, treatment with 10150 does not significantly protect tumors from the cell killing effects of radiation therapy. This combined with other studies that have shown that 10150 significantly prevents radiation induced normal tissue injury suggests that 10150 has the potential to achieve protection of normal tissue without protection of tumor tissue. Additionally, it appears the down-regulation of radiation induced HIF-1 expression and VEGF and up-regulation of IL-4 may provide additional anti-tumor effects. Thus, 10150, through its down-regulation of VEGF, may inhibit formation of blood vessels required for tumor re-growth, while protecting normal tissues from damage induced by radiation and chemotherapy.
Future Development Plans
We are leveraging the significant investment made by U.S. government agencies to develop this promising compound for use in oncology indications, where it would be used in combination with chemotherapy and radiation therapy, and is currently in development for use as both a therapeutic and prophylactic drug. Data has already been published showing that 10150 does not interfere with the therapeutic benefit of radiation therapy in prostate and lung cancer preclinical studies.
In 2015, we expect to initiate a safety study in non-small cell lung cancer patients receiving chemotherapy and radiotherapy. Upon the successful completion of the Phase I study and approval of a protocol by the FDA and the appropriate IRBs, we expect to begin a Phase II study in cancer patients receiving radiation therapy. A specific indication for the phase 2 study has not yet been chosen, however, we are considering esophageal, prostate, head and neck and NSCLC patients.
Competition
There are currently three drugs approved for the treatment of the side effects of radiation therapy. We do not believe that any of these drugs directly competes with 10150 in terms of mechanism of action or targeted therapeutic benefit when used in combination with radiation therapy.
63
Amifostine (Ethyol®) is approved by the FDA as a radioprotector. Amifostine (Ethyol) is marketed by MedImmune, Inc. for use in reduction of chemotherapy-induced kidney toxicity associated with repeated administration of cisplatin in patients with advanced ovarian cancer and radiation-induced xerostomia (damage to the salivary gland) in patients undergoing post-operative radiation treatment for head and neck cancer. MedImmune, Inc. is studying Amifostine in other indications of radiation therapy. KepivanceTM (palifermin) is marketed by Amgen, Inc. for use in the treatment of severe oral mucositis (mouth sores) in patients with hematologic (blood) cancers who are undergoing high-dose chemotherapy followed by bone transplant. Amgen, Inc. is also studying Kepivance as an antimucositis agent in patients with head and neck cancer, non-small cell lung cancer and colon cancer. Salagen Tablets (pilocarpine hydrochloride) is marketed by Eisai Pharmaceuticals in the United States as a treatment for the symptoms of xerostomia induced by radiation therapy in head and neck cancer patients. In addition, there are many drugs under development to treat the side effects of radiation therapy.
Funding Options
Substantially all of our costs associated with the CMC and toxicology necessary for the oncology indications have been covered by the BARDA Contract. We expect such costs to continue to be covered by the BARDA Contract unless BARDA chooses not to exercise its options under the BARDA Contract. In such a circumstance, we would need to raise additional capital, or partner with another firm, in order to complete the non-clinical and safety programs related to Lung-ARS noted above. We will need to internally fund the human efficacy programs in oncology, as well as any non-clinical studies that may be necessary for specific oncology indications. We expect that a portion of the net proceeds from the 2015 Securities Placement will be used to fund part of the future costs of this program that are not funded by the BARDA Contract, including funds for an initial trial in human patients receiving therapeutic radiation for cancer. We may still raise capital through other sources even if BARDA exercises additional options under the BARDA Contract.
10150 Clinical Development Program to Date
10150 has been tested for safety, tolerability and pharmacokinetics with no serious or clinically significant adverse effects observed. To date, 38 patients have received 10150 in three clinical trials designed to test the safety and tolerability of the drug candidate.
In September 2005, we completed a multi-center, double-blind, randomized, placebo-controlled, Phase I clinical trial. This escalating-dose study was conducted to evaluate the safety, tolerability and pharmacokinetics of 10150 administered by twice daily subcutaneous injections in patients with ALS.
In the Phase Ia study, 4-5 patients diagnosed with ALS were placed in a dosage cohort (3 or 4 receiving 10150 and 1 receiving placebo). Each dose cohort was evaluated at a separate clinical center. In total, seven separate cohorts were evaluated in the study, and 25 ALS patients received 10150. Based upon an analysis of the data, it was concluded that single doses of 10150 ranging from 3 mg to 75 mg were safe and well tolerated. In addition, no serious or clinically significant adverse clinical events were reported, nor were there any significant laboratory abnormalities. Based upon extensive cardiovascular monitoring (i.e., frequent electrocardiograms and continuous Holter recordings for up to 48 hours following dosing), there were no compound-related cardiovascular abnormalities.
The most frequently reported adverse events in this Phase I clinical trial were injection site reactions, followed by dizziness and headache. Adverse events were primarily mild in severity, and approximately one-half of the events were considered to have a possible relationship to the study medication. In addition, no clinically meaningful findings were noted in the safety, laboratory, vital sign, the Unified Parkinson's disease Rating Scale ("UPDRS"), functional ALS, or electro cardiogram ("ECG") data. All cohorts exhibited dose-related peak plasma drug concentrations and consistent disappearance half-lives.
In October 2006, we completed a multi-center, double-blind, randomized, placebo-controlled, Phase Ib clinical trial. This multiple dose study was conducted to evaluate the safety, tolerability and pharmacokinetics of 10150 administered by subcutaneous injection and infusion pump in patients with ALS. Under the multiple dose protocol, three groups of six ALS patients (four receiving 10150 and two receiving placebo) were enrolled.
64
The Phase Ib study was conducted at five academic clinical ALS centers. Male and female ALS patients, 18 to 70 years of age, who were ambulatory (with the use of a walker or cane, if needed) and capable of orthostatic blood pressure assessments were enrolled in the study. Clinical signs/symptoms, laboratory values, cardiac assessments and pharmacokinetics (PK) were performed.
Based upon an analysis of the data, it was concluded that multiple doses of 10150 for a period of six and one half consecutive days in the amount of 40 mg per day, 60 mg per day and 2 mg per kilogram per day were safe and well tolerated. No serious or clinically significant adverse events were reported or observed. The most frequent adverse events related to study compound were injection site observations related to compound delivery. There were no significant laboratory abnormalities. Based upon extensive cardiovascular monitoring (i.e., frequent electrocardiograms and continuous Holter recordings throughout the six and one half days of dosing), there were no compound-related cardiovascular abnormalities.
Pharmacokinetic findings from the Phase Ib study to data are as follows:
| · | Increases in Cmax and AUC (0-8) appear to correlate with increases in dose, but the correlation is not strong. |
| · | The mean Cmax for the 40 mg cohort was 1,735 ng/mL; 2,315 ng/mL for the 60 mg cohort and 1,653 ng/mL for the 2 mg/kg cohort. |
| · | There were probable linear correlations between both Cmax and AUC(0-8) and dose based on body weight. |
| · | The terminal half-life (a measurement of the time period for which a compound stays in the body) as determined from Day 7 data was approximately 8 to 9 hours. |
| · | Steady-state occurs within three days of multiple dosing. There was no evidence for a third longer half-life that would be associated with long term accumulation. Thus, compound accumulation is not expected beyond the third day with multiple dosing. |
| · | From 48 hours to the end of the infusion, the plasma concentrations of 10150 during the infusion showed little variability, indicating a smoother delivery of the drug than with twice-daily injections. |
During 2008, we completed a follow-on Phase I open label compassionate use multiple dose study of 10150 in a patient diagnosed with progressive and debilitating amyotrophic ALS. The study was conducted at the University of California, Los Angeles by Martina Wiedau-Pazos, M.D., and was designed to evaluate the safety and efficacy of 10150 in an ALS patient over an extended period of time. The patient received a subcutaneous injection of 75mg of 10150 two times each day for 34 days. Efficacy and safety data was monitored for the duration of the study. The primary objective of this study was to assess the clinical efficacy of 10150 with respect to the patient's baseline assessment of functional status. Secondary objectives included the assessments of muscle strength, respiratory function, quality of life and safety. The patent's baseline efficacy results were an ALS Functional Rating Scale ("ALSFRS-R") rating of 19, Muscle strength Manual Muscle Testing Scale ("MMTS") of 68 and a forced vital capacity ("FVC") of 30%. The patient's results after 2 months were an ALSFRS-R rating of 22, a MMTS rating of 86 and an FVC of 28%. It should be noted that the subject began using breathing assistance (BiPAP) approximately two weeks after the study started. The patient discontinued treatment due to nausea and moderately increased liver transaminases. Other drug-associated adverse events included mild skin irritation at the injection site and mild urine discoloration.
AEOL 10150 in Idiopathic Pulmonary Fibrosis
Overview
Idiopathic pulmonary fibrosis ("IPF") is a progressive, fibrosing interstitial pneumonia of unknown cause characterized by the development of scar tissue that thickens the lining of the lungs and generally leads to death due to inhibition of lung function. IPF affects approximately 128,000 people in the United States with 48,000 new cases and 40,000 deaths. It usually affects older individuals, those in the sixth to seventh decade, and is a severe disease with a variable clinical course and a high mortality rate. Approximately 2/3 of IPF sufferers will die within 5 years of diagnosis. The median survival after diagnosis is 3 to 4 years.
65
Standard of care for IPF is treatment with anti-inflammatory drugs such as corticosteroids although there is no evidence that these treatments are effective in increasing long-term survival. Supplemental oxygen may improve patient quality of life, but has no effect on the course of the disease. In some instances, lung transplantation may be considered. On October 15, 2014, the FDA approved Esbtriet® (pirfenidone), manufactured by Intermune, and Ofev® (nintedanib), manufactured by Boehringer Ingelheim, for the treatment of IPF. We believe that there is still a significant need for additional and more effective treatments for IPF.
Pre-clinical Studies
Whole Thorax Irradiation ("WTLI") is a validated model for IPF in animals. The efficacy studies under the BARDA Contract in monkey and mice were conducted using a WTLI model. Those studies demonstrated a significant reduction in inflammation and fibrosis in animals treated with AEOL 10150. Additionally, during 2015, researchers at National Jewish Health conducted efficacy studies of 10150 using a Bleomycin induced fibrosis model. These studies also demonstrated a significant reduction in fibrosis in animals treated with 10150, and provided further support for the drug's potential as a therapy for patients with pulmonary fibrosis, including IPF.
Development Plans
In March 2015, the Company received notice from the Office of Orphan Products Development at the U.S. Food & Drug Administration (FDA) granting Orphan Drug Designation for AEOL 10150 "for treatment of idiopathic pulmonary fibrosis." Orphan Drug Designation entitles the sponsor to a seven-year marketing exclusivity period, clinical protocol assistance with the FDA, as well as federal grants and tax credits. In October 2015, the Company announced the formation of a Pulmonary Fibrosis Clinical Advisory Board ("PFCAB") led by Kevin Brown, MD, Professor and Vice Chair, Department of Medicine, at National Jewish Health. Upon resolution of the clinical hold with the FDA for the Lung ARS indication, the Company plans to meet with the Respiratory division of the FDA and file an IND to initiate phase 1 studies of 10150 in patients with pulmonary fibrosis.
Competition
Esbriet® and Ofev® were approved by the FDA for the treatment of IPF on October 15, 2014. Esbriet® and Ofev® are also approved for use in Europe, Canada, Japan, South Korea and other countries. The effectiveness of Esbriet® and Ofev® are, however, limited and we believe that there is still a significant need for more effective treatments. There are a number of potential treatments for IPF in advanced development by multiple biotechnology and pharmaceuticals. Many of these potential treatments are monoclonal antibody therapies. We believe that 10150 exploits a novel mechanism of action in the treatment for IPF. In general, however, we believe that there will be substantial, well-funded competition for the development of new IPF treatments.
Funding
Although the initial data supporting a program in IPF was largely generated under the BARDA Contract, we are uncertain as to whether funding for human safety and efficacy studies will be available from that source. However, as with radiation oncology, manufacturing, toxicology and safety work under the contract can also be used in the development of 10150 for IPF. We expect that a portion of the net proceeds from the 2015 Securities Placement will be used to fund part of the future costs of this program that are not funded by the BARDA Contract, including funds for an initial trial in patients with IPF. The Company will also seek non-dilutive capital sources and pursue potential partnership opportunities to advance the IPF program.
AEOL 11114B, AEOL 11203 and AEOL 11207 in Parkinson's' Disease
Overview
We have evaluated three compounds, 11114, AEOL 11203 ("11203") and AEOL 11207 ("11207"), for the treatment of Parkinson's disease with funding from MJFF. Parkinson's disease is a common neurodegenerative disorder, second in occurrence among these disorders only to Alzheimer's disease. According to the National Parkinson Foundation, Parkinson's affects as many as one million people in the United States, with approximately 60,000 new cases diagnosed in the United States each year.
66
Parkinson's specifically involves the progressive destruction of the nerves that secrete dopamine and control the basal ganglia, an area of the brain involved in the regulation of movement. Dopamine turnover has been shown to elevate the levels of ROS in the brain. In addition, a street-drug contaminant has appeared that can cause Parkinsonism in drug abusers. The compound N-methyl-4-phenyl-1, 2, 3, 6tetrahydropyridine ("MPTP") has been identified in underground laboratory preparations of a potent analog of meperidine (Demerol). MPTP-containing powder, sometimes sold as a new "synthetic heroin," can be dissolved in water and administered intravenously or taken by the intranasal route. MPTP has been documented to produce irreversible chronic Parkinson symptoms in drug abusers. Agents such as MPTP overproduce ROS in the basal ganglia. Therefore, ROS mediated neuronal dysfunction may play a key role in the development of Parkinson's disease. Symptoms of this disease include tremors, rigidity and bradykinesia (i.e., slowness of movement). In the more advanced stages, it can cause fluctuations in motor function, sleep problems and various neuro-psychiatric disorders. A biological hallmark of Parkinson's disease is a reduction in brain dopamine levels. Preventing or slowing the destruction of brain cells that lead to the depletion of dopamine levels in the brain is an important therapeutic approach for the treatment of this disease.
Pre-clinical studies
Data developed by our scientists and Dr. Manisha Patel at the University of Colorado Health Sciences Center and Department of Medicine, indicate that when administered orally, 11207 is greater than 80% bioavailable, meaning that it is readily absorbed and reaches both the circulatory system and the brain in sufficient amounts to demonstrate biological activity. Data developed with 11207 in a widely used animal model of Parkinson's disease (the "MPTP model") showed that when administered orally, 11207 crosses the blood brain barrier and protected dopamine neurons in a dose-dependent manner. Further data suggest that the compound has a half- life (a measurement of the time period for which a compound stays in the body) of about 3 days in both the circulatory system and the brain, and that prior to stopping administration of the compound, the levels of 11207 in both the circulatory system and brain reach a steady state (a valuable measurement of when the levels of the drug in the body remain substantially constant, neither increasing nor decreasing) after 2 days of dosing. Data have also been developed that indicate that when dosing of 11207 is stopped, the compound is excreted from the body.
In September 2010, Manisha Patel of the University of Colorado was informed by the Michael J. Fox Foundation that she had been awarded a supplement to her grant for the synthesis of additional quantities of 11114 and 11203; for the completion of the evaluation of 11114 and 11203's effects on MPTP toxicity (TH+ cells in substantia nigra), and behavioral testing and accumulation of manganese after chronic dosing.
Dr. Patel's research under the supplement established the maximum tolerated dose, pharmacokinetics, dosing paradigm, drug stability, and drug protein binding and efficacy for 11114. The data from the efficacy work performed show that daily or every other day dosing with the compound protects against oxidative stress, improves performance on behavioral tests, and mitigates dopamine depletion in the 6-hyroxydopamine model of Parkinson's disease in restorative manner. These results validate 11114's efficacy in the MPTP mouse model and suggest that the drug can be given after symptom onset after a 6-hydorydopamine lesion in rats to achieve neuroprotection. Further, administration of 11114 every other day reduced oxidative stress and protected against behavioral deficits and loss of striatal dopaminergic neurons in the Substantia Nigra Pars Compacta ("SNPc"). Parkinson's disease is characterized by the death of neurons in the SNPc. Currently available treatments for Parkinson's lessen the symptoms for some time, but there remains a great need for more effective therapies, such as 11114, that have shown the potential to protect against the loss of dopaminergic neurons. 11114 readily crosses the blood brain barrier, and daily or every other day treatment prevents oxidative stress and protects against oxidative stress, neuron loss and behavioral deficits in both a rat and mouse model of Parkinson's disease.
Prior to receiving the funding for this program, we filed a new composition of matter and use patent for AEOL11114. In October 2015, the Company received notice of allowance of a composition of matter patent from the European Patent Office.
67
Future Development Plans
We plan to initiate the necessary pre-clinical work in the first quarter of calendar 2016 that will allow us to file an IND and initiate Phase 1 studies in Parkinson's disease in 2017
Funding Options
We expect that a portion of the net proceeds from the 2015 Securities Placement will be used to fund part of the future costs of this program, including amounts to fund the pre-clinical work necessary to file an IND for 11114. We will also seek non-dilutive capital sources and pursue potential partnership opportunities to fund the human clinical studies of 11114 for Parkinson's disease.
Contracts and Grants
We seek to advance development of our drug candidates through external funding arrangements. We may slow down development programs or place them on hold during periods that are not covered by external funding. We have received external funding awards for the development of 10150 as an MCM for Lung-ARS, GI-ARS, mustard gas and chlorine gas exposure from the NIH.
BARDA Contract; Background and Recent Developments
In December 2009, we were informed by BARDA that we had been chosen to submit a full proposal for funding of our Lung-ARS program from its current stage through FDA approval, based on a summary "white paper" submitted by us earlier in 2009. We submitted a full proposal in February 2010. We were notified in July 2010 that our proposal had been chosen by BARDA, and then entered into negotiations for a development contract with the agency.
On February 11, 2011, we signed a five-year, cost-plus contract with BARDA for the development of 10150 as a MCM against Lung-ARS. BARDA is the government agency responsible for the advanced development and purchase of medical countermeasures for chemical, biological, radiological and nuclear threats. The contract fully funds the advanced development of 10150 through approval by the FDA under 21 CFR Part 314 Subpart I and Part 601 Subpart H (the "Animal Rule.") The Animal Rule allows for approval of drugs using only animal studies when human clinical trials cannot be conducted ethically.
We were awarded approximately $10.4 million in the base period of the contract. On April 16, 2012, we announced that BARDA had exercised two options under the BARDA Contract worth approximately $9.1 million. On September 17, 2013, we announced that BARDA had exercised $6.0 million in additional contract options. On May 07, 2014, we announced that BARDA had exercised a Contract Modification worth approximately $1.8 million. On June 26, 2015, we announced that BARDA had exercised $3.0 million in additional contract options under its advanced research and development contract for AEOL 10150. The June option exercise brings the total exercised contract value to date to approximately $30.3 million. We may receive up to an additional $88.1 million in options exercisable over the remaining years of the contract. If all of the options are exercised by BARDA, the total value of the contract would be approximately $118.4 million.
Activities under the contract to date include animal efficacy studies, animal model development with radiation survival curve studies, dosing studies, bulk drug manufacturing, bulk drug and final drug product manufacturing, validation testing, compliance studies, stability studies, ADME and metabolic studies, genotoxicity studies and the filing of an orphan drug status application and a fast track designation application with the FDA.
Following the commencement of the BARDA Contract, we entered into a series of agreements with various parties in furtherance of our efforts under the BARDA Contract, which are described below.
On February 18, 2011, we entered into a Research and Manufacturing Agreement with Johnson Matthey Pharmaceutical Materials, Inc. (d/b/a Johnson Matthey Pharma Services) ("JMPS"), pursuant to which we engaged JMPS to, among other things, assess and develop a reliable separations or manufacturing process for certain chemical compounds as required by us and to perform such additional work as may be required or agreed upon by the parties and to manufacture compounds for us. Each project performed by JMPS under the agreement will have a detailed project description and separate fee agreement based on the nature and duration of the project and the specific services to be performed by JMPS. The term of the agreement with JMPS will continue until February 10, 2016 or the date on which all projects under the agreement have been completed or terminated.
68
On February 14, 2012, the Aeolus team presented the results and deliverables that had been produced during the first twelve months under the base period of the BARDA Contract at an "In-Progress Review" meeting with BARDA, and requested the exercise of additional contract options, which contain additional key items required in the advanced development of 10150.
On February 15, 2012, we announced that we entered into a contract modification and no-cost extension with BARDA. The modification and extension allowed us to continue operating under the base period of the contract awarded in February 2011, and restructured the timing and components of the options that could be awarded under the remaining four years of the agreement. The changes did not impact the total potential value of the contract, which remains at approximately $118.4 million. The contract restructure was driven by our ability to generate cost savings in the base year contract, and to allow BARDA to better manage contract options to expedite development program.
On April 16, 2012, we announced that BARDA had exercised two contract options worth approximately $9.1 million. BARDA's exercise of the options was in response to the presentation of the deliverables and progress made under the contract at the meeting on February 14, 2012. Among the key items in the options BARDA exercised are animal efficacy studies, mechanism of action research and manufacturing and process validation work. All of these items build off of work successfully completed during the first twelve months of the contract base period. The contract is designed to produce the data necessary for an approval under the FDA "Animal Rule" and for a potential Emergency Use Authorization (EUA). An approval or EUA would allow the federal government to buy 10150 for the Strategic National Stockpile under Project Bioshield. Project Bioshield is designed to accelerate the research, development, purchase and availability of effective medical countermeasures for the Strategic National Stockpile.
On November 7, 2012, we and the Office of Research and Development of the University of Maryland, Baltimore ("UMB") entered into a Sub-award Agreement, pursuant to which we engaged UMB to, among other things, perform mouse studies supporting the licensure of 10150. Prior to this agreement, our mouse studies had been conducted at Duke University. In 2012, the research team at Duke responsible for conducting the studies moved to UMB. The Sub-award Agreement is a fixed fee agreement inclusive of all direct and indirect costs. As a result of the contract modification and no-cost extension with BARDA mentioned above, the term of the Sub-award Agreement will continue through at least February 10, 2016.
On July 29, 2013, Aeolus presented the results and deliverables that had been produced during the first 28 months of the contract at an "In-Progress Review" meeting with BARDA, and requested the exercise of additional contract options.
On September 17, 2013 we announced that BARDA had exercised $6.0 million in additional options under the contract. The options that BARDA exercised will fund our IND filing for AEOL-10150 as a treatment for Lung-ARS, additional animal efficacy studies designed to optimize timing and duration of dosing and the continued development of large-scale GMP manufacturing capability to meet potential future demand. When combined with our ongoing studies in non-human primates and our completed work in GMP manufacturing development, these options will help Aeolus meet the requirements for a pre-EUA filing for AEOL-10150 in 2016.
On May 07, 2014, we announced that BARDA had exercised a Contract Modification worth approximately $1,778,000. The Contract Modification allowed Aeolus to reconcile actual costs incurred with billings under the original provisional indirect billing rate. It established a new provisional indirect billing rate and placed a cap on the current and future provisional indirect billing rates.
On September 4, 2014 we announced positive results from a large study in rhesus macaque monkeys funded by the BARDA Contract. In the study, 10150 was administered beginning 24 hours after exposure to 10.74 Gray of whole thorax radiation. Survival at six months post-exposure in the 60-day treatment group was 50%, compared to 25% survival in the radiation only group. On May 4, 2015, we reported the complete results, including secondary endpoints, from the same study. The study found that administration of 10150 for 60 days beginning 24 hours after exposure to 10.74 Gy of radiation:
69
| · | Increased overall survival from 25% in the untreated control group to 50% |
| · | Increased mean and median overall survival time |
| · | Increased mean and median survival time in subjects that did not survive to 180 days |
| · | Increased time to onset of increased respiratory rate, a clinical measure of lung injury |
| · | Decreased mortality in subjects with elevated respiratory rate |
| · | Decreased wet lung weight in all animals, suggesting less parenchymal damage and edema |
| · | Increased Sp02, a measure of compensated lung function |
| · | Diminished radiographic evidence of pneumonitis and fibrosis during the later stages of the study (days 90 -180) |
In addition, a new approach to investigating lipids, metabolites and proteins in pathophysiological process, matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) was employed in the study to measure potential biomarkers for lung injury in tissue samples from study subject. Analysis using MALDI-MSI showed that the molecular profile for the naïve (un-irradiated) lung is statistically distinct from irradiated lung and treatment with 10150 shifts the molecular profile back towards the naïve lung. Two prospective biomarkers found in irradiated, damaged lung tissue were not detectable in either naïve or 10150-treated samples.
On September 22, 2014, we received notice from the FDA that the Phase I safety study in healthy normal volunteers proposed in its IND for Lung-ARS had been placed on clinical hold. This notice will delay BARDA funding for the clinical study and its initiation until the FDA has reviewed the Company's response and removed the clinical hold.
On April 23, 2015, the Company executed a Modification of Contract (the "Modification") with BARDA. The purpose of the Modification is to add funding in the amount of $1,021,000 for the purpose of funding ongoing murine efficacy, pharmacokinetic and ADME studies and project management.
On June 26, 2015 we announced that BARDA had exercised one option under the BARDA Contract worth approximately $1,989,000 for the purpose of funding laboratory work suggested by the FDA as a pathway to removing the clinical hold on the IND. The funds will also support ongoing project management.
As of September 30, 2015, the total contract value exercised by BARDA under the BARDA Contract is $30.3 million.
Aeolus' Anti-Inflammatory/Anti-Infective Program
We established this program to explore to create novel compounds that both reduce inflammation and combat anti-biotic resistant bacteria, with the goal of addressing unmet medical needs in commercial applications, such as cystic fibrosis, while also potentially providing novel approaches to treating major threats to public health from drug resistant bacteria such as burkholderia cepacia ("BC"), pseudomonas aeruginosa ("PA") and others listed as "Biggest Threats" by the Center for Disease Control and Prevention. We have achieved our initial research objectives and begun to extend our preclinical accomplishments into non-clinical studies, clinical trials and drug development programs.
Our anti-inflammatory/anti-infective program is designed to:
| · | Utilize a selective detoxification pathway that preserves host defense and limits inflammation |
| · | Create and develop stable, small molecules so that they: |
| o | Maintain pathogen killing with less off target damage, |
| o | Provide faster resolution of inflammation, |
| o | Decrease irreversible injury and oxidative stress, and |
| o | Produce candidates that are orally available, stable and inexpensive to manufacture. |
70
Our most advanced anti-infective compound, AEOL 20415, is an orally-available, small molecule that has shown the ability to reduce inflammation and to kill bacteria. A patent application for 20415 was filed in April 2015, and in-vitro studies have been completed to demonstrate 20415's activity against BC and PA. A mouse study testing 20415's anti-infective and anti-inflammatory ability should be completed in the first quarter of 2015. If the data confirms the positive in-vitro activity, we intend to initiate the pre-clinical work necessary to file an IND for this compound with an initial indication of Cystic Fibrosis. We expect that a portion of the net proceeds from the 2015 Securities Placement will be used to fund the pre-clinical work necessary to file an IND for 20415. We will also seek non-dilutive capital sources and pursue potential partnership opportunities to fund the human clinical studies of 20415.
Collaborative and Licensing Arrangements
Duke Licenses
Pursuant to our license agreements with Duke, we have obtained exclusive worldwide rights from Duke to products using antioxidant technology and compounds developed by Dr. Irwin Fridovich and other scientists at Duke. The license from Duke covers, among other items, AEOL11203, AEOL11207 and some of the intellectual property related to 10150. We are obligated under the licenses to pay Duke royalties ranging in the low single digits of net product sales during the term of the Duke licenses, and we must make payments upon the occurrence of certain development milestones in an aggregate amount of up to $2,000,000. In addition, we are obligated under the Duke licenses to pay patent filing, prosecution, maintenance and defense costs. The Duke licenses are terminable by Duke in the event of breach by us and otherwise expire when the last licensed patent expires.
National Jewish Medical and Research Center and National Jewish Health
We have obtained an exclusive worldwide license from the National Jewish Medical and Research Center ("NJMRC") to develop, make, use and sell products using proprietary information and technology developed under a previous Sponsored Research Agreement within the field of antioxidant compounds and related discoveries. The license from NJMC covers, among other items, the composition of matter for 10150 and some use patents related to alkylating and vesicant agents. We must make milestone payments to the NJMRC in an aggregate amount of up to $250,000 upon the occurrence of certain development milestones. Our royalty payment obligations to the NJMRC under this license agreement are in the low single digits of net product sales. We are also obligated to pay patent filing, prosecution, maintenance and defense costs. This NJMRC license agreement is terminable by the NJMRC in the event of breach and otherwise expires when the last licensed patent expires.
In 2009, we obtained an additional exclusive worldwide license from National Jewish Health to develop, make, use and sell products using proprietary information and technology developed at NJH related to certain compounds as an MCM against mustard gas exposure. Under this license agreement, we must make milestone payments to NJH in an aggregate amount of up to $500,000 upon the occurrence of certain development milestones. In addition, we must make royalty payments to NJH under this license agreement ranging in the low-single digits as a percentage of all sublicensing fees, milestone payments and sublicense royalties that we receive from sublicenses granted by us pursuant to this license agreement. We are also obligated to pay patent filing, prosecution, maintenance and defense costs. This NJH license agreement is terminable by NJH in the event of breach and otherwise expires when the last licensed patent expires.
In April 2015, we obtained an exclusive worldwide license from National Jewish Health to develop, make, use and sell products using proprietary information and technology developed at NJH with funding from the Company for the treatment of infection caused by bacterial, viral and fungal pathogens. Under this license agreement, we must make milestone payments to NJH in an aggregate amount of up to $1,000,000 upon the occurrence of certain development milestones. In addition, we must make royalty payments to NJH under this license agreement ranging in the low-single digits as a percentage of all net sales, sublicensing fees, milestone payments and sublicense royalties that we receive from sublicenses granted by us pursuant to this license agreement. We are also obligated to pay patent filing, prosecution, maintenance and defense costs. This NJH license agreement is terminable by NJH in the event of breach and otherwise expires when the last licensed patent expires.
71
University of Colorado
In April 2015, we obtained an exclusive worldwide license from UC to develop, make, use and sell products using proprietary information and technology developed at UC for the treatment of Parkinson's disease or any other indication. There are no milestone payments under the licensing agreement. In addition, we must make royalty payments to UC under this license agreement ranging in the low-single digits as a percentage of all net sales, sublicensing fees, milestone payments and sublicense royalties that we receive from sublicenses granted by us pursuant to this license agreement. We are also obligated to pay patent filing, prosecution, maintenance and defense costs. This UC license agreement is terminable by UC in the event of breach and otherwise expires when the last licensed patent expires.
Research and Development Expenditures
Expenditures for research and development activities were $3,509,000 and $6,966,000 during the years ended September 30, 2015 and 2014, respectively. Research and development expenses for fiscal 2015 and 2014 related primarily to the advancement of our lead compound, 10150.
Manufacturing
We currently do not have the capability to manufacture any of our drug candidates on a commercial scale. Materials for non-clinical and clinical studies are produced under contract with third parties. To date, we have partnered with JMPS for the manufacture of our active pharmaceutical ingredients. JMPS is a 200 year-old company that is a global supplier of active pharmaceutical ingredients, fine chemicals and other specialty chemical products and services to a wide range of chemical and pharmaceutical industry customers and industrial and academic research organizations. JMPS is a leader in the manufacture of metal-based pharmaceutical products.
Commercialization
If BARDA elects to procure 10150 following a pre-EUA application, as described above, or after FDA approval, it may be possible for us to generate significant sales revenue without the need to raise significant funds to build a commercial organization. Depending on the size of those procurements, and assuming the successful development and FDA approval of our compounds in other, non-biodefense indications, we may have sufficient financial resources to internally fund the building of a commercial organization. However, in the event procurements from BARDA are not made, and assuming successful development and FDA approval of one or more of our compounds, to successfully commercialize our catalytic antioxidant programs, we must seek corporate partners with expertise in commercialization or develop this expertise internally. However, we may not be able to successfully commercialize our catalytic antioxidant technology, either internally or through collaboration with others.
Marketing
Our potential catalytic antioxidant products are being developed for large therapeutic markets. We believe these markets are best approached by partnering with established biotechnology or pharmaceutical companies that have broad sales and marketing capabilities. We are pursuing collaborations of this type as part of our search for development partners. However, we may not be able to enter into any marketing arrangements for any of our products on satisfactory terms or at all.
Biodefense Industry
Market Overview
The market for biodefense countermeasures has grown dramatically as a result of the increased awareness of the threat of global terror activity in the wake of the September 11, 2001 terrorist attacks. The U.S. government is the principal source of worldwide biodefense spending. Most U.S. government spending on biodefense programs is in the form of development funding from NIAID, BARDA and the Department of Defense ("DoD") and procurements of countermeasures by BARDA, the CDC and the DoD. The U.S. government is now the largest source of development and procurement funding for academic institutions and biotechnology companies conducting biodefense research or developing vaccines and immunotherapies directed at potential agents of bioterror or biowarfare.
72
We analyze the biodefense market in three segments; the United States civilian market, United States military market and non U.S. markets, with the U.S. government funding representing the vast majority of the worldwide market.
| · | U.S. Civilian: The U.S. civilian market includes funds to protect the U.S. population from biological agents and is largely funded by the Project BioShield Act of 2004 ("Project BioShield"). Project BioShield is the U.S. government's largest biodefense initiative. Project BioShield was extended through the Pandemic All Hazards and Preparedness Reauthorization Act of 2013, which authorized BARDA to administer a Special Reserve Fund of $2.8 billion for MCM procurement. |
| · | U.S. Military: The DoD is responsible for the development and procurements of countermeasures for the military segment, which focuses on providing protection for military personnel and civilians who are on active duty. |
| · | Non-U.S. Markets: Non-U.S. markets address protection against biowarfare agents for both civilians and military personnel in foreign countries. We anticipate that foreign countries will want to procure biodefense products as they are developed and validated by procurements by the U.S. government. |
Project BioShield and the Pandemic and All-Hazards Preparedness Act
Project BioShield became law in 2004 and authorizes procurements of countermeasures for chemical, biological, radiological and nuclear attacks for the SNS, which is a national repository of medical assets and countermeasures designed to provide federal, state and local public health agencies with medical supplies needed to treat those affected by terrorist attacks, natural disasters, industrial accidents and other public health emergencies. Project BioShield provided appropriations of $5.6 billion to be expended over ten years into a Special Reserve Fund.
The Pandemic and All-Hazards Preparedness Act ("PAHPA"), passed in 2006, established BARDA as the agency responsible for awarding procurement contracts for biomedical countermeasures and providing development funding for advanced research and development in the biodefense arena, and supplements the funding available under Project BioShield for chemical, biological, radiological and nuclear countermeasures, and provides funding for infectious disease pandemics. Funding for BARDA is provided by annual appropriations by Congress. Congress also appropriates annual funding for the CDC for procurements of medical assets and countermeasures for the SNS and for NIAID to conduct biodefense research. This appropriation funding supplements amounts available under Project BioShield.
The Pandemic and All-Hazards Preparedness Reauthorization Act ("PAHPRA"), passed in 2013, extended the programs started under Project BioShield and PAHPA. PAHPRA authorized a $2.8 billion Special Reserve Fund to be administered by BARDA for the purpose of procuring MCMs for the SNS. Currently, the U.S. government may, at its discretion, purchase critical biodefense products for the SNS prior to FDA approval after the filing of a pre-EUA application.
On an ongoing basis we monitor notices for requests for proposal, grants and other potential sources of government funding that could potentially support the development of our drug candidates. Nevertheless, changes in government budgets, priorities and agendas as well as political pressures could result in a reduction in overall government financial support for the biodefense sector in general and/or specifically the drug candidates we are developing. At any time, the U.S. government may be forced or choose to reduce or delay spending in the biodefense field, which could decrease the likelihood of future government contract awards, the likelihood that the government will exercise its right to extend any of its existing contracts and/or the likelihood that the government would procure products from us.(For further information, see "Risk Factors — Risks Related to Our Dependence on U.S. Government Grants and Contracts — Most of our immediately foreseeable future revenues are contingent upon grants and contracts from the U.S. government and we may not achieve sufficient, if any, revenues from these agreements to attain profitability.") As a result, further development of our drug candidates and ultimate product sales to the government, if any, could be delayed or stopped altogether.
73
Legislation and Regulation Related to Bioterrorism Counteragents
Because some of our drug candidates are intended for the treatment of diseases that may result from acts of bioterrorism, they may be subject to the specific legislation and regulation described below.
Project BioShield
Project BioShield provides expedited procedures for bioterrorism related procurements and awarding of research grants, making it easier for HHS to quickly commit funds to countermeasure projects. Project BioShield relaxes procedures under the Federal Acquisition Regulation for procuring property or services used in performing, administering or supporting biomedical countermeasure research and development. In addition, if the Secretary of HHS deems that there is a pressing need, Project BioShield authorizes the Secretary to use an expedited award process, rather than the normal peer review process, for grants, contracts and cooperative agreements related to biomedical countermeasure research and development activity.
Under Project BioShield, the Secretary of HHS, with the concurrence of the Secretary of the Department of Homeland Security ("DHS"), and upon the approval of the President, can contract to purchase unapproved countermeasures for the SNS in specified circumstances. Congress is notified of a recommendation for a stockpile purchase after Presidential approval. Project BioShield specifies that a company supplying the countermeasure to the SNS is paid on delivery of a substantial portion of the countermeasure. To be eligible for purchase under these provisions, the Secretary of HHS must determine that there are sufficient and satisfactory clinical results or research data, including data, if available, from preclinical and clinical trials, to support a reasonable conclusion that the countermeasure will qualify for approval or licensing within eight years. Project BioShield also allows the Secretary of HHS to authorize the emergency use of medical products that have not yet been approved by the FDA. To exercise this authority, the Secretary of HHS must conclude that:
| · | the agent for which the countermeasure is designed can cause serious or life-threatening disease; |
| · | the product may reasonably be believed to be effective in detecting, diagnosing, treating or preventing the disease; |
| · | the known and potential benefits of the product outweigh its known and potential risks; and |
| · | there is no adequate alternative to the product that is approved and available. |
Although this provision permits the Secretary of HHS to circumvent the FDA approval process, its use would be limited to emergency circumstances.
Safety Act
The Support Anti-Terrorism by Fostering Effective Technologies Act enacted by the U.S. Congress in 2002 (the "Safety Act") creates product liability limitations for qualifying anti-terrorism technologies for claims arising from or related to an act of terrorism. In addition, the Safety Act provides a process by which an anti-terrorism technology may be certified as an "approved product" by the DHS and therefore entitled to a rebuttable presumption that the government contractor defense applies to sales of the product. The government contractor defense, under specified circumstances, extends the sovereign immunity of the United States to government contractors who manufacture a product for the government. Specifically, for the government contractor defense to apply, the government must approve reasonably precise specifications, the product must conform to those specifications and the supplier must warn the government about known dangers arising from the use of the product.
Public Readiness and Emergency Preparedness Act
The Public Readiness and Emergency Preparedness Act enacted by Congress in 2005 (the "PREP Act") provides immunity for manufacturers from all claims under state or federal law for "loss" arising out of the administration or use of a "covered countermeasure." However, injured persons may still bring a suit for "willful misconduct" against the manufacturer under some circumstances. "Covered countermeasures" include security countermeasures and "qualified pandemic or epidemic products." For these immunities to apply, the Secretary of HHS must issue a declaration in cases of public health emergency or "credible risk" of a future public health emergency. We cannot predict whether Congress will fund the relevant PREP Act compensation programs; or whether the necessary prerequisites for immunity would be triggered with respect to our drug candidates.
74
Competition
General
Competition in the pharmaceutical industry is intense and we expect it to increase. Technological developments in our field of research and development occur at a rapid rate and we expect competition to intensify as advances in this field are made. We will be required to continue to devote substantial resources and efforts to research and development activities. Our most significant competitors, among others, are fully integrated pharmaceutical companies and more established biotechnology companies, which have substantially greater financial, technical, sales, marketing and human resources than we do. These companies may succeed in developing and obtaining regulatory approval for competitive products more rapidly than we can for our drug candidates. In addition, competitors may develop technologies and products that are, or are perceived as being, cheaper, safer or more effective than those being developed by us or that would render our technology obsolete.
In addition, we face significant competition for U.S. government funding for both development and procurements of medical countermeasures for biological, chemical and nuclear threats, diagnostic testing systems and other emergency preparedness countermeasures. The U.S. federal government has currently allocated a significant amount of research funding to the development of countermeasures against the effects of radiation exposure. As a result, there are many drug candidates and products under development as a possible countermeasure against the effects of radiation exposure.
We expect that important competitive factors in our potential product markets will be the relative speed with which we and other companies can develop products, complete the clinical testing and approval processes, and supply commercial quantities of a competitive product to the market. With respect to clinical testing, competition might result in a scarcity of clinical investigators and patients available to test our potential products, which could delay development.
Antioxidants
Several companies have explored the therapeutic potential of antioxidant compounds in numerous indications. Historically, most of these companies have focused on engineered versions of naturally occurring antioxidant enzymes, but with limited success, perhaps because the large size of these molecules makes delivery into the cells difficult. Antioxidant drug research continues at a rapid pace despite previous clinical setbacks.
Patents and Proprietary Rights
We currently license rights to our potential products from third parties. We generally seek patent protection in the United States and other jurisdictions for the potential products and proprietary technology licensed from these third parties. The process for preparing and prosecuting patents is lengthy, uncertain and costly. Patents may not issue on any of the pending patent applications owned by us or licensed by us from third parties. Even if patents issue, the claims allowed might not be sufficiently broad to protect our technology or provide us protection against competitive products or otherwise be commercially valuable. Patents issued to or licensed by us could be challenged, invalidated, infringed, circumvented or held unenforceable. Even if we successfully defend our patents for our products, the costs of defense can be significant.
As of December 20, 2015, our catalytic antioxidant small molecule technology base is described in 12 issued United States patents and five United States pending patent applications. These patents and patent applications belong in whole or in part to Duke or the NJH and are licensed to us. These patents and patent applications cover soluble manganic porphyrins as antioxidant molecules as well as targeted compounds obtained by coupling such antioxidant compounds to molecules that bind to specific extracellular elements. The pending U.S. patent applications and issued U.S. patents include composition of matter claims and method claims for several series of compounds. Corresponding international patent applications have been filed, 88 of which have issued, and one of which has been allowed as of December 20, 2013. Our 12 issued U.S. patents will expire between 2015 and 2023.
75
On November 22, 2013, we filed a provisional patent application with the United States Patent and Trademark Office entitled "Synthesis and Formulation of Porphyrin Compounds." The filing was for brand new patents for synthesis, formulation and pharmaceutical composition of 10150 and other porphyrin compounds. If granted, these new patents could substantially extend the term of patent protection for 10150 beyond the current composition of matter claims.
On November 21, 2014, a patent application was filed under the Patent Cooperation Treaty (PCT) with the United States Receiving Office (US/RO), thereby preserving the right to seek patent protection in all PCT contracting States.
On April 15, 2015, we announced that we had filed a provisional patent application with the United States Patent Office for a new series of compounds demonstrating anti-microbial and anti-inflammatory action. The compounds were developed by Aeolus in collaboration with Brian J. Day, PhD at National Jewish Health in Denver, Colorado as a result of an Aeolus-sponsored research grant. We also announced that we have obtained a worldwide, exclusive license to develop the compounds from NJH.
On November 2, 2015 we announced that we had received notice of allowance of a composition of matter patent for 11114 from the European Patent Office. This new compound has demonstrated neuro-protective activity in models of Parkinson's disease and other indications. The compounds were invented by Aeolus in collaboration with Brian J. Day, PhD at National Jewish Health and Manisha Patel, PhD at the University of Colorado, Anschutz Medical Campus, Department of Pharmaceutical Sciences in collaboration with Aeolus Pharmaceuticals, and have been tested and developed by Dr. Patel under two research grants from MJFF. Aeolus has obtained worldwide, exclusive licenses to develop the compounds from National Jewish Health and the University of Colorado.
In addition to patent protection, we rely upon trade secrets, proprietary know-how and technological advances that we seek to protect, in part, through confidentiality agreements with our collaborative partners, employees and consultants. Our employees and consultants are required to enter into agreements providing for confidentiality and the assignment of rights to inventions made by them while in our service. We also enter into non-disclosure agreements to protect our confidential information furnished to third parties for research and other purposes.
Government Regulation
Our research and development activities and the manufacturing and marketing of our future products are subject to regulation by numerous governmental agencies in the United States and in other countries. The FDA and comparable agencies in other countries impose mandatory procedures and standards for the conduct of clinical trials and the production and marketing of products for diagnostic and human therapeutic use. Before obtaining regulatory approvals for the commercial sale of any of our products under development, we must demonstrate through preclinical studies and clinical trials that the product is safe and efficacious for use in each target indication. The results from preclinical studies and early clinical trials might not be predictive of results that will be obtained in large-scale testing. Our clinical trials might not successfully demonstrate the safety and efficacy of any products or result in marketable products.
The United States system of drug approvals is considered to be the most rigorous in the world. It takes an over ten years for a drug candidate to move through the clinical and approval phases in the United States according to a November 2014 study by the Tufts Center for the Study of Drug Development. Only five in 5,000 drug candidates that enter preclinical testing make it to human testing and only one of those five is approved for commercialization. On average, it costs a company $1.4 billion in out of pocket costs to get one new drug candidate from the laboratory to United States patients according to the report. In addition, time costs, expected returns that investors forego while the drug is in development, averaged $1.2 billion.
76
The steps required by the FDA before new drug products may be marketed in the United States include:
| · | completion of preclinical studies; |
| · | the submission to the FDA of a request for authorization to conduct clinical trials on an IND, which must become effective before clinical trials may commence; |
| · | adequate and well-controlled Phase I clinical trials, which typically involves normal, healthy volunteers. The tests study a drug candidate's safety profile, including the safe dosage range. The studies also determine how a drug is absorbed, distributed, metabolized and excreted as well as the duration of its action; |
| · | adequate and well-controlled Phase II clinical trials which typically involve treating patients with the targeted disease with the drug candidate to assess a drug's effectiveness; |
| · | adequate and well-controlled Phase III clinical trials involving a larger population of patients with the targeted disease are treated with the drug candidate to confirm efficacy of the drug candidate in the treatment of the targeted indication and to identify adverse events; |
| · | submission to the FDA of an NDA; and |
| · | review and approval of the NDA by the FDA before the product may be shipped or sold commercially. |
In addition to obtaining FDA approval for each product, each product manufacturing establishment must be registered with the FDA and undergo an inspection prior to the approval of an NDA. Each manufacturing facility and its quality control and manufacturing procedures must also conform and adhere at all times to the FDA's current good manufacturing practices ("cGMP") regulations. In addition to preapproval inspections, the FDA and other government agencies regularly inspect manufacturing facilities for compliance with these requirements. Manufacturers must expend substantial time, money and effort in the area of production and quality control to ensure full technical compliance with these standards.
Preclinical testing includes laboratory evaluation and characterization of the safety and efficacy of a drug and its formulation. Preclinical testing results are submitted to the FDA as a part of an IND, which must become effective prior to commencement of clinical trials. Clinical trials are typically conducted in three sequential phases following submission of an IND. Phase I represents the initial administration of the drug to a small group of humans, either patients or healthy volunteers, typically to test for safety (adverse effects), dosage tolerance, absorption, distribution, metabolism, excretion and clinical pharmacology, and, if possible, to gain early evidence of effectiveness. Phase II involves studies in a small sample of the actual intended patient population to assess the efficacy of the drug for a specific indication, to determine dose tolerance and the optimal dose range and to gather additional information relating to safety and potential adverse effects. Once an investigational drug is found to have some efficacy and an acceptable safety profile in the targeted patient population, Phase III studies are initiated to further establish clinical safety and efficacy of the therapy in a broader sample of the general patient population, in order to determine the overall risk-benefit ratio of the drug and to provide an adequate basis for any physician labeling. During all clinical studies, we must adhere to good clinical practices ("GCPs") standards. The results of the research and product development, manufacturing, preclinical studies, clinical studies and related information are submitted in an NDA to the FDA.
The process of completing clinical testing and obtaining FDA approval for a new drug is likely to take a number of years and require the expenditure of substantial resources. If an application is submitted, there can be no assurance that the FDA will review and approve the NDA. Even after initial FDA approval has been obtained, further studies, including post-market studies, might be required to provide additional data on safety and will be required to gain approval for the use of a product as a treatment for clinical indications other than those for which the product was initially tested and approved. Also, the FDA will require post-market reporting and might require surveillance programs to monitor the side effects of the drug. Results of post-marketing programs might limit or expand the further marketing of the products. Further, if there are any modifications to the drug, including changes in indication, manufacturing process, labeling or a change in manufacturing facility, an NDA supplement might be required to be submitted to the FDA.
The rate of completion of any clinical trials will be dependent upon, among other factors, the rate of patient enrollment. Patient enrollment is a function of many factors, including the size of the patient population, the nature of the trial, the availability of alternative therapies and drugs, the proximity of patients to clinical sites and the eligibility criteria for the study. Delays in planned patient enrollment might result in increased costs and delays, which could have a material adverse effect on us.
77
Failure to comply with applicable FDA requirements may result in a number of consequences that could materially and adversely affect us. Failure to adhere to approved trial standards and GCPs in conducting clinical trials could cause the FDA to place a clinical hold on one or more studies which would delay research and data collection necessary for product approval. Noncompliance with GCPs could also have a negative impact on the FDA's evaluation of an NDA. Failure to adhere to GMPs and other applicable requirements could result in FDA enforcement action and in civil and criminal sanctions, including but not limited to fines, seizure of product, refusal of the FDA to approve product approval applications, withdrawal of approved applications, and prosecution.
Whether or not FDA approval has been obtained, approval of a product by regulatory authorities in foreign countries must be obtained prior to the commencement of marketing of the product in those countries. The requirements governing the conduct of clinical trials and product approvals vary widely from country to country, and the time required for approval might be longer or shorter than that required for FDA approval. Although there are some procedures for unified filings for some European countries, in general, each country at this time has its own procedures and requirements. There can be no assurance that any foreign approvals would be obtained.
In addition to the regulatory framework for product approvals, we and our collaborative partners must comply with laws and regulations regarding occupational safety, laboratory practices, the use, handling and disposition of radioactive materials, environmental protection and hazardous substance control, and other local, state, federal and foreign regulation. The impact of such regulation upon us cannot be predicted and could be material and adverse.
In addition, because some of our drug candidates are intended for the treatment of diseases that may result from acts of bioterrorism, they may be subject to the specific legislation and regulation described above under "Project BioShield and the Pandemic and All-Hazards Preparedness Act" and "Legislation and Regulation Related to Bioterrorism Counteragents."
Regulations Regarding Government Contracting
We are a government contractor in the United States and we may become a government contractor elsewhere which would mean that we would be subject to various statutes and regulations that govern procurements of goods and services by agencies of the United States and other countries, including the Federal Acquisition Regulation. These governing statutes and regulations can impose stricter penalties than those normally applicable to commercial contracts, such as criminal and civil damages liability and suspension and debarment from future government contracting. In addition, pursuant to various statutes and regulations, our government contracts may be subject to unilateral termination or modification by the government for convenience in the United States and elsewhere, detailed auditing requirements and accounting systems, statutorily controlled pricing, sourcing and subcontracting restrictions and statutorily mandated processes for adjudicating contract disputes.
Hazardous Materials and Select Agents
Our development and manufacturing processes involve the use of hazardous materials, including chemicals and radioactive materials, and produce waste products. Accordingly, we are subject to federal, state and local laws and regulations governing the use, manufacturing, storage, handling and disposal of these materials. In addition to complying with environmental and occupational health and safety laws, we must comply with special regulations relating to biosafety administered by the CDC, HHS and the DoD.
Other Regulations
In the United States and elsewhere, the research, manufacturing, distribution, sale and promotion of drug and biological products are subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services; other divisions of HHS, such as the Office of Inspector General: the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice and state and local governments. For example, sales, marketing and scientific and educational grant programs must comply with the anti-kickback and fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provisions of the Health Insurance Portability and Accountability Act and similar state laws. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws.
78
CPEC, LLC
We were previously developing bucindolol for the treatment of heart failure, but development was discontinued in 1999. Commercial rights to bucindolol are owned by CPEC, LLC, a limited liability company ("CPEC"), of which we own 35% and Endo Pharmaceuticals (formerly Indevus Pharmaceuticals), Inc. owns 65%.
ARCA biopharma, Inc. is developing bucindolol under the trade name Gencaro as a beta-blocker and mild vasodilator for the treatment of atrial fibrillation in patients with heart failure and left ventricular dysfunction. ARCA biopharma, Inc. is headquartered in Broomfield, Colorado. The future development of Gencaro is uncertain. In the event that Gencaro is approved for sale, however, we may be owed royalties on sales. There can be no guarantees, however, that Gencaro will ever be approved or sold or achieve sufficient revenues to generate royalties.
Employees
At February 2, 2016, we had four full-time employees and no part time employees. None of our employees is represented by a labor union. In addition to our employees, we utilize a team of consultants and subcontractors to perform key functions for us.
79
MANAGEMENT
Information Regarding Directors
The following sets forth the names and ages (as of December 31, 2015) of our directors, and certain other information about them.
|
Name of Director
|
Age as of
December 31, 2015
|
Director Since
|
||
|
David C. Cavalier
|
46
|
April 2004
|
||
|
John M. Farah, Jr., Ph.D.
|
63
|
October 2005
|
||
|
Amit Kumar, Ph.D.
|
52
|
June 2004
|
||
|
Chris A. Rallis
|
62
|
June 2004
|
||
|
John M. Clerici
|
45
|
May 2013
|
||
|
Mitchell D. Kaye, J.D.
|
47
|
May 2013
|
||
|
Jeffrey A. Scott, M.D.
|
58
|
May 2013
|
David C. Cavalier has been the Chairman of our Board since April 30, 2004, and became our full time employee in November 2009. In June 2013, he became Chief Financial Officer. Since 2001, he has been a Principal and the Chief Operating Officer of Xmark Opportunity Partners, LLC, and a manager of a family of private investment funds. From 1995 to 1996, Mr. Cavalier worked for Tiger Real Estate, a $785 million private investment fund sponsored by Tiger Management Corporation. Mr. Cavalier began his career in 1994 in the Investment Banking Division of Goldman, Sachs & Co. working on debt and equity offerings for public and private real estate companies. He received a B.A. from Yale University and an M.Phil. from Oxford University.
John M. Farah, Jr., Ph.D. has been an independent director of ours since October 2005 and a member of our audit committee. He is managing director of a private consultancy serving biopharma clients in the US and abroad and recently, Dr. Farah affiliated with a strategic consultancy to provide market insight and value proposition analysis for clients about their products. From 2008 to 2010, Dr. Farah was an independent director of GenSpera, Inc. (GNSZ), a publicly-traded pharmaceutical development stage company. Dr. Farah was a Vice President at Cephalon, Inc., a biopharmaceutical company, from 1992 until 2011 when the company was integrated into Teva Pharmaceuticals. Dr. Farah led Cephalon's headquarter team of an international business unit with oversight of strategic product registrations, operations and sales abroad; he was latterly responsible for key Asia Pacific markets coordinating corporate product support for third party distributors and licensees. Dr. Farah joined Cephalon to manage scientific affairs which enhanced the company's R&D initiatives. As a senior team member of worldwide business development from 1997 to 2003, he promoted and negotiated R&D and commercial alliances with multinational and regional pharmaceutical firms. From 2003 until 2011, Dr. Farah led worldwide product export with P&L responsibilities for third party product sales, and in 2006, focused on strategic growth and commercial success in Asia and the Americas ex-US. In addition to his responsibilities for business development and regional international revenues, Dr. Farah oversaw successful patent litigation in Europe and Latin America. From 2008 until Cephalon's acquisition by Teva, he was treasurer and a director of the company's political action committee. From 1986 to 1992, Dr. Farah was a research investigator at GD Searle (now Pfizer) in nervous system and immunoinflammatory disease programs. His training included postdoctoral research at the National Institutes of Health (NIH, NINCDS) following his Ph.D. in physiology from the Uniformed Services University. He holds a B.S. in zoology from the University of Maryland and a B.H.A. from New College of California. We believe that Dr. Farah should serve as a director of our Company because of his extensive career in the pharmaceutical industry and international experience. Dr. Farah's past experience negotiating research partnerships, product licensing and academic collaborations are a valuable contribution to our Board and his current client-based experience allows him to provide additional insight to our Board in considering and approving these types of partnerships for the Company.
80
Amit Kumar, Ph.D. is currently President and CEO of Geo Fossil Fuels, a private biotech and energy company. He is also Chairman of the Board of Ascent Solar Technologies, a publicly-held solar energy company. From September 2001 to June 2010, Dr. Kumar was President and Chief Executive Officer of CombiMatrix Corporation, a publicly-held biotechnology company and director from September 2000 to June 2012. Previously, Dr. Kumar was Vice President of Life Sciences of Acacia Research Corp. From January 1999 to February 2000, Dr. Kumar was the founding President and CEO of Signature BioSciences, Inc., a life science company developing technology for advanced research in genomics, proteomics and drug discovery. From January 1998 to December 1999, Dr. Kumar was an Entrepreneur in Residence with Oak Investment Partners, a venture capital firm. From October 1996 to January 1998, Dr. Kumar was a Senior Manager at Idexx Laboratories, Inc., a biotechnology company. From October 1993 to September 1996, he was Head of Research & Development for Idetek Corporation, which was later acquired by Idexx Laboratories, Inc. Dr. Kumar received his B.A. in Chemistry, with honors, from Occidental College. After joint studies at Stanford University and the California Institute of Technology, he received his Ph.D. from the California Institute of Technology in 1991. He also completed a post-doctoral fellowship at Harvard University from 1991 to 1993. We believe that Dr. Kumar should serve as a director of our Company in light of his experience serving as an officer and on the board of directors of a number of publicly-held companies, as well as his past venture capital and capital-raising experience. Dr. Kumar's experience in scientific research and development is also a valuable contribution to our Board, particularly during deliberations and discussions relating to research and development matters.
Chris A. Rallis has been an executive-in-residence at Pappas Ventures, a life science venture capital firm since January 2008. Previously, Mr. Rallis was the President and Chief Executive Officer of ImmunoBiosciences, Inc. ("IBI"), a vaccine technology company located in Raleigh, North Carolina from April 2006 through June 2007. Prior to joining IBI, Mr. Rallis served as an executive in residence (part time) for Pappas Ventures, and as a consultant for Duke University and Panacos Pharmaceuticals, Inc. Mr. Rallis is the former President and Chief Operating Officer and director of Triangle Pharmaceuticals, Inc., which was acquired by Gilead Sciences in January 2003 for approximately $465 million. Prior to assuming the role of President and COO in March 2000, he was Executive Vice President, Business Development and General Counsel. While at Triangle, Mr. Rallis participated in 11 equity financings generating gross proceeds of approximately $500 million. He was also primarily responsible for all business development activities which included a worldwide alliance with Abbott Laboratories and the in-licensing of ten compounds. Before joining Triangle in 1995, Mr. Rallis served in various business development and legal management roles with Burroughs Wellcome Co. over a 13-year period, including Vice President of Strategic Planning and Business Development. Mr. Rallis also serves on the boards of Fennec Pharmaceuticals Inc. , a publicly-held biopharmaceutical company located in Research Triangle Park, NC and Tenax Therapeutics, Inc., a publicly-held biopharmaceutical company located in Morrisville, NC. Mr. Rallis serves on the audit committees of both boards and chairs the audit committee at Adherex. Mr. Rallis received his A.B. degree in economics from Harvard College and a J.D. from Duke University. We believe that Mr. Rallis should serve as a director of our Company in light of his experience serving as an executive officer of, and participating in a number of equity financings for, other pharmaceutical companies. Mr. Rallis' experiences in development activities and strategic alliances are valuable to Board deliberations. In addition, his venture capital consulting experience allows him to contribute additional insight to the Board in refining our Company's business strategies and commercial objectives.
John M. Clerici is a founding Principal of Tiber Creek Partners, LLC, a company focused on providing scientific and business counseling to biotechnology companies seeking to use non-dilutive capital from the U.S. and foreign governments and from non-governmental organizations. Mr. Clerici is also a Partner in the government contracts practice at McKenna Long & Aldridge LLP. For over 14 years, Mr. Clerici has been at the forefront of the creation of the public health preparedness sector, including helping large pharmaceutical and emerging biotechnology companies develop creative approaches to access non-dilutive capital to fund the development of biotechnology for emerging disease and engineered threats. Since 1999, Mr. Clerici has assisted over three dozen companies in obtaining nearly $4 billion in funding for research, development and procurement of public health countermeasures from the Federal government, which includes the majority of the awards made under Project Bioshield, the U.S. Government's initiative for preparing the United States against a bioterrorist attack. Prior to joining McKenna Long & Aldridge LLP, Mr. Clerici was a judge advocate with the U.S. Air Force where, among other assignments, he advised the Air Force Research Laboratory on the procurement of technology from research institutions throughout the United States, Europe and Asia. Mr. Clerici earned his Juris Doctor from the University of North Carolina at Chapel Hill in 1995. He did his undergraduate work at the Catholic University of America, graduating summa cum laude. We believe that Mr. Clerici should serve as a director of our Company in light of his over 14 years of experience in working with the US Government in public health preparedness. We believe he will contribute significantly to our medical countermeasure development programs.
81
Mitchell D. Kaye, J.D. is a Managing Director at Biotechnology Value Fund, a biotechnology investment fund. Previously, Mr. Kaye was founder of MedClaims Liaison, LLC and served as its Chief Executive Officer from 2009 to 2013. MedClaims is a consumer advocacy business that works on behalf of families in managing reimbursement disputes with medical providers and insurance companies. From 2008-2010, Mr. Kaye was a Managing Director with Navigant Capital Advisors, a financial and strategic advisory services firm, and Head of Navigant's Financial Institutions Restructuring Solutions Team (FIRST). While at Navigant, Mr. Kaye led numerous high profile engagements on behalf of investment funds and investors. Previously, as a successful entrepreneur in the asset management industry, Mr. Kaye launched two highly profitable asset management companies. Mr. Kaye was the founding member of Xmark Opportunity Partners, LLC, an investment fund exclusively focused on investments in publicly traded life sciences companies, and has served as a member of the management committee since 2001. Mr. Kaye established a venerable reputation as an activist investor, taking influential stakes in numerous companies, forcing changes at the boards of directors and management team levels, and guiding the sale of several of his portfolio companies to the benefit of shareholders. In 1996, Mr. Kaye began his career as a founding member of Brown Simpson Asset Management, LLC (Brown Simpson), an investment fund that was at the foreground of private placement investing in the public markets. Brown Simpson's life sciences investment unit produced a value weighted cash-on-cash return in excess of 100% during the life of the fund. During his career, Mr. Kaye has consummated over 100 transactions as a lead investor, structured over a billion dollars in debt and equity-linked transactions, and taken an active role in the management of numerous portfolio companies. Mr. Kaye has served on the boards of several private and public companies, and also served on the board of the New York Alzheimer's Association. From September 2007 until the company's unwinding in June 2009, Mr. Kaye served on the board of directors of Genaera Corporation, a biopharmaceutical company that was listed on the Nasdaq Capital Market. Mr. Kaye received his BA from Wesleyan University, and his Juris Doctorate from Northwestern University School of Law. We believe that Mr. Kaye should serve as a director of our Company in light of his experience in business development and financing biotech companies, which we believe will be invaluable as we begin to generate efficacy data from our animal efficacy and cancer studies.
Jeffrey A. Scott, M.D. is a Board Certified Medical Oncologist and is currently the President and Chief Medical Officer of Integra Connect, a healthcare company involved in Revenue Cycle Management, Population Health and Pharmaceutical Services. Prior to this Dr. Scott was the General Manager/Senior Vice President for P4 Healthcare, a division of Cardinal Health Specialty Solutions, which is a division of Cardinal Health. He was also a member of Cardinal Health's Operating Committee. Prior to the 2010 sale of P4 Healthcare to Cardinal Health, Dr. Scott was the Founder, President and Chief Executive Officer of P4 Healthcare, since its inception in 2006. P4 Healthcare was a multimedia Healthcare Marketing and Education Company with a focus in Oncology. From 1998 to 2002, Dr. Scott served as the National Medical Director and President of the International Oncology Network (ION), a network of more than 4,000 U.S. private practice oncologists headquartered in Baltimore, Maryland. In 2002, ION became a subsidiary of Amerisource Bergen Corporation upon its sale. Dr. Scott continued to serve as President and General Manager for ION until 2005. Dr. Scott was a practicing physician, Founding Partner and Chief Financial Officer of Georgia Cancer Specialists located in Atlanta, Georgia from 1990 to 2000. During Dr. Scott's tenure as Chief Financial Officer of Georgia Cancer Specialists, the physician practice had over $100 million in revenue and Dr. Scott was responsible for development of financial programs of practice after the merger and corporate buyout by Phymatrix. Also at the Georgia Cancer Specialists, Dr. Scott took responsibility for the development of an extensive clinical research program. From 1998 to 2000, he also served as Medical Chief of Staff at Emory Northlake Regional Medical Center in Atlanta, Georgia. Dr. Scott's biotechnology experience includes his role as a Consultant to NexStar Pharmaceuticals, Inc. ("NexStar") of Boulder, Colorado. Prior to NexStar's 1999 merger with Gilead Sciences, Inc., it was engaged in the discovery, development, manufacture and commercialization of products to treat serious and life-threatening illnesses. As a consultant to NexStar, Dr. Scott was responsible for assisting and educating the sales force in dealing with physician networks and consulting with investment advisors regarding potential investments in other biotechnology companies. Dr. Scott's educational background includes a B.S. degree in Microbiology from the University of Michigan, Ann Arbor, Michigan, a medical education at Wayne State University, Detroit, Michigan, and a fellowship in Oncology at University of Texas Health Sciences, San Antonio, Texas. Dr. Scott has Board Certifications from the American Board of Internal Medicine, Internal Medicine, September 1987, and the American Board of Internal Medicine, Medical Oncology, November 1989. Dr. Scott has served on the board of directors of Biovest International, Inc. (OTCQB: BVTI) since March 2004. We believe that Dr. Scott should serve as a director of our Company in light of his experience as an oncologist, drug developer and senior executive in one of the leading pharmaceutical distribution companies, which we believe will be invaluable as we begin to move forward with our AEOL 10150 oncology development program and begin to formulate our strategies for making our compound available in the most efficient and effective way for biodefense purposes.
82
Executive Officers
Our executive officers and their ages as of December 31, 2015 were as follows:
|
Name
|
Age
|
Position(s)
|
|||||
|
David Cavalier
|
46
|
Chairman of the Board, Chief Financial Officer
|
|||||
|
John L. McManus
|
51
|
President and Chief Executive Officer
|
|||||
David C. Cavalier. Mr. Cavalier's background is described in "Information Regarding Directors" above.
John L. McManus. Mr. McManus began as a consultant to Aeolus in June 2005 as President. He became employed as our President and Chief Operating Officer in July 2006 and was appointed President and Chief Executive Officer in March 2007. Mr. McManus, who received his degree in business administration from the University of Southern California in 1986, is the founder and president of McManus Financial Consultants, Inc. ("MFC"), which provides strategic, financial and investor relations advice to senior managements and boards of directors of public companies, including advice on mergers and acquisitions. These companies have a combined value of over $25 billion. He has served as president of MFC since 1997. In addition, Mr. McManus previously served as Vice President, Finance and Strategic Planning to Spectrum Pharmaceuticals, Inc. (NASDAQ: SPPI), where he had primary responsibility for restructuring Spectrum's operations and finances, including the design of strategic and financial plans to enhance Spectrum's corporate focus, and leading the successful implementation of these plans and relisting of the Company on NASDAQ. The implementation of these plans led to an increase in Spectrum's market value from $1 million to more than $125 million at the time of Mr. McManus' departure.
Family Relationships and Orders, Judgments and Decrees
There is no family relationship between any of our officers or directors. There are no orders, judgments, or decrees of any governmental agency or administrator, or of any court of competent jurisdiction, revoking or suspending for cause any license, permit or other authority to engage in the securities business or in the sale of a particular security or temporarily or permanently restraining any of our officers or directors from engaging in or continuing any conduct, practice or employment in connection with the purchase or sale of securities, or convicting such person of any felony or misdemeanor involving a security, or any aspect of the securities business or of theft or of any felony. Nor are any of the officers or directors of any corporation or entity affiliated with us so enjoined.
Code of Ethics
We have adopted a code of ethics that applies to our principal executive officer, principal financial officer, principal accounting officer and persons performing similar functions. We have posted the text of Code of Ethics on our Internet website at www.aeoluspharma.com. A copy of the Code of Ethics can also be obtained free of charge by writing to David Cavalier, Aeolus Pharmaceuticals, Inc., 26361 Crown Valley Parkway, Suite 150 Mission Viejo, CA 92691.
Audit Committee
The Board has established an Audit Committee in accordance with section 3(a)(58)(A) of the Exchange Act.
83
COMPENSATION OF DIRECTORS
The following table sets forth information for the fiscal year ended September 30, 2015 regarding the compensation of our directors.
Director Compensation
|
Name
|
Fees Earned or
Paid in Cash
|
Option Awards(1)
|
All Other
Compensation
|
Total
|
||||||||||||
|
David C. Cavalier
|
—
|
$
|
—
|
—
|
$
|
—
|
||||||||||
|
John M. Farah, Jr., Ph.D.
|
—
|
21,752
|
—
|
21,752
|
||||||||||||
|
Amit Kumar, Ph.D.
|
—
|
21,752
|
—
|
21,752
|
||||||||||||
|
Chris A. Rallis
|
—
|
21,752
|
—
|
21,752
|
||||||||||||
|
John M. Clerici
|
—
|
16,314
|
—
|
16,314
|
||||||||||||
|
Mitchell D. Kaye
|
—
|
—
|
—
|
—
|
||||||||||||
|
Jeffrey Scott
|
—
|
16,314
|
—
|
16,314
|
||||||||||||
| (1) | The amounts in the "Option Awards" column reflect the aggregate grant date fair value of awards for grants of options to each listed director in fiscal 2015, computed in accordance with Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) Topic 718. These amounts do not represent the actual amounts paid to or realized by the directors during fiscal 2015. The fair value of the options was determined at the date of the grant using the Black-Scholes option pricing model with the following weighted average assumptions: (i) dividend yield: 0%; (ii) unvested forfeiture rate: 5.35%; (iii) expected volatility: 137%; (iv) risk-free interest rate: 1.7%; and (v) expected option life after shares are vested: 5.27 years. We use a straight line method of amortization of stock-based compensation. |
All directors are reimbursed for expenses incurred in connection with each board or committee meeting attended. In addition, the Board adopted the following compensation program for the outside members of the Board on March 7, 2013, effective beginning March 7, 2013:
· Each non-executive Board member shall be eligible to receive a nonqualified stock option of 75,000 shares per year issued at the beginning of each year. The option exercise prices shall be equal to the closing price of the Common Stock on the grant date. The options shall have 10-year terms and vest, as long as the director remains on the Board, on a monthly basis over a 12-month period beginning on the date of grant. Unvested options expire upon resignation or termination from the Board.
· In addition, each Audit Committee member shall be eligible to receive a nonqualified stock option of 25,000 shares per year issued at the beginning of each year. The option exercise prices shall be equal to the closing price of the Common Stock on the grant date. The options shall have 10-year terms and vest, as long as the director remains on the Board, on a monthly basis over a 12-month period beginning on the date of grant. Unvested options expire upon resignation or termination from the Board.
Outstanding Equity Awards for Directors as of September 30, 2015
The following table sets forth information regarding unexercised stock options for each Director outstanding as of September 30, 2015. We have not awarded stock grants or other equity incentive awards and as such have not made any disclosures regarding such awards.
84
|
Name
|
Number of
Securities
Underlying
Unexercised Options
Exercisable
|
Number of
Securities
Underlying
Unexercised
Options
Unexercisable
|
Equity Incentive
Plan Awards:
Number of
Securities Underlying
Unexercised Unearned
Options
|
|||||||||
|
David C. Cavalier
|
132,750
|
—
|
—
|
|||||||||
|
John M. Farah, Jr., Ph.D.
|
565,757
|
8,334
|
—
|
|||||||||
|
Amit Kumar, Ph.D.
|
596,916
|
8,334
|
—
|
|||||||||
|
Chris A. Rallis
|
596,916
|
8,334
|
—
|
|||||||||
|
John M. Clerici
|
218,750
|
6,250
|
—
|
|||||||||
|
Mitchell D. Kaye
|
150,000
|
—
|
—
|
|||||||||
|
Jeffrey A. Scott, M.D.
|
218,750
|
6,250
|
—
|
|||||||||
85
EXECUTIVE COMPENSATION
The following table sets forth all compensation earned for the fiscal year ended September 30, 2015 and 2014, by its principal executive officer, principal financial officer, and its one other executive officer who served in such capacity as of the end of fiscal 2015, collectively referred to as the "Named Executive Officers".
Summary Compensation Table
|
Annual Compensation
|
All Other
|
||||||||||||||||||||
|
Name and Principal
|
Fiscal
|
Option
|
Compensation
|
||||||||||||||||||
|
Position(s)
|
Year
|
Salary ($)
|
Bonus ($)
|
Awards ($) (1)
|
($)
|
Total ($)
|
|||||||||||||||
|
John L. McManus
|
2015
|
$
|
446,925
|
—
|
$
|
57,444
|
$
|
—
|
$
|
504,369
|
|||||||||||
|
President and
|
2014
|
$
|
433,908
|
—
|
$
|
344,242
|
—
|
$
|
778,150
|
||||||||||||
|
Chief Executive Officer
|
|||||||||||||||||||||
|
David C. Cavalier
|
2015
|
$
|
363,127
|
—
|
$
|
—
|
$
|
—
|
$
|
363,127
|
|||||||||||
|
Chairman of the Board and Chief Financial
|
2014
|
$
|
352,550
|
—
|
$
|
—
|
$
|
—
|
$
|
352,550
|
|||||||||||
|
Officer
|
|||||||||||||||||||||
| (1) | The amounts in the "Option Awards" column reflect the aggregate grant date fair value of awards for grants of options to each listed Named Executive Officer, computed in accordance with FASB ASC Topic 718. These amounts do not represent the actual amounts paid to or realized by any of the Named Executive Officers during fiscal 2015 or fiscal 2014. The fair value of the options was determined at the date of the grant using the Black-Scholes option pricing model with the following weighted average assumptions: (i) dividend yield: 0%; (ii) unvested forfeiture rate: 11.65%; (iii) expected volatility: 125.49%; (iv) risk-free interest rate: 1.64%; and (v) expected option life after shares are vested: 5.27 years. We use a straight line method of amortization of stock-based compensation. |
Grants of Plan Based Awards During the Fiscal Year Ended September 30, 2015
The following table summarizes all option grants during the fiscal year ended September 30, 2015 to the Named Executive Officers. Each of these options was granted pursuant to the 2004 Plan.
|
Name
|
Grant Date
|
All Other Option Awards:
Number of Securities
Underlying Options (#)(1)
|
Exercise or
Base Price
Exercise Price
of Option
Awards
|
Grant Date
Fair Value of
Option
Awards
(2)
|
|||||||||
|
John L. McManus
|
3/4/2015
|
250,000
|
$
|
0.27
|
$
|
57,444
|
|||||||
|
President and Chief Executive Officer
|
|||||||||||||
| (1) | The option grant vests on a monthly basis for twelve months with a ten-year term, subject to earlier termination upon certain events. |
| (2) | The amounts in the "Grant Date Fair Value of Option Awards" column reflect the aggregate grant date fair value of awards for grants of options to Mr. McManus in fiscal 2015, computed in accordance with FASB ASC Topic 718. These amounts do not represent the actual amounts paid to or realized by Mr. McManus during fiscal 2015. |
86
Outstanding Equity Awards as of September 30, 2015
The following table sets forth information regarding outstanding unexercised stock options for each of the Named Executive Officers as of September 30, 2015. We have not awarded stock grants or other equity incentive awards and as such have not made any disclosures regarding such awards.
|
Name
|
Number of
Securities
Underlying
Unexercised
Options
Exercisable
|
Number of
Securities
Underlying
Unexercised
Options
Unexercisable
|
Option Awards
Equity Incentive Plan
Awards: Number of
Securities Underlying
Unexercised Unearned
Options
|
Option
Exercise
Price
|
Option
Expiration
Date
|
||||||||||||||||
|
John L. McManus
|
10,000
|
—
|
—
|
$
|
1.15
|
10/31/2015
|
|||||||||||||||
|
10,000
|
—
|
—
|
$
|
1.03
|
11/30/2015
|
||||||||||||||||
|
10,000
|
—
|
—
|
$
|
0.95
|
12/30/2015
|
||||||||||||||||
|
10,000
|
—
|
—
|
$
|
0.89
|
1/31/2016
|
||||||||||||||||
|
10,000
|
—
|
—
|
$
|
0.90
|
2/28/2016
|
||||||||||||||||
|
10,000
|
—
|
—
|
$
|
0.80
|
3/31/2016
|
||||||||||||||||
|
10,000
|
—
|
—
|
$
|
0.75
|
4/28/2016
|
||||||||||||||||
|
10,000
|
—
|
—
|
$
|
0.60
|
5/31/2016
|
||||||||||||||||
|
10,000
|
—
|
—
|
$
|
0.81
|
6/30/2016
|
||||||||||||||||
|
250,000
|
—
|
—
|
$
|
0.75
|
7/14/2016
|
||||||||||||||||
|
250,000
|
—
|
—
|
$
|
0.90
|
7/13/2017
|
||||||||||||||||
|
250,000
|
—
|
—
|
$
|
0.32
|
7/14/2013
|
||||||||||||||||
|
1,000,000
|
—
|
—
|
$
|
0.30
|
5/6/2019
|
||||||||||||||||
|
250,000
|
—
|
—
|
$
|
0.39
|
7/30/2019
|
||||||||||||||||
|
250,000
|
—
|
—
|
$
|
0.40
|
7/14/2020
|
||||||||||||||||
|
1,500,000
|
—
|
—
|
$
|
0.40
|
7/29/2020
|
||||||||||||||||
|
250,000
|
—
|
—
|
$
|
0.40
|
7/14/2021
|
||||||||||||||||
|
250,000
|
—
|
—
|
$
|
0.28
|
7/14/2022
|
||||||||||||||||
|
2,000,000
|
—
|
—
|
$
|
0.40
|
3/4/2023
|
||||||||||||||||
|
250,000
|
—
|
—
|
$
|
0.26
|
3/4/2024
|
||||||||||||||||
|
125,000
|
125,000
|
(1
|
)
|
$
|
0.27
|
3/4/2025
|
|||||||||||||||
|
David Cavalier
|
30,000
|
—
|
—
|
$
|
0.85
|
9/12/2016
|
|||||||||||||||
|
30,000
|
—
|
—
|
$
|
0.55
|
7/27/2017
|
||||||||||||||||
|
27,750
|
—
|
—
|
$
|
0.40
|
12/11/2013
|
||||||||||||||||
|
3,750
|
—
|
—
|
$
|
0.29
|
2/5/2019
|
||||||||||||||||
|
11,250
|
—
|
—
|
$
|
0.33
|
3/26/2019
|
||||||||||||||||
|
3,750
|
—
|
—
|
$
|
0.38
|
4/30/2019
|
||||||||||||||||
|
11,250
|
—
|
—
|
$
|
0.35
|
6/4/2019
|
||||||||||||||||
|
15,000
|
—
|
—
|
$
|
0.39
|
7/30/2019
|
||||||||||||||||
(1) Options vest at a rate of approximately 20,833 per month from the grant date for twelve months, provided that John McManus is an employee or consultant of the Company on the applicable vesting date. In the event of a sale of the Company, through a merger or otherwise, all of the options shall be fully vested and immediately exercisable.
Option Exercises and Stock Vested During the Fiscal Year Ended September 30, 2015
No stock options were exercised by any Named Executive Officer during the fiscal year ended September 30, 2015.
We had no stock awards outstanding as of or for the year ended September 30, 2015.
Employment Agreement with John McManus
On March 4, 2013, we and John McManus entered into an amended and restated employment agreement (the "Restated Agreement"). Under the Amended Employment Agreement, Mr. McManus continues to serve as President, Chief Executive Officer and Chief Operating Officer of the Company. Pursuant to the agreement, Mr. McManus is paid $ 36,159 per month.
87
Under the Amended Employment Agreement, Mr. McManus will be entitled to receive an option to purchase at least 250,000 shares of the Company's common stock with an exercise price equal to the closing price of the Company's common stock on the date of grant. In addition, the Amended Employment Agreement provides that, on the date of the agreement, Mr. McManus shall be granted an option to purchase 2,000,000 shares of the Company's common stock with an exercise price equal to the closing price of the Company's common stock on the date of grant. In each case, the options shall vest at a monthly rate of 8.33% following the date of grant, subject to Mr. McManus remaining employed with the Company. The Amended Employment Agreement also provides that, upon a Change in Control of the Company (as defined in the Amended Employment Agreement), all of the stock options granted to Mr. McManus will fully vest and become immediately exercisable. In addition, if the Company signs a Corporate Partnership, as defined in the Amended Employment Agreement, of if there is a Change in Control, Mr. McManus is entitled to receive a bonus of not less than $250,000 upon the execution and delivery of a definitive and enforceable agreement.
The current term of the Amended Employment Agreement is through March 4, 2015 unless terminated earlier. Pursuant to the Amended Employment Agreement, if (A) the Company terminates Mr. McManus' employment without "Cause" (as defined in the Amended Employment Agreement), and the Company has not provided Mr. McManus with a Non-Renewal Notice, or (B) Mr. McManus terminates his employment for "Good Reason" (as defined in the Amended Employment Agreement), in either case subject to Mr. McManus' agreement to release any claims against the Company, Mr. McManus will be entitled to receive a cash severance, payable over 12 months, equal to: (i) Mr. McManus' effective base salary at the time of termination, plus (ii) the average of the annual bonus(es) paid to Mr. McManus, if any, during the two full years immediately preceding the year in which the termination occurs. If the Company provides Mr. McManus with a Non-Renewal Notice, other than as a result of Mr. McManus' death, disability or termination for "Cause", subject to Mr. McManus' agreement to release any claims against the Company, Mr. McManus will be entitled to receive a cash severance equal to: (x) Mr. McManus' effective base salary at the time of termination, plus (y) the average of the annual bonus(es) paid to Mr. McManus, if any, during the two full years immediately preceding the year in which the termination occurs, less (z) the salary payable to Mr. McManus under the Amended Employment Agreement for the remainder of his employment term, which amount will be payable in equal monthly installments following the end of the employment term until the one-year anniversary of the date of the Non-Renewal Notice.
Consulting Arrangements
Prior to June 30, 2011, McManus & Company, Inc. ("M&C"), which is owned by Mr. John McManus, provided us with administrative, accounting and financial consulting services. In addition, M&C also provided us with our corporate headquarters, facilities management and the outsourcing of the administrative, accounting, finance and accounting functions. Pursuant to an agreement with M&C, we paid M&C a monthly consulting payment of $25,000. During fiscal 2012 and 2011, we paid M&C $0 and $180,000, respectively, in consulting fees pursuant to services rendered by M&C under the agreement. The agreement terminated on June 30, 2011.
Separation Agreements
We did not enter into any separation agreements during fiscal 2015.
Payments Upon Termination or Change of Control
We have an employment agreement with Mr. John McManus, which provides for payments to Mr. McManus upon termination of employment or a change of control of Aeolus under specified circumstances. For information regarding the specific circumstances that would trigger payments and the provision of benefits, the manner in which payments and benefits would be provided and conditions applicable to the receipt of payments and benefits, see "—Employment Agreement with John McManus."
The following tables set forth information regarding potential payments and benefits that each Named Executive Officer who was serving as an executive officer on September 30, 2015 would receive upon termination of employment or consulting arrangement or a change of control of Aeolus under specified circumstances, assuming that the triggering event occurred on September 30, 2015.
88
Summary of Potential Payments Upon Termination or Change of Control
|
Termination without Cause
|
Voluntary
Resignation
|
||||||||
|
Value of Options
|
|||||||||
|
Cash
|
Value of
|
with Accelerated
|
Cash
|
||||||
|
Name
|
Payments(1)
|
Benefits(2)
|
Vesting
|
Payments
|
|||||
|
John L. McManus
|
|
$446,925
|
|
$26,451
|
|
$—
|
(3)
|
—
|
|
____________________
| (1) | This amount reflects a lump sum payment equal to the remaining term of the Named Executive Officer's employment agreement with the Company, assuming notice of termination was given on September 30, 2015. |
| (2) | The amounts in this column reflect the estimated value of health, dental, life and disability insurance that would be provided to the Named Executive Officer pursuant to his employment agreement with the Company. |
| (3) | Pursuant to the Named Executive Officer's employment agreement with the Company, in the event the Named Executive Officer was terminated without cause on September 30, 2015, options to purchase 1,000,000 shares would have vested. The amounts in this column are calculated based on the difference between $0.28, the closing market price per share of the Common Stock on September 30, 2015, the last trading day of fiscal year 2013, and the exercise price per share of $0.40 for the options subject to accelerated vesting. |
|
Immediately upon a Change of
Control
|
Termination without Cause in Connection
with a Change of Control
|
|||||||||||||||||||||||||||
|
Name
|
Cash
Payments(4)
|
Value of
Options
with
Accelerated
Vesting
|
Cash
Payments(6)
|
Value of
Benefits(7)
|
Value of
Options with
Accelerated
Vesting
|
|||||||||||||||||||||||
|
John L. McManus
|
$
|
250,000
|
$
|
—
|
(5
|
)
|
$
|
683,908
|
$
|
26,451
|
$
|
—
|
(5
|
)
|
||||||||||||||
____________________
| (4) | The amounts in this column reflect the lump sum payment payable upon a change of control pursuant to the Named Executive Officer's employment agreement with the Company in effect on September 30, 2015 assuming a change of control of the Company occurred on September 30, 2015. |
| (5) | Pursuant to the 2004 Plan, all outstanding options shall vest in connection with a change of control of the Company. The amounts in this column are calculated based on the difference between $0.25, the closing market price per share of the Common Stock on September 30, 2015, the last trading day of fiscal year 2015, and the $0.40 exercise price per share of the 1,000,000 options subject to accelerated vesting. |
| (6) | The amounts in this column reflect the lump sum payment payable pursuant to a termination upon a change of control pursuant to the Named Executive Officer's employment agreement with the Company in effect on September 30, 2015 assuming a change of control of the Company occurred on September 30, 2015. |
| (7) | The amounts in this column reflect the estimated value of health, dental, life and disability insurance that would be provided to the Named Executive Officer pursuant to his employment agreement with the Company for the period from March 4, 2015 to March 4, 2016. |
Summary of Actual Payments Upon Termination of Employment
No payments were made to any Named Executive Officer in connection with a termination of employment during fiscal 2015.
89
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Aeolus has adopted a policy that all transactions between Aeolus and its executive officers, directors and other affiliates must be approved by a majority of the members of the Board and by a majority of the disinterested members of the Board, and must be on terms no less favorable to Aeolus than could be obtained from unaffiliated third parties.
September 2015 Bridge Loan Financing
On September 29, 2015, we received funding in the form of convertible promissory notes (the "BVF Notes") from Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P. One of our directors, Mitchell Kaye, is a Managing Director of Biotechnology Value Fund.
The BVF Notes had an aggregate principal balance of $1,000,000, accrue interest at a rate of 6% per annum and have a scheduled maturity date of September 28, 2016 (the "Maturity Date"). The terms of the BVF Notes provided that the outstanding principal and accrued interest on the BVF Notes shall automatically convert into Company equity securities issued in a Qualified Financing (as defined below) at a conversion rate carrying a 15% discount to the lowest price per share (or share equivalent) issued in a Qualified Financing (an "Automatic Conversion"). If, prior to the Maturity Date, the Company enters into an agreement pertaining to a Corporate Transaction (as defined below) and the BVF Notes had not been previously converted pursuant to an Automatic Conversion, then, the outstanding principal balance and unpaid accrued interest of the BVF Notes shall automatically convert in whole into the right of the holder to receive, in lieu of any other payment and in cancellation of the BVF Notes, an amount in cash upon closing of the Corporate Transaction (as defined below) equal to two times the outstanding principal amount of the BVF Notes.
For purposes of the foregoing: "Qualified Financing" means a bona fide new money equity securities financing on or before the Maturity Date with total proceeds to the Company of not less than four million dollars; and "Corporate Transaction" means a sale, lease or other disposition of all or substantially all of the Company's assets or a consolidation or merger of the Company with or into any other corporation or other entity or person, or any other corporate reorganization, in which the stockholders of the Company immediately prior to such consolidation, merger or reorganization own less than fifty percent (50%) of the voting power of the surviving entity immediately after such consolidation, merger or reorganization.
In connection with the 2015 Securities Placement, the BVF Notes were converted pursuant to an Automatic Conversion and the holders of the BVF Notes received the securities described below under "BVF Convert."
2015 Securities Placement
On December 10, 2015, we entered into securities purchase agreements with certain accredited investors to sell and issue (i) an aggregate of 10,215,275 common units issued at a purchase price of $0.22 per unit, and (ii) 4,500 preferred stock units issued to Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P., for an aggregate purchase price of $4.5 million, resulting in aggregate gross proceeds to the Company of approximately $6.75 million (the "2015 Securities Placement"). Each common unit consisted of one share of the Company's common stock and a five year warrant to purchase one share of the Company's common stock, subject to adjustment. The preferred stock units collectively consisted of (i) 4,500 shares of our Series C Preferred Stock that are collectively convertible into an aggregate of 20,454,546 shares of the Company's common stock and (ii) warrants to purchase an aggregate of 20,454,546 shares of our common stock, in each case subject to adjustment. Each of the foregoing warrants has an initial exercise price of $0.22 per share.
Following the completion of the 2015 Securities Placement, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of our common stock and warrants to purchase an additional 5,414,402 shares of our common stock at an exercise price per share of $0.22 subject to adjustment (the "BVF Convert"). As a result, the BVF Notes are no longer outstanding as of the date of this prospectus. As described above, one of our directors, Mitchell Kaye, is a Managing Director of Biotechnology Value Fund.
2013 Warrant Repricing, Exercise and Lockup Agreement
90
Effective February 15, 2013, Aeolus and each of Xmark JV Investment Partners ("JV Partners"), Xmark Opportunity Fund, Ltd. ("Opportunity Ltd.") and Xmark Opportunity Fund, L.P. ("Opportunity LP" and, together with JV Partners and Opportunity Ltd., the "Xmark Entities") entered into a Warrant Repricing, Exercise and Lockup Agreement (the "Xmark Warrant Agreement") pursuant to which the Company agreed to reduce the exercise price of outstanding warrants to purchase an aggregate of up to 59,149,000 shares of our common stock held by the Xmark Entities (the "Xmark Warrants") to $0.01 per share. Prior to the entry into the Xmark Warrant Agreement, the exercise price of the Xmark Warrants covering an aggregate of 55.4 million shares of Aeolus' common stock was $0.28 per share, and the exercise price covering an aggregate of 3.8 million shares of Aeolus' common stock was $0.50 per share. In consideration for the reduction of the exercise price of the Xmark Warrants, each of the Xmark Entities agreed to immediately exercise all of the Xmark Warrants by cashless exercise. The Xmark Warrant Agreement also provides that the Xmark Entities will not transfer the shares issuable upon exercise of the Xmark Warrants (the "Xmark Warrant Shares") until the Company either (i) declares a cash dividend on its common stock or otherwise makes a cash distribution or (ii) effects a Change of Control, subject in each case to the terms of the Xmark Warrant Agreement. Xmark Opportunity Partners, LLC ("Opportunity Partners") is the investment manager of JV Partners and the sole member of the investment manager of each of Opportunity Ltd. and Opportunity LP, and, as such, possesses the sole power to vote and the sole power to direct the disposition of all securities of the Company held by each of the Xmark Entities. Mitchell Kaye and David C. Cavalier, the Co-Managing Members of Xmark Capital Partners, LLC, a Delaware limited liability company, the Managing Member of Opportunity Partners, share voting and dispositive power with respect to all securities of the Company beneficially owned by Opportunity Partners. The foregoing description of the Xmark Warrant Agreement (and the transactions effected thereunder) does not purport to be complete and is qualified in its entirety by reference to the Xmark Warrant Agreement, a copy of which is attached as Exhibit 10.4 to Form 8-K filed with the SEC on February 19, 2013.
Director Independence
After review of all relevant transactions or relationships between each director, or any of his family members, and the Company, our senior management and its independent registered public accounting firm, the Board of Directors has affirmatively determined that all of our directors are independent directors within the meaning of the applicable Nasdaq Stock Market, LLC ("Nasdaq") listing standards, as currently in effect, excluding Mr. Cavalier.
91
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
Security Ownership of Certain Beneficial Owners and Management
The following tables set forth certain information regarding the ownership of shares of Aeolus' Common Stock and Series B Preferred as of the close of business on December 31, 2015, by:
| · | each person known by Aeolus to beneficially own more than 5% of the outstanding shares of each class of our stock; |
| · | each of our directors; |
| · | each of our Named Executive Officers (as defined under "Executive Compensation" above); and |
| · | all of our directors and executive officers as a group |
|
Preferred Stock
|
Common Stock
|
|||||||||||||||||
|
Identity of Owner or Group (1)(2)
|
Beneficially
Owned
|
Percentage
Owned
|
Beneficially
Owned(4)
|
Percentage
Owned(5)
|
||||||||||||||
|
Directors:
|
||||||||||||||||||
|
David C. Cavalier
|
-
|
-
|
97,064,694
|
(6)
|
64.0%
|
|||||||||||||
|
John M. Farah, Jr., Ph.D. (7)
|
-
|
-
|
607,424
|
*
|
||||||||||||||
|
Mitchell D. Kaye, J.D. (8)
|
-
|
-
|
97,081,944
|
64.1%
|
||||||||||||||
|
Amit Kumar, Ph.D. (7)
|
-
|
-
|
638,583
|
*
|
||||||||||||||
|
John M. Clerici (7)
|
-
|
-
|
250,000
|
*
|
||||||||||||||
|
Chris A. Rallis (7)
|
-
|
-
|
638,583
|
*
|
||||||||||||||
|
Jeffrey A. Scott, M.D. (7)
|
-
|
-
|
250,000
|
*
|
||||||||||||||
|
Named Executive Officers:
|
||||||||||||||||||
|
John L. McManus (9)
|
-
|
-
|
6,569,467
|
4.2%
|
||||||||||||||
|
David C. Cavalier (6)
|
-
|
-
|
97,064,694
|
*
|
||||||||||||||
|
All directors and executive officers as a group (8 persons)
|
-
|
-
|
106,468,751
|
(10)
|
66.6%
|
|||||||||||||
|
Greater than 5% Stockholders:
|
||||||||||||||||||
|
Elan Corporation, plc
|
526,080
|
100.0
|
%(3)
|
526,080
|
(11)
|
*
|
||||||||||||
|
Lincoln House
|
||||||||||||||||||
|
Lincoln Place
|
||||||||||||||||||
|
Dublin 2, Ireland
|
||||||||||||||||||
|
BVF Partners, L.P. and its affiliates
|
4,500
|
100.0
|
%(3)
|
15,205,106
|
(12)
|
9.99%
|
(13)
|
|||||||||||
|
900 N. Michigan Avenue,
Suite 1100
Chicago, IL 60611
|
||||||||||||||||||
|
Xmark Opportunity Partners, LLC and its affiliates
|
-
|
-
|
97,214,694
|
(14)
|
64.0%
|
|||||||||||||
|
90 Grove Street
|
||||||||||||||||||
|
Ridgefield, CT 06877
|
||||||||||||||||||
* Less than one percent
(1) Unless otherwise indicated, the address of all the owners is: c/o Aeolus Pharmaceuticals, Inc., 26361 Crown Valley Parkway, Suite 150, Mission Viejo, California 92691.
(2) This table is based upon information supplied by our executive officers, directors and principal stockholders and Schedule 13Ds and 13Gs, as amended, filed with the SEC. Unless otherwise indicated in the footnotes to this table and subject to community property laws where applicable, we believe that each of the stockholders named in this table has sole voting and investment power with respect to the shares indicated as beneficially owned.
(3) Percent of shares beneficially owned by the person listed above is calculated by dividing the number of shares of each class of preferred stock beneficially owned by that person by the number of shares of each class of preferred stock outstanding. Elan Corporation, plc owns 100% of the outstanding shares of Series B Preferred Stock. BVF Partners, L.P. and its affiliates own 100% of the outstanding shares of Series C Preferred Stock.
(4) The number of shares of common stock beneficially owned includes any shares issuable pursuant to stock options or warrants that are currently exercisable or may be exercised within 60 days after December 31, 2015 as well as shares of preferred stock convertible into common stock. Shares issuable pursuant to such options or warrants and shares issuable upon conversion of such preferred stock are deemed outstanding for computing the ownership percentage of the person holding such options but are not deemed to be outstanding for computing the ownership percentage of any other person.
92
(5) Applicable percentages are based on 151,559,745 shares outstanding on December 31, 2015, plus the number of shares such stockholder can acquire within 60 days after December 31, 2015. All percentages are rounded.
(6) Consists of 132,750 shares of Common Stock issuable upon exercise of options held by David C. Cavalier; 29,095,831 shares of Common Stock beneficially owned by Xmark Opportunity Fund, L.P., a Delaware limited partnership ("Opportunity LP") (including 957,326 shares held by Goodnow Capital L.L.C. (L.P.)); 63,680,083 shares of Common Stock owned by Xmark Opportunity Fund, Ltd., a Cayman Islands exempted company ("Opportunity Ltd") (including 2,475,490 shares held by Goodnow Capital L.L.C. (Ltd.)); 1,508,567 shares of Common Stock owned by Xmark JV Investment Partners, LLC, a Delaware limited liability company ("JV Partners"); and another 2,647,463 shares of Common Stock owned by Goodnow Capital, L.L.C. ("Goodnow"), a Delaware limited liability company that are not reflected above. Mr. Cavalier shares voting and dispositive power over the shares listed above (other than shares subject to options) with Mr. Kaye.
(7) Consists solely of shares of Common Stock issuable upon exercise of options.
(8) Consists of 150,000 shares of Common Stock issuable upon exercise of options held by Mitchell D. Kaye. Also consists of shares beneficially owned as referenced in footnote 6 above, including: 29,095,831 shares of Common Stock beneficially owned by Opportunity LP; 63,680,083 shares of Common Stock beneficially owned by Opportunity Ltd; 1,508,567 shares of Common Stock beneficially owned by JV Partners; and 2,647,463 shares of Common Stock owned by Goodnow. Mr. Kaye shares voting and dispositive power over these shares with Mr. Cavalier. Mr. Kaye does not possess voting or investment power over Aeolus securities held or beneficially owned by BVF Partners, L.P. and its affiliates, as described below in footnote 12. Mr. Kaye is a Managing Director of Biotechnology Value Fund, L.P. which is affiliated with BVF Partners, L.P.
(9) Consists of 70,300 shares owned directly, 10,000 shares owned directly by Mr. McManus' spouse and 6,489,167 shares issuable upon exercise of options.
(10) Consists of shares of Common Stock beneficially owned by our directors and the following executive officers: Mr. McManus and Mr. Cavalier. See footnotes (5), (6), (7), and (9) above, the shares held by Opportunity LP, Opportunity Ltd, JV Partners and Goodnow, which are deemed to be beneficially owned by Messrs. Cavalier and Kaye have been counted only once for purposes of this calculation. Consists of 9,456,507 shares subject to options.
(11) Consists of 526,080 shares of Common Stock which were issuable upon conversion of an aggregate of 526,080 shares of Series B Preferred Stock as of the close of business on the Record Date.
(12) Consists of 14,561,580 shares of issued and outstanding Common Stock owned directly by the entities listed below plus 643,526 shares of Common Stock that may be acquired upon the exercise of certain warrants held by such entities up to the exercise and conversion limitations described in footnote 13. In light of such exercise and conversion limitations, the beneficial ownership of the number of shares of Common Stock listed in the table does not reflect all Common Stock subject to certain warrants and Series C Preferred Stock presented below that are held by affiliates of BVF Partners, L.P. that are listed below. The following beneficial ownership information is presented for informational purposes assuming no such limitation on the exercise of warrants or conversion of Series C Preferred Stock.
As of December 31, 2015, (i) Biotechnology Value Fund, L.P. ("BVF") beneficially owned 31,544,723 shares of Common Stock, including 15,557,560 shares of Common Stock issuable upon the exercise of certain warrants held by it and 8,677,273 shares of common stock that may be acquired upon conversion of Series C Preferred Stock, (ii) Biotechnology Value Fund II, L.P. ("BVF2") beneficially owned 18,501,586 shares of Common Stock, including 9,104,300 shares of Common Stock issuable upon the exercise of certain warrants held by it and 5,213,636 shares of common stock that may be acquired upon conversion of Series C Preferred Stock, (iii) BVF Investments, L.L.C. ("BVLLC") beneficially owned 352,980 shares of Common Stock, (iv) Investment 10, L.L.C. ("ILL10") beneficially owned 6,184,885 shares of Common Stock, including 3,056,638 shares of Common Stock issuable upon the exercise of certain warrants held by it and 1,186,364 shares of common stock that may be acquired upon conversion of Series C Preferred Stock, (v) MSI BVF SPV, L.L.C. ("MSI BVF") beneficially owned 6,128,172 shares of Common Stock, including 3,064,086 shares of Common Stock issuable upon the exercise of certain warrants held by it and 2,290,909 shares of common stock that may be acquired upon conversion of Series C Preferred Stock, and (vi) Biotechnology Value Trading Fund OS, L.P. ("Fund OS") beneficially owned 6,172,728 shares of Common Stock, including 3,086,364 shares of Common Stock issuable upon the exercise of certain warrants held by it and 3,086,364 shares of common stock that may be acquired upon conversion of Series C Preferred Stock.
BVF Partners L.P. ("Partners"), as the general partner of BVF and BVF2, the manager of BVLLC, the investment adviser of ILL10, the attorney-in-fact of MSI BVF and the investment manager of Fund OS, may be deemed to beneficially own 68,885,074 shares of Common Stock, including 54,323,494 shares of Common Stock currently issuable upon the exercise of certain warrants and conversion of Series C Preferred Stock, beneficially owned in the aggregate by BVF, BVF2, BVLLC, ILL10, MSI BVF, and Fund OS.
BVF Inc., as the general partner of Partners, may be deemed to beneficially own the 68,885,074 shares of Common Stock, including 54,323,494 shares of Common Stock currently issuable upon the exercise of certain warrants and conversion of Series C Preferred Stock, beneficially owned by Partners.
Mark N. Lampert, as a director and officer of BVF Inc., may be deemed to beneficially own the 68,885,074 shares of Common Stock, including 54,323,494 shares of Common Stock currently issuable upon the exercise of certain warrants, beneficially owned by BVF Inc.
1 Note to BVF: Please fill in the blanks.
93
The foregoing should not be construed in and of itself as an admission by any of Partners, BVF Inc. or Mark N. Lampert as to beneficial ownership of any shares of Common Stock owned by BVF, BVF2, BVLLC, ILL10, MSI BVF, and Fund OS. Each of Partners, BVF Inc. and Mr. Lampert disclaims beneficial ownership of the shares of Common Stock beneficially owned by BVF, BVF2, BVLLC, ILL10, MSI BVF, and Fund OS and this filing shall not be construed as an admission that any such person or entity is the beneficial owner of any such securities.
(13) The terms of the warrants and Series C Preferred Stock held by BVF, BVF2, BVLLC, ILL10, MSI BVF, and Fund OS each contain an issuance limitation prohibiting the holder from exercising such warrants and converting such Series C Preferred Stock to the extent that, after giving effect to such exercise or conversion thereof, the holder, including any of its affiliates, would beneficially own more than 9.99% of the Common Stock of the Company then issued and outstanding.
(14) Consists of 132,750 shares of Common Stock issuable upon exercise of options held by David C. Cavalier; 150,000 shares of Common Stock issuable upon exercise of options held by Mitchell D. Kaye; 29,095,831 shares of Common Stock owned by Xmark Opportunity Fund, L.P., a Delaware limited partnership ("Opportunity LP"); 63,680,083 shares of Common Stock owned by Xmark Opportunity Fund, Ltd., a Cayman Islands exempted company ("Opportunity Ltd"); 1,508,567 shares of Common Stock owned by Xmark JV Investment Partners, LLC, a Delaware limited liability company ("JV Partners"); and 2,647,463 shares of Common Stock owned by Goodnow Capital, L.L.C. ("Goodnow"), a Delaware limited liability company. Mr. Cavalier shares voting and dispositive power over these shares with Mr. Kaye.
94
SELLING STOCKHOLDERS
We are registering the shares of common stock identified in the table below in order to permit the selling stockholders to offer the shares for resale from time to time. Of the 73,432,471 shares of common stock listed, the following securities were issued in the 2015 Securities Placement: (i) 15,629,676 shares of common stock, (ii) 20,454,546 shares of common stock issuable upon conversion of 4,500 shares of Series C Preferred Stock that were issued in the 2015 Securities Placement and (iii) 37,298,249 shares of common stock issuable upon exercise of warrants which shares and warrants were issued in connection with the 2015 Securities Purchase. We also issued 50,000 shares of our common stock issuable upon the exercise of warrants in 2014. The shares and warrants were issued in reliance on Section 4(a)(2) of the Securities Act, and Rule 506 promulgated thereunder. For additional information regarding the shares offered pursuant to this prospectus, please see "Description of the Shares Included in the Prospectus" beginning on page 28 of this prospectus.
Each of Laidlaw & Company (UK) Ltd. And Newport Coast Securities, Inc. are broker-dealers registered with the Financial Industry Regulatory Authority or FINRA, and also selling stockholders in this offering. All other selling stockholders who are broker-dealers received their warrants to purchase common stock as underwriting compensation for their services as placement agents in the 2015 Securities Placement.
The following table sets forth, as of the date of this prospectus: (1) the name of the stockholder for whom we are registering shares under this registration statement; (2) the number of shares of our common stock owned by the stockholder prior to this offering; (3) the number of shares of our common stock being offered pursuant to this prospectus; and (4) the amount and the percentage (if 1% or more) of the class to be owned by such stockholder after completion of the offering. The percentage of outstanding common stock owned upon completion of the offering is calculated based on 151,559,745 shares of common stock issued and outstanding as of December 31, 2015. We have reflected beneficial ownership and selling stockholder information in the table below without giving effect to issuance limitations set forth within the warrants and the Series C Preferred Stock; these limitations prohibit the holder from exercising or converting such securities in certain circumstances. See "Security Ownership of Certain Beneficial Owners and Management," (footnote 13) and "Description of the Shares Included in This Prospectus." We prepared this table based on the information supplied to us by the selling stockholders named in the table and we have not sought to verify such information.
95
|
Name and Address of Selling Stockholder
|
Common Stock Owned Prior to Offering(1)
|
Common Stock
Being Offered
Pursuant to this
Prospectus
|
Common Stock
Owned Upon
Completion of
Offering (1)(2)
|
Percentage of
Common Stock
Owned Upon
Completion of
Offering
|
|
Albert & Yvonne Tjan Family Trust
8 Woodlawn
Irvine, CA 92620
|
200,568 (3)
|
200,568
|
0
|
*
|
|
Anna Belle Ambrose
577 Tower Blvd.
Powell, WY 82435
|
181,818 (4)
|
181,818
|
0
|
*
|
|
K. Tucker Andersen
61 Above All Rd.
Warren, CT 06754
|
909,090 (5)
|
909,090
|
0
|
*
|
|
Stephen Ball
2001 Los Amigos St.
La Canada Flintridge, CA 91011
|
181,818 (6)
|
181,818
|
0
|
*
|
|
Biotechnology Value Fund, L.P.
One Sansome Street, 30th Floor San Francisco, CA 94104
|
31,544,723 (7)(55) (56)
|
22,632,504
|
8,912,219
|
5.72%
|
96
|
Biotechnology Value Fund II, L.P.
One Sansome Street, 30th Floor San Francisco, CA 94104
|
18,501,586 (8)(55) (56)
|
13,464,752
|
5,036,834
|
3.27%
|
|
Biotechnology Value Trading Fund OS, L.P.
One Sansome Street, 30th Floor San Francisco, CA 94104
|
6,172,728 (9)(55) (56)
|
6,172,728
|
0
|
*
|
|
Linda Bonner
18916 E. Briargate Ln.
Parker, CO 80134
|
90,910 (10)
|
90,910
|
0
|
*
|
|
Brio Capital Master Fund LTD.
c/o Brio Capital Management, LLC
100 Merrick Road, Suite 401W
Rockville Centre, NY 11570-4800
|
5,939,331(11)
|
4,545,456
|
0
|
*
|
|
Bruno J. Casatelli
8862 Bloomfield Blvd
Sarasota, FL 34238
|
409,080 (12)
|
409,080
|
0
|
*
|
|
Chase Family Trust
3730 Valley Vista Rd.
Bonita, CA 91902
|
90,910 (13)
|
90,910
|
0
|
*
|
|
Timothy L. Dewey
102 Marie Drive
Box 648
Cimarron, KS 67835
|
190,908 (14)
|
190,908
|
0
|
*
|
|
Eric Figge
4 Lakeridge
Dove Canyon, CA 92679
|
180,000 (15)
|
180,000
|
0
|
*
|
|
James Galvin
2 Via Marin
San Clemente, CA 92676
|
90,908 (16)
|
90,909
|
0
|
*
|
|
Sally Garcia
3029 Treefern Dr.
Duarte, CA 91010
|
72,728 (17)
|
72,728
|
0
|
*
|
|
Dean Glau
6615 S. Piney Creek Circle
Centennial, CO 80016
|
45,454 (18)
|
45,454
|
0
|
*
|
97
|
Donald E. Goodin
620 Steeple Chase Road
Glasgow, KY 42141
|
100,000
(19)
|
100,000
|
0
|
*
|
|
Michael Halperin
1901 Skycrest Drive #2
Walnut, CA 94595
|
145,454 (20)
|
145,454
|
0
|
*
|
|
Kenneth H. Hancock
1831 Hillcrest Lane
Selina, KS 67401
|
454,540 (21)
|
454,540
|
0
|
*
|
|
HS Contrarian Investment, LLC
68 Fiesta Way
Fort Lauderdale, FL 33301
|
1,136,364 (22)
|
1,136,364
|
0
|
*
|
|
Intracoastal Capital, LLC
245 Palm Trail
Delray Beach, FL 33483
|
1,818,182 (23)
|
1,818,182
|
0
|
*
|
|
Investment 10, L.L.C.
One Sansome Street, 30th Floor San Francisco, CA 94104
|
6,184,885 (24)(55) (56)
|
3,339,740
|
2,845,145
|
1.86%
|
|
Iroquois Capital Investment Group LLC
205 E. 42nd St., 20th Floor
New York, NY 10017
|
90,910 (25)
|
90,910
|
0
|
*
|
|
Iroquois Master Fund LTD.
c/o Iroquois Capital Management LLC
205 E. 42nd St., 20th Floor
New York, NY 10017
|
227,274 (26)
|
227,274
|
0
|
*
|
|
Douglas E. Jasek
1515 Old Trail Court
Sugarland Texas 77479
|
727,272 (27)
|
727,272
|
0
|
*
|
|
Joseph M. Tosti Revocable Trust U/A 6-15-93
15615 Alton Prwy. #210
Irvine, CA 92618
|
181,818 (28)
|
181,818
|
0
|
*
|
|
Lee Keyte
3402 Altura Ave.
La Cresenta, CA 91214
|
136,364 (29)
|
136,364
|
0
|
*
|
|
Kingsbrook Opportunities
Master Fund LP
c/o Kingsbrook Partners LP
689 Fifth Avenue, 12th Floor
New York, NY 10022
|
2,000,000 (30)
|
2,000,000
|
0
|
*
|
98
|
Laidlaw & Co.
546 5th Avenue, 5th Floor
New York, NY 10036
|
986,589 (31)
|
986,589
|
0
|
*
|
|
Lincoln Park Capital Fund, LLC
C/O Lincoln Park Capital
440 N. Wells Street, Suite 410
Chicago, IL 60654
|
2,272,734 (32)
|
2,272,734
|
0
|
*
|
|
Henry Lindenmann
577 Tower Blvd.
Powell, WY 82435
|
45,454 (33)
|
45,454
|
0
|
*
|
|
Gregory McCloskey
138 White Flower
Irvine, CA 92603
|
83,863 (34)
|
83,863
|
0
|
*
|
|
James W. Mosich and Carmella Mosich
Living Trust U/A 10/1/84
2 Camino Platino
San Clemente, CA 92673
|
45,454 (35)
|
45,454
|
0
|
*
|
|
MSI BVF SPV, L.L.C.
One Sansome Street, 30th Floor San Francisco, CA 94104
|
6,128,172 (36)(55)(56)
|
6,128,172
|
0
|
*
|
|
Newport Coast Securities, Inc.
Newport Coast Securities
180 Maiden Lane, 17th Floor
New York, NY 10038
|
56,859 (37)
|
56,859
|
0
|
*
|
|
Michael A. Parimucha
2621 Oakgrove Ave
St. Augustine, FL 32092
|
227,260 (38)
|
227,260
|
0
|
*
|
|
Michael Pezzuolo
1332 Cerritos Dr.
Laguna Beach, CA 92651
|
45,454 (39)
|
45,454
|
0
|
*
|
|
Jeffery Ploen
6222 S. Blackhawk Ct.
Centennial, CO 80111
|
200,000 (40)
|
200,000
|
0
|
*
|
|
Harry Radie
24292 Bellerive Cir
Laguna Niguel, Ca 92677
|
88,945 (41)
|
88,945
|
0
|
*
|
|
Randall L. Payne Kathy S. Payne JTWROS
445 Janan Ct.
Conway, AR 72034
|
545,454 (42)
|
545,454
|
0
|
*
|
|
Daniel Rudden
5460 South Quebec St.
Greenwood Village, CO 80111
|
200,000 (43)
|
200,000
|
0
|
*
|
|
Savoy International Corporation
565 5th Avenue, Floor 30
New York, NY 10017
|
681,820(44)
|
681,820
|
0
|
*
|
99
|
R. Douglas Shearer
PO Box 515
515 Admirals Circle
Pine Beach, NJ 08741
|
454,546 (45)
|
454,546
|
0
|
*
|
|
Alva Staples
6705 E. Dorado Place
Greenwood Village, CO 80111
|
227,272 (46)
|
227,272
|
0
|
*
|
|
Ralph Tenebruso
7 High Point Road
Holmdel, NJ 07733
|
56,859 (47)
|
56,859
|
0
|
*
|
|
James Terwilliger
2014 Oak Springs Drive
Cordova, TN 38016
|
11,365 (48)
|
11,365
|
0
|
*
|
|
Thomas Green/Ardell Family Trust
4 Shadeyside
Trabuco Canyon, CA 92679
|
477,273 (49)
|
477,273
|
0
|
*
|
|
Albert Tjan
8 Woodlawn
Irvine, CA 92620
|
354,546 (50)
|
354,546
|
0
|
*
|
|
Joseph Tosti
15615 Alton Prwy. #210
Irvine, CA 92618
|
136,364 (51)
|
136,364
|
0
|
*
|
|
Jeff Stockstill
9471 Old Plantation Cove
Germantown, TN 38139
|
11,365 (52)
|
11,365
|
0
|
*
|
|
Mulhern Family Revocable Trust
1885 Mesa Ridge Ave
Westlake Village, CA 91362
|
227,272 (53)
|
227,272
|
0
|
*
|
|
Dina Lyaskovets
1024 Bayside Drive, #217 Newport Beach, CA 92660
|
50,000 (54)
|
50,000
|
0
|
*
|
|
TOTAL
|
91,620,544
|
73,432,471
|
15,205,106 (55) (56)
|
9.99% (56)
|
____________
* Less than one percent
(1) Includes shares of common stock issuable upon exercise of warrants held by any holder hereunder as well as issuable upon the conversion of Series C Preferred Stock. For the purposes hereof, we assume the issuance of all shares issuable upon exercise of warrants or any conversion of Series C Preferred Stock and disregard any limitations on the exercise of warrants or any conversion of Series C Preferred Stock that may otherwise apply. See "Description of Shares" included in this prospectus.
(2) Assumes the sale by the selling stockholders of all of the shares of common stock available for resale under this prospectus and disregards any limitations on the exercise of warrants or any conversion of Series C Preferred Stock that may otherwise apply. See "Description of Shares" included in this prospectus.
(3) Consists of 100,284 shares of common stock held directly by Albert & Yvonne Tjan Family Trust and 100,284 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(4) Consists of 90,909 shares of common stock held directly by Anna Belle Ambrose and 90,909 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
100
(5) Consists of 454,545 shares of common stock held directly by K. Trucker Andersen and 454,545 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(6) Consists of 90,909 shares of common stock held directly by Stephen Ball and 90,909 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(7) Consists of 7,309,890 shares held directly by Biotechnology Value Fund, L.P., 2,638,979 shares of common stock issuable upon exercise of warrants issued in the BVF Convert, 8,677,273 shares of common stock issuable upon conversion of 1,909 shares of Series C Preferred Stock that were issued in the 2015 Securities Placement, and 8,677,273 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement, 4,241,308 shares of common stock issuable upon exercise of warrants issued in prior placements, and disregards any limitations on the exercise of warrants or any conversion of Series C Preferred Stock.
(8) Consists of 4,183,650 shares held directly by Biotechnology Value Fund, L.P. II, 1,518,740 shares of common stock issuable upon exercise of warrants issued in the BVF Convert, 5,213,636 shares of common stock issuable upon conversion of 1,147 shares of Series C Preferred Stock that were issued in the 2015 Securities Placement, and 5,213,636 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement, 2,371,924 shares of common stock issuable upon exercise of warrants issued in prior placements,and disregards any limitations on the exercise of warrants or any conversion of Series C Preferred Stock.
(9) Consists of 3,086,364 shares of common stock issuable to Biotechnology Value Trading Fund OS, L.P. upon conversion of 679 shares of Series C Preferred Stock that were issued in the 2015 Securities Placement, and 3,086,364 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement, and disregards any limitations on the exercise of warrants or any conversion of Series C Preferred Stock.
(10) Consists of 45,455 shares of common stock held directly by Linda Bonner and 45,455 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(11) Consists of 2,466,603 shares of common stock held directly by Brio Capital Master Fund LTD., 2,272,728 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement, and 1,200,000 shares of common stock issuable upon exercise of warrants issued in prior placements.
(12) Consists of 204,540 shares of common stock held directly by Bruno J. Casatelli and 204,540 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(13) Consists of 45,455 shares of common stock held directly by Chase Family Trust and 45,455 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(14) Consists of 95,454 shares of common stock held directly by Timothy L. Dewey and 95,454 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(15) Consists of 90,000 shares of common stock held directly by Eric Figge and 90,000 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(16) Consists of 45,454 shares of common stock held directly by James Galvin and 45,454 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(17) Consists of 36,364 shares of common stock held directly by Sally Garcia and 36,364 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(18) Consists of 22,727 shares of common stock held directly by Dean Glau and 22,727 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(19) Consists of 50,000 shares of common stock held directly by Donald E. Goodin and 50,000 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(20) Consists of 72,727 shares of common stock held directly by Michael Halperin and 72,727 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
101
(21) Consists of 227,270 shares of common stock held directly by Kenneth H. Hancock and 227,270 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(22) Consists of 568,182 shares of common stock held directly by HS Contrarian Investment, LLC and 568,182 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(23) Consists of 909,091 shares of common stock held directly by Intracoastal Capital, LLC and 909,091 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement. Mitchell P. Kopin ("Mr. Kopin") and Daniel B. Asher ("Mr. Asher"), each of whom are managers of Intracoastal Capital LLC ("Intracoastal"), have shared voting control and investment discretion over the securities reported herein that are held by Intracoastal. As a result, each of Mr. Kopin and Mr. Asher may be deemed to have beneficial ownership (as determined under Section 13(d) of the Exchange Act) of the securities reported herein that are held by Intracoastal. Mr. Asher, who is a manager of Intracoastal, is also a control person of a broker-dealer. As a result of such common control, Intracoastal may be deemed to be an affiliate of a broker-dealer. Intracoastal acquired the Common Stock being registered hereunder in the ordinary course of business, and at the time of the acquisition of the Common Stock and warrants described herein Intracoastal did not have any arrangements or understandings with any person to distribute such securities.
(24) Consists of 1,941,883 shares held directly by Investment 10, L.L.C., 483,506 shares of common stock issuable upon exercise of warrants issued in the BVF Convert, 1,186,364 shares of common stock issuable upon conversion of 261 shares of Series C Preferred Stock that were issued in the 2015 Securities Placement, and 1,186,364 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement, 1,386,768 shares of common stock issuable upon exercise of warrants issued in prior placements, and disregards any limitations on the exercise of warrants or any conversion of Series C Preferred Stock.
(25) Consists of 45,455 shares of common stock held directly by Iroquois Capital Investment Group LLC and 45,455 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(26) Consists of 113,637 shares of common stock held directly by Iroquois Master Fund LTD. and 113,637 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(27) Consists of 363,636 shares of common stock held directly by Douglas E. Jasek and 363,636 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(28) Consists of 90,909 shares of common stock held directly by Joseph M. Tosti Revocable Trust and 90,909 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(29) Consists of 68,182 shares of common stock held by Lee Keyte and 68,182 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(30) Consists of 1,000,000 shares of common stock held by Kingsbrook Opportunities Master Fund LP and 1,000,000shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(31) Consists of 986,589 shares of common stock issuable upon exercise of warrants issued as 2015 Placement Agent Warrants. Laidlaw & Co. (UK) Ltd. is a FINRA registered broker-dealer and is deemed an underwriter in this offering.
(32) Consists of 1,136,367 shares of common stock held directly by Lincoln Park Capital Fund, LLC and 1,136,367 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(33) Consists of 22,727 shares of common stock held directly by Henry Lindenmann and 22,727 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(34) Consists of 40,909 shares of common stock held directly by Gregory McCloskey, 40,909 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement, and 2,045 shares of common stock issuable upon exercise of warrants issued as 2015 Placement Agent Warrants. Mr. McCloskey is an employee of Newport Coast Securities, Inc.
(35) Consists of 22,727 shares of common stock held directly by James W. Mosich and Carmella Mosich
Living Trust U/A 10/1/84 and 22,727 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(36) Consists of 773,177 shares held directly by MSI BVF SPV, L.L.C., 773,177 shares of common stock issuable upon exercise of warrants issued in the BVF Convert, 2,290,909 shares of common stock issuable upon conversion of 504 shares of Series C Preferred Stock that were issued in the 2015 Securities Placement, and 2,290,909 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement, and disregards any limitations on the exercise of warrants or any conversion of Series C Preferred Stock.
102
(37) Consists of 56,859 shares of common stock issuable to Newport Coast Securities, Inc. upon exercise of warrants issued as 2015 Placement Agent Warrants. Newport Coast Securities, Inc. is a FINRA registered broker-dealer and is deemed an underwriter in this offering.
(38) Consists of 113,630 shares of common stock held directly by Michael A. Parimucha and 113,630 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(39) Consists of 22,727 shares of common stock held directly by Michael Pezzuolo and 22,727 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(40) Consists of 100,000 shares of common stock held directly by Jeffery Ploen and 100,000 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(41) Consists of 88,945 shares of common stock issuable to Harry Radie upon exercise of warrants issued as 2015 Placement Agent Warrants. Mr. Radie is an employee of Newport Coast Securities, Inc.
(42) Consists of 272,727 shares of common stock held directly by Randall L. Payne Kathy S. Payne JTWROS and 272,727 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(43) Consists of 100,000 shares of common stock held directly by Daniel Rudder and 100,000 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(44) Consists of 340,910 shares of common stock held directly by Savoy International Corporation and 340,910 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(45) Consists of 227,273 shares of common stock held directly by R. Douglas Shearer and 227,273 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(46) Consists of 113,636 shares of common stock held directly by Alva Staples and 113,636 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(47) Consists of 56,859 shares of common stock issuable to Ralph Tenebruso upon exercise of warrants issued as 2015 Placement Agent Warrants. Mr. Tenebruso is an employee of Newport Coast Securities, Inc.
(48) Consists of 11,365 shares of common stock issuable to James Terwilliger upon exercise of warrants issued as 2015 Placement Agent Warrants. Mr. Terwilliger is an employee of Newport Coast Securities, Inc.
(49) Consists of 238,637 shares of common stock held directly by Thomas Green/Ardell Family Trust and 238,637 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(50) Consists of 177,273 shares of common stock held directly by Albert Tjan and 177,273 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(51) Consists of 68,182 shares of common stock held directly by Joseph Tosti and 68,182 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
(52) Consists of 11,365 shares of common stock issuable to Jeff Stockstill upon exercise of warrants issued as 2015 Placement Agent Warrants. Mr. Stockstill is an employee of Newport Coast Securities, Inc.
(53) Consists of 113,636 shares of common stock held directly by Mulhern Family Revocable Trust and 113,636 shares of common stock issuable upon exercise of warrants issued in the 2015 Securities Placement.
103
(54) Consists of 50,000 shares of our common stock issuable upon exercise of warrants that we issued in 2014. For the avoidance of doubt, the securities owned by Dina Lyaskovets were not issued in the 2015 Securities Placement.
(55) The beneficial ownership and selling stockholder information in the table above is presented assuming no limitation on the exercise of warrants and conversion of Series C Preferred Stock described below in footnote 56. Without giving effect to such limitations, as of December 31, 2015, (i) Biotechnology Value Fund, L.P. ("BVF") beneficially owned 31,544,723 shares of Common Stock, including 15,557,560 shares of Common Stock issuable upon the exercise of certain warrants held by it, (ii) Biotechnology Value Fund II, L.P. ("BVF2") beneficially owned 18,501,586 shares of Common Stock, including 9,104,300 shares of Common Stock issuable upon the exercise of certain warrants held by it, (iii) BVF Investments, L.L.C. ("BVLLC") beneficially owned 352,980 shares of Common Stock, (iv) Investment 10, L.L.C. ("ILL10") beneficially owned 6,184,885 shares of Common Stock, including 3,056,638 shares of Common Stock issuable upon the exercise of certain warrants held by it, (v) MSI BVF SPV, L.L.C. ("MSI BVF") beneficially owned 6,128,172 shares of Common Stock, including 3,064,086 shares of Common Stock issuable upon the exercise of certain warrants held by it, and (vi) Biotechnology Value Trading Fund OS, L.P. ("Fund OS") beneficially owned 6,172,728 shares of Common Stock, including 3,086,364 shares of Common Stock issuable upon the exercise of certain warrants held by it.
BVF Partners L.P. ("Partners"), as the general partner of BVF and BVF2, the manager of BVLLC and the investment adviser of ILL10, may be deemed to beneficially own 68,885,074 shares of Common Stock, including 54,323,494 shares of Common Stock currently issuable upon the exercise of certain warrants and conversion of Series C Preferred Stock, beneficially owned in the aggregate by BVF, BVF2, BVLLC, ILL10, and MSI BVF.
BVF Inc., as the general partner of Partners, may be deemed to beneficially own the 68,885,074 shares of Common Stock, including 54,323,494 shares of Common Stock currently issuable upon the exercise of certain warrants, beneficially owned by Partners.
Mark N. Lampert, as a director and officer of BVF Inc., may be deemed to beneficially own the 68,885,074 shares of Common Stock, including 54,323,494 shares of Common Stock currently issuable upon the exercise of certain warrants, beneficially owned by BVF Inc.
The foregoing should not be construed in and of itself as an admission by any of Partners, BVF Inc. or Mark N. Lampert as to beneficial ownership of any shares of Common Stock owned by BVF, BVF2, BVLLC, ILL10, and MSI BVF. Each of Partners, BVF Inc. and Mr. Lampert disclaims beneficial ownership of the shares of Common Stock beneficially owned by BVF, BVF2, BVLLC, ILL10, and MSI BVF and this filing shall not be construed as an admission that any such person or entity is the beneficial owner of any such securities.
(56) The warrants and Series C Preferred Stock issued in the 2015 Securities Placement each contain an issuance limitation prohibiting the holder from exercising such warrants and converting such Series C Preferred Stock to the extent that, after giving effect to such exercise or conversion thereof, the holder, including any of its affiliates, would beneficially own more than 9.99% of the Common Stock of the Company then issued and outstanding.
104
DESCRIPTION OF CAPITAL STOCK
As of December 31, 2015, we were authorized to issue up to 200,000,000 shares of common stock and 10,000,000 shares of preferred stock under our Amended and Restated Certificate of Incorporation. The preferred stock is divided into three series: 1,250,000 shares of preferred stock are designated "Series A Convertible Preferred Stock," 1,600,000 shares of preferred stock are designated "Series B Convertible Preferred Stock" and 5,000 share of preferred stock are designated "Series C Convertible Preferred Stock"
Common Stock
As of December 31, 2015, we had 151,559,745 shares of common stock outstanding. As of December 31, 2015, there were 12,572,591 shares of common stock issuable upon the exercise of outstanding stock options and 53,247,877 shares of common stock issuable upon the exercise of warrants to purchase common stock.
Holders of shares of common stock are entitled to one vote per share on all matters to be voted upon by the stockholders and are not entitled to cumulate votes for the election of directors. Subject to preferences that may be applicable to any outstanding shares of preferred stock, including our Series B Convertible Preferred Stock, holders of shares of common stock are entitled to receive ratably such dividends, if any, as may be declared from time to time by our board of directors out of funds legally available therefor. In the event of liquidation, dissolution or winding up of our company, the holders of shares of common stock are entitled to share ratably in all assets remaining after payment of liabilities, subject to prior distributions rights applicable to any outstanding shares of preferred stock. Shares of common stock have no preemptive, conversion or other subscription rights, and there are no redemption or sinking fund provisions applicable to the common stock.
Preferred Stock
As of December 31, 2015, there were (i) issued and outstanding 526,080 shares of Series B Convertible Preferred Stock, (ii) issued and outstanding 4,500 shares of Series C Preferred Stock and (iii) 896,037 shares of Series B Convertible Preferred stock issuable upon the exercise of warrants to purchase Series B Convertible Preferred Stock. As of December 31, 2015, no shares of Series A Convertible Preferred Stock were issued and outstanding.
Under our Amended and Restated Certificate of Incorporation, our board of directors has the authority to issue preferred stock in one or more series and to fix the rights, preferences, privileges and restrictions, including the dividend, conversion, voting, redemption (including sinking fund provisions), and other rights, liquidation preferences, and the number of shares constituting any series and the designations of such series, without any further vote or action by our stockholders.
Because the terms of the preferred stock may be fixed by our board of directors without stockholder action, the preferred stock could be issued quickly with terms calculated to defeat a proposed takeover of our company or to make the removal of our management more difficult. Under certain circumstances this could have the effect of decreasing the market price of our common stock.
Series B Convertible Preferred Stock
All shares of Series B Convertible Preferred Stock and warrants to purchase shares of Series B Convertible Preferred Stock currently are owned by Elan Corporation plc. The Series B Convertible Preferred Stock is non-voting stock. Each share of Series B Convertible Preferred Stock is convertible into one shares of our common stock, provided that no conversion may be effected that would result in the holders of Series B Convertible Preferred Stock owning more than 9.9% of our common stock on a fully converted to common stock basis. If we pay a cash dividend on our common stock, we also must pay the same dividend on an as converted basis on the Series B Convertible Preferred Stock.
Series C Convertible Preferred Stock
105
All shares of Series C Preferred Stock are owned by Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P. The Series C Preferred Stock is non-voting stock. Each share of Series C Preferred Stock are collectively convertible into an aggregate of shares of our common stock and (ii) warrants to purchase shares of our common stock, in each case subject to adjustment, provided that no conversion may be effected that would result in the holders of Series C Convertible Preferred Stock owning more than 9.99% of our common stock on a fully converted to common stock basis. Dividends on the Series C Preferred Stock are due whenever dividends are due on the Company's common stock on an as-if-converted basis, but shall be subordinate to any dividends due to holders of the Company's Series B Convertible Preferred Stock as a result of such common stock dividends. The Series C Preferred Stock shall also be junior to the Series B Convertible Preferred Stock in the event of liquidation of the Company.
Warrants
Effective February 15, 2013, the Company and each of Xmark JV Investment Partners, LLC, Xmark Opportunity Fund, Ltd. and Xmark Opportunity Fund, L.P. (collectively, the "Xmark Entities") entered into a Warrant Repricing, Exercise and Lockup Agreement (the "Xmark Warrant Agreement") pursuant to which the Company agreed to reduce the exercise price of outstanding warrants to purchase an aggregate of up to 59,149,999 shares of Common Stock held by the Xmark Entities (the "Xmark Warrants") to $0.01 per share. In consideration for the reduction of the exercise price of the Xmark Warrants, each of the Xmark Entities agreed to immediately exercise all of the Xmark Warrants. The Xmark Warrant Agreement also provides that the Xmark Entities will not transfer the shares issuable upon exercise of the Xmark Warrants (the "Xmark Warrant Shares") until the Company either (i) declares a cash dividend on its common stock or otherwise makes a cash distribution or (ii) effects a Change of Control, subject in each case to the terms of the Xmark Warrant Agreement.
As of December 31, 2015, warrants to purchase an aggregate of 53,247,877 shares of common stock were outstanding with a weighted average exercise price of $0.24 per share. Details of the warrants for common stock outstanding at December 31, 2015 are as follows:
|
Number of Shares
|
Exercise Price
|
Expiration Date
|
|||||
|
50,000
|
$
|
0.50
|
May 2016
|
||||
|
50,000
|
$
|
0.50
|
July 2016
|
||||
|
50,000
|
$
|
1.00
|
July 2016
|
||||
|
50,000
|
$
|
1.50
|
July 2016
|
||||
|
50,000
|
$
|
2.00
|
July 2016
|
||||
|
50,000
|
$
|
2.50
|
July 2016
|
||||
|
1,337,627
|
$
|
0.40
|
March 2017
|
||||
|
325,000
|
$
|
0.40
|
April 2017
|
||||
|
300,000
|
$
|
0.258
|
June 2017
|
||||
|
50,000
|
$
|
0.26
|
June 2017
|
||||
|
140,000
|
$
|
0.35
|
October 2017
|
||||
|
12,205,000
|
$
|
0.25
|
February 2018
|
||||
|
1,242,000
|
$
|
0.25
|
March 2018
|
||||
|
50,000
|
$
|
0.49
|
January 2020
|
||||
|
37,298,250
|
$
|
0.22
|
December 2020
|
||||
|
53,247,877
|
|||||||
As of December 31, 2015, one warrant to purchase an aggregate of 896,037 shares of preferred stock was outstanding. The warrant has an exercise price of $0.01 per share and expires in February 2016.
Section 203 of the Delaware Corporation Law
Section 203 of the General Corporation Law of the State of Delaware (the "DGCL") prevents an "interested stockholder" (defined in Section 203 of the DGCL, generally, as a person owning 15% or more of a corporation's outstanding voting stock), from engaging in a "business combination" (as defined in Section 203 of the DGCL) with a publicly-held Delaware corporation for three years following the date such person became an interested stockholder, unless:
106
| · | before such person became an interested stockholder, the board of directors of the corporation approved the transaction in which the interested stockholder became an interested stockholder or approved the business combination; |
| · | upon consummation of the transaction that resulted in the interested stockholder's becoming an interested stockholder, the interested stockholder owns at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced (excluding stock held by directors who are also officers of the corporation and by employee stock plans that do not provide employees with the rights to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer); or |
| · | following the transaction in which such person became an interested stockholder, the business combination is approved by the board of directors of the corporation and authorized at a meeting of stockholders by the affirmative vote of the holders of two-thirds of the outstanding voting stock of the corporation not owned by the interested stockholder. |
Our certificate of incorporation expressly provides that the provisions of Section 203 of the DGCL do not apply. Consequently, a person or entity wishing to acquire control of our company would not have to comply with the director or stockholder approvals required by Section 203. This could make a takeover of our company easier even if the takeover were not approved by the board of directors or opposed by the stockholders as not being in their best interests.
Limitation of Liability
Section 145 of the DGCL provides a detailed statutory framework covering indemnification of officers and directors against liabilities and expenses arising out of legal proceedings brought against them by reason of their being or having been directors or officers. Section 145 generally provides that a director or officer of a corporation:
| · | shall be indemnified by the corporation for all expenses of such legal proceedings when he is successful on the merits; |
| · | may be indemnified by the corporation for the expenses, judgments, fines and amounts paid in settlement of such proceedings (other than a derivative suit), even if he is not successful on the merits, if he acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful; and |
| · | may be indemnified by the corporation for the expenses of a derivative suit (a suit by a stockholder alleging a breach by a director or officer of a duty owed to the corporation), even if he is not successful on the merits, if he acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation. |
The indemnification discussed in clauses two and three above may be made only upon a determination that indemnification is proper because the applicable standard of conduct has been met. Such a determination may be made by a majority of a quorum of disinterested directors, independent legal counsel, the stockholders or a court of competent jurisdiction. The indemnification discussed in clause three above may not apply, however, if the director or officer is adjudged liable for negligence or misconduct in the performance of his duties to the corporation, unless a corporation determines that despite such adjudication, but in view of all the circumstances, he is entitled to indemnification.
Article Sixth of our certificate of incorporation provides in substance that, to the fullest extent permitted by the DGCL as it now exists or as amended, each director and officer shall be indemnified against reasonable costs and expenses, including attorney's fees, and any liabilities which he may incur in connection with any action to which he may be made a party by reason of his being or having been a director or officer of our company. The indemnification provided by our certificate of incorporation is not deemed exclusive of or intended in any way to limit any other rights to which any person seeking indemnification may be entitled. Section 102(b)(7) of the DGCL permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, except for liability
107
| · | for any breach of the director's duty of loyalty to the corporation or its stockholders, |
| · | for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, |
| · | under Section 174 of the DGCL, or |
| · | for any transaction from which the director derived an improper personal benefit. |
Article Eighth of our certificate of incorporation provides for the elimination of personal liability of a director for breach of fiduciary duty, as permitted by Section 102(b)(7) of the DGCL. We maintain liability insurance on our officers and directors against liabilities that they may incur in such capacities. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling our company pursuant to the foregoing provisions, we have been informed that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Anti-Takeover Effects
Bylaws
Our Bylaws are designed to make it difficult for a third party to acquire control of us, even if a change of control would be beneficial to stockholders. Our Bylaws do not permit any person other than the board of directors or certain executive officers to call special meetings of the stockholders. In addition, we must receive a stockholders' proposal for an annual meeting within a specified period for that proposal to be included on the agenda. Because stockholders do not have the power to call meetings and are subject to timing requirements in submitting stockholder proposals for consideration at an annual or special meeting, any third-party takeover not supported by the board of directors would be subject to significant delays and difficulties.
No Cumulative Voting
The DGCL provides that stockholders are denied the right to cumulate votes in the election of directors unless our amended and restated certificate of incorporation provides otherwise. Our amended and restated certificate of incorporation does not provide for cumulative voting.
Undesignated Preferred Stock
The authority that is possessed by our Board of Directors to issue preferred stock could potentially be used to discourage attempts by third parties to obtain control of our company through a merger, tender offer, proxy contest or otherwise by making such attempts more difficult or more costly. Our Board of Directors may issue preferred stock with voting rights or conversion rights that, if exercised, could adversely affect the voting power of the holders of common stock.
Authorized but Unissued Shares
Our authorized but unissued shares of common stock and preferred stock will be available for future issuance without stockholder approval. We may use additional shares for a variety of purposes, including future offerings to raise additional capital, to fund acquisitions and as employee compensation. The existence of authorized but unissued shares of common stock and preferred stock could render more difficult or discourage an attempt to obtain control of us by means of a proxy contest, tender offer, merger or otherwise.
108
The combination of the provisions summarized above will make it more difficult for our stockholders to replace our Board of Directors as well as for another party to obtain control of us by replacing our Board of Directors. Therefore, these provisions may have the effect of deterring hostile takeovers or delaying changes in our control or management. These provisions are intended to enhance the likelihood of continued stability in the composition of our Board of Directors and in the policies they implement, and to discourage certain types of transactions that may involve an actual or threatened change of our control. These provisions are designed to reduce our vulnerability to an unsolicited acquisition proposal. The provisions also are intended to discourage certain tactics that may be used in proxy fights. However, such provisions could have the effect of discouraging others from making tender offers for our shares and, as a consequence, they also may inhibit fluctuations in the market price of our shares that could result from actual or rumored takeover attempts. Such provisions may also have the effect of preventing changes in our management.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is American Stock Transfer and Trust Company.
109
PLAN OF DISTRIBUTION
The selling stockholders, which as used herein includes donees, pledgees, transferees or other successors-in-interest selling shares of common stock or interests in shares of common stock received after the date of this prospectus from a selling stockholder as a gift, pledge, partnership distribution or other transfer, may, from time to time, sell, transfer or otherwise dispose of any or all of their shares of common stock or interests in shares of common stock on any stock exchange, market or trading facility on which the shares are traded or in private transactions. These dispositions may be at fixed prices, at prevailing market prices at the time of sale, at prices related to the prevailing market price, at varying prices determined at the time of sale, or at negotiated prices.
The selling stockholders may use any one or more of the following methods when disposing of shares or interests therein:
| – | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| – | block trades in which the broker-dealer will attempt to sell the shares as agent, but may position and resell a portion of the block as principal to facilitate the transaction; |
| – | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| – | an exchange distribution in accordance with the rules of the applicable exchange; |
| – | privately negotiated transactions; |
| – | short sales effected after the date the registration statement of which this prospectus is a part is declared effective by the Commission; |
| – | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
– broker-dealers may agree with the selling stockholders to sell a specified number of such shares at a stipulated price per share;
| – | a combination of any such methods of sale; and |
| – | any other method permitted by applicable law. |
The selling stockholders may, from time to time, pledge or grant a security interest in some or all of the shares of common stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of common stock, from time to time, under this prospectus, or under an amendment to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act amending the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus. The selling stockholders also may transfer the shares of common stock in other circumstances, in which case the transferees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
In connection with the sale of our common stock or interests therein, the selling stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the common stock in the course of hedging the positions they assume. The selling stockholders may also sell shares of our common stock short and deliver these securities to close out their short positions, or loan or pledge the common stock to broker-dealers that in turn may sell these securities. The selling stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or the creation of one or more derivative securities which require the delivery to such broker-dealer or other financial institution of shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
110
The aggregate proceeds to the selling stockholders from the sale of the common stock offered by them will be the purchase price of the common stock less discounts or commissions, if any. Each of the selling stockholders reserves the right to accept and, together with their agents from time to time, to reject, in whole or in part, any proposed purchase of common stock to be made directly or through agents. We will not receive any of the proceeds from this offering. Upon any exercise of the warrants by payment of cash, however, we will receive the exercise price of the warrants.
The selling stockholders also may resell all or a portion of the shares in open market transactions in reliance upon Rule 144 under the Securities Act of 1933, provided that they meet the criteria and conform to the requirements of that rule.
The selling stockholders and any underwriters, broker-dealers or agents that participate in the sale of the common stock or interests therein may be "underwriters" within the meaning of Section 2(11) of the Securities Act. Any discounts, commissions, concessions or profit they earn on any resale of the shares may be underwriting discounts and commissions under the Securities Act. Selling stockholders who are "underwriters" within the meaning of Section 2(11) of the Securities Act will be subject to the prospectus delivery requirements of the Securities Act.
Each selling stockholder has advised us that they have not entered into any written or oral agreements, understandings or arrangements with any underwriter or broker-dealer regarding the sale of the resale shares. There is no underwriter or coordinating broker acting in connection with the proposed sale of the resale shares by the selling stockholders.
To the extent required, the shares of our common stock to be sold, the names of the selling stockholders, the respective purchase prices and public offering prices, the names of any agents, dealer or underwriter, any applicable commissions or discounts with respect to a particular offer will be set forth in an accompanying prospectus supplement or, if appropriate, a post-effective amendment to the registration statement that includes this prospectus.
In order to comply with the securities laws of some states, if applicable, the common stock may be sold in these jurisdictions only through registered or licensed brokers or dealers. In addition, in some states the common stock may not be sold unless it has been registered or qualified for sale or an exemption from registration or qualification requirements is available and is complied with.
We have advised the selling stockholders that the anti-manipulation rules of Regulation M under the Exchange Act may apply to sales of shares in the market and to the activities of the selling stockholders and their affiliates. In addition, to the extent applicable we will make copies of this prospectus (as it may be supplemented or amended from time to time) available to the selling stockholders for the purpose of satisfying the prospectus delivery requirements of the Securities Act. The selling stockholders may indemnify any broker-dealer that participates in transactions involving the sale of the shares against certain liabilities, including liabilities arising under the Securities Act.
We have agreed to indemnify the selling stockholders against certain liabilities, including liabilities under the Securities Act and state securities laws, relating to the registration of the shares offered by this prospectus.
We have agreed with each selling stockholder to keep the registration statement of which this prospectus constitutes a part effective until the earlier of (1) such time as all of the shares covered by this prospectus have been disposed of pursuant to the registration statement or Rule 144 of the Securities Act or (2) the date on which the shares may be sold by such selling stockholder without volume restrictions pursuant to Rule 144 of the Securities Act.
111
LEGAL MATTERS
Drinker Biddle & Reath LLP, Philadelphia, Pennsylvania, passed upon certain legal matters for us in connection with the offered securities.
EXPERTS
The consolidated financial statements included in this prospectus and elsewhere in the registration statement have been included in this prospectus in reliance upon the report of Grant Thornton LLP, independent registered public accountants, upon the authority of said firm as experts in accounting and auditing.
ADDITIONAL INFORMATION
We have filed with the SEC a Registration Statement on Form S-1 under the Securities Act with respect to the securities offered in this prospectus. This prospectus, which forms a part of such registration statement, does not contain all of the information set forth in the registration statement or the exhibits and schedules filed with it. When we make references in this prospectus to any of our agreements or other documents, the references are not necessarily complete and you should refer to the exhibits filed with the registration statement for copies of the actual agreements or other documents.
We are subject to the information and periodic reporting requirements of the Exchange Act and in accordance therewith file reports, proxy statements and other information with the SEC. Such reports, proxy statements, other information, and a copy of the registration statement and its exhibits may be inspected by anyone without charge and copies of these materials may be obtained upon the payment of the fees prescribed by the SEC, at the Public Reference Room maintained by the SEC at 100 F Street, N.E., Washington, D.C. 20549, on official business days during the hours of 10 a.m. to 3 p.m. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The registration statement and the reports, proxy statements and other information filed by us are also available through the SEC's website on the World Wide Web at www.sec.gov, as well as on our website at www.aeoluspharma.com.
112
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
We disclose important information to you by referring you to documents that we have previously filed with the SEC or documents that we will file with the SEC in the future. The information incorporated by reference is considered to be part of this prospectus, and information in documents that we file later with the SEC will automatically update and supersede information in this prospectus. We incorporate by reference the documents listed below into this prospectus, and any future filings made by us with the SEC under Section 13(a), 13(c), 14 or 15(d) or the Exchange Act until we close this offering. We hereby incorporate by reference our Annual Report on Form 10-K filed with the SEC on December 18, 2015.
Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus is modified or superseded for purposes of the prospectus to the extent that a statement contained in this prospectus or in any other subsequently filed document which also is or is deemed to be incorporated by reference herein modifies or supersedes such statement. Any statement so modified or superseded does not, except as so modified or superseded, constitute a part of this prospectus.
You may request a copy of these filings, at no cost, by written or oral request made to us at the following address or telephone number:
Aeolus Pharmaceuticals, Inc.
26361 Crown Valley Parkway, Suite 150
Mission Viejo, CA 92691
(212) 380-1210
113
INDEX TO FINANCIAL STATEMENTS
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|||
|
Consolidated Balance Sheets – As of September 30, 2015 and 2014
|
F-3
|
|||
|
Consolidated Statements of Operations – For the fiscal years ended September 30, 2015 and 2014
|
F-4
|
|||
|
Consolidated Statements of Stockholders' Equity (Deficit) – For the fiscal years ended September 30, 2015 and 2014
|
F-5
|
|||
|
Consolidated Statements of Cash Flows – For the fiscal years ended September 30, 2015 and 2014
|
F-6
|
|||
|
Notes to Consolidated Financial Statements
|
F-7
|
|||
AEOLUS PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
(DOLLARS IN THOUSANDS)
CONSOLIDATED BALANCE SHEETS
(DOLLARS IN THOUSANDS)
|
|
September 30,
|
|||||||
|
|
2015
|
2014
|
||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$
|
94
|
$
|
1,517
|
||||
|
Accounts receivable
|
1,585
|
1,559
|
||||||
|
Deferred subcontractor cost
|
21
|
426
|
||||||
|
Prepaid expenses and other current assets
|
45
|
46
|
||||||
|
Total current assets
|
1,745
|
3,548
|
||||||
|
Investment in CPEC LLC
|
32
|
32
|
||||||
|
Total assets
|
$
|
1,777
|
$
|
3,580
|
||||
|
LIABILITIES AND STOCKHOLDERS' (DEFICIT) EQUITY
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable and accrued expenses
|
$
|
1,598
|
$
|
1,552
|
||||
|
Deferred revenue
|
22
|
443
|
||||||
|
Note payable to shareholders, net of debt discount of $273
|
727
|
—
|
||||||
|
Note payable to shareholders redemption liability
|
275
|
—
|
||||||
|
Total current liabilities
|
2,622
|
1,995
|
||||||
|
Total liabilities
|
2,622
|
1,995
|
||||||
|
Commitments and Contingencies (Notes E and K)
|
||||||||
|
Stockholders' equity:
|
||||||||
|
Preferred stock, $0.01 par value per share, 10,000,000 shares authorized:
|
||||||||
|
Series A nonredeemable convertible preferred stock, 1,250,000 shares authorized as of September 30, 2015 and 2014, respectively; no shares issued and outstanding as of September 30, 2015 and 2014, respectively
|
—
|
—
|
||||||
|
Series B nonredeemable convertible preferred stock, 1,600,000 and 1,600,000 shares authorized as of September 30, 2015 and 2014, respectively; 526,080 and 526,080 shares issued and outstanding as of September 30, 2015 and 2014, respectively
|
5
|
5
|
||||||
|
Common stock, $0.01 par value per share, 200,000,000 shares authorized; 135,930,068 and 135,850,068 shares issued and outstanding at September 30, 2015 and 2014, respectively
|
1,359
|
1,359
|
||||||
|
Additional paid-in capital
|
184,421
|
184,223
|
||||||
|
Accumulated deficit
|
(186,630
|
)
|
(184,002
|
)
|
||||
|
Total stockholders' (deficit) equity
|
(845
|
)
|
1,585
|
|||||
|
Total liabilities and stockholders' (deficit) equity
|
$
|
1,777
|
$
|
3,580
|
||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
AEOLUS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(IN THOUSANDS, EXCEPT PER SHARE DATA)
CONSOLIDATED STATEMENTS OF OPERATIONS
(IN THOUSANDS, EXCEPT PER SHARE DATA)
|
Fiscal Year Ended September 30,
|
||||||||
|
2015
|
2014
|
|||||||
|
Revenue:
|
||||||||
|
Contract revenue
|
$
|
3,111
|
$
|
9,631
|
||||
|
Costs and expenses:
|
||||||||
|
Research and development
|
3,509
|
6,966
|
||||||
|
General and administrative
|
2,228
|
2,745
|
||||||
|
Total costs and expenses
|
5,737
|
9,711
|
||||||
|
Loss from operations
|
(2,626
|
)
|
(80
|
)
|
||||
|
Interest expense
|
2
|
—
|
||||||
|
Net loss
|
$
|
(2,628
|
)
|
$
|
(80
|
)
|
||
|
Net loss attributable to common stockholders – basic
|
$
|
(2,628
|
)
|
$
|
(80
|
)
|
||
|
Net loss attributable to common stockholders – diluted
|
$
|
(2,628
|
)
|
$
|
(80
|
)
|
||
|
Basic net loss per common share
|
$
|
(0.02
|
)
|
$
|
0.00
|
|||
|
Diluted net loss per common share
|
$
|
(0.02
|
)
|
$
|
0.00
|
|||
|
Weighted average common shares outstanding:
|
||||||||
|
Basic
|
135,883
|
134,667
|
||||||
|
Diluted
|
135,883
|
134,667
|
||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
AEOLUS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT)
(Dollars in thousands)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT)
(Dollars in thousands)
|
Series B Preferred Stock
|
Common Stock
|
Additional
|
Accumulated
|
Total Stockholders'
|
||||||||||||||||||||||||
|
Shares
|
Par Value
|
Shares
|
Par Value
|
Paid-in Capital
|
Deficit
|
Equity (Deficit)
|
||||||||||||||||||||||
|
Balance at September 30, 2013
|
526,080
|
$
|
5
|
134,550,068
|
$
|
1,346
|
$
|
183,276
|
$
|
(183,922
|
)
|
$
|
705
|
|||||||||||||||
|
Exercise of warrants
|
—
|
—
|
1,300,000
|
13
|
312
|
—
|
325
|
|||||||||||||||||||||
|
Issuance of warrants to consultants
|
—
|
—
|
—
|
—
|
9
|
—
|
9
|
|||||||||||||||||||||
|
Stock-based compensation
|
—
|
—
|
—
|
—
|
626
|
—
|
626
|
|||||||||||||||||||||
|
Net loss for the fiscal year ended September 30, 2014
|
—
|
—
|
—
|
—
|
—
|
(80
|
)
|
(80
|
)
|
|||||||||||||||||||
|
Balance at September 30, 2014
|
526,080
|
5
|
135,850,068
|
1,359
|
184,223
|
(184,002
|
)
|
1,585
|
||||||||||||||||||||
|
Exercise of warrants
|
—
|
—
|
80,000
|
—
|
20
|
—
|
20
|
|||||||||||||||||||||
|
Issuance of warrants to consultants
|
—
|
—
|
—
|
—
|
14
|
—
|
14
|
|||||||||||||||||||||
|
Stock-based compensation
|
—
|
—
|
—
|
—
|
164
|
—
|
164
|
|||||||||||||||||||||
|
Net loss for the fiscal year end September 30, 2015
|
—
|
—
|
—
|
—
|
—
|
(2,628
|
)
|
(2,628
|
)
|
|||||||||||||||||||
|
Balance at September 30, 2015
|
526,080
|
$
|
5
|
135,930,068
|
$
|
1,359
|
$
|
184,421
|
$
|
(186,630
|
)
|
$
|
(845
|
)
|
||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
AEOLUS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(IN THOUSANDS)
|
Fiscal Year Ended September 30,
|
||||||||
|
2015
|
2014
|
|||||||
|
Cash flows from operating activities:
|
||||||||
|
Net loss
|
$
|
(2,628
|
)
|
$
|
(80
|
)
|
||
|
Adjustments to reconcile net loss to net cash (used in) provided by operating activities:
|
||||||||
|
Accrued interest
|
2
|
—
|
||||||
|
Noncash compensation
|
178
|
635
|
||||||
|
Change in assets and liabilities:
|
||||||||
|
Accounts receivable
|
(26
|
)
|
(1,189
|
)
|
||||
|
Deferred subcontractor cost
|
405
|
230
|
||||||
|
Prepaid expenses and other current assets
|
1
|
(7
|
)
|
|||||
|
Accounts payable and accrued expenses
|
46
|
973
|
||||||
|
Deferred revenue
|
(421
|
)
|
(239
|
)
|
||||
|
Net cash (used in) provided by operating activities
|
(2,443
|
)
|
323
|
|||||
|
|
||||||||
|
Cash flows from financing activities:
|
||||||||
|
Proceeds from exercise of common stock warrants
|
20
|
325
|
||||||
|
Proceeds from issuance of note payable to shareholders
|
1,000
|
—
|
||||||
|
Net cash provided by financing activities
|
1,020
|
325
|
||||||
|
|
||||||||
|
Net (decrease) increase in cash and cash equivalents
|
(1,423
|
)
|
648
|
|||||
|
|
||||||||
|
Cash and cash equivalents at beginning of year
|
1,517
|
869
|
||||||
|
Cash and cash equivalents at end of year
|
$
|
94
|
$
|
1,517
|
||||
|
|
||||||||
|
Supplemental disclosure of non-cash financing activities:
|
||||||||
|
Note payable to shareholders redemption liability
|
$
|
275
|
$
|
—
|
||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
AEOLUS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
SEPTEMBER 30, 2015 and 2014
A. Organization, Business and Summary of Significant Accounting Policies
Organization
The accompanying audited consolidated financial statements include the accounts of Aeolus Pharmaceuticals, Inc. and its wholly-owned subsidiary, Aeolus Sciences, Inc. (collectively "we," "us," "Company" or "Aeolus"). All significant intercompany accounts and transactions have been eliminated in consolidation. Aeolus is a Delaware corporation. The Company's primary operations are located in Mission Viejo, California.
Business
Aeolus is developing a new class of broad-spectrum, catalytic antioxidant compounds based on technology discovered at Duke University and National Jewish Health. The Company's lead compound, 10150, is a metalloporphyrin specifically designed to neutralize reactive oxygen and nitrogen species. The Company is developing 10150 as a medical countermeasure against the pulmonary effects of radiation exposure under a contract ("BARDA Contract") valued at up to $118.4 million with the Biomedical Advanced Research and Development Authority ("BARDA"), a division of the Department of Health and Human Services ("HHS"). Additionally, Aeolus receives development support from the National Institutes of Health ("NIH") for development of the compound as a medical countermeasure against radiation and chemical exposure.
B. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements include the accounts of Aeolus and its wholly owned subsidiary. All significant intercompany accounts and transactions have been eliminated. The Company uses the equity method to account for its 35% ownership interest in CPEC, which is further discussed in Note D.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Such estimates include revenue recognition, allowance for doubtful accounts and stock-based compensation. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company invests available cash in short-term bank deposits. Cash and cash equivalents include investments with maturities of three months or less at the date of purchase. The carrying value of cash and cash equivalents approximate their fair market value at September 30, 2015 and 2014 due to their short-term nature. Also, the Company maintains cash balances with financial institutions in excess of federally insured limits. The Company does not anticipate any losses with such cash balances.
F-7
Significant customers and accounts receivable
For the year ended September 30, 2015, the Company's only customer was BARDA. For the year ended September 30, 2015, revenues from BARDA comprised 100% of total revenues. As of September 30, 2015, the Company's receivable balances were comprised 100% from this customer. Unbilled accounts receivable, included in accounts receivable, totaling $589,000 as of September 30, 2015 relate to work that has been performed, though invoicing has not yet occurred. All of the unbilled receivables are expected to be billed and collected within the next year. Accounts receivable are stated at invoice amounts and consist primarily of amounts due from HHS as well as amounts due under reimbursement contracts with other government entities and non-government and philanthropic organizations. If necessary, the Company records a provision for doubtful receivables to allow for any amounts which may be unrecoverable. This provision is based upon an analysis of the Company's prior collection experience, customer creditworthiness and current economic trends. As of September 30, 2015 and 2014, an allowance for doubtful accounts was not recorded as the collection history from the Company's customers indicated that collection was probable.
Concentrations of credit risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents and accounts receivable. The Company places its cash and cash equivalents with high quality financial institutions. Management believes that the financial risks associated with its cash and cash equivalents and investments are minimal. Because accounts receivable consist entirely of amounts due from the U.S. federal government agencies, management deems there to be minimal credit risk.
Revenue Recognition
Aeolus recognizes revenue in accordance with the authoritative guidance for revenue recognition. Revenue is recognized when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller's price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured.
The BARDA Contract is classified as a "cost-plus-fixed-fee" contract. Aeolus recognizes government contract revenue in accordance with the authoritative guidance for revenue recognition including the authoritative guidance specific to federal government contracts. Reimbursable costs under the contract primarily include direct labor, subcontract costs, materials, equipment, travel, and indirect costs.
Aeolus estimates subcontractor costs, materials and related revenue for some vendors when invoices are not received timely. We receive regular updates from our subcontractors regarding estimated completion of individual projects. Management evaluates the status of each project with respect to budgeted work completed, actual work completed, and cost of actual work completed. We are required to provide BARDA with monthly reports in addition to our bi-weekly conference calls with BARDA regarding the progress of each project.
In addition, we receive a fixed fee on consultant labor and subcontract labor under the BARDA Contract, which is unconditionally earned as allowable costs are incurred and is not contingent on success factors. Reimbursable costs under this BARDA Contract, including the fixed fee, are generally recognized as revenue in the period the reimbursable costs are incurred and become billable.
Fair Value of Financial Instruments
The carrying amounts of our short-term financial instruments, which include cash and cash equivalents, accounts receivable, accounts payable, accrued liabilities, debt, and redemption liability, approximate their fair values due to their short maturities.
Fair Value Measurements
The Company adopted Accounting Standards Codification ("ASC") Topic 820, Fair Value Measurements and Disclosures, for financial and non-financial assets and liabilities.
F-8
ASC 820 discusses valuation techniques, such as the market approach (comparable market prices), the income approach (present value of future income or cash flow) and the cost approach (cost to replace the service capacity of an asset or replacement cost). The Company utilizes the market approach. The statement utilizes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. The following is a brief description of those three levels:
| · | Level 1: Observable inputs such as quoted prices (unadjusted) in active markets for identical assets or liabilities. |
| · | Level 2: Inputs other than quoted prices that are observable for the asset or liability, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active. |
| · | Level 3: Unobservable inputs that reflect the reporting entity's own assumptions. |
The redemption liability, discussed further at Note F. Debt, is measured at fair market value on a recurring basis as of September 30, 2015 and is summarized below (in thousands):
|
Fair value at September 30, 2015
|
||||||||||
|
Level 1
|
Level 2
|
Level 3
|
||||||||
|
$
|
275
|
$
|
—
|
$
|
—
|
|||||
The following table summarizes, as of September 30, 2015, the redemption liability activity subject to Level 1 inputs which are measured on a recurring basis:
|
Fair value measurement of redemption liability
|
||||
|
Balance at September 30, 2014
|
$
|
—
|
||
|
Issuance of convertible promissory notes with redemption feature
|
275
|
|||
|
Change in fair value of redemption liability
|
—
|
|||
|
Balance at September 30, 2015
|
$
|
275
|
||
Research and Development
Research and development costs are expensed in the period incurred.
Leases
The Company leases office space and office equipment under month to month operating lease agreements. For the years ended September 30, 2015 and 2014, total rent expense was approximately $42,000 and $42,000, respectively.
Income Taxes
The Company recognizes liabilities or assets for the deferred tax consequences of temporary differences between the tax bases of assets or liabilities and their reported amounts in the financial statements. These temporary differences will result in taxable or deductible amounts in future years when the reported amounts of the assets or liabilities are recovered or settled. A valuation allowance is established when management determines that it is more likely than not that all or a portion of a deferred tax asset will not be realized. Management evaluates the Company's ability to realize its net deferred tax assets on a quarterly basis and valuation allowances are provided, as necessary. During this evaluation, management reviews its forecasts of income in conjunction with other positive and negative evidence surrounding the Company's ability to realize its deferred tax assets to determine if a valuation allowance is required. Adjustments to the valuation allowance will increase or decrease the Company's income tax provision or benefit. Management also applies the relevant guidance to determine the amount of income tax expense or benefit to be allocated among continuing operations, discontinued operations, and items charged or credited directly to stockholders' equity (deficit).
F-9
A tax position must meet a minimum probability threshold before a financial statement benefit is recognized. The minimum threshold is a tax position that is more likely than not to be sustained upon examination by the applicable taxing authority, including resolution of any related appeals or litigation process, based on the technical merits of the position. The Company recognizes interest and penalties related to uncertain tax positions in income tax expense.
Net Income (Loss) Per Common Share
The Company computes basic net income (loss) per weighted average share attributable to common stockholders using the weighted average number of shares of common stock outstanding during the period. The Company computes diluted net income (loss) per weighted average share attributable to common stockholders using the weighted average number of shares of common and dilutive potential common shares outstanding during the period. Potential common shares outstanding consist of stock options, warrants and convertible preferred stock using the treasury stock method and are excluded if their effect is anti-dilutive. Diluted weighted average common shares did not include any incremental shares for the fiscal year ended September 30, 2015 and 2014. Diluted weighted average common shares excluded incremental shares from common and preferred shares and warrants of approximately 29,536,000 and 29,048,000 for the fiscal year 2015 and 2014, respectively, due to their anti-dilutive effect.
|
In thousands, except per share data
|
||||||||
|
Fiscal Year Ended September 30,
|
||||||||
|
2015
|
2014
|
|||||||
|
Numerator:
|
||||||||
|
Net income (loss)
|
$
|
(2,628
|
)
|
$
|
(80
|
)
|
||
|
Net income attributable to participating securities
|
—
|
—
|
||||||
|
Net income (loss) attributable to common stockholders – basic
|
$
|
(2,628
|
)
|
$
|
(80
|
)
|
||
|
Net income (loss)
|
$
|
(2,628
|
)
|
$
|
(80
|
)
|
||
|
Less gain (loss) on warrant liability for participating common warrants
|
—
|
—
|
||||||
|
Net income (loss) attributable to common stockholders – diluted
|
$
|
(2,628
|
)
|
$
|
(80
|
)
|
||
|
Denominator:
|
||||||||
|
Weighted-average shares used in computing net income (loss) per share attributable to common stockholders – basic
|
135,883
|
134,667
|
||||||
|
Effect of potentially dilutive securities:
|
||||||||
|
Common stock warrants
|
—
|
—
|
||||||
|
Convertible preferred warrants
|
—
|
—
|
||||||
|
Convertible preferred stock
|
—
|
—
|
||||||
|
Common stock options
|
—
|
—
|
||||||
|
Non-participating common stock warrants
|
—
|
—
|
||||||
|
Weighted-average shares used in computing net income (loss) per share attributable to common stockholders – diluted
|
135,883
|
134,667
|
||||||
|
Basic net income (loss) per common share
|
$
|
(0.02
|
)
|
$
|
0.00
|
|||
|
Diluted net income (loss) per common share
|
$
|
(0.02
|
)
|
$
|
0.00
|
|||
F-10
Accounting for Stock-Based Compensation
The Company recognizes stock-based compensation expense in the statement of operations based upon the fair value of the equity award amortized over the vesting period.
Segment Reporting
The Company currently operates in one segment.
C. Liquidity
The Company completed a financing in December 2015, which is discussed further at Note H. Subsequent event. The primary use of our cash from operations is to further develop our various drug compounds and depending on the results of that development we may need to raise additional capital in the future.
D. Investments
Investment in CPEC LLC
The Company uses the equity method to account for its 35% ownership interest in CPEC. CPEC had $91,000 of net assets at each of September 30, 2015 and 2014. Aeolus' 35% share of CPEC's net assets, which is approximately $32,000, is included in other assets and the Company has no operations or activities unrelated to the out licensing of bucindolol. The other 65% is owned by Endo Pharmaceuticals.
E. Commitments
The Company acquires assets still in development and enters into license and research and development arrangements with third parties that often require milestone and royalty payments to the third party contingent upon the occurrence of certain future events linked to the success of the asset in development. Milestone payments may be required, contingent upon the successful achievement of an important point in the development life-cycle of the pharmaceutical product (e.g., approval of the product for marketing by a regulatory agency). If required by the arrangement, the Company may also be required to make royalty payments based upon a percentage of the net sales of the pharmaceutical product in the event that regulatory approval for marketing is obtained. Because of the contingent nature of these payments, they are not included in the table of contractual obligations. No milestones have been met, nor have any payments been made, as of September 30, 2015.
We are also obligated to pay patent filing, prosecution, maintenance and defense costs, if any, for the intellectual property the Company has licensed from National Jewish Health ("NJH"), National Jewish Medical and Research Center (the "NJMRC") and Duke University.
These arrangements may be material individually, and in the unlikely event that milestones for multiple products covered by these arrangements were reached in the same period, the aggregate charge to expense could be material to the results of operations in any one period. In addition, these arrangements often give Aeolus the discretion to unilaterally terminate development of the product, which would allow Aeolus to avoid making the contingent payments; however, Aeolus is unlikely to cease development if the compound successfully achieves clinical testing objectives.
F. Debt
Convertible Promissory Notes
September 29, 2015, the Company received funding in exchange for issuance of convertible promissory notes (the "BVF Notes") from Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P.
The BVF Notes have an aggregate principal balance of $1,000,000, accrue interest at a rate of 6% per annum and have a scheduled maturity date of September 28, 2016 (the "Maturity Date"). The outstanding principal and accrued interest on the BVF Notes shall automatically convert into Company equity securities issued in a Qualified Financing (as defined below) at a conversion rate carrying a 15% discount to the lowest price per share (or share equivalent) issued in a Qualified Financing (an "Automatic Conversion"). If, prior to the Maturity Date, the Company enters into an agreement pertaining to a Corporate Transaction (as defined below) and the Note has not been previously converted pursuant to an Automatic Conversion, then, the outstanding principal balance and unpaid accrued interest of the Note shall automatically convert in whole into the right of the holder to receive, in lieu of any other payment and in cancellation of the Note, an amount in cash upon closing of the Corporate Transaction equal to two times the outstanding principal amount of the Note.
F-11
For purposes of the foregoing: "Qualified Financing" means a bona fide new money equity securities financing on or before the Maturity Date with total proceeds to the Company of not less than four million dollars; and "Corporate Transaction" means a sale, lease or other disposition of all or substantially all of the Company's assets or a consolidation or merger of the Company with or into any other corporation or other entity or person, or any other corporate reorganization, in which the stockholders of the Company immediately prior to such consolidation, merger or reorganization own less than fifty percent (50%) of the voting power of the surviving entity immediately after such consolidation, merger or reorganization.
As of September 30, 2015, the $1,000,000 principal balance of the BVF Notes is recorded in the financial statements at face value and is net with a discount of $273,000 as a result of separating the fair value of the Qualified Financing redemption discount ("Redemption Feature") of 15% on the price per share in the BVF Notes. The Redemption Feature qualifies as a derivative and is subject to fair value treatment. The Redemption Feature is amortized over the expected life of the derivative, and the amortization expense is presented net with the interest expense from the BVF Notes, yielding an effective interest rate of 40% that is different than the 6% stated in the BVF Notes.
On December 11, 2015, following the completion of a Qualified Financing described above, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of the Company's common stock and warrants to purchase an additional 5,414,402 shares of the Company's common stock at an exercise price per share of $0.22 subject to adjustment. As a result, the BVF Notes were no longer outstanding as of that date.
G. Stockholders' Equity (Deficit)
Basis of Presentation
Preferred Stock
The Certificate of Incorporation of the Company authorizes the issuance of up to 10,000,000 shares of Preferred Stock, at a par value of $0.01 per share, of which 1,250,000 shares are designated Series A Convertible Preferred Stock and 1,600,000 shares are designated Series B Convertible Preferred Stock. The Board of Directors has the authority to issue Preferred Stock in one or more series, to fix the designation and number of shares of each such series, and to determine or change the designation, relative rights, preferences, and limitations of any series of Preferred Stock, without any further vote or action by the stockholders of the Company.
As of September 30, 2015 and 2014, 526,080 shares of Series B Stock were outstanding, respectively. There are no shares of Series A Convertible Preferred Stock issued or outstanding.
With respect to dividend rights and rights upon liquidation, winding up and dissolution, the Series B Stock ranks pari passu with the common stock. Subject to any rights of senior stock, holders of Series B Stock are entitled to receive dividends or distributions as, when and if declared by the Board of Directors. In the event the Board of Directors declares a dividend or distribution with respect to the outstanding common stock, the holders of Series B Stock are entitled to receive the amount of dividends per share in the same form payable on the common stock based on the largest number of shares of common stock issuable upon conversion of the outstanding Series B Stock. In the event of a liquidation, winding up or dissolution of the Company, subject to any rights of senior stock, the holders of Series B Stock are entitled to receive, pari passu with the holders of the common stock, the assets of the Company based on the largest number of shares of common stock issuable upon conversion of the outstanding Series B Stock.
F-12
Each share of Series B Stock is convertible into one share of common stock. The Series B Stock can be converted into common stock at any time upon the election of the holders of the Series B Stock except to the extent such conversion would result in the holders of Series B Stock owning in the aggregate more than 9.99% of the outstanding common stock.
The Series B Stock is not entitled to vote on any matter submitted to the vote of holders of the common stock except that the Company must obtain the approval of a majority of the outstanding shares of Series B Stock to either amend the Company's Certificate of Incorporation in a manner that would adversely affect the Series B Stock (including by creating an additional class or series of stock with rights that are senior or pari passu to the Series B Stock) or change the rights of the holders of the Series B Stock in any other respect.
On December 10, 2015, the Company entered into securities purchase agreements with certain accredited investors to sell and issue 4,500 preferred stock units issued to Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P., for an aggregate purchase price of $4.5 million, resulting in aggregate gross proceeds to the Company of approximately $6.75 million. The preferred units collectively consist of (i) 4,500 shares of Series C Preferred Stock of the Company that are collectively convertible into an aggregate of 20,454,546 common shares and (ii) warrants to purchase an aggregate of 20,454,546 Common Shares, in each case subject to adjustment. The warrants have an initial exercise price of $0.22 per share. The warrants may not be exercised until after 90 days following the date of issuance. The Series C Preferred Stock and warrants contain provisions restricting the conversion or exercise of such securities in circumstances where such event would result in the holder and its affiliates to beneficially own in excess of 9.99% of the Company's outstanding common stock.
Common Stock
On December 10, 2015, the Company entered into securities purchase agreements with certain accredited investors to sell and issue (i) an aggregate of 10,215,275 common units issued at a purchase price of $0.22 per unit. Each common unit consists of one share of the Company's common stock and a five year warrant to purchase one share of the Company's common stock, subject to adjustment. The warrants may not be exercised until after 90 days following the date of issuance. The warrants contain provisions restricting the conversion or exercise of such securities in circumstances where such event would result in the holder and its affiliates to beneficially own in excess of 9.99% of the Company's outstanding common stock.
On September 29, 2015, the Company received funding in the form of convertible promissory notes (the "BVF Notes") from Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P. The BVF Notes have an aggregate principal balance of $1,000,000, accrue interest at a rate of 6% per annum and have a scheduled maturity date of September 28, 2016. The outstanding principal and accrued interest on the BVF Notes will automatically convert into Company equity securities, provided a Qualified Financing of not less than $4,000,000 occurs.
On December 11, 2015, following the completion of a Qualified Financing described above, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of the Company's common stock and warrants to purchase an additional 5,414,402 shares of the Company's common stock at an exercise price per share of $0.22 subject to adjustment. As a result, the BVF Notes were no longer outstanding as of that date.
Dividends
The Company has never paid a cash dividend on its common stock and does not anticipate paying cash dividends on its common stock in the foreseeable future. If we pay a cash dividend on our common stock, we also must pay the same dividend on an as converted basis on our Series B preferred stock.
F-13
Warrants
As of September 30, 2015, warrants to purchase an aggregate of 15,949,627 shares of common stock were outstanding. Details of the warrants for common stock outstanding at September 30, 2015 were as follows:
|
Number of Shares
|
Exercise Price
|
Expiration Date
|
|||||
|
50,000
|
$
|
0.50
|
May 2016
|
||||
|
50,000
|
$
|
0.50
|
July 2016
|
||||
|
50,000
|
$
|
1.00
|
July 2016
|
||||
|
50,000
|
$
|
1.50
|
July 2016
|
||||
|
50,000
|
$
|
2.00
|
July 2016
|
||||
|
50,000
|
$
|
2.50
|
July 2016
|
||||
|
1,337,627
|
$
|
0.40
|
March 2017
|
||||
|
325,000
|
$
|
0.40
|
April 2017
|
||||
|
300,000
|
$
|
0.258
|
June 2017
|
||||
|
50,000
|
$
|
0.26
|
June 2017
|
||||
|
140,000
|
$
|
0.35
|
October 2017
|
||||
|
12,205,000
|
$
|
0.25
|
February 2018
|
||||
|
1,242,000
|
$
|
0.25
|
March 2018
|
||||
|
50,000
|
$
|
0.49
|
January 2020
|
||||
|
15,949,627
|
|||||||
As of September 30, 2015, one warrant to purchase an aggregate of 896,037 shares of preferred stock was outstanding.
As of September 30, 2014, warrants to purchase an aggregate of 16,029,627 shares of common stock were outstanding. Details of the warrants for common stock outstanding at September 30, 2014 were as follows:
|
Number of Shares
|
Exercise Price
|
Expiration Date
|
|||||
|
50,000
|
$
|
0.38
|
April 2015
|
||||
|
50,000
|
$
|
0.50
|
May 2016
|
||||
|
50,000
|
$
|
0.50
|
July 2016
|
||||
|
50,000
|
$
|
1.00
|
July 2016
|
||||
|
50,000
|
$
|
1.50
|
July 2016
|
||||
|
50,000
|
$
|
2.00
|
July 2016
|
||||
|
50,000
|
$
|
2.50
|
July 2016
|
||||
|
1,337,627
|
$
|
0.40
|
March 2017
|
||||
|
325,000
|
$
|
0.40
|
April 2017
|
||||
|
300,000
|
$
|
0.258
|
June 2017
|
||||
|
50,000
|
$
|
0.26
|
June 2017
|
||||
|
140,000
|
$
|
0.35
|
October 2017
|
||||
|
12,285,000
|
$
|
0.25
|
February 2018
|
||||
|
1,242,000
|
$
|
0.25
|
March 2018
|
||||
|
16,029,627
|
|||||||
As of September 30, 2014, one warrant to purchase an aggregate of 896,037 shares of preferred stock was outstanding.
F-14
Below is a summary of warrant activity for the last two fiscal years ended September 30:
|
Weighted Average
|
|||||||||||||
|
Remaining
|
Aggregate
|
||||||||||||
|
Number
|
Exercise
|
Contractual
|
Intrinsic
|
||||||||||
|
of Shares
|
Price
|
Term (in years)
|
Value
|
||||||||||
|
Outstanding at 9/30/2013
|
18,775,664
|
$
|
0.29
|
4.1 years
|
$
|
693,340
|
|||||||
|
Granted
|
50,000
|
$
|
0.26
|
$
|
-
|
||||||||
|
Exercised
|
(1,300,000
|
)
|
$
|
0.25
|
$
|
318,000
|
|||||||
|
Cancelled
|
(600,000
|
)
|
$
|
0.86
|
$
|
-
|
|||||||
|
Forfeited
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Outstanding at 9/30/2014
|
16,925,664
|
$
|
0.27
|
3.1 years
|
$
|
215,048
|
|||||||
|
Granted
|
50,000
|
$
|
0.49
|
$
|
-
|
||||||||
|
Exercised
|
(80,000
|
)
|
$
|
0.25
|
$
|
10,975
|
|||||||
|
Cancelled
|
(50,000
|
)
|
$
|
0.38
|
$
|
-
|
|||||||
|
Forfeited
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Outstanding at 9/30/2015
|
16,845,664
|
$
|
0.27
|
2.2 years
|
$
|
206,626
|
|||||||
|
Exercisable at 9/30/2015
|
16,845,664
|
$
|
0.27
|
2.2 years
|
$
|
206,626
|
|||||||
H. Subsequent event
On December 10, 2015, the Company entered into securities purchase agreements with certain accredited investors to sell and issue (i) an aggregate of 10,215,275 common units issued at a purchase price of $0.22 per unit, and (ii) 4,500 preferred stock units issued to existing investors, Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P., for an aggregate purchase price of $4.5 million, resulting in aggregate gross proceeds to the Company of approximately $6.75 million
Each common unit consists of one share of the Company's common stock and a five year warrant to purchase one share of the Company's common stock, subject to adjustment. The preferred units collectively consist of (i) 4,500 shares of Series C Convertible Preferred Stock of the Company that are collectively convertible into an aggregate of 20,454,546 common shares and (ii) warrants to purchase an aggregate of 20,454,546 Common Shares, in each case subject to adjustment. The warrants have an initial exercise price of $0.22 per share. The warrants may not be exercised until after 90 days following the date of issuance. The Series C Stock and warrants contain provisions restricting the conversion or exercise of such securities in circumstances where such event would result in the holder and its affiliates to beneficially own in excess of 9.99% of the Company's outstanding common stock.
On September 29, 2015, the Company received funding in the form of convertible promissory notes (the "BVF Notes") from Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P. The BVF Notes have an aggregate principal balance of $1,000,000, accrue interest at a rate of 6% per annum and have a scheduled maturity date of September 28, 2016. The outstanding principal and accrued interest on the BVF Notes will automatically convert into Company equity securities, provided a Qualified Financing of not less than $4 million occurs.
On December 11, 2015, following the completion of a Qualified Financing described above, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of the Company's common stock and warrants to purchase an additional 5,414,402 shares of the Company's common stock at an exercise price per share of $0.22 subject to adjustment. As a result, the BVF Notes were no longer outstanding as of that date.
I. Stock-Based Compensation
As an integral component of a management and employee retention program designed to motivate, retain and provide incentive to the Company's management, employees and key consultants, the Board of Directors approved the 2004 Stock Incentive Plan (the "2004 Plan") and reserved 25,000,000 shares of common stock for issuance under the 2004 Plan. As of September 30, 2015, 12,835,409 shares were available to be granted under the 2004 Plan. The exercise price of the incentive stock options ("ISOs") granted under the 2004 Plan must not be less than the fair market value of the common stock as determined on the date of the grant. The options may have a term up to 10 years. Options typically vest immediately or up to one year following the date of the grant.
F-15
Below is a summary of stock option activity for the last two fiscal years ended September 30:
|
Weighted Average
|
|||||||||||||
|
Remaining
|
Aggregate
|
||||||||||||
|
Number
|
Exercise
|
Contractual
|
Intrinsic
|
||||||||||
|
of Shares
|
Price
|
Term (in years)
|
Value
|
||||||||||
|
Outstanding at 9/30/2013
|
11,214,898
|
$
|
0.52
|
6.7 years
|
$
|
2
|
|||||||
|
Granted
|
775,000
|
$
|
0.26
|
$
|
-
|
||||||||
|
Exercised
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Cancelled
|
(393,307
|
)
|
$
|
2.64
|
$
|
-
|
|||||||
|
Forfeited
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Outstanding at 9/30/2014
|
11,596,591
|
$
|
0.43
|
6.1 years
|
$
|
1
|
|||||||
|
Granted
|
775,000
|
$
|
0.27
|
$
|
-
|
||||||||
|
Exercised
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Cancelled
|
(207,000
|
)
|
$
|
0.90
|
$
|
-
|
|||||||
|
Forfeited
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Outstanding at 9/30/2015
|
12,164,591
|
$
|
0.42
|
5.4 years
|
$
|
1
|
|||||||
|
Exercisable at 9/30/2015
|
12,002,089
|
$
|
0.42
|
5.4 years
|
$
|
1
|
|||||||
Stock options granted to consultants during fiscal year 2015 and 2014 were fully vested when issued or vested over a twelve month period. Stock option expense for stock options granted to consultants was zero and $11,000 for fiscal year 2015 and 2014, respectively. For the fiscal years 2015 and 2014, all stock options were issued at or above fair market value of a share of common stock. The weighted-average grant-date fair value of options granted during fiscal years 2015 and 2014 was $0.24 and $0.26, respectively.
A summary of the status of non-vested shares for the fiscal years ended September 30 was:
|
Weighted Average
|
||||||||
|
Number
|
Grant-Date
|
|||||||
|
of Shares
|
Fair Value
|
|||||||
|
Unvested at September 30, 2013
|
1,482,286
|
$
|
0.37
|
|||||
|
Granted
|
775,000
|
$
|
0.22
|
|||||
|
Vested
|
(2,088,534
|
)
|
$
|
0.33
|
||||
|
Forfeited
|
-
|
$
|
-
|
|||||
|
Unvested at September 30, 2014
|
168,752
|
$
|
0.22
|
|||||
|
Granted
|
775,000
|
$
|
0.24
|
|||||
|
Vested
|
(781,250
|
)
|
$
|
0.24
|
||||
|
Forfeited
|
-
|
$
|
-
|
|||||
|
Unvested at September 30, 2015
|
162,502
|
$
|
0.23
|
|||||
The total unrecognized compensation expense for outstanding stock options was $24,000 as of September 30, 2015, which will be recognized over a weighted average period of five months. The total fair value of shares vested during fiscal years 2015 and 2014 was $156,000 and $688,000, respectively.
F-16
The details of stock options for the fiscal year ended September 30, 2015 are as follows:
|
Options Outstanding
|
Options Exercisable
|
|||||||||||||||||||||||||
|
Weighted
|
Weighted
|
|||||||||||||||||||||||||
|
Number
|
Weighted
|
Average
|
Number
|
Weighted
|
Average
|
|||||||||||||||||||||
|
Range of
|
Outstanding at
|
Average
|
Remaining
|
Exercisable at
|
Average
|
Remaining
|
||||||||||||||||||||
|
Exercise
|
September 30,
|
Exercise
|
Contractual
|
September 30,
|
Exercise
|
Contractual
|
||||||||||||||||||||
|
Prices
|
2015
|
Price
|
Life (in years)
|
2015
|
Price
|
Life (in years)
|
||||||||||||||||||||
|
$
|
0.23-$0.30
|
3,087,500
|
$
|
0.28
|
6.37
|
2,924,998
|
$
|
0.28
|
6.20
|
|||||||||||||||||
|
$
|
0.31-$0.40
|
6,551,500
|
$
|
0.39
|
5.83
|
6,551,500
|
$
|
0.39
|
5.83
|
|||||||||||||||||
|
$
|
0.41-$0.50
|
502,000
|
$
|
0.45
|
6.25
|
502,000
|
$
|
0.45
|
6.25
|
|||||||||||||||||
|
$
|
0.51-$0.60
|
982,250
|
$
|
0.59
|
3.71
|
982,250
|
$
|
0.59
|
3.71
|
|||||||||||||||||
|
$
|
0.61-$0.70
|
60,000
|
$
|
0.68
|
1.03
|
60,000
|
$
|
0.68
|
1.03
|
|||||||||||||||||
|
$
|
0.71-$0.80
|
366,750
|
$
|
0.75
|
1.75
|
366,750
|
$
|
0.75
|
1.75
|
|||||||||||||||||
|
$
|
0.81-$0.90
|
570,591
|
$
|
0.87
|
1.26
|
570,591
|
$
|
0.87
|
1.26
|
|||||||||||||||||
|
$
|
0.91-$1.00
|
14,000
|
$
|
0.95
|
0.25
|
14,000
|
$
|
0.95
|
0.25
|
|||||||||||||||||
|
$
|
1.01-$1.19
|
30,000
|
$
|
1.10
|
0.22
|
30,000
|
$
|
1.10
|
0.22
|
|||||||||||||||||
|
12,164,591
|
$
|
0.42
|
5.43
|
12,002,089
|
$
|
0.42
|
5.38
|
|||||||||||||||||||
Stock-based compensation expense recognized in the statement of operations is as follows (in thousands):
|
For the fiscal year ended September 30,
|
||||||||
|
2015
|
2014
|
|||||||
|
Research and Development Expenses
|
$
|
—
|
$
|
11
|
||||
|
General and Administrative Expenses
|
178
|
624
|
||||||
|
Total Stock-based Compensation Expense
|
$
|
178
|
$
|
635
|
||||
The fair value of the options associated with the above compensation expense was determined at the date of the grant using the Black-Scholes option pricing model with the following weighted average assumptions:
|
For the fiscal year ended September 30,
|
||||||||
|
2015
|
2014
|
|||||||
|
Dividend yield
|
0
|
%
|
0
|
%
|
||||
|
Unvested forfeiture rate
|
5.35
|
%
|
0
|
%
|
||||
|
Expected volatility
|
137
|
%
|
122
|
%
|
||||
|
Risk-free interest rate
|
1.70
|
%
|
1.54
|
%
|
||||
|
Expected option life after shares are vested
|
5.27 years
|
5.27 years
|
||||||
J. Income Taxes
As of September 30, 2015 and 2014, the Company had federal net operating loss ("NOL") carry-forwards of $115,515,000 and $113,321,000, respectively and state operating loss carry-forwards of $38,916,000 and $35,244,000, respectively. The use of these federal and state NOL carry-forwards might be subject to limitation under the rules regarding a change in stock ownership as determined by the Internal Revenue Code (the "Code"). The Company may have had a change of control under Section 382 of the Code during fiscal 2004 and 2006; however, a complete analysis of the limitation of the NOL carry-forwards will not be completed until the time the Company projects it will be able to utilize such NOLs. The federal net operating and the state net operating losses began to expire in 2010. Additionally, the Company had federal research and development carry-forwards as of September 30, 2015 and 2014 of $4,127,000 and $3,990,000, respectively. The Company had state research and development carry-forwards as of September 30, 2015 and 2014 of $1,452,000 and $1,316,000, respectively.
F-17
Significant components of the Company's deferred tax assets at September 30, 2015 and 2014 consisted of the following (in thousands):
|
2015
|
2014
|
|||||||
|
Accrued payroll related liabilities
|
$
|
1,036
|
$
|
1,103
|
||||
|
Depreciation and amortization
|
586
|
704
|
||||||
|
Total deferred tax assets
|
1,622
|
1,807
|
||||||
|
State taxes
|
(113
|
)
|
(126
|
)
|
||||
|
Total deferred tax liabilities
|
(113
|
)
|
(126
|
)
|
||||
|
Net deferred tax assets
|
1,509
|
1,681
|
||||||
|
Valuation allowance for deferred assets
|
(1,509
|
)
|
(1,681
|
)
|
||||
|
Net deferred tax asset
|
$
|
—
|
$
|
—
|
||||
Due to the uncertainty surrounding the realization of the favorable tax attributes in future tax returns, all of the deferred tax assets have been fully offset by a valuation allowance. The change in the valuation allowance is primarily a result of the net operating loss carry-forwards.
Taxes computed at the statutory federal income tax rate of 34% are reconciled to the provision for income taxes as follows (dollars in thousands):
|
2015
|
2014
|
|||||||
|
Effective income tax rate
|
0
|
%
|
0
|
%
|
||||
|
United States Federal income tax at statutory rate
|
$
|
(891
|
)
|
$
|
(25
|
)
|
||
|
State income taxes (net of federal benefit)
|
1
|
1
|
||||||
|
NQSO forfeiture
|
234
|
—
|
||||||
|
Prior year deferred true up
|
39
|
(280
|
)
|
|||||
|
Change in valuation reserves
|
(153
|
)
|
96
|
|||||
|
FIN 48
|
883
|
354
|
||||||
|
Other
|
(111
|
)
|
(144
|
)
|
||||
|
Provision for income taxes
|
$
|
2
|
$
|
2
|
||||
F-18
At September 30, 2015, we had unrecognized tax benefits of $5,085,000, all of which would impact the effective tax rate if recognized, however a valuation allowance would be recorded against this amount. During fiscal 2014, unrecognized tax benefits increased to $4,859,000 as a result of the tax positions taken in prior years and current year. No interest and penalties have been provided for with respect to the unrecognized tax benefits.
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows:
|
Amount
|
||||
|
(in thousands)
|
||||
|
Unrecognized tax benefits at October 1,
|
$
|
4,859
|
||
|
Additions for tax positions related to current year
|
226
|
|||
|
Additions/reductions for tax positions taken in prior years
|
||||
|
Settlements
|
—
|
|||
|
Lapse of Limitations
|
—
|
|||
|
Unrecognized tax benefits at September 30, 2015
|
$
|
5,085
|
||
The Company's federal income tax returns for the tax years 2011 to 2013 remain open to examination. The Company's California income tax returns for the tax years 2010 to 2013 remain open to examination.
A reconciliation of the current year tax provision is as follows:
|
Current – Federal
|
$
|
—
|
||
|
Current – State
|
2
|
|||
|
2
|
||||
|
Deferred - Federal
|
134
|
|||
|
Deferred – State
|
38
|
|||
|
172
|
||||
|
Less: Valuation Allowance
|
(172
|
)
|
||
|
—
|
||||
|
Total current and deferred
|
$
|
2
|
F-19
K. Contingencies
Duke Licenses
The Company has obtained exclusive worldwide licenses (the "Duke Licenses") from Duke University ("Duke") to develop, make, have made, use and sell products using certain technology in the field of free radical and antioxidant research, developed by certain scientists at Duke. Future discoveries in the field of antioxidant research from these scientists' laboratories at Duke are also covered by the Duke Licenses. The Duke Licenses require the Company to use its best efforts to pursue development of products using the licensed technology and compounds. These efforts are to include the manufacture or production of products for testing, development and sale. Aeolus is also obligated to use its best efforts to have the licensed technology cleared for marketing in the United States by the U.S. Food and Drug Administration and in other countries in which Aeolus intends to sell products using the licensed technology. Aeolus will pay royalties to Duke on net product sales during the terms of the Duke Licenses, and milestone payments upon certain regulatory approvals and annual sales levels. In addition, Aeolus is obligated under the Duke Licenses to pay all or a portion of patent prosecution, maintenance and defense costs. Unless earlier terminated, the Duke Licenses continue until the expiration of the last to expire issued patent on the licensed technology.
National Jewish Medical and Research Center Agreements
Aeolus has an exclusive worldwide license ("NJH License") from National Jewish Health to develop, make, have made, use and sell products using certain technology developed by certain scientists at NJH. The NJH License requires Aeolus to use commercially reasonable efforts to diligently pursue the development and government approval of products using the licensed technology. Aeolus will be obligated to pay royalties to NJH on net product sales during the term of the NJH License and a milestone payment upon regulatory approval, if obtained. In addition, Aeolus is obligated under the NJH License to pay all or a portion of patent prosecution, maintenance and defense costs. Unless earlier terminated, the NJH License continues until the expiration of the last to expire issued patent on the licensed technology.
F-20
L. Agreements
Elan Corporation, plc
In May 2002, the Company entered into a collaboration transaction with affiliates of Elan Corporation, plc for the development of the Company's catalytic antioxidant compounds as a treatment for tissue damage from cancer radiation and chemotherapy. Although Elan and the Company terminated this collaboration in January 2003, the Company will pay Elan a royalty on net sales of its catalytic antioxidant products sold, if any, for the prevention and treatment of radiation-induced and chemotherapy-induced tissue damage.
M. Litigation
From time to time we are subject to various lawsuits and claims with respect to matters arising out of the normal course of our business. Due to the uncertainty of the ultimate outcome of these matters, the impact on future financial results is not subject to reasonable estimates.
N. Quarterly Financial Data (unaudited)
|
First
Quarter
|
Second
Quarter
|
Third
Quarter
|
Fourth
Quarter
|
Total
Year
|
||||||||||||||||
|
(in thousands, except per share amounts)
|
||||||||||||||||||||
|
Fiscal 2015
|
||||||||||||||||||||
|
Total revenue
|
$
|
925
|
$
|
1,189
|
$
|
63
|
$
|
934
|
$
|
3,111
|
||||||||||
|
Net income (loss)
|
$
|
(698
|
)
|
$
|
(712
|
)
|
$
|
(784
|
)
|
$
|
(434
|
)
|
$
|
(2,628
|
)
|
|||||
|
Net income available to stockholders – Basic
|
$
|
(698
|
)
|
$
|
(712
|
)
|
$
|
(784
|
)
|
$
|
(434
|
)
|
$
|
(2,628
|
)
|
|||||
|
Net income available to stockholders – Diluted
|
$
|
(698
|
)
|
$
|
(712
|
)
|
$
|
(784
|
)
|
$
|
(434
|
)
|
$
|
(2,628
|
)
|
|||||
|
Basic net income (loss) per common share attributable to common stockholders
|
$
|
(0.01
|
)
|
$
|
(0.01
|
)
|
$
|
(0.01
|
)
|
$
|
0.00
|
$
|
(0.02
|
)
|
||||||
|
Diluted net income (loss) per common share attributable to common stockholders
|
$
|
(0.01
|
)
|
$
|
(0.01
|
)
|
$
|
(0.01
|
)
|
$
|
0.00
|
$
|
(0.02
|
)
|
||||||
|
Fiscal 2014
|
||||||||||||||||||||
|
Total revenue
|
$
|
793
|
$
|
1,438
|
$
|
4,983
|
$
|
2,417
|
$
|
9,631
|
||||||||||
|
Net income (loss)
|
$
|
(695
|
)
|
$
|
(437
|
)
|
$
|
1,576
|
$
|
(524
|
)
|
$
|
(80
|
)
|
||||||
|
Net income available to stockholders – Basic
|
$
|
(695
|
)
|
$
|
(437
|
)
|
$
|
1,576
|
$
|
(524
|
)
|
$
|
(80
|
)
|
||||||
|
Net income available to stockholders – Diluted
|
$
|
(695
|
)
|
$
|
(437
|
)
|
$
|
1,576
|
$
|
(524
|
)
|
$
|
(80
|
)
|
||||||
|
Basic net income (loss) per common share attributable to common stockholders
|
$
|
(0.01
|
)
|
$
|
0.00
|
$
|
0.01
|
$
|
0.00
|
$
|
0.00
|
|||||||||
|
Diluted net income (loss) per common share attributable to common stockholders
|
$
|
(0.01
|
)
|
$
|
0.00
|
$
|
0.01
|
$
|
0.00
|
$
|
0.00
|
|||||||||
O. Recently Issued Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board that are adopted by us as of the specified effective date. Unless otherwise discussed, we believe that the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption.
F-21
In May 2014, the Financial Accounting Standards Board ("FASB") issued Accounting Standards Update ("ASU") 2015-14 Revenue, which deferred the effective date of ASU 2014-09 Revenue from Contracts with Customers (ASC 606), which updates the principles for recognizing revenue. ASU 2014-09 also amends the required disclosures of the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers. ASU 2014-09 is now effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period. The Company is evaluating the potential impacts of the new standard on its existing revenue recognition policies and procedures.
In August 2014, the FASB issued ASU 2014-15, Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern. ASU 2014-15 requires that an entity's management evaluate whether there are conditions or events that raise substantial doubt about the entity's ability to continue as a going concern within one year after the date that the financial statements are issued. ASU 2014-15 is effective for annual periods beginning after December 15, 2016 and for interim periods thereafter. The Company is evaluating the potential impacts of this new standard on its quarterly reporting process.
F-22
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth the costs and expenses to be paid in connection with the sale of shares being registered, all of which we will pay. All amounts, other than the SEC registration fee are estimates.
|
SEC registration fee
|
$
|
1,627.03
|
||
|
Legal fees and expenses
|
35,000.00
|
*
|
||
|
Accounting fees and expenses
|
25,000.00
|
|||
|
Total
|
$
|
61,627.03
|
Item 14. Indemnification of Directors and Officers
Section 145 of the Delaware General Corporation Law (the "DGCL") generally provides that a director or officer of a corporation (i) shall be indemnified by the corporation for all expenses of such legal proceedings when he or she is successful on the merits, (ii) may be indemnified by the corporation for the expenses, judgments, fines and amounts paid in settlement of such proceedings (other than a derivative suit), even if he or she is not successful on the merits, if he or she acts in good faith and in a manner he or she reasonably believes to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceedings, had no reasonable cause to believe his or her conduct was unlawful, and (iii) may be indemnified by the corporation for the expenses of a derivative suit (a suit by a stockholder alleging a breach by a director or officer of a duty owed to the corporation), even if he or she is not successful on the merits, if he or she acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interest of the corporation. No indemnification may be made under clause (iii) above, however, if the director or officer is adjudged liable for negligence or misconduct in the performance of his or her duties to the corporation, unless a corporation determines that, despite such adjudication, but in view of all the circumstances, he or she is entitled to indemnification. The indemnification described in clauses (ii) and (iii) above may be made upon a determination that indemnification is proper because the applicable standard of conduct has been met. Such a determination may be made by a majority of a quorum of disinterested directors, independent legal counsel, the stockholders or a court of competent jurisdiction.
The registrant's certificate of incorporation and Bylaws provide in substance that, to the fullest extent permitted by Delaware law as it now exists or as amended, each director and officer shall be indemnified against reasonable costs and expenses, including attorneys' fees and any liabilities which he or she may incur in connection with any action to which he or she may be made a party by reason or his or her being or having been a director or officer of the registrant or any of its affiliated enterprises. The indemnification provided by the registrant's Bylaws is not deemed exclusive of or intended in any way to limit any other rights to which any person seeking indemnification may be entitled.
Section 102(b)(7) of the DGCL permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, except for liability (i) for any breach of the director's duty of loyalty to the corporation or its stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (iii) under Section 174 of the DGCL, or (iv) for any transaction from which the director derived an improper personal benefit. The registrant's Certificate of Incorporation provides for the elimination of personal liability of a director for breach of fiduciary duty, as permitted by Section 102(b)(7) of the DGCL.
II-1
We have directors' and officers' liability insurance which provides, subject to certain policy limits, deductible amounts and exclusions, coverage for all persons who have been, are or may in the future be, directors or officers of Aeolus Pharmaceuticals, Inc., against amounts which such persons may pay resulting from claims against them by reason of their being such directors or officers during the policy period for certain breaches of duty, omissions or other acts done or wrongfully attempted or alleged. Such policies provide coverage in certain situations where we cannot directly provide indemnification under the DGCL.
Item 15. Recent Sales of Unregistered Securities
The following is a summary of transactions by us from January 1, 2013 through the date of this registration statement involving sales of our securities that were not registered under the Securities Act of 1933, as amended, or the Securities Act.
1. On December 10, 2015, we entered into securities purchase agreements with certain accredited investors to sell and issue (i) an aggregate of 10,215,275 common units issued at a purchase price of $0.22 per unit, and (ii) 4,500 preferred stock units issued to Biotechnology Value Fund, L.P. and other affiliates of BVF Partners, L.P., for an aggregate purchase price of $4.5 million, resulting in aggregate gross proceeds to the Company of approximately $6.75 million (the "2015 Securities Placement"). Each common unit consists of one share of the Company's common stock and a five year warrant to purchase one share of the Company's common stock, subject to adjustment. The preferred units collectively consist of (i) 4,500 shares of Series C Preferred Stock of the Company that are collectively convertible into an aggregate of 20,454,546 common shares and (ii) warrants to purchase an aggregate of 20,454,546 Common Shares, in each case subject to adjustment. The warrants have an initial exercise price of $0.22 per share.
2. On September 29, 2015, the Company received funding in the form of convertible promissory notes (the "BVF Notes") from Biotechnology Value Fund, L.P. and certain other affiliates of BVF Partners, L.P. The BVF Notes (i) has an aggregate principal balance of $1,000,000, (ii) accrued interest at a rate of 6% per annum, (iii) had a scheduled maturity date of September 28, 2016 and (iv) were subject to automatic conversion into Company equity securities, provided a qualified financing of not less than $4 million occurred. On December 11, 2015, following the completion of the 2015 Securities Placement, the principal and accrued interest amounts under the BVF Notes were converted into 5,414,402 shares of the Company's common stock and warrants to purchase an additional 5,414,402 shares of the Company's common stock at an exercise price per share of $0.22 subject to adjustment. As a result, the BVF Notes were no longer outstanding as of December 11, 2015.
3. On February 19, 2013 and March 4, 2013, we entered into a Securities Purchase Agreement, which we refer to as the Purchase Agreement, with certain accredited investors to sell and issue to such investors an aggregate of approximately 14,462,000, which we refer to as the Units, at a purchase price of $0.25 per unit, resulting in aggregate gross proceeds to us of approximately $3.6 million, we refer to this transaction throughout the prospectus as the 2015 Securities Placement. Each Unit consists of (i) one share of common stock and (ii) a five year warrant to purchase one share of our common stock. The warrants in the 2015 Securities Placement have an initial exercise price of $0.25 per share. In addition, we paid a cash fee of 7% and issued a warrant equal to 5% of the number of units purchased by certain investors to Ladenburg Thalmann & Co. Inc., as co-placement agent, and we paid a cash fee of 13% and issued a warrant equal to 10% of the number of units purchased by certain investors to Neidiger, Tucker, Bruner, Inc., as co-placement agents.
Unless otherwise disclosed above, the offerings of the securities above were exempt from registration under Section 4(a)(2) of the Securities Act, and Regulation D promulgated thereunder. In each instance, we had a reasonable belief that, among other things, the purchasers had access to information concerning our operations and financial condition, that the purchasers acquired the securities for their own account and not with a view to the distribution thereof, and that each purchaser was an "accredited investor" as such term is defined in Regulation D promulgated under the Securities Act. In addition, there was no general solicitation or general advertising related to any of such offerings.
Other than as disclosed above, we did not employ any underwriters, placement agents, brokers, finders or financial advisors in connection with any of the transactions set forth above.
II-2
Item 16. Exhibits and Financial Statement Schedules
(a) Exhibits
The exhibits set forth commencing on page II-8 are incorporated herein by reference.
(b) Financial Statement Schedules
All schedules have been omitted because the information required to be presented in them is not applicable or is shown in the financial statements or related notes.
Item 17. Undertakings
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the "Calculation of Registration Fee" table in the effective registration statement;
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(b) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
(c) The undersigned registrant hereby undertakes that:
(1) That for purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this Registration Statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this Registration Statement as of the time it was declared effective.
II-3
(2) That for the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-4
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this Amendment No. 1 to the Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Mission Viejo, California, on the 12th day of February, 2016.
|
AEOLUS PHARMACEUTICALS, INC.
|
||
|
By:
|
/s/ John L. McManus
|
|
|
John McManus
President and Chief Executive Officer
|
||
Pursuant to the requirements of the Securities Act of 1933, as amended, this Amendment No. 1 to the Registration Statement on Form S-1 has been signed below by the following persons in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||||||
|
/s/ John L. McManus
|
President and Chief Executive Officer
(principal executive officer)
|
|||||||
|
John L. McManus
|
February 12, 2016
|
|||||||
|
/s/ David C. Cavalier
|
Chief Financial Officer and
|
|||||||
|
David C. Cavalier
|
Director (Chairman)
|
February 12, 2016
|
||||||
|
*
|
||||||||
|
John M. Farah, Jr., Ph.D.
|
Director
|
February 12, 2016
|
||||||
|
*
|
||||||||
|
John M. Clerici
|
Director
|
February 12, 2016
|
||||||
|
*
|
||||||||
|
Amit Kumar, Ph.D.
|
Director
|
February 12, 2016
|
||||||
|
Mitchell D. Kaye, J.D.
|
Director
|
February 12, 2016
|
||||||
|
*
|
||||||||
|
Chris A. Rallis
|
Director
|
February 12, 2016
|
||||||
|
*
|
||||||||
|
Jeffrey A. Scott, M.D.
|
Director
|
February 12, 2016
|
||||||
|
*By:
|
/s/ John L. McManus
|
|
| Attorney in fact |
EXHIBIT INDEX
|
|
|
Incorporated by Reference To
|
|
|||||||
|
Exhibit
Number
|
Description of Document
|
Registrant's
Form
|
Date Filed with the SEC
|
Exhibit
Number
|
Filed
Herewith
|
|||||
|
2.1
|
Agreement and Plan of Merger and Reorganization dated September 16, 2003 between Incara, Inc. and Incara Pharmaceuticals Corporation
|
S-4
|
09/19/03
|
2.1
|
|
|||||
|
3.1
|
Amended and Restated Certificate of Incorporation
|
10-K
|
12/31/12
|
3.1
|
||||||
|
3.2
|
Certificate of Designation for Series C Preferred Stock
|
8-K
|
12/15/15
|
10.1
|
||||||
|
3.3
|
Bylaws
|
8-K
|
10/27/15
|
3.1
|
||||||
|
4.1
|
Form of Common Stock Certificate
|
10-Q
|
08/11/04
|
4.1
|
|
|||||
|
4.2
|
Form of Series B Preferred Stock Certificate
|
S-4
|
09/19/03
|
4.8
|
|
|||||
|
4.3
|
Form of Warrant to Purchase Common Stock dated June 5, 2006.
|
8-K
|
06/06/06
|
10.3
|
|
|||||
|
4.4
|
Registration Rights Agreement dated May 22, 2007 by and among the Company and each of the Purchasers whose names appear on the Schedule attached thereto.
|
8-K
|
5/23/07
|
4.1
|
|
|||||
|
4.5
|
Registration Rights Agreement dated October 6, 2009 by and among the Company and the investors whose names appear on the signature pages thereof.
|
8-K
|
10/06/09
|
4.1
|
|
|||||
|
4.6
|
Form of Warrant to Purchase Common Stock dated May 22, 2007.
|
8-K
|
5/23/07
|
10.2
|
|
|||||
|
4.7
|
Form of Warrant to Purchase Common Stock
|
8-K
|
10/06/09
|
10.2
|
|
|||||
|
4.8
|
Registration Rights Agreement dated September 16, 2003 among Incara Pharmaceuticals Corporation, Incara, Inc. and Goodnow Capital, L.L.C.
|
S-4
|
09/19/03
|
10.101
|
||||||
|
4.9
|
Registration Rights Agreement dated August 11, 2010 by and among Aeolus Pharmaceuticals, Inc. and the investors listed therein
|
8-K
|
8/12/10
|
4.1
|
||||||
|
4.10
|
Registration Rights Agreement dated March 4, 2013 by and among Aeolus Pharmaceuticals, Inc. and the investors listed therein
|
8-K
|
03/06/13
|
10.2
|
|
|||||
|
4.11
|
Form of Warrant to Purchase Common Stock dated March 4, 2013.
|
8-K
|
03/06/13
|
10.3
|
||||||
|
4.12
|
Warrant Repricing, Exercise and Lockup Agreement dated February 19, 2013 by and among the Company, Xmark JV Investment Partners, LLC and affiliates
|
8-K
|
02/19/13
|
10.4
|
||||||
|
4.13
|
Form of Series C Preferred Stock Certificate
|
10-K
|
12/18/15
|
4.13
|
||||||
|
4.14
|
Form of Warrant
|
8-K
|
12/15/15
|
10.2
|
||||||
|
Opinion of Drinker Biddle & Reath
|
**
|
|||||||||
|
10.1*
|
License Agreement between Duke University and Aeolus Pharmaceuticals, Inc., dated July 21, 1995
|
S-1
|
12/08/95
|
10.4
|
|
|||||
|
10.2
|
Amended and Restated Limited Liability Company Agreement of CPEC LLC dated July 15, 1999, among CPEC LLC, Intercardia, Inc. and Interneuron Pharmaceuticals, Inc.
|
8-K
|
07/23/99
|
10.42
|
|
|||||
|
10.3
|
Assignment, Assumption and License Agreement dated July 15, 1999, between CPEC LLC and Intercardia, Inc.
|
8-K
|
07/23/99
|
10.43
|
|
|||||
II-5
|
|
|
Incorporated by Reference To
|
|
|||||||
|
Exhibit
Number
|
Description of Document
|
Registrant's
Form
|
Date Filed with the SEC
|
Exhibit
Number
|
Filed
Herewith
|
|||||
|
10.4*
|
License Agreement dated January 19, 2001 between Incara Pharmaceuticals Corporation and Incara Development, Ltd.
|
10-Q
|
02/13/01
|
10.59
|
|
|||||
|
10.5*
|
License Agreement dated January 19, 2001 between Elan Corporation, plc, Elan Pharma International Ltd. and Incara Development, Ltd.
|
10-Q
|
02/13/01
|
10.60
|
|
|||||
|
10.6
|
Registration Rights Agreement dated December 21, 2000 among Incara Pharmaceuticals Corporation, Elan International Services, Ltd. and Elan Pharma International Ltd.
|
10-Q
|
02/13/01
|
10.62
|
|
|||||
|
10.7
|
Agreement and Amendment, effective as of January 22, 2001, by and among Incara Pharmaceuticals Corporation, Elan International Services, Ltd. and Elan Pharma International Limited
|
10-Q
|
05/14/01
|
10.64
|
|
|||||
|
10.8
|
Second Agreement and Amendment, effective as of January 22, 2001, by and among Incara Pharmaceuticals Corporation, Elan International Services, Ltd. and Elan Pharma International Limited
|
10-Q
|
05/14/01
|
10.65
|
|
|||||
|
10.9
|
Third Agreement and Amendment, effective as of January 22, 2001, by and among Incara Pharmaceuticals Corporation, Elan International Services, Ltd. and Elan Pharma International Limited
|
8-K
|
06/01/01
|
10.66
|
|
|||||
|
10.10
|
Agreement and Fourth Amendment, effective February 13, 2002, by and among Incara Pharmaceuticals Corporation, Elan International Services, Ltd., Elan Pharma International Limited and Elan Pharmaceutical Investments III, Ltd.
|
10-Q
|
02/14/02
|
10.75
|
|
|||||
|
10.11*
|
License Agreement dated June 25, 1998 between Duke University and Aeolus Pharmaceuticals, Inc.
|
10-Q
|
05/15/02
|
10.82
|
|
|||||
|
10.12*
|
License Agreement dated May 7, 2002 between Duke University and Aeolus Pharmaceuticals, Inc.
|
10-Q
|
05/15/02
|
10.83
|
|
|||||
|
10.13*
|
License Agreement dated November 17, 2000 between National Jewish Medical and Research Center and Aeolus Pharmaceuticals, Inc.
|
10-Q
|
02/13/01
|
10.56
|
|
|||||
|
10.14
|
Exclusive License Agreement, dated January 15, 2009, by and between the Company and National Jewish Health
|
10-Q
|
05/16/11
|
10.7
|
||||||
|
10.15*
|
Securities Purchase Agreement dated as of May 15, 2002, among Incara Pharmaceuticals Corporation, Aeolus Pharmaceuticals, Inc., Elan Pharma International Limited and Elan International Services, Ltd.
|
8-K/A
|
07/03/02
|
10.84
|
|
|||||
|
10.16*
|
Development and Option Agreement dated May 15, 2002, among Elan Pharma International Limited, Incara Pharmaceuticals Corporation and Aeolus Pharmaceuticals, Inc.
|
8-K/A
|
07/03/02
|
10.85
|
|
|||||
|
10.17
|
Amended and Restated Registration Rights Agreement dated as of May 15, 2002, among Incara Pharmaceuticals Corporation, Elan International Services, Ltd. and Elan Pharma International Limited
|
8-K/A
|
07/03/02
|
10.86
|
|
II-6
|
|
|
Incorporated by Reference To
|
|
|||||||
|
Exhibit
Number
|
Description of Document
|
Registrant's
Form
|
Date Filed with the SEC
|
Exhibit
Number
|
Filed
Herewith
|
|||||
|
10.18
|
Amendment No. 1 to License Agreement dated May 14, 2002, between Aeolus Pharmaceuticals, Inc. and Duke University (amending License Agreement dated July 21, 1995)
|
8-K/A
|
07/03/02
|
10.87
|
|
|||||
|
10.19
|
Amendment No. 1 to License Agreement dated May 14, 2002, between Aeolus Pharmaceuticals, Inc. and Duke University (amending License Agreement dated June 25, 1998)
|
8-K/A
|
07/03/02
|
10.88
|
|
|||||
|
10.20
|
Amendment No. 1 to License Agreement dated May 14, 2002, between Aeolus Pharmaceuticals, Inc. and National Jewish Medical and Research Center (amending License Agreement dated November 17, 2000)
|
8-K/A
|
07/03/02
|
10.89
|
|
|||||
|
10.21*
|
Subaward Agreement, dated March 16, 2011, by and between the Company and the Office of Research and Development of the University of Maryland, Baltimore
|
10-Q
|
05/16/11
|
10.4
|
|
|||||
|
10.22
|
Letter dated May 17, 2004 from Elan International Services, Limited and Elan Pharma International Limited to Incara Pharmaceuticals Corporation
|
10-Q
|
08/11/04
|
10.106
|
|
|||||
|
10.23+
|
Aeolus Pharmaceuticals, Inc. 1994 Stock Option Plan, as amended
|
10-Q
|
08/11/04
|
10.109
|
|
|||||
|
10.24+
|
Aeolus Pharmaceuticals, Inc. Amended and Restated 2004 Stock Incentive Plan
|
14-C
|
|
11/16/12
|
D
|
|
||||
|
10.25+
|
Amended and Restated Employment Agreement dated July 30, 2010 between Aeolus Pharmaceuticals, Inc. and John L. McManus
|
8-K
|
08/02/10
|
10.4
|
||||||
|
10.26+
|
Letter Agreement dated July 10, 2006 between Aeolus Pharmaceuticals, Inc. and McManus & Company, Inc.
|
8-K
|
07/10/06
|
10.2
|
|
|||||
|
10.27+
|
Form of Indemnity Agreement
|
10-K
|
12/27/11
|
10.27
|
|
|||||
|
10.28
|
Terms of Outside Director Compensation
|
10-K
|
12/17/04
|
10.114
|
|
|||||
|
10.29+
|
Form of Incentive Stock Option Agreement
|
10-Q
|
02/08/05
|
10.115
|
|
|||||
|
10.30+
|
Form of Nonqualified Stock Option Agreement
|
10-Q
|
02/08/05
|
10.116
|
|
|||||
|
10.31
|
Subscription Agreement dated June 5, 2006 by and between the Company and the investors whose names appear on the signature pages thereof.
|
8-K
|
06/06/06
|
10.1
|
|
|||||
|
10.32
|
Board Observer Letter dated June 5, 2006 by and among the Company and Efficacy Biotech Master Fund Ltd.
|
8-K
|
06/06/06
|
10.6
|
|
|||||
|
10.33+
|
Consulting Agreement, dated December 1, 2010, between Aeolus Pharmaceuticals, Inc. and Brian J. Day
|
8-K
|
12/03/10
|
10.1
|
|
|||||
|
10.34*
|
Sponsored Research Agreement (Non-Clinical), dated April 12, 2011, by and between the Company and Duke University
|
10-Q
|
05/16/11
|
10.5
|
|
|||||
|
10.35
|
Securities Purchase Agreement dated August 11, 2010 by and among Aeolus Pharmaceuticals, Inc. and the investors listed therein
|
8-K
|
8/12/10
|
10.1
|
|
II-7
|
|
Incorporated by Reference To
|
|
|||||
|
Exhibit
Number
|
Description of Document
|
Registrant's
Form
|
Date Filed with the SEC
|
Exhibit
Number
|
Filed
Herewith
|
||
|
10.36
|
Form of Warrant pursuant to Securities Purchase Agreement dated August 11, 2010 by and among Aeolus Pharmaceuticals, Inc. and the investors listed therein
|
8-K
|
8/12/10
|
10.2
|
|
||||||||
|
10.37
|
Convertible Promissory Note dated February 7, 2007 issued by Aeolus Pharmaceuticals, Inc. to Elan Pharma International Ltd.
|
S-1
|
06/04/07
|
10.43
|
|
||||||||
|
10.38
|
Amendment No. 1 To Convertible Promissory Note dated February 7, 2009 by and between Aeolus Pharmaceuticals, Inc. and Elan Pharma International Limited
|
8-K
|
3/16/09
|
10.1
|
|
||||||||
|
10.39+
|
Form of Restricted Share Award Agreement
|
S-8 POS
|
3/31/08
|
99.2
|
|
||||||||
|
10.40
|
Securities Purchase and Exchange Agreement dated October 6, 2009 by and among the Company and the investors whose names appear on the signature pages thereof
|
8-K
|
10/06/09
|
10.1
|
|
||||||||
|
10.41
|
Amendment Agreement to the Securities Purchase and Exchange Agreement, dated December 24, 2009, by and among the Company and the investors whose names appear on the signature pages thereof
|
8-K
|
12/28/09
|
10.1
|
|||||||||
|
10.42+
|
Intentionally Omitted
|
8-K
|
02/16/11
|
10.1
|
|||||||||
|
10.43*
|
Contract No. HHSO100201100007C, dated February 11, 2011, by and between the Company and the U.S. Department of Health and Human Services Biomedical Advanced Research and Development Authority
|
10-Q
|
05/16/11
|
10.1
|
|||||||||
|
10.44*
|
Research and Manufacturing Agreement, dated February 18, 2011 (the "JMPS Agreement"), by and between the Company and Johnson Matthey Pharmaceutical Materials, Inc. (d/b/a Johnson Matthey Pharma Services).
|
10-Q
|
05/16/11
|
10.2
|
|||||||||
|
10.45*
|
Appendix 2 to the JMPS Agreement, dated February 18, 2011
|
10-Q
|
8/14/12
|
10.4
|
|||||||||
|
10.46*
|
Appendix 3 to the JMPS Agreement, dated April 30, 2012
|
10-Q
|
8/14/12
|
10.5
|
|||||||||
|
10.47*
|
Appendix 4 to the JMPS Agreement, dated April 30, 2012
|
10-Q
|
8/14/12
|
10.6
|
|||||||||
|
10.48*
|
Appendix 5 to the JMPS Agreement, dated April 30, 2012
|
10-Q
|
8/14/12
|
10.7
|
|||||||||
|
10.49*
|
Appendix 6 to the JMPS Agreement, dated April 30, 2012
|
10-Q
|
8/14/12
|
10.8
|
|||||||||
|
10.50*
|
General Management Consulting Assignment, dated February 23, 2011, by and between the Company and Booz Allen Hamilton Inc.
|
10-Q
|
05/16/11
|
10.3
|
|||||||||
|
10.51
|
Form of Securities Purchase Agreement by and among the Company and the investors whose names appear on the signature pages thereof
|
8-K
|
4/5/12
|
10.1
|
|||||||||
|
10.52
|
Form of Registration Rights Agreement by and among the Company and the investors party thereto
|
8-K
|
4/5/12
|
10.2
|
|||||||||
|
10.53
|
Form of Warrant issued to investors in March and April 2012
|
8-K
|
4/5/12
|
10.3
|
|||||||||
|
10.54
|
Amended and Restated Employment Agreement by and between the Company and John L. McManus
|
8-K
|
03/05/13
|
10.1
|
|||||||||
II-8
|
|
Incorporated by Reference To
|
|
||||
|
Exhibit
Number
|
Description of Document
|
Registrant's
Form
|
Date Filed with the SEC
|
Exhibit
Number
|
Filed
Herewith
|
|
|
10.54
|
Amended and Restated Employment Agreement by and between the Company and John L. McManus
|
8-K
|
03/05/13
|
10.1
|
|||||||
|
10.55
|
Form of Registration Rights Agreement
|
8-K
|
12/15/15
|
10.2
|
|||||||
|
21.1
|
List of Subsidiaries
|
10-K
|
12/18/15
|
21.1
|
|
||||||
|
Consent of Grant Thornton LLP, Independent Registered Public Accounting Firm
|
|
|
|
|
X
|
||||||
|
101.INS†
|
XBRL Instance Document
|
X
|
|||||||||
|
101.SCH†
|
XBRL Taxonomy Extension Schema Document
|
X
|
|||||||||
|
101.CAL†
|
XBRL Taxonomy Extension Calculation Linkbase Document
|
X
|
|||||||||
|
101.DEF†
|
XBRL Taxonomy Extension Definition Linkbase Document
|
X
|
|||||||||
|
101.LAB†
|
XBRL Taxonomy Extension Label Linkbase Document
|
X
|
|||||||||
|
101.PRE†
|
XBRL Taxonomy Extension Presentation Linkbase Document
|
X
|
|||||||||
* The Company has received confidential treatment of certain portions of this agreement which have been omitted and filed separately with the U.S. Securities and Exchange Commission.
** To be filed by amendment
+ Indicates management contract or compensatory plan or arrangement.
† Attached as Exhibit 101 to this report are documents formatted in XBRL (Extensible Business Reporting Language). Users of this data are advised that, pursuant to Rule 406T of Regulation S-T, the interactive data file is deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, is deemed not filed for purposes of Section 18 of the Exchange Act and is otherwise not subject to liability under these sections.
II-9