Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
þ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2014
or
¨ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission File Number 000-55133
ACUCELA INC. |
(Exact name of registrant as specified in its charter) |
Washington | 02-0592619 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) | |
1301 SECOND AVENUE, SUITE 4200 SEATTLE, WASHINGTON | 98101 | |
(Address of principal executive offices) | (Zip Code) | |
(206) 805-8300
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, No Par Value Per Share
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by a check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ¨ | Accelerated filer ¨ | Non-accelerated filer þ | Smaller reporting company ¨ | |||
(Do not check if a smaller reporting company) | ||||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
The aggregate market value of the common stock held by non-affiliates of the registrant outstanding as of June 30, 2014 (the last business day of the registrant's most recently completed second fiscal quarter), based upon the closing price of the common stock on June 30, 2014 as reported on the Mothers Market of the Tokyo Stock Exchange, was approximately $138,370,000. For purposes of this disclosure, shares of common stock held by each officer and director of the registrant and by persons who hold more than 5% of the outstanding shares of common stock (or his or her affiliate) have been treated as shares held by affiliates. This treatment of affiliate status is not necessarily a conclusive determination of affiliate status or other purposes.
As of March 30, 2015, the registrant had outstanding 35,809,467 shares of common stock.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant's information statement relating to the registrant's special meeting of shareholders, to be held on or about May 1, 2015, are incorporated by reference into Part III of this Annual Report on Form 10-K where indicated.
ACUCELA INC.
FORM 10-K
For the fiscal year ended December 31, 2014
INDEX
PART I | ||
ITEM 1. | ||
ITEM 1A. | ||
ITEM 1B. | ||
ITEM 2. | ||
ITEM 3. | ||
ITEM 4. | ||
PART II | ||
ITEM 5. | ||
ITEM 6. | ||
ITEM 7. | ||
ITEM 7A. | ||
ITEM 8. | ||
ITEM 9. | ||
ITEM 9A. | ||
ITEM 9B. | ||
PART III | ||
ITEM 10. | ||
ITEM 11. | ||
ITEM 12. | ||
ITEM 13. | ||
ITEM 14. | ||
PART IV | ||
ITEM 15. | ||
PART I
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (“Report”), including the “Management's Discussion and Analysis of Financial Condition and Results of Operations,” contains forward-looking statements regarding future events and our future results that are based on our current expectations, estimates, forecasts, and projections about our business, our results of operations, the industry in which we operate and the beliefs and assumptions of our management. Words such as “expects,” “anticipates,” “targets,” “goals,” “projects,” “would,” “could,” “intends,” “plans,” “believes,” “seeks” and “estimates,” variations of these words, and similar expressions are intended to identify those forward-looking statements. These forward-looking statements are only predictions and are subject to risks, uncertainties and assumptions that are difficult to predict. Therefore, actual results may differ materially and adversely from those expressed in any forward-looking statements. Factors that might cause or contribute to such differences include, but are not limited to, those discussed in this Report under the section entitled “Risk Factors” in Item 1A of Part I and elsewhere herein, and in other reports we file with the SEC. While forward-looking statements are based on reasonable expectations of our management at the time that they are made, you should not rely on them. We undertake no obligation to revise or update publicly any forward-looking statements for any reason, whether as a result of new information, future events or otherwise, except as may be required by law.
ITEM 1. BUSINESS
Overview
Acucela Inc., a Washington corporation, is a clinical-stage biotechnology company that specializes in discovering and developing novel drug candidates to potentially treat and slow the progression of sight-threatening ophthalmic diseases impacting millions of individuals worldwide. Emixustat hydrochloride ("emixustat"), our lead investigational visual cycle modulation ("VCM") compound, is designed to reduce retinal toxins and preserve the integrity of retinal tissue in patients suffering from geographic atrophy ("GA") associated with dry age-related macular degeneration ("AMD"). We intend to expand our ophthalmology pipeline by focusing on various degenerative retinal diseases, glaucoma, and dry eye.
Emixustat is currently being evaluated in a Phase 2b/3 study for GA associated with dry AMD. Currently, there are no U.S. Food and Drug Administration (FDA) approved therapies to treat any form of dry AMD, including GA associated with dry AMD. We are co-developing emixustat under our co-development and collaboration agreement, or the Emixustat Agreement, with Otsuka Pharmaceutical Co., Ltd., or Otsuka. Pursuant to the Emixustat Agreement, we and Otsuka have agreed to develop and commercialize emixustat and/or other back-up compounds for the treatment of dry AMD and other potential ophthalmic indications that the parties agree to pursue under the terms of the agreement.
We completed our initial public offering ("IPO") in February 2014. We manage our operations and allocate resources as a single reporting segment. Financial information about our segment and geographic areas is incorporated herein by reference to Note 1 of Notes to Financial Statements in this Annual Report. In addition, financial information regarding our operations, assets and liabilities, including our total net revenue and net income (loss) for the years ended December 31, 2014, 2013 and 2012 and our total assets as of December 31, 2014 and 2013, is included in our Financial Statements in this Annual Report.
Age-Related Macular Degeneration ("AMD")
The retina is a thin layer of tissue on the inside back wall of the eye which contains millions of light-sensitive cells and neurosensory cells that receive and organize visual information. The retina sends this information to the brain via the optic nerve, resulting in vision. Retinal diseases can affect the area of the retina that serves the central vision (the macula and fovea at the center of the macula). Retinal degenerative diseases are a worldwide leading cause of blindness. According to visiongain: Macular Degeneration and other Retinal Diseases World Drug Market 2014-2024, the market for retinal pharmaceuticals was $5.1 billion in 2012 and is expected grow to approximately $16.6 billion by 2024. The sub-market for dry AMD is estimated to be approximately $2.5 billion in 2024.
AMD is a retinal disease that can cause patients to experience reduced central vision and lead to significant and irreversible loss of central vision in severe cases. The disease exists in two forms, dry AMD and wet AMD. As individuals with AMD age, the disease may gradually destroy the fine central vision needed to see objects clearly and to perform common daily tasks, such as reading and driving. In particular, the advanced stages of dry AMD, GA and wet AMD, are leading causes of vision loss and blindness among persons age 50 years and older in the United States.
Glaucoma
Glaucoma is a progressive optic neuropathy that leads to irreversible damage to retinal ganglion cells. The damage may lead to diminished visual function and blindness, especially when not adequately treated. Since there is no known cure for glaucoma, the principal goal of therapy is to prevent further progression and preserve visual function once therapy is initiated. In 2012, we conducted a Phase 1/2 clinical trial in patients with glaucoma or ocular hypertension in the United States for OPA-6566, an adenosine A2a receptor agonist, under a development and collaboration agreement, or the Glaucoma Agreement, and our co-development partner, Otsuka, is currently evaluating next steps for the program.
Dry Eye Syndrome
Dry eye syndrome, or keratoconjunctivitis sicca, is a chronic and potentially debilitating condition involving ocular surface damage and symptoms of irritation that result from a decline in the quality or quantity of tears. Dry eye has multiple causes and consequences, ranging in severity from mild discomfort to debilitating vision loss and pain. The first line treatment for dry eye syndrome is typically the use of ophthalmic lubricants (artificial tears) which are available as OTC products. Over the last years therapeutic prescription products like Restasis in the United States and Hyalein in Japan have gained importance and market share.
EMIXUSTAT HYDROCHLORIDE ("emixustat")
Market for Emixustat. According to MarketScope, 2014 Report on the Retinal Pharmaceuticals & Biologics Market, GA represents a vast unmet medical need impacting an estimated nine million people worldwide in 2014, and Marketscope estimates that this number will grow to about 10.3 million by 2019. Currently, there are no FDA-approved therapies to treat any form of dry AMD, including GA associated with dry AMD.
Investigational Product Candidate. Emixustat has been formulated as an orally-administered pill and is designed to directly modulate utilization of vitamin A in the visual cycle through inhibition of a rate-limiting enzyme called retinal pigment epithelium protein 65 (RPE65). In preclinical studies, emixustat has been shown to protect the retina from light mediated damage and reduce levels of vitamin A-based toxins, such as A2E. A2E is a known cytotoxin that has been implicated in the pathogenesis of GA associated with dry AMD. It is theorized that reducing the biosynthesis of vitamin A-based toxins may slow the progression of GA lesions.
Development Status. Our investigational new drug application, or IND, for the GA associated with dry AMD indication was submitted to the U.S. Food and Drug Administration, or FDA, in May 2008. In 2010, the FDA granted fast track designation based on the lack of available therapies and the chronically debilitating nature of the disease. Fast track status is anticipated to facilitate a more rapid new drug application (NDA) review process compared to conventional FDA review processes.
Emixustat has been generally well tolerated in all five completed Phase 1 studies (four single dose studies with doses ranging from 2 to 75 mg, and one multiple dose study with doses ranging from 5 to 40 mg once daily for 14 days) and a Phase 2a multi-center, randomized, double-masked, placebo-controlled pharmacodynamic study with doses ranging from 2 to 10 mg once daily for up to 90 days.
In the first quarter of 2013, a multi-center, randomized, double-masked dose-ranging Phase 2b/3 study was initiated to evaluate the safety and efficacy of emixustat compared to a placebo in patients with GA associated with dry AMD. Enrollment of 508 subjects was completed in the first quarter of 2014. The objectives of the study are to assess treatment effects for differences in:
• | the rate of GA progression between treatment and control groups as measured by fundus autofluorescence imaging; |
• | the development of wet AMD; and |
• | safety and tolerability. |
On May 8, 2014, we announced an update on our emixustat clinical development program. Based on recommendations from the FDA, we are continuing the ongoing Phase 2b/3 study through the original 24-month treatment duration without access to interim results and, depending on the results of the Phase 2b/3 study, conduct at least one additional confirmatory Phase 3 clinical trial in patients with GA associated with dry AMD. The FDA's recommendations were not based on any data reviews related to the emixustat program.
4
Collaboration with Otsuka. Under the terms of the Emixustat Agreement:
• | Otsuka paid us a $5 million initial license fee; |
• | Otsuka funded $40 million of all development costs incurred prior to completion of Phase 2 clinical trials in the United States (or similar trials in other North American countries) and has agreed to share equally with us in the ongoing development costs in excess of $40 million that are incurred pursuant to a development plan covering the development of emixustat for dry AMD and any other potential ophthalmic indications selected for investigation by a joint development committee, or JDC, established under the agreement; |
• | Otsuka has agreed to loan funds to us for the payment of our share of development costs in excess of $40 million; |
• | Otsuka paid us $15 million over a three-year period to fund a cooperative research program between the parties pursuant to an agreed research plan covering development of emixustat for ophthalmic indications other than dry AMD and research and development of emixustat’s back-up compounds; |
• | If our clinical trials of the products and compounds developed under the agreement are successful and other conditions are satisfied, Otsuka has agreed to co-promote those products with us in North America and will share with us in profits and losses. If we do not elect to participate in co-promotion in one or more North American countries, Otsuka has agreed to solely commercialize products developed under the agreement in those countries pursuant to an exclusive license and will pay us royalties of 19% to 23% on net sales, depending on the amount of net sales; |
• | Otsuka will have sole development and commercialization rights to the products and compounds developed under the agreement in selected countries in Asia, the Pacific, the Middle East and North Africa, which we refer to as Otsuka’s sole territory, and will pay us a royalty of 2% on net sales; |
• | We will have sole development and commercialization rights to the products and compounds developed under the agreement in Europe and South America, and other regions or countries outside North America and Otsuka’s sole territory, subject to Otsuka’s right to first negotiate for these rights to the extent we intend to license them to a third-party, and have agreed to pay Otsuka a royalty of 2% on net sales; |
• | If the emixustat JDC does not approve emixustat for development for any additional ophthalmologic indications in our shared territory, then each party will have sole development and commercialization rights in its sole territory; |
• | Otsuka is potentially obligated to pay us up to $77.5 million in milestone payments, excluding the initial license fee and milestone payments made to date, based on the achievement of certain development goals and regulatory submissions and approvals; and |
• | Otsuka is potentially obligated to pay us up to $175 million in milestone payments based upon attainment of agreed upon sales levels of emixustat. |
If we elect to co-promote emixustat under the agreement with respect to one or more North American countries, our share of profits and losses from those sales will range between 35% and 50% for each country depending upon the participation level we choose at the time of our election. Prior to the submission of an NDA for emixustat, we intend to exercise our co-promotion option for emixustat at the participation level of 50%. The payment of royalties based on net sales of emixustat extends, on a country-by-country basis, until the later of 10 years after commercial launch and the expiration of patent rights covering the manufacture, importation, use or sale of emixustat in each particular country. The royalty rate on net sales payable by a party in a country is reduced on a country-by-country basis to the extent that the patent rights in a particular country expire. The royalty rate is reduced by 50% if a generic form of emixustat is marketed in that country and such generic form represents a total prescription unit volume of at least 20%. We have licensed to Otsuka our rights under our U.S. and foreign patents and applications.
Otsuka has the right to terminate this agreement prior to its expiration or renewal for any reason upon six months’ prior notice. Otsuka also has the right to terminate this agreement:
• | upon 14 days’ prior notice in the event that Otsuka decides not to bear its share of any further development costs in excess of $40 million incurred in connection with Phase 2 clinical trials or any further development costs incurred in connection with Phase 3 clinical trials, and terminates the collaboration in its entirety; |
• | upon 14 days’ prior notice in the event Otsuka decides to terminate the agreement after considering the results of a Phase 2 or Phase 3 clinical trial; and |
• | if we experience a change in control, excluding our IPO. |
Otsuka may also terminate the collaboration relationship if:
5
• | Dr. Kubota is no longer one of our employees prior to certain regulatory approvals from the FDA; |
• | Dr. Kubota no longer serves as our chief executive officer; |
• | in Otsuka’s reasonable judgment, Dr. Kubota ceases to act as Co-Chair of the emixustat JDC; or |
• | in Otsuka’s reasonable judgment, Dr. Kubota ceases to act as an ongoing and active member of our development team. |
We have consulted with Otsuka regarding our transition of the chief executive officer role from Dr. Kubota to Brian O’Callaghan. We are currently discussing with Otsuka to amend the Emixustat Agreement to remove the clauses permitting Otsuka to terminate the agreement due to one of the provisions relating to Dr. Kubota above. Otsuka has given us verbal assurances that they do not intend to exercise their right to terminate the Emixustat Agreement based on Dr. Kubota’s changed role.
If the collaboration is terminated at Otsuka’s election or due to our insolvency, a material uncured breach of the agreement, our experiencing a change in control, Otsuka may continue to develop, manufacture and commercialize the products and compounds developed under the agreement and, except in the case of a termination due to a change in our control, the milestone and royalty payment obligations under the agreement will be reduced by 50%. If either party elects to terminate the collaboration for any reason (other than Acucela’s election to terminate the agreement as a result of a material uncured breach of the agreement by Otsuka) and we continue to develop and commercialize emixustat, we would be required to pay royalties to Otsuka based on net sales of emixustat following such termination.
Under the agreement, we have primary responsibility for the implementation of all development activities in North America through the completion of Phase 3 clinical trials and responsibility for all regulatory matters in North America until the parties have initiated the NDA process with the FDA. Otsuka has primary responsibility for the preparation and filing of the NDA and other approvals necessary to commercialize therapies resulting from the emixustat compound, or an approved backup compound, in North America. If we elect to co-promote emixustat under the agreement, Otsuka will manage operations and accounting with respect to all products resulting from our agreement in North America and all sales of these products shall be transacted in Otsuka’s name.
Our Strategy
Our goal is to develop an innovative portfolio of ophthalmology products. Key elements of our strategy to achieve this goal are to:
• | Work collaboratively with Otsuka on the development of emixustat. The emixustat Phase 2b/3 clinical trial is continuing as scheduled. In addition, our co-development partner, Otsuka, is currently evaluating next steps for OPA-6566, an adenosine A2a receptor agonist for the potential treatment of glaucoma. |
• | Initiate partnering efforts for emixustat in Acucela’s territories. In anticipation of topline data from the emixustat Phase 2b/3 study in mid-2016, we plan to initiate our out-licensing efforts under the Emixustat Agreement, primarily for Europe. |
• | Leverage expertise in VCM. Our VCM-based molecules are designed to specifically target rod photoreceptor cells within the retina to potentially treat and slow the progression of certain retinal diseases. We are evaluating the potential to develop emixustat for additional indications such as diabetic retinopathy (DR) or diabetic macular edema (DME). We are planning to initiate and complete preclinical animal model studies in 2015 to evaluate the potential for development of emixustat in DR. In addition, we are committed to further enhancing our patent portfolio. |
• | Build an ophthalmic product pipeline through internal research, M&A, and additional partnering or in-licensing opportunities. We intend to deploy capital in 2015 to fund our internal drug discovery and development efforts, as well as license or otherwise acquire the rights to potential new ophthalmic product candidates. |
Intellectual Property
We believe a strong patent portfolio is critical to our success. We aggressively seek patent protection for our technology. We also rely upon unpatented proprietary technology and know-how because, in some cases, our interest would be better served by reliance on trade secrets or confidentiality agreements than by patents. We have built a portfolio of 114 issued patents and 175 pending patent applications as of December 31, 2014. As of December 31, 2014, this portfolio includes 17 issued patents and 14 pending applications in the United States and six issued patent and nine pending patent applications in Japan. The following is a description of our intellectual property portfolio.
6
Visual Cycle Modulation. For our lead VCM-based product candidate, emixustat, we have one issued U.S. patent (U.S. Patent No. 7,982,071) and four pending U.S. patent applications. The issued patent will expire on or around 2029. Outside the United States, we have a total of eleven issued patents, as well as approximately 50 pending foreign counterparts. If issued, these patents will cover compositions of matter and methods of using emixustat and would expire between 2028 and 2033.
In addition patents and patent applications covering our VCM technology related to emixustat, we have twelve other issued U.S. patents and eight pending U.S. patent applications relating to VCM. Outside the United States, we have approximately 40 granted patents and approximately 90 pending foreign counterparts in Europe and other countries. If issued, these patents will cover compositions of matter and methods of using these compositions. If issued, these patents would expire between 2028 and 2034.
Pursuant to the Emixustat Agreement, we hold a non-exclusive, worldwide, fully paid-up license to make, have made, use, sell, offer for sale and import certain products based on VCM that are developed by employees of Otsuka. This license is subject to certain restrictions but is irrevocable except in the event of termination of the Emixustat Agreement.
OPA-6566. In September 2010, we entered into a development and collaboration agreement, or the Glaucoma Agreement, with Otsuka to develop and commercialize OPA-6566. OPA-6566 is the subject of one U.S. patent assigned to Otsuka (U.S. Patent No. 7,834,002) and the subject of two additional patent applications filed by Otsuka at the U.S. Patent and Trademark Office. The issued patent will expire on or around 2025. If issued, the patents resulting from the two pending applications would expire on or around 2025 and 2030, respectively.
Pursuant to the Glaucoma Agreement, until we exercise our right to co-develop and co-promote products based on OPA-6566, we hold a non-exclusive, royalty-free, fully paid-up license in the United States to certain patents directed to OPA-6566 and related proprietary know-how controlled by Otsuka.
Other Technologies. We intend to continue to invest in our internal drug discovery and development programs and actively seek to license or otherwise acquire the rights to potential new drugs to expand or enhance our product pipeline. For example, in May 2012, we acquired the ophthalmic intellectual property rights to fenretinide, a retinol-binding protein antagonist, and its related compounds from ReVision Therapeutics, in order to further strengthen our leadership position in VCM technology, particularly as it relates to dry AMD.
Competition
The pharmaceutical and biotechnology industries are intensely competitive, and our product candidates, if commercialized, would compete with potential or existing drugs and therapies. In addition, there are many pharmaceutical companies, biotechnology companies, public and private universities, government agencies and research organizations actively engaged in research and development of products targeting the same markets as our product candidates. Many of these organizations have substantially greater financial, technical, manufacturing and marketing resources than we have. Moreover, physicians frequently prescribe legally available therapies for uses that are not described in the product’s labeling and that differ from those tested in clinical studies and approved by the FDA or similar regulatory authorities in other countries. These unapproved, or “off-label,” uses are common across medical specialties and may represent a potential source of competition to our product candidates. Our ability to compete successfully will depend largely on our ability to:
• | design and develop products that are superior to other products in the market; |
• | attract and retain qualified scientific, product development and commercial personnel; |
• | obtain patent and/or other intellectual property protection for our product candidates and technologies; |
• | obtain required regulatory approvals; and |
• | successfully collaborate in the design, development and commercialization of new products. |
We expect to compete on, among other things, product efficacy and safety, time to market, price, extent of adverse side effects and the basis and convenience of treatment procedures.
Geographic Atrophy. There are no FDA-approved treatments for any form of dry AMD, including GA. In response to this unmet medical need, new investigational product candidates are being evaluated that would compete with emixustat if emixustat is approved for GA associated with dry AMD. Product candidates under development include neuroprotective agents, anti-inflammatory compounds, small interfering RNA molecules, complement inhibitors and other drugs that seek to preserve photoreceptors and the RPE. Key investigational compounds include Roche/Genentech’s lampalizumab, an intravitreal injection; GlaxoSmithKline’s GSK933776, an intravenously administered anti-amyloid immunotherapy drug candidate; MacuCLEAR’s MC1101, an antihypertensive drug being developed as an eye drop; and Allergan's brimonidine implant.
7
Glaucoma. In the United States, there are a number of drugs that have been approved for the reduction of intraocular pressure (IOP) in patients with ocular hypertension or open angle glaucoma or are under development for this indication. Each of these drugs is a potential competitor to OPA-6566 if it were to be approved for the reduction of IOP associated with glaucoma. Key product classes that are most commonly used are prostaglandins, such as Xalatan, and beta-blockers, such as timolol. Moreover, a large number of generic versions of drugs target elevated IOP and may have market penetration and price advantages over OPA-6566. In addition to these approved treatments, a number of investigational compounds are being evaluated that would compete with OPA-6566 if OPA-6566 would be approved to treat glaucoma: Aerie Pharmaceuticals, Inc.'s AR 13324, a Rho kinase / Norepinephrine Transporter inhibitor; Valeant / Bausch & Lomb and NicOx S.A.'s latanoprostene bunod, a modified latanoprost compound; OphthaliX’s CF101, an adenosine A3 agonist; and Inotek Pharmaceuticals’ trabodenoson, an adenosine A1 receptor agonist.
Dry Eye Syndrome. In the United States and worldwide there are numerous ophthalmic lubricants (artificial tears) available as OTC products. The only FDA approved therapeutic product in the United States is Restasis (cyclosporine) ophthalmic emulsion. Several other formulations of cyclosporine are in different stages of development. Shire’s liftegrast has finished Phase 3 studies and an NDA has been filed with the FDA. Mimetogen’s D-3 is presently undergoing Phase 3 studies. Several other products have failed studies and not been submitted or approved.
Sales and Marketing
As we continue to evolve as an organization and further develop our product candidates, we plan to evaluate our commercialization plan and strategy to optimize the market potential for our product candidates.
We have the right to co-promote emixustat with Otsuka in North America. Otsuka owns the exclusive development and commercialization rights in selected countries in Asia, the Pacific, the Middle East and North Africa ("Otsuka's sole territory"). We will receive a royalty on net sales in Otsuka's sole territory. We retain sole development and commercialization rights in Europe, South America and other regions and countries outside of North America and Otsuka's sole territory, and subject to Otsuka's right to first negotiate the rights in these territories, will pay Otsuka a royalty on net sales. As of the date of this filing, we intend to co-promote emixustat with Otsuka in North America and we will further define our commercial strategy in our own territories.
Research and Development
During the years ended December 31, 2014, 2013, and 2012, we expended approximately $25.6 million, $36.4 million, and $31.6 million, respectively, on research and development activities.
Manufacturing and Supply
We rely on third parties to manufacture emixustat in accordance with the FDA’s current good manufacturing practice, or cGMP guidelines, for research, development, preclinical and clinical trials, and do not expect to establish our own manufacturing capability. We maintain relationships with third-party manufacturers for the development of formulations and potential large-scale production of our product candidates upon agreement of Otsuka and us. Otsuka is responsible for the manufacture and supply of OPA-6566.
We have no contractual commitments to any manufacturers for future manufacturing though we believe that there are several manufacturing sources available on commercially reasonable terms to meet our clinical and any future commercial production requirements.
Government and Other Regulation
General Overview
Government authorities in the United States and other countries extensively regulate, among other things, the research, development, testing, quality, efficacy, safety (pre- and post-marketing), manufacturing, labeling, storage, record-keeping, advertising, promotion, export, import, marketing and distribution of pharmaceutical products.
United States
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act (FFDCA) and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable FDA or other requirements may subject a company to a variety of administrative or judicial sanctions, such as the
8
FDA’s refusal to approve pending applications, imposing a clinical hold, issuing enforcement letters, such as warning letters, initiating seizure of products or injunctive relief, imposing partial or total suspension of production, withdrawing the product from the market, assessing fines or civil penalties, or initiating criminal prosecution.
FDA approval is required before any new drug, such as a new chemical entity, or a new dosage form, new use or new route of administration of a previously approved product, can be marketed in the United States. The process required by the FDA before a new drug product may be marketed in the United States generally involves:
• | completion of preclinical laboratory and animal testing and formulation studies in compliance with the FDA’s good laboratory practice, or GLP, regulations; |
• | submission to the FDA of an Investigational New Drug (IND) application for human clinical testing which must become effective before human clinical trials may begin in the United States; |
• | approval by an independent institutional review board, or IRB, at each clinical trial site before each trial may be initiated; |
• | completion of adequate and well-controlled human clinical trials in accordance with good clinical practices, or GCP, to establish the safety and efficacy of the proposed product candidate for each intended use; |
• | satisfactory completion of an FDA pre-approval inspection of the facility or facilities at which the product is manufactured to assess compliance with cGMP, to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and purity; |
• | submission to the FDA of a new product application, or New Drug Application (NDA), or supplemental NDA; |
• | satisfactory completion of an FDA advisory committee review, if applicable; |
• | payment of user fees, if applicable; and |
• | FDA review and approval of the NDA. |
The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all. Preclinical tests include laboratory evaluation of product chemistry, formulation, stability and toxicity, as well as animal studies to assess the characteristics and potential safety and efficacy of the product. The results of preclinical tests, together with manufacturing information, analytical data and a proposed clinical trial protocol and other information, are submitted as part of an IND to the FDA. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions relating to one or more proposed clinical trials and places the clinical trial on a clinical hold, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND does not automatically result in FDA authorization to commence clinical trials. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Even if the IND becomes effective and the trial proceeds without initial FDA objection, FDA may stop the trial at a later time if it has concerns, such as unacceptable safety risks arise.
Further, an independent review board, or IRB, that has jurisdiction over each clinical site planning to participate in the clinical trial must review and approve the plan for any clinical trial and informed consent information for subjects before the trial commences at that site and it must monitor the study until completed. The FDA, the IRB, or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk or for failure to comply with the IRB’s requirements, or may impose other conditions.
As a separate amendment to an IND, a sponsor may submit a request for a Special Protocol Assessment, or SPA, from the FDA. Under the SPA procedure, a sponsor may seek the FDA’s agreement on the proposed design and size of a clinical trial intended to form the primary basis for determining a product’s efficacy. Upon specific request by a sponsor, the FDA will evaluate the protocol and respond to a sponsor’s questions regarding, among other things, primary efficacy endpoints, trial conduct and data analysis within 45 days of receipt of the request with the goal of reaching an agreement that the Phase 3 trial protocol design, clinical endpoints, and statistical analyses are acceptable to support regulatory approval of the product candidate with respect to effectiveness for the indication studied. Under an SPA, the FDA agrees to not later alter its position with respect to adequacy of the design, execution, or analyses of the clinical trial intended to form the primary basis of an effectiveness claim in an NDA, without the sponsor’s agreement, unless the FDA identifies a substantial scientific issue essential to determining the safety or efficacy of the product after testing begins that might change the agency's initial decision. Moreover, any change to a study protocol after agreement with the FDA is reached can invalidate an SPA. Agreements and
9
disagreements between the FDA and the sponsor regarding an SPA are documented by the FDA in an SPA letter to the sponsor or in the minutes of a meeting between the sponsor and the FDA.
Clinical trials involve the administration of the investigational new product to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Sponsors of clinical trials generally must register and report, at the National Institutes of Health-maintained website ClinicalTrials.gov, key parameters of certain clinical trials. For purposes of an NDA submission and approval, human clinical trials are typically conducted in the following sequential phases, which may overlap or be combined:
• | Phase 1: The product is initially introduced into healthy human subjects or patients and tested for safety, dose tolerance, absorption, metabolism, distribution and excretion and, if possible, to gain an early indication of its effectiveness. |
• | Phase 2: The product is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more extensive clinical trials. |
• | Phase 3: These are commonly referred to as pivotal studies. When Phase 2 trials demonstrate that a dose range of the product appears to be effective and has an acceptable safety profile, trials are undertaken in large patient populations to further evaluate dosage, to obtain additional evidence of clinical efficacy and safety in an expanded patient population at multiple, geographically-dispersed clinical trial sites, to establish the overall risk-benefit relationship of the product and to provide adequate information for the labeling of the product. |
• | Phase 4: In some cases, the FDA may conditionally approve an NDA for a product candidate on the sponsor’s agreement to conduct additional clinical trials to further assess the product’s safety and effectiveness. |
The FDA closely monitors the progress of each phase of clinical testing and may, at its discretion, reevaluate, alter, suspend or terminate testing based on data accumulated to that point and its assessment of the risk/benefit relationship to the patient. Additional clinical testing may be required for special classes of patients, e.g., geriatric patients, pediatric patients, or patients with renal impairment. The results of product development, preclinical studies and clinical trials are submitted to the FDA as part of a NDA. NDAs must also contain extensive information relating to the product’s pharmacology, chemistry, manufacturing and controls, proposed labeling, and risk/benefit relationship of the drug to the patient, among other things.
For some products, especially controlled substances, the FDA may require a risk evaluation and mitigation strategy, or REMS, which could include measures imposed by the FDA such as prescribing restrictions, requirements for post-marketing studies or certain restrictions on distribution and use. In determining whether a REMS is necessary, the FDA must consider the size of the population most likely to use the drug, the seriousness of the disease or condition to be treated, the expected benefit of the drug, the duration of treatment, the seriousness of known or potential adverse events and whether or not the drug is a new chemical entity. If the FDA determines a REMS is necessary, the sponsor must propose the REMS plan at the time of approval. A REMS may be required to include various elements, such as a medication guide or patient package insert, a communication plan to educate health providers of the drug’s risks, limitation on who may prescribe or dispense the drug or other measures that the FDA deems necessary to assure the safe use of the drug. In addition, the REMS must include a timetable to assess the strategy at 18 months, three years, and seven years after the strategy’s approval. The FDA may also impose a REMS requirement on a drug already on the market if the FDA determines, based on new safety information, that a REMS is necessary to ensure that the drug’s benefits outweigh its risks.
Under federal law, the submission of most NDAs is additionally subject to a substantial application user fee, and the manufacturer and/or sponsor under an approved NDA are also subject to annual product and establishment user fees. Under the Prescription Drug User Fee Act, or PDUFA, the applicant must pay a fee which is substantial (i.e., in excess of $2.0 million) and increases every year. The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information and is subject to payment of additional user fees. The resubmitted application is also subject to review before the FDA accepts it for filing.
10
Once the submission has been accepted for filing, the FDA begins an in-depth substantive review. Under the Prescription Drug User Fee Act, or PDUFA, the FDA agrees to specific performance goals for NDA review time through a two-tiered classification system, Standard Review and Priority Review. Standard Review NDAs have a goal of being completed within a ten-month timeframe. A Priority Review designation is given to products that offer major advances in treatment, or provide a treatment where no adequate therapy exists. The goal for completing a Priority Review is six months.
The review process may be extended by the FDA for three additional months to consider certain information or obtain clarification regarding information already provided in the submission. The FDA may refer applications for novel products or products which present difficult questions of safety or efficacy to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA may inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP, for pharmaceuticals, and if applicable, Quality System Regulation or QSR requirements, for medical devices, and are adequate to assure consistent production of the product within required specifications. Additionally, the FDA will typically inspect one or more clinical sites to assure compliance with GCP before approving an NDA.
After the FDA evaluates the NDA and, in some cases, the related manufacturing facilities, it may issue an approval letter or a Complete Response Letter, or CRL, to indicate that the review cycle for an application is complete and that the application is not ready for approval. CRLs generally outline the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when the deficiencies have been addressed to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications.
Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems are identified after the product reaches the market. In addition, the FDA may require post-approval testing, including Phase 4 studies, and surveillance programs or new product labeling to monitor the effect of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs. Products may be marketed only for the approved indications and in accordance with the provisions of the approved label, and, even if the FDA approves a product, it may limit the approved indications for use for the product or impose other conditions, including labeling or distribution restrictions or other risk-management mechanisms, such as a Black Box Warning, which highlights a specific warning (typically life-threatening), or a REMS program. Further, if there are any modifications to the product, including changes in indications, labeling, or manufacturing processes or facilities, a company may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require the company to develop additional data or conduct additional preclinical studies and clinical trials, before any such change may be made.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to product listing, recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and generally require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance, as well as oversight of third-party vendors to ensure regulatory compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
• | restrictions on the marketing or manufacturing of the product or complete withdrawal of the product from |
11
the market;
• | fines, warning letters or holds on post-approval clinical trials; |
• | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
• | product seizure or detention, or refusal to permit the import or export of products; or |
• | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. While physicians may prescribe for unapproved, or off-label, uses, manufacturers may only promote for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies, such as the U.S. Department of Justice and state regulatory bodies, actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off label uses may be subject to significant liability, both on the federal and state levels, including significant fines or exclusion from participation in healthcare programs.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution, including a drug pedigree which tracks the distribution of prescription drugs.
The FDA may require post-approval studies and clinical trials if the FDA finds that scientific data, including information regarding related drugs, deem it appropriate. The purpose of such studies would be to assess a known serious risk or signals of serious risk related to the drug or to identify an unexpected serious risk when available data indicate the potential for a serious risk. The FDA may also require a labeling change if it becomes aware of new safety information that it believes should be included in the labeling of a drug.
With respect to post-market product information dissemination, advertising and promotion, the FDA imposes a number of complex regulations, which include, among others, standards for healthcare professional-directed and direct-to-consumer advertising, off-label discussions, industry-sponsored scientific and educational activities, and promotional activities involving the internet. The FDA has very broad enforcement authority under the FFDCA, and failure to abide by these regulations can result in penalties, including the issuance of an enforcement letter, such as a warning letter directing entities to correct deviations from FDA standards, a requirement that future advertising and promotional materials are pre-cleared by the FDA, and state and federal civil and criminal investigations and prosecutions.
Fast Track Designation
The FDA’s “fast track” program is intended to facilitate the development and to expedite the review of drugs that are intended for the treatment of a serious or life-threatening condition and that demonstrate the potential to address unmet medical needs. Fast track designation permits the FDA to initiate review of sections of an NDA before the application is complete. This so called “rolling review” is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant has paid the applicable user fees, half of which are due at the time of filing and half of which are due at the time of approval. The FDA’s PDUFA review clock does not begin until the complete application is submitted. Additionally, the fast track designation may be withdrawn by the FDA if it believes that the designation is no longer supported by emerging data, or the designated development program is no longer being pursued.
Other Regulatory Considerations
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of our drug product candidates, some of the U.S. patents covering our product candidates may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND, and the submission date of an NDA or BLA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved product is eligible for the extension and the application for extension must be made prior to expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for
12
restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant application.
Market exclusivity provisions under the FFDCA also can delay the submission or the approval of certain applications. The FFDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity, non-infringement or unenforceability. The FFDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an approved NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving competitor products for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA submitted under section 501(b)(1) of the FFDCA.
Third-Party Payor Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of product candidates for which we obtain regulatory approval. In the United States as well as in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend, in part, on coverage and reimbursement decisions made by third-party payors. In the United States, third-party payors include the government (i.e., Medicare and Medicaid programs) as well as private health insurers.
Payors processes for determining whether to provide coverage for a particular product are often separate from the processes for setting the reimbursement rate for the product. Payors may limit coverage to drug products on an approved list, or formulary, which might not include all of the FDA-approved drug products for a particular indication. In addition, payors are increasingly making coverage determinations by examining the medical necessity and cost-effectiveness of medical products. Thus, we may need to conduct expensive pharmacoeconomic studies, outside and apart from the studies we conduct for obtaining FDA approval, in order to demonstrate the medical necessity and cost-effectiveness of our products. Payors may determine that our product candidates are not medically necessary or cost-effective and as a result, may decide not to cover our products. Additionally, even if payors decide to cover our drug product, they may not approve adequate reimbursement which enables us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Several significant laws have been enacted in the United States which affect the pharmaceutical industry. For example, as a result of the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA), a Medicare prescription drug benefit (Medicare Part D) became effective at the beginning of 2006. Medicare is the federal health insurance program for people who are 65 or older, certain younger people with disabilities, and people with End-Stage Renal Disease. Medicare coverage and reimbursement for some of the costs of prescription drugs may increase demand for any products for which we receive FDA approval. However, we would be required to sell products to Medicare beneficiaries through entities called “prescription drug plans,” which will likely seek to negotiate discounted prices for our products.
As another example, in March 2010, the President of the United States signed into law the Patient Protection and Affordable Care Act and the Healthcare and Education Reconciliation Act of 2010 (collectively referred to as the “ACA”). The ACA made significant changes to the U.S. healthcare system, such as imposing new requirements on health insurers, expanding the number of individuals covered by health insurance, modifying healthcare reimbursement and delivery systems, and establishing new requirements designed to prevent fraud and abuse. In addition, provisions in the ACA promote the development of new payment and healthcare delivery systems, such as the Medicare Shared Savings Program, bundled payment initiatives and the Medicare pay for performance initiatives.
The ACA and the related regulations, guidances and court decisions have had, and will continue to have, a significant impact on the pharmaceutical industry. In addition to the general reforms briefly described above, provisions of the ACA directly address drugs. For example, the ACA:
• | Increases the minimum level of Medicaid rebates payable by manufacturers of brand-name drugs from 15.1% to 23.1%; |
• | Requires Medicaid rebates for covered outpatient drugs to be extended to Medicaid managed care organizations; |
13
• | Requires manufacturers of drugs covered under Medicare Part D to participate in a coverage gap discount program, under which they must agree to offer a 50% point-of-sale discount off negotiated prices of applicable brand drugs to eligible Medicare beneficiaries during their coverage gap period; and |
• | Imposes a non-deductible annual fee on pharmaceutical manufacturers or importers who sell "branded prescription drugs" to specified federal government programs. |
Federal, state and local governments in the United States continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Future legislation and regulations could further limit payments for pharmaceuticals, such as the product candidates that we are developing. In addition, court decisions have the potential to affect coverage and reimbursement for prescription drugs. It is unclear whether future legislation, regulations or court decisions will affect the demand for our product candidates once commercialized.
The Physician Payment Sunshine Act
The ACA includes the Physician Payment Sunshine Act (the “Act”). The Act requires extensive tracking of physician and teaching hospital payments and other transfers of value, and public reporting of the payment data. The Centers for Medicare and Medicaid Services (“CMS”) released a final rule on February 1, 2013 implementing the Act provisions and clarified the scope of the reporting obligations, requiring manufacturers to begin tracking on August 1, 2013 and reporting data to CMS by March 31, 2014. Although we are not independently subject to the Act, we are required to provide information to Otsuka who is subject to the reporting requirements of the Act. Failure to comply with the reporting obligations may result in civil monetary penalties.
Foreign Regulations
Foreign regulatory systems, although varying from country to country, include risks similar to those associated with FDA regulations in the United States.
Under the European Union regulatory system, applications for drug approval may be submitted either in a centralized or decentralized procedure. Under the centralized procedure, a single marketing application to the EMA leads to an approval granted by the European Commission, which permits marketing of the product throughout the European Union (currently 28 member states), as well as in Iceland, Liechtenstein and Norway. The centralized procedure is mandatory for new chemical entities, biotech and orphan drug products and products to treat AIDS, cancer, diabetes and neuro-degenerative disorder, auto-immune diseases, other immune dysfunctions and viral diseases. Products that constitute a significant therapeutic, scientific or technical innovation or which are in the interests of patients at the European Union community level may also be submitted under this procedure. Our product would potentially qualify for this procedure as a product that constitutes a significant therapeutic, scientific or technical innovation and is a new chemical entity. The Decentralized Procedure provides for mutual recognition of nationally approved decisions and is used for products that do not comply with the requirements for the centralized procedure. Under the decentralized procedure, the holders of national marketing authorization in one of the countries within the European Union may submit further applications to other countries within the European Union, who will be requested to recognize the original authorization based on an assessment report provided by the country in which marketing authorization is held.
Employees
As of December 31, 2014, we had 49 employees employed in the areas of research, clinical development and operations and administration. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We have never experienced any employment-related work stoppages and consider relations with our employees to be good.
Available Information
Our corporate headquarters are located at 1301 Second Avenue, Suite 4200, Seattle, Washington 98101-3805 and our telephone number is (206) 805-8300. We maintain a website at www.acucela.com. The contents of our website are not incorporated into, or otherwise to be regarded as part of, this Annual Report on Form 10-K.
We make available our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or Section 15(d) of the Securities Exchange Act of 1934, as amended, free of charge on our website at ir.acucela.com, as soon as reasonably practicable after they are electronically filed with or furnished to the Securities and Exchange Commission or SEC. Additionally, copies of materials filed by us with the SEC may be accessed at the SEC's Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549 or at www.sec.gov. For information about the SEC's Public Reference Room, contact 1-800-SEC-0330.
14
ITEM 1A. RISK FACTORS
An investment in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below and all other information contained in this Annual Report on Form 10-K, including our financial statements and the related notes, before making a decision to invest in our common stock. Our business, operating results, financial condition or prospects could be materially and adversely affected by any of these risks and uncertainties. In that case, the trading price of our common stock could decline and you might lose all or part of your investment. In addition, the risks and uncertainties discussed below are not the only ones we face. Our business, operating results, financial performance or prospects could also be harmed by risks and uncertainties not currently known to us or that we currently do not believe are material. In assessing the risks and uncertainties described below, you should also refer to the other information contained in this Form 10-K before making a decision to invest in our common stock.
Risks Related to Our Business and Industry
If Dr. Kubota and SBI are successful in their efforts to replace our board of directors, our business, including our collaborations with Otsuka, may be significantly harmed.
On January 28, 2015, we received a letter from SBI Holdings, Inc., or SBI, the parent company of several of our shareholders, demanding that we hold a special meeting of our shareholders for the purposes of removing our current board members, other than Dr. Kubota, and electing a slate of directors proposed by SBI. In connection with this demand, SBI granted Dr. Kubota an irrevocable proxy over the shares held collectively by SBI, giving Dr. Kubota voting authority over shares representing over 50% of our outstanding stock. On March 3, 2015, SBI and Dr. Kubota filed a lawsuit in Washington state court seeking an order compelling us to hold a special shareholder meeting on or before April 28, 2015, and to provide our shareholders written notice of that meeting as soon as practicable but no later than March 31, 2015.
On March 13, 2015, the Washington state superior court presiding over this lawsuit, issued an order requiring us to hold a special shareholders meeting no later than May 1, 2015 and to give notice of such a meeting as soon as practicable. In response to the hearing, we have announced that we plan to hold the special shareholders meeting on May 1, 2015 (Pacific Daylight Time) at our headquarters and have set a record date for shareholders of record as March 19, 2015 (Pacific Daylight Time). If Dr. Kubota votes all shares over which he has voting authority in favor of the business to be brought before the special meeting, then our current members of our board of directors (other than Dr. Kubota) will be replaced by nominees proposed by SBI.
Changes on our board of directors and changes in our senior management (if enacted by a new board of directors) may be disruptive to our business and, before and during the transition period, there may be uncertainty among investors, employees and our collaboration partner concerning our future direction and performance. Any such disruption or uncertainty could have a material adverse impact on our results of operations and financial condition and the market price of our common stock. For instance, if Dr. Kubota and SBI are able to replace our board of directors, we may have difficulty retaining and motivating our key management and scientific staff, most of whom have indicated support for our current board of directors. In addition, Otsuka has recently indicated that if SBI gains control of the Company, then Otsuka will view that as a significant challenge to our collaborations with Otsuka. Consequently, we cannot assure you that Otsuka will not elect to exercise its rights to terminate the existing collaboration agreements if the current board of directors is replaced.
We do not have any products that are approved for commercial sale.
To date, we have not generated any product revenue and have funded our operations through proceeds from our February 2014 IPO, private sales of our equity and debt securities and from our various collaboration agreements with Otsuka, primarily the co-development and collaboration agreement relating to the development and commercialization of emixustat which we refer to as the Emixustat Agreement. We will not receive revenues from sales of emixustat or any other product candidates unless we succeed, either independently or with third parties, in developing and obtaining regulatory approval and marketing drugs with commercial potential. We may never succeed in these activities, and may not generate sufficient revenues to continue our business operations.
Revenues from research and development activities in collaboration with Otsuka and Otsuka’s funding of our portion of development costs under the Emixustat Agreement represented all of our revenues during the year ended December 31, 2014, and the loss of these revenues would adversely affect our business.
Revenues from research and development activities under our collaboration agreements with Otsuka have been our only source of revenues in 2014, 2013, and 2012, and we expect they will continue to have a significant impact on our results
15
of operations in future years. As explained further below, in 2013, Otsuka terminated a co-development agreement with us under which we derived significant revenues from clinical programs. It would be difficult to replace Otsuka as a collaboration partner, and the revenues derived from research and development activities on behalf of Otsuka and Otsuka’s funding of our portion of development costs under the Emixustat Agreement. Accordingly, the loss of Otsuka as a collaboration partner would have a material adverse effect on our business. In addition, any publicity associated with the loss of Otsuka as a collaboration partner could harm our reputation. Otsuka can terminate its collaboration agreements with us on relatively short notice in various circumstances, such as our material breach or insolvency, changes in control of us or, in the case of the Emixustat Agreement, a decision by Otsuka to discontinue funding development costs after considering the results of a Phase 2 or Phase 3 clinical trial, and also for any reason upon six months’ prior notice. Otsuka currently has the right to terminate the collaboration agreements because Dr. Kubota is no longer serving as our chief executive officer and, while we intend to enter into an amendment to the collaboration agreements to remove this termination right, we cannot assure you that Otsuka will not elect to exercise this right. For more information regarding the termination rights under each of our collaboration agreements with Otsuka, see “Business—EMIXUSTAT—Collaboration with Otsuka”.
In addition, Otsuka’s interests may differ from ours in relation to development of the product candidates under our collaboration agreements with them due to changes in management, priorities or its strategic focus. For example, in September 2013, Otsuka terminated its agreement with us to co-develop rebamipide for the treatment of dry eye syndrome in the United States due to the fact that the primary endpoints were not met in the Phase 3 clinical trial in the United States. As a result, the associated clinical trials were suspended and our development activities halted. Losing the support and focus of Otsuka would adversely affect the development and commercialization of the product candidates under our collaboration agreements. Our revenues and operating results would suffer and we may need to curtail or cease operations if, among other things, Otsuka terminates the Emixustat Agreement or otherwise fails to fund development costs for any reason.
We incurred losses in the last fiscal year, and will continue to incur losses in the future.
We incurred a net loss of $2.0 million during the year ended December 31, 2014, and as of December 31, 2014, we had an accumulated deficit of $5.5 million. We expect to incur net losses for the next several years as we continue to develop emixustat and any other product development candidates, and over the long-term if we expand our research and development programs and acquire or in-license products, technologies or businesses that are complementary to our own. As a result of these losses, we may exhaust our financial resources and be unable to complete the development of our product development candidates. Since 2008, revenues from research and development activities under our collaboration agreements with Otsuka have been our only source of revenues. In the second half of 2011, Otsuka began funding our portion of the development costs under the Emixustat Agreement and we record these advances as revenues in our financial statements. We are contingently obligated to repay these advances, plus interest, with a portion of any revenues we generate, if any, from the commercialization of products under the Emixustat Agreement in future periods. For a detailed discussion of our financial condition and results of operations, see "Management's Discussion and Analysis of Financial Condition and Results of Operations". If revenue does not increase, our operating results will be negatively affected. If we fail to raise capital as needed, we may need to curtail operations or cease operations in the future. There can be no assurances that there will be adequate financing available to us in the future on acceptable terms, or at all.
Our long-term prospects are dependent on emixustat and we cannot be certain that it will achieve success in clinical trials, regulatory approval or be successfully commercialized.
We have invested a significant portion of our time and financial resources in the development of emixustat, the lead product candidate emerging from our internally-developed VCM compounds. VCM is an emerging technology and its long-term effect is unknown, and thus, there can be no assurance that our product candidate will achieve regulatory approval. Clinical development is a long, expensive and uncertain process and subject to delays or additional requirements. For instance, based on guidance from the FDA in April 2014, we now expect to conduct at least one additional confirmatory Phase 3 clinical trial in patients with GA associated with dry AMD after we complete our current 24-month emixustat study. We may also encounter delays or rejections based on our inability to enroll enough patients to complete our clinical trials in a timely manner. It may take several years and require the expenditure of substantial resources to complete the testing of a product candidate, and failure can occur at any stage of testing. For example:
• | interim results of clinical or non-clinical studies may not necessarily predict their final results, and acceptable results in early studies might not be seen in later studies, in large part because earlier phases of studies are often conducted in smaller groups of patients than later studies, and without the same trial design features, such as randomized controls and long-term patient follow-up and analysis; |
• | product candidates that appear promising at early stages of development may ultimately fail for a number of reasons, including the possibility that the product candidates may be ineffective, less effective than products or product candidates of our competitors or cause harmful side effects; |
16
• | any clinical or non-clinical test may fail to produce results satisfactory to the FDA or foreign regulatory authorities; |
• | clinical and non-clinical data can be interpreted in different ways, which could delay, limit or prevent regulatory approval; |
• | negative or inconclusive results from a non-clinical study or clinical trial or adverse medical events during a clinical trial could cause a non-clinical study or clinical trial to be repeated or a program to be terminated, even if other studies or trials relating to the program are successful; |
• | the FDA can place a clinical hold on a trial if, among other reasons, it finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury; |
• | we may encounter delays or rejections based on changes in regulatory agency policies during the period in which we develop a drug or the period required for review of any application for regulatory agency approval; and |
• | our clinical trials may not demonstrate the safety and efficacy of any product candidates or result in marketable products. |
Even if clinical trials and testing are successful in the future, we believe the process of completing clinical trials and submitting NDAs to the FDA for approval will take several years and require the expenditure of substantial resources. If we are required to conduct additional clinical trials or other studies of emixustat beyond those that we currently contemplate, if we are unable to successfully complete our clinical trials or other studies or if the results of these trials or studies are not positive or are only modestly positive, we may be delayed or unsuccessful in obtaining marketing approval for emixustat. Additionally, we may not be able to obtain marketing approval or we may obtain approval for indications that are not as broad as intended. Our product development costs will also increase if we experience delays in testing or approvals. Significant clinical trial delays could allow our competitors to bring products to market before we do and impair our ability to commercialize products.
Even if clinical trials are successful in the future, our growth prospects could be adversely affected and we may need to curtail or cease operations if we do not pursue the optimal commercialization strategy for us under our collaboration agreement.
As a part of our collaboration strategy, we seek to retain an option to choose either to obtain commercialization rights once emixustat or any other product candidates reach a later stage of development or to receive royalties from future product sales. Under our collaboration agreements with Otsuka, we have exclusive co-promotion rights in certain regions of the world. If we choose to not exercise these rights, we would be entitled to receive royalties on net sales of emixustat and we would not be entitled to any royalties or other payments based on the commercialization of OPA-6566. In general, we must make the decision to exercise these rights before the completion of clinical trials, based on our prediction of future sales and costs and other information. It is possible that the commercialization strategy we choose to pursue will ultimately be less beneficial to us than the other commercialization strategies available to us, which could harm our prospects for growth and could cause us to curtail or cease operations. We may choose a commercialization strategy that results in a less optimal outcome for a variety of reasons, including our failure to accurately predict future sales and costs or our inability to pay the exercise fees and milestone payments.
Our decision to license emixustat to Otsuka means that we no longer have complete control over how emixustat is developed and commercialized and how it may be perceived in the marketplace, and we may lose an even greater degree of control over how it is commercialized if we fail to make our co-promotion election under the terms of the Emixustat Agreement. In addition, unless and until we exercise co-promotion rights with respect to OPA-6566, we will have limited or no control over how the product candidate is developed and commercialized. Even if we exercise commercialization rights for these product candidates, we will depend, in part, on the efforts of Otsuka to commercialize any successfully-developed product candidates and will not have control over a number of key elements relating to the commercialization of those product candidates. Otsuka may fail to effectively commercialize emixustat or any other product candidates under our collaboration agreements with them for a variety of reasons, including because it may not regard our programs as significant for its own businesses, because it does have sufficient resources or decides not to devote necessary resources.
The pharmaceutical market is intensely competitive. Even if we are successful in obtaining approval of emixustat or any other product candidates, we may be unable to compete effectively with existing drugs, new treatment methods and new technologies.
The pharmaceutical market is intensely competitive and rapidly changing. Many large pharmaceutical and biotechnology companies, academic institutions, governmental agencies and other public and private research organizations are pursuing the development of novel therapies for the same indications that we are targeting or expect to target.
17
If emixustat is approved for GA associated with dry AMD, we anticipate that it would potentially compete with lampalizumab (Genentech/Novartis), which is presently undergoing two global Phase 3 studies. We are also aware of a number of new drugs and other therapeutic candidates under development for the treatment of AMD (including dry AMD), glaucoma, and dry eye, including several for each of these disease categories by some of the larger pharmaceutical companies. For a listing of competing therapies or products and therapies under development, see "Business—Competition."
Many of our competitors have:
• | much greater financial, technical and human resources than we have at every stage of discovery, development, manufacture and commercialization of products; |
• | more extensive experience in non-clinical testing, conducting clinical trials, obtaining regulatory approvals, manufacturing and marketing pharmaceutical products; |
• | product candidates that are based on previously tested or accepted technologies; |
• | products that have been approved or are in late stages of development; and |
• | collaboration arrangements in our target markets with leading companies and research institutions. |
Our competitors may succeed in obtaining patent protection, receiving FDA approval or commercializing drugs for the same indications before we do. Any competing drugs may be more effective or marketed and sold more effectively than any products we develop. Moreover, physicians frequently prescribe therapies for uses that are not described in the product’s labeling and that differ from those tested in clinical studies and approved by the FDA or similar regulatory authorities in other countries. These unapproved, or “off-label,” uses are common across medical specialties and may represent a potential source of competition to emixustat or any other product candidates. Competitive therapies, including surgical procedures and medical devices, may make any drug products we develop obsolete or noncompetitive before we can recover the expenses of developing and commercializing them.
Market acceptance of emixustat and other products we develop in the future may be limited.
The commercial success of the products for which we may obtain marketing approval from the FDA or other regulatory authorities will depend upon the acceptance of these products by the medical community and third-party payors as clinically useful, cost-effective and safe. Even if a potential product displays a favorable efficacy and safety profile in clinical trials, market acceptance of the product will not be known until after it is commercially launched. We expect that many of the products we develop will be based upon mechanisms new to the market. For example, emixustat is a small-molecule compound within the phenylalkylamine chemical family. To date, no such small-molecule compound has been approved as a pharmaceutical by the FDA. As a result, it may be more difficult for us to achieve market acceptance of our products, particularly the first products that we introduce to the market. Our efforts to educate the medical community about these potentially novel approaches may require greater resources than would be typically required for products based on previously tested or accepted technologies.
Our operating results may fluctuate in the future, which could cause our stock price to decline.
Our quarterly and annual results of operations may fluctuate in the future due to a variety of factors, many of which are outside of our control. The revenues we generate, if any, and our operating results will be affected by numerous factors, including, but not limited to:
• | the development status of emixustat and, particularly, the timing of any milestone payments to be paid or received by us under our collaboration agreements; |
• | the incurrence of clinical expenses that could fluctuate significantly from period to period; |
• | the unpredictable effects of collaborations during these periods; |
• | the timing of our satisfaction of applicable regulatory requirements; |
• | the rate of expansion of our clinical development and other internal development efforts; |
• | the effect of competing technologies and products and market developments; and |
• | general and industry-specific economic conditions. |
If our operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any fluctuations in our operating results and cash flows may, in turn, cause the price of our stock to fluctuate substantially. We believe that comparisons of our historical financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
18
We have limited in-house sales and marketing capabilities and will need to invest significantly to develop these capabilities if our product candidates are successfully developed.
In order to exploit any commercialization opportunities under our agreements with Otsuka, we believe we will need to develop our own sales and marketing infrastructure to facilitate sales of our product candidates. We cannot assure you that we will be able to do this on a timely basis or at all. The failure to do so would harm our ability to generate product revenues. To the extent we cannot or choose not to use internal resources for the marketing, sales or distribution of any product candidates in the United States. or elsewhere, we intend to rely on collaboration partners, such as Otsuka, or licensees. We may be unable to establish or maintain such relationships. To the extent that we depend on collaboration partners or other third parties for marketing, sales and distribution, any revenues we receive will depend upon their efforts. Such efforts may not be successful, and we will not be able to control the amount and timing of resources that our licensees or collaborators or other third parties devote to our products.
In addition, to the extent we utilize such relationships for marketing, sales or distribution of any approved products outside of the United States, we will be subject to additional risks related to entering into international business relationships, such as reduced protection for intellectual property rights; unexpected changes in tariffs, trade barriers and regulatory requirements; economic weakness, including inflation, or political instability in particular foreign economies and markets; foreign taxes, including withholding of payroll taxes; and foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country.
We may not be successful in our efforts to expand our portfolio of product candidates.
We are seeking to expand our portfolio of product candidates beyond emixustat through internal development and by partnering with other pharmaceutical or biotechnology companies.
A significant portion of our internal research program involves unproven technologies. Research programs to identify new disease targets and product candidates require substantial technical, financial and human resources whether or not we ultimately identify any candidates. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development for a number of reasons, including without limitation:
• | the research methodology used may not be successful in identifying potential product candidates; |
• | potential product candidates may, on further study, be shown to have harmful side effects or other characteristics that indicate they are unlikely to be effective drugs; or |
• | we may fail to obtain intellectual property protection for our product candidates and technologies. |
We may attempt to license or acquire product candidates and be unable to do so for a number of reasons. In particular, the licensing and acquisition of pharmaceutical products is a competitive area. A number of more established companies are also pursuing strategies to license or acquire products in the ophthalmic field. These established companies may have a competitive advantage over us due to their size, financial resources and greater clinical development and commercialization capabilities. Other factors that may prevent us from licensing or otherwise acquiring suitable product candidates include the following:
• | we may be unable to license or acquire the relevant intellectual property rights on terms that would allow us to make an appropriate return from the product; |
• | companies that perceive us to be their competitors may be unwilling to assign or license their product rights to us; or |
• | we may be unable to identify suitable products or product candidates within our areas of expertise. |
Relying on third-party manufacturers may result in delays in our clinical trials and product introductions.
We have limited experience in, and we do not own facilities for, manufacturing any product candidates, and we do not intend to develop facilities for the manufacture of product candidates for clinical trials or commercial purposes in the foreseeable future. We will depend on Otsuka for the manufacture and supply of product candidates subject to our collaboration agreements. Otsuka is contracting with third-party manufacturers to produce, in collaboration with Otsuka and us, our product candidates for clinical trials. While there are likely competitive sources available to manufacture our product candidates, entering into new arrangements may cause delays and additional expenditures, which we cannot estimate with certainty.
There are risks inherent in pharmaceutical manufacturing that could affect the ability of our third-party manufacturers to meet our requirements, which could result in unusable products and cause delays in our development process and our clinical trials. We need to contract with manufacturers who can comply with FDA-mandated current good manufacturing practices, or cGMPs, and comparable requirements of foreign regulatory bodies on an on-going basis. If we
19
receive necessary regulatory approval for any product candidate, we also expect to rely on third parties, including our collaboration partners, to produce materials required for commercial production. Should we experience difficulties in obtaining and maintaining adequate manufacturing capacity, our ability to successfully develop and commercialize our products could be adversely impacted.
A failure of any of our third-party manufacturers to perform their obligations in a timely manner or establish and follow cGMPs with proper documentation may result in significant delays in clinical trials or in obtaining regulatory approval of product candidates or the ultimate launch of our products into the market. These failures could cause delays and other problems resulting in a material adverse effect on our business, financial condition and results of operations. If we are required to change manufacturers for any reason, we may incur significant costs and be required to devote significant time to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines.
We are dependent on our management team, particularly Brian O'Callaghan, our chief executive officer and president, and if we are unable to retain and motivate our key management and scientific staff, our drug development programs may be delayed and we may be unable to successfully develop or commercialize our product candidates.
We are dependent upon the continued services of our executive officers and other key personnel, particularly Brian O'Callaghan, our chief executive officer and president. The relationships that our collaboration team members have cultivated with Otsuka make us particularly dependent upon certain employees, including Mr. O'Callaghan and many of our collaboration team leaders, remaining employed by us. In addition, Otsuka has the right to terminate our collaboration agreements with them if Dr. Kubota is no longer serving as our chief executive officer. We have consulted with Otsuka regarding our transition of the chief executive officer role from Dr. Kubota to Brian O’Callaghan and are currently discussing with Otsuka to amend the Emixustat Agreement to remove the clauses permitting Otsuka to terminate the agreement if Dr. Kubota is no longer serving as our chief executive officer. Otsuka has given us verbal assurances that they do not intend to exercise their right to terminate the Emixustat Agreement based on Dr. Kubota’s changed role.
As we acquire or obtain rights to develop and commercialize new product candidates, our success will depend on our ability to attract, retain and motivate highly qualified management and scientific personnel to manage the development of these new product candidates. We face competition for experienced scientists and other technical and professional personnel from numerous companies and academic and other research institutions. Competition for qualified personnel is particularly intense in the Seattle, Washington area, where very few people possess the skills and expertise we need to develop and commercialize our product candidates. Our operating history and the uncertainties attendant to being a clinical-stage biotechnology company with limited capital resources could limit our ability to attract and retain personnel.
Mr. O'Callaghan and each of our key management and scientific personnel may terminate his or her employment at any time. If we lose any of our key management personnel, we may be unable to find replacements suitable to us or Otsuka, and our business would be harmed as a result. For instance, if Dr. Kubota and SBI are able to replace our board of directors, we may have difficulty retaining and motivating our key management and scientific staff, most of whom have indicated support for our current board of directors. In addition, if we cannot continue to attract and retain, on acceptable terms, the qualified personnel necessary for the continued development of our business, we may be unable to sustain our operations or grow.
If any products we develop become subject to third-party reimbursement practices, unfavorable pricing regulations or healthcare reform initiatives, our business could be harmed.
The availability and levels of reimbursement by governmental and other third-party payors of healthcare services affect the market for our potential products. These payors continually attempt to contain or reduce the costs of healthcare by challenging the prices charged for medical products and services. In the United States, we will need to obtain approvals for payment for our potential products from private insurers, including managed care organizations, and from the Medicare program. We cannot be sure that reimbursement will be available for any future product candidates and reimbursement amounts, if any, may reduce the demand for, or the price of, our future products.
Obtaining approvals from governmental and other third-party payors of healthcare services can be a time-consuming and expensive process. Because our product candidates are under development, we are unable at this time to determine the level or method of reimbursement. Our business would be materially adversely affected if we do not receive approval for reimbursement of our product candidates from Medicare or private insurers on a timely or satisfactory basis. The Medicare program can deny coverage of a particular drug on the basis of the drug not being “reasonable and necessary” for Medicare beneficiaries. Limitations on coverage could also be imposed at the local Medicare carrier level or by fiscal intermediaries. Our business could be materially adversely affected if the Medicare program, local Medicare carriers or fiscal intermediaries were to make such a determination and deny or limit the reimbursement of our potential products, including the procedures under which they are administered, if any. Our business could also be adversely affected if private insurers,
20
including managed care organizations, the Medicare program or other reimbursing bodies or payors limit the indications for which our potential products will be reimbursed.
There have been, and likely will continue to be, legislative and regulatory proposals in the United States and in some foreign jurisdictions directed at broadening the availability of health care and containing or lowering the cost of health care. We cannot predict the initiatives that may be adopted in the future. These legislative and/or regulatory changes may negatively impact the reimbursement for drug products, following approval, and thus affect our ability to sell our products profitably. The continuing efforts of government and other third-party payors of healthcare services to contain or reduce costs of health care may adversely affect:
• | the demand for any drug products for which we may obtain regulatory approval; |
• | our ability to set a price or achieve a rate of reimbursement that we believe is fair for our product candidates; |
• | our ability to generate revenue and achieve or maintain profitability; |
• | the level of taxes that we are required to pay; and |
• | our access to capital. |
We need to grow the size of our organization, and we may experience difficulties in managing this growth.
As our commercialization plans and strategies develop, we will need to expand the size of our employee base for managerial, operational, sales, marketing, financial, human resources and other functional areas. Competition for these employees is intense and we may be unable to hire additional qualified personnel in a timely manner and on reasonable terms. While we attempt to provide competitive compensation packages to attract and retain key personnel, many of our competitors are likely to have greater resources and more experience than we have, and we have had difficulty competing successfully for key personnel. Our ability to do so may be impeded by business disruptions and uncertainty caused by the unsolicited special meeting demand and director replacement proposals made by SBI. Any future growth would impose significant added responsibilities on members of management, including the need to recruit, hire, retain, motivate and integrate additional employees. Also, our management may have to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. Our future financial performance and our ability to commercialize our product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
We face the risk of product liability claims and may not be able to obtain insurance, and we may have exposure to significant contingent liabilities.
Our business exposes us to the risk of product liability claims that are inherent in the development, manufacturing, testing and marketing of drugs and related products. If the use of one or more of our products harms people, we may be subject to costly and damaging product liability claims. We have product liability insurance that covers our clinical trials up to a $10 million annual aggregate limit. We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for any of the products that we may develop. Insurance coverage is increasingly expensive and may not be available on reasonable terms, if at all. We may be unable to obtain or maintain adequate protection against potential liabilities. If we are unable to obtain insurance at acceptable cost or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which may materially and adversely affect our business and financial position.
We may need additional financing, which may be difficult to obtain. Our failure to obtain necessary financing or doing so on unattractive terms could adversely affect our development programs and other operations.
As of December 31, 2014, we had cash, cash equivalents and investments of $187.8 million and working capital of $99.2 million. We believe that our cash, cash equivalents and investments, together with Otsuka’s funding of our share of development costs under the Emixustat Agreement and interest income, will be sufficient to meet our working capital and capital expenditure requirements for at least the next twelve months. However, our future working capital and capital expenditure requirements will depend on many factors, including:
• | the success of our collaborations with Otsuka to develop and commercialize product candidates; |
• | the scope and results of our clinical trials; |
• | advancement of other product candidates into development; |
• | potential acquisition or licensing of other products or technologies; |
• | the timing of, and the costs involved in, obtaining regulatory approvals; |
• | the cost of manufacturing activities; |
21
• | the cost of commercialization activities, including product marketing, sales and distribution; |
• | the costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims and other patent-related costs, including litigation costs and the results of such litigation; and |
• | our ability to establish and maintain additional collaborative arrangements. |
Additional financing may not be available to us when we need it or it may not be available on favorable terms. If we are unable to obtain adequate financing on a timely basis, we may be required to significantly curtail one or more of our development, licensing or acquisition programs. We could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies, product candidates or products which we would otherwise pursue on our own. If we raise additional funds by issuing equity securities or securities convertible into equity, our then-existing shareholders will experience dilution and the terms of any new equity securities or securities convertible into equity may have preferences over our common stock.
We are an “emerging growth company,” and any decision on our part to comply only with certain reduced disclosure requirements applicable to emerging growth companies could make our common stock less attractive to investors.
We are an emerging growth company, as defined in the Jumpstart Our Business Startups Act, and, for as long as we continue to be an emerging growth company, we may choose to take advantage of certain exemptions from various reporting requirements applicable to other public companies, including, but not limited to, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years, although, if the market value of our common stock that is held by non-affiliates exceeds $700 million as of any June 30 before the end of that five-year period, we would cease to be an emerging growth company as of the following December 31. We cannot predict if investors will find our common stock less attractive if we choose to rely on these exemptions. If some investors find our common stock less attractive as a result of any choices to reduce future disclosure, there may be a less active trading market for our common stock and our stock price may be more volatile.
Regulatory Risks
We may not be able to obtain regulatory approval for any of the products resulting from our development efforts. Failure to obtain these approvals could materially harm our business.
All of the products we are developing or may develop in the future will require additional research or development. None of our product candidates have received regulatory approval for marketing in the United States and failure to receive such approvals on one or more of our product candidates could materially harm our business. We will be required to obtain an effective Investigational New Drug Application, or IND, prior to initiating new human clinical trials in the United States, and must submit a New Drug Application, or NDA, to obtain marketing approval prior to commercializing our products in the United States This process is expensive, highly uncertain and lengthy, often taking a number of years until a product is approved for marketing in the United States, if at all. For instance, based on guidance from the FDA in April 2014, we now expect to conduct at least one additional confirmatory Phase 3 clinical trial in patients with GA associated with dry AMD after we complete our current 24-month emixustat study. Approval policies or regulations may change and the FDA and other comparable foreign regulatory authorities have substantial discretion in the drug approval process, including the ability to delay, limit, or deny approval of a product candidate for many reasons, such as:
• | such authorities may disagree with the design or implementation of our, Otsuka’s, or any of our future development partners’ clinical trials; |
• | we, Otsuka, or any of our future development partners may be unable to demonstrate to the satisfaction of the FDA or other regulatory authorities that a product candidate is safe and effective for any indication or its clinical and other benefits outweigh any safety risk; |
• | such authorities may not accept clinical data from trials which are conducted at clinical facilities or in countries where the standard of care is potentially different from the United States; |
• | the results of clinical trials may not demonstrate the safety or efficacy required by such authorities for approval; |
• | such authorities may disagree with our interpretation of data from preclinical studies or clinical trials or the use of results from antibody studies that served as precursors to our current drug candidates; |
• | such authorities may find deficiencies in the manufacturing processes or facilities of third-party manufacturers with which we, Otsuka, or any of our future development partners contract for clinical and commercial supplies; and |
22
• | the approval policies or regulations of such authorities may significantly change in a manner rendering our, Otsuka’s, or any of our future development partners’ clinical data insufficient for approval. |
With respect to foreign markets, approval procedures vary among countries and, in addition to the aforementioned risks, can involve additional product testing, administrative review periods and agreements with pricing authorities. In addition, events raising questions about the safety of certain marketed pharmaceuticals may result in increased cautiousness by the FDA and comparable foreign regulatory authorities in reviewing new drugs based on safety, efficacy or other regulatory considerations and may result in significant delays in obtaining regulatory approvals. Any delay in obtaining, or inability to obtain, applicable regulatory approvals would prevent us, Otsuka, or any of our future development partners from commercializing our product candidates.
We may need to successfully address a number of technological challenges in order to complete the development of our product candidates. Success in early clinical trials does not mean that later clinical trials will be successful because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety or efficacy despite having progressed through initial clinical testing. Companies frequently suffer significant setbacks in advanced clinical trials, even after earlier clinical trials have shown promising results. In addition, our product candidates may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining regulatory approval or prevent or limit commercial use, which could have a material adverse effect on our business. Accordingly, there can be no assurance that the FDA and other regulatory authorities will approve any product that we develop.
The “fast track” designation may not actually lead to a faster regulatory review or approval process.
If a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to address unmet medical needs for this condition, the drug sponsor may apply for FDA “fast track” designation. The fast track classification does not apply to the product alone, but applies to the combination of the product and the specific indication or indications for which it is being studied. The FDA’s fast track program is designed to facilitate the clinical development and evaluation of the drug’s safety and efficacy for the fast track indication or indications. NDAs or marketing applications filed by sponsors of products in fast track development may qualify for expedited review under policies or procedures offered by the FDA, but the fast track designation does not assure such qualification. Although we have obtained a fast track designation from the FDA for emixustat for the treatment of dry macular degeneration (geographic atrophy), we may not experience a faster review or approval compared to conventional FDA process and we do not expect FDA approval to be granted for at least several years, if at all. Our fast track designation may be withdrawn by the FDA if it believes that the designation is no longer supported by data from our clinical development program. Our fast track designation does not guarantee that we will qualify for or be able to take advantage of the expedited review procedures. For instance, based on guidance from the FDA in April 2014, we plan to continue our current emixustat study through the original 24-month treatment duration without any access to interim results.
Our products could be subject to restrictions or withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements, or if we experience problems with our products, when and if any of them are approved.
Any product for which we obtain marketing approval will be subject to continual requirements, review and periodic inspections by the FDA and other regulatory bodies. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed. The approval may contain conditions or requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Later discovery of previously unknown problems with our products, manufacturer or manufacturing processes, or failure to comply with regulatory requirements, may result in:
• | voluntary or mandatory recall; |
• | withdrawal of the products from the market; |
• | restrictions on such products or manufacturing processes; |
• | fines; |
• | suspension of regulatory approvals; |
• | product seizure; and |
• | injunctions or the imposition of civil or criminal penalties. |
We may be slow to adapt, or we may never adapt, to changes in existing regulatory requirements or adoption of new regulatory requirements or policies.
23
Failure to obtain regulatory approval in foreign jurisdictions would prevent us from marketing our products outside of the United States.
We and Otsuka may market emixustat in our respective exclusive territories. To market emixustat in foreign jurisdictions, we or Otsuka must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies from country to country and can involve additional testing and documentation. The time required to obtain approval may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We and Otsuka may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market. The failure to obtain these approvals could materially adversely affect our business, financial condition and results of operations.
Risks Relating to Intellectual Property and Other Legal Matters
If our efforts to protect the proprietary nature of the intellectual property related to our products are not adequate, we may be unable to compete effectively in our markets.
The strength of our patents in the biotechnology and pharmaceutical fields involves complex legal and scientific questions and can be uncertain. In addition to the rights we have obtained from Otsuka relating to some of our product candidates, we rely upon intellectual property we own relating to our product candidates, including patents, patent applications and trade secrets. For a detailed discussion of our patent rights, see “Business—Intellectual Property.” Our patent applications may be challenged or fail to result in issued patents and our existing or future patents may be too narrow to prevent third parties from developing or designing around these patents. Further, there is no uniform, global policy regarding the subject matter and scope of claims allowable in pharmaceutical patents and the criteria applied by patent offices throughout the world to grant patents are not always predictable or uniform.
In general, there is no guarantee that the patents we file or in-license will be granted or that the patents we hold would be found valid and enforceable if challenged. Third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed or invalidated. Additionally, we may lose our rights to the patents and patent applications we license in the event of a breach or termination of the collaboration agreement. Manufacturers may also seek to obtain approval to sell generic versions of our product candidates prior to the expiration of the relevant licensed patents. If the sufficiency of the breadth or strength of protection provided by the patents we license with respect to our product candidates is threatened, it could dissuade others from collaborating with us to develop, and threaten our ability to commercialize, our other product candidates. Further, if we encounter delays in our clinical trials or our development activities are otherwise impeded, the period of time during which we could market our product candidates under patent protection would be reduced. Once the patent life has expired for a product, we may be subject to competition from generic pharmaceutical companies.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
We rely on trade secret protection and confidentiality agreements to protect certain proprietary know-how that is not patentable, for processes for which patents are difficult to enforce and for any other elements of our development processes with respect to emixustat and our other product candidates that involve proprietary know-how, information and technology that is not covered by patent applications. While we maintain physical security of our premises and physical and electronic security of our information technology systems, security measures may be breached and we may not have adequate remedies to protect the confidentiality of our proprietary information and know-how. Our systems and external backup measures may also be vulnerable to damage or breach due to natural disasters or other unexpected events.
While we require all of our employees, consultants, advisors and any third-parties who have access to our proprietary know-how, information and technology to enter into confidentiality agreements, we cannot be certain that this know-how, information and technology will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. These agreements may be terminated or breached, and we may not have adequate remedies for any such termination or breach. Furthermore, to the extent we develop our operations outside the United States, these agreements may not provide meaningful protection for our trade secrets and know-how in the event of unauthorized use or disclosure as the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States. Additionally, to the extent we license certain proprietary know-how, information or technologies from third-parties, including our collaboration partners, we also rely on such third-parties to adopt and enforce policies to protect such proprietary know-how, information and technology.
24
Third-party claims of intellectual property infringement may prevent or delay our discovery, development and commercialization efforts with respect to emixustat and our other product candidates.
Our commercial success depends, in part, on avoiding infringement of the patents and proprietary rights of third parties. Third parties may assert that we are employing their proprietary technology without authorization. Although we are not currently aware of any litigation or other proceedings or third-party claims of intellectual property infringement related to emixustat, the biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may allege that our activities infringe their patents or that we are employing their proprietary technology without authorization. We may not have identified all the patents, patent applications or published literature that affect our business either by blocking our ability to commercialize our product, by preventing the patentability of one or more aspects of our products or those of our licensors or by covering the same or similar technologies that may affect our ability to market our product. In addition, third parties may obtain patents in the future and claim that use of our product candidates or technologies infringes upon these patents. Furthermore, parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, obtain one or more licenses from third parties or pay royalties, or we may be enjoined from further developing or commercializing our product candidates and technologies.
We may become involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To the extent we rely on third parties for our proprietary rights, we will have limited control over the protection and defense of such rights. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings brought by the U.S. Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patents and patent applications or those of our collaborators or licensors. An unfavorable outcome could require us to cease using the technology or to attempt to license rights to it from the prevailing party. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distraction of our management and other employees.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
If we use hazardous or biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Our research and development activities involve the controlled use of potentially hazardous substances, including chemical and biological materials. In addition, our operations produce hazardous waste products. Federal, state and local laws and regulations in the United States govern the use, manufacture, storage, handling and disposal of hazardous materials. Although we believe that our procedures for use, handling, storing and disposing of these materials comply with legally prescribed standards, we may incur significant additional costs to comply with applicable laws in the future. Also, even if we are in compliance with applicable laws, we cannot completely eliminate the risk of contamination or injury resulting from hazardous materials and we may incur liability as a result of any such contamination or injury. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources. We do not have any insurance for liabilities arising from hazardous materials. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts, which could harm our business, operating results and financial condition.
Risks Related to Ownership of Our Common Stock
The price of our common stock may be volatile, and you could lose all or part of your investment.
25
The market prices for securities of biotechnology and pharmaceutical companies in general, and clinical-stage companies in particular, have been highly volatile and may continue to be highly volatile in the future. Volatility in the market price for a particular company’s stock has often been unrelated or disproportionate to the operating performance of that company. Market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance. The following factors, in addition to the other factors described in this “Risk Factors” section, may have a significant impact on the market price of our common stock:
• | the development status of our product candidates, including results of our clinical trials; |
• | market conditions or trends related to the biotechnology and pharmaceutical industries, or the market in general; |
• | announcements of technological innovations, new commercial products or other material events by our competitors or us; |
• | disputes or other developments concerning our proprietary rights; |
• | changes in, or failure to meet, securities analysts’ or investors’ expectations of our financial performance; |
• | additions or departures of key personnel, particularly Mr. O'Callaghan, or members of our board of directors; |
• | discussions of our business, products, financial performance, prospects or stock price by the financial and scientific press or securities analysts or lack of analyst coverage; |
• | public concern as to the safety of drugs and drug delivery techniques; |
• | regulatory developments in the United States, Japan and other foreign countries; |
• | changes in health care payment systems, including developments in price control legislation; or |
• | general economic and political factors, including wars, terrorism, natural disasters and political unrest. |
In the past, following periods of volatility in the market price of a particular company’s securities, securities class action litigation has often resulted. We may become subject to this type of litigation, which is often extremely expensive and diverts management’s attention, even if such litigation is ultimately concluded in a manner favorable to us.
As a public company, we are subject to additional financial and other reporting and corporate governance requirements that may be difficult for us to satisfy, will raise our costs and may divert resources and management attention from operating our business.
As a public company and particularly after we cease to be an “emerging growth company,” we have incurred and will incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, we are subject to reporting and corporate governance requirements, including the listing standards of the Mothers market of the Tokyo Stock Exchange ("TSE") and the provisions of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and the regulations promulgated thereunder, which impose significant new compliance obligations upon us. Our management and other personnel need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations have increased and will continue to increase our legal, accounting and financial compliance costs and have made and will continue to make some activities more time-consuming and costly. For example, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or to incur substantial costs to maintain the same or similar coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors or our board committees or as executive officers.
In order to continue to meet the listing standard of the Mothers market of the TSE, the adequacy of our internal control over financial reporting must be assessed by management each year. We are required to test operating effectiveness of our internal control over financial reporting on a periodic basis in accordance with Section 404 of the Sarbanes-Oxley Act. If we are unable to comply with Section 404, management might be unable to certify, and our independent registered public accounting firm might not be able to report on, the adequacy of our internal control over financial reporting. If we are unable to maintain adequate internal control over financial reporting, we might be unable to report our financial information on a timely basis and might suffer adverse regulatory consequences or violate TSE listing standards. There could also be a negative reaction in the financial markets due to a loss of investor confidence in us and the reliability of our financial statements.
Furthermore, investor perceptions of our company may suffer if deficiencies are found, and this could cause a decline in the market price of our stock. Irrespective of compliance with Section 404, any failure of our internal controls could have a material adverse effect on our stated results of operations and harm our reputation. If we are unable to implement these
26
changes effectively or efficiently, it could harm our operations, financial reporting or financial results and could result in an adverse opinion on internal control from our independent registered public accounting firm.
Any future issuance of equity or debt securities by us may adversely affect the rights or value of our previously issued common stock.
If we raise additional capital through the issuance of equity or securities convertible into equity, further dilution to our then existing shareholders will result and new investors could have rights superior to the rights of our existing shareholders. If we raise additional funds through the issuance of debt securities, these securities could have rights that are senior to holders of our common stock and could contain covenants that restrict our operations. In addition, the terms of future financings may restrict our ability to raise additional capital, which could delay or prevent the further development or commercialization of our product candidates.
Because the principal trading market for our shares is the Mothers market of the TSE, the corporate governance rules of the major U.S. stock exchanges will not apply to us. As a result, our governance practices may differ from those of a company listed on such U.S. exchanges.
Our governance practices need not comply with certain New York Stock Exchange and NASDAQ corporate governance standards, including:
• | the requirement that a majority of our board of directors consists of independent directors; |
• | the requirement that we have an audit committee that is composed entirely of independent directors with a written charter addressing the committee’s purpose and responsibilities; and |
• | the requirement that we have a compensation committee that is composed entirely of independent directors with a written charter addressing the committee’s purpose and responsibilities. |
There can be no assurance that we will voluntarily comply with any of the foregoing requirements. Accordingly, you may not have the same protections afforded to stockholders of companies that are subject to such corporate governance requirements.
A limited number of shareholders will have the ability to influence the outcome of director elections and other matters requiring shareholder approval.
As of March 19, 2015, Dr. Kubota, our largest shareholder, as an individual, and our directors and executive officers and their affiliates, as a group, beneficially own approximately 28.6% and 28.8% of our outstanding common stock, respectively. On January 28, 2015, we received a letter from SBI disclosing that SBI has granted Dr. Kubota the right to vote 7,752,425 shares held by SBI at any annual or special meeting of our shareholders at which members of our board are to be elected (see “Risk Factors - If Dr. Kubota and SBI are successful in their efforts to replace our board of directors, our business, including our collaborations with Otsuka, may be significantly harmed). Dr. Kubota's right to vote these shares expires on July 28, 2015. We have announced that we plan to hold the special shareholders meeting on May 1, 2015 (Pacific Daylight Time) at our headquarters and have set a record date for shareholders of record as March 19, 2015 (Pacific Daylight Time). If Dr. Kubota votes all shares over which he has voting authority in favor of the business to be brought before the special meeting, then our current members of our board of directors (other than Dr. Kubota) will be replaced by nominees proposed by SBI.
In addition, Dr. Kubota and SBI, or other large shareholders, could exert substantial influence over matters requiring approval by our shareholders, including electing directors and approving mergers, acquisitions or other business combination transactions. This concentration of ownership may discourage, delay or prevent a change of control of our company, which could deprive our shareholders of an opportunity to receive a premium for their stock as part of a sale of our company and might reduce our stock price. These actions may be taken even if they are opposed by our other shareholders.
Anti-takeover provisions under Washington law could make an acquisition of us more difficult and affect the market price of our common stock.
Because we are incorporated in the state of Washington, we are governed by the provisions of Chapter 23B.19 of the Washington Business Corporation Act, which prohibits certain business combinations between us and certain significant shareholders unless specified conditions are met. These provisions may also have the effect of delaying or preventing a change of control of our company, even if this change of control would benefit our shareholders.
We do not expect to pay dividends in the foreseeable future. As a result, you must rely on stock appreciation for any return on your investment.
27
We do not anticipate paying cash dividends on our common stock in the foreseeable future. Any payment of cash dividends will also depend on our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. Accordingly, you will have to rely on capital appreciation, if any, to earn a return on your investment in our common stock. Furthermore, we may in the future become subject to contractual restrictions on, or prohibitions against, the payment of dividends.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
Our headquarters are in Seattle, Washington, where we lease approximately 38,723 square feet of office space that we use for general and administrative purposes. This lease will expire on either November 30, 2021 or February 22, 2022. We lease approximately 17,488 square feet of laboratory and office space in Bothell, Washington that we use for laboratory, research and development and general and administrative purposes. This lease will expire on February 28, 2017. This lease also provides us an option to terminate the lease ten months early and an option to extend for an additional three-year term.
We believe our space is adequate for our current needs.
ITEM 3. LEGAL PROCEEDINGS
From time to time, we may be subject to legal proceedings and claims in the ordinary course of business. We are not currently a party to any material legal proceedings, and to our knowledge none is threatened. There can be no assurance that future legal proceedings arising in the ordinary course of business or otherwise will not have a material adverse effect on our financial position, results of operations or cash flows.
On January 28, 2015, we received a letter from SBI Holdings, Inc., or SBI, the parent company of several of our shareholders, demanding that we hold a special meeting of our shareholders for the purposes of removing our current board members, other than Dr. Kubota, and electing a slate of directors proposed by SBI. In connection with this demand, SBI granted Dr. Kubota an irrevocable proxy over the shares held collectively by SBI, giving Dr. Kubota voting authority over shares representing over 50% of our outstanding stock. On March 3, 2015, SBI and Dr. Kubota filed a lawsuit in Washington state court seeking an order compelling us to hold a special shareholder meeting on or before April 28, 2015, and to provide our shareholders written notice of that meeting as soon as practicable but no later than March 31, 2015.
On March 13, 2015, the Washington state superior court presiding over this lawsuit, issued an order requiring us to hold a special shareholders meeting no later than May 1, 2015 and to give notice of such a meeting as soon as practicable. In response to the hearing, we have announced that we plan to hold the special shareholders meeting on May 1, 2015 (Pacific Daylight Time) at our headquarters and have set a record date for shareholders of record as March 19, 2015 (Pacific Daylight Time).
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock has been listed on the Mothers Market of the Tokyo Stock Exchange under the code "4589" since February 13, 2014. Prior to that date, there was no public trading market for our common stock. Our IPO was priced at ¥1800, or approximately $17.65, per share on February 13, 2014. On March 19, 2015, the closing price for our common stock as reported on the Mothers Market of the Tokyo Stock Exchange was ¥683, or approximately $5.70, per share.
28
The following table sets forth, for the periods indicated, the reported high and low sales prices per share of our common stock as reported on the Mothers Board of the Tokyo Stock Exchange.
Year Ended December 31, 2014 | |||||||
High | Low | ||||||
First quarter (from February 13, 2014) | $ | 21.03 | $ | 13.31 | |||
Second quarter | $ | 15.07 | $ | 4.84 | |||
Third quarter | $ | 8.99 | $ | 6.11 | |||
Fourth quarter | $ | 7.41 | $ | 4.77 | |||
Holders of Common Stock
As of March 19, 2015, there were 29 holders of record of our common stock. As many of our shares of common stock are held by brokers and other institutions on behalf of shareholders, we are unable to estimate the total number of beneficial holders of our common stock represented by these record holders.
Dividends
We have not declared or paid a cash dividend on our capital stock and do not intend to pay cash dividends for the foreseeable future. All dividends are subject to the approval of our board of directors. Any future determinations to pay dividends on our capital stock would depend on our results of operations, our financial condition and liquidity requirements, restrictions that may be imposed by applicable law or our contracts, and any other factors that our board of directors in its sole discretion may consider relevant in declaring a dividend.
Stock Performance Graph
The following shall not be deemed "filed" for purposes of Section 18 of the Exchange Act, or otherwise subject to the liabilities under that Section and shall not be incorporated by reference into any of our other filings under the Exchange Act or the Securities Act, except to the extent we specifically incorporate it by reference into such filing.
The following graph compares the cumulative total return on our common stock with that of the Japan TOPIX Index and the NASDAQ Biotechnology Index from February 13, 2014 (the date our common stock commenced trading on the Mothers Market of the Tokyo Stock Exchange) and December 31, 2014. The chart assumes $100 was invested at the close of market on February 13, 2014 in our common stock, and assumes the reinvestment of any dividends. The stock price performance on the following graph is not necessarily indicative of future stock price performance.
The closing price of our common stock on December 30, 2014, the last trading day of fiscal 2014, was ¥710 or $5.89 per share.
29
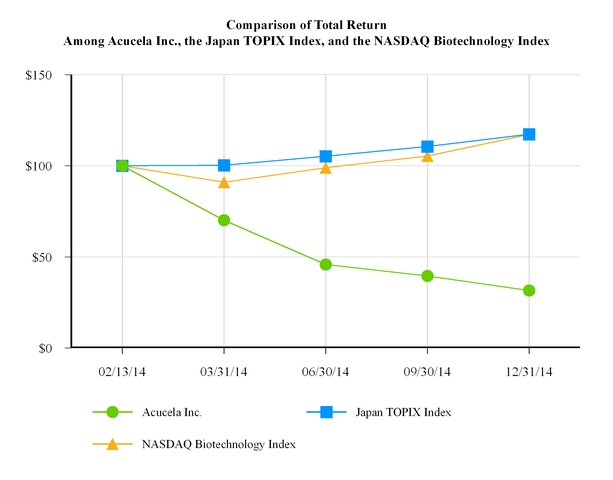
Base Period | Quarter Ending | ||||||||||||||
Company / Index | 2/13/2014 | 3/31/2014 | 6/30/2014 | 9/30/2014 | 12/31/2014 | ||||||||||
Acucela Inc. | $ | 100 | $ | 70.11 | $ | 45.81 | $ | 39.56 | $ | 31.49 | |||||
Japan TOPIX Index | 100 | 100.26 | 105.24 | 110.55 | 117.32 | ||||||||||
Nasdaq Biotechnology Index | 100 | 90.95 | 98.96 | 105.38 | 117.17 | ||||||||||
Recent Sales of Unregistered Securities
None.
Use of Proceeds
In February 2014, we closed our IPO of 9,200,000 shares of our common stock, at a price to the public of approximately $17.72 per share. The offer and sale of all of the shares in the IPO were registered under the Securities Act pursuant to a registration statement on Form S-1 (File No. 333-192900), which was declared effective by the SEC on February 3, 2014. The offering closed on February 13, 2014. We raised approximately $142.0 million from the IPO, net of underwriters’ discounts and commissions, and offering expenses. In addition, upon the closing of the IPO, $12.0 million of outstanding principal underlying convertible notes that we issued to affiliates of SBI Holdings, Inc. in May 2006 automatically converted into 3,636,365 shares of common stock, after an intermediate conversion into Series C preferred shares. The number of shares issued upon conversion was determined by dividing the principal of the note by $3.30.
As of December 31, 2014, we have used approximately $2.9 million of the net proceeds of our IPO for the following purposes:
• | approximately $1.0 million in research and development expenditures, including the development of emixustat in Europe as well as other VCM-based product candidates and |
• | approximately $1.9 million in general and administrative expenditures. |
30
To date, we have not used any proceeds of our IPO for sales and marketing expenditures. The amounts and timing of our actual expenditures will depend on numerous factors, including the success of our studies, changes in business strategy, the amount of cash used in or generated by our operations and the extent to which we exercise co-promotion rights under our collaboration agreements.
There has been no material change in the planned use of proceeds from our IPO as described in our final prospectus filed with the SEC on January 31, 2014 pursuant to Rule 424(b) under the Securities Act. Pending the uses described, we maintain the cash received in money market funds, as well as corporate debt securities, commercial paper, and certificates of deposit.
Share Repurchases
During the three months ended December 31, 2014, there were no purchases of shares of common stock made by, or on behalf of, the Company or any "affiliated purchaser," as defined by Rule 10b-18 of the Securities Exchange Act of 1934.
31
ITEM 6. SELECTED FINANCIAL DATA
You should read the selected financial data set forth below in conjunction with the information in Item 7, "Management's Discussion and Analysis of Financial Condition and Results of Operations," and our audited financial statements and the related notes thereto appearing elsewhere in this Annual Report on Form 10-K, which are incorporated herein by reference. Historical results are not necessarily indicative of future results.
Years Ended December 31, | |||||||||||
Statements of Operations Data: | 2014 | 2013 | 2012 | ||||||||
(in thousands, except per share data) | |||||||||||
Revenue from collaborations | $ | 35,396 | $ | 52,947 | $ | 46,424 | |||||
Expenses: | |||||||||||
Research and development | 25,582 | 36,405 | 31,604 | ||||||||
General and administrative | 10,002 | 9,548 | 7,787 | ||||||||
Total expenses | 35,584 | 45,953 | 39,391 | ||||||||
Income (loss) from operations | (188 | ) | 6,994 | 7,033 | |||||||
Other income (expense), net: | |||||||||||
Interest income | 519 | 122 | 27 | ||||||||
Interest expense | (15 | ) | (116 | ) | (138 | ) | |||||
Other income (expense), net | 37 | 182 | (97 | ) | |||||||
Income before income tax | 353 | 7,182 | 6,825 | ||||||||
Income tax expense | (2,359 | ) | (2,883 | ) | (2,647 | ) | |||||
Net income (loss) | $ | (2,006 | ) | $ | 4,299 | $ | 4,178 | ||||
Net income attributable to participating securities | — | 3,138 | 3,056 | ||||||||
Net income (loss) attributable to common shareholders | $ | (2,006 | ) | $ | 1,161 | $ | 1,122 | ||||
Net income (loss) per share attributable to common shareholders, basic | $ | (0.06 | ) | $ | 0.10 | $ | 0.09 | ||||
Weighted average shares used to compute net income (loss) per share attributable to common shareholders, basic | 32,869 | 11,964 | 11,901 | ||||||||
Net income (loss) attributable to common shareholders per share, diluted | $ | (0.06 | ) | $ | 0.09 | $ | 0.09 | ||||
Weighted average shares used to compute net income (loss) per share attributable to common shareholders, diluted | 32,869 | 12,355 | 12,158 | ||||||||
As of December 31, | |||||||
Balance Sheet Data: | 2014 | 2013 | |||||
(in thousands) | |||||||
Cash, cash equivalents and investments | $ | 187,819 | $ | 32,419 | |||
Working capital | 99,158 | 19,412 | |||||
Total assets | 196,966 | 54,048 | |||||
Contingently convertible debt, related party (including current portion) | — | 12,000 | |||||
Convertible preferred stock | — | 28,209 | |||||
Accumulated deficit | (5,466 | ) | (3,460 | ) | |||
Total shareholders’ equity | 184,363 | 31,124 | |||||
32
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This Report, including the “Management's Discussion and Analysis of Financial Condition and Results of Operations,” contains forward-looking statements regarding future events and our future results that are based on our current expectations, estimates, forecasts, and projections about our business, our results of operations, the industry in which we operate and the beliefs and assumptions of our management. Words such as “expects,” “anticipates,” “targets,” “goals,” “projects,” “would,” “could,” “intends,” “plans,” “believes,” “seeks” and “estimates,” variations of these words, and similar expressions are intended to identify those forward-looking statements. These forward-looking statements are only predictions and are subject to risks, uncertainties and assumptions that are difficult to predict. Therefore, actual results may differ materially and adversely from those expressed in any forward-looking statements. Factors that might cause or contribute to such differences include, but are not limited to, those discussed in this Report under the section entitled “Risk Factors” in Item 1A of Part I and elsewhere herein, and in other reports we file with the SEC. While forward-looking statements are based on reasonable expectations of our management at the time that they are made, you should not rely on them. We undertake no obligation to revise or update publicly any forward-looking statements for any reason, whether as a result of new information, future events or otherwise, except as may be required by law.
Overview
Acucela Inc., a Washington corporation, is a clinical-stage biotechnology company that specializes in discovering and developing novel drug candidates to potentially treat and slow the progression of sight-threatening ophthalmic diseases impacting millions of individuals worldwide. Emixustat hydrochloride ("emixustat"), our lead investigational visual cycle modulation ("VCM") compound, is designed to reduce retinal toxins and preserve the integrity of retinal tissue in patients suffering from geographic atrophy ("GA") associated with dry age-related macular degeneration ("AMD"). We intend to expand our ophthalmology pipeline by focusing on various degenerative retinal diseases, glaucoma, and dry eye.
Emixustat is currently being evaluated in a Phase 2b/3 study for GA associated with dry AMD. Currently, there are no U.S. Food and Drug Administration (FDA) approved therapies to treat any form of dry AMD, including GA associated with dry AMD. We are co-developing emixustat under our co-development and collaboration agreement, or the Emixustat Agreement, with Otsuka Pharmaceutical Co., Ltd., or Otsuka. Pursuant to the Emixustat Agreement, we and Otsuka have agreed to develop and commercialize emixustat and/or other back-up compounds for the treatment of dry AMD and other potential ophthalmic indications that the parties agree to pursue under the terms of the agreement.
We completed our initial public offering ("IPO") in February 2014. We manage our operations and allocate resources as a single reporting segment. Financial information about our segment and geographic areas is incorporated herein by reference to Note 1 of Notes to Financial Statements in this Annual Report. In addition, financial information regarding our operations, assets and liabilities, including our total net revenue and net income (loss) for the years ended December 31, 2014, 2013 and 2012 and our total assets as of December 31, 2014 and 2013, is included in our Financial Statements in this Annual Report.
Recent Developments
2015 Strategic Plan
In addition to our continued development of emixustat for the treatment of GA associated with dry AMD, in late 2014, we launched a new strategic plan focused on leveraging our internal research and development efforts, our expertise in VCM and pursuing external partnerships, in-licensing and M&A opportunities to develop certain of our proprietary preclinical compounds and compounds we in-license to create therapies for glaucoma, dry eye and various other retinal diseases. We believe that a significant market opportunity exists for these potential therapies, based on the large and growing worldwide ophthalmic pharmaceutical market and our belief that currently available therapies for these indications are inadequate. We anticipate that these therapies will be developed independently, and our development expenditures on these programs will not be funded by collaborative partners. As a consequence, we expect that our total research and development expenses will increase and that we will incur net losses from our operating activities for the upcoming fiscal year.
Our strategy includes continued collaboration with Otsuka to successfully develop emixustat; initiating out-licensing efforts under the Emixustat Agreement, primarily for Europe; leveraging our expertise in VCM by evaluating the potential to develop emixustat for additional indications such as DR or DME; and continued expansion of our ophthalmic product pipeline through internal research, M&A and additional partnering or in-licensing opportunities.
Management Changes
33
On December 22, 2014, we announced that Brian O'Callaghan was appointed our Chief Executive Officer (CEO), effective January 1, 2015. On March 24, 2015, Dewey H. Blocker, Jr. was appointed our Vice President Finance, Treasurer and Secretary. Mr. Blocker will serve as our interim principal accounting officer and interim principal financial officer. In connection with Mr. Blocker's appointment, Mr. O'Callaghan resigned the positions of Interim Chief Financial Officer, Treasurer and Secretary. Mr. O'Callaghan continues to serve as our CEO and President.
Special Shareholders Meeting
On January 28, 2015, we received a letter from SBI Holdings, Inc. (SBI), the parent company of several of our shareholders, demanding that we hold a special meeting of our shareholders for the purposes of removing our current board members, other than Dr. Kubota, and electing a slate of directors proposed by SBI. In connection with this demand, SBI granted Dr. Kubota an irrevocable proxy over the shares held collectively by SBI, giving Dr. Kubota voting authority over shares representing over 50% of our outstanding stock. On March 3, 2015, SBI and Dr. Kubota filed a lawsuit in Washington state court seeking an order compelling us to hold a special shareholder meeting on or before April 28, 2015, and to provide our shareholders written notice of that meeting as soon as practicable but no later than March 31, 2015. On March 13, 2015, the Washington state superior court presiding over this lawsuit, issued an order requiring us to hold a special shareholders meeting no later than May 1, 2015 and to give notice of such a meeting as soon as practicable. Thereafter, we announced that we plan to hold the special shareholders meeting on May 1, 2015 (Pacific Daylight Time) at our headquarters and have set a record date for shareholders of record as March 19, 2015 (Pacific Daylight Time). If Dr. Kubota votes all shares over which he has voting authority in favor of the business to be brought before the special meeting, then our current members of our board of directors (other than Dr. Kubota) will be replaced as nominees proposed by SBI.
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations are based on our financial statements which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of our financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues, costs and expenses and related disclosures. On an ongoing basis, we evaluate these estimates and judgments, including those described below. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results and experiences may differ materially from these estimates. To the extent that there are differences between our estimates and actual results, our future financial statement presentation, financial condition, results of operations and cash flows will be affected. We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
Revenue Recognition. One of our business strategies is to enter into collaboration agreements with pharmaceutical companies for the development and commercialization of product candidates. The terms of the agreements may include nonrefundable license fees, funding of research and development activities, payments based upon achievement of development milestones, payments based upon achievement of regulatory and revenue milestones, and product sales or royalties on product sales. We recognize revenue when four basic criteria have been met: (a) persuasive evidence of an arrangement exists; (b) delivery has occurred or services rendered; (c) the fee is fixed or determinable; and (d) collectability is reasonably assured. Amounts received prior to satisfying these criteria, such as advanced payments received for our share of the funding of activities under the Emixustat Agreement, are recorded as deferred revenue.
Multiple Element Arrangements. Our collaboration agreements are multiple element arrangements which were analyzed to identify the deliverables included in the agreements and determine if the deliverables qualify as separate units of accounting. Deliverables are considered a separate unit of accounting when all of the following criteria are met: (i) the delivered item has value to the customer on a standalone basis and (ii) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially in our control. There are no rights of return in any of our collaboration agreements.
Arrangement consideration is allocated to the separate units of accounting based on their relative selling price. We follow a hierarchy to determine the selling price for each unit of accounting, determining first if there is vendor-specific objective evidence, or VSOE, of fair value (the price at which the goods or services are regularly sold by us on a standalone basis). If VSOE of fair value is not available, third-party evidence, or TPE, of vendors selling similar goods or services to similarly situated customers on a standalone basis is used to establish fair value. If neither VSOE nor TPE of fair value exists, we use our best estimate of the selling price, or BESP, for that unit of accounting. Our BESP represents the price at which we would transact if the unit of accounting were sold by us regularly on a standalone basis. In accordance with our hierarchical
34
approach, we determined that neither VSOE nor TPE were available and relied on BESP to establish fair value for the various units of accounting under our collaboration agreements.
Once the selling price for each unit of accounting has been established, the consideration received is allocated among the units of accounting based on their relative selling price, and the applicable revenue recognition criteria are applied to each of the separate units. The amount of arrangement consideration allocable to a delivered item that is a separate unit of accounting is limited to arrangement consideration that is fixed or determinable. Payments that are contingent upon the occurrence of future events that are not exclusively within our control are excluded from the allocable arrangement consideration until the contingency is resolved
When we have continuing performance obligations, revenue is recognized using one of two methods, a proportional performance model or a time-based method. When we are able to estimate the total amount of services under a unit of accounting and such performance obligations are provided on a best-efforts basis, revenue is recognized using a proportional performance model. Costs incurred to date compared to total expected costs are used to determine proportional performance, as this is considered to be representative of the delivery of outputs. Changes in estimates of total expected costs are accounted for prospectively as a change in estimates. When we cannot reasonably estimate the total amount of service that is to be performed, but can reasonably estimate when the performance obligation ceases or becomes inconsequential, a time-based method is used to recognize revenue. Under the time-based method, revenue is recognized ratably over the estimated performance period of the unit of accounting, but not before the removal of any contingencies. If we cannot reasonably estimate when our performance obligation either ceases or becomes inconsequential and perfunctory, then revenue is deferred until the time at which we can reasonably estimate when the performance obligation ceases or becomes inconsequential and perfunctory. Revenue is then recognized over the remaining estimated period of performance. Significant judgment is required in determining the level of effort required and the period over which we are expected to complete our performance obligations under each unit of accounting.
Substantive Milestone Payments. Our collaboration agreements contain substantive milestones. A substantive milestone is defined as an event that meets the following conditions: (i) there is substantive uncertainty on the date the arrangement is entered into about whether the event will be achieved; (ii) achievement of the event is based in whole, or in part, on either our performance or a specific outcome resulting from our performance; and (iii) achievement of the event results in additional payment due to us. For a milestone to be considered substantive, the payment associated with its achievement must have all of the following characteristics: (i) relate solely to our past performance; (ii) be reasonable, relative to all of the deliverables and payment terms in the arrangement; and (iii) be commensurate with either our effort required to achieve the milestone or the enhanced value of the delivered item(s) as a result of the milestone achievement.
Substantive milestone payments are recognized upon achievement of the milestone only if all of the previous conditions are met and the milestone payments are nonrefundable. Determination as to whether a payment meets the aforementioned conditions involves our judgment. We have evaluated the nature of our arrangements and have elected to make a policy election to apply the milestone method where appropriate for our arrangements.
If any of the aforementioned conditions are not met, the resulting payment would not be considered a substantive milestone, and therefore, the resulting payment would be determined to be part of the allocable arrangement consideration and would be recognized as revenue as such performance obligations are performed under either the proportional performance or time-based methods, as applicable, and in accordance with the policies as described above.
Funded Development. As the Emixustat Agreement contains elements of funded development, we evaluated the agreement to determine if our obligation to Otsuka should be accounted for as a liability to repay a loan or as an obligation to perform contractual services. To conclude that a liability to repay a loan does not exist, the transfer of the financial risk involved with research and development from us to Otsuka must be substantive and genuine. We determined that our obligation to Otsuka should be accounted for as an obligation to perform contractual services because repayment depends solely on the results of development having future economic benefit. Consequently, amounts received from Otsuka for our share of development costs under the arrangement are recognized as revenue. Through December 31, 2014, we had recognized revenue of approximately $49.7 million, which amounts are contingently repayable as described above. As of December 31, 2014, the contingently repayable funding has accrued $2.5 million of interest, which is also contingently repayable under the same terms as the funding.
Research and Development and Clinical Trial Accounting. Research and development expenses include salaries, fees paid to external service providers and contract research organizations to conduct research and development activities, laboratory supplies, license fees, consulting fees and travel. Research and development costs are expensed as incurred. Certain indirect expenses are allocated between research and development, and general and administrative expenses as appropriate.
35
We record prepaid assets and accrued liabilities related to our clinical trials, associated with contract research organizations, clinical trial investigators and other vendors, based upon amounts paid and the estimated amount of work completed on each clinical trial or other activity. The financial terms of agreements vary from vendor to vendor and may result in uneven payment flows. As such, if we have advanced funds exceeding our estimate of the work completed, we record a prepaid asset. If our estimate of the work completed exceeds the amount paid, an accrued liability is recorded. All such costs are charged to research and development expenses based on these estimates or on actual costs incurred. Our estimates may or may not match the actual services performed by the organizations as determined by patient enrollment levels and related activities. We monitor patient enrollment levels and related activities to the extent possible through internal reviews, correspondence and discussions with our contract research organizations and review of contractual terms. However, if we have incomplete or inaccurate information, we may underestimate or overestimate activity levels associated with various clinical trials at a given point in time. In this event, we could record significant research and development expenses in future periods when the actual level of activities becomes known. To date, we have not experienced material changes in these estimates. Additionally, we do not expect material adjustments to research and development expenses to result from changes in the nature and level of clinical trial activity and related expenses that are currently subject to estimation. In the future, as we expand our clinical trial activities, we expect to have increased levels of research and development costs that will be subject to estimation.
Income Taxes. We recognize deferred tax assets and liabilities for the expected future tax consequences of events that have already been recognized in our financial statements or tax returns. Excess tax benefits associated with stock option exercises and other equity awards are credited to shareholders' equity. Deferred tax liabilities and assets are based on the difference between financial statement carrying amounts and the tax basis of assets and liabilities, operating loss, and tax credit carryforwards and are measured using enacted tax rates expected to be in effect in the years the differences or carryforwards are anticipated to be recovered or settled. A valuation allowance is established when we believe that it is more likely than not that benefits of the deferred tax assets will not be realized. We recorded a partial valuation allowance of $2.3 million against our deferred tax assets due to expected future losses as a result of our new strategic plan. The Company had no uncertain tax positions as of December 31, 2014, 2013, or 2012, and we do not anticipate any significant changes in our unrecognized tax benefits over the next twelve months. Interest and penalties, if applicable, are recorded as tax expense.
Tax Loss Carryforwards. The tax benefit associated with the utilization of loss carryforwards was $0.6 million, $0.8 million, and $3.0 million in the years ended December 31, 2014, 2013, and 2012, As of December 31, 2014 and 2013, we had research and development tax credit carryforwards of $1.1 million and $0.7 million, respectively. The carryforwards and credits are available to offset future tax liabilities. The research and development (R&D) tax credits begin to expire in 2027. The annual limitation may result in the expiration of carryforwards before they can be utilized.
Stock-Based Compensation. Stock-based compensation cost is estimated at the grant date based on the award’s fair value and is recognized on a straight line basis as expense, less estimated forfeitures, over the requisite service period, which is generally the vesting period. The fair value of stock options under our equity-based incentive plans (the "Equity Plans") are calculated using the Black-Scholes-Merton (“BSM”) option pricing model. The BSM model requires various judgmental assumptions regarding volatility and expected option life. If any of the assumptions used in the BSM model change significantly, stock-based compensation expense for new awards may differ materially from that recorded for existing awards.
Equity awards to nonexecutive employees generally vest and become exercisable over a four-year period. Equity awards to executives generally vest and become exercisable over a five-year period. In 2014, we began granting restricted stock unit awards to employees. Restricted stock unit awards generally vest and are expensed over a four-year period.
Description of Operating Accounts
Revenue from collaborations to date has been generated primarily by our research and development activities based on the collaboration and license agreements with Otsuka. Our revenue primarily consists of reimbursement from Otsuka for our fees paid to external service providers in connection with the collaboration agreements, Otsuka’s funding of our portion of development costs under the Emixustat Agreement, payment from Otsuka for our development services provided by our personnel, for services rendered as part of our collaborative research program, an initial license fee as part of the Emixustat Agreement, and milestone payments. We expect any revenue we generate will fluctuate from quarter to quarter as a result of the nature and timing of the compounds under development.
Research and development expenses incurred to date have substantially focused on developing therapies for sight-threatening diseases. Since entering into our collaboration agreements with Otsuka, our efforts have been principally directed towards fulfilling our commitments thereunder. We recognize research and development expenses as they are incurred and these costs primarily consist of fees paid to consultants, contract research organizations, independent monitors of our clinical trials, and parties acquiring and evaluating data in conjunction with our clinical trials, including all related fees such as investigator grants, patient screening, lab work and data compilation and statistical analysis; costs related to production of clinical materials, including fees paid to contract manufacturers; costs related to compliance with FDA regulatory requirements;
36
consulting fees paid to third parties involved in research and development activities; compensation and related expenses for personnel in research and development functions; and an allocated portion of certain general and administrative costs. We expect our research and development expenses to increase in absolute dollars as we continue to develop our product candidates and as we continue with our discovery research activities.
General and administrative expenses consist primarily of compensation for employees in executive and administrative functions, including finance, accounting, and human resources. Other significant costs include facilities costs and professional fees for accounting and legal services, including legal services associated with obtaining and maintaining patents. We expect other general and administrative expenses to increase in absolute dollars as we incur additional costs related to the growth of our business, including our intellectual property portfolio, and as we assume the reporting requirements and compliance obligations of a public company.
Interest income consists primarily of interest earned on our cash, cash equivalents and short-term and long-term investments.
Interest expense consisted primarily of interest expense incurred on our contingently convertible debt.
Other income (expense) consists primarily of foreign exchange gains/losses incurred on transactions occurring in Japan, gain or losses from the disposal of fixed assets or other miscellaneous items.
Income tax benefit (expense) consists primarily of the provision of a partial valuation allowance related to deferred tax assets for which we do not anticipate future realization, and taxes incurred on income, partially offset by the utilization of our carryforwards including our NOL or R&D tax credit, less taxes incurred on income.
Results of Operations
Year ended December 31, 2014 compared to the year ended December 31, 2013
REVENUE FROM COLLABORATIONS
Our clinical programs consist of the following categories: Proprietary, which includes emixustat (program under the Emixustat Agreement); and In-Licensed, which includes rebamipide, Otsuka's proprietary compound for treatment of dry eye, which was the subject of a now-terminated agreement between us and Otsuka referred to as the Rebamipide Agreement and OPA-6566, Otsuka's proprietary compound for treatment of glaucoma, which was the subject of a development and collaboration agreement with Otsuka, referred to as the Glaucoma Agreement.
The following table presents revenues for clinical programs (in thousands, except percentages):
Years Ended December 31, | 2013 to 2014 $ Change | 2013 to 2014 % Change | ||||||||||||
2014 | 2013 | |||||||||||||
Emixustat | $ | 35,364 | $ | 39,186 | $ | (3,822 | ) | (9.8 | )% | |||||
Rebamipide | 25 | 12,271 | (12,246 | ) | (99.8 | )% | ||||||||
OPA-6566 | 7 | 1,490 | (1,483 | ) | (99.5 | )% | ||||||||
Total | $ | 35,396 | $ | 52,947 | $ | (17,551 | ) | (33.1 | )% | |||||
Proprietary
Emixustat. Revenues from clinical programs under the Emixustat Agreement decreased by $3.8 million for the year ended December 31, 2014, or 9.8%, as compared to the prior year. The decrease in the year ended December 31, 2014 was due to the receipt of a $5.0 million milestone payment associated with the initiation of the Phase 2b/3 clinical trial in the prior year. We did not receive a milestone payment during 2014.
In-Licensed
Rebamipide. Revenues under the Rebamipide Agreement decreased by $12.2 million for the year ended December 31, 2014, or 99.8%, as compared to the prior year. In September 2013, Otsuka terminated the Rebamipide Agreement for the reason that the primary end points were not met in the Phase 3 clinical trial.
OPA-6566. Revenues under the Glaucoma Agreement decreased by $1.5 million for the year ended December 31, 2014, or 99.5%, as compared to prior year, due to the completion of the Phase 1/2 clinical study.
37
We do not expect to generate significant revenue from collaborations related to in-licensed clinical programs with Otsuka for the foreseeable future.
OPERATING EXPENSES
Research and development
We prepare financial information related to our clinical programs and internal research program for various purposes. Our clinical programs consist of the following categories: Proprietary, which includes emixustat (program under the Emixustat Agreement); In-Licensed, which includes rebamipide (program under the terminated Rebamipide Agreement) and OPA-6566 (program under the Glaucoma Agreement); and Internal Research, which consists of costs and expenses associated with our internal research activities related primarily to our VCM compounds.
The following table presents our research and development expenses for clinical programs and internal research programs: (in thousands, except percentages):
Years Ended December 31, | 2013 to 2014 $ Change | 2013 to 2014 % Change | ||||||||||||
2014 | 2013 | |||||||||||||
Emixustat | $ | 24,509 | $ | 25,537 | $ | (1,028 | ) | (4.0 | )% | |||||
Rebamipide | 15 | 7,081 | (7,066 | ) | (99.8 | )% | ||||||||
OPA-6566 | 8 | 1,232 | (1,224 | ) | (99.4 | )% | ||||||||
Internal Research | 1,050 | 2,555 | (1,505 | ) | (58.9 | )% | ||||||||
Total | $ | 25,582 | $ | 36,405 | $ | (10,823 | ) | (29.7 | )% | |||||
Proprietary
Emixustat. Research and development expense related to clinical programs under the Emixustat Agreement decreased $1.0 million for the year ended December 31, 2014, or 4.0%, as compared to the prior year. The decrease was due to timing and fewer research and development activities in the year ended December 31, 2014.
In-Licensed
Rebamipide. Research and development expense under the Rebamipide Agreement decreased $7.1 million in the year ended December 31, 2014, or 99.8%, as compared to the prior year, due to the termination of the agreement in September 2013. In September 2013, Otsuka terminated the Rebamipide Agreement for the reason that the primary end points were not met in the Phase 3 clinical trial.
OPA-6566. Research and development under the Glaucoma Agreement decreased $1.2 million in the year ended December 31, 2014, or 99.4%, compared to prior year, due to completion of the Phase 1/2 study for OPA-6566.
We do not expect to incur significant research and development expenses related to in-licensed clinical programs with Otsuka for the foreseeable future.
Internal Research. Research and development expense under our internal research activities for the year ended December 31, 2014 decreased $1.5 million, or 58.9%, compared to the prior year due to our strategic restructuring. In October 2013, we announced a plan to reduce expenses, including a workforce reduction, as a result of the termination of the Rebamipide Agreement. The plan resulted in a reduction in force of approximately 35% of our total workforce, or approximately 30 employees, effective January 1, 2014.
In addition to our continued development of emixustat for the treatment of GA associated with dry AMD, in late 2014, we launched a new strategic plan focused on leveraging our internal research and development efforts, our expertise in VCM and pursuing external partnerships, in-licensing and M&A opportunities to develop certain of our proprietary preclinical compounds and compounds we in-license to create therapies for glaucoma, dry eye and various other retinal diseases. We believe that a significant market opportunity exists for these potential therapies, based on the large and growing worldwide ophthalmic pharmaceutical market and our belief that currently available therapies for these indications are inadequate. We anticipate that these therapies will be developed independently, and our development expenditures on these programs will not be funded by collaborative partners. As a consequence, we expect that our total research and development expenses will increase and that we will incur net losses from our operating activities for the upcoming fiscal year.
General and administrative
38
General and administrative expense increased $0.5 million from $9.5 million to $10.0 million in the year ended December 31, 2014, or 4.8%, related to increased corporate legal and accounting costs related to being a public company. We expect general and administrative expenses to increase in 2015 as we invest in infrastructure and due to planned business development activities.
Interest income
Interest income for the year ended December 31, 2014 increased $0.4 million from $0.1 million to $0.5 million in the year ended December 31, 2014, compared to prior year, due to interest earned on proceeds from our IPO.
Interest expense
Interest expense for the year ended December 31, 2014 decreased $0.1 million from $0.1 million to $0 in the year ended December 31, 2014, compared to prior year, due to the conversion of contingently convertible debt, which converted in connection with our IPO.
Income tax expense
Income tax expense for the year ended December 31, 2014 totaled approximately $2.4 million. Income tax expense was approximately $2.9 million for the year ended December 31, 2013. This represented effective tax rates of 667.7% and 39.8% in 2014 and 2013, respectively. The difference between the U.S. federal statutory rate of 34% and our effective tax rates in 2014 was due primarily to the provision of a partial valuation allowance related to deferred tax assets for which we do not anticipate future realization and permanent differences in book and tax earnings for stock options, meals and entertainment, and other miscellaneous items. We recorded a partial valuation allowance of $2.3 million against our deferred tax assets due to expected future losses as a result of our new strategic plan. In 2013, there were no individual items representing a greater than 5% impact on the effective tax rate.
Year ended December 31, 2013 compared to the year ended December 31, 2012
REVENUE FROM COLLABORATIONS
Revenue from collaborations totaled approximately $52.9 million in the year ended December 31, 2013, representing an increase of approximately $6.5 million, or 14.1%, as compared to the prior year. The increase in revenues was due primarily to a $19.9 million increase in revenues from development services under the Emixustat Agreement related to increased activity in the Phase 2b/3 clinical trial, including a milestone payment of $5.0 million associated with the initiation of the Phase 2b/3 clinical trial, partially offset by a $6.6 million decrease in revenue under the Glaucoma Agreement due to a completion in October 2012 of the Phase 1/2 clinical trial and a $6.7 million decrease in revenue under the Rebamipide Agreement due to completion in May 2013 of the Phase 3 clinical trial.
The following table presents revenues for clinical and other programs (in thousands, except percentages):
Years Ended December 31, | 2012 to 2013 $ Change | 2012 to 2013 % Change | ||||||||||||
2013 | 2012 | |||||||||||||
Emixustat | $ | 39,186 | $ | 19,328 | $ | 19,858 | 102.7 | % | ||||||
Rebamipide | 12,271 | 18,988 | (6,717 | ) | (35.4 | )% | ||||||||
OPA-6566 | 1,490 | 8,108 | (6,618 | ) | (81.6 | )% | ||||||||
Total | $ | 52,947 | $ | 46,424 | $ | 6,523 | 14.1 | % | ||||||
Proprietary
Emixustat. Revenues from clinical programs under the Emixustat Agreement increased by $19.9 million for the year ended December 31, 2013, or 102.7%, as compared to the prior year. The increase was due primarily to increased activity related to the preparation, initiation, and execution of the Phase 2b/3 clinical trial, as well as a milestone payment of $5.0 million associated with the initiation of that trial. There was no milestone received in 2012.
In-Licensed
Rebamipide. Revenues under the Rebamipide Agreement decreased due to termination of the Rebamipide Agreement in September 2013, partially offset by the recognition of $2.0 million of deferred revenue. In September 2013, Otsuka terminated the Rebamipide Agreement for the reason that the primary end points were not met in the Phase 3 clinical
39
trial. The expiration of refund provisions, resulting from the termination of this agreement, triggered the recognition of $2.0 million of the deferred revenue.
OPA-6566. The decrease in revenues under the Glaucoma Agreement was due to the completion of the Phase 1/2 clinical study in October 2012.
OPERATING EXPENSES
Research and development
Research and development expense for the year ended December 31, 2013 totaled approximately $36.4 million, representing an increase of approximately $4.8 million, or 15.2%, as compared to the prior year. The increase was due to higher research and development expense of $11.7 million associated with emixustat due to the initiation and conduct of the Phase 2b/3 clinical trial. The increase was partially offset by a decrease of $4.3 million in research and development expense, related to clinical programs, under the Glaucoma Agreement due to the completion of the Phase 1/2 study for OPA-6566 and a decrease of $2.7 million in research and development expense due to the termination of the Rebamipide Agreement in September 2013. The expenses related to internal research were materially consistent with the prior period.
The following table presents our research and development expenses for clinical programs and internal research programs were as follows (in thousands, except percentages):
Years Ended December 31, | 2012 to 2013 $ Change | 2012 to 2013 % Change | ||||||||||||
2013 | 2012 | |||||||||||||
Emixustat | $ | 25,537 | $ | 13,707 | $ | 11,830 | 86.3 | % | ||||||
Rebamipide | 7,081 | 9,823 | (2,742 | ) | (27.9 | )% | ||||||||
OPA-6566 | 1,232 | 5,559 | (4,327 | ) | (77.8 | )% | ||||||||
Internal Research | 2,555 | 2,515 | 40 | 1.6 | % | |||||||||
Total | $ | 36,405 | $ | 31,604 | $ | 4,801 | 15.2 | % | ||||||
Proprietary
Emixustat. Research and development expense related to clinical programs under the Emixustat Agreement increased $11.8 million for the year ended December 31, 2013, or 86.3%, as compared to the prior year. The increase was due primarily to development activity associated with the initiation and execution of the Phase 2b/3 study for emixustat. We also spent $0.1 million on development of emixustat outside of the Emixustat Agreement.
In-Licensed
Rebamipide. Research and development expense related to clinical programs under the Rebamipide Agreement decreased $2.7 million in the year ended December 31, 2013, or 27.9%, as compared to the prior year, due to the termination of the Rebamipide Agreement in September 2013. In September 2013, Otsuka terminated the Rebamipide Agreement for the reason that the primary end points were not met in the Phase 3 clinical trial.
OPA-6566. Research and development expense related to clinical programs under the Glaucoma Agreement decreased $4.3 million in the year ended December 31, 2013, or 77.8%, as compared to the prior year, due to the completion in October 2012 of the Phase 1/2 study for OPA-6566.
Internal Research. Research and development expense under our discovery research activities for the year ended December 31, 2013 was materially consistent with the same period for the prior year.
General and administrative
General and administrative expenses for the year ended December 31, 2013 totaled approximately $9.5 million, representing an increase of approximately $1.8 million, or 22.6%, as compared to the prior year. The increase was due primarily to stock and related compensation granted to our CEO pursuant to his employment agreement, with a total expense of $0.8 million and increased recruiting and related expenses. Compensation and benefit expense, including stock-based compensation expenses of $0.7 million, also increased due to higher headcount.
Income tax expense
40
Income tax expense for the year ended December 31, 2013 totaled approximately $2.9 million. Income tax expense was approximately $2.6 million for the year ended December 31, 2012. This represented effective tax rates of 39.8% and 38.8% in 2013 and 2012, respectively. The difference between the U.S. federal statutory rate of 34% and our effective tax rate was due primarily to differences in book and tax earnings, with no individual items representing a greater than 5% impact on effective tax rate.
Liquidity and Capital Resources
Prior to our IPO, we funded our operations primarily from the issuance of convertible preferred stock and contingently convertible debt and, since 2009, from cash generated from operations. Historically, our need for cash has been limited due to Otsuka’s funding of development activities and our receipt of milestone payments from Otsuka.
In addition to our continued development of emixustat for the treatment of GA associated with dry AMD, in late 2014, we launched a new strategic plan focused on leveraging our internal research and development efforts, our expertise in VCM and pursuing external partnerships, in-licensing and M&A opportunities to develop certain of our proprietary preclinical compounds and compounds we in-license to create therapies for glaucoma, dry eye and various other retinal diseases. We believe that a significant market opportunity exists for these potential therapies, based on the large and growing worldwide ophthalmic pharmaceutical market and our belief that currently available therapies for these indications are inadequate. We anticipate that these therapies will be developed independently, and our development expenditures on these programs will not be funded by collaborative partners. As a consequence, we expect that our total research and development expenses will increase and that we will incur net losses from our operating activities for the upcoming fiscal year.
On February 13, 2014, upon the closing of our initial public offering, we issued and sold 9,200,000 shares of common stock at approximately $17.72 per share and received net proceeds of $142.0 million (after underwriting discounts and commissions and offering costs). As a result of the IPO, all preferred stock and contingently convertible debt converted to common stock.
As of December 31, 2014 and 2013, we had cash, cash equivalents and investments of $187.8 million and $32.4 million, respectively. Cash and cash equivalents include all short-term, highly liquid investments with an original maturity of three months or less at the date of purchase. As of December 31, 2014, cash equivalents consist of money market funds and certificates of deposit. Short-term investments as of December 31, 2014 and 2013 were comprised of commercial paper, corporate debt securities, and certificates of deposit. Investments with maturities between three months and one year at the date of purchase are classified as short-term investments. Amounts on deposit with third-party financial institutions may exceed the applicable Federal Deposit Insurance Corporation and the Securities Investor Protection Corporation insurance limits, as applicable.
The following table shows a summary of our cash flows for the years ended December 31, 2014, 2013, and 2012 (in thousands):
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cash flows provided by operating activities | $ | 8,844 | $ | 7,246 | $ | 11,246 | |||||
Cash flows used in investing activities | (152,334 | ) | (6,581 | ) | (3,743 | ) | |||||
Cash flows provided by (used in) financing activities | 148,274 | (3,310 | ) | (624 | ) | ||||||
Cash Flows From Operating Activities
Operating activities generated $8.8 million, $7.2 million, and $11.2 million of cash and cash equivalents for the years ended December 31, 2014, 2013, and 2012, respectively. In 2014, cash inflow was primarily the result of a decrease in accounts receivable of $5.0 million related to timing of collections, a decrease in deferred tax assets of $2.3 million related to the establishment of a partial valuation allowance in 2014, an increase in deferred revenue of $6.2 million, offset by net loss of $2.0 million, a decrease in accrued liabilities of $2.4 million and a decrease in accrued compensation of $1.6 million. In 2013, cash inflow was primarily the result of $4.3 million of net income, partially offset by increases in accounts receivable of $1.6 million and decreased in deferred revenue of $2.6 million, plus decreases in deferred tax assets of $2.3 million and increases in accrued liabilities of $2.7 million. In 2012, cash inflow was primarily the result of $4.2 million of net income, adjusted by decreases in deferred tax assets of $2.4 million and accounts receivable of $3.6 million, and an increase in accrued liabilities of $1.3 million, partially offset by a decrease in accounts payable of $1.9 million.
Cash Flows From Investing Activities
41
Net cash used in investing activities in 2014, 2013, and 2012 was $152.3 million, $6.6 million, and $3.7 million, respectively. These changes were primarily the result of net purchases of marketable securities.
Cash Flows From Financing Activities
Net cash provided by financing activities in 2014 was $148.3 million and consisted of net proceeds from our IPO and stock option exercises. In 2013 and 2012 the cash outflow was primarily the result of deferred costs associated with our IPO.
Recent Developments
On March 24, 2015, our Board of Directors approved the February 24, 2015 resolutions of the Compensation Committee of our Board of Directors (the “Committee”) regarding the payment of a discretionary bonus of $515,520 to Mr. O’Callaghan, our CEO and President, on April 15, 2015, provided Mr. O’Callaghan remains chief executive officer as of March 31, 2015, in lieu of the performance-based bonus in the same amount provided for under his employment agreement, dated October 14, 2014.
If Mr. O’Callaghan’s employment is terminated without Cause or terminates for Good Reason (as such terms are defined in his employment agreement), he will be entitled to receive 18 months of salary, up to 18 months of the premiums for him and his family to obtain health benefit coverage provided under our COBRA program, and a pro-rated portion of his annual bonus (“the CEO severance amounts”). On March 24, 2015, our Board of Directors approved the creation of a segregated interest-bearing account (to be identified as Restricted Cash on the balance sheet as of March 31, 2015) equal to the CEO severance amounts. The funds will remain the property of the Company until amounts are due to Mr. O’Callaghan under the terms of his amended employment agreement.
On March 24, 2015, our Board of Directors approved the terms of a Severance and Change in Effective Control Agreement to be entered into with each member of our management team and certain other employees. The Severance and Change in Effective Control Agreement provides that if the employee terminates for any reason or for no reason (including disability), voluntarily resigns for good reason (as defined in the agreement), or the employee dies, and the termination occurs within the six month period following a Qualifying Change in Effective Control, the employee will be entitled to an amount equal to the sum of six months of his or her monthly base salary, plus 50% of the employee’s annual target bonus for 2015, plus the premiums required to continue the employee’s group health care coverage for a period of six months following termination, which will be “grossed up” to cover taxes. In addition, upon a qualifying termination, the employee will receive an additional 12 months of vesting on any outstanding equity awards. A “Qualifying Change in Effective Control” is defined to mean that a majority of members of our Board is replaced by directors whose appointment or election is not endorsed by a majority of the members of our Board before the date of the appointment or election, which qualifies as a change in effective control under U.S. treasury regulations. The agreements terminate December 31, 2015 or the employee’s termination (unless the termination is within six months following a Qualifying Change in Effective Control).
On February 24, 2015, the Committee approved the creation of a pool of $600,000 to be distributed at the discretion of the Committee to employees who remain employed with us on December 31, 2015. Allocations of the pool will not be determined until the fourth quarter of 2015. All employees, other than Mr. O’Callaghan, are eligible to receive payments from this pool.
We believe that cash from operations and our existing cash, cash equivalents and investment balances will be sufficient to fund our ongoing operating activities, working capital, capital expenditures and other capital requirements for at least the next 12 months. Our future capital requirements will depend on many factors, including our rate of revenue growth, the expansion of our research and development activities, the timing and extent of our elections to co-promote product candidates under our collaboration agreements with Otsuka, and the timing of achievement of milestones under our collaboration agreements with Otsuka. Although we are not currently a party to any agreement or letter of intent regarding potential investments in, or acquisitions of, complementary businesses, applications or technologies, we may enter into these types of arrangements, which could require us to seek additional equity or debt financing.
Contractual Obligations and Commitments
The following is a summary of our contractual obligations as of December 31, 2014 (in thousands):
42
Payments Due by Period | |||||||||||||||||||||||||||
2015 | 2016 | 2017 | 2018 | 2019 | Thereafter | Total | |||||||||||||||||||||
Operating lease obligations | $ | 1,324 | $ | 1,295 | $ | 997 | $ | 968 | $ | 1,007 | $ | 2,039 | $ | 7,630 | |||||||||||||
Total | $ | 1,324 | $ | 1,295 | $ | 997 | $ | 968 | $ | 1,007 | $ | 2,039 | $ | 7,630 | |||||||||||||
Co-Development and Co-Promotion Options
The Emixustat Agreement provides us the right to co-promote with Otsuka in countries within our shared territory of North America. If we elect to co-promote, we will be responsible for a specified portion, ranging from 25% to 50%, of certain obligations in accordance with the agreement. The Glaucoma Agreement provides us the right to co-develop and co-promote OPA-6566. If we elect to co-develop and co-promote OPA-6566, we are obligated to pay an opt-in fee ranging from $10 million to $55 million depending on the timing and participation level of co-development and co-promotion.
We currently intend to exercise our co-promotion rights with respect to emixustat. We are unable to estimate with certainty the timing or future costs we will incur if we exercise our co-promotion option. The Glaucoma Agreement also provides for potential milestone payments to Otsuka of up to $75 million, based upon various clinical and sales objectives for the treatment of glaucoma.
Contingently Repayable Advances
Under the Emixustat Agreement, Otsuka has agreed to advance funds to us, which are secured by our interest in net profits and royalty payments and in our entire interests in ownership of the related emixustat compound and its backup compounds, certain pharmaceutical formulations developed under the Emixustat Agreement that contain one of those compounds, and the underlying intellectual property rights. Amounts may be advanced under this arrangement only to fund our share of development costs under the Emixustat Agreement. Any advances bear interest at the three month LIBOR rate plus three percent. These advances are exclusively payable out of:
• | 50% of either our share of net profits, as described in the Emixustat Agreement, generated from collaboration product sales in North America or, if applicable, 50% of the royalty payable to us for those sales; |
• | 50% of the net profits, as described in the Emixustat Agreement, we generate from collaboration product sales outside North America and Otsuka’s sole territory; and |
• | 50% of the consideration, as described in the Emixustat Agreement, we receive from the sale or license of collaboration compounds and collaboration products developed under the agreement outside North America and Otsuka’s sole territory. |
The percentages above will increase to 75% if we have not repaid the advances within five years of the first commercial sale of a product based on emixustat in North America. There are no financial covenant requirements under our arrangement with Otsuka. There were outstanding advances, including accrued interest, of $58.5 million and $32.9 million under this arrangement as of December 31, 2014 and 2013, respectively.
Off-Balance Sheet Transactions
To date, we have not had any relationships with unconsolidated entities or financial partnerships, such as entities referred to as structured finance or special purpose entities, which are established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Beginning in 2011, we have been sharing the development costs under the Emixustat Agreement, with Otsuka funding our share of these development costs, subject to our agreement to repay them from our share of the profits, if any, or sale or licensing proceeds, if any, from the commercialization of emixustat. Through December 31, 2014 and 2013, we recognized cumulative revenue of approximately $49.7 million and $32.4 million, respectively, under the agreement as described above. As of December 31, 2014 and 2013, the contingently repayable borrowing has accrued $2.5 million and $1.2 million, respectively, of interest, which is also contingently repayable under the same terms as the borrowing.
43
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to financial market risks, including changes in the market values of our debt investments and interest rates.
Financial market risk: Our exposure to market risk results primarily from interest rate changes on our debt securities. We do not invest in financial instruments or their derivatives for trading or speculative purposes. The three primary goals that guide our investment decisions, with the first being the most important, are: preservation of principal, fulfillment of liquidity needs, and balancing pre-tax return and portfolio risk. These objectives are achieved through specific guidelines around maturity parameters, credit quality and allowable investments. Our investment portfolio at December 31, 2014 was well-diversified and included corporate debt securities, commercial paper, certificates of deposits and money market funds. We consider the market value, default, and liquidity risks of our investments to be low at December 31, 2014.
Interest rate risk: We continually review our debt securities in order to ensure our portfolio is appropriately balanced as part of our overall interest rate risk management strategy, and through this process we consider both short-term and long-term factors in the U.S. and global financial markets in making adjustments to our tolerable exposure to interest rate risk. At December 31, 2014, all of the debt securities that we held were fixed-rate earning instruments that carry a degree of interest rate risk. Fixed-rate securities may have their fair market value adversely impacted due to a rise in interest rates. We may suffer losses in principal if we are forced to sell securities that have declined in market value due to changes in interest rates. At December 31, 2014, our cash and cash equivalents of $18.8 million were primarily held in money market funds, and our short-term investment balances of $85.0 million were held in corporate debt securities, commercial paper and certificates of deposit. At December 31, 2014, our long-term investment balances of $84.0 million were held in certificates of deposit and corporate debt securities. We consider the interest rate risk for our cash equivalents and marketable fixed-income securities held at December 31, 2014 to be low. A hypothetical increase in interest rates of 1% as of December 31, 2014 would result in an adverse change in the fair value of our investment portfolio of approximately $1.6 million. For further detail on our cash, cash equivalent and investment holdings, please see “Note 4: Cash and Cash Equivalents and Investments” of the Notes to Financial Statements in Part II Item 8 of our annual report.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our financial statements, together with related notes, are listed in Item 15(a) and included herein beginning on page F-1.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management evaluated, with the participation of Brian O'Callaghan and Dewey H. Blocker, Jr., our chief executive officer and interim principal financial officer, respectively, the effectiveness of our disclosure controls and procedures as of the period covered by this Annual Report on Form 10-K. Based on this evaluation, Mr. O'Callaghan and Mr. Blocker have concluded that our disclosure controls and procedures were effective as of December 31, 2014 to ensure that information we are required to disclose in reports that we file or submit under the Securities Exchange Act of 1934 is accumulated and communicated to our management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure, and that such information is recorded, processed, summarized, and reported within the time periods specified in the Securities and Exchange Commission rules and forms.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f). Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future
44
periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of management, including Mr. O'Callaghan and Mr. Blocker, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control – Integrated Framework (2013 framework) issued by the Committee of the Sponsoring Organizations of the Treadway Commission to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with U.S. GAAP.
Based on our evaluation under the framework in Internal Control – Integrated Framework (2013 framework), our management concluded that our internal control over financial reporting was effective as of December 31, 2014.
Ernst & Young LLP has audited the effectiveness of our internal control over financial reporting as of December 31, 2014 and its report is included below.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2014 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Acucela Inc.
We have audited Acucela Inc.'s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). Acucela Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Acucela Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets as of December 31, 2014 and 2013 and the related statements of operations, comprehensive income (loss), shareholders’ equity and cash flows for each of the three years in the period ended December 31, 2014 of Acucela Inc. and our report dated March 30, 2015 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Seattle, Washington
March 30, 2015
ITEM 9B. OTHER INFORMATION
CEO Bonus
On March 24, 2015, our Board of Directors approved the February 24, 2015 resolutions of the Compensation Committee of our Board of Directors (the “Committee”) regarding the payment of a discretionary bonus of $515,520 to Mr. O’Callaghan, our CEO and President, on April 15, 2015, provided Mr. O’Callaghan remains chief executive officer as of March 31, 2015, in lieu of the performance-based bonus in the same amount provided for under his employment agreement, dated October 14, 2014.
CEO Severance Escrow
On March 24, 2015 we and Brian O'Callaghan entered into an amendment to Mr. O'Callaghan's employment agreement. If Mr. O’Callaghan is terminated without Cause or for Good Reason (as such terms are defined in his employment agreement), he will be entitled to receive 18 months of salary, up to 18 months of the premiums for his and his family to obtain health benefit coverage provided under our then-available COBRA program, and a pro-rated portion of his annual bonus.
The foregoing description of Mr. O'Callaghan's employment agreement does not purport to be complete, and is qualified in its entirety by reference to the full text of his employment agreement, a copy of which was filed as Exhibit 10.01 to our current report on Form 8-K on October 17, 2014 and the amendment to his employment agreement, a copy of which is filed as Exhibit 10.19 hereto, and is incorporated herein by reference.
In connection with the amendment to Mr. O'Callaghan's employment agreement, on March 24, 2015, our Board of Directors approved the creation of a segregated interest-bearing account. We intend to deposit the full amount payable to Mr. O'Callaghan as severance under his employment agreement into this account. These funds will remain our property until due to Mr. O’Callaghan under the terms of his amended employment agreement and the related escrow agreement we intend to enter into.
Severance and Change in Effective Control Agreements
On March 24, 2015, our Board of Directors approved the terms of a Severance and Change in Effective Control Agreement to be entered into with each member of our management team and certain other employees. The Severance and Change in Effective Control Agreement provides that if the employee terminates for any reason or for no reason (including disability), voluntarily resigns for good reason (as defined in the agreement), or the employee dies, and the termination occurs within the six month period following a Qualifying Change in Effective Control, the employee will be entitled to an amount equal to the sum of six months of his or her monthly base salary, plus 50% of the employee’s annual target bonus for 2015, plus the premiums required to continue the employee’s group health care coverage for a period of six months following termination, which will be “grossed up” to cover taxes. In addition, upon a qualifying termination, the employee will receive an additional 12 months of vesting on any outstanding equity awards. A “Qualifying Change in Effective Control” is defined to mean that a majority of members of our Board is replaced by directors whose appointment or election is not endorsed by a majority of the members of our Board before the date of the appointment or election, which qualifies as a change in effective control under U.S. treasury regulations. The agreements terminate upon the earlier of December 31, 2015 or the employee’s termination (unless the termination is within six months following a Qualifying Change in Effective Control).
The foregoing description of the Severance and Change in Effective Control Agreement does not purport to be complete, and is qualified in its entirety by reference to the full text of the agreement, a copy of which is filed as Exhibit 10.20 hereto, and is incorporated herein by reference.
Amendments to Equity Incentive Plans and Share-based Awards
On March 24, 2015, our Board approved amendments to outstanding equity awards granted under our 2002 Stock Option and Restricted Stock Plan, our 2012 Equity Incentive Plan, and our 2014 Equity Incentive Plan. The letter agreement amending awards under our 2002 Stock Option and Restricted Stock Plan is filed as Exhibit 10.23 hereto and the letter agreement amending awards under our 2012 Equity Incentive Plan is filed as Exhibit 10.22 hereto. Our 2014 Equity Incentive Plan, as amended, is filed as Exhibit 10.21 hereto. The amendments provide that in the event of an applicable qualifying termination under these plans, any unvested portion of the awards under these plans will become immediately vested. In addition, a holder of awards under these plans will have up to 12 months following termination in order to exercise their awards.
The foregoing descriptions of the amendments to our 2002 Stock Option and Restricted Stock Plan, our 2012 Equity Incentive Plan, and our 2014 Equity Incentive Plan do not purport to be complete, and are qualified in their entirety by reference to the full text of the letter agreement amending awards under our 2002 Stock Option and Restricted Stock Plan filed as Exhibit 10.23 hereto, the letter agreement amending awards under our 2012 Equity Incentive Plan filed as Exhibit 10.22 hereto, and our 2014 Equity Incentive Plan, as amended, filed as Exhibit 10.21 hereto. These exhibits are incorporated herein by reference.
In addition, our employees, executive officers and non-employee members of our Board will be permitted to exercise their awards up to twelve months after their termination.
Future grants under our 2014 Equity Incentive Plan will include these provisions as well.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item is incorporated by reference to the information statement relating to the special meeting of shareholders to be filed with the Securities and Exchange Commission (SEC) within 120 days of the fiscal year ended December 31, 2014.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item is incorporated by reference to the information statement relating to the special meeting of shareholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2014.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item is incorporated by reference to the information statement relating to the special meeting of shareholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2014.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE
The information required by this Item is incorporated by reference to the information statement relating to the special meeting of shareholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2014.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The audit committee has retained Ernst & Young LLP to serve as our independent registered public accounting firm to conduct an audit of our financial statements for 2015, and the Board has directed that our management submit the selection of Ernst & Young LLP for ratification by the shareholders at the Annual Meeting. In retaining Ernst & Young LLP, the audit committee considered carefully Ernst & Young LLP’s performance for us in that capacity since its retention in 2009, its independence with respect to the services to be performed and its general reputation for adherence to professional auditing standards.
Selection of our independent registered public accounting firm is not required to be submitted to a vote of the shareholders for ratification. The Sarbanes-Oxley Act of 2002 requires the audit committee to be directly responsible for the appointment, compensation and oversight of the audit work of the independent registered public accounting firm. The Board is, however, submitting this matter to the shareholders as a matter of good corporate practice. If the shareholders fail to vote on an advisory basis in favor of ratifying this selection, the audit committee will reconsider whether to retain Ernst & Young LLP, and may retain that firm or another firm without re-submitting the matter to our shareholders. Even if the shareholders vote on an advisory basis in favor of ratifying the appointment, the audit committee, in its discretion, may direct the appointment of a different independent registered public accounting firm at any time during the year if it determines that such a change would be in the best interests of us and our shareholders.
Representatives of Ernst & Young LLP are expected to be present at the Annual Meeting. They will have the opportunity to make a statement if they desire to do so and are expected to be available to respond to appropriate questions.
Services and Fees
The following table lists the fees for services rendered by Ernst & Young LLP for 2014 and 2013 (in thousands):
Services | 2014 | 2013 | ||||||
Audit Fees | $ | 650 | $ | 1,820 | ||||
Audit-Related Fees | — | — | ||||||
Tax Fees | 76 | 13 | ||||||
All Other Fees | — | — | ||||||
Total | $ | 726 | $ | 1,833 | ||||
_____________________
Audit Fees. Consists of fees associated with the audit of our annual financial statements, the reviews of our interim financial statements, and the issuance of consents and comfort letters in connection with registration statements.
Audit-Related Fees. Consists of fees for assurance and related services that are reasonably related to the performance of the audit or review of our financial statements, including accounting consultations, and are not reported under “Audit Fees.”
Tax Fees. Consists of fees for tax compliance, tax advice and tax planning.
All Other Fees. Consists of fees associated with permitted corporate finance assistance and permitted advisory services, none of which were provided by our independent auditors during the last two fiscal years.
Ernst & Young LLP to date has not performed any non-audit services for us, other than tax related services.
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-Audit Services of Independent Registered Public Accounting Firm
Our audit committee’s policy is to pre-approve all audit and permissible non-audit services provided by the independent registered public accounting firm. These services may include audit services, audit-related services, tax services and other services. Pre-approval is detailed as to the particular service or category of services and is generally subject to a specific budget. The independent registered public accounting firm and management are required to periodically report to the audit committee regarding the extent of services provided by the independent registered public accounting firm in accordance with this pre-approval, and the fees for the services performed to date.
All of the services relating to the fees described in the table above were approved by our audit committee.
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) The following documents are filed as part of this report:
(1) Financial Statements and Report of Independent Auditors
(2) Financial Statement Schedules
All schedules are omitted as the required information is inapplicable or the information is presented in the Financial Statements or Notes to Financial Statements under Item 8.
(3) Exhibits
See Exhibit Index following the signature page of this report.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized on.
ACUCELA INC. | ||
Dated: | March 30, 2015 | |
By: | /s/ Brian O'Callaghan | |
Brian O'Callaghan | ||
Chief Executive Officer and President | ||
(Principal Executive Officer) | ||
Dated: | March 30, 2015 | |
BY: | /s/ Dewey H. Blocker, Jr. | |
Dewey H. Blocker, Jr. | ||
Vice President of Finance | ||
(Principal Financial Officer and Principal Accounting Officer) | ||
KNOW ALL PERSONS BY THESE PRESENTS, that each individual whose signature appears below constitutes and appoints Brian O'Callaghan, his or her true and lawful attorneys-in-fact each acting alone, with full power of substitution and re-substitution, for him or her and in his or her name, place and stead in any and all capacities to sign any or all amendments to this report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact, or their substitutes, each acting alone, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
Name | Title | Date | ||
/s/ Brian O'Callaghan | Chief Executive Officer and President | |||
Brian O'Callaghan | (Principal Executive Officer) | March 30, 2015 | ||
/s/ Dewey H. Blocker, Jr. | Vice President Finance | |||
Dewey H. Blocker, Jr. | (Principal Financial Officer and Principal Accounting Officer) | March 30, 2015 | ||
/s/ Peter A. Kresel | ||||
Peter A. Kresel | Director | March 30, 2015 | ||
/s/Ryo Kubota | ||||
Ryo Kubota | Director | March 30, 2015 | ||
/s/ Glen Y. Sato | ||||
Glen Y. Sato | Director | March 30, 2015 | ||
/s/ Michael T. Schutzler | ||||
Michael T. Schutzler | Director | March 30, 2015 | ||
/s/ Brian O'Callaghan | ||||
Brian O’Callaghan | Director | March 30, 2015 | ||
51
The following exhibits are filed herewith or are incorporated by reference to exhibits previously filed with the SEC:
Incorporated by Reference | ||||||
Exhibit Number | Filed Herewith | Form | File No. | Exhibit | Filing Date | |
3.01 | Form of Amended and Restated Articles of Incorporation. | S-1 | 333-192900 | 3.02 | 12/17/2013 | |
3.02 | Form of Restated Bylaws. | S-1 | 333-192900 | 3.04 | 12/17/2013 | |
4.01 | Form of Common Stock certificate. | S-1 | 333-192900 | 4.01 | 12/17/2013 | |
4.02 | Amended and Restated Investors’ Rights Agreement dated May 31, 2006 by and among the Company and certain of its shareholders. | S-1 | 333-192900 | 4.02 | 12/17/2013 | |
4.03 | Letter agreement dated December 9, 2013 between the Company and entities affiliated with SBI. | S-1/A | 333-192900 | 4.03 | 1/30/2014 | |
10.01* | Form of Indemnity Agreement. | S-1 | 333-192900 | 10.01 | 12/17/2013 | |
10.02* | 2002 Stock Option/Restricted Stock Plan. | S-1 | 333-192900 | 10.02 | 12/17/2013 | |
10.03* | 2012 Equity Incentive Plan and forms of stock option agreement and stock option exercise agreement. | S-1 | 333-192900 | 10.03 | 12/17/2013 | |
10.04* | 2014 Equity Incentive Plan and forms of notice of stock option grant, stock option agreement, notice of restricted stock award, restricted stock agreement, notice of restricted stock unit award, restricted stock unit agreement, notice of stock appreciation right award, and stock appreciation right award agreement. | S-1 | 333-192900 | 10.04 | 12/17/2013 | |
10.05 | Lease Agreement by and between Nexus Canyon Park, LLC and the Company dated February 13, 2006. | S-1 | 333-192900 | 10.05 | 12/17/2013 | |
10.06 | First Amendment to Lease by and between Nexus Canyon Park, LLC and the Company dated August 15, 2011. | S-1 | 333-192900 | 10.06 | 12/17/2013 | |
10.07 | Lease Agreement by and between The Northwestern Mutual Life Insurance Company and the Company dated June 22, 2010. | S-1 | 333-192900 | 10.07 | 12/17/2013 | |
10.08* | Employment Agreement by and between Dr. Ryo Kubota, M.D. and the Company dated April 18, 2005, as amended. | S-1 | 333-192900 | 10.08 | 12/17/2013 | |
10.09† | Co-Development and Commercialization Agreement by and between Otsuka Pharmaceutical Co., Ltd. and the Company dated September 4, 2008, as amended and supplemented. | S-1 | 333-192900 | 10.09 | 12/17/2013 | |
10.10* | Consulting Agreement by and between Peter Kresel and the Company dated January 1, 2012, as amended. | S-1 | 333-192900 | 10.10 | 12/17/2013 | |
10.11† | Development and Collaboration Agreement by and between Otsuka Pharmaceutical Co., Ltd. and the Company dated September 15, 2010, as amended. | S-1 | 333-192900 | 10.11 | 12/17/2013 | |
10.12 | Agreement for Stock Purchase by and between Otsuka Pharmaceutical Co., Ltd. and the Company dated September 15, 2010. | S-1 | 333-192900 | 10.11 | 12/17/2013 | |
10.13† | Sublease Agreement by and between the Boeing Company and the Company dated June 2014. | 8-K | 000-55133 | 10.13 | 7/1/2014 | |
10.14* | Terms of Separation and Consultancy, dated September 17, 2014, between the Company and David L. Lowrance. | 8-K | 000-55133 | 10.01 | 9/19/2014 | |
10.15* | Seventh Amendment to Employment Agreement, dated September 16, 2014, between the Company and Ryo Kubota. | 8-K | 000-55133 | 10.02 | 9/19/2014 | |
10.16 | Second Amendment to Lease Agreement between Nexus Canyon Park LLC and the Company effective as of September 1, 2014 | 8-K | 000-55133 | 10.01 | 9/22/2014 | |
10.17* | Employment Agreement, between the Company and Brian O'Callaghan, dated September 6, 2014. | 8-K | 000-55133 | 10.01 | 10/17/2014 | |
10.18* | Letter Agreement between Acucela, Inc. and Dewey H. Blocker, Jr., dated March 24, 2015 | 8-K | 000-55133 | 10.01 | 3/26/2015 | |
10.19* | First Amendment to Employment Agreement between the Company and Brian O'Callaghan, dated March 25, 2015 | X | ||||
10.20* | Form of Severance and Change in Effective Control Agreement | X | ||||
10.21* | 2014 Equity Incentive Plan (As Amended) and forms of notice of stock option grant and stock option agreement | X | ||||
10.22* | 2012 Equity Incentive Plan Letter | X | ||||
10.23* | 2002 Stock Option Plan/Restricted Stock Plan Letter | X | ||||
23.01 | Consent of Independent Registered Public Accounting Firm. | X | ||||
24.01 | Power of Attorney (included on Page 51). | X | ||||
31.1 | Certification of Principal Executive Officer Pursuant to Rule 13-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | X | ||||
52
31.2 | Certification of Principal Financial Officer Pursuant to Rule 13-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | X | ||||
32.1 | Certification of Principal Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | X | ||||
32.2 | Certification of Principal Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | X | ||||
101.INS | XBRL Instance Document | |||||
101.SCH** | XBRL Taxonomy Schema Linkbase Document | |||||
101.CAL** | XBRL Taxonomy Calculation Linkbase Document | |||||
101.DEF** | XBRL Taxonomy Definition Linkbase Document | |||||
101.LAB** | XBRL Labels Linkbase Document | |||||
101.PRE** | XBRL Presentation Linkbase Document | |||||
† | The Company has omitted portions of the referenced exhibit and filed such exhibit separately with the Securities and Exchange Commission pursuant to a grant of confidential treatment under Rule 406 promulgated under the Securities Act | |||||
* | Indicates management contract or compensatory plan or arrangement. | |||||
** | Pursuant to applicable securities laws and regulations, the Registrant is deemed to have complied with the reporting obligation relating to the submission of interactive data files in such exhibits and is not subject to liability under any anti-fraud provisions of the federal securities laws as long as the Registrant has made a good faith attempt to comply with the submission requirements and promptly amends the interactive data files after becoming aware that the interactive data files fail to comply with the submission requirements. In accordance with Rule 406T of Regulation S-T, the information in these exhibits is furnished and deemed not filed or part of a registration statement or the Prospectus for purposes of sections 11 or 12 of the Securities Act of 1933, as amended, is deemed not filed for purposes of section 18 of the Exchange Act of 1934, and otherwise is not subject to liability under these sections. | |||||
53
ACUCELA INC.
INDEX TO FINANCIAL STATEMENTS
Page | ||
F-1
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Acucela Inc.
We have audited the accompanying balance sheets of Acucela Inc. as of December 31, 2014 and 2013, and the related statements of operations, comprehensive income (loss), shareholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Acucela Inc. at December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Acucela Inc.'s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 30, 2015 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Seattle, Washington
March 30, 2015
F-2
ACUCELA INC.
BALANCE SHEETS
(in thousands)
December 31, | |||||||
2014 | 2013 | ||||||
Assets | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 18,778 | $ | 13,994 | |||
Investments | 85,008 | 14,947 | |||||
Accounts receivable from collaborations | 5,285 | 10,262 | |||||
Deferred tax asset | 61 | 1,114 | |||||
Prepaid expenses and other current assets | 2,582 | 1,964 | |||||
Total current assets | 111,714 | 42,281 | |||||
Property and equipment, net | 742 | 1,112 | |||||
Long-term investments | 84,033 | 3,478 | |||||
Long-term deferred tax asset | 42 | 1,280 | |||||
Deferred offering costs | — | 5,548 | |||||
Other assets | 435 | 349 | |||||
Total assets | $ | 196,966 | $ | 54,048 | |||
Liabilities and shareholders’ equity | |||||||
Current liabilities: | |||||||
Current maturities of contingently convertible debt, related party | $ | — | $ | 12,000 | |||
Accounts payable | 441 | 754 | |||||
Accrued liabilities | 4,176 | 6,579 | |||||
Accrued compensation | 1,683 | 3,269 | |||||
Deferred revenue from collaborations | 6,231 | — | |||||
Deferred rent and lease incentives | 25 | 267 | |||||
Total current liabilities | 12,556 | 22,869 | |||||
Commitments and contingencies (Note 13) | |||||||
Long-term deferred rent, lease incentives, and others | 47 | 55 | |||||
Total long-term liabilities | 47 | 55 | |||||
Shareholders’ equity: | |||||||
Convertible preferred stock | |||||||
Series A, no par value, no shares authorized as of December 31, 2014 and 2,734 shares authorized as of December 31, 2013; issued and outstanding, no shares as of December 31, 2014 and 2,734 shares as of December 31, 2013 | — | 2,051 | |||||
Series B, no par value, no shares authorized as of December 31, 2014 and 17,900 shares authorized as of December 31, 2013; issued and outstanding, no shares as of December 31, 2014 and 17,900 shares as of December 31, 2013 | — | 13,387 | |||||
Series C, no par value, no shares authorized as of December 31, 2014 and 31,818 shares authorized as of December 31, 2013; issued and outstanding, no shares as of December 31, 2014 and 11,807 shares as of December 31, 2013 | — | 12,771 | |||||
Common stock, no par value, 100,000 shares authorized as of December 31, 2014 and 60,000 shares authorized as of December 31, 2013; issued and outstanding, 35,809 shares as of December 31, 2014 and 11,971 shares as of December 31, 2013 | 186,589 | 3,654 | |||||
Additional paid-in capital | 3,601 | 2,728 | |||||
Accumulated other comprehensive loss | (361 | ) | (7 | ) | |||
Accumulated deficit | (5,466 | ) | (3,460 | ) | |||
Total shareholders’ equity | 184,363 | 31,124 | |||||
Total liabilities and shareholders’ equity | $ | 196,966 | $ | 54,048 | |||
F-3
ACUCELA INC.
STATEMENTS OF OPERATIONS
(in thousands, except per share data)
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Revenue from collaborations | $ | 35,396 | $ | 52,947 | $ | 46,424 | |||||
Expenses: | |||||||||||
Research and development | 25,582 | 36,405 | 31,604 | ||||||||
General and administrative | 10,002 | 9,548 | 7,787 | ||||||||
Total expenses | 35,584 | 45,953 | 39,391 | ||||||||
Income (loss) from operations | (188 | ) | 6,994 | 7,033 | |||||||
Other income (expense), net: | |||||||||||
Interest income | 519 | 122 | 27 | ||||||||
Interest expense | (15 | ) | (116 | ) | (138 | ) | |||||
Other income (expense), net | 37 | 182 | (97 | ) | |||||||
Total other income (expense), net | 541 | 188 | (208 | ) | |||||||
Income before income tax | 353 | 7,182 | 6,825 | ||||||||
Income tax expense | (2,359 | ) | (2,883 | ) | (2,647 | ) | |||||
Net income (loss) | (2,006 | ) | 4,299 | 4,178 | |||||||
Net income attributable to participating securities | — | 3,138 | 3,056 | ||||||||
Net income (loss) attributable to common shareholders | $ | (2,006 | ) | $ | 1,161 | $ | 1,122 | ||||
Net income (loss) per share attributable to common shareholders | |||||||||||
Basic | $ | (0.06 | ) | $ | 0.10 | $ | 0.09 | ||||
Diluted | $ | (0.06 | ) | $ | 0.09 | $ | 0.09 | ||||
Weighted average shares used to compute net income (loss) per share attributable to common shareholders: | |||||||||||
Basic | 32,869 | 11,964 | 11,901 | ||||||||
Diluted | 32,869 | 12,355 | 12,158 | ||||||||
See accompanying notes to financial statements.
F-4
ACUCELA INC.
STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(in thousands)
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Net income (loss) | $ | (2,006 | ) | $ | 4,299 | $ | 4,178 | ||||
Other comprehensive income (loss): | |||||||||||
Net unrealized gain (loss) on securities, net of tax of $48, $3 and $0 | (354 | ) | (7 | ) | 6 | ||||||
Comprehensive income (loss) | $ | (2,360 | ) | $ | 4,292 | $ | 4,184 | ||||
See accompanying notes to financial statements.
F-5
ACUCELA INC.
STATEMENTS OF SHAREHOLDERS’ EQUITY
(in thousands)
Convertible Preferred Stock | Additional Paid-In Capital | Accumulated Other Comprehensive Loss | Accumulated Deficit | Total | |||||||||||||||||||||||||||||||||||||||
Series A | Series B | Series C | Common Stock | ||||||||||||||||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||||||
Balance at December 31, 2011 | 2,734 | $ | 2,051 | 17,900 | $ | 13,387 | 11,807 | $ | 12,771 | 11,899 | $ | 3,133 | $ | 1,441 | $ | (6 | ) | $ | (11,937 | ) | $ | 20,840 | |||||||||||||||||||||
Stock-based compensation | — | — | — | — | — | — | — | — | 524 | — | — | 524 | |||||||||||||||||||||||||||||||
Common stock issued in connection with stock option exercises | — | — | — | — | — | — | 5 | 2 | — | — | — | 2 | |||||||||||||||||||||||||||||||
Common stock issued in connection with the restricted stock purchase agreement | — | — | — | — | — | — | 6 | 57 | — | — | — | 57 | |||||||||||||||||||||||||||||||
Net income | — | — | — | — | — | — | — | — | — | — | 4,178 | 4,178 | |||||||||||||||||||||||||||||||
Unrealized gain on marketable securities available for sale | — | — | — | — | — | — | — | — | — | 6 | — | 6 | |||||||||||||||||||||||||||||||
Balance at December 31, 2012 | 2,734 | 2,051 | 17,900 | 13,387 | 11,807 | 12,771 | 11,910 | 3,192 | 1,965 | — | (7,759 | ) | 25,607 | ||||||||||||||||||||||||||||||
Stock-based compensation | — | — | — | — | — | — | — | — | 667 | — | — | 667 | |||||||||||||||||||||||||||||||
Excess net tax benefit related to share-based awards | — | — | — | — | — | — | — | — | 96 | — | — | 96 | |||||||||||||||||||||||||||||||
Common stock issued in connection with stock option exercises | — | — | — | — | — | — | 30 | 6 | — | — | — | 6 | |||||||||||||||||||||||||||||||
Common stock issued in connection with the restricted stock purchase agreement | — | — | — | — | — | — | 31 | 456 | — | — | — | 456 | |||||||||||||||||||||||||||||||
Net income | — | — | — | — | — | — | — | — | — | — | 4,299 | 4,299 | |||||||||||||||||||||||||||||||
Unrealized loss on marketable securities available for sale | — | — | — | — | — | — | — | — | — | (7 | ) | — | (7 | ) | |||||||||||||||||||||||||||||
Balance at December 31, 2013 | 2,734 | 2,051 | 17,900 | 13,387 | 11,807 | 12,771 | 11,971 | 3,654 | 2,728 | (7 | ) | (3,460 | ) | 31,124 | |||||||||||||||||||||||||||||
Common stock issued in connection with IPO offering, net of issuance costs of $7,093 | — | — | — | — | — | — | 9,200 | 142,044 | — | — | — | 142,044 | |||||||||||||||||||||||||||||||
Common stock issued in connection with conversion of convertible preferred stock upon IPO | (2,734 | ) | (2,051 | ) | (17,900 | ) | (13,387 | ) | (11,807 | ) | (12,771 | ) | 10,814 | 28,209 | — | — | — | — | |||||||||||||||||||||||||
Common stock issued in connection with conversion of contingently convertible notes upon IPO | — | — | — | — | — | — | 3,636 | 12,000 | — | — | — | 12,000 | |||||||||||||||||||||||||||||||
Stock-based compensation | — | — | — | — | — | — | — | — | 516 | — | — | 516 | |||||||||||||||||||||||||||||||
Excess net tax benefit related to IPO costs | — | — | — | — | — | — | — | — | 421 | — | — | 421 | |||||||||||||||||||||||||||||||
Excess net tax provision related to share-based awards | — | — | — | — | — | — | — | — | (64 | ) | — | — | (64 | ) | |||||||||||||||||||||||||||||
Common stock issued in connection with stock option exercises | — | — | — | — | — | — | 188 | 682 | — | — | — | 682 | |||||||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | — | — | — | (2,006 | ) | (2,006 | ) | |||||||||||||||||||||||||||||
Unrealized loss on marketable securities available for sale | — | — | — | — | — | — | — | — | — | (354 | ) | — | (354 | ) | |||||||||||||||||||||||||||||
Balance at December 31, 2014 | — | $ | — | — | $ | — | — | $ | — | 35,809 | $ | 186,589 | $ | 3,601 | $ | (361 | ) | $ | (5,466 | ) | $ | 184,363 | |||||||||||||||||||||
See accompanying notes to financial statements.
F-6
ACUCELA INC.
STATEMENTS OF CASH FLOWS
(in thousands)
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cash flows from operating activities | |||||||||||
Net income (loss) | $ | (2,006 | ) | $ | 4,299 | $ | 4,178 | ||||
Adjustments to reconcile net income (loss) to net cash provided by operating activities: | |||||||||||
Depreciation and amortization | 501 | 531 | 464 | ||||||||
Amortization of deferred financing costs | — | — | 10 | ||||||||
Loss from the disposal of fixed assets | — | — | 79 | ||||||||
Stock-based compensation | 516 | 1,123 | 581 | ||||||||
Amortization of premium/discount on marketable securities | 1,175 | 332 | 107 | ||||||||
Deferred taxes | 2,349 | 2,274 | 2,434 | ||||||||
Excess net tax provision related to share-based awards | (64 | ) | — | — | |||||||
Changes in operating assets and liabilities: | |||||||||||
Accounts receivable from collaborations | 4,977 | (1,562 | ) | 3,551 | |||||||
Prepaid expenses and other current assets | (197 | ) | (443 | ) | (1,037 | ) | |||||
Accounts payable | (313 | ) | (102 | ) | (1,949 | ) | |||||
Accrued liabilities | (2,403 | ) | 2,650 | 1,315 | |||||||
Accrued compensation | (1,586 | ) | 856 | 778 | |||||||
Deferred rent and lease incentives | (250 | ) | (264 | ) | 48 | ||||||
Deferred revenue from collaborations | 6,231 | (2,570 | ) | 570 | |||||||
Other assets | (86 | ) | 122 | 117 | |||||||
Net cash provided by operating activities | 8,844 | 7,246 | 11,246 | ||||||||
Cash flows from investing activities | |||||||||||
Purchases of marketable securities available for sale | (201,134 | ) | (23,217 | ) | (15,580 | ) | |||||
Maturities of marketable securities available for sale | 48,931 | 17,136 | 12,163 | ||||||||
Additions to property and equipment | (131 | ) | (500 | ) | (326 | ) | |||||
Net cash used in investing activities | (152,334 | ) | (6,581 | ) | (3,743 | ) | |||||
Cash flows from financing activities | |||||||||||
Proceeds from issuance of common stock | 149,819 | 6 | 2 | ||||||||
Restricted investments income | — | — | (13 | ) | |||||||
Excess net tax benefit related to share-based awards | — | 96 | — | ||||||||
Payments for deferred offering costs | (1,545 | ) | (3,412 | ) | (613 | ) | |||||
Net cash used in financing activities | 148,274 | (3,310 | ) | (624 | ) | ||||||
(Decrease) increase in cash and cash equivalents | 4,784 | (2,645 | ) | 6,879 | |||||||
Cash and cash equivalents—beginning of year | 13,994 | 16,639 | 9,760 | ||||||||
Cash and cash equivalents—end of year | $ | 18,778 | $ | 13,994 | $ | 16,639 | |||||
Supplemental disclosure | |||||||||||
Cash paid for interest | $ | — | $ | — | $ | 420 | |||||
Cash paid for income taxes | 60 | 828 | 151 | ||||||||
Unpaid deferred offering costs | 5,548 | 937 | — | ||||||||
Restriction of investments as collateral | — | (5,759 | ) | 5,750 | |||||||
Conversion of convertible preferred stock upon IPO | 28,209 | — | — | ||||||||
Conversion of contingently convertible debt, related party, upon IPO | 12,000 | — | — | ||||||||
See accompanying notes to financial statements.
F-7
ACUCELA INC.
NOTES TO FINANCIAL STATEMENTS
1. Business and Basis of Presentation
Business
Acucela Inc. (“we,” “our” and “us”) is a clinical-stage biotechnology company that specializes in discovering and developing novel drug candidates to potentially treat and slow the progression of sight-threatening ophthalmic diseases affecting millions of individuals worldwide. In 2008, we and Otsuka Pharmaceutical Co., Ltd. (“Otsuka”) entered into a definitive agreement to co-develop emixustat hydrochloride ("emixustat"), our lead compound for dry AMD. Emixustat is in Phase 2b/3 clinical development in the United States.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (“GAAP”) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from these estimates.
Segments
We operate in one segment, pharmaceutical product development. All of our significant assets are located in the United States. During the years ended December 31, 2014, 2013, and 2012, all revenue was generated in the United States.
2. Significant Accounting Policies
Revenue Recognition
Our business strategy includes entering into collaboration agreements with pharmaceutical companies for the development and commercialization of our product candidates. The terms of the agreements may include nonrefundable license fees, funding of research and development activities, payments based upon achievement of development milestones, payments based upon achievement of revenue milestones, or royalties on product sales. We recognize revenue when four basic criteria have been met: (a) persuasive evidence of an arrangement exists; (b) delivery has occurred or services rendered; (c) the fee is fixed or determinable; and (d) collectability is reasonably assured.
Revenue recognized for the years ended December 31, 2014, 2013, and 2012 consists entirely of amounts derived from our collaboration agreements with Otsuka (See Note 11).
Multiple Element Arrangements
Our collaboration agreements are multiple element arrangements that must be analyzed to identify the deliverables included in the agreements and determine if the deliverables qualify as separate units of accounting. Deliverables are considered a separate unit of accounting when all of the following criteria are met: (i) the delivered item has value to the customer on a standalone basis and (ii) if the arrangement includes a general right of return relative to the delivered item, and the delivery or performance of the undelivered item is considered probable and substantially within our control. There are no rights of return in our collaboration agreements.
Arrangement consideration is allocated to the separate units of accounting based on their relative selling prices. We follow a hierarchy to determine the selling price for each unit of accounting, determining first if there is vendor-specific objective evidence (“VSOE”) of fair value (the price at which the goods or services are regularly sold by us on a standalone basis). If VSOE of fair value is not available, third-party evidence (“TPE”) of vendors selling similar goods or services to similarly situated customers on a standalone basis is used to establish fair value. If neither VSOE nor TPE of fair value exists, we use our best estimate of the selling price (“BESP”) for that unit of accounting. Our BESP represents the price at which we would transact if we regularly sold the unit of accounting on a standalone basis.
We consider market conditions and entity-specific factors when estimating the selling price. Once the selling price for each unit of accounting has been established, the consideration received is allocated among the units of accounting based on their relative selling price, and the applicable revenue recognition criteria are applied to each of the separate units. The amount
F-8
of arrangement consideration allocable to a delivered item that is a separate unit of accounting is limited to arrangement consideration that is fixed or determinable. Payments that are contingent upon the occurrence of future events that are not exclusively within our control are excluded from the allocable arrangement consideration until the contingency is resolved.
When we have continuing performance obligations, revenue is recognized using one of two methods. Where we are able to estimate the total amount of services under a unit of accounting and such performance obligations are provided on a best-efforts basis, revenue is recognized using a proportional performance model. Costs incurred to date compared to total expected costs are used to determine proportional performance, as this is considered to be representative of the delivery of outputs. Changes in estimates of total expected costs are accounted for prospectively as a change in estimates. When we cannot reasonably estimate the total amount of service that is to be performed, but can reasonably estimate when the performance obligation ceases or becomes inconsequential, a time-based method is used to recognize revenue. Under the time-based method, revenue is recognized ratably over the estimated performance period of the unit of accounting, but not before the removal of any contingencies. If we cannot reasonably estimate when our performance obligation either ceases or becomes inconsequential and perfunctory, then revenue is not recognized until we can reasonably estimate when the performance obligation ceases or becomes inconsequential and perfunctory. Revenue is then recognized over the remaining estimated period of performance. Significant management judgment is required in determining the level of effort required and the period over which we are expected to complete our performance obligations under each unit of accounting.
Substantive Milestone Payments
Our collaboration agreements contain substantive milestones. A substantive milestone is defined as an event that meets the following conditions: (i) there is substantive uncertainty on the date the arrangement is entered into about whether the event will be achieved; (ii) achievement of the event is based in whole, or in part, on either our performance or a specific outcome resulting from our performance; and (iii) achievement of the event results in additional payment due to us. For a milestone to be considered substantive, the payment associated with our achievement must have all of the following characteristics: (i) relate solely to our past performance; (ii) be reasonable relative to all of the deliverables and payment terms in the arrangement; and (iii) be commensurate with either our effort required to achieve the milestone or the enhanced value of the delivered item(s) as a result of the milestone achievement.
Substantive milestone payments are recognized as revenue upon achievement of the milestone only if all of the previous conditions are met and the milestone payments are nonrefundable. Determination as to whether a payment meets the aforementioned conditions involves management’s judgment. If any of the aforementioned conditions are not met, the resulting payment would not be considered a substantive milestone and, therefore, the resulting payment would be determined to be part of the allocable arrangement consideration and would be recognized as revenue as such performance obligations are performed under either the proportional performance or time-based methods, as applicable, and in accordance with the policies as described above.
Deferred Revenue
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue.
Cash and Cash Equivalents and Investments
We consider investments in highly liquid instruments purchased with an original maturity at purchase of three months or less to be cash equivalents. The amounts are recorded at cost, which approximates fair value. Our cash equivalents consist of money market funds and certificates of deposit, and money market funds and corporate debt securities, at December 31, 2014 and 2013, respectively.
We have classified our entire investment portfolio, which consists of corporate debt securities, commercial paper and certificates of deposit, as available-for-sale. Available-for-sale securities are stated at fair value as of each balance sheet date based on market quotes, and unrealized gains and losses are reflected as a net amount under the caption of accumulated other comprehensive loss. Premiums or discounts arising at acquisition are amortized into earnings.
We periodically evaluate whether declines in fair values of our investments below their cost are other-than-temporary. This evaluation consists of several qualitative and quantitative factors regarding the severity and duration of the unrealized loss, as well as whether it is more likely than not that we will hold the investment until recovery of its amortized cost basis. Realized gains and losses are calculated using the specific identification method. Realized gains and losses and declines in value judged to be other-than-temporary are recorded within the statements of income under the caption other income (expense).
F-9
We consider an investment with a maturity greater than twelve months from the balance sheet date as long-term and a maturity less than twelve months as short-term at the balance sheet date.
Accounts Receivable
Our accounts receivable, as of December 31, 2014 and 2013, consist of amounts due from our collaborations with Otsuka. There was no allowance for doubtful accounts for the periods presented, as we believe all outstanding amounts will be paid based on our contractual arrangements with Otsuka and history of successful collections thereunder and collateral is not required.
Property and Equipment
Property and equipment are recorded at cost, less accumulated depreciation. We provide for depreciation of equipment on a straight-line basis over an estimated useful life of five years, except leasehold improvements which are amortized on a straight-line basis over the shorter of the lease term or estimated useful life of the assets.
Expenditures for maintenance and repairs are expensed as incurred.
Long-lived assets held for use are subject to an impairment assessment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. If the carrying value is no longer recoverable based upon the undiscounted future cash flows of the asset, the amount of the impairment is the difference between the carrying amount and the fair value of the asset. We have recorded no impairment charges for the periods presented.
Fair Value
We measure and report at fair value our cash equivalents and investment securities. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability, an exit price, in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value maximize the use of observable inputs and minimize the use of unobservable inputs.
The carrying amounts reflected in the balance sheets for accounts receivable and accounts payable approximate fair value due to their short-term nature.
Stock-Based Compensation
Stock-based compensation cost is estimated at the grant date based on the award’s fair value and is recognized on a straight line basis as expense, less estimated forfeitures, over the requisite service period, which is generally the vesting period. The fair value of stock options under our equity-based incentive plans (the "Equity Plans") are calculated using the Black-Scholes-Merton (“BSM”) option pricing model. The BSM model requires various judgmental assumptions regarding volatility and expected option life. If any of the assumptions used in the BSM model change significantly, stock-based compensation expense for new awards may differ materially from that recorded for existing awards.
Equity awards to nonexecutive employees generally vest and become exercisable over a four-year period. Equity awards to executives generally vest and become exercisable over a five-year period. In 2014, we began granting restricted stock unit awards to employees.
Research and Development Costs
Research and development costs include salaries, fees paid to external service providers and contract research organizations to conduct research and development activities, laboratory supplies, license fees, consulting fees, and travel. Research and development costs are expensed as incurred.
Deferred Offering Costs
External costs we incurred directly attributable to our public offering were deferred and recorded as noncurrent assets and were offset against the proceeds of our 2014 initial public offering.
401(k) Retirement Plan
We sponsor an employee retirement plan under Section 401(k) of the Internal Revenue Code of 1986, as amended. All employees who meet minimum eligibility requirements are eligible to participate in the plan. Beginning in 2015, we match 50% on the first 6% of employee contributions made to the plan. In January 2015, the Board of Directors also authorized a non-elective discretionary employer contribution of $0.4 million which vests over a four-year period, based on employee hire dates.
F-10
Income Taxes
We recognize deferred tax assets and liabilities for the expected future tax consequences of events that have already been recognized in the financial statements or tax returns. Excess tax benefits associated with stock option exercises and other equity awards are credited to stockholders' equity. Deferred tax liabilities and assets are based on the difference between financial statement carrying amounts and the tax basis of assets and liabilities, operating loss, and tax credit carryforwards and are measured using enacted tax rates expected to be in effect in the years the differences or carryforwards are anticipated to be recovered or settled. A valuation allowance is established when we believe that it is more likely than not that benefits of the deferred tax assets will not be realized.
3. Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board issued Accounting Standards Update ("ASU") 2014-09, Revenue from Contracts with Customers. This ASU amends the existing accounting standards for revenue recognition. Under the new revenue recognition model, a company should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. The ASU is effective for annual reporting periods beginning after December 15, 2016, including interim periods within that reporting period. Early adoption is not permitted. The amendment may be applied retrospectively to each prior period presented or retrospectively with the cumulative effect recognized as of the date of initial application. We are currently evaluating the transition alternatives and impact on our financial statements.
4. Cash and Cash Equivalents and Investments
Cash, cash equivalents and investments at December 31, 2014 and 2013 include all cash, money market funds, corporate debt securities, commercial paper and certificates of deposit. We consider our investments as available-for-sale. Available-for-sale securities are stated at fair value. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. To increase the comparability of fair value measures, the following hierarchy prioritizes the inputs to valuation methodologies used to measure fair value:
Level 1—Quoted prices in active markets for identical assets and liabilities,
Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities, and
Level 3—Unobservable inputs in which there is little or no market data available, which requires us to develop our own assumptions.
We measure the fair value of money market funds based on quoted prices in active markets for identical assets or liabilities. All other financial instruments were valued either based on recent trades of securities in inactive markets or based on quoted market prices of similar instruments and other significant inputs derived from or corroborated by observable market data. We did not hold any financial instruments categorized as Level 3 as of December 31, 2014 or 2013.
Cash and cash equivalents and investments as of December 31, 2014 and 2013 consisted of the following (in thousands):
F-11
December 31, 2014 | |||||||||||||||
Amortized Cost | Gross Unrealized | Fair Value | |||||||||||||
Holding Gains | Holding Losses | ||||||||||||||
Cash | $ | 767 | $ | — | $ | — | $ | 767 | |||||||
Level 1 Securities: | |||||||||||||||
Money market funds | 17,771 | — | — | 17,771 | |||||||||||
Level 2 Securities: | |||||||||||||||
Commercial paper | 15,992 | 2 | (1 | ) | 15,993 | ||||||||||
Corporate debt securities | 131,586 | — | (398 | ) | 131,188 | ||||||||||
Certificates of deposit | 22,115 | 4 | (19 | ) | 22,100 | ||||||||||
$ | 188,231 | $ | 6 | $ | (418 | ) | $ | 187,819 | |||||||
December 31, 2013 | |||||||||||||||
Amortized Cost | Gross Unrealized | Fair Value | |||||||||||||
Holding Gains | Holding Losses | ||||||||||||||
Cash | $ | 868 | $ | — | $ | — | $ | 868 | |||||||
Level 1 Securities: | |||||||||||||||
Money market funds | 12,501 | — | — | 12,501 | |||||||||||
Level 2 Securities: | |||||||||||||||
Commercial paper | 1,099 | 1 | — | 1,100 | |||||||||||
Corporate debt securities | 12,101 | — | (4 | ) | 12,097 | ||||||||||
Municipal bonds | 625 | — | — | 625 | |||||||||||
Certificates of deposit | 5,235 | 2 | (9 | ) | 5,228 | ||||||||||
$ | 32,429 | $ | 3 | $ | (13 | ) | $ | 32,419 | |||||||
As of December 31, 2014, $5.2 million of certificates of deposit and $78.8 million of corporate debt securities mature in greater than one year, but less than two years. There were no investments which were in an unrealized loss position for a period of twelve months or more. All other investment securities held at December 31, 2014 mature within 12 months.
Market values were determined for each individual security in the investment portfolio. The declines in value of certain of these investments are primarily related to changes in interest rates and are considered to be temporary in nature. We evaluate, among other things, the duration and extent to which the fair value of a security is less than its cost, the financial condition of the issuer, and our intent to sell, or whether it is more likely than not we will be required to sell the security before recovery of the amortized cost basis. We do not consider these investments to be other-than-temporarily impaired as of December 31, 2014.
5. Property and Equipment
Property and equipment as of December 31, 2014 and 2013 consist of the following (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Laboratory equipment | $ | 2,910 | $ | 2,844 | |||
Leasehold improvements | 1,812 | 1,812 | |||||
Office furniture and equipment | 497 | 457 | |||||
5,219 | 5,113 | ||||||
Less accumulated depreciation and amortization | (4,477 | ) | (4,001 | ) | |||
Property and equipment, net | $ | 742 | $ | 1,112 | |||
F-12
6. Contingently Convertible Debt with Related Party
The contingently convertible debt was automatically converted into 3,636,365 shares of common stock upon the closing of our 2014 IPO. The number of shares issued upon conversion was determined by dividing the principal of the note by $3.30, which was subject to adjustment for any subsequent recapitalizations, stock combinations, stock dividends, or stock splits.
SBI Holdings, Inc. (a related party), the holder of the notes and one of our shareholders, received payment for interest, at approximately 1% average interest rate, on the unsecured promissory notes of $0.4 million in the year ended December 31, 2012.
7. Shareholders’ Equity
Common Stock
Our articles of incorporation, as amended and restated, authorize us to issue 100,000,000 shares of common stock without par value.
In February 2014, upon the closing of our IPO, all shares of our outstanding convertible preferred stock automatically converted into 10,813,867 shares of common stock. We issued 9,200,000 shares of common stock for aggregate proceeds of $142.0 million from the IPO, net of underwriters’ discounts and commissions, and offering expenses. In addition, upon the closing of the IPO, $12.0 million of outstanding principal underlying convertible notes that we issued to affiliates of SBI Holdings, Inc. in May 2006 automatically converted into 3,636,365 shares of common stock, after an intermediate conversion into Series C preferred shares. The number of shares issued upon conversion was determined by dividing the principal of the note by $3.30.
Changes in Accumulated Other Comprehensive Loss (in thousands):
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Beginning balance | $ | (7 | ) | $ | — | $ | (6 | ) | |||
Current period other comprehensive gain (loss), net of tax | (354 | ) | (7 | ) | 6 | ||||||
Ending balance | $ | (361 | ) | $ | (7 | ) | $ | — | |||
The changes in accumulated other comprehensive loss relate to unrealized holding gains and losses in available-for-sale securities.
8. Stock-Based Compensation
Equity-Based Incentive Plan
The Board has adopted and approved equity-based incentive plans (the “Equity Plans”) which provide for the issuance of nonqualified and incentive stock options to employees, board members, and consultants to acquire shares of common stock. The Equity Plans also allow for the issuance of restricted stock and restricted stock units ("RSUs").
As of December 31, 2014, 2,329,629 shares of common stock were reserved to be issued in conjunction with the Equity Plans. On January 1, 2015, an additional 1,432,358 shares were reserved for issuance under the evergreen provision of the 2014 Equity Incentive Plan. On January 27, 2015, we granted 762,480 options, including 712,480 options to our CEO, and 414,060 RSUs, including 356,410 RSUs which were granted to our CEO. On February 24, 2015, we granted 7,100 RSUs. On March 24, 2015, we granted 40,000 options to our Vice President Finance and 31,400 RSUs, including 20,000 RSUs which were granted to our Vice President Finance. All equity awards were granted under our 2014 Equity Plan.
The term of each option is ten years. Equity awards to nonexecutive employees generally vest and become exercisable over a four-year period, with 25% vesting after one year and 25% vesting annually thereafter. Equity awards to
F-13
executives generally vest and become exercisable over a five-year period, with 20% vesting after one year and 20% vesting annually thereafter. The option agreements include restrictions on the sale of shares acquired by the exercise of options. We currently use authorized and unissued shares to satisfy share award exercises or restricted stock unit vesting events.
Amendments to Equity Incentive Plans and Share-based Awards
On March 24, 2015, our Board approved amendments to outstanding equity awards granted under our 2002 Stock Option and Restricted Stock Plan, our 2012 Equity Incentive Plan, and our 2014 Equity Incentive Plan to our employees, executive officers and non-employee members of our Board. The amendments provide that for employees and executive officers, if we undergo a change in control and their employment is terminated without Cause or for Good Reason (as such terms are defined in the severance and change in control agreement), then any unvested portion of the awards held by employees and executives will become immediately vested.
In addition, our employees, executive officers and non-employee members of our Board will be permitted to exercise their awards up to twelve months after their termination. We expect to recognize incremental stock-based compensation expense in the first quarter of 2015 related to the modification of these awards.
Future grants under our 2014 Equity Incentive Plan will include these provisions as well.
During 2014, the following activity occurred under the Company's Equity Plans:
Stock options: | Options | Weighted Average Exercise Price | Weighted Average Grant Date Fair Value | Intrinsic Value (in thousands) | Weighted Average Remaining Contractual Term (in years) | |||||||||||
Unexercisable, December 31, 2013 | 284,779 | $ | 10.92 | $ | 5.87 | |||||||||||
Outstanding, December 31, 2013 | 696,878 | 7.21 | ||||||||||||||
Granted | 101,000 | 7.78 | 5.00 | |||||||||||||
Exercised | (187,007 | ) | 3.64 | 2.24 | ||||||||||||
Forfeited | (167,699 | ) | 8.27 | 4.86 | ||||||||||||
Expired | (67,216 | ) | 8.42 | 4.86 | ||||||||||||
Outstanding, December 31, 2014 | 375,956 | $ | 8.45 | $ | 397 | 6.5 | ||||||||||
Exercisable, December 31, 2014 | 263,929 | $ | 6.18 | $ | 381 | 5.8 | ||||||||||
Unexercisable, December 31, 2014 | 112,027 | $ | 13.78 | $ | 7.25 | |||||||||||
RSUs: | Shares | Weighted Average Grant Date Fair Value | ||||||||||||||
Outstanding, December 31, 2013 | — | |||||||||||||||
Granted | 10,000 | $ | 7.46 | |||||||||||||
Forfeited | — | |||||||||||||||
Expired | — | |||||||||||||||
Outstanding, December 31, 2014 | 10,000 | $ | 7.46 | |||||||||||||
Supplemental information is presented below
Supplemental Information | 2014 | 2013 | 2012 | ||||||||
Stock options: | |||||||||||
Weighted average grant date fair value per share - granted | $ | 5.00 | $ | 8.09 | $ | 5.59 | |||||
Weighted average grant date fair value per share - vested | $ | 5.26 | $ | 4.52 | $ | 3.48 | |||||
Weighted average grant date fair value per share - forfeited | $ | 4.86 | $ | 4.89 | $ | 2.81 | |||||
Total fair value of options vested (in thousands) | $ | 482 | $ | 625 | $ | 482 | |||||
Total intrinsic value of options exercised (in thousands) | $ | 886 | $ | 307 | $ | 47 | |||||
Scheduled vesting for outstanding restricted stock units at December 31, 2014 is as follows:
F-14
RSUs | 2015 | 2016 | 2017 | 2018 | 2019 | Thereafter | Total | |||||||||||||
Scheduled vesting (shares) | 3,400 | 2,500 | 2,500 | 1,600 | — | — | 10,000 | |||||||||||||
As of December 31, 2014, there was $0.7 million and $0.1 million of unrecognized compensation cost related to nonvested options and RSUs granted under the Equity Plans, respectively. That cost is expected to be recognized over a weighted average period of 2.7 years and 3.4 years, respectively.
The fair value of stock options granted for the years ended December 31, 2014, 2013, and 2012 was calculated using the Black-Scholes option-pricing model and applied the following assumptions:
Years ended December 31, | |||||
2014 | 2013 | 2012 | |||
Risk-free interest rate | 1.5%–2.5% | 1.2%–2.1% | 1.0% | ||
Expected term | 6.2 years | 6.3 years | 6.3 years | ||
Dividend yield | —% | —% | —% | ||
Expected volatility | 70% | 50% | 50%–65% | ||
Risk-Free Interest Rate. We base the risk-free interest rate used in our option-pricing model on the implied yield currently available on U.S. Treasury issued with an equivalent term. Where the expected term of our stock-based awards does not correspond with the term for which an interest rate is quoted, we perform a straight-line interpolation to determine the rate from the available term maturities.
Expected Term. The expected term used in our option-pricing model represents the period that our stock-based awards are expected to be outstanding and is determined based on the simplified method. The simplified method uses a simple average of the vesting and original contractual terms of the option. We use the simplified method to determine the expected option term, since our stock option exercise experience does not provide a reasonable basis upon which to estimate the expected option term.
Dividend Yield. We have never paid cash dividends and have no present intention to pay cash dividends in the future. Accordingly, the expected dividend used in our option-pricing model is zero.
Expected Volatility. The volatility factor used in our option-pricing model is estimated using comparable public company stock volatility.
Stock-based compensation is included in our statements of operations as follows (amounts in thousands):
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Research and development | $ | 221 | $ | 414 | $ | 247 | |||||
General and administrative | 295 | 709 | 334 | ||||||||
Total | $ | 516 | $ | 1,123 | $ | 581 | |||||
Former CEO Equity Agreement
We have an employment agreement with Dr. Ryo Kubota. Until our IPO, we were obligated pursuant to the employment agreement to grant stock options or allow Dr. Kubota to purchase restricted shares of common stock as often as necessary to maintain Dr. Kubota’s equity position in our company equal to at least 51% of our issued and outstanding voting common stock on an as-converted basis. The shares under these awards are subject to repurchase provisions that lapse quarterly over 36 months from the date of grant. The purchase price of each stock grant could be made by an interest-bearing full recourse promissory note. Per the terms of the employment agreement, we were obligated to periodically pay Dr. Kubota cash bonuses consisting of principal and interest amounts due under such promissory notes and additional taxes incurred by Dr. Kubota as a result of receiving such bonuses, if any. Compensation expense related to this arrangement is included in general and administrative expense.
During the years ended December 31, 2013 and 2012, in connection with stock option exercises, we entered into a restricted stock purchase agreement with Dr. Kubota for the issuance of 31,452 and 5,504 shares, respectively, of our common stock in exchange for a three-year promissory note in the total amount of $0.5 million and $0.1 million, respectively. Concurrent with the execution of a restricted stock purchase agreement, we paid a bonus to Dr. Kubota, which was in turn used
F-15
to repay the promissory note and as compensation for taxes associated with the award. During the years ended December 31, 2013 and 2013, we recorded approximately $0.8 million and $0.1 million, respectively, in compensation expense in connection with the award. This arrangement terminated with the completion of our IPO in 2014.
9. Strategic Restructuring
In October 2013, we announced a plan to reduce expenses, including a workforce reduction, as a result of the termination of the Rebamipide Agreement (see Note 11). The plan resulted in a reduction in force of approximately 35% of our total workforce, or approximately 30 employees, effective January 1, 2014.
As a result of this workforce reduction, we recorded a $1.0 million charge in 2013 in general and administrative expense, related to severance, other termination benefits, and outplacement services. The cash outlays related to this charge primarily took place in the first six months of 2014 and activities were complete as of June 30, 2014. The following table summarizes the utilization of the restructuring liability (in thousands):
Severance and Other Termination Benefits | |||
Balance, December 31, 2013 | $ | 966 | |
Adjustments | (8 | ) | |
Cash payments | (958 | ) | |
Balance, December 31, 2014 | $ | — | |
10. Income Taxes
Deferred tax assets arise from temporary differences between financial and tax reporting. We have established a valuation allowance because it is more likely than not that the deferred tax assets will expire before we are able to realize their benefits or the future deductibility is uncertain. Periodically, the valuation allowance is reviewed and adjusted based on management’s assessments the realizability of deferred tax assets. During the year ended December 31, 2014, we recorded a partial valuation allowance of $2.3 million against our deferred tax assets due to expected future losses as a result of our new strategic plan. Our Board of Directors approved a new strategic plan that includes commencement of development of certain proprietary preclinical programs or in-license opportunities. Because these opportunities will be developed independently, our development expenditures on these programs will not be funded by collaborative partners and we expect that our total research and development expenses will increase and that we will incur net losses from our operating activities.
F-16
Deferred tax assets are as follows (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Deferred tax assets: | |||||||
Research and development tax credit carryforwards | $ | 925 | $ | 714 | |||
Compensation | 774 | 1,077 | |||||
Deferred rent | 25 | 110 | |||||
Alternative minimum tax credits | 334 | 363 | |||||
Property and equipment | 224 | 124 | |||||
Unrealized losses | 142 | 6 | |||||
Other | 3 | — | |||||
Total deferred tax asset | $ | 2,427 | $ | 2,394 | |||
Less: Valuation allowance | (2,324 | ) | — | ||||
$ | 103 | $ | 2,394 | ||||
Reported as: | |||||||
Current deferred tax asset | $ | 61 | $ | 1,114 | |||
Long-term deferred tax asset | 42 | 1,280 | |||||
$ | 103 | $ | 2,394 | ||||
For the year ended December 31, 2012, we had taxable income that was offset by the utilization of the NOL.
The components of the tax expense (benefit) are as follows (in thousands):
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Federal: | |||||||||||
Current | $ | (79 | ) | $ | 575 | $ | 169 | ||||
Deferred | 2,400 | 2,295 | 2,478 | ||||||||
2,321 | 2,870 | 2,647 | |||||||||
State: | |||||||||||
Current | $ | 19 | $ | 34 | $ | — | |||||
Deferred | 19 | (21 | ) | — | |||||||
38 | 13 | — | |||||||||
Total | $ | 2,359 | $ | 2,883 | $ | 2,647 | |||||
The tax benefit associated with the utilization of loss carryforwards was $0.6 million, $0.8 million, and $3.0 million in the years ended December 31, 2014, 2013 and 2012, respectively.
The reconciliation of the statutory federal income tax rate to our effective income tax rate is as follows:
F-17
Years Ended December 31, | ||||||||
2014 | 2013 | 2012 | ||||||
Statutory rate | 34.0 | % | 34.0 | % | 34.0 | % | ||
State income taxes | 6.8 | — | — | |||||
Stock compensation | (7.8 | ) | — | — | ||||
Other permanent items | 5.5 | — | — | |||||
Meals and entertainment | 7.1 | — | — | |||||
Return to provision items | (6.0 | ) | — | — | ||||
Valuation allowance | 631.7 | — | — | |||||
Loss carryforward adjustment | — | 3.7 | — | |||||
Other, net | (3.6 | ) | 2.1 | 4.8 | ||||
Effective tax rate | 667.7 | % | 39.8 | % | 38.8 | % | ||
As of December 31, 2014, we had research and development tax credit carryforwards of $1.1 million. The carryforwards are available to offset future tax liabilities. The research and development tax credits begin to expire in 2027.
Approximately $0.2 million of the research and development tax credit carryforward relates to tax deductible stock-based compensation in excess of amounts recognized for financial statement purposes. To the extent that research and development tax credit carryforwards, if realized, relate to stock-based compensation, the resulting tax benefits will be recorded to shareholders' equity, rather than to results of operations.
We file our income tax return in the U.S. federal jurisdiction. We are no longer subject to U.S. federal tax examinations by tax authorities for the years before 2009. However, the Internal Revenue Service (“IRS”) could adjust certain unused tax attributes carried forward from tax years prior to 2009.
We recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such a position are measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. However, there are no material unrecognized tax benefits as of December 31, 2014, 2013, and 2012. Furthermore, we do not anticipate any significant changes in our unrecognized tax benefits over the next 12 months.
We recognize interest and penalties related to our liabilities for uncertain tax positions in income tax expense. However, during the years ended December 31, 2014, 2013, and 2012, we did not have any accrued interest or penalties associated with any unrecognized tax benefits.
11. Collaboration and License Agreements
Emixustat Collaboration
In 2008, we entered into a definitive agreement with Otsuka to co-develop and commercialize emixustat, our compound, for the dry form of AMD and for other potential indications in the United States, Canada, and Mexico (“Shared Territory”). Under the agreement, we retained all rights in Europe, South America, Central America, the Caribbean, and Africa (“Acucela Territory”), and Otsuka acquired the exclusive development and commercialization rights to the compound in Asia, the Middle East, and selected markets in the rest of the world (“Otsuka Territory”). Otsuka paid us a $5.0 million nonrefundable up-front license fee upon its entry into the agreement.
Under the agreement, Otsuka agreed to fund all development activities in the Shared Territory through Phase 2, up to $40.0 million. If the Phase 2 development costs exceed $40.0 million, Otsuka could have, at its sole discretion, either (i) terminated the agreement or (ii) continued the agreement and share equally with us all Phase 2 development costs in excess of $40.0 million. In 2012, the cost of development activities exceeded $40.0 million and Otsuka agreed to continue the agreement and equally share development costs with us. Phase 3 costs are to be shared equally by Otsuka and us under the agreement. In addition, under the agreement, we have the potential to receive development milestones totaling $82.5 million.
F-18
The co-development portion of the agreement is governed by a Joint Development Committee (“JDC”). We may earn development milestones as follows:
i. | Initial Indication—$55.0 million |
a. | $5.0 million upon initiation of a Phase 2b/3 clinical trial in the United States (received in the year ending December 31, 2013) |
b. | $5.0 million upon initiation of a Phase 3 clinical trial in the United States, or the filing of a New Drug Application (“NDA”) with the FDA in the United States, if a second Phase 3 clinical trial is not needed |
c. | $15.0 million upon filing of a NDA with the FDA in the United States |
d. | $20.0 million upon receipt of approval by the FDA of an NDA in the United States |
e. | $10.0 million upon receipt of approval by the regulatory authority of a marketing approval application in Japan |
ii. | Second Indication—$27.5 million |
a. | $5.0 million upon initiation of a Phase 3 clinical trial in the United States |
b. | $7.5 million upon filing of an NDA with the FDA in the United States |
c. | $10.0 million upon receipt of approval by the FDA of an NDA in the United States |
d. | $5.0 million upon receipt of approval by the regulatory authority of a marketing approval application in Japan |
Under the agreement, Otsuka will fund our share of the Phase 2 and Phase 3 development costs in the form of a secured promissory note. The promissory note provides that (a) interest will accrue daily and be calculated on the basis of 360 days per year and be payable on all amounts advanced to us from the date of advance until paid in full; (b) unpaid interest will compound annually; and (c) the applicable interest rate will be adjusted quarterly to reflect the then-effective rate equal to the three-month London InterBank Offered Rate (“LIBOR”) in the “Money Rates” column of The Wall Street Journal as of the first business day of each calendar quarter, plus 3%; and (d) all amounts are payable in U.S. dollars. The agreement includes a security interest agreement that grants Otsuka a first priority interest on our interests in net profits and royalty payments, and on our interests in ownership of the related collaboration compounds and collaboration products and the underlying intellectual property rights, both in the Shared Territory and the Acucela Territory.
The loan is repayable only in the event that proceeds are generated by any future product sales under the collaboration agreement or by the sale or license of collaboration compounds and collaboration products developed under the agreement outside North America and Otsuka’s sole territory.
As the agreement contains elements of funded development, we evaluated the agreement to determine if our obligation to Otsuka under the secured promissory note should be accounted for as a liability to repay a loan or as an obligation to perform contractual services. To conclude that a liability to repay a loan does not exist, the transfer of the financial risk involved with research and development from us to Otsuka must be substantive and genuine. We have determined that our obligation to Otsuka should be accounted for as an obligation to perform contractual services because repayment depends solely on the results of development having future economic benefit. Consequently, amounts received from Otsuka for our share of development costs under the agreement are recognized as revenue. Through the years ended December 31, 2014, 2013, and 2012, we had recognized cumulative revenue of approximately $49.7 million, $32.4 million, and $15.4 million respectively, which is contingently repayable as described above. As of December 31, 2014 and 2013, the contingently repayable funding has accrued $2.5 million and $1.2 million of interest, respectively, which is contingently repayable along with the above.
Upon commercialization, we may exercise our option to co-promote in the Shared Territory on a country-by-country basis. In markets where we opt to co-promote the product, Otsuka and we equally share all expenses and profits from sales of the product in the Shared Territory. If we do not elect to co-promote in a country or countries in the Shared Territory, Otsuka shall pay us royalties on the annual aggregate net sales of collaboration products in the country or countries in the Shared Territory for which we did not elect to participate in co-promotion. Each party shall pay the other party a royalty of 2% on annual aggregate net sales of collaboration products in their sole territories. In addition, we have the potential to receive net sales milestones totaling $175.0 million. The co-promotion arrangement is governed by a Joint Commercialization Committee (“JCC”). The milestones are as follows:
i. | All Indications |
a. | $25.0 million upon reaching $250.0 million in aggregate annual worldwide sales of all collaboration products |
F-19
b. | $50.0 million upon reaching $500.0 million in aggregate annual worldwide sales of all collaboration products |
c. | $100.0 million upon reaching $1.0 billion in aggregate annual worldwide sales of all collaboration products |
The agreement also includes a three-year research program (the “Research Program”), the purpose of which was to identify a second indication for the lead collaborative compound and to conduct development on a backup compound for the collaborative compound. During the three years of the Research Program, which ended in 2011, Otsuka paid us $5.0 million per year, payable on a quarterly basis. The agreement also provides Otsuka with a right of first negotiation to license new compounds discovered or developed by us (independent of the collaboration activities) during the agreement term.
Our agreement with Otsuka is a multiple element arrangement, and we have determined that the elements within the arrangement consist of the license, the Research Program, and research and development services.
The license granted to Otsuka was determined to be a separate unit of accounting because it has value to Otsuka on a standalone basis. Because Otsuka may license and develop the intellectual property independent of the development or research program services that are to be provided by us, we conducted a net present value valuation of the license, and it was determined that the estimated standalone selling price for the license at inception of the agreement exceeded the arrangement consideration received for the license fee. Since the value assigned to a delivered element cannot exceed the arrangement consideration, the arrangement consideration of $5.0 million due and paid upon execution of the agreement was assigned to the license.
We have determined that the activities associated with development meet the criterion for a separate unit of accounting, since these services have value to Otsuka on a standalone basis. BESP is based on the Company’s analysis of the value of the services provided and consideration of the fees charged by third party vendors for similar development services and represent our BESP. Revenue from development efforts is recognized as services are performed. In the years ended December 31, 2014, 2013 and 2012, we recognized $35.4 million, $39.2 million and $19.3 million, respectively, of revenue associated with development activities.
We have determined that the activities associated with the Research Program meet the criterion for a separate unit of accounting, since these services have value to Otsuka on a standalone basis. The types of services contemplated under the Research Program may be performed by a third party. We have determined that the fees charged for the research services are competitive with the price other third-party vendors charge for similar research services. Our BESP of the selling price for the Research Program was $15.0 million, which equals the agreement consideration. Revenue from the research activities was recognized under a proportional performance model.
We evaluated the development and net sales milestones in the arrangement and determined that they each meet the criteria of a milestone under ASC 605-28, Revenue Recognition-Milestone Method. We recognize consideration that is contingent upon achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. During the year ended December 31, 2013, we received and recognized as revenue the $5.0 million milestone payment associated with the initiation of the Phase 2b/3 clinical trial. No development or net sales milestones were achieved during the years ended December 31, 2014 or 2012.
OPA-6566 Collaboration
In 2010, Otsuka and we entered into a definitive agreement to develop OPA-6566, Otsuka’s proprietary compound for the treatment of glaucoma. The agreement grants us an opt-in right to co-develop and co-promote OPA-6566 in the United States. Until we exercise our opt-in right, Otsuka will have responsibility for directing development activities and costs. Upon our exercise of the opt-in right, Otsuka will grant us additional opt-in rights, which include: (1) the right to co-develop and co-promote OPA-6566 for ophthalmological indications in the United States other than for glaucoma; (2) the right to co-develop and co-promote new formulations of OPA-6566 for glaucoma in the United States; and (3) a right of first negotiation to co-develop and co-promote other adenosine A2a receptor agonist compounds for the treatment of ophthalmologic diseases in the United States.
We evaluated the agreement and determined that the development activities under the agreement represented the only deliverable under the arrangement. Revenue from development activities is recognized as services are performed. During the years ended December 31, 2014, 2013 and 2012, we recognized $0, $1.5 million, and $8.1 million respectively, of revenues in performance of the agreement.
Rebamipide Collaboration
F-20
In 2008, Otsuka and we entered into a definitive agreement to co-develop rebamipide, Otsuka’s proprietary compound for the treatment of dry eye. Under the agreement, the parties agreed to collaborate in the clinical development efforts for rebamipide in the United States. Otsuka paid us a $2.0 million up-front payment and, under the agreement, we had the potential to receive clinical development milestones and royalties on net sales of the product in the United States and the European Union. Under the agreement, Otsuka was responsible for all clinical development and commercialization expenses.
We evaluated the agreement and determined that the clinical development activities represented the only deliverable under the arrangement. Revenue from clinical development efforts is recognized as services are performed. During the years ended December 31, 2014, 2013 and 2012 we recognized $0, $12.3 million, and $14.0 million, respectively, of revenue associated with the rebamipide clinical development activities. We evaluated the development milestones under the agreement and determined that they each meet the criteria of a substantive milestone. We recognize consideration that is contingent upon achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. No development milestones were achieved during the year ended December 31, 2014. In 2012, we received and recognized as revenue the $5.0 million milestone payment associated with the initiation of the Phase 3 clinical trial.
In September 2013, Otsuka elected to end its rebamipide co-development agreement with us. As a result, we recognized as revenue, in the year ended December 31, 2013, a $2.0 million upfront payment from Otsuka that had been deferred due to refund provisions. These refund provisions expired with the end of the co-development agreement.
Continued Involvement of the Former CEO
The Company’s two remaining collaboration arrangements with Otsuka require the continuing involvement of our Chairman and Founder and former CEO, Dr. Ryo Kubota. In the event of the departure of Dr. Kubota from the Company or a change in his role or responsibilities with the Company, the arrangements are subject to termination, at the option of Otsuka. The Company has consulted with Otsuka regarding its transition of the chief executive officer role from Dr. Kubota to Brian O'Callaghan. The Company is currently discussing with Otsuka to amend the Emixustat Agreement to remove the clauses permitting Otsuka to terminate the agreement due to one of the provisions relating to Dr. Kubota. Otsuka has given the Company verbal assurances that they do not intend to exercise their right to terminate the Emixustat Agreement based on Dr. Kubota's changed role. For each agreement, this provision expires upon the approval of the NDA for the first indication in the United States.
12. Net Income (Loss) Per Share
Net income (loss) per share attributable to common shareholders is presented in conformity with the two-class method required for participating securities for periods in which we have net income. Prior to the IPO, all series of convertible preferred stock were considered to be participating securities, as the holders were entitled to participate in any dividends prior and in preference to dividends declared or paid on the common stock. Undistributed earnings allocated to these participating securities were subtracted from net income in determining net income attributable to common shareholders.
Immediately prior to the closing of our IPO, all outstanding shares of preferred stock were converted to common. We issued 9,200,000 shares of common stock in the IPO. In addition, 3,636,365 shares of common stock were issued upon the conversion of the contingently convertible debt held by a related party. As a result, as of December 31, 2014, common stock is our only outstanding class of capital stock.
Basic net income (loss) per share is calculated by dividing net income attributable to common shareholders by the weighted average number of shares outstanding for the period. Diluted net income (loss) per share is calculated by dividing net income (loss) attributable to common shareholders by the weighted average number of shares of the common stock outstanding and other dilutive securities outstanding during the period. The potential dilutive shares of our common stock include the exercise of outstanding stock options that are dilutive and restricted stock units.
The following tables reconcile the numerator and denominator used to calculate diluted net income per share for the periods presented (in thousands):
F-21
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Numerator: | |||||||||||
Net income (loss) attributable to common shareholders | $ | (2,006 | ) | $ | 1,161 | $ | 1,122 | ||||
Denominator: | |||||||||||
Weighted average shares outstanding—basic | 32,869 | 11,964 | 11,901 | ||||||||
Dilutive effect of stock options and RSUs | — | 391 | 257 | ||||||||
Weighted average shares outstanding—diluted | 32,869 | 12,355 | 12,158 | ||||||||
For the year ended December 31, 2014, 185,551 stock options and RSUs were excluded from the calculation of diluted net income (loss) per share because the impact was anti-dilutive.
13. Commitments and Contingencies
Leases
We lease laboratory and corporate office space under operating leases. On June 26, 2014, we entered into an agreement for the lease of approximately 38,723 square feet of office space in our headquarters building in Seattle, Washington. The term of the lease commenced on January 1, 2015 and, subject to the terms of the lease, will expire on either November 30, 2021 or February 22, 2022. We received a tenant leasehold improvement allowance of $1.2 million in connection with the commencement of this lease in 2015.
On September 19, 2014, we entered into an amendment of our lease agreement for the lease of approximately 17,488 square feet of laboratory and office space in Bothell, Washington. The amendment extended the expiration date of the original lease from February 28, 2015 to February 28, 2017 and reduced the basic annual rent to approximately $0.4 million, subject to adjustment pursuant to the terms of the lease. The lease agreement includes an option to terminate the lease ten months early.
Lease incentives are recognized as deferred rent liabilities and amortized to rent expense over the term of the lease. Minimum rent payments under operating leases are recognized on a straight-line basis over the term of the lease, including any periods of free rent and reduced rent.
Future minimum lease payments under operating leases (with initial or remaining lease terms in excess of one year) as of December 31, 2014 are as follows (in thousands):
Payments Due by Period | |||||||||||||||||||||||||||
2015 | 2016 | 2017 | 2018 | 2019 | Thereafter | Total | |||||||||||||||||||||
Operating lease obligations | $ | 1,324 | $ | 1,295 | $ | 997 | $ | 968 | $ | 1,007 | $ | 2,039 | $ | 7,630 | |||||||||||||
Total | $ | 1,324 | $ | 1,295 | $ | 997 | $ | 968 | $ | 1,007 | $ | 2,039 | $ | 7,630 | |||||||||||||
Rent expense was $1.0 million, $1.0 million, and $0.8 million, respectively, in the years ended December 31, 2014, 2013, and 2012, respectively.
Litigation
From time to time, we may be subject to legal proceedings and claims in the ordinary course of business. We are not currently a party to any material legal proceedings, and to our knowledge none is threatened. There can be no assurance that future legal proceedings arising in the ordinary course of business or otherwise will not have a material adverse effect on our financial position, results of operations or cash flows.
14. Related-Party Transactions
Peter Kresel, M.B.A., a member of the Board, received payments from us for consulting services and reimbursement of direct expenses. This consulting relationship terminated in January 2014. Mr. Kresel’s payments for
F-22
consulting services and expense reimbursements were $0, $0.3 million, and $0.2 million during 2014, 2013, and 2012 respectively.
SBI Holdings, Inc. (a related party), one of our shareholders, was the holder for our contingently convertible debt. In connection with our IPO, the contingently convertible debt automatically converted into 3,636,365 shares of common stock(see Note 6).
15. Quarterly Information (Unaudited)
The following tables set forth our unaudited quarterly statement of operations data for each of the last eight quarters in the period ended December 31, 2014. The unaudited quarterly statement of operations data below have been prepared on the same basis as the audited financial statements included elsewhere in this Form 10-K and reflect all necessary adjustments, consisting only of normal recurring adjustments, that we believe are necessary for a fair statement of this information. The results of historical quarters are not necessarily indicative of the results of operations for a full year or any future period:
March 31 | June 30 | September 30 | December 31 | ||||||||||||
(In thousands, except per share amounts) | |||||||||||||||
2014 | |||||||||||||||
Revenue from collaborations | $ | 10,546 | $ | 9,086 | $ | 8,119 | $ | 7,645 | |||||||
Net income (loss) | 54 | 71 | (1,536 | ) | (1) | (595 | ) | ||||||||
Net income (loss) attributable to common shareholders | 54 | 71 | (1,536 | ) | (595 | ) | |||||||||
Basic net income (loss) per share attributable to common shareholders | $ | — | $ | — | $ | (0.04 | ) | $ | (0.02 | ) | |||||
Diluted net income (loss) per share attributable to common shareholders | $ | — | $ | — | $ | (0.04 | ) | $ | (0.02 | ) | |||||
(1) In the third quarter of 2014, we recorded a valuation allowance against our deferred tax assets of $1.6 million due to expected future losses as a result of our new strategic plan.
March 31 | June 30 | September 30 | December 31 | ||||||||||||
(In thousands, except per share amounts) | |||||||||||||||
2013 | |||||||||||||||
Revenue from collaborations | $ | 15,980 | $ | 11,023 | $ | 14,692 | $ | 11,252 | |||||||
Net income (loss) | 3,711 | (20 | ) | 1,420 | (812 | ) | |||||||||
Net income (loss) attributable to common shareholders | 999 | (20 | ) | 382 | (812 | ) | |||||||||
Basic net income (loss) per share attributable to common shareholders | $ | 0.08 | $ | — | $ | 0.04 | $ | (0.07 | ) | ||||||
Diluted net income (loss) per share attributable to common shareholders | $ | 0.08 | $ | — | $ | 0.03 | $ | (0.07 | ) | ||||||
16. Subsequent Events
Special Shareholders Meeting
On January 28, 2015, we received a letter from SBI Holdings, Inc. (SBI), the parent company of several of our shareholders, demanding that we hold a special meeting of our shareholders for the purposes of removing our current board members, other than Dr. Kubota, and electing a slate of directors proposed by SBI. In connection with this demand, SBI granted Dr. Kubota an irrevocable proxy over the shares held collectively by SBI, giving Dr. Kubota voting authority over shares representing over 50% of our outstanding stock. On March 3, 2015, SBI and Dr. Kubota filed a lawsuit in Washington
state court seeking an order compelling us to hold a special shareholder meeting on or before April 28, 2015, and to provide our shareholders written notice of that meeting as soon as practicable but no later than March 31, 2015. On March 13, 2015, the Washington state superior court presiding over this lawsuit, issued an order requiring us to hold a special shareholders meeting no later than May 1, 2015 and to give notice of such a meeting as soon as practicable. We have announced that we plan to hold the special shareholders meeting on May 1, 2015 (Pacific Daylight Time) at our headquarters and have set a record date for shareholders of record as March 19, 2015 (Pacific Daylight Time).
CEO Bonus
On March 24, 2015, our Board of Directors approved the February 24, 2015 resolutions of the Compensation Committee of our Board of Directors (the “Committee”) regarding the payment of a discretionary bonus of $515,520 to Mr. O’Callaghan, our CEO and President, on April 15, 2015, provided Mr. O’Callaghan remains chief executive officer as of March 31, 2015, in lieu of the performance-based bonus in the same amount provided for under his employment agreement, dated October 14, 2014.
CEO Severance Escrow
Under Mr. O'Callaghan's employment agreement, dated October 14, 2014, if Mr. O’Callaghan’s employment is terminated without Cause or terminates for Good Reason (as such terms are defined in his employment agreement), he will be entitled to receive 18 months of salary, up to 18 months of the premiums for him and his family to obtain health benefit coverage provided under our COBRA program, and a pro-rated portion of his annual bonus (“the CEO severance amounts”). On March 24, 2015, our Board of Directors approved the creation of a segregated interest-bearing account equal to the CEO severance amounts. The funds will remain the property of the Company until amounts are due to Mr. O’Callaghan under the terms of his amended employment agreement.
Severance and Change in Effective Control Agreements
On March 24, 2015, our Board of Directors approved the terms of a Severance and Change in Effective Control Agreement to be entered into with each member of our management team and certain other employees. The Severance and Change in Effective Control Agreement provides that if the employee terminates for any reason or for no reason (including disability), voluntarily resigns for good reason (as defined in the agreement), or the employee dies, and the termination occurs within the six month period following a Qualifying Change in Effective Control, the employee will be entitled to an amount equal to the sum of six months of his or her monthly base salary, plus 50% of the employee’s annual target bonus for 2015, plus the premiums required to continue the employee’s group health care coverage for a period of six months following termination, which will be “grossed up” to cover taxes. In addition, upon a qualifying termination, the employee will receive an additional 12 months of vesting on any outstanding equity awards. A “Qualifying Change in Effective Control” is defined to mean that a majority of members of our Board is replaced by directors whose appointment or election is not endorsed by a majority of the members of our Board before the date of the appointment or election, which qualifies as a change in effective control under U.S. treasury regulations. The agreements terminate upon the earlier of December 31, 2015 or the employee’s termination (unless the termination is within six months following a Qualifying Change in Effective Control).
Retention Pool
On February 24, 2015, the Committee approved the creation of a pool of $600,000 to be distributed at the discretion of the Committee to employees who remain employed with us on December 31, 2015. Allocations of the pool will not be determined until the fourth quarter of 2015. All employees, other than Mr. O’Callaghan, are eligible to receive payments from this pool.