Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Power of The Dream Ventures Inc | Financial_Report.xls |
| EX-32 - EXHIBIT 32.2 - Power of The Dream Ventures Inc | ex322.htm |
| EX-32 - EXHIBIT 32.1 - Power of The Dream Ventures Inc | ex321.htm |
| EX-31 - EXHIBIT 31.2 - Power of The Dream Ventures Inc | ex312.htm |
| EX-31 - EXHIBIT 31.1 - Power of The Dream Ventures Inc | ex311.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
Q ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended: December 31, 2014
or
¨ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
| POWER OF THE DREAM VENTURES, INC. |
| (Exact name of registrant as specified in its charter) |
| Delaware | 001-52289 | 51-0597895 |
|
(State or other jurisdiction of incorporation or organization) |
(Commission File Number) |
(I.R.S. Employer Identification Number) |
| 1095 Budapest, Soroksari, ut 94-96, Hungary |
| (Address of principal executive offices, including zip code) |
| +36-1-456-6061 |
| (Registrant’s telephone number, including area code) |
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act: Common Stock, $0.001 par value
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes þ No ¨
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the last 90 days.
Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. Yes ¨ No þ
| 1 |
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large Accelerated Filer ¨ | Accelerated Filer ¨ |
| Non-Accelerated Filer ¨ | Smaller reporting company þ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
Issuer’s revenues for its most recent fiscal year were approximately $0.
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant on June 29, 2014, based on a closing price of $0.10 was approximately $3,500,000 As of March 31, 2015, the registrant had 46,500,896 shares of its common stock, par value $0.001 per share, outstanding.
Documents Incorporated By Reference: None.
| 2 |
POWER OF THE DREAM VENTURES, INC.
FOR THE FISCAL YEAR ENDED
DECEMBER 31, 2014
TABLE OF CONTENTS
| Page | ||
| PART I | ||
| Item 1. | Business. | 5 |
| Item 1A. | Risk Factors. | 34 |
| Item 1B. | Unresolved Staff Comments. | 56 |
| Item 2. | Properties. | 56 |
| Item 3. | Legal Proceedings. | 56 |
| Item 4. | Mine Safety Disclosures. | 57 |
| PART II | ||
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. | 57 |
| Item 6. | Selected Financial Data. | 58 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results Of Operations. | 58 |
| Item 7A. | Quantitative And Qualitative Disclosures About Market Risk. | 62 |
| Item 8. | Financial Statements and Supplementary Data. | 62 |
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. | 62 |
| Item 9A. | Controls and Procedures. | 62 |
| Item 9B. | Other Information. | 63 |
| PART III | ||
| Item 10. | Directors, Executive Officers and Corporate Governance. | 64 |
| Item 11. | Executive Compensation. | 65 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. | 66 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. | 67 |
| Item 14. | Principal Accounting Fees and Services. | 67 |
| PART IV | ||
| Item 15. | Exhibits, Financial Statement Schedules. | 68 |
| 3 |
FORWARD LOOKING STATEMENTS
Included in this Form 10-K are “forward-looking” statements, as well as historical information. Although we believe that the expectations reflected in these forward-looking statements are reasonable, we cannot assure you that the expectations reflected in these forward-looking statements will prove to be correct. Our actual results could differ materially from those anticipated in forward-looking statements as a result of certain factors, including matters described in the section titled “Risk Factors.” Forward-looking statements include those that use forward-looking terminology, such as the words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “project,” “plan,” “will,” “shall,” “should,” and similar expressions, including when used in the negative. Although we believe that the expectations reflected in these forward-looking statements are reasonable and achievable, these statements involve risks and uncertainties and we cannot assure you that actual results will be consistent with these forward-looking statements. We undertake no obligation to update or revise these forward-looking statements, whether to reflect events or circumstances after the date initially filed or published, to reflect the occurrence of unanticipated events or otherwise.
| 4 |
PART I
Item 1. Business.
Power of the Dream Ventures, Inc., a Delaware corporation (“PDV”, “We” or the “Company”) is a Hungarian-based holding company focused on technology acquisition and development enabling the delivery of revolutionary concepts and ready to market products to the international market place. We develop, acquire, license, or co-develop technologies that typically originate in Hungary that are in prototype stage based on existing patents; in prototype stage prior to patenting; existing products that require expansion capital to commercialize; emerging science and high-technology research projects that require help in patenting, developing the product and marketing, university spin-off technologies and ideas from the very early stages of what represents “disruptive technologies.”
We were incorporated in Delaware on August 17, 2006, under the name Tia V, Inc. Since inception, and prior to our acquisition of Vidatech on April 10, 2007, we were engaged solely in organizational efforts and obtaining initial financing. Our sole business purpose was to identify, evaluate and complete a business combination with an operating company.
On April 10, 2007, we completed our acquisition of Vidatech, Kft. (also known as Vidatech Technological Research and Development LLC) a limited liability company formed under the laws of the Republic of Hungary. Vidatech is a company formed for the purpose of investing in, acquiring, developing, licensing, and commercializing technologies developed in Hungary. In furtherance of its business, Vidatech provides research and development services to the companies from which it acquires technologies or participation interests in such technologies.
Through Vidatech, we aim to provide pro-active support for idea, research, start-up and expansion-stage technology companies having rights to technologies or intellectual properties which we believe to be potentially commercially viable, by offering a range of services designed to encourage and protect the continuing development and eventual commercialization of those technologies.
OVERVIEW
The Company currently focuses on the operations of its wholly-owned subsidiary, Genetic Immunity, which is a clinical-stage biotechnology company focused on the discovery, development and commercialization of a new class of immunotherapeutic biologics ("Immune Therapies" or "Therapeutic Vaccines") for the treatment of chronic viral infections, cancer and allergy. Our Immune Therapies are designed to intensify or boost specific immune responses to modify or control these presently incurable diseases. The Company’s main focus is completing Genetic Immunity’s clinical trials program and on commercialization tasks related to the Company’s lead product candidate DermaVir therapeutic HIV vaccine.
PRODUCTS
Our Lead Product Candidate – DermaVir HIV-specific Immune Therapy
| 1. | Summary of DermaVir Development Milestones |
We have been developing DermaVir for the treatment of HIV/AIDS. DermaVir is a patented Immune Therapy which, based on the Food and Drug Administration (“FDA”) classification, is a combination of our original biologic product (DermaVir) and our medical device (DermaPrep). DermaVir is administered topically with DermaPrep. DermaVir’s Active Pharmaceutical Ingredient (“API”) is a pDNA expressing the broadest HIV antigen repertoires and Virus-like Particles (“VLP”).
We demonstrated in Phase I and Phase II clinical trials that DermaVir exhibits its specific pharmacologic effects by intensifying the patient’s own immune system to kill HIV-infected cells. Our clinical trials in approximate 70 HIV-infected patients demonstrated an excellent safety and tolerability in all doses. We found the optimal DermaVir dose for boosting HIV-specific T cell immune responses and demonstrated killing of HIV-infected cells by showing a statistically significant reduction of HIV-RNA in DermaVir treated patients compared to placebo.
| 5 |
DermaVir has a novel immunological mechanism of action different from antiretroviral drugs that kill the virus but cannot kill the infected cells. During effective Highly Active Antiretroviral Therapy (“HAART”), HIV-infected cells remain in the reservoirs. Even HAART intensification could not eliminate infected cells, decrease reservoirs or improve the immune system to fight against the virus. We believe that DermaVir treatments will reduce the amount of HIV-infected cells remaining in the reservoirs of HAART patients. This could provide additional treatment benefits by reducing the number of antiretroviral drugs that patients must take.
“Proof of concept” for the immunological and antiretroviral activities of our products was demonstrated in non-human primates with AIDS. This data showed that repeated DermaVir immunization resulted in a cumulative boosting of the HIV-specific cellular immunity without causing significant toxicities or adverse effects. After successful reconstitution of the monkey’s SIV-specific immune system, DermaVir immunized monkeys controlled their virus after interruption of HAART.
Among our animal studies, the non-human primate model is superior to other commonly used animal models (e.g., mice, rats, rabbits) because the disease progression and response to treatments in non-human primates is similar to the human disease. We believe that DermaVir has the potential to be the first Immune Therapeutic agent designed for the immune mediated control of viral replication by cytotoxic killing of HIV-infected cells. Currently, there are no approved Immune Therapy products on the market for HIV.
DermaVir was found to be safe and well tolerated at all doses in all clinical studies. A single treatment with DermaVir takes 20 minutes to apply, and left on the skin for three hours after which the patches are removed.. Our Phase I clinical results indicated an increase in the number of long-lived HIV-specific precursor T cells in all treated individuals in a dose-dependent manner. DermaVir-induced T cells remained in the body even after a one-year follow up, albeit at a lower level. These findings suggest that DermaVir boosted HIV-specific T cells similarly to that seen in primates. A separate Phase I/II study conducted by our collaborators in the US further confirmed the safety of repeated DermaVir vaccinations in combination with HAART (ACTG5176). A Phase II clinical trial with DermaVir in Germany on a drug naïve patient population (36 HIV-infected individuals) also demonstrated excellent safety and antiretroviral efficacy. Viral load suppression by DermaVir vaccinations occurs slowly, similarly to cancer vaccines as predicted by its mechanism of action, and that is different from antiretroviral drugs that kill the virus but do not kill HIV-infected cells.
Although our clinical development program is not yet final, and the marketing approval has not yet been obtained, we believe that our preclinical and human studies performed to date support the Proof of Concept, which is a relationship between DermaVir-induced antiretroviral immune responses and clinical benefit. We believe that DermaVir may be most effective if it is initially administered every six weeks to intensify HIV-specific immunity then at every regular doctor’s visit (every three months) to maintain the immunity.
| 2. | Our DermaVir Family for Personalized Treatment |
DermaVir is based on HIV subtype-B that represents the majority of HIV infection within the major marketplaces including North America and Western Europe. In addition, we have developed an initial DermaVir Product Portfolio to provide optimal treatment for every HIV-infected patient by considering the diversity of the infected population.
HIV is a highly variable virus. We found that the therapeutic effectiveness of DermaVir depends on the HIV sequence replicating in the patient and the patient’s genetic background. DermaVir contains related HIV-specific antigen sequences characteristic for the different Clades (subtypes) of the virus. To ensure the optimal therapeutic benefit for every patient, the treating physician will enter the information on the patient’s HIV sequences and genetic background (HLA) into our eMINER software. After analyzing the data, eMINER will select the optimal DermaVir product from the DermaVir Product Family and also calculate the specific T cell epitopes that will play a role in the immune boosting of the individual patient. The treating physician can then make an educated decision for personalized treatment of the patient and could also prescribe the proper immune monitoring based on the predicted T cell epitopes.
| 6 |
The targeted epidemics and our initial Product Portfolio are summarized in the Table below. According to UNAIDS, subtype-B represents approximately four million individuals (Europe, North, Central and South America, Caribbean, Australia). Our other product candidates can provide optimized treatment options for an additional 24.7 million HIV-infected individuals (2009 UNAIDS Report and HIV-1 Global Distribution, IAVI report Aug 2003).
| Present DermaVir Product Family | Targeted epidemics | Infected population |
| DermaVir B | Europe, America, Caribbean, Australia | 4 million |
| DermaVir C | Sub-Saharan Africa, South-Asia | 22.5 million |
| DermaVir BC | China | 0.77 million |
| DermaVir BF | South-America | 1.4 million |
| DermaVir AB | Eastern Europe | 10,000 |
We are pursuing the development of a DermaVir Product Family based on subtypes for the following reasons:
| · | DermaVir Product Family provides an individually-optimized Immune Therapy to boost the immune system against the specific HIV replicating in the patients. The availability of subtype-specific products permits “personalized treatment” with DermaVir Product Family. HIV subtype of the infected individual is diagnosed with available sequencing of the virus. The patient’s genetic background (HLA type) can also be diagnosed with routine methods. The subtype or recombinant form (HIV diversity) plus the patient’s genetic background for antigen processing (patient diversity) together define the best-matching Product from the Family. Based on these data our eMINER can suggest the personalized treatment for the patient. |
| · | DermaVir Product Family also supports different pricing in the various markets. Differential pricing of drugs manufactured by the same company has resulted in the illegal re-importation of the cheaper products to North America and Western Europe. The subtype-optimized DermaVir products will, by medical necessity, differ in HIV subtypes by geographical markets, product supplied to developing countries should not be diverted to other markets. |
| · | We intend to seek additional patent protection for the API in our subtype-specific DermaVir Product Family and method for treatment, also within the target regions. This will allow the granting of manufacturing and marketing licenses to specific countries based on unmet medical needs while providing additional protection against re-importation of third country-manufactured products to the United States and E.U. |
| · | Based on our technology platform, manufacturing and control of the DermaVir Product Family will not require additional methods or technology. The same manufacturing technology, control methods and facility can be used for cGMP manufacturing of all our products. |
| i. | Non-Clinical Studies |
Prior to human trials, regulatory agencies require the demonstration of their quality, safety and efficacy initially in appropriate animal models. To demonstrate the mechanism of action, first we established in human primary cells in vitro and in macaques ex vivo that DermaVir-expressing dendritic cells prime naïve T cells and induce both HIV-specific helper and cytotoxic T cells. Preclinical animal studies conducted in mice, rabbits and macaques consistently demonstrated that in vivo DermaVir immunization targets the pDNA immunogen to the dendritic cells of the lymph nodes that express the pDNA-encoded antigens. Importantly, immune responses following in vivo DermaVir immunization were shown to be similar to ex vivo immunization with cultured dendritic cells “DCs”. Both ex vivo and in vivo DermaVir immunizations employed lymph node dendritic cells, expressed similar amounts of pDNA-derived antigens, and induced Th1-type antigen-specific CD4 T helper and CD8 cytotoxic T cells. The induction of Th1-type cellular immune responses by DermaVir was also confirmed by delayed-type hypersensitivity skin reaction (DTH) tests. Additional experiments showed that in vivo DermaVir immunization leads to the induction of antigen-specific memory/precursor T cells. Recent animal studies have demonstrated that generation of highly protective immunity against smallpox required a live virus delivered via skin scarification. For DermaVir we used our platform technology, the modern version of the smallpox vaccination approach by employing a pDNA nanomedicine expressing complex VLP+ instead of a live virus and DermaPrep administration instead of skin scarification.
| 7 |
To study the safety of DermaVir vaccination, repeated immunization studies in good laboratory practice “GLP” were performed in swine and rabbits. These preclinical studies demonstrated that the major side effect of DermaVir vaccinations was erythema caused primarily by the DermaPrep administration. The erythema was transient and provided a mild inflammation, a desired side effect resulting in the activation of the Langerhans cells to seek and capture DermaVir nanoparticles applied under the patch.
To study the reproductive toxicity of DermaVir another repeated dose study GLP was performed in Wistar rats from the premating period through implantation. No significant differences between DermaVir treated and control animals were observed in body weights, food consumption, estrus cycle, fertility and mating index, or mating days until Day 0 postcoitus. Because no adverse effects were seen, the Maximum Tolerated Dose (MTD) cannot be determined, but is considered to be greater than 0.33 mg pDNA.
We have assessed the in vivo stability of our nanomedicine product. We could visualize the nanoparticles in the epidermis by two-photon microscopy over 120 minutes and demonstrate that DermaPrep skin preparation is essential for the effective penetration of the nanomedicine into the epidermis. The half-life of the nanomedicine in the lymph nodes was 11-12 days. The half-life of the nanoparticles in the lymph nodes depends on the migration of nanoparticle-containing Langerhans cells to the lymph nodes, intracellular degradation of pDNA, cytotoxic killing of antigen-presenting dendritic cells and clearance of dendritic cells.
The immunogenicity of DermaVir was investigated in uninfected and Simian Immunodeficiency Virus (“SIV”)-infected rhesus macaques with a DermaVir product formulated with a relevant plasmid DNA construct based on Simian Human Immunodeficiency Virus. The API of DermaVir used in all macaque studies expresses a VLP+ with the gag and pol genes homologous to SIV and an HIV env. The method for topical administration and the dose per skin surface in the macaque studies were similar to human trials. Proof of Concept efficacy studies in chronically SIV-infected macaques, some of them with AIDS, suggested that repeated DermaVir immunizations, alone or in combination with antiretroviral drugs, result in viral load reduction and survival benefit (see data in Lisziewicz et al AIDS 2005). DermaVir administered in combination with HAART boosted SIV-specific T cells that possessed significant antiretroviral activity demonstrated by maintenance of undetectable viral load after interruption of HAART in both chronically infected and late stage macaques. DermaVir treatment alone doubled the median survival time of infected macaques. These primate experiments provided the rationale to investigate repeated DermaVir immunizations in combination with HAART in HIV-infected human subjects.
| ii. | Summary of the Clinical Trials to Date |
As DermaVir associated clinical benefits were first seen in primates receiving HAART, the first two human studies were designed for the treatment of HIV-infected subjects receiving stable, fully suppressive HAART.
GIHU004 Trial to Evaluate the Tolerability and Safety of DermaVir in HIV-Infected Subject Currently Under Treatment with HAART (EudraCT Number 2004-001585-41, ClinicalTrials.gov ID NCT00712530).
The objective of the trial was to provide data on the safety, tolerability and immunogenicity of one DermaVir treatment in individuals with chronic HIV-1 infection treated with fully suppressive HAART.
| 8 |
This Phase I clinical study demonstrates that DermaVir was found to be safe and well tolerated at all doses. Vaccination did not affect HIV RNA (all subjects remained less than 50 copies/mL) and CD4+ counts. However, this therapeutic vaccination broadened and significantly increased the HIV-specific memory/precursor T cell pool, measured as the PHPC count, in a dose-dependent manner. The durability of vaccine-induced T cell responses was demonstrated after one year following a single vaccination in all treated individuals, albeit in significantly lower quantities (Lisziewicz et al. CROI2008 - Poster #715). These findings suggest that DermaVir boosted HIV-specific T cells similarly to that seen in primates.
Trial ACTG A5176. A Phase I/II, Randomized, Double-Blind Study to Evaluate the Safety, Tolerability, and Immunogenicity of DermaVir, in HIV-1-Infected Subjects Currently Under Treatment with HAART. (ClinicalTrials.gov ID NCT00270205 ).
The trial, conducted under a US Investigational New Drug “IND” application was sponsored by the US government (NIAID, NIH) enrolled 26 HIV-infected individuals on fully suppressive HAART to three cohorts in four leading HIV clinics located in the US. Based on the design, dose escalation to the last cohort implied that no subject in the current or lower dose cohort experienced a primary safety endpoint (i.e., no more than one subject in any cohort exhibited an adverse event greater than Grade 3).
The primary objective of this randomized, double-blind, placebo-controlled trial was to evaluate the safety and tolerability of multiple DermaVir treatments. Secondary objectives were to explore the immunogenicity of DermaVir for the treatment of individuals with chronic HIV-1 infection and HAART-induced durable reduction of viral replication.
The trial population comprised 26 HIV-infected men and women 18 to 50 years of age. HAART regimens were fully reduced within the 12 weeks prior to trial entry (Screening plasma HIV-1 level of less than 50 copies/mL, and CD4+ cell count greater than 350 cells/mm3 within 12 weeks prior to trial entry and nadir CD4+ cell count greater than 250 cells/mm3). The total treatment duration was 61 weeks. Each cohort’s treatment schedule was administered over a 13-week period, with an additional 48 weeks of follow-up for safety evaluations.
Twenty-six subjects were enrolled in the study. The primary endpoint was any possibly or definitely vaccine-related grade ≥3 adverse event (AE) appearing up to 28 days after the final study vaccination. No primary safety endpoints or AE-related study treatment changes / discontinuations have occurred. AE incidence was similar across groups (Rodriguez B. et al. XVIII International AIDS Conference, 2010 - Abstract # A-240-0111-10145). DermaVir administration was associated with a trend toward greater HIV-specific, predominantly central memory T-cell responses. The intermediate DermaVir dose tended to show the greatest immunogenicity, consistent with previous studies in different HIV-infected patient populations.
Phase II Trial
Repeated DermaVir immunizations in chronically infected macaques in the absence of HAART transiently suppressed virus replication leading to improvement of median survival time from 18 to 38 weeks compared to no treatment. These primate experiments provided the rationale to investigate repeated DermaVir immunizations prior to initiation of HAART in HIV-infected individuals. As DermaVir immunizations in combination with HAART did not show any product- or administration-related AEs higher than grade 2, we developed a Phase II protocol to evaluate the safety and to test the immunogenicity and antiretroviral efficacy of repeated DermaVir immunizations.
GIEU-006: Randomized, Placebo-Controlled, Multi-Center Trial to Evaluate the Safety, Tolerability, Immunogenicity, and Antiretroviral Activity of DermaVir in Treatment-Naïve HIV-1-Infected Patients (EudraCT number: 2007-001955-20, ClinicalTrials.gov ID NCT00711230)
This study was conducted in Germany in accordance with European Medicines Agency (“EMA”) approval and Current Good Clinical Practices “cGCP”. GIEU-006, a Phase II randomized, placebo-controlled, dose-finding, double-blinded study was conducted to assess the safety, tolerability, immunogenicity, and antiretroviral activity of DermaVir therapeutic vaccine, for the treatment of antiretroviral therapy naïve adults with HIV-infection.
| 9 |
The study population comprised of 36 antiretroviral treatment naïve HIV-infected men and women 18-50 years of age. The patients were randomized into one of the six treatment arms to receive either DermaVir or placebo. Four immunizations were administered over an 18-week period with an identical follow up schedule continuing until week 24; patients were followed for an additional 24 weeks for safety and immunogenicity evaluations. An additional 234-week safety follow up is being performed (every 26 weeks) including virology, immunology, chemistry and hematology assessments and physical examinations.
Immunizations were done on days 0, 42, 84, and 126. The total number of patches that a patient received throughout the study is 8, 16, or 32 in the low, medium, and high dose arms, respectively. The same skin sites were used for all immunizations.
The primary endpoint of the study was safety at week 24. Adverse events solely attributed to DermaPrep and were analyzed separately in the Local Reaction Assessment.
Out of the 36, 34 subjects have reached week 24 (visit 10), the time point of the analysis of primary and secondary endpoints of the study. The data of the two subjects that did not reach visit 10 were used according to the statistical section of the protocol. One of the subjects got arrested after visit 7 and discontinued from the study; the other subject decided to start antiretroviral therapy despite a stable CD4 count greater than 600/µl.
We did not observe any greater than grade 2 adverse events including signs/symptoms, lab toxicities, and/or clinical events possibly or definitely related to study treatment any time from the first day of study treatment until 42 days after the last DermaVir administration. There was no premature discontinuation of immunizations at the patient’s request for reasons having to do with the effects, or the perceived effects, of DermaVir or the DermaPrep administration procedure.
No patients required termination from any further immunizations due to toxicity attributable to DermaVir product, or due to tolerability failure. No patients experienced Grade 3 or higher toxicity to be discussed regardless of study treatment attribution.
The overall adverse event profile of the treatment was found to be consisting of grade 1 and grade 2 events. Most of the events were judged by the investigators to be not related to the study treatment. Only one grade 2 adverse events judged as possibly related to treatment (Positive Gaenslen's Test) in the 0.2 mg DermaVir group. We did not find any significant differences between study arms and treatment groups.
| iii. | Pivotal Trials |
We are currently evaluation the requirements of a Phase IIB pivotal trial in order to supply the FDA with all relevant data the agency needs in order to grant a conditional marketing approval, or an accelerated approval. We intend to conduct this Phase IIB trial in 2015 and have results available in the first half of 2016.
A Phase IV trials following marketing approval will be necessary if we intend to further study the efficacy of DermaVir in different patient populations. Presentations and publications of such trials are essential to successful marketing of DermaVir. Therefore, our strategy will be to obtain marketing approval within the shortest period of time and consequently continue the clinical trials for the establishment of a large safety and efficacy database. Please note that such a database on DermaVir is expected to facilitate the approval of pipeline products since the differences between products will be only in the sequence of the plasmid DNA.
| 10 |
| iv. | Our DermaVir Product Development and Registration Strategy |
Our clinical development and market approval strategy is focusing on immune intensification with DermaVir in Conditional Marketing Approval. We believe that patients treated with such Immune Intensification will maintain an undetectable HIV-RNA level and have an increased HIV-specific T cell frequency. Achieving optimal T cell responses and elimination the majority of HIV-infected cells from the reservoir might lead to remission (functional cure) in some HIV-infected people. These people would stop their antiretroviral drug treatment (if any) and receive only immune boosting DermaVir treatments during regular doctor visits.
In addition to the foregoing reasons, we believe that the treatment for HIV/AIDS with DermaVir may be desirable to physicians and patients for the following reasons:
| · | Effectiveness based upon boosted natural immunity; |
| · | Delayed disease progression; |
| · | No interference with current or future drug-treatment options; |
| · | No systemic toxicities; |
| · | Infrequent administration of a patch (only for three hours) during regular office visits; |
| · | No fear of the side effects and acquiring resistance of anti-HIV drugs; and |
| · | Reimbursement by insurance. |
According to Datamonitor (Future of HIV Market 2011), some patients may prefer a once-monthly administration (even intravenous drug infusion in the doctor’s office) over daily oral HIV treatment, which makes topical DermaVir treatment very attractive.
We believe that DermaVir, when approved, may meet the needs of these market opportunities due to favorable safety, tolerability and efficacy characteristics in comparison to currently prescribed drugs. The major toxicities of DermaVir are not systemic, but limited to local skin reactions. It is conveniently administered during regular doctor visits. The efficacy is based on the induction of HIV specific precursor T cells that are long lasting. DermaVir is expected to be introduced to the market for the treatment of HIV in combination with the presently available drugs since none of them can reconstitute HIV-specific immunity.
DermaVir has the potential to be the first Immune Therapy registered for the treatment of HIV/AIDS. To reach this objective we will use a strategy combining the experiences of registration of antiretroviral drugs and biologics. We have worked closely with the FDA on studies required for both the Biologics Master File “BMF” and the Investigational New Drug “IND” applications and will continue this effort. The FDA has expressed the desire to work with the pharmaceutical industry in the development of useful immune-based therapies for HIV/AIDS that may contribute to the body’s own defense against HIV and improve clinical outcome over drug therapy alone. We intend to develop effective and safe Immune Therapy products to delay HIV disease in HIV infected individuals not treated with antiretroviral drugs.
We had a preliminary meeting with the European Medicines Agency (“EMA”) to discuss the clinical development plan of DermaVir. EMA ensured us to fully support the development of DermaVir and suggested to organize an official Scientific Advisory Meeting to obtain approval for our clinical development and Chemistry, Manufacturing and Controls (“CMC”) strategy. We intend to propose the development of DermaVir for immune intensification in frame of a Conditional Marketing Approval. During the treatments we intend to use eMINER to match the patient with the best vaccine selected from our DermaVir Product Portfolio. After the DermaVir induction phase (3 treatments every 6 weeks) we expect DermaVir boosting of HIV-specific immunity compared to placebo. This is followed by the DermaVir maintenance phase (4 treatments every year). We believe that the maintenance not only maintains the high immune responses in the patients but also decreases the number of infected cells in the reservoir. This will be clinically demonstrated by maintenance of undetectable HIV-RNA after decreasing the number of drugs used in the HAART regimen. The primary endpoint of the pivotal trial will be non-inferiority between DermaVir + 1 Drug and HAART (standard of care). Additional treatment benefits include less toxicity in the DermaVir arm and lower pill burden.
| 11 |
We believe that we will get approval for our DermaVir Product Family together with our biomarker strategy and that regulatory agencies will not ask us to conduct separate clinical development programs with the different members in our DermaVir Product Family. The size of DermaVir Product Family initially will be less than 10, which is less than we envision to be expanded after emergence of new HIV variants, because it will provide optimal immunogenicity for selected HIV-infected patients, no difference in toxicities and all members in the DermaVir Product Family is produced by the same manufacturing and control methods. The future of HIV therapy, similar to several other indications, is already moving to personalized treatment. Pharmacogenetics screening is already used prior to initiation of some drug treatment in the US and EU (Datamonitor, Future of HIV Market 2011). We envision that our personalized DermaVir Immune Therapy will combine the scientific knowledge and our product portfolio to select the safest and most effective treatment of all HIV-infected patients.
According to EMA regulations, investigational drugs treating HIV infection with appropriate phase II clinical results, a favorable toxicity profile and good rationale for efficacy may receive Conditional Marketing Approval. Based on our clinical results with DermaVir parallel to the Conditional Marketing Approval process, we are planning to apply for Breakthrough Therapy Designation at the FDA.
Our Immune Therapy Technology Platform
The human immune system is well prepared to fight against diseases and most of the time does it successfully. In people with chronic diseases the immune system fights against such diseases but often cannot prevail. To live a better and longer life people boost their immune systems with vitamins, dietary supplements and herbs. We designed our Immune Therapy technology to specifically boost the patients’ own the immune system to focus on the target disease and win the battle.
The disease-modifying efficacy of our Immune Therapeutic products is based on the intensification or boosting of Th1-type cellular immunity. Based on the available preclinical and clinical data, we expect from this type of immune intensification to provide a therapeutic benefit that cannot be achieved by traditional drugs in patients with chronic infectious diseases, cancer or allergies.
There are four principal components that comprise our comprehensive Immune Therapy platform technology which are designed to work in conjunction to treat and/or control chronic infectious diseases, cancer and allergy, as follows:
| · | Our proprietary Active Pharmaceutical Ingredients (“API”): We have been designing (ANTIGENeering) our APIs to be specific, safe and effective. It consists of a single plasmid DNA (pDNA) immunogen ANTIGENeered to express several antigens, to contain molecular safety features and to release Virus Like Particles (VLP+) in the body of the patients. API with these crucial properties is not feasible with protein or peptide antigens. |
| · | Nanomedicine - our proprietary product platform: Our products are referred here as the “nanomedicine” according to their unique biophysical and biological features. Our APIs are formulated with our novel polymer excipient to synthetic “pathogen-like” nanoparticles. The nanomedicine formulation is essential to achieve potent antigen expression from the pDNA and antigen presentation by dendritic cells. This is critical for the intensification of antigen-specific immune responses in people with chronic diseases. |
| · | DermaPrep - our topical administration platform: We have developed the DermaPrep medical device for targeted in vivo delivery of our nanomedicine products to the dendritic cells of the lymph nodes via the Langerhans cells. We obtained the marketing approval in the European Union (CE Mark) for our DermaPrep device. |
| 12 |
| · | IT - Applied information technology: IT innovations have been supporting the discovery, development, manufacturing, and personalized treatment processes of Genetic Immunity. These include the rational antigen design, clinical trial and data management and matching the patients with the optimal Immune Therapy Product. |
Each of these components, as well as our lead product DermaVir, which has been developed using this platform technology, is described below in detail.
Our Proprietary Active Pharmaceutical Ingredients (“API”)
We have been designing (ANTIGENeering) our APIs to be specific, safe and effective. It consists of a single plasmid DNA (pDNA) immunogen ANTIGENeered to express several antigens, to contain molecular safety features and to release Virus Like Particles (“VLP+”) in the body of the patients. API with these crucial properties is not feasible with protein or peptide antigens.
In all our products, the specificity of immune boosting is determined by the nucleotide sequence of the pDNA that encodes several antigens. These antigens are specific to the causative agent of the disease. For chronic infectious diseases the recombinant antigens are derived from the target virus or intracellular bacteria to boost the immune system to eliminate the infected cells. For cancer, the antigens are derived from genes that specifically expressed in the cancer cells to boost the immune system to kill the cancer cells. For allergy, the antigens are derived from recombinant allergens to boost the immune system and balance the pathogenic immune responses.
We refer to our proprietary process for designing, preparing and testing pDNA-encoded antigens as “ANTIGENeering”. The API discovery program with ANTIGENeering technology supports a large proprietary product portfolio with long patent life.
Our Immune Therapies elicit potent Th1-type antigen-specific CD8 and CD4 T cell responses. The broadest antigen repertoire needs to be ANTIGENeered to boost polyclonal antigen-specific T cells to successfully fight the target disease.
Main steps of the ANTIGENeering process are:
| · | Establishment of an in silico database by collecting relevant scientific and patent information for the target disease; |
| · | Safety profile design of the API to completely eliminate antigen-related dangerous features (e.g., viral integration and replication) by molecular modifications using our proprietary ANTIGENeering software; |
| · | Immunological profile design of the API to express broad specificity CD4 and CD8 T cell epitopes using our proprietary eMINER software; and |
| · | In vitro testing of the new API candidates for antigen expression and antigen characterization with our standardized methods. |
The first example is the API in our lead DermaVir product. It is a single pDNA immunogen representing the broadest antigen repertoire among HIV vaccine candidates. This pDNA was ANTIGENeered for the regulated expression of thirteen complete and two non-functional HIV protein antigens. These proteins self-assemble into VLP+ structurally resembling the wild type HIV. We have introduced multiple irreversible safety features by genetic modifications including the complete impairment of integration, reverse transcription, the function of Nef and the 3’LTR. Our epitope analysis, validated with experimentally proven epitopes, predicted that DermaVir Product could present over 3,000 high-affinity T cell epitopes. In silico prediction of high-affinity epitopes revealed the importance of encoding multiple antigens in the pDNA.
The Figure below illustrate the mechanism
of action of our API in the DermaVir
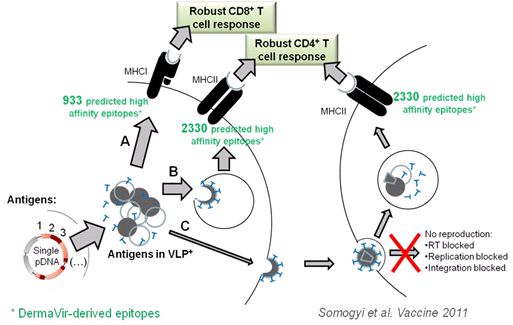
| 13 |
We developed an in silico antigen-specific T cell epitope prediction method for reliable modeling of the immunological potential of our API candidates. Our analysis demonstrated that HIV protein antigens have important differences in the ability to boost CD4 and CD8 T cell responses and revealed the importance of including both structural and regulatory proteins in an HIV vaccine. The other unique advantage of encoding the above mentioned broad antigen repertoire is the formation of VLP+ that further boosts the immune responses.
We have also developed the specification and validated quality control methods for the API. We have licensed a pDNA manufacturing process suitable for in house API manufacturing (1g scale) and also identified several contract manufacturers capable of producing cGMP quality API according to our specification. We have also built our own GMP certified facility to manufacture our API and excipients to mitigate the dependency from the Contract Manufacturing Organization (“CMO”) and to deliver the necessary material for preclinical and clinical trials.
Nanomedicine – Our Proprietary Product Platform
Our products are referred here as the “nanomedicines” according to their unique biophysical and biological features. Our APIs are formulated with a novel polymer excipient to synthetic “pathogen-like” nanoparticles. The nanomedicine formulation is essential to achieve potent antigen expression from the pDNA (“API”) and antigen presentation by dendritic cells. This is critical for the intensification of antigen-specific immune responses in people with chronic diseases.
Our nanomedicine products consist of a pDNA (API) core that covered with a synthetic polymer (polyethylenimine mannose, PEIm). These synthetic “pathogen-like” nanoparticles have the size and the shape of spherical viruses that naturally evolved to deliver nucleic acids to the cells. These nanoparticles deliver the pDNA to the cells similarly to viruses, but much safer because replication cannot occur. The nanoparticles mimic the size, surface properties, cellular entry by endocytosis, endosomal escape, and gene expression of pathogens (eg, viruses), thus allowing direct targeting of the antigen presenting cells of the immune system. The table below compares the properties of our nanoparticles to one of the pathogens (viruses):
| Features | Our “pathogen-like” nanomedicines | Pathogens (virus) |
| Structure | Synthetic polymer coat (PEIm) and pDNA | Protein coat and genetic material |
| Size | 70 to 300 nm | 50 to 500 nm |
| Surface properties | Sugar residues | Glycoproteins |
| Entry into the cell | Endocytosis | Endocytosis and other mechanisms |
|
Delivery of genetic material to the nucleus |
1. Efficient endosomal escape promoted by PEIm buffering capacity (acts as proton sponge) 2. Intra-cytoplasmic trafficking determined by the degree of association of PEIm and pDNA |
Viral protein-mediated mechanisms 1. efficient endosomal escape
2. intra-cytoplasmic trafficking |
| Gene expression |
Authentic protein expression profile, Virus-like Particle (VLP+) assembly |
Authentic virus protein expression profile, virion assembly |
The formation of “pathogen-like” nanoparticles is required not only for the targeting of dendritic cells, but also to protect the API from cellular degradation. These properties are essential for the efficient expression of the pDNA in dendritic cells of the lymphoid organs, which is required for the boosting of antigen specific T-cells and directing the immune responses to Th1-type. A naked pDNA without formulation to nanoparticles is susceptible to degradation both in extra- and intracellular space. Therefore, the biological activity of our nanomedicines is superior to the technologies which use pDNA without protection. Furthermore, to increase the efficiency of our nanomedicines we have inserted in our technology some targeting elements to ensure that the nanomedicines are targeted to the Langerhans/dendritic cells (mannosylation) and the API arrives at the place of antigen expression (nuclear targeting).
| 14 |
We have identified the key steps of the mechanism of action, supported by in vitro and in vivo experiments and developed a nanomedicine formulation that is optimal for both the intracellular stability and shelf life of our products. We have discovered a new biophysical property, the degree of association (measured by hyperchromicity) that determines the ability of the nanomedicine to escape from the endosome and to release the pDNA at the nuclear site. This parameter, which is in direct relation with the biological activity of the nanomedicine, can be controlled by the means of CMC methods like choosing the optimal ionic strength or pH. Optimizing the structure of these nanomedicines was warranted to improve their biological activity, develop stable formulations, and design an efficient, reproducible manufacturing technology.
The Figure below illustrates the
experimentally-proven mechanism of action of our nanomedicine products inside the cells.
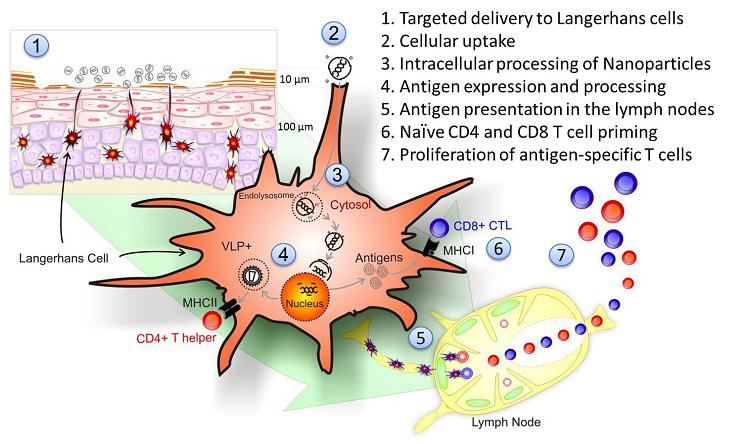
We have performed the detailed physico-chemical characterization of the nanomedicine according to the regulatory guidelines using our validated assays, and systematically investigated the variability of nanomedicine components and their relationship with the structure, in vitro biological activity and stability of nanomedicine (Tőke et al. IJP 2010). The potent antigen expression of the nanoparticles depends on their size, their entry through endocytosis, their ability to escape from the endosome and the release of the plasmid DNA cargo at the nucleus.
We have not only exploited this novel formulation but also implemented “Quality-by-Design” (“QbD”) in our biologic product development, manufacturing and control processes. The FDA encourages companies to implement the concept of QbD into their processes. The focus of QbD is that quality should be built into a product with thorough characterization of the product and understanding the processes by which it is developed and manufactured along with a knowledge of the risks involved in manufacturing and how best to mitigate those risks. We have been developing commercial manufacturing processes and validated quality control methods based on our QbD processes and thorough knowledge on our nanomedicine.
DermaPrep – Our Topical Administration Platform
We have developed the DermaPrep medical device for targeted in vivo delivery of our nanomedicine products to the dendritic cells of the lymph nodes via the Langerhans cells. We obtained the Marketing Approval in the European Union (CE Mark) for our DermaPrep medical device.
Provenge of Dendreon Corporation (“Provenge”) is the only antigen-specific Immune Therapy that is approved by the FDA. Provenge employs dendritic cells for antigen presentation to boost cellular immunity and eliminate prostate cancer cells. A prostate cancer-specific antigen is targeted to the patient’s own dendritic cells during the ex vivo manufacturing processes that Provenge developed for personalized treatment. Earlier, we have successfully utilized a similar ex vivo technology to target our nanomedicine into dendritic cells and boosted potent cellular immunity.
Dendritic cell targeting is very important for the efficacy of antigen-specific Immune Therapies. To improve the above described logistically cumbersome ex vivo immunization approach we developed DermaPrep the first dendritic cell-targeting administration device. Unlike that ex vivo technology, our Immune Therapy products can be manufactured in sizeable batches and administered topically with DermaPrep. One immunization procedure takes about 20 minutes and after three hours the patch is removed by the patient. Our filed patent application covers the new medical device for topical administration of liquid formulations. The figure below illustrates our in vivo Immune Therapy compared to the ex vivo technology of our competitors:
| 15 |
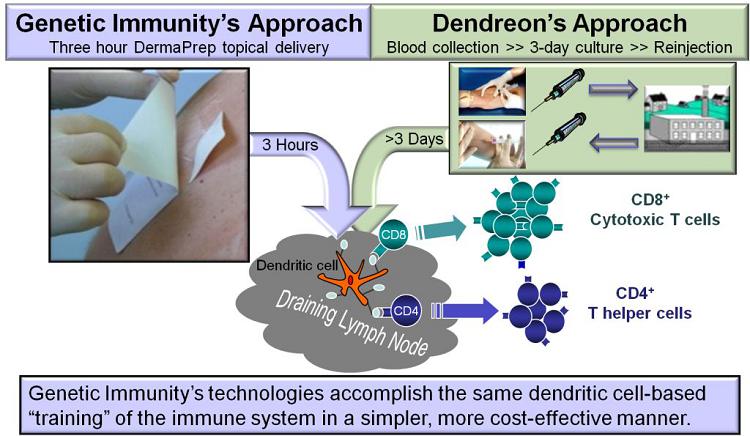
DermaPrep employs a skin preparation method that interrupts the stratum corneum facilitating the epidermal penetration of our nanomedicine product applied on the skin surface under our specially designed patch. The Langerhans cells, located in the skin under our patch, then look for pathogens and 800,000 of them has the chance to capture our “pathogen-like” nanomedicine and travel to the local lymph nodes. Here Langerhans cells mature to antigen-presenting dendritic cells. These cells secrete cytokines to induce Th1-polarized antigen-specific precursor/memory T cells that further differentiate into circulating effector or killer cells.
Several preclinical and clinical trials consistently demonstrated that DermaPrep administration was safe, well-tolerated, no greater than grade 2 events ever occurred. The main adverse event of DermaPrep vaccination is mild and transient erythema. The efficiency and reproducibility of the DermaPrep administration procedure was proven in the biodistribution study conducted on different cohorts of rabbits, where more than 50% of the absorbed nanomedicine was targeted to the lymph nodes.
Based on communication with the EMA and the FDA, we understood that there are currently no similar topical administration devices on the market. We obtained the Marketing Approval in the European Union (CE Mark) for our DermaPrep device.
IT - Applied Information Technology
IT innovations have been supporting the discovery, development, manufacturing, and personalized treatment processes of Genetic Immunity. These include the rational antigen design, clinical trial and data management, and matching the patients with the optimal Immune Therapy Product. We utilize the latest IT and physics including semantic web tools, ultrafast femtosecond laser technologies and embed the gathered knowledge in a broad portfolio of innovative, patient tailored therapeutics.
Our clinical development processes are supported by e-CRF solutions for managing Clinical Trials and eCTD conform solutions to optimize the clinical development processes, minimize the errors and time to submit different applications and reports to regulatory agencies word-wide. Our IT team also provides logistic support for the clinical trials by auditing the sites and identifying potential errors of measurements’ methods and errors in data collection and evaluation.
An electronic Labor Management System (“e-LMS”) supporting our processes in CMC provides a competitive advantage in product development by saving time and manpower. e-LMS supports our quality control tests using computerized Standard Operating Procedures (“SOP”). We electronically select the SOP and the required materials and equipment from our storage. In some cases, experimental data are captured directly from the equipment and stored in our databases. Deviations from protocols are registered and the results are calculated and summarized in a table format. Detailed reports, required by regulatory agencies, can be either printed and filed or stored electronically. Our IT team continuously improves the software solutions to meet our emerging requirement.
The API discovery and documentation process is presently supported by two of our proprietary softwares. The ANTIGENeering software ‘collect and mine’ research data from different sources and publications to support the pre-decision making and to ensure safe and specific antigen design. The eMINER software is utilized for the determination of the immunological potential (high affinity epitopes) on different human leukocyte antigen (“HLA”) alleles. With greater than 85% probability, we can in silico predict the cellular immune responses of patients treated with our Immune Therapeutic products. We envision that the eMINER will help doctors to match patients with optimal Products and design specific immune diagnostic tools.
Using the above-described tools we have established a DermaVir Product Family to provide every HIV-infected individual an optimal Immune Therapy. HIV is a highly variable virus. We found that the therapeutic effectiveness of DermaVir depends on HIV sequence replicating in the patients and the patients’ genetic background. Our DermaVir Product Portfolio is a family of products containing related HIV-specific antigen sequences characteristic for the different Clades (subtypes) of the virus. To ensure the optimal therapeutic benefit for every patient the treating physician will enter the diagnostic information on the patient’s HIV sequences and genetic background (HLA) into our eMINER software. After analyzing the data the eMINER will select the optimal DermaVir product from the DermaVir Product Family and also calculate the specific T cell epitopes that will play a role in the immune boosting of the individual patient. This way the treating physician can then make an educated decision for selecting the optimal Product for personalized treatment for the patient and could also prescribe the immune monitoring.
| 16 |
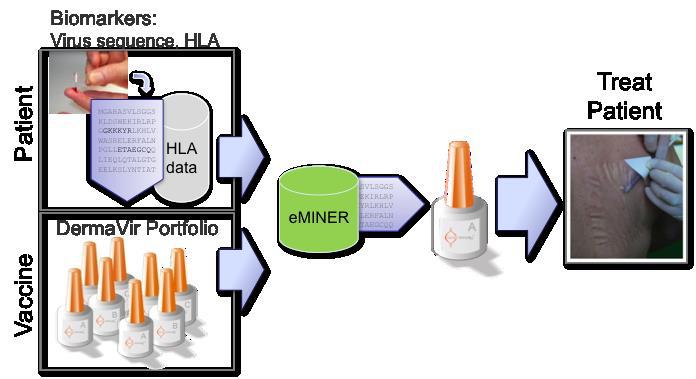
Our competitive IT team consists of informaticians and mathematicians who are dedicated to support our business agenda. They are essential part of our patient-driven technology platform, providing us innovative solution to daily challenges including data capture, evaluation and storage of experimental data, website design, literature search, 3D visualization of the skin penetration of our nanomedicines based on fiber-integrated 2-photon microscopy and auditing the data management and data quality on clinical sites. We believe that our integration of bioinformatics and mathematics into our everyday workflow will significantly improve competitiveness because it provides efficient, accessible and high quality data and information collection and distribution throughout the entire research and development and commercialization processes. Our IT team develops project-based, customized services to improve upon the efficiency of our work being performed and to save time and eliminate errors caused by insufficient data management.
Pipeline Product Candidate
Our pipeline product candidate with animal proof of concept, for the treatment of Human Papilloma Virus (“HPV”) infection, is referred to as “HuPaDerm” and our allergen-specific immunotherapy is referred to as “DermAll”. We partnered with the German Cancer Research Center located in Heidelberg (“DKFZ”) to develop HuPaDerm. DKFZ is a world leading research center in tumor virology and Harald zur Hausen was awarded the Nobel Medicine Prize for his work on HPV caused cancer of the cervix. HuPaDerm animal trials are performed in Heidelberg. DermAll has decreased the allergic symptoms by balancing allergen-specific immune responses in mice experiments.
DermAll
Allergic diseases are caused by an immune response induced by different allergens. We believe that our DermAll products will suppress pathogenic immune responses in allergic people by boosting allergen-specific immune responses. This new mechanism is expected to restore a balanced immune response that is normally characteristic for people who do not have any allergic disease. The DermAll Product Portfolio will target several different allergies. The first target will be Egg Allergy (“EA”), since we have already collected preclinical animal data in this DermAll indication suggesting that DermAll-EA treatment can suppress allergic sneezing by balancing immunity. We expect clinical efficacy of specific Immune Therapy that not only suppress allergic rhinitis symptoms but also prevent progression to asthma or atopic dermatitis.
For allergic diseases Immune Therapy offers a vastly different treatment option with disease-modifying potential to traditional symptomatic treatments. In 2009, Grazax became the first Immune Therapy in Europe to gain approval as a ‘disease modifying treatment,’ representing a significant step towards the possibility of a cure. The major limitations of present Immune Therapy approaches include serious safety concerns and cost of treatment, therefore conventional symptomatic treatments continue to be the more popular option (Datamonitor 2010). DermAll Immune Therapy addresses both limitations using our proprietary API design to obtain products with clinically proven excellent safety features, and cost effective manufacturing technology.
| 17 |
HuPaDerm
Development of HuPaDerm Product Portfolio is targeting the treatment of the leading cause of cervical cancer as well as papillomatosis, genital warts and later cancer caused by the different HPVs. Our development strategy for the HuPaDerm Product Portfolio has already been discussed with the EMA. We partnered with DKFZ to develop the HuPaDerm product. DKFZ is a world leading research center in tumor virology. Harald zur Hausen was awarded the Nobel Medicine Prize for his work on HPV caused cancer of the cervix. Zur Hausen, former Scientific Director of DKFZ, is recognized for finding that cervical cancer is caused by viral infections. His research made it possible to develop a vaccine against one of the most frequent cancers in women. Zur Hausen shared the Nobel Prize for Medicine with Françoise Barré-Sinoussi and Luc Montagnier for discovering HIV, the virus that causes AIDS. HuPaDerm animal trials are performed in Heidelberg. DermAll decreased the allergic symptoms by balancing allergen-specific immune responses in mice experiments.
Immune Therapy for Cancer
ProstaDerm Immune Therapy against virus-based prostate cancer
We have also considered developing ProstaDerm Immune Therapy to provide an in vivo alternative to Provenge that is manufactured ex vivo from the patient’s blood. Xenotropic murine leukemia virus-related virus (“XMRV”) is an authentic, newly recognized human retrovirus first identified in prostate cancer tissue. Studies have detected XMRV at different rates in prostate cancer cases (up to 27%) and in patients with chronic fatigue syndrome (CFS; up to 67%) (Silverman et al. Nature 2010). Therefore a pDNA developed for expressing XMRV antigens might effectively be used for the treatment of XMRV-associated prostate cancer.
Immune Therapy against melanoma
Melanoma is a malignant tumor of melanocytes, pigment producing skin cells. Melanoma is less common than other skin cancers. However, it is much more dangerous and causes the majority (75%) of deaths related to skin cancer (Jerant et al. AAFP 2000). This cancer has well characterized and specific biomarker proteins (melanoma-associated antigens, MAGE) therefore melanoma is a possible target indication for our Immune Therapy Platform Technology. ANTIGENeering of a pDNA specific for these antigens could result in a product candidate for patients suffering from melanoma.
Immune Therapies for Emerging Infectious Diseases
A potential extension of our technology could be utilized to develop treatments for infectious diseases like Chlamydia, hepatitis C and B viruses and West Nile Virus where T-cell responses can be directed against cells harboring the intracellular pathogens. Opportunities are under consideration for initiating the development of some of these products at a later stage.
Immune Therapy against West Nile Virus
We are working together with a European Consortium (WINGs) to develop a prophylactic and therapeutic vaccine against the West Nile virus. The project is led by the Fraunhofer Institute IZI in Leipzig, Germany, and is funded from a $3.5M European Union grant.
Our role in the project is to provide the nanoformulation technology for the vaccine candidates and the transdermal delivery technology for the administration of the vaccine. As industrial partner of the consortium we are also responsible for the clinical development and the commercialization if the animal studies are successful.
GRANTS
In 2005 we received a $1 million grant from the EU, which funded Phase I clinical trials of DermaVir in Hungary. Since being awarded a $7 million grant from the Hungarian government in 2005, we have been applying the funds to develop product manufacturing technology in Hungary, build a pilot manufacturing facility and develop pipelines in collaboration with Hungarian academic institutions pursuant to a Consortium Agreement establishing the Vaccine Therapy Cluster, a collaboration between the Company and the University of Szeged. As a result, our product development, manufacturing, information technology, and clinical operations have been established in Budapest. The business, financial and commercial operations remain in the U.S. In 2009, the DVCLIN01 consortium we lead received an additional grant of $4.5 million to perform a Phase II clinical study with DermaVir in Germany. The results of this study were presented at the AIDS International Conference in Vienna July, 2010. In 2010, the FIBERSCN consortium led by R&D Ultrafast Lasers Kft. received a grant of $3 million to investigate DermaVir immunization technology with fiber integrated 2-photon microscopy. We investigated the penetration of DermaVir to the skin and uptake by epidermal Langerhans cells. The West Nile Shield project (“Wings”) consortium led by Fraunhofer Institute won a $3.5 million grant to develop novel approach for prophylactic and therapeutic vaccines against West Nile Virus. Both the FIBERSCN and WINGS grants have been successfully completed, and result have been publiched.
| 18 |
Using the above funding, we have developed improved formulation and scalable manufacturing technology for our biological product portfolio; conducted preclinical animal studies to demonstrate that our proprietary technology is suitable for additional product development including DermAll and ChlamyDerm; developed regulatory strategy for obtaining Marketing Approval for our DermaVir Product Portfolio as personalized Immune Therapy; and designed an allergen-specific Immune Therapy Product Portfolio.
INTELLECTUAL PROPERTY
Protection of our intellectual property and proprietary technology is a strategic priority for our business. We rely on a combination of license agreements and patent, trademark, copyright and trade secret laws along with institutional know-how and continuing technological advancement to develop and maintain our competitive position. Our ability to protect and use our intellectual property rights in the continued development and commercialization of our technologies and products, operate without infringing the rights of others, and prevent others from infringing our rights, is crucial to our continued success. We will be able to protect our products and technologies from unauthorized use by third parties only to the extent that they are covered by valid and enforceable patents, trademarks or copyrights, or are effectively maintained as trade secrets, know-how or other proprietary information. Our policy is to seek U.S. and international patent protection for technological developments that we believe will enhance the market position of our product candidates and methods of using our product candidates.
For additional DermaVir Product Family we plan to file a new patent application for the API that would protect our DermaVir Products until 2031 when it is issued. Filing the new patent application on API designed with our ANTIGENeering technology is our strategy for all the pipeline products.
To supplement our patent portfolio, we also depend on the skill, knowledge and experience of our scientific and technical personnel, as well as that of our advisors, consultants, and other contractors. To help protect our proprietary know-how and inventions for which patents may be difficult to enforce, we rely on trade secret protection and confidentiality agreements to protect our interests. We require all employees, consultants, advisors, and other contractors to enter into confidentiality agreements that prohibit disclosure of confidential information and, where applicable, require disclosure and assignment to us of ideas, developments, discoveries and inventions important to our business.
MARKET OPPORTUNITIES
Therapeutic Vaccine Market for HIV/AIDS
According to the Joint United Nations Programme on HIV/AIDS (“UNAIDS”), about 33.3 million people are living with HIV/AIDS in the world, including 2.5 million children, with 2.3 million of them in North America and Europe. During 2009, 100,000 patients were newly infected and 35,000 patients were reported to have died from AIDS in North America and Europe. The rise in prevalence has been driven by a number of factors, including a growing number of heterosexual transmissions, immigration from countries with a high HIV prevalence, and a growth in high-risk sexual behavior. There is strong evidence of resurgent HIV epidemics among men who have sex with men in North America and in Western Europe. Immigrants living with HIV have become a growing feature of the epidemics in several countries in Europe. Heterosexual transmission accounts for about half of the people newly infected with HIV in Central Europe (2010 UNAIDS Report). There are significant emerging markets including Eastern Europe, Russia, India and China with significant income and are expected to take actions to slow down their fast growing HIV epidemics. Therefore, additional market opportunities for HIV/AIDS treatments are projected to emerge.
| 19 |
The patient population on HAART is already demanding simplification of treatment that can be achieved by combining several drugs into one pill. The development of novel original antiretroviral drugs will be more and more expensive and more difficult to get approved. In 2009, the HIV market generated sales of about $12 billion and is expected to grow up to $15.6 billion in 2015 (Datamonitor HIV market forecast (06/2010)). This growth has been driven by the launch of several new fixed-dose combinations such as Truvada and Epzicom™, and a new class of antiretrovirals known as the entry inhibitors. The combination of several drugs in one pill improves the tolerability of HAART.
When DermaVir enters the market, there will be greater than 28 generic antiretroviral drugs on the market providing treating physicians a diverse source of inexpensive and potent drugs for managing their patients. According to recent treatment recommendations, doctors will start treating their patients earlier with HAART and as a result, more HIV-infected patients will live a longer life. We expect that by 2016 a significantly larger patient population will be treated with HAART due to the inexpensive and potent generics. These patients will be eligible for DermaVir immune intensification and that might further increase the potential market for DermaVir.
Market opportunity for DermaVir
Market demand for DermaVir will be driven by the demand for (1) a cure and (2) simplified treatment options by HIV-infected patients. Such patients seek safe drug sparing treatment options that fully control their virus, decrease of toxicities, immune boosting and cure of HIV disease. We believe that the DermaVir immune intensification will be the first on the market offering significant therapeutic benefit over HAART and a potential cure (remission) for some of the patients who achieved the maximal immune intensification to control HIV disease. Due to decreasing costs and better access to generic HAART, the treated patient population will significantly grow. The new state of the art treatment will be DermaVir in combination with generic HAART. Doctors will use our eMINER to match patients with their optimally effective DermaVir selected from our product portfolio and prescribe the personalized diagnostic assay to monitor when the HAART can be simplified by dropping one, two or even all three components. We believe that the new, potent and expensive antiretroviral drugs will be very important for salvage and will be used for patients failing generic HAART.
We believe that after market entry, DermaVir Immune Therapy will be the new state of the art treatment. Since HIV drugs decrease immune responses and do not provide a cure, DermaVir immune boosting could be added to every HAART-treated patients. The minimum effect of DermaVir would be to boost the patients’ own immune system’s responses to eliminate infected cells from the body. But additional benefits expected for DermaVir treated patients are the decrease in the number of drugs and consequently such patient will suffer less toxicity. DermaVir treatment will provide a hope for cure; in some DermaVir-treated patients, immune boosting can result in remission allowing the patient to lead a healthy life in the absence of HIV drug treatment (for some years). If the HIV rebounds because the immune system cannot fight any longer, the patient resume to take the drugs and his/her life will not be in danger.
As a new state of the art immune boosting therapy, DermaVir treatment should be cost effective. Saving on drugs and cost of treatment could support insurance reimbursement. It is conservative to estimate that annual DermaVir treatment will be lower than the cost of HAART. We believe this can represent up to over $5 billion DermaVir sales for the seven major markets per year, based on 30% market penetration. In the event competitors for a cure enter into the market this figure will decrease. DermaVir is one of the best-characterized HIV therapeutic vaccine product candidates (solid randomized and controlled Phase II clinical data, commercial manufacturing methods and controls) and may have the highest chance to obtain first marketing approval.
| 20 |
We believe that in the not too distant future DermaVir will have significant sales outside of the major markets based on the high unmet need. These markets include the 22 million people, including children, in sub-Saharan Africa. However, developing countries such as Brazil, China, India and Russia could become significant markets.
The global HIV drug market size is expected to grow to $15.6 billion by 2015 with approximately 35 million people infected with the virus. Of this amount, Europe represents approximately 2.3 million HIV infected people. Russia, our next potential approval target, represents another 1 million infected people.
Initially we will aim to treat 5% of the European population, or approximately 130,000 individuals (of the 2.3 million total infected.) Current HAART based treatments cost approximately 12,000 EURO a year per patient. Based on our initial pricing model, and assuming a 5% market penetration, this represents a EURO 1 billion market opportunity per year for DermaVir in Europe alone.
Competition
In the field of HIV treatment, several immune therapies are under development because there is a substantial unmet medical need to find new treatment options for patients who have developed resistance to the current antiviral drugs and for patients who cannot tolerate life-long daily drug treatment. A description of such immune therapies follows:
| i. | Broad-Spectrum Immune Therapies |
The rationale of broad-spectrum Immune Therapy originated from the successful use of interferon-alpha for the treatment of another chronic infectious disease, hepatitis C. The best available current treatment for chronic hepatitis C (peg-interferon alpha plus ribavirin) leads to an overall sustained response rate of approximately 70%. In HIV infection, however, no viral load suppression after current drug treatment interruption can be achieved. The current, broad-spectrum immune therapies described below are not specific to any disease:
| · | Hemispherx Biopharma is evaluating the safety and activity of orally administered low dose interferon alfa to modulate general, nonspecific immune responses in subjects infected with HIV who have not yet demonstrated clinical symptoms of immune suppression. |
| · | Interleukin-2 (IL-2, Aldesleukin, Proleukin) is made by Novartis (Chiron) and Amgen. This drug is currently approved for the treatment of certain types of blood cell cancers (e.g., specific types of leukemia). It is considered an experimental drug for HIV-related therapy because the FDA has not yet evaluated it for the HIV indication. Two large efficacy trials in HIV patients (SILCAAT and ESPRIT) were conducted recently. However, despite a substantial and sustained increase in the CD4+ cell count, as compared with antiretroviral therapy alone, Interleukin-2 plus antiretroviral therapy yielded no clinical benefit in either study. |
| · | CYT107, Cytheris’s recombinant human Interleukin-7 developed as a general immune system booster. The company has four ongoing Phase I/II or Phase II studies with CYT107. According to the company’s latest press release, dated March 1, 2011, which reported the results of INSPIRE2 Phase IIa study, CYT107 was able to reconstitute CD4 T-cells in chronically HIV-1 infected patients whose CD4 T-cell counts remained low despite treatment with anti-retroviral-therapies (HAART). |
| · | Inovio has been developing a new DNA administration technology, called Electroporation, for the intracellular delivery of various immunotherapeutic DNA solutions. Resultant gene expression matched or exceeded that achieved using viral or lipid delivery methods, without side effects that hamper these approaches. The device is being tested in Phase I/II and Phase II trials. |
| ii. | HIV-Specific Immune Therapies |
The rationale for HIV-specific immune therapies is to amplify HIV-specific immune responses present during chronic infection to control viral replication. Such antiviral immune responses provide the balance between infection and the immune system and in some cases the immune system wins over the infection (e.g., most of the non-progressors), but in the majority of the cases, HIV overwhelms the immune system resulting in disease progression and AIDS. Immune Therapies have been developed for viral, parasitic, cancer, and allergic diseases. Antigen-specific immune therapeutic approaches are (1) based on antibodies that specifically bind the target antigens, and (2) T-cells that can eliminate HIV-infected cells from the body. Our competitors are the most promising therapeutic vaccines according to the Datamonitor report: R&D Trends: HIV (Reference Code: HC00083-007), published in March 2011. Each of our competitor’s products described below are ex vivo or injected by needle and not topically delivered like DermaVir:
| 21 |
Phase II Immune Therapy candidates
| · | Vical, in collaboration with the NIH and Merck & Co., is developing a DNA vaccine, VRC DNA/rA, for the treatment and prevention of HIV using its naked DNA gene delivery technology. The vaccine is currently in Phase II clinical development (NCT00865566, 2011; www.clinicaltrials.gov) (Vical, 2011) |
| · | FIT-06 is a DNA vaccine consisting of the Clade B HIV nef gene, under development by FIT Biotech using its GTU nuclear anchoring technology, for the treatment and prophylaxis of HIV/AIDS. It has completed Phase II trials in South Africa (Fit Biotech, 2010) |
| · | AGS-004 is one of a series of autologous dendritic cell vaccines under development by Argos Therapeutics for the treatment of HIV infection. The vaccines are pulsed with amplified mRNA from infected cells to stimulate an immune response. |
| · | The Vaccine Research Center at the US NIAID is collaborating with GenVec on the development of an HIV DNA vaccine (VRC-HIVADV014-00VP). The vaccine consists of an adenovirus-5 vector expressing modified HIV-1 genes. It is being evaluated alone and in combination with a plasmid DNA vaccine prime. Vaccine candidates are evaluated as therapeutic and prophylactic AIDS vaccines (GenVec, 2011a). It has been evaluated in several Phase I/II clinical trials (GenVec, 2011b) |
| · | In October 2010 Bionor Pharma reported that the primary endpoints of the placebo-controlled study of Vacc-4x (NCT 00659789, 2010, www.clinicaltrials.gov) had not been met. However, the decision was made to resume the development of the vaccine candidate due to a statistical significant treatment difference regarding viral load, the secondary endpoint (Bionor Pharma 2010). Bionor Pharma is currently seeking partnering opportunities. It is also developing a follow-up vaccine Vacc-C5, targeting immune activation associated with HIV infection that is expected to enter clinical trials in 2011 (Bionor Pharma 2011). |
| · | V-1 Immunitor (V1) is a therapeutic AIDS vaccine formulated as an oral pill comprising heat-inactivated HIV antigens derived from pooled blood of HIVpositive donors that is being developed in Russia and Thailand (Bourinbaiar et al., 2010) |
Phase I/II Immune Therapy candidates
| · | TUTI-16 is a water-soluble, fully synthetic, self-adjuvanting lipopeptide vaccine developed by Thymon (Goldstein and Chicca, 2010). A US Phase I/II clinical trial was completed in February 2011 (NCT00848211, 2011; www.clinicaltrials.gov). A second Phase I/II clinical trial, NCT01144026, is currently recruiting participants |
| · | A dendritic cell vaccine is developed by Esteve, consisting of autologous monocyte-derived dendritic cells pulsed with heat inactivated autologous HIV-1. Phase I/II results were reported in 2011 (Garcia et al., 2011) |
| · | Virax’s HIV program is focused on the development of a therapy for early stage HIV infection using an immunotherapeutic approach. VIR201 is a recombinant fowl pox-virus technology designed to co-express genes for immunogenic but highly conserved parts of the HIV in conjunction with interferon gamma. The objective is to amplify the T-cell-mediated immune response of the human immune system to the HIV virus. Results from a Phase I/IIa trial were reported in October 2010 (Virax, 2010) |
| 22 |
Phase I Immune Therapy Candidates
| · | PENNVAX-B is an HIV env clade B-specific/gag/pol DNA vaccine for the prevention and treatment of HIV/AIDS. The vaccine was licensed from the University of Pennsylvania. As part of the deal Inovio has worldwide commercialization rights for vaccine and delivery technology. Pennvax-B has completed a US Phase Ib study (NCT00775424, 2011; www.clinicaltrials.gov) (Inovio, 2011) |
| · | A pDNA vaccine delivered via electroporation in a co-formulation with a plasmid containing IL-12, which acts as a molecular adjuvant is developed by Profectus BioSciences. The vaccine is currently in a US Phase I clinical trial (NCT01266616, 2011; www.clinicaltrials.gov) |
| · | EP-1090 is a DNA vaccine developed by Inovio and VaxOnco in In Phase I clinical trial since 2007 (NCT00532974, 2007; www.clinicaltrials.gov) |
| · | GX-120 is a DNA vaccine developed by Dong-A Pharmaceuticals and Genexine. GX-120 contains four plasmids expressing gag, pol, env and a mutant IL-12 (IL-12N222L). It is in a Korean Phase I trial (Genexine, 2010) |
| · | NAcGM3/VSSP is a Ganglioside-based vaccine developed by the Center of Molecular Immunology Havanna, Cuba and Recombio. The N-acetylated ganglioside GM3 vaccine (NGcGM3) is combined with Neisseria OMP vesicle-based very small proteoliposomes (VSSP) as carrier. It is in two Phase I trials in Argentina and Cuba for the treatment of HIV/AIDS (Recombio, 2011) |
Advantages of DermaVir Over Antiretroviral Drugs
HAART requires the combination of three drugs selected from minimum two different classes. It has been effective in decreasing morbidity and mortality associated with HIV infection. However, full life expectancy of HIV-infected people cannot be restored even with optimal care. It has been demonstrated that HIV-infected people treated with optimal HAART live 12 years shorter than those without HIV.
HAART does not provide a cure for HIV/AIDS. Several recent studies demonstrated that HAART intensification with drugs of additional classes (e.g. integrase inhibtor) leads to no change of viremia or reservoirs measured by HIV DNA. In addition, long-term HAART decreases HIV-specific immune responses that could negatively affect the clearance of the reservoir. Viremia persist in the reservoirs even after 7 year of fully suppressive HAART (ABBOTT 720 study), therefore eradication of HIV with HAART does not seems to be feasible.
An alternative strategy to cure HIV disease would be to induce remission, similar to cancer model. A recently completed study has demonstrated (presented at CROI2010)an inverse correlation between the frequency of HIV-specific T cells in the gut and the size of the reservoir. This is the first evidence suggesting that boosting of the patients’ own immune system might decrease the number of HIV-infected cells in the reservoirs.
We have shown that DermaVir boosts cellular immunity to kill HIV-infected cells and functions differently from antiretroviral drugs that kill the virus. We believe that DermaVir treatment can reduce the amount of HIV-infected cells remaining in the reservoirs in patients treated by HAART. This novel activity of DermaVir could provide additional treatment benefits by reducing the number of antiretroviral drugs that patients must take daily. We envision that after successful reconstitution of the HIV-specific immune system by DermaVir, immune control of HIV might be achieved. DermaVir could change the HIV treatment paradigm by giving patients an option for a safe interruption of life-long drug treatment by boosting their own immune system.
Achieving optimal T cell responses and elimination the majority of HIV-infected cells from the reservoirs could lead to remission in some HIV-infected people. These people would be able to stop their antiretroviral drug treatment and receive immune boosting DermaVir treatments during regular doctor visits.
| 23 |
In contrast to antiretroviral drugs, DermaVir treatment provides a hope for patients for a functional cure. We do not believe that eradication of integrated HIV can be achieved by any treatments. However, several viruses are successfully controlled by a healthy immune system, including HIV in some exceptional people; therefore remission (functional cure) is an achievable treatment objective. Based on our findings, we believe that DermaVir, by boosting HIV-specific cellular immunity in patients treated with HAART, could effectively kill HIV-infected cells and decrease the size of the reservoirs. After repeated DermaVir treatments a significant intensification of HIV-specific immunity and the elimination of the majority of infected cells from the reservoirs could be achieved. We believe that the hope for a cure that HAART cannot provide for HIV-infected people will be an option with DermaVir when used initially in combination with HAART to eliminate both the virus and the infected cells.
DermaVir sales could be favored and not limited by Generic Antiretroviral Drugs, which would be a significant competitive advantage compared to original or new antiretrovirals. These original drugs could be more expensive than the generics and as a result their sales could be limited to patients who are not responding to generic drugs. Therefore, effective generic treatment options could limit the profit of a new HIV drugs entering into the market. In contrast, we believe that DermaVir sales will be independent of original or generic drugs used in HAART.
We believe that the DermaVir Immune Therapy might be used for immune intensification of most of the HAART treated people. According to recent treatment recommendations, doctors will start treating their patients with HAART earlier and consequently more HIV-infected patients will have a longer life expectancy. We believe that a significantly larger patient population will be treated with HAART and DermaVir due to the market entry of inexpensive and potent generics.
An additional benefit of DermaVir will be what we believe to be a favorable price. We believe that the cost of DermaVir manufacturing will be manageable on a per dose level and that will allow us to sell the product for a competitive price. We estimate that the price of DermaVir in the seven major markets might be similar to original antiretrovirals (e.g. $12,000 per year). The cheapest price of DermaVir might be in sub-Saharan Africa (e.g. $500 per year), which is similar to generic HAART.
We believe that DermaVir immune intensification will be the first option on the market offering a significant therapeutic benefit over HAART and a potential functional cure (remission) for some of the patients who achieve maximal immune intensification to control the HIV disease. We believe DermaVir will be used in conjunction with generic HAART. We believe that doctors will use our eMINER application to match patients with their optimally effective DermaVir selected from our product portfolio and prescribe the personalized diagnostic assay to monitor when HAART can be simplified by dropping one, two or even all three components. We believe that the new, potent and expensive antiretroviral drugs will be very important for salvage therapy and will be used for patients failing generic HAART.
OUR BUSINESS STRATEGY
Our business objective is to develop and commercialize DermaVir and our pipeline products in order to become one of the leading Immune Therapy companies. We believe that our unique combination of product candidates, our preclinical and clinical Proof of Concept and our nanomedicine manufacturing technology that beside the patents provides additional long-term protection against genetic competitors, supports such an ambitious objective. It is our intention to obtain the funding required for the growth of the Company from the public market.
Up to date we have focused on R&D and proof of concept for our Technology Platform. From now on, besides adding to our team talents and experts essential to develop our products, we intend to expand our internal management and business development team.
SALES AND MARKETING
We currently have no internal sales or distribution capabilities. In order to successfully commercialize any of our products, we must either internally develop sales, marketing and distribution capabilities, or make arrangements with third parties to perform such services. For the foreseeable future, we may not be able to establish marketing and sales capabilities internally or hire a large enough number of sales personnel with appropriate expertise to market and sell our products, if the products are approved by appropriate regulatory authorities. Therefore, we may need to enter into an alliance with a major pharmaceutical company or other third party to market and sell our products, which we may be unable to do.
| 24 |
EFFECT OF EXISTING OR PROBABLE GOVERNMENTAL REGULATION
We plan to market our products following approval on a global basis. The approval and selling of our products are subject to various regulatory and governmental oversight bodies. Depending on the regulatory category of each product, such as biological product, medical device or drug, separate regulations apply and could vary on a market-by-market basis.
FDA Regulation
We believe that our products are defined as combination products consisting of two or more regulated components, a biologic and a medical device. In the U.S., a combination product usually is assigned by the FDA to one of the agency’s centers, such as the Center for Biologics Evaluation & Research (“CBER”) or the Center for Device and Radiological Health (“CDRH”) ,with the chosen center taking the lead in pre-marketing review and approval of the combination product. Other FDA centers also may review the product in regard to matters that are within their expertise. The FDA selects the lead center based on an assessment of the combination product’s “primary mode of action.” Some products also may require approval or clearance from more than one FDA center.
To determine which FDA center or centers will review a combination product submission, companies may submit a request for assignment to the FDA. Those requests may be handled formally or informally. In some cases, jurisdiction may be determined informally based on FDA experience with similar products. However, informal jurisdictional determinations are not binding on the FDA. Companies also may submit a formal Request for Designation to the FDA’s Office of Combination Products. The Office of Combination Products will review the request and make its jurisdictional determination within 60 days of receiving a Request for Designation. We are currently discussing the potential regulatory pathway for DermaVir with the FDA. Based on those discussions to date, we believe that the product will be reviewed by the CBER and that the DermaPrep may be reviewed by the CDRH either in consultation with CBER as part of the Biologics License Application (“BLA”) or separately as a medical device.
Domestic Regulation of Our Products and Business
The testing, manufacturing, and potential labeling, advertising, promotion, distribution, import and marketing of our product candidates are subject to extensive regulation by governmental authorities in the U.S. and in other countries. In the U.S., the FDA, under the Public Health Service Act, the Federal Food, Drug and Cosmetic Act, and its implementing regulations, regulates biologics and medical device products.
The labeling, advertising, promotion, marketing and distribution of biopharmaceuticals, or biologics and medical devices also must be in compliance with the FDA and U.S. Federal Trade Commission (“FTC”) requirements which include, among others, standards and regulations for off-label promotion, industry sponsored scientific and educational activities, promotional activities involving the internet, and direct-to-consumer advertising. The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the issuance of a warning letter directing us to correct deviations from regulatory standards and enforcement actions that can include seizures, injunctions and criminal prosecution. Recently, promotional activities for FDA-regulated products of other companies have been the subject of enforcement action brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the federal Lanham Act and similar state laws, competitors and others can initiate litigation relating to advertising claims. In addition, we are required to meet regulatory requirements in countries outside the U.S., which can change rapidly with relatively short notice.
| 25 |
The FDA has broad post-market and regulatory enforcement powers. Manufacturers of biologics and medical devices are subject to unannounced inspections by the FDA to determine compliance with applicable regulations, and these inspections may include the manufacturing facilities of some of our subcontractors. Failure by manufacturers or their suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities. Potential FDA enforcement actions include:
- warning letters;
- civil or criminal penalties, fines and/or injunctions;
- product seizures or detentions;
- import or export bans or restrictions;
- voluntary or mandatory product recalls and related publicity requirements;
- suspension or withdrawal of regulatory approvals;
- total or partial suspension of production; and
- refusal to approve pending applications for marketing approval of new products or supplements to approved applications.
In addition, other government authorities influence the success of our business, including the availability of adequate reimbursement from third party payors, including government programs such as Medicare and Medicaid. Medicare and Medicaid reimbursement policies can also influence corresponding policies of private insurers and managed care providers, which can further affect our business.
Biologics Regulation
Biological products must satisfy the requirements of the Public Health Services Act and its implementing regulations. In order for a biologic product to be legally marketed in the U.S., the product must have a BLA approved by the FDA.
The BLA Approval Process
The steps for obtaining FDA approval of a BLA to market a biopharmaceutical, or biologic product in the U.S. include:
| · | completion of preclinical laboratory tests, animal studies and formulation studies under the FDA’s GLP regulations; |
| · | submission to the FDA of an IND application, for human clinical testing, which must become effective before human clinical trials may begin and which must include Institutional Review Board (“IRB”) approval at each clinical site before the trials may be initiated; |
| · | performance of adequate and well-controlled clinical trials in accordance with GCP to establish the safety, purity, and potency of the product for each indication; |
| · | submission to the FDA of a BLA, which contains detailed information about the chemistry, manufacturing and controls for the product, reports of the outcomes of the clinical trials, and proposed labeling and packaging for the product; |
| · | the FDA’s acceptance of the BLA for filing; |
| · | for any biological product containing an active ingredient not previously approved, automatic referral to an appropriate advisory committee for review prior to approval, unless the FDA decides otherwise; |
| · | satisfactory review of the contents of the BLA by the FDA, including the satisfactory resolution of any questions raised during the review or by the advisory committee, if applicable; |
| · | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMP regulations, to assure that the facilities, methods and controls are adequate to ensure the product’s identity, strength, quality and purity; and |
| 26 |
| · | FDA approval of the BLA. |
Preclinical studies include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. We submitted the results of these preclinical studies, together with manufacturing information and analytical data to the FDA in our Biologic Master File.
An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about issues such as the conduct of the trials as outlined in the IND. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed.
Clinical trials are subject to extensive monitoring, recordkeeping and reporting requirements. Clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial sites and must comply with FDA regulations, including but not limited to those relating to GCP. Adverse events must be reported and investigated timely. To conduct a clinical trial, a company is also required to obtain the patients’ informed consent in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations. The sponsor, the FDA or the IRB could suspend a clinical trial at any time for various reasons, including a belief that the risks to trial subjects outweigh the anticipated benefits. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each site at which the trial is conducted must approve the protocol and any amendments. Foreign studies performed under an IND must meet the same requirements that apply to U.S. studies. The FDA will accept a foreign clinical trial not conducted under an IND only if the trial is well-designed, well-conducted, performed by qualified investigators in accordance with international principles for GCP, and conforms to the ethical principles contained in the Declaration of Helsinki, or with the laws and regulations of the country in which the research was conducted, whichever provides greater protection of the human subjects. The FDA, however, has substantial discretion in deciding whether to accept data from foreign non-IND clinical trials.
Clinical trials involving biopharmaceutical products are typically conducted in three sequential phases. The phases may overlap or be combined. A fourth, or post-approval, phase may include additional clinical trials. These phases generally include the following:
| · | Phase I. Phase I clinical trials involve the initial introduction of the medicine into human subjects to determine the adverse effects associated with increasing doses. |
| · | Phase II. Phase II clinical trials usually involve studies in a limited patient population to evaluate the efficacy of the medicine for specific, targeted indications, to determine dosage tolerance and optimal dosage, and to identify possible adverse effects and safety risks. |
| · | Phase III. If the medicine is found to be potentially effective and to have an acceptable safety profile in Phase II (or sometimes Phase I) trials, the clinical trial program will be expanded to further demonstrate clinical efficacy, optimal dosage and safety within an expanded patient population at geographically dispersed clinical trial sites. |
| · | Post-Approval (Phase IV). Post-approval clinical trials are required of or agreed to by a sponsor as a condition of, or subsequent to marketing approval. Further, if the FDA becomes aware of new safety information about an approved product, it is authorized to require post approval trials of the biological product. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of biologics approved under accelerated approval regulations. If the FDA approves a product while a company has ongoing clinical trials that were not necessary for approval, a company may be able to use the data from these clinical trials to meet all or part of any Phase IV clinical trial requirement. These clinical trials are often referred to as Phase III/IV post approval clinical trials. Failure to promptly conduct Phase IV clinical trials could result in withdrawal of approval for products approved under accelerated approval regulations. |
| 27 |
Clinical testing may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted under an IND and may, at its discretion, reevaluate, alter, suspend, or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. The FDA or the sponsor may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request that additional pre-clinical studies or clinical trials be conducted as a condition to product approval. Additionally, new government requirements may be established that could delay or prevent regulatory approval of our products under development. Furthermore, IRBs have the authority to suspend clinical trials in their respective institutions at any time for a variety of reasons, including safety issues.
Certain information about clinical trials, including a description of the trial, participation criteria, location of trial sites, and contact information, is required to be sent to the NIH for inclusion in a publicly-assessable database. Sponsors also are subject to certain state laws imposing requirements to make publicly available certain information on clinical trial results. In addition, the FDA Amendments Act of 2007 directs the FDA to issue regulations that will require sponsors to submit to the NIH the results of certain controlled clinical trials, other than Phase I studies.
Assuming successful completion of the required clinical testing, the results of the preclinical studies and of the clinical trials, together with other detailed information, including information on the chemistry, manufacture and composition of the product, are submitted to the FDA in the form of a BLA requesting approval to market the product for one or more indications. In most cases, the BLA must be accompanied by a substantial user fee. The FDA will initially review the BLA for completeness before it accepts the BLA for filing. There can be no assurance that the submission will be accepted for filing or that the FDA may not issue a Refuse to File (“RTF”). If a RTF is issued, there is opportunity for dialogue between the sponsor and the FDA in an effort to resolve all concerns. If the BLA submission is accepted for filing, the FDA will begin an in-depth review of the BLA to determine, among other things, whether a product is safe and effective for its intended use and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. If the biological product contains a new active ingredient not previously approved, the BLA automatically will be referred to an appropriate advisory committee for review prior to approval of the biological product, unless the FDA decides otherwise and specifies such reasons in a complete response letter to the sponsor. The FDA, however, is not bound by the opinion of the advisory committee.
Companies also may seek fast track designation for their products. Fast track products are those that are intended for the treatment of a serious or life-threatening condition and that demonstrate the potential to address unmet medical needs for such a condition. If awarded, the fast track designation applies to the product only for the indication for which the designation was received. Fast track products are eligible for two means of potentially expediting product development and FDA review of BLAs. First, a fast track product may be approved on the basis of either a clinical endpoint or a surrogate endpoint that is reasonably likely to predict clinical benefit. Approvals of this kind may be subject to requirements for appropriate post-approval studies to validate the surrogate endpoint or otherwise confirm the effect on the clinical endpoint, and to certain other conditions. Second, if the FDA determines after review of preliminary clinical data submitted by the sponsor that a fast track product may be effective, it may begin review of portions of a BLA before the sponsor submits the complete BLA, thereby accelerating the date on which review of a portion of the BLA can begin. There can be no assurance that any of our other product candidates will receive designation as fast track products. And even if they are designated as fast track products, we cannot assure you that our product candidates will be reviewed or approved more expeditiously for their fast track indications than would otherwise have been the case or will be approved promptly, or at all. Furthermore, the FDA can revoke fast track status at any time.
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-approval clinical trials to verify and further define the drug’s clinical benefit and safety profile. There can be no assurance that any of our product candidates will receive accelerated approval. Even if accelerated approval is granted, the FDA may withdraw such approval if the sponsor fails to conduct the required post-approval clinical trials, or if the post-approval clinical trials fail to confirm the early benefits seen during the accelerated approval process.
| 28 |
Fast-Track designation and accelerated approval should be distinguished from priority review although products awarded fast track status may also be eligible for priority review. Products regulated by the CBER may receive priority review if they provide significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious or life-threatening disease. Products awarded priority review are given abbreviated review goals by the agency. Under the Prescription Drug User Fee Act of 2007, the agency has agreed to the performance goal of reviewing products awarded priority review within six months, whereas products under standard review receive a ten-month target. The review process, however, is often significantly extended by FDA requests for additional information or clarification regarding information already provided in the submission. Priority review is requested at the time the BLA is submitted, and the FDA makes a decision as part of the agency’s review of the application for filing. We plan to seek priority review for DermaVir but cannot guarantee that the FDA will grant the designation and cannot predict if awarded, what impact, if any, it will have on the review time for approval of our product.
If granted, Fast-Track designation, accelerated approval, and priority review may expedite the approval process, but they do not change the standards for approval.
Before approving a BLA, the FDA will generally inspect the facility or the facilities at which the finished product and its components are manufactured to ensure compliance with cGMP. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it will either issue “not approvable” letter or an “approvable” letter. A “not approvable” letter means that the FDA refuses to approve the application because the BLA or manufacturing facilities do not satisfy the regulatory criteria for approval. An “approvable” letter means that the FDA considers the BLA and manufacturing facilities to be favorable, but the letter will outline the deficiencies and provide the applicant with an opportunity to submit additional information or data to address the deficiencies. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. Separate approval is required for each proposed indication. If we want to expand the use of an approved product, we will have to design additional clinical trials, submit the trial designs to the FDA for review and complete those trials successfully.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. Data obtained from clinical activities are not always conclusive, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions, such as post approval studies, on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Requirements
After regulatory approval of a product is obtained, companies are required to comply with a number of post-approval requirements relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping. For example, as a condition of approval of a BLA, the FDA may require post-approval testing and surveillance to monitor the product’s safety or efficacy. In addition, holders of an approved BLA are required to keep extensive records, to report certain adverse reactions and production deviations and problems to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for their products. If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities, or previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
| 29 |
Specifically, our products could be subject to voluntary recall if we or the FDA determine, for any reason, that our products pose a risk of injury or are otherwise defective. Moreover, the FDA can order a mandatory recall if there is a reasonable probability that our device would cause serious adverse health consequences or death. In addition, the FDA could suspend the marketing of or withdraw a previously approved product from the market upon receipt of newly discovered information regarding the drug’s safety or effectiveness.
Medical Device Regulation
With respect to medical devices, the FDA has implemented numerous regulations to ensure that medical products distributed domestically or internationally are safe and effective for their intended uses. These include:
| · | product listing and establishment registration, which helps facilitate FDA inspections and other regulatory action; |
| · | the FDA’s QSR, which requires manufacturers, including third party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process; |
| · | labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label use or indications; |
| · | clearance or approval of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use; |
| · | medical device reporting regulations, which require that manufacturers comply with FDA requirements to report if their device may have caused or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the device or a similar device were to recur; |
| · | post-approval restrictions or conditions, including post-approval trial commitments; |
| · | post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device; and |
| · | notices of correction or removal and recall regulations. |
FDA’s Premarket Clearance and Approval Requirements
Unless an exemption applies, or if the FDA determines otherwise, before we can commercially distribute medical devices in the U.S., we must obtain, depending on the type of device, either prior 510(k) clearance or Premarket Approval (“PMA”) from the FDA. The FDA classifies medical devices into one of three classes:
| · | Class I devices (e.g., low risk devices), which are subject to only general regulatory oversight (e.g., labeling, medical devices reporting, and prohibitions against adulteration and misbranding) and, in some cases, to the 510(k) premarket clearance requirements; |
| · | Class II devices (e.g., greater risk devices), generally requiring 510(k) premarket clearance before they may be commercially marketed in the U.S.; and |
| · | Class III devices (e.g., greatest risk devices), consisting of devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a predicate device, generally requiring submission of a PMA supported by clinical trial data. |
| 30 |
All of our product candidates are currently in the development stage. DermaVir, our lead candidate, is currently undergoing clinical evaluation. We are currently developing our regulatory strategies with respect to which regulatory pathway will be necessary to obtain clearance or approval of the DermaPrep component, if medical device clearance or approval is required at all.
510(k) Clearance Pathway
When a 510(k) clearance is required, we must submit a premarket notification demonstrating that our proposed device is substantially equivalent to a previously cleared 510(k) device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of PMAs. By regulation, the FDA is required to clear or deny a 510(k) premarket notification within 90 days of submission of the application. As a practical matter, clearance may take longer. The FDA may require further information, including clinical data, to make a determination regarding substantial equivalence.
Any modification to a 510(k)-cleared device that would constitute a major change in its intended use, or any change that could significantly affect the safety or effectiveness of the device, requires a new 510(k) clearance and may even, in some circumstances, require a PMA, if the change raises complex or novel scientific issues or the product has a new intended use. The FDA requires every manufacturer to make the determination regarding the need for a new 510(k) submission in the first instance, but the FDA may review any manufacturer’s decision. We have modified our devices since they received the FDA clearance. If the FDA were to disagree with any of our determinations that changes did not require a new 510(k), it could require us to cease marketing and distribution and/or recall the modified device until 510(k) clearance or PMA approval is obtained. If the FDA requires us to seek 510(k) clearance or PMA approval for any modifications, we may be required to cease marketing and/or recall the modified device, if already in distribution, until 510(k) clearance or PMA approval is obtained and we could be subject to significant regulatory fines or penalties.
There is no guarantee that the FDA will grant 510(k) clearance or PMA approval of our future products, if necessary, and failure to obtain necessary clearances or approvals for our future products would adversely affect our ability to grow our business. Delays in receipt or failure to receive clearances or approvals, the loss of previously received clearances or approvals, or the failure to comply with existing or future regulatory requirements could reduce our sales, profitability and future growth prospects.
PMA Pathway
A PMA must be submitted to the FDA if the device cannot be cleared through the 510(k) process. A PMA must be supported by extensive data, including but not limited to, technical, preclinical, clinical trials, manufacturing and labeling to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device for its intended use. No device that we are marketing to date has required premarket approval. During the review period, the FDA will typically request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The FDA may or may not accept the panel’s recommendation. In addition, the FDA will generally conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with the FDA’s QSR.
New PMAs or PMA supplements are required for modifications that affect the safety or effectiveness of the device, including, for example, certain types of modifications to the device’s indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require as extensive clinical data or the convening of an advisory panel. There is no guarantee that the FDA will grant PMA approval of our future products, if necessary, and failure to obtain necessary approvals for our future products would adversely affect our ability to grow our business. Delays in receipt or failure to receive approvals, the loss of previously received approvals, or the failure to comply with existing or future regulatory requirements could reduce our sales, profitability and future growth prospects.
| 31 |
Clinical Trials
Clinical trials are generally required to support a PMA application and are sometimes required for 510(k) clearance. Such trials generally require an investigational device exemption application (“IDE”), approved in advance by the FDA for a specified number of patients and trial sites, unless the product is deemed a nonsignificant risk device eligible for more abbreviated IDE requirements. Clinical trials are subject to extensive monitoring, recordkeeping and reporting requirements. Clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial sites and must comply with FDA regulations, including but not limited to those relating to GCPs. To conduct a clinical trial, we are also required to obtain the patients’ informed consent in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations. We, the FDA or the IRB could suspend a clinical trial at any time for various reasons, including a belief that the risks to trial subjects outweigh the anticipated benefits. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA approval to market the product in the U.S. Similarly, in Europe the clinical trial must be approved by a local ethics committee and in some cases, including studies with high-risk devices, by the ministry of health in the applicable country.
Pervasive and Continuing Regulation
After a device is placed on the market, medical device companies are required to comply with regulations relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping. If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities, or previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with QSR and other aspects of regulatory compliance.
Specifically, our products could be subject to voluntary recall if we or the FDA determine, for any reason, that our products pose a risk of injury or are otherwise defective. Moreover, the FDA can order a mandatory recall if there is a reasonable probability that our device could cause serious adverse health consequences or death. In addition, the FDA could suspend the marketing of or withdraw a previously cleared or approved product from the market upon receipt of newly discovered information regarding the drug’s safety or effectiveness.
Health Care Regulation
Fraud and Abuse
Other government authorities influence the success of our business, including the availability of adequate reimbursement from third party payors, including government programs such as Medicare and Medicaid. Medicare and Medicaid reimbursement policies can also influence corresponding policies of private insurers and managed care providers, which can further affect our business.
We may directly or indirectly be subject to various federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback laws. In particular, the federal healthcare programs anti-kickback statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for a good or service, or the furnishing, arranging for or recommending a good or service, for which payment may be made in whole or part under federal healthcare programs, such as the Medicare and Medicaid programs. The anti-kickback statute is broad and prohibits or restricts many arrangements and practices that are lawful in businesses outside of the healthcare industry. In implementing the statute, the U.S. Department of Health and Human Services’ Office of Inspector General has issued a series of regulations, known as the “safe harbors,” which began in July 1991. These safe harbors set forth provisions that, if all their applicable requirements are met, will assure healthcare providers and other parties that they will not be prosecuted under the federal anti-kickback statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy all requirements of an applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the Office of Inspector General. Penalties for violations of the federal anti-kickback statute include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal healthcare programs.
| 32 |
The Federal False Claims Act prohibits persons from knowingly filing or causing to be filed a false claim to, or the knowing use of false statements to obtain payment from, the federal government. Certain types of lawsuits filed under the False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government. These individuals, sometimes known as “relators” or, more commonly, as “whistleblowers,” may share in any amounts paid by the entity to the government in fines or settlement. The number of filings of qui tam actions has increased significantly in recent years, causing more healthcare companies to have to defend a False Claim action. If an entity is determined to have violated the federal False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties ranging from $5,500 to $11,000 for each separate false claim. Various states have also enacted similar laws modeled after the federal False Claims Act which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
HIPAA created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government sponsored programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items, or services. A violation of this statute is a felony and may result in fines or imprisonment.
If any of our operations are found to have violated or be in violation of any of the laws described above and other applicable state and federal fraud and abuse laws, we may be subject to penalties, among them being civil and criminal penalties, damages, fines, exclusion from government healthcare programs, and the curtailment or restructuring of our operations.
Health Information
U.S. federal and state laws protect the confidentiality of certain health information, in particular individually identifiable information such as medical records, and restrict the use and disclosure of that protected information. At the federal level, HIPAA protects health information by regulating its use and disclosure, including for research purposes. Failure of a HIPAA “covered entity” (such as a hospital or academic medical center) to comply with HIPAA could constitute a violation of federal law, subject to civil and criminal penalties. We are not directly subject to the HIPAA rules as a “covered entity,” however, and under HIPAA we would be permitted to obtain information from our customers under certain conditions, such as when relevant to our responsibilities for overseeing the quality, safety, or effectiveness of our products. Nevertheless, because conduct by a person that may not be prosecuted directly under HIPAA’s criminal provisions could potentially be prosecuted under aiding and abetting or conspiracy laws, we are unable to determine whether our actions could be subject to prosecution in the event of an impermissible disclosure of data to us.
In addition, many state laws apply to the use and disclosure of health information, which could affect the manner in which we conduct our research and development, as well as other aspects of our operations. Moreover, such laws are not necessarily preempted by HIPAA and its rules, in particular those state laws that afford greater privacy protection to the individual than HIPAA and/or that protect certain categories of information, such as HIV/AIDS-related information. Such state health information confidentiality laws typically have their own penalty provisions, which could be applied in the event of an unlawful action regarding health information.
| 33 |
International Government Regulation
International sales of biological substances and medical devices are subject to foreign government regulations, which vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA licensure clearance or approval, and the requirements may differ.
The E.U., which consists of 25 of the major countries in Europe, has adopted numerous directives and standards regulating the design, manufacture, clinical trials, labeling, and adverse event reporting for medical devices. Other countries, such as Switzerland, have voluntarily adopted laws and regulations that mirror those of the E.U. with respect to medical devices. Devices that comply with the requirements of a relevant directive will be entitled to bear CE conformity marking and, accordingly, can be commercially distributed throughout the member states of the E.U., and other countries that comply with or mirror these directives. The method of assessing conformity varies depending on the type and class of the product, but normally involves a combination of self- assessment by the manufacturer and a third party assessment by a “Notified Body,” an independent and neutral institution appointed to conduct conformity assessment. This third party assessment consists of an audit of the manufacturer’s quality system and technical review and testing of the manufacturer’s product. An assessment by a Notified Body in one country within the E.U. is required in order for a manufacturer to commercially distribute the product throughout the E.U. In addition, compliance with voluntary harmonizing standards International Standards Organization 9001 and International Standards Organization 13845 issued by the International Organization for Standards establishes the presumption of conformity with the essential requirements for a CE marking.
ESTIMATE OF THE AMOUNT SPENT ON RESEARCH AND DEVELOPMENT
Research and development expenses were $0 and $0 in 2014 and 2013, respectively. We are focused on regulatory issues, including the delivery of data from all clinical trials to date. Currently no research and development is required.
COSTS AND EFFECTS OF ENVIRONMENTAL COMPLIANCE
The Company has not spent any sums on environmental compliance and does not expect to be required to spend any sums on environmental compliance in the future.
EMPLOYEES
We currently have eight (8) employees, of which two (2) are full-time and three (5) are part-time.
AVAILABLE INFORMATION
We file electronically with the U.S. Securities and Exchange Commission (SEC) our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. We make available on our website at www.powerofthedream.com, free of charge, copies of these reports as soon as reasonably practicable after filing these reports with, or furnishing them to, the SEC. The public can also obtain materials that we file with the SEC through the SEC’s website at http://www.sec.gov or at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. Information on the operation of the Public Reference Room is available by calling the SEC at 800-SEC-0330.
Item 1A. Risk Factors.
You should carefully consider the risks described below, together with all of the other information included in this report, in considering our business and prospects. The risks and uncertainties described below are not the only ones facing the Company. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations. The occurrence of any of the following risks could harm our business, financial condition or results of operations.
| 34 |
Risks Related to the Our Business and Industry
WE HAVE A LIMITED OPERATING HISTORY, WHICH MAKES YOUR EVALUATION OF OUR BUSINESS DIFFICULT. WE HAVE NEVER BEEN PROFITABLE DUE TO OUR DEVELOPMENT STAGE ACTIVITIES AND MAY NEVER BE PROFITABLE.
Our future is dependent upon our ability to obtain financing and upon future profitable operations from the commercialization of our product candidates. Our success and ability to generate revenue or be profitable will depend on our ability to, among other things, undertake pre-clinical development studies and clinical trials; apply for and receive regulatory approvals for conduct of clinical trials and for marketing of our products when development has been completed; formulate and manufacture products; and conduct sales and marketing activities. These factors raise substantial doubt that we will be able to continue as a going concern. From April 26, 2006 (inception) through December 31, 2014, we incurred aggregate losses of $44,854,780 and anticipate incurring additional losses for at least the next twelve (12) months.
Our auditors have expressed substantial doubt about our ability to continue as a going concern.
As of December 31, 2014, we had $1,524 of cash and $8,529,030 in current obligations. In addition, we anticipate our new operating expenses to be between $3,000,000 - $5,000,000 for year 2015. We do not have sufficient resources to satisfy our current obligations nor to fund our planned operations for the next 12 months. We will require additional financing to sustain our operations and without it we will not be able to continue operations.
Our ability to obtain additional funding will determine our ability to continue as a going concern. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Management believes the Company has enough existing cash and cash equivalent and can raise adequate capital to keep the Company functioning through December 31, 2015. However, the need may arise, to raise additional capital if we want to support our future development programs, including the completion of additional clinical trials. Our funding requirements may also increase or change as a result of many factors, including delays in development activities, underestimates of budget items, unanticipated cash requirements, limitation of development of new potential products, future product opportunities with collaborators, future licensing opportunities and future business combinations. No assurance can be given that the Company can obtain additional working capital, or if obtained, that such funding will not cause substantial dilution to shareholders of the Company. If the Company is unable to raise additional funds, if needed, it may not be able to (i) complete planned pre-clinical studies and clinical trials, (ii) complete construction of a manufacturing facility, or (iii) obtain conditional marketing approval for DermaVir from the EMA and/or other regulatory authorities. Being a development stage company, the Company is subject to all the risks inherent in the establishment of a new enterprise and the marketing and manufacturing of a new product, many of which risks are beyond the control of the Company. All of the factors discussed above raise substantial doubt about the Company’s ability to continue as a going concern.
Our future is dependent upon our ability to obtain financing. If we do not obtain such financing, we may be forced to discontinue product development, curtail operations, reduce or forego sales and marketing efforts and lose attractive business opportunities. We have no arrangements or agreements with any person regarding any potential future financings.
WE CURRENTLY DO NOT HAVE, AND MAY NEVER DEVELOP OR ACQUIRE ANY COMMERCIALIZED PRODUCTS FOR DISTRIBUTION AND SALE.
We currently do not have any
commercialized product(s) or any significant source of revenue. We have devoted
significant financial resources to research and
development of our product
candidates, but have not yet
commercialized any of
them. As a result, we have never
generated any revenue from product sales. Until and
unless we receive approval from the
EMA, FDA and foreign regulatory authorities
to market a product
candidate, we
cannot sell any product and will
have no product
revenues.
| 35 |
Physicians and patients may not accept and use our Immune Therapies.
Even if the EMA/FDA and/or foreign agencies approve one of our Immune Therapies for marketing, physicians and patients may not accept and use it. Acceptance and use of our products will depend upon a number of factors, including perceptions by the health care community, including physicians, about their safety and effectiveness, their cost-effectiveness relative to competing products, availability of reimbursement for our products from government or other healthcare payors, and the effectiveness of marketing and distribution efforts by us, our licensees and distributors, if any.
Because we expect sales of our current lead product candidate, if approved for marketing, to generate substantially all of our product revenues for the foreseeable future, the failure of this product to find market acceptance would harm our business and could require us to seek additional financing or take other measures, possibly retrenching.
Our ability to generate product revenues will be diminished if our products sell for inadequate prices or patients are unable to obtain adequate reimbursement.
Our ability to commercialize our products, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from government and health administration authorities, private health maintenance organization, health insurers, and other payors.
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payors, including Medicare, routinely challenge prices charged. Government and other healthcare payors increasingly attempt to contain health care costs by limiting both coverage and reimbursement. In the U.S., there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to market and sell our product candidates profitably. Although we cannot predict the full effect on our business of the implementation of any legislation, we believe that legislation that reduces reimbursement for our products could adversely impact how much or under what circumstances healthcare providers will prescribe or administer our products. This could materially and adversely impact our business by reducing our ability to generate revenue, raise capital, obtain additional collaborators and market our products. In addition, we believe the increasing emphasis on managed care in the U.S. has and will continue to put pressure on the price and usage of pharmaceutical products, which may adversely impact product sales.
Our product development programs depend upon third party researchers and we cannot control the amount of timing or resources they devote to our programs or the quality of their performance.
We depend in large part upon independent investigators and collaborators, such as universities, medical institutions, and clinical research organizations, to conduct our basic research, animal studies, pre-clinical studies and clinical trials and, in certain cases, to develop a product for its target indication. Although we contract with these collaborators, they are not our employees, and we cannot control the amount or timing of resources they devote to our programs. They may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking them ourselves. If outside collaborators fail to devote sufficient time and resources to our programs, or if their performance is substandard, the approval of our FDA and EMA applications, and our introduction of new products, if any, will be delayed. These collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators were to assist our competitors at our expense, our competitive position would be harmed.
| 36 |
We currently rely on third parties to manufacture and supply us with the raw materials that we assemble to make up our Product candidates, and any interruptions in the manufacture or supply of these raw materials could harm our business.
We currently rely on a limited number of vendors to supply raw materials for our product candidates, and the loss of one of these parties could significantly harm our business. For the clinical trials we have conducted up to date we relied on Althea Technologies, Inc., a DNA manufacturing company located in the U.S., as the source for our supply of pDNA, the API in DermaVir. We have also relied on PolyPlus Transfection SA, a manufacturer in France, as the source for our clinical supply of the raw material polyethylenimine mannose (“PEIm”) used in DermaVir. DermaPrep was manufactured by Tapemark, a medical device manufacturing company located in the U.S. The finished DermaVir product was manufactured at the clinical pharmacy prior to DermaVir immunization of patients.
In order to obtain the raw materials in the required quality and quantity we contacted several alternative CMOs. We tested the quality of pDNAs contract manufactured by Aldevron and WGX, two DNA manufacturing companies located in the U.S. Both pDNA materials could fulfill our quality criteria. To mitigate the risk on manufacturing CMOs (1) we licensed pDNA manufacturing technology from Aldevron for the high quality purification of our pDNA and (2) developed our own chemical synthesis for the manufacturing of PEIm. We also developed a commercial manufacturing method for our finished products. Therefore, based on our own technology our cGMP manufacturing facility could be built and we could be independent from any CMO to produce enough material for clinical trials. However, to build and staff such facility will be costly and time consuming. Therefore, if either of our selected CMO fails to provide us with sufficient quantities, we may not be able to obtain an alternative supply on a timely or commercially acceptable basis. Even if we are able to reach an agreement with back-up suppliers, a change in supplier may require comparability testing, including clinical tests, to demonstrate that the new raw material will not substantially change the finished Product. Furthermore, even if an alternative source is available, there can be no assurance that such alternative source would be capable of producing the raw materials in sufficient quantities to meet our total requirements. Any such interruption would disrupt our ability to manufacture the DermaVir and could have a material adverse effect on our business.
We identified Stokvis Tapes Group a Hungarian manufacturer with worldwide presence specialized for patches for health industry. Stokvis presented us a sample batch of DermaPrep to demonstrate their capability to manufacture our DermaPrep medical device. The sample batch fulfilled our quality criteria. We plan to contract Stokvis to deliver DermaPrep for clinical trials. This company would also have the capability and capacity to produce our device also in commercial quantity.
If there is a significant increase in the price of raw materials or DermaPrep, our business could be seriously harmed. We currently have no supply contracts beyond current clinical trials. We may not be able to contract with third parties to provide the API and DermaPrep beyond current clinical trials on acceptable terms and at commercially reasonable prices, if at all. The number of third parties with the expertise, required regulatory approvals, and facilities to supply the API and the finished Product on a commercial scale is limited. Even if we locate third parties that can provide commercial supplies, the FDA and comparable foreign regulators must approve these manufacturers’ facilities and processes prior to our use, which would require new testing and compliance inspections. Any of these factors could cause us to delay or suspend regulatory submissions, required approvals or commercialization of DermaVir. They could also entail higher costs, and could result in our being unable to commercialize DermaVir successfully. Furthermore, if our contract manufacturers fail to deliver the required commercial quantities of bulk raw materials on a timely basis and at commercially reasonable prices, and if we were unable to find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volumes and quality, and on a timely basis, we are likely to be unable to meet the demand for DermaVir, if marketing approval is obtained, and we would lose potential revenue.
WE DO NOT CURRENTLY HAVE SALES, MARKETING AND DISTRIBUTION CAPABILITIES, AND WE MAY BE UNABLE TO EFFECTIVELY SELL, MARKET AND DISTRIBUTE OUR PRODUCT CANDIDATES IN THE FUTURE, WHICH WOULD HAVE AN ADVERSE EFFECT ON OUR BUSINESS.
| 37 |
We currently have no internal sales or distribution capabilities. In order to successfully commercialize any of our product candidates, we must either internally develop sales, marketing and distribution capabilities or make arrangements with third parties to perform these services. For the foreseeable future, we may not be able to establish marketing and sales capabilities internally or hire a sufficient number of sales personnel with appropriate expertise to market and sell our products when the products are approved by appropriate regulatory authorities. Therefore, we may need to enter into an alliance with a major pharmaceutical company or other third party to market and sell our products, which we may be unable to do. Even if we are able to identify one or more acceptable collaborators to perform these services for us, we may not be able to enter into collaborative arrangements on favorable terms, or at all.
If we enter into collaborative arrangements for the marketing or sale of our products, our product revenues are likely to be lower than if we directly marketed and sold our products. In addition, any revenues we receive would depend upon the efforts of our collaborators, which may not be adequate due to lack of attention or resource commitments, management turnover, and/or change of strategic focus, business combinations or other factors outside our control. Depending on the terms of our collaboration, the remedies we have against an underperforming collaborator may be limited. If we were to terminate the relationship, it could be difficult or impossible to find a replacement collaborator on acceptable terms, if at all.
If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues, and our business will suffer.
The HIV/AIDS market for our lead product, DermaVir, is characterized by intense competition and rapid technological advances. If any of our product candidates receive marketing approval, it will compete with a number of existing and future drugs and Immune Therapies developed, manufactured and marketed by others. The extent to which DermaVir or any of our other Product candidates achieves market acceptance will depend in part on our ability to compete with these other companies. Competition in the pharmaceutical and biotechnology industries is intense and has been accentuated by the rapid pace of technology development. If our competitors develop and commercialize products faster than we do, or develop and commercialize products that are superior to our product candidates, our commercial opportunities will be reduced or eliminated. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products fail to capture and maintain market share, we may not achieve sufficient product revenues, and our business will suffer.
We compete against larger, fully integrated pharmaceutical companies, smaller companies that collaborate with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Companies that currently sell HIV/AIDS products include Bristol-Myers Squibb Company, F. Hoffmann-La Roche Ltd., Gilead Sciences, Inc., GlaxoSmithKline plc., Merck & Co., Inc., Novartis AG, Pfizer Inc., Sanofi-Aventis U.S. LLC. Alternative technologies are being developed to improve upon or replace current products for the treatment of HIV/AIDS, several of which are in clinical trials or are awaiting marketing approval. In addition, companies pursuing different but related fields represent substantial competition. Many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs and have substantially greater financial resources than we do. Additionally, they have significantly greater experience in developing drugs, undertaking pre-clinical studies and human clinical trials, obtaining EMA and FDA and other regulatory approvals and formulating, manufacturing, launching, marketing and selling drugs. These organizations also compete with us to attract qualified personnel, license proprietary technology that is competitive with the technology we are developing, attract funding, and attract parties for acquisitions, joint ventures or other collaborations.
We may be exposed to liability claims associated with using hazardous materials and chemicals.
Our research and development activities involve the controlled use of hazardous materials and chemicals. Although we believe our safety procedures for using, storing, handling, and disposing of these materials comply with applicable environmental laws and regulations, we cannot eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we could be held liable for resulting damages, which could materially adversely affect our business, financial condition and results of operations. In addition, laws and regulations governing the use, manufacture, storage, handling, and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations.
| 38 |
We may incur substantial liabilities and be required to limit commercialization of our products in response to product liability lawsuits.
Our proposed products could be the subject of product liability claims. A failure of our product candidates to function as anticipated, whether as a result of the design of these products, unanticipated health consequences or side effects, or misuse or mishandling by third parties of such products, could result in injury. We may be held liable if serious adverse reactions from the use of our product candidates occur either in clinical trials or in subsequent marketing. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Our inability to obtain sufficient product liability insurance at acceptable cost against claims could prevent or inhibit the commercialization of biopharmaceutical products we develop, alone or with collaborators. We currently carry clinical trial insurance but do not carry product liability insurance. We, or any collaborators, may not be able to obtain insurance at reasonable cost, if at all. Agreements with future collaborators entitling us to indemnification against losses may not be adequate if claims arise. We may also be required to indemnify collaborators against losses arising out of product liability claims.
THERE ARE RISKS ASSOCIATED WITH OUR PROPOSED OPERATIONS IN HUNGARY.
Special risks may be associated with our efforts to undertake operations in the Republic of Hungary. Such operations will be subject to political, economic and other uncertainties, including among other things, import, export and transportation regulations, tariffs, taxation policy, including royalty and tax increases and retroactive tax claims, exchange controls, currency fluctuations and other uncertainties arising out of the Republic of Hungary’s sovereignty over our operations.
FLUCTUATION IN THE VALUE OF THE HUNGARIAN FORINT RELATIVE TO OTHER CURRENCIES MAY HAVE A MATERIAL ADVERSE EFFECT ON OUR BUEINSS AND/OR AN INVESTMENT IN OUR SHARES.
We maintain our books in local currency: U.S. dollars for Power of the Dream and for Genetic Immunity, Inc. in the United States and the Hungarian Forint for Vidatech and Genetic Immunity, Kft in Hungary. Our operations are conducted primarily outside of the United States through Vidatech and Genetic Immunity, Kft., our wholly-owned subsidiaries. As a result, fluctuations in currency exchange rates may significantly affect our sales, profitability and financial position when the foreign currencies, primarily the Hungarian Forint, of our international operations are translated into U.S. dollars for financial reporting. During 2014, the Hungarian Forint has fluctuated between HUF 260 and HUF 290 to the U.S. dollar. In addition, we are also subject to currency fluctuation risk with respect to certain foreign currency denominated receivables and payables. Although we cannot predict the extent to which currency fluctuations may or will affect our business and financial position, there is a risk that such fluctuations will have an adverse impact on our sales, profits and financial position. Because differing portions of our revenues and costs are denominated in foreign currency, movements could impact our margins by, for example, decreasing our foreign revenues when the dollar strengthens and not correspondingly decreasing our expenses. We do not currently hedge our currency exposure. In the future, we may engage in hedging transactions to mitigate foreign exchange risk.
OUR MANAGEMENT TEAM HAS LIMITED EXPERIENCE IN PUBLIC COMPANY MATTERS, WHICH COULD IMPAIR OUR ABILITY TO COMPLY WITH LEGAL AND REGULATORY REQUIREMENTS.
| 39 |
Our management team has had limited experience managing a U.S. public company, which could impair our ability to comply with legal and regulatory requirements, such as the Sarbanes-Oxley Act of 2002 and applicable federal securities laws, including filing on a timely basis required reports and other required information. Our management may not be able to implement programs and policies in an effective and timely manner that adequately responds to increased legal or regulatory compliance and reporting requirements imposed by such laws and regulations. Our failure to comply with such laws and regulations could lead to the imposition of fines and penalties and further result in the deterioration of our business.
INVESTOR CONFIDENCE AND THE MARKET PRICE OF OUR SHARES MAY BE ADVERSELY IMPACTED BY OUR ADVERSE OPINION ON THE EFFECTIVENESS OF OUR INTERNAL CONTROLS OVER OUR FINANCIAL REPORTING.
We are subject to the reporting requirements of the U.S. Securities and Exchange Commission, or SEC. The SEC, as directed by Section 404 of the U.S. Sarbanes-Oxley Act of 2002, adopted rules requiring public companies to include a report of management on their internal control structure and procedures for financial reporting in their annual reports on Form 10-K that contain an assessment by management of the effectiveness of their internal controls over financial reporting. These requirements first applied to our annual report on Form 10-KSB for the fiscal year ending on December 31, 2007. For the past several years we have reported ineffective internal controls over financial reporting due to material weaknesses related to insufficient staffing and lack of top level reviews. We have been unable to remediate these weaknesses due our limited resources. Our failure to implement effective controls could harm our operating results. Any failure to implement effective controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our reporting obligations. Even if our management concludes that our internal controls over financial reporting are effective, our independent registered public accounting firm may issue a report that is qualified if it is not satisfied with our controls or the level at which our controls are documented, designed, operated or reviewed, or if it interprets the relevant requirements differently from us. In the event that we are unable to have effective internal controls, investors and others may lose confidence in the reliability of our financial statements and our ability to obtain equity or debt financing as needed could suffer.
CONCENTRATION OF OWNERSHIP AMONG OUR DIRECTORS, EXECUTIVE OFFICERS AND PRINCIAPL SHAREHOLDERS MAY PRESENT NEW INVESTORS FROM INFLUENCE SIGNIFICANT CORPORATE DECISIONS.
Based upon beneficial ownership as of December 31, 2014, our directors, executive officers and holders of more than 5% of our common stock, alone or together with their affiliates own, in the aggregate, approximately 12% of our outstanding shares of common stock. Our executive officer, Viktor Rozsnyay, and Peter Boros, Chief Executive Officer of iGlue, Inc., own 100% of our Series A Preferred Stock, which gives them majority voting power. As a result, these shareholders, will be able to exercise a controlling influence over matters requiring shareholder approval, including the election of directors and approval of significant corporate transactions, and will have significant control over our management and policies. Some of these persons or entities may have interests that are different from yours. For example, these shareholders may support proposals and actions with which you may disagree or which are not in your interests. The concentration of ownership could delay or prevent a change in control of our company or otherwise discourage a potential acquirer from attempting to obtain control of our company, which in turn could reduce the price of our common stock. In addition, these shareholders, some of whom have representatives sitting on our Board of Directors, could use their voting influence to maintain our existing management and directors in office, delay or prevent changes of control of our company, or support or reject other management and board proposals that are subject to stockholder approval, such as amendments to our employee stock plans and approvals of significant financing transactions.
WE DEPEND ON KEY EMPLOYEES,
AND IF WE FAIL TO ATTRACT AND RETAIN EMPLOYEES WITH EXPERTISE REQUIRED FOR OUR BUSINESS, WE CANNOT GROW OR ACHIEVE PROFITABILITY.
We are dependent on our executive officers and key employees, primarily Viktor Rozsnyay, our Chief Executive Officer of Power of the Dream, Ildiko Rozsa, Chief Financial Officer of Power of the Dream and Peter Boros a member of the board of directors. We do not have “key person” life insurance policies for any of our officers. The loss of the technical knowledge and management and industry expertise of any of our key personnel could result in delays in product development, loss of customers and sales, if any, and diversion of management resources, which could adversely affect our operating results. In addition, we rely on clinical and regulatory advisors to assist us in formulating the development strategy. Our advisors have other jobs and commitments, and may be subject to non-disclosure obligations that may limit their availability to work with us.
| 40 |
If we are unable to hire qualified personnel, our ability to grow our business may be harmed. We must hire and retain skilled employees in a tight labor market.
Attracting and retaining qualified personnel is critical to our success. We need to hire additional qualified personnel with expertise in basic and clinical research, government regulation, formulation and manufacturing, and sales and marketing. We will require additional scientific personnel in many fields, some of which are addressed by relatively few companies. As a result, depending upon the success and the timing of clinical trials, we may experience difficulty in hiring and retaining highly skilled employees.
We compete for qualified individuals with numerous pharmaceutical and biotechnology companies, universities and other research institutions, some of which are more established than we are and have the ability to pay more cash compensation than we do. Competition for such individuals, particularly in Budapest, Hungary, is intense, and we cannot be certain that our search for such personnel will be successful. If we are unable to hire and retain skilled scientists, our business, financial condition, operating results and future prospects could be materially adversely affected.
If we are unable to obtain adequate insurance, our financial condition could be adversely affected in the event of uninsured or inadequately insured loss or damage. Our ability to effectively recruit and retain qualified officers and directors could also be adversely affected if we experience difficulty in obtaining adequate directors’ and officers’ liability insurance.
We may not be able to obtain insurance policies on terms affordable to us that would adequately insure our business and property against damage, loss or claims by third parties. To the extent our business or property suffers any damages, losses or claims by third parties, which are not covered or adequately covered by insurance, our financial condition may be materially adversely affected.
We may be unable to maintain sufficient insurance as a public company to cover liability claims made against our officers and directors. If we are unable to adequately insure our officers and directors, we may not be able to retain or recruit qualified officers and directors to manage us.
OUR COMPLIANCE WITH CHANGING LAWS AND RULES REGARDING CORPORATE GOVERNANCE AND PUBLIC DISCLOSURE MAY RESULT IN ADDITIONAL EXPENSES TO US WHICH, IN TURN, MAY ADVERSELY AFFECT OUR ABILITY TO CONTINUE OUR OPERATIONS.
Keeping abreast of, and in compliance with, changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, new SEC regulations and, in the event we are ever approved for listing on either NASDAQ or a registered exchange, NASDAQ and stock exchange rules, will require an increased amount of management attention and external resources. We intend to continue to invest all reasonably necessary resources to comply with evolving standards, which may result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities. This could have a materially adverse effect on our ongoing operations.
| 41 |
Risks Related to Regulatory Environment
NONE OF OUR PRODUCT CANDIDATES HAVE BEEN APPROVED BY E.U., U.S., AND/OR WORLDWIDE REGULATORY AUTHORITIES FOR COMMERCIAL SALE, AND WE MAY NEVER RECEIVE SUCH APPROVALS. OBTAINING AND MAINTAINING THE NECESSARY E.U. OR U.S. AND/OR WORLDWIDE MARKETING APPROVALS FOR OUR PRODUCT CANDIDATES MAY BE TIME CONSUMING, DIFFICULT, COMPLEX AND COSTLY, PARTICULARLY SINCE OUR LEAD PRODUCT AND OTHER PRODUCT CANDIDATES ARE COMBINATION PRODUCTS BASED ON NOVEL MECHANISMS OF ACTION. IF WE FAIL TO OBTAIN AND MAINTAIN THE NECESSARY APPROVALS, WE WILL BE UNABLE TO COMMERCIALIZE OUR PRODUCT CANDIDATES.
None of our product candidates have been approved by the E.U. or U.S. or worldwide regulatory authorities for commercial sale, and we may never receive such approvals. All of our product candidates are in pre-commercial stages, and we do not expect any of our product candidates to be commercially available for several years, if at all. Government regulations in the E.U. or the U.S. and other countries have a significant impact on our business and affect research and development, manufacture and marketing of our products. We will need EMA and FDA approval to commercialize our product candidates in the E.U, the U.S., and approvals from similar foreign regulatory authorities to commercialize our product candidates in other such jurisdictions. In order to obtain EMA and FDA marketing approval of a product candidate, we must submit to the EMA and the FDA a Biologics License Application (a “BLA”) demonstrating, to the satisfaction of the EMA and the FDA and other regulatory authorities, that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant laboratory research and animal tests, which are referred to as pre- clinical studies, as well as adequate and well-controlled human tests, which are referred to as clinical trials. These studies and trials may be time consuming, difficult and costly, and we cannot predict whether our research and clinical approaches will result in products the EMA, the FDA and other regulatory authorities consider safe for humans and effective for indicated uses. The EMA and the FDA has substantial discretion, to be exercised in the public interest, in the approval process, and may either refuse to accept our application, or may decide after review that our data are insufficient to allow approval.
Certain clinical trials for DermaVir will be conducted in Europe. Although the FDA may accept a foreign clinical trial to support approval of a BLA, the trial must be well- designed, well-conducted, performed by qualified investigators in accordance with international principles for Good Clinical Practice (“GCP”), and must conform to the ethical principles contained in the Declaration of Helsinki, or with the laws and regulations of the country in which the research was conducted, whichever provides greater protection of the human subjects. The EMA and the FDA has substantial discretion in deciding whether to accept data from our foreign clinical trials, and if the agency does not accept such data, our product development and commercialization efforts for the DermaVir or our other product candidates could be curtailed or significantly delayed.
Once we submit our BLA, there can be no assurance that the BLA will be accepted for filing or that the FDA may not issue a Refusal to File (“RTF”), if it believes the filing is inadequate or incomplete. Even if the FDA accepts the BLA for filing, data obtained from pre-clinical studies and clinical trials are susceptible to varying interpretations. If the FDA disagrees with our interpretation and does not accept or approve our application, it may require us to conduct additional pre-clinical studies, clinical trials, or manufacturing studies and submit that data before it will reconsider our application, or require us to perform post-approval studies. Delays in obtaining regulatory approvals will most likely delay commercialization of, and our ability to derive product revenues from, our product candidates, impose costly procedures on us, and diminish the competitive advantages that we may otherwise enjoy.
Our lead product candidate, DermaVir, and our other product candidates, are immune therapies, which utilize a novel mechanism of action. Therefore, regulatory agencies may lack experience with them, which may lengthen the regulatory review process, increase our development costs and delay or prevent our commercialization of DermaVir and our other Immune Therapy Products under development. To date, the EMA nor the FDA has not approved for commercial sale in the U.S. any Immune Therapy product designed to stimulate an immune response against HIV. Consequently, there is no precedent for the successful commercialization of products based on our technologies in this area.
| 42 |
For our lead candidate, we intend to seek fast track designation, and conditional marketing approval, and priority review designation, all of which are various regulatory processes intended to accelerate drug development, review, or approval for any Product candidates. Fast track designation is a regulatory mechanism intended to facilitate the development and expedite the review of products intended for the treatment of serious or life- threatening conditions and that demonstrate the potential to address an unmet medical need for such a condition. Accelerated marketing approval is reserved for a product that demonstrates a meaningful therapeutic advantage over existing treatments or shows the potential to address an unmet medical need in a serious or life-threatening condition. In some cases, a product receiving accelerated approval may be approved on the basis of a surrogate measure of benefit, in which case further clinical trials (as post-approval commitments) are generally required to further define the safety and efficacy of the product. Priority review designation would allow DermaVir to be reviewed in a shorter time frame. There can be no assurance, however, that the FDA will grant DermaVir fast track designation or accelerated approval or will award the BLA priority review. Further, these regulatory processes do not change the standards for approval and do not guarantee that our product will be approved.
We have limited experience in developing our Immune Therapy products, which consist of nanoparticles assembled from plasmid DNA and a polymer with special properties. Due to their special properties, nanomedicine formulations may pose different issues than non-nanoscale products. It is unknown whether the nanoscale size of these products may magnify safety and effectiveness issues. We also do not know whether the use of nanoscale materials may change the regulatory status of our products. The FDA may require us to conduct additional toxicology tests and clinical trials to confirm the safety and effectiveness of our nanomedicine Product candidates, which would impact our development plan.
Even if we comply with all EMA and FDA requests for data and information, the EMA or the FDA may ultimately determine, under its statutory authority, to reject one or more of our BLAs. We cannot be certain that we will ever obtain regulatory approval or clearance for any product candidate. Failure to obtain EMA and/or FDA approval or clearance of any of our principal product candidates will severely undermine our business by reducing our number of saleable products and, therefore, corresponding product revenues. Even if our product candidates obtain marketing approval, the EMA and the FDA may impose restrictions on the approved indications or distribution of the product. Also, the EMA and the FDA might approve one or more of our product candidates for marketing but may also approve competitors’ products with characteristics that offer their own treatment advantages.
Our lead product also is a combination product and includes a new medical device that may require medical device clearance or approval separate from the biologics approval. This clearance or approval will need to be obtained from a group within the EMA and the FDA, which may be different from the group acting on the BLA. This makes the approval process more complex and could delay approval.
In foreign jurisdictions, approval from the appropriate regulatory, pricing and reimbursement authorities must be obtained before commercialization and marketing of our Products can begin. These processes generally include all of the risks associated with EMA and FDA procedures. Pursuing foreign approvals will be time-consuming and expensive. Regulations vary among countries, and foreign authorities may require different or additional clinical trials than those required to obtain EMA and FDA approval.
Our Product candidates, including our lead Product candidate, are combination products comprised of a biologic and medical device component. We cannot be sure how the EMA and the FDA will regulate our product candidates. The EMA and the FDA may require us to obtain marketing clearance and approval from two different EMA and FDA centers. The review of combination products is often more complex and more time consuming than the review of product candidates under the jurisdiction of only one center within the EMA and the FDA.
| 43 |
Our product candidates require EMA and FDA authorization by means of an approval or clearance prior to marketing. Some of our product candidates, including DermaVir, will likely be regulated as combination products. For a combination product, the EMA and the FDA must determine which center or centers within the EMA and the FDA will review the product candidate and under what legal authority the product candidate will be reviewed. We are currently developing our regulatory strategies with respect to which regulatory pathway will be necessary to obtain clearance or approval of the DermaPrep component of DermaVir, if medical device clearance or approval is required at all. Based on discussions with the FDA we believe that the biologic component of DermaVir will be reviewed by the Center for Biologics Evaluation and Research (“CBER”) and that the DermaPrep device may be reviewed by the Center for Devices and Radiological Health (“CDRH”) either in consultation with CBER as part of the BLA or separately as a medical device. The process of obtaining FDA marketing clearance or approval is lengthy, expensive, and uncertain, and we cannot be sure that our biologic-device combination product candidates, or any other product candidates, will be cleared or approved in a timely fashion, or at all. In addition, the review of combination products is often more complex and more time consuming than the review of a product candidate under the jurisdiction of only one center within the FDA. We cannot be sure that the FDA will not select to have our combination products reviewed and regulated by only one FDA center and/or different legal authority, in which case the path to regulatory approval would be different and could be more lengthy and costly. If the FDA does not approve or clear our product candidates in a timely fashion or at all, our business and financial condition will be adversely affected.
We have only limited experience in regulatory affairs, and some of our product candidates are based on novel mechanisms of action. These factors may affect our ability or the time we require to obtain necessary marketing approvals.
We have only limited experience in preparing and filing the applications necessary to gain marketing approvals. Moreover, some of our product candidates are based on novel mechanisms of action that have not been extensively tested in humans. The regulatory requirements governing these types of product candidates may be less well defined or more rigorous than for conventional products. As a result, we may experience a longer regulatory process in connection with obtaining marketing approvals of any products that we develop.
Our product candidates will remain subject to ongoing regulatory requirements even if they receive marketing approval, and if we fail to comply with these requirements, we could lose these approvals, and the sales of any approved commercial products could be suspended.
Even if we receive approval to market a particular product candidate, the product will remain subject to extensive regulatory requirements, including requirements relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, product deviation reporting, distribution and recordkeeping. Even if marketing approval is granted, it may contain requirements for costly post-approval testing and surveillance to monitor the safety or efficacy of the product, which could negatively impact us or our collaboration partners by reducing revenues or increasing expenses, and cause the approved product candidate not to be commercially viable. In addition, as clinical experience with a biologic, drug or medical device expands after approval or clearance, typically because it is used by a greater number and more diverse group of patients after approval than during clinical trials, side effects and other problems may be observed after approval that were not seen or anticipated during pre-approval clinical trials or other studies. Any adverse effects observed after marketing of a product could result in limitations on the use of or withdrawal of such product from the marketplace. Absence of long-term safety data may also limit the approved uses of our products, if any. If we fail to comply with the regulatory requirements of the EMA and the FDA and other applicable E.U., U.S. and foreign regulatory authorities, or previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks, including the following:
| · | restrictions on the products, manufacturers or manufacturing process; |
| 44 |
| · | civil or criminal penalties, fines and/or injunctions; |
| · | unanticipated expenditures to address or defend such actions; |
| · | product seizures or detentions; |
| · | orders for physician notification or device repair, replacement or refund; |
| · | import or export bans or restrictions; |
| · | voluntary or mandatory product recalls and related publicity requirements; |
| · | suspension or withdrawal of regulatory clearances and approvals; |
| · | total or partial suspension of production; and |
| · | refusal to approve pending applications for marketing approval or clearance of new products or supplements to approved applications. |
If any of these actions were to occur it would harm our reputation and cause our product sales and profitability to suffer and might prevent us from generating revenue. Furthermore, our key component suppliers may not currently be or might not continue to be in compliance with all applicable regulatory requirements, which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
In addition, if the EMA and the FDA becomes aware of new post-approval safety information, the EMA and FDA is authorized to require us to conduct post approval studies, including clinical trials to investigate known serious risks or signals of serious risks, or identifying unexpected serious risks. If we are required to conduct such studies, we must submit periodic status reports to the EMA and FDA. Failure to conduct such post-approval studies in a timely manner may result in substantial civil fines.
If we or our collaborators are slow to adapt, or are unable to adapt, to changes in existing regulatory requirements or adoption of new regulatory requirements or policies, marketing approval for our product candidates may be lost or cease to be achievable, resulting in decreased revenue from milestones, product sales or royalties, which would have a material adverse effect on our results of operations.
We may be subject to fines, penalties or injunctions if we are determined to be promoting the use of our products for unapproved or “off-label” uses.
Our promotional materials and training methods must comply with EMA and FDA and other applicable laws and regulations. If the EMA and FDA determines that our promotional materials or training constitutes promotion of an unapproved use of our product candidates, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and adoption of the products would be impaired.
If the third parties on which we rely to conduct our clinical trials and to assist us with pre-clinical development do not perform as contractually required or expected, we may not be able to obtain regulatory approval for or commercialize our products.
We do not have the ability to independently conduct some of our pre-clinical studies and clinical trials for our products and we must rely on third parties, contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct such studies. Although we rely on third parties to conduct our pre- clinical studies and clinical trials, we are not relieved from the responsibility of ensuring that such studies and trials are conducted in accordance with regulatory requirements. Some pre-clinical studies must comply with current Good Laboratory Practice (“cGLP”) requirements. Clinical trials must be conducted in accordance with the investigational plan and protocol, as well as current Good Clinical Practice (“cGCP”) requirements for conducting, monitoring, maintaining records, and reporting adverse events and the results of clinical trials to ensure, among other things, that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties to conduct pre-clinical studies and clinical trials does not relieve us of the responsibility for complying with regulatory obligations. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize, our products on a timely basis, if at all, and our business, operating results and prospects may be adversely affected. Furthermore, our third party clinical trial investigators may be delayed in conducting our clinical trials for reasons outside of their control.
| 45 |
We and our contract manufacturers are subject to significant regulation with respect to manufacturing of our products.
All of those involved in the preparation of a biologic or medical device for clinical trials or commercial sale, including our existing third party suppliers and manufacturers, are subject to extensive regulation. The finished product and its components used for commercial sale or in clinical trials must be manufactured in accordance with current Good Manufacturing Practices (“cGMP”), a series of complex regulations and recommendations in guidance documents. The medical device cGMPs are codified in the Quality System Regulation (“QSR”) promulgated by the FDA. The cGMP and QSR requirements govern manufacturing processes and procedures and the implementation and operation of quality systems to control and assure the quality of investigational products and products approved for commercial distribution. Our facilities and quality systems and the facilities and quality systems of our third party suppliers and manufacturers may be subject to a pre-approval inspection for compliance with the applicable regulations as a condition of FDA approval of DermaVir or any of our other potential products. In addition, the FDA or the European Medicines Agency (“EMA”) may, at any time, audit or inspect a manufacturing facility involved with the preparation of our products, its components, or our other product candidates or the associated quality systems for compliance with the regulations applicable to the activities being conducted. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulation occurs independent of such an inspection or audit, we or the FDA may require remedial measures that may be costly and/or time consuming for us or a third party to implement and that may include the temporary or permanent suspension of a clinical trial or commercial sales or the temporary or permanent closure of a facility. Material violations of cGMP or QSR requirements could result in regulatory sanctions and, in severe cases, could result in a mandated closure of our or our third party’s facilities. Any such remedial measures imposed upon us or third parties with whom we contract could harm our business.
Our products may in the future be subject to product recalls that could harm our reputation, business and financial results.
The EMA, FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies, safety issues, or defects in design or manufacture. Manufacturers may, under their own initiative, recall a product if any material deficiency, or safety issue in a product is found. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our financial condition and results of operations. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the EMA and FDA could take enforcement action against us if such recall is the result of failure(s) to comply with applicable regulatory requirements, or if we fail to report the recalls within required timeframes.
FAILURE TO OBTAIN MARKETING APPROVAL IN ADDITIONAL FOREIGN JURISDICTIONS WILL PREVENT US FROM EXPANDING THE COMMERCIALIZATION OF OUR PRODUCTS ABROAD.
| 46 |
We intend to market our products in a number of international markets. In order to market our products in foreign jurisdictions we must obtain separate regulatory approvals. The approval procedure varies among jurisdictions and can involve substantial additional testing. Approval or clearance by the EMA and the FDA does not ensure approval by regulatory authorities in other jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign jurisdictions or by the FDA. The foreign regulatory approval process may include all of the risks associated with obtaining EMA or FDA approval in addition to other risks. In addition, the time required to obtain foreign approval may differ from that required to obtain EMA and FDA approval and we may not obtain foreign regulatory approvals on a timely basis, if at all.
FEDERAL REGULATORY REFORMS MAY ADVERSELY AFFECT OUR ABILITY TO SELL OUR PRODUCTS PROFITABLY.
From time to time, legislation is drafted and introduced in the European Union and in the United States Congress (“Congress”) that could significantly change the statutory provisions governing the marketing approval, manufacture and marketing of a regulated product. In addition, EMA and FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted or EMA and FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be.
Without limiting the generality of the foregoing, Congress has recently enacted, and the President has signed into law, the Food and Drug Administration Amendments Act of 2007 (the “FDA Amendments”). The FDA Amendments require certain information about clinical trials for biological products, including a description of the trial, participation criteria, location of trial sites, and contact information, to be sent to the National Institutes of Health (“NIH”) for inclusion in a publicly-assessable database. We also will be required to submit to the NIH the results of certain clinical trials, other than Phase I studies, for our biological products. Once implemented, compliance with those regulations may require us to take additional steps in the development and manufacture of our products and labeling. These steps may require additional resources and could be costly.
In addition, Congress could take action to create an abbreviated regulatory pathway for biological products. If this were to occur, then our Products may be subject to competition from follow-on versions.
The regulatory climate for follow-on versions of biological products approved under a BLA in the U.S. remains uncertain. Currently, there is no established statutory or regulatory pathway for the abbreviated approval of follow-on or “generic” versions of biological products approved under a BLA. However, members of Congress recently introduced legislation to establish a legal pathway to approve follow-on versions of approved BLA products. We are uncertain as to when any such process may be adopted or how such a process would relate to our intellectual property rights, but any such process could have a material effect on the prospects of our products.
Risks Related to Biologics Regulation
The DermaVir is the only Product we are currently testing in clinical trials and it may never be successfully commercialized if these clinical trials are not successful.
Our lead product candidate, DermaVir, is the only product we are testing in clinical trials and it may never be successfully commercialized if these clinical trials are not successful. The clinical trials required by the EMA and the FDA for DermaVir are long and complex. Currently, this product candidate is completed Phase II clinical testing on a limited number of subjects as described below.
It also is possible the FDA or the EMA will require us to conduct more extensive non-clinical studies than we currently anticipate before they will consider our products for marketing approval. Some of our future trials also may involve treatment for durations that are significantly longer than those we have tested thus far. The longer-term trials could reveal safety or other issues that could adversely affect marketing approval. We may need to commit substantial time and additional resources in order to conduct further clinical trials before we can submit a BLA with respect to any of these product candidates. We cannot predict with any certainty if or when we might submit a BLA for regulatory approval of any of our product candidates.
| 47 |
CLINICAL TRIALS ARE TIME-CONSUMING, DIFFICULT AND COSTLY TO DESIGN AND IMPLEMENT.
Before we can obtain marketing approval for the commercial sale of any of our product candidates, we and our collaborators are required to complete extensive human clinical trials to demonstrate, to the satisfaction of the EMA, the FDA and other regulatory authorities, that our product candidates are safe and effective for use in humans. In addition, if we choose to make claims of superiority over currently marketed competitive products, we must substantiate those claims with scientific evidence from prospectively designed head-to-head clinical trials. Human clinical trials are expensive and difficult to design and implement, in part because the science behind them is complex and they are therefore subject to rigorous regulatory requirements. In the event we incorrectly design or carry out clinical trials or those clinical trials fail to demonstrate statistically clinical significance, we are not likely to be able to obtain EMA and FDA approval for our product candidates. Further, the medical, regulatory and commercial environment for biopharmaceutical products, or biologics, changes quickly and often in ways we may not be able to accurately predict. The clinical trial process is also time-consuming. We estimate that clinical trials of our lead product candidate, the DermaVir, will take at least several more years to complete. Furthermore, as failure can occur at any stage, we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed or suspended by the sponsor, regulatory authorities, and ethics committees based on many factors, including the following:
| · | changes in regulatory requirements; |
| · | delays in obtaining regulatory authorizations to commence a clinical trial; |
| · | delays in identifying and reaching agreement on acceptable terms with prospective clinical trial sites; |
| · | delay or failure in obtaining Institutional Review Board (“IRB”) review and approval of the clinical trial protocol; |
| · | unforeseen safety issues and side effects; |
| · | determination of dosing issues; |
| · | lack of effectiveness in clinical trials; |
| · | slower than expected patient recruitment; |
| · | inability to monitor patients adequately during or after treatment; |
| · | inability or unwillingness of medical investigators to follow our clinical protocols and current cGCP requirements; |
| · | inability to maintain a sufficient supply of the investigational product to support the trials; |
| · | suspension or termination of clinical trials for noncompliance with regulatory requirements; and |
| · | changes in clinical care protocols and standards of care within the institutions in which our trials take place. |
A number of companies in the biologics and medical device industry have suffered significant setbacks in advanced clinical trials despite promising results in earlier trials. For the same reason, we may be unable to develop marketable products.
Delays in patient enrollment for clinical trials could increase costs and delay any regulatory approvals.
The rate of completion of our clinical trials will depend on the rate of patient enrollment. There may be substantial competition to enroll patients in clinical trials for our products in development. This competition has delayed clinical trials of other biopharmaceutical and drug development companies. In addition, recent improvements in existing drug therapy, particularly for medical products for the treatment of HIV/AIDS, may make it more difficult for us to enroll patients in our clinical trials as the patient population may choose to (i) remain with existing drug therapies, (ii) enroll in clinical trials sponsored by other companies, or (iii) switch to alternative therapies. Delays in patient enrollment can result in increased development costs and delays in regulatory approvals.
| 48 |
The results of our clinical trials may not support our Product candidate claims or may result in the discovery of adverse side effects.
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our Product candidate claims or that the EMA, FDA or foreign authorities will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe for humans or that they are effective for the proposed indicated uses, which could cause us to abandon a product candidate and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our BLAs and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s profile. In addition, our clinical trials for the DermaVir involve a relatively small patient population. Because of the small sample size, their results may not be indicative of future results.
In any drug development program, it is typically during the pivotal large Phase III clinical trials, when large numbers of patients are tested, that the most reliable information on side effects is collected. We have not reached this development stage yet, and we cannot accurately predict if pivotal clinical trials with our lead Product DermaVir, will reveal unexpected side effects. Occurrence of any side effect could delay or terminate further development and hamper or prevent regulatory approval or marketing of our product.
We will need to demonstrate that the DermaVir PRODUCT manufactured at our own facility is comparable to the Product used in clinical trials.
In addition to increased production efforts, we may make manufacturing changes to the components or to the manufacturing process for DermaVir. These changes could result in delays in the development or marketing approval of the DermaVir and in reduction or interruption of commercial sales, if the product is approved, any of which could materially harm our business. We will be required to demonstrate product comparability for each manufacturing site. The FDA and EMA may require additional testing beyond what we propose.
We intend to rely on results of pre-clinical studies and clinical trials performed using the form of the Product candidate produced using the prior formulation or production method or at the prior scale. Depending upon the type and degree of differences between manufacturing processes or component substitutions for a Product candidate, we may be required to conduct additional studies or clinical trials to demonstrate that the new method(s), substitute component or product candidate is sufficiently similar to the previously produced material.
We have no commercial or other large-scale manufacturing experience, and if we are unsuccessful in developing commercially viable technologies to enable us to manufacture the finished Product ourselves in sufficient qualities and quantities at a competitive price to meet projected commercial demand, our business would be harmed.
| 49 |
To be successful, our product candidates must be capable of being manufactured in sufficient quantities, in compliance with regulatory requirements and at a marketable cost. We have no commercial or other large-scale manufacturing experience. We currently rely on third parties for certain aspects of the clinical trial manufacture of DermaVir and its components and for our other Product candidates. We are in the process of developing and implementing manufacturing methods and building a pilot cGMP compliant facility in Hungary to manufacture larger quantities of our Immune Therapy Products. We must hire and train a significant number of employees and comply with applicable regulations for our facilities, which are extensive. There currently are no contract manufacturers that have already manufactured the final DermaVir product or any other product candidate. Therefore, if we encounter delays or difficulties with our suppliers and/or cannot manufacture the finished Product ourselves, we may not be able to conduct clinical trials as planned or to meet demand for DermaVir, if it is approved, any of which could harm our business.
The EMA, FDA and regulatory agencies in other countries periodically inspect manufacturing facilities, including third parties who manufacture our raw materials for us. The FDA or EMA may believe that neither we nor our suppliers have enough experience making these Products or the raw materials, and may subject those manufacturers and/or us to increased scrutiny. Biopharmaceutical manufacturing facilities must comply with cGMP standards, requiring usually an investment of substantial funds, time and effort to ensure full compliance with these standards and make quality products. We do not have control over our suppliers’ compliance with these requirements. Failure to comply with regulatory requirements can result in sanctions, fines, delays, suspensions of approvals, seizures or recalls of products, operating restrictions, manufacturing interruptions, costly corrective actions, injunctions, adverse publicity against us and our products, and criminal prosecution.
If any of our product candidates receives marketing approval, we expect to manufacture the final product using our technology and our production facility in Hungary. If we are unable to comply with cGMP standards and other government regulations, the qualification of our facility could be a lengthy process, and there may not be adequate alternatives to meet our needs. As a result, we may not be able to obtain the necessary raw materials or final product in the future on a timely basis, if at all. This would negatively affect our business.
Risks Related to Medical Device Regulations
If we fail to obtain regulatory approvals and clearances, or experience significant delays in obtaining, FDA clearances or approvals for future products or product enhancements, our ability to commercially distribute and market these products could suffer.
If required by the FDA to obtain a 510(k) clearance or approval of a Premarket Approval (“PMA”), the process of obtaining regulatory clearances or approvals to market a medical device can be costly and time consuming, and we may not be able to obtain these clearances or approvals on a timely basis, if at all. In particular, the FDA permits commercial distribution of a new medical device only after the device has received clearance under Section 510(k) of the Federal Food, Drug and Cosmetic Act, or is the subject of an approved PMA unless the device is specifically exempt from those requirements. The FDA will clear marketing of a lower risk medical device through the 510(k) process if the manufacturer demonstrates that the new product is substantially equivalent to other 510(k)-cleared products. High risk devices deemed to pose the greatest risk, such as life-sustaining, life-supporting, or implantable devices, or devices not deemed substantially equivalent to a previously cleared device, require the approval of a PMA. The PMA process is more costly, lengthy and uncertain than the 510(k) clearance process. A PMA application must be supported by extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the device for its intended use.
We are currently developing our regulatory strategies with respect to which regulatory pathway will be necessary to obtain clearance or approval of the DermaPrep component of DermaVir, if medical device clearance or approval is required at all. DermaVir, as well as our other product candidates, are Immune Therapies, which utilize a novel mechanism of action. To date, the FDA has not cleared or approved for commercial sale in the U.S. any Immune Therapy product designed to stimulate an immune response against HIV. Consequently, there is no precedent for the successful commercialization of products based on our technologies in this area. If the FDA requires us to submit a 510(k) or PMA for DermaPrep, we may experience significant delays in obtaining marketing approval of our product candidates, if at all. If we fail to obtain such approvals, or experience significant delays in obtaining, such approvals for future products or product enhancements, our ability to commercially distribute and market these products could suffer.
| 50 |
If our medical device products cause or contribute to a death or a serious injury, or malfunction in certain ways, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA’s medical device reporting regulations, medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. If we fail to report these events to the FDA within the required timeframes, or at all, the FDA could take enforcement action against us. Any such adverse event involving our products also could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
Risks Related to Health Care Regulations
WE MAY BE SUBJECT, DIRECTLY OR INDIRECTLY, TO FEDERAL AND STATE HEALTHCARE FRAUD AND ABSUSE LAWS AND REGULATIONS AND COULD FACE SUBSTANTIAL PENALITIES IF WE ARE UNABLE TO FULLY COMPLY WITH SUCH LAWS.
While we do not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third party payors, many healthcare laws and regulations apply to our business. For example, we could be subject to healthcare fraud and abuse and patient privacy regulation and enforcement by both the federal government and the states in which we conduct our business. The healthcare laws and regulations that may affect our ability to operate include:
| · | the federal
healthcare programs’
Anti-Kickback Law,
which
prohibits, among
other things,
persons or entities
from soliciting, receiving,
offering or
providing remuneration,
directly or
indirectly,
in return for or
to induce
either the
referral of
an individual
for,
or the purchase
order or
recommendation of, any
item or services
for which
payment may
be made under
a federal
healthcare program
such as the Medicare
and
Medicaid programs; |
| · | federal false
claims laws
which prohibit,
among other
things, individuals
or entities from
knowingly presenting, or
causing to
be presented,
claims for
payment from
Medicare, Medicaid,
or other
third party payors that
are false or
fraudulent, and which
may apply
to entities like
us to the
extent that
our interactions with customers
may affect their
billing or coding
practices; |
| · | the federal Health
Insurance Portability and
Accountability Act
of 1996 (“HIPAA”)
which established new federal
crimes for
knowingly and willfully
executing a
scheme to
defraud any healthcare
benefit program or
making false statements
in connection
with the
delivery of or
payment for
healthcare benefits,
items or
services, as well
as imposing
certain requirements
relating to the
privacy, security and
transmission
of individually
identifiable health
information;
and |
| · | state law equivalents of each of the federal laws mentioned on the preceding page, such as anti- kickback and false claims laws which may apply to items or services reimbursed by any third party payor, including commercial insurers, and state laws governing the privacy of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
| 51 |
The biopharmaceuticals industry is, and in recent years has been, under heightened scrutiny as the subject of government investigations and enforcement actions involving manufacturers who allegedly offered unlawful inducements to potential or existing customers in an attempt to procure their business, including arrangements with physician consultants. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from the Medicare and Medicaid programs, and the curtailment or restructuring of our operations. Any penalties, damages, fines, exclusions, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. The risk of our being found in violation of these laws is increased by the fact that many of these laws are broad and their provisions are open to a variety of interpretations. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
Risks Related to Intellectual Property
If we, or the third parties from whom we license intellectual property, are unable to secure and maintain patent or other intellectual property protection for the intellectual property contained in our Products, our ability to compete will be harmed.
Our commercial success depends, in part, on obtaining patent and other intellectual property protection for our products, methods, processes and other technologies. The patent positions of biopharmaceutical companies, including ours, can be highly uncertain and involve complex and evolving legal and factual questions. If we, or the third parties from whom we license intellectual property, fail to obtain adequate patent or other intellectual property protection for intellectual property contained in our products, methods, processes and other technologies or if any protection is reduced or eliminated, others could use our intellectual property, resulting in harm to our competitive business position. In addition, patent and other intellectual property protection may not provide us with a competitive advantage against competitors that devise ways of making competitive products without infringing any patents that we own or have rights to.
As of December 31, 2014, we have filed multiple patent applications, including U.S. and E.U. patent applications, as well as rights under foreign patents and patent applications. . U.S. patents and patent applications may be subject to interference proceedings and U.S. patents may be subject to reexamination proceedings in the U.S. Patent and Trademark Office. Foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent offices. Any of these proceedings could result in either loss of the patent or denial of the patent application, or loss or reduction in the scope of one or more of the claims of the patent or patent application. Changes in either patent laws or in interpretations of patent laws may also diminish the value of our intellectual property or narrow the scope of our protection. Interference, reexamination and opposition proceedings may be costly and time-consuming, and we, or the third parties from whom we license intellectual property, may be unsuccessful in defending against such proceedings. Thus, any patents that we own or license may provide limited or no protection against competitors. In addition, our pending patent applications and those we may file in the future may have claims narrowed during prosecution or may not result in patents being issued. Even if any of our pending or future applications are issued, they may not provide us with adequate protection or any competitive advantages. Our ability to develop additional patentable products, methods, processes and other technologies is also uncertain.
Non-payment or delay in payment of patent fees or annuities, whether intentional or unintentional, may also result in the loss of patents or patent rights important to our business. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as do the laws of the U.S., particularly in the field of biopharmaceuticals.
| 52 |
If we are unable to prevent unauthorized use or disclosure of our proprietary trade secrets and unpatented know-how, our ability to compete will be harmed.
Proprietary trade secrets, copyrights, trademarks and unpatented know-how are also very important to our business. We rely on a combination of trade secrets, copyrights, trademarks, confidentiality agreements and other contractual provisions and measures to protect certain aspects of our products, methods, processes and other technologies, especially where we do not believe that patent protection is appropriate or obtainable. To this end, we require all of our employees, consultants, advisors and contractors to enter into confidentiality and, where applicable, grant-back agreements. However, these measures may not be adequate to safeguard our proprietary intellectual property. Our employees, consultants, advisors and contractors may unintentionally or willfully disclose our confidential information to competitors. In addition, confidentiality agreements may be unenforceable or may not provide an adequate remedy in the event of unauthorized disclosure. Enforcing a claim that a third party illegally obtained and is using our trade-secrets is expensive and time consuming, and the outcome is unpredictable. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of products developed by our employees, consultants, advisors and contractors that we consider proprietary. As a result, third parties may be able to use our proprietary employees, consultants, advisors and contractors, and our ability to compete in the market would be harmed.
We could become subject to patent and other intellectual property litigation that could be costly, result in the diversion of management’s attention, require us to pay damages and force us to discontinue selling our products.
The biopharmaceutical industry is characterized by competing intellectual property and a substantial amount of litigation over patent and other intellectual property rights. Determining whether a product infringes a patent involves complex legal and factual issues, and the outcome of a patent litigation action is often uncertain. We have not conducted an extensive search of patents issued to third parties, and no assurance can be given that third party patents containing claims covering our products, parts of our products, technology or methods do not exist, have not been filed, or could not be filed or issued. Because of the number of patents issued and patent applications filed in our areas, our competitors or other third parties may assert that our products and the methods we employ in the use of our products are covered by U.S. or foreign patents held by them. In addition, because patent applications can take many years to issue and because publication schedules for pending applications vary by jurisdiction, there may be applications now pending of which we are unaware, and which may result in issued patents which our current or future products infringe. Also, because the claims of published patent applications can change between publication and patent grant, there may be published patent applications that may ultimately issue with claims that we infringe. There could also be existing patents that one or more of our products or parts may infringe and of which we are unaware. In certain situations, we or third parties from whom we license intellectual property may determine that it is in our best interests or their best interests to voluntarily challenge a third party’s products or patents in litigation or other proceedings, including patent interferences or reexaminations. As a result, we may become involved in unwanted litigations and actions that could be costly, result in diversion of managements’ attention, require us to pay damages and force us to discontinue selling our products.
Infringement actions and other intellectual property claims and proceedings, whether with or without merit, may cause us to incur substantial costs and could place a significant strain on our financial resources, divert the attention of management from our business and harm our reputation. Some of our competitors may be able to sustain the costs of complex patent or intellectual property litigation more effectively than we can because they have substantially greater resources.
| 53 |
We cannot be certain that we will successfully defend against allegations of infringement of third party patents and intellectual property rights. In the event that we become subject to a patent infringement or other intellectual property lawsuit and if the other party’s patents or other intellectual property were upheld as valid and enforceable and we were found to infringe the other party’s patents or violate the terms of a license to which we are a party, we could be required to pay damages. We could also be prevented from selling our products unless we could obtain a license to use the products, methods, processes or other technologies covered by such patents or were able to redesign our products, methods, processes and other technologies to avoid infringement. A license may not be available at all or on commercially reasonable terms or we may not be able to redesign our products, methods, processes and other technologies to avoid infringement. Modification of our products or development of new products could require us to conduct clinical trials and to revise our filings with the FDA and other regulatory bodies, which would be time-consuming and expensive. In these circumstances, we may be unable to sell our products at competitive prices or at all, our business and operating results could be harmed and our stock price may decline. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
We may be subject to damages resulting from claims that our employees or we have wrongfully used or disclosed alleged trade secrets of their former employers.
Some of our employees were previously employed at universities or other enterprises. We could, in the future, be subject to claims that these employees, or we, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending against such claims, a court could order us to pay substantial damages and prohibit us from using products, methods, processes and other technologies that are essential to our products, if such products, methods, processes and other technologies are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. In addition, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain potential products, which could severely harm our business. Even if we are successful in defending against these claims, such litigation could result in substantial costs and be a distraction to management.
Risks Related to Our Common Stock
Our common stock is quoted on the OTCBB which may have an unfavorable impact on our stock price and liquidity.
Our common stock is quoted on the OTCBB. The OTCBB is a significantly more limited market than the New York Stock Exchange or the NASDAQ Stock Market. The quotation of our shares on the OTCBB may result in a less liquid market available for existing and potential stockholders to trade shares of our common stock, could depress the trading price of our common stock and could have a long-term adverse impact on our ability to raise capital in the future.
BECAUSE OUR BUSINESS ASSETS, DIRECTORS AND OFFICERS ARE LOCATED OUTSIDE OF THE UNITED STATES, OUR SHAREHOLDERS MAY BE LIMITED IN ABILITY TO ENFORCE CIVIL ACTIONS AGAINST OUR ASSETS OR OUR DIRECTORS AND OFFICERS.
We are incorporated under the laws of Delaware but because we are headquartered in Hungary, all of our officers and members of the Board reside in Hungary. Therefore our shareholders may have difficulty enforcing civil liabilities under the U.S. federal securities laws against our officers and directors. Because some of our assets are located outside the U.S., it may be difficult for an investor to succeed in an action, for any reason, against us or any of our directors or officers through U.S. jurisdictions. If an investor was able to obtain a judgment against us or any of our directors or officers in a U.S. court based on U.S. securities or other laws, it may be difficult to enforce such judgment in Hungary. We are uncertain as to the enforceability, in original actions in Hungarian courts, of liability based upon the U.S. federal securities laws and as to the enforceability in Hungarian courts of judgments of U.S. courts obtained in actions based upon the civil liability provisions of the U.S. federal securities laws.
| 54 |
WE MAY CONDUCT FURTHER OFFERINGS IN THE FUTURE, IN WHICH CASE YOUR PERCENTAGE INTEREST IN OUR COMPANY WILL BE DILUTED.
Since inception, we have relied on sales of our common stock to fund our operations. We may conduct further offerings in the future to finance our current projects or to finance subsequent projects that we decide to undertake. If common stock is issued in return for additional funds, the price per share could be lower than that paid by our current shareholders. We anticipate continuing to rely on equity sales of our common stock in order to fund our business operations. If we issue additional stock, your percentage interest in us will be diluted and the value of your stock could be reduced.
WE MAY ISSUE PREFERRED STOCK WHICH MAY HAVE GREATER RIGHTS THAN OUR COMMON STOCK.
We are permitted in our Certificate of Incorporation to issue up to 10,000,000 shares of preferred stock. We currently have 2,000,000 shares of Series A Preferred Shares, 1,000,000 shares of Series B Preferred Shares, 1,000,000 shares of Series C Preferred Shares, 1,000,000 shares of Series D Preferred Shares and 1,045,000 shares of Series E Preferred Shares issued and outstanding; however, we can issue shares of our preferred stock in one or more series and can set the terms of the preferred stock without seeking any further approval from our common shareholders. Any preferred stock that we issue may rank ahead of our common stock in terms of dividend priority or liquidation premiums and may have greater voting rights than our common stock. In addition, such preferred stock may contain provisions allowing it to be converted into shares of common stock, which could dilute the value of common stock to current shareholders and could adversely affect the market price, if any, of our common stock.
THE APPLICATION OF THE “PENNY STOCK” RULES COULD ADVERSELY AFFECT THE MARKET PRICE OF OUR COMMON SHARES AND INCREASE YOUR TRANSACTION COSTS TO SELL THOSE SHARES.
The Securities and Exchange Commission (the “SEC”) has adopted rule 3a51-1 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, Rule 15g-9 requires:
- that a broker or dealer approve a person’s account for transactions in penny stocks;
- the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased; and
- that a broker or dealer approve a person’s account for transactions in penny stocks.
In order to approve a person’s account
- obtain financial information and investment experience objectives of the person; and
- make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks.
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form:
| 55 |
- sets forth the basis on which the broker or dealer made the suitability determination; and
- that the broker or dealer received a signed, written agreement from the investor prior to the transaction.
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of our common stock and cause a decline in the market value of our stock.
AS AN ISSUER OF “PENNY STOCK,” THE PROTECTION PROVIDED BY THE FEDERAL SECURITIES LAWS RELATING TO FORWARD LOOKING STATEMENTS DOES NOT APPLY TO US.
Although federal securities laws provide a safe harbor for forward-looking statements made by a public company that files reports under the federal securities laws, this safe harbor is not available to issuers of penny stocks. As a result, the Company will not have the benefit of this safe harbor protection in the event of any legal action based upon a claim that the material provided by the Company contained a material misstatement of fact or was misleading in any material respect because of the Company’s failure to include any statements necessary to make the statements not misleading. Such an action could hurt our financial condition.
WE HAVE NOT PAID DIVIDENDS IN THE PAST AND DO NOT EXPECT TO PAY DIVIDENDS FOR THE FORESEEABLE FUTURE. ANY RETURN ON INVESTMENT MAY BE LIMITED TO THE VALUE OF OUR COMMON STOCK.
No cash dividends have been paid on the Company’s common stock. We expect that any income received from operations will be devoted to our future operations and growth. The Company does not expect to pay cash dividends in the near future. Payment of dividends would depend upon our profitability at the time, cash available for those dividends, and other factors as the Company’s board of directors may consider relevant. If the Company does not pay dividends, the Company’s common stock may be less valuable because a return on an investor’s investment will only occur if the Company’s stock price appreciates.
Item 1B. Unresolved Staff Comments.
Not applicable.
Item 2. Property.
The Company currently leases a 1,000 square foot office located at 1095 Budapest, Soroksari ut 94-96, Hungary, for approximately $1,100 per month. We also lease research and development facilities for Genetic Immunity, consisting of approximately 6,500 square feet, for approximately $12,500 a month, located at 1045 Budapest, Berlini ut 47-49.
Item 3. Legal Proceedings.
The Company’s wholly-owned subsidiary, Genetic Immunity, has initiated a lawsuit against the National Development Agency of Hungary. In 2009, Genetic Immunity successfully applied for and won an 800 million Hungarian forint (approximately $4,000,000) research grant to conduct dust mite allergy vaccine research. Subsequently, the agency refused to sign the grant and denied payment of funds. The company initiated a lawsuit to request that the court reinstate the contract that resulted in a grant award. On February 13, 2013, an appeals court ruled that the court could not reinstate the contract between Genetic Immunity and the National Development Agency as the contract has been validly in existence since its original execution in 2009. As such Genetic Immunity was ordered to pay 52,500,000 HUF (approximately $250,000) in lawsuit costs. However, simultaneously, the judgment stated that the company is entitled to receive the original grant amount, interest payment and/or damages since the original grant contract is valid. On February 18, 2013, we filed a new petition to have the courts establish the damages and awards that Genetic Immunity can receive based on the appeals court’s ruling. We are currently seeking 4,800,000,000 HUF (approximately $17 million) for damages sustained. On November 18, 2013 we have entered into a six month suspension of said lawsuit, as agreed with the government, in hopes of negotiating an out of court settlement. As out of court settlement discussion were unsuccessful we resumed the law suite. Because of the continued change in the government agency responsible for our claim there has been no significant advancement in the case. The next court date is scheduled for April 2nd, 2015.
| 56 |
Other than the matter described above, we are not currently involved in any litigation that we believe could have a materially adverse effect on our financial condition or results of operations. There is no action, suit, proceeding, inquiry or investigation before or by any court, public board, government agency, self-regulatory organization or body pending or, to the knowledge of the executive officers of our Company or any of our subsidiaries, threatened against or affecting our Company, our common stock, any of our subsidiaries or of our Company’s or our Company’s subsidiaries’ officers or directors in their capacities as such, in which an adverse decision could have a material adverse effect.
Item 4. Mine Safety Disclosures.
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
(a) Market Information
The Company’s Common Stock is quoted on the OTCBB under the under the symbol “PWRV.” The following table sets forth the quarterly high and low sale prices for our common shares for the last two completed fiscal years and the subsequent interim periods. Quotations on the OTCBB reflect bid and ask quotations, may reflect inter-dealer prices, without retail markup, markdown or commission, and may not represent actual transactions.
| Quarter ended | High | Low | ||||||
| March 31, 2015 | $ | 0.06 | $ | 0.04 | ||||
| December 31, 2014 | $ | 0.10 | $ | 0.06 | ||||
| September 30, 2014 | $ | 0.10 | $ | 0.06 | ||||
| June 30, 2014 | $ | 0.10 | $ | 0.06 | ||||
| March 31, 2014 | $ | 0.10 | $ | 0.05 | ||||
| December 31, 2013 | $ | 0.12 | $ | 0.04 | ||||
| September 30, 2013 | $ | 0.10 | $ | 0.04 | ||||
| June 30, 2013 | $ | 0.16 | $ | 0.05 | ||||
| March 31, 2013 | $ | 0.33 | $ | 0.30 | ||||
(b) Holders
As of March 31, 2015, there were approximately 98 holders of record of our common stock. The common stock figure does not take into account those shareholders whose certificates are held in the name of broker-dealers or other nominees.
(c) Dividends
The Company issued a dividend to our shareholders of record on December 23, 2011, in the form of restricted common stock of iGlue, Inc. (“iGlue”). Each shareholder received 0.05 shares of restricted common stock of iGlue. A total of 3,115,015 shares of iGlue’s common stock were distributed on February 15, 2012. The Company still holds 2,884,986 shares of iGlue’s common stock and warrants to purchase shares of iGlue’s common stock.
We have never paid any cash dividends on our common shares, and we do not anticipate that we will pay any dividends with respect to those securities in the foreseeable future. Our current business plan is to retain any future earnings to finance the expansion development of our business.
(d) Securities Authorized for Issuance under Equity Compensation Plan
We currently have no Equity Compensation Plan in effect.
Transfer Agent
Our transfer agent is Fidelity Transfer Company, 8915 S. 700 E. Suite 102, Sandy, UT 84070.
Recent Sales of Unregistered Securities
On November 28, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 170,000 shares of its Series E Preferred Stock to five unaffiliated private investors for aggregate proceeds of $165,600.
| 57 |
In September, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 300,000 shares of its Series E Preferred Stock to seven unaffiliated private investors for aggregate proceeds of $300,000.
On May 28, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 200,000 shares of its Series E Preferred Stock to two unaffiliated private investors for aggregate proceeds of $100,000.
On April 28, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 100,000 shares of its Series C Preferred Stock to one unaffiliated private investor for aggregate proceeds of $27,949.
On April 7, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 50,000 shares of its Series E Preferred Stock to one unaffiliated private investor for aggregate proceeds of $50,000.
On February 19, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 200,000 shares of its Series E Preferred Stock to two unaffiliated private investors for aggregate proceeds of $200,000. The holders of Series E Preferred Stock are entitled to receive dividends equal to the amount that would be received if each one share of Series E Preferred stock was fully converted into twelve and one half shares of the common stock. Each one share of the Series E Preferred has voting rights equal to five votes of common Stock. Each share of Series E Preferred stock is convertible into 10 shares of common stock.
Rule 10B-18 Transactions
During the years ended December 31, 2014, there were no repurchases of the Company’s common stock by the Company.
Item 6. Selected Financial Data.
Not applicable.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
THE FOLLOWING DISCUSSION OF OUR PLAN OF OPERATION AND RESULTS OF OPERATIONS SHOULD BE READ IN CONJUNCTION WITH THE FINANCIAL STATEMENTS AND RELATED NOTES TO THE FINANCIAL STATEMENTS INCLUDED ELSEWHERE IN THIS REPORT. THIS DISCUSSION CONTAINS FORWARD-LOOKING STATEMENTS THAT RELATE TO FUTURE EVENTS OR OUR FUTURE FINANCIAL PERFORMANCE. THESE STATEMENTS INVOLVE KNOWN AND UNKNOWN RISKS, UNCERTAINTIES AND OTHER FACTORS THAT MAY CAUSE OUR ACTUAL RESULTS, LEVELS OF ACTIVITY, PERFORMANCE OR ACHIEVEMENTS TO BE MATERIALLY DIFFERENT FROM ANY FUTURE RESULTS, LEVELS OF ACTIVITY, PERFORMANCE OR ACHIEVEMENTS EXPRESSED OR IMPLIED BY THESE FORWARD-LOOKING STATEMENTS. THESE RISKS AND OTHER FACTORS INCLUDE, AMONG OTHERS, THOSE LISTED UNDER “FORWARD-LOOKING STATEMENTS” AND “RISK FACTORS” AND THOSE INCLUDED ELSEWHERE IN THIS REPORT.
Overview
Power of the Dream Ventures, Inc. is one of Hungary’s leading technology holding companies. We identify, invest in and acquire technologies originating in Hungary we deem to be internationally marketable. We currently hold equity interest in iGlue, Inc. a US public company. Through our wholly owned subsidiary, Genetic Immunity, a biotechnology company we acquired in October of 2012, we now focus on the commercialization tasks related to Genetic Immunity’s lead product candidate therapeutic HIV vaccine called DermaVir.
| 58 |
We are looking to generate revenue through the sale of Genetic Immunity’s therapeutic vaccines upon US, European and global marketing approvals, and through the sale of our equity interest in iGlue.
Since inception through December 31, 2014, we had a deficit accumulated during the development stage of $44,854,780 and net cash used in operations of $4,352,933. To date all of our funding has been provided through the sale of our common and preferred stock. We believe we can raise sufficient additional financing through the sale of our common stock to allow us to continue operations and execute our business plan to the end of 2015. The Company also anticipates receiving a minimum of $4 million and a maximum of $16 million from a lawsuit initiated against the National Development Agency in Hungary for breach of contract for a previously awarded grant. Should the need arise, we plan to raise additional funds, both from U.S. and international investors. We believe that through the acquisition of Genetic Immunity and the company’s lead product candidate DermaVir, we will be in a position to start generating revenue in 2016. We believe that our ownership of one of the world’s first, clinically proven, therapeutic HIV vaccine will prove attractive to both private and institutional investors, making fundraising a less strenuous process than before. We also believe that as Hungary’s only US public company that is actively engaged in Hungarian intellectual property acquisition (like Genetic Immunity and iGlue,) we are an even more attractive investment candidate.
If necessary, we will raise additional capital in a manner that is the least dilutive to our shareholders yet at the same time serves our development, commercialization and operating needs. We anticipate one round of fundraising in 2015 as we move closer to commercializing DermaVir. By the end of 2015, we will seek conditional marketing approval for DermaVir in the United States.
Even though we believe our public status will allow us to raise additional capital, if needed, no assurance can be given that we can in fact obtain additional working capital, or if obtained, that such funding will not cause substantial dilution to shareholders of the Company. If we are unable to raise additional funds, we may be forced to change or delay our contemplated marketing and business plan.
In 2015 we anticipate spending approximately $3,000,000 - $5.000.000 on DermaVir’s regulatory approval, commercialization and general administrative expenses. We believe these funds will become available to us from proceeds obtained from our lawsuit against the Hungarian Development Agency as described above in Item 3.
Being a development stage company, we are subject to all the risks inherent in the establishment of a new enterprise and the marketing and manufacturing of new products, many of which risks are beyond the control of the Company.
Over the course of the next 12 months we will focus on accelerating the development and potential commercialization of Genetic Immunity’s DermaVir HIV vaccine. We plan on applying for a Conditional Marketing Authorization (“CMA”) with the EMA, and pursuing similar available venues in the United States. We believe given DermaVir’s safety and efficacy profile and recent changes in the regulatory landscape, the time is ripe for these initiatives.
Although we believe that DermaVir can become a significant new product in the treatment of HIV, there can be no assurance that we will be able to complete the required regulatory processes. If we are unable to complete regulatory permits within the next 12 months we will be required to raise additional capital to support our operations until such time when we can maintain operations from revenue(s) realized upon marketing authorization.
Upon regulatory approval of DermaVir, we anticipate making substantial equipment purchases to expand our GMP manufacturing faculty and to increase our work force to fully implement commercialization tasks.
| 59 |
If we are unable to raise additional financing there is substantial doubt about our ability to continue as a going concern. The condensed consolidated financial statements included elsewhere in this prospectus do not include any adjustments relating to the recoverability of recorded asset amounts that might be necessary as a result of the above uncertainty.
Results of Operations
For the Year Ended December 31, 2014 Compared to the Year Ended December 31, 2013
| For the Year Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Net sales | $ | - | - | |||||
| Gross profit | $ | - | - | |||||
| General and administrative expenses | $ | 1,255,316 | 738,975 | |||||
| Loss from operations | $ | (31,087,049) | (1,164,332) | |||||
| Interest Expenses and Exchange Gains | $ | (749,380) | (629,625) | |||||
| Net loss | $ | (31,836,441) | (1,794,718) | |||||
Revenues
During the years ended December 31, 2014 and 2013, respectively, the Company had no revenue.
Research and development
For the year ended December 31, 2014, research and development expenses were $0 as compared to $41,812 for the year ended December 31, 2013.
General, selling and administrative expenses
For the year ended December 31, 2014, general, selling and administrative expenses were $1,255,316 as compared to $738,975 for the year ended December 31, 2013. The increase in general, selling and administrative expenses is attributable to higher consulting and administrative expenses.
Loss from Operations
Loss from operations for the year ended December 31, 2014, was $31,087,049, as compared to $1,164,332 for the year ended December 31, 2013. The primary increase in loss from operations is attributable to the impairment of intangibles. .
While the company continues to believe that the value of the patents associated with DermaVir will be recoverable through the eventual marketing and sale of DermaVir, we have determined that due to our continued limited success in raising the necessary amount of capital to bring DermaVir or any other product to market, our previous cash flow projections, as well as the discount rate used in our estimate of fair value, needed to be revised to reflect the uncertainty as to whether we will ultimately realize any cash flows from these patents. As a result, we recognized an impairment charge of $29,570,912 in 2014 related to our acquired patent intangibles.
| 60 |
Net Loss
Net loss for year ended December 31, 2014, was $31,836,441 or loss per share of $0.69, as compared to $1,794,718 or loss per share of $0.04, for the comparable year ended December 31, 2013.
Liquidity and Capital Resources
The following table summarizes total current assets, liabilities and working capital at December 31, 2014, compared to December 31, 2013:
December 31, 2014 | December 31, 2013 | Increase/Decrease | ||||||||||
| Current Assets | $ | 101,397 | $ | 262,992 | $ | (161,595 | ) | |||||
| Current Liabilities | $ | 8,529,030 | $ | 8,240,680 | $ | 288,350 | ||||||
| Working Capital (Deficit) | $ | (8,427,633 | ) | $ | (7,977,688 | ) | $ | (449,945 | ) | |||
At December 31, 2014, we had a working capital deficit of $8,427,633, as compared to a working capital deficit of $7,977,688, at December 31, 2013, an increase of $449,945. The increase in deficit is mainly attributed due to the increase in accrued interest.
Net cash used in operating activities for the year ended December 31, 2014 and 2013 was $(1,782,845) and $(175,521), respectively. The net loss for the years ended December 31, 2014 and 2013 was $31,836,441 and $1,794,718, respectively. The Company’s cash used in operations increased due to financing from accounts payable.
Net cash obtained through all financing activities for the years ended December 31, 2014 was $1,020,513, as compared to $500,326 for the year ended December 31, 2013. The company raised $843,549 through private placements of our preferred stock.
Off-Balance Sheet Arrangements
We have no significant known off balance sheet arrangements.
Critical Accounting Policies
We prepare our consolidated financial statements in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements require the use of estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amount of revenues and expenses during the reporting period. Our management periodically evaluates the estimates and judgments made. Management bases its estimates and judgments on historical experience and on various factors that are believed to be reasonable under the circumstances. Actual results may differ from these estimates as a result of different assumptions or conditions.
The methods, estimates, and judgment we use in applying our most critical accounting policies have a significant impact on the results we report in our financial statements. The SEC has defined “critical accounting policies” as those accounting policies that are most important to the portrayal of our financial condition and results, and require us to make our most difficult and subjective judgments, often as a result of the need to make estimates of matters that are inherently uncertain. Based upon this definition, our most critical estimates relate to the fair value of warrant liabilities. We also have other key accounting estimates and policies, but we believe that these other policies either do not generally require us to make estimates and judgments that are as difficult or as subjective, or it is less likely that they would have a material impact on our reported results of operations for a given period. For additional information see Note 2, “Summary of Significant Accounting Policies” in the notes to our reviewed financial statements appearing elsewhere in this report. Although we believe that our estimates and assumptions are reasonable, they are based upon information presently available, and actual results may differ significantly from these estimates.
| 61 |
Going Concern
We have limited working capital and no revenues from sales of products, services, or licensing. During the fiscal year ended December 31, 2014, our operating expenses continued to be greater than our revenues. These factors have caused our accountants to express substantial doubt about our ability to continue as a going concern. The accompanying financial statements do not include any adjustment that might be necessary if we are unable to continue as a going concern.
Our ability to continue as a going concern has caused the Board of Directors to continue to look for sources of investment capital, and investigate merger and acquisition opportunities. We will look to further diversify our holdings and sources of cash flow until we can support our operation from revenue.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
We do not hold any derivative instruments and do not engage in any hedging activities.
Item 8. Financial Statements and Supplementary Data.
Our financial statements are contained in pages F-1 through F-24 which appear at the end of this Annual Report.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
(a) Evaluation of Disclosure and Control Procedures
Based on their evaluation as of the end of the period covered by this Annual Report on Form 10-K, our principal executive officer and principal financial officer have concluded that our disclosure controls and procedures (as defined in Rules 13a-15(c) and 15d-15(e) under the Exchange Act) are not effective to ensure that information required to be disclosed by us in report that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the U.S. Securities and Exchange Commission’s rules and forms and to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate to allow timely decisions regarding required disclosure due to material weakness in our internal control over financial reporting discussed more fully below.
(b) Management’s Assessment of Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in the Exchange Act Rules 13a-15(f). A system of internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
| 62 |
Under the supervision and with the participation of management, including the principal executive officer and the principal financial officer, the Company’s management has evaluated the effectiveness of its internal control over financial reporting as of December 31, 2014, based on the criteria established in a report entitled “Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission” and the interpretive guidance issued by the Commission in Release No. 34-55929. Based on this evaluation, the Company’s management has evaluated and concluded that as of December 31, 2014, the Company’s internal control over financial reporting was not effective due to the existence of material weakness as described below.
A material weakness is a control deficiency, or combination of control deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements would not be prevented or detected on a timely basis. As of December 31, 2014, we have concluded that our internal control over financial reporting was ineffective. The Company’s assessment identified certain material weaknesses which are set forth below:
Functional Controls and Segregation of Duties
Because of the company’s limited resources, there are limited controls over information processing, and no internal controls over the accuracy, completeness and authorization of transactions.
There is an inadequate segregation of duties consistent with control objectives. Our Company’s management is composed of a small number of individuals resulting in a situation where limitations on segregation of duties exist. In order to remedy this situation we would need to hire additional staff to provide greater segregation of duties. Currently, it is not feasible to hire additional staff to obtain optimal segregation of duties. Management will reassess this matter in the following year to determine whether improvement in segregation of duty is feasible.
There is a lack of top level reviews in place to review targets, product development, joint ventures or financing. All major business decisions are carried out by the officers with board of director approval when needed.
Accordingly, as the result of identifying the above material weaknesses we have concluded that these control deficiencies resulted in a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis by the Company’s internal controls.
Management believes that the material weaknesses set forth above were the result of the scale of our operations and are intrinsic to our small size. Management believes these weaknesses did not have a material effect on our financial results and intends to take remedial actions upon receiving funding for the Company’s business operations.
This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm pursuant to temporary rules of the SEC that permit the Company to provide only management’s report herein.
(c) Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act, during our most recently completed fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information.
| 63 |
None.
PART III
Item 10. Directors, Executive Officers, and Corporate Governance.
The following table and biographical summaries set forth information, including principal occupation and business experience, about our directors and executive officers at April 15, 2013:
| Name | Age | Position | Officer and/or Director Since | |||
| Viktor Rozsnyay | 44 | Chairman, Chief Executive Officer, President | April 2007 | |||
| Ildiko Rozsa | 38 | Chief Financial Officer | October 2007 | |||
| Peter Boros | 38 | Director | May 2013 |
Viktor Rozsnyay, age 44
Viktor Rozsnyay has served as the Chairman, President and Chief Executive Officer of the Company since April 5, 2007. From April 2006 to present, Mr. Rozsnyay has been a Manager of Vidatech Kft., a Hungarian company focused on the acquisition of technologies developed in Hungary, of which he was a founder; Vidatech is now our wholly owned Hungarian subsidiary. From 2004 to 2006 Mr. Rozsnyay was engaged in researching and establishing the foundation of Vidatech. From 2001 to 2004, he was the Founder and Chief Operating Officer of 10Charge, Inc., an ISSO R&D spin-off company formed to commercialize 10 minute battery charging technology, a product Mr. Rozsnyay and his former partners invented and patented. 10Charge, Inc. was a full SEC reporting company until it was acquired by US investors. Mr. Rozsnyay was the primarily person responsible for organizing and completing a reverse merger for 10Charge, Inc. Prior thereto, Mr. Rozsnyay was the Founder and Managing Director of ISSO R&D Kft., an aerospace research company based in the Republic of Hungary, pursuing advanced aerospace propulsion research. Mr. Rozsnyay attended the Jozsef Katona Technical College in Budapest, graduating in 1989.
Mr. Rozsnyay’s unique expertise in Hungary of US public company operations and requirements and as our Founder and CEO make him the perfect and only choice to fulfill the Chairman of the Board position.
Ildiko Rozsa, age 41
Ildiko Rozsa has served as the Chief Financial Officer of the Company since October 24, 2007. Mrs. Rozsa is a qualified statutory accountant, and completted her PhD studies at the Budapest Technical and Economic University in Business Science in 2013. Ms. Rozsa graduated at the College of Finance and Accountancy, later obtaining her mastered degree (MBA) at the Budapest University of Economics. Ms. Rozsa is a qualified member of ACCA. From 1995 through 1996 she was audit assistant at Price Waterhouse’s Budapest Audit Department. From 1997 through 2002 Ms. Rozsa was Finance and Accounting Director at Vivendi Telecom Hungary. From 2002 through 2004 Ms. Rozsa was Chief Financial Officer at Bacardi-Martini Hungary Kft In 2004 she founded and became Managing Director, RIBZ Consulting where she works for multinational clients on IFRS and US GAAP projects, on privatization engagements. Ildiko is an Assistant Professor at the Budapest Technical University, Economic Faculty.
Peter Boros, age 38
Mr. Peter Boros, age 37, currently serves as Chief Executive Officer of iGlue, Inc., a Hungarian-based development stage software company, where he is responsible for the day to day operations of the company. Since July 1, 2007, Mr. Boros has served as the Commercial Director of Co-Op Inc., a retail chain in Hungary. From 2000 to 2007, Mr. Boros was the manager of the Sales and Marketing Department of Co-Op Inc. Additionally, since 2006, Mr. Boros was the owner and operator of Co-Op Gyor, Inc., a grocery chain which was sold in 2011. Mr. Boros currently serves as member of the board of directors of iGlue, Inc.
| 64 |
Family Relationships
There are no family relationships among our directors, executive officers, or persons nominated or chosen by the Company to become directors or executive officers. None of our directors or executive officers or their respective immediate family members or affiliates are indebted to us.
Committees of the Board of Directors
We have not established any committees, including an audit committee, a compensation committee or a nominating committee. The Board currently acts as our audit committee. At the present time, we believe that our Board is capable of analyzing and evaluating our financial statements and understanding internal controls and procedures for financial reporting.
Legal Proceedings
There are no material proceedings to which any director or officer, or any associate of any such director or officer, is a party that is adverse to our Company or any of our subsidiaries or has a material interest adverse to our Company or any of our subsidiaries. No director or executive officer has been a director or executive officer of any business which has filed a bankruptcy petition or had a bankruptcy petition filed against it during the past ten years. No director or executive officer has been convicted of a criminal offense or is the subject of a pending criminal proceeding during the past ten years. No director or executive officer has been the subject of any order, judgment or decree of any court permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities or banking activities during the past ten years. No director or officer has been found by a court to have violated a federal or state securities or commodities law during the past ten years.
Compliance with Section 16(A) of the Exchange Act
Section 16(a) of the Exchange Act requires the Company’s directors, executive officers and persons who beneficially own 10% or more of a class of securities registered under Section 12 of the Exchange Act to file reports of beneficial ownership and changes in beneficial ownership with the SEC. Directors, executive officers and greater than 10% stockholders are required by the rules and regulations of the SEC to furnish the Company with copies of all reports filed by them in compliance with Section 16(a).
Based solely on our review of certain reports filed with the Securities and Exchange Commission pursuant to Section 16(a) of the Securities Exchange Act of 1934, as amended, the reports required to be filed with respect to transactions in our common stock during the fiscal year ended December 31, 2014, were not timely.
Code of Ethics
We have not yet adopted a code of ethics because we wanted to complete our constitution of the Board prior to adopting such code of ethics to allow the entire Board to review and approve our code of ethics.
Item 11. Executive Compensation.
The following summary compensation table sets forth all compensation awarded to, earned by, or paid to the named executive officers paid by us during the periods ended December 31, 2014, 2012 and 2013.
| 65 |
| Name And Principal Position |
Year | Salary ($) |
Bonus ($) |
Stock Awards ($) |
Option Awards ($) |
Non-Equity Incentive Plan Compen- sation ($) |
All Other Compen- sation ($) |
Total ($) | ||||||||||||||||||||||||
| Viktor Rozsnyay | 2014 | $ | 8,400 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 8,400 | |||||||||||||||||
| Chief Executive Officer & President | 2013 | $ | 8,400 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 8,400 | |||||||||||||||||
| 2012 | $ | 8,400 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 8,400 | ||||||||||||||||||
| Ildiko Rozsa | 2014 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | |||||||||||||||||
| Chief Financial Officer | 2013 | 0 | 0 | 0 | 0 | 0 | 0 | $ | 0 | |||||||||||||||||||||||
| 2012 | 0 | 0 | 15,000 | 0 | 0 | 0 | $ | 15,000 | ||||||||||||||||||||||||
Outstanding Equity Awards at December 31, 2014
As of December 31, 2014, there were no outstanding equity awards.
Director Compensation
During the year, ended December 31, 2014, the Company did not pay any cash fees or expenses to our directors for serving on the Board.
Employment Agreements
We currently do not have any employee agreements.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth, as of March 31, 2015, the number of shares of our common stock owned by (i) each person who is known by us to own of record or beneficially five percent (5%) or more of our outstanding shares, (ii) each of our directors, (iii) each of our executive officers and (iv) all of our directors and executive officers as a group. Unless otherwise indicated, each of the persons listed below has sole voting and investment power with respect to the shares of our common stock beneficially owned.
| Name and Address |
Beneficial Relationship to Company |
Outstanding Common Stock |
Percentage of Ownership of Common Stock (1) |
||||||
| Directors and Named Executive Officers | |||||||||
| Viktor Rozsnyay(3) | Chief Executive Officer, President, Chairman | 1,874,300 | 4 | %(2) | |||||
| Ildiko Rozsa (4) | Chief Financial Officer | 570,000 | 1.22 | % | |||||
| Peter Boros (5) | Director | 1,159,907 | 2.5 | % | |||||
| Officers and Directors as a Group (3 persons) | - | 2,444,300 | 8.4 | % | |||||
| 66 |
* less than 1%
| (1) | Based on 46,500,896 shares of common stock outstanding as of March 31, 2015. |
| (2) | This percentage does not include shares of Preferred Stock. |
| (3) | Mr. Rozsnyay owns 1,000,000 shares of our Series A Preferred Stock and 1,874,300 shares of our common stock. His mailing address is 2049 Diosd, Ligetszepe ut 54, Hungary. |
| (4) | Ms. Rozsa owns 570,000 shares of our common stock. Ms. Rozsa’s mailing address is 1066 Budapest, Terez Krt. 22, Hungary. | |
| (5) | Peter Boros owns 1,000,000 shares of our Series A Preferred stock, 1,000,000 shares of our Series D Preferred stock and 1,159,907 shares of our common stock. Mr. Boros’s address is Gyor, Ybl seteny 23, Hungary. |
Securities Authorized for Issuance under Equity Compensation Plans
The Company currently does not have an equity compensation plans in effect.
Changes in Control
We are not aware of any arrangements that may result in “changes in control” as that term is defined by the provisions of Item 403(c) of Regulation S-K.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Related Transactions
During the year ended December 31, 2014, there were no related transactions required to be reported under Item 404 of Regulation S-K.
Director Independence
On an annual basis, each director and executive officer will be obligated to disclose any transactions with the Company in which a director or executive officer, or any member of his or her immediate family, have a direct or indirect material interest in accordance with Item 407(a) of Regulation S-K. Following completion of these disclosures, the Board will make an annual determination as to the independence of each director using the current standards for “independence” that satisfy both the criteria for the Nasdaq and the American Stock Exchange.
As of December 31, 2014, the Board determined that none of our directors are independent under these standards.
Item 14. Principal Accounting Fees and Services.
The following table sets forth the aggregate fees billed for each of the last two fiscal years for professional services rendered by the principal accountant for the audit of the Company's annual financial statements and review of financial statements included in the Company's Form 10-Q quarterly reports or services that are normally provided by the accountant in connection with statutory and regulatory filings or engagements for those fiscal years.
| 2014 | 2013 | |||||||
| Audit Fees | $ | 71,360 | $ | 34,227 | ||||
| Audit-Related Fees | $ | 0 | $ | 0 | ||||
| Tax Fees | $ | 0 | $ | 0 | ||||
| All Other Fees | $ | 0 | $ | 0 | ||||
| TOTAL | $ | 71,360 | $ | 34,227 | ||||
We do not have an audit committee. Our entire board of directors pre-approves all services provided by our independent auditors.
| 67 |
PART IV
Item 15. Exhibits, Financial Statement Schedules.
| Exhibit No. | Description | |
| 23.1 | Consent of BDO Hungary, Ltd.* | |
| 31.1 | Certification by the Principal Executive Officer of Registrant pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Rule 13a-14(a) or Rule 15d-14(a)). * | |
| 31.2 | Certification by the Principal Financial Officer of Registrant pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Rule 13a-14(a) or Rule 15d-14(a)). * | |
| 32.1 | Certification by the Principal Executive Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. * | |
| 32.2 | Certification by the Principal Executive Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. * | |
| 101.INS | XBRL Instance Document ** | |
| 101.SCH | XBRL Taxonomy Extension Schema ** | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase ** | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase ** | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase ** | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase ** | |
* filed herewith
** In accordance with Regulation S-T, the XBRL-related information on Exhibit No. 101 to this Annual Report on Form 10-K shall be deemed “furnished” herewith and not “filed.”
| 68 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| POWER OF THE DREAM VENTURES, INC. | |||||
| Date: March 31, 2015 | By: | /s/ Viktor Rozsnyay | |||
| Name: Viktor Rozsnyay | |||||
|
Title: Chief Executive Officer (Principal Executive Officer) |
|||||
| Date: March 31, 2015 | By: | /s/ Ildiko Rozsa | |||
| Name: Ildiko Rozsa | |||||
|
Title: Chief Financial Officer (Principal Financial Officer)
|
|||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | Position | Date | ||
| /s/ Viktor Rozsnyay | Chief Executive Officer, President and Chairman | March 31, 2015 | ||
| Viktor Rozsnyay | ||||
| /s/ Ildiko Rozsa | Chief Financial Officer | March 31, 2015 | ||
| Ildiko Rozsa | ||||
| /s/ Peter Boros | Director | March 31, 2015 | ||
| Peter Boros | ||||
| 69 |
Index to Financial Statements
| Page No. | |
| Report of Independent Registered Public Accounting Firm | F-1 |
| Consolidated Balance Sheet at December 31, 2014 | F-2 |
| Consolidated Statements of Operations and Comprehensive Income for the years ended December 31, 2014 and 2013 | F-3 |
| Consolidated Statements of Deficiency in Stockholders’ Equity for the two years ended December 31, 2014 | F-4 to F-10 |
| Consolidated Statements of Cash Flows for the years ended December 31, 2014 and 2013 | F-11 |
| Notes to Consolidated Financial Statements | F-12 to F-40 |
| 70 |
Report of Independent Public Accounting Firm
Board of Directors and Stockholders
Power of the Dream Ventures, Inc. (formerly Tia V)
(a development stage company)
Budapest, Hungary
We have audited the accompanying balance sheets of Power of the Dream Ventures, Inc. (formerly Tia V), a development stage company, as of December 31, 2014 and 2013 and the related statements of operations, stockholders’ equity, and cash flows for each of the two years in the period then ended and the period from inception (April 26, 2006) to December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Power of the Dream Ventures, Inc. (formerly Tia V) at December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the two years in the period then ended and the period from inception (April 26, 2006) through December 31, 2014, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
March 31, 2015
BDO Hungary Audit Ltd.
1103 Budapest, Kőér utca 2/A
Registration number: 002387
| Zsuzsanna Jasper | Ferenc Baumgartner | |
| Managing Director |
Certified Auditor Chamber registration No.: 002955 |
| F-1 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED BALANCE SHEET
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
Amounts in USD
| Notes | December 31, 2014 | December 31, 2013 | ||||||||||
| ASSETS | ||||||||||||
| Current Assets | ||||||||||||
| Cash | $ | 1,524 | $ | 22,922 | ||||||||
| Other receivables | 3 | 81,048 | 217,419 | |||||||||
| Inventories | 18,825 | 22,651 | ||||||||||
| Total Current Assets | 101,397 | 262,992 | ||||||||||
| Intangibles | 5 | 315,327 | 30,055,381 | |||||||||
| Fixed assets, net | 5 | 92,043 | 189,194 | |||||||||
| Total Assets | 508,767 | 30,507,567 | ||||||||||
| LIABILITIES | ||||||||||||
| Current Liabilities | ||||||||||||
| Accounts payable and accrued and other liabilities | 9 | $ | 4,465,149 | $ | 4,083,294 | |||||||
| Grants received | 10 | 1,170,492 | 1,432,042 | |||||||||
| Capital leases payable, current portion | 7 | 50,622 | 55,784 | |||||||||
| Short term loans from related parties | 144,045 | 86,899 | ||||||||||
| Liability from warrants | 8 | 63,638 | 87,502 | |||||||||
| Note payable | 6 | 330,964 | 154,000 | |||||||||
| Short term loans | 11 | 2,304,120 | 2,341,159 | |||||||||
| Total Current Liabilities | 8,529,030 | 8,240,680 | ||||||||||
| Long term liabilities | ||||||||||||
| Capital leases payable, less current portion | 7 | — | 35,692 | |||||||||
| Total Long Term Liabilities | — | 35,692 | ||||||||||
| Stockholders’ Equity | ||||||||||||
| Preferred stock, $0.001 par value, 10,000,000 shares authorized, 5,920,000 and 4,900,000 issued and outstanding | 8 | 5,920 | 4,900 | |||||||||
| Common stock, $.001 par value; 100,000,000 shares authorized, 46,700,896 and 46,500,896 shares issued and outstanding | 8 | 46,701 | 46,501 | |||||||||
| Additional Paid-In Capital Deficit accumulated during development stage Other Comprehensive Income | 36,246,114 (44,854,780) 535,782 | 35,403,285 (13,018,339) (205,152) | ||||||||||
| Unearned Compensation | — | — | ||||||||||
| Total Stockholders’ Equity | (8,020,263 | ) | 22,231,195 | |||||||||
| Total liabilities and stockholders’ equity | 508,767 | 30,507,567 | ||||||||||
| F-2 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF OPERATIONS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Amounts in USD
| Notes |
For the
year ended |
For the
year ended |
For the Period from April 26, 2006 (date of inception) to December 31, 2014 |
|||||
| Net Sales | $ - | $ - | $ 5,833 | |||||
| Cost of Sales | - | - | (3,711) | |||||
| Gross margin | - | - | 2,122 | |||||
| Material expenses | 29,548 | 34,073 | 175,522 | |||||
| General administration | 12 | 1,255,316 | 738,975 | 10,150,548 | ||||
| Research and development | 4 | - | 41,812 | 866,619 | ||||
| Personnel expenses | 89,489 | 271,114 | 1,055,093 | |||||
| Depreciation and amortization | 5 | 94,810 | 145,219 | 802,091 | ||||
| Grants received | 10 | - | (150,196) | (276,979) | ||||
| Impairment of intangibles | 5 | 29,570,912 | - | 29,570,912 | ||||
| Other expenses, net | 13 | 46,974 | 83,335 | 760,932 | ||||
| Operating expenses | 31,087,049 | 1,164,332 | 43,104,738 | |||||
| Loss from operations | (31,087,049) | (1,164,332) | (43,102,616) | |||||
| Interest income and exchange gains |
(749,380) |
(629,625) |
(1,547,736) | |||||
|
Loss before income taxes |
(31,836,429) |
(1,793,957) |
(44,650,352) |
|||||
| Income taxes | (12) | (761) | (3,964) | |||||
|
Net loss |
(31,836,441) |
(1,794,718) |
$ (44,654,316) |
|||||
|
Basic loss per share Diluted loss per share Weighted average number of shares outstanding – Basic Weighted average number of shares outstanding – Diluted |
$(0.69) $(0.69)
46,500,896
46,500,896 |
$(0.04) $(0.04)
44,909,013
44,909,013 |
| F-3 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| Common Shares | Stocks Amount | Accumulated Deficit During Developmental Stage | Additional Paid In Capital | Other Comprehensive Income | Unearned Compensation | Total | Comprehensive Income/ (Loss) | |||||||||||||||||||||||||
| Issuance of common stock | 33,300,000 | $ | 3,330 | $ | 10,670 | $ | 14,000 | |||||||||||||||||||||||||
| Contributed Capital | 96,100 | 96,100 | ||||||||||||||||||||||||||||||
| Currency Translation Adjustment | $ | 4,151 | 4,151 | 4,151 | ||||||||||||||||||||||||||||
| Net loss for the period | $ | (35,100 | ) | (35,100 | ) | (35,100 | ) | |||||||||||||||||||||||||
| Balance at December 31, 2006 | 33,300,000 | 3,330 | (35,100 | ) | 106,770 | 4,151 | — | 79,151 | (30,949 | ) | ||||||||||||||||||||||
| Contributed Capital | 53,735 | 53,735 | ||||||||||||||||||||||||||||||
| Recapitalization upon Reverse Merger on April 10, 2007 (See Note 1) | 2,500,000 | 250 | (250,763 | ) | (250,513 | ) | ||||||||||||||||||||||||||
| Private placement of shares at $0.34 per share (See Note 10) | 2,250,000 | 225 | 764,775 | 765,000 | ||||||||||||||||||||||||||||
| Shares issued for services at $0.34 per share (See Note 10) | 1,875,000 | 188 | 637,313 | $ | (467,501 | ) | 170,000 | |||||||||||||||||||||||||
| Shares issued for research and development at $0.34 per share (See Note 10) | 100,000 | 10 | 33,990 | 34,000 | ||||||||||||||||||||||||||||
| Private placement at $2.5 per share (See Note 10) | 104,000 | 10 | 259,990 | 260,000 | ||||||||||||||||||||||||||||
| Shares issued for stock based compensation at $2.5 per share (See Note 10) | 1,036,000 | 104 | 2,589,896 | (2,590,000 | ) | — | ||||||||||||||||||||||||||
| Amortization of Unearned Compensation | 1,124,932 | 1,124,932 | ||||||||||||||||||||||||||||||
| Currency Translation Adjustment | (14,001 | ) | (14,001 | ) | (14,001 | ) | ||||||||||||||||||||||||||
| Net loss for the period | (1,992,472 | ) | (1,992,472 | ) | (1,992,472 | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2007 | 41,165,000 | $ | 4,117 | $ | (2,278,335 | ) | $ | 4,446,469 | $ | (9,850 | ) | $ | (1,932,569 | ) | $ | 229,832 | $ | (2,006,473 | ) | |||||||||||||
| F-4 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| Common Shares | Stocks Amount | Accumulated Deficit During Developmental Stage | Additional Paid In Capital | Other Comprehensive Income | Unearned Compensation | Total | Comprehensive Income/ (Loss) | |||||||||||||||||||||||||
| Balance at December 31, 2007 | 41,165,000 | $ | 4,117 | $ | (2,278,335 | ) | $ | 4,446,469 | $ | (9,850 | ) | $ | (1,932,569 | ) | $ | 229,832 | ||||||||||||||||
| Private placement of shares at $3.25 per share (See Note 10) | 32,500 | 3 | 105,622 | 105,625 | ||||||||||||||||||||||||||||
| Shares issued for services at $0.7 per share (See Note 10) | 306,570 | 31 | 214,568 | (214,599 | ) | — | ||||||||||||||||||||||||||
| Shares issued for services at $0.75 per share (See Note 10) | 1,500,000 | 150 | 1,124,850 | (1,125,000 | ) | — | ||||||||||||||||||||||||||
| Shares issued for services at $1.35 per share (See Note 10) | 111,111 | 11 | 149,989 | (150,000 | ) | — | ||||||||||||||||||||||||||
| Private placement of shares at $0.4 per share (See Note 10) | 2,500,000 | 250 | 999,750 | 1,000,000 | ||||||||||||||||||||||||||||
| Shares issued for Standby Equity Distribution Agreement at $0.4 per share | 2,000,000 | 200 | (200 | ) | — | |||||||||||||||||||||||||||
| Amortization of Unearned Compensation | 3,018,710 | 3,018,710 | ||||||||||||||||||||||||||||||
| Currency Translation Adjustment | 36,465 | 36,465 | 36,465 | |||||||||||||||||||||||||||||
| Net loss for the period | (3,958,212 | ) | (3,958,212 | ) | (3,958,212 | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2008 | 47,615,181 | $ | 4,762 | $ | (6,236,547 | ) | $ | 7,041,048 | $ | 26,615 | $ | (403,458 | ) | $ | 432,420 | $ | (3,921,747 | ) | ||||||||||||||
| Shares issued for stock based compensation at $0.4 per share (See Note 8) | 700,000 | 70 | 279,930 | (280,000 | ) | — | ||||||||||||||||||||||||||
| Private placement for $0.20 per share | 111,110 | 11 | 22,211 | 22,222 | ||||||||||||||||||||||||||||
| Private placement for $0.16 per share | 175,000 | 18 | 27,982 | 28,000 | ||||||||||||||||||||||||||||
| Shares issued for services at $0.4 per share (See Note 8 | 250,000 | 25 | 99,975 | (60,000 | ) | 40,000 | ||||||||||||||||||||||||||
| Amortization of Unearned Compensation | 728,458 | 728,458 | ||||||||||||||||||||||||||||||
| Currency Translation Adjustment | 17,823 | 17,823 | 17,823 | |||||||||||||||||||||||||||||
| Net loss for the period | (1,518,077 | ) | (1,518,077 | ) | (1,518,077 | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2009 | 48,851,291 | $ | 4,886 | $ | (7,754,624 | ) | $ | 7,471,146 | $ | 44,438 | $ | (15,000 | ) | $ | (249,154 | ) | $ | (1,500,254 | ) | |||||||||||||
| F-5 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| Common Shares | Stocks Amount | Accumulated Deficit During Developmental Stage | Additional Paid In Capital | Other Comprehensive Income | Unearned Compensation | Total | Comprehensive Income/ (Loss) | |||||||||||||||||||||||||
| Balance at December 31, 2009 | 48,851,291 | $ | 4,886 | $ | (7,754,624 | ) | $ | 7,471,146 | $ | 44,438 | $ | (15,000 | ) | $ | (249,154 | ) | ||||||||||||||||
| Private placement for $0.12 and $0.1 per share | 1,597,500 | 159 | 159,542 | 159,701 | ||||||||||||||||||||||||||||
| Private placement for $0.5 per share (See Note 8) | 70,000 | 7 | 34,993 | 35,000 | ||||||||||||||||||||||||||||
| Shares issued for services at $0.5 per share (See Note 8) | 1,027,000 | 100 | 513,400 | (513,500 | ) | — | ||||||||||||||||||||||||||
| Shares issued for stock based compensation at $0.1 per share (See Note 8) | 540,000 | 54 | 53,946 | (54,000 | ) | — | ||||||||||||||||||||||||||
| Shares issued for services at $0.1 per share (See Note 8) | 20,000 | 2 | 1,998 | (2,000 | ) | — | ||||||||||||||||||||||||||
| Exercise of warrant | 3,226 | 3 | 1,613 | 1,616 | ||||||||||||||||||||||||||||
| Amortization of Unearned Compensation | 200,528 | 200,528 | ||||||||||||||||||||||||||||||
| Currency Translation Adjustment | (38,874 | ) |
| (38,874 | ) | (38,874 | ) | |||||||||||||||||||||||||
| Net loss for the period | (588,910 | ) | (588,910 | ) | (588,910 | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2010 | 52,109,017 | $ | 5,211 | $ | (8,343,534 | ) | 8,236,638 | $ | 5,564 | $ | (383,972 | ) | $ | (480,093 | ) | $ | (627,784 | ) | ||||||||||||||
| F-6 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| Preferred Shares | Common Shares | Stocks Amount | Accumulated Deficit During Developmental Stage | Additional Paid In Capital | Other Comprehensive Income | Unearned Compensation | Total | |||||||||||||||||||||||||||||
| Balance at December 31, 2010 | — | 52,109,017 | $ | 5,211 | $ | (8,343,534 | ) | 8,236,638 | $ | 5,564 | $ | (383,972 | ) | $ | (480,093 | ) | $ | (627,784 | ) | |||||||||||||||||
| Private placement at $0.29 per share (see Note 8) | 196,489 | 20 | 56,962 | 56,982 | ||||||||||||||||||||||||||||||||
| Conversion of note at $0.29 per share | 2,400,000 | 240 | 69,760 | 70,000 | ||||||||||||||||||||||||||||||||
| Private placement at $0.16 per share (see Note 8) | 1,727,013 | 172 | 276,150 | 276,322 | ||||||||||||||||||||||||||||||||
| Private placement at $0.12 per share (see Note 8) | 17,825 | 2 | 2,135 | 2,137 | ||||||||||||||||||||||||||||||||
| Shares issued for services per share (See Note 8) | 1,970,000 | 197 | 457,691 | (457,888 | ) | — | ||||||||||||||||||||||||||||||
| Shares issued for stock based compensation at $0.15 per share (See Note 8) | 540,000 | 54 | 80,946 | (81,000 | ) | — | ||||||||||||||||||||||||||||||
| Private placement at $0.16 per share (see Note 8) | 27,700 | 3 | 4,429 | 4,432 | ||||||||||||||||||||||||||||||||
| Conversion of note at $0.01 per share | 3,500,000 | 350 | 19,650 | 20,000 | ||||||||||||||||||||||||||||||||
| Cancellation of common stock (and issue of preferred stock) | 2,000,000 | (24,000,000 | ) | — | ||||||||||||||||||||||||||||||||
| Amortization of Unearned Compensation | 781,458 | 781,458 | ||||||||||||||||||||||||||||||||||
| Currency Translation Adjustment | (13,791 | ) |
| (13,791 | ) | (13,791 | ) | |||||||||||||||||||||||||||||
| Net loss for the period | (1,251,035 | ) | (1,251,035 | ) | (1,251,035 | ) | ||||||||||||||||||||||||||||||
| Balance at December 31, 2011 | 2,000,000 | 38,488,044 | $ | 6,249 | $ | (9,594,569 | ) | 9,204,361 | $ | (8,227 | ) | $ | (141,402 | ) | $ | (533,588 | ) | $ | (1,264,826 | ) | ||||||||||||||||
| F-7 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| Preferred Shares | Common Shares | Stocks Amount | Accumulated Deficit During Developmental Stage | Additional Paid In Capital | Other Comprehensive Income | Unearned Compensation | Total | |||||||||||||||||||||||||||||
| Balance at December 31, 2011 | 2,000,000 | 38,488,044 | $ | 6,249 | $ | (9,594,569 | ) | 9,204,361 | $ | (8,227 | ) | $ | (141,402 | ) | $ | (533,588 | ) | $ | (1,264,826 | ) | ||||||||||||||||
| Conversion of common shares to preferred shares (change in value) | (2,200 | ) | 2,200 | — | ||||||||||||||||||||||||||||||||
| Shares issued for services | 135,000 | 14 | 21,586 | 21,600 | ||||||||||||||||||||||||||||||||
| Private placement at $0.30 per share (see Note 8) | 42,200 | 4 | 12,654 | 12,658 | ||||||||||||||||||||||||||||||||
| Amortization of Unearned Compensation | 197,328 | 197,328 | ||||||||||||||||||||||||||||||||||
| Change of par value from $0.0001 to $0.001 | 36,598 | (36,598 | ) | — | ||||||||||||||||||||||||||||||||
| Shares issued for services | 1,930,000 | 1,930 | 178,240 | (132,850 | ) | 47,320 | ||||||||||||||||||||||||||||||
| Conversion of loan to stocks | 1,900,000 | 1,900 | 74,100 | 76,000 | ||||||||||||||||||||||||||||||||
| Acquisition of Genetic Immunity | 1,000,000 | 1,000 | 50,000 | 25,023,620 | 25,074,620 | |||||||||||||||||||||||||||||||
| Currency Translation Adjustment | (37,272 | ) | (37,272 | ) | (37,272 | ) | ||||||||||||||||||||||||||||||
| Net loss for the period | (1,679,052 | ) | (1,679,052 | ) | (1,679,052 | ) | ||||||||||||||||||||||||||||||
| Balance at December 31, 2012 | 3,000,000 | 42,495,244 | $ | 45,495 | $ | (11,223,621 | ) | 34,480,163 | $ | (45,499 | ) | $ | (76,924 | ) | $ | 23,179,614 | $ | (1,716,324 | ) | |||||||||||||||||
| F-8 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| Preferred Shares | Common Shares | Stocks Amount | Accumulated Deficit During Developmental Stage | Additional Paid In Capital | Other Comprehensive Income | Unearned Compensation | Total | |||||||||||||||||||||||||||||
| Balance at December 31, 2012 | 3,000,000 | 42,495,244 | $ | 45,495 | $ | (11,223,621 | ) | 34,480,163 | $ | (45,499 | ) | $ | (76,924 | ) | $ | 23,179,614 | $ | (1,716,324 | ) | |||||||||||||||||
| Cancellation of shares for services | (250,000 | ) | (250 | ) | (34,775 | ) | 35,025 | — | ||||||||||||||||||||||||||||
| Amortization of Unearned Compensation | 41,899 | 41,899 | ||||||||||||||||||||||||||||||||||
| Shares issued for services | 100,000 | 100 | 19,800 | — | 19,900 | |||||||||||||||||||||||||||||||
| Shares issued to prior owner | 1,000,000 | 1,000 | 159,000 | 160,000 | ||||||||||||||||||||||||||||||||
| Issuance of shares to settle payable | 655,652 | 656 | 43,344 | 44,000 | ||||||||||||||||||||||||||||||||
| Private placement | 2,500,000 | 2,500 | 130,000 | 132,500 | ||||||||||||||||||||||||||||||||
| Private placement | 900,000 | 900 | 269,100 | 270,000 | ||||||||||||||||||||||||||||||||
| Private Placement | 1,000,000 | 1000 | 336,653 | 337,653 | ||||||||||||||||||||||||||||||||
| Currency Translation Adjustment | (159,653 | ) | (159,653 | ) | (159,653 | ) | ||||||||||||||||||||||||||||||
| Net profit for the period | (1,794,718 | ) | (1,794,718 | ) | (1,794,718 | ) | ||||||||||||||||||||||||||||||
| Balance at December 31, 2013 | 4,900,000 | 46,500,896 | $ | 51,401 | $ | (13,018,339 | ) | 35,403,285 | $ | (205,152 | ) | $ | — | $ | 22,231,195 | $ | (1,954,371 | ) | ||||||||||||||||||
| 9 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| Preferred Shares | Common Shares | Stocks Amount | Accumulated Deficit During Developmental Stage | Additional Paid In Capital | Other Comprehensive Income | Total | Comprehensive Income/(loss) | ||||
| Balance at December 31, 2013 |
4,900,000 |
46,500,896 | $51,401 | $(13,018,339) | 35,403,285 | $(205,152) | $22,231,195 |
$(1,954,371) | |||
| Private placement | 1,020,000 | 1,020 | 842,529 | 843,549 | |||||||
| Currency Translation Adjustment | 740,934 | 740,934 |
740,934 | ||||||||
| Conversion of debt | 200,000 | 200 | 300 |
|
500 | ||||||
| Net loss for the period | (31,836,441) | (31,836,441) |
(31,836,441) | ||||||||
| Balance at December 31, 2014 |
|
5,920,000 |
46,700,896 | $52,621 | $(44,854,780) | 36,246,114 | $535,782 | $(8,020,263) |
$(31,095,507) |
| F-10 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF CASH FLOWS
Amounts in USD
|
For the year ended December 31, 2014 |
For the year ended December 31, 2013 |
Cumulative from April 26, 2006 (date of inception) to December 31, 2014 | ||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||
| Net loss | $(31,836,441) | $(1,794,718) | $(44,654,316) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||
| Amortization of stock-based compensation | - | 256,824 | 6,308,238 | |||
| Recapitalization under reverse merger | - | - | (250,763) | |||
| Impairment of intangibles | 29,570,912 | 29,570,912 | ||||
| Issue of shares for services | - | 19,900 | 229,900 | |||
| Issue of shares for research and development | - | - | 34,000 | |||
| Liability from warrant | (21,557) | 87,502 | 63,638 | |||
| Reversal of grant accrual | - | (114,732) | (245,306) | |||
| Depreciation and amortization | 94,810 | 145,219 | 802,091 | |||
| (2,192,276) | (1,400,005) | (8,141,606) | ||||
| Changes in operating assets and liabilities: | ||||||
| (Increase) Decrease in other current assets | 140,197 | 114,599 | (99,873) | |||
|
(Decrease) Increase in accounts payable and accrued liabilities |
269,234 | 927,633 | 3,888,546 | |||
| Net cash used in operating activities | (1,782,845) | (357,773) | (4,352,933) | |||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||
| Purchase of fixed assets | - | (206,216) | (702,413) | |||
| Net cash used in investing activities | - | (206,216) | (702,413) | |||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||
| Proceeds from stockholders | - | (4,985) | 86,899 | |||
| Exercise of warrant | - | - | 1,616 | |||
| Proceeds from sale of preferred stock | 843,549 | 1,451,202 | ||||
| Proceeds from sale of common stock | - | 687,563 | 2,650,407 | |||
| Proceeds from issuance of notes | 176,964 | - | 330,964 | |||
| Net cash from financing activities | 1,020,513 | 682,578 | 4,521,088 | |||
| Effect of exchange rate changes on cash | 740,934 | (159,653) | 535,782 | |||
| Net (decrease) increase in cash | (21,398) | (41,064) | 1,524 | |||
| Cash at beginning of period | 22,922 | 63,986 | - | |||
| Cash at end of period | $1,524 | $22,922 | $1,524 | |||
| Supplemental disclosure of cash flow information: | ||||||
| Non-cash investing and financing transactions | ||||||
| Issuance of shares for services | $- | $201,770 | $4,256,158 | |||
|
Issuance of shares for liabilities assumed under reverse merger |
- | - | $250,513 | |||
| Issuance of stock based compensation shares | - | - | $2,590,000 | |||
| Purchase of fixed assets through the assumption of capital lease obligations | - | 406,558 | $406,558 | |||
| Cash paid for: | ||||||
| Interest | - | - | 40,980 | |||
| Taxes | - | - | - |
| F-11 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 1 - GENERAL INFORMATION
Power of the Dream Ventures, Inc., f/k/a “Tia V, Inc.” (“PDV” or the “Company”) was incorporated in Delaware on August 17, 2006, with the objective to acquire, or merge with, an operating business.
Reverse merger
PDV entered into and consummated a Securities Exchange Agreement (“Exchange Agreement”) on April 10, 2007. Under the terms of the Exchange Agreement, PDV acquired all the outstanding equity interests of Vidatech, Kft. (also known as Vidatech Technological Research and Development LLC) a limited liability company formed under the laws of the Republic of Hungary, (“Vidatech”) in exchange for 33,300,000 shares of PDV’s common stock, and Vidatech thereby became a wholly-owned Hungarian subsidiary of PDV. PDV is governed by the law of the State of Delaware, and its wholly-owned subsidiary, Vidatech, is governed by the law of the Republic of Hungary. PDV and Vidatech are herein collectively referred to as the “Company.”
Following the acquisition the former stockholders of Vidatech owned a majority of the issued and outstanding common stock of PDV and the management of Vidatech controlled the Board of Directors of PDV and its wholly-owned Hungarian subsidiary Vidatech. Therefore the acquisition has been accounted for as a reverse merger (the “Reverse Merger”) with Vidatech as the accounting acquirer of PDV. The accompanying consolidated financial statements of the Company reflect the historical results of Vidatech, and the consolidated results of operations of PDV subsequent to the acquisition date. In connection with the Exchange Agreement, PDV adopted the fiscal year end of Vidatech as December 31.
All reference to shares and per share amounts in the accompanying consolidated financial statements have been restated to reflect the aforementioned shares exchange.
Genetic Immunity
On October 2, 2012 the Company acquired Genetic Immunity, Inc., a US corporation domiciled in the state of Delaware. Genetic Immunity is an immunotherapeutic vaccine company whose lead product candidate DermaVir has passed Phase II clinical trials.
Through the acquisitions Genetic Immunity became 100% wholly owned subsidiary. The acquisition was completed for 1,000,000 shares of Series B Preferred stock, convertible into 40 million shares of common stock beginning on January 1, 2014.
The Company filed an 8-K with the SEC disclosing this transaction on October 4, 2012.
The acquisition of Genetic Immunity, Inc. has been accounted for as a business combination whereby the purchase price was allocated to intangible assets based on their fair values of the acquisition date. A summary of the purchase price consideration and related purchase price allocation are shown below:
Purchase price (at par value) $1.000
Fair value adjustment of shares 25,072,621
Total purchase price 25,073,621
The fair value of the shares equals the fair value of Genetic Immunity, Inc. at the acquisition date. The fair value of the company was estimated by an independent valuation company and the valuation was carried out on a discounted cash flow basis. The Company issued 1,000,000 shares of Series B Preferred stock which is not traded publicly and no market price could be estimated. As a result of this, the fair value of the shares was evaluated by a valuation company. The value of the shares is $25.07362 per share.
| F-12 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 1 - GENERAL INFORMATION (Continued)
Purchase price allocation
| Cash | $ | 357,276 | ||
| Inventories | 13,843 | |||
| Other receivables | 306,731 | |||
| Intangibles | 30,111,909 | |||
| Fixed assets | 418,483 | |||
| Accounts payable and accrued liabilities | (1,893,833 | ) | ||
| Grants received | (1,677,348 | ) | ||
| Short term loans to related parties | (154,631 | ) | ||
| Bridge loan | (2,334,326 | ) | ||
| Other liabilities | (73,483 | ) | ||
| Total purchase price | 25,074,621 | |||
In December 2014, the Company prepared an impairment test to test the recoverability of intangibles. As a result of the impairment test, the Company accounted for an impairment expense of $29,570,912.
Business
The Company is engaged in the acquisition, development, licensing and commercialization of and the investment in, directly or through business acquisitions, technologies developed in Hungary. In furtherance of its business, the Company provides research and development services to the companies, inventors from whom it acquires technologies or participation interests in technologies. A goal of the Company is to support research and development activities and to sell the products of inventions to the technological market.
From inception through December 31, 2014, the Company primarily focused on the raising of capital. As of December 31, 2014, the Company originally managed seven technologies: RiverPower, the Buresch Inventions (Desalination and H2O gas technology,) the Kalmar inventions (FireSAFE fire-proofing liquid; technology for utilizing communal waste as a concrete additive; technology for repairing potholes with the use of recycled plastics; technology for neutralizing red mud; biodegradable deicing solution and PVC shielded electric cable recycling technology), and an equity interest in ‘iGlue, Inc (Ticker: IGLU)’, a company formed to develop next generation semantic internet based search engine and content organizer applications. As of December 31, 2014 the Company has discontinued efforts to develop the Buresch inventions, the Kalmar inventions, RiverPower and the Toth Telescope. (see Note 4). As of December 31, 2014, the Company has only realized limited revenues from the now discontinued TothTelescope project and has not realized any revenues from the other, now discontinued inventions. As a result, the accompanying consolidated financial statements have been presented on a development stage basis.
| F-13 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
The Kalmar Inventions: FireSAFE, HardCrete concrete from communal waste additive; Recycled plastic pothole filler; PVC covered electrical cable and radial tire recycling technology; red mud neutralization; and environmentally friendly salt free de-icing solution.
As of December 31, 2011 the Company elected to discontinue development work on all of these technologies. The rational behind this decision rests with availability of resources, the time that would be needed to develop said inventions, the potential return the Company could realize on a partial or full exit from these investments and international completion.
TothTelescope
On July 15, 2009 the Company’s exclusive distributor agreement expired with Attila Toth, Inventor of the TothTelescope. After extensive review of the technology, the available market and hurdles associated with manufacturing and distributing the TothTelescope, we have elected not to renew the distribution agreement and discontinue the project. Since inception only 10 telescopes were sold. Overall invested capital was returned from commissions earned on these sales.
Yorkville SEDA
On October 8, 2008, Power of the Dream Ventures, Inc. (the “Company”) entered into a Standby Equity Distribution Agreement (the “Standby Equity Distribution Agreement”) with YA Global Investments, L.P. (the “Investor”). Pursuant to the terms of the Standby Equity Distribution Agreement, the Company (a) agreed to issue and sell to the Investor up to $5,000,000 of shares of the Company’s common stock, par value $0.0001 per share (the “Common Stock”) in tranches of equity, based upon a specified discount to the market price of the Common Stock, calculated over the five trading days following notice by the Company of an election to sell shares; and (b) issued to the Investor a warrant (the “Warrant”) to purchase 4,027,386 shares of Common Stock at the exercise price per share of $0.29. The Warrant is not part of the commitment shares issued by the company to the investor. The investor must purchase the shares underlying the Warrant. The Warrant price was determined based upon the highest Bid price on the day of the closing of the agreement.
In connection with the Standby Equity Distribution Agreement, the Company entered into a Registration Rights Agreement with the Investor (the “Registration Rights Agreement”) pursuant to which the Company agreed to register for resale the shares of Common Stock that may be purchased by the Investor pursuant to the Standby Equity Distribution Agreement, the shares of Common Stock issuable upon exercise of the Warrant and 2,000,000 shares of Common Stock (the “Commitment Shares”) issued to the Investor as a commitment fee pursuant to the terms of the Standby Equity Distribution Agreement. The commitment fee is recorded by decreasing additional paid in capital.
Financing from this transaction will be used by the Company for the continued development of its current technologies, commercialization of same, the acquisition of new technologies and for general corporate expenses.
The Company filed the details of this transaction on Form 8-K with the Commission on October 14, 2008.
On September 24, 2010 the Company notified YA Global Investments in writing of its intention to terminate the Standby Equity Distribution Agreement (SEDA).
In response to this letter YA Global informed the company in a letter dated October 5, 2010 that the SEDA agreement will terminate on or about October 15, 2010. YA Global also informed the company that even though the SEDA agreement is terminated its associated Warrant to purchase 4,027,386 shares of Our common stock remains in effect until it expires on October 8, 2013.
On October 18, 2010 the company received notice from YA global for the exercise of 50,000 warrant shares on a cashless basis. This cashless exercise resulted in the issuance of 3,226 shares of Common stock to YA Global. Following this exercise YA Global has 3,977,386 warrant shares available for purchase.
| F-14 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
iGlue Software via Equity position in iGlue, Inc.
From 2007 to 2011 we funded the development of iGlue, the cornerstone of the current technology portfolio. The financial commitment to in4, Ltd, the company behind iGlue, amounted to a total of approximately $600,000 for years 2007 through 2010. The total chronological events of iGlue are:
- On August 2, 2007 Vidatech Kft., the Company’s wholly owned Hungarian subsidiary, entered into a joint development agreement with the inventors/creators of iGlue, an online content organization and search application based on semantic web technology. In the transaction Vidatech received 30% equity in the newly formed in4 Kft (http://www.in4.hu), the owner and exclusive developer of iGlue (http://www.iglue.com), for HUF 900 thousand (approximately USD 5,000). Up until May 31, 2009, Vidatech was expected to provide up to HUF 48 million (approximately USD 271,000) in member loans to in4 Kft. to cover the development expense of iGlue.
- As of December 31, 2008 the Company has provided this amount to in4, Ltd.
- As part of the agreement, Vidatech had the option of converting its HUF 48 million member loan into an additional 10% equity in in4 Kft. on or before the end of May, 2009. As of January 8, 2009 the Company has elected to covert our member loan into the additional 10% equity in in4, Ltd., resulting in Vidatech owning 40% of equity.
- On May 17th, 2010, the Company transferred its equity ownership from its Hungarian subsidiary Vidatech, Kft. to Power of the Dream Ventures. The transfer was initiated to prepare for its eventual exit from iGlue.
- Also on May 17th, 2010, the Company arranged for a new investor to provide further funds for the continued development of iGlue, in the amount of HUF 110 million (approximately USD 550,000.) Prior to this event, up to May 17th, 2010 the Company has provided an additional HUF 64million ($310,000) as member loan to in4 Ltd. to finance development work, bringing our total investment to date to HUF 112,000,000 (approximately $600,000). As part of the new round of fund raising the Company agreed to convert its HUF 60 million in member loan into 3% equity. Following this conversion the new investor received 10% in in4, Ltd., of which the Company provided 1.5% and Peter Vasko, Founder of in4 limited provided 8.5%. Following the fund raising the new equity positions in in4 changed to Peter Vasko owning 51.5% (down from the pre-transaction 60%), Power of the Dream Ventures owning 38.5% (down from the pre-transaction 40%) and the new investor owning 10% equity.
- On May 19 of 2011 a new member joined in4, Ltd, Zoltan Siklosi who received 3% equity in the company for services rendered. Following his entry the new equity structure of the company changed to Peter Vasko owning 48.5% percent, Gyorgy Markos owning 10%, Zoltan Siklosi owning 3% and Power of the Dream Ventures owning 38.5%.
- Subsequently, to take iGlue to the next level, the Company organized a going public roadmap for the company. As part of this process, on November 3, 2011, in4, Ltd signed a Definitive Share Exchange Agreement to merge with Hardwired Interactive (HDWR) a fully reporting public company. As part of this agreement all holders of in4, Ltd equity received common and preferred stock in the newly public entity. As part of the transaction we received 600,000 shares of Series B Hardwired Interactive common stock convertible into 6,000,000 shares of common stock upon a reverse stock split. The Company also received warrants to purchase a total of three million shares of post split common stock, one million shares each at $5 dollars, $7 dollars and $9 dollars per share.
- On November 23, 2011 the Company announced plans to provide a dividend, in the form of iGlue common stock, to PWRV shareholders of record on December 23, 2011. Each PWRV share was to receive 0.05 share of iGlue common stock in the dividend. Upon approval from FINRA this dividend was paid out to PWRV shareholders on February 15, 2012. Following this distribution the Company still holds 2,884,986 shares of iGlue common stock and warrants to purchase a total of three million addition shares.
| F-15 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
Basis of presentation
The accompanying consolidated financial statements have been prepared by the Company pursuant to the rules and regulations of the SEC. Certain information and footnote disclosures normally included in financial statements prepared in accordance with accounting principles generally accepted in the United States of America for financial information have been condensed or omitted pursuant to such rules and regulations. In the opinion of management, the accompanying financial statements include all adjustments (consisting of normal recurring accruals) considered necessary to make the financial statements not misleading as of and for the period ended December 31, 2014 and for the period from April 26, 2006 (date of inception) to December 31, 2014.
Going Concern and Management’s Plan
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America, which contemplate the continuation of the Company as a going concern and assume realization of assets and the satisfaction of liabilities in the normal course of business. The Company has incurred losses from operations since inception. Management anticipates incurring additional losses in 2015. Further, the Company may incur additional losses thereafter, depending on its ability to generate revenues from the licensing or sale of its technologies and products, or to enter into any or a sufficient number of joint ventures. The Company has minimal revenue to date. There is no assurance that the Company can successfully commercialize any of its technologies and products and realize any revenues therefore. The Company’s technologies and products have never been utilized on a large-scale commercial basis and there is no assurance that any of its technologies or products will receive market acceptance. There is no assurance that the Company can continue to identify and acquire new technologies.
Since inception through December 31, 2014, the Company had an accumulated deficit of $44,854,780 and net cash used in operations of $4,352,933. However, management of the Company believes that future funding from the private placement of the Company’s common shares will allow them to continue operations and execute its business plan.
During 2014 and 2013 the Company raised a total of $843,549 and $607,653, respectively through private placements of common and preferred stock. These funds were used to finance operations, to reduce Genetic Immunity debt, and to finance Genetic Immunity’s regulatory process towards a marketing approval application. Additional funds used for these tasks, in the amount of $337,657 were previously provided in the prior years by the director, Peter Boros, who elected in November of 2013 to convert said loan into 1,000,000 shares of our Series D Preferred Stock.
On December 31, 2014 the Company entered into a Promissory Note with a private person provided $22,964, in loans to the Company. The note bears interest at 10%, is due in one year.
On July 28, 2014, on August 4, 2014, and on 14 August, 2014 the Company entered into three Promissory Notes with a private person provided $24,000, $55,000 and $75,000, respectively, in loans to the Company. The note bears interest at 10%, is due in one year.
On March 16, 2011 the Company entered into a Convertible Promissory Note with Infinite Funding LLC whereby Infinite Funding provided $76,000 dollars in loans to the Company. The note bears interest at 10%, is due December 5, 2011, and is convertible into shares of common stock at a conversion price of $0.04. On December 4, 2011 we received an extension to this note to December 31, 2012. The note was converted to 1,900,000 common shares at September 30, 2012.
On July 19, 2011 the Company entered into a Convertible Promissory Note with Infinite Funding LLC whereby Infinite Funding provided $49,000 dollars in loans to the Company. The note bears interest at 12%.
| F-16 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
Management believes the Company has adequate capital to keep the Company functioning through December 31, 2015. The need may arise, in the normal course of business, to raise additional capital if we want to accelerate development work, for the acquisition of additional technologies, or to meet unforeseen financial needs. No assurance can be given that the Company can obtain additional working capital, or if obtained, that such funding will not cause substantial dilution to shareholders of the Company. If the Company is unable to raise additional funds, if needed, it may be forced to change or delay its contemplated marketing and business plan. Being a development stage company, the Company is subject to all the risks inherent in the establishment of a new enterprise and the marketing and manufacturing of a new product, many of which risks are beyond the control of the Company. All of the factors discussed above raise substantial doubt about the Company’s ability to continue as a going concern.
These consolidated financial statements do not include any adjustments relating to the recoverability of recorded asset amounts that might be necessary as a result of the above uncertainty.
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
The significant accounting policies adopted in preparation of the consolidated financial statements are set out below.
Principles of Consolidation
The consolidated financial statements include the accounts of PDV and its wholly-owned Hungarian subsidiary, Vidatech. All significant intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates:
The preparation of the financial statements in conformity with GAAP requires management to make estimates, judgments and assumptions that affect amounts reported herein. Management believes that such estimates, judgments and assumptions are reasonable and appropriate. However, due to the inherent uncertainty involved, actual results may differ from those based upon management’s judgments, estimates and assumptions. Critical accounting policies requiring the use of estimates are depreciation and amortization and share-based payments
Revenue Recognition:
Sales are recognized when there is evidence of a sales agreement, the delivery of the goods or services has occurred, the sales price is fixed or determinable and collectability is reasonably assured, generally upon shipment of product to customers and transfer of title under standard commercial terms. Sales are measured based on the net amount billed to a customer. Generally there are no formal customer acceptance requirements or further obligations. Customers do not have a general right of return on products shipped therefore no provisions are made for return.
Accounts Receivable and Allowance for Doubtful Accounts:
Accounts receivable are stated at historical value, which approximates fair value. The Company does not require collateral for accounts receivable. Accounts receivable are reduced by an allowance for amounts that may be uncollectible in the future. This estimated allowance is determined by considering factors such as length of time accounts are past due, historical experience of write offs, and customers’ financial condition.
| F-17 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
Inventories:
Inventories are stated at the lower of cost, determined based on weighted average cost or market. Inventories are reduced by an allowance for excess and obsolete inventories based on management’s review of on-hand inventories compared to historical and estimated future sales and usage.
Fixed assets:
Fixed assets are stated at cost or fair value for impaired assets. Depreciation and amortization is computed principally by the straight-line method. Asset amortization charges are recorded for long lived assets. In the related periods, no asset impairment charges were accounted for. Depreciation is recorded commencing the date the assets are placed in service and is calculated using the straight line basis over their estimated useful lives.
The estimated useful lives of the various classes of long-lived assets are approximately 3-7 years.
Pensions and Other Post-retirement Employee benefits:
In Hungary, pensions are guaranteed and paid by the government or by pension funds, therefore no pensions and other post-retirement employee benefit costs or liabilities are to be calculated and accounted by the Company.
Product warranty:
The Company accrues for warranty obligations for products sold based on management estimates, with support from sales, quality and legal functions, of the amount that eventually will be required to settle such obligations. At December 31, 2014, the Company had no warranty obligations in connection with products sold.
Advertising costs:
Advertising and sales promotion expenses are expensed as incurred.
Research and development and Investment and Advances to Non-Consolidated Entities:
In accordance with ASC 730-10-25 “Accounting for Research and Development Costs,” all research and development (“R&D”) costs are expensed when they are incurred, unless they are reimbursed under specific contracts. Assets used in R&D activity, such as machinery, equipment, facilities and patents that have alternative future use either in R&D activities or otherwise are capitalized. In connection with investments and advances in development-stage technology entities in which the company owns or controls less than a 50% voting interest, (see Note 4) where repayment from such entity is based on the results of the research and development having future economic benefit, the investment and advances are accounted for as costs incurred by the Company as research and development in accordance with ASC 730-20-25 “Research and Development Arrangements”.
Income taxes:
The Company accounts for income taxes in accordance with ASC 740-10-25, “Accounting for Income Taxes”. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Valuation allowances are provided against deferred tax assets to the extent that it is more likely than not that the deferred tax assets will not be realized.
| F-18 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
Comprehensive Income (Loss):
ASC 220-10-25, “Accounting for Comprehensive Income,” establishes standards for reporting and disclosure of comprehensive income and its components (including revenues, expenses, gains and losses) in a full set of general-purpose financial statements. The items of other comprehensive income that are typically required to be disclosed are foreign currency items, minimum pension liability adjustments, and unrealized gains and losses on certain investments in debt and equity securities. Accumulated other comprehensive gain, at December 31, 2014 is $535,782.
Translation of Foreign Currencies:
The U.S. dollar is the functional currency for all of the Company’s businesses, except its operations in Hungary. Foreign currency denominated assets and liabilities for this unit is translated into U.S. dollars based on exchange rates prevailing at the end of each period presented, and revenues and expenses are translated at average exchange rates during the period presented. The effects of foreign exchange gains and losses arising from these translations of assets and liabilities are included as a component of equity, under other comprehensive income.
Loss per Share:
Under ASC 260-10-45, “Earnings Per Share”, basic loss per common share is computed by dividing the loss applicable to common stockholders by the weighted average number of common shares assumed to be outstanding during the period of computation. Diluted loss per common share is computed using the weighted average number of common shares and, if dilutive, potential common shares outstanding during the period. There were no common stock equivalents or potentially dilutive securities outstanding during the years ended December 31, 2014 and 2013, respectively. Accordingly, the weighted average number of common shares outstanding for the years ended December 31, 2014 and 2013, respectively, is the same for purposes of computing both basic and diluted net income per share for such years. At December 31, 2014 all warrants were excluded from the diluted EPS calculation according to management estimate because they are anti-dilutive.
Business Segment:
ASC 280-10-45, “Disclosures About Segments of an Enterprise and Related Information,” establishes standards for the way public enterprises report information about operating segments in annual consolidated financial statements and requires reporting of selected information about operating segments in interim financial statements regarding products and services, geographical areas and major customers. The Company has determined that under ASC 280-10-45, there are no operating segments since substantially all business operations, assets and liabilities are in Hungarian geographic segment.
Share-Based Payments:
In accordance with ASC 718-10 “Share-Based Payment” all forms of share-based payment (“SBP”) awards including shares issued under employee stock purchase plans, stock options, restricted stock and stock appreciation rights result in a cost that is measured at fair value on the awards’ grant date, based on the estimated number of awards that are expected to vest. As the Company did not issue any employee SBP prior to September 30, 2007, there is no compensation cost recognized in the accompanied consolidated financial statements.
The Company accounts for equity instruments issued to non-employees in accordance with the provisions of ASC 505-50, “Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services,” which requires that such equity instruments are recorded at their fair value on the measurement date. The measurement of stock-based compensation is subject to periodic adjustment as the underlying equity instrument vests. Non-employee stock-based compensation charges are amortized over the vesting period or period of performance of the services.
| F-19 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
Recent Accounting Pronouncements:
On June 13, 2014, the FASB issued ASU 2014-101( “Elimination of Certain Financial Reporting Requirements, including an Amendment to Variable Interest Entities Guidance in ASC Topic 810, Consolidation”) to eliminate the concept of a development stage entity (“DSE”) from U.S. GAAP. This change rescinds certain financial reporting requirements that have historically applied to DSEs and is intended to result in cost-savings for affected entities, such as certain start-up or research and development entities. In addition, ASU 2014-10 introduces new disclosure requirements about the reporting entity’s risks and uncertainties. ASU 2014-101 is effective prospectively for fiscal years, and interim periods within those years, beginning after December 15, 2014, with an option for the early adoption. The adoption of the standard will not have a material impact on our financial position or results of operations.
In August 2014, the Financial Accounting Standards Board (‘‘FASB”) issued Accounting Standards Update (“ASU”) 2014- 15, Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern , which defines management’s responsibility to assess an entity’s ability to continue as a going concern, and to provide related footnote disclosures if there is substantial doubt about its ability to continue as a going concern. The pronouncement is effective for annual reporting periods ending after December 15, 2016 with early adoption permitted. The adoption of this guidance is not expected to have a material impact on the Company’s financial statements.
There were various other updates recently issued, most of which represented technical corrections to the accounting literature or application to specific industries and are not expected to a have a material impact on the Company's consolidated financial position, results of operations or cash flows.
| F-20 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 3 - OTHER RECEIVABLES
December 31, 2014 | December 31, 2013 | |||||||
| Taxes reclaimable | $ | 39,570 | $ | 10,940 | ||||
| Advances given | 40,840 | 178,075 | ||||||
| Other | 638 | 28,404 | ||||||
| Total | $ | 81,048 | $ | 217,419 | ||||
Given advances contain advances to lawyers on the legal case of Genetic Immunity on its legal case in progress. The Company accounted for impairment on advances given in the amount of $61,392 and $92,721 during 2014 and 2013, respectively.
NOTE 4 - RESEARCH AND DEVELOPMENT (“R&D”)
In August, 2008, the Company entered into an agreement with a Hungarian individual to establish FireLESS Kft (FireLESS). FireLESS’s business is focused on acquiring the appropriate licenses and certificates to internationally market FireSAFE and will work with local and international fire agencies to test and establish usability baselines for FireSAFE. The Company is a minority shareholder in FireLESS with 30% voting rights, which operates under independent management.
In August, 2007, the Company entered into an agreement with two Hungarian individuals to establish In4 Kft (“in4). in4’s business is focused on software development and information technology purposes. Originally the Company was a minority shareholder in in4 with 30% voting rights. In4 Ltd. operates under independent management.
In August, 2007, the Company also entered into a loan commitment agreement with in4. According to the agreement the Company has committed a loan of approximately $271,000 to in4. The loan amount has been provided by May 31, 2009. In November of 2008 the Company transferred to in4 the entire loan amount, upon which the Company elected to covert the loan into an additional 10% equity in in4. As of January 15, 2009 the Company maintains a minority 40% equity in in4 Ltd. Since capitalization of the loan to equity, as of December 31, 2009 the Company has provided an additional $310,000 loan to in4. During 2010 a new investor entered in4 Ltd, and the share of the Company decreased to 38.5%.
Since the repayment of loans, advances and other investment is contingent on the results of the R&D of iGlue having future economic benefit, management has expensed the Company's investment in in4 and in FireLESS of approximately $5,000 and $900, respectively, and loans to in4 of approximately $310,000 as R&D in the accompanying condensed consolidated statements of operations, in accordance with ASC 730-10-25 "Research and Development Arrangements".
| F-21 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 5 - INTANGIBLES AND FIXED ASSETS
Intangibles and Net property and equipment consisted of the followings at December 31, 2014 and 2013:
| Intangibles | Machinery and other equipments | Vehicles | Total Net property and equipment | |||
| Gross book value | ||||||
| at December 31, 2012 | 30,115,115 | 599,765 | 319,833 | 919,598 | ||
| Corrections | 480,491 | 376,769 | 376,769 | |||
| Additions | 11,329 | 1,471 | - | 1,471 | ||
| Disposals | - | (324,837) | - | (324,837) | ||
| Change in FX rates | 5,251 | 24,266 | 7,948 | 32,214 | ||
| at December 31, 2013 | 30,612,186 | 677,434 | 327,781 | 1,005,215 | ||
| Additions | - | - | - | - | ||
| Impairments | (29,570,912) | - | - | - | ||
| Change in FX rates | (154,878) | (133,832) | (55,378) | (189,210) | ||
| at December 31, 2014 | 886,396 | 543,602 | 272,403 | 816,005 | ||
| Accumulated depreciation and amortization | ||||||
| at December 31, 2012 | 22,274 | 172,722 | 243,707 | 416,429 | ||
| Corrections | 480,491 | 376,770 | 376,770 | |||
| Additions | 50,914 | 79,744 | 18,489 | 98,233 | ||
| Disposals | - | (95,121) | - | (95,121) | ||
| Change in FX rates | 3,126 | 13,654 | 6,056 | 19,710 | ||
| at December 31, 2013 | 556,805 | 547,769 | 268,252 | 816,021 | ||
| Additions | 49,004 | 45,806 | - | 45,806 | ||
| Disposals | - | - | - | - | ||
| Change in FX rates | (34,740) | (92,544) | (45,321) | (137,865) | ||
| at December 31, 2014 | 571,069 | 501,031 | 222,931 | 723,962 | ||
| Net book values at | ||||||
| December 31, 2012 | 30,092,841 | 427,043 | 76,126 | 503,169 | ||
| December 31, 2013 | 30,055,381 | 129,665 | 59,529 | 189,194 | ||
| December 31, 2014 | 315,327 | 42,571 | 49,472 | 92,043 |
| F-22 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 5 - INTANGIBLES AND FIXED ASSETS
The net book value of fixed assets under capital lease amount to $49,472 and $59,529 at December 31, 2014 and at December 31, 2013, respectively.
While the company continues to believe that the value of the patents associated with DermaVir will be recoverable through the eventual marketing and sale of DermaVir, it was determined that due to the continued limited success in raise the necessary amount of capital to bring DermaVir or any other product to market, the Company’s previous cash flow projections, as well as the discount rate used in the estimate of fair value, needed to be revised to reflect the uncertainty as to whether the Company will ultimately realize any cash flows from these patents. As a result, the Company recognized an impairment charge of $29,570,912 in 2014 related to our acquired patent intangibles.
NOTE 6 - NOTE PAYABLE
Mary Passalaqua
On October 20, 2011 the Company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $125,000 owned. As second step in this conversion process Mary Passalaqua requested the conversion of $20,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 3,500,000 shares of common. Upon conversion of the $20,000 our outstanding Promissory Note balance will be $105,000 plus accrued interest.
On August 5, 2011 the company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $195,000 owned. As first step in this conversion process Mary Passalaqua requested the conversion of $70,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 2,400,000 shares of common. Upon conversion of the $70,000 outstanding Promissory Note balance will be $125,000 plus accrued interest.
On April 10, 2007, in connection with reverse merger (See Note 1), the Company assumed a note payable of $250,000 to a former stockholder, Mary Passalaqua originally with one year maturity at April 5, 2008. The note has been expanded by one year to April 5, 2009 with the same conditions. As such note payable was issued immediately prior to the reverse merger, such issuance was recorded as additional compensation by the Company prior to the reverse merger. Accordingly, such compensation is reflected in the accompanying consolidated balance sheet as the accumulated deficit of the Company, and will not be reflected in the Statement of operations, as such compensation expense was structured as an expense prior to the recapitalization.
In November, 2008 the Company settled $35,000 from the outstanding liability, and in May of 2009 the Company settled another $20,000 from the liability. The note payable bears interest at the prime rate (3.25% at December 31, 2014). Interest expense in connection with such note amounted to $55,829 and $52,417 for the year ended at December 31, 2014 and at December 31, 2013, respectively, and was accrued and included in accounts payable and accrued liabilities in the accompanying consolidated balance sheet.
| F-23 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 6 - NOTE PAYABLE (Continued)
Infinite Funding
On July 19, 2011 the Company entered into a Promissory Note with Infinite Funding, Inc whereby Infinite Funding provided $49,000 dollars in loans to the Company. The Note bears interest at 12% per annum.
On March 16, 2011 the Company entered into a Convertible Promissory Note with Infinite Funding LLC whereby Infinite Funding provided $76,000 dollars in loans to the Company. The note bears interest at 10%, is due December 5, 2011, and is convertible into shares of common stock at a conversion price of $0.05. On December 4, 2011 the Company received an extension to this note to December 31, 2012. On September 30, 2012, the loan was converted into 1,900,000 common stocks at a conversion price of $0.04 per share.
Other loans
On December 31, 2014 the Company entered into a Promissory Note with a private person provided $22,964, in loans to the Company. The note bears interest at 10%, is due in one year.
On July 28, 2014, on August 4, 2014, and on 14 August, 2014 the Company entered into three Promissory Notes with a private person provided $24,000, $55,000 and $75,000 dollars, respectively, in loans to the Company. The note bears interest at 10%, is due in one year.
| F-24 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 7 - CAPITAL LEASES PAYABLE
In August, 2007, the Company entered into capital lease agreements on 3 vehicles for management purposes. The maturity of the lease is 60 months and is denominated in CHF. Installments and interest is due on a monthly basis. In December, 2007, the Company entered into additional capital lease agreements on 2 vehicles for management purposes. The maturity of the lease is 72 months and is denominated in CHF. Installments and interest is due on a monthly basis.
In December 2008, the Company settled the capital lease agreements on 2 vehicles and replaced them with new agreements for additional 2 vehicles, totaling 5 capital lease agreements at December 31, 2008. The maturity of the new leases varies from 60 to 72 months and are denominated in EUR and CHF. Installments and interests are due on a monthly basis.
In September 2009, the Company closed a capital lease agreement for one vehicle. The related asset has been sold.
NOTE 8 - STOCKHOLDERS’ EQUITY
On October 2, 2012 the Company acquired Genetic Immunity, Inc., a US corporation domiciled in the state of Delaware. Genetic Immunity is an immunotherapeutic vaccine company whose lead product candidate DermaVir has passed Phase II clinical trials.
Through the acquisitions Genetic Immunity became 100% wholly owned subsidiary. The acquisition was completed for 1,000,000 shares of Series B Preferred stock, convertible into 40 million shares of common stock beginning on January 1, 2014.
On October 26, 2011 the Company entered into an equity exchange agreement with each of Messrs. Viktor Rozsnyay and Daniel Kun, Jr. Mr. Rozsnyay is President, CEO and Chairman of the Board of Directors of the Company and Mr. Kun is its Vice-President. Each Agreement is identical and provides in summary form as follows: each of Messrs. Rozsnyay and Kun will deliver twelve million (12,000,000) shares of their shares of common stock of the Company to the Company for cancellation immediately. The Agreement provides that the Exchange Shares so delivered will be returned to the treasury of the Company as unauthorized shares of common stock and will become available for subsequent issuance by the Company from time to time. In return for the surrender of the Exchange Shares, the Company will issue and deliver two million (2,000,000) restricted shares of a new class of Series A Preferred shares, 1,000,000 shares each to Mr. Rozsnyay and Mr. Kun. The Company attached as an Exhibit the Series A preferred agreements.
| F-25 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
Private placements
On November 28, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 170,000 shares of its Series E Preferred Stock to five unaffiliated private investors for aggregate proceeds of $165,600.
In September, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 300,000 shares of its Series E Preferred Stock to seven unaffiliated private investors for aggregate proceeds of $300,000.
On May 28, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 200,000 shares of its Series E Preferred Stock to two unaffiliated private investors for aggregate proceeds of $100,000.
On April 28, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 100,000 shares of its Series C Preferred Stock to one unaffiliated private investor for aggregate proceeds of $27,949.
On April 7, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 50,000 shares of its Series E Preferred Stock to one unaffiliated private investor for aggregate proceeds of $50,000.
On February 19, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 200,000 shares of its Series E Preferred Stock to two unaffiliated private investors for aggregate proceeds of $200,000. The holders of Series E Preferred Stock are entitled to receive dividends equal to the amount that would be received if each one share of Series E Preferred stock was fully converted into twelve and one half shares of the common stock. Each one share of the Series E Preferred has voting rights equal to five votes of common Stock. Each share of Series E Preferred stock is convertible into 10 shares of common stock.
On November 1, 2013, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,000,000 shares of its Series D preferred stock to one affiliated private investor, Peter Boros, Director of the company, for aggregate proceeds of $337,653. This amount was previously provided to the company by Mr. Boros as a loan, and on November 1 Mr. Boros elected to convert this loan into aforementioned preferred stock.
On October 17, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 250,000 shares of its Series C Preferred Stock to one unaffiliated private investors for aggregate proceeds of $75,000.
On September 26, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 650,000 shares of its Series C Preferred Stock to two unaffiliated private investors for aggregate proceeds of $195,000.
On September 25, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 1,000,000 shares of its common stock to one unaffiliated private investors for aggregate proceeds of $80,000.
Series C Preferred Stocks can be converted with a completed form of conversion notice into 20 shares of common stock. The holders of Series C Preferred shall be entitled to receive dividends on a fully converted basis. Each one share of Series C Preferred shall be entitled to the dividend granted to each one share of common stock multiplied by twenty. Dividends on Series C Preferred must be delivered and paid to the holders not later than five business days after each specified payment date of the dividend.
| F-26 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
On May 9, 2013, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 100,000 shares of its common stock at $0.035 per share to an unaffiliated private investor for aggregate proceeds of $3,500.
On April 3, 2013, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,400,000 shares of its common stock at $0.035 per share to an unaffiliated private investor for aggregate proceeds of $49,000.
On June 13, 2012, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 42,200 shares of its common stock at $0.3 per share to an unaffiliated private investor for aggregate proceeds of $12,658.
During the first half of 2012, the Company received stock advance from an unaffiliated private investor for aggregate proceeds of $67,153.
On November 14, 2011, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 27,700 shares of its common stock at $0.16 per share to an unaffiliated private investor for aggregate proceeds of $4,432.
On September 2, 2011, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,727,013 shares of its common stock at $0.16 per share to two unaffiliated private investors for aggregate proceeds of $276,322.
On August 29, 2011, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 17,825 shares of its common stock at $0.12 per share to one unaffiliated private investor for aggregate proceeds of $2,137.
During the second quarter of 2011, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 196,489 shares of its common stock at $0.29 for aggregate proceeds of $56,983.
During the third quarter of 2010, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 70,000 shares of its common stock at $0.50 for aggregate proceeds of $35,000.
During the first half of 2010, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,470,000 shares of its common stock at $0.10 for aggregate proceeds of $147,000, and sold 62,500 shares of its common stock at $0.08 for proceeds of $5,000.
In February 2010, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 65,000 shares of its common stock at $0.12 for aggregate proceeds of $7,701.
In November 2009, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 175,000 shares of its common stock at $0.16 and 111,110 shares of its common stock at $0.20 per share for aggregate proceeds of $50,222.
In October 2008, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 2,500,000 shares of its common stock at $0.4 per share for aggregate proceeds of $1,000,000.
In February of 2008, shares of common stock of the Company have been approved by FINRA for quotation and trading on the Over The Counter Bulletin Board (OTCBB) under the ticker symbol PWRV. Trading commenced in the Company’s securities on the OTCBB beginning on February 21, 2008.
| F-27 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
In January 2008, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 32,500 shares of its common stock at $3.25 per share for aggregate proceeds of $105,625.
In October 2007, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 104,000 shares of its common stock at $2.50 per share for aggregate proceeds of $260,000. The Company also entered into a Registration Rights Agreement, pursuant to which it agreed that as soon as practicable from the Offering Termination Date, as defined in the Registration Rights Agreement, it would file a registration statement with the SEC covering the resale of the shares of the Company’s common stock that are issuable pursuant to this private placement. There are no stipulated damages outlined in the Registration Rights Agreement for failure to file within the agreed upon time frame. Under such agreement, the holder is entitled to exercise all rights granted by law, including recovery of damages under this agreement.
The Company filed Form SB-2 a registration statement with SEC on November 14, 2007, which was approved on January 30, 2008.
Consulting agreements
During November 2012, the Company entered into agreements with four unaffiliated professionals for one year business consulting services. According to the agreement the professionals provide consulting and rental services to the Company for 12 months. In connection with these services, the Company issued to them 1,930,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of contracting date which is the market price on the commitment date $0.06 and $0.19 per share in the total amount of $180,170 and the related expense was recorded under general administration.
On April 11, 2012 the Company entered into a Consultation Services agreement with Stockvest for IR services and issued a total of 135,000 shares. As of May 17, 2012 this agreement has been terminated.
During October 2011, the Company entered into agreements with three unaffiliated professionals for one year business consulting services. According to the agreement the professionals provide consulting services to the Company for 12 months. In connection with these services, the Company issued to them 640,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of contracting date which is the market price on the commitment date $0.11 and $0.09 per share in the total amount of $61,200 and the related expense was recorded under general administration.
On September 27, 2011, the Company entered into agreements with two unaffiliated professionals for one year business consulting services. According to the agreement the professionals provide consulting services to the Company for 12 months. In connection with these services, the Company issued to them 100,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of contracting date which is the market price on the commitment date $0.14 per share in the total amount of $14,000 and the related expense was recorded under general administration.
During April and May of 2011, the Company entered into agreements with five unaffiliated professionals for one year business consulting services. According to the agreement the professionals provide consulting services to the Company for 12 months. In connection with these services, the Company issued to them 505,000 shares of the Company’s common stock.
These share issuances were recorded at the fair value of contracting date which is the market price on the commitment date, ranging from $0.34 to $0.48 per share in the total amount of $190,900 and the related expense was recorded under general administration.
| F-28 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
On February 7, 2011 the Company entered into a one year business consultation agreement with an unaffiliated person who is to provide general business development and incubation consultation. In exchange for his services the Company issued a onetime payment of 215,000 shares of our common stock, which vested immediately. These share issuances were recorded at $0.3 per share in the total amount of $64,500 and the related expense was recorded under general administration.
On February 9, 2011 the Company entered into a one year business consultation agreement with an unaffiliated person who is to provide services in term of potential business opportunities in Poland. In exchange for his services we issued a onetime payment of 435,000 shares of our common stock, which vested immediately. These share issuances were recorded at $0.24 per share in the total amount of $104,400 and the related expense was recorded under general administration.
On February 24, 2011 the Company entered into a one year business consultation agreement with an unaffiliated person who is to provide general business development and incubation consultation. In exchange for his services we issued a onetime payment of 75,000 shares of our common stock, which vested immediately. These share issuances were recorded at $0.3 per share in the total amount of $22,500 and the related expense was recorded under general administration.
The consulting agreements are entered for a 12 months period. According to the agreements the consultants will provide general business consulting services. As consideration for such services, the Company issued an aggregate of 1,970,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of commitment date in the total amount of $457,888 in accordance with measurement date principles prescribed under ASC 505-50 and ASC 718-10. The Company is amortizing the fair value of the shares over the term of the agreement to stock-based compensation expense, which amounted to $316,098 for the period ended December 31, 2014, respectively and $316,098 for the period from April 26, 2006 (date of inception) to December 31, 2014, in accordance with ASC 505-50 and ASC 718-10.
During the October and November 2010, the Company entered into agreements with several unaffiliated professionals for consulting services. According to the agreement the professionals provide consulting services to the Company in 2010. In connection with these services, the Company issued to them 874,000 shares of the Company’s common stock. These share issuances were recorded at $0.5 per share in the total amount of $437,000 and the related expense was recorded under general administration.
In September 2010, the Company entered into an agreement with four unaffiliated professionals for consulting services. According to the agreement the professionals provided consulting services to the Company in 2010. In connection with these services, the Company issued to them 153,000 shares of the Company’s common stock. These share issuances were recorded at $0.5 per share in the total amount of $76,500 and the related expense was recorded under general administration.
The consulting agreements are entered for a 12 months period. According to the agreements the consultants will provide general business consulting services. As consideration for such services, the Company issued an aggregate of 1,027,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of commitment date ($0.5 per share) in the total amount of $513,500 in accordance with measurement date principles prescribed under ASC 505-50 and ASC 718-10. The Company is amortizing the fair value of the shares over the term of the agreement to stock-based compensation expense, which amounted to $312,972 and $200,528 for the periods ended December 31, 2014 and 2013, respectively and $513,500 for the period from April 26, 2006 (date of inception) to December 31, 2014, in accordance with ASC 505-50 and ASC 718-10.
In June 2010, the Company entered into an agreement with two unaffiliated professionals for consulting services. According to the agreement the professionals provided consulting services to the Company in 2010. In connection with these services, the Company issued to them 20,000 shares of the Company’s common stock. These share issuances were recorded at $0.1 per share in the total amount of $2,000 and the related expense was recorded under general administration.
| F-29 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
In April 2009, the Company entered into an agreement with two unaffiliated professionals for consulting services. According to the agreement the professionals provided consulting services to the Company in 2009. In connection with these services, the Company issued to them 250,000 shares of the Company’s common stock. These share issuances were recorded at $0.4 per share in the total amount of $100,000 and the related expense was recorded under general administration.
In June 2007, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 2,250,000 shares of its common stocks at $0.34 per share for a total subscription receivable of $765,000. The Company also entered into a Registration Rights Agreement, pursuant to which it agreed that as soon as practicable from the Offering Termination Date, as defined in the Registration Rights Agreement, it would file a registration statement with the SEC covering the resale of the shares of the Company’s common stock that are issuable pursuant to this private placement. There are no stipulated damages outlined in the Registration Rights Agreement for failure to file within the agreed upon time frame. Under such agreement, the holder is entitled to exercise all rights granted by law, including recovery of damages under this agreement. In June 2007, the Company entered into five consulting agreements with five consultants for 12 to 24 month periods. According to the agreements the consultants will provide general business consulting services. As consideration for such services, the Company issued an aggregate of 1,375,000 shares of the Company’s common stock. These share issuances were recorded at $0.34 per share in the total amount of $467,501 in accordance with measurement date principles prescribed under ASC 505-50 and ASC 718-10. The Company is amortizing the fair value of the shares over the term of the agreement to stock-based compensation expense, which amounted to $0 for the period ended December 31, 2014 and 2013, respectively and $467,501 for the period from April 26, 2006 (date of inception) to December 31, 2014, in accordance with ASC 505-50 and ASC 718-10.
In April 2007, the Company entered into an agreement with two professionals for legal services. According to the agreement the professionals provided legal services to the Company in 2007. In connection with these services, the Company issued to them 500,000 shares of the Company’s common stock. These share issuances were recorded at $0.34 per share in the total amount of $170,000 and the related expense was recorded under general administration.
In connection with the ITA in 2007 (See Note 4), the Company issued 100,000 shares of the Company’s common stock to the Inventors. These shares issuance were recorded at fair value of $0.34 per share in the total amount of $34,000. The cost of the related invention was recorded as research and development expense.
Other agreements
On October 8, 2008, Power of the Dream Ventures, Inc. (the “Company”) entered into a Standby Equity Distribution Agreement (the “Standby Equity Distribution Agreement”) with YA Global Investments, L.P. (the “Investor”). Pursuant to the terms of the Standby Equity Distribution Agreement, the Company (a) agreed to issue and sell to the Investor up to $5,000,000 of shares of the Company’s common stock, par value $0.0001 per share (the “Common Stock”) in tranches of equity, based upon a 7% discount to the market price of the Common Stock, calculated over the five trading days following notice by the Company of an election to sell shares; and (b) issued to the Investor a warrant (the “Warrant”) to purchase 4,027,386 shares of Common Stock at the exercise price per share of $0.29. The Warrant is not part of the commitment shares issued by the company to the investor.
The investor must purchase the shares underlying the Warrant. The Warrant price was determined based upon the highest Bid price on the day of the closing of the agreement. The YA Global is required to exercise the warrant upon notice by PDV of an election to have the warrants exercised at a fix price of $0.29 per common stock.
| F-30 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
In connection with the Standby Equity Distribution Agreement, the Company entered into a Registration Rights Agreement with the Investor (the “Registration Rights Agreement”) pursuant to which the Company agreed to register for resale the shares of Common Stock that may be purchased by the Investor pursuant to the Standby Equity Distribution Agreement, the shares of Common Stock issuable upon exercise of the Warrant and 2,000,000 shares of Common Stock (the “Commitment Shares”) issued to the Investor as a commitment fee pursuant to the terms of the Standby Equity Distribution Agreement. The 2,000,000 shares issued as a commitment fee were valued at $0.4 per share or $800,000 based on the fair value at issuance date. The $800,000 commitment fee has been debited against additional paid in capital in accordance with the provisions of Staff Accounting Bulletin Topic 5A .
On September 24, 2010 the Company notified YA Global Investments in writing of its intention to terminate the Standby Equity Distribution Agreement (SEDA).
In response to this letter YA Global informed the company in a letter dated October 5, 2010 that the SEDA agreement will terminate on or about October 15, 2010. YA Global also informed the company that even though the SEDA agreement is terminated its associated Warrant to purchase 4,027,386 shares of common stock remains in effect until it expires on October 8, 2013.
On October 18, 2010 the company received notice from YA global for the exercise of 50,000 warrant shares on a cashless basis. This cashless exercise resulted in the issuance of 3,226 shares of Common stock to YA Global. Following this exercise YA Global has 3,977,386 warrant shares available for purchase.
On September 10, 2012 YA Global notified the company that according to the terms of the Warrant Agreement there should have been a proportional reduction in the Warrant’s exercise price to the price of a private placement that was completed at a lower price than the $0.29 exercise price. YA Global and the company have agreed to modify the Warrant exercise price from the original $0.29 cents to $0.10 cents.
At the same time YA Global also notified the company of their intention to sell the Warrant in a private transaction to an unaffiliated accredited investor. This transaction was contemplated on September 10, 2012. The new owner, Bluestar Consulting, now has the right to acquire, on or before October 8, 2013, 3,977,386 share of common stock at a per share exercise price of $0.10. The terms of the warrants are such that the exercise price was lowered due to a subsequent round of financing, from $0.29 per share to the price the subsequent investors paid, $0.10 per share. This down round protection feature causes the warrant to fail the criteria in ASC 815-40-15-7 (EITF 07-5) which would permit them to be classified as equity. Consequently, the warrants are recorded at fair value and classified as liabilities and marked to fair value each period. The warrant is extended to October 8, 2018.
| F-31 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
The fair value of the liability using Black-Scholes valuation at December 31, 2014 is $63,638, which is classified at short term liabilities. The fair value of the liability is established as a Level 3 fair value measurement. The following table shows the movement in fair value along with the change of warrant conditions:
| December 31, 2014 | Book value | Fair value |
| Financial liabilities | ||
| Liabilities measured at fair value 63,638 | 63,638 | |
| December 31, 2013 | Book value | Fair value |
| Financial liabilities | ||
| Liabilities measured at fair value 87,502 | 87,502 | |
| Fair value of warrant per share | December 31, 2014 | December 31, 2013 |
| Per share | $0.016 | $0.022 |
The inputs used in fair value estimation are:
current share price at December 31, 2014: $0.0621
exercise price $0.1
expected time to expiry (years): 3.75
risk free rate 3.25 (FED prime rate)
expected dividend yield: 0
volatility: 50%
The change in the amount of warrant liability is accounted as other expense.
Reconciliation of warrant valuation:
Balance at December 31, 2013: $87,402
Decrease in fair value of share (23,764)
Balance at December 31, 2014: $63,638
On May 17, 2008 the Company entered into an agreement with Wakabayashi Fund LLC in order to arrange financing for working capital as an intermediary. Wakabayashi Fund LLC provided capital funding services including serving as an investment banking liaison and acted as capital consultant for a six month period. The Company issued 111,111 shares of restricted common stock upfront at $1.35 per share, the market price of the stock on the commitment date of the agreement. Additionally, the Company agreed to pay for the capital funding services 7% success fee. These share issuances were recorded at $1.35 per share in the total amount of $150,000 in accordance with measurement date principles prescribed under ASC 505-50. The Company is amortizing the fair value of the shares in general and administration expenses over the term of the agreement to stock-based compensation expense, which amounted to $0 for the period ended December 31, 2014 and $150,000 for the period from April 26, 2006 (date of inception) to December 31, 2014, in accordance with ASC 505-50. As of November 17, 2008 this agreement has been terminated without any funds raised.
| F-32 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
On April 18, 2008 the Company entered an agreement with RedChip Companies Inc. and Partner Media4Equity Inc. for an investor relationship program for a period of 12 months. The Company secured and delivered 306,570 restricted common shares with a market price of $0.70 for a 12 months period in connection with RedChip investor relationship services. The compensation for Media4Equity services was the delivery of 1,500,000 restricted common shares. These share issuances were recorded at $0.75 per share, the market price of the stock on the commitment date of the agreement, for a total amount of $1,125,000 in accordance with measurement date principles prescribed under ASC 505-50. The Company is amortizing the fair value of the shares in general and administration expenses over the term of the agreement to stock-based compensation expense, which amounted to $0 for the periods ended December 31, 2014 and December 31, 2013, respectively and $1,339,599 for the period from April 26, 2006 (date of inception) to December 31, 2014, in accordance with ASC 505-50.
Stock based compensations
On January 1, 2011, the Company entered into a restricted stock agreement with Ildiko Rozsa, who is to serve as the Chief Financial Officer of the Company. As part of the agreement Ms. Rozsa was granted 100,000 shares of the Company’s restricted common stock of which 25,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Rozsa is employed by the Company.
On January 1, 2011, the Company entered into a restricted stock agreement with Szilvia Toth, the Chief Accounting Officer of the Company. As part of the agreement Ms. Toth was granted 100,000 shares of restricted common stock, of which 25,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Toth is employed by the Company.
On January 1, 2011, the Company entered into a restricted stock agreement with Imre Eotvos, the Technology Assistant of the Company. As part of the agreement Mr. Eotvos was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Eotvos is employed by the Company.
On January 1, 2011, the Company entered into a restricted stock agreement with Daniel Kun, Jr., who is to serve as Secretary and Vice President of the company on a going forward basis. As part of the agreement Mr. Kun was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Kun is employed by the Company.
On January 1, 2011, the Company entered into a restricted stock agreement with Gene Guhne, who is to serve as Director of the company on a going forward basis. As part of the agreement Mr. Guhne was granted 100,000 shares of restricted common stock, which will vest on equal installments of 25,000 shares quarterly, at the end of each quarter, so long as Mr. Guhne is employed by the Company.
As consideration for the above services, the Company issued an aggregate of 540,000 shares of the Company’s common stock. These share issuances were recorded at $0.15 per share in the total amount of $81,000 in accordance with measurement date principles prescribed under FAS 123 (R).
During the second quarter of 2010, the Company entered into a restricted stock agreement with Ildiko Rozsa, who is to serve as the Chief Financial Officer of the Company. As part of the agreement Ms. Rozsa was granted 100,000 shares of the Company’s restricted common stock of which 25,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Rozsa is employed by the Company.
During the second quarter of 2010, the Company entered into a restricted stock agreement with Szilvia Toth, the Chief Accounting Officer of the Company. As part of the agreement Ms. Toth was granted 100,000 shares of restricted common stock, of which 25,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Toth is employed by the Company.
| F-33 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
During the second quarter of 2010, the Company entered into a restricted stock agreement with Imre Eotvos, the Technology Assistant of the Company. As part of the agreement Mr. Eotvos was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Eotvos is employed by the Company.
During the second quarter of 2010, the Company entered into a restricted stock agreement with Daniel Kun, Jr., who is to serve as Secretary and Vice President of the company on a going forward basis. As part of the agreement Mr. Kun was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Kun is employed by the Company.
During the second quarter of 2010, the Company entered into a restricted stock agreement with Gene Guhne, who is to serve as Director of the company on a going forward basis. As part of the agreement Mr. Guhne was granted 100,000 shares of restricted common stock, which will vest on equal installments of 25,000 shares quarterly, at the end of each quarter, so long as Mr. Guhne is employed by the Company.
As consideration for the above services, the Company issued an aggregate of 540,000 shares of the Company’s common stock. These share issuances were recorded at $0.1 per share in the total amount of $54,000 in accordance with measurement date principles prescribed under FAS 123 (R).
On February 5, 2009, the Company entered into a restricted stock agreement with Ildiko Rozsa, who is to serve as the Chief Financial Officer of the Company. As part of the agreement Ms. Rozsa was granted 120,000 shares of the Company’s restricted common stock of which 30,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Rozsa is employed by the Company.
On February 5, 2009, the Company entered into a restricted stock agreement with Szilvia Toth, the Chief Accounting Officer of the Company. As part of the agreement Ms. Toth was granted 120,000 shares of restricted common stock, of which 30,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Toth is employed by the Company.
On February 5, 2009, the Company entered into a restricted stock agreement with Mihaly Zala, the Chief Technology Officer of the Company. As part of the agreement Mr. Zala was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Zala is employed by the Company. As of July 15, 2009 Mr. Zala is no longer employed by the Company, therefore 60,000 shares of the 120,000 granted to him were cancelled and returned to the authorized and unissued stock of the company.
On February 5, 2009, the Company entered into a restricted stock agreement with Imre Eotvos, the Technology Assistant of the Company. As part of the agreement Mr. Eotvos was granted 200,000 shares of restricted common stock, which will vest on equal installments of 50,000 shares quarterly, at the end of each quarter, so long as Mr. Eotvos is employed by the Company.
On February 5, 2009, the Company entered into a restricted stock agreement with Daniel Kun, Jr., who is to serve as Secretary and Vice President of the company on a going forward basis. As part of the agreement Mr. Kun was granted 200,000 shares of restricted common stock, which will vest on equal installments of 50,000 shares quarterly, at the end of each quarter, so long as Mr. Kun is employed by the Company.
As consideration for the above services, the Company issued an aggregate of 700,000 shares of the Company’s common stock. These share issuances were recorded at $0.4 per share in the total amount of $280,000 in accordance with measurement date principles prescribed under FAS 123 (R).
| F-34 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
On October 24, 2007, the Company entered into a restricted stock agreement with Ildiko Rozsa, who is to serve as the Chief Financial Officer of the Company. As part of the agreement Ms. Rozsa was granted 250,000 shares of the Company’s restricted common stock of which 100,000 shares are vested upon grant and 30,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Rozsa is employed by the Company.
On October 1, 2007, the Company entered into a restricted stock agreement with Szilvia Toth, the Chief Accounting Officer of the Company. As part of the agreement Ms. Toth was granted 100,000 shares of restricted common stock of which 50,000 shares are vested upon grant and 10,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Toth is employed by the Company.
On October 24, 2007, the Company entered into a restricted stock agreement with Mihaly Zala, the Chief Technology Officer of the Company. As part of the agreement Mr. Zala was granted 150,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Zala is employed by the Company.
On October 24, 2007, the Company entered into a restricted stock agreement with Imre Eotvos, the Technology Assistant of the Company. As part of the agreement Mr. Eotvos was granted 25,000 shares of restricted common stock, which will vest on equal installments of 5,000 shares quarterly, at the end of each quarter, so long as Mr. Eotvos is employed by the Company.
On October 1, 2007, the Company entered into a restricted stock agreement with Sandorne Juhasz, who provides payroll accounting services to the Company on a subcontracting basis. As part of the agreement Ms. Juhasz was granted 11,000 shares of restricted common stock, of which 8,000 is will vest upon grant and 750 shares will vest quarterly, at the end of each quarter, so long as Ms. Juhasz is employed by the Company.
On October 24, 2007, the Company entered into a restricted stock agreement with Daniel Kun, Jr., who is to serve as Secretary and Vice President of the company on a going forward basis. As part of the agreement Mr. Kun was granted 250,000 shares of restricted common stock, which will vest on equal installments of 50,000 shares quarterly, at the end of each quarter, so long as Mr. Kun is employed by the Company.
On October 24, 2007, the Company entered into a restricted stock agreement with Viktor Rozsnyay, who is serving as President and Chief Executive Officer of the Company. As part of the agreement Mr. Rozsnyay was granted 250,000 shares of restricted common stock, which will vest on equal installments of 50,000 shares quarterly, at the end of each quarter, so long as Mr. Rozsnyay is employed by the Company.
As consideration for the above services for the employment of the above 7 persons, the Company issued an aggregate of 1,036,000 shares of the Company’s common stock. These share issuances were recorded at $2.5 per share in the total amount of $2,590,000 in accordance with measurement date principles prescribed under ASC 505-50 and ASC 718-10. The Company is amortizing the fair value of the shares over the term of the agreement to stock-based compensation expense, which amounted to $0 for the period ended December 31, 2014 and $2,590,000 for the period from April 26, 2006 (date of inception) to December 31, 2014, in accordance with ASC 505-50 and ASC 718-10.
| F-35 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
Reverse merger
On August 5, 2011 the company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $195,000 owned. As first step in this conversion process Mary Passalaqua requested the conversion of $70,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 2,400,000 shares of common. On October 20, 2011, Mary Passalaqua requested the conversion of additional $20,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 3,500,000 shares of common.
On October 20, 2011 the Company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $125,000 owned. As second step in this conversion process Mary Passalaqua requested the conversion of $20,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 3,500,000 shares of common. Upon conversion of the $20,000 our outstanding Promissory Note balance will be $105,000 plus accrued interest.
On April 10, 2007, PDV entered into a reverse merger transaction with Vidatech. In connection with the merger 2,500,000 shares of PDV common stock remained outstanding and PDV issued 33,300,000 shares of its common stock for all the outstanding common stock of Vidatech. As a result of this transaction, the former stockholders of Vidatech became the controlling stockholders of PDV. Accordingly, the reverse merger has been accounted for as a recapitalization of Vidatech.
In May, 2006, the Company entered into a short term loan agreement with its Chief Executive Officer, Viktor Rozsnyay, for approximately $96,100 with a maturity of April 30, 2007. On December 28, 2006 Mr. Rozsnyay elected to convert the loan into equity, which is recorded as additional paid in capital.
In March, 2007, the Company entered into a short term loan agreement with its Chief Financial Officer, Daniel Kun Jr., for approximately $53,735 with a maturity of March 31, 2007. On March 31, 2007 Mr. Kun elected to convert the loan into equity, which is recorded as additional paid in capital.
At December 31, 2014 there are no non-vested restricted stock awards.
The weighted average grant-date fair value of restricted stock awards granted for the year ended December 31, 2014 and 2013 were $0. The total fair value of shares vested during the year ended December 31, 2014 and 2013 was $0.
| F-36 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 9 - ACCOUNTS PAYABLE, ACCRUED AND OTHER LIABILITIES
December 31, 2014 | December 31, 2013 | |||||||
| Accounts payable and accrued expenses | $ | 1,672,947 | $ | 1,868,534 | ||||
| Interest accruals | 2,792,202 | 2,214,760 | ||||||
| Total | $ | 4,465,149 | $ | 4,083,294 | ||||
In 2007, Genetic Immunity conducted a US$2.0 million convertible debenture bridge in anticipation of a reverse merger which was not consummated due to then current economic and market conditions. The debenture was due August 2009. The US$1.1 million in face value of debentures is in receivership with the Small Business Investment Company (“SBIC”) The remaining US$900,000 of face value of the debenture is held by individuals.
NOTE 10 - GRANTS RECEIVED
During 2007 and 2008, Genetic Immunity gained several government grants from the Hungarian state in order to finance the research and development projects. The grants are payable in arrears to 4 projects of the Company: DERMAVI_, DVCLIN01, FIBERSC2 and WINGS. The Company accrued for the grants received and it is reversed to the income statement along with the occurrence of the related expenses. Grants are payable in accordance with the milestone, not annually. Audited financial statements had to be disclosed to State Office as settlement of each milestone. Theses audits have been performed by the statutory auditor of the Hungarian Subsidiary. WINGS is an EU FP7 international consortium grant financed by the European Union.
DERMAVI_ is successfully closed in 2009. DVCLIN01 supported the Phase II clinical development of DermaVir. This grant was closed at the end of 2013.. FIBERSC2 supports the investigation of DermaVir mechanism of action with novel fiber integrated microscopy. Originally scheduled for completion in February of 2014, this grant has been extended to August of 2014. WINGS supports the development of West Nile Virus vaccine with DermaVir technology. The grant ended in February of 2014, the Company has filed the required closing documents, this grant has been closed, the related liability is released at the official close at year end.
| F-37 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 11 - SHORT TERM LOANS
December 31, 2014 | December 31, 2013 | |||||||
| Bridge Loan | $ | 2,000,000 | $ | 2,000,000 | ||||
| Bridge loan payable provision | — | — | ||||||
| RIGHT loan | 274,120 | 311,159 | ||||||
| Bryan Sanders loan | 30,000 | 30,000 | ||||||
| Total | 2,304,120 | 2,341,159 | ||||||
Bridge Loan
On August 29, 2007, the Company entered into a Bridge Loan agreement as a Senior Secured Convertible Debenture for an amount of $2,000,000. The loan providers are the followings:
| Name | Bridge Amount |
| SBIC Investors | |
| Trident | $1,100,000 |
| Non-SBIC Investors | |
| Andy Browder | 25,000 |
| Proto Investments | 250,000 |
| Ford Sasser | 25,000 |
| Danny Vela | 25,000 |
| Steve Walton | 150,000 |
| Hilton Wilson | 25,000 |
| Will Wilson | 250,000 |
| Rodman & Renshaw | 150,000 |
| $2,000,000 |
| F-38 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 11 - SHORT TERM LOANS (Continued)
The loan bears an annual interest rate of 12%. According to the loan agreement, the Company settles the loan principal on the earlier of August 29, 2009 or consummation of a qualifying transaction other than reverse merger effected by the Company in completion of or in connection with a Public Offering. In case of late payment, the Company is liable to pay 18% late interest fee. The accumulated interest payable, along with the late payment interest is accrued in accrued expenses (see Note 6).
As of the printing of this document, the US$1.1 million in face value of debentures was in receivership with the U.S. SBA. The remaining US$900,000.00 of face value of the debenture is held by individuals.
NOTE 12 - GENERAL AND ADMINISTRATION
General and administration expenses consisted of the followings at December 31, 2014 and at December 31, 2013:
|
December 31, 2014 |
December 31, 2013 | ||
| Stock based compensation | - | - | |
| Stock based Consultant services | - | 201,899 | |
| Other | 1,255,316 | 537,076 | |
| Total | 1,255,316 | 738,975 |
| F-39 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2014 AND 2013
NOTE 13 - OTHER EXPENSES
Other expenses include income from the revaluation of liability for warrants for Bluestar Consulting in the amount of $23,864.
NOTE 14 - SUBSEQUENT EVENTS
On March 11, 2015, we issued a total of 1,000,000 shares of our restricted our common stock to one unaffiliated entity, Berlini Kft in liu of payment of Geentic Immunity’s laboratory facilities.
Also on March 11, 2015, we issued a total of 200,000 shares of our restricted our common stock to the Company’s Transfer Agent, Fidelity Transfer Company, for services rendered.
| F-40 |