Attached files
| file | filename |
|---|---|
| 8-K - 8-K - AMICUS THERAPEUTICS, INC. | a16-1606_18k.htm |
| EX-99.1 - EX-99.1 - AMICUS THERAPEUTICS, INC. | a16-1606_1ex99d1.htm |
| EX-99.2 - EX-99.2 - AMICUS THERAPEUTICS, INC. | a16-1606_1ex99d2.htm |
Exhibit 99.3
3rd Annual Dermatology Summit SD-101 for Epidermolysis Bullosa (EB) Jay Barth, MD, Chief Medical Officer

Safe Harbor This presentation will contain, “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995 relating to preclinical and clinical development of Amicus’ candidate drug products, the timing and reporting of results from preclinical studies and clinical trials evaluating Amicus’ candidate drug products, financing plans, and the projected cash position for the Company. Words such as, but not limited to, “look forward to,” “believe,” “expect,” “anticipate,” “estimate,” “intend,” “potential,” “plan,” “targets,” “likely,” “may,” “will,” “would,” “should” and “could,” and similar expressions or words identify forward-looking statements. Such forward-looking statements are based upon current expectations that involve risks, changes in circumstances, assumptions and uncertainties. The inclusion of forward-looking statements should not be regarded as a representation by Amicus that any of its plans will be achieved. Any or all of the forward-looking statements in this press release may turn out to be wrong. They can be affected by inaccurate assumptions Amicus might make or by known or unknown risks and uncertainties. For example, with respect to statements regarding the goals, progress, timing and outcomes of discussions with regulatory authorities, and in particular the timing of an NDA submission for migalastat monotherapy, and the potential goals, progress, timing and results of preclinical studies and clinical trials, actual results may differ materially from those set forth in this release due to the risks and uncertainties inherent in the business of Amicus, including, without limitation: the potential that results of clinical or pre-clinical studies indicate that the product candidates are unsafe or ineffective; the potential that it may be difficult to enroll patients in our clinical trials; the potential that regulatory authorities may not grant or may delay approval for our product candidates; the potential that preclinical and clinical studies could be delayed because we identify serious side effects or other safety issues; the potential that we will need additional funding to complete all of our studies and, our dependence on third parties in the conduct of our clinical studies. Further, the results of earlier preclinical studies and/or clinical trials may not be predictive of future results. With respect to statements regarding projections of the Company’s cash position, actual results may differ based on market factors and the Company’s ability to execute its operational and budget plans. In addition, all forward looking statements are subject to other risks detailed in our Annual Report on Form 10-K for the year ended December 31, 2014 and Form 10-Q for the quarter ended June 30, 2015. You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. All forward-looking statements are qualified in their entirety by this cautionary statement, and Amicus undertakes no obligation to revise or update this news release to reflect events or circumstances after the date hereof. This caution is made under the safe harbor provisions of Section 21E of the Private Securities Litigation Reform Act of 1995.

Epidermolysis Bullosa (EB) Multiple genes cause disease which results in fragility of skin and can also affect internal organs Diagnosed from infancy to adulthood Severe blistering, open wounds and scarring in response to minor friction to the skin Disfiguring, excruciatingly painful, and can be fatal Given lack of treatment, any reduction in disease symptoms would be considered meaningful 30,000 – 40,000 diagnosed patients in major global regions Rare, Devastating, Connective Tissue Disorder with No Approved Treatments 1 Third party market research
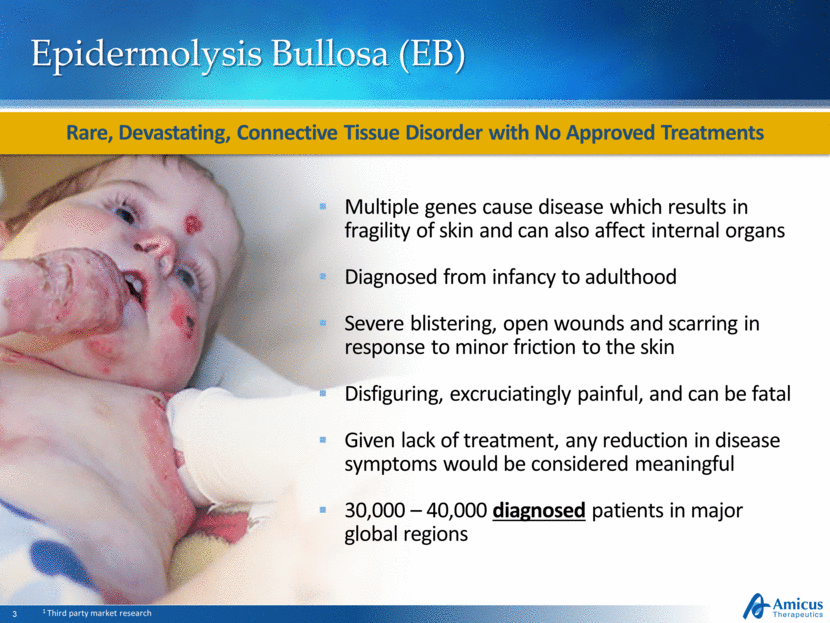
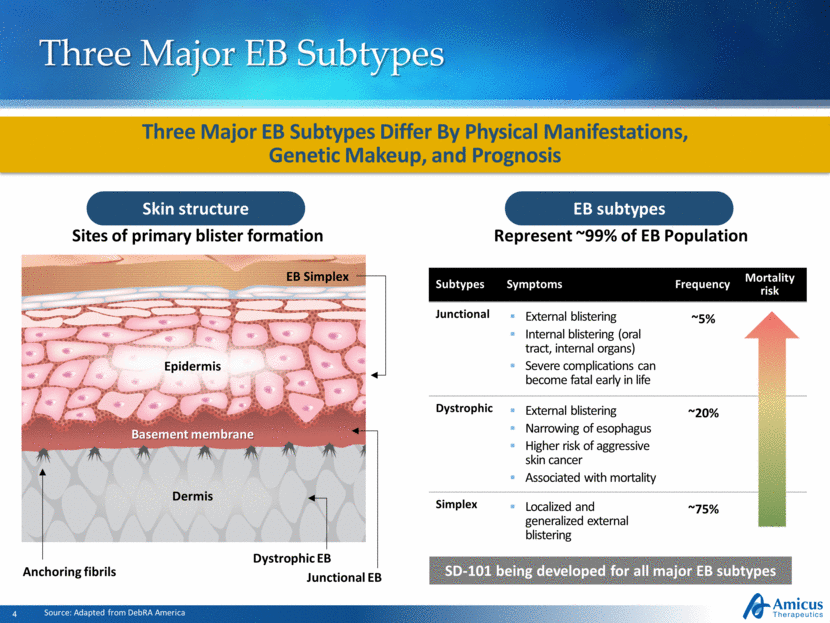
Subtypes Symptoms Frequency Mortality risk Junctional External blistering Internal blistering (oral tract, internal organs) Severe complications can become fatal early in life ~5% Dystrophic External blistering Narrowing of esophagus Higher risk of aggressive skin cancer Associated with mortality ~20% Simplex Localized and generalized external blistering ~75% Three Major EB Subtypes EB subtypes Source: Adapted from DebRA America Skin structure Sites of primary blister formation EB Simplex Junctional EB Dystrophic EB Basement membrane Anchoring fibrils Epidermis Dermis Three Major EB Subtypes Differ By Physical Manifestations, Genetic Makeup, and Prognosis Represent ~99% of EB Population SD-101 being developed for all major EB subtypes
SD-101 Overview Patented High Concentration Allantoin with Breakthrough Therapy Designation Novel, Proprietary Topical Cream Promotes Healing of Wounds in EB and is Differentiated by Applicability for All Major EB Subtypes *Margraf and Covey 1977; Meixell and Mecca 1966; Settle 1969; Flesch 1958; Fisher 1981; Cajkovac et al., 1992; Medda 1976 Active Ingredient & ROA Proposed Indication Development Phase Proposed MOA* Formulation Proprietary topical cream containing 6% allantoin, applied to entire body once daily All major EB subtypes (Simplex, Dystrophic, Junctional) Phase 3 registration study (SD-005) ongoing Aids inflammatory response, bactericidal effects, loosens protein bridges, promotes collagen Patented formulation to deliver high concentration in highly stable, soluble form

Phase 2b (Study 003) Design 48 EB patients (age ≥ 6 months)* - 1:1:1 Randomization - Daily Topical Application Placebo (n=17) SD-101 6% (n=15) Open-Label SD-101 (6%) 3-Month Double-Blind Treatment Period Assessments: 0, 14, 30, 60, 90 Days Optional Extension (SD-004) Primary Efficacy Endpoint: Target Wound Healing at Month 1 Baseline wound: Chronic (≥ 21 days), size 5-50 cm2 SD-101 3% (n=16) *Initial Disease Severity: Mean target lesion size (cm2) 14.0 (range 5-39); mean lesional BSA: 19.4% (range 0.4-48%); mean wound age (days): 182 (range 21-1,639) EB Subtypes enrolled: Simplex (n=11), Recessive Dystrophic (n=29), and Junctional (n=8) 42/44 patients entered extension study

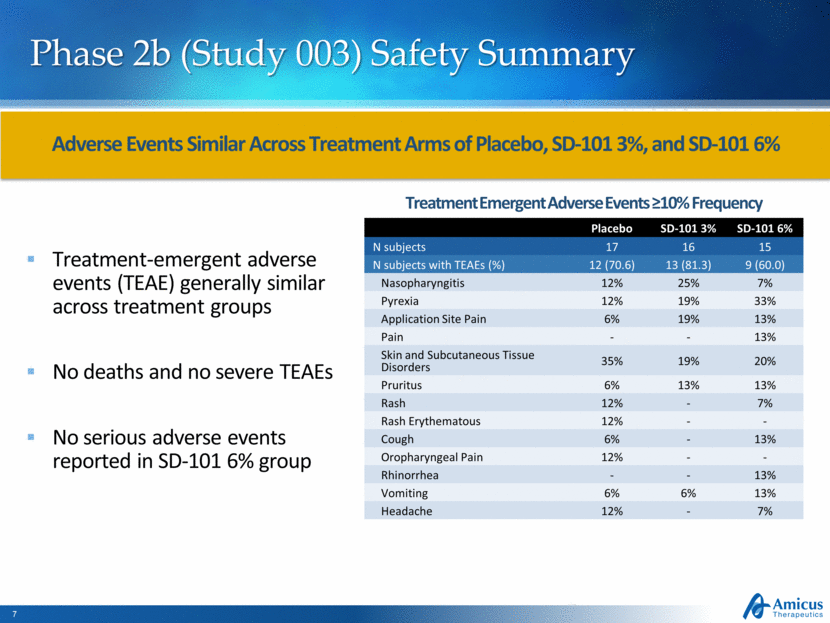
Phase 2b (Study 003) Safety Summary Treatment-emergent adverse events (TEAE) generally similar across treatment groups No deaths and no severe TEAEs No serious adverse events reported in SD-101 6% group Adverse Events Similar Across Treatment Arms of Placebo, SD-101 3%, and SD-101 6% Placebo SD-101 3% SD-101 6% N subjects 17 16 15 N subjects with TEAEs (%) 12 (70.6) 13 (81.3) 9 (60.0) Nasopharyngitis 12% 25% 7% Pyrexia 12% 19% 33% Application Site Pain 6% 19% 13% Pain - - 13% Skin and Subcutaneous Tissue Disorders 35% 19% 20% Pruritus 6% 13% 13% Rash 12% - 7% Rash Erythematous 12% - - Cough 6% - 13% Oropharyngeal Pain 12% - - Rhinorrhea - - 13% Vomiting 6% 6% 13% Headache 12% - 7% Treatment Emergent Adverse Events ≥10% Frequency
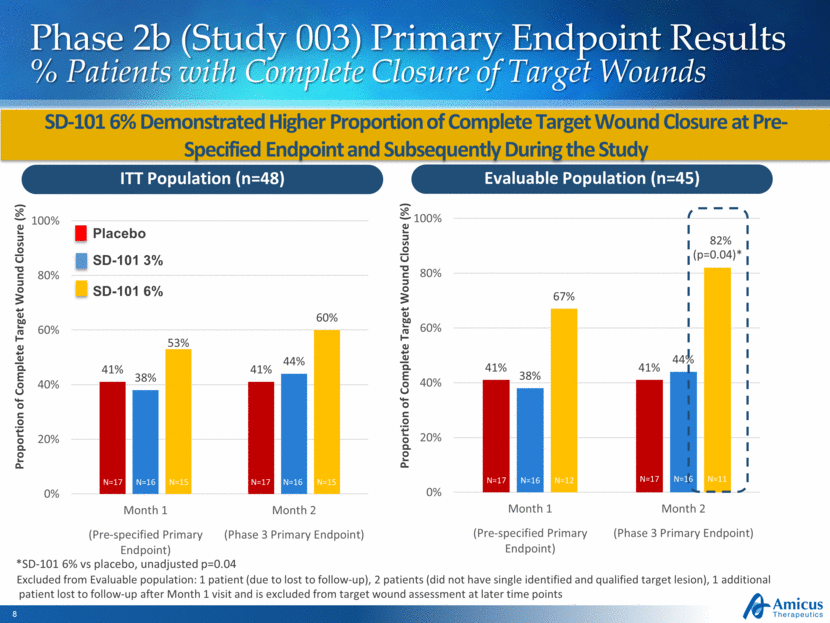
Phase 2b (Study 003) Primary Endpoint Results % Patients with Complete Closure of Target Wounds SD-101 6% Demonstrated Higher Proportion of Complete Target Wound Closure at Pre-Specified Endpoint and Subsequently During the Study *SD-101 6% vs placebo, unadjusted p=0.04 Excluded from Evaluable population: 1 patient (due to lost to follow-up), 2 patients (did not have single identified and qualified target lesion), 1 additional patient lost to follow-up after Month 1 visit and is excluded from target wound assessment at later time points ITT Population (n=48) Evaluable Population (n=45) (p=0.04)* Proportion of Complete Target Wound Closure (%) Proportion of Complete Target Wound Closure (%) N=17 N=16 N=15 N=17 N=16 N=15 N=17 N=16 N=12 N=17 N=16 N=11 Placebo SD-101 3% SD-101 6% 41% 41% 38% 44% 67% 82% 0% 20% 40% 60% 80% 100% Month 1 Month 2 (Pre-specified Primary Endpoint) (Phase 3 Primary Endpoint) 41% 41% 38% 44% 53% 60% 0% 20% 40% 60% 80% 100% Month 1 Month 2 (Pre-specified Primary Endpoint) (Phase 3 Primary Endpoint)
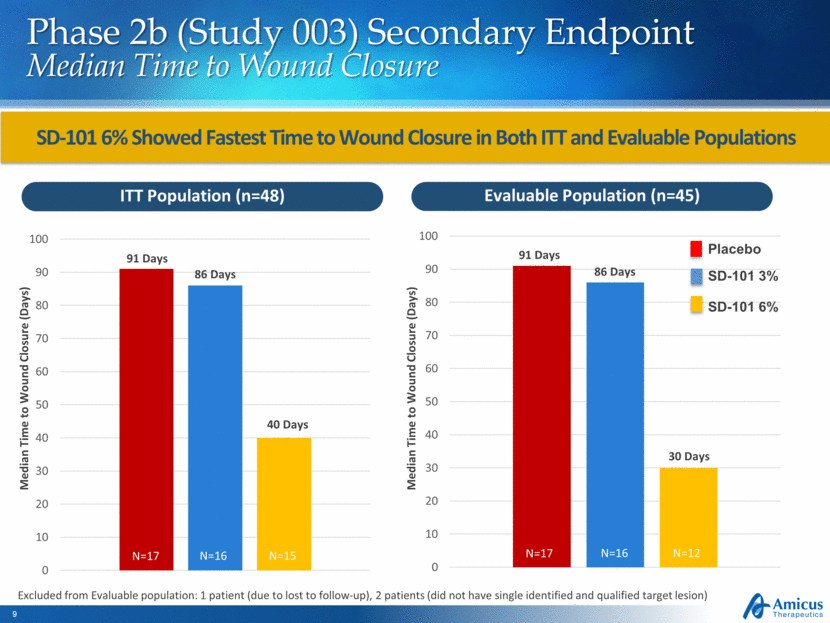
Phase 2b (Study 003) Secondary Endpoint Median Time to Wound Closure SD-101 6% Showed Fastest Time to Wound Closure in Both ITT and Evaluable Populations ITT Population (n=48) Evaluable Population (n=45) Median Time to Wound Closure (Days) Median Time to Wound Closure (Days) 91 Days 91 Days 86 Days 86 Days 40 Days 30 Days Excluded from Evaluable population: 1 patient (due to lost to follow-up), 2 patients (did not have single identified and qualified target lesion) Placebo SD-101 3% SD-101 6% N=17 N=16 N=15 N=17 N=16 N=12 0 10 20 30 40 50 60 70 80 90 100 0 10 20 30 40 50 60 70 80 90 100
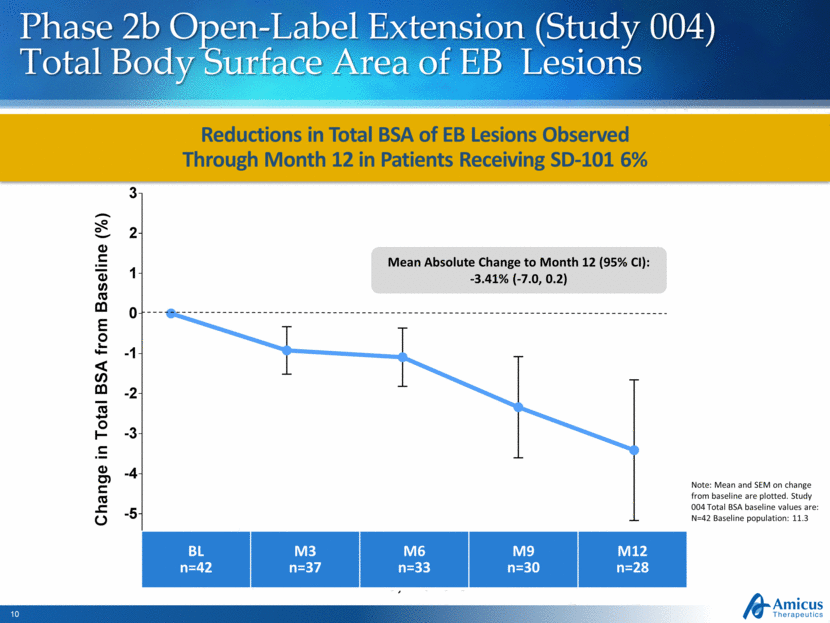
Reductions in Total BSA of EB Lesions Observed Through Month 12 in Patients Receiving SD-101 6% Phase 2b Open-Label Extension (Study 004) Total Body Surface Area of EB Lesions Note: Mean and SEM on change from baseline are plotted. Study 004 Total BSA baseline values are: N=42 Baseline population: 11.3 BL n=42 M3 n=37 M6 n=33 M9 n=30 M12 n=28 Mean Absolute Change to Month 12 (95% CI): -3.41% (-7.0, 0.2) 3 6 9 12 -5 -4 -3 -2 -1 0 1 2 3 Baseline Time, Months C h a n g e i n T o t a l B S A f r o m B a s e l i n e ( % )
Phase 2b (Study 003): Results Summary and Key Learnings SD-101 6% concentration selected for Phase 3 study based on Phase 2b dose response Placebo response minimized by analyzing subgroup of patients with wounds ≥ 10 cm2 Complete target wound closure SD-101 6% - 50% (n= 4) vs. Placebo - 12.5% (n=8) at Month 2 Phase 2b results used to calculate appropriate sample size in Phase 3 study p ≤ 0.05 if treatment difference ~17% or greater Wound closure at Month 2 (versus Month 1) is optimal time to measure primary endpoint Increases ability to distinguish SD-101 vs placebo Endpoint accepted by FDA and EU regulators Defined approval pathway with Phase 3 study design based on EMA and FDA feedback Phase 2b Learnings Informed Dose Selection, Patient Population, and Primary Endpoint for Phase 3 Trial

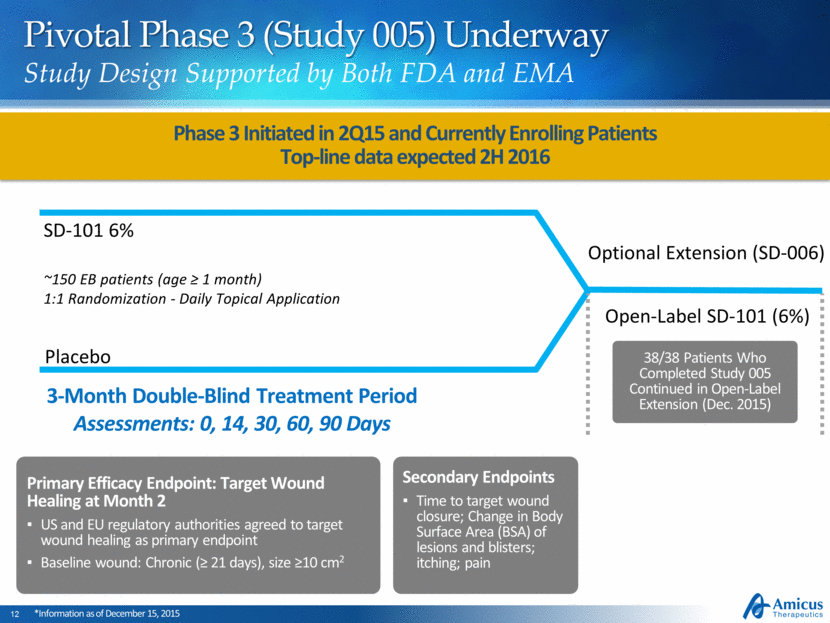
Pivotal Phase 3 (Study 005) Underway Study Design Supported by Both FDA and EMA ~150 EB patients (age ≥ 1 month) 1:1 Randomization - Daily Topical Application Placebo SD-101 6% Open-Label SD-101 (6%) 3-Month Double-Blind Treatment Period Assessments: 0, 14, 30, 60, 90 Days Optional Extension (SD-006) Primary Efficacy Endpoint: Target Wound Healing at Month 2 US and EU regulatory authorities agreed to target wound healing as primary endpoint Baseline wound: Chronic (≥ 21 days), size ≥10 cm2 Phase 3 Initiated in 2Q15 and Currently Enrolling Patients Top-line data expected 2H 2016 *Information as of December 15, 2015 Secondary Endpoints Time to target wound closure; Change in Body Surface Area (BSA) of lesions and blisters; itching; pain 38/38 Patients Who Completed Study 005 Continued in Open-Label Extension (Dec. 2015)
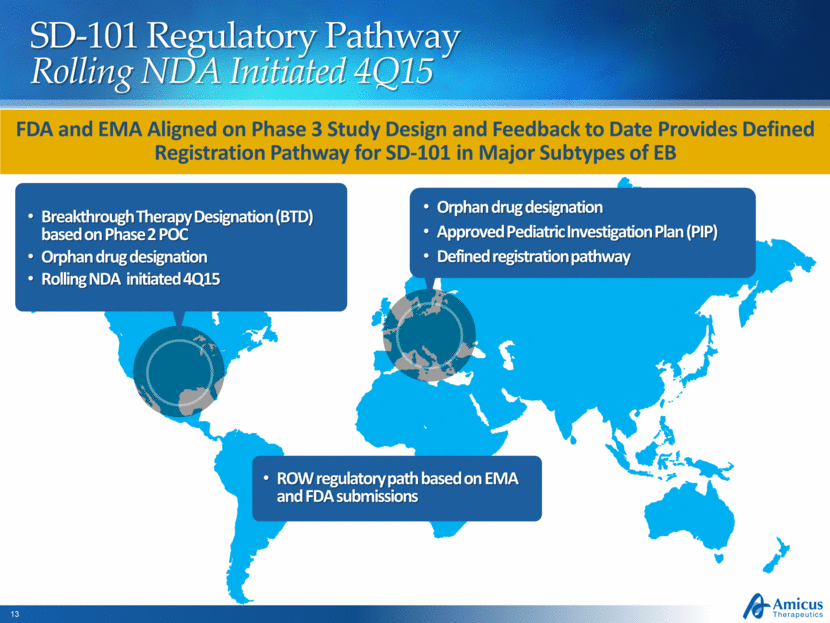
SD-101 Regulatory Pathway Rolling NDA Initiated 4Q15 Breakthrough Therapy Designation (BTD) based on Phase 2 POC Orphan drug designation Rolling NDA initiated 4Q15 Orphan drug designation Approved Pediatric Investigation Plan (PIP) Defined registration pathway FDA and EMA Aligned on Phase 3 Study Design and Feedback to Date Provides Defined Registration Pathway for SD-101 in Major Subtypes of EB ROW regulatory path based on EMA and FDA submissions
3rd Annual Dermatology Summit SD-101 for Epidermolysis Bullosa (EB) Jay Barth, MD, Chief Medical Officer