Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - AYTU BIOPHARMA, INC | d85936dex321.htm |
| EX-23.1 - EX-23.1 - AYTU BIOPHARMA, INC | d85936dex231.htm |
| EX-31.1 - EX-31.1 - AYTU BIOPHARMA, INC | d85936dex311.htm |
| EX-31.2 - EX-31.2 - AYTU BIOPHARMA, INC | d85936dex312.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended June 30, 2015
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 333-146542
AYTU BIOSCIENCE, INC.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 47-0883144 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification Number) |
| 373 Inverness Parkway Suite 206 Englewood, Colorado |
80112 | |
| (Address of principal executive offices) | (Zip Code) |
(720) 437-6580
(Registrant’s telephone number, including area code)
373 Inverness Parkway, Suite 200
Englewood, CO 80112
(Former name, former address and former fiscal year, if changed since last report)
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act
Common Stock, par value $.0001 per share
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No x
Indicate by a check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K x
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | x | |||
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of common stock held by non-affiliates of the Registrant as of December 31, 2014 was $4,349,344 based on the closing price of $2.31 as of that date.
Indicate the number of shares outstanding of each of the Registrant’s classes of common stock, as of the latest practicable date:
As of September 1, 2015, 14,259,681 shares of common stock were outstanding.
Table of Contents
This Annual Report on Form 10-K refers to trademarks, such as Aytu, Zertane, Vyrix, RedoxSYS, MiOXSYS, ProstaScint and Luoxis which are protected under applicable intellectual property laws and are our property or the property of our subsidiaries. This Form 10-K also contains trademarks, service marks, copyrights and trade names of other companies which are the property of their respective owners. Solely for convenience, our trademarks and tradenames referred to in this Form 10-K may appear without the ® or ™ symbols, but such references are not intended to indicate in any way that we will not assert, to the fullest extent under applicable law, our rights to these trademarks and tradenames.
Unless otherwise indicated or unless the context otherwise requires, references in this Form 10-K to the “Company,” “Aytu,” “we,” “us,” or “our” are to Aytu BioScience, Inc; and references to “Ampio” are to Ampio Pharmaceuticals, Inc., our parent company.
2
Table of Contents
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS AND INDUSTRY DATA
Forward Looking Statements
This Annual Report on Form 10-K, or Annual Report, includes forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, or the Exchange Act. All statements other than statements of historical facts contained in this Annual Report, including statements regarding our anticipated future clinical and regulatory events, future financial position, business strategy and plans and objectives of management for future operations, are forward-looking statements. Forward looking statements are generally written in the future tense and/or are preceded by words such as “may,” “will,” “should,” “forecast,” “could,” “expect,” “suggest,” “believe,” “estimate,” “continue,” “anticipate,” “intend,” “plan,” or similar words, or the negatives of such terms or other variations on such terms or comparable terminology. Such forward-looking statements include, without limitation, statements regarding the anticipated start dates, durations and completion dates, as well as the potential future results, of our ongoing and future clinical trials, the anticipated designs of our future clinical trials, anticipated future regulatory submissions and events, the potential future commercialization of our product candidates, our anticipated future cash position and future events under our current and potential future collaborations. These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including without limitation the risks described in “Risk Factors” in Part I, Item 1A of this Annual Report. These risks are not exhaustive. Other sections of this Annual Report include additional factors that could adversely impact our business and financial performance. Moreover, we operate in a very competitive and rapidly changing environment. New risk factors emerge from time to time and it is not possible for our management to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. We cannot assure you that the events and circumstances reflected in the forward-looking statements will be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. We assume no obligation to update or supplement forward-looking statements.
We obtained statistical data, market and product data, and forecasts used throughout this Form 10-K from market research, publicly available information and industry publications. While we believe that the statistical data, industry data and forecasts and market research are reliable, we have not independently verified the data, and we do not make any representation as to the accuracy of the information.
3
Table of Contents
AYTU BIOSCIENCE, INC.
We are a specialty healthcare company concentrating on developing and commercializing products with an initial focus on urological conditions. We are focused primarily on the urological disorders market and initially sexual dysfunction, urological cancers and male infertility. We are currently focused on commercializing our ProstaScint® product, which we plan to launch through a focused commercial infrastructure in the U.S. while developing corporate relationships outside the U.S. to launch ProstaScint in major healthcare markets around the world. We acquired ProstaScint in May 2015 from Jazz Pharmaceuticals. ProstaScint, which is approved by the U.S. Food and Drug Administration, or FDA, is a marketed biologic imaging agent specifically indicated for the diagnostic staging of prostate cancer patients. We plan to launch the RedoxSYS® oxidation-reduction potential system, or the RedoxSYS System, into the global research market while developing numerous clinical applications for this potential first-in-class diagnostic device, including an application for the detection of infertility in semen. We have developed a RedoxSYS product line extension called MiOXSYS™ intended for use in male infertility assessment, for which this product is undergoing clinical studies. Further, we are entering late-stage development of our lead therapeutic candidate, Zertane, which is being studied in premature ejaculation.
Our product candidate Zertane is in clinical development for the treatment of premature ejaculation. The premature ejaculation market in the U.S. and Europe is expected to reach over $1.3 billion in annual sales by 2017, representing a projected increase of 10.3% from 2010. According to recent published analyses, premature ejaculation, or PE, is a highly prevalent male sexual dysfunction affecting 20-30% of men worldwide. Based on internal market research and published reports, we believe that PE is up to 1.5-times more prevalent than erectile dysfunction, or ED. Currently, there are no FDA-approved prescription products in the United States to treat PE, and to our knowledge, only two other prescription products have been approved elsewhere in the world. Treatment options for PE have traditionally included antidepressant drugs prescribed “off label,” topical numbing medications, and cognitive behavior therapy or counseling, all of which have had limited effectiveness in treating the disorder. PE therefore represents an area of significant unmet medical need.
By virtue of our recent acquisition of ProstaScint, we are now commercial stage and generating sales for this FDA-approved prostate cancer imaging agent. As prostate cancer is a condition commonly diagnosed and treated by urologists, ProstaScint complements our urology-focused product pipeline. Prostate cancer is the most common cancer among men in the United States, with an estimated 218,000 annual cases (as of 2010). Further, more than 2,200,000 men were alive with some history of prostate cancer in 2006, and over 30,000 U.S. men die each year from the disease. The effect of prostate cancer on healthcare economics is substantial, which makes the need for accurate disease staging critical for treatment and management strategies. The U.S. market for the diagnosis and screening of prostate cancer is expected to total $17.4 billion by 2017, compound annual growth rate, or CAGR of 7.5%.
We are actively developing the global market for the RedoxSYS System across a range of applications. Specifically, we have begun commercializing RedoxSYS System for research use through direct selling, distribution partners, and academic collaborators. Over the past 18 months, we have engaged in over 60 trials around the world whereby prominent researchers are implementing oxidation-reduction potential as a marker in both chronic and acute illnesses and disorders in both clinical research as well as basic science research.
Through our extensive network of researchers, the RedoxSYS System has demonstrated the potential to have broad clinical applications. Studies are now underway at a major U.S. university in the area of male infertility. Thus, we have developed the MiOXSYS System to uniquely detect oxidation-reduction potential in semen specimens. Male infertility is prevalent, underserved, and oxidative stress is widely implicated in its pathophysiology. The global male infertility market is expected to grow to over $300 million by 2020 with a CAGR of nearly 5% from 2013 to 2020. Oxidative stress is broadly implicated in the pathophysiology of oxidative stress, yet very few diagnostic tools exist to effectively measure oxidative stress levels in men. However, antioxidants are widely available and recommended to infertile men. With the introduction of the MiOXSYS System, we believe for the first time there will be an easy and effective diagnostic tool to assess degree of oxidative stress, sperm motility and morphology, and potentially enabling the monitoring of patients’ responses to antioxidant therapy as a treatment regimen for infertility.
The technology underpinning the RedoxSYS and MiOXSYS systems was developed by Luoxis Diagnostics, Inc. in the two years immediately preceding the merger between Luoxis, Vyrix Pharmaceuticals, Inc., and us (under our former name of Rosewind Corporation) in April 2015. Upon the consummation of the merger, the RedoxSYS System and MiOXSYS System became our assets. Prior to the incorporation of Luoxis, the predecessor technologies that are now incorporated into these products were developed by the research team of Dr. David Bar-Or when he was at DMI BioSciences, Inc. and subsequently at Ampio Pharmaceuticals, Inc.
4
Table of Contents
Corporate History
We were incorporated as Rosewind Corporation on August 9, 2002 in the State of Colorado.
Vyrix Pharmaceuticals, Inc., or Vyrix, was incorporated under the laws of the State of Delaware on November 18, 2013 and was wholly owned by Ampio Pharmaceuticals, Inc. (NYSE MKT: AMPE), or Ampio, immediately prior to the completion of the Merger (defined below). Vyrix was previously a carve-out of the sexual dysfunction therapeutics business, including the late-stage men’s health product candidates, Zertane and Zertane-ED, from Ampio, which carve out was announced in December 2013. Luoxis Diagnostics, Inc., or Luoxis, was incorporated under the laws of the State of Delaware on January 24, 2013 and was majority owned by Ampio immediately prior to the completion of the Merger. Luoxis was focused on initially developing and advancing the RedoxSYS System. The MiOXSYS System was developed following the completed development of the RedoxSYS System.
On March 20, 2015, Rosewind formed Rosewind Merger Sub V, Inc. and Rosewind Merger Sub L, Inc., each a wholly-owned subsidiary formed for the purpose of the Merger, and on April 16, 2015, Rosewind Merger Sub V, Inc. merged with and into Vyrix and Rosewind Merger Sub L, Inc. merged with and into Luoxis, and Vyrix and Luoxis became subsidiaries of Rosewind. Immediately thereafter, Vyrix and Luoxis merged with and into Rosewind with Rosewind as the surviving corporation (herein referred to as the Merger).
Concurrent with the closing of the Merger, Rosewind abandoned its pre-merger business plans to develop a sailing school, and we now solely pursue the specialty healthcare market, focusing on urological related conditions, including the business of Vyrix and Luoxis.
On June 8, 2015, we (i) reincorporated as a domestic Delaware corporation under Delaware General Corporate Law and changed our name from Rosewind Corporation to Aytu BioScience, Inc., and (ii) effected a reverse stock split in which each common stock holder received one share of common stock for each every 12.174 shares outstanding (herein referred to as the Reverse Stock Split). All share and per share amounts in this Annual Report have been adjusted to reflect the effect of the Reverse Stock Split.
Products
Zertane for Premature Ejaculation
Our premature ejaculation product candidate, Zertane, is a specifically formulated orally disintegrating tablet, or ODT, of tramadol hydrochloride patented for the on-demand treatment of PE. Zertane is being developed utilizing a regulatory pathway pursuant to Section 505(b)(2) of the Food, Drug and Cosmetic Act, as amended, or the FDCA, as the active ingredient is already well characterized for the treatment of pain, and we are relying on the FDA’s finding of safety of tramadol hydrochloride to support its use in a new indication, PE, at a lower dose. If we receive marketing approval for Zertane, we believe it will be the first commercial product approved by the FDA for PE.
Effective August 18, 2014 the Drug Enforcement Administration, or DEA, began classifying tramadol as a Schedule IV substance, including its salts, isomers, and salts of isomers. Tramadol has a low potential for abuse relative to the drugs or substances in Schedule III and has a currently accepted medical use in treatment in the U.S. for moderate to moderately severe pain. We expect the same classification for Zertane regardless of the indication, including premature ejaculation. As a result of this classification, manufacturers are required to print “C-IV” on the labeling of commercial containers of tramadol. It is unclear as to what extent the FDA and/or the DEA would expect manufacturers of tramadol-containing products to further control its distribution or monitor its use. However, we do not expect this classification to adversely affect the commercial potential for Zertane considering that tramadol has a long history of use and a low potential for abuse.
The method of use patents covering the use of tramadol for premature ejaculation were originally developed and held by Ampio (our parent predecessor company). However, these patents and all related intellectual property were assigned to Vyrix when it was established as an Ampio subsidiary. Following the Merger of Vyrix, Luoxis and us, the patents were transferred and are now solely owned by us.
ProstaScint for Prostate Cancer Imaging
A key part of our strategy is to identify, acquire, license, or otherwise promote marketed, complementary urology assets in order to establish a commercial footprint and generate revenues for already-approved or near-term medical products. To that end we acquired ProstaScint from Jazz Pharmaceuticals shortly after the Merger. ProstaScint received FDA approval on October 28, 1996 and was initially marketed by Cytogen Corporation.
ProstaScint (capromab pendetide) is a radio-labeled monoclonal antibody, which is a biologic product that targets a specific antigen. ProstaScint targets Prostate Specific Membrane Antigen (PSMA), a protein uniquely expressed by prostate tissue. Indium (In 111) is attached to the proprietary, mouse-derived antibody. The radiolabeled antibody is infused into the patient and is taken up
5
Table of Contents
by prostate cancer cells which can be detected and visualized with single-photon emission tomography (SPECT). ProstaScint has been shown to be clinically effective in determining the course of treatment for a patient who has had a prostatectomy and/or has suspected metastasis (spread of the cancer cells beyond the prostate). Further, ProstaScint has demonstrated efficacy in newly diagnosed patients classified as high-risk or with recurrent prostate cancer.
Multiple clinical studies have been conducted in the United States and published in peer-reviewed publications. These studies consistently demonstrate substantial clinical efficacy of ProstaScint in staging prostate cancer patients and specifically identifying whether the cancer is confined to the prostate or has metastasized to other parts of the body. Through more accurate clinical staging and identification of metastatic prostate cancer, clinicians are able to better direct therapeutic interventions and improve outcomes.
RedoxSYS System for Research Use
We completed the development of the RedoxSYS System during the two years preceding the Merger. In 2014, we received ISO 13485 certification, demonstrating our compliance with global quality standards in medical device manufacturing. This enables the launch of the RedoxSYS System into the research market around the world. We also received a CE marking in Europe and Health Canada clearance to begin the market development of the RedoxSYS System as a clinical diagnostic in Europe, Canada, and elsewhere around the world where CE marking is recognized. We launched sales efforts into the research market in late 2014 and since that time have already placed the RedoxSYS System at a number of prominent research centers in the United States, Europe, Israel, Japan, Taiwan, Singapore and Korea.
MiOXSYS System for Male Infertility
As part of our strategy to develop future clinical applications of the RedoxSYS System, we have conducted initial studies in male reproductive health. Male infertility is a significant medical condition in which oxidative stress is well known to play a substantial role. As such, we believe developing a clinical application to assess oxidative stress levels with the uniquely designed and programmed MiOXSYS System for semen analysis represents a significant commercial opportunity. Oxidative stress is well established as a leading contributing factor to male infertility. Further, a significant proportion of male infertility remains unexplained in part because of the lack of standardized tests available to clinicians and researchers to assess oxidative stress in semen and seminal plasma. This lack of standardization has resulted in poor implementation of semen and plasma analysis around the world. Further, currently available tests are cumbersome, time consuming to perform, and costly.
We have conducted initial proof-of-concept clinical studies in male infertility with a leading research center in the United States, which demonstrates that oxidation-reduction potential effectively measures oxidative stress levels in semen and seminal plasma – and that these levels strongly correlate with established markers of infertility. Semen analysis studies are routinely conducted to assess causes of infertility, so we expect clinicians and oxidative stress researchers to readily integrate the MiOXSYS System into routine use upon the completion of more extensive studies and regulatory clearance for this use. Additional studies are now in the late planning stages that will evaluate the MiOXSYS System’s performance in the detection of oxidative stress levels and correlations with key semen parameters in both healthy and infertile males. The MiOXSYS System must receive 510(k) de novo clearance from the FDA before we can market it for clinical use in the United States. Of the $300 million male infertility market projected for 2020, the North American, Middle Eastern, and Asia Pacific markets dominate due to prevalence, awareness of treatment, and availability of treatment resources. Thus, it is important that we have already established distribution relationships and direct access to major oxidative stress researchers in many of these important markets.
An attractive aspect of the reproductive health market relates to reimbursement, as infertility treatments and the associated diagnostic tests are generally paid directly by patients. The current infertility treatments and associated diagnostics typically cost in excess of $10,000 per treatment cycle, so the addition of a moderately priced oxidative stress test would consume nominal relative costs while providing specific, actionable information needed to improve the oxidative status of infertile patients. The current infertility treatments include antioxidant supplements and lifestyle modifications that lower oxidative stress (e.g., smoking cessation, exercise, dietary changes, etc.), so the measurements reported by the MiOXSYS System could effectively guide treatment in the infertile patients.
We have an extensive range of intellectual property across our primary assets, Zertane, MiOXSYS, and RedoxSYS. We have patent protection in the United States and several other large markets worldwide. Specifically, we have numerous patents issued and pending for the RedoxSYS/MiOXSYS systems and their use in the U.S., Europe, Israel, and major markets in Asia inclusive of Japan, Korea, China, and the Middle East. Further, we have patent protection in the United States and several other large markets worldwide for the use of tramadol hydrochloride to treat PE. We also have intellectual property specifically covering Zertane-ED, our product candidate to treat comorbid premature ejaculation and erectile dysfunction, or ED, that has issued patents in several large markets worldwide and is pending in the United States. However, we are not actively developing Zertane-ED at this time.
6
Table of Contents
Strategy
Key elements of our strategy include:
| • | Develop a pipeline of therapeutics and diagnostics focused on urological conditions, with a focus on the initiation and completion of two Phase 3 clinical trials for Zertane in the United States and the development of worldwide commercialization and marketing partnerships. |
| • | Commercialize FDA-approved ProstaScint for the staging of both newly diagnosed high-risk and recurrent prostate cancer patients. We plan to commercialize ProstaScint in the U.S. and in key markets around the world. |
| • | Establish the RedoxSYS System initially as a research tool and establish MiOXSYS as a clinical diagnostic device with application in male infertility. |
| • | Acquire established marketed products and late-stage development assets within our core urology focus. |
Product Pipeline
Our Lead Therapeutic Product Candidate – Zertane
Our lead therapeutic product candidate, Zertane, is a specifically formulated orally disintegrating tablet, or ODT, of tramadol hydrochloride patented for the on-demand treatment of PE. Zertane is being developed utilizing a regulatory pathway pursuant to Section 505(b)(2) of the Food, Drug and Cosmetic Act, as amended, or the FDCA, as the active ingredient is already well characterized for the treatment of pain, and we are relying on the FDA’s finding of safety of tramadol hydrochloride to support its use in a new indication, PE, at a lower dose. If we receive marketing approval for Zertane, we believe it will be the first commercial product approved by the FDA for PE.
There are three main types of NDAs, which are covered by Section 505 of the FDCA: (1) an application that contains full reports of investigations of safety and efficacy (Section 505(b)(1)); (2) an application that contains full reports of investigations of safety and effectiveness but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the application has not obtained a right of reference (Section 505(b)(2)); and (3) an application that contains information to show that the proposed product is identical in active ingredient, dosage form, strength, route of administration, labeling, quality, performance characteristics, and intended use, among other things, to a previously approved product (Section 505 (j)). Section 505(b)(2) expressly permits the FDA to rely, for approval of an NDA, on data not developed by the applicant. In the preIND briefing meeting with Ampio and in June 2012, the FDA agreed that our NDA may be submitted under Section 505(b)(2). As such, we intend to rely on studies published in the scientific literature and reference FDA-approved NDAs for tramadol-containing products (NDAs 21-693, 20-281 and 21-692) to support the safety and efficacy demonstrated in our clinical program. Relying on Section 505(b)(2) is advantageous because we or our collaborators may not be required (i) to perform the full range of safety and efficacy trials that is otherwise required to secure approval of a new drug, and (ii) obtain a “right of reference” from the applicant that obtained approval of the previously approved drug. However, a Section 505(b)(2) application must support the proposed change of the previously approved drug by including necessary and adequate information, as determined by the FDA, and the FDA may still require us to perform a portion or the full range of safety and efficacy trials. There can be no assurance that we would be successful under any Section 505(b)(2) application.
According to recent published analyses, PE is a highly prevalent male sexual dysfunction affecting 20-30% of men worldwide. Based on internal market research and published reports, we believe that PE is up to 1.5-times more prevalent than erectile dysfunction. Currently, there are no FDA-approved prescription products in the United States to treat PE, and to our knowledge, only one oral prescription product has been approved elsewhere in the world. Treatment options for PE have traditionally included antidepressant drugs prescribed “off label,” topical numbing medications, and cognitive behavior therapy or counseling, all of which have had limited effectiveness in treating the disorder. PE therefore represents an area of significant unmet medical need.
By virtue of significant development work performed by a previous corporate partner, Zertane has already been evaluated outside the United States in two Phase 1 clinical trials, two Phase 2 clinical trials and two Phase 3 clinical trials. This development work has demonstrated a favorable safety and efficacy profile of Zertane in men with PE and helped inform the design and endpoints of the Phase 3 clinical trials we will need to obtain FDA approval. Furthermore, the safety and pharmacology of the drug substance in Zertane, tramadol hydrochloride, is well characterized, which we believe will eliminate the need for us to conduct additional pre-clinical studies and safety trials. We believe we are well positioned to initiate Phase 3 clinical trials with Zertane in the United States. Upon completion of the trials, if successful, we plan to submit a New Drug Application, or NDA, and subsequently market Zertane in the United States, if approved.
Our strategy for Zertane is focused on the initiation and completion of the Phase 3 clinical program in the United States and the development of worldwide commercialization and marketing partnerships. We expect to finalize clinical development of Zertane, seek FDA marketing approval and – if approved—commercialize the product candidate in the United States either directly or via
7
Table of Contents
partnerships. While we have not yet submitted an IND for Zertane to the FDA that would allow us to initiate the planned Phase 3 trial, we expect to submit the IND in the second half of 2015. We will seek partnerships to commercialize Zertane in the major markets outside of the United States. We already have partnerships in place to market Zertane in South Korea (Daewoong Pharmaceutical Co., Ltd.) and Brazil (FBM Farma Industria Farmaceutica), which could provide near-term revenue for us if, working with our partners, we are able to successfully obtain regulatory approval in those countries. In addition, we recently entered into an agreement with Endo Ventures Limited, which recently acquired Paladin Labs Inc., or Paladin, a leading Canadian specialty pharmaceutical company, to provide exclusive rights to market, sell and distribute Zertane in Canada, the Republic of South Africa, certain countries in Sub-Saharan Africa, Colombia and Latin America.
Market Opportunity
PE Market
PE is a significant unmet medical need in the United States and worldwide as it causes significant emotional distress for affected men and their partners. According to an article published in European Urology in 2010 and a survey published in the Journal of the American Medical Association (JAMA) in 1999, PE is a highly prevalent male sexual dysfunction affecting 20-30% of men worldwide. However, most prevalence data on PE is based on patient surveys, which are inherently subjective, and therefore some of the men surveyed may not have PE as it is defined by major medical societies. In a 2007 study published in European Urology, the incidence of PE was assessed via a web-based survey of 12,133 men ages 18-70 in the United States, Germany and Italy. In this survey, 2,754 or 22.7% of the men reported that they suffer from PE. The vast majority (87.9%) of men with PE wished that they had more control over time to climax. Additionally, a majority (57.6%) of the men surveyed reported that they would seek medical treatment if they knew that a pill to control ejaculation were available. We believe men would also ask their doctor about treatment options if their partner suggested it. Additional primary company market research indicated that over 66% of patients that see a urologist for PE were self-referred, which we believe further demonstrates that PE is a condition for which patients are actively seeking treatment.
Presently, there are no approved prescription pharmaceuticals in the United States to treat PE and only two pharmaceuticals known to be approved elsewhere in the world. Current “off label” or unapproved therapies used to treat PE carry with them unwanted side effects and inconsistent or limited effectiveness. Topical over-the-counter, or OTC, options are not preferred due to route of administration and may also have an impact on partner satisfaction. Oral therapeutics, specifically selective serotonin reuptake inhibitors, or SSRIs, carry potentially significant side effects; the most notable of which is diminished libido. PDE-5 inhibitors have been prescribed “off label” for PE but have not demonstrated efficacy. Outside of oral or topical therapeutics, non-medical options include behavioral therapy and relationship counseling, both of which can be time consuming and stressful and frequently ineffective for men and their partners.
We believe patients and their partners are generally dissatisfied with existing pharmacologic and non-pharmacologic treatments for PE. Based on primary market research commissioned by us, which included discussions with a cross section of clinicians that treat patients with PE, we believe that there are significant issues with existing PE treatments demonstrating a real need for a safe and effective, FDA-approved product to treat PE that does not have a ramp-up period.
Our lead product candidate, Zertane for premature ejaculation, contains 89 mg tramadol hydrochloride in an orally dissolving tablet, or ODT. Tramadol hydrochloride is a well-established, centrally acting synthetic analgesic and has been used for more than 30 years as a treatment for moderate to severe pain. The drug and its active metabolite (M1, O-desmethyltramadol) act as an opiate agonist, apparently by selective activity at the µ-receptor. Although the mechanism by which tramadol hydrochloride delays ejaculation has not been identified, numerous laboratory studies have shown that tramadol hydrochloride also acts as an N-methyl-D-aspartate receptor antagonist, 5-hydroxytryptamine type 2C receptor antagonist, 5 nicotinic acetylcholine receptor antagonist, M1 and M3 muscarinic acetylcholine receptor antagonist, and a serotonin and norepinephrine modulator. It is possible that one or a combination of these effects leads to a delay in ejaculation. The relative contribution of tramadol hydrochloride versus its M1 metabolite to delay ejaculation is unknown. However, the metabolite is six times more potent than the parent drug in producing analgesia in animal models and 200 times more potent in µ-receptor binding. As a pain medication, tramadol hydrochloride has been associated with certain adverse effects including dizziness, nausea, constipation, vertigo, headache, vomiting and drowsiness. However, we intend that our labeling for Zertane, if regulatory approval is obtained, will suggest “as required” dosing before sexual intercourse and not to exceed one tablet per day. Based on previous clinical studies, we believe that limiting the dosing to no more than once per day will minimize any side effects.
Our Marketed, FDA-Approved Prostate Imaging Product – ProstaScint
On May 20, 2015, we acquired ProstaScint from Jazz Pharmaceuticals. ProstaScint is already approved by the FDA and is generating revenues. As such, we expect to launch a commercial infrastructure in order to support increased sales and distribution of ProstaScint in the U.S. ProstaScint, or capromab pendetide, is a radio-labeled monoclonal antibody, which is a biologic product that targets a specific antigen. ProstaScint targets Prostate Specific Membrane Antigen (PSMA), a protein uniquely expressed by prostate tissue. A
8
Table of Contents
radioactive substance called Indium In 111 is attached to the proprietary, mouse-derived antibody. The radiolabeled antibody is infused into the patient and is taken up by prostate cancer cells which can be detected and visualized with a special nuclear medicine scan (single-photon emission tomography, or SPECT). ProstaScint has been shown to be clinically effective in determining the course of treatment for a patient who has had a prostatectomy and/or has suspected metastasis (spread of the cancer cells beyond the prostate). Further, ProstaScint has demonstrated efficacy in newly diagnosed patients classified as High Risk or with recurrent prostate cancer. In addition to U.S. approval ProstaScint has also been approved by Health Canada.
ProstaScint fills an important medical need in the detection of a common illness facing a significant number of men in the U.S. and around the world. Prostate cancer is the most common cancer among men in the United States, with an estimated 218,000 annual cases (as of 2010). Further, more than 1,800,000 men are alive with some history of prostate cancer, and over 30,000 U.S. men die each year from the disease. The effect of prostate cancer on healthcare economics is substantial, which makes the need for accurate disease staging critical for treatment and management strategies. The U.S. market for the diagnosis and screening of prostate cancer is expected to total $17.4 billion in 2017, a CAGR of 7.5%.
ProstaScint has several unique selling features that we believe will enable significant sales growth and regular use by healthcare providers diagnosing and treating prostate cancer. ProstaScint is the only imaging agent that specifically targets prostate cancer cells and demonstrates high sensitivity, specificity, and accuracy. In multiple clinical studies researchers have shown that when SPECT/CT scans were used in patients pre-treated with ProstaScint, ProstaScint imaging was highly sensitive in detecting prostate cancer and significantly predictive of 10-year biochemical disease free survival in prostate cancer patients (86.6% vs. 65.5%; p=0.0014). Importantly, ProstaScint is already approved by the FDA with a history of sales in the U.S. Additionally, the American Cancer Society specifically recognizes ProstaScint by name in current prostate cancer diagnosis guidelines.
Summary of Select ProstaScint Clinical Studies
Multiple clinical studies have been conducted in the United States and published in peer-reviewed publications. These studies consistently demonstrate substantial clinical efficacy of ProstaScint in staging prostate cancer patients and specifically identifying whether the cancer is confined to the prostate or has metastasized to other parts of the body. Through more accurate clinical staging and identification of metastatic prostate cancer, clinicians are able to better direct therapeutic interventions and improve outcomes. A brief summary of key clinical findings for ProstaScint from select studies are summarized below.
| Principal Investigator(s)/ Primary Authors |
Publication |
Patient Population |
Conclusion/Results | |||
| Ellis RJ et al. | Int. J. Radiation Oncology Biol. Phy. (2010) | Patients presenting for primary radiotherapy having a clinical diagnosis of localized primary prostate cancer; Patients evaluated for tumor stage using conventional staging and SPECT/CT (N=239) | SPECT/CT imaging with ProstaScint pre-treatment was significantly predictive of 10-year biochemical disease-free survival (86.6% vs. 65.5%; p=0.0014) | |||
| Haseman MK et al. | Urology (2007) | Men with prostate cancer who underwent imaging with ProstaScint pretreatment; Patients were divided according to the presence or absence of central abdominal uptake(CAU) (N=341) | SPECT/CT imaging with ProstaScint pretreatment effectively predicted death rates among patients with central abdominal uptake (CAU), and demonstrated that prostate cancer-specific death rates were 10 times higher in patients identified with ProstaScint as having central abdominal uptake (p=0.005). | |||
| Ellis RJ et al. | Brachytherapy (2005) | Men with prostate cancer of all risk categories who underwent imaging with ProstaScint pretreatment; patients were divided into low, intermediate, and high risk and underwent brachytherapy (N=239) | SPECT/CT imaging with ProstaScint pretreatment effectively predicted biochemical disease recurrence regardless of the patient’s risk category; 7-year outcomes data from brachytherapy patients with treatment based on the ProstaScint scan showed a significant difference in biochemical disease-free survival. | |||
9
Table of Contents
Radiation oncology experts have published numerous papers expressing the potential for expanded use of ProstaScint in prostate cancer imaging due to advances in imaging technologies since its FDA approval in 1996. Since the early 2000s significantly greater image resolution has been enabled due to the advent of dual head cameras (and improved imaging in general) along with the use of co-registered images where radiologists now combine the images of SPECT and computerized tomography (CT) or magnetic resonance imaging (MRI).
Our Marketed Research Instrument – The RedoxSYS System
Our leading diagnostic product candidate, the RedoxSYS System, is now fully developed for research use. RedoxSYS is a novel, portable device that measures oxidation-reduction potential, or ORP, a global measure of oxidative stress. This system is the first and only system that measures ORP in biologic specimens to provide a complete measure of redox balance, which is broadly implicated across a wide range of both acute and chronic conditions. To date, Canadian and European regulators have characterized RedoxSYS System as Class II medical device and regulated them accordingly. Classification of a medical device as Class II in Europe and Canada indicates that the device is generally regarded as posing medium risk, and non-invasive medical devices that come into contact with injured skin are generally classified as Class II. As we have conducted initial validation studies with the RedoxSYS System across a range of conditions and obtained a CE marking in Europe and Health Canada clearance to begin the market development of the RedoxSYS System as a clinical diagnostic in Europe, Canada, and elsewhere around the world where CE marking is recognized, we are now initiating commercialization for use of the RedoxSYS System as a research tool. By employing a focused commercial infrastructure and a focused network of distributors around the world, we believe we can efficiently penetrate the academic and industry-based research centers who study oxidative stress. With this growth in the research market, we intend to develop clinical applications for the RedoxSYS System.
Our strategy for the RedoxSYS System is to continue deployment of this system at leading academic centers around the world, develop research collaborations with key opinion leaders in oxidative stress research, identify clinical applications for the platform, and aggressively pursue infertility studies to establish efficacy of the system in this setting of care. In 2013 and 2014, we deployed the RedoxSYS System around the world in the development of numerous future clinical applications. While many areas of study have been undertaken, we have focused research resources on high-value areas where significant medical needs remain unmet. Given our initial orientation around trauma, the studies completed thus far have focused on large conditions related to critical care. These initial studies demonstrated the initial clinical validation for the RedoxSYS System and represent substantial opportunities as growth applications and markets following initial entry into the research and infertility markets. As our direct commercial efforts will focus on male infertility, we expect that other areas of clinical use would be pursued through partnerships with corporate partners established in these non-urological clinical channels. A significant potential opportunity that has presented promise through our research is the application of ORP in the study of male infertility. ORP represents a unique approach to assessing oxidative stress in male infertility as it relates to semen analysis, and early clinical studies have been conducted. We are now beginning larger-scale clinical studies with a globally recognized U.S. university in male infertility. As such we have developed a RedoxSYS System line extension called MiOXSYS specifically designed and programmed to detect oxidative stress in semen specimens. Oxidative stress is widely assessed in semen analysis laboratories as part of male infertility assessment, and we believe the MiOXSYS System, if proven effective, will provide a simple, comprehensive solution to oxidative stress detection and management of antioxidant and lifestyle intervention in this underserved market.
Future Products
We plan to augment our core development and commercial assets through efficient identification of complementary therapeutics, devices, and diagnostics related to urological disorders. We intend to seek assets that are near commercial stage or already generating revenues. Further, we intend to seek to acquire products through asset purchases, licensing, co-development, or collaborative commercial arrangements (co-promotions, co-marketing, etc.).
Our management team has extensive experience across a wide range of business development activities and have in-licensed or acquired products from large, mid-sized, and small enterprises in the United States and abroad. Through an assertive product and business development approach, we expect that we will rapidly advance our internal products as well as externally sourced assets.
10
Table of Contents
Business Overview
We are a specialty healthcare company concentrating on developing and commercializing products with an initial focus on urological related conditions. We are focused primarily on the urological disorders market and specifically sexual dysfunction, urological cancers and male infertility. We are currently focused on commercializing our ProstaScint® product, which we plan to launch through a focused commercial infrastructure in the U.S. while developing corporate relationships outside the U.S. to launch ProstaScint in major healthcare markets around the world. We acquired ProstaScint in May 2015 from Jazz Pharmaceuticals. ProstaScint, which is approved by the U.S. Food and Drug Administration, or FDA, is a marketed biologic imaging agent specifically indicated for the diagnostic staging of prostate cancer patients. We have launched the RedoxSYS® oxidation-reduction potential system, or the RedoxSYS System, into the global research market and are developing numerous clinical applications for this potential first-in-class diagnostic device. Additionally, we have developed the MiOXSYS System as a diagnostic device for the detection of infertility in semen analysis. Further, we plan to enter Phase 3 clinical development for our lead therapeutic candidate, Zertane, which is being studied in premature ejaculation.
The premature ejaculation market in the U.S. and Europe is expected to reach over $1.3 billion in annual sales in 2015, representing an increase of 10.3% since 2010. According to recent published analyses, premature ejaculation, or PE, is a highly prevalent male sexual dysfunction affecting 20-30% of men worldwide. Based on internal market research and published reports, we believe that PE is up to 1.5-times more prevalent than erectile dysfunction, or ED. Currently, there are no FDA-approved prescription products in the United States to treat PE, and to our knowledge, only two other prescription products have been approved elsewhere in the world. Treatment options for PE have traditionally included antidepressant drugs prescribed “off label,” topical numbing medications, and cognitive behavior therapy or counseling, all of which have had limited effectiveness in treating the disorder. PE therefore represents an area of significant unmet medical need. In addition, approximately 32% of the more than 12,000 men with PE surveyed in a 2007 study published in European Urology also suffered from ED. Accordingly, we believe that a combination product candidate to treat both PE and ED represents another significant worldwide market opportunity for us.
By virtue of our recent acquisition of ProstaScint, we are now commercial stage and generating sales for this FDA-approved prostate cancer imaging agent. As prostate cancer is a condition commonly diagnosed and treated by urologists, ProstaScint complements our urology-focused product pipeline. Prostate cancer is the most common cancer among men in the United States, with an estimated 218,000 annual cases (as of 2010). Further, more than 1,800,000 men are alive with some history of prostate cancer, and over 30,000 U.S. men die each year from the disease. The effect of prostate cancer on healthcare economics is substantial, which makes the need for accurate disease staging critical for treatment and management strategies. The U.S. market for the diagnosis and screening of prostate cancer is expected to total $17.4 billion in 2017, a CAGR of 7.5%.
We are actively developing the global market for the RedoxSYS System across a range of applications. Specifically, we have begun commercializing the RedoxSYS System for research use through direct selling, distribution partners, and academic collaborators. Over the past 18 months, we have engaged in over 60 trials around the world whereby prominent researchers are implementing oxidation-reduction potential, or ORP, as a marker in both chronic and acute illnesses and disorders in both clinical research as well as basic science research.
Through our extensive network of researchers, the RedoxSYS System has demonstrated the potential to have broad clinical applications. Studies are now underway at a major U.S. university in the area of male infertility. As such, we developed the MiOXSYS System as a line extension to RedoxSYS to specifically assess oxidative stress in semen as a tool to assess male infertility. Male infertility is prevalent, underserved, and oxidative stress is widely implicated in its pathophysiology. As such, we expect to bolster our research focus in this area with the MiOXSYS System to complement our focus on urologic conditions. The global male infertility market is expected to grow to over $300 million by 2020 with a CAGR of nearly 5% from 2013 to 2020. Oxidative stress is broadly implicated in the pathophysiology of male infertility, yet very few diagnostic tools exist to effectively measure oxidative stress levels in men. However, antioxidants are widely available and recommended to infertile men. With the introduction of the MiOXSYS System, we believe for the first time there will be an easy and effective diagnostic tool to assess degree of oxidative stress and monitor patients’ responses to antioxidant therapy.
11
Table of Contents
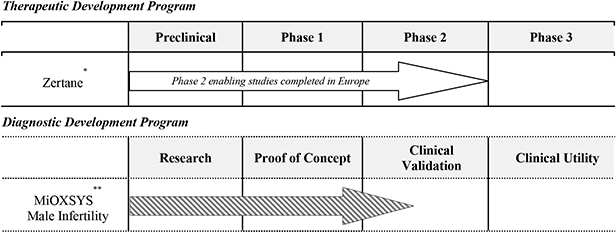
| * | 505(b)(2) regulatory pathway |
| ** | 510(k) de novo regulatory pathway |
Zertane
Our premature ejaculation product candidate, Zertane, is a specifically formulated orally disintegrating tablet, or ODT, of tramadol hydrochloride patented for the on-demand treatment of PE. Zertane is being developed utilizing a regulatory pathway pursuant to Section 505(b)(2) of the FDCA, as the active ingredient is already well characterized for the treatment of pain, and we are relying on the FDA’s finding of safety of tramadol hydrochloride to support its use in a new indication, PE, at a lower dose. If we receive marketing approval for Zertane, we believe it will be the first commercial product approved by the FDA for PE.
By virtue of significant development work performed by a previous partner of Ampio, Zertane has already been evaluated outside the United States in two Phase 1 clinical trials, two Phase 2 clinical trials and two Phase 3 clinical trials. The two Phase 1 safety trials were conducted to characterize the concentration of tramadol hydrochloride in plasma after oral administration of a single Zertane ODT (89 mg) in healthy volunteers. Two randomized, placebo-controlled, blinded Phase 2 clinical trials were conducted in a total of 102 patients. These trials evaluated doses of tramadol hydrochloride between 25 and 120 mg in male subjects with PE. Two placebo-controlled, randomized and double-blind Phase 3 clinical trials were conducted in Europe to investigate tramadol hydrochloride 62 mg and 89 mg ODT for the treatment of PE when taken as needed between two and eight hours before a sexual event. This development work has demonstrated a favorable safety and efficacy profile of Zertane in men with PE. Furthermore, the safety and pharmacology of the drug substance in Zertane, tramadol hydrochloride, is well characterized, which should eliminate the need for us to conduct additional pre-clinical studies and safety trials. Based on guidance received at a recent consultation meeting with the FDA, we believe we are well positioned to initiate a Phase 3 clinical program with Zertane in the United States. Specifically, we believe the meeting clarified specific aspects of the trial, including the primary efficacy endpoints and patient inclusion and exclusion criteria. The FDA agreed to review a draft study protocol in advance of us submitting our Investigational New Drug application, or IND. Upon completion of the program, if successful, we plan to submit a New Drug Application, or NDA, and subsequently market Zertane in the United States, if approved.
ProstaScint
On May 20, 2015, we acquired ProstaScint® from Jazz Pharmaceuticals. ProstaScint Kit, or capromab pendetide (ProstaScint), is a radio-labeled monoclonal antibody, which is a biologic product that targets a specific antigen. ProstaScint targets Prostate Specific Membrane Antigen (PSMA), a protein uniquely expressed by prostate tissue. A radioactive substance called Indium In 111 is attached to the proprietary, mouse-derived antibody. The radiolabeled antibody infused into the patient and is taken up by prostate cancer cells which can be detected and visualized with a special nuclear medicine scan (single-photon emission tomography, or SPECT). ProstaScint has been shown to be clinically effective in determining the course of treatment for a patient who has had a prostatectomy and/or has suspected metastasis (spread of the cancer cells beyond the prostate). Further, ProstaScint has demonstrated efficacy in patients classified as High Risk or with recurrent prostate cancer. ProstaScint has been approved by the FDA and Health Canada, and significant clinical data exist demonstrating the significant predictive value in prostate cancer staging.
According to the American Cancer Society prostate cancer is the most common cancer among men in the United States, with an estimated 218,000 annual cases (as of 2010). Further, more than 1,800,000 men are alive with some history of prostate cancer, and over 30,000 U.S. men die each year from the disease. The effect of prostate cancer on healthcare economics is substantial, which makes the need for accurate disease staging critical for treatment and management strategies. The U.S. market for the diagnosis and screening of prostate cancer is expected to total $17.4 billion in 2017, a CAGR of 7.5%. Importantly, ProstaScint is the only FDA-
12
Table of Contents
approved radiopharmaceutical (for use in radioimmunoscintigraphy) specifically indicated for prostate cancer screening and is specifically highlighted in the American Cancer Society practice guidelines for prostate cancer screening and staging. A radiopharmaceutical is a radioactive chemical or pharmaceutic preparation, labeled with a radionuclide in varying concentrations, which is used as a diagnostic or therapeutic agent. ProstaScint is labeled with the radionuclide indium in 111 and is used as an imaging agent utilizing SPECT (single-photon emission computed tomography) imaging. Radioimmunoscintography is an imaging procedure involving the administration of radioactively-labeled monoclonal antibodies to detect specific affected cells in a diagnostic procedure. Monoclonal antibodies are proteins specifically genetically engineered from a clone of a particular type of cell (a B cell) and programmed to enable a specific effect on a cellular target.
RedoxSYS System for Research Use
We completed the development of the RedoxSYS System during the two years preceding the Merger. In 2014, we received ISO 13485 certification, demonstrating our compliance with global quality standards in medical device manufacturing. This enabled the launch of the RedoxSYS System into the research market around the world. We also received a CE marking in Europe and Health Canada clearance to begin the market development of the RedoxSYS System as a clinical diagnostic in Europe, Canada, and elsewhere around the world where CE marking is recognized. We launched sales efforts into the research market in late 2014 and since that time have already placed the RedoxSYS System at a number of prominent research centers in the United States, Europe, and Israel. These research placements are depicted below.
Prominent U.S. Research Centers
13
Table of Contents
Prominent Centers in Europe and Israel
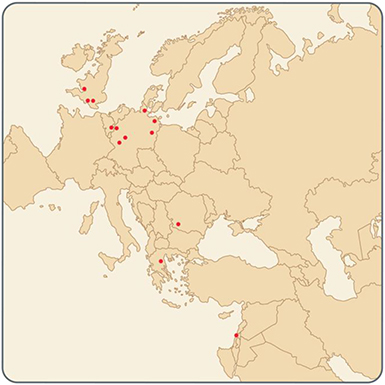
|
• Johannes Gutenberg-Universität Mainz (Mainz, Germany) • Uniklinik RWTH Aachen (Aachen, Germany) • Universitätsklinikum Frankfurt (Frankfurt, Germany) • Universitätsklinikum Rostock (Rostock, Germany) • Charité – Universitätsmedizin Berlin (Berlin, Germany) • Universitätsklinikum Schleswig-Holstein (Kiel, Germany) • Cardiff University (Cardiff, Wales, UK) • University College London (London, England) • Imperial College London (London, England) • Southampton University (Southampton, England) • University of Liège (Wallonia, Belgium) • Universitatea de Medicină şi Farmacie din Craiova (Craiova, Romania) • University of Thessaly (Larissa, Greece) • Western Galilee Hospital (Nahariya, Israel) • Eliachar Research Laboratory (Nahariya, Israel) |
We expect to leverage these research relationships and build numerous applications in areas where researchers are studying oxidative stress. Currently, there are no available research platforms that measure oxidation-reduction potential in biologic fluids (i.e., blood, plasma, serum, semen, seminal fluid, cerebrospinal fluid, tissue, and cells). While oxidative stress is commonly studied in research settings around the world (both academia and industry), the current assessment methods are incomplete, time consuming, and often impractical for assessing oxidative stress completely. To position the RedoxSYS System effectively in the research market, we have placed key personnel in the United States, Europe, and Asia to develop direct research business relationships as well as distribution networks. From these networks, we expect to realize initial product revenues for the RedoxSYS System for research applications in 2015.
MiOXSYS System for Male Infertility
As part of our strategy to develop future clinical applications of the RedoxSYS System, we have conducted initial studies in reproductive health. Male infertility is a significant medical condition in which oxidative stress is well known to play a substantial role. As such, we believe developing a specific instrument platform (called the MiOXSYS System) to assess oxidative stress levels in semen and seminal fluid represents a significant commercial opportunity. Oxidative stress is well established as a leading contributing factor to male infertility. Further, a significant proportion of male infertility remains unexplained in part because of the lack of standardized tests available to clinicians and researchers to assess oxidative stress in semen and plasma. This lack of standardization has resulted in poor implementation of semen and plasma analysis around the world.
We have conducted proof-of-concept studies in male infertility with the MiOXSYS System with a leading research center in the United States, which demonstrate that oxidation-reduction potential effectively measures oxidative stress levels in semen and seminal plasma. Semen analysis studies are routinely conducted to assess causes of infertility, so we expect clinicians and oxidative stress researchers to readily integrate the MiOXSYS System into routine clinical use. Additional studies are now underway to evaluate the MiOXSYS System’s performance in the detection of oxidative stress levels in semen in healthy and infertile males. Of the approximately $300 million male infertility market, the North American market and Asia Pacific market dominate due to prevalence, awareness of treatment, and availability of treatment resources. Thus, it is important that we have already begun to establish distribution relationships and direct access to major oxidative stress researchers and clinicians.
An attractive aspect of the reproductive health market relates to reimbursement as infertility treatments and the associated diagnostic tests are generally paid directly by patients. The current infertility treatments could cost in excess of $10,000 per treatment cycle, so the addition of a moderately priced oxidative stress test would consume nominal relative costs while providing specific, actionable information needed to improve the oxidative status of infertile patients. The current infertility treatments include antioxidant supplements and lifestyle modifications that lower oxidative stress (e.g., smoking cessation, exercise, dietary changes, etc.), so the measurements reported by the MiOXSYS System could effectively guide treatment in the infertile patients.
14
Table of Contents
We have an extensive range of intellectual property across our two primary, near-term assets. We have patent protection in the United States and several other large markets worldwide for the oxidation-reduction potential products and product candidates. Further, we have patent protection in the United States and several other large markets worldwide for the use of tramadol hydrochloride to treat PE. We also have intellectual property specifically covering Zertane-ED, and methods of using Zertane-ED to treat comorbid PE and ED that has issued patents in several large markets worldwide and is pending in the United States.
Our Strategy
Key elements of our strategy include:
| • | Develop a pipeline of therapeutics and diagnostics focused on urological conditions, with a focus on the initiation and completion of two Phase 3 clinical trials for Zertane in the United States and the development of worldwide commercialization and marketing partnerships. |
Our lead therapeutic product candidate is Zertane, a Phase 3-ready oral product for the treatment of PE. Zertane is in advanced clinical studies, and we have already completed pivotal study-enabling studies in Europe that demonstrate favorable efficacy and safety in more than 600 patients with PE. We intend to initiate and complete Phase 3 clinical trials in order to submit an NDA to obtain FDA approval for Zertane.
We intend to maximize the value of Zertane in the United States via either securing a licensing arrangement or commercializing it ourselves. We also expect to seek collaboration agreements to commercialize Zertane in the rest of world, or ROW, markets. We already have such agreements in place to commercialize Zertane in South Korea and Brazil, Canada, the Republic of South Africa, certain countries in Sub-Saharan Africa, Colombia and Latin America which will – if approved – provide royalty and milestone-based revenue for us.
| • | Commercialize FDA-approved ProstaScint for the staging of both newly diagnosed high-risk and recurrent prostate cancer patients. We plan to commercialize ProstaScint in the U.S. and in key markets around the world. |
Because ProstaScint is already approved by the FDA, we expect to launch a commercial infrastructure in order to support increased sales and distribution of ProstaScint in the U.S. In addition to U.S. approval, ProstaScint has also been approved by Health Canada. ProstaScint fills an important medical need in the detection of a common illness facing a significant number of men in the U.S. and around the world. Prostate cancer is the most common cancer among men in the United States, with an estimated 218,000 annual cases (as of 2010). Further, more than 1,800,000 men are alive with some history of prostate cancer, and over 30,000 U.S. men die each year from the disease.
ProstaScint has several unique selling features that we believe will enable significant sales growth and regular use by healthcare providers diagnosing and treating prostate cancer. ProstaScint is the only imaging agent that specifically targets prostate cancer cells and demonstrates high sensitivity, specificity, and accuracy. In multiple clinical studies researchers have shown that when SPECT/CT scans were used in patients pre-treated with ProstaScint, ProstaScint imaging was highly sensitive in detecting prostate cancer and significantly predictive of 10-year biochemical disease free survival in prostate cancer patients (86.6% vs. 65.5%; p=0.0014). Additionally, the American Cancer Society specifically recognizes ProstaScint by name in current prostate cancer diagnosis guidelines.
| • | Establish the RedoxSYS System as a research tool and expand its application to other indications with a focus on male infertility and adjacent applications. |
Our lead diagnostic product candidate is the MiOXSYS System, which is currently being studied in clinical trials at a major U.S. academic center to evaluate its utility in measuring oxidative stress in infertile males. If successful, these clinical trials are expected to pave the way for our MiOXSYS System to enter the male infertility market around the world.
The RedoxSYS System is expected to be commercialized initially as a research tool, and pending the outcome of our clinical studies – oxidative stress as a key indicator of male infertility – be commercialized for additional indications, if approved. Internationally, we intend to commercialize the RedoxSYS System for multiple potential indications where oxidative stress is implicated, if approved. The United States will be our primary focus in terms of commercialization opportunities. However, we intend to leverage ROW opportunities to drive incremental revenue and adoption of our RedoxSYS System as well as to facilitate research to develop further redox-modulated application for our RedoxSYS System. We intend to utilize distribution partners in Europe, Asia, and elsewhere to launch and grow sales in both research and diagnostic applications. We have established research collaborations with over 20 leading
15
Table of Contents
academic centers around the world. Further, the RedoxSYS System has been used in numerous clinical studies supported by pharmaceutical companies. As oxidative stress is widely studied in both academic and industry settings, the research application of the RedoxSYS System spans a wide range of applications in clinical research, basic science research, and health and wellness research. We seek, either directly or through distributors, to commercialize the RedoxSYS System across a broad range of potential customers. To date, we have signed distribution agreements for the research market with four companies, including EuroBio in France, Una Health in the United Kingdom, THP Medical in Austria, and KYS Technologies in Taiwan. We expect to significantly expand our distribution network throughout 2015 to enable entry into major markets in Europe and Asia. We plan to commercialize directly in the United States. We expect these research collaborations will generate new findings and potential clinical applications for the RedoxSYS System. In turn, we believe this development of new data and publications will enable us to pursue adjunct conditions where oxidative stress is implicated.
| • | Acquire established marketed products and late-stage development assets within our focus. |
In order to diversify our product portfolio and create more value, we intend to seek to acquire complementary products or product candidates to develop and/or commercialize including marketed assets. Initially, the focus will be on acquiring products or product candidates for urological conditions but we will opportunistically consider other products or product candidates based on their ability to create value and complement our focus. We plan to pursue product acquisitions, inclusive of therapeutics, diagnostics, and devices, which will be evaluated for their strategic fit and potential for near-term and/or accretive value to us. In May 2015, we acquired ProstaScint as part of this strategy.
Our Product Pipeline
Our Lead Therapeutic Product Candidate – Zertane
Our lead therapeutic product candidate, Zertane, is a specifically formulated orally disintegrating tablet, or ODT, of tramadol hydrochloride patented for the on-demand treatment of PE. Zertane is being developed utilizing a regulatory pathway pursuant to Section 505(b)(2) of the FDCA, as the active ingredient is already well characterized for the treatment of pain, and we are relying on the FDA’s finding of safety of tramadol hydrochloride to support its use in a new indication, PE, at a lower dose. If we receive marketing approval for Zertane, we believe it will be the first commercial prescription product approved by the FDA for PE.
According to recent published analyses, PE is a highly prevalent male sexual dysfunction affecting 20-30% of men worldwide. Based on internal market research and published reports, we believe that PE is up to 1.5-times more prevalent than erectile dysfunction. Currently, there are no FDA-approved prescription products in the United States to treat PE, and to our knowledge, only one oral prescription product has been approved elsewhere in the world. Treatment options for PE have traditionally included antidepressant drugs prescribed “off label,” topical numbing medications, and cognitive behavior therapy or counseling, all of which have had limited effectiveness in treating the disorder. PE therefore represents an area of significant medical need.
By virtue of significant development work performed by a previous partner of Ampio, Zertane has already been evaluated outside the United States in two Phase 1 clinical trials, two Phase 2 clinical trials and two Phase 3 clinical trials. This development work has demonstrated a favorable safety and efficacy profile of Zertane in men with PE and helped inform the design and endpoints of the Phase 3 clinical trials we will need to obtain FDA approval. Furthermore, the safety and pharmacology of the drug substance in Zertane, tramadol hydrochloride, is well characterized, which we believe will eliminate the need for us to conduct additional pre-clinical studies and safety trials. We believe we are well positioned to initiate Phase 3 clinical trials with Zertane in the United States. Upon completion of the trials, if successful, we plan to submit an NDA to the FDA and subsequently market Zertane in the United States, if approved.
Our strategy for Zertane is focused on the initiation and completion of the Phase 3 clinical program in the United States and the development of worldwide commercialization and marketing partnerships. We expect to finalize clinical development of Zertane, seek FDA marketing approval and – if approved—commercialize the product candidate in the United States either directly or via partnerships. We will seek partnerships to commercialize Zertane in rest of world, or ROW, markets. We already have partnerships in place to market Zertane in South Korea and Brazil, which could provide near-term revenue for us if, working with our partners, we are able to successfully obtain regulatory approval in those countries. In addition, we recently entered into an agreement with Endo Ventures Limited, which recently acquired Paladin Labs Inc., or Paladin, a leading Canadian specialty pharmaceutical company, to provide exclusive rights to market, sell and distribute Zertane in Canada, the Republic of South Africa, certain countries in Sub-Saharan Africa, Colombia and Latin America. We also intend to build awareness of PE in the United States with the intention of paving the way for successful product introduction and initiate pre-clinical work on Zertane-ED as a potential combination treatment for PE and ED. We expect to use a similar clinical and commercial approach for Zertane-ED and we have a clinical development collaboration in place with our South Korean partner for the combination product candidate when we elect to actively pursue its development.
16
Table of Contents
PE Market Opportunity
What is PE?
Medical literature contains several one-dimensional and multi-dimensional operational definitions of PE. The Diagnostic and Statistical Manual of Mental Disorders defines PE as persistent or recurrent ejaculation with minimal sexual stimulation before, on, or shortly after penetration and before the person wishes it, with the disturbance causing marked distress or interpersonal difficulty. Alternately, the International Society for Sexual Medicine, or the ISSM, has adopted a new and evidence-based definition for lifelong PE: “premature ejaculation is a male sexual dysfunction characterized by ejaculation which always or nearly always occurs prior to or within a minute of vaginal penetration; and inability to delay ejaculation on all or nearly all vaginal penetrations; and negative personal consequences, such as distress, bother, frustration and/or the avoidance of sexual intimacy.”
PE may be classified as lifelong (primary) or acquired (secondary). Lifelong PE is characterized by onset from the first sexual experience and remains a problem throughout life. Acquired PE is characterized by either a gradual or subtle onset, with ejaculation being considered normal before onset of PE. Time to ejaculation is short but not usually as fast as in lifelong (less than 1 minute in almost all encounters). We believe acquired PE and/or situational PE suggests a psychological cause and behavioral therapy and/or relationship counseling may be the most appropriate initial therapy. In contrast, we believe lifelong PE suggests a biogenic cause and pharmacologic treatment is the most appropriate initial therapy.
Ejaculation latency, most commonly quantified using intravaginal ejaculation latency time, or IELT, is a dominant component of PE assessment in clinical trials. IELT is defined as the time between vaginal intromission and intravaginal ejaculation. Although a standard cut-off for ejaculatory latency does not exist, it has been suggested that an IELT of 2 minutes or less may serve as an adequately sensitive criterion for defining PE and some studies have used IELT values from 1 to 2 minutes for defining PE. In a pre-IND meeting with FDA, we agreed to use an IELT of less than or equal to 1 minute as one of the enrollment criteria for our planned Phase 3 clinical trials.
However, IELT is not the sole criteria used to diagnose PE. We believe diagnosis of PE should also include a subject’s perceived control over ejaculation, as well as distress and interpersonal difficulty due to the condition. The need to objectively assess and diagnose PE beyond IELT has led to the development of several questionnaires within the male sexual dysfunction medical community including the Premature Ejaculation Diagnostic Tool (PEDT), Premature Ejaculation Profile, or PEP, the Index of Premature Ejaculation (IPE), and the Male Sexual Health Questionnaire Ejaculatory Dysfunction (MSHQ-EjD). In collaboration with the FDA, Ampio has also developed a modified version of PEP known as the Patient Outcome for Premature Ejaculation, referred to as the POPE.
Prevalence of PE
PE is a significant medical need in the United States and worldwide as it causes significant emotional distress for affected men and their partners. According to an article published in European Urology in 2010 and a survey published in the Journal of the American Medical Association (JAMA) in 1999, PE is a highly prevalent male sexual dysfunction affecting 20-30% of men worldwide. However, most prevalence data on PE is based on patient surveys, which are inherently subjective, and therefore some of the men surveyed may not have PE as it is defined by the medical societies noted above. In a 2007 study published in European Urology, the incidence of PE was assessed via a web-based survey of 12,133 men ages 18-70 in the United States, Germany and Italy. In this survey, 2,754 or 22.7%, of the men reported that they suffer from PE. The vast majority (87.9%) of men with PE wished that they had more control over time to climax. Additionally, a majority (57.6%) of the men surveyed reported that they would seek medical treatment if they knew that a pill to control ejaculation were available. We believe men would also ask their doctor about treatment options if their partner suggested it. Additional primary company market research by a prominent pharmaceutical market research consulting firm commissioned by Ampio indicated that over 66% of patients that see a urologist for PE were self-referred, which we believe further demonstrates that PE is a condition for which patients are actively seeking treatment.
Existing therapies do not satisfy the significant PE market need
Presently, there are no approved prescription pharmaceuticals in the United States to treat PE and only two pharmaceuticals known to be approved elsewhere in the world. Current “off label” or unapproved therapies used to treat PE carry with them unwanted side effects and inconsistent or limited effectiveness. Topical over-the-counter, or OTC, options are not preferred due to route of administration and may also have an impact on partner satisfaction. Oral therapeutics, specifically selective serotonin reuptake inhibitors, or SSRIs, carry potentially significant side effects most notably of which is diminished libido. PDE-5 inhibitors have been prescribed “off label” for PE but have not demonstrated efficacy. Outside of oral or topical therapeutics, non-medical options include behavioral therapy and relationship counseling, both of which can be time consuming and stressful and frequently ineffective for men and their partners.
17
Table of Contents
The following table illustrates the current methods used to treat patients with PE and the associated issues based upon our sponsored market research:
| Pharmacologic Treatment |
Issues | |
| Tricyclic antidepressants * |
Fatigue, nausea, dizziness, dry mouth, hypotension | |
| Short-acting SSRIs |
Nausea, diarrhea, headache, dizziness, risk of suicidal ideation | |
| Long-acting SSRIs * |
ED, decreased libido, fatigue, nausea, increased perspiration | |
| Topical desensitizing agents |
Messy, numbing of vagina, skin irritation | |
| PDE-5 inhibitors * |
Ineffectiveness, headache, flushing, nausea | |
| Non-Pharmacologic Treatment |
Issues | |
| “Stop-Start” strategy |
Requires an understanding partner | |
| “Squeeze” method |
Requires an understanding partner | |
| Psychological therapy |
Time consuming for patients; costly, long-term benefits unknown | |
| Pharmacologic Treatment |
Issues | |
| Relationship counseling |
Time consuming, costly, requires an understanding partner | |
| * | NOT approved for treatment of PE. |
We believe patients and their partners are generally dissatisfied with existing pharmacologic and non-pharmacologic treatments for PE. Based on primary market research by a prominent pharmaceutical market research consulting firm commissioned by Ampio, which previously owned the rights to Zertane and is our largest stockholder, which included discussions with a cross section of clinicians that treat patients with PE, we believe that there are significant issues with existing PE treatments demonstrating a real need for a safe and effective, FDA-approved product to treat PE that does not have a ramp-up period. This primary market research supports that ideal product characteristics include:
| • | effectiveness; |
| • | fewer side effects than anti-depressants, such as SSRIs, that are used “off-label” for PE; and |
| • | quick onset/on-demand usage. |
Zertane
Our lead therapeutic product candidate, Zertane for premature ejaculation, contains 89 mg tramadol hydrochloride in an orally dissolving tablet, or ODT. Tramadol hydrochloride is a well-established, centrally acting synthetic analgesic and has been used for more than 30 years as a treatment for moderate to severe pain. The drug and its active metabolite (M1, O-desmethyltramadol) act as an opiate agonist, apparently by selective activity at the µ-receptor. Although the mechanism by which tramadol hydrochloride delays ejaculation has not been identified, numerous laboratory studies have shown that tramadol hydrochloride also acts as an N-methyl-D-aspartate receptor antagonist, 5-hydroxytryptamine type 2C receptor antagonist, 5 nicotinic acetylcholine receptor antagonist, M1 and M3 muscarinic acetylcholine receptor antagonist, and a serotonin and norepinephrine modulator. It is possible that one or a combination of these effects leads to a delay in ejaculation. The relative contribution of tramadol hydrochloride versus its M1 metabolite to delay ejaculation is unknown. However, the metabolite is six times more potent than the parent drug in producing analgesia in animal models and 200 times more potent in µ-receptor binding. As a pain medication, tramadol hydrochloride has been associated with certain adverse effects including dizziness, nausea, constipation, vertigo, headache, vomiting and drowsiness. However, we intend that our labeling for Zertane, if regulatory approval is obtained, will suggest “as required” dosing before sexual intercourse and not to exceed one tablet per day. Based on previous clinical studies, we believe that limiting the dosing to no more than once per day will minimize any side effects.
As an alternative to antidepressant and anxiolytic medication, tramadol hydrochloride has been recognized as a potential therapy for treatment of PE, after an association was observed between its use and improvements in ejaculation latency time. The combination of completed clinical trials, primarily performed in Europe, planned clinical trials in the United States, and literature are intended to establish the safety and efficacy of Zertane in the treatment of the condition and standardize the dose and dosing regimen.
18
Table of Contents
History of Zertane
Dr. David Bar-Or discovered the utility of tramadol hydrochloride for the treatment of PE in June 1999, and this discovery and accompanying intellectual property were the property of DMI BioSciences, Inc., or DMI BioSciences. DMI BioSciences conducted two Phase 2 clinical studies in male subjects with PE using a pharmacy-compounded gelatin capsule preparation. A proof-of-concept Phase 2 study was initiated in March 2003, and final patient assessments were on July 7, 2003. A dose-ranging study Phase 2 study was initiated in September 2004 and final patient assessments were in October 2005. In 2007, DMI BioSciences licensed the worldwide rights to tramadol hydrochloride for PE to Biovail Laboratories International, or Biovail.
Biovail had previously entered into a product development and licensing agreement with Ethypharm S.A., or Ethypharm, in 2002, which had developed an ODT formulation of tramadol hydrochloride using its proprietary FLASHTAB technology. In this agreement, Biovail acquired the rights to develop and market Ethypharm’s tramadol hydrochloride ODT product for use in the management of pain. In March of 2004, Biovail submitted a Section 505(b)(2) NDA (US NDA 21,639) to FDA for market registration of tramadol hydrochloride ODT 50 mg (RYBIX) for the management of moderate to moderately severe pain, and this NDA was granted approval in May 2005.
Biovail decided not to launch the RYBIX 50 mg ODT product in the United States, and in 2009, signed an NDA agreement with Ethypharm pursuant to which Ethypharm acquired all the rights to RYBIX 50 mg ODT and Biovail acquired the rights to develop, manufacture and market a FLASHTAB ODT product containing tramadol hydrochloride for the symptomatic treatment of PE. In this agreement, Biovail assigned to Ethypharm all of Biovail’s rights, title and interest in and to the approved NDA for RYBIX, the regulatory documentation and product rights to enable Ethypharm to develop and market RYBIX in the United States for the management of pain. In addition, the agreement granted Biovail the right to reference and use all data, regulatory filings and regulatory communication, including the approved NDA for RYBIX, the product rights (including any information, data, know-how, formulas, assays, or intellectual property contained in the approved NDA) and any and all related regulatory documentation that would be relevant for the purposes of developing, manufacturing or marketing the tramadol hydrochloride ODT product for PE.
Using the newly acquired FLASHTAB technology and manufacturing processes obtained in the NDA agreement with Ethypharm, Biovail developed two strengths of tramadol hydrochloride ODT (62 and 89 mg) for use in four NDA-enabling clinical trials: two Phase 1 pharmacokinetic/bioavailability and two Phase 3 placebo-controlled pivotal trials. These trials were initiated in the latter half of 2009. In July 2010, Biovail terminated the clinical trials in Europe as a direct result of a merger with Valeant Pharmaceuticals International, or Valeant, which was announced in June 2010 but was finalized in September 2010. Following the merger, Valeant adopted a new research and development model and began identifying product development programs that did not align with this new R&D model. In connection with this new R&D model, Valeant decided to terminate Biovail’s licensing agreement for tramadol hydrochloride for PE and also to terminate the ongoing Phase 3 studies. The Valeant model did not include compounds that had any regulatory or clinical risk (i.e., were still in development) and instead was focused solely on “commercially ready” products.
Biovail withdrew from its licensing agreement with DMI Biosciences and the worldwide rights to tramadol hydrochloride for PE reverted back to DMI Biosciences, which was acquired in March 2011 by Ampio. In December of 2011, the Ethypharm-Biovail NDA Agreement was transferred to Ampio in an asset purchase from Valeant, providing Ampio access to all data, regulatory filings and rights to develop, manufacture and market tramadol hydrochloride ODT for PE. Ampio completed clinical study reports for the European Phase 3 studies and submitted tramadol hydrochloride 89 mg ODT for marketing authorization in Australia.
In December 2013, Ampio spun out the assets and agreements associated with its sexual dysfunction treatment portfolio, including Zertane, to its then wholly-owned subsidiary Vyrix. We acquired Vyrix in April 2015.
Clinical Data
Six European clinical trials have been completed with Zertane: two Phase 1 trials in healthy volunteers and two Phase 2 and two Phase 3 trials in men with lifelong PE. The two Phase 1 safety trials were conducted to characterize the concentration of tramadol hydrochloride in plasma after oral administration of a single Zertane ODT (89 mg) in healthy volunteers.
Two randomized, placebo-controlled, blinded Phase 2 clinical trials were conducted in a total of 102 patients. The first of these showed that a single 25-mg dose of conventionally formulated immediate release tramadol hydrochloride (i.e., immediate-release gelatin capsules) was safe, well-tolerated and prolonged time to ejaculation in some, but not all, patients. The second trial evaluated three higher doses of tramadol hydrochloride: 65 mg, 85 mg and 120 mg. In this trial, a clear dose response was seen for both efficacy and safety, leading DMI Biosciences to conclude that the optimal tramadol hydrochloride dose to treat PE was likely to be in the range 60-90 mg.
Two placebo-controlled, randomized and double-blind Phase 3 clinical trials were conducted in Europe to investigate tramadol hydrochloride 62 mg and 89 mg ODT for the treatment of PE when taken as needed between two and eight hours before a sexual event. A total of 677 patients were randomized in the trials and received either the 62 or 89 mg ODT or a matching placebo ODT. Our
19
Table of Contents
claim that Zertane was efficacious in the Phase 3 trials is based on at least one of the two doses resulting in statistically significant improvements in both IELT and PEP measures (i.e., co-primary endpoints) from baseline to the end of the trial. Using IELT in combination with a self-report questionnaire (e.g., PEP or POPE) has gained acceptance as meaningful measures of pharmacoactivity and efficacy in the scientific and regulatory communities. The results of the Phase 3 trials suggest that tramadol hydrochloride 89 mg ODT is consistently more effective as a treatment for PE than tramadol hydrochloride 62 mg ODT. In accordance with the definitions from the clinical trial protocols, only the tramadol hydrochloride 89 mg ODT dose satisfied the claim for effective treatment of PE in both Phase 3 trials.
The following table summarizes the six prior clinical trials of Zertane.
| Trial Name |
Phase |
Demographic |
Enrollment |
Design |
Duration of |
Zertane |
Noteworthy Findings | |||||||
| BVF-324-101 (June 26, 2009 to July 5, 2009/ Biovail*) |
1 | Healthy males and females (18-55 yr) |
20 | Comparative | N/A | 89 | Overall systemic exposure of tramadol hydrochloride and 2 metabolites were similar following 89 mg ODT and the reference 89 mg tramadol hydrochloride solution.
13 subjects experienced a total of 37 adverse events (AEs) during the study. Most common AEs were GI disorders (nausea) and central nervous system disorders (sleepiness, dizziness, headache). | |||||||
| BVF-324-102 (July 14, 2009 to July 30, 2009/ Biovail*) |
1 | Healthy males (19-55 yr) |
424 | Alcohol interaction study |
N/A | 89 | There was no significant difference in the peak and total systemic exposures of tramadol hydrochloride compared to when tramadol hydrochloride 89 mg ODT was taken with water, or with either strength of alcohol.
Tramadol hydrochloride was well tolerated with alcohol. 7 subjects experienced 11 AEs, most frequently decreased blood pressure and dizziness. | |||||||
20
Table of Contents
| Trial Name |
Phase |
Demographic |
Enrollment |
Design |
Duration of |
Zertane |
Noteworthy Findings | |||||||
| KNL 40237 (March 13, 2003 to July 7, 2003/ DMI**) |
2 | Men with PE (18-70 yr) |
37 | Double-blind, randomized placebo- controlled |
3 weeks | 25 | Treatment with tramadol hydrochloride 25 mg had no statistically different effect than placebo in subjects with a baseline IELT of 2 minutes or less (n=30; P=0.4560) or in a subpopulation of subjects with a baseline IELT of 1 minute or less (n=19; P=0.1796).
46 AEs emerged during treatment (28 with tramadol hydrochloride and 18 with placebo). There were no deaths, serious or other significant AEs. All AEs were of mild intensity. Most common AEs were GI disorders (nausea) and central nervous system disorders (headache). | |||||||
| KNL 40491 (September 28, 2004 to October 18, 2005/ DMI**) |
2 | Men with PE (18-70 yr) |
68 | Double-blind, randomized placebo- controlled |
12 weeks | 65, 85, 120 | Tramadol hydrochloride significantly increased the median IELT compared to both Baseline and placebo. Statistically significant increases in IELT compared to Baseline were observed to be 3.0-fold (P=0.0013), 4.2-fold (P<0.0001), and 5.1-fold (P<0.0001) for the 65-, 85-, and 120-mg dose levels, respectively.
Doses of 65 and 85 mg were generally well tolerated. 120 mg was less well tolerated with adverse events known to be associated with tramadol hydrochloride (headache, dizziness, somnolence, insomnia) and related to sexual function (penile hypoaesthesia, anorgasmia, and ED). | |||||||
21
Table of Contents
| Trial Name |
Phase |
Demographic |
Enrollment |
Design |
Duration of |
Zertane |
Noteworthy Findings | |||||||
| BVF-324-301 (August 17, 2009 to September 9, 2010/ Biovail*) |
3 | Men with PE (18-65 yr) |
221 | Double-blind, randomized placebo- controlled |
12 weeks | 62, 89 | There was a statistically significant change in IELT from Baseline to the end of the study for tramadol hydrochloride ODT 89 mg compared to placebo (p = 0.002). The significant difference was apparent at the first visit during the double-blind treatment period and each visit thereafter (p < 0.05 for all Visits). Tramadol hydrochloride 62 mg and 89 mg ODT demonstrated statistically significant improvements in PEP measures compared with placebo.
During the double-blind treatment or open-label extension periods, 21 subjects experienced at least 1 treatment-emergent AE. There were no deaths or serious AEs during the study. | |||||||
| BVF-324-302 (September 30, 2009 to August 23, 2010/ Biovail*) |
3 | Men with PE (18-65 yr) |
456 | Double-blind, randomized placebo- controlled |
12 weeks | 62, 89 | There was a statistically significant change in IELT from Baseline to the end of the study for tramadol hydrochloride ODT 62 and 89 mg compared to placebo (p = 0.01 and p = 0.02). Tramadol hydrochloride 62 mg and 89 mg ODT demonstrated statistically significant improvements in both IELT and PEP measures compared with placebo.
A total of 28 out of 399 subjects (7.5%) experienced at least 1 treatment-emergent AE during the double-blind or open-label treatment period. No subject died or experienced a severe AE. | |||||||
| Total |
896 | |||||||||||||
| * | Biovail Laboratories International, SRL |
| ** | DMI Biosciences, Inc. |
22
Table of Contents
Planned Phase 3 Clinical Program in United States
In light of the estimated size of the U.S. market opportunity for Zertane, FDA approval is a critical value driver for us in the near term. We believe we are well positioned to quickly move into NDA-enabling Phase 3 clinical development in order to complete and submit our Section 505(b)(2) NDA for Zertane. Ampio met with the FDA in a pre-IND meeting on June 20, 2012 and we subsequently met with the FDA on October 23, 2014. As a result of these meetings, we have been able to define the number and key aspects of necessary trials to progress Zertane to an NDA that may be accepted for filing by the FDA; furthermore, the results from the European trials will inform the design, endpoints and inclusion criteria for the U.S. Phase 3 program as well as recommendations by FDA:
| • | Patients will be those with a subset of disease symptoms described by the ISSM Ad Hoc Committee for the Definition of PE; |
| • | Two tramadol hydrochloride doses (62 and 89 mg) will be evaluated; |
| • | The patient reported outcome questionnaire, or PEP, from the European Phase 3 trials, was modified to capture the most clinically significant aspects of PE from the ISSM definition – now termed Patient Outcomes in Premature Ejaculation, or POPE – and this 4-question questionnaire will be validated during the first trial; |
| • | Key efficacy assessments will include intravaginal ejaculation latency time, or IELT, and the subject’s frustration or bother due to PE; and |
| • | Total enrollment for each trial will be approximately 350 – 400 subjects. |
As in the Phase 3 trials conducted in Europe, co-primary endpoints will be used for determination of efficacy. Both improvement in IELT, which will be captured by the partner in a blinded diary, and PE-related frustration or bother, which will be assessed after each sexual intercourse attempt as well as at the final study visit by a single question in the POPE, will be evaluated as co-primary endpoints to determine efficacy.
We currently expect to file the IND for Zertane in the second half of 2015. We are prepared to commence our Phase 3 clinical program soon after the IND becomes effective because our supplies are already manufactured and packaged and only require appropriate clinical labeling of the product candidate. Importantly, our manufacturing process has already been validated at the site of commercial manufacture.
ROW Opportunity
Beyond seeking U.S. NDA approval, we plan to leverage partnerships to gain approval and eventual marketing authorization for Zertane in several key markets around the world. We already have partnerships in place with Daewoong Pharmaceuticals Co., Ltd. in South Korea and FBM Industria Farmaceutica Ltda. in Brazil, thereby providing potential royalty and milestone-based revenue if, working with our partners, we are successfully able to obtain regulatory approval in those countries. In addition, our recent agreement with Paladin provides Paladin with exclusive rights to market, sell and distribute Zertane in Canada, the Republic of South Africa, certain countries in Sub-Saharan Africa, Colombia and Latin America. Before marketing any products in Brazil and South Korea, approval must be received from the Brazilian Health Surveillance Agency (Anvisa) and the Ministry of Food and Drug Safety (MFDS, previously the Korean Food and Drug Administration), respectively. We intend to leverage the expertise of our local collaborators in these jurisdictions to navigate the regulatory requirements and help determine an efficient and effective pathway to bringing Zertane to market. If required regulatory approvals are obtained, partnerships in ROW markets will potentially provide additional revenue to us and help to establish the Zertane brand and the role of tramadol hydrochloride in PE around the world. Such clinical experience will be useful as we seek to impact treatment guidelines for PE and gather post-marketing data to better inform our U.S. NDA submission.
Commercial Strategy
U.S. Commercial Strategy
Given the population and the anticipated pricing, the United States represents the largest PE opportunity and we will seek to maximize the market potential for Zertane in one or more of the following ways:
| • | License U.S. promotional rights to Zertane to an established pharmaceutical company – We will seek to secure a milestone and royalty-based agreement with a company to market Zertane to high-value U.S. prescribers. Potential partners may already have a commercial presence in urology (the single largest prescriber population for PE drugs) as well as primary care physicians (the single largest category of physicians in the United States). Alternatively, we may attempt to secure agreements with multiple partners to promote Zertane to different specialties: e.g., one for urology and one for primary care physicians. We may also consider divesting Zertane as an alternative to a licensing agreement. |
| • | Commercialize Zertane via our own commercial infrastructure –Our management has experience with all aspects of commercialization and we may deploy a specialized sales force to initiate promotion of Zertane specifically to urologists. We may expand our commercial presence into primary care and other relevant physician specialties. Alternatively we may deploy a sales force of our own focused on urologists, while also seeking a partner for the larger primary care segment. |
23
Table of Contents
ROW Commercial Strategy
As stated previously, we intend to leverage partnerships to market Zertane around the world. We will seek to collaborate in the largest viable markets with companies that are well positioned to maximize sales of Zertane. Considerations for collaborations will include the prospective collaborator’s presence in the therapeutic category of men’s health, its ability to invest in the regulatory and commercialization activities necessary, and its commitment to Zertane, our management team and economic terms. Countries will be prioritized for partnering based on viability of Zertane in the market, including but not limited to regulatory pathway (i.e., the amount of regulatory work required for the partner to submit for approval); market conditions (markets that accept sexual health products); economic factors (pricing); and market size, as well as deal teams, including but not limited to royalty and/or milestone based economic terms; upfront payments based on market; and commitment to marketing and/or regulatory investments.
We already have entered into collaboration agreements in South Korea and Brazil, which represent two of the larger ROW market opportunities. In addition, our recent agreement with Paladin provides Paladin with exclusive rights to market, sell and distribute Zertane in Canada, the Republic of South Africa, certain countries in Sub-Saharan Africa, Colombia and Latin America. We will seek to enter into similar arrangements in additional countries with additional companies in order to capitalize on the sizable opportunity for Zertane, and later, Zertane-ED.
ProstaScint Acquisition
Overview
On May 20, 2015, we acquired ProstaScint® from Jazz Pharmaceuticals. ProstaScint Kit, or capromab pendetide, is a radio-labeled monoclonal antibody, which is a biologic product that targets a specific antigen. ProstaScint targets Prostate Specific Membrane Antigen, or PSMA, a protein uniquely expressed by prostate tissue. A radioactive substance called Indium In 111 is attached to the proprietary, mouse-derived antibody. The radiolabeled antibody infused into the patient and is taken up by prostate cancer cells which can be detected and visualized with a special nuclear medicine scan (single-photon emission tomography, or SPECT). ProstaScint has been shown to be clinically effective in determining the course of treatment for a patient who has had a prostatectomy and/or has suspected metastasis (spread of the cancer cells beyond the prostate). Further, ProstaScint has demonstrated efficacy in patients classified as High Risk or with recurrent prostate cancer. ProstaScint has been approved by the FDA and Health Canada, and significant clinical data exist demonstrating the significant predictive value in prostate cancer staging.
Prostate Cancer Market
According to the American Cancer Society prostate cancer is the most common cancer among men in the United States, with an estimated 218,000 annual cases (as of 2010). Further, more than 1,800,000 men are alive with some history of prostate cancer, and over 30,000 U.S. men die each year from the disease. The effect of prostate cancer on healthcare economics is substantial, which makes the need for accurate disease staging critical for treatment and management strategies. The U.S. market for the diagnosis and screening of prostate cancer is expected to total $17.4 billion in 2017, a CAGR of 7.5%. Importantly, ProstaScint is the only FDA-approved radiopharmaceutical (for use in radioimmunoscintigraphy) specifically indicated for prostate cancer screening and is specifically highlighted in the American Cancer Society practice guidelines for prostate cancer screening and staging.
Prostate cancer is classified into four stages based on severity: Stage 1 – 4. Stage 3 is considered “High Risk” and Stage 4 is when cancer has become metastatic. Radioimmunoscintigraphy has been established as diagnostic to stage cancer malignancy and one of the most widespread clinical uses has been for the detection of prostate cancer.
ProstaScint Clinical Data
Multiple clinical studies have been conducted in the United States and published in peer-reviewed publications that consistently demonstrate substantial clinical efficacy of ProstaScint in staging prostate cancer patients and specifically identify whether the cancer is confined to the prostate or has metastasized to other parts of the body. Through more accurate clinical staging and identification of metastatic prostate cancer, clinicians are able to better direct therapeutic interventions and improve outcomes. A brief summary of key clinical findings for ProstaScint from select studies are summarized below.
24
Table of Contents
| Principal Investigator(s)/ Primary Authors |
Publication |
Patient Population |
Conclusion/Results | |||
| Ellis RJ et al. | Int. J. Radiation Oncology Biol. Phy. (2010) | Patients presenting for primary radiotherapy having a clinical diagnosis of localized primary prostate cancer; Patients evaluated for tumor stage using conventional staging and SPECT/CT (N=239) | SPECT/CT imaging with ProstaScint pre-treatment was significantly predictive of 10-year biochemical disease-free survival (86.6% vs. 65.5%; p=0.0014) | |||
| Haseman MK et al. | Urology (2007) | Men with prostate cancer who underwent imaging with ProstaScint pretreatment; Patients were divided according to the presence or absence of central abdominal uptake(CAU) (N=341) | SPECT/CT imaging with ProstaScint pretreatment effectively predicted death rates among patients with central abdominal uptake (CAU), and demonstrated that prostate cancer-specific death rates were 10 times higher in patients identified with ProstaScint as having central abdominal uptake (p=0.005). | |||
| Ellis RJ et al. | Brachytherapy (2005) | Men with prostate cancer of all risk categories who underwent imaging with ProstaScint pretreatment; patients were divided into low, intermediate, and high risk and underwent brachytherapy (N=239) | SPECT/CT imaging with ProstaScint pretreatment effectively predicted biochemical disease recurrence regardless of the patient’s risk category; 7-year outcomes data from brachytherapy patients with treatment based on the ProstaScint scan showed a significant difference in biochemical disease-free survival. | |||
Radiation oncology experts have published numerous papers expressing the potential for expanded use of ProstaScint in prostate cancer imaging due to advances in imaging technologies since the product’s initial approval. Since the early 2000s significantly greater image resolution has been enabled due to the advent of dual head cameras (and improved imaging in general) along with the use of co-registered images where radiologists now combine the images of SPECT and computerized tomography, or CT, or magnetic resonance imaging, or MRI. Because of these factors we believe there is significant commercial opportunity for ProstaScint.
ProstaScint Product Information
ProstaScint is provided as a two-vial kit which contains all of the non-radioactive ingredients necessary to produce a single unit dose for administration by intravenous injection. The ProstaScint vial contains 0.5 mg of capromab pendetide in 1 mL of sodium phosphate buffered saline solution adjusted to pH 6; a sterile, pyrogen-free, clear, colorless solution that may contain some translucent particles. The vial of sodium acetate buffer contains 82 mg of sodium acetate in 2 mL of Water for Injection adjusted to pH 5-7 with glacial acetic acid; it is a sterile, pyrogen-free, clear, and colorless solution. Neither solution contains a preservative. Each kit also includes one sterile 0.22 µm Millex® GV filter, prescribing information, and two identification labels. It may also be helpful in conjunction with other scans (CT or MRI) for higher risk patients, by detecting lymph nodes in the abdomen that are involved with prostate cancer cells, but may still appear falsely normal on CT or MRI scans.
The procedure to administer ProstaScint is as follows: the patient is given an intravenous, or IV, infusion of the monoclonal antibody, and 30 minutes later, a scan is performed. A second scan is done between 96 and 120 hours (4-5 days) after the infusion. The first scan (on the day of the infusion) takes approximately 1 hour, while the second scan takes approximately 2.5 hours.
25
Table of Contents
ProstaScint Uses
ProstaScint is indicated as a diagnostic imaging agent in newly-diagnosed patients with biopsy-proven prostate cancer, thought to be clinically-localized after standard diagnostic evaluation (e.g. chest x-ray, bone scan, CT scan, or MRI), who are at high-risk for pelvic lymph node metastases. It is not indicated in patients who are not at high risk.
ProstaScint is also indicated as a diagnostic imaging agent in post-prostatectomy patients with a rising PSA and a negative or equivocal standard metastatic evaluation in whom there is a high clinical suspicion of occult metastatic disease. The imaging performance of Indium In 111 ProstaScint following radiation therapy has not been studied.
The information provided by Indium In 111 ProstaScint imaging should be considered in conjunction with other diagnostic information. Scans that are positive for metastatic disease should be confirmed histologically in patients who are otherwise candidates for surgery or radiation therapy unless medically contraindicated. Scans that are negative for metastatic disease should not be used in lieu of histological confirmation. ProstaScint is not indicated as a screening tool for carcinoma of the prostate nor for re-administration for the purpose of assessment of response to treatment.
Our Novel Research Instrument – The RedoxSYS System
Our leading diagnostic product candidate, the RedoxSYS System, is now fully developed for research use. The RedoxSYS System was developed by Luoxis Diagnostics in the two years immediately preceding the Merger between Luoxis, Vyrix, and us in April 2015. Upon the consummation of the Merger, the RedoxSYS System became our asset. Prior to the incorporation of Luoxis, the predecessor technologies that are now incorporated into the RedoxSYS System were developed by the research team of Dr. David Bar-Or through a predecessor company, DMI BioSciences, Inc. that was acquired by Ampio.
The RedoxSYS System is a novel, portable device that measures oxidation-reduction potential, or ORP, a global measure of oxidative stress. The RedoxSYS System is the first and only system that measures ORP in biologic specimens to provide a complete measure of redox balance, which is broadly implicated across a wide range of both acute and chronic conditions. To date, Canadian and European regulators have characterized the RedoxSYS System as Class II medical device and regulated them accordingly. Classification of a medical device as Class II in Europe and Canada indicates that the device is generally regarded as posing medium risk, and non-invasive medical devices that come into contact with injured skin are generally classified as Class II. As we have conducted initial validation studies with the RedoxSYS System across a range of conditions and obtained a CE marking in Europe and Health Canada clearance to begin the market development of the RedoxSYS System as a clinical diagnostic in Europe, Canada, and elsewhere around the world where CE marking is recognized, we are now initiating commercialization for use of the RedoxSYS System as a research tool. By employing a focused commercial infrastructure and a growing network of distributors around the world, we believe we can efficiently penetrate the academic and industry-based research centers who study oxidative stress. With this growth in the research market, we intend to develop clinical applications for the RedoxSYS System.
Our strategy for the RedoxSYS System is to continue deployment of this system at leading academic centers around the world, develop research collaborations with key opinion leaders in oxidative stress research, identify clinical applications for the platform, and aggressively pursue infertility studies to establish efficacy of the system in this setting of care. Our plans include introduction of the RedoxSYS System to researchers in the United States through a direct commercial effort while engaging with distributors in major markets around the world, including Canada, Europe, and Asia (with a focus on Japan, Korea, Taiwan, Singapore and Malaysia).
Oxidative Stress and the Male Infertility Market Opportunity
An early potential opportunity that has presented promise through our research is the application of ORP in the study of male infertility. ORP represents a unique approach to assessing oxidative stress in male infertility, and early proof-of-concept studies have been conducted. We are now beginning clinical studies with a globally recognized U.S.-based university in male infertility. Oxidative stress is widely assessed in male infertility laboratories, and we believe the RedoxSYS System, if proven effective, will provide a simple, comprehensive solution to oxidative stress detection and management of antioxidant and lifestyle intervention in this underserved market. Given the preliminary results obtained from clinical trials conducted in semen analysis, we believe significant opportunity exists to launch a derivative product of RedoxSYS to specifically assess oxidative stress in the context of male infertility. Accordingly, we developed the MiOXSYS System as a uniquely designed and programmed device specific to the detection of oxidation-reduction potential in semen and seminal fluid.
Oxidative stress plays a pivotal role in the pathogenesis of male infertility. The presence of excess levels of reactive oxygen species, or ROS, is associated with both the structural and functional integrity of sperm. Moreover, these increased levels of ROS – the increase of oxidative stress – directly interfere with capacitation and fertilization. It is widely estimated that total infertility is driven by male infertility in half of the cases, and the major factors influencing the rise in infertility among men are the change in lifestyle, increasing age, and environmental effects.
26
Table of Contents
Prevalence of Male Infertility
Of all sexually active adults, 12-15% are infertile and male infertility is the sole cause or contributing factor 50% of the time. The global male infertility market is large and growing. The market is expected to grow to more than $300 million globally by 2020, with a CAGR of nearly 5% from 2013 to 2020. Despite the prevalence of male infertility, difficulties remain in effectively diagnosis root causes. Oxidative stress assessment is considered a standard practice but due to various factors is not effectively used routinely.
Potential Role of ORP in Male Infertility
Oxidation-reduction potential is defined in the published literature as follows:
“ORP in a biological system is an integrated measure of the balance between total oxidants and reductants. In plasma, many constituents contribute to the ORP. Reactive oxygen species (ROS), such as the superoxide ion, hydroxyl radical, hydrogen peroxide, nitric oxide, peroxynitrite, transition metal ions, and hypochlorous acid, contribute to the oxidative potential. Plasma reductants include thiols, vitamin C, tocopherol, ß-carotene, lycopene, uric acid, bilirubin, and flavinoids. Enzymes such as superoxide dismutase, or SOD, catalase, and glutathione peroxidase, are involved in the conversion of ROS into less reactive species. ORP monitoring of plasma represents a single measurement that integrates the overall quantitative balance among the oxidants and reductants of the system.”
Given that ORP represents a single, global measure of oxidative stress in a biological system, we believe the potential for ORP to serve as a standardized marker in semen analysis and other aspects of infertility assessment is significant. A major limitation of oxidative stress assays relates to the fact that there is poor standardization in testing. As many factors contribute to oxidative stress (e.g., free radical proliferation, antioxidant depletion, DNA damage, etc.), it is important to have an integrated measure that combines all known and unknown oxidants and reductants in the respective system into one measurement. We believe ORP is an integrated measure of oxidative stress that can be easily and quickly measured with the RedoxSYS System.
Current assays are incomplete and only
approximate global redox balance
|
|
||
| Unlike current redox assays, Oxidation-Reduction Potential incorporates all (known and unknown) oxidants and reductants that contribute to global redox imbalance. | ||
In the context of infertility, having an integrated value representing all relevant biologic constituents contributing to oxidative stress will enable simple, robust analysis in a four-minute test.
Existing infertility assessment tools do not satisfy the clinical need
There are various techniques in use to assess semen in cases of male infertility. The most commonly implemented techniques involve DNA fragmentation, oxidative stress analysis, microscopic examination, sperm penetration assays, sperm agglutination, computer assisted semen analysis, and others. The currently available oxidative stress analysis tools are widely considered expensive and cumbersome to use in routine clinical practice. In both developed countries as well as in the developing world, expensive analysis tools and recurring reagent expenses make routine testing nearly impossible to implement with regularity.
27
Table of Contents
Market Opportunity for the RedoxSYS System
We believe the market opportunity for the RedoxSYS System is significant as scientists implicate oxidative stress in numerous diseases and acute conditions. Our initial focus of commercializing the RedoxSYS System for use in research enables us to rapidly build a base of clinical validation and utility data across a range of illnesses. As such, we expect to generate early revenues from oxidative stress researchers implementing the RedoxSYS System into their routine oxidative stress research programs while generating published clinical data demonstrating ORP’s usefulness in disease monitoring and prognostic assessment. From a clinical perspective, we plan to continue our research efforts in male infertility while expanding future applications in acute and chronic illnesses.
The global male infertility market is in excess of $300 million, while the broader in vitro diagnostics, or IVD, market is expected to grow to more than $80 billion by 2017. With a substantial base of conditions for which the MiOXSYS System may present utility, we expect to realize significant revenue potential from this first-in-class system.
Through our collaborative research efforts our scientific team has identified several diagnoses in which we believe the ORP technology derives distinct values that may be useful in better identifying a patient’s risk for the development of more severe illness – beyond reproductive health and infertility. Further, we believe the ORP technology has the potential to better stratify patients according to clinical characteristics detected uniquely by ORP measurements. Specific diseases where the RedoxSYS System has been initially studied include trauma, critical illness, stroke, heart failure, diabetes, and pregnancy. Multiple other clinical areas exist where the system could be studied and potentially applied.
Diagnostics serve a key role in the health value chain by influencing the quality of patient care, health outcomes and downstream resource requirements across a wide range of clinical conditions. From consumer-friendly at-home pregnancy and glucose monitoring tests to more complex automated laboratory-based systems, these tests are often first-line health decision tools. While diagnostics comprise less than 5% of hospital costs and about 1.6% of all Medicare costs, their findings are commonly believed to influence as much as 60-70% of health care decision-making. The value of diagnostics accrues not only to clinicians and patients, but to health care managers, third-party payors and quality assurance organizations that use diagnostic performance to measure and improve health care quality.
ORP is a tightly controlled measurement, much like the vital signs routinely measured in medical practice—temperature, heart rate, respiratory rate, blood pressure and oxygen saturation of blood. Abnormal changes in oxidation-reduction potential are closely associated with poor outcomes in critically ill patients, including traumatic brain injury, multi-trauma injury, stroke, sepsis, and pneumonia. Rapid results are essential for optimal treatment adjustments in critical care areas such as emergency and intensive care departments.
ORP results may also help determine which patients are at high risk of early readmission at hospital discharge, especially patients with heart attack, heart failure, stroke, and pneumonia, and this represents an incremental assessment not afforded by currently available diagnostic tests.
Numerous scientific studies confirm the clinical value of measuring oxidative stress. Recently, a large assortment of blood and cell tests have been used in research studies to measure separate biomarkers of oxidative stress, such as lipid peroxidation, protein oxidation and total antioxidants, but currently many of these separate biomarker test results are needed to start to assess total oxidative stress. Despite the importance of assessing oxidative stress, we are not aware of any practical or efficient method for measuring these oxidative stress biomarkers in a point-of-care setting. Oxidative stress is often a marker for inflammation, which in turn indicates the presence of disease-related processes or developing conditions across a wide array of diseases.
The worldwide IVD market was $44 billion in 2010 and is projected to grow to $82 billion by 2017. The IVD market includes all laboratory and hospital-based sales and home-use, or over-the counter, product sales covering immunoassays, clinical chemistry, microbiology, hematology, histology/cytology, point-of-care testing, and over-the-counter diagnostics. The professional point-of-care testing worldwide market is a significant opportunity and was in excess of $5 billion in 2009 and is expected to grow to almost $7 billion by 2014.
Importantly, critical care (an application for which the RedoxSYS System is targeted) constituted 14% ($700 million) of sales in 2009 with expected CAGR of 5%, and cardiac markers constituted 9% ($435 million) of sales in 2009 with expected CAGR of 8%.
28
Table of Contents
Background on the RedoxSYS System
We believe the RedoxSYS System is the first and only research and clinical diagnostic platform that provides an accurate, easy, and complete real-time assessment of redox status. Through the development and commercialization of the RedoxSYS System, we are pioneering the true measurement of redox potential, also called oxidation-reduction potential, a novel measure of oxidative stress, which has been implicated in numerous critical injuries, illnesses, and chronic conditions with significant appreciation of the role of oxidative stress in infertility. We believe the RedoxSYS platform has a broad range of potential in both research and clinical settings where no other methods exist to measure oxidation-reduction potential in biologic specimens.
Through research directed by Dr. David Bar-Or and Raphael Bar-Or at Ampio, we believe we have developed the first and only clinical diagnostic device that measures ORP in biologic specimens (i.e., plasma, serum, semen, seminal plasma, cells, tissues, cell cultures, and others). This device, called the RedoxSYS System, rapidly measures ORP and antioxidant capacity, a global marker of antioxidant reserve and concentration to provide a global picture of oxidative status. The system uses a small, bench top analyzer in conjunction with small, disposable sensors that work electrochemically without any liquid reagents or difficult sample preparation. The RedoxSYS System has been CE marked and approved for use in Europe and Canada, and we are now collaborating with major medical centers around the world for its use in research. The RedoxSYS System is an easy-to-use diagnostic device that can be used in various research settings. Further, this portable device can be decentralized to multiple research sites and enable real-time reporting of oxidative stress values at the patient’s point of care or in a clinical laboratory setting. We received ISO 13485:2003 medical device certification in 2014.
The RedoxSYS System has been developed over an 18-year period by Dr. David Bar-Or, Raphael Bar-Or, and their colleagues, and the research and development work has focused on both the technical and clinical development of various applications.
The RedoxSYS System Overview
The RedoxSYS System is comprised of two distinct, patented components that enable a system capable of measuring the ORP and antioxidant capacity of a biological fluid: an analyzer and sensor strips. In mechanical terms, ORP is defined as the potential between a working electrode and a reference electrode at equilibrium. The RedoxSYS System has been specifically studied in human whole blood, serum, plasma, semen, and other biological fluids. The RedoxSYS System measures two distinct elements to determine a patient’s oxidation reduction potential:
| • | Static ORP – the standard potential between a working electrode and a reference electrode with no driving current (or extremely small current). This is proportional to the balance of redox agents and is what is classically defined as ORP. Low ORP values mean that the biological sample is in the normal range of oxidative stress. Higher than normal ORP values means that the biological sample is in a higher oxidation state. |
| • | Capacity – the measure of antioxidant reserve available in the body’s system. High capacity values mean that the biological sample has levels of antioxidant reserves. Lower than normal capacity values means that the biological sample has below normal antioxidant reserves. |
The RedoxSYS Analyzer
The RedoxSYS analyzer is a portable, lightweight desktop platform that may be used in a clinical or research laboratory or near a patient care area. The analyzer is a small device that accepts an inserted sensor that has collected a small specimen as obtained by traditional specimen collection procedures. The analyzer is battery powered and equipped with a custom 5 lead strip connector. The reader consists of a Galvanostat analog circuit with greater than 1012 MHz input impedance.
The analyzer contains a 10 MHz external crystal (internal 4X PLL for 40 MHz operation), and a programming/serial header is externally accessible. The device has internal power/heart-beat indicator LED, primary storage of 128Mbit (16Mbyte) SPI Flash (3.3V) (Bulk data storage), and secondary storage of 2Mbit (256Kbyte) SPI FRAM (3.3V) (Hi-Speed Storage).
The RedoxSYS analyzer contains a user-friendly interface that is flexibly designed to accommodate multiple endpoints depending upon the specific clinical condition being considered. The interface is LCD, 16x2, with a white backlight, variable delay auto-off time-out. Two status LED indicators are visible through front panel mounted lenses. Further, the reader contains three DPDT push-button switches (Left, Center, Right), power on button(s) for battery mode operation, switch usage switch, audible alerts, strip detection, and test completion signals.
Further, the RedoxSYS analyzer enables data transfer, has USB serial communication, and is configured for data download to a connected PC.
The RedoxSYS analyzer’s power management consists of an external 5VDC power jack with input capacitance and filtering, a boost converter supplied by external 5VDC power or internal Li-Ion battery, and provides main 5VDC digital board supply. The reader functions with or without the battery connected. The battery lasts in excess of 24 hours with continuous operation to enable prolonged use outside of a laboratory setting.
29
Table of Contents
Image of the RedoxSYS Analyzer


The RedoxSYS Sensor Strips
The ORP sensor strips, via standard biological specimen collection techniques, receive 20-40 microliters of a specimen from which the ORP clinical analysis is performed. The ORP sensor strips are small, disposable, and biocompatible and consist of a ceramic substrate and a five-lead configuration. Significant intellectual property surrounds the design, construct, and electrochemical algorithms associated with the sensors.
Image of the RedoxSYS Sensor Strips

Clinical Studies
“Clinical Studies” for Male Infertility – The MiOXSYS System Opportunity
We have begun clinical studies in male infertility with a prominent, U.S.-based university medical center utilizing the newly developed MiOXSYS System. If successful, we anticipate we will demonstrate the MiOXSYS System’s ability to measure oxidative stress in semen samples as an adjunctive measure of infertility. These studies are summarized below:
| Aim |
Hypothesis |
Methods and Procedures |
Expected Outcome | |||
| 1 |
The MiOXSYS System will be able to detect oxidation-reduction potential (ORP) in fresh seminal ejaculates | 1. Standardize the instrument for inter/intra observer and assay variability
2. Standardize the system for fresh ejaculate and seminal plasma measurements
3. Test the ability of the system to measure the effect of exogenous ROS in semen
4. Establish the effect of time on ORP measurements |
The assumption that MiOXSYS System can measure ORP in semen shall be confirmed | |||
30
Table of Contents
| Aim |
Hypothesis |
Methods and Procedures |
Expected Outcome | |||
| 2 |
ORP values can be detected in fresh and frozen semen samples | 1. Compare the effect of rapid freezing on the ORP of the seminal ejaculate and seminal plasma
2. The effect of exogenous ROS addition to the ORP and capacitance of both ejaculate and seminal plasma after rapid freezing
3. Establish the effect of different subzero storage temperatures and storage time on ORP measurement in frozen seminal ejaculate and seminal plasma samples
4. Comparing the effect of storage time after rapid freezing on the ORP of both the seminal ejaculate and seminal plasma |
It shall be confirmed that ORP can be measured in both fresh and frozen semen samples and that n differences will exist | |||
| 3 |
Establish ORP values in ejaculates of healthy men. | 1. Establish ORP values in seminal ejaculates of healthy men with normal semen parameters i.e normal concentration, motility and morphology according to WHO, 2010
2. Establish the effect of varying sperm concentration 0, 10, 20, 40 and 100 X 106/ mL on ORP levels (concentration response). |
ORP values in ejaculates of healthy men will be established | |||
| 4 |
Establish ORP values in ejaculates of with different clinical characteristics. | 1. Establish the ORP values in seminal ejaculates in infertile men in the absence or presence of leukocytes (Endtz negative and Endtz positive) samples.
2. Establishing ORP in healthy normozoospermic donors and infertile men with an abnormal spermiogram
3. Examine effect of abstinence on ORP in semen samples from normozoospermic donors
4. ORP levels in infertile men with varicocele |
Specific and different ORP values will be established in the ejaculates from men with different pathologies | |||
| 5 |
The ORP values correlate with ROS-TAC scores in ejaculates of healthy men. | 1. Comparative study between ORP and. ROS–TAC scores as measured by chemiluminescence and total antioxidant capacity |
A positive correlation will be found between ORP values and ROS-TAC scores | |||
Results from these clinical studies could validate that the MiOXSYS System can be used for detection of ORP in human ejaculates and seminal plasma without requiring large sample volumes. Furthermore, it can show that the results obtained are comparable to the current parameters measured. This could validate the use of the MiOXSYS System in the clinical andrology setting as a tool to provide real time information on oxidative status. If successful, these studies will provide substantial clinical validation for use of the MiOXSYS System in male infertility and enable a specific clinical application in the field of andrology and male infertility.
Clinical Study for Other Indications
Beyond male infertility, we have generated robust clinical data with ORP. As much of our initial research efforts focused on trauma, research beginning in 2014 has expanded into research and clinical applications in the areas of:
| • | Stroke; |
| • | Sepsis; |
| • | Ventilator-Associated Pneumonia; |
| • | Athletic exertion; |
| • | Hemodialysis; |
| • | Post cardiac surgery; |
31
Table of Contents
| • | Congestive heart failure; |
| • | Hip fracture; |
| • | Frailty; and |
| • | Diabetic Kidney Disease. |
Additionally, we have conducted numerous pre-clinical studies looking at various neurodegenerative conditions including Parkinson’s disease and multiple sclerosis. We have active research in place with over 20 key opinion leaders around the world with over 80 studies in various stages or planning or execution.
In 2013 and 2014, we deployed the RedoxSYS System around the world in the development of numerous future clinical applications. While many areas of study have been undertaken, we have focused research resources on high-value areas where significant medical needs remain unmet. Given our initial orientation around trauma, the studies completed thus far have focused on large conditions related to critical care. These initial studies demonstrated the initial clinical validation for the RedoxSYS and MiOXSYS systems and represent substantial opportunities as growth applications and markets following initial entry into the research and infertility markets.
We have initiated over 80 studies across a range of study types, ranging from studies in trauma, liver disease, diabetes, cardiac conditions, wellness/exercise, and fertility. From this research has presented unique opportunities to deploy the RedoxSYS System to users in multiple markets around the world in various conditions. With a CE marking now in place and research instruments now manufactured and ready for use, we are positioned to begin commercializing the RedoxSYS System.
Select Published Clinical Trials with Oxidation-Reduction Potential
Significant research has been performed on the ORP diagnostic platform, and numerous peer-reviewed publications demonstrate the various considerations made in the development of this application in a clinical setting. Further, the research conducted to date demonstrates the clinical relevance of ORP as a diagnostic marker in trauma, the development of transfusion-related acute lung injury, or TRALI, via blood transfusions, and other conditions. Over the past 20 years, Ampio/DMI Bioscience employees, Dr. Bar-Or, Raphael Bar-Or, Leonard Rael, and their colleagues, have employed the resources of two Level 1 trauma centers in the state of Colorado. Specific, select studies reporting on the clinical role of ORP as it relates to trauma, acute lung injury related to blood transfusions, and traumatic brain injury include:
32
Table of Contents
| Study Summary |
Phase |
N |
Demographic |
Outcome/Findings | ||||
| Oxidation-Reduction Potential in Trauma Patients | Clinical Proof of Concept | Healthy = 39
Trauma = 10 |
Trauma patients admitted to a Level 1 trauma center | Plasma ORP in multi-trauma patients increased during the first few days of hospitalization and approached normal ORP levels upon discharge | ||||
| Effect of Storage on Oxidative Biomarkers on Packed Red Blood Cells | Clinical Proof of Concept | 10 patient specimens | Critically ill patients with a risk of developing transfusion-related acute lung injury | Oxidation-reduction potential significantly increased (p < 0.05) in the day 42 sample versus the day 1 sample. The oxidation of human serum albumin increased by 63.6% during the storage time. | ||||
| Plasma Oxidation-Reduction Potential and Protein Oxidation in Traumatic Brain Injury | Clinical Proof of Concept | Non-head injury trauma patients = 26
Moderate ITBI patients = 18
Healthy patients = 22 |
Isolated traumatic brain injury | Admission plasma ORP was significantly elevated in all traumatized patients compared to controls. Maximum ORP was detected on day 6 for severe ITBI and non-head injury traumatized patients. Maximum ORP values were significantly higher p<0.05) in the severe ITBI group compared to the non-head injury group | ||||
| Oxidation-reduction potential in trauma patients | Clinical Proof of Concept | Trauma patients = 39
Healthy patients = 10 |
Critically injured patients | The presence of an oxidative environment in the plasma of the critically injured as measured by ORP. ORP can differentiate the degree of oxidative stress based on the severity of the trauma and degree of inflammation | ||||
Regulatory Pathway
We achieved ISO 13485: 2003 in early 2014 following the successful development of a compliant medical device quality system. Following the issuance of our ISO certification, we were awarded a CE marking for the RedoxSYS System, which has enabled initial market development in Europe and markets that accept a CE marking. We also received Health Canada clearance, and we are now working with multiple centers in Canada in research projects. In the United States, we intend to pursue 510k de novo clearance with the FDA for the MiOXSYS System.
33
Table of Contents
Commercial Strategy
U.S. Commercial Strategy
If the clinical studies to measure oxidative stress in male infertility are successful, we expect to pursue that intended use for the MiOXSYS System via the FDA 510k de novo pathway. If cleared for the intended use, we intend to seek to commercialize MiOXSYS System as a new tool for the assessment of oxidative stress in infertility in men. We envision pursuing a direct sales effort to high priority urology/andrology laboratories, infertility clinics and reference centers across the United States. We have identified the primary, influential centers in the United States and believe our commercial deployment will be efficient through a focused sales and marketing effort. We intend to seek to sell the MiOXSYS System into individual centers and laboratories but will focus our revenue model on the repeat ordering of the disposable, single use MiOXSYS sensor strips. We expect to realize a substantial gross margin and profit margin on the basis of low cost of goods sold on both components of the system. We envision an average selling price for the disposable sensors of approximately $25-$40. We envision selling the MiOXSYS analyzers for $5,500 but will also pursue an instrument rental agreement model with minimum disposable sensor purchase requirements.
We also intend to leverage our urology commercialization efforts with other products with a focus on urology centers, infertility clinics, and reproductive health laboratories around the United States.
We believe a focused sales force at the onset of commercialization will enable effective representation of our products and penetration of the reproductive health market. Our sales efforts into the research markets will be enabled initially through a full-time business development professional who will focus on collaborative research and research sales to major oxidative stress centers in the United States. We expect identical pricing in the research market as we intend to pursue in the clinical diagnostics markets.
ROW Commercial Strategy
We intend to undertake a similar strategy outside the United States for the RedoxSYS and MiOXSYS systems while complementing our efforts in infertility and research with adjunct applications in critical care conditions. To efficiently execute across our strategy, we intend to utilize a network of established distributors in the target markets in Europe and Asia. We have already engaged with distributors in three European countries and one Asian country, while many other potential distributors are in advanced stages of discussions with us. We anticipate slightly reduced pricing outside the United States for the disposable sensors given the anticipated lower pricing observed ex-U.S. for diagnostic and research products.
Government Regulation
Approval Process for Pharmaceutical Products
FDA Approval Process for Pharmaceutical Products
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act, or the FDC Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Pharmaceutical product development in the United States typically involves the performance of satisfactory nonclinical, also referred to as pre-clinical, laboratory and animal studies under the FDA’s Good Laboratory Practice, or GLP, regulation, the development and demonstration of manufacturing processes, which conform to FDA mandated current good manufacturing requirements, or cGMP, including a quality system regulating manufacturing, the submission and acceptance of an IND application, which must become effective before human clinical trials may begin in the United States, obtaining the approval of Institutional Review Boards, or IRBs, at each site where we plan to conduct a clinical trial to protect the welfare and rights of human subjects in clinical trials, adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought, and the submission to the FDA for review and approval of an NDA. Satisfaction of FDA requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease.
Pre-clinical tests generally include laboratory evaluation of a product candidate, its chemistry, formulation, stability and toxicity, as well as certain animal studies to assess its potential safety and efficacy. Results of these pre-clinical tests, together with chemistry, manufacturing controls and analytical data and the clinical trial protocol, which details the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated, along with other requirements must be submitted to the FDA as part of an IND, which must become effective before human clinical trials can begin. The entire clinical trial and its protocol must be in compliance with what are referred to as good clinical practice, or GCP, requirements. The term, GCP, is used to refer to various FDA laws and regulations, as well as international scientific standards intended to protect the rights, health and safety of patients, define the roles of clinical trial sponsors and assure the integrity of clinical trial data.
34
Table of Contents
An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the intended conduct of the trials and imposes what is referred to as a clinical hold. Pre-clinical studies generally take several years to complete, and there is no guarantee that an IND based on those studies will become effective, allowing clinical testing to begin. In addition to FDA review of an IND, each medical site that desires to participate in a proposed clinical trial must have the protocol reviewed and approved by an independent IRB or Ethics Committee, or EC. The IRB considers, among other things, ethical factors, and the selection and safety of human subjects. Clinical trials must be conducted in accordance with the FDA’s GCP requirements. The FDA and/or IRB may order the temporary, or permanent, discontinuation of a clinical trial or that a specific clinical trial site be halted at any time, or impose other sanctions for failure to comply with requirements under the appropriate entity jurisdiction.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1 clinical trials, a product candidate is typically introduced either into healthy human subjects or patients with the medical condition for which the new drug is intended to be used. The main purpose of the trial is to assess a product candidate’s safety and the ability of the human body to tolerate the product candidate. Phase 1 clinical trials generally include less than 50 subjects or patients. During Phase 2 trials, a product candidate is studied in an exploratory trial or trials in a limited number of patients with the disease or medical condition for which it is intended to be used in order to: (i) further identify any possible adverse side effects and safety risks, (ii) assess the preliminary or potential efficacy of the product candidate for specific target diseases or medical conditions, and (iii) assess dosage tolerance and determine the optimal dose for Phase 3 trials. Phase 3 trials are generally undertaken to demonstrate clinical efficacy and to further test for safety in an expanded patient population with the goal of evaluating the overall risk-benefit relationship of the product candidate. Phase 3 trials are generally designed to reach a specific goal or endpoint, the achievement of which is intended to demonstrate the candidate product’s clinical efficacy and adequate information for labeling of the approved drug.
There are three main types of NDAs, which are covered by Section 505 of the FDC Act: (1) an application that contains full reports of investigations of safety and efficacy (Section 505(b)(1)); (2) an application that contains full reports of investigations of safety and effectiveness but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the application has not obtained a right of reference (Section 505(b)(2)); and (3) an application that contains information to show that the proposed product is identical in active ingredient, dosage form, strength, route of administration, labeling, quality, performance characteristics, and intended use, among other things, to a previously approved product (Section 505(j)). Section 505(b)(2) expressly permits the FDA to rely, for approval of an NDA, on data not developed by the applicant. In the pre-IND briefing meeting with Ampio and in June 2012, the FDA agreed that our NDA may be submitted under Section 505(b)(2). As such, we intend to rely on studies published in the scientific literature and reference FDA-approved NDAs for tramadol-containing products (NDAs 21-693, 20-281 and 21-692) to support the safety and efficacy demonstrated in our clinical program.
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the U.S. The NDA must include the results of all pre-clinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA is substantial. Under federal law, the submission of most NDAs is additionally subject to a substantial application user fee, currently exceeding $2.3 million and the manufacturer and/or sponsor under an approved NDA are also subject to annual product and establishment user fees, currently approximately $0.1 million per product and $0.6 million per establishment. These fees are typically increased annually.
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the FDA’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs. Most such applications for standard review drug products are reviewed within ten months; most applications for priority review drugs are reviewed in six months. Priority review can be applied to drugs that the FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission. The FDA may also refer applications for novel drug products, or drug products which present difficult questions of safety or efficacy, to an advisory committee—typically a panel that includes clinicians and other experts—for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with cGMP is satisfactory and the NDA contains data that provide substantial evidence that the drug is safe and effective in the indication studied.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks.
35
Table of Contents
REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
The Hatch-Waxman Act
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant’s product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an abbreviated new drug application, or ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, pre-clinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book that: 1) the required patent information has not been filed; 2) the listed patent has expired; 3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or 4) the listed patent is invalid or will not be infringed by the new product. A certification that the new product will not infringe the already approved product’s listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA application also will not be approved until any non-patent exclusivity listed in the Orange Book for the referenced product has expired. Federal law provides a period of five years following approval of a drug containing no previously approved active ingredients during which ANDAs for generic versions of those drugs cannot be submitted, unless the submission contains a Paragraph IV challenge to a listed patent—in which case the submission may be made four years following the original product approval. Federal law provides for a period of three years of exclusivity during which FDA cannot grant effective approval of an ANDA based on the approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage form, route of administration or combination, or for a new use; the approval of which was required to be supported by new clinical trials conducted by, or for, the applicant.
Post-Approval Regulation
Even if a product candidate receives regulatory approval, the approval is typically limited to specific clinical indications. Further, even after regulatory approval is obtained, subsequent discovery of previously unknown problems with a product may result in restrictions on its use or even complete withdrawal of the product from the market. Any FDA-approved products manufactured or distributed by us are subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse events or experiences. Further, drug manufacturers and their subcontractors are required to register their establishments with the FDA and state agencies, and are subject to periodic inspections by the FDA and state agencies for compliance with cGMP, which impose rigorous procedural and documentation requirements upon us and our contract manufacturers. We cannot be certain that we or our present or future contract manufacturers or suppliers will be able to comply with cGMP regulations and other FDA regulatory requirements. Failure to comply with these requirements may result in, among other things, total or partial suspension of production activities, failure of the FDA to grant approval for marketing, and withdrawal, suspension, or revocation of marketing approvals.
36
Table of Contents
If the FDA approves one or more of our product candidates, we and the contract manufacturers we use for manufacture of clinical supplies and commercial supplies must provide certain updated safety and efficacy information. Product changes, as well as certain changes in the manufacturing process or facilities where the manufacturing occurs or other post-approval changes may necessitate additional FDA review and approval. The labeling, advertising, promotion, marketing and distribution of a drug or biologic product or medical devices, also must be in compliance with FDA and Federal Trade Commission, or FTC, requirements which include, among others, standards and regulations for direct-to-consumer advertising, off-label promotion, industry sponsored scientific and educational activities, and promotional activities involving the Internet. The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the issuance of a warning letter directing us to correct deviations from regulatory standards and enforcement actions that can include seizures, fines, injunctions and criminal prosecution.
Approval Process for Medical Devices
In the United States, the FDCA, FDA regulations and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. The FDA regulates the design, manufacturing, servicing, sale and distribution of medical devices, including molecular diagnostic test kits and instrumentation systems. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Unless an exemption applies, each medical device we wish to distribute commercially in the United States will require marketing authorization from the FDA prior to distribution. The two primary types of FDA marketing authorization applicable to a device are premarket notification, also called 510(k) clearance, and premarket approval, also called PMA approval. The type of marketing authorization is generally linked to the classification of the device. The FDA classifies medical devices into one of three classes (Class I, II or III) based on the degree of risk the FDA determines to be associated with a device and the level of regulatory control deemed necessary to ensure the device’s safety and effectiveness. Devices requiring fewer controls because they are deemed to pose lower risk are placed in Class I or II. Class I devices are deemed to pose the least risk and are subject only to general controls applicable to all devices, such as requirements for device labeling, premarket notification and adherence to the FDA’s current Good Manufacturing Practices, or cGMP, known as the Quality System Regulations, or QSR. Class II devices are intermediate risk devices that are subject to general controls and may also be subject to special controls such as performance standards, product-specific guidance documents, special labeling requirements, patient registries or post-market surveillance. Class III devices are those for which insufficient information exists to assure safety and effectiveness solely through general or special controls and include life sustaining, life-supporting or implantable devices, devices of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury.
Most Class I devices and some Class II devices are exempted by regulation from the 510(k) clearance requirement and can be marketed without prior authorization from the FDA. Some Class I devices that have not been so exempted and Class II devices are eligible for marketing through the 510(k) clearance pathway. By contrast, devices placed in Class III generally require PMA approval or 510(k) de novo clearance prior to commercial marketing. The PMA approval process is more stringent, time-consuming and expensive than the 510(k) clearance process, however, the 510(k) clearance process has also become increasingly stringent and expensive. The FDA has provided initial guidance to us that the RedoxSYS System is appropriate for the 510(k) clearance process, likely through the de novo pathway.
510(k) Clearance. To obtain 510(k) clearance for a medical device, an applicant must submit a premarket notification to the FDA demonstrating that the device is “substantially equivalent” to a device legally marketed in the United States that is not subject to PMA approval, commonly known as the “predicate device.” A device is substantially equivalent if, with respect to the predicate device, it has the same intended use and has either (i) the same technological characteristics or (ii) different technological characteristics and the information submitted demonstrates that the device is as safe and effective as a legally marketed device and does not raise different questions of safety or effectiveness. A showing of substantial equivalence sometimes, but not always, requires clinical data. Generally, the 510(k) clearance process can exceed 90 days and may extend to a year or more.
Application fees must accompany medical device submissions. Such fees under the Medical Device User Fees Act, or MDUFA, for 2015 are approximately $258,000 for a full fee application and approximately $5,000 for a 510(k). Fees are adjusted annually.
There are also establishment registration and reporting fees of approximately $4,000 and $9,000, respectively.
After a device has received 510(k) clearance for a specific intended use, any change or modification that significantly affects its safety or effectiveness, such as a significant change in the design, materials, method of manufacture or intended use, may require a new 510(k) clearance or PMA approval and payment of an FDA user fee. The determination as to whether or not a modification could significantly affect the device’s safety or effectiveness is initially left to the manufacturer using available FDA guidance; however, the FDA may review this determination to evaluate the regulatory status of the modified product at any time and may require the manufacturer to cease marketing and recall the modified device until 510(k) clearance or PMA approval is obtained. The manufacturer may also be subject to significant regulatory fines or penalties.
37
Table of Contents
Before we can submit a medical device for 510(k) clearance, we may have to perform a series of generally short studies over a period of months, including method comparison, reproducibility, interference and stability studies to ensure that users can perform the test successfully. Some of these studies may take place in clinical environments, but are not usually considered clinical trials. For PMA submissions, we would generally be required to conduct a longer clinical trial over a period of years that supports the clinical utility of the device and how the device will be used.
Although clinical investigations of most devices are subject to the investigational device exemption, or IDE, requirements, clinical investigations of diagnostic tests, including our products and products under development, are generally exempt from the IDE requirements. Thus, clinical investigations by intended users for intended uses of our products generally do not require the FDA’s prior approval but may require approval of an Institutional Review Board, or IRB, and written informed consent by the patient, provided the clinical evaluation testing is non-invasive, does not require an invasive sampling procedure that presents a significant risk, does not intentionally introduce energy into the subject and is not used as a diagnostic procedure without confirmation by another medically established test or procedure. In addition, our products must be labeled per FDA regulations “for research use only- RUO” or “for investigational use only-IUO,” and distribution controls must be established to assure that our products distributed for research, method comparisons or clinical evaluation studies are used only for those purposes.
Regulation after FDA Clearance or Approval
Any devices we manufacture or distribute pursuant to clearance or approval by the FDA are subject to pervasive and continuing regulation by the FDA and certain state agencies. We are required to adhere to applicable regulations setting forth detailed cGMP requirements, as set forth in the QSR, which include, among other things, testing, control and documentation requirements. Noncompliance with these standards can result in, among other things, fines, injunctions, civil penalties, recalls or seizures of products, total or partial suspension of production, refusal of the government to grant 510(k) clearance or PMA approval of devices, withdrawal of marketing approvals and criminal prosecutions, fines and imprisonment. Our contract manufacturers’ facilities operate under the FDA’s cGMP requirements.
Foreign Regulatory Approval
Outside of the United States, our ability to market our product candidates will be contingent also upon our receiving marketing authorizations from the appropriate foreign regulatory authorities, whether or not FDA approval has been obtained. The foreign regulatory approval process in most industrialized countries generally encompasses risks similar to those we will encounter in the FDA approval process. The requirements governing conduct of clinical trials and marketing authorizations, and the time required to obtain requisite approvals, may vary widely from country to country and differ from those required for FDA approval.
In the European Union, we are required under the European Medical Device Directive (Council Directive 93/42/EEC) to affix the CE mark to certain of our products in order to sell the products in member countries of the European Union. The CE mark is an international symbol that represents adherence to certain essential principles of safety and effectiveness mandated in the European Medical Device Directive, which are referred to as the “essential requirements”. Once affixed, the CE mark enables a product to be sold within the European Economic Area, or EEA, which is composed of the 28 member states of the EU plus Norway, Iceland and Liechtenstein as well as other countries that accept the CE mark.
To demonstrate compliance with the essential requirements, we must undergo a conformity assessment procedure which varies according to the type of medical device and its classification. Except for low risk medical devices (Class I with no measuring function and which are not sterile) where the manufacturer can issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the essential requirements of the Medical Devices Directive, a conformity assessment procedure requires the intervention of an organization accredited by a member state of the EEA to conduct conformity assessments, or a notified body. Depending on the relevant conformity assessment procedure, the notified body would typically audit and examine the technical file and the quality system for the manufacture, design and final inspection of our devices. The notified body issues a CE certificate of Conformity following successful completion of a conformity assessment procedure conducted in relation to the medical device and its manufacturer and their conformity with the essential requirements. This certificate entitles the manufacturer to affix the CE mark to its medical devices after having prepared and signed a related EC Declaration of Conformity.
If we modify our devices we may need to apply for permission to affix the CE mark to the modified product. Additionally, we may need to apply for a CE mark for any new products that we may develop in the future. Certain products regulated as medical devices according to EC-Directives are subject to vigilance requirements for reporting of adverse events.
We will be subject to additional regulations in other countries in which we market, sell and import our products, including Canada. We or our distributors must receive all necessary approvals or clearance prior to marketing and/or importing our products in those markets.
38
Table of Contents
The International Standards Organization, or ISO, promulgates internationally recognized standards, including those for the requirements of quality systems. To support ISO certifications, surveillance audits are conducted by a notified body yearly and recertification audits every three years that assess continued compliance with the relevant ISO standards.
Other Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, in the United States, the Centers for Medicare & Medicaid Services, other divisions of the Department of Health and Human Services, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments. In the United States, sales, marketing and scientific/educational programs must also comply with state and federal fraud and abuse laws. Pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990 and more recent requirements in the Health Care Reform Law, as amended by the Health Care and Education Affordability Reconciliation Act, or ACA. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. The handling of any controlled substances must comply with the U.S. Controlled Substances Act and Controlled Substances Import and Export Act. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive recordkeeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines, imprisonment or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of product approvals, or refusal to allow a firm to enter into supply contracts, including government contracts. In addition, even if a firm complies with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw product approval. Prohibitions or restrictions on sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
U.S. Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and other specific aspects of the FDA approval of our drug candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, if any of our NDA’s are approved, we intend to apply for restoration of patent term for one of our currently owned or licensed patents to add patent life beyond the current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA.
39
Table of Contents
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain marketing applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity, or NCE. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. Recently, the FDA stated that it may change its interpretation of 5-year NCE exclusivity determinations to apply to each drug substance in a fixed-combination drug product, not for the drug product as a whole. If this change is implemented, for example, a fixed-combination drug product that contains a drug substance with a single, new active moiety would be eligible for 5 year NCE exclusivity, even if the fixed-combination also contains a drug substance with a previously approved active moiety. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a Section 505(b)(2) NDA submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovator drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the active agent for the original indication or condition of use. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the pre-clinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness. Orphan drug exclusivity, as described above, may offer a seven-year period of marketing exclusivity, except in certain circumstances. Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
Reimbursement
We do not anticipate that the sales of two of our product candidates (Zertane and the RedoxSYS System), once approved for sale, will be heavily dependent upon reimbursement by third-party payors. Traditionally, sales of pharmaceutical products that are not “life style” indications depend, in part, on the extent to which products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly reducing reimbursements for medical products and services. ProstaScint is dependent upon reimbursement for continued use in the U.S. market, and ProstaScint does have a reimbursement code as assigned by the American Medical Association. ProstaScint is currently reimbursed by Medicare, Medicaid, and various private health plans. However, reimbursement is not universally available throughout the United States for ProstaScint.
Lack of third-party reimbursement for our product candidate or a decision by a third-party payor to not cover our product candidates could reduce physician usage of the product candidate and have a material adverse effect on our sales, results of operations and financial condition.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
DEA Regulation
Zertane, because it contains tramadol, will be regulated as a “controlled substance” as defined in the Controlled Substances Act of 1970, or CSA, and the U.S. Drug Enforcement Agency’s, or DEA, implementing regulations, which establish registration, security, recordkeeping, reporting, storage, distribution, importation, exportation, inventory, quota and other requirements administered by the DEA. These requirements are directly applicable to us and also applicable to our manufacturers and to distributors, prescribers and dispensers of Zertane. The DEA regulates the handling of controlled substances through a closed chain of distribution. This control extends to the equipment and raw materials used in their manufacture and packaging in order to prevent loss and diversion into illicit channels of commerce.
40
Table of Contents
The DEA regulates controlled substances as Schedule I, II, III, IV or V substances. Schedule I substances by definition have no established medicinal use, and may not be marketed or sold in the United States. A pharmaceutical product may be listed as Schedule II, III, IV or V.
We expect that Zertane will be listed by the DEA as Schedule IV controlled substances under the CSA. Consequently, any importation of API for Zertane, as well as the manufacture, shipping, storage, sales and use of Zertane, will be subject to a high degree of regulation. Also, distribution and dispensing of these drugs are highly regulated.
Annual registration is required for any facility that manufactures, distributes, dispenses, imports or exports any controlled substance. The registration is specific to the particular location, activity and controlled substance schedule. For example, separate registrations are needed for import and manufacturing, and each registration will specify which schedules of controlled substances are authorized. Similarly, separate registrations are also required for separate facilities.
The DEA typically inspects a facility to review its security measures prior to issuing a registration and on a periodic basis. Reports must also be made for thefts or losses of any controlled substance, and to obtain authorization to destroy any controlled substance. In addition, special permits and notification requirements apply to imports and exports of narcotic drugs.
The DEA establishes annually an aggregate quota for how much of a controlled substance may be produced in total in the United States based on the DEA’s estimate of the quantity needed to meet legitimate scientific and medicinal needs. The DEA may adjust aggregate production quotas and individual production and procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments. Our or our manufacturers’ quotas of an active ingredient may not be sufficient to meet commercial demand or complete clinical trials. Any delay, limitation or refusal by the DEA in establishing our or our manufacturers’ quota for controlled substances could delay or stop our clinical trials or product launches, which could have a material adverse effect on our business, financial position and results of operations.
To enforce these requirements, the DEA conducts periodic inspections of registered establishments that handle controlled substances. Failure to maintain compliance with applicable requirements, particularly as manifested in loss or diversion, can result in administrative, civil or criminal enforcement action that could have a material adverse effect on our business, results of operations and financial condition. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate administrative proceedings to revoke those registrations. In some circumstances, violations could result in criminal proceedings.
Individual states also independently regulate controlled substances. We and our manufacturers will be subject to state regulation on distribution of these products, including, for example, state requirements for licensures or registration.
Intellectual Property
The current Vyrix patent portfolio consists of 79 issued patents and nine pending applications worldwide. The portfolio primarily consists of three families filed in the United States and throughout the world. The first family includes 30 issued patents for the use of tramadol to treat premature ejaculation. The standard 20-year expiration for patents in this family is in 2022. The other two families are for the use of a combination of tramadol and a phosphodiesterase inhibitor to treat comorbid premature ejaculation and erectile dysfunction and to treat sexual dysfunction side effects associated with administration of tramadol. These two families include issued patents in Europe, Australia, Canada, China, Mexico, New Zealand, Japan, the Philippines and South Africa and pending applications in the United States, Brazil, China, India, Japan, Korea, and the Philippines. The standard 20-year expiration for patents in these families is in 2028.
The current RedoxSYS/MiOXSYS patent portfolio consists of 14 issued patents and 56 pending applications worldwide. The portfolio primarily consists of four families filed in the United States and throughout the world. The first family includes four issued patents and five pending applications with claims directed to the measurement of the ORP of a patient sample to evaluate various conditions. The standard 20-year expiration for patents in this family is in 2028. The second family includes two pending United States applications, and nine pending applications worldwide with claims directed to the measurement of the ORP capacity of a patient sample to evaluate various conditions. The standard 20-year expiration for patents in this family is in 2033. The third family includes eight issued patents and 18 pending applications with claims directed to devices and methods for the measurement of ORP and ORP capacity. The standard 20-year expiration for patents in this family is in 2032. The fourth family includes one pending United States application and 16 pending applications worldwide with claims directed to multiple layer gel test strip measurement devices and methods of making for use in measuring ORP and ORP capacity. The standard 20-year expiration for patents in this family is in 2033.
ProstaScint is protected by significant trade secrets, manufacturing know how related to the production of the product’s base monoclonal antibody, and a highly protected proprietary cell line.
41
Table of Contents
We also maintain trade secrets and proprietary know-how that we seek to protect through confidentiality and nondisclosure agreements. These agreements may not provide meaningful protection or adequate remedies in the event of unauthorized use or disclosure of confidential and proprietary information. If we do not adequately protect our trade secrets and proprietary know-how, our competitive position and business prospects could be materially harmed.
We expect to seek United States and foreign patent protection for drug and diagnostic products we discover, as well as therapeutic and diagnostic products and processes. We expect also to seek patent protection or rely upon trade secret rights to protect certain other technologies which may be used to discover and characterize drugs and diagnostic products and processes, and which may be used to develop novel therapeutic and diagnostic products and processes.
The patent positions of companies such as ours involve complex legal and factual questions and, therefore, their enforceability cannot be predicted with any certainty. Our issued and licensed patents, and those that may be issued to us in the future, may be challenged, invalidated or circumvented, and the rights granted under the patents or licenses may not provide us with meaningful protection or competitive advantages. Our competitors may independently develop similar technologies or duplicate any technology developed by us, which could offset any advantages we might otherwise realize from our intellectual property. Furthermore, even if our product candidates receive regulatory approval, the time required for development, testing, and regulatory review could mean that protection afforded us by our patents may only remain in effect for a short period after commercialization. The expiration of patents or license rights we hold could adversely affect our ability to successfully commercialize our pharmaceutical drugs or diagnostics, thus harming our operating results and financial position.
We will be able to protect our proprietary intellectual property rights from unauthorized use by third parties primarily to the extent that such rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. If we must litigate to protect our intellectual property from infringement, we may incur substantial costs and our officers may be forced to devote significant time to litigation-related matters. The laws of certain foreign countries do not protect intellectual property rights to the same extent as do the laws of the United States.
Our pending patent applications, or those we may file or license from third parties in the future, may not result in patents being issued. Until a patent is issued, the claims covered by an application for patent may be narrowed or removed entirely, thus depriving us of adequate protection. As a result, we may face unanticipated competition, or conclude that without patent rights the risk of bringing product candidates to market exceeds the returns we are likely to obtain. We are generally aware of the scientific research being conducted in the areas in which we focus our research and development efforts, but patent applications filed by others are maintained in secrecy for at least 18 months and, in some cases in the United States, until the patent is issued. The publication of discoveries in scientific literature often occurs substantially later than the date on which the underlying discoveries were made. As a result, it is possible that patent applications for products similar to our drug or diagnostic products and product candidates may have already been filed by others without our knowledge.
The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights, and it is possible that our development of products and product candidates could be challenged by other pharmaceutical or biotechnology companies. If we become involved in litigation concerning the enforceability, scope and validity of the proprietary rights of others, we may incur significant litigation or licensing expenses, be prevented from further developing or commercializing a product or product candidate, be required to seek licenses that may not be available from third parties on commercially acceptable terms, if at all, or subject us to compensatory or punitive damage awards. Any of these consequences could materially harm our business.
Competition
The healthcare industry is highly competitive and subject to significant and rapid technological change as researchers learn more about diseases and develop new technologies and treatments. Significant competitive factors in our industry include product efficacy and safety; quality and breadth of an organization’s technology; skill of an organization’s employees and its ability to recruit and retain key employees; timing and scope of regulatory approvals; government reimbursement rates for, and the average selling price of, products; the availability of raw materials and qualified manufacturing capacity; manufacturing costs; intellectual property and patent rights and their protection; and sales and marketing capabilities.
We cannot assure you that any of our products that we successfully develop will be clinically superior or scientifically preferable to products developed or introduced by our competitors.
PE has traditionally been treated by behavioral or psychosexual therapy, antidepressant drugs, such as SSRIs, or topical desensitizing agents, all of which have significant drawbacks. Behavioral and psychosexual therapy as a treatment for PE requires an understanding partner and can be frustrating, embarrassing, time consuming and expensive, among other things. Antidepressant drugs are sometimes prescribed “off-label” and have numerous shortcomings, including side effects such as nausea, headaches, drop of libido, and ED, need for chronic dosing, ramp up periods, variable responses and unwanted drug-drug interactions. Topical agents, including lidocaine-based products affect spontaneity, can numb a partner and be messy.
42
Table of Contents
Dapoxetine (brand name Priligy, owned by Furiex Pharmaceuticals, Inc, which is owned by Actavis plc, is currently the only approved oral prescription drug to treat PE, with approval in several European countries. Priligy is not approved in the United States. In addition, we are aware of a topical product in late-stage development for PE by Plethora Solutions referred to as PSD502 and studies of Botox for the treatment of PE are being conducted in the United States. These products – if approved in the United States – would represent competition and alternative choices for physicians and potential patients.
In addition, generic tramadol hydrochloride is available in the United States and abroad for treatment of pain. Although the generic drug is not available in the same dosage as Zertane for treatment of PE, it is possible that physicians could prescribe the generic version of the drug “off label” for the treatment of PE instead of Zertane, if Zertane is approved for commercialization. Patients could use generic tramadol hydrochloride dosages that are either higher or lower than what will be approved for Zertane or they could attempt to split dosages to arrive at the dosages approved for Zertane. While any such “off label” use of generic tramadol hydrochloride for treatment of PE may constitute infringement of the Vyrix patent portfolio, liability in that circumstance would be at the level of the physician or the patient making enforcement difficult or impractical.
Currently, there are several FDA approved imaging techniques for cancer in general, however there is only one specifically targeting prostate cancer—ProstaScint. The other imaging methods are F18-fluorodeoxyglucose (F18-FDG), C11-Acetate, and C11-Choline. The primary advantage of these methods is that they all use PET imaging, a technique with better resolution than SPECT. The use of PET is also a disadvantage, however, since it uses radiolabels with short half-lives necessitating the need for a local or on-site cyclotron to generate the labels. The half-life of fluorine-18 (F18) and of carbon-11 (C11) are approximately 110 and 20 minutes, respectively. The radiolabel used by ProstaScint is Indium-11, with a half-life of about 2-3 days. This longer time period allows the radiolabel to be made remotely and shipped to the imaging facility; however it does use SPECT as the imaging modality.
As indicated, ProstaScint is the only radio-imaging marker that is specific for prostate cancer. ProstaScint is based on radiolabeling the antibody against prostate specific membrane antigen, or PSMA, a protein express by prostate cells. This specificity for prostate cells is what allows ProstaScint to detect the metastases of prostate cancer regardless of location. The mechanism of labeling for F18-FDG, C11-Acetate, and C11-Choline is the intracellular accumulation of these markers in cancer cells, due to the fact that cancer cells typically have a higher cellular metabolism than non-cancerous cells. Thus, these markers can accumulate in any type of cancer cell with a high metabolism, unfortunately prostate cancer cells tend to have a lower cellular metabolism resulting in higher false positives attributed to hyperplasia and prostatitis.
In a meta-analysis of 21 studies evaluating accuracy, sensitivity, specificity, positive/negative predictive values, ProstaScint using combined SPECT/CT imaging was comparable to PET/CT imaging based on F18-FDG and C11-Choline.
There are other oxidative stress diagnostic tests available throughout the world, although none are approved in the United States for clinical use. Diagnostic systems that are marketed for clinical use outside the United States include the FRAS 4 system (H&D srl), FREE Carpe Diem (Diacron International), and the FORM and FORMPlus systems (Callegari srl). These systems are used in both research and clinical settings but do not generate significant sales in the clinical setting. If approved in the United States for clinical use, these systems could present competition to the RedoxSYS System.
Our competitors may also succeed in obtaining FDA or other regulatory approvals for their product candidates more rapidly than we are able to do, which could place us at a significant competitive disadvantage or deny us marketing exclusivity rights. Market acceptance of our product candidates will depend on a number of factors, including: (i) potential advantages over existing or alternative therapies or tests, (ii) the actual or perceived safety of similar classes of products, (iii) the effectiveness of sales, marketing, and distribution capabilities, and (iv) the scope of any approval provided by the FDA or foreign regulatory authorities.
Although we believe our product candidates possess attractive attributes, we cannot assure you that our product candidates will achieve regulatory or market acceptance, or that we will be able to compete effectively in the pharmaceutical drug markets. If our product candidates fail to gain regulatory approvals and acceptance in their intended markets, we may not generate meaningful revenues or achieve profitability.
Research and Development
Our strategy is to minimize fixed overhead by outsourcing much of our research and development activities. We believe we will benefit from Ampio’s research and development experience as well as regulatory expertise. Additionally, we intend to utilize consultants with domain experience for research, development and regulatory guidance.
43
Table of Contents
We have consulting agreements in place with two such companies who are actively participating with us on the impending clinical trials for Zertane. When we focus on Zertane-ED in the future, as planned, we intend to collaborate on pre-clinical studies and clinical trials with our partner in Korea, Daewoong Pharmaceuticals Co.
Our RedoxSYS System has been developed in conjunction with numerous medical device and diagnostic development consultants. Further, we have relationships with regulatory consultants who are actively assisting in the development of our regulatory strategy with the FDA. To complement our internal clinical research efforts with the RedoxSYS System, we have engaged with numerous universities around the world to identify and develop research and clinical applications for the RedoxSYS System. Through these engagements we have access to data and analyses that enable us to develop new uses for the RedoxSYS and MiOXSYS systems. Additionally, we have formal research agreements in place with two prominent U.S.-based universities and one prominent European university for which we are paying a research fee.
Manufacturing
Our business strategy is to use cGMP compliant contract manufacturers for the manufacture of clinical supplies as well as for commercial supplies if required by our commercialization plans, and to transfer manufacturing responsibility to our collaboration partners when possible.
We are party to a 10-year supply agreement with an established manufacturer of tramadol hydrochloride for Zertane. Importantly, product supply has been produced for our planned clinical trials for Zertane.
We have acquired a two-year supply of ProstaScint through our asset purchase agreement with Jazz Pharmaceuticals. Further, we intend to transfer the manufacturing of ProstaScint to a new contract manufacturer, and have initiated discussions with the contract manufacturer and expect to sign an agreement whereby we will transfer the production to a similar facility operated by the new contract manufacturer. Accordingly, we also expect to put a supply agreement in place for an extended duration.
We have completed the technical development of the RedoxSYS System by engaging contract development and manufacturing companies in the United States. We secured supply and quality agreements with manufacturers for both the RedoxSYS and MiOXSYS instruments as well as the RedoxSYS and MiOXSYS sensor strips. Both manufacturers hold long-standing ISO 13485:2003 certifications and are established medical device manufacturers. Both manufacturers have high volume manufacturing capacity such that production volumes can be easily scaled. Both manufacturers have been audited by our quality engineers and are fully compliant.
Employees
As of August 1, 2015, we had 9 full-time employees and utilized the services of a number of consultants on a temporary basis. Overall, we have not experienced any work stoppage and do not anticipate any work stoppage in the foreseeable future. None of our employees is subject to a collective bargaining agreement. Management believes that relations with our employees are good.
Available Information
Our principal executive offices are located at 373 Inverness Parkway, Suite 206, Englewood, Colorado 80112 USA, and our phone number is (720) 437-6580.
We maintain a website on the internet at http://aytubio.com. We make available free of charge through our website, by way of a hyperlink to a third-party site that includes filings we make with the SEC website (www.sec.gov), our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports electronically filed or furnished pursuant to Section 15(d) of the Exchange Act. The information on our website is not, and shall not be deemed to be, a part of this Annual Report on Form 10-K or incorporated into any other filings we make with the SEC. In addition, the public may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, N.E., Washington D.C., 20549. Information on the operation of the Public Reference Room may be obtained by calling the SEC at 1-800-SEC-0330.
Code of Ethics
We currently have not adopted a written code of ethics that applies to our officers, directors and employees, including our principal executive officer and principal accounting officer. Our Board of Directors intends to adopt such a formal code of ethics when it deems appropriate based on the size of our operations and personnel.
44
Table of Contents
| Item 1A. | Risk Factors |
Investing in our common stock includes a high degree of risk. You should consider carefully the specific factors discussed below, together with all of the other information contained in this Annual Report. If any of the following risks actually occurs, our business, financial condition, results of operations and future prospects would likely be materially and adversely affected. This could cause the market price of our common stock to decline and could cause you to lose all or part of your investment.
Risks Related to Our Financial Condition and Capital Requirements
We have limited operating history, have incurred losses, and can give no assurance of profitability.
We are a clinical-stage healthcare company with a limited operating history. We have not generated material revenue to date and are not profitable, and have incurred losses in each year since our inception. Our net loss for the years ended June 30, 2015 and 2014 was $7.7 million and $5.6 million, respectively. We have not demonstrated the ability to be a profit-generating enterprise, and without significant financing, there is substantial doubt about our ability to continue as a going concern. We expect to incur substantial losses for the foreseeable future. Our ability to generate revenue is uncertain, and we may never achieve profitability. We have a very limited operating history on which investors can evaluate our potential for future success. Potential investors should evaluate us in light of the expenses, delays, uncertainties, and complications typically encountered by early-stage healthcare businesses, many of which will be beyond our control. These risks include the following:
| • | U.S. regulatory approval of our product candidates; |
| • | foreign regulatory approval of our products and product candidates; |
| • | lack of sufficient capital; |
| • | uncertain market acceptance of our products and product candidates; |
| • | unanticipated problems, delays, and expense relating to product development and implementation; |
| • | lack of intellectual property; |
| • | competition; and |
| • | technological changes. |
As a result of our limited operating history, and the increasingly competitive nature of the markets in which we compete, our historical financial data, which, prior to April 16, 2015, consists of allocations of expenses from Ampio, is of limited value in anticipating future operating expenses. Our planned expense levels will be based in part on our expectations concerning future operations, which is difficult to forecast accurately based on our stage of development. We may be unable to adjust spending in a timely manner to compensate for any unexpected budgetary shortfall.
We have not received any material revenues from the commercialization of our products or product candidates and might not receive significant revenues from the commercialization of products and our product candidates in the near term. Even though ProstaScint is an approved drug that we are marketing, we only acquired it in May 2015 and have limited experience on which to base the revenue we could expect to receive. To obtain revenues from our products and product candidates, we must succeed, either alone or with others, in a range of challenging activities, including expanding markets for our existing products and completing clinical trials of our product candidates, obtaining positive results from the clinical trials, achieving marketing approval for these product candidates, manufacturing, marketing and selling our existing products and those products for which we, or our collaborators, may obtain marketing approval, satisfying any post-marketing requirements and obtaining reimbursement for our products from private insurance or government payors. We, and our collaborators, if any, may never succeed in these activities and, even if we do, or one of our collaborators does, we may never generate revenues that are sufficient enough for us to achieve profitability.
We may need to raise additional funding, which may not be available on acceptable terms, or at all. Failure to obtain necessary capital when needed may force us to delay, limit or terminate our product expansion and development efforts or other operations.
We are currently advancing our product candidates through clinical development. Developing product candidates is expensive, lengthy and risky, and we expect our research and development expenses to increase substantially in connection with our ongoing activities, particularly as we advance Zertane into two planned Phase 3 clinical trials in the United States and develop the RedoxSYS System for additional applications. In addition, we are expending resources to expand the market for ProstaScint, which might not be successful or might take longer and be more expensive than anticipated.
45
Table of Contents
As of June 30, 2015, our cash and cash equivalents were $7.4 million. Our operating plan may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. In any event, we will require additional capital to obtain regulatory approval for, and to commercialize, our product candidates. Raising funds in the current economic environment, as well our lack of operating history, may present additional challenges. Even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to expand any existing product or develop and commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our stockholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our shares to decline. The sale of additional equity or convertible securities would dilute all of our stockholders. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results and prospects.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any product candidate or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
If we do not obtain the capital necessary to fund our operations, we will be unable to successfully expand, develop, obtain regulatory approval of, and commercialize, our products and product candidates.
The development of pharmaceutical products, medical diagnostics and medical devices is capital-intensive. We anticipate we may require additional financing to continue to fund our operations. Our future capital requirements will depend on, and could increase significantly as a result of, many factors including:
| • | progress in, and the costs of, our pre-clinical studies and clinical trials and other research and development programs; |
| • | the scope, prioritization and number of our research and development programs; |
| • | the achievement of milestones or occurrence of other developments that trigger payments under any collaboration agreements we obtain; |
| • | the costs of securing manufacturing arrangements for commercial production; |
| • | the costs of establishing or contracting for sales and marketing capabilities for any existing products and if we obtain regulatory clearances to market our product candidates; |
| • | the extent to which we are obligated to reimburse, or entitled to reimbursement of, clinical trial costs under future collaboration agreements, if any; and |
| • | the costs involved in filing, prosecuting, enforcing and defending patent claims and other intellectual property rights |
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through collaboration arrangements, sales of our securities, debt financings, or by out-licensing one or more of our product candidates. Dislocations in the financial markets have generally made equity and debt financing more difficult to obtain, and may have a material adverse effect on our ability to meet our fundraising needs. We cannot be certain that additional funding will be available to us on acceptable terms, if at all. If funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our technologies, research or development programs or our commercialization efforts. Additional funding, if obtained, may significantly dilute existing shareholders if that financing is obtained through issuing equity or instruments convertible into equity.
We will incur increased costs associated with, and our management will need to devote substantial time and effort to, compliance with public company reporting and other requirements.
As a public company, we will incur significant legal, accounting and other expenses that Vyrix and Luoxis did not incur as private companies. In addition, the rules and regulations of the SEC and any national securities exchange to which we may be subject in the future impose numerous requirements on public companies, including requirements relating to our corporate governance practices,
46
Table of Contents
with which we will need to comply. Further, we will be required to, among other things, file annual, quarterly and current reports with respect to our business and operating results. Based on currently available information and assumptions, we estimate that we will incur approximately $500,000 in expenses on an annual basis as a direct result of the requirements of being a publicly traded company. Our management and other personnel will need to devote substantial time to gaining expertise regarding operations as a public company and compliance with applicable laws and regulations, and our efforts and initiatives to comply with those requirements could be expensive.
Risks Related to Product Development, Regulatory Approval and Commercialization
We cannot be certain that we will be able to obtain regulatory approval for, or successfully commercialize, any of our product candidates.
We may not be able to develop our current or any future product candidates. Our product candidates will require substantial additional clinical development, testing, and regulatory approval before we are permitted to commence commercialization. The clinical trials of our product candidates are, and the manufacturing and marketing of our product candidates will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries where we intend to test and, if approved, market any product candidate. Before obtaining regulatory approvals for the commercial sale of any product candidate, we must demonstrate through pre-clinical testing and clinical trials that the product candidate is safe and effective for use in each target indication. This process can take many years and may include post-marketing studies and surveillance, which will require the expenditure of substantial resources. Of the large number of drugs in development in the U.S., only a small percentage successfully completes the FDA regulatory approval process and is commercialized. Accordingly, even if we are able to obtain the requisite financing to continue to fund our development and clinical programs, we cannot assure you that any of our product candidates will be successfully developed or commercialized.
We are not permitted to market a product in the U.S. until we receive approval of a New Drug Application, or an NDA, for that product from the FDA, or in any foreign countries until we receive the requisite approval from such countries. Obtaining approval of an NDA is a complex, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval of any product candidate for many reasons, including, among others:
| • | we may not be able to demonstrate that a product candidate is safe and effective to the satisfaction of the FDA; |
| • | the results of our clinical trials may not meet the level of statistical or clinical significance required by the FDA for marketing approval; |
| • | the FDA may disagree with the number, design, size, conduct or implementation of our clinical trials; |
| • | the FDA may require that we conduct additional clinical trials; |
| • | the FDA may not approve the formulation, labeling or specifications of any product candidate; |
| • | the clinical research organizations, or CROs, that we retain to conduct our clinical trials may take actions outside of our control that materially adversely impact our clinical trials; |
| • | the FDA may find the data from pre-clinical studies and clinical trials insufficient to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks, such as the risk of drug abuse by patients or the public in general; |
| • | the FDA may disagree with our interpretation of data from our pre-clinical studies and clinical trials; |
| • | the FDA may not accept data generated at our clinical trial sites; |
| • | if an NDA, if and when submitted, is reviewed by an advisory committee, the FDA may have difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional pre-clinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; |
| • | the FDA may require development of a Risk Evaluation and Mitigation Strategy, or REMS, as a condition of approval or post-approval; |
| • | the FDA may not approve the manufacturing processes or facilities of third-party manufacturers with which we contract; or |
| • | the FDA may change its approval policies or adopt new regulations. |
These same risks apply to applicable foreign regulatory agencies from whom we may seek approval for any of our product candidates.
47
Table of Contents
Any of these factors, many of which are beyond our control, could jeopardize our ability to obtain regulatory approval for and successfully market any product candidate. Moreover, because a substantial portion of our business is dependent upon our existing product candidates, any such setback in our pursuit of regulatory approval would have a material adverse effect on our business and prospects.
Favorable results in the prior clinical trials of Zertane outside of the United States may not be predictive of the results in our planned Phase 3 clinical trials of Zertane in the United States or the designs of our Phase 3 clinical trials may be inadequate for FDA approval.
A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in earlier-stage development. The prior clinical trials of Zertane showed favorable safety and efficacy data; however, we will have different enrollment criteria in our planned Phase 3 clinical trials. In the Phase 2 clinical trials, we were able to enroll patients utilizing a broader definition of PE.
Ejaculation latency, most commonly quantified using intravaginal ejaculation latency time, or IELT, is a dominant component of PE assessment in clinical trials. IELT is defined as the time between vaginal intromission and intravaginal ejaculation. Although a standard cut-off for ejaculatory latency does not exist, it has been suggested that an IELT of two minutes or less may serve as an adequately sensitive criterion for defining PE and some studies have used IELT values from one to two minutes for defining PE. In a pre-IND meeting with FDA, we agreed to use an IELT of less than or equal to 1 minute as one of the enrollment criteria for our planned Phase 3 clinical trials. The previous European Phase 3 trials allowed for an IELT of less than two minutes however a significant proportion of enrollees had an IELT of one minute or less. In our planned Phase 3 clinical trials, we will be utilizing the definition of lifelong PE adopted by the International Society for Sexual Medicine, or the ISSM: “premature ejaculation is a male sexual dysfunction characterized by ejaculation which always or nearly always occurs prior to or within a minute of vaginal penetration; and inability to delay ejaculation on all or nearly all vaginal penetrations; and negative personal consequences, such as distress, bother, frustration and/or the avoidance of sexual intimacy.” As a result, we may encounter difficulty enrolling a sufficient number of patients in a timely fashion and we may not observe a similarly favorable safety and efficacy profile as our prior clinical trials.
In addition, Ampio obtained guidance from the FDA on our planned Phase 3 trials at a pre-IND meeting held in June 2012, including information to help us define the target patient population, select co-primary endpoints and design an acceptable patient-reported outcome measure. As a result of direction provided at the meeting, along with the existing data from six clinical trials of Zertane conducted outside the U.S. to date, we believe we are positioned to advance Zertane into Phase 3 clinical trials in the United States. However, we can provide no assurance that the FDA will not change its guidance about our planned Phase 3 clinical trials and require us to significantly modify the design of, or endpoints for, our planned clinical trials. Any change in the guidance we have received could delay and/or render more expensive our planned Phase 3 trial for Zertane.
We were not involved in any of the prior clinical studies for Zertane and are relying on the data collected from those prior clinical trials by various third parties, including a previous partner of our majority stockholder, Ampio Pharmaceuticals. Dr. David Bar-Or (now the Chief Scientific Officer of Ampio Pharmaceuticals) discovered the utility of tramadol hydrochloride for the treatment of PE in June 1999, and this discovery and accompanying intellectual property were at that time the property of DMI BioSciences, Inc., or DMI BioSciences. DMI BioSciences conducted various clinical trials, prior to licensing the worldwide rights to tramadol hydrochloride for PE to Biovail Laboratories International, or Biovail. Biovail also conducted several clinical trials and began two Phase 3 clinical trials, which trials were completed by Ampio upon its acquisition of DMI Biosciences, to whom the rights had reverted. This lack of prior involvement may have a negative impact on our understanding of these prior clinical trials and the design of our planned Phase 3 trial.
If we fail to successfully acquire new products, we may lose market position.
Acquiring new products will be an important factor in our sales growth, including products that already have been developed and found market acceptance. If we fail to identify existing or emerging consumer markets and trends and to acquire new products, we will not develop a strong revenue source to help pay for our development activities as well as possible acquisitions. This would delay our business plan, which could have a negative adverse effect on our business and prospects.
If we do not secure collaborations with strategic partners to test, commercialize and manufacture product candidates, we may not be able to successfully develop products and generate meaningful revenues.
A key aspect of our current strategy is to selectively enter into collaborations with third parties to conduct clinical testing, as well as to commercialize and manufacture product candidates. If we are able to identify and each an agreement with one or more collaborators, our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements. Collaboration agreements typically call for milestone payments that depend on
48
Table of Contents
successful demonstration of efficacy and safety, obtaining regulatory approvals, and clinical trial results. Collaboration revenues are not guaranteed, even when efficacy and safety are demonstrated. The current economic environment may result in potential collaborators electing to reduce their external spending, which may prevent us from developing our product candidates.
Even if we succeed in securing collaborators, the collaborators may fail to develop or effectively commercialize products using our product candidates. Collaborations involving our product candidates pose a number of risks, including the following:
| • | collaborators may not have sufficient resources or may decide not to devote the necessary resources due to internal constraints such as budget limitations, lack of human resources, or a change in strategic focus; |
| • | collaborators may believe our intellectual property is not valid or is unenforceable or the product candidate infringes on the intellectual property rights of others; |
| • | collaborators may dispute their responsibility to conduct development and commercialization activities pursuant to the applicable collaboration, including the payment of related costs or the division of any revenues; |
| • | collaborators may decide to pursue a competitive product developed outside of the collaboration arrangement; |
| • | collaborators may not be able to obtain, or believe they cannot obtain, the necessary regulatory approvals; |
| • | collaborators may delay the development or commercialization of our product candidates in favor of developing or commercializing their own or another party’s product candidate; or |
| • | collaborators may decide to terminate or not to renew the collaboration for these or other reasons. |
As a result, collaboration agreements may not lead to development or commercialization of our product candidates in the most efficient manner or at all. For example, our former collaborator that licensed Zertane conducted clinical trials which we believe demonstrated efficacy in treating PE, but the collaborator undertook a merger that we believe altered its strategic focus and thereafter terminated the collaboration agreement. The Merger also created a potential conflict with a principal customer of the acquired company, which sells a product to treat premature ejaculation in certain European markets.
Collaboration agreements are generally terminable without cause on short notice. Once a collaboration agreement is signed, it may not lead to commercialization of a product candidate. We also face competition in seeking out collaborators. If we are unable to secure collaborations that achieve the collaborator’s objectives and meet our expectations, we may be unable to advance our product candidates and may not generate meaningful revenues.
We or our strategic partners may choose not to continue an existing product or choose not to develop a product candidate at any time during development, which would reduce or eliminate our potential return on investment for that product.
At any time and for any reason, we or our strategic partners may decide to discontinue the development or commercialization of a product or product candidate. If we terminate a program in which we have invested significant resources, we will reduce the return, or not receive any return, on our investment and we will have missed the opportunity to have allocated those resources to potentially more productive uses. If one of our strategic partners terminates a program, we will not receive any future milestone payments or royalties relating to that program under our agreement with that party.
Our product candidates are expected to undergo clinical trials that are time-consuming and expensive, the outcomes of which are unpredictable, and for which there is a high risk of failure. If clinical trials of our product candidates fail to satisfactorily demonstrate safety and efficacy to the FDA and other regulators, we or our collaborators may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of these product candidates.
Pre-clinical testing and clinical trials are long, expensive and unpredictable processes that can be subject to extensive delays. We cannot guarantee that any clinical studies will be conducted as planned or completed on schedule, if at all. It may take several years to complete the pre-clinical testing and clinical development necessary to commercialize a drug or biologic, and delays or failure can occur at any stage. Interim results of clinical trials do not necessarily predict final results, and success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials even after promising results in earlier trials and we cannot be certain that we will not face similar setbacks. The design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. An unfavorable outcome in one or more trials would be a major set-back for that product candidate and for us. Due to our limited financial resources, an unfavorable outcome in one or more trials may require us to delay, reduce the scope of, or eliminate one or more product development programs, which could have a material adverse effect on our business, prospects and financial condition and on the value of our common stock.
49
Table of Contents
In connection with clinical testing and trials, we face a number of risks, including:
| • | a product candidate is ineffective, inferior to existing approved medicines, unacceptably toxic, or has unacceptable side effects; |
| • | patients may die or suffer other adverse effects for reasons that may or may not be related to the product candidate being tested; |
| • | the results may not confirm the positive results of earlier testing or trials; and |
| • | the results may not meet the level of statistical significance required by the FDA or other regulatory agencies to establish the safety and efficacy of the product candidate. |
The results of pre-clinical studies do not necessarily predict clinical success, and larger and later-stage clinical trials may not produce the same results as earlier-stage clinical trials. Frequently, product candidates developed by pharmaceutical companies have shown promising results in early pre-clinical studies or clinical trials, but have subsequently suffered significant setbacks or failed in later clinical trials. In addition, clinical trials of potential products often reveal that it is not possible or practical to continue development efforts for these product candidates.
If we do not successfully complete pre-clinical and clinical development, we will be unable to market and sell products derived from our product candidates and generate revenues. Even if we do successfully complete clinical trials, those results are not necessarily predictive of results of additional trials that may be needed before an NDA may be submitted to the FDA. Although there are a large number of drugs and biologics in development in the United States and other countries, only a small percentage result in the submission of an NDA to the FDA, even fewer are approved for commercialization, and only a small number achieve widespread physician and consumer acceptance following regulatory approval. If our clinical trials are substantially delayed or fail to prove the safety and effectiveness of our product candidates in development, we may not receive regulatory approval of any of these product candidates and our business, prospects and financial condition will be materially harmed.
Delays, suspensions and terminations in our clinical trials could result in increased costs to us and delay or prevent our ability to generate revenues.
Human clinical trials are very expensive, time-consuming, and difficult to design, implement and complete. We currently expect clinical trials of our therapeutic product candidates could take up to 24 months to complete, but the completion of trials for these candidates may be delayed for a variety of reasons, including delays in:
| • | demonstrating sufficient safety and efficacy to obtain regulatory approval to commence a clinical trial; |
| • | reaching agreement on acceptable terms with prospective contract research organizations and clinical trial sites; |
| • | validating test methods to support quality testing of the drug substance and drug product; |
| • | obtaining sufficient quantities of the drug substance; |
| • | manufacturing sufficient quantities of a product candidate; |
| • | obtaining approval of an Investigational New Drug application, or IND, from the FDA; |
| • | obtaining institutional review board approval to conduct a clinical trial at a prospective clinical trial site; |
| • | determining dosing and clinical design and making related adjustments; and |
| • | patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical trial sites, the availability of effective treatments for the relevant disease and the eligibility criteria for the clinical trial. |
The commencement and completion of clinical trials for our product candidates may be delayed, suspended or terminated due to a number of factors, including:
| • | lack of effectiveness of product candidates during clinical trials; |
| • | adverse events, safety issues or side effects relating to the product candidates or their formulation; |
| • | inability to raise additional capital in sufficient amounts to continue clinical trials or development programs, which are very expensive; |
| • | the need to sequence clinical trials as opposed to conducting them concomitantly in order to conserve resources; |
| • | our inability to enter into collaborations relating to the development and commercialization of our product candidates; |
50
Table of Contents
| • | failure by us or our collaborators to conduct clinical trials in accordance with regulatory requirements; |
| • | our inability or the inability of our collaborators to manufacture or obtain from third parties materials sufficient for use in pre-clinical studies and clinical trials; |
| • | governmental or regulatory delays and changes in regulatory requirements, policy and guidelines, including mandated changes in the scope or design of clinical trials or requests for supplemental information with respect to clinical trial results; |
| • | failure of our collaborators to advance our product candidates through clinical development; |
| • | delays in patient enrollment, variability in the number and types of patients available for clinical trials, and lower-than anticipated retention rates for patients in clinical trials; |
| • | difficulty in patient monitoring and data collection due to failure of patients to maintain contact after treatment; |
| • | a regional disturbance where we or our collaborative partners are enrolling patients in our clinical trials, such as a pandemic, terrorist activities or war, or a natural disaster; and |
| • | varying interpretations of our data, and regulatory commitments and requirements by the FDA and similar foreign regulatory agencies. |
Many of these factors may also ultimately lead to denial of an NDA for a product candidate. If we experience delay, suspensions or terminations in a clinical trial, the commercial prospects for the related product candidate will be harmed, and our ability to generate product revenues will be delayed.
In addition, we may encounter delays or product candidate rejections based on new governmental regulations, future legislative or administrative actions, or changes in FDA policy or interpretation during the period of product development. If we obtain required regulatory approvals, such approvals may later be withdrawn. Delays or failures in obtaining regulatory approvals may result in:
| • | varying interpretations of data and commitments by the FDA and similar foreign regulatory agencies; and |
| • | diminishment of any competitive advantages that such product candidates may have or attain. |
Furthermore, if we fail to comply with applicable FDA and other regulatory requirements at any stage during this regulatory process, we may encounter or be subject to:
| • | diminishment of any competitive advantages that such product candidates may have or attain; |
| • | delays or termination in clinical trials or commercialization; |
| • | refusal by the FDA or similar foreign regulatory agencies to review pending applications or supplements to approved applications; |
| • | product recalls or seizures; |
| • | suspension of manufacturing; |
| • | withdrawals of previously approved marketing applications; and |
| • | fines, civil penalties, and criminal prosecutions. |
We or our collaborators intend to seek FDA approval for some of our product candidates using an expedited process established by the FDA. If we, or our collaborators, are unable to secure clearances to use expedited development pathways from the FDA for certain of our drug product candidates, we, or they, may be required to conduct additional pre-clinical studies or clinical trials beyond those that we, or they, contemplate, which could increase the expense of obtaining, and delay the receipt of, necessary marketing approvals and of any product revenues.
Assuming successful completion of clinical trials, we expect to submit NDAs to the FDA at various times in the future under Section 505(b)(2) of the Food, Drug and Cosmetic Act, as amended, or the FDCA. NDAs submitted under this section are eligible to receive FDA approval by relying in part on the FDA’s findings of safety and efficacy for a previously approved drug. We specifically intend to do this for Zertane. The FDA’s 1999 guidance on Section 505(b)(2) applications states that new indications for a previously approved drug, a new combination product, a modified active ingredient, or changes in dosage form, strength, formulation, and route of administration of a previously approved product are encompassed within the Section 505(b)(2) NDA process. Relying on Section 505(b)(2) is advantageous because we or our collaborators may not be required (i) to perform the full range of safety and efficacy trials that is otherwise required to secure approval of a new drug, and (ii) obtain a “right of reference” from the applicant that obtained approval of the previously approved drug. However, a Section 505(b)(2) application must support the proposed change of the previously approved drug by including necessary and adequate information, as determined by the FDA, and the FDA may still require us to perform a portion or the full range of safety and efficacy trials. There can be no assurance that we would be successful under any Section 505(b)(2) application.
51
Table of Contents
The approval process outside the United States varies among countries and may limit our ability to develop, manufacture and sell our products internationally. Failure to obtain marketing approval in international jurisdictions would prevent our product candidates from being marketed abroad.
In order to market and sell our products in the European Union and many other jurisdictions, we, and our collaborators, must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and may involve additional testing. We may conduct clinical trials for, and seek regulatory approval to market, our product candidates in countries other than the United States. Depending on the results of clinical trials and the process for obtaining regulatory approvals in other countries, we may decide to first seek regulatory approvals of a product candidate in countries other than the United States, or we may simultaneously seek regulatory approvals in the United States and other countries. If we or our collaborators seek marketing approval for a product candidate outside the United States, we will be subject to the regulatory requirements of health authorities in each country in which we seek approval. With respect to marketing authorizations in Europe, we will be required to submit a European Marketing Authorization Application, or MAA, to the European Medicines Agency, or EMA, which conducts a validation and scientific approval process in evaluating a product for safety and efficacy. The approval procedure varies among regions and countries and may involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval.
Obtaining regulatory approvals from health authorities in countries outside the United States is likely to subject us to all of the risks associated with obtaining FDA approval described above. In addition, marketing approval by the FDA does not ensure approval by the health authorities of any other country, and approval by foreign health authorities does not ensure marketing approval by the FDA.
Even if we, or our collaborators, obtain marketing approvals for our product candidates, the terms of approvals and ongoing regulation of our products may limit how we or they market our products, which could materially impair our ability to generate revenue.
Even if we receive regulatory approval for a product candidate, this approval may carry conditions that limit the market for the product or put the product at a competitive disadvantage relative to alternative therapies. For instance, a regulatory approval may limit the indicated uses for which we can market a product or the patient population that may utilize the product, or may be required to carry a warning in its labeling and on its packaging. Products with boxed warnings are subject to more restrictive advertising regulations than products without such warnings. These restrictions could make it more difficult to market any product candidate effectively. Accordingly, assuming we, or our collaborators, receive marketing approval for one or more of our product candidates, we, and our collaborators expect to continue to expend time, money and effort in all areas of regulatory compliance.
Any of our products and product candidates for which we, or our collaborators, obtain marketing approval in the future could be subject to post-marketing restrictions or withdrawal from the market and we, and our collaborators, may be subject to substantial penalties if we, or they, fail to comply with regulatory requirements or if we, or they, experience unanticipated problems with our products following approval.
Any of our approved products and product candidates for which we, or our collaborators, obtain marketing approval, as well as the manufacturing processes, post-approval studies and measures, labeling, advertising and promotional activities for such products, among other things, are or will be subject to continual requirements of and review by the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping. Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, including the FDA requirement to implement a REMS to ensure that the benefits of a drug or biological product outweigh its risks.
The FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of a product. The FDA and other agencies, including the Department of Justice, closely regulate and monitor the post-approval marketing and promotion of products to ensure that they are manufactured, marketed and distributed only for the approved indications and in accordance with the provisions of the approved labeling. The FDA imposes stringent restrictions on manufacturers’ communications regarding off-label use and if we, or our collaborators, do not market any of our product candidates for which we, or they, receive marketing approval for only their approved indications, we, or they, may be subject to warnings or enforcement action for off-label marketing. Violation of the FDCA and other statutes, including the False Claims Act, relating to the promotion and advertising of prescription drugs may lead to investigations or allegations of violations of federal and state health care fraud and abuse laws and state consumer protection laws.
52
Table of Contents
If we do not achieve our projected development and commercialization goals in the timeframes we announce and expect, the commercialization of our product candidates may be delayed, and our business will be harmed.
We sometimes estimate for planning purposes the timing of the accomplishment of various scientific, clinical, regulatory and other product development objectives. These milestones may include our expectations regarding the commencement or completion of scientific studies and clinical trials, the submission of regulatory filings, or commercialization objectives. From time to time, we may publicly announce the expected timing of some of these milestones, such as the initiation or completion of an ongoing clinical trial, the initiation of other clinical programs, receipt of marketing approval, or a commercial launch of a product. The achievement of many of these milestones may be outside of our control. All of such milestones are based on a variety of assumptions which may cause the timing of achievement of the milestones to vary considerably from our estimates, including:
| • | our available capital resources or capital constraints we experience; |
| • | the rate of progress, costs and results of our clinical trials and research and development activities, including the extent of scheduling conflicts with participating clinicians and collaborators, and our ability to identify and enroll patients who meet clinical trial eligibility criteria; |
| • | our receipt of approvals from the FDA and other regulatory agencies and the timing thereof; |
| • | other actions, decisions or rules issued by regulators; |
| • | our ability to access sufficient, reliable and affordable supplies of compounds used in the manufacture of our product candidates; |
| • | the efforts of our collaborators with respect to the commercialization of our products; and |
| • | the securing of, costs related to, and timing issues associated with, product manufacturing as well as sales and marketing activities. |
If we fail to achieve announced milestones in the timeframes we announce and expect, the commercialization of our product candidates may be delayed and our business, prospects and results of operations may be harmed.
We rely on third parties to conduct our clinical trials and perform data collection and analysis, which may result in costs and delays that prevent us from successfully commercializing product candidates.
We rely, and will rely in the future, on medical institutions, clinical investigators, contract research organizations, contract laboratories, and collaborators to perform data collection and analysis and others to carry out our clinical trials. Our development activities or clinical trials conducted in reliance on third parties may be delayed, suspended, or terminated if:
| • | the third parties do not successfully carry out their contractual duties or fail to meet regulatory obligations or expected deadlines; |
| • | we replace a third party; or |
| • | the quality or accuracy of the data obtained by third parties is compromised due to their failure to adhere to clinical protocols, regulatory requirements, or for other reasons. |
Third party performance failures may increase our development costs, delay our ability to obtain regulatory approval, and delay or prevent the commercialization of our product candidates. While we believe that there are numerous alternative sources to provide these services, in the event that we seek such alternative sources, we may not be able to enter into replacement arrangements without incurring delays or additional costs.
In addition, for Zertane, we are currently using, and relying on, single suppliers and single manufacturers for drug supply for our planned Phase 3 clinical trials and our commercial products. Although there are potential alternative suppliers and manufacturers for Zertane if need be, we have not qualified these vendors to date. If we were required to change vendors, it could result in a failure to meet regulatory requirements or projected timelines and necessary quality standards for successful manufacturing of the various required lots of material for our development and commercialization efforts, any of which could have an adverse effect on our business, prospects and financial condition.
Even if collaborators with which we contract in the future successfully complete clinical trials of our product candidates, those product candidates may not be commercialized successfully for other reasons.
Even if we contract with collaborators that successfully complete clinical trials for one or more of our product candidates, those candidates may not be commercialized for other reasons, including:
| • | failure to receive regulatory clearances required to market them as drugs; |
53
Table of Contents
| • | being subject to proprietary rights held by others; |
| • | being difficult or expensive to manufacture on a commercial scale; |
| • | having adverse side effects that make their use less desirable; or |
| • | failing to compete effectively with products or treatments commercialized by competitors. |
Relying on third-party manufacturers may result in delays in our clinical trials, product introductions and product supply.
Developing and commercializing new medicines and devices entails significant risks and expenses. Our clinical trials may be delayed if third-party manufacturers are unable to assure a sufficient quantity of the drug product to meet our study needs. If our clinical trials are delayed, our commercialization efforts may be impeded, or our costs may increase.
Once regulatory approval is obtained, a marketed product and its manufacturer are subject to continual review. The discovery of previously unknown problems with a product or manufacturer may result in restrictions on the product, manufacturer or manufacturing facility, including withdrawal of the product from the market. Any manufacturers with which we contract are required to operate in accordance with FDA-mandated current good manufacturing practices, or cGMPs. A failure of any of our contract manufacturers to establish and follow cGMPs and to document their adherence to such practices could lead to significant delays in the launch of our products candidates into the market or in the continued supply of any product after approval. Failure by third-party manufacturers to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, revocation or suspension of marketing approval for any products granted pre-market approvals, seizures or recalls of products, operating restrictions, and criminal prosecutions.
Further, if any manufacture needs to be replaced or new or increased product supplies obtained, we might not be able to locate and engage a new manufacturer on a timely basis or at all. Any of these events could have a material adverse effect on sales of an approved product, which could negatively impact our financial condition and our business.
We face substantial competition from companies with considerably more resources and experience than we have, which may result in others discovering, developing, receiving approval for, or commercializing products before or more successfully than us.
Many of our potential competitors have substantially greater financial, technical, personnel and marketing resources than we do. In addition, many of these competitors have significantly greater resources devoted to product development and pre-clinical research. Our ability to compete successfully will depend largely on our ability to:
| • | expand the market for any approved products; |
| • | successfully commercialize our product candidates alone or with commercial partners; |
| • | discover and develop product candidates that are superior to other products in the market; |
| • | obtain required regulatory approvals; |
| • | attract and retain qualified personnel; and |
| • | obtain patent and/or other proprietary protection for our product candidates |
Established pharmaceutical companies devote significant financial resources to discovering, developing or licensing novel compounds that could make our products and product candidates obsolete. Our competitors may obtain patent protection, receive FDA approval, and commercialize medicines before us. Other companies are or may become engaged in the discovery of compounds that may compete with the product candidates we are developing.
While no oral medication has been approved by the FDA for PE, Priligy (dapoxetine) has been approved in some countries. Many commonly prescribed oral medications may delay orgasm and be prescribed alone or in combination with other treatments. These medications include antidepressants, treatments for erectile dysfunction and tramadol for the treatment of pain. We also are aware of topical products which are over-the-counter, or OTC, monograph products for premature ejaculation which include brands such as Promescent (Absorption Pharmaceuticals), a topical spray approved by the FDA in 2013, EjectDelay (Innovus Pharma) and PreBoost (Aspen Park Pharmaceuticals), all of which would compete with Zertane.
For the RedoxSYS System and ProstaScint, we also compete with companies that design, manufacture and market already existing and new in-vitro diagnostics systems and tests.
We anticipate that we will face increased competition in the future as new companies enter the market with new technologies and our competitors improve their current products. One or more of our competitors may offer technology superior to ours and render our technology obsolete or uneconomical. Most of our current competitors, as well as many of our potential competitors, have greater name recognition, more substantial intellectual property portfolios, longer operating histories, significantly greater resources to invest
54
Table of Contents
in new technologies, more substantial experience in new product development, greater regulatory expertise, more extensive manufacturing capabilities and the distribution channels to deliver products to customers. If we are not able to compete successfully, we may not generate sufficient revenue to become profitable.
Any new product that competes with a currently-approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and/or safety in order to address price competition and be commercially successful. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
Even if any of our product candidates are commercialized, they may not be accepted by physicians, patients, or the medical community in general.
Even if the medical community accepts a product as safe and efficacious for its indicated use, physicians may choose to restrict the use of the product if we or any collaborator is unable to demonstrate that, based on experience, clinical data, side-effect profiles and other factors, our product is preferable to any existing medicines or treatments. We cannot predict the degree of market acceptance of any product candidate that receives marketing approval, which will depend on a number of factors, including, but not limited to:
| • | the demonstration of the clinical efficacy and safety of the product; |
| • | the approved labeling for the product and any required warnings; |
| • | the advantages and disadvantages of the product compared to alternative treatments; |
| • | our and any collaborator’s ability to educate the medical community about the safety and effectiveness of the product; |
| • | the reimbursement policies of government and third-party payors pertaining to the product; and |
| • | the market price of our product relative to competing treatments. |
In the case of Zertane, tramadol hydrochloride is a well-established centrally acting synthetic analgesic that has been used for more than 30 years as a treatment for moderate to severe pain. As an opioid, tramadol hydrochloride has been associated with certain adverse effects including dizziness, nausea, constipation, vertigo, headache, vomiting and drowsiness. As a result, physicians may be reluctant to prescribe Zertane to treat premature ejaculation.
Zertane will, and our other product candidates may, contain controlled substances, the manufacture, use, sale, importation, exportation, prescribing and distribution of which are subject to regulation by the DEA.
Before we can commercialize Zertane, and potentially our other product candidates, the DEA will need to determine the controlled substance schedule, taking into account the recommendation of the FDA. This may be a lengthy process that could delay our marketing of a product candidate and could potentially diminish any regulatory exclusivity periods for which we may be eligible. Zertane will, and our other product candidates may, if approved, be regulated as “controlled substances” as defined in the Controlled Substances Act of 1970, or CSA, and the implementing regulations of the DEA, which establish registration, security, recordkeeping, reporting, storage, distribution, importation, exportation, inventory, quota and other requirements administered by the DEA. These requirements are applicable to us, to our third-party manufacturers and to distributors, prescribers and dispensers of our product candidates. The DEA regulates the handling of controlled substances through a closed chain of distribution. This control extends to the equipment and raw materials used in their manufacture and packaging, in order to prevent loss and diversion into illicit channels of commerce. A number of states and foreign countries also independently regulate these drugs as controlled substances.
The DEA regulates controlled substances as Schedule I, II, III, IV or V substances. Schedule I substances by definition have no established medicinal use, and may not be marketed or sold in the United States. A pharmaceutical product may be listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest risk of abuse and Schedule V substances the lowest relative risk of abuse among such substances.
We expect that Zertane will, and our other product candidates may, be listed by the DEA as Schedule IV controlled substances under the CSA. Consequently, the manufacturing, shipping, storing, selling and using of the products will be subject to a high degree of regulation. Also, distribution, prescribing and dispensing of these drugs are highly regulated.
Annual registration is required for any facility that manufactures, distributes, dispenses, imports or exports any controlled substance. The registration is specific to the particular location, activity and controlled substance schedule.
Because of their restrictive nature, these laws and regulations could limit commercialization of our product candidates containing controlled substances. Failure to comply with these laws and regulations could also result in withdrawal of our DEA registrations, disruption in manufacturing and distribution activities, consent decrees, criminal and civil penalties and state actions, among other consequences.
55
Table of Contents
Generic tramadol hydrochloride is available in the United States and abroad for treatment of pain. Although the generic drug is not available in the same dosage or formulation as Zertane for treatment of PE, it is possible that physicians could prescribe the generic version of the drug “off label” for the treatment of PE instead of Zertane, which would adversely affect our business.
Although Zertane is a specifically formulated, unique dosage of tramadol hydrochloride, generic tramadol hydrochloride is commercially available in the United States and abroad for treatment of pain. Although the generic drug is not available in the same dosage or form as Zertane for treatment of PE, it is possible that physicians could prescribe the generic version of the drug “off label” for the treatment of PE instead of Zertane, which would adversely affect our business. Patients could use generic tramadol hydrochloride dosages that are either higher or lower than what may be approved for Zertane or they could attempt to split dosages to arrive at reasonably similar dosages approved for Zertane. While any such “off label” use of generic tramadol hydrochloride for treatment of PE may constitute infringement of our patent portfolio, liability in that circumstance would be at the level of the physician or the patient making enforcement difficult or impractical.
Government restrictions on pricing and reimbursement, as well as other healthcare payor cost-containment initiatives, may negatively impact our ability to generate revenues if we obtain regulatory approval to market a product.
The continuing efforts of the government, insurance companies, managed care organizations and other payors of health care costs to contain or reduce costs of health care may adversely affect one or more of the following:
| • | our or our collaborators’ ability to set a price we believe is fair for our products, if approved; |
| • | our ability to generate revenues and achieve profitability; and |
| • | the availability of capital. |
The 2010 enactments of the Patient Protection and Affordable Care Act, or PPACA, and the Health Care and Education Reconciliation Act, or the Health Care Reconciliation Act, are expected to significantly impact the provision of, and payment for, health care in the United States. Various provisions of these laws have only recently taken effect or have yet to take effect, and are designed to expand Medicaid eligibility, subsidize insurance premiums, provide incentives for businesses to provide health care benefits, prohibit denials of coverage due to pre-existing conditions, establish health insurance exchanges, and provide additional support for medical research. Amendments to the PPACA and/or the Health Care Reconciliation Act, as well as new legislative proposals to reform healthcare and government insurance programs, along with the trend toward managed healthcare in the United States, could influence the purchase of medicines and medical devices and reduce demand and prices for our products and product candidates, if approved. This could harm our or our collaborators’ ability to market any products and generate revenues. Although we do not expect to receive significant revenues from reimbursement of our products by commercial third-party payors and government payors, cost containment measures that health care payors and providers are instituting and the effect of further health care reform could significantly reduce potential revenues from the sale of any of our products and product candidates approved in the future, and could cause an increase in our compliance, manufacturing, or other operating expenses. In addition, in certain foreign markets, the pricing of prescription drugs and devices is subject to government control and reimbursement may in some cases be unavailable. We believe that pricing pressures at the federal and state level, as well as internationally, will continue and may increase, which may make it difficult for us to sell any approved product at a price acceptable to us or any of our future collaborators.
In addition, in some foreign countries, the proposed pricing for a drug or medical c device must be approved before it may be lawfully marketed. The requirements governing pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. A member state may require that physicians prescribe the generic version of a drug instead of our approved branded product. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products or product candidates. Historically, pharmaceutical products launched in the European Union do not follow price structures of the United States and generally tend to have significantly lower prices.
Our products and product candidates may cause undesirable side effects that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other regulatory authorities. For example, adverse events associated with tramadol use in general and observed in the PE clinical trials included: gastrointestinal, or GI disorders (nausea) and central nervous system disorders (sleepiness, dizziness, headache) and decreased blood pressure.
56
Table of Contents
Further, if a product candidate receives marketing approval and we or others identify undesirable side effects caused by the product after the approval, or if drug abuse is determined to be a significant problem with an approved product, a number of potentially significant negative consequences could result, including:
| • | regulatory authorities may withdraw or limit their approval of the product; |
| • | regulatory authorities may require the addition of labeling statements, such as a “boxed” warning or a contraindication; |
| • | we may be required to change the way the product is distributed or administered, conduct additional clinical trials or change the labeling of the product; |
| • | we may decide to remove the product from the marketplace; |
| • | we could be sued and held liable for injury caused to individuals exposed to or taking the product; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidate and could substantially increase the costs of commercializing an affected product or product candidates and significantly impact our ability to successfully commercialize or maintain sales of our product or product candidates and generate revenues.
ProstaScint may prove to be difficult to effectively commercialize following our acquisition of the product from Jazz Pharmaceuticals.
Various commercial, regulatory, and manufacturing factors may impact our ability to maintain or grow revenues following our acquisition of ProstaScint in May 2015. Specifically, we may encounter difficulty by virtue of:
| • | our inability to secure continuing prescribing of ProstaScint by current or previous users of the product; |
| • | our inability to effectively transfer and scale manufacturing as needed to maintain an adequate commercial supply; |
| • | our inability to gain regulatory clearance required as the new distributor of the product in the U.S. and elsewhere where we seek to commercialize ProstaScint; |
| • | our inability to adequately resource a manufacturing site transfer, which, we expect, will be required in order to guarantee ongoing commercial supply; |
| • | reimbursement and medical policy changes that may adversely affect the pricing, profitability or commercial appeal of ProstaScint; and |
| • | our inability to effectively identify and align with commercial partners outside the United States, or the inability of those selected partners to gain the required regulatory, reimbursement, and other approvals needed to enable commercial success of ProstaScint. |
We may use hazardous chemicals and biological materials in our business. Any claims relating to improper handling, storage or disposal of these materials could be time consuming and costly.
Our research and development processes may involve the controlled use of hazardous materials, including chemicals and biological materials. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from these materials. We may be sued for any injury or contamination that results from our use or the use by third parties of these materials, and our liability may exceed any insurance coverage and our total assets. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous materials and specified waste products, as well as the discharge of pollutants into the environment and human health and safety matters. Compliance with environmental laws and regulations may be expensive and may impair our research and development efforts. If we fail to comply with these requirements, we could incur substantial costs, including civil or criminal fines and penalties, clean-up costs or capital expenditures for control equipment or operational changes necessary to achieve and maintain compliance. In addition, we cannot predict the impact on our business of new or amended environmental laws or regulations or any changes in the way existing and future laws and regulations are interpreted and enforced.
57
Table of Contents
The regulatory clearance or approval process is expensive, time consuming and uncertain, and the failure to obtain and maintain required clearances or approvals could prevent us from broadly commercializing the MiOXSYS System for clinical use.
The MiOXSYS System is subject to 510(k) clearance by the FDA prior to its marketing for commercial use in the United States, and to regulatory approvals required by foreign governmental entities prior to its marketing outside the United States. In addition, any changes or modifications to a device that has received regulatory clearance or approval that could significantly affect its safety or effectiveness, or would constitute a major change in its intended use, may require the submission of a new application for 510(k) clearance, pre-market approval, or foreign regulatory approvals. The 510(k) clearance and pre-market approval processes, as well as the process of obtaining foreign approvals, can be expensive, time consuming and uncertain. It generally takes from four to twelve months from submission to obtain 510(k) clearance, and from one to three years from submission to obtain pre-market approval; however, it may take longer, and 510(k) clearance or pre-market approval may never be obtained. We have limited experience in filing FDA applications for 510(k) clearance and pre-market approval. In addition, we are required to continue to comply with applicable FDA and other regulatory requirements even after obtaining clearance or approval. There can be no assurance that we will obtain or maintain any required clearance or approval on a timely basis, or at all. Any failure to obtain or any material delay in obtaining FDA clearance or any failure to maintain compliance with FDA regulatory requirements could harm our business, financial condition and results of operations.
Our financial results will depend on the acceptance among hospitals, third-party payors and the medical community of our products and product candidates.
Our future success depends on the acceptance by our target customers, third-party payors and the medical community that our products and product candidates are reliable, safe and cost-effective. Many factors may affect the market acceptance and commercial success of our products and product candidates, including:
| • | our ability to convince our potential customers of the advantages and economic value our products and product candidates over existing technologies and products; |
| • | the relative convenience and ease of our products and product candidates over existing technologies and products; |
| • | the introduction of new technologies and competing products that may make our products and product candidates a less attractive for our target customers; |
| • | our success in training medical personnel on the proper use of our products and product candidates; |
| • | the willingness of third-party payors to reimburse our target customers that adopt our products and product candidates; |
| • | the acceptance in the medical community of our products and product candidates; |
| • | the extent and success of our marketing and sales efforts; and |
| • | general economic conditions |
If third-party payors do not reimburse our customers for the products we may sell or if reimbursement levels are set too low for us to sell one or more of our products at a profit, our ability to sell those products and our results of operations will be harmed.
While ProstaScint is already FDA-approved and generating revenues in the U.S., the product may not continue to receive physician, hospital, or laboratory acceptance, or it may not maintain adequate reimbursement from third party payors. Additionally, even if one of our product candidates is approved and reaches the market, the product may not achieve physician, hospital, or laboratory acceptance, or it may not obtain adequate reimbursement from third party payors. We expect to sell our products and product candidates to target customers substantially all of whom receive reimbursement for the health care services they provide to their patients from third-party payors, such as Medicare, Medicaid, other domestic and foreign government programs, private insurance plans and managed care programs. Reimbursement decisions by particular third-party payors depend upon a number of factors, including each third-party payor’s determination that use of a product is:
| • | a covered benefit under its health plan; |
| • | appropriate and medically necessary for the specific indication; |
| • | cost effective; and |
| • | neither experimental nor investigational. |
Third-party payors may deny reimbursement for covered products if they determine that a medical product was not used in accordance with cost-effective diagnosis methods, as determined by the third-party payor, or was used for an unapproved indication. Third-party payors also may refuse to reimburse for procedures and devices deemed to be experimental.
Obtaining coverage and reimbursement approval for a product from each government or third- party payor is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our potential product to each government or third-party payor. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. In addition, eligibility for coverage does not imply that any product will be covered and reimbursed in all cases or reimbursed at a rate that allows our potential customers to make a profit or even cover their costs.
58
Table of Contents
Third-party payors are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement for medical products and services. Levels of reimbursement may decrease in the future, and future legislation, regulation or reimbursement policies of third-party payors may adversely affect the demand for and reimbursement available for any product or product candidate, which in turn, could negatively impact pricing. If our customers are not adequately reimbursed for our products, they may reduce or discontinue purchases of our products, which would result in a significant shortfall in achieving revenue expectations and negatively impact our business, prospects and financial condition.
Manufacturing risks and inefficiencies may adversely affect our ability to produce our products.
As part of the acquisition of ProstaScint from Jazz Pharmaceuticals, we expect to resume the relationship with the third-party manufacturer of ProstaScint. We expect to transition the site of manufacture within the current manufacturer’s qualified sites. During this transition we may not be able to supply sufficient quantities and on a timely basis, while maintaining product quality, acceptable manufacturing costs and complying with regulatory requirements, such as quality system regulations. In addition, we expect to engage third parties to manufacture components of the RedoxSYS and MiOXSYS systems. For any future product, we expect to use third-party manufacturers because we do not have our own manufacturing capabilities. In determining the required quantities of any product and the manufacturing schedule, we must make significant judgments and estimates based on inventory levels, current market trends and other related factors. Because of the inherent nature of estimates and our limited experience in marketing any products, there could be significant differences between our estimates and the actual amounts of product we require. If we do not effectively transition sites with our manufacturing and development partners to enable to production scale of ProstaScint, or if we do not secure collaborations with manufacturing and development partners to enable production to scale of the RedoxSYS and MiOXSYS systems, we may not be successful in selling ProstaScint or in commercializing the RedoxSYS and MiOXSYS systems in the event we receive regulatory approval of the MiOXSYS System. If we fail in similar endeavors for future products, we may not be successful in establishing or continuing the commercialization of our products and product candidates.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured these components ourselves, including:
| • | reliance on third parties for regulatory compliance and quality assurance; |
| • | possible breaches of manufacturing agreements by the third parties because of factors beyond our control; |
| • | possible regulatory violations or manufacturing problems experienced by our suppliers; and |
| • | possible termination or non-renewal of agreements by third parties, based on their own business priorities, at times that are costly or inconvenient for us. |
Further, if we are unable to secure the needed financing to fund our internal operations, we may not have adequate resources required to effectively and rapidly transition our site of ProstaScint manufacture. We may not be able to meet the demand for the RedoxSYS System if one or more of any third-party manufacturers is not able to supply us with the necessary components that meet our specifications. It may be difficult to find alternate suppliers for any of our products or product candidates in a timely manner and on terms acceptable to us.
Any third-party manufacturers we engage are subject to various governmental regulations, and we may incur significant expenses to comply with, and experience delays in our product commercialization as a result of these regulations.
The manufacturing processes and facilities of third-party manufacturers we engage are required to comply with the federal Quality System Regulation, or QSR, which covers procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of devices. The FDA enforces the QSR through periodic unannounced inspections of manufacturing facilities. Any inspection by the FDA could lead to additional compliance requests that could cause delays in our product commercialization. Failure to comply with applicable FDA requirements, or later discovery of previously unknown problems with the manufacturing processes and facilities of third-party manufacturers we engage, including the failure to take satisfactory corrective actions in response to an adverse QSR inspection, can result in, among other things:
| • | administrative or judicially imposed sanctions; |
| • | injunctions or the imposition of civil penalties; |
| • | recall or seizure of the product in question; |
| • | total or partial suspension of production or distribution; |
| • | the FDA’s refusal to grant pending future clearance or pre-market approval; |
| • | withdrawal or suspension of marketing clearances or approvals; |
| • | clinical holds; |
59
Table of Contents
| • | warning letters; |
| • | refusal to permit the export of the product in question; and |
| • | criminal prosecution. |
Any of these actions, in combination or alone, could prevent us from marketing, distributing or selling the RedoxSYS System or ProstaScint or any other approved product, and would likely harm our business.
In addition, a product defect or regulatory violation could lead to a government-mandated or voluntary recall by us. We believe the FDA would request that we initiate a voluntary recall if a product was defective or presented a risk of injury or gross deception. Regulatory agencies in other countries have similar authority to recall drugs or devices because of material deficiencies or defects in design or manufacture that could endanger health. Any recall would divert our management attention and financial resources, expose us to product liability or other claims, and harm our reputation with customers.
We have limited experience in sales and marketing and may be unable to successfully commercialize our products and product candidates.
As a company, we have limited marketing, sales and distribution experience and capabilities. Our ability to achieve profitability depends on attracting and retaining customers for our products and product candidates, and building brand loyalty. To successfully perform sales, marketing, distribution and customer support functions, we will face a number of risks, including:
| • | our ability to attract and retain skilled support team, marketing staff and sales force necessary to increase the market for our approved products and to commercialize and gain market acceptance for our product candidates; |
| • | the ability of our sales and marketing team to identify and penetrate the potential customer base; and |
| • | the difficulty of establishing brand recognition and loyalty for our diagnostic products. |
In addition, we may seek to enlist one or more third parties to assist with sales, distribution and customer support globally or in certain regions of the world. If we do seek to enter into these arrangements, we may not be successful in attracting desirable sales and distribution partners, or we may not be able to enter into these arrangements on favorable terms, or at all. If our sales and marketing efforts, or those of any third-party sales and distribution partners, are not successful, our currently approved products may not achieve increased market acceptance and our product candidates may not gain market acceptance, which would materially impact our business and operations.
We face intense competition from established and new companies in the in-vitro diagnostics field.
We compete with companies that design, manufacture and market already-existing and new in-vitro diagnostics systems and tests. We anticipate that we will face increased competition in the future as new companies enter the market with new technologies and our competitors improve their current products. One or more of our competitors may offer technology superior to ours and render our technology obsolete or uneconomical. Most of our current competitors, as well as many of our potential competitors, have greater name recognition, more substantial intellectual property portfolios, longer operating histories, significantly greater resources to invest in new technologies, more substantial experience in new product development, greater regulatory expertise, more extensive manufacturing capabilities and the distribution channels to deliver products to customers. If we are not able to compete successfully, we may not generate sufficient revenue to become profitable.
Our future growth depends, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability will depend, in part, on our ability to commercialize our products and product candidates in foreign markets for which we intend to primarily rely on collaboration with third parties. If we commercialize our products or product candidates in foreign markets, we would be subject to additional risks and uncertainties, including:
| • | our inability to directly control commercial activities because we are relying on third parties; |
| • | the burden of complying with complex and changing foreign regulatory, tax, accounting and legal requirements; |
| • | different medical practices and customs in foreign countries affecting acceptance in the marketplace; |
| • | import or export licensing requirements; |
| • | longer accounts receivable collection times; |
| • | longer lead times for shipping; |
| • | language barriers for technical training; |
60
Table of Contents
| • | reduced protection of intellectual property rights in some foreign countries, and related prevalence of generic alternatives to our products; |
| • | foreign currency exchange rate fluctuations; |
| • | our customers’ ability to obtain reimbursement for our products in foreign markets; and |
| • | the interpretation of contractual provisions governed by foreign laws in the event of a contract dispute. |
Foreign sales of our products or product candidates could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs.
We are subject to various regulations pertaining to the marketing of our products.
We are subject to various federal and state laws pertaining to healthcare fraud and abuse, including prohibitions on the offer of payment or acceptance of kickbacks or other remuneration for the purchase of our products, including inducements to potential patients to request our products and services. Additionally, any product promotion educational activities, support of continuing medical education programs, and other interactions with health-care professionals must be conducted in a manner consistent with the FDA regulations and the Anti-Kickback Statute. The Anti-Kickback Statute prohibits persons or entities from knowingly and willfully soliciting, receiving, offering or providing remuneration, directly or indirectly, to induce either the referral of an individual, or the furnishing, recommending, or arranging for a good or service, for which payment may be made under a federal healthcare program such as the Medicare and Medicaid programs. Violations of the Anti-Kickback Statute can also carry potential federal False Claims Act liability. Additionally, many states have adopted laws similar to the Anti-Kickback Statute. Some of these state prohibitions apply to referral of patients for healthcare items or services reimbursed by any third-party payer, not only the Medicare and Medicaid programs, and do not contain identical safe harbors. These and any new regulations or requirements may be difficult and expensive for us to comply with, may adversely impact the marketing of our existing products or delay introduction of our product candidates, which may have a material adverse effect on our business, operating results and financial condition.
Intellectual Property Risks Related to Our Business
Our ability to compete may decline if we do not adequately protect our proprietary rights.
Our commercial success depends on obtaining and maintaining proprietary rights to our products and product candidates as well as successfully defending these rights against third-party challenges. We will only be able to protect our products and product candidates from unauthorized use by third parties to the extent that valid and enforceable patents, or effectively protected trade secrets, cover them. Our ability to obtain patent protection for our products and product candidates is uncertain due to a number of factors, including:
| • | we may not have been the first to make the inventions covered by pending patent applications or issued patents; |
| • | we may not have been the first to file patent applications for our products and product candidates; |
| • | others may independently develop identical, similar or alternative products, compositions or devices and uses thereof; |
| • | our disclosures in patent applications may not be sufficient to meet the statutory requirements for patentability; |
| • | any or all of our pending patent applications may not result in issued patents; |
| • | we may not seek or obtain patent protection in countries that may eventually provide us a significant business opportunity; |
| • | any patents issued to us may not provide a basis for commercially viable products, may not provide any competitive advantages, or may be successfully challenged by third parties; |
| • | our compositions, devices and methods may not be patentable; |
| • | others may design around our patent claims to produce competitive products which fall outside of the scope of our patents; or |
| • | others may identify prior art or other bases which could invalidate our patents. |
Even if we have or obtain patents covering our products and product candidates, we may still be barred from making, using and selling them because of the patent rights of others. Others may have filed, and in the future may file, patent applications covering products that are similar or identical to ours. There are many issued U.S. and foreign patents relating to chemical compounds, therapeutic products, diagnostic devices, and some of these relate to our products and product candidates. These could materially affect our ability to sell our products and develop our product candidates. Because patent applications can take many years to issue, there may be currently pending applications unknown to us that may later result in issued patents that our products and product candidates may infringe. These patent applications may have priority over patent applications filed by us.
61
Table of Contents
Obtaining and maintaining a patent portfolio entails significant expense and resources. Part of the expense includes periodic maintenance fees, renewal fees, annuity fees, various other governmental fees on patents and/or applications due in several stages over the lifetime of patents and/or applications, as well as the cost associated with complying with numerous procedural provisions during the patent application process. We may or may not choose to pursue or maintain protection for particular inventions. In addition, there are situations in which failure to make certain payments or noncompliance with certain requirements in the patent process can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If we choose to forgo patent protection or allow a patent application or patent to lapse purposefully or inadvertently, our competitive position could suffer.
Legal actions to enforce our patent rights can be expensive and may involve the diversion of significant management time. In addition, these legal actions could be unsuccessful and could also result in the invalidation of our patents or a finding that they are unenforceable. We may or may not choose to pursue litigation or other actions against those that have infringed on our patents, or used them without authorization, due to the associated expense and time commitment of monitoring these activities. If we fail to protect or to enforce our intellectual property rights successfully, our competitive position could suffer, which could harm our business, prospects, financial condition and results of operations.
Pharmaceutical and medical device patents and patent applications involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent positions of pharmaceutical and medical device companies can be highly uncertain and involve complex legal and factual questions. The interpretation and breadth of claims allowed in some patents covering pharmaceutical compositions may be uncertain and difficult to determine, and are often affected materially by the facts and circumstances that pertain to the patented compositions and the related patent claims. The standards of the United States Patent and Trademark Office, or USPTO, are sometimes uncertain and could change in the future. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to re-examination proceedings, post-grant review and/or inter partes review in the USPTO. Foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, re-examination, post-grant review, inter partes review and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
In addition, changes in or different interpretations of patent laws in the United States and foreign countries may permit others to use our discoveries or to develop and commercialize our technology and products and product candidates without providing any compensation to us, or may limit the number of patents or claims we can obtain. The laws of some countries do not protect intellectual property rights to the same extent as U.S. laws and those countries may lack adequate rules and procedures for defending our intellectual property rights.
If we fail to obtain and maintain patent protection and trade secret protection of our products and product candidates, we could lose our competitive advantage and competition we face would increase, reducing any potential revenues and adversely affecting our ability to attain or maintain profitability.
Developments in patent law could have a negative impact on our business.
From time to time, the United States Supreme Court, other federal courts, the United States Congress or the USPTO may change the standards of patentability and any such changes could have a negative impact on our business.
In addition, the Leahy-Smith America Invents Act, or the America Invents Act, which was signed into law in 2011, includes a number of significant changes to U.S. patent law. These changes include a transition from a “first-to-invent” system to a “first-to-file” system, changes the way issued patents are challenged, and changes the way patent applications are disputed during the examination process. These changes may favor larger and more established companies that have greater resources to devote to patent application filing and prosecution. The USPTO has developed regulations and procedures to govern the full implementation of the America Invents Act, and many of the substantive changes to patent law associated with the America Invents Act, and, in particular, the first-to-file provisions, became effective on March 16, 2013. Substantive changes to patent law associated with the America Invents Act may affect our ability to obtain patents, and if obtained, to enforce or defend them. Accordingly, it is not clear what, if any, impact the America
62
Table of Contents
Invents Act will ultimately have on the cost of prosecuting our patent applications, our ability to obtain patents based on our discoveries and our ability to enforce or defend any patents that may issue from our patent applications, all of which could have a material adverse effect on our business.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to patent protection, because we operate in the highly technical field of discovery and development of therapies and medical devices, we rely in part on trade secret protection in order to protect our proprietary technology and processes. However, trade secrets are difficult to protect. We expect to enter into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific and commercial collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. These agreements also generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us.
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, provide adequate protection for our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. Trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our competitive position could be harmed.
We may not be able to enforce our intellectual property rights throughout the world.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to pharmaceuticals and medical devices. This could make it difficult for us to stop the infringement of some of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property.
Third parties may assert ownership or commercial rights to inventions we develop.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. We have or expect to have written agreements with collaborators that provide for the ownership of intellectual property arising from our collaborations. These agreements provide that we must negotiate certain commercial rights with collaborators with respect to joint inventions or inventions made by our collaborators that arise from the results of the collaboration. In some instances, there may not be adequate written provisions to address clearly the resolution of intellectual property rights that may arise from a collaboration. If we cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from our use of a third-party collaborator’s materials where required, or if disputes otherwise arise with respect to the intellectual property developed with the use of a collaborator’s samples, we may be limited in our ability to capitalize on the market potential of these inventions. In addition, we may face claims by third parties that our agreements with employees, contractors, or consultants obligating them to assign intellectual property to us are ineffective, or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such inventions. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property, or may lose our exclusive rights in that intellectual property. Either outcome could have an adverse impact on our business.
63
Table of Contents
Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
We might employ individuals who were previously employed at universities or other biopharmaceutical or medical device companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
A dispute concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be time consuming and costly, and an unfavorable outcome could harm our business.
There is significant litigation in the pharmaceutical and medical device industries regarding patent and other intellectual property rights. While we are not currently subject to any pending intellectual property litigation, and are not aware of any such threatened litigation, we may be exposed to future litigation by third parties based on claims that our products or product candidates infringe the intellectual property rights of others. If our development and commercialization activities are found to infringe any such patents, we may have to pay significant damages or seek licenses to such patents. A patentee could prevent us from using the patented drugs, compositions or devices. We may need to resort to litigation to enforce a patent issued to us, to protect our trade secrets, or to determine the scope and validity of third-party proprietary rights. From time to time, we may hire scientific personnel or consultants formerly employed by other companies involved in one or more areas similar to the activities conducted by us. Either we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of prior affiliations. If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources, regardless of whether we win or lose. We may not be able to afford the costs of litigation. Any adverse ruling or perception of an adverse ruling in defending ourselves against these claims could have a material adverse impact on our cash position and stock price. Any legal action against us or our collaborators could lead to:
| • | payment of damages, potentially treble damages, if we are found to have willfully infringed a party’s patent rights; |
| • | injunctive or other equitable relief that may effectively block our ability to further develop, commercialize, and sell products; or |
| • | we or our collaborators having to enter into license arrangements that may not be available on commercially acceptable terms, if at all, all of which could have a material adverse impact on our cash position and business, prospects and financial condition. As a result, we could be prevented from commercializing our products and product candidates. |
Risks Related to Our Organization, Structure and Operation
Ampio controls us, including having the ability to control the election of our directors, and its interests may conflict with or differ from your interests as stockholders.
As of June 30, 2015, Ampio owned 81.5% of our outstanding common stock. If Ampio were to choose so, as a result of its stock ownership, Ampio may be able to influence our management and affairs and control all matters submitted to our stockholders for approval, including the election of directors and approval of any merger, consolidation, or sale of all or substantially all of our assets or other major corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control of our company and might affect the market price of our common stock. This control may delay, deter or prevent acts that would be favored by our other stockholders, as the interests of Ampio may not always coincide with our interests or the interests of our other stockholders. For example, Ampio may seek to cause us to take courses of action that, in its judgment, could enhance its investment in us, but which might involve risks to our other stockholders or adversely affect us or our other stockholders.
We may be unable to achieve some or all of the benefits that we expect to achieve from our separation from Ampio.
As a stand-alone, independent public company, we believe that our business will benefit from, among other things, allowing our management to design and implement corporate policies and strategies that are based primarily on the characteristics of our business, allowing us to focus our financial resources wholly on our own operations and implement and maintain a capital structure designed to meet our own specific needs. However, as a result of our separation from Ampio in April 2015 there is a risk that we may be more susceptible to market fluctuations and other adverse events than we would have been if we were still a part of Ampio. We may not be able to achieve some or all of the benefits that we expect to achieve as a stand-alone healthcare company or such benefits may be delayed or may not occur at all. For example, there can be no assurance that analysts and investors will place a greater value on our company as a stand-alone healthcare company than on our business as a part of Ampio.
64
Table of Contents
Our historical financial information as a business conducted by Ampio may not be representative of our results as an independent public company.
The historical financial information included or incorporated herein does not necessarily reflect what our financial position, operating results or cash flows would have been had we been an independent entity during the historical periods presented. The historical costs and expenses reflected in our financial statements include amounts for certain corporate functions historically provided by Ampio, including costs of finance and other administrative services, and income taxes. These expense allocations were developed on the basis of what we and Ampio considered to be reasonable prices for the utilization of services provided or the benefits received by us. The historical financial information in our audited financial statements may not be indicative of what our results of operations, financial position, changes in equity and cash flows would have been had we been a separate stand-alone entity during the periods presented or will be in the future. We have not made adjustments to reflect many significant changes that will occur in our cost structure, funding and operations as a result of our separation from Ampio, including changes in our employee base, changes in our tax structure, potential increased costs associated with reduced economies of scale and increased costs associated with being a publicly traded, stand-alone company, such as audit fees, directors and officers insurance costs and compliance costs, nor have we made offsetting adjustments to reflect the benefits of this offering, as these factors are presently difficult to quantify. These same risks will apply to the financial information of the ProstaScint business when it is included in our financial statements.
We may have received better terms from unaffiliated third parties than the terms we received in our agreements with Ampio.
The agreements related to our separation from Ampio, including the assignment and assumption agreement, services agreement and the other agreements, were negotiated in the context of our separation from Ampio while we were still part of Ampio and, accordingly, may not reflect terms that would have resulted from arm’s-length negotiations among unaffiliated third parties. The terms of the agreements we negotiated in the context of our separation related to, among other things, allocation of assets, liabilities, rights, indemnifications and other obligations among Ampio and us. We may have received better terms from third parties because third parties may have competed with each other to win our business. Our sole director is also a member of the Ampio board.
Our ability to operate our business effectively may suffer if we or Ampio terminate our services agreement, or if we are unable to establish on a cost-effective basis our own administrative and other support functions in order to operate as a stand-alone company after the expiration or termination of our services agreement with Ampio.
Prior to the Merger, we relied on administrative and other resources of Ampio to operate our business. We have entered into a services agreement to retain the ability for specified periods to use certain Ampio resources. We may elect to continue this agreement for an indefinite period of time. Any decision by us to terminate this agreement would be approved by disinterested members of our management and board of directors under our procedures regarding related party transactions. After the termination of this agreement, we would need to create our own administrative and other support systems or contract with third parties to replace Ampio’s services. These services may not be provided at the same level, and we may not be able to obtain the same benefits that we received prior to the separation. These services may not be sufficient to meet our needs, and if our agreement with Ampio is terminated, we may not be able to replace these services at all or obtain these services at prices and on terms as favorable as we currently have with Ampio. Any failure or significant downtime in our own administrative systems or in Ampio’s administrative systems during the transitional period could result in unexpected costs, impact our results or prevent us from paying our suppliers or employees and performing other administrative services on a timely basis.
Adverse developments at Ampio could negatively impact our company.
We acquired the businesses of Vyrix (Zertane) and Luoxis (the RedoxSYS and MiOXSYS systems) from Ampio in the Merger. In addition, Joshua Disbrow and Jarrett Disbrow held executive positions at Ampio and/or its subsidiaries prior to the Merger. Further, at June 30, 2015, Ampio owned approximately 81.5% of our outstanding common stock. As a result, negative developments, including negative publicity, at Ampio could be imputed to our company and have an adverse impact on our business and prospects, including our ability to raise capital or enter into collaborations, and on the price of our common stock.
Third parties may seek to hold us responsible for liabilities of Ampio that we did not assume in our agreements.
In connection with our separation from Ampio, Ampio has generally agreed to retain all liabilities that did not historically arise from our business. Third parties may seek to hold us responsible for Ampio’s retained liabilities. Under our agreements with Ampio, Ampio has agreed to indemnify us for claims and losses relating to these retained liabilities. However, if those liabilities are significant and we are ultimately liable for them, we cannot assure you that we will be able to recover the full amount of our losses from Ampio.
65
Table of Contents
Any disputes that arise between us and Ampio with respect to our past and ongoing relationships could harm our business operations.
Disputes may arise between Ampio and us in a number of areas relating to our past and ongoing relationships, including:
| • | intellectual property, technology and business matters, including failure to make required technology transfers and failure to comply with non-compete provisions applicable to Ampio and us; |
| • | labor, tax, employee benefit, indemnification and other matters arising from our separation from Ampio; |
| • | distribution and supply obligations; |
| • | employee retention and recruiting; |
| • | business combinations involving us; |
| • | sales or distributions by Ampio of all or any portion of its ownership interest in us; |
| • | the nature, quality and pricing of transitional services Ampio has agreed to provide us; and |
| • | business opportunities that may be attractive to both Ampio and us. |
We may not be able to resolve any potential conflicts, and even if we do, the resolution may be less favorable than if we were dealing with an unaffiliated party.
The agreements we have entered into with Ampio may be amended upon agreement between the parties. While we are controlled by Ampio, Ampio may be able to require us to agree to amendments to these agreements that may be less favorable to us than the original terms of the agreements.
Some of our management may have conflicts of interest because of their ownership of Ampio common stock, options to acquire Ampio common stock and positions with Ampio.
Our sole director and three of our executive officers own Ampio common stock and options to purchase Ampio common stock. In addition, our sole director is also the Chief Executive Officer and a director of Ampio and our Chief Financial Officer is also the Chief Financial Officer of Ampio. Ownership of Ampio common stock and options to purchase Ampio common stock by our director and officers and the presence of a director of Ampio on our board of directors could create, or appear to create, conflicts of interest with respect to matters involving both us and Ampio. For example, corporate opportunities may arise that are applicable or complementary to both of our businesses and that each business would be free to pursue, such as the potential acquisition of a particular business or technology. However, we do not believe that Ampio intends to acquire businesses that are focused on urological disorders. We have not established at this time any procedural mechanisms to address actual or perceived conflicts of interest of these individuals and expect that our board of directors, in the exercise of its fiduciary duties, will determine how to address any actual or perceived conflicts of interest on a case-by-case basis. If any corporate opportunity arises and if our sole director or officers do not pursue it on our behalf, we may not become aware of, and may potentially lose, a significant business opportunity.
We will need to develop and expand our company, and we may encounter difficulties in managing this development and expansion, which could disrupt our operations.
As of June 30, 2015, we had nine full-time employees, and in connection with being a public company, we expect to increase our number of employees and the scope of our operations. To manage our anticipated development and expansion, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Also, our management may need to divert a disproportionate amount of its attention away from its day-to-day activities and devote a substantial amount of time to managing these development activities. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. This may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. The physical expansion of our operations may lead to significant costs and may divert financial resources from other projects, such as the planned expanded commercialization of our approved products and the development of our product candidates. If our management is unable to effectively manage our expected development and expansion, our expenses may increase more than expected, our ability to generate or increase our revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to expand the market for our approved products and develop our product candidates, if approved, and compete effectively will depend, in part, on our ability to effectively manage the future development and expansion of our company.
66
Table of Contents
We depend on key personnel and attracting qualified management personnel and our business could be harmed if we lose personnel and cannot attract new personnel.
Our success depends to a significant degree upon the technical and management skills of our officers and key personnel, including in particular those of Joshua Disbrow, our Chief Executive Officer, and Jarrett Disbrow, our Chief Operating Officer. The loss of the services of any of these individuals would likely have a material adverse effect on us. Our success also will depend upon our ability to attract and retain additional qualified management, marketing, technical, and sales executives and personnel. We do not maintain key person life insurance for any of our officers or key personnel. The loss of any of our key executives, or the failure to attract, integrate, motivate, and retain additional key personnel could have a material adverse effect on our business.
We compete for such personnel against numerous companies, including larger, more established companies with significantly greater financial resources than we possess. There can be no assurance that we will be successful in attracting or retaining such personnel, and the failure to do so could have a material adverse effect on our business, prospects, financial condition, and results of operations.
Product liability and other lawsuits could divert our resources, result in substantial liabilities and reduce the commercial potential of our product candidates.
The risk that we may be sued on product liability claims is inherent in the development and commercialization of pharmaceutical and medical device products. Side effects of, or manufacturing defects in, products that we develop and commercialized could result in the deterioration of a patient’s condition, injury or even death. Once a product is approved for sale and commercialized, the likelihood of product liability lawsuits increases. Claims may be brought by individuals seeking relief for themselves or by individuals or groups seeking to represent a class. These lawsuits may divert our management from pursuing our business strategy and may be costly to defend. In addition, if we are held liable in any of these lawsuits, we may incur substantial liabilities and may be forced to limit or forgo further commercialization of the affected products.
We may be subject to legal or administrative proceedings and litigation other than product liability lawsuits which may be costly to defend and could materially harm our business, financial condition and operations.
Although we expect to maintain general liability, clinical trial liability and product liability insurance, this insurance may not fully cover potential liabilities. In addition, inability to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product or other legal or administrative liability claims could prevent or inhibit the commercial production and sale of any of our products and product candidates that receive regulatory approval, which could adversely affect our business. Product liability claims could also harm our reputation, which may adversely affect our collaborators’ ability to commercialize our products successfully.
In order to satisfy our obligations as a public company, we may need to hire additional qualified accounting and financial personnel with appropriate public company experience in the event that we no longer utilize the finance and administrative functions of Ampio.
As a public company, we must establish and maintain effective disclosure and financial controls. We may need to hire additional accounting and financial personnel with appropriate public company experience and technical accounting knowledge, and it may be difficult to recruit and maintain such personnel. Even if we are able to hire appropriate personnel, our existing operating expenses and operations will be impacted by the direct costs of their employment and the indirect consequences related to the diversion of management resources from product development efforts.
Our internal computer systems, or those of our third-party contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Despite the implementation of security measures, our internal computer systems and those of our third-party contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we do not believe that we have experienced any such system failure, accident, or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a loss of clinical trial data for our product candidates which could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications or other data or applications relating to our technology or product candidates, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and the further development of our product candidates could be delayed.
We may acquire businesses or products, or form strategic alliances, in the future, and we may not realize the benefits of such acquisitions.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and
67
Table of Contents
company culture. We may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected synergies to justify the transaction. These risks apply to our acquisition of ProstaScint in May 2015.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
As of June 30, 2015, we had federal net operating loss carryforwards of approximately $17.1 million. The available net operating losses, if not utilized to offset taxable income in future periods, will begin to expire in 2031 and will completely expire in 2034. Under the Internal Revenue Code of 1986, as amended (the “Code”) and the regulations promulgated thereunder, including, without limitation, the consolidated income tax return regulations, various corporate changes could limit our ability to use our net operating loss carryforwards and other tax attributes (such as research tax credits) to offset our income. Were Ampio’s equity ownership interest in our Company to fall below 80% and we were to be deconsolidated from Ampio’s consolidated federal income tax group, certain of our net operating loss carryforwards may not be available to us and we would not be able to use them to offset our U.S. federal taxable income. In the event of such a deconsolidation, it is possible that certain other tax attributes and benefits resulting from U.S. federal income tax consolidation may no longer be available to us. Our Company and Ampio do not have a tax sharing agreement that could mitigate the loss of net operating losses and other tax attributes resulting from a deconsolidation or our incurrence of liability for the taxes of other members of the consolidated group by reason of the joint and several liability of group members. In addition to the deconsolidation risk, an “ownership change” (generally a 50% change (by value) in equity ownership over a three-year period) under Section 382 of the Code could limit our ability to offset, post-change, our U.S. federal taxable income. Section 382 of the Code imposes an annual limitation on the amount of post-ownership change taxable income a corporation may offset with pre-ownership change net operating loss carryforwards and certain recognized built-in losses. Either the deconsolidation or the ownership change scenario could result in increased future tax liability to us.
Risks Related to Securities Markets and Investment in our Common Stock
There is not now, and there may never be, an active, liquid and orderly trading market for our common stock.
There is not now, nor has there been since our inception, any substantial trading activity in our common stock or a market for shares of our common stock, and an active trading market for our shares may never develop or be sustained. As a result, investors in our common stock must bear the economic risk of holding those shares for an indefinite period of time. Although our common stock is quoted on the OTCQB, an over-the-counter quotation system, trading of our common stock is extremely limited and sporadic and at very low volumes. We do not now, and may not in the future, meet the initial listing standards of any national securities exchange, and we presently anticipate that our common stock will continue to be quoted on the OTCQB or another over-the-counter quotation system in the foreseeable future. In those venues, our stockholders may find it difficult to obtain accurate quotations as to the market value of their shares of our common stock, and may find few buyers to purchase their stock and few market makers to support its price. As a result of these and other factors, you may be unable to resell your shares of our common stock at or above the price for which you purchased them, or at all. Further, an inactive market may also impair our ability to raise capital by selling additional equity in the future, and may impair our ability to enter into strategic partnerships or acquire companies or products by using shares of our common stock as consideration.
Our share price is volatile and may be influenced by numerous factors, some of which are beyond our control.
The trading price of our common stock is likely to be highly volatile, and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this report, these factors include:
| • | the products or product candidates we acquire for commercialization; |
| • | the product candidates we seek to pursue, and our ability to obtain rights to develop, commercialize and market those product candidates; |
| • | our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; |
| • | actual or anticipated adverse results or delays in our clinical trials; |
| • | our failure to commercialize our product candidates, if approved; |
| • | unanticipated serious safety concerns related to the use of any of our product candidates; |
| • | adverse regulatory decisions; |
68
Table of Contents
| • | additions or departures of key scientific or management personnel; |
| • | changes in laws or regulations applicable to our product candidates, including without limitation clinical trial requirements for approvals; |
| • | disputes or other developments relating to patents and other proprietary rights and our ability to obtain patent protection for our product candidates; |
| • | our dependence on third parties, including CROs and scientific and medical advisors; |
| • | failure to meet or exceed any financial guidance or expectations regarding development milestones that we may provide to the public; |
| • | actual or anticipated variations in quarterly operating results; |
| • | failure to meet or exceed the estimates and projections of the investment community; |
| • | overall performance of the equity markets and other factors that may be unrelated to our operating performance or the operating performance of our competitors, including changes in market valuations of similar companies; |
| • | conditions or trends in the healthcare, biotechnology and pharmaceutical industries; |
| • | introduction of new products offered by us or our competitors; |
| • | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
| • | our ability to maintain an adequate rate of growth and manage such growth; |
| • | issuances of debt or equity securities; |
| • | sales of our common stock by us or our stockholders in the future, or the perception that such sales could occur; |
| • | trading volume of our common stock; |
| • | ineffectiveness of our internal control over financial reporting or disclosure controls and procedures; |
| • | general political and economic conditions; |
| • | effects of natural or man-made catastrophic events; and |
| • | other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and the stocks of small-cap healthcare, biotechnology and pharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors,” could have a dramatic and material adverse impact on the market price of our common stock.
FINRA sales practice requirements may limit a stockholder’s ability to buy and sell our stock.
The Financial Industry Regulatory Authority, or FINRA, has adopted rules requiring that, in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative or low-priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA has indicated its belief that there is a high probability that speculative or low-priced securities will not be suitable for at least some customers. If these FINRA requirements are applicable to us or our common stock, they may make it more difficult for broker-dealers to recommend that at least some of their customers buy our common stock, which may limit the ability of our stockholders to buy and sell our common stock and could have an adverse effect on the market for and price of our common stock.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and any trading volume could decline.
Any trading market for our common stock that may develop will depend in part on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on us or our business. If no securities or industry analysts commence coverage of our company, the trading price for our stock would be negatively affected. If securities or industry analysts initiate coverage, and one or more of those analysts downgrade our stock or
69
Table of Contents
publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and any trading volume to decline.
We may have material liabilities that have not been discovered as a result of the Merger.
As a result of the Merger, the former business plan and management of Rosewind have been abandoned and replaced with the business and management team of Vyrix and Luoxis. Prior to the Merger, there were no relationships or other connections among the businesses or individuals associated with Rosewind and Vyrix and Luoxis. As a result, we may have material liabilities that have not been discovered as a result of the Merger. We could experience losses as a result of any such undisclosed liabilities that are discovered, which could materially harm our business, prospects and financial condition. Although the Merger Agreement contains customary representations and warranties from Rosewind concerning its assets, liabilities, financial condition and affairs, there may be limited or no recourse against Rosewind’s pre-Merger stockholders or principals in the event those representations prove to be untrue. As a result, our stockholders will bear some, or all, of the risks relating to any such unknown or undisclosed liabilities.
We may be exposed to additional risks as a result of “going public” by means of a merger transaction.
We may be exposed to additional risks because the business of Vyrix and Luoxis has become a public company through a “reverse merger” transaction. There has been increased focus by government agencies on transactions such as the Merger in recent years, and we may be subject to increased scrutiny by the SEC and other government agencies and holders of our securities as a result of the completion of that transaction. Additionally, our “going public” by means of a reverse merger transaction may make it more difficult for us to obtain coverage from securities analysts of major brokerage firms following the Merger because there may be little incentive to those brokerage firms to recommend the purchase of our common stock. The occurrence of any such event could cause our business or stock price to suffer.
Because we became public by means of a “reverse merger,” it may be more difficult to list on a national exchange such as the NASDAQ, NYSE or NYSE MKT.
It may be more difficult to list on a major exchange because we have conducted a reverse merger. In 2011, the SEC approved new rules of the three major U.S. listing markets that toughen the standards that companies going public through a reverse merger must meet to become listed on those exchanges. Under the rules, NASDAQ, NYSE and NYSE MKT impose more stringent listing requirements for companies that become public through a reverse merger. Specifically, the rules prohibit a reverse merger company from applying to list on either the NASDAQ, NYSE or NYSE MKT until:
| • | the company has completed a one-year “seasoning period” by trading in the U.S. over the counter market or on another regulated U.S. or foreign exchange following the reverse merger, and filed all required reports with the SEC, including audited financial statements; and |
| • | the company maintains the requisite minimum share price for a sustained period, and for at least 30 of the 60 trading days, immediately prior to its listing application and the exchange’s decision to list. |
It is possible for a reverse merger company to be exempt from these special requirements, but only if a listing is in connection with a substantial, firm commitment underwritten public offering.
We have a substantial number of shares of authorized but unissued capital stock, and if we issue additional shares of our capital stock in the future, our existing stockholders will be diluted.
Our Certificate of Incorporation authorize the issuance of up to 300,000,000 shares of our common stock and up to 50,000,000 shares of preferred stock with the rights, preferences and privileges that our Board of Directors may determine from time to time. As of June 30, 2015, we had 14,259,681 shares of our common stock issued and outstanding, which represents 4.75% of our total authorized shares. In addition to capital raising activities, which we expect to continue to pursue in order to raise the funding we will need in order to continue our operations, other possible business and financial uses for our authorized capital stock include, without limitation, future stock splits, acquiring other companies, businesses or products in exchange for shares of our capital stock, issuing shares of our capital stock to partners or other collaborators in connection with strategic alliances, attracting and retaining employees by the issuance of additional securities under our equity compensation plans, or other transactions and corporate purposes that our Board of Directors deems are in the best interest of our company. Additionally, shares of our capital stock could be used for anti-takeover purposes or to delay or prevent changes in control or our management. Any future issuances of shares of our capital stock may not be made on favorable terms or at all, they may not enhance stockholder value, they may have rights, preferences and privileges that are superior to those of our common stock, and they may have an adverse effect on our business or the trading price of our common stock. The issuance of any additional shares of our common stock will reduce the book value per share and may contribute to a reduction in the market price of the outstanding shares of our common stock. Additionally, any such issuance will reduce the proportionate ownership and voting power of all of our current stockholders.
70
Table of Contents
Sales of a substantial number of shares of our common stock in the public market, or the perception that such sales could occur, could cause our stock price to fall.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market after the legal restrictions on resale discussed in this prospectus lapse or after those shares become registered for resale pursuant to an effective registration statement, the trading price of our common stock could decline. As of June 30, 2015, a total of 14,259,681 shares of our common stock were outstanding. Of those shares, only approximately 1.2% are currently freely tradable, without restriction, in the public market. We have registered for resale under the Securities Act an aggregate of 2,463,080 shares of our common stock held by persons who are not affiliates of Ampio and those shares are freely tradable without restriction, except for shares held by our affiliates, and any sales of those shares or any perception in the market that such sales may occur could cause the trading price of our common stock to decline. Additionally, an aggregate of 12,957,032 shares subject to lock-up agreements entered into in connection with the Merger became or will become freely tradable upon the phased expiration of the lock-up agreements on June 30, 2015 (327,348 shares), April 16, 2017 (3,910,133 shares), October 16, 2017 (2,906,517 shares), April 16, 2018 (2,906,517 shares) and October 16, 2018 (2,906,517 shares).
We also have outstanding convertible notes that we issued in July and August 2015, in the aggregate principal amount of approximately $5.2 million. The notes are convertible at any time in a noteholder’s discretion into that number of shares of our common stock equal in an amount equal to 120% of the number of shares of common stock calculated by dividing the then outstanding principal and accrued interest by $4.63. A holder of notes will be obligated to convert on the terms of our next public offering of our stock resulting in proceeds to us of at least $5,000,000 in gross proceeds (excluding indebtedness converted in such financing) prior to the maturity date of the notes, referred to as a Qualified Financing. The principal and accrued interest under the notes will automatically convert into a number of shares of such equity securities of our company sold in such financing equal to 120% of the principal and accrued interest under such note divided by the lesser of (i) the lowest price paid by an investor in such financing or (ii) $4.63. In the event that we sell equity securities to investors at any time while the notes are outstanding in a financing transaction that is not a Qualified Financing, then the noteholders will have the option to convert in whole the outstanding principal and accrued interest as of the closing of such financing into a number of shares of our capital stock in an amount equal to 120% of the number of such shares calculated by dividing the outstanding principal and accrued interest by the lesser of (a) the lowest cash price per share paid by purchasers of shares in such financing, or (b) $4.63. If these notes are converted, the shares of common stock issuable upon conversion could be sold. The perception of such issuance and sale could negatively impact the price of our common stock.
In addition, shares of common stock that are reserved for future issuance under our future equity incentive plans will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, Rule 144 and Rule 701 under the Securities Act, and any future registration of such shares under the Securities Act. If these additional shares of common stock are sold, or if it is perceived that they will be sold, in the public market, the trading price of our common stock could decline.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plan or otherwise, could result in dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To raise capital, we may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors in a prior transaction may be materially diluted by subsequent sales. Additionally, any such sales may result in material dilution to our existing stockholders, and new investors could gain rights, preferences and privileges senior to those of holders of our common stock. Further, any future sales of our common stock by us or resales of our common stock by our existing stockholders could cause the market price of our common stock to decline. Any future grants of options, warrants or other securities exercisable or convertible into our common stock, or the exercise or conversion of such shares, and any sales of such shares in the market, could have an adverse effect on the market price of our common stock.
Some provisions of our charter documents and applicable Delaware law may discourage an acquisition of us by others, even if the acquisition may be beneficial to some of our stockholders.
Provisions in our Certificate of Incorporation and Amended and Restated Bylaws, as well as certain provisions of Delaware law, could make it more difficult for a third-party to acquire us, even if doing so may benefit some of our stockholders. These provisions include:
| • | the authorization of 50,000,000 shares of “blank check” preferred stock, the rights, preferences and privileges of which may be established and shares of which may be issued by our Board of Directors at its discretion from time to time and without stockholder approval; |
71
Table of Contents
| • | limiting the removal of directors by the stockholders; |
| • | allowing for the creation of a staggered board of directors; |
| • | eliminating the ability of stockholders to call a special meeting of stockholders; and |
| • | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings. |
These provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management. In addition, we are subject to Section 203 of the Delaware General Corporation Law, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder, unless such transactions are approved by the board of directors. This provision could have the effect of discouraging, delaying or preventing someone from acquiring us or merging with us, whether or not it is desired by or beneficial to our stockholders.
Any provision of our Certificate of Incorporation or Bylaws or of Delaware law that is applicable to us that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock in the event that a potentially beneficial acquisition is discouraged, and could also affect the price that some investors are willing to pay for our common stock.
The elimination of personal liability against our directors and officers under Delaware law and the existence of indemnification rights held by our directors, officers and employees may result in substantial expenses.
Our Certificate of Incorporation and our Bylaws eliminate the personal liability of our directors and officers to us and our stockholders for damages for breach of fiduciary duty as a director or officer to the extent permissible under Delaware law. Further, our Certificate of Incorporation and our Bylaws and individual indemnification agreements we intend to enter with each of our directors and executive officers provide that we are obligated to indemnify each of our directors or officers to the fullest extent authorized by the Delaware law and, subject to certain conditions, advance the expenses incurred by any director or officer in defending any action, suit or proceeding prior to its final disposition. Those indemnification obligations could expose us to substantial expenditures to cover the cost of settlement or damage awards against our directors or officers, which we may be unable to afford. Further, those provisions and resulting costs may discourage us or our stockholders from bringing a lawsuit against any of our current or former directors or officers for breaches of their fiduciary duties, even if such actions might otherwise benefit our stockholders.
We do not intend to pay cash dividends on our capital stock in the foreseeable future.
We have never declared or paid any dividends on our common stock and do not anticipate paying any dividends in the foreseeable future. Any future payment of cash dividends in the future would depend on our financial condition, contractual restrictions, solvency tests imposed by applicable corporate laws, results of operations, anticipated cash requirements and other factors and will be at the discretion of the our Board of Directors. Our stockholders should not expect that we will ever pay cash or other dividends on our outstanding capital stock.
Our common stock is subject to the “penny stock” rules of the SEC and the trading market in the securities is limited, which makes transactions in the stock cumbersome and may reduce the value of an investment in the stock.
The SEC has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require:
| • | that a broker or dealer approve a person’s account for transactions in penny stocks; and |
| • | the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased. |
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must:
| • | obtain financial information and investment experience objectives of the person; and |
| • | make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks. |
72
Table of Contents
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form sets forth:
| • | the basis on which the broker or dealer made the suitability determination; and |
| • | that the broker or dealer received a signed, written agreement from the investor prior to the transaction. |
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of common stock and cause a decline in the market value of stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
Pursuant to our services agreements with Ampio, we use a portion of Ampio’s office space as our office space, which is located in Englewood, Colorado. We have also opened a 1,333 square foot office in Raleigh, North Carolina for which the lease runs until July 31, 2018. Ampio’s lease expires in 2024, and our cost for the office space is included in our quarterly payment under the services agreements. We expect to establish a new office in the Englewood, Colorado area in the near future. We believe our current office space is sufficient to meet our current needs.
On August 19, 2015, Aytu entered into a 37 month non-cancellable operating lease for new office space effective September 1, 2015. The new lease has initial base rent of $8,500 per month beginning in October 2015, with the total base rent over the term of the lease of approximately $318,000 which includes rent abatements. We recognize rental expense of the facility on a straight-line basis over the term of the lease. Differences between the straight-line net expenses on rent payments are classified as liabilities between current deferred rent and long-term deferred rent.
| Item 3. | Legal Proceedings |
We are currently not party to any material legal or administrative proceedings and are not aware of any material pending or threatened legal or administrative proceedings in which we will become involved.
| Item 4. | Mine Safety Disclosures. |
Not applicable.
73
Table of Contents
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Data
Our common stock has been quoted on the OTCQB under the symbol “AYTU.” The following table sets forth the range of bid and asked closing quotations for our common stock on the OTCQB for the periods shown. The quotations represent inter-dealer prices without retail markup, markdown or commission, and may not necessarily represent actual transactions.
| Fiscal Year ended June 30, 2014 |
High | Low | ||||||
| First Quarter |
$ | 3.04 | $ | 2.43 | ||||
| Second Quarter |
$ | 4.26 | $ | 2.43 | ||||
| Third Quarter |
$ | 6.09 | $ | 3.17 | ||||
| Fourth Quarter |
$ | 10.35 | $ | 2.45 | ||||
| Fiscal Year ended June 30, 2015 |
High | Low | ||||||
| First Quarter |
$ | 2.45 | $ | 2.01 | ||||
| Second Quarter |
$ | 2.31 | $ | 2.31 | ||||
| Third Quarter |
$ | 3.04 | $ | 1.95 | ||||
| Fourth Quarter |
$ | 11.81 | $ | 2.43 | ||||
On September 1, 2015, the closing price as reported on the OTCQB of our common stock was $4.75. As of September 1, 2015, there were 127 holders of record of our common stock.
We have not paid any cash dividends on our common stock and our Board of Directors presently intends to continue a policy of retaining earnings, if any, for use in our operations. The declaration and payment of dividends in the future, of which there can be no assurance, will be determined by the Board of Directors in light of conditions then existing, including earnings, financial condition, capital requirements and other factors. Delaware law prohibits us from declaring dividends where, if after giving effect to the distribution of the dividend:
| • | we would not be able to pay our debts as they become due in the usual course of business; or |
| • | our total assets would be less than the sum of our total liabilities plus the amount that would be needed to satisfy the rights of stockholders who have preferential rights superior to those receiving the distribution. |
Except as set forth above, there are no restrictions that currently materially limit our ability to pay dividends or which we reasonably believe are likely to limit materially the future payment of dividends on common stock.
Our Board of Directors has the right to authorize the issuance of preferred stock, without further stockholder approval, the holders of which may have preferences over the holders of our common stock as to payment of dividends.
Equity Compensation Plan Information
In June 2015, our shareholders approved the adoption of a stock and option award plan (the “2015 Plan”), under which 10,000,000 shares were reserved for future issuance under restricted stock awards, options, and other equity awards. The 2015 Plan permits grants of equity awards to employees, directors and consultants. The following table displays equity compensation plan information as of June 30, 2015.
74
Table of Contents
| Plan Category |
Number of Securities to be Issued upon Exercise of Outstanding Options (a) |
Weighted-Average Exercise Price of Outstanding Options (b) |
Number of Securities Remaining Available for Issuance under Equity Compensation Plans (Excluding Securities Reflected in Column (a)) (c) |
|||||||||
| Equity compensation plans approved by security holders |
— | $ | — | 10,000,000 | ||||||||
| Equity compensation plans not approved by security holders |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
— | $ | — | 10,000,000 | ||||||||
|
|
|
|
|
|
|
|||||||
In connection with our private placement of approximately $5.2 million of convertible notes in July and August 2015, we issued to the placement agent a warrant to purchase 58,040 shares of our common stock. The placement agent warrant has a term of five years, will have an exercise price equal to 100% of the price per share at which equity securities are sold in our next equity financing, and provides for cashless exercise. The warrant was issued subsequent to June 30, 2015 and therefore no shares are reflected in the table above. This warrant was not approved by our stockholders.
| Item 6. | Selected Financial Data |
Not applicable.
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes appearing elsewhere in this Annual Report. Some of the information contained in this discussion and analysis, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. You should read the “Risk Factors” section of this Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a specialty healthcare company concentrating on developing and commercializing products with an initial focus on urological indications and related conditions. We are focused primarily on the urological disorders market and specifically sexual dysfunction, urological cancers and male infertility.
Through a multi-step reverse triangular merger, on April 16, 2015, Vyrix Pharmaceuticals, Inc. (“Vyrix”) and Luoxis Diagnostics, Inc. (“Luoxis”) merged with and into our Company (herein referred to as the Merger) and we abandoned our pre-merger business plans to solely pursue the specialty healthcare market, including the business of Vyrix and Luoxis. On June 8, 2015, we reincorporated as a domestic Delaware corporation under Delaware General Corporate Law and changed our name from Rosewind Corporation to Aytu BioScience, Inc., and effected a reverse stock split in which each common stock holder received one share of common stock for each every 12.174 shares outstanding (herein referred to as the Reverse Stock Split). All share and per share amounts in this Annual Report have been adjusted to reflect the effect of the Reverse Stock Split.
In May 2015, we entered into and closed on an asset purchase agreement with Jazz Pharmaceuticals, Inc., pursuant to which we purchased assets related to Jazz Pharmaceuticals’ product known as ProstaScint® (capromab pendetide), including certain intellectual property and contracts, and the product approvals, inventory and work in progress (together, the “ProstaScint Business”), and assumed certain of Jazz Pharmaceuticals’ liabilities, including those related to product approvals and the sale and marketing of ProstaScint. The purchase price consists of the upfront payment of $1.0 million. We also agreed to pay an additional $500,000 payable within five days after transfer for the ProstaScint-related product inventory and $227,000 payable on September 30, 2015 (which represents a portion of certain FDA fees). We also will pay 8% on its net sales made after October 31, 2017, payable up to a maximum aggregate payment of an additional $2.5 million.
75
Table of Contents
To date, we have financed operations through a combination of private and public debt and equity financings including the net proceeds from the private placement of stocks as well as a convertible note. Although it is difficult to predict our liquidity requirements, based upon our current operating plan, we believe we will have sufficient cash to meet our projected operating requirements for at least the next 12 months. See “Liquidity and Capital Resources.”
We have not received any significant revenues from the commercialization of our product candidates and do not expect to receive significant revenues from the commercialization of our product candidates in the near term. However, we have recognized limited revenue from ProstaScint sales. We have incurred accumulated net losses since our inception, and as of June 30, 2015, we had a deficit accumulated of $18.4 million. Our net loss $7.7 million for the year ended June 30, 2015 and $5.6 million for the year ended June 30, 2014.
Significant Accounting Policies and Estimates
Basis of Presentation
These historical financial statements prior to April 16, 2015 include the financial statements of Vyrix from its inception in November 2013, combined with the carve-out financial statements related to the Vyrix Acquired Assets from March 23, 2011, the date Ampio originally acquired the Vyrix assets (the “Vyrix Acquired Assets”) through its merger with DMI BioSciences, Inc. (“BioSciences”) and the financial statements of Luoxis from its inception in January 2013, combined with the carve-out financial statements related to Luoxis.
The carve-out financial statements present the statements of financial position of Vyrix and Luoxis and the Vyrix Acquired Assets and the statement of operations and cash flows for purposes of presenting complete comparative stand-alone financial statements in accordance with Regulation S-X, Article 3, General Instructions to Financial Statements, and Staff Accounting Bulletin Topic 1-B1, Costs Reflected in Historical Financial Statements. Historically, financial statements have not been prepared for Vyrix and Luoxis, as they were not held in a separate legal entity. Although Vyrix and Luoxis have not been segregated as a separate legal entity, related revenues, direct costs and expenses, assets and liabilities have historically been segregated on Ampio’s books. In addition, we allocated corporate overhead costs based on a review of specific labor and other overhead expenses and a reasonable estimate of activities related to Vyrix and Luoxis. Allocated labor and other overhead totaled $264,000 in 2015 and $253,000 in 2014. We also prepared a calculation of income tax expense and deferred income tax assets and liabilities on a “separate return” basis (see Note 4 – Income Taxes). These financial statements do not include a carve-out for cash as the operations have historically been funded by Ampio. The historical carve-out financial statements may not be indicative of the future results of Vyrix and Luoxis as a stand-alone entities.
“We”, “us” and “our” as referred to herein include Vyrix and Luoxis, collectively.
Our activities, being primarily research and development, have not generated significant revenue to date.
Cash and Cash Equivalents
We consider all highly liquid instruments purchased with an original maturity of three months or less to be cash equivalents. Cash equivalents consist primarily of money market fund investments. Our investment policy is to preserve principal and maintain liquidity. We periodically monitor our positions with, and the credit quality of the financial institutions with which we invest. Periodically, throughout the year, we have maintained balances in excess of federally insured limits.
Revenue Recognition
License Agreements and Royalties
Payments received upon signing of license agreements are for the right to use the license and are deferred and amortized over the lesser of the license term or patent life of the licensed drug. Milestone payments relate to obtaining regulatory approval, cumulative sales targets, and other projected milestones recognized at the time the milestones are achieved. Royalties will be recognized as revenue when earned.
Product & Service Sales
We recognize revenue from product and service sales when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable and collectability is reasonably assured.
Fixed Assets
Fixed assets are recorded at cost and after being placed in service, are depreciated using the straight-line method over estimated useful lives.
76
Table of Contents
In-Process Research and Development
In-process research and development (“IPRD”) relates to the Zertane product and clinical trial data acquired in connection with the 2011 acquisition of BioSciences. The $7,500,000 recorded was based on an independent, third party appraisal of the fair value of the assets acquired. IPRD is considered an indefinite-lived intangible asset and its fair value will be assessed annually and written down if impaired. Once the Zertane product obtains regulatory approval and commercial production begins, IPRD will be reclassified to an intangible that will be amortized over its estimated useful life. If we decided to abandon the Zertane product, the IPRD would be expensed.
Patents
Costs of establishing patents, consisting of legal and filing fees paid to third parties, are expensed as incurred. The fair value of the Zertane patents, determined by an independent third party appraisal, is $500,000. The Zertane patents were acquired in connection with the 2011 acquisition of BioSciences and the cost is being amortized over the remaining U.S. patent lives of approximately 11 years. The cost of the RedoxSYS/MiOXSYS patents was $380,000 and is being amortized over the remaining U.S. patent lives of approximately 15 years.
Use of Estimates
The preparation of financial statements in accordance with Generally Accepted Accounting Principles in the United States of America (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosures of contingent assets and liabilities as of the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. Significant items subject to such estimates and assumptions include valuation allowances, stock-based compensation, warrant valuation, purchase price allocation, valuation of contingent consideration, sales returns and allowances, useful lives of fixed assets and assumptions in evaluating impairment of definite and indefinite lived assets. Actual results could differ from these estimates.
Income Taxes
We are included in the consolidated tax returns of Ampio. Our taxes are computed and reported on a “separate return” basis for these financial statements. Deferred taxes are provided on an asset and liability method whereby deferred tax assets are recognized for deductible temporary differences and operating loss and tax credit carry forwards and deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and their tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. Deferred tax assets and liabilities are adjusted for the effects of changes in tax laws and rates on the date of enactment.
The amount of income taxes and related income tax positions taken would be subject to audits by federal and state tax authorities if we filed these taxes on a separate basis. We have adopted accounting guidance for uncertain tax positions which provides that in order to recognize an uncertain tax benefit, the taxpayer must be more likely than not of sustaining the position, and the measurement of the benefit is calculated as the largest amount that is more than 50% likely to be realized upon settlement with the taxing authority. We believe that we have no material uncertain tax positions. Our policy is to record a liability for the difference between the benefits that are both recognized and measured pursuant to the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification(“ASC”) 740-10, Accounting for Uncertainty in Income Taxes—an interpretation of FASB Statement No. 109 (“ASC 740-10”) and tax position taken or expected to be taken on the tax return. Then, to the extent that the assessment of such tax positions changes, the change in estimate is recorded in the period in which the determination is made. We report tax-related interest and penalties as a component of income tax expense. During the periods reported, our management has concluded that no significant tax position requires recognition under ASC 740-10.
Stock-Based Compensation
We account for share based payments by recognizing compensation expense based upon the estimated fair value of the awards on the date of grant. We determine the estimated grant fair value using the Black-Scholes option pricing model and recognizes compensation costs ratably over the period of service using the graded method.
Research and Development
Research and development costs are expensed as incurred with expenses recorded in the respective periods.
Fair Value of Financial Instruments
The carrying amounts of financial instruments, including cash and cash equivalents, accounts payable and other current assets and other liabilities are carried at cost which approximates fair value due to the short maturity of these instruments.
77
Table of Contents
Impairment of Long-Lived Assets
Aytu routinely performs an annual evaluation of the recoverability of the carrying value of its long-lived assets to determine if facts and circumstances indicate that the carrying value of assets or intangible assets may be impaired and if any adjustment is warranted. Based on its evaluation as of June 30, 2015 and 2014, respectively, no impairment existed for long-lived assets.
Newly Issued Accounting Pronouncements
In June 2015, the FASB issued Accounting Standards Update (“ASU”) 2015-10, “Technical Corrections and Improvements”. The amendments represent changes to clarify the codification, correct unintended application of guidance, or make minor improvements to the codification that are not expected to have a significant effect on current accounting practice or create a significant administrative cost. In addition, some of the amendments will make the codification easier to understand and easier to apply by eliminating inconsistencies, providing needed clarifications, and improving the presentation of guidance in the codification. The amendments that require transition guidance are effective for all entities for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2015. Early adoption is permitted, including adoption in an interim period. All other amendments will be effective upon issuance. We are evaluating the impact of ASU 2015-10 on its financial statements.
In April 2015, the FASB issued ASU 2015-03, “Interest—Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs.” The update requires debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of the related debt liability instead of being presented as an asset. Debt disclosures will include the face amount of the debt liability and the effective interest rate. The update requires retrospective application and represents a change in accounting principle. The update is effective for fiscal years beginning after December 15, 2015. Early adoption is permitted for financial statements that have not been previously issued. We are evaluating the impact of ASU 2015-03 on its financial statements.
In January 2015, the FASB issued ASU 2015-01, “Extraordinary and Unusual Items (Subtopic 225-20): Simplifying Income Statement Presentation by Eliminating the Concept of Extraordinary Items.” The purpose of this amendment is to eliminate the concept of extraordinary items. As a result, an entity will no longer be required to separately classify, present and disclose extraordinary events and transactions. The amendment is effective for annual reporting periods beginning after December 15, 2015 and subsequent interim periods with early application permitted. We are evaluating the impact the adoption of ASU 2015-01 will have on its financial statements.
In August 2014, the FASB issued ASU No. 2014-15, “Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern” (“ASU 2014-15”). ASU 2014-15 is intended to define management’s responsibility to evaluate whether there is substantial doubt about an organization’s ability to continue as a going concern and to provide related footnote disclosures. The amendments in this ASU are effective for reporting periods beginning after December 15, 2016, with early adoption permitted. We are evaluating the impact the adoption of ASU 2014-15 will have on its financial statements.
In May 2014, the FASB issued ASU 2014-09 regarding ASC Topic 606, “Revenue from Contracts with Customers”. The standard provides principles for recognizing revenue for the transfer of promised goods or services to customers with the consideration to which the entity expects to be entitled in exchange for those goods or services. The guidance will be effective for annual reporting periods beginning after December 15, 2017, with early adoption permitted but not prior to the original public organization effective date of December 15, 2016. We are evaluating the accounting, transition and disclosure requirements of the standard and cannot currently estimate the financial statement impact of adoption.
Results of Operations—June 30, 2015 Compared to June 30, 2014
Results of operations for the year ended June 30, 2015 (“2015”) and the year ended June 30, 2014 (“2014”) reflected losses of approximately $7,723,000 and $5,579,000, respectively.
Revenue
We have not generated significant revenue in our operating history. The total revenue recognized during 2015 and 2014 was $262,000 and $59,000, respectively. Revenue is earned from the amortization of advanced payments received on our license agreements. In 2012, we received a payment of $500,000 for our license agreement of Zertane with a Korean pharmaceutical company. This payment was deferred and is being recognized over 10 years. In 2014, we received a payment of $250,000 for our license agreement of Zertane with a Canadian-based supplier. This payment was deferred and is being recognized over seven years.
The $176,000 product and service revenue recognized in 2015 represents sales of our ProstaScint product and the RedoxSYS System.
78
Table of Contents
Expenses
Research and Development
Research and development costs consist of clinical trials and sponsored research, labor, stock-based compensation, sponsored research – related party and consultants and other. These costs relate solely to research and development without an allocation of general and administrative expenses and are summarized as follows:
| Years Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Clinical trials and sponsored research |
$ | 2,244,000 | $ | 3,411,000 | ||||
| Labor |
411,000 | 195,000 | ||||||
| Stock-based compensation |
517,000 | 244,000 | ||||||
| Sponsored research - related party |
204,000 | 126,000 | ||||||
| Consultants and other |
47,000 | 83,000 | ||||||
|
|
|
|
|
|||||
| $ | 3,423,000 | $ | 4,059,000 | |||||
|
|
|
|
|
|||||
Comparison of Years Ended June 30, 2015 and 2014
Research and development expenses decreased $636,000, or 15.7%, in 2015 over 2014. This was due primarily to decreased costs in clinical trial and sponsored research related to the Zertane trials. We expect research and development expenses to increase in 2016 as compared to the 2015 level due to ramping up the Zertane trials.
General and Administrative
General and administrative expenses consist of personnel costs for employees in executive, business development and operational functions and director fees; stock-based compensation; patents and intellectual property; professional fees including legal, auditing and accounting; occupancy, travel and other including rent, governmental and regulatory compliance, insurance, investor/public relations and professional subscriptions. These costs are summarized as follows:
| Years Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Labor |
$ | 979,000 | $ | 557,000 | ||||
| Stock-based compensation |
500,000 | 256,000 | ||||||
| Patent costs |
488,000 | 582,000 | ||||||
| Professional fees |
1,189,000 | 238,000 | ||||||
| Occupancy, travel and other |
1,227,000 | 714,000 | ||||||
|
|
|
|
|
|||||
| $ | 4,383,000 | $ | 2,347,000 | |||||
|
|
|
|
|
|||||
Comparison of Years Ended June 30, 2015 and 2014
General and administrative costs increased $2,036,000, or 86.7%, in 2015 over 2014. The increase in professional fees, labor costs and stock-based compensation primarily relates to increased costs related to the Merger and associated SEC filings and legal expenses, professional staffing, bonuses earned and stock options granted as well as the continuing vesting of stock option awards granted in previous years. Occupancy, travel and other increased due to the additional travel to commercialize the RedoxSYS System. We expect general and administrative expenses to increase in 2016 due to the expected overall growth of our company.
Net Cash Used in Operating Activities
During 2015, our operating activities used $6.6 million in cash. The use of cash was approximately $1,089,000 lower than the net loss due primarily to non-cash charges for stock-based compensation, depreciation and amortization, amortization of prepaid research and development related party, an increase in accounts payable and an increase in contingent consideration related to the ProstaScint asset purchase. Cash used in operating activities also included a $24,000 deferred tax benefit and $561,000 decrease in payable to Ampio.
During 2014, our operating activities used $5.5 million in cash. The use of cash was approximately $77,000 lower than the net loss due primarily to non-cash charges for stock-based compensation, depreciation and amortization and an increase in related party payable. Cash used in operating activities also included a $814,000 deferred tax benefit, a $497,000 increase in prepaid expenses and a $465,000 increase in research and development - related party.
79
Table of Contents
Net Cash Used in Investing Activities
During 2015, cash was used to acquire ProstaScint as well as deposits for office space.
During 2014, cash was used to purchase fixed assets.
Net Cash from Financing Activities
Net cash provided by financing activities in 2015 was $12.4 million which reflects a $7.4 million loan from Ampio which was later converted to stock, $5 million stock subscription payment from Ampio, $27,000 paid out to Luoxis option holders pursuant to the Merger and $20,000 paid out for liabilities pursuant to the Merger.
Net cash provided by financing activities in 2014 was $5.2 million which reflects a $4.6 million loan from Ampio and $637,000 in contributions from Ampio.
Contractual Obligations and Commitments
Commitments and contingencies are described below and summarized by the following table as of June 30, 2015:
| Total | 2016 | 2017 | 2018 | 2019 | 2020 | Thereafter | ||||||||||||||||||||||
| Management fee |
$ | 1,800,000 | $ | 360,000 | $ | 360,000 | $ | 360,000 | $ | 360,000 | $ | 360,000 | $ | — | ||||||||||||||
| ProstaScint Inventory Transfer |
500,000 | 500,000 | — | — | — | — | — | |||||||||||||||||||||
| Sponsored research agreement with related party |
350,000 | 70,000 | 70,000 | 70,000 | 70,000 | 70,000 | — | |||||||||||||||||||||
| Clinical research and trial obligations |
329,000 | 329,000 | — | — | — | — | — | |||||||||||||||||||||
| Manufacturing |
133,000 | 133,000 | — | — | — | — | — | |||||||||||||||||||||
| Office Lease |
110,000 | 35,000 | 36,000 | 36,000 | 3,000 | — | — | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| $ | 3,222,000 | $ | 1,427,000 | $ | 466,000 | $ | 466,000 | $ | 433,000 | $ | 430,000 | $ | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Office Lease
In June 2015, we entered into a 37 month operating lease. This lease has initial base rent is $2,900 a month, with total base rent over the term of the lease of approximately $112,000. We recognize rental expense of the facility on a straight-line basis over the term of the lease. Differences between the straight-line net expenses on rent payments are classified as liabilities between current deferred rent and long-term deferred rent.
Aytu Manufacturing and Commercial Development
We entered into agreements with manufacturing companies to build its RedoxSYS and MiOXSYS systems. The current remaining commitment is $133,000.
Clinical Research and Trial Obligations
In connection with the Zertane clinical trials and RedoxSYS/MiOXSYS research studies, the remaining commitment is $329,000.
Sponsored Research Agreement with Related Party
We entered into a Sponsored Research Agreement with Trauma Research LLC (“TRLLC”), a related party, in June 2013. Under the terms of the Sponsored Research Agreement, TRLLC agreed to work collaboratively in advancing the RedoxSYS System diagnostic platform through research and development efforts. The Sponsored Research Agreement may be terminated without cause by either party on 30 days’ notice.
ProstaScint Inventory Transfer Fee
We are obligated to pay $500,000 for the ProstaScint-related product inventory upon the inventory transfer in July 2015.
Management Fee
In July 2015, we entered into agreements with Ampio whereby we agreed to pay Ampio $30,000 per month for shared overhead which includes costs related to the shared facility, corporate staff, and other miscellaneous overhead expenses. These agreements will be in effect until they are terminated in writing by both parties.
80
Table of Contents
Liquidity and Capital Resources
We have not generated significant revenue as our primary activities are focused on research and development, advancing our primary product candidates, and raising capital. As of June 30, 2015, we had cash and cash equivalents totaling $7.4 million available to fund our operations and $1.2 million in accounts payable. Based upon the Ampio commitment to fund our operations with another $5.0 million and the sale of convertible notes in July and August 2015, we believe we have adequate capital to continue operations through calendar year 2016 and into calendar year 2017. This projection is based on a number of assumptions that may prove to be wrong, and we could exhaust our available cash and cash equivalents earlier than presently anticipated. We intend to seek additional capital within the next 12 months to expand our clinical development activities for Zertane and RedoxSYS/MiOXSYS as well as building out a sales force for our ProstaScint product. In addition, we intend to evaluate the capital markets from time to time to determine when to raise additional capital in the form of equity, convertible debt or otherwise, depending on market conditions relative to our need for funds at such time, and we will seek to raise additional capital during the next 12 months at such time as we conclude that such capital is available on terms that we consider to be in the best interests of our company and our stockholders.
We have prepared a budget for 2015 which reflects cash requirements for fixed, on-going expenses such as payroll, legal and accounting, patents and overhead at an average cash burn rate of approximately $400,000 to $500,000 per month. Additional funds are planned for regulatory approvals, clinical trials, outsourced research and development and commercialization consulting. Accordingly, it will be necessary to raise additional capital and/or enter into licensing or collaboration agreements. At this time, we expect to satisfy our future cash needs through Ampio’s contributions to us and private or public sales of our securities or debt financings. We cannot be certain that financing will be available to us on acceptable terms, or at all. Over the last three years, volatility in the financial markets has adversely affected the market capitalizations of many bioscience companies and generally made equity and debt financing more difficult to obtain. This volatility, coupled with other factors, may limit our access to additional financing.
As part of our plan to raise capital, during July and August 2015, we sold in a private placement convertible promissory notes with an aggregate principal amount of $5.2 million. The notes are our unsecured obligation. Unless earlier converted, the notes will mature 18 months from their respective dates of issuance which will be on January 22, February 11 and February 28, 2017, with an option to extend up to six months at our discretion (provided that in the event we exercise such extension option, the then applicable interest rate shall increase by 2% for such extension period). We do not have the right to prepay the notes prior to the maturity date. Interest will accrue on the notes in the following amounts: (i) 8% simple interest per annum for the first six months and (ii) 12% simple interest per annum thereafter if not converted during the first six months. If there has not been a registration statement on Form S-1 filed with the SEC for the registration of the shares of common stock underlying the notes by the expiration of the first six-month period then (a) the interest rate will increase to 14% for the remainder of the period in which the notes remain outstanding and (b) any notes held by officers and directors of our Company will be subordinated to the remaining notes. Interest will accrue, is payable with the principal upon maturity, conversion or acceleration of the notes and may be paid in kind or in cash, in our sole discretion. The notes are convertible at any time in a noteholder’s discretion into that number of shares of our common stock equal in an amount equal to 120% of the number of shares of common stock calculated by dividing the then outstanding principal and accrued interest by $4.63. A holder of notes will be obligated to convert on the terms of our next public offering of our stock resulting in proceeds to us of at least $5,000,000 in gross proceeds (excluding indebtedness converted in such financing) prior to the maturity date of the notes (a “Qualified Financing”). The principal and accrued interest under the notes will automatically convert into a number of shares of such equity securities of our Company sold in such financing equal to 120% of the principal and accrued interest under such note divided by the lesser of (i) the lowest price paid by an investor in such financing or (ii) $4.63. In the event that we sell equity securities to investors at any time while the notes are outstanding in a financing transaction that is not a Qualified Financing, then the noteholders will have the option to convert in whole the outstanding principal and accrued interest as of the closing of such financing into a number of shares of our capital stock in an amount equal to 120% of the number of such shares calculated by dividing the outstanding principal and accrued interest by the lesser of (i) the lowest cash price per share paid by purchasers of shares in such financing, or (ii) $4.63.
If we cannot raise adequate additional capital in the future when we require it, we will be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. We also may be required to relinquish greater or all rights to product candidates at an earlier stage of development or on less favorable terms than we would otherwise choose. This may lead to impairment or other charges, which could materially affect our balance sheet and operating results.
Off Balance Sheet Arrangements
We do not have off-balance sheet arrangements, financings, or other relationships with unconsolidated entities or other persons, also known as “variable interest entities.”
81
Table of Contents
Impact of Inflation
In general, we believe that our operating expenses can be negatively impacted by increases in the cost of clinical trials due to inflation and rising health care costs.
| Item | 7A. Quantitative and Qualitative Disclosures about Market Risks |
Not applicable.
| Item 8. | Financial Statements and Supplementary Data |
The financial statements required by this item are identified in Item (a)(1) of Part IV and begin at page F-1 of this Annual Report on Form 10-K and are incorporated herein by reference.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
Effective on April 16, 2015 and with the approval of our Board of Directors, we dismissed HJ & Associates, LLC, or HJ, as our independent registered public accounting firm engaged to audit our financial statements.
The report issued by HJ dated November 26, 2014 relating to its audits of our balance sheets as of August 31, 2014 and 2013, and the related statements of operations, changes in stockholders’ equity (deficit) and cash flows for each of the fiscal years then ended, contained an explanatory paragraph stating that there was substantial doubt about our ability to continue as a going concern. Other than as disclosed above, such reports did not contain an adverse opinion or disclaimer of opinion and were not qualified as to uncertainty, audit scope or accounting principles.
Our decision to dismiss HJ is not the result of any disagreement between us and HJ on matters of accounting principles or practices, financial statement disclosure or auditing scope or procedures. During our two most recent fiscal years, there were no disagreements with HJ on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedures, which disagreements, if not resolved to the satisfaction of HJ, would have caused HJ to make a reference to the subject matter of the disagreement in connection with its reports. Pursuant to the rules of the SEC applicable to smaller reporting companies, HJ was not required to provide an attestation as to the effectiveness of our internal control over financial reporting for any period since our inception.
Other than as disclosed above, there were no reportable events (as that term is defined in Item 304(a)(1)(v) of Regulation S-K) during our two most recent fiscal years. Our Board of Directors discussed the subject matter referred to above with HJ. We authorized HJ to respond fully and without limitation to all requests of our successor accountant concerning all matters related to the annual and interim periods audited and reviewed by HJ, including with respect to the subject matter of any reportable event.
We provided HJ with a copy of the above disclosures and requested that HJ furnish a letter addressed to the SEC stating whether or not it agrees with the above statements, and, if not, stating the respects in which it does not agree. A copy of the letter dated April 22, 2015, is filed as Exhibit 16.1 to our Current Report on Form 8-K filed on April 22, 2015.
Effective on April 16, 2015 and with the approval of our Board of Directors, we have engaged EKS&H LLLP, or EKS&H, as our new independent registered public accounting firm. EKS&H was engaged by Vyrix and Luoxis prior to the Merger to audit their financial statements for the years ended June 30, 2014 and 2013 and the related statements of operations, changes in stockholders’ equity and cash flows for each of the years then ended June 30, 2014 and 2013.
During our two most recent fiscal years and through the date of our engagement of EKS&H, neither we nor anyone on our behalf consulted with EKS&H regarding either (i) the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered with respect to our financial statements, and no written report or oral advice was provided to us by EKS&H that was an important factor considered by us in reaching a decision as to any accounting, auditing or financial reporting issue; or (ii) any matter that was the subject of a disagreement (as that term is defined in Item 304(a)(1)(iv) of Regulation S-K promulgated under the Securities Act and the related instructions) or a reportable event (as that term is defined in Item 304(a)(1)(v) of Regulation S-K) relating to our company.
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Our management is responsible for establishing and maintaining adequate “disclosure controls and procedures,” as such term is defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934 (the “Exchange Act”), that are designed to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. Our management has not assessed the effectiveness of our disclosure controls as of June 30, 2015.
82
Table of Contents
In accordance with the SEC’s guidance under Section 214.01 of the SEC’s Compliance and Disclosure Interpretations on Regulation S-K, we did not conduct an assessment of our disclosure controls because we completed the Merger with Luoxis and Vyrix on April 16, 2015, very shortly before our fiscal year end. As a result of the Merger, our business as well as management and operations significantly changed. Given those changes, and the short period of time that had elapsed since the Merger, we were unable to fully implement disclosure controls and to conduct an assessment of our disclosure controls.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as such term is defined in Rules 13a-15(f) under the Exchange Act). Our management has not assessed the effectiveness of our internal control over financial reporting as of June 30, 2015.
In accordance with the SEC’s guidance under Section 215.02 of the SEC’s Compliance and Disclosure Interpretations on Regulation S-K, we did not conduct an assessment of our internal control over financial reporting because we completed the Merger with Luoxis and Vyrix on April 16, 2015, very shortly before our fiscal year end. As a result of the Merger, our business as well as management and operations significantly changed. Given those changes, and the short period of time that had elapsed since the Merger, we were unable to fully implement internal controls and to conduct an assessment of our internal controls over financial reporting. Therefore, this annual report does not include a report of management’s assessment regarding internal control over financial reporting. We will be required to make our first assessment of our internal control over financial reporting for the year ending June 30, 2016.
This Annual Report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our independent registered public accounting firm pursuant to rules of the SEC that permit us to provide only management’s report in this annual report.
Changes in Internal Control over Financial Reporting
There were no changes in our internal controls over financial reporting that occurred during the fourth quarter of our fiscal year 2015 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information |
None.
83
Table of Contents
| Item 10. | Directors and Executive Officers, and Corporate Governance |
The following table sets forth the names and ages of all of our directors and executive officers as of June 30, 2015. Our Board of Directors is currently comprised of one member, who is elected annually to serve for one year or until his successor is duly elected and qualified, or until his earlier resignation or removal. Executive officers serve at the discretion of the Board of Directors and are appointed by the Board of Directors. Each of the director and executive officers listed below joined us upon the closing of the Merger on April 16, 2015.
| Name |
Age |
Position | ||
| Joshua R. Disbrow |
40 | Chief Executive Officer | ||
| Jarrett T. Disbrow |
40 | Chief Operating Officer | ||
| Gregory A. Gould |
49 | Chief Financial Officer, Secretary, and Treasurer | ||
| Michael Macaluso |
63 | Director |
The following is a biographical summary of the experience of our executive officers and directors during the past five years, and an indication of directorships held by the director in other companies subject to the reporting requirements under the federal securities law.
Executive Officers
Joshua R. Disbrow—Chief Executive Officer
Joshua R. Disbrow has been employed by us since April 16, 2015. Prior to the closing of the Merger, Mr. Disbrow was the Chief Executive Officer of Luoxis since January 2013. Mr. Disbrow was also the Chief Operating Officer of Ampio since December 2012. Prior to joining Ampio, he served as the Vice President of Commercial Operations at Arbor Pharmaceuticals, a specialty pharmaceutical company, from May 2007 through October 2012. He joined Arbor as that company’s second full-time employee. Mr. Disbrow led the company’s commercial efforts from inception to the company’s acquisition in 2010 and growth to over $127 million in net sales in 2011. By the time Mr. Disbrow departed Arbor in late 2012, he had led the growth of the commercial organization to comprise over 150 people in sales, marketing sales training, managed care, national accounts, and other commercial functions. Mr. Disbrow has spent over 17 years in the pharmaceutical, diagnostic and medical device industries and has held positions of increasing responsibility in sales, marketing, sales management, commercial operations and commercial strategy. Prior to joining Arbor, Mr. Disbrow served as Regional Sales Manager with Cyberonics, Inc., a medical device company focused on neuromodulation therapies from June 2005 through April 2007. Prior to joining Cyberonics he was the Director of Marketing at LipoScience, an in vitro diagnostics company. Mr. Disbrow holds an MBA from Wake Forest University and BS in Management from North Carolina State University.
Jarrett T. Disbrow, Ph.D.—Chief Operating Officer
Jarrett Disbrow has been employed by us since April 16, 2015. Prior to the closing of the Merger, Mr. Disbrow was the Chief Executive Officer of Vyrix since November 2013. Mr. Disbrow joined Vyrix from Eurus Pharma LLC, or Eurus Pharma, where he held the position of general manager from 2011 to 2013. Prior to joining Eurus Pharma, Mr. Disbrow was the founder, president and chief executive officer of Arbor Pharmaceuticals, Inc., or Arbor Pharmaceuticals from 2006 to 2010. Following Arbor Pharmaceuticals’ acquisition in 2010, Mr. Disbrow remained with the company as vice president of commercial development. Prior to founding Arbor Pharmaceuticals in 2006, he was head of marketing for Accentia Biopharmaceuticals, Inc. from 2002 to 2006. Mr. Disbrow began his career with GlaxoWellcom, Inc. (now GlaxoSmithKline plc) from 1997 to 2001, where he held positions of increasing responsibility in sales and later marketing. Mr. Disbrow received a BS in business management from North Carolina State University in Raleigh, NC.
Gregory A. Gould —Chief Financial Officer, Secretary, and Treasurer
Gregory A. Gould has been our Chief Financial Officer since April 16, 2015. Mr. Gould is also the Chief Financial Officer of Ampio where he has been employed since June 2014. Prior to joining Ampio, he provided financial and operational consulting services to the biotech industry through his consulting company, Gould LLC from April 2012 until June 2014. Mr. Gould was Chief Financial Officer, Treasurer and Secretary of SeraCare from November 2006 until the company was sold to Linden Capital Partners in April 2012. During the period from July 2011 until April 2012 Mr. Gould also served as the Interim President and Chief Executive Officer
84
Table of Contents
of SeraCare Life Sciences. Mr. Gould has held several other executive positions at publicly traded life sciences companies including the Chief Financial Officer role at Atrix Laboratories, Inc., an emerging specialty pharmaceutical company focused on advanced drug delivery. During Mr. Gould’s tenure at Atrix he was instrumental in the negotiation and sale of the company to QLT, Inc. for over $855 million. He also played a critical role in the management of several licensing agreements including the global licensing agreement with Sanofi-Synthelabo of the Eligard® products. Mr. Gould was the Chief Financial Officer at Colorado MedTech, Inc., a publicly traded medical device design and manufacturing company where he negotiated the transaction to sell the company to KRG Capital Partners. Mr. Gould began his career as an auditor with Arthur Andersen, LLP. He currently serves on the board of directors of CytoDyn, Inc., a publicly traded drug development company pursuing anti-viral agents for the treatment of HIV. Mr. Gould graduated from the University of Colorado with a BS in Business Administration and is a Certified Public Accountant.
Non-Executive Directors
Michael Macaluso—Director
Michael Macaluso has been a member of our Board of Directors since April 16, 2015. Mr. Macaluso is also the Chief Executive Officer of Ampio where he founded Life Sciences and has been a member of the board of directors of Life Sciences, Ampio’s predecessor, since its inception. Mr. Macaluso has also been a member of our Board of Directors since the merger with Chay Enterprises in March 2010 and our Chief Executive Officer since January 9, 2012. Mr. Macaluso was appointed president of Isolagen, Inc. (AMEX: ILE) and served in that position from June 2001 to August 2001, when he was appointed chief executive officer. In June 2003, Mr. Macaluso was re-appointed as president of Isolagen and served as both chief executive officer and president until September 2004. Mr. Macaluso also served on the board of directors of Isolagen from June 2001 until April 2005. From October 1998 until June 2001, Mr. Macaluso was the owner of Page International Communications, a manufacturing business. Mr. Macaluso was a founder and principal of International Printing and Publishing, a position Mr. Macaluso held from 1989 until 1997, when he sold that business to a private equity firm.
Mr. Macaluso’s experience in executive management and marketing within the pharmaceutical industry, monetizing company opportunities, and corporate finance led to the conclusion of our Board of Directors that he should serve as a director of our company in light of our business and structure.
Family Relationships
Jarrett T. Disbrow, our Chief Operating Officer, is the brother of Joshua R. Disbrow, our Chief Executive Officer. There are no other family relationships among or between any of our current or former executive officers and directors.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors, executive officers and holders of more than 10% of our common stock to file with the SEC initial reports of ownership and reports of changes in the ownership of our common stock and other equity securities. Such persons are required to furnish us copies of all Section 16(a) filings. Based solely upon a review of the copies of the forms furnished to us, we believe that our officers, directors and holders of more than 10% of our common stock complied with all applicable filing requirements during the fiscal year ended June 30, 2015.
Meetings
During the year ended June 30, 2015, there were held (i) four meetings of the Board of Directors, (ii) one meeting of the Audit Committee, (iii) no meetings of the Compensation Committee, and (iv) no meetings of the Nominating and Governance Committee. No incumbent director attended fewer than seventy-five percent (75%) of the aggregate of (1) the total number of meetings of the Board, and (2) the total number of meetings held by all committees of the Board during the period that such director served.
85
Table of Contents
Annual Meeting Attendance and Shareholder Communications
We currently do not have a policy concerning director attendance at annual meetings.
We have not implemented a formal policy or procedure by which our shareholders can communicate directly with our Board of Directors. Nevertheless, every effort will be made to ensure that the views of shareholders are heard by the Board of Directors or individual directors, as applicable, and that appropriate responses are provided to shareholders in a timely manner. We believe that we are responsive to shareholder communications, and therefore have not considered it necessary to adopt a formal process for shareholder communications with our Board. During the upcoming year, our Board will continue to monitor whether it would be appropriate to adopt such a policy. Communications will be distributed to the Board, or to any individual director or directors as appropriate, depending on the facts and circumstances outlined in the communications. Items that are unrelated to the duties and responsibilities of the Board may be excluded, such as:
| • | junk mail and mass mailings |
| • | resumes and other forms of job inquiries |
| • | surveys |
| • | solicitations or advertisements. |
In addition, any material that is unduly hostile, threatening, or illegal in nature may be excluded, provided that any communication that is excluded will be made available to any outside director upon request.
Leadership Structure of the Board
The Board of Directors does not currently have a policy on whether the same person should serve as both the chief executive officer and chairman of the board or, if the roles are separate, whether the chairman should be selected from the non-employee directors or should be an employee. The Board believes that it should have the flexibility to make these determinations at any given point in time in the way that it believes best to provide appropriate leadership for us at that time.
Risk Oversight
The Board oversees risk management. Generally, the Board oversees risks that may affect our business as a whole, including operational matters; our accounting and financial reporting processes, our financial statements, internal controls and other accounting and related matters; certain risks related to compensation programs; and certain corporate governance risks. Management is responsible for implementing the risk management strategy and developing policies, controls, processes and procedures to identify and manage risks.
Board Committees
Due to the small size of our company, our recent refocus of our business following the Merger, and the fact that our Board of Directors consists of only one member, our Board of Directors has not established an Audit Committee, a Compensation Committee or a Nominating and Governance Committee. Our Board intends to attract other directors in the future and establish Audit, Compensation and Nominating and Governance Committees when appropriate, given the size of the Board.
| Item 11. | Executive Compensation |
Executive Compensation
In accordance with Item 402 of Regulation S-K promulgated by the SEC, we are required to disclose certain information regarding the makeup of and compensation for our company’s directors, former directors and named executive officers, in certain cases for each of the last three completed fiscal years. On April 16, 2015, we acquired Luoxis and Vyrix in the Merger. Because our sole director was a director on the boards of directors of Luoxis and Vyrix, and our named executive officers were, prior to the April 16, 2015, employed by Luoxis and Vyrix, we are providing past compensation information concerning such director and executive officers with respect to Luoxis and Vyrix.
Compensation of Directors
In establishing director compensation, our Board is guided by the following goals:
| • | compensation should consist of a combination of cash and equity awards that are designed to fairly pay the directors for work required for a company of our size and scope; |
| • | compensation should align the directors’ interests with the long-term interests of stockholders; and |
| • | compensation should assist with attracting and retaining qualified directors. |
86
Table of Contents
Jarrett T. Disbrow, who served as a member of Vyrix’s board of directors during 2014, did not receive any compensation, equity awards or non-equity awards for his service as a director, although Mr. Disbrow did receive compensation in 2014 from and with respect to his employment with Vyrix. James B. Wiegand, who served as the sole director of Rosewind in 2014, did not receive any compensation, equity awards or non-equity awards for his service as a director. Mr. Wiegand was appointed President, Chief Executive Officer and Secretary and a director of Rosewind on August 9, 2002. He resigned from all of his positions with us on April 16, 2015.
We have not yet established a compensation package for our director, Michael Macaluso, our non-employee and sole director, and future non-employee directors other than reimbursement of expenses incurred in connection with their service as director.
The following table provides information regarding all compensation paid to non-employee directors of Vyrix and Luoxis during the fiscal year ended June 30, 2015.
| Fees Earned or | Stock Option | All Other | ||||||||||||||
| Name | Paid in Cash | Awards (1) | Compensation | Total | ||||||||||||
| Micael Macaluso (2) |
$ | — | $ | 198,000 | $ | — | $ | 198,000 | ||||||||
| Gary V. Cantrell (3) |
$ | — | $ | — | $ | — | $ | — | ||||||||
| John A. Donofrio Jr (4) |
$ | — | $ | — | $ | — | $ | — | ||||||||
| Nicholas J. Leb (5) |
$ | — | $ | — | $ | — | $ | — | ||||||||
| (1) | This column reflects the aggregate grant date fair value computed in accordance with Financial Accounting Standards Board, or “FASB”, issued Accounting Standards Update, or “ASC”, Topic 718. |
| (2) | Michael Macaluso was appointed a director of Luoxis in January 2013 and a director of Vyrix in November 2013. In connection with the Merger, he resigned from the boards of Luoxis and Vyrix and was appointed a director of Aytu upon the closing of the Merger on April 16, 2015. |
| (3) | Gary V. Cantrell was appointed a director of Vyrix in February 2014. In connection with the Merger, he resigned from this position on April 16, 2015 as we continue to assess the appropriate corporate governance structure. |
| (4) | John A. Donofrio Jr. was appointed a director of Vyrix in February 2014. In connection with the Merger, he resigned from this position on April 16, 2015 as we continue to assess the appropriate corporate governance structure. |
| (5) | Nicholas J. Leb was appointed a director of Vyrix in February 2014. In connection with the Merger, he resigned from this position on April 16, 2015 as we continue to assess the appropriate corporate governance structure. |
87
Table of Contents
Executive Officer Compensation
From fiscal year 2012 to the completion of the Merger on April 16, 2015, no compensation was earned by or paid to James B. Wiegand, the former President, Chief Financial Officer and Secretary of Rosewind.
The following table sets forth all cash compensation earned, as well as certain other compensation paid or accrued for the years ended June 30, 2015 and 2014 to each of the following named executive officers.
Summary Compensation of Named Executive Officers
| Name and Principal Position |
Year | Salary ($) |
Bonus ($) |
Stock Award ($) |
Option Award ($) (1) |
Non-Equity Incentive Plan Compensation ($) |
Change in Pension Value and Nonqualified Deferred Compensation Earnings ($) |
All Other Compensation ($) |
Total ($) |
|||||||||||||||||||||||||||
| Joshua R. Disbrow (2) |
||||||||||||||||||||||||||||||||||||
| Chief Executive Officer |
2015 | 246,000 | 202,500 | — | 198,000 | — | — | — | 646,500 | |||||||||||||||||||||||||||
| since December 2012 |
2014 | 245,000 | 227,500 | — | — | — | — | — | 472,500 | |||||||||||||||||||||||||||
| Jarrett T. Disbrow (3) |
||||||||||||||||||||||||||||||||||||
| Chief Operating Officer, |
2015 | 218,000 | 5,000 | — | — | — | — | — | 223,000 | |||||||||||||||||||||||||||
| Secretary and Treasurer |
2014 | 140,000 | 5,000 | — | 223,000 | — | — | — | 368,000 | |||||||||||||||||||||||||||
| Gregory A. Gould (4) |
||||||||||||||||||||||||||||||||||||
| Chief Financial Officer, |
2015 | — | — | — | 66,000 | — | — | — | 66,000 | |||||||||||||||||||||||||||
| since June 2014 |
2014 | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Vaughan Clift, M.D. (5) |
||||||||||||||||||||||||||||||||||||
| Former Chief Medical Officer |
2015 | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| 2014 | — | — | — | 65,000 | — | — | — | 65,000 | ||||||||||||||||||||||||||||
| (1) | Option awards are reported at fair value at the date of grant. See Item 15 of Part IV, “Notes to the Financial Statements – Note 7 – Equity Instruments.” These pre merger awards were cancelled in April 2015. |
| (2) | Joshua R. Disbrow received a salary increase to $250,000 effective April 16, 2015 when he was appointed Chief Executive Officer of Aytu. |
| (3) | Jarrett T. Disbrow received a salary increase to $250,000 effective April 16, 2015 when he was appointed Chief Operating Officer of Aytu. |
| (4) | Mr. Gould was appointed to Chief Financial Officer, Secretary and Treasurer effective April 16, 2015. His compensation expense is part of the shared service agreement with Ampio. |
| (5) | Dr. Clift resigned from his position as Chief Medical Officer of Vyrix on March 31, 2015, prior to the Merger. |
Our executive officers are reimbursed by us for any out-of-pocket expenses incurred in connection with activities conducted on our behalf.
Outstanding Equity Awards
As of June 30, 2015, there were no outstanding equity awards.
88
Table of Contents
Grants of Plan-Based Awards
The following table sets forth certain information regarding grants of plan-based awards to the Named Executive Officers as of June 30, 2015:
| All Other Option | ||||||||||||||||
| Awards: Number of | Grant Date Fair | |||||||||||||||
| Securities | Exercise Price of | Value of Option | ||||||||||||||
| Underlying Options | Option Awards | Awards | ||||||||||||||
| Name | Grant Date | (#) | ($/Share) | ($)(1) | ||||||||||||
| Named Exective Officers |
||||||||||||||||
| Joshua R Disbrow |
8/11/2014 | 150,000 | (2) | $ | 1.60 | $ | 198,000 | |||||||||
| Gregory A. Gould |
8/11/2014 | 50,000 | (2) | $ | 1.60 | $ | 66,000 | |||||||||
| (1) | The amounts reported in this column represent the aggregate grant date fair value computed in accordance with FASB ASC 718, excluding the effect of any estimated forfeitures and may not correspond to the actual value that will be realized by the named executive officer. |
| (2) | These Luoxis options were accelerated and cancelled in connection with the Merger. Because the consideration paid to these holders of common stock of Luoxis was less than the exercise price of such options, no amount was paid to the option holder in connection with the cancellation. |
Employment Agreements
We entered into an employment agreement with Joshua Disbrow in connection with his employment as our Chief Executive Officer. The agreement is for a term of 24 months beginning on April 16, 2015, subject to termination by us with or without Cause or as a result of officer’s disability, or by the officer with or without Good Reason (as discussed below). Mr. Disbrow is entitled to receive $250,000 in annual salary, plus a discretionary performance bonus with a target of 125% of his base salary and 600,000 stock options with 50% vesting upon grant and the remainder vesting on the following two anniversaries of the grant date. Mr. Disbrow is also eligible to participate in the benefit plans maintained by us from time to time, subject to the terms and conditions of such plans.
We entered into an employment agreement with Jarrett Disbrow, our Chief Operating Officer, in connection with his employment with us. The agreement is for a term of 24 months beginning on April 16, 2015, subject to termination by us with or without Cause or as a result of the officer’s disability, or by the officer with or without Good Reason (as discussed below). Mr. Disbrow is entitled to receive $250,000 in annual salary, plus a discretionary performance bonus with a target of 125% of his base salary and 600,000 stock options with 50% vesting upon grant and the remainder vesting on the following two anniversaries of the grant date. Mr. Disbrow is also eligible to participate in the benefit plans maintained by us from time to time, subject to the terms and conditions of such plans.
Payments Provided Upon Termination for Good Reason or Without Cause
Pursuant to the employment agreements, in the event Mr. Joshua Disbrow’s or Mr. Jarrett Disbrow’s employment is terminated without Cause by us or either officer terminates his employment with Good Reason, we will be obligated to pay him any accrued compensation and a lump sum payment equal to two times his base salary in effect at the date of termination, as well as continued participation in the health and welfare plans for up to two years. All vested stock options shall remain exercisable from the date of termination until the expiration date of the applicable award. So long as a Change in Control is not in effect, then all options which are unvested at the date of termination Without Cause or for Good Reason shall be accelerated as of the date of termination such that the number of option shares equal to 1/24th the number of option shares multiplied by the number of full months of such officer’s employment shall be deemed vested and immediately exercisable by the officer. Any unvested options over and above the foregoing shall be cancelled and of no further force or effect, and shall not be exercisable by such officer.
“Good Reason” means, without the officer’s written consent, there is:
| • | a material reduction in the officer’s overall responsibilities or authority, or scope of duties (it being understood that the occurrence of a Change in Control shall not, by itself, necessarily constitute a reduction in the officer’s responsibilities or authority); |
| • | a material reduction of the level of the officer’s compensation (excluding any bonuses) (except where there is a general reduction applicable to the management team generally, provided, however, that in no case may the base salary be reduced below certain specified amounts); or |
| • | a material change in the principal geographic location at which the officer must perform his services. |
89
Table of Contents
“Cause” means:
| • | conviction of, or entry of a plea of guilty to, or entry of a plea of nolo contendere with respect to, any crime, other than a traffic violation which is a misdemeanor; |
| • | willful malfeasance or willful misconduct by the officer in connection with his employment; |
| • | gross negligence in performing any of his duties; |
| • | willful and deliberate violation of any of our policies; |
| • | unintended but material breach of any written policy applicable to all employees adopted by us which is not cured to the reasonable satisfaction of the board; |
| • | unauthorized use or disclosure of any proprietary information or trade secrets of us or any other party as to which the officer owes an obligation of nondisclosure as a result of the officer’s relationship with us; |
| • | willful and deliberate breach of his obligations under the employment agreement; or |
| • | any other material breach by officer of any of his obligations which is not cured to the reasonable satisfaction of the board. |
The severance benefits described above are contingent on each officer executing a general release of claims.
Payments Provided Upon a Change in Control
Pursuant to the employment agreements, in the event of a Change in Control of us, all stock options, restricted stock and other stock-based grants granted or may be granted in the future by us to the officers will immediately vest and become exercisable.
“Change in Control” means: the occurrence of any of the following events:
| • | the acquisition by any individual, entity, or group (within the meaning of Section 13(d)(3) or 14(d)(2) of the Exchange Act) (the “Acquiring Person”), other than us, or any of our Subsidiaries, of beneficial ownership (within the meaning of Rule 13d-3- promulgated under the Exchange Act) of 50% or more of the combined voting power or economic interests of the then outstanding voting securities of us entitled to vote generally in the election of directors (excluding any issuance of securities by us in a transaction or series of transactions made principally for bona fide equity financing purposes); or; |
| • | the acquisition of us by another entity by means of any transaction or series of related transactions to which we are party (including, without limitation, any stock acquisition, reorganization, merger or consolidation but excluding any issuance of securities by us in a transaction or series of transactions made principally for bona fide equity financing purposes ) other than a transaction or series of related transactions in which the holders of the voting securities of us outstanding immediately prior to such transaction or series of related transactions retain, immediately after such transaction or series of related transactions, as a result of shares in us held by such holders prior to such transaction or series of related transactions, at least a majority of the total voting power represented by the outstanding voting securities of us or such other surviving or resulting entity (or if we or such other surviving or resulting entity is a wholly-owned subsidiary immediately following such acquisition, its parent); or |
| • | the sale or other disposition of all or substantially all of the assets of us in one transaction or series of related transactions. |
Payments Provided Upon Termination for Cause or Without Good Reason, Death or Disability
Pursuant to the employment agreements, in the event we end the officer’s employment for Cause, if such officer resigns as an employee for reasons other than an event of Good Reason, such officer dies or disability occurs, then we shall pay to the officer the accrued compensation but shall have no obligation to pay the officer any amount, whether for salary, benefits, bonuses, or other compensation or expense reimbursements of any kind, accruing after the end of the employment, and such rights shall, except as otherwise required by law or pursuant to the applicable award agreement or plan, be forfeited immediately upon the end of the employment. For the sake of clarity, any stock options, restricted stock or other equity compensation shall, to the extent vested on the date of resignation without Good Reason, the date we end the employment for Cause, or the date of the officer’s death or disability, remain outstanding and exercisable to the extent provided in the applicable award agreement or plan, by the officer or his personal representative or executor.
| Receipient and Benefit |
Cause; Without good reason |
Without Cause: Good reason |
Death; Disability |
Change in Control |
||||||||||||
| Joshua Disbrow |
||||||||||||||||
| Salary |
— | $ | 500,000 | — | — | |||||||||||
| Stock Options |
— | — | — | — | ||||||||||||
| Value of health benefits provided after termination (1) |
— | 56,510 | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
— | $ | 556,510 | — | — | |||||||||||
| Jarrett Disbrow |
||||||||||||||||
| Salary |
— | $ | 500,000 | — | — | |||||||||||
| Stock Options |
— | — | — | — | ||||||||||||
| Value of health benefits provided after termination (1) |
— | 56,510 | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
— | $ | 556,510 | — | — | |||||||||||
| (1) | The value of such benefits is determined based on the estimated cost of providing health benefits to the Named Executive Officer for a period of two years. |
90
Table of Contents
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The following table sets forth information with respect to the beneficial ownership of our common stock as of August 31, 2015 for:
| • | each beneficial owner of more than 5% of our outstanding common stock; |
| • | each of our director and named executive officers; and |
| • | all of our directors and executive officers as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include common stock that can be acquired within 60 days of June 30, 2015. The percentage ownership information shown in the table is based upon 14,259,681 shares of common stock outstanding as of June 30, 2015.
Except as otherwise indicated, all of the shares reflected in the table are shares of common stock and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
In computing the number of shares of common stock beneficially owned by a person and the percentage ownership of that person, we deemed outstanding shares of common stock subject to options and warrants held by that person that are immediately exercisable or exercisable within 60 days of June 30, 2015. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Beneficial ownership representing less than 1% is denoted with an asterisk (*). The information in the table below is based on information known to us or ascertained by us from public filings made by the stockholders. Except as otherwise indicated in the table below, addresses of the director, executive officers and named beneficial owners are in care of Aytu BioScience, Inc., 373 Inverness Parkway, Suite 200, Englewood, Colorado 80112.
| Name of Beneficial Owner |
Number of Shares Beneficially Owned |
Percentage of Shares Beneficially Owned |
||||||
| 5% Stockholders: |
||||||||
| Ampio Pharmaceuticals, Inc. (1) |
11,626,068 | 81.50 | % | |||||
| Directors and Named Executive Officers: |
||||||||
| Joshua R. Disbrow (2) |
— | — | ||||||
| Jarrett T. Disbrow (3) |
— | — | ||||||
| Gregory A. Gould |
— | — | ||||||
| Michael Macaluso |
— | — | ||||||
| All directors and executive officers as a group (four persons) |
— | — | ||||||
| (1) | The address of Ampio Pharmaceuticals, Inc. is 373 Inverness Parkway, Suite 200, Englewood, CO 80112. |
| (2) | 558,567 shares are held by an irrevocable trust for estate planning in which Mr. Disbrow is a beneficiary. Mr. Disbrow does not have or share investment control over the shares held by the trust, Mr. Disbrow is not the trustee of the trust (nor is any member of Mr. Disbrow’s immediate family) and Mr. Disbrow does not have or share the power to revoke the trust. As such, under Rule 16a-8(b) and related rules, Mr. Disbrow does not have beneficial ownership over the shares purchased and held by the trust. |
| (3) | 558,567 shares are held by an irrevocable trust for estate planning in which Mr. Disbrow is a beneficiary. Mr. Disbrow does not have or share investment control over the shares held by the trust, Mr. Disbrow is not the trustee of the trust (nor is any member of Mr. Disbrow’s immediate family) and Mr. Disbrow does not have or share the power to revoke the trust. As such, under Rule 16a-8(b) and related rules, Mr. Disbrow does not have beneficial ownership over the shares purchased and held by the trust. |
91
Table of Contents
| Item 13. | Certain Relationships, Related Transactions, and Director Independence |
Related Party Transactions
We describe below all transactions and series of similar transactions, other than compensation arrangements, during the last three fiscal years, to which we were a party or will be a party, in which:
| • | the amounts involved exceeded or will exceed $120,000; and |
| • | any of our directors, executive officers or holders of more than 5% of our capital stock, or any member of the immediate family of the foregoing persons, had or will have a direct or indirect material interest. |
Merger
On April 16, 2015, pursuant to the Merger Agreement entered into among Rosewind, Luoxis, Vyrix and two subsidiaries of Rosewind created solely for the purposes of the Merger, and which did not survive the Merger, the Merger occurred in two stages.
In the first stage, each of Vyrix and Luoxis merged with one of Rosewind’s merger subsidiaries. Vyrix and Luoxis survived these mergers. The outstanding shares of stock of Vyrix and the outstanding shares of stock of Luoxis were converted into the right to receive shares of our common stock. The Vyrix stock and the Luoxis stock were each converted at an exchange factor. The exchange factor for each of them was determined upon the basis of a relative value opinion obtained by Ampio, the parent company of Vyrix and Luoxis. The outstanding shares of Rosewind’s merger subsidiary that merged with Vyrix were converted into shares of Vyrix as the surviving corporation. The outstanding shares of Rosewind’s merger subsidiary that merged with Luoxis were converted into shares of Luoxis as the surviving corporation. After completion of the first stage, Vyrix and Luoxis became subsidiaries of Rosewind.
In the second stage, which occurred on the same day as the first stage, each of Vyrix and Luoxis merged with Rosewind with Rosewind surviving. The first and second stage mergers are referred to collectively as the “Merger.”
Concurrently with the Merger:
| • | The board of directors of Rosewind, whose sole member was James Wiegand, increased the number of directors by one, and appointed Michael Macaluso to fill the vacancy created by that increase. James Wiegand resigned from the board immediately thereafter. The board of directors of Rosewind, whose sole member is Michael Macaluso, then appointed Joshua Disbrow as Chief Executive Officer, Jarrett Disbrow as Chief Operating Officer and Gregory A. Gould as our Chief Financial Officer, Secretary and Treasurer. |
| • | Ampio purchased 4,761,787 shares of our common stock for (i) issuance to Rosewind of a promissory note of Ampio in the principal amount of $10,000,000, maturing on the first anniversary of the Merger; (ii) cancellation of indebtedness of Luoxis to Ampio in the amount of $8,000,000; and (iii) cancellation of indebtedness of Vyrix to Ampio in the amount of $4,000,000. |
| • | James Wiegand entered into a consulting agreement with us with a one year duration, providing for compensation of $50,000 to him. |
| • | Each of James Wiegand and Michael Wiegand executed a release in our favor. |
| • | Each of Ampio, James Wiegand, Michael Wiegand, a trust affiliated with Joshua Disbrow and a trust affiliated with Jarrett Disbrow entered into a lock-up agreement with us agreeing not to sell its shares of our company for two years (except for the one with Ampio, more than three years). The lock-up agreements other than the one with Ampio release 25% of the shares subject to it on or prior to June 30, 2015. The Ampio lock-up agreement terminates upon a change-in-control event of either our company or Ampio. Each other lock-up agreement terminates upon a change-of-control event of our company. |
| • | Joshua Disbrow entered into an employment agreement with us. |
| • | Jarrett Disbrow entered into an employment agreement with us. |
| • | The sailing boat owned by Rosewind was transferred to James Wiegand upon the closing of the Merger in exchange for cancellation of indebtedness owing to James Wiegand in the amount of approximately $30,000 (being the approximate value of the sailing boat). We paid James Wiegand $19,963 in May 2015 for accrued interest on this indebtedness and other expense that he had incurred prior to the Merger being completed. |
92
Table of Contents
Rosewind
As of August 31, 2014, Rosewind has a secured promissory note to the sole officer and director for $30,985 for working capital. The loan carries a 6% interest rate, matures on demand and is secured by the sailing vessel. Accrued interest payable on the loan totaled $17,607 as of August 31, 2014.
For the year ended August 31, 2014, the sole officer of Rosewind contributed services valued at $3,690. This amount has been booked to additional paid in capital.
On March 3, 2015, Rosewind accepted a cash investment from two irrevocable trusts for estate planning of which Joshua Disbrow and Jarrett Disbrow are beneficiaries. None of such persons have or share investment control over our shares held by such trusts. None of such persons, nor members of their respective immediate families, are trustees of such trusts. None of such persons have or share power to revoke such trusts. Accordingly, under Rule 16a-8(b) and related rules, none of such persons has beneficial ownership over our shares purchased and held by such trusts.
Luoxis and Vyrix
Ampio Loan Agreements
In November 2013, Vyrix entered into a loan agreement with Ampio. Pursuant to the loan agreement, Ampio agreed to lend Vyrix up to an aggregate amount of $3,000,000 through cash advances of up to $500,000 each. Unpaid principal amounts under the loan agreement bear simple interest at the “Applicable Federal Rate” for long-term obligations prescribed under Section 1274(d) of the Internal Revenue Code of 1986, as amended (or any successor provision with similar applicability). The initial term of this loan agreement is for one year, subject to automatic extension of successive one-year terms. Vyrix may repay any outstanding balance at any time without penalty. Ampio has an option of converting any balance outstanding under the loan agreement into shares of Vyrix common stock at the fair market value per share of Vyrix common stock, as determined by the Ampio board of directors, as of such conversion date. As of June 30, 2014, the amount advanced was $1,600,000 with interest rates from 3.11%-3.32%. On April 16, 2015, in connection with the closing of the Merger, Ampio released Vyrix from its then outstanding obligation of $4,000,000 under the loan agreement as consideration of its share purchase, and the loan agreement was terminated.
In March 2014, Luoxis entered into a loan agreement with Ampio. Pursuant to the loan agreement, Ampio agreed to lend Luoxis $3,000,000. Unpaid principal amounts under the loan agreement bear simple interest at the “Applicable Federal Rate” for long-term obligations prescribed under Section 1274(d) of the Internal Revenue Code of 1986, as amended (or any successor provision with similar applicability). The initial term of this loan agreement is for one year, subject to automatic extension of successive one-year terms. Luoxis may repay any outstanding balance at any time without penalty. Ampio has an option of converting any balance outstanding under the loan agreement into shares of Luoxis common stock at the fair market value per share of Luoxis common stock, as determined by the Ampio board of directors, as of such conversion date. As of June 30, 2014, the amount advanced was $3,000,000 with interest rates from 3.11% - 3.32%. On April 16, 2015, in connection with the closing of the Merger, Ampio released Luoxis from its then outstanding obligation of $8,000,000 under the loan agreement as consideration of its share purchase, and the loan agreement was terminated.
On April 16, 2015, Ampio received 4,761,787 shares of common stock of Aytu for (i) issuance to Aytu of a promissory note from Ampio in the principal amount of $10,000,000, maturing on the first anniversary of the Merger, (ii) cancellation of indebtedness of Luoxis to Ampio in the amount of $8,000,000; and (iii) cancellation of indebtedness of Vyrix to Ampio in the amount of $4,000,000.
Services Agreements
In January 2013, Luoxis entered into a services agreement with Ampio whereby Ampio provides corporate overhead services and a shared facility with Luoxis in exchange for $15,000 per month. The amount can be modified in writing upon the consent of both parties. The agreement may be terminated at any time by either party. In January 2014, Vyrix entered into a services agreement with Ampio whereby Ampio provides corporate overhead services to Vyrix in exchange for $7,000 per month. The amount can be modified in writing upon the consent of both parties. The agreement may be terminated at any time by either party. Both agreements were assigned to us upon the closing of the Merger.
In July 2015, Aytu entered into agreements with Ampio whereby Aytu agreed to pay Ampio $30,000 per month for shared overhead which includes costs related to the shared facility, corporate staff, and other miscellaneous overhead expenses. These agreements will be in effect until they are terminated in writing by both parties.
93
Table of Contents
Sponsored Research Agreement
In June 2013, Luoxis entered into a sponsored research agreement with TRLLC, an entity controlled by Ampio’s director and Chief Scientific Officer, Dr. Bar-Or. The agreement, which was amended in September 2013 and provides for Luoxis to pay $6,000 per month to TRLLC in consideration for services related to research and development of Luoxis’ RedoxSYS System. In March 2014, Luoxis also agreed to pay a sum of $615,000 which is being amortized over the contractual term of 60.5 months and is divided between current and long-term on the balance sheet; this amount has been paid in full. This agreement is set to expire March 2019 and cannot be terminated prior to March 2017.
Review, Approval or Ratification of Transactions with Related Persons
Due to the small size of our company, we do not at this time have a formal written policy regarding the review of related party transactions, and rely on our Board of Directors to review, approve or ratify such transactions and identify and prevent conflicts of interest. Our Board of Directors reviews any such transaction in light of the particular affiliation and interest of any involved director, officer or other employee or stockholder and, if applicable, any such person’s affiliates or immediate family members. Management aims to present transactions to our Board of Directors for approval before they are entered into or, if that is not possible, for ratification after the transaction has occurred. If our Board of Directors finds that a conflict of interest exists, then it will determine the appropriate action or remedial action, if any. Our Board of Directors approves or ratifies a transaction if it determines that the transaction is consistent with our best interests and the best interest of our stockholders.
Director Independence
Our common stock is not listed on any exchange. Consequently no exchange rules regarding director independence are applicable to us. Audit Committee members must satisfy the independence criteria set forth in Rule 10A-3 under the Securities Exchange Act of 1934, as amended, for listed companies. In order to be considered to be independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors or any other board committee: (1) accept, directly or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries; or (2) be an affiliated person of the listed company or any of its subsidiaries.
Our sole director, Michael Macaluso, is not independent under the definition of either the NYSE or Nasdaq due to the payments we make to Ampio under the services agreements with Luoxis and Vyrix. However, even if our common stock was listed on either NYSE or Nasdaq, we would be exempt from the director independence rules (other than those applicable to audit committees) due to exemptions for listed companies that are “controlled companies,” which are companies of which more than 50% of the voting securities are held by one individual or entity. Because of Ampio beneficially owns 81.5% of our common stock, we would be a “controlled company.”
| Item 14. | Principal Accountant Fees and Services |
EKS&H LLLP has served as our independent auditors since April 2015 and has been appointed by the Board of Directors to continue as our independent auditors for the fiscal year ended June 30, 2015.
The following table presents aggregate fees for professional services rendered by our independent registered public accounting firm, EKS&H LLLP for the audit of our annual financial statements for the respective periods.
| Year Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Audit fees (1) |
70,000 | 55,000 | ||||||
| Audit-related fees (2) |
23,100 | 106,381 | ||||||
| Tax fees (3) |
975 | — | ||||||
|
|
|
|
|
|||||
| Total Fees |
94,075 | 161,381 | ||||||
|
|
|
|
|
|||||
| (1) | Audit fees are comprised of annual audit fees and quarterly review fees. |
| (2) | Audit-related fees for fiscal year 2015 were comprised of fees related to registration statements and consultation fees. The 2014 fees were related to an S-1 that was filed by Vyrix prior to the Merger. |
| (3) | Tax fees are comprised of tax compliance, preparation and consultation fees. |
94
Table of Contents
Policy on Pre-Approval of Services of Independent Registered Public Accounting Firm
Our Board of Directors has responsibility for appointing, setting compensation and overseeing the work of the independent registered public accounting firm. In recognition of this responsibility, the Board of Directors pre-approves all audit and permissible non-audit services provided by the independent registered public accounting firm although, it has no written policy on this matter. Prior to engagement of the independent registered public accounting firm for the following year’s audit, management will submit to the Board of Directors for approval a description of services expected to be rendered during that year for each of following four categories of services:
Audit services include audit work performed in the preparation and audit of the annual financial statements, review of quarterly financial statements, reading of annual, quarterly and current reports, as well as work that generally only the independent auditor can reasonably be expected to provide, such as the provision of consents and comfort letters in connection with the filing of registration statements.
Audit-related services are for assurance and related services that are traditionally performed by the independent auditor, including due diligence related to mergers and acquisitions and special procedures required to meet certain regulatory requirements.
Tax services consist principally of assistance with tax compliance and reporting, as well as certain tax planning consultations.
Other services are those associated with services not captured in the other categories. We generally do not request such services from our independent auditor.
Prior to the engagement, the Board of Directors pre-approves these services by category of service. The fees are budgeted, and the Board of Directors requires the independent registered public accounting firm and management to report actual fees versus the budget periodically throughout the year by category of service. During the year, circumstances may arise when it may become necessary to engage the independent registered public accounting firm for additional services not contemplated in the original pre-approval. In those instances, the Board of Directors requires specific pre-approval before engaging the independent registered public accounting firm.
95
Table of Contents
| Item 15. | Exhibits and Financial Statement Schedules |
| (a)(1) | Financial Statements |
The following documents are filed as part of this Form 10-K, as set forth on the Index to Financial Statements found on
page F-1.
| • | Report of Independent Registered Public Accounting Firm |
| • | Balance Sheets as of June 30, 2015 and 2014 |
| • | Statements of Operations for the years ended June 30, 2015 and 2014 |
| • | Statements of Stockholders’ Equity (Deficit) for the years ended June 30, 2015 and 2014 |
| • | Statements of Cash Flows for the years ended June 30, 2015 and 2014 |
| • | Notes to the Financial Statements |
| (a)(2) | Financial Statement Schedules |
Not Applicable.
| (a)(3) | Exhibits |
| Exhibit No. |
Description |
Registrant’s |
Date |
Exhibit |
Filed | |||||
| 2.1 |
Agreement and Plan of Merger among Rosewind, Luoxis, Vyrix, two major stockholders of Rosewind and two subsidiaries of Rosewind, dated as of April 16, 2015 | 8-K | 4/22/15 | 2.1 | ||||||
| 2.2 |
Certificate of Merger | 8-K | 4/22/15 | 2.2 | ||||||
| 3.1 |
Certificate of Incorporation | 8-K | 6/09/15 | 3.1 | ||||||
| 3.2 |
Bylaws | 8-K | 6/09/15 | 3.2 | ||||||
| 4.1 |
Form of Convertible Note issued in 2015 Convertible Note Financing | 8-K | 7/24/15 | 4.1 | ||||||
| 4.2 |
Form of Placement Agent Warrant issued in 2015 Convertible Note Financing | 8-K | 7/24/15 | 4.2 | ||||||
| 10.1† |
Form of Indemnification Agreement, to be entered into between the Registrant and its directors and officers | 8-K | 4/22/15 | 10.1 | ||||||
| 10.2† |
Employment Agreement between the Registrant and Joshua R. Disbrow, dated as of April 16, 2015 | 8-K | 4/22/15 | 10.2 | ||||||
| 10.3† |
Employment Agreement between the Registrant and Jarrett Disbrow, dated as of April 16, 2015 | 8-K | 4/22/15 | 10.3 | ||||||
| 10.4# |
Asset Purchase Agreement between the Registrant (as assigned to it by Ampio/Vyrix) and Valeant International (Barbados) SRL, effective as of December 2, 2011 | 8-K/A | 6/08/15 | 10.4 | ||||||
| 10.5# |
Manufacturing and Supply Agreement between the Registrant (as assigned to it by Ampio/Vyrix) and Ethypharm S.A., dated September 10, 2012 | 8-K/A | 6/08/15 | 10.5 | ||||||
| 10.6 |
License, Development and Commercialization Agreement between the Registrant (as assigned to it by Ampio/Vyrix) and Daewoong Pharmaceuticals Co., Ltd., effective as of August 23, 2011 (incorporated by reference to Exhibit 10.1 of Ampio Pharmaceutical’s Form 8-K/A filed October 5, 2011; File No. 001-25182) | |||||||||
96
Table of Contents
| Exhibit No. |
Description |
Registrant’s |
Date |
Exhibit |
Filed | |||||
| 10.7# |
Distribution Agreement between the Registrant (as assigned to it by Ampio/Vyrix) and FBM Industria Farmaceutica, Ltda., dated as of March 1, 2012 | 8-K/A | 6/08/15 | 10.7 | ||||||
| 10.8# |
Distribution and License Agreement between the Registrant (as assigned to it by Ampio/Vyrix) and Endo Ventures Limited, dated April 9, 2014 | 8-K/A | 6/08/15 | 10.8 | ||||||
| 10.9# |
Sponsored Research Agreement between the Registrant (as assigned to it by Ampio/Luoxis) and Trauma Research LLC, dated September 1, 2009 | 8-K/A | 6/08/15 | 10.9 | ||||||
| 10.10# |
Addendum No. 4 to Sponsored Research Agreement between the Registrant (as assigned to it by Ampio/Luoxis) and Trauma Research LLC, dated March 17, 2014 | 8-K | 5/27/15 | 10.14 | ||||||
| 10.11 |
Promissory Note issued by Ampio to the Registrant on April 16, 2015 | 8-K | 4/22/15 | 10.11 | ||||||
| 10.12 |
Subscription Agreement between the Registrant and Ampio, dated April 16, 2015 | 8-K | 4/22/15 | 10.12 | ||||||
| 10.13 |
Voting Agreement between the Registrant and Ampio, dated April 21, 2015 (incorporated by reference to Exhibit 10.1 to Ampio’s Form 8-K filed April 22, 2015; File No. 001-35182) | |||||||||
| 10.14 |
Asset Purchase Agreement between Jazz Pharmaceuticals, Inc. and Rosewind Corporation, dated May 20, 2015 | 8-K | 5/27/15 | 10.14 | ||||||
| 10.15 |
Aytu BioScience 2015 Stock Option and Incentive Plan | S-1 | 7/01/15 | 10.15 | ||||||
| 10.17 |
Form of Note Purchase Agreement for 2015 Convertible Note Financing | 8-K | 7/24/15 | 10.17 | ||||||
| 16.1 |
Letter from HJ & Associates, LLC, dated April 22, 2015 | 8-K | 4/22/15 | 16.1 | ||||||
| 23.1 |
Consent of EKS&H LLLP, Independent Registered Public Accounting Firm. | X | ||||||||
| 31.1 |
Certificate of the Chief Executive Officer of Aytu BioScience, Inc. pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | X | ||||||||
| 31.2 |
Certificate of the Chief Financial Officer of Aytu BioScience, Inc. pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | X | ||||||||
| 32.1 |
Certificate of the Chief Executive Officer and the Chief Financial Officer of Aytu BioScience, Inc. pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |||||||||
| 101* |
XBRL (extensible Business Reporting Language). The following materials from Aytu BioScience, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2014 formatted in XBRL: (i) the Balance Sheets, (ii) the Statements of Operations, (iii) the Statements of Stockholders’ Equity (Deficit), (iv) the Statements of Cash Flows, and (v) the Notes to the Financial Statements. | X* | ||||||||
| † | Indicates is a management contract or compensatory plan or arrangement. |
| # | The company has received confidential treatment of certain portions of this agreement. These portions have been omitted and filed separately with the Securities and Exchange Commission pursuant to a confidential treatment request. |
| * | Due to SEC EDGAR system error, XBRL may not be included herewith, and if it is not, it will be filed as soon as practicable. |
97
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities and Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| AYTU BIOSCIENCE, INC. | ||||||
| Date: September 28, 2015 |
By: | /s/ Joshua R. Disbrow | ||||
| Joshua R. Disbrow President and Chief Executive Officer (Principal Executive Officer) | ||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant in the capacities indicated, on September 28, 2015.
| Signature | Title | |
| /s/ Joshua R. Disbrow |
President and Chief Executive Officer (Principal Executive Officer) | |
| Joshua R. Disbrow |
||
| /s/ Gregory A. Gould |
Chief Financial Officer (Principal Financial and Accounting Officer) | |
| Gregory A. Gould |
||
| /s/ Michael Macaluso |
Director | |
| Michael Macaluso |
||
98
Table of Contents
INDEX TO THE FINANCIAL STATEMENTS
AYTU BIOSCIENCE, INC.
| Page | ||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Aytu Bioscience, Inc.
Englewood, Colorado
We have audited the accompanying balance sheets of Aytu Bioscience, Inc. (the “Company”) as of June 30, 2015 and 2014, and the related statements of operations, stockholders’ equity, and cash flows for each of the periods then ended. The Company’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Aytu Bioscience, Inc. as of June 30, 2015 and 2014, and the results of its operations and its cash flows for each of the periods then ended, in conformity with accounting principles generally accepted in the United States of America.
/s/ EKS&H LLLP
September 28, 2015
Denver, Colorado
F-2
Table of Contents
AYTU BIOSCIENCE, INC.
| June 30, | ||||||||
| 2015 | 2014 | |||||||
| Assets | ||||||||
| Current assets |
||||||||
| Cash and cash equivalents |
$ | 7,353,061 | $ | 2,639,650 | ||||
| Accounts receivable |
157,058 | — | ||||||
| Inventory |
39,442 | — | ||||||
| Prepaid expenses |
370,888 | 521,322 | ||||||
| Prepaid research and development—related party (Note 8) |
121,983 | 121,983 | ||||||
| Deferred tax asset |
41,427 | 18,897 | ||||||
|
|
|
|
|
|||||
| Total current assets |
8,083,859 | 3,301,852 | ||||||
|
|
|
|
|
|||||
| Fixed assets, net (Note 2) |
29,706 | 57,246 | ||||||
| Developed technology, net |
780,125 | — | ||||||
| Customer contracts, net |
711,000 | — | ||||||
| Trade names, net |
79,000 | — | ||||||
| Goodwill |
74,000 | — | ||||||
| In-process research and development |
7,500,000 | 7,500,000 | ||||||
| Patents, net |
628,776 | 699,563 | ||||||
| Long-term portion of prepaid research and development—related party (Note 8) |
335,454 | 457,438 | ||||||
| Deposits |
4,886 | — | ||||||
|
|
|
|
|
|||||
| 10,142,947 | 8,714,247 | |||||||
|
|
|
|
|
|||||
| Total assets |
$ | 18,226,806 | $ | 12,016,099 | ||||
|
|
|
|
|
|||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current liabilities |
||||||||
| Accounts payable and accrued liabilities |
$ | 1,196,817 | $ | 649,503 | ||||
| Accrued liabilities—related party (Note 8) |
— | 150,000 | ||||||
| Accrued compensation |
196,503 | — | ||||||
| Deferred revenue |
85,714 | 85,714 | ||||||
| Payable to Ampio |
— | 561,059 | ||||||
| Notes to Ampio |
— | 4,600,000 | ||||||
| Interest payable to Ampio |
— | 46,002 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
1,479,034 | 6,092,278 | ||||||
| Contingent consideration |
664,000 | — | ||||||
| Long-term deferred revenue |
425,893 | 511,607 | ||||||
| Noncurrent deferred tax liability |
41,427 | 42,807 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
2,610,354 | 6,646,692 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies (Note 5) |
||||||||
| Stockholders’ equity |
||||||||
| Preferred Stock, par value $.0001; 50,000,000 shares authorized; none issued |
— | — | ||||||
| Common Stock, par value $.0001; 300,000,000 shares authorized; shares issued and outstanding 14,259,681 in 2015 and 7,901,426 in 2014 |
1,426 | 790 | ||||||
| Additional paid-in capital |
38,996,367 | 16,026,554 | ||||||
| Ampio stock subscription |
(5,000,000 | ) | — | |||||
| Accumulated deficit |
(18,381,341 | ) | (10,657,937 | ) | ||||
|
|
|
|
|
|||||
| Total equity |
15,616,452 | 5,369,407 | ||||||
|
|
|
|
|
|||||
| Total liabilities and equity |
$ | 18,226,806 | $ | 12,016,099 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements.
F-3
Table of Contents
AYTU BIOSCIENCE, INC.
| Years Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Product and service revenue |
$ | 176,068 | $ | — | ||||
| License revenue |
85,714 | 58,929 | ||||||
|
|
|
|
|
|||||
| Total revenue |
261,782 | 58,929 | ||||||
|
|
|
|
|
|||||
| Expenses |
||||||||
| Cost of sales |
88,109 | — | ||||||
| Research and development |
3,219,361 | 3,933,619 | ||||||
| Research and development—related party (Note 8) |
203,992 | 125,587 | ||||||
| General and administrative |
4,382,640 | 2,346,557 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(7,632,320 | ) | (6,346,834 | ) | ||||
| Interest (expense) income |
(114,994 | ) | (45,553 | ) | ||||
|
|
|
|
|
|||||
| Net loss, before income tax |
(7,747,314 | ) | (6,392,387 | ) | ||||
| Deferred income tax benefit |
23,910 | 813,697 | ||||||
|
|
|
|
|
|||||
| Net loss |
$ | (7,723,404 | ) | $ | (5,578,690 | ) | ||
|
|
|
|
|
|||||
| Weighted average number of Aytu common shares outstanding |
9,207,917 | 6,949,476 | ||||||
|
|
|
|
|
|||||
| Basic and diluted Aytu net loss per common share |
$ | (0.84 | ) | $ | (0.80 | ) | ||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements.
F-4
Table of Contents
AYTU BIOSCIENCE, INC.
Statements of Stockholders’ Equity
| Common Stock | Parent’s | Additional paid-in |
Ampio | Accumulated | Total Stockholders’ |
|||||||||||||||||||||||
| Shares | Amount | Equity | capital | Stock Subscription | Deficit | Equity | ||||||||||||||||||||||
| Balance—June 30, 2013 |
5,437,158 | $ | 544 | $ | 10,471,515 | $ | 4,418,385 | $ | — | $ | (5,079,247 | ) | $ | 9,811,197 | ||||||||||||||
| Investment from Ampio in Vyrix |
— | — | 637,210 | — | — | — | 637,210 | |||||||||||||||||||||
| Issuance of common stock in exchange for Vyrix Aquired Assets |
2,464,268 | 246 | (11,108,725 | ) | 11,108,479 | — | — | — | ||||||||||||||||||||
| Stock-based compensation |
— | — | — | 499,690 | — | — | 499,690 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (5,578,690 | ) | (5,578,690 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance—June 30, 2014 |
7,901,426 | 790 | — | 16,026,554 | — | (10,657,937 | ) | 5,369,407 | ||||||||||||||||||||
| Ampio stock subscription payment |
2,164,448 | 216 | — | 9,999,784 | (10,000,000 | ) | — | — | ||||||||||||||||||||
| Issurance of common stock to Ampio in exchange for Aytu debt |
2,597,339 | 260 | — | 11,999,740 | — | — | 12,000,000 | |||||||||||||||||||||
| Ampio stock subscription payment |
— | — | — | — | 5,000,000 | — | 5,000,000 | |||||||||||||||||||||
| Liabilities paid pursuant to the merger |
— | — | — | (20,013 | ) | — | — | (20,013 | ) | |||||||||||||||||||
| Luoxis options paid-out pursuant to the merger |
— | — | — | (27,476 | ) | — | — | (27,476 | ) | |||||||||||||||||||
| Reverse merger |
1,596,468 | 160 | — | (160 | ) | — | — | — | ||||||||||||||||||||
| Stock-based compensation |
— | — | — | 1,017,938 | — | — | 1,017,938 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (7,723,404 | ) | (7,723,404 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance—June 30, 2015 |
14,259,681 | $ | 1,426 | $ | — | $ | 38,996,367 | $ | (5,000,000 | ) | $ | (18,381,341 | ) | $ | 15,616,452 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
The accompanying notes are an integral part of these financial statements.
F-5
Table of Contents
AYTU BIOSCIENCE, INC.
| Years Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Cash flows from operating activities |
||||||||
| Net loss |
$ | (7,723,404 | ) | $ | (5,578,690 | ) | ||
| Stock-based compensation expense |
1,017,938 | 499,690 | ||||||
| Depreciation and amortization |
118,202 | 97,476 | ||||||
| Amortization of prepaid research and development—related party (Note 8) |
121,984 | 35,579 | ||||||
| Deferred taxes |
(23,910 | ) | (813,697 | ) | ||||
| Increase in accounts receivable |
(157,058 | ) | — | |||||
| Increase in inventory |
(39,442 | ) | — | |||||
| Decrease (increase) in prepaid expenses, other |
150,434 | (497,322 | ) | |||||
| Increase in prepaid research and development—related party (Note 8) |
(150,000 | ) | (465,000 | ) | ||||
| (Decrease) increase in interest payable to Ampio |
(46,002 | ) | 46,002 | |||||
| Increase in accounts payable |
547,314 | 421,870 | ||||||
| Increase in accrued compensation |
196,503 | — | ||||||
| (Decrease) increase in payable to Ampio |
(561,059 | ) | 561,059 | |||||
| (Decrease) increase in deferred revenue |
(85,714 | ) | 191,071 | |||||
|
|
|
|
|
|||||
| Net cash used in operating activities |
(6,634,214 | ) | (5,501,962 | ) | ||||
|
|
|
|
|
|||||
| Cash flows used in investing activities |
||||||||
| Deposits |
(4,886 | ) | — | |||||
| Purchase of ProstaScint Business |
(1,000,000 | ) | — | |||||
| Purchase of fixed assets |
— | (9,298 | ) | |||||
|
|
|
|
|
|||||
| Net cash used in investing activities |
(1,004,886 | ) | (9,298 | ) | ||||
|
|
|
|
|
|||||
| Cash flows from financing activities |
||||||||
| Ampio stock subscription payment |
5,000,000 | — | ||||||
| Proceeds from convertible note from Ampio converted to stock |
7,400,000 | 4,600,000 | ||||||
| Luoxis option payout pursuant to the merger |
(27,476 | ) | — | |||||
| Liabilities paid out pursuant to the merger |
(20,013 | ) | — | |||||
| Contribution from Ampio |
— | 637,210 | ||||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
12,352,511 | 5,237,210 | ||||||
|
|
|
|
|
|||||
| Net change in cash and cash equivalents |
4,713,411 | (274,050 | ) | |||||
| Cash and cash equivalents at beginning of period |
2,639,650 | 2,913,700 | ||||||
|
|
|
|
|
|||||
| Cash and cash equivalents at end of period |
$ | 7,353,061 | $ | 2,639,650 | ||||
|
|
|
|
|
|||||
| Non-cash transactions: |
||||||||
| Ampio stock subscription |
$ | 5,000,000 | $ | — | ||||
| Ampio unpaid debt converted to stock, received prior to 2015 |
$ | 4,600,000 | $ | — | ||||
| Contingent consideration related to the ProstaScint purchase |
$ | 664,000 | $ | — | ||||
| Issuance of common stock in exchange for Vyrix acquired assets |
$ | — | $ | 6,803,356 | ||||
| Related party research and development liability included in prepaid research and development—related party |
$ | — | $ | 150,000 | ||||
The accompanying notes are an integral part of these financial statements.
F-6
Table of Contents
AYTU BIOSCIENCE, INC
Notes to the Financial Statements
Note 1 – Business, Acquisition of Assets and Basis of Presentation
Business/Acquisition of Assets
Aytu BioScience, Inc. (“Aytu” or the “Company”) was incorporated as Rosewind Corporation on August 9, 2002 in the State of Colorado. Aytu was re-incorporated in the state of Delaware on June 8, 2015. Aytu is a specialty healthcare company concentrating on developing and commercializing products focused primarily on the urological disorders market, specifically sexual dysfunction, urological cancer and male infertility.
Basis of Presentation
Aytu’s current business was formed through a reverse triangular merger (the “Merger”) in which Luoxis Diagnostics, Inc. (“Luoxis”) and Vyrix Pharmaceuticals, Inc. (“Vyrix”) merged into Rosewind Corporation in a multi-step merger on April 16, 2015. These historical financial statements prior to April 16, 2015 include the combined financial statements of Vyrix from its inception in November 2013, combined with the carve-out financial statements related to Vyrix assets acquired in the Merger (the “Vyrix Acquired Assets”) from March 23, 2011, the date, its parent company Ampio Pharmaceuticals, Inc. (“Ampio”) originally acquired the Vyrix Acquired Assets through its merger with DMI BioSciences, Inc. (“BioSciences”) and the financial statements of Luoxis from its inception in January 2013, combined with the carve-out financial statements related to Luoxis.
The carve-out financial statements present the statements of financial position of Vyrix and Luoxis and the Vyrix Acquired Assets and the statements of operations and cash flows for purposes of presenting complete comparative stand-alone financial statements in accordance with Regulation S-X, Article 3, General Instructions to Financial Statements, and Staff Accounting Bulletin Topic 1-B1, Costs Reflected in Historical Financial Statements. Historically, financial statements have not been prepared for Vyrix and Luoxis, as they were not held in a separate legal entities. Although Vyrix and Luoxis have not been segregated as a separate legal entity, related revenues, direct costs and expenses, assets and liabilities have historically been segregated on Ampio’s books. In addition, the Company allocated corporate overhead costs based on a review of specific labor and other overhead expenses and a reasonable estimate of activities related to Vyrix and Luoxis. Allocated labor and other overhead totaled $264,000 in 2015 and $253,000 in 2014. The Company also prepared a calculation of income tax expense and deferred income tax assets and liabilities on a “separate return” basis (see Note 4 – Income Taxes). These financial statements do not include a carve-out for cash as the operations have historically been funded by Ampio. The historical carve-out financial statements may not be indicative of the future results of Vyrix and Luoxis as a stand-alone entities.
The “Company” as referred to in the notes to these financial statements includes Vyrix and Luoxis, collectively.
The Company’s activities, being primarily research and development, have not generated significant revenue to date.
As of June 30, 2015, Ampio is the majority shareholder of 81.5% of Aytu’s outstanding common stock.
On June 8, 2015, in connection with the reincorporation as a Delaware corporation, we effected a reverse stock split in which each common stock holder received one share of common stock for each every 12.174 shares then outstanding (the “Reverse Stock Split”). All share and per share amounts in this Annual Report have been adjusted to reflect the effect of the Reverse Stock Split.
Business Combination—ProstaScint
In May 2015, Aytu entered into and closed on an asset purchase agreement with Jazz Pharmaceuticals, Inc. (the “Seller”). Pursuant to the agreement, Aytu purchased assets related to the Seller’s product known as ProstaScint® (capromab pendetide), including certain intellectual property and contracts, and the product approvals, inventory and work in progress (together, the “ProstaScint Business”), and assumed certain of the Seller’s liabilities, including those related to product approvals and the sale and marketing of ProstaScint.
The purchase price consists of the upfront payment of $1.0 million. Aytu also agreed to pay an additional $500,000 payable within five days after transfer for the ProstaScint-related product inventory and $227,000 payable on September 30, 2015 (which represents a portion of certain FDA fees). Aytu also will pay 8% as contingent consideration on its net sales made after October 31, 2017, payable up to a maximum aggregate payment of an additional $2.5 million. The contingent consideration was valued at $664,000 using a discounted cash flow. The total fair value consideration for the purchase was $2.4 million.
F-7
Table of Contents
The Company’s allocation on consideration transferred for ProstaScint as of the purchase date May 20, 2015 is as follows:
| Estimated Fair Value |
||||
| Tangible assets |
$ | 727,000 | ||
| Intangible assets |
1,590,000 | |||
| Goodwill |
74,000 | |||
|
|
|
|||
| Total assets acquired |
$ | 2,391,000 | ||
|
|
|
|||
The intangible assets will be amortized over a ten year period.
Future amortization from the year ended June 30, 2015 is as follows:
| 2016 |
$ | 159,000 | ||
| 2017 |
159,000 | |||
| 2018 |
159,000 | |||
| 2019 |
159,000 | |||
| 2020 |
159,000 | |||
| Thereafter |
775,000 | |||
|
|
|
|||
| $ | 1,570,000 | |||
|
|
|
Pro Forma Information
The unaudited pro-forma results presented below include the effects of the ProstaScint acquisition as if it has been consummated as of July 1, 2013, with adjustments to give effect to pro forma events that are directly attributable to the acquisition which includes adjustments related to the amortization of acquired intangible assets. The unaudited pro forma results do not reflect any operating efficiency or potential cost savings which may result from the consolidation of ProstaScint. Accordingly, these unaudited pro forma results are presented for informational purposes only and are not necessarily indicative of what the actual results of operation of the combined company would have been if the acquisition had occurred at the beginning of the period presented nor are they indicative of future results of operations and are not necessarily indicative of either future results of operations or results that might have been achieved had the acquisition been consummated as of July 1, 2013.
F-8
Table of Contents
| Years ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Total revenue |
$ | 1,371,106 | $ | 1,736,139 | ||||
|
|
|
|
|
|||||
| Expenses |
||||||||
| Cost of sales—ProstaScint |
1,818,690 | 2,054,786 | ||||||
| Research and development |
3,065,626 | 3,933,619 | ||||||
| Research and development—related party (Note 8) |
156,988 | 125,587 | ||||||
| General and administrative |
4,417,884 | 2,388,665 | ||||||
| Amortization and impairment of intangible assets |
131,989 | 100,000 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(8,220,071 | ) | (6,866,518 | ) | ||||
| Interest (expense) income |
(114,994 | ) | (45,553 | ) | ||||
|
|
|
|
|
|||||
| Net loss, before income tax |
(8,335,065 | ) | (6,912,071 | ) | ||||
| Deferred income tax benefit |
23,910 | 813,697 | ||||||
|
|
|
|
|
|||||
| Net loss |
$ | (8,311,155 | ) | $ | (6,098,374 | ) | ||
|
|
|
|
|
|||||
Note 2 – Summary of Significant Accounting Policies
Cash and Cash Equivalents
Aytu considers all highly liquid instruments purchased with an original maturity of three months or less to be cash equivalents. Cash equivalents consist primarily of money market fund investments. Aytu’s investment policy is to preserve principal and maintain liquidity. The Company periodically monitors its positions with, and the credit quality of the financial institutions with which it invests. Periodically, throughout the year, Aytu has maintained balances in excess of federally insured limits.
Revenue Recognition
License Agreements and Royalties
Payments received upon signing of license agreements are for the right to use the license and are deferred and amortized over the lesser of the license term or patent life of the licensed drug. Milestone payments relate to obtaining regulatory approval, cumulative sales targets, and other projected milestones and are recognized at the time the milestones are achieved. Royalties will be recognized as revenue when earned.
Product & Service Sales
Aytu recognizes revenue from product and service sales when there is persuasive evidence that an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable and collectability is reasonably assured.
Estimated Sales Returns and Allowances
Aytu records estimated reductions in revenue for potential returns of products by customers. As a result, management must make estimates of potential future product returns and other allowances related to current period product revenue. In making such estimates, management analyzes historical returns, current economic trends and changes in customer demand and acceptance of our products. If management were to make different judgments or utilize different estimates, material differences in the amount of the Company’s reported revenue could result.
Accounts Receivable
Accounts receivable are recorded at their net realized value. Aytu evaluates collectability of accounts receivable on a quarterly basis and records a valuation allowance accordingly. As of June 30, 2015 and 2014, no allowance for doubtful accounts has been recorded.
Inventories
Inventories are recorded at the lower of cost or market, with cost determined on a first-in, first-out basis. Aytu periodically reviews the composition of its inventories in order to identify obsolete, slow-moving or otherwise unsaleable items. If unsaleable items are observed and there are no alternate uses for the inventory, Aytu will record a write-down to net realizable value in the period that the impairment is first recognized.
F-9
Table of Contents
When future commercialization is considered probable and the future economic benefit is expected to be realized, based on management’s judgment, Aytu capitalizes pre-launch inventory costs prior to regulatory approval. A number of factors are taken into consideration, including the current status in the regulatory approval process, potential impediments to the approval process, such as safety or efficacy, anticipated research and development initiatives that could impact the indication in which the compound will be used, viability of commercialization and marketplace trends. For product candidates that have not been approved by the FDA, inventory used in clinical trials is expensed at the time of production and recorded as research and development expense. For products that have been approved by the FDA, inventory used in clinical trials is expensed at the time the inventory is packaged for the clinical trial. Prior to receiving FDA approval, costs related to purchases of the active pharmaceutical ingredient and the manufacturing of the product candidate are recorded as research and development expense.
Fixed Assets
Fixed assets are recorded at cost. After being placed in service, the fixed assets are depreciated using the straight-line method over estimated useful lives. Fixed assets consist of the following:
| Estimated | June 30, | |||||||||
| Useful Lives in years |
2015 | 2014 | ||||||||
| Lab equipment |
3 - 5 | 90,000 | 90,000 | |||||||
| Less accumulated depreciation |
(60,000 | ) | (33,000 | ) | ||||||
|
|
|
|
|
|||||||
| Fixed assets, net |
$ | 30,000 | $ | 57,000 | ||||||
|
|
|
|
|
|||||||
Aytu recorded the following depreciation expense in the respective periods:
| Year Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Depreciation expense |
$ | 27,000 | $ | 27,000 | ||||
In-Process Research and Development
In-process research and development (“IPRD”) relates to the Company’s Zertane product and clinical trial data acquired in connection with the 2011 acquisition of BioSciences. The $7,500,000 recorded was based on an independent, third party appraisal of the fair value of the assets acquired. IPRD is considered an indefinite-lived intangible asset and its fair value will be assessed annually and written down if impaired. Once the Zertane product obtains regulatory approval and commercial production begins, IPRD will be reclassified to an intangible that will be amortized over its estimated useful life. If the Company decided to abandon the Zertane product, the IPRD would be expensed.
F-10
Table of Contents
Patents
Costs of establishing patents, consisting of legal and filing fees paid to third parties, are expensed as incurred. The fair value of the Zertane patents, determined by an independent third party appraisal, is $500,000. The Zertane patents were acquired in connection with the 2011 acquisition of BioSciences and are being amortized over the remaining U.S. patent lives of approximately 11 years which expires in March 2022. The cost of the Luoxis patents was $380,000 when they were acquired in connection with the 2013 formation of Luoxis and is being amortized over the remaining U.S. patent lives of approximately 15 years which expires in March 2028. Patents consist of the following:
| June 30, | ||||||||
| 2015 | 2014 | |||||||
| Patents |
$ | 880,000 | $ | 880,000 | ||||
| Less accumulated armortization |
(251,000 | ) | (180,000 | ) | ||||
|
|
|
|
|
|||||
| Patents, net |
$ | 629,000 | $ | 700,000 | ||||
|
|
|
|
|
|||||
Aytu recorded the following amortization expense in the respective periods:
| Year Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Amortization expense |
$ | 71,000 | $ | 70,000 | ||||
Future amortization from the year ended June 30, 2015 is as follows:
| 2016 |
$ | 71,000 | ||
| 2017 |
71,000 | |||
| 2018 |
71,000 | |||
| 2019 |
71,000 | |||
| 2020 |
71,000 | |||
| Thereafter |
274,000 | |||
|
|
|
|||
| $ | 629,000 | |||
|
|
|
F-11
Table of Contents
Business Combinations
The Company accounts for its business acquisitions under the acquisition method of accounting as indicated in the Financial Accounting Standards Board’s (“FASB”) Accounting Standards Codification (“ASC”) 805, “Business Combinations”, which requires the acquiring entity in a business combination to recognize the fair value of all assets acquired, liabilities assumed, and any non-controlling interest in the acquire; and establishes the acquisition date as the fair value measurement point. Accordingly, the Company recognizes assets acquired and liabilities assumed in business combinations, including contingent assets and liabilities and non-controlling interest in the acquiree, based on the fair value estimates as of the date of acquisition. In accordance with ASC 805, the Company recognizes and measures goodwill as of the acquisition date, as the excess of the fair value of the consideration paid over the fair value of the identified net assets acquired.
Goodwill
The ProstaScint purchase price allocation was based upon an analysis of the fair value of the assets and liabilities acquired from Jazz Pharmaceuticals. The final purchase price may be adjusted up to one year from the date of the acquisition. Identifying the fair value of the tangible and intangible assets and liabilities acquired required the use of estimates by management, and were based upon currently available data, as noted below.
The Company allocated the excess of purchase price over the identifiable intangible and net tangible assets to goodwill. Such goodwill is not deductible for tax purposes and represents the value placed on entering new markets and expanding market share.
The Company tests its goodwill for impairment annually, or whenever events or changes in circumstances indicate an impairment may have occurred, by comparing the carrying value to its implied fair value. Impairment may result from, among other things, deterioration in the performance of the acquired business, adverse market conditions, adverse changes in applicable laws or regulations and a variety of other circumstances. If the Company determines that an impairment has occurred, it is required to record a write-down of the carrying value and charge the impairment as an operating expense in the period the determination is made. In evaluating the recoverability of the carrying value of goodwill, the Company must make assumptions regarding estimated future cash flows and other factors to determine the fair value of the acquired assets. Changes in strategy or market conditions could significantly impact those judgments in the future and require an adjustment to the recorded balances. The goodwill was recorded as part of the acquisition of ProstaScint that occurred on May 20, 2015. There was no impairment of goodwill for the year ended June 30, 2015.
Use of Estimates
The preparation of financial statements in accordance with Generally Accepted Accounting Principles in the United States of America (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosures of contingent assets and liabilities as of the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. Significant items subject to such estimates and assumptions include valuation allowances, stock-based compensation, warrant valuation, purchase price allocation, valuation of contingent consideration, sales returns and allowances, useful lives of fixed assets and assumptions in evaluating impairment of definite and indefinite lived assets. Actual results could differ from these estimates.
Income Taxes
Aytu is included in the consolidated tax returns of Ampio. Aytu’s taxes are computed and reported on a “separate return” basis for these financial statements. Deferred taxes are provided on an asset and liability method whereby deferred tax assets are recognized for deductible temporary differences and operating loss and tax credit carry forwards and deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and their tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. Deferred tax assets and liabilities are adjusted for the effects of changes in tax laws and rates on the date of enactment.
The amount of income taxes and related income tax positions taken would be subject to audits by federal and state tax authorities if Aytu filed these taxes on a separate basis. The Company has adopted accounting guidance for uncertain tax positions which provides that in order to recognize an uncertain tax benefit, the taxpayer must be more likely than not of sustaining the position, and the measurement of the benefit is calculated as the largest amount that is more than 50% likely to be realized upon settlement with the taxing authority. The Company believes that it has no material uncertain tax positions. The Company’s policy is to record a liability for the difference between the benefits that are both recognized and measured pursuant to FASB ASC 740-10, “Accounting for Uncertainty in Income Taxes—an interpretation of FASB Statement No. 109” (“ASC 740-10”) and tax position taken or expected to be taken on the tax return. Then, to the extent that the assessment of such tax positions changes, the change in estimate is recorded in the period in which the determination is made. The Company reports tax-related interest and penalties as a component of income tax expense. During the periods reported, management of the Company has concluded that no significant tax position requires recognition under ASC 740-10.
F-12
Table of Contents
Stock-Based Compensation
Aytu accounts for share based payments by recognizing compensation expense based upon the estimated fair value of the awards on the date of grant. The Company determines the estimated grant fair value using the Black-Scholes option pricing model and recognizes compensation costs ratably over the period of service using the graded method.
Research and Development
Research and development costs are expensed as incurred with expenses recorded in the respective period.
Fair Value of Financial Instruments
The carrying amounts of financial instruments, including cash and cash equivalents, accounts payable and other current assets and other liabilities are carried at cost which approximates fair value due to the short maturity of these instruments.
Impairment of Long-Lived Assets
Aytu routinely performs an annual evaluation of the recoverability of the carrying value of its long-lived assets to determine if facts and circumstances indicate that the carrying value of assets or intangible assets may be impaired and if any adjustment is warranted. Based on its evaluation as of June 30, 2015 and 2014, respectively, no impairment existed for long-lived assets.
Newly Issued Accounting Pronouncements
In June 2015, the FASB issued Accounting Standards Update (“ASU”) 2015-10, “Technical Corrections and Improvements”. The amendments represent changes to clarify the codification, correct unintended application of guidance, or make minor improvements to the codification that are not expected to have a significant effect on current accounting practice or create a significant administrative cost. In addition, some of the amendments will make the codification easier to understand and easier to apply by eliminating inconsistencies, providing needed clarifications, and improving the presentation of guidance in the codification. The amendments that require transition guidance are effective for all entities for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2015. Early adoption is permitted, including adoption in an interim period. All other amendments will be effective upon issuance. The Company is evaluating the impact of ASU 2015-10 on its financial statements.
In April 2015, the FASB issued ASU 2015-03, “Interest—Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs.” The update requires debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of the related debt liability instead of being presented as an asset. Debt disclosures will include the face amount of the debt liability and the effective interest rate. The update requires retrospective application and represents a change in accounting principle. The update is effective for fiscal years beginning after December 15, 2015. Early adoption is permitted for financial statements that have not been previously issued. The Company is evaluating the impact of ASU 2015-03 on its financial statements.
In January 2015, the FASB issued ASU 2015-01, “Extraordinary and Unusual Items (Subtopic 225-20): Simplifying Income Statement Presentation by Eliminating the Concept of Extraordinary Items.” The purpose of this amendment is to eliminate the concept of extraordinary items. As a result, an entity will no longer be required to separately classify, present and disclose extraordinary events and transactions. The amendment is effective for annual reporting periods beginning after December 15, 2015 and subsequent interim periods with early application permitted. The Company is evaluating the impact the adoption of ASU 2015-01 will have on its financial statements.
In August 2014, the FASB issued ASU No. 2014-15, “Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern” (“ASU 2014-15”). ASU 2014-15 is intended to define management’s responsibility to evaluate whether there is substantial doubt about an organization’s ability to continue as a going concern and to provide related footnote disclosures. The amendments in this ASU are effective for reporting periods beginning after December 15, 2016, with early adoption permitted. The Company is evaluating the impact the adoption of ASU 2014-15 will have on its financial statements.
In May 2014, the FASB issued ASU 2014-09 regarding ASC Topic 606, “Revenue from Contracts with Customers”. The standard provides principles for recognizing revenue for the transfer of promised goods or services to customers with the consideration to which the entity expects to be entitled in exchange for those goods or services. The guidance will be effective for annual reporting periods beginning after December 15, 2017, with early adoption permitted but not prior to the original public organization effective date of December 15, 2016. The Company is evaluating the accounting, transition and disclosure requirements of the standard and cannot currently estimate the financial statement impact of adoption.
F-13
Table of Contents
Note 3 – License Agreement/Revenue Recognition
During 2011, Ampio entered into a license, development and commercialization agreement with a major Korean pharmaceutical company which was assigned to Vyrix when it was formed in 2013. The agreement grants the pharmaceutical company exclusive rights to market Zertane in South Korea for the treatment of premature ejaculation (“PE”) and for a combination drug to be developed, utilizing Zertane and an erectile dysfunction drug. Upon signing of the agreement, Ampio received a $500,000 upfront payment, the net proceeds of which were $418,000 after withholding of Korean tax. The upfront payment has been deferred and is being recognized as license revenue over a ten year period. Milestone payments of $3,200,000 may be earned and recognized contingent upon achievement of regulatory approvals and cumulative net sales targets, which may take several years. In addition, Aytu may earn a royalty based on 25% of net sales, as defined, if the royalty exceeds the transfer price of the Zertane product. No royalties have been earned to date.
In April 2014, Vyrix entered into a Distribution and License Agreement (the “Paladin Agreement”) with Endo Ventures Limited, which recently acquired Paladin Labs Inc. (“Paladin”), whereby Paladin has exclusive rights to market, sell and distribute Zertane in Canada, the Republic of South Africa, certain countries in Sub Saharan Africa, Colombia and Latin America. The Paladin Agreement expires on a country by country basis upon the later of fifteen years after the first commercial sale of the product in that country or expiration of market exclusivity for Zertane in that country. Paladin paid $250,000 to Vyrix upon signing the Paladin Agreement and is obligated to make milestone payments aggregating up to $3,025,000 based upon achieving Canadian and South African product regulatory approval and achieving specific sales goals. The upfront payment has been deferred and is being recognized as license revenue over a seven year period. In addition, the Paladin Agreement provides that Paladin pay royalties based on sales volume.
Note 4 – Income Taxes
As previously discussed in Note 2 – Summary of Significant Accounting Policies, the Company is included in Ampio’s consolidated tax returns. For purposes of these financial statements, the Company’s taxes are computed and reported on a “separate return” basis. Ampio and Aytu do not have a tax sharing agreement. Accordingly, certain tax attributes, e.g., net operating loss carryforwards, reflected in these financial statements, may or may not be available to Aytu. In the event that Ampio’s ownership interest in Aytu falls below 80% and Aytu is deconsolidated from Ampio’s consolidated income tax return, the net operating loss carryforwards originated prior to the incorporation of Vyrix and Luoxis would no longer be available to Aytu and the related deferred income tax asset would be removed and recorded as a deemed dividend to the parent, Ampio.
Income tax benefit resulting from applying statutory rates in jurisdictions in which Aytu is taxed (Federal and State of Colorado) differs from the income tax provision (benefit) in the Aytu’s financial statements. The following table reflects the reconciliation for the respective periods:
| Years Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Benefit at federal statutory rate |
(34.00 | )% | (34.00 | )% | ||||
| State, net of federal income tax benefit |
(2.79 | )% | (2.89 | )% | ||||
| Stock-based compensation |
5.51 | % | 1.84 | % | ||||
| Change in valuation allowance |
30.95 | % | 22.29 | % | ||||
| Other |
0.03 | % | 0.03 | % | ||||
|
|
|
|
|
|||||
| Effective tax rate |
(0.30 | )% | (12.73 | )% | ||||
|
|
|
|
|
|||||
F-14
Table of Contents
Deferred income taxes arise from temporary differences in the recognition of certain items for income tax and financial reporting purposes. The approximate tax effects of significant temporary differences which comprise the deferred tax assets and liabilities are as follows for the respective periods:
| 2015 | 2014 | |||||||
| Current deferred income tax asset: |
||||||||
| Deferred revenue short-term |
$ | 32,000 | $ | 32,000 | ||||
| Accrued expenses |
73,000 | — | ||||||
| Valuation allowance |
(64,000 | ) | (13,000 | ) | ||||
|
|
|
|
|
|||||
| Total current deferred income tax asset |
41,000 | 19,000 | ||||||
|
|
|
|
|
|||||
| Long-term deferred income tax assets (liabilities): |
||||||||
| Net operating loss carryforward |
6,337,000 | 3,847,000 | ||||||
| Section 197 intangible |
453,000 | 482,000 | ||||||
| Deferred revenue long-term |
158,000 | 190,000 | ||||||
| Share-based compensation expense |
— | 80,000 | ||||||
| Acquired in-process research and development |
(2,779,000 | ) | (2,779,000 | ) | ||||
| Less: Valuation allowance |
(4,210,000 | ) | (1,863,000 | ) | ||||
|
|
|
|
|
|||||
| Total long-term deferred income tax assets (liabilities) |
(41,000 | ) | (43,000 | ) | ||||
|
|
|
|
|
|||||
| Total deferred income tax assets (liabilities) |
$ | — | $ | (24,000 | ) | |||
|
|
|
|
|
|||||
Aytu has recorded income tax benefits in its statements of operations since inception, stemming from its operating losses, and is expected to incur operating losses for the foreseeable future. During the year ended June 30, 2015, the net deferred tax liability was reduced to zero based upon the operating losses, thus Aytu established a valuation allowance offsetting any future net deferred tax asset. As such, Aytu would no longer record income tax benefits in its results of operations after the year ended June 30, 2015 because management is currently unable to conclude that it is more likely than not that a benefit will be realized.
In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income, carry back opportunities and tax planning strategies in making the assessment. The Company believes it is more likely than not it will realize the benefits of these deductible differences, net of the valuation allowance provided.
The Company has federal net operating loss carryforwards of approximately $17.1 million and $10.4 million as of June 30, 2015 and June 30, 2014, respectively that, subject to limitation, may be available in future tax years to offset taxable income. The available net operating losses, if not utilized to offset taxable income in future periods, will begin to expire in 2031 through 2034. Net operating loss carryforwards are subject to examination in the year they are utilized regardless of whether the tax year in which they are generated has been closed by statute. The amount subject to disallowance is limited to the NOL utilized. Accordingly, the Company may be subject to examination for prior NOLs generated as such NOLs are utilized.
As of June 30, 2015 and 2014, the Company has no liability for gross unrecognized tax benefits or related interest and penalties.
Aytu has made its best estimates of certain income tax amounts included in the financial statements. Application of the Company’s accounting policies and estimates, however, involves the exercise of judgment and use of assumptions as to future uncertainties and, as a result, could differ from these estimates. In arriving at its estimates, factors the Company considers include how accurate the estimates or assumptions have been in the past, how much the estimates or assumptions have changed and how reasonably likely such changes may have a material impact. Aytu has been historically included in the Ampio consolidated tax return. Under the general statute of limitations, the Company would not be subject to federal or Colorado income tax examinations for years prior to 2011 and 2010, respectively. However, given the net operating losses generated since inception, all tax years since inception are subject to examination.
F-15
Table of Contents
Note 5 – Commitments and Contingencies
Commitments and contingencies are described below and summarized by the following table as of June 30, 2014:
| Total | 2016 | 2017 | 2018 | 2019 | 2020 | Thereafter | ||||||||||||||||||||||
| Management fee |
$ | 1,800,000 | $ | 360,000 | $ | 360,000 | $ | 360,000 | $ | 360,000 | $ | 360,000 | $ | — | ||||||||||||||
| ProstaScint Inventory Transfer |
500,000 | 500,000 | — | — | — | — | — | |||||||||||||||||||||
| Sponsored research agreement with related party |
350,000 | 70,000 | 70,000 | 70,000 | 70,000 | 70,000 | — | |||||||||||||||||||||
| Clinical research and trial obligations |
329,000 | 329,000 | — | — | — | — | — | |||||||||||||||||||||
| Manufacturing |
133,000 | 133,000 | — | — | — | — | — | |||||||||||||||||||||
| Office Lease |
110,000 | 35,000 | 36,000 | 36,000 | 3,000 | — | — | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| $ | 3,222,000 | $ | 1,427,000 | $ | 466,000 | $ | 466,000 | $ | 433,000 | $ | 430,000 | — | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Management Fee
In July 2015, Aytu entered into agreements with Ampio whereby Aytu agreed to pay Ampio $30,000 per month for shared overhead which includes costs related to the shared facility, corporate staff, and other miscellaneous overhead expenses. These agreements will be in effect until they are terminated in writing by both parties.
ProstaScint Inventory Transfer Fee
Aytu is obligated to pay $500,000 for the ProstaScint-related product inventory upon the inventory transfer in July 2015.
Sponsored Research Agreement with Related Party
Aytu entered into a Sponsored Research Agreement with Trauma Research LLC (“TRLLC”), a related party, in June 2013. Under the terms of the Sponsored Research Agreement, TRLLC agreed to work collaboratively in advancing the RedoxSYS System diagnostic platform through research and development efforts. The Sponsored Research Agreement may be terminated without cause by either party on 30 days’ notice.
Clinical Research and Trial Obligations
In connection with the Zertane clinical trials and RedoxSYS research studies, the remaining commitment is $329,000.
Aytu Manufacturing and Commercial Development
Aytu entered into agreements with manufacturing companies to build its RedoxSYS system. The current remaining commitment is $133,000.
Office Lease
In June 2015, Aytu entered into a 37 month operating lease. This lease has initial base rent of $2,900 a month, with total base rent over the term of the lease of approximately $112,000. The Company recognizes rental expense of the facility on a straight-line basis over the term of the lease. Differences between the straight-line net expenses on rent payments are classified as liabilities between current deferred rent and long-term deferred rent. Rent expense for the respective periods is as follows:
| Years Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Rent expense |
$ | 51,000 | $ | 11,000 | ||||
Note 6 – Common Stock
Capital Stock
At June 30, 2015 and 2014, Aytu had 300 million shares of common stock authorized with a par value of $0.0001 per share and 50 million shares of preferred stock authorized with a par value of $0.0001 per share.
F-16
Table of Contents
Note 7 – Equity Instruments
Options
Prior to the Merger, Aytu had two approved stock option plans (Luoxis 2013 Stock Option Plan and Vyrix 2013 Stock Option Plan), pursuant to which Aytu had reserved a total of 1,718,828 million shares of common stock, both of which were terminated on April 16, 2015 upon the closing of the Merger.
The Luoxis options that were in the money and all outstanding Vyrix options issued under the 2013 Option Plans were accelerated and cancelled in connection with the Merger. Option holders received a cash payment per option share equal to the difference between the consideration payable per share of common stock pursuant to the Merger and the exercise price of the option, if the consideration paid to holders of common stock was less than the exercise price of such options, no amount was paid to the option holder in connection with the cancellation. The cash payment during the period ended June 30, 2015 was $27,000. The company recognized compensation of $422,000 and $189,000 related to the Luoxis and Vyrix options that had accelerated vesting as of the Merger date.
The Luoxis options that were not paid out were terminated pursuant to the terms of the 2013 Luoxis Option Plan. The Company treated these options as pre-vesting forfeitures and $433,000 of previously recognized compensation was reversed.
Pursuant to the Luoxis 2013 Stock Option Plan, 1,102,761 shares of its common stock were reserved for issuance. The fair value of the options was calculated using the Black-Scholes option pricing model. In order to calculate the fair value of the options, certain assumptions are made regarding components of the model, including the estimated fair value of the underlying common stock, risk-free interest rate, volatility, expected dividend yield and expected option life. Changes to the assumptions could cause significant adjustments to valuation. Aytu estimates the expected term based on the average of the vesting term and the contractual term of the options. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of the grant for treasury securities of similar maturity. The assumptions are as follows:
| Years Ended June 30, | ||||
| 2015 | 2014 | |||
| Expected volatility |
79% - 108% | 79% - 82% | ||
| Risk free interest rate |
1.62% - 2.09% | 0.75% - 1.53% | ||
| Expected term (years) |
5.5 - 7.0 | 5.0 - 6.5 | ||
| Dividend yield |
0% | 0% | ||
Stock option activity is as follows:
| Number of Options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding June 30, 2013 |
396,994 | $ | 4.53 | 9.96 | $ | 1,272,000 | ||||||||||
| Granted |
33,083 | $ | 4.53 | |||||||||||||
| Exercised |
— | $ | — | |||||||||||||
| Forfeited/Cancelled |
— | $ | — | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding June 30, 2014 |
430,077 | $ | 4.53 | 9.01 | $ | 1,374,000 | ||||||||||
| Granted |
195,189 | $ | 7.25 | |||||||||||||
| Exercised |
— | $ | — | |||||||||||||
| Forfeited/Cancelled |
(625,266 | ) | $ | 5.40 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding June 30, 2015 |
— | $ | — | |||||||||||||
|
|
|
|||||||||||||||
| Exercisable at June 30, 2015 |
— | $ | — | |||||||||||||
|
|
|
|||||||||||||||
| Available for grant at June 30, 2015 |
— | |||||||||||||||
|
|
|
|||||||||||||||
F-17
Table of Contents
Pursuant to the Vyrix 2013 Stock Option Plan, 616,067 shares of its common stock were reserved for issuance. The fair value of the options was calculated using the Black-Scholes option pricing model. In order to calculate the fair value of the options, certain assumptions are made regarding components of the model, including the estimated fair value of the underlying common stock, risk-free interest rate, volatility, expected dividend yield and expected option life. Changes to the assumptions could cause significant adjustments to valuation. Aytu estimates the expected term based on the average of the vesting term and the contractual term of the options. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of the grant for treasury securities of similar maturity. In accordance with the Vyrix 2013 Stock Option Plan, no additional options were granted during the year-ended June 30, 2015. The assumptions are as follows:
| Year Ended June 30, | ||
| 2014 | ||
| Expected volatility |
63% - 76% | |
| Risk free interest rate |
0.90% - 2.02% | |
| Expected term (years) |
5.0 - 6.5 | |
| Dividend yield |
0% |
Stock option activity is as follows:
| Number of Options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding June 30, 2013 |
— | $ | — | — | $ | — | ||||||||||
| Granted |
117,053 | $ | 5.68 | |||||||||||||
| Exercised |
— | $ | — | |||||||||||||
| Forfeited/Cancelled |
— | $ | — | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding June 30, 2014 |
117,053 | $ | 5.68 | 9.54 | $ | 417,000 | ||||||||||
| Granted |
— | $ | — | |||||||||||||
| Exercised |
— | $ | — | |||||||||||||
| Forfeited/Cancelled |
(117,053 | ) | $ | 5.68 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding June 30, 2015 |
— | $ | — | |||||||||||||
|
|
|
|||||||||||||||
| Exercisable at June 30, 2015 |
— | $ | — | |||||||||||||
|
|
|
|||||||||||||||
| Available for grant at June 30, 2015 |
— | |||||||||||||||
|
|
|
|||||||||||||||
F-18
Table of Contents
Stock-based compensation expense related to the fair value of stock options was included in the statements of operations as research and development expenses and general and administrative expenses as set forth in the table below. Aytu determined the fair value as of the date of grant using the Black-Scholes option pricing model and expenses the fair value ratably over the vesting period. The following table summarizes stock-based compensation expense for the years ended June 30 2015 and 2014:
| Years Ended June 30, | ||||||||
| 2015 | 2014 | |||||||
| Research and development expenses |
||||||||
| Stock options |
||||||||
| Luoxis |
$ | 427,000 | $ | 206,000 | ||||
| Vyrix |
92,000 | 38,000 | ||||||
| General and administrative expenses |
||||||||
| Stock options |
||||||||
| Luoxis |
316,000 | 152,000 | ||||||
| Vyrix |
183,000 | 104,000 | ||||||
|
|
|
|
|
|||||
| $ | 1,018,000 | $ | 500,000 | |||||
|
|
|
|
|
|||||
| Unrecognized expense at June 30, 2015 |
||||||||
| Luoxis |
$ | — | ||||||
| Vyrix |
$ | — | ||||||
| Weighted average remaining years to vest |
||||||||
| Luoxis |
— | |||||||
| Vyrix |
— | |||||||
On June 1, 2015, Aytu’s stockholders approved the 2015 Stock Option and Incentive Plan (the “2015 Plan”), which provides for the award of stock options, stock appreciation rights, restricted stock and other equity awards for up to an aggregate of 10,000,000 shares of common stock. The shares of common stock underlying any awards that are forfeited, canceled, reacquired by Aytu prior to vesting, satisfied without any issuance of stock, expire or are otherwise terminated (other than by exercise) under the 2015 Plan will be added back to the shares of common stock available for issuance under the 2015 Plan. As of September 28, 2015, no grants have been made under the 2015 Plan.
Warrants
Aytu issued warrants in conjunction with its 2013 private placement. A summary of all warrants is as follows:
| Number of Warrants |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life |
||||||||||
| Outstanding June 30, 2013 |
102,613 | $ | 4.53 | 4.41 | ||||||||
| Outstanding June 30, 2014 |
102,613 | $ | 4.53 | 3.92 | ||||||||
| Outstanding June 30, 2015 |
102,613 | $ | 4.53 | 2.92 | ||||||||
These warrants were valued using the Black-Scholes option pricing model. In order to calculate the fair value of the warrants, certain assumptions were made regarding components of the model, including the closing price of the underlying common stock, risk-free interest rate, volatility, expected dividend yield, and expected life. Changes to the assumptions could cause significant adjustments to valuation. The Company estimated a volatility factor utilizing a weighted average of comparable published volatilities of peer companies. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of the grant for treasury securities of similar maturity. The offering costs and the additional paid-in capital for the warrants associated with the common stock offering were valued at $313,000 using the Black-Scholes valuation methodology.
F-19
Table of Contents
Note 8 – Related Party Transactions
Ampio Loan Agreement
In November 2013, Vyrix entered into a loan agreement with Ampio. Pursuant to the loan agreement, Ampio agreed to lend Vyrix up to an aggregate amount of $3,000,000 through cash advances of up to $500,000 each. Unpaid principal amounts under the loan agreement bear simple interest at the “Applicable Federal Rate” for long-term obligations prescribed under Section 1274(d) of the Internal Revenue Code of 1986, as amended (or any successor provision with similar applicability). The initial term of this loan agreement is for one year, subject to automatic extension of successive one-year terms. Vyrix may repay any outstanding balance at any time without penalty. Ampio has an option of converting any balance outstanding under the loan agreement into shares of Vyrix common stock at the fair market value per share of Vyrix common stock, as determined by the Ampio board of directors, as of such conversion date. As of June 30, 2014, the amount advanced was $1,600,000 with interest rates from 3.11%-3.32%. On April 16, 2015, in connection with the closing of the Merger, Ampio released Vyrix from its then outstanding obligation of $4,000,000 under the loan agreement as consideration of its share purchase, and the loan agreement was terminated.
In March 2014, Luoxis entered into a loan agreement with Ampio. Pursuant to the loan agreement, Ampio agreed to lend Luoxis $3,000,000. Unpaid principal amounts under the loan agreement bear simple interest at the “Applicable Federal Rate” for long-term obligations prescribed under Section 1274(d) of the Internal Revenue Code of 1986, as amended (or any successor provision with similar applicability). The initial term of this loan agreement is for one year, subject to automatic extension of successive one-year terms. Luoxis may repay any outstanding balance at any time without penalty. Ampio has an option of converting any balance outstanding under the loan agreement into shares of Luoxis common stock at the fair market value per share of Luoxis common stock, as determined by the Ampio board of directors, as of such conversion date. As of June 30, 2014, the amount advanced was $3,000,000 with interest rates from 3.11%—3.32%. On April 16, 2015, in connection with the closing of the Merger, Ampio released Luoxis from its then outstanding obligation of $8,000,000 under the loan agreement as consideration of its share purchase, and the loan agreement was terminated.
On April 16, 2015, Ampio received 4,761,787 shares of common stock of Aytu for (i) issuance to Aytu of a promissory note from Ampio in the principal amount of $10,000,000, maturing on the first anniversary of the Merger, (ii) cancellation of indebtedness of Luoxis to Ampio in the amount of $8,000,000; and (iii) cancellation of indebtedness of Vyrix to Ampio in the amount of $4,000,000.
Services Agreement
The Company has service agreements with Ampio which are described in Note 5.
Sponsored Research Agreement
In June 2013, Luoxis entered into a sponsored research agreement with TRLLC, an entity controlled by Ampio’s director and Chief Scientific Officer, Dr. Bar-Or. The agreement, which was amended in January 2015 and provides for Luoxis (now Aytu) to pay $6,000 per month to TRLLC in consideration for services related to research and development of the Oxidation Reduction Potential platform. In March 2014, Luoxis also agreed to pay a sum of $615,000 which is being amortized over the contractual term of 60.5 months and is divided between current and long-term on the balance sheet; this amount has been paid in full. This agreement is set to expire March 2019 and cannot be terminated prior to March 2017.
Note 9 – Litigation
As of June 30, 2015, Aytu was not party to any legal matters or claims, and none of its property is subject to any legal proceedings. In the future Aytu may become party to legal matters and claims arising in the ordinary course of business, the resolution of which it does not anticipate would have a material adverse impact on its financial position, results of operations or cash flows.
Note 10 – Employee Benefit Plan
Aytu allows its employees to participate in Ampio’s 401(k) plan. The plan allows participants to contribute a portion of their salary, subject to eligibility requirements and annual IRS limits. Aytu does not match employee contributions.
Note 11 – Subsequent Event
During July and August 2015, Aytu closed on note purchase agreements with institutional and high net worth individual investors for the purchase and sale of convertible promissory notes with an aggregate principal amount of $5.2 million. The sale of the notes was pursuant to a private placement.
F-20
Table of Contents
Aytu intends to use the net proceeds of the offering to conduct clinical studies for both Zertane® and RedoxSYS™ and for working capital to begin commercializing FDA-approved ProstaScint®, as well as general corporate purposes.
The notes are an unsecured obligation. Unless earlier converted, the notes will mature 18 months from their respective dates of issuance which will be on January 22, February 11 and February 28, 2017, with an option to extend up to six months at our discretion (provided that in the event Aytu exercises such extension option, the then applicable interest rate shall increase by 2% for such extension period). Aytu does not have the right to prepay the notes prior to the maturity date. Interest will accrue on the notes in the following amounts: (i) 8% simple interest per annum for the first six months and (ii) 12% simple interest per annum thereafter if not converted during the first six months. If there has not been a registration statement on Form S-1 filed with the SEC for the registration of the shares of common stock underlying the notes by the expiration of the first six-month period then (a) the interest rate will increase to 14% for the remainder of the period in which the notes remain outstanding and (b) any notes held by officers and directors of the Company will be subordinated to the remaining notes. Interest will accrue, is payable with the principal upon maturity, conversion or acceleration of the notes and may be paid in kind or in cash, in Aytu’s sole discretion.
The notes are convertible at any time in a noteholder’s discretion into that number of shares of Aytu common stock equal in an amount equal to 120% of the number of shares of common stock calculated by dividing the then outstanding principal and accrued interest by $4.63. A holder of notes will be obligated to convert on the terms of Aytu’s next public offering of its stock resulting in proceeds to it of at least $5,000,000 in gross proceeds (excluding indebtedness converted in such financing) prior to the maturity date of the notes (a “Qualified Financing”). The principal and accrued interest under the notes will automatically convert into a number of shares of such equity securities of the Company sold in such financing equal to 120% of the principal and accrued interest under such note divided by the lesser of (i) the lowest price paid by an investor in such financing or (ii) $4.63. In the event that Aytu sells equity securities to investors at any time while the notes are outstanding in a financing transaction that is not a Qualified Financing, then the noteholders will have the option to convert in whole the outstanding principal and accrued interest as of the closing of such financing into a number of shares of Aytu capital stock in an amount equal to 120% of the number of such shares calculated by dividing the outstanding principal and accrued interest by the lesser of (i) the lowest cash price per share paid by purchasers of shares in such financing, or (ii) $4.63.
Newbridge Securities Corporation, Member FINRA/SIPC, through LifeTech Capital, acted as sole placement agent for the institutional portion of the offering. Aytu sold the balance of the notes to individuals and entities with whom Aytu has an established relationship. For notes sold by the placement agent, Aytu paid the placement agent 8% of the gross proceeds of notes sold by the placement agent and a warrant to purchase shares of Aytu’s common stock equal to 8% of the gross proceeds of the notes sold by the placement agent divided by the price per share at which equity securities are sold in Aytu’s next equity financing, in addition to a previously paid non-refundable retainer fee of $20,000. The placement agent warrant has a term of five years, will have an exercise price equal to 100% of the price per share at which equity securities are sold in Aytu’s next equity financing, and provides for cashless exercise.
On August 19, 2015, Aytu entered into a 37 month non-cancellable operating lease for new office space effective September 1, 2015. The new lease has initial base rent of $8,500 per month beginning in October 2015, with the total base rent over the term of the lease of approximately $318,000 which includes rent abatements. The Company recognizes rental expense of the facility on a straight-line basis over the term of the lease. Differences between the straight-line net expenses on rent payments are classified as liabilities between current deferred rent and long-term deferred rent.
F-21