Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - SELLAS Life Sciences Group, Inc. | Financial_Report.xls |
| EX-31.1 - EXHIBIT 31.1 - SELLAS Life Sciences Group, Inc. | gale-20150331xex311.htm |
| EX-32.1 - EXHIBIT 32.1 - SELLAS Life Sciences Group, Inc. | gale-20150331xex321.htm |
| EX-31.2 - EXHIBIT 31.2 - SELLAS Life Sciences Group, Inc. | gale-20150331xex312.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
________________________________
FORM 10-Q
________________________________
(Mark One)
ý | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended March 31, 2015
OR
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 001-33958
________________________________
Galena Biopharma, Inc.
(Exact name of registrant as specified in its charter)
________________________________
Delaware | 20-8099512 | |
(State of incorporation) | (I.R.S. Employer Identification No.) | |
4640 SW Macadam Ave., Suite 270, Portland, OR 97239
(Address of principal executive office) (Zip code)
Registrant’s telephone number: (855) 855-4253
________________________________
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter time that the registrant was required to submit and post such files). Yes ý No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act (Check one):
Large accelerated filer | o | Accelerated filer | ý | |||
Non-accelerated filer | o | (Do not check if a smaller reporting company) | Smaller reporting company | o | ||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act): o Yes ý No
As of April 30, 2015, Galena Biopharma, Inc. had outstanding 161,715,398 shares of common stock, $0.0001 par value per share, exclusive of treasury shares.
GALENA BIOPHARMA, INC.
FORM 10-Q — QUARTER ENDED MARCH 31, 2015
INDEX
Part No. | Item No. | Description | Page No. | ||
I | |||||
1 | |||||
Condensed Consolidated Balance Sheets as of March 31, 2015 (unaudited) and December 31, 2014 | |||||
Condensed Consolidated Statements of Comprehensive Loss for the three months ended March 31, 2015 and 2014 | |||||
Condensed Consolidated Statement of Stockholders' Equity for the three months ended March 31, 2015 | |||||
Condensed Consolidated Statements of Cash Flows for the three months ended March 31, 2015 and 2014 | |||||
2 | |||||
3 | |||||
4 | |||||
II | |||||
1 | Legal Proceedings | ||||
1A | Risk Factors | ||||
6 | |||||
EX-31.1 | |||||
EX-31.2 | |||||
EX-32.1 | |||||
1
PART I
ITEM 1. FINANCIAL STATEMENTS
GALENA BIOPHARMA, INC.
CONDENSED CONSOLIDATED BALANCE SHEETS
(Amounts in thousands, except share and per share data)
March 31, 2015 | December 31, 2014 | ||||||
(Unaudited) | |||||||
ASSETS | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 52,860 | $ | 23,650 | |||
Restricted cash | 200 | 200 | |||||
Accounts receivable, net | 1,060 | 1,839 | |||||
Inventories | 637 | 655 | |||||
Prepaid expenses | 2,564 | 2,680 | |||||
Total current assets | 57,321 | 29,024 | |||||
Equipment and furnishings, net | 528 | 555 | |||||
In-process research and development | 12,864 | 12,864 | |||||
Abstral rights, net | 14,387 | 14,533 | |||||
Zuplenz rights | 8,101 | 8,101 | |||||
GALE-401 rights | 9,255 | 9,255 | |||||
Goodwill | 6,069 | 6,069 | |||||
Deposits and other assets | 82 | 87 | |||||
Total assets | $ | 108,607 | $ | 80,488 | |||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 3,293 | $ | 2,271 | |||
Accrued expenses and other current liabilities | 12,149 | 15,669 | |||||
Fair value of warrants potentially settleable in cash | 14,528 | 5,383 | |||||
Current portion of long-term debt | 3,994 | 3,910 | |||||
Total current liabilities | 33,964 | 27,233 | |||||
Deferred tax liability | 5,053 | 5,053 | |||||
Contingent purchase price consideration | 6,972 | 6,651 | |||||
Long-term debt, net of current portion | 3,534 | 4,492 | |||||
Total liabilities | 49,523 | 43,429 | |||||
Commitments and contingencies | |||||||
Stockholders’ equity: | |||||||
Preferred stock, $0.0001 par value; 5,000,000 shares authorized; no shares issued and outstanding | — | — | |||||
Common stock, $0.0001 par value; 200,000,000 shares authorized, 158,736,552 shares issued and 158,061,552 shares outstanding at March 31, 2015; 130,146,341 shares issued and 129,471,341 shares outstanding at December 31, 2014 | 15 | 12 | |||||
Additional paid-in capital | 288,936 | 256,377 | |||||
Accumulated deficit | (226,018 | ) | (215,481 | ) | |||
Less treasury shares at cost, 675,000 shares | (3,849 | ) | (3,849 | ) | |||
Total stockholders’ equity | 59,084 | 37,059 | |||||
Total liabilities and stockholders’ equity | $ | 108,607 | $ | 80,488 | |||
See accompanying notes to condensed consolidated financial statements.
2
GALENA BIOPHARMA, INC.
CONDENSED CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(Amounts in thousands, except share and per share data)
(Unaudited)
Three Months Ended March 31, | |||||||
2015 | 2014 | ||||||
Net revenue | $ | 2,750 | $ | 2,173 | |||
Costs and expenses: | |||||||
Cost of revenue (excluding amortization of certain acquired intangible assets) | 393 | 331 | |||||
Research and development | 5,910 | 6,770 | |||||
Selling, general, and administrative | 7,427 | 6,830 | |||||
Amortization of certain acquired intangible assets | 146 | 91 | |||||
Total costs and expenses | 13,876 | 14,022 | |||||
Operating loss | (11,126 | ) | (11,849 | ) | |||
Non-operating income (expense): | |||||||
Change in fair value of warrants potentially settleable in cash | 1,152 | 9,792 | |||||
Interest income (expense), net | (242 | ) | (314 | ) | |||
Other income (expense) | (321 | ) | (165 | ) | |||
Total non-operating income (expense), net | 589 | 9,313 | |||||
Net loss | $ | (10,537 | ) | $ | (2,536 | ) | |
Net loss per common share: | |||||||
Basic and diluted net loss per share | $ | (0.08 | ) | $ | (0.02 | ) | |
Weighted-average common shares outstanding: basic and diluted | 136,054,864 | 116,244,209 | |||||
See accompanying notes to condensed consolidated financial statements.
3
GALENA BIOPHARMA, INC.
CONDENSED CONSOLIDATED STATEMENT OF STOCKHOLDERS' EQUITY
(Amounts in thousands, except share amounts)
(Unaudited)
Common Stock | Additional Paid-In Capital | Accumulated Deficit | Treasury Stock | Total | ||||||||||||||||||
Shares Issued | Amount | |||||||||||||||||||||
Balance at December 31, 2014 | 130,146,341 | $ | 12 | $ | 256,377 | $ | (215,481 | ) | $ | (3,849 | ) | $ | 37,059 | |||||||||
Issuance of common stock | 28,504,839 | 3 | 42,118 | — | — | 42,121 | ||||||||||||||||
Common stock warrants issued in connection with March 2015 common stock offering | — | — | (10,296 | ) | — | — | (10,296 | ) | ||||||||||||||
Issuance of common stock in connection with employee stock purchase plan | 85,372 | — | 110 | — | — | 110 | ||||||||||||||||
Stock-based compensation for directors and employees | — | — | 627 | — | — | 627 | ||||||||||||||||
Net loss | — | — | — | (10,537 | ) | — | (10,537 | ) | ||||||||||||||
Balance at March 31, 2015 | 158,736,552 | $ | 15 | $ | 288,936 | $ | (226,018 | ) | $ | (3,849 | ) | $ | 59,084 | |||||||||
See accompanying notes to condensed consolidated financial statements.
4
GALENA BIOPHARMA, INC.
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Amounts in thousands)
(Unaudited)
For the Three Months Ended March 31, | |||||||
2015 | 2014 | ||||||
Cash flows from operating activities: | |||||||
Net loss | $ | (10,537 | ) | $ | (2,536 | ) | |
Adjustment to reconcile net loss to net cash used in operating activities: | |||||||
Depreciation and amortization expense | 264 | 232 | |||||
Allowance for doubtful accounts | — | — | |||||
Non-cash stock-based compensation | 627 | 1,623 | |||||
Change in fair value of common stock warrants | (1,151 | ) | (9,792 | ) | |||
Change in fair value of contingent consideration | 321 | 166 | |||||
Changes in operating assets and liabilities: | |||||||
Accounts receivable | 779 | 2,217 | |||||
Inventories | 18 | 28 | |||||
Prepaid expenses and other assets | 121 | 74 | |||||
Accounts payable | 1,022 | (1,086 | ) | ||||
Accrued expenses and other current liabilities | (3,020 | ) | 1,026 | ||||
Net cash used in operating activities | (11,556 | ) | (8,048 | ) | |||
Cash flows from investing activities: | |||||||
Cash paid for acquisition of Zuplenz rights | (500 | ) | — | ||||
Cash paid for acquisition of GALE-401 rights | — | (2,010 | ) | ||||
Cash paid for purchase of equipment and furnishings | (18 | ) | (22 | ) | |||
Net cash used in investing activities | (518 | ) | (2,032 | ) | |||
Cash flows from financing activities: | |||||||
Net proceeds from issuance of common stock | 42,121 | — | |||||
Net proceeds from exercise of stock options | — | 4,059 | |||||
Proceeds from exercise of warrants | — | 10,611 | |||||
Proceeds from common stock issued in connection with ESPP | 110 | 53 | |||||
Principle payments on long-term debt | (947 | ) | (3 | ) | |||
Net cash provided by financing activities | 41,284 | 14,720 | |||||
Net decrease in cash and cash equivalents | 29,210 | 4,640 | |||||
Cash and cash equivalents at the beginning of period | 23,650 | 47,787 | |||||
Cash and cash equivalents at end of period | $ | 52,860 | $ | 52,427 | |||
Supplemental disclosure of cash flow information: | |||||||
Cash received during the periods for interest | $ | 2 | $ | 5 | |||
Cash paid during the periods for interest | $ | 166 | $ | 211 | |||
Supplemental disclosure of non-cash investing and financing activities: | |||||||
Fair value of warrants issued in connection with common stock recorded as cost of equity | $ | 10,296 | $ | — | |||
Reclassification of warrant liabilities upon exercise | $ | — | $ | 26,808 | |||
5
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
1. Business and Basis of Presentation
Overview
Galena Biopharma, Inc. (“we,” “us,” “our,” “Galena” or the “company”) is a biopharmaceutical company focused on developing and commercializing innovative, targeted oncology therapeutics that address major medical needs across the full spectrum of cancer care. Galena’s development portfolio ranges from mid- to late-stage clinical assets, including a robust immunotherapy program led by NeuVax™ (nelipepimut-S) currently in an international, Phase 3 clinical trial. The company’s commercial drugs include Abstral® (fentanyl) Sublingual Tablets and Zuplenz® (ondansetron) Oral Soluble Film. Collectively, our clinical and commercial strategy focuses on identifying and advancing therapeutic opportunities to improve cancer care, from direct treatment of the disease to the reduction of its debilitating side-effects.
Novel Cancer Immunotherapies
Our targeted cancer immunotherapy approach is based upon preventing recurrence of cancer, which is becoming increasingly important as the number of cancer survivors continues to grow. Once a patient’s tumor becomes metastatic, the outcome is most often fatal, making the prevention of recurrence a potentially critical component of overall patient care. Our programs primarily target patients in the adjuvant (after-surgery) setting who have relatively healthy immune systems, but may still have undetected minimal residual disease.
Our therapies utilize immunologically active peptides combined with the immune adjuvant, recombinant human granulocyte macrophage-colony stimulating factor (rhGM-CSF), and work by harnessing the patient’s own immune system to seek out and attack any residual cancer cells. Using peptide immunogens has many potential clinical advantages, including a favorable safety profile, since these drugs may lack the toxicities typical of most cancer therapies. They also have the potential to evoke long-lasting protection through activation of the immune system and convenient, intradermal mode of delivery. We are currently engaged in multiple clinical trials with NeuVax™ (nelipepimut-S) and GALE-301, or Folate Binding Protein (FBP), targeting the prevention of recurrence in breast, gastric, ovarian and endometrial cancers.
NeuVax™ (nelipepimut-S)
NeuVax™ (nelipepimut-S), our lead product candidate, is a targeted cancer immunotherapy and is being developed for the prevention of cancer recurrence in human epidermal growth factor receptor (HER2) expressing cancers. NeuVax is the immunodominant nonapeptide derived from the extracellular domain of the HER2 protein, a well-established target for therapeutic intervention in breast and gastric carcinomas. The NeuVax vaccine is combined with GM-CSF for injection under the skin, or intradermal administration. Data has shown that an increased presence of circulating tumor cells (CTCs) may predict Disease Free Survival (DFS) and Overall Survival (OS) - suggesting a dormancy of isolated micrometastases, which, over time, may lead to recurrence. After binding to the HLA A2 or A3 molecules on antigen presenting cells, the nelipepimut-S sequence stimulates specific cytotoxic T lymphocyte (CTLs). These activated CTLs recognize, neutralize and destroy, through cell lysis, HER2 expressing cancer cells, including undetected occult cancer cells and micrometastatic foci. The nelipepimut immune response can also generate CTLs to other immunogenic peptides through inter- and intra-antigenic epitope spreading.
We have multiple trials currently ongoing for NeuVax. For our pivotal, fully enrolled, Phase 3 PRESENT (Prevention of Recurrence in Early-Stage, Node-Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) trial, NeuVax is targeting the 30,000-40,000 of the 230,000 female breast cancer patients annually diagnosed in the U.S. who are at a higher risk of their breast cancer recurring, which we refer to as “disease recurrence,” after achieving “no evidence of disease” (NED) status, (or becoming a “survivor”) with standard-of-care therapy (surgery, chemotherapy, radiation). These high-risk patients have a particular molecular signature and disease status: HER2 IHC 1+/2+ (oncoprotein associated with aggressive tumor growth), node positive (disease present in the axillary lymph nodes prior to surgery), and HLA A2/A3 (human leukocyte antigen from A2/A3 patients who have the same loci of genes which represents approximately 65% of population). Up to 25% of resectable, node-positive breast cancer patients, having no radiographic evidence of disease following surgery and adjuvant chemo/radiation therapy, are expected to relapse within three years following diagnosis. The prognosis upon recurrence is very poor. These cancer patients presumably still had isolated, undetected tumor CTCs that led to a recurrence of cancer in the breast (local recurrence) or in another location (metastatic disease).
6
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
We currently have a number of ongoing or planned clinical trials designed to expand the clinical and geographic footprint of NeuVax:
• | Phase 3 Ongoing: Our Phase 3 PRESENT (Prevention of Recurrence in Early- Stage, Node-Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) study is enrolling HER2 1+ and 2+ patients under a Special Protocol Assessment (SPA) granted by the U.S. Food and Drug Administration (FDA). The multinational, multicenter, randomized, double-blinded PRESENT trial is fully enrolled with trial sites in North America, Western and Eastern Europe, and Israel. Additional information on the study can be found at www.neuvax.com. |
• | Phase 2b Ongoing: A randomized, multicenter, investigator-sponsored, 300 patient Phase 2b clinical trial is enrolling HER2 1+/2+ node-positive and high-risk node-negative breast cancer patients to study NeuVax in combination with trastuzumab (Herceptin®; Genentech/Roche) in the adjuvant setting. This trial is partially funded by Genentech/Roche. |
• | Phase 2 Ongoing: An investigator-sponsored trial is ongoing to study NeuVax in combination with Herceptin. The study will enroll 100 patients who are neoadjuvantly treated node positive and negative HER2 IHC 3+ patients, not achieving a pathological complete response (pCR) or adjuvantly treated node positive HER2 IHC 3+ patients. Partial funding for this trial comes from the Department of Defense (DoD) through the Congressionally Directed Medical Research Program (CDMRP) via legislation known as the Defense Appropriations Act. The grant was awarded under a Breast Cancer Research Program (BCRP) Breakthrough Award given to the lead investigator for the trial. |
• | Phase 2 Planned: In January 2014, we partnered with Dr. Reddy’s Laboratories, Ltd. in India for the commercialization of NeuVax in that region. Dr. Reddy’s is responsible for running a Phase 2 gastric cancer trial of NeuVax in India that is expected to initiate in 2016. |
GALE-301 (folate binding protein or FBP)
Our second immunotherapy product candidate, GALE-301, targets folate binding protein receptor-alpha, a well-validated therapeutic target, which is highly over-expressed (20-80 fold) in ovarian, endometrial and breast cancers. GALE-301 is an immunogenic peptide and can stimulate CTLs to recognize and destroy FBP-expressing cancer cells. GALE-301 consists of an FBP peptide combined with GM-CSF, and is currently in a Phase 2a clinical trial for the prevention of recurrence in patients with ovarian and endometrial cancers. Current treatments for these diseases are principally with chemotherapeutic agents and patients suffer a high recurrence rate; and, most patients relapse with an extremely poor prognosis. Preliminary promising results from the Phase 2a clinical trial of GALE-301 were presented in November 2014 at the Society for Immunotherapy of Cancer conference and showed a 38% reduction in relative risk of recurrence, and that the agent was well-tolerated with primarily Grade 1 and 2 toxicities and elicited a strong in vivo immune response. We expect to present top line data from the Phase 2a trial mid-year 2015.
Hematology
GALE-401 (anagrelide controlled release (CR))
In January 2014, we announced the acquisition of the worldwide rights to anagrelide controlled release (CR), which we renamed GALE-401, through our acquisition of Mills Pharmaceuticals, LLC. GALE-401 contains the active ingredient anagrelide, an FDA-approved product, for the treatment of patients with myeloproliferative neoplasms (MPNs) to lower abnormally elevated platelet levels. The currently available immediate release (IR) version of anagrelide causes adverse events that are believed to be dose and plasma concentration dependent. Therefore, reducing the maximum concentration (Cmax) is hypothesized to reduce the side effects, but preserve efficacy.
7
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
Multiple Phase 1 studies in 98 healthy subjects have shown GALE-401 reduces the Cmax of anagrelide following oral administration, appears to be well tolerated at the doses administered, and to be capable of reducing platelet levels. The Phase 1 program provided the desired PK/PD (pharmacokinetic/pharmacodynamic) profile to enable the initiation of the ongoing Phase 2 proof-of-concept trial. The Phase 2 trial enrolled 18 patients in the United States for the treatment of thrombocytosis, or elevated platelet counts in patients with MPNs. Phase 2 top-line safety and efficacy data will be presented this year. Based on a regulatory meeting with the FDA, Galena believes a 505(b)(2) regulatory filing is an acceptable pathway for development and potential approval of GALE-401, with the reference drug Agrylin® (anagrelide; Shire Pharmaceuticals).
Commercial Capabilities
Abstral® (fentanyl) Sublingual Tablets
Our first commercial product, Abstral® (fentanyl) Sublingual Tablets, is an important treatment option for inadequately controlled breakthrough cancer pain (BTcP), which affects more than 50% of all cancer patients. Abstral is approved by the FDA, and is a sublingual (under the tongue) tablet for the management of breakthrough pain in patients with cancer, 18 years of age and older, who are already receiving, and who are tolerant to, opioid therapy for their persistent baseline cancer pain. The Abstral formulation delivers the analgesic power and increased bioavailability of micronized fentanyl in a convenient sublingual tablet that is designed to dissolve under the tongue in seconds and provide relief of breakthrough pain in minutes. Abstral is a transmucosal immediate release fentanyl (TIRF) product with product class oversight by the TIRF Risk Evaluation and Mitigation Strategy (REMS) access program. Abstral is manufactured for us by contract manufacturers and we distribute and sell Abstral in the U.S. through our commercial organization.
Zuplenz® (ondansetron) Oral Soluble Film
In July 2014 we expanded our commercial portfolio through the licensing of our second commercial product, Zuplenz® (ondansetron) Oral Soluble Film, from MonoSol Rx, LLC. Zuplenz is approved by the FDA in adult patients for the prevention of highly and moderately emetogenic chemotherapy-induced nausea and vomiting (CINV), radiotherapy-induced nausea and vomiting (RINV), and post-operative nausea and vomiting (PONV). Zuplenz is also approved in pediatric patients treated with moderately emetogenic CINV. Nausea and vomiting are two of the most common side-effects experienced by post-surgery patients and patients receiving chemotherapy or radiation. It is estimated that up to 90% of chemotherapy and up to 80% of radiotherapy patients will experience CINV and RINV, respectively.
Zuplenz utilizes MonoSol’s proprietary PharmFilm® technology, an oral soluble film that dissolves on the tongue in less than 30 seconds. Zuplenz eliminates the burden of swallowing pills during periods of emesis, may be advantageous for patients with oral irritation, and may increase patient adherence and the patient's ability to keep the medication down without vomiting. The active pharmaceutical ingredient in Zuplenz, ondansetron, belongs to a class of medications called serotonin 5-HT3 receptor antagonists and works by blocking the action of serotonin, a natural substance that may cause nausea and vomiting. Ondansetron is the most widely prescribed drug in this class of anti-emetics, and used broadly across the oncology spectrum. MonoSol will exclusively manufacture Zuplenz for us for sale in the U.S. through our commercial organization.
8
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
Basis of Presentation and Significant Accounting Policies
The accompanying consolidated financial statements included herein have been prepared by Galena pursuant to the rules and regulations of the Securities and Exchange Commission (SEC). Unless the context otherwise indicates, references in these notes to the “company,” “we,” “us” or “our” refer to Galena, our wholly owned subsidiary, Apthera, Inc., or “Apthera,” and our wholly owned subsidiary, Mills Pharmaceuticals, LLC or "Mills."
Uses of Estimates in Preparation of Financial Statements — The preparation of these consolidated financial statements in accordance with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ materially from those estimates.
Principles of Consolidation — The consolidated financial statements include the accounts of Galena and its wholly owned subsidiaries. All material intercompany accounts have been eliminated in consolidation.
Reclassifications — Certain prior year amounts have been reclassified to conform to current year presentation. These reclassifications had no effect on net loss per share.
Cash and Cash Equivalents — The company considers all highly liquid debt instruments with an original maturity of 90 days or less to be cash equivalents. Cash equivalents consist primarily of amounts invested in money market accounts and demand deposits.
Restricted Cash — Restricted cash consists of certificates of deposit on hand with the company’s financial institutions as collateral for its corporate credit cards.
Fair Value of Financial Instruments — The carrying amounts reported in the balance sheet for cash equivalents, accounts receivable, accounts payable, and capital leases approximate their fair values due to their short-term nature and market rates of interest.
Accounts Receivable - The company maintains credit limits for all customers based upon several factors, including but not limited to financial condition and stability, payment history, published credit reports and use of credit references. Management performs analysis to evaluate accounts receivables to ensure recorded amounts reflect estimated net realizable value. An allowance for doubtful accounts is established based on the Company's best estimate of the amount of probably credit losses in the Company's accounts receivable. As of March 31, 2015 and December 31, 2014 the allowance for doubtful accounts was $160,000.
Inventories — Inventories are stated at the lower of cost or market value and are determined using the first-in, first-out ("FIFO") method. Inventories consist of Abstral work-in-process and finished goods. The company has entered into manufacturing and supply agreements for the manufacture and packing of Abstral finished goods. As of March 31, 2015, the company had inventories of $637,000, consisting of $455,000 of work-in-process and $182,000 of finished goods. As of December 31, 2014, the company had inventories of $655,000 consisting of $455,000 of work-in-process and $200,000 of finished goods.
Equipment and Furnishings — Equipment and furnishings are stated at cost and depreciated using the straight-line method based on the estimated useful lives (generally three to five years) of the related assets.
9
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
Goodwill and Intangible Assets — Goodwill and indefinite-lived intangible assets are not amortized but are tested annually for impairment at the reporting unit level, or more frequently if events and circumstances indicate impairment may have occurred. Factors the company considers important that could trigger an interim review for impairment include, but are not limited to, the following:
•Significant changes in the manner of its use of acquired assets or the strategy for its overall business;
•Significant negative industry or economic trends;
•Significant decline in stock price for a sustained period; and
•Significant decline in market capitalization relative to net book value.
Goodwill and other intangible assets with indefinite lives are evaluated for impairment first by a qualitative assessment to determine the likelihood of impairment. If it is determined that impairment is more likely than not, the company will then proceed to the two step impairment test. The first step is to compare the fair value of the reporting unit to the carrying amount of the reporting unit. If the carrying amount exceeds the fair value, a second step must be followed to calculate impairment. Otherwise, if the fair value of the reporting unit exceeds the carrying amount, the goodwill is not considered to be impaired as of the measurement date. In its review of the carrying value of the goodwill for its single reporting unit and its indefinite-lived intangible assets, the company determines fair values of its goodwill using the market approach, and its indefinite-lived intangible assets using the income approach.
Intangible assets not considered indefinite-lived are reviewed for impairment when facts or circumstances suggest that the carrying value of these assets may not be recoverable. The company’s policy is to identify and record impairment losses, if necessary, on intangible assets when events and circumstances indicate that the assets might be impaired and the undiscounted cash flows estimated to be generated by those assets are less than the carrying amounts.
The company performed its review for impairment using the qualitative assessment for both goodwill and indefinite-lived intangible assets, as well as assets not considered to be indefinite-lived, and has determined that there has been no impairment to these assets as of March 31, 2015.
Revenue Recognition - The company recognizes revenue from the sale of Abstral. Revenue is recognized when (i) persuasive evidence of an arrangement exists, (ii) delivery has occurred and title has passed, (iii) the price is fixed or determinable and (iv) collectability is reasonably assured.
We sell Abstral product in the United States to wholesale pharmaceutical distributors and retail pharmacies, or our "customers," subject to rights of return. We recognize Abstral product sales at the time title transfers to our customer, and provide allowances for estimated future product returns, prompt pay discounts, wholesaler discounts, rebates, chargebacks, patient assistance program benefits and other deductions as needed. The company is required to make significant judgments and estimates in determining some of these allowances. If actual results differ from its estimates, the company will be required to make adjustments to these allowances in the future.
Returns - The company estimates future returns based on historical return information, as well as information regarding prescription information and sell-through trends, in relation to the estimated amount of product in the sales channels and product expiration dates. The allowance for returns is recorded as a reduction to revenue in the period in which the revenue is recognized, with a corresponding allowance against accounts receivable.
Prompt Pay Discounts - As an incentive for prompt payment, the company offers a cash discount to customers, which is generally 2% of gross sales. The company expects that all customers will comply with the contractual terms to earn the discount. The company records prompt pay discounts as a reduction to revenue in the period in which the revenue is recognized, with a corresponding allowance against accounts receivable.
10
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
Wholesaler Discounts - The company offers discounts on sales to wholesalers and distributors based on contractually determined rates. The company accrues the discount as a reduction of receivables due from the wholesalers upon shipment to the respective wholesale distributors and retail pharmacies and recognizes the discount as a reduction of revenue in the same period the related revenue is recognized.
Rebates - The company participates in certain rebate programs, which provide discounted prescriptions to members of certain managed care organizations, group purchasing organizations and specialty pharmacies. Under these rebate programs, the company pays the rebates generally two to three months after the end of the quarter in which prescriptions subject to the rebate are filled. The company estimates and accrues these rebates based on current contract prices, historical and estimated future percentages of product sold to qualifying member pharmacies and estimated levels of inventory in the distribution channel. Rebates are recognized as a reduction to revenue in the period that the related revenue is recognized, with a corresponding liability in accrued expenses and other current liabilities.
Chargebacks - The company provides discounts primarily to authorized users of the Federal Supply Schedule (FSS) of the General Services Administration under an FSS contract negotiated by the Department of Veterans Affairs and various organizations under Medicaid or Medicare contracts and regulations. These entities purchase products from the wholesale distributors at a discounted price, and the wholesale distributors then charge back to the company the difference between the current retail price and the price the entity paid for the product. The company estimates and accrues chargebacks based on estimated wholesaler inventory levels, current contract prices and historic chargeback activity. Chargebacks are recognized as a reduction of revenue in the period the related revenue is recognized, with a corresponding allowance against accounts receivable.
Patient Assistance Programs - The company offers discount card programs to patients for Abstral in which patients receive discounts on their Abstral prescriptions that are reimbursed by the company. The company estimates the total amount that will be recognized based on a percentage of actual redemption applied to inventory in the distribution and retail channel and recognizes the discount as a reduction of revenue and as an other current liability (see Note 4) in the same period the related revenue is recognized.
Acquisitions and In-Licensing — For all in-licensed products and technologies, we perform an analysis to determine whether we hold a variable interest or a controlling financial interest in a variable interest entity. On the basis of our interpretations and conclusions, we determine whether the acquisition falls under the purview of variable interest entity accounting and if so, consider the necessity to consolidate the acquisition. As of March 31, 2015, we determined there were no variable interest entities required to be consolidated.
We also perform an analysis to determine if the assets and liabilities acquired in an acquisition qualify as a "business." The excess of the purchase price over the fair value of the net assets acquired can only be recognized as goodwill in a business combination. The Company completes its valuation analysis no later than twelve months from the date of the acquisition.
Contingent Purchase Price Consideration — Contingent consideration in business combinations is recorded at the estimated fair value as of the acquisition date. The fair value of the contingent consideration is re-measured at each reporting period with any adjustments in fair value included in our consolidated statement of comprehensive loss.
Patents and Patent Application Costs — Although the company believes that its patents and underlying technology have continuing value, the amount of future benefits to be derived from the patents is uncertain. Patent costs are, therefore, expensed as incurred.
Share-based Compensation — The company follows the provisions of the FASB ASC Topic 718, “Compensation — Stock Compensation” (“ASC 718”), which requires the measurement and recognition of compensation expense for all stock-based payment awards made to employees, non-employee directors, and consultants, including stock options and warrants. Stock compensation expense based on the grant date fair value estimated in accordance with the provisions of ASC 718 is recognized as an expense over the requisite service period.
11
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
For stock options and warrants granted as consideration for services rendered by non-employees, the company recognizes compensation expense in accordance with the requirements of FASB ASC Topic 505-50 (“ASC 505-50”), “ Equity Based Payments to Non- Employees.” Non-employee option and warrant grants that do not vest immediately upon grant are recorded as an expense over the vesting period. At the end of each financial reporting period prior to vesting, the value of these options and warrants, as calculated using the Black-Scholes option-pricing model, is re-measured using the fair value of the company’s common stock and the non-cash compensation recognized during the period is adjusted accordingly. Since the fair market value of options and warrants granted to non-employees is subject to change in the future, the amount of the future compensation expense will include fair value re-measurements until the stock options are fully vested.
Research and Development Expenses — Research and development costs are expensed as incurred. Included in research and development costs are wages, benefits and other operating costs, facilities, supplies, external services and overhead related to our research and development departments, and clinical trial expenses.
Clinical trial expenses include direct costs associated with contract research organizations (CROs), as well as patient-related costs at sites at which our trials are being conducted.
Direct costs associated with our CROs are generally payable on a time and materials basis, or when certain enrollment and monitoring milestones are achieved. Expense related to a milestone is recognized in the period in which the milestone is achieved or in which we determine that it is more likely than not that it will be achieved.
The invoicing from clinical trial sites can lag several months. We accrue these site costs based on our estimate of upfront set-up costs upon the screening of the first patient at each site, and the patient related costs based on our knowledge of patient enrollment status at each site.
Income Taxes — The company recognizes liabilities or assets for the deferred tax consequences of temporary differences between the tax basis of assets or liabilities and their reported amounts in the financial statements in accordance with FASB ASC 740-10, “Accounting for Income Taxes” (“ASC 740-10”). These temporary differences will result in taxable or deductible amounts in future years when the reported amounts of the assets or liabilities are recovered or settled. ASC 740-10 requires that a valuation allowance be established when management determines that it is more likely than not that all or a portion of a deferred asset will not be realized. The company evaluates the realizability of its net deferred income tax assets and valuation allowances as necessary, at least on an annual basis. During this evaluation, the company reviews its forecasts of income in conjunction with other positive and negative evidence surrounding the realizability of its deferred income tax assets to determine if a valuation allowance is required. Adjustments to the valuation allowance will increase or decrease the company’s income tax provision or benefit. The recognition and measurement of benefits related to the company’s tax positions requires significant judgment, as uncertainties often exist with respect to new laws, new interpretations of existing laws, and rulings by taxing authorities. Differences between actual results and the company’s assumptions or changes in the company’s assumptions in future periods are recorded in the period they become known.
There was no income tax expense or benefit for the three months periods ended March 31, 2015 and 2014. We continue to maintain a full valuation allowance against our net deferred tax assets.
Concentrations of Credit Risk — Financial instruments that potentially subject the company to significant concentrations of credit risk consist principally of cash and cash equivalents. The company maintains cash balances in several accounts with two banks, which at times are in excess of federally insured limits. As of March 31, 2015, the company’s cash equivalents were invested in money market mutual funds. The company’s investment policy does not allow investment in any debt securities rated less than “investment grade” by national ratings services. The company has not experienced any losses on its deposits of cash and cash equivalents. The company maintains significant cash and cash equivalents at two financial institutions that are in excess of federally insured limits.
Comprehensive Loss — Comprehensive loss consists of our net loss.
12
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
2. Business Combinations
On July 17, 2014 the Company entered into a definitive license and supply agreement with MonoSol Rx, LLC (MonoSol) for the U.S. commercial rights to Zuplenz® (ondansetron) Oral Soluble Film (Zuplenz), an FDA approved product. The transaction was accounted for as a business combination under the acquisition method of accounting based on Accounting Standards Codification 805, "Business Combinations." Accordingly, the assets acquired and liabilities assumed were recorded at fair value. As of the issuance date of the condensed consolidated financial statements for the quarter ended March 31, 2015, the Company had not finalized the valuation of the acquired assets and liabilities for the transaction. Specifically, more complete information for credit memos for expiring channel inventory will be required prior to finalizing business combination accounting.
The following table summarizes the purchase price consideration and allocation of purchase price:
Total Acquisition Date Fair Value | ||||
Purchase price consideration: | ||||
Cash and cash equivalents | $ | 3,556 | ||
Common stock | 2,482 | |||
Liabilities assumed: | ||||
Contingent consideration | 240 | |||
Credit memos for expiring channel inventory | 1,995 | |||
Total consideration | $ | 8,273 | ||
Asset acquired: | ||||
Zuplenz rights | $ | 8,101 | ||
Goodwill | 172 | |||
Fair value of assets acquired | $ | 8,273 | ||
The above contingent consideration represents a risk adjusted net present value relating to cash payments on achievement of certain milestones.
3. Fair Value Measurements
The company follows ASC 820, “Fair Value Measurements and Disclosures,” (“ASC 820”) for the company’s financial assets and liabilities that are re-measured and reported at fair value at each reporting period, and are re-measured and reported at fair value at least annually using a fair value hierarchy that is broken down into three levels. Level inputs are defined as follows:
Level 1 — quoted prices in active markets for identical assets or liabilities.
Level 2 — other significant observable inputs for the assets or liabilities through corroboration with market data at the measurement date.
Level 3 — significant unobservable inputs that reflect management’s best estimate of what market participants would use to price the assets or liabilities at the measurement date.
13
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
The company categorized its cash equivalents as Level 1. The valuations for Level 1 were determined based on a “market approach” using quoted prices in active markets for identical assets. Valuation of these assets does not require a significant degree of judgment. The company categorized its warrants potentially settleable in cash as Level 2 inputs. The warrants are measured at market value on a recurring basis and are being marked to market each quarter-end until they are completely settled. The warrants are valued using an appropriate pricing model, using assumptions consistent with our application of ASC 718. The contingent purchase price consideration is categorized as Level 3 inputs and is measured at its estimated fair value on a recurring basis and is adjusted at each quarter-end until it is completely settled. The contingent purchase price consideration is valued based on the expected timing of milestones, the expected probability of success for each milestone and discount rates based on a corporate debt interest rate index publicly issued.
The following tables present information about our assets and liabilities measured at fair value on a recurring basis in the condensed consolidated balance sheets (in thousands):
Description | March 31, 2015 | Quoted Prices In Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Unobservable Inputs (Level 3) | |||||||||||
Assets: | |||||||||||||||
Cash equivalents | $ | 48,960 | $ | 48,960 | $ | — | $ | — | |||||||
Total assets measured and recorded at fair value | $ | 48,960 | $ | 48,960 | $ | — | $ | — | |||||||
Liabilities: | |||||||||||||||
Warrants potentially settleable in cash | $ | 14,528 | $ | — | $ | 14,528 | $ | — | |||||||
Contingent purchase price consideration | 6,972 | — | — | 6,972 | |||||||||||
Total liabilities measured and recorded at fair value | $ | 21,500 | $ | — | $ | 14,528 | $ | 6,972 | |||||||
Description | December 31, 2014 | Quoted Prices In Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Unobservable Inputs (Level 3) | |||||||||||
Assets: | |||||||||||||||
Cash equivalents | $ | 19,477 | $ | 19,477 | $ | — | $ | — | |||||||
Total assets measured and recorded at fair value | $ | 19,477 | $ | 19,477 | $ | — | $ | — | |||||||
Liabilities: | |||||||||||||||
Warrants potentially settleable in cash | $ | 5,383 | $ | — | $ | 5,383 | $ | — | |||||||
Contingent purchase price consideration | 6,651 | — | — | 6,651 | |||||||||||
Total liabilities measured and recorded at fair value | $ | 12,034 | $ | — | $ | 5,383 | $ | 6,651 | |||||||
The company did not transfer any financial instruments into or out of Level 3 classification during the three months ended March 31, 2015 or 2014. A reconciliation of the beginning and ending Level 3 liabilities for the three months ended March 31, 2015 is as follows (in thousands):
Fair Value Measurements Using Significant Unobservable Inputs (Level 3) | |||
Balance, January 1, 2015 | $ | 6,651 | |
Change in the estimated fair value of the contingent purchase price consideration | 321 | ||
Balance at March 31, 2015 | $ | 6,972 | |
14
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
The fair value of the contingent purchase price consideration is measured at the end of each reporting period using Level 3 inputs in a probability-weighted, discounted cash-outflow model. The significant unobservable assumptions include the probability of achieving each milestone, the date we expect to reach the milestone, and a determination of present value factors used to discount future expected cash outflows.
4. Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities consist of the following (in thousands):
March 31, 2015 | December 31, 2014 | ||||||
Clinical trial costs | $ | 4,728 | $ | 6,967 | |||
Credit memos for expiring Zuplenz channel inventory | 1,995 | 1,995 | |||||
Professional fees | 1,596 | 860 | |||||
Patient assistance programs and rebates | 1,428 | 2,444 | |||||
Compensation and related benefits | 1,373 | 2,198 | |||||
Royalties | 738 | 408 | |||||
Zuplenz milestone payments | 240 | 740 | |||||
Interest expense | 51 | 57 | |||||
Accrued expenses and other current liabilities | $ | 12,149 | $ | 15,669 | |||
5. Long-term Debt
On May 8, 2013 we entered into a loan and security agreement with Oxford Finance LLC, as collateral agent, and related lenders under which we borrowed the first tranche of $10 million (the "Loan"). The Loan payment terms include 12 months of interest-only payments at the fixed coupon rate of 8.45%, followed by 30 months of amortization of principal and interest until maturity in November 2016. In connection with the Loan, we paid the lender a 1% cash facility fee and a 5.5% cash final payment and granted to the lenders seven-year warrants to purchase up to 182,186 shares of our common stock at an exercise price of $2.47, which equaled a 20-day average market price of our common stock prior to the date of the grant.
6. Legal Proceedings, Commitments and Contingencies
Legal Proceedings
In early 2014, several purported shareholder derivative complaints were filed against our company, as nominal defendant, and certain of our officers and directors in the Circuit Court of Oregon for the County of Multnomah, the U.S. District Court for the District of Oregon, and the Delaware Court of Chancery. On April 11, 2014, the derivative complaints pending in the U.S. District Court for the District of Oregon were consolidated in the matter of In Re Galena Biopharma, Inc. Derivative Litigation, No. 3:14-cv-382-SI (D. Or.), and on August 25, 2014, the lead plaintiffs filed a consolidated amended complaint. On July 21, 2014, all of the derivative complaints pending in the Delaware Court of Chancery were consolidated in the matter of In re Galena Biopharma, Inc. Stockholder Derivative Litigation, Consolidated C.A. No. 9715-VCN (Del. Ch.). On February 10, 2015, the lead plaintiffs in the derivative complaints pending in the Delaware Court of Chancery voluntarily dismissed their action without prejudice. As a result of this dismissal, and at the recommendation of the special litigation committee of the board established on July 21, 2014 to investigate the derivative claims, on February 26, 2015 our board of directors disbanded the special litigation committee.
The operative complaints allege, among other things, breaches of fiduciary duties and abuse of control by the officers and directors in connection with public statements purportedly issued by us or on our behalf and sales of our common stock by certain of our officers and directors in January and February of 2014, improper stock-option grants, and excessive compensation of our non-employee directors.
15
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
Also, five purported securities class action complaints filed in the U.S. District Court for the District of Oregon have been consolidated into a single action, In re Galena Biopharma, Inc. Securities Litigation, No. 3:14-cv-367-SI (D. Or.), and a lead plaintiff has been appointed. On October 31, 2014, the lead plaintiff filed a consolidated amended complaint, which alleges, among other things, that our company and certain of our officers and directors violated the federal securities laws by making materially false and misleading statements and omissions in press releases and in filings with the SEC arising out of the same circumstances that are the subject of the derivative actions described above, and which alleges that certain of our officers and directors sold company stock while in possession of material non-public information.
We intend to vigorously defend against and seek resolution to the foregoing claims. At March 31, 2015, we have not recorded any liabilities with respect to the claims in our consolidated financial statements. We believe that claims are covered under our liability insurance, and we have notified our insurance carriers of the claims. The insurers have responded by requesting additional information and by reserving their rights under the policies, including the rights to deny coverage under various policy exclusions. Subject to their reservation of rights, we are being reimbursed by our insurer for substantially all legal fees relating to our defense of the claims.
We are aware that the SEC is investigating certain matters relating to the use of certain outside investor-relations professionals by us and other public companies. We have been in contact with the SEC staff through our counsel and are cooperating with the investigation.
Litigation is inherently uncertain, and there is no assurance as to the outcome of the matters described above. We could incur substantial unreimbursed legal fees, settlements, judgments, and other expenses in connection with these or other legal and regulatory proceedings that may not qualify for coverage under, or may exceed the limits of, our applicable directors and officers liability insurance policies and could have a material adverse effect on our financial condition, liquidity, and results of operations. These matters also may distract the time and attention of our officers and directors or divert our other resources away from our ongoing commercial and development programs. An unfavorable outcome in any of these matters could damage our business and reputation or result in additional claims or proceedings against us.
We have received a Paragraph IV certification notice from Actavis Pharma, Inc. and related companies (Actavis) contending that the patents held by Orexo for Abstral that are listed in the Orange Book (U.S. Patents 6,759,059, 6,761,910 and 7,910,132, which expire in September 2019), are invalid, unenforceable and/or will not be infringed by the manufacture, use, or sale of a generic form of Abstral. In response to these notices, Orexo filed suit against Actavis to defend their patent rights, which we license from Orexo. We are obligated under our contract with Orexo to absorb 88% of the legal costs associated with defending Abstral patents. We intend to work with Orexo to continue to vigorously enforce intellectual property rights relating to any future challenges the Abstral product.
Commitments
The company acquires assets still in development and enters into research and development arrangements with third parties that often require milestone and royalty payments based on the progress of the asset through development stages. Milestone payments may be required, for example, upon approval of the product for marketing by a regulatory agency. In certain agreements, the company is required to make royalty payments based upon a percentage of the sales.
These arrangements may be material individually, and in the unlikely event that milestones for multiple products covered by these arrangements were reached in the same period, the aggregate charge to expense could be material to the results of operations. In addition, these arrangements often give the company the discretion to unilaterally terminate development of the product, which would allow the company to avoid making the contingent payments; however, the company is unlikely to cease development if the compound successfully achieves clinical testing objectives.
The company applies the disclosure provisions FASB ASC Topic 460 (“ASC 460”), “ Guarantor’s Accounting and Disclosure Requirements for Guarantees, Including Indirect Guarantees of Indebtedness of Others ”, to its agreements that contain guarantee or indemnification clauses. The company provides (i) indemnifications of varying scope and size to certain investors and other parties for certain losses suffered or incurred by the indemnified party in connection with various types of third-party claims and (ii) indemnifications of varying scope and size to officers and directors against third party claims arising from the services they provide to us. These indemnifications give rise only to the disclosure provisions of ASC 460. To date, the company has not incurred costs as a result of these obligations and does not expect to incur material costs in the future. Accordingly, the company has not accrued any liabilities in its financial statements related to these indemnifications.
16
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
We have received a Paragraph IV certification notice from Actavis Pharma, Inc. and related companies (Actavis) contending that the patents held by Orexo for Abstral that are listed in the Orange Book (U.S. Patents 6,759,059, 6,761,910 and 7,910,132, which expire in September 2019), are invalid, unenforceable and/or will not be infringed by the manufacture, use, or sale of a generic form of Abstral. In response to these notices, Orexo filed suit against Actavis to defend their patent rights, which we license from Orexo. We are obligated under our contract with Orexo to absorb 88% of the legal costs associated with defending Abstral patents. We intend to work with Orexo to continue to vigorously enforce intellectual property rights relating to any future challenges the Abstral product.
7. Stockholders’ Equity
Preferred Stock — The company has authorized up to 5,000,000 shares of preferred stock, $0.0001 par value per share, for issuance. The preferred stock will have such rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences, as shall be determined by the company’s Board of Directors upon its issuance. To date, the company has not issued any preferred shares.
Common Stock — The company has authorized up to 200,000,000 shares of common stock, $0.0001 par value per share, for issuance.
November 2014 Purchase Agreement with Lincoln Park Capital, LLC - On November 18, 2014, the company entered into a purchase agreement with Lincoln Park Capital, LLC (LPC), pursuant to which the company has the right to sell to LPC up to $50 million in shares of the company's common stock, subject to certain limitations and conditions over the 36 month term of the purchase agreement. Pursuant to the purchase agreement, LPC initially purchased 2.5 million shares of the company's common stock at $2.00 per share and the company issued 631,221 shares of common stock to LPC as a commitment fee, which was recorded as a cost of capital. As a result of this initial issuance, the company received initial net proceeds of $4.9 million, after deducting commissions and other offering expenses. In addition to LPC’s initial purchase of our common stock under the purchase agreement, during the first quarter of 2015, we received net proceeds of $4.4 million from LPC’s subsequent purchases of a total of 2.7 million shares of our common stock, excluding the commitment fee shares.
At Market Issuance Sales Agreements - On May 24, 2013 the Company entered into At Market Issuance Sales Agreements (ATM) with MLV & Co. LLC and Maxim Group LLC (the Agents). From time to time during the term of the ATM, we may issue and sell through the Agents, shares of our common stock, and the Agents collect a fee equal to 3% of the gross proceeds from the sale of shares, up to a total limit of $20 million in gross proceeds. The ATM is available to the company until it is terminated by the Agents or the company. During the first quarter of 2015, we received $2.3 million in net proceeds from the sale of 1.4 million shares of our common stock through the ATM. There were no sales of our common stock under the ATM in 2013.
March 2015 Underwritten Public Offering - On March 18, 2015 the company closed an underwritten public offering of 24,358,974 units at a price to the public of $1.56 per unit for gross proceeds of $38 million (the "March 2015 Offering"). Each unit consists of one share of common stock, and a warrant to purchase 0.50 of a share of common stock at an exercise price of $2.08 per share. The March 2015 Offering included an over-allotment option for the underwriters to purchase an additional 3,653,846 shares of common stock and/or warrants to purchase up to 1,826,923 shares of common stock. On March 18, 2015, the underwriters exercised their over-allotment option to purchase warrants to purchase an aggregate of 1,826,923 shares of common stock. On April 10, 2015, the underwriters exercised their over-allotment option to purchase 3,653,846 shares of common stock for additional net proceeds of $5.4 million. The total net proceeds of the March 2015 Offering, including the exercise of the over-allotment option to purchase the warrants, were $35.4 million, after deducting underwriting discounts and commissions and offering expenses payable by the company. Including the underwriters exercise of the over-allotment option to purchase the additional shares in April 2015, the total net proceeds were $40.8 million.
17
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
Shares of common stock for future issuance are reserved for as follows (in thousands):
As of March 31, 2015 | ||
Warrants outstanding | 22,546 | |
Stock options outstanding | 10,683 | |
Options reserved for future issuance under the Company’s 2007 Incentive Plan | 794 | |
Shares reserved for future issuance under the Employee Stock Purchase Plan | 674 | |
Total reserved for future issuance | 34,697 | |
8. Warrants
The following is a summary of warrant activity for the three months ended March 31, 2015 (in thousands):
March 2015 Warrants | September 2013 Warrants | December 2012 Warrants | April 2011 Warrants | March 2011 Warrants | March 2010 Warrants | Consultant and Oxford Warrants | Total | ||||||||||||||||
Outstanding, January 1, 2015 | — | 3,973 | 3,031 | 615 | 176 | 25 | 720 | 8,540 | |||||||||||||||
Issued | 14,006 | — | — | — | — | — | — | 14,006 | |||||||||||||||
Outstanding, March 31, 2015 | 14,006 | 3,973 | 3,031 | 615 | 176 | 25 | 720 | 22,546 | |||||||||||||||
Expiration | March 2020 | September 2018 | December 2017 | April 2017 | March 2016 | March 2016 | Varies 2014-2020 | ||||||||||||||||
Warrants consist of warrants potentially settleable in cash, which are liability-classified warrants, and equity-classified warrants.
Warrants classified as liabilities
Liability-classified warrants consist of warrants to purchase common stock issued in connection with equity financings in March 2015, September 2013, December 2012, April 2011, March 2011, and March 2010. These warrants are potentially settleable in cash and were determined not to be indexed to our common stock.
The estimated fair value of outstanding warrants accounted for as liabilities is determined at each balance sheet date. Any decrease or increase in the estimated fair value of the warrant liability since the most recent balance sheet date is recorded in the condensed consolidated statement of comprehensive loss as other income (expense). The fair value of the warrants is estimated using an appropriate pricing model with the following inputs:
As of March 31, 2015 | |||||||||||||||||||||||
March 2015 Warrants | September 2013 Warrants | December 2012 Warrants | April 2011 Warrants | March 2011 Warrants | March 2010 Warrants | ||||||||||||||||||
Strike price | $ | 2.08 | $ | 2.50 | $ | 1.83 | $ | 0.65 | $ | 0.65 | $ | 2.02 | |||||||||||
Expected term (years) | 4.97 | 3.47 | 2.73 | 2.06 | 0.93 | 0.99 | |||||||||||||||||
Volatility % | 73.29 | % | 75.54 | % | 77.39 | % | 75.05 | % | 65.09 | % | 67.34 | % | |||||||||||
Risk-free rate % | 1.36 | % | 1.00 | % | 0.80 | % | 0.58 | % | 0.24 | % | 0.26 | % | |||||||||||
18
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
As of December 31, 2014 | |||||||||||||||||||
September 2013 Warrants | December 2012 Warrants | April 2011 Warrants | March 2011 Warrants | March 2010 Warrants | |||||||||||||||
Strike price | $ | 2.50 | $ | 1.90 | $ | 0.65 | $ | 0.65 | $ | 2.15 | |||||||||
Expected term (years) | 3.72 | 2.98 | 2.31 | 1.18 | 1.24 | ||||||||||||||
Volatility % | 75.60 | % | 76.85 | % | 78.24 | % | 77.38 | % | 77.12 | % | |||||||||
Risk-free rate % | 1.30 | % | 1.09 | % | 0.80 | % | 0.32 | % | 0.35 | % | |||||||||
The expected volatility assumptions are based on the company's implied volatility in combination with the implied volatilities of similar publicly traded entities. The expected life assumption is based on the remaining contractual terms of the warrants. The risk-free rate is based on the zero coupon rates in effect at the time of valuation. The dividend yield used in the pricing model is zero, because the company has no present intention to pay cash dividends.
The changes in fair value of the warrant liability for the three months ended March 31, 2015 were as follows (in thousands):
March 2015 Warrants | September 2013 Warrants | December 2012 Warrants | April 2011 Warrants | March 2011 Warrants | March 2010 Warrants | Total | |||||||||||||||||||||
Warrant liability, January 1, 2015 | $ | — | $ | 2,560 | $ | 2,027 | $ | 625 | $ | 163 | $ | 8 | $ | 5,383 | |||||||||||||
Fair value of warrants issued | 10,296 | — | — | — | — | — | 10,296 | ||||||||||||||||||||
Change in fair value of warrants | (266 | ) | (435 | ) | (334 | ) | (85 | ) | (27 | ) | (4 | ) | (1,151 | ) | |||||||||||||
Warrant liability, March 31, 2015 | $ | 10,030 | $ | 2,125 | $ | 1,693 | $ | 540 | $ | 136 | $ | 4 | $ | 14,528 | |||||||||||||
Warrants classified as equity
Equity-classified warrants consist of warrants issued in connection with consulting services provided to us. Additionally, on May 8, 2013 as a part of our Loan financing, we granted Oxford Financial LLC warrants to purchase up to 182,186 shares of common stock at an exercise price of $2.47, which equaled to the 20-day average market price of our common stock prior to the date of the grant. The warrants were valued using the Black Scholes model as described in Note 8, below. The fair value assumptions for the grant included a volatility of 75.34%, expected term of seven years, risk free rate of 1.20%, and a dividend rate of 0.00%. The fair value of the warrants granted was $1.93 per share. These warrants are recorded in equity at fair value upon issuance, and not as liabilities, and are not subject to adjustment to fair value in subsequent reporting periods.
9. Stock-Based Compensation
Options to Purchase Shares of Common Stock — The company follows the provisions ASC 718, which requires the measurement and recognition of compensation expense for all share-based payment awards made to employees, non-employee directors, including employee stock options. Stock compensation expense based on the grant date fair value estimated in accordance with the provisions of ASC 718 is recognized as an expense over the requisite service period.
For stock options and warrants granted in consideration for services rendered by non-employees, the company recognizes compensation expense in accordance with the requirements of ASC Topic 505-50. Non-employee option and warrant grants that do not vest immediately upon grant are recorded as an expense over the vesting period. At the end of each financial reporting period prior to vesting, the value of these options and warrants, as calculated using the Black-Scholes option-pricing model, is re-measured using the fair value of the company’s common stock and the non-cash compensation recognized during the period is adjusted accordingly. Since the fair market value of options and warrants granted to non-employees is subject to change in the future, the amount of the future compensation expense will include fair value re-measurements until the stock options and warrants are fully vested.
19
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
The following table summarizes the components of stock-based compensation expense in the condensed consolidated statements of comprehensive loss for the three months ended March 31, 2015 and 2014, respectively (in thousands):
Three Months Ended March 31, | |||||||
2015 | 2014 | ||||||
Research and development | $ | 78 | $ | 160 | |||
Selling, general, and administrative | 549 | 1,524 | |||||
Total stock-based compensation | $ | 627 | $ | 1,684 | |||
The company uses the Black-Scholes option-pricing model and the following weighted-average assumptions to determine the fair value of all its stock options granted:
Three Months Ended March 31, | |||||
2015 | 2014 | ||||
Risk free interest rate | 1.41 | % | 2.06 | % | |
Volatility | 74.62 | % | 78.53 | % | |
Expected lives (years) | 6.25 | 6.25 | |||
Expected dividend yield | 0.00 | % | 0.00 | % | |
The weighted-average fair value of options granted during the three months ended March 31, 2015 was $1.16 per share.
The company’s expected common stock price volatility assumption is based upon the company's own implied volatility in combination with the implied volatility of a basket of comparable companies. The expected life assumptions for employee grants were based upon the simplified method provided for under ASC 718-10, which averages the contractual term of the company’s options of ten years with the average vesting term of four years for an average of six years. The expected life assumptions for non-employees were based upon the contractual term of the option. The dividend yield assumption is zero, because the company has never paid cash dividends and presently has no intention to do so. The risk-free interest rate used for each grant was also based upon prevailing short-term interest rates. The company has estimated an annualized forfeiture rate of 15% for options granted to its employees, 8% for options granted to senior management and zero for non-employee directors. The company will record additional expense if the actual forfeitures are lower than estimated and will record a recovery of prior expense if the actual forfeiture rates are higher than estimated.
As of March 31, 2015, there was $5,411,000 of unrecognized compensation cost related to outstanding options that is expected to be recognized as a component of the company’s operating expenses over a weighted-average period of 2.98 years.
As of March 31, 2015, an aggregate of 16,500,000 shares of common stock were reserved for issuance under the company’s 2007 Incentive Plan, including 10,683,000 shares subject to outstanding common stock options granted under the plan and 794,000 shares available for future grants. The administrator of the plan determines the times when an option may become exercisable. Vesting periods of options granted to date have not exceeded four years. The options will expire, unless previously exercised, no later than ten years from the grant date.
20
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
The following table summarizes option activity of the company:
Total Number of Shares (In Thousands) | Weighted Average Exercise Price | Aggregate Intrinsic Value (In Thousands) | ||||||||
Outstanding at January 1, 2015 | 8,590 | $ | 3.25 | $ | — | |||||
Granted | 2,431 | 1.75 | — | |||||||
Exercised | — | — | — | |||||||
Cancelled | (338 | ) | 2.71 | — | ||||||
Outstanding at March 31, 2015 | 10,683 | $ | 2.93 | $ | 478 | |||||
Options exercisable at March 31, 2015 | 5,768 | $ | 3.50 | $ | 413 | |||||
The aggregate intrinsic values of outstanding and exercisable options at March 31, 2015 were calculated based on the closing price of the company’s common stock as reported on The NASDAQ Capital Market on March 31, 2015 of $1.39 per share. The aggregate intrinsic value equals the positive difference between the closing fair market value of the company’s common stock and the exercise price of the underlying options.
Note 10. Other Income (Expense)
Other income (expense) is summarized as follows (in thousands):
Three Months Ended March 31, | |||||||
2015 | 2014 | ||||||
Change in fair value of the contingent purchase price liability | $ | (321 | ) | $ | (166 | ) | |
Miscellaneous other income (expense) | — | 1 | |||||
Total other income (expense) | $ | (321 | ) | $ | (165 | ) | |
11. Net Loss Per Share
The company accounts for and discloses net loss per common share in accordance with FASB ASC Topic 260 “Earnings per Share.” Basic net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted-average number of common shares outstanding. Diluted net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted-average number of common shares that would have been outstanding during the period assuming the issuance of common shares for all potential dilutive common shares outstanding. Potential common shares consist of shares issuable upon the exercise of stock options and warrants.
The following table sets forth the potentially dilutive common shares excluded from the calculation of net loss per common share because their inclusion would be anti-dilutive (in thousands):
Three Months Ended March 31, | |||||
2015 | 2014 | ||||
Warrants to purchase common stock | 22,546 | 9,560 | |||
Options to purchase common stock | 10,683 | 9,848 | |||
Total | 33,229 | 19,408 | |||
21
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
12. License Agreements
As part of its business, the company enters into licensing agreements with third parties that often require milestone and royalty payments based on the progress of the licensed assets through development and commercial stages. Milestone payments may be required, for example, upon approval of the product for marketing by a regulatory agency, and the company may be required to make royalty payments based upon a percentage of net sales of the product. The expenditures required under these arrangements in any period may be material and are likely to fluctuate from period to period.
These arrangements sometimes permit the company to unilaterally terminate development of the product and thereby avoid future contingent payments; however, the company is unlikely to cease development if the compound successfully achieves clinical testing objectives.
In conjunction with the acquisition of NeuVaxTM, the company acquired rights and assumed obligations under a license agreement among Apthera and The University of Texas M. D. Anderson Cancer Center (“MDACC”) and The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. (“HJF”) which grants exclusive worldwide rights to a U.S. patent covering the nelipepimut-S peptide and several U.S. and foreign patents and patent applications covering methods of using the peptide as a vaccine. Under the terms of this license, we are required to pay an annual maintenance fee of $200,000, a milestone payment of $200,000 upon commencing the Phase 3 PRESENT trial of NeuVax and other clinical milestone payments, as well as royalty payments based on sales of NeuVax or other therapeutic products developed from the licensed technologies.
Effective December 3, 2012, we entered into a license and supply agreement with ABIC Marketing Limited, a subsidiary of Teva Pharmaceuticals (“ABIC”), under which we granted ABIC exclusive rights to seek marketing approval in Israel for our NeuVax product candidate for intradermal injection for the treatment of breast cancer following its approval by the FDA or the European Medicines Agency, and to market, sell and distribute NeuVax in Israel assuming such approval is obtained. ABIC’s rights also include a right of first refusal in Israel for all future indications for which NeuVax may be approved. Under the license and supply agreement, ABIC will assume responsibility for regulatory registration of NeuVax in Israel, provide financial support for local development, and commercialize the product in the region in exchange for making royalty payments to us based on future sales of NeuVax. ABIC also agrees in the license and supply agreement to purchase from us all supplies of NeuVax at a price determined according to a specified formula.
On March 18, 2013, we acquired Abstral® (fentanyl) Sublingual Tablets for sale and distribution in the United States from Orexo AB (ORX.ST), an emerging specialty pharmaceutical company based in Sweden. Abstral has been approved by the U.S. Food and Drug Administration (FDA) and is a transmucosal immediate-release fentanyl (TIRF) product.
Under our agreement with Orexo, we assumed responsibility for the U.S. commercialization of Abstral and for all regulatory and reporting matters in the U.S. We also agreed to establish and maintain through 2015 a specified minimum commercial field force to market, sell and distribute Abstral and to use commercially reasonable efforts to reach the specified sales milestones. Orexo is entitled to reacquire the U.S. rights to Abstral from us for no consideration if we breach our obligations to establish and maintain the requisite sales force throughout the marketing period. We expect to maintain our sales efforts beyond this date. We officially launched U.S. commercial sales of Abstral in October 2013.
In exchange for the U.S. rights to Abstral, (1) we paid Orexo $10 million upfront, and a $5 million milestone payment in cash in October 2013 upon the approval by the FDA of a specified U.S. manufacturer of Abstral; and (2) we agreed to pay to Orexo: (a) three one-time future cash milestone payments based on our net sales of Abstral; and (b) a low double-digit royalty on future net sales. No further milestone or royalty payments will be due after the date on which all claims of the last remaining licensed patents expire (currently 2019) or become invalidated by a governmental agency.
On January 12, 2014, we acquired worldwide rights to anagrelide controlled release (CR) formulation, which we renamed GALE-401, through our acquisition of Mills Pharmaceuticals, LLC ("Mills") (see Note 5), and Mills became a wholly owned subsidiary. GALE-401 contains the active ingredient anagrelide, an FDA-approved product that has been in use since the late 1990s for the treatment of essential thrombocythemia (ET). Mills holds an exclusive license to develop and commercialize anagrelide CR formulation, pursuant to a license agreement with BioVascular, Inc. Under the terms of the license agreement, Mills has agreed to pay BioVascular, Inc. a mid-to-low single digit royalty on net revenue from the sale of licensed products as well as future cash milestone payments based on the achievement of specified regulatory milestones. Mills is also responsible for patent prosecution and maintenance.
22
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
On July 17, 2014, we entered into a definitive license and supply agreement with MonoSol Rx, LLC (MonoSol) for the U.S. commercial rights to Zuplenz® (ondansetron) Oral Soluble Film, an FDA-approved product for the prevention of highly and moderately emetogenic chemotherapy-induced nausea and vomiting (CINV), radiotherapy-induced nausea and vomiting (RINV), and post-operative nausea and vomiting (PONV). In exchange for the U.S. rights to Zuplenz, and in connection with the effectiveness of the license and transfer to us of the New Drug Application (NDA) for Zuplenz, we paid MonoSol a total of $5 million in cash and shares of our common stock in 2014 and $0.5 million in cash in 2015. In addition to these payments, we will pay MonoSol $0.25 million within 30 days after MonoSol’s payment of applicable fees relating to the notice of allowance by the United States Patent and Trademark Office of a U.S. patent with composition claims covering Zuplenz that extend beyond 2028, (ii) future cash milestone payments based on our achievement of specified “net sales” of Zuplenz, and (iii) a low double-digit royalty on future “net sales.”
Under the terms of the license agreement, we assumed responsibility for the commercialization of Zuplenz and for all regulatory and reporting matters in the U.S. We also agreed in the license and supply agreement to use our best commercial efforts to begin commercializing Zuplenz in the U.S. in accordance with a joint commercialization plan to be established by the company and MonoSol. We also agreed that, until net sales of Zuplenz exceed a specified minimum amount or a competing product has been approved by the FDA and is placed into the market for sale, we will maintain a specified minimum number of field sales force personnel on specified terms.
Under the license and supply agreement, MonoSol has the exclusive right to supply all of our requirements for Zuplenz, subject to certain conditions.
13. Significant Customers and Concentration of Credit Risk
The company is engaged in the business of developing and commercializing pharmaceutical products. As of March 31, 2015, the Company had one active commercial product, Abstral, available in six dosing strengths, and all sales reported are in the United States.
Sales to the following customers represented 10% or more of net revenue during at least one of the periods are presented as follows:
Three Months Ended March 31, | ||||||
Customer | 2015 | 2014 | ||||
Customer A | 33 | % | 51 | % | ||
Customer B | 25 | % | 14 | % | ||
Customer C | 20 | % | 12 | % | ||
Customer D | 20 | % | 4 | % | ||
Customer E | 1 | % | 15 | % | ||
The following customers represented 10% or more of total accounts receivable as of at least one of the balance sheet dates presented:
March 31, 2015 | December 31, 2014 | |||||
Customer | (Unaudited) | |||||
Customer A | 48 | % | 63 | % | ||
Customer B | 19 | % | 1 | % | ||
Customer C | 11 | % | 5 | % | ||
Customer D | 19 | % | 3 | % | ||
Customer E | 2 | % | 21 | % | ||
23
GALENA BIOPHARMA, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS - Continued
(Unaudited)
In addition to concentrations in our customer base, concentrations also exist with respect to the dispensing pharmacies to which our customers sell Abstral. For example 57% and 61% of our prescriptions, at wholesaler acquisition cost, were dispensed by our top 3 pharmacies for the three month periods ending March 31, 2015 and 2014, respectively.
14. Subsequent Events
The company evaluated all events or transactions that occurred after March 31, 2015 up through the date these financial statements were issued. Other than as disclosed elsewhere in the notes to the condensed consolidated financial statements, the company did not have any material recognizable or unrecognizable subsequent events.
24
ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
In this section, "Galena," “we,” “our,” “ours” and “us” refer to Galena Biopharma, Inc. and its consolidated subsidiaries, Apthera, Inc., or “Apthera,” and Mills Pharmaceuticals, LLC, or "Mills."
This management’s discussion and analysis of financial condition as of March 31, 2015 and results of operations for the three months ended March 31, 2015 and 2014, respectively, should be read in conjunction with management’s discussion and analysis of financial condition and results of operations included in our Annual Report on Form 10-K for the year ended December 31, 2014 which was filed with the SEC on March 5, 2015.
The discussion and analysis below includes certain forward-looking statements related to the commercialization of our products in the U.S., our future financial condition and results of operations and potential for profitability, the sufficiency of our cash resources, our ability to obtain additional equity or debt financing, possible partnering or other strategic opportunities for the development of our products, as well as other statements related to the progress and timing of our product commercialization and development activities, present or future licensing, collaborative or financing arrangements or that otherwise relate to future periods, which are all forward-looking statements as defined by the Private Securities Litigation Reform Act of 1995. These statements represent, among other things, the expectations, beliefs, plans and objectives of management and/or assumptions underlying or judgments concerning the future financial performance and other matters discussed in this document. The words “may,” “will,” “should,” “plan,” “believe,” “estimate,” “intend,” “anticipate,” “project,” and “expect” and similar expressions are intended to connote forward-looking statements. All forward-looking statements involve certain risks, including the uncertainties and other factors described in our Annual Report on Form 10-K for the year ended December 31, 2014 that could cause our actual commercialization efforts, financial condition and results of operations, and business prospects and opportunities to differ materially from these expressed in, or implied by, those forward-looking statements. We caution investors not to place significant reliance on the forward-looking statements contained in this report. These statements, like all statements in this report, speak only as of the date of this report (unless another date is indicated) and we undertake no obligation to update or revise forward-looking statements.
25
Overview
Galena Biopharma, Inc. (“we,” “us,” “our,” “Galena” or the “company”) is a biopharmaceutical company focused on developing and commercializing innovative, targeted oncology therapeutics that address major medical needs across the full spectrum of cancer care. Galena’s development portfolio ranges from mid- to late-stage clinical assets, including a robust immunotherapy program led by NeuVax™ (nelipepimut-S) currently in an international, Phase 3 clinical trial. The company’s commercial drugs include Abstral® (fentanyl) Sublingual Tablets and Zuplenz® (ondansetron) Oral Soluble Film. Collectively, our clinical and commercial strategy focuses on identifying and advancing therapeutic opportunities to improve cancer care, from direct treatment of the disease to the reduction of its debilitating side-effects.
We are seeking to build value for shareholders through pursuit of the following objectives:
• | Develop novel cancer immunotherapies to address unmet medical needs through the use of peptide based vaccines targeting well-established tumor antigens in the adjuvant, minimum residual disease setting, in high risk patients who are more likely to benefit from treatment via immunotherapy. Our immunotherapy programs currently seek to significantly decrease the risk of disease recurrence in breast cancer, gastric cancer, endometrial and ovarian cancers. |
• | Expand our development pipeline by enhancing the potential clinical and geographic footprint of our technologies. We can accomplish this through the initiation of additional clinical trials as well as through acquisition of additional development stage products in related oncology indications. We also seek to leverage valuable partnerships and collaborations, as well as investigator-sponsored trial arrangements, to maximize the scope of potential clinical opportunities in a cost effective and efficient manner. |
• | Maintain commercial capabilities to sell, market, and distribute oncology related pharmaceutical products in the U.S. through our established commercial infrastructure. This commercial strategy creates the opportunity to generate accretive cash flows to support our development programs, and also provides future leverage to support the potential commercialization of our clinical stage technologies in one of the world's largest economic markets. |
The chart below summarizes the current status of our pipeline: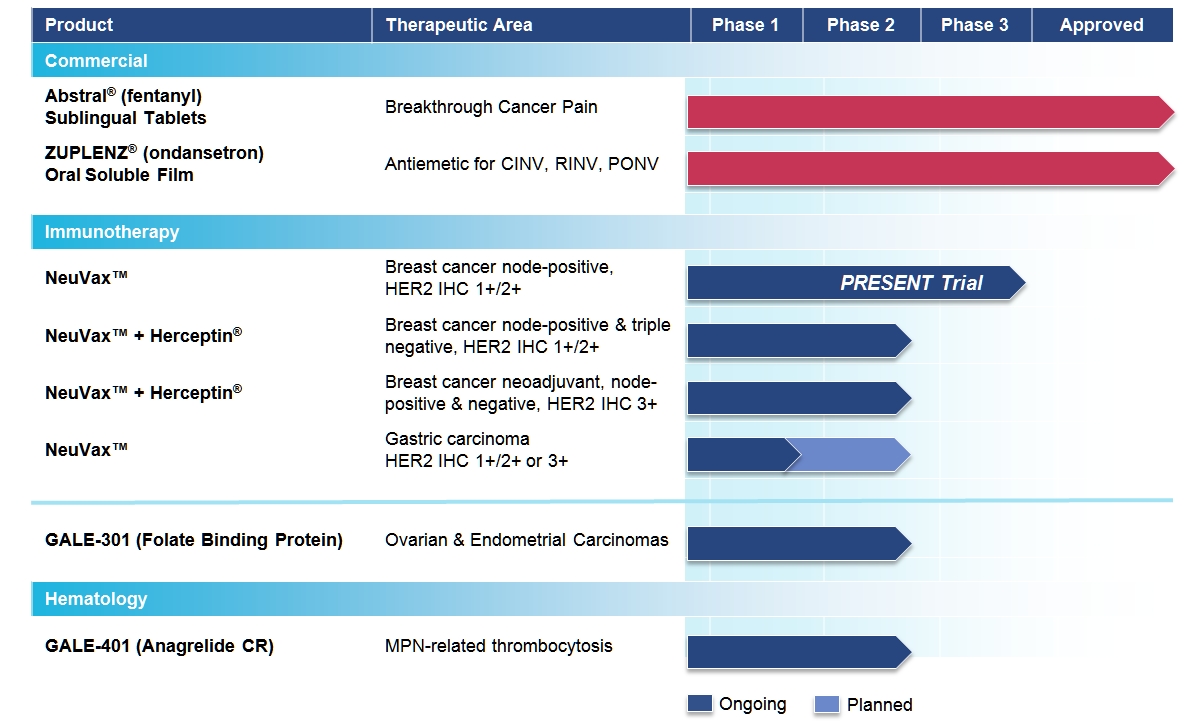
26
Novel Cancer Immunotherapies
Our targeted cancer immunotherapy approach is based upon preventing recurrence of cancer, which is becoming increasingly important as the number of cancer survivors continues to grow. Once a patient’s tumor becomes metastatic, the outcome is most often fatal, making the prevention of recurrence a potentially critical component of overall patient care. Our programs primarily target patients in the adjuvant (after-surgery) setting who have relatively healthy immune systems, but may still have minimal undetected residual disease.
Our therapies utilize immunologically active peptides combined with the immune adjuvant, recombinant human granulocyte macrophage-colony stimulating factor (rhGM-CSF), and work by harnessing the patient’s own immune system to seek out and attack any residual cancer cells. Using peptide immunogens has many potential clinical advantages, including a favorable safety profile, since these drugs may lack the toxicities typical of most cancer therapies. They also have the potential to evoke long-lasting protection through activation of the immune system via a convenient, intradermal mode of delivery. We are currently engaged in multiple clinical trials with NeuVax™ (nelipepimut-S) and GALE-301, or Folate Binding Protein (FBP), targeting the prevention of recurrence in breast, gastric, ovarian and endometrial cancers.
NeuVax™ (nelipepimut-S)
NeuVax™ (nelipepimut-S), our lead product candidate, is a targeted cancer immunotherapy and is being developed for the prevention of cancer recurrence in human epidermal growth factor receptor (HER2) expressing cancers. NeuVax is the immunodominant nonapeptide derived from the extracellular domain of the HER2 protein, a well-established target for therapeutic intervention in breast and gastric carcinomas. The NeuVax vaccine is combined with GM-CSF for injection under the skin, or intradermal administration. Data has shown that an increased presence of circulating tumor cells (CTCs) may predict Disease Free Survival (DFS) and Overall Survival (OS) - suggesting a dormancy of isolated micrometastases, which, over time, may lead to recurrence. After binding to the HLA A2 or A3 molecules on antigen presenting cells, the nelipepimut-S sequence stimulates specific cytotoxic T lymphocyte (CTLs). These activated CTLs recognize, neutralize and destroy, through cell lysis, HER2 expressing cancer cells, including undetected occult cancer cells and micrometastatic foci. The nelipepimut immune response can also generate CTLs to other immunogenic peptides through inter- and intra-antigenic epitope spreading.
Breast Cancer: According to the National Cancer Institute, over 230,000 women in the U.S. are diagnosed with breast cancer annually. While improved diagnostics and targeted therapies have decreased breast cancer mortality in the U.S., metastatic breast cancer remains incurable. Approximately 75% of breast cancer patients have tissue test positive for some increased amount of the HER2 receptor, which is associated with disease progression and decreased survival. Only approximately 20% to 30% of all breast cancer patients - those with HER2 immunohistochemistry (IHC) 3+ disease, or IHC 2+ and fluorescence in situ hybridization (FISH) positive - have an approved treatment option available. This leaves the majority of breast cancer patients with low-to-intermediate HER2 IHC 1+/2+ ineligible for targeted therapy and without an effective treatment option to prevent cancer recurrence.
We have multiple trials currently ongoing for NeuVax. For our pivotal, fully enrolled, Phase 3 PRESENT (Prevention of Recurrence in Early-Stage, Node-Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) trial, NeuVax is targeting the 30,000-40,000 of the 230,000 female breast cancer patients annually diagnosed in the U.S. who are at a higher risk of their breast cancer recurring, which we refer to as “disease recurrence,” after achieving “no evidence of disease” (NED) status, (or becoming a “survivor”) with standard-of-care therapy (surgery, chemotherapy, radiation). These high-risk patients have a particular molecular signature and disease status: HER2 IHC 1+/2+ (oncoprotein associated with aggressive tumor growth), node positive (disease present in the axillary lymph nodes prior to surgery), and HLA A2/A3 (human leukocyte antigen from A2/A3 patients who have the same loci of genes which represents approximately 65% of population). Up to 25% of resectable, node-positive breast cancer patients, having no radiographic evidence of disease following surgery and adjuvant chemo/radiation therapy, are expected to relapse within three years following diagnosis. The prognosis upon recurrence is very poor. These cancer patients presumably still had isolated, undetected tumor CTCs that led to a recurrence of cancer in the breast (local recurrence) or in another location (metastatic disease).
27
Gastric Cancer: Gastric cancer (also known as stomach cancer) is a disease in which the cells forming the inner lining of the stomach become abnormal and start to divide uncontrollably, forming a cancerous tumor mass. Cancer can develop in any of the five sections of the stomach. Symptoms and outcomes of the disease will vary depending on the location of the cancer. Stomach cancer is one of the leading causes of cancer deaths in several areas of the world, most notably Republic of Korea and other Asian countries. Annually, almost one million people will be diagnosed worldwide with stomach cancer and over 700,000 will die from the disease. More than 90% of stomach cancers are caused by adenocarcinomas, malignant cancers that originate in glandular tissues. Overexpression of the HER2 receptor occurs in approximately 20% of gastric and gastro-esophageal junction adenocarcinomas, predominantly those of the intestinal type. Overall, only approximately 28% of patients with stomach cancer live at least five years following diagnosis and new adjuvant treatments are needed to prevent disease recurrence.
We currently have a number of ongoing or planned clinical trials designed to expand the clinical and geographic footprint of NeuVax:
• | Phase 3 Ongoing: Our Phase 3 PRESENT (Prevention of Recurrence in Early- Stage, Node-Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) study is enrolling HER2 1+ and 2+ patients under a Special Protocol Assessment (SPA) granted by the U.S. Food and Drug Administration (FDA). The multinational, multicenter, randomized, double-blinded PRESENT trial is fully enrolled with trial sites in North America, Western and Eastern Europe, and Israel. Additional information on the study can be found at www.neuvax.com. |
• | Phase 2b Ongoing: A randomized, multicenter, investigator-sponsored, 300 patient Phase 2b clinical trial is enrolling HER2 1+/2+ node-positive and high-risk node-negative breast cancer patients to study NeuVax in combination with trastuzumab (Herceptin®; Genentech/Roche) in the adjuvant setting. This trial is partially funded by Genentech/Roche. |
• | Phase 2 Ongoing: An investigator-sponsored trial is ongoing to study NeuVax in combination with Herceptin. The study will enroll 100 patients who are neoadjuvantly treated node positive and negative HER2 IHC 3+ patients, not achieving a pathological complete response (pCR) or adjuvantly treated node positive HER2 IHC 3+ patients. Partial funding for this trial comes from the Department of Defense (DoD) through the Congressionally Directed Medical Research Program (CDMRP) via legislation known as the Defense Appropriations Act. The grant was awarded under a Breast Cancer Research Program (BCRP) Breakthrough Award given to the lead investigator for the trial. |
• | Phase 2 Planned: In January 2014, we partnered with Dr. Reddy’s Laboratories, Ltd. in India for the commercialization of NeuVax in that region. Dr. Reddy’s is responsible for running a Phase 2 gastric cancer trial of NeuVax in India that is expected to initiate in 2016. |
GALE-301 (folate binding protein or FBP)
Our second immunotherapy product candidate, GALE-301, targets folate binding protein receptor-alpha, a well-validated therapeutic target, which is highly over-expressed (20-80 fold) in ovarian, endometrial and breast cancers. GALE-301 is an immunogenic peptide and can stimulate CTLs to recognize and destroy FBP-expressing cancer cells. GALE-301 consists of an FBP peptide combined with GM-CSF, and is currently in a Phase 2a clinical trial for the prevention of recurrence in patients with ovarian and endometrial cancers. Current treatments for these diseases are principally with chemotherapeutic agents and patients suffer a high recurrence rate; and, most patients relapse with an extremely poor prognosis. Preliminary promising results from the Phase 2a clinical trial of GALE-301 were presented in November 2014 at the Society for Immunotherapy of Cancer conference and showed a 38% reduction in relative risk of recurrence, and that the agent was well-tolerated with primarily Grade 1 and 2 toxicities and elicited a strong in vivo immune response. We expect to present top line data from the Phase 2a trial mid-year 2015.
28
Ovarian and Endometrial Cancer: Ovarian cancer occurs in more than 22,000 patients per year in the U.S. and is the most lethal gynecologic cancer. Despite the incidence of ovarian cancer being only approximately 10% of that of breast cancer, the number of patients who die from ovarian cancer is nearly 50% that of breast cancer. Due to the lack of specific symptoms, the majority of ovarian cancer patients are diagnosed at later stages of the disease. For their treatment, these patients typically have their tumors surgically debulked to minimal residual disease, and then are treated with platinum- and/or taxane-based chemotherapy. While most patients respond to this treatment regimen and become clinically free-of-disease, the majority of these patients will relapse, and once the disease recurs, the treatment options and successes drop dramatically. Endometrial cancer is the most common gynecologic cancer and occurs in more than 46,000 women with more than 8,000 deaths in the U.S. annually. There are two basic types of endometrial cancer: endometrioid and papillary serous. The latter has a much more aggressive clinical course and the majority of these patients will die of this form of the disease.
Hematology
GALE-401 (anagrelide controlled release (CR))
In January 2014, we announced the acquisition of the worldwide rights to anagrelide controlled release (CR), which we renamed GALE-401, through our acquisition of Mills Pharmaceuticals, LLC. GALE-401 contains the active ingredient anagrelide, an FDA-approved product, for the treatment of patients with myeloproliferative neoplasms (MPNs) to lower abnormally elevated platelet levels. The currently available immediate release (IR) version of anagrelide causes adverse events that are believed to be dose and plasma concentration dependent. Therefore, reducing the maximum concentration (Cmax) is hypothesized to reduce the side effects, but preserve efficacy.
Multiple Phase 1 studies in 98 healthy subjects have shown GALE-401 reduces the Cmax of anagrelide following oral administration, appears to be well tolerated at the doses administered, and to be capable of reducing platelet levels. The Phase 1 program provided the desired PK/PD (pharmacokinetic/pharmacodynamic) profile to enable the initiation of the ongoing Phase 2 proof-of-concept trial. The Phase 2 trial enrolled 18 patients in the United States for the treatment of thrombocytosis, or elevated platelet counts in patients with MPNs. Phase 2 top-line safety and efficacy data will be presented this year. Based on a regulatory meeting with the FDA, Galena believes a 505(b)(2) regulatory filing is an acceptable pathway for development and potential approval of GALE-401, with the reference drug Agrylin® (anagrelide; Shire Pharmaceuticals).
Myeloproliferative neoplasms: MPNs are a closely related group of hematological malignancies in which the bone marrow cells that produce the body's blood cells develop and function abnormally. The main myeloproliferative neoplasms are Polycythemia Vera (PV), Essential Thrombocythemia (ET), Primary Myelofibrosis (PMF), and Chronic Myelogenous Leukemia (CML), all of which are associated with high platelet counts. The MPNs are progressive blood cancers that can strike anyone at any age, and for which there is no known cure.
Commercial Capabilities
Abstral® (fentanyl) Sublingual Tablets
Our first commercial product, Abstral® (fentanyl) Sublingual Tablets, is an important treatment option for inadequately controlled breakthrough cancer pain (BTcP), which affects more than 50% of all cancer patients. Abstral is approved by the FDA, and is a sublingual (under the tongue) tablet for the management of breakthrough pain in patients with cancer, 18 years of age and older, who are already receiving, and who are tolerant to, opioid therapy for their persistent baseline cancer pain. The Abstral formulation delivers the analgesic power and increased bioavailability of micronized fentanyl in a convenient sublingual tablet that is designed to dissolve under the tongue in seconds and provide relief of breakthrough pain within minutes. Abstral is a transmucosal immediate release fentanyl (TIRF) product with product class oversight by the TIRF Risk Evaluation and Mitigation Strategy (REMS) access program. Abstral is manufactured for us by contract manufacturers and we distribute and sell Abstral in the U.S. through our commercial organization.
29
Zuplenz® (ondansetron) Oral Soluble Film
In July 2014 we expanded our commercial portfolio through the licensing of our second commercial product, Zuplenz® (ondansetron) Oral Soluble Film, from MonoSol Rx, LLC. Zuplenz is approved by the FDA in adult patients for the prevention of highly and moderately emetogenic chemotherapy-induced nausea and vomiting (CINV), radiotherapy-induced nausea and vomiting (RINV), and post-operative nausea and vomiting (PONV). Zuplenz is also approved in pediatric patients treated with moderately emetogenic CINV. Nausea and vomiting are two of the most common side-effects experienced by post-surgery patients and patients receiving chemotherapy or radiation. It is estimated that up to 90% of chemotherapy and up to 80% of radiotherapy patients will experience CINV and RINV, respectively.
Zuplenz utilizes MonoSol’s proprietary PharmFilm® technology, an oral soluble film that dissolves on the tongue in less than 30 seconds. Zuplenz eliminates the burden of swallowing pills during periods of emesis, may be advantageous for patients with oral irritation, and may increase patient adherence and the patient's ability to keep the medication down without vomiting. The active pharmaceutical ingredient in Zuplenz, ondansetron, belongs to a class of medications called serotonin 5-HT3 receptor antagonists and works by blocking the action of serotonin, a natural substance that may cause nausea and vomiting. Ondansetron is the most widely prescribed drug in this class of anti-emetics, and used broadly across the oncology spectrum. MonoSol will exclusively manufacture Zuplenz for us for sale in the U.S. through our commercial organization.
30
Recent Operational Developments (in reverse chronological order)
NeuVax Phase 3 PRESENT Over-Enrollment Completed - We announced the completion of enrollment in our NeuVax Phase 3 PRESENT clinical trial.
On April 14, 2015 we announced the completion of enrollment in the NeuVax™ (nelipepimut-S) Phase 3 PRESENT (Prevention of Recurrence in Early-Stage, Node-Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) clinical trial. NeuVax is a first-in-class, HER2-directed cancer immunotherapy under evaluation to prevent breast cancer recurrence after standard of care treatment in the adjuvant setting. As anticipated, we over-enrolled the trial by 7.7% with a total of 758 patients now in the intent-to-treat (ITT) population. The protocol for the PRESENT trial, being conducted under an FDA approved Special Protocol Assessment (SPA), called for 700 patients; and, the Company expects this higher number of ITT patients will increase the confidence in both the timing and quality of the statistics and the final outcome of the trial. The primary endpoint is currently expected to be reached in 2018, after the last patient dosed reaches her 36th month of treatment, or a total of 141 events (recurrence or death) occur, whichever comes later.
Expanded Patient Population in NeuVax Combination Trial - We announced the expansion of the patient population in the NeuVax and trastuzumab combination trial in HER2 1+/2+ patients to include HLA A24+ or HLA-A26+ patients.
On March 26, 2015, we announce that human leukocyte antigen (HLA) - A24+ or HLA-A26+ women are now eligible for enrollment into the ongoing Phase 2b clinical trial with NeuVax™ (nelipepimut-S) in combination with trastuzumab (Herceptin®; Genentech/Roche). The trial evaluates node positive and triple negative, node negative breast cancer patients with immunohistochemistry (IHC) HER2 1+/2+ expressing tumors who are disease-free after standard of care therapy.
Enrolled 700th Patient in NeuVax Phase 3 PRESENT Clinical Trial - We announced enrollment of the 700th patient in our Phase 3 PRESENT clinical trial.
On February 9, 2015, we announced the enrollment of the 700th patient in the NeuVax™ (nelipepimut-S) Phase 3 PRESENT (Prevention of Recurrence in Early-Stage, Node-Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) clinical trial. Seven hundred is the patient enrollment target as defined by the PRESENT Phase 3 clinical trial protocol.
31
Results of Operations for the Three Months Ended March 31, 2015 and 2014
For the three months ended March 31, 2015, our net loss was approximately $10.5 million compared with a net loss of $2.5 million for the three months ended March 31, 2014. The net loss increased by $8.0 million, or approximately 315%, primarily attributable to a $9.8 million non-cash gain on our warrant liability for the three months ended March 31, 2014 compared to a $1.2 million non-cash gain on our warrant liability for the three months ended March 31, 2015. Additional reasons are discussed below.
Abstral is our first commercial product and revenue was recorded for the first time during the year ended December 31, 2013. We acquired our second commercial product, Zuplenz, in the third quarter of 2014 and are expected to launch the product in the second quarter of 2015. There was no gross or net revenue from the sale of Zuplenz in the first quarter of 2015 or in 2014. For these reasons, we expect our future results of operations may differ materially from our historical results. We expect to continue to incur significant costs and expenses in connection with our commercial operations in the U.S. and must achieve higher net sales before realizing a profit from the sale and distribution of our commercial products. Further analysis of the changes and trends in our operating results are discussed below.
Net Revenue
The company recognizes revenue from the sale of Abstral to wholesale pharmaceutical distributors and retail pharmacies, net of product-related discounts, allowances, product returns, rebates, chargebacks, and patient assistance benefits, as applicable.
Net revenue for the three months ended March 31, 2015 and 2014, was as follows (in thousands):
Three Months Ended March 31, | ||||||||||
2015 | 2014 | % Change | ||||||||
Net revenue | $ | 2,750 | $ | 2,173 | 27 | % | ||||
Gross revenue, gross to net deductions, and net revenue for the three months ended March 31, 2015 and 2014 were as follows (in thousands):
Three Months Ended March 31, | |||||||||||||
2015 | 2014 | ||||||||||||
$ | % of Gross Revenue | $ | % of Gross Revenue | ||||||||||
Gross revenue | $ | 4,204 | 100 | % | $ | 4,863 | 100 | % | |||||
Gross to net deductions | 1,454 | 35 | % | 2,690 | 55 | % | |||||||
Net revenue | $ | 2,750 | 65 | % | $ | 2,173 | 45 | % | |||||
Net revenue for the three months ended March 31, 2015 increased 27% compared to the three months ended March 31, 2014. The increase was a result of a 36% improvement in the gross to net deductions, partially offset by a 14% decrease in gross revenue. In March of 2014 we launched the Galena Patient Services (GPS) program, a full service support program designed to navigate patient access to Abstral that is coordinated through a third party vendor. Along with the launch of GPS, we also made changes to our patient assistance program (PAP) to reduce the use of free product vouchers and rely more heavily upon an expedited prior authorization process. The success of the GPS program has resulted in a significant improvement in our gross to net deductions as a percent of gross revenue.
Despite a decrease in gross revenues, overall prescription demand increased 8% for the three months ended March 31, 2015 compared to three months ended March 31, 2014. More importantly, non-voucher prescriptions increased 113% for the three months ended March 31, 2015 compared to the three months ended March 31, 2014, as a direct result of the successful implementation of our GPS program and the changes to our PAP program. The decrease in our gross to net deduction is attributable to the reduction in our prescriptions being filled with vouchers. Our current expectations for 2015 net revenue from the sale of our commercial products is approximately $15 million to $18 million, but there is no assurance we will achieve our
32
expectations. We acquired our second commercial product, Zuplenz, in the third quarter of 2014 and are expected to launch the product in the second quarter of 2015.
Cost of Revenue and Amortization of Certain Acquired Intangible Assets
Cost of revenue and amortization of certain acquired intangible assets for the three months ended March 31, 2015 and 2014, respectively, were as follows (dollars in thousands):
Three Months Ended March 31, | |||||||||||||
2015 | % of net revenue | 2014 | % of net revenue | ||||||||||
Cost of revenue (excluding amortization of certain acquired intangible assets: | |||||||||||||
Abstral royalties | $ | 330 | 12 | % | $ | 259 | 12 | % | |||||
Direct product costs and related overhead | 19 | 1 | % | 29 | 1 | % | |||||||
Other cost of revenue | 44 | 2 | % | 43 | 2 | % | |||||||
Total cost of revenue | $ | 393 | 14 | % | $ | 331 | 15 | % | |||||
Amortization of certain acquired intangible assets | $ | 146 | 5 | % | $ | 91 | 4 | % | |||||
Cost of revenue, which excludes the amortization of certain acquired intangible assets, was $0.4 million and $0.3 million for the three months ended March 31, 2015 and 2014, respectively. Variable cost of revenue includes the royalty due to Orexo and product costs. Product related overhead and other cost of revenue are fixed in nature, and will decrease or increase as a percentage of net revenue as net revenue increases or decreases, respectively. Cost of revenue improved 7% as a percent of net revenue, which is attributable to the 27% increase in net revenue for the three months ended March 31, 2015 compared to three months ended March 31, 2014.
Amortization of certain acquired intangible assets was $146,000 and $91,000 for the three months ended March 31, 2015 and 2014, respectively. Amortization of certain acquired intangible assets is a non-cash variable cost based on net revenue during the period.
Research and Development Expense
Research and development expense consists primarily of clinical trial expenses, compensation-related costs for our employees dedicated to research and development activities, compensation paid to our Scientific Advisory Board (“SAB”) members, and licensing fees and patent prosecution costs. Research and development expense for the three months ended March 31, 2015 and 2014, respectively, was as follows (dollars in thousands):
Three Months Ended March 31, | ||||||||||
2015 | 2014 | % Change | ||||||||
Research and development expense | $ | 5,910 | $ | 6,770 | (13 | )% | ||||
The 13% decrease for the three months ended March 31, 2015 compared to the three months ended March 31, 2014 in research and development expense was primarily related to the completion of our enrollment efforts for the Phase 3 PRESENT clinical trial. We expect research and development expense related to our PRESENT trial to decrease throughout 2015 as we transition to the monitoring and follow-up phase. The expected decrease in costs could be partially offset by the increase in research and development expense related to enrollment efforts in our NeuVax combination clinical trials, ongoing trial costs related to our GALE-301 Phase 2 clinical trial, and completion of our Phase 2 clinical trial of the GALE-401 program.
33
Selling, General and Administrative Expense
Selling, general and administrative expense includes compensation-related costs for our employees dedicated to sales and marketing, general and administrative activities, legal fees, audit and tax fees, consultants and professional services, and general corporate expenses. Selling, general and administrative expense for the three months ended March 31, 2015 and 2014, respectively, was as follows (dollars in thousands):
Three Months Ended March 31, | ||||||||||
2015 | 2014 | % Change | ||||||||
Selling, general and administrative expense | $ | 7,427 | $ | 6,830 | 9 | % | ||||
The 9% increase for the three months ended March 31, 2015 compared to the three months ended March 31, 2014 in selling, general and administrative expense was driven by the ramp up of our commercial operations leading into the launch of Zuplenz, the expansion of our management team and infrastructure needed to support the expanded commercial operations and development pipeline, as well as non-recurring legal and other professional fees incurred during the period, partially offset by a decrease in our non-cash stock-based compensation expense of $0.6 million and $1.7 million for the three months ended March 31, 2015 and 2014, respectively.
Non-Operating Income
Non-operating income for the three months ended March 31, 2015 and 2014, respectively, was as follows (dollars in thousands):
Three Months Ended March 31, | ||||||||||
2015 | 2014 | % Change | ||||||||
Non-operating income | $ | 589 | $ | 9,313 | (94 | )% | ||||
The decrease to our non-operating income during the three months ended March 31, 2015 compared to the three months ended March 31, 2014 was primarily due to a decrease in the change in fair value of warrants accounted for as liabilities. This decrease in the estimated fair value of our warrant liabilities was primarily due to the decrease in our common stock price, which is one of the most impactful inputs into the pricing model we use to estimate the fair value of our warrant liabilities. The decrease to our non-operating income (expense) during the three months ended March 31, 2015 was primarily due to a decrease in the fair value of warrants accounted for as liabilities, which was attributable to the decrease in our common stock price.
Income Taxes
For the three months ended March 31, 2015 and 2014, there was no income tax benefit or expense recognized.
34
Liquidity and Capital Resources
We had cash and cash equivalents of approximately $52.9 million as of March 31, 2015, compared with $23.7 million as of December 31, 2014.
The increase of approximately $29.2 million in our cash and cash equivalents from December 31, 2014 to March 31, 2015 was attributable primarily to $42.2 million in net proceeds from issuance of common stock and warrants to purchase common stock, partially offset by $11.6 million used in operating activities, $0.9 million in payments on long-term debt, and $0.5 million for a milestone related to our acquisition of U.S. rights to Zuplenz.
On March 18, 2015, we announced the closing of our underwritten public offering of 24,358,974 shares of common stock and 12,179,487 warrants to purchase our common stock at an exercise price of $2.08. The underwriters also exercised their over-allotment option to purchase warrant to purchase an aggregate of 1,826,923 shares of our common stock. On April 10, 2015 the underwriters exercised their option to purchase an additional 3,653,846 shares of common stock for additional net proceeds of $5.4 million. The total net proceeds to us were approximately $40.8 million.
We expect to continue to incur operating losses as we continue to commercialize Abstral, launch commercialization of Zuplenz in the U.S. and advance our product candidates through the drug development and regulatory process. We will need to generate significant revenue to achieve profitability and may never do so. In the absence of revenue from the commercialization of Abstral, Zuplenz or our product candidates, our potential sources of operational funding are proceeds from the sale of equity and funded research and development payments and payments received under partnership and collaborative agreements.
We believe that our existing cash and cash equivalents, along with revenue from our commercial operations, should be sufficient to fund our operations for at least one year. This projection is based on our current planned operations and revenue expectations and is subject to changes in our plans and uncertainties inherent in our business, and we may need to seek to replenish our existing cash and cash equivalents sooner than we project. There is no guarantee that we will generate sufficient revenue from the sale of Abstral and Zuplenz to become profitable or that any debt, additional equity or other funding will be available to us on acceptable terms, or at all. If we fail to generate adequate revenue or obtain additional funding when needed, we would be forced to scale back, or terminate, our operations or to seek to merge with or to be acquired by another company.
Net Cash Flow from Operating Activities
Net cash used in operating activities was approximately $11.6 million for the three months ended March 31, 2015, compared with $8.0 million for the three months ended March 31, 2014. The increase in cash used in operating activities of corresponds to the year-over-year changes in selling, general and administrative expenses and research and development expenses noted above.
Net Cash Flow from Investing Activities
Net cash used in investing activities was $0.5 million for the three months ended March 31, 2015, compared with $2.0 million for the three months ended March 31, 2014. The decrease was due to the $2.1 million initial payment for GALE-401 rights during the three months ended March 31, 2014 compared to the $0.5 million milestone payment for U.S. rights to Zuplenz during the three months ended March 31, 2015.
Net Cash Flow from Financing Activities
Net cash provided by financing activities was $41.3 million for the three months ended March 31, 2015, compared with $14.7 million for the three months ended March 31, 2014. The increase was primarily attributable to $42.2 million in net proceeds from the issuance of common stock and warrants to purchase common stock during the three months ended March 31, 2015 compared to net proceeds of common stock warrant exercises of $10.6 million and net proceeds of common stock option exercises of $4.1 million during the three months ended March 31, 2014.
35
Off-Balance Sheet Arrangements
We have not entered into any off-balance sheet financing arrangements other than operating leases.
Critical Accounting Policies and Estimates
In our Annual Report on Form 10-K for the year ended December 31, 2014, we disclosed our critical accounting policies and estimates upon which our financial statements are derived. There have been no changes to these policies since December 31, 2014 that are not included in Note 1 of the accompanying condensed consolidated financial statements for the three months ended March 31, 2015. Readers are encouraged to read our Annual Report on Form 10-K in conjunction with this report.
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
The primary objective of our investment activities is to preserve capital. We do not utilize hedging contracts or similar instruments.
We are exposed to certain market risks relating primarily to interest rate risk on our cash and cash equivalents and risks relating to the financial viability of the institutions which hold our capital and through which we have invested our funds. We manage the latter risks by investing primarily in money market mutual funds.
In addition, we are exposed to foreign currency exchange rate fluctuations relating to payments we make to certain vendors and suppliers and license partners using foreign currencies. We do not hedge against foreign currency risks. Consequently, changes in exchange rates could adversely affect our operating results and stock price. Such losses have not been significant to date.
ITEM 4. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
As of the end of the period covered by this quarterly report on Form 10-Q, our principal executive officer and our principal accounting officer (the “Certifying Officers”), evaluated the effectiveness of our disclosure controls and procedures. Disclosure controls and procedures are controls and procedures designed to reasonably assure that information required to be disclosed in our reports filed under the Securities Exchange Act of 1934 (the “Exchange Act”), such as this quarterly report on Form 10-Q, is recorded, processed, summarized and reported within the time periods specified in the SEC rules and forms. Disclosure controls and procedures are also designed to reasonably assure that such information is accumulated and communicated to our management, including the Certifying Officers, as appropriate to allow timely decisions regarding required disclosure. Based on these evaluations, the Certifying Officers have concluded, that, as of the end of the period covered by this quarterly report on Form 10-Q:
(a) | our disclosure controls and procedures were effective to provide reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act was recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms; and |
(b) | our disclosure controls and procedures were effective to provide reasonable assurance that material information required to be disclosed by us in the reports we file or submit under the Exchange Act was accumulated and communicated to our management, including the Certifying Officers, as appropriate to allow timely decisions regarding required disclosure. |
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting that occurred during the three months ended March 31, 2015 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
36
PART II
ITEM 1. LEGAL PROCEEDINGS
In early 2014, several purported shareholder derivative complaints were filed against our company, as nominal defendant, and certain of our officers and directors in the Circuit Court of Oregon for the County of Multnomah, the U.S. District Court for the District of Oregon, and the Delaware Court of Chancery. On April 11, 2014, the derivative complaints pending in the U.S. District Court for the District of Oregon were consolidated in the matter of In Re Galena Biopharma, Inc. Derivative Litigation, No. 3:14-cv-382-SI (D. Or.), and on August 25, 2014, the lead plaintiffs filed a consolidated amended complaint. On July 21, 2014, all of the derivative complaints pending in the Delaware Court of Chancery were consolidated in the matter of In re Galena Biopharma, Inc. Stockholder Derivative Litigation, Consolidated C.A. No. 9715-VCN (Del. Ch.). On February 10, 2015, the lead plaintiffs in the derivative complaints pending in the Delaware Court of Chancery voluntarily dismissed their action without prejudice. As a result of this dismissal, and at the recommendation of the special litigation committee of the board established on July 21, 2014 to investigate the derivative claims, on February 26, 2015 our board of directors disbanded the special litigation committee.
The operative complaints allege, among other things, breaches of fiduciary duties and abuse of control by the officers and directors in connection with public statements purportedly issued by us or on our behalf and sales of our common stock by our officers and directors in January and February of 2014, improper stock-option grants, and excessive compensation of our non-employee directors.
Also, five purported securities class action complaints filed in the U.S. District Court for the District of Oregon have been consolidated into a single action, In re Galena Biopharma, Inc. Securities Litigation, No. 3:14-cv-367-SI (D. Or.), and a lead plaintiff has been appointed. On October 31, 2014, the lead plaintiff filed a consolidated amended complaint, which alleges, among other things, that our company and certain of our officers and directors violated the federal securities laws by making materially false and misleading statements and omissions in press releases and in filings with the SEC arising out of the same circumstances that are the subject of the derivative actions described above, and which alleges that certain of our officers and directors sold company stock while in possession of material non-public information.
We intend to vigorously defend against and seek resolution to the foregoing claims. At March 31, 2015, we have not recorded any liabilities with respect to the claims in our consolidated financial statements. We believe that claims are covered under our liability insurance, and we have notified our insurance carriers of the claims. The insurers have responded by requesting additional information and by reserving their rights under the policies, including the rights to deny coverage under various policy exclusions. Subject to their reservation of rights, we are being reimbursed by our insurer for substantially all legal fees relating to our defense of the claims.
We are aware that the SEC is investigating certain matters relating to the use of certain outside investor-relations professionals by us and other public companies. We have been in contact with the SEC staff through our counsel and are cooperating with the investigation.
Litigation is inherently uncertain, and there is no assurance as to the outcome of the matters described above. We could incur substantial unreimbursed legal fees, settlements, judgments, and other expenses in connection with these or other legal and regulatory proceedings that may not qualify for coverage under, or may exceed the limits of, our applicable directors and officers liability insurance policies and could have a material adverse effect on our financial condition, liquidity, and results of operations. These matters also may distract the time and attention of our officers and directors or divert our other resources away from our ongoing commercial and development programs. An unfavorable outcome in any of these matters could damage our business and reputation or result in additional claims or proceedings against us.
37
ITEM 1A. RISK FACTORS
In addition the risk factors included in our Annual Report on Form 10-K for the year ended December 31, 2014 filed with the SEC, you should consider the following new or updated risk factors:
Our products are subject to ongoing and continued regulatory review, which may result in significant expense and adversely affect our commercialization activities.
Even after U.S. regulatory approval for a product, the FDA may still impose significant restrictions on the approved indicated uses for which the product may be marketed or on the conditions of approval. For example, a product’s approval may contain requirements for potentially costly post-approval studies and surveillance, including Phase 4 clinical trials, to monitor the safety and efficacy of the product. We will also be subject to ongoing FDA obligations and continued regulatory review with respect to the manufacturing, processing, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for our products. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMPs, good clinical practices and good laboratory practices.
In the case of Abstral, we and our contract manufacturers are also subject to ongoing DEA regulatory obligations, including, among other things, annual registration renewal, security, recordkeeping, theft and loss reporting, periodic inspection and annual quota allotments for the raw material for commercial production of Abstral. In addition, manufacturers of drug products and its facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. If we or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where, or processes by which, the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturer or us, including requiring product recall, notice to physicians, withdrawal of the product from the market or suspension of manufacturing.
In November 2014, we were inspected by the FDA and received a Form FDA 483 containing observations from that inspection. The Form FDA 483 noted certain deficiencies pertaining to the manufacture and post-marketing activities for Abstral and Zuplenz. These observations related to the establishment and formal designation of a quality unit, the formal written documentation of responsibilities and procedures applicable to the quality unit, the development of written procedures for the evaluation and reporting to the FDA of post marketing adverse drug experiences, and the periodic submission to the FDA of non-alert adverse drug experiences for Abstral. The issues noted in the Form FDA 483 had previously been identified and addressed by our management as part of an internal review of our systems, practices and procedures governing the areas of vendor oversight, quality, and regulatory compliance. Our response to the Form FDA 483 described the corrective actions that we have taken and will continue to address the FDA’s observations. However, in April of 2015, the FDA issued a written warning letter based on subset of the issues identified in the Form FDA 483, specifically regarding our our overall pharmacovigilance process and our adverse events reporting systems. We responded to the warning letter, advising the FDA of the corrective actions we are taking to address all the matters covered in the warning letter.
If we or the manufacturing facilities for our products fail to comply with applicable regulatory requirements, a regulatory agency may:
• | impose restrictions on the marketing or manufacturing of our products, suspend or withdraw product approvals or revoke necessary licenses; |
• | issue warning letters, show cause notices or untitled letters describing alleged violations, which may be publicly available; |
• | commence criminal investigations and prosecutions; |
• | impose injunctions, suspensions or revocations of necessary approvals or other licenses; |
• | impose fines or other civil or criminal penalties; |
• | deny or reduce quota allotments for the raw material for commercial production of our products; |
• | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
38
• | seize our products or require us to initiate a product recall. |
In addition, our product labeling, advertising and promotion are subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription drug products. In particular, a drug product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling, although the FDA does not regulate the prescribing practices of physicians. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution.
The FDA’s regulations, policies or guidance may change and new or additional statutes or government regulations may be enacted that could further restrict or regulate post-approval activities relating to Abstral. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action. If we are not able to achieve and maintain regulatory compliance, we may not be permitted to market our products, which would adversely affect our ability to generate revenue and achieve or maintain profitability.
We may not be able to increase our authorized shares, and as a result, may not be able to obtain sufficient financing develop our product candidates.
We believe that our existing cash and cash equivalents, along with revenue from Abstral sales, should be sufficient to fund our operations for at least one year. This projection is based on our current planned operations and revenue expectations and is subject to changes in our plans and uncertainties inherent in our business, and we may need to seek to replenish our existing cash and cash equivalents sooner than we project. In the future, we may be dependent on obtaining further financing from third parties in order to maintain our operations and to meet our financial obligations. Based on the number of common shares outstanding, along with the shares reserved for the exercise of warrants, stock option and stock based compensation programs, we will need our stockholders approval to amend our Certificate of Incorporation to increase the number of shares authorized. We cannot assure that we will obtain such approval, or that additional funding to maintain our operations and to meet our obligations to our licensors will be available to us in the future on acceptable terms, or at all. If we fail to obtain additional funding when needed, we would be forced to scale back, or terminate, our operations, or to seek to merge with or to be acquired by another company.
ITEM 6. EXHIBITS
Exhibit Number | Description | ||
31.1 | Sarbanes-Oxley Act Section 302 Certification of Mark W.Schwartz, Ph.D. | ||
31.2 | Sarbanes-Oxley Act Section 302 Certification of Ryan M. Dunlap. | ||
32.1 | Sarbanes-Oxley Act Section 906 Certification of Mark W. Schwartz, Ph.D., and Ryan M. Dunlap. | ||
101.INS | XBRL Instance Document. | ||
101.SCH | XBRL Taxonomy Extension Schema. | ||
101.CAL | XBRL Taxonomy Extension Calculation. | ||
101.DEF | XBRL Taxonomy Extension Definition. | ||
101.LAB | XBRL Taxonomy Extension Label. | ||
101.PRE | XBRL Taxonomy Extension Presentation. | ||
101.PRE | XBRL Taxonomy Extension Presentation. | ||
* | Indicates a management contract or compensatory plan or arrangement. |
39
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
GALENA BIOPHARMA, INC. | |||
By: | /s/ Mark W. Schwartz | ||
Mark W. Schwartz, Ph.D. | |||
President and Chief Executive Officer | |||
Date: May 7, 2015 | |||
By: | /s/ Ryan M. Dunlap | ||
Ryan M. Dunlap | |||
Vice President and Chief Financial Officer | |||
Date: May 7, 2015 | |||
40