Attached files
| file | filename |
|---|---|
| 8-K - 8-K - ARDELYX, INC. | d30457d8k.htm |
| EX-99.2 - EX-99.2 - ARDELYX, INC. | d30457dex992.htm |
Exhibit 99.1
Ardelyx, Inc.
Overview
We are a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of innovative, minimally-systemic therapeutic drugs that work exclusively in the gastrointestinal, or GI, tract to treat cardio-renal, GI and metabolic diseases. We have developed a proprietary drug discovery and design platform enabling us, in a rapid and cost-efficient manner, to discover and design novel drug candidates. Utilizing our platform, we discovered and designed our lead product candidate, tenapanor, which in a Phase 2b clinical study has demonstrated the ability to improve the symptoms of constipation-predominant irritable bowel syndrome, or IBS-C. We expect to initiate a Phase 3 clinical program to evaluate tenapanor in the treatment of IBS-C in the fourth quarter 2015. In a separate Phase 2b clinical trial, tenapanor demonstrated the ability to treat hyperphosphatemia, or elevated serum phosphorus, in patients on dialysis with end-stage renal disease, or ESRD. We expect to initiate a Phase 2b clinical trial to evaluate dosing regimens of tenapanor for the treatment of hyperphosphatemia in these patients in the fourth quarter 2015. We are developing another drug candidate, RDX022, for the treatment of hyperkalemia, or elevated serum potassium, in patients with chronic kidney disease, or CKD, and in patients with heart failure, or HF. In the fourth quarter 2015, we expect to begin a Phase 1 clinical trial in healthy adults evaluating the safety and pharmacodynamic, or biological activity, of RDX022. We intend to pursue a 505(b)(2) regulatory pathway for RDX022, and we expect to advance RDX022 into a Phase 3 clinical program as early as the second half of 2016. We have several other drug candidates in earlier stages of research and development focused in cardio-renal, GI and metabolic diseases including RDX002, which we have licensed to Sanofi S.A., or Sanofi, for the treatment of hyperphosphatemia, RDX009, a secretagogue of glucagon-like peptide-1, or GLP-1, and glucagon-like peptide-2, or GLP-2, and RDX013, a potassium secretagogue.
Our Strategy
We are a biotechnology company with a clinical pipeline of drug candidates in the fields of cardio-renal, GI and metabolic diseases. We have two product candidates, tenapanor and RDX022, which we expect to advance into late-stage clinical development by the second half of 2016. We intend to use our cash, including the proceeds from our recent private placement and from our at-the-market offering pursuant to a Registration Statement on Form S-3, to develop these two drugs in Phase 3 clinical programs.
We also expect to use our proprietary drug discovery and design platform to discover new minimally-systemic drug candidates that prevent and treat important diseases in these same therapeutic areas of cardio-renal, GI and metabolic diseases. To date, all of our drug candidates have resulted from research completed by the Company.
If our drug candidates are approved for marketing by the FDA, we expect to participate in the commercialization of our products with our own specialty-based sales force, initially targeting gastroenterologists and nephrologists. Our executive management team, and in particular our President and Chief Executive Officer, Michael Raab, has extensive experience in developing and commercializing therapeutic drugs for the CKD and ESRD markets, and we expect to leverage this expertise in a manner that will allow us to create and retain the most value from our marketed products.
Our Pipeline
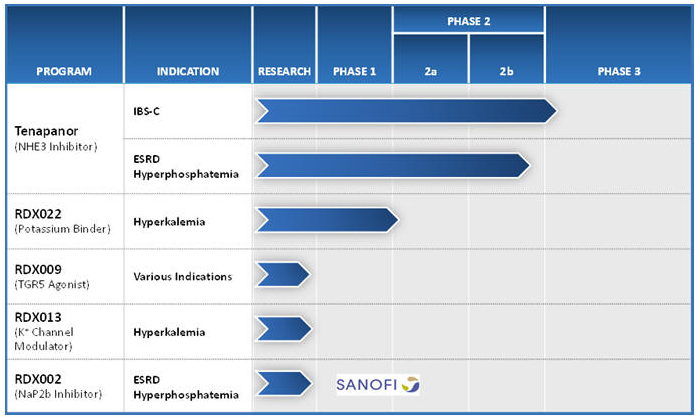
Tenapanor is a minimally-absorbed, small molecule that acts locally in the GI tract to inhibit the sodium transporter NHE3 and reduce sodium and phosphorus uptake from the gut. We announced in the second quarter of 2015 that we regained the worldwide development and commercialization rights for tenapanor and a related portfolio of our NHE3 compounds from AstraZeneca AB, or AstraZeneca. In human studies of orally-administered tenapanor, the drug was detected in the blood in only 0.7% of more than 3,000 collected serum samples, and even in those, at very low levels (< 1.5 ng/mL). We have evaluated tenapanor in fourteen human clinical studies in over 1,000 individuals to date.
In October 2014, we announced results from a Phase 2b clinical trial evaluating tenapanor in IBS-C patients. At the twice daily 50 mg dose of tenapanor, the study met its primary efficacy endpoint of an increase in the complete spontaneous bowel movement, or CSBM, responder rate. Most secondary endpoints, including abdominal pain, the overall responder rate and other abdominal and IBS-C symptoms, demonstrated statistically significant and clinically meaningful improvements. We plan to initiate a Phase 3 clinical program in IBS-C patients in the fourth quarter of 2015.
In February 2015, we announced results from a Phase 2b clinical trial evaluating tenapanor for the treatment of hyperphosphatemia in CKD patients on dialysis. In the study, there was a statistically significant dose-related decrease in serum phosphate levels for tenapanor-treated patients compared to patients receiving placebo (p=0.012). It was noted, however, that the rate of diarrhea and the rate of discontinuations due to diarrhea were higher than expected in the 30mg once daily and 30mg twice daily dose groups. We plan to initiate a Phase 2b clinical trial in the fourth quarter of 2015 to evaluate the optimal dosing regimen for tenapanor for the treatment of hyperphosphatemia in dialysis patients.
We are also developing RDX022 for the treatment of hyperkalemia in CKD and heart failure patients. RDX022 is a polymer being developed to treat hyperkalemia. RDX022 is designed with improved chemical and physical properties as well as formulation improvements that may allow for a more palatable dosage form than other treatments for hyperkalemia. We will be pursuing a 505b(2) regulatory pathway in the United States for RDX022, allowing for a faster path to approval by referencing the literature and the U.S. Food and Drug Administration’s previous findings of safety and effectiveness of the referenced drug product. We expect to supplement this approach with nonclinical and Phase 3 clinical data on RDX022 to provide information for inclusion in the product label. We have commenced and plan to conduct early stage clinical trials with RDX022 during 2015 including a Phase 1 clinical trial in healthy adults evaluating the safety and pharmacodynamic activity of RDX022. Assuming the successful completion of those trials, we expect to initiate a Phase 3 clinical program to evaluate RDX022 for treatment in hyperkalemia patients as early as the second half of 2016.
Other product candidates in our pipeline include the following:
| • | RDX013 Program: We are continuing to research RDX013, a small molecule drug candidate for hyperkalemia. This agent, a potential potassium secretagogue, is intended to enhance potassium secretion or prevent potassium absorption with a much lower pill burden than potassium binders and may provide significant advantages as a stand-alone agent or used in combination with the potassium binders, including RDX022. |
| • | RDX009 Program: Our focus is the discovery and development of minimally-absorbed TGR5 agonists that stimulate GLP-1 and GLP-2 secretion for various indications. |
| • | NaP2b Program: We have discovered novel NaP2b inhibitors for the treatment of hyperphosphatemia in CKD patients on dialysis by inhibiting the active absorption of phosphorus. In February 2014, we entered into an option and license agreement with Sanofi under which we granted Sanofi an exclusive worldwide license to conduct research utilizing our small molecule NaP2b inhibitors. Sanofi has the option to obtain an exclusive license to develop, manufacture and commercialize our NaP2b inhibitors. Under our arrangement, Sanofi is responsible for all of the costs and expenses for research and preclinical activities and, should it exercise its option, for the development and commercialization efforts under the program, while we retain an option to co-promote licensed products in the United States. |
Our Proprietary Drug Discovery and Design Platform
Our platform, comprised of proprietary know-how and drug discovery and design tools provides us with a competitive advantage in drug development. This platform enables us, in a rapid and cost-efficient manner, to discover and design novel drug candidates that work exclusively in the GI tract to treat cardio-renal, GI and metabolic diseases. By targeting receptors and transporters localized in the GI tract, we can modulate important functions of the gut, such as absorption of specific nutrients and minerals, or the gut’s various hormonal functions, to treat and prevent diseases while avoiding systemic toxicities.
The pillars of our drug discovery and design platform include the following:
| • | Medicinal Chemistry to develop minimally-systemic products: our medicinal chemistry team has developed the tools, expertise and approach to ensure that our small molecule drug candidates are potent, have appropriate drug properties, are able to be readily manufactured, and are minimally-systemic, a property that we believe is not generally targeted for most other drug programs; |
| • | In vivo pharmacology: Our in vivo pharmacology group has developed animal models and expertise in-house that allow it to rapidly assess the minimally-systemic nature of our drug candidates as well as test the hypotheses that our drug candidates can treat and prevent diseases and conditions in our targeted therapeutic fields; |
| • | GI Informatics: With genic and proteomic informatics tools, we have identified over 3,800 human GI tract-specific RNA transcripts and proteins on the inner surface of the gut, many of which we believe may be drug targets. We intend to become the experts in human and rodent gut physiology and the molecular pathways that lead to disease in our targeted therapeutic areas; |
| • | In vitro expertise: We have developed a cell-culture system that simulates gut tissue. We call this component of our discovery platform Ardelyx Primary Enterocyte and Colonocyte Culture System, or APECCS. APECCS involves the biopsy of various segments of the gut and the growth of those cells under proprietary conditions to maintain, to the extent possible, the integrity and functionality of the various cell types and substructures. We have developed this into a miniaturized format that allows us to utilize it for cell based drug screening. In addition to using APPECS in the design of our small molecule drug candidates, we use the APECCS technology to measure epithelial transport of ions and nutrients and to screen compounds to identify potential disease modulators such as inhibitors or activators using phenotypic screening. APECCS has the potential to allow us to identify novel targets, mechanisms of action and physiology as well as provide us an early understanding of how identified compounds may interact with specific gut tissues. In addition, we believe that APECCS may also provide us with a clear path to translate cell-based observations into in vivo rodent models and ultimately into human clinical studies. |
Our Markets
Constipation-Predominant Irritable Bowel Syndrome, or IBS-C
IBS-C is a GI disorder in which abdominal pain or discomfort is associated with constipation, which significantly affects the health and quality of life of affected patients. It is unknown what causes IBS-C. There is no specific test or biomarker for IBS-C and therefore its presence is diagnosed by symptoms and by eliminating other disorders. IBS-C is very similar to chronic constipation and it is clinically distinguished by a significant pain component.
Numerous treatments exist for the constipation component of IBS-C, many of which are over-the-counter. We are aware of two prescription products marketed for IBS-C, Linzess (linaclotide) marketed by Ironwood Pharmaceuticals and Actavis and Amitiza (lubiprostone) marketed by Sucampo and Takeda. In Phase 3 clinical trials of Linzess in IBS-C patients, up to 20% more patients receiving Linzess than placebo reached the primary endpoint, overall responder rate, indicating a significant response during 6 out of 12 weeks of treatment. In these studies, Linzess caused diarrhea in up to 17% more patients than placebo.
We believe that tenapanor may offer a significant benefit over currently marketed drugs like Amitiza and Linzess, due in part to the potential to adjust the dose and/or dose frequency of tenapanor in order to optimize its efficacy and minimize diarrhea. The data we have generated in both animal and human studies have suggested that the effect of tenapanor for the treatment of IBS-C can be modulated by adjusting its dose and dose frequency.
Based on reports in the literature regarding the prevalence of IBS in the U.S. population and the percentage of individuals who have IBS-C as opposed to other forms of IBS, we estimate that approximately 1.4% of the U.S. population has IBS-C, or about 4.4 million individuals, and that approximately 1.0 million of those patients have been diagnosed with IBS-C. Additionally, we estimate there are about 6.6 million IBS-C patients in Europe and about 3.4 million in Japan. The per-patient economic burden of IBS-C is estimated to be $1,500 to $7,500 per year in direct costs and $800 to $7,700 per year in indirect costs, implying the total burden in the United States is $2 billion to $15 billion.
Hyperphosphatemia
CKD is the progressive deterioration of renal function that can occur over several months or years. The symptoms of worsening kidney function are nonspecific, and can include having less energy, reduced appetite, dry itchy skin, swollen feet and ankles, or generally just not feeling well. If the deterioration continues and is not halted by either changes in lifestyle or with the assistance of pharmacological intervention, the disease will likely cause significant cardiovascular morbidity, and can progress to ESRD, the final stage of CKD, where kidney function will be lost entirely.
Current management of ESRD includes hemodialysis, and peritoneal dialysis as a means to filter toxins from the blood once kidneys have failed. Unless this intervention occurs, kidney failure results in the accumulation of waste products that may ultimately cause death. Hemodialysis, the most common form of dialysis, generally requires a patient to visit a dialysis center at least three times per week for a three- to five-hour session, significantly reducing quality of life.
Phosphorus, a vital element required for most cellular processes, is present in almost every food in the Western diet, and, in individuals with normal kidney function, any excess dietary phosphorus is efficiently removed by the kidney and excreted in urine. In adults with functioning kidneys, normal serum phosphorus levels are 2.6 to 3.8 mg/dL. With kidney failure, elevated phosphorus becomes a toxin and is diagnosed as hyperphosphatemia when serum phosphorus levels are greater than 5.0 mg/dL. Although patients with ESRD rely on dialysis to eliminate toxins, phosphorus is not readily removed by the procedure and other means of managing phosphorus levels must be employed.
In ESRD, excess levels of phosphorus have been shown to lead to an increase in cardiovascular disease risk, as well as increases in serum FGF-23, an important serum endocrine hormone that regulates phosphorus metabolism, and elevated parathyroid hormone, also known as secondary hyperparathyroidism. These endocrine changes in ESRD patients are a concern as elevated parathyroid hormone leads to the development of renal osteodystrophy, a condition of abnormal bone growth characterized by brittle bones.
Since dialysis is unable to efficiently eliminate excess phosphorus, ESRD patients are put on restrictive low phosphorus diets and are prescribed medications called phosphate binders, the only pharmacologic interventions currently marketed for the treatment of hyperphosphatemia. Binders are a collection of drugs whose function is to bind, or absorb, dietary phosphorus and are taken in conjunction with meals and snacks. They include calcium, iron or lanthanum, a rare-earth metal, which bind to and precipitate with dietary phosphate in the GI tract. The goal is for patients to eliminate the precipitated phosphorus in their stool. A limitation of this approach is the systemic excess absorption of calcium, iron or lanthanum, resulting in side effects and other unintended consequence for ESRD patients. In an effort to eliminate these unwanted side effects, non-absorbed exchange resins, such as sevelamer, were developed to bind to phosphate in the GI tract and to be eliminated in stool.
Safety and tolerability have been significant concerns with many approved phosphate binders. The more common side effects of approved phosphate binders include long-term vascular calcification, nausea and vomiting, diarrhea or constipation and ileus or disruption of the normal propulsive ability of the GI tract.
ESRD patients take on average 10-14 oral medications each day, and they are severely restricted in their fluid intake. In addition, to control their serum phosphorus, their phosphate binder-related pill burden is significant, typically consisting of nine or more pills a day. The amount of phosphate a binder can remove is limited by its binding capacity, and therefore, increasing the dose, and the pill burden, of the binder is the only way to increase the amount of phosphate being bound and excreted. As a result, prescribed binder doses are intolerable for many patients.
According to the most recent data available from the U.S. Renal Data System, in 2012 there were 408,711 patients on hemodialysis in the United States. Additionally, according to the European ERA-EDTA Registry 2012 Annual Report and a study in 2010 by the Japanese Society for Dialysis Therapy, there were approximately 280,000 patients on hemodialysis in Europe and about 250,000 in Japan. We estimate, based on phosphate binder utilization, the only approved therapies for hyperphosphatemia, that there are approximately 280,000, 225,000 and 220,000 ESRD patients with hyperphosphatemia in the United States, Europe and Japan, respectively.
Because many ERSD patients with hyperphosphatemia are unable to lower serum phosphorus levels to below 5.5 mg/dL with currently marketed phosphate binders, we believe there is a significant opportunity for new agents with new mechanisms, demonstrated efficacy, a strong safety profile, and significantly lower pill burden.
We believe that tenapanor, if approved, has the potential to have the lowest pill burden among any currently marketed hyperphosphatemia drugs, with milligram rather than gram quantities dosed once or twice daily. In addition, we may evaluate whether tenapanor has the potential to be used in combination with phosphate binders for those patients who cannot achieve adequate phosphate control with a single agent.
Hyperkalemia
Hyperkalemia is defined as the presence of blood potassium levels greater than 5.0mEq/L. Normal levels are 3.5 to 5.0 mEq/L. When hyperkalemia is severe, or above 7.0mEq/L, there is a significantly increased risk of death because of the potential for heart conductance problems.
Hyperkalemia can be caused by a variety of sources. Kidney disease can result in the build-up of potassium in the blood. Also, certain drugs such as the common blood pressure medications known as RAAS inhibitors, or inhibitors of the renin-angiotensin-aldosterone system, can cause hyperkalemia. RAAS inhibitors, though quite effective for controlling blood pressure, are often significantly reduced in patients, such as in those with CKD and congestive heart failure, or CHF, whose potassium levels are elevated because of the fear that elevated potassium can cause significantly worse problems than hypertension including sudden cardiac arrest in severe cases.
According to a retrospective observational study of a national cohort of 246,000 veterans cared for in the Veterans Health Administration (Einhorn et al, 2009), about 21% and 42% of patients with CKD Stage 3b and Stage 4, respectively, had a hyperkalemic event during a 12-month period suggesting that hyperkalemia affects about 900,000 individuals with CKD Stage 3b or Stage 4 in the United States. According to the United States Renal Data System 2014 Atlas of CKD & ESRD, over 50% of CKD Stage 3b and Stage 4 patients are prescribed RAAS inhibitors because of their efficacy in controlling hypertension and success in slowing the clinical course of CKD. Additionally, according to the American Heart Association, 5.7 million Americans are living today with heart failure. Our proprietary research suggests that up to 16%, or approximately 900,000, of these patients had hyperkalemia during a 12-month period. Over half of heart failure patients are prescribed RAAS inhibitors.
Despite the success of RAAS inhibitors in both of these populations, several published guidelines have suggested that physicians should reduce and possibly discontinue RAAS inhibitors in order to manage the risk of hyperkalemia in CKD and heart failure patients. The alternative medications used to control hypertension, including diuretics and calcium channel blockers, are significantly less effective than RAAS inhibitors, particularly in patients with failing kidneys and severe hypertension. According to Market Dynamix: Hyperkalemia recently released by Spherix Global Insights, cardiologists reported that of the patients who would benefit from RAAS inhibition, up to 38% of patients with heart failure and up to 55% of patients with both heart failure and CKD are being administered a sub-optimal dose or none at all, and nephrologists reported that at least one-third of patients who would benefit from RAAS inhibition receive a sub-optimal dose or none at all. We believe there is a strong medical need for new medications that control hyperkalemia in order to allow for continued use of RAAS inhibitors to control hypertension in these patient populations.
An additional market not currently addressed by any product on the market is hyperkalemia in ESRD patients on dialysis. Our proprietary research suggests that up to 48% of patients on dialysis have at least one intervention for hyperkalemia during a 12-month period despite being dialyzed, resulting in a 7% mortality rate. This suggests up to 200,000 patients with ESRD on dialysis that could benefit from an agent that treats hyperkalemia.
Corporate Information
We were founded in October 2007 as a Delaware corporation under the name Nteryx, Inc. Our principal executive offices are located at 34175 Ardenwood Blvd., Suite 200, Fremont, CA 94555, and our telephone number is (510) 745-1700. Our website address is www.ardelyx.com. The information on, or that can be accessed through, our website is not part of this prospectus. We have included our website address as an inactive textual reference only.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year following the fifth anniversary of the completion of our initial public offering of common stock (December 31, 2019), (2) the last day of the fiscal year in which we have total annual gross revenue of at least $1.0 billion, (3) the last day of the fiscal year in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million as of the prior June 30th, and (4) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.