Attached files
| file | filename |
|---|---|
| EX-21.1 - EX-21.1 - Aralez Pharmaceuticals Ltd | a15-16894_4ex21d1.htm |
| EX-23.3 - EX-23.3 - Aralez Pharmaceuticals Ltd | a15-16894_4ex23d3.htm |
| EX-23.2 - EX-23.2 - Aralez Pharmaceuticals Ltd | a15-16894_4ex23d2.htm |
As filed with the Securities and Exchange Commission on August 7, 2015
Registration No. 333-
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
ARALEZ PHARMACEUTICALS LIMITED
(Exact name of Registrant as Specified in Its Charter)
|
Ireland |
|
2834 |
|
Not Applicable |
|
(State or Other Jurisdiction of |
|
(Primary Standard Industrial |
|
(IRS Employer |
|
Incorporation or Organization) |
|
Classification Code Number) |
|
Identification No.) |
56 Fitzwilliam Square
Dublin 2, Ireland
(+353 1 905-3581)
(Address, Including Zip Code, and Telephone Number, Including Area Code, of Registrant’s Principal Executive Offices)
Adrian Adams
Chief Executive Officer
Aralez Pharmaceuticals Limited
1414 Raleigh Road, Suite 400
Chapel Hill, North Carolina 27517
(919) 913-1030
(Name, Address, Including Zip Code, and Telephone Number, Including Area Code, of Agent for Service)
Copies to:
Andrew P. Gilbert, Esq.
David C. Schwartz, Esq.
DLA Piper LLP (US)
51 John F. Kennedy Parkway
Short Hills, New Jersey 07078
(973) 520-2550
Approximate date of commencement of proposed sale to the public: As soon as practicable after this registration statement becomes effective.
If any of the securities being registered on this form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box.x
If this form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.o
If this form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.o
If this form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.o
Indicate by check mark whether registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (check one):o Large accelerated filer, x Accelerated filer, o Non-accelerated filer (do not check if a smaller reporting company) or o Smaller reporting company.
CALCULATION OF REGISTRATION FEE
|
Title of Each Class of |
|
Amount |
|
Proposed |
|
Proposed |
|
Amount of |
| |||
|
Ordinary shares, $0.001 nominal value per share |
|
7,861,636 |
|
$ |
11.67 |
|
$ |
91,745,292 |
|
$ |
10,661 |
|
(1) The ordinary shares will be offered for resale by the selling shareholders pursuant to the prospectus contained herein. Pursuant to Rule 416 under the Securities Act of 1933, as amended, this Registration Statement also covers any additional shares that may be offered or issued in connection with any stock split, stock dividend or similar transaction.
(2) Pursuant to Rule 457(c) under the Securities Act, the offering price and registration fee are computed based on the average of the high and low prices reported for POZEN Inc.’s common stock traded on The NASDAQ Global Select Market on August 3, 2015. Aralez Pharmaceuticals plc will be the successor issuer to POZEN Inc. pursuant to the transactions described herein.
(3) The registration fee of $10,661 is being paid at the time of this filing.
The information in this prospectus is not complete and may be changed. The selling shareholders may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities, and selling shareholders are not soliciting offers to buy these securities, in any jurisdiction where the offer or sale is not permitted.
PRELIMINARY PROSPECTUS, SUBJECT TO COMPLETION, DATED AUGUST 7, 2015
Prospectus
7,861,636 Shares
ARALEZ PHARMACEUTICALS PLC
Ordinary Shares
This prospectus relates to the resale of up to 7,861,636 of our ordinary shares, which may be offered for sale from time to time by the selling shareholders named in this prospectus. Our ordinary shares covered by this prospectus (the “Shares”) are to be issued by us to the selling shareholders upon conversion of the convertible notes to be issued in a private placement pursuant to a Debt Facility Agreement, dated as of June 8, 2015, as more fully described in this prospectus.
The selling shareholders may from time to time sell, transfer or otherwise dispose of any or all of their Shares in a number of different ways and at varying prices. We will not receive any proceeds from the sale of Shares by the selling shareholders.
We will be the successor issuer to POZEN Inc. (“Pozen”) pursuant to the transactions described in this prospectus. Shares of Pozen are listed on The NASDAQ Global Select Market under the symbol “POZN”. Our ordinary shares will be listed on The NASDAQ Stock Market LLC (“NASDAQ”) under the symbol “ARLZ” and on the Toronto Stock Exchange (“TSX”) under the symbol “ARZ” following the closing of the merger with Tribute Pharmaceuticals Canada Inc. (“Tribute”), as described in more detail in this prospectus. Shares of Pozen will be delisted from the NASDAQ Global Select Market following the commencement of trading of our ordinary shares.
Investing in our ordinary shares involves risks. You should carefully consider the risks that we have described in “Risk Factors” relating to Pozen and Tribute beginning on page 5 and page 25 of this prospectus, respectively, and under similar headings in any amendments or supplements to this prospectus, before investing in the Shares.
Neither the Securities and Exchange Commission nor any state securities commission, nor any securities regulatory authority in Canada or Ireland, has approved or disapproved of these securities or passed on the adequacy or accuracy of this prospectus or determined that this proxy statement/prospectus is accurate or complete. Any representation to the contrary is a criminal offense. This proxy statement/prospectus does not constitute an offer to buy or sell, or a solicitation of an offer to buy or sell, any securities, or a solicitation of a proxy, in any jurisdiction to or from any person to whom it is unlawful to make any such offer or solicitation in such jurisdiction. For the avoidance of doubt, the accompanying proxy statement/prospectus is not intended to be and is not a prospectus for the purposes of the Companies Act 2014 of Ireland (the ‘‘Companies Act’’), the Prospectus (Directive 2003/71/EC) Regulations 2005 of Ireland or the Prospectus Rules of Ireland, and the Central Bank of Ireland has not approved this document.
You should rely only on the information contained in this prospectus or any prospectus supplement or amendment. Neither we nor the selling shareholders have authorized anyone to provide you with different information. The selling shareholders are not making an offer of their Shares in any state where such offer is not permitted.
The date of this prospectus is 2015.
|
|
2 |
| |
|
|
2 |
| |
|
|
5 |
| |
|
|
36 |
| |
|
|
37 |
| |
|
|
38 |
| |
|
|
39 |
| |
|
|
41 |
| |
|
|
43 |
| |
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
|
49 |
|
|
|
66 |
| |
|
|
104 |
| |
|
|
104 |
| |
|
|
108 |
| |
|
|
140 |
| |
|
|
142 |
| |
|
|
144 |
| |
|
|
145 |
| |
|
|
149 |
| |
|
|
157 |
| |
|
|
161 |
| |
|
|
161 |
| |
|
|
162 |
| |
|
INDEX TO FINANCIAL STATEMENTS |
|
F-1 |
|
This prospectus relates to the resale of an aggregate of 7,861,636 ordinary shares of Aralez Pharmaceuticals plc by the shareholders listed herein under the Section entitled Selling Shareholders. Pursuant to the Debt Facility Agreement dated June 8, 2015 (the “Facility Agreement”) by and among POZEN Inc. (“Pozen” or the “Company”), Aralez Pharmaceuticals Limited (“we”, “us”, “our”, “Aralez” or “Parent”) (or a wholly-owned subsidiary of the Parent, depending on whether certain conditions of the Facility Agreement occur) (the “Borrower”), Deerfield Private Design Fund III, L.P. (“Deerfield Private Design”), Deerfield International Master Fund, L.P. (“Deerfield International”), Deerfield Partners, L.P. (“Deerfield Partners” together with Deerfield Private Design and Deerfield International, the “Selling Shareholders”), following the consummation of the transactions contemplated by the Agreement and Plan of Merger and Arrangement dated as of June 8, 2015 (the “merger agreement”), among Tribute, Aguono Limited (which was renamed Aralez Pharmaceuticals Limited and which, prior to the effectiveness of the merger, will re-register as a public limited company incorporated in Ireland and be renamed as Aralez Pharmaceuticals plc), Trafwell Limited (which was renamed Aralez Pharmaceutical Holdings Limited) (“Ltd2”), ARLZ US Acquisition Corp. (“US Merger Sub”), ARLZ CA Acquisition Corp. (“Can Merger Sub”) and Pozen, the Borrower will borrow from the Selling Shareholders up to an aggregate principle amount of $275 million, of which $75 million will be in the form of a 2.5% senior secured convertible promissory note due six years from issuance and convertible into Parent Shares at a conversion price of $9.54 per share (the “Convertible Notes”), issued and sold by Borrower to Deerfield Private Design or its registered assigns, upon the terms and conditions of the Facility Agreement. Pursuant to the Facility Agreement, the Selling Shareholders will receive an aggregate of 7,861,636 ordinary shares of Aralez upon conversion of the Convertible Notes.
No person has been authorized to give any information or make any representation concerning us, the Selling Shareholders or the Shares to be registered hereunder (other than as contained in this prospectus) and, if any such other information or representation is given or made, you should not rely on it as having been authorized by us or the Selling Shareholders. You should not assume that the information contained in this prospectus is accurate as of any date other than the date on the front cover of this prospectus or as otherwise set forth in this prospectus.
The Selling Shareholders are offering the Shares only in jurisdictions where such issuances are permitted. The distribution of this prospectus and the sale of the Shares in certain jurisdictions may be restricted by law. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the distribution of this prospectus and the sale of the Shares outside the United States. This prospectus does not constitute, and may not be used in connection with, an offer to sell, or a solicitation of an offer to buy, the Shares by any person in any jurisdiction in which it is unlawful for such person to make such an offer or solicitation.
Background of Aralez Pharmaceuticals plc
Upon completion of the merger, Aralez will become the successor issuer to Pozen. Pozen formed Aralez to serve as the new parent company of Pozen and Tribute following consummation of the merger and plan of arrangement. Pozen was incorporated in the State of Delaware on September 25, 1996 and is operating in a single reportable segment. Pozen has been a pharmaceutical company committed to transforming medicine that transforms lives. Since inception, the Company has focused its efforts on developing products which can provide improved efficacy, safety or patient convenience in the treatment of acute and chronic pain and pain related conditions and has developed a portfolio of integrated aspirin therapies. Historically, the Company has entered into collaboration agreements to commercialize its product candidates. The Company’s licensing revenues include upfront payments, additional payments if and when certain milestones in the product’s development or commercialization are reached, and the eventual royalty payments based on product sales.
The Company has decided to retain ownership of its lead product candidates, the proprietary, investigational, coordinated-delivery tablets combining immediate-release omeprazole, a proton pump inhibitor, or PPI, and enteric coated, or EC, aspirin in a single tablet, now known as YOSPRALA 81/40 and 325/40 (aspirin/omeprazole delayed release tablets) (“PA” or “YOSPRALA”).
On June 8, 2015, Pozen and Tribute agreed to a business combination under the terms of the merger agreement. In order to effect the transactions contemplated by the merger agreement, US Merger Sub, an indirect subsidiary of Parent, will be merged with and into Pozen (the “merger”). Pozen will be the surviving corporation and, through the merger, will become an indirect wholly owned subsidiary of Parent. The merger of Pozen into US Merger Sub will be effected under Delaware law so that Pozen will be reorganized into a holding company structure. In accordance with the merger agreement, Can Merger Sub will offer to acquire, and will acquire, all of the outstanding Tribute common shares pursuant to a court approved plan of arrangement in Canada in the manner provided for by the merger agreement (the “arrangement”). The Parent Shares (as defined below) to be issued to Tribute shareholders in the arrangement are not being registered pursuant to this registration statement. Upon completion of the arrangement, Tribute will also become an indirect wholly owned subsidiary of Parent. Upon completion, the merger and the arrangement do not constitute a change of control of Pozen.
As a result of the merger, each share of Pozen common stock will be converted into the right to receive from Parent one ordinary share of Parent, $0.001 nominal value per share (the “Parent Shares”) (the “merger consideration”) for each share of Pozen common stock that they own as of the record date for the special meeting of Pozen stockholders to vote on the proposals set forth in the Form S-4 (as defined below) (the “Pozen special meeting”). The Parent Shares to be issued to Pozen stockholders pursuant to the merger are not being registered pursuant to this registration statement. Pursuant to the arrangement, each outstanding
Tribute common share will be exchanged for 0.1455 Parent Shares. Upon completion of the merger and arrangement, current Pozen stockholders will own approximately 66% of the outstanding Parent Shares, and current Tribute shareholders will own approximately 34% of the outstanding Parent Shares before giving effect to (i) any exercise of outstanding options and warrants or the vesting and delivery of shares underlying restricted stock units (“RSUs”) of either company and (ii) the Parent Shares to be issued to new investors pursuant to the equity and debt financings described below. It is expected that Parent Shares will be listed and traded on NASDAQ under the symbol “ARLZ” and on the “TSX” under the symbol “ARZ”. The terms and conditions of the merger and the arrangement are contained in the merger agreement. After giving effect to the transactions and the proposed Parent equity financing and debt financing, the former stockholders of Pozen as a group will hold Parent Shares constituting approximately 49% of the outstanding Parent Shares and the former shareholders of Tribute as a group will hold Parent Shares constituting approximately 28% of the outstanding Parent Shares.
On July 20, 2015, Parent filed with the Securities and Exchange Commission (the “SEC”) a registration statement on Form S-4, which includes a preliminary proxy statement of Pozen and also constitutes a preliminary prospectus of Parent (the “Form S-4”), in connection with the proposed business combination between Pozen and Tribute. The Form S-4 has not yet been declared effective by the SEC.
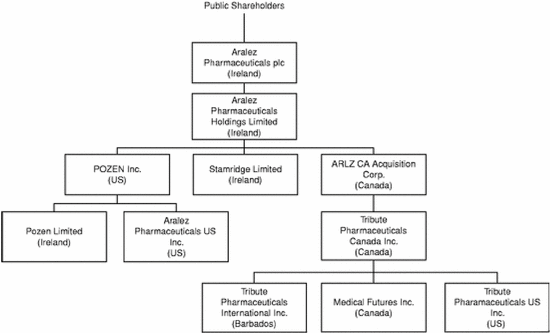
Below is a complete corporate chart of Parent and its subsidiaries immediately following the effective time of the merger and the arrangement:
Equity Financing
On June 8, 2015, Pozen executed the Subscription Agreement by and among Purchaser, Tribute, Parent, Pozen and the Selling Shareholders. Pursuant to the Subscription Agreement, Parent will sell to Purchaser and the Selling Shareholders up to $75 million of the Parent Shares in a private placement at a purchase price of $7.20 per Parent Share. The Subscription Agreement provides that Parent will prepare and file two registration statements with the SEC on such form as may be required to effect a registration of the Parent Shares issued under the Subscription Agreement within 60 days of the date of the signing of the Subscription Agreement and for certain other registration rights for each of Purchaser and the Investors under the Securities Act and the rules and regulations thereunder, or any similar successor statute, and applicable state securities laws.
Debt Financing
On June 8, 2015, Pozen also executed a Debt Facility Agreement (the “Facility Agreement”) among Pozen, the Parent (or a wholly-owned subsidiary of the Parent, depending on whether certain conditions of the Facility Agreement occur) (the “Borrower”), Deerfield Private Design, Deerfield International, Deerfield Partners, and the other lender parties thereto (together with Deerfield Private Design, Deerfield International, and Deerfield Partners, the “Lenders”). Pursuant to the Facility Agreement, the Borrower will borrow from the Lenders up to an aggregate principal amount of $275 million, of which (i) $75 million will be in the form of a 2.5% senior secured convertible promissory note due six years from issuance and convertible into Parent Shares at a conversion price of $9.54 per share (the “Convertible Notes”), issued and sold by Borrower to Deerfield Private Design or its registered assigns, upon the
terms and conditions of the Facility Agreement, and (ii) up to an aggregate principal amount of $200 million with an interest rate of 12.5%, which will be made available for Permitted Acquisitions (as defined in the Facility Agreement), will be in the form of secured promissory notes issued and sold by the Borrower to the Lenders (the “Acquisition Notes”), evidencing the Acquisition Loans, upon the terms and conditions and subject to the limitations set forth in the Acquisition Notes, all subject to the terms and conditions of the Facility Agreement.
In connection with the Facility Agreement, on June 8, 2015, the Lenders and Parent entered into a Registration Rights Agreement (the “Registration Rights Agreement”). Pursuant to the Registration Rights Agreement, Parent has agreed to prepare and file with the SEC a Registration Statement on Form S-3, or other such form as required to effect a registration of the Parent Shares issued or issuable upon conversion of or pursuant to the Convertible Notes (the “Registerable Securities”), covering the resale of the Registerable Securities and such indeterminate number of additional ordinary shares as may become issuable upon conversion of or otherwise pursuant to the Convertible Notes to prevent dilution resulting from certain corporate actions. Such Registration Statement must be filed within 45 calendar days following the date of issuance of the Convertible Notes. In the event the SEC does not permit all of the Registerable Securities to be included in the Registration Statement or if the Registerable Securities are not otherwise included in a Registration Statement filed under the Registration Rights Agreement, the Parent has agreed to file an additional Registration Statement by no later than the Additional Filing Deadline (as defined in the Registration Rights Agreement) covering the resale of all Registerable Securities not already covered by an existing and effective Registration Statement for an offering to be made on a continuous basis pursuant to Rule 415 of the Securities Act of 1933, as amended (the “Securities Act”). The Registration Rights Agreement also provides for piggy-back registration, subject to the terms and conditions of the Registration Rights Agreement. The ordinary shares offered hereby by the Selling Shareholder will be issued upon conversion of the Convertible notes to be issued pursuant to the Facility Agreement.
Investing in our ordinary shares involves a high degree of risk. You should carefully consider the following risk factors and all other information contained in this prospectus, in addition to the other information contained in this prospectus. You should also read and consider the risks associated with the business of Tribute and the risks associated with the business of Pozen, because these risks will also affect Aralez. The risks associated with the business of Tribute can be found in Tribute’s Annual Report on Form 10-K for the fiscal year ended December 31, 2014, and Tribute’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2015, under the heading “Risk Factors”. The risks associated with the business of Pozen can be found in Pozen’s Annual Report on Form 10-K for the fiscal year ended December 31, 2014, and Pozen’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2015, under the heading “Risk Factors.” See the section entitled “Where You Can Find More Information” beginning on page [·] of this prospectus.
Risk Factors Related to the Pozen Business
We have incurred losses since inception and we may continue to incur losses for the foreseeable future. Product revenue for products which we license out is dependent upon the commercialization efforts of our partners, including the sales and marketing efforts of AstraZeneca AB (“AstraZeneca”) and Horizon Pharma Inc. (“Horizon”) relating to VIMOVO®.
We have incurred significant losses since our inception. As of March 31, 2015, our accumulated deficit was approximately $96.9 million. Our ability to receive product revenue from the sale of products is dependent on a number of factors, principally the development, regulatory approval and successful commercialization of our product candidates. We expect that the amount of our operating losses will fluctuate significantly from quarter to quarter principally as a result of increases and decreases in our development efforts and the timing and amount of payments that we may receive from others. We expect to continue to incur significant operating losses associated with our research and development efforts and do not know the amount or timing of product revenue we will receive as a result of sales of VIMOVO by AstraZeneca and Horizon or our other product candidates, including PA. If our licensed products do not perform well in the marketplace our royalty revenue will impacted and our business could be materially harmed.
Our primary current source of revenue is the royalty payments that we may receive pursuant to our collaboration agreement with AstraZeneca. We have received all regulatory milestone payments under our collaboration agreement with AstraZeneca. On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understand that AstraZeneca will instead now focus on those countries where the product has shown growth and which AstraZeneca believes have the greatest potential for future growth. On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. As a result, royalty revenues for sales of VIMOVO in the United States will be received from Horizon. On July 28, 2014, Horizon announced that it had been verbally notified by CVS Caremark and Express Scripts, Inc. that VIMOVO would no longer be on their formularies and will be placed on their exclusion lists effective January 1, 2015. Horizon estimates that approximately 20-30% of VIMOVO prescriptions could be impacted by these decisions.
We depend heavily on the success of our product candidates, which may never be approved for commercial use. If we are unable to develop, gain approval of or commercialize those product candidates, we will never receive revenues from the sale of our product candidates.
We anticipate that for the foreseeable future our ability to achieve profitability will be dependent on the successful commercialization of VIMOVO and, if approved, sales of our PA product candidates. If we fail to gain timely approval to commercialize our products from the United States Food and Drug Administration (the “FDA”) and other foreign regulatory bodies, we will be unable to generate the revenue we will need to build our business. These approvals may not be granted on a timely basis, if at all, and even if and when they are granted, they may not cover all of the indications for which we seek approval. For example, absent the availability of such a lower dose formulation in the market if PA32540 is approved, the FDA indicated that it may limit the indication for PA32540 to use in post coronary artery bypass graft surgery (CABG) with a treatment duration not to exceed one year. On April 25, 2014, we received a Complete Response Letter (the “CRL”) from the FDA because of deficiencies noted during an inspection of a supplier of an active ingredient used in the manufacture of the PA products, which has delayed approval of our NDA. On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. Satisfactory resolution of these deficiencies is required before this application may be approved. The active ingredient supplier has informed us that they received a warning letter relating to the Form 483 inspection deficiencies and have submitted a plan of corrective actions to address the matters raised in the warning letter to FDA. There were no clinical or safety deficiencies noted with respect to either PA32540 or PA8140 and no other deficiencies were noted in the CRL. Final agreement on the draft product labeling is also pending. There can be no assurance that our PA products will be approved by the FDA and, if so approved, for the expected indication.
Many factors could negatively affect our ability to obtain regulatory approval for our product candidates. For example, in October 2008, the FDA informed us that it was conducting an internal review of the acceptability of using endoscopic gastric ulcers as a primary endpoint in clinical trials. Reduction of endoscopic gastric ulcers was the primary endpoint in our Phase 3 trials for VIMOVO and the primary endpoint in the ongoing Phase 3 trials for our PA32540 product. In late January 2009, the FDA informed us that it had completed its internal discussions and that there was no change to previous agreements that gastric ulcer incidence was an acceptable primary endpoint for our clinical programs. The FDA decided to obtain further advice on this issue and held an Advisory Board meeting on November 4, 2010 to discuss the adequacy of endoscopically documented gastric ulcers as an outcome measure to evaluate drugs intended to prevent gastrointestinal complications of non-steroidal anti-inflammatory drugs, including aspirin. The Advisory Board voted in favor (8-4) that endoscopically-documented gastric/duodenal ulcers are an adequate primary efficacy endpoint for evaluating products intended to prevent non-steroidal anti-inflammatory drug (NSAID)-associated upper gastrointestinal (UGI) toxicity, which vote supports the clinical design of the pivotal Phase 3 trials conducted for VIMOVO and PA32540. However, there can be no assurance that FDA will continue to accept the recommendation of the Advisory Board or will not decide to reassess the acceptability of this endpoint in the future.
In addition to the inability to obtain regulatory approval, many other factors could negatively affect the success of our efforts to develop and commercialize our product candidates, including those discussed in the risk factors that follow as well as negative, inconclusive or otherwise unfavorable results from any studies or clinical trials, such as those that we obtained with respect to MT 500, which led to our decision to discontinue development of that product candidate in 2002.
Changes in regulatory approval policy or statutory or regulatory requirements, or in the regulatory environment, during the development period of any of our product candidates may result in delays in the approval, or rejection, of the application for approval of one or more of our product candidates. If we fail to obtain approval, or are delayed in obtaining approval, of our product candidates, our ability to generate revenue will be severely impaired.
The process of drug development and regulatory approval for product candidates takes many years, during which time the FDA’s interpretations of the standards against which drugs are judged for approval may evolve or change. The FDA can also change its approval policies based upon changes in laws and regulations. In addition, the FDA can decide, based on its then current approval policies, any changes in those policies and its broad discretion in the approval process, to weigh the benefits and the risks of every drug candidate. As a result of any of the foregoing, the FDA may decide that the data we submit in support of an application for approval of a drug candidate are insufficient for approval. For example, in October 2008, the FDA has informed us that it was conducting an internal review of the acceptability of using endoscopic gastric ulcers as a primary endpoint in clinical trials. Reduction of endoscopic gastric ulcers was the primary endpoint in our Phase 3 trials for VIMOVO (formerly referred to as PN 400) and the primary endpoint in our on-going Phase 3 trials for PA32540. In late January 2009, the FDA informed us that it had completed its internal discussions and that there was no change to previous agreements that gastric ulcer incidence was an acceptable primary endpoint for our clinical programs. The FDA decided to obtain further advice on this issue and held an Advisory Board meeting on November 4, 2010 to discuss the adequacy of endoscopically documented gastric ulcers as an outcome measure to evaluate drugs intended to prevent gastrointestinal complications of non-steroidal anti-inflammatory drugs, including aspirin. The Advisory Board voted in favor (8-4) that endoscopically-documented gastric/duodenal ulcers are an adequate primary efficacy endpoint for evaluating products intended to prevent NSAID-associated UGI toxicity, which vote supports the clinical design of the pivotal Phase 3 trials conducted for VIMOVO and PA32540. However, there can be no assurance that FDA will accept the recommendation of the Advisory Board or will not decide to reassess the acceptability of this endpoint in the future.
Changes in policy or interpretation may not be the subject of published guidelines and may therefore be difficult to evaluate. For example, in February 2012, the FDA requested we demonstrate the bioequivalence of PA32540 to EC aspirin 325 mg, with respect to acetylsalicylic acid in an additional Phase 1 study. EC products such as PA32540 and EC aspirin 325 mg have highly variable pharmacokinetics. Based on our analyses, we believed that the results demonstrated bioequivalence, but the FDA did not agree. However, the FDA did agree that the results from this Phase 1 study, together with additional information that was submitted by us in the NDA for the product, constitutes sufficient data to support the establishment of a clinical and pharmacological bridge between the product and EC aspirin 325 mg. There can be no assurance that our PA products will be approved by the FDA and, if so approved, for the expected indications.
As another example, the FDA has not recently published guidelines for the approval of new migraine therapies, and we have had to rely on periodic guidance from the FDA obtained (in conversations) informally and in other meetings, the content of which may be subject to significant modification over the period of a drug’s development program. There is also the risk that we and the FDA may interpret such guidance differently. The FDA made several changes to the omeprazole label that relate, in part, to the agency’s concern regarding certain reported adverse events in patients taking long term PPI such as omeprazole. For example, with VIMOVO, in dosage and administration, the label states to use the lowest effective dose for the shortest duration consistent with individual patient treatment goals. There is a risk that further omeprazole safety issue may arise in the future that could impact FDA’s benefit/risk assessment of the dose or duration of PPI in subjects requiring long-term PPI use.
Further, additional information about the potential risks of marketed drugs may affect the regulatory approval environment, or the FDA’s approval policies, for new product candidates. For example, in February 2005 an advisory committee convened by the FDA met to address the potential cardiovascular risks of COX-2 selective NSAIDs and related drugs in response to disclosures made about possible adverse effects from the use of some of these drugs. On April 7, 2005, the FDA issued a Public Health Advisory, or the Advisory, based, in part, upon the recommendations of the advisory committee. The Advisory stated that it would require that manufacturers of all prescription products containing NSAIDs provide warnings regarding the potential for adverse cardiovascular events as well as life-threatening gastrointestinal events associated with the use of NSAIDs. Moreover, subsequent to the FDA advisory committee meeting in February 2005, the FDA has indicated that long-term studies evaluating cardiovascular risk will be required for approval of new NSAID products that may be used on an intermittent or chronic basis. Long-term cardiovascular safety studies were not required at for FDA approval of our VIMOVO. However, we cannot guarantee that such studies will not be required in the future if new information about naproxen safety concerns becomes available. We will continue to evaluate and review with the FDA its expectations and recommendations regarding the efficacy and safety requirements and study design necessary to support approval of NDAs for product candidates we may develop that contain NSAIDs.
If we, or our current or future collaborators, do not obtain and maintain required regulatory approvals for one or more of our product candidates, we will be unable to commercialize those product candidates. Further, if we are delayed in obtaining or unable to obtain, any required approvals, or our contract manufacturers are unable to manufacture and supply product for sale, our collaborators may terminate, or be entitled to terminate, their agreements with us or reduce or eliminate their payments to us under these agreements or we may be required to pay termination payments under these agreements.
Our product candidates under development are subject to extensive domestic and foreign regulation. In the U.S., an NDA or supplement must be filed with respect to each indication for which marketing approval of a product is sought. Each NDA, in turn, requires the successful completion of preclinical, toxicology, genotoxicity and carcinogenicity studies, as well as clinical trials demonstrating the safety and efficacy of the product for that particular indication. We may not receive regulatory approval of any of the NDAs that we file with the FDA or of any approval applications we may seek in the future outside the U.S.
The FDA regulates, among other things, the development, testing, manufacture, safety, efficacy, record keeping, labeling, storage, approval, advertisement, promotion, sale and distribution of pharmaceutical products in the U.S. In order to market our products abroad, we must comply with extensive regulation by foreign governments. If we are unable to obtain and maintain FDA and foreign government approvals for our product candidates, we, alone or through our collaborators, will not be permitted to sell them. Failure to obtain regulatory approval for a product candidate will prevent us from commercializing that product candidate. Except for Treximet®, which was approved for commercial sale in the U.S. on April 15, 2008, and VIMOVO, which was approved for commercial sale in the U.S. on April 30, 2010 and has been approved in a number of additional countries in the rest of the world, none of our other product candidates are approved for sale in the U.S. or any foreign market and they may never be approved. For example, we received two approvable letters relating to our NDA for Treximet which communicated the FDA’s concerns that delayed marketing approval. An approvable letter, now called a CRL, is an official notification from the FDA that contains conditions that must be satisfied prior to obtaining final U.S. marketing approval. In June 2006, we received the first approvable letter in which the FDA requested additional safety information on Treximet, and in August 2007, we received a second approvable letter in which the FDA requested that we address their concern about the potential implications from one preclinical in vitro chromosomal aberration study in which a signal for genotoxicity was seen for the combination of naproxen sodium and sumatriptan. We have also previously received not-approvable letters from the FDA relating to our NDAs for MT 100 and MT 300. On April 25, 2014, we received a CRL advising that the review of our NDA is completed and questions remain that preclude the approval of the NDA for our PA32540 and PA8140 product candidates. Specifically, an inspection of the manufacturing facility of an active ingredient supplier of ours concluded with certain inspection deficiencies. On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. The active ingredient supplier has informed us that they received a warning letter relating to the Form 483 inspection deficiencies and have submitted a plan of corrective actions to address the matters raised in the warning letter to FDA. Satisfactory resolution of these deficiencies is required before this application may be approved.
Further, our current or future collaboration agreements may terminate, or require us to make certain payments to our collaborators, or our collaborators may have the right to terminate their agreements with us or reduce or eliminate their payments to us under these agreements, based on our inability to obtain, or delays in obtaining, regulatory approval for our product candidates or our contract manufacturers’ inability to manufacture our products or to supply the sufficient quantities of our products to meet market demand. For example, under our PN collaboration agreement with AstraZeneca, AstraZeneca had the right to terminate the agreement if certain delays occurred or specified development and regulatory objectives were not met. For example, this termination right could have been triggered by AstraZeneca if the FDA had determined that endoscopic gastric ulcers were no longer an acceptable endpoint and we had been required to conduct additional trials which would have delayed NDA approval for VIMOVO. Both AstraZeneca, Horizon and Pernix Therapeutics Holdings, Inc. have the right to terminate their respective agreements with us upon a 90 day notice for any reason. Further, if we or our contract manufacturers do not maintain required regulatory approvals, or our contract manufacturers are unable to manufacture our product or to supply sufficient quantities of our products to meet market demand, we may not be able to commercialize our products.
Approval of a product candidate may be conditioned upon certain limitations and restrictions as to the drug’s use, such as a possible warning that the FDA may require in the PA32540 label regarding the concomitant use of PA32540 and Plavix®, or upon the conduct of further studies, and is subject to continuous review. The FDA has indicated that, absent the availability of such a lower dose formulation in the market if PA32540 is approved, that it may limit the indication for PA32540 to use in CABG with a treatment duration not to exceed one year. We believe that the FDA is concerned that without a formulation containing a lower dose of aspirin, physicians will not have a full range of dosing options available to prescribe in accordance with current cardiovascular treatment guidelines, which recommend doses of 81 mgs or 162 mgs of aspirin for most indications and we followed the FDA’s suggestion to seek approval for a lower dose formulation of the product containing 81 mg of EC aspirin as part of our NDA for PA32540. However, there can be no assurance that the FDA will approve a lower dose formulation of the product or will allow a broader indication for PA32540. There can be no assurance that our PA products will be approved by the FDA and, if so approved, for the expected indications. The FDA may also require us to conduct additional post-approval studies. These post-approval studies may include carcinogenicity studies in animals or further human clinical trials. The later discovery of previously unknown problems with the product, manufacturer or manufacturing facility may result in criminal prosecution, civil penalties, recall or seizure of products, or total or partial suspension of production, as well as other regulatory action against our product candidates or us. If approvals are withdrawn for a product, or if a product is seized or recalled, we would be unable to sell that product and therefore would not receive any revenues from that product.
We and our contract manufacturers are required to comply with the applicable FDA current Good Manufacturing Practices, or cGMP, regulations, which include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. Further, manufacturing facilities must be approved by the FDA before they can be used to manufacture our product candidates, and are subject to additional FDA inspection. On April 25, 2014, we announced that we had received a CRL from the FDA with respect to the NDA for our PA32540 and PA8140 product candidates. In the CRL, the FDA noted that, during an inspection of the manufacturing facility of an active ingredient supplier, inspection deficiencies were found. Satisfactory resolution of deficiencies noted by the field investigator is required before the NDA may be approved. On May 9, 2014, our active ingredient supplier submitted a response to the FDA addressing the inspection deficiencies and subsequently submitted an update to its initial response. On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. The active ingredient supplier has informed us that they received a warning letter relating to the Form 483 inspection deficiencies and have submitted a plan of corrective actions to address the matters raised in the warning letter to FDA. Satisfactory resolution of these deficiencies is required before this application may be approved. There were no clinical or safety deficiencies noted with respect to either PA32540 or PA8140 and no other deficiencies were noted in the CRL.
Manufacturing facilities may also be subject to state regulations. We, or our third-party manufacturers, may not be able to comply with cGMP regulations or other FDA regulatory requirements, or applicable state regulations, or may not be able to successfully manufacture our products which could result in a delay or an inability to manufacture the products. If we or our partners wish or need to identify an alternative manufacturer, delays in obtaining FDA approval of the alternative manufacturing facility could cause an interruption in the supply of our products.
Labeling and promotional activities are subject to scrutiny by the FDA and state regulatory agencies and, in some circumstances, the Federal Trade Commission, or FTC. FDA enforcement policy prohibits the marketing of unapproved products as well as the marketing of approved products for unapproved, or off-label, uses. These regulations and the FDA’s interpretation of them may limit our or our partners’ ability to market products for which we gain approval. Failure to comply with these requirements can result in federal and state regulatory enforcement action. Further, we may not obtain the labeling claims we or our partners believe are necessary or desirable for the promotion of our product candidate. As part of the CRLs received in connection with our PA32540 and PA8140 products, FDA indicated that the final agreement on draft product labeling remains pending.
If third parties challenge the validity of the patents or proprietary rights of our marketed products or assert that we have infringed their patents or proprietary rights, we may become involved in intellectual property disputes and litigation that would be costly and time consuming and could negatively impact the commercialization of our products that we develop or acquire. We have received a Paragraph IV Notice Letters notifying us of the filing of ANDAs with the FDA for approval to market a generic version of VIMOVO. We previously received Paragraph IV Letters notifying us of the filing of Abbreviated New Drug Applications (ANDAs) with the FDA for approval to market a generic version of Treximet and those cases have been concluded. We filed patent infringement lawsuits in response to these ANDAs that has led and will continue to lead to costly and time consuming patent litigation.
The intellectual property rights of pharmaceutical companies, including us, are generally uncertain and involve complex legal, scientific and factual questions. Our success in developing and commercializing pharmaceutical products may depend, in part,
on our ability to operate without infringing on the intellectual property rights of others and to prevent others from infringing on our intellectual property rights. There has been substantial litigation regarding patents and other intellectual property rights in the pharmaceutical industry. For example, third parties seeking to market generic versions of branded pharmaceutical products often file ANDAs with the FDA, containing a certification stating that the ANDA applicant believes that the patents protecting the branded pharmaceutical product are invalid, unenforceable and/or not infringed. Such certifications are commonly referred to as paragraph IV certifications.
We, AstraZeneca and Horizon are also engaged in Paragraph IV litigation with several generic pharmaceutical companies with respect to patents listed in the Orange Book with respect to VIMOVO currently pending in the United States District Court for the District of New Jersey and in an inter partes review (IPR) brought by Dr. Reddy’s with respect to the ‘285 patent and in two IPRs brought by the Coalition for Affordable Drugs (CFAD). We and AstraZeneca are also engaged in a proceeding in Canada with Mylan Canada which is seeking approval of its generic version of VIMOVO in Canada prior to the expiration of our Canadian patent.
Litigation can be time consuming and costly and we cannot predict with certainty the outcome. If we are unsuccessful in any of the above-described proceedings and the FDA approves a generic version of one of our marketed products, such an outcome would have a material adverse effect on sales of such product, our business and our results of operations.
Our reliance on collaborations with third parties to develop and commercialize our products is subject to inherent risks and may result in delays in product development and lost or reduced revenues, restricting our ability to commercialize our products and adversely affecting our profitability.
With respect to the products we have licensed, we depend upon collaborations with third parties to develop these product candidates and we depend substantially upon third parties to commercialize these products. As a result, our ability to develop, obtain regulatory approval of, manufacture and commercialize our existing and possibly future product candidates depends upon our ability to maintain existing, and enter into and maintain new, contractual and collaborative arrangements with others. We also engage, and may in the future to continue to engage, contract manufacturers and clinical trial investigators.
In addition, the identification of new compounds or product candidates for development has led us in the past, and may continue to require us, to enter into license or other collaborative agreements with others, including pharmaceutical companies and research institutions. Such collaborative agreements for the acquisition of new compounds or product candidates would typically require us to pay license fees, make milestone payments and/or pay royalties. Furthermore, these agreements may result in our revenues being lower than if we developed our product candidates ourselves and in our loss of control over the development of our product candidates.
Contractors or collaborators may have the right to terminate their agreements with us after a specified notice period for any reason or upon a default by us or reduce their payments to us under those agreements on limited or no notice and for no reason or reasons outside of our control. For example, we had a collaboration agreement with Desitin Arzneimittel GmbH, or Desitin, for the development and commercialization of MT 400 for the 27 countries of the European Union, as well as Switzerland and Norway, but on February 27, 2013, we received written notice from Desitin that it was terminating the license agreement due to reimbursement uncertainty for MT 400 in Germany, a major market for Desitin in the territory. We can also mutually agree with our collaborators to terminate the agreements. For example, on November 29, 2014, we executed a termination agreement with sanofi-aventis U.S. LLC (“Sanofi U.S.” or “Sanofi”) terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi U.S. were terminated and all rights to the products licensed to Sanofi U.S. under the agreement reverted to us. In December 2014, we received a mutual termination letter from Cilag GmbH International (“Cilag”), a division of Johnson & Johnson, terminating our then-current License Agreement with Cilag, for the exclusive development and commercialization of MT 400 in Brazil, Colombia, Ecuador and Peru.
Collaborators may also decide not to continue marketing our products in certain countries of the territory or to assign their rights under our agreement to third parties. For example, we had a collaboration with GlaxoSmithKline (“GSK”) for the development and commercialization of certain triptan combinations using our MT 400 technology, including Treximet, in the U.S. and GSK entered into an agreement to divest of all of its rights, title and interest to develop, commercialize and sell the licensed products in the U.S. to Pernix . In addition, on May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understood that AstraZeneca would instead focus on those countries where the product has shown growth and which AstraZeneca believed had the greatest potential for future growth. On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon.
Contractors or collaborators may have the right to reduce their payments to us under those agreements. For example, Pernix, and AstraZeneca and Horizon have the right to reduce the royalties on net sales of products payable to us under their respective agreements if generic competitors enter the market and attain a pre-determined share of the market for products marketed under the agreements, or if they must pay a royalty to one or more third parties for rights it licenses from those third parties to commercialize
products marketed under the agreements. AstraZeneca was also entitled to terminate its agreement with us if certain delays occur or specified development or regulatory objectives were not met. This termination could have been triggered by AstraZeneca if in January 2009, the FDA had determined that endoscopic gastric ulcers were no longer an acceptable endpoint and we had been required to conduct additional trials which would have delayed NDA approval for VIMOVO.
If our current or future licensees exercise termination rights they may have, or if these license agreements terminate because of delays in obtaining regulatory approvals, or for other reasons, and we are not able to establish replacement or additional research and development collaborations or licensing arrangements, we may not be able to develop and/or commercialize our product candidates. Moreover, any future collaborations or license arrangements we may enter into may not be on terms favorable to us. A further risk we face with our collaborations is that business combinations and changes in the collaborator or their business strategy may adversely affect their willingness or ability to complete their obligations to us.
Our current or any future collaborations or license arrangements ultimately may not be successful. Our agreements with collaborators typically allow them discretion in electing whether to pursue various regulatory, commercialization and other activities or with respect to the timing of the development, such as our agreement with GSK, which was assigned to Pernix, under which GSK determined, among other things, the exact formulation and composition of the product candidates using our MT 400 technology for use in the Treximet clinical trials. Similarly, under our agreement with AstraZeneca, AstraZeneca had the right to manufacture clinical trial material itself or through a third party.
If any collaborator were to breach its agreement with us or otherwise fail to conduct collaborative activities in a timely or successful manner, the pre-clinical or clinical development or commercialization of the affected product candidate or research program would be delayed or terminated. Any delay or termination of clinical development or commercialization, such as the delay in FDA approval we experienced as a result of approvable letters we received from the FDA in June 2006 and August 2007 related to our Treximet NDA, or a delay in FDA approval of VIMOVO which could have occurred if the FDA determined in January 2009 that endoscopic gastric ulcers were no longer an acceptable primary endpoint in clinical trials and we were required to conduct additional clinical trials for the product, would delay or possibly eliminate our potential product revenues. Further, our collaborators may be able to exercise control, under certain circumstances, over our ability to protect our patent rights under patents covered by the applicable collaboration agreement. For example, under our collaboration agreements with GSK, now assigned to Pernix, Horizon and AstraZeneca, GSK, Horizon and AstraZeneca each has the first right to enforce our patents under their respective agreements and would have exclusive control over such enforcement litigation. GSK elected not to exercise its first right to prosecute infringement suits against third parties that submitted ANDAs to the FDA for approval to market a generic version of Treximet tablets and we filed suit against these companies in the United States District Court for the Eastern District of Texas. The cases have been concluded. On the other hand, AstraZeneca has elected to its first right to prosecute infringement suits against Dr. Reddy’s, Lupin, Anchen, Watson and Mylan, each of which submitted an ANDA to the FDA for approval to market a generic version of VIMOVO. We and AstraZeneca filed suit against Dr. Reddy’s, Lupin, Actavis and Mylan in the United States District Court for the District of New Jersey. As part of Horizon’s purchase of all of AstraZeneca’s rights, title and interest to develop, commercialize and sell VIMOVO in the United States, Horizon has assumed AstraZeneca’s right to lead the above-described Paragraph IV litigation and IPR proceedings relating to VIMIVO.
Other risks associated with our collaborative and contractual arrangements with others include the following:
· we may not have day-to-day control over the activities of our contractors or collaborators;
· our collaborators may fail to defend or enforce patents they own on compounds or technologies that are incorporated into the products we develop with them;
· third parties may not fulfill their regulatory or other obligations;
· we may not realize the contemplated or expected benefits from collaborative or other arrangements; and
· disagreements may arise regarding a breach of the arrangement, the interpretation of the agreement, ownership of proprietary rights, clinical results or regulatory approvals.
These factors could lead to delays in the development of our product candidates and/or the commercialization of our products or reduction in the milestone payments we receive from our collaborators, or could result in our not being able to commercialize our products. Further, disagreements with our contractors or collaborators could require or result in litigation or arbitration, which would be time-consuming and expensive. Our ultimate success may depend upon the success and performance on the part of these third parties. If we fail to maintain these relationships or establish new relationships as required, development of our product candidates and/or the commercialization of our products will be delayed or may never be realized.
A collaborator may withdraw support or cease to perform work on our product candidates if the collaborator determines to develop its own competing product candidate or other product candidates instead.
We have entered into collaboration and license agreements, and may continue to enter into such agreements, with companies that may have products or may develop new product candidates that compete or may compete with our product candidates or which have greater commercial potential. If one of our collaborators should decide that the product or a product candidate that the collaborator is developing would be more profitable for the collaborator than our product candidate covered by the collaboration or license agreement, the collaborator may withdraw support for our product candidate or may cease to perform under our agreement. On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understood that AstraZeneca would instead focus on those countries where the product had shown growth and which AstraZeneca believed had the greatest potential for future growth. On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. In addition, GSK divested of all of its rights, title and interest to develop, commercialize and sell MT 400 products, including Treximet, in the U.S. to Pernix on August 20, 2014.
In the event of a termination of the collaborator’s agreement upon such cessation of performance, we may need to negotiate an agreement with another collaborator in order to continue the development and commercialization efforts for the product candidate. If we were unsuccessful in negotiating another agreement, we might have to cease development activities of the particular product candidate. For example, our development and commercialization agreements with Horizon and AstraZeneca are subject to this risk. Under the terms of our agreement with AstraZeneca and Horizon, either party has the right to terminate the agreement by notice in writing to the other party upon or after any material breach of the agreement by the other party, if the other party has not cured the breach within 90 days after written notice to cure has been given, with certain exceptions. The parties also can terminate the agreement for cause under certain defined conditions. In addition, AstraZeneca can terminate the agreement, at any time, at will, for any reason or no reason, in its entirety or with respect to countries outside the U.S., upon 90 days’ notice. If terminated at will, AstraZeneca will owe us a specified termination payment or, if termination occurs after the product is launched, AstraZeneca may, at its option, under and subject to the satisfaction of conditions specified in the agreement, elect to transfer the product and all rights to us. However, under the circumstance above, or similar circumstance, we may need to enter into a new development and commercialization agreement and may need to start the development process all over again. If we were able to negotiate a new development and commercialization agreement to develop our technology, which is not certain, or if we decide to commercialize the products previously partnered by ourselves, we would face delays and redundant expenses in that development.
We need to maintain current agreements and enter into additional agreements with third parties that possess sales, marketing and distribution capabilities, or establish internally the capability to perform these functions, in order to successfully market and sell our future drug products.
We have no sales or distribution personnel or capabilities at the present time. If we are unable to maintain current collaborations or enter into additional collaborations with established pharmaceutical or pharmaceutical services companies to provide those capabilities as required, we may not be able to successfully commercialize our products. To the extent that we enter into marketing and sales agreements with third parties, such as our agreement with Horizon to sell VIMOVO in the United States and AstraZeneca to sell VIMOVO outside the United States, our revenues, if any, will be affected by the sales and marketing efforts of those third parties. If our licensed products do not perform well in the marketplace our royalty revenue will impacted and our business could be materially harmed. For example, on July 28, 2014, Horizon announced that it had been verbally notified by CVS Caremark and Express Scripts, Inc. that VIMOVO would no longer be on their formularies and will be placed on their exclusion lists effective January 1, 2015. We have been notified that Horizon estimates that approximately 20-30% of VIMOVO prescriptions could be impacted by these decisions.
We refined our strategy and decided to retain control of our PA product candidates for cardiovascular indications through the clinical development and pre-commercialization stage. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016. Our business and operations model is evolving. On June 1, 2015, our board of directors (the “Board”) appointed Adrian Adams, our new Chief Executive Officer and Andrew I. Koven, our new President and Chief Business Officer, each of whom has experience creating, leading and expanding pharmaceutical companies with marketing and sales capabilities. If the proposed business combination with Tribute is approved by our stockholders and Tribute’s shareholders and such transaction and concurrent equity and debt financings are closed, we will become a specialty pharmaceutical company focused on cardiovascular and pain therapies with a diversified portfolio of marketed products and developmental stage product candidates primarily in the United States and Canada.
We need to conduct preclinical, toxicology, genotoxicity and carcinogenicity and other safety studies, and clinical trials for our product candidates. Any negative or unanticipated results, unforeseen costs or delays in the conduct of these studies or trials, or the need to conduct additional studies or trials or to seek to persuade the FDA to evaluate the results of a study or trial in a different manner, could cause us to discontinue development of a product candidate or reduce, delay or eliminate our receipt of potential revenues for one or more of our product candidates and adversely affect our ability to achieve profitability.
Generally, we must demonstrate the efficacy and safety of our product candidates before approval to market can be obtained from the FDA or the regulatory authorities in other countries. Our existing and future product candidates are and will be in various stages of clinical development. Depending upon the type of product candidate and the stage of the development process of a product candidate, we will need to complete preclinical, toxicology, genotoxicity and carcinogenicity and other safety studies, as well as clinical trials, on these product candidates before we submit marketing applications in the United States and abroad. These studies and trials can be very costly and time-consuming. For example, long-term cardiovascular safety studies, such as those the FDA has indicated will be required for approval of certain product candidates containing NSAIDs, typically take approximately three years. In addition, we rely on third parties to perform significant aspects of our studies and clinical trials, introducing additional sources of risk into our development programs.
It should be noted that the results of any of our preclinical and clinical trial testing are not necessarily predictive of results we will obtain in subsequent or more extensive clinical trials or testing. This may occur for many reasons, including, among others, differences in study design, including inclusion/exclusion criteria, the variability of patient characteristics, including patient symptoms at the time of study treatment, the larger scale testing of patients in later trials, or differences in formulation or doses of the product candidate used in later trials. For example, our results from the first of our two Phase 3 pivotal clinical trials of Treximet differed from the results of our second Phase 3 clinical trial and from the Phase 2 proof-of-concept trial of MT 400 that we conducted prior to entering into our collaboration with GSK. Whereas in the Phase 2 trial statistical significance was reached at two hours over placebo in the relief of all associated symptoms of migraine (nausea, photophobia and phonophobia), in the first Phase 3 study Treximet failed to achieve statistical significance at two hours compared to placebo in the relief of nausea. In the second Phase 3 pivotal clinical trial, Treximet demonstrated superiority over the individual components measured by sustained pain-free response (p<0.001 vs. naproxen; p=0.009 vs. sumatriptan) and met all other regulatory endpoints versus placebo.
The successful completion of any of our clinical trials depends upon many factors, including the rate of enrollment of patients. If we are unable to recruit sufficient clinical patients during the appropriate period, we may need to delay our clinical trials and incur significant additional costs. We also rely on the compliance of our clinical trial investigators with FDA regulatory requirements and noncompliance can result in disqualification of a clinical trial investigator and data that are unusable. In addition, the FDA or Institutional Review Boards may require us to conduct additional trials or delay, restrict or discontinue our clinical trials on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Further, even though we may have completed all clinical trials for a product candidate that were planned for submission in support of an application, we may be required to conduct additional clinical trials, studies or investigations or to submit additional data to support our marketing applications. For example, in February, 2012, the FDA requested an additional Phase 1 study to assess the bioequivalence of PA32540 to EC aspirin 325 mg with respect to ASA. EC products such as PA32540 and EC aspirin 325 mg have highly variable pharmacokinetics, making bioequivalence difficult to demonstrate using traditional methods and standards. The FDA made a preliminary review of the study results and the Company’s summary analyses and, based on its preliminary assessment of the information available to it at the time, the FDA did not agree that bioequivalence of PA32540 to EC aspirin 325 mg was demonstrated.
In addition, we and/or our marketing or development partners may determine that pre-approval marketing support studies should be conducted. Unanticipated adverse outcomes of such studies, including recognition of certain risks to human subjects, could a have material impact on the approval of filed or planned market applications or could result in limits placed on the marketing of the product. We may also determine from time to time that it would be necessary to seek to provide justification to the FDA or other regulatory agency that would result in evaluation of the results of a study or clinical trial in a manner that differs from the way the regulatory agency initially or customarily evaluated the results, as was the case with the Phase 1 study to assess the bioequivalence of PA32540 to EC aspirin 325 mg described in the preceding paragraph. In addition, we may have unexpected results in our preclinical or clinical trials or other studies that require us to reconsider the need for certain studies or trials or cause us to discontinue development of a product candidate. For example, in reviewing our NDA for Treximet, the FDA expressed concern about the potential implications from one preclinical in-vitro chromosomal aberration study, one of four standard genotoxicity assays, in which a possible genotoxicity signal was seen for the combination of naproxen sodium and sumatriptan. Further, additional information about potential drug-drug interactions may restrict the patient population for our products, thus limiting the potential market and our potential revenue. For example, recent scientific publications contain conflicting data regarding a possible interaction between clopidogrel (Plavix), a widely prescribed anti-platelet agent, and proton pump inhibitor products, and its impact on cardiovascular outcomes. If the clinical relevance of the possible interaction is unresolved by the time PA32540 and PA8140 enters the marketplace, even if the interaction is later proven definitively to have no clinical impact on cardiovascular outcomes, the market potential of the product may be reduced.
Once submitted, an NDA requires FDA approval before the product described in the application can be distributed or commercialized. Even if we determine that data from our clinical trials, toxicology, genotoxicity carcinogenicity and other safety studies are positive, we cannot assure you that the FDA, after completing its analysis, will not determine that the trials or studies should have been conducted or analyzed differently, and thus reach a different conclusion from that reached by us, or request that further trials, studies or analyses be conducted. For example, the FDA requested additional safety information on Treximet in the approvable letter we received in June 2006 relating to our NDA for Treximet, which required conduct of additional studies, and in August 2007, we received a second approvable letter in which the FDA raised an additional concern about the potential implications from one preclinical in vitro chromosomal aberration study, one of four standard genotoxicity assays, in which a genotoxicity signal was seen for the combination of naproxen sodium and sumatriptan.
The FDA may also require data in certain subpopulations, such as pediatric use, or, if such studies were not previously completed, may require long-term carcinogenicity studies, prior to NDA approval, unless we can obtain a waiver of such a requirement. We face similar regulatory hurdles in other countries to those that we face in the U.S.
Our costs associated with our human clinical trials vary based on a number of factors, including:
|
· |
the order and timing of clinical indications pursued; |
|
|
|
|
· |
the extent of development and financial support from collaborative parties, if any; |
|
|
|
|
· |
the need to conduct additional clinical trials or studies; |
|
|
|
|
· |
the number of patients required for enrollment; |
|
|
|
|
· |
the difficulty of obtaining sufficient patient populations and clinicians; |
|
|
|
|
· |
the difficulty of obtaining clinical supplies of our product candidates; and |
|
|
|
|
· |
governmental and regulatory delays. |
We currently depend and will in the future depend on third parties to manufacture our product candidates. If these manufacturers fail to meet our requirements or any regulatory requirements, the product development and commercialization of our product candidates will be delayed.
We do not have, and have no plans to develop, the internal capability to manufacture either clinical trial or commercial quantities of products that we may develop or have under development. We rely upon third-party manufacturers and our partners to supply us with our product candidates. We also need supply contracts to sell our products commercially. On December 19, 2011, we entered into a Supply Agreement and a related Capital Agreement with Patheon Pharmaceuticals Inc. (“Patheon”) pursuant to which Patheon has agreed to manufacture, and we have agreed to purchase, a specified percentage of the Company’s requirements of PA32540 for sale in the United States. The Supply Agreement and Capital Agreement were amended on July 10, 2013 to, among other things, expressly incorporate the Company’s PA8140 product candidate into the Supply Agreement and to replace the schedule of the Capital Agreement which lists dedicated and non-dedicated capital equipment and facility modifications to be funded in whole or in part by the Company, with a new updated schedule reflecting the parties’ current assumptions regarding the need for and timing of capital equipment expenditures. We also rely on third parties to supply the active ingredients and other ingredients used in the manufacture of our products. Failure of such ingredient suppliers to comply with regulatory requirements can impact our ability to obtain approval of our products or our ability to supply the market with our products after approval. For example, On April 25, 2014, we announced that we had received a CRL from the FDA with respect to the NDA for our PA32540 and PA8140 products. In the CRL, the FDA noted that, during an inspection of the manufacturing facility of an active ingredient supplier, inspection deficiencies were found. Satisfactory resolution of deficiencies noted by the field investigator is required before the NDA may be approved. On May 9, 2014, our active ingredient supplier submitted a response to the FDA addressing the inspection deficiencies and subsequently submitted an update to its initial response. On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. Satisfactory resolution of these deficiencies is required before this application may be approved. The active ingredient supplier has informed us that they received a warning letter relating to the Form 483 inspection deficiencies and have submitted a plan of corrective actions to address the matters raised in the warning letter to FDA. There were no clinical or safety deficiencies noted with respect to either PA32540 or PA8140 and no other deficiencies were noted in the CRL.
There is no guarantee that manufacturers that enter into commercial supply contracts with us will be financially viable entities going forward, or will not otherwise breach or terminate their agreements with us. If we do not have the necessary commercial supply contracts, or if Patheon is, or any of our future contract manufacturers are unable to satisfy our requirements or meet any regulatory requirements, and we are required to find alternative sources of supply, there may be additional costs and delays in product
development and commercialization of our product candidates or we may be required to comply with additional regulatory requirements.
If our competitors develop and commercialize products faster than we do or if their products are superior to ours, our commercial opportunities will be reduced or eliminated.
Our product candidates will have to compete with existing and any newly developed drugs in our therapeutic areas for any newly developed product candidates for the treatment of other diseases. There are also likely to be numerous competitors developing new products to treat other diseases and conditions for which we may seek to develop products in the future, which could render our products and product candidates or technologies obsolete or non-competitive. For example, our primary competitors will likely include large pharmaceutical companies, biotechnology companies, universities and public and private research institutions. The competition for VIMOVO and any other PN products that may be developed and receive regulatory approval will come from the oral NSAID market, or more specifically the traditional non-selective NSAIDs (such as naproxen and diclofenac), traditional NSAID/gastroprotective agent combination products or combination product packages (such as Arthrotec® and Prevacid® NapraPAC™), combinations of NSAIDs and PPIs taken as separate pills and the only remaining COX-2 inhibitor, Celebrex®. The competition for our PA product candidates for which we have conducted studies for secondary prevention of cardiovascular events will come from aspirin itself as well as other products used for secondary prevention. AstraZeneca, with whom we collaborated in the development of VIMOVO, has publicly announced that it has obtained regulatory approval for a combination product containing aspirin and esomeprazole in Europe and has also filed a NDA with the FDA for such product, and for which the FDA issued a CRL declining approval. AstraZeneca has stated that it is evaluating the CRL and will continue discussions with the FDA to determine next steps. This product has entered the European market and, if it enters the U.S. market, will compete with our PA cardiovascular product candidates.
Based upon their drug product and pipeline portfolios and the overall competitiveness of our industry, we believe that we face, and will continue to face, intense competition from other companies for securing collaborations with pharmaceutical companies, establishing relationships with academic and research institutions, and acquiring licenses to proprietary technology. Our competitors, either alone or with collaborative parties, may also succeed with technologies or products that are more effective than any of our current or future technologies or products. Many of our actual or potential competitors, either alone or together with collaborative parties, have substantially greater financial resources, and almost all of our competitors have larger numbers of scientific and administrative personnel than we do.
Many of these competitors, either alone or together with their collaborative parties, also have significantly greater resources to or experience in:
|
· |
developing product candidates; |
|
|
|
|
· |
undertaking preclinical testing and human clinical trials; |
|
|
|
|
· |
obtaining FDA and other regulatory approvals of product candidates; and |
|
|
|
|
· |
manufacturing and marketing products. |
Accordingly, our actual or potential competitors may succeed in obtaining patent protection, receiving FDA or other regulatory approval or commercializing products where we cannot or before we do. Any delays we encounter in obtaining regulatory approvals for our product candidates, such as we experienced as a result of the approvable letters we received from the FDA in June 2006 and August 2007 relating to the NDA for Treximet, as a result of the not-approvable letters we received from the FDA on MT 100 and MT 300, and as a result of the CRLs we received from the FDA relating to the NDA for PA32540 and PA8140 on April 25, 2014 and December 16, 2014, increase this risk. Our competitors may also develop products or technologies that are superior to those that we are developing, and render our product candidates or technologies obsolete or non-competitive. If we cannot successfully compete with new or existing products, our marketing and sales will suffer and we may not ever receive any revenues from sales of products or may not receive sufficient revenues to achieve profitability.
If we are unable to protect our patents or proprietary rights, or if we are unable to operate our business without infringing the patents and proprietary rights of others, we may be unable to develop our product candidates or compete effectively.
The pharmaceutical industry places considerable importance on obtaining patent and trade secret protection for new technologies, products and processes. Our success will depend, in part, on our ability, and the ability of our licensors, to obtain and to keep protection for our products and technologies under the patent laws of the United States and other countries, so that we can stop others from using our inventions. Our success also will depend on our ability to prevent others from using our trade secrets. In addition, we must operate in a way that does not infringe, or violate, the patent, trade secret and other intellectual property rights of other parties.
We cannot know how much protection, if any, our patents will provide or whether our patent applications will issue as patents. The breadth of claims that will be allowed in patent applications cannot be predicted and neither the validity nor
enforceability of claims in issued patents can be assured. If, for any reason, we are unable to obtain and enforce valid claims covering our products and technology, we may be unable to prevent competitors from using the same or similar technology or to prevent competitors from marketing identical products. For example, if we are unsuccessful in protecting our patents in the litigation against Dr. Reddy’s, Lupin, Actavis, and Mylan or other companies who may file ANDAs for VIMOVO, such companies could market a generic version of the product prior to the expiration of our and AstraZeneca’s patents.
In addition, due to the extensive time needed to develop, test and obtain regulatory approval for our products, any patents that protect our product candidates may expire early during commercialization. This may reduce or eliminate any market advantages that such patents may give us.
In certain territories outside the U.S., our issued patents may be subject to opposition by competitors within a certain time after the patent is issued. Such opposition proceedings and related appeals may not be resolved for several years, and may result in the partial or total revocation of the issued patent. For example, in October 2005 oppositions were filed against our issued European patent for MT 400 by Merck & Co., Inc. and Almirall Prodesfarma asserting that the European patent should not have been granted. As a result of these oppositions and subsequent proceedings, the European Patent Office found that claims relating to combinations of sumatriptan and naproxen for the treatment of migraine were valid. However, broader claims relating to certain other 5-HT 1B/1D agonists and long-acting NSAIDs were held to be insufficiently supported by the presently available technical evidence. In addition, in April 2011, oppositions were also filed against our issued European patent for VIMOVO and our PA products by Chatfield Laboratories and Strawman Limited asserting that the European patent should not have been granted. Strawman Limited subsequently withdrew from the opposition. Following oral proceedings, the Opposition Division of the European Patent Office found that claims relating to the combination of PPIs and NSAIDs are valid. Chatfield Laboratories did not appeal this decision.
We may need to submit our issued patents for amendment or reissue if we determine that any claims within our patents should not have been issued. While such a submission may be based on our view that only specified claims should not have been granted to us, there can be no assurance that a patent examiner will not determine that additional claims should not have been granted to us. Such was the case with one of our patents covering MT 100, which we submitted for reissue after determining that certain specified claims that are not central to our protection of MT 100 should not have been issued. In April 2006, we received an office action on the reissue application and, consistent with our decision not to devote further resources to the development of this product in the U.S., the reissue application was abandoned in January 2007.
We may need to license rights to third party patents and intellectual property to continue the development and marketing of our product candidates. If we are unable to acquire such rights on acceptable terms, our development activities may be blocked and we may be unable to bring our product candidates to market.
We may enter into litigation to defend ourselves against claims of infringement, assert claims that a third party is infringing one or more of our patents, protect our trade secrets or know-how, or determine the scope and validity of others’ patent or proprietary rights. For example, we filed patent infringement lawsuits against Par, Alphapharm, Teva, Dr. Reddy’s and Sun in the federal court in the Eastern District of Texas in connection with their respective ANDA submissions to the FDA containing Paragraph IV certifications for approval to market sumatriptan 85 mg/naproxen sodium 500 mg tablets, a generic version of Treximet tablets, before the expiration of our patents. Further, we and AstraZeneca filed a patent infringement lawsuit against Dr. Reddy’s, Lupin, Actavis and Mylan in the federal court in the District of New Jersey in connection with their respective ANDA submissions to the FDA containing a Paragraph IV certification for approval to market (a generic version of VIMOVO tablets, before the expiration of our and AstraZeneca’s patents. Dr. Reddy’s and CFAD have also challenged the validity of certain patents covering VIMOVO in IPR proceedings before the Patent Trials and Appeal Board (“PTAB”). We and AstraZeneca are also engaged in a proceeding in Canada with Mylan ULC which is seeking approval of its generic version of VIMOVO in Canada prior to the expiration of our Canadian patent. With respect to some of our product candidates, under certain circumstances, our development or commercialization collaborators have the first right to enforce our patents and would have exclusive control over such enforcement litigation. For example, under our collaboration agreements with GSK and AstraZeneca, GSK and AstraZeneca each has the first right to enforce our patents under their respective agreements. GSK advised us that it elected not to exercise its first right to bring an infringement suit against Par, Alphapharm, Teva, and Dr. Reddy’s, and Sun each of which submitted ANDAs to the FDA for approval to market a generic version of Treximet tablets, while AstraZeneca has exercised its first right to bring an infringement suit against Dr. Reddy’s Lupin, Actavis and Mylan, each of which submitted an ANDA to the FDA for approval to market a generic version of VIMOVO tablets and AstraZeneca Canada has exercised its first right to defend the proceeding in Canada against Mylan ULC. As part of Horizon’s purchase of all of AstraZeneca’s rights, title and interest to develop, commercialize and sell VIMOVO in the United States, Horizon has assumed AstraZeneca’s right to lead the above-described Paragraph IV litigation relating to VIMIVO. Horizon is also leading the defense in the IPRs brought by Dr. Reddy’s and CFAD.
If we are found to infringe the patent rights of others, then we may be forced to pay damages in an amount that might irreparably harm our business and/or be prevented from continuing our product development and marketing activities. Additionally, if we or our development or commercialization collaborator seek to enforce our patents and are unsuccessful, we may be subject to claims for bringing a failed enforcement action, including claims alleging various forms of antitrust violations (both state and federal)
and unfair competition. If we are found to be liable for such claims, then we may be forced to pay damages in an amount that might irreparably harm our business and/or be prevented from continuing our product development and commercialization activities. Even if we are successful in defending any such claims of infringement or in asserting claims against third parties, such litigation is expensive, may have a material effect on our operations, and may distract management from our business operations. Regardless of its eventual outcome, any lawsuit that we enter into may consume time and resources that would impair our ability to develop and market our product candidates.
We have entered into confidentiality agreements with our employees, consultants, advisors and collaborators. However, these parties may not honor these agreements and, as a result, we may not be able to protect our rights to unpatented trade secrets and know-how. Others may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets and know-how. Also, many of our scientific and management personnel were previously employed by competing companies. As a result, such companies may allege trade secret violations and similar claims against us.
Our products may not be accepted by the market.
The commercial success of our product candidates depends upon the acceptance of these products in the marketplace. Even if a product displays a favorable efficacy and safety profile in clinical trials, market acceptance of a product will not be known until after it is launched and a product may not generate the revenues that we anticipate. The degree of market acceptance will depend upon a number of factors, including:
|
· |
the acceptance by physicians and third-party payors of VIMOVO and YOSPRALA, if and when approved, as an alternative to other therapies; |
|
|
|
|
· |
the receipt and timing of regulatory approvals; |
|
|
|
|
· |
the availability of third-party reimbursement; |
|
|
|
|
· |
the indications for which the product is approved; |
|
|
|
|
· |
the rate of adoption by healthcare providers; |
|
|
|
|
· |
the rate of product acceptance by target patient populations; |
|
|
|
|
· |
the price of the product relative to alternative therapies; |
|
|
|
|
· |
the availability of alternative therapies; |
|
|
|
|
· |
the extent and effectiveness of marketing efforts by our collaborators, and third-party distributors and agents; |
|
|
|
|
· |
the existence of adverse publicity regarding our products or similar products; and |
|
|
|
|
· |
the extent and severity of side effects as compared to alternative therapies. |
If we or our commercialization partners do not receive adequate third-party reimbursements for our future products, our revenues and profitability will be reduced.
Our ability to commercialize our product candidates successfully will depend, in part, on the extent to which reimbursement for the costs of such products and related treatments will be available from government health administration authorities, such as Medicare and Medicaid in the U.S., private health insurers and other organizations. Significant uncertainty exists as to the reimbursement status of a newly approved healthcare product. Adequate third-party coverage may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product research and development. If adequate coverage and reimbursement levels are not provided by government and third-party payors for use of our products, our products may fail to achieve market acceptance. For example, on July 28, 2014, Horizon announced that it had been verbally notified by CVS Caremark and Express Scripts, Inc. that VIMOVO would no longer be on their formularies and will be placed on their exclusion lists effective January 1, 2015. Horizon estimates that approximately 20-30% of VIMOVO prescriptions could be impacted by these decisions.
Our future revenues, profitability and access to capital will be affected by the continuing efforts of governmental and private third-party payors to contain or reduce the costs of healthcare through various means. We expect that a number of federal, state and foreign proposals will seek to control the cost of drugs through governmental regulation. We are unsure of the form that any healthcare reform legislation may take or what actions federal, state, foreign and private payors may take in response to any proposed reforms. Therefore, we cannot predict the effect of any implemented reform on our business.
Legislative or regulatory reform of the healthcare system may affect our ability to sell our products profitably.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could impact our ability to sell our products profitably. On March 23, 2010, President Obama signed into law the Patient Protection and Affordable Care Act, or PPACA, which includes a number of health care reform provisions and requires most U.S. citizens to have health insurance. PPACA increased the minimum Medicaid drug rebates for pharmaceutical companies, expands the 340B drug discount program, and makes changes to affect the Medicare Part D coverage gap, or “donut hole.” The law also revises the definition of “average manufacturer price” for reporting purposes which could increase the
amount of the Company’s Medicaid drug rebates to states. The law also imposed a significant annual fee on companies that manufacture or import branded prescription drug products. Substantial new provisions affecting compliance also have been added, which may require modification of business practices with health care practitioners.
The reforms imposed by the law will significantly impact the pharmaceutical industry; however, the full effects of PPACA cannot be known until these provisions are fully implemented and the Centers for Medicare & Medicaid Services and other federal and state agencies issue applicable regulations or guidance. Moreover, in the coming years, additional changes could be made to governmental healthcare programs that could significantly impact the success of our products, and we could be adversely affected by current and future health care reforms.
If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
The testing and marketing of pharmaceutical products entail an inherent risk of product liability. Product liability claims might be brought against us by consumers, healthcare providers, pharmaceutical companies or others selling our future products. If we cannot successfully defend ourselves against such claims, we may incur substantial liabilities or be required to limit the commercialization of our products. We have product liability insurance that covers our commercialized product and human clinical trials in an amount equal to up to $10 million annual aggregate limit with a $0.1 million deductible per claim. The amount of insurance that we currently hold may not be adequate to cover all liabilities that may occur. However, insurance coverage is becoming increasingly expensive, and no assurance can be given that we will be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. We will explore, on an on-going basis, expanding our insurance coverage related to the sale of our future marketed products when we obtain marketing approval for such products and commercial sales of such products begin. However, we may not be able to obtain commercially reasonable product liability insurance for any products approved for marketing. If a plaintiff brings a successful product liability claim against us in excess of our insurance coverage, if any, we may incur substantial liabilities and our business may be harmed or fail.
We may need additional funding and may not have access to capital. If we are unable to raise capital when needed, we may need to delay, reduce or eliminate our product development or commercialization efforts.
In the future, we may need to raise additional funds to execute our evolving business strategy. We have incurred losses from operations since inception and we may continue to incur additional operating losses. Our actual capital requirements will depend upon numerous factors, including:
|
· |
the progress of our research and development programs; |
|
|
|
|
· |
the progress of preclinical studies, clinical and other testing or the need conduct additional trials, studies or other testing; |
|
|
|
|
· |
the time and cost involved in obtaining any regulatory approvals; |
|
|
|
|
· |
the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
|
|
|
|
· |
the effect of competing technological and market developments; |
|
|
|
|
· |
the timing of our receipt, if any, of milestone payments and royalties under collaborative agreements; |
|
|
|
|
· |
the effect of changes and developments in, or termination of, our collaborative, license and other relationships; |
|
|
|
|
· |
the terms and timing of any additional collaborative, license and other arrangements that we may establish; and |
|
|
|
|
· |
our ability to commercialize or arrange for the commercialization of our product candidates. |
Our operating expenses for the three months ended March 31, 2015 totaled $4.2 million, including non-cash compensation expense of $0.5 million related to stock options and other stock-based awards. For fiscal years 2012 through 2014, our average annual operating expenses (including average non-cash deferred compensation of $2.9 million) were $24.6 million. As of March 31, 2015, we had an aggregate of $43.9 million in cash and cash equivalents. We expect that our operating expenses for 2015 and 2016 will exceed the net level of our operating expenses in 2014. We believe that we will have sufficient cash reserves and cash flow to maintain our planned level of business activities, through 2015. However, our anticipated cash flow includes continued receipt of royalty revenue from Horizon and AstraZeneca’s sale of VIMOVO but does not include any additional milestone or royalty payments. In addition, our expenses might increase during that period beyond currently expected levels if we decide to, or any regulatory agency requires us to, conduct additional clinical trials, studies or investigations for any of our product candidates, including in connection with the agency’s consideration, or reconsideration, of our regulatory filings for our product candidates. We are planning to commercialize our PA product candidates in the United States without a commercial partner and our expenses will increase relative to prior years as we move from a development company which licenses its product candidates to other companies towards a fully integrated, specialty pharmaceutical company. If our projected revenues decrease, we may need to raise additional capital.
If our projected expenses increase for our product candidates currently in development, if we expand our studies for additional indications for our PA product candidates or new product candidates, of if we commercialize our product candidates ourselves then, as a result of these or other factors, we may need to raise additional capital.
If the proposed business combination with Tribute is not consummated and such transaction and concurrent equity and debt financings do not close, we will need to raise capital to commercialize YOSPRALA and to implement our plan to become a fully integrated, specialty pharmaceutical company. We may be unable to raise additional equity funds when we desire to do so due to unfavorable market conditions in our industry or generally, or due to other unforeseen developments in our business. Further, we may not be able to find sufficient debt or equity funding, if at all, on acceptable terms. If we cannot, we may need to delay, reduce or eliminate research and development programs and therefore may not be able to execute our business strategy. Further, to the extent that we obtain additional funding through collaboration and licensing arrangements, it may be necessary for us to give up valuable rights to our development programs or technologies or grant licenses on terms that may not be favorable to us.
The sale by us of additional equity securities or the expectation that we will sell additional equity securities may have an adverse effect on the price of our common stock.
We depend on key personnel and may not be able to retain these employees or recruit additional qualified personnel, which would harm our research and development and commercialization efforts.
We are highly dependent on the efforts of our key management and scientific personnel, especially Adrian Adams, our Chief Executive Officer, and Andrew I. Koven, our President and Chief Business Officer. If we should lose the services of Mr. Adams or Mr. Koven, or are unable to replace the services of our other key personnel who may leave the Company, or if we fail to recruit other key scientific and commercial personnel, we may be unable to achieve our business objectives. There is intense competition for qualified scientific and commercial personnel. We may not be able to continue to attract and retain the qualified personnel necessary for developing our business. Furthermore, our future success may also depend in part on the continued service of our other key management personnel and our ability to recruit and retain additional personnel, as required by our business.
New and changing corporate governance and public disclosure requirements add uncertainty to our compliance policies and increase our costs of compliance.
Changing laws, regulations and standards relating to accounting, corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, other SEC regulations, and the NASDAQ Global Market rules, are creating uncertainty for companies like ours. These laws, regulations and standards may lack specificity and are subject to varying interpretations. Their application in practice may evolve over time, as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs of compliance as a result of ongoing revisions to such corporate governance standards.
In particular, our efforts to comply with Section 404 of the Sarbanes-Oxley Act of 2002 and the related regulations regarding our required assessment of our internal controls over financial reporting and our external auditors’ audit of that assessment requires the commitment of significant financial and managerial resources. We consistently assess the adequacy of our internal controls over financial reporting, remediate any control deficiencies that may be identified, and validate through testing that our controls are functioning as documented. While we do not anticipate any material weaknesses, the inability of management to assess our internal controls over financial reporting as effective could result in adverse consequences to us, including, but not limited to, a loss of investor confidence in the reliability of our financial statements, which could cause the market price of our stock to decline. The existence of this or one or more other material weaknesses or significant deficiencies in our internal control over financial reporting could result in errors in our financial statements, and substantial costs and resources may be required to rectify any internal control deficiencies. Although we continually review and evaluate internal control systems to allow management to report on the sufficiency of our internal controls, we cannot assure you that we will not discover weaknesses in our internal control over financial reporting. Any such weakness or failure to remediate any existing material weakness could materially adversely affect our ability to comply with applicable financial reporting requirements and the requirements of our various agreements.
We are committed to maintaining high standards of corporate governance and public disclosure, and our efforts to comply with evolving laws, regulations and standards in this regard have resulted in, and are likely to continue to result in, increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities. In addition, the laws, regulations and standards regarding corporate governance may make it more difficult for us to obtain director and officer liability insurance. Further, our board members, Chief Executive Officer, President and Chief Financial Officer could face an increased risk of personal liability in connection with their performance of duties. As a result, we may face difficulties attracting and retaining qualified board members and executive officers, which could harm our business. If we fail to comply with new or changed laws, regulations or standards of corporate governance, our business and reputation may be harmed.
Risks Related to Commercialization of our Product Candidates
We continue to evaluate the commercial opportunities for our current product candidates in connection with our development of a worldwide commercialization strategy. We expect to commercialize YOSPRALA ourselves in the United States and may pursue commercial opportunities for our future products ourselves. If we are unable to develop sales and marketing capabilities on our own, or through partner acquisition, we will not be able to fully exploit the commercial potential of our future products and the costs of pursuing such a strategy may have a material adverse impact on our results of operations.
We continue to evaluate the commercial opportunities for our product candidates in connection with our development of a worldwide commercialization strategy. We decided to retain ownership of our PA product candidates through the clinical development and pre-commercialization stage and our former chief commercial officer developed the commercialization strategy for these products and conducted all the required pre-commercialization activities in the United States. On November 29, 2014, we executed a termination agreement with Sanofi U.S. terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi U.S. were terminated and all rights to the products licensed to Sanofi U.S. under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi U.S. relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. Our business and operations model is evolving. On June 1, 2015, our Board appointed Adrian Adams, as our new Chief Executive Officer and Andrew I. Koven, as our new President and Chief Business Officer, each of whom has experience creating, leading and expanding pharmaceutical companies with marketing and sales capabilities. If the proposed business combination with Tribute is approved by our stockholders and Tribute’s shareholders and such transaction and concurrent equity and debt financings are closed, we will become a specialty pharmaceutical company focused on cardiovascular and pain therapies with a diversified portfolio of marketed products and developmental stage product candidates in the United States and Canada. We plan to make significant expenditures to secure commercial resources to sell YOSPRALA and the Tribute products and to expand our marketing capabilities to support our anticipated growth. Any failure or extended delay in the expansion of our sales and marketing capabilities or inability to effectively operate in the marketplace alone or together with our partners could adversely impact our business. There can be no assurance that our sales and marketing efforts will generate significant revenues and costs of pursuing such a strategy may have a material adverse impact on our results of operations. Events or factors that may inhibit or hinder our commercialization efforts include:
|
· |
developing our own commercial team or playing a role in the commercialization with a partner will be expensive and time-consuming and could result in high cash burn or reduced profitability; |
|
|
|
|
· |
failure to acquire sufficient or suitable personnel to establish, oversee, or implement our commercialization strategy; |
|
|
|
|
· |
failure to recruit, train, oversee and retain adequate numbers of effective sales and marketing personnel; |
|
|
|
|
· |
failure to develop a commercial strategy ourselves or together with partners that can effectively reach and persuade adequate numbers of physicians to prescribe our products; |
|
|
|
|
· |
our or our partners’ inability to secure reimbursement at a reasonable price; |
|
|
|
|
· |
unforeseen costs and expenses associated with creating or acquiring and sustaining an independent commercial organization; |
|
|
|
|
· |
incurrence of costs in advance of anticipated revenues and subsequent failure to generate sufficient revenue to offset additional costs; and |
|
|
|
|
· |
our ability to fund our commercialization efforts alone or together with our partners on terms acceptable to us, if at all. |
As we pursue commercialization of YOSPRALA and other opportunities for our future products ourselves, failure to comply with the laws governing the marketing and sale of such products may result in regulatory agencies taking action against us and/or our partners, which could significantly harm our business.
We retained ownership of our PA product candidates through the clinical development and pre-commercialization stage and our chief commercial officer developed the commercialization strategy for these products and conducted pre-commercialization activities in the United States. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. Our business model is continuing to evolve. Our Board has appointed a new Chief Executive and a new President and Chief Business Officer who have experience creating and leading pharmaceutical companies with marketing and sales capabilities. Our business and operations model is evolving. On June 1, 2015, our Board appointed Adrian Adams as our new Chief Executive Officer and Andrew I. Koven as our new President and Chief Business Officer, each of whom has experience creating, leading and expanding pharmaceutical companies with marketing and sales capabilities. If the proposed business combination with Tribute is approved by our stockholders and Tribute’s shareholders and such transaction and concurrent equity and debt financings are closed, we will become a specialty pharmaceutical company focused on cardiovascular and pain therapies with a diversified portfolio of marketed products and developmental stage product candidates in the United States and Canada. As we pursue commercialization of YOSPRALA and other product candidates we will be subject to a large body of legal and regulatory requirements. In particular, there
are many federal, state and local laws that we will need to comply with if we become engaged in the marketing, promoting, distribution and sale of pharmaceutical products. The FDA extensively regulates, among other things, promotions and advertising of prescription drugs. In addition, the marketing and sale of prescription drugs must comply with the Federal fraud and abuse laws, which are enforced by the Office of the Inspector General of the Division, or OIG, of the Department of Health and Human Services. These laws make it illegal for anyone to give or receive anything of value in exchange for a referral for a product or service that is paid for, in whole or in part, by any federal health program. The federal government can pursue fines and penalties under the Federal False Claims Act which makes it illegal to file, or induce or assist another person in filing, a fraudulent claim for payment to any governmental agency. Because, as part of our and/or our partners commercialization efforts, we or our partners may provide physicians with samples we will be required to comply with the Prescription Drug Marketing Act, or PDMA, which governs the distribution of prescription drug samples to healthcare practitioners. Among other things, the PDMA prohibits the sale, purchase or trade of prescription drug samples. It also sets out record keeping and other requirements for distributing samples to licensed healthcare providers.
In addition, we will need to comply with the body of laws comprised of the Medicaid Rebate Program, the Veterans’ Health Care Act of 1992 and the Deficit Reduction Act of 2005. This body of law governs product pricing for government reimbursement and sets forth detailed formulas for how we must calculate and report the pricing of our products so as to ensure that the federally funded programs will get the best price. Moreover, many states have enacted laws dealing with fraud and abuse, false claims, the distribution of prescription drug samples and gifts and the calculation of best price. These laws typically mirror the federal laws but in some cases, the state laws are more stringent than the federal laws and often differ from state to state, making compliance more difficult. We expect more states to enact similar laws, thus increasing the number and complexity of requirements with which we would need to comply.
Compliance with this body of laws is complicated, time consuming and expensive. We cannot assure you that we will be in compliance with all potentially applicable laws and regulations. Even minor, inadvertent irregularities can potentially give rise to claims that the law has been violated. Failure to comply with all potentially applicable laws and regulations could lead to penalties such as the imposition of significant fines, debarment from participating in drug development and marketing and the exclusion from government-funded healthcare programs. The imposition of one or more of these penalties could adversely affect our revenues and our ability to conduct our business as planned.
In addition, the Federal False Claims Act allows any person to bring suit alleging the false or fraudulent submission of claims for payment under federal programs and other violations of the statute and to share in any amounts paid by the entity to the government in fines or settlement. Such suits, known as qui tam actions, have increased significantly in recent years and have increased the risk that companies like us may have to defend a false claim action. We could also become subject to similar false claims litigation under state statutes. If we are unsuccessful in defending any such action, such action may have a material adverse effect on our business, financial condition and results of operations.
Risks Related to the Proposed Transactions
The merger agreement is subject to conditions and could be terminated in accordance with its terms and the transactions contemplated thereby may not be completed.
The merger agreement contains a number of conditions that must be satisfied or waived to complete the transactions contemplated thereby. Those conditions include, among others: receipt of the Pozen stockholder approval, receipt of Tribute shareholder approval, court approval of the transactions under the arrangement, expiration or termination of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended (the “HSR Act”), if applicable, absence of any law or order preventing or prohibiting completion of the transactions contemplated under the merger agreement and no governmental authority instituting any proceeding seeking to enjoin or prohibit the completion of the transactions contemplated under the merger agreement, effectiveness of the registration statement of which the Form S-4, is a part, approval of the Parent Shares to be issued in the transactions contemplated under the merger agreement for listing on NASDAQ and the TSX, the continued accuracy of the representations and warranties of both parties, subject to specified materiality standards, and the performance in all material respects by both parties of their covenants and agreements. If the transactions contemplated under the merger agreement are not completed by January 31, 2016, either Pozen or Tribute may choose not to proceed with the transactions and terminate the merger agreement. No assurance can be given that all of the conditions to the closing of the transactions contemplated under the merger agreement will be satisfied or, if they are, as to the timing of such satisfaction. In addition, Pozen or Tribute may elect to terminate the merger agreement in certain other circumstances, including, but not limited to, a tax termination event, and the parties can mutually decide to terminate the merger agreement at any time prior to the completion of the transaction, before or after the Pozen stockholder approval or Tribute shareholder approval.
Obtaining required approvals necessary to satisfy the conditions to the completion of the transactions contemplated under the merger agreement may delay or prevent completion of such transactions.
The transactions contemplated under the merger agreement are subject to closing conditions, which include the expiration or termination of the waiting period under the HSR Act, if applicable, obtaining of the adoption of the merger agreement by affirmative vote or content of Pozen stockholders holding a majority of the Pozen common stock outstanding and entitled to vote (the “Pozen stockholder approval”) as well as obtaining the affirmative vote of at least 662/3 % of the votes cast on the arrangement resolution by Tribute shareholders present in person or represented by proxy at the Tribute meeting of shareholders (the “Tribute shareholder approval”).
Under the HSR Act and the rules and regulations promulgated thereunder by the FTC, Pozen and Tribute may be required to submit notifications and certain documents and information to the FTC and the Antitrust Division of the Department of Justice (the “Antitrust Division”), and to observe a statutory waiting period, before completing the transactions. If notifications and submissions are required from Pozen and Tribute under the HSR Act, following those submissions the FTC or the Antitrust Division may open an investigation, issue a request for additional information and documentary materials, extend the statutory waiting period, or seek to prevent, delay, or otherwise restrain the completion of the transactions under the antitrust laws. No assurances can be given that the FTC or the Antitrust Division will not seek to take one or more of these steps. Similarly, no assurances can be given that the Pozen stockholders or Tribute shareholders will approve the transactions or that the other conditions to closing of the transactions will be satisfied. In the event Pozen’s stockholders approve the merger, but Tribute’s shareholders do not approve the arrangement, or if Pozen’s stockholders do not vote to approve the issuance by Parent of Parent Shares pursuant to the Subscription Agreement and the Facility Agreement, the transactions contemplated by the merger agreement will not close.
Under the Competition Act (Canada) (the “Competition Act”), Pozen and Tribute may be required to submit notifications and certain documents and information to the Canadian Competition Bureau, and to observe a statutory waiting period, before completing the transactions. If notifications and submissions are required from Pozen and Tribute under the Competition Act, following those submissions the Canadian Competition Bureau (the “Competition Bureau”) may open an investigation, issue a request for additional information and documentary materials, extend the statutory waiting period, or seek to prevent, delay, or otherwise restrain the completion of the transactions contemplated by the merger agreement under applicable laws. No assurances can be given that the Competition Bureau will not seek to take one or more of these steps.
Certain changes in the U.S. federal tax laws on or before the closing date of the merger agreement could jeopardize the consummation of the transactions contemplated by the merger agreement.
Pozen and/or Tribute are permitted to terminate the merger agreement if, prior to the closing date of the merger agreement, there is (i) a change in U.S. federal tax law (whether or not such change in law is yet effective) or any official interpretations thereof as set forth in published guidance by the U.S. Treasury Department or the Internal Revenue Service (the “IRS”) (other than IRS news releases) (whether or not such change in official interpretation is yet effective) or (ii) a bill that would implement such a change that has been passed by the United States House of Representatives and the United States Senate and for which the time period for the President of the United States to sign or veto such bill has not yet elapsed, in any such case, that, as a result of consummating the transactions contemplated by the merger agreement, in the opinion of nationally recognized U.S. tax counsel, would have a material adverse effect, including causing Parent to be treated as a United States domestic corporation for United States federal income tax purposes, as further specified in the merger agreement.
Aralez’s status as a foreign corporation for U.S. federal tax purposes could be affected by IRS action or a change in U.S. tax law.
Although Parent will be incorporated in Ireland, the IRS may assert that Parent should be treated as a U.S. corporation (and, therefore, a U.S. tax resident) for U.S. federal income tax purposes pursuant to Section 7874 of the Internal Revenue Code (the “Code”). A corporation generally is considered a tax resident in the jurisdiction of its organization or incorporation for U.S. federal income tax purposes. Because Parent is an Irish incorporated entity, generally it would be classified as a foreign corporation (and, therefore, a non-U.S. tax resident) under these rules. Section 7874 of the Code provides an exception pursuant to which a foreign incorporated entity may, in certain circumstances, be treated as a U.S. corporation for U.S. federal income tax purposes.
Under Section 7874 of the Code, Parent would be treated as a foreign corporation for U.S. federal income tax purposes if the former stockholders of Pozen own (within the meaning of Section 7874 of the Code) less than 80% (by both vote and value) of Parent Shares by reason of holding shares in Pozen (the “ownership test”). The Pozen stockholders are expected to own less than 80% (by both vote and value) of the Parent Shares after the transactions contemplated under the merger agreement by reason of their ownership of shares of Pozen common stock. As a result, under current law, Parent is expected to be treated as a foreign corporation for U.S. federal income tax purposes. However, there can be no assurance that the IRS will agree with the position that the ownership test is satisfied. There is limited guidance regarding the application of Section 7874 of the Code, including with respect to the provisions regarding the application of the ownership test. Pozen’s obligation to complete the transactions is conditional upon its receipt of the Section 7874 opinion from DLA Piper LLP (US), counsel to the registrant (“DLA Piper”), dated as of the closing date of the merger agreement and subject to certain qualifications and limitations set forth therein, to the effect that Section 7874 of the Code existing regulations promulgated thereunder, and official interpretation thereof as set forth in published guidance should not apply in such a manner so as to cause Parent to be treated as a U.S. corporation for U.S. federal income tax purposes from and after the closing date. However, an opinion of tax counsel is not binding on the IRS or a court. Therefore, there can be no assurance that the IRS will not take a position contrary to DLA Piper’s Section 7874 opinion or that a court will not agree with the IRS in the event of litigation.
Failure to complete the transactions contemplated under the merger agreement could negatively impact the stock price and the future business and financial results of Pozen.
If the transactions contemplated under the merger agreement are not completed, the ongoing business of Pozen may be materially and adversely affected and, without realizing any of the benefits of having completed the transactions, Pozen will be subject to a number of risks, including the following:
· Pozen will be required to pay certain costs relating to the transactions, including legal, accounting, investment banking, filing, and other fees and mailing, financial printing and other expenses, whether or not the transactions contemplated under the merger agreement are completed, and Pozen may be required to pay Tribute a termination fee of up to $3.5 million in the event the merger agreement is terminated under certain conditions;
· the current price of Pozen common stock may reflect a market assumption that the transactions contemplated under the merger agreement will occur, meaning that a failure to complete the transactions could result in a material decline in the price of Pozen common stock;
· Pozen may experience negative reactions from its customers, regulators and employees;
· the merger agreement places certain restrictions on the conduct of Pozen’s business prior to completion of the transactions contemplated thereunder. Such restrictions, the non-compliance of which is subject to the consent of Tribute, may prevent Pozen from making certain acquisitions or taking certain other specified actions during the pendency of the transactions; and
· matters relating to the transactions contemplated under the merger agreement (including integration planning) have required and will continue to require substantial commitments of time and resources by Pozen management, which could otherwise have been devoted to day-to-day operations and other opportunities that may have been beneficial to Pozen as an independent company. In addition to the above risks, Pozen may be required to pay to Tribute a termination fee, which may materially adversely affect Pozen’s financial results.
In addition to the above risks, Pozen may be required to pay to Tribute a termination fee, which may materially adversely affect Pozen’s financial results. If the transactions contemplated under the merger agreement are not completed, these risks may materialize and may materially and adversely affect Pozen’s business, financial results and share price.
Pozen stockholders will have a reduced ownership and voting interest after the merger and will exercise less influence over management.
Pozen stockholders currently have the right to vote in the election of the Board and on other matters affecting Pozen. Upon the completion of the merger, each Pozen stockholder that receives Parent Shares will become a shareholder of Parent, with a percentage ownership of Parent that is smaller than such stockholder’s percentage ownership of Pozen. As of the date of the proxy statement/prospectus on Form S-4, as filed on July 20, 2015 by Parent, the former stockholders of Pozen as a group will receive Parent Shares in the merger constituting approximately 66% of the outstanding Parent Shares immediately after the transactions contemplated under the merger agreement. After giving effect to the transactions and the proposed Parent equity financing and debt financing, the former stockholders of Pozen as a group will hold Parent Shares constituting approximately 49% of the outstanding Parent Shares and the former Tribute shareholders as a group will hold Parent Shares constituting approximately 28% of the Parent Shares. Because of this, Pozen stockholders will have less influence on the management and policies of Parent than they now have on the management and policies of Pozen. The relative ownership of Parent Shares by current Pozen stockholders and current Tribute shareholders referred to above is on an economic basis, and does not represent the analysis under Section 7874 of the Code, discussed throughout the Form S-4, as to whether, following the merger, former stockholders of Pozen will own less than 80% (by both vote and value) of Parent Shares.
Pozen and Tribute may fail to realize all of the anticipated benefits of the transactions or those benefits may take longer to realize than expected. The combined company may also encounter significant difficulties in integrating the two businesses.
The ability of Parent to realize the anticipated benefits of the transactions contemplated under the merger agreement will depend, to a large extent, on the combined company’s ability to integrate the businesses of Pozen and Tribute. The combination of two independent businesses is a complex, costly and time-consuming process. As a result, Parent will be required to devote significant management attention and resources to integrating their business practices and operations. The integration process may disrupt the businesses and, if implemented ineffectively, would restrict the realization of the full expected benefits. The failure to meet the challenges involved in integrating the two businesses and to realize the anticipated benefits of the transaction could cause an interruption of, or a loss of momentum in, the activities of the combined company and could adversely affect the results of operations of the combined company.
In addition, the overall integration of the businesses may result in material unanticipated problems, expenses, liabilities, competitive responses, loss of customer relationships and diversion of management’s attention. The difficulties of combining the operations of the companies include, among others:
· diversion of management’s attention to integration matters;
· difficulties in achieving anticipated cost savings, synergies, business opportunities and growth prospects from the combination of the businesses of Pozen and Tribute;
· difficulties in the integration of operations and systems;
· conforming standards, controls, procedures and accounting and other policies, business cultures and compensation structures between the two companies;
· difficulties in the assimilation of employees;
· difficulties in managing the expanded operations of a significantly larger and more complex company;
· challenges in keeping existing customers and obtaining new customers;
· potential unknown liabilities or larger liabilities than projected, adverse consequences and unforeseen increased expenses associated with the merger; and
· coordinating a geographically dispersed organization.
Many of these factors will be outside the control of Pozen, Tribute or Parent, and any one of them could result in increased costs, decreases in the amount of expected revenues and diversion of management’s time and energy, which could materially impact the business, financial condition and results of operations of the combined company. In addition, even if the operations of the businesses of Pozen and Tribute are integrated successfully, the full benefits of the transactions may not be realized, including the synergies, cost savings or sales or growth opportunities that are expected. These benefits may not be achieved within the anticipated time frame, or at all. Additional unanticipated costs may be incurred in the integration of the businesses of Pozen and Tribute. All of these factors could cause dilution to the earnings per share of the combined company, decrease or delay the expected accretive effect of the transactions and negatively impact the price of Parent Shares following the transactions contemplated under the merger agreement. As a result, we cannot assure you that the combination of Pozen and Tribute will result in the realization of the full benefits anticipated from the transactions.
The benefits described in the Form S-4, are also subject to a variety of other factors, many of which are beyond Pozen’s and Tribute’s ability to control, such as changes in the rate of economic growth in jurisdictions in which the combined company will do business, the financial performance of the combined business in various jurisdictions, currency exchange rate fluctuations, and significant changes in trade, monetary or fiscal policies, including changes in interest rates, and tax law of the jurisdictions in which the combined company will do business. The impact of these factors, individually and in the aggregate, is difficult to predict, in part because the occurrence of the events or circumstances described in such factors may be interrelated, and the impact to the combined company of the occurrence of any one of these events or circumstances could be compounded or, alternatively, reduced, offset, or more than offset, by the occurrence of one or more of the other events or circumstances described in such factors.
The transactions contemplated under the merger agreement may not be accretive and may cause dilution to Parent’s earnings per share, which may negatively affect the market price of Parent Shares following the transactions.
Although Parent currently anticipates that the transactions contemplated under the merger agreement will be accretive to earnings per share from and after the transactions, this expectation is based on preliminary estimates which may change materially. Parent expects to issue approximately 67.26 million Parent Shares in connection with the completion of the transactions. The issuance of these new Parent Shares could have the effect of depressing the market price of Parent Shares. In addition, Parent could also encounter additional transaction-related costs or other factors such as the failure to realize all of the benefits anticipated in the transactions contemplated by the merger agreement. All of these factors could cause dilution to Parent’s earnings per share or decrease or delay the expected accretive effect of the transactions and cause a decrease in the market price of Parent Shares following the transactions.
Transfers of Shares, other than by means of the transfer of book-entry interests in the Depository Trust Company (“DTC”) or by means of the transfer of book entry interests in Clearing and Depositary Services Inc. (“CDS”) may be subject to Irish stamp duty.
It is anticipated that, for the majority of transfers of Shares, there will not be any Irish stamp duty. Transfers of Shares effected by means of the transfer of book-entry interests in DTC or CDS should not be subject to Irish stamp duty. However, if you hold your Shares directly rather than beneficially through DTC or CDS, any transfer of your Shares could be subject to Irish stamp duty (currently at the rate of 1% of the higher of the price paid or the market value of the shares acquired). A shareholder who directly holds shares may transfer those shares into his or her own broker account to be held through DTC or CDS (or vice versa) without giving rise to Irish stamp duty, provided that there is no change in the beneficial ownership of the shares as a result of the transfer and the transfer is not in contemplation of a sale of the shares by a beneficial owner to a third party.
Payment of Irish stamp duty is generally a legal obligation of the transferee. The potential for stamp duty could adversely affect the price of your shares. See the section entitled “Irish Tax Considerations—Stamp Duty” beginning on page [·] of this registration statement.
Dividends paid in respect of the Shares may be subject to Irish dividend withholding tax.
In certain limited circumstances, dividend withholding tax (currently at a rate of 20%) may arise in respect of dividends paid on Shares. A number of exemptions from dividend withholding tax exist, such that shareholders resident in European Union member states (other than Ireland) or other countries with which Ireland has signed a double tax treaty, which would include the U.S. or Canada, should generally be entitled to exemptions from dividend withholding tax provided that the appropriate documentation is in place. See “Irish Tax Considerations—Withholding Tax on Dividends” beginning on page [·] of this registration statement and, in particular, please note the requirement to complete certain dividend withholding tax forms in order to qualify for many of the exemptions.
It is expected that shareholders resident in the U.S. who hold their shares through DTC may not be subject to dividend withholding tax if the addresses of the beneficial owners of such shares in the records of the brokers holding such shares are recorded as being in the U.S. (and such brokers have further transmitted the relevant information to a qualifying intermediary appointed by us).
Factors That May Affect Aralez’s Shareholders
The Pozen and Tribute stock prices have been volatile, which may result in significant losses to Aralez’s shareholders.
There has been significant volatility in the market prices of biotechnology companies’ securities. Various factors and events may have a significant impact on the market price of Aralez’s shareholders. These factors include:
|
· |
fluctuations in our operating results and revenues generated by our marketed products; |
|
|
|
|
· |
announcements of technological innovations, acquisitions or licensing of therapeutic products or product candidates by us or our competitors; |
|
|
|
|
· |
published reports by securities analysts; |
|
|
|
|
· |
positive or negative progress with our clinical trials or with regulatory approvals of our product candidates; |
|
|
|
|
· |
commercial success of VIMOVO and our other product candidates in the marketplace once approved; |
|
· |
governmental regulation, including reimbursement policies; |
|
|
|
|
· |
developments in patent or other proprietary rights; |
|
|
|
|
· |
developments in our relationships with collaborative partners; |
|
|
|
|
· |
announcements by our collaborative partners regarding our products or product candidates; |
|
|
|
|
· |
developments in new or pending litigation; |
|
|
|
|
· |
public concern as to the safety and efficacy of our products; and |
|
|
|
|
· |
general market conditions. |
The trading price of our common stock has been, and could continue to be, subject to wide fluctuations in response to these factors, including the sale or attempted sale of a large amount of our common stock into the market. From October 16, 2000, when our common stock began trading on The NASDAQ National Market (now known as The NASDAQ Global Market), through August 3, 2015, the high and low sales prices of our common stock ranged from $2.15 to $21.88. Broad market fluctuations may also adversely affect the market price of our common stock.
Sales of substantial amounts of our common stock in the public market by us or our largest stockholders could depress our stock price.
We have not sold shares of common stock in a public offering since our initial public offering in October 2000. Accordingly, we have a relatively small number of shares that are traded in the market. Prior to the merger and contemplated financings, approximately 9% of our outstanding shares are beneficially held by John Plachetka, our former Chairman, President and Chief Executive Officer. Additionally, we believe, based upon our review of public filings by certain stockholders and other publicly available information, an aggregate of approximately 25% of Pozen’s outstanding shares are held by three other stockholders, with one stockholder beneficially owning greater than 10% of our outstanding shares. Following the merger and the equity and debt financing, Deerfield Private Design and its affiliates will own approximately % of Aralez. Any sales of substantial amounts of our common stock in the public market, including sales or distributions of shares by our large stockholders, or the perception that such sales or distributions might occur, could harm the market price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. For example, our executive officers may sell shares pursuant to Rule 10b5-1 trading plans. Further, stockholders’ ownership will be diluted if we raise additional capital by issuing equity securities, and will be the case if the merger with Tribute is approved by our shareholders and the proposed equity and debt financing closes.
Anti-takeover provisions in Parent’s memorandum and articles of association and under Irish law could prevent or delay transactions that our stockholders may favor and may prevent shareholders from changing the direction of Parent’s business or management.
Provisions of Parent’s memorandum and articles of association may discourage, delay or prevent a merger or acquisition that Parent’s shareholders may consider favorable, including transactions in which shareholders might otherwise receive a premium for your shares, and may also frustrate or prevent any attempt by shareholders to change the direction or management of Parent. For example, these provisions:
· authorize the issuance of “blank check” preferred stock without any need for action by shareholders;
· provide for a classified board of directors with staggered three-year terms;
· require supermajority stockholder approval to effect various amendments to our memorandum and articles of association;
· prohibit shareholder action by written consent; and
· establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted on by shareholders at shareholder meetings.
Any transaction in which a third party seeks to acquire 30% or more of the voting rights of Aralez and other acquisitions of Aralez securities will be governed by the Irish Takeover Panel Act 1997 (the “Takeover Panel Act”) and the Irish Takeover Rules 2007, as amended (“Irish Takeover Rules”) which will be regulated by the Irish Takeover Panel (the “Panel”). The “General Principles” of the Irish Takeover Rules and certain important aspects of the Irish Takeover Rules are described more fully in the section entitled “Description of Share Capital” on page [·].
The rights of our shareholders may differ from the rights typically offered to shareholders of a U.S. corporation and these differences may make our ordinary shares less attractive to investors.
We are incorporated under Irish law and, therefore, certain of the rights of holders of our shares are governed by Irish law, including the provisions of the Irish Companies Acts, and by our articles of association. These rights differ in certain respects from the rights of shareholders in typical U.S. corporations and these differences may make our ordinary shares less attractive to investors. The principal differences include the following:
· under Irish law, dividends may only be declared if we have, on an individual entity basis, profits available for distribution, within the meaning of the Irish Companies Acts;
· under Irish law, each shareholder generally has preemptive rights to subscribe on a proportionate basis to any issuance of shares. Under U.S. law, shareholders generally do not have preemptive rights unless specifically granted in the certificate of incorporation or otherwise Pre-emption rights may be disapplied under Irish law for renewable five year periods by Irish companies by way of a provision in their articles of association or special resolution of their shareholders, which is an option we expect to avail ourselves of prior to the consummation of this offering;
· under Irish law, certain matters require the approval of holders of 75% of the votes cast at a general meeting of our shareholders, including amendments to our articles of association. This may make it more difficult for us to complete certain types of corporate transactions deemed advisable by our Board. Under U.S. law, generally only majority shareholder approval is required to amend the certificate of incorporation or to approve other significant transactions;
· under Irish law, a bidder seeking to acquire us would need, on a tender offer, to receive shareholder acceptance in respect of 80% of our outstanding shares. If this 80% threshold is not achieved in the offer, under Irish law, the bidder cannot complete a “second step merger” to obtain 100% control of us. Accordingly, tender of 80% of our outstanding shares will likely be a condition in a tender offer to acquire us, not 50% as is more common in tender offers for corporations organized under U.S. law; and
· under Irish law, shareholders may be required to disclose information regarding their equity interests upon our request, and the failure to provide the required information could result in the loss or restriction of rights attaching to the shares, including prohibitions on the transfer of the shares, as well as restrictions on voting, dividends and other payments. Comparable provisions generally do not exist under U.S. law.
Parent does not expect to pay dividends for the foreseeable future, and you must rely on increases in the trading price of the Parent Shares for returns on your investment.
Parent has never paid cash dividends on Parent Shares and does not expect to pay dividends in the immediate future. Parent anticipates that it will retain all earnings, if any, to support its operations. Any future determination as to the payment of dividends will, subject to Irish legal requirements, be at the sole discretion of Parent’s board of directors and will depend on Parent’s financial condition, results of operations, capital requirements and other factors Parent’s board of directors deems relevant. Holders of Parent Shares must rely on increases in the trading price of their shares for returns on their investment in the foreseeable future. In addition, the Facility Agreement prohibits Parent from making any cash dividend or distributing any of its assets, including its intangibles, to any of its shareholders in such capacity or its affiliates, subject to certain exceptions. The Facility Agreement also includes restrictions on Parent from incurring liens and undertaking indebtedness, subject to certain exceptions, which limitations may further impact the ability of Parent to pay any future dividends.
Parent will seek Irish High Court approval of the creation of distributable reserves. Parent expects this will be forthcoming, but cannot guarantee this.
Under Irish law, dividends may only be paid and share repurchases and redemptions must generally be funded only out of distributable reserves, which Parent will not have immediately following the closing. The creation of distributable reserves of Parent involves a reduction in Parent’s Parent Share premium account, which requires the approval of the Irish High Court and, in connection with seeking such court approval, the approval of Pozen and Tribute stockholders is being sought. Parent is not aware of any reason why the Irish High Court would not approve the creation of distributable reserves in this manner; however, the issuance of the required order is a matter for the discretion of the Irish High Court. There will also be no guarantee that the approvals by Pozen and Tribute stockholders will be obtained. In the event that distributable reserves of Parent are not created, no distributions by way of dividends, share repurchases or otherwise will be permitted under Irish law until such time as the group has created sufficient distributable reserves from its business activities.
Risk Factors Related to the Tribute Business
If Tribute loses its license from any licensors, Tribute may be unable to continue a substantial part of its business.
Tribute has licensed certain assets, including certain intellectual property, marketing authorizations and related data, and medical commercial and technical information, used in a substantial part of Tribute’s business. Such license agreement, may be terminated by the licensor if Tribute is in breach of its obligations under, or fails to perform any terms of, the agreement and fails to cure that breach. If such license agreements is terminated, then Tribute may lose its rights to utilize the intellectual property and other assets covered by that agreement to manufacture, market, promote, distribute and sell the licensed products, which may prevent Tribute from continuing a substantial part of its business and may result in a material and serious adverse effect on Tribute’s financial condition, results of operations and any prospects for growth.
Tribute’s failure to successfully discover, acquire, license or develop and market additional product candidates or approved products would impair Tribute’s ability to grow.
As part of Tribute’s growth strategy, it intends to acquire, license or develop and market additional products and product candidates. Tribute is pursuing various therapeutic opportunities through its pipeline. The product candidates to which Tribute allocates its resources may not end up being successful. In addition, because Tribute’s internal research capabilities are limited, it may depend upon pharmaceutical and biotechnology and other researchers to sell or license products or technology to it. The success of this strategy depends partly upon Tribute’s ability to identify, select, discover, license and/or acquire promising pharmaceutical product candidates and products for Canada and elsewhere. Failure of this strategy would impair Tribute’s ability to grow.
The process of proposing, negotiating and implementing a license or acquisition of a product candidate or approved product is lengthy and complex. Other companies, including some with substantially greater financial, marketing and sales resources, may compete with Tribute for the license or acquisition of product candidates and approved products. Tribute may devote resources to potential acquisitions or in-licensing opportunities that are never completed, or Tribute may fail to realize the anticipated benefits of such efforts. Tribute may not be able to acquire the rights to additional product candidates on terms that Tribute finds acceptable, or at all.
In addition, future acquisitions may entail numerous operational and financial risks, including:
· exposure to unknown liabilities;
· disruption of Tribute’s business and diversion of management’s time and attention to develop acquired products or technologies;
· incurrence of substantial debt, dilutive issuances of securities or depletion of cash to pay for acquisitions;
· higher than expected acquisition and integration costs;
· difficulty in combining the operations and personnel of any acquired businesses with Tribute’s operations and personnel;
· increased amortization expenses;
· impairment of relationships with key suppliers or customers of any acquired businesses due to changes in management and ownership; and
· inability to motivate key employees of any acquired businesses.
Further, any product candidate that Tribute acquires may require additional development efforts prior to commercial sale, including extensive clinical testing and approval by applicable regulatory authorities. All product candidates are prone to risks of failure typical of pharmaceutical product development, including the possibility that a product candidate will not be shown to be sufficiently safe and effective for approval by applicable regulatory authorities.
Tribute may not be able to compete with treatments now being developed and marketed, or which may be developed and marketed in the future by other companies.
Tribute’s products and licensed products will compete with existing and new therapies and treatments and numerous pharmaceutical and biotechnology companies, hospitals, research organizations, individual scientists and nonprofit organizations are engaged in the development of alternatives to Tribute’s technologies and products. Some of these companies have greater research and development capabilities, experience, manufacturing, marketing, financial and managerial resources than does Tribute. Collaborations or mergers between large pharmaceutical or biotechnology companies with competing drugs and technologies could enhance Tribute’s competitors’ financial, marketing and other resources. Developments by other drug companies could make Tribute’s products or technologies uncompetitive or obsolete. Accordingly, Tribute’s competitors may succeed in developing competing drugs or technologies, obtaining regulatory approval for products or gaining market acceptance more rapidly than Tribute can.
If government programs and insurance companies do not agree to pay for or reimburse patients for Tribute’s pharmaceutical products and licensed products, Tribute’s success will be impacted.
Sales of Tribute’s products and licensed products will depend in part on the availability of reimbursement by third-party payers such as government health administration authorities, private health insurers and other organizations. Third-party payers often challenge the price and cost-effectiveness of medical products and services. Governmental approval of health care products does not guarantee that these third-party payers will pay, or pay in full, for the products. Legislation and regulations affecting the pricing of pharmaceuticals may change before Tribute’s products or licensed products are approved for marketing and any such changes could further limit reimbursement. The availability and amount of such reimbursement could adversely affect Tribute’s results of operations and financial condition.
Tribute and its partners are subject to extensive Canadian, U.S. and foreign government regulation, including the requirement of approval before products may be manufactured or marketed.
Tribute, its present and future collaboration partners, and the drug product candidates developed by, or licensed to, Tribute or in collaboration with partners are subject to extensive regulation by governmental authorities in Canada, the U.S. and other countries. Failure to comply with applicable requirements could result in, among other things, any of the following actions: warning letters, fines and other civil penalties, unanticipated expenditures, delays in approving or refusal to approve a product candidate, product recall or seizure, interruption of manufacturing or clinical trials, operating restrictions, injunctions and criminal prosecution.
The product candidates of Tribute and its partners cannot be marketed in Canada, the U.S. or any other jurisdiction without regulatory approval from a regulatory authority such as Health Canada or the FDA (“Regulatory Authority”). Obtaining regulatory approval from a Regulatory Authority requires substantial time, effort, and financial resources, and may be subject to both expected and unforeseen delays, including, without limitation, citizen’s petitions or other filings with the Regulatory Authority, and there can be no assurance that any approval will be granted on a timely basis, if at all, or that delays will be resolved favorably or in a timely manner by any Regulatory Authority. If Tribute’s or its partners’ product candidates are not approved in a timely fashion, or are not approved at all, Tribute’s business and financial condition may be adversely affected.
In addition, both before and after regulatory approval, Tribute, its collaboration partners and its product candidates are subject to numerous requirements by Regulatory Authorities covering, among other things, testing, manufacturing, quality control, labeling, advertising, promotion, distribution and export. These requirements may change and additional government regulations may be promulgated that could affect Tribute, its collaboration partners or its product candidates. Tribute cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in Canada, the U.S. or abroad.
Such laws and regulations could hinder or prevent Tribute from successfully developing and commercializing Bezalip® SR (bezafibrate) for the United States market pursuant to its exclusive license, or any other product candidates. Tribute could fail to successfully obtain the required approvals for its two currently unmarketed products, including bilastine, which is pending registration from Health Canada and MycoVaTM, which has not been filed with Health Canada. There can be no assurance that neither Tribute nor any of its partners will be required to incur significant costs to comply with such laws and regulations in the future or that such laws or regulations will not have a material adverse effect upon Tribute’s business.
Even if regulatory approvals are obtained for Tribute’s products and licensed products, such products will be subject to ongoing regulatory review. If Tribute or a partner fails to comply with continuing Canadian, U.S. and foreign regulations, the approvals to market Tribute’s products and licensed products could be lost and Tribute’s business would be materially adversely affected.
Following any initial Health Canada, FDA or foreign regulatory approval of any drugs Tribute or a partner may develop, such drugs, will continue to be subject to regulatory review, including the review of adverse drug experiences, safety reports and clinical results that are reported after such drugs are made available to patients. This would include results from any post marketing tests or vigilance required as a condition of approval. The manufacturer and manufacturing facilities used to make any drug candidates will also be subject to periodic review and inspection by regulatory authorities, including Health Canada and/or the FDA. The discovery of any new or previously unknown problems with the product, manufacturer or facility may result in restrictions on the drug or manufacturer or facility, including withdrawal of the drug from the market. Marketing, advertising and labeling also will be subject to regulatory requirements and continuing regulatory review. The failure to comply with applicable continuing regulatory requirements may result in fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and other adverse consequences.
Materials necessary to manufacture Tribute’s products and licensed products may not be available on commercially reasonable terms, or at all, which may result in reduced revenues due to product shortages.
Tribute relies on third-party manufacturers to manufacture its products and licensed products. Most of Tribute’s third-party suppliers purchase, on its behalf, the materials necessary to produce the finished, final product for sale including the active pharmaceutical ingredients (“APIs”) and other such materials necessary to produce finished, saleable products for the commercial distribution of Tribute’s products and licensed products. In the event that suppliers of a product, ingredient or any materials Tribute needs to manufacture or package its products or licensed products are not available or not for sale at the time Tribute needs such ingredient or material in order to meet Tribute’s required delivery schedule or on commercially reasonable terms, then Tribute could be at risk of a product shortage or stock-out. Tribute relies on its suppliers in many cases to ensure the adequate supply of ingredients, APIs and packaging material and for the timely delivery of orders placed by Tribute. Should Tribute experience a shortage in supply of a product, licensed product, or API, any material sales of such product or licensed product could be harmed or reduced and Tribute’s ability to generate revenues from such product or licensed product may be impaired.
Tribute’s product candidates, products and licensed products may not gain acceptance or continued acceptance among physicians, patients and the medical community, thereby limiting Tribute’s potential to generate revenues.
The degree of market acceptance or continued market acceptance of Tribute’s product candidates (if approved for commercial sale by a Regulatory Authority), products or licensed products by physicians, healthcare professionals and third-party payers, and Tribute’s profitability and growth, will depend on a number of factors, including:
|
· |
demonstration of efficacy; |
|
|
|
|
· |
changes in the practice guidelines and the standard of care for the targeted indication; |
|
|
|
|
· |
relative convenience and ease of administration; |
|
|
|
|
· |
the prevalence and severity of any adverse side effects; |
|
|
|
|
· |
budget impact of adoption of Tribute’s product or licensed product on relevant drug formularies and the availability, cost and potential advantages of alternative treatments, including less expensive generic drugs; |
|
|
|
|
· |
pricing and cost effectiveness, which may be subject to regulatory control; |
|
|
|
|
· |
effectiveness of Tribute’s or any of its partners’ sales and marketing strategies; |
|
|
|
|
· |
the final product labeling or product insert required by Regulatory Authorities; and |
|
|
|
|
· |
the availability of adequate third-party insurance coverage or reimbursement. |
If any product candidate or product that Tribute acquires, licenses or develops does not provide a treatment regimen that is as beneficial as, or is perceived as being as beneficial as, the current standard of care or otherwise does not provide patient benefit, that product candidate (if approved for commercial sale by a Regulatory Authority), product or licensed product likely will not achieve market acceptance or continued market acceptance. Tribute’s ability to effectively promote and sell any approved products will also depend on pricing and cost-effectiveness, including Tribute’s ability to produce a product at a competitive price and Tribute’s ability to obtain sufficient third-party coverage or reimbursement. If any product candidate, product or licensed product is approved but does not achieve an adequate level of acceptance, or continued acceptance, by physicians, patients and third-party payers, Tribute’s ability to generate revenues from that product or licensed product would be substantially reduced. In addition, Tribute’s efforts to educate the medical community and third-party payers on the benefits of Tribute’s product candidates, products or licensed products may require significant resources, may be constrained by Regulatory Authority rules and policies on product promotion and may never be successful.
Guidelines and recommendations published by various organizations can impact the use of Tribute’s products and licensed products.
Government agencies promulgate regulations and guidelines directly applicable to Tribute and to its products and licensed products. In addition, professional societies, practice management groups, private health and science foundations and organizations involved in various diseases from time to time may also publish guidelines or recommendations to the health care and patient communities. Recommendations of government agencies or these other groups or organizations may relate to such matters as usage, dosage, route of administration and use of concomitant therapies. Recommendations or guidelines suggesting the reduced use of Tribute’s products or licensed products or the use of competitive or alternative products that are followed by patients and health care providers could result in decreased use of Tribute’s products and licensed products.
Tribute has limited manufacturing experience or resources, and Tribute must incur significant costs to develop this expertise or rely on third parties to manufacture Tribute’s products and licensed products.
Tribute relies on several contract manufacturers for Tribute’s supply of products and licensed products. There are risks inherent in pharmaceutical manufacturing that could affect the ability of Tribute’s contract manufacturers to meet Tribute’s delivery time requirements or provide adequate amounts of material to meet Tribute’s needs. Included in these risks are synthesis and purification failures and contamination during the manufacturing process, which could result in unusable product and cause delays in Tribute’s development process, as well as additional expense to Tribute. To fulfill Tribute’s requirements, if any, Tribute may also need to secure alternative suppliers for Tribute’s products or licensed products. In addition to the manufacture of certain of Tribute’s products and licensed products, Tribute may have additional manufacturing requirements related to the technology required for any of Tribute’s products or licensed products. In some cases, the delivery technology Tribute utilizes is highly specialized or proprietary, and for technical and legal reasons, Tribute may have access to only one or a limited number of potential manufacturers for such delivery technology. Failure by these manufacturers to properly formulate Tribute’s products or licensed products for delivery could also result in unusable product and cause delays in Tribute’s discovery and development process, as well as additional expense to Tribute.
The manufacturing process for any products based on Tribute’s technologies that Tribute or its partners may develop is subject to regulatory approvals from Regulatory Authorities and compliance with ongoing regulatory requirements, and together with Tribute’s partners Tribute needs to contract with manufacturers who can meet all applicable regulatory guidelines and requirements. In addition, if Tribute receives the necessary regulatory approval for any product candidate, it also expects to rely on third parties, including its commercial partners, to produce materials required for commercial supply. Tribute may experience difficulty in obtaining adequate manufacturing capacity for its needs. If Tribute is unable to obtain or maintain contract manufacturing for its product candidates, products or licensed products, or to do so on commercially reasonable terms, Tribute may not be able to successfully develop and commercialize its products or licensed products.
To the extent that Tribute enters into manufacturing arrangements with third parties, Tribute will depend on these third parties to perform its obligations in a timely manner and consistent with regulatory requirements, including those related to quality control and quality assurance. The failure of a third-party manufacturer to perform its obligations as expected could adversely affect Tribute’s business in a number of ways, including:
|
· |
Tribute may not be able to initiate or continue pre-clinical and clinical trials of products or licensed products that are under development; |
|
|
|
|
· |
Tribute may be delayed in submitting regulatory applications, or receiving regulatory approvals, for Tribute’s product candidates; |
|
|
|
|
· |
Tribute may lose the cooperation of Tribute’s partners; |
|
|
|
|
· |
Tribute’s products or licensed products could be the subject of inspections by Regulatory Authorities; |
|
|
|
|
· |
Tribute may be required to cease distribution or recall some or all batches of Tribute’s products or licensed products; and |
|
· |
Tribute potentially may be unable to meet commercial demands for Tribute’s products or licensed products. |
If a third-party manufacturer with whom Tribute contracts fails to perform its obligations, Tribute may be forced to manufacture the materials itself, for which Tribute may not have the capabilities or resources, or enter into an agreement with a different third-party manufacturer, which Tribute may not be able to do on reasonable terms, if at all or within acceptable timelines. In some cases, the technical skills required to manufacture Tribute’s product or licensed product may be unique to the original manufacturer and Tribute may have difficulty transferring such skills to a back-up or alternate supplier, or Tribute may be unable to transfer such skills. In addition, if Tribute is required to change manufacturers for any reason, Tribute will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer could negatively affect Tribute’s ability to develop product candidates in a timely manner or within budget. Furthermore, a manufacturer may possess technology related to the manufacture of Tribute’s product candidate that such manufacturer owns independently. This would increase Tribute’s reliance on such manufacturer or require Tribute to obtain a license from such manufacturer in order to have another third party manufacture Tribute’s products or licensed products.
Tribute’s products and licensed products may become obsolete.
The pharmaceutical industry is characterized by rapidly changing markets, technology, emerging industry standards and frequent introduction of new products. The introduction of new products embodying new technologies, including new manufacturing processes and the emergence of new industry standards may render Tribute’s products and licensed products obsolete, less competitive or less marketable. The process of developing Tribute’s products and licensed products is extremely complex and requires significant continuing development efforts and third party commitments. Tribute’s failure to develop new products and the obsolescence of existing products could adversely affect Tribute’s business.
Tribute may be unable to anticipate changes in Tribute’s potential customer requirements that could make Tribute’s existing products and licensed products obsolete. Tribute’s success will depend, in part, on its ability to continue to enhance Tribute’s existing products, develop new products that address the increasing sophistication and varied needs of the market, and respond to technological advances and emerging industry standards and practices on a timely and cost-effective basis. The development of Tribute’s products and licensed products entails significant technical and business risks. Tribute may not be successful in adapting to evolving customer or medical requirements or preferences or emerging industry standards.
Tribute faces competition in its markets from a number of large and small companies, some of which have greater financial, research and development, production and other resources than Tribute has.
Tribute’s products and licensed products face competition from products which may be used as an alternative or substitute therefor. In addition, Tribute competes with several large companies in the healthcare industry. To the extent these companies, or new entrants into the market, offer comparable products at lower or similar prices, Tribute’s business could be adversely affected. Tribute’s competitors can be expected to continue to improve the design and performance of their products and to introduce new products with competitive performance characteristics. There can be no assurance that Tribute will have sufficient resources to maintain Tribute’s current competitive position.
Tribute uses certain intellectual property that it licenses from third parties. If Tribute does not comply with those licenses, Tribute could lose its rights under them.
Tribute relies, in part, on licenses to use certain intellectual property that is important to Tribute’s business, and Tribute does not own the patents or other intellectual property that underlies those licenses. Tribute’s rights to use the products and technologies claimed in the licensed patents are subject to Tribute abiding by the terms of those licenses and the licensors not terminating them. Tribute believes it is currently in material compliance with all requirements of those licenses. In certain cases, Tribute does not control the filing, prosecution or maintenance of the patent rights to which Tribute holds licenses and may rely upon Tribute’s licensors to prosecute infringement of those rights. The scope of Tribute’s rights under its licenses may be subject to dispute by its licensors or third parties.
It is difficult and costly to protect Tribute’s proprietary rights, and Tribute may not be able to ensure their protection.
Tribute’s commercial success depends in part on obtaining and maintaining patent protection and trade secret protection of its products and licensed products, and the methods used to manufacture them, as well as successfully defending Tribute’s patents and licensed patents against third-party challenges. Tribute will only be able to protect its products from unauthorized making, using, selling, offer to sell or importation by third parties to the extent that Tribute or its licensors have rights under valid and enforceable patents or trade secrets that cover these activities.
As of the date of this prospectus, Tribute has five issued United States patents, two Canadian patents and a patent in each of Australia, China, the European Community and Japan. Additionally, as of the date of this prospectus Tribute has two pending foreign patent applications covering high dose patents. Some of Tribute’s patents, however, will expire as early as May 15, 2017.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in pharmaceuticals and biotechnology patents has emerged to date in Canada and the United States. The pharmaceutical and biotechnology patent situation outside of Canada and the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in Canada and the United States and other countries may diminish the value of Tribute’s intellectual property. Accordingly, Tribute cannot predict the breadth of claims that may be allowed or enforced in Tribute’s patents and licensed patents or in third-party patents.
Issued patents and patents issuing from pending applications may be challenged, invalidated or circumvented. Moreover, the United States Leahy-Smith America Invents Act, enacted in September 2011, brought significant changes to the U.S. patent system, which include a change to a “first to file” system from a “first to invent” system and changes to the procedures for challenging issued patents and disputing patent applications during the examination process, among other things. The effects of these changes on Tribute’s patent portfolio and business have yet to be determined, as the final substantive provisions of the America Invents Act took effect on March 16, 2013. The United States Patent and Trademark Office (the “USPTO”) only recently finalized the rules relating to these changes and the courts have yet to address the new provisions. These changes could increase the costs and uncertainties surrounding the prosecution of Tribute’s patent applications and the enforcement or defense of its patent rights. Additional uncertainty may result from legal precedent handed down by the United States Court of Appeals for the Federal Circuit and United States Supreme Court as they determine legal issues concerning the scope and construction of patent claims and inconsistent interpretation of patent laws by the lower courts. Accordingly, Tribute cannot ensure that any of its pending patent applications will result in issued patents, or even if issued, predict the breadth of the claims upheld in its and other companies’ patents. Given that the degree of future protection for Tribute’s proprietary rights is uncertain, Tribute cannot ensure that it was the first to invent the inventions covered by its pending patent applications, it was the first to file patent applications for these inventions, the patents it has obtained are valid and enforceable, and any proprietary products or technologies it develops will be patentable.
In addition, unauthorized parties may attempt to copy or otherwise obtain and use Tribute’s products or technology. Monitoring unauthorized use of Tribute’s intellectual property is difficult, and Tribute cannot be certain that the steps it has taken will prevent unauthorized use of its products or technologies, particularly in certain foreign countries where the local laws may not protect Tribute’s proprietary rights as fully as in the United States. Moreover, third parties could use or practice Tribute’s products or technologies in territories where Tribute does not have patent protection. Such third parties may then try to import into the United States or other territories products, or information leading to potentially competing products, made using Tribute’s products or technologies in countries where Tribute does not have patent protection for those products or technologies. If competitors are able to use Tribute’s products or technologies, Tribute’s ability to compete effectively could be harmed. Moreover, others may independently develop and obtain patents for products or technologies that are similar to or superior to Tribute’s products or technologies. If that happens, Tribute may need to license these products or technologies, and it may not be able to obtain licenses on reasonable terms, if at all, which could harm Tribute’s business.
The degree of future protection for Tribute’s proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect Tribute’s rights or permit it to gain or keep its competitive advantage. For example:
|
· |
others may be able to make compounds that are competitive with Tribute’s product candidates but that are not covered by the claims of Tribute’s patents; |
|
|
|
|
· |
Tribute might not have been the first to make the inventions covered by Tribute’s pending patent applications; |
|
|
|
|
· |
Tribute might not have been the first to file patent applications for these inventions; |
|
|
|
|
· |
others may independently develop similar or alternative technologies or duplicate any of Tribute’s technologies; |
|
|
|
|
· |
it is possible that Tribute’s pending patent applications will not result in issued patents; |
|
|
|
|
· |
Tribute may not develop additional proprietary technologies that are patentable; or |
|
|
|
|
· |
the patents of others may have an adverse effect on Tribute’s business. |
Tribute also may rely on trade secrets to protect its technology, especially where Tribute does not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. While Tribute uses reasonable efforts to protect its trade secrets, Tribute’s employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose Tribute’s information to competitors. Enforcing a claim that a third party illegally obtained and is using Tribute’s trade secrets, is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside of Canada and the United States are sometimes less willing to protect trade secrets. Moreover, Tribute’s competitors may independently develop equivalent knowledge, methods and know-how.
Tribute may incur substantial costs as a result of litigation or other proceedings relating to enforcing its patent and other intellectual property rights, including its licensed intellectual property rights, and Tribute may be unable to protect Tribute’s rights to, or to use, its technology or products.
If Tribute chooses to litigate against someone else from using the inventions claimed in Tribute’s patents or licensed patents, that individual or company has the right to ask the court to rule that these patents or licensed patents are invalid and/or should not be enforced against that third party. Proving patent infringement may be difficult, especially where it is possible to manufacture a product by multiple processes. Although Tribute believes that it has conducted its patent prosecution in accordance with the duty of candor and in good faith, the outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. These lawsuits are expensive and would consume time and other resources even if Tribute were successful in stopping the infringement of these patents. In addition, there is a risk that the court will decide that Tribute’s patents or licensed patents are not valid and that Tribute does not have the right to stop the other party from using the inventions. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, Tribute would not be able to exclude others from practicing the inventions claimed therein. Such a loss of patent protection could have a material adverse impact on Tribute’s business. There is also the risk that, even if the validity of Tribute’s patents or licensed patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe Tribute’s rights to these patents. Even if Tribute’s patent rights are found to be valid and enforceable, patent claims that survive litigation may not cover commercially valuable products or prevent competitors from importing or marketing products similar to Tribute’s products or licensed products, or using manufacturing processes or manufacturing components similar to those used to produce Tribute’s products or licensed products.
Although Tribute believes that it, or its licensors, as applicable, has or have obtained assignments of patent rights from all inventors, if an inventor did not adequately assign their patent rights to Tribute, or Tribute’s licensors, as applicable, a third party could obtain a license to the patent from such inventor. This could preclude Tribute from enforcing its patent or licensed patent against such third party.
Because some patent applications in Canada and the United States may be maintained in secrecy until the patents are issued, patent applications in Canada and the United States and many foreign jurisdictions are typically not published until eighteen months after filing, and publications in the scientific literature often lag behind actual discoveries, Tribute cannot be certain that others have not filed patent applications for technology or products covered by its issued or licensed patents or its pending applications or that Tribute (or its licensor(s), as applicable) was the first to invent the technology or products. Tribute’s competitors may have filed, and may in the future file, patent applications covering technology or products similar to Tribute’s. Some of Tribute’s competitors may be able to sustain the costs of complex patent litigation more effectively than Tribute can because they have substantially greater resources. Any such patent application may have priority over Tribute’s patent applications and could further require it to obtain rights to issued patents covering such technologies or products. If another party has filed a United States patent application on inventions similar to Tribute’s, Tribute may have to participate in an interference proceeding declared by the USPTO, to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, resulting in a loss of Tribute’s United States patent position with respect to such inventions.
Litigation or other proceedings or third-party claims of intellectual property infringement could require Tribute to spend significant time and money and could prevent Tribute from commercializing its products or technologies or impact Tribute’s stock price.
Tribute’s commercial success also depends in part on not infringing patents and proprietary rights of third parties, and not breaching any licenses or other agreements that it has entered into with regard to its technologies, products and business. Tribute cannot ensure that patents have not been issued to third parties that could block Tribute’s or its partners’ ability to obtain patents or to operate as Tribute would like. There may be patents in some countries that, if valid, may block Tribute’s ability to make, use or sell its products or licensed products in those countries, or import its products or licensed products into those countries, if Tribute is unsuccessful in circumventing or acquiring the rights to these patents. There also may be claims in patent applications filed in some countries that, if granted and valid, also may block Tribute’s ability to commercialize products or processes in these countries if Tribute is unable to circumvent or license them.
The pharmaceutical and biotechnology industries are characterized by frequent and extensive litigation regarding patents and other intellectual property rights. Many companies have employed intellectual property litigation as a way to gain a competitive advantage. Tribute’s involvement in litigation, interferences, opposition proceedings or other intellectual property proceedings inside and outside of the United States, to defend its intellectual property rights, and licensed intellectual property rights, or as a result of alleged infringement of the rights of others, may divert management time from focusing on business operations and could cause Tribute to spend significant amounts of money. Some of Tribute’s competitors may have significantly greater resources and, therefore, they are likely to be better able to sustain the cost of complex patent or intellectual property litigation than could Tribute. The uncertainties associated with litigation could have a material adverse effect on Tribute’s ability to raise the funds necessary to continue its business or to enter into additional collaborations with others. Furthermore, any potential intellectual property litigation also could force Tribute or its partners to do one or more of the following:
|
· |
stop selling, incorporating or using products that use the intellectual property at issue; |
|
|
|
|
· |
obtain from the third party asserting its intellectual property rights a license to sell or use the relevant technology or product, which license may not be available on reasonable terms, if at all; or |
|
|
|
|
· |
redesign those products or processes that use any allegedly infringing technology, or relocate the operations relating to the allegedly infringing technology to another jurisdiction, which may result in significant cost or delay to Tribute, or which could be technically infeasible. |
A third party may claim that Tribute is using inventions covered by the third party’s patent rights and may go to court to stop Tribute from engaging in its normal operations and activities, including making or selling Tribute’s product candidates or licensed products. These lawsuits are costly and could affect Tribute’s results of operations and divert the attention of managerial and technical personnel. There is a risk that a court would decide that Tribute is infringing the third party’s patents and would order Tribute to stop the activities covered by the patents or licensed patents. In addition, there is a risk that a court will order Tribute to pay the other party damages for having violated the other party’s patents. The pharmaceutical industry has produced a proliferation of patents, and it is not always clear to industry participants, including Tribute, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If Tribute is sued for patent infringement, Tribute would need to demonstrate that its products or licensed products, as applicable, or methods of use either do not infringe the patent claims of the relevant patent and/or that the patent claims are invalid, and Tribute may not be able to do this. Proving invalidity, in particular, is difficult since it requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
Tribute may not be able to enforce its intellectual property rights throughout the world.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States and Canada. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection. This could make it difficult for Tribute to stop the infringement of its patents and licensed patents or misappropriation of its other intellectual property rights. Proceedings to enforce Tribute’s patent rights in foreign jurisdictions could result in substantial costs and divert Tribute’s efforts and attention from other aspects of its business. Accordingly, Tribute’s efforts to protect its intellectual property rights in such countries may be inadequate.
If Tribute’s products or technologies are stolen, misappropriated or reverse engineered, others could use Tribute’s products or licensed products to produce competing products or technologies.
Third parties, including Tribute’s partners, contract manufacturers, contractors and others involved in Tribute’s business often have access to Tribute’s products, licensed products, and technologies. If Tribute’s products, licensed products or technologies were stolen, misappropriated or reverse engineered, they could be used by other parties that may be able to reproduce Tribute’s products, licensed products, or technologies for their own commercial gain. If this were to occur, it would be difficult for Tribute to challenge this type of use, especially in countries with limited intellectual property protection.
If product liability lawsuits are successfully brought against Tribute, Tribute may incur substantial liabilities and may be required to limit commercialization of certain products.
Tribute faces an inherent risk of product liability lawsuits related to Tribute’s products and licensed products. Currently, Tribute is not aware of any anticipated product liability claims with respect to Tribute’s products or licensed products. In the future, an individual may bring a liability claim against Tribute if one of its products or licensed products causes, or merely appears to have caused, an injury. If Tribute cannot successfully defend itself against the product liability claim, it may incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
|
· |
decreased demand for Tribute’s products or licensed products; |
|
|
|
|
· |
injury to Tribute’s reputation; |
|
|
|
|
· |
withdrawal of clinical trial participants; |
|
|
|
|
· |
costs of related litigation; |
|
|
|
|
· |
initiation of investigations by regulators; |
|
|
|
|
· |
substantial monetary awards to patients or other claimants; |
|
|
|
|
· |
distraction of management’s attention from Tribute’s primary business; |
|
|
|
|
· |
product recalls; |
|
|
|
|
· |
loss of revenue; and |
|
|
|
|
· |
the inability to commercialize Tribute’s product candidates, products or licensed products. |
Tribute’s current insurance coverage may prove insufficient to cover any liability claims brought against Tribute. In addition, because of the increasing costs of insurance coverage, Tribute may not be able to maintain insurance coverage at a reasonable cost or obtain insurance coverage that will be adequate to satisfy liabilities that may arise.
Obtaining and maintaining Tribute’s patent protection depends on compliance with various procedural, document submissions, fee payments and other requirements imposed by governmental patent agencies on Tribute and certain parties from which Tribute licenses products, and Tribute’s patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
Confidentiality agreements with employees and others may not adequately prevent disclosure of Tribute’s trade secrets and other proprietary information and may not adequately protect Tribute’s intellectual property, which could limit Tribute’s ability to compete.
Because Tribute operates in the highly confidential environment, it relies in part on trade secret protection in order to protect Tribute’s proprietary trade secrets and unpatented know-how. However, trade secrets are difficult to protect, and Tribute cannot be certain that others will not develop the same or similar technologies on their own. Tribute has taken steps, including entering into confidentiality agreements with Tribute’s employees, consultants, outside scientific collaborators, sponsored researchers and other advisors, to protect Tribute’s trade secrets and unpatented know-how. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by Tribute during the course of the party’s relationship with Tribute. Tribute also typically obtains agreements from these parties which provide that inventions conceived by the party in the course of rendering services to Tribute will be Tribute’s exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to Tribute. Enforcing a claim that a party illegally obtained and is using Tribute’s trade secrets or know-how is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside Canada and the United States may be less willing to protect trade secrets or know-how. The failure to obtain or maintain trade secret protection could adversely affect Tribute’s competitive position.
Tribute may be subject to claims that Tribute’s employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the pharmaceutical industry, Tribute employs individuals who were previously employed at other pharmaceutical companies, including Tribute’s competitors or potential competitors. Although no claims against Tribute are currently pending, Tribute may be subject to claims that these employees or Tribute has inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if Tribute is successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated in this prospectus by reference contain “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements include, but are not limited to, statements regarding the transactions, the financing of the transactions, Pozen and Tribute’s expected future performance (including, but not limited to, any projections or prospective financial information, expected results of operations and financial guidance), and the our future financial condition, operating results, strategy and plans. Statements including words such as “believes,” “expects,” “anticipates,” “intends,” “estimates,” “plan,” “will,” “may,” “look forward,” “intend,” “guidance,” “future” or similar expressions are forward-looking statements. Because these statements reflect Pozen and Tribute’s current views, expectations and beliefs concerning future events, these forward-looking statements involve risks and uncertainties. Although Pozen and Tribute believe that these forward-looking statements and information are based upon reasonable assumptions and expectations, readers should not place undue reliance on them, or any other forward-looking statements or information in prospectus. Investors should note that many factors, as more fully described in the documents filed by Tribute with the SEC, including under the heading “Risk Factors” in Tribute’s Form 10-K, 10-Q and Form 8-K filings, as applicable, as well as the securities regulators in Canada on the System for Electronic Document Analysis and Retrieval and as otherwise enumerated herein or therein, and by Pozen with the SEC, including under the heading “Risk Factors” in Pozen’s Form 10-K, 10-Q and Form 8-K filings, as applicable, and as otherwise enumerated herein or therein, could affect future financial results and could cause actual results to differ materially from those expressed in forward-looking statements contained in this communication. Important factors that, individually or in the aggregate, could cause actual results to differ materially from expected and historical results include, but are not limited to:
· the failure to receive the required Pozen stockholder approval, the Tribute shareholder approval and the approval of applicable government and regulatory authorities (and the terms of those approvals, including, but not limited to, the Ontario Superior Court of Justice (Commercial List) with respect to the arrangement);
· the risk that a condition to closing the transactions may not be satisfied or waived;
· the ultimate outcome and results of integrating the operations of Pozen and Tribute, the ultimate outcome of our operating strategy and the ultimate ability to realize synergies and the magnitude of such synergies;
· the effects of the business combination of Pozen and Tribute, including the combined company’s future financial condition, operating results, strategy and plans;
· our ability to achieve significant upside potential for shareholders by obtaining approval of product candidates, including YOSPRALA, and by accelerating the growth of the products of the combined company;
· our ability to acquire new products or companies on terms acceptable to us;
· our ability to sustain and grow revenues and cash flow from operations in its markets and to maintain and grow its customer base, the need for innovation and the related capital expenditures and the unpredictable economic conditions in the United States, Canada, Ireland and other markets;
· the impact of competition from other market participants;
· the development and commercialization of new products, including YOSPRALA;
· the effects of governmental regulation on our business or potential business combination transactions;
· changes in tax laws or interpretations that could increase our consolidated tax liabilities, including, if the transaction is consummated, changes in tax laws that would result in us being treated as a domestic corporation for United States federal tax purposes;
· the availability and access, in general, of funds to meet our debt obligations prior to or when they become due and to fund its operations and necessary capital expenditures, either through (i) cash on hand, (ii) free cash flow or (iii) access to the capital or credit markets; and
· our ability to comply with all covenants under existing credit facilities, any violation of which, if not cured in a timely manner, could trigger a default of its other obligations under cross-default provisions.
Other unknown or unpredictable factors could also have material adverse effects on our future results, performance or achievements. All forward-looking statements attributable to us, to Pozen or Tribute or any person acting on either of their behalf are expressly qualified in their entirety by this cautionary statement. We, Pozen and Tribute do not assume any obligation to publicly update any forward-looking statements, whether as a result of new information, future developments or otherwise, except as may be required under applicable securities law.
On June 8, 2015, Pozen and Tribute agreed to a business combination under the terms of the merger agreement. In order to effect the transactions contemplated by the merger agreement, US Merger Sub, an indirect subsidiary of Parent, will be merged with and into Pozen. Pozen will be the surviving corporation and, through the merger, will become an indirect wholly owned subsidiary of Parent. The merger of Pozen into US Merger Sub will be effected under Delaware law so that Pozen will be reorganized into a holding company structure. In accordance with the merger agreement, Can Merger Sub will offer to acquire, and will acquire, all of the outstanding Tribute common shares pursuant to a court approved plan of arrangement in Canada in the manner provided for by the merger agreement. The Parent Shares to be issued to Tribute shareholders in the arrangement are not being registered pursuant to this registration statement. The Parent Shares to be issued to Pozen stockholders in the merger are not being registered pursuant to this registration statement. Upon completion of the arrangement, Tribute will also become an indirect wholly owned subsidiary of Parent. Upon completion, the merger and the arrangement do not constitute a change of control of Pozen.
As of August 3, 2015, Pozen had approximately 32,734,343 shares of common stock issued and outstanding. As a result of the merger, each share of Pozen common stock will be converted into the right to receive from Parent one Parent Share for each share of Pozen common stock that they own as of the record date for the Pozen special meeting. Pursuant to the arrangement, each outstanding Tribute common share will be exchanged for 0.1455 Parent Shares. Upon completion of the merger and arrangement, current Pozen stockholders will own approximately 66% of the outstanding Parent Shares, and current Tribute shareholders will own approximately 34% of the outstanding Parent Shares before giving effect to (i) any exercise of outstanding options and warrants or the vesting and delivery of shares underlying RSUs of either company and (ii) the Parent Shares to be issued to new investors pursuant to the equity and debt financings described below. It is expected that Parent Shares will be listed and traded on NASDAQ under the symbol “ARLZ” and on the “TSX” under the symbol “ARZ”. The terms and conditions of the merger and the arrangement are contained in the merger agreement. After giving effect to the transactions and the proposed Parent equity financing and debt financing, the former stockholders of Pozen as a group will hold Parent Shares constituting approximately 49% of the outstanding Parent Shares and the former shareholders of Tribute as a group will hold Parent Shares constituting approximately 28% of the outstanding Parent Shares.
On July 20, 2015, Parent filed with the SEC the Form S-4 in connection with the proposed business combination between Pozen and Tribute. The Form S-4 has not yet been declared effective by the SEC.
We will not receive any proceeds from the sale of any Shares by the Selling Shareholders.
The Selling Shareholders will receive all of the net proceeds from the sale of any Shares offered by them under this prospectus. The Selling Shareholders will pay any underwriting discounts and commissions and expenses incurred by the Selling Shareholders for brokerage, accounting, tax, legal services or any other expenses incurred by the Selling Shareholders in disposing of these shares. We will bear all other costs, fees and expenses incurred in effecting the registration of the Shares covered by this prospectus.
MARKET FOR COMMON EQUITY AND RELATED SHAREHOLDER MATTERS
Pozen’s common stock is currently listed on NASDAQ under the ticker symbol “POZN”. Pozen’s common stock began trading on NASDAQ on October 16, 2000. The following table sets forth the quarterly range of high and low reported sale prices of Pozen’s common stock as reported on NASDAQ:
|
|
|
High |
|
Low |
| ||
|
2013 |
|
|
|
|
| ||
|
First quarter |
|
$ |
6.49 |
|
$ |
5.11 |
|
|
Second quarter |
|
$ |
5.56 |
|
$ |
4.50 |
|
|
Third quarter |
|
$ |
5.99 |
|
$ |
4.92 |
|
|
Fourth quarter |
|
$ |
9.90 |
|
$ |
5.35 |
|
|
2014 |
|
|
|
|
| ||
|
First quarter |
|
$ |
8.99 |
|
$ |
7.37 |
|
|
Second quarter |
|
$ |
9.73 |
|
$ |
7.56 |
|
|
Third quarter |
|
$ |
9.59 |
|
$ |
5.96 |
|
|
Fourth quarter |
|
$ |
9.71 |
|
$ |
7.07 |
|
|
2015 |
|
|
|
|
| ||
|
First quarter |
|
$ |
8.16 |
|
$ |
6.56 |
|
|
Second quarter |
|
$ |
12.69 |
|
$ |
6.38 |
|
|
Third quarter (through August 3, 2015) |
|
$ |
12.37 |
|
$ |
9.85 |
|
Approximate Number of Equity Security Holders
The closing sale price on NASDAQ for Pozen’s common stock on August 3, 2015 was $11.62 per share. As of August 3, 2015, Pozen had approximately 72 registered holders of record of its shares of common stock. Because shares of Pozen’s common stock are held by depositaries, brokers and other nominees, the number of beneficial holders of its shares is larger than the number of stockholders.
Trading Information Following Consumation of the Proposed Transactions
As of August 3, 2015, Pozen had approximately 32,734,343 shares of common stock issued and outstanding. As a result of the merger, each share of Pozen common stock will be converted into the right to receive from Parent one Parent Share for each share of Pozen common stock that they own as of the record date for the Pozen special meeting. Pursuant to the arrangement, each outstanding Tribute common share will be exchanged for 0.1455 Parent Shares. Upon completion of the merger and arrangement, current Pozen stockholders will own approximately 66% of the outstanding Parent Shares, and current Tribute shareholders will own approximately 34% of the outstanding Parent Shares before giving effect to (i) any exercise of outstanding options and warrants or the vesting and delivery of shares underlying RSUs of either company and (ii) the Parent Shares to be issued to new investors pursuant to the equity and debt financings described below. After giving effect to the transactions and the proposed Parent equity financing and debt financing, the former stockholders of Pozen as a group will hold Parent Shares constituting approximately 49% of the outstanding Parent Shares and the former shareholders of Tribute as a group will hold Parent Shares constituting approximately 28% of the outstanding Parent Shares.
SELECTED HISTORICAL FINANCIAL DATA OF POZEN
The following tables present selected historical financial data for Pozen as of and for the fiscal years ended December 31, 2014, 2013, 2012, 2011 and 2010 and as of and for the three months ended March 31, 2015 and 2014. The statement of operations data for each of the three years in the period ended December 31, 2014 and the balance sheet data as of December 31, 2014 and 2013 have been obtained from Pozen's audited financial statements contained in its Annual Report on Form 10-K for the fiscal year ended December 31, 2014, which appears elsewhere in this prospectus. The statements of operations data for the years ended December 31, 2011 and 2010 and the balance sheet data as of December 31, 2012, 2011 and 2010 have been obtained from Pozen's audited financial statements for such years, which have not been incorporated into this document by reference. The financial data as of March 31, 2015 and for the three months ended March 31, 2015 and 2014 have been obtained from Pozen's unaudited condensed financial statements included in its Quarterly Report on Form 10-Q for the three months ended March 31, 2015.
The information set forth below is not necessarily indicative of future results and should be read together with the other information contained in Pozen's Annual Report on Form 10-K for the fiscal year ended December 31, 2014 and Pozen's Quarterly Report on Form 10-Q for the three months ended March 31, 2015, including "Management's Discussion and Analysis of Financial Condition and Results of Operations" and the financial statements and related notes therein. See the section entitled "Where You Can Find More Information" beginning on page 162 of this prospectus.
|
|
|
For the Year Ended December 31, |
|
For the Three |
| |||||||||||||||||
|
|
|
2010 |
|
2011 |
|
2012 |
|
2013 |
|
2014 |
|
2014 |
|
2015 |
| |||||||
|
|
|
(in thousands, except per share data) |
| |||||||||||||||||||
|
Statement of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Revenue: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Sale of royalty rights, net of costs |
|
$ |
— |
|
$ |
71,870 |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
Licensing revenue |
|
68,417 |
|
15,081 |
|
5,349 |
|
10,322 |
|
32,394 |
|
7,549 |
|
4,404 |
| |||||||
|
Development revenue |
|
132 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
| |||||||
|
Total revenue |
|
68,549 |
|
86,951 |
|
5,349 |
|
10,322 |
|
32,394 |
|
7,548 |
|
4,404 |
| |||||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Sales, general and administrative |
|
23,755 |
|
21,752 |
|
19,024 |
|
17,161 |
|
10,078 |
|
2,823 |
|
3,262 |
| |||||||
|
Research and development |
|
22,651 |
|
23,020 |
|
11,867 |
|
9,945 |
|
5,740 |
|
1,828 |
|
983 |
| |||||||
|
Total operating expenses |
|
46,406 |
|
44,772 |
|
30,891 |
|
27,106 |
|
15,818 |
|
4,651 |
|
4,245 |
| |||||||
|
Interest and other income |
|
929 |
|
161 |
|
259 |
|
76 |
|
3,099 |
|
7 |
|
(186 |
) | |||||||
|
Net income (loss) attributable to common stockholders |
|
$ |
23,072 |
|
$ |
42,340 |
|
$ |
(25,283 |
) |
$ |
(16,708 |
) |
$ |
19,675 |
|
$ |
2,905 |
|
$ |
(27 |
) |
|
Basic net income (loss) per common share |
|
$ |
0.77 |
|
$ |
1.41 |
|
$ |
(0.84 |
) |
$ |
(0.55 |
) |
$ |
0.63 |
|
$ |
0.09 |
|
$ |
0.00 |
|
|
Shares used in computing basic net income (loss) per common share |
|
29,880 |
|
29,925 |
|
30,092 |
|
30,450 |
|
31,360 |
|
30,744 |
|
32,259 |
| |||||||
|
Diluted net income per common share |
|
$ |
0.76 |
|
$ |
1.40 |
|
$ |
(0.84 |
) |
$ |
(0.55 |
) |
$ |
0.60 |
|
0.09 |
|
0.00 |
| ||
|
Shares used in computing diluted net income per common share |
|
30,246 |
|
30,296 |
|
30,092 |
|
30,450 |
|
32,811 |
|
32,489 |
|
32,259 |
| |||||||
|
|
|
December 31, |
|
As of and for the |
| ||||||||||||||
|
|
|
2010 |
|
2011 |
|
2012 |
|
2013 |
|
2014 |
|
March 31, 2015 |
| ||||||
|
|
|
(in thousands) |
|
|
| ||||||||||||||
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Cash, cash equivalents and short-term investments |
|
$ |
64,091 |
|
$ |
119,620 |
|
$ |
87,314 |
|
$ |
32,828 |
|
$ |
40,582 |
|
$ |
43,905 |
|
|
Total assets |
|
69,698 |
|
121,553 |
|
89,597 |
|
35,334 |
|
50,454 |
|
48,847 |
| ||||||
|
Total liabilities |
|
9,070 |
|
16,055 |
|
5,519 |
|
17,546 |
|
3,713 |
|
1,543 |
| ||||||
|
Accumulated deficit |
|
(116,927 |
) |
(74,588 |
) |
(99,871 |
) |
(116,579 |
) |
(96,904 |
) |
(96,932 |
) | ||||||
|
Total stockholders’ equity |
|
60,628 |
|
105,498 |
|
84,077 |
|
17,789 |
|
46,741 |
|
47,304 |
| ||||||
SELECTED HISTORICAL FINANCIAL DATA OF TRIBUTE
The following tables present selected historical financial data for Tribute as of and for the fiscal years ended December 31, 2014, 2013, 2012, 2011 and 2010 and as of and for the three months ended March 31, 2015 and 2014. The statement of operations data for each of the five years in the period ended December 31, 2014 and the balance sheet data as of December 31 have been obtained from Tribute's audited financial statements for such years and, with respect to the periods ended December 31, 2014, 2013 and 2012, from its Annual Report on Form 10-K for the fiscal year ended December 31, 2014, which appear elsewhere in this prospectus. The financial data as of March 31, 2015 and for the three months ended March 31, 2015 and 2014 have been obtained from Tribute's unaudited condensed financial statements included in its Quarterly Report on Form 10-Q for the three months ended March 31, 2015.
The information set forth below is not necessarily indicative of future results and should be read together with the other information contained in Tribute's Annual Report on Form 10-K for the fiscal year ended December 31, 2014 and Tribute's Quarterly Report on Form 10-Q for the three months ended March 31, 2015, including the "Management's Discussion and Analysis of Financial Condition and Results of Operations" and the consolidated financial statements and related notes therein. See the section entitled "Where You Can Find More Information" beginning on page 162 of this prospectus.
|
|
|
For the Year Ended December 31, |
|
Three Months Ended |
| |||||||||||||||||
|
|
|
2010 |
|
2011 |
|
2012 |
|
2013 |
|
2014 |
|
2014 |
|
2015 |
| |||||||
|
Statement of Operation Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Licensed domestic product net sales |
|
$ |
— |
|
$ |
572,272 |
|
$ |
8,322,945 |
|
$ |
8,598,385 |
|
$ |
9,106,038 |
|
$ |
2,276,383 |
|
$ |
2,521,080 |
|
|
Other domestic product sales |
|
1,879,554 |
|
1,977,167 |
|
2,494,359 |
|
3,366,374 |
|
6,127,968 |
|
734,779 |
|
2,601,622 |
| |||||||
|
International product sales |
|
835,381 |
|
1,306,215 |
|
1,525,479 |
|
1,277,678 |
|
1,619,372 |
|
463,978 |
|
469,595 |
| |||||||
|
Royalty and licensing revenues |
|
2,022,383 |
|
14,227 |
|
— |
|
197,924 |
|
18,414 |
|
18,414 |
|
— |
| |||||||
|
Total Revenue |
|
4,737,318 |
|
3,869,881 |
|
12,342,783 |
|
13,440,361 |
|
16,871,792 |
|
3,493,554 |
|
5,592,297 |
| |||||||
|
Cost of Sales |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Licensor sales and distribution fees |
|
— |
|
484,480 |
|
5,916,845 |
|
5,844,494 |
|
5,902,034 |
|
1,413,043 |
|
1,452,064 |
| |||||||
|
Cost of products sold |
|
947,069 |
|
932,755 |
|
1,220,716 |
|
1,541,662 |
|
1,787,584 |
|
345,864 |
|
575,246 |
| |||||||
|
Write down of inventories |
|
195,488 |
|
26,117 |
|
36,345 |
|
56,935 |
|
53,099 |
|
— |
|
— |
| |||||||
|
Total Cost of Sales |
|
1,142,557 |
|
1,443,352 |
|
7,173,906 |
|
7,443,091 |
|
7,742,717 |
|
1,758,907 |
|
2,027,310 |
| |||||||
|
Gross Profit |
|
3,594,761 |
|
2,426,529 |
|
5,168,877 |
|
5,997,270 |
|
9,129,075 |
|
1,734,647 |
|
3,564,987 |
| |||||||
|
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Selling, general and administrative |
|
2,488,278 |
|
3,034,740 |
|
8,870,609 |
|
9,489,579 |
|
10,149,854 |
|
3,222,661 |
|
3,325,922 |
| |||||||
|
Amortization of assets |
|
49,720 |
|
77,951 |
|
718,981 |
|
1,245,846 |
|
1,511,021 |
|
290,352 |
|
621,623 |
| |||||||
|
Total Operating Expenses |
|
2,537,998 |
|
3,112,691 |
|
9,589,590 |
|
10,735,425 |
|
11,660,875 |
|
3,513,013 |
|
3,947,545 |
| |||||||
|
Loss from Operations |
|
1,056,763 |
|
(686,162 |
) |
(4,420,713 |
) |
(4,738,155 |
) |
(2,531,800 |
) |
(1,778,366 |
) |
(382,558 |
) | |||||||
|
Non Operating Income (expenses) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Gain on derivative instrument |
|
— |
|
— |
|
— |
|
— |
|
(167,511 |
) |
200,000 |
|
— |
| |||||||
|
Retirement payout |
|
(401,000 |
) |
— |
|
— |
|
— |
|
— |
|
— |
|
— |
| |||||||
|
Change in warrant liability |
|
(10,048 |
) |
214,280 |
|
247,486 |
|
(399,217 |
) |
283,305 |
|
(1,411,774 |
) |
(2,695,600 |
) | |||||||
|
Cost of extending warrant |
|
— |
|
— |
|
(135,157 |
) |
— |
|
— |
|
— |
|
— |
| |||||||
|
Change in fair value of contigent consideration |
|
— |
|
(57,996 |
) |
79,724 |
|
— |
|
— |
|
— |
|
— |
| |||||||
|
Research and Development |
|
(115,471 |
) |
(49,977 |
) |
(21,402 |
) |
— |
|
— |
|
— |
|
— |
| |||||||
|
Loss on disposal of equipment |
|
(15,308 |
) |
(259,636 |
) |
— |
|
— |
|
— |
|
— |
|
— |
| |||||||
|
Acquisition and restructuring |
|
— |
|
(671,112 |
) |
— |
|
— |
|
— |
|
— |
|
— |
| |||||||
|
Loss on disposal of intangible asset |
|
— |
|
— |
|
— |
|
(161,200 |
) |
— |
|
— |
|
— |
| |||||||
|
Loss on extinguishment of debt |
|
— |
|
— |
|
— |
|
(620,835 |
) |
— |
|
— |
|
— |
| |||||||
|
Unrealized foreign currency exchange on debt |
|
— |
|
— |
|
— |
|
(340,553 |
) |
(1,641,238 |
) |
— |
|
(1,433,456 |
) | |||||||
|
Accretion expense |
|
— |
|
(6,888 |
) |
(140,154 |
) |
(103,775 |
) |
(167,555 |
) |
(31,118 |
) |
(73,999 |
) | |||||||
|
Interest income |
|
10,772 |
|
18,910 |
|
13,940 |
|
3,559 |
|
59,586 |
|
372 |
|
125 |
| |||||||
|
Interest expense |
|
— |
|
— |
|
(253,143 |
) |
(527,079 |
) |
(1,441,729 |
) |
(267,292 |
) |
(595,975 |
) | |||||||
|
Net Loss for the period |
|
525,708 |
|
(1,498,581 |
) |
(4,629,419 |
) |
(6,887,255 |
) |
(5,606,942 |
) |
(3,288,178 |
) |
(5,181,463 |
) | |||||||
|
Deferred income tax recovery |
|
— |
|
976,800 |
|
1,209,300 |
|
314,900 |
|
— |
|
— |
|
— |
| |||||||
|
Current income tax recovery |
|
— |
|
— |
|
71,153 |
|
— |
|
— |
|
— |
|
— |
| |||||||
|
Unrealized gain(loss) on derivative instrument, net of tax |
|
— |
|
— |
|
— |
|
(38,156 |
) |
— |
|
(103,488 |
) |
43,400 |
| |||||||
|
Net loss and comprehensive loss for the period |
|
$ |
525,708 |
|
$ |
(521,781 |
) |
$ |
(3,348,966 |
) |
$ |
(6,610,511 |
) |
$ |
(5,606,942 |
) |
$ |
(3,391,666 |
) |
$ |
(5,138,063 |
) |
|
Basic net income (loss) per common share |
|
$ |
0.02 |
|
$ |
(0.02 |
) |
$ |
(0.09 |
) |
$ |
(0.13 |
) |
$ |
(0.08 |
) |
$ |
(0.06 |
) |
$ |
(0.05 |
) |
|
Shares used in computing basic net income (loss) per common share |
|
23,767,369 |
|
25,706,000 |
|
39,167,419 |
|
49,169,414 |
|
71,940,005 |
|
51,420,127 |
|
96,685,405 |
| |||||||
|
Diluted net income (loss) per common share |
|
$ |
0.02 |
|
$ |
(0.02 |
) |
$ |
(0.09 |
) |
$ |
(0.13 |
) |
$ |
(0.08 |
) |
$ |
(0.06 |
) |
$ |
(0.05 |
) |
|
Shares used in computing diluted net income per common share |
|
23,767,369 |
|
25,706,000 |
|
39,167,419 |
|
49,169,414 |
|
71,940,005 |
|
51,420,127 |
|
96,685,405 |
| |||||||
|
|
|
For the Year Ended December 31, |
|
Three Months Ended |
| |||||||||||||||||
|
|
|
2010 |
|
2011 |
|
2012 |
|
2013 |
|
2014 |
|
2014 |
|
2015 |
| |||||||
|
Balance Sheet Data |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Cash and Cash equivalents |
|
$ |
4,352,285 |
|
$ |
2,227,973 |
|
$ |
2,283,868 |
|
$ |
2,813,472 |
|
$ |
3,505,791 |
|
$ |
3,073,341 |
|
$ |
5,550,766 |
|
|
Total Assets |
|
7,280,594 |
|
19,208,435 |
|
20,828,532 |
|
20,034,541 |
|
53,079,740 |
|
20,765,355 |
|
55,823,501 |
| |||||||
|
Total Liabilities |
|
1,132,123 |
|
5,835,574 |
|
9,094,408 |
|
12,134,428 |
|
22,739,009 |
|
15,889,808 |
|
26,432,955 |
| |||||||
|
Accumulated deficit |
|
(3,852,809 |
) |
(4,374,590 |
) |
(7,723,556 |
) |
(14,295,911 |
) |
(19,902,853 |
) |
(17,584,089 |
) |
(25,084,316 |
) | |||||||
|
Total stockholders’ equity |
|
6,148,471 |
|
13,372,861 |
|
11,734,124 |
|
7,900,113 |
|
30,340,731 |
|
4,875,547 |
|
29,390,546 |
| |||||||
SELECTED UNAUDITED PRO FORMA FINANCIAL DATA
The following selected unaudited pro forma condensed combined financial data (the "selected pro forma data") gives effect to the business combination of Pozen and Tribute and the other transactions described in the section entitled "Unaudited Pro Forma Condensed Combined Financial Information" beginning on page F-90 of this prospectus, as well as the acquisition of Medical Futures Inc. (“MFI”) by Tribute (the “MFI Acquisition”), which closed on June 16, 2015. The transactions have been reflected in the unaudited pro forma condensed combined financial statements as being accounted for under the acquisition method in accordance with ASC 805, Business Combination, with Pozen treated as the accounting acquirer; and the MFI Acquisition has been reflected in the unaudited pro forma condensed combined financial statements in accordance with ASC 805 with Tribute treated as the accounting acquirer. The selected unaudited pro forma condensed combined balance sheet data as of March 31, 2015 give effect to the merger as if it had occurred on March 31, 2015. The selected unaudited pro forma condensed combined statement of operations data for the year ended December 31, 2014 and for the three months ended March 31, 2015 give effect to the merger as if it had occurred on January 1, 2014.
The selected pro forma data have been derived from, and should be read in conjunction with, the more detailed unaudited pro forma condensed combined financial information (the "pro forma financial statements") of the combined company appearing elsewhere in this prospectus and the accompanying notes to the pro forma financial statements. In addition, the pro forma financial statements were based on, and should be read in conjunction with, the historical financial statements and related notes of Pozen and the historical consolidated financial statements of Tribute and MFI for the applicable periods which appear elsewhere in this prospectus. See the sections entitled "Unaudited Pro Forma Condensed Combined Financial Information" and "Where You Can Find More Information" beginning on pages F-90 and 162, respectively, of this prospectus for additional information. The selected pro forma data have been presented for informational purposes only and are not necessarily indicative of what the combined company's financial position or results of operations actually would have been had the acquisition been completed as of the dates indicated. In addition, the selected pro forma data do not purport to project the future financial position or operating results of the combined company. Also, as explained in more detail in the accompanying notes to the pro forma financial statements, the preliminary fair values of assets acquired and liabilities assumed reflected in the selected pro forma data are subject to adjustment and may vary significantly from the fair values that will be recorded upon completion of the merger.
|
|
|
Year ended |
|
Three months ended |
| ||
|
Revenues |
|
|
|
|
| ||
|
Royalty and licensing revenue |
|
$ |
32,410,910 |
|
$ |
4,404,463 |
|
|
Licensed domestic product net sales |
|
8,247,339 |
|
2,036,276 |
| ||
|
Other domestic product sales |
|
14,498,312 |
|
3,866,861 |
| ||
|
International product sales |
|
1,466,665 |
|
379,292 |
| ||
|
Total Revenues |
|
56,623,226 |
|
10,686,892 |
| ||
|
Cost of Sales |
|
|
|
|
| ||
|
Licensor sales and distribution fees |
|
5,345,472 |
|
1,172,832 |
| ||
|
Cost of products sold |
|
5,999,640 |
|
1,263,072 |
| ||
|
Write down of inventories |
|
48,092 |
|
— |
| ||
|
Total cost of sales |
|
11,393,204 |
|
2,435,904 |
| ||
|
Gross Profit |
|
45,230,022 |
|
8,250,988 |
| ||
|
Operating expenses |
|
|
|
|
| ||
|
Sales, general, and administrative |
|
23,754,893 |
|
7,054,227 |
| ||
|
Research and development |
|
5,739,848 |
|
983,511 |
| ||
|
Amortization |
|
9,434,864 |
|
2,356,357 |
| ||
|
Total operating expenses |
|
38,929,605 |
|
10,394,094 |
| ||
|
Non-operating income (expense) |
|
|
|
|
| ||
|
Change in warrant liability |
|
256,589 |
|
(2,177,236 |
) | ||
|
Interest expense |
|
(362,280 |
) |
— |
| ||
|
Interest income |
|
97,067 |
|
101 |
| ||
|
Other non-operating income |
|
1,346,154 |
|
(1,338,431 |
) | ||
|
Total other income (expense) |
|
1,337,531 |
|
(3,515,566 |
) | ||
|
Income before taxes |
|
7,637,947 |
|
(5,658,672 |
) | ||
|
Income tax expense (benefit) |
|
(862,582 |
) |
(205,757 |
) | ||
|
Net Income (loss) attributable to common stockholders |
|
$ |
8,500,530 |
|
$ |
(5,452,915 |
) |
|
Basic net income (loss) per share |
|
$ |
0.17 |
|
$ |
(0.10 |
) |
|
Shares used in computing basic net income (loss) per share |
|
51,139,659 |
|
52,039,362 |
| ||
|
Diluted net income (loss) per share |
|
$ |
0.17 |
|
$ |
(0.10 |
) |
|
Shares used in computing diluted net income (loss) per share |
|
52,571,262 |
|
52,039,362 |
| ||
Selected Unaudited Pro Forma Condensed Combined Balance Sheet
|
|
|
As of March 31, 2015 |
| |
|
Total assets |
|
$ |
301,319,270 |
|
|
Total liabilities |
|
$ |
63,393,637 |
|
|
Total shareholders’ equity |
|
$ |
237,925,632 |
|
COMPARATIVE PER SHARE DATA
The following tables set forth certain historical, pro forma and pro forma equivalent per share financial information for Tribute common shares and shares of Pozen common stock. The unaudited pro forma and pro forma equivalent per share financial information gives effect to the combination of Pozen and Tribute (along with the MFI Acquisition) as if the transactions had occurred on March 31, 2015.
Presented below are Tribute’s and Pozen’s historical per share data for the three months ended March 31, 2015 and the year ended December 31, 2014 and unaudited condensed combined pro forma per share data for the three months ended March 31, 2015 and the year ended December 31, 2014. The historical book value per share is computed by dividing total stockholders’ equity (deficit) by the number of shares of common stock outstanding at the end of the period. The pro forma earnings per Tribute common share is computed by dividing the pro forma net income by the pro forma weighted average number of shares outstanding. The pro forma book value per share of the combined company is computed by dividing total pro forma stockholders’ equity by the pro forma number of shares of common stock outstanding at the end of the period. The Pozen unaudited pro forma equivalent data is calculated by multiplying the combined unaudited pro forma data amounts by the merger consideration ratio of per share of Pozen common stock. The pro forma information described below includes certain adjustments and assumptions regarding the combined company after giving effect to the transactions.
The following information should be read in conjunction with (i) the audited financial statements of Tribute, which are included elsewhere in this prospectus, (ii) the audited financial statements of Pozen which are included elsewhere in this prospectus and (iii) the financial information contained in the sections entitled “Unaudited Pro Forma Condensed Combined Financial Information,” “Selected Historical Financial Data of Pozen,” and “Selected Historical Financial Data of Tribute” beginning on pages [·], [·] and [·], respectively, of this prospectus. The unaudited pro forma information below is presented for informational purposes only and is not necessarily indicative of the operating results or financial position that would have occurred if the transactions had been completed as of the periods presented, nor is it necessarily indicative of the future operating results or financial position of the combined company. In addition, the unaudited pro forma information does not purport to indicate balance sheet data or results of operations data as of any future date or for any future period. Tribute
has not declared or paid any cash dividends on Tribute common shares, and Pozen has not declared or paid any regular dividends on shares of Pozen common stock.
|
|
|
As of and for the |
|
As of and for the |
| ||
|
Combined Unaudited Pro Forma Data |
|
|
|
|
| ||
|
Net loss per Pozen share |
|
|
|
|
| ||
|
Diluted |
|
$ |
(0.10 |
) |
$ |
0.17 |
|
|
Basic |
|
$ |
(0.10 |
) |
$ |
0.17 |
|
|
Book value per Pozen share |
|
$ |
4.57 |
|
— |
| |
|
|
|
As of and for the |
|
As of and for the |
| ||
|
Tribute Unaudited Pro Forma Data |
|
|
|
|
| ||
|
Net loss per Tribute share |
|
|
|
|
| ||
|
Diluted |
|
$ |
(0.02 |
) |
$ |
0.02 |
|
|
Basic |
|
$ |
(0.02 |
) |
$ |
0.02 |
|
|
Book value per Tribute share |
|
$ |
0.67 |
|
— |
| |
COMPARATIVE PER SHARE MARKET PRICE DATA AND DIVIDEND INFORMATION
It is expected that Parent Shares will be listed and traded on NASDAQ under the symbol “ARLZ” and on the TSX under the symbol “ARZ”. Pozen common stock is currently listed and traded on NASDAQ under the symbol “POZN”. Tribute common shares are currently listed on TSXV under the symbol “TRX” and on the OTCQX under the symbol “TBUFF”. The following table sets forth, for the calendar quarters indicated, the high and low sales prices of Pozen common stock and Tribute common shares, each as reported on NASDAQ, the OTCQX and the TSXV, respectively. On June 30, 2015, there were 113,719,683 Tribute common shares outstanding and 32,708,898 shares of Pozen common stock outstanding. On , 2015, the record date for the Pozen special meeting, there were shares of Pozen common stock outstanding. Tribute has not declared or paid any cash dividends on Tribute common shares, and, except for the December 30, 2013 $1.75 per share special cash distribution (representing a surplus of corporate cash and accounted for as a return of capital to stockholders) Pozen has not declared or paid any regular dividends on shares of Pozen common stock.
|
|
|
Pozen(1) |
|
Tribute(1)(2) |
|
Tribute(3)(4)(5) |
| ||||||||||||
|
|
|
High |
|
Low |
|
High |
|
Low |
|
High |
|
Low |
| ||||||
|
For the quarterly period ended: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
2012 |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
First Quarter |
|
$ |
6.15 |
|
$ |
3.96 |
|
$ |
0.70 |
|
$ |
0.35 |
|
$ |
— |
|
$ |
— |
|
|
Second Quarter |
|
$ |
8.12 |
|
$ |
5.53 |
|
$ |
0.66 |
|
$ |
0.40 |
|
$ |
— |
|
$ |
— |
|
|
Third Quarter |
|
$ |
6.95 |
|
$ |
5.71 |
|
$ |
0.52 |
|
$ |
0.25 |
|
$ |
— |
|
$ |
— |
|
|
Fourth Quarter |
|
$ |
6.80 |
|
$ |
4.81 |
|
$ |
0.48 |
|
$ |
0.31 |
|
$ |
— |
|
$ |
— |
|
|
2013 |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
First Quarter |
|
$ |
6.49 |
|
$ |
5.02 |
|
$ |
0.43 |
|
$ |
0.32 |
|
$ |
— |
|
$ |
— |
|
|
Second Quarter |
|
$ |
5.56 |
|
$ |
4.26 |
|
$ |
0.45 |
|
$ |
0.34 |
|
$ |
— |
|
$ |
— |
|
|
Third Quarter |
|
$ |
5.99 |
|
$ |
4.92 |
|
$ |
0.75 |
|
$ |
0.35 |
|
$ |
— |
|
$ |
— |
|
|
Fourth Quarter |
|
$ |
9.90 |
|
$ |
5.35 |
|
$ |
0.50 |
|
$ |
0.34 |
|
$ |
— |
|
$ |
— |
|
|
2014 |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
First Quarter |
|
$ |
8.99 |
|
$ |
7.37 |
|
$ |
0.63 |
|
$ |
0.32 |
|
$ |
— |
|
$ |
— |
|
|
Second Quarter |
|
$ |
9.73 |
|
$ |
7.56 |
|
$ |
1.10 |
|
$ |
0.44 |
|
$ |
0.98 |
|
$ |
0.60 |
|
|
Third Quarter |
|
$ |
9.59 |
|
$ |
5.96 |
|
$ |
0.78 |
|
$ |
0.43 |
|
$ |
0.85 |
|
$ |
0.47 |
|
|
Fourth Quarter |
|
$ |
9.71 |
|
$ |
7.07 |
|
$ |
0.59 |
|
$ |
0.41 |
|
$ |
0.67 |
|
$ |
0.45 |
|
|
2015 |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
First Quarter |
|
$ |
8.16 |
|
$ |
6.56 |
|
$ |
0.81 |
|
$ |
0.43 |
|
$ |
1.02 |
|
$ |
0.51 |
|
|
Second Quarter |
|
$ |
12.69 |
|
$ |
6.38 |
|
$ |
1.86 |
|
$ |
0.65 |
|
$ |
2.33 |
|
$ |
0.82 |
|
(1) Currency denoted in U.S. dollars.
(2) Per share price reported on the OTCQX, trading under the symbol “TBUFF”.
(3) Tribute began trading on the TSXV effective May 27, 2014.
(4) Per share price reported on the TSXV, trading under the symbol “TRX”.
(5) Currency denoted in Canadian dollars.
The following table represents the closing prices of Pozen common stock and Tribute common shares on (i) June 5, 2015, the last trading day before the public announcement of the merger agreement, (ii) June 8, 2015, the trading day on which Pozen and Tribute announced the merger, and (iii) July 15, 2015, the last practicable trading day prior to the mailing of the Form S-4.
|
Date |
|
Pozen |
|
Tribute |
|
Tribute |
| |||
|
June 5, 2015 |
|
$ |
7.55 |
|
$ |
1.05 |
|
$ |
1.30 |
|
|
June 8, 2015 |
|
$ |
8.98 |
|
$ |
1.17 |
|
$ |
1.50 |
|
|
July 15, 2015 |
|
$ |
10.96 |
|
$ |
1.51 |
|
$ |
1.98 |
|
(1) Price in U.S. dollars.
(2) Price in Canadian dollars.
MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of Pozen’s financial condition and results of Pozen’s operations in conjunction with Pozen’s consolidated financial statements and the notes to those statements included elsewhere in this prospectus. This discussion contains forward-looking statements reflecting Pozen’s current expectations that involve risks and uncertainties. Pozen’s actual results and the timing of events may differ materially from those contained in these forward-looking statements due to a number of factors, including those discussed in the section entitled “Risk Factors” and elsewhere in this prospectus.
Statements in the following discussion may include forward-looking statements. These forward-looking statements involve risks and uncertainties. See “Information Regarding Forward-Looking Statements” for additional discussion of these factors and risks.
Except as otherwise noted, all references in this Management’s Discussion and Analysis to “we”, “us”, “our”, or the “Company” refer to Pozen.
Overview
We are a pharmaceutical company focused on transforming medicines that can transform lives. Historically, we have operated a business model that has focused on the following:
· developing innovative products that address unmet medical needs in the marketplace;
· obtaining patents for those innovative ideas which we believe have value in the marketplace;
· utilizing a small group of talented employees to develop those ideas by working with strategic outsource partners;
· developing a regulatory pathway with the appropriate agency; and
· determining how best to commercialize our products.
The success of our business is highly dependent on the marketplace value of our ideas and the related patents we obtain, our ability to obtain approval to sell the developed products from the required regulatory agencies, and our ability to successfully commercialize our products. Under our earlier business model, we hired experts with strong project management skills in the specific disciplines we believed were important to maintain within our company. We contracted with and manage strong outsource partners as we complete the necessary development work, permitting us to avoid incurring the cost of buying or building laboratories, manufacturing facilities or clinical research operation sites. This allowed us to control our annual expenses, but to utilize “best in class” resources as required. We decided to retain ownership of our PA product candidates for cardiovascular indications which contain a combination of a proton pump inhibitor and EC aspirin in a single tablet, through the clinical development and pre-commercialization stage and our former chief commercial officer was responsible for developing the commercialization strategy for these products and conducting all the required pre-commercialization activities.
On September 3, 2013 we entered into an exclusive license agreement with Sanofi U.S., for the commercialization of PA8140 and PA32540, now known as YOSPRALA. Under the terms of the agreement, Sanofi U.S. had exclusive rights to commercialize all PA combinations that contain 325 mg or less of EC aspirin in the United States.
On April 25, 2014, we received a CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude the approval of the NDA in its then-current form. Specifically, an inspection of the manufacturing facility of an active ingredient supplier of ours concluded with certain inspection deficiencies. Satisfactory resolution of these deficiencies is required before the NDA may be approved. There were no clinical or safety deficiencies noted with respect to either PA8140 or PA32540 and no other deficiencies were noted in CRL. On June 30, 2014, we resubmitted the NDA for PA32540 and PA8140 to the FDA and the FDA notified us that the new action fee date is December 30, 2014. On May 9, 2014, our active ingredient supplier submitted a response to the FDA addressing the inspection deficiencies and subsequently submitted an update to its initial response. On November 29, 2014, we executed a termination agreement with Sanofi U.S. terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi U.S. were terminated and all rights to the products licensed to Sanofi U.S. under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi U.S. relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products.
On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. Satisfactory resolution of these deficiencies is required before this application may be approved. There were no clinical or safety deficiencies noted with respect to either PA32540 or PA8140 and no other deficiencies were noted in the CRL. Final agreement on the draft product labeling is also pending.
FDA regulations allowed us to request a Type A meeting with the FDA to discuss the next steps required to gain approval of our NDA. The FDA granted the Type A meeting, which was held in late January 2015. At the meeting, representatives from the FDA’s Office of Compliance stated that the active ingredient supplier’s responses to the 483 inspectional observations submitted in May 2014 were still under review and the Office of Compliance would be communicating with the supplier in the coming weeks. The active ingredient supplier has informed Pozen that they received a warning letter relating to the Form 483 inspection deficiencies. They have submitted a plan of corrective actions to address the matters raised in the warning letter to FDA. We have had continuing discussions with FDA about these issues and the impact on our NDA and with our active ingredient supplier regarding the corrective actions it is taking to address the inspectional observations at its facility. We are also working toward securing and seeking FDA’s approval for an alternative back up supplier.
In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
Our commercialization strategy for PA outside the United States was to secure relationships with one or more strong commercial partners with relevant expertise to commercialize our future products globally. With respect to future products we may develop, we had decided that we will no longer commit substantial resources to further drug development without a partner who agrees to pay the full cost of that development. Consistent with this model, we reduced our research and development staff and other costs and expenses as our PA development program activities wind down.
Our business and operations model is evolving. On June 1, 2015, our Board appointed a new Chief Executive Officer and a new President and Chief Business Officer, each of whom has experience creating, leading and expanding pharmaceutical companies with marketing and sales capabilities. If the proposed business combination with Tribute is approved by our stockholders and Tribute’s shareholders and such transaction and concurrent equity and debt financings are closed, we will become a specialty pharmaceutical company focused on cardiovascular and pain therapies with a diversified portfolio of marketed products and developmental stage product candidates in the United States and Canada.
Proposed Business Combination with Tribute.
On June 8, 2015, we entered into the merger agreement among Tribute, Parent, Ltd2, US Merger Sub, and Can Merger Sub.
The merger agreement provides for, among other things, a business combination whereby we will merge with US Merger Sub. As a result of the merger, the separate corporate existence of US Merger Sub will cease and we will continue as the surviving corporation. On the date of the closing of the merger, we will become an indirect wholly owned subsidiary of Parent. In accordance with the merger agreement, Can Merger Sub will offer to and acquire, and will acquire, all of the outstanding shares of Tribute in the manner provided for by the merger agreement. Following completion of the Arrangement, Tribute will also become an indirect wholly-owned subsidiary of Parent. The boards of directors of each party, including Tribute, Parent, US Merger Sub, Can Merger Sub and us have unanimously approved the merger agreement, determined that the merger and arrangement, upon the terms and subject to the conditions set forth in the merger agreement, is in the best interests of its respective companies and stockholders, and have declared the advisability of the merger and arrangement and the execution of the merger agreement.
Upon consummation of the merger and arrangement, each outstanding share of our common stock will be converted into the right to receive from Parent one Parent Share, $0.001 nominal value per share, and each outstanding share of Tribute common stock will be converted into the right to receive from Parent or Can Merger Sub 0.1455 Parent Shares. Upon completion of the merger and arrangement, our stockholders will own approximately 66% of the outstanding Parent Shares, and current Tribute shareholders will own approximately 34% of the outstanding Parent Shares before giving effect to (i) any exercise of outstanding options and warrants or the vesting and delivery of shares underlying restricted stock units of either company and (ii) the ordinary shares of Parent to be issued to new investors pursuant to the Subscription Agreement and the ordinary shares of Parent issuable upon conversion of the Convertible Notes to be issued pursuant to the Facility Agreement. After giving effect to the transactions and the proposed Parent equity financing and debt financing, the former stockholders of Pozen as a group will hold Parent Shares constituting approximately 49% of the outstanding Parent Shares and the former shareholders of Tribute as a group will hold Parent Shares constituting approximately 28% of the outstanding Parent Shares.
The completion of the merger and arrangement is subject to the approval of our stockholders and the shareholders of Tribute. In addition, the merger and the arrangement are subject to other customary closing conditions, including, among others, (i) the expiration or termination of the applicable waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended, and the obtaining of approval under Canada’s Competition Act (to the extent required under applicable law), (ii) the declaration by the SEC of the effectiveness of the Form S-4 registering the Parent Shares to be issued in connection with the merger, (iii) the approval of the listing on
NASDAQ and the TSX of the Parent Shares to be issued in connection with the merger and arrangement, and (iv) the conditions to closing the equity and debt financings described below having been met or waived.
On June 8, 2015, we also executed the Facility Agreement among the Borrower and the Lenders. Pursuant to the Facility Agreement, the Borrower will borrow from the Lenders up to an aggregate principal amount of $275 million, of which (i) $75 million of Convertible Notes, issued and sold by Borrower to Deerfield Private Design or its registered assigns, upon the terms and conditions of the Facility Agreement, and (ii) up to an aggregate principal amount of $200 million, which will be made available for “Permitted Acquisitions (a) deferred in the Facility Agreement),” of Acquisition Notes, evidencing the Acquisition Loans, upon the terms and conditions and subject to the limitations set forth in the Acquisition Notes, all subject to the terms and conditions of the Facility Agreement.
On June 8, 2015, we also executed the Subscription Agreement by and among Purchaser, Tribute, Parent, us and the Investors. Pursuant to the Subscription Agreement, Parent will sell to Purchaser and the Investors up to $75 million of the Parent Shares in a private placement at a purchase price of $7.20 per Parent Share. The Subscription Agreement provides that the Company will prepare and file two registration statements with the SEC on such form as may be required to effect a registration of the Parent Shares issued under the Subscription Agreement within 60 days of the date of the signing of the Subscription Agreement and for certain other registration rights for each of Purchaser and the Investors under the Securities Act and the rules and regulations thereunder, or any similar successor statute, and applicable state securities laws.
A description of the merger agreement, the Facility Agreement and the Subscription Agreement, as well as other agreements related to the merger and financing transactions is set forth in a Form 8-K we filed with the SEC on June 8, 2015 and copies of these agreements are attached as exhibits to such Form 8-K. The foregoing description of these agreements does not purport to be complete and is qualified in its entirety by reference to the full text of the agreements.
Treximet
We have previously developed Treximet® in collaboration with GlaxoSmithKline, or GSK. Treximet is the brand name for the product combining sumatriptan 85 mg, formulated with RT Technology™ and naproxen sodium 500 mg in a single tablet designed for the acute treatment of migraine. On April 15, 2008, the FDA approved Treximet for the acute treatment of migraine attacks with or without aura in adults. Upon receipt of FDA approval, GSK notified us of its intention to launch the product and Treximet was available in pharmacies in May 2008.
On November 23, 2011, we entered into a purchase and sale agreement, or the Purchase Agreement, with CPPIB Credit Investments Inc., or CII, pursuant to which we sold, and CII purchased, our right to receive future royalty payments arising from U.S. sales of MT 400, including Treximet. Under the Purchase Agreement, we received $75 million and will receive a 20% interest in any royalties received by CII relating to the period commencing on April 1, 2018.
On May 13, 2014, we, or GSK, CII and Pernix entered into certain agreements in connection with GSK’s divestiture of all of its rights, title and interest to develop, commercialize and sell Treximet in the U.S. to Pernix. Upon the closing of the divestiture on August 20, 2014, GSK assigned the Product Development and Commercialization Agreement executed as of June 11, 2003 between us and GSK, the Treximet Agreement, to Pernix. Immediately following the closing of the divestiture, Amendment No. 1 to the Treximet Agreement, or Amendment No.1, between us and Pernix became effective. Amendment No. 1, among other things, amends the royalty provisions of the Agreement to provide for a guaranteed quarterly minimum royalty payable to CII of $4 million for the calendar quarters commencing on January 1, 2015 and ending on March 31, 2018 and requires that Pernix continue certain of GSK’s ongoing development activities and to undertake certain new activities, for which we will provide reasonable assistance. Amendment No. 1 also eliminates restrictions in the Agreement on our right to develop and commercialize certain dosage forms of sumatriptan/naproxen combinations outside of the United States and permits Pozen to seek approval for these combinations on the basis of the approved NDA for Treximet. Pernix also issued us a warrant to purchase 500,000 shares of Pernix common stock at an exercise price equal to $4.28, the closing price of Pernix common stock as listed on the NASDAQ Global Market on May 13, 2014. In the first quarter of 2015, the Company sold the warrant for $2,479,000. Lastly, we, GSK, Pernix and CII executed a letter agreement whereby we expressly consented to the assignment by GSK and the assumption by Pernix of the Treximet Agreement. On July 30, the parties entered into Amendment No. 2 to the Treximet Agreement which will permit Pernix’s Irish affiliate to which Pernix will assigns its rights to further assign the Agreement without our prior written consent as collateral security for the benefit of the lenders which financed the acquisition. Amendment No. 2 became effective upon the closing of the divestiture, which occurred on August 20, 2014.
VIMOVO®
We have developed VIMOVO with AstraZeneca. VIMOVO (formerly referred to as PN 400) is the brand name for a proprietary fixed dose combination of the PPI esomeprazole magnesium with the NSAID naproxen in a single tablet. On April 30, 2010, the FDA approved VIMOVO for the relief of the signs and symptoms of osteoarthritis, or OA, rheumatoid arthritis, or RA, and ankylosing spondylitis, or AS, and to decrease the risk of developing gastric ulcers in patients at risk of developing NSAID-associated gastric ulcers.
In August 2006, we entered into an exclusive global collaboration and license agreement with AstraZeneca to co-develop and commercialize VIMOVO, which agreement was amended in September 2007 and October 2008. We began the Phase 3 program in September 2007. As part of the program, we conducted two Phase 3 pivotal trials of VIMOVO in patients who are at risk for developing NSAID-associated gastric ulcers, the primary endpoint for which was the reduction in endoscopic gastric ulcers.
The NDA for VIMOVO was submitted on June 30, 2009 and was accepted for filing by FDA in August 2009. Pozen received a $10.0 million milestone payment from AstraZeneca in September 2009 for the achievement of such milestone. Earlier, in May 2010, we had received a $20.0 million milestone payment when we received FDA approval of VIMOVO. In October 2009, AstraZeneca submitted a Marketing Authorization Application, or MAA, for VIMOVO in the European Union, or EU, via the Decentralized Procedure, or DCP, and has filed for approval in a number of other countries which are not covered by the DCP. On October 11, 2010, we announced with AstraZeneca that VIMOVO had received positive agreement for approval in 23 countries across the EU following all 22 “Concerned Member States” agreeing with the assessment of the Netherlands Health Authority (MEB), acting as the “Reference Member State” for the DCP. We received a $25.0 million milestone payment when pricing and reimbursement for VIMOVO was granted in the United Kingdom.
On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understand that AstraZeneca will instead now focus on those countries where the product has shown growth and which AstraZeneca believes have the greatest potential for future growth.
On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. As required by the terms of our agreement with AstraZeneca, we gave our consent to AstraZeneca’s divestiture of such rights to Horizon because we believed that Horizon’s expertise in commercializing products for pain and inflammatory disease, including a similar combination product containing a NSAID and a gastroprotective agent, make it an excellent partner to maximize the potential of VIMOVO in the United States. We were also able to negotiate a guaranteed annual minimum royalty in the United States in the amount of $5 million in calendar year 2014, and a guaranteed annual minimum royalty amount of $7.5 million each calendar year thereafter, provided that the patents owned by us which cover VIMOVO are in effect and no generic forms of VIMOVO are in the marketplace.
On July 28, 2014, Horizon announced that it had been verbally notified by CVS Caremark and Express Scripts, Inc. that VIMOVO would no longer be on their formularies and will be placed on their exclusion lists effective January 1, 2015. We saw a decline in VIMOVO prescriptions in the first quarter of 2015 of approximately 20% versus the fourth quarter of 2014 and our quarterly royalty declined to $3.2 million, well ahead of our minimum quarterly royalty of $1.875 million.
We, AstraZeneca and Horizon are also engaged in Paragraph IV litigation with several generic pharmaceutical companies with respect to patents listed in the Orange Book with respect to VIMOVO currently pending in the United States District Court for the District of New Jersey and in an IPR brought by Dr. Reddy’s with respect to the ‘285 patent and in two IPRs brought by CFAD. We and AstraZeneca are also engaged in a proceeding in Canada with Mylan ULC which is seeking approval of its generic version of VIMOVO in Canada prior to the expiration of our Canadian patent.
Our Principal Product Candidates
Our PA product candidates, containing a PPI and aspirin, have completed clinical development testing in the United States. Our PA product candidates, now known as YOSPRALA, are excluded from our agreement with AstraZeneca. We met with the FDA to discuss the overall development program requirements for PA32540 for the secondary prevention of cardiovascular and cerebrovascular disease in patients at risk for gastric ulcers. An investigational new drug application, or IND, was filed in the fourth quarter of 2007. We completed a study which demonstrated that the (SA) component of PA32540 was bioequivalent to the reference drug, EC aspirin. We filed a Special Protocol Assessment, or SPA, with the FDA for the design of the Phase 3 studies for the product, the primary endpoint for which is the reduction in the cumulative incidence of endoscopic gastric ulcers.
Based upon the FDA’s earlier confirmation that endoscopic gastric ulcers were an acceptable primary endpoint, in October 2009, we began two pivotal Phase 3 and one long-term safety study for PA32540 for the cardiovascular indication. The primary endpoint of the pivotal studies, which included approximately 500 subjects per study, was a significant reduction in the cumulative incidence of gastric ulcers following administration of PA32540 vs. 325 mg EC aspirin over the six-month treatment period. The primary endpoint was met in both studies. Additionally, the studies met their key secondary endpoints, including a reduction in gastroduodenal ulceration as well as a reduction in discontinuation due to upper gastrointestinal adverse events in subjects taking PA32540 compared to 325 mg enteric-coated aspirin.
In February 2012, the FDA requested an additional Phase 1 study to assess the bioequivalence of PA32540 to EC aspirin 325 mg with respect to acetylsalicylic acid, or ASA. After the Company completed the requested bioequivalence study, the FDA made a preliminary review of the study results and the Company’s summary analyses and, based on its preliminary assessment of the information available to it at the time, the FDA did not agree that bioequivalence of PA32540 to EC aspirin 325 mg was demonstrated. The Company then submitted to the FDA additional information and analyses from the requested bioequivalence study, as well as other relevant pharmacokinetic, clinical pharmacology, and in vitro dissolution data as a Briefing Document in support of a request for a Type A meeting with the FDA. At the Type A meeting held in August 2012, the FDA confirmed that, although it believes bioequivalence of PA32540 to EC aspirin 325 mg, was not strictly established in our bioequivalence study according to the predetermined criteria, the results from this study, together with additional information that was submitted by the Company in the NDA, constitutes sufficient data to support the establishment of a clinical and pharmacological bridge between the product and EC aspirin 325 mg. The FDA indicated that it would make a final determination during the NDA review. The FDA also indicated that a similar strategy to bridge to the reference listed drug, inclusive of a new, single pharmacokinetic study, could be utilized for a low dose version of PA32540 (currently PA8140). The Company conducted this study with the low dose version against the EC aspirin 81mg. Based on the predetermined criteria acceptable to the FDA, the study demonstrated that PA8140 is bioequivalent to EC aspirin 81mg using criteria for highly variable drugs and had comparable bioavailability.
During a pre-submission meeting with respect to its NDA for PA32540 in April 2012, the FDA suggested that the Company also seek approval for a lower dose formulation of the product containing 81 mg of enteric coated aspirin as part of its NDA for PA32540. Absent the availability of such a lower dose formulation in the market if PA32540 is approved, the FDA indicated that it may limit the indication for PA32540 to use in CABG with treatment duration not to exceed one year. During the Type A meeting held in August 2012, the FDA confirmed its preference to have both PA32540 and a lower dose version available in the market so that physicians can have both a low and high dose option available, and agreed that, if both dosage strengths were included in the NDA and subsequently approved, the indications for both will be consistent with the full range of indications described in the current aspirin monograph.
We had generated some clinical pharmacology data and chemical, manufacturing and controls (CMC) data for a product which contains 81 mg of enteric coated aspirin and 40 mg of omeprazole in a single tablet known as PA8140. The Company filed this existing data, together with additional CMC data to be generated and evidence from the scientific literature relating to the ulcerogenic risk of 81 mg of aspirin with the FDA. At this time, we do not intend to conduct Phase 3 clinical trials for PA8140. The data package submitted for PA8140 was similar to that used to gain approval for a lower dosage form of VIMOVO containing 375 mg of naproxen. We have no assurance such data will be sufficient for the FDA to approve PA8140 or to allow a broader indication for PA32540. The FDA will make a final determination with respect to the approvability of and indications for PA32540 and PA8140.
Generation of additional data with respect to PA8140 and incorporation of data into the NDA for PA32540 delayed submission of the NDA from the original planned submission date in the third quarter of 2012. The NDA was filed for both products in March 2013 and in May 2013 the FDA accepted the NDA for review. The FDA assigned a user fee date of January 24, 2014. As part of our continuing discussions with the FDA concerning the NDA for PA32540 and PA8140 tablets, we decided to conduct an additional comparative Phase 1 pharmacokinetic study to determine the pharmacokinetic profile of the omeprazole component of PA 8140 tablets and compare it to that of PA32540 tablets. We submitted study information and data to the FDA as it became available during the conduct of the study and FDA reviewed such information and data from the study when submitted. The final study report for the study was submitted to the FDA in accordance with our agreed timeline. FDA informed us that the Company’s user fee date was April 25, 2014. On April 25, 2014, we received a CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude the approval of the NDA in its current form. Specifically, an inspection of the manufacturing facility of an active ingredient supplier of ours concluded with certain inspection deficiencies. Satisfactory resolution of these deficiencies is required before the NDA may be approved. There were no clinical or safety deficiencies noted with respect to either PA8140 or PA32540 and no other deficiencies were noted in CRL.
On June 30, 2014, we resubmitted the NDA for PA32540 and PA8140 to the FDA and the FDA notified us that the new action fee date is December 30, 2014. On May 9, 2014, our active ingredient supplier submitted a response to the FDA addressing the inspection deficiencies and subsequently submitted an update to its initial response. On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. Satisfactory resolution of these deficiencies is
required before this application may be approved. There were no clinical or safety deficiencies noted with respect to either PA32540 or PA8140 and no other deficiencies were noted in the CRL. Final agreement on the draft product labeling is also pending. We continue to assist the FDA compliance division with their review. FDA regulations allowed us to request a Type A meeting with the FDA to discuss the next steps required to gain approval of our NDA. The FDA granted the Type A meeting which was held in late January 2015. At the meeting, representatives from the FDA’s Office of Compliance stated that the active ingredient supplier’s responses to the 483 inspectional observations submitted in May 2014 were still under review and the Office of Compliance would be communicating with the supplier in the coming weeks. The active ingredient supplier has informed Pozen that they received a warning letter relating to the Form 483 inspection deficiencies. They have submitted a plan of corrective actions to address the matters raised in the warning letter to FDA. We have had continuing discussions with FDA about these issues and the impact on our NDA and with our active ingredient supplier regarding the corrective actions it is taking to address the inspectional observations at its facility.
To assess whether a similar interaction occurs between clopidogrel and PA32540, which contains immediate release omeprazole, we completed two Phase 1 drug-drug interaction studies to evaluate the ex-vivo platelet aggregation effects of PA32540 plus clopidogrel. In the first study, we evaluated ex-vivo platelet aggregation of PA32540 plus clopidogrel when dosed at the same time or dosed 10 hours apart compared to aspirin 325 mg plus clopidogrel dosed together. When PA32540 and clopidogrel were dosed together, data from the study showed a mean 36.7% platelet inhibition compared to a mean 44.0% platelet inhibition when aspirin and clopidogrel were dosed together suggesting a drug-drug interaction based on the study’s pre-specified primary analysis. When PA32540 and clopidogrel were dosed 10 hours apart, data from the study indicate no ex-vivo drug-drug interaction based on the study’s pre-specified primary analysis. In the second Phase 1 study, we evaluated ex-vivo platelet aggregation of PA32540 plus clopidogrel dosed 10 hour apart compared to a current standard of care of Plavix + Prilosec 40 mg + EC ASA 81 mg dosed together. Subjects on PA32450 demonstrated significantly greater inhibition of platelet aggregation than subjects on standard of care. The clinical relevance of these ex vivo platelet data on cardiovascular events is not known. No further Phase 1 studies on the clopidogrel-PPI interaction are planned. FDA assessment of available data on the drug-drug interaction may result in the inclusion of a warning, similar to that in the current Prilosec label, against the concomitant use of PA32540 and Plavix.
We are also continuing to evaluate how best to commercialize the PA product candidates and programs. We are evaluating the regulatory requirements to obtain an indication for PA for the secondary prevention of colorectal neoplasia. In January 2010, we received input from the FDA with respect to the development requirements for a possible indication in colorectal neoplasia. Further discussions are being considered. We are also evaluating the possibility of developing another dosage strength of PA containing 650 mg of EC aspirin and 20 mg of omeprazole (PA65020) for the treatment of osteoarthritis and similar conditions and we met with the FDA in December 2010 to obtain input with respect to the regulatory requirements to obtain such an indication. The FDA advised us that we will need to demonstrate a reduction in gastric ulcers compared to aspirin alone in a replicate Phase 3 program similar to that we performed for VIMOVO, in addition to a study to establish efficacy of the product in the treatment of OA.
We met with a European country that served as the Reference Member State for approval of VIMOVO in Europe to obtain guidance on a clinical development program for approval of PA65020 in the Europe. We were advised that Phase 3 endoscopic trials demonstrating a reduction in gastric ulcers would not be required in addition to an osteoarthritis efficacy study since omeprazole’s actions on gastric ulcers has been well characterized. Instead, Phase 1 studies assessing pharmacokinetics and gastric acid suppression could be used to support the benefit of omeprazole as a component of PA65020.
We have also received Scientific Advice from the MEB in the Netherlands, which has agreed to be the Reference Member State in a decentralized filing procedure, regarding the development program required for the approval in the EU of PA tablets, including a lower dosage form containing 100 mg of aspirin and 40 mg of omeprazole (PA10040) for the secondary prevention of cardiovascular disease. The MEB agreed that a full Phase 3 clinical development program for PA10040, to demonstrate the reduction of endoscopic gastric ulcers vs. EC aspirin, would not be necessary. Instead, a Phase 1 pharmacodynamic study comparing gastric pH control for PA10040 vs. EC omeprazole 20 mg, along with a study to demonstrate bioequivalence of PA10040 to a currently marketed EC aspirin product using ASA as the analyte would be sufficient. Study PA10040-101 demonstrated that PA10040 had comparable bioavailability and is bioequivalent to an EU reference listed EC aspirin 100 mg. Study PA10040-102 demonstrated that PA10040 provides gastric pH greater than 4 control for a percent time over 24 hours of 47%. Furthermore, compared to Losec 20 mg (EC omeprazole 20 mg), PA10040 produced a similar level of 24-hour pH control ((p=0.02).
A pre-submission follow up meeting with the MEB was held in July 2014. We proposed seeking approval of PA10040 only since the ASA component in that dosage form matches the clinical practice for use of ASA in the EU. Approval of PA10040 would be based on specific studies conducted with the product, as well as data from the entire PA program conducted in the United States. Based on discussions at the meeting, the MEB agreed that the MAA filing can proceed without any further clinical studies and that the Netherlands would be the lead country for the decentralized review process. The MEB also requested that a complete risk-benefit
analysis of PA10040 for the intended cardiovascular patient population and proposed indication should be included as part of the MAA submission.
We have incurred significant losses since our inception and have not yet generated significant revenue from product sales. As of March 31, 2015, our accumulated deficit was approximately $96.9 million. We record revenue under the following categories: royalty revenues and licensing revenues. Our licensing revenues include upfront payments upon contract signing, additional payments if and when certain milestones in the product’s development or commercialization are reached, while the royalty payments are based on product sales. Our historical operating losses have resulted principally from our research and development activities, including clinical trial activities for our product candidates and sales, general and administrative expenses. Research and development expenses include salaries and benefits for personnel involved in our research and development activities and direct development costs, which include costs relating to the formulation and manufacturing of our product candidates, costs relating to preclinical studies, including toxicology studies, and clinical trials, and costs relating to compliance with regulatory requirements applicable to the development of our product candidates. Since inception, our research and development expenses have represented approximately 62% of our total operating expenses. For the three months ended March 31, 2015, our research and development expenses represented approximately 23% of our total operating expenses.
Operating losses may be incurred over the next several years as we complete the development and seek regulatory approval for our product candidates, develop other product candidates, conduct pre-commercialization activities, and acquire and/or develop product portfolios in other therapeutic areas. Our results may vary depending on many factors, including:
· The progress of our PA product candidates and our other product candidates in the clinical and regulatory process;
· The ability of Horizon and AstraZeneca to successfully commercialize VIMOVO in the United States and outside the United States, respectively, and our ability to successfully commercialize our PA product candidates;
· The establishment of potential new collaborations and progress and/or maintenance of our existing collaborations for the development and commercialization of any of our product candidates;
· Our ability to successfully defend our regulatory market exclusivity and patent rights against generic challenges and to succeed in obtaining extensions of such exclusivity for which we may be eligible;
· Our ability to commercialize our products either ourselves or with commercial partners in a highly regulated and extremely competitive marketplace;
· The possible acquisition and/or in-licensing, and development of our therapeutic product candidates; and
· The ability of us to integrate our business with that of Tribute if the merger is consummated.
We have entered into collaborations and may continue to enter into additional collaborations with established pharmaceutical or pharmaceutical services companies to commercialize and manufacture our product candidates once approved. We decided to retain control of our PA product candidates for cardiovascular indications through the clinical development and pre-commercialization stage. To that end, our chief commercial officer evaluated the commercial opportunities for these product candidates and developed a worldwide commercial strategy, which enabled us to conduct pre-commercialization activities prior to licensing these products to commercial partners. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
Our business and operations model is evolving. On June 1, 2015, our Board appointed a new Chief Executive Officer, Adrian Adams and a new President and Chief Business Officer, Andrew Koven, each of whom has experience creating, leading and expanding pharmaceutical companies with marketing and sales capabilities. If the proposed business combination with Tribute is approved by our stockholders and Tribute’s shareholders and concurrent equity and debt financings are closed, we will become a specialty pharmaceutical company focused on cardiovascular and pain therapies with a diversified portfolio of marketed products and developmental stage product candidates in the United States and Canada.
Our ability to generate revenue in the near term is dependent upon our ability, alone or with collaborators, to achieve the milestones set forth in our collaboration agreements, to enter into additional collaboration agreements, to successfully develop product candidates, to obtain regulatory approvals and to successfully manufacture and commercialize our future products. These milestones are earned when we have satisfied the criteria set out in our revenue recognition footnote accompanying the financial statements included in our Annual Report on Form 10-K, filed with the SEC on March 11, 2015 which appear elsewhere in this prospectus. These payments generate large non-recurring revenue that will cause large fluctuations in quarterly and annual profit and loss.
Our principal executive office is located at 1414 Raleigh Road, Suite 400, Chapel Hill, North Carolina 27517, and our telephone number is (919) 913-1030. Our website address is www.POZEN.com. The information on our website is not incorporated into this prospectus and should not be considered to be a part of this prospectus. We have included our website address as an inactive textual reference only.
Status and Expenses Related to Our Approved Products and Product Candidates
There follows a brief discussion of the status of the development of our approved products and our product candidates, as well as the costs relating to our development activities. Our direct research and development expenses were $0.5 million for the three months ended March 31, 2015 and $1.1 million for the three months ended March 31, 2014. Our research and development expenses that are not direct development costs consist of personnel and other research and development departmental costs and are not allocated by product candidate. We generally do not maintain records that allocate our employees’ time by the projects on which they work and, therefore, are unable to identify costs related to the time that employees spend on research and development by product candidate. Total compensation and benefit costs for our personnel involved in research and development were $0.4 million for the three months ended March 31, 2015 and $0.7 million for the three months ended March 31, 2014. Total compensation included $0.1 million and $0.1 million charge for non-cash compensation for stock option expense for the three months ended March 31, 2015 and March 31, 2014, respectively. Other research and development department costs were $0.1 million for the six months ended March 31, 2015 and less than $0.1 million for the three months ended March 31, 2014.
Treximet/MT400
On April 15, 2008, the FDA approved Treximet for the acute treatment of migraine attacks with or without aura in adults. GSK notified us of its intention to launch the product and Treximet was available in pharmacies in May 2008. As part of our NDA program for Treximet, we conducted five Phase 1 trials, two Phase 3 pivotal trials, and one 12-month open label safety trial using a formulation of Treximet developed by GSK. The Phase 3 pivotal trials, including the endpoints required to evaluate Treximet, were designed to demonstrate superiority to placebo for relief of pain and the associated symptoms of migraine (nausea, photophobia and phonophobia) at two hours. Additionally, the program was designed to demonstrate that each component makes a contribution to the efficacy of Treximet (the “combination drug rule” that the FDA requires of all combination products). The efficacy endpoint for the combination was sustained pain free, which is defined as improvement from moderate or severe pain to no pain at two hours and remaining at no pain through twenty four hours without the use of rescue medicine. Further, GSK has conducted market support studies for Treximet, including evaluations in a pediatric population. As required by the terms of our agreement with GSK, we transferred ownership of the NDA and other regulatory filings for Treximet to GSK on May 14, 2008, and GSK took responsibility for all ongoing regulatory obligations for the product, including post marketing clinical trial requirements.
Since inception we have incurred total direct development costs of $26.5 million associated with the development of our MT 400 and Treximet programs. Our direct development costs do not include the cost of research and development personnel or any allocation of our overhead expenses.
On May 13, 2014, we, GSK, CII and Pernix, entered into certain agreements in connection with GSK’s divestiture of all of its rights, title and interest to develop, commercialize and sell Treximet in the U.S. to Pernix. Upon the closing of the divestiture on August 20, 2014, GSK assigned the Treximet Agreement to Pernix. Immediately following the closing of the divestiture, Amendment No. 1 between us and Pernix became effective. Amendment No. 1, among other things, amends the royalty provisions of the Agreement to provide for a guaranteed quarterly minimum royalty of $4 million for the calendar quarters commencing on January 1, 2015 and ending on March 31, 2018 and requires that Pernix continue certain of GSK’s ongoing development activities and to undertake certain new activities, for which we will provide reasonable assistance. Amendment No. 1 also eliminates restrictions in the Agreement on our right to develop and commercialize certain dosage forms of sumatriptan/naproxen combinations outside of the United States and permits Pozen to seek approval for these combinations on the basis of the approved NDA for Treximet. Pernix also granted us a warrant to purchase 500,000 shares of Pernix common stock at an exercise price of $4.28, the closing price of Pernix common stock as reported on the NASDAQ Global Market on May 13, 2014. In the first quarter of 2015, the Company sold the warrant for $2,479,000.
Lastly, we, GSK, Pernix and CII executed a letter agreement whereby we expressly consented to the assignment by GSK and the assumption by Pernix of the Treximet Agreement. On July 30, the parties entered into Amendment No. 2 to the Treximet Agreement which will permit Pernix’s Irish affiliate to which Pernix will assign its rights to further assign the Agreement without our prior written consent as collateral security for the benefit of the lenders which financed the acquisition. Amendment No. 2 became effective upon closing of the divestiture, which occurred on August 20, 2014.
PN/VIMOVO Program
Under our PN program, we completed formulation development and clinical studies for several combinations of a PPI and a NSAID in a single tablet intended to provide effective management of pain and inflammation associated with chronic conditions such as osteoarthritis, and intended to have fewer gastrointestinal complications compared to a NSAID taken alone in patients at risk for developing NSAID associated gastric ulcers. We entered into an exclusive,worldwide (except for Japan) collaboration agreement with AstraZeneca on August 1, 2006 and which was amended in September 2007 and October 2008 relating to the development and commercialization of our PN products. Our agreement with AstraZeneca covered the development and commercialization of proprietary fixed dose combinations of the PPI esomeprazole magnesium with the NSAID naproxen in a single tablet. The initial product developed under the agreement, VIMOVO (formerly PN 400), was approved by the FDA on April 30, 2010 for the relief of the signs and symptoms of OA, RA and AS and to decrease the risk of developing gastric ulcers in patients at risk of developing NSAID-associated gastric ulcers.
The NDA for VIMOVO was submitted on June 30, 2009 and was accepted for filing in August 2009. We received a $10.0 million milestone payment from AstraZeneca in September 2009 for the achievement of such milestone. On April 30, 2010, VIMOVO was approved by FDA for the relief of the signs and symptoms of OA, RA and AS and to decrease the risk of developing gastric ulcers in patients at risk of developing NSAID-associated gastric ulcers. We received a $20.0 million milestone payment from AstraZeneca in May 2010 in connection with such approval. As required by the terms of our agreement with AstraZeneca, we transferred ownership of the NDA and other regulatory filings for VIMOVO to AstraZeneca on June 1, 2010, and AstraZeneca now has responsibility for all ongoing regulatory obligations for the product in the U.S., including post marketing clinical trial requirements, in addition to responsibility for all regulatory obligations outside the U.S.
Under our agreement with AstraZeneca, AstraZeneca had responsibility for the development program for PN products outside the U.S., including interactions with regulatory agencies. In October 2009, AstraZeneca submitted a MAA for VIMOVO in the EU via the DCP and has filed for approval in a number of other countries which are not covered by the DCP. On October 11, 2010, we announced with AstraZeneca that VIMOVO had received positive agreement for approval in 39 countries across the EU following all 22 Concerned Member States agreeing with the assessment of the Netherlands Health Authority, acting as the Reference Member State for the DCP. We received a $25.0 million milestone payment when pricing and reimbursement for VIMOVO was granted in the United Kingdom. Other Member States are now pursuing pricing and reimbursement and national approvals. Earlier, in May 2010, we had received a $20.0 million milestone payment when we received FDA approval of VIMOVO. As of the end of December 31, 2013, VIMOVO has been filed for regulatory approval in 81 countries and approved in 71 countries.
On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understand that AstraZeneca will instead now focus on those countries where the product has shown growth and which AstraZeneca believes have the greatest potential for future growth. We continue to assess the financial impact this decision will have on our royalty revenue. On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. As required by the terms of our agreement with AstraZeneca, we gave our consent to AstraZeneca’s divestiture of such rights to Horizon because we believed that Horizon’s expertise in commercializing products for pain and inflammatory disease, including a similar combination product containing a NSAID and a gastroprotective agent, make it an excellent partner to maximize the potential of VIMOVO in the United States. We were also able to negotiate a guaranteed annual minimum royalty in the United States in the amount of $5.0 million in calendar year 2014, and a guaranteed annual minimum royalty amount of $7.5 million each calendar year thereafter, provided that the patents owned by us which cover VIMOVO are in effect and no generic forms of VIMOVO are in the marketplace. On July 28, 2014, Horizon announced that it had been verbally notified by CVS Caremark and Express Scripts, Inc. that VIMOVO would no longer be on their formularies and will be placed on their exclusion lists effective January 1, 2015. We saw a decline in VIMOVO prescriptions in Q1 2015 of approximately 20% versus the fourth quarter of 2014 and our quarterly royalty declined to $3.2 million, well ahead of our minimum quarterly royalty of $1.875 million.
Since inception we have incurred total direct development cost of $96.2 million associated with the development of our PN program of which $57.1 million was funded by development revenue from AstraZeneca. Our direct development costs do not include the cost of research and development personnel or any allocation of our overhead expense.
PA Program
As part of our PA program, we are developing a PPI and aspirin in a single tablet. Similar to the PN program, our PA product candidate is intended to induce fewer gastrointestinal complications compared to an aspirin taken alone in patients at risk for developing aspirin associated gastric ulcers. Our PA product candidates are covered under the same patent as PN, but we retained all rights to this program through the clinical development and pre-commercialization stage.
Our PA product candidates, PA32540 and PA8140, have completed clinical development testing in the United States. Based upon the FDA’s earlier confirmation that endoscopic gastric ulcers were an acceptable primary endpoint, in October 2009, we began two pivotal Phase 3 and one long-term safety study for PA32540 for the cardiovascular indication. The primary endpoint was met in both studies. Additionally, the studies met their key secondary endpoints, including a reduction in gastroduodenal ulceration as well as a reduction in discontinuation due to upper gastrointestinal adverse events in subjects taking PA32540 compared to 325 mg EC aspirin.
In February 2012, the FDA requested an additional Phase 1 study to assess the bioequivalence of PA32540 to EC aspirin 325 mg with respect to ASA. After the Company completed the requested bioequivalence study, the FDA has made a preliminary review of the study results and the Company’s summary analyses and, based on its preliminary assessment of the information available to it at the time, the FDA did not agree that bioequivalence of PA32540 to EC aspirin 325 mg was demonstrated. We then submitted to the FDA additional information and analyses from the requested bioequivalence study, as well as other relevant pharmacokinetic, clinical pharmacology, and in vitro dissolution data as a Briefing Document in support of a request for a Type A meeting with the FDA. At the Type A meeting held in August 2012, the FDA confirmed that, although it believes bioequivalence of PA32540 to EC aspirin 325 mg, was not strictly established in our bioequivalence study according to the predetermined criteria, the results from this study, together with additional information that will be submitted by us in the NDA, constitutes sufficient data to support the establishment of a clinical and pharmacological bridge between the product and EC aspirin 325 mg. The FDA indicated that it would make a final determination during the NDA review. FDA also indicated that a similar strategy to bridge to the reference listed drug, inclusive of a new, single pharmacokinetic study, could be utilized for a low dose version of PA32540 (PA8140). We have conducted this study with the low dose version against the EC aspirin 81 mg. Based on the predetermined criteria acceptable to the FDA, the study demonstrated that PA8140 is bioequivalent to EC aspirin using criteria for highly variable drugs and had comparable bioavailability.
During a pre-submission meeting with respect to its NDA for PA32540 in April 2012, the FDA suggested that the Company also seek approval for a lower dose formulation of the product containing 81 mg of EC aspirin as part of its NDA for PA32540. We intended to seek an indication for the secondary prevention of cardiovascular disease in patients at risk for gastric ulcers. During the Type A meeting held in August 2012, the FDA has confirmed its preference to have both PA32540 and a lower dose version available in the market so that physicians can have both a low and high dose option available, and agreed that, if both dosage strengths were included in the NDA and subsequently approved, the indications for both will be consistent with the full range of indications described in the current aspirin monograph.
We had generated some clinical pharmacology data and chemical, manufacturing and controls, or CMC, data for a product which contains 81 mg of EC aspirin and 40 mg of omeprazole in a single tablet known as PA8140. We filed this existing data, together with additional CMC data to be generated and evidence from the scientific literature relating to the ulcerogenic risk of 81 mg of aspirin with the FDA. At this time, the Company does not intend to conduct Phase 3 clinical trials for PA8140. The data package submitted for PA8140 was similar to that used to gain approval for a lower dosage form of VIMOVO containing 375 mg of naproxen. We have no assurance such data will be sufficient for the FDA to approve PA8140 or to allow a broader indication for PA32540. The FDA will make a final determination with respect to the approvability of and indications for PA32540 and PA8140.
Generation of additional data with respect to PA8140 and incorporation of data into the NDA for PA32540 delayed submission of the NDA from the original planned submission date in the third quarter of 2012. The NDA was filed for both products in March 2013 and in May 2013 the FDA accepted the NDA for review. The FDA assigned a user fee date of January 24, 2014. As part of our continuing discussions with the FDA concerning the NDA for PA32540 and PA8140 tablets, we decided to conduct an additional comparative Phase 1 pharmacokinetic study to determine the pharmacokinetic profile of the omeprazole component of PA8140 tablets and compare it to that of PA32540 tablets. We submitted study information and data to the FDA as it became available during the conduct of the study and FDA agreed to review such information and data from the study when submitted. The final study report for the study was submitted to the FDA in accordance with our agreed timeline. FDA has informed us that the Company’s user fee date was April 25, 2014. On April 25, 2014, we received a CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude the approval of the NDA in its current form. Specifically, an inspection of the manufacturing facility of an active ingredient supplier of ours concluded with certain inspection deficiencies. Satisfactory resolution of these deficiencies is required before the NDA may be approved. There were no clinical or safety deficiencies noted with respect to either PA8140 or PA32540 and no other deficiencies were noted in CRL. On June 30, 2014, we resubmitted the NDA for PA32540 and PA8140 to the FDA and the FDA notified us that the new action fee date is December 30, 2014. On May 9, 2014, our active ingredient supplier submitted a response to the FDA addressing the inspection deficiencies and subsequently submitted an update to its initial response.
On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. Satisfactory resolution of these deficiencies is required before this application may be approved. There were no clinical or safety deficiencies noted with respect to either PA32540 or PA8140 and no other deficiencies were noted in the CRL. Final agreement
on the draft product labeling is also pending. We continue to assist the FDA compliance division with their review. FDA regulations allow us to request a Type A meeting with the FDA to discuss the next steps required to gain approval of our NDA. The FDA granted the Type A meeting, which was held in late January 2015. At the meeting, representatives from the FDA’s Office of Compliance stated that the active ingredient supplier’s responses to the 483 inspectional observations submitted in May 2014 were still under review, but that the review had been placed on a fast track and the Office of Compliance would be communicating with the supplier in the coming weeks. The active ingredient supplier has informed Pozen that they received a warning letter relating to the Form 483 inspection deficiencies. They have submitted a plan of corrective actions to address the matters raised in the warning letter to FDA. We have had continuing discussions with FDA about these issues and the impact on our NDA and with our active ingredient supplier regarding the corrective actions it is taking to address the inspectional observations at its facility. We are also working toward securing and seeking FDA’s approval for an alternative back up supplier.
We are also continuing to evaluate how best to commercialize the PA product candidates and programs. We are evaluating the regulatory requirements to obtain an indication for PA for the secondary prevention of colorectal neoplasia. In January 2010, we received input from the FDA with respect to the development requirements for a possible indication in colorectal neoplasia. Further discussions are being considered. We are also evaluating the possibility of developing another dosage strength of PA containing 650 mg of EC aspirin and 20 mg of omeprazole (PA65020) for the treatment of osteoarthritis and similar conditions and we met with the FDA in December 2010 to obtain input with respect to the regulatory requirements to obtain such an indication. The FDA advised us that we will need to demonstrate a reduction in gastric ulcers compared to aspirin alone in a replicate Phase 3 program similar to that we performed for VIMOVO, in addition to a study to establish efficacy of the product in the treatment of osteoarthritis.
We recently met with a European country that served as the reference member state for approval of VIMOVO in Europe to obtain guidance on a clinical development program for approval of PA65020 in the Europe. We were advised that Phase 3 endoscopic trials demonstrating a reduction in gastric ulcers would not be required in addition to an osteoarthritis efficacy study since omeprazole’s actions on gastric ulcers has been well characterized. Instead, Phase 1 studies assessing pharmacokinetics and gastric acid suppression could be used to support the benefit of omeprazole as a component of PA65020.
We have also received Scientific Advice from the MEB in the Netherlands, which has agreed to be the Reference Member State in a decentralized filing procedure, regarding the development program required for the approval in the EU of PA tablets, including a lower dosage form containing 100 mg of aspirin and 40 mg of omeprazole (PA10040) for the secondary prevention of cardiovascular disease. The MEB agreed that a full Phase 3 clinical development program for PA10040, to demonstrate the reduction of endoscopic gastric ulcers vs. EC aspirin, would not be necessary. Instead, a Phase 1 pharmacodynamic study comparing gastric pH control for PA10040 vs. EC omeprazole 20 mg, along with a study to demonstrate bioequivalence of PA10040 to a currently marketed EC aspirin product using ASA as the analyte would be sufficient. Study PA10040-101 demonstrated that PA10040 had comparable bioavailability and is bioequivalent to an EU reference listed EC aspirin 100 mg. Study PA10040-102 demonstrated that PA10040 provides gastric pH greater than 4 control for a percent time over 24 hours of 47%. Furthermore, compared to Losec 20 mg (EC omeprazole 20 mg), PA10040 produced a similar level of 24-hour pH control ((p=0.02).
A pre-submission follow up meeting with the MEB was held in July 2014. We proposed seeking approval of PA10040 only since the ASA component in that dosage form matches the clinical practice for use of ASA in the EU. Approval of PA10040 would be based on specific studies conducted with the product, as well as data from the entire PA program conducted in the United States. Based on discussions at the meeting, the MEB agreed that the MAA filing can proceed without any further clinical studies and that the Netherlands would be the lead country for the decentralized review process. The MEB also requested that a complete risk-benefit analysis of PA10040 for the intended cardiovascular patient population and proposed indication should be included as part of the MAA submission.
We cannot reasonably estimate or know the amount or timing of the costs necessary to continue development and/or complete the development of any PA product candidates we may seek to develop or when, if and to what extent we will receive cash inflows from any PA products. The additional costs that may be incurred include expenses relating to clinical trials and other research and development activities and activities necessary to obtain regulatory approvals. We decided to retain control of our PA product candidates for cardiovascular indications through the clinical development and pre-commercialization stage. On September 3, 2013 we entered into a License and Collaboration Agreement with Sanofi U.S. to commercialize our PA product candidates containing an immediate release proton pump inhibitor and 325 mg or less of delayed release or EC aspirin in the United States. Even though the License and Collaboration Agreement was terminated on November 29, 2014, we believe we were able to negotiate more favorable terms with Sanofi U.S. for rights to commercialize the products in the United States than we had licensed the product candidates at an earlier stage in development and will be able to achieve more favorable terms with other partners outside the United States if we are successful in licensing PA products in other territories in the future. Our business and operations model is evolving. On June 1, 2015, our Board appointed a new Chief Executive Officer and a new President and Chief Business Officer, each of whom has experience creating, leading and expanding pharmaceutical companies with marketing and sales capabilities. If the proposed
business combination with Tribute is approved by our stockholders and Tribute’s shareholders and such transaction and concurrent equity and debt financings are closed, we will become a specialty pharmaceutical company focused on cardiovascular and pain therapies with a diversified portfolio of marketed products and developmental stage product candidates in the United States and Canada.
We have incurred direct development costs associated with the development of our PA program of $0.5 million during the three months ended March 31, 2015. From inception through March 31, 2015, we incurred total direct development cost of $75.1 million associated with the development of our PA program. Our direct development costs do not include the cost of research and development personnel or any allocation of our overhead expenses.
Collaborative Arrangements
We have entered into and may continue to enter into collaborations with established pharmaceutical or pharmaceutical services companies to develop, commercialize and/or manufacture our product candidates. Our existing collaborations are described below.
GlaxoSmithKline (GSK)
In June 2003, we signed an agreement with GSK for the development and commercialization of proprietary combinations of a triptan (5-HT1B/1D agonist) and a long-acting NSAID. The combinations covered by the agreement are among the combinations of MT 400. Under the terms of the agreement, GSK has exclusive rights in the U.S. to commercialize all combinations which combine GSK’s triptans, including Imitrex® (sumatriptan succinate) or Amerge® (naratriptan hydrochloride), with a long-acting NSAID. We were responsible for development of the first combination product, while GSK provided formulation development and manufacturing. Pursuant to the terms of the agreement, we received $25.0 million in initial payments from GSK following termination of the waiting period under the Hart-Scott-Rodino notification program and the issuance of a specified patent. In May 2004, we received a $15.0 million milestone payment as a result of our commencement of Phase 3 clinical trial activities. In October 2005, we received a $20.0 million milestone payment upon the FDA’s acceptance for review of the NDA for Treximet, the trade name for the product. On April 26, 2008, we received, from GSK, $20.0 million in milestone payments which were associated with the approval of, and GSK’s intent to commercialize, Treximet. In addition, Pernix, as assignee of GSK will pay two sales performance milestones totaling $80.0 million if certain sales thresholds are achieved. Up to an additional $10.0 million per product is payable upon achievement of milestones relating to other products. GSK will pay royalties on all net sales of marketed products until at least the expiration of the last to expire issued applicable patent (October 2, 2025) based upon the scheduled expiration of currently issued patents. GSK may reduce, but not eliminate, the royalty payable to us if generic competitors attain a pre-determined share of the market for the combination product, or if GSK owes a royalty to one or more third parties for rights it licenses from such third parties to commercialize the product. The agreement terminates on the date of expiration of all royalty obligations unless earlier terminated by either party for a material breach or by GSK at any time upon 90 days’ written notice to us for any reason or no reason. GSK has the right, but not the obligation, to bring, at its own expense, an action for infringement of certain patents by third parties. If GSK does not bring any such action within a certain time frame, we have the right, at our own expense, to bring the appropriate action. With regard to certain other patent infringements, we have the sole right to bring an action against the infringing third party. Each party generally has the duty to indemnify the other for damages arising from breaches of each party’s representations, warranties and obligations under the agreement, as well as for gross negligence or intentional misconduct. We also have a duty to indemnify GSK for damages arising from our development and manufacture of MT 400 prior to the effective date of the agreement, and each party must indemnify the other for damages arising from the development and manufacture of any combination product after the effective date.
On November 23, 2011, we entered into the Purchase Agreement, with CII, pursuant to which we sold, and CII purchased, our right to receive future royalty payments arising from U.S. sales of MT 400, including Treximet.
On May 13, 2014, we, GSK, CII and Pernix, entered into certain agreements in connection with GSK’s divestiture of all of its rights, title and interest to develop, commercialize and sell Treximet® in the U.S. to Pernix. Upon the closing of the divestiture on August 20, 2014, GSK assigned the Treximet Agreement to Pernix. Immediately following the closing of the divestiture, Amendment No. 1 between us and Pernix became effective. Amendment No. 1, among other things, amends the royalty provisions of the Agreement to provide for a guaranteed quarterly minimum royalty of $4 million for the calendar quarters commencing on January 1, 2015 and ending on March 31, 2018 and requires that Pernix continue certain of GSK’s ongoing development activities and to undertake certain new activities, for which we will provide reasonable assistance. Amendment No. 1 also eliminates restrictions in the Agreement on our right to develop and commercialize certain dosage forms of sumatriptan/naproxen combinations outside of the United States and permits Pozen to seek approval for these combinations on the basis of the approved NDA for Treximet. Pernix has also granted us a warrant to purchase 500,000 shares of Pernix common stock at an exercise price equal to$4.28 per share, the closing price of Pernix common stock as reported on the NASDAQ Global Market on May 13, 2014. In the first quarter of 2015, the Company sold the warrant for $2,479,000. Lastly, we, GSK, Pernix and CII executed a letter agreement whereby we expressly consented to the assignment by GSK and the assumption by Pernix of the Treximet Agreement. On July 30, the parties entered into Amendment No. 2
to the Treximet Agreement which will permit Pernix’s Irish affiliate to which Pernix assigned its rights to further assign the Agreement without our prior written consent as collateral security for the benefit of the lenders which financed the acquisition. Amendment No. 2 became effective upon the closing of the divestiture on August 20, 2014.
AstraZeneca AB (AstraZeneca)/ Horizon Pharma Inc.
In August 2006, we entered into a collaboration and license agreement dated as of August 1, 2006 and effective September 7, 2006 with AstraZeneca, a Swedish corporation, regarding the development and commercialization of proprietary fixed dose combinations of the PPI esomeprazole magnesium with the NSAID naproxen, in a single tablet for the management of pain and inflammation associated with conditions such as OA and RA in patients who are at risk for developing NSAID associated gastric ulcers, as amended, the “Original Agreement”. Under the terms of the Original Agreement, we granted to AstraZeneca an exclusive, fee-bearing license, in all countries of the world except Japan, under our patents and know-how relating to combinations of gastroprotective agents and NSAIDs (other than aspirin and its derivatives). Pursuant to the terms of the agreement, we received an upfront license fee of $40.0 million from AstraZeneca following termination of the waiting period under the Hart-Scott-Rodino notification program.
We retained responsibility for the development and filing of the NDA for the product in the U.S. AstraZeneca is responsible for all development activities outside the U.S., as well as for all manufacturing, marketing, sales and distribution activities worldwide. We agreed to bear all expenses related to certain specified U.S. development activities. All other development expenses, including all manufacturing-related expenses, will be paid by AstraZeneca. The agreement established joint committees with representation of both us and AstraZeneca to manage the development and commercialization of the product. The committees operate by consensus, but if consensus cannot be reached, we generally will have the deciding vote with respect to development activities required for marketing approval of the product in the U.S. and AstraZeneca generally will have the deciding vote with respect to any other matters.
In September 2007, we agreed with AstraZeneca to amend the Original Agreement effective as of September 6, 2007. Under the terms of the amendment, AstraZeneca has agreed to pay us up to $345.0 million, in the aggregate, in milestone payments upon the achievement of certain development, regulatory and sales events. In September 2007 we received a $10.0 million payment upon execution of the amendment and a $20.0 million payment in recognition of the achievement of the primary endpoints for the PN400-104 study, a study which compared acid suppression of different doses of VIMOVO (formerly PN 400), and achievement of the interim results of the PN200-301 study, a six month comparative trial of PN 200 as compared to EC naproxen in patients requiring chronic NSAID therapy, meeting mutually agreed success criteria. In May 2010, we received a $20.0 million payment for the NDA approval of VIMOVO. We also received an additional $25.0 million milestone in December 2010 when VIMOVO received approval (including pricing and reimbursement approval) in a major ex-U.S. market and up to $260.0 million will be paid as sales performance milestones if certain aggregate sales thresholds are achieved.
The amendment also revised the royalty rates we were to have received under the Original Agreement. Prior to the effective date of the amendment, under the terms of the Original Agreement, we were to receive a royalty based on annual net sales by AstraZeneca, its affiliates or sublicensees during the royalty term. The royalty rate varied based on the level of annual net sales of products made by AstraZeneca, its affiliates and sublicensees, ranging from the mid-single digits to the mid-teens. Under the amendment, we receive a flat, low double digit royalty rate during the royalty term on annual net sales of products made by AstraZeneca, its affiliates and sublicensees, in the U.S. and royalties ranging from the mid-single digits to the high-teens on annual net sales of products made by AstraZeneca, its affiliates and sublicensees outside of the U.S. The amendment also revised the rate of reduction to the royalty rate based upon loss of market share due to generic competition inside and outside of the U.S. to account for the new royalty structure. Our right to receive royalties from AstraZeneca for the sale of such products under the collaboration and license agreement, as amended, expires on a country-by-country basis upon the later of (a) expiration of the last-to-expire of certain patent rights relating to such products in that country, and (b) ten years after the first commercial sale of such products in such country.
We further amended the Original Agreement effective October 1, 2008 to shorten the timing of AstraZeneca’s reimbursement obligation for certain development expenses incurred by us under the agreement and to update the description of the target product profile studies (as defined in the agreement) for VIMOVO.
On March 31, 2015 we have receivables of $4.4 million related to VIMOVO royalty revenue, $3.2 million related to U.S. sales and $1.2 million related to ROW sales. The agreement, unless earlier terminated, will expire upon the payment of all applicable royalties for the products commercialized under the agreement. Either party has the right to terminate the agreement by notice in writing to the other party upon or after any material breach of the agreement by the other party, if the other party has not cured the breach within 90 days after written notice to cure has been given, with certain exceptions. The parties also can terminate the agreement for cause under certain defined conditions. In addition, AstraZeneca can terminate the agreement, at any time, at will, for any reason or no reason, in its entirety or with respect to countries outside the U.S., upon 90 days’ notice. If terminated at will, AstraZeneca will owe us a specified termination payment or, if termination occurs after the product is launched, AstraZeneca may, at
its option, under and subject to the satisfaction of conditions specified in the agreement, elect to transfer the product and all rights to us.
On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understand that AstraZeneca will instead now focus on those countries where the product has shown growth and which AstraZeneca believes have the greatest potential for future growth.
On September 16, 2013, we and AstraZeneca entered into another amendment to the Original Agreement which made clarifications to certain intellectual property provisions of the Original Agreement to clarify that AstraZeneca’s rights under those provisions do not extend to products which contain ASA. On September 16, 2013, we and AstraZeneca also executed a letter agreement whereby we agreed that in the event that AstraZeneca divested its rights and obligations to market VIMOVO in the United States to a third party, AstraZeneca would be relieved of its obligations under the Original Agreement with respect to the United States as of the effective date of such divestiture, including its obligation under the Original Agreement to guarantee the performance of such assignee and/or sublicensee.
On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. In connection with this divestiture, on November 18, 2013 we and AstraZeneca entered into an Amended and Restated Collaboration and License Agreement for the United States, the “U.S. Agreement,” and an Amended and Restated License and Collaboration Agreement for Outside the United States, the “ROW Agreement,” which agreements collectively amend and restate the Original Agreement. AstraZeneca has assigned the U.S. Agreement to Horizon in connection with the divestiture with our consent.
We and Horizon also entered into Amendment No. 1 to the U.S. Agreement which, among other things, amends the royalty provisions of the U.S. Agreement to provide for a guaranteed annual minimum royalty amount of $5 million in calendar year 2014, and a guaranteed annual minimum royalty amount of $7.5 million each calendar year thereafter, provided that the patents owned by us which cover VIMOVO are in effect and no generic forms of VIMOVO are in the marketplace. Amendment No. 1 also provides that Horizon has assumed AstraZeneca’s right to lead the on-going Paragraph IV litigation relating to VIMIVO currently pending in the United States District Court for the District of New Jersey and will assume all patent-related defense costs relating to such litigation, including reimbursement up to specified amounts of the cost of any counsel retained by us, amends certain time periods for Horizon’s delivery of quarterly sales reports to Pozen, and provides for quarterly update calls between the parties to discuss VIMOVO’s performance and Horizon’s commercialization efforts.
Further, we, AstraZeneca and Horizon executed a letter agreement whereby Pozen expressly consented to the assignment by AstraZeneca and the assumption by Horizon of the U.S. Agreement. In addition, the letter agreement establishes a process for AstraZeneca and Horizon to determine if sales milestones set forth in the Original Agreement are achieved on a global basis and other clarifications and modifications required as a result of incorporating the provisions of the Original Agreement into the U.S. Agreement and the ROW Agreement or as otherwise agreed by the parties.
We, AstraZeneca and Horizon are also engaged in Paragraph IV litigation with several generic pharmaceutical companies with respect to patents listed in the Orange Book with respect to VIMOVO currently pending in the United States District Court for the District of New Jersey and in an IPR brought by Dr. Reddy’s with respect to the ‘285 patent and two IPRs brought by CFAD. We and AstraZeneca are also engaged in a proceeding in Canada with Mylan ULC which is seeking approval of its generic version of VIMOVO in Canada prior to the expiration of our Canadian patent.
sanof-aventis U.S. LLC (Sanofi U.S.)
On September 3, 2013, we entered into a license and collaboration agreement with Sanofi U.S. with respect to the commercialization of our PA products in the United States. On November 29, 2014, we executed a termination agreement with Sanofi U.S. terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi U.S. were terminated and all rights to the products licensed to Sanofi U.S. under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi U.S. relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products in the United States. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
Patheon Pharmaceuticals Inc. (Patheon)
On December 19, 2011, we entered into a Manufacturing Services Agreement, or the Supply Agreement, and a related Capital Expenditure and Equipment Agreement, or the Capital Agreement, relating to the manufacture of PA32450. Under the terms of the Supply Agreement, Patheon has agreed to manufacture, and we have agreed to purchase, a specified percentage of the Company’s requirements of the PA32540 for sale in the United States. The term of the Supply Agreement extends until December 31st of the fourth year after we notify Patheon to begin manufacturing services under the Supply Agreement, or the Initial Term, and will automatically renew thereafter for periods of two years, unless terminated by either party upon eighteen months’ written notice prior to the expiration of the Initial Term or 12 months’ written notice prior to the expiration of any renewal term. In addition to usual and customary termination rights which allow each party to terminate the Supply Agreement for material, uncured breaches by the other party, we can terminate the Supply Agreement upon 30 days’ prior written notice if a governmental or regulatory authority takes any action or raises any objection that prevents us from importing, exporting, purchasing or selling PA32540 or if it is determined that the formulation or sale of PA32540 infringes any patent rights or other intellectual property rights of a third party. We can also terminate the Supply Agreement upon 24 months’ prior written notice if we license, sell, assign or otherwise transfer any rights to commercialize PA32540 to a third party. The Supply Agreement contains general and customary commercial supply terms and conditions, as well as establishing pricing for bulk product and different configurations of packaged product, which pricing will be adjusted annually as set forth in the Supply Agreement. Under the terms of the Capital Agreement, we will be responsible for the cost of purchasing certain equipment specific to the manufacture of PA32540, the cost of which, based on current volume projections, is expected to be less than $150,000. If additional equipment and facility modifications are required to meet our volume demands for PA32540, we may be required to contribute to the cost of such additional equipment and facility modifications, up to a maximum of approximately $2.5 million in the aggregate.
The Supply Agreement and Capital Agreement were amended on July 10, 2013. The First Amendment to the Manufacturing and Services Agreement (the “Amendment to the Supply Agreement”) expressly incorporates the Company’s PA8140 product candidate into the Supply Agreement. The Amendment to the Supply Agreement also clarifies that the manufacturing services contemplated by the Supply Agreement include the manufacture of validation batches, but the placing of an order for such validation batches will not trigger the Commencement Date of the Initial Term (each as defined in the Supply Agreement), updates pricing for the Company’s PA32540 product candidate and a incorporates a new pricing schedule for PA8140, as well as other conforming changes to the Supply Agreement. The First Amendment to the Capital Expenditure and Equipment Agreement (the “Amendment to the Capital Agreement”), replaces the existing Schedule A of the Capital Agreement, which lists dedicated and non-dedicated capital equipment and facility modifications to be funded in whole or in part by the Company, with a new updated schedule which reflects the parties’ current assumptions regarding the need for and timing of capital equipment expenditures based upon Patheon’s current and anticipated production capacity and current volume projections for the PA32540 and PA8140. Under the terms of the Capital Agreement, the Company was previously required to contribute to the cost of such additional capital equipment and facility modifications, up to a maximum of approximately $2.5 million in the aggregate. Pursuant to the terms of the Amendment to the Capital Agreement, the parties have agreed to reduce the amount of such maximum expenditure to approximately $1.2 million dollars in light of the revised capacity and volume assumptions.
Results of Operations
Three months ended March 31, 2015 compared to the three months ended March 31, 2014
Net (loss) income per share: Net loss attributable to common stockholders for the three months ended March 31, 2015 was $(27,358), or $0.00 per share, on a diluted basis, as compared to net income of $2.9 million, or $0.09 per share, on a diluted basis, for the three months ended March 31, 2014. The net loss for the three months ended March 31, 2015 included a $0.5 million, or $0.01 per share charge for non-cash stock-based compensation expense as compared to $0.8 million, or $0.02 per share for the same period of 2014.
Revenue: We recognized total revenue of $4.4 million for the three months ended March 31, 2015 as compared to total revenue of $7.5 million for the three months ended March 31, 2014. The decrease in revenue was primarily due to a decrease of $3.0 million in amortization of PA licensing revenue from receipt of $15.0 million upfront fee for the PA agreement with Sanofi U.S. Licensing revenue for the three months ended March 31, 2015 consisted of $4.4 million of royalty revenue compared to $4.5 million of royalty revenue and $3.0 million of other licensing revenue for the three months ended March 31, 2014. Our licensing and collaboration agreements have terms that include upfront payments upon contract signing and additional payments if and when certain milestones in the product development or related milestones are achieved. All upfront payments have been recognized as of December 31, 2014. Substantive milestone payments are recognized as revenue upon completion of the contractual events.
Research and development: Research and development expenses decreased by $0.8 million to $1.0 million for the three months ended March 31, 2015, as compared to the same period of 2014. The decrease was due primarily to a decrease in direct development costs for our PA program and in departmental costs, as compared to the same period of 2014. Direct development costs for the PA program decreased by $0.6 million to $0.5 million, primarily due to the completion of the clinical trial activities and other
product development activities during the three months ended March 31, 2015. Other direct departmental costs and departmental expenses decreased by $0.2 million primarily due to decreased personnel costs, as compared to the same period of 2014. We have included in our research and development total expenses the departmental personnel costs associated with our research and development activities and direct costs associated with pharmaceutical development, clinical trials, toxicology activities and regulatory matters.
Sales, general and administrative: Sales, general and administrative expenses increased by $0.4 million to $3.3 million for the three months ended March 31, 2015, as compared to the same period of 2014. The increase reflects patent defense litigation expense, as well as increases in ongoing business development and public company expenses, as compared to the same period of 2014. Sales, general and administrative expenses consisted primarily of the costs of administrative personnel, facility infrastructure, business development expenses, and public company activities.
Other income (loss): Interest and bond amortization income was $7,400 for the three months ended March 31, 2014 and a loss of $0.2 million related to the sale of the Pernix warrant for the three months ended March 31, 2015.
Income Taxes
We estimate an annual effective tax rate of 0% for the year ended December 31, 2015, and our effective tax rate was 0% for the three month period ended March 31, 2015. However, the actual effective rate may vary depending upon actual licensing fees and milestone payments received, specifically the pre-tax book income for the year, and other factors. Income taxes have been accounted for using the liability method in accordance with FASB ASC 740. Since our inception, we have incurred substantial cumulative losses and may incur substantial and recurring losses in future periods. The utilization of these loss carryforwards to reduce future income taxes will depend on the Company’s ability to generate sufficient taxable income prior to the expiration of the loss carryforwards. In addition, the maximum annual use of net operating loss and research credit carryforwards is limited in certain situations where changes occur in stock ownership.
We currently file income tax returns in the U.S. federal jurisdiction, and the state of North Carolina. We are no longer subject to federal or North Carolina income tax examinations by tax authorities for years before 2010. However, the loss carryforwards generated prior to 2010 may still be subject to change, if we subsequently begin utilizing these losses in a year that is open under statute and subject to federal or North Carolina income tax examinations by tax authorities.
At March 31, 2015, we had no unrecognized tax benefits that would reduce the Company’s effective tax rate if recognized. We recognize any interest and penalties accrued related to unrecognized tax benefits as income tax expense. During the three months ended March 31, 2015 and 2014, there were no such interest and penalties.
Liquidity and Capital Resources
At March 31, 2015, cash, cash equivalents and investments in warrants totaled $43.9 million, an increase of $0.6 million compared to December 31, 2014. The $0.6 million increase in cash and investments resulted from the receipt of $5.6 million in VIMOVO royalty payments and $2.5 million from the sale of the Pernix warrant. This is offset by payments of $4.8 million in operating expenses and decrease in investments of $2.7 million for the sale of the Pernix warrant. Our cash is invested in money market funds that invest primarily in commercial paper and certificates of deposit guaranteed by banks.
Pozen received $5.6 million in operating cash during the three months ended March 31, 2015 pursuant to the terms of our collaboration agreements with AstraZeneca, Sanofi U.S. and Horizon. In addition, our balance sheet included a $4.4 million accounts receivable for royalties under the AstraZeneca and Horizon agreements.
Based upon the indirect method of presenting cash flow, cash provided by operating activities totaled $0.7 million and cash used in operating activities totaled $6.1 million for the three months ended March 31, 2015 and March 31, 2014, respectively. Net cash provided by investing activities totaled $2.5 million during the three months ended March 31, 2015 and there were no investing activities during the three months ended March 31, 2014. Net cash provided by financing activities during the three months ended March 31, 2015 totaled $0.1 million and $0.4 million for the three months ended March 31, 2014. Cash required for our operating activities during 2015, as compared to our 2014 requirements, is projected to decrease as a result of decreased development activities but may increase if we elect to undertake increased pre-commercialization activities. During the three months ended March 31, 2015 and March 31, 2014 we recorded non-cash stock-based compensation expense of $0.5 million and $0.8 million, respectively, associated with the grant of stock options and restricted stock units.
As of March 31, 2015, we had $43.9 million in cash and cash equivalents. Our anticipated cash
flow includes continued receipt of royalty revenue from Horizon and AstraZeneca’s sale of VIMOVO. In addition, our expenses might increase during that period beyond currently expected levels if we decide to, or any regulatory agency requires us to, conduct additional clinical trials, studies or investigations for any of our product candidates, including in connection with the agency’s consideration, or reconsideration, of our regulatory filings for our product candidates. Our decision to make a cash distribution in December 2013, resulted from the determination that we had surplus corporate cash, based on the decision not to undertake future development programs without a partner.
As part of our ongoing assessment of our business and liquidity needs, we regularly assess available funding options and will consider available funding opportunities as they arise. We consider our current royalty stream as cash assets that could be monetized to accelerate the expected cash flow. We also could sell shares of common stock in the future to fund additional development or pre-commercialization activities and increase our working capital. Any additional equity financing may be dilutive to stockholders, and debt financing, if available, may involve restrictive covenants.
Our business and operations model is evolving. On June 1, 2015, our Board appointed Adrian Adams as our new Chief Executive Officer and Andrew I. Koven as our new President and Chief Business Officer, each of whom has experience creating, leading and expanding pharmaceutical companies with marketing and sales capabilities. If the proposed business combination with Tribute is approved by our stockholders and Tribute’s shareholders and such transaction and concurrent equity and debt financings are closed, we will become a specialty pharmaceutical company focused on cardiovascular and pain therapies with a diversified portfolio of marketed products and developmental stage product candidates in the United States and Canada.
Our forecast of the period of time through which we expect that our financial resources will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary as a result of a number of factors. Our future capital requirements will depend on many factors, including:
· the number and progress of our clinical trials and other trials and studies;
· our success, or any delays, in obtaining, regulatory approval of our product candidates and success in, and manner of, commercializing our products;
· the success of our existing collaborations and our ability to establish additional collaborations;
· the extent to which we acquire or invest in businesses, technologies or products;
· costs incurred to enforce and defend our patent claims and other intellectual rights;
· costs incurred in the defense of our VIMOVO patent against generic companies that have filed ANDAs with the FDA to market the product prior to the expiration of our and AstraZeneca’s patents: and
· our ability to consummate the combination with Tribute and the proposed equity and debt financings.
Overview of Pozen
Except as otherwise noted, all references in the section “Business-Overview of Pozen” to “we”, “us”, “our”, or the “Company” refer to Pozen.
We are a pharmaceutical company focused on transforming medicines that can transform lives. Historically, we have operated a business model that has focused on the following:
· developing innovative products that address unmet medical needs in the marketplace;
· obtaining patents for those innovative ideas which we believe have value in the marketplace;
· utilizing a small group of talented employees to develop those ideas by working with strategic outsource partners;
· developing a regulatory pathway with the appropriate agency; and
· determining how best to commercialize our products.
The success of our business is highly dependent on the marketplace value of our ideas and the related patents we obtain, our ability to obtain approval to sell the developed products from the required regulatory agencies, and our ability to successfully commercialize our products. Under our earlier business model, we hired experts with strong project management skills in the specific disciplines we believed were important to maintain within our company. Historically, we contracted with and manage strong outsource partners as we complete the necessary development work, permitting us to avoid incurring the cost of buying or building laboratories, manufacturing facilities or clinical research operation sites. This allowed us to control our annual expenses, but to utilize “best in class” resources as required.
Our business and operations model is evolving. On June 1, 2015, our Board appointed Adrian Adams as our new Chief Executive Officer and Andrew I. Koven as our new President and Chief Business Officer, each of whom has experience creating, leading and expanding pharmaceutical companies with marketing and sales capabilities. If the proposed business combination with Tribute is approved by our stockholders and Tribute’s shareholders and such transaction and concurrent equity and debt financings are closed, we will become a specialty pharmaceutical company focused on cardiovascular and pain therapies with a diversified portfolio of marketed products and developmental stage product candidates in the United States and Canada.
We decided to retain ownership of our proprietary, investigational, coordinated-delivery tablets combining immediate-release omeprazole, a proton pump inhibitor, or PPI, and EC aspirin in a single tablet, now known as YOSPRALA, including PA8140 and PA32540 through the clinical development and pre-commercialization stage and our chief commercial officer was responsible for developing the commercialization strategy for these products and conducting all the required pre-commercialization activities. On September 3, 2013, we entered into an exclusive license agreement with sanofi-aventis U.S. LLC, or Sanofi U.S., for the commercialization YOSPRALA. On November 29, 2014, we executed a termination agreement with Sanofi U.S. terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi U.S. were terminated and all rights to the products licensed to Sanofi U.S. under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi U.S. relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
Overview of Our Lead Product Candidate, YOSPRALA
On September 3, 2013 we entered into an exclusive license agreement with Sanofi U.S., for the commercialization of PA8140 and PA32540, now known as YOSPRALA. Under the terms of the agreement, Sanofi U.S. had exclusive rights to commercialize all PA combinations that contain 325 mg or less of EC aspirin in the United States.
On April 25, 2014, we received a CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude the approval of the NDA in its then-current form. Specifically, an inspection of the manufacturing facility of an active ingredient supplier of ours concluded with certain inspection deficiencies. Satisfactory resolution of these deficiencies is required before the NDA may be approved. There were no clinical or safety deficiencies noted with respect to either PA8140 or PA32540 and no other deficiencies were noted in CRL. On June 30, 2014, we resubmitted the NDA for PA32540 and PA8140 to the FDA and the FDA notified us that the new action fee date is December 30, 2014. On May 9, 2014, our active ingredient supplier submitted a response to the FDA addressing the inspection deficiencies and subsequently submitted an update to its initial response. On November 29, 2014, we executed a termination agreement with Sanofi U.S. terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi U.S. were terminated and all rights to the products licensed to Sanofi U.S. under the agreement reverted to us. The termination agreement
further provides for the transfer of specified commercial know-how developed by Sanofi U.S. relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products.
On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. Satisfactory resolution of these deficiencies is required before this application may be approved. There were no clinical or safety deficiencies noted with respect to either PA32540 or PA8140 and no other deficiencies were noted in the CRL. Final agreement on the draft product labeling is also pending.
FDA regulations allowed us to request a Type A meeting with the FDA to discuss the next steps required to gain approval of our NDA. The FDA granted the Type A meeting, which was held in late January 2015. At the meeting, representatives from the FDA’s Office of Compliance stated that the active ingredient supplier’s responses to the 483 inspectional observations submitted in May 2014 were still under review and the Office of Compliance would be communicating with the supplier in the coming weeks. The active ingredient supplier has informed Pozen that they received a warning letter relating to the Form 483 inspection deficiencies. They have submitted a plan of corrective actions to address the matters raised in the warning letter to FDA. We have had continuing discussions with FDA about these issues and the impact on our NDA and with our active ingredient supplier regarding the corrective actions it is taking to address the inspectional observations at its facility. We are also working toward securing and seeking FDA’s approval for an alternative back up supplier.
In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
Treximet
We have previously developed Treximet® in collaboration with GlaxoSmithKline, or GSK. Treximet is the brand name for the product combining sumatriptan 85 mg, formulated with RT Technology™ and naproxen sodium 500 mg in a single tablet designed for the acute treatment of migraine. On April 15, 2008, the FDA approved Treximet for the acute treatment of migraine attacks with or without aura in adults. Upon receipt of FDA approval, GSK notified us of its intention to launch the product and Treximet was available in pharmacies in May 2008.
On November 23, 2011, we entered into a purchase and sale agreement, or the Purchase Agreement, with CPPIB Credit Investments Inc. or CII, pursuant to which we sold, and CII purchased, our right to receive future royalty payments arising from U.S. sales of MT 400, including Treximet. Under the Purchase Agreement, we received $75 million and will receive a twenty percent (20%) interest in any royalties received by CII relating to the period commencing on April 1, 2018.
On May 13, 2014, we, Glaxo Group Limited, d/b/a GlaxoSmithKline, or GSK, CII and Pernix entered into certain agreements in connection with GSK’s divestiture of all of its rights, title and interest to develop, commercialize and sell Treximet® in the U.S. to Pernix. Upon the closing of the divestiture on August 20, 2014, GSK assigned the Product Development and Commercialization Agreement executed as of June 11, 2003 between us and GSK, the Treximet Agreement, to Pernix. Immediately following the closing of the divestiture, Amendment No. 1 to the Treximet Agreement, or Amendment No.1, between us and Pernix became effective. Amendment No. 1, among other things, amends the royalty provisions of the Agreement to provide for a guaranteed quarterly minimum royalty payable to CII of $4 million for the calendar quarters commencing on January 1, 2015 and ending on March 31, 2018 and requires that Pernix continue certain of GSK’s ongoing development activities and to undertake certain new activities, for which we will provide reasonable assistance. Amendment No. 1 also eliminates restrictions in the Agreement on our right to develop and commercialize certain dosage forms of sumatriptan/naproxen combinations outside of the United States and permits POZEN to seek approval for these combinations on the basis of the approved NDA for Treximet. Pernix has also issued us a warrant to purchase 500,000 shares of Pernix common stock at an exercise price equal to $4.28, the closing price of Pernix common stock as listed on the NASDAQ Global Market on May 13, 2014. The common stock underlying the warrants will be registered by Pernix with the SEC and the warrant is exercisable from August 20, 2014, the closing date of the Divestiture, until February 28, 2018. Because the warrant has not been registered by Pernix with the SEC, the Company cannot sell or transfer the warrant in reliance upon Rule 144 until after November 13, 2014 when the Company meets certain holding requirements. The warrant is valued using the Black-Scholes valuation model. Under the terms of the warrant, the Company may elect to receive the number of shares equal to the value of the Pernix shares less $4.28 divided by the fair market value of one share. At December 31, 2014 this would have been 272,098 shares
valued at $2.6 million. Lastly, we, GSK, Pernix and CII executed a letter agreement whereby we expressly consented to the assignment by GSK and the assumption by Pernix of the Treximet Agreement. On July 30, the parties entered into Amendment No. 2 to the Treximet Agreement which will permit Pernix’s Irish affiliate to which Pernix will assigns its rights to further assign the Agreement without our prior written consent as collateral security for the benefit of the lenders which financed the acquisition. Amendment No. 2 became effective upon the closing of the divestiture on August 20, 2014.
VIMOVO
We have developed VIMOVO with AstraZeneca. VIMOVO (formerly referred to as PN 400) is the brand name for a proprietary fixed dose combination of the PPI esomeprazole magnesium with the NSAID naproxen in a single tablet. On April 30, 2010, the FDA approved VIMOVO for the relief of the signs and symptoms of OA RA and AS, and to decrease the risk of developing gastric ulcers in patients at risk of developing NSAID-associated gastric ulcers.
In August 2006, we entered into an exclusive global collaboration and license agreement with AstraZeneca to co-develop and commercialize VIMOVO, which agreement was amended in September 2007 and October 2008. We began the Phase 3 program in September 2007. As part of the program, we conducted two Phase 3 pivotal trials of VIMOVO in patients who are at risk for developing NSAID-associated gastric ulcers, the primary endpoint for which was the reduction in endoscopic gastric ulcers.
The NDA for VIMOVO was submitted on June 30, 2009 and was accepted for filing by FDA in August 2009. POZEN received a $10.0 million milestone payment from AstraZeneca in September 2009 for the achievement of such milestone. Earlier, in May 2010, we had received a $20.0 million milestone payment when we received FDA approval of VIMOVO. In October 2009, AstraZeneca submitted a MAA for VIMOVO in the EU via the DCP and has filed for approval in a number of other countries which are not covered by the DCP. On October 11, 2010, we announced with AstraZeneca that VIMOVO had received positive agreement for approval in 23 countries across the EU following all 22 Concerned Member States agreeing with the assessment of the MEB, acting as the Reference Member State for the DCP. We received a $25.0 million milestone payment when pricing and reimbursement for VIMOVO was granted in the United Kingdom.
On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understand that AstraZeneca will instead now focus on those countries where the product has shown growth and which AstraZeneca believes have the greatest potential for future growth.
On November 18, 2013, AstraZeneca and Horizon Pharma Inc., or “Horizon,” entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. As required by the terms of our agreement with AstraZeneca, we gave our consent to AstraZeneca’s divestiture of such rights to Horizon because we believed that Horizon’s expertise in commercializing products for pain and inflammatory disease, including a similar combination product containing a NSAID and a gastroprotective agent, make it an excellent partner to maximize the potential of VIMOVO in the United States. We were also able to negotiate a guaranteed annual minimum royalty in the United States in the amount of $5 million in calendar year 2014, and a guaranteed annual minimum royalty amount of $7.5 million each calendar year thereafter, provided that the patents owned by us which cover VIMOVO are in effect and no generic forms of VIMOVO are in the marketplace.
On July 28, 2014, Horizon announced that it had been verbally notified by CVS Caremark and Express Scripts, Inc. that VIMOVO would no longer be on their formularies and will be placed on their exclusion lists effective January 1, 2015. We have been informed that Horizon estimates that approximately 20-30% of VIMOVO prescriptions could be impacted by these decisions. While Horizon believes it has a strategy to mitigate the effect on VIMOVO sales, POZEN’s royalty revenue from Net Sales of VIMOVO beginning in 2015 may be negatively affected, although we will continue to receive a guaranteed annual minimum royalty of $7.5 million as described above.
Our Principal Product Candidates
Our PA product candidates, containing a PPI and aspirin, have completed clinical development testing in the United States. Our PA product candidates, now known as YOSPRALA), are
excluded from our agreement with AstraZeneca. We met with the FDA to discuss the overall development program requirements for PA32540 for the secondary prevention of cardiovascular and cerebrovascular disease in patients at risk for gastric ulcers. An investigational new drug application, or IND, was filed in the fourth quarter of 2007. We completed a study which demonstrated that the (SA) component of PA32540 was bioequivalent to the reference drug, EC aspirin. We filed a SPA with the FDA for the design of the Phase 3 studies for the product, the primary endpoint for which is the reduction in the cumulative incidence of endoscopic gastric ulcers.
Based upon the FDA’s earlier confirmation that endoscopic gastric ulcers were an acceptable primary endpoint, in October 2009, we began two pivotal Phase 3 and one long-term safety study for PA32540 for the cardiovascular indication. The primary endpoint of the pivotal studies, which included approximately 500 subjects per study, was a significant reduction in the cumulative incidence of gastric ulcers following administration of PA32540 vs. 325 mg EC aspirin over the six-month treatment period. The primary endpoint was met in both studies. Additionally, the studies met their key secondary endpoints, including a reduction in gastroduodenal ulceration as well as a reduction in discontinuation due to upper gastrointestinal adverse events in subjects taking PA32540 compared to 325 mg EC aspirin.
In February 2012, the FDA requested an additional Phase 1 study to assess the bioequivalence of PA32540 to EC aspirin 325 mg with respect to ASA. After the Company completed the requested bioequivalence study, the FDA made a preliminary review of the study results and the Company’s summary analyses and, based on its preliminary assessment of the information available to it at the time, the FDA did not agree that bioequivalence of PA32540 to EC aspirin 325 mg was demonstrated. The Company then submitted to the FDA additional information and analyses from the requested bioequivalence study, as well as other relevant pharmacokinetic, clinical pharmacology, and in vitro dissolution data as a Briefing Document in support of a request for a Type A meeting with the FDA. At the Type A meeting held in August 2012, the FDA confirmed that, although it believes bioequivalence of PA32540 to EC aspirin 325 mg, was not strictly established in our bioequivalence study according to the predetermined criteria, the results from this study, together with additional information that will be submitted by the Company in the NDA, constitutes sufficient data to support the establishment of a clinical and pharmacological bridge between the product and EC aspirin 325 mg. The FDA indicated that it would make a final determination during the NDA review. The FDA also indicated that a similar strategy to bridge to the reference listed drug, inclusive of a new, single pharmacokinetic study, could be utilized for a low dose version of PA32540 (currently PA8140). The Company conducted this study with the low dose version against the EC aspirin 81mg. Based on the predetermined criteria acceptable to the FDA, the study demonstrated that PA8140 is bioequivalent to EC aspirin 81mg using criteria for highly variable drugs and had comparable bioavailability.
During a pre-submission meeting with respect to its NDA for PA32540 in April 2012, the FDA suggested that the Company also seek approval for a lower dose formulation of the product containing 81 mg of EC aspirin as part of its NDA for PA32540. Absent the availability of such a lower dose formulation in the market if PA32540 is approved, the FDA indicated that it may limit the indication for PA32540 to use in CABG with treatment duration not to exceed one year. During the Type A meeting held in August 2012, the FDA confirmed its preference to have both PA32540 and a lower dose version available in the market so that physicians can have both a low and high dose option available, and agreed that, if both dosage strengths were included in the NDA and subsequently approved, the indications for both will be consistent with the full range of indications described in the current aspirin monograph.
We had generated some clinical pharmacology data and CMC data for a product which contains 81 mg of EC aspirin and 40 mg of omeprazole in a single tablet known as PA8140. The Company filed this existing data, together with additional CMC data to be generated and evidence from the scientific literature relating to the ulcerogenic risk of 81 mg of aspirin with the FDA. At this time, we do not intend to conduct Phase 3 clinical trials for PA8140. The data package submitted for PA8140 was similar to that used to gain approval for a lower dosage form of VIMOVO containing 375 mg of naproxen. We have no assurance such data will be sufficient for the FDA to approve PA8140 or to allow a broader indication for PA32540. The FDA will make a final determination with respect to the approvability of and indications for PA32540 and PA8140.
Generation of additional data with respect to PA8140 and incorporation of data into the NDA for PA32540 delayed submission of the NDA from the original planned submission date in the third quarter of 2012. The NDA was filed for both products in March 2013 and in May 2013 the FDA accepted the NDA for review. The FDA assigned a user fee date of January 24, 2014. As part of our continuing discussions with the FDA concerning the NDA for PA32540 and PA8140 tablets, we decided to conduct an additional comparative Phase 1 pharmacokinetic study to determine the pharmacokinetic profile of the omeprazole component of PA 8140 tablets and compare it to that of PA32540 tablets. We submitted study information and data to the FDA as it became available during the conduct of the study and FDA reviewed such information and data from the study when submitted. The final study report for the study was submitted to the FDA in accordance with our agreed timeline. FDA informed us that the Company’s user fee date was April 25, 2014. On April 25, 2014, we received a CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude the approval of the NDA in its current form. Specifically, an inspection of the manufacturing facility of an active ingredient supplier of ours concluded with certain inspection deficiencies. Satisfactory resolution of these deficiencies is required before the NDA may be approved. There were no clinical or
safety deficiencies noted with respect to either PA8140 or PA32540 and no other deficiencies were noted in CRL. On June 30, 2014, we resubmitted the NDA for PA32540 and PA8140 to the FDA and the FDA notified us that the new action fee date is December 30, 2014. On May 9, 2014, our active ingredient supplier submitted a response to the FDA addressing the inspection deficiencies and subsequently submitted an update to its initial response. On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. Satisfactory resolution of these deficiencies is required before this application may be approved. There were no clinical or safety deficiencies noted with respect to either PA32540 and PA8140 and no other deficiencies were noted in the CRL. Final agreement on the draft product labeling is also pending. We continue to assist the FDA compliance division with their review. FDA regulations allow us to request a Type A meeting with the FDA to discuss the next steps required to gain approval of our NDA. The FDA granted the Type A meeting which was held in late January 2015. At the meeting, representatives from the FDA’s Office of Compliance stated that the active ingredient supplier’s responses to the 483 inspectional observations submitted in May 2014 were still under review and the Office of Compliance would be communicating with the supplier in the coming weeks. The active ingredient supplier has informed POZEN that they received a warning letter relating to the Form 483 inspection deficiencies. They are evaluating what additional corrective actions may be required to address the matters raised in the warning letter. We will continue to provide assistance to our active ingredient supplier in taking corrective actions to address the inspectional observations at its facility.
To assess whether a similar interaction occurs between clopidogrel and PA32540, which contains immediate release omeprazole, we completed two Phase 1 drug-drug interaction studies to evaluate the ex-vivo platelet aggregation effects of PA32540 plus clopidogrel. In the first study, we evaluated ex-vivo platelet aggregation of PA32540 plus clopidogrel when dosed at the same time or dosed 10 hours apart compared to aspirin 325 mg plus clopidogrel dosed together. When PA32540 and clopidogrel were dosed together, data from the study showed a mean 36.7% platelet inhibition compared to a mean 44.0% platelet inhibition when aspirin and clopidogrel were dosed together suggesting a drug-drug interaction based on the study’s pre-specified primary analysis. When PA32540 and clopidogrel were dosed 10 hours apart, data from the study indicate no ex-vivo drug-drug interaction based on the study’s pre-specified primary analysis. In the second Phase 1 study, we evaluated ex-vivo platelet aggregation of PA32540 plus clopidogrel dosed 10 hour apart compared to a current standard of care of Plavix + Prilosec 40 mg + EC ASA 81 mg dosed together. Subjects on PA32450 demonstrated significantly greater inhibition of platelet aggregation than subjects on standard of care. The clinical relevance of these ex vivo platelet data on cardiovascular events is not known. No further Phase 1 studies on the clopidogrel-PPI interaction are planned. FDA assessment of available data on the drug-drug interaction may result in the inclusion of a warning, similar to that in the current Prilosec label, against the concomitant use of PA32540 and Plavix.
We are also continuing to evaluate how best to commercialize the PA product candidates and programs. We are evaluating the regulatory requirements to obtain an indication for PA for the secondary prevention of colorectal neoplasia. In January 2010, we received input from the FDA with respect to the development requirements for a possible indication in colorectal neoplasia. Further discussions are being considered. We are also evaluating the possibility of developing another dosage strength of PA containing 650 mg of EC aspirin and 20 mg of omeprazole (PA65020) for the treatment of OA and similar conditions and we met with the FDA in December 2010 to obtain input with respect to the regulatory requirements to obtain such an indication. The FDA advised us that we will need to demonstrate a reduction in gastric ulcers compared to aspirin alone in a replicate Phase 3 program similar to that we performed for VIMOVO, in addition to a study to establish efficacy of the product in the treatment of OA.
We met with a European country that served as the reference member state for approval of VIMOVO in Europe to obtain guidance on a clinical development program for approval of PA65020 in the Europe. We were advised that Phase 3 endoscopic trials demonstrating a reduction in gastric ulcers would not be required in addition to an OA efficacy study since omeprazole’s actions on gastric ulcers has been well characterized. Instead, Phase 1 studies assessing pharmacokinetics and gastric acid suppression could be used to support the benefit of omeprazole as a component of PA65020.
We have also received Scientific Advice from the Medicines Evaluation Board, or MEB in the Netherlands, which has agreed to be the Reference Member State in a decentralized filing procedure, regarding the development program required for the approval in the EU of PA tablets, including a lower dosage form containing 100 mg of aspirin and 40 mg of omeprazole (PA10040) for the secondary prevention of cardiovascular disease. The MEB agreed that a full Phase 3 clinical development program for PA10040, to demonstrate the reduction of endoscopic gastric ulcers vs. EC aspirin, would not be necessary. Instead, a Phase 1 pharmacodynamic study comparing gastric pH control for PA10040 vs.EC omeprazole 20 mg, along with a study to demonstrate bioequivalence of PA10040 to a currently marketed EC aspirin product using ASA as the analyte would be sufficient. Study PA10040-101 demonstrated that PA10040 had comparable bioavailability and is bioequivalent to an EU reference listed EC aspirin 100 mg. Study PA10040-102 demonstrated that PA10040 provides gastric pH greater than 4 control for a percent time over 24 hours of 47%. Furthermore, compared to Losec 20 mg (EC omeprazole 20 mg), PA10040 produced a similar level of 24-hour pH control (p=0.02).
A pre-submission follow up meeting with the MEB was held in July 2014. We proposed seeking approval of PA10040 only since the ASA component in that dosage form matches the clinical practice for use of ASA in the EU. Approval of PA10040 would be
based on specific studies conducted with the product, as well as data from the entire PA program conducted in the United States. Based on discussions at the meeting, the MEB agreed that the MAA filing can proceed without any further clinical studies and that the Netherlands would be the lead country for the decentralized review process. The MEB also requested that a complete risk-benefit analysis of PA10040 for the intended cardiovascular patient population and proposed indication should be included as part of the MAA submission.
We have incurred significant losses since our inception and have not yet generated significant revenue from product sales. As of December 31, 2014, our accumulated deficit was approximately $96.9 million. We record revenue under the following categories: royalty revenues and licensing revenues. Our licensing revenues include upfront payments upon contract signing, additional payments if and when certain milestones in the product’s development or commercialization are reached, while the royalty payments are based on product sales. Our historical operating losses have resulted principally from our research and development activities, including clinical trial activities for our product candidates and sales, general and administrative expenses. Research and development expenses include salaries and benefits for personnel involved in our research and development activities and direct development costs, which include costs relating to the formulation and manufacturing of our product candidates, costs relating to preclinical studies, including toxicology studies, and clinical trials, and costs relating to compliance with regulatory requirements applicable to the development of our product candidates. Since inception, our research and development expenses have represented approximately 64% of our total operating expenses. For the fiscal year ended December 31, 2014, our research and development expenses represented approximately 36% of our total operating expenses.
Operating losses may be incurred over the next several years as we complete the development and seek regulatory approval for our product candidates, develop other product candidates, conduct pre-commercialization activities, and acquire and/or develop product portfolios in other therapeutic areas. Our results may vary depending on many factors, including:
· The progress of our PA product candidates and our other product candidates in the clinical and regulatory process;
· The ability of Horizon and AstraZeneca to successfully commercialize VIMOVO in the United States and outside the United States, respectively, and our ability to successfully commercialize our PA product candidates in the United States;
· The establishment of new collaborations and progress and/or maintenance of our existing collaborations for the development and commercialization of any of our product candidates;
· Our ability to successfully defend our regulatory market exclusivity and patent rights against generic challenges and to succeed in obtaining extensions of such exclusivity for which we may be eligible;
· Our ability to commercialize our products with commercial partners in a highly regulated and extremely competitive marketplace; and
· The possible acquisition and/or in-licensing, and development of our therapeutic product candidates.
We do not currently have internal commercialization or manufacturing capabilities. We have entered into collaborations and may continue to enter into additional collaborations with established pharmaceutical or pharmaceutical services companies to commercialize and manufacture our product candidates once approved. We decided to retain control of our PA product candidates for cardiovascular indications through the clinical development and pre-commercialization stage. To that end, our chief commercial officer evaluated the commercial opportunities for these product candidates and developed a worldwide commercial strategy, which enabled us to conduct pre-commercialization activities prior to licensing these products to commercial partners. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
Our ability to generate revenue in the near term is dependent upon our ability, alone or with collaborators, to achieve the milestones set forth in our collaboration agreements, to enter into additional collaboration agreements, to successfully develop product candidates, to obtain regulatory approvals and to successfully manufacture and commercialize our future products. These milestones are earned when we have satisfied the criteria set out in our revenue recognition footnote accompanying the financial statements included in our Annual Report on Form 10-K, filed with the SEC on March 6, 2014, as amended by Amendment No. 1 to our Annual Report on Form 10-K/A, filed with the SEC on September 22, 2014. These payments generate large non-recurring revenue that will cause large fluctuations in quarterly and annual profit and loss.
Arthritis Market Overview
Arthritis means joint inflammation and the term is used to describe the pain, stiffness and/or swelling in the joints of the body where one or more bones are joined by tendons and muscles. An arthritic joint is one that may have varying degrees of inflammation and possibly destruction of the joint cartilage, which normally provides a smooth surface enabling adjacent bones to move and glide on each other during normal motion.
The most common type of arthritis is called OA and is more common with advancing age. OA is one of the most frequent causes of physical disability among adults. It is estimated that by 2030, 20% of Americans who are over the age of 65 years, or approximately 70 million people, will be at risk for OA. People with OA usually have joint pain and limited movement. Unlike some other forms of arthritis, OA affects only the joints. This condition is also sometimes called degenerative joint disease. OA primarily affects the joint cartilage. Healthy cartilage allows bones to glide over one another and absorbs energy from the shock of physical movement. However, with OA, the surface layer of cartilage breaks down and wears away. This allows the bony surface under the cartilage to rub together, causing, pain, swelling, and loss of motion of the joint. Over time, affected joints may lose their normal shape. Also, bone spurs, small growths called osteophytes, may grow on the edges of the joint. Thus bits of bone or cartilage can break off and float inside the joint space, causing more pain and possible damage.
The second most common form of arthritis, RA, may affect not only the joints, but organs of the body as well. RA is recognized as a systemic disease that involves responses of the immune system that play a role in the inflammation that affects joints and other organs. RA may begin at a younger age than does OA. Often patients with RA will require medications not only to treat the pain of arthritis, but drugs which modulate the immune system to control inflammation in other parts of the body.
Non-steroidal anti-inflammatory drugs, or NSAIDs, both over-the-counter (“OTC”) and prescription, are commonly taken to manage the pain of backache, OA, RA, headache and other painful conditions. In 2012, approximately 100 million prescriptions were dispensed for oral anti-arthritis NSAIDs for the management of pain. Prescription sales of oral anti-arthritis NSAIDs in the U.S. in 2011 were approximately $3.0 billion. In spite of their widespread use and apparent safety, according to the Agency for Healthcare Research and Quality Statistical Brief released in December 2008, in 2006, there were approximately 16,300 deaths and 500,000 hospitalizations with a primary diagnosis of upper gastrointestinal, or GI, bleeding costing approximately $2 billion. The most common underlying conditions of GI bleeding were gastric, duodenal, peptic, or gastroduodenal ulcers or perforations, conditions frequently associated with NSAID use. We are responding to this unmet medical need to provide a “safer NSAID” through development of our PN product candidates for the treatment of conditions such as OA in patients who are at risk for developing NSAID-associated gastric ulcers.
PN Program
Under our PN program, we completed formulation development and clinical studies for several combinations of a PPI and a NSAID in a single tablet intended to provide effective management of pain and inflammation associated with chronic conditions such as OA, and intended to have fewer gastrointestinal complications compared to a NSAID taken alone in patients at risk for developing NSAID associated gastric ulcers. We entered into an exclusive ,worldwide (except for Japan) collaboration agreement with AstraZeneca on August 1, 2006 and which was amended in September 2007 and October 2008 relating to the development and commercialization of our PN products. Our agreement with AstraZeneca covered the development and commercialization of proprietary fixed dose combinations of the PPI esomeprazole magnesium with the NSAID naproxen in a single tablet. The initial product developed under the agreement, VIMOVO (formerly PN 400), was approved by the FDA on April 30, 2010 for the relief of the signs and symptoms of OA, RA and AS and to decrease the risk of developing gastric ulcers in patients at risk of developing NSAID-associated gastric ulcers.
The NDA for VIMOVO was submitted on June 30, 2009 and was accepted for filing in August 2009. We received a $10.0 million milestone payment from AstraZeneca in September 2009 for the achievement of such milestone. On April 30, 2010, VIMOVO was approved by FDA for the relief of the signs and symptoms of OA, RA and AS and to decrease the risk of developing gastric ulcers in patients at risk of developing NSAID-associated gastric ulcers. We received a $20.0 million milestone payment from AstraZeneca in May 2010 in connection with such approval. As required by the terms of our agreement with AstraZeneca, we transferred ownership of the NDA and other regulatory filings for VIMOVO to AstraZeneca on June 1, 2010, and AstraZeneca now has responsibility for all ongoing regulatory obligations for the product in the U.S., including post marketing clinical trial requirements, in addition to responsibility for all regulatory obligations outside the U.S.
Under our agreement with AstraZeneca, AstraZeneca had responsibility for the development program for PN products outside the U.S., including interactions with regulatory agencies. In October 2009, AstraZeneca submitted a MAA for VIMOVO in the EU via the DCP and has filed for approval in a number of other countries which are not covered by the DCP. On October 11, 2010, we announced with AstraZeneca that VIMOVO had received positive agreement for approval in 39 countries across the EU following all 22 Concerned Member States agreeing with the assessment of the Netherlands Health Authority, acting as the Reference Member State for the DCP. We received a $25.0 million milestone payment when pricing and reimbursement for VIMOVO was
granted in the United Kingdom. Other Member States are now pursuing pricing and reimbursement and national approvals. Earlier, in May 2010, we had received a $20.0 million milestone payment when we received FDA approval of VIMOVO. As of the end of December 31, 2013, VIMOVO has been filed for regulatory approval in 81 countries and approved in 71 countries.
On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understand that AstraZeneca will instead now focus on those countries where the product has shown growth and which AstraZeneca believes have the greatest potential for future growth. We continue to assess the financial impact this decision will have on our royalty revenue. On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. As required by the terms of our agreement with AstraZeneca, we gave our consent to AstraZeneca’s divestiture of such rights to Horizon because we believed that Horizon’s expertise in commercializing products for pain and inflammatory disease, including a similar combination product containing a NSAID and a gastroprotective agent, make it an excellent partner to maximize the potential of VIMOVO in the United States. We were also able to negotiate a guaranteed annual minimum royalty in the United States in the amount of $5.0 million in calendar year 2014, and a guaranteed annual minimum royalty amount of $7.5 million each calendar year thereafter, provided that the patents owned by us which cover VIMOVO are in effect and no generic forms of VIMOVO are in the marketplace. On July 28, 2014, Horizon announced that it had been verbally notified by CVS Caremark and Express Scripts, Inc. that VIMOVO would no longer be on their formularies and will be placed on their exclusion lists effective January 1, 2015. We have been informed that Horizon estimates that approximately 20-30% of VIMOVO prescriptions could be impacted by these decisions. While Horizon believes it has a strategy to mitigate the effect on VIMOVO sales, POZEN’s royalty revenue from Net Sales of VIMOVO beginning in 2015 may be negatively affected, although we will continue to receive a guaranteed annual minimum royalty of $7.5 million as described above.
Since inception we have incurred total direct development cost of $96.2 million associated with the development of our PN program of which $57.1 million was funded by development revenue from AstraZeneca. Our direct development costs do not include the cost of research and development personnel or any allocation of our overhead expense.
Cardiovascular Market Overview
Cardiovascular disease, or CVD, is a broad term used to describe a range of common diseases that affect the heart or blood vessels. Many common conditions fall under the definition of CVD, including coronary artery disease, heart attack, heart failure, high blood pressure and stroke. In fact, the term “cardiovascular disease” is often used interchangeably with heart disease because both terms refer to diseases of the heart of arteries. Despite recent advances in medical research, cardiovascular disease, including heart attack and stroke is still the leading killer of men and women in the United States. It is also the most costly cause of death in men and women in the United States, according to the American Heart Association, or AHA.
An estimated 80 million American adults, or one in three, have one or more types of CVD, and 24 million have been identified as secondary prevention patients (post-event patients who have suffered one or more cardiovascular or cerebrovascular events). It is estimated that CVD causes one in every three deaths in the United States. Approximately every 25 seconds, someone in the United States suffers a coronary event with one related to death each minute.
Coronary artery disease is caused by atherosclerosis and often develops into angina pectoris and myocardial infarction (MI). The condition caused about 375,000 deaths in 2011 and remains the leading single cause of death in America today. Roughly 15.4 million have a history of MI and/or angina.
This year, approximately 620,000 American will have a new coronary attack, and approximately 295,000 will have a recurrent attack. It is estimated that an additional 150,000 silent myocardial incidents occur each year. Each year, approximately 795,000 people experience a new or recurrent stroke. Approximately 610,000 of these are first heart attacks, and 185,000 are recurrent attacks. On average, every 40 seconds, someone in the United States has a stroke. Direct and indirect costs related to the condition are projected to exceed $163 billion annually.
Aspirin therapy has become the standard of care for reducing an individual’s risk of a second heart attack or stroke. Studies have found that a daily aspirin regimen for people who have experienced a previous heart attack reduces the risk of a second heart attack by about one-third. Aspirin has been incorporated into the American Heart Association’s clinical guidelines for the secondary prevention of cardiovascular events. In accordance with these guidelines, approximately 24 million Americans should be taking aspirin for secondary prevention of cardiovascular events.
Although the CVD benefits of aspirin are well established, the use of aspirin is associated with the risk of upper gastrointestinal bleeding, or, UGIB. The use of aspirin is associated with a 2- to 4- fold increased risk of UGIB. In addition, aspirin use for CVD is an important cause of gastrointestinal bleeding-related death. The use of the proton pump inhibitors, or PPIs, such as
omeprazole can significantly reduce the risk of upper gastrointestinal bleeding. The American College of Cardiology with the AHA issued a Clinical Expert Consensus in 2008 recommending PPIs as preferred agents for the therapy and prophylaxis of aspirin-associated gastrointestinal injury.
PA Program
As part of our PA program, we are developing a PPI and aspirin in a single tablet. Similar to the PN program, our PA product candidate is intended to induce fewer gastrointestinal complications compared to an aspirin taken alone in patients at risk for developing aspirin associated gastric ulcers. Our PA product candidates are covered under the same patent as PN, but we have retained all rights to this program through the clinical development and pre-commercialization stage. On September 3, 2013 we entered into a License and Collaboration Agreement with Sanofi U.S. to commercialize our PA product candidates containing an immediate release proton pump inhibitor and 325 mg or less of delayed release or EC aspirin in the United States. On November 29, 2014, we executed a termination agreement with Sanofi U.S. terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi U.S. were terminated and all rights to the products licensed to Sanofi U.S. under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi U.S. relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
Our PA product candidates, PA32540 and PA8140, have completed clinical development testing in the United States. Based upon the FDA’s earlier confirmation that endoscopic gastric ulcers were an acceptable primary endpoint, in October 2009, we began two pivotal Phase 3 and one long-term safety study for PA32540 for the cardiovascular indication. The primary endpoint was met in both studies. Additionally, the studies met their key secondary endpoints, including a reduction in gastroduodenal ulceration as well as a reduction in discontinuation due to upper gastrointestinal adverse events in subjects taking PA32540 compared to 325 mg EC aspirin.
In February 2012, the FDA requested an additional Phase 1 study to assess the bioequivalence of PA32540 to EC aspirin 325 mg with respect to ASA. After the Company completed the requested bioequivalence study, the FDA has made a preliminary review of the study results and the Company’s summary analyses and, based on its preliminary assessment of the information available to it at the time, the FDA did not agree that bioequivalence of PA32540 to EC aspirin 325 mg was demonstrated. We then submitted to the FDA additional information and analyses from the requested bioequivalence study, as well as other relevant pharmacokinetic, clinical pharmacology, and in vitro dissolution data as a briefing document in support of a request for a Type A meeting with the FDA. At the Type A meeting held in August 2012, the FDA confirmed that, although it believes bioequivalence of PA32540 to EC aspirin 325 mg, was not strictly established in our bioequivalence study according to the predetermined criteria, the results from this study, together with additional information that will be submitted by us in the NDA, constitutes sufficient data to support the establishment of a clinical and pharmacological bridge between the product and EC aspirin 325 mg. The FDA indicated that it would make a final determination during the NDA review. FDA also indicated that a similar strategy to bridge to the reference listed drug, inclusive of a new, single pharmacokinetic study, could be utilized for a low dose version of PA32540 (PA8140). We have conducted this study with the low dose version against the EC aspirin 81 mg. Based on the predetermined criteria acceptable to the FDA, the study demonstrated that PA8140 is bioequivalent to EC aspirin using criteria for highly variable drugs and had comparable bioavailability.
During a pre-submission meeting with respect to its NDA for PA32540 in April 2012, the FDA suggested that the Company also seek approval for a lower dose formulation of the product containing 81 mg of EC aspirin as part of its NDA for PA32540. We intended to seek an indication for the secondary prevention of cardiovascular disease in patients at risk for gastric ulcers. During the Type A meeting held in August 2012, the FDA has confirmed its preference to have both PA32540 and a lower dose version available in the market so that physicians can have both a low and high dose option available, and agreed that, if both dosage strengths were included in the NDA and subsequently approved, the indications for both will be consistent with the full range of indications described in the current aspirin monograph.
We had generated some clinical pharmacology data and, CMC data for a product which contains 81 mg of EC aspirin and 40 mg of omeprazole in a single tablet known as PA8140. We filed this existing data, together with additional CMC data to be generated and evidence from the scientific literature relating to the ulcerogenic risk of 81 mg of aspirin with the FDA. At this time, the Company does not intend to conduct Phase 3 clinical trials for PA8140. The data package submitted for PA8140 was similar to that used to gain approval for a lower dosage form of VIMOVO containing 375 mg of naproxen. We have no assurance such data will be sufficient for the FDA to approve PA8140 or to allow a broader indication for PA32540. The FDA will make a final determination with respect to the approvability of and indications for PA32540 and PA8140.
Generation of additional data with respect to PA8140 and incorporation of data into the NDA for PA32540 delayed submission of the NDA from the original planned submission date in the third quarter of 2012. The NDA was filed for both products in March 2013 and in May 2013 the FDA accepted the NDA for review. The FDA assigned a user fee date of January 24, 2014. As part of our continuing discussions with the FDA concerning the NDA for PA32540 and PA8140 tablets, we decided to conduct an additional comparative Phase 1 pharmacokinetic study to determine the pharmacokinetic profile of the omeprazole component of PA8140 tablets and compare it to that of PA32540 tablets. We submitted study information and data to the FDA as it became available during the conduct of the study and FDA agreed to review such information and data from the study when submitted. The final study report for the study was submitted to the FDA in accordance with our agreed timeline. FDA has informed us that the Company’s user fee date was April 25, 2014. On April 25, 2014, we received a CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude the approval of the NDA in its current form. Specifically, an inspection of the manufacturing facility of an active ingredient supplier of ours concluded with certain inspection deficiencies. Satisfactory resolution of these deficiencies is required before the NDA may be approved. There were no clinical or safety deficiencies noted with respect to either PA8140 or PA32540 and no other deficiencies were noted in CRL. On June 30, 2014, we resubmitted the NDA for PA32540 and PA8140 to the FDA and the FDA notified us that the new action fee date was December 30, 2014. On May 9, 2014, our active ingredient supplier submitted a response to the FDA addressing the inspection deficiencies and subsequently submitted an update to its initial response. On December 17, 2014, we received a second CRL from the FDA advising that the review of our NDA is completed and questions remain that preclude approval of the NDA in its current form. In this CRL, the FDA used identical wording to that of the first CRL. Satisfactory resolution of these deficiencies is required before this application may be approved. There were no clinical or safety deficiencies noted with respect to either PA32540 and PA8140 and no other deficiencies were noted in the CRL. Final agreement on the draft product labeling is also pending. FDA regulations allow us to request a Type A meeting with the FDA to discuss the next steps required to gain approval of our NDA. The FDA granted the Type A meeting, which was held in late January 2015. At the meeting, representatives from the FDA’s Office of Compliance stated that the active ingredient supplier’s responses to the 483 inspectional observations submitted in May 2014 were still under review, but that the review had been placed on a fast track and the Office of Compliance would be communicating with the supplier in the coming weeks. The active ingredient supplier has informed POZEN that they received a warning letter relating to the Form 483 inspection deficiencies. They are evaluating what additional corrective actions may be required to address the matters raised in the warning letter. We will continue to provide assistance to our active ingredient supplier in taking corrective actions to address the inspectional observations at its facility.
We are also continuing to evaluate how best to commercialize the PA product candidates and programs. We are evaluating the regulatory requirements to obtain an indication for PA for the secondary prevention of colorectal neoplasia. In January 2010, we received input from the FDA with respect to the development requirements for a possible indication in colorectal neoplasia. Further discussions are being considered. We are also evaluating the possibility of developing another dosage strength of PA containing 650 mg of EC aspirin and 20 mg of omeprazole (PA65020) for the treatment of OA and similar conditions and we met with the FDA in December 2010 to obtain input with respect to the regulatory requirements to obtain such an indication. The FDA advised us that we will need to demonstrate a reduction in gastric ulcers compared to aspirin alone in a replicate Phase 3 program similar to that we performed for VIMOVO, in addition to a study to establish efficacy of the product in the treatment of OA.
We also met with a European country that served as the reference member state for approval of VIMOVO in Europe to obtain guidance on a clinical development program for approval of PA65020 in the Europe. We were advised that Phase 3 endoscopic trials demonstrating a reduction in gastric ulcers would not be required in addition to an OA efficacy study since omeprazole’s actions on gastric ulcers has been well characterized. Instead, Phase 1 studies assessing pharmacokinetics and gastric acid suppression could be used to support the benefit of omeprazole as a component of PA65020.
We have also received scientific advice from the MEB in the Netherlands, which has agreed to be the Reference Member State in a decentralized filing procedure, regarding the development program required for the approval in the EU of PA tablets, including a lower dosage form containing 100 mg of aspirin and 40 mg of omeprazole (PA10040) for the secondary prevention of cardiovascular disease. The MEB agreed that a full Phase 3 clinical development program for PA10040, to demonstrate the reduction of endoscopic gastric ulcers vs. EC aspirin, would not be necessary. Instead, a Phase 1 pharmacodynamic study comparing gastric pH control for PA10040 vs. EC omeprazole 20 mg, along with a study to demonstrate bioequivalence of PA10040 to a currently marketed EC aspirin product using ASA as the analyte would be sufficient. Study PA10040-101 demonstrated that PA10040 had comparable bioavailability and is bioequivalent to an EU reference listed EC aspirin 100 mg. Study PA10040-102 demonstrated that PA10040 provides gastric pH greater than 4 control for a percent time over 24 hours of 47%. Furthermore, compared to Losec 20 mg (EC omeprazole 20 mg), PA10040 produced a similar level of 24-hour pH control ((p=0.02).
A pre-submission follow up meeting with the MEB was held in July 2014. We proposed seeking approval of PA10040 only since the ASA component in that dosage form matches the clinical practice for use of ASA in the EU. Approval of PA10040 would be based on specific studies conducted with the product, as well as data from the entire PA program conducted in the United States. Based on discussions at the meeting, the MEB agreed that the MAA filing can proceed without any further clinical studies and that the
Netherlands would be the lead country for the decentralized review process. The MEB also requested that a complete risk-benefit analysis of PA10040 for the intended cardiovascular patient population and proposed indication should be included as part of the MAA submission.
We cannot reasonably estimate or know the amount or timing of the costs necessary to continue development and/or complete the development of any PA product candidates we may seek to develop or when, if and to what extent we will receive cash inflows from any PA products. The additional costs that may be incurred include expenses relating to clinical trials and other research and development activities and activities necessary to obtain regulatory approvals. We have refined our strategy and decided to retain control of our PA product candidates for cardiovascular indications through the clinical development and pre-commercialization stage and then seek strong commercial partners to maximize the potential of these product candidates. On September 3, 2013 we entered into a License and Collaboration Agreement with Sanofi U.S. to commercialize our PA product candidates containing an immediate release proton pump inhibitor and 325 mg or less of delayed release or EC aspirin in the United States. Even though the License and Collaboration Agreement was terminated on November 29, 2014, we believe we were able to negotiate more favorable terms with Sanofi U.S. for rights to commercialize the products in the United States than we had licensed the product candidates at an earlier stage in development and will be able to achieve more favorable terms with other partners outside the United States if we are successful in licensing PA products in other territories in the future.
We have incurred direct development costs associated with the development of our PA program of $3.2 million during the fiscal year ended December 31, 2014. Since inception we incurred total direct development cost of $74.7 million associated with the development of our PA program. Our direct development costs do not include the cost of research and development personnel or any allocation of our overhead expenses.
Migraine Market Overview
Migraine is characterized by recurring attacks of throbbing headache pain, often associated with visual, auditory or gastrointestinal disturbances. Attacks range from mild to severe and can last from 4 hours to 72 hours. In the most severe attacks, migraine sufferers are unable to pursue basic daily activities. According to the American Council for Headache Education, migraines afflict 25 million to 30 million people in the U.S. alone. As many as 6% of all men and up to 18% of all women experience a migraine headache at some time in their life. While the precise mechanism of migraine is unknown, researchers believe migraine attacks are caused by acute inflammation surrounding selected blood vessels in the head. The average migraine sufferer experiences the first attack during the early teen years, and the attacks generally continue throughout adulthood.
Not all migraine attacks are of the same severity. Consequently, a variety of oral, injectable, and intranasal therapies are used to treat different types of migraine attacks. Many patients use a personal, individually developed, step-care approach to treat their attacks. Attacks are often treated initially with simple OTC analgesics, particularly if the patient is unable to determine if the attack is a migraine or some other type of headache. If OTC remedies are unsuccessful, patients often turn to more potent prescription drugs, including narcotics, analgesic/narcotic drug combinations and triptans.
Triptans are the family of drugs most commonly prescribed for the treatment of migraine attacks. Triptans have demonstrated the ability to treat migraines by constricting blood vessels in the brain. Although triptans can be effective in treating migraine symptoms, they are often associated with significant side effects and other disadvantages that include:
· the occurrence of cardiovascular related events, including chest pain/discomfort, throat discomfort and warm/cold sensations;
· the potential for other serious cardiovascular events, including death;
· difficulty in producing sustained benefits with a single dose in a majority of patients;
· the occurrence of nausea and dizziness during treatment; and
· the need for cardiovascular evaluations from physicians before initially prescribing triptans to patients with cardiovascular disease risk factors.
Despite these shortcomings, according to IMS Health’s IMS National Sales Perspective™, or IMS, in 2011 total triptan sales in the U.S. were approximately $1.7 billion. Sumatriptan is the leading triptan product. There are currently three types of sumatriptan formulations commercially available: oral, intranasal and injectable. An oral triptan is often the physician’s first choice as a prescription treatment for migraine pain. Intranasal triptans are often prescribed for patients requiring faster relief than oral drugs can provide or who cannot take oral medications. For the most severe attacks, patients sometimes use an injectable form of a triptan.
MT 400/Treximet
On April 15, 2008, the FDA approved Treximet for the acute treatment of migraine attacks with or without aura in adults. GSK notified us of its intention to launch the product and Treximet was available in pharmacies in May 2008. As part of our NDA program for Treximet, we conducted five Phase 1 trials, two Phase 3 pivotal trials, and one 12-month open label safety trial using a
formulation of Treximet developed by GSK. The Phase 3 pivotal trials, including the endpoints required to evaluate Treximet, were designed to demonstrate superiority to placebo for relief of pain and the associated symptoms of migraine (nausea, photophobia and phonophobia) at two hours. Additionally, the program was designed to demonstrate that each component makes a contribution to the efficacy of Treximet (the “combination drug rule” that the FDA requires of all combination products). The efficacy endpoint for the combination was sustained pain free, which is defined as improvement from moderate or severe pain to no pain at two hours and remaining at no pain through twenty four hours without the use of rescue medicine. Further, GSK has conducted market support studies for Treximet, including evaluations in a pediatric population. As required by the terms of our agreement with GSK, we transferred ownership of the NDA and other regulatory filings for Treximet to GSK on May 14, 2008, and GSK took responsibility for all ongoing regulatory obligations for the product, including post marketing clinical trial requirements.
Since inception we have incurred total direct development costs of $26.5 million associated with the development of our MT 400 and Treximet programs. Our direct development costs do not include the cost of research and development personnel or any allocation of our overhead expenses.
On November 23, 2011, we entered into the Purchase Agreement with CII, pursuant to which we sold, and CII purchased, our right to receive future royalty payments arising from U.S. sales of MT 400, including Treximet.
On March 21, 2011, we entered into a license agreement with Cilag, a division of Johnson & Johnson, for the exclusive development and commercialization of MT 400 in Brazil, Colombia, Ecuador and Peru. On December 22, 2014, we entered into a mutual termination letter with Cilag. In accordance with the terms of the termination letter the agreement terminated on January 21, 2015. There was no dispute between the parties regarding the license agreement. At our request, for a period of two years after termination, Cilag has agreed to negotiate in good faith commercially reasonable terms of a supply agreement whereby Cilag would supply us or our licensees, with MT400 for a period equal to the shorter of (i) two (2) years; or (ii) until we establish an alternative supplier. We recognized approximately $257,300 in licensing revenue in the fourth quarter of as a result of this termination.
On May 13, 2014, we, GSK, CII and Pernix, entered into certain agreements in connection with GSK’s divestiture of all of its rights, title and interest to develop, commercialize and sell Treximet in the U.S. to Pernix. Upon the closing of the divestiture on August 20, 2014, GSK assigned the Treximet Agreement to Pernix. Immediately following the closing of the divestiture, Amendment No. 1 between us and Pernix became effective. Amendment No. 1, among other things, amends the royalty provisions of the Agreement to provide for a guaranteed quarterly minimum royalty of $4 million for the calendar quarters commencing on January 1, 2015 and ending on March 31, 2018 and requires that Pernix continue certain of GSK’s ongoing development activities and to undertake certain new activities, for which we will provide reasonable assistance. Amendment No. 1 also eliminates restrictions in the Agreement on our right to develop and commercialize certain dosage forms of sumatriptan/naproxen combinations outside of the United States and permits POZEN to seek approval for these combinations on the basis of the approved NDA for Treximet. Pernix has also granted us a warrant to purchase 500,000 shares of Pernix common stock at an exercise price of $4.28, the closing price of Pernix common stock as reported on the NASDAQ Global Market on May 13, 2014. The common stock underlying the warrants will be registered by Pernix with the SEC and will be exercisable from August 20, 2014, the closing date of the divestiture until February 28, 2018. Lastly, we, GSK, Pernix and CII executed a letter agreement whereby we expressly consented to the assignment by GSK and the assumption by Pernix of the Treximet Agreement. On July 30, the parties entered into Amendment No. 2 to the Treximet Agreement which will permit Pernix’s Irish affiliate to which Pernix will assign its rights to further assign the Agreement without our prior written consent as collateral security for the benefit of the lenders which financed the acquisition. Amendment No. 2 became effective upon closing of the divestiture on August 20, 2014.
Collaborative Arrangements
We have entered into and may continue to enter into collaborations with established pharmaceutical or pharmaceutical services companies to develop, commercialize and/or manufacture our product candidates. Our existing collaborations are described below.
GlaxoSmithKline (GSK)
In June 2003, we signed an agreement with GSK for the development and commercialization of proprietary combinations of a triptan (5-HT1B/1D agonist) and a long-acting NSAID. The combinations covered by the agreement are among the combinations of MT 400. Under the terms of the agreement, GSK has exclusive rights in the U.S. to commercialize all combinations which combine GSK’s triptans, including Imitrex® (sumatriptan succinate) or Amerge® (naratriptan hydrochloride), with a long-acting NSAID. We were responsible for development of the first combination product, while GSK provided formulation development and manufacturing. Pursuant to the terms of the agreement, we received $25.0 million in initial payments from GSK following termination of the waiting period under the Hart-Scott-Rodino notification program and the issuance of a specified patent. In May 2004, we received a $15.0 million milestone payment as a result of our commencement of Phase 3 clinical trial activities. In October 2005, we received a $20.0
million milestone payment upon the FDA’s acceptance for review of the NDA for Treximet, the trade name for the product. On April 26, 2008, we received, from GSK, $20.0 million in milestone payments which were associated with the approval of, and GSK’s intent to commercialize, Treximet. In addition, GSK will pay us two sales performance milestones totaling $80.0 million if certain sales thresholds are achieved. Up to an additional $10.0 million per product is payable upon achievement of milestones relating to other products. GSK will pay us royalties on all net sales of marketed products until at least the expiration of the last to expire issued applicable patent (October 2, 2025) based upon the scheduled expiration of currently issued patents. GSK may reduce, but not eliminate, the royalty payable to us if generic competitors attain a pre-determined share of the market for the combination product, or if GSK owes a royalty to one or more third parties for rights it licenses from such third parties to commercialize the product. The agreement terminates on the date of expiration of all royalty obligations unless earlier terminated by either party for a material breach or by GSK at any time upon 90 days’ written notice to us for any reason or no reason. Among the contract breaches that would entitle us to terminate the agreement is GSK’s determination not to further develop or to launch the combination product under certain circumstances. GSK has the right, but not the obligation, to bring, at its own expense, an action for infringement of certain patents by third parties. If GSK does not bring any such action within a certain time frame, we have the right, at our own expense, to bring the appropriate action. With regard to certain other patent infringements, we have the sole right to bring an action against the infringing third party. Each party generally has the duty to indemnify the other for damages arising from breaches of each party’s representations, warranties and obligations under the agreement, as well as for gross negligence or intentional misconduct. We also have a duty to indemnify GSK for damages arising from our development and manufacture of MT 400 prior to the effective date of the agreement, and each party must indemnify the other for damages arising from the development and manufacture of any combination product after the effective date.
On November 23, 2011, we entered into the Purchase Agreement, with CII, pursuant to which we sold, and CII purchased, our right to receive future royalty payments arising from U.S. sales of MT 400, including Treximet.
On May 13, 2014, we, GSK, CII and Pernix, entered into certain agreements in connection with GSK’s divestiture of all of its rights, title and interest to develop, commercialize and sell Treximet in the U.S. to Pernix. Upon the closing of the divestiture on August 20, 2014, GSK assigned the Treximet Agreement to Pernix. Immediately following the closing of the divestiture, Amendment No. 1 between us and Pernix became effective. Amendment No. 1, among other things, amends the royalty provisions of the Agreement to provide for a guaranteed quarterly minimum royalty of $4 million for the calendar quarters commencing on January 1, 2015 and ending on March 31, 2018 and requires that Pernix continue certain of GSK’s ongoing development activities and to undertake certain new activities, for which we will provide reasonable assistance. Amendment No. 1 also eliminates restrictions in the Agreement on our right to develop and commercialize certain dosage forms of sumatriptan/naproxen combinations outside of the United States and permits Pozen to seek approval for these combinations on the basis of the approved NDA for Treximet. Pernix has also granted us a warrant to purchase 500,000 shares of Pernix common stock at an exercise price equal to$4.28 per share, the closing price of Pernix common stock as reported on the NASDAQ Global Market on May 13, 2014. The common stock underlying the warrants have been registered by Pernix with the SEC and was exercisable from the August 20, 2014, the closing date of the divestiture until February 28, 2018. If the Divestiture had not been consummated, the warrants would have been null and void. Because the warrant has not been registered by Pernix with the SEC, the Company cannot sell or transfer the warrant in reliance upon Rule 144 until after November 13, 2014 when the Company meets certain holding requirements. Lastly, we, GSK, Pernix and CII executed a letter agreement whereby we expressly consented to the assignment by GSK and the assumption by Pernix of the Treximet Agreement. On July 30, the parties entered into Amendment No. 2 to the Treximet Agreement which will permit Pernix’s Irish affiliate to which Pernix assigned its rights to further assign the Agreement without our prior written consent as collateral security for the benefit of the lenders which financed the acquisition. Amendment No. 2 became effective upon the closing of the divestiture on August 20, 2014.
AstraZeneca AB (AstraZeneca)/Horizon Pharma Inc. (Horizon)
In August 2006, we entered into the Original Agreement. Under the terms of the Original Agreement, we granted to AstraZeneca an exclusive, fee-bearing license, in all countries of the world except Japan, under our patents and know-how relating to combinations of gastroprotective agents and NSAIDs (other than aspirin and its derivatives). Pursuant to the terms of the agreement, we received an upfront license fee of $40.0 million from AstraZeneca following termination of the waiting period under the Hart-Scott-Rodino notification program.
We retained responsibility for the development and filing of the NDA for the product in the U.S. AstraZeneca is responsible for all development activities outside the U.S., as well as for all manufacturing, marketing, sales and distribution activities worldwide. We agreed to bear all expenses related to certain specified U.S. development activities. All other development expenses, including all manufacturing-related expenses, will be paid by AstraZeneca. The agreement established joint committees with representation of both
us and AstraZeneca to manage the development and commercialization of the product. The committees operate by consensus, but if consensus cannot be reached, we generally will have the deciding vote with respect to development activities required for marketing approval of the product in the U.S. and AstraZeneca generally will have the deciding vote with respect to any other matters.
In September 2007, we agreed with AstraZeneca to amend the Original Agreement effective as of September 6, 2007. Under the terms of the amendment, AstraZeneca has agreed to pay us up to $345.0 million, in the aggregate, in milestone payments upon the achievement of certain development, regulatory and sales events. In September 2007 we received a $10.0 million payment upon execution of the amendment and a $20.0 million payment in recognition of the achievement of the primary endpoints for the PN400-104 study, a study which compared acid suppression of different doses of VIMOVO (formerly PN 400), and achievement of the interim results of the PN200-301 study, a six month comparative trial of PN 200 as compared to EC naproxen in patients requiring chronic NSAID therapy, meeting mutually agreed success criteria. In May 2010, we received a $20.0 million payment for the NDA approval of VIMOVO. We also received an additional $25.0 million milestone in December 2010 when VIMOVO received approval (including pricing and reimbursement approval) in a major ex-U.S. market and up to $260.0 million will be paid as sales performance milestones if certain aggregate sales thresholds are achieved.
The amendment also revised the royalty rates we were to have received under the Original Agreement. Prior to the effective date of the amendment, under the terms of the Original Agreement, we were to receive a royalty based on annual net sales by AstraZeneca, its affiliates or sublicensees during the royalty term. The royalty rate varied based on the level of annual net sales of products made by AstraZeneca, its affiliates and sublicensees, ranging from the mid-single digits to the mid-teens. Under the amendment, we receive a flat, low double digit royalty rate during the royalty term on annual net sales of products made by AstraZeneca, its affiliates and sublicensees, in the U.S. and royalties ranging from the mid-single digits to the high-teens on annual net sales of products made by AstraZeneca, its affiliates and sublicensees outside of the U.S. The amendment also revised the rate of reduction to the royalty rate based upon loss of market share due to generic competition inside and outside of the U.S. to account for the new royalty structure. Our right to receive royalties from AstraZeneca for the sale of such products under the collaboration and license agreement, as amended, expires on a country-by-country basis upon the later of (a) expiration of the last-to-expire of certain patent rights relating to such products in that country, and (b) ten years after the first commercial sale of such products in such country.
We further amended the Original Agreement effective October 1, 2008 to shorten the timing of AstraZeneca’s reimbursement obligation for certain development expenses incurred by us under the agreement and to update the description of the target product profile studies (as defined in the agreement) for VIMOVO.
On December 31, 2014 we have receivables of $5.6 million related to VIMOVO royalty revenue, $4.3 million related to U.S. sales and $1.3 million related to ROW sales. The agreement, unless earlier terminated, will expire upon the payment of all applicable royalties for the products commercialized under the agreement. Either party has the right to terminate the agreement by notice in writing to the other party upon or after any material breach of the agreement by the other party, if the other party has not cured the breach within 90 days after written notice to cure has been given, with certain exceptions. The parties also can terminate the agreement for cause under certain defined conditions. In addition, AstraZeneca can terminate the agreement, at any time, at will, for any reason or no reason, in its entirety or with respect to countries outside the U.S., upon 90 days’ notice. If terminated at will, AstraZeneca will owe us a specified termination payment or, if termination occurs after the product is launched, AstraZeneca may, at its option, under and subject to the satisfaction of conditions specified in the agreement, elect to transfer the product and all rights to us.
On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understand that AstraZeneca will instead now focus on those countries where the product has shown growth and which AstraZeneca believes have the greatest potential for future growth.
On September 16, 2013, we and AstraZeneca entered into another amendment to the Original Agreement which made clarifications to certain intellectual property provisions of the Original Agreement to clarify that AstraZeneca’s rights under those provisions do not extend to products which contain acetyl salicylic acid. On September 16, 2013, we and AstraZeneca also executed a letter agreement whereby we agreed that in the event that AstraZeneca divested its rights and obligations to market VIMOVO in the United States to a third party, AstraZeneca would be relieved of its obligations under the Original Agreement with respect to the United States as of the effective date of such divestiture, including its obligation under the Original Agreement to guarantee the performance of such assignee and/or sublicensee.
On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. In connection with this divestiture, on November 18, 2013 we and AstraZeneca entered into an Amended and Restated Collaboration and
License Agreement for the United States, the “U.S. Agreement” and an Amended and Restated License and Collaboration Agreement for Outside the United States, the “ROW Agreement” which agreements collectively amend and restate the Original Agreement. AstraZeneca has assigned the U.S. Agreement to Horizon in connection with the Divestiture with our consent.
We and Horizon also entered into Amendment No. 1 to the U.S. Agreement which, among other things, amends the royalty provisions of the U.S. Agreement to provide for a guaranteed annual minimum royalty amount of $5 million in calendar year 2014, and a guaranteed annual minimum royalty amount of $7.5 million each calendar year thereafter, provided that the patents owned by us which cover VIMOVO are in effect and no generic forms of VIMOVO are in the marketplace. Amendment No. 1 also provides that Horizon has assumed AstraZeneca’s right to lead the on-going Paragraph IV litigation relating to VIMIVO currently pending in the United States District Court for the District of New Jersey and will assume all patent-related defense costs relating to such litigation, including reimbursement up to specified amounts of the cost of any counsel retained by us, amends certain time periods for Horizon’s delivery of quarterly sales reports to POZEN, and provides for quarterly update calls between the parties to discuss VIMOVO’s performance and Horizon’s commercialization efforts.
Further, the Company, AstraZeneca and Horizon executed a letter agreement whereby POZEN expressly consented to the assignment by AstraZeneca and the assumption by Horizon of the U.S. Agreement. In addition, the letter agreement establishes a process for AstraZeneca and Horizon to determine if sales milestones set forth in the Original Agreement are achieved on a global basis and other clarifications and modifications required as a result of incorporating the provisions of the Original Agreement into the U.S. Agreement and the ROW Agreement or as otherwise agreed by the parties.
On June 13, 2011, we and AstraZeneca received a Paragraph IV Notice Letter from Lupin informing us that Lupin had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the POZEN and the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, the ‘466 patent and, each of which are assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Lupin’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Lupin and, accordingly, we and AstraZeneca filed suit against Lupin on July 25, 2011 in the United States District Court for the District of New Jersey. On November 19, 2014, an amended complaint was filed in which the 504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, all assigned to AstraZeneca or its affiliates, were not asserted against Lupin. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On September 19, 2011, we and AstraZeneca received Paragraph IV Notice Letter from Anchen informing us that Anchen had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, the ‘085 patent, the ‘070 patent, and the ‘466 patent. The patents are among those listed with respect to VIMOVO in the Orange Book and expire at various times between 2018 and 2023. Anchen’s Paragraph IV Notice Letter asserts that its generic product will not infringe those patents or that those patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Anchen and, accordingly, we and AstraZeneca filed suit against Anchen on October 28, 2011 in the United States District Court for the District of New Jersey. On October 4, 2013, Anchen filed an amendment to its ANDA seeking to change its Paragraph IV certification to a Paragraph III. It is unclear when or if the FDA will enter Anchen’s amendment. On October 25, 2013, Anchen filed a Motion to Dismiss the case against it, based on its proposed re-certification. On November 18, 2013, we and AstraZeneca filed an Opposition to Anchen’s Motion to Dismiss. On June 11, 2014, the court granted Anchen’s Motion and dismissed the case against them.
On November 20, 2012 we and AstraZeneca received a Paragraph IV Notice Letter from Dr. Reddy’s, informing us that Dr. Reddy’s had filed a second ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the POZEN and the 504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, the ‘466 patent and, each of which are assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Dr. Reddy’s second Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Dr. Reddy’s on its second ANDA filing and, accordingly, we and AstraZeneca filed suit against Dr. Reddy’s on January 4, 2013, in the United States District Court for the District of New Jersey. On April 15, 2013 a Stipulation of Partial Dismissal was filed which sought dismissal of all infringement claims relating to the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent (which are each assigned to AstraZeneca), as well as Dr. Reddy’s defenses and counterclaims relating to those patents. On April 18, 2013, the District Court issued the Stipulation and Order dismissing with prejudice those claims and defenses. On June 28, 2013 we and AstraZeneca filed a Motion for Summary Judgment relating to the second ANDA filing asserting that U.S. Patent No. 6,926,907 is not invalid. On August 12, 2013, DRL filed an opposition to the Motion for Summary Judgment. On March 28, 2014, the District Court denied the Motion. On October 11, 2013, DRL filed a Motion for Summary Judgment asserting that the product which is the subject matter of its second ANDA does not infringe the ‘907 patent. On November 4, 2013, POZEN and AstraZeneca filed a Motion for an Order Denying DRL’s Motion for Summary Judgment Pursuant to Rule 56(d) and an Opposition to DRL’s Motion. On May 29, 2014, the court
issued an order denying DRL’s motion. This case was consolidated with the originally filed Dr. Reddy’s case and is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On March 29, 2013, we and AstraZeneca received a Paragraph IV Notice Letter from Watson, now Actavis, informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the Company and the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Watson’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Watson. On May 10, 2013, we and AstraZeneca filed a patent infringement lawsuit against Watson in the U.S. District Court of New Jersey. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On May 16, 2013, Pozen and AstraZeneca received a Paragraph IV Notice Letter from Mylan informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the Company and the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Mylan’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Mylan. On June 28, 2013, we and AstraZeneca filed a patent infringement lawsuit against Mylan in the U.S. District Court of New Jersey. On February 13, 2015, the Court entered a joint stipulation of dismissal of counts related to certain patents, dismissing claims related to the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On October 15, 2013, the United States Patent Office issued the ‘285 patent. The ‘285 patent, entitled “Pharmaceutical compositions for the coordinated delivery of NSAIDs” and assigned to Pozen, is related to the ‘907 patent. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement of the ‘285 patent and, accordingly, on October 23, 2013, we, and AstraZeneca filed patent infringement lawsuits against DRL, Lupin, Watson and Mylan in the U.S. District Court of New Jersey alleging that their ANDA products infringe the ‘285 patent. On November 8, 2013, we, and AstraZeneca filed a Motion to Amend the Complaint in the actions against DRL, Lupin, Watson and Mylan or, in the alternative, to consolidate the actions involving the ‘285 patent with the existing consolidated action. DRL, Lupin, Watson and Mylan have each filed answers to the respective amended complaints, thus adding claims relating to the ‘285 patent against each of the Defendants to the consolidated case.
As part of Horizon’s purchase of all of AstraZeneca’s rights, title and interest to develop, commercialize and sell VIMOVO in the United States, Horizon has assumed AstraZeneca’s right to lead the above-described Paragraph IV litigation relating to VIMIVO currently pending in the United States District Court for the District of New Jersey and has assumed patent-related defense costs relating to such litigation, including reimbursement up to specified amounts of the cost of any counsel retained by us. On December 12, 2013, Horizon filed Motions to Join under Fed.R.Civ.Proc. 25(c) as a co-plaintiff in each of the above referenced actions and the consolidated action. On January 31, 2014 and February 2, 2014, the Court granted Horizon’s motions.
sanof-aventis U.S. LLC
On September 3, 2013, we entered into a license and collaboration agreement with Sanofi U.S. On November 29, 2014, we executed a termination agreement with Sanofi U.S. terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi U.S. were terminated and all rights to the products licensed to Sanofi U.S. under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi U.S. relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products in the United States. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
In consideration for the rights granted to Sanofi U.S. under the license agreement, Sanofi U.S. paid us an upfront payment of $15 million. The upfront payment was amortized and recognized as revenue, $4 million during the year ended December 31, 2013 and the remaining $11 million during the year ended December 31, 2014.
Cilag GmbH International (Cilag)
On March 21, 2011, we entered into a license agreement with Cilag, a division of Johnson & Johnson, for the exclusive development and commercialization of MT 400 in Brazil, Colombia, Ecuador and Peru. On December 22, 2014, we entered into a
mutual termination letter with Cilag. In accordance with the terms of the termination letter the agreement terminated on January 21, 2015. There was no dispute between the parties regarding the license agreement and, at our request, for a period of two years after termination, Cilag has agreed to negotiate in good faith commercially reasonable terms of a supply agreement whereby Cilag would supply us or our licensees, with MT400 for a period equal to the shorter of (i) two (2) years; or (ii) until we establish an alternative supplier. We recognized approximately $257,300 in licensing revenue in the fourth quarter of as a result of this termination.
Patheon Pharmaceuticals Inc. (Patheon)
On December 19, 2011, we entered into the Supply Agreement and the Capital Agreement relating to the manufacture of PA32450. Under the terms of the Supply Agreement, Patheon has agreed to manufacture, and we have agreed to purchase, a specified percentage of the Company’s requirements of the PA32540 for sale in the United States. The term of the Supply Agreement extends until December 31st of the fourth year after the Initial Term, and will automatically renew thereafter for periods of two years, unless terminated by either party upon eighteen months’ written notice prior to the expiration of the Initial Term or twelve months’ written notice prior to the expiration of any renewal term. In addition to usual and customary termination rights which allow each party to terminate the Supply Agreement for material, uncured breaches by the other party, we can terminate the Agreement upon 30 days’ prior written notice if a governmental or regulatory authority takes any action or raises any objection that prevents us from importing, exporting, purchasing or selling PA32540 or if it is determined that the formulation or sale of PA32540 infringes any patent rights or other intellectual property rights of a third party. We can also terminate the Supply Agreement upon 24 months’ prior written notice if we license, sell, assign or otherwise transfer any rights to commercialize PA32540 in the Territory to a third party. The Supply Agreement contains general and customary commercial supply terms and conditions, as well as establishing pricing for bulk product and different configurations of packaged product, which pricing will be adjusted annually as set forth in the Supply Agreement. Under the terms of the Capital Agreement, we will be responsible for the cost of purchasing certain equipment specific to the manufacture of PA32540, the cost of which, based on current volume projections, is expected to be less than $150,000. If additional equipment and facility modifications are required to meet our volume demands for PA32540, we may be required to contribute to the cost of such additional equipment and facility modifications, up to a maximum of approximately $2.5 million in the aggregate.
The Supply Agreement and Capital Agreement were amended on July 10, 2013. The Amendment to the Supply Agreement expressly incorporates the Company’s PA8140 product candidate into the Supply Agreement. The Amendment to the Supply Agreement also clarifies that the manufacturing services contemplated by the Supply Agreement include the manufacture of validation batches, but the placing of an order for such validation batches will not trigger the Commencement Date of the Initial Term, updates pricing for the Company’s PA32540 product candidate and a incorporates a new pricing schedule for PA8140, as well as other conforming changes to the Supply Agreement. The Amendment to the Capital Agreement, replaces the existing Schedule A of the Capital Agreement, which lists dedicated and non-dedicated capital equipment and facility modifications to be funded in whole or in part by the Company, with a new updated schedule which reflects the parties’ current assumptions regarding the need for and timing of capital equipment expenditures based upon Patheon’s current and anticipated production capacity and current volume projections for the PA32540 and PA8140. Under the terms of the Capital Agreement, the Company was previously required to contribute to the cost of such additional capital equipment and facility modifications, up to a maximum of approximately $2.5 million in the aggregate. Pursuant to the terms of the Amendment to the Capital Agreement, the parties have agreed to reduce the amount of such maximum expenditure to approximately $1.2 million dollars in light of the revised capacity and volume assumptions.
Manufacturing
We currently have no manufacturing capability.
To date, we have entered into arrangements with third-party manufacturers for the supply of formulated and packaged clinical trial materials. We have also entered into a Supply Agreement and a related Capital Agreement with Patheon for the manufacture of PA32450 and PA8140 for sale in the United States. We believe our current supplier agreements should be sufficient to meet our commercial supply needs for PA32540 and PA8140 in the United States. Under our agreements with GSK, AstraZeneca and Horizon, it is the obligation of our partners to obtain commercial supplies of products developed under those agreements. Use of third-party manufacturers enables us to focus on our clinical development activities, minimize fixed costs and capital expenditures and gain access to advanced manufacturing process capabilities and expertise.
Competition
Competition for VIMOVO
The competition for VIMOVO comes from the oral anti-arthritic market, or more specifically the traditional non-selective NSAIDs (such as naproxen and diclofenac), traditional NSAID/gastroprotective agent combination products or combination product packages (such as Arthrotec® and Prevacid® NapraPAC™) and the only remaining COX-2 inhibitor, Celebrex®. The U.S. prescription market for oral solid NSAIDs was approximately $2.9 billion in 2011, of which 62% was accounted for by Celebrex, according to IMS. This market is continuing to undergo significant change, due to the voluntary withdrawal of Vioxx® by Merck & Co. in September 2004, the FDA-ordered withdrawal of Bextra® by Pfizer in April 2005 and the issuance of a Public Health Advisory by the FDA in April 2005 stating that it would require that manufacturers of all prescription products containing NSAIDs provide warnings regarding the potential for adverse cardiovascular events as well as life-threatening gastrointestinal events associated with the use of NSAIDs. Moreover, subsequent to the FDA advisory committee meeting in February 2005 that addressed the safety of NSAIDs, and, in particular, the cardiovascular risks of COX-2 selective NSAIDs, the FDA has indicated that long-term studies evaluating cardiovascular risk will be required for approval of new NSAID products that may be used on an intermittent or chronic basis. However, based on a meeting with the FDA in September 2005, we believe, although we cannot guarantee, that long-term cardiovascular safety studies may not be required at this time for FDA approval of our PN product candidates containing naproxen.
Competition for PA Products
The pharmaceutical and biopharmaceutical industries are intensely competitive and are characterized by rapid technological progress. Certain pharmaceutical and biopharmaceutical companies and academic and research organizations currently engage in, or have engaged in, efforts related to the discovery and development of new medicines for the treatment of migraine symptoms. Significant levels of research in chemistry and biotechnology occur in universities and other nonprofit research institutions. These entities have become increasingly active in seeking patent protection and licensing revenues for their research results. They also compete with us in recruiting skilled scientific talent.
Our ability to compete successfully will be based on our ability to create and maintain scientifically advanced technology, develop proprietary products, attract and retain scientific personnel, obtain patent or other protection for our products, obtain required regulatory approvals and manufacture and successfully market our products either alone or through outside parties. Some of our competitors have substantially greater financial, research and development, manufacturing, marketing and human resources and greater experience than we do in product discovery, development, clinical trial management, FDA regulatory review, manufacturing and marketing, which may enable them to compete more effectively than we can.
Treatment for the secondary prevention of cardiovascular and cerebrovascular disease typically consists of multiple prescription and over-the-counter drugs, including statins, anti-hypertensives and anti-platelet agents. Competition for PA will come from the prescription anti-platelet market as well as OTC aspirin and gastro-protective agents. An estimated 24 million Americans fall within the guidelines for chronic anti-platelet therapy as set forth by the American Heart Association. Prescription anti-platelet therapies include PLAVIX (clopidogrel) and generics, EFFIENT (prasugrel) and BRILINTA (ticagrelor). In 2011, prior to loss of market exclusivity, PLAVIX sales exceeded $9 billion worldwide. Because OTC aspirin is used to treat many conditions, including pain and inflammation, identifying the portion of sales attributable to anti-platelet therapy is difficult.
Patents and Proprietary Information
We have obtained and intend to actively seek to obtain, when appropriate, protection for our products and proprietary technology by means of U.S. and foreign patents, trademarks and contractual arrangements. In addition, we rely upon trade secrets and contractual agreements to protect certain of our proprietary technology and products.
We have issued U.S. patents and pending U.S. patent applications, as well as pending foreign patent applications or issued foreign patents, relating to our marketed products and product candidates. We also have U.S. and foreign patent applications pending relating to novel product concepts. There can be no assurance that our patent applications will issue as patents or, with respect to our issued patents, that they will provide us with significant protection. The following provides a general description of our patent portfolio and is not intended to represent an assessment of claim limitations or claim scope.
MT 400/Treximet
We have four issued U.S. patents with claims relating to methods, compositions and therapeutic packages involving the use of certain NSAIDs and 5-HT receptor agonists in treating patients with migraine. Outside of the U.S., we have issued patents in Australia, Canada, Europe, Hong Kong and Japan. The expected expiration date of the issued patents relating to MT 400 is August 14, 2017. We expect that patents issued from pending patents related to MT 400 will also expire in August 2017.
Oppositions were filed against the issued European patent in October 2005 by Merck & Co., Inc. and Almirall Prodesfarma asserting that the European patent should not have been granted. We filed a response to these oppositions and the Opposition Division of the European Patent Office called for oral proceedings. During the oral proceedings and in the written opinion subsequently
provided, the European Patent Office found that claims relating to combinations of sumatriptan and naproxen for the treatment of migraine were valid. However, broader claims relating to certain other 5-HT1B/1D agonists and long-acting NSAIDs were held to be insufficiently supported by the presently available technical evidence.
We also have an issued U.S. patent with claims relating to formulations of MT 400 which, we expect to expire in October 2025. We have additional pending U.S. and foreign patent applications with claims directed to formulations of MT 400 which, if issued, we expect to expire in 2027.
PN/PA
We have issued patents in the U.S., Australia, Canada, Europe, Mexico and Eurasia, with claims directed to certain compositions containing a combination of acid inhibitors, including PPIs, and NSAIDs. The issued patents also have claims to treatment methods involving the use of such compositions. We have pending U.S. and foreign patent applications that also have claims to compositions containing acid inhibitors and NSAIDS and to various treatment methods involving such compositions. The issued U.S. patents and related U.S. patent applications are expected to expire on February 28, 2023.
Oppositions were filed against the issued European patent in April 2011 by Chatfield Pharmaceuticals and Strawman Ltd. asserting that the European patent should not have been granted. We filed for a response to these oppositions and the Opposition Division of the European Patent Office called for oral proceedings. Strawman Ltd. Subsequently withdrew from the opposition proceedings. During the proceedings in December 2012, the European Patent Office found that claims relating to combination of PPIs and NSAIDS were valid. Chatfield may appeal the decision by giving notice within 60 days of the date on which the Opposition Division issues its written decisions. The European patent will expire in May 2022, but we have obtained supplement protection certificates (SPCs) for VIMOVO which extend to October 25, 2025, and we expect to apply for SPCs for PA upon approval. We expect that patents outside of the U.S. and Europe, as well as additional patents which issue from the pending foreign patent applications, to expire on May 31, 2022.
We, together with AstraZeneca, have filed joint patent applications relating to VIMOVO. We expect any patents which issue from these applications to expire in 2029 and 2030. We have filed additional patent applications related to PA. We expect any patents which issue from these applications to expire between 2030 and 2032.
We, AstraZeneca and Horizon are engaged in Paragraph IV litigation with several generic pharmaceutical companies with respect to patents listed in the Orange Book with respect to VIMOVO currently pending in the United States District Court for the District of New Jersey which is described on page 17 of our Annual Report on Form 10-K, filed with the SEC on March 11, 2015.
Government Regulation
The FDA and comparable regulatory agencies in foreign countries impose substantial requirements on the clinical development, manufacture and marketing of pharmaceutical product candidates. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record-keeping, approval and promotion of our product candidates. All of our product candidates will require regulatory approval before commercialization. In particular, therapeutic product candidates for human use are subject to rigorous preclinical and clinical testing and other requirements of the Federal Food, Drug and Cosmetic Act, or FFDCA, implemented by the FDA, as well as similar statutory and regulatory requirements of foreign countries. Obtaining these marketing approvals and subsequently complying with ongoing statutory and regulatory requirements is costly and time-consuming. Any failure by us or our collaborators, licensors or licensees to obtain, or any delay in obtaining, regulatory approvals or in complying with other regulatory requirements could adversely affect the commercialization of product candidates then being developed by us and our ability to receive product or royalty revenues.
The steps required before a new drug product candidate may be distributed commercially in the U.S. generally include:
· conducting appropriate preclinical laboratory evaluations of the product candidate’s chemistry, formulation and stability and preclinical studies in animals to assess the potential safety and efficacy of the product candidate;
· submitting the results of these evaluations and tests to the FDA, along with manufacturing information and analytical data, in an IND;
· initiating clinical trials under the IND and addressing any safety or regulatory concerns of the FDA;
· obtaining approval of Institutional Review Boards, or IRBs, to introduce the drug into humans in clinical studies;
· conducting adequate and well-controlled human clinical trials that establish the safety and efficacy of the product candidate for the intended use, typically in the following three sequential, or slightly overlapping stages:
Phase 1: The product is initially introduced into human subjects or patients and tested for safety, dose tolerance, absorption, metabolism, distribution and excretion;
Phase 2: The product candidate is studied in patients to identify possible adverse effects and safety risks, to determine dosage tolerance and the optimal dosage, and to collect some efficacy data;
Phase 3: The product candidate is studied in an expanded patient population at multiple clinical study sites, to confirm efficacy and safety at the optimized dose, by measuring primary and secondary endpoints established at the outset of the study;
· submitting the results of preclinical studies, and clinical trials as well as chemistry, manufacturing and control information on the product candidate to the FDA in a NDA; and
· obtaining FDA approval of the NDA prior to any commercial sale or shipment of the product candidate.
This process can take a number of years and require substantial financial resources. Each NDA must be accompanied by a user fee, pursuant to the requirements of the Prescription Drug User Fee Act, or PDUFA, and its amendments. The FDA adjusts the PDUFA user fees on an annual basis. According to the FDA’s fee schedule, effective on October 1, 2013 for the fiscal year 2014, the user fee for an application requiring clinical data, such as an NDA, is $2,169,100. PDUFA also imposes an annual product fee for each marketed prescription drug ($104,060), and an annual establishment fee ($554,600) on facilities used to manufacture prescription drugs and biologics. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. However, there are no waivers for product or establishment fees.
The results of preclinical studies and initial clinical trials are not necessarily predictive of the results from large-scale clinical trials, and clinical trials may be subject to additional costs, delays or modifications due to a number of factors, including the difficulty in obtaining enough patients, clinical investigators, product candidate supply and financial support.
Even after FDA approval has been obtained, further studies, including post-marketing studies, may be required. Results of post-marketing studies may limit or expand the further marketing of the products. If we propose any modifications to a product, including changes in indication, manufacturing process, manufacturing facility or labeling, a supplement to our NDA may be required to be submitted to the FDA and approved.
The FDA may also require testing and surveillance programs to monitor the effect of approved product candidates that have been commercialized, and the agency has the power to prevent or limit further marketing of a product candidate based on the results of these post-marketing programs. Upon approval, a product candidate may be marketed only in those dosage forms and for those indications approved in the NDA.
The status of the NDAs we have submitted to the FDA for Treximet, VIMOVO and our PA product candidates is discussed above in “MT400/Treximet,” “PN/VIMOVO Program,” and “Status of Our Product Candidates and Exploratory Programs” — PA Program”.
In addition to obtaining FDA approval for each indication to be treated with each product candidate, each domestic product candidate manufacturing establishment must register with the FDA, list its product with the FDA, comply with the applicable cGMP regulations, which include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation, and permit and pass manufacturing plant inspections by the FDA. Moreover, the submission of applications for approval may require additional time to complete manufacturing stability studies. Foreign establishments manufacturing product for distribution in the U.S. also must list their product candidates with the FDA and comply with cGMP regulations. They are also subject to periodic inspection by the FDA or by local authorities under agreement with the FDA.
Any product candidates manufactured or distributed by us pursuant to FDA approvals are subject to extensive continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the product candidate. In addition to continued compliance with standard regulatory requirements, the FDA may also require post-marketing testing and surveillance to monitor the safety and efficacy of the marketed product. Adverse experiences with the product candidate must be reported to the FDA. Product approvals may be affected and even withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product are discovered following approval.
The FFDCA also mandates that products be manufactured consistent with cGMP regulations. In complying with the cGMP regulations, manufacturers must continue to spend time, money and effort in production, record keeping, quality control, and auditing to ensure that the marketed product meets applicable specifications and other requirements. The FDA periodically inspects manufacturing facilities to ensure compliance with cGMP regulations. Failure to comply subjects the manufacturer to possible FDA action, such as warning letters, suspension of manufacturing, seizure of the product, voluntary recall of a product or injunctive action, as well as possible civil penalties. We currently rely on, and intend to continue to rely on, third parties to manufacture our compounds and product candidates. These third parties will be required to comply with cGMP regulations.
Products manufactured in the U.S. for distribution abroad will be subject to FDA regulations regarding export, as well as to the requirements of the country to which they are shipped. These latter requirements are likely to cover the conduct of clinical trials, the submission of marketing applications, and all aspects of manufacturing and marketing. Such requirements can vary significantly from country to country.
We and our contractors are also subject to various federal, state and local laws, rules, regulations and policies relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and the use of and disposal of hazardous or potentially hazardous substances used in connection with our research work. Although we believe that safety procedures employed for handling and disposing of such materials comply with current federal, state and local laws, rules, regulations and policies, the risk of accidental injury or contamination from these materials cannot be entirely eliminated.
Before a medicinal product can be supplied in the EU, it must first be granted a marketing authorization. There are three routes by which this may be achieved: the centralized procedure whereby a single European license is granted by the European Commission permits the supply of the product in question throughout the EU or the decentralized, or DC, or mutual recognition procedures, or MRP, through which the views of one national authority (Reference Member State, or RMS) are “recognized” by other authorities (Concerned Member States) when conducting their reviews; the DC applies if the medicinal product in question has not yet received a marketing authorization in any member state at the time of the application whereas the MRP applies to a currently approved medicinal product. These latter two processes lead to individual licenses in each member state for the supply of products in that country only. The centralized route is compulsory for biotechnology products and is optional for certain so-called “high technology” products and products containing entirely new active substances. All products which are not authorized by the centralized route must be authorized by the DC or MRP unless the product is designed for use in a single country in which case a National Application can be made.
In making an application for a new medicinal product not governed compulsorily by the centralized procedure, typically use will be made of the DC although the MRP would be used if a marketing authorization were first secured in an RMS. The procedural steps for the DC and the MRP are governed by Directive 2001/83/EC, as amended, and are described in the Notice to Applicants, Volume 2A Chapter 2 - Mutual Recognition (updated version - November 2005). The procedures provide for set time periods for each process (DC - 120 days; MRP — 90 days) but if consensus is not reached between all the CMS and the RMS in that time, the application is referred to arbitration through the Co-ordination Group for Mutual Recognition and Decentralized Procedures, or CMD, with referral to the Committee for Human Medicinal Products, or CHMP. If a referral is made, the procedure is suspended; marketing of the product would only be possible in the RMS in the case of an MRP. The opinion of the CMD/CHMP, which is binding, could support or reject the objections or alternatively reach a compromise position acceptable to all EU countries concerned. The arbitration procedure may require the delivery of additional data. Once granted, any MAA remains subject to pharmacovigilance and all competent authorities have the power to vary, suspend or revoke an MAA on grounds of safety.
The extent of U.S. and foreign government regulation which might result from future legislation or administrative action cannot be accurately predicted. For example, in the U.S., although the Food and Drug Administration Modernization Act of 1997, or FDAMA, modified and created requirements and standards under the FFDCA with the intent of facilitating product development and marketing, the FDA is still in the process of developing regulations implementing FDAMA and the more recent Food and Drug Administration Amendments Act of 2007, or FDAAA. FDA has been actively implementing drug safety plans called Risk Evaluation and Mitigation Strategies, or REMS, as authorized by FDAAA, as a condition of drug approval, or after initial marketing, if FDA becomes aware of new safety data about the drug. These and other legislative initiatives may impose additional regulatory requirements on us, and may impact approval of our drugs or our marketing plans. The actual effect of these and other developments on our own business is uncertain and unpredictable.
Corporate Information
We were incorporated in Delaware on September 25, 1996. Our principal offices are located in the Exchange Office Building at 1414 Raleigh Road, Suite 400, Chapel Hill, NC 27517. Our telephone number is (919) 913-1030. We maintain a website at www.POZEN.com and make available free of charge through this website our Annual Reports on Form 10-K, our Quarterly Reports on Form 10-Q, our Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (the “Exchange Act”) as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. We also similarly make available, free of charge on our website, the reports filed with the SEC by our executive officers, directors and 10% stockholders pursuant to Section 16 under the Exchange Act as soon as reasonably practicable after copies of those filings are provided to us by those persons. We are not including the information contained at www.POZEN.com, or at any other Internet address as part of, or incorporating it by reference into, this registration statement.
In addition, we make available on our website (i) the charters for the committees of our Board, including the Audit Committee, Compensation Committee and Nominating/Corporate Governance Committee, and (ii) our Code of Business Conduct and Ethics governing our directors, officers and employees. We intend to disclose on our website any amendments to, or
waivers from, our Code of Business Conduct and Ethics that are required to be disclosed pursuant to the rules of the SEC and the NASDAQ Global Market.
Employees
As of August 1, 2015, Pozen had a total of 22 full-time employees. All of Pozen’s current employees are based at its headquarters in Chapel Hill, North Carolina. Of Pozen’s 22 employees, 12 hold advanced degrees, including three with an M.D., Pharm.D. or Ph.D. degree.
Overview of Tribute
Tribute is a Canadian specialty pharmaceutical company with a primary focus on the acquisition, licensing, development and promotion of healthcare products in Canada and the United States. Tribute targets several therapeutic areas in Canada with a particular interest in products for the treatment of neurology, pain, dermatology, endocrinology, cardiology and gastro-intestinal disorders.
Tribute markets Cambia® (diclofenac potassium for oral solution), Bezalip® SR (bezafibrate), Soriatane® (acitretin), NeoVisc® (1.0% sodium hyaluronate solution), Uracyst® (sodium chondroitin sulfate solution 2%), Fiorinal®, Fiorinal® C, Visken®, Viskazide® and Collatamp® G in the Canadian market. Additionally, NeoVisc and Uracyst are commercially available and are sold globally through various international partnerships. Tribute also has the U.S. rights to Fibricor® (fenofibric acid) and its related authorized generic. In addition, Tribute has the exclusive rights to develop and commercialize Bezalip SR in the U.S. and has the exclusive right to sell bilastine, a product licensed from Faes Farma for the treatment of allergic rhinitis and chronic idiopathic urticaria (hives), in Canada. The exclusive license is inclusive of prescription and non-prescription rights for bilastine, as well as adult and pediatric presentations in Canada. This product is subject to receiving Canadian regulatory approval. In addition to the foregoing, pursuant to Tribute’s recently completed MFI Acquisition, Tribute’s product portfolio in Canada has been further diversified through the addition of 13 marketed products including; Durela® (tramadol extended release), Proferrin® (heme-iron polypeptide), Iberogast®, Moviprep®, Normacol®, Resultz®, Pegalax®, Balanse®, Balanse® Kids, Diaflor™, Mutaflor®, Purfem®, Onypen® and one product recently approved by Health Canada but not launched (ibSium®) and two pipeline products (Octasa® and BedBugz™), both of which are pending submission to Health Canada.
Tribute markets its products in Canada through its own sales force and currently has licensing agreements for the distribution of NeoVisc® and Uracyst® in over 20 countries, and continues to expand this footprint. Tribute’s focus on business development is very comprehensive including product acquisitions, company acquisitions, in-licensing and out-licensing for immediate impact on its revenue stream, as well as product development for future growth and stability.
Tribute’s management team has a strong track record of success in senior management positions from companies such as Wyeth, Syntex/Roche, Astra-Zeneca, Amgen, Bayer, Novartis, Johnson & Johnson, Nautilus Neurosciences and Biovail. The team has extensive operational and business development experience in the Canadian and United States markets and international business experience overseas.
Tribute was incorporated under the Business Corporations Act (Ontario) on November 14, 1994 under the name “Stellar International Inc.” On January 1, 2005 Tribute changed its name from Stellar International Inc. to Stellar Pharmaceuticals Inc. and on January 1, 2013 Tribute changed its name from Stellar Pharmaceuticals Inc. to Tribute Pharmaceuticals Canada Inc. On December 1, 2011, Stellar Pharmaceuticals Inc. acquired 100% of the outstanding shares of the then privately held Tribute Pharmaceuticals Canada Ltd. and Tribute Pharma Canada Inc. In 2015, Tribute opened its international subsidiary Tribute Pharmaceuticals International Inc. in Barbados and its US subsidiary, Tribute Pharmaceuticals US Inc., in Charlotte, North Carolina. Tribute maintains two facilities in Canada including its head office located at 151 Steeles Ave. East, Milton, Ontario, Canada L9T 1Y1, Tribute’s operations facility at 544 Egerton Street, London, Ontario, Canada N5W 3Z8. Additionally, Tribute’s US office is at 2015 Ayrsley Town Blvd, Suite 202 Charlotte, NC 28273 and in Barbados at Suite 203, Building Number 8, Harbour Road, St. Michael. Tribute’s main telephone number is 905-876-1118 in Milton and (519) 434-1540 in Londonits facsimile number is (519) 434-4382 and its e-mail address is info@tributepharma.com and its website is www.tributepharma.com. We are not including the information contained at www.tributepharma.com, or at any other Internet address as part of, or incorporating it by reference into, this registration statement.
2014 Highlights
During the second year of commercial launch, Cambia® (diclofenac potassium for oral solution) achieved consistent quarter-over-quarter growth throughout 2014 as illustrated below in Table 1. Furthermore, Cambia has continued to obtain coverage on many private insurance payor open plans in Canada. Cambia® is now widely available to Canadian patients.
In 2014, Tribute continued to expand its product portfolio which, combined with increased Cambia® sales, enabled it to increase revenues over 2013. Tribute is actively seeking to add more products to its sales portfolio in Canada, which will be supported by Tribute’s well-established sales force. Tribute is also seeking further growth from its internally developed proprietary products NeoVisc and Uracyst in countries where it does not yet have distribution agreements. Tribute has also begun looking for accretive, niche product opportunities in the U.S.
For the twelve month period ended December 31, 2014, total revenues from all sources increased by 25.5% or $3,431,400 to $16,871,800 compared to $13,440,400 in 2013, with licensed domestic product net sales contributing $507,600 or an increase of 5.9% over 2013, other domestic product sales contributing $2,761,600 up 82% over 2013 and an increase in international product sales contributing $341,700 up 26.7% compared to 2013.
Income from operations for the three month period ended December 31, 2014, was $582,100 compared to a loss of $1,310,900 for the same period in 2013, an improvement of 144.7% and income from operations excluding amortization of assets for the three month period ended December 31, 2014 was $1,209,500 compared to a loss of $1,033,800 for the same period in 2013, an improvement of 217.0%.
On May 13, 2014, Tribute announced the signing of an exclusive license agreement that grants Tribute the exclusive right to sell the Faes Farma SA product bilastine for the treatment of allergic rhinitis and chronic idiopathic urticaria (hives) in Canada, following receipt of regulatory approval from Health Canada for such product. The exclusive license is inclusive of prescription and non-prescription rights for bilastine, as well as adult and paediatric presentations in Canada.
On September 25, 2014 Tribute announced that it had received an additional patent from the USPTO for intellectual property that is central to one of Tribute’s lead products, Uracyst (a sterile sodium chondroitin sulfate solution, 2%, which is used in the treatment of interstitial cystitis / bladder pain syndrome (“IC/BPS”)). Tribute’s fourth Uracyst patent was issued as United States Patent No. 8,778,908 and is entitled “Cystitis Treatment with High Dose Chondroitin Sulfate.” This patent relates to the treatment of IC/BPS by instillation of a unit dose of chondroitin sulfate that is at least 350 mg or more, most preferably 400 mg, of chondroitin sulfate and 20 mL of an aqueous buffer. Unlike the earlier patents which covered how Uracyst is used, this patent covers the product itself. This patent provides market exclusivity through to 2024.
On October 2, 2014 Tribute announced that it had acquired the Canadian rights to Fiorinal, Fiorinal C, Visken and Viskazide from Novartis AG and Novartis Pharma AG (“Novartis Pharma”, collectively, with Novartis AG, “Novartis”) for $32 million which was paid in cash on closing. Combined net sales of such products during the twelve month period ended August 31, 2014, were approximately $10.8 million. Fiorinal and Fiorinal C are indicated for the relief of tension-type headache and Visken/Viskazide for the treatment of mild-moderate hypertension. Visken is also indicated for the prevention of angina pectoris.
On October 30, 2014 Tribute announced that it had received a patent approval from the European Patent Office for intellectual property central to one of Tribute’s lead products, Uracyst/Uropol® (a sterile sodium chondroitin sulfate solution, 2%), for the treatment of interstitial cystitis by instillation into the bladder of a patient. Treatments of other bladder disorders are also covered by the patent, including glycosaminoglycan (“GAG”) deficient forms of cystitis or GAG-deficiency, such as, accompanying chronic urinary tract infection, radiation-induced cystitis, chemical-induced cystitis or hemorrhagic cystitis. The European patent coverage extends to drug compositions comprising a unit dose of chondroitin sulfate in an amount of at least 400 mgs and an aqueous vehicle. This patent will run until February 18, 2024.
Q1- 2015 Highlights
IMS Health, an audited third party provider of sales data, reported a 10.8% increase in total prescriptions written for Cambia® during the three months ended March 31, 2015 compared to the three months ended December 31, 2014, or a 106.0% increase when comparing Q1 2015 and Q1 2014.
Products
Approved & Marketed Products
Cambia®
Cambia (diclofenac potassium for oral solution) was licensed from Nautilus Neurosciences, Inc. (“Nautilus”) in November 2010. Cambia® was approved by the FDA in June 2009 and is currently marketed by Depomed in the U.S. Cambia® was approved by Health Canada in March 2012 and was commercially launched to specialists in Canada in October 2012 and broadly to all primary care physicians in February 2013. The market for prescription migraine products in Canada is valued at approximately $126 million based on IMS Health data.
Cambia is a NSAID and the only prescription NSAID available and approved for the acute treatment of migraine attacks with or without aura in adults 18 years of age or older. Cambia is available as an oral solution in individual packets each designed to deliver a 50mg dose when mixed in water. Cambia is the only approved prescription NSAID available that was studied and proven to be an effective treatment for migraine according to guidelines published in September 2013 by the International Headache Society that reached statistically significant results for all four co-primary endpoints including: pain free response at two hours; nausea free; photophobia free (sensitivity to light); and phonophobia free (sensitivity to sound). In addition, Cambia provides fast migraine pain relief within 30 minutes of dosing due in part to the significant benefits of the proprietary Dynamic Buffering Technology™ (“DBT”). DBT provides for enhanced drug absorption and bioavailability. In fasting
volunteers, measurable plasma levels were observed within five minutes of dosing with Cambia. Peak plasma levels were achieved at approximately 15 minutes, with a range of approximately 10 to 40 minutes. The use of some NSAIDs has been associated with an increased incidence of cardiovascular adverse events such as myocardial infarction, stroke or thrombotic events. The risk may increase with duration of use and patients should only take this medication as prescribed by a physician.
Migraine Treatment Options: There are a number of different treatment options for migraine in Canada. Acute migraine treatment options can be broken down to three main categories: (i) triptans or 5-HT1 receptor agonists (e.g. sumatriptan, rizatriptan); (ii) ergot alkaloids (e.g., ergotamine, dihydroergotamine); and (iii) NSAIDs (Cambia). Triptans may cause dizziness, nausea, weakness and chest discomfort and should not be used by patients with heart disease, uncontrolled high blood pressure, blood vessel disease or who have a history of stroke. Ergots may cause chest pain, tingling or burning sensations, nausea, vomiting, and cramps. Furthermore, ergots may reduce blood flow to the extremities (hands and feet) and may lead to tissue damage. Ergots should also not be used by anyone with heart disease, uncontrolled high blood pressure or blood vessel disease. NSAIDs such as Cambia may increase the incidence of cardiovascular adverse events such as myocardial infarction, stroke or thrombotic events, gastrointestinal adverse events such as peptic/duodenal ulceration, perforation and gastrointestinal bleeding and are contraindicated in the third trimester of pregnancy.
In September 2013 the Canadian Neurological Sciences Federations issued revised Canadian Headache Society Guidelines for Acute Drug Therapy for Migraine Headaches through the Canadian Journal of Neurological Sciences. Cambia (diclofenac potassium for oral solution) was acknowledged as a potential first line therapy, with a fast onset of action and having a strong recommendation, high quality evidence and recommended for the acute treatment of migraine.
Migraine in Canada: The annual prescription migraine market in Canada is valued at approximately $126 million. Management estimates that four million women and one million men suffer from migraine headaches in Canada and that 60 percent of those with migraine have one or more attacks per month while 25 percent of those with migraine have at least one attack per week. One Canadian study found that those with migraine lose 6.5 days of work each year resulting from their migraine. According to a study conducted by Pryse Phillip, et al, published by the Canadian Journal of Neurological Sciences in 1992, they estimate that 7,000,000 working days are lost annually in Canada due to migraine and that direct and indirect cost in the workplace due to migraine is estimated at $500 million annually. It was also found that 48% of all women suffering from migraine have never consulted a physician for their headaches.
Competitive Analysis: It is estimated that half of all people suffering from migraine in Canada never seek help from a physician but rather self-treat their condition with OTC medications such as Aspirin® (Bayer), acetaminophen (Tylenol®) and OTC NSAID’s such as ibuprofen (Advil®) and naproxen sodium (Aleve®). The main prescription pharmacological agents used to treat acute migraine includes the triptan class of drugs or 5-HT1 receptor agonists as they are known and these products include sumatriptan (Imitrex®), rizatriptan (Maxalt®), zolmitriptan (Zomig®), almotriptan (Axert®), naratriptan (Amerge®), eletriptan (Relpax®) and frovatriptan (Frova®). There are also the ergot alkaloids such as ergotamine (Cafergot®) and dihydroergotamine (Migrinal®) used in some cases as are narcotics such as meperidine (Demerol) and the combination drug of aspirin, butalbital, and caffeine (Fiorinal®). In spite of a number of possible treatment options for treating migraines, many of these treatments are without a formal indication from Health Canada. Tribute considers the competitive market as the triptans class, which currently sells approximately $126 million annually in Canada.
Bezalip SR
Bezalip SR (bezafibrate) is a well-established pan-peroxisome proliferator-activated receptor (pan-PPAR) activator. Bezalip SR, used to treat hyperlipidemia, has over 25 years of therapeutic use globally with a good safety profile. Bezalip SR helps lower low-density lipoprotein cholesterol (LDL-C) and triglycerides while raising high-density lipoprotein cholesterol (HDL-C) levels. It also improves insulin sensitivity and reduces blood glucose levels, which in combination with the cholesterol effects may significantly lower the incidence of cardiovascular events and development of diabetes in patients with features of metabolic syndrome. Bezalip SR is under license from Actavis Group PTC ehf and is sold exclusively in Canada by Tribute. Tribute also has the exclusive development and licensing rights to Bezalip SR in the U.S. and recently filed an investigational new drug that received clearance from the FDA in the U.S. The initial target indication being pursued in the U.S. is for severe hypertriglyceridemia. Bezalip SR is contraindicated in patients with hepatic and renal impairment, pre-existing gallbladder disease, hypersensitivity to bezafibrate, or pregnancy or lactation.
Bezalip SR is currently approved in more than 40 countries worldwide, however is currently not approved in the U.S. According to a third party the U.S. fibrate and prescription fish oil market is estimated at nearly $2.5 billion in 2014. Upon approval, should such an approval be obtained, Tribute would enjoy a five year market exclusivity period from the FDA extended to all new chemical entities.
Hyperlipidemia Treatment Options: Hyperlipidemia, or high cholesterol, is a very common chronic condition and is characterized by an excess of fatty substances called lipids, mainly cholesterol and triglycerides, in the blood. It is also called hyperlipoproteinemia because these fatty substances travel in the blood attached to proteins. This is the only way that these fatty substances can remain dissolved while in circulation. Hyperlipidemia, in general, can be divided into two subcategories:
· Hypercholesterolemia, in which there is a high level of cholesterol; and
· Hypertriglyceridemia, in which there is a high level of triglycerides, the most common form of fat.
Competitive Analysis: Cholesterol-lowering drugs in Canada include: statins, niacin, bile-acid resins, fibric acid derivatives (fibrates), and cholesterol absorption inhibitors. All classes of cholesterol-lowering medicines are most effective when combined with increased exercise and a low-fat, high-fiber diet. The statin class includes some of the largest-selling prescription products in the world (Lipitor®, Zocor®, Crestor®, etc.). Statins dominate single-agent prescribing for the treatment of lipid disorders. The niacin (nicotinic acid — vitamin B3) class includes brands such as Niaspan®, which work primarily on increasing HDL cholesterol. The fibrates class of cholesterol lowering treatments is composed of three competing molecules: gemfibrozil (Lopid®), bezafibrate (Bezalip SR), and fenofibrate (Lipidil® in Canada or Tricor® in the U.S.). Clinical studies have demonstrated that bezafibrate, the active ingredient in Bezalip SR was shown to be very effective in lowering high levels of triglycerides, raising HDL cholesterol and lowering LDL cholesterol. As of the end of 2014, management estimates the annual fibrate market in Canada to be approximately $37 million.
Soriatane®
Soriatane (acitretin) is chemically known as acitretin, and is indicated for the treatment of severe psoriasis (including erythrodermic and pustular types) and other disorders of keratinization. Soriatane is a retinoid, an aromatic analog of vitamin A.
Soriatane was approved in Canada in 1994 and is the first and only oral retinoid indicated for psoriasis. Soriatane is often used when milder forms of psoriasis treatments like topical steroids, emollients and topical tar-based therapies have failed.
Soriatane should be reserved for patients unresponsive to, or intolerant of standard treatment. In addition, Soriatane should only be prescribed by physicians knowledgeable in the use of systemic retinoids. Soriatane is teratogenic (can cause birth defects) and should not be used by women who are pregnant or who are planning to become pregnant during, or within three years after stopping, treatment of Soriatane.
Psoriasis Treatment Options: There are a number of different treatment options for psoriasis. Typically, topical agents are used for mild disease, phototherapy for moderate disease and oral systemic agents and biologicals for more severe disease.
The three main traditional systemic treatments are methotrexate, cyclosporine and retinoids. Unlike Soriatane, methotrexate and cyclosporine are immunosuppressant drugs. Methotrexate may cause a decrease in the number of blood cells made by bone marrow, may cause liver damage, lung damage, damage to the lining of the mouth, stomach or intestines and may increase the risk of developing lymphoma (cancer that begins in the cells of the immune system), among other serious side effects. Methotrexate may also cause serious or life-threatening skin reactions. Cyclosporines may cause side effects that could be very serious, such as high blood pressure and kidney and liver problems. It may also reduce the body’s ability to fight infections.
Competitive Analysis: Severe psoriasis is a condition that involves more than 10% of the body area or is physically, occupationally or psychologically disabling. Soriatane will typically be used in combination with other drugs such as topical steroids, emollients or tar-based therapies. Soriatane is most effective for treating psoriasis when it is used with phototherapy. Soriatane is sometimes used with biologic agents such as etanercept (Enbrel®), adalimumab (Humira®) or infliximab (Remicade®) and may also be prescribed in rotation with cyclosporine or methotrexate. Biologic therapies such as Enbrel® Humira® and Remicade® are effective in treating severe forms of the disease, but are very expensive and sometimes not reimbursed by government or other private drug plans. Cyclosporine and methotrexate are also oral agents that are often used for severe forms of psoriasis. The market for moderate to severe psoriasis in Canada is estimated by management to be greater than $200 million for 2014.
Collatamp G
Tribute acquired the exclusive Canadian licensing rights for Collatamp G (restorable gentamicin-collagen haemostat) from Theramed Corporation in June 2012. EUSA Pharma (“EUSA”) owns the worldwide rights (except U.S.) to Collatamp G for the implant indication and licensed the product to Theramed in 2008. Collatamp G, approved by Health Canada on August 1, 2007 and launched in Canada in 2008, is a fully resorbable gentamicin-collagen haemostat, used as a surgical implant for haemostasis and local delivery of high doses of gentamicin. The market in which Collatamp G competes in Canada is estimated at $20 million based on best estimates from management.
Collatamp G is indicated for the local haemostasis of capillary, parenchymatous and seeping haemorrhages in areas with a high risk of infection and has been shown to reduce post-operative infections across a range of surgical disciplines, including a reduction of 53% in a large randomized controlled study in cardiac surgery. Based on internal data, Collatamp G is currently used in over 100 hospitals and surgical centers across Canada and is approved or used in over 50 countries throughout the world.
Collatamp G contains gentamicin sulphate at a locally effective dose and has been shown to be efficacious in the treatment and prevention of post-operative acquired infection across many surgical interventions including: cardiac surgery, gastro-intestinal surgery, vascular surgery and orthopaedic surgery.
Collatamp G is a unique product within the surgical field as it is the only product in Canada that combines a hemostatic device with a locally acting antibiotic.
Competitive Analysis: There are a number of haemostatic agents on the market in Canada and gentamycin is available as an intravenous drug but Collatamp G is unique in that it combines a haemostat with the antibiotic gentamycin in a topical, collagen matrix.
NeoVisc and NeoVisc Single Dose
NeoVisc is a proprietary product developed by Tribute and is used for the temporary replacement of synovial fluid in osteoarthritic joints. It is available as a triple-dosed, 2 mL pre-filled syringe of sterile 1.0% sodium hyaluronate solution and a single dose 6 mL pre-filled syringe of sterile 1.0% sodium hyaluronate solution. NeoVisc is classified in Canada by the Therapeutic Products Directorate (“TPD”) as a “medical device” under the Medical Devices Regulations of the Food and Drugs Act (Canada). NeoVisc is administered by intra-articular injection, by injecting the product directly into the affected joint and may be administered either as a single 6 mL injection or three 2 mL injections given over a two week period. Injections are typically repeated every six to eight months thereafter and dependent on the patient’s response. The market for NeoVisc® in Canada is estimated at $25 million based on management estimates.
This type of treatment, referred to as viscosupplementation, is a well-established treatment for OA of the knee, having gained Canadian approval in 1992 and United States approval in 1997. Viscosupplementation has also been used since the mid-1980s in many European markets. Replacing or supplementing the joint fluid provides symptomatic relief from the pain of OA of the knee for up to 4 to 12 months before repeated injections are required. In late 2003, the first single dose product was launched in Canada and by 2009 there were four single dose therapies available in the Canadian market, including NeoVisc single dose. Single dose products like NeoVisc offer convenience of a single injection but the clinical effect typically is shorter in duration than the triple dose administration.
Osteoarthritis and Treatment Options: OA is the most common form of chronic arthritis worldwide and is a key cause of pain and disability in older adults. According to the Arthritis Society of Canada, OA affects about 10% of the adult population. OA of the knee, about twice as common as OA of the hip, is becoming an increasingly prevalent condition with the aging population. OA risk factors include injury, prior joint inflammation, abnormalities of joint shape, and obesity. OA is a degenerative and sometimes painful disease that is associated with long term wear on weight-bearing joints. The market for OA is expected to grow significantly in future years as the average age of the population increases.
Current OA strategies and treatment goals include:
1. patient education;
2. physical therapy;
3. OTC analgesics;
4. NSAID, such as diclofenac and COX2 inhibitors such as Celebrex;
5. intra-articular viscosupplements, such as NeoVisc;
6. intra-articular steroids — corticosteroids are also used to treat inflammation associated with OA;
7. opioids; and
8. joint replacement — surgical replacement with artificial joints.
Products such as NeoVisc provide a non-pharmacological option in obtaining symptomatic improvements by supplementing the synovial fluid in the affected joint. NeoVisc can also be used in conjunction with other treatment strategies like physical therapy, OTC medications and NSAIDs, and as a result may reduce the amount of medication required and potentially delay joint replacement.
Competitive Analysis: There are a number of competitive viscosupplements to NeoVisc in Canada for both NeoVisc and NeoVisc Single Dose, including Sanofi’s products Synvisc® and Synvisc® One. The competitive landscape in the United States and other international markets is now very similar to the Canadian market. NeoVisc is an effective, technically engineered, highly purified, high molecular weight linear format, free of any avian peptides and available in a single or triple dose presentation. Furthermore, NeoVisc is the only marketed viscosupplement manufactured and packaged in Canada and marketed by a Canadian company.
Uracyst (Uropol)
Uracyst, developed by Tribute, is used in the treatment of certain forms of interstitial cystitis (“IC”) and non-common cystitis. Uracyst is a sterile 2.0% sodium chondroitin sulfate solution available in a 20 mL vial. This product is instilled by catheter directly into a patient’s bladder. Management is unaware of any reliable global market estimates of the IC market, but based on U.S. data, it has estimated that the global market is valued between $300 - $400 million. One of the difficulties in estimating the market size is the number of products used off-label as part of a multi-modal approach to the treatment of IC.
Uracyst provides symptomatic relief for patients suffering from GAG deficient cystitis such as IC and non-common cystitis (including radiation-induced cystitis and hemorrhagic cystitis) by supplementing and replenishing deficiencies in the glycosaminoglycan lining of the bladder wall. This GAG lining acts as a protective barrier between urine and the bladder wall. It protects the bladder wall against irritants and toxins (e.g., micro crystals, carcinogens and acid) in the urine and serves as an important defense mechanism against bacterial adherence. Many researchers believe that a large number of IC patients (over 70%) have “leaky” or deficient GAG layers in their bladder.
Uracyst is typically instilled weekly for six weeks, then once a month until symptoms resolve. Because these types of cystitis are typically chronic diseases of no known cause, patients will usually require re-treatment after a variable period of time when symptoms recur.
Tribute has been issued patents in the United States, Europe, China, Japan, Australia and Canada for the use of Uracyst treatments and has international patents pending. Uracyst is classified in Canada by TPD as a medical device under the Medical Devices Regulations of the Food and Drugs Act (Canada).
Interstitial Cystitis and Treatment Options: IC is a chronic inflammation of the bladder wall and is often associated with painful symptoms of the lower abdomen. Unlike common cystitis, IC is not caused by bacteria and does not respond to conventional antibiotic therapy. IC can affect people of any age, race or sex, but is more frequently diagnosed in women.
IC causes some or all of the following symptoms:
· Frequency: Day and/or night frequency of urination (up to 60 times a day in severe cases). In early or very mild cases, frequency is sometimes the only symptom;
· Urgency: Pain, pressure or spasms may also accompany the sensation of having to urinate immediately;
· Pain: Can be abdominal, urethral or vaginal. Pain is also frequently associated with sexual intercourse; and
· Other: Some patients also report experiencing symptoms such as muscle and joint pain, migraines, allergic reactions, colon and stomach problems, as well as the more common symptoms of IC described above.
At present, there is neither a cure for IC nor is there an effective treatment which works for everyone. The following treatments have been used to relieve the symptoms of IC in some people: (i) diet, (ii) bladder distention, (iii) instilled dimethyl sulfoxide (“DMSO”), heparin or hyaluronic acid, (iv) anti-inflammatory drugs, (v) antispasmodic drugs, (vi) antihistamines and (vii) muscle relaxants.
In severe cases, several types of surgery have been performed including bladder augmentation and urinary diversion. Products available for treating IC vary in their effectiveness. Most work for short periods of time and generally, are effective in about 30% to 40% of patients. Some therapies can take up to six months of active treatment before patients start to show symptomatic improvement.
Competitive Analysis: The treatment of IC is a relatively small niche market. Because of low efficacy rates and relatively expensive treatment costs for competitive products, management believes the treatment of IC remains an unsatisfied market with no dominant competitive product. Ortho McNeil Pharmaceutical, Inc. a competitor in the IC market has marketed Elmiron® (pentosan polysulfate sodium) in Canada since 1993. Elmiron is used as an oral GAG replenishment therapy. Side effects reported from the use of Elmiron include hair loss, diarrhea and mild to extreme gastrointestinal discomfort.
Fiorinal/Fiorinal C
Fiorinal and Fiorinal C (ASA, caffeine and butalbital tablets and capsules; ASA, caffeine, butalbital and codeine capsules) were acquired from Novartis in October 2014. Fiorinal and Fiorinal C were originally approved by Health Canada in 1970 for the relief of tension-type headache. The market for prescription tension-type headache products in Canada is estimated by management to be $30-40 million annually.
Fiorinal is a fixed dose combination drug which combines the analgesic properties of ASA, with the anxiolytic and muscle relaxant properties of butalbital, and the central nervous system stimulant properties of caffeine. Fiorinal C expands on the properties of Fiorinal with the additional analgesic effect of codeine. Fiorinal and Fiorinal C are the only prescription products indicated for relief of tension type headaches. Fiorinal and Fiorinal C are currently marketed in hard gelatin capsules
containing 330mg ASA, 40mg caffeine, 50mg butalbital, and in the case of Fiorinal C, the addition or 15mg or 30mg of codeine. Codeine and butalbital are both habit-forming and potentially abusable. Consequently, the extended use of Fiorinal or Fiorinal C is not recommended. Fiorinal and Fiorinal C are associated with exacerbation of headache (medication overuse headaches) in susceptible patients. Repeated use of Fiorinal and Fiorinal C can lead to “rebound” headaches as each dose wears off. With repeated doses physical and psychological dependence can develop. In addition to dependence, butalbital-containing products can lead to tolerance, and at higher doses can produce withdrawal symptoms after discontinuation.
Tension-Type Headache in Canada: The annual prescription tension-type headache market in Canada is valued at approximately $30 to $40 million. Tension-type headaches are the most common type of headache and are caused by muscle tightening in the back of the neck or scalp. These headaches are typically triggered by emotional stress, fatigue or depression. There are two classifications of tension-type headache; episodic tension headaches, which occur randomly and less frequently; and, chronic tension headaches, which may occur daily or continually and the intensity of the pain may vary during a 24-hour cycle. Tension headaches differ from migraine headaches due to the lack of aura, photophobia, phonophobia and/or nausea.
Competitive Analysis: Tension-type headache may be treated with OTC NSAIDs like Tylenol®, Advil®, Aleve®, or Aspirin®. Prescription NSAIDs may also be used, such as Naprosyn®, Anaprox®, Toradol®, as well as prescription analgesic/opiate combinations like Percocet®, Tylenol with codeine, and Fiorinal/Fiorinal C. In spite of a number of possible treatment options for treating tension-type headaches, all of these treatments are without a formal indication from Health Canada. Tribute considers the competitive market as the prescription NSAID and prescription analgesic/opiate combination class, which has an estimated tension-type headache value of $30-40 million annually in Canada. The OTC market for tension-type headache is estimated to be exponentially larger given the large patient population, however the true value is extremely difficult to determine considering the broad range of indications OTC NSAIDs are used for.
Visken/Viskazide
Visken and Viskazide (pindolol tablets; pindolol and hydrochlorothiazide fixed dose combination tablets) were acquired from Novartis in October 2014. Visken and Viskazide were originally approved by Health Canada in 1978 and 1983, respectively. Visken is indicated for the treatment of mild to moderate hypertension (high blood pressure) as well as the prophylaxis (prevention) of angina pectoris. Viskazide is indicated for the maintenance therapy of patients with hypertension who require pindolol and hydrochlorothiazide in the dosage and ratios present in Viskazide®. Both products are considered beta-blockers. According to IMS Health, an audited third party provider of sales data, the market for beta-blockers in Canada is approximately $135 million annually.
Visken (pindolol) is a beta-adrenergic-receptor-blocking agent which possesses partial agonist activity (intrinsic sympathomimetic activity - I.S.A.). Visken is usually used in combination with other drugs, particularly a thiazide diuretic. However, it may be used alone as an initial agent in those patients in whom, in the judgment of the physician, treatment should be started with a beta-blocker rather than a diuretic. The mechanism of the antihypertensive effect of Visken has not been established. Among the factors that may be involved are: a) competitive ability to antagonize catecholamine-induced tachycardia at the beta-receptor sites in the heart, thus decreasing cardiac output; b) a reduction in total peripheral resistance; c) inhibition of the vasomotor centres; and, d) inhibition of renin release by the kidneys. The mechanism of the antianginal effect of Visken has not been established. Visken may reduce the oxygen requirement of the heart at any level of effort by blocking catecholamine induced increases in the heart rate, systolic blood pressure, and the velocity and extent of myocardial contraction. However, oxygen requirements may be increased by such actions as increases in left ventricular fibre length, end diastolic pressure and the systolic ejection period. When the net effect is beneficial in patients with angina, it manifests itself during exercise or stress by delaying the onset of pain and reducing the incidence and severity of anginal attacks.
Viskazide combines the antihypertensive activity of two agents: a beta-adrenergic receptor-blocking agent (pindolol) and a diuretic (hydrochlorothiazide). Hydrochlorothiazide increases excretion of sodium and chloride in approximately equivalent amounts, and may cause a simultaneous, usually minimal, loss of bicarbonate. Natriuresis is usually accompanied by some loss of potassium. The mechanism of the antihypertensive effect of thiazides may be related to the excretion and redistribution of body sodium. Hydrochlorothiazide usually does not decrease normal blood pressure. The combination of pindolol with thiazide-like diuretics has been shown to be compatible and generally more effective than either of the drugs used alone in reducing elevated blood pressure. Special caution should be exercised when administering Visken or Viskazide to patients with a history of heart failure. Patients with angina should be warned against abrupt discontinuation of Visken or Viskazide. There have been reports of severe exacerbation of angina, and of myocardial infarction or ventricular arrhythmias occurring in patients with angina pectoris, following abrupt discontinuation of beta-blocker therapy. Various skin rashes and conjunctival xerosis have been reported with beta-blockers, including Visken and Viskazide. Sinus bradycardia may occur with the use of Visken due to unopposed vagal activity remaining after blockade of Beta 1-adrenergic receptors.
Hypertension and Beta-Blockers in Canada: The annual prescription beta-blocker market in Canada is valued at approximately $135 million. According to CHEP (Canadian Hypertension Education Program) more than one in five adult Canadians has hypertension and
the lifetime risk of developing hypertension is approximately 90%. CHEP also states that over 90% of Canadians with hypertension have additional cardiovascular risk factors, including an unhealthy diet, high dietary sodium intake, tobacco use, physical inactivity, abdominal obesity, dyslipidemia, and dysglycemia. Blood pressure and other cardiovascular risk factors can be improved by following a healthy diet, engaging in regular physical activity, moderating alcohol consumption, reducing dietary sodium, avoiding tobacco exposure and managing high stress levels. Most people with hypertension require lifestyle changes and pharmacotherapy to achieve recommended blood pressure targets. Diuretics like hydrochlorothiazide are often required for “resistant” hypertension.
Competitive Analysis: Hypertension is typically treated with a variety of therapeutic agents including diuretics, beta-blockers, calcium channel blockers, angiotensin II receptor blockers (ARBs), and angiotensin-converting-enzyme inhibitors (ACE inhibitors). CHEP guidelines recommend initial monotherapy with one of the agents listed above. If blood pressure targets are not achieved with monotherapy, then polytherapy utilizing a combination of the agents listed above is recommended. Other products in the beta-blocker class include; propranolol (Inderal® - Pfizer/Wyeth), metoprolol (Lopressor® - Novartis), carvedilol (Coreg® - GSK), labetalol (Trandate® - Paladin Labs (ENDO International plc)), timolol (Blocadren® - Merck), bisoprolol (Monocor® - Biovail/Valeant) and the recently launched nebivolol (Bystolic® - Actavis/Forest). Tribute considers the competitive market as the beta-blocker and beta-blocker diuretic fixed dose combination class, which has an estimated value of $135 million annually in Canada.
Fibricor
Fibricor (fenofibric acid tablets) U.S. rights were acquired from a wholly owned step down subsidiary of Sun Pharmaceutical Industries Ltd. in May 2015. Fibricor was originally approved by the FDA in 2009 and was subsequently launched by Mutual Pharmaceutical Company, Inc. Fibricor is indicated as adjunctive therapy to diet for treatment of severe hypertriglyceridemia (TG > 500 mg/dL) and as adjunctive therapy to diet to reduce elevated low-density lipoprotein cholesterol (LDL-C), total cholesterol (Total-C), triglycerides (TG), and apolipoprotein B (Apo B), and to increase high-density lipoprotein cholesterol (HDL-C) in patients with primary hypercholesterolemia or mixed dyslipidemia.
Fibricor is a unique and patent protected fenofibric acid formulation that competes in the ~US$2.5 billion triglyceride lowering medication market. Fibricor contains the lowest dose of fenofibric acid or fenofibrate available in the U.S., consisting of 105mg and 35mg tablet presentations. The product is protected by four patents extending out to August 20, 2027.
The MFI Products
Durela (tramadol HCI extended release capsules) is indicated for the management of moderate to moderately severe pain in adults who require continuous treatment for several days or more. Durela is available in a unique, patent protected formulation, consisting of an immediate release tablet, encapsulated with controlled release beads, providing for a unique drug release profile and once a day dosing. MFI licensed Durela from Cipher Pharmaceuticals Inc. in September 2011 and has marketed the product in Canada since 2012.
Proferrin is a unique heme iron polypeptide formulated in a tablet. Heme is a naturally sourced form of iron derived from bovine hemoglobin and is used to prevent and treat those at risk of iron deficiency. Each Proferrin tablet contains the equivalent of 11mg of elemental iron. Proferrin has a unique mechanism of action which results in optimal iron uptake, minimal side effects and equal efficacy with or without food. MFI originally licensed Proferrin from Colorado Biolabs, Inc. in December 2006.
Resultz (50% isopropyl myristate topical solution) is indicated for the treatment of head lice infestations in individuals two years and older. Resultz is a unique, non-toxic/pesticide free, patent protected, topical solution which is available without a prescription in pharmacies across Canada. MFI assumed the Canadian license to Resultz from Piedmont Pharmaceuticals LLC in August 2012.
The Balanse portfolio consists of probiotic combinations formulated in capsules or chewable tablets, used in the daily maintenance of a healthy and normal functioning digestive system.
Iberogast was originally launched by MFI in 2006. Iberogast is distributed by MFI in Canada under an exclusive license from Steigerwald Arzneimittelwerk GmbH, which was subsequently acquired by Bayer in 2013. Iberogast has been available in Europe since 1960, and plays a leading role in the treatment of irritable stomach and irritable bowel syndrome.
Pegalax was launched by MFI in 2009 and is indicated for the relief of occasional constipation and is available as a powder for solution in 17g single use sachets. The main active ingredient in Pegalax is polyethylene glycol 3350. Polyethylene glycol 3350 is in a class of medications called osmotic laxatives.
Purfem was launched by MFI in 2010 under an exclusive license from Bifodan A/S. Purfem is a vaginal suppository ovule which contains two probiotic ingredients. Purfem is indicated for preventing and treating vaginal itching, burning, dryness, odor and discharge caused by an imbalance of the vaginal microbial flora. Purfem is a unique product which competes in the vaginal fungicide market. As a probiotic, Purfem is the only product in this category which does not contain a fungicide such as
clotrimazole, miconazole, terconazole or nystatin. Purfem provides women with a unique, non-pharmacological, treatment option for bacterial vaginosis and yeast infections.
Mutaflor was launched by MFI in 2010 under an exclusive Canada and US license from Pharma Zentrale GmbH. Mutaflor is indicated for patients who need to normalize stool frequency and consistency in diarrhea and constipation. Furthermore, it is for patients who experience abdominal cramps and pain. Mutaflor has been shown to be effective in patients with ulcerative colitis, Crohn’s disease, pouchitis, collagenous colitis, irritable bowel syndrome, chronic constipation, diverticulitis and many other related disorders through over 80 years of clinical experience. The key active ingredient in Mutaflor is the probiotic Escherichia coli Nissle 1917, formulated into EC capsules for oral administration. Mutaflor is used as an adjunct to existing treatments for all of the above mentioned GI and lower abdominal disorders.
Diaflor was recently launched in the second quarter of 2015, was licensed from Cerbios-Pharma SA helps to reduce the duration of diarrhea in adults over the age of 18. Diaflor antagonizes enteric pathogens through biological competition, acidification of the gut environment, production of biologically active substances and modulation of the immune system to reduce the duration and severity of diarrhea, prevents the depletion of intestinal flora during antibiotic therapy and reconstitutes intestinal flora (“good bacteria”). Diaflor has a fast onset of action and significantly shortens the duration and severity of diarrhea, rapid elimination of diarrhea and related clinical disturbances and complete improvement and recovery faster in patients treated with Diaflor versus placebo.
Onypen was launched by MFI in 2011 under an exclusive license from YouMedical B.V. in the Netherlands. Onypen is a non-pharmacologic treatment for onychomycosis or nail fungal infections.
Moviprep, launched in Canada in 2011, is distributed under license from Norgine B.V. from the Netherlands. Moviprep is a gastrointestinal lavage used as a bowel cleanser prior to colonoscopy in adults over 18 years of age. The key active ingredient in Movi-Prep is Polyethylene Glycol 3350 (PEG 3350). The clinical efficacy derives from the osmotic action of PEG 3350, sodium sulphate, ascorbic acid and sodium ascorbate acting in concert. Due to the sequestration of water by PEG 3350, these ingredients exert a synergistic action, increasing the final osmolality to levels greater than would be theoretically calculated from the individual components.
Normacol is distributed under license from Norgine B.V. from the Netherlands. Normacol is a natural source fibre brand for the gentle relief of occasional constipation. The product is approved as an NPN in Canada and is available over the counter. Normacol would compete with other natural source fibre brands.
Product Development Strategy
Tribute is an specialty pharmaceutical company with a primary focus on the acquisition, licensing, development and promotion of healthcare products in Canada and the United States. In addition to growing the business in Canada, Tribute is also building revenues through out-licensing its current products to international markets, the cost of such activities is borne directly by the customers.
Tribute’s future product development efforts will be focused initially on developing strategic partners to assist Tribute in gaining regulatory approval in the U.S. and other key international markets for NeoVisc and Uracyst.
Tribute obtained FDA clearance for an investigational new drug related to Bezalip SR in October 2013. The fibrate and prescription fish oil market alone is estimated at approximately $2.5 billion annually in the U.S. and Tribute will explore all possibilities to obtain a market authorization for Bezalip SR in the U.S. including, but not limited to, identifying a development and commercialization partner for Bezalip SR. In March 2014, Tribute entered into an agreement with JSB-Partners (“JSB”), a global life sciences advisor, to support Tribute in finding a co-development and commercial partner for Tribute’s Bezalip SR (bezafibrate) in the U.S.
Sales and Marketing
Tribute’s sales and marketing strategy is focused on the organic growth of existing marketed products through several key activities. First, Tribute’s sales force ensures that it targets known prescribers of its medications or medications that compete with its products. Tribute creates demand by providing customers with reliable and trustworthy information from credible sources and by coordinating and facilitating continuing health education events in targeted areas. Second, Tribute supports its products by providing physicians and other healthcare practitioners with quality patient care materials. And third, Tribute ensures that its products are easy to purchase through all major wholesalers and distributors in Canada and manages its supply chain efficiently to ensure that it can meet demand.
Tribute considers its sales force to be very experienced and well trained. All of Tribute’s representatives have experience from other pharmaceutical companies including many of the largest companies in the industry. Additionally, Tribute offers its representatives a competitive incentive plan based on the achievement of results.
Manufacturing
Tribute currently outsources the manufacturing of its proprietary products to pharmaceutical manufacturing facilities operated by third party contractors. These facilities are in compliance with applicable Health Canada, TPD division medical device guidelines and cGMP regulations. Tribute believes these facilities have sufficient excess capacity at present to meet Tribute’s short and long term objectives. A significant interruption in the supply of any of Tribute’s products could impair the successful marketing of such products.
Tribute’s licensed products are manufactured by authorized, third-party, contract manufacturing organizations in various places throughout the world. Tribute’s manufacturers are all cGMP compliant and approved fabricators of pharmaceutical products according to Health Canada.
The manufacture of Tribute’s products involves the handling and use of substances that are subject to various environmental laws and regulations that impose limitations on the discharge of pollutants into the soil, air and water, and establish standards for their storage and disposal. Tribute believes that the manufacturers of its products are in material compliance with such environmental laws and regulations.
The sale and use of Tribute’s products entails risk of product liability and Tribute presently carries product liability insurance. There can be no assurance that, despite testing by Tribute, as well as testing by regulatory agencies, defects will not be found in new products after commencement of commercial shipments. The occurrence of such defects could result in the loss of, or delay in, market acceptance of Tribute’s products, which could have a material adverse effect on Tribute. Furthermore, litigation, regardless of its outcome, could result in substantial costs to Tribute, divert management’s attention and resources from Tribute’s operations, and result in negative publicity that might impair Tribute’s on-going marketing efforts.
Tribute is responsible for secondary packaging of its proprietary products at its London, Ontario facility. Tribute’s licensed products are packaged by its third party contract manufactures.
Tribute’s products are distributed in Canada by a third-party logistics provider, which provides warehousing, distribution, customer service and accounts receivable directly to Tribute.
Tribute, as a common practice for all of its products, maintains several months of inventory (including raw materials and finished goods) at any given time to mitigate against any risks of potential manufacturing disruptions. Tribute did not experience any product disruptions of any significance in 2014.
The Industry
The pharmaceutical industry is highly competitive and is characterized by rapidly changing technology. Tribute believes that competition in its markets is based on, among other things, product safety, efficacy, convenience of dosing, reliability, availability and price. The market is dominated by a small number of highly-concentrated global competitors, many of which boast substantially greater resources than Tribute. Given the size and scope of the competition, there can be no assurance that Tribute will maintain or grow its current market position in its therapeutic areas, or that developments by others will not render Tribute’s products or technologies non-competitive or obsolete. Also, many current and potential competitors of Tribute may have greater name and brand recognition, or may enjoy more extensive customer relationships that could be leveraged to gain market share to Tribute’s detriment. Tribute is unaware of any competitors who may be able to complete the regulatory approval process more rapidly than Tribute and therefore may achieve market entry ahead of Tribute’s products.
In order to maintain and improve its position in the industry, Tribute is dedicated to enhancing its current products, developing or acquiring new products and product extensions, and implementing a comprehensive domestic and international sales and distribution marketing strategy. If Tribute is not able to compete effectively against current and future competitors, such failure may result in fewer customer orders, reduced gross margins and profitability and loss of market share, any of which would materially adversely affect Tribute.
Competition
Tribute faces product competition from companies marketing competing pharmaceutical products and medical devices in Canada and potentially on new products that could be launched in the future. The introduction of generic products of Tribute’s products as well as lower priced, similar competing products could have a profound impact on Tribute’s existing business.
See also “Item 1. Business — Products” in Tribute’s Annual Report on Form 10-K for the period ended December 31, 2014, filed with the SEC on March 3, 2015, for a discussion of the other products that specifically compete with Tribute’s products.
Competitive Strengths
Management believes that Tribute maintains a high level of competitive advantage within its chosen therapeutic areas over other Canadian companies or multi-national subsidiaries seeking to license or acquire products in Canada. These include:
· a well trained and skilled sales force and employees;
· expertise in marketing new and existing products;
· its ability to obtain regulatory approvals for new and existing products in Canada and abroad;
· expertise in business development including sourcing, evaluation, negotiation and ability to complete business transactions to acquire or license new products for Canada;
· the ability to offer cost-effective pricing while maintaining acceptable gross profit margins with many of its products;
· the implementation and development of lifecycle management strategies;
· clear and defendable patents for certain of its products; and
· the ability to obtain reasonable reimbursement and good pricing in Canada.
REGULATORY, QUALITY ASSURANCES, SAFETY AND MEDICAL INFORMATION
Tribute currently utilizes a combination of internal and outsourced resources to address all of its quality assurance, regulatory affairs, pharmacovigilance and medical information needs. Tribute’s London, Ontario facility maintains a Health Canada Drug Establishment License and is ISO 13485 approved. Tribute is compliant with all regulatory guidelines and reporting obligations as of the issuance of this report.
Canadian Regulatory Overview
The TPD is the Canadian federal authority that regulates, evaluates and monitors the safety, effectiveness, and quality of drugs, medical devices, biologics and other therapeutic products available to Canadians. The TPD is part of Health Canada. The TPD’s regulatory process for review, approval and regulatory oversight of products is similar to the regulatory process conducted by the FDA in the United States, the European Medicines Agency (“EMA”) in the EU, and other regulatory agencies around the world.
Prior to being given market authorization for a product, a manufacturer must present substantive scientific evidence of a product’s safety, efficacy and quality as required by the Food and Drugs Act (Canada) and associated regulations. This information is submitted in the form of a New Drug Submission (“NDS”) in Canada.
The TPD performs a thorough review of the submitted information, sometimes using external consultants and advisory committees, to evaluate the potential benefits and risks of a drug. If, at the completion of the review, the conclusion is that the patient benefits outweigh the risks associated with the drug, the drug is issued a Notice of Compliance (“NOC”) and a Drug Identification Number (“DIN”), which permits the manufacturer to market the drug in Canada.
Currently, the process for the review of a drug typically takes an average of one to two years from the time that a manufacturer submits an NDS until the TPD approves a drug. The average time to approval varies but on average takes about 15 to 18 months.
All establishments engaged in the fabrication, packaging/labeling, importation, distribution, wholesale and operation of a testing laboratory relating to drugs are required to hold a Drug Establishment License unless expressly exempted under the Food and Drugs Act (Canada) and associated regulations. The basis for the issuance of a Drug Establishment License is compliance with cGMP as determined by inspection. Foreign sites whose products are being imported into Canada are also required to demonstrate cGMP compliance.
Regulatory obligations and oversight continue following the initial market approval. The manufacturer must report any new information received concerning serious side effects, including the failure of a drug to produce the desired effect. The manufacturer must also notify the TPD of any new safety issues that it becomes aware of after the launch of a product.
Canadian Reimbursement Overview
After regulatory approval is received for a prescription drug, it can be sold to the public in accordance with prescription pharmaceutical regulations. Revenues are generated from prescription drug sales in Canada through one of three sources:
· Cash: Patients will pay “out of pocket” at their sole expense. It is estimated that 10% of all prescription dollars spent in Canada come from cash purchases.
· Private Insurance: Approximately 45% of prescription dollars spent in Canada are reimbursed via third-party private insurers, under plans generally provided by patients’ employers. Patients may be reimbursed a percentage of the cost of covered drugs
minus deductibles or co-pays. The availability for reimbursement of drugs varies according to the type of reimbursement plan designed by the insurance company. There are a number of private insurers operating in Canada that provide employee plans to private and public sector employers.
· Government Drug Plans: Government drug plans cover the cost of nearly 45% of prescription dollars spent in Canada, and generally serve patients over the age of 65 or patients for whom the cost of medications represents a significant financial burden such as families receiving social assistance. Each provincial government pays the cost of drugs that are listed on their own provincial formulary.
After regulatory approval of a drug is granted, approval for reimbursement is typically sought from provincial governments and private insurance companies. Until provincial and private reimbursement is approved, the product is sold only via cash purchases. Decisions to list drugs for reimbursement on private and government formularies vary widely depending on the drug, indications, competitive products and price.
Hospital products or products dispensed in the hospital are treated differently in Canada. All medications taken while in a hospital are fully reimbursed by the provincial governments. If a patient leaves the hospital and is prescribed a drug to be taken at home, this prescription would be paid for either by cash, private insurance or public insurance plans.
Common Drug Review (CDR)
The CDR was implemented in 2003 to provide formulary listing recommendations for new drugs to participating publicly-funded federal, provincial and territorial drug benefit plans in Canada.
The CDR consists of:
· a systematic review of the available clinical evidence and a review of the pharmacoeconomic data for the drug; and
· a listing recommendation made by the Canadian Expert Drug Advisory Committee.
Based on the targeted timeframes of the CDR, a review should be completed approximately 20 to 26 weeks following receipt of a manufacturer’s submission, after which recommendations are made to participating drug plans.
At the provincial and territorial level, products are reviewed on the basis of their cost-effectiveness, comparable utility to other similar products, projected utilization and cost implications to the publicly-funded drug budget. Each submission is reviewed but there is wide variance in the formulary decisions and the time taken to make such decisions. Provinces and territories may utilize the recommendations of the CDR or perform their own analysis.
Presently, all provinces and territories except Quebec use the CDR recommendations in their assessment, but make their formulary decisions independently from the CDR. In many provinces, the formulary committee may grant “restricted or limited use approvals” for a drug as a means of regulating the size of the patient population eligible for reimbursement for the cost of the drug and by encouraging physicians to use older generation products first before prescribing newer, sometimes more costly medications.
Patent and Proprietary Protection
Tribute is able to protect its technology from unauthorized use by third parties only to the extent that it is covered by valid and enforceable patents or is effectively maintained as a trade secret or is protected by confidentiality agreements. Accordingly, patents or other proprietary rights are an essential element of Tribute’s business.
Tribute currently has patents issued for Uracyst that include a low dose patent in both the United States (Patent No. 6,083,933 —- issued 07/04/2000 and expiring 04/19/2019) and Canada (Patent No. 2,269,260 — issued 12/31/2002 and expiring 04/16/2019). In addition, Tribute has approved patents for its high dose product in Australia (Patent No. AU 2004212650 — issued 11/05/2009 and expiring 02/18/2024), Canada (Patent No. 2,515,512 — issued 07/10/2012 and expiring 02/18/2024), China (Patent No. ZL200480006467.1 — issued 05/26/2010 and expiring 02/18/2024), Europe (Patent No. 1,603,578 — issued 10/29/2014 and expiring 02/18/2024), the United States (Patent No. US 7,772,210 — issued 08/10/2010, Patent No. US 8,084,441 — issued 12/27/2011, Patent No. 8,334,276 — issued 12/18/2012, and Patent No. 8,778,908 — issued 07/15/2014, each expiring 02/19/2023), and Japan (Patent JP 4,778,888 — issued 07/08/2011 and expiring 02/18/2024). Jurisdictions with patents pending related to the high dose Uracyst include: India, and Israel. Uracyst is classified as a medical device in all countries in which it currently has approval.
Tribute has rights to patents on Cambia through its licensing agreement with Depomed, Inc. (previously Nautilus Neuroscience) (Patent No. 2,254,144 expiring 05/15/2017) and there is also one patent pending for Cambia in Canada.
Tribute has rights to patents covering bilastine through its licensing agreement with Faes Farma SA (Patent Nos. 2,206,754 issued 01/23/2007 and expiring 06/03/2017; and 2,484,460 issued 09/29/2009 and expiring 04/19/2022).
Tribute has U.S. patents covering Fibricor (Patent No.7569612 issued on 08/04/2009 and expiring 08/20/2027; Patent No. 7741373 issued on 06/22/2010 and expiring 08/20/2027; Patent No. 7741374 issued on 06/22/2010 and expiring 08/20/2027; and Patent No. 7915247 issued on 03/29/2011 and expiring 08/20/2027).
Tribute has rights to a Canadian patent covering Durela through its licensing agreement with Cipher Pharmaceutical Inc. (Patent No. 2495463 issued on 07/14/2011 and expiring on 10/24/2022).
Tribute has rights to a Canadian patent covering Moviprep through its licensing agreement with Norgine B.V. (Patent No. 2502103 issued on 04/07/2009 and expiring on 10/24/2023).
Tribute has rights to Canadian patents covering Resultz and Bedbugz through its licensing agreement with Piedmont Pharmaceuticals LLC (Patent No. 2484183 issued on 06/14/2011 and expiring on 04/14/2023; and Patent Application No. 2497145 which if granted will expire on 08/27/2023).
While trade secret protection is an essential element of Tribute’s business and it has taken security measures to protect its proprietary information and trade secrets, Tribute cannot give assurance that its unpatented proprietary technology will afford it significant commercial protection. Tribute seeks to protect its trade secrets by entering into confidentiality agreements with third parties, employees and consultants. Tribute’s employees and consultants also sign agreements requiring that they assign to Tribute their interests in intellectual property arising from their work for Tribute. All employees sign an agreement not to engage in any conflicting employment or activity during their employment with Tribute and not to disclose or misuse its confidential information. However, it is possible that these agreements may be breached or invalidated, and if so, there may not be an adequate corrective remedy available. Accordingly, Tribute cannot ensure that employees, consultants or third parties will not breach the confidentiality provisions in Tribute’s contracts, infringe or misappropriate Tribute’s trade secrets and other proprietary rights or that measures Tribute is taking to protect its proprietary rights will be adequate.
Where deemed appropriate, Tribute files patent applications for products or technologies which it owns or in respect of which it has acquired a license and, if necessary, then further developed to make such technologies marketable. Such applications may cover composition of matter, the production of active ingredients and their novel applications and may be filed globally or in select territories that Tribute may intend to commercialize its products. Licensed products often include rights to the intellectual property (“IP”) of the licensor.
Tribute retains independent patent counsel where appropriate. Management of Tribute believes that the use of outside patent specialists ensures prompt filing of patent applications, as well as the ability to access specialists in various areas of patents and patent law to ensure complete patent filings.
The patent position relating to medical devices and drug development is uncertain and involves many complex legal, scientific and factual questions. While Tribute intends to protect its valuable proprietary information and believes that certain of its information is novel and patentable, there can be no assurance that: (i) any patent application owned by, or licensed to, Tribute will issue to patent in all or any countries; (ii) proceedings will not be commenced seeking to challenge Tribute’s patent rights or that such challenges will not be successful; (iii) proceedings taken against a third party for infringement of patent rights will be successful; (iv) processes or products of Tribute will not infringe upon the patents of third parties; or (v) the scope of patents issued to, or licensed by, Tribute will successfully prevent third parties from developing similar and competitive products. The cost of litigation to uphold the validity and prevent infringement of the patents owned by, or licensed to, Tribute may be significant.
Issues may arise with respect to claims of others to rights in the patents or patent applications owned by or licensed to Tribute. As the industry expands and more patents are issued, the risk increases that Tribute’s processes and products may give rise to claims that they infringe the patents of others. Actions could be brought against Tribute or its commercial partners claiming damages or an accounting of profits and seeking to enjoin them from clinically testing, manufacturing and marketing the affected product or process. If any such action were successful, in addition to any potential liability for damages, Tribute or its commercial partners could be required to obtain a license in order to continue to manufacture or market the affected product or use the affected process. There can be no assurance that Tribute or its commercial partners could prevail in any such action or that any license required under any such patent would be made available or, if available, would be available on acceptable terms. If no license is available, Tribute’s ability to commercialize its products may be negatively affected. There may be significant litigation in the industry regarding patents and other intellectual property rights and such litigation could consume substantial resources. If required, Tribute may seek to negotiate licenses under competitive or blocking patents, which it believes are required for it to commercialize its products.
Although the scope of patent protection ultimately afforded by the patents and patent applications owned by or licensed to Tribute is difficult to quantify, management of Tribute believes that such patents will afford adequate protection for it to ensure exclusivity in the conduct of its business operations as described in this prospectus. Tribute also intends to rely upon trade secrets, confidential and unpatented proprietary know-how, and continuing technological innovation to develop and maintain its competitive position. To protect these rights, Tribute whenever possible requires all employees and consultants to enter into confidentiality agreements with Tribute. There can be no assurance, however, that these agreements will provide meaningful protection for Tribute’s trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Further, in the absence of patent protection, Tribute’s business may be adversely affected by competitors who independently develop substantially equivalent or superior technology.
The existence, scope and duration of patent protection vary among Tribute’s products and among the different countries where Tribute’s products may be sold. They may also change over the course of time as patents are granted or expire, or become extended, modified or revoked.
Pricing and Reimbursement
As pressures for cost containment increase, particularly in Canada, the United States and the EU, there can be no assurance that the prices Tribute can charge for its products will be as favorable as historical pharmaceutical product prices. Reimbursement by government, private insurance organizations, and other healthcare payers has become increasingly important, as has the listing of new products on large formularies, such as those of pharmaceutical benefit providers and group buying organizations. The failure of one or more products to be included on formulary lists, or to be reimbursed by government or private insurance organizations, could have a negative impact on Tribute’s results of operation and financial condition.
Product Pricing Regulation on Certain Patented Drug Products
Patented drug products in Canada are subjected to the regulation of the Patented Medicine Prices Review Board (“PMPRB”). The PMPRB’s objective is to ensure that prices of patented products in Canada are not excessive. For new patented products, the price in Canada is limited to either the cost of existing drugs sold in Canada or the median of prices for the same drug sold in other specified industrial countries. For existing patented products, prices cannot increase by more than the Consumer Price Index. The PMPRB monitors compliance through a review of the average transaction price of each patented drug product as reported by Tribute over a recurring six-month reporting period.
The PMPRB does not approve prices for drug products in advance of their introduction to the market. The PMPRB provides guidelines from which companies like Tribute set their prices at the time they launch their products. All patented pharmaceutical products introduced in Canada are subject to the post-approval, post launch scrutiny of the PMPRB. Since the PMPRB does not pre-approve prices for a patented drug product in Canada, there may be risk involved in the determination of an allowable price selected for a patented drug product at the time of introduction to the market by Tribute launching such products in Canada. If the PMPRB does not agree with the pricing assumptions chosen by such company introducing a new drug product, the price chosen could be challenged by the PMPRB and, if it is determined that the price charged is excessive, the price of the product may be reduced and a fine may be levied against Tribute for any amount deemed to be in excess of the allowable price determined. Drug products that have no valid patents are not subject to review by the PMPRB.
License Agreements
On December 1, 2011, Tribute acquired 100% of the outstanding shares of Tribute Pharmaceuticals Canada Ltd. and Tribute Pharma Canada Inc. Included in this transaction were the following license agreements:
On June 30, 2008, Tribute signed a Sales, Marketing and Distribution Agreement with Actavis Group PTC ehf (“Actavis”) to perform certain sales, marketing, distribution, finance and other general management services in Canada in connection with the importation, marketing, sales and distribution of Bezalip SR and Soriatane (the “Actavis Products”). On January 1, 2010, a first amendment was signed with Actavis to grant Tribute the right and obligation to more actively market and promote the Actavis Products in Canada. On March 31, 2011, a second amendment was signed with Actavis that extended the term of the agreement, modified the terms of the agreement and increased Tribute’s responsibilities to include the day-to-day management of regulatory affairs, pharmacovigilance and medical information relating to the Actavis Products. Tribute pays Actavis a sales and distribution fee up to an annual base-line net sales forecast plus an incremental fee for incremental net sales above the base-line. Tribute agreed and fulfilled a marketing budget for the first three years of not less than $3,750,000. On May 4, 2011, Tribute signed a Product Development and Profit Share Agreement with Actavis to develop, obtain regulatory approval of and market Bezalip SR in the U.S. Tribute shall pay US$5,000,000 to Actavis within 30 days of receipt of the regulatory approval to market Bezalip SR in the U.S.
On November 9, 2010, Tribute signed a license agreement with Nautilus Neurosciences, Inc. (“Nautilus”) for the exclusive rights to develop, register, promote, manufacture, use, market, distribute and sell Cambia in Canada. On August 11, 2011, Tribute and
Nautilus executed the first amendment to the license agreement and on September 30, 2012 executed the second amendment to the license agreement. Aggregate payments of US$1,000,000 were issued under this agreement, which included an upfront payment to Nautilus upon the execution of the agreement and an amount payable upon the first commercial sale of the product. These payments have been included in intangible assets and will be amortized over the life of the license agreement, as amended. Up to US$6,000,000 in additional one-time performance based sales milestones, based on a maximum of six different sales tiers, are payable over time, due upon achieving annual net sales ranging from US$2,500,000 to US$20,000,000 in the first year of the achievement of the applicable milestone. Royalty rates are tiered and payable at rates ranging from 22.5% to 25.0% of net sales.
Subsequent to Tribute’s acquisition of Tribute Pharmaceuticals Canada Ltd. and Tribute Pharma Canada Inc. additional license agreements, as described herein, have been executed:
On May 13, 2014, Tribute entered into an exclusive license and supply agreement with Faes Farma, S.A. (“Faes”), a Spanish pharmaceutical company, for the exclusive right to sell bilastine, a product for the treatment of allergic rhinitis and chronic idiopathic urticaria (hives) in Canada. The exclusive license is inclusive of prescription and non-prescription rights for bilastine, as well as adult and paediatric presentations in Canada. Sales of bilastine are subject to receiving regulatory approval from Health Canada. Payment for the licensing rights is based on an initial fee of €250,000 ($373,576) with the remaining milestone payments based on the achievement of specific events, including the approval of bilastine from Health Canada and net sales milestones.
On October 2, 2014, Tribute entered into an asset purchase agreement (the “Asset Purchase Agreement”) with Novartis (together with Tribute, the “Parties”) pursuant to which Tribute acquired from Novartis the Canadian rights to manufacture, market, promote, distribute and sell Fiorinal, Fiorinal C, Visken and Viskazide for the relief of pain from headache and for the treatment of cardiovascular conditions (in this paragraph defined as the “Novartis Products”), as well as certain other assets relating to the Novartis Products, including certain intellectual property, marketing authorizations and related data, medical, commercial and technical information, and the partial assignment of certain manufacturing and supply agreements and tenders with third parties (the “Acquired Assets”). Tribute also assumed certain liabilities arising out of the Acquired Assets and the Licensed Assets (as described below) after the acquisition, including product liability claims or intellectual property infringement claims by third parties relating to the sale of the Novartis Products by Tribute in Canada. In connection with the acquisition of the Acquired Assets, and pursuant to the terms of the Asset Purchase Agreement, Tribute concurrently entered into a license agreement with Novartis AG, Novartis Pharma AG and Novartis Pharmaceuticals Canada Inc. (the “License Agreement”, and, together with the Asset Purchase Agreement, the “Agreements”). Pursuant to the terms of the License Agreement, the Novartis entities agreed to license to Tribute certain assets relating to the Novartis Products, including certain intellectual property, marketing authorizations and related data, and medical, commercial and technical information (the “Licensed Assets”). Tribute concurrently entered into a supply agreement with Novartis Pharma AG, pursuant to which Novartis Pharma AG agreed to supply Tribute with the requirements of Novartis Products for sale for a transition period until Tribute is able to transfer the marketing authorizations to Tribute. The consideration paid for the Acquired Assets and the Licensed Assets was $32,000,000 in cash.
On May 21, 2015, Tribute Pharmaceuticals International Inc., a wholly-owned subsidiary of Tribute entered into an asset purchase agreement with Mutual Pharmaceutical Company, Inc. and Sun Pharmaceutical Industries, Inc., pursuant to which Tribute International acquired from Sun Pharma the U.S. rights to manufacture, market, promote, distribute and sell Fibricor and its related authorized generic (“AG”), as well as certain other assets relating to the Fibricor and the AG, including certain inventory, documentation, licenses, approvals, consents, franchises, authorizations, and security clearances of, to, from or with any governmental entity relating to Fibricor and the AG (that are assignable), and all intellectual property used in connection with Sun Pharma’s commercialization of Fibricor and the AG. Tribute International also agreed to assume liabilities relating to the transaction after the closing date.
Fibricor is indicated as adjunctive therapy to diet for treatment of severe hypertriglyceridemia (TG > 500 mg/dL) and as adjunctive therapy to diet to reduce elevated low-density lipoprotein cholesterol (LDL-C), total cholesterol (Total-C), triglycerides (TG), and apolipoprotein B (Apo B), and to increase high-density lipoprotein cholesterol (HDL-C) in patients with primary hypercholesterolemia or mixed dyslipidemia.
The consideration to be paid for Fibricor and AG is US$10 million, with US$5 million paid on the closing date, US$2 million payable 180 days from the closing date, and US$3 million payable 365 days from the closing date. Tribute funded the payment due on the closing date with cash from a concurrent financing. In addition, Tribute International agreed to make certain milestone payments and royalty payments to Sun Pharma based on Tribute International’s achievement of certain net sales figures.
On June 16, 2015, Tribute entered into a share purchase agreement with the shareholders of MFI pursuant to which Tribute acquired all of the outstanding shares of MFI (the “MFI Shares”). The consideration paid for the MFI Shares was comprised of (1) Cdn$8.3 million in cash on closing, (2) Cdn$5 million through the issuance of 3,723,008 common shares of Tribute, (3) Cdn$5 million in the form of a one-year term promissory note bearing interest at 8% annually convertible in whole or in part at the holder’s option at any time during the term into 2,813,778 common shares of Tribute at a conversion rate of Cdn$1.77
per common share (subject to adjustment in certain events), and (4) future contingent cash milestone payments totaling Cdn$6 million that will be paid upon the achievement of certain conditions, including the receipt of regulatory approval for MFI’s two pipeline products described below and certain required consents.
This acquisition diversifies Tribute’s product portfolio in Canada through the addition of 13 marketed products (Durela, Proferrin, Iberogast, Moviprep, Normacol, Resultz, Pegalax, Balanse, Balanse Kids, Balanse Diaflor, Purfem, and Onypen) and 2 pipeline products (Octasa and BedBugz, both of which are pending submission to Health Canada).
Other Laws and Regulations
Tribute’s operations are or may be subject to various federal, provincial, state and local laws, regulations and recommendations relating to the marketing of products and relationships with treating physicians, data protection, safe working conditions, laboratory and manufacturing practices, patient safety, the export of products to certain countries and the purchase, storage, movement, use and disposal of hazardous or potentially hazardous substances. Although Tribute believes its safety procedures comply with the standards prescribed by federal, provincial, state and local regulations, the risk of contamination, injury or other accidental harm cannot be eliminated completely. In the event of an accident, Tribute could be held liable for any damages that result. The amount of such damages could have a materially adverse effect on Tribute’s results of operations and financial condition.
Employees
Tribute currently employs forty-nine employees including 46 full-time, one part-time and two contract employees. 33 of these employees are in sales and marketing and the remainder are in management and administration positions. Tribute may add additional staff in areas that its management may feel is necessary for the successful operation of Tribute.
Customers
During the year ended December 31, 2014, Tribute had three significant pharmaceutical wholesale customers that account for 60.1% (McKesson Pharmaceutical — 33.3%, Kohl & Frisch — 11.8% and Shoppers Drug Mart Inc. - 15.0%) of Tribute’s sales. This is normal and customary in the Canadian pharmaceutical business. These are well known and highly respected customers that have a solid track record of paying all outstanding amounts owing on time and Tribute does not anticipate that this will materially change in 2015.
Since March 2002, our corporate facilities have been located in 17,000 square feet in the Exchange Office Building in Chapel Hill, North Carolina under a lease commencing in March 2002 and expiring in 2010. We have exercised our option to renew this lease for an additional five year and seven month term, terminating on September 30, 2015, and we have an additional option to renew the extended term for one additional three year period. We believe that the Exchange Office Building facility is adequate for our current needs and that suitable additional or alternative space will be available in the future on commercially reasonable terms. On July 15, 2015, the Company signed a six-month extension to its lease, adding approximately $52,000 to its lease commitments.
On March 14, 2011, we and AstraZeneca received a Paragraph IV Notice Letter from Dr. Reddy’s informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent in 2023. The ‘907 patent is assigned to POZEN and listed with respect to VIMOVO in the Orange Book. On September 19, 2011, Dr. Reddy’s amended its ANDA to include a Paragraph IV certification against the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, which are assigned to AstraZeneca or its affiliates and listed in the Orange Book, with respect to VIMOVO. The patents listed in the Orange Book which are owned by AstraZeneca or its affiliates expire at various times between 2014 and 2018. AstraZeneca advised us that it elected to exercise its first right to prosecute the infringement suit against Dr. Reddy’s. Accordingly, we and AstraZeneca filed suit against Dr. Reddy’s on April 21, 2011 in the United States District Court for the District of New Jersey, asserting only the ‘907 patent against Dr. Reddy’s. An amended complaint was filed on October 28, 2011 to include the AstraZeneca patents. On December 19, 2012, the District Court conducted a pre-trial “Markman” hearing to determine claim construction. On May 1, 2012, the Court issued a Markman Order construing the claim terms disputed by the parties. On April 15, 2013 a Stipulation of Partial Dismissal was filed which sought dismissal of all infringement claims relating to the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent (each of which is assigned to AstraZeneca), as well as Dr. Reddy’s defenses and counterclaims relating to those patents. On April 18, 2013, the District Court issued a Stipulation and Order dismissing with prejudice those claims and defenses. The first Dr. Reddy’s case is considered the lead case and has been consolidated with the actions described below for the purpose of pre-trial and discovery. A scheduling order for this case, and all of the consolidated cases, was issued by the Court on June 27, 2014. Fact discovery closed in the consolidated case on November 20, 2014 and expert discovery closed on June 25, 2015. In view of the retirement of presiding Judge Pisano, on February 9, 2015, the consolidated cases were reassigned to Judge Mary L. Cooper.
On June 13, 2011, we and AstraZeneca received a Paragraph IV Notice Letter from Lupin informing us that Lupin had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to POZEN and the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, the ‘466 patent, each of which is assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Lupin’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca advised us that it elected to exercise its first right to prosecute the infringement suit against Lupin and, accordingly, we and AstraZeneca filed suit against Lupin on July 25, 2011 in the United States District Court for the District of New Jersey. On November 19, 2014, an amended complaint was filed in which the 504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, all assigned to AstraZeneca or its affiliates, were not asserted against Lupin. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On September 19, 2011, we and AstraZeneca received Paragraph IV Notice Letter from Anchen informing us that Anchen had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, the ‘085 patent, the ‘070 patent, and the ‘466 patent. The patents are among those listed with respect to VIMOVO in the Orange Book and expire at various times between 2018 and 2023. Anchen’s Paragraph IV Notice Letter asserts that its generic product will not infringe those patents or that those patents are invalid or unenforceable. AstraZeneca advised us that it elected to exercise its first right to
prosecute the infringement suit against Anchen and, accordingly, we and AstraZeneca filed suit against Anchen on October 28, 2011 in the United States District Court for the District of New Jersey. On October 4, 2013, Anchen filed an amendment to its ANDA seeking to change its Paragraph IV certification to a Paragraph III. It is unclear when or if the FDA will enter Anchen’s amendment. On October 25, 2013, Anchen filed a Motion to Dismiss the case against it, based on its proposed re-certification. On November 18, 2013, we and AstraZeneca filed an Opposition to Anchen’s Motion to Dismiss. On June 11, 2014, the Court granted Anchen’s Motion and dismissed the case against them.
On November 20, 2012 we and AstraZeneca received a Paragraph IV Notice Letter from Dr. Reddy’s, informing us that Dr. Reddy’s had filed a second ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to POZEN and the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, the ‘466 patent, each of which is assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Dr. Reddy’s second Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca advised us that it elected to exercise its first right to prosecute the infringement suit against Dr. Reddy’s on its second ANDA filing and, accordingly, we and AstraZeneca filed suit against Dr. Reddy’s on January 4, 2013, in the United States District Court for the District of New Jersey. On April 15, 2013 a Stipulation of Partial Dismissal was filed which sought dismissal of all infringement claims relating to the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent (which are each assigned to AstraZeneca), as well as Dr. Reddy’s defenses and counterclaims relating to those patents. On April 18, 2013, the District Court issued the Stipulation and Order dismissing with prejudice those claims and defenses. On June 28, 2013 we and AstraZeneca filed a Motion for Summary Judgment relating to the second ANDA filing asserting that U.S. Patent No. 6,926,907 is not invalid. On August 12, 2013, Dr. Reddy’s filed an opposition to the Motion for Summary Judgment. On March 28, 2014, the District Court denied the Motion. On October 11, 2013, Dr. Reddy’s filed a Motion for Summary Judgment asserting that the product which is the subject matter of its second ANDA does not infringe the ‘907 patent. On November 4, 2013, we and AstraZeneca filed a Motion for an Order Denying Dr. Reddy’s Motion for Summary Judgment Pursuant to Rule 56(d) and an Opposition to Dr. Reddy’s Motion. On May 29, 2014, the Court issued an order denying Dr. Reddy’s Motion. On July 9, 2015, Dr. Reddy’s renewed its Motion for Summary Judgment that the product which is the subject matter of its second ANDA does not infringe the ‘907 patent. This case was consolidated with the originally filed Dr. Reddy’s case and is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On March 29, 2013, we and AstraZeneca received a Paragraph IV Notice Letter from Watson Laboratories, Inc.–Florida, or Watson, now Actavis, informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the Company and the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which is assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Watson’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca advised us that it elected to exercise its first right to prosecute the infringement suit against Watson. On May 10, 2013, we and AstraZeneca filed a patent infringement lawsuit against Watson in the U.S. District Court of New Jersey. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On May 16, 2013, we and AstraZeneca received a Paragraph IV Notice Letter from Mylan informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the Company and the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which is assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Mylan’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca advised us that it elected to exercise its first right to prosecute the infringement suit against Mylan. On June 28, 2013, we and AstraZeneca filed a patent infringement lawsuit against Mylan in the U.S. District Court of New Jersey. On February 13, 2015, the Court entered a joint stipulation of dismissal of counts related to certain patents, dismissing claims related to the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which is
assigned to AstraZeneca or its affiliates. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On October 15, 2013, the United States Patent Office issued the ‘285 patent. The ‘285 patent, entitled “Pharmaceutical compositions for the coordinated delivery of NSAIDs” and assigned to POZEN, is related to the ‘907 patent. AstraZeneca advised us that it elected to exercise its first right to prosecute the infringement of the ‘285 patent and, accordingly, on October 23, 2013, we, and AstraZeneca filed patent infringement lawsuits against Dr. Reddy’s, Lupin, Watson and Mylan in the U.S. District Court of New Jersey alleging that their ANDA products infringe the ‘285 patent. On November 8, 2013, we, and AstraZeneca filed a Motion to Amend the Complaint in the actions against Dr. Reddy’s, Lupin, Watson and Mylan or, in the alternative, to consolidate the actions involving the ‘285 patent with the existing consolidated action. Dr. Reddy’s, Lupin, Watson and Mylan have each filed answers to the respective amended complaints, thus adding claims relating to the ‘285 patent against each of the Defendants to the consolidated case.
As part of the purchase of all of AstraZeneca’s rights, title and interest to develop, commercialize and sell VIMOVO in the United States, Horizon Pharma Inc., or Horizon, has assumed AstraZeneca’s right to lead the above-described Paragraph IV litigation relating to VIMOVO currently pending in the United States District Court for the District of New Jersey and has assumed patent-related defense costs relating to such litigation, including reimbursement up to specified amounts of the cost of any counsel retained by us. On December 12, 2013, Horizon filed Motions to Join under Fed.R.Civ.Proc. 25(c) as a co-plaintiff in each of the above referenced actions and the consolidated action. On January 31, 2014 and February 2, 2014, the Court granted Horizon’s motions.
On October 7, 2014, the United States Patent Office issued United States Patent No. 8,852.636 (the “‘636 patent”). The ‘636 patent, entitled “Pharmaceutical compositions for the coordinated delivery of NSAIDs” and assigned to POZEN, is related to the ‘907 and ‘285 patents. On October 14, 2014, the United States Patent Office issued United States Patent No. 8,858,996 (the “‘996 patent”). The ‘996 patent, entitled “Pharmaceutical compositions for the coordinated delivery of NSAIDs” and assigned to POZEN, is also related to the ‘907 and ‘285 patents. On October 21, 2014, the United States Patent Office issued United States Patent No. 8,865,190 (the “‘190 patent”) ‘190 patent”. The ‘190 patent, entitled “Pharmaceutical compositions for the coordinated delivery of NSAIDs” and assigned to POZEN, is related to the ‘907 and ‘285 patents. Horizon advised us that it elected to exercise its first right to prosecute the infringement of the ‘636, ‘996 and ‘190 patents and, accordingly, on May 13, 2015, we, and Horizon filed patent infringement lawsuits against Dr. Reddy’s, Lupin, Actavis and Mylan in the U.S. District Court of New Jersey alleging that their ANDA products infringe the ‘636 and ‘996 patents. On June 18, 2013, we, and Horizon filed an Amended Complaint in the actions against Dr. Reddy’s, Lupin, Watson and Mylan, adding the ‘190 patent to the case. The cases are in the initial phase, and no schedules have been set.
On February 24, 2015, Dr. Reddy’s filed a Petition for IPR of the ‘285 patent with the PTAB of the U.S. Patent and Trademark Office. We and Horizon filed a Preliminary Response on July 12, 2015. The PTAB has three months in which to institute or deny the IPR proceeding. If the PTAB decides to institute the IPR proceeding, Dr. Reddy’s will have the opportunity to challenge the validity of the ‘285 patent in whole or in part before the PTAB via a patent validity trial. We and Horizon intend to defend the validity of the ‘285 patent in both the IPR and district court settings.
On May 21, 2015, the CFAD filed a Petition for IPR of the ‘907 patent with the PTAB of the USPTO. We and Horizon may file an optional Preliminary Response on September 6, 2015. Upon receipt of such a Preliminary Response, the PTAB has three months in which to institute or deny the IPR proceeding. If the PTAB decides to institute the IPR proceeding, CFAD will have the opportunity to challenge the validity of the ‘907 patent in whole or in part before the PTAB via a patent validity trial. We and Horizon intend to defend the validity of the ‘907 patent in both the IPR and district court settings.
On June 5, 2015, CFAD filed a Petition for IPR of the ’285 patent with the PTAB of the U.S. Patent and Trademark Office. We and Horizon may file an optional Preliminary Response on September 19, 2015. Upon receipt of such a Preliminary Response, the PTAB has three months in which to institute or deny the IPR proceeding. If the PTAB decides to institute the IPR proceeding, CFAD will have the opportunity to challenge the validity of the ‘285 patent in whole or in part before the PTAB via a patent validity trial. We and Horizon intend to defend the validity of the ‘285 patent in both the IPR and district court settings.
In Canada, on January 20, 2015, AstraZeneca Canada received a Notice of Allegation from Mylan Canada informing that Mylan Canada has filed an ANDS in Canada for approval of its naproxen/esomeprazole magnesium tablets and alleging non-infringement of some of the claims and invalidity of the ‘098 patent. AstraZeneca Canada is the licensee pursuant to a Collaboration Agreement, and the ‘098 patent is listed in respect of AstraZeneca Canada’s VIMOVO products. A Notice of Allegation in Canada is similar to a Paragraph IV Notice Letter in the United States, and in response, we and AstraZeneca Canada commenced a proceeding in the Federal Court of Canada in relation to the ‘098 patent on March 5, 2015. The Canadian proceeding is summary in nature and expected to be completed before March 5, 2017. The current schedule as approved by the Court provides for the service of affidavit evidence of AstraZeneca Canada and POZEN by September 11, 2015 and affidavit evidence of Mylan Canada by January, 8, 2016. The parties are to complete cross-examinations on the affidavit evidence by April 29, 2016. The Written Records for the hearing are to be served by AstraZeneca and us by July 4, 2016 and by Mylan Canada by September 2, 2016. A hearing date has not yet been set; however the parties are to seek the Court’s assistance in setting a hearing date in November 2016. The proceeding will decide whether approval for Mylan Canada’s naproxen/esomeprazole magnesium tablets will be prohibited until the expiry of the ‘098 patent because none of Mylan Canada’s allegations in respect of the ‘098 patent are justified; the proceeding will not finally decide ‘098 patent validity or infringement. The ‘098 patent expires on May 31, 2022.
On April 24, 2015, we and Horizon received a third Paragraph IV Notice Letter from Dr. Reddy’s informing us that it had amended its Paragraph IV certifications made with respect to its second ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO. Dr. Reddy’s amended certifications relate to the ‘285 patent, the ‘636 patent and the ‘996 patent which are all assigned to the Company. The patents are listed with respect to VIMOVO in the Orange Book and expire in 2022. Dr. Reddy’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. We and Horizon are in the process of assessing the nature and merits of Dr. Reddy’s claims.
On June 1, 2015, we and Horizon received a second Paragraph IV Notice Letter from Actavis informing us that it had amended its Paragraph IV certifications made in its ANDA seeking regulatory approval to market a generic version of VIMOVO. Actavis’s amended certifications relate to the ‘636, the ‘996 patents and United States Patent No. 8,945,621 (the “‘621 patent”), which are all assigned to the Company. The patents are listed with respect to VIMOVO in the Orange Book and expire in 2022 or 2031. Actavis’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable.
On July 17, 2015, we and Horizon received a second Paragraph IV Notice Letter from Lupin informing us that it had amended its Paragraph IV certifications made in its ANDA seeking regulatory approval to market a generic version of VIMOVO. Lupin’s amended certifications relate to the ‘636 and ‘996 patents which are each assigned to the Company. The patents are listed with respect to VIMOVO in the Orange Book and expire in 2022 or 2031. Lupin’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. We and Horizon are in the process of assessing the nature and merits of Lupin’s claims.
As with any litigation proceeding, we cannot predict with certainty the patent infringement suit against Dr. Reddy’s, Lupin, Mylan and Watson relating to a generic version of VIMOVO. We will have to incur expenses in connection with the lawsuits relating to VIMOVO, which may be substantial. In the event of an adverse outcome or outcomes, our business could be materially harmed. Moreover, responding to and defending pending litigation will result in a significant diversion of management’s attention and resources and an increase in professional fees.
Executive Officers and Directors
The following table sets forth names, ages and positions of the Pozen directors and executive officers as of August 1, 2015:
|
Name (1) |
|
Age |
|
Current Position |
|
|
|
|
|
|
|
New Executive Officers: |
|
|
|
|
|
Adrian Adams |
|
64 |
|
Chief Executive Officer and Director |
|
Andrew I. Koven |
|
58 |
|
President and Chief Business Officer |
|
Scott Charles |
|
41 |
|
Senior Vice President, Finance |
|
Jennifer Armstrong |
|
45 |
|
Executive Vice President, Human Resources and Administration |
|
Mark Glickman |
|
50 |
|
Chief Commercial Officer |
|
Eric L. Trachtenberg |
|
42 |
|
Deputy General Counsel |
|
|
|
|
|
|
|
Legacy Executive Officers: |
|
|
|
|
|
William L. Hodges, CPA |
|
60 |
|
Senior Vice President, Finance and Administration, Chief Financial Officer |
|
John G. Fort, MD, MBA |
|
60 |
|
Chief Medical Officer |
|
Dennis L. McNamara |
|
49 |
|
Senior Vice President, Chief Business Operations Officer |
|
Gilda M. Thomas, JD |
|
60 |
|
Senior Vice President and General Counsel |
|
|
|
|
|
|
|
Directors: |
|
|
|
|
|
Kenneth Lee(2)(3) |
|
67 |
|
Director |
|
Arthur Kirsch(2)(3)(4) |
|
63 |
|
Director and Chairman |
|
Seth Rudnick(2)(3)(4) |
|
66 |
|
Director |
|
Neal Fowler(3)(4) |
|
53 |
|
Director |
(1) Geraldine Lillis and Andrew Ryan, currently non-employee directors of Aralez, will resign upon consummation of the merger. We anticipate that, except as noted below, the new executive officers and directors shall serve in their current roles upon consummation of the merger.
(2) Member of our audit committee.
(3) Member of our compensation committee.
(4) Member of our nominating and corporate governance committee
Executive Officers
Adrian Adams has been Pozen’s Chief Executive Officer and director since June 2015. Prior to joining the Company, Mr. Adams served as a consultant to Pozen from April 2, 2015 to May 31, 2015. Previously, Mr. Adams served as Chief Executive Officer and President and as a director of Auxilium Pharmaceuticals Inc., a specialty biopharmaceutical company, from December 2011 until January 2015, when it was acquired by Endo International plc. Prior to joining Auxilium, from September 2011 to November 2011, Mr. Adams served as Chairman and Chief Executive Officer of Neurologix, Inc., a company focused on development of multiple innovative gene therapy development programs. Before Neurologix, Mr. Adams served as President and Chief Executive Officer of Inspire Pharmaceuticals, Inc., a specialty pharmaceutical company, from February 2010 until May 2011 when it was acquired by Merck & Co., Inc. Previously, Mr. Adams served as President and Chief Executive Officer of Sepracor Inc., a specialty pharmaceutical company, from March 2007 and May 2007, respectively, until February 2010 at which time Sepracor was acquired by Dainippon Sumitomo Pharma Co., Ltd. Prior to his appointment as Chief Executive Officer of Sepracor, Mr. Adams served as its Chief Operating Officer. Prior to joining Sepracor, Mr. Adams served as the President and Chief Executive Officer of Kos Pharmaceuticals, Inc., a specialty pharmaceutical company, from 2002 until its acquisition by Abbott Laboratories in December 2006. Mr. Adams has also held general management and senior international and national marketing positions at SmithKline Beecham, Novartis and ICI (now part of AstraZeneca). Mr. Adams has served as chairman of the board of directors of AcelRx Pharmaceuticals, Inc. since February 2013 and recently served on the board of directors of Amylin Pharmaceuticals, Inc. from October 2007 to August 2012. Mr. Adams graduated from the Royal Institute of Chemistry at Salford University in the U.K. Mr. Adams serves as Chairman of the Board of AcelRx Pharmaceuticals and recently served as a director of Amylin Pharmaceuticals.
Mr. Adams has also been a director of Pozen since June 2015. Mr. Adams’ position as Chief Executive Officer of Pozen, along with his many years of service in the pharmaceutical industry in chief executive positions, enables him to provide important insights regarding the operations of Pozen and the pharmaceutical industry generally, including finance, marketing, strategic planning, and senior management personnel matters.
Andrew I. Koven has been Pozen’s President and Chief Business Officer since June 2015. Prior to joining Pozen, Mr. Koven served as Chief Administrative Officer and General Counsel of Auxilium Pharmaceuticals Inc., a specialty biopharmaceutical company, from February 2012 until January 2015, when it was acquired by Endo International plc. Prior to that, from September 2011 to November 2011, Mr. Koven served as President and Chief Administrative Officer and a member of the board of directors of Neurologix, Inc., a company focused on development of multiple innovative gene therapy development programs. Before Neurologix, Mr. Koven served as Executive Vice President and Chief Administrative and Legal Officer of Inspire Pharmaceuticals, Inc., a
specialty pharmaceutical company, from July 2010 until May 2011 when it was acquired by Merck & Co., Inc. Previously, Mr. Koven served as Executive Vice President, General Counsel and Corporate Secretary of Sepracor Inc., a specialty pharmaceutical company, from March 2007 until February 2010 when it was acquired by Dainippon Sumitomo Pharma Co., Ltd. Prior to joining Sepracor, Mr. Koven served as Executive Vice President, General Counsel and Corporate Secretary of Kos Pharmaceuticals, Inc., a specialty pharmaceutical company, from August 2003 until its acquisition by Abbott Laboratories in December 2006. Mr. Koven began his career in the pharmaceutical industry first as an Assistant General Counsel and then as Associate General Counsel at Warner-Lambert Company from 1993 to 2000, followed by his role as Senior Vice President and General Counsel at Lavipharm Corporation from 2000 to 2003. From 1986 to 1992 he was a corporate associate at Cahill, Gordon & Reindel in New York. From 1992 to 1993 he served as Counsel, Corporate and Investment Division, at The Equitable Life Assurance Society of the U.S.
Scott Charles has been Pozen’s Senior Vice President Finance since July 2015. Mr. Charles will become Chief Financial Officer of Aralez upon consummation of the transactions. Prior to joining Pozen, Mr. Charles served as the Vice President of Finance and Treasurer at Ikaria, Inc., a critical care pharmaceutical company from April 2008 to July 2015. From April 2002 to March 2008, Mr. Charles worked at Reliant Pharmaceuticals, Inc. in various finance functions, culminating with serving as the Vice President of Finance and Treasurer from April 2006 to March 2008. Prior to that, he was a Manager of Assurance and Business Advisory Services at Arthur Andersen, LLP. He holds a Bachelor of Science degree in Business Administration from Bucknell University and is a Certified Public Accountant.
Jennifer Armstrong joined Pozen in June 2015 as Executive Vice President, Human Resources and Administration. Previously, she served as Senior Vice President of Human Resources at Auxilium Pharmaceuticals, Inc., a specialty biopharmaceutical company, from July 2009 to March 2015. Prior to that, she served at Senior Vice President of Human Resources and Corporate Communications at Genaera Corporation, a specialty biopharmaceutical company, from January 1998 to May 2009. On June 12, 2009, Genaera Corporation transferred all of its assets and liabilities to the Genaera Liquidating Trust and filed a Certificate of Dissolution with the Delaware Secretary of State pursuant to the Plan of Complete Liquidation and Dissolution adopted at a special meeting of stockholders. Ms. Armstrong holds a Masters degree in Arts Administration and a Bachelors degree in Corporate Communications, both from Drexel University.
Mark Glickman has been Chief Commercial Officer of Pozen since June 2015. Mr. Glickman previously served as Executive Vice President of Sales and Marketing for Auxilium Pharmaceuticals, a specialty biopharmaceutical company, from February 2012 to February 2015. From February 2009 to February 2012, he served as Vice President in the medical device division at Otsuka America Pharmaceutical, Inc., a pharmaceutical and medical device company and a subsidiary of Otsuka America, Inc. Prior to Otsuka, Mr. Glickman served as Senior Vice President of Sales and Marketing at Oscient Pharmaceuticals Corp., a commercial-stage pharmaceutical company, from September 2007 to September 2009. Before joining Oscient, from May 2007 to September 2007, Mr. Glickman served as Vice President of Sales at Bayer Healthcare’s Diabetes Care Division. From 2001 to 2007, he held various positions at Kos Pharmaceuticals, including Director of Marketing, Regional Sales Director and Vice President of Sales. Mr. Glickman started his pharmaceutical career at Bristol-Myers Squibb where he was responsible for the marketing of cardiovascular products, including the blockbuster Plavix. Mr. Glickman holds a Master of Business Administration degree from New York University.
Eric L. Trachtenberg joined Pozen in June 2015 as Deputy General Counsel. Mr. Trachtenberg will become General Counsel of Aralez upon consummation of the transactions. Prior to joining Pozen, Mr. Trachtenberg most recently served as Deputy General Counsel at Auxilium Pharmaceuticals, Inc., a specialty biopharmaceutical company, from May 2012 through its acquisition by Endo Pharmaceuticals in February 2015. Prior to Auxilium, he was Vice President, General Counsel and Corporate Secretary of Enobia Pharma, Inc., from April 2011 to April 2012, and managed all legal aspects of Enobia’s sale to Alexion Pharmaceuticals. Prior to that, Mr. Trachtenberg served as Vice President and Associate General Counsel of Sepracor Inc. and remained in that position with Sunovion Pharmaceuticals Inc. following the acquisition of Sepracor by Dainippon Sumitomo Pharma. Mr. Trachtenberg also held a Senior Counsel position at Kos Pharmaceuticals, Inc. before its acquisition by Abbott. Mr. Trachtenberg began his career as an Associate at Blank Rome LLP. He holds a Bachelor of Science degree in Management from Tulane University and a Juris Doctorate and Master of Business Administration degree from Temple University.
William L. Hodges joined Pozen in August 2004 as Senior Vice President of Finance and Administration and Chief Financial Officer. Mr. Hodges began his career in the pharmaceutical industry with Burroughs Wellcome Co. in 1985. In 1991, he moved to London and worked in Group Finance for the Wellcome Foundation, Ltd. Mr. Hodges worked on mergers and acquisitions and was Regional Controller for Northern Europe and Japan. In 1993, he returned to Burroughs Wellcome in North Carolina as Director of Procurement. Mr. Hodges was Vice President, Corporate Planning and Business Support at GlaxoWellcome before being appointed acting Senior Vice President and CFO for the fifteen months leading up to the merger between GlaxoWellcome plc and SmithKline Beecham plc which was completed in December of 2000. Prior to joining Pozen, Mr. Hodges was Senior Vice President and CFO of Pergo, Inc. located in Raleigh, North Carolina. Mr. Hodges received his B.S. from the University of North Carolina at Chapel Hill and is a Certified Public Accountant.
Dennis L. McNamara has been Senior Vice President, Chief Business Operations Officer since June 2015. Previously, from January 2014 through May 2015, he was Senior Vice President and Chief Business Officer. Mr. McNamara joined Pozen in December 1998 as Vice President of Business Development. Prior to joining Pozen, Mr. McNamara held positions in business development with private and publicly-traded development stage biotechnology companies including AlphaVax, Sequana Therapeutics and Apex Bioscience, and in pharmaceutical sales with Abbott Laboratories. Before joining the pharmaceutical industry Mr. McNamara conducted receptor pharmacology research at the University of North Carolina. Mr. McNamara earned his M.B.A. from the University of Michigan and an A.B. degree from Duke University.
Gilda M. Thomas joined Pozen in January 2007 as Senior Vice President and General Counsel. Prior to joining Pozen, Ms. Thomas was Vice President, General Counsel and company secretary at EMD Pharmaceuticals, Inc., an affiliate of Merck KGaA, Darmstadt, Germany from July 2001 to December 2006. Prior to joining EMD, she spent 14 years at Burroughs Wellcome Co., which merged into Glaxo Welcome, Inc. At Glaxo Wellcome Ms. Thomas was Associate General Counsel responsible for the 13 member corporate section of the legal department. Ms. Thomas received her J.D. from Harvard Law School, a M.S. from Simmons College and a B.A. degree from Wellesley College.
John G. Fort, M.D. joined Pozen in July 2007 as Chief Medical Officer. Prior to joining Pozen, Dr. Fort was Vice President, Medical Affairs at Adolor Corporation from 2004 until 2007. Dr. Fort held positions with Pfizer Inc., including Vice President, Medical Affairs, and was Vice President, Arthritis and Pain at G.D. Searle & Co., Monsanto Corporation from September 1994 to December 2003. Prior to joining the pharmaceutical industry, he was an Associate Professor of Medicine at Thomas Jefferson University, Division of Rheumatology. Dr. Fort received his M.D. from the University of Valencia Faculty of Medicine and is board certified in internal medicine with a subspecialty certification in rheumatology.
Non-Employee Directors
Kenneth B. Lee, Jr. has been the lead independent director of Pozen since 2005. Independent consultant since June 2002 and general partner of Hatteras Venture Partners (formerly Hatteras BioCapital. LLC and BioVista Capital, LLC), the general partner of Hatteras BioCapital Fund, L.P., a venture capital fund focusing on life sciences companies, since 2003. President of A.M. Pappas & Associates, a venture capital firm, between January 2002 and June 2002. Partner of Ernst & Young LLP from 1982 through 2000. Partner of Ernst & Young Corporate Finance LLC from 2000 to 2001. Managing Director of Ernst & Young’s Health Sciences Corporate Finance Group from 2000 to 2001. Mr. Lee serves on the board of Biocryst Pharmaceuticals, Inc., a public company, for which he serves as chairman of the audit committee and chairman of the finance committee. He is also a director of Clinverse, Inc. and Clinipace Worldwide, two privately held companies. Previously, he served on the boards of CV Therapeutics, Inc., for which he served as lead independent director and chair of the audit committee and a member of the compensation committee, Abgenix, Inc., for which he served on the audit committee, OSI Pharmaceuticals, for which he served as a member of the audit committee, Inspire Pharmaceuticals Inc., for which he served as chairman of the board of directors, chair of the audit committee and a member of the compensation committee and finance committee, and Maxygen, Inc., for which he served as chairman of the audit committee and a member of the nominating/ governance committee and the compensation committee. Mr. Lee served as a member of the executive committee of the Board of the North Carolina Biotechnology Industry Organization and as a member of the board of Ibiliti, a nonprofit organization dedicated to building and expanding networks of resources for advanced medical technology companies.
Mr. Lee brings his extensive accounting and financial background to the Pozen Board, as well as expertise in the life sciences industry from his experience as a general partner of several venture capital funds specializing in life sciences. He has also served and is serving on the boards and audit committees of several public pharmaceutical companies similar in size to the Company, including serving as Chairman of the Board of Biocryst Pharmaceuticals, Inc. Mr. Lee is also a co-founder of the National Conference on Biotechnology Ventures.
Arthur S. Kirsch has been Senior Advisor, GCA Savvian, LLC (formerly Perseus Group, LLC), an investment bank, since June 2005. Mr. Kirsch is a founding member and Managing Director of Vector Securities, LLC, an investment and merchant banking firm, from 2001 to May 2005. He was a Managing Director and Head of Healthcare Research and Capital Markets of Prudential Vector Healthcare Group, a unit of Prudential Securities, Inc., a full-service brokerage firm, from 1999 to 2001. Mr. Kirsch was the Director, Equity Research of Vector Securities International, Inc., an investment banking firm, from 1995 to 1999. He currently serves as a director of PhysioSonics, Inc., a privately held company developing noninvasive neurological products.
Mr. Kirsch has over 25 years of experience working in the equity capital markets and has extensive knowledge of the healthcare and life sciences field. Mr. Kirsch, who has spent the majority of his career in investment banking with a focus on the healthcare industry, brings both financial and industry expertise to the Board.
Seth A. Rudnick, M.D. has been venture partner and previously general partner at Canaan Partners, a venture capital firm, since 1998, from which he is now retired. Formerly, Dr. Rudnick was the Chief Executive Officer and Chairman of CytoTherapeutics Inc., a company developing stem cell-based therapies. He helped found and served as the Head of Research and Development for Ortho Biotech, a division of Johnson & Johnson focusing on cancer and chronic illnesses from 1991 to 1998. He currently serves on the boards of directors of the following privately held biotechnology companies: Chimerix, Inc., Meryx Pharmaceuticals, for which he serves as Chairman, Liquidia Technologies, Inc., for which he serves as Chairman, and G1 Therapeutics, for which he serves as Executive Chairman. Dr. Rudnick also serves on the Board of Square 1, a public company. Currently he is a Clinical Adjunct Professor of Medicine at University of North Carolina, Chapel Hill.
Dr. Rudnick brings deep operational experience in the pharmaceutical and biotechnology industries acquired through a variety of senior research and development positions in several large and mid-size pharmaceutical companies and as Chief Executive Officer, and Chairman of CytoTherapeutics, Inc., Chairman of Liquidia Technologies, Inc., Executive Chairman of GI Therapeutics, and Chairman of Meryx Pharmaceuticals. Dr. Rudnick retired from Canaan Partners, a global venture capital firm with significant investments in the healthcare sector, where he served as general and now a venture partner since 1998, which has provided him with significant experience in and insight into life sciences investments.
Neal F. Fowler has been Chief Executive Officer of Liquidia Technologies, Inc., a privately held biotechnology company since 2008 and Chief Executive Officer of Envisia Technologies, a privately held biotechnology company, since 2013. Mr. Fowler was the President of Centocor, Inc., a subsidiary of Johnson & Johnson from 2006 to 2008. President of Ortho-McNeil Neurologics, Inc., a subsidiary of Johnson & Johnson from 2004 to 2006 and Franchise Vice President-CNS from 2001 to 2004. He held various positions at Eli Lilly and Company from 1988 to 2001, including Area Director, Primary Care Division, Director U.S. Cardiovascular Business Unit, Cardiovascular Product Manager, Operations Manager, Southwest Area, Manager Medical Device and Diagnostics, Associate, Marketing Plans, Endocrinology, Associate, Business Development/New Product Planning, Oncology, and Retail Sales Representative.
Mr. Fowler brings his extensive background in the pharmaceutical industry acquired through a variety of marketing and general manager positions at several large pharmaceutical companies. He is currently chief executive officer at Liquidia Technologies, Inc. and Envisia Technologies, positions which have provided him with experience in running an emerging growth company.
Our executive officers are elected by, and serve at the discretion of, our Board. There are no family relationships among any of our executive officers or directors.
Board Composition
Our Board currently consists of five members. Our certificate of incorporation provides that our Board be divided into three classes serving staggered three-year terms, as follows:
· Class I, which consists of Messrs. Fowler and Kirsch, and whose term will expire at our annual meeting of stockholders to be held in 2016;
· Class II, which consists of Messrs. Lee and Adams, and whose term will expire at our annual meeting of stockholders to be held in 2017; and
· Class III, which consists of Dr. Rudnick, and whose term will expire at our annual meeting of stockholders to be held in 2018.
Following consummation of the transactions, our Board will be comprised of nine members, consisting of our Chief Executive Officer, five directors appointed by Pozen, two directors appointed by Tribute, and one director appointed pursuant to the terms in the merger agreement.
Director Independence
Our Board has determined that each of the members of the Board, with the exception of Mr. Adrian Adams, who serves as our Chief Executive Officer, is independent as that term is defined under the applicable independence listing standards of the NASDAQ Global Market.
Committees of the Board
Our Board currently has three standing committees: an Audit Committee, a Compensation Committee and a Nominating/Corporate Governance Committee. These committees, their principal functions and their respective memberships are described below.
Audit Committee
The current members of the Audit Committee are Mr. Kirsch, who serves as Chairman, Mr. Lee and Dr. Rudnick. Each of the members of the Audit Committee is independent as defined by the applicable NASDAQ listing standards and the SEC, rules applicable to audit committee members. Our Board has determined that each also qualifies as an audit committee financial expert as defined by the SEC.
The Audit Committee was established in accordance with section 3(a)(58)(A) of the Exchange Act. The Audit Committee oversees our financial reporting process and system of internal control over financial reporting, and selects and oversees the performance of, and approves in advance the services provided by, our independent auditors. The Audit Committee provides an open avenue of communication among our independent auditors, financial and senior management and the Board. The Audit Committee meets regularly with our independent auditors without management present, and from time to time with management in separate private sessions, to discuss any matters that the Audit Committee or these individuals believe should be discussed privately with the Audit Committee, including any significant issues or disagreements that may arise concerning our accounting practices or financial statements. The Audit Committee also oversees our whistleblower policy for receiving and handling complaints or concerns regarding accounting, internal accounting controls or auditing matters. In addition, the Audit Committee assists the Board in its oversight role by receiving periodic reports regarding our risk and control environment.
The Audit Committee held four meetings during the year ended December 31, 2014. A copy of the Audit Committee’s charter is posted on our website at www.POZEN.com.
Nominating/Corporate Governance Committee
The current members of the Nominating/Corporate Governance Committee are Dr. Rudnick, who serves as Chairman, Mr. Fowler, and Mr. Kirsch. Each of the members of the Nominating/Corporate Governance Committee is independent as defined by the applicable NASDAQ listing standards.
The Nominating/Corporate Governance Committee assists the Board in fulfilling its responsibilities regarding the oversight of the composition of the Board and other corporate governance matters. Among its other duties, the Nominating/Corporate Governance Committee: (i) evaluates nominees and reviews the qualifications of individuals eligible to stand for election and reelection as directors and makes recommendations to the Board on this matter; (ii) oversees compliance with our Code of Business Conduct and Ethics; (iii) reviews and approves related party transactions; (iv) recommends and advises the Board on certain other corporate governance matters; and (v) oversees the Board’s performance evaluation process. The Nominating/Corporate Governance Committee does not have a specific policy with regard to the consideration of diversity in identifying director nominees. However, our Nominating/Corporate Governance Committee values diversity on our Board and considers the diversity of the professional experience, education and skills, as well as diversity of origin, in identifying director nominees.
The Nominating/Corporate Governance Committee held three meetings during the year ended December 31, 2014. A copy of the Nominating/Corporate Governance Committee’s charter is posted on our website at www.POZEN.com.
Compensation Committee
The current members of the Compensation Committee are Mr. Lee, Mr. Kirsch, Dr. Rudnick and Mr. Fowler. Mr. Lee serves as Chairman of the Compensation Committee. Each of the current members of the Compensation Committee is independent as defined by the applicable NASDAQ listing standards.
Decisions regarding the compensation of our executive officers are made by the Compensation Committee. The Compensation Committee’s principal responsibilities include reviewing POZEN’s overall compensation philosophy and the adequacy and market competitiveness of our compensation plans and programs, evaluating the Company’s compensation policies and practices to determine whether these policies and practices create incentives for a particular employee group to take actions which could put the Company at undue risk, evaluating the performance of and reviewing and approving compensation for our executive officers, evaluating and recommending director compensation, and reviewing and discussing with management the Compensation Discussion and Analysis included in the Form S-4. The Compensation Committee also administers our equity-based and other incentive plans, including assuming responsibility for granting, or delegating as appropriate the authority for granting, and making decisions with respect to, awards under our equity compensation and other incentive plans.
To assist in its efforts to meet the objectives and responsibilities outlined above, the Compensation Committee has retained an executive compensation consultant. During 2014, the Compensation Committee retained Radford, an Aon Hewitt Company, or Radford, a nationally known executive compensation and benefits consulting firm, to advise it on various matters related to executive and director compensation and compensation programs. Radford may also from time to time advise management, with the Compensation Committee’s consent. Radford was hired by and reports to the Compensation Committee. Pursuant to its charter, the Compensation Committee has the power to hire and fire such consultants and to engage other advisors. A human resources consultant retained by management also provides information and support to the Compensation Committee as requested.
The Compensation Committee values the input of our stockholders regarding compensation decisions. In 2013, the Compensation Committee commissioned a third party to contact institutional stockholders that collectively owned greater than 50% of the non-affiliated shares in an effort to understand any concerns they had regarding our executive compensation program. In 2014, Mr. Lee, the Chairman of the Compensation Committee, also contacted certain institutional shareholders to continue this dialogue and sent a letter to the stockholders describing certain steps taken by the Board to address stockholder concerns, a copy of which was included in the Company’s 2014 proxy statement. As a result of these efforts, and with advice from Radford, in February 2014 our Board adopted new policies relating to new stock ownership guidelines for our Chief Executive Officer and new policies relating to equity retention and incentive payment clawback applicable to all executive officers. The Committee also made the 2014 annual equity grants for certain of our executive officers more specifically performance-based by allocating a portion of the annual equity award to performance-based grants that vest on significant corporate achievements.
The Compensation Committee held 10 meetings during the year ended December 31, 2014. A copy of the Compensation Committee’s charter is posted on our website at www.POZEN.com.
Compensation Committee Interlocks and Insider Participation
None of our executive officers serves as a member of the Board or compensation committee, or other committee serving an equivalent function, of any other entity that has one or more of its executive officers serving as a member of our Board or Compensation Committee. None of the members of our Compensation Committee has ever been our employee or one of our officers.
Code of Business Ethics and Conduct
We have adopted a Code of Business Conduct and Ethics that applies to our employees (including our principal executive officer, chief financial officer and other members of our finance and administration department) and our directors. Our Code of Business Conduct and Ethics is posted on our website at www.POZEN.com. In addition, we intend to post on our website all disclosures that are required by law or NASDAQ Stock Market listing standards concerning any amendments to, or waivers from, any provision of our Code of Business Conduct and Ethics.
Director Compensation
Discussed in the following paragraphs and tables is the compensation paid to the non-employee directors who serve on our Board. Directors who are also our employees do not receive any additional compensation for their service as directors of the Company.
Cash Compensation. We reimburse each non-employee director for out-of-pocket expenses incurred in connection with attending Board and Board committee meetings and otherwise in connection with service as a director. We also pay each non-employee director the following retainer fees:
· an annual retainer of $40,000;
· an annual retainer for Board committee Chairs, as follows: $12,000 for service as Chair of the Nominating/Corporate Governance Committee; $17,500 for service as Chair of the Compensation Committee; and $25,000 for service as Chair of the Audit Committee; and
· an annual retainer for Board committee members (other than committee Chairs), as follows: $8,000 for service on the Nominating/Corporate Governance Committee; $10,000 for service on the Compensation Committee; and $12,500 for service on the Audit Committee.
All retainers are payable quarterly and pro-rated for service of less than a full quarter; retainers may be reduced if a director fails to attend at least 75% of all required Board and committee meetings. No compensation is paid to directors for attendance at individual Board or Board committee meetings.
Equity Compensation.
· Upon his or her initial election to the Board, 14,000 RSUs. This initial grant vests one-third annually over three years, subject to continued service as a director.
· On the date of each annual meeting of stockholders, an amount of RSUs with a market value as of the grant date equal to $80,000. The RSUs vest on the earlier of the one-year anniversary of the grant or the date of the next annual stockholder meeting, subject in either case to the director’s continued service on the Board at that date.
Equity grants awarded pursuant to this director compensation program are granted under and subject to the terms and conditions of the POZEN Inc. 2010 Equity Compensation Plan (the “2010 Plan” or the “Plan”), including without limitation the terms providing for acceleration of vesting upon a change of control. All stock options are granted at an exercise price per share equal to the closing price of our common stock, as reported on NASDAQ, on the date of grant, have a ten-year term and are exercisable for a period of up to three years following the date the director’s service on the Board terminates, to the extent vested as of such date, but not beyond the expiration of the ten-year term.
The Board has adopted a non-employee director stock ownership guideline of shares equal in value to three times the annual director retainer of $40,000, to be acquired over a five year period. Directors are strongly encouraged to hold their shares of Pozen stock while they serve on the Board.
The following table further summarizes the compensation paid by us to our non-employee directors during the 2014 fiscal year. Except as noted below, all of our directors are paid at the same rate. The differences among directors in the table below are a function of additional compensation for chairing a committee and/or serving on one or more committees.
|
Name |
|
Fees Earned or |
|
Stock |
|
Option |
|
Non-Equity |
|
Change in |
|
All Other |
|
Total |
| |||
|
Neal F. Fowler |
|
$ |
56,333 |
|
$ |
79,996 |
|
— |
|
— |
|
— |
|
— |
|
$ |
136,329 |
|
|
Arthur S. Kirsch |
|
$ |
79,250 |
|
$ |
79,996 |
|
— |
|
— |
|
— |
|
— |
|
$ |
159,246 |
|
|
Kenneth B. Lee, Jr. |
|
$ |
67,917 |
|
$ |
79,996 |
|
— |
|
— |
|
— |
|
— |
|
$ |
147,913 |
|
|
Martin Nicklasson, Ph.D. |
|
$ |
29,375 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
$ |
29,375 |
| |
|
Seth A. Rudnick, M.D. |
|
$ |
69,292 |
|
$ |
79,996 |
|
— |
|
— |
|
— |
|
— |
|
$ |
149,288 |
|
(1) Consists of the following:
a. Neal F. Fowler: four quarterly payments toward 2014 annual fees, including a 2014 annual retainer of $40,000 and $16,333 for service as a member of one or more Board Committees.
b. Arthur S. Kirsch: four quarterly payments toward 2014 annual fees, including a 2014 annual retainer of $40,000, $22,917 for service as Chair of the Audit Committee and $16,333 for service as a member of one or more Board Committees.
c. Kenneth B. Lee, Jr: four quarterly payments toward 2014 annual fees, including a 2014 annual retainer of $40,000, $16,548 for serving as Chairman of the Compensation Committee and $11,369 for service as a member of one or more Board Committees.
d. Martin Nicklasson: two quarterly payments toward 2014 annual fees, including a 2014 annual retainer of $40,000 and $9,375 for service as a member of one or more Board Committees. Mr. Nicklasson did not stand for re-election and retired from the Board as of the 2014 Shareholder’s Meeting.
e. Seth A. Rudnick: four quarterly payments toward 2014 annual fees, including a 2014 annual retainer of $40,000, $12,000 for serving as Chairman of the Governance Committee and $17,292 for service as a member of one or more Board Committees.
(2) The amounts included in this column are the dollar amounts representing the full grant date fair value of each restricted stock unit award calculated in accordance with the Financial Accounting Standards Board Accounting Standards Codification Topic 718, or FASB ASC Topic 718. At December 31, 2014, each director held awards of 9,291 RSUs, all of which had been granted on June 6, 2014 and vest on the earlier of the one-year anniversary of the grant or the date of the next annual stockholder meeting (the 2015 Annual Meeting). For information on the valuation assumptions used in calculating this amount, see Note 6 to Pozen’s audited financial statements included in the Annual Report on Form 10-K for the fiscal year ended December 31, 2014, as filed with the SEC.
The following table lists the number of outstanding options held by each of the directors as of December 31, 2014, and provides additional information concerning the options granted to these directors during 2014, each of which was granted at an exercise price equal to the closing price of Pozen’s common stock as reported by NASDAQ on the respective date of grant. Options granted prior to 2007 vest annually over four years and annual option grants after 2006 vest on the earlier of the one-year anniversary of the grant or the date of the next annual stockholders meeting. The “Grant Date Fair Value” dollar amounts represent the full grant date fair value of each stock option award calculated in accordance with FASB ASC Topic 718 and do not represent the actual value that may be recognized by the directors upon option exercise. For information on the valuation assumptions used in calculating this amount, see Note 6 to Pozen’s audited financial statements included in the Annual Report on Form 10-K for the fiscal year ended December 31, 2014, as filed with the SEC.
|
Name |
|
Options |
|
Options |
|
Date of |
|
2014 Option |
|
2014 Option |
|
Grant Date |
|
|
Neal F. Fowler |
|
45,804 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
Arthur S. Kirsch |
|
67,179 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
Kenneth B. Lee, Jr. |
|
67,179 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
Martin Nicklasson, Ph.D. |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
Seth A. Rudnick, M.D. |
|
39,697 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
EXECUTIVE COMPENSATION
Compensation Discussion and Analysis
This CD&A explains our compensation program as it pertains to our named executive officers. Our named executive officers for the fiscal year ended December 31, 2014 consist of our former Chief Executive Officer, Chief Financial Officer, and the three most highly compensated executive officers (other than the former Chief Executive Officer and Chief Financial Officer) who were serving as executive officers as of December 31, 2015. John R. Plachetka served as our President and Chief Executive Officer and Chief Scientific Officer through his retirement in June 2015. For purposes of this CD&A, we refer to these persons as our “executive officers.” Our discussion focuses on compensation and practices relating to our most recently completed fiscal year.
Executive Summary of 2014 Performance
We are a pharmaceutical company focused on transforming medicine that can transform lives. We have operated a unique business model that focuses on (i) developing innovative products that meet unmet medical needs in the marketplace; (ii) obtaining patents for those innovative ideas which we believe have value in the marketplace; (iii) utilizing a small group of talented employees to develop those ideas by working with strategic outsource partners; (iv) developing a regulatory pathway with the appropriate agency; and (v) determining how best to commercialize our products.
We establish corporate goals at the beginning of every calendar year which are reviewed and approved by our Board. Our goals are designed to drive long term value for our stockholders, such as obtaining approval for our product candidates, which can take many years, obtaining partners to commercialize our approved products, or managing our expenses. Our employees, including our named executive officers, then develop individual performance goals which support these corporate goals. At the end of each year, the Compensation Committee assesses the Company’s achievement against these goals to establish pools for annual cash and equity incentives. A description of the process for granting individual cash incentives and equity and other long-term incentive compensation for our executive officers is described on pages [·] through [·] of this prospectus.
2014 Corporate Goals
Our corporate goals for 2014 were:
· obtain approval of the NDA from the FDA for PA8140 and PA32540;
· execute one new license for a Pozen product, or negotiate a significant change in an existing license;
· manage to a net cash burn of <$8 million, before any PA payments; and
· execute the 2014 steps of the Strategic Plan.
All of the goals were achieved except for the goal to obtain approval of the NDA for PA8140 and PA32540 from the FDA by the end of 2014. As a result, the Compensation Committee decided to fund the bonus pool for cash bonuses and equity grants at 75% of target level. A description of our efforts to achieve each goal is described below.
On April 25, 2014 we announced that we had received a CRL from the FDA denying approval of the NDA for our PA8140 and PA32540 product candidates. In the CRL, FDA noted that, during an inspection of the manufacturing facility of an active ingredient supplier completed the day of our PDUFA date, inspection deficiencies were found. Satisfactory resolution of deficiencies noted by the field investigator is required before the NDA can be approved. No safety or efficacy issues with respect to the PA8140 and PA32540 product candidates were noted. We worked with the active ingredient supplier to resolve the deficiencies, including sending employees and third party consultants to assess progress and assisting the supplier with drafting and providing updates to the FDA. Despite these efforts, the FDA did not complete its review of the response of the active ingredient supplier to the inspection deficiencies prior to our new PDUFA date and on December 16, 2014, we received a second CRL from the FDA which contained identical wording with respect to inspection deficiencies of our active ingredient supplier. As a result of the Company’s failure to meet its goal of obtaining approval of the NDA for PA8140 and PA32540 by the end of 2014, the Compensation Committee decided to fund the bonus and equity pool at 75% of target level.
During 2014, we negotiated significant changes to an existing license agreement with GSK for rights to commercialize Treximet (sumatriptan/naproxen sodium) which achieved a second corporate goal. As a condition of our consent to GSK’s assignment of the Product Development and Commercialization Agreement for Treximet to Pernix, the restrictions on our right to develop and commercialize certain additional dosage forms of sumatriptan/naproxen combinations outside of the United States were eliminated and we can now seek approval for these combinations on the basis of the approved U.S. NDA. We also obtained Pernix’s commitment to continue certain of GSK’s ongoing development activities and to undertake certain new activities. Lastly, we received a warrant to purchase 500,000 shares of Pernix common stock at an exercise
price equal to $4.28, the closing market price on May 9, 2014. In addition to negotiating favorable changes in the license agreement for Treximet, we negotiated a mutual termination agreement with Sanofi U.S. for the commercialization of our PA8140 and PA32540 investigational products which provided for the transfer of specified commercial know-how developed by Sanofi U.S. relating to the PA products to us and allows us and any future collaborators to use this know-how to commercialize the products.
For the majority of 2014, our strategy was to collect the royalty and milestone cash flows from our investments in development and approved pharmaceutical products and to distribute as much cash to our stockholders as was prudent through future distributions and dividends. We accomplished this by reducing expenses to an appropriate level to efficiently and effectively run the business of working with our partners, licensing any unpartnered assets to third parties, and collecting the royalties and milestones under our existing licensing agreements. Our corporate goal of managing the 2014 net cash burn in furtherance of this strategy was achieved through strict control of expenses and royalty revenues that exceeded our forecast.
During 2014, we continued to execute our strategic plan in support of the above-described strategy and this goal was also deemed achieved by the Compensation Committee based upon the completion of activities. As a result of the termination of our license agreement with Sanofi U.S. for the PA8140 and PA32540 investigational products, we are currently evaluating all strategic options available to us now that we have full ownership of the PA products in the United States.
Given our overall business model and strategy described above, we do not believe that our incentive programs encourage short-term risky behavior because the performance criteria on which our incentive programs are primarily based are longer-term strategic and corporate goals designed to reward our executives for outstanding corporate performance, including success in progressing our product development programs and our out-licensing activities, both of which can take many years.
Responsibility; Philosophy; Objectives
The Compensation Committee of our Board, which is comprised solely of independent directors and “outside directors” as determined under Section 162(m) of the Code and the applicable Treasury Regulations, is responsible for our executive compensation program. The Compensation Committee reviews and determines its independence using factors set forth in applicable SEC and NASDAQ rules on an annual basis. The Compensation Committee receives staff support from members of our management. In addition, the Compensation Committee directly engages Radford Survey + Consulting, or Radford, a leading compensation consultant, to assist the Committee in the performance of its duties. As part of its 2014 review of the Company’s compensation programs, the Committee engaged Radford to assist with several compensation-related projects, including an assessment of our director compensation program and to confirm that its 2014 compensation decisions were in line with industry norms. Other than services provided to the Committee, Radford did not perform any services for the Company or any of its management in 2014.
The Compensation Committee reviews and approves all compensation paid to our executive officers and is responsible for determining the most appropriate total executive compensation principles that govern such compensation. These principles are based on our business strategy and business model and are designed to be competitive with our peer group of companies and consistent with stockholder interests without encouraging unnecessary or excessive short-term risk. In accordance with its charter, the Compensation Committee’s responsibilities include reviewing and approving our overall compensation philosophy and the adequacy and market effectiveness of our compensation plans and programs; evaluating the performance of and reviewing and approving total compensation for our executive officers; and administering our equity-based and other incentive programs.
We are committed to providing competitive levels of compensation to our employees, including our executive officers, to ensure that we are able to recruit, retain and motivate the high caliber talent we require in our unique business model. Our business model includes a significant amount of outsourcing and we therefore need smart, talented, experienced business managers in each area of expertise to be successful. We believe it is important that our employees be given the opportunity to be well rewarded for strong performance against goals that support individual development and our future success. In determining the total compensation for our executive officers, the Compensation Committee’s aim is to provide compensation that assists us in meeting these objectives. The Compensation Committee seeks to maintain compensation that is in overall conformance with sound market practices and comparable to and competitive with the compensation packages of executives of similar companies, while recognizing individual and organizational performance.
We rely on survey data and information on compensation paid by comparable companies gathered by our compensation consultant, Radford to benchmark our executive compensation programs. Radford conducts an independent review of the peer group selection criteria and specific companies at the Committee’s request. In selecting peer companies, the Compensation Committee considers a number of factors, including whether a potential peer has products on the market, whether a potential peer has executive positions of similar scope of responsibility, as well as whether investors might consider such company as a peer when considering investments in the Company. The Compensation Committee also considers the peer group criteria used by groups such as Institutional Shareholder Services (ISS) and Glass Lewis for making comparisons. In selecting the peer companies, we believe that the Company’s market cap and the fact that it has products on the market sold by licensees are the two most critical criteria for making pay comparisons. Because the institutional investor advisory firms do not limit the peer group review to companies with products on the market and with similar business models, we have found that there is only limited overlap between our peer group and those used by the institutional investor advisory firms.
The companies below were identified by Radford in 2013 as the Pozen peer group for purposes of compensation benchmarking and remain unchanged in 2014.
|
AMAG Pharmaceuticals |
ImmunoGen |
|
BioDelivery Sciences International |
LifeVantage |
|
Cempra |
Momenta Pharmaceuticals |
|
Cryolife |
Repligen |
|
Cumberland Pharmaceuticals |
SciClone Pharmaceuticals |
|
Dendreon |
Spectrum Pharmaceuticals |
|
Depomed |
Sucampo Phama |
|
DURECT |
Zogenix |
|
Dyax |
|
These companies were selected based on the following criteria:
· Market Capitalization: range of 50% to 200% of the Company’s then current valuation, approximately $100M to $500M.
· Publicly traded biopharmaceuticals/biotherapeutics companies with a product on the market, with consideration for the therapeutic area.
· Location: predominately east coast (as available).
Radford has served as an advisor to the Compensation Committee since 2008 in connection with the compensation decisions for the executive officers. As part of its review of 2014 compensation, the Compensation Committee consulted with Radford to conduct an assessment of competitiveness of the director compensation program which was used in adopting changes to Board compensation effective June 1, 2014 and to inform its 2014 compensation decisions and confirm that the decisions were in line with industry norms. Other than services provided to the Compensation Committee, Radford did not perform any services for the Company or any of its management in 2014.
What We Reward
Our executive compensation program is designed to reward achievement of annual and long-term corporate goals, as well as individual goals that are supportive of our corporate goals and strategic objectives. Our executive officers establish and submit annual corporate goals for the year to our Board for approval. These annual business goals are based on calendar year objectives that are specific and measurable, and align with our longer term strategic plan. The goals represent important corporate achievements and value drivers of Pozen, and generally involve progressing specific product candidates in the product development pipeline, achieving product regulatory milestones, achieving financial targets or progressing corporate strategic activities. The Compensation Committee evaluates the achievement of these goals, along with completion of other strategic activities and individual performance, and uses its discretion to determine annual adjustments to compensation and annual awards for our executive officers. The Compensation Committee recognizes that internal, external and other extraordinary factors may lead to adjustments of corporate efforts that may not be reflected in our annual Board-approved corporate goals; therefore, the Compensation Committee uses its judgment in completing a thorough review of annual corporate and personal performance before the annual awards are approved.
Our compensation program is designed to provide higher levels of pay when executive and organizational performance exceeds the performance standards. Likewise, individual and organizational performance that falls short of the approved standards will result in payments and overall compensation that are at the lower end of competitive market targets. Our compensation programs are designed not only to reward past performance, but to provide incentives for continued high levels of executive performance, particularly through the multi-year vesting of our equity awards. We also consider the use of special performance based programs for longer term, key objectives, such as the PA32540 equity program which was implemented in 2011 and the PA8140 equity program which was implemented in 2012. Individual executives are reviewed annually to assess performance against their goals. We are guided by the overarching principle that the highest comparative levels of compensation should be paid to our highest performing executives.
We believe our approach to goal setting assists in mitigating excessive risk-taking that could harm our value or reward poor judgment by our executives. Several features of our programs reflect sound risk management practices. We believe we have allocated our compensation among base salary and short and long-term compensation target opportunities in such a way as to not encourage excessive risk-taking. In addition, under the 2010 Plan, we may provide a mix of equity award instruments that includes performance based equity awards, full value awards as well as the multi-year vesting of our equity awards, which will also mitigate risk and properly account for the time horizon of risk.
We believe that the mix of salary and potentially significant variable cash and equity-based incentives that we employ in our executive compensation program motivates our executive officers to work to build long-term value for our stockholders. We also believe that all employees should be owners of the Company and all of our executive officers are stockholders. The executive officers beneficially own 14.7% of outstanding shares of the Company, which creates alignment with the stockholders. The Compensation Committee believes that, based on its evaluation, the compensation paid to our executive officers, as reported in this CD&A and the compensation tables included in this prospectus, is fair and reasonable.
Role of Stockholder Say-on-Pay Votes
We provide our stockholders with the opportunity to cast an annual, nonbinding advisory vote on executive compensation (a “say-on-pay proposal”). At the Annual Meetings of Stockholders held on June 4, 2014 and June 10, 2015, approximately 77% and 53%, respectively, of the votes cast on the say-on-pay proposal were voted in favor of the proposal.
The Compensation Committee considers the outcome of the Company’s say-on-pay votes when making future compensation decisions for the named executive officers.
Role of Executive Officers in Determining Executive Compensation
The Compensation Committee is responsible for making all compensation decisions for our executive officers. During 2014, Dr. Plachetka, our former CEO, reviewed the performance of each of our other executive officers and made recommendations regarding their compensation to the Compensation Committee. The annual goal setting process for our executive officers other than our CEO involves establishing performance criteria supportive of our annual corporate goals and includes elements of participation and refinement by our executive officers, with final agreement by our CEO. Each executive officer’s goals are designed to require significant effort, cooperation and effectiveness in business plan execution in order to achieve the performance standards. In evaluating our executive officers other than the CEO, the Compensation Committee relies in part on the input and recommendations of our CEO. In evaluating our CEO’s compensation, the Compensation Committee considers, among other factors, an annual self-assessment submitted by our CEO, as well as a thorough review of corporate performance. Our CEO is not present during the Compensation Committee’s deliberations or determinations of his compensation.
Elements of Compensation
The primary components of our executive compensation program are:
· base salary;
· annual cash incentives;
· long-term incentives; and
· benefits.
In addition, employment agreements with each of our executive officers provide for potential payments upon certain terminations of employment and upon a change of control of our company. Each of the four principal elements of our executive compensation program is discussed in the following paragraphs. The employment agreements are described in the narrative accompanying the Summary Compensation Table and Grants of Plan-Based Awards Table that are included in this prospectus and the section of this prospectus beginning on page [·] entitled “Potential Payments on Termination and Change of Control”. The Compensation Committee believes that each of these compensation elements complements the others and that together they serve to achieve our compensation objectives.
In compensating our CEO and our other executive officers, the Compensation Committee seeks to ensure stockholder alignment by providing competitive base salaries; annual performance-based cash incentives; and longer-term awards under our equity-based incentive programs that are all targeted at the median of the peer group. The Compensation Committee, in conjunction with management, continues to review the level of current equity compensation and alternative equity compensation strategies to determine if changes or alternatives are more appropriate given Pozen’s stage of development and changes to the competitive landscape. As noted above, the Compensation Committee is considering the most appropriate employee retention vehicles, including making all or a portion of the annual equity grants awarded to employees and executive officers performance-based.
Although all of our full time, regular salaried employees are eligible to receive cash bonuses and equity-based compensation, the Compensation Committee places a higher percentage of our CEO’s and other executive officers’ total compensation at risk, as they have greater responsibility for and a more direct impact on overall corporate results. The compensation tables included in this prospectus detail a three-year average fixed pay versus variable compensation splits of approximately 21% - / 79% for our former CEO to 49% - / 51% for the other executive officers. In making decisions that result in this allocation, the Compensation Committee relies upon advice from Radford, its independent compensation consultant. The Compensation Committee, when determining allocation for 2014, also considered the fact that our former CEO served Pozen in many roles, including as President, CEO and Chairman. Dr. Plachetka is one of three founders of the Company was with the Company for 19 years. His expertise in many areas and unique skill set allows him to perform multiple roles, which allowed the Company to forego several additional senior level positions at a considerable cost savings. In addition to his responsibilities as CEO, President and Chairman, Dr. Plachetka led our technology and
scientific development efforts as Chief Scientific Officer, and our investor relations activities, and also played a key role in our business development activities, ongoing product creation and development, and in our interactions with the FDA and other regulatory bodies with respect to our product development programs. He was also a named inventor on each of the Company’s issued patents and patent applications and was essential in the defense of our patents against generic competitors.
Three-Year Average Fixed Pay Versus Variable Compensation
|
|
|
|

Base salary
We believe that the base salary of our CEO and other executive officers should provide a level of assured cash compensation that is commensurate with their senior professional status and career accomplishments. Accordingly, their base salaries are designed to be competitive with similar positions within the biopharmaceutical industry. In addition to the peer group analyses undertaken by the Compensation Committee as described above, we participate in and subscribe to the Radford Global Life Sciences Survey, which includes data from nearly 600 participating companies. The Compensation Committee relies on these tools to set base salaries for our executive officers that are benchmarked to similar roles in the peer group.
Base salary adjustments include a combination of cost-of-living and merit increases, based on the executive’s performance of his or her key responsibilities and duties, and are approved, communicated, and implemented in March of each year to allow for evaluation of the entire year, including the Company’s financial performance. The Compensation Committee considers each executive officer’s self-assessment of annual performance in its base salary review process and takes into account the CEO’s assessment of and recommendations with respect to each of the other executive officers. In addition, the Committee considers the market pay practices for the individual jobs.
In March 2014, based on the Radford Global Life Sciences Survey data provided by our outside compensation consultants, the Compensation Committee’s evaluation of the Company’s and each executive officer’s individual performance (as described under Annual cash incentives below), the Compensation Committee awarded Dr. Plachetka an increase in his base salary of approximately 3.0 % over his base salary in 2013. Dr. Fort, Mr. Hodges, Ms. Thomas and Mr. McNamara were also granted salary increases of approximately 3.0%. The 3.0% range used for salary adjustments is in line with survey data to which we subscribe. These increases were in line with the increases provided to the broader employee population.
Annual cash incentives
The Compensation Committee’s practice is to award annual cash incentives to our CEO and our other executive officers on a discretionary basis based on a review of corporate and individual performance objectives. Our executive officers have the opportunity to earn an annual cash incentive that is calculated as a percentage of the executive’s annual base salary. Dr. Plachetka’s target annual cash incentive level, as specified in his employment agreement, is 65% of base salary. The annual cash incentive target level for each of the other executive officers for 2014 is 40% of base salary. Annual cash incentive targets were set based upon advice from the Compensation Committee’s independent consultants. Annual cash incentives are approved, communicated and paid in March of each year in recognition of the achievement of goals and other contributions during the previous year to allow for evaluation of the entire year, including the Company’s financial performance. If warranted in special circumstances, individual one-time discretionary bonuses may also be awarded during the course of the year.
In considering annual cash incentives, the Compensation Committee evaluates the annual performance of the CEO and each of the other executive officers, focusing on the executive’s performance in his or her area or areas of functional responsibility as well as the achievement of our annual corporate goals and other significant corporate accomplishments. With respect to the executive officers other than the CEO, the annual cash incentive is also based on achievement of the executive’s individual goals for the year, which may include individual development goals designed to facilitate professional growth and succession planning. As former Chief Executive Officer, President and Chief Scientific Officer of the Company, the Board believed it was appropriate for Dr. Plachetka’s individual goals to mirror the overall corporate goals of the Company. Therefore, for 2014, Dr. Plachetka’s individual goals and the Company’s
goals, as described in the Executive Summary, were identical. The corporate goals are set forth in our Executive Summary on page 116 above. The corporate goals are not assigned specific weightings. The Compensation Committee also takes into account the recommendations of the CEO in determining the annual cash incentives for our other executive officers. Annual cash incentives are utilized to drive annual performance based upon the establishment and agreement of annual goals. The level of the annual cash incentive may also be impacted by other accomplishments during the year.
For 2014, the Compensation Committee awarded the Company credit for achievement of 75% of its corporate goals. A major corporate goal, to gain approval of the NDA for PA32540 and PA8140 by the end of the calendar year, was not achieved. After receiving a CRL in April 2014, which sited inspection deficiencies found during an inspection of the manufacturing facility of an active ingredient supplier, we worked with the active ingredient supplier to resolve the deficiencies, including sending employees and third party consultants to assess progress and assisting the supplier with drafting and providing updates to the FDA. Despite these efforts, the FDA did not complete its review of the response of the active ingredient supplier to the inspection deficiencies prior to our new PDUFA date and on December 16, 2014, we received a second CRL from the FDA which contained identical wording to that of the first CRL with respect to inspection deficiencies of our active ingredient supplier. All of the remaining 2014 corporate goals were achieved. We negotiated significant changes to an existing license agreement as a condition of our consent to GSK’s assignment of the Product Development and Commercialization Agreement for Treximet to Pernix, including the elimination of the restrictions on our right to develop and commercialize certain additional dosage forms of sumatriptan/naproxen combinations outside of the United States, allowing us to seek approval for these combinations on the basis of the approved U.S. NDA, and obtaining Pernix’s commitment to continue certain of GSK’s ongoing development activities and to undertake certain new activities. We also negotiated consideration consisting of a warrant to purchase 500,000 shares of Pernix common stock at an exercise price equal to $4.28, the closing market price on May 9, 2014. Our corporate goal of managing the 2014 net cash burn to $8 million or less was achieved through strict control of expenses. During 2014, we also continued to execute our strategic plan in and this goal was also deemed achieved by the Compensation Committee based upon the completion of activities.
Based on the foregoing assessment of performance, the Compensation Committee awarded Dr. Plachetka an annual cash incentive of $382,700 which represented 75% of his targeted cash incentive opportunity. The cash incentive was awarded in recognition of Dr. Plachetka’s contributions and leadership during the year, including his multiple roles as Chairman, President, CEO and principal scientific innovator, his key role in regulatory interactions and business development activities, ongoing product creation and development, and patent applications and patent defense. The Compensation Committee considered a variety of factors in awarding the cash incentive, including Dr. Plachetka’s scientific contributions to the progression of the PA322540 and PA8140 development program, his lead role in the Company’s interactions with the FDA with respect to PA32540 and PA8140 and with our active ingredient supplier whose inspection deficiencies resulted in the Company’s receipt of two CRLs, his key role in the negotiation with Pernix, and his role as a named inventor on three new patents issued 2014.
Other executive officers were awarded annual cash incentives for fiscal 2014 performance, as follows:
|
William L. Hodges |
|
$ |
108,225 |
|
(75% of target opportunity) |
|
|
John G. Fort |
|
$ |
114,000 |
|
(75% of target opportunity) |
|
|
Gilda M. Thomas |
|
$ |
101,025 |
|
(75% of target opportunity) |
|
|
Dennis L. McNamara |
|
$ |
78,000 |
|
(75% of target opportunity) |
|
The Compensation Committee approved these cash incentive awards in recognition of the accomplishment of all but one of the corporate goals, as well as the individual executive officer’s performance in his or her areas of functional responsibility and accomplishment of individual goals.
For Mr. Hodges, his functional responsibilities included managing the investor relations and public relations functions, providing management information to the Board, managing the financial and accounting function, leading the annual strategic planning process and ensuring accurately and timely filing of required SEC documents. The Compensation Committee determined that Mr. Hodges achieved all of his goals in 2014; however, because the Company did not achieve its goal to obtain approval of its NDA for PA32540 and PA8140, the pool for annual cash incentives was not fully funded, and Mr. Hodges and the other executive officers were paid at 75% of their target opportunity. One of his primary individual goals for 2014, with a weighting of 40%, related to managing the Company’s financial resources efficiently and effectively to accomplish the Company’s strategic objectives while ensuring a net cash burn of less than $8 million, which was also a corporate goal. This goal was deemed achieved by the Compensation Committee as evidenced by the Company’s increased cash balance at the end of 2014. An additional goal for Mr. Hodges, with a weighting of 50%, related to execute the 2014 steps of the strategic plan. This goal was deemed achieved by the Compensation Committee based upon the completion of the activities. A final goal, weighted at 10%, related to improving the percentage of stockholders who voted in favor of the Company’s say-on-pay proposal in the 2014 proxy. Mr. Hodges achieved this goal by planning and participating in outreach to shareholders to understand their concerns regarding the Company’s compensation practices, assisting in improving proxy disclosure to better explain the Company’s compensation practices, working with Mr. Lee, Chairman of the Compensation Committee, to draft a letter to stockholders, and assisting in the creation and implementation of new
corporate governance policies. These efforts resulted in approximately 77% of stockholders voting in favor of the Company’s say-on-pay proposal in proxy, up from approximately 54% in 2013.
For Dr. Fort, his functional responsibilities included serving as the Company’s Chief Medical Officer, managing overall safety aspects of the Company’s clinical studies, reviewing and approving all documents requiring medical review and interpretation, establishing and maintaining contacts with key opinion leaders, and serving as the functional head of clinical pharmacology. The Compensation Committee determined that Dr. Fort achieved all of his goals in 2014; however, because the Company did not achieve its goal to obtain approval of its NDA for PA32540 and PA8140, the pool for annual cash incentives was not fully funded, and Dr. Fort and the other executive officers, were paid at 75% of their target opportunity. One of Dr. Fort’s goals for 2014, weighted at 60%, was to conduct all clinical and other development activities required to obtain approval of PA32540 and PA8140. The Committee deemed that this goal was achieved by Dr. Fort based upon his contributions to the completion of a required Phase 1 study for PA8140 on time and within budget, his review and coordination of efforts to resolve the inspection deficiencies at the Company’s active ingredient supplier, his review and assistance with validation activities, and his lead role in the labeling negotiations for the PA products with FDA. Another goal for Dr. Fort, weighted at 20%, was to support the business development activities relating to the license of the PA product candidates, which he met by providing medical and scientific input into marketing and business development activities as required, and representing the Company in discussions with potential strategic partners with respect to scientific, clinical and regulatory matters. A final goal for Dr. Fort, weighted at 20% was to complete the development of PA10040 for commercialization outside of the United States and to support licensing activities for the product. Dr. Fort achieved this goal by his contributions to the conduct of a required Phase 1 study on time and within budget. He also met with the Medicines Evaluation Board in the Netherlands to discuss requirements for filing a regulatory submission for the product and reviewed and prepared documents required for submission of a MAA.
For Ms. Thomas, her functional responsibilities included drafting, reviewing and structuring agreements in support of all business and corporate activities; coordinating our compliance activities; and providing legal support to all business development and strategic alliance initiatives. The Compensation Committee determined that Ms. Thomas achieved all of her goals in 2014; however, because the Company did not achieve its goal to obtain approval of its NDA for PA32540 and PA8140, the pool for annual cash incentives was not fully funded, and Ms. Thomas and the other executive officers, were paid at 75% of their target opportunity. One of Ms. Thomas’ primary individual goals for 2014, weighted at 35%, was to provide all required legal support for the corporate goal to execute a new license or to negotiate a significant change in an existing license. She achieved this objective by providing legal support in the negotiations with Pernix, which negotiation resulted in a number of benefits to the Company, including a warrant to purchase 500,000 shares of Pernix common stock and the elimination of restrictions on the Company’s ability to market certain formulations of sumatriptan and naxproxen sodium outside of the United States. She also assisted in the negotiating of a mutual termination agreement with Sanofi U.S. which permitted the company to obtain access to certain commercial know how generated by Sanofi U.S. A second goal, weighted at 25%, related to the Paragraph IV litigation in with respect to VIMOVO and Treximet. She achieved this goal by, among other things, managing outside counsel, providing input on all court filings, participating in the development of and approving litigation strategy, assisting in the preparation of deposition witnesses, and serving as the Company’s representative at Court-required settlement conferences. A third goal, also weighted at 30%, was to implement a continuing reduction in force to meet the Company’s current business needs, which goal was deemed achieved by the Compensation Committee based upon the completion of specified activities. A final goal, weighted at 10%, related to improving the percentage of stockholders who voted in favor of the Company’s say-on-pay proposal in the 2014 proxy. She achieved this goal by assisting Mr. Hodges with stockholder outreach and the creation of new corporate governance policies, and by drafting proxy disclosure which better explained the Company’s compensation practices.
For Mr. McNamara, his functional responsibilities include executing corporate development activities, primarily to secure partners to commercialize the Pozen portfolio of products, interfacing with strategic advisors on corporate development initiatives, managing strategic alliances, leading analytics for projecting sales of commercialized products and financial modeling to support transactions, directing and assisting in the enforcement of intellectual property rights by outside counsel and licensees; and leading the commercial analysis and identification of potential intellectual property protection for novel product concepts. The Compensation Committee determined that Mr. McNamara achieved all of his goals in 2014; however, because the Company did not achieve its goal to obtain approval of its NDA for PA32540 and PA8140, the pool for annual cash incentives was not fully funded, and Mr. McNamara and the other executive officers, were paid at 75% of their target opportunity. One of Mr. McNamara’s primary individual goals for 2014, weighted at 40%, was to execute a new license for a Pozen product or to negotiate a significant change in an existing license agreement. He achieved this objective by leading the business negotiations with Pernix, which provided a number of benefits to the Company, including a warrant to purchase 500,000 shares of Pernix common stock and the elimination of restrictions on the Company’s ability to market certain formulations of sumatriptan and naxproxen sodium outside of the United States. He also worked with Ms. Thomas to negotiate the mutual termination agreement with Sanofi on terms favorable to the Company. A second goal relating to alliance management, weighted at 40%, was deemed achieved by the Compensation Committee based on completion of specified activities. A final goal, weighted at 20%, was to obtain at least one new patent for a Pozen product, which Mr. McNamara achieved by managing outside patent counsel and counsel for the Company’s licensee and advancing the Company’s patent prosecution efforts. As a result of his efforts, four new patents were issued and one patent application was allowed which application, when issued, will substantially extend the patent life of one of the Company’s products.
Equity and other long-term incentive compensation
As described above, stock-based incentives are a key component of our executive compensation program and have historically been provided to all of our full-time employees. Employee ownership is a core value of our operating culture, and we and the Compensation Committee believe that stock ownership encourages our executives to create value for our shareholders over the long term, and promotes retention and affiliation with the Company by allowing our employees to share in our long-term success while aligning employee and executive interests with those of our stockholders. To reflect our commitment to employee ownership, the Board has adopted stock ownership guidelines for the CEO of 6x times base salary, as well as a stock retention policy for all named executive officers requiring such officers to retain at least 50% of the total equity credited from grants of equity awards (net of amounts required to pay taxes and exercise prices) while such individual remains a named executive officer. As of [DATE], Dr. Plachetka owned shares of the Company with a value greater than forty-five times his 2014 base pay and all other executives have retained well in excess of the 50% minimum required acquired equity.
Equity awards are awarded annually after careful review of corporate and individual performance. If the corporate goals are achieved, the equity pool is funded at the target level for all employees. The Compensation Committee also evaluates the corporate and individual performance of the CEO and other named executive officers and awards annual equity grants based upon performance and evaluation of market practices of the peer companies. We have traditionally vested these awards over four years to include a retention element to the awards. As discussed above, the Committee is evaluating future equity awards and retention as part of its overall review of compensation, given the company’s strategic direction.
In certain circumstances, the Compensation Committee may determine that non-equity long-term incentives are preferable to equity-based awards. For example, due in part to his significant ownership of our stock, the Compensation Committee has determined that long-term incentive awards to our CEO may include a non-equity component, or may be paid partly or wholly in cash, as determined by the Committee.
Stock options and other long-term equity incentive awards are made under our 2010 Plan. Stock options generally have a ten-year term and vest over a number of years based on continued employment. Vesting for service based stock options awarded to our executive officers has typically been 25% annually over four years from the date of grant. Our stock options are granted at an exercise price equal to the closing price of our common stock on the date of grant. Accordingly, the actual value an executive will realize is tied to future stock appreciation and is therefore aligned with corporate performance and stockholder returns. We have more recently used restricted stock units for annual and performance-based awards to ensure all employees, including our executive officers, are true owners of the Company.
Each year, the Compensation Committee determines the level of long-term incentive award opportunity to be provided to our executive officers. In determining the target opportunity and amount of the awards, the Compensation Committee evaluates factors that contribute to overall corporate growth and development and to increasing long-term stockholder value, such as progression of our drug development pipeline, licensing deals, regulatory approval, stock price movement relative to our peers, execution of and/or progress toward fulfilling our long-term strategic plan, as well as the executive’s performance and contribution to our annual and long-term strategic goals, and each executive officer’s achievement of his or her individual goals and objectives, which are the same goals and objectives which serve as the basis for the award of annual cash incentives described above. The Compensation Committee may, at its discretion, consider both the achievement of the annual Board-approved corporate goals and other significant corporate accomplishments during the year. For our executive officers other than the CEO, the Compensation Committee also takes into account the recommendations of the CEO in determining the amount of the grant to each executive officer.
Until this year, these long-term incentive were granted in March of the following year after performance for the last completed fiscal year has been evaluated. In accordance with that practice, after reviewing 2013 performance, in March 2014, our executive officers were granted restricted stock units, and our former CEO was granted a long-term incentive award consisting of a mix of cash and RSUs. In April 2014, the Compensation Committee granted an additional amount of RSUs to our executive officers, other than Dr. Plachetka, bringing the total amount of restricted stock units granted to the target level recommended by Radford and tied vesting of such additional amounts to the payment of certain milestones under the license agreement with Sanofi U.S. These incentive awards are described more fully below and are reflected in the Summary Compensation Table and the Grants of Plan-Based Award Table included in this prospectus.
With respect to 2014 performance, the Compensation Committee decided to return to a schedule in which long-term incentives are granted at the end of December after evaluation of performance for the calendar year. In accordance with this practice, on December 31, 2014, our executive officers were granted restricted stock units, and our CEO was granted a long-term incentive award consisting of a mix of cash and restricted stock units. The long-term incentive awards granted in December 2014 based on 2014 performance have been disclosed in our Section 16 filings.
2014 Long-Term Incentive Awards. In March 2014, after review of the Company’s progress on its key strategic objectives and in accordance with the principles outlined above, the Compensation Committee awarded our CEO a long-term incentive award of $ 850,000 which vests 33% per year on March 15th over three years, and 98,039 restricted stock units, which vest 25% per year on March 15th over four years. During 2013, Dr. Plachetka filled the roles of Chief Executive Officer, Chairman of the Board, President and Chief Scientific Officer, as well as being an inventor on all of Pozen’s patents. The Compensation Committee considered these factors and the Dr. Plachetka’s significant contributions to the Company in 2013 in approving the award. The Compensation Committee evaluated Dr. Plachetka’s significant contribution to the progress our PA product candidates culminating in the filing of
the NDA for PA32540 and PA8140 in March 2013 and establishing the bioavailability of the aspirin component of PA10040, which is a critical step in completing the submission of a MAA in the EU for PA10040, as well has his key role in the negotiation of an exclusive license agreement with Sanofi U.S. for the commercialization of PA8140 and PA32540 in the United States. The Compensation Committee also considered his role as a named inventor on one new U.S. patent issued in 2013. Based upon progress on the key strategic objectives, long term compensation was awarded at 100% of target. Mr. Hodges, Dr. Fort and Ms. Thomas were awarded 20,000 RSUs and Mr. McNamara was awarded 15,000 RSUs in March 2014, which was at target level.
Procedures and Policies for Granting Equity-based Awards
As described above, the Compensation Committee approves the grant of all stock options and other awards to our former CEO and other executive officers, as well as to the non-employee members of our Board. New-hire grants for our executive officers are approved by the Compensation Committee prior to employment and are granted on the date of hire. Annual equity awards to our executive officers, as well as to all employees, are granted in mid-March, following the year under review in order to allow more time to review the entire year, including the financial results of the Company. In cases where equity awards are granted as a result of certain material achievements, such grants are issued no earlier than two days after the public announcement of the material information. In all cases, stock options are granted at exercise prices equal to the closing price of our stock as reported on NASDAQ on the date of grant.
Under our 2010 Plan, the Compensation Committee may determine that an equity award is considered “qualified performance compensation” under Section 162(2) of the Code if certain criteria set forth in the 2010 Plan are met. As permitted under our 2010 Plan, the Compensation Committee has delegated to our CEO the authority to grant up to a specified aggregate number of stock options in two circumstances:
· option and RSU grants to non-executive officer employees in connection with their year-end performance reviews; and
· initial option and RSU grants to new non-executive officer employees upon commencement of employment in accordance with a specified schedule of numbers of options per grant, based on hiring position.
These options are granted at an exercise price equal to the closing price of our common stock on the grant date and on vesting and other terms consistent with standard forms of option agreement approved for use under our 2010 Plan. Any grants at levels above the schedule or otherwise not on such authorized terms must be approved by the Compensation Committee.
Benefits; Perquisites
Benefits offered to our executive officers serve as a safety net of protection against financial catastrophes that can result from illness, disability or death. Benefits offered to our executive officers are substantially the same as those offered to all of our regular full-time employees.
We maintain a 401(k) plan for our employees, including our executive officers, to encourage our employees to save some portion of their cash compensation for their eventual retirement. Pursuant to a discretionary employer match, in 2013 we matched all employee contributions at 50% up to the IRS imposed limit. The IRS maximum allowable contribution in 2014 was $17,500 with an additional $5,500 allowed for employees who are 50 years old or older. We also increase our employees’ base salary, including our executive officers’, for the cost of group long-term disability insurance coverage to allow the premium to be employee paid, and provide a group life insurance benefit in a coverage amount equal to two times the employee’s annual base salary. Our former CEO participated in these programs on the same terms and conditions as our executive officers and other employees and received $11,500 in contributions in his 401(k) plan as shown in the “All Other Compensation” in the Summary Compensation Table included in this prospectus.
Perquisites
We provided certain additional perquisites to our former CEO which were negotiated at the time Dr. Plachetka became CEO. These perks included the payment of life and disability insurance premiums above the level provided to our other employees, and reimbursement of certain expenses associated with our former CEO’s tax and estate planning. The aggregate compensation value of these benefits was $52,444 in 2014, and is shown in the “All Other Compensation” column in the Summary Compensation Table included in this prospectus.
Post-employment Benefits
We do not offer post-employment health or life insurance to our executive officers other than to the extent such benefits are payable pursuant to their employment agreements as described below under “Severance and Change of Control Benefits”.
Severance and Change of Control Benefits
We believe that providing reasonable severance benefits to our executive officers upon a change of control event or in the context of termination by us without cause or by the executive for good reason (as defined in their employment agreements) is an important part of maintaining a competitive executive compensation program and contributes to our ability to attract and retain high
quality executives. In part, this reflects our recognition that it may be difficult for a senior executive to find a comparable position in a relatively short period of time following termination of employment. We also believe that providing reasonable protections to our executive officers in the event of a change of control is helpful in aligning our executives’ interests with those of our stockholders in the event a potential change of control situation should occur.
We maintain certain plans and have entered into employment agreements with our executive officers that require that we provide severance and related benefits in the event of a termination of employment or a change of control. In connection with negotiating these provisions in our executives’ employment agreements, the Compensation Committee received advice from its consultants as to practices and levels of such benefits among comparable companies. These provisions and benefits, as well as an estimate of the dollar value of these benefits that would be payable to our executive officers under specified assumed conditions, are described in the section of this prospectus beginning on page [·] entitled “Potential Payments on Termination and Change of Control.”
Tax and Accounting Implications
In setting elements of compensation, the Compensation Committee considers the impact of the following tax and accounting provisions:
· Section 162(m). In making compensation decisions, the Compensation Committee is mindful of the potential impact of Section 162(m) of the Code, which generally disallows a tax deduction to public companies for certain compensation over $1 million paid in any year to its chief executive officer and its three most highly compensated executive officers (other than its chief executive officer and chief financial officer). Qualifying performance-based compensation is not subject to this deduction limit if certain requirements are met. The Compensation Committee generally seeks, where feasible, to structure the incentive compensation granted to our executive officers in a manner that is intended to minimize or eliminate the impact of Section 162(m) of the Code. However, the Compensation Committee may elect to make awards that are subject to the Section 162(m) deduction limit, such as time-based restricted stock units or cash awards, when it believes that such awards are appropriate to attract and retain top-quality executives or otherwise achieve our compensation objectives.
· Section 409A. Section 409A of the Code, which governs the form and timing of payment of deferred compensation, generally changes the tax rules that affect most forms of deferred compensation that were not earned and vested prior to 2005. It also expands the types of compensation that are considered deferred compensation subject to these regulations. Section 409A imposes sanctions, including a 20% penalty and an interest penalty, on the recipient of deferred compensation that does not comply with Section 409A. The Compensation Committee takes into account the potential implications of Section 409A of the Code in determining the form and timing of compensation awarded to our executives.
· Sections 280G and 4999. Pre-2009 employment agreements provide for tax protection in the form of a gross-up payment to reimburse the executive for certain excise taxes imposed under Section 4999 of the Code as well as additional taxes resulting from such reimbursement. Section 4999 of the Code imposes a 20% excise tax on each executive who receives “excess parachute payments” in connection with a change of control, and Section 280G disallows the tax deduction to the company of any amount of an excess parachute payment that is contingent on a change of control. Payments as a result of a change of control that exceed three times the executive’s base amount (the average annualized taxable compensation for the five preceding years) may be considered excess parachute payments, and the excise tax is imposed on the parachute payments that exceed the executive’s base amount. The intent of the tax gross-up is to provide a benefit without a tax penalty to our executives whose employment terminates in connection with a change of control. The Compensation Committee considered the adverse tax liabilities imposed by Sections 280G and 4999, as well as other competitive factors, when it structures pre-2009 post-termination benefits for our executive officers. In any agreements executed after January 1, 2009, the gross-up payment has been eliminated.
· Accounting Rules. Various rules under generally accepted accounting principles determine the manner in which grants for equity-based and other compensation are accounted for in our financial statements. We record compensation expenses with respect to equity awards in accordance with FASB ASC Topic 718. Among the factors it considers when making compensation decisions for our executive officers, the Compensation Committee takes into account the accounting treatment under FASB ASC Topic 718 of equity-based and alternative forms of compensation.
EXECUTIVE COMPENSATION TABLES
Summary Compensation Table
The following table summarizes the total compensation paid to or earned by, or with regard to stock awards and options, the grant date fair value of such awards granted during the fiscal years ended December 31, 2014, 2013 and 2012 to our named executive officers.
|
|
|
|
|
|
|
|
|
Stock |
|
Option |
|
Non-Equity |
|
All Other |
|
|
| |||||||
|
|
|
|
|
Salary |
|
Bonus |
|
Awards |
|
Awards |
|
Incentive Plan |
|
Compensation |
|
|
| |||||||
|
Name and Principal Position |
|
Year |
|
($) |
|
($) (1) |
|
($) (2) |
|
($) (2) |
|
Compensation |
|
($) (3) |
|
Total ($) |
| |||||||
|
John R. Plachetka, Pharm.D., |
|
2014 |
|
$ |
609,620 |
|
— |
|
$ |
1,433,212 |
|
— |
|
$ |
1,783,150 |
(4) |
$ |
63,944 |
(5) |
$ |
3,889,926 |
| ||
|
Former President and |
|
2013 |
|
$ |
591,877 |
|
— |
|
$ |
424,997 |
|
$ |
— |
|
$ |
1,657,700 |
(4) |
$ |
51,990 |
(5) |
$ |
2,726,564 |
| |
|
Chief Executive Officer, |
|
2012 |
|
$ |
574,521 |
|
— |
|
$ |
698,754 |
|
568,548 |
|
$ |
938,160 |
(4) |
$ |
74,300 |
(5) |
$ |
2,854,283 |
| ||
|
Chief Scientific Officer |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
William L. Hodges, |
|
2014 |
|
$ |
363,602 |
|
— |
|
$ |
402,000 |
|
— |
|
$ |
108,225 |
(6) |
$ |
11,500 |
|
$ |
885327 |
| ||
|
Chief Financial Officer, |
|
2013 |
|
$ |
353,065 |
|
— |
|
$ |
121,400 |
|
$ |
— |
|
$ |
140,100 |
(6) |
$ |
11,500 |
|
$ |
626,065 |
| |
|
Senior Vice President, |
|
2012 |
|
$ |
342,827 |
|
— |
|
$ |
179,290 |
|
75,500 |
|
$ |
136,000 |
(6) |
$ |
11,250 |
|
$ |
744,867 |
| ||
|
Finance and Administration |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
John G. Fort, M.D., |
|
2014 |
|
$ |
382,998 |
|
— |
|
$ |
402,000 |
|
— |
|
$ |
114,000 |
(6) |
$ |
11,500 |
|
$ |
910,498 |
| ||
|
Chief Medical Officer |
|
2013 |
|
$ |
371,960 |
|
— |
|
$ |
121,400 |
|
$ |
— |
|
$ |
147,600 |
(6) |
$ |
11,500 |
|
$ |
652,460 |
| |
|
|
|
2012 |
|
$ |
361,223 |
|
— |
|
$ |
174,570 |
|
67,950 |
|
$ |
143,300 |
(6) |
$ |
11,500 |
|
$ |
758,293 |
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
Gilda M. Thomas |
|
2014 |
|
$ |
339,648 |
|
— |
|
$ |
402,000 |
|
— |
|
$ |
101,025 |
(6) |
$ |
11,500 |
|
$ |
854,173 |
| ||
|
Senior Vice President, |
|
2013 |
|
$ |
329,811 |
|
$ |
10,000 |
(7) |
$ |
121,400 |
|
$ |
— |
|
$ |
130,800 |
(6) |
$ |
11,500 |
|
$ |
603,511 |
|
|
General Counsel |
|
2012 |
|
$ |
320,273 |
|
— |
|
$ |
179,290 |
|
75,500 |
|
$ |
126,900 |
(6) |
$ |
11,250 |
|
$ |
713,213 |
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
Dennis L. McNamara |
|
2014 |
|
$ |
262,522 |
|
— |
|
$ |
361,500 |
|
— |
|
$ |
78,000 |
(6) |
$ |
8,750 |
|
$ |
710,772 |
| ||
|
Chief Business Officer |
|
2013 |
|
$ |
225,332 |
|
— |
|
$ |
24,280 |
|
$ |
— |
|
$ |
44,630 |
(6) |
$ |
8,750 |
|
$ |
302,992 |
| |
|
|
|
2012 |
|
$ |
216,973 |
|
— |
|
$ |
60,388 |
|
60,000 |
|
$ |
53,700 |
(6) |
$ |
8,750 |
|
$ |
399,561 |
| ||
(1) Reflects discretionary bonuses accrued during the indicated year.
(2) The amounts included in this column are the dollar amounts representing the full grant date fair value of each stock option or RSU award, as applicable, calculated in accordance with FASB ASC Topic 718 and do not represent the actual value that may be recognized by the named executive officers upon option exercise or settlement of the RSU award. For information on the valuation assumptions used in calculating this amount, see Note 6 to Pozen’s audited financial statements included in the Annual Report on Form 10-K for the fiscal year ended December 31, 2014, as filed with the SEC. For the performance-based RSU and options awarded under the PA32540 and PA8140 equity programs, the grant date fair value reported in the table is based on the probable outcome of the performance goals and assuming full achievement of the performance goals.
(3) For each named executive officer, the amounts shown in this column reflect an employer matching contribution to 401(k) plan.
(4) Under certain circumstances, the Compensation Committee may determine that non-equity incentive awards are preferable to equity-based awards. Included here are amounts that were earned based on performance objectives identified at the beginning of the performance period in 2014, 2013, 2012 and 2011, and also amounts earned as long-term cash incentive awards (“LTIA”) granted for 2014, 2013 and 2012. Included in the 2014, the cash performance award was $295,650 and the LTIAs were $850,000 on March 15th and $637,500 on December 31st. For 2013 the cash performance award was $382,700 and the LTIA was $1,275,000. For 2012 the cash performance award was $371,500 and the LTIA was $566,660. Each individual LTIA grant has a payout over a three-year time-based vesting schedule. The 2014 LTIAs vests one-third per year beginning on the first anniversary of one award’s March 15, 2014 grant date and one-third per year beginning on the first anniversary of one award’s December 31, 2014 grant date. The 2013 LTIA vests one-third per year beginning on the first anniversary of the award’s March 15, 2013 grant date, the 2012 LTIA vests one-third per year beginning on the first anniversary of the award’s March 15, 2012 grant. Consequently, the amounts included in this column may not reflect the compensation expense recognized by Pozen for financial statement reporting purposes for the fiscal years ended December 31, 2014, December 31, 2013 and December 31, 2012. The terms of the long-term incentive program are described on page 123 under the heading “Equity and other long-term incentive compensation” and on page 127 under the heading “Employment and other Agreements.”
(5) This amount includes the following:
· 2014: $11,500 in employer matching contribution to 401(k) plan; $17,763 for payment of supplemental life and disability insurance premiums; $11,946 for reimbursement of employment agreement related legal fees and expenses for tax, estate and financial planning services, and $22,735 for the related tax gross-up.
· 2013: $11,500 in employer matching contribution to 401(k) plan; $16,353 for payment of supplemental life and disability insurance premiums; $6,584 for reimbursement of employment agreement related legal fees and expenses for tax, estate and financial planning services, and $17,553 for the related tax gross-up.
· 2012: $11,250 in employer matching contribution to 401(k) plan; $14,948 for payment of supplemental life and disability insurance premiums; $21,337 for reimbursement of employment agreement related legal fees and expenses for tax, estate and financial planning services, and $26,765 for the related tax gross-up.
(6) This amount represents the amount that was earned based on performance objectives identified at the beginning of the performance period in 2014, 2013 and 2012.
(7) This amount represents a 2013 bonus paid to Ms. Thomas upon the execution of the Sanofi U.S. license agreement.
Grants of Plan-Based Awards in 2014
The following table provides additional information about awards granted to our named executive officers in 2014.
|
Name |
|
Award |
|
Grant Date |
|
Date of |
|
Estimated |
|
Estimated |
|
All Other |
|
All Other |
|
Exercise or |
|
Grant Date |
| |||
|
John R. Plachetka, Pharm.D. |
|
AIC |
|
— |
|
— |
|
$ |
405,983 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
LTI |
|
— |
|
— |
|
$ |
1,700,000 |
|
— |
|
— |
|
— |
|
$ |
— |
|
$ |
— |
|
|
|
|
RSU |
|
3/15/2014 |
|
3/15/2014 |
|
— |
|
— |
|
98,039 |
|
— |
|
$ |
8.10 |
|
$ |
794,116 |
| |
|
|
|
|
|
12/31/2014 |
|
12/31/2014 |
|
|
|
|
|
79,887 |
|
|
|
8.00 |
|
639,096 |
| |||
|
William L. Hodges |
|
AIC |
|
— |
|
— |
|
$ |
148,608 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
RSU |
|
3/15/2014 |
|
3/15/2014 |
|
— |
|
— |
|
20,000 |
|
— |
|
$ |
8.10 |
|
$ |
162,000 |
| |
|
|
|
|
|
12/31/2014 |
|
12/31/2014 |
|
|
|
|
|
30,000 |
|
|
|
$ |
8.00 |
|
$ |
240,000 |
| |
|
John G. Fort |
|
AIC |
|
— |
|
— |
|
$ |
156,593 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
RSU |
|
3/15/2014 |
|
3/15/2014 |
|
— |
|
— |
|
20,000 |
|
— |
|
$ |
8.10 |
|
$ |
162,000 |
| |
|
|
|
|
|
12/31/2014 |
|
12/31/2014 |
|
|
|
|
|
30,000 |
|
|
|
$ |
8.00 |
|
$ |
240,000 |
| |
|
Gilda M. Thomas |
|
AIC |
|
— |
|
— |
|
$ |
138,739 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
RSU |
|
3/15/2014 |
|
3/15/2014 |
|
— |
|
— |
|
20,000 |
|
— |
|
$ |
8.10 |
|
$ |
162,000 |
| |
|
|
|
|
|
12/31/2014 |
|
12/31/2014 |
|
|
|
|
|
30,000 |
|
|
|
$ |
8.00 |
|
$ |
240,000 |
| |
|
Dennis L. McNamara |
|
AIC |
|
— |
|
— |
|
$ |
106,470 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
RSU |
|
3/15/2014 |
|
3/15/2014 |
|
— |
|
— |
|
15,000 |
|
— |
|
$ |
8.10 |
|
$ |
121,500 |
| |
|
|
|
|
|
12/31/2014 |
|
12/31/2014 |
|
|
|
|
|
30,000 |
|
|
|
$ |
8.00 |
|
$ |
240,000 |
| |
(1) Award types are as follows: AIC is an annual incentive cash award, LTI is a long-term incentive cash award, OPT is a stock option and RSU is a restricted stock unit.
(2) Each annual cash incentive award amount represents the individual’s current salary multiplied by their target bonus opportunity. The target long-term incentive cash award for Dr. Plachetka can be allocated, at the discretion of the Compensation Committee, among cash, stock options and RSUs.
(3) The RSU awards on March 15, 2014 and December 31, 2014 were granted under our 2010 Plan and vests in four equal annual installments, with an initial vesting date of March 15, 2015 and December 31, 2015, respectively. Vesting may accelerate in the event of a change of control, in accordance with the terms of our 2010 Plan, and the stock units are payable in shares of common stock, to the extent vested, when Dr. Plachetka ceases to be employed by, or provide service to Pozen.
(4) The amounts included in this column are the dollar amounts representing the full grant date fair value of each option calculated in accordance with FASB ASC TOPIC 718 and do not represent the actual value that may be recognized by the named executive officers upon option exercise. For information on the valuation assumptions used in calculating this amount, see Note 6 to Pozen’s audited financial statements included in the Annual Report on Form 10-K for the fiscal year ended December 31, 2014, as filed with the SEC.
Employment and other Agreements
During 2014, each of our named executive officers was employed pursuant to employment agreements with us. Each employment agreement specifies, among other things, the named executive officer’s initial base salary, bonus opportunity, entitlement to participate in our benefits plans and post-termination benefits and obligations. The post-employment benefits are described in the section entitled “Potential Payments on Termination and Change of Control” beginning on page [ ].
Dr. Plachetka’s agreement, which became effective on March 14, 2006, has an initial term of three years and automatically renews for successive one-year periods thereafter unless either party provides at least six months’ notice of its intention not to renew the agreement. Under the agreement, Dr. Plachetka is entitled to an annual base salary of at least $462,000 effective as of January 1, 2006. Annual increases, if any, are to be made based on performance and in the sole discretion of our Board or the Compensation Committee. Under the terms of the agreement, Dr. Plachetka is eligible to receive an annual cash incentive bonus, based on performance, payable in the discretion of the Compensation Committee, with a targeted amount of 65% of Dr. Plachetka’s annual base salary. Dr. Plachetka is also eligible to receive annual awards under a long-term incentive program with a target value of $1,700,000 for the first year of the agreement, subject to annual review by the Compensation Committee. Awards under the long-term incentive program are based on performance and made in the discretion of the Compensation Committee. The agreement also provides for the payment by the Company of certain life and disability insurance premiums and the reimbursement of certain estate, tax and legal expenses relating to the agreement, and expenses relating to the establishment and administration of a Rule 10b5-1 securities selling program, incurred by Dr. Plachetka.
Dr. Plachetka retired as Chairman, President and Chief Executive Officer and resigned as a director, effective June 1, 2015. Dr. Plachetka will remain an employee of the Company for 90 days following May 29, 2015 (the "Signature Date"). Dr. Plachetka will receive certain benefits in connection with his retirement under the terms of a Separation and General Release Agreement (the "Separation Agreement"). Under the terms of the Separation Agreement, Dr. Plachetka will continue to be paid his full compensation and benefits for 90 days following the Signature Date (the "Separation Date"). Subsequently, Dr. Plachetka will receive certain severance benefits, including the continuation of his base salary at the current rate for a period of 24 months and a lump sum payment of two times the average annual bonus actually awarded to him over the prior two years. He will also receive reimbursement of the actual cost of continuing his health and dental benefits under COBRA for the 18 months following the Separation Date. The Separation Agreement also states that Dr. Plachetka is eligible to receive payment of an amount equal to the portion of his long term cash incentive awards that would have become vested on the next vesting date if he had not retired. Subject to certain conditions, all equity awards previously granted to Dr. Plachetka under the Company’s 2000 Equity Compensation Plan and the 2010 Plan, that remain unvested at the Separation Date will be deemed fully vested at the Separation Date. The Separation Agreement also requires the exercise period for all outstanding options held by Dr. Plachetka to be extended so that they terminate on the date that is the earlier of the second anniversary of the Separation Date or the date on which such options otherwise expire. Dr. Plachetka is also eligible to receive additional payments totaling up to $1.5 million, provided the release has become effective. The Separation Agreement also provides for special performance-based compensation to Dr. Plachetka in recognition of his efforts to secure approval of YOSPRALA by the FDA. As of the Separation Date, Dr. Plachetka was be granted nonqualified stock options with a grant date fair value of $1 million subject to performance-based vesting as set forth in the Separation Agreement. In addition, Dr. Plachetka is eligible to receive a cash bonus of up to $708,334 if YOSPRALA approval is obtained from the FDA within certain time frames set forth in the Separation Agreement, in lieu of certain forfeited Long Term Incentive Plan awards.
Our employment agreements with Mr. Hodges, Dr. Fort, Ms. Thomas and Mr. McNamara have initial terms of one year. Each agreement automatically renews for successive one-year terms after the expiration of the initial term, unless either party to the agreement terminates the agreement. The agreements specify initial annual base salary amounts that are subject in each case to performance and merit-based increases, as determined by the Compensation Committee. The executives are eligible to receive annual bonuses of up to 40% of base salary, to be awarded as determined by and in the discretion of the Compensation Committee.
The employment agreements for certain of the executive officers (except those described below) provide for the following severance payments upon a termination by Pozen without “cause” and by the executive for “good reason”:
· 12 months base salary;
· COBRA continuation for 12 months; and
· average annual cash incentive awarded over the previous two years.
Under Pozen’s recently adopted severance plan, any Pozen executives who were employed at March 31, 2015, however, are entitled to enhanced severance benefits equal to 12 months base salary, COBRA continuation for 18 months and a lump sum payment equal to their target annual cash incentive to be awarded in the year of termination in the event that the executive is terminated without cause by Pozen. These severance benefits will only be paid if the executive officer complies with the confidentiality, development, assignment, and one-year non-solicitation, non-competition and non-disparagement covenants set forth in the executive officer’s employment agreement, and signs a release and waiver of claims in favor of Pozen and its affiliates. Mr. Hodges, Mr. Barnhardt and Ms. Thomas are entitled to a gross-up of excess parachute payments within the meaning of Section 280G of the Code if there is a change of control and the severance benefits exceed the 280G limit. The merger is not considered a “change of control” for purposes of Section 280G of Code, so this provision is not applicable.
Outstanding Equity Awards at December 31, 2014
The following table summarizes the equity awards we have made to our named executive officers that had not been exercised and remained outstanding as of December 31, 2014.
|
|
|
Option Awards |
|
Stock Awards |
| |||||||||||||||||
|
Name |
|
Number |
|
Number of |
|
Equity |
|
Option |
|
Option |
|
Number of |
|
Market |
|
Equity |
|
Equity |
| |||
|
John R. Plachetka, Pharm.D. |
|
206,131 |
|
— |
|
— |
|
$ |
8.62 |
|
1/3/2016 |
(3) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
35,271 |
|
— |
|
— |
|
$ |
13.83 |
|
1/3/2017 |
(4) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
107,040 |
|
— |
|
— |
|
$ |
8.36 |
|
3/14/2018 |
(5) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
62,053 |
|
— |
|
— |
|
$ |
11.83 |
|
5/6/2018 |
(6) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
49,151 |
|
— |
|
— |
|
$ |
4.64 |
|
3/13/2019 |
(7) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
165,198 |
|
— |
|
— |
|
$ |
5.33 |
|
3/15/2020 |
(8) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
52,439 |
|
— |
|
$ |
3.77 |
|
3/15/2021 |
(9) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
32,744 |
(10) |
$ |
261,950 |
(11) |
— |
|
— |
| ||
|
|
|
10,179 |
|
— |
|
5,089 |
(12) |
$ |
1.98 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
2,756 |
(12) |
$ |
6,229 |
(13) | ||
|
|
|
— |
|
229,964 |
(14) |
— |
|
$ |
3.87 |
|
3/15/2022 |
(14) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
12,404 |
(15) |
$ |
78,021 |
(13) | ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
62,035 |
(16) |
$ |
496,278 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
98,039 |
(17) |
$ |
784,312 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
79,887 |
(18) |
$ |
639,096 |
(11) |
— |
|
— |
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||
|
William L. Hodges |
|
50 |
|
— |
|
— |
|
$ |
8.62 |
|
1/3/2016 |
(3) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
109,936 |
|
— |
|
— |
|
$ |
13.84 |
|
1/3/2017 |
(4) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
20,277 |
(6) |
— |
|
— |
|
$ |
11.83 |
|
5/6/2018 |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
45,807 |
|
— |
|
— |
|
$ |
4.64 |
|
3/13/2019 |
(7) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
40,921 |
|
— |
|
— |
|
$ |
5.33 |
|
3/15/2020 |
(8) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
37,493 |
|
12,498 |
|
— |
|
$ |
3.77 |
|
3/15/2021 |
(9) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
5,089 |
(12) |
$ |
1.98 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
2,755 |
(12) |
$ |
6,226 |
(13) | ||
|
|
|
— |
|
30,537 |
|
— |
|
$ |
3.87 |
|
3/15/2022 |
(14) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
12,404 |
(15) |
$ |
78,021 |
(13) | ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
17,720 |
(19) |
$ |
141,760 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
20,000 |
(20) |
$ |
160,000 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
30,000 |
(21) |
$ |
240,000 |
(11) |
— |
|
— |
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||
|
John G. Fort |
|
48,860 |
|
— |
|
— |
|
$ |
15.73 |
|
7/16/2017 |
(22) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
7,329 |
|
— |
|
— |
|
$ |
8.36 |
|
3/14/2018 |
(5) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
18,322 |
(6) |
— |
|
— |
|
$ |
11.83 |
|
5/6/2018 |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
18,322 |
|
— |
|
— |
|
$ |
4.64 |
|
3/13/2019 |
(7) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
36,645 |
(8) |
— |
|
— |
|
$ |
5.33 |
|
3/15/2020 |
(8) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
13,742 |
|
— |
|
$ |
3.77 |
|
3/15/2021 |
(9) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
5,089 |
(12) |
$ |
1.98 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
2,755 |
(12) |
$ |
6,226 |
(13) | ||
|
|
|
— |
|
27,484 |
|
— |
|
$ |
3.87 |
|
3/15/2022 |
(14) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
12,404 |
(15) |
$ |
78,021 |
(13) | ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
17,720 |
(19) |
$ |
141,760 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
20,000 |
(20) |
$ |
160,000 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
30,000 |
(21) |
$ |
240,000 |
(11) |
— |
|
— |
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||
|
Gilda M. Thomas |
|
48,860 |
|
— |
|
— |
|
$ |
13.25 |
|
1/8/2017 |
(22) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
48,860 |
|
— |
|
— |
|
$ |
8.36 |
|
3/14/2018 |
(5) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
18,811 |
(6) |
— |
|
— |
|
$ |
1.83 |
|
5/6/2018 |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
61,075 |
|
— |
|
— |
|
$ |
4.64 |
|
3/13/2019 |
(7) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
61,075 |
|
— |
|
— |
|
$ |
5.33 |
|
3/15/2020 |
(8) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
41,226 |
|
13,742 |
|
— |
|
$ |
3.77 |
|
3/15/2021 |
(9) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
10,179 |
|
— |
|
5,089 |
(12) |
$ |
1.98 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
2,755 |
(12) |
$ |
6,226 |
(13) | ||
|
|
|
— |
|
30,537 |
|
— |
|
$ |
3.87 |
|
3/15/2022 |
(14) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
12,404 |
(15) |
$ |
78,021 |
(13) | ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
17,720 |
(19) |
$ |
141,760 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
20,000 |
(20) |
$ |
160,000 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
30,000 |
(21) |
$ |
240,000 |
(11) |
— |
|
— |
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||
|
Dennis L. McNamara |
|
6,107 |
|
— |
|
— |
|
$ |
8.62 |
|
1/3/2016 |
(3) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
21,376 |
|
— |
|
— |
|
$ |
8.45 |
|
8/4/2016 |
(23) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
30,537 |
|
— |
|
— |
|
$ |
13.83 |
|
1/3/2017 |
(4) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
7,817 |
|
— |
|
— |
|
$ |
8.36 |
|
3/14/2018 |
(5) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
6,840 |
|
— |
|
— |
|
$ |
11.83 |
|
5/6/2018 |
(6) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
9,772 |
|
— |
|
— |
|
$ |
4.64 |
|
3/13/2019 |
(7) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
12,215 |
|
— |
|
— |
|
$ |
5.33 |
|
3/15/2020 |
(8) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
2,443 |
|
2,443 |
|
— |
|
$ |
3.77 |
|
3/15/2020 |
(9) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
2,036 |
(12) |
$ |
1.98 |
|
— |
|
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
1,101 |
(12) |
$ |
2,488 |
(13) | ||
|
|
|
— |
|
4,886 |
|
— |
|
$ |
3.87 |
|
3/15/2022 |
(14) |
— |
|
— |
|
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
4,961 |
(15) |
$ |
31,205 |
(13) | ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
3,544 |
(19) |
$ |
28,352 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
15,000 |
(20) |
$ |
120,000 |
(11) |
— |
|
— |
| ||
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
30,000 |
(21) |
$ |
240,000 |
(11) |
— |
|
— |
| ||
|
(1) |
Each of these options was granted under our 2010 Plan or 2000 Equity Compensation Plan, has a 10-year term and vests and becomes exercisable in four equal annual installments, with the initial vesting date occurring on the one-year anniversary of the respective date of grant, unless a specific footnote indicates differently. |
|
(2) |
The exercise price of each of the options included in this table is equal to the closing price of Pozen’s common stock as reported by NASDAQ on the respective date of grant. |
|
(3) |
The option award vests 25% per year beginning on the first anniversary of the option’s 1/3/2006 grant date. |
|
(4) |
The option award vests 25% per year beginning on the first anniversary of the option’s 1/3/2007 grant date. |
|
(5) |
The option award vests 25% per year beginning on the first anniversary of the option’s 3/14/2008 grant date. |
|
(6) |
These options were granted under our 2000 Equity Compensation Plan, pursuant to an incentive program (the “PN incentive program”), to all of the Company’s employees, including its executive officers, and have a 10-year term. 25% vested in August 2009 upon the acceptance by the FDA of the NDA for VIMOVO. The remaining 75% of the options granted vested in April 2010 upon the receipt by the Company of an action letter from the FDA indicating approval of the NDA for VIMOVO. |
|
(7) |
The option award vests 25% per year beginning on the first anniversary of the option’s 3/13/2009 grant date. |
|
(8) |
The option award vests 25% per year beginning on the first anniversary of the option’s 3/15/2010 grant date. |
|
(9) |
The option award vests 25% per year beginning on the first anniversary of the option’s 3/15/2011 grant date. |
|
(10) |
Represents the unvested portion of 130,975 restricted stock units awarded to Dr. Plachetka in March 2011. The RSUs vest in four equal annual installments, commencing with an initial vesting date of March 15, 2012, and in the event of a change of control, in accordance with the terms of our 2000 Equity Compensation Plan. The shares of common stock represented by the RSUs, once vested, are payable when Dr. Plachetka ceases to be employed by or perform services for Pozen. No dividends are payable on the RSUs; however, the RSUs will be appropriately adjusted in the event of a stock split, stock dividend or other change in capitalization of Pozen. |
|
(11) |
Calculated by multiplying the closing market price of Pozen’s common stock on December 31, 2013 by the unvested number of RSUs. |
|
(12) |
Each of these options and stock awards was granted under our 2010 Plan, has a 10-year term and vest in accordance with the following schedule: (a) one-half (1/2) upon first cycle NDA approval of PA32540 (otherwise 25% upon NDA approval after first cycle), and (b) one-half (1/2) upon execution of a significant partnering transaction for PA32540 in a major territory (this performance condition was achieved in September 2013 with the execution of the Sanofi U.S. agreement), subject in each case to continued employment or service to the Company. |
|
(13) |
The amounts included in this column are the dollar amounts representing the full grant date fair value of each option and RSU award calculated in accordance with FASB ASC TOPIC 718 and do not represent the actual value that may be recognized by the named executive officers upon option exercise or settlement of RSUs. For information on the valuation assumptions used in calculating this amount, see Note 6 to Pozen’s audited financial statements included in the Annual Report on Form 10-K for the fiscal year ended December 31, 2012, as filed with the SEC. |
|
(14) |
The option award vests 50% per year beginning on the third anniversary of the option’s 3/15/2012 grant date. |
|
(15) |
Each of these options and stock awards was granted under our 2010 Plan, has a 10-year term and vest in accordance with the following schedule: (a) 1/2 upon the acceptance by the FDA of the filing of a NDA for a low dose PA product, currently PA8140 and (b) 1/2 upon approval by the FDA of an NDA for a low dose PA product, currently PA8140. |
|
(16) |
Represents the unvested portion of 82,713 restricted stock units awarded to Dr. Plachetka in March 2013. The RSUs vest in four equal annual installments, commencing with an initial vesting date of March 15, 2014, and in the event of a change of control, in accordance with the terms of our 2000 Equity Compensation Plan. The shares of common stock represented by the RSUs, once vested, are payable when Dr. Plachetka ceases to be employed by or perform services for Pozen. No dividends are payable on the RSUs; however, the RSUs will be appropriately adjusted in the event of a stock split, stock dividend or other change in capitalization of Pozen. |
|
(17) |
Represents the unvested portion of 98,039 RSUs awarded to Dr. Plachetka in March 2014. The RSUs vest in four equal annual installments, commencing with an initial vesting date of March 15, 2015, and in the event of a change of control, in accordance with the terms of our 2000 Equity Compensation Plan. The shares of common stock represented by the RSUs, once vested, are payable when Dr. Plachetka ceases to be employed by or perform services for Pozen. No dividends are payable on the restricted stock units; however, the restricted stock units will be appropriately adjusted in the event of a stock split, stock dividend or other change in capitalization of Pozen. |
|
(18) |
Represents the unvested portion of 79,887 RSUs awarded to Dr. Plachetka in December 2014. The RSUs vest in four equal annual installments, commencing with an initial vesting date of January 1, 2016, and in the event of a change of control, in accordance with the terms of our 2000 Equity Compensation Plan. The shares of common stock represented by the RSUs, once vested, are payable when Dr. Plachetka ceases to be employed by or perform services for Pozen. No dividends are payable on the RSUs; however, the RSUs will be appropriately adjusted in the event of a stock split, stock dividend or other change in capitalization of Pozen. |
|
(19) |
The RSU award vests 25% per year beginning on the first anniversary of the option’s 3/15/2013 grant date. |
|
(20) |
The RSU award vests 25% per year beginning on the first anniversary of the option’s 3/15/2014 grant date. |
|
(21) |
The RSU award vests 25% per year beginning on the first anniversary of the option’s 12/31/14 grant date. |
|
(22) |
The option award vests 25% per year beginning on the first anniversary of the option’s 1/8/2007 grant date. |
|
(23) |
The option award vests 25% per year beginning on the first anniversary of the option’s 8/4/2006 grant date. |
Option Exercises and Stock Vested in 2014 Fiscal Year
The following table provides information regarding our named executive officers’ exercise of stock options and vesting of restricted stock awards during the year ended December 31, 2014.
|
|
|
Option Awards |
|
Stock Awards |
| ||||||
|
Name |
|
Number |
|
Value |
|
Number |
|
Value |
| ||
|
John R. Plachetka, Pharm.D. |
|
500,000 |
|
$ |
1,714,395 |
|
91,592 |
|
$ |
794,100 |
|
|
William L. Hodges |
|
320,177 |
|
$ |
731,995 |
|
11,812 |
|
$ |
102,410 |
|
|
John G. Fort, M.D. |
|
13,725 |
|
$ |
80,154 |
|
11,222 |
|
$ |
97,295 |
|
|
Gilda M. Thomas |
|
— |
|
— |
|
11,812 |
|
$ |
102,410 |
| |
|
Dennis L. McNamara |
|
30,537 |
|
$ |
96,496 |
|
2,126 |
|
$ |
18,432 |
|
|
(1) |
Calculated based upon the closing market price or sale price of Pozen’s common stock on the respective date of exercise less the exercise price of each share. |
|
(2) |
Represents the value of RSUs that vested during 2014. Calculated by multiplying the number of shares represented by the RSUs by the closing market price of Pozen’s common stock on the vesting date. |
Pension Benefits for 2014 Fiscal Year
The table disclosing the value of accumulated benefits under and other information concerning defined benefit plans during the year is omitted because we do not have a defined benefit plan for our named executive officers or other employees. The only retirement plan available to our named executive officers in 2013 was our 401(k) plan which is available to all employees.
Nonqualified Deferred Compensation for 2014 Fiscal Year
The table disclosing contributions to and aggregate earnings under or distributions from nonqualified defined contribution or other deferred compensation plans is omitted because we do not have any such nonqualified deferred compensation plans.
Potential Payments on Termination and Change of Control
Upon termination of employment or a change of control, our named executive officers are entitled to certain compensation and benefits under the terms of their employment agreements, as well as other plans and arrangements provided by us. The terms of the employment agreements for Dr. Plachetka, Mr. Hodges and Ms. Thomas contain provisions providing for a tax gross up in the event that any severance payment or benefit would constitute an “excess parachute payment” within the meaning of Section 280G of the Internal Revenue Code. Executive employment agreements executed after January 1, 2009, including those executed by Dr. Fort and Mr. McNamara, our Senior Vice President and Chief Business Officer, effective January 1, 2014, as well as Mr. Adams, our newly appointed Chief Executive Officer, do not contain this provision. The tables below list the potential compensation payable to our executive officers under various hypothetical termination scenarios. The discussion and the amounts shown in the tables assume that the termination or change of control took place on December 31, 2014 (and thus include amounts earned through such time), and assume that the price per share of our stock was the closing market price on December 31, 2014 ($8.00 per share). The amounts shown are estimates of the amounts that would be paid out to the executive officers. The amounts that the executive officers would receive in an actual termination or change of control can only be determined at the time the event occurs.
John R. Plachetka
The following table describes the potential payments upon termination or a change of control for John R. Plachetka, Pharm.D., our President and Chief Executive Officer (CEO).
|
Executive Benefits |
|
Termination |
|
Termination |
|
Death or |
|
Non- |
|
Change of |
|
Change of |
|
Change of |
| |||||||
|
Compensation: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Salary Continuation (1x or 2x) (1) |
|
$ |
— |
|
$ |
1,256,806 |
|
$ |
— |
|
$ |
— |
|
$ |
628,403 |
|
$ |
1,256,806 |
|
$ |
— |
|
|
Bonus (1x or 2x) (2) |
|
$ |
— |
|
$ |
678,350 |
|
$ |
339,175 |
|
$ |
— |
|
$ |
339,175 |
|
$ |
678,350 |
|
$ |
— |
|
|
Stock Options — Accelerated |
|
$ |
— |
|
$ |
696,693 |
(3) |
$ |
— |
|
$ |
— |
|
$ |
1,171,568 |
(4) |
$ |
1,171,568 |
(4) |
$ |
1, 171,568 |
(4) |
|
Restricted Stock Units |
|
$ |
— |
|
$ |
783,231 |
(3) |
$ |
— |
|
$ |
— |
|
$ |
2,181,640 |
(5) |
$ |
2,181,640 |
(5) |
$ |
2,181,640 |
(5) |
|
LTIP (6),(7) |
|
$ |
— |
|
$ |
1,109,722 |
(6) |
$ |
— |
|
$ |
— |
|
$ |
2,313,889 |
(7) |
$ |
2,313,889 |
(7) |
$ |
2,313,889 |
(7) |
|
Benefits and Perquisites |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Health Care Continuation (8) |
|
$ |
— |
|
$ |
27,911 |
|
$ |
— |
|
$ |
— |
|
$ |
27,911 |
|
$ |
27,911 |
|
$ |
— |
|
|
280G Tax Gross Up (9) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
3,310,002 |
|
$ |
— |
|
|
(1) |
Annual 2013 base salary is $628,403. |
|
(2) |
The bonus component is based on the annual cash incentive for 2014 and 2013 and excludes special bonuses made during those years. The reported amount is calculated as the average annual cash incentive awarded over the previous two years. The average of the annual cash incentive paid in 2014 ($295,650) and 2013 ($371,500). |
|
(3) |
This amount represents options or RSUs that would otherwise vest in 2015. The aggregate value reported is based on the spread between the closing stock market price of $8.00 on December 31, 2014 and the exercise price of the options. |
|
(4) |
Pursuant to our 2010 Plan, unless the Compensation Committee determines otherwise, upon a change of control all awards vest as of the change of control date. This number assumes that all outstanding unvested options held as of December 31, 2014 would vest. The aggregate value reported is based on the spread between the closing stock market price of $8.00 on December 31, 2014 and the exercise prices of the option grants. |
|
(5) |
This number assumes that all outstanding unvested RSUs held as of December 31, 2014 would vest (see note 4 above). The reported value for the RSUs is equal to the grants with underlying shares times the closing market stock price of $8.00 on December 31, 2014. |
|
(6) |
This number assumes that the tranches from the Long Term Incentive Cash Awards that would otherwise vest in 2015 become vested. |
|
(7) |
The number assumes that all remaining tranches from his Long Term Incentive Cash Awards vest upon a change in control. |
|
(8) |
Dr. Plachetka is entitled to continue participation in our health and dental plan for 18 months after termination, or, alternatively Pozen will reimburse him for its share of COBRA premiums for such health and dental benefits for a period of 18 months. The reported amount assumes that we will pay 100% of the employee premium and 50% of the dependent premium in effect at December 31, 2014 for 18 months. |
|
(9) |
See the narrative that follows these tables for a discussion of the tax gross-up benefit payable to Dr. Plachetka. The reported numbers assume a December 31, 2014 closing stock price of $8.00. |
|
(10) |
As described above under the heading “Employment and Other Agreements” on page 127, Dr. Plachetka retired effective June 1, 2015. The payments and compensation he received or will be receiving as a result of his termination of employment are those he became entitled to under his employment agreement in connection with a termination of employment without cause as described above with the following adjustments: (1) additional lump sum payments totaling $1,500,000; (2) COBRA premium reimbursement for 24 months; (3) an extension of his option exercise period for all outstanding options; (4) full vesting of all equity awards granted under the 2000 Equity Compensation Plan and the 2010 Plan; (5) a nonqualified option grant with a grant date fair value of $1 million subject to performance-based vesting as set forth in the Separation Agreement; and (6) a cash bonus of up to $708,334 if certain performance targets are met within certain time frames set forth in the Separation Agreement. |
William L. Hodges
The following table describes the potential payments upon termination or a change of control of Pozen for William L. Hodges, Senior Vice President and Chief Financial Officer.
|
Executive Benefits |
|
Termination |
|
Termination |
|
Death or |
|
Non- |
|
Change of |
|
Change of |
| ||||||
|
Compensation: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Salary (1) |
|
$ |
— |
|
$ |
373,787 |
|
$ |
— |
|
$ |
— |
|
$ |
373,787 |
|
$ |
— |
|
|
Bonus (2) |
|
$ |
— |
|
$ |
122,113 |
|
$ |
— |
|
$ |
— |
|
$ |
122,113 |
|
$ |
— |
|
|
Stock Options — Accelerated (3) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
178,984 |
|
$ |
178,984 |
|
|
Restricted Stock Units (4) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
541,760 |
|
$ |
541,760 |
|
|
Benefits and Perquisites |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Health Care Continuation (5) |
|
$ |
— |
|
$ |
18,607 |
|
$ |
— |
|
$ |
— |
|
$ |
18,607 |
|
$ |
— |
|
|
280G Tax Gross Up (6) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
(1) |
Annual 2013 base salary is $373,787. |
|
(2) |
The bonus component is based on the annual cash incentive for 2014 and 2013 and excludes special bonuses made during those years. The reported amount is calculated as the average annual cash incentive awarded over the previous two years. The average of the annual cash incentive paid in 2014 ($295,650) and 2013 ($136,000). |
|
(3) |
Pursuant to our 2010 Plan, unless the Compensation Committee determines otherwise, upon a change of control all awards vest as of the change of control date. This number assumes that all outstanding unvested options held as of December 31, 2014 would vest. The aggregate value reported is based on the spread between the closing stock market price of $8.00 on December 31, 2014 and the exercise prices of the option grants. |
|
(4) |
This number assumes that all outstanding unvested RSUs held as of December 31, 2014 would vest (see note 3 above). The reported value for the RSUs is equal to the grants with underlying shares times the closing market stock price of $8.00 on December 31, 2014. |
|
(5) |
Mr. Hodges is entitled to continue participation in our health and dental plan for the shorter of one year or until he obtains comparable coverage from another employer after termination. The reported amount assumes we will pay 100% of the employee premium and 50% of the dependent premium in effect at December 31, 2014 for 12 months. |
|
(6) |
See the narrative that follows these tables for a discussion of the tax gross-up benefit payable to Mr. Hodges. The reported number assumes a December 31, 2014 closing stock price of $8.00. Based on such closing stock price and the terms and conditions of Mr. Hodges’ employment agreement, the calculated 280G payment is zero. |
John G. Fort, M.D.
The following table describes the potential payments upon termination or a change of control of Pozen for John G. Fort, M.D., Chief Medical Officer.
|
Executive Benefits |
|
Termination |
|
Termination |
|
Death or |
|
Non- |
|
Change of |
|
Change of |
| ||||||
|
Compensation: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Salary (1) |
|
$ |
— |
|
$ |
393,872 |
|
$ |
— |
|
$ |
— |
|
$ |
393,872 |
|
$ |
— |
|
|
Bonus (2) |
|
$ |
— |
|
$ |
128,650 |
|
$ |
— |
|
$ |
— |
|
$ |
128,650 |
|
$ |
— |
|
|
Stock Options — Accelerated (3) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
171,638 |
|
$ |
171,638 |
|
|
Restricted Stock Units (4) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
541,760 |
|
$ |
541,760 |
|
|
Benefits and Perquisites |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Health Care Continuation (5) |
|
$ |
— |
|
$ |
17,223 |
|
$ |
— |
|
$ |
— |
|
$ |
17,223 |
|
$ |
— |
|
|
(1) |
Annual 2013 base salary is $393,872. |
|
(2) |
The bonus component is based on the annual cash incentive for 2014 and 2013 and excludes special bonuses made during those years. The reported amount is calculated as the average annual cash incentive awarded over the previous two years. The average of the annual cash incentive paid in 2014 ($114,000) and 2013 ($143,300). |
|
(3) |
Pursuant to our 2010 Plan, unless the Compensation Committee determines otherwise, upon a change of control all awards vest as of the change of control date. This number assumes that all outstanding unvested options held as of December 31, 2014 would vest. The aggregate value reported is based on the spread between the closing stock market price of $8.00 on December 31, 2014 and the exercise prices of the option grants. |
|
(4) |
This number assumes that all outstanding unvested RSUs held as of December 31, 2014 would vest (see note 3 above). The reported value for the RSUs is equal to the grants with underlying shares times the closing market stock price of $8.00 on December 31, 2014. |
|
(5) |
Dr. Fort is entitled to continue participation in our health and dental plan for the shorter of one year or until he obtains comparable coverage from another employer after termination. The reported amount assumes we will pay 100% of the employee premium and 50% of the dependent premium in effect at December 31, 2014 for 12 months. |
Gilda M. Thomas
The following table describes the potential payments upon termination or a change of control of Pozen for Gilda M. Thomas, Senior Vice President, General Counsel.
|
Executive Benefits |
|
Termination |
|
Termination |
|
Death or |
|
Non- |
|
Change of |
|
Change of |
| ||||||
|
Compensation: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Salary (1) |
|
$ |
— |
|
$ |
348,964 |
|
$ |
— |
|
$ |
— |
|
$ |
348,964 |
|
$ |
— |
|
|
Bonus (2) |
|
$ |
— |
|
$ |
113,963 |
|
$ |
— |
|
$ |
— |
|
$ |
113,963 |
|
$ |
— |
|
|
Stock Options — Accelerated (3) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
184,246 |
|
$ |
184,246 |
|
|
Restricted Stock Units (4) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
541,760 |
|
$ |
541,760 |
|
|
Benefits and Perquisites |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Health Care Continuation (5) |
|
$ |
— |
|
$ |
18,607 |
|
$ |
— |
|
$ |
— |
|
$ |
18,607 |
|
$ |
— |
|
|
280G Tax Gross Up(6) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
(1) |
Annual 2013 base salary is $348,964. |
|
(2) |
The bonus component is based on the annual cash incentive for 2014 and 2013 and excludes special bonuses made during those years. The reported amount is calculated as the average annual cash incentive awarded over the previous two years. The average of the annual cash incentive paid in 2014 ($101,025) and 2013 ($126,900). |
|
(3) |
Pursuant to our 2010 Plan, unless the Compensation Committee determines otherwise, upon a change of control all awards vest as of the change of control date. This number assumes that all outstanding unvested options held as of December 31, 2014 would vest. The aggregate value reported is based on the spread between the closing stock market price of $8.00 on December 31, 2014 and the exercise prices of the option grants. |
|
(4) |
This number assumes that all outstanding unvested RSUs held as of December 31, 2014 would vest (see note 3 above). The reported value for the RSUs is equal to the grants with underlying shares times the closing market stock price of $8.00 on December 31, 2014. |
|
(5) |
Ms. Thomas is entitled to continue participation in our health and dental plan for the shorter of one year or until he obtains comparable coverage from another employer after termination. The reported amount assumes we will pay 100% of the employee premium and 50% of the dependent premium in effect at December 31, 2014 for 12 months. |
|
(6) |
See the narrative that follows these tables for a discussion of the tax gross-up benefit payable to Ms. Thomas. The reported number assumes a December 31, 2014 closing stock price of $8.00. Based on such closing stock price and the terms and conditions of Ms. Thomas’ employment agreement, the calculated 280G payment is zero. |
Dennis L. McNamara
The following table describes the potential payments upon termination or a change of control of Pozen for Dennis L. McNamara, Senior Vice President and Chief Business Officer.
|
Executive Benefits |
|
Termination |
|
Termination |
|
Death or |
|
Non- |
|
Change of |
|
Change of |
| ||||||
|
Compensation: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Salary (1) |
|
$ |
— |
|
$ |
267,800 |
|
$ |
— |
|
$ |
— |
|
$ |
267,800 |
|
$ |
— |
|
|
Bonus (2) |
|
$ |
— |
|
$ |
50,290 |
|
$ |
— |
|
$ |
— |
|
$ |
50,290 |
|
$ |
— |
|
|
Stock Options — Accelerated (3) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
30,513 |
|
$ |
30,513 |
|
|
Restricted Stock Units (4) |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
388,352 |
|
$ |
388,352 |
|
|
Benefits and Perquisites |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Health Care Continuation (5) |
|
$ |
— |
|
$ |
11,685 |
|
$ |
— |
|
$ |
— |
|
$ |
11,685 |
|
$ |
— |
|
|
(1) |
Annual 2013 base salary is $267,800. |
|
(2) |
The bonus component is based on the annual cash incentive for 2014 and 2013 and excludes special bonuses made during those years. The reported amount is calculated as the average annual cash incentive awarded over the previous two years. The average of the annual cash incentive paid in 2014 ($50,290) and 2013 ($44,630). |
|
(3) |
Pursuant to our 2010 Plan, unless the Compensation Committee determines otherwise, upon a change of control all awards vest as of the change of control date. This number assumes that all outstanding unvested options held as of December 31, 2014 would vest. The aggregate value reported is based on the spread between the closing stock market price of $8.00 on December 31, 2014 and the exercise prices of the option grants. |
|
(4) |
This number assumes that all outstanding unvested RSUs held as of December 31, 2014 would vest (see note 3 above). The reported value for the RSUs is equal to the grants with underlying shares times the closing market stock price of $8.00 on December 31, 2014. |
|
(5) |
Mr. McNamara is entitled to continue participation in our health and dental plan for the shorter of one year or until he obtains comparable coverage from another employer after termination. The reported amount assumes we will pay 100% of the employee premium and 50% of the dependent premium in effect at December 31, 2014 for 12 months. |
Base Compensation and Bonuses
Former Chief Executive Officer
Pursuant to his employment agreement, upon a termination without cause or a voluntary termination by John R. Plachetka, our former CEO, for good reason (each as defined in his employment agreement), he is entitled to continue to receive annual salary for a period of two years following such termination. He is also entitled to receive a lump sum bonus equal to two times the average of the annual cash incentives paid to him in the two prior years. Upon termination due to death or disability, he is entitled to receive a lump sum bonus equal to a prorated amount of the average annual cash incentives paid to him in the two prior years. Upon a termination by our CEO for good reason in connection with a change of control, he is entitled to receive annual salary for a period of one year following such termination and a lump sum bonus equal to the average of the annual cash incentives paid to him in the two prior years.
As a result of Dr. Plachetka’s retirement described under section entitled “Employment and Other Agreements” on page [ ], he received or will be receiving the payments pursuant to the terms of the Separation Agreement described under the section entitled “Potential Payments on Termination and Change of Control” beginning on page [ ].
Other Named Executive Officers
Pursuant to their employment agreements, upon a termination without cause or a voluntary termination by the executive for good reason (each as defined in their employment agreements), whether or not in connection with a change of control, our other current named executive officers are entitled to a severance payment equal to one year’s base salary plus the average annual cash incentives paid to them over the preceding two years subject to the terms of Pozen’s recently adopted severance policy which provides for certain enhanced severance benefits.
Accelerated Vesting of Options and Other Stock-Based Awards
2010 Plan
Under the change of control provisions of our 2010 Plan, unless the Compensation Committee determines otherwise, all outstanding options and stock appreciation rights, including those held by our named executive officers, will automatically accelerate and become fully exercisable, the restrictions and conditions on all outstanding stock awards will immediately lapse, and all stock units, dividend equivalents and other stock-based awards will become fully vested and will be paid at their target value or in such greater amounts as the Compensation Committee may determine. The Compensation Committee may also take certain other actions as provided in the 2010 Plan, including determining that outstanding options and stock appreciation rights that are not exercised will be assumed by, or replaced with comparable options or rights by, the surviving corporation (or a parent or subsidiary of the surviving corporation), and other outstanding grants that remain in effect after the change of control will be converted to similar grants of the surviving corporation or a parent or subsidiary of the surviving corporation).
For purposes of the 2010 Plan, a change of control is generally defined to include any of the following:
|
· |
if a person, entity or affiliated group (with certain exceptions) acquires more than 50% of our then outstanding voting securities; | ||
|
|
|
|
|
|
· |
if we merge into another entity unless the holders of our voting shares immediately prior to the merger have at least 50% of the combined voting power of the securities in the merged entity or its parent; | ||
|
|
|
|
|
|
· |
if we sell or dispose of all or substantially all of our assets; | ||
|
|
|
|
|
|
· |
if we are liquidated or dissolved; or | ||
|
|
|
|
|
|
· |
if a majority of the Board have been members of the Board for less than one year, unless the election or nomination for election of each new Director who was not a director at the beginning of such one year period was approved by a vote of at least two-thirds of the directors then still in office who were directors at the beginning of such period. |
Former Chief Executive Officer
Stock Options. Under his employment agreement, in the event of a termination of employment without cause by Pozen, or voluntary termination by the former CEO for good reason, Dr. Plachetka will be entitled to accelerated vesting of any stock options which would otherwise vest during the 12 month period following the termination. For purposes of this analysis, based upon the assumed December 31, 2014 termination date, he would be entitled to vest upon such termination in any options which would otherwise vest prior to December 31, 2015. However, in the event of a change of control on December 31, 2013, in accordance with the terms of the 2010 Plan, unless the Compensation Committee determined otherwise, all of the CEO’s options would become fully vested.
All of these options must be exercised within 90 days, or in the case of options granted in 2009 and after one year, of the former CEO’s termination of employment according to the terms of the applicable stock option agreements.
Restricted Stock Units. In 2007 and thereafter, our former CEO was granted RSUs under our Second Amended and Restated Pozen Inc. 2000 Equity Compensation Plan, the predecessor plan to our 2010 Plan, payable in shares of common stock, to the extent vested, when the former CEO terminates his employment with or service to Pozen. Upon a change of control, in accordance with the terms of the grants, unless the Compensation Committee determines otherwise, all of the former CEO’s RSUs would become fully vested.
Other Named Executive Officers
In the event of a change of control on December 31, 2014, in accordance with the terms of our 2010 Plan, unless the Compensation Committee determined otherwise, all of these executive officers’ options would become fully vested. All of these options must be exercised within 90 days, or in the case of options granted in 2009 and later one year of the executive’s termination of employment according to the terms of the applicable stock option agreements.
General Release
Under the terms of our employment agreements with our named executive officers, payment of severance compensation and benefits upon termination of the executive’s employment without cause by Pozen, the executive’s voluntary termination for good reason or termination of the executive’s employment for good reason in connection with a change of control are subject to and conditioned upon the executive’s signing a general release of claims against Pozen.
Termination without Cause or Upon Non-Renewal of Term and Termination for Good Reason
Our named executive officers other than our former CEO will be entitled to certain benefits as described in the tables above if the executive officer’s employment is terminated by Pozen for reasons other than cause or by the executive officer for good reason.
Named Executive Officers Other than Former CEO
For our other named executive officers, a termination is for cause if the executive:
|
· |
commits an illegal or dishonest act that is materially detrimental to Pozen; | ||
|
|
|
|
|
|
· |
fails to carry out his assigned duties, which remains uncorrected 30 days after receiving written notice; | ||
|
|
|
|
|
|
· |
fails to comply with the policies or directives of the Board; | ||
|
|
|
|
|
|
· |
violates the terms and conditions of the employment agreement or his or her nondisclosure, inventions and non-solicitation agreement; or | ||
|
|
|
|
|
|
· |
violates company harassment or discrimination policies. |
These executives may terminate their employment for good reason if:
|
· |
Pozen breaches its obligations under the executive’s employment agreement; | ||
|
|
|
|
|
|
· |
the executive’s duties and responsibilities are substantially reduced or diminished; | ||
|
|
|
|
|
|
· |
the executive’s office is relocated to a location more than fifty miles from the current location; or | ||
|
|
|
|
|
|
· |
a change of control occurs and the executive gives notification of his or her intention to terminate his employment (see discussion below). |
Our other named executive officers will be entitled to certain benefits as described in the tables above if, within 60 days following the change of control event, the executive officer provides us with a notice of the officer’s intent to terminate his employment, and the effective date of such termination is not less than 60 days after the date of the notice. For purposes of these executives’ employment agreements, a change of control has the same meaning as under our 2000 Equity Compensation Plan, the predecessor plan to the 2010 Plan. Our other executive officers will receive no severance benefits based solely on termination by non-renewal of their employment agreements at the end of their respective terms.
280G Tax Gross-up
Other Named Executive Officers
Upon a change of control of Pozen, Mr. Hodges and Ms. Thomas may be subject to certain excise taxes pursuant to Section 280G of the Code. They are entitled to a full reimbursement by Pozen of any excise taxes that are imposed upon them as a result of the change of control, any income and excise taxes imposed on them as a result of Pozen’s reimbursement of the excise tax amount and any additional income and excise taxes that are imposed on them as a result of this reimbursement for excise or income taxes. For purposes of the 280G calculation reflected in the preceding table, it is assumed that no amounts will be discounted as attributable to reasonable compensation and no value will be attributed to Mr. Hodges or Ms. Thomas executing a noncompetition agreement. The payment of the 280G tax gross-up will be payable to them for any excise tax incurred regardless of whether their employment is terminated. The executive employment agreements of Dr. Fort and Mr. McNamara, which were executed after January 1, 2009, do not contain provisions entitling them to full reimbursement by Pozen for all excise taxes that are imposed on them under Section 280G of the Code.
Employment Agreement with Adrian Adams
Adrian Adams was appointed our Chief Executive Officer on May 31, 2015. Under the terms of Mr. Adams’ employment agreement effective May 31, 2015, he is entitled to (i) a base salary of $700,000, with annual increases, if any, to be made based on performance and in the sole discretion of the Board, (ii) an annual cash bonus, based on performance, payable in the discretion of the Compensation Committee, with a targeted amount of 100% of base salary, (iii) annual equity awards under our equity compensation plan with a target value of not less than 225% of his base salary, and (iv) a one-time sign-on equity award in the form of 1,944,888 RSUs. He will also receive a tax equalization payment for any taxes imposed by Section 4985 of the Code. In addition, Mr. Adams’ employment agreement provides for benefits if his employment is terminated under certain circumstances. In the event Pozen terminates Mr. Adams’ employment without cause, if he voluntarily terminates his employment for good reason, in the event of his death, or if he or Pozen terminate his employment due to disability, Mr. Adams will receive (i) accrued but unpaid base salary and vacation through the date of termination; (ii) a lump sum equal to 24 months of base salary; (iii) a lump sum equal to two times the greater of (x) the average annual bonus paid over the previous two years or (y) the annual bonus paid the year preceding the year in which his termination of employment occurs; (iv) continued medical and life insurance benefits at the same cost as Mr. Adams would be required to pay as an active employee for a period of 24 months following the termination date; and (v) acceleration of the vesting of all equity and equity-based awards that would otherwise vest in the next 24 month period. In the event that, within 12 months of a change in control of Pozen, Mr. Adams terminates his employment for good reason or Pozen terminates Mr. Adams’ employment without cause, Mr. Adams will receive (i) accrued but unpaid base salary and vacation through the date of termination; (ii) a lump sum equal to 36 months of base salary; (iii) a lump sum equal to three times the greater of (x) the average annual bonus paid over the previous two years or (y) the annual bonus paid the year preceding the year in which his termination of employment occurs; (iv) continued medical and life insurance benefits at the same cost as Mr. Adams would be required to pay as an active employee for a period of 36 months following the termination date, and (v) immediate and full vesting of all outstanding unvested equity awards. In the event of a change in control, Mr. Adams will not be entitled to indemnification with respect to excise taxes under Section 4999 of the Code. Instead, any payments to Mr. Adams that would be subject to the excise tax will be reduced to the level at which the excise tax will not be applied unless Mr. Adams would be in a better net after-tax position by receiving the full payments and paying the excise tax.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT OF POZEN COMMON STOCK
The following table sets forth information known to Pozen concerning the beneficial ownership of Pozen common stock as of June 30, 2015, for:
· each person known by Pozen to beneficially own 5% or more of the outstanding shares of Pozen common stock;
· each of Pozen’s directors;
· each of Pozen’s named executive officers; and
· all of Pozen’s directors and current executive officers as a group.
Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. In computing the number of shares of Pozen common stock beneficially owned by a person and the percentage ownership of that person, shares of Pozen common stock that could be issued upon the exercise of outstanding options and warrants held by that person that are currently exercisable or that will be exercisable within days of June 30, 2015 are considered outstanding. These shares, however, are not considered outstanding as of June 30, 2015 when computing the percentage ownership of each other person.
Except as indicated in the footnotes to this table and pursuant to state community property laws, each Pozen stockholder named in the table has sole voting and investment power for the shares shown as beneficially owned by them. Percentage of ownership is based on 32,708,898 shares of Pozen common stock outstanding on June 30, 2015.
|
Name and Address of Beneficial Owner |
|
Number of Shares |
|
Percentage |
|
|
John R. Plachetka, Pharm.D. |
|
3,796,475 |
(1) |
11.3 |
% |
|
PAR Investment Partners, L.P. |
|
3,863,699 |
(2) |
11.80 |
% |
|
BlackRock, Inc. |
|
2,601,301 |
(3) |
8.0 |
% |
(1) This amount reflects ownership by Silver Hill Investments, LLC, John R. Plachetka and Clare A. Plachetka and certain affiliated entities, and consists of (i) 1,157,808 shares owned by Silver Hill Investments, LLC, which is 50% owned by the Family Trust under the John R. Plachetka Irrevocable Trust (the “JRP Family Trust”), 40% owned by John R. Plachetka through his assignee, the Revocable Declaration of Trust, John R. Plachetka, Trustee (the “JRP Revocable Trust”), and 10% owned by his wife, Clare A. Plachetka, through her assignee, the Clare A. Plachetka Revocable Declaration of Trust, Clare A. Plachetka, Trustee (the “CAP Revocable Trust”); (ii) 1,554,102 shares owned by the JRP Revocable Trust; (iii) 221,910 shares owned by the CAP Revocable Trust; (iv) 22,631 shares owned by the JRP Family Trust; and (v) 37,580 shares held by John R. Plachetka (vi) 802,444 shares of common stock issuable pursuant to options granted to John R. Plachetka exercisable within 60 days. This number does not include 773,013 shares of common stock issuable pursuant to RSUs granted to John R. Plachetka. John R. Plachetka and Clare A. Plachetka claim shared voting and dispositive power as to the shares set forth in (i), (iii) and (iv) above.
On May 29, 2015, Dr. Plachetka also entered into a Voting Agreement with the Company (the “Voting Agreement”), pursuant to which, among other matters, he granted to the Company an irrevocable proxy with respect to all the shares directly and indirectly owned by him for a term of three years. With respect to a possible merger, sale or other transfer, directly or indirectly and whether in one or a series of transactions, of all or a significant portion of the assets or securities of the Company or any extraordinary corporate transaction, regardless of the form or structure of such transaction, in each case if and to the extent adopted or approved by the Board of Directors and recommended to the Company’s stockholders for adoption or approval, Dr. Plachetka and his affiliates agreed to (i) appear at such meeting or otherwise cause the Shares to be counted as present thereat for purposes of calculating a quorum; and (ii) vote (or cause to be voted) or deliver a written consent (or cause a written consent to be delivered) covering all of the Shares that such Stockholder shall be entitled to so vote in favor of adoption and approval of the Potential Transaction.
The Voting Agreement applies to all shares of the Company’s common stock directly and indirectly owned or beneficially held by Dr. Plachetka, as well as additional shares of the Company’s common stock acquired by him during its term. Dr. Plachetka is also subject to certain restrictions on the shares of the Company’s common stock directly and indirectly owned by him.
(2) Based on information disclosed on a report on Schedule 13G/A filed with the SEC on December 11, 2014 with respect to ownership as of December 1, 2014 by PAR Investment Partners, L.P., PAR Group, L.P. and PAR Capital Management, Inc., each of PAR Group, L.P. and PAR Capital Management, Inc. are general partners of PAR Investment Partners, L.P.
(3) Based on information disclosed on a report on Schedule 13G/A filed with the SEC on January 23, 2015 with respect to ownership as of December 31, 2014 by BlackRock, Inc. as parent company of BlackRock Institutional Trust Company, N.A., BlackRock Fund Advisors, BlackRock Asset Management Canada Limited, BlackRock Advisors, LLC and BlackRock Investment Management, LLC.
|
Name of Beneficial Owner(1) |
|
Number of Shares |
|
Percentage |
|
|
Adrian Adams |
|
— |
(2) |
* |
|
|
Jennifer Armstrong |
|
— |
(3) |
* |
|
|
John G. Fort, M.D. |
|
131,104 |
(4) |
* |
|
|
Neal F. Fowler |
|
71,095 |
(5) |
* |
|
|
Mark Glickman |
|
— |
(6) |
* |
|
|
William L. Hodges |
|
289,238 |
(7) |
* |
|
|
Arthur S. Kirsch |
|
107,470 |
(8) |
* |
|
|
Andrew I. Koven |
|
— |
(9) |
* |
|
|
Kenneth B. Lee, Jr. |
|
79,073 |
(10) |
* |
|
|
Dennis L. McNamara |
|
195,306 |
(11) |
* |
|
|
Seth A. Rudnick, M.D. |
|
47,501 |
(12) |
* |
|
|
Gilda M. Thomas |
|
157,624 |
(13) |
* |
|
|
Eric L. Trachtenberg |
|
— |
(14) |
* |
|
|
All current directors, director nominees and executive officers as a group (13 persons) |
|
1,078,411 |
(15) |
3.2 |
% |
*
Less than 1%
(1) Unless otherwise set forth herein, the street address of the named beneficial owners is c/o POZEN Inc., Suite 400, 1414 Raleigh Road, Chapel Hill, North Carolina 27517.
(2) Does not include 1,944,888 shares issuable pursuant to RSUs previously granted.
(3) Does not include 21,853 shares issuable pursuant to RSUs previously granted.
(4) Includes 82,756 shares of common stock issuable pursuant to options exercisable within 60 days.
(5) Includes 45,804 shares of common stock issuable pursuant to options exercisable within 60 days, but does not include 9,390 shares issuable pursuant to RSUs previously granted.
(6) Does not include 29,137 shares issuable pursuant to RSUs previously granted.
(7) Includes 159,885 shares of common stock issuable pursuant to options exercisable within 60 days.
(8) Includes 67,179 shares of common stock issuable pursuant to options exercisable within 60 days, but does not include 9,390 shares issuable pursuant to RSUs previously granted.
(9) Does not include 1,476,674 shares issuable pursuant to RSUs previously granted.
(10) Includes 6,107 shares of common stock issuable pursuant to options exercisable within 60 days, but does not include 9,390 shares issuable pursuant to RSUs previously granted.
(11) Includes 101,993 shares of common stock issuable pursuant to options exercisable within 60 days.
(12) Does not include 9,390 shares issuable pursuant to RSUs previously granted.
(13) Includes 94,953 shares of common stock issuable pursuant to options exercisable within 60 days.
(14) Does not include 25,000 shares issuable pursuant to RSUs previously granted.
(15) Includes 558,677 shares of common stock issuable pursuant to options exercisable within 60 days. This number does not include 1,944,888 shares of common stock issuable pursuant to RSUs held by Mr. Adams; 1,476,674 shares of common stock issuable pursuant to RSUs held by Mr. Koven; an aggregate of 75,990 shares of common stock issuable pursuant to RSUs held by Ms. Armstrong, Mr. Glickman and Mr. Trachtenberg, nor does it include an aggregate of 37,560 shares of common stock issuable pursuant to RSUs previously granted and held by other directors, each of which hold 9,390 shares individually.
This prospectus covers the public resale of the Shares owned by the Selling Shareholders named below. Such Selling Shareholders may from time to time offer and sell pursuant to this prospectus any or all of the Shares they own. The Selling Shareholders, however, make no representations that the shares will be offered for sale. The table below presents information regarding the Selling Shareholders and the Shares that each such Selling Shareholder may offer and sell from time to time under this prospectus.
The following table sets forth:
· the name of each Selling Shareholder;
· the number of Shares beneficially owned by each Selling Shareholder prior to the sale of the Shares covered by this prospectus;
· the number of Shares that may be offered by each Selling Shareholder pursuant to this prospectus;
· the number of Shares to be beneficially owned by each Selling Shareholder following the sale of any Shares covered by this prospectus; and
· the percentage of our issued and outstanding ordinary shares to be owned by each Selling Shareholder following the sale of any Shares covered by this prospectus (based on ordinary shares of Aralez issued and outstanding as of , 2015, immediately following the merger and contemplated equity and convertible note financings by Aralez).
All information with respect to ownership of our ordinary shares by the Selling Shareholders has been furnished by or on behalf of the Selling Shareholders, is as of August 3, 2015, and assumes effectiveness of the merger and consummation of the equity and debt financings.
Because the Selling Shareholders identified in the table may sell some or all of the Shares owned by them which are included in this prospectus, and because there are currently no agreements, arrangements or understandings with respect to the sale of any of the Shares, no estimate can be given as to the number of Shares available for resale hereby that will be held by the Selling Shareholders upon termination of this offering. In addition, the Selling Shareholders may have sold, transferred or otherwise disposed of, or may sell, transfer or otherwise dispose of, at any time and from time to time, the ordinary shares they hold in transactions exempt from the registration requirements of the Securities Act after the date on which they acquire the shares pursuant to the Facility Agreement. We have, therefore, assumed for the purposes of the following table, that the Selling Shareholders will sell all of the Shares which will be issued to each of them pursuant to the Facility Agreement and which are covered by this prospectus.
|
Selling Shareholder |
|
Ordinary Shares |
|
Number of |
|
Number of |
|
% of class |
|
|
Deerfield Private Design Fund III, L.P. |
|
4,625,263 |
(1)(4) |
3,930,818 |
|
694,445 |
|
— |
% |
|
Deerfield International Master Fund, L.P. |
|
2,590,148 |
(2)(4) |
2,201,258 |
|
388,890 |
|
— |
% |
|
Deerfield Partners, L.P. |
|
2,035,116 |
(3)(4) |
1,729,560 |
|
305,556 |
|
— |
% |
(1) Comprised of 3,930,818 ordinary shares issuable upon conversion of the notes and 694,445 ordinary shares. The provisions of the convertible notes beneficially owned restrict the exercise or conversion of such securities to the extent that, upon such exercise or conversion, the number of shares then beneficially owned by the holder and its affiliates and any other person or entities with which such holder would constitute a Section 13(d) “group” would exceed 9.985% of the total number of shares then outstanding (the “Ownership Cap”). Accordingly, notwithstanding the number of shares reported, Deerfield Private Design Fund, III, L.P., Deerfield International Master Fund, L.P. and Deerfield Partners, L.P. (collectively the “Deerfield Funds”) disclaim beneficial ownership of the shares underlying such convertible notes to the extent beneficial ownership of such shares would cause the Deerfield Funds, in the aggregate, to exceed the Ownership Cap.
(2) Comprised of 2,201,258 ordinary shares issuable upon conversion of the notes and 388,890 ordinary shares. The provisions of the convertible notes beneficially owned restrict the exercise or conversion of such securities to the extent that, upon such exercise or conversion, the number of shares then beneficially owned by the holder and its affiliates and any other person or entities with which such holder would constitute a Section 13(d) “group” would exceed 9.985% of Ownership Cap. Accordingly, notwithstanding the number of shares reported, the Deerfield Funds disclaim beneficial ownership of the shares underlying such convertible notes to the extent beneficial ownership of such shares would cause the Deerfield Funds, in the aggregate, to exceed the Ownership Cap.
(3) Comprised of 1,729,560 ordinary shares issuable upon conversion of the notes and 305,556 ordinary shares. The provisions of the convertible notes beneficially owned restrict the exercise or conversion of such securities to the extent that, upon such exercise or conversion, the number of shares then beneficially owned by the holder and its affiliates and any other person or entities with which such holder would constitute a Section 13(d) “group” would exceed 9.985% of the Ownership Cap. Accordingly, notwithstanding the number of shares reported, the Deerfield Funds disclaim beneficial ownership of the shares underlying such convertible notes to the extent beneficial ownership of such shares would cause the Deerfield Funds, in the aggregate, to exceed the Ownership Cap.
(4) James E. Flynn, with an address at 780 Third Avenue, 37 Floor, New York, New York 10017 has voting and disposition power over these securities.
The Selling Shareholders identified in this prospectus under “Selling Shareholders” may offer and sell from time to time the ordinary shares covered by this prospectus. We will not receive any of the proceeds from the offering of the ordinary shares by the Selling Shareholders. In connection with the Subscription Agreement, we agreed to register the ordinary shares covered by this prospectus.
We are registering the ordinary shares covered by this prospectus to permit holders to conduct public secondary trading of these shares from time to time after the date of this prospectus. We have agreed, among other things, to bear all reasonable expenses, other than underwriting discounts and commissions, incurred in connection with the registrations, filings or qualifications of the ordinary shares covered by this prospectus, including, without limitation, all registration, listing and qualification fees, printers and accounting fees, and the fees and disbursements of counsel for the Company.
The Selling Shareholders may sell all or a portion of the ordinary shares beneficially owned by them and offered hereby from time to time:
· directly; or
· through underwriters, broker-dealers or agents, who may receive compensation in the form of discounts, commissions or concessions from the Selling Shareholders and/or from the purchasers of the ordinary shares for whom they may act as agent.
The ordinary shares may be sold from time to time in one or more transactions at:
· fixed prices, which may be changed;
· prevailing market prices at the time of sale;
· varying prices determined at the time of sale; or
· negotiated prices.
These prices will be determined by the holders of the securities or by agreement between these holders and underwriters or dealers who may receive fees or commissions in connection with the sale. The aggregate proceeds to the Selling Shareholders from the sale of the ordinary shares offered by them hereby will be the purchase price of the ordinary shares less discounts and commissions, if any. The sales described above may be effected in transactions on any national securities exchange or quotation service on which the ordinary shares may be listed or quoted at the time of sale.
These transactions may include block transactions or crosses. Crosses are transactions in which the same broker acts as an agent on both sides of the trade.
In connection with sales of the ordinary shares under this prospectus, the Selling Shareholders may enter into hedging transactions with broker-dealers. These broker-dealers may in turn engage in short sales of the ordinary shares, short and deliver the ordinary shares to close out such short positions, or loan or pledge the ordinary shares to broker-dealers that may in turn sell such shares. The Selling Shareholders may pledge or grant a security interest in all or a portion of the ordinary shares that it owns and, if it defaults in the performance of its secured obligations, the pledgees or secured parties may offer and sell the ordinary shares from time to time pursuant to this prospectus. The Selling Shareholders may also transfer and donate ordinary shares in other circumstances, in which case the transferees, donees, pledgees or other successors in interest will be Selling Shareholders for the purposes of this prospectus.
The Selling Shareholders and any underwriters, broker-dealers or agents that participate in the sale of the ordinary shares may be deemed to be “underwriters” within the meaning of the Securities Act. Any commissions they receive and any profits on any resale of the ordinary shares may be deemed to be underwriting discounts and commissions under the Securities Act. Neither we nor any Selling Shareholder can presently estimate the amount of any such compensation. Selling Shareholders who are “underwriters” within the meaning of Section 2(11) of the Securities Act will be subject to the prospectus delivery requirements of the Securities Act.
It is expected that our outstanding ordinary shares will be listed and traded on NASDAQ under the symbol “ARLZ” and on the TSX under the symbol “ARZ”. We cannot guarantee that any trading market will develop for the ordinary shares. Even if a market does develop for the ordinary shares, the market may not be maintained.
The ordinary shares offered under this prospectus may be sold in some states only through registered or licensed brokers or dealers. In addition, in some states the securities may not be sold unless they have been registered or qualified for sale or an exemption from registration or qualification requirements is available and is complied with.
To our knowledge, there are currently no plans, arrangements or understandings between any Selling Shareholders and any underwriter, broker-dealer or agent regarding the sale of the ordinary shares by the Selling Shareholders. Selling Shareholders may choose not to sell any, or less than all, of the ordinary shares offered pursuant to this prospectus. In addition, we cannot assure you that a Selling Shareholder will not transfer, devise or gift the ordinary shares by means other than as described in this prospectus. In addition, any ordinary shares covered by this prospectus which qualify for sale pursuant to Rule 144 under Rule 144A of the Securities Act may be sold under Rule 144 or Rule 144A rather than pursuant to this prospectus. The Selling Shareholders may agree to indemnify any agent, dealer or broker-dealer that participates in transactions involving sales of the ordinary shares against certain liabilities, including liabilities arising under the Securities Act.
Upon notification to us by a Selling Shareholder that any material arrangement has been entered into with a broker-dealer for the sale of securities through a block trade, special offering, exchange distribution or secondary distribution or a purchase by a broker-dealer, a supplement to this prospectus will be filed, if required, pursuant to Rule 424(b) under the Securities Act, disclosing (i) the name of each such Selling Shareholder and of the participating brokers-dealer(s), (ii) the amount of securities involved, (iii) the price at which such securities were sold, (iv) the commissions paid or discounts or concessions allowed to such brokers-dealer(s), where applicable, (v) that such brokers-dealer(s) did not conduct any investigation to verify the information set out in this prospectus and (vi) other facts material to the transaction.
The ordinary shares will be issued and sold to the Selling Shareholders upon completion of the transactions contemplated by the merger agreement and the arrangement on or prior to January 31, 2016 or such later date as may be agreed to in writing by the parties, in a transaction exempt from the registration requirements of Section 5 of the Securities Act.
The Selling Shareholders and any other person participating in such distribution will be subject to the Exchange Act. The Exchange Act rules include, without limitation, Regulation M, which may limit the timing of purchases and sales of the ordinary shares by the Selling Shareholders and any such other person. In addition, Regulation M of the Exchange Act may restrict the ability of any person engaged in the distribution of the ordinary shares to engage in market-making activities with respect to the ordinary shares being distributed for a period of up to five business days prior to the commencement of distribution. This may affect the marketability of the ordinary shares and the ability of any person or entity to engage in market-making activities with respect to the ordinary shares.
We will pay all expenses of the shelf registration statement. Each Selling Shareholder will be required to bear the expenses of any broker’s commission, agency fee or underwriter’s discount or commission.
Other than the executive and director compensation arrangements, including the employment, termination of employment and change in control arrangements, discussed above under “Executive Compensation,” the indemnification arrangements with our executive officers and directors discussed above under “Executive Compensation,” and the registration rights described below under “Description of Capital Stock,” we have not entered into any transactions to which we have been or are a party, in which the amount involved in the transaction exceeded or will exceed $120,000, and in which any of our directors, executive officers or beneficial holders of more than 5% of our capital stock, or any immediate family member of, or person sharing the household with, any of these individuals, had or will have a direct or indirect material interest.
Review, Approval or Ratification of Transactions with Related Parties
As provided in the audit committee charter, the audit committee of our board of directors must review and approve in advance any related party transaction. We intend to adopt a related party transaction policy in the future.
It is our intention to ensure that all transactions between us and our officers, directors and principal stockholders and their affiliates, are approved by the audit committee of our board of directors, and are on terms no less favorable to us than those that we could obtain from unaffiliated third parties.
Immediately following the closing of the transactions, the authorized share capital of Aralez will consist of:
· 1,000,000,000 ordinary shares, $0.001 nominal value per share;
· 25,000 euro deferred shares, €1.00 nominal value per share; and
· 35,000,000 preferred shares, $0.001 nominal value per share.
Our board of directors is authorized, without shareholder approval, to issue additional shares of our capital stock once authorized to do so by the memorandum and articles of association or by an ordinary resolution adopted by the shareholders at a general meeting. The authorization may be granted for a maximum period of five years, at which point it must be renewed by the shareholders by an ordinary resolution.
Our memorandum and articles of association authorize our board of directors to issue ordinary or preferred shares without shareholder approval for a period of five years from the date of adoption of the articles of association.
The following description summarizes certain important terms of our share capital. Because it is only a summary, it does not contain all the information that may be important to you. For complete information, please see our memorandum and articles of association that will be in effect upon the completion of this offering, which are filed as exhibits to the registration statement of which this prospectus is a part, and to the applicable provisions of Irish law.
Dividend Rights
Our memorandum and articles of association authorize our directors to declare dividends without shareholder approval to the extent they appear justified by profits. Our board of directors may also recommend a dividend to be approved and declared by the shareholders at a general meeting and may direct that the payment be made by distribution of assets, shares or cash. No dividend issued may exceed the amount recommended by the directors. Dividends may be declared and paid in the form of cash or non-cash assets and may be paid in dollars or any other currency.
Voting Rights
Each shareholder is entitled to one vote for each Share that he or she holds as of the record date for a meeting of shareholders. Irish law requires approval of certain matters by “special resolution” of the shareholders at a general meeting. A special resolution requires the approval of not less than 75% of the votes of shareholders cast at a general meeting at which a quorum is present. Ordinary resolutions, by contrast, require a simple majority of the votes shareholders cast at a general meeting at which a quorum is present. Our memorandum and articles of association provide for a classified board of directors, to be divided into three classes with staggered three-year terms. Only one class of directors will be elected at each annual meeting of our shareholders, with the other classes continuing for the remainder of their respective three-year term. The term of the initial Class I directors shall terminate on the date of the 2016 annual general meeting; the term of the initial Class II directors shall terminate on the date of the 2017 annual general meeting; and the term of the initial Class III directors shall terminate on the date of the 2018 annual general meeting. At each annual general meeting of members beginning in 2016, successors to the class of directors whose term expires at that annual general meeting shall be elected for a three-year term.
No Preemptive or Similar Rights
Under Irish law, certain statutory pre-emption rights apply by default in favor of shareholders where shares are to be issued for cash. However, we have opted out of these pre-emption rights in our memorandum and articles of association as permitted under Irish law. Because Irish law requires this opt-out to be renewed every five years by special resolution, this opt-out must be so renewed in accordance with Irish statutory requirements if it is to remain effective. If the opt-out is not renewed, shares issued for cash must be offered to existing shareholders on a pro rata basis to their existing shareholding before the shares may be issued to any new shareholders.
Fully Paid and Non-assessable
All of shares of capital stock to be issued following the effectiveness of the Form S-4 will be fully paid and will not be subject to calls for any additional payments (non-assessable).
Registration Rights
In connection with the Facility Agreement, on June 8, 2015, the Lenders and Parent entered into the Registration Rights Agreement. Pursuant to the Registration Rights Agreement, Parent agreed to prepare and file with the SEC a Registration Statement on Form S-3, or other such form as required to effect a registration of the Registerable Securities, covering the resale of the Registerable Securities and such indeterminate number of additional ordinary shares as may become issuable upon conversion of or otherwise pursuant to the Convertible Notes to prevent dilution resulting from certain corporate actions. Such registration statement
must be filed within 45 calendar days following the date of issuance of the Convertible Notes. In the event the SEC does not permit all of the Registerable Securities to be included in the registration statement or if the Registerable Securities are not otherwise included in a registration statement filed under the Registration Rights Agreement, the Parent has agreed to file an additional registration statement by no later than the Additional Filing Deadline covering the resale of all Registerable Securities not already covered by an existing and effective registration statement for an offering to be made on a continuous basis pursuant to Rule 415 of the Securities Act. The Registration Rights Agreement also provides for piggy-back registration, subject to the terms and conditions of the Registration Rights Agreement.
The Subscription Agreement provides that Pozen shall prepare and cause to be filed with the SEC two registration statements on such form as may be required to effect a registration of the Parent Shares issued under the Subscription Agreement within 60 days of the date of the signing of the Subscription Agreement and for certain other registration rights for each of Purchaser and the Investors under the Securities Act and the rules and regulations thereunder, or any similar successor statute, and applicable state securities laws.
Provisions of our memorandum and articles of association may discourage, delay or prevent a merger or acquisition that our shareholders may consider favorable, including transactions in which shareholders might otherwise receive a premium for your shares, and may also frustrate or prevent any attempt by shareholders to change the direction or management of Aralez. For example, these provisions:
· authorize the issuance of “blank check” preferred stock without any need for action by shareholders;
· provide for a classified board of directors with staggered three-year terms;
· require supermajority stockholder approval to effect various amendments to our memorandum and articles of association;
· prohibit shareholder action by written consent; and
· establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted on by shareholders at shareholder meetings.
Any transaction in which a third party seeks to acquire 30% or more of the voting rights of Aralez and other acquisitions of Aralez securities will be governed by the Takeover Panel Act and the Irish Takeover Rules which will be regulated by the Panel. The “General Principles” of the Irish Takeover Rules and certain important aspects of the Irish Takeover Rules are described below.
General Principles
The Takeover Rules are built on the following General Principles which will apply to any transaction regulated by the Panel:
(a) in the event of an offer, all holders of security of the target company should be afforded equivalent treatment and, if a person acquires control of a company, the other holders of securities must be protected;
(b) the holders of the securities in the target company must have sufficient time and information to enable them to reach a properly informed decision on the offer; where it advises the holders of securities, the board of the target company must give its views on the effects of implementation of the offer on employment, conditions of employment and the locations of the target company’s places of business;
(c) the board of the target company must act in the interests of the company as a whole and must not deny the holders of securities the opportunity to decide on the merits of the offer;
(d) false markets must not be created in the securities of the target company, the bidder or of any other company concerned by the offer in such a way that the rise or fall of the prices of the securities becomes artificial and the normal functioning of the markets is distorted;
(e) a bidder must announce an offer only after ensuring that he or she can fulfill in full, any cash consideration, if such is offered, and after taking all reasonable measures to secure the implementation of any other type of consideration;
(f) a target company must not be hindered in the conduct of its affairs for longer than is reasonable by an offer for its securities; and
(g) a substantial acquisition of securities (whether such acquisition is to be effected by one transactions or a series of transaction) shall take place only at an acceptable speed and shall be subject to adequate and timely disclosure.
Mandatory Bid
Under certain circumstances, a person who acquires shares or other voting rights in Aralez may be required under the Irish Takeover Rules to make a mandatory cash offer for the remaining outstanding shares in Aralez at a price not less than the highest price paid for the shares by the acquirer (or any parties acting in concert with the acquirer) during the previous 12 months. This mandatory bid requirement is triggered if an acquisition of shares would increase the aggregate holding of an acquirer (including the holdings of any parties acting in concert with the acquirer) to shares representing 30% or more of the voting rights in Aralez, unless the Panel otherwise consents. An acquisition of shares by a person holding (together with its concert parties) shares representing between 30% and 50% of the voting rights in Aralez would also trigger the mandatory bid requirement if, after giving effect to the acquisition, the percentage of the voting rights held by that person (together with its concert parties) would increase by 0.05% within a 12-month period. Any person (excluding any parties acting in concert with the holder) holding shares representing more than 50% of the voting rights of a company is not subject to these mandatory offer requirements in purchasing additional securities.
Voluntary Bid; Requirements to Make a Cash Offer and Minimum Price Requirements
If a person makes a voluntary offer to acquire outstanding Aralez shares, the offer price must be no less than the highest price paid for Aralez shares by the bidder or its concert parties during the three-month period prior to the commencement of the offer period. The Panel has the power to extend the “look back” period to 12 months if the Panel, taking into account the General Principles, believes it is appropriate to do so.
If the bidder or any of its concert parties has acquired Aralez shares (i) during the period of 12 months prior to the commencement of the offer period which represent more than 10% of the total Aralez shares or (ii) at any time after the commencement of the offer period, the offer must be in cash (or accompanied by a full cash alternative) and the price per Aralez share must not be less than the highest price paid by the bidder or its concert parties during, in the case of (i), the 12-month period prior to the commencement of the offer period and, in the case of (ii), the offer period. The Panel may apply this rule to a bidder who, together with its concert parties, has acquired less than 10% of the total Aralez shares in the 12-month period prior to the commencement of the offer period if the Panel, taking into account the General Principles, considers it just and proper to do so. An offer period will generally commence from the date of the first announcement of the offer or proposed offer.
Substantial Acquisition Rules
Except in certain circumstances, an acquisition or series of acquisitions of shares or rights over shares representing 10% or more of the voting rights of Aralez is prohibited, if such acquisition(s), when aggregated with shares or rights already held, would result in the acquirer holding 15% or more but less than 30% of the voting rights of Aralez and such acquisitions are made within a period of seven days. These rules also require accelerated disclosure of acquisitions of shares or rights over shares relating to such holdings.
Frustrating Action
Under the Irish Takeover Rules, Parent’s board of directors is not permitted to take any action which might frustrate an offer for the shares of Aralez once Aralez’s board of directors has received an approach which may lead to an offer or has reason to believe an offer is imminent, subject to certain exceptions.
Potentially frustrating actions such as (i) the issue of shares, options or convertible securities, (ii) material acquisitions or disposals, (iii) entering into contracts other than in the ordinary course of business or (iv) any action, other than seeking alternative offers, which may result in frustration of an offer, are prohibited during the course of an offer or at any time during which the board has reason to believe an offer is imminent. Exceptions to this prohibition are available where:
· the action is approved by Aralez shareholders at a general meeting; or
· the Panel has given its consent, where:
· it is satisfied the action would not constitute frustrating action;
· Aralez shareholders that hold 50% of the voting rights state in writing that they approve the proposed action and would vote in favor of it at a general meeting;
· the action is taken in accordance with a contract entered into prior to the announcement of the offer; or
· the decision to take such action was made before the announcement of the offer and either has been at least partially implemented or is in the ordinary course of business.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Computershare. The transfer agent’s address is 1290 6th Avenue, 9th Floor, New York, New York 10104.
Listing
It is expected that our Shares will be listed and traded on NASDAQ under the symbol “ARLZ” and on the TSX under the symbol “ARZ”.
CERTAIN U.S. FEDERAL INCOME TAX CONSIDERATIONS
The following is a general discussion of the material U.S. federal income tax consequences of the ownership and disposition of the Shares that may be relevant to U.S. holders (as defined below) of such shares. Except where noted, this discussion deals only with Shares held as capital assets within the meaning of Section 1221 of the Code (generally, property held for investment). As used herein, the term “U.S. holder” means a beneficial owner of Shares that is for U.S. federal income tax purposes:
· a citizen or resident of the United States;
· a corporation or other entity taxable as a corporation for U.S. federal income tax purposes created or organized in or under the laws of the United States, any state thereof or the District of Columbia;
· an estate the income of which is subject to U.S. federal income taxation regardless of its source; or
· a trust if it (i) is subject to the primary supervision of a court within the United States and one or more “U.S. persons” (as defined in U.S. Treasury regulations) have the authority to control all substantial decisions of the trust or (ii) has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a U.S. person.
This discussion does not address all aspects of U.S. federal taxation that may be relevant to a particular holder in light of that holder’s particular circumstances or to holders subject to special treatment under the U.S. federal income tax laws, including without limitation:
· non-U.S. holders;
· dealers in securities;
· tax-exempt organizations;
· life insurance companies;
· holders who hold shares of Shares as part of a hedge, appreciated financial position, straddle, constructive sale, conversion transactions or other risk reduction transaction;
· holders who purchase or sell securities as part of a wash sale for tax purposes;
· holders who acquired Shares pursuant to the exercise of employee options or otherwise as compensation;
· traders in securities that elect to use a mark-to-market method of accounting for securities holdings;
· holders liable for alternative minimum tax;
· holders that actually or constructively own 10% or more of our voting stock; or
· holders whose functional currency is not the U.S. dollar.
The discussion below is based upon the provisions of the Code, its legislative history, existing and proposed regulations, published rulings and court decisions, all as currently in effect, as well as the current income tax treaty between Ireland and the U.S. (the “Tax Treaty”). These laws are subject to change, possibly on a retroactive basis. No ruling is intended to be sought from the IRS with respect to the transactions described herein, and
there can be no assurance that the IRS or a court will not take a contrary position regarding the tax consequences described herein.
This discussion does not address the tax treatment of partnerships (or entities or arrangements that are treated as partnerships for U.S. federal income tax purposes) or persons that Shares through partnerships or other pass-through entities for U.S. federal income tax purposes. If a partnership, including any entity or arrangement treated as a partnership for U.S. federal income tax purposes, holds Shares, the U.S. federal income tax treatment of a partner in such partnership will generally depend upon the status of the partner and the activities of the partnership. If you are a partner of a partnership holding Shares, you should consult your tax advisors regarding the particular tax consequences of the transactions to you.
Subject to the discussion below under “Passive Foreign Investment Company Rules,” this discussion assumes that we are a foreign corporation that is not, and will not become, a passive foreign investment company, or PFIC, as described below.
THIS DISCUSSION IS NOT A COMPLETE ANALYSIS OF ALL THE POTENTIAL U.S. FEDERAL INCOME TAX CONSEQUENCES RELATED TO THE TRANSACTIONS. IN ADDITION, THIS DISCUSSION DOES NOT ADDRESS ANY STATE, LOCAL OR FOREIGN CONSEQUENCES OF THE TRANSACTIONS OR ANY U.S. FEDERAL TAX CONSEQUENCES OF THE TRANSACTIONS OTHER THAN U.S. FEDERAL INCOME TAX CONSEQUENCES, SUCH AS ESTATE AND GIFT TAX CONSEQUENCES. YOU SHOULD CONSULT YOUR OWN TAX ADVISOR REGARDING THE U.S. FEDERAL, STATE AND LOCAL AND OTHER TAX CONSEQUENCES OF THE TRANSACTIONS TO YOU IN LIGHT OF YOUR OWN PARTICULAR CIRCUMSTANCES, AS WELL AS ANY CONSEQUENCES ARISING UNDER THE LAWS OF ANY OTHER TAXING JURISDICTION. IN PARTICULAR, YOU SHOULD CONFIRM YOUR STATUS AS A U.S. HOLDER ELIGIBLE FOR THE TAX TREATY WITH YOUR ADVISOR AND SHOULD DISCUSS ANY POSSIBLE CONSEQUENCES OF FAILING TO QUALIFY AS SUCH.
Taxation of Distributions
Subject to the PFIC rules discussed below, the gross amount of cash distributions on Shares (including any withheld Irish taxes) will be taxable as dividends to the extent paid out of our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Such income (including any withheld Irish taxes) will be includable in such holder’s gross income as ordinary income on the day actually or constructively received by the holder. For U.S. corporate holders, such dividends generally will not be eligible for the dividends-received deduction, except for a portion of certain dividends received by a corporate U.S. holder that owns 10% of all Shares (measured by both vote and value).
With respect to non-corporate U.S. holders, certain dividends received from a qualified foreign corporation may be subject to reduced rates of taxation (“qualified dividend income”). A qualified foreign corporation includes a foreign corporation that is eligible for the benefits of a comprehensive income tax treaty with the United States that the U.S.
Treasury Department determines to be satisfactory for these purposes and which includes an exchange of information provision. The U.S. Treasury Department has determined that the Tax Treaty meets these requirements. However, a foreign corporation is also treated as a qualified foreign corporation with respect to dividends paid by that corporation on shares that are readily tradable on an established securities market in the United States. U.S. Treasury Department guidance indicates that the Shares, which are expected to be listed on the NASDAQ and TSX, will be considered readily tradable on an established securities market in the United States, but there can be no assurance that the Shares will be considered readily tradable on an established securities market. Non-corporate holders that do not meet a minimum holding period requirement during which they are not protected from the risk of loss or that elect to treat the dividend income as “investment income” pursuant to Section 163(d)(4) of the Code (dealing with the deduction for investment interest expense) will not be eligible for the reduced rates of taxation applicable to qualified dividend income regardless of our status as a qualified foreign corporation. In addition, the rate reduction will not apply to dividends if the recipient of a dividend is obligated to make related payments with respect to positions in substantially similar or related property. This disallowance applies even if the minimum holding period has been met.
Distributions in excess of our current and accumulated earnings and profits, as determined for U.S. federal income tax purposes, will be treated as a non-taxable return of capital to the extent of the U.S. holder’s basis in the Shares, and thereafter as capital gain.
Subject to certain limitations, any Irish tax withheld on dividends paid with respect to Shares in accordance with the Tax Treaty and paid over to Ireland will be eligible for credit or deduction against the U.S. holder’s U.S. federal income tax liability, but special complex rules apply in determining the foreign tax credit limitation with respect to dividends that are subject to the preferential tax rates. U.S. holders are urged to consult their own tax advisors to determine eligibility. Subject to the discussion below regarding Section 904(h) of the Code, dividends generally will be foreign source income and will, depending on the U.S. holder’s circumstances, be either “passive” or “general” income for purposes of computing the foreign tax credit allowable to such holder.
Under Section 904(h) of the Code, dividends paid by a foreign corporation that is treated as 50% or more owned, by vote or value, by U.S. persons may be treated as U.S. source income (rather than foreign source income) for foreign tax credit purposes, to the extent the foreign corporation earns U.S. source income. In most circumstances, U.S. holders would be able to choose the benefits of Section 904(h)(10) of the Code and elect to treat dividends that would otherwise be U.S. source dividends as foreign source dividends, but in such a case, the foreign tax credit limitations would be separately determined with respect to such “resourced” income. In general, therefore, the application of Section 904(h) of the Code may adversely affect a U.S. holder’s ability to use foreign tax credits.
Since the Shares are expected to be listed on the NASDAQ and the TSX, we may be treated as 50% or more owned by U.S. persons for purposes of Section 904(h) of the Code. U.S.
holders are strongly urged to consult their own tax advisors regarding the possible impact if Section 904(h) of the Code should apply.
Distributions of Shares to a U.S. holder with respect to Shares that are made as part of a pro rata distribution to all of our shareholders generally will not be subject to U.S. federal income tax.
Sale or Other Taxable Disposition of Shares
Subject to the PFIC rules discussed below, a U.S. holder that sells or otherwise disposes of Shares will recognize capital gain or loss for U.S. federal income tax purposes equal to the difference between the amount realized and such holder’s tax basis in the Shares. Capital gain of a noncorporate U.S. holder is generally taxed at preferential rates where the property is held for more than one year. The gain or loss will generally be income or loss from sources within the United States for foreign tax credit limitation purposes.
If the consideration received for the Shares is paid in foreign currency, the amount realized will be the U.S. dollar value of the payment received translated at the spot rate of exchange on the date of disposition. A U.S. holder may realize additional gain or loss upon the subsequent sale or disposition of such currency, which will generally be treated as U.S. source ordinary income or loss. If the Shares are treated as traded on an established securities market and the relevant holder is either a cash basis taxpayer or an accrual basis taxpayer who has made a special election (which must be applied consistently from year to year and cannot be changed without the consent of the IRS), such holder will determine the U.S. dollar value of the amount realized in a foreign currency by translating the amount received at the spot rate of exchange on the settlement date of the disposition. If the Shares are not treated as traded on an established securities market, or the relevant U.S. holder is an accrual basis taxpayer that is not eligible to or does not elect to determine the amount realized using the spot rate on the settlement date, such U.S. holder will recognize foreign currency gain or loss to the extent of any difference between the U.S. dollar amount realized on the date of disposition (as determined above) and the U.S. dollar value of the currency received at the spot rate on the settlement date. Any such foreign currency gain or loss will generally be U.S. source ordinary income or loss.
Passive Foreign Investment Company Rules
In general, we will be a PFIC with respect to a U.S. holder if, for any taxable year in which such holder held Shares:
· at least 75% of our gross income for the taxable year is passive income; or
· at least 50% of the value, determined on the basis of a quarterly average, of our assets is attributable to assets that produce or are held for the production of passive income.
Passive income generally includes dividends, interest, royalties, rents (other than certain rents and royalties derived in the active conduct of a trade or business), annuities and gains from assets that produce passive income. Assets that produce or are held for the production of passive income may include cash, even if held as working capital or raised in a public offering, marketable securities and other assets that may produce passive income. The average value of a corporation’s assets for this purpose, in the case of a corporation whose shares are publicly traded for the taxable year, generally is the average of their fair market value at the end of each quarter. In determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account. If a foreign corporation owns at least 25% by value of the stock of another corporation, the foreign corporation is treated for purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation, and as receiving directly its proportionate share of the other corporation’s income.
Based on the nature of our business, the projected composition of our income and the projected composition and estimated fair market values of our assets, we do not expect to be a PFIC in the 2015 taxable year. However, there can be no assurances in this regard, or that the IRS will agree with our conclusion. In addition, there can be no assurances regarding our PFIC status in one or more subsequent years to the extent that our activities change, and our United States counsel expresses no opinion with respect to our PFIC status in the 2015 taxable year, and also expresses no opinion with respect to our predictions or past determinations regarding our PFIC status in the past or in the future.
If we are treated as a PFIC in any taxable year during which a U.S. holder owns Shares, such U.S. holder that does not make a mark-to-market election, as described below, will be subject to special rules with respect to:
· any gain realized on the sale or other disposition of such holder’s Shares; and
· any excess distribution made to such holder (generally, any distributions during a single taxable year that are greater than 125% of the average annual distributions received in respect of Shares during the three preceding taxable years or, if shorter, the holder’s holding period for the Shares).
Under these rules:
· the gain or excess distribution will be allocated ratably over such holder’s holding period for the Shares;
· the amount allocated to the taxable year in which such holder realized the gain or excess distribution will be taxed as ordinary income;
· the amount allocated to each prior year, with certain exceptions, will be taxed at the highest tax rate in effect for that year; and
· the interest charge generally applicable to underpayments of tax will be imposed in respect of the tax attributable to each such year.
Special rules apply for calculating the amount of the foreign tax credit with respect to excess distributions by a PFIC.
A U.S. holder who owns shares in a PFIC that are treated as marketable stock may make a mark-to-market election. If this election is made, the U.S. holder will not be subject to the PFIC rules described above. Instead, in general, the holder will include as ordinary income each year the excess, if any, of the fair market value of the shares at the end of the taxable year over such holder’s adjusted basis in the shares. These amounts of ordinary income will not be eligible for the favorable tax rates applicable to qualified dividend income or long-term capital gains. Such holder will also be allowed to take an ordinary loss in respect of the excess, if any, of the adjusted basis of the shares over their fair market value at the end of the taxable year (but only to the extent of the net amount of previously included income as a result of the mark-to-market election). A holder’s basis in the shares will be adjusted to reflect any such income or loss amounts.
A U.S. holder’s Shares will be treated as stock in a PFIC if we are a PFIC at any time during such holder’s holding period in the Shares, even if we are not currently a PFIC. For purposes of this rule, a U.S. holder that makes a mark-to-market election with respect to such holder’s Shares will be treated as having a new holding period in such Shares beginning on the first day of the first taxable year beginning after the last taxable year for which the mark-to-market election applies.
In addition, notwithstanding any election made with regard to Shares, dividends that a U.S. holder receives from us will not constitute qualified dividend income if we are a PFIC either in the taxable year of the distribution or the preceding taxable year. Dividends that a U.S. holder receives that do not constitute qualified dividend income are not eligible for taxation at the preferential rates applicable to qualified dividend income. Instead, such holder must include the gross amount of any such dividend paid by us out of our accumulated earnings and profits (as determined for United States federal income tax purposes) in such holder’s gross income, and it will be subject to tax at rates applicable to ordinary income.
A U.S. holder that owns Shares during any year that we are a PFIC with respect to such holder may be required to file IRS Form 8621.
Medicare Tax
A U.S. holder that is an individual or estate, or a trust that does not fall into a special class of trusts that is exempt from such tax, is subject to a 3.8% tax on the lesser of (1) the U.S. holder’s “net investment income” for the relevant taxable year and (2) the excess of the U.S. holder’s modified adjusted gross income for the taxable year over a certain threshold (which in the case of individuals is between $125,000 and $250,000, depending on the individual’s circumstances). A U.S. holder’s net investment income generally includes its dividend income and its net gains from the disposition of shares, unless such dividend income or net gains are derived in the ordinary course of the conduct of a trade or business (other than a trade or business that consists of certain passive or trading activities). U.S.
holders that are individuals, estates or trusts are urged to consult their tax advisors regarding the applicability of the Medicare tax to income and gains in respect of an investment in Shares.
Information with Respect to Foreign Financial Assets
Owners of “specified foreign financial assets” with an aggregate value in excess of $50,000 (and in some circumstances, a higher threshold) may be required to file an information report with respect to such assets with their tax returns. Specified foreign financial assets may include financial accounts maintained by foreign financial institutions, as well as the following, but only if they are not held in accounts maintained by financial institutions: (i) stocks and securities issued by non-United States persons, (ii) financial instruments and contracts held for investment that have non-United States issuers or counterparties, and (iii) interests in foreign entities. U.S. holders are urged to consult their tax advisors regarding the application of this reporting requirement to their ownership of the shares.
Backup Withholding and Information Reporting
For a noncorporate U.S. holder, information reporting requirements, on IRS Form 1099, generally will apply to:
· dividend payments or other taxable distributions made to such holder within the United States; and
· the payment of proceeds to such holder from the sale of Shares effected at a U.S. office of a broker.
Additionally, backup withholding may apply to such payments to a noncorporate U.S. holder that:
· fails to provide an accurate taxpayer identification number;
· is notified by the IRS that such holder has failed to report all interest and dividends required to be shown on the holder’s federal income tax returns; or
· in certain circumstances, fails to comply with applicable certification requirements.
Payment of the proceeds from the sale of Shares effected at a foreign office of a broker generally will not be subject to information reporting or backup withholding. However, a sale of Shares that is effected at a foreign office of a broker will be subject to information reporting and backup withholding if:
· the proceeds are transferred to an account maintained by in the United States;
· the payment of proceeds or the confirmation of the sale is mailed to a U.S. address; or
· the sale has some other specified connection with the United States as provided in U.S. Treasury regulations,
unless the broker does not have actual knowledge or reason to know that the holder is a U.S. person and the documentation requirements described above are met or the holder otherwise establish an exemption.
In addition, a sale of Shares effected at a foreign office of a broker will be subject to information reporting if the broker is:
· a U.S. person;
· a controlled foreign corporation for U.S. tax purposes;
· a foreign person 50% or more of whose gross income is effectively connected with the conduct of a U.S. trade or business for a specified three-year period; or
· a foreign partnership, if at any time during its tax year:
· one or more of its partners are U.S. persons who in the aggregate hold more than 50% of the income or capital interest in the partnership, or
· such foreign partnership is engaged in the conduct of a U.S. trade or business,
unless the broker does not have actual knowledge or reason to know that the holder is a U.S. person and the documentation requirements described above are met or the holder otherwise establish an exemption. Backup withholding will apply if the sale is subject to information reporting and the broker has actual knowledge that the holder is a United States person.
A holder generally may obtain a refund of any amounts withheld under the backup withholding rules that exceed such holder’s income tax liability by filing a refund claim with the IRS.
Scope of Discussion
The following is a summary of the material Irish tax considerations for certain beneficial owners of our ordinary shares. The summary does not purport to be a comprehensive description of all of the tax considerations that may be relevant to each of the shareholders. The summary is based upon Irish tax laws and the practice of the Irish Revenue Commissioners in effect on the date of this registration statement. Changes in law and/or administrative practice may result in alteration of the tax considerations described below.
The summary does not constitute tax advice and is intended only as a general guide. The summary is not exhaustive and shareholders should consult their own tax advisors about the Irish tax consequences (and tax consequences under the laws of other relevant jurisdictions) of the acquisition, ownership and disposal of their ordinary shares. The summary applies only to shareholders who will own ordinary shares as capital assets and does not apply to other categories of shareholders, such as dealers in securities, trustees, insurance companies, collective investment schemes, pension funds or shareholders who have, or who are deemed to have, acquired their ordinary shares by virtue of an Irish office or employment (performed or carried on in Ireland).
Irish Tax on Chargeable Gains
Shareholders who are neither resident nor ordinarily resident in Ireland for Irish tax purposes and do not hold their shares in connection with a trade carried on by such shareholders through an Irish branch or agency will not be within the charge to Irish tax on chargeable gains on the disposal of their ordinary shares.
Shareholders who are resident or ordinarily resident for tax purposes in Ireland, or who hold their shares in connection with a trade carried on by such holder in Ireland through a branch or agency may, subject to the availability of any exemptions and reliefs, be subject to Irish tax on chargeable gains on a disposal of their ordinary shares. Such shareholders should consult their own tax advisors as to the Irish tax consequences of such a disposal.
Stamp Duty
The rate of stamp duty (where applicable) on transfers of shares of Irish incorporated companies is 1% of the price paid or the market value of the shares acquired, whichever is greater. Where Irish stamp duty arises it is generally a liability of the transferee.
Irish stamp duty may, depending on the manner in which the ordinary shares are held, be payable in respect of transfers of ordinary shares.
Shares Held Through DTC or CDS
The statements on stamp duty in this and the following section on transfer of shares through, into and out of DTC and CDS are based on the position as confirmed in the past by the Irish Revenue Commissioners in respect of transfers of shares in Irish incorporated companies such as Aralez. Similar confirmations from the Irish Revenue Commissioners will be sought by Aralez.
A transfer of our ordinary shares effected by means of the transfer of book entry interests in DTC or CDS will not be subject to Irish stamp duty.
On the basis that most of our ordinary shares are expected to be held through DTC or CDS, most transfers of our ordinary shares should be exempt from Irish stamp duty.
Shares Held Outside of DTC or CDS Transferred Into or Out of DTC or CDS
A transfer of our ordinary shares where any party to the transfer holds such shares outside of DTC or CDS may be subject to Irish stamp duty. Shareholders wishing to transfer their shares into (or out of) DTC or CDS may do so without giving rise to Irish stamp duty provided:
· there is no change in the beneficial ownership of such shares as a result of the transfer; and
· the transfer into (or out of) DTC or CDS is not effected in contemplation of a subsequent sale of such shares by a beneficial owner to a third party.
Due to the potential Irish stamp duty charge on transfers of our ordinary shares, it is strongly recommended that shareholders hold their shares through DTC or CDS (or through a broker who in turn holds such shares through
DTC or CDS).
Withholding Tax on Dividends
To the extent that we make dividend payments (or other returns to shareholders that are treated as “distributions” for Irish tax purposes), it should be noted that such distributions will, in the absence of one of many exemptions, be subject to Irish dividend withholding tax (“DWT”), currently at a rate of 20%.
For DWT purposes, a distribution includes any distribution that may be made by us to our shareholders, including cash dividends, non-cash dividends and additional shares taken in lieu of a cash dividend.
Where an exemption does not apply in respect of a distribution made to a particular shareholder, we are responsible for withholding DWT prior to making such distribution.
General Exemptions
The following is a general overview of the scenarios where it is possible for us to make payments of dividends without deduction of DWT.
Irish domestic law provides that a non-Irish resident shareholder is not subject to DWT on dividends received from us if such shareholder is beneficially entitled to the dividend and is either:
· a person (not being a company) resident for tax purposes in a “relevant territory” (including the U.S. and Canada) and is neither resident nor ordinarily resident in Ireland (for a list of relevant territories for DWT purposes see Annex D to the Form S-4);
· a company resident for tax purposes in a relevant territory, provided such company is not under the control, whether directly or indirectly, of a person or persons who is or are resident in Ireland;
· a company, wherever resident, that is controlled, directly or indirectly, by persons resident in a relevant territory and who is or are (as the case may be) not controlled by, directly or indirectly, persons who are not resident in a relevant territory;
· a company, wherever resident, whose principal class of shares (or those of its 75% direct or indirect parent) is substantially and regularly traded on a stock exchange in Ireland, on a recognized stock exchange in a relevant territory or on such other stock exchange approved by the Irish Minister for Finance; or
· a company, wherever resident, that is wholly owned, directly or indirectly, by two or more companies where the principal class of shares of each of such companies is substantially and regularly traded on a stock exchange in Ireland, on a recognized stock exchange in a relevant territory or on such other stock exchange approved by the Irish Minister for Finance;
and provided, in all cases noted above, we or, in respect of shares held through DTC or CDS, any qualifying intermediary appointed by us, have received from the shareholder, where required, the relevant Irish Revenue Commissioners DWT forms, which are referred to in this registration statement as “DWT forms,” prior to the payment of the dividend. In practice, in order to ensure sufficient time to process the receipt of relevant DWT forms, the shareholder where required should furnish the relevant DWT forms to:
· its broker (and the relevant information should be further transmitted to any qualifying intermediary appointed by us) before the record date for the dividend (or such later date before the dividend payment date as may be notified to the shareholder by the broker) if its shares are held through DTC or CDS, or
· our transfer agent, Computershare Trust Company, N.A., at least seven business days before the record date for the dividend if its shares are held outside of DTC or CDS.
Links to the various DWT forms are available at: http://www.revenue.ie/en/tax/dwt/forms/index.html. Such forms are generally valid, subject to a change in circumstances, until December 31 of the fifth year after the year in which such forms were completed.
For non-Irish resident shareholders who cannot avail themselves of one of Ireland’s domestic law exemptions from DWT, it may be possible for such shareholders to rely on the provisions of a double tax treaty to which Ireland is party to reduce the rate of DWT.
Shares Held by U.S. Resident Shareholders
The statements on DWT in this section on shares held by U.S. resident shareholders are based on the position as confirmed in the past by the Irish Revenue Commissioners in respect of shares held through DTC. Similar confirmations will be sought by Aralez, including in respect of shares held through CDS.
It is expected that dividends paid in respect of our ordinary shares that are owned by U.S. residents and held through DTC or CDS may not be subject to DWT provided the addresses of the beneficial owners of such shares in the records of the broker holding such shares are in the U.S. It is strongly recommended that such shareholders ensure that their information is properly recorded by their brokers (so that such brokers can further transmit the relevant information to a qualifying intermediary appointed by us).
Dividends paid in respect of our ordinary shares that are held outside of DTC and are owned by residents of the U.S., will not be subject to DWT if such shareholders satisfy the conditions of one of the exemptions referred to above under the heading “Irish Tax Considerations—General Exemptions,” including the requirement to furnish the appropriate and valid DWT form and/or a completed IRS Form 6166 to our transfer agent, [Computershare Trust Company, N.A.], to confirm their U.S. residence at least seven business days before the record date for the dividend.
If any shareholder who is resident in the U.S. receives a dividend from which DWT has been withheld, the shareholder should generally be entitled to apply for a refund of such DWT from the Irish Revenue Commissioners provided the shareholder is beneficially entitled to the dividend.
Shares Held by Residents of “Relevant Territories” Other Than the U.S.
Shareholders who are residents of relevant territories, other than the U.S., such as Canada, must satisfy the conditions of one of the exemptions referred to above under the section entitled “Irish Tax Considerations—General Exemptions” beginning on page [·] of this registration statement, including the requirement to furnish valid DWT forms, in order to receive dividends without suffering DWT. If such shareholders hold their shares through DTC or CDS, they must provide the appropriate DWT forms to their brokers (so that such brokers can further transmit the relevant information to a qualifying intermediary appointed by us) before the record date for the dividend (or such later date before the dividend payment date as may be notified to the shareholder by the broker). If such shareholders hold their shares outside of DTC or CDS, they must provide the appropriate DWT forms to our transfer agent, [Computershare Trust Company, N.A.], at least seven business days before the record date for the dividend.
If any shareholder who is resident in a relevant territory receives a dividend from which DWT has been withheld, the shareholder may be entitled to a refund of such DWT from the Irish Revenue Commissioners provided the shareholder is beneficially entitled to the dividend.
Shares Held by Residents of Ireland
Most Irish resident or ordinarily resident shareholders (other than Irish resident companies that have completed the appropriate DWT forms) will be subject to DWT in respect of dividends paid on their ordinary shares.
Shareholders who are residents of Ireland, but are entitled to receive dividends without DWT, must complete the appropriate DWT forms and provide them to their brokers (so that such brokers can further transmit the relevant information to a qualifying intermediary appointed by us) before the record date for the dividend (or such later date before the dividend payment date as may be notified to the shareholder by the broker) (in the case of shares held through DTC or CDS), or to our transfer agent, Computershare Trust Company, N.A., at least seven business days before the record date for the dividend (in the case of shares held outside of DTC or CDS).
Shareholders who are resident or ordinarily resident in Ireland or are otherwise subject to Irish tax should consult their own tax advisors.
Shares Held by Other Persons
Shareholders who do not fall within any of the categories specifically referred to above may nonetheless fall within other exemptions from DWT. If any shareholders are exempt from DWT, but receive dividends subject to DWT, such shareholders may apply for a refund of such DWT from the Irish Revenue Commissioners.
Qualifying Intermediary
Prior to paying any dividend, we will put in place an agreement with an entity that is recognized by the Irish Revenue Commissioners as a “qualifying intermediary” and which will provide for certain arrangements relating to distributions in respect of our ordinary shares that are held through DTC or CDS (the “deposited securities”). The agreement will provide that the qualifying intermediary shall distribute or otherwise make available to the relevant nominee of the depository, any cash dividend or other cash distribution with respect to the deposited securities after we deliver or causes to be delivered to the qualifying intermediary the cash to be distributed.
The qualifying intermediary will be responsible for determining where shareholders reside and whether they have provided the required U.S. tax information and/or the required DWT forms, as applicable.
Income Tax on Dividends Paid on Shares
Irish income tax may arise for certain persons in respect of dividends received from Irish resident companies. A shareholder who is neither resident nor ordinarily resident in Ireland and who is entitled to an exemption from DWT generally has no liability to Irish income tax or the universal social charge on a dividend from us unless he or she holds his or her ordinary shares through a branch or agency in Ireland through which a trade is carried on.
A shareholder who is neither resident nor ordinarily resident in Ireland and who is not entitled to an exemption from DWT generally has no additional liability to Irish income tax or to the universal social charge unless he or she holds his or her ordinary shares through a branch or agency in Ireland through which a trade is carried on. The DWT deducted by us discharges the liability to Irish income tax.
A shareholder who is neither resident nor ordinarily resident in Ireland and is a resident of a relevant territory or otherwise exempt from Irish DWT but who receives dividends subject to DWT should be able to make a reclaim of the DWT from the Irish Revenue Commissioners unless he or she holds his or her ordinary shares through a branch or agency in Ireland through which a trade is carried on.
Irish resident or ordinarily resident shareholders may be subject to Irish tax and/or the universal social charge and/or Pay Related Social Insurance on dividends received from us. Such shareholders should consult their own tax advisors.
Capital Acquisitions Tax
Irish capital acquisitions tax (“CAT”) comprises principally gift tax and inheritance tax. CAT could apply to a gift or inheritance of our ordinary shares irrespective of the place of residence, ordinary residence or domicile of the parties. This is because our shares are regarded as property situated in Ireland as our share register must be held in Ireland. The person who receives the gift or inheritance has primary liability for CAT.
CAT is currently levied at a rate of 33% above certain tax-free thresholds. The appropriate tax-free threshold is dependent upon (i) the relationship between the donor and the donee and (ii) the aggregation of the values of previous taxable gifts and inheritances received by the donee from persons within the same group threshold. Gifts and inheritances passing between spouses are exempt from CAT. Children have a tax-free threshold of €225,000 in respect of taxable gifts or inheritances received from their parents. There is also a “small gift exemption” from CAT whereby the first €3,000 of the taxable value of all taxable gifts taken by a donee from any one donor, in each calendar year, is exempt from CAT and is also excluded from any future aggregation. This exemption does not apply to an inheritance. Shareholders should consult their own tax advisors as to whether CAT is creditable or deductible in computing any domestic tax liabilities.
THE IRISH TAX CONSIDERATIONS SUMMARIZED ABOVE ARE FOR GENERAL INFORMATION ONLY. EACH SHAREHOLDER SHOULD CONSULT HIS OR HER TAX ADVISOR AS TO THE PARTICULAR CONSEQUENCES THAT MAY APPLY TO SUCH SHAREHOLDER.
A&L Goodbody will provide an opinion regarding the validity of the Shares to be offered by this prospectus.
The financial statements of POZEN Inc. at December 31, 2014 and 2013, and for each of the three years in the period ended December 31, 2014, included in this Prospectus and Registration Statement, and the effectiveness of POZEN Inc.’s internal control over financial reporting as of December 31, 2014, have been audited by Ernst &Young LLP, independent registered public accounting firm, as set forth in their report thereon appearing elsewhere herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
The financial statements of Tribute appearing in Tribute’s Annual Report on Form 10-K for the year ended December 31, 2014, have been audited by McGovern, Hurley, Cunningham, LLP, an independent registered public accounting firm, as set forth in their reports thereon, incorporated by reference therein, and incorporated herein by reference. Such financial statements are incorporated herein by reference in reliance upon such reports given on the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
Pozen files annual, quarterly and current reports, proxy statements and other information with the SEC. Tribute files annual, quarterly and current reports and other information with the SEC. You may read and copy any document that Aralez, Pozen and Tribute file at the SEC’s public reference room at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the public reference room. The SEC also maintains an Internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC, including Pozen, Tribute and Aralez following the completion of the transactions. The SEC’s Internet site can be found at http://www.sec.gov.
POZEN INC
|
|
Page |
|
F-2 | |
|
|
|
|
Financial Statements as of December 31, 2014 and 2013 and for the Three Years in the Period then Ended |
|
|
F-4 | |
|
F-5 | |
|
F-6 | |
|
F-7 | |
|
F-8 | |
|
|
|
|
Financial Statements (Unaudited) for the Three Months Ended March 31, 2015 |
|
|
F-30 | |
|
F-31 | |
|
F-32 | |
|
F-33 |
TRIBUTE PHARMACEUTICALS, INC.
|
|
Page |
|
F-45 | |
|
|
|
|
Financial Statements (Audited) for the Two Year Period Ended December 31, 2014 |
|
|
F-46 | |
|
F-47 | |
|
F-48 | |
|
F-49 | |
|
F-50 | |
|
|
|
|
Condensed Financial Statements (Unaudited) for the Three Months Ended March 31, 2015 |
|
|
F-75 | |
|
Condensed Interim Statements of Operations, Comprehensive Loss and Deficit |
F-76 |
|
F-77 | |
|
F-78 | |
|
|
|
|
ARALEZ PHARMACEUTICALS PLC UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL INFORMATION | |
|
| |
|
F-90 | |
|
Statement of Operations for the Year Ended December 31, 2014 |
F-91 |
|
Statement of Operations for the Three Months Ended March 31, 2015 |
F-92 |
|
F-93 | |
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of POZEN Inc.
We have audited the accompanying balance sheets of POZEN Inc. as of December 31, 2014 and 2013, and the related statements of comprehensive income (loss), shareholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of POZEN Inc. at December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), POZEN Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 11, 2015 expressed an unqualified opinion thereon.
|
|
/s/ Ernst & Young LLP |
Raleigh, North Carolina
March 11, 2015
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of POZEN, Inc.
We have audited POZEN Inc. internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). POZEN Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, POZEN Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of POZEN Inc. as of December 31, 2014 and 2013 and the related statements of comprehensive income (loss), stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2014 and our report March 12, 2015 expressed an unqualified opinion thereon.
|
|
/s/ Ernst & Young LLP |
Raleigh, North Carolina
March 11, 2015
POZEN Inc.
|
|
|
December 31, |
| ||||
|
|
|
2014 |
|
2013 |
| ||
|
ASSETS |
|
|
|
|
| ||
|
Current assets: |
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
40,582,415 |
|
$ |
32,827,732 |
|
|
Investments in warrants |
|
2,678,773 |
|
— |
| ||
|
Accounts receivable |
|
5,629,209 |
|
1,673,000 |
| ||
|
Prepaid expenses and other current assets |
|
583,061 |
|
794,665 |
| ||
|
Total current assets |
|
49,473,458 |
|
35,295,397 |
| ||
|
Property and equipment, net of accumulated depreciation |
|
27,382 |
|
38,979 |
| ||
|
Noncurrent deferred tax asset |
|
952,900 |
|
— |
| ||
|
Total assets |
|
$ |
50,453,740 |
|
$ |
35,334,376 |
|
|
|
|
|
|
|
| ||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
| ||
|
Current liabilities: |
|
|
|
|
| ||
|
Accounts payable |
|
$ |
606,948 |
|
$ |
1,500,671 |
|
|
Accrued compensation |
|
1,899,456 |
|
3,132,468 |
| ||
|
Accrued expenses |
|
253,624 |
|
1,655,212 |
| ||
|
Deferred revenue |
|
— |
|
11,257,300 |
| ||
|
Current deferred tax liability |
|
952,900 |
|
— |
| ||
|
Total current liabilities |
|
3,712,928 |
|
17,545,651 |
| ||
|
|
|
|
|
|
| ||
|
Preferred stock, $0.001 par value; 10,000,000 shares authorized, issuable in series, of which 90,000 shares are designated Series A Junior Participating Preferred Stock, none outstanding |
|
— |
|
— |
| ||
|
Common stock, $0.001 par value, 90,000,000 shares authorized; 32,221,397 and 30,677,437 shares issued and outstanding at December 31, 2014 and December 31, 2013, respectively |
|
32,221 |
|
30,677 |
| ||
|
Additional paid-in capital |
|
143,613,024 |
|
134,337,213 |
| ||
|
Accumulated deficit |
|
(96,904,433 |
) |
(116,579,165 |
) | ||
|
Total stockholders’ equity |
|
46,740,812 |
|
17,788,725 |
| ||
|
|
|
|
|
|
| ||
|
Total liabilities and stockholders’ equity |
|
$ |
50,453,740 |
|
$ |
35,334,376 |
|
See accompanying Notes to Financial Statements.
POZEN Inc.
Statements of Comprehensive Income (Loss)
|
|
|
Year ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
2012 |
| |||
|
Royalty and licensing revenue: |
|
$ |
32,394,232 |
|
$ |
10,322,000 |
|
$ |
5,349,000 |
|
|
Operating expenses: |
|
|
|
|
|
|
| |||
|
Sales, general and administrative |
|
10,078,771 |
|
17,160,810 |
|
19,024,164 |
| |||
|
Research and development |
|
5,739,848 |
|
9,945,049 |
|
11,866,554 |
| |||
|
|
|
|
|
|
|
|
| |||
|
Total operating expenses |
|
15,818,619 |
|
27,105,859 |
|
30,890,718 |
| |||
|
Interest and other income |
|
3,099,119 |
|
75,560 |
|
258,697 |
| |||
|
Net income (loss) attributable to common stockholders |
|
19,674,732 |
|
(16,708,299 |
) |
(25,283,021 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Change in unrealized gains/(loss) on marketable Securities |
|
— |
|
3,253 |
|
14,388 |
| |||
|
Comprehensive income (loss) |
|
$ |
19,674,732 |
|
$ |
(16,705,046 |
) |
$ |
(25,268,633 |
) |
|
|
|
|
|
|
|
|
| |||
|
Basic net income (loss) per common share |
|
$ |
0.63 |
|
$ |
(0.55 |
) |
$ |
(0.84 |
) |
|
Shares used in computing basic net income (loss) per common share |
|
31,359,867 |
|
30,449,721 |
|
30,091,985 |
| |||
|
Diluted net income (loss) per common share |
|
$ |
0.60 |
|
$ |
(0.55 |
) |
$ |
(0.84 |
) |
|
Shares used in computing diluted net income (loss) per common share |
|
32,810,587 |
|
30,449,721 |
|
30,091,985 |
| |||
See accompanying Notes to Financial Statements.
POZEN Inc.
Statements of Stockholders’ Equity
|
|
|
|
|
|
|
Accumulated Other |
|
|
|
Total |
| |||||
|
|
|
Common |
|
Additional |
|
Comprehensive |
|
Accumulated |
|
Stockholders’ |
| |||||
|
|
|
Stock |
|
Paid-In Capital |
|
Income |
|
Deficit |
|
Equity |
| |||||
|
Balance at December 31, 2011 |
|
$ |
29,975 |
|
$ |
180,073,755 |
|
$ |
(17,641 |
) |
$ |
(74,587,845 |
) |
$ |
105,498,244 |
|
|
Exercise of common stock options |
|
253 |
|
1,306,106 |
|
— |
|
— |
|
1,306,359 |
| |||||
|
Payments related to net settlement of stock awards |
|
— |
|
(188,528 |
) |
— |
|
— |
|
(188,528 |
) | |||||
|
Issuance of common stock upon vesting of restricted stock |
|
94 |
|
(94 |
) |
— |
|
— |
|
— |
| |||||
|
Stock-based compensation |
|
— |
|
2,729,920 |
|
— |
|
— |
|
2,729,920 |
| |||||
|
Net loss |
|
— |
|
— |
|
— |
|
(25,283,021 |
) |
(25,283,021 |
) | |||||
|
Other comprehensive income |
|
— |
|
— |
|
14,388 |
|
— |
|
14,388 |
| |||||
|
Balance at December 31, 2012 |
|
30,322 |
|
183,921,159 |
|
(3,253 |
) |
(99,870,866 |
) |
84,077,362 |
| |||||
|
Exercise of common stock options |
|
151 |
|
661,823 |
|
— |
|
— |
|
661,974 |
| |||||
|
Payments related to net settlement of stock awards |
|
— |
|
(522,439 |
) |
— |
|
— |
|
(522,439 |
) | |||||
|
Issuance of common stock upon vesting of restricted stock |
|
204 |
|
(204 |
) |
— |
|
— |
|
— |
| |||||
|
Distribution to shareholders |
|
— |
|
(53,685,512 |
) |
— |
|
— |
|
(53,685,512 |
) | |||||
|
Stock-based compensation |
|
— |
|
3,962,386 |
|
— |
|
— |
|
3,962,386 |
| |||||
|
Net loss |
|
— |
|
— |
|
— |
|
(16,708,299 |
) |
(16,708,299 |
) | |||||
|
Other comprehensive income |
|
— |
|
— |
|
3,253 |
|
— |
|
3,253 |
| |||||
|
Balance at December 31, 2013 |
|
30,677 |
|
134,337,213 |
|
— |
|
(116,579,165 |
) |
17,788,725 |
| |||||
|
Exercise of common stock options |
|
1,484 |
|
7,587,445 |
|
— |
|
— |
|
7,588,929 |
| |||||
|
Payments related to net settlement of stock awards |
|
— |
|
(192,536 |
) |
— |
|
— |
|
(192,536 |
) | |||||
|
Issuance of common stock upon vesting of restricted stock |
|
60 |
|
(60 |
) |
— |
|
— |
|
— |
| |||||
|
Stock-based compensation |
|
— |
|
1,880,962 |
|
— |
|
— |
|
1,880,962 |
| |||||
|
Net income |
|
— |
|
— |
|
— |
|
19,674,732 |
|
19,674,732 |
| |||||
|
Balance at December 31, 2014 |
|
$ |
32,221 |
|
$ |
143,613,024 |
|
$ |
— |
|
$ |
(96,904,433 |
) |
$ |
46,740,812 |
|
See accompanying Notes to Financial Statements.
POZEN Inc.
|
|
|
Year ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
2012 |
| |||
|
Operating Activities |
|
|
|
|
|
|
| |||
|
Net income (loss) |
|
$ |
19,674,732 |
|
$ |
(16,708,299 |
) |
$ |
(25,283,021 |
) |
|
Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: |
|
|
|
|
|
|
| |||
|
Depreciation |
|
18,933 |
|
29,413 |
|
45,251 |
| |||
|
Loss on disposal of fixed assets |
|
— |
|
5,205 |
|
1,535 |
| |||
|
Bond amortization income |
|
— |
|
63,389 |
|
1,520,071 |
| |||
|
Gain on investments in warrants |
|
(2,678,773 |
) |
— |
|
— |
| |||
|
Noncash compensation expense |
|
1,880,962 |
|
3,962,386 |
|
2,729,920 |
| |||
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
| |||
|
Accounts receivable |
|
(3,956,209 |
) |
(321,000 |
) |
(222,000 |
) | |||
|
Prepaid expenses and other current assets |
|
211,604 |
|
63,758 |
|
(158,097 |
) | |||
|
Accounts payable and other accrued expenses |
|
(3,528,323 |
) |
1,026,201 |
|
(4,782,946 |
) | |||
|
Deferred revenue |
|
(11,257,300 |
) |
11,000,000 |
|
— |
| |||
|
Net cash provided by (used in) operating activities |
|
365,626 |
|
(878,947 |
) |
(26,149,287 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Investing activities |
|
|
|
|
|
|
| |||
|
Purchase of equipment |
|
(7,336 |
) |
(1,652 |
) |
(15,821 |
) | |||
|
Purchase of investments |
|
— |
|
— |
|
(35,922,138 |
) | |||
|
Sale and maturities of investments |
|
— |
|
18,838,000 |
|
24,395,000 |
| |||
|
Net cash (used in) provided by investing activities |
|
(7,336 |
) |
18,836,348 |
|
(11,542,959 |
) | |||
|
Financing activities |
|
|
|
|
|
|
| |||
|
Proceeds from issuance of common stock |
|
7,588,929 |
|
661,974 |
|
1,306,359 |
| |||
|
Distribution to shareholders |
|
— |
|
(53,685,512 |
) |
— |
| |||
|
Payments related to net settlement of stock-based awards |
|
(192,536 |
) |
(522,439 |
) |
(188,528 |
) | |||
|
Net cash provided by (used in) financing activities |
|
7,396,393 |
|
(53,545,977 |
) |
1,117,831 |
| |||
|
Net increase (decrease) in cash and cash equivalents |
|
7,754,683 |
|
(35,588,576 |
) |
(36,574,415 |
) | |||
|
Cash and cash equivalents at beginning of year |
|
32,827,732 |
|
68,416,308 |
|
104,990,723 |
| |||
|
Cash and cash equivalents at end of year |
|
$ |
40,582,415 |
|
$ |
32,827,732 |
|
$ |
68,416,308 |
|
See accompanying Notes to Financial Statements.
POZEN Inc.
1. Significant Accounting Policies
General
POZEN Inc. (“we” or “POZEN” or the “Company”) was incorporated in the State of Delaware on September 25, 1996 and is operating in a single reportable segment. The Company has been a pharmaceutical company committed to transforming medicine that transforms lives. Since inception, the Company has focused its efforts on developing products which can provide improved efficacy, safety or patient convenience in the treatment of acute and chronic pain and pain related conditions and has developed a portfolio of integrated aspirin therapies. Historically, the Company has entered into collaboration agreements to commercialize its product candidates. The Company’s licensing revenues include upfront payments, additional payments if and when certain milestones in the product’s development or commercialization are reached, and the eventual royalty payments based on product sales.
We decided to retain ownership of our PA product candidates which contain a combination of a proton pump inhibitor and enteric coated aspirin in a single tablet through the clinical development and pre-commercialization stage and our chief commercial officer was responsible for developing the commercialization strategy for these products and conducting all the required pre-commercialization activities. On September 3, 2013, we entered into an exclusive license agreement with sanofi-aventis U.S. LLC, or Sanofi US, for the commercialization of POZEN’s proprietary, investigational, coordinated-delivery tablets combining immediate-release omeprazole, a proton pump inhibitor, or PPI, and enteric-coated, or EC, aspirin in a single tablet, now known as YOSPRALA 81/40 and 325/40 (“PA” or “YOSPRALA”), including PA8140 and PA32540. On November 29, 2014, we executed a termination agreement with Sanofi US terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi US were terminated and all rights to the products licensed to Sanofi US under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi US relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
With respect to future products we may develop, we have decided that we will no longer commit substantial resources to further drug development without a partner who agrees to pay the full cost of that development. Consistent with this model, we have reduced our R&D staff and other costs and expenses as our PA development program activities wind down and intend to continue to reduce staff that is no longer required to support our then current business activities. Our board of directors and management team continue to explore potential ways to return value to our stockholders. On November 21, 2013, our Board of Directors declared a special cash distribution of $1.75 per share to all stockholders of record as of the close of business on December 11, 2013, with a payment date of December 30, 2013. This distribution represented a surplus of corporate cash and is accounted for as a return of capital to stockholders. We are committed to return as much cash to our stockholders as is prudent and may consider other cash distributions in the future.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts in the financial statements and accompanying notes. Actual results could differ from the estimates and assumptions used.
Revenue Recognition
Revenue for the fiscal years ended December 31, 2014, 2013 and 2012 consisted of the following royalty revenue and other licensing revenue:
|
|
|
For the year ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
2012 |
| |||
|
Royalty Revenue |
|
$ |
21,136,932 |
|
$ |
6,322,000 |
|
$ |
4,849,000 |
|
|
Other licensing revenue |
|
11,257,300 |
|
4,000,000 |
|
500,000 |
| |||
|
Total licensing revenue |
|
$ |
32,394,232 |
|
$ |
10,322,000 |
|
$ |
5,349,000 |
|
With regard to royalty revenues, the Company’s licensing agreements have terms that include royalty payments based on the manufacture, sale or use of the Company’s products or technology. VIMOVO® (naproxen and esomeprazole magnesium) delayed release tablets royalty revenue has been recognized when earned, as will any other future royalty revenues. For VIMOVO or those future arrangements where royalties are reasonably estimable, the Company recognizes revenue based on estimates of royalties earned during the applicable period and reflects in future revenue any differences between the estimated and actual royalties. These estimates are based upon information reported to the Company by its collaboration partners. During the fiscal years ended December 31, 2014, December 31, 2013, and December 31, 2012 the Company recognized $21.1 million, $6.3 million, and $4.8 million, respectively, for VIMOVO royalty revenue.
Also, with regard to the licensing revenues, the Company’s licensing agreements have had terms that include upfront payments upon contract signing and additional payments if and when certain milestones in the product’s development or commercialization are reached. Historically, the non-refundable portion of upfront payments received under the Company’s existing agreements is deferred by the Company upon receipt and recognized on a straight-line basis over periods ending on the anticipated date of regulatory approvals, as specified in the agreements relating to the product candidates, or the conclusion of any obligation on the part of the Company. If regulatory approvals or other events relating to our product candidates are accelerated, delayed or not ultimately obtained, then the amortization of revenues for these products is prospectively accelerated or reduced accordingly. Milestone payments along with the refundable portions of up-front payments are recognized as licensing revenue upon the achievement of specified milestones if (i) the milestone is substantive in nature and the achievement of the milestone was not reasonably assured at the inception of the agreement; and (ii) the fees are non-refundable. Any milestone payments received prior to satisfying these revenue recognition criteria are recorded as deferred revenue.
In September 2013, the Company announced the signing of an exclusive license agreement its PA products, including, PA8140 and PA32540, in the United States. to commercialize all PA combinations that contain 325 mg or less of enteric-coated aspirin in the United States. On November 29, 2014, we executed a termination agreement with Sanofi US terminating the license. As of the termination date, all licenses granted to Sanofi US were terminated and all rights to the products licensed to Sanofi US under the agreement reverted to us. The Company received an upfront payment of $15.0 million which is included within the license revenue in the accompanying statements of comprehensive income (loss). The revenue for the fiscal years ended December 31, 2014 and December 31, 2013 was $11.0 million and $4.0 million, respectively.
On March 21, 2011, the Company entered into a license agreement with Cilag GmbH International (“Cilag”) a division of Johnson & Johnson, for the exclusive development and commercialization of MT 400 in Brazil, Colombia, Ecuador and Peru. Cilag’s upfront payment of $257,300 was deferred until the licensing agreement’s termination and is included in other licensing revenue for the fiscal year ended December 31, 2014.
Cash, Cash Equivalents, Investments and Concentration of Credit Risk
Cash is invested in open-ended money market mutual funds, interest-bearing investment-grade debt securities and insured bank deposits. Cash is restricted to the extent of a $42,000 letter of credit in compliance with the terms of the Company’s office lease. The Company considers all highly liquid investments with maturities of 90 days or less when purchased to be cash equivalents.
The Company invests in high-credit quality investments in accordance with its investment policy, which attempts to minimize the possibility of loss. However, cash and cash equivalents include financial instruments that potentially subject the Company to a concentration of credit risk. Cash and cash equivalents are of a highly liquid nature and are held with high credit quality financial institutions and money market mutual fund managers. Cash held directly with financial institutions is insured up to $250,000 per account and any excess amounts are uninsured.
Cash is also held in insured bank deposits through a cash management program that offers a bank network ensuring full FDIC insurance on all deposits. Approximately 55% of the Company’s cash and cash equivalents are held in fully insured bank deposits and approximately 45% by money market mutual fund managers.
In connection with its acquisition of all rights, title and interest to develop, commercialize and sell Treximet® (sumatriptan / naproxen sodium) from GlaxoSmithKline (“GSK”), Pernix Therapeutics Holdings, Inc. (“Pernix”) issued the Company a warrant to purchase 500,000 shares of Pernix common stock at an exercise price of $4.28 (the closing price of Pernix common stock as listed on the NASDAQ Global Market on May 13, 2014). The common stock underlying the warrants will be registered by Pernix with the Securities and Exchange Commission and the warrant is exercisable from August 20, 2014, the closing date of the acquisition, until February 28, 2018. We are valuing the warrant using the Black-Sholes option valuation model.
The warrant also provides for cashless exercise whereby the holder may elect to receive the number of shares of Pernix common stock equal to the number of shares being exercised multiplied by the fair market value of one share of Pernix common stock, less $4.28 (the exercise price) divided by the fair market value of one share of Pernix common stock. Assuming a cashless exercise at December 31, 2014, this would have resulted in 272,098 shares of Pernix common stock valued at $2.6 million. Because the warrant has not been registered by Pernix with the Securities and Exchange Commission, the Company cannot sell or transfer the warrant in reliance upon Rule 144 until after November 13, 2014 when the Company meets certain holding requirements. In November 2014 Pernix submitted a filing to register the underlying shares with the Securities and Exchange Commission but as of December 31, 2014 this had not been completed and, therefore, upon exercise of the warrant the Company is restricted from transferring or selling these shares until such time as such filing is declared effective or an exemption from registration is otherwise met.
The following table sets forth our financial instruments carried at fair value as of December 31, 2014 and December 31, 2013:
|
|
|
Financial Instruments |
| ||||
|
|
|
Carried at Fair Value |
| ||||
|
|
|
December 31, |
|
December |
| ||
|
Assets: |
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
40,582,415 |
|
$ |
32,827,732 |
|
|
Investments in Pernix warrants |
|
2,678,773 |
|
— |
| ||
|
Total cash and investments |
|
$ |
43,261,188 |
|
$ |
32,827,732 |
|
Fair Value Measurements
Financial instruments consist of cash and cash equivalents, short-term investments, accounts receivable and accounts payable. The carrying values of these amounts, other than the investment in warrants, approximate the fair value due to their short-term nature.
A part of its acquisition of Treximet® (sumatriptan / naproxen sodium) from GlaxoSmithKline (GSK), Pernix Therapeutics Holdings, Inc. (Pernix) granted POZEN a warrant to purchase 500,000 shares of Pernix common stock at an exercise price of $4.28. The common stock underlying the warrants was registered by Pernix with the Securities and Exchange Commission and is exercisable from August 20, 2014, the closing date of the acquisition, until February 28, 2018. The warrant is valued at $2,678,773 using Black-Sholes valuation model discounted for the warrant’s lack of marketability and liquidity.
Short-term investments gains consisted of the investment in warrants valuation of $2,740,800 on August 20, 2014, with a mark to market adjustment of ($62,027) at December 31, 2014 and a net December 31, 2014 short-term gain of $2,678,773.
The Company defines fair value (“FV”) as the price that would be received to sell an asset or paid to transfer a liability (“the exit price”) in an orderly transaction between market participants at the measurement date.
The FV hierarchy for inputs maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. The Company uses the following hierarchy of inputs to measure FV:
· Level 1 - quoted prices in active markets for identical assets and liabilities.
· Level 2 - observable inputs other than quoted prices in active markets for identical assets and liabilities, including quoted prices in active markets for instruments that are similar or quoted prices in markets that are not active for identical or similar instruments and model-derived valuations in which all significant inputs and value drivers are observable in active markets.
· Level 3 - unobservable inputs that are significant to the overall valuation, for which there is little or no market data available and which require the Company to develop its own assumptions.
The Company values investments using the most observable inputs available that are current as of the measurement date and classifies them according to the lowest level of inputs used. Observable inputs are inputs that market participants would use in pricing the asset or liability developed from market data obtained from independent sources. Unobservable inputs are inputs that reflect the Company’s judgment concerning the assumptions that market participants would use in pricing the asset or liability developed from the best information available under the circumstances.
The financial assets for which we perform recurring measurements are cash equivalents and investments in warrants. As of December 31, 2014, financial assets utilizing Level 1 inputs included cash equivalents. Financial assets utilizing Level 2 inputs included investments in warrants.
Fair value is a market-based measure considered from the perspective of a market participant who holds the asset or owes the liability rather than an entity-specific measure. Therefore, even when market assumptions are not readily available, our own assumptions are set to reflect those that market participants would use in pricing the asset or liability at the measurement date.
Our Level 1 valuations are based on the market approach and consist primarily of quoted prices for identical items on active securities exchanges. Our Level 2 valuations may also use the market approach and are based on significant other observable inputs such as quoted prices for financial instruments not traded on a daily basis. We did not rely on Level 3 input for valuation of our securities at December 31, 2014.
The following table sets forth our financial instruments carried at fair value within the fair value hierarchy and using the lowest level of input as of December 31, 2014:
|
|
|
Financial Instruments |
| ||||||||||
|
|
|
Carried at Fair Value |
| ||||||||||
|
|
|
Quoted prices |
|
Significant |
|
Significant |
|
|
| ||||
|
|
|
(Level 1) |
|
(Level 2) |
|
(Level 3) |
|
Total |
| ||||
|
Assets: |
|
|
|
|
|
|
|
|
| ||||
|
Cash and cash equivalents |
|
$ |
40,582,415 |
|
$ |
— |
|
$ |
— |
|
$ |
40,582,415 |
|
|
Investment in warrants |
|
— |
|
2,678,773 |
|
— |
|
2,678,773 |
| ||||
|
Total cash and investments |
|
$ |
40,582,415 |
|
$ |
2,678,773 |
|
$ |
— |
|
$ |
43,261,188 |
|
The following table sets forth our financial instruments carried at FV within the ASC 820 hierarchy and using the lowest level of input as of December 31, 2013:
|
|
|
Financial Instruments |
| ||||||||||
|
|
|
Carried at Fair Value |
| ||||||||||
|
|
|
Quoted prices |
|
Significant |
|
Significant |
|
Total |
| ||||
|
Cash and cash equivalents |
|
$ |
32,827,732 |
|
$ |
— |
|
$ |
— |
|
$ |
32,827,732 |
|
|
Short-term investments |
|
— |
|
— |
|
— |
|
— |
| ||||
|
Total |
|
$ |
32,827,732 |
|
$ |
— |
|
$ |
— |
|
$ |
32,827,732 |
|
The Company targets investment principally in Level 1 and Level 2 cash equivalents and financial instruments and records them at FV. The Company expects that the carrying values of cash equivalents will approximate FV because of their short maturities.
Equipment
Equipment consists primarily of computer hardware and software and furniture and fixtures and is recorded at cost. Depreciation is computed using an accelerated method over the estimated useful lives of the assets ranging from five to seven years. Accumulated depreciation at December 31, 2014 and 2013 totaled $0.7 million.
Research and Development Costs, Including Clinical Trial Expenses
Research and development costs are charged to operations as incurred. The Company has included in research and development expenses the personnel costs associated with research and development activities and costs associated with pharmaceutical development, clinical trials, toxicology activities, and regulatory matters.
Interest and Other Income
Interest and bond amortization income was $43,100 and $138,900 for the fiscal years ended December 31, 2014 and 2013, respectively. Other income also included short-term investments gains consisting of the investment in warrants with an initial valuation of $2,740, 800 on August 20, 2014, with a mark to market adjustment of ($62,027) at December 31, 2014 and a net of $377,269 related to the disgorgement of short-swing profits arising from trades by a POZEN stockholder under Section 16(b) of the Securities and Exchange Act of 1934.
Taxes
Income taxes are computed using the asset and liability approach, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the Company’s financial statements or tax returns. In estimating future tax consequences, the Company generally considers all expected future events other than enactment of changes in tax law or rates. If it is more likely than not that some or all of a deferred tax asset will not be realized, the Company records a valuation allowance.
Net Income (Loss) Per Share
Basic and diluted net income or loss per common share amounts have been computed using the weighted-average number of shares of common stock outstanding for the fiscal year ended December 31, 2014 and 2013. During the fiscal years ended December 31, 2014 and 2013, the Company had potential common stock equivalents related to its outstanding stock options. These potential common stock equivalents were not included, if the effect would have been antidilutive. The Company has excluded the impact of any shares which might be issued under the Rights Plan, detailed below, from the earnings per share calculation because the Rights are not exercisable since the specified contingent future event has not occurred.
Reconciliation of denominators for basic and diluted earnings per share computations:
|
|
|
Years ended December 31, |
| ||||
|
|
|
2014 |
|
2013 |
|
2012 |
|
|
Basic weighted average shares outstanding |
|
31,359,867 |
|
30,449,721 |
|
30,091,985 |
|
|
Effect of dilutive employee and director awards |
|
1,450,720 |
|
— |
|
— |
|
|
Diluted weighted-average shares outstanding and assumed conversions |
|
32,810,587 |
|
30,449,721 |
|
30,091,985 |
|
Patent Costs
The Company expenses patent costs, including legal expenses, in the period in which they are incurred. Patent expenses are included in general and administrative expenses in the Company’s Statements of Comprehensive Income (Loss).
Stock-Based Compensation
Stock-based compensation cost is estimated at the grant date based on the fair value of the award and is recognized as expense over the requisite service period of the award. The fair value of restricted stock awards is
determined by reference to the fair market value of our common stock on the date of grant. We use the Black-Scholes model to value service condition and performance condition option awards. For awards with only service conditions and graded-vesting features, we recognize compensation cost on a straight-line basis over the requisite service period. For awards with performance conditions granted we recognize compensation cost over the expected period to achieve the performance conditions, provided achievement of the performance conditions are deemed probable.
Contingencies
We, AstraZeneca and Horizon are engaged in Paragraph IV litigation with several generic pharmaceutical companies with respect to patents listed in the Orange Book with respect to VIMOVO currently pending in the United States District Court for the District of New Jersey which is described on page 17 of this Form 10-K.
On March 14, 2011, we and AstraZeneca received a Paragraph IV Notice Letter from Dr. Reddy’s informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent in 2023. The ‘907 patent is assigned to POZEN and listed with respect to VIMOVO in the Orange Book. On September 19, 2011, Dr. Reddy’s amended its ANDA to include a Paragraph IV certification against the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, which are assigned to AstraZeneca or its affiliates and listed in in the Orange Book, with respect to VIMOVO. The patents listed in the Orange Book which are owned by AstraZeneca or its affiliates expire at various times between 2014 and 2018. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Dr. Reddy’s. Accordingly, we and AstraZeneca filed suit against Dr. Reddy’s on April 21, 2011 in the United States District Court for the District of New Jersey, asserting only the ‘907 patent against Dr. Reddy’s. An amended complaint was filed on October 28, 2011 to include the AstraZeneca patents. On December 19, 2012, the District Court conducted a pre-trial “Markman” hearing to determine claim construction. On May 1, 2012, the Court issued a Markman Order construing the claim terms disputed by the parties. On April 15, 2013 a Stipulation of Partial Dismissal was filed which sought dismissal of all infringement claims relating to the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent (which are each assigned to AstraZeneca), as well as Dr. Reddy’s defenses and counterclaims relating to those patents. On April 18, 2013, the District Court issued a Stipulation and Order dismissing with prejudice those claims and defenses. The first Dr. Reddy’s case is considered the lead case and has been consolidated with the actions described below for the purpose of pre-trial and discovery. A scheduling order for this case, and all of the consolidated cases, was issued by the Court on June 27, 2014. Fact discovery closed in the consolidated case on November 20, 2015. Expert discovery is ongoing and set to close May 21, 2015. In view of the upcoming retirement of presiding Judge Pisano, on February 9, 2015, the consolidated cases were reassigned to Judge Mary L. Cooper.
On June 13, 2011, we and AstraZeneca received a Paragraph IV Notice Letter from Lupin informing us that Lupin had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the POZEN and the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, the ‘466 patent and, each of which are assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Lupin’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Lupin and, accordingly, we and AstraZeneca filed suit against Lupin on July 25, 2011 in the United States District Court for the District of New Jersey. On November 19, 2014, an amended complaint was filed in which the 504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, all assigned to AstraZeneca or its affiliates, were not asserted against Lupin. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On September 19, 2011, we and AstraZeneca AB received Paragraph IV Notice Letter from Anchen informing us that Anchen had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, the ‘085 patent, the ‘070 patent, and the ‘466 patent. The patents are among those listed with respect to VIMOVO in the Orange Book and expire at various times between 2018 and 2023. Anchen’s Paragraph IV Notice Letter asserts that its generic product will not infringe those patents or that those patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Anchen and, accordingly, we and AstraZeneca filed suit against
Anchen on October 28, 2011 in the United States District Court for the District of New Jersey. On October 4, 2013, Anchen filed an amendment to its ANDA seeking to change its Paragraph IV certification to a Paragraph III. It is unclear when or if the FDA will enter Anchen’s amendment. On October 25, 2013, Anchen filed a Motion to Dismiss the case against it, based on its proposed re-certification. On November 18, 2013, we and AstraZeneca filed an Opposition to Anchen’s Motion to Dismiss. On June 11, 2014, the Court granted Anchen’s Motion and dismissed the case against them.
On November 20, 2012 we and AstraZeneca AB received a Paragraph IV Notice Letter from Dr. Reddy’s, informing us that Dr. Reddy’s had filed a second ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the POZEN and the 504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, the ‘466 patent and, each of which are assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Dr. Reddy’s second Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Dr. Reddy’s on its second ANDA filing and, accordingly, we and AstraZeneca filed suit against Dr. Reddy’s on January 4, 2013, in the United States District Court for the District of New Jersey. On April 15, 2013 a Stipulation of Partial Dismissal was filed which sought dismissal of all infringement claims relating to the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent (which are each assigned to AstraZeneca), as well as Dr. Reddy’s defenses and counterclaims relating to those patents. On April 18, 2013, the District Court issued the Stipulation and Order dismissing with prejudice those claims and defenses. On June 28, 2013 we and AstraZeneca filed a Motion for Summary Judgment relating to the second ANDA filing asserting that U.S. Patent No. 6,926,907 is not invalid. On August 12, 2013, DRL filed an opposition to the Motion for Summary Judgment. On March 28, 2014, the District Court denied the Motion. On October 11, 2013, DRL filed a Motion for Summary Judgment asserting that the product which is the subject matter of its second ANDA does not infringe the ‘907 patent. On November 4, 2013, POZEN and AstraZeneca filed a Motion for an Order Denying DRL’s Motion for Summary Judgment Pursuant to Rule 56(d) and an Opposition to DRL’s Motion. On May 29, 2014, the Court issued an order denying DRL’s Motion. This case was consolidated with the originally filed Dr. Reddy’s case and is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On March 29, 2013, we and AstraZeneca received a Paragraph IV Notice Letter from Watson, now Actavis, informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the Company and the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Watson’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Watson. On May 10, 2013, we and AstraZeneca filed a patent infringement lawsuit against Watson in the U.S. District Court of New Jersey. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On May 16, 2013, POZEN and AstraZeneca AB received a Paragraph IV Notice Letter from Mylan informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the Company and the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Mylan’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Mylan. On June 28, 2013, we and AstraZeneca filed a patent infringement lawsuit against Mylan in the U.S. District Court of New Jersey. On February 13, 2015, the Court entered a joint stipulation of dismissal of counts related to certain patents, dismissing claims related to the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On October 15, 2013, the United States Patent Office issued the ‘285 patent. The ‘285 patent, entitled “Pharmaceutical compositions for the coordinated delivery of NSAIDs” and assigned to POZEN, is related to the ‘907 patent. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement of the ‘285 patent and, accordingly, on October 23, 2013, we, and AstraZeneca filed patent infringement lawsuits against DRL, Lupin, Watson and Mylan in the U.S. District Court of New Jersey alleging that their ANDA products infringe the ‘285 patent. On November 8, 2013, we, and AstraZeneca filed a Motion to Amend the Complaint in the actions against DRL, Lupin, Watson and Mylan or, in the alternative, to consolidate the actions involving the ‘285 patent with the existing consolidated action. DRL, Lupin, Watson and Mylan have each filed answers to the respective amended complaints, thus adding claims relating to the ‘285 patent against each of the Defendants to the consolidated case.
As part of Horizon’s purchase of all of AstraZeneca’s rights, title and interest to develop, commercialize and sell VIMOVO in the United States, Horizon has assumed AstraZeneca’s right to lead the above-described Paragraph IV litigation relating to VIMIVO currently pending in the United States District Court for the District of New Jersey and has assumed patent-related defense costs relating to such litigation, including reimbursement up to specified amounts of the cost of any counsel retained by us. On December 12, 2013, Horizon filed Motions to Join under Fed.R.Civ.Proc. 25(c) as a co-plaintiff in each of the above referenced actions and the consolidated action. On January 31, 2014 and February 2, 2014, the Court granted Horizon’s motions.
As with any litigation proceeding, we cannot predict with certainty the patent infringement suit against Dr. Reddy’s, Lupin, Mylan and Watson relating to a generic version of VIMOVO. We have incurred an aggregate of $17.5 million in legal fees through the fiscal year ended December 31, 2014. Furthermore, we will have to incur additional expenses in connection with the lawsuits relating to VIMOVO, which may be substantial. In the event of an adverse outcome or outcomes, our business could be materially harmed. Moreover, responding to and defending pending litigation will result in a significant diversion of management’s attention and resources and an increase in professional fees.
New Accounting Pronouncements
Revenue from Contracts with Customers
In May 2014, the FASB issued new accounting rules related to revenue recognition for contracts with customers requiring revenue recognition based on the transfer of promised goods or services to customers in an amount that reflects consideration the Company expects to be entitled to in exchange for goods or services. The new rules supersede prior revenue recognition requirements and most industry-specific accounting guidance. The new rules will be effective for the Company in the first quarter of 2017 with either full retrospective or modified retrospective application required. The Company does not expect the adoption of the new accounting rules to have a material impact on the Company’s financial condition, results of operations or cash flows.
2. License Agreements
We have entered into and may continue to enter into collaborations with established pharmaceutical or pharmaceutical services companies to develop, commercialize and/or manufacture our product candidates. Our existing collaborations are described below.
GlaxoSmithKline (GSK)
In June 2003, we signed an agreement with GSK for the development and commercialization of proprietary combinations of a triptan (5-HT1B/1D agonist) and a long-acting NSAID. The combinations covered by the agreement are among the combinations of MT 400. Under the terms of the agreement, GSK has exclusive rights in the U.S. to commercialize all combinations which combine GSK’s triptans, including Imitrex® (sumatriptan succinate) or Amerge® (naratriptan hydrochloride), with a long-acting NSAID. We were responsible for development of the first combination product, while GSK provided formulation development and manufacturing. Pursuant to the terms of the agreement, we received $25.0 million in initial payments from GSK following termination of the waiting period under the Hart-Scott-Rodino notification program and the issuance of a specified patent. In May 2004, we received a $15.0 million milestone payment as a result of our commencement of Phase 3 clinical trial activities. In October 2005, we received a $20.0 million milestone payment upon the FDA’s acceptance for review of the NDA for Treximet, the trade name for the product. On April 26, 2008, we received, from GSK, $20.0 million in milestone payments which were associated with the approval of, and GSK’s intent to commercialize, Treximet. In addition, GSK will pay us two sales performance milestones totaling $80.0 million if certain sales thresholds are achieved. Up to an additional $10.0 million per product is payable upon achievement of milestones relating to other products. GSK will pay us royalties on all net sales of marketed products until at least the expiration of the last to expire issued applicable patent (October 2, 2025) based upon the scheduled expiration of currently issued patents. GSK may reduce, but not eliminate, the royalty payable to us if generic competitors attain a pre-determined share of the market for the combination product, or if GSK owes a royalty to one or more third parties for rights it licenses from such third parties to commercialize the product. The agreement terminates on the date of expiration of all royalty obligations unless earlier terminated by either party for a material breach or by GSK at any time upon ninety (90) days’ written notice to us for any reason or no reason. Among the contract breaches that would entitle us to terminate the agreement is GSK’s determination not to further develop or to launch the combination product under certain circumstances. GSK has the right, but not the obligation, to bring, at its own expense, an action for infringement of certain patents by third parties. If GSK does not bring any such action within a certain time frame, we have the right, at our own expense, to bring the appropriate action. With regard to certain other patent infringements, we have the sole right to bring an action against the infringing third party. Each party generally has the duty to indemnify the other for damages arising from breaches of each party’s representations, warranties and obligations under the agreement, as well as for gross negligence or intentional misconduct. We also have a duty to indemnify GSK for damages arising from our development and manufacture of MT 400 prior to the effective date of the agreement, and each party must indemnify the other for damages arising from the development and manufacture of any combination product after the effective date.
On November 23, 2011, we entered into the Purchase Agreement, with CII, pursuant to which we sold, and CII purchased, our right to receive future royalty payments arising from U.S. sales of MT 400, including Treximet.
On May 13, 2014, we, GSK, CII and Pernix, entered into certain agreements in connection with GSK’s divestiture of all of its rights, title and interest to develop, commercialize and sell Treximet® in the U.S. to Pernix. Upon the closing of the divestiture on August 20, 2014, GSK assigned the Treximet Agreement to Pernix. Immediately following the closing of the divestiture, Amendment No. 1 between us and Pernix became effective. Amendment No. 1, among other things, amends the royalty provisions of the Agreement to provide for a guaranteed quarterly minimum royalty of $4 million for the calendar quarters commencing on January 1, 2015 and ending on March 31, 2018 and requires that Pernix continue certain of GSK’s ongoing development activities and to undertake certain new activities, for which we will provide reasonable assistance. Amendment No. 1 also eliminates restrictions in the Agreement on our right to develop and commercialize certain dosage forms of sumatriptan/naproxen combinations outside of the United States and permits POZEN to seek approval for these combinations on the basis of the approved NDA for Treximet. Pernix has also granted us a warrant to purchase 500,000 shares of Pernix common stock at an exercise price equal to$4.28, the closing price of Pernix common stock as reported on the NASDAQ Global Market on May 13, 2014. The common stock underlying the warrants will be registered by Pernix with the Securities and Exchange Commission and will be exercisable from the August 20, 2014, the closing date of the divestiture until February 28, 2018. If the Divestiture is not consummated, the warrants will be null and void. Because the warrant has not been registered by Pernix with the Securities and Exchange Commission, the Company cannot sell or transfer the warrant in reliance upon Rule 144 until after November 13, 2014 when the Company meets certain holding requirements. Lastly, we, GSK, Pernix and CII executed a letter agreement whereby we expressly consented to the assignment by GSK and the assumption by
Pernix of the Treximet Agreement. On July 30, the parties entered into Amendment No. 2 to the Treximet Agreement which will permit Pernix’s Irish affiliate to which Pernix assigned its rights to further assign the Agreement without our prior written consent as collateral security for the benefit of the lenders which financed the acquisition. Amendment No. 2 became effective upon the closing of the divestiture on August 20, 2014.
AstraZeneca AB (AstraZeneca)
In August 2006, we entered into a collaboration and license agreement dated as of August 1, 2006 and effective September 7, 2006 with AstraZeneca, a Swedish corporation, regarding the development and commercialization of proprietary fixed dose combinations of the PPI esomeprazole magnesium with the NSAID naproxen, in a single tablet for the management of pain and inflammation associated with conditions such as osteoarthritis and rheumatoid arthritis in patients who are at risk for developing NSAID associated gastric ulcers, as amended, the “Original Agreement”. Under the terms of the Original Agreement, we granted to AstraZeneca an exclusive, fee-bearing license, in all countries of the world except Japan, under our patents and know-how relating to combinations of gastroprotective agents and NSAIDs (other than aspirin and its derivatives). Pursuant to the terms of the agreement, we received an upfront license fee of $40.0 million from AstraZeneca following termination of the waiting period under the Hart-Scott-Rodino notification program.
We retained responsibility for the development and filing of the NDA for the product in the U.S. AstraZeneca is responsible for all development activities outside the U.S., as well as for all manufacturing, marketing, sales and distribution activities worldwide. We agreed to bear all expenses related to certain specified U.S. development activities. All other development expenses, including all manufacturing-related expenses, will be paid by AstraZeneca. The agreement established joint committees with representation of both us and AstraZeneca to manage the development and commercialization of the product. The committees operate by consensus, but if consensus cannot be reached, we generally will have the deciding vote with respect to development activities required for marketing approval of the product in the U.S. and AstraZeneca generally will have the deciding vote with respect to any other matters.
In September 2007, we agreed with AstraZeneca to amend the Original Agreement effective as of September 6, 2007. Under the terms of the amendment, AstraZeneca has agreed to pay us up to $345.0 million, in the aggregate, in milestone payments upon the achievement of certain development, regulatory and sales events. In September 2007 we received a $10.0 million payment upon execution of the amendment and a $20.0 million payment in recognition of the achievement of the primary endpoints for the PN400-104 study, a study which compared acid suppression of different doses of VIMOVO (formerly PN 400), and achievement of the interim results of the PN200-301 study, a six month comparative trial of PN 200 as compared to EC naproxen in patients requiring chronic NSAID therapy, meeting mutually agreed success criteria. In May 2010, we received a $20.0 million payment for the NDA approval of VIMOVO. We also received an additional $25.0 million milestone in December 2010 when VIMOVO received approval (including pricing and reimbursement approval) in a major ex-U.S. market and up to $260.0 million will be paid as sales performance milestones if certain aggregate sales thresholds are achieved.
The amendment also revised the royalty rates we were to have received under the Original Agreement. Prior to the effective date of the amendment, under the terms of the Original Agreement, we were to receive a royalty based on annual net sales by AstraZeneca, its affiliates or sublicensees during the royalty term. The royalty rate varied based on the level of annual net sales of products made by AstraZeneca, its affiliates and sublicensees, ranging from the mid-single digits to the mid-teens. Under the amendment, we receive a flat, low double digit royalty rate during the royalty term on annual net sales of products made by AstraZeneca, its affiliates and sublicensees, in the U.S. and royalties ranging from the mid-single digits to the high-teens on annual net sales of products made by AstraZeneca, its affiliates and sublicensees outside of the U.S. The amendment also revised the rate of reduction to the royalty rate based upon loss of market share due to generic competition inside and outside of the U.S. to account for the new royalty structure. Our right to receive royalties from AstraZeneca for the sale of such products under the collaboration and license agreement, as amended, expires on a country-by-country basis upon the later of (a) expiration of the last-to-expire of certain patent rights relating to such products in that country, and (b) ten years after the first commercial sale of such products in such country.
We further amended the Original Agreement effective October 1, 2008 to shorten the timing of AstraZeneca’s reimbursement obligation for certain development expenses incurred by us under the agreement and to update the description of the target product profile studies (as defined in the agreement) for VIMOVO.
On December 31, 2014 we accrued $5.6 million of VIMOVO royalty revenue, $4.3 million related to U.S. sales and $1.3 million related to ROW sales. The agreement, unless earlier terminated, will expire upon the payment of all applicable royalties for the products commercialized under the agreement. Either party has the right to terminate the agreement by notice in writing to the other party upon or after any material breach of the agreement by the other party, if the other party has not cured the breach within 90 days after written notice to cure has been given, with certain exceptions. The parties also can terminate the agreement for cause under certain defined conditions. In addition, AstraZeneca can terminate the agreement, at any time, at will, for any reason or no reason, in its entirety or with respect to countries outside the U.S., upon 90 days’ notice. If terminated at will, AstraZeneca will owe us a specified termination payment or, if termination occurs after the product is launched, AstraZeneca may, at its option, under and subject to the satisfaction of conditions specified in the agreement, elect to transfer the product and all rights to us.
On May 3, 2013, AstraZeneca informed us that, after a strategic business review, it had decided to cease promotion and sampling of VIMOVO by the end of the third quarter of 2013 in certain countries, including the U.S. and all countries in Europe, other than Spain and Portugal, which have pre-existing contractual relationships with third parties. We understand that AstraZeneca will instead now focus on those countries where the product has shown growth and which AstraZeneca believes have the greatest potential for future growth.
On September 16, 2013, we and AstraZeneca entered into another amendment to the Original Agreement which made clarifications to certain intellectual property provisions of the Original Agreement to clarify that AstraZeneca’s rights under those provisions do not extend to products which contain acetyl salicylic acid. On September 16, 2013, we and AstraZeneca also executed a letter agreement whereby we agreed that in the event that AstraZeneca divested its rights and obligations to market VIMOVO in the United States to a third party, AstraZeneca would be relieved of its obligations under the Original Agreement with respect to the United States as of the effective date of such divestiture, including its obligation under the Original Agreement to guarantee the performance of such assignee and/or sublicensee.
On November 18, 2013, AstraZeneca and Horizon entered into certain agreements in connection with AstraZeneca’s divestiture of all of its rights, title and interest to develop, commercialize and sell VIMOVO in the United States to Horizon. In connection with this divestiture, on November 18, 2013 we and AstraZeneca entered into an Amended and Restated Collaboration and License Agreement for the United States, the “U.S. Agreement,” and an Amended and Restated License and Collaboration Agreement for Outside the United States, the “ROW Agreement,” which agreements collectively amend and restate the Original Agreement. AstraZeneca has assigned the U.S. Agreement to Horizon in connection with the Divestiture with our consent.
We and Horizon also entered into Amendment No. 1 to the U.S. Agreement which, among other things, amends the royalty provisions of the U.S. Agreement to provide for a guaranteed annual minimum royalty amount of $5 million in calendar year 2014, and a guaranteed annual minimum royalty amount of $7.5 million each calendar year thereafter, provided that the patents owned by us which cover VIMOVO are in effect and no generic forms of VIMOVO are in the marketplace. Amendment No. 1 also provides that Horizon has assumed AstraZeneca’s right to lead the on-going Paragraph IV litigation relating to VIMIVO currently pending in the United States District Court for the District of New Jersey and will assume all patent-related defense costs relating to such litigation, including reimbursement up to specified amounts of the cost of any counsel retained by us, amends certain time periods for Horizon’s delivery of quarterly sales reports to POZEN, and provides for quarterly update calls between the parties to discuss VIMOVO’s performance and Horizon’s commercialization efforts.
Further, the Company, AstraZeneca and Horizon executed a letter agreement whereby POZEN expressly consented to the assignment by AstraZeneca and the assumption by Horizon of the U.S. Agreement. In addition, the letter agreement establishes a process for AstraZeneca and Horizon to determine if sales milestones set forth in the Original Agreement are achieved on a global basis and other clarifications and modifications required as a result of incorporating the provisions of the Original Agreement into the U.S. Agreement and the ROW Agreement or as otherwise agreed by the parties.
sanof-aventis U.S. LLC
On September 3, 2013, we entered into an exclusive license and collaboration agreement with Sanofi US for .the commercialization of products containing a combination of immediate release omeprazole and 325 mg or less of delayed release aspirin, including PA32540 and PA8140 in the United States. On November 29, 2014, we executed a termination agreement with Sanofi US terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi US were terminated and all rights to the products licensed to Sanofi US under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi US relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products.
Cilag GmbH International (Cilag)
On March 21, 2011, we entered into a license agreement with Cilag, a division of Johnson & Johnson, for the exclusive development and commercialization of MT 400 in Brazil, Colombia, Ecuador and Peru. In December 2014 we received an executed, mutual termination from Cilag. There was no dispute between the parties regarding the license agreement and, at our request, for a period of two years after termination, Cilag has agreed to negotiate in good faith commercially reasonable terms of a supply agreement whereby Cilag would supply us or our licensees, with MT400 for a period equal to the shorter of (i) two (2) years; or (ii) until we establish an alternative supplier. We recognized approximately $257,300 in licensing revenue in the fourth quarter of as a result of this termination that had previously been recorded as deferred revenue.
Patheon Pharmaceuticals Inc. (Patheon)
On December 19, 2011, we entered into a Manufacturing Services Agreement, or the Supply Agreement, and a related Capital Expenditure and Equipment Agreement, or the Capital Agreement, relating to the manufacture of PA32450. Under the terms of the Supply Agreement, Patheon has agreed to manufacture, and we have agreed to purchase, a specified percentage of the Company’s requirements of the PA32540 for sale in the United States. The term of the Supply Agreement extends until December 31st of the fourth year after the we notify Patheon to begin manufacturing services under the Supply Agreement, or the Initial Term, and will automatically renew thereafter for periods of two years, unless terminated by either party upon eighteen months’ written notice prior to the expiration of the Initial Term or twelve months’ written notice prior to the expiration of any renewal term. In addition to usual and customary termination rights which allow each party to terminate the Supply Agreement for material, uncured breaches by the other party, we can terminate the Agreement upon thirty (30) days’ prior written notice if a governmental or regulatory authority takes any action or raises any objection that prevents us from importing, exporting, purchasing or selling PA32540 or if it is determined that the formulation or sale of PA32540 infringes any patent rights or other intellectual property rights of a third party. We can also terminate the Supply Agreement upon twenty-four (24) months’ prior written notice if we license, sell, assign or otherwise transfer any rights to commercialize PA32540 in the Territory to a third party. The Supply Agreement contains general and customary commercial supply terms and conditions, as well as establishing pricing for bulk product and different configurations of packaged product, which pricing will be adjusted annually as set forth in the Supply Agreement. Under the terms of the Capital Agreement, we will be responsible for the cost of purchasing certain equipment specific to the manufacture of PA32540, the cost of which, based on current volume projections, is expected to be less than $150,000. If additional equipment and facility modifications are required to meet our volume demands for PA32540, we may be required to contribute to the cost of such additional equipment and facility modifications, up to a maximum of approximately $2.5 million in the aggregate.
The Supply Agreement and Capital Agreement were amended on July 10, 2013. The First Amendment to the Manufacturing and Services Agreement (the “Amendment to the Supply Agreement”) expressly incorporates the Company’s PA8140 product candidate into the Supply Agreement. The Amendment to the Supply Agreement also clarifies that the manufacturing services contemplated by the Supply Agreement include the manufacture of validation batches, but the placing of an order for such validation batches will not trigger the Commencement Date of the Initial Term (each as defined in the Supply Agreement), updates pricing for the Company’s PA32540 product candidate and a incorporates a new pricing schedule for PA8140, as well as other conforming changes to the Supply Agreement. The First Amendment to the Capital Expenditure and Equipment Agreement (the “Amendment to
the Capital Agreement”), replaces the existing Schedule A of the Capital Agreement, which lists dedicated and non-dedicated capital equipment and facility modifications to be funded in whole or in part by the Company, with a new updated schedule which reflects the parties’ current assumptions regarding the need for and timing of capital equipment expenditures based upon Patheon’s current and anticipated production capacity and current volume projections for thePA32540 and PA8140. Under the terms of the Capital Agreement, the Company was previously required to contribute to the cost of such additional capital equipment and facility modifications, up to a maximum of approximately $2.5 million in the aggregate. Pursuant to the terms of the Amendment to the Capital Agreement, the parties have agreed to reduce the amount of such maximum expenditure to approximately $1.2 million dollars in light of the revised capacity and volume assumptions.
3. Stockholders’ Equity
Shares Reserved for Future Issuance
In January 2005, the Company approved a stockholder rights plan (the “Rights Plan”), pursuant to which the Company entered into a Rights Agreement dated January 12, 2005 with StockTrans, Inc., as Rights Agent, and the Company declared a dividend of a right to acquire one preferred share purchase right (a “Right”) for each outstanding share of the Company’s Common Stock, $0.001 par value per share, to stockholders of record at the close of business on January 28, 2005. Generally, the Rights only are triggered and become exercisable if a person or group acquires beneficial ownership of 15 percent or more of the Company’s common stock or announces a tender offer for 15 percent or more of the Company’s common stock. The Rights Plan is similar to plans adopted by many other publicly-traded companies. The effect of the Rights Plan is to discourage any potential acquirer from triggering the Rights without first convincing POZEN’s Board of Directors that the proposed acquisition is fair to, and in the best interest of, the shareholders and the Company. The provisions of the Plan will substantially dilute the equity and voting interest of any potential acquirer unless the Board of Directors determines that the proposed acquisition is in the best interest of the shareholders. In connection with the Plan, the Company designated 90,000 shares of its authorized Preferred Stock as Series A Junior Participating Preferred Stock. Each Right, if and when exercisable, will entitle the registered holder to purchase from the Company one one-thousandth of a share of Series A Junior Participating Preferred Stock, $0.001 par value per share, at a purchase price of $80.00 for each one one-thousandth of a share, subject to adjustment. Each holder of a Right (except for the Acquiring Person (as defined in the Rights Plan), whose Rights will be null and void upon such event) shall thereafter have the right to receive, upon exercise, that number of shares of Common Stock of the Company (or, in certain circumstances, cash, property or other securities of the Company) which equals the exercise price of the Right divided by 50% of the current market price (as defined in the Rights Agreement) per share of Common Stock at the date of the occurrence of such event. The Rights can be terminated by POZEN’s Board of Directors and are subject to optional redemption by the Company at $0.001 per Right. The Rights Plan has a 10-year term and contains provisions requiring a periodic review and evaluation by the Board of Directors
If there is any change in the number or kind of shares of company stock outstanding or if the value of outstanding shares of company stock is substantially reduced as a result of an extraordinary dividend or distribution, the Company’s 2010 Stock Option Plan requires that an equitable adjustment be made to all outstanding grants to preclude dilution of rights and benefits under the plan. Therefore, as a result of the December 31, 2013 cash dividend distribution, a dividend equivalent was provided to all outstanding grants. The adjustments were in the form of additional RSUs to RSU holders or an adjustment to both the outstanding number of options and their strike price; all adjustments were made in compliance with Sections 409A and 424 of the Internal Revenue Code. In addition, the 2010 Stock Option Plan provides for an adjustment to the number of common shares available for grant under the stock option plan. Therefore, as a result of the December 31, 2013 cash dividend distribution, the number of common shares available for grant was adjusted by 416,971 shares and that increase is reflected in the table below.
At December 31, 2014, shares of our common stock reserved for future issuance are as follows:
|
Common shares available for grant under stock option plans |
|
2,474,430 |
|
|
Common shares issuable pursuant to options and restricted stock units granted under equity compensations plans |
|
3,817,920 |
|
|
Rights Plan shares issuable as Series A Junior Participating Preferred Stock |
|
90,000 |
|
|
Total Reserved |
|
6,382,350 |
|
4. Accrued Expenses
Accrued expenses consist of the following at December 31:
|
|
|
2014 |
|
2013 |
| ||
|
Research and development costs |
|
$ |
55,227 |
|
$ |
1,025,995 |
|
|
Other |
|
198,397 |
|
629,217 |
| ||
|
|
|
$ |
253,624 |
|
$ |
1,655,212 |
|
5. Income Taxes
The Company did not record a provision for income taxes during the years ended December 31, 2014, 2013 and 2012.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets are as follows at December 31:
|
($ in thousands) |
|
2014 |
|
2013 |
| ||
|
Current |
|
|
|
|
| ||
|
Deferred income tax assets |
|
|
|
|
| ||
|
Other current assets |
|
$ |
662 |
|
$ |
1,080 |
|
|
Less valuation allowance |
|
(647 |
) |
(1,080 |
) | ||
|
Total net deferred income tax assets, current |
|
$ |
15 |
|
$ |
— |
|
|
|
|
|
|
|
| ||
|
Deferred income tax liabilities |
|
|
|
|
| ||
|
Investment in warrants |
|
(968 |
) |
— |
| ||
|
Total net deferred income taxes, current |
|
$ |
(953 |
) |
$ |
— |
|
|
|
|
|
|
|
| ||
|
Non-current |
|
|
|
|
| ||
|
Deferred income tax assets (liabilities) |
|
|
|
|
| ||
|
Tax loss carryforwards |
|
$ |
20,840 |
|
$ |
25,909 |
|
|
Research and development credits |
|
13,987 |
|
13,992 |
| ||
|
Equity compensation and other |
|
6,683 |
|
7,549 |
| ||
|
Total gross deferred income taxes, non-current |
|
41,510 |
|
47,450 |
| ||
|
Less valuation allowance |
|
(40,557 |
) |
(47,450 |
) | ||
|
Total net deferred income taxes, non-current |
|
$ |
953 |
|
$ |
— |
|
|
|
|
|
|
|
| ||
|
Total net deferred income taxes |
|
$ |
— |
|
$ |
— |
|
At December 31, 2014 and 2013, the Company had federal net operating loss carryforwards of approximately $53 million and $66.8 million respectively, state net economic loss carryforwards of approximately $78 million and $82.9 million respectively, and research and development credit carryforwards of approximately $14 million and $14 million, respectively. The federal and state net operating loss carryforwards begin to expire in 2028 and 2015, respectively, and the research and development credit carryforwards begin to expire in 2018. For financial reporting purposes, a valuation allowance has been recognized to offset the deferred tax assets related to the carryforwards, based on the Company’s assessment regarding the realizability of these deferred tax assets in future periods. Of the total decrease in valuation allowance of $7.3 million, a decrease of $7.3 million was allocable to current operating activities. The utilization of the loss carryforwards to reduce future income taxes will depend on the Company’s ability to generate sufficient taxable income prior to the expiration of the loss carryforwards. In
addition, the maximum annual use of net operating loss and research credit carryforwards is limited in certain situations where changes occur in stock ownership. The recognized tax benefit related to net operating loss carryforwards was approximately $4.8M, $0, and $0 for the years ended December 31, 2014, 2013 and 2012, respectively.
The research and development credit, which had previously expired on December 31, 2011, was reinstated as part of the American Taxpayer Relief Act of 2012 enacted on January 2, 2013. This legislation retroactively reinstated and extended the credit from the previous expiration date through December 31, 2013. As a result, the Company adjusted its deferred tax assets in 2013 for both the 2013 and 2012 research and development credits, which resulted in an increase to the deferred tax assets and a corresponding increase to the valuation allowance of $0.02 million and $0.11 million, respectively.
On July 23, 2013, North Carolina enacted House Bill 998, which reduced the corporate income tax rate from 6.9% in 2013 to 6% in 2014 and to 5% in 2015. As a result of the new enacted tax rate, the Company adjusted its deferred tax assets in 2013 by applying the lower rate, which resulted in a decrease to the deferred tax assets and a corresponding decrease to the valuation allowance of approximately $0.03 million.
The actual income tax benefit (expense) for the years ended December 31, 2014, 2013 and 2012, differed from the amounts computed by applying the U.S. federal tax rate of 35% to income (loss) before taxes as a result of the following:
|
($ in thousands) |
|
2014 |
|
2013 |
|
2012 |
| |||
|
(Loss) income before income tax |
|
$ |
19,675 |
|
$ |
(16,708 |
) |
$ |
(25,283 |
) |
|
Federal tax rate |
|
35 |
% |
35 |
% |
35 |
% | |||
|
Federal income tax provision at statutory rate |
|
6,886 |
|
(5,848 |
) |
(8,849 |
) | |||
|
State tax provision |
|
224 |
|
(215 |
) |
(343 |
) | |||
|
|
|
7,110 |
|
(6,063 |
) |
(9,192 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Decrease (increase) in income tax benefit resulting from: |
|
|
|
|
|
|
| |||
|
Research and development credits |
|
4 |
|
66 |
|
— |
| |||
|
Non-deductible expenses and other |
|
177 |
|
302 |
|
409 |
| |||
|
Change in state tax rate |
|
35 |
|
966 |
|
— |
| |||
|
Change in valuation allowance |
|
(7,326 |
) |
4,729 |
|
8,783 |
| |||
|
Income tax expense |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
The Company had gross unrecognized tax benefits of approximately $0.5 million as of January 1, 2014. As of December 31, 2014, the total gross unrecognized tax benefits were approximately $0.5 million and of this total, none would reduce the Company’s effective tax rate if recognized. The Company does not anticipate a significant change in total unrecognized tax benefits or the Company’s effective tax rate due to the settlement of audits or the expiration of statutes of limitations within the next 12 months.
The Company’s policy for recording interest and penalties associated with tax audits is to record them as a component of provision for income taxes. The Company has not recorded any interest or penalty since adoption of FASB ASC 740-10.
The Company has analyzed its filing positions in all significant federal, state and foreign jurisdictions where it is required to file income tax returns, as well as open tax years in these jurisdictions. With few exceptions, the Company is no longer subject to US Federal and state and local tax examinations by tax authorities for years before 2011, although carryforward attributes that were generated prior to 2011 may still be adjusted upon examination by the Internal Revenue Service (IRS) if they either have been or will be used in a future period. No income tax returns are currently under examination by taxing authorities.
Rollforward of gross unrecognized tax positions:
|
|
|
($ in thousands) |
| |
|
Gross tax liability at January 1, 2014 |
|
$ |
538 |
|
|
|
|
|
| |
|
Additions/Decreases for tax positions of prior years |
|
(1 |
) | |
|
Additions/Decreases for tax positions of the current year |
|
— |
| |
|
|
|
|
| |
|
Gross tax liability at December 31, 2014 |
|
$ |
537 |
|
6. Equity Compensation Plans
In 1996, the Company established a Stock Option Plan (the “Option Plan”) and authorized the issuance of options to attract and retain quality employees and to allow such employees to participate in the growth of the Company. In June 2000, the stockholders approved the POZEN Inc. 2000 Equity Compensation Plan (the “2000 Plan”) and the 2000 Plan became effective upon the completion of the Company’s initial public offering in October 2000, after which time no further grants were made under the Option Plan. In May 2004, the stockholders approved an amendment to and restatement of the 2000 Plan. The amendment to the 2000 Plan provided for an increase in the number of shares of common stock authorized for issuance under the 2000 Plan from 3,000,000 to 5,500,000 shares. In June 2007, the stockholders approved the amendment and restatement of the 2000 Plan to, among other things, increase the number of shares authorized for issuance under the 2000 Plan to 6,500,000 shares and continue the various performance criteria for use in establishing specific vesting targets for certain awards. In June 2010, stockholders approved the POZEN Inc. 2010 Equity Compensation Plan, (“the 2010 Plan”), a successor incentive compensation plan to the 2000 Plan which was merged with and into the 2010 Plan and all grants outstanding under the 2000 Plan were issued or transferred under the 2010 Plan.
The 2010 Plan provides for grants of incentive stock options, nonqualified stock options, stock awards, and other stock-based awards, such as restricted stock units and stock appreciation rights (“SARs”), to employees, non-employee directors, and consultants and advisors who perform services for us and our subsidiaries. The 2010 Plan authorizes up to 7,452,327 shares of common stock for issuance, which includes 2,000,000 shares of our common stock which were in excess of the number of shares previously reserved under the 2000 Plan. The maximum number of shares for which any individual may receive grants in any calendar year is 1,000,000 shares. The Compensation Committee of the Board of Directors, which administers the 2010 Plan, will determine the terms and conditions of options, including when they become exercisable. Neither our Board nor the Committee can amend the 2010 Plan or options previously granted under the Plan to permit a repricing of options or SARs, without prior stockholder approval. If options granted under the 2010 Plan expire or are terminated for any reason without being exercised, or if stock awards, performance units, or other stock-based awards are forfeited or otherwise terminate, the shares of common stock underlying the grants will again be available for awards granted under the 2010 Plan.
If there is any change in the number or kind of shares of company stock outstanding or if the value of outstanding shares of company stock is substantially reduced as a result of a spinoff or the Company’s payment of an extraordinary dividend or distribution, the 2010 Plan requires that an equitable adjustment be made to all outstanding grants to preclude dilution of rights and benefits under the plan. Consequently, as a result of the December 31, 2013 cash dividend distribution, a dividend equivalent totaling 987,000 shares was provided to all outstanding grants. The adjustments were in the form of additional RSUs to RSU holders or an adjustment to both the outstanding number of options and their strike price, in compliance with Sections 409A and 424 of the Internal Revenue Code.
Our Statements of Comprehensive Income (Loss) for the fiscal years ended December 31, 2014, 2013 and 2012 include the following stock-based compensation expense:
|
|
|
Years ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
2012 |
| |||
|
Research and development |
|
$ |
295,631 |
|
$ |
765,526 |
|
$ |
461,118 |
|
|
Sales, general and administrative |
|
1,585,331 |
|
3,196,860 |
|
2,268,802 |
| |||
|
Total expense |
|
$ |
1,880,962 |
|
$ |
3,962,386 |
|
$ |
2,729,920 |
|
Unrecognized stock-based compensation expense, including time-based options, performance-based options and restricted stock awards, expected to be recognized over an estimated weighted-average amortization period of 2.0 years, was $4.1 million at December 31, 2014.
Time-Based Stock Awards
No new time-based awards were granted during the year ended December 31, 2014. Previously, the fair value of each time-based award was estimated on the date of grant using the Black-Scholes option valuation model, which used the assumptions described below. Our weighted-average assumptions used in the Black-Scholes valuation model for equity awards with time-based vesting provisions granted for the years ended December 31, 2013 and 2012 are shown in the following table:
|
|
|
2013 |
|
2012 |
|
|
Expected volatility |
|
63.7% |
|
68.0–72.3% |
|
|
Expected dividends |
|
0% |
|
0% |
|
|
Expected terms |
|
6.0 Years |
|
6.0 Years |
|
|
Risk-free interest rate |
|
1.25% |
|
0.91–1.33% |
|
|
Weighted average grant date fair value |
|
$5.35 |
|
$4.87 |
|
For the years ended December 31, 2013 and 2012, the expected volatility rate was estimated based on an equal weighting of the historical volatility of POZEN common stock over approximately a six-year period. For the years ended December 31, 2013 and 2012, the expected term was based upon average historical terms to exercise. The risk-free interest rate was based on six-year U.S. Treasury securities. The pre-vesting forfeiture rates used of the years ended December 31, 2013 and 2012 were based on historical rates. We adjust the estimated forfeiture rate based upon actual experience.
A summary of the time-based stock awards as of December 31, 2014, and changes during the year ended December 31, 2014, are as follows:
|
Time-Based Stock Awards |
|
Underlying |
|
Weighted- |
|
Average |
|
Aggregate |
| ||
|
Outstanding at December 31, 2013 |
|
4,315 |
|
$ |
6.82 |
|
4.3 |
|
$ |
8,553 |
|
|
Granted |
|
— |
|
— |
|
|
|
|
| ||
|
Exercised |
|
(1,479 |
) |
5.34 |
|
|
|
|
| ||
|
Forfeited or expired |
|
(495 |
) |
8.52 |
|
|
|
|
| ||
|
Outstanding at December 31, 2014 |
|
2,341 |
|
7.39 |
|
4.1 |
|
$ |
4,382 |
| |
|
Exercisable at December 31, 2014 |
|
1,865 |
|
$ |
8.29 |
|
3.4 |
|
$ |
2,402 |
|
|
Vested or expected to vest at December 31, 2014 |
|
2,270 |
|
$ |
7.39 |
|
4.1 |
|
$ |
4,248 |
|
The aggregate intrinsic value of options outstanding represents the pretax value (the period’s closing market price, less the exercise price, times the number of in-the-money options) that would have been received by all option holders had they exercised their options at the end of the period. The exercise price of stock options granted during the years ended December 31, 2014, 2013 and 2012 was equal to the market price of the underlying common stock on the grant date. A total of 1,479,000 stock options were exercised during the year ended December 31, 2014 with an intrinsic value of $4.6 million, a total of 138,562 stock options were exercised during the year ended December 31, 2013 with an intrinsic value of $589,000 and a total of 252,398 stock options were exercised during the year ended December 31, 2012 with an intrinsic value of $304,000. The fair value of shares vested during the years ended December 31, 2014, 2013 and 2012 were $1.1 million, $0.6 million and $0.4 million, respectively.
A summary of the time-based nonvested awards as of December 31, 2014, and changes during the year ended December 31, 2014, are as follows:
|
|
|
Underlying |
|
Weighted- |
| |
|
Nonvested outstanding at December 31, 2013 |
|
760 |
|
$ |
4.06 |
|
|
Granted |
|
— |
|
— |
| |
|
Forfeited or expired |
|
(7 |
) |
3.87 |
| |
|
Vested |
|
(276 |
) |
4.47 |
| |
|
Nonvested outstanding at December 31, 2014 |
|
477 |
|
$ |
3.85 |
|
Restricted Stock and Restricted Stock Units
For the years ended December 31, 2014, 2013 and 2012, the Company recognized $1.0 million, $1.2 million and $1.0 million, respectively, in compensation expense related to restricted stock units.
A summary of the restricted stock awards as of December 31, 2014, and changes during the year ended December 31, 2014, are as follows:
|
|
|
Underlying |
|
Weighted- |
| |
|
Restricted stock outstanding at December 31, 2013 |
|
747 |
|
$ |
6.22 |
|
|
Granted |
|
450 |
|
8.32 |
| |
|
Vested and released |
|
(84 |
) |
5.32 |
| |
|
Forfeited or expired |
|
(4 |
) |
5.91 |
| |
|
Restricted stock outstanding at December 31, 2014 |
|
1,109 |
|
$ |
7.14 |
|
As of December 31, 2014 there was an aggregate $3.8 million of unrecognized compensation expense related to unvested restricted stock units. There were 627,000 unvested restricted stock units outstanding at December 31, 2014, 523,000 unvested restricted stock units outstanding at December 31, 2013, and 430,000 unvested restricted stock units outstanding at December 31, 2012. The total fair value of restricted stock that vested during the years ended December 31, 2014, 2013 and 2012 was $726,000, $863,000and $920,000, respectively.
Performance-Based Awards
In May 2008, pursuant to an incentive program (the “PN incentive program”) approved by the Compensation Committee of the Board of Directors of the Company, stock options were granted to all of the Company’s employees, including its executive officers, to purchase an aggregate of 281,433 shares of common stock with an exercise price of $14.45 per share. In September 2008, additional stock options were granted under the PN incentive program, to purchase 11,700 shares of common stock at an exercise price of $10.82 per share. The stock options have a ten-year term and have an exercise price equal to the closing sale price of the Company’s common stock, as reported on the NASDAQ Global Market, on the date immediately preceding the date of grant. Twenty-five percent (25%) of the PN incentive program options granted vested in 2009, upon completion of the performance goal and the remaining seventy-five percent (75%) of the options granted vested in 2010 upon the completion of the remaining performance goals. The fair value of the performance-based options granted under the PN incentive program was estimated as of the grant date using the Black-Scholes option valuation model without consideration of the performance measures. The options also include provisions that require satisfactory employee performance prior to vesting. Additionally, 20,000 options were granted to an executive officer in May 2008 under the PN incentive plan, with similar grant and exercise terms. The Company recognized compensation costs for these awards over the expected service period.
In October 2011, pursuant to an incentive program (the “PA32540 incentive program”) approved by the Compensation Committee of the Board of Directors of the Company, stock options were granted to all of the
Company’s employees, including its executive officers, to purchase an aggregate of 453,960 shares of common stock. The underlying stock options and RSUs were performance-based and focus on the successful completion of certain value-enhancing events for the Company’s Yosprala product candidate. The stock options have a ten-year term and have an exercise price equal to the closing sale price of the Company’s common stock, as reported on the NASDAQ Global Market, on the date immediately preceding the date of grant. The underlying stock options and RSUs vest in accordance with the following schedule: (a) one-third (1/3) upon the acceptance of the filing of a new drug application (the “NDA”) for Yosprala, assuming the NDA filing is made prior to December 31, 2012, (b) one-third (1/3) upon first cycle NDA approval of Yosprala (otherwise 16.5% upon NDA approval after first cycle), and (c) one-third (1/3) upon execution of a significant partnering transaction for Yosprala in a major territory as determined by the Compensation Committee of the Company, in its sole discretion, at the time of such transaction, subject in each case to continued employment or service to the Company.
During a pre-submission meeting with respect to its NDA for Yosprala in April 2012, the FDA suggested that the Company also seek approval for a lower dose formulation of the product containing 81 mg of enteric coated aspirin as part of its NDA for Yosprala. The Company decided to include data and information relating to a lower dose formulation in its NDA. Generation of additional data with respect to lower dose formulation of Yosprala and incorporation of data into the NDA for Yosprala would delay submission of the NDA from the original planned submission date.
Therefore, in October 2012, the Compensation Committee granted performance-based incentive awards (the “PA8140 incentive program”) both to compensate the employees for the expected loss of value under the PA32540 Incentive Program, as well as to provide additional incentive to employees to complete the value-added activities required for submission and approval of the lower dose product. The Compensation Committee granted an aggregate of 208,740 restricted stock units to various employees of the Company, including 105,000 restricted stock units granted to the Company’s named executive officers. The restricted stock units were performance-based and vest in accordance with the following schedule: (a) one-half (1/2) upon the acceptance by the FDA of the filing of an NDA for a lower dose Yosprala product candidate, and (b) one-half (1/2) upon approval by the FDA of an NDA for a lower dose Yosprala product candidate. In 2012, 132,883 options were forfeited in acknowledgement that certain performance goals would not be met under the PA32540 incentive program.
In April 2014, the Compensation Committee granted an aggregate of 73,000 restricted stock units to various employees of the Company, including 65,000 restricted stock units granted to the Company’s named executive officers. The restricted stock units were performance-based and vest in accordance with the following schedule: (i) 50% upon receipt of the milestone payment by Sanofi US under the License and Collaboration Agreement, dated as of September 3, 2013 (the “Agreement”) to be received upon approval by the U.S. Food and Drug Administration of the PA product candidates; and (ii) 50% upon receipt of the milestone payment by Sanofi US upon achievement of commercial readiness (as defined in the Agreement). The entire award was forfeited in 2014 upon the termination of the Sanofi US agreement. In 2014, a total of 177,818 options were forfeited in acknowledgement that certain performance goals would not be met under the PA32540 and PA8140 incentive programs.
During the twelve months ended December 31, 2014, in acknowledgement that certain performance goals would not be met under the PA32540 and PA8140 incentive programs and as a result of the forfeitures and accompanying prior expense reversals, there was a net negative expense of $11,000 recorded related to the achievement of vesting criteria for performance-based awards under the PA32540 and PA8140 incentive programs. As of December 31, 2014, there was $6,000 in unrecognized compensation expense related to performance-based awards granted under the PA32540 and PA8140 incentive programs.
A summary of the performance-based stock awards as of December 31, 2014, and changes during the fiscal year ended December 31, 2014, are as follows:
|
|
|
Underlying |
|
Weighted- |
| |
|
Performance-based outstanding at December 31, 2013 |
|
540 |
|
$ |
6.76 |
|
|
Granted |
|
73 |
|
7.89 |
| |
|
Exercised |
|
(46 |
) |
1.99 |
| |
|
Forfeited or expired |
|
(199 |
) |
5.77 |
| |
|
Performance-based outstanding at December 31, 2014 |
|
368 |
|
$ |
8.12 |
|
The December 31, 2014 amount is expected to be recognized at the time of the grant vesting over the period ending in second quarter 2015. Under the PA32540 and PA8140 incentive programs, there were 139,000 unvested performance-based options outstanding at December 31, 2014. No performance-based awards vested during the twelve months ended December 31, 2014 and December 31, 2012. A total of 231,000 performance-based awards vested during the twelve months ended December 31, 2013. There were 229,000 vested performance-based options outstanding at December 31, 2014. The total value of performance-based awards that vested during the year ended December 31, 2014, 2013 and 2012 was $0.0, $1.0 million and $0, respectively. There were 199,000 awards forfeited during the twelve months ended December 31, 2014, 37,000 awards forfeited during the twelve months ended December 31, 2013, and 204,123 awards forfeited during the year ended December 31, 2012. A total of 46,000 performance-based awards were exercised during the year ended December 31, 2014, 162,000 performance-based awards were exercised during the year ended December 31, 2013 and no performance-based awards were exercised during the years ended December 31, 2012. At December 31, 2014, the performance-based options had an intrinsic value of $1.3 million and a remaining weighted contractual life of 5.2 years.
7. Leases
The Company leases its office space and certain equipment under cancellable and noncancellable operating lease agreements. Rent expense incurred by the Company was approximately $419,000, $419,000 and $419,000, for the fiscal years ended December 31, 2014, 2013 and 2012, respectively. At December 31, 2014 noncancellable future minimum lease payments for operating leases totaled $0.4 million, all relating to the 2015 lease agreement.
On February 16, 2009, the Company modified certain terms to our existing lease agreement, dated November 21, 2001, relating to approximately 17,009 square feet of office space located at Exchange Office Building, Chapel Hill, North Carolina. Under the terms of the modification, the lease term was extended for an additional 5 years and 7 months, terminating on September 30, 2015. The modification also provides the Company with a reduced notice period of 7 months for renewals of the lease. The Company is also entitled to a 3-year lease extension option available at the end of the term and a first offer right on available space located within the Exchange Office Building property. As a result of entering into the modification, the Company’s noncancellable future minimum lease payments for operating leases increased by approximately $2.7 million over the lease term. The Company is recognizing rent expense on a straight-line basis over the term of the lease which resulted in a deferred rent balance of $62,600 at December 31, 2014.
8. Retirement Savings Plan
The Company has adopted a defined contribution 401(k) plan (the “Plan”) covering substantially all employees who are at least 21 years of age. Based upon management’s discretion, the Company may elect to make contributions to the Plan. During the fiscal years ended December 31, 2014, 2013 and 2012, the Company made contributions of $141,887 and $191,582 and $224,420 respectively, to the Plan.
9. Summary of Operations by Quarters (Unaudited)
|
|
|
2014 |
| ||||||||||
|
|
|
1st Quarter |
|
2nd Quarter |
|
3rd Quarter |
|
4th Quarter |
| ||||
|
Revenue |
|
|
|
|
|
|
|
|
| ||||
|
Licensed revenue |
|
$ |
7,548,676 |
|
$ |
7,419,306 |
|
$ |
7,539,741 |
|
$ |
9,886,509 |
|
|
Total revenue |
|
7,548,676 |
|
7,419,306 |
|
7,539,741 |
|
9,886,509 |
| ||||
|
Operating expenses |
|
4,651,396 |
|
4,426,615 |
|
3,628,176 |
|
3,112,431 |
| ||||
|
Income before income tax expense |
|
2,904,691 |
|
2,999,457 |
|
6,752,169 |
|
7,018,415 |
| ||||
|
Income tax expense |
|
— |
|
— |
|
— |
|
— |
| ||||
|
Net income attributable to common stockholders |
|
$ |
2,904,691 |
|
$ |
2,999,457 |
|
$ |
6,752,169 |
|
$ |
7,018,415 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
Basic net income per common share |
|
$ |
0.09 |
|
$ |
0.10 |
|
$ |
0.21 |
|
$ |
0.22 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
Diluted net income per common share |
|
$ |
0.09 |
|
$ |
0.09 |
|
$ |
0.20 |
|
$ |
0.21 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
Shares used in computing basic net income per common share |
|
30,743,966 |
|
31,022,557 |
|
31,589,192 |
|
32,083,752 |
| ||||
|
|
|
|
|
|
|
|
|
|
| ||||
|
Shares used in computing diluted net income per common share |
|
32,489,969 |
|
32,604,123 |
|
32,949,779 |
|
33,353,631 |
| ||||
|
|
|
|
|
|
|
|
|
|
| ||||
|
Comprehensive income |
|
$ |
2,904,691 |
|
$ |
2,999,457 |
|
$ |
6,752,169 |
|
$ |
7,018,415 |
|
|
|
|
2013 |
| ||||||||||
|
|
|
1st Quarter |
|
2nd Quarter |
|
3rd Quarter |
|
4th Quarter |
| ||||
|
Revenue |
|
|
|
|
|
|
|
|
| ||||
|
Licensed revenue |
|
$ |
1,415,000 |
|
$ |
1,651,000 |
|
$ |
2,583,000 |
|
$ |
4,673,000 |
|
|
Total revenue |
|
1,415,000 |
|
1,651,000 |
|
2,583,000 |
|
4,673,000 |
| ||||
|
Operating expenses |
|
7,217,983 |
|
5,654,378 |
|
7,364,190 |
|
6,869,308 |
| ||||
|
Loss before income tax benefit |
|
(5,777,932 |
) |
(3,987,996 |
) |
(4,767,193 |
) |
(2,175,178 |
) | ||||
|
|
|
|
|
|
|
|
|
|
| ||||
|
Income tax expense |
|
— |
|
— |
|
— |
|
— |
| ||||
|
Net loss attributable to common stockholders |
|
$ |
(5,777,932 |
) |
$ |
(3,987,996 |
) |
$ |
(4,767,193 |
) |
$ |
(2,175,178 |
) |
|
|
|
|
|
|
|
|
|
|
| ||||
|
Basic and diluted net loss per common share |
|
$ |
(0.19 |
) |
$ |
(0.13 |
) |
$ |
(0.16 |
) |
$ |
(0.07 |
) |
|
|
|
|
|
|
|
|
|
|
| ||||
|
Shares used in computing basic and diluted net loss per common share |
|
30,336,398 |
|
30,403,670 |
|
30,476,562 |
|
30,353,631 |
| ||||
|
|
|
|
|
|
|
|
|
|
| ||||
|
Comprehensive Loss |
|
$ |
(5,774,679 |
) |
$ |
(3,987,996 |
) |
$ |
(4,767,193 |
) |
$ |
(2,175,178 |
) |
Because of the method used in calculating per share data, the quarterly per share data will not necessarily add to the per share data as computed for the year.
POZEN Inc.
(Unaudited)
|
|
|
March 31, |
|
December |
| ||
|
ASSETS |
|
|
|
|
| ||
|
Current assets: |
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
43,904,945 |
|
$ |
40,582,415 |
|
|
Investments in warrants |
|
— |
|
2,678,773 |
| ||
|
Accounts receivable |
|
4,405,319 |
|
5,629,209 |
| ||
|
Prepaid expenses and other current assets |
|
511,650 |
|
583,061 |
| ||
|
Total current assets |
|
48,821,914 |
|
49,473,458 |
| ||
|
Property and equipment, net of accumulated depreciation |
|
24,963 |
|
27,382 |
| ||
|
Noncurrent deferred tax asset |
|
— |
|
952,900 |
| ||
|
Total assets |
|
$ |
48,846,877 |
|
$ |
50,453,740 |
|
|
|
|
|
|
|
| ||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
| ||
|
Current liabilities: |
|
|
|
|
| ||
|
Accounts payable |
|
$ |
202,164 |
|
$ |
606,948 |
|
|
Accrued compensation |
|
510,761 |
|
1,899,456 |
| ||
|
Accrued expenses |
|
829,921 |
|
253,624 |
| ||
|
Current deferred tax liability |
|
— |
|
952,900 |
| ||
|
Total current liabilities |
|
1,542,846 |
|
3,712,928 |
| ||
|
|
|
|
|
|
| ||
|
|
|
|
|
|
| ||
|
Preferred stock, $0.001 par value; 10,000,000 shares authorized, issuable in series, of which 90,000 shares are designated Series A Junior Participating Preferred Stock, none outstanding |
|
— |
|
— |
| ||
|
Common stock, $0.001 par value, 90,000,000 shares authorized; 32,322,057 and 32,221,397 shares issued and outstanding at March 31, 2015 and December 31, 2014, respectively |
|
32,322 |
|
32,221 |
| ||
|
Additional paid-in capital |
|
144,203,500 |
|
143,613,024 |
| ||
|
Accumulated deficit |
|
(96,931,791 |
) |
(96,904,433 |
) | ||
|
Total stockholders’ equity |
|
47,304,031 |
|
46,740,812 |
| ||
|
Total liabilities and stockholders’ equity |
|
$ |
48,846,877 |
|
$ |
50,453,740 |
|
See accompanying Notes to Financial Statements.
POZEN Inc.
STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(Unaudited)
|
|
|
Three months ended March |
| ||||
|
|
|
2015 |
|
2014 |
| ||
|
Revenue: |
|
|
|
|
| ||
|
Licensing revenue |
|
$ |
4,404,463 |
|
$ |
7,548,676 |
|
|
|
|
|
|
|
| ||
|
Operating expenses: |
|
|
|
|
| ||
|
Selling, general and administrative |
|
3,262,320 |
|
2,823,518 |
| ||
|
Research and development |
|
983,511 |
|
1,827,878 |
| ||
|
Total operating expenses |
|
4,245,831 |
|
4,651,396 |
| ||
|
|
|
|
|
|
| ||
|
Interest and other (loss) income |
|
(185,990 |
) |
7,411 |
| ||
|
(Loss) income before income tax expense |
|
(27,358 |
) |
2,904,691 |
| ||
|
Income tax expense |
|
— |
|
— |
| ||
|
Net (loss) income attributable to common stockholders |
|
$ |
(27,358 |
) |
$ |
2,904,691 |
|
|
Basic net (loss) income per common share |
|
$ |
0.00 |
|
$ |
0.09 |
|
|
Shares used in computing basic net(loss) income per common share |
|
32,259,570 |
|
30,743,966 |
| ||
|
Diluted net (loss) income per common share |
|
$ |
0.00 |
|
$ |
0.09 |
|
|
Shares used in computing diluted net (loss) income per common share |
|
32,259,570 |
|
32,489,969 |
| ||
|
Comprehensive (loss) income |
|
$ |
(27,358 |
) |
$ |
2,904,691 |
|
See accompanying Notes to Financial Statements.
POZEN Inc.
(Unaudited)
|
|
|
Three months ended |
| ||||
|
|
|
2015 |
|
2014 |
| ||
|
Operating activities |
|
|
|
|
| ||
|
Net (loss) income |
|
$ |
(27,358 |
) |
$ |
2,904,691 |
|
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
| ||
|
Depreciation |
|
4,971 |
|
4,955 |
| ||
|
Loss on sale of warrants |
|
199,373 |
|
— |
| ||
|
Noncash compensation expense |
|
476,517 |
|
756,035 |
| ||
|
Changes in operating assets and liabilities: |
|
|
|
|
| ||
|
Accounts receivable |
|
1,223,890 |
|
(2,875,676 |
) | ||
|
Prepaid expenses and other current assets |
|
71,411 |
|
83,618 |
| ||
|
Accounts payable and other accrued expenses |
|
(1,217,182 |
) |
(3,931,384 |
) | ||
|
Deferred Revenue |
|
— |
|
(3,000,000 |
) | ||
|
|
|
|
|
|
| ||
|
Net cash provided by (used in) operating activities |
|
731,622 |
|
(6,057,761 |
) | ||
|
|
|
|
|
|
| ||
|
Investing activities |
|
|
|
|
| ||
|
Purchase of equipment |
|
(2,552 |
) |
— |
| ||
|
Proceeds from sale of warrants |
|
2,479,400 |
|
— |
| ||
|
|
|
|
|
|
| ||
|
Net cash provided by investing activities |
|
2,476,848 |
|
— |
| ||
|
|
|
|
|
|
| ||
|
Financing activities |
|
|
|
|
| ||
|
Proceeds from issuance of common stock |
|
285,022 |
|
559,962 |
| ||
|
Payments related to net settlement of stock-based awards |
|
(170,962 |
) |
(186,483 |
) | ||
|
Net cash provided by financing activities |
|
114,060 |
|
373,479 |
| ||
|
|
|
|
|
|
| ||
|
Net increase (decrease) in cash and cash equivalents |
|
3,322,530 |
|
(5,684,282 |
) | ||
|
Cash and cash equivalents at beginning of period |
|
40,582,415 |
|
32,827,732 |
| ||
|
Cash and cash equivalents at end of period |
|
$ |
43,904,945 |
|
$ |
27,143,450 |
|
See accompanying Notes to Financial Statements.
POZEN Inc.
(Unaudited)
1. Significant Accounting Policies
General
POZEN Inc. (“we” or “POZEN” or the “Company”) was incorporated in the State of Delaware on September 25, 1996 and is operating in a single reportable segment. The Company has been a pharmaceutical company committed to transforming medicine that transforms lives. Since inception, the Company has focused its efforts on developing products which can provide improved efficacy, safety or patient convenience in the treatment of acute and chronic pain and pain related conditions and has developed a portfolio of integrated aspirin therapies. Historically, the Company has entered into collaboration agreements to commercialize its product candidates. The Company’s licensing revenues include upfront payments, additional payments if and when certain milestones in the product’s development or commercialization are reached, and the eventual royalty payments based on product sales.
We decided to retain ownership of our PA product candidates which contain a combination of a proton pump inhibitor and enteric coated aspirin in a single tablet through the clinical development and pre-commercialization stage and our chief commercial officer was responsible for developing the commercialization strategy for these products and conducting all the required pre-commercialization activities. On September 3, 2013, we entered into an exclusive license agreement with sanofi-aventis U.S. LLC, or Sanofi US, for the commercialization of POZEN’s proprietary, investigational, coordinated-delivery tablets combining immediate-release omeprazole, a proton pump inhibitor, or PPI, and enteric-coated, or EC, aspirin in a single tablet, now known as YOSPRALA 81/40 and 325/40 (“PA” or “YOSPRALA”), including PA8140 and PA32540. On November 29, 2014, we executed a termination agreement with Sanofi US terminating the license agreement for PA. As of the termination date, all licenses granted to Sanofi US were terminated and all rights to the products licensed to Sanofi US under the agreement reverted to us. The termination agreement further provides for the transfer of specified commercial know-how developed by Sanofi US relating to the PA products to us and allows us, and any future collaborators, to use this know-how to commercialize the products. We are currently evaluating all strategic options available to us now that we have full ownership of the PA products. In light of the warning letter sent to our active ingredient supplier and of the time requirements necessary to complete an assessment of its strategic options, and to properly prepare the market for the launch of our YOSPRALA product candidates, we believe the products will be available for commercialization in 2016.
With respect to future products we may develop, we have decided that we will no longer commit substantial resources to further drug development without a partner who agrees to pay the full cost of that development. Consistent with this model, we have reduced our R&D staff and other costs and expenses as our PA development program activities wind down and intend to continue to reduce staff that is no longer required to support our then current business activities. Our board of directors and management team continue to explore potential ways to return value to our stockholders. On November 21, 2013, our Board of Directors declared a special cash distribution of $1.75 per share to all stockholders of record as of the close of business on December 11, 2013, with a payment date of December 30, 2013. This distribution represented a surplus of corporate cash and is accounted for as a return of capital to stockholders. We are committed to return as much cash to our stockholders as is prudent and may consider other cash distributions in the future.
Basis of Presentation
The accompanying financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America for interim financial reporting and the instructions to Form 10-Q and do not include all of the information and footnotes required for complete financial statements. In the opinion of the Company’s management, all adjustments (consisting of normal recurring adjustments) considered necessary for a fair presentation of the results for the interim periods have been included. Operating results for the three months ended March 31, 2015 are not necessarily indicative of the results for the year ending December 31, 2015 or future periods. The accompanying financial statements should be read in conjunction with the Company’s audited financial
statements and related notes included in the Company’s Annual Report on Form 10-K filed on March 11, 2015 and available on the website of the United States Securities and Exchange Commission (www.sec.gov). The accompanying balance sheet as of December 31, 2014 has been derived from the audited balance sheet as of that date included in the Form 10-K.
2. Summary of Significant Accounting Policies
Use of Estimates— The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts in the financial statements and accompanying notes. Actual results could differ from the estimates and assumptions used.
Accrued expenses, including contracted costs— Significant management judgments and estimates must be made and used in connection with accrued expenses, including those related to contract costs, such as costs associated with clinical trials. Specifically, the Company must make estimates of costs incurred to date but not yet paid for or not yet invoiced in relation to contracted, external costs. The Company analyzes the progress of product development, clinical trial and related activities, invoices received, amounts paid, and budgeted costs when evaluating the adequacy of the accrued liability for these related costs.
The Company believes that its current assumptions and other considerations used to estimate accrued expenses for the period are appropriate. However, determining the date on which certain contract services commence, the extent of services performed on or before a given date and the cost of such, paid and unpaid, involves subjective judgments and estimates and often must be based upon information provided by third parties. In the event that management does not identify certain contract costs which have begun to be incurred or under- or over-estimates the extent of services performed or the costs of such services, management adjusts costs during the period in which the information becomes available.
Accrued costs related to product development and operating activities, including clinical trials, based upon the progress of these activities covered by the related contracts, invoices received and estimated costs totaled $0.8 million at March 31, 2015 and $0.9 million at March 31, 2014. The variance, at each of these ending periods, between the actual expenses incurred and the estimated expenses accrued was not material or significant.
Revenue Recognition— The Company records revenue under the following categories: sale of royalty rights and, licensing revenues consisting of royalty revenues and other licensing revenues.
With regard to royalty revenues, the Company’s licensing agreements have terms that include royalty payments based on the manufacture, sale or use of the Company’s products or technology. VIMOVO® (naproxen and esomeprazole magnesium) delayed release tablets royalty revenue has been recognized when earned, as will any other future royalty revenues. For VIMOVO or those future arrangements where royalties are reasonably estimable, the Company recognizes revenue based on estimates of royalties earned during the applicable period and reflects in future revenue any differences between the estimated and actual royalties. These estimates are based upon information reported to the Company by its collaboration partners. During the three months ended March 31, 2015 and March 31, 2014 the Company recognized $4.4 million and $4.5 million, respectively, for VIMOVO royalty revenue.
Also, with regard to the licensing revenues, the Company’s licensing agreements have had terms that include upfront payments upon contract signing and additional payments if and when certain milestones in the product’s development or commercialization are reached. Historically, the non-refundable portion of upfront payments received under the Company’s existing agreements is deferred by the Company upon receipt and recognized on a straight-line basis over periods ending on the anticipated date of regulatory approvals, as specified in the agreements relating to the product candidates, or the conclusion of any obligation on the part of the Company. If regulatory approvals or other events relating to our product candidates are accelerated, delayed or not ultimately obtained, then the amortization of revenues for these products is prospectively accelerated or reduced accordingly. Milestone payments along with the refundable portions of up-front payments are recognized as licensing revenue upon the achievement of specified milestones if (i) the milestone is substantive in nature and the achievement of the
milestone was not reasonably assured at the inception of the agreement; and (ii) the fees are non-refundable. Any milestone payments received prior to satisfying these revenue recognition criteria are recorded as deferred revenue.
In September 2013, the Company announced the signing of an exclusive license agreement its PA products, including, PA8140 and PA32540, in the United States to commercialize all PA combinations that contain 325 mg or less of enteric-coated aspirin in the United States. On November 29, 2014, we executed a termination agreement with Sanofi US terminating the license. As of the termination date, all licenses granted to Sanofi US were terminated and all rights to the products licensed to Sanofi US under the agreement reverted to us. The Company received an upfront payment of $15.0 million which was included within the license revenue and was completely amortized by the end of the 2014 fiscal year. The revenue for the three months ended March 31, 2014 was $3.0 million.
On March 21, 2011, the Company entered into a license agreement with Cilag GmbH International (“Cilag”) a division of Johnson & Johnson, for the exclusive development and commercialization of MT 400 in Brazil, Colombia, Ecuador and Peru. Cilag’s upfront payment of $257,300 was deferred until the licensing agreement’s termination and was included in other licensing revenue for the fiscal year ended December 31, 2014.
Cash, Cash Equivalents, Investments and Concentration of Credit Risk — Cash is invested in open-ended money market mutual funds, interest-bearing investment-grade debt securities and insured bank deposits. Cash is restricted to the extent of a $42,000 letter of credit in compliance with the terms of the Company’s office lease. The Company considers all highly liquid investments with maturities of 90 days or less when purchased to be cash equivalents.
The Company invests in high-credit quality investments in accordance with its investment policy, which attempts to minimize the possibility of loss. However, cash and cash equivalents include financial instruments that potentially subject the Company to a concentration of credit risk. Cash and cash equivalents are of a highly liquid nature and are held with high credit quality financial institutions and money market mutual fund managers. Cash held directly with financial institutions is insured up to $250,000 per account and any excess amounts are uninsured. Cash is also held in insured bank deposits through a cash management program that offers a bank network ensuring full FDIC insurance on all deposits. Approximately 87% of the Company’s cash and cash equivalents are held in fully insured bank deposits and approximately 13% by money market mutual fund managers.
In connection with its acquisition of all rights, title and interest to develop, commercialize and sell Treximet® (sumatriptan / naproxen sodium) from GlaxoSmithKline (“GSK”), Pernix Therapeutics Holdings, Inc. (“Pernix”) issued the Company a warrant to purchase 500,000 shares of Pernix common stock at an exercise price of $4.28 (the closing price of Pernix common stock as listed on the NASDAQ Global Market on May 13, 2014). The common stock underlying the warrant will be registered by Pernix with the Securities and Exchange Commission and the warrant is exercisable from August 20, 2014, the closing date of the acquisition, until February 28, 2018.
The warrant was sold in the first quarter of 2015 and the Company received $2,479,400 from the sale. The Company recognized a loss of $199,373 in the March 31, 2015 financial statements.
The following table sets forth our financial instruments carried at fair value as of March 31, 2015 and December 31, 2014:
|
|
|
Financial Instruments |
| ||||
|
|
|
Carried at Fair Value |
| ||||
|
|
|
March 31, |
|
December |
| ||
|
Assets: |
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
43,904,945 |
|
$ |
40,582,415 |
|
|
Investments in Pernix warrants |
|
— |
|
2,678,773 |
| ||
|
Total cash and investments |
|
$ |
43,904,945 |
|
$ |
43,261,188 |
|
Fair Value of Financial Instruments
Financial instruments consist of cash and cash equivalents, short-term investments, accounts receivable and accounts payable. The carrying values of these amounts approximate the fair value due to their short-term nature.
Fair Value Measurement
The Company defines fair value (“FV”) as the price that would be received to sell an asset or paid to transfer a liability (“the exit price”) in an orderly transaction between market participants at the measurement date. The FV hierarchy for inputs maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. The Company uses the following hierarchy of inputs to measure FV:
· Level 1 - quoted prices in active markets for identical assets and liabilities.
· Level 2 - observable inputs other than quoted prices in active markets for identical assets and liabilities, including quoted prices in active markets for instruments that are similar or quoted prices in markets that are not active for identical or similar instruments and model-derived valuations in which all significant inputs and value drivers are observable in active markets.
· Level 3 - unobservable inputs that are significant to the overall valuation, for which there is little or no market data available and which require the Company to develop its own assumptions.
The Company values investments using the most observable inputs available that are current as of the measurement date and classifies them according to the lowest level of inputs used. Observable inputs are inputs that market participants would use in pricing the asset or liability developed from market data obtained from independent sources. Unobservable inputs are inputs that reflect the Company’s judgment concerning the assumptions that market participants would use in pricing the asset or liability developed from the best information available under the circumstances.
The financial assets for which we perform recurring measurements are cash equivalents and investments in warrants. As of March 31, 2015, financial assets utilizing Level 1 inputs included cash equivalents. Financial assets utilizing Level 2 inputs included investments in warrants.
Fair value is a market-based measure considered from the perspective of a market participant who holds the asset or owes the liability rather than an entity-specific measure. Therefore, even when market assumptions are not readily available, our own assumptions are set to reflect those that market participants would use in pricing the asset or liability at the measurement date.
Our Level 1 valuations are based on the market approach and consist primarily of quoted prices for identical items on active securities exchanges. Our Level 2 valuations may also use the market approach and are based on significant other observable inputs such as quoted prices for financial instruments not traded on a daily basis. We did not rely on Level 3 input for valuation of our securities at March 31, 2015.
Stock-based Compensation— Stock-based compensation cost is estimated at the grant date based on the fair value of the award and is recognized as expense over the requisite service period of the award. The fair value of restricted stock awards is determined by reference to the fair market value of our common stock on the date of grant. We use the Black-Scholes model to value service condition and performance condition option awards. For awards with only service conditions and graded-vesting features, we recognize compensation cost on a straight-line basis over the requisite service period. For awards with performance conditions granted we recognize compensation cost over the expected period to achieve the performance conditions, provided achievement of the performance conditions are deemed probable.
Our Statements of Comprehensive (Loss) Income for the three months ended March 31, 2015 and 2014 include the following stock-based compensation expense:
|
|
|
Three months ended March 31, |
| ||||
|
|
|
2015 |
|
2014 |
| ||
|
Research and development |
|
$ |
55,104 |
|
$ |
141,182 |
|
|
General and administrative |
|
421,413 |
|
614,853 |
| ||
|
Total expense |
|
$ |
476,517 |
|
$ |
756,035 |
|
Unrecognized stock-based compensation expense, including time-based options, performance-based options and restricted stock awards, expected to be recognized over an estimated weighted-average amortization period of 2.0 years, was $3.7 million at March 31, 2015.
Stock Plans
In 1996, the Company established a Stock Option Plan (the “Option Plan”) and authorized the issuance of options to attract and retain quality employees and to allow such employees to participate in the growth of the Company. In June 2000, the stockholders approved the POZEN Inc. 2000 Equity Compensation Plan (the “2000 Plan”) and the 2000 Plan became effective upon the completion of the Company’s initial public offering in October 2000, after which time no further grants were made under the Option Plan. In May 2004, the stockholders approved an amendment to and restatement of the 2000 Plan. The amendment to the 2000 Plan provided for an increase in the number of shares of common stock authorized for issuance under the 2000 Plan from 3,000,000 to 5,500,000 shares. In June 2007, the stockholders approved the amendment and restatement of the 2000 Plan to, among other things, increase the number of shares authorized for issuance under the 2000 Plan to 6,500,000 shares and continue the various performance criteria for use in establishing specific vesting targets for certain awards. In June 2010, stockholders approved the POZEN Inc. 2010 Equity Compensation Plan, (“the 2010 Plan”), a successor incentive compensation plan to the 2000 Plan which was merged with and into the 2010 Plan and all grants outstanding under the 2000 Plan were issued or transferred under the 2010 Plan.
The 2010 Plan provides for grants of incentive stock options, nonqualified stock options, stock awards, and other stock-based awards, such as restricted stock units and stock appreciation rights (“SARs”), to employees, non-employee directors, and consultants and advisors who perform services for us and our subsidiaries. The 2010 Plan authorizes up to 7,452,327 shares of common stock for issuance, which includes 2,000,000 shares of our common stock which were in excess of the number of shares previously reserved under the 2000 Plan. The maximum number of shares for which any individual may receive grants in any calendar year is 1,000,000 shares. The Compensation Committee of the Board of Directors, which administers the 2010 Plan, will determine the terms and conditions of options, including when they become exercisable. Neither our Board nor the Committee can amend the 2010 Plan or options previously granted under the Plan to permit a repricing of options or SARs, without prior stockholder approval. If options granted under the 2010 Plan expire or are terminated for any reason without being exercised, or if stock awards, performance units, or other stock-based awards are forfeited or otherwise terminate, the shares of common stock underlying the grants will again be available for awards granted under the 2010 Plan.
As a result of a December 31, 2013 cash dividend distribution, a dividend equivalent totaling 987,000 shares was provided to all outstanding grants. The adjustments were in the form of additional RSUs to RSU holders or an adjustment to both the outstanding number of options and their strike price, in compliance with Sections 409A and 424 of the Internal Revenue Code.
Time-Based Stock Awards
No new time-based awards were granted during the three months ended March 31, 2015 and March 31, 2014. Previously, the fair value of each time-based award was estimated on the date of grant using the Black-Scholes option valuation model. Historically, the expected volatility rate was estimated based on an equal weighting of the historical volatility of POZEN common stock over approximately a six-year period, the expected term was based upon average historical terms to exercise and the risk-free interest rate was based on six-year U.S. Treasury securities. The pre-vesting forfeiture rates used were also based on historical rates. We adjust the estimated forfeiture rate based upon actual experience.
A summary of the time-based stock awards as of March 31, 2015, and changes during the three months ended March 31, 2015, are as follows:
|
Time-Based Stock Awards |
|
Underlying |
|
Weighted- |
|
Average |
|
Aggregate |
| ||
|
Outstanding at December 31, 2014 |
|
2,341 |
|
$ |
7.39 |
|
4.1 |
|
$ |
4,382 |
|
|
Granted |
|
— |
|
— |
|
|
|
|
| ||
|
Exercised |
|
(65 |
) |
4.09 |
|
|
|
|
| ||
|
Forfeited or expired |
|
— |
|
— |
|
|
|
|
| ||
|
Outstanding at March 31, 2015 |
|
2,277 |
|
7.48 |
|
3.8 |
|
$ |
3,796 |
| |
|
Exercisable at March 31, 2015 |
|
2,095 |
|
$ |
7.79 |
|
3.5 |
|
$ |
3,098 |
|
|
Vested or expected to vest at March 31, 2015 |
|
2,249 |
|
$ |
7.48 |
|
3.8 |
|
$ |
3,751 |
|
The aggregate intrinsic value of options outstanding represents the pretax value (the period’s closing market price, less the exercise price, times the number of in-the-money options) that would have been received by all option holders had they exercised their options at the end of the period. A total of 64,821 stock options were exercised during the three months ended March 31, 2015 with an intrinsic value of $228,000, and a total of 101,315 stock options were exercised during the three months ended March 31, 2014 with an intrinsic value of $284,000.
A summary of the time-based nonvested awards as of March 31, 2015, and changes during the three months ended March 31, 2015, are as follows:
|
|
|
Underlying |
|
Weighted- |
| |
|
|
|
|
|
|
| |
|
Nonvested outstanding at March 31, 2014 |
|
477 |
|
$ |
3.85 |
|
|
Granted |
|
— |
|
— |
| |
|
Forfeited or expired |
|
— |
|
— |
| |
|
Vested |
|
(296 |
) |
4.47 |
| |
|
Nonvested outstanding at March 31, 2015 |
|
181 |
|
$ |
3.87 |
|
Restricted Stock and Restricted Stock Units
For the quarters ended March 31, 2015 and 2014, the Company recognized $374,000 and $255,000, respectively, in compensation expense related to restricted stock units.
A summary of the restricted stock awards as of March 31, 2015, and changes during the three months ended March 31, 2015, are as follows:
|
|
|
Underlying |
|
Weighted- |
| |
|
|
|
|
|
|
| |
|
Restricted stock outstanding at December 31, 2014 |
|
1,109 |
|
$ |
7.14 |
|
|
Granted |
|
— |
|
— |
| |
|
Vested and released |
|
(49 |
) |
7.17 |
| |
|
Forfeited or expired |
|
— |
|
— |
| |
|
Restricted stock outstanding at March 31, 2015 |
|
1,060 |
|
$ |
7.14 |
|
As of March 31, 2015 there was an aggregate $3.5 million of unrecognized compensation expense related to unvested restricted stock units. There were 522,000 unvested restricted stock units outstanding at March 31, 2015 and 364,000 unvested restricted stock units outstanding at March 31, 2014. The total fair value of restricted stock that vested during the quarters ended March 31, 2015 and 2014 was $363,000 and $491,000, respectively.
Performance-Based Awards
In May 2008, pursuant to an incentive program (the “PN incentive program”) approved by the Compensation Committee of the Board of Directors of the Company, stock options were granted to all of the Company’s employees, including its executive officers, to purchase an aggregate of 281,433 shares of common stock with an exercise price of $14.45 per share. In September 2008, additional stock options were granted under the PN incentive program, to purchase 11,700 shares of common stock at an exercise price of $10.82 per share. The stock options have a ten-year term and have an exercise price equal to the closing sale price of the Company’s common stock, as reported on the NASDAQ Global Market, on the date immediately preceding the date of grant. Twenty-five percent (25%) of the PN incentive program options granted vested in 2009, upon completion of the performance goal and the remaining seventy-five percent (75%) of the options granted vested in 2010 upon the completion of the remaining performance goals. The fair value of the performance-based options granted under the PN incentive program was estimated as of the grant date using the Black-Scholes option valuation model without consideration of the performance measures. The options also include provisions that require satisfactory employee performance prior to vesting. Additionally, 20,000 options were granted to an executive officer in May 2008 under the PN incentive plan, with similar grant and exercise terms. The Company recognized compensation costs for these awards over the expected service period.
In October 2011, pursuant to an incentive program (the “PA32540 incentive program”) approved by the Compensation Committee of the Board of Directors of the Company, stock options were granted to all of the Company’s employees, including its executive officers, to purchase an aggregate of 453,960 shares of common stock. The underlying stock options and RSUs were performance-based and focus on the successful completion of certain value-enhancing events for the Company’s Yosprala product candidate. The stock options have a ten-year term and have an exercise price equal to the closing sale price of the Company’s common stock, as reported on the NASDAQ Global Market, on the date immediately preceding the date of grant. The underlying stock options and RSUs vest in accordance with the following schedule: (a) one-third (1/3) upon the acceptance of the filing of a new drug application (the “NDA”) for Yosprala, assuming the NDA filing is made prior to December 31, 2012, (b) one-third (1/3) upon first cycle NDA approval of Yosprala (otherwise 16.5% upon NDA approval after first cycle), and (c) one-third (1/3) upon execution of a significant partnering transaction for Yosprala in a major territory as determined by the Compensation Committee of the Company, in its sole discretion, at the time of such transaction, subject in each case to continued employment or service to the Company.
During a pre-submission meeting with respect to its NDA for Yosprala in April 2012, the FDA suggested that the Company also seek approval for a lower dose formulation of the product containing 81 mg of enteric coated aspirin as part of its NDA for Yosprala. The Company decided to include data and information relating to a lower dose formulation in its NDA. Generation of additional data with respect to lower dose formulation of Yosprala and incorporation of data into the NDA for Yosprala would delay submission of the NDA from the original planned submission date.
Therefore, in October 2012, the Compensation Committee granted performance-based incentive awards (the “PA8140 incentive program”) both to compensate the employees for the expected loss of value under the PA32540 Incentive Program, as well as to provide additional incentive to employees to complete the value-added activities required for submission and approval of the lower dose product. The Compensation Committee granted an aggregate of 208,740 restricted stock units to various employees of the Company, including 105,000 restricted stock units granted to the Company’s named executive officers. The restricted stock units were performance-based and vest in accordance with the following schedule: (a) one-half (1/2) upon the acceptance by the FDA of the filing of an NDA for a lower dose Yosprala product candidate, and (b) one-half (1/2) upon approval by the FDA of an NDA for a lower dose Yosprala product candidate. In 2012, 132,883 options were forfeited in acknowledgement that certain performance goals would not be met under the PA32540 incentive program.
In April 2014, the Compensation Committee granted an aggregate of 73,000 restricted stock units to various employees of the Company, including 65,000 restricted stock units granted to the Company’s named executive officers. The restricted stock units were performance-based and vest in accordance with the following schedule: (i) 50% upon receipt of the milestone payment by Sanofi US under the License and Collaboration Agreement, dated as of September 3, 2013 (the “Agreement”) to be received upon approval by the U.S. Food and Drug Administration of the PA product candidates; and (ii) 50% upon receipt of the milestone payment by Sanofi US upon achievement of commercial readiness (as defined in the Agreement). The entire award was forfeited in 2014 upon the termination of the Sanofi US agreement. In 2014, a total of 177,818 options were forfeited in
acknowledgement that certain performance goals would not be met under the PA32540 and PA8140 incentive programs.
As of March 31, 2015, there was $4,000 in unrecognized compensation expense related to performance-based awards granted under the PA32540 and PA8140 incentive programs.
A summary of the performance-based stock awards as of March 31, 2015, and changes during the three months ended March 31, 2015, are as follows:
|
|
|
Underlying |
|
Weighted- |
| |
|
|
|
|
|
|
| |
|
Performance-based outstanding at December 31, 2014 |
|
368 |
|
$ |
8.12 |
|
|
Granted |
|
— |
|
— |
| |
|
Exercised |
|
(10 |
) |
1.98 |
| |
|
Forfeited or expired |
|
— |
|
— |
| |
|
Performance-based outstanding at March 31, 2015 |
|
358 |
|
$ |
8.29 |
|
The March 31, 2015 amount is expected to be recognized, at the time of the grant vesting, over the period ending in fourth quarter 2015. Under the PA32540 and PA8140 incentive programs, there were 139,000 unvested performance-based options outstanding at March 31, 2015. No performance-based awards vested during the three months ended March 31, 2015 and March 31, 2014. There were 289,000 vested performance-based options outstanding at March 31, 2014. No awards forfeited during the three months ended March 31, 2015 and 11,000 awards forfeited during the three months ended March 31, 2014. A total of 10,000 performance-based awards were exercised during the three months ended March 31, 2015 and 6,000 performance-based awards were exercised during the three months ended March 31, 2014. At March 31, 2015, the performance-based options had an intrinsic value of $1.2 million and a remaining weighted contractual life of 4.9 years.
Net Income (Loss) Per Share— Basic and diluted net income or loss per common share amounts have been computed using the weighted-average number of shares of common stock outstanding for the three months ended March 31, 2015 and 2014. During the three months ended March 31, 2015 and 2014, the Company had potential common stock equivalents related to its outstanding stock options and restricted stock units. These outstanding stock options and restricted stock units were awarded under the Company’s stock option plans and they have vested or may vest to the option holder upon the completion of predetermined service periods or performance criteria. Vested awards are eligible for conversion into common stock. These potential common stock equivalents were not included in diluted net loss per common share amounts, during the three months ended March 31, 2015, since the effect would have been antidilutive. The Company has excluded the impact of any shares which might be issued under the Rights Plan, detailed below, from the earnings per share calculation because the Rights are not exercisable since the specified contingent future event has not occurred.
Reconciliation of denominators for basic and diluted earnings per share computations:
|
|
|
Three months ended March |
| ||
|
|
|
2015 |
|
2014 |
|
|
Basic weighted average shares outstanding |
|
32,259,570 |
|
30,743,966 |
|
|
Effect of dilutive employee and director awards |
|
— |
|
1,746,003 |
|
|
Diluted weighted-average shares outstanding and assumed conversions |
|
32,259,570 |
|
32,489,969 |
|
If there is any change in the number or kind of shares of company stock outstanding or if the value of outstanding shares of company stock is substantially reduced as a result of an extraordinary dividend or distribution, the Company’s 2010 Stock Option Plan requires that an equitable adjustment be made to all outstanding grants to
preclude dilution of rights and benefits under the plan. Therefore, as a result of the December 31, 2013 cash dividend distribution, a dividend equivalent was provided to all outstanding grants. The adjustments were in the form of additional RSUs to RSU holders or an adjustment to both the outstanding number of options and their strike price; all adjustments were made in compliance with Sections 409A and 424 of the Internal Revenue Code. In addition, the 2010 Stock Option Plan provides for an adjustment to the number of common shares available for grant under the stock option plan. Therefore, as a result of the December 31, 2013 cash dividend distribution, the number of common shares available for grant was adjusted by 416,971 shares and that increase is reflected in the table below.
At March 31, 2015, shares of our common stock reserved for future issuance are as follows:
|
Common shares available for grant under stock option plans |
|
2,474,430 |
|
|
Common shares issuable pursuant to options and restricted stock units granted under equity compensations plans |
|
3,694,404 |
|
|
|
|
|
|
|
Total Reserved |
|
6,168,834 |
|
Leases—On February 16, 2009, the Company modified certain terms to our existing lease agreement, dated November 21, 2001, relating to approximately 17,009 square feet of office space located at Exchange Office Building, Chapel Hill, North Carolina. Under the terms of the modification, the lease term was extended for an additional 5 years and 7 months, terminating on September 30, 2015. The modification also provides the Company with a reduced notice period of 7 months for renewals of the lease. The Company is also entitled to a 3-year lease extension option available at the end of the term and a first offer right on available space located within the Exchange Office Building property. As a result of entering into the modification, the Company’s noncancellable future minimum lease payments for operating leases increased by approximately $2.7 million over the lease term. The Company is recognizing rent expense on a straight-line basis over the term of the lease which resulted in a deferred rent balance of $42,900 at March 31, 2015.
New Accounting Pronouncements— Revenue from Contracts with Customers: In May 2014, the FASB issued new accounting rules related to revenue recognition for contracts with customers requiring revenue recognition based on the transfer of promised goods or services to customers in an amount that reflects consideration the Company expects to be entitled to in exchange for goods or services. The new rules supersede prior revenue recognition requirements and most industry-specific accounting guidance. The new rules will be effective for the Company in the first quarter of 2017 with either full retrospective or modified retrospective application required. The Company does not expect the adoption of the new accounting rules to have a material impact on the Company’s financial condition, results of operations or cash flows.
Contingencies—On March 14, 2011, we and AstraZeneca received a Paragraph IV Notice Letter from Dr. Reddy’s Laboratories, Ltd and Dr. Reddy’s Laboratories, Inc., collectively, Dr. Reddy’s informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent in 2023. The ‘907 patent is assigned to POZEN and listed with respect to VIMOVO in the Orange Book. On September 19, 2011, Dr. Reddy’s amended its ANDA to include a Paragraph IV certification against the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, which are assigned to AstraZeneca or its affiliates and listed in in the Orange Book, with respect to VIMOVO. The patents listed in the Orange Book which are owned by AstraZeneca or its affiliates expire at various times between 2014 and 2018. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Dr. Reddy’s. Accordingly, we and AstraZeneca filed suit against Dr. Reddy’s on April 21, 2011 in the United States District Court for the District of New Jersey, asserting only the ‘907 patent against Dr. Reddy’s. An amended complaint was filed on October 28, 2011 to include the AstraZeneca patents. On December 19, 2012, the District Court conducted a pre-trial “Markman” hearing to determine claim construction. On May 1, 2012, the Court issued a Markman Order construing the claim terms disputed by the parties. On April 15, 2013 a Stipulation of Partial Dismissal was filed which sought dismissal of all infringement claims relating to the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent (which are each assigned to AstraZeneca), as well as Dr. Reddy’s defenses and counterclaims relating to those patents. On April 18, 2013, the District Court issued a Stipulation and Order dismissing with prejudice those claims and defenses. The first Dr. Reddy’s case is considered the lead case
and has been consolidated with the actions described below for the purpose of pre-trial and discovery. A scheduling order for this case, and all of the consolidated cases, was issued by the Court on June 27, 2014. Fact discovery closed in the consolidated case on November 20, 2014. Expert discovery is ongoing and set to close May 21, 2015. In view of the retirement of presiding Judge Pisano, on February 9, 2015, the consolidated cases were reassigned to Judge Mary L. Cooper.
On June 13, 2011, we and AstraZeneca received a Paragraph IV Notice Letter from Lupin Ltd., or Lupin informing us that Lupin had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the POZEN and the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, the ‘466 patent and, each of which are assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Lupin’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Lupin and, accordingly, we and AstraZeneca filed suit against Lupin on July 25, 2011 in the United States District Court for the District of New Jersey. On November 19, 2014, an amended complaint was filed in which the 504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, all assigned to AstraZeneca or its affiliates, were not asserted against Lupin. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On September 19, 2011, we and AstraZeneca received Paragraph IV Notice Letter from Anchen Pharmaceuticals, Inc., or Anchen informing us that Anchen had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, the ‘085 patent, the ‘070 patent, and the ‘466 patent. The patents are among those listed with respect to VIMOVO in the Orange Book and expire at various times between 2018 and 2023. Anchen’s Paragraph IV Notice Letter asserts that its generic product will not infringe those patents or that those patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Anchen and, accordingly, we and AstraZeneca filed suit against Anchen on October 28, 2011 in the United States District Court for the District of New Jersey. On October 4, 2013, Anchen filed an amendment to its ANDA seeking to change its Paragraph IV certification to a Paragraph III. It is unclear when or if the FDA will enter Anchen’s amendment. On October 25, 2013, Anchen filed a Motion to Dismiss the case against it, based on its proposed re-certification. On November 18, 2013, we and AstraZeneca filed an Opposition to Anchen’s Motion to Dismiss. On June 11, 2014, the Court granted Anchen’s Motion and dismissed the case against them.
On November 20, 2012 we and AstraZeneca received a Paragraph IV Notice Letter from Dr. Reddy’s, informing us that Dr. Reddy’s had filed a second ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the POZEN and the 504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, the ‘466 patent and, each of which are assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Dr. Reddy’s second Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Dr. Reddy’s on its second ANDA filing and, accordingly, we and AstraZeneca filed suit against Dr. Reddy’s on January 4, 2013, in the United States District Court for the District of New Jersey. On April 15, 2013 a Stipulation of Partial Dismissal was filed which sought dismissal of all infringement claims relating to the ‘504 patent, the ‘085 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent (which are each assigned to AstraZeneca), as well as Dr. Reddy’s defenses and counterclaims relating to those patents. On April 18, 2013, the District Court issued the Stipulation and Order dismissing with prejudice those claims and defenses. On June 28, 2013 we and AstraZeneca filed a Motion for Summary Judgment relating to the second ANDA filing asserting that U.S. Patent No. 6,926,907 is not invalid. On August 12, 2013, Dr. Reddy’s filed an opposition to the Motion for Summary Judgment. On March 28, 2014, the District Court denied the Motion. On October 11, 2013, Dr. Reddy’s filed a Motion for Summary Judgment asserting that the product which is the subject matter of its second ANDA does not infringe the ‘907 patent. On November 4, 2013, POZEN and AstraZeneca filed a Motion for an Order Denying Dr. Reddy’s Motion for Summary Judgment Pursuant to Rule 56(d) and an Opposition to Dr. Reddy’s Motion. On May 29, 2014, the Court issued an order denying Dr. Reddy’s Motion. This case was consolidated with the originally filed Dr. Reddy’s case and is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On March 29, 2013, we and AstraZeneca received a Paragraph IV Notice Letter from Watson Laboratories, Inc. — Florida, or Watson, now Actavis, informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the Company and the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Watson’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Watson. On May 10, 2013, we and AstraZeneca filed a patent infringement lawsuit against Watson in the U.S. District Court of New Jersey. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On May 16, 2013, we and AstraZeneca received a Paragraph IV Notice Letter from Mylan Pharmaceuticals Inc., or Mylan informing us that it had filed an ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO before the expiration of the ‘907 patent, which is assigned to the Company and the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The patents are listed with respect to VIMOVO in the Orange Book and expire at various times between 2014 and 2023. Mylan’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement suit against Mylan. On June 28, 2013, we and AstraZeneca filed a patent infringement lawsuit against Mylan in the U.S. District Court of New Jersey. On February 13, 2015, the Court entered a joint stipulation of dismissal of counts related to certain patents, dismissing claims related to the ‘504 patent, the ‘085 patent, the ‘424 patent, the ‘872 patent, the ‘070 patent, and the ‘466 patent, each of which assigned to AstraZeneca or its affiliates. The case is currently consolidated for discovery and pretrial purposes with the first filed Dr. Reddy’s case.
On October 15, 2013, the United States Patent Office issued the ‘285 patent. The ‘285 patent, entitled “Pharmaceutical compositions for the coordinated delivery of NSAIDs” and assigned to POZEN, is related to the ‘907 patent. AstraZeneca has advised us that it has elected to exercise its first right to prosecute the infringement of the ‘285 patent and, accordingly, on October 23, 2013, we, and AstraZeneca filed patent infringement lawsuits against Dr. Reddy’s , Lupin, Watson and Mylan in the U.S. District Court of New Jersey alleging that their ANDA products infringe the ‘285 patent. On November 8, 2013, we, and AstraZeneca filed a Motion to Amend the Complaint in the actions against Dr. Reddy’s , Lupin, Watson and Mylan or, in the alternative, to consolidate the actions involving the ‘285 patent with the existing consolidated action. Dr. Reddy’s, Lupin, Watson and Mylan have each filed answers to the respective amended complaints, thus adding claims relating to the ‘285 patent against each of the Defendants to the consolidated case.
As part of the purchase of all of AstraZeneca’s rights, title and interest to develop, commercialize and sell VIMOVO in the United States, Horizon Pharma Inc., or Horizon, has assumed AstraZeneca’s right to lead the above-described Paragraph IV litigation relating to VIMOVO currently pending in the United States District Court for the District of New Jersey and has assumed patent-related defense costs relating to such litigation, including reimbursement up to specified amounts of the cost of any counsel retained by us. On December 12, 2013, Horizon filed Motions to Join under Fed.R.Civ.Proc. 25(c) as a co-plaintiff in each of the above referenced actions and the consolidated action. On January 31, 2014 and February 2, 2014, the Court granted Horizon’s motions.
On February 24, 2015, Dr. Reddy’s filed a Petition for Inter Partes Review (“IPR”) of the’285 patent with the Patent Trials and Appeals Board (“PTAB”) of the U.S. Patent and Trademark Office. On March 13, 2015, the PTAB issued its Notice of Filing Date which included a determination that Dr. Reddy’s Petition met the minimal formal filing requirements, thereby according the IPR a February 24, 2015 filing date. We and Horizon may file an optional Preliminary Response which, pursuant to the Notice of Filing Date, is due three months from the date of that notice. Upon receipt of such a Preliminary Response, the PTAB has another three months in which to institute or deny the IPR proceeding. If the PTAB decides to institute the IPR proceeding, Dr. Reddy’s will have the opportunity to challenge the validity of the ‘285 patent in whole or in part before the PTAB via a patent validity trial. We and Horizon intend to defend the validity of the ‘285 patent in both the IPR and district court settings.
In Canada, on January 20, 2015, AstraZeneca Canada Inc. (“AstraZeneca Canada”) received a Notice of Allegation from Mylan Pharmaceuticals ULC (“Mylan ULC”), informing that Mylan ULC has filed an Abbreviated New Drug Submission (ANDS) in Canada for approval of its naproxen/esomeprazole magnesium tablets and alleging non-infringement of some of the claims and invalidity of POZEN’s Canadian Patent No. 2,449,098 (“098) AstraZeneca Canada is the licensee pursuant to a Collaboration Agreement, and the 098 patent is listed in respect of AstraZeneca Canada’s VIMOVO products. A notice of Allegation is similar to a Paragraph IV Notice Letter, and in response, AstraZeneca Canada and POZEN as the patentee commenced a proceeding in the Federal Court of Canada in relation to the 098 patent on March 5, 2015. The Canadian proceeding is summary in nature and expected to be completed before March 5, 2017. The proceeding will decide whether approval for Mylan ULC’s naproxen/esomeprazole magnesium tablets will be prohibited until the expiry of the 098 patent because none of Mylan ULC’s allegations in respect of the 098 patent are justified; the proceeding will not finally decide 098 patent validity or infringement. The 098 patent expires on May 31, 2022.
On April 24, 2015, we and Horizon received a third Paragraph IV Notice Letter from Dr. Reddy’s informing us that it had amended its Paragraph IV certifications made with respect to its second ANDA with the FDA seeking regulatory approval to market a generic version of VIMOVO. Dr. Reddy’s amended certifications relate to the ‘285 patent, and United State Patent Nos. 8,852,636 (“the ‘636 patent”) and 8,858,996 (“the ‘996 patent”) which are all assigned to the Company. The patents are listed with respect to VIMOVO in the Orange Book and expire in 2022 or 2023. Dr. Reddy’s Paragraph IV Notice Letter asserts that its generic product will not infringe the listed patents or that the listed patents are invalid or unenforceable. POZEN and Horizon are in the process of assessing the nature and merits of Dr. Reddy’s claims.
As with any litigation proceeding, we cannot predict with certainty the patent infringement suit against Dr. Reddy’s, Lupin, Mylan and Watson relating to a generic version of VIMOVO. We have incurred, from the inception of these matters, an aggregate of $17.7 million in legal fees through the three months ended March 31, 2015. Furthermore, we will have to incur additional expenses in connection with the lawsuits relating to VIMOVO, which may be substantial. In the event of an adverse outcome or outcomes, our business could be materially harmed. Moreover, responding to and defending pending litigation will result in a significant diversion of management’s attention and resources and an increase in professional fees.
McGovern, Hurley, Cunningham, LLP
Chartered Accountants
|
|
|
2005 Sheppard Avenue East, Suite 300 | |
|
|
|
Toronto, Ontario | |
|
|
|
M2J 5B4, Canada | |
|
|
|
Phone |
416-496-1234 |
|
|
|
Fax |
416-496-0125 |
|
|
|
Web |
www.mhc-ca.com |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Tribute Pharmaceuticals Canada Inc.
We have audited the accompanying balance sheets of Tribute Pharmaceuticals Canada Inc. (the “Company”) as of December 31, 2014 and 2013, and the related statements of changes in shareholders’ equity, operations and comprehensive (loss), and cash flows for each of the years in the two-year period ended December 31, 2014. Tribute Pharmaceuticals Canada Inc.’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Tribute Pharmaceuticals Canada Inc. as of December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2014, in conformity with accounting principles generally accepted in the United States of America.
|
|
|
McGOVERN, HURLEY, CUNNINGHAM, LLP |
|
|
|
|
|
|
|
/s/ McGovern, Hurley, Cunningham, LLP |
|
|
|
|
|
|
|
Chartered Accountants |
|
|
|
|
|
|
|
Licensed Public Accountants |
|
TORONTO, Canada |
|
|
|
|
|
|
|
February 27, 2015 |
|
|
A member of UHY International, a network of Independent accounting and consulting firms
TRIBUTE PHARMACEUTICALS CANADA INC.
(Expressed in Canadian dollars)
|
|
|
As at |
|
As at |
| ||
|
ASSETS |
|
|
|
|
| ||
|
Current |
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
3,505,791 |
|
$ |
2,813,472 |
|
|
Accounts receivable, net of allowance of $nil (2013 - $nil) (Note 19d) |
|
2,145,319 |
|
591,766 |
| ||
|
Inventories (Note 4) |
|
1,037,387 |
|
1,044,831 |
| ||
|
Taxes recoverable |
|
130,623 |
|
651,791 |
| ||
|
Loan receivable |
|
15,814 |
|
15,814 |
| ||
|
Prepaid expenses and other receivables (Note 5) |
|
187,279 |
|
165,886 |
| ||
|
Current portion of debt issuance costs, net (Note 9) |
|
128,134 |
|
91,100 |
| ||
|
Total current assets |
|
7,150,347 |
|
5,374,660 |
| ||
|
Property, plant and equipment, net (Note 6) |
|
1,012,285 |
|
1,089,919 |
| ||
|
Intangible assets, net (Note 7) |
|
40,958,870 |
|
9,717,173 |
| ||
|
Goodwill (Note 8) |
|
3,599,077 |
|
3,599,077 |
| ||
|
Debt issuance costs, net (Note 9) |
|
359,161 |
|
253,712 |
| ||
|
Total assets |
|
$ |
53,079,740 |
|
$ |
20,034,541 |
|
|
LIABILITIES |
|
|
|
|
| ||
|
Current |
|
|
|
|
| ||
|
Accounts payable and accrued liabilities |
|
$ |
4,344,606 |
|
$ |
3,284,756 |
|
|
Current portion of long term debt (Note 9) |
|
1,319,030 |
|
204,700 |
| ||
|
Warrant liability (Note 10c) |
|
3,107,880 |
|
2,966,714 |
| ||
|
Other current liability (Note 20) |
|
— |
|
38,156 |
| ||
|
Total current liabilities |
|
8,771,516 |
|
6,494,326 |
| ||
|
Long term debt (Note 9) |
|
13,967,493 |
|
5,640,102 |
| ||
|
Total liabilities |
|
22,739,009 |
|
12,134,428 |
| ||
|
Contingencies and commitments (Notes 9 and 13) |
|
|
|
|
| ||
|
|
|
|
|
|
| ||
|
SHAREHOLDERS’ EQUITY |
|
|
|
|
| ||
|
Capital Stock |
|
|
|
|
| ||
|
AUTHORIZED |
|
|
|
|
| ||
|
Unlimited Non-voting, convertible redeemable and retractable preferred shares with no par value |
|
|
|
|
| ||
|
Unlimited Common shares with no par value |
|
|
|
|
| ||
|
ISSUED (Note 10a) |
|
|
|
|
| ||
|
Common shares 94,476,238 (2013 — 51,081,238) |
|
41,182,630 |
|
19,947,290 |
| ||
|
Additional paid-in capital options (Note 10b) |
|
2,713,605 |
|
2,286,890 |
| ||
|
Warrants (Note 10c) |
|
6,347,349 |
|
— |
| ||
|
Accumulated other comprehensive loss |
|
— |
|
(38,156 |
) | ||
|
Deficit |
|
(19,902,853 |
) |
(14,295,911 |
) | ||
|
Total shareholders’ equity |
|
30,340,731 |
|
7,900,113 |
| ||
|
Total liabilities and shareholders’ equity |
|
53,079,740 |
|
$ |
20,034,541 |
| |
See accompanying notes to the financial statements.
TRIBUTE PHARMACEUTICALS CANADA INC.
STATEMENTS OF CHANGES IN SHAREHOLDERS’ EQUITY
(Expressed in Canadian dollars)
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
|
|
|
Number of |
|
Common |
|
Warrants |
|
Additional |
|
Accumulated |
|
Deficit |
|
|
|
|
# |
|
$ |
|
$ |
|
$ |
|
$ |
|
$ |
|
|
BALANCE, January 1, 2013 |
|
39,610,042 |
|
17,589,957 |
|
— |
|
1,867,723 |
|
— |
|
(7,723,556 |
) |
|
Units issued (Note 10a) |
|
11,471,196 |
|
4,713,787 |
|
— |
|
— |
|
— |
|
— |
|
|
Options issued to employees and directors (Note 10b) |
|
— |
|
— |
|
— |
|
419,167 |
|
— |
|
— |
|
|
Broker warrants — valuation allocation (Note 10a) |
|
— |
|
(172,986 |
) |
— |
|
— |
|
— |
|
— |
|
|
Common share purchase warrants — valuation (Note 10c) |
|
— |
|
(1,746,503 |
) |
— |
|
— |
|
— |
|
— |
|
|
Share issuance costs (Note 10a) |
|
— |
|
(436,965 |
) |
— |
|
— |
|
— |
|
— |
|
|
Unrealized loss on derivative instrument |
|
— |
|
— |
|
— |
|
— |
|
(38,156 |
) |
— |
|
|
Net loss for the year |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(6,572,355 |
) |
|
December 31, 2013 |
|
51,081,238 |
|
19,947,290 |
|
— |
|
2,286,890 |
|
(38,156 |
) |
(14,295,911 |
) |
|
Units issued (Note 10a) |
|
42,895,000 |
|
30,026,500 |
|
— |
|
— |
|
— |
|
— |
|
|
Common shares issued for services (Note 10a) |
|
500,000 |
|
211,812 |
|
— |
|
426,715 |
|
— |
|
— |
|
|
Options issued to employees and directors (Note 10b) |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
Broker warrants — valuation allocation (Note 10a) |
|
— |
|
(1,177,468 |
) |
1,177,468 |
|
— |
|
— |
|
— |
|
|
Common share purchase warrants — valuation (Note 10c) |
|
— |
|
(5,169,881 |
) |
5,169,881 |
|
— |
|
— |
|
— |
|
|
Share issuance costs (Note 10a) |
|
— |
|
(2,655,623 |
) |
— |
|
— |
|
— |
|
— |
|
|
Unrealized gain on derivative instrument (Note 20) |
|
— |
|
— |
|
— |
|
— |
|
38,156 |
|
— |
|
|
Net loss for the year |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(5,606,942 |
) |
|
December 31, 2014 |
|
94,476,238 |
|
41,182,630 |
|
6,347,349 |
|
2,713,605 |
|
— |
|
(19,902,853 |
) |
See accompanying notes to financial statements.
TRIBUTE PHARMACEUTICALS CANADA INC.
COMPREHENSIVE LOSS
(Expressed in Canadian dollars)
For the Years Ended December 31,
|
|
|
2014 |
|
2013 |
| ||
|
Revenues |
|
|
|
|
| ||
|
Licensed domestic product net sales |
|
$ |
9,106,038 |
|
$ |
8,598,385 |
|
|
Other domestic product sales |
|
6,127,968 |
|
3,366,374 |
| ||
|
International product sales |
|
1,619,372 |
|
1,277,678 |
| ||
|
Royalty and licensing revenues |
|
18,414 |
|
197,924 |
| ||
|
Total revenues (Notes 14 and 17) |
|
16,871,792 |
|
13,440,361 |
| ||
|
Cost of sales |
|
|
|
|
| ||
|
Licensor sales and distribution fees |
|
5,902,034 |
|
5,844,494 |
| ||
|
Cost of products sold |
|
1,787,584 |
|
1,541,662 |
| ||
|
Write down of inventories |
|
53,099 |
|
56,935 |
| ||
|
Total cost of sales |
|
7,742,717 |
|
7,443,091 |
| ||
|
Gross Profit |
|
9,129,075 |
|
5,997,270 |
| ||
|
Expenses |
|
|
|
|
| ||
|
Selling, general and administrative (Notes 10b, 13c, 13e, 15 and 18) |
|
10,149,854 |
|
9,489,579 |
| ||
|
Amortization |
|
1,511,021 |
|
1,245,846 |
| ||
|
Total operating expenses |
|
11,660,875 |
|
10,735,425 |
| ||
|
(Loss) from operations |
|
(2,531,800 |
) |
(4,738,155 |
) | ||
|
Non-operating income (expenses) |
|
|
|
|
| ||
|
Change in warrant liability (Note 10c) |
|
283,305 |
|
(399,217 |
) | ||
|
Loss on disposal of intangible asset (Note 7) |
|
— |
|
(161,200 |
) | ||
|
Loss on extinguishment of loan (Note 9) |
|
— |
|
(620,835 |
) | ||
|
Unrealized foreign currency exchange on debt (Note 18) |
|
(1,641,238 |
) |
(340,553 |
) | ||
|
Loss on derivative instrument (Note 20) |
|
(167,511 |
) |
— |
| ||
|
Accretion expense (Note 9) |
|
(167,555 |
) |
(103,775 |
) | ||
|
Interest expense |
|
(1,441,729 |
) |
(527,079 |
) | ||
|
Interest income |
|
59,586 |
|
3,559 |
| ||
|
Loss and comprehensive loss before tax |
|
(5,606,942 |
) |
(6,887,255 |
) | ||
|
Deferred income tax recovery (Note 16) |
|
— |
|
314,900 |
| ||
|
Net (loss) for the year |
|
(5,606,942 |
) |
(6,572,355 |
) | ||
|
Unrealized loss on derivative instrument, net of tax (Note 20) |
|
— |
|
(38,156 |
) | ||
|
Total comprehensive loss |
|
$ |
(5,606,942 |
) |
$ |
(6,610,511 |
) |
|
Loss Per Share (Note 11) |
|
|
|
|
| ||
|
- Basic |
|
$ |
(0.08 |
) |
$ |
(0.13 |
) |
|
- Diluted |
|
$ |
(0.08 |
) |
$ |
(0.13 |
) |
|
Weighted Average Number of Common Shares Outstanding |
|
|
|
|
| ||
|
- Basic |
|
71,940,005 |
|
49,169,414 |
| ||
|
- Diluted |
|
71,940,005 |
|
49,169,414 |
| ||
See accompanying notes to the financial statements.
TRIBUTE PHARMACEUTICALS CANADA INC.
(Expressed in Canadian dollars)
For the Years Ended December 31,
|
|
|
2014 |
|
2013 |
| ||
|
Cash flows from (used in) operating activities |
|
|
|
|
| ||
|
Net (loss) |
|
$ |
(5,606,942 |
) |
$ |
(6,572,355 |
) |
|
Items not affecting cash: |
|
|
|
|
| ||
|
Deferred income tax recovery |
|
— |
|
(314,900 |
) | ||
|
Amortization |
|
1,541,326 |
|
1,288,509 |
| ||
|
Change in warrant liability (Note 10c) |
|
(283,305 |
) |
399,217 |
| ||
|
Stock-based compensation (Note 10b) |
|
426,715 |
|
419,167 |
| ||
|
Unrealized foreign currency loss |
|
1,641,238 |
|
340,553 |
| ||
|
Paid in common shares for services (Note 10a) |
|
211,812 |
|
— |
| ||
|
Accretion expense |
|
167,555 |
|
103,775 |
| ||
|
Loss on disposal of intangible asset (Note 7) |
|
— |
|
161,200 |
| ||
|
Loss of extinguishment of loan (Note 9) |
|
— |
|
620,835 |
| ||
|
Change in non-cash operating assets and liabilities (Note 12) |
|
13,516 |
|
(1,643,044 |
) | ||
|
Cash flows (used in) operating activities |
|
(1,888,085 |
) |
(5,197,043 |
) | ||
|
Cash flows (used in) investing activities |
|
|
|
|
| ||
|
Additions to property, plant and equipment |
|
(12,759 |
) |
(26,795 |
) | ||
|
Cash paid for intangible assets |
|
(32,573,815 |
) |
(33,345 |
) | ||
|
Cash flows (used in) investing activities |
|
(32,586,574 |
) |
(60,140 |
) | ||
|
Cash flows from (used in) financing activities |
|
|
|
|
| ||
|
Financing costs deferred |
|
(225,684 |
) |
(305,227 |
) | ||
|
Long term debt repayment (Note 9) |
|
— |
|
(3,386,630 |
) | ||
|
Long term debt issued (Note 9) |
|
8,801,241 |
|
6,084,437 |
| ||
|
Payment of contingent liabilities |
|
— |
|
(460,000 |
) | ||
|
Units issued (Note 10a) |
|
30,026,500 |
|
4,713,787 |
| ||
|
Debt extinguishment costs (Note 9) |
|
— |
|
(348,420 |
) | ||
|
Share issuance costs (Note 10a) |
|
(2,655,623 |
) |
(436,966 |
) | ||
|
Cash flows from financing activities |
|
35,946,434 |
|
5,860,981 |
| ||
|
Changes in cash and cash equivalents |
|
1,471,775 |
|
603,798 |
| ||
|
Change in cash due to changes in foreign exchange |
|
(779,456 |
) |
(74,194 |
) | ||
|
Cash and cash equivalents, beginning of year |
|
2,813,472 |
|
2,283,868 |
| ||
|
Cash and cash equivalents, end of year |
|
$ |
3,505,791 |
|
$ |
2,813,472 |
|
See accompanying notes to the financial statements.
TRIBUTE PHARMACEUTICALS CANADA INC.
NOTES TO THE FINANCIAL STATEMENTS
(Expressed in Canadian dollars)
DECEMBER 31, 2014 AND 2013
1. DESCRIPTION OF BUSINESS
Tribute Pharmaceuticals Canada Inc. (“Tribute Pharmaceuticals” or the “Company”) is an emerging Canadian specialty pharmaceutical company with a primary focus on the acquisition, licensing, development and promotion of healthcare products in Canada. The Company targets several therapeutic areas in Canada with a particular interest in products for the treatment of neurology, pain, urology, dermatology and endocrinology/cardiology. In addition to developing and selling healthcare products in Canada, Tribute also sells products globally through a number of international partners.
Tribute Pharmaceuticals current portfolio consists of ten marketed products in Canada, including: NeoVisc® (Triple and Single Dose), Uracyst®, Bezalip® SR, Soriatane®, Cambia®, Fiorinal®, Fiorinal® C, Visken®, Viskazide® and Collatamp® G. NeoVisc® and Uracyst® are also sold in several countries globally through strategic partners of the Company. Tribute also has an exclusive license for the development and commercialization of Bezalip® SR (bezafibrate) for the U.S. market and an exclusive license for the development and commercialization of bilastine in Canada.
2. ACQUISITIONS AND GOODWILL
Asset Purchase Agreement
On October 2, 2014, the Company entered into an asset purchase agreement (the “Asset Purchase Agreement”) with Novartis AG and Novartis Pharma AG (collectively, “Novartis,” and together with the Company, the “Parties”) pursuant to which the Company acquired from Novartis the Canadian rights to manufacture, market, promote, distribute and sell Fiorinal®, Fiorinal® C, Visken® and Viskazide® for the relief of pain from headache and for the treatment of cardiovascular conditions (the “Products”), as well as certain other assets relating to the Products, including certain intellectual property, marketing authorizations and related data, medical, commercial and technical information, and the partial assignment of certain manufacturing and supply agreements and tenders with third parties (the “Acquired Assets”). The Company also assumed certain liabilities arising out of the Acquired Assets and the Licensed Assets (as described below) after the acquisition, including product liability claims or intellectual property infringement claims by third parties relating to the sale of the Products by the Company in Canada. In connection with the acquisition of the Acquired Assets, and pursuant to the terms of the Asset Purchase Agreement, the Company concurrently entered into a license agreement with Novartis AG, Novartis Pharma AG and Novartis Pharmaceuticals Canada Inc. (the “License Agreement”, and, together with the Asset Purchase Agreement, the “Agreements”). Pursuant to the terms of the License Agreement, the Novartis entities agreed to license to the Company certain assets relating to the Products, including certain intellectual property, marketing authorizations and related data, and medical, commercial and technical information (the “Licensed Assets”). The Company concurrently entered into a supply agreement with Novartis Pharma AG (the “Supply Agreement”), pursuant to which Novartis Pharma AG agreed to supply the Company with the requirements of Products for sale for a transition period until the Company is able to transfer the marketing authorizations to the Company. The consideration paid for the Acquired Assets and the Licensed Assets was $32,000,000 in cash.
The transaction was accounted for as an asset acquisition. Costs incurred to complete the acquisition were $117,521, which were capitalized to the cost of the assets acquired. The fair value of the acquired identifiable net assets was $32,117,521, which was allocated between the product rights acquired (Visken® and Fiorinal®). The fair value of the acquired identifiable net assets was allocated as follows:
|
Fiorinal® Product Line |
|
$ |
29,922,888 |
|
|
Visken® Product Line |
|
2,194,633 |
| |
|
|
|
$ |
32,117,521 |
|
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
These financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America on a basis consistent with that of the prior year.
a) CASH AND CASH EQUIVALENTS
Cash and cash equivalents include cash and all highly liquid investments purchased with an original maturity of three months or less at the date of purchase. Cash and cash equivalents are held with three major financial institutions in Canada. As at December 31, 2013 and 2014, the Company did not have any cash equivalents.
b) ACCOUNTS RECEIVABLE
The Company routinely assesses the recoverability of all material trade and other receivables to determine their collectability by considering factors such as historical experience, credit quality, the age of the accounts receivable balances, and current economic conditions that may affect a customer’s ability to pay.
c) REVENUE RECOGNITION
The Company recognizes revenue when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the price is fixed or determinable, and collectability is reasonably assured. License fees which are comprised of initial fees and milestone payments are recognized upon achievement of the milestones, provided the milestone is meaningful, and provided that collectability is reasonably assured and other revenue recognition criteria are met. Milestone payments are recognized into income upon the achievement of the specified milestones when the Company has no further involvement or obligation to perform services, as related to that specific element of the arrangement. Up-front fees and other amounts received in excess of revenue recognized are recorded as deferred revenues.
Revenues from the sale of products, net of trade discounts, returns and allowances, are recognized when legal title to the goods has been passed to the customer and collectability is reasonably assured. Revenues associated with multiple-element arrangements are attributed to the various elements, if certain criteria are met, including whether the delivered element has standalone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered elements. Non-refundable up-front fees for the transfer of methods and technical know-how, not requiring the Company to perform additional research or development activities or other significant future performance obligations, are recognized upon delivery of the methods and technical know-how.
Royalty revenue is recognized when the Company has fulfilled the terms in accordance with the contractual agreement and has no material future obligation, other than inconsequential and perfunctory support, as would be expected under such agreements and the amount of the royalty fee is determinable and collection is reasonably assured.
A customer is obligated to pay for products sold to it within a specified number of days from the date that title to the products is transferred to the customer. The Company’s standard terms typically range from 0.5% to 2% discount, 15 to 20 days net 30 from the date of invoice.
The Company has a product returns policy on some of its products, which allows the customer to return pharmaceutical products that have expired, for full credit, provided the expired products are returned within twelve months from the expiration date.
Transfer of title occurs and risk of ownership passes to a customer at the time of shipment or delivery, depending on the terms of the agreement with a particular customer. The sale price of the Company’s products is substantially fixed or determinable at the date of sale based on purchase orders generated by a customer and accepted by the Company. A customer’s obligation to pay the Company for products sold to it is not contingent upon the resale of those products. The Company recognizes revenues for the sale of products from the date the title to the products is transferred to the customer.
In connection with the Asset Purchase Agreement (Note 2), the Company entered into a transition services and supply agreement with Novartis to facilitate the seamless and efficient transfer of products to the Company. The agreement required that Novartis continue to manufacture and distribute products until the Company obtained the necessary marketing authorizations to allow it to take over these functions as principal. Novartis provided the Company with a monthly reconciliation of revenues, cost of goods, and marketing and selling expenses for which the Company then billed Novartis for the net amount receivable. The Company relied on the financial information provided by Novartis to estimate the amounts due under this agreement. Based on the terms of this arrangement and the guidance per ASC 605-45 regarding agency relationships, for the period of this arrangement the Company recorded revenues relating to the Asset Purchase Agreement on a net basis in the statement of operations, net of cost of goods and marketing and selling expenses.
d) INVENTORIES
Inventories are valued at the lower of cost and net realizable value with cost being determined on a first-in, first-out basis. Cost is determined to be purchase cost for raw materials and the production cost (materials, labor and indirect manufacturing cost) for work-in-process and finished goods. Throughout the manufacturing process, the related production costs are recorded within inventory.
e) PROPERTY, PLANT AND EQUIPMENT
Property, plant and equipment are stated at cost. The Company periodically evaluates whether current facts or circumstances indicate that the carrying value of such assets to be held and used may not be recoverable. The Company reviews its long-term assets, such as fixed assets to be held and used or disposed of, for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. If the sum of the expected undiscounted cash flows is less than the carrying amount of the asset, an impairment loss is recognized in the amount by which the carrying amount of the asset exceeds its fair value. The basis of amortization and estimated useful lives of these assets are provided for as follows:
|
Asset Classification |
|
Amortization Method |
|
Useful Life |
|
Building |
|
Straight-line |
|
20 years |
|
Computer and office equipment |
|
Straight-line |
|
5 years |
|
Leasehold improvements |
|
Straight-line over the lease term |
|
5 years |
|
Manufacturing equipment |
|
Straight-line & activity based |
|
5 to 10 years |
|
Warehouse equipment |
|
Straight-line |
|
5 to 10 years |
|
Packaging equipment |
|
Activity based |
|
5 to 10 years |
Activity based amortization is based on the number of uses for each asset in that category.
f) GOODWILL AND INTANGIBLE ASSETS
Goodwill represents the excess of acquisition cost over the fair value of the net assets of the acquired businesses. Goodwill has an indefinite useful life and is not amortized, but instead tested for impairment annually. Intangible assets include patents, product rights, a licensing asset and licensing agreements.
Patents represent capitalized legal costs incurred in connection with applications for patents. In-process patents pending are not amortized. All patents subject to amortization are amortized on a straight line basis over an estimated useful life of up to 17 years. The Company regularly evaluates patents and applications for impairment or abandonment, at which point the Company charges the remaining net book value to expenses. The licensing asset represents amounts paid for exclusive Canadian licensing rights to develop, register, promote, manufacture, use, market, distribute and sell pharmaceutical products. The licensing agreements represent the fair value assigned to licensing agreements acquired. The licensing asset and licensing agreement are amortized over the remaining life of the agreement, upon product approval or over their estimated useful lives ranging from 4 years to 25 years. See Note 7.
The Company evaluates the recoverability of amortizable intangible assets for possible impairment whenever events or circumstances indicate that the carrying amount of such assets may not be recoverable. Recoverability of these
assets is measured by a comparison of the carrying amounts to the future undiscounted cash flows the assets are expected to generate. If such review indicates that the carrying amount of intangible assets is not recoverable, the carrying amount of such assets is reduced to fair value. The Company has not recorded any impairment charge during the years presented.
When assessing goodwill impairment, the Company assesses qualitative factors first to determine whether the existence of events or circumstances leads to a determination that it is more likely than not that the fair value of the reporting unit is less than its carrying amount. If, after assessing the totality of events or circumstances, the Company determines it is not more likely than not that the fair value of the reporting unit is less than its carrying amount, then the two-step impairment test is not performed. In the event that there are qualitative factors which indicate that the carrying amount is greater than the fair value of the reporting unit, then the two step impairment approach is performed.
The first step, identifying a potential impairment, compares the fair value of the reporting unit with its carrying amount. If the carrying amount exceeds its fair value, the second step would need to be performed; otherwise, no further step is required. The second step, measuring the impairment loss, compares the implied fair value of the goodwill with the carrying amount of the goodwill. Any excess of the goodwill carrying amount over the applied fair value is recognized as an impairment loss, and the carrying value of goodwill is written down to fair value. As of December 31, 2014 and 2013, no impairment of goodwill has been identified.
g) USE OF ESTIMATES
The preparation of these financial statements has required management to make estimates and assumptions that affect the amounts of assets and liabilities and disclosure of contingent liabilities and the revenue and expenses recorded. On an ongoing basis, the Company evaluates its estimates, including those related to provision for doubtful accounts, inventories, accrued liabilities, accrued returns, discounts and rebates, derivative instruments, income taxes, stock based compensation, revenue recognition, goodwill, intangible assets, contingent consideration and the estimated useful lives of property, plant and equipment and intangible assets. The Company bases its estimates on historical experiences and on various other assumptions believed to be reasonable under the circumstances.
Actual results could differ from those estimates. As adjustments become necessary, they are recorded in the statement of operations and comprehensive loss in the period in which they become known. Such adjustments could be material.
h) DEFERRED INCOME TAXES
The Company accounts for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements.
Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax results in deferred tax assets and liabilities is recognized in income in the period that includes the enactment date. The Company records net deferred tax assets to the extent management believe these assets will more likely than not be realized. In making such determination, all available positive and negative evidence is utilized, including scheduled reversals of deferred tax liabilities, projected future taxable income, tax planning strategies and recent financial operations. In the event a determination is made that the Company would be able to realize deferred income tax assets in the future in excess of the net recorded amount, an adjustment to the valuation allowance would be made, which would reduce the provision for income taxes.
Tax benefits from an uncertain tax position may be recognized when it is more likely than not that the position will be sustained upon examination, including resolution of any related appeals or litigation processes, based on the technical merits. Income tax positions must meet a more-likely-than-not recognition threshold to be recognized. This interpretation also provides guidance on measurement, de-recognition, classification, interest and penalties, accounting in interim periods, disclosure and transition.
i) STOCK-BASED CONSIDERATION
The Company uses the fair value based method of accounting for all its stock-based compensation in accordance with FASB Accounting Standards Codification (“ASC”) ASC 718 “Compensation — Stock Compensation”. The estimated fair value of the options that are ultimately expected to vest based on performance related conditions, as well as the options that are expected to vest based on future service, is recorded over the option’s requisite service period and charged to stock-based compensation. In determining the amount of options that are expected to vest, the Company takes into account, voluntary termination behavior as well as trends of actual option forfeitures.
Stock options and warrants which are indexed to a factor which is not a market, performance or service condition, in addition to the Company’s share price, are classified as liabilities and re-measured at each reporting date based on the Black-Scholes option pricing model with a charge to operations, until the date of settlement. Some warrants have been reflected as a liability as they are indexed to a factor which is not a market performance or service condition.
j) FOREIGN CURRENCY TRANSACTIONS AND TRANSLATION
Monetary assets and liabilities are translated into Canadian dollars, which is the functional currency of the Company, at the year-end exchange rate, while foreign currency revenues and expenses are translated at the exchange rate in effect on the date of the transaction. The resultant gains or losses are included in the statement of operations and comprehensive loss. Non-monetary items are translated at historical rates.
k) RESEARCH AND DEVELOPMENT
Research and development costs are expensed as incurred. The approved refundable portion of the tax credits are netted against the related expenses. Non-refundable investment tax credits are recorded in the period when reasonable assurance exists that the Company has complied with the terms and conditions required for approval of the tax credit and it is more likely than not that the Company will realize the benefits of these tax credits against the deferred taxes. Refundable investment tax credits are recorded in the period when reasonable assurance exists that the Company has complied with the terms and conditions required for approval of the tax credit and it is more likely than not that the Company will collect it. At December 31, 2014, the Company had no outstanding refundable tax credits (2013 - nil).
l) COMPREHENSIVE INCOME
Comprehensive income is defined as the change in equity during a period related to transactions and other events and circumstances from non-owner sources. It includes all changes in equity during a period except those resulting from investments by owners and distributions to owners.
m) EARNINGS (LOSS) PER SHARE
FASB ASC Section 260, “Earnings (Loss) Per Share”, requires presentation of both basic and diluted earnings (loss) per share (EPS) with a reconciliation of the numerator and denominator of the basic EPS computation to the numerator and denominator of the diluted EPS computation. Basic EPS excludes dilution. Diluted EPS reflects the potential dilution that could occur if securities or other contracts to issue shares were exercised or converted into shares that would then share in the earnings.
Basic earnings (loss) per share are computed based on the weighted average number of common shares outstanding each year. The diluted loss per share is not presented when the effect is anti-dilutive.
n) ACQUISITIONS
The accounting for acquisitions requires extensive use of estimates and judgments to measure the fair value of the identifiable tangible and intangible assets acquired, including license agreement assets and liabilities assumed. Additionally, the Company must determine whether an acquired entity is considered to be a business or a set of net
assets, because the excess of the purchase price over the fair value of net assets acquired can only be recognized as goodwill in a business combination.
o) CONTINGENT CONSIDERATION
Contingent consideration liabilities represent future amounts the Company may be required to pay in conjunction with various business combinations. The ultimate amount of future payments is based on specified future criteria, such as sales performance and the achievement of certain future development, regulatory and sales milestones. The Company estimates the fair value of the contingent consideration liabilities related to sales performance using the income approach, which involves forecasting estimated future net cash flows and discounting the net cash flows to their present value using a risk-adjusted rate of return. The Company estimates the fair value of the contingent consideration liabilities related to the achievement of future development and regulatory milestones by assigning an achievement probability to each potential milestone and discounting the associated cash payment to its present value using a risk-adjusted rate of return. The Company evaluates its estimates of the fair value of contingent consideration liabilities on a periodic basis. Any changes in the fair value of contingent consideration liabilities are included in the Company’s statements of operations.
p) FAIR VALUE MEASUREMENTS
The Company applies fair value accounting for all financial assets and liabilities and non-financial assets and liabilities that are recognized or disclosed at fair value in the financial statements on a recurring basis. The Company defines fair value as the price that would be received from selling an asset or paid to transfer a liability in orderly transaction between market participants at the measurement date. When determining the fair value measurements for assets and liabilities, which are required to be recorded at fair value, the Company considers the principal or most advantageous market in which the Company would transact and the market-based risk measurements or assumptions that market participants would use in pricing the asset or liability, such as risks inherent in valuation techniques, transfer restrictions and credit risk. Fair value is estimated by applying the following hierarchy, which prioritizes the inputs used to measure fair value into three levels and bases the categorization within the hierarchy upon the lowest level of input that is available and significant to the fair value measurement:
|
Level 1 - |
Quoted prices in active markets for identical assets or liabilities. |
|
Level 2 - |
Observable inputs other than quoted prices in active markets for identical assets and liabilities, quoted prices for identical or similar assets or liabilities in inactive markets, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
|
Level 3 - |
Inputs that are generally unobservable and typically reflect management’s estimate of assumptions that market participants would use in pricing the asset or liability. |
The Company’s valuation techniques used to measure the fair value of money market funds and certain marketable equity securities were derived from quoted prices in active markets for identical assets or liabilities. The valuation techniques used to measure the fair value of all other financial instruments, all of which have counterparties with high credit ratings, were valued based on quoted market prices or model driven valuations using significant inputs derived from or corroborated by observable market data.
In accordance with the fair value accounting requirements, companies may choose to measure eligible financial instruments and certain other items at fair value. The Company has not elected the fair value option for any eligible financial instruments.
The carrying amounts of the Company’s financial assets and liabilities including cash and cash equivalents, accounts receivable, loan receivable, accounts payable and accrued liabilities are approximate of their fair values due to the short maturity of these instruments. The fair value of the long term debt is estimated based on quoted market prices and interest rates.
The Company’s equity-linked financial instruments reflected as warrant liability on the balance sheet represent financial liabilities classified as Level 2 as per ASU 2009-05. As required by the guidance, assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. The fair value of the warrant liability which is not traded in an active market has been determined using the Black-Scholes option pricing model based on assumptions that are supported by observable market conditions. The estimated fair value of the contingent non-cash consideration was based on the Company’s stock price.
q) ACCOUNTING STANDARDS NOT YET ADOPTED
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606): Revenue from Contracts with Customers, which guidance in this update will supersede the revenue recognition requirements in Topic 605, Revenue Recognition, and most industry-specific guidance when it becomes effective. ASU No. 2014-09 affects any entity that enters into contracts with customers to transfer goods or services or enters into contracts for the transfer of nonfinancial assets unless those contracts are within the scope of other standards. The core principal of ASU No. 2014-09 is that a company will recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. In doing so, companies will need to use more judgment and make more estimates than under current guidance. These may include identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligation. ASU No. 2014-09 is effective for annual reporting periods beginning after December 15, 2016, including interim periods within that reporting period, which will be the Company’s fiscal year 2017 (or January 1, 2017), and entities can transition to the standard either retrospectively or as a cumulative-effect adjustment as of the date of adoption. Early adoption is prohibited. The Company is currently in the process of evaluating the impact of adoption of ASU No. 2014-09 on its consolidated financial statements and related disclosures.
4. Inventories
|
|
|
December |
|
December |
| ||
|
Raw materials |
|
$ |
290,197 |
|
$ |
236,444 |
|
|
Finished goods |
|
399,830 |
|
418,635 |
| ||
|
Packaging materials |
|
70,870 |
|
56,007 |
| ||
|
Work in process |
|
276,490 |
|
333,745 |
| ||
|
|
|
$ |
1,037,387 |
|
$ |
1,044,831 |
|
During the year ended December 31, 2014, the Company assessed its inventory and determined that $53,099 of its on-hand inventory would not be used prior to its potential useful life (2013 - $56,935). Therefore, $26,241 (2013 - $1,710) of finished goods, $21,581 (2013 - $34,972) of raw materials and $5,277 (2013 - $20,253) of packaging materials were written off during the year.
5. Prepaid Expenses and Other Receivables
|
|
|
December |
|
December |
| ||
|
Prepaid operating expenses and other receivables |
|
$ |
180,304 |
|
$ |
140,986 |
|
|
Manufacturing deposits |
|
— |
|
18,825 |
| ||
|
Interest receivable on loan receivables |
|
6,975 |
|
6,075 |
| ||
|
|
|
$ |
187,279 |
|
$ |
165,886 |
|
6. Property, Plant and Equipment
|
|
|
December 31, 2014 |
| |||||||
|
|
|
Cost |
|
Accumulated |
|
Net |
| |||
|
Land |
|
$ |
90,000 |
|
$ |
— |
|
$ |
90,000 |
|
|
Building |
|
618,254 |
|
300,798 |
|
317,456 |
| |||
|
Leasehold improvements |
|
10,359 |
|
4,662 |
|
5,697 |
| |||
|
Office equipment |
|
61,308 |
|
52,124 |
|
9,184 |
| |||
|
Manufacturing equipment |
|
1,103,525 |
|
602,667 |
|
500,858 |
| |||
|
Warehouse equipment |
|
17,085 |
|
17,085 |
|
— |
| |||
|
Packaging equipment |
|
111,270 |
|
62,744 |
|
48,526 |
| |||
|
Computer equipment |
|
142,873 |
|
102,309 |
|
40,564 |
| |||
|
|
|
$ |
2,154,674 |
|
$ |
1,142,389 |
|
$ |
1,012,285 |
|
|
|
|
December 31, 2013 |
| |||||||
|
|
|
Cost |
|
Accumulated |
|
Net |
| |||
|
Land |
|
$ |
90,000 |
|
$ |
— |
|
$ |
90,000 |
|
|
Building |
|
618,254 |
|
269,886 |
|
348,368 |
| |||
|
Leasehold improvements |
|
10,359 |
|
2,590 |
|
7,769 |
| |||
|
Office equipment |
|
61,308 |
|
48,299 |
|
13,009 |
| |||
|
Manufacturing equipment |
|
1,103,525 |
|
576,862 |
|
526,663 |
| |||
|
Warehouse equipment |
|
17,085 |
|
16,737 |
|
348 |
| |||
|
Packaging equipment |
|
111,270 |
|
51,700 |
|
59,570 |
| |||
|
Computer equipment |
|
130,114 |
|
85,922 |
|
44,192 |
| |||
|
|
|
$ |
2,141,915 |
|
$ |
1,051,996 |
|
$ |
1,089,919 |
|
During the year ended December 31, 2014, the Company disposed of $nil (2013 - $nil) in property, plant and equipment.
During the year ended December 31, 2014, the Company recorded total amortization of tangible assets of $90,391 (2013 - $96,252), which was recorded as $15,728 (2013 - $19,842) to cost of goods sold, $14,576 (2013 - $22,812) to inventory and the remaining $60,087 (2013 - $53,598) was recorded to amortization expense on the statements of operations and comprehensive loss.
7. Intangible Assets
|
|
|
December 31, 2014 |
| |||||||
|
|
|
Cost |
|
Accumulated |
|
Net Carrying |
| |||
|
Patents |
|
$ |
351,754 |
|
$ |
53,242 |
|
$ |
298,512 |
|
|
Licensing asset |
|
1,005,820 |
|
174,084 |
|
831,736 |
| |||
|
Licensing agreements |
|
10,377,325 |
|
2,345,049 |
|
8,032,276 |
| |||
|
Product rights |
|
32,117,521 |
|
321,175 |
|
31,796,346 |
| |||
|
|
|
$ |
43,852,420 |
|
$ |
2,893,550 |
|
$ |
40,958,870 |
|
|
|
|
December 31, 2013 |
| |||||||
|
|
|
Cost |
|
Accumulated |
|
Net |
| |||
|
Patents |
|
$ |
268,786 |
|
$ |
39,562 |
|
$ |
229,224 |
|
|
Licensing asset |
|
1,005,820 |
|
96,713 |
|
909,107 |
| |||
|
Licensing agreements |
|
10,004,000 |
|
1,425,158 |
|
8,578,842 |
| |||
|
|
|
$ |
11,278,606 |
|
$ |
1,561,433 |
|
$ |
9,717,173 |
|
The Company recorded a loss of $161,200 on intangible assets during the year ended December 31, 2013 due to the termination of a promotion and marketing agreement, which was acquired as part of a business combination in 2012.
Amortization expense of intangible assets for the years ended December 31, 2014 and 2013 were $1,332,118 and $1,038,152, respectively.
The Company has patents pending of $45,392 and licensing agreements of $373,325 at December 31, 2014 (2013 - $112,902 and $nil, respectively) not currently being amortized.
The licensing asset consists of capitalized payments to third party licensors related to the achievement of regulatory approvals to commercialize products in specified markets and up-front payments associated with royalty obligations for products.
The Company tests for impairment of definite-lived intangible assets when events or circumstances indicate that the carrying value of the assets may not be recoverable. No such triggering events were identified in 2014 and 2013, and therefore no impairment loss was recognized during those periods.
Estimated future amortization expense of intangible assets at December 31, 2014 is as follows:
|
|
|
Amount |
| |
|
2015 |
|
$ |
2,630,448 |
|
|
2016 |
|
2,630,503 |
| |
|
2017 |
|
2,630,309 |
| |
|
2018 |
|
2,630,272 |
| |
|
2019 |
|
2,299,919 |
| |
|
Thereafter |
|
27,718,699 |
| |
|
|
|
$ |
40,540,150 |
|
8. Goodwill
The goodwill relates to the Company’s acquisition of Tribute Pharmaceuticals Canada Ltd and Tribute Pharma Canada Inc. and an agreement with Theramed Corporation. The Company has evaluated the goodwill during the fourth quarter and has determined that there is no impairment of the values at December 31, 2014 and 2013. The Company has completed a quantitative goodwill assessment and concluded that there were no indications of impairment.
9. Long Term Debt and Debt Issuance Costs
On May 11, 2012, the Company entered into a loan and security agreement (the “MidCap Loan Agreement”) with MidCap Funding III, LLC (the “Lender” or “MidCap”) for a 36 month term loan that was due May 11, 2015. The term loan allowed for a total advancement of US$6,000,000 ($6,381,600). An amount of US$3,500,000 ($3,482,150) was drawn on execution of the MidCap Loan Agreement and the remainder was available to be advanced if the Company raised an amount of not less than US$6,000,000 ($6,381,600) from any combination of: an equity issuance; upfront payments associated with a pharmaceutical partnership; or upfront payments in conjunction with the acquisition or in-licensing of pharmaceutical products. The availability of advancements of the remainder of the loan expired on March 31, 2013. The MidCap Loan Agreement was secured by all assets of the Company and contained customary covenants that, among other things, generally restricted the Company’s ability to incur additional indebtedness. The Loan Agreement included a financial covenant to raise not less than US$3,000,000 ($3,190,800) by March 31, 2013 in the form of an equity raise or cash from an upfront payment associated with a pharmaceutical partnership, which was completed prior to March 31, 2013 (Note 10 a). The first six (6) payments were interest-only, with principal and interest payments due monthly thereafter. Interest was calculated at the higher of 4% or the thirty (30) day London Inter Bank Offered Rate (“LIBOR”) plus 7%. Pursuant to the below Credit Agreement, the MidCap Loan Agreement was repaid in full.
On August 8, 2013, SWK Funding LLC (“SWK”), a wholly-owned subsidiary of SWK Holdings Corporation, entered into a credit agreement (the “Credit Agreement”) with the Company pursuant to which SWK provided to the Company a term loan in the principal amount of US$6,000,000 ($6,381,600) (the “Loan”) which was increased, as per the terms of the Credit Agreement, by an additional US$2,000,000 ($2,211,000) at the Company’s request on February 4, 2014. SWK served as the agent under the Credit Agreement.
On October 1, 2014 (the “Amendment Closing Date”), the Company entered into the First Amendment to the Credit Agreement and Guarantee (the “First Amendment”, and, together with the Credit Agreement, the “Amended Credit Agreement”) with SWK. The Amended Credit Agreement provides for a multi-draw term loan to the Company for up to a maximum amount of US$17,000,000 ($19,721,700) (the “Loan Commitment Amount”). On the Amendment Closing Date, SWK advanced the Company an additional amount equal to US$6,000,000 ($6,724,800) pursuant to the terms of a promissory note executed on the Amendment Closing Date (the “October 2014 Note”). The October 2014 Note is for a total principal amount of US$14,000,000 ($16,241,400) (comprised of US$8,000,000 ($8,592,600) advanced under the Credit Agreement and the additional US$6,000,000 ($6,724,800) advanced on October 1, 2014) due and payable on December 31, 2018 (the “Term Loan Maturity Date”). In addition, an
origination fee of US$120,000 ($124,172) was paid to SWK and treated as a discount to the carrying value of the Loan.
Interest and principal under the Loan will be paid by a revenue based payment (“Revenue Based Payment”) that is charged on quarterly revenues of the Company, applied in the following priority (i) first, to the payment of all fees, costs, expenses and indemnities due and owing to SWK under the Amended Credit Amended Agreement, (ii) second, to the payment of all fees, costs, expenses and indemnities due and owing to the lenders under the Credit Agreement, (iii) third, to the payment of all accrued but unpaid interest until paid in full; and (iv) fourth, for each payment date on or after payment date in April 2015, to the payment of all principal under the Loan up to a maximum of US$1,000,000 ($1,116,100) in respect of any fiscal quarter. All amounts applied under the Revenue Based Payment will be made to each lender according to its pro-rata share of the Loan. The lenders will be entitled to certain additional payments in connection with repayments of the Loan, both on maturity and in connection with a prepayment or partial prepayment. Pursuant to the terms of the Amended Credit Agreement, the Company entered into a Guaranty and Collateral Agreement granting the lenders a security interest in substantially all of the Company’s assets (the “Collateral”). The Amended Credit Agreement contains customary affirmative and negative covenants for credit facilities of its type, including but not limited to, limiting the Company’s ability to pay dividends or make any distributions, incur additional indebtedness, grant additional liens, engage in any other line of business, make investments, merge, consolidate or sell all or substantially all of its assets and enter into transactions with related parties. The Amended Credit Agreement also contains certain financial covenants, including, but not limited to, certain minimum net sales requirements and a requirement to maintain at least $1,000,000 of unencumbered liquid assets at the end of each fiscal quarter. The Amended Credit Agreement includes customary events of default, including but not limited to, failure to pay principal, interest or fees when due, failure to comply with covenants, default under certain other indebtedness, certain insolvency or bankruptcy events, the occurrence of certain material judgments, the institution of any proceeding by a government agency or a change of control of the Company. The obligations under the Amended Credit Agreement to repay the Loan may be accelerated upon the occurrence of an event of default under the Amended Credit Agreement. A 4% agent fee on the above mentioned transaction was paid on the amounts borrowed above US$3,500,000 ($4,060,350) up to US$8,000,000 ($9,280,800).
The Loan accrues interest at an annual rate of 11.5% plus LIBOR Rate (as defined in the Amended Credit Agreement), with LIBOR Rate being subject to a minimum floor of 2%, such that the minimum interest rate is 13.5%. In the event of a change of control, a merger or a sale of all or substantially all of the Company’s assets, the Loan shall be due and payable.
In connection with the Loan the Company issued to SWK 755,794 common share purchase warrants with each warrant entitling SWK to acquire one common share in the capital of the Company at an exercise price of US$0.5954 ($0.6907), at any time prior to August 8, 2020. The grant date fair value of the warrants was $445,794, which was recorded as warrant liability, with an equal amount recorded as a discount to the carrying value of the Loan. In addition, an origination fee of US$120,000 ($124,172) was paid to SWK and treated as a discount to the carrying value of the loan.
Upon receipt of the additional US$2,000,000 ($2,211,000) of the Loan, the Company issued to SWK 347,222 common share purchase warrants with a grant date fair value of the warrants of $120,914. Each warrant entitles SWK to acquire one common share in the capital of the Company at an exercise price of US$0.432 ($0.501), at any time on or prior to February 4, 2021.
Upon receipt of the additional US$6,000,000 ($6,724,800) of the Loan, the Company issued to SWK 740,000 common share purchase warrants with a grant date fair value of the warrants of $303,557. Each warrant entitles SWK to acquire one common share in the capital of the Company at an exercise price of US$0.70 ($0.8121), at any time on or prior to October 1, 2019. In addition, an origination fee of US$90,000 ($100,872) was paid to SWK and treated as a discount to the carrying value of the loan.
The discount to the carrying value of the Loan is being amortized as a non-cash interest expense over the term of the Loan using the effective interest rate method. The grant date fair value of the warrants issued to SWK was determined using the Black-Scholes model with the following weighted average assumptions: expected volatility of 123%, a risk-free interest rate of 1.90%, an expected life of 6.2 years, and no expected dividend yield.
During the year ended December 31, 2014, the Company accreted $167,555 (2013 - $103,755) in non-cash accretion expense in connection with the long term loans, which is included in accretion expense on the statements of operations and comprehensive loss.
Upon repayment of the MidCap loan all financing fees and legal costs associated with the MidCap loan not yet amortized were expensed to loss on extinguishment of loan on the statements of operations and comprehensive loss. These costs, including an exit fee of US$240,000, amount to $620,835.
During 2014, the Company incurred US$203,389 ($225,858) (2013 - US$294,971 ($303,374)) in financing fees and legal costs related to closing the Credit Agreement and recorded US$80,000 ($92,808) (2013 - US$60,000 ($63,816)) related to an exit fee payable to SWK upon the retirement of the Loan. These fees and costs were classified as debt issuance costs on the balance sheets. These assets are being amortized as a non-cash interest expense over the term of the outstanding Loan using the effective interest rate method. During the year ended December 31, 2014, the Company amortized $118,814 (2013 — $154,097) in non-cash interest expense, which is included in amortization expense on the statements of operations and comprehensive loss.
During the year ended December 31, 2014, the Company made no principal payments (2013 - US$3,281,250 ($3,386,630)) and interest payments of US$1,090,500 ($1,207,262) (2013 — US$409,653 ($422,341)) under the MidCap Financial LLC loan agreement (now repaid) and SWK Credit Agreement and Amended Credit Agreement. The Company has estimated the following revenue-based principal and interest payments over the next four years ended December 31 based on the assumption that only the minimum revenue requirements will be met under the Amended Credit Agreement:
|
|
|
Principal Payments |
|
Interest Payments |
|
|
2015 |
|
$US1,136,997($1,319,030) |
|
$US1,847,086($2,142,804) |
|
|
2016 |
|
$US1,454,476($1,687,338) |
|
$US1,670,898($1,938,409) |
|
|
2017 |
|
$US1,666,664($1,933,497) |
|
$US1,451,475($1,683,856) |
|
|
2018 |
|
$US9,741,863($11,301,535) |
|
$US1,206,058($1,399,148) |
|
10. Capital Stock
a) Common Shares
During the year ended December 31, 2013, the Company completed two private placement offerings in which 11,471,196 units were issued for gross proceeds of US$4,595,000 ($4,713,787). As a part of the private placements, the Company issued 11,362,500 units at a price of US$0.40 ($0.46) per unit and granted 11,362,500 common share purchase warrants to the participants. Each unit consisted of one common share of the Company’s stock and one-half of one Series A common share purchase warrant (a “Series A Warrant”) and one-half of one Series B common share purchase warrant (a “Series B Warrant”). Each whole Series A Warrant entitles the holder thereof to acquire one common share of the Company at any time during the period ending 24 months after the date of issuance at a price of US$0.50 ($0.58) per common share. Each whole Series B Warrant entitles the holder thereof to acquire one common share of the Company at a price of US$0.60 ($0.70) per share at any time during the period ending 60 months after the date of issuance. The terms of the Series B Warrants provide the Company with a right to call the Series B Warrants at a price of US$0.001 per warrant if certain conditions are met including the common shares trading at a volume weighted average price for 20 out of 30 consecutive trading days at a price which exceeds US$1.20 (subject to adjustment for stock splits, recapitalizations and other corporate transactions) with average daily volume during such period of at least US$30,000. The remaining 108,696 units were issued at a price of US$0.46 ($0.53) per unit. Each unit consists of one common share of the Company’s stock and one warrant exercisable at any time during the period ending 60 months after the date of the issuance at a price of US$0.55 ($0.64).
Directors, officers and individuals related to directors purchased 6,046,196 units for gross proceeds of US$2,425,000 ($2,485,625) pursuant to this private placement.
In connection with the private placement, the Company paid cash commissions of US$248,219 ($252,101) and issued 345,188 Series A broker warrants and 345,187 Series B broker warrants valued at US$168,491 ($172,986). Each Series A broker warrant entitles the holder to purchase one common share at an exercise price of US$0.50
($0.58) for a period of twenty four months. Each Series B broker warrant entitles the holder to purchase one common share at an exercise price of $0.60 ($0.70) for a period of 60 months after the date of issuance. Total other issuance costs associated with the private placements were $184,856. The Series B broker warrants also contain a call right similar to the Series B Warrant described above.
During the year ended December 31, 2014, the Company completed a public offering in which 42,895,000 units (“Units”) were issued at a price of $0.70 per Unit for gross proceeds of $30,026,500. Each Unit consisted of one common share of the Company’s stock and one-half of one common share purchase warrant. Each whole warrant entitles the holder to acquire one common share of the Company at a price per share of $0.90 at any time on or before July 15, 2016. As part of the public offering, the Company issued 21,447,500 common share purchase warrants to the purchasers.
In connection with the public offering, the Company paid cash commissions to the syndicate of underwriters of $2,251,988 and issued an aggregate of 3,217,125 non-transferable broker warrants valued at $1,177,468. See Note 10 (c). Each broker warrant entitles the holder to purchase one Unit at an exercise price of $0.70 at any time on or before July 15, 2016. Total other issuance costs associated with the public offering were $403,636.
During the year ended December 31, 2014, the Company issued 500,000 common shares to a consultant for services and recorded $211,812 as paid-in common shares based on the fair market value of the common shares at the date of issuance.
b) Stock Based Compensation
The Company’s stock-based compensation program (“Plan”) includes stock options in which some options vest based on continuous service, while others vest based on performance conditions such as profitability and sales goals. For those equity awards that vest based on continuous service, compensation expense is recorded over the service period from the date of grant. For performance-based awards, compensation expense is recorded over the remaining service period when the Company determines that achievement is probable.
During the year ended December 31, 2014, there were 1,827,985 options granted to officers, employees and consultants of the Company (2013 — 1,173,250). The exercise price of 1,107,985 of these options is $0.40, with one-eighth vesting quarterly over two years on each of March 31, June 30, September 30 and December 31, in 2015 and 2016, upon achieving certain financial objectives. Since stock-based compensation is recognized only for those awards that are ultimately expected to vest, the Company has applied an estimated forfeiture rate (based on historical experience and projected employee turnover) to unvested awards for the purpose of calculating compensation expense. The grant date fair value of these options was estimated as $0.33 using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 123%; expected risk free interest rate of 1.61%; and expected term of 5 years.
In addition, during the year ended December 31, 2014, 200,000 options were granted (2013 — nil) with an exercise price of $0.42 and fully vested on January 3, 2015 (Note 15). The grant date fair value of these options was estimated as $0.39 using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 123%; expected risk free interest rate of 1.69%; and expected term of 5 years.
In addition, during the year ended December 31, 2014, 20,000 options were granted with an exercise price of $0.61, with 10,000 vesting on November 30, 2014 and the remaining 10,000 vesting over two years on each of March 31, June 30, September 30 and December 31, in 2015 and 2016. The grant date fair value of these options was estimated as $0.52 using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 123%; expected risk free interest rate of 1.50%; and expected term of 5 years.
During the year ended December 31, 2014, 500,000 options were granted to a consultant for services (2013 — nil) with an exercise price of $0.57. Of these options, 25% vested on February 15, 2015, 25% will vest on August 15, 2015, the remaining 50% vest one-eighth quarterly over two years, starting in the first quarter following the achievement of certain financial objectives. The fair value of these options was estimated as $0.42 using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 123%; expected risk free interest rate of 1.66%; and expected term of 4.62 years.
For the year ended December 31, 2014, the Company recorded $426,714 (2013 — $419,168) as additional paid in capital for options issued to directors, officers, employees and consultants based on continuous service. Included in this amount is $153,334 for options issued to consultants for services (Note 15). This expense was recorded as selling, general and administrative expense on the statements of operations and comprehensive loss. Due to termination of employment and non-achievement of performance-based awards, 817,830 options were removed from the number of options issued during the year ended December 31, 2014 (year ended December 31, 2013 — 560,917).
The Company uses the Black-Scholes option-pricing model to estimate the grant date fair value of stock options with the following weighted average assumptions:
|
|
|
2014 |
|
2013 |
|
|
Risk-free interest rate |
|
1.60 |
% |
1.76 |
% |
|
Expected life |
|
5 years |
|
5 years |
|
|
Expected volatility |
|
123 |
% |
123 |
% |
|
Expected dividend yield |
|
0 |
% |
0 |
% |
The Company’s computation of expected volatility for the years ended December 31, 2014 and 2013 is based on the Company’s market close price over the period equal to the expected life of the options. The Company’s computation of expected life reflects actual historical exercise activity and assumptions regarding future exercise activity of unexercised, outstanding options.
The Company’s expected dividend yield is 0%, since there is no history of paying dividends and there are no plans to pay dividends. The Company’s risk-free interest rate is the Canadian Treasury Bond rate for the period equal to the expected term.
The maximum number of options that may be issued under the Plan is floating at an amount equivalent to 10% of the issued and outstanding common shares, or 9,447,624 as at December 31, 2014 (2013 — 5,108,124). The total remaining options available for granting under the plan at December 31, 2014 was 4,612,634 (2013 — 1,283,289).
The total number of options outstanding as at December 31, 2014 was 4,834,991 (2013 — 3,824,835). The weighted average grant date fair value of the options granted during the year ended December 31, 2014, was $0.38 (2013 - $0.35).
The activities in options outstanding are as noted below:
|
|
|
Number of |
|
Weighted |
| |
|
Balance, December 31, 2012 |
|
3,212,502 |
|
0.65 |
| |
|
Granted |
|
1,173,250 |
|
0.43 |
| |
|
Forfeited |
|
(560,917 |
) |
0.50 |
| |
|
Balance, December 31, 2013 |
|
3,824,835 |
|
$ |
0.60 |
|
|
Granted |
|
1,827,986 |
|
0.45 |
| |
|
Forfeited |
|
(817,830 |
) |
0.55 |
| |
|
Balance, December 31, 2014 |
|
4,834,991 |
|
$ |
0.55 |
|
When employees or non-employees exercise their stock options, the capital stock is credited by the sum of the consideration paid together with the related portion previously credited to additional paid-in capital when stock-based compensation costs were recorded.
As at December 31, 2014, the Company had 2,713,921 (2013 — 2,260,253) vested options. As at December 31, 2014, the number of unvested options expected to vest (including the impact of expected forfeitures) had been estimated at 2,121,070 (2013 — 1,564,582) with a weighted average contractual life of 3.7 years (2013 — 3.8 years) and exercise price of $0.486 (2013 - $0.497). As at December 31, 2014, the total fair value of future expense to be recorded in subsequent periods (assuming no forfeiture occurs) is $339,314 (2013 - $296,186). The weighted average time remaining for these options to vest is 1.71 years (2013 — 1.5 years).
As at December 31, 2014, the aggregate intrinsic value of outstanding options was $209,700 (2013 - $nil) and the aggregate intrinsic value of exercisable options was $80,848 (2013 - $nil) based on the Company’s closing common share price on the OTCQX under the trading symbol TBUFF of US$0.46 ($0.53) (2013 - US$0.36 ($0.38).
The Company recognizes compensation expense for the fair values of stock options using the graded vesting method over the requisite service period for the entire award.
The following table presents information relating to stock options outstanding and exercisable at December 31, 2014.
|
|
|
|
|
Options Outstanding |
|
|
|
Options Exercisable |
| ||||||
|
Range of |
|
Number |
|
Weighted |
|
Weighted |
|
Number |
|
Weighted |
|
Weighted |
| ||
|
$0.30 to $0.49 |
|
1,788,242 |
|
3.42 |
|
$ |
0.40 |
|
714,000 |
|
$ |
0.42 |
|
2.94 |
|
|
$0.50 to $0.69 |
|
2,513,749 |
|
2.59 |
|
0.57 |
|
1,489,921 |
|
0.58 |
|
1.95 |
| ||
|
$0.90 to $1.09 |
|
510,000 |
|
0.50 |
|
0.95 |
|
510,000 |
|
0.95 |
|
0.50 |
| ||
|
|
|
4,834,991 |
|
2.68 |
|
$ |
0.55 |
|
2,713,921 |
|
$ |
0.60 |
|
1.94 |
|
c) Warrants
As at December 31, 2014, the following compensation warrants were outstanding:
Warrant Liability
|
Expiration Date |
|
Warrants |
|
Weighted Average Exercise Price |
|
Fair Value |
|
Fair Value |
| ||
|
May 11, 2017 |
|
750,000 |
|
US$0.43 ($0.50) |
|
$ |
227,090 |
|
$ |
223,356 |
|
|
February 27, 2015 |
|
4,429,688 |
|
US$0.50 ($0.58) |
|
$ |
184,999 |
|
$ |
518,256 |
|
|
February 27, 2018 |
|
4,429,687 |
|
US$0.60 ($0.70) |
|
$ |
1,310,414 |
|
$ |
1,286,216 |
|
|
March 5, 2015 |
|
1,253,000 |
|
US$0.50 ($0.58) |
|
$ |
56,691 |
|
$ |
146,596 |
|
|
March 5, 2018 |
|
1,253,000 |
|
US$0.60 ($0.70) |
|
$ |
372,123 |
|
$ |
363,825 |
|
|
March 11, 2015 |
|
343,750 |
|
US$0.50 ($0.58) |
|
$ |
17,547 |
|
$ |
49,723 |
|
|
March 11, 2018 |
|
343,750 |
|
US$0.60 ($0.70) |
|
$ |
102,089 |
|
$ |
99,812 |
|
|
August 8, 2018 |
|
755,794 |
|
US$0.5954 ($0.6907) |
|
$ |
334,060 |
|
$ |
245,982 |
|
|
September 20, 2018 |
|
108,696 |
|
US$0.55 ($0.64) |
|
$ |
36,442 |
|
$ |
32,948 |
|
|
February 4, 2021 |
|
347,222 |
|
US$0.4320 ($0.5012) |
|
$ |
160,319 |
|
$ |
— |
|
|
October 1, 2021 |
|
740,000 |
|
US$0.70 ($0.81) |
|
$ |
306,106 |
|
$ |
— |
|
|
|
|
14,754,587 |
|
US$0.55 ($0.64) |
|
$ |
3,107,880 |
|
$ |
2,966,714 |
|
On May 11, 2012, the Company granted 750,000 warrants in connection with a loan agreement with MidCap Financial LLC, at an exercise price of US$0.56 ($0.65). Subsequently, the pro rata exercise price of the 750,000 warrants described above was adjusted due to the exercise rate of the 755,794 common share purchase warrants being issued to SWK during 2013. The effect of this pro rata change was a new warrant exercise price of US$0.43 ($0.50). The fair value of these warrants fluctuates based on the current stock price, volatility, the risk free interest rate, time remaining until expiry and changes in the exchange rate between the U.S. and Canadian dollar. The fair value of the warrant liability at the date of grant of the 750,000 warrants was $312,000 and was estimated using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 124%; risk free interest rate of 1.48%; and expected term of 5 years.
In connection with the SWK Credit Agreement the Company issued to SWK 755,794 common share purchase warrants with each warrant entitling SWK to acquire one common share in the capital of the Company at an exercise price of US$0.5954 ($0.6907), at any time prior to August 8, 2020. The fair value of the warrant liability at the date of grant was $445,012 and was estimated using the Black-Scholes option pricing model, based on the following
assumptions: expected dividend yield of 0%; expected volatility of 128%; risk free interest rate of 2.14%; and expected term of 7 years.
In connection with the private placement offerings completed during the year ended December 31, 2013, the Company granted an aggregate of 12,161,571 share purchase warrants to the participants each exercisable into one common share as follows: 6,026,438 at US$0.50 ($0.58) exercisable on or before March 11, 2015 and 6,026,437 at US$0.60 ($0.70) exercisable on or before March 11, 2018. The exercise price of the 12,052,875 warrants is denominated in U.S. dollars while the Company’s functional and reporting currency is the Canadian dollar. As a result, the fair value of the warrants fluctuates based on the current stock price, volatility, the risk free interest rate, time remaining until expiry and changes in the exchange rate between the U.S. and Canadian dollar. The fair value of the warrant liability at the date of grant for these warrants was $1,896,679 and was estimated using the Black-Scholes option pricing model, based on the following weighted average assumptions: expected dividend yield of 0%; expected volatility of 117.4%; risk free interest rate of 1.16%; and expected term of 3.5 years. The remaining 108,696 share purchase warrants are exercisable on or before September 20, 2018 at US$0.55 ($0.64). The fair value of the warrant liability at the date of grant for these warrants was $22,810 and was estimated using the Black-Scholes option pricing model, based on the following weighted average assumptions: expected dividend yield of 0%; expected volatility of 130.0%; risk free interest rate of 1.89%; and expected term of 5 years.
In connection with the additional US$2,000,000 ($2,211,000) loan from SWK described in Note 9, the Company issued SWK 347,222 common share purchase warrants with each warrant entitling SWK to acquire one common share in the capital of the Company at an exercise price of US$0.432 ($0.5012), at any time on or prior to February 4, 2021. The fair value of the warrant liability at the date of grant was $120,914 and was estimated using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 117%; risk free interest rate of 1.85%; and expected term of 7 years.
In connection with the additional US$6,000,000 ($6,724,800) loan described in Note 9, the Company issued SWK 740,000 common share purchase warrants with each warrant entitling SWK to acquire one common share in the capital of the Company at an exercise price of US$0.70 ($0.8121), at any time on or prior to October 1, 2019. The fair value of the warrant liability at the date of grant was $303,557 and was estimated using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 122%; risk free interest rate of 1.56%; and expected term of 5 years.
ASC 815 “Derivatives and Hedging” indicates that warrants with exercise prices denominated in a different currency other than an entity’s functional currency should not be classified as equity. As a result, these warrants have been treated as derivatives and recorded as liabilities carried at their fair value, with period-to-period changes in the fair value recorded as a gain or loss in the statements of operations and comprehensive loss. The Company treated the compensation warrants as a liability upon their issuance.
As at December 31, 2014, the fair value of the warrant liability of $3,107,880 (2013 - $2,966,714) was estimated using the Black-Scholes option pricing model based on the following weighted average assumptions: expected dividend yield of 0% (2013 — 0%) expected volatility of 88% (2013 — 114%) risk-free interest rate of 1.22% (2013 — 1.58%) and expected term of 2.18 years (2013 — 2.94 years).
This model requires management to make estimates of the expected volatility of its common shares, the expected term of the warrants and interest rates. The risk free interest rate is based on the Canadian Treasury Bond rate. The Company has not paid dividends and does not expect to pay dividends in the foreseeable future. The expected term of the warrants is the contractual term of the warrants upon initial recognition.
For the year ended December 31, 2014, the Company recorded a gain of $283,305 (2013 — a loss of $399,217) as change in warrant liability on the statement of operations and comprehensive loss.
Warrants — Equity
|
Expiration Date |
|
Number of |
|
Weighted |
|
Grant Date |
| ||
|
July 15, 2016 |
|
21,447,500 |
|
$ |
0.90 |
|
$ |
5,169,881 |
|
|
July 15, 2016 |
|
3,217,125 |
|
$ |
0.70 |
|
$ |
1,177,468 |
|
|
|
|
24,664,625 |
|
$ |
0.87 |
|
$ |
6,347,349 |
|
In connection with the public offering completed during the year ended December 31, 2014, the Company issued 21,447,500 share purchase warrants to the purchasers, each exercisable into one common share of the Company at $0.90, exercisable at any time on or prior to July 15, 2016. In addition, the Company granted 3,217,125 non-transferable broker warrants, each exercisable into a Unit of the Company, at an exercise price of $0.70 exercisable at any time on or prior to July 15, 2016. Each Unit consists of one common share of the Company and one-half of one common share purchase warrant with each whole warrant entitling the holder to acquire one common share of the Company at a price of $0.90. The fair value of the warrants and broker warrants at the date of grant was $6,347,349 and was estimated using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 99%; risk free interest rate of 1.12%; and expected term of 2 years.
11. Loss Per Share
The treasury stock method assumes that proceeds received upon the exercise of all warrants and options outstanding in the period is used to repurchase the Company’s shares at the average share price during the period. The diluted loss per share is not computed when the effect of such calculation is anti-dilutive. In years when losses are reported, the weighted-average number of common shares outstanding excludes common stock equivalents because their inclusion would be anti-dilutive. Potentially dilutive securities, which were not included in diluted weighted average shares for the years ended December 31, 2014 and 2013 consist of outstanding stock options (4,834,991 and 3,824,835, respectively) and outstanding warrants (39,419,212 at December 31, 2014 and 13,667,365 at December 31, 2013).
The following table sets forth the computation of loss per share:
|
|
|
December 31 |
| ||||
|
|
|
2014 |
|
2013 |
| ||
|
Numerator: |
|
|
|
|
| ||
|
Net loss available to common shareholders |
|
$ |
(5,606,942 |
) |
$ |
(6,572,355 |
) |
|
Denominator: |
|
|
|
|
| ||
|
Weighted average number of common shares outstanding |
|
71,940,005 |
|
49,169,414 |
| ||
|
Effect of dilutive common shares |
|
— |
|
— |
| ||
|
Diluted weighted average number of common shares outstanding |
|
71,940,005 |
|
49,169,414 |
| ||
|
Loss per share — basic and diluted |
|
$ |
(0.08 |
) |
$ |
(0.13 |
) |
12. Changes in Non-Cash Operating Assets and Liabilities
Changes in non-cash balances related to operations are as follows:
|
|
|
December 31 |
| ||||
|
|
|
2014 |
|
2013 |
| ||
|
Accounts receivable |
|
$ |
(1,553,553 |
) |
$ |
613,321 |
|
|
Inventories |
|
7,444 |
|
(44,274 |
) | ||
|
Prepaid expenses and other receivables |
|
(21,393 |
) |
(46,976 |
) | ||
|
Taxes recoverable |
|
521,168 |
|
(390,391 |
) | ||
|
Accounts payable and accrued liabilities |
|
1,059,850 |
|
(1,774,724 |
) | ||
|
|
|
$ |
13,516 |
|
$ |
(1,643,044 |
) |
Included in accounts payable and accrued liabilities at the year ended December 31, 2014, is an amount related to patents and licenses of $31,655 (2013 - $14,365).
During the year ended December 31, 2014, there was $1,207,262 (2013 - $422,341) in interest paid and $nil in taxes paid (2013 — $nil).
During the year ended December 31, 2014, there was $118,815 (2013 - $63,816) of non-cash debt issuance costs (see Note 9) expensed as amortization of assets.
During the year ended December 31, 2014, 1,087,222 warrants (2013 - 755,794) were issued and valued at $424,471 (2013 - $445,794) in regards to the additional USD$8,000,000 ($8,935,800) advanced under the Credit Agreement and the Amended Credit Agreement (see Note 9).
During the year ended December 31, 2014, broker warrants were issued and valued at $1,177,468 in regards to the public offering that was completed (Note 10a) (2013 - $172,986).
13. Contingencies and Commitments
a) License Agreements
On December 1, 2011, the Company acquired 100% of the outstanding shares of Tribute Pharmaceuticals Canada Ltd. and Tribute Pharma Canada Inc. (“Tribute”). Included in this transaction were the following license agreements:
On June 30, 2008, Tribute signed a Sales, Marketing and Distribution Agreement with Actavis Group PTC ehf (“Actavis”) to perform certain sales, marketing, distribution, finance and other general management services in Canada in connection with the importation, marketing, sales and distribution of Bezalip® SR and Soriatane® (the “Actavis Products”). On January 1, 2010, a first amendment was signed with Actavis to grant the Company the right and obligation to more actively market and promote the Actavis Products in Canada. On March 31, 2011, a second amendment was signed with Actavis that extended the term of the agreement, modified the terms of the agreement and increased the Company’s responsibilities to include the day-to-day management of regulatory affairs, pharmacovigilance and medical information relating to the Actavis Products. The Company pays Actavis a sales and distribution fee up to an annual base-line net sales forecast plus an incremental fee for incremental net sales above the base-line. On May 4, 2011, the Company signed a Product Development and Profit Share Agreement with Actavis to develop, obtain regulatory approval of and market Bezalip SR in the U.S. The Company is required to pay US$5,000,000 ($5,800,500) to Actavis within 30 days of receipt of the regulatory approval to market Bezalip SR in the U.S.
On November 9, 2010, the Company signed a license agreement with Nautilus Neurosciences, Inc. (“Nautilus”) for the exclusive rights to develop, register, promote, manufacture, use, market, distribute and sell Cambia® in Canada. On August 11, 2011, the Company and Nautilus executed the first amendment to the license agreement and on September 30, 2012 executed the second amendment to the license agreement. Aggregate payments of US$1,000,000 ($1,005,820) were issued under this agreement, which included an upfront payment to Nautilus upon the execution of the agreement and an amount payable upon the first commercial sale of the product. These payments have been included in intangible assets and will be amortized over the life of the license agreement, as amended. Up to US$6,000,000 ($6,960,600) in additional one-time performance based sales milestones, based on a maximum of six different sales tiers, are payable over time, due upon achieving annual net sales ranging from US$2,500,000 ($2,900,250) to US$20,000,000 ($23,202,000) in the first year of the achievement of the applicable milestone. Royalty rates are tiered and payable at rates ranging from 22.5% to 27.5% of net sales.
On December 30, 2011, the Company signed a license agreement to commercialize MycoVa in Canada. As of September 30, 2014, this product has not been filed with Health Canada and to-date no upfront payments have been paid. Within 10 days of execution of a manufacturing agreement, the Company shall pay an up-front license fee of $200,000. Upon Health Canada approval of MycoVa, the Company shall pay $400,000. Sales milestones payments of $250,000 each are based on the achievement of aggregate net sales in increments of $5,000,000. Royalties are payable at rates ranging from 20% to 25% of net sales.
On May 13, 2014, the Company entered into an exclusive license and supply agreement with Faes Farma, S.A. (“Faes”), a Spanish pharmaceutical company, for the exclusive right to sell bilastine, a product for the treatment of allergic rhinitis and chronic idiopathic urticaria (hives) in Canada. The exclusive license is inclusive of prescription and non-prescription rights for bilastine, as well as adult and paediatric presentations in Canada. Sales of bilastine are subject to receiving regulatory approval from Health Canada. Payment for the licensing rights is based on an initial fee of €250,000 ($368,337), these payments have been included in intangible assets and will be amortized over the life of the license agreement. Any remaining milestone payments based on the achievement of specific
events, including regulatory and sales milestones of up to $3,558,813 (€1,466,600 ($2,058,813) and $1,500,000) are payable over time, beginning with an approval for bilastine from Health Canada. Milestones are payable upon attainment of cumulative net sales targets, up to net sales of $60,000,000. The license agreement is also subject to certain minimum purchase obligations upon regulatory approval and commercial sale of the product.
b) Executive Termination Agreements
The Company currently has employment agreements with the provision of termination and change of control benefits with officers and executives of the Company. The agreements for the officers and executives provide that in the event that any of their employment is terminated during the initial term (i) by the Company for any reason other than just cause or death; (ii) by the Company because of disability; (iii) by the officer or executive for good reason; or (iv) following a change of control, the officers and executives shall be entitled to the balance of the remuneration owing for the remainder of the initial term of up to an aggregate amount of $247,200 as of December 31, 2014 (2013 - $792,200) or if a change of control occurs subsequent to the initial term, while the officers or executives are employed on an indefinite basis, a lump sum payment of up to an aggregate amount of $2,072,200 (based on current base salaries). See Note 21b.
c) Consultant Royalty Agreements
The Company has consultant royalty agreements in place for several of its international license agreements. These agreements involve royalty payments to be issued to the consultants who assisted in locating the licensee who signed the license agreements with the Company.
The royalty payments issued to consultants include 10% of the upfront fees received from the licensee and 10% of any future milestone payments received. No royalties on license fees were paid for the year ended December 31, 2014 (2013 - $nil). In addition, royalty payments on product sales are also based on 4% to 5% of the total sales of Uracyst® at a declining rate of 1% per year over a three to five year period, declining to a 1% rate effective in the final year. The expenses recorded in regards to royalty fees on product sales for the year ended December 31, 2014 were $5,084 (2013 - $1,593). These amounts have been recorded as royalty expense in selling, general and administrative expenses on the statements of operations and comprehensive (loss).
d) Manufacturing Agreements
During 2014 and 2013, the Company’s NeoVisc® product was manufactured at Therapure Biopharma Inc. in Mississauga, Ontario, Canada and Uracyst® was manufactured by Jubilant HollisterStier, Inc. in Kirkland, Quebec, Canada. Under the terms of these agreements the Company is obligated to make payments for batches to be manufactured within the one year termination notification period.
e) Lease Obligations
The Company presently leases office and warehouse equipment under operating leases. For the year ended December 31, 2014, expenses related to these leases were $2,213 (2013 - $2,213). These amounts have been recorded as rent expense in selling, general and administrative expenses on the statements of operations and comprehensive (loss).
On September 1, 2012, the Company entered into a five year operating lease for its head office. For the year ended December 31, 2014, expenses related to this lease were $98,667 (2013 - $96,000).
As at December 31, 2014, minimum operating lease payments under these leases are as follows:
|
|
|
Total |
|
2015 |
|
2016 |
|
2017 |
| ||||
|
Operating lease obligations |
|
$ |
279,343 |
|
$ |
105,468 |
|
$ |
104,542 |
|
$ |
69,333 |
|
14. Significant Customers
During the year ended December 31, 2014, the Company had three significant pharmaceutical wholesale customers (2013 — two) that represented 60.1% (2013 — 58.2%) of product sales.
The Company believes that its relationships with these customers are satisfactory.
15. Related Party Transactions
During the years ended December 31, 2014 and 2013, the Company granted 200,000 and nil, respectively, options to purchase common shares of the Company, to LMT Financial Inc. (“LMT”), a company beneficially owned by a director and former interim officer of the Company, and his spouse for consulting services. For the year ended December 31, 2014, the Company recorded $77,333 as a non-cash expense. During the year ended December 31, 2013, the Company recorded and paid to LMT an aggregate of $60,000. These amounts have been recorded as selling, general and administrative expense in the statements of operations and comprehensive loss.
See Notes 10b and 13b.
16. Income Taxes
Rate reconciliation: A reconciliation of income tax (benefit) expense computed at the statutory income tax rate included in the statements of operations and comprehensive loss follows:
Income tax expense (benefit) is comprised of:
|
|
|
2014 |
|
2013 |
| ||
|
Income tax expense (benefit) at statutory rate at 26.5% (2013- 26.5%) |
|
$ |
(1,485,800 |
) |
$ |
(1,825,100 |
) |
|
Adjusted for: |
|
|
|
|
| ||
|
Impact on legislated changes in tax rates |
|
— |
|
28,400 |
| ||
|
Change in valuation allowance |
|
2,045,100 |
|
1,611,100 |
| ||
|
Share issue costs |
|
(1,070,400 |
) |
(409,200 |
) | ||
|
Non-deductible expenses |
|
241,500 |
|
484,900 |
| ||
|
Other |
|
269,600 |
|
109,900 |
| ||
|
Deferred income tax (recovery) |
|
$ |
— |
|
$ |
— |
|
Deferred tax assets and liabilities reflect losses carry-forward, the cumulative carry-forward pool of scientific research and experimental development (“SR&ED”) expenditures and the net effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and their corresponding tax basis. Significant components of net deferred tax assets are listed below:
Components of deferred income tax assets and liabilities:
|
|
|
2014 |
|
2013 |
| ||
|
Benefit of net operating losses carry-forward |
|
$ |
3,438,300 |
|
$ |
2,796,800 |
|
|
Book values of property, plant and equipment and intangible assets in excess of tax bases |
|
192,100 |
|
58,900 |
| ||
|
Benefit of SR&ED expenditures |
|
476,600 |
|
476,600 |
| ||
|
Share issue costs |
|
985,700 |
|
345,200 |
| ||
|
Non-refundable tax credits |
|
341,300 |
|
341,300 |
| ||
|
License agreements |
|
(2,030,000 |
) |
(2,273,000 |
) | ||
|
Long-term debt |
|
290,700 |
|
— |
| ||
|
Valuation allowance |
|
(3,694,700 |
) |
(1,745,800 |
) | ||
|
|
|
$ |
— |
|
$ |
— |
|
A valuation allowance was provided against certain deferred tax assets at December 31, 2014 and 2013, because the realization of the asset remains not determinable.
At December 31, 2013, the Company had non-capital losses carry-forward for income tax purposes in the amount of $10,553,600. At December 31, 2014, $1,013,100 of the non-capital losses carry-forward expired. The remaining losses, which may be applied against future years’ taxable income, expire as follows.
|
2026 |
|
$ |
231,900 |
|
|
2027 |
|
85,400 |
| |
|
2028 |
|
53,700 |
| |
|
2030 |
|
755,300 |
| |
|
2031 |
|
1,994,900 |
| |
|
2032 |
|
2,071,000 |
| |
|
2033 |
|
4,348,300 |
| |
|
2034 |
|
3,434,400 |
| |
|
|
|
$ |
12,974,900 |
|
Tax years 2008 through 2014 remain open to examination by the taxing jurisdictions to which the Company is subject. The Company has not been notified by any taxing jurisdictions of any proposed or planned examination.
The Company has non-refundable tax credits as at December 31, 2014 of $341,300 (2013 - $341,300).
The cumulative carry-forward pool of scientific research and experimental development (SR&ED) expenditures as at December 31, 2014 applicable to future years, with no expiry date, is $1,798,300 (2013 - $1,798,300). The tax credits have a full valuation allowance on them as they do not meet the more-likely-than-not test.
17. Segmented Information
The Company is a specialty pharmaceutical company with a primary focus on the acquisition, licensing, development and promotion of healthcare products in Canada. The Company targets several therapeutic areas in Canada, but has a particular interest in products for the treatment of pain, dermatology and endocrinology/cardiology. The Company also sells Uracyst® and NeoVisc® internationally through a number of strategic partnerships. Currently, all of the Company’s manufacturing assets are located in Canada. All direct sales take place in Canada. Licensing arrangements have been obtained to distribute and sell the Company’s products in various countries around the world.
Revenue for the years ended December 31, 2014 and 2013 includes products sold in Canada and international sales of products through licensing agreements. Revenue earned is as follows:
|
|
|
December 31 |
| ||||
|
|
|
2014 |
|
2013 |
| ||
|
Product sales: |
|
|
|
|
| ||
|
Canadian sales |
|
$ |
15,193,221 |
|
$ |
11,918,105 |
|
|
International sales |
|
1,619,372 |
|
1,277,678 |
| ||
|
Other revenue |
|
40,785 |
|
46,654 |
| ||
|
Total |
|
$ |
16,853,378 |
|
$ |
13,242,437 |
|
|
Royalty revenues |
|
18,414 |
|
197,924 |
| ||
|
Total revenues |
|
$ |
16,871,792 |
|
$ |
13,440,361 |
|
The Company currently sells its own products and is in-licensing other products in Canada. In addition, revenues include products which the Company out-licenses throughout most countries in Europe, the Caribbean, Austria, Germany, Italy, Lebanon, Kuwait, Malaysia, Portugal, Romania, Spain, South Korea, Turkey, Egypt, Hong Kong and the United Arab Emirates. The operations reflected in the statements of operations and comprehensive (loss) includes the Company’s activity in these markets.
18. Foreign Currency Gain (Loss)
The Company enters into foreign currency transactions in the normal course of business. Expenses incurred in currencies other than Canadian dollars are therefore subject to gains or losses due to fluctuations in these currencies. As at December 31, 2014, the Company held cash of $1,319,013 (US$1,135,304 and €1,387) in denominations other than in Canadian dollars (2013 - $1,211,602 (US$1,134,686 and €747)); had accounts receivables of $319,764 (US$67,125 and €172,313) denominated in foreign currencies (2013 - $258,027 (US$51,395 and €138,964); had accounts payable and accrued liabilities of $32,857 (US$26,125 and €1,816) denominated in foreign currencies (2013 — $115,373 (US$72,693 and €25,969)); warrant liability of $3,107,880 (US$2,682,994) (2013 - $2,966,714 (US$2,789,315)); and long term debt of $16,2541,400 (US$14,000,000) (2013 - $6,381,600 (US$6,000,000)). For the year ended December 31, 2014, the Company had a foreign currency loss of $762,801, (2013 — gain of $227,227). These amounts have been included in selling, general and administrative expenses in the statements of operations and comprehensive loss.
19. Financial Instruments
(a) Financial assets and liabilities — fair values
The carrying amounts of cash and cash equivalents, accounts receivable, certain other current assets, accounts payables and accrued liabilities are a reasonable estimate of their fair values because of the short maturity of these instruments.
Warrant liability and other current liability are financial liability where fluctuations in market rates will affect the fair value of these financial instruments.
Cash equivalents and other current liability are classified as Level 2 financial instruments within the fair value hierarchy.
(b) Derivative liability — warrant liability
In connection with various financing arrangements, the Company has granted warrants to purchase up to 14,754,587 common shares of the Company as disclosed in Note 10c. The warrants have a weighted average exercise price of US$0.55 ($0.64). The warrants expire at dates ranging from February 27, 2015 to February 4, 2021. The warrants are accounted for as derivative liabilities because the exercise price is denominated in a currency other than the Company’s functional currency.
The table below summarizes the fair value of the Company’s financial liabilities measured at fair value:
|
|
|
Fair Value at |
|
Fair Value Measurement Using |
| ||||||||
|
|
|
2014 |
|
Level 1 |
|
Level 2 |
|
Lev el 3 |
| ||||
|
Derivative liability - Warrants |
|
$ |
3,107,880 |
|
$ |
— |
|
$ |
— |
|
$ |
3,107,880 |
|
|
|
|
Fair Value at |
|
Fair Value Measurement Using |
| ||||||||
|
|
|
2013 |
|
Level 1 |
|
Level 2 |
|
Level 3 |
| ||||
|
Derivative liability - Warrants |
|
$ |
2,966,714 |
|
$ |
— |
|
$ |
— |
|
$ |
2,966,714 |
|
The table below sets forth a summary of changes in the fair value of the Company’s Level 3 financial liabilities (warrant derivative liability) for the years ended December 31, 2014 and December 31, 2013:
|
|
|
December |
|
December |
| ||
|
Balance at beginning of year |
|
$ |
2,966,714 |
|
$ |
202,213 |
|
|
Additions to derivative instruments, recognized as a discount to the carrying value of long term debt on the balance sheet |
|
424,471 |
|
445,794 |
| ||
|
Additions to derivative instruments, recognized as a reallocation from common shares |
|
— |
|
1,919,490 |
| ||
|
Change in fair value, recognized in earnings as Change in warrant liability |
|
(283,305 |
) |
399,217 |
| ||
|
Balance at the end of the year |
|
$ |
3,107,880 |
|
$ |
2,966,714 |
|
The following is quantitative information about significant unobservable inputs (level 3) for the Company as of December 31, 2014.
|
Liability Category |
|
Fair Value |
|
Valuation Technique |
|
Unobservable Input |
|
Input |
| |
|
Warrant Liability |
|
$ |
3,107,880 |
|
Black-Scholes valuation model |
|
Volatility |
|
88 |
% |
The following represents the impact on fair value measurements to changes in unobservable inputs:
|
Unobservable Inputs |
|
Increase in Inputs Increase in Valuation |
|
Decreases in Inputs Increase in Valuation |
|
|
Volatility |
|
Increase |
|
Decrease |
|
These instruments were valued using pricing models that incorporate the price of a common share (as quoted on the relevant over-the-counter trading market in the U.S.), volatility, risk free rate, dividend rate and estimated life. The Company computed the value of the warrants using the Black-Scholes model. There were no transfers of assets or liabilities between Level 1, Level 2, or Level 3 during the years ended December 31, 2014 and December 31, 2013.
The following are the key weighted average assumptions used in connection with this computation:
|
|
|
December 31, 2014 |
|
December 31, 2013 |
|
|
Number of shares underlying the warrants |
|
14,754,587 |
|
13,667,365 |
|
|
Fair market value of the warrant |
|
$US0.18 ($0.21) |
|
$US0.20($0.22) |
|
|
Exercise price |
|
$US0.55 ($0.64) |
|
$US0.55($0.58) |
|
|
Expected volatility |
|
88 |
% |
114 |
% |
|
Risk-free interest rate |
|
1.22 |
% |
1.58 |
% |
|
Expected dividend yield |
|
0 |
% |
0 |
% |
|
Expected warrant life (years) |
|
2.18 |
|
2.94 |
|
(c) Liquidity risk
The Company generates sufficient cash from operating and financing activities to fund its operations and fulfill its obligations as they become due. The Company has sufficient funds available through its cash, cash equivalents, and financing arrangements, should its cash requirements exceed cash generated from operations to cover financial liability obligations. The Company’s investment policy is to invest excess cash resources into highly liquid short-term investments purchased with an original maturity of three months or less with tier one financial institutions. As at December 31, 2014, there were no restrictions on the flow of these funds nor have any of these funds been committed in any way, except as outlined in the detailed notes.
In the normal course of business, management considers various alternatives to ensure that the Company can meet some of its operating cash flow requirements through financing activities, such as private placements of the Company’s common shares, preferred stock offerings and offerings of debt and convertible debt instruments as well as through merger or acquisition opportunities. Management may also consider strategic alternatives, including strategic investments and divestitures. As future operations may be financed out of funds generated from financing activities, the Company’s ability to do so is dependent on, among other factors, the overall state of capital markets and investor appetite for investments in the pharmaceutical industry and the Company’s securities in particular. Should the Company elect to satisfy its cash commitments through the issuance of securities, by way of either private placement or public offering or otherwise, there can be no assurance that its efforts to obtain such additional funding will be successful, or achieved on terms favorable to the Company or its existing shareholders. If adequate funds are not available on terms favorable to the Company, it may have to reduce substantially or eliminate expenditures such as promotion, marketing or production of its current or proposed products, or obtain funds through other sources such as divestiture or monetization of certain assets or sublicensing (where permitted) of certain rights to certain of its technologies or products.
(d) Concentration of credit risk and major customers
The Company considers its maximum credit risk to be $2,161,133 (2013 - $607,580). This amount is the total of the following financial assets: accounts receivable and loan receivable. The Company’s cash and cash equivalents are held through various high grade financial institutions.
The Company is exposed to credit risk from its customers and continually monitors its customers’ credit. It establishes the provision for doubtful accounts based upon the credit risk applicable to each customer. In line with other pharmaceutical companies, the Company sells its products through a small number of wholesalers and retail pharmacy chains in addition to hospitals, pharmacies, physicians and other groups. Note 14 discloses the significant customer details and the Company believes that the concentrations on the Company’s customers are considered normal for the Company and its industry.
As at December 31, 2014, the Company had two customers which made up 65.7% of the outstanding accounts receivable in comparison to three customers which made up 38.4% at December 31, 2013. As at December 31, 2014, 12.2% of the outstanding accounts receivable was related to product sales related to one wholesale account and 53.5% was related to an amount owing related to the product sales associated with the Novartis transition period (Note 2). As at December 31, 2013 all outstanding accounts receivables were related to product sales, of which $63,722 or 10.8% was related to one wholesale account and $163,220 or 27.6% was related to two international customers.
(e) Foreign exchange risk
The Company principally operates within Canada; however, a portion of the Company’s revenues, expenses, and current assets and liabilities, are denominated in United States dollars and the EURO. The Company’s long term debt is repayable in U.S. dollars, which exposes the Company to foreign exchange risk due to changes in the value of the Canadian dollar. As at December 31, 2014, a 5% change in the foreign exchange rate would increase/decrease the long term debt balance by $700,000 and would increase/decrease both interest expense and net loss by approximately $72,100 for the year ended December 31, 2014. As at December 31, 2014, a 5% change in the foreign exchange rate would increase/decrease the warrant liability balance by $155,400 and would increase/decrease both changes in warrant liability and net loss by $155,400 for the twelve month period ended December 31, 2014.
(f) Interest rate risk
The Company is exposed to interest rate fluctuations on its cash and cash equivalents as well as its long term debt. At December 31, 2014, the Company had an outstanding long term debt balance of US$14,000,000 ($16,241,400), which bears interest annually at a rate of 11.5% plus the LIBOR Rate with the LIBOR Rate being subject to a minimum floor of 2%, such that that minimum interest rate is 13.5%, which may expose the Company to market risk due to changes in interest rates. For the year ended December 31, 2014, a 1% increase in interest rates would increase interest expense and net loss by approximately $162,414. However, based on current LIBOR interest rates, which are currently under the minimum floor set at 2% and based on historical movements in LIBOR rates, the Company believes a near-term change in interest rates would not have a material adverse effect on the financial position or results of operations.
20. Derivative Financial Instruments
The Company enters into foreign currency contracts with financial institutions to reduce the risk that its cash flows and earnings will be adversely affected by foreign currency exchange rate fluctuations. In accordance with the Company’s current foreign exchange rate risk management policy, this program is not designated for trading or speculative purposes. The Company had no foreign currency contracts in place at December 31, 2014 (2013 — $5,000,000 notional principal with a fair value of ($38,156)). The Company has determined foreign currency call options to be Level 2 within the fair value hierarchy.
The Company recognizes derivative instruments as either assets or liabilities in the accompanying balance sheets at fair value.
The Company initially reports any gain or loss on the effective portion of the cash flow hedge as a component of other comprehensive income and subsequently reclassifies to the statements of operations when the hedged transaction occurs.
Valuation techniques used to measure fair value are intended to maximize the use of observable inputs and minimize the use of unobservable inputs.
The notional principal amounts provide one measure of the transaction volume outstanding as of December 31, 2014 and 2013, and do not represent the amount of the Company’s exposure to market loss. The estimates of fair value are based on applicable and commonly used pricing models using prevailing financial market information as of December 31, 2014 and 2013. The amounts ultimately realized upon settlement of these financial instruments, together with the gains and losses on the underlying exposures, will depend on actual market conditions during the remaining life of the instruments.
21. Comparative Figures
Certain comparative figures in 2013 have been reclassified to conform to the current year’s presentation. The reclassified information is as follows:
Reclassification of Operating and Non-operating income (expenses)
|
|
|
As |
|
Reclassified |
|
Adjusted |
| |||
|
Expenses |
|
|
|
|
|
|
| |||
|
Selling, general and administrative |
|
$ |
9,830,132 |
|
$ |
(340,553 |
) |
$ |
9,489,579 |
|
|
Amortization |
|
1,245,846 |
|
— |
|
1,245,846 |
| |||
|
Total operating expenses |
|
11,075,978 |
|
(340,553 |
) |
10,735,425 |
| |||
|
|
|
(5,078,708 |
) |
(340,553 |
) |
(4,738,155 |
) | |||
|
Non-operating income (expenses) |
|
|
|
|
|
|
| |||
|
Change in warrant liability |
|
(399,217 |
) |
— |
|
(399,217 |
) | |||
|
Loss on disposal of intangible asset |
|
(161,200 |
) |
— |
|
(161,200 |
) | |||
|
Loss on extinguishment of loan |
|
(620,835 |
) |
— |
|
(620,835 |
) | |||
|
Unrealized foreign currency exchange on debt |
|
— |
|
(340,553 |
) |
(340,553 |
) | |||
|
Accretion expense |
|
(103,775 |
) |
— |
|
(103,755 |
) | |||
|
Interest expense |
|
(527,079 |
) |
— |
|
(527,079 |
) | |||
|
Interest income |
|
3,559 |
|
— |
|
3,559 |
| |||
|
Loss and comprehensive loss before tax |
|
(6,887,255 |
) |
— |
|
(6,887,255 |
) | |||
|
Deferred income tax recovery |
|
314,900 |
|
— |
|
314,900 |
| |||
|
Net (loss) for the year |
|
$ |
(6,572,355 |
) |
$ |
— |
|
$ |
(6,572,355 |
) |
22. Subsequent Events
a) Employee Stock Options
On January 29, 2015, the Company granted 3,158,903 options to directors, officers, employees and consultants of the Company. The weighted average exercise price of these options is $0.62. Of these options 2,908,903 were granted to directors, officers and employee’s as performance based options and will vest one eighth at the end of each fiscal quarter following the date of grant, commencing on March 31, 2016, upon achieving certain financial objectives. In addition, 200,000 options were issued to a consultant and will vest on January 4, 2016. The remaining 50,000 options will vest equally over four quarters commencing on April 30, 2015. The options have a term of five years.
b) Executive Termination Agreements
The Company entered into new employment agreements with its officers and executives which became effective January 1, 2015 and include provisions of termination and change of control benefits. The agreements for the officers and executives provide that in the event that any of their employment is terminated during the term of the
agreement (i) by the Company for any reason other than just cause or death; (ii) by the Company because of disability; (iii) by the officer or executive for good reason; or (iv) following a change of control, the officers and executives shall be entitled to remuneration of a lump sum payment of up to an aggregate amount of $1,510,480 (based on current base salaries).
c) Warrants Exercised
As at February 25, 2015, the Company issued 4,612,500 common shares of the Company, for warrants exercised at an exercise price of US$0.50 per common share, for aggregate proceeds of US$2,306,250 ($2,833,644) (Note 10a and 10c). Of these, 2,656,250 common shares were issued to directors of the Company.
TRIBUTE PHARMACEUTICALS CANADA INC.
CONDENSED INTERIM BALANCE SHEETS
(Expressed in Canadian dollars)
(Unaudited)
|
|
|
As at |
|
As at |
| ||
|
ASSETS |
|
|
|
|
| ||
|
Current |
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
5,550,766 |
|
$ |
3,505,791 |
|
|
Accounts receivable, net of allowance of $nil (December 31, 2014 - $nil) (Note 16 d) |
|
3,124,207 |
|
2,145,319 |
| ||
|
Inventories (Note 2) |
|
1,193,968 |
|
1,037,387 |
| ||
|
Taxes recoverable |
|
119,062 |
|
130,623 |
| ||
|
Loan receivable |
|
15,814 |
|
15,814 |
| ||
|
Prepaid expenses and other receivables (Note 3) |
|
227,722 |
|
187,279 |
| ||
|
Current portion of debt issuance costs, net (Note 6) |
|
139,758 |
|
128,134 |
| ||
|
Other current asset (Note 17) |
|
43,400 |
|
— |
| ||
|
Total current assets |
|
10,414,697 |
|
7,150,347 |
| ||
|
Property, plant and equipment, net (Note 4) |
|
978,956 |
|
1,012,285 |
| ||
|
Intangible assets, net (Note 5) |
|
40,464,367 |
|
40,958,870 |
| ||
|
Goodwill |
|
3,599,077 |
|
3,599,077 |
| ||
|
Debt issuance costs, net (Note 6) |
|
366,404 |
|
359,161 |
| ||
|
Total assets |
|
$ |
55,823,501 |
|
$ |
53,079,740 |
|
|
LIABILITIES |
|
|
|
|
| ||
|
Current |
|
|
|
|
| ||
|
Accounts payable and accrued liabilities |
|
$ |
4,287,067 |
|
$ |
4,344,606 |
|
|
Current portion of long term debt (Note 6) |
|
1,525,656 |
|
1,319,030 |
| ||
|
Warrant liability (Note 7 c) |
|
5,359,374 |
|
3,107,880 |
| ||
|
Total current liabilities |
|
11,172,097 |
|
8,771,516 |
| ||
|
Long term debt (Note 6) |
|
15,260,858 |
|
13,967,493 |
| ||
|
Total liabilities |
|
26,432,955 |
|
22,739,009 |
| ||
|
Contingencies and commitments (Notes 6 and 10) |
|
|
|
|
| ||
|
SHAREHOLDERS’ EQUITY |
|
|
|
|
| ||
|
Capital Stock |
|
|
|
|
| ||
|
AUTHORIZED |
|
|
|
|
| ||
|
Unlimited Non-voting, convertible redeemable and retractable preferred shares with no par value |
|
|
|
|
| ||
|
Unlimited Common shares with no par value |
|
|
|
|
| ||
|
ISSUED (Note 7 a) |
|
|
|
|
| ||
|
Common shares 99,907,488 (December 31, 2014 — 94,476,238) |
|
45,022,189 |
|
41,182,630 |
| ||
|
Additional paid-in capital options (Note 7 b) |
|
3,061,924 |
|
2,713,605 |
| ||
|
Warrants (Note 7 c) |
|
6,347,349 |
|
6,347,349 |
| ||
|
Accumulated other comprehensive income (Note 17) |
|
43,400 |
|
— |
| ||
|
Deficit |
|
(25,084,316 |
) |
(19,902,853 |
) | ||
|
Total shareholders’ equity |
|
29,390,546 |
|
30,340,731 |
| ||
|
Total liabilities and shareholders’ equity |
|
$ |
55,823,501 |
|
$ |
53,079,740 |
|
See accompanying notes to the condensed interim financial statements.
TRIBUTE PHARMACEUTICALS CANADA INC.
CONDENSED INTERIM STATEMENTS OF OPERATIONS,
COMPREHENSIVE LOSS AND DEFICIT
(Expressed in Canadian dollars)
(Unaudited)
|
|
|
For the Three Month |
| ||||
|
|
|
2015 |
|
2014 |
| ||
|
Revenues |
|
|
|
|
| ||
|
Licensed domestic product net sales |
|
$ |
2,521,080 |
|
$ |
2,276,383 |
|
|
Other domestic product sales |
|
2,601,622 |
|
734,779 |
| ||
|
International product sales |
|
469,595 |
|
463,978 |
| ||
|
Royalty and licensing revenues |
|
— |
|
18,414 |
| ||
|
Total revenues (Notes 11 and 14) |
|
5,592,297 |
|
3,493,554 |
| ||
|
Cost of Sales |
|
|
|
|
| ||
|
Licensor sales and distribution fees |
|
1,452,064 |
|
1,413,043 |
| ||
|
Cost of products sold |
|
575,246 |
|
345,864 |
| ||
|
Total Cost of Sales |
|
2,027,310 |
|
1,758,907 |
| ||
|
Gross Profit |
|
3,564,987 |
|
1,734,647 |
| ||
|
Expenses |
|
|
|
|
| ||
|
Selling, general and administrative (Notes 7 b, 12 and 15) |
|
3,325,922 |
|
3,222,661 |
| ||
|
Amortization of assets |
|
621,623 |
|
290,352 |
| ||
|
Total operating expenses |
|
3,947,545 |
|
3,513,013 |
| ||
|
Loss from operations |
|
(382,558 |
) |
(1,778,366 |
) | ||
|
Non-operating income (expenses) |
|
|
|
|
| ||
|
Gain on derivative instrument (Note 17) |
|
— |
|
200,000 |
| ||
|
Change in warrant liability (Notes 7 c and 16 b) |
|
(2,695,600 |
) |
(1,411,774 |
) | ||
|
Unrealized foreign currency exchange on debt (Note 17) |
|
(1,433,456 |
) |
— |
| ||
|
Accretion expense (Note 6) |
|
(73,999 |
) |
(31,118 |
) | ||
|
Interest income |
|
125 |
|
372 |
| ||
|
Interest expense |
|
(595,975 |
) |
(267,292 |
) | ||
|
Net loss for the period |
|
$ |
(5,181,463 |
) |
$ |
(3,288,178 |
) |
|
Unrealized gain loss on derivative instrument, net of tax (Note 17) |
|
43,400 |
|
(103,488 |
) | ||
|
Net loss and comprehensive (loss) for the period |
|
$ |
(5,138,063 |
) |
$ |
(3,391,666 |
) |
|
Deficit, beginning of period |
|
(19,902,853 |
) |
(14,295,911 |
) | ||
|
Deficit, end of period |
|
$ |
(25,084,316 |
) |
$ |
(17,584,089 |
) |
|
Loss per share (Note 8) — Basic and diluted |
|
$ |
(0.05 |
) |
$ |
(0.06 |
) |
|
Weighted Average Number of Common Shares — Basic |
|
96,685,405 |
|
51,420,127 |
| ||
|
Weighted Average Number of Common Shares - Diluted |
|
96,685,405 |
|
51,420,127 |
| ||
See accompanying notes to the condensed interim financial statements.
TRIBUTE PHARMACEUTICALS CANADA INC.
CONDENSED INTERIM STATEMENTS OF CASH FLOWS
(Expressed in Canadian dollars)
(Unaudited)
For the Periods Ended March 31,
|
|
|
For Three Month |
| ||||
|
|
|
2015 |
|
2014 |
| ||
|
Cash flows from (used in) operating activities |
|
|
|
|
| ||
|
Net loss |
|
$ |
(5,181,463 |
) |
$ |
(3,288,178 |
) |
|
Items not affecting cash: |
|
|
|
|
| ||
|
Amortization |
|
644,458 |
|
295,128 |
| ||
|
Changes in warrant liability (Note 7 c) |
|
2,695,600 |
|
1,411,774 |
| ||
|
Stock-based compensation (Note 7 b) |
|
348,320 |
|
117,133 |
| ||
|
Accretion expense |
|
73,999 |
|
31,118 |
| ||
|
Paid-in common shares for services |
|
— |
|
211,812 |
| ||
|
Change in non-cash operating assets and liabilities (Note 9) |
|
(1,221,890 |
) |
(796,497 |
) | ||
|
Cash flows (used in) operating activities |
|
(2,640,976 |
) |
(2,017,710 |
) | ||
|
Cash flows from (used in) investing activities |
|
|
|
|
| ||
|
Additions to property, plant and equipment |
|
(4,972 |
) |
(4,353 |
) | ||
|
Increase in patents and licensing agreements |
|
(85,210 |
) |
(16,593 |
) | ||
|
Cash flows (used in) investing activities |
|
(90,182 |
) |
(20,946 |
) | ||
|
Cash flows from (used in) financing activities |
|
|
|
|
| ||
|
Financing costs deferred |
|
— |
|
(128,181 |
) | ||
|
Long term debt issued (Note 6) |
|
— |
|
2,211,000 |
| ||
|
Common shares issued (Note 7 a) |
|
3,395,453 |
|
— |
| ||
|
Cash flows from financing activities |
|
3,395,453 |
|
2,082,819 |
| ||
|
Changes in cash and cash equivalents |
|
664,295 |
|
44,163 |
| ||
|
Change in cash and cash equivalents due to changes in foreign exchange |
|
1,380,680 |
|
215,706 |
| ||
|
Cash and cash equivalents, beginning of period |
|
3,505,791 |
|
2,813,472 |
| ||
|
Cash and cash equivalents, end of period |
|
$ |
5,550,766 |
|
$ |
3,073,341 |
|
See accompanying notes to the condensed interim financial statements.
TRIBUTE PHARMACEUTICALS CANADA INC.
NOTES TO THE CONDENSED INTERIM FINANCIAL STATEMENTS
(Expressed in Canadian dollars)
(Unaudited)
1. Basis of Presentation
These unaudited condensed interim financial statements should be read in conjunction with the annual financial statements for Tribute Pharmaceuticals Canada Inc.’s (“Tribute” or the “Company”) most recently completed fiscal year ended December 31, 2014. These unaudited condensed interim financial statements do not include all disclosures required in annual financial statements, but rather are prepared in accordance with recommendations for interim financial statements in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”). These unaudited condensed interim financial statements have been prepared using the same accounting policies and methods as those used by the Company in the annual audited financial statements for the year ended December 31, 2014, except when disclosed below.
The unaudited condensed interim financial statements contain all adjustments (consisting of only normal recurring adjustments) which are necessary to present fairly the financial position of the Company as at March 31, 2015, and the results of its operations for the three month periods ended March 31, 2015 and 2014 and its cash flows for the three month periods ended March 31, 2015 and 2014. Note disclosures have been presented for material updates to the information previously reported in the annual audited financial statements.
a) Estimates
The preparation of these financial statements has required management to make estimates and assumptions that affect the amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of the revenues and expenses during the reporting period. On an ongoing basis, the Company evaluates its estimates, including those related to provision for doubtful accounts, accrued liabilities, income taxes, stock based compensation, revenue recognition, intangible assets and derivative financial instruments. The Company bases its estimates on historical experiences and on various other assumptions believed to be reasonable under the circumstances. Actual results could differ from those estimates. As adjustments become necessary, they are reported in earnings in the period in which they become known.
2. Inventories
|
|
|
March 31, |
|
December |
| ||
|
Raw materials |
|
$ |
322,450 |
|
$ |
290,197 |
|
|
Finished goods |
|
450,736 |
|
399,830 |
| ||
|
Packaging materials |
|
74,557 |
|
70,870 |
| ||
|
Work in process |
|
346,225 |
|
276,490 |
| ||
|
|
|
$ |
1,193,968 |
|
$ |
1,037,387 |
|
3. Prepaid Expenses and Other Receivables
|
|
|
March 31, |
|
December |
| ||
|
Prepaid operating expenses |
|
$ |
220,747 |
|
$ |
180,304 |
|
|
Interest receivable on loan receivables |
|
6,975 |
|
6,975 |
| ||
|
|
|
$ |
227,722 |
|
$ |
187,279 |
|
4. Property, Plant and Equipment
|
|
|
March 31, 2015 |
| |||||||
|
|
|
Cost |
|
Accumulated |
|
Net |
| |||
|
Land |
|
$ |
90,000 |
|
$ |
— |
|
$ |
90,000 |
|
|
Building |
|
618,254 |
|
308,526 |
|
309,728 |
| |||
|
Leasehold improvements |
|
10,359 |
|
5,180 |
|
5,179 |
| |||
|
Office equipment |
|
61,308 |
|
53,024 |
|
8,284 |
| |||
|
Manufacturing equipment |
|
1,103,524 |
|
623,971 |
|
479,553 |
| |||
|
Warehouse equipment |
|
17,085 |
|
17,085 |
|
— |
| |||
|
Packaging equipment |
|
111,270 |
|
66,319 |
|
44,951 |
| |||
|
Computer equipment |
|
147,844 |
|
106,583 |
|
41,261 |
| |||
|
|
|
$ |
2,159,644 |
|
$ |
1,180,688 |
|
$ |
978,956 |
|
|
|
|
December 31, 2014 |
| |||||||
|
|
|
Cost |
|
Accumulated |
|
Net |
| |||
|
Land |
|
$ |
90,000 |
|
$ |
— |
|
$ |
90,000 |
|
|
Building |
|
618,254 |
|
300,798 |
|
317,456 |
| |||
|
Leasehold improvements |
|
10,359 |
|
4,662 |
|
5,697 |
| |||
|
Office equipment |
|
61,308 |
|
52,124 |
|
9,184 |
| |||
|
Manufacturing equipment |
|
1,103,525 |
|
602,667 |
|
500,858 |
| |||
|
Warehouse equipment |
|
17,085 |
|
17,085 |
|
— |
| |||
|
Packaging equipment |
|
111,270 |
|
62,744 |
|
48,526 |
| |||
|
Computer equipment |
|
142,873 |
|
102,309 |
|
40,564 |
| |||
|
|
|
$ |
2,154,674 |
|
$ |
1,142,389 |
|
$ |
1,012,285 |
|
5. Intangible Assets
|
|
|
March 31, 2015 |
| |||||||
|
|
|
Cost |
|
Accumulated |
|
Net |
| |||
|
Patents |
|
$ |
436,964 |
|
$ |
6,463 |
|
$ |
374,501 |
|
|
Licensing asset |
|
1,005,820 |
|
193,427 |
|
812,393 |
| |||
|
Licensing agreements |
|
10,377,325 |
|
2,575,022 |
|
7,802,303 |
| |||
|
Product rights |
|
32,117,521 |
|
642,351 |
|
31,475,170 |
| |||
|
|
|
$ |
43,937,630 |
|
$ |
3,473,263 |
|
$ |
40,464,367 |
|
|
|
|
December 31, 2014 |
| |||||||
|
|
|
Cost |
|
Accumulated |
|
Net |
| |||
|
Patents |
|
$ |
351,754 |
|
$ |
53,242 |
|
$ |
298,512 |
|
|
Licensing asset |
|
1,005,820 |
|
174,084 |
|
831,736 |
| |||
|
Licensing agreements |
|
10,377,325 |
|
2,345,049 |
|
8,032,276 |
| |||
|
Product rights |
|
32,117,521 |
|
321,175 |
|
31,796,346 |
| |||
|
|
|
$ |
43,852,420 |
|
$ |
2,893,550 |
|
$ |
40,958,870 |
|
Amortization expense of intangible assets for the three month period ended March 31, 2015 was $579,712 (2014 - $252,185).
The Company has patents pending of $45,942 at March 31, 2015 (December 31, 2014 - $45,392) and licensing agreements of $373,325 (December 31, 2014 - $373,325) not currently being amortized.
6. Long Term Debt and Debt Issuance Costs
On August 8, 2013, SWK Funding LLC (“SWK”), a wholly-owned subsidiary of SWK Holdings Corporation, entered into a credit agreement (the “Credit Agreement”) with the Company and SWK pursuant thereto, provided to the Company a term loan in the principal amount of US$6,000,000 ($6,381,600) which was increased, as per the terms of the Credit Agreement, by an additional US$2,000,000 ($2,211,000) at the Company’s request on February 4, 2014. SWK served as the agent under the Credit Agreement.
On October 1, 2014 (the “Amendment Closing Date”), the Company entered into the First Amendment to the Credit Agreement and Guarantee (the “First Amendment,” and together with the Credit Agreement, the “Amended Credit Agreement”) with SWK. The Amended Credit Agreement provides for a multi-draw term loan to the Company for up to a maximum amount of US$17,000,000 ($21,561,100) (the “Loan Commitment Amount”). On the Amendment Closing Date, SWK advanced the Company an additional amount equal to US$6,000,000 ($6,724,800) pursuant to the terms of a promissory note executed on the Amendment Closing Date (the “October 2014 Note”). The October 2014 Note is for a total principal amount of US$14,000,000 ($17,756,200) (the “Loan”) (comprised of US$8,000,000 ($8,592,600) advanced under the Credit Agreement and the additional US$6,000,000 ($6,724,800) advanced on October 1, 2014) due and payable on December 31, 2018 (the “Term Loan Maturity Date”).
The Loan accrues interest at an annual rate of 11.5% plus the Libor Rate (as defined in the Amended Credit Agreement), with the Libor Rate being subject to a minimum floor of 2%, such that the minimum interest rate is 13.5%. In the event of a change of control, a merger or a sale of all or substantially all of the Company’s assets, the Loan shall be due and payable.
The discount to the carrying value of the Loan is being amortized as a non-cash interest expense over the term of the Loan using the effective interest rate method.
During the three month period ended March 31, 2015, the Company accreted $73,999 (2014 - $31,118) in non-cash accretion expense in connection with the long term loans, which is included in accretion expense on the condensed interim statements of operations, comprehensive loss and deficit.
Legal fees and costs associated with the Loan Commitment Amount were classified as debt issuance costs on the balance sheet. These assets are being amortized as a non-cash interest expense over the term of the outstanding Loan using the effective interest rate method. During the three month period ended March 31, 2015, the Company amortized $26,446 (2014 — $24,764) in non-cash interest expense, which is included in amortization expense on the condensed interim statements of operations, comprehensive loss and deficit.
During the three month period ended March 31, 2015, the Company made no principal payments (year ended December 31, 2014 - $nil) and interest payments of US$477,750 ($606,474) (year ended December 31, 2014 — US$1,090,500 ($1,207,262)) under the Credit Agreement and Amended Credit Agreement. The Company has estimated the following revenue-based principal and interest payments over the next four years ended December 31
based on the assumption that only the minimum revenue requirements will be met under the Amended Credit Agreement:
|
|
|
Principal Payments |
|
Interest Payments |
|
|
2015 |
|
US$1,136,997($1,442,053) |
|
US$1,390,095($1,763,057) |
|
|
2016 |
|
US$1,454,476($1,844,712) |
|
US$1,704,169($2,161,398) |
|
|
2017 |
|
US$1,666,664($2,113,830) |
|
US$1,489,600($1,889,260) |
|
|
2018 |
|
US$9,741,863($12,355,605) |
|
US$1,428,903($1,812,278) |
|
7. Capital Stock
(a) Common Shares
During the three month period ended March 31, 2015, the Company issued 5,431,250 common shares on the exercise of 5,431,250 common share purchase warrants exercised at an average exercise price of US$0.50 ($0.625) for gross proceeds of US$2,715,600 ($3,395,453).
|
|
|
Number of |
|
Amount |
| |
|
Common Shares |
|
|
|
|
| |
|
Balance, December 31, 2014 |
|
94,476,238 |
|
$ |
41,182,630 |
|
|
Warrants exercised |
|
5,431,250 |
|
3,395,453 |
| |
|
Fair value of warrants exercised |
|
— |
|
444,106 |
| |
|
Balance, March 31, 2015 |
|
99,907,488 |
|
$ |
45,022,189 |
|
(b) Stock Based Compensation
The Company’s stock-based compensation program (“Plan”) includes stock options in which some options vest based on continuous service, while others vest based on performance conditions such as profitability and sales goals. For those equity awards that vest based on continuous service, compensation expense is recorded over the service period from the date of grant. For performance-based awards, compensation expense is recorded over the remaining service period when the Company determines that achievement is probable.
During the three month period ended March 31, 2015, there were 3,158,903 options, granted to officers, employees and consultants of the Company (2014 — 1,298,245). The exercise price of 2,908,903 of these options is $0.62, vesting quarterly one-eighth over two years on each of March 31, June 30, September 30 and December 31, in 2016 and 2017. Of these options 864,000 are time-based, while the remaining 2,044,903 are based upon achieving certain financial objectives. Since stock-based compensation is recognized only for those awards that are ultimately expected to vest, the Company has applied an estimated forfeiture rate (based on historical experience and projected employee turnover) to unvested awards for the purpose of calculating compensation expense. The grant date fair value of these options was estimated as $0.51 using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 121%; expected risk free interest rate of 0.61%; and expected term of 5 years.
In addition, 200,000 options were granted with an exercise price of $0.62 and will fully vest on January 4, 2016 (Note 12). The grant date fair value of these options was estimated as $0.43 using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 121%; expected risk free interest rate of 0.87%; and expected term of 5 years.
The remaining 50,000 options were granted with an exercise price of $0.62, with one quarter vesting over one year on each of April 29, July 29, October 29 in 2015 and January 29, 2016. The grant date fair value of these options was estimated as $0.52 using the Black-Scholes option pricing model, based on the following assumptions: expected dividend yield of 0%; expected volatility of 122%; expected risk free interest rate of 0.87%; and expected term of 5 years.
For the three month period ended March 31, 2015, the Company recorded $348,320 (2014 — $117,133) as additional paid in capital for options issued to directors, officers, employees and consultants based on continuous service. Included in this amount is $171,759 for options issued to consultants for services (Note 12). This expense was recorded as selling, general and administrative expense on the condensed interim statements of operations, comprehensive loss and deficit. Due to termination of employment and non-achievement of performance-based awards, 129,587 options were removed from the number of options issued during the three month period ended March 31, 2015 (year ended December 31, 2014 — 817,830).
The activities in additional paid in-capital options are as follows:
|
|
|
Amount |
| |
|
Balance, December 31, 2014 |
|
$ |
2,713,605 |
|
|
Expense recognized for options issued to employees |
|
176,560 |
| |
|
Expense recognized for options issued to consultants |
|
171,759 |
| |
|
Balance, March 31, 2015 |
|
$ |
3,061,924 |
|
The total number of options outstanding as at March 31, 2015 was 7,864,307 (December 31, 2014 — 4,834,991). The weighted average grant date fair value of the options granted during the three month period ended March 31, 2015, was $0.51 (2014 - $0.34).
The maximum number of options that may be issued under the Plan is floating at an amount equivalent to 10% of the issued and outstanding common shares, or 9,990,749 as at March 31, 2015 (December 31, 2014 — 9,447,624).
(c) Warrants
As at March 31, 2015, the following warrants were outstanding:
Warrant Liability
|
Expiration Date |
|
Number |
|
Weighted Average |
|
Fair |
|
Fair |
| ||
|
May 11, 2017 |
|
750,000 |
|
US$0.43($0.55) |
|
$ |
430,905 |
|
$ |
227,090 |
|
|
February 27, 2015 |
|
— |
|
US$0.50($0.63) |
|
$ |
— |
|
$ |
184,999 |
|
|
February 27, 2018 |
|
4,429,687 |
|
US$0.60($0.76) |
|
$ |
2,522,559 |
|
$ |
1,310,414 |
|
|
March 5, 2015 |
|
— |
|
US$0.50($0.63) |
|
$ |
— |
|
$ |
56,691 |
|
|
March 5, 2018 |
|
1,253,000 |
|
US$0.60($0.76) |
|
$ |
713,542 |
|
$ |
372,123 |
|
|
March 11, 2015 |
|
— |
|
US$0.50($0.63) |
|
$ |
— |
|
$ |
17,547 |
|
|
March 11, 2018 |
|
343,750 |
|
US$0.60($0.76) |
|
$ |
195,754 |
|
$ |
102,089 |
|
|
August 8, 2018 |
|
755,794 |
|
US$0.5954($0.7551) |
|
$ |
588,564 |
|
$ |
334,060 |
|
|
September 20, 2018 |
|
108,696 |
|
US$0.55($0.70) |
|
$ |
67,275 |
|
$ |
36,442 |
|
|
February 4, 2021 |
|
347,222 |
|
US$0.4320($0.5479) |
|
$ |
281,404 |
|
$ |
160,319 |
|
|
October 1, 2021 |
|
740,000 |
|
US$0.70($0.89) |
|
$ |
559,371 |
|
$ |
306,106 |
|
|
|
|
8,728,149 |
|
US$0.59($0.74) |
|
$ |
5,359,374 |
|
$ |
3,107,880 |
|
ASC 815 “Derivatives and Hedging” indicates that warrants with exercise prices denominated in a currency other than an entity’s functional currency should not be classified as equity. As a result, these warrants have been treated as derivatives and recorded as liabilities carried at their fair value, with period-to-period changes in the fair value recorded as a gain or loss in the condensed interim statements of operations, comprehensive loss and deficit. The Company treated the compensation warrants as a liability upon their issuance. The warrant liability is classified as Level 3 within the fair value hierarchy (see Note 16(b)).
As at March 31, 2015, the fair value of the aggregate warrant liability of $5,359,374 (December 31, 2014 - $3,107,880) was estimated using the Black-Scholes option pricing model based on the following weighted average assumptions: expected dividend yield of 0% (December 31, 2014 — 0%) expected volatility of 100% (December 31, 2014 — 88%) risk-free interest rate of 0.78% (December 31, 2014 — 1.22%) and expected term of 3.32 years (December 31, 2014 — 2.18 years).
Warrants — Equity
|
Expiration Date |
|
Number of |
|
Weighted |
|
Grant Date |
| ||
|
July 15, 2016 |
|
21,447,500 |
|
$ |
0.90 |
|
$ |
5,169,881 |
|
|
July 15, 2016 |
|
3,217,125 |
|
$ |
0.70 |
|
$ |
1,177,468 |
|
|
|
|
24,664,625 |
|
$ |
0.90 |
|
$ |
6,347,349 |
|
8. Loss Per Share
The treasury stock method assumes that proceeds received upon the exercise of all warrants and options outstanding in the period is used to repurchase the Company’s shares at the average share price during the period. The diluted loss per share is not computed when the effect of such calculation is anti-dilutive. In periods when losses are reported, the weighted-average number of common shares outstanding excludes common stock equivalents because their inclusion would be anti-dilutive. Potentially dilutive securities, which were not included in diluted weighted average shares for the three month periods ended March 31, 2015 and 2014, consisted of outstanding stock options (7,864,307 and 5,109,330, respectively) and outstanding warrant grants (33,392,774 and 14,014,587, respectively).
The following table sets forth the computation of loss per share:
|
|
|
For the Three Month |
| ||||
|
|
|
2015 |
|
2014 |
| ||
|
Numerator: |
|
|
|
|
| ||
|
Net loss available to common shareholders |
|
$ |
(5,181,463 |
) |
$ |
(3,288,178 |
) |
|
Denominator: |
|
|
|
|
| ||
|
Weighted average number of common shares |
|
96,685,405 |
|
51,420,127 |
| ||
|
Effect of dilutive common shares |
|
— |
|
— |
| ||
|
Diluted weighted average number of common shares outstanding |
|
96,685,405 |
|
51,420,127 |
| ||
|
Loss per share — basic and diluted |
|
$ |
(0.05 |
) |
$ |
(0.06 |
) |
9. Statement of Cash Flows
Changes in non-cash balances related to operations are as follows:
|
|
|
For the Three Months |
| ||||
|
|
|
2015 |
|
2014 |
| ||
|
Accounts receivable |
|
$ |
(978,888 |
) |
$ |
(1,065,085 |
) |
|
Inventories |
|
(156,581 |
) |
(59,101 |
) | ||
|
Prepaid expenses and other receivables |
|
(40,443 |
) |
(9,036 |
) | ||
|
Taxes recoverable |
|
11,561 |
|
529,381 |
| ||
|
Accounts payable and accrued liabilities |
|
(57,539 |
) |
(192,656 |
) | ||
|
|
|
$ |
(1,221,890 |
) |
$ |
(796,497 |
) |
Included in accounts payable and accrued liabilities at March 31, 2015, is an amount related to patents and licenses of $2,605 (December 31, 2014 - $31,655).
During the three month period ended March 31, 2015, there was $606,474 (2014 - $267,291) in interest paid and $nil in taxes paid (2014 — $nil).
During the three month period ended March 31, 2015, there was $26,446 (2014 - $24,764) of non-cash debt issuance costs (see Note 6) expensed as amortization of assets.
10. Contingencies and Commitments
The Company has royalty, licensing and manufacturing agreements that have remained in effect for the Company during the quarter. In addition, there were no material changes to the lease agreements during the period.
(a) License Agreements
On December 1, 2011, the Company acquired 100% of the outstanding shares of Tribute Pharmaceuticals Canada Ltd. and Tribute Pharma Canada Inc. Included in this transaction were the following license agreements:
On June 30, 2008, Tribute signed a Sales, Marketing and Distribution Agreement with Actavis Group PTC ehf (“Actavis”) to perform certain sales, marketing, distribution, finance and other general management services in Canada in connection with the importation, marketing, sales and distribution of Bezalip® SR and Soriatane® (the “Actavis Products”). On January 1, 2010, a first amendment was signed with Actavis to grant the Company the right and obligation to more actively market and promote the Actavis Products in Canada. On March 31, 2011, a second amendment was signed with Actavis that extended the term of the agreement, modified the terms of the agreement and increased the Company’s responsibilities to include the day-to-day management of regulatory affairs,
pharmacovigilance and medical information relating to the Actavis Products. The Company pays Actavis a sales and distribution fee up to an annual base-line net sales forecast plus an incremental fee for incremental net sales above the base-line. On May 4, 2011, the Company signed a Product Development and Profit Share Agreement with Actavis to develop, obtain regulatory approval of and market Bezalip SR in the U.S. The Company shall pay US$5,000,000 ($6,341,500) to Actavis within 30 days of receipt of the regulatory approval to market Bezalip SR in the U.S.
On November 9, 2010, the Company signed a license agreement with Nautilus Neurosciences, Inc. (“Nautilus”) for the exclusive rights to develop, register, promote, manufacture, use, market, distribute and sell Cambia® in Canada. On August 11, 2011, the Company and Nautilus executed the first amendment to the license agreement and on September 30, 2012 executed the second amendment to the license agreement. Aggregate payments of US$1,000,000 ($1,005,820) were issued under this agreement, which included an upfront payment to Nautilus upon the execution of the agreement and an amount payable upon the first commercial sale of the product. These payments have been included in intangible assets and will be amortized over the life of the license agreement, as amended. Up to US$6,000,000 ($7,609,800) in additional one-time performance based sales milestones, based on a maximum of six different sales tiers, are payable over time, due upon achieving annual net sales ranging from US$2,500,000 ($3,170,750) to US$20,000,000 ($25,366,000) in the first year of the achievement of the applicable milestone. Royalty rates are tiered and payable at rates ranging from 22.5% to 25.0% of net sales.
On December 30, 2011, the Company signed a license agreement with Apricus Bioscience, Inc. to commercialize MycoVa in Canada. As of March 31, 2015, this product has not been filed with Health Canada and to-date no upfront payments have been paid. Within 10 days of execution of a manufacturing agreement, the Company shall pay an up-front license fee of $200,000. Upon Health Canada approval of MycoVa, the Company shall pay $400,000. Sales milestones payments of $250,000 each are based on the achievement of aggregate net sales in increments of $5,000,000. Royalties are payable at rates ranging from 20% to 25% of net sales.
On May 13, 2014, the Company entered into an exclusive license and supply agreement with Faes Farma, S.A. (“Faes”), a Spanish pharmaceutical company, for the exclusive right to sell bilastine, a product for the treatment of allergic rhinitis and chronic idiopathic urticaria (hives) in Canada. The exclusive license is inclusive of prescription and non-prescription rights for bilastine, as well as adult and paediatric presentations in Canada. Sales of bilastine are subject to receiving regulatory approval from Health Canada. Payment for the licensing rights is based on an initial fee of €250,000 ($368,337), these payments have been included in intangible assets and will be amortized over the life of the license agreement. Any remaining milestone payments based on the achievement of specific events, including regulatory and sales milestones of up to $3,497,949 (€1,466,600 ($1,997,949) and $1,500,000) are payable over time, beginning with an approval for bilastine from Health Canada. Thereafter, milestones are payable upon attainment of cumulative net sales targets, up to net sales of $60,000,000. The license agreement is also subject to certain minimum purchase obligations upon regulatory approval and commercial sale of product.
(b) Executive Termination Agreements
The Company currently has employment agreements with the provision of termination and change of control benefits with officers and executives of the Company. The agreements for the officers and executives provide that in the event that any of their employment is terminated during the term (i) by the Company for any reason other than just cause or death; (ii) by the Company because of disability; (iii) by the officer or executive for good reason; or (iv) following a change of control, the officers and executives shall be entitled to an aggregate amount of $1,979,540 as of March 31, 2015 (December 31, 2014 - $247,200) or if a change of control occurs while the officers or executives are employed on an indefinite basis, a lump sum payment of up to an aggregate amount of $3,499,562 (based on current base salaries) (December 31, 2014 - $2,072,200).
11. Significant Customers
During the three month period ended March 31, 2015, the Company had three significant wholesale customers (2014 — three) that represented 69.8% (2014 — 65.6%) of product sales.
The Company believes that its relationship with these customers is satisfactory.
12. Related Party Transactions
During the three month period ended March 31, 2015 the Company granted 200,000 (2014 - 200,000) stock options to LMT Financial Inc. (“LMT”), a company beneficially owned by a director and former interim officer of the
Company, and his spouse for consulting services. For the three month period ended March 31, 2015, the Company recorded $49,147 (2014 - $16,222) as a non-cash expense. These amounts have been recorded as selling, general and administrative expense in the condensed interim statements of operations, comprehensive loss and deficit.
13. Income Taxes
The Company has no taxable income under Canadian Federal and Provincial tax laws for the three month period ended March 31, 2015 and 2014. The Company has non-capital loss carry-forwards at March 31, 2015 totaling approximately $13,953,400, which may be offset against future taxable income. If not utilized, the loss carry-forwards will expire between 2015 and 2034. The cumulative carry-forward pool of SR&ED expenditures as at March 31, 2015, that may be offset against future taxable income, with no expiry date, is $1,798,300.
The non-refundable portion of the tax credits as at March 31, 2015 was $341,300.
14. Segmented Information
The Company is a specialty pharmaceutical company with a primary focus on the acquisition, licensing, development and promotion of healthcare products in Canada. The Company targets several therapeutic areas in Canada, but has a particular interest in products for the treatment of pain, dermatology and endocrinology/cardiology. The Company also sells Uracyst® and NeoVisc® internationally through a number of strategic partnerships. Currently, all of the Company’s manufacturing assets are located in Canada. All direct sales take place in Canada. Licensing arrangements have been obtained to distribute and sell the Company’s products in various countries around the world.
Revenue for the three month periods ended March 31, 2015 and 2014 includes products sold in Canada and international sales of products through licensing agreements. Revenue earned is as follows:
|
|
|
For the Three Month |
| ||||
|
|
|
2015 |
|
2014 |
| ||
|
Product sales: |
|
|
|
|
| ||
|
Domestic sales |
|
$ |
5,109,509 |
|
$ |
2,999,912 |
|
|
International sales |
|
469,595 |
|
463,978 |
| ||
|
Other revenue |
|
13,193 |
|
11,250 |
| ||
|
Total |
|
$ |
5,592,297 |
|
$ |
3,475,140 |
|
|
Royalty revenues |
|
$ |
— |
|
$ |
18,414 |
|
|
Total revenues |
|
$ |
5,592,297 |
|
$ |
3,493,554 |
|
The Company currently sells its own products and is in-licensing other products in Canada. In addition, revenues include products which the Company out-licenses throughout most countries in Europe, the Caribbean, Austria, Germany, Italy, Lebanon, Kuwait, Malaysia, Portugal, Romania, Spain, South Korea, Turkey, Egypt, Hong Kong and the United Arab Emirates. The operations reflected in the condensed interim statements of operations, comprehensive loss and deficit includes the Company’s activity in these markets.
15. Foreign Currency Gain (Loss)
The Company enters into foreign currency transactions in the normal course of business. Expenses incurred in currencies other than Canadian dollars are therefore subject to gains or losses due to fluctuations in these currencies. As at March 31, 2015, the Company held cash of $5,326,943 (US$4,129,010 and €66,152) in denominations other than in Canadian dollars (December 31, 2014 - $1,319,013 (US$1,135,304 and €1,387)); had accounts receivables of $389,930 (US$101,157 and €192,052) denominated in foreign currencies (December 31, 2014 - $319,764 (US$67,125 and €172,313); had accounts payable and accrued liabilities of $230,109 (US$109,785 and €66,703) denominated in foreign currencies (December 31, 2014 — $32,857 (US$26,125 and €1,816)); warrant liability of $5,359,374 (US$4,225,636) (December 31, 2014 - $3,107,880 (US$2,682,994)); and long term debt of $17,756,200 (US$14,000,000) (December 31, 2014 - $16,241,400 (US$14,000,000)). For the three month period ended March 31, 2015, the Company had a foreign currency loss of $1,179,509 (2014 —$196,066). These amounts have been included in selling, general and administrative expenses in the condensed interim statements of operations, comprehensive loss and deficit.
16. Financial Instruments
(a) Financial assets and liabilities — fair values
The carrying amounts of cash and cash equivalents, accounts receivable, certain other current assets, accounts payables and accrued liabilities are a reasonable estimate of their fair values because of the short maturity of these instruments.
Warrant liability and other current asset/liabilities are financial assets/liabilities where fluctuations in market rates will affect the fair value of these financial instruments. The Company uses the following hierarchy for determining and disclosing the fair value of financial instruments by valuation technique:
Level 1: quoted prices in active markets for identical assets or liabilities.
Level 2: other techniques for which all inputs which have a significant effect on the recorded fair value are observable, either directly or indirectly.
Level 3: techniques which use inputs which have a significant effect on the recorded fair value that are not based on observable market data.
Cash equivalents and other current asset/liabilities are classified as Level 2 financial instruments within the fair value hierarchy.
(b) Derivative liability — warrant liability
In connection with various financing arrangements, the Company has granted warrants to purchase up to 8,728,149 common shares of the Company as disclosed in Note 7c. The warrants have a weighted average exercise price of US$0.59 ($0.74). The warrants expire at dates ranging from May 11, 2017 to October 1, 2021. The warrants are accounted for as derivative liabilities because the exercise price is denominated in a currency other than the Company’s functional currency.
The table below summarizes the fair value of the Company’s financial liabilities measured at fair value:
|
|
|
Fair Value |
|
Fair Value Measurement Using |
| ||||||||
|
|
|
2015 |
|
Level 1 |
|
Level 2 |
|
Level 3 |
| ||||
|
Derivative liability - Warrants |
|
$ |
5,359,374 |
|
$ |
— |
|
$ |
— |
|
$ |
5,359,374 |
|
|
|
|
Fair Value |
|
Fair Value Measurement Using |
| ||||||||
|
|
|
31, 2014 |
|
Level 1 |
|
Level 2 |
|
Level 3 |
| ||||
|
Derivative liability - Warrants |
|
$ |
3,107,880 |
|
$ |
— |
|
$ |
— |
|
$ |
3,107,880 |
|
The table below sets forth a summary of changes in the fair value of the Company’s Level 3 financial liabilities (warrant derivative liability) for the periods ended March 31, 2015 and December 31, 2014:
|
|
|
Three |
|
Year |
| ||
|
Balance at beginning of period |
|
$ |
3,107,880 |
|
$ |
2,966,714 |
|
|
Additions (deletions) to derivative instruments |
|
(444,106 |
) |
424,471 |
| ||
|
Change in fair market value, recognized in earnings as Change in warrant liability |
|
2,695,600 |
|
(283,305 |
) | ||
|
Balance end of period |
|
$ |
5,359,374 |
|
$ |
3,107,880 |
|
The following is quantitative information about significant unobservable inputs (Level 3) for the Company as of March 31, 2015.
|
Liability Category |
|
Fair Value |
|
Valuation Technique |
|
Unobservable Input |
|
Input |
| |
|
Warrant Liability |
|
$ |
5,359,374 |
|
Black-Scholes valuation model |
|
Volatility |
|
100 |
% |
The following represents the impact on fair value measurements to changes in unobservable inputs:
|
Unobservable Inputs |
|
Increase in Inputs Increase in Valuation |
|
Decreases in Inputs Increase in Valuation |
|
Volatility |
|
Increase |
|
Decrease |
These instruments were valued using pricing models that incorporate the price of a common share (as quoted on the relevant over-the-counter trading market in the U.S.), volatility, risk free rate, dividend rate and estimated life. The Company computed the value of the warrants using the Black-Scholes model. There were no transfers of assets or liabilities between Level 1, Level 2, or Level 3 during the periods ended March 31, 2015 and December 31, 2014.
The following are the key weighted average assumptions used in connection with this computation:
|
|
|
Three Months Period |
|
Year Ended |
|
|
Number of shares underlying the warrants |
|
8,728,149 |
|
14,754,587 |
|
|
Fair market value of the stock |
|
US$0.48 $(0.61) |
|
US$0.18 $(0.21) |
|
|
Exercise price |
|
US$0.59 $(0.75) |
|
US$0.55 $(0.64) |
|
|
Expected volatility |
|
100% |
|
88% |
|
|
Risk-free interest rate |
|
0.78% |
|
1.22% |
|
|
Expected dividend yield |
|
0% |
|
0% |
|
|
Expected warrant life (years) |
|
3.32 |
|
2.18 |
|
(c) Liquidity risk
The Company generates sufficient cash from operating and financing activities to fund its operations and fulfill its obligations as they become due. The Company has sufficient funds available through its cash, cash equivalents, and financing arrangements, should its cash requirements exceed cash generated from operations to cover financial liability obligations. The Company’s investment policy is to invest excess cash resources into highly liquid short-term investments purchased with an original maturity of three months or less with tier one financial institutions. As at March 31, 2015, there were no restrictions on the flow of these funds nor have any of these funds been committed in any way, except as outlined in the detailed notes.
In the normal course of business, management considers various alternatives to ensure that the Company can meet some of its operating cash flow requirements through financing activities, such as private placements of our common stock, preferred stock offerings and offerings of debt and convertible debt instruments as well as through merger or acquisition opportunities. Management may also consider strategic alternatives, including strategic investments and divestitures. As future operations may be financed out of funds generated from financing activities, the Company’s ability to do so is dependent on, among other factors, the overall state of capital markets and investor appetite for investments in the pharmaceutical industry and our securities in particular. Should the Company elect to satisfy its cash commitments through the issuance of securities, by way of either private placement or public offering or otherwise, there can be no assurance that its efforts to obtain such additional funding will be successful, or achieved on terms favorable to the Company or its existing shareholders. If adequate funds are not available on terms favorable to the Company, it may have to reduce substantially or eliminate expenditures such as promotion, marketing or production of its current or proposed products, or obtain funds through other sources such as divestiture or monetization of certain assets or sublicensing (where permitted) of certain rights to certain of its technologies or products.
(d) Concentration of credit risk and major customers
The Company considers its maximum credit risk to be $3,140,021 (December 31, 2014 - $2,161,133). This amount is the total of the following financial assets: accounts receivable and loan receivable. The Company’s cash and cash equivalents are held through various high grade financial institutions.
The Company is exposed to credit risk from its customers and continually monitors its customers’ credit. It establishes the provision for doubtful accounts based upon the credit risk applicable to each customer. In line with
other pharmaceutical companies, the Company sells its products through a small number of wholesalers and retail pharmacy chains in addition to hospitals, pharmacies, physicians and other groups. Note 11 discloses the significant customer details and the Company believes that the concentrations on the Company’s customers are considered normal for the Company and its industry.
As at March 31, 2015, the Company had three customers which made up 67.1% of the outstanding accounts receivable in comparison to two customers which made up 65.7% at December 31, 2014. As at March 31, 2015, 28.5% (December 31, 2014 — 12.2%) of the outstanding accounts receivable was related to product sales related to two wholesale accounts (December 31, 2014 — one wholesale account) and 38.6% (December 31, 2014 — 53.5%) was related to an amount owing related to the product sales.
(e) Foreign exchange risk
The Company principally operates within Canada; however, a portion of the Company’s revenues, expenses, and current assets and liabilities, are denominated in United States dollars and the EURO. The Company’s long term debt is repayable in U.S. dollars, which exposes the Company to foreign exchange risk due to changes in the value of the Canadian dollar. As at March 31, 2015, a 5% change in the foreign exchange rate would increase/decrease the long term debt balance by $700,000 and would increase/decrease both interest expense and net loss by approximately $29,800 for the three month period ended March 31, 2015. As at March 31, 2015, a 5% change in the foreign exchange rate would increase/decrease the warrant liability balance by $268,000 and would increase/decrease both changes in warrant liability and net loss by $268,000 for the three month period ended March 31, 2015.
(f) Interest rate risk
The Company is exposed to interest rate fluctuations on its cash and cash equivalents as well as its long term debt. At March 31, 2015, the Company had an outstanding long term debt balance of US$14,000,000 ($17,756,200), which bears interest annually at a rate of 11.5% plus the Libor Rate with the Libor Rate being subject to a minimum floor of 2%, such that that minimum interest rate is 13.5%, which may expose the Company to market risk due to changes in interest rates. For the three month period ended March 31, 2015, a 1% increase in interest rates would increase interest expense and net loss by approximately $44,400. However, based on current LIBOR interest rates, which are currently under the minimum floor set at 2% and based on historical movements in LIBOR rates, the Company believes a near-term change in interest rates would not have a material adverse effect on the financial position or results of operations.
17. Derivative Financial Instruments
The Company enters into foreign currency contracts with financial institutions to reduce the risk that its cash flows and earnings will be adversely affected by foreign currency exchange rate fluctuations. In accordance with the Company’s current foreign exchange rate risk management policy, this program is not designated for trading or speculative purposes.
The Company recognizes derivative instruments as either assets or liabilities in the accompanying balance sheets at fair value.
During the three month period ended March 31, 2015, the Company entered into foreign currency call options designated as cash flow hedges to hedge certain forecasted expenses related to its loan obligation denominated in United States Dollars. The notional principal of the foreign currency call option to purchase US$3,500,000 was $4,397,400 at July 23, 2015.
The Company initially reports any gain or loss on the effective portion of the cash flow hedge as a component of other comprehensive income and subsequently reclassifies to the statements of operations when the hedged transaction occurs.
Valuation techniques used to measure fair value are intended to maximize the use of observable inputs and minimize the use of unobservable inputs.
The Company has determined the foreign currency call option to be Level 2. The fair value of the foreign currency call option at March 31, 2015 was a gain of $43,400 (December 31, 2014 — $nil), and is reported in other current asset/liability in the accompanying balance sheets. During the three month period ended March 31, 2015, the Company had not settled any foreign exchange contracts (2014 - recognized a gain of $200,000).
At March 31, 2015 and December 31, 2014, the notional principal and fair value of the Company’s outstanding foreign currency derivative financial instruments were as follows:
|
|
|
March 31, 2015 |
|
December 31, 2014 |
| ||||||||
|
|
|
Notional Principal |
|
Fair |
|
Notional Principal |
|
Fair |
| ||||
|
Foreign currency sold — call options |
|
USD$ |
3,500,000 |
|
$ |
43,400 |
|
USD$ |
— |
|
$ |
— |
|
The notional principal amounts provide one measure of the transaction volume outstanding as of March 31, 2015 and December 31, 2014, and do not represent the amount of the Company’s exposure to market loss. The estimates of fair value are based on applicable and commonly used pricing models using prevailing financial market information as of March 31, 2015 and December 31, 2014. The amounts ultimately realized upon settlement of these financial instruments, together with the gains and losses on the underlying exposures, will depend on actual market conditions during the remaining life of the instruments.
18. Subsequent Events
Subsequent to March 31, 2015, the Company issued 700,000 common shares in connection with the exercise of 620,000 common share purchase warrants and 80,000 compensation options exercised at a weighted average exercise price of $0.88 per common share, for aggregate proceeds of $614,000. In addition, the Company issued 40,000 common share purchase warrants on the exercise of 80,000 compensation options. Each such warrant has an exercise price of $0.90 and an expiry date of July 15, 2016 (Note 7c).
Aralez Pharmaceuticals plc
UNAUDITED PRO FORMA CONDENSED COMBINED BALANCE SHEET
As of March 31, 2015
|
|
|
|
|
Tribute |
|
|
|
|
|
|
|
|
| |||||||||||||||||||
|
|
|
Historical |
|
Historical |
|
Historical |
|
MFI |
|
|
|
Pro Forma |
|
Pro Forma |
|
Accounting |
|
Pro Forma |
|
|
|
Pro Forma |
| |||||||||
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Current assets: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Cash and cash equivalents |
|
$ |
43,904,945 |
|
$ |
5,550,766 |
|
$ |
61,574 |
|
$ |
917,348 |
|
5a |
|
$ |
6,529,688 |
|
$ |
5,164,069 |
|
$ |
— |
|
$ |
(39,230,032 |
) |
7a |
|
$ |
9,838,983 |
|
|
Accounts receivable |
|
4,405,319 |
|
3,124,207 |
|
1,065,973 |
|
— |
|
|
|
4,190,180 |
|
3,313,846 |
|
— |
|
— |
|
|
|
7,719,165 |
| |||||||||
|
Inventories |
|
— |
|
1,193,968 |
|
1,747,916 |
|
— |
|
|
|
2,941,884 |
|
2,326,618 |
|
— |
|
1,186,290 |
|
7b |
|
3,512,908 |
| |||||||||
|
Prepaid expenses and other assets |
|
511,650 |
|
545,756 |
|
462,228 |
|
78,481 |
|
5b |
|
1,086,465 |
|
859,241 |
|
— |
|
1,430,870 |
|
7c |
|
2,801,761 |
| |||||||||
|
Total current assets |
|
48,821,914 |
|
10,414,697 |
|
3,337,691 |
|
995,829 |
|
|
|
14,748,217 |
|
11,663,775 |
|
— |
|
(36,612,872 |
) |
|
|
23,872,817 |
| |||||||||
|
Property and equipment, net |
|
24,963 |
|
978,956 |
|
190,808 |
|
— |
|
|
|
1,169,764 |
|
925,120 |
|
— |
|
— |
|
|
|
950,083 |
| |||||||||
|
Intangible assets, net |
|
— |
|
40,464,367 |
|
1,413,574 |
|
(1,413,574 |
) |
5c |
|
40,464,367 |
|
32,001,649 |
|
— |
|
98,015,735 |
|
7e |
|
130,017,384 |
| |||||||||
|
Goodwill |
|
— |
|
3,599,077 |
|
— |
|
24,694,731 |
|
5c |
|
28,293,808 |
|
22,376,441 |
|
— |
|
124,102,545 |
|
7d |
|
146,478,986 |
| |||||||||
|
Debt issuance costs, net |
|
— |
|
366,404 |
|
— |
|
838,945 |
|
5d |
|
1,205,349 |
|
953,262 |
|
— |
|
(953,262 |
) |
7f |
|
— |
| |||||||||
|
Total assets |
|
$ |
48,846,877 |
|
$ |
55,823,501 |
|
$ |
4,942,073 |
|
$ |
25,115,931 |
|
|
|
$ |
85,881,505 |
|
$ |
67,920,247 |
|
$ |
— |
|
$ |
184,552,146 |
|
|
|
$ |
301,319,270 |
|
|
LIABILITIES AND STOCKHOLDERS EQUITY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Current liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Accounts payable |
|
$ |
202,164 |
|
$ |
4,287,067 |
|
$ |
1,545,526 |
|
$ |
— |
|
|
|
$ |
5,832,593 |
|
$ |
4,612,764 |
|
$ |
(3,469,895 |
) |
$ |
— |
|
|
|
$ |
1,345,033 |
|
|
Warrant liability |
|
— |
|
5,359,374 |
|
— |
|
— |
|
|
|
5,359,374 |
|
4,238,515 |
|
— |
|
11,503,172 |
|
7g |
|
15,741,686 |
| |||||||||
|
Current portion of long term debt |
|
— |
|
1,525,656 |
|
1,954,685 |
|
10,545,315 |
|
5e |
|
14,025,656 |
|
11,092,330 |
|
— |
|
(11,092,330 |
) |
7i |
|
— |
| |||||||||
|
Accrued compensation |
|
510,761 |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
348,033 |
|
— |
|
|
|
858,794 |
| |||||||||
|
Accrued expenses |
|
829,921 |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
3,121,862 |
|
18,385,746 |
|
7h |
|
22,337,530 |
| |||||||||
|
Contingent consideration |
|
— |
|
— |
|
— |
|
5,695,500 |
|
5f |
|
5,695,000 |
|
4,503,948 |
|
— |
|
1,977,150 |
|
7j |
|
6,481,098 |
| |||||||||
|
Promissory note |
|
— |
|
— |
|
— |
|
5,000,000 |
|
5g |
|
5,000,000 |
|
3,954,300 |
|
— |
|
— |
|
|
|
3,954,300 |
| |||||||||
|
Total current liabilities |
|
1,542,846 |
|
11,172,097 |
|
3,500,211 |
|
21,240,315 |
|
|
|
35,912,623 |
|
28,401,857 |
|
— |
|
20,773,738 |
|
|
|
50,718,441 |
| |||||||||
|
Long-term debt |
|
— |
|
15,260,858 |
|
535,151 |
|
— |
|
|
|
15,796,009 |
|
12,492,432 |
|
— |
|
(12,069,202 |
) |
7i |
|
423,230 |
| |||||||||
|
Deferred tax liabilities |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
12,251,967 |
|
7k |
|
12,251,967 |
| |||||||||
|
Total liabilities |
|
1,542,846 |
|
26,432,955 |
|
4,035,362 |
|
21,240,315 |
|
|
|
51,708,632 |
|
40,894,289 |
|
— |
|
20,956,503 |
|
|
|
63,393,637 |
| |||||||||
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Stockholders’ Equity: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Common stock |
|
32,322 |
|
45,022,189 |
|
350,001 |
|
(346,278 |
) |
5h |
|
45,025,912 |
|
35,609,193 |
|
— |
|
(35,591,001 |
) |
7l |
|
50,514 |
| |||||||||
|
Additional paid-in capital |
|
144,203,500 |
|
3,061,924 |
|
— |
|
4,996,277 |
|
5i |
|
8,058,201 |
|
6,372,909 |
|
— |
|
209,259,357 |
|
7m |
|
359,835,766 |
| |||||||||
|
Retained earnings/(Accumulated deficit) |
|
(96,931,791 |
) |
(25,084,316 |
) |
556,710 |
|
(774,383 |
) |
5j |
|
(25,301,989 |
) |
(20,010,331 |
) |
— |
|
(12,025,769 |
) |
7n |
|
(128,967,891 |
) | |||||||||
|
Warrants |
|
— |
|
6,347,349 |
|
— |
|
— |
|
|
|
6,347,349 |
|
5,019,864 |
|
— |
|
1,987,379 |
|
7o |
|
7,007,243 |
| |||||||||
|
Accumulated other comprehensive (loss) income |
|
— |
|
43,400 |
|
— |
|
— |
|
|
|
43,400 |
|
34,323 |
|
— |
|
(34,323 |
) |
7p |
|
— |
| |||||||||
|
Total stockholders’ equity |
|
47,304,031 |
|
29,390,546 |
|
906,711 |
|
3,875,616 |
|
|
|
34,172,873 |
|
27,025,958 |
|
— |
|
163,595,643 |
|
|
|
237,925,632 |
| |||||||||
|
Total liabilities and stockholders’ equity |
|
$ |
48,846,877 |
|
$ |
55,823,501 |
|
$ |
4,942,073 |
|
$ |
25,115,931 |
|
|
|
$ |
85,881,505 |
|
$ |
67,920,247 |
|
$ |
— |
|
$ |
184,552,146 |
|
|
|
$ |
301,319,270 |
|
See accompanying notes to unaudited Pro Forma Condensed Combined Financial Statements
Aralez Pharmaceuticals plc
UNAUDITED PRO FORMA CONDENSED COMBINED STATEMENT OF OPERATIONS
Year Ended December 31, 2014
|
|
|
|
|
Tribute |
|
|
|
|
|
|
|
|
| ||||||||||||||||||
|
|
|
Historical |
|
Historical |
|
Historical |
|
MFI |
|
|
|
Pro Forma |
|
Pro Forma |
|
Accounting |
|
Pro Forma |
|
|
|
Pro Forma |
| ||||||||
|
Revenues |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
|
Royalty and licensing revenue |
|
$ |
32,394,232 |
|
18,414 |
|
$ |
— |
|
$ |
— |
|
|
|
$ |
18,414 |
|
$ |
16,678 |
|
$ |
— |
|
$ |
— |
|
|
|
$ |
32,410,910 |
|
|
Licensed domestic product net sales |
|
— |
|
9,106,038 |
|
— |
|
— |
|
|
|
9,106,038 |
|
8,247,339 |
|
— |
|
— |
|
|
|
8,247,339 |
| ||||||||
|
Other domestic product sales |
|
— |
|
6,127,968 |
|
9,879,885 |
|
— |
|
|
|
16,007,853 |
|
14,498,312 |
|
— |
|
— |
|
|
|
14,498,312 |
| ||||||||
|
International product sales |
|
— |
|
1,619,372 |
|
— |
|
— |
|
|
|
1,619,372 |
|
1,466,665 |
|
— |
|
— |
|
|
|
1,466,665 |
| ||||||||
|
Total Revenues |
|
32,394,232 |
|
16,871,792 |
|
9,879,885 |
|
— |
|
|
|
26,751,677 |
|
24,228,994 |
|
— |
|
— |
|
|
|
56,623,226 |
| ||||||||
|
Cost of Sales |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
|
Licensor sales and distribution fees |
|
— |
|
5,902,034 |
|
— |
|
— |
|
|
|
5,902,034 |
|
5,345,472 |
|
— |
|
— |
|
|
|
5,345,472 |
| ||||||||
|
Cost of products sold |
|
— |
|
1,787,584 |
|
4,836,729 |
|
— |
|
|
|
6,624,313 |
|
5,999,640 |
|
— |
|
— |
|
|
|
5,999,640 |
| ||||||||
|
Write down of inventories |
|
— |
|
53,099 |
|
— |
|
— |
|
|
|
53,099 |
|
48,092 |
|
— |
|
— |
|
|
|
48,092 |
| ||||||||
|
Total cost of sales |
|
— |
|
7,742,717 |
|
4,836,729 |
|
— |
|
|
|
12,579,446 |
|
11,393,204 |
|
— |
|
— |
|
|
|
11,393,204 |
| ||||||||
|
Gross Profit |
|
32,394,232 |
|
9,129,075 |
|
5,043,156 |
|
— |
|
|
|
14,172,231 |
|
12,835,790 |
|
— |
|
— |
|
|
|
45,230,022 |
| ||||||||
|
Operating expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
|
Sales, general, and administrative |
|
10,078,771 |
|
10,149,854 |
|
4,950,203 |
|
— |
|
|
|
15,100,057 |
|
13,676,122 |
|
— |
|
— |
|
|
|
23,754,893 |
| ||||||||
|
Research and development |
|
5,739,848 |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
— |
|
|
|
5,739,848 |
| ||||||||
|
Amortization |
|
— |
|
1,511,021 |
|
207,365 |
|
(188,971 |
) |
5k |
|
1,529,415 |
|
1,385,191 |
|
(107,610 |
) |
8,157,283 |
|
7r |
|
9,434,864 |
| ||||||||
|
Total operating expenses |
|
15,818,619 |
|
11,660,875 |
|
5,157,568 |
|
(188,971 |
) |
|
|
16,629,472 |
|
15,061,313 |
|
(107,610 |
) |
8,157,283 |
|
|
|
38,929,605 |
| ||||||||
|
Non-operating income (expense) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
|
Change in warrant liability |
|
— |
|
283,305 |
|
— |
|
— |
|
|
|
283.305 |
|
256,589 |
|
— |
|
— |
|
|
|
256,589 |
| ||||||||
|
Interest expense |
|
— |
|
(1,441,729 |
) |
(49,948 |
) |
(350,052 |
) |
5l |
|
(1,841,729 |
) |
(1,668,054 |
) |
(107,610 |
) |
1,413,384 |
|
7s |
|
(362,280 |
) | ||||||||
|
Interest income |
|
43,100 |
|
59,586 |
|
— |
|
— |
|
|
|
59,586 |
|
53,967 |
|
— |
|
— |
|
|
|
97,067 |
| ||||||||
|
Other non-operating income |
|
3,056,019 |
|
(1,976,304 |
) |
(79,144 |
) |
— |
|
|
|
(2,055,448 |
) |
(1,861,619 |
) |
— |
|
151,755 |
|
7s |
|
1,346,154 |
| ||||||||
|
Total other income (expense) |
|
3,099,119 |
|
(3,075,142 |
) |
(129,092 |
) |
(350,052 |
) |
|
|
(3,554,286 |
) |
(3,219,117 |
) |
(107,610 |
) |
1,565,138 |
|
|
|
1,337,531 |
| ||||||||
|
Income before taxes |
|
19,674,732 |
|
(5,606,942 |
) |
(243,504 |
) |
(161,081 |
) |
|
|
(6,011,527 |
) |
(5,444,640 |
) |
— |
|
(6,592,145 |
) |
|
|
7,637,947 |
| ||||||||
|
Income tax expense (benefit) |
|
— |
|
— |
|
107 |
|
(42,686 |
) |
5m |
|
(42,579 |
) |
(38,564 |
) |
|
|
(824,018 |
) |
7t |
|
(862,582 |
) | ||||||||
|
Net Income (loss) attributable to common stockholders |
|
19,674,732 |
|
(5,606,942 |
) |
(243,611 |
) |
(118,395 |
) |
|
|
(5,968,948 |
) |
(5,406,076 |
) |
— |
|
(5,768,127 |
) |
|
|
8,500,530 |
| ||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Basic net income (loss) per share |
|
$ |
0.63 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7u |
|
$ |
0.17 |
| ||||||
|
Shares used in computing basic net income (loss) per share |
|
31,359,867 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7u |
|
51,139,659 |
| ||||||||
|
Diluted net income (loss) per share |
|
$ |
0.60 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7u |
|
$ |
0.17 |
| ||||||
|
Shares used in computing diluted net income (loss) per share |
|
32,810,587 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7u |
|
52,571,262 |
| ||||||||
See accompanying Notes to Unaudited Pro Forma Condensed Combined Financial Information.
Aralez Pharmaceuticals plc
UNAUDITED PRO FORMA CONDENSED COMBINED STATEMENT OF OPERATIONS
Three Months Ended March 31, 2015
|
|
|
|
|
Tribute |
|
|
|
|
|
|
|
|
| |||||||||||||||||||
|
|
|
Historical |
|
Historical |
|
Historical |
|
MFI |
|
|
|
Pro Forma |
|
Pro Forma |
|
Accounting |
|
Pro Forma |
|
|
|
Pro Forma |
| |||||||||
|
Revenues |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Royalty and licensing revenue |
|
$ |
4,404,463 |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
|
$ |
4,404,463 |
|
|
Licensed domestic product net sales |
|
— |
|
2,521,080 |
|
— |
|
— |
|
|
|
2,521,080 |
|
2,036,276 |
|
— |
|
— |
|
|
|
2,036,276 |
| |||||||||
|
Other domestic product sales |
|
— |
|
2,601,622 |
|
2,185,874 |
|
— |
|
|
|
4,787,496 |
|
3,866,861 |
|
— |
|
— |
|
|
|
3,866,861 |
| |||||||||
|
International product sales |
|
— |
|
469,595 |
|
— |
|
— |
|
|
|
469,595 |
|
379,292 |
|
— |
|
— |
|
|
|
379,292 |
| |||||||||
|
Total Revenues |
|
4,404,463 |
|
5,592,297 |
|
2,185,874 |
|
— |
|
|
|
7,778,171 |
|
6,282,429 |
|
— |
|
— |
|
|
|
10,686,892 |
| |||||||||
|
Cost of Sales |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Licensor sales and distribution fees |
|
— |
|
1,452,064 |
|
— |
|
— |
|
|
|
1,452,064 |
|
1,172,832 |
|
— |
|
— |
|
|
|
1,172,832 |
| |||||||||
|
Cost of products sold |
|
— |
|
575,246 |
|
988,542 |
|
— |
|
|
|
1,563,788 |
|
1,263,072 |
|
— |
|
— |
|
|
|
1,263,072 |
| |||||||||
|
Total cost of sales |
|
— |
|
2,027,310 |
|
988,542 |
|
— |
|
|
|
3,015,852 |
|
2,435,904 |
|
— |
|
— |
|
|
|
2,435,904 |
| |||||||||
|
Gross Profit |
|
4,404,463 |
|
3,564,987 |
|
1,197,332 |
|
— |
|
|
|
4,762,319 |
|
3,846,525 |
|
— |
|
— |
|
|
|
8,250,988 |
| |||||||||
|
Operating expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Sales, general, and administrative |
|
3,262,320 |
|
3,325,922 |
|
1,613,668 |
|
— |
|
|
|
4,939,590 |
|
3,989,707 |
|
— |
|
(197,800 |
) |
7q |
|
7,054,227 |
| |||||||||
|
Research and development |
|
983,511 |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
— |
|
|
|
983,511 |
| |||||||||
|
Amortization |
|
— |
|
621,623 |
|
50,891 |
|
(47,276 |
) |
5k |
|
625,238 |
|
505,005 |
|
(21,360 |
) |
1,872,712 |
|
7s |
|
2,356,357 |
| |||||||||
|
Total operating expenses |
|
4,245,831 |
|
3,947,545 |
|
1,664,559 |
|
(47,276 |
) |
|
|
5,564,828 |
|
4,494,712 |
|
(21,360 |
) |
1,674,912 |
|
|
|
10,394,094 |
| |||||||||
|
Non-operating income (expense) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
Charge in warrant liability |
|
— |
|
(2,695,600 |
) |
— |
|
— |
|
|
|
(2,695,600 |
) |
(2,177,236 |
) |
— |
|
— |
|
|
|
(2,177,236 |
) | |||||||||
|
Interest expense |
|
— |
|
(595,975 |
) |
(13,033 |
) |
13,033 |
|
5l |
|
(595,975 |
) |
(481,369 |
) |
(21,360 |
) |
502,729 |
|
7s |
|
— |
| |||||||||
|
Interest income |
|
— |
|
125 |
|
— |
|
— |
|
|
|
125 |
|
101 |
|
— |
|
— |
|
|
|
101 |
| |||||||||
|
Other non-operating income (expense) |
|
(185,990 |
) |
(1,507,455 |
) |
6,638 |
|
— |
|
|
|
(1,500,817 |
) |
(1,212,210 |
) |
— |
|
59,769 |
|
7s |
|
(1,338,431 |
) | |||||||||
|
Total other income (expense) |
|
(185,990 |
) |
(4,798,905 |
) |
(6,395 |
) |
13,033 |
|
|
|
(4,792,267 |
) |
(3,870,714 |
) |
(21,360 |
) |
562,498 |
|
|
|
(3,515,566 |
) | |||||||||
|
Income before taxes |
|
(27,358 |
) |
(5,181,463 |
) |
(473,622 |
) |
60,309 |
|
|
|
(5,594,776 |
) |
(4,518,901 |
) |
— |
|
(1,112,414 |
) |
|
|
(5,658,672 |
) | |||||||||
|
Income tax expense (benefit) |
|
— |
|
— |
|
(98,569 |
) |
15,982 |
|
5m |
|
(82,587 |
) |
(66,706 |
) |
|
|
(139,052 |
) |
7t |
|
(205,757 |
) | |||||||||
|
Net Income (loss) attributable to common stockholders |
|
(27,358 |
) |
(5,181,463 |
) |
(375,053 |
) |
44,327 |
|
|
|
(5,512,189 |
) |
(4,452,195 |
) |
— |
|
(973,362 |
) |
|
|
(5,452,915 |
) | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
|
Basic net income (loss) per share |
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7u |
|
$ |
(0.10 |
) | |||||||
|
Shares used in computing basic net income (loss) per share |
|
32,259,570 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7u |
|
52,039,362 |
| |||||||||
|
Diluted net income (loss) per share |
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7u |
|
$ |
(0.10 |
) | |||||||
|
Shares used in computing diluted net income (loss) per share |
|
32,259,570 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7u |
|
52,039,362 |
| |||||||||
See accompanying Notes to Unaudited Pro Forma Condensed Combined Financial Information.
NOTES TO UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL STATEMENTS
1. DESCRIPTION OF TRANSACTIONS
The Merger and Arrangement: On June 8, 2015, POZEN Inc. (“Pozen”) and Tribute Pharmaceuticals Canada Inc. (“Tribute”) agreed to a business combination under the terms of the Agreement and Plan of Merger and Arrangement, among Tribute, Aguono Limited (which was renamed Aralez Pharmaceuticals Limited and which, prior to the merger effective time, as defined in the merger agreement, will re-register as a public limited company incorporated in Ireland and be renamed as Aralez Pharmaceuticals plc) (“Parent”), Trafwell Limited (which was renamed Aralez Pharmaceutical Holdings Limited) (“Ltd2”), ARLZ US Acquisition Corp. (“US Merger Sub”), ARLZ CA Acquisition Corp. (“Can Merger Sub”) and Pozen, dated as of June 8, 2015 (the “merger agreement”). In order to effect the transactions contemplated by the merger agreement, US Merger Sub, an indirect subsidiary of Parent, will be merged with and into Pozen (the “merger”). Pozen will be the surviving corporation and, through the merger, will become an indirect wholly-owned subsidiary of Parent. The merger of Pozen into US Merger Sub will be effected under Delaware law so that Pozen will be reorganized into a holding company structure. In accordance with the merger agreement, Can Merger Sub will offer to acquire, and will acquire, all of the outstanding Tribute common shares pursuant to a court approved plan of arrangement in Canada in the manner provided for by the merger agreement (the “arrangement”). Upon completion of the arrangement, Tribute will also become an indirect wholly-owned subsidiary of Parent. Upon completion, this transaction does not constitute a change of control of Pozen. The merger and the arrangement are collectively referred to as the “transactions.”
As a result of the merger, each share of Pozen common stock will be converted into the right to receive from Parent one ordinary share of Parent, $0.001 nominal value per share (the “Parent Shares”) (the “merger consideration”) for each share of Pozen common stock that they own as of the record date. Pursuant to the arrangement, each outstanding Tribute common share will be converted into the right to receive from Parent 0.1455 Parent Shares.
The transactions value the entire issued and to be issued share capital of Tribute at approximately $198.1 million at a closing share price of $10.89 on July 14, 2015 (the most recent practicable date used for preparation of the pro forma condensed combined financial information) and an exchange ratio of 0.1455. The value of the consideration that Tribute shareholders will receive when the transactions are completed will ultimately be based on the closing date share price of Parent’s stock on the closing date and could materially change.
At the closing time, each outstanding Tribute warrant will entitle its respective holders the right to purchase 0.1455 fully paid and non-assessable Parent Shares for no additional consideration beyond that set out in the respective Tribute warrant. Each Tribute compensation option, which, prior to the transactions, entitled the holder to purchase one Tribute common share and one-half of one Tribute warrant, will entitle its respective holders to purchase 0.1455 fully paid and non-assessable Parent shares, as well as 0.1455 one-half warrants for Parent shares, for no additional consideration beyond that set out in the respective compensation option certificate. For the purposes of these pro forma statements, it has been assumed that Tribute stock options will be cancelled and converted into Tribute common shares and converted into the right to receive .1455 Parent Shares for each Tribute common share. The warrants, employee stock options, and compensation options will be outstanding, fully vested and exercisable at any time.
Pozen will include as consideration $18.8 million for the fair value of the awards including (i) $7.0 million related to equity-classified warrants; (ii) $1.3 million related to compensation options; (iii) $3.3 million related to employee stock options that were vested prior to the transactions; and
(iv) $7.2 million related to employee stock options for which vesting was accelerated as a result of automatic change in control provisions within the respective employee’s employment agreements, which related to pre-combination services. Pozen will also recognize an $11.5 million increase to the fair value of the Tribute warrant liability acquired in the transactions, which related to warrants denominated in a currency other than Pozen’s functional currency, which is the U.S. dollar. This conversion is not expected to result in incremental value to the share/option holders; however if it is determined that the exchange results in incremental value at the acquisition date, Pozen would recognize post-combination expense.
Pozen estimates that it will recognize post-combination compensation expense of $2.1 million as a one-time charge for the portion of the Tribute employee stock options for which vesting was accelerated based upon discretionary change in control provisions in the Tribute stock option plan, and as a result of the merger agreement, which related to services not provided as of the date of this prospectus. This post-combination compensation expense has been excluded from the unaudited pro forma condensed combined statement of operations as they reflect charges directly attributable to the acquisition that will not have a continuing impact on Pozen’s operations; however, it has been reflected in retained earnings, net of tax of $0.3 million on the unaudited pro forma balance sheet.
In addition, certain executive officers of Tribute will be automatically entitled to receive severance compensation per their executive employment agreements upon a change of control, regardless of whether the executive’s employment is terminated. Pozen will recognize an assumed liability of $1.8 million for such arrangements as part of the accounting for the transactions.
Pursuant to the merger agreement, except for awards held by certain new employees, each outstanding Pozen non-qualified or incentive stock option will become vested and converted into an option for one Parent Share. Each outstanding restricted stock unit will become vested and convert into one Parent Share. The converted awards will relate to a number of Parent Shares equal to the number of Pozen shares subject to the corresponding pre-conversion award and will continue to have, subject to applicable law, the same terms and conditions that were applicable to the corresponding pre-conversion Pozen award (including repurchase rights, as applicable). The $3.6 million of compensation cost associated with the accelerated vesting of Pozen awards has been excluded from the unaudited pro forma condensed combined statement of operations as they reflect charges directly attributable to the transactions that will not have a continuing impact on Parent’s operations; however, it has been reflected in retained earnings, net of tax of $0.5 million on the unaudited pro forma balance sheet. Pozen estimates that all current outstanding non-qualified options will be exercised into Parent Shares, while the incentive stock options will remain outstanding and exercisable into Parent Shares following the transactions. However, for the purposes of the pro forma financial statements, such exercise of non-qualified options has not been reflected because an amount is not factually supportable at the time of the filing of this prospectus.
Upon completion of the transactions, Pozen stockholders will own approximately 66% of the outstanding Parent Shares, and current Tribute shareholders will own approximately 34% of the outstanding Parent Shares before giving effect to (i) any exercise of outstanding options and warrants or the vesting and delivery of shares underlying restricted stock units of either company and (ii) the Parent Shares to be issued to new investors pursuant to the Subscription Agreement (defined below)
and the Parent Shares issuable upon conversion of the Convertible Notes to be issued pursuant to the Facility Agreement (defined below).
MFI Acquisition: On June 16, 2015, Tribute acquired Medical Futures Inc. (“MFI”) in a transaction valued at $26.1 million (CAD). Financial terms of the deal include the payment of: $8.5 million (CAD) in cash on closing; $5 million (CAD) through the issuance of 3,723,008 Tribute common shares; and, $5 million (CAD) in the form of a one year unsecured promissory note bearing interest at 8% annually convertible at the holder’s option at any time during the term into 2,813,778 Tribute common shares (which will be converted into the right to receive 409,405 of Parent Shares); and future contingent cash milestone payments totaling $5.7 million (CAD) based on attainment of certain conditions expected shortly after the close of this transaction. In addition, on the receipt of each regulatory approval for MFI’s two pipeline products (or upon the occurrence of a change of control of Tribute), MFI will receive a payment of $1.25 million (CAD) per product. Tribute also entered in to a debenture agreement (“Tribute debenture’) of $12.5 million (CAD) which was necessary to complete its acquisition of MFI.
Facility Agreement: On June 8, 2015, Pozen executed the Facility Agreement among the Parent (or a wholly-owned subsidiary of the Parent, depending on whether certain conditions of the Facility Agreement occur) (the “Borrower”), Tribute, Deerfield Private Design Fund III, L.P. (“Deerfield Private Design”), Deerfield International Master Fund, L.P. (“Deerfield International”), and Deerfield Partners, L.P. (“Deerfield Partners”), and the other lender parties thereto (together with Deerfield Private Design, Deerfield International, and Deerfield Partners, the “Lenders”).
Pursuant to the Facility Agreement, the Borrower may borrow from the Lenders up to an aggregate principle amount of $275 million, of which (i) $75 million will be in the form of a 2.5% senior secured convertible promissory note due six years from issuance and convertible into Parent Shares at a conversion price of $9.54 per share (the “Convertible Notes”), issued and sold by Borrower to Deerfield Private Design or its registered assigns, upon the terms and conditions of the Facility Agreement, and (ii) up to an aggregate principal amount of $200 million, which will be made available for permitted acquisitions, will be in the form of secured promissory notes issued and sold by the Borrower to the Lenders (the “Acquisition Notes”), evidencing the Acquisition Loans, upon the terms and conditions and subject to the limitations set forth in the Acquisition Notes, all subject to the terms and conditions of the Facility Agreement.
The proceeds from the Facility Agreement will be used for working capital needs, general corporate purposes and for future acquisitions. This Facility Agreement is not directly attributable to the transactions or the MFI Acquisition; as such no pro forma adjustments have been made in relation to this Facility Agreement in these unaudited pro forma condensed combined financial statements.
Share Subscription Agreement: On June 8, 2015, Pozen executed a Share Subscription Agreement (the “Subscription Agreement”) among QLT Inc., a corporation existing under the laws of the Province of British Columbia, Canada (“Purchaser”), Tribute, Parent, and the following investors thereto: Deerfield Private Design; Deerfield International; Deerfield Partners; EcoR1 Capital Fund, L.P.; EcoR1 Capital Fund Qualified, L.P.; Broadfin Healthcare Master Fund, Ltd; JW Partners, LP; and JW Opportunities Fund, LLC (each, an “Investor” and together, the “Investors”). Pursuant to the Subscription Agreement, Parent will sell to Purchaser and the Investors up to $75 million of the Parent Shares in a private placement at a purchase price of $7.20 per Parent Share. The Subscription
Agreement provides that Pozen shall prepare and cause to be filed with the SEC two registration statements on Form S-3 or such form as may be required to effect a registration of the Parent Shares issued under the Subscription Agreement within 60 days of the date of the signing of the Subscription Agreement and for certain other registration rights for each of Purchaser and the Investors under the Securities Act and the rules and regulations thereunder, or any similar successor statute, and applicable state securities laws.
The issuance of Parent Shares in connection with this Subscription Agreement is not directly attributable to the transactions or the MFI Acquisition; as such no pro forma adjustments have been made in relation to this Subscription Agreement in these unaudited pro forma condensed combined financial statements.
2. BASIS OF PRESENTATION
The unaudited pro forma condensed combined financial statements were prepared in accordance with generally accepted accounting principles in the United States (“U.S. GAAP”) and pursuant to U.S. Securities and Exchange Commission Regulation S-X Article 11, and present the pro forma financial position and results of operations of the consolidated companies based upon the historical information after giving effect to the transactions and MFI Acquisition and adjustments described in these footnotes. The unaudited pro forma condensed combined balance sheet is presented as if the transactions and MFI Acquisition had occurred on March 31, 2015; and the unaudited pro forma condensed combined statement of operations for the year ended December 31, 2014 and the three month period ended March 31, 2015 is presented as if the transactions and MFI Acquisition had occurred on January 1, 2014.
The historical results of Pozen, Tribute and MFI for the year ended December 31, 2014 have been derived from their respective audited financial statements. The historical results of Pozen, Tribute and MFI as of and for the three months ended March 31, 2015 have been derived from unaudited financial information.
In addition, each of Tribute and MFI have historically reported its financial statements in its local currency, the Canadian dollar (“CAD”); in order to present the unaudited pro forma condensed combined financial statements in U.S. dollars, the pro forma financial information for Tribute, which reflects the MFI Acquisition has been translated to U.S. dollars using the spot rate of $0.79 as of March 31, 2015 for the balance sheet and average rates of $0.91 and $0.81 for the statements of operations for the twelve months ended December 31, 2014 and three months ended March 31, 2015, respectively.
Adjustments have also been recorded to the historical financial statements to reclassify financial statement line items as necessary. See Note 3, “Accounting Policies and Reclassifications.”
The transactions have been reflected in the unaudited pro forma condensed combined financial statements as being accounted for under the acquisition method in accordance with ASC 805, Business Combination, with Pozen treated as the accounting acquirer; and the MFI Acquisition has been reflected in the unaudited pro forma condensed combined financial statements in accordance with ASC 805 with Tribute treated as the accounting acquirer. Under the acquisition method, the total estimated purchase price is calculated as described in Note 4 and Note 6 for the MFI Acquisition and transactions, respectively. In accordance with ASC 805, the assets acquired and the liabilities assumed
have been measured at fair value based on various preliminary estimates. These estimates are based on key assumptions related to the transactions and MFI Acquisition, including reviews of publicly disclosed information for other acquisitions in the industry, historical experience, data that was available through the public domain and Pozen’s due diligence review of Tribute’s business. Due to the fact that the unaudited pro forma condensed combined financial information has been prepared based on preliminary estimates, the final amounts recorded for the transactions and MFI Acquisition may differ materially from the information presented herein. These estimates are subject to change pending further review of the fair value of assets acquired and liabilities assumed. In addition, the final determination of the recognition and measurement of the identified assets acquired and liabilities assumed will be based on the fair market value of actual net tangible and intangible assets and liabilities of Tribute at the closing date.
For purposes of measuring the estimated fair value, where applicable, of the assets acquired and the liabilities assumed as reflected in the unaudited pro forma condensed combined financial information, Pozen has applied the guidance in ASC 820, Fair Value Measurements and Disclosures, which we refer to as ASC 820, which establishes a framework for measuring fair value. In accordance with ASC 820, fair value is an exit price and is defined as “the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date.” Under ASC 805, acquisition-related transaction costs and acquisition-related restructuring charges are not included as components of consideration transferred but are accounted for as expenses in the period in which the costs are incurred. For the periods presented, neither Pozen nor Tribute incurred material transaction costs related to the transactions.
The unaudited pro forma condensed combined financial information does not reflect ongoing cost savings that Parent expects to achieve as a result of the transactions or the costs necessary to achieve these costs savings or synergies.
3. ACCOUNTING POLICIES AND RECLASSIFICATIONS
Pozen performed certain procedures for the purpose of identifying any material differences in significant accounting policies between Pozen and Tribute, and any accounting adjustments that would be required in connection with adopting uniform policies. Procedures performed by Pozen involved a review of Tribute’s publicly disclosed summary of significant accounting policies, including those disclosed in Tribute’s Annual Report for the year ended December 31, 2014 and preliminary discussion with Tribute’s management regarding Tribute’s significant accounting policies to identify material adjustments. Pozen expects to engage in additional discussion with Tribute’s management to continue to evaluate the impact of Tribute’s accounting policies on its historical results after completion of the transactions. As a result of that review, management may identify differences that, when conformed, could have a material impact on this unaudited pro forma condensed combined financial information.
In addition, the historical consolidated financial statements of Tribute presented herein have been adjusted by condensing certain line items related to “prepaid expenses and other assets”; by reclassifying certain line items in order to conform to Pozen’s financial statement presentation; these reclassifications are reflected in the column “Accounting Policies and Reclassifications.”
The reclassification adjustments on the unaudited pro forma balance sheet pertain to the reclassification of certain balances of Tribute and MFI from “Accounts payable and other accrued expenses” into “Accounts Payable,” “Accrued Compensation” and “Accrued Expenses.”
The reclassification adjustments on the unaudited pro forma statements of operations pertain to the reclassification of amortization of deferred financing fees from the “Amortization” line item into “Interest Expense.”
4. MFI ACQUISITION—PRELIMINARY CONSIDERATION TRANSFERRED AND PRELIMINARY FAIR VALUE OF NET ASSETS ACQUIRED
The MFI Acquisition has been accounted for using the acquisition method of accounting in accordance with ASC 805, which requires, among other things, that the assets acquired and liabilities assumed be recognized at their acquisition date fair values, with any excess of the consideration transferred over the estimated fair values of the identifiable net assets acquired recorded as goodwill. In addition, ASC 805 establishes that the common stock issued to effect the MFI Acquisition be measured at the closing date of the MFI Acquisition at the then-current market price.
The following is a summary of the consideration transferred by Tribute for the acquisition of MFI (in $CAD):
|
Cash consideration |
|
$ |
8,492,868 |
|
|
Issuance of 3,723,008 shares of Tribute common stock |
|
5,000,000 |
| |
|
Issuance of Promissory Note |
|
5,000,000 |
| |
|
Preliminary estimate of fair value of contingent consideration |
|
5,695,000 |
| |
|
Repayment of MFI indebtedness |
|
1,954,685 |
| |
|
Total consideration transferred |
|
$ |
26,142,553 |
|
The fair value of consideration transferred of $26.1 million (CAD) consisted of $8.5 million (CAD) in cash, $5 million (CAD) through the issuance of 3,723,008 Tribute common shares; and, $5 million (CAD) in the form of a one year unsecured promissory note bearing interest at 8% annually convertible at the holder’s option at any time during the term into 2,813,778 Tribute common shares. In addition, the consideration also included future contingent cash milestone payments totaling $5.7 million (CAD) based on attainment of certain conditions expected shortly after the close of this transaction and $2.0 million (CAD) associated with the payoff of MFI’s bank indebtedness. At the close of the MFI Acquisition, the fair value of the contingent consideration related to regulatory approval of two of MFI’s pipeline products was deemed to be immaterial.
The following is a summary of the preliminary estimated fair values of the net assets acquired ($CAD):
|
Total estimated consideration transferred |
|
$ |
26,142,553 |
|
|
Working capital(i) |
|
1,792,165 |
| |
|
Property, plant, and equipment |
|
190,808 |
| |
|
Other noncurrent liabilities |
|
(535,151 |
) | |
|
Net assets acquired |
|
1,447,822 |
| |
|
Goodwill(ii) |
|
$ |
24,694,731 |
|
(i) Working capital consists of current assets less current liabilities.
(ii) Includes other intangible assets that have not been allocated a portion of the consideration transferred at the date of this prospectus.
Tribute has made preliminary allocation estimates of the fair value of net assets acquired in the MFI Acquisition. The valuations will consist of physical appraisals, discounted cash flow analyses, or other appropriate valuation techniques to determine the fair value of the assets acquired and liabilities assumed. The amounts allocated to assets acquired and liabilities assumed in the MFI Acquisition could differ materially from the preliminary amounts presented in these unaudited pro forma condensed combined financial statements. A decrease in the fair value of assets acquired or an increase in the fair value of liabilities assumed in the MFI Acquisition from those preliminary valuations presented in these unaudited pro forma condensed combined financial statements would result in a dollar-for-dollar corresponding increase in the amount of goodwill that will result from the MFI Acquisition. In addition, if the value of the acquired assets is higher than the preliminary indication, it may result in higher amortization and depreciation expense than is presented in these unaudited pro forma condensed combined financial statements.
5. MFI ACQUISITION—PRELIMINARY PRO FORMA ADJUSTMENTS
The preliminary pro forma adjustments included in the unaudited pro forma condensed combined financial statements related to the MFI Acquisition are as follows:
(a) Cash and cash equivalents—Adjustment reflects the preliminary net adjustment to cash in connection with the MFI Acquisition (in $CAD):
|
Cash portion of MFI Acquisition consideration |
|
$ |
(8,492,868 |
) |
|
Repayment of MFI indebtedness |
|
(1,954,685 |
) | |
|
Payment of transaction related expenses |
|
(296,154 |
) | |
|
Proceeds from Tribute debenture, net of issuance costs |
|
11,661,055 |
| |
|
Pro forma adjustment to cash and cash equivalents |
|
$ |
917,348 |
|
(b) Prepaid expenses and other assets—Represents tax receivable due to the tax benefit obtained as a result of the of transaction expenses incurred by Tribute for the MFI Acquisition.
(c) Intangible assets—Elimination of historical intangible assets of MFI. For purposes of these unaudited condensed combined pro forma financial statements, the identifiable intangible assets acquired by Tribute in the MFI Acquisition have been reflected as goodwill, as management of Tribute has not completed its valuation of such identifiable intangible assets. In conjunction with the transactions, Pozen has reflected a preliminary estimate of acquired identifiable intangible assets related to the combined business of Tribute and MFI.
(d) Debt issuance costs, net—Adjustment relates to the capitalization of $0.8 million (CAD) of debt issuance costs recognized in connection with the $12.5 million (CAD) Tribute debenture, which will be capitalized on the unaudited pro forma balance sheet and amortized over the life of the underlying debt instrument.
(e) Current portion of long term debt—Adjustment reflects the $12.5 million (CAD) Tribute debenture issued to complete the MFI Acquisition, partially offset by the payoff of the MFI bank loan of $2.0 million (CAD).
(f) Contingent consideration—Adjustment relates to the estimated fair value of the contingent consideration that Tribute management expects to pay the selling shareholders of MFI for future milestone.
(g) Promissory note—Reflects the promissory note that was issued by Tribute as part of its consideration transferred for MFI Acquisition.
(h) Common shares—Reflects the elimination of MFI’s historical common share balance of $0.4 million (CAD), partially offset by the par value of shares issued in connection with the MFI Acquisition of $4.0 thousand (CAD).
(i) APIC—Reflects total value of common shares issued in connection with the MFI Acquisition of $5.0 million (CAD), less the par value of common shares of $4.0 thousand (CAD).
(j) Accumulated deficit—Adjustment reflects the transaction costs incurred by Tribute to complete the MFI Acquisition of $0.3 million (CAD), net of tax of $0.1 million (CAD) and the elimination of MFI’s historical retained earnings of $0.6 million (CAD).
(k) Amortization—Adjustment reflects the elimination of amortization expenses related to the historical intangible assets of MFI.
(l) Interest expense—Adjustment reflects the interest recognized in connection with the promissory note issued of $0.4 million (CAD), partially offset by the elimination of the interest expense associated with the bank loan that was repaid in connection with the MFI Acquisition of $50.0 thousand (CAD) for the year ended December 31, 2014. For the three months ended March 31, 2015, the adjustment relates to the elimination of the interest expense associated with the bank loan that was repaid in connection with the MFI Acquisition of $13.0 thousand (CAD); no additional interest expense was recognized for the promissory note as the note had a term of one year. In addition, no interest expense was recorded related to the debenture that was issued as part of the MFI acquisition as the debenture was repaid in connection with the Tribute acquisition.
(m) Income tax benefit—Adjustment reflects the income tax impacts of the pro forma adjustments made to the pro forma statement of operations, using the Canadian statutory tax rate of 26.5%.
6. MERGER AND ARRANGEMENT—PRELIMINARY CONSIDERATION TRANSFERRED AND PRELIMINARY FAIR VALUE OF NET ASSETS ACQUIRED
The transactions have been accounted for using the acquisition method of accounting in accordance with ASC 805, which requires, among other things, that the assets acquired and liabilities assumed be recognized at their acquisition date fair values, with any excess of the consideration transferred over the estimated fair values of the identifiable net assets acquired recorded as goodwill. In addition, ASC 805 establishes that the common stock issued to effect the transactions be measured at the closing date of the transactions at the then-current market price.
Based on (1) the closing price of Pozen’s common stock of $10.89 per share on July 14, 2015, (2) the number of Tribute common shares outstanding as of June 30, 2015, (3) the number of stock options, compensation options and warrants outstanding as at June 30, 2015, and (4) Tribute’s outstanding indebtedness to be repaid upon a change of control, the total estimated consideration to be transferred would approximate $238.4 million (using the most recent practicable dates prior to the filing of the Form S-4). Changes in the share price of Pozen’s common stock, or
changes in the number of outstanding Tribute common shares, stock options, compensation options and warrants of Tribute could result in material differences in the consideration and, thus, the result of the related transactions. At the effective time, each outstanding Tribute common share will be exchanged for 0.1455 Parent Shares.
The following is a preliminary estimate of the consideration to be transferred by Pozen (in U.S. dollars)
|
Preliminary estimate of fair value of Parent Shares issued(i) |
|
$ |
198,110,326 |
|
|
Preliminary estimate of fair value of equity instruments(ii) |
|
18,817,997 |
| |
|
Repayment of Tribute indebtedness(iii) |
|
24,663,259 |
| |
|
Total consideration transferred |
|
$ |
241,591,582 |
|
(i) Represents the conversion of each of Tribute’s common shares outstanding at June 30, 2015 (125,030,578) at a conversion rate of 0.1455 with a value of $10.89 at July 14, 2015.
(ii) The table below summarizes the Tribute equity instruments included within purchase consideration:
|
Type of award |
|
Number of |
|
Parent shares |
|
Fair value |
|
|
Equity classified warrants |
|
6,857,962 |
|
997,833 |
|
7,007,243 |
|
|
Compensation options |
|
1,307,706 |
|
190,271 |
|
1,332,447 |
|
|
Employee stock options vested prior to the close of the transactions |
|
2,074,600 |
|
301,854 |
|
3,287,193 |
|
|
Employee stock options for which vesting was accelerated pursuant to automatic change in control provisions |
|
4,538,426 |
|
660,341 |
|
7,191,114 |
|
|
Total |
|
14,778,694 |
|
2,150,300 |
|
18,817,997 |
|
(iii) Represents repayment of Tribute indebtedness associated with SWK loan in amount of $14.4 million and payoff of Tribute debenture for $10.3 million including accrued interest and other costs.
The estimated value of the consideration does not purport to represent the actual value of the total consideration that will be received by Tribute’s shareholders when the transactions are complete. In accordance with US GAAP, the fair value of the equity securities issued as part of the consideration will be measured at the closing date at the then-current market price. This requirement will likely result in a per share value component different from the $10.89 per share on July 14, 2015 assumed in the calculation, and that difference may be material. For example, an increase and decrease of 10% in the price of Pozen’s common stock on the closing date of the transactions from the price of Pozen stock assumed in these unaudited pro forma condensed combined financial statements would change the value of the consideration by approximately $22.1 million and $21.0 million, respectively, which would be reflected as an equivalent increase or decrease to goodwill.
The following is a summary of the preliminary estimated fair values of the net assets (in US dollars):
|
Total estimated consideration transferred |
|
$ |
241,591,582 |
|
|
Working capital(i) |
|
3,022,373 |
| |
|
Property, plant, and equipment |
|
925,120 |
| |
|
Intangible assets |
|
130,017,384 |
| |
|
Other liabilities |
|
(38,852,281 |
) | |
|
Net assets acquired |
|
95,112,596 |
| |
|
Goodwill |
|
$ |
146,478,986 |
|
(i) Working capital consists of current assets less Accounts payable, accrued compensation, and accrued expenses.
Pozen has made preliminary allocation estimates based on limited access to information and will not have sufficient information to make final allocations until after completion of the transactions. The final determination of the accounting for the business combination is anticipated to be completed as soon as practicable after completion of the transactions. Pozen anticipates that the valuations of the acquired assets and liabilities will include, but not be limited to inventory, property, plant, and equipment, developed products, and in-process research and development. The valuations will consist of physical appraisals, discounted cash flow analyses, or other appropriate valuation techniques to determine the fair value of the assets acquired and liabilities assumed.
The final consideration, and amounts allocated to assets acquired and liabilities assumed in the transactions could differ materially from the preliminary amounts presented in these unaudited pro forma condensed combined financial statements. A decrease in the fair value of assets acquired or an increase in the fair value of liabilities assumed in the transactions from those preliminary valuations presented in these unaudited pro forma condensed combined financial statements would result in a dollar-for-dollar corresponding increase in the amount of goodwill that will result from the transactions. In addition, if the value of the acquired assets is higher than the preliminary indication, it may result in higher amortization and depreciation expense than is presented in these unaudited pro forma condensed combined financial statements.
7. MERGER AND ARRANGEMENT—PRELIMINARY PRO FORMA ADJUSTMENTS
The preliminary pro forma adjustments included in the unaudited pro forma condensed combined financial statements related to the transactions are as follows:
(a) Cash and cash equivalents—Adjustment reflects the preliminary net adjustment to cash in connection with the transactions (in US dollars):
|
Repayment of Tribute SWK Loan, including fees(i) |
|
$ |
(14,382,079 |
) |
|
Repayment of Tribute debenture, including redemption fee(ii) |
|
(10,281,180 |
) | |
|
Transaction expenses to be incurred by Pozen(iii) |
|
(11,125,859 |
) | |
|
Transaction expenses to be incurred by Tribute(iii) |
|
(3,440,913 |
) | |
|
Pro forma adjustment to cash and cash equivalents |
|
(39,230,032 |
) | |
Components of the adjustment (i) a decrease in cash related to the repayment of Tribute’s SWK loan, including an early payment premium of $0.8 million; (ii) repayment of Tribute’s debenture incurred in connection with the MFI Acquisition of $10.3 million, including an early payment premium of $0.4 million and (iii) estimated transaction related expenses of $14.6 million, consisting of $11.1 million and $3.4 million to be incurred by Pozen and Tribute, respectively.
(b) Inventories—Adjustment reflects the preliminary estimated fair value adjustment of $1.2 million to total inventory acquired in the transactions. As the raw materials inventory was assumed to be at market value, the preliminary adjustment is related to finished goods inventory. The preliminary fair value of finished goods inventory to be acquired in the transactions was determined based on an analysis of estimated future selling prices, costs of disposal, and gross profit on disposal costs. The unaudited pro forma combined statements of operations do not reflect the impact of this preliminary inventory increase in cost of sales as such amounts are directly attributable to the transactions and will not have a continuing impact on the combined results.
(c) Prepaid expenses and other assets—Adjustment reflects the preliminary estimate of deferred tax asset of $0.1 million and a $1.4 million current tax receivable recognized based on the various pro forma adjustments (refer to Note 7(k) for further details), partially offset by the $0.1 million write off of the short term portion of Tribute’s deferred financing costs related to the loan with SWK.
(d) Goodwill—Adjustment reflects the preliminary estimated adjustment to goodwill as a result of the transactions. Goodwill represents the excess of the consideration transferred over the preliminary fair value of the assets acquired and liabilities assumed as described in Note 6. The goodwill will not be amortized, but instead will be tested for impairment at least annually and whenever events or circumstances have occurred that may indicate a possible impairment exists. In the event management determines that the value of goodwill has become impaired, Pozen will incur an accounting charge for the amount of the impairment during the period in which the determination is made. The goodwill is attributable to the expected synergies of the combined business operations, new growth opportunities, and the acquired assembled and trained workforce of Tribute. The goodwill is not expected to be deductible for tax purposes. The preliminary pro forma adjustment to goodwill is calculated as follows (in US dollars):
|
Consideration transferred |
|
$ |
241,591,582 |
|
|
Less: Fair value of net assets to be acquired |
|
95,112,596 |
| |
|
Total estimated goodwill |
|
146,478,986 |
| |
|
Less: Tribute pro forma goodwill amounts |
|
22,376,441 |
| |
|
Pro forma adjustment to goodwill |
|
$ |
124,102,545 |
|
(e) Intangible assets—Adjustment reflects the preliminary fair market value related to the change in fair value of identifiable intangible assets acquired in the transactions. The preliminary fair market
value was determined using a market approach. The preliminary amounts assigned to the identifiable intangible assets are as follows (in US dollars):
|
Intangible Asset |
|
Preliminary |
| |
|
Tribute developed products |
|
$ |
73,549,980 |
|
|
IPR&D products |
|
32,425,260 |
| |
|
MF developed products |
|
20,087,844 |
| |
|
MF IPR&D products |
|
3,954,300 |
| |
|
Total |
|
130,017,384 |
| |
|
Less: Tribute pro forma intangible assets amounts |
|
(32,001,649 |
) | |
|
Pro forma adjustment to intangible assets |
|
$ |
98,015,735 |
|
(f) Debt issuance costs, net—Adjustment reflects the write off of the long term potion of deferred financing costs related to the SWK Loan.
(g) Warrant liability—Adjustment reflects an increase to the fair value of the Tribute warrant liability assumed in the transactions, which related to warrants denominated in a currency other than Pozen’s functional currency, which is US dollars.
(h) Other accrued liabilities—Adjustment reflects the $16.9 million related to the make-whole payment of excise tax for each director and executive officer of Pozen that is expected to be paid out and $1.8 million of severance payments that will be paid out to certain executive officers of Tribute upon a change in control. This is offset by a $0.3 million reduction for the payoff of accrued interest in association with the SWK loan which was repaid.
(i) Current and long-term debt—The current portion of the long term debt relates to the repayment of the Tribute debenture that was issued in connection with the MFI Acquisition of $9.9 million and the repayment of the current portion of the SWK Loan of $1.2 million. The adjustment related to the long term debt relates to the repayment of the long term portion of the SWK Loan of $12.1 million.
(j) Contingent consideration—Adjustment to reflect the estimated fair value of contingent consideration from the MFI Acquisition related to the attainment of regulatory approval of two of MFI’s pipeline products, which becomes due upon a change in control of Tribute.
(k) Deferred income taxes—Adjustment reflects the deferred income tax effects of the preliminary pro forma adjustments made to the pro forma balance sheet, using the Irish statutory tax rate of 12.5%, primarily as indicated in the table below (in US dollars):
|
|
|
Adjustment to |
|
Current |
|
Non-Current |
|
|
Estimated fair value adjustment of identifiable intangible assets acquired |
|
98,015,735 |
|
— |
|
12,251,967 |
|
|
Estimated fair value adjustment of inventory acquired |
|
1,186,290 |
|
148,286 |
|
— |
|
|
Estimated tax impact of Pozen transactions costs |
|
N/A |
|
(973,513 |
) |
— |
|
|
Estimated tax impact of post-combination expense related to the payment of unvested equity awards in connection with the transactions |
|
N/A |
|
(262,814 |
) |
— |
|
|
Estimated tax impact of accelerated vesting of Pozen equity instruments |
|
|
|
(453,358 |
) |
— |
|
|
Total adjustments to deferred tax liabilities (assets) |
|
|
|
(1,541,399 |
) |
12,251,967 |
|
(l) Common shares—Adjustment relates to the elimination of the Tribute pro forma historical common shares of $35.6 million offset by the par value of common shares issued in the transactions of $18.2 thousand.
(m) APIC—The preliminary unaudited pro forma adjustment to capital in excess of par is calculated as follows:
|
Fair value of Tribute stock options exchanged for Parent Shares using the exchange ratio |
|
$ |
12,580,821 |
|
|
Accelerated vesting of Pozen RSU’s, Non-qualified options, and Incentive stock options |
|
3,626,864 |
| |
|
Fair value of Tribute warrants and compensation options exercisable for Parent Shares, using the exchange ratio |
|
1,332,447 |
| |
|
Capital in excess of par from the Tribute acquisition (125,030,578 Parent Shares issued at $10.89, less par value) |
|
198,092,134 |
| |
|
Less: Tribute pro forma historical equity |
|
(6,372,909 |
) | |
|
Pro forma adjustment to additional paid-in-capital |
|
$ |
209,259,357 |
|
(n) Retained earnings/(accumulated deficit)—The preliminary unaudited pro forma adjustment is calculated as follows:
|
Estimated fees and expenses expected to be incurred by Pozen related to the transactions of $28.0 million, and net of tax of $1.0 million |
|
$ |
(27,022,894 |
) |
|
Post combination expense of $2.1 million related to unvested equity awards upon completion of the transactions, net of tax of $0.3 million |
|
(1,839,700 |
) | |
|
Compensation expense of $3.6 million related to unvested Pozen equity awards for which vesting will accelerate upon completion of the transactions, net of tax of $0.5 million |
|
(3,173,506 |
) | |
|
Less: Tribute pro forma historical equity |
|
20,010,331 |
| |
|
Pro forma adjustment to retained earnings/(accumulated deficit) |
|
$ |
(12,025,769 |
) |
The estimated fees and expenses and post-combination compensation expense associated with the payment of accelerated equity awards have been excluded from the unaudited pro forma condensed combined statements of operations as they reflect charges directly attributable to the transactions that will not have a continuing impact on Parent’s operations.
(o) Warrants—Reflects the elimination for the historical warrant balance offset by the fair value of Parent warrants issued in exchange for Tribute warrants denominated in $USD as a result of the transactions.
(p) Accumulated other comprehensive loss—The preliminary unaudited pro forma adjustment to accumulated other comprehensive loss eliminates Tribute’s pro forma historical accumulated other comprehensive loss of $34.3 thousand.
(q) Sales, general, and administrative expenses—Adjustment reflects the removal of transaction expenses incurred related to the transactions. These expenses are one-time and non-recurring and have therefore been thus removed for the purposes of the pro forma statements of operations.
(r) Amortization of intangibles assets—Adjustment reflects the preliminary adjustment to the amortization expense associated with the fair value of the identifiable intangible assets acquired in the transactions of $8.2 million and $1.9 million for the year ended December 31, 2014 and the three months ended March 31, 2015, respectively.
The preliminary amortization expense for the intangible assets acquired in the transactions is as follows:
|
Intangible Asset |
|
Estimated |
|
Preliminary |
|
Amortization Expense |
|
Amortization Expense |
| |||
|
Tribute developed products |
|
10 |
|
$ |
73,549,980 |
|
$ |
7,354,998 |
|
$ |
1,838,750 |
|
|
IPR&D products |
|
|
|
32,425,260 |
|
N/A |
|
N/A |
| |||
|
MF developed products |
|
10 |
|
20,087,844 |
|
2,008,784 |
|
502,196 |
| |||
|
MF IPR&D products |
|
|
|
3,954,300 |
|
N/A |
|
N/A |
| |||
|
Total |
|
|
|
130,017,384 |
|
9,363,782 |
|
2,340,946 |
| |||
|
Less: Tribute pro forma amounts |
|
|
|
(32,001,649 |
) |
(1,206,499 |
) |
(468,233 |
) | |||
|
Pro forma adjustment |
|
|
|
$ |
98,015,735 |
|
$ |
8,157,283 |
|
$ |
1,872,712 |
|
The estimated fair value of amortizable intangible assets is expected to be amortized on a straight-line basis over the estimated useful lives. The amortizable lives reflect the periods over which the assets are expected to provide material economic benefit. With other assumptions held constant, a 10% increase in the fair value adjustment for amortizable intangible assets would increase annual pro forma amortization by approximately $0.9 million. In addition, with other assumptions held constant, a one year change in the estimated useful lives developed products would change annual amortization expense by approximately $0.7 million.
(s) Interest expense—Adjustment reflects the removal of the interest expense, amortization expense of deferred financing fees, and accretion expense associated with the repayment of the SWK Loan.
(t) Income tax expense (benefit)—Adjustment reflects the income tax impacts of the pro forma adjustments made to the pro forma statement of operations using the Irish statutory tax rate of 12.5%.
(u) Basic and diluted net income per common share—The unaudited pro forma adjustment to shares outstanding used in the calculation of basic and diluted earnings per share is calculated as follows (in shares):
|
|
|
Year ended |
|
Three months ended |
| ||
|
Basic EPS |
|
|
|
|
| ||
|
Historical Pozen weighted average Pozen shares outstanding |
|
31,359,867 |
|
32,259,570 |
| ||
|
Parent Shares to be issued to Tribute shareholders(i) |
|
18,191,949 |
|
18,191,949 |
| ||
|
Tribute options converted to Parent shares(ii) |
|
962,195 |
|
962,195 |
| ||
|
Pozen accelerated vesting of RSU’s(iii) |
|
625,648 |
|
625,648 |
| ||
|
Total weighted average shares outstanding |
|
51,139,659 |
|
52,039,362 |
| ||
|
Pro forma net income available to common shareholders |
|
8,500,530 |
|
(5,452,915 |
) | ||
|
Basic earnings per share |
|
$ |
0.17 |
|
$ |
(0.10 |
) |
|
|
|
Year ended |
| |
|
Diluted EPS(iv) |
|
|
| |
|
Weighted average shares outstanding for basic EPS |
|
51,139,659 |
| |
|
Parent Shares to be issued to Tribute shareholders based on assumed exercise of Tribute warrants and compensation options(v) |
|
574,608 |
| |
|
Conversion of MFI promissory note(vi) |
|
409,405 |
| |
|
Pozen stock options(vii) |
|
447,591 |
| |
|
Total weighted average shares outstanding |
|
52,571,262 |
| |
|
Pro forma net income available to common shareholders(viii) |
|
8,794,530 |
| |
|
Diluted earnings per share |
|
$ |
0.17 |
|
(i) Represents a total of 125,030,578 Tribute common shares converted at the ratio of 0.1455. This share amount is inclusive of the 3,723,008 shares issued by Tribute for the MFI Acquisition.
(ii) Represents Tribute stock options exchangeable for Tribute common shares and converted into Parent shares at an exchange ratio of 0.1455 at the closing.
(iii) As a result of the transactions, RSU’s held by Pozen employees will accelerate and be converted into shares of Parent.
(iv) Since Pozen is in a net loss position for the three months ended March 31, 2015, it has excluded the effect of the 1) assumed exercise of Tribute equity instruments 2) conversion
of the MFI promissory note and 3) the exercise of Pozen stock options in the diluted net loss per share calculations as their effects would have been anti-dilutive.
(v) Represents the total dilutive effect of the assumed exercise of Tribute warrants and compensation options using the treasury stock method. This includes an amount of 3,949,194 shares issuable on the exercise of warrants and compensation options (converted at a 0.1455 ratio)
(vi) As stated in Note 4, the conversion of the MFI promissory note would result in the issuance of 409,405 Parent Shares.
(vii) Represents Pozen stock options which will vest upon the close of the transactions and be outstanding Parent options.
(viii) This was calculated based on pro forma net income available to shareholders of $8.5 million plus an add back on interest expense related to the MFI promissory note of $0.4 million, net of tax of $0.1 million, that would be deemed to have converted into common shares at January 1, 2014.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 13. Other Expenses of Issuance and Distribution.
The following table sets forth the costs and expenses to be paid by us, other than underwriting discounts and commissions, in connection with the sale of the ordinary shares being registered hereby. All amounts are estimates except for the SEC registration fee, the FINRA filing fee and the filing fee.
|
|
|
Amount Paid |
| |
|
SEC registration fee |
|
$ |
5,651 |
|
|
Printing and engraving |
|
* |
| |
|
Legal fees and expenses |
|
* |
| |
|
Accounting fees and expenses |
|
* |
| |
|
Blue sky fees and expenses |
|
* |
| |
|
Transfer agent and registrar fees and expenses |
|
* |
| |
|
Miscellaneous |
|
* |
| |
|
Total |
|
$ |
* |
|
* To be updated by amendment.
ITEM 14. Indemnification of Directors and Officers.
We are incorporated under the laws Ireland. Under Irish law, a company may not exempt its directors from liability for negligence or a breach of duty. However, where a breach of duty has been established, directors may be statutorily exempted by an Irish court from personal liability for negligence or breach of duty if, among other things, the court determines that they have acted honestly and reasonably, and that they may fairly be excused as a result.
Pursuant to our memorandum and articles of association, our directors and secretary are indemnified to the extent permitted by the Companies Act. We may indemnify our directors or secretary only if the indemnified party receives a favorable judgment in respect of the liability, or where an Irish court determines that the director or the secretary acted honestly and reasonably and ought fairly to be excused, or the proceedings are otherwise disposed of without any finding or admission of any material breach of duty on the part of the director or secretary, or in which the director or secretary is acquitted. This restriction in the Companies Act does not apply to executives who are not our directors or the secretary. Any provision for indemnification to a greater extent is void under Irish law, whether contained in a memorandum and articles of association or any contract between the director and the Irish company.
Our memorandum and articles of association also contain indemnification and expense advancement provisions for current or former executives who are not our directors or secretary, except no indemnification shall be made in respect of any claim, issue or matter as to which such person shall have been adjudged to be liable for fraud or dishonesty in the performance of her or her duty to the company.
We indemnify our directors and certain officers, as well as individuals serving as directors or officers of its subsidiaries, pursuant to indemnification agreements.
ITEM 16. Exhibits and Financial Statement Schedules.
(a) Exhibits.
|
Exhibit |
|
Exhibit Title |
|
1.1 |
|
Underwriting Agreement, dated June 25, 2014, among Tribute, Dundee Securities Ltd. and Mackie Research Capital Corporation.* |
|
|
|
|
|
1.2 |
|
Agency Agreement, dated as of June 16, 2015, by and among Tribute Pharmaceuticals Canada Inc., and KES 7 Capital Inc., Bloom Burton & Co. Ltd, Mackie Research Capital Corporation, Laurentian Bank Securities Inc. and Dundee Capital Markets Ltd.* |
|
|
|
|
|
1.3 |
|
Agency Agreement, dated as of May 21, 2015, by and among Tribute, Dundee Securities Ltd., KES 7 Capital Inc. and Bloom Burton & Co. Inc.* |
|
|
|
|
|
2.1 |
|
Agreement and Plan of Merger and Arrangement, dated as of June 8, 2015, among Pozen, Tribute, Parent, Ltd2, US Merger Sub and Can Merger Sub (filed as Exhibit 2.1 to Pozen’s Current Report on Form 8-K filed June 11, 2015).* |
|
|
|
|
|
2.2 |
|
Share Purchase Agreement, dated December 1, 2011, by and among Stellar Pharmaceuticals Inc., Elora Financial Management Inc., Mary-Ann Harris, Rob Harris and Scott Langille.* |
|
|
|
|
|
2.3 |
|
Asset Purchase Agreement, dated October 2, 2014, by and among Novartis AG, Novartis Pharma AG and Tribute. †* |
|
|
|
|
|
2.4 |
|
License Agreement, dated as of October 2, 2014, by and among Novartis AG, Novartis Pharma AG, Novartis Pharmaceuticals Canada Inc. and Tribute. †* |
|
|
|
|
|
2.5 |
|
Share Purchase Agreement, dated as of June 16, 2015, by and among Tribute Pharmaceuticals Canada Inc. and the shareholders of Medical Futures Inc. †* |
|
|
|
|
|
2.6 |
|
Asset Purchase Agreement, dated as of May 21, 2015, by and among Tribute, Mutual Pharmaceutical Company, Inc. and Sun Pharmaceutical Industries, Inc. †* |
|
|
|
|
|
3.1 |
|
Certificate of Incorporation of Aguono Limited, dated May 12, 2015 (filed as Exhibit 3.1 to the Registrant’s Registration Statement on Form S-4 filed July 20, 2015). |
|
|
|
|
|
3.2 |
|
Memorandum of Association of Aguono Limited, dated as of May 12, 2015, and Articles of Association of Aguono Limited, dated as of May 12, 2015 (filed as Exhibit 3.2 to the Registrant’s Registration Statement on Form S-4 filed July 20, 2015). |
|
3.3 |
|
Certificate of Incorporation on change of name of Aralez Pharmaceuticals Limited (f/k/a Aguono Limited), dated June 16, 2015 (filed as Exhibit 3.3 to the Registrant’s Registration Statement on Form S-4 filed July 20, 2015). |
|
|
|
|
|
3.4 |
|
Memorandum of Association of Aralez Pharmaceuticals Limited, dated as of June 5, 2015, and Articles of Association of Aralez Pharmaceuticals Limited, dated as of June 5, 2015 (filed as Exhibit 3.4 to the Registrant’s Registration Statement on Form S-4 filed July 20, 2015). |
|
|
|
|
|
3.5 |
|
Memorandum and Articles of Association of Aralez Pharmaceuticals Public Limited Company (amended , 2015).* |
|
|
|
|
|
4.1 |
|
Specimen Ordinary Share certificate.* |
|
|
|
|
|
4.2 |
|
Form of Series A/B Warrant, dated February 27, 2013.* |
|
|
|
|
|
4.3 |
|
Form of Series A/B Warrant, dated March 5, 2013.* |
|
|
|
|
|
4.4 |
|
Form of Series A/B Warrant, dated March 11, 2013.* |
|
|
|
|
|
4.5 |
|
Warrant, dated May 11, 2012, issued to Midcap Funding III, LLC.* |
|
|
|
|
|
4.6 |
|
Amended and Restated Warrant, dated February 27, 2013, issued to Midcap Funding III, LLC.* |
|
|
|
|
|
4.7 |
|
Promissory Note, dated August 8, 2013, issued to SWK Funding LLC.* |
|
|
|
|
|
4.8 |
|
Warrant, dated August 8, 2013, issued to SWK Funding LLC.* |
|
|
|
|
|
4.9 |
|
Promissory Note, dated February 4, 2014, issued to SWK Funding LLC.* |
|
|
|
|
|
4.1 |
|
Warrant, dated February 4, 2014, issued to SWK Funding LLC.* |
|
|
|
|
|
4.11 |
|
Warrant Indenture, dated July 15, 2014, between Tribute and Equity Financial Trust Company.* |
|
|
|
|
|
4.12 |
|
Promissory Note issued on June 16, 2015. †* |
|
|
|
|
|
4.13 |
|
Form of Debenture for the Debenture Offering that closed on June 16, 2015.* |
|
|
|
|
|
5.1 |
|
Opinion of A&L Goodbody regarding the legality of the securities being registered.* |
|
|
|
|
|
10.1 |
|
Executive Employment Agreement with William L. Hodges dated August 3, 2004 (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q filed October 27, 2004). + |
|
|
|
|
|
10.2 |
|
First Amendment to Executive Employment Agreement with William L. Hodges, dated September 28, 2007 (filed as Exhibit 10.5 to Pozen’s Quarterly Report on Form 10-Q filed November 5, 2007). + |
|
|
|
|
|
10.3 |
|
Lease Agreement between The Exchange at Meadowmont LLC and the Registrant dated as of November 21, 2001 (filed as Exhibit 10.21 to Pozen’s Annual Report on Form 10-K filed April 1, 2002). |
|
|
|
|
|
10.4 |
|
Product Development and Commercialization Agreement dated June 11, 2003 between the Registrant and Glaxo Group Ltd. (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q filed August 12, 2003 and Form 10-Q/A filed November 8. 2004).† |
|
|
|
|
|
10.5 |
|
License Agreement dated June 11, 2003 between the Registrant and Glaxo Group Ltd. (filed as Exhibit 10.2 to Pozen’s Quarterly Report on Form 10-Q filed August 12, 2003 and Quarterly Report on Form 10-Q/A filed November 8. 2004).† |
|
|
|
|
|
10.6 |
|
Collaboration and License Agreement dated September 3, 2003 between the Registrant and Valeant Pharmaceuticals NA (formerly Xcel Pharmaceuticals, Inc.) (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q filed November 6, 2003 and Quarterly Report on Form 10-Q/A filed November 8. 2004).† |
|
|
|
|
|
10.7 |
|
Restricted Stock Unit Agreement dated May 4, 2004 between Registrant and John R. Plachetka (filed as Exhibit 10.4 to Pozen’s Quarterly Report on Form 10-Q filed July 30, 2004).+ |
|
|
|
|
|
10.8 |
|
First Amendment, dated September 28, 2007, to Restricted Stock Unit Agreement, dated May 4, 2004, between Registrant and John R. Plachetka (filed as Exhibit 10.2 to Pozen’s Quarterly Report on Form 10-Q filed November 5, 2007).+ |
|
|
|
|
|
10.9 |
|
Collaboration and License Agreement dated August 1, 2006 between the Registrant and AstraZeneca AB (filed as Exhibit 10.1 to Pozen’s Quarterly Report on From 10-Q filed November 3, 2006).† |
|
|
|
|
|
10.10 |
|
Amendment No. 1 to the Collaboration and License Agreement, dated September 6, 2007, between the Registrant and AstraZeneca AB (filed as Exhibit 10.8 to Pozen’s Quarterly Report on Form 10-Q filed November 5, 2007). † |
|
|
|
|
|
10.11 |
|
Side Letter dated September 19, 2006 Re: Collaboration and License Agreement dated as of August 1, 2006 by and between the Registrant and AstraZeneca AB (filed as 10.2 to Pozen’s Quarterly Report on From 10-Q filed November 3, 2006).† |
|
|
|
|
|
10.12 |
|
Side Letter Agreement, dated October 1, 2007, between the Registrant and AstraZeneca, AB (filed as Exhibit 10.9 to Pozen’s Quarterly Report on Form 10-Q filed November 5, 2007). † |
|
|
|
|
|
10.13 |
|
Long-Term Cash Incentive Award Agreement between the Registrant and John R. Plachetka dated February 14, 2007 (filed as Exhibit 10.4 to Pozen’s Quarterly Report on Form 10-Q filed May 3, 2007).+ |
|
|
|
|
|
10.14 |
|
Amendment No. 2 to the Collaboration and License Agreement, dated October 1, 2008, between the registrant and AstraZeneca, AB (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q filed November 4, 2008). † |
|
10.15 |
|
Lease Modification Agreement No. 1, dated as of February 16, 2009, by and between the Registrant and The Exchange at Meadowmont LLC (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K filed on February 17, 2009). |
|
|
|
|
|
10.16 |
|
Executive Employment Agreement, dated as of December 10, 2009, between the Company and John G. Fort, M.D. (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K, filed on December 11, 2009).+ |
|
|
|
|
|
10.17 |
|
Purchase and Sale Agreement, dates as November 23, 2011, by and between POZEN Inc. and CPPIB Credit Investments Inc. (filed as Exhibit 10.37 to the Registrants Annual Report on Form 10-K filed March 9. 2012). |
|
|
|
|
|
10.18 |
|
Manufacturing Services Agreement, dates as December 19, 2011, by and between POZEN Inc. and Patheon Pharmaceuticals, Inc.†(filed as Exhibit 10.38 to the Registrants Amendment No.1 to the Annual Report on Form 10-K, filed June 29,2012). |
|
|
|
|
|
10.19 |
|
Capital Expenditure and Equipment Agreement, dates as of December 19, 2011, by and between POZEN Inc. and Patheon Pharmaceuticals, Inc. (filed is Exhibit 10.39 to the Registrants Amendment No.11 to Annual Report on Form 10-K, filed June 29,2012). |
|
|
|
|
|
10.20 |
|
First Amendment to Manufacturing Services Agreement, between Patheon Pharmaceuticals Inc., and POZEN Inc., dated as of July 10, 2013 (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q, filed August 7, 2013).† |
|
|
|
|
|
10.21 |
|
First Amendment to Capital Expenditure and Equipment Agreement, between Patheon Pharmaceuticals Inc., and POZEN Inc., dated as of July 10, 2013 (filed as Exhibit 10.2 to Pozen’s Quarterly Report on Form 10-Q, filed August 7, 2013).† |
|
|
|
|
|
10.22 |
|
License and Development Agreement, dated as of May 7, 2012, by and between POZEN Inc. and DESITIN Arzneimittel GmbH (filed as Exhibit 10.1 to Registrants Quarterly Report on Form 10-Q, filed on August 8, 2012). |
|
|
|
|
|
10.23 |
|
Amendment No. 3 to the Collaboration and License Agreement between POZEN Inc. and AstraZeneca AB, dated as of September 16, 2013 (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q, filed August 7, 2013).† |
|
|
|
|
|
10.24 |
|
Letter Agreement among POZEN Inc., AstraZeneca AB and Horizon Pharma U.S.A. Inc. dated as of November 18, 2013 (filed as Exhibit 10.43 to Pozen’s Annual Report on Form 10-K, filed March 6, 2014). † |
|
|
|
|
|
10.25 |
|
Amendment No. 1 to Amended and Restated Collaboration and License Agreement for the United States by and between POZEN Inc. and Horizon Pharma U.S.A. Inc. dated as of November 18, 2013 (filed as Exhibit 10.44 to Pozen’s Annual Report on Form 10-K, filed March 6, 2014). † |
|
|
|
|
|
10.26 |
|
Amended and Restated Collaboration and License Agreement for the United States by and between POZEN Inc. and AstraZeneca AB dated as of November 18, 2013 (filed as Exhibit 10.45 to Pozen’s Annual Report on Form 10-K, filed March 6, 2014). † |
|
|
|
|
|
10.27 |
|
Amended and Restated Collaboration and License Agreement for outside of the United States by and between POZEN Inc. and AstraZeneca AB dated as of November 18, 2013 (filed as Exhibit 10.46 to Pozen’s Annual Report on Form 10-K, filed March 6, 2014). † |
|
|
|
|
|
10.28 |
|
Letter Agreement, dated as of May 13, 2014, by and among POZEN Inc., CPPIB Credit Investments, Inc. Pernix Therapeutics Holdings, Inc. and Glaxo Group Limited (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q, filed November 6, 2014). |
|
|
|
|
|
10.29 |
|
First Amendment to Product Development and Commercialization Agreement, dated as of May 13, 2014, by and between POZEN Inc. and Pernix Therapeutics Holdings, Inc. (filed as Exhibit 10.2 to Pozen’s Quarterly Report on Form 10-Q, filed November 6, 2014). |
|
|
|
|
|
10.30 |
|
Second Amendment to Product Development and Commercialization Agreement dated as of July 30, 2014, by and between POZEN Inc. and Pernix Therapeutics Holdings, Inc. (filed as Exhibit 10.3 to Pozen’s Quarterly Report on Form 10-Q, filed November 6, 2014). |
|
|
|
|
|
10.31 |
|
Facility Agreement, dated as of June 8, 2015, among the Borrower, Pozen, Tribute and the Lenders (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K filed June 11, 2015). |
|
10.32 |
|
Registration Rights Agreement, dated as of June 8, 2015, among Parent and the Lenders (filed as Exhibit 10.2 to Pozen’s Current Report on Form 8-K filed June 11, 2015). |
|
|
|
|
|
10.33 |
|
Share Subscription Agreement, dated as of June 8, 2015, among Pozen, Purchaser, Tribute, Parent and the Investors (filed as Exhibit 10.3 to Pozen’s Current Report on Form 8-K filed June 11, 2015). |
|
|
|
|
|
10.34 |
|
Executive Employment Agreement between Pozen and Adrian Adams dated May 31, 2015 (filed as Exhibit 10.3 to Pozen’s Current Report on Form 8-K filed June 3, 2015).+ |
|
|
|
|
|
10.35 |
|
Executive Employment Agreement between Pozen and Andrew I. Koven dated May 31, 2015(filed as Exhibit 10.4 to Pozen’s Current Report on Form 8-K filed June 3, 2015). + |
|
|
|
|
|
10.36 |
|
Executive Employment Agreement between Pozen and Mark Glickman dated June 22, 2015.* + |
|
|
|
|
|
10.37 |
|
Executive Employment Agreement between Pozen and Eric Trachtenberg dated June 22, 2015.* + |
|
|
|
|
|
10.38 |
|
Executive Employment Agreement between Pozen and Jennifer Armstrong dated June 22, 2015.* + |
|
|
|
|
|
10.39 |
|
Executive Employment Agreement between Pozen and Scott Charles dated July 27, 2015.* + |
|
|
|
|
|
10.40 |
|
Separation and General Release Agreement between Pozen and John R. Plachetka, dated May 29, 2015 (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K filed June 3, 2015). + |
|
|
|
|
|
10.41 |
|
Voting Agreement between Pozen and John R. Plachetka, dated May 29, 2015 (filed as Exhibit 10.2 to Pozen’s Current Report on Form 8-K filed June 3, 2015). + |
|
|
|
|
|
10.42 |
|
POZEN Inc. Employee Severance Plan and Summary Plan Description (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K filed June 25, 2015). + |
|
|
|
|
|
10.43 |
|
Form of Pozen Retention Agreement (filed as Exhibit 10.2 to Pozen’s Current Report on Form 8-K filed June 25, 2015).+ |
|
|
|
|
|
10.44 |
|
Aralez Pharmaceuticals plc 2015 Long-Term Incentive Plan.* |
|
|
|
|
|
21.1 |
|
Subsidiaries of Registrant. |
|
|
|
|
|
23.1 |
|
Consent of Ernst & Young LLP, independent registered public accounting firm for Pozen. |
|
|
|
|
|
23.2 |
|
Consent of McGovern, Hurley, Cunningham, LLP, independent auditors for Tribute. |
|
|
|
|
|
23.3 |
|
Consent of A&L Goodbody (included in Exhibit 5.01).* |
|
|
|
|
|
24.1 |
|
Power of Attorney (see signature page hereto).* |
* To be filed by amendment.
** Filed herewith.
+ Compensation Related Contract.
† Confidential treatment requested. Confidential materials omitted and filed separately with Securities and Exchange Commission.
(b) Financial Statement Schedules.
All financial statement schedules have been omitted because they are either inapplicable or the required information has been given in the Registrant’s consolidated financial statements or the notes thereto.
Item 17. Undertakings.
The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(5) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act, and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by the controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
SIGNATURES
Pursuant to the requirements of the Securities Act, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized in the city of Dublin, Ireland, on August 7, 2015.
|
|
Aralez Pharmaceuticals Limited | ||
|
|
|
| |
|
|
By: |
/s/ ADRIAN ADAMS | |
|
|
|
Name: |
Adrian Adams |
|
|
|
Title: |
Chief Executive Officer |
BE IT KNOWN BY THESE PRESENTS: That each person whose name is signed hereto has made, constituted and appointed, and does hereby make, constitute and appoint Adrian Adams and William L. Hodges, his or her true and lawful attorney-in-fact and agent, with full power of substitution, for him or her and in his or her name, place and stead, in any and all capacities, to affix his or her signature as director or officer or both, as the case may be, of the registrant, to any and all registration statements and amendments thereto (including post-effective amendments) and to file the same, with all exhibits thereto, and other documents in connection therewith, and to file with the Securities and Exchange Commission, granting unto each such attorney-in-fact full power and authority to do and perform every act and thing whatsoever necessary to be done in the premises, as fully as he or she might or could do if personally present, hereby ratifying and confirming all that each such attorney-in-fact shall lawfully do or cause to be done by virtue hereof. Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed by the following persons in the capacities and on the dates indicated below.
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ ADRIAN ADAMS |
|
Chief Executive Officer (Principal Executive Officer) |
|
August 7, 2015 |
|
Adrian Adams |
|
|
| |
|
|
|
|
|
|
|
/s/ WILLIAM L. HODGES |
|
Chief Financial Officer (Principal Financial and Accounting Officer), Director |
|
August 7, 2015 |
|
William L. Hodges |
| |||
|
|
|
|
|
|
|
/s/ GERALDINE LILLIS |
|
Director |
|
August 7, 2015 |
|
Geraldine Lillis |
| |||
|
|
|
|
|
|
|
/s/ ANDREW RYAN |
|
Director |
|
August 7, 2015 |
|
Andrew Ryan |
|
EXHIBIT INDEX
|
Exhibit |
|
Exhibit Title |
|
1.1 |
|
Underwriting Agreement, dated June 25, 2014, among Tribute, Dundee Securities Ltd. and Mackie Research Capital Corporation.* |
|
|
|
|
|
1.2 |
|
Agency Agreement, dated as of June 16, 2015, by and among Tribute Pharmaceuticals Canada Inc., and KES 7 Capital Inc., Bloom Burton & Co. Ltd, Mackie Research Capital Corporation, Laurentian Bank Securities Inc. and Dundee Capital Markets Ltd.* |
|
|
|
|
|
1.3 |
|
Agency Agreement, dated as of May 21, 2015, by and among Tribute, Dundee Securities Ltd., KES 7 Capital Inc. and Bloom Burton & Co. Inc.* |
|
|
|
|
|
2.1 |
|
Agreement and Plan of Merger and Arrangement, dated as of June 8, 2015, among Pozen, Tribute, Parent, Ltd2, US Merger Sub and Can Merger Sub (filed as Exhibit 2.1 to Pozen’s Current Report on Form 8-K filed June 11, 2015).* |
|
|
|
|
|
2.2 |
|
Share Purchase Agreement, dated December 1, 2011, by and among Stellar Pharmaceuticals Inc., Elora Financial Management Inc., Mary-Ann Harris, Rob Harris and Scott Langille.* |
|
|
|
|
|
2.3 |
|
Asset Purchase Agreement, dated October 2, 2014, by and among Novartis AG, Novartis Pharma AG and Tribute. †* |
|
|
|
|
|
2.4 |
|
License Agreement, dated as of October 2, 2014, by and among Novartis AG, Novartis Pharma AG, Novartis Pharmaceuticals Canada Inc. and Tribute. †* |
|
|
|
|
|
2.5 |
|
Share Purchase Agreement, dated as of June 16, 2015, by and among Tribute Pharmaceuticals Canada Inc. and the shareholders of Medical Futures Inc. †* |
|
|
|
|
|
2.6 |
|
Asset Purchase Agreement, dated as of May 21, 2015, by and among Tribute, Mutual Pharmaceutical Company, Inc. and Sun Pharmaceutical Industries, Inc. †* |
|
|
|
|
|
3.1 |
|
Certificate of Incorporation of Aguono Limited, dated May 12, 2015 (filed as Exhibit 3.1 to the Registrant’s Registration Statement on Form S-4 filed July 20, 2015). |
|
|
|
|
|
3.2 |
|
Memorandum of Association of Aguono Limited, dated as of May 12, 2015, and Articles of Association of Aguono Limited, dated as of May 12, 2015 (filed as Exhibit 3.2 to the Registrant’s Registration Statement on Form S-4 filed July 20, 2015). |
|
|
|
|
|
3.3 |
|
Certificate of Incorporation on change of name of Aralez Pharmaceuticals Limited (f/k/a Aguono Limited), dated June 16, 2015 (filed as Exhibit 3.3 to the Registrant’s Registration Statement on Form S-4 filed July 20, 2015). |
|
|
|
|
|
3.4 |
|
Memorandum of Association of Aralez Pharmaceuticals Limited, dated as of June 5, 2015, and Articles of Association of Aralez Pharmaceuticals Limited, dated as of June 5, 2015 (filed as Exhibit 3.4 to the Registrant’s Registration Statement on Form S-4 filed July 20, 2015). |
|
|
|
|
|
3.5 |
|
Memorandum and Articles of Association of Aralez Pharmaceuticals Public Limited Company (amended , 2015).* |
|
|
|
|
|
4.1 |
|
Specimen Ordinary Share certificate.* |
|
|
|
|
|
4.2 |
|
Form of Series A/B Warrant, dated February 27, 2013.* |
|
|
|
|
|
4.3 |
|
Form of Series A/B Warrant, dated March 5, 2013.* |
|
|
|
|
|
4.4 |
|
Form of Series A/B Warrant, dated March 11, 2013.* |
|
|
|
|
|
4.5 |
|
Warrant, dated May 11, 2012, issued to Midcap Funding III, LLC.* |
|
|
|
|
|
4.6 |
|
Amended and Restated Warrant, dated February 27, 2013, issued to Midcap Funding III, LLC.* |
|
|
|
|
|
4.7 |
|
Promissory Note, dated August 8, 2013, issued to SWK Funding LLC.* |
|
|
|
|
|
4.8 |
|
Warrant, dated August 8, 2013, issued to SWK Funding LLC.* |
|
|
|
|
|
4.9 |
|
Promissory Note, dated February 4, 2014, issued to SWK Funding LLC.* |
|
|
|
|
|
4.1 |
|
Warrant, dated February 4, 2014, issued to SWK Funding LLC.* |
|
|
|
|
|
4.11 |
|
Warrant Indenture, dated July 15, 2014, between Tribute and Equity Financial Trust Company.* |
|
|
|
|
|
4.12 |
|
Promissory Note issued on June 16, 2015. †* |
|
|
|
|
|
4.13 |
|
Form of Debenture for the Debenture Offering that closed on June 16, 2015.* |
|
|
|
|
|
5.1 |
|
Opinion of A&L Goodbody regarding the legality of the securities being registered.* |
|
|
|
|
|
10.1 |
|
Executive Employment Agreement with William L. Hodges dated August 3, 2004 (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q filed October 27, 2004). + |
|
|
|
|
|
10.2 |
|
First Amendment to Executive Employment Agreement with William L. Hodges, dated September 28, 2007 (filed as Exhibit 10.5 to Pozen’s Quarterly Report on Form 10-Q filed November 5, 2007). + |
|
|
|
|
|
10.3 |
|
Lease Agreement between The Exchange at Meadowmont LLC and the Registrant dated as of November 21, 2001 (filed as Exhibit 10.21 to Pozen’s Annual Report on Form 10-K filed April 1, 2002). |
|
|
|
|
|
10.4 |
|
Product Development and Commercialization Agreement dated June 11, 2003 between the Registrant and Glaxo Group Ltd. (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q filed August 12, 2003 and Form 10-Q/A filed November 8. 2004).† |
|
|
|
|
|
10.5 |
|
License Agreement dated June 11, 2003 between the Registrant and Glaxo Group Ltd. (filed as Exhibit 10.2 to Pozen’s Quarterly Report on Form 10-Q filed August 12, 2003 and Quarterly Report on Form 10-Q/A filed November 8. 2004).† |
|
|
|
|
|
10.6 |
|
Collaboration and License Agreement dated September 3, 2003 between the Registrant and Valeant Pharmaceuticals NA (formerly Xcel Pharmaceuticals, Inc.) (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q filed November 6, 2003 and Quarterly Report on Form 10-Q/A filed November 8. 2004).† |
|
|
|
|
|
10.7 |
|
Restricted Stock Unit Agreement dated May 4, 2004 between Registrant and John R. Plachetka (filed as Exhibit 10.4 to Pozen’s Quarterly Report on Form 10-Q filed July 30, 2004).+ |
|
|
|
|
|
10.8 |
|
First Amendment, dated September 28, 2007, to Restricted Stock Unit Agreement, dated May 4, 2004, between Registrant and John R. Plachetka (filed as Exhibit 10.2 to Pozen’s Quarterly Report on Form 10-Q filed November 5, 2007).+ |
|
|
|
|
|
10.9 |
|
Collaboration and License Agreement dated August 1, 2006 between the Registrant and AstraZeneca AB (filed as Exhibit 10.1 to Pozen’s Quarterly Report on From 10-Q filed November 3, 2006).† |
|
|
|
|
|
10.10 |
|
Amendment No. 1 to the Collaboration and License Agreement, dated September 6, 2007, between the Registrant and AstraZeneca AB (filed as Exhibit 10.8 to Pozen’s Quarterly Report on Form 10-Q filed November 5, 2007). † |
|
10.11 |
|
Side Letter dated September 19, 2006 Re: Collaboration and License Agreement dated as of August 1, 2006 by and between the Registrant and AstraZeneca AB (filed as 10.2 to Pozen’s Quarterly Report on From 10-Q filed November 3, 2006).† |
|
|
|
|
|
10.12 |
|
Side Letter Agreement, dated October 1, 2007, between the Registrant and AstraZeneca, AB (filed as Exhibit 10.9 to Pozen’s Quarterly Report on Form 10-Q filed November 5, 2007). † |
|
|
|
|
|
10.13 |
|
Long-Term Cash Incentive Award Agreement between the Registrant and John R. Plachetka dated February 14, 2007 (filed as Exhibit 10.4 to Pozen’s Quarterly Report on Form 10-Q filed May 3, 2007).+ |
|
|
|
|
|
10.14 |
|
Amendment No. 2 to the Collaboration and License Agreement, dated October 1, 2008, between the registrant and AstraZeneca, AB (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q filed November 4, 2008). † |
|
|
|
|
|
10.15 |
|
Lease Modification Agreement No. 1, dated as of February 16, 2009, by and between the Registrant and The Exchange at Meadowmont LLC (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K filed on February 17, 2009). |
|
|
|
|
|
10.16 |
|
Executive Employment Agreement, dated as of December 10, 2009, between the Company and John G. Fort, M.D. (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K, filed on December 11, 2009).+ |
|
|
|
|
|
10.17 |
|
Purchase and Sale Agreement, dates as November 23, 2011, by and between POZEN Inc. and CPPIB Credit Investments Inc. (filed as Exhibit 10.37 to the Registrants Annual Report on Form 10-K filed March 9. 2012). |
|
|
|
|
|
10.18 |
|
Manufacturing Services Agreement, dates as December 19, 2011, by and between POZEN Inc. and Patheon Pharmaceuticals, Inc.†(filed as Exhibit 10.38 to the Registrants Amendment No.1 to the Annual Report on Form 10-K, filed June 29,2012). |
|
|
|
|
|
10.19 |
|
Capital Expenditure and Equipment Agreement, dates as of December 19, 2011, by and between POZEN Inc. and Patheon Pharmaceuticals, Inc. (filed is Exhibit 10.39 to the Registrants Amendment No.11 to Annual Report on Form 10-K, filed June 29,2012). |
|
|
|
|
|
10.20 |
|
First Amendment to Manufacturing Services Agreement, between Patheon Pharmaceuticals Inc., and POZEN Inc., dated as of July 10, 2013 (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q, filed August 7, 2013).† |
|
|
|
|
|
10.21 |
|
First Amendment to Capital Expenditure and Equipment Agreement, between Patheon Pharmaceuticals Inc., and POZEN Inc., dated as of July 10, 2013 (filed as Exhibit 10.2 to Pozen’s Quarterly Report on Form 10-Q, filed August 7, 2013).† |
|
|
|
|
|
10.22 |
|
License and Development Agreement, dated as of May 7, 2012, by and between POZEN Inc. and DESITIN Arzneimittel GmbH (filed as Exhibit 10.1 to Registrants Quarterly Report on Form 10-Q, filed on August 8, 2012). |
|
|
|
|
|
10.23 |
|
Amendment No. 3 to the Collaboration and License Agreement between POZEN Inc. and AstraZeneca AB, dated as of September 16, 2013 (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q, filed August 7, 2013).† |
|
|
|
|
|
10.24 |
|
Letter Agreement among POZEN Inc., AstraZeneca AB and Horizon Pharma U.S.A. Inc. dated as of November 18, 2013 (filed as Exhibit 10.43 to Pozen’s Annual Report on Form 10-K, filed March 6, 2014). † |
|
|
|
|
|
10.25 |
|
Amendment No. 1 to Amended and Restated Collaboration and License Agreement for the United States by and between POZEN Inc. and Horizon Pharma U.S.A. Inc. dated as of November 18, 2013 (filed as Exhibit 10.44 to Pozen’s Annual Report on Form 10-K, filed March 6, 2014). † |
|
|
|
|
|
10.26 |
|
Amended and Restated Collaboration and License Agreement for the United States by and between POZEN Inc. and AstraZeneca AB dated as of November 18, 2013 (filed as Exhibit 10.45 to Pozen’s Annual Report on Form 10-K, filed March 6, 2014). † |
|
|
|
|
|
10.27 |
|
Amended and Restated Collaboration and License Agreement for outside of the United States by and between POZEN Inc. and AstraZeneca AB dated as of November 18, 2013 (filed as Exhibit 10.46 to Pozen’s Annual Report on Form 10-K, filed March 6, 2014). † |
|
|
|
|
|
10.28 |
|
Letter Agreement, dated as of May 13, 2014, by and among POZEN Inc., CPPIB Credit Investments, Inc. Pernix |
|
|
|
Therapeutics Holdings, Inc. and Glaxo Group Limited (filed as Exhibit 10.1 to Pozen’s Quarterly Report on Form 10-Q, filed November 6, 2014). |
|
|
|
|
|
10.29 |
|
First Amendment to Product Development and Commercialization Agreement, dated as of May 13, 2014, by and between POZEN Inc. and Pernix Therapeutics Holdings, Inc. (filed as Exhibit 10.2 to Pozen’s Quarterly Report on Form 10-Q, filed November 6, 2014). |
|
|
|
|
|
10.30 |
|
Second Amendment to Product Development and Commercialization Agreement dated as of July 30, 2014, by and between POZEN Inc. and Pernix Therapeutics Holdings, Inc. (filed as Exhibit 10.3 to Pozen’s Quarterly Report on Form 10-Q, filed November 6, 2014). |
|
|
|
|
|
10.31 |
|
Facility Agreement, dated as of June 8, 2015, among the Borrower, Pozen, Tribute and the Lenders (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K filed June 11, 2015). |
|
|
|
|
|
10.32 |
|
Registration Rights Agreement, dated as of June 8, 2015, among Parent and the Lenders (filed as Exhibit 10.2 to Pozen’s Current Report on Form 8-K filed June 11, 2015). |
|
|
|
|
|
10.33 |
|
Share Subscription Agreement, dated as of June 8, 2015, among Pozen, Purchaser, Tribute, Parent and the Investors (filed as Exhibit 10.3 to Pozen’s Current Report on Form 8-K filed June 11, 2015). |
|
|
|
|
|
10.34 |
|
Executive Employment Agreement between Pozen and Adrian Adams dated May 31, 2015 (filed as Exhibit 10.3 to Pozen’s Current Report on Form 8-K filed June 3, 2015).+ |
|
|
|
|
|
10.35 |
|
Executive Employment Agreement between Pozen and Andrew I. Koven dated May 31, 2015(filed as Exhibit 10.4 to Pozen’s Current Report on Form 8-K filed June 3, 2015). + |
|
|
|
|
|
10.36 |
|
Executive Employment Agreement between Pozen and Mark Glickman dated June 22, 2015.* + |
|
|
|
|
|
10.37 |
|
Executive Employment Agreement between Pozen and Eric Trachtenberg dated June 22, 2015.* + |
|
|
|
|
|
10.38 |
|
Executive Employment Agreement between Pozen and Jennifer Armstrong dated June 22, 2015.* + |
|
|
|
|
|
10.39 |
|
Executive Employment Agreement between Pozen and Scott Charles dated July 27, 2015.* + |
|
|
|
|
|
10.40 |
|
Separation and General Release Agreement between Pozen and John R. Plachetka, dated May 29, 2015 (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K filed June 3, 2015). + |
|
|
|
|
|
10.41 |
|
Voting Agreement between Pozen and John R. Plachetka, dated May 29, 2015 (filed as Exhibit 10.2 to Pozen’s Current Report on Form 8-K filed June 3, 2015). + |
|
|
|
|
|
10.42 |
|
POZEN Inc. Employee Severance Plan and Summary Plan Description (filed as Exhibit 10.1 to Pozen’s Current Report on Form 8-K filed June 25, 2015). + |
|
|
|
|
|
10.43 |
|
Form of Pozen Retention Agreement (filed as Exhibit 10.2 to Pozen’s Current Report on Form 8-K filed June 25, 2015). + |
|
|
|
|
|
10.44 |
|
Aralez Pharmaceuticals plc 2015 Long-Term Incentive Plan.* |
|
|
|
|
|
10.45 |
|
Amended and Restated Stock Option Plan effective June 22, 2011. +* |
|
|
|
|
|
10.45 |
|
Executive Employment Agreement, effective January 1, 2015, by and between Tribute and Rob Harris. +* |
|
|
|
|
|
10.47 |
|
Executive Employment Agreement, effective January 1, 2015, by and between Tribute and Scott Langille. +* |
|
|
|
|
|
10.48 |
|
Amended and Restated Executive Employment Agreement, effective January 1, 2015, by and between Tribute and Janice Clarke. +* |
|
|
|
|
|
10.49 |
|
Consulting Agreement, effective January 1, 2015, by and between Tribute and LMT Financial Inc. +* |
|
|
|
|
|
10.50 |
|
Form of Securities Purchase Agreement, dated February 27, 2013.* |
|
|
|
|
|
10.51 |
|
Form of Registration Rights Agreement, dated February 27, 2013.* |
|
|
|
|
|
10.52 |
|
Form of Securities Purchase Agreement, dated March 5, 2013.* |
|
|
|
|
|
10.53 |
|
Form of Registration Rights Agreement, dated March 5, 2013.* |
|
|
|
|
|
10.54 |
|
Form of Securities Purchase Agreement, dated March 11, 2013.* |
|
|
|
|
|
10.55 |
|
Form of Registration Rights Agreement, dated March 11, 2013.* |
|
|
|
|
|
10.56 |
|
Credit Agreement, dated August 8, 2013, by and between Tribute and SWK Funding LLC. †* |
|
|
|
|
|
10.57 |
|
First Amendment to Credit Agreement and Guarantee, dated October 1, 2014, by and between Tribute and SWK Funding LLC. †* |
|
|
|
|
|
10.58 |
|
Guarantee and Collateral Agreement, dated August 8, 2013 by and between Tribute and SWK Funding LLC.* |
|
|
|
|
|
10.59 |
|
Intellectual Property Security Agreement, dated August 8, 2013, by and between Tribute and SWK Funding LLC.* |
|
|
|
|
|
10.60 |
|
Form of Tribute Support Agreement.* |
|
|
|
|
|
21.1 |
|
Subsidiaries of Registrant. |
|
|
|
|
|
23.1 |
|
Consent of Ernst & Young LLP, independent registered public accounting firm for Pozen. |
|
|
|
|
|
23.2 |
|
Consent of McGovern, Hurley, Cunningham, LLP, independent auditors for Tribute. |
|
|
|
|
|
23.3 |
|
Consent of A&L Goodbody (included in Exhibit 5.01).* |
|
|
|
|
|
24.1 |
|
Power of Attorney (see signature page hereto).* |
* To be filed by amendment.
** Filed herewith.
+ Compensation Related Contract.
† Confidential treatment requested. Confidential materials omitted and filed separately with Securities and Exchange Commission.