Attached files
| file | filename |
|---|---|
| EX-31.2 - EX-31.2 - VERACYTE, INC. | a2223500zex-31_2.htm |
| EX-32.2 - EX-32.2 - VERACYTE, INC. | a2223500zex-32_2.htm |
| EX-32.1 - EX-32.1 - VERACYTE, INC. | a2223500zex-32_1.htm |
| EX-23.1 - EX-23.1 - VERACYTE, INC. | a2223500zex-23_1.htm |
| EX-31.1 - EX-31.1 - VERACYTE, INC. | a2223500zex-31_1.htm |
| EX-23.2 - EX-23.2 - VERACYTE, INC. | a2223500zex-23_2.htm |
| EX-10.26 - EX-10.26 - VERACYTE, INC. | a2223500zex-10_26.htm |
| EXCEL - IDEA: XBRL DOCUMENT - VERACYTE, INC. | Financial_Report.xls |
Use these links to rapidly review the document
TABLE OF CONTENTS
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| (Mark One) | ||
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2014 |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the transition period from to |
||
Commission File Number 001-36156
VERACYTE, INC.
(Exact Name of Registrant as Specified in its Charter)
| Delaware (State or Other Jurisdiction of Incorporation or Organization) |
20-5455398 (I.R.S. Employer Identification Number) |
7000 Shoreline Court, Suite 250
South San Francisco, California 94080
(Address of Principal Executive Offices, Including Zip Code)
(650) 243-6300
(Registrant's Telephone Number, Including Area Code)
Securities Registered Pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered | |
|---|---|---|
| Common Stock, par value $0.001 per share | The NASDAQ Stock Market LLC |
Securities Registered Pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer ý (Do not check if a smaller reporting company) |
Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
As of June 30, 2014, the aggregate market value of voting and non-voting common stock held by non-affiliates of the registrant was approximately $122.4 million, based on the closing price of the common stock as reported on The NASDAQ Global Market for that date.
The number of shares of the registrant's Common Stock outstanding as of March 2, 2015 was 22,551,242.
DOCUMENTS INCORPORATED BY REFERENCE
Item 10 (as to directors and Section 16(a) Beneficial Ownership Reporting Compliance), 11, 12, 13 and 14 of Part III incorporate by reference information from the registrant's proxy statement to be filed with the Securities and Exchange Commission in connection with the solicitation of proxies for the registrant's 2015 Annual Meeting of Stockholders to be held on May 18, 2015.
This report contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. When used in this report, the words "expects," "anticipates," "intends," "estimates," "plans," "believes," "continuing," "ongoing," and similar expressions are intended to identify forward-looking statements. These are statements that relate to future events and include, but are not limited to, the factors that may impact our financial results; our expectations regarding revenue; our expectations with respect to our future research and development, general and administrative and selling and marketing expenses and our anticipated uses of our funds; our expectations regarding capital expenditures; our anticipated cash needs and our estimates regarding our capital requirements; our need for additional financing; potential future sources of cash; our business strategy and our ability to execute our strategy; our ability to achieve and maintain reimbursement from third-party payers at acceptable levels; our belief that our published evidence provides a basis for inclusion of our Afirma GEC test in practice guidelines; the estimated size of the global markets for our tests and our future tests; the potential benefits of our tests and any future tests we may develop to patients, physicians and payers; the factors we believe drive demand for and reimbursement of our tests; our ability to sustain or increase demand for our tests; our intent to expand into other clinical areas; our ability to develop new tests, including tests for lung cancer and interstitial lung disease, and the timeframes for development or commercialization; our ability to get our data and clinical studies accepted in peer-reviewed publications; our dependence on and the terms of our agreements with Genzyme and TCP, and on other strategic relationships, and the success of those relationships; our beliefs regarding our laboratory capacity; the applicability of clinical results to actual outcomes; our expectations regarding our international expansion, including entering new international markets and the timing thereof; the occurrence, timing, outcome or success of clinical trials or studies; the ability of our tests to impact treatment decisions; our beliefs regarding our competitive position; our ability to compete with potential competitors; our compliance with federal, state and international regulations; the potential impact of regulation of our tests by the FDA or other regulatory bodies; the impact of new or changing policies, regulation or legislation, or of judicial decisions, on our business; our ability to comply with the requirements of being a public company; the impact of seasonal fluctuations and economic conditions on our business; our belief that we have taken reasonable steps to protect our intellectual property; the impact of accounting pronouncements and our critical accounting policies, judgments, estimates, models and assumptions on our financial results; and anticipated trends and challenges in our business and the markets in which we operate.
Forward-looking statements are based on our current plans and expectations and involve risks and uncertainties which could cause actual results to differ materially. These risks and uncertainties include, but are not limited to, those risks discussed in Part I, Item 1A of this report, as well as risks and uncertainties related to: our limited operating history and history of losses since inception; our ability to increase usage of and reimbursement for the Afirma GEC and any other tests we may develop, including Percepta; our dependence on a limited number of payers for a significant portion of our revenue; the complexity, time and expense associated with billing and collecting for our test; current and future laws, regulations and judicial decisions applicable to our business, including potential regulation by the FDA or by regulatory bodies outside of the United States; changes in legislation related to the U.S. healthcare system; our dependence on strategic relationships, collaborations and co-promotion arrangements; unanticipated delays in research and development efforts; our ability to develop and commercialize new products and the timing of commercialization; our ability to successfully enter new product or geographic markets; our ability to conduct clinical studies and the outcomes of such clinical studies; the applicability of clinical results to actual outcomes; trends and challenges in our business; our ability to compete against other companies and products; our ability to protect our intellectual property; and our ability to obtain capital when needed. These forward-looking statements speak only as of the date hereof. We expressly
1
disclaim any obligation or undertaking to update any forward-looking statements contained herein to reflect any change in our expectations with regard thereto or any change in events, conditions or circumstances on which any such statement is based.
When used in this report, all references to "Veracyte," the "company," "we," "our" and "us" refer to Veracyte, Inc.
Veracyte, Afirma, Percepta, the Veracyte logo and the Afirma logo are our trademarks or registered trademarks. We also refer to trademarks of other corporations or organizations in this report.
This annual report contains statistical data and estimates that we obtained from industry publications and reports. These publications typically indicate that they have obtained their information from sources they believe to be reliable, but do not guarantee the accuracy and completeness of their information. Some data contained in this annual report is also based on our internal estimates. Although we have not independently verified the third-party data, we are responsible for its inclusion in the annual report and believe it to be reasonable.
Overview
We are a molecular diagnostics company, pioneering the field of molecular cytology with genomic solutions that resolve diagnostic ambiguity and enable physicians to make more informed treatment decisions at an early stage in patient care. We believe that diagnostic ambiguity is a significant problem impacting our healthcare system, resulting in hundreds of thousands of patients undergoing unnecessary, invasive procedures and wasting billions of healthcare dollars each year. We target diseases in which large numbers of patients undergo invasive and costly diagnostic procedures that could be avoided with a more accurate diagnosis from a cytology sample taken preoperatively. By improving diagnosis preoperatively, we help patients avoid unnecessary invasive procedures and surgeries while reducing healthcare costs. We are focused on the continued growth of our endocrinology franchise and the entry into our second clinical indication, pulmonology, in 2015. Together, we believe these two market opportunities offer a near-term estimated addressable market of over $2 billion.
In 2011, we launched our first commercial solution, the Afirma® Thyroid FNA Analysis, for use in thyroid cancer diagnosis. Our offering centers on our proprietary Afirma Gene Expression Classifier, or GEC, which is used to resolve diagnostic ambiguity among the more than 525,000 patients who undergo fine needle aspiration, or FNA, biopsies each year in the United States to assess potentially cancerous thyroid nodules. The GEC helps physicians reduce the number of unnecessary surgeries by employing a proprietary 142-gene signature to preoperatively determine whether thyroid nodules previously classified by cytopathology as indeterminate can be reclassified as benign. As of March 2015, we have received nearly 160,000 FNA samples and have performed more than 30,000 GEC tests to resolve indeterminate cytopathology results, helping an estimated 15,000 patients avoid unnecessary surgery and reducing healthcare costs by an estimated $200 million. We launched our first product extension—the Afirma Malignancy Classifiers—in 2014, which comprise genomic tests for medullary thyroid cancer, or MTC, and BRAF V600E mutation status. These genomic tests are intended to preoperatively inform physicians' choice of thyroid surgery when surgery is needed. We believe Afirma offers the most comprehensive solution for the assessment and management of patients with thyroid nodules. We estimate our addressable thyroid nodule diagnostic market opportunity today is approximately $500 million per year in the United States, and we believe that there is an estimated $300 million additional market opportunity for the Afirma GEC internationally.
The Afirma GEC is supported by multiple, peer-reviewed published studies, including a prospective, multicenter, double-blind clinical validation study published in The New England Journal of Medicine in 2012, which suggests that the test can reduce the number of unnecessary surgeries by 50%. As of March 2015, the GEC is recommended in leading practice guidelines and is covered for 145 million patient lives in the United States, including through Medicare and many commercial insurance plans. Additionally,
2
Afirma is contracted for nearly 100 million lives, making us an in-network provider for payers including Medicare, UnitedHealthcare and Cigna.
We market our Afirma solution through our dedicated specialty sales force and through a co-promotion agreement with Genzyme, a subsidiary of Sanofi, which targets the same endocrinologist customers with Thyrogen®. Since the beginning of 2014, we have more than tripled the size of our internal sales force, enabling us to further drive market penetration and expansion for Afirma, in the physician office, or ambulatory practice setting, as well as in institutional accounts and integrated delivery networks, or IDNs. We now offer models that are proven to be effective in each of these market segments and believe we are positioned to continue to drive growth in both. We estimate our addressable thyroid nodule diagnostic market opportunity today is approximately $500 million per year in the United States, and we believe that there is an estimated $300 million additional market opportunity for the Afirma GEC internationally. To date, substantially all of our revenue has been derived from customers we serve in the United States. Our revenue has increased from $11.6 million in 2012, to $21.9 million in 2013 and $38.2 million in 2014.
In September 2014, we acquired Allegro Diagnostics Corp., based in Maynard, Massachusetts, to accelerate our entry into pulmonology, our second planned clinical area. We intend to enter the lung cancer diagnostics market by mid-2015 with the Percepta™ Bronchial Genomic Classifier, a gene expression test utilizing the novel "field of injury" genomic technology, which is designed to resolve diagnostic ambiguity among the approximately 250,000 patients in the United States who undergo bronchoscopy each year to assess potentially cancerous lung nodules. Currently, approximately 40% of these procedures produce non-diagnostic results, which can lead to unnecessary invasive procedures for a definitive diagnosis. We believe the market opportunity for this test is between $350 million and $400 million in the United States, depending on the value we can extract for out test. Percepta has been validated in two multicenter, prospective clinical studies, with data presented on the AEGIS I study at the American Thoracic Society International Conference in 2014 and additional data expected to be released in 2015. We estimate that the number of bronchoscopies—and non-diagnostic results—could expand significantly in the next two to three years as, beginning in early 2015, more than eight million Americans at high risk for lung cancer have become eligible for annual screening through new Affordable Care Act and Medicare coverage.
We plan to expand our footprint in pulmonology in 2016 with the launch of a product designed to preoperatively identify idiopathic pulmonary fibrosis, or IPF, among patients presenting with a suspected interstitial lung disease, or ILD. Our IPF test will target pulmonologists, the same physicians we will address with Percepta, and will also be used to test cytology samples obtained through bronchoscopy. IPF is the most common form of ILD, a group of diseases characterized by chronic, progressive scarring of the lungs, and is often difficult to distinguish from other ILDs or lung conditions. Currently, we believe that many of the estimated 175,000 to 200,000 patients in the United States and Europe who present with suspected ILDs each year may endure months of incorrect or missed diagnoses, undergoing invasive, risky and expensive diagnostic surgeries, or receiving suboptimal treatment. The need for improved IPF diagnosis becomes increasingly important given the availability of new therapies to halt or slow progression of this often fatal disease's progression, which were approved by the Food and Drug Administration, or FDA, in late 2014. In May 2014, we presented proof-of-concept data at the American Thoracic Society International Conference demonstrating the ability of a molecular classifier to accurately identify IPF cases using tissue samples. We expect to present data demonstrating our test's performance on bronchoscopy samples in 2015. We estimate the addressable market for our IPF test to be over $500 million in the United States and Europe.
We believe additional clinical areas offer opportunities for future expansion of our molecular cytology franchise beyond endocrinology and pulmonology. In determining new clinical areas to enter, we will focus on diseases in which a large number of patients undergo invasive and costly diagnostic procedures that could be avoided with a more accurate diagnosis from a cytology sample taken preoperatively. We intend
3
to leverage our demonstrated core capabilities in research and development, clinical development, and managed care and reimbursement to expand our business into other clinical areas of unmet need, either through internal development or through acquisition.
Our Strategy
We believe the market opportunities are significant and have focused our strategic objectives around these three growth vectors:
- •
- Accelerate the Growth of Afirma in Endocrinology. We
expect to continue to invest in driving the adoption of Afirma and expanding our base of prescribing physicians, both in the community physician office market as well as in institutional settings. We
plan to continue to leverage our relationship with Genzyme in the U. S. market and pursue select international markets for entry where attractive regulations and reimbursement exists. We plan to use
our inclusion in guidelines and the extensive library of published evidence on Afirma to date, coupled with our core expertise in managed care, claims adjudication, and billing, to drive broader
coverage determinations, particularly with the Blue Cross and Blue Shield family of payers, as well as additional in-network contracts, to expand reimbursement and smooth our path to profitability.
- •
- Expand into Pulmonology through the Launch of
Percepta. We believe our molecular cytology strategy addresses several unmet clinical needs in pulmonology. Through the acquisition of
Allegro, we plan to launch the Percepta Bronchial Genomic Classifier for lung cancer diagnosis by mid-2015. Our ability to efficiently integrate the company and technologies, including the completion
of all analytical verification studies, has accelerated our launch timing. Using our existing infrastructure, we plan to commence testing of patient samples in our CLIA-certified laboratory by
mid-year and continue to build out the body of evidence that will be required to gain Medicare reimbursement as well as commercial payer coverage. We believe this lung cancer test will serve as the
foundational application to expand our molecular cytology platform within the pulmonology vertical where we plan to introduce a second product to improve the diagnosis of patients suspicious for ILD,
specifically IPF.
- •
- Further Expand our Franchise into Additional
Indications. We intend to leverage our demonstrated core capabilities in research and development, clinical development, and managed
care and reimbursement to expand our business into other clinical areas of unmet need, either through internal development or through acquisition. For each clinical area we target, we deploy a proven
strategy comprised of four key pillars:
- •
- Inform the Right Clinical Question. We focus on developing
genomic tests that answer a relevant clinical question and that, when used at the optimal point in the diagnostic pathway, provide physicians with information that can significantly alter physician
decision-making, enabling patients to avoid unnecessary invasive and costly procedures. We then work with key opinion leaders and other clinicians to understand the performance criteria that will be
needed for a new test to give physicians confidence to change clinical-care decisions. Only when we have pinpointed this information do we then deploy the appropriate science to develop the test.
- •
- Develop Proprietary Science and Validate in Well-designed Clinical Trials. Once we know the parameters of the test we need to develop to change patient care, we apply rich, broad-based genomic science based on our expertise in biomarker discovery and algorithm development. We utilize proprietary technology, intellectual property and scientific know-how to extract rich genomic information from tiny cytology samples, sometimes with only nanogram quantities of biological material, to answer our target clinical question. We then conduct prospective, blinded, multicenter clinical validation studies and seek to obtain publication in peer-reviewed journals to establish the clinical performance of our test.
4
- •
- Demonstrate Clear Value. We build into our commercialization
strategy the steps that will be needed to prove that our tests do indeed change clinical practice and provide healthcare cost savings. To do this, we design and initiate clinical utility and
cost-effectiveness studies early in the process so that we will be able to quickly and efficiently demonstrate value to physicians and payers.
- •
- Achieve Coverage and Reimbursement Success. By developing the clinical evidence for our tests, which is then published in peer-reviewed publications, we create compelling evidence for our tests to be included in clinical practice guidelines, helping to establish a new routine standard of care. We believe guideline inclusion, along with the capabilities we have built in managed care and claims adjudication, is key to successful payer coverage, contracts and reimbursement. Our team combines expertise in advocating for positive coverage decisions with specific insights into what tactical steps will maximize reimbursement from each payer. As a result, we have developed detailed knowledge of the intricacies of specific payer practices and requirements, which informs our strategy across indication selection, clinical study design, marketing and sales.
Limitations of Disease Diagnosis Today
Surgical pathology has long been part of the standard of care for diagnosis of numerous complex diseases, including many types of cancer and lung diseases. Patient samples collected from surgeries allow multiple slices, or sections, of the tissue to be stained, permitting a pathologist to use a microscope to evaluate the shape and structure of the cells in question to diagnose the sample. However, surgical pathology by definition requires an invasive procedure. Cytopathology, or the analysis of small numbers of cells using minimally invasive methods (which we refer to as cytology samples), is designed to provide a pathologic diagnosis using a small biopsy. It is often the first step in the diagnostic process because it offers a less-invasive and cost-effective alternative to surgery. However, cytology samples are often small and non-uniform, which can make definitive diagnoses difficult. The high rate of ambiguity in diagnosis using cytology samples today results in many patients undergoing other subsequent invasive procedures, often including surgery, to obtain an accurate diagnosis.
The role of genomic information in medical practice is evolving rapidly and has affected the diagnosis of disease as well as treatment decisions. Over the past decade, molecular diagnostic tests that analyze genomic material from surgical tissue samples have emerged as an important complement to evaluations performed by pathologists. Information at the molecular level enables one to understand more fully the makeup and specific subtype of disease to improve diagnosis. In many cases, the genomic information derived from these samples can help guide treatment decisions as part of the standard of care. However, due to limitations of available technologies, many of these molecular tests require relatively large quantities of tissue with specific levels of cellularity, which most often must be obtained through an invasive surgical procedure.
Cytology samples offer a more attractive alternative for early, less invasive and less costly diagnosis. These samples are commonly obtained using minimally invasive methods, such as FNA biopsies, washings, brushings, lavages or bronchoscopy biopsies, from which to diagnose various diseases. Physicians typically collect these without performing surgery, and therefore have the potential to offer a lower cost and less invasive approach to disease diagnosis. Cytology samples, however, are challenging for both traditional cytopathology, as well as molecular cytology, due to the small amount of cellular material obtained in the collection process and the often non-uniform nature of the collected tissue. The high rate of ambiguity in diagnosis on cytology samples today results in many patients undergoing other subsequent invasive procedures, often including surgery, to obtain an accurate diagnosis.
5
Extracting clinically meaningful genomic information from these small, heterogeneous cytology samples offers the potential to reduce ambiguity in diagnosis prior to surgery and inform treatment decisions at a much lower cost to the healthcare system.
Our Solutions
We are pioneering the field of molecular cytology with genomic solutions that resolve diagnostic ambiguity and enable physicians to make more informed treatment decisions at an early stage in patient care. We target diseases in which a large number of patients undergo invasive and costly diagnostic procedures that could be avoided with a more accurate diagnosis from a cytology sample taken preoperatively. In contrast to molecular diagnostics developed for surgical tissue, our solutions solve many of the technical challenges associated with generating analytically valid and clinically relevant genomic information from very small, heterogeneous cytology samples. By improving diagnosis before surgery, we help patients avoid unnecessary invasive procedures while reducing healthcare costs.
Our molecular cytology products are designed to deliver a number of benefits to physicians, payers and patients, including a reduction of unnecessary surgeries, lower healthcare costs, and actionable information from a single patient visit with a single diagnostic procedure.
Our initial focus is on the clinical areas of endocrinology, where we have made significant inroads to date, and pulmonology, which we expect to enter by mid-2015. Together, we believe these two market opportunities offer a near-term estimated addressable market of over $2 billion.
Endocrinology
We entered the endocrinology market in January 2011 with our Afirma Thyroid FNA Analysis, which is included in leading practice guidelines and gaining market share in thyroid cancer diagnosis. Our offering centers on our proprietary Afirma GEC, which is used to resolve diagnostic ambiguity among the more than 525,000 patients who undergo FNA procedures each year to assess thyroid nodules that are potentially cancerous. We launched our first product extension—the Afirma Malignancy Classifiers—comprising tests for MTC and BRAF V600E gene mutation status to provide results that might preoperatively inform surgery selection for those patients who need surgery.
As of March 2015, we have received nearly 160,000 FNA samples and have performed more than 30,000 GEC tests to resolve indeterminate cytopathology results, helping an estimated 15,000 patients avoid unnecessary surgery and reducing healthcare costs by an estimated $200 million. The Afirma GEC is covered as a medically necessary test for 145 million lives, including through Medicare and many commercial payers including UnitedHealthcare, Cigna, Aetna, Humana and leading Blue Cross and/or Blue Shield plans such as Highmark, Horizon Blue Cross, and Blue Shield of California. Afirma is contracted for nearly 100 million lives, making us an in-network provider for payers including Medicare, UnitedHealthcare and Cigna, which facilitates adoption. In February 2015, we obtained a separate CPT code, or Current Procedural Terminology code, for the Afirma GEC, which we believe will facilitate our progress with payer coverage contracts, and reimbursement. The new code became effective March 1, 2015.
We estimate that, of the total number of FNAs performed annually, we have a market share of approximately 20% among office-based practices and 10% among institutional accounts.
Pulmonology
We have two products in the pipeline that address unmet clinical needs in the diagnosis of lung cancer and ILDs, specifically IPF. Our lung cancer test, the Percepta Bronchial Genomic Classifier, is designed to help resolve diagnostic ambiguity among the approximately 250,000 patients each year who undergo bronchoscopy to determine if lung nodules are benign or cancerous. Our solution is intended to identify
6
patients with non-diagnostic bronchoscopy results whose nodules are at low risk of being cancerous so that these patients can avoid surgery and be monitored with low-dose computed tomography, or LDCT, screening instead. We plan to launch Percepta by mid-2015 and expect to see meaningful revenue from the test in 2017.
We believe our introduction of Percepta will facilitate the subsequent launch of our IPF test, which will target the same customer, pulmonologists, and will similarly be run on cytology samples obtained through bronchoscopy. Our IPF test is in development and intended to preoperatively identify patients with IPF among those presenting with a suspected ILD, so that these patients can obtain an accurate diagnosis and proper treatment sooner—without the need for invasive surgery. We are collaborating with more than 25 clinical sites in the United States and Europe to develop our IPF test, which we plan to launch in 2016.
The Endocrinology Market
Our Afirma solution addresses the large and growing thyroid nodule diagnostic market, which is burdened with significant ambiguity in cytopathology, offering the potential to reduce the rate of surgery needed to diagnose and subsequently treat thyroid cancers.
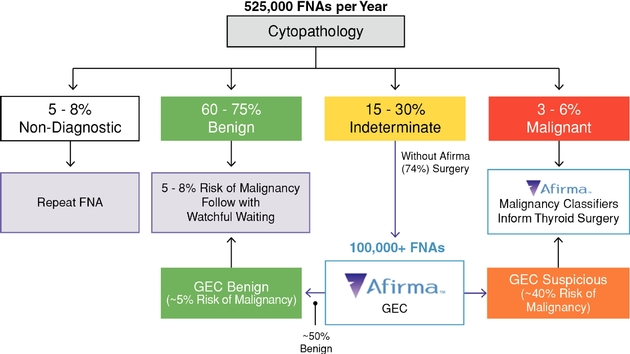
Thyroid cancer is the fastest growing cancer in the United States, according to the American Cancer Society, and evaluation of thyroid nodules—the most common indicator of thyroid cancer—is rapidly increasing the number of thyroid FNAs conducted. Approximately 525,000 thyroid FNAs were performed in the United States in 2011, which is more than double the number of FNAs performed in 2006. We estimate our addressable thyroid nodule diagnostic market opportunity today is approximately $500 million per year in the United States, consisting of an estimated $100 million in cytopathology testing, $350 million in Afirma GEC tests performed on indeterminate cytopathology samples and an additional $40 million related to our Afirma Malignancy Classifiers. Our estimates are based on the product of FNA volumes and the estimated reimbursement per test for both cytology and the Afirma GEC, not our list price at which we bill. We believe that there is an estimated $300 million additional market opportunity for the Afirma GEC internationally.
7
The biology of thyroid cells is complex. Approximately 15% to 30% of thyroid nodule FNAs performed in the United States are deemed indeterminate following cytopathology review, meaning they cannot be diagnosed as definitively benign or malignant by cytopathology alone. Because the risk of malignancy in such patients ranges from 20% to 30%, clinical practice guidelines have traditionally recommended that most of these patients undergo surgery to remove all or part of the thyroid for a definitive diagnosis. Following surgery, however, 70% to 80% of these patients prove to have benign nodules, meaning the surgery was unnecessary. We estimate each surgery costs $15,000 on average. Additionally, such surgeries have a complication rate of 2% to 10%, and most patients subsequently require lifelong thyroid hormone replacement therapy.
We estimate that approximately 3,500 endocrinologists specialize in thyroid disease and perform FNAs. We also serve other specialists, including radiologists and ear, nose and throat, or ENT, physicians who similarly perform FNAs. Approximately 60% of FNAs are performed in ambulatory, or community-based, practices, with the remaining 40% conducted in institutional settings, comprised of both academic centers and integrated delivery networks, which are networks of facilities and providers that work together to offer a continuum of care to a specific geographic area or market. While endocrinologists generally diagnose patients and refer them to surgery when necessary, endocrinologists do not perform the surgeries themselves. Institutions, which influence standard of care, typically have cytopathology laboratories on-site, to which the institutions' endocrinologists submit patient samples for review. Additional stakeholders that may be involved in the decision-making process in institutions include radiologists, pathologists and, occasionally, administration. We offer Afirma to institutional customers as an option following their internal cytopathology testing, and receive orders for the Afirma GEC only and/or the Malignancy Classifiers. This approach represents a higher margin opportunity versus in the community-practice setting where we also conduct the lower margin cytopathology assessment.
Afirma Thyroid FNA Analysis
Launched in 2011, the Afirma Thyroid FNA Analysis is our comprehensive offering for thyroid nodule assessment. The solution centers on our proprietary Afirma GEC to resolve indeterminate FNA results, based on cytopathology, so that patients whose nodules are actually benign can avoid unnecessary diagnostic surgery and undergo routine monitoring instead. The Afirma GEC is a 142-gene signature that is proven in multiple peer-reviewed, published studies to identify benign nodules with a high level of accuracy among those deemed indeterminate by cytopathology. Data suggest the test can enable unnecessary surgeries to be reduced by approximately 50%. Our comprehensive solution also includes our Afirma Malignancy Classifiers—comprised of tests for medullary thyroid cancer, a rare and aggressive form of thyroid cancer, and BRAF V600E gene mutational status, which is often predictive for papillary thyroid cancer—which were launched in May 2014 to preoperatively help inform selection of surgery when surgery is needed, minimizing the need for patients to undergo an additional "completion surgery." The MTC test result is included as part of the patient report when an Afirma GEC is performed on any FNA that is indeterminate by cytopathology. Physicians can also order it separately for use on FNAs that are malignant by cytopathology. The BRAF test is performed when ordered specifically by the physician on either GEC suspicious or malignant by cytopathology FNAs.
The Afirma Thyroid FNA Analysis includes initial cytopathology to optimize utilization of the Afirma GEC, ensuring that the test is used appropriately and without the need for patients to return for a repeat FNA procedure. We offer Afirma through two models, designed to meet the needs of both our community-practice and institutional physician customers.
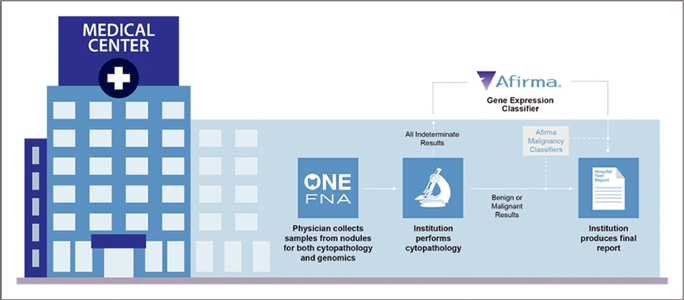
Model for Community Practices
Our model allows community-based physicians to implement Afirma in their practice without any meaningful changes to their workflow. Samples for both cytopathology and the Afirma GEC are collected during one FNA procedure using well-accepted and widely-used techniques. Customers send both the
8
cytopathology and the Afirma GEC samples overnight to our CLIA-certified laboratory in Austin, Texas. After we accession the samples into our laboratory information system, the Afirma GEC samples are stored in a freezer while the cytopathology samples are prepared and stained for review by Thyroid Cytopathology Partners, or TCP, a specialized cytopathology practice in Austin, Texas, that provides professional diagnoses on these samples. When cytopathology results are indeterminate, we send the stored sample to our CLIA-certified laboratory in South San Francisco, California, where we perform the Afirma GEC and/or Malignancy Classifiers. Results are provided to the ordering physician via a comprehensive report that provides cytopathology results and identifies the Afirma GEC results as either "benign" or "suspicious" for malignancy and the Afirma Malignancy Classifiers as "positive" or "negative."
Approximately 14% to 17% of thyroid FNA biopsies from TCP have been classified as indeterminate and have been reflexed to the GEC. This rate is at the low end of the 15% to 30% range cited in the 2009 American Thyroid Association Guidelines, suggesting TCP's specialized focus on thyroid cytopathology offers results that are more consistent with those of academic settings. Through our relationship with TCP, the high quality of care historically only accessible to patients in academic settings is now broadly available. By using a large, high-volume, thyroid-specialized pathology practice to offer consistent cytopathology analysis, we can optimize quality and manage appropriate utilization, ensuring that the Afirma GEC is not run on cytologically benign or malignant samples, or where the FNA contains insufficient cellular material for diagnosis. We believe our ability to manage utilization is attractive to payers looking to capture the value we promise in patient care.
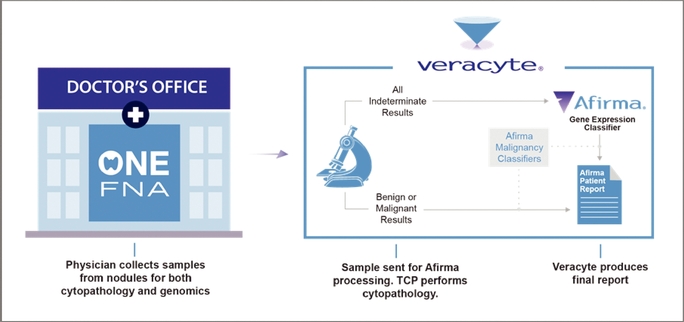
Afirma-enabled Model for Institutional Customers
Academic and hospital-based customers as well as integrated delivery networks typically perform their own cytopathology analysis and then only send us samples for Afirma GEC testing when the cytopathology result is indeterminate. We also receive samples to perform the Afirma Malignancy Classifiers either in addition to the GEC or for patients with a suspicious for malignancy result by cytopathology.
Physicians in these institutional settings generally conduct cytopathology in their own laboratory. With Afirma, the physician collects the FNA sample for GEC testing at the same time the FNA sample is collected for cytopathology review. The GEC test sample is preserved until the cytopathology results are processed. When the cytopathology result is reported, the preserved FNA sample is sent overnight to our CLIA laboratory for testing, using the Afirma GEC when the result is indeterminate and/or using the Malignancy Classifier analysis for suspicious samples.
9
Whether the final result is rendered by cytopathology alone or a combination of cytopathology and genomic testing, physicians receive an actionable answer based on samples collected in a single patient visit.
Our Afirma Growth Strategy
Our business growth is predominantly driven by growth of the Afirma GEC. Key initiatives include:
- •
- Position Afirma as a Comprehensive Solution for Managing Patients with Thyroid
Nodules. We believe that Afirma offers a unique, comprehensive solution for patients to avoid unnecessary surgeries and for cost
savings. Our service models for both community practices as well as institutions provides a comprehensive assessment, preoperatively, on a single FNA collected on the first patient visit. We are
strengthening our messaging of this value proposition on behalf of patients.
- •
- Expanded Sales Force. We have more than tripled the size
of our internal sales force during 2014, enabling us to further drive market penetration and expansion for Afirma, in both the ambulatory physician practice setting as well as in institutional
accounts and integrated delivery networks. Additionally, the Genzyme sales force complements our efforts in each sales territory, whereby they generate and qualify leads and provide ongoing
maintenance and support of existing accounts.
- •
- Strengthen Marketing Programs. We support our sales
efforts with comprehensive marketing initiatives that include medical education, speakers programs for physicians to share their experience with Afirma, as well as more traditional promotional
campaigns targeting both endocrinologists and patients who have been diagnosed with a thyroid nodule. Together with Genzyme, we are expanding our messages to include patient education, targeting media
outlets to educate and inform patients about Afirma.
- •
- Drive Payer Coverage and Contracts. Many physicians typically require a test to have broad coverage and be offered by a service provider that has in-network status before they will offer it to their patients. We will continue our efforts to advance payer coverage decisions and contracts to facilitate rapid adoption of Afirma among ordering physicians. With Medicare and most of the leading commercial payers covering Afirma, as well as several Blue Cross and Blue Shield plans, we intend to focus our efforts on obtaining coverage from remaining "Blues" plans. Additionally, we are expanding our resources to negotiate and secure in-network contracts which we believe will facilitate adoption as well as provide more predictable reimbursement and revenue.
10
- •
- Selective International Expansion. We intend to create a network of international partners, including Genzyme in certain markets, to offer Afirma around the world. We consider local practice patterns and local reimbursement to determine countries to enter, with special focus early on in countries where cash-based health care practices exist with local hospitals.
Development of the Afirma Gene Expression Classifier and Malignancy Classifiers
We used a whole-genome approach to develop the Afirma GEC, identifying gene expression patterns that could best identify a benign thyroid nodule signature in thyroid FNA samples diagnosed as indeterminate by cytopathology. We utilized microarray technology to perform whole-genome analyses on hundreds of thyroid samples, producing a rich database of more than one billion genomic measurements of thyroid biology. We initially measured mRNA expression in over 247,000 transcripts before selecting the target genes to be measured. We acquired large numbers of FNA samples taken at endocrinology practices across the United States in the early development of the Afirma GEC. Because thyroid cancer is a complex disease with multiple, sometimes rare, subtypes, this approach provided the diversity of clinical samples that would be encountered both during clinical validation and in commercial practice. Our scientists then developed machine-learning algorithms using sophisticated statistical approaches to distill the large amount of genomic data and to address FNA sample variability, dilution effects and RNA quantity and quality challenges. The development of the Afirma GEC first on thyroid surgical tissue and then on thyroid FNA samples was first published in 2010 in the Journal of Clinical Endocrinology and Metabolism. Using our extensive thyroid-genomic database derived from the whole-genome discovery work that led to the GEC, which we believe to be the largest single data set for thyroid conditions, we developed the Afirma Malignancy Classifiers as an extension to the GEC.
Published Evidence for Afirma
More than a dozen studies and review articles have been published in peer-reviewed journals supporting Afirma. Following is an overview of the key studies.
Clinical Validation
Preoperative Diagnosis of Benign Thyroid Nodules with Indeterminate Cytology (The New England Journal of Medicine, 2012)
In this study, which was sponsored by us and conducted with the support of institutional research grants from us, our Afirma GEC exhibited a negative predictive value, or NPV, of 95% for indeterminate results in the atypia or follicular lesion of undetermined significance category (AUS/FLUS) and 94% for indeterminate results in the suspicious for follicular or Hürthle cell neoplasm category (SFN/SHN) and reclassified as benign over half of the true benign FNA samples that had indeterminate cytopathology diagnoses, which the authors defined to include any results suspicious for malignancy in addition to AUS/FLUS and SFN/SHN. This pivotal validation study employed a prospective, multicenter, double-blind study design to validate the accuracy of preoperative Afirma GEC benign results compared to post-operative expert pathology review. It was the second prospective multicenter study validating the Afirma GEC approach. The study supported the consideration of a more conservative approach than surgery for most patients with thyroid nodules that are cytologically indeterminate but benign according to Afirma GEC results.
This large multicenter study included 49 academic and community practices across 26 states over 19 months. The study involved patients with ultrasonographically confirmed thyroid nodules one centimeter or larger in diameter. 4,812 thyroid FNA samples were prospectively collected from 3,789 patients. In the independent validation set of 265 nodules that were indeterminate by cytopathology, 85 were subsequently determined malignant by surgical pathology, equivalent to a 32% risk of malignancy. The Afirma GEC correctly identified 78 of the 85 malignant nodules as suspicious, a 92% sensitivity
11
(95% confidence interval, or CI, 84 to 97). The Afirma GEC achieved a 52% specificity (95% CI 44 to 59) and reclassified as benign over half of the true benign FNA samples that had indeterminate cytopathology diagnoses. The authors concluded that a benign Afirma GEC result has a post-test probability of malignancy that is similar to the probability for operated nodules with cytologically benign features on an FNA, making watchful waiting a safe and effective clinical option for these patients.
Molecular Classification of Thyroid Nodules using High- Dimensionality Genomic Data (Journal of Clinical Endocrinology and Metabolism, 2010)
In this study, which we sponsored, our FNA trained classifier exhibited an NPV of 96% on a modest sized test set of FNA samples, demonstrating an NPV similar to operated nodules with benign FNA cytology. In this study, the authors defined indeterminate results to include any cytological results suspicious for malignancy in addition to AUS/FLUS and SFN/SHN. This prospective, multicenter, double-blind study was the first study on an independent modest-sized set of FNA samples to clinically validate the gene expression classifier approach. In addition, this study demonstrated that even with substantial degradation of RNA and in the presence of blood, in some cases with dilution of up to 80%, the GEC correctly recognized benign nodules and did not miss malignancy in the majority of FNA samples.
The GEC was prospectively validated on an independent test set of 48 FNA samples, one-half of which had indeterminate cytopathology. The GEC exhibited an NPV of 96% and a specificity of 84%. The reference gold standard in this outcome study was the post-operative determination of whether the thyroid nodule was benign or malignant by expert endocrine surgical pathologists who were blinded to the GEC results. The authors concluded that the GEC performance and validation conducted on an independent validation set demonstrated a high enough specificity to reclassify over half of indeterminate FNAs as benign and that the observed NPV indicated that those nodules classified as benign by the GEC carry a similar risk of malignancy as a benign diagnosis by thyroid nodule FNA cytopathology alone.
Clinical Utility
The Impact of Benign Gene Expression Classifier Test Results on the Endocrinologist-patient Decision to Operate in Patients with Thyroid Nodules with Indeterminate Fine Needle Aspiration Cytopathology (Thyroid, 2012)
This study, which was sponsored by us and supported with institutional research grants, found that approximately one surgery was avoided for every two GECs run on thyroid FNAs with indeterminate cytopathology, which the authors defined to include any results suspicious for malignancy in addition to AUS/FLUS and SFN/SHN. This study evaluated the clinical utility of the Afirma GEC in a multicenter, cross-sectional survey of the endocrinologists' decision to operate on patients with a cytopathology indeterminate FNA and a benign Afirma GEC result. The study reviewed the first 2,040 GEC tests performed on samples that were classified as indeterminate by cytopathology, of which the Afirma GEC reclassified 52.3% of these results as benign. In the study, a cohort of 51 endocrinologists (46 community based; five academic based) at 21 practice sites in 11 states completed case report forms on whether surgery was recommended for their Afirma benign patients. Of 368 unique patients (395 cytopathology indeterminate FNAs) for whom data was collected, physicians and patients opted for watchful waiting in lieu of diagnostic thyroid surgery 92.4% of the time when the Afirma GEC result reclassified the patient's indeterminate nodule as benign. Surgery was performed on only 7.6% (95% CI 5.1 to 10.8) of patients, compared to the 74% historic rate of surgery on indeterminate thyroid nodules previously reported by Thyroid in 2011, a 90% relative reduction in the decision to operate (p < 0.001). Additionally, this 7.6% rate of surgery is similar to the 9.0% rate of surgery associated with cytology benign FNA results and reflects other factors considered by physicians, including the size and growth rate of the nodule, the presence of other suspicious or malignant nodules, and other symptoms. The study demonstrates the effect of the GEC on clinical decision making for patients with indeterminate thyroid nodules.
12
Multicenter Clinical Experience with the Afirma Gene Expression Classifier (Journal of Clinical Endocrinology and Metabolism, 2014)*
This study sought to determine how use of the Afirma GEC affects clinical practice in a real-world environment. Researchers at five academic centers followed all thyroid nodule patients who were tested with the Afirma GEC following indeterminate biopsy results based on cytopathology between 2010 and 2013. Among the 339 patients with indeterminate thyroid nodules, the Afirma GEC identified 174 (51%) as benign and, of these, 71 patients were followed clinically for an average of nine months. Of these 71 patients, only one cancer was identified over the course of the study, confirming a high NPV for the Afirma GEC of over 95%, which is similar to the malignancy risk of a benign cytopathology result. These findings reaffirm data from the initial validation trial published previously in The New England Journal of Medicine. The study also supports previous findings regarding the clinical utility of the Afirma GEC, as only 6% of patients with nodules identified as benign by our test underwent surgery.
Cost-effectiveness
Cost-effectiveness of a Novel Molecular Test for Cytologically Indeterminate Thyroid Nodules (Journal of Clinical Endocrinology and Metabolism, 2011) ©The Endocrine Society*
This clinical study was conducted by researchers from the Johns Hopkins University School of Medicine. Supported with a research grant from us, the authors found that use of the GEC can potentially avoid almost three-fourths of currently performed surgeries in patients with benign nodules but indeterminate cytopathology results, which the authors defined to include any results suspicious for malignancy in addition to AUS/FLUS and SFN/SHN.
Researchers modeled the direct cost savings of utilizing the Afirma GEC in clinical practice. They developed a 16-state Markov decision model based upon the 2009 American Thyroid Association Guidelines for the treatment of adult patients with thyroid nodules with an FNA cytopathology indeterminate diagnosis. The decision model was based on clinical validation study results and expert opinion though model variables necessarily require a substantial degree of judgment. One million patient simulations were run through the decision model to represent five years of treatment and follow-up for patients who first presented with cytologically indeterminate thyroid nodules. Utilization of the Afirma GEC yielded an estimated direct cost savings of $1,453 and an increase of 0.07 quality adjusted life years, or QALYs, per patient, a modest increase in the quality of life. A Monte Carlo simulation of 10,000 trials testing the sensitivity of all variables across a range of values resulted in the Afirma GEC being both less costly and more effective in improving care quality 92.5% of the time. A Monte Carlo simulation is the repeated sampling of random outcomes to predict likely outcomes. Additionally, the authors found no difference in cancers left untreated between the current care paradigm of sending patients with indeterminate nodules to surgery versus clinical observation following a benign Afirma GEC result. The authors concluded that if the GEC were to be universally adopted in routine clinical practice in the United States, every year 74% fewer surgeries would be performed on patients with benign nodules that cytopathology would have classified as indeterminate.
- *
- A
co-author of this study is a consultant and member of our clinical advisory board, and owned shares of our common stock at the time of the study.
- *
- A co-author of this study is a consultant and member of our clinical advisory board, and owned shares of our common stock at the time of the study. This study was conducted with the support of institutional research grants by us.
13
The cost savings estimate in the Johns Hopkins model was based on an estimated 14% rate of surgery on a benign Afirma GEC nodule, which rate is almost double the 7.6% and 6.3% subsequently reported in prior studies published in Thyroid and the Journal of Clinical Endocrinology and Metabolism. Based on the rate of surgery on GEC benign nodules reported in Thyroid, this study found that each Afirma GEC test would save approximately $2,600.
Analytical Validity
Analytical Performance Verification of a Molecular Diagnostic for Cytology-Indeterminate Thyroid Nodules (Journal of Clinical Endocrinology and Metabolism, 2012)
This study evaluated the Afirma GEC's ability to provide a robust, accurate and reproducible assay result on patient samples. The findings showed that the RNA content in an FNA sample that is preserved in our proprietary FNAProtect is stable for up to six days at room temperature with no changes in RNA yield or quality. Additionally, the Afirma GEC results were found to be stable over the range of shipping conditions expected in community practice. Analytic sensitivity studies demonstrated tolerance to variation in RNA input (5-25ng) and to the dilution of malignant FNA material down to 20%. Analytic specificity studies using malignant samples mixed with blood up to 83% and genomic DNA up to 30% demonstrated negligible assay interference with respect to false-negative results, although benign FNA samples mixed with relatively high proportions of blood demonstrated a potential for false-positive results. The Afirma GEC results were shown to be reproducible across operators, runs, reagent lots, and in inter-laboratory comparisons (standard deviation of 0.158 for scores on a >6 unit scale), demonstrating the highest level of evidence for analytic validity based on the Evaluation of Genomic Applications in Practice and Prevention, or EGAPP, criteria. Analytical sensitivity, analytical specificity, robustness, and quality control of the Afirma GEC were successfully demonstrated.
Machine Learning from Concept to Clinic: Reliable Detection of BRAF V600E DNA Mutations in Thyroid Nodules Using High-Dimensional RNA Expression Data (Pacific Symposium on Biocomputing, 2015)
This study, which was sponsored by us and supported with institutional research grants, demonstrated the analytical and clinical validity of the Afirma BRAF test, one of our Afirma Malignancy Classifiers, and confirms that the RNA-based classifier detects the BRAF V600E gene mutation with high diagnostic accuracy. In the study, researchers evaluated 535 FNA samples using both the Afirma RNA-based classifier and a sensitive, standard PCR DNA-based test. The Afirma BRAF RNA-based classifier accurately determined the presence or absence of the BRAF V600E gene mutation with equal performance, but with a lower non-diagnostic rate, than the DNA-based test (7.6% vs. 24.5%).
Additionally, strong clinical validation data demonstrating the ability of the Afirma MTC test to accurately identify cases of medullary thyroid cancer, which were missed by cytopathology alone, were presented at the American Association of Clinical Endocrinologists, or AACE, 23rd Annual Scientific & Clinical Congress in May 2014 and are expected to be published in 2015.
Afirma in Practice Guidelines
We believe inclusion of diagnostic tests in clinical practice guidelines is essential to drive their broad adoption and reimbursement. In January 2013, the National Comprehensive Cancer Network, or NCCN, modified its thyroid cancer guidelines to recommend that physicians consider molecular testing in lieu of diagnostic surgery for patients with cytopathology indeterminate thyroid nodules, provided that the molecular test predicts a risk of malignancy comparable to the risk of malignancy of a benign cytopathology result. Based on published evidence, the Afirma GEC meets these criteria. In July 2014, the NCCN further modified its guidelines to include the Afirma GEC by name. Additionally, UpToDate, a leading evidence-based clinical decision support resource for physicians, recommended the Afirma GEC in
14
its February 2013 review. Finally, based on preliminary American Thyroid Association, or ATA, guidelines on the management of thyroid nodules, which were presented in June 2014, we believe the Afirma GEC is positioned well as a "rule out" test to meet the ATA's proposed inclusion criteria for using molecular testing to help patients avoid unnecessary surgery following an indeterminate cytopathology result. The ATA is expected to finalize its guidelines in 2015.
Afirma Marketing and Sales
Marketing
We employ diverse marketing programs to inform key stakeholders of the value of our Afirma solution in order to drive adoption and reimbursement. As part of our marketing strategy, we educate physicians, healthcare professionals and managed care executives about our unique value proposition, which is supported by numerous peer-reviewed publications demonstrating the analytical and clinical validity, clinical utility and cost-effectiveness of Afirma. We primarily achieve this through national and regional clinical meetings focused on thyroid and endocrine disease and disorders. We also sponsor physician speaker programs and continuing medical education where both academic and community physicians educate their peers on the benefits of Afirma. Genzyme jointly participates in our marketing and medical education activities.
In May 2014, we introduced a comprehensive promotional campaign targeting endocrinologists, which highlights the patient benefits of Afirma—primarily its ability to help avoid unnecessary surgeries using information derived from a single FNA procedure. We expanded this campaign to focus on a patient audience while still highlighting the patient experience for physicians. The campaign's centerpiece, www.afirma.com, serves as the digital home for a an inbound marketing campaign for patients diagnosed with a thyroid nodule that includes paid search, search engine optimization, advertising in physician offices, and outreach to patient advocacy organizations. To support the consumer campaign, a robust physician campaign includes sales aids, medical conference promotion, print and online advertising and direct mail promotion.
Sales
We market our Afirma solution through our dedicated specialty sales force and through a co-promotion agreement with Genzyme Corporation, which targets the same endocrinologist customers with Thyrogen. We estimate that approximately 3,500 endocrinologists specialize in thyroid disease and perform FNAs to determine whether a thyroid nodule is malignant for cancer or benign. We also serve other specialists, including radiologists and ENT physicians, who also perform FNAs. We estimate that 60% of FNAs are collected in the physician office ambulatory setting and 40% in institutions and integrated delivery networks. In the early years of commercialization of Afirma, our success was attributed to our ability to gain adoption in the ambulatory setting where the physician alone can make a decision to use Afirma. In 2014 we refined our "Afirma-enabled" model in institutions, which involves a more complex sales process due to the multiple stakeholders within the institutions that participate in the decision to adopt Afirma. We believe servicing both models is important to our future growth.
We entered 2015 with a significantly expanded team of 26 sales professionals (versus eight at the beginning of 2014) that focuses on driving Afirma adoption and GEC test volume among both community-based and institutional customers, with the continued engagement of the Genzyme sales force. The Genzyme sales force focuses on lead development and ongoing support and maintenance of existing and new customers. The two teams work hand in hand, territory by territory to maximize our penetration and retention of the business. We have designed sales goals and financial incentives to align the interests of all sales representatives, regardless of company affiliation, to drive Afirma adoption and growth. In our Amended and Restated U.S. Co-Promotion Agreement, or Amended Agreement, effective January 2015, we reduced the fees we pay to Genzyme from 32% to 15% of Afirma cash revenue and invested the savings
15
in fees in our expanded internal team. We believe this more robust sales footprint positions us well for continued Afirma growth, particularly with institutional accounts.
In 2014 we entered our first international market in Brazil through a partnership with Fleury Health and Medicine, one of the largest diagnostics organizations in Brazil. Through our amended ex-U.S. Co-Promotion Agreement with Genzyme, we anticipate collaborating on additional international market entries in 2015.
Our Product Pipeline
We are continuously evaluating substantial unmet clinical needs in large, addressable markets where we can leverage our molecular cytology platform to commercialize comprehensive solutions that improve quality of life for patients by reducing unnecessary surgeries and costs. Today, minimally invasive cytology biopsies are routinely collected from numerous organs such as breast, cervix, endometrium and others. Similar to thyroid and lung, these often generate ambiguous results that lead to invasive procedures including surgery.
We are now applying our proven molecular cytology approach to other disease areas, beginning with lung diseases, which are often challenging to diagnose without surgery or other invasive procedures. The following graphic shows our progress across our product pipeline:
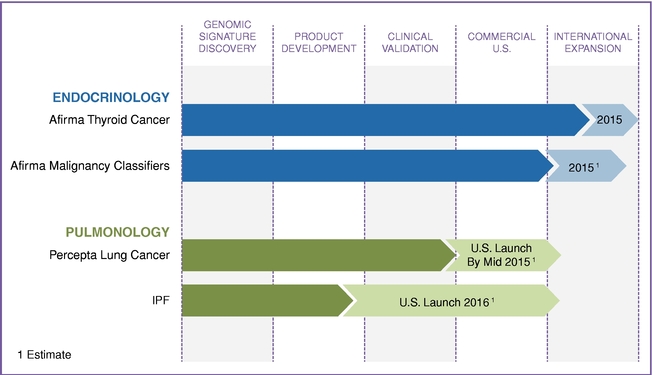
Developing new products is a lengthy and complex process, and is subject to numerous risks and uncertainties. We may not be able to commercialize on a timely basis, or at all, products we are developing. If we are not able to do so, our business and our ability to generate revenue could be harmed.
Pulmonology
Pulmonology represents a significant opportunity for molecular cytology, given the inherent challenges in diagnosing lung cancer and lung diseases, which are difficult to access without invasive procedures.
16
Lung Cancer Diagnostic Market
Lung cancer is the leading cause of cancer deaths in the United States, with an estimated 224,000 new diagnoses and 160,000 deaths in 2014. Approximately 250,000 patients with suspected lung cancer currently undergo bronchoscopy each year in the United States to assess lung nodules or lesions that are suspicious for lung cancer. Bronchoscopy, a procedure typically performed in an outpatient setting, enables the physician to visualize and collect cells from the patient's lung airways and is considered safer than other, more invasive sampling methods, such as transthoracic needle biopsy, or TTNB, or surgery.
Approximately 40% of bronchoscopies produce non-diagnostic results, meaning that malignancy was not found—but cannot be ruled out—in approximately 100,000 patients each year in the United States. This results from difficulty in accessing small and/or peripheral nodules with bronchoscopy devices. This leaves physicians with the dilemma of whether to direct these patients to surgery or other invasive procedures to obtain a diagnosis, which are risky and costly at over $20,000 per surgery, or to actively monitor the patients with imaging techniques.
Beginning in early 2015, more than eight million Americans at high-risk for lung cancer are eligible for annual screening with LDCT through new coverage requirements for private insurers as part of the Affordable Care Act, and through Medicare. This screening requirement resulted from the National Lung Screening Trial, a landmark 2011 government study, which found that annual screening using newer LDCT scans reduced lung cancer deaths by 20% among older current and former smokers. These findings had subsequently prompted the U.S. Preventive Services Task Force to recommend annual LDCT screening for people at high risk of lung cancer due to their age (from 55 to 80 years old) and history of smoking the equivalent of a pack a day for 30 years. While annual screening is expected to save many lives through early detection, it is anticipated to also find many lung nodules that prove to be benign, which has raised concerns that many patients will be unnecessarily subjected to invasive, risky and expensive procedures just to get a diagnosis.

A test that could improve the accuracy of bronchoscopy results could help identify patients who are at low risk of lung cancer and can thus avoid unnecessary invasive diagnostic procedures and be monitored with CT scans instead. We believe the market opportunity for such a test is between $350 million and $400 million in the United States, based on the current number of bronchoscopies performed to evaluate
17
lung nodules that are suspicious for cancer. The number of patients screened for lung cancer—and the number of non-diagnostic bronchoscopies—could expand significantly in the next two to three years as the screening programs are implemented.
Percepta Bronchial Genomic Classifier
The Percepta Bronchial Genomic Classifier is a gene expression test designed to identify patients with lung nodules who are at low risk of cancer following a non-diagnostic bronchoscopy so that they may be monitored with CT surveillance without the need for invasive procedures or surgery to obtain a sample from the nodule directly. The test comprises a 23-gene molecular classifier that utilizes "field of injury" technology to detect molecular changes that occur in the epithelial cells lining the lung's respiratory tract in response to smoking—the cause of 85-90% of lung cancers. These changes can be detected in cytologically normal airway cells and have been shown to correlate with the presence of malignancy or disease processes from distant sites in the lung. This field of injury genomic technology plays a key role in our positioning of Percepta at the point in the clinical pathway following a bronchoscopy procedure that yields non-diagnostic results. By resolving ambiguity following a bronchoscopy, we believe our test results can potentially help physicians and patients avoid an invasive surgical procedure as the next step in achieving diagnostic results.
We estimate that approximately 4,000 physicians perform bronchoscopies in the United States, of which approximately 80% are pulmonologists. The remaining bronchoscopies are performed by thoracic surgeons, general surgeons and other subspecialty physicians. Most bronchoscopies are performed in hospitals and the majority of those for lung cancer diagnosis take place in the hospital outpatient setting. The primary decision maker for Percepta will be the pulmonologist, although other physicians involved in the diagnostic work-up for lung cancer will also be involved, including the pathologist, thoracic surgeon, oncologist and radiologist.
Development of Percepta Bronchial Genomic Classifier
We gained Percepta and its underlying technology and intellectual property through the acquisition of Allegro. Early work published in Nature Medicine in 2007 demonstrated how gene expression alterations in cytologically normal large-airway epithelial cells of current and former smokers could serve as a lung cancer diagnostic. Percepta was developed using a training set of 299 patients, a subset of patients enrolled in the AEGIS, or Airway Epithelium Gene Expression in the Diagnosis of Lung Cancer, trials, designed as prospective, observational, cohort studies of current and former cigarette smokers with lung nodules suspicious for cancer, who were undergoing bronchoscopy as part of their diagnostic work-up. Samples were collected at medical centers around the country using standard cytopathology brushings during bronchoscopy. The microarray-based gene expression algorithm was derived using genes associated with lung cancer and with three clinical covariates, including gender, tobacco use and smoking history, as well as age, and then applying logistical regression modeling techniques to lock a classifier that could accurately predict cancer status.
Clinical Evidence for Percepta
Clinical Validation
The performance of Percepta has been demonstrated in studies enrolling over 1,000 patients from more than 30 centers in domestic and international sites, including two prospective clinical validation studies. Results of the first clinical validation study, the AEGIS I study on a cohort of 298 patients, were presented at the American Thoracic Society, or ATS, 2014 International Conference in May 2014 and demonstrated that the test was highly accurate, with an NPV of greater than 90%, in identifying patients at low risk of lung cancer following a non-diagnostic bronchoscopy result. In the study, all patients were followed longitudinally to the point of diagnosis of either primary lung cancer, using pathology results as
18
the gold standard, or benign disease. The second validation study, AEGIS II, involved 341 patients and demonstrated similar results. We expect these data will be presented at a scientific meeting in 2015. Data from the combined AEGIS I and AEGIS II clinical validation studies, along with data describing the algorithm's development and initial clinical validation, are expected to be submitted for publication in 2015.
Additional Evidence Development
Analytical verification studies are underway to document the robust laboratory process transferred from Allegro to Veracyte's CLIA laboratory. We plan to build out the remaining body of evidence to demonstrate how the test changes clinical care decisions once we commercialize the test by mid-2015. Finally, we have cost-effectiveness studies underway to demonstrate the test's value to payers.
Practice Guidelines
Several existing guidelines cover the management of patients undergoing a diagnostic workup for lung cancer. In 2013, the American College of Chest Physicians, or ACCP, released comprehensive guidelines for the diagnosis and management of lung cancer, updating their 2007 guidelines. NCCN also publishes guidelines for lung cancer screening and management of non-small cell lung cancer and small cell lung cancer. These guidelines are updated on a more frequent basis. Both organizations' recommendations advise on when to proceed to a biopsy. However, there is little guidance on what to do after a non-diagnostic bronchoscopy. Our internal research suggests that physicians vary widely in how they proceed with these patients. For example, some physicians take all of these patients to surgery or TTNB, while others are more conservative and place them on CT surveillance. ACCP guidelines place non-diagnostic patients at an intermediate risk of malignancy, thus implying that pulmonologists should treat these patients as they would any other intermediate-risk patient. Current guidelines, however, do not provide definitive guidance on what to do for this group. We believe that Percepta can change this diagnostic paradigm by offering evidence-based medicine to further guide how to manage "intermediate-risk" patients, identifying those who are at low risk for lung cancer so they can be followed with CT surveillance rather than moving on to additional invasive diagnostic procedures.
Marketing and Sales
We plan to enter the market with a small, targeted pulmonary product specialist field force, under the Percepta Early Access Program, while we complete the remaining studies needed to build out our library of evidence. Once these studies are completed and published in peer-reviewed journals, we will pursue coverage decisions by Medicare as well as commercial payers. We plan to initially target sites with high volumes of bronchoscopies for lung cancer, both in academic and community settings.
Similar to our approach with Afirma, the physicians will collect the samples for Percepta at the same time that the original bronchoscopy is conducted. This approach will help ensure that Percepta fits smoothly into physicians' workflow, minimizing complexity for our customers and optimizing utilization of our test to maximize patient benefit and cost savings.
Interstitial Lung Diseases Diagnostic Market
The market for an ILD diagnostic, and particularly IPF, represents another large opportunity to improve preoperative diagnosis, helping to reduce the need for unnecessary invasive procedures and associated costs. The physician specialist for our IPF product is also the pulmonologist, leveraging our pulmonology channel which we plan to enter this year with Percepta.
IPF is the most common and most deadly form of ILD, a diagnostic category comprising more than 200 diverse lung disorders characterized by progressive scarring of the lungs. An estimated 175,000 to 200,000 patients in the United States and major European countries present with suspected ILDs each
19
year. IPF and other ILDs are often similar in symptoms and appearance, making them challenging for physicians to distinguish from each other.
This uncertainty can result in incorrect or missed diagnoses; invasive, risky and expensive diagnostic surgeries costing over $40,000 per surgery; and/or suboptimal treatment. In addition, patients diagnosed with IPF who actually have another, less-serious ILD could be erroneously told that they have a deadly disease with a very poor prognosis and may be subjected to inadequate and/or potentially harmful treatment. The need for improved IPF diagnosis is increasingly important with the recent availability of new therapies for IPF in the United States and Europe, pirfenidone and nintedanib, that slow IPF progression, and with numerous additional drugs under development with the potential to slow or reverse IPF-related lung damage.
IPF diagnosis is typically made by a multidisciplinary team, or MDT, comprised of pulmonologists, radiologists and pathologists, based on a thorough clinical work-up combined with the presence of a specific pattern called usual interstitial pneumonia, or UIP, from high-resolution computed tomography, or HRCT, or from a pathology diagnosis made from a tissue sample collected from a surgical procedure. These UIP patterns are often difficult to distinguish, and even experienced radiologists and pathologists may not agree on the diagnosis. Additionally, many patients live in areas where an MDT is not available. When an IPF diagnosis is uncertain by HRCT, diagnostic surgery is considered the best approach; however, lung surgery is invasive, risky and expensive and many patients are too sick to undergo surgery.
A genomic test that could resolve diagnostic ambiguity found in the majority of patients presenting with potential IPF or another ILD could enable many patients to be diagnosed and treated appropriately, sooner, and without the need for diagnostic surgery. Our research suggests that clinicians see the need for a genomic test that could provide greater confidence in making an IPF or other ILD diagnosis. We estimate the addressable market for our IPF test to be over $500 million in the United States and Europe.
Our IPF Test
We are developing a molecular test to enable less-invasive, more accurate and less costly diagnosis of IPF using cytology samples obtained through bronchoscopy. Our IPF test is intended to replace diagnostic surgery by providing valuable, objective information that will enable the MDT to make more accurate diagnoses earlier. We expect to launch our test in 2016.
Our molecular classifier is designed to identify patients with pathology patterns that correspond with IPF versus those typically associated with other ILDs. In May 2014, we presented preliminary data at the ATS 2014 International Conference demonstrating the ability of our molecular classifier, developed using a whole-genome approach and microarray and deep RNA and DNA sequencing on tissue samples, to accurately distinguish IPF from other ILDs. We are now optimizing the test for use on bronchoscopy samples.
To this end, we are working with more than 25 clinical sites in the United States and internationally to prospectively collect hundreds of patient samples for use in developing—and later, in validating—our test under our BRAVE protocols. Our intent is to obtain samples that represent all types of cases and associated clinical annotations, which we believe our classifier will be exposed to once commercialized. We have formed a "virtual" MDT of world-renowned experts in pulmonology, radiology and pathology to establish "clinical truth" against which we are developing and measuring our test's performance. We expect to present initial clinical data demonstrating the performance of our IPF test on bronchoscopy samples at a scientific meeting in 2015.
20
Third-party Relationships
Genzyme
We began our co-promotion partnership with Genzyme, a subsidiary of Sanofi, in January 2012 by executing a co-promotion agreement. Genzyme is an established leader in endocrinology globally, developing and commercializing Thyrogen (thyrotropin alfa for injection) in the United States and over 42 countries worldwide. Thyrogen is an adjunctive diagnostic agent used in follow up of patients with well differentiated thyroid cancer, and an adjunctive treatment for ablation or destruction of thyroid remnants in patients who have had their thyroid removed for the treatment of well-differentiated thyroid cancer. We manage the relationship through a steering committee that oversees certain tactical and strategic planning activities.
Under the 2012 agreement, Genzyme paid us a $10.0 million upfront fee and we are required to pay Genzyme a co promotion fee that was equal to a percentage of our U.S. cash receipts from the sale of the Afirma GEC test, which fee varied over time. We record the Genzyme co-promotion fees, net of amortization related to the upfront fee, within selling and marketing expense in our statements of operations.
On November 7, 2014, we signed an Amended and Restated U.S. Co-Promotion Agreement, or Amended Agreement. Under the Amended Agreement, the co-promotion fees payable to Genzyme as a percentage of U.S. cash receipts from the sale of the Afirma GEC test were reduced from 32% to 15% beginning January 1, 2015, and the earliest either party may terminate the Amended Agreement for convenience is July 1, 2016. Our Amended Agreement with Genzyme expires in January 2027.
On February 13, 2015, we entered into an Ex-U.S. Co-Promotion Agreement, or Ex-U.S. Agreement, with Genzyme for the co-exclusive promotion of the Afirma GEC test in two countries outside the United States: Brazil and Singapore. We also granted Genzyme, for a limited period of time, an exclusive right of first negotiation to enter into an agreement with us for the promotion of the Afirma GEC test in three additional countries: Canada, the Netherlands and Italy. Further, upon mutual agreement, the parties may add additional countries (other than the United States) to the Ex-U.S. Agreement. The term of the Ex-U.S. Agreement commenced January 1, 2015 and continues until December 31, 2019 with extension of the agreement possible upon agreement of the parties. Country specific terms have been established under the Ex-U.S. Agreement for Brazil and Singapore. Pursuant to these terms, we will pay Genzyme 25% of cash receipts from the sale of the Afirma GEC test in Brazil and Singapore over a five-year period commencing January 1, 2015. Beginning in the fourth year of the agreement, if we terminate the agreement for convenience with respect to Brazil, we may be required to pay a termination fee contingent on the number of GEC billable results generated. This will give us optionality to drive new models in each market and more efficiently solidify the foundation of an international brand presence, retaining our Genzyme relationship in markets that make sense.
TCP
We rely on Thyroid Cytopathology Partners, P.A. to provide cytopathology professional diagnoses on thyroid FNA samples pursuant to a pathology services agreement. We originally entered into the pathology services agreement in November 2010 with Brazos Valley Pathology, P.A. D/B/A Reitpath, which assigned the contract to TCP in May 2011. In December 2012, we further amended the pathology services agreement. Pursuant to the agreement, as amended in full, TCP has the exclusive right to provide the cytopathology diagnoses on FNA samples that are referred to us as part of the Afirma solution at a fixed price per test with volume discounts. TCP can terminate the agreement upon our failure to pay any amounts due under the contract, and either we or TCP can terminate the agreement upon the insolvency of the other party, breach of the agreement by the other party, termination or breach of the service terms or the suspension or termination of the necessary regulatory licenses and approvals needed to perform the FNA diagnoses. TCP is co-located in a portion of our facilities in Austin, Texas and reimburses us for a portion of our actual out-of-pocket rental and related operating expense costs. Our agreement with TCP is effective until December 31, 2015 and thereafter automatically renews every year unless either party provides notice of intent not to renew at least twelve months prior to the end of the then-current term.
21
Reimbursement
Revenue for Afirma Thyroid FNA Analysis comes from several sources, including commercial third-party payers, such as insurance companies and health maintenance organizations, government payers, such as Medicare and Medicaid, and patients. We believe that reimbursement for our lung products will be derived from similar sources but with a greater proportion coming from Medicare and potentially Medicaid due to the age of the patient population.
Payer Landscape for Afirma
Reimbursement for Afirma is comprised of cytopathology, the GEC and/or the Malignancy Classifiers when these tests are performed as part of our comprehensive solution. To date, a high percentage of FNA samples received are accessioned for cytopathology and for which we bill both the technical and professional component using established CPT codes. Under our "Afirma-enabled" model, which is used predominantly by our institutional customers, reimbursement is sought for the Afirma GEC and/or the Malignancy Classifiers. We bill payers directly for the GEC and the Malignancy Classifiers using either a unique code or a miscellaneous code.
Effective January 2012, Palmetto GBA, the regional Medicare administrative contractor, or MAC, that handled claims processing for Medicare services with jurisdiction at that time, issued coverage and payment determinations for the GEC. Their review determined that the Afirma GEC met their criteria for analytical and clinical validity, and clinical utility as a reasonable and necessary Medicare benefit. This coverage decision provided approximately 50 million Medicare participants with access to the Afirma GEC. In mid-September 2013, Noridian Administrative Services succeeded Palmetto as the MAC for our region. Noridian continues to reimburse under our unique Z code originally established by Palmetto. On a five year rotational basis, Medicare requests bids for its regional MAC services. Any future changes in the MAC processing or coding for Medicare claims for the Afirma GEC or for future products could result in a change in the coverage or reimbursement rates for such products, or the loss of coverage.
Collectively, as of March 2015, we have 145 million lives under positive medical coverage policies for the Afirma GEC including from Medicare (January 2012) and leading commercial insurers, including UnitedHealthcare (April 2013), Aetna (June 2013), Humana (July 2013), Cigna (December 2013) and several leading Blue Cross and/or Blue Shield plans, including Highmark, Horizon Blue Cross, and Blue Shield of California (all 2014). We have nearly 100 million lives under contract for the Afirma GEC, which establishes us as an in-network provider and helps facilitate adoption. However, payers may suspend or discontinue reimbursement at any time, may require or increase co-payments from patients, or may reduce the reimbursement rates paid to us. Any such actions could have a negative effect on our revenue.
Dependence on Certain Third-party Payers
We rely on a small number of third-party payers for a significant portion of our revenue. Reimbursement on behalf of patients covered by Medicare accounted for 26%, 32% and 34% of our revenue for the years ended December 31, 2014, 2013 and 2012, respectively. UnitedHealthcare accounted for 18%, 18% and 12% of our revenue for the years ended December 31, 2014, 2013 and 2012, respectively. Aetna accounted for 11%, 9% and 13% of our revenue for the years ended December 31, 2014, 2013 and 2012, respectively. The loss of one or more of these payers would have a negative effect on our business and our revenue.
22
Reimbursement Strategy
We employ a multi-pronged strategy designed to achieve broad coverage and reimbursement for our tests:
- •
- Meet the Evidence Standards Necessary to Be Consistent with Leading Clinical
Guidelines. We believe inclusion in leading clinical practice guidelines plays a critical role in payers' coverage decisions. The data
published on the Afirma GEC to date is consistent with the recommendations of the widely-recognized NCCN clinical practice guidelines. We believe that our data provides compelling evidence for
inclusion in the American Thyroid Association and the American Association of Clinical Endocrinologists guidelines as well.
- •
- Execute an Internal Managed Care and Claims Adjudication Function as Part of Our Core Business
Operations. We believe that obtaining adequate and widespread reimbursement is a critical factor in our long-term success. We employ a team of in-house claims processing and
reimbursement specialists who work with payers, physician practices and patients to obtain maximum reimbursement. In parallel, a managed care team collaborates with our reimbursement specialists to
ensure our payer outreach strategy reacts and anticipates the changing needs of our customer base. Our customer service team is an integral part of our reimbursement strategy, working with physician
practices and patients to navigate the claims process.
- •
- Cultivate a Network of Key Opinion Leaders. Key opinion
leaders are able to influence clinical practice by publishing research and determining whether new tests should be integrated into practice guidelines. We collaborate with key opinion leaders early in
the development process to ensure our clinical studies are designed and executed in a way that clearly demonstrates the benefits of our tests to physicians and payers. Ongoing studies to support real
world experience with our tests are a key component of our efforts to maintain collaborations with physician influencers.
- •
- Compile a Growing Library of Peer-reviewed Studies that Demonstrate the Test Is Effective. To date, several peer-reviewed articles and review papers have been published and have helped support our efforts aimed at widespread adoption and reimbursement of Afirma. In each disease area we pursue, we intend to conduct studies in order to develop similar supporting literature.
Research and Development
Our technology platform offers a number of key attributes, which are applicable to Afirma, Percepta and our subsequent products:
- •
- Core Expertise in Broad-based Genomic Analysis. Our team
of bioinformatics and computational scientists possess extensive knowledge of both existing computational methods as well as the capacity to develop proprietary methods as needed for algorithm design.
We demonstrated our ability to make sense of large amounts of genomic data with machine learning algorithms in the development of the GEC.
- •
- Proprietary Capabilities in Analyzing Small, Heterogeneous Cytology Samples. We have developed proprietary technology, intellectual property and know-how for optimized methods for extraction and analysis of nanogram quantities of RNA from small biopsy samples. Although others can extract RNA from these small biopsies, we believe their process has not been optimized and scaled for high-throughput clinical testing and large-scale clinical development studies involving amplification and hybridization to high-density microarrays. Our process uses commercially available reagents and instruments with our own proprietary process and protocols, which results in RNA extraction from the range of FNAs used in our clinical development studies and our commercial laboratory test.
23
- •
- Precision and Reproducibility. We have in place standard
operating procedures governing reagents, materials, instruments and controls and extensive experience from numerous verification studies performed for the GEC. We are applying the same high-quality
control methods that were developed for our reagents and processes, along with our proprietary software for automation, sample tracking, data quality control and statistical analysis, to our
development process in interstitial lung disease and expect to do so for other diseases in the future.
- •
- Technology Agnostic Discovery Platform. We are not reliant on specific formats and are able to take advantage of a multitude of genomic technologies in developing future tests. When we developed the GEC in 2008, microarray technologies were a cost-effective discovery technology compared to other approaches that were nascent at the time. More recently, the rapid cost reductions achieved in next generation sequencing platforms has allowed us to pursue our whole genome approach to biomarker discovery using a range of technologies, including gene expression and DNA methylation, as well as DNA and RNA sequencing.
Laboratory Operations
Our laboratory operations are headquartered at our CLIA-certified laboratory in South San Francisco, California, where we perform all Afirma GEC testing and plan to perform our Percepta testing and subsequent tests in pulmonology. Our customers ship samples for cytopathology assessment to our CLIA-registered laboratory in Austin, Texas. Once received, samples are processed through our automated accessioning system, prepared for cytopathology review, and delivered to TCP for cytopathology diagnosis. If cytopathology results are indeterminate, the sample is transferred to South San Francisco where we perform GEC testing. Institutions and other clients using our Afirma-enabled model ship the samples for Afirma GEC and/or the Afirma Malignancy Classifiers directly to our South San Francisco laboratory. Percepta samples will be shipped directly to South San Francisco. Our South San Francisco facility is responsible for quality assurance oversight, licensing and regulation compliance and maintenance for both of our laboratories to ensure data integrity and consistent, validated processes.
We believe we have sufficient laboratory capacity to process Afirma tests, including our Afirma Malignancy Classifiers, as well as Percepta and our IPF test.
Quality Assurance
Our quality assurance function oversees the quality of our laboratories as well as the quality systems used in research and development, client services, billing operations and sales and marketing. We have established a quality system implementation and maintenance, document control, supplier qualification, corrective or preventive actions oversight, and employee training processes that we believe achieves excellence in operations across the entire business. We continuously monitor and improve our quality over time and believe our implementation of these processes has supported our achievement of product performance, customer satisfaction and retention and a philosophy of continuous improvement.
Competition
We believe the principal competitive factors in the thyroid cancer market include:
- •
- quality and strength of clinical validation and utility data;
- •
- confidence in diagnostic results backed by our analytical verification data;
- •
- the extent of reimbursement;
- •
- inclusion in practice guidelines;
- •
- cost-effectiveness; and
24
- •
- ease of use.
We believe we compete favorably on the factors described above with our Afirma solution.
Our principal competition for Afirma comes from traditional methods used by physicians to diagnose thyroid cancer. Practice guidelines in the United States have historically recommended that patients with indeterminate diagnoses from cytopathology results be considered for surgery to remove all or part of the thyroid to rule out cancer. This practice has been the standard of care in the United States, as well as in many international markets, for many years, and we will need to educate physicians about the benefits of our test in order to change clinical practice.
We also face competition from commercial laboratories, such as Laboratory Corporation of America Holdings, Quest Diagnostics Incorporated and Sonic Healthcare USA with strong infrastructure to support the commercialization of diagnostic services. We face potential competition from companies such as Illumina, Inc. and Thermo Fisher Scientific Inc., both of which have announced their intention to enter the clinical diagnostics market. Other potential competitors include companies that develop diagnostic products, such as Roche Diagnostics, a division of Roche Holding Ltd, Siemens AG, Qiagen N.V. and Rosetta Genomics Ltd. We also face competition from companies and academic institutions that use next generation sequencing technology or other methods to measure mutational markers such as BRAF and KRAS along with numerous other mutations. In the future, we may also face competition from companies developing new products or technologies that are able to compete with Afirma's high negative predictive value to rule out cancer.
With Percepta, we anticipate facing potential competition from the companies and institutions listed above, but additionally from companies developing approaches for assessing malignancy risk in patients with lung nodules using alternative samples, such as blood-based tests or sputums, which are performed early in the diagnostic paradigm.
Competitors may develop their own versions of our solution in countries where we do not have patents or where our intellectual property rights are not recognized and compete with us in those countries, including encouraging the use of their solution by physicians in other countries.
Many of our potential competitors have widespread brand recognition and substantially greater financial, technical and research and development resources and selling and marketing capabilities than we do. Others may develop products with prices lower than ours that could be viewed by physicians and payers as functionally equivalent to our solution, or offer solutions at prices designed to promote market penetration, which could force us to lower the list price of our solutions and affect our ability to achieve profitability. If we are unable to change clinical practice in a meaningful way or compete successfully against current and future competitors, we may be unable to increase market acceptance and sales of our products, which could prevent us from increasing our revenue or achieving profitability and could cause our stock price to decline. As we add new tests and services, we will face many of these same competitive risks for these new tests.
Intellectual Property
In order to remain competitive, we must develop and maintain protection of the proprietary aspects of our technologies. To that end, we rely on a combination of patents, copyrights and trademarks, as well as contracts, such as confidentiality, invention assignment and licensing agreements. We also rely upon trade secret laws to protect unpatented know-how and continuing technological innovation. In addition, we have what we consider to be reasonable security measures in place to maintain confidentiality. Our intellectual property strategy is intended to develop and maintain our competitive position.
We have five issued patents which expire between 2029 and 2032 related to methods used in the Afirma diagnostic platform, in addition to seven pending U.S. utility patent applications, two U.S. provisional applications and one PCT application. Some of these U.S. utility patent applications have
25
pending foreign counterparts. We also exclusively licensed intellectual property, including rights to three pending U.S. utility patent applications in the thyroid space that would expire between 2030 and 2033 once issued, related to methods that are used in the Afirma diagnostic test, some of which have foreign counterparts.
In the lung diagnostic space, we exclusively licensed intellectual property rights to 19 pending patent applications and two issued patents in the United States and abroad. Patents issuing from the licensed portfolio will expire between 2024 and 2028. In addition, we own four pending patent applications, a PCT application, and a provisional U.S. application related to our lung cancer test under development, Percepta, as well as a PCT application and two provisional U.S. applications related to our interstitial lung disease test under development. Any patents granted from the current lung cancer patent applications will expire from 2032 to 2034 and those from the interstitial lung disease patent applications will expire from 2034 to 2036.
We intend to file additional patent applications in the United States and abroad to strengthen our intellectual property rights; however, our patent applications (including the patent applications listed above) may not result in issued patents in a timely fashion or at all, and we cannot assure investors that any patents that have issued or might issue will protect our technology. We may receive notices of claims of potential infringement from third parties in the future.
We hold registered trademarks in the United States for "Veracyte" and "Afirma," and the Veracyte and Afirma logos. We also hold registered trademarks in various jurisdictions outside of the United States.
We require all employees and technical consultants working for us to execute confidentiality agreements, which provide that all confidential information received by them during the course of the employment, consulting or business relationship be kept confidential, except in specified circumstances. Our agreements with our research employees provide that all inventions, discoveries and other types of intellectual property, whether or not patentable or copyrightable, conceived by the individual while he or she is employed by us are assigned to us. We cannot provide any assurance, however, that employees and consultants will abide by the confidentiality or assignment terms of these agreements. Despite measures taken to protect our intellectual property, unauthorized parties might copy aspects of our technology or obtain and use information that we regard as proprietary.
Regulation
Clinical Laboratory Improvement Amendments of 1988, or CLIA
As a clinical reference laboratory, we are required to hold certain federal, state and local licenses, certifications and permits to conduct our business. Under CLIA, we are required to hold a certificate applicable to the type of laboratory examinations we perform and to comply with standards covering personnel, facilities administration, quality systems and proficiency testing.
We have current certificates under CLIA to perform testing at each of our locations. To renew our CLIA certificates, we are subject to survey and inspection every two years to assess compliance with program standards. The regulatory and compliance standards applicable to the testing we perform may change over time, and any such changes could have a material effect on our business.
If one of our clinical reference laboratories is out of compliance with CLIA requirements, we may be subject to sanctions such as suspension, limitation or revocation of our CLIA certificate, as well as directed plan of correction, state on-site monitoring, civil money penalties, civil injunctive suit or criminal penalties. We must maintain CLIA compliance and certification to be eligible to bill for diagnostic services provided to Medicare beneficiaries. If we were to be found out of compliance with CLIA requirements and subjected to sanction, our business could be harmed.
26
U.S. Food and Drug Administration: Diagnostic Kits
Diagnostic kits, including collection systems, which are sold and distributed through interstate commerce are regulated as medical devices by the FDA. Devices subject to FDA regulation must undergo premarket review prior to commercialization unless the device is of a type exempted from such review. In addition, manufacturers of medical devices must comply with various regulatory requirements under the Federal Food, Drug, and Cosmetic Act, or FDC Act, and implementing regulations promulgated under that Act. Entities that fail to comply with FDA requirements may be subject to issuance of notice of observations, untitled or warning letters, and can be liable for criminal or civil penalties, such as recalls, import detentions, seizures, or injunctions, including orders to cease manufacturing.
The FDC Act classifies medical devices into one of three categories based on the risks associated with the device and the level of control necessary to provide reasonable assurance of safety and effectiveness. Class I devices are deemed to be low risk and are subject to the fewest regulatory controls. Many Class I devices are exempt from FDA premarket review requirements. For Class II devices, the FDA generally requires clearance through the premarket notification, or 510(k) clearance process. Class III devices are generally the highest risk devices and are subject to the highest level of regulatory control to provide reasonable assurance of the device's safety and effectiveness. Class III devices must typically be approved by the FDA before they are marketed.
Generally, establishments that manufacture or distribute devices, including manufacturers, repackagers and relabelers, specification developers, and initial importers, are required to register their establishments with the FDA and provide the FDA a list of the devices that they handle at their facilities.
After a device is placed on the market, numerous regulatory requirements apply. These include: all of the relevant elements of the Quality System Regulation, or QSR, labeling regulations, restrictions on promotion and advertising, the Medical Device Reporting regulation (which requires that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur), and the Reports of Corrections and Removals regulation (which requires manufacturers to report certain recalls and field actions to the FDA).
The FDA has issued a regulation outlining specific requirements for "specimen transport and storage containers." "Specimen transport and storage containers" are medical devices "intended to contain biological specimens, body waste, or body exudate during storage and transport" so that the specimen can be used effectively for diagnostic examination. A specimen transport and storage container is a Class I device. It is subject to MDR requirements, the reporting of corrections and removals, registration and listing. It is exempt from premarket review and from QSR requirements, except for recordkeeping and complaint handling requirements, so long as no sterility claims are made. Our facility is registered with the FDA as a specification developer, which means that we can sell the collection system under our own name and outline the specifications used to make the collection system, but a third party assembles the collection system for us. The container we provide for collection and transport of FNA samples from a physician to our clinical reference laboratory is listed with the FDA as a Class I medical device and is subject to regulation by the FDA. We also plan to list our sample collection containers for use with our lung cancer and IPF tests with the FDA as Class I medical devices. If the FDA were to determine that our sample collection containers are a Class II medical device, we would be required to obtain FDA clearance to use the container.
The FDA enforces the requirements described above by various means, including inspection and market surveillance. If the FDA finds a violation, it can institute a wide variety of enforcement actions, ranging from an Untitled Letter or Warning Letter to more severe sanctions such as:
- •
- fines, injunctions, and civil penalties;
- •
- recall or seizure of products;
27
- •
- operating restrictions, partial suspension or total shutdown of production; and
- •
- criminal prosecution.
Federal Oversight of Laboratory Developed Tests and Research Use Only Products
Clinical laboratory tests like Afirma are regulated under CLIA, as administered by the Centers for Medicare & Medicaid Services, or CMS, as well as by applicable state laws. Clinical laboratory tests that are developed and validated by a laboratory for its own use, which are referred to as laboratory developed tests, or LDTs, currently are generally not subject to FDA regulation, although reagents, instruments, software or components provided by third parties and used to perform LDTs may be subject to regulation. We believe that the Afirma GEC is an LDT. FDA currently exercises its enforcement discretion for LDTs. In October 2014, the FDA published draft guidance documents describing the framework by which they might regulate LDTs. The framework is similar to the guidance they issued previously. The comment period ended in February 2015. There is no timeframe in which the FDA must issue final guidance documents.
Some of the materials we use for Afirma and that we may use for future products, such as Percepta, are for research use only, or RUO. An RUO product is not intended for human clinical use and must be labeled "For Research Use Only. Not for use in diagnostic procedures." RUOs are a separate regulatory category and are not considered medical devices. They are therefore not subject to the FDA regulatory requirements discussed above. They cannot make any claims related to safety, effectiveness, or diagnostic utility or be intended for human clinical diagnostic or prognostic use. In November 2013, the FDA issued guidance regarding "Commercially Distributed In-Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only."
We cannot predict the ultimate form or impact of any such RUO, LDT or other guidance and the potential effect on our solutions or materials used to perform our diagnostic services. While we qualify all materials used in our diagnostic services according to CLIA regulations, we cannot be certain that the FDA might not promulgate rules or issue guidance documents that could affect our ability to purchase materials necessary for the performance of our diagnostic services. Should any of the reagents obtained by us from vendors and used in conducting our diagnostic services be affected by future regulatory actions, our business could be adversely affected by those actions, including increasing the cost of service or delaying, limiting or prohibiting the purchase of reagents necessary to perform the service.
We cannot provide any assurance that FDA regulation, including premarket review, will not be required in the future for our diagnostic services, whether through additional guidance or regulations issued by the FDA, new enforcement policies adopted by the FDA or new legislation enacted by Congress. Legislative proposals addressing oversight of LDTs were introduced in recent years, and we expect that new legislative proposals will be introduced from time to time. It is possible that legislation could be enacted into law or regulations or guidance could be issued by the FDA which may result in new or increased regulatory requirements for us to continue to offer our diagnostic services or to develop and introduce new services.
If premarket review, including approval, is required, our business could be negatively affected until such review is completed and clearance to market or approval is obtained, and the FDA could require that we stop selling our diagnostic services pending premarket clearance or approval. If our diagnostic services are allowed to remain on the market but there is uncertainty about the legal status of our services, if we are required by the FDA to label them investigational, or if labeling claims the FDA allows us to make are limited, order levels may decline and reimbursement may be adversely affected. The regulatory process may involve, among other things, successfully completing additional clinical studies and submitting a premarket notification or filing a PMA application with the FDA. If premarket review is required by the FDA, there can be no assurance that our diagnostic services will be cleared or approved on a timely basis, if at all, nor can there any be assurance that labeling claims will be consistent with our current claims or
28
adequate to support continued adoption of and reimbursement for our solution. Ongoing compliance with FDA regulations would increase the cost of conducting our business, and subject us to heightened requirements of the FDA and penalties for failure to comply with these requirements. We may also decide voluntarily to pursue FDA premarket review of our diagnostic services if we determine that doing so would be appropriate.
Health Insurance Portability and Accountability Act
Under the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, the Department of Health and Human Services, or HHS, has issued regulations to protect the privacy and security of protected health information used or disclosed by health care providers, such as us. HIPAA also regulates standardization of data content, codes and formats used in health care transactions and standardization of identifiers for health plans and providers. Penalties for violations of HIPAA regulations include civil and criminal penalties.
We have developed and implemented policies and procedures designed to comply with these regulations. The requirements under these regulations may change periodically and could have an effect on our business operations if compliance becomes substantially more costly than under current requirements.
In addition to federal privacy regulations, there are a number of state laws governing confidentiality of health information that are applicable to our business. The U.S. Department of Commerce, the European Commission and the Swiss Federal Data Protection and Information Commissioner have agreed on a set of data protection principles and frequently asked questions, referred to as the Safe Harbor Principles, to enable U.S. companies to satisfy the requirement under European Union and Swiss law that adequate protection is given to personal information transferred from the European Union or Switzerland to the United States. The European Commission and Switzerland have also recognized the Safe Harbor Principles as providing adequate data protection.
New laws governing privacy may be adopted in the future as well. We have taken steps to comply with health information privacy requirements to which we are aware that we are subject. However, we can provide no assurance that we are or will remain in compliance with diverse privacy requirements in all of the jurisdictions in which we do business. Failure to comply with privacy requirements could result in civil or criminal penalties, which could have a materially adverse effect on our business.
Federal and State Physician Self-referral Prohibitions
We are subject to the federal physician self-referral prohibitions, commonly known as the Stark Law, and to similar restrictions under California's Physician Ownership and Referral Act, or PORA. Together these restrictions generally prohibit us from billing a patient or any governmental or private payer for any diagnostic services when the physician ordering the service, or any member of such physician's immediate family, has an investment interest in or compensation arrangement with us, unless the arrangement meets an exception to the prohibition.
Both the Stark Law and PORA contain an exception for compensation paid to a physician for personal services rendered by the physician. We have compensation arrangements with a number of physicians for personal services, such as speaking engagements and consulting activities. We have structured these arrangements with terms intended to comply with the requirements of the personal services exception to Stark and PORA.
However, we cannot be certain that regulators would find these arrangements to be in compliance with Stark, PORA or similar state laws. We would be required to refund any payments we receive pursuant to a referral prohibited by these laws to the patient, the payer or the Medicare program, as applicable.
29
Sanctions for a violation of the Stark Law include the following:
- •
- denial of payment for the services provided in violation of the prohibition;
- •
- refunds of amounts collected by an entity in violation of the Stark Law;
- •
- a civil penalty of up to $15,000 for each service arising out of the prohibited referral;
- •
- possible exclusion from federal healthcare programs, including Medicare and Medicaid; and
- •
- a civil penalty of up to $100,000 against parties that enter into a scheme to circumvent the Stark Law's prohibition.
These prohibitions apply regardless of the reasons for the financial relationship and the referral. No finding of intent to violate the Stark Law is required for a violation. In addition, knowing violations of the Stark Law may also serve as the basis for liability under the Federal False Claims Act.
Further, a violation of PORA is a misdemeanor and could result in civil penalties and criminal fines. Finally, other states have self-referral restrictions with which we have to comply that differ from those imposed by federal and California law. While we have attempted to comply with the Stark Law, PORA and similar laws of other states, it is possible that some of our financial arrangements with physicians could be subject to regulatory scrutiny at some point in the future, and we cannot provide assurance that we will be found to be in compliance with these laws following any such regulatory review.
Federal and State Anti-kickback Laws
The Federal health care program Anti-kickback Law makes it a felony for a person or entity, including a laboratory, to knowingly and willfully offer, pay, solicit or receive remuneration, directly or indirectly, in order to induce business that is reimbursable under any federal health care program. A violation of the Anti-kickback Law may result in imprisonment for up to five years and fines of up to $250,000 in the case of individuals and $500,000 in the case of organizations. Convictions under the Anti-kickback Law result in mandatory exclusion from federal health care programs for a minimum of five years. In addition, HHS has the authority to impose civil assessments and fines and to exclude health care providers and others engaged in prohibited activities from Medicare, Medicaid and other federal health care programs. Actions which violate the Anti-kickback Law also incur liability under the Federal False Claims Act, which prohibits knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to the U.S. Government.
Although the Anti-kickback Law applies only to federal health care programs, a number of states, including California, have passed statutes substantially similar to the Anti-kickback Law pursuant to which similar types of prohibitions are made applicable to all other health plans and third- party payers. Both California's fee-splitting statute, Business and Professions Code Section 650, and its Medi-Cal anti-kickback statute, Welfare and Institutions Code Section 14107.2, have been interpreted by the California Attorney General and California courts in substantially the same way as HHS and the courts have interpreted the Anti-kickback Law. A violation of Section 650 is punishable by imprisonment and fines of up to $50,000. A violation of Section 14107.2 is punishable by imprisonment and fines of up to $10,000.
Federal and state law enforcement authorities scrutinize arrangements between health care providers and potential referral sources to ensure that the arrangements are not designed as a mechanism to induce patient care referrals or induce the purchase or prescribing of particular products or services. The law enforcement authorities, the courts and Congress have also demonstrated a willingness to look behind the formalities of a transaction to determine the underlying purpose of payments between health care providers and actual or potential referral sources. Generally, courts have taken a broad interpretation of the scope of the Anti-kickback Law, holding that the statute may be violated if merely one purpose of a payment arrangement is to induce referrals or purchases.
30
In addition to statutory exceptions to the Anti-kickback Law, regulations provide for a number of safe harbors. If an arrangement meets the provisions of a safe harbor, it is deemed not to violate the Anti-kickback Law. An arrangement must fully comply with each element of an applicable safe harbor in order to qualify for protection. There are no regulatory safe harbors to California's Section 650.
Among the safe harbors that may be relevant to us is the discount safe harbor. The discount safe harbor potentially applies to discounts provided by providers and suppliers, including laboratories, to physicians or institutions. If the terms of the discount safe harbor are met, the discounts will not be considered prohibited remuneration under the Anti-kickback Law. California does not have a discount safe harbor. However, as noted above, Section 650 has generally been interpreted consistent with the Anti-kickback Law.
The personal services safe harbor to the Anti-kickback Law provides that remuneration paid to a referral source for personal services will not violate the Anti-kickback Law provided all of the elements of that safe harbor are met. One element is that if the agreement is intended to provide for the services of the physician on a periodic, sporadic or part-time basis, rather than on a full-time basis for the term of the agreement, the agreement specifies exactly the schedule of such intervals, their precise length, and the exact charge for such intervals. Our personal services arrangements with some physicians may not meet the specific requirement of this safe harbor that the agreement specify exactly the schedule of the intervals of time to be spent on the services because the nature of the services, such as speaking engagements, does not lend itself to exact scheduling and therefore meeting this element of the personal services safe harbor is impractical. Failure to meet the terms of the safe harbor does not render an arrangement illegal. Rather, the government may evaluate such arrangements on a case-by-case basis, taking into account all facts and circumstances.
While we believe that we are in compliance with the Anti-kickback Law and Section 650, there can be no assurance that our relationships with physicians, academic institutions and other customers will not be subject to investigation or challenge under such laws. If imposed for any reason, sanctions under the Anti-kickback Law and Section 650 could have a negative effect on our business.
Other Federal and State Fraud and Abuse Laws
In addition to the requirements discussed above, several other health care fraud and abuse laws could have an effect on our business. For example, provisions of the Social Security Act permit Medicare and Medicaid to exclude an entity that charges the federal health care programs substantially in excess of its usual charges for its services. The terms "usual charge" and "substantially in excess" are ambiguous and subject to varying interpretations.
Further, the Federal False Claims Act prohibits a person from knowingly submitting a claim, making a false record or statement in order to secure payment or retaining an overpayment by the federal government. In addition to actions initiated by the government itself, the statute authorizes actions to be brought on behalf of the federal government by a private party having knowledge of the alleged fraud. Because the complaint is initially filed under seal, the action may be pending for some time before the defendant is even aware of the action. If the government is ultimately successful in obtaining redress in the matter or if the plaintiff succeeds in obtaining redress without the government's involvement, then the plaintiff will receive a percentage of the recovery. Finally, the Social Security Act includes its own provisions that prohibit the filing of false claims or submitting false statements in order to obtain payment. Violation of these provisions may result in fines, imprisonment or both, and possible exclusion from Medicare or Medicaid programs. California has an analogous state false claims act applicable to all payers, as do many other states; however, we may not be aware of all such rules and statutes and cannot provide assurance that we will be in compliance with all such laws and regulations.
31
International
Many countries in which we may offer Afirma in the future have anti-kickback regulations prohibiting providers from offering, paying, soliciting or receiving remuneration, directly or indirectly, in order to induce business that is reimbursable under any national health care program. In situations involving physicians employed by state-funded institutions or national health care agencies, violation of the local anti-kickback law may also constitute a violation of the United States Foreign Corrupt Practices Act, or FCPA.
The FCPA prohibits any U.S. individual, business entity or employee of a U.S. business entity to offer or provide, directly or through a third party, including any potential distributors we may rely on in certain markets, anything of value to a foreign government official with corrupt intent to influence an award or continuation of business or to gain an unfair advantage, whether or not such conduct violate local laws. In addition, it is illegal for a company that reports to the SEC to have false or inaccurate books or records or to fail to maintain a system of internal accounting controls. We will also be required to maintain accurate information and control over sales and distributors' activities that may fall within the purview of the FCPA, its books and records provisions and its anti-bribery provisions.
The standard of intent and knowledge in the Anti-Bribery cases is minimal—intent and knowledge are usually inferred from that fact that bribery took place. The accounting provisions do not require intent. Violations of the FCPA's anti-bribery provisions for corporations and other business entities are subject to a fine of up to $2 million and officers, directors, stockholders, employees, and agents are subject to a fine of up to $100,000 and imprisonment for up to five years. Other countries, including the United Kingdom and other OECD Anti-Bribery Convention members, have similar anti-corruption regulations, such as the United Kingdom Bribery Act.
When marketing our tests outside of the United States, we may be subject to foreign regulatory requirements governing human clinical testing, prohibitions on the import of tissue necessary for us to perform our tests or restrictions on the export of tissue imposed by countries outside of the United States or the import of tissue into the United States, and marketing approval. These requirements vary by jurisdiction, differ from those in the United States and may in some cases require us to perform additional pre-clinical or clinical testing. In many countries outside of the United States, coverage, pricing and reimbursement approvals are also required.
California Laboratory Licensing
In addition to federal certification requirements of laboratories under CLIA, licensure is required and maintained for our South San Francisco clinical reference laboratory under California law. Such laws establish standards for the day-to-day operation of a clinical reference laboratory, including the training and skills required of personnel and quality control. In addition, California laws mandate proficiency testing, which involves testing of specimens that have been specifically prepared for the laboratory.
If our clinical reference laboratory is out of compliance with California standards, the California Department of Health Services, or DHS, may suspend, restrict or revoke our license to operate our clinical reference laboratory, assess substantial civil money penalties, or impose specific corrective action plans. Any such actions could materially affect our business. We maintain a current license in good standing with DHS. However, we cannot provide assurance that DHS will at all times in the future find us to be in compliance with all such laws.
32
New York Laboratory Licensing
Because we receive specimens from New York State, our clinical reference laboratories are required to be licensed by New York, under New York laws and regulations, which establish standards for:
- •
- day-to-day operation of a clinical laboratory, including training and skill levels required of laboratory personnel;
- •
- physical requirements of a facility;
- •
- equipment; and
- •
- validation and quality control.
New York law also mandates proficiency testing for laboratories licensed under New York state law, regardless of whether such laboratories are located in New York. If a laboratory is out of compliance with New York statutory or regulatory standards, the New York State Department of Health, or DOH, may suspend, limit, revoke or annul the laboratory's New York license, censure the holder of the license or assess civil money penalties. Statutory or regulatory noncompliance may result in a laboratory's operator being found guilty of a misdemeanor under New York law. DOH also must approve the LDT before the test is offered in New York. Should we be found out of compliance with New York laboratory requirements, we could be subject to such sanctions, which could harm our business. We maintain a current license in good standing with DOH for our South San Francisco and Austin laboratories. We cannot provide assurance that the DOH will at all times find us to be in compliance with applicable laws.
Other States' Laboratory Licensing
In addition to New York and California, other states including Florida, Maryland, Pennsylvania and Rhode Island, require licensing of out-of- state laboratories under certain circumstances. We have obtained licenses from states where we believe we are required to be licensed, and believe we are in compliance with applicable licensing laws.
From time to time, we may become aware of other states that require out- of-state laboratories to obtain licensure in order to accept specimens from the state, and it is possible that other states do have such requirements or will have such requirements in the future. If we identify any other state with such requirements or if we are contacted by any other state advising us of such requirements, we intend to comply with such requirements.
Corporate Practice of Medicine
Numerous states, including California and Texas, have enacted laws prohibiting corporations such as us from practicing medicine and employing or engaging physicians to practice medicine. These laws are designed to prevent interference in the medical decision-making process by anyone who is not a licensed physician. This prohibition is generally referred to as the prohibition against the corporate practice of medicine. Violation of this prohibition may result in civil or criminal fines, as well as sanctions imposed against us or the professional through licensing proceedings. The pathologists who review and classify thyroid FNA cytopathology results for Afirma are employed by Thyroid Cytopathology Partners, a Texas professional association, pursuant to services agreement between us and TCP. Pursuant to the agreement, we pay TCP a monthly fee on a per FNA basis, and TCP manages and supervises the pathologists who perform the cytopathology services as a component of Afirma. TCP is managed by Pathology Resources Consultants, or PRC, which provides management and other services to medical practitioners. We have entered into a services agreement with PRC in connection with our arrangement with TCP, pursuant to which we engaged PRC exclusively to manage the pathology services being provided by TCP. Our agreement with PRC is effective until December 31, 2015 and thereafter automatically renews every year unless either party provides notice of intent not to renew at least twelve months prior to the end of the then-current term.
33
Employees
At December 31, 2014, we had 167 employees, of which 28 work in laboratory operations, 29 in research and development and clinical development, 37 in selling and marketing, 73 in general and administrative including 41 in billing and client services, 11 in information technology, four in human resources, and one in quality and regulatory affairs. None of our employees are the subject of collective bargaining arrangements, and our management considers its relationships with employees to be good.
Environmental Matters
Our operations require the use of hazardous materials (including biological materials) which subject us to a variety of federal, state and local environmental and safety laws and regulations. Some of these regulations provide for strict liability, holding a party potentially liable without regard to fault or negligence. We could be held liable for damages and fines as a result of our, or others', business operations should contamination of the environment or individual exposure to hazardous substances occur. We cannot predict how changes in laws or new regulations will affect our business, operations or the cost of compliance.
Raw Materials and Suppliers
We procure reagents, equipment, chips and other materials we use to perform our tests from sole suppliers such as NuGEN Technologies, Inc., Affymetrix, Inc. and Thermo Fischer Scientific, Inc. We also purchase components used in our collection kits from sole-source suppliers. Some of these items are unique to these suppliers and vendors. In addition, we utilize a sole source to assemble and distribute our sample collection kits. While we have developed alternate sourcing strategies for these materials and vendors, we cannot be certain whether these strategies will be effective or whether alternative sources will be available when we need them. If these suppliers can no longer provide us with the materials we need to perform the tests and for our collection kits, if the materials do not meet our quality specifications, or if we cannot obtain acceptable substitute materials, an interruption in test processing could occur and we may not be able to deliver patient reports, and thus our business would be negatively affected.
Legal Proceedings
From time to time, we may be party to lawsuits in the ordinary course of business. We are currently not a party to any legal proceedings.
Available Information
We were incorporated in Delaware as Calderome, Inc. in August 2006. Calderome operated as an incubator until early 2008. We changed our name to Veracyte, Inc. in March 2008. Our principal executive offices are located at 7000 Shoreline Court, Suite 250, South San Francisco, California 94080 and our telephone number is (650) 243-6300. Our website address is www.veracyte.com. The information contained on, or that can be accessed through, our website is not part of this annual report on Form 10-K.
We make available free of charge on our website our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission, or SEC. The reports are also available at www.sec.gov.
34
Risks Related to Our Business
We are an early-stage company with a history of losses, and we expect to incur net losses for the foreseeable future and may never achieve or sustain profitability.
We have incurred net losses since our inception. For the years ended December 31, 2014 and 2013, we had a net loss of $29.4 million and $25.6 million, respectively, and we expect to incur additional losses in the future. From inception through December 31, 2014, we had an accumulated deficit of $115.0 million. We may never achieve revenue sufficient to offset our expenses. Over the next several years, we expect to continue to devote substantially all of our resources to increase adoption of, and reimbursement for, Afirma, as well as our lung cancer test, Percepta, which we expect to launch by mid-2015, as well as the development of additional tests we plan to commercialize, including our test for Idiopathic Pulmonary Fibrosis, or IPF. We may never achieve or sustain profitability, and our failure to achieve and sustain profitability in the future could cause the market price of our common stock to decline.
Our financial results depend solely on sales of Afirma, and we will need to generate sufficient revenue from this and other diagnostic solutions to grow our business.
All of our revenue have been derived from the sale of Afirma, which we commercially launched in January 2011. For the foreseeable future, we expect to derive substantially all of our revenue from sales of Afirma. We are planning to launch our first product in pulmonology for lung cancer, Percepta, by mid-2015, and our efforts may not be successful. In addition, we are in various stages of research and development for other diagnostic solutions that we may offer, but there can be no assurance that we will be able to identify other diseases that can be effectively addressed with our molecular cytology platform or, if we are able to identify such diseases, whether or when we will be able to successfully commercialize these solutions. If we are unable to increase sales and expand reimbursement for Afirma, or successfully develop and commercialize Percepta and other solutions, our revenue and our ability to achieve and sustain profitability would be impaired, and the market price of our common stock could decline.
We depend on a few payers for a significant portion of our revenue and if one or more significant payer stops providing reimbursement or decreases the amount of reimbursement for our tests, our revenue could decline.
Revenue for tests performed on patients covered by Medicare, Aetna and UnitedHealthcare was 26%, 11% and 18%, respectively, of our revenue for the year ended December 31, 2014, compared with 32%, 9% and 18%, respectively, in the year ended December 31, 2013. The percentage of our revenue derived from significant payers is expected to fluctuate from period to period as our revenue increases, as additional payers provide reimbursement for our tests or if one or more payers were to stop reimbursing for our tests. Effective January 2012, Palmetto GBA, the regional Medicare administrative contractor, or MAC, that handled claims processing for Medicare services with jurisdiction at that time, issued coverage and payment determinations for the GEC. On a five-year rotational basis, Medicare requests bids for its regional MAC services. Any future changes, in the MAC processing or coding for Medicare claims for the Afirma GEC could result in a change in the coverage or reimbursement rates for such products, or the loss of coverage.
In late 2014, we entered into contracts with Cigna and UnitedHealthcare that establish in-network allowable rates of reimbursement for our tests. However, payers may suspend or discontinue reimbursement at any time, may require or increase co-payments from patients, or may reduce the reimbursement rates paid to us. Any such actions could have a negative effect on our revenue.
35
If payers do not provide reimbursement, rescind or modify their reimbursement policies or delay payments for our tests, or if we are unable to successfully negotiate additional reimbursement contracts, our commercial success could be compromised.
Physicians may not order our tests unless payers reimburse a substantial portion of the test price. There is significant uncertainty concerning third-party reimbursement of any test incorporating new technology, including the Afirma GEC and Malignancy Classifiers as well as Percepta, which we plan to launch in mid-2015. Reimbursement by a payer may depend on a number of factors, including a payer's determination that these tests are:
- •
- not experimental or investigational;
- •
- pre-authorized and appropriate for the specific patient;
- •
- cost-effective;
- •
- supported by peer-reviewed publications; and
- •
- included in clinical practice guidelines.
Since each payer makes its own decision as to whether to establish a coverage policy or enter into a contract to reimburse our test, seeking these approvals is a time-consuming and costly process.
We do not have a contracted rate of reimbursement with most payers. Without a contracted rate for reimbursement, our claims are often denied upon submission, and we must appeal the claims. The appeals process is time consuming and expensive, and may not result in payment. In cases where there is not a contracted rate for reimbursement, there is typically a greater patient co-insurance or co-payment requirement which may result in further delay or decreased likelihood of collection.
We expect to continue to focus substantial resources on increasing adoption of and coverage and reimbursement for Afirma. We believe it may take several years to achieve coverage and contracted reimbursement with a majority of third-party payers. However, we cannot predict whether, under what circumstances, or at what payment levels payers will reimburse for our test. In addition, the Afirma Malignancy Classifiers, launched in May 2014, and any other new products we may develop in the future, including Percepta, may require that we expend substantial time and resources in order to obtain reimbursement. Our failure to establish broad adoption of and reimbursement for our products, or our inability to maintain existing reimbursement from payers, will negatively impact our ability to generate revenue and achieve profitability, as well as our future prospects and our business.
We may experience limits on our revenue if physicians decide not to order Afirma.
If we are unable to create or maintain demand for Afirma in sufficient volume, we may not become profitable. To generate demand, we will need to continue to educate physicians about the benefits and cost-effectiveness of Afirma through published papers, presentations at scientific conferences and one-on-one education by our sales force. In addition, our ability to obtain and maintain adequate reimbursement from third-party payers will be critical to generating revenue.
Several existing guidelines and historical practices in the United States regarding indeterminate thyroid nodule FNA results recommend a full or partial surgical thyroidectomy in most cases. Accordingly, physicians may be reluctant to order a diagnostic solution that may suggest surgery is unnecessary where some current guidelines and historical practice have typically led to such procedures. Moreover, our diagnostic services are performed at a specialized clinical reference laboratory rather than by a pathologist in a local laboratory, so pathologists may be reluctant to support our services. In addition, guidelines for the diagnosis and treatment of thyroid nodules may subsequently be revised to recommend another type of treatment protocol, and these changes may result in medical practitioners deciding not to use Afirma. These facts may make physicians reluctant to convert to using or continuing to use Afirma, which could limit our ability to generate revenue and our ability to achieve profitability. To the extent international
36
markets have existing practices and standards of care that are different than those in the United States, we may face challenges with the adoption of Afirma outside the United States.
The success of our relationship with Genzyme to co-promote Afirma may have a significant effect on our business.
We sell Afirma in the United States through our internal sales team and through our Amended and Restated U.S. Co-Promotion Agreement with Genzyme Corporation, or Amended Agreement. Under the Amended Agreement, we are required to pay Genzyme a co-promotion fee that is equal to a percentage of our cash receipts from the sale of the Afirma GEC test. The percentage is currently set at 15% beginning on January 1, 2015. Our agreement with Genzyme expires in 2027. We have also granted Genzyme a right of first offer to co-promote any future thyroid cancer product that we commercialize. If Genzyme does not commit the necessary resources to market and sell the Afirma GEC test to the level of our expectations, or if they terminate the agreement, we may not realize the benefits of this relationship and our ability to generate revenue in the future may be harmed. If our agreement with Genzyme were terminated, we would have to hire additional sales personnel to support the growth of Afirma and any other thyroid product we had previously agreed to co-promote with Genzyme.
On February 13, 2015, we entered into an Ex-U.S. Co-Promotion Agreement, or Ex-U.S. Agreement, with Genzyme for the co-exclusive promotion of the Afirma GEC test in two countries outside the United States: Brazil and Singapore. We also granted Genzyme, for a limited period of time, an exclusive right of first negotiation to enter into an agreement with us for the promotion of the Afirma GEC test in three additional countries: Canada, the Netherlands and Italy. Further, upon mutual agreement, the parties may add additional countries (other than the United States) to the Ex-U.S. Agreement. The term of the Ex-U.S. Agreement commenced January 1, 2015 and continues until December 31, 2019 with extension of the agreement possible upon agreement of the parties. Country specific terms have been established under this agreement for Brazil and Singapore. We will pay Genzyme 25% of cash receipts from the sale of the Afirma GEC test in Brazil and Singapore over a five-year period commencing January 1, 2015. If Genzyme does not commit the necessary resources to market and sell the Afirma GEC test outside the U.S. to the level of our expectations, or if they terminate the agreement, we may not realize the benefits of this relationship and our ability to generate revenue in the future may be harmed.
Due to how we recognize revenue, our quarterly operating results are likely to fluctuate.
We recognize a significant portion of our revenue upon the earlier of receipt of third-party payer notification of payment or when cash is received. We have little visibility as to when we will receive payment for our diagnostic test, and we must appeal negative payment decisions, which delays collections. For tests performed where we have an agreed upon reimbursement rate or we are able to make a reasonable estimate of reimbursement at the time delivery is complete, such as in the case of Medicare and certain other payers, we recognize the related revenue upon delivery of a patient report to the prescribing physician based on the established billing rate less contractual and other adjustments to arrive at the amount that we expect to collect. We determine the amount we expect to collect based on a per payer, per contract or agreement basis. In situations where we are not able to make a reasonable estimate of reimbursement, we recognize revenue upon the earlier of receipt of third-party notification of payment or when cash is received. Upon ultimate collection, the amount received from Medicare and other payers where reimbursement was estimated is compared to previous estimates and the contractual allowance is adjusted accordingly. These factors will likely result in fluctuations in our quarterly revenue. Should we recognize revenue from payers on an accrual basis and later determine the judgments underlying estimated reimbursement change, or were incorrect at the time we accrued such revenue, our financial results could be negatively impacted in future quarters. As a result, comparing our operating results on a period-to-period basis may not be meaningful. You should not rely on our past results as an indication of our future performance. In addition, these fluctuations in revenue may make it difficult for us, research analysts and investors to accurately forecast our revenue and operating results. If our revenue or operating results fall below expectations, the price of our common stock would likely decline.
37
We rely on sole suppliers for some of the reagents, equipment, chips and other materials used to perform our tests, and we may not be able to find replacements or transition to alternative suppliers.
We rely on sole suppliers, such as NuGEN Technologies, Inc., Affymetrix, Inc. and Thermo Fischer Scientific, Inc., for critical supply of reagents, equipment, chips and other materials that we use to perform our tests. We also purchase components used in our collection kits from sole-source suppliers. Some of these items are unique to these suppliers and vendors. In addition, we utilize a sole source to assemble and distribute our sample collection kits. While we have developed alternate sourcing strategies for these materials and vendors, we cannot be certain whether these strategies will be effective or the alternative sources will be available when we need them. If these suppliers can no longer provide us with the materials we need to perform the tests and for our collection kits, if the materials do not meet our quality specifications, or if we cannot obtain acceptable substitute materials, an interruption in test processing could occur and we may not be able to deliver patient reports. Any such interruption may significantly affect our future revenue, cause us to incur higher costs, and harm our customer relations and reputation. In addition, in order to mitigate these risks, we maintain inventories of these supplies at higher levels than would be the case if multiple sources of supply were available. If our test volume decreases, we may hold excess inventory with expiration dates that occur before use.
We depend on a specialized cytopathology practice to perform the cytopathology component of Afirma, and our ability to perform our diagnostic solution would be harmed if we were required to secure a replacement.
We rely on Thyroid Cytopathology Partners, P.A., or TCP, to provide cytopathology professional diagnoses on thyroid FNA samples pursuant to a pathology services agreement. Pursuant to this agreement, TCP has the exclusive right to provide the cytopathology diagnoses on FNA samples at a fixed price per test. We have also agreed to allow TCP to co-locate in a portion of our facilities in Austin, Texas. Our agreement with TCP is effective through December 31, 2015 and thereafter automatically renews every year unless either party provides notice of intent not to renew at least 12 months prior to the end of the then-current term.
If TCP were not able to support our current test volume or future increases in test volume or to provide the quality of services we require, or if we are unable to agree on commercial terms and our relationship with TCP were to terminate, our business would be harmed until we are able to secure the services of another cytopathology provider. There can be no assurance that we would be successful in finding a replacement that would be able to conduct cytopathology diagnoses at the same volume or with the same high-quality results as TCP. Locating another suitable cytopathology provider could be time consuming and would result in delays in processing tests until a replacement was fully integrated with our test processing operations.
If we are unable to support demand for Afirma or any of our future products or solutions, our business could suffer.
As demand for Afirma grows, and as we commercialize new products such as Percepta, we will need to continue to scale our testing capacity and processing technology, expand customer service, billing and systems processes and enhance our internal quality assurance program. We will also need additional certified laboratory scientists and other scientific and technical personnel to process higher volumes of our tests. We cannot assure you that any increases in scale, related improvements and quality assurance will be successfully implemented or that appropriate personnel will be available. Failure to implement necessary procedures, transition to new processes or hire the necessary personnel could result in higher costs of processing tests, quality control issues or inability to meet demand. There can be no assurance that we will be able to perform our testing on a timely basis at a level consistent with demand, or that our efforts to scale our operations will not negatively affect the quality of test results. If we encounter difficulty meeting market demand or quality standards, our reputation could be harmed and our future prospects and our business could suffer.
38
If the FDA were to begin regulating our tests, we could incur substantial costs and delays associated with trying to obtain premarket clearance or approval.
Clinical laboratory tests like Afirma are regulated under the Clinical Laboratory Improvement Amendments of 1988, or CLIA, as well as by applicable state laws. Most laboratory developed tests, or LDTs, are not currently subject to FDA regulation, although reagents, instruments, software or components provided by third parties and used to perform LDTs may be subject to regulation. Although the FDA has never defined what qualifies as an LDT, we believe that Afirma is an LDT. FDA currently exercises its enforcement discretion for LDTs. In October 2014, the FDA published draft guidance documents describing the framework by which they might regulate LDTs. The framework is similar to the guidance they issued previously. There is no timeframe in which the FDA must issue final guidance documents.
If the FDA requires us to seek clearance or approval to offer Afirma or any of our future products for clinical use, including Percepta, we may not be able to obtain such approvals on a timely basis, or at all. If premarket review is required, our business could be negatively impacted if we are required to stop selling our products pending their clearance or approval or the launch of any new products that we develop could be delayed by new requirements. The cost of conducting clinical trials and otherwise developing data and information to support premarket applications may be significant. In addition, future regulation by the FDA could subject our business to further regulatory risks and costs. Failure to comply with applicable regulatory requirements of the FDA could result in enforcement action, including receiving untitled or warning letters, fines, injunctions, or civil or criminal penalties. In addition, we could be subject to a recall or seizure of current or future products, operating restrictions, partial suspension or total shutdown of production. Any such enforcement action would have a material adverse effect on our business, financial condition and operations. In addition, our sample collection container is classified as a Class I medical device and is listed with the FDA. If the FDA was to determine that it is a Class II medical device, we would be required to file a 510(k) application and obtain FDA clearance to use the container, which could be time consuming and expensive.
Some of the materials we use for Afirma and that we may use for future products, such as Percepta, are labeled for research use only. In November 2013, the FDA finalized guidance regarding the sale and use of products labeled for research or investigational use only. Among other things, the guidance advises that the FDA continues to be concerned about distribution of research-investigational-use only products intended for clinical diagnostic use and that the manufacturer's objective intent for the product's intended use will be determined by examining the totality of circumstances, including advertising, instructions for clinical interpretation, presentations that describe clinical use, and specialized technical support, surrounding the distribution of the product in question. The FDA has advised that if evidence demonstrates that a product is inappropriately labeled for research or investigational use only, the device would be misbranded and adulterated within the meaning of the Federal Food, Drug and Cosmetic Act. Some of the reagents, instruments, software or components obtained by us from suppliers for use in Afirma and the Afirma Malignancy Classifier are currently labeled as research-use only products. If the FDA were to undertake enforcement actions, some of our suppliers may cease selling research-use only products to us, and any failure to obtain an acceptable substitute could significantly and adversely affect our business, financial condition and results of operations, including increasing the cost of testing or delaying, limiting or prohibiting the purchase of reagents, instruments, software or components necessary to perform testing.
If we are unable to compete successfully, we may be unable to increase or sustain our revenue or achieve profitability.
Our principal competition for Afirma comes from traditional methods used by physicians to diagnose thyroid cancer. Practice guidelines in the United States have historically recommended that patients with indeterminate diagnoses from cytopathology results be considered for surgery to remove all or part of the
39
thyroid to rule out cancer. This practice has been the standard of care in the United States for many years, and we need to educate physicians about the benefits of Afirma to change clinical practice.
We also face competition from commercial laboratories, such as Laboratory Corporation of America Holdings, Quest Diagnostics Incorporated and Sonic Healthcare USA with strong infrastructure to support the commercialization of diagnostic services. We face potential competition from companies such as Illumina, Inc. and Thermo Fisher Scientific Inc., both of which have announced their intention to enter the clinical diagnostics market. Other potential competitors include companies that develop diagnostic products, such as Roche Diagnostics, a division of Roche Holding Ltd, Siemens AG, Qiagen N.V. and Rosetta Genomics Ltd. We also face competition from companies and academic institutions that use next generation sequencing technology or other methods to measure mutational markers such as BRAF and KRAS along with numerous other mutations. In the future, we may also face competition from companies developing new products or technologies that are able to compete with Afirma's high negative predictive value to rule out cancer.
In addition, competitors may develop their own versions of our solution in countries where we do not have patents or where our intellectual property rights are not recognized and compete with us in those countries, including encouraging the use of their solution by physicians in other countries.
To compete successfully we must be able to demonstrate, among other things, that our diagnostic test results are accurate and cost effective, and we must secure a meaningful level of reimbursement for our products.
Many of our potential competitors have widespread brand recognition and substantially greater financial, technical and research and development resources, and selling and marketing capabilities than we do. Others may develop products with prices lower than ours that could be viewed by physicians and payers as functionally equivalent to our solution, or offer solutions at prices designed to promote market penetration, which could force us to lower the list price of our solution and affect our ability to achieve profitability. If we are unable to change clinical practice in a meaningful way or compete successfully against current and future competitors, we may be unable to increase market acceptance and sales of our products, which could prevent us from increasing our revenue or achieving profitability and could cause the market price of our common stock to decline. As we add new tests and services, we will face many of these same competitive risks for these new tests.
The loss of members of our senior management team or our inability to attract and retain key personnel could adversely affect our business.
Our success depends largely on the skills, experience and performance of key members of our executive management team and others in key management positions. The efforts of each of these persons together will be critical to us as we continue to develop our technologies and test processes and focus on our growth. If we were to lose one or more of these key employees, we may experience difficulties in competing effectively, developing our technologies and implementing our business strategy.
In addition, our research and development programs and commercial laboratory operations depend on our ability to attract and retain highly skilled scientists. We may not be able to attract or retain qualified scientists and technicians in the future due to the intense competition for qualified personnel among life science businesses, particularly in the San Francisco Bay Area. Our success in the development and commercialization of advanced diagnostics requires a significant medical and clinical staff to conduct studies and educate physicians and payers on the merits of our tests in order to achieve adoption and reimbursement. We are in a highly competitive industry to attract and retain this talent. As a public company located in the San Francisco Bay Area, we face intense competition for highly skilled finance and accounting personnel. If we are unable to attract and retain finance and accounting personnel experienced in public company financial reporting, we risk being unable to close our books and file our public documents on a timely basis. Additionally, our success depends on our ability to attract and retain qualified
40
sales people. During 2014, we significantly expanded our sales force for Afirma. There can be no assurance that they will be successful in maintaining and growing the business. As we plan to further increase our sales channels for new tests such as Percepta, we may have difficulties locating and recruiting additional sales personnel or retaining qualified salespeople, which could cause a delay or decline in the rate of adoption of our tests. Finally, our business requires specialized capabilities in reimbursement, billing, finance, and other areas and there may be a shortage of qualified individuals. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that could adversely affect our ability to support our research and development, clinical laboratory, sales and reimbursement, billing and finance efforts. All of our employees are at-will, which means that either we or the employee may terminate their employment at any time. We do not carry key man insurance for any of our employees.
We may be unable to manage our future growth effectively, which could make it difficult to execute our business strategy.
In addition to the need to scale our testing capacity, future growth, including our transition to a multi-product company with international operations, will impose significant added responsibilities on management, including the need to identify, recruit, train and integrate additional employees with the necessary skills to support the growing complexities of our business. In addition, rapid and significant growth may place strain on our administrative, financial and operational infrastructure. Our ability to manage our business and growth will require us to continue to improve our operational, financial and management controls, reporting systems and procedures. We have implemented an internally developed data warehouse, which is critical to our ability to track our diagnostic services and patient reports delivered to physicians, as well as to support our financial reporting systems. The time and resources required to optimize these systems is uncertain, and failure to complete optimization in a timely and efficient manner could adversely affect our operations. Additionally, growth will require us to expand and move our South San Francisco operations by 2016. Any move to a new facility could require us to re-certify our laboratory in South San Francisco. This could disrupt our business and will require the investment of resources. If we are unable to manage our growth effectively, it may be difficult for us to execute our business strategy and our business could be harmed.
Billing for our diagnostic tests is complex, and we must dedicate substantial time and resources to the billing process to be paid.
Billing for clinical laboratory testing services is complex, time consuming and expensive. Depending on the billing arrangement and applicable law, we bill various payers, including Medicare, insurance companies and patients, all of which have different billing requirements. We generally bill third-party payers for our diagnostic tests and pursue reimbursement on a case-by-case basis where pricing contracts are not in place. To the extent laws or contracts require us to bill patient co-payments or co-insurance, we must also comply with these requirements. We may also face increased risk in our collection efforts, including potential write-offs of doubtful accounts and long collection cycles, which could adversely affect our business, results of operations and financial condition.
Several factors make the billing process complex, including:
- •
- differences between the list price for our tests and the reimbursement rates of payers;
- •
- compliance with complex federal and state regulations related to billing Medicare;
- •
- disputes among payers as to which party is responsible for payment;
- •
- differences in coverage and in information and billing requirements among payers;
- •
- the effect of patient co-payments or co-insurance;
- •
- changes to billing codes used for our tests;
41
- •
- incorrect or missing billing information; and
- •
- the resources required to manage the billing and claims appeals process.
Standard industry billing codes, known as CPT codes, that we use to bill for cytopathology do not exist for our proprietary molecular diagnostic tests. Therefore, until such time that we are awarded and are able to use a designated CPT code specific to our tests, we use "miscellaneous" codes for claim submissions. These codes can change over time. When codes change, there is a risk of an error being made in the claim adjudication process. These errors can occur with claims submission, third-party transmission or in the processing of the claim by the payer. Claim adjudication errors may result in a delay in payment processing or a reduction in the amount of the payment received. Coding changes, therefore, may have an adverse effect on our revenues. Even when we receive a designated CPT code specific to our tests, there can be no assurance that payers will recognize these codes in a timely manner or that the process to transitioning to such a code will not result in errors or delays in payments.
As we introduce new tests, such as Percepta, we will need to add new codes to our billing process as well as our financial reporting systems. Failure or delays in effecting these changes in external billing and internal systems and processes could negatively affect our revenue and cash flow.
Additionally, our billing activities require us to implement compliance procedures and oversight, train and monitor our employees, challenge coverage and payment denials, assist patients in appealing claims, and undertake internal audits to evaluate compliance with applicable laws and regulations as well as internal compliance policies and procedures. Payers also conduct external audits to evaluate payments, which add further complexity to the billing process. If the payer makes an overpayment determination, there is a risk that we may be required to return some portion of prior payments we have received. These billing complexities, and the related uncertainty in obtaining payment for our diagnostic solution, could negatively affect our revenue and cash flow, our ability to achieve profitability, and the consistency and comparability of our results of operations.
We rely on a third-party to transmit claims to payers, and any delay in transmitting claims could have an adverse effect on our revenue.
While we manage the overall processing of claims, we rely on a third-party provider to transmit the actual claims to payers based on the specific payer billing format. We have previously experienced delays in claims processing when our third-party provider made changes to its invoicing system, and again when it did not submit claims to payers within the timeframe we require. Additionally, coding for diagnostic tests may change, and such changes may cause short-term billing errors that may take significant time to resolve. If claims are not submitted to payers on a timely basis or are erroneously submitted, or if we are required to switch to a different provider to handle claim submissions, we may experience delays in our ability to process these claims and receipt of payments from payers, which would have an adverse effect on our revenue and our business.
Developing new products involves a lengthy and complex process, and we may not be able to commercialize on a timely basis, or at all, other products we are developing.
We have enhancements to our current Afirma offering and other diagnostic solutions under development that will require us to devote considerable resources to research and development. There can be no assurance that we will be able to identify other diseases that can be effectively addressed with our molecular cytology platform. In addition, if we identify such diseases, we may not be able to develop products with the diagnostic accuracy necessary to be clinically useful and commercially successful. We may face challenges obtaining sufficient numbers of samples to validate a genomic signature for a molecular diagnostic product. We are in the process of developing tests for lung cancer and interstitial lung disease, specifically IPF. Our planned lung cancer test, Percepta, has been clinically validated in three independent clinical trials including two multi-center, prospective studies, but we must complete analytical verification
42
and studies required to transfer it to our CLIA-certified laboratory prior to commercial launch. This test may not be successfully transferred to the laboratory as planned and launched by mid-2015, and our product for interstitial lung diseases may not be fully developed and introduced as planned in 2016.
In order to develop and commercialize diagnostic tests, we need to:
- •
- expend significant funds to conduct substantial research and development;
- •
- conduct successful analytical and clinical studies;
- •
- scale our laboratory processes to accommodate new tests; and
- •
- build the commercial infrastructure to market and sell new products.
Our product development process involves a high degree of risk and may take several years. Our product development efforts may fail for many reasons, including:
- •
- failure to identify a genomic signature in biomarker discovery;
- •
- inability to secure sufficient numbers of samples at an acceptable cost and on an acceptable timeframe to conduct analytical and
clinical studies; or
- •
- failure of clinical validation studies to support the effectiveness of the test.
Typically, few research and development projects result in commercial products, and success in early clinical studies often is not replicated in later studies. At any point, we may abandon development of a product candidate or we may be required to expend considerable resources repeating clinical studies, which would adversely affect the timing for generating potential revenue from a new product and our ability to invest in other products in our pipeline. If a clinical validation study fails to demonstrate the prospectively defined endpoints of the study or if we fail to sufficiently demonstrate analytical validity, we might choose to abandon the development of the product, which could harm our business. In addition, competitors may develop and commercialize competing products or technologies faster than us or at a lower cost.
We may acquire businesses or assets, form joint ventures or make investments in other companies or technologies that could harm our operating results, dilute our stockholders' ownership, increase our debt or cause us to incur significant expense.
We acquired Allegro Diagnostics in September 2014, and we may pursue additional acquisitions of complementary businesses or assets, as well as technology licensing arrangements as part of our business strategy. We also may pursue strategic alliances that leverage our core technology and industry experience to expand our offerings or distribution, or make investments in other companies. To date, we have limited experience with respect to acquisitions and the formation of strategic alliances and joint ventures. We may not be able to integrate acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. In addition, we may not realize the expected benefits of our recent acquisition of Allegro or any businesses we may acquire in the future. Any acquisitions made by us also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could harm our operating results. Integration of acquired companies or businesses we may acquire in the future also may require management resources that otherwise would be available for ongoing development of our existing business. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance, joint venture or investment.
To finance any acquisitions or investments, we may choose to issue shares of our stock as consideration, which would dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may not be able to acquire other companies for stock. Alternatively, it may be necessary for us to raise additional funds for these activities through public or private financings. Additional funds
43
may not be available on terms that are favorable to us, or at all. If these funds are raised through the sale of equity or convertible debt securities, dilution to our stockholders could result. Our current loan and security agreement contains covenants that could limit our ability to sell debt securities or obtain additional debt financing arrangements.
If we are unable to develop products to keep pace with rapid technological, medical and scientific change, our operating results and competitive position could be harmed.
In recent years, there have been numerous advances in technologies relating to diagnostics, particularly diagnostics that are based on genomic information. These advances require us to continuously develop our technology and to work to develop new solutions to keep pace with evolving standards of care. Our solutions could become obsolete unless we continually innovate and expand our product offerings to include new clinical applications. If we are unable to develop new products or to demonstrate the applicability of our products for other diseases, our sales could decline and our competitive position could be harmed.
If we fail to comply with federal, state and foreign laboratory licensing requirements, we could lose the ability to perform our tests or experience disruptions to our business.
We are subject to CLIA, a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. CLIA regulations mandate specific standards in the areas of personnel qualifications, administration, and participation in proficiency testing, patient test management and quality assurance. CLIA certification is also required in order for us to be eligible to bill state and federal healthcare programs, as well as many private third-party payers. To renew these certifications, we are subject to survey and inspection every two years. Moreover, CLIA inspectors may make random inspections of our clinical reference laboratories. If we relocate either of our facilities, we would be required to undergo certification at our new facility in order to offer our tests.
We are also required to maintain state licenses to conduct testing in our laboratories. California, New York, Texas, among other states' laws, require that we maintain a license and establishes standards for the day-to-day operation of our clinical reference laboratories, including the training and skills required of personnel and quality control matters. In addition, both of our clinical reference laboratories are required to be licensed on a test-specific basis by New York State. We have received approval for the Afirma tests we currently offer, but will need to obtain approval for Percepta and any tests we may offer in the future. New York law also mandates proficiency testing for laboratories licensed under New York state law, regardless of whether such laboratories are located in New York. Several other states require that we hold licenses to test samples from patients in those states. Other states may have similar requirements or may adopt similar requirements in the future. If we were to lose our CLIA certificate or California license for our South San Francisco laboratory, whether as a result of revocation, suspension or limitation, we would no longer be able to perform the GEC, which would eliminate our primary source of revenue and harm our business. If we were to lose our CLIA certificate for our Austin laboratory, we would need to move the receipt and storage of FNAs, as well as the slide preparation for cytopathology, to South San Francisco, which could result in a delay in processing tests during that transition and increased costs. If we were to lose our licenses issued by New York or by other states where we are required to hold licenses, we would not be able to test specimens from those states. New tests we may develop may be subject to new approvals by regulatory bodies such as New York State, and we may not be able to offer our new tests until such approvals are received.
Finally, we may be subject to regulation in foreign jurisdictions as we pursue offering our tests internationally. Other limitations, such as prohibitions on the import of tissue necessary for us to perform our tests or restrictions on the export of tissue imposed by countries outside of the United States or the import of tissue into the United States, may constrain our ability to offer tests internationally in the future.
44
Changes in healthcare policy, including legislation reforming the U.S. healthcare system, may have a material adverse effect on our financial condition and operations.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, collectively, the PPACA, enacted in March 2010, makes changes that are expected to significantly affect the pharmaceutical and medical device industries and clinical laboratories. Beginning in 2013, each medical device manufacturer must pay a sales tax in an amount equal to 2.3% of the price for which such manufacturer sells its medical devices that are listed with the FDA. The FDA has asserted that clinical laboratory tests such as Afirma are medical devices. However, consistent with the FDA's policy of exercising enforcement discretion for LDTs, Afirma is not currently listed as a medical device with the FDA. We cannot assure you that the tax will not be extended to services such as ours in the future if Afirma were to be regulated as a device. The PPACA also mandates a reduction in payments for clinical laboratory services paid under the Medicare Clinical Laboratory Fee Schedule, or CLFS, of 1.75% for the years 2011 through 2015 and a productivity adjustment to the CLFS which would affect our cytopathology billings.
Other significant measures contained in the PPACA include, for example, coordination and promotion of research on comparative clinical effectiveness of different technologies and procedures, initiatives to revise Medicare payment methodologies, such as bundling of payments across the continuum of care by providers and physicians, and initiatives to promote quality indicators in payment methodologies. The PPACA also includes significant new fraud and abuse measures, including required disclosures of financial arrangements with physician customers, lower thresholds for violations and increasing potential penalties for such violations. In addition, the PPACA establishes an Independent Payment Advisory Board, or IPAB, to reduce the per capita rate of growth in Medicare spending. The IPAB has broad discretion to propose policies to reduce expenditures, which may have a negative effect on payment rates for services. The IPAB proposals may affect payments for clinical laboratory services beginning in 2016 and for hospital services beginning in 2020. We are monitoring the effect of the PPACA to determine the trends and changes that may be necessitated by the legislation, any of which may potentially affect our business.
In addition to the PPACA, the effect of which on our business cannot presently be fully quantified, various healthcare reform proposals have also emerged from federal and state governments. For example, in February 2012, Congress passed the Middle Class Tax Relief and Job Creation Act of 2012, which in part resets the clinical lab payment rates on the Medicare CLFS by 2% in 2013. In addition, a further reduction of 2% is anticipated from implementation of the automatic expense reductions (sequester) under the Budget Control Act of 2011, which is legislated to be in effect for dates of service on or after April 1, 2013 until fiscal year 2024. Reductions resulting from the Congressional sequester are applied to total claims payment made; however, they do not currently result in a rebasing of the negotiated or established Medicare or Medicaid reimbursement rates.
State legislation on reimbursement applies to Medicaid reimbursement and Managed Medicaid reimbursement rates within that state. Some states have passed or proposed legislation that would revise reimbursement methodology for clinical laboratory payment rates under those Medicaid programs. Recent changes to reimbursement methodologies have not changed the payment rate for Afirma; however, we cannot predict whether future healthcare initiatives will be implemented at the federal or state level or in countries outside of the United States in which we may do business, or the effect any future legislation or regulation will have on us. The taxes imposed by the new federal legislation, cost reduction measures and the expansion in the role of the U.S. government in the healthcare industry may result in decreased revenue, lower reimbursement by payers for our tests or reduced medical procedure volumes, all of which may adversely affect our business, financial condition and results of operations. In addition, sales of our tests outside the United States will subject our business to foreign regulatory requirements and cost-reduction measures, which may also change over time.
45
Ongoing calls for deficit reduction at the Federal government level and reforms to programs such as the Medicare program to pay for such reductions may affect the pharmaceutical, medical device and clinical laboratory industries. In particular, recommendations by the Simpson-Bowles Commission called for the combination of Medicare Part A (hospital insurance) and Part B (physician and ancillary service insurance) into a single co-insurance and co-payment structure. Currently, clinical laboratory services are excluded from the Medicare Part B co-insurance and co-payment as preventative services. Combining Parts A and B may require clinical laboratories to collect co-payments from patients which may increase our costs and reduce the amount ultimately collected.
In April 2014, the President signed the Protecting Access to Medicare Act of 2014, or PAMA, which included a substantial new payment system for clinical laboratory tests under the CLFS. Under PAMA, laboratories that receive the majority of their Medicare revenues from payments made under the CLFS would report, beginning January 1, 2016, and then on an every three year basis thereafter (or annually for "advanced diagnostic laboratory tests"), private payer payment rates and volumes for their tests. CMS will use the rates and volumes reported by laboratories to develop Medicare payment rates for the tests equal to the volume-weighted median of the private payer payment rates for the tests. The payment rates calculated under PAMA will be effective starting January 1, 2017. Any reductions to payment rates resulting from the new methodology are limited to 10% per test per year in each of the years 2017 through 2019 and to 15% per test per year in each of 2020 through 2022. Although CMS has not yet issued regulations to implement PAMA, we believe our Afirma GEC as well as Percepta would be considered an advanced diagnostic laboratory test. Further rule-making from CMS will define the time period and data elements evaluated on an annual basis to set reimbursement rates for tests like ours.
PAMA also requires CMS to issue unique Health Care Common Procedure Coding System, or HCPCS codes to advanced diagnostic laboratory tests by January 1, 2016 for tests that were paid under the Medicare program prior to passage of the Act. In March 2015, we were issued a unique Tier1 CPT code that could impact reimbursement of the Afirma GEC in the future. Under the PAMA, new advanced diagnostic laboratory tests paid by Medicare after the date of passage of PAMA will also receive unique HCPCS codes impacting private payer reimbursement of future tests we may commercialize.
PAMA codified coverage rules for laboratory tests by requiring any local coverage determination to be made following the established procedures for development and appeals of local coverage determinations. PAMA also authorizes CMS to consolidate coverage policies for clinical laboratory tests among one to four laboratory-specific MACs. These same contractors may also be designated to process claims if CMS determines that such a model is appropriate.
In addition to changes adopted by PAMA, in 2013 CMS announced plans to bundle payments for clinical laboratory tests together with other services performed during hospital outpatient visits under the Hospital Outpatient Prospective Payment System. CMS exempted molecular diagnostic tests from this packaging provision at that time. It is possible that this exemption could be removed by CMS in future rule making, which might result in lower reimbursement for tests performed in this setting.
We may experience limits on our revenue if patients decide not to use our tests.
Some patients may decide not to use our tests because of price, all or part of which may be payable directly by the patient if the patient's insurer denies reimbursement in full or in part. There is a growing trend among insurers to shift more of the cost of healthcare to patients in the form of higher co-payments or premiums, and this trend is accelerating which puts patients in the position of having to pay more for our tests. Implementation of provisions of the PPACA has also resulted in increases in premiums and reductions in coverage for some patients. If the United States Supreme Court finds parts of PPACA to be unconstitutional, patients may be unable to afford our tests due to changes in their insurance coverage. These events may result in patients delaying or forgoing medical checkups or treatment due to their inability to pay for our tests, which could have an adverse effect on our revenue.
46
Complying with numerous statutes and regulations pertaining to our business is an expensive and time-consuming process, and any failure to comply could result in substantial penalties.
Our operations are subject to other extensive federal, state, local, and foreign laws and regulations, all of which are subject to change. These laws and regulations currently include, among others:
- •
- the Federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which established comprehensive federal standards
with respect to the privacy and security of protected health information and requirements for the use of certain standardized electronic transactions, and amendments to HIPAA under the Health
Information Technology for Economic and Clinical Health Act, which strengthen and expand HIPAA privacy and security compliance requirements, increase penalties for violators, extend enforcement
authority to state attorneys general, and impose requirements for breach notification;
- •
- Medicare billing and payment regulations applicable to clinical laboratories;
- •
- the Federal Anti-Kickback Statute, which prohibits knowingly and willfully offering, paying, soliciting, or receiving remuneration,
directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing, arranging for, or recommending of an item or service that is reimbursable, in whole or in
part, by a federal health care program;
- •
- the Federal Stark physician self-referral law (and state equivalents), which prohibits a physician from making a referral for certain
designated health services covered by the Medicare program, including laboratory and pathology services, if the physician or an immediate family member has a financial relationship with the entity
providing the designated health services, unless the financial relationship falls within an applicable exception to the prohibition;
- •
- the Federal Civil Monetary Penalties Law, which prohibits, among other things, the offering or transfer of remuneration to a Medicare
or state health care program beneficiary if the person knows or should know it is likely to influence the beneficiary's selection of a particular provider, practitioner, or supplier of services
reimbursable by Medicare or a state health care program, unless an exception applies;
- •
- the Federal False Claims Act, which imposes liability on any person or entity that, among other things, knowingly presents, or causes
to be presented, a false or fraudulent claim for payment to the federal government;
- •
- other federal and state fraud and abuse laws, such as anti-kickback laws, prohibitions on self-referral, fee-splitting restrictions,
prohibitions on the provision of products at no or discounted cost to induce physician or patient adoption, and false claims acts, which may extend to services reimbursable by any third-party payer,
including private insurers;
- •
- the prohibition on reassignment of Medicare claims, which, subject to certain exceptions, precludes the reassignment of Medicare
claims to any other party;
- •
- the rules regarding billing for diagnostic tests reimbursable by the Medicare program, which prohibit a physician or other supplier
from marking up the price of the technical component or professional component of a diagnostic test ordered by the physician or other supplier and supervised or performed by a physician who does not
"share a practice" with the billing physician or supplier;
- •
- state laws that prohibit other specified practices related to billing such as billing physicians for testing that they order, waiving
co-insurance co-payments, deductibles, and other amounts owed by patients, and billing a state Medicaid program at a price that is higher than what is charged to other payers; and
- •
- the Foreign Corrupt Practices Act of 1977, and other similar laws, which apply to our international activities.
47
We have adopted policies and procedures designed to comply with these laws and regulations. In the ordinary course of our business, we conduct internal reviews of our compliance with these laws. Our compliance is also subject to governmental review. The growth of our business and sales organization and our expansion outside of the United States may increase the potential of violating these laws or our internal policies and procedures. We believe that we are in material compliance with all statutory and regulatory requirements, but there is a risk that one or more government agencies could take a contrary position. These laws and regulations are complex and are subject to interpretation by the courts and by government agencies. If one or more such agencies alleges that we may be in violation of any of these requirements, regardless of the outcome, it could damage our reputation and adversely affect important business relationships with third parties, including managed care organizations and other commercial third-party payers. Any action brought against us for violation of these or other laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management's attention from the operation of our business. If our operations are found to be in violation of any of these laws and regulations, we may be subject to any applicable penalty associated with the violation, including civil and criminal penalties, damages and fines, we could be required to refund payments received by us, and we could be required to curtail or cease our operations. Any of the foregoing consequences could seriously harm our business and our financial results.
International expansion of our business exposes us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the United States.
Our business strategy includes international expansion in select countries, and may include developing and maintaining physician outreach and education capabilities outside of the United States, establishing agreements with laboratories, and expanding our relationships with international payers. Doing business internationally involves a number of risks, including:
- •
- multiple, conflicting and changing laws and regulations such as tax laws, privacy laws, export and import restrictions, employment
laws, regulatory requirements and other governmental approvals, permits and licenses;
- •
- failure by us to obtain regulatory approvals where required for the use of our solution in various countries;
- •
- complexities associated with managing multiple payer reimbursement regimes, government payers or patient self-pay systems;
- •
- logistics and regulations associated with shipping tissue samples, including infrastructure conditions and transportation delays;
- •
- challenges associated with establishing laboratory partners, including proper sample collection techniques, inventory management,
sample logistics, billing and promotional activities;
- •
- limits on our ability to penetrate international markets if we are not able to process tests locally;
- •
- financial risks, such as longer payment cycles, difficulty in collecting from payers, the effect of local and regional financial
crises, and exposure to foreign currency exchange rate fluctuations;
- •
- natural disasters, political and economic instability, including wars, terrorism, and political unrest, outbreak of disease, boycotts,
curtailment of trade and other business restrictions; and
- •
- regulatory and compliance risks that relate to maintaining accurate information and control over activities that may fall within the purview of the Foreign Corrupt Practices Act of 1977, its books and records provisions or its anti-bribery provisions.
Any of these factors could significantly harm our future international expansion and operations and, consequently, our revenue and results of operations.
48
If we are sued for product liability or errors and omissions liability, we could face substantial liabilities that exceed our resources.
The marketing, sale and use of Afirma or future tests, such as Percepta, could lead to product liability claims if someone were to allege that the tests failed to perform as they were designed. We may also be subject to liability for errors in the results we provide to physicians or for a misunderstanding of, or inappropriate reliance upon, the information we provide. Our Afirma GEC is performed on FNA samples that are diagnosed as indeterminate by standard cytopathology review. We report results as benign or suspicious to the prescribing physician. Under certain circumstances, we might report a result as benign that later proves to have been malignant. This could be the result of the physician having poor nodule sampling in collecting the FNA, performing the FNA on a different nodule than the one that is malignant or failure of the GEC to perform as intended. We may also be subject to similar types of claims related to our Afirma Malignancy Classifiers as well as tests we may develop in the future, including Percepta which may classify a patient as low risk for lung cancer who is later found to have a malignancy. A product liability or errors and omissions liability claim could result in substantial damages and be costly and time consuming for us to defend. Although we maintain product liability and errors and omissions insurance, we cannot assure you that our insurance would fully protect us from the financial impact of defending against these types of claims or any judgments, fines or settlement costs arising out of any such claims. Any product liability or errors and omissions liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any product liability lawsuit could cause injury to our reputation or cause us to suspend sales of our products and solutions. The occurrence of any of these events could have an adverse effect on our business and results of operations.
If our laboratory in South San Francisco becomes inoperable due to an earthquake or either of our laboratories becomes inoperable for any other reason, we will be unable to perform our testing services and our business will be harmed.
We perform all of the Afirma GEC testing at our laboratory in South San Francisco, California. Our laboratory in Austin, Texas accepts and stores substantially all FNA samples pending transfer to our California laboratory for Afirma GEC processing. We plan to commence testing for Percepta in our South San Francisco laboratory as well. The equipment we use to perform our tests would be costly to replace and could require substantial lead time to replace and qualify for use. Either of our facilities may be harmed or rendered inoperable by natural or man-made disasters, including earthquakes, flooding and power outages, which may render it difficult or impossible for us to perform our testing services for some period of time or to receive and store samples. The inability to perform our tests for even a short period of time may result in the loss of customers or harm our reputation, and we may be unable to regain those customers in the future. Although we maintain insurance for damage to our property and the disruption of our business, this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, if at all.
If we cannot enter into new clinical study collaborations, our product development and subsequent commercialization could be delayed.
In the past, we have entered into clinical study collaborations, and our success in the future depends in part on our ability to enter into additional collaborations with highly regarded institutions. This can be difficult due to internal and external constraints placed on these organizations. Some organizations may limit the number of collaborations they have with any one company so as to not be perceived as biased or conflicted. Organizations may also have insufficient administrative and related infrastructure to enable collaborations with many companies at once, which can extend the time it takes to develop, negotiate and implement a collaboration. Additionally, organizations often insist on retaining the rights to publish the clinical data resulting from the collaboration. The publication of clinical data in peer-reviewed journals is a
49
crucial step in commercializing and obtaining reimbursement for our diagnostic tests, and our inability to control when and if results are published may delay or limit our ability to derive sufficient revenue from them.
If we use hazardous materials in a manner that causes contamination or injury, we could be liable for resulting damages.
We are subject to federal, state and local laws, rules and regulations governing the use, discharge, storage, handling and disposal of biological material, chemicals and waste. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, remediation costs and any related penalties or fines, and any liability could exceed our resources or any applicable insurance coverage we may have. The cost of compliance with these laws and regulations may become significant, and our failure to comply may result in substantial fines or other consequences, and either could negatively affect our operating results.
Our inability to raise additional capital on acceptable terms in the future may limit our ability to develop and commercialize new solutions and technologies and expand our operations.
We expect capital expenditures and operating expenses to increase over the next several years as we expand our infrastructure, commercial operations and research and development activities. We may seek to raise additional capital through equity offerings, debt financings, collaborations or licensing arrangements. Additional funding may not be available to us on acceptable terms, or at all. If we raise funds by issuing equity securities, dilution to our stockholders could result. Any equity securities issued also may provide for rights, preferences or privileges senior to those of holders of our common stock. The terms of debt securities issued or borrowings could impose significant restrictions on our operations. The incurrence of additional indebtedness or the issuance of certain equity securities could result in increased fixed payment obligations and could also result in restrictive covenants, such as limitations on our ability to incur additional debt or issue additional equity, limitations on our ability to acquire or license intellectual property rights, and other operating restrictions that could adversely affect our ability to conduct our business. In addition, the issuance of additional equity securities by us, or the possibility of such issuance, may cause the market price of our common stock to decline. In the event that we enter into collaborations or licensing arrangements to raise capital, we may be required to accept unfavorable terms. These agreements may require that we relinquish or license to a third-party on unfavorable terms our rights to technologies or product candidates that we otherwise would seek to develop or commercialize ourselves, or reserve certain opportunities for future potential arrangements when we might be able to achieve more favorable terms. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more research and development programs or selling and marketing initiatives. In addition, we may have to work with a partner on one or more of our products or market development programs, which could lower the economic value of those programs to our company.
Security breaches, loss of data and other disruptions to us or our third-party service providers could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of our business, we and our third-party service providers collect and store sensitive data, including legally protected health information, personally identifiable information about our patients, credit card information, intellectual property, and our proprietary business and financial information. We manage and maintain our applications and data utilizing a combination of on-site systems, managed data center systems and cloud-based data center systems. We face a number of risks relative to our protection of, and our service providers' protection of, this critical information, including loss of
50
access, inappropriate disclosure and inappropriate access, as well as risks associated with our ability to identify and audit such events.
The secure processing, storage, maintenance and transmission of this critical information is vital to our operations and business strategy, and we devote significant resources to protecting such information. Although we take measures to protect sensitive information from unauthorized access or disclosure, our information technology and infrastructure may be vulnerable to attacks by hackers or viruses or otherwise breached due to employee error, malfeasance or other activities. While we are not aware of any such attack or breach, if such event would occur and cause interruptions in our operations, our networks would be compromised and the information we store on those networks could be accessed by unauthorized parties, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, such as HIPAA, and regulatory penalties. Unauthorized access, loss or dissemination could also disrupt our operations, including our ability to process tests, provide test results, bill payers or patients, process claims and appeals, provide customer assistance services, conduct research and development activities, collect, process and prepare company financial information, provide information about our tests and other patient and physician education and outreach efforts through our website, manage the administrative aspects of our business and damage our reputation, any of which could adversely affect our business.
In addition, the interpretation and application of consumer, health-related and data protection laws in the United States, Europe and elsewhere are often uncertain, contradictory and in flux. It is possible that these laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. Complying with these various laws could cause us to incur substantial costs or require us to change our business practices, systems and compliance procedures in a manner adverse to our business.
If we cannot license rights to use technologies on reasonable terms, we may not be able to commercialize new products in the future.
In the future, we may license third-party technology to develop or commercialize new products. In return for the use of a third-party's technology, we may agree to pay the licensor royalties based on sales of our solutions. Royalties are a component of cost of revenue and affect the margins on our solutions. We may also need to negotiate licenses to patents and patent applications after introducing a commercial product. Our business may suffer if we are unable to enter into the necessary licenses on acceptable terms, or at all, if any necessary licenses are subsequently terminated, if the licensors fail to abide by the terms of the license or fail to prevent infringement by third parties, or if the licensed patents or other rights are found to be invalid or unenforceable.
If we are unable to protect our intellectual property effectively, our business would be harmed.
We rely on patent protection as well as trademark, copyright, trade secret and other intellectual property rights protection and contractual restrictions to protect our proprietary technologies, all of which provide limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. If we fail to protect our intellectual property, third parties may be able to compete more effectively against us and we may incur substantial litigation costs in our attempts to recover or restrict use of our intellectual property.
We apply for and in-license patents covering our products and technologies and uses thereof, as we deem appropriate, however we may fail to apply for patents on important products and technologies in a timely fashion or at all, or we may fail to apply for patents in potentially relevant jurisdictions. We have five issued patents which expire between 2029 and 2032 related to methods used in the Afirma diagnostic platform, in addition to seven pending United States utility patent applications, two United States
51
provisional applications and one PCT application. Some of these United States utility patent applications have pending foreign counterparts. We also exclusively licensed intellectual property, including rights to three pending United States utility patent applications in the thyroid space that would expire between 2030 and 2033 once issued, related to methods that are used in the Afirma diagnostic test, some of which have foreign counterparts . In the lung diagnostic space, we exclusively licensed intellectual property rights to 19 pending patent applications and two issued patents in the United States and abroad. Patents issuing from the licensed portfolio will expire between 2024 and 2028. In addition, we own four pending patent applications, a PCT application, and a provisional U.S. application related to our lung cancer test under development, Percepta, as well as a PCT application and two provisional United States applications related to our interstitial lung disease test under development. Any patents granted from the current lung cancer patent applications will expire from 2032 to 2034 and those from the interstitial lung disease patent applications will expire from 2034 to 2036. It is possible that none of our pending patent applications will result in issued patents in a timely fashion or at all, and even if patents are granted, they may not provide a basis for intellectual property protection of commercially viable products, may not provide us with any competitive advantages, or may be challenged and invalidated by third parties. It is possible that others will design around our current or future patented technologies. We may not be successful in defending any challenges made against our patents or patent applications. Any successful third-party challenge to our patents could result in the unenforceability or invalidity of such patents and increased competition to our business. The outcome of patent litigation can be uncertain and any attempt by us to enforce our patent rights against others may not be successful, or, if successful, may take substantial time and result in substantial cost, and may divert our efforts and attention from other aspects of our business.
The patent positions of life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies' patents has emerged to date in the United States or elsewhere. Courts frequently render opinions in the biotechnology field that may affect the patentability of certain inventions or discoveries, including opinions that may affect the patentability of methods for analyzing or comparing DNA.
In particular, the patent positions of companies engaged in the development and commercialization of genomic diagnostic tests, like the Afirma GEC, Malignancy Classifiers and Percepta, are particularly uncertain. Various courts, including the U.S. Supreme Court, have rendered decisions that affect the scope of patentability of certain inventions or discoveries relating to certain diagnostic tests and related methods. These decisions state, among other things, that patent claims that recite laws of nature (for example, the relationship between blood levels of certain metabolites and the likelihood that a dosage of a specific drug will be ineffective or cause harm) are not themselves patentable. What constitutes a law of nature is uncertain, and it is possible that certain aspects of genomic diagnostics tests would be considered natural laws. Accordingly, the evolving case law in the United States may adversely affect our ability to obtain patents and may facilitate third-party challenges to any owned and licensed patents.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and we may encounter difficulties protecting and defending such rights in foreign jurisdictions. The legal systems of many other countries do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biotechnology, which could make it difficult for us to stop the infringement of our patents in such countries. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Changes in either the patent laws or in interpretations of patent laws in the United States or other countries may diminish the value of our intellectual property. We cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents. We may not develop additional proprietary products, methods and technologies that are patentable.
52
In addition to pursuing patents on our technology, we take steps to protect our intellectual property and proprietary technology by entering into agreements, including confidentiality agreements, non-disclosure agreements and intellectual property assignment agreements, with our employees, consultants, academic institutions, corporate partners and, when needed, our advisors. Such agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements, and we may not be able to prevent such unauthorized disclosure. If we are required to assert our rights against such party, it could result in significant cost and distraction.
Monitoring unauthorized disclosure is difficult, and we do not know whether the steps we have taken to prevent such disclosure are, or will be, adequate. If we were to enforce a claim that a third-party had illegally obtained and was using our trade secrets, it would be expensive and time consuming, and the outcome would be unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets.
We may also be subject to claims that our employees have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers, or to claims that we have improperly used or obtained such trade secrets. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights and face increased competition to our business. A loss of key research personnel work product could hamper or prevent our ability to commercialize potential products, which could harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Further, competitors could attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. Others may independently develop similar or alternative products and technologies or replicate any of our products and technologies. If our intellectual property does not adequately protect us against competitors' products and methods, our competitive position could be adversely affected, as could our business.
We have not registered certain of our trademarks, including Afirma and Percepta, in all of our potential markets. If we apply to register these trademarks, our applications may not be allowed for registration in a timely fashion or at all, and our registered trademarks may not be maintained or enforced. In addition, opposition or cancellation proceedings may be filed against our trademark applications and registrations, and our trademarks may not survive such proceedings. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
To the extent our intellectual property offers inadequate protection, or is found to be invalid or unenforceable, we would be exposed to a greater risk of direct competition. If our intellectual property does not provide adequate coverage of our competitors' products, our competitive position could be adversely affected, as could our business. Both the patent application process and the process of managing patent disputes can be time consuming and expensive.
We may be involved in litigation related to intellectual property, which could be time-intensive and costly and may adversely affect our business, operating results or financial condition.
We may receive notices of claims of direct or indirect infringement or misappropriation or misuse of other parties' proprietary rights from time to time. Some of these claims may lead to litigation. We cannot assure you that we will prevail in such actions, or that other actions alleging misappropriation or misuse by us of third-party trade secrets, infringement by us of third-party patents and trademarks or other rights, or the validity of our patents, trademarks or other rights, will not be asserted or prosecuted against us.
53
We might not have been the first to make the inventions covered by each of our pending patent applications and we might not have been the first to file patent applications for these inventions. To determine the priority of these inventions, we may have to participate in interference proceedings, derivation proceedings, or other post-grant proceedings declared by the United States Patent and Trademark Office that could result in substantial cost to us. No assurance can be given that other patent applications will not have priority over our patent applications. In addition, recent changes to the patent laws of the United States allow for various post-grant opposition proceedings that have not been extensively tested, and their outcome is therefore uncertain. Furthermore, if third parties bring these proceedings against our patents, we could experience significant costs and management distraction.
Litigation may be necessary for us to enforce our patent and proprietary rights or to determine the scope, coverage and validity of the proprietary rights of others. The outcome of any litigation or other proceeding is inherently uncertain and might not be favorable to us, and we might not be able to obtain licenses to technology that we require on acceptable terms or at all. Further, we could encounter delays in product introductions, or interruptions in product sales, as we develop alternative methods or products. In addition, if we resort to legal proceedings to enforce our intellectual property rights or to determine the validity, scope and coverage of the intellectual property or other proprietary rights of others, the proceedings could be burdensome and expensive, even if we were to prevail. Any litigation that may be necessary in the future could result in substantial costs and diversion of resources and could have a material adverse effect on our business, operating results or financial condition.
As we move into new markets and applications for our products, incumbent participants in such markets may assert their patents and other proprietary rights against us as a means of slowing our entry into such markets or as a means to extract substantial license and royalty payments from us. Our competitors and others may now and, in the future, have significantly larger and more mature patent portfolios than we currently have. In addition, future litigation may involve patent holding companies or other adverse patent owners who have no relevant product revenue and against whom our own patents may provide little or no deterrence or protection. Therefore, our commercial success may depend in part on our non-infringement of the patents or proprietary rights of third parties. Numerous significant intellectual property issues have been litigated, and will likely continue to be litigated, between existing and new participants in our existing and targeted markets and competitors may assert that our products infringe their intellectual property rights as part of a business strategy to impede our successful entry into or growth in those markets. Third parties may assert that we are employing their proprietary technology without authorization. In addition, our competitors and others may have patents or may in the future obtain patents and claim that making, having made, using, selling, offering to sell or importing our products infringes these patents. We could incur substantial costs and divert the attention of our management and technical personnel in defending against any of these claims. Parties making claims against us may be able to obtain injunctive or other relief, which could block our ability to develop, commercialize and sell products, and could result in the award of substantial damages against us. In the event of a successful claim of infringement against us, we may be required to pay damages and ongoing royalties, and obtain one or more licenses from third parties, or be prohibited from selling certain products. We may not be able to obtain these licenses on acceptable terms, if at all. We could incur substantial costs related to royalty payments for licenses obtained from third parties, which could negatively affect our financial results. In addition, we could encounter delays in product introductions while we attempt to develop alternative methods or products to avoid infringing third-party patents or proprietary rights. Defense of any lawsuit or failure to obtain any of these licenses could prevent us from commercializing products, and the prohibition of sale of any of our products could materially affect our business and our ability to gain market acceptance for our products.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could
54
be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
In addition, our agreements with some of our customers, suppliers or other entities with whom we do business require us to defend or indemnify these parties to the extent they become involved in infringement claims, including the types of claims described above. We could also voluntarily agree to defend or indemnify third parties in instances where we are not obligated to do so if we determine it would be important to our business relationships. If we are required or agree to defend or indemnify third parties in connection with any infringement claims, we could incur significant costs and expenses that could adversely affect our business, operating results, or financial condition.
Risks Related to Being a Public Company
We will continue to incur increased costs and demands on management as a result of compliance with laws and regulations applicable to public companies, which could harm our operating results.
As a public company, we will continue to incur significant legal, accounting, consulting and other expenses that we did not incur as a private company, including costs associated with public company reporting requirements. In addition, the Sarbanes-Oxley Act of 2002 and the Dodd-Frank Act of 2010, as well as rules implemented by the Securities and Exchange Commission, or the SEC, and The NASDAQ Stock Market, impose a number of requirements on public companies, including with respect to corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance and disclosure obligations. Moreover, these rules and regulations have and will continue to increase our legal, accounting and financial compliance costs and make some activities more complex, time-consuming and costly. We also expect that it will continue to be expensive for us to maintain director and officer liability insurance.
If we are unable to implement and maintain effective internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our reported financial information and the market price of our common stock may be negatively affected.
As a public company, we are required to maintain internal control over financial reporting and to report any material weaknesses in such internal control. Section 404 of the Sarbanes-Oxley Act of 2002 requires that we evaluate and determine the effectiveness of our internal control over financial reporting and, beginning with this annual report for the year ending December 31, 2014, provide a management report on our internal controls on an annual basis. If we have material weaknesses in our internal control over financial reporting, we may not detect errors on a timely basis and our financial statements may be materially misstated. We have only recently compiled the systems, processes and documentation necessary to comply with Section 404 of the Sarbanes-Oxley Act. We will need to maintain and enhance these processes and controls as we grow, and we will require additional management and staff resources to do so. Additionally, even if we conclude our internal controls are effective for a given period, we may in the future identify one or more material weaknesses in our internal controls, in which case our management will be unable to conclude that our internal control over financial reporting is effective. Moreover, when we are no longer an emerging growth company, our independent registered public accounting firm will be required to issue an attestation report on the effectiveness of our internal control over financial reporting. Even if our management concludes that our internal control over financial reporting is effective, our independent registered public accounting firm may conclude that there are material weaknesses with respect to our internal controls or the level at which our internal controls are documented, designed, implemented or reviewed.
55
If we are unable to conclude that our internal control over financial reporting is effective, or when we are no longer an emerging growth company, if our auditors were to express an adverse opinion on the effectiveness of our internal control over financial reporting because we had one or more material weaknesses, investors could lose confidence in the accuracy and completeness of our financial disclosures, which could cause the price of our common stock to decline. Irrespective of compliance with Section 404, any failure of our internal control over financial reporting could have a material adverse effect on our reported operating results and harm our reputation. Internal control deficiencies could also result in a restatement of our financial results.
We are an emerging growth company and may elect to comply with reduced public company reporting requirements applicable to emerging growth companies, which could make our common stock less attractive to investors.
We are an emerging growth company, as defined under the Securities Act of 1933, or the Securities Act. We will remain an emerging growth company until December 31, 2018, although if our revenue exceeds $1 billion in any fiscal year before that time, we would cease to be an emerging growth company as of the end of that fiscal year. In addition, if the market value of our common stock that is held by non-affiliates exceeds $700 million as of the last business day of our second fiscal quarter of any fiscal year before the end of that five-year period, we would cease to be an emerging growth company as of December 31 of that year. As an emerging growth company, we may choose to take advantage of exemptions from various reporting requirements applicable to certain other public companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, reduced financial statement and financial-related disclosures, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirement of holding a nonbinding advisory vote on executive compensation and obtaining stockholder approval of any golden parachute payments not previously approved by our stockholders. We cannot predict whether investors will find our common stock less attractive if we choose to rely on any of these exemptions. If some investors find our common stock less attractive as a result of any choices to reduce future disclosure we may make, there may be a less active trading market for our common stock and our stock price may be more volatile.
Risks Related to Our Common Stock
Our stock price may be volatile, and you may not be able to sell shares of our common stock at or above the price you paid.
Prior to our initial public offering in October 2013, there was no public market for our common stock, and an active and liquid public market for our stock may not develop or be sustained. In addition, the trading price of our common stock is likely to continue to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. These factors include:
- •
- actual or anticipated variations in our and our competitors' results of operations;
- •
- announcements by us or our competitors of new products, commercial relationships or capital commitments;
- •
- changes in reimbursement by current or potential payers;
- •
- issuance of new securities analysts' reports or changed recommendations for our stock;
- •
- periodic fluctuations in our revenue, due in part to the way in which we recognize revenue;
- •
- actual or anticipated changes in regulatory oversight of our products;
- •
- developments or disputes concerning our intellectual property or other proprietary rights;
- •
- commencement of, or our involvement in, litigation;
56
- •
- announced or completed acquisitions of businesses or technologies by us or our competitors;
- •
- any major change in our management; and
- •
- general economic conditions and slow or negative growth of our markets.
In addition, the stock market in general, and the market for stock of life sciences companies and other emerging growth companies in particular, has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors may seriously affect the market price of our common stock, regardless of our actual operating performance. These fluctuations may be even more pronounced in the trading market for our stock for some period of time following our initial public offering, especially if substantial investors sell large blocks of stock, particularly if the trading volume in our stock remains low. In addition, in the past, following periods of volatility in the overall market and the market price of a particular company's securities, securities class action litigation has often been instituted against these companies. This litigation, if instituted against us, could result in substantial costs and a diversion of our management's attention and resources.
If securities or industry analysts issue an adverse opinion regarding our stock or do not publish research or reports about our company, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that equity research analysts publish about us and our business. We do not control these analysts or the content and opinions or financial models included in their reports. Securities analysts may elect not to provide research coverage of our company, and such lack of research coverage may adversely affect the market price of our common stock. The price of our common stock could also decline if one or more equity research analysts downgrade our common stock or if those analysts issue other unfavorable commentary or cease publishing reports about us or our business. If one or more equity research analysts cease coverage of our company, we could lose visibility in the market, which in turn could cause our stock price to decline.
Insiders have substantial control over us and will be able to influence corporate matters.
As of March 2, 2015, directors and executive officers and their affiliates beneficially owned, in the aggregate, 62% of our outstanding capital stock. As a result, these stockholders will be able to exercise significant influence over all matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions, such as a merger or other sale of our company or its assets. This concentration of ownership could limit stockholders' ability to influence corporate matters and may have the effect of delaying or preventing a third-party from acquiring control over us.
Anti-takeover provisions in our charter documents and under Delaware law could discourage, delay or prevent a change in control and may affect the trading price of our common stock.
Provisions in our restated certificate of incorporation and our amended and restated bylaws may have the effect of delaying or preventing a change of control or changes in our management. Our restated certificate of incorporation and amended and restated bylaws include provisions that:
- •
- authorize our board of directors to issue, without further action by the stockholders, up to 5.0 million shares of undesignated
preferred stock;
- •
- require that any action to be taken by our stockholders be effected at a duly called annual or special meeting and not by written
consent;
- •
- specify that special meetings of our stockholders can be called only by our board of directors, our chairman of the board, or our chief executive officer;
57
- •
- establish an advance notice procedure for stockholder approvals to be brought before an annual meeting of our stockholders, including
proposed nominations of persons for election to our board of directors;
- •
- establish that our board of directors is divided into three classes, Class I, Class II and Class III, with each
class serving staggered terms;
- •
- provide that our directors may be removed only for cause;
- •
- provide that vacancies on our board of directors may, except as otherwise required by law, be filled only by a majority of directors
then in office, even if less than a quorum;
- •
- specify that no stockholder is permitted to cumulate votes at any election of directors; and
- •
- require a super-majority of votes to amend certain of the above-mentioned provisions.
In addition, we are subject to the provisions of Section 203 of the Delaware General Corporation Law regulating corporate takeovers. Section 203 generally prohibits us from engaging in a business combination with an interested stockholder subject to certain exceptions.
We have never paid dividends on our capital stock, and we do not anticipate paying dividends in the foreseeable future.
We have never paid dividends on any of our capital stock and currently intend to retain any future earnings to fund the growth of our business. In addition, our loan and security agreement restricts our ability to pay cash dividends on our common stock and we may also enter into credit agreements or other borrowing arrangements in the future that will restrict our ability to declare or pay cash dividends on our common stock. Any determination to pay dividends in the future will be at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements, general business conditions and other factors that our board of directors may deem relevant. As a result, capital appreciation, if any, of our common stock will be the sole source of gain for the foreseeable future.
58
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We currently lease 24,000 square feet of office and laboratory space at our headquarters in South San Francisco, California, under a lease that expires in March 2016, with an option for us to extend the lease for an additional three years. We also lease approximately 10,400 square feet of office and laboratory space in Austin, Texas, under a lease that expires in July 2018, with an option for us to extend the lease for an additional five years.
We are not currently a party to any material legal proceedings. We may from time to time become involved in legal proceedings arising in the ordinary course of business.
ITEM 4. MINE SAFETY DISCLOSURE
Not applicable.
EXECUTIVE OFFICERS OF THE REGISTRANT
Our executive officers and their ages and positions as of March 2, 2015, are as set forth below:
Name
|
Age | Position | |||
|---|---|---|---|---|---|
Bonnie H. Anderson |
57 | President, Chief Executive Officer and Director | |||
Julie A. Brooks |
69 | General Counsel and Secretary | |||
Shelly D. Guyer |
54 | Chief Financial Officer | |||
Christopher M. Hall |
46 | Chief Operating Officer | |||
Bonnie H. Anderson has served as our Chief Executive Officer and as a member of our board of directors since February 2008. In August 2013, she was appointed as our President. Prior to joining us, Ms. Anderson was an independent strategic consultant from April 2006 to January 2008, including as a strategic consultant for us from July 2007 to January 2008. Ms. Anderson was a Vice President at Beckman Coulter, Inc., a manufacturer of biomedical testing instrument systems, tests and supplies, from September 2000 to March 2006. She currently serves as an Emeritus member of the Board of Trustees of the Keck Graduate Institute of Applied Life Sciences. Ms. Anderson holds a B.S. in Medical Technology from Indiana University of Pennsylvania.
Julie A. Brooks has served as our General Counsel and Secretary since March 2014. Prior to joining us, Ms. Brooks was a legal consultant for Auxogyn, Inc., a women's health company, from September 2013 to December 2013. From June 2013 to September 2013, Ms. Brooks served as Vice President, General Counsel for Bayer HealthCare LLC, which acquired Conceptus, Inc., a medical device company, in June 2013, where she served as Executive Vice President, General Counsel and Secretary from November 2009 through June 2013. Previously, from November 2007 through October 2009, Ms. Brooks was Senior Vice President, General Counsel and Secretary of Perlegen Sciences, a genomics company. Ms. Brooks has also held executive roles with a number of medical device, healthcare IT, eCommerce and healthcare services companies, including Virgin HealthCare, Access Health and Westmark International. Ms. Brooks holds a B.A. in Comparative Literature and an M.B.A. from the University of Washington, a J.D. from Santa Clara University and a Masters of Law in Taxation from Georgetown University Law Center.
Shelly D. Guyer has served as our Chief Financial Officer since April 2013 and served as our Secretary from April 2013 to March 2014. Prior to joining us, Ms. Guyer served as Chief Financial Officer and Executive Vice President of Finance and Administration of iRhythm Technologies, Inc., a medical device
59
and service company, from April 2008 to December 2012. From March 2006 to August 2007, Ms. Guyer served as Vice President of Business Development and Investor Relations of Nuvelo Inc., a biopharmaceutical company. Prior to joining Nuvelo, Ms. Guyer worked at J.P. Morgan Securities and its predecessor companies for over 17 years, serving in a variety of roles including in healthcare investment banking. Ms. Guyer holds an A.B. in Politics from Princeton University and an M.B.A. from the Haas School of Business at the University of California, Berkeley.
Christopher M. Hall has served as our Chief Operating Officer since September 2014. Mr. Hall served as our Chief Commercial Officer from March 2010 to September 2014. Prior to joining us, Mr. Hall served as Chief Business Officer of Celera Corporation, a diagnostics company focusing on personalized disease management, from October 2008 to February 2010. From August 2002 to February 2010, Mr. Hall served in various executive and senior positions at Berkeley HeartLab, Inc., a cardiovascular disease management company that was acquired by Celera in October 2007, including Chief Clinical Operations Officer and Vice President of Marketing. Mr. Hall holds a B.A. in Economics and Political Science from DePauw University and an M.B.A. from Harvard University.
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock commenced trading under the symbol "VCYT" on The NASDAQ Global Market under the symbol "VCYT" on October 30, 2013. Prior to that time, there was no public market for our common stock. Our common stock in our initial public offering priced at $13.00 per share. The following table sets forth the high and low sales prices of our common stock, on a per share basis, as reported by The NASDAQ Global Market, for the periods indicated:
| |
High | Low | |||||
|---|---|---|---|---|---|---|---|
2014 |
|||||||
Fourth Quarter |
$ | 9.85 | $ | 6.01 | |||
Third Quarter |
$ | 17.92 | $ | 9.22 | |||
Second Quarter |
$ | 18.01 | $ | 12.24 | |||
First Quarter |
$ | 19.00 | $ | 13.76 | |||
2013 |
|||||||
Fourth Quarter (from October 30, 2013) |
$ | 14.80 | $ | 10.88 | |||
As of March 2, 2015, there were approximately 39 holders of record of our common stock. However, because many of our outstanding shares are held in accounts with brokers and other institutions, we have more beneficial owners.
Dividend Policy
We have never declared or paid dividends on our common stock and do not expect to pay dividends on our common stock for the foreseeable future. Instead, we anticipate that all of our earnings in the foreseeable future will be used for the operation and growth of our business. Any future determination to declare dividends will be subject to the discretion of our board of directors and will depend on various factors, including applicable laws, our results of operations, financial condition, future prospects, and any other factors deemed relevant by our board of directors. In addition, the terms of our loan and security agreement restricts our ability to pay dividends on our common stock, and we may also enter into credit agreements or other borrowing arrangements in the future that will further restrict our ability to declare or pay dividends on our common stock.
60
Stock Performance Graph
The following information is not deemed to be "soliciting material" or to be "filed" with the Securities and Exchange Commission or subject to Regulation 14A or 14C under the Securities Exchange Act of 1934 or to the liabilities of Section 18 of the Securities Exchange Act of 1934, and will not be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Securities Exchange Act of 1934, except to the extent we specifically incorporate it by reference into such a filing.
The graph below shows the cumulative total stockholder return (change in stock price plus reinvested dividends) assuming the investment of $100.00 on the date specified in each of our common stock, The NASDAQ Global Market Index, and the NASDAQ Biotechnology Index for the period commencing on October 30, 2013 (the first day of trading of our common stock) and ending on December 31, 2014. The comparisons in the table are required by the Securities and Exchange Commission and are not intended to forecast or be indicative of future performance of our common stock.
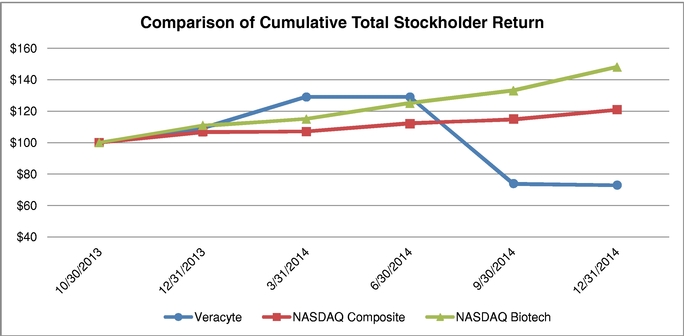
| |
October 30, 2013 |
December 31, 2013 |
March 31, 2014 |
June 30, 2014 |
September 30, 2014 |
December 31, 2014 |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Veracyte, Inc. |
$ | 100.00 | $ | 109.00 | $ | 129.00 | $ | 129.00 | $ | 74.00 | $ | 73.00 | |||||||
NASDAQ Market Index |
$ | 100.00 | $ | 107.00 | $ | 107.00 | $ | 112.00 | $ | 115.00 | $ | 121.00 | |||||||
NASDAQ Biotechnology Index |
$ | 100.00 | $ | 111.00 | $ | 115.00 | $ | 125.00 | $ | 133.00 | $ | 148.00 | |||||||
Use of Proceeds
On November 4, 2013, we completed an initial public offering, or IPO, of our common stock. In connection with our IPO, we issued and sold 5,100,351 shares of common stock at a price to the public of $13.00 per share. As a result of the IPO, we received $59.2 million in net proceeds, after deducting underwriting discounts and commissions of $4.6 million and offering expenses of $2.5 million payable by us.
We registered the shares under the Securities Act on a Registration Statement on Form S-1 (Registration No. 333-191282), or the Registration Statement, which was filed on September 20, 2013 and
61
declared effective on October 29, 2013, and on a Registration Statement on Form S-1 (Registration No. 333-1919782), which was filed on October 30, 2013 and was immediately effective.
From the date of the initial closing of the IPO through December 31, 2014, we have used approximately $39 million of the net proceeds from the sale of these securities to fund our operations, to make capital expenditures, in connection with the acquisition of Allegro, for working capital and for other general corporate purposes.
Other than approximately $8.7 million we spent in connection with the acquisition of Allegro, there has been no material change in the planned use of proceeds from our IPO as described in the Registration Statement. We invested the remainder of funds received in a short-term money market account primarily consisting of U.S. Treasury reserves.
Sales of Unregistered Securities
In June 2013 we issued to a lender a warrant to purchase up to 49,602 shares of Series C convertible preferred stock with an exercise price of $7.56 per share. Upon the drawdown of a term loan, the warrant became exercisable for 24,801 shares. In November 2013, in connection with our IPO, the warrant automatically became exercisable for 24,801 shares of common stock at an exercise price of $7.56 per share. The lender exercised the warrant with respect to 24,801 shares through a cashless exercise in March 2014, resulting in the issuance of 13,739 shares of our common stock. The transaction was exempt from registration under the Securities Act of 1933 in reliance on Section 4(2) or Regulation D as transactions by an issuer not involving a public offering.
ITEM 6. SELECTED FINANCIAL DATA
The information set forth below should be read in conjunction with "Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations" and our financial statements and related notes included elsewhere in this annual report. The selected consolidated balance sheet data at December 31, 2014, 2013, 2012 and 2011 and the selected consolidated statements of operations data for each of the years ended December 31, 2014, 2013, 2012 and 2011 have been derived from our audited
62
consolidated financial statements that are included elsewhere in this report. The financial data included in this report are historical and are not necessarily indicative of results to be expected in any future period.
| |
Year Ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | 2011 | |||||||||
| |
(In thousands except share and per share data and FNAs received) |
||||||||||||
Consolidated Statements of Operations Data: |
|||||||||||||
Revenue |
$ | 38,190 | $ | 21,884 | $ | 11,628 | $ | 2,645 | |||||
Operating expenses: |
|||||||||||||
Cost of revenue(1) |
16,606 | 12,607 | 7,584 | 2,925 | |||||||||
Research and development(1) |
9,804 | 7,810 | 6,608 | 6,680 | |||||||||
Selling and marketing(1) |
21,932 | 12,540 | 8,447 | 2,934 | |||||||||
General and administrative(1) |
18,854 | 12,100 | 7,918 | 5,372 | |||||||||
| | | | | | | | | | | | | | |
Total operating expenses(1) |
67,196 | 45,057 | 30,557 | 17,911 | |||||||||
| | | | | | | | | | | | | | |
Loss from operations |
(29,006 | ) | (23,173 | ) | (18,929 | ) | (15,266 | ) | |||||
Interest expense |
(439 | ) | (233 | ) | — | — | |||||||
Other income (expense), net |
72 | (2,174 | ) | 280 | 821 | ||||||||
| | | | | | | | | | | | | | |
Net loss |
$ | (29,373 | ) | $ | (25,580 | ) | $ | (18,649 | ) | $ | (14,445 | ) | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Net loss per common share, basic and diluted |
$ | (1.36 | ) | $ | (6.15 | ) | $ | (28.68 | ) | $ | (24.90 | ) | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Shares used in computing net loss per common share, basic and diluted |
21,639,374 | 4,158,664 | 650,333 | 580,061 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Other Operating Data: |
|||||||||||||
FNAs received |
65,848 | 49,670 | 25,890 | 6,402 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
- (1)
- Includes employee stock-based compensation as follows:
| |
Year Ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | 2011 | |||||||||
| |
(In thousands) |
||||||||||||
Cost of revenue |
$ | 51 | $ | 34 | $ | 26 | $ | 32 | |||||
Research and development |
790 | 250 | 131 | 130 | |||||||||
Selling and marketing |
707 | 169 | 111 | 77 | |||||||||
General and administrative |
2,000 | 794 | 407 | 227 | |||||||||
| | | | | | | | | | | | | | |
Total stock-based compensation |
$ | 3,548 | $ | 1,247 | $ | 675 | $ | 466 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Consolidated Balance Sheets Data:
| |
As of December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | 2011 | |||||||||
| |
(In thousands) |
||||||||||||
Cash and cash equivalents |
$ | 35,014 | $ | 71,220 | $ | 14,002 | $ | 7,566 | |||||
Working capital |
26,203 | 61,019 | 7,390 | 6,707 | |||||||||
Total assets |
64,839 | 79,630 | 19,067 | 10,451 | |||||||||
Convertible preferred stock |
— | — | 63,372 | 49,296 | |||||||||
Accumulated deficit |
(115,022 | ) | (85,649 | ) | (60,069 | ) | (41,420 | ) | |||||
Total stockholders' equity (deficit) |
41,374 | 56,443 | (58,471 | ) | (40,766 | ) | |||||||
63
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of financial condition and results of operations should be read together with the consolidated financial statements and the related notes included in Item 8 of Part II of this Annual Report on Form 10-K. This discussion and analysis contains certain forward-looking statements that involve risks and uncertainties. Our actual results may differ materially from those discussed below. Factors that could cause or contribute to such differences include, but are not limited to, those identified below and those set forth under the section entitled "Risk Factors" in Item 1A, and other documents we file with the Securities and Exchange Commission. Historical results are not necessarily indicative of future results.
Overview
We are a diagnostics company pioneering the field of molecular cytology, focusing on genomic solutions that resolve diagnostic ambiguity and enable physicians to make more informed treatment decisions at an early stage in patient care. By improving preoperative diagnostic accuracy, we aim to help patients avoid unnecessary invasive procedures while reducing healthcare costs. Our first commercial solution, the Afirma Thyroid FNA Analysis, or Afirma, centers on the proprietary Gene Expression Classifier, or GEC, to resolve ambiguity in diagnosis and is becoming a new standard of care in thyroid nodule assessment. The GEC helps physicians reduce the number of unnecessary surgeries by approximately 50% by employing a proprietary 142-gene signature to preoperatively identify benign thyroid nodules among those deemed indeterminate by cytopathology alone. We have demonstrated the clinical utility and cost effectiveness of the GEC in multiple studies published in peer-reviewed journals and established the clinical validity of the GEC in a study published in The New England Journal of Medicine in 2012. The comprehensive Afirma offering also includes cytopathology testing and the Afirma Malignancy Classifiers, launched in May 2014. Since we commercially launched Afirma in January 2011, we have received nearly 160,000 FNA samples for evaluation using Afirma and performed more than 30,000 GECs to resolve indeterminate cytopathology results.
We are expanding our molecular cytology franchise into other clinical areas of unmet need, focusing first on difficult-to-diagnose lung diseases, where current diagnostic ambiguity frequently requires invasive, risky and costly procedures to obtain a definitive diagnosis. Through our acquisition of Allegro Diagnostics Corp., or Allegro, on September 16, 2014, we plan to launch our first pulmonology product by mid-2015, aimed at improving the risk stratification of patients with lung nodules that are suspicious for cancer. Our proprietary technology has been developed to help physicians determine which patients with non-diagnostic bronchoscopy results can be safely monitored with routine CT scans versus an invasive surgical biopsy.
Our second pulmonology pipeline product is intended to help patients with suspected interstitial lung diseases, or ILDs, specifically idiopathic pulmonary fibrosis, or IPF, obtain an accurate diagnosis without surgery. ILDs present a significant challenge for diagnosis today without invasive surgical biopsy, leaving many patients with ambiguous diagnoses that can lead to suboptimal or even harmful treatment. We are developing a genomic test, which we currently plan to introduce in 2016, to improve the diagnosis of IPF from the other ILDs.
On November 7, 2014, we signed an Amended and Restated U.S. Co-Promotion Agreement, or Amended Agreement, with Genzyme that reduced the co-promotion fees we owe to Genzyme from 32% to 15% beginning January 1, 2015. The Amended Agreement expires in January 2027. On February 13, 2015, we entered into an Ex-U.S. Co-Promotion Agreement, or Ex-U.S. Agreement, with Genzyme for the co-exclusive promotion of the Afirma GEC test in two countries outside the United States: Brazil and Singapore. We also granted Genzyme, for a limited period of time, an exclusive right of first negotiation to enter into an agreement with us for the promotion of the Afirma GEC test in three additional countries: Canada, the Netherlands and Italy. Further, upon mutual agreement, the parties may add additional
64
countries (other than the United States) to the Ex-U.S. Agreement. The term of the agreement commences January 1, 2015 and continues until December 31, 2019, with extension of the agreement possible upon agreement of the parties.
We increased the list price billed for the GEC from $4,275 to $4,875 per test in January 2014, while the list price billed for routine cytopathology remained at $490 per test. We obtained Medicare coverage for the GEC effective in January 2012 and contracted reimbursement at an agreed upon rate of $3,200. We received positive coverage determinations from UnitedHealthcare and Cigna in 2013 and in late 2014 signed contracts with these payers establishing in-network allowable rates for both our GEC and cytopathology tests. We have also received positive coverage determinations from numerous other commercial payers and, as of March 2015, the GEC is covered by payers representing 145 million covered lives. Contracted and reimbursement rates vary by payer.
We recognized revenue of $38.2 million, $21.9 million and $11.6 million in the years ended December 31, 2014, 2013 and 2012, respectively. Revenue increased by 75%, 88% and 340% for the years ended December 31, 2014, 2013 and 2012, respectively. We incurred a net loss of $29.4 million, $25.6 million and $18.6 million for the years ended December 31, 2014, 2013 and 2012, respectively. As of December 31, 2014, we had an accumulated deficit of $115.0 million.
Factors Affecting Our Performance
The Number of FNAs We Receive and Test
The growth in our business is tied to the number of FNAs we receive and the number of GECs performed. Approximately 91% of FNAs we receive are for the Afirma solution, which consists of cytopathology, and if the cytopathology result is indeterminate, the GEC is performed. The remaining approximate 9% of FNAs are received from centers performing cytopathology in their institution where the cytopathology result is indeterminate and we perform the GEC only. The rate at which adoption occurs in these two settings will cause these two percentages to fluctuate over time. Approximately 1% to 2% of the FNA samples we receive for cytopathology have insufficient cellular material from which to render a cytopathology diagnosis. We only bill the technical component, including slide preparation, for these tests. For results that are benign or suspicious/malignant by cytopathology, we bill for these services when we issue the report to the physician. If the cytopathology result is indeterminate, defined as atypia/follicular lesions of undetermined significance (AUS/FLUS) or suspicious for FN/HCN, we perform the GEC. Historically, approximately 14%-17% of samples we have received for the Afirma solution have yielded indeterminate results by cytopathology. Approximately 5%-10% of the samples for GEC testing have insufficient ribonucleic acid, or RNA, from which to render a result. The GEC can be reported as Benign, Suspicious or No Result. We bill for the GEC Benign and GEC Suspicious results only. After the GEC is completed, we issue the cytopathology report for the indeterminate results as well as the GEC report, and then bill for both of these tests. We incur costs of collecting and shipping the FNAs and a portion of the costs of performing tests where we cannot ultimately issue a patient report. Because we cannot bill for all samples received, the number of FNAs received does not directly correlate to the total number of patient reports issued and the amount billed.
Continued Adoption of and Reimbursement for Afirma
To date, only a small number of payers have reimbursed us for Afirma at full list price. Revenue growth depends on both our ability to achieve broader reimbursement at increased levels from third-party payers and to expand our base of prescribing physicians and increase our penetration in existing accounts. Because some payers consider the GEC experimental and investigational, we may not receive payment on many tests and payments may not be at acceptable levels compared to what we have billed. We expect our revenue growth will increase as more payers make a positive coverage decision and as payers enter into contracts with us, which should enhance our accrued revenue and collections. To drive increased adoption
65
of Afirma, we have increased our internal sales force in high-volume geographies domestically during 2014 and plan to continue to do so into 2015, along with increasing our marketing efforts. We have also hired institutional channel managers to focus on the institutional segment, which accounts generally send us only GECs. If we are unable to expand the base of prescribing physicians and penetration within these accounts at an acceptable rate, or if we are not able to execute our strategy for increasing reimbursement, we may not be able to effectively increase our revenue.
How We Recognize Revenue
A significant portion of our revenue is recognized upon the earlier of receipt of third-party notification of payment or when cash is received. For Medicare and certain other payers where we have an agreed upon reimbursement rate or we are able to make a reasonable estimate of reimbursement at the time delivery is complete, we recognize the related revenue on an accrual basis. Until we have contracts with or can make a reasonable estimate of reimbursement from a larger number of payers, we will recognize a large portion of our revenue upon the earlier of notification of payment or when cash is received. Additionally, as we commercialize new products, we will need to contract with or be able to make a reasonable estimate of reimbursement for each payer for each new product offering prior to being able to recognize the related revenue on an accrual basis. Because the timing and amount of cash payments received from payers is difficult to predict, we expect that our revenue will fluctuate significantly in any given quarter. In addition, even if we begin to accrue larger amounts of revenue related to Afirma, when we introduce new products we do not expect we will be able to recognize revenue from new products on an accrual basis for some period of time. This may result in continued fluctuations in our revenue.
Revenue recognized when cash is received was $25.7 million, $14.6 million and $7.5 million for the years ended December 31, 2014, 2013 and 2012, respectively. Revenue recognized on an accrual basis was $12.5 million, $7.3 million and $4.1 million for the years ended December 31, 2014, 2013 and 2012, respectively.
As of December 31, 2014, amounts billed in the last 12 months at list price, for tests processed which were not recognized as revenue upon delivery of a patient report because our accrual revenue recognition criteria were not met and for which we have not received notification of payment, collected cash or written off as uncollectible, totaled $47.1 million.
As of December 31, 2013, amounts billed in the last 12 months at list price, for tests processed which were not recognized as revenue upon delivery of a patient report because our accrual revenue recognition criteria were not met and for which we have not received notification of payment, collected cash or written off as uncollectible, totaled $32.4 million. Of this amount, we recognized revenue of $7.3 million in the year ended December 31, 2014, when cash was received.
Although primarily all cash we receive is collected within 12 months of the date the test is billed, we cannot provide any assurance as to when, if ever, or to what extent any of these amounts will be collected. Notwithstanding our efforts to obtain payment for these tests, payers may deny our claims, in whole or in part, and we may never receive revenue from previously performed but unpaid tests. Revenue from these tests, if any, may not be equal to the billed amount due to a number of factors, including differences in reimbursement rates, the amounts of patient co-payments and co-insurance, the existence of secondary payers and claims denials.
We incur expense for tests in the period in which the test is conducted and recognize revenue for tests in the period in which our revenue recognition criteria are met. Accordingly, any revenue that we recognize as a result of cash collection in respect of previously performed but unpaid tests will favorably impact our liquidity and results of operations in future periods.
66
Impact of Genzyme Co-promotion Agreement
The $10.0 million fee we received from Genzyme under the Co-Promotion Agreement dated as of January 18, 2012 is being amortized over the estimated useful life based on the provisions of the agreement, and is recorded as a reduction to selling and marketing expenses. We amortized $2.3 million, $2.5 million and $2.4 million of the $10.0 million in the years ended December 31, 2014, 2013 and 2012, respectively, and these offsets to expense are included in selling and marketing expense in our consolidated statements of operations and comprehensive loss. The 2012 agreement required that we pay a certain percentage of our cash receipts from the sale of the Afirma GEC test to Genzyme, which percentage decreased over time. The percentage was 40% from January 2013 through February 2014, 32% from February 2014 through December 2014, and decreased to 15% in January 2015 (upon execution of the Amended and Restated U.S. Co-Promotion Agreement, dated as of November 7, 2014, with Genzyme). Our co-promotion fees were $12.0 million, $8.6 million and $5.5 million in the years ended December 31, 2014, 2013 and 2012, respectively, and are included in selling and marketing expenses in our consolidated statements of operations and comprehensive loss.
On August 12, 2014, we signed a binding Letter of Agreement with Genzyme to amend the terms of the 2012 agreement. On November 7, 2014, we signed an Amended and Restated U.S. Co-Promotion Agreement, or Amended Agreement, with Genzyme. Under the Amended Agreement, the co-promotion fees Genzyme will receive as a percentage of U.S. cash receipts from the sale of the Afirma GEC test were reduced from 32% to 15% beginning January 1, 2015. Further, we have agreed to assume more responsibilities for sales and marketing activities. Either party may terminate the agreement for convenience with six months prior notice, however, neither party can terminate the agreement for convenience prior to June 30, 2016. Our agreement with Genzyme expires in January 2027.
On February 13, 2015, we entered into an Ex-U.S. Co-Promotion Agreement, or Ex-U.S. Agreement, with Genzyme for the co-exclusive promotion of the Afirma GEC test in two countries outside the United States: Brazil and Singapore. We also granted Genzyme, for a limited period of time, an exclusive right of first negotiation to enter into an agreement with us for the promotion of the Afirma GEC test in three additional countries: Canada, the Netherlands and Italy. Further, upon mutual agreement, the parties may add additional countries (other than the United States) to the Ex-U.S. Agreement. The term of the agreement commenced January 1, 2015 and continues until December 31, 2019, with extension of the agreement possible upon agreement of the parties. Country specific terms have been established under this agreement for Brazil and Singapore. We will pay Genzyme 25% of cash receipts from the sale of the Afirma GEC test in Brazil and Singapore over a five-year period commencing January 1, 2015. Beginning in the fourth year of the agreement, if we terminate the agreement for convenience in Brazil, we may be required to pay a termination fee contingent on the number of GEC billable results generated.
Development of Additional Products
We rely on sales of Afirma to generate all of our revenue. In May 2014, we commercially launched our Afirma Malignancy Classifiers, which we believe will enhance our Afirma Thyroid FNA Analysis as a comprehensive way to manage thyroid nodule patients and serve our current base of prescribing physicians. We also plan to pursue development of products for additional diseases to increase and diversify our revenue. For example, in September 2014 we acquired Allegro and with it their molecular diagnostic lung cancer test designed to help physicians determine which patients with lung nodules who have had a non-diagnostic bronchoscopy result are at low risk for cancer and can thus be safely monitored with CT scans rather than undergoing invasive procedures. We plan to launch Percepta, the test acquired from Allegro, by mid-2015. Additionally, we are pursuing a solution for interstitial lung disease, or ILD, that will offer an alternative to surgery by developing a genomic signature to classify samples collected through less invasive bronchoscopy techniques. Accordingly, we expect to continue to invest heavily in research and development in order to expand the capabilities of our solutions and to develop additional
67
products. Our success in developing new products will be important in our efforts to grow our business by expanding the potential market for our products and diversifying our sources of revenue.
Timing of Our Research and Development Expenses
We deploy state-of-the-art and costly genomic technologies in our biomarker discovery experiments, and our spending on these technologies may vary substantially from quarter to quarter. We also spend a significant amount to secure clinical samples that can be used in discovery and product development as well as clinical validation studies. The timing of these research and development activities is difficult to predict, as is the timing of sample acquisitions. If a substantial number of clinical samples are acquired in a given quarter or if a high-cost experiment is conducted in one quarter versus the next, the timing of these expenses can affect our financial results. We conduct clinical studies to validate our new products as well as on-going clinical studies to further the published evidence to support our commercialized test, Afirma. As these studies are initiated, start-up costs for each site can be significant and concentrated in a specific quarter. Spending on research and development, for both experiments and studies, may vary significantly by quarter depending on the timing of these various expenses.
Historical Seasonal Fluctuations in FNA Volume and Collections
Our business is subject to fluctuations in the number of FNA samples received for both cytopathology and GEC testing throughout the year as a result of physician practices being closed for holidays or endocrinology and thyroid-related industry meetings which are widely attended by our prescribing physicians. Like other companies in our field, vacations by physicians and patients tend to negatively affect our volumes more during the summer months and during the end of year holidays compared to other times of the year. Additionally, we may receive fewer FNAs in the winter months due to severe weather if patients are not able to visit their doctor's office. Our reimbursed rates and cash collections are also subject to seasonality. Medicare normally makes adjustments in its fee schedules at the beginning of the year which may affect our reimbursement. Additionally, some plans reset their deductibles at the beginning of each year which means that patients early in the year are responsible for a greater portion of the cost of our tests, and we have lower collection rates from individuals than from third-party payers. Later in the year, particularly in the fourth quarter, we experience improved payment results as third-party payers tend to clear pending claims toward year end. This trend historically has increased our cash collections in the fourth quarter. The effects of these seasonal fluctuations in prior periods may have been obscured by the growth of our business.
Financial Overview
Revenue
Through December 31, 2014, all of our revenue have been derived from the sale of Afirma. Our solution to date has been delivered primarily to physicians in the United States. We generally invoice third-party payers upon delivery of a patient report to the prescribing physician. As such, we take the assignment of benefits and the risk of collection from the third-party payer and individual patients. Our
68
third-party payers in excess of 10% of revenue and their related revenue as a percentage of total revenue were as follows:
| |
Year Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Medicare |
26 | % | 32 | % | 34 | % | ||||
Aetna |
11 | % | 9 | % | 13 | % | ||||
United Healthcare |
18 | % | 18 | % | 12 | % | ||||
| | | | | | | | | | | |
|
55 | % | 59 | % | 59 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
As the number of payers reimbursing for Afirma increases, the percentage of revenue derived from Medicare and other significant third-party payers has changed and will continue to change as a percentage of total revenue.
For tests performed where we have an agreed upon reimbursement rate or we are able to make a reasonable estimate of reimbursement at the time delivery is complete, such as in the case of Medicare and certain other payers, we recognize the related revenue upon delivery of a patient report to the prescribing physician based on the established billing rate less contractual and other adjustments to arrive at the amount that we expect to collect. We determine the amount we expect to collect based on a per payer, per contract or agreement basis. The expected amount is typically lower than, if applicable, the agreed upon reimbursement amount due to several factors, such as the amount of patient co-payments, the existence of secondary payers and claim denials. In other situations, where we are not able to make a reasonable estimate of reimbursement, we recognize revenue upon the earlier of receipt of third-party payer notification of payment or when cash is received. Upon ultimate collection, the amount received from Medicare and commercial payers where reimbursement was estimated is compared to previous estimates and the contractual allowance is adjusted accordingly. Our ability to increase our revenue will depend on our ability to penetrate the market, obtain positive coverage policies from additional third-party payers, obtain reimbursement and/or enter into contracts with additional third-party payers, and increase reimbursement rates for tests performed. Finally, should we recognize revenue from payers on an accrual basis and later determine the judgments underlying estimated reimbursement change, our financial results could be negatively impacted in future quarters.
Cost of Revenue
The components of our cost of revenue are materials and service costs, including cytopathology testing services, stock-based compensation expense, direct labor costs, equipment and infrastructure expenses associated with testing samples, shipping charges to transport samples, and allocated overhead including rent, information technology, equipment depreciation and utilities. Costs associated with performing tests are recorded as the test is processed regardless of whether and when revenue is recognized with respect to that test. As a result, our cost of revenue as a percentage of revenue may vary significantly from period to period because we do not recognize all revenue in the period in which the associated costs are incurred. We expect cost of revenue in absolute dollars to increase as the number of tests we perform increases. However, we expect that the cost per test will decrease over time due to leveraging fixed costs, efficiencies we may gain as test volume increases and from automation, process efficiencies and other cost reductions. As we introduce new tests, initially our cost of revenue will be high and will increase disproportionately our aggregate cost of revenue until we achieve efficiencies in processing these new tests.
69
Research and Development
Research and development expenses include costs incurred to develop our technology, collect clinical samples and conduct clinical studies to develop and support our products. These costs consist of personnel costs, including stock-based compensation expense, prototype materials, laboratory supplies, consulting costs, costs associated with setting up and conducting clinical studies at domestic and international sites, and allocated overhead including rent, information technology, equipment depreciation and utilities. We expense all research and development costs in the periods in which they are incurred. We expect our research and development expenses will increase in future periods as we continue to invest in research and development activities related to developing additional products and evaluating various platforms. We expect that in the next 12 months the increase in research and development expenses will be for the continued development and support of Afirma and other new products and programs under development, including our lung cancer and ILD programs. Specifically, we plan to: increase the body of clinical evidence to support Afirma; incur research and development expenses associated with analytical verification and clinical utility studies to support the commercialization of Percepta; and incur expenses associated with clinical validation studies in our ILD program.
Selling and Marketing
Selling and marketing expenses consist of personnel costs, including stock-based compensation expense, direct marketing expenses, consulting costs, and allocated overhead including rent, information technology, equipment depreciation and utilities. In addition, up-front co-promotion fees paid to Genzyme, net of amortization, are included in selling and marketing expenses. In November 2014, we amended the co-promotion agreement with Genzyme. As a result of this amendment, we expect our selling and marketing expenses for Afirma to remain relatively flat over the next year. While we expect that our personnel costs will increase as we take on more sales and marketing responsibilities related to Afirma, we expect these increases will be offset by the lower rate we are required to pay Genzyme under the agreement beginning in January 2015. In 2015, we also expect to incur selling and marketing expenses as a result of investments in our lung product portfolio. Therefore, we believe total selling and marketing expenses will increase in 2015.
General and Administrative
General and administrative expenses include executive, finance and accounting, human resources, legal, billing and client services, and quality and regulatory functions. These expenses include personnel costs, including stock-based compensation expense, audit and legal expenses, consulting costs, costs associated with being a public company, and allocated overhead including rent, information technology, equipment depreciation and utilities. The year ended December 31, 2014 also includes transaction costs related to the acquisition of Allegro in September 2014, including charges for merger related severance and bonuses. We expect our general and administration expenses will increase over the next 12 months as we expand our billing group to support anticipated increased demand for our tests, hire more personnel in accounting and finance, incur increasing expenses related to the documentation of our internal controls in connection with compliance with Section 404 of the Sarbanes-Oxley Act of 2002, and incur greater legal costs for patent prosecution and for public company compliance and general corporate purposes.
Interest Expense
Interest expense is attributable to our borrowings under our loan and security agreement.
70
Other Income (Expense), Net
Other income (expense), net, for the year ended December 31, 2014 consists primarily of sublease rental income, interest income received from payers and from our cash equivalents, partially offset by amortization of debt issuance costs.
Other income (expense), net, in the years ended December 31, 2013 and 2012 also included the change in the fair value of the preferred stock liability associated with our obligation to issue additional shares of Series C convertible preferred stock. We determined that the liability to issue additional Series C convertible preferred stock at a future date was a freestanding instrument that should be accounted for as a liability. Accordingly, we recorded a liability related to this instrument at the time of the initial close in November 2012, and we re-measured the liability at each reporting period with the corresponding gain or loss from the adjustment recorded as other income (expense), net, through the issuance of the final Series C tranche in June 2013, at which time the preferred stock liability was extinguished.
In addition, other income (expense), net, in the year ended December 31, 2013 included changes in value of the preferred stock warrant liability issued in connection with our draw-down of borrowings under the loan and security agreement in June 2013. We recorded a liability related to this warrant and re-measured the liability at each reporting period with the corresponding gain or loss from the adjustment recorded as other income (expense), net. The preferred stock warrant liability was converted into a warrant to purchase our common stock upon the completion of our initial public offering (IPO) in November 2013. This warrant was exercised through a cashless exercise in March 2014.
Critical Accounting Polices and Estimates
Our management's discussion and analysis of our financial condition and results of operations is based on our audited financial statements, which have been prepared in accordance with United States generally accepted accounting principles, or U.S. GAAP. The preparation of the financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenue generated and expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material. We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management's judgments and estimates.
Revenue Recognition
Our revenue is generated from the provision of diagnostic services using the Afirma solution. Our service is completed upon the delivery of test results to the prescribing physician which triggers the billing for the service. We recognize revenue related to billings for Medicare and commercial payers on an accrual basis, net of contractual adjustments, when we can reasonably estimate reimbursement. These contractual adjustments represent the difference between the list price (the billing rate) and the reimbursement rate for each payer. Upon ultimate collection, the amount received from Medicare and commercial payers where reimbursement was estimated is compared to previous estimates and the contractual allowance is adjusted accordingly. Until a contract has been negotiated with a commercial payer or governmental program, the Afirma solution may or may not be covered by these entities' existing reimbursement policies. In addition, patients do not enter into direct agreements with us that commit them to pay any portion of the cost of the tests in the event that their insurance declines to reimburse us. In the absence of an agreement or other clearly enforceable legal right to demand payment from the patient, the related revenue is only recognized upon the earlier of payment notification, if applicable, or cash receipt.
71
For all services performed, we consider whether or not the following revenue recognition criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; and a reasonable estimate of reimbursement can be made.
Persuasive evidence of an arrangement exists and delivery is deemed to have occurred upon delivery of a patient report to the prescribing physician. The assessment of whether a reasonable estimate of reimbursement can be made requires significant judgment by management. Where our judgment indicates a reasonable estimate of reimbursement can be made, we recognize revenue upon delivery of the patient report. Some patients have out-of- pocket costs for amounts not covered by their insurance carrier, and we may bill the patient directly for these amounts in the form of co-payments and co-insurance in accordance with their insurance carrier and health plans. Some payers may not cover the GEC as ordered by the prescribing physician under their reimbursement policies. We pursue reimbursement from such patients on a case-by-case basis.
In the absence of contracted reimbursement coverage or the ability to reasonably estimate reimbursement, we recognize revenue upon the earlier of receipt of third-party payer notification of payment or when cash is received.
We use judgment in determining if we are able to make a reasonable estimate of reimbursement. We also use judgment in estimating the amounts we expect to collect by payer. Our judgments will continue to evolve in the future as we continue to gain payment experience with third-party payers and patients.
Allowance for Doubtful Accounts
We estimate an allowance for doubtful accounts against our individual accounts receivable based on estimates of expected payment consistent with historical payment experience. Our allowance for doubtful accounts is evaluated on a regular basis and adjusted when trends or significant events indicate that a change in estimate is appropriate. Accounts receivable are written off against the allowance when the appeals process is exhausted or when there is other substantive evidence that the account will not be paid.
Business Combination
We account for acquisitions using the acquisition method of accounting which requires the recognition of tangible and identifiable intangible assets acquired and liabilities assumed at their estimated fair values as of the business combination date. We allocate any excess purchase price over the estimated fair value assigned to the net tangible and identifiable intangible assets acquired and liabilities assumed to goodwill. Transaction costs are expensed as incurred in general and administrative expenses. Results of operations and cash flows of acquired companies are included in our operating results from the date of acquisition.
Goodwill
We review goodwill for impairment on an annual basis or more frequently if events or circumstances indicate that it may be impaired. Our goodwill evaluation is based on both qualitative and quantitative assessments regarding the fair value of goodwill relative to its carrying value. We have determined that we operate in a single segment and have a single reporting unit associated with the development and commercialization of diagnostic products. In the event we determine that it is more likely than not the carrying value of the reporting unit is higher than its fair value, quantitative testing is performed comparing recorded values to estimated fair values. If impairment is present, the impairment loss is measured as the excess of the recorded goodwill over its implied fair value. We perform our annual evaluation of goodwill during the fourth quarter of each fiscal year.
72
Intangible Assets
Our intangible assets are comprised of acquired in-process research and development, or IPR&D. The fair value of IPR&D acquired through a business combination is capitalized as an indefinite-lived intangible asset until the completion or abandonment of the related research and development activities. IPR&D is tested for impairment annually or when events or circumstances indicate that the fair value may be below the carrying value of the asset. If and when research and development is complete, the associated assets would then be amortized over their estimated useful lives.
Impairment of Long-lived Assets
We review long-lived and indefinite lived assets other than goodwill for impairment on an annual basis or whenever events or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. We recognize an impairment loss when the total of estimated future undiscounted cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. Impairment, if any, would be assessed using discounted cash flows or other appropriate measures of fair value.
Derivative Liability
We account for derivative financial instruments as either equity or liabilities based upon the characteristics and provisions of each instrument. We recorded the preferred stock liability incurred in connection with our Series C convertible preferred stock and the preferred stock warrant liability related to the issuance of a warrant for Series C convertible preferred stock, each as a derivative financial instrument liability at their fair value on the date of issuance, and we re-measured them on each subsequent balance sheet date. The changes in fair value were recognized as a gain or loss from the adjustment to other income (expense), net, in the statements of operations and comprehensive loss. We estimated the fair value of this liability using option-pricing models that include assumptions for future financings, expected volatility, expected life, yield and risk-free interest rate. The preferred stock liability was extinguished in June 2013. The warrant to purchase Series C convertible preferred stock was converted into a warrant to purchase our common stock as of the closing of our IPO and was exercised through a cashless exercise in March 2014.
Deferred Tax Assets
We file U.S. federal income tax returns and tax returns in California, Texas and other states.
As of December 31, 2014 and December 31, 2013, our gross deferred tax assets were $43.4 million and $32.8 million, respectively. The deferred tax assets were primarily comprised of federal and state tax net operating loss and tax credit carryforwards. Utilization of the net operating loss and tax credit carryforwards may be subject to annual limitation due to historical or future ownership percentage change rules provided by the Internal Revenue Code of 1986, and similar state provisions. The annual limitation may result in the expiration of certain net operating loss and tax credit carryforwards before their utilization.
We are required to reduce our deferred tax assets by a valuation allowance if it is more likely than not that some or all of our deferred tax assets will not be realized. We must use judgment in assessing the potential need for a valuation allowance, which requires an evaluation of both negative and positive evidence. The weight given to the potential effect of negative and positive evidence should be commensurate with the extent to which it can be objectively verified. In determining the need for and amount of our valuation allowance, if any, we assess the likelihood that we will be able to recover our deferred tax assets using historical levels of income, estimates of future income and tax planning strategies. As a result of historical cumulative losses and, based on all available evidence, we believe it is more likely than not that our recorded net deferred tax assets will not be realized. Accordingly, we recorded a
73
valuation allowance against all of our net deferred tax assets at December 31, 2014 and 2013. We will continue to maintain a full valuation allowance on our net deferred tax assets until there is sufficient evidence to support the reversal of all or some portion of this allowance.
Stock-based Compensation
We recognize stock-based compensation cost for only those shares underlying stock options that we expect to vest on a straight-line basis over the requisite service period of the award. We estimate the fair value of stock options using a Black-Scholes valuation model, which requires the input of highly subjective assumptions, including the option's expected term and stock price volatility. In addition, judgment is also required in estimating the number of stock-based awards that are expected to be forfeited. Forfeitures are estimated based on historical experience at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The assumptions used in calculating the fair value of share-based payment awards represent management's best estimates, but these estimates involve inherent uncertainties and the application of management's judgment. As a result, if factors change and we use different assumptions, our stock-based compensation expense could be materially different in the future.
Results of Operations
Comparison of the Years Ended December 31, 2014 and 2013
| |
Year Ended December 31, |
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Dollar Change |
% Change |
|||||||||||
| |
2014 | 2013 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Revenue |
$ | 38,190 | $ | 21,884 | $ | 16,306 | 75 | % | |||||
Operating expense: |
|||||||||||||
Cost of revenue |
16,606 | 12,607 | 3,999 | 32 | % | ||||||||
Research and development |
9,804 | 7,810 | 1,994 | 26 | % | ||||||||
Selling and marketing |
21,932 | 12,540 | 9,392 | 75 | % | ||||||||
General and administrative |
18,854 | 12,100 | 6,754 | 56 | % | ||||||||
| | | | | | | | | | | | | | |
Total operating expenses |
67,196 | 45,057 | 22,139 | 49 | % | ||||||||
| | | | | | | | | | | | | | |
Loss from operations |
(29,006 | ) | (23,173 | ) | (5,833 | ) | 25 | % | |||||
Interest expense |
(439 | ) | (233 | ) | (206 | ) | N/A | ||||||
Other income (expense), net |
72 | (2,174 | ) | 2,246 | N/A | ||||||||
| | | | | | | | | | | | | | |
Net loss and comprehensive loss |
$ | (29,373 | ) | $ | (25,580 | ) | $ | (3,793 | ) | 15 | % | ||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Revenue
Revenue increased $16.3 million, or 75%, for the year ended December 31, 2014 compared to the same period in 2013. The increase was primarily as a result of increased collections which resulted from realizing higher reimbursement rates from payers as well as from increased volume due to increased adoption of Afirma and increased percentage of samples for the GEC test only.
For the year ended December 31, 2014 compared to the same period in 2013, revenue recognized when cash was received increased by approximately $11.1 million, or 76%, to $25.7 million, reflecting increased adoption and collections. For the year ended December 31, 2014 compared to the same period in 2013, revenue recognized on an accrual basis increased by approximately $5.2 million, or 71% to $12.5 million, primarily reflecting increased adoption and to a lesser extent, additional payers meeting our revenue recognition criteria.
74
Cost of revenue
Cost of revenue increased $4.0 million, or 32%, for the year ended December 31, 2014 compared to the same period in 2013. The increase was primarily due to an increase in variable costs that are directly related to the increase in the number of FNAs received, offset in part by continuing refinements in our testing process and economies of scale related to the increase in FNAs processed. FNAs received increased 16,178, or 33%, to 65,848 in the year ended December 31, 2014.
Research and development
Comparison of the years ended December 31, 2014 and 2013 is as follows:
| |
Year Ended December 31, |
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Dollar Change |
% Change |
|||||||||||
| |
2014 | 2013 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Research and development expense: |
|||||||||||||
Personnel related expense |
$ | 4,534 | $ | 3,875 | $ | 659 | 17 | % | |||||
Stock-based compensation expense |
790 | 250 | 540 | 216 | % | ||||||||
Direct R&D expense |
2,734 | 1,685 | 1,049 | 62 | % | ||||||||
Other expense |
1,746 | 2,000 | (254 | ) | –13 | % | |||||||
| | | | | | | | | | | | | | |
Total |
$ | 9,804 | $ | 7,810 | $ | 1,994 | 26 | % | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Research and development expense increased $2.0 million, or 26%, for the year ended December 31, 2014 compared to the same period in 2013. The increase in personnel related expense was primarily due to a 38% increase in headcount at December 31, 2014 as compared to December 31, 2013. The increase in stock-based compensation expense reflects option grants to new and existing employees. The increase in direct R&D expense was due primarily to the timing of genome sequencing expenses and other laboratory expenses. The decrease in other expense was due primarily to $530,000 in licensing fees to secure thyroid intellectual property in 2013, partially offset by an increase in consulting and recruiting fees.
Selling and marketing
Comparison of the years ended December 31, 2014 and 2013 is as follows:
| |
Year Ended December 31, |
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Dollar Change |
% Change |
|||||||||||
| |
2014 | 2013 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Selling and marketing expense: |
|||||||||||||
Genzyme co-promotion expense, net |
$ | 9,733 | $ | 6,084 | $ | 3,649 | 60 | % | |||||
Personnel related expense |
8,121 | 4,291 | 3,830 | 89 | % | ||||||||
Stock-based compensation expense |
707 | 169 | 538 | 318 | % | ||||||||
Direct marketing expense |
1,544 | 982 | 562 | 57 | % | ||||||||
Other expense |
1,827 | 1,014 | 813 | 80 | % | ||||||||
| | | | | | | | | | | | | | |
Total |
$ | 21,932 | $ | 12,540 | $ | 9,392 | 75 | % | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Selling and marketing expense increased $9.4 million, or 75%, for the year ended December 31, 2014 compared to the same period in 2013. The increase in Genzyme co-promotion expense, net, reflects growth in cash collections, partially offset by a reduction in the co-promotion percentage rate payable to Genzyme in 2014 as compared to 2013 under the 2012 co-promotion agreement. The increase in personnel related expense were primarily due to a 107% increase in headcount of our sales force at December 31, 2014 as compared to December 31, 2013. The increase in stock-based compensation expense reflects option grants
75
to new and existing employees. The increase in direct marketing expense was due primarily to increased marketing and promotional materials and market research and consultants. The increase in other expense was primarily due to an increase in information technology and facilities expenses that were related to sales and marketing activities.
In November 2014, we entered into an Amended and Restated U.S. Co-Promotion Agreement, or Amended Agreement, with Genzyme. As a result of the Amended Agreement, we expect our selling and marketing expenses for Afirma to remain flat over the next year. While we expect that our personnel costs will increase as we take on more sales and marketing responsibilities related to Afirma, we expect these increases will be offset by the lower rate we are required to pay Genzyme under the Amended Agreement beginning in January 2015. In 2015, we also expect to incur selling and marketing expenses as a result of investments in our lung product portfolio. Therefore, we believe total selling and marketing expenses will increase in 2015.
General and administrative
Comparison of the years ended December 31, 2014 and 2013 is as follows:
| |
Year Ended December 31, |
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Dollar Change |
% Change |
|||||||||||
| |
2014 | 2013 | |||||||||||
| |
(In thousands) |
|
|||||||||||
General and administrative expense: |
|||||||||||||
Personnel related expense |
$ | 9,563 | $ | 6,454 | $ | 3,109 | 48 | % | |||||
Stock-based compensation expense |
2,000 | 794 | 1,206 | 152 | % | ||||||||
Professional fees expense |
4,525 | 2,645 | 1,880 | 71 | % | ||||||||
Rent and other facilities expense |
1,504 | 1,477 | 27 | 2 | % | ||||||||
Other expense |
1,262 | 730 | 532 | 73 | % | ||||||||
| | | | | | | | | | | | | | |
Total |
$ | 18,854 | $ | 12,100 | $ | 6,754 | 56 | % | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
General and administrative expense increased $6.8 million, or 56%, for the year ended December 31, 2014 compared to the same period in 2013. The increase in personnel related expense was primarily due to a 32% increase in headcount at December 31, 2014 as compared to December 31, 2013 and to acquisition costs of $1.2 million for bonus and severance paid to Allegro employees. The increase in stock-based compensation expense was primarily due to option grants to new and existing employees. The increase in professional fees includes higher audit, legal and other corporate expenses including insurance, associated with operating as a public company for the full year. In addition, professional fees included Allegro acquisition costs of approximately $0.3 million for audit, legal and valuation services. The increase in other expense was due primarily to an increase in consulting expense of approximately $0.9 million, including approximately $0.2 million for the Allegro acquisition. Other expense also included fees for our billing system and postage which increased as a result of increased FNA volume, and tax/license fees which increased as a result of being a public company. These other expenses were largely offset by decreases in computer and facilities allocations as a result of increased headcount in other functions.
Interest expense
Interest expense increased $206,000 for the year ended December 31, 2014 compared to the same period in 2013 primarily due to higher interest expense associated with our loan which was outstanding for the full year in 2014 and only half a year in 2013.
76
Other income (expense), net
Other income (expense), net, increased $2.2 million for the year ended December 31 2014, compared to the same period in 2013 primarily due to the one-time $2.1 million expense related to the increase in the fair value of the preferred stock liability associated with our obligation to issue additional shares of Series C convertible preferred stock, and an $86,000 expense related to the increase in the fair value of the preferred stock warrant liability in the year ended December 31, 2013. Other income (expense), net, for 2014 consisted primarily of sublease rental income of $86,000, interest income received from payers and from our cash equivalents of $31,000, partially offset by amortization of debt issuance costs of $44,000.
Comparison of the Years Ended December 31, 2013 and 2012
| |
Year Ended December 31, |
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Dollar Change |
% Change |
|||||||||||
| |
2013 | 2012 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Revenue |
$ | 21,884 | $ | 11,628 | $ | 10,256 | 88 | % | |||||
Operating expense: |
|||||||||||||
Cost of revenue |
12,607 | 7,584 | 5,023 | 66 | % | ||||||||
Research and development |
7,810 | 6,608 | 1,202 | 18 | % | ||||||||
Selling and marketing |
12,540 | 8,447 | 4,093 | 48 | % | ||||||||
General and administrative |
12,100 | 7,918 | 4,182 | 53 | % | ||||||||
| | | | | | | | | | | | | | |
Total operating expenses |
45,057 | 30,557 | 14,500 | 47 | % | ||||||||
| | | | | | | | | | | | | | |
Loss from operations |
(23,173 | ) | (18,929 | ) | (4,244 | ) | 22 | % | |||||
Interest expense |
(233 | ) | — | (233 | ) | N/A | |||||||
Other income (expense), net |
(2,174 | ) | 280 | (2,454 | ) | N/A | |||||||
| | | | | | | | | | | | | | |
Net loss and comprehensive loss |
$ | (25,580 | ) | $ | (18,649 | ) | $ | (6,931 | ) | 37 | % | ||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Revenue
Revenue increased $10.3 million, or 88%, for the year ended December 31, 2013 compared to the same period in 2012 primarily as a result of a $7.2 million increase in commercial revenue from increased reimbursement and collections and a $3.1 million increase in Medicare revenue as a result of increased Afirma adoption.
For the year ended December 31, 2013 compared to the same period in 2012, revenue recognized when cash was received increased by approximately $7.1 million, or 95%, to $14.6 million, reflecting increased adoption and collections. For the year ended December 31, 2013 compared to the same period in 2012, revenue recognized on an accrual basis increased by approximately $3.2 million, or 78%, to $7.3 million.
Cost of revenue
Cost of revenue increased $5.0 million, or 66%, for the year ended December 31, 2013 compared to the same period in 2012. This increase was primarily due to a $4.7 million, or 77%, increase in variable costs that are directly related to the increase in the number of FNAs received, offset in part by continuing refinements in our testing process and economies of scale related to the increase in FNAs. FNAs received increased 23,780, or 92%, to 49,670 in the year ended December 31, 2013.
77
Research and development
Comparison of the years ended December 31, 2013 and 2012 is as follows:
| |
Year Ended December 31, |
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Dollar Change |
% Change |
|||||||||||
| |
2013 | 2012 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Research and development expense: |
|||||||||||||
Personnel related expense |
$ | 3,875 | $ | 3,236 | $ | 639 | 20 | % | |||||
Stock-based compensation expense |
250 | 131 | 119 | 91 | % | ||||||||
Direct R&D expense |
1,685 | 1,504 | 181 | 12 | % | ||||||||
Other expense |
2,000 | 1,737 | 263 | 15 | % | ||||||||
| | | | | | | | | | | | | | |
Total |
$ | 7,810 | $ | 6,608 | $ | 1,202 | 18 | % | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Research and development expense increased $1.2 million, or 18%, for the year ended December 31, 2013 compared to the same period in 2012. This increase was primarily due to a $0.6 million increase in personnel related expenses related to a 24% increase in headcount, and a $0.5 million increase in licensing expenses, included above in other expense, to secure intellectual property to augment our existing thyroid patent portfolio, offset by a decrease in other expenses.
Selling and marketing
Comparison of the years ended December 31, 2013 and 2012 is as follows:
| |
Year Ended December 31, |
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Dollar Change |
% Change |
|||||||||||
| |
2013 | 2012 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Selling and marketing expense: |
|||||||||||||
Genzyme co-promotion expense, net |
$ | 6,084 | $ | 3,094 | $ | 2,990 | 97 | % | |||||
Personnel related expense |
4,291 | 3,824 | 467 | 12 | % | ||||||||
Stock-based compensation expense |
169 | 111 | 58 | 52 | % | ||||||||
Direct marketing expense |
982 | 591 | 391 | 66 | % | ||||||||
Other expense |
1,014 | 827 | 187 | 23 | % | ||||||||
| | | | | | | | | | | | | | |
Total |
$ | 12,540 | $ | 8,447 | $ | 4,093 | 48 | % | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Selling and marketing expense increased $4.1 million, or 48%, for the year ended December 31, 2013 compared to the same period in 2012. This increase was primarily due to a $3.0 million increase in net expense recognized under our co-promotion agreement with Genzyme, reflecting growth in cash collections, partially offset by amortization of the deferred fee, a $0.5 million increase in personnel related expense related to a 28% increase in headcount, a $0.5 million increase in marketing and promotional materials, and a $0.1 million increase in consulting expenses.
78
General and administrative
Comparison of the years ended December 31, 2013 and 2012 is as follows:
| |
Year Ended December 31, |
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Dollar Change |
% Change |
|||||||||||
| |
2013 | 2012 | |||||||||||
| |
(In thousands) |
|
|||||||||||
General and administrative expense: |
|||||||||||||
Personnel related expense |
$ | 6,454 | $ | 4,661 | $ | 1,793 | 38 | % | |||||
Stock-based compensation expense |
794 | 407 | 387 | 95 | % | ||||||||
Professional fees expense |
2,645 | 1,089 | 1,556 | 143 | % | ||||||||
Rent and other facilities expense |
1,477 | 1,202 | 275 | 23 | % | ||||||||
Other expense |
730 | 559 | 171 | 31 | % | ||||||||
| | | | | | | | | | | | | | |
Total |
$ | 12,100 | $ | 7,918 | $ | 4,182 | 53 | % | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
General and administrative expense increased $4.2 million, or 53%, for the year ended December 31, 2013 compared to the same period in 2012. This increase was primarily due to a $1.8 million increase in personnel related expenses related to a 63% increase in headcount, a $0.4 million increase in stock-based compensation expense primarily related to 2013 option grants, an increase in professional fees primarily due to non-capitalizable IPO related audit and legal services of $1.4 million, an increase in rent and other facilities expenses primarily due to the opening of the Austin, Texas facility of $0.4 million, and a $0.2 million increase in insurance expenses related to higher premiums associated with being a public company.
Interest expense
Interest expense increased $0.2 million for the year ended December 31, 2013 compared to the same period in 2012. Interest expense of $0.2 million for the year ended December 31, 2013 is interest incurred on the initial June 2013 drawdown of $5.0 million under our loan and security agreement. We did not have any debt in the same period in 2012.
Other income (expense), net
Other income (expense), net, decreased $2.5 million in the year ended December 31, 2013 compared to the same period in 2012. The decrease was primarily related to a $2.4 million increase in the fair value of the preferred stock liability from a gain of $0.3 million in 2012 to a loss of $2.1 million in 2013, and a $0.1 million increase in the fair value of the preferred stock warrant liability. As the preferred stock liability was extinguished in 2013, and the preferred stock warrant liability was converted into a warrant to purchase our common stock upon the completion of the IPO in 2013, any related expenses will not carry forward to future periods.
Liquidity and Capital Resources
We have incurred net losses since our inception. For the years ended December 31, 2014, 2013 and 2012, we had a net loss of $29.4 million, $25.6 million and $18.6 million, respectively, and we expect to incur additional losses in 2015 and in future years. As of December 31, 2014, we had an accumulated deficit of $115.0 million. We may never achieve revenue sufficient to offset our expenses. As of December 31, 2014, we had $35.0 million in cash and cash equivalents. We believe our existing cash and cash equivalents as of December 31, 2014 and our revenue from the sale of Afirma in 2015 will be sufficient to meet our anticipated cash requirements for at least the next 12 months.
Since inception, we have received $154.0 million in net proceeds from various sources to finance our operations, including net proceeds of $78.6 million from sales of our preferred stock, net proceeds of
79
$59.2 million from our IPO, $10.0 million from the Genzyme co-promotion agreement, net borrowings of $4.9 million under our loan and security agreement, and $1.3 million from the exercise of stock options.
In June 2013, we entered into a loan and security agreement, the Original Loan, with a financial institution. The Original Loan provided for term loans of up to an aggregate of $10.0 million. On entering the Original Loan, we drew down an initial $5.0 million term loan. We opted not to draw the remaining $5.0 million and the option to do so expired in March 2014. We were required to repay the outstanding principal in 30 equal installments beginning 18 months after the date of the borrowing and the loan was due in full in June 2017. The Original Loan had an interest rate of 6.06% per annum, carried prepayment penalties of 2.25% and 1.50% for prepayment within one and two years, respectively, and 0.75% thereafter.
In December 2014, we amended certain terms and conditions of the Original Loan, which we refer to as the Amended Loan. The Amended Loan provides for term loans of up to $15.0 million in aggregate, in three tranches of $5.0 million each. We borrowed $5.0 million under the first tranche in December 2014 and used the funds for repayment of the $5.0 million in principal outstanding under the Original Loan, in a cashless transaction. In addition, we paid the accrued but unpaid interest of $14,000 due on the Original Loan and the related end-of-term payment of $110,000. The Amended Loan waived the prepayment premium of $75,000 under the Original Loan and reduced the end-of-term payment of $225,000 under the Original Loan to $110,000. The second $5.0 million tranche under the Amended Loan is available through December 31, 2015, and we may borrow the third $5.0 million tranche any time through June 30, 2016 after achieving the third tranche revenue milestone as defined in the Amended Loan.
Under the Amended Loan, we are required to repay the outstanding principal in 24 equal installments beginning 24 months after the date of the borrowing and the loan is due in full in December 2018. The first tranche of the Amended Loan bears interest at a rate of 5.00% per annum, and the Amended Loan carries prepayment penalties of 2.00% and 1.00% for prepayment within one and two years, respectively. In connection with the Amended Loan, we paid approximately $45,000 in third-party fees.
Loans drawn under the Original Loan and the Amended Loan were used for working capital and general corporate purposes. Our obligations under the Amended Loan are secured by a security interest on substantially all of our assets, excluding our intellectual property and certain other assets. The Amended Loan contains customary conditions to borrowing, events of default, and covenants, including covenants limiting our ability to dispose of assets, undergo a change in control, merge with or acquire other entities, incur debt, incur liens, pay dividends or other distributions to holders of our capital stock, repurchase stock and make investments, in each case subject to certain exceptions. The Amended Loan also allows the lender to call the debt in the event there is a material adverse change in our business or financial condition. We are required to be in compliance with a minimum liquidity or minimum revenue covenant. As of December 31, 2014, we were in compliance with the financial covenants.
In connection with the draw-down of the initial $5.0 million term loan under the Original Loan, we issued the lender a warrant to purchase 24,801 shares of our common stock upon completion of the IPO. The lender exercised the warrant through a cashless exercise in March 2014, resulting in the issuance of 13,739 shares of common stock at an exercise price of $7.56 per share.
On September 16, 2014, we acquired Allegro via a merger with Full Moon Acquisition, Inc., or Full Moon, our wholly-owned subsidiary. Allegro was a privately-held company based in Maynard, Massachusetts, focused on the development of genomic tests to improve the preoperative diagnosis of lung cancer. In conjunction with the merger, we issued 964,377 shares of our common stock, paid $2.7 million in cash, settled in cash outstanding indebtedness of Allegro totaling $4.3 million, and paid severance and bonus to Allegro personnel of $1.2 million.
We expect that our near- and longer-term liquidity requirements will continue to consist of selling and marketing expenses, research and development expenses, working capital, and general corporate expenses
80
associated with the growth of our business. However, we may also use cash to acquire or invest in complementary businesses, technologies, services or products that would change our cash requirements. If we are not able to generate revenue to finance our cash requirements, we will need to finance future cash needs primarily through public or private equity offerings, debt financings, borrowings or strategic collaborations or licensing arrangements. If we raise funds by issuing equity securities, dilution to stockholders may result. Any equity securities issued may also provide for rights, preferences or privileges senior to those of holders of our common stock. If we raise funds by issuing debt securities, these debt securities would have rights, preferences and privileges senior to those of holders of our common stock. The terms of debt securities or borrowings could impose significant restrictions on our operations. If we raise funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our technologies or products, or grant licenses on terms that are not favorable to us. The credit market and financial services industry have in the past, and may in the future, experience periods of upheaval that could impact the availability and cost of equity and debt financing. If we are not able to secure additional funding when needed, on acceptable terms, we may have to delay, reduce the scope of or eliminate one or more research and development programs or selling and marketing initiatives. In addition, we may have to work with a partner on one or more of our product or market development programs, which could lower the economic value of those programs to us.
The following table summarizes our cash flows for the years ended December 31, 2014, 2013 and 2012:
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
| |
(in thousands) |
|||||||||
Cash used in operating activities |
$ | (27,632 | ) | $ | (19,159 | ) | $ | (7,167 | ) | |
Cash used in investing activities |
(9,010 | ) | (1,282 | ) | (1,462 | ) | ||||
Cash provided by financing activities |
436 | 77,659 | 15,065 | |||||||
Cash Flows from Operating Activities
Cash used in operating activities for the year ended December 31, 2014 was $27.6 million. The net loss of $29.4 million includes non-cash charges of $2.3 million in amortization of the deferred fee received from Genzyme, offset primarily by $3.5 million of stock-based compensation expense, $1.2 million of depreciation and amortization, $0.2 million in amortization of debt discount and issuance costs and debt balloon interest expense, and $0.1 million of bad debt expense. The increase in net operating assets of $0.9 million was primarily due to a $2.0 million increase in accounts receivable due to increases in Afirma adoption and new payers for whom revenue is recognized on an accrual basis, a $1.1 million increase in supplies inventory due to the increased volume of testing performed and a strategic decision to increase our inventory on hand, offset by a $2.2 million net increase in accounts payable and accrued liabilities resulting from the timing of payments.
Cash used in operating activities for the year ended December 31, 2013 was $19.2 million. The net loss of $25.6 million was offset by non-cash charges of $2.1 million for the change in the value of the preferred stock liability, $2.5 million in amortization of the deferred fee received from Genzyme, $1.2 million of stock based compensation, $1.0 million of depreciation and amortization, $0.1 million of bad debt expense, a $0.1 million charge for the change in value of the preferred stock warrant liability, and $0.1 million for non-cash interest on the outstanding debt. The increase in net changes in assets and liabilities of $4.3 million was primarily due to a $7.2 million increase in accounts payable and accrued liabilities due to timing of payments offset by a $2.9 million increase in assets, including a $0.7 million increase in prepaid expenses due primarily to increased public company related prepaid insurance premiums, a $1.5 million increase in supply inventory due to the increase in volume of testing performed, and a $0.7 million increase in accounts receivable due to increased revenues from Medicare.
81
Cash used in operating activities for the year ended December 31, 2012 was $7.2 million. The net loss of $18.6 million was offset by non-cash charges of $0.9 million of stock- and equity-based compensation, $0.7 million for depreciation and amortization, $0.3 million for the change in value of the preferred stock liability and $0.2 million of bad debt expense. The increase in net operating assets of $12.3 million was primarily due to the $10.0 million deferred payment from Genzyme, of which we amortized $2.4 million as of December 31, 2012. Accounts payable and accrued liabilities increased $3.9 million due to the growth in our operations and the timing of our payments. Accounts receivable increased by $0.6 million due to the increase in accrued revenue in 2012 as we had only begun to sell Afirma in 2011. In addition, there was a $0.8 million increase in supplies inventory related to increased test demand.
Cash Flows from Investing Activities
Cash used in investing activities was $9.0 million, $1.3 million and $1.5 million in the years ended December 31, 2014, 2013 and 2012, respectively. We acquired Allegro in September 2014 for net cash of approximately $6.9 million and we restricted the use of $70,000 of cash as of December 31, 2014 to cover hold-back liabilities associated with the acquisition. We purchased laboratory equipment, software and leasehold improvements of $2.0 million, $1.3 million and $1.5 million for the years ended December 31, 2014, 2013 and 2012, respectively.
Cash Flows from Financing Activities
Cash provided by financing activities for the year ended December 31, 2014 of $0.4 million consisted of $0.7 million we received from the exercise of options to purchase our common stock, offset by $0.1 million of IPO-related disbursements and a $0.1 million end-of-term payment on our Original Loan.
Cash provided by financing activities for the year ended December 31, 2013 of $77.7 million consisted of the receipt of $59.3 million in net proceeds from the issuance of common stock in connection with our IPO, the receipt of $12.9 million in net proceeds from the sale of our convertible preferred stock, net borrowings of $4.9 million under the Original Loan and $0.6 million from the exercise of options to purchase our common stock.
Cash provided by financing activities for the year ended December 31, 2012 of $15.1 million consisted of the receipt of $15.0 million in net proceeds from the sale of our convertible preferred stock and $0.1 million from the exercise of options to purchase our common stock.
Contractual Obligations
The following table summarizes certain contractual obligations as of December 31, 2014 (in thousands):
| |
Payments Due by Period | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Less than 1 Year |
1 to 3 Years |
3 to 5 Years |
More than 5 Years |
Total | |||||||||||
Operating lease obligations |
$ | 989 | $ | 635 | $ | 130 | $ | — | $ | 1,754 | ||||||
Long-term debt obligations |
— | 2,437 | 2,563 | — | 5,000 | |||||||||||
Interest on debt |
242 | 451 | 71 | — | 764 | |||||||||||
Supplies purchase commitments |
715 | — | — | — | 715 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total |
$ | 1,946 | $ | 3,523 | $ | 2,764 | $ | — | $ | 8,233 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
In February 2010, we entered into a non-cancelable lease agreement for our headquarters and laboratory space in South San Francisco, California. The lease expires in March 2016.
In November 2012, we entered into a non-cancelable lease agreement commencing February 2013 for our laboratory and office space in Austin, Texas. The lease expires in July 2018.
82
In May 2010 and February 2013, we entered into a non-cancelable purchase commitment with two suppliers to purchase a minimum quantity of supplies.
Off-balance Sheet Arrangements
We have not entered into any off-balance sheet arrangements.
JOBS Act Accounting Election
We are an emerging growth company, as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update, or ASU, No. 2014-09, Revenue from Contracts with Customers, requiring an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers. The updated standard will replace most existing revenue recognition guidance in U.S. GAAP when it becomes effective and permits the use of either the retrospective or cumulative effect transition method. Early adoption is not permitted. The updated standard becomes effective for us in the first quarter of fiscal 2017. We have not yet selected a transition method and are currently evaluating the effect that the updated standard may have on our financial statements.
In August 2014, FASB issued Accounting Standards Update No. 2014-15, Presentation of Financial Statements Going Concern—Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern. The amendments require management to assess an entity's ability to continue as a going concern by incorporating and expanding upon certain principles that are currently in U.S. auditing standards. Specifically, the amendments: (1) provide a definition of the term substantial doubt; (2) require an evaluation every reporting period including interim periods; (3) provide principles for considering the mitigating effect of management's plans; (4) require certain disclosures when substantial doubt is alleviated as a result of consideration of management's plans; (5) require an express statement and other disclosures when substantial doubt is not alleviated; and (6) require an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). ASU 2014-15 will be effective for annual periods ending after December 15, 2016 and interim periods within annual periods beginning after December 15, 2016 with early adoption permitted. ASU 2014-15 will be effective for us beginning with our annual report for fiscal 2016 and interim periods thereafter. We have not yet determined the effect of the adoption of this standard on our consolidated financial statements.
83
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to market risks in the ordinary course of our business. These risks primarily relate to interest rates. We had cash and cash equivalents of $35.0 million as of December 31, 2014 which consisted of bank deposits and money market funds. Such interest-bearing instruments carry a degree of risk; however, a hypothetical 10% change in interest rates during any of the periods presented would not have had a material impact on our audited consolidated financial statements.
84
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Veracyte, Inc.
Index to Consolidated Financial Statements
85
Report of Independent Registered Public Accounting Firm
The
Board of Directors and Stockholders
Veracyte, Inc.
We have audited the accompanying consolidated balance sheet of Veracyte, Inc. as of December 31, 2014, and the related consolidated statement of operations and comprehensive loss, convertible preferred stock and stockholders' equity (deficit), and cash flows for the year ended December 31, 2014. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company's internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Veracyte, Inc. at December 31, 2014, and the results of its operations and its cash flows for the year ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young LLP
Redwood
City, California
March 24, 2015
86
Report of Independent Registered Public Accounting Firm
To
the Board of Directors and Stockholders of
Veracyte, Inc.
In our opinion, the balance sheet as of December 31, 2013 and the related statements of operations and comprehensive loss, of convertible preferred stock and stockholders' deficit, and of cash flows for each of two years in the period ended December 31, 2013 present fairly, in all material respects, the financial position of Veracyte, Inc. at December 31, 2013, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2013 in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits. We conducted our audits of these statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers LLP
San
Jose, California
March 20, 2014
87
VERACYTE, INC.
Consolidated Balance Sheets
(In thousands, except share and per share amounts)
| |
As of December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Assets |
|||||||
Current assets: |
|||||||
Cash and cash equivalents |
$ | 35,014 | $ | 71,220 | |||
Accounts receivable, net of allowance of $84 and $107 as of December 31, 2014 and 2013 |
3,050 | 1,143 | |||||
Supplies inventory |
3,696 | 2,567 | |||||
Prepaid expenses and other current assets |
1,218 | 1,477 | |||||
Deferred tax asset |
300 | — | |||||
Restricted cash |
70 | — | |||||
| | | | | | | | |
Total current assets |
43,348 | 76,407 | |||||
Property and equipment, net |
4,161 | 2,952 | |||||
In-process research and development |
16,000 | — | |||||
Goodwill |
1,057 | — | |||||
Restricted cash |
118 | 118 | |||||
Other assets |
155 | 153 | |||||
| | | | | | | | |
Total assets |
$ | 64,839 | $ | 79,630 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Liabilities and Stockholders' Equity |
|||||||
Current liabilities: |
|||||||
Accounts payable |
$ | 7,397 | $ | 5,294 | |||
Accrued liabilities |
7,851 | 7,594 | |||||
Deferred Genzyme co-promotion fee |
1,897 | 2,500 | |||||
| | | | | | | | |
Total current liabilities |
17,145 | 15,388 | |||||
Long-term debt |
4,923 | 4,899 | |||||
Deferred tax liability |
300 | — | |||||
Deferred rent, net of current portion |
149 | 286 | |||||
Deferred Genzyme co-promotion fee, net of current portion |
948 | 2,614 | |||||
| | | | | | | | |
Total liabilities |
23,465 | 23,187 | |||||
| | | | | | | | |
Commitments and contingencies (Note 7) |
|||||||
Stockholders' equity: |
|||||||
Preferred stock, $0.001 par value; 5,000,000 shares authorized, 0 shares issued and outstanding as of December 31, 2014 and 2013 |
— | — | |||||
Common stock, $0.001 par value; 125,000,000 shares authorized, 22,523,529 and 21,143,313 shares issued and outstanding as of December 31, 2014 and 2013, respectively |
23 | 21 | |||||
Additional paid-in capital |
156,373 | 142,071 | |||||
Accumulated deficit |
(115,022 | ) | (85,649 | ) | |||
| | | | | | | | |
Total stockholders' equity |
41,374 | 56,443 | |||||
| | | | | | | | |
Total liabilities and stockholders' equity |
$ | 64,839 | $ | 79,630 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
88
VERACYTE, INC.
Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except share and per share amounts)
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Revenue |
$ | 38,190 | $ | 21,884 | $ | 11,628 | ||||
Operating expenses: |
||||||||||
Cost of revenue |
16,606 | 12,607 | 7,584 | |||||||
Research and development |
9,804 | 7,810 | 6,608 | |||||||
Selling and marketing |
21,932 | 12,540 | 8,447 | |||||||
General and administrative |
18,854 | 12,100 | 7,918 | |||||||
| | | | | | | | | | | |
Total operating expenses |
67,196 | 45,057 | 30,557 | |||||||
| | | | | | | | | | | |
Loss from operations |
(29,006 | ) | (23,173 | ) | (18,929 | ) | ||||
Interest expense |
(439 | ) | (233 | ) | — | |||||
Other income (expense), net |
72 | (2,174 | ) | 280 | ||||||
| | | | | | | | | | | |
Net loss and comprehensive loss |
$ | (29,373 | ) | $ | (25,580 | ) | $ | (18,649 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per common share, basic and diluted |
$ | (1.36 | ) | $ | (6.15 | ) | $ | (28.68 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Shares used to compute net loss per common share, basic and diluted |
21,639,374 | 4,158,664 | 650,333 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
89
VERACYTE, INC.
Consolidated Statements of Convertible Preferred Stock and Stockholders' Equity (Deficit)
(In thousands, except share and per share amounts)
| |
Convertible Preferred Stock |
|
|
|
|
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Common Stock | |
|
|
||||||||||||||||||
| |
Additional Paid-in Capital |
Accumulated Deficit |
Total Stockholders' Equity (Deficit) |
|||||||||||||||||||
| |
Shares | Amount | Shares | Amount | ||||||||||||||||||
Balance at December 31, 2011 |
45,147,999 | $ | 49,296 | 594,941 | $ | 1 | $ | 653 | $ | (41,420 | ) | $ | (40,766 | ) | ||||||||
Issuance of Series C convertible preferred stock in November and December 2012 at $1.89 per share, net of issuance costs of $63 and $861 preferred stock liability |
7,936,508 | 14,076 | — | — | — | — | — | |||||||||||||||
Common stock issued on exercise of stock options |
— | — | 72,743 | — | 76 | — | 76 | |||||||||||||||
Stock-based compensation expense (employee) |
— | — | — | — | 590 | — | 590 | |||||||||||||||
Stock-based compensation expense (non-employee) |
— | — | — | — | 85 | — | 85 | |||||||||||||||
Equity-based compensation |
— | — | — | — | 193 | — | 193 | |||||||||||||||
Net loss and comprehensive loss |
— | — | — | — | — | (18,649 | ) | (18,649 | ) | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2012 |
53,084,507 | 63,372 | 667,684 | 1 | 1,597 | (60,069 | ) | (58,471 | ) | |||||||||||||
Issuance of Series C convertible preferred stock in June 2013 at $1.89 per share, net of issuance costs of $53 |
6,904,761 | 12,997 | — | — | — | — | — | |||||||||||||||
Extinguishment of preferred stock liability |
— | 2,653 | — | — | — | — | — | |||||||||||||||
Issuance of common stock on exercise of stock options |
— | — | 377,966 | — | 552 | — | 552 | |||||||||||||||
Issuance of common stock in initial public offering, net of discounts and commissions of $4,642 and issuance costs of $2,507 |
— | — | 5,100,351 | 5 | 59,151 | — | 59,156 | |||||||||||||||
Conversion of preferred stock into common stock upon initial public offering |
(59,989,268 | ) | (79,022 | ) | 14,997,312 | 15 | 79,007 | — | 79,022 | |||||||||||||
Reclassification of preferred stock warrant liability into additional paid-in capital upon initial public offering |
— | — | — | — | 261 | — | 261 | |||||||||||||||
Stock-based compensation expense (employee) |
— | — | — | — | 1,041 | — | 1,041 | |||||||||||||||
Stock-based compensation expense (non-employee) |
— | — | — | — | 206 | — | 206 | |||||||||||||||
Equity-based compensation |
— | — | — | — | 259 | — | 259 | |||||||||||||||
Common stock subject to repurchase |
— | — | — | — | (3 | ) | — | (3 | ) | |||||||||||||
Net loss and comprehensive loss |
— | — | — | — | — | (25,580 | ) | (25,580 | ) | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 |
— | — | 21,143,313 | 21 | 142,071 | (85,649 | ) | 56,443 | ||||||||||||||
Issuance of common stock on exercise of stock options |
— | — | 402,100 | 1 | 674 | — | 675 | |||||||||||||||
Issuance of common stock on cashless exercise of stock warrant |
— | — | 13,739 | — | — | — | — | |||||||||||||||
Common stock subject to repurchase |
— | — | — | — | 3 | — | 3 | |||||||||||||||
Issuance of common stock for acquisition |
— | — | 964,377 | 1 | 10,077 | — | 10,078 | |||||||||||||||
Stock-based compensation expense (employee) |
— | — | — | — | 3,388 | — | 3,388 | |||||||||||||||
Stock-based compensation expense (non-employee) |
— | — | — | — | 160 | — | 160 | |||||||||||||||
Net loss and comprehensive loss |
— | — | — | — | — | (29,373 | ) | (29,373 | ) | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2014 |
— | — | 22,523,529 | $ | 23 | $ | 156,373 | $ | (115,022 | ) | $ | 41,374 | ||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
90
VERACYTE, INC.
Consolidated Statements of Cash Flows
(In thousands)
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Operating activities |
||||||||||
Net loss |
$ | (29,373 | ) | $ | (25,580 | ) | $ | (18,649 | ) | |
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||
Depreciation and amortization |
1,175 | 999 | 706 | |||||||
Bad debt expense |
54 | 109 | 225 | |||||||
Genzyme co-promotion fee amortization |
(2,269 | ) | (2,500 | ) | (2,386 | ) | ||||
Stock-based compensation |
3,548 | 1,247 | 675 | |||||||
Equity-based compensation |
— | — | 259 | |||||||
Amortization of debt discount and issuance costs |
97 | 56 | — | |||||||
Interest on debt balloon payment |
81 | 42 | — | |||||||
Change in value of preferred stock liability |
— | 2,070 | (278 | ) | ||||||
Change in value of preferred stock warrant liability |
— | 86 | — | |||||||
Changes in operating assets and liabilities: |
||||||||||
Accounts receivable |
(1,961 | ) | (683 | ) | (565 | ) | ||||
Supplies inventory |
(1,129 | ) | (1,517 | ) | (771 | ) | ||||
Prepaid expenses and current other assets |
(38 | ) | (722 | ) | (191 | ) | ||||
Other assets |
(46 | ) | 24 | (119 | ) | |||||
Accounts payable |
1,874 | 3,348 | 1,348 | |||||||
Accrued liabilities and deferred rent |
355 | 3,862 | 2,579 | |||||||
Deferred Genzyme co-promotion fee |
— | — | 10,000 | |||||||
| | | | | | | | | | | |
Net cash used in operating activities |
(27,632 | ) | (19,159 | ) | (7,167 | ) | ||||
| | | | | | | | | | | |
Investing activities |
||||||||||
Purchases of property and equipment |
(2,024 | ) | (1,332 | ) | (1,462 | ) | ||||
Cash remitted for acquisition, net of cash received |
(6,916 | ) | — | — | ||||||
Change in restricted cash |
(70 | ) | 50 | — | ||||||
| | | | | | | | | | | |
Net cash used in investing activities |
(9,010 | ) | (1,282 | ) | (1,462 | ) | ||||
| | | | | | | | | | | |
Financing activities |
||||||||||
Proceeds from the issuance of long-term debt, net of debt issuance costs |
— | 4,877 | — | |||||||
Payment of end-of-term debt obligation |
(110 | ) | — | — | ||||||
Proceeds from issuance of redeemable convertible preferred stock, net of issuance costs |
— | 12,945 | 14,989 | |||||||
Proceeds from issuance of common stock in initial public offering, gross |
— | 66,304 | — | |||||||
Commissions and issuance costs relating to the initial public offering |
(129 | ) | (7,019 | ) | — | |||||
Proceeds from the exercise of common stock options |
675 | 552 | 76 | |||||||
| | | | | | | | | | | |
Net cash provided by financing activities |
436 | 77,659 | 15,065 | |||||||
| | | | | | | | | | | |
Net increase (decrease) in cash and cash equivalents |
(36,206 | ) | 57,218 | 6,436 | ||||||
Cash and cash equivalents at beginning of period |
71,220 | 14,002 | 7,566 | |||||||
| | | | | | | | | | | |
Cash and cash equivalents at end of period |
$ | 35,014 | $ | 71,220 | $ | 14,002 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Supplementary cash flow information of non-cash investing and financing activities: |
||||||||||
Fair value of common stock issued for acquisition |
$ | 10,078 | — | — | ||||||
Non-cash issuance of long-term debt |
5,000 | — | — | |||||||
Non-cash repayment of long-term debt |
(5,000 | ) | — | — | ||||||
Purchases of property and equipment included in accounts payable and accrued liabilities |
383 | $ | 25 | $ | 109 | |||||
Non-cash purchases of property and equipment |
— | 257 | — | |||||||
Preferred stock liability |
— | — | 861 | |||||||
Transfer of preferred stock liability to equity |
— | 2,653 | — | |||||||
Convertible preferred stock issuance costs included in accounts payable |
— | — | 52 | |||||||
Preferred stock warrants |
— | 175 | — | |||||||
Conversion of preferred stock warrant liability to common stock warrants |
— | 261 | — | |||||||
Issuance of common stock from the non-cash exercise of common stock warrants |
187 | — | — | |||||||
Conversion of convertible preferred stock to common stock |
— | 79,022 | — | |||||||
IPO costs included in accounts payable and accrued liabilities |
— | 129 | — | |||||||
Cash paid for interest on debt |
307 | 132 | — | |||||||
Transfer of equity-based compensation from liabilities to equity |
— | 259 | 193 | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
91
VERACYTE, INC.
Notes to Consolidated Financial Statements
1. Organization and Description of Business
Veracyte, Inc. (the "Company") was incorporated in the state of Delaware on August 15, 2006 as Calderome, Inc. Calderome operated as an incubator until early 2008. On March 4, 2008, the Company changed its name to Veracyte, Inc. Veracyte is a diagnostics company pioneering the field of molecular cytology to improve patient outcomes and lower healthcare costs. The Company specifically targets diseases that often require invasive procedures for an accurate diagnosis—diseases where many healthy patients undergo costly interventions that ultimately prove unnecessary. The Company improves the accuracy of diagnosis at an earlier stage of patient care by deriving clinically actionable genomic information from cytology samples.
The Company's first commercial solution, the Afirma® Thyroid FNA Analysis, includes as its centerpiece the Gene Expression Classifier ("GEC"). The GEC helps physicians reduce the number of unnecessary surgeries by employing a proprietary 142-gene signature to preoperatively determine whether thyroid nodules previously classified by cytopathology as indeterminate can be reclassified as benign. The comprehensive offering also includes cytopathology testing and the Afirma Malignancy Classifiers, launched in May 2014. The Company markets and sells Afrma through a co-promotion agreement with Genzyme Corporation, a subsidiary of Sanofi.
On September 16, 2014, the Company acquired Allegro Diagnostics Corp. ("Allegro") to accelerate its entry into pulmonology, the Company's second planned clinical area. Allegro was a privately-held company based in Maynard, Massachusetts, focused on the development of genomic tests to improve the preoperative diagnosis of lung cancer. See Note 4. The Company intends to enter the lung cancer diagnostics market by mid-2015 with the Percepta™ Bronchial Genomic Classifier ("Percepta"), a test designed to resolve diagnostic ambiguity among the approximately 250,000 patients in the United States who undergo bronchoscopy each year to assess potentially cancerous lung nodules.
The Company's operations are based in South San Francisco, California and Austin, Texas, and it operates in one segment in the United States.
Initial Public Offering
On November 4, 2013, the Company completed an initial public offering ("IPO") of its common stock. In connection with its IPO, the Company issued and sold 5,100,351 shares of common stock at a price to the public of $13.00 per share. As a result of the IPO, the Company received $59.2 million in net proceeds, after deducting underwriting discounts and commissions of $4.6 million and offering expenses of $2.5 million payable by the Company. In connection with the IPO, the Company's outstanding shares of convertible preferred stock were automatically converted into 14,997,312 shares of common stock.
2. Summary of Significant Accounting Policies
Basis of Presentation
The Company's consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States ("GAAP"). The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiary. All intercompany accounts and transactions have been eliminated in consolidation.
92
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Use of Estimates
The preparation of the consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Significant items subject to such estimates include: revenue recognition; contractual allowances; allowance for doubtful accounts; the useful lives of property and equipment; the recoverability of long-lived assets; the estimation of the fair value of intangible assets; the determination of fair value of the Company's common stock prior to the Company's IPO; stock options; preferred stock liability; income tax uncertainties, including a valuation allowance for deferred tax assets; and contingencies. The Company bases these estimates on historical and anticipated results, trends, and various other assumptions that the Company believes are reasonable under the circumstances, including assumptions as to future events. These estimates form the basis for making judgments about the carrying values of assets and liabilities and recorded revenue and expenses that are not readily apparent from other sources. Actual results could differ from those estimates and assumptions.
Liquidity
The Company has incurred net losses since its inception and expects to incur additional losses in 2015 and in future years. As of December 31, 2014, the Company had an accumulated deficit of $115.0 million. The Company may never achieve revenue sufficient to offset its expenses. The Company believes its cash and cash equivalents of $35.0 million as of December 31, 2014 and its revenue from the sale of Afirma in 2015 will be sufficient to meet its anticipated cash requirements for at least the next 12 months.
If the Company is not able to generate revenue to finance its cash requirements, the Company will need to finance future cash needs primarily through public or private equity offerings, debt financings, borrowings or strategic collaborations or licensing arrangements. If the Company is not able to secure additional funding when needed, on acceptable terms, it may have to delay, reduce the scope of or eliminate one or more research and development programs or selling and marketing initiatives which may have a material adverse effect on the Company's business, results of operations, financial condition and/or its ability to fund its scheduled obligations on a timely basis or at all.
Concentrations of Credit Risk and Other Risks and Uncertainties
The Company's cash and cash equivalents are deposited with one major financial institution in the United States, as required by the loan and security agreement discussed in Note 8. Deposits in this institution may exceed the amount of insurance provided on such deposits. The Company has not experienced any losses on its deposits of cash and cash equivalents.
Several of the components of the Company's sample collection kit and test reagents are obtained from single-source suppliers. If these single-source suppliers fail to satisfy the Company's requirements on a timely basis, it could suffer delays in being able to deliver its diagnostic solution, a possible loss of revenue, or incur higher costs, any of which could adversely affect its operating results.
The Company is also subject to credit risk from its accounts receivable related to its sales of Afirma. The Company generally does not perform evaluations of customers' financial condition and generally does not require collateral.
93
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Through December 31, 2014, all of the Company's revenue have been derived from the sale of Afirma. The Company's solution to date has been delivered primarily to physicians in the United States. The Company's third-party payers in excess of 10% of revenue and their related revenue as a percentage of total revenue were as follows:
| |
Year Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Medicare |
26 | % | 32 | % | 34 | % | ||||
Aetna |
11 | % | 9 | % | 13 | % | ||||
United Healthcare |
18 | % | 18 | % | 12 | % | ||||
| | | | | | | | | | | |
|
55 | % | 59 | % | 59 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
As the number of payers reimbursing for Afirma increases, the percentage of revenue derived from Medicare and other significant third-party payers has changed and will continue to change as a percentage of total revenue.
The Company's significant third-party payers and their related accounts receivable balance at December 31, 2014 and 2013 as a percentage of total accounts receivable are as follows:
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Medicare |
64 | % | 78 | % | |||
Aetna |
12 | % | — | ||||
United Healthcare |
14 | % | 3 | % | |||
No other third-party payer represented more than 10% of the Company's accounts receivable balances for these periods.
Cash Equivalents
Cash equivalents consist of short-term, highly liquid investments with original maturities of three months or less from the date of purchase. Cash equivalents consist primarily of amounts invested in a money market account primarily consisting of U.S. Treasury reserves.
Restricted Cash
The Company reserved $70,000 in cash as of December 31, 2014 to cover liabilities associated with the acquisition of Allegro as discussed in Note 4. This restricted cash is included in current assets on the Company's consolidated balance sheet.
The Company had long-term deposits of $118,000 as of December 31, 2014 and 2013, restricted from withdrawal and held by a bank in the form of collateral for letters of credit. The balance for each period consists of a letter of credit totaling $118,000 held as security for the lease of the Company's office space in South San Francisco, California. This restricted cash is included in long-term assets on the Company's consolidated balance sheets.
94
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Allowance for Doubtful Accounts
The Company estimates an allowance for doubtful accounts against its individual accounts receivable based on estimates of expected reimbursement consistent with historical payment experience in relation to the amounts billed. Bad debt expense is included in general and administrative expense on the Company's statements of operations and comprehensive loss. Accounts receivable are written off against the allowance when there is substantive evidence that the account will not be paid.
The balance of allowance for doubtful accounts as of December 31, 2014 and 2013, including charges to bad debt expense and write-offs, net of recoveries, was as follows:
| |
As of December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
| |
(In thousands) |
||||||
Beginning balance |
$ | 107 | $ | 222 | |||
Charged to expense |
54 | 109 | |||||
Write-offs, net of recoveries |
(77 | ) | (224 | ) | |||
| | | | | | | | |
Ending balance |
$ | 84 | $ | 107 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Supplies Inventory
Supplies inventory consists of test reagents and other consumables used in the sample collection kits and in cytopathology and GEC test processing and are valued at the lower of cost or market value. Cost is determined using actual costs on a first-in, first-out basis.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation and amortization. Depreciation is computed using the straight-line method over the estimated useful lives of the assets, generally between three and five years. Leasehold improvements are amortized using the straight-line method over the shorter of the estimated useful life of the asset or the term of the lease. Maintenance and repairs are charged to expense as incurred, and improvements and betterments are capitalized. When assets are retired or otherwise disposed of, the cost and accumulated depreciation are removed from the balance sheet and any resulting gain or loss is reflected in the statements of operations and comprehensive loss in the period realized.
Internal-use Software
The Company capitalizes costs incurred in the application development stage to design and implement the software used in the tracking and reporting of laboratory activity. Costs incurred in the development of application software are capitalized and amortized over an estimated useful life of three years on a straight line basis. The total cost, accumulated depreciation and net book value was $927,000, $330,000 and $597,000, respectively, as of December 31, 2014, and was $482,000, $195,000 and $287,000, respectively, as of December 31, 2013, and are included in property and equipment in the Company's consolidated balance sheets. During the years ended December 31, 2014 and 2013, the Company
95
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
capitalized $445,000 and $212,000, respectively, of software development costs. Amortization expense totaled $135,000, $108,000 and $47,000 in the years ended December 31, 2014, 2013 and 2012, respectively.
Business Combination
The Company accounts for acquisitions using the acquisition method of accounting which requires the recognition of tangible and identifiable intangible assets acquired and liabilities assumed at their estimated fair values as of the business combination date. The Company allocates any excess purchase price over the estimated fair value assigned to the net tangible and identifiable intangible assets acquired and liabilities assumed to goodwill. Transaction costs are expensed as incurred in general and administrative expenses. Results of operations and cash flows of acquired companies are included in the Company's operating results from the date of acquisition.
Goodwill
Goodwill, derived from the Company's acquisition of Allegro, is reviewed for impairment on an annual basis or more frequently if events or circumstances indicate that it may be impaired. The Company's goodwill evaluation is based on both qualitative and quantitative assessments regarding the fair value of goodwill relative to its carrying value. The Company has determined that it operates in a single segment and has a single reporting unit associated with the development and commercialization of diagnostic products. In the event the Company determines that it is more likely than not the carrying value of the reporting unit is higher than its fair value, quantitative testing is performed comparing recorded values to estimated fair values. If impairment is present, the impairment loss is measured as the excess of the recorded goodwill over its implied fair value. The Company performs its annual evaluation of goodwill during the fourth quarter of each fiscal year. There was no impairment for the year ended December 31, 2014.
Intangible Assets
The Company's intangible assets are comprised of acquired in-process research and development, or IPR&D. The fair value of IPR&D acquired through a business combination is capitalized as an indefinite-lived intangible asset until the completion or abandonment of the related research and development activities. IPR&D is tested for impairment annually or when events or circumstances indicate that the fair value may be below the carrying value of the asset. There was no impairment for the year ended December 31, 2014. If and when research and development is complete, the associated assets would then be amortized over their estimated useful lives.
Impairment of Long-lived Assets
The Company annually reviews long-lived and indefinite lived assets other than goodwill for impairment or whenever events or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. The Company recognizes an impairment loss when the total of estimated future undiscounted cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. Impairment, if any, would be assessed using discounted cash flows or other appropriate measures of fair value. There were no impairments for the years ended December 31, 2014 and 2013.
96
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Bonus Accruals
The Company accrues for liabilities under discretionary employee and executive bonus plans. These estimated compensation liabilities are based on progress against corporate objectives approved by the Board of Directors, compensation levels of eligible individuals, and target bonus percentage levels. The Board of Directors and the Compensation Committee of the Board of Directors review and evaluate the performance against these objectives and ultimately determine what discretionary payments are made. The Company accrued $1.1 million as of December 31, 2014 and 2013 for liabilities associated with these employee and executive bonus plans which are included in accrued liabilities in the Company's consolidated balance sheets.
Fair Value of Financial Instruments
The carrying amounts of certain financial instruments including cash and cash equivalents, accounts receivable, prepaid expenses and other current assets, accounts payable and accrued liabilities approximate fair value due to their relatively short maturities.
Revenue Recognition
The Company's revenue is generated from the provision of diagnostic services using the Afirma solution. The Company's service is completed upon the delivery of test results to the prescribing physician which triggers the billing for the service. The Company recognizes revenue related to billings for Medicare and commercial payers on an accrual basis, net of contractual adjustments, when a reasonable estimate of reimbursement can be made. These contractual adjustments represent the difference between the list price (the billing rate) and the reimbursement rate for each payer. Upon ultimate collection, the amount received from Medicare and commercial payers where reimbursement was estimated is compared to previous estimates and the contractual allowance is adjusted accordingly. Until a contract has been negotiated with a commercial payer or governmental program, the Afirma solution may or may not be covered by these entities' existing reimbursement policies. In addition, patients do not enter into direct agreements with the Company that commit them to pay any portion of the cost of the tests in the event that their insurance declines to reimburse the Company. In the absence of an agreement with the patient or other clearly enforceable legal right to demand payment from the patient, the related revenue is only recognized upon the earlier of payment notification, if applicable, or cash receipt.
For all services performed, the Company considers whether or not the following revenue recognition criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; and a reasonable estimate of reimbursement can be made.
Persuasive evidence of an arrangement exists and delivery is deemed to have occurred upon delivery of a patient report to the prescribing physician. The assessment of whether a reasonable estimate of reimbursement can be made requires significant judgment by management. Where management's judgment indicates a reasonable estimate of reimbursement can be made, revenue is recognized upon delivery of the patient report. Some patients have out-of-pocket costs for amounts not covered by their insurance carrier, and the Company may bill the patient directly for these amounts in the form of co-payments and co-insurance in accordance with their insurance carrier and health plans. Some payers may not cover the Company's GEC as ordered by the prescribing physician under their reimbursement policies. The Company pursues reimbursement from such patients on a case-by-case basis. In the absence
97
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
of contracted reimbursement coverage or the ability to reasonably estimate reimbursement, the Company recognizes revenue upon receipt of third-party payer notification of payment or when cash is received.
Revenue recognized when cash is received was $25.7 million, $14.6 million and $7.5 million for the years ended December 31, 2014, 2013 and 2012, respectively. Revenue recognized on an accrual basis was $12.5 million, $7.3 million and $4.1 million for the years ended December 31, 2014, 2013 and 2012, respectively.
Cost of Revenue
Cost of revenue is expensed as incurred and includes material and service costs, cytopathology testing services performed by a third-party pathology group, stock-based compensation expense, direct labor costs, equipment and infrastructure expenses associated with testing samples, shipping charges to transport samples, and allocated overhead including rent, information technology, equipment depreciation and utilities.
Research and Development
Research and development costs are charged to operations as incurred. Research and development costs include, but are not limited to, payroll and personnel-related expenses, stock-based compensation expense, prototype materials, laboratory supplies, consulting costs, costs associated with setting up and conducting clinical studies at domestic and international sites, and allocated overhead including rent, information technology, equipment depreciation and utilities.
Income Taxes
The Company accounts for income taxes under the liability method. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized.
The Company assesses all material positions taken in any income tax return, including all significant uncertain positions, in all tax years that are still subject to assessment or challenge by relevant taxing authorities. The Company's assessment of an uncertain tax position begins with the initial determination of the position's sustainability and is measured at the largest amount of benefit that is more-likely-than-not of being realized upon ultimate settlement. As of each balance sheet date, unresolved uncertain tax positions must be reassessed, and the Company will determine whether (i) the factors underlying the sustainability assertion have changed and (ii) the amount of the recognized tax benefit is still appropriate. The recognition and measurement of tax benefits requires significant judgment. Judgments concerning the recognition and measurement of a tax benefit may change as new information becomes available.
Stock-based Compensation
Stock-based compensation expense for equity instruments issued to employees is measured based on the grant-date fair value of the awards. The fair value of each employee stock option is estimated on the date of grant using the Black-Scholes option-pricing model. The Company recognizes compensation costs on a straight-line basis for all employee stock based compensation awards that are expected to vest over
98
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
the requisite service period of the awards, which is generally the awards' vesting period. Forfeitures are required to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
Equity awards issued to non-employees are valued using the Black-Scholes option-pricing model and are subject to re-measurement as the underlying equity awards vest.
Net Loss per Common Share
Basic net loss per common share is calculated by dividing net loss attributable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration of common stock equivalents. Diluted net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted-average number of common share equivalents outstanding for the period determined using the treasury stock method. Potentially dilutive securities consisting of options and warrants to purchase common stock are considered to be common stock equivalents and were excluded from the calculation of diluted net loss per common share because their effect would be anti-dilutive for all periods presented.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board ("FASB") issued Accounting Standards Update ("ASU") No. 2014-09, Revenue from Contracts with Customers, requiring an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers. The updated standard will replace most existing revenue recognition guidance in GAAP when it becomes effective and permits the use of either the retrospective or cumulative effect transition method. Early adoption is not permitted. The updated standard becomes effective for the Company in the first quarter of fiscal 2017. The Company has not yet selected a transition method and is currently evaluating the potential effect of the updated standard on its financial statements.
In August 2014, FASB issued Accounting Standards Update No. 2014-15, Presentation of Financial Statements Going Concern—Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern. The amendments require management to assess an entity's ability to continue as a going concern by incorporating and expanding upon certain principles that are currently in U.S. auditing standards. Specifically, the amendments: (1) provide a definition of the term substantial doubt; (2) require an evaluation every reporting period including interim periods; (3) provide principles for considering the mitigating effect of management's plans; (4) require certain disclosures when substantial doubt is alleviated as a result of consideration of management's plans; (5) require an express statement and other disclosures when substantial doubt is not alleviated; and (6) require an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). ASU 2014-15 will be effective for annual periods ending after December 15, 2016 and interim periods within annual periods beginning after December 15, 2016 with early adoption permitted. ASU 2014-15 will be effective for the Company beginning with its annual report for fiscal 2016 and interim periods thereafter. The Company has not yet determined the effect of the adoption of this standard on the Company's consolidated financial statements.
99
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
3. Net Loss Per Common Share
The following table presents the calculation of basic and diluted net loss per common share for the years ended December 31, 2014, 2013 and 2012 (in thousands, except share and per share amounts):
| |
Year Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Net loss |
$ | (29,373 | ) | $ | (25,580 | ) | $ | (18,649 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Shares used to compute net loss per common share, basic and diluted |
21,639,374 | 4,158,664 | 650,333 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per common share, basic and diluted |
$ | (1.36 | ) | $ | (6.15 | ) | $ | (28.68 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The following outstanding common stock equivalents have been excluded from diluted net loss per common share for the years ended December 31, 2014 and 2013 because their inclusion would be anti-dilutive:
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Shares of common stock subject to outstanding options |
3,249,469 | 2,359,287 | |||||
Shares of common stock issuable upon exercise of warrants |
— | 24,801 | |||||
| | | | | | | | |
Total shares of common stock equivalents |
3,249,469 | 2,384,088 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
4. Business Combination
On September 16, 2014, the Company acquired Allegro via a merger with Full Moon Acquisition, Inc., a wholly-owned subsidiary of the Company. Allegro was a privately-held company based in Maynard, Massachusetts, focused on the development of genomic tests to improve the preoperative diagnosis of lung cancer. Allegro merged with Full Moon, (the "Merger"), with Allegro surviving the Merger as a wholly-owned subsidiary of the Company. At the effective time of the Merger, each share of the common stock of Full Moon issued and outstanding immediately prior to the effective time of the Merger was automatically converted into one share of common stock of Allegro and represented the only outstanding common stock of Allegro at the effective time of the Merger; all previously issued and outstanding shares of common stock of Allegro were canceled. The Series A preferred stock of Allegro issued and outstanding immediately prior to the effective time of the Merger was canceled and automatically converted into the right to receive a total of 964,377 shares of the Company's common stock and $2.7 million in cash. Outstanding indebtedness of Allegro totaling $4.3 million was settled in cash by the Company on the effective date of the Merger. All outstanding stock options under Allegro's equity incentive plan were canceled.
The acquisition of Allegro is expected to accelerate the Company's molecular diagnostics business into the pulmonology diagnostics market. Allegro's lung cancer test is designed to help physicians determine which patients with lung nodules who have had a non-diagnostic bronchoscopy result are at low risk for cancer and can thus be safely monitored with CT scans rather than undergoing invasive procedures. The Company plans to launch its lung cancer test in the middle of 2015.
100
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
4. Business Combination (Continued)
The Merger was accounted for using the acquisition method of accounting with the Company treated as the accounting acquirer. The purchase price was allocated based on the estimated fair value of the assets acquired and liabilities assumed at the date of the acquisition.
The Company incurred approximately $0.5 million in acquisition-related costs related to the Merger, which primarily consisted of legal, accounting and valuation-related expenses. In addition, the Company incurred $1.2 million related to transaction bonuses and severance payments to former Allegro employees associated with the Merger. These expenses were recorded in general and administrative expense in the accompanying consolidated statements of operations and comprehensive loss. Total expenses and net loss associated with the acquired Allegro business in the Company's consolidated statements of operations and comprehensive loss were not separately identifiable due to the integration with the Company's operations.
The acquisition consideration was comprised of (in thousands):
Stock |
$ | 10,078 | ||
Cash |
2,725 | |||
Payment of outstanding indebtedness |
4,290 | |||
| | | | | |
Total acquisition consideration |
$ | 17,093 | ||
| | | | | |
| | | | | |
| | | | | |
The stock consideration of $10.1 million was determined based on the closing price of the Company's common stock on September 16, 2014 ($10.45 per share).
The fair value of the assets acquired and liabilities assumed at the closing date of the Merger are summarized below (in thousands):
Cash and cash equivalents |
$ | 29 | ||
Prepaid expenses |
3 | |||
Other current assets |
13 | |||
In-process research and development ("IPR&D") |
16,000 | |||
Goodwill |
1,057 | |||
Accrued liabilities |
(9 | ) | ||
| | | | | |
Total net assets acquired |
$ | 17,093 | ||
| | | | | |
| | | | | |
| | | | | |
The fair value of IPR&D was determined using the multi-period excess earnings method of the income approach, which estimates the economic benefits of the IPR&D over multiple time periods by identifying the cash flows associated with the use of the asset, based on forecasts prepared by management, and deducting a periodic charge reflecting a fair return for the use of contributory assets. The forecasted cash flows were discounted based on a discount rate of 18.5%. The discount rate represents the Company's weighted average return on assets and was benchmarked against the internal rate of return and cost of capital of guideline publicly traded companies. The fair value of the IPR&D was capitalized as of the closing date of the Merger and is subsequently accounted for as an indefinite-lived intangible asset, tested for impairment at least annually, until completion or abandonment of the associated research and development activities. Once complete, amortization of the acquired IPR&D asset into earnings will commence. The Company estimates that the acquired IPR&D asset will have a useful life of less than 20 years after taking into consideration expected use of the asset, legal or regulatory provisions that may limit or extend the life of the asset, as well as the effects of obsolescence and other economic factors.
101
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
4. Business Combination (Continued)
Goodwill, which represents the purchase price in excess of the fair value of net assets acquired, is not expected to be deductible for income tax purposes. This goodwill is reflective of the value derived from the expected acceleration of the Company's entry into the pulmonology market.
Pro Forma Financial Information (Unaudited)
The following pro forma financial information is based on the historical financial statements of the Company and presents the Company's results as if the Merger had occurred as of January 1, 2013 (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Revenue |
$ | 38,190 | $ | 21,884 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Net loss |
$ | (29,090 | ) | $ | (28,605 | ) | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The pro forma results present the combined historical results of operations with adjustments to reflect one-time charges including:
- •
- The reversal of costs related to transaction bonuses and other payments to employees and acquisition-related expenses directly related
to the Merger of $2.2 million for the year ended December 31, 2014; and
- •
- the elimination of interest expense related to Allegro indebtedness of $2.3 million and $4.5 million for the years ended December 31, 2014 and 2013, respectively.
The pro forma information presented does not purport to present what the actual results would have been had the Merger actually occurred on January 1, 2013, nor is the information intended to project results for any future period.
5. Balance Sheet Components
Property and Equipment, Net
Property and equipment consisted of the following (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Leasehold improvements |
$ | 788 | $ | 779 | |||
Laboratory equipment |
4,199 | 2,946 | |||||
Computer equipment |
875 | 645 | |||||
Software, including software developed for internal use |
1,353 | 901 | |||||
Furniture and fixtures |
197 | 189 | |||||
Construction-in-process |
739 | 307 | |||||
| | | | | | | | |
Total property and equipment, at cost |
8,151 | 5,767 | |||||
Accumulated depreciation and amortization |
(3,990 | ) | (2,815 | ) | |||
| | | | | | | | |
Total property and equipment, net |
$ | 4,161 | $ | 2,952 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
102
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
5. Balance Sheet Components (Continued)
Depreciation and amortization expense was $1,175,000, $999,000 and $706,000 for the years ended December 31, 2014, 2013 and 2012, respectively, and was recorded in the statements of operations and comprehensive loss as follows (in thousands):
| |
Year Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Cost of revenue |
$ | 677 | $ | 593 | $ | 401 | ||||
Research and development |
187 | 179 | 184 | |||||||
Selling and marketing |
101 | 54 | 46 | |||||||
General and administrative |
210 | 173 | 75 | |||||||
| | | | | | | | | | | |
Total depreciation and amortization expense |
$ | 1,175 | $ | 999 | $ | 706 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Accrued Liabilities
Accrued liabilities consisted of the following (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Accrued compensation expenses |
$ | 2,673 | $ | 1,962 | |||
Accrued Genzyme co-promotion fees |
3,309 | 4,915 | |||||
Accrued other |
1,869 | 717 | |||||
| | | | | | | | |
Total accrued liabilities |
$ | 7,851 | $ | 7,594 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
6. Fair Value Measurements
The Company records its financial assets and liabilities at fair value. The carrying amounts of certain financial instruments of the Company, including cash and cash equivalents, prepaid expenses and other current assets, accounts payable and accrued liabilities, approximate fair value due to their relatively short maturities. The carrying value of debt approximates its fair value because the interest rate approximates market rates that the Company could obtain for debt with similar terms. The accounting guidance for fair value provides a framework for measuring fair value, clarifies the definition of fair value, and expands disclosures regarding fair value measurements. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the reporting date. The accounting guidance establishes a three-tiered hierarchy, which prioritizes the inputs used in the valuation methodologies in measuring fair value as follows:
- •
- Level I: Inputs which include quoted prices in active markets for identical assets and liabilities.
- •
- Level II: Inputs other than Level I that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
103
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
6. Fair Value Measurements (Continued)
- •
- Level III: Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The fair value of the Company's financial assets, which consist only of money market funds, was $33.2 million and $70.0 million as of December 31, 2014 and December 31, 2013, respectively, and are Level I assets as described above.
The Company has no Level III liabilities as of December 31, 2014 and 2013. The following table sets forth the changes in the fair value of the Company's Level III financial liabilities, which consisted of a preferred stock liability during 2013, which were measured on a recurring basis (in thousands):
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Beginning balance |
$ | — | $ | 583 | |||
Change in fair value of preferred stock liability recorded as other expense, net |
— | 2,070 | |||||
Settlement of preferred stock liability |
— | (2,653 | ) | ||||
Fair value of preferred stock warrant liability |
— | 175 | |||||
Change in fair value of preferred stock warrant liability recorded as other expense, net |
— | 86 | |||||
Conversion of preferred stock warrant liability |
— | (261 | ) | ||||
| | | | | | | | |
Ending balance |
$ | — | $ | — | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
In November 2012, the Company recorded a preferred stock liability as investors received the right to purchase from the Company, on the same terms, additional shares of Series C convertible preferred stock, in a second tranche. As the investors held a majority of the board seats, the decision to complete the second tranche was deemed to be outside the control of the Company. The preferred stock liability was valued using the option-pricing method, which resulted in an initial fair value of $0.9 million for the Company's obligation to sell the convertible preferred stock. In June 2013, the Company settled the preferred stock liability upon completion of the sale of the second tranche of Series C convertible preferred stock. Immediately prior to settlement, the Company revalued the preferred stock liability to $2.7 million and recorded other expense of $2.1 million related to the change in value of the liability through that date. The preferred stock liability was valued using the option-pricing method with the following assumptions: 100% probability of success of the second tranche, fair value of Series C preferred stock of $2.39, a term of 0.003 years and expected volatility of 36.4%.
7. Commitments and Contingencies
Operating Leases
The Company leases its headquarters and South San Francisco laboratory facilities under a non-cancelable lease agreement that expires March 31, 2016. The Company provided security deposits in the form of irrevocable standby letters of credit secured with restricted cash deposits at the Company's primary bank. The Company deposited $118,000 in restricted cash accounts as collateral for the lease which is included in restricted cash in the Company's consolidated balance sheets as of December 31, 2014 and 2013.
104
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
7. Commitments and Contingencies (Continued)
The Company also leases laboratory space in Austin, Texas. The lease expires on July 31, 2018. The Company provided a cash security deposit of $75,000, which is included in other assets in the Company's consolidated balance sheets as of December 31, 2014 and 2013.
Future minimum lease payments under non-cancelable operating leases as of December 31, 2014 are as follows (in thousands):
Year Ending December 31,
|
Amounts | |||
|---|---|---|---|---|
2015 |
$ | 989 | ||
2016 |
413 | |||
2017 |
222 | |||
2018 |
130 | |||
Thereafter |
— | |||
| | | | | |
Total minimum lease payments |
$ | 1,754 | ||
| | | | | |
| | | | | |
| | | | | |
The Company recognizes rent expense on a straight-line basis over the non-cancelable lease period. Facilities rent expense was $852,000, $840,000 and $711,000 for the years ended December 31, 2014, 2013 and 2012, respectively.
Supplies Purchase Commitments
The Company had a non-cancelable purchase commitment with two suppliers to purchase a minimum quantity of supplies for approximately $715,000 at December 31, 2014, all of which is expected to be paid in early 2015.
Debt Obligations
See Note 8, Debt.
Contingencies
From time to time, the Company may be involved in legal proceedings arising in the ordinary course of business. The Company believes there is no litigation pending that could have, individually or in the aggregate, a material adverse effect on the financial position, results of operations or cash flows.
8. Debt
In June 2013, the Company entered into a loan and security agreement ("Original Loan") with a financial institution to fund its working capital and other general corporate needs. The Original Loan provided for term loans of up to $10.0 million in aggregate. The Company drew down $5.0 million in funds under the agreement in June 2013, and did not draw the remaining $5.0 million on or before the expiration date of March 31, 2014. The Company was required to repay the outstanding principal in 30 equal installments beginning 18 months after the date of the borrowing and was due in full in June 2017. The Original Loan had an interest rate of 6.06% per annum, carried prepayment penalties of 2.25% and 1.50% for prepayment within one and two years, respectively, and 0.75% thereafter.
In December 2014, the Company amended certain terms and conditions of the Original Loan ("Amended Loan"). The Amended Loan provides for term loans of up to $15.0 million in aggregate, in
105
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
8. Debt (Continued)
three tranches of $5.0 million each. The Company borrowed $5.0 million under the first tranche in December 2014 and used the funds for repayment of the $5.0 million in principal outstanding under the Original Loan, in a cashless transaction. In addition, the Company paid the accrued but unpaid interest of $14,000 due on the Original Loan and the related end-of-term payment of $110,000. The Amended Loan waived the prepayment premium of $75,000 under the Original Loan and reduced the end-of-term payment of $225,000 under the Original Loan to $110,000. The second $5.0 million tranche under the Amended Loan is available through December 31, 2015 and the Company may borrow the third $5.0 million tranche any time through June 30, 2016 after achieving the third tranche revenue milestone as defined in the Amended Loan.
The carrying value of the debt approximates its fair value because the interest rate approximates market rates that the Company could obtain for debt with similar terms. Under the Amended Loan borrowing, the Company is required to repay the outstanding principal in 24 equal installments beginning 24 months after the date of the borrowing and is due in full in December 2018. The first tranche of the Amended Loan bears interest at a rate of 5.00% per annum. The Amended Loan carries prepayment penalties of 2.00% and 1.00% for prepayment within one and two years, respectively, and no prepayment penalty thereafter. In connection with the Amended Loan, the Company paid approximately $45,000 in third-party fees.
The Amended Loan results in a debt modification under ASC 470-50, Modifications and Extinguishments, as the change in present value of the remaining cash flows associated with the Original Loan and Amended Loan are not substantial.
As of December 31, 2014 and 2013, the net debt obligation was as follows (in thousands):
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Debt and unpaid accrued end-of term payment |
$ | 5,003 | $ | 5,042 | |||
Unamorized note discount |
(80 | ) | (143 | ) | |||
| | | | | | | | |
Net debt obligation |
$ | 4,923 | $ | 4,899 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Future principal payments under the Amended Loan are as follows (in thousands):
Year ending December 31:
|
|
|||
|---|---|---|---|---|
2015 |
$ | — | ||
2016 |
— | |||
2017 |
2,437 | |||
2018 |
2,563 | |||
| | | | | |
Total |
$ | 5,000 | ||
| | | | | |
| | | | | |
| | | | | |
The obligation at December 31, 2014 includes an end-of-term payment of $237,500, representing 4.75% of the total outstanding principal balance, which accretes over the life of the loan as interest expense. As a result of the debt discount and the end-of-term payment, the effective interest rate for the loan differs from the contractual rate.
106
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
8. Debt (Continued)
Interest expense on the debt was as follows (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Nominal interest |
$ | 296 | $ | 158 | |||
Amortization of debt discount |
62 | 33 | |||||
End-of-term payment interest |
81 | 42 | |||||
| | | | | | | | |
Total |
$ | 439 | $ | 233 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Upon execution of the Original Loan, the Company issued the financial institution a warrant to purchase shares of Series C convertible preferred stock at $7.56 per share. At the time of issuance, the aggregate fair value of the warrant for the 24,801 shares exercisable under the warrant was $175,000. The fair value of the warrant was deducted from total proceeds, resulting in a debt discount to be amortized to interest expense over 48 months, through the maturity date of the Original Loan, using the effective interest rate method, and was recorded as a preferred stock warrant liability. The warrant was converted to a warrant to purchase the Company's common stock upon the completion of the Company's IPO. See Note 9.
The Company's obligations under the Amended Loan are secured by a security interest in substantially all of its assets, excluding its intellectual property and certain other assets. The Amended Loan contains customary conditions related to borrowing, events of default, and covenants, including covenants limiting the Company's ability to dispose of assets, undergo a change in control, merge with or acquire other entities, incur debt, incur liens, pay dividends or other distributions to holders of its capital stock, repurchase stock and make investments, in each case subject to certain exceptions. The Amended Loan also allows the lender to call the debt in the event there is a material adverse change in the Company's business or financial condition. The Company is required to be in compliance with a minimum liquidity or minimum revenue covenant. As of December 31, 2014, the Company was in compliance with the financial covenants.
9. Convertible Preferred Stock Warrant
In June 2013, in conjunction with the execution of the Original Loan, as discussed in Note 8, the Company issued to the lender a warrant to purchase up to 49,602 shares of Series C convertible preferred stock with an exercise price of $7.56 per share. Upon the draw-down of the $5.0 million term loan, the related warrant became exercisable for 24,801 shares. In November 2013, in connection with the Company's IPO, the warrant automatically became exercisable for 24,801 shares at an exercise price of $7.56 per share. The lender exercised the warrant with respect to 24,801 shares through a cashless exercise in March 2014, resulting in the issuance of 13,739 shares of the Company's common stock.
The fair value of the then currently exercisable portion of the warrant in the amount of $175,000 was recorded as a preferred stock warrant liability upon issuance and was subject to re-measurement at each reporting period up to the closing date of the IPO when the Series C preferred stock converted into common stock. The fair value of the warrant upon issuance was calculated using the Black-Scholes option-pricing model with the following assumptions: Series C preferred stock value of $2.40 per share, contractual term of 7.3 years, risk-free interest rate of 2.1%, expected volatility of 73.7%, and expected
107
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
9. Convertible Preferred Stock Warrant (Continued)
dividend yield of 0%. Just prior to the closing of the IPO, the fair value of the warrant was approximately $261,000, and was calculated using the Black-Scholes option-pricing model with the following assumptions: Series C preferred stock value of $13.14 per share, contractual term of 7.0 years, risk-free interest rate of 2.0%, expected volatility of 81.4%, and expected dividend yield of 0%. The change in the fair value of approximately $86,000 was reported as an expense for the year ended December 31, 2013 and was included in other income (expense), net, in the statements of operations and comprehensive loss. The warrant was converted into a warrant to purchase common stock upon the completion of the IPO in 2013, and was reclassified to additional paid- in-capital in the Company's consolidated balance sheet.
10. Convertible Preferred Stock
In November 2012, the Company recorded a preferred stock liability as the investors received the right to purchase from the Company, on the same terms, additional shares of Series C convertible preferred stock, in a second tranche. As the investors held a majority of the board seats, the decision to complete the second tranche was deemed to be outside the control of the Company. The preferred stock liability was valued using the option-pricing method with the following assumptions: 100% probability of success of the second tranche, fair value of Series C preferred stock of $1.78, a term of 0.67 years and expected volatility of 44%. This resulted in an initial fair value of $0.9 million for the Company's obligation to sell the convertible preferred stock. At December 31, 2012, the Company revalued the preferred stock liability to $0.6 million, and recorded the $0.3 million valuation decrease to other income (expense), net, in the Company's statements of operations and comprehensive loss. In June 2013, the Company revalued the preferred stock liability to $2.7 million and recorded the $2.1 million valuation increase to other income (expense), net, in the Company's consolidated statements of operations and comprehensive loss. In June 2013, the $2.7 million liability was settled upon the issuance of the second tranche of Series C convertible preferred stock and was reclassified to additional paid-in-capital in the Company's consolidated balance sheets.
On November 4, 2013, the Company completed its IPO. In connection with the IPO, the Company's 59,989,268 outstanding shares of convertible preferred stock were automatically converted into 14,997,312 shares of common stock.
11. Stockholders' Equity
Common Stock
The Company's Restated Certificate of Incorporation authorizes the Company to issue 125,000,000 shares of common stock with a par value of $0.001 per share. The holder of each share of common stock shall have one vote for each share of stock. The common stockholders are also entitled to receive dividends whenever funds and assets are legally available and when declared by the Board of Directors, subject to the prior rights of holders of all series of convertible preferred stock outstanding. No dividends have been declared as of December 31, 2014.
108
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
11. Stockholders' Equity (Continued)
As of December 31, 2014 and 2013, the Company had reserved shares of common stock for issuance as follows:
| |
December 31, 2014 |
December 31, 2013 |
|||||
|---|---|---|---|---|---|---|---|
Options issued and outstanding |
3,249,469 | 2,359,287 | |||||
Options available for grant under stock option plans |
1,341,252 | 1,787,802 | |||||
Common stock warrants issued and outstanding |
— | 24,801 | |||||
| | | | | | | | |
Total |
4,590,721 | 4,171,890 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Preferred Stock
The Company's Restated Certificate of Incorporation authorizes the Company to issue 5,000,000 shares of preferred stock with a par value of $0.001 per share. No shares were issued and outstanding at December 31, 2014 or 2013.
12. Stock Incentive Plans
Stock Option Plans
In February 15, 2008, the Company adopted the 2008 Stock Plan (the "2008 Plan"). The 2008 Plan provides for the granting of options to purchase common stock and common stock to employees, directors and consultants of the Company. The Company may grant incentive stock options ("ISOs"), non-statutory stock options ("NSOs") or restricted stock under the 2008 Plan. ISOs may only be granted to Company employees (including directors who are also considered employees). NSOs and restricted stock may be granted to Company employees, directors and consultants. Options may be granted for terms of up to ten years from the date of grant, as determined by the Board of Directors, provided however, that with respect to an ISO granted to a person who owns stock representing more than 10% of the voting power of all classes of stock of the Company, the term shall be for no more than five years from the date of grant. The exercise price of options granted must be at a price no less than 100% of the estimated fair value of the shares on the date of grant, as determined by the Board of Directors, provided however, that with respect to an ISO granted to an employee who at the time of grant of such option owns stock representing more than 10% of the voting power of all classes of stock of the Company, the exercise price shall not be less than 110% of the estimated fair value of the shares on the date of grant. Options granted to newly hired employees generally vest over four years (generally 25% after one year and monthly thereafter). Options granted to employees as part of annual bonus compensation are generally fully vested at the grant date.
On October 2, 2013, the Company adopted the 2013 Stock Incentive Plan (the "2013 Plan"). The 2013 Plan was subsequently approved by the Company's stockholders and became effective on November 4, 2013, immediately before the closing of the Company's IPO. Following the effectiveness of the 2013 Plan, no additional options will be granted under the 2008 Plan. An aggregate of 1,700,000 shares were initially reserved for issuance under the 2013 Plan. In addition, to the extent that any awards outstanding or subject to vesting restrictions under the 2008 Plan are subsequently forfeited or terminated for any reason before being exercised or settled, the shares of common stock reserved for issuance pursuant to such awards as of the closing of the IPO will become available for issuance under the 2013 Plan. The remaining shares available for grant under the 2008 Plan became available for issuance under the 2013 Plan upon the closing
109
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
12. Stock Incentive Plans (Continued)
of the IPO. On the first day of each year from 2014 to 2023, the 2013 Plan authorizes an annual increase of the lesser of 4% of outstanding shares on the last day of the immediately preceding fiscal year or a lesser amount as determined by the Company's Board of Directors. As of December 31, 2014, 1,341,252 shares were available for future issuance under the 2013 Plan.
Pursuant to the 2013 Plan, stock options, restricted shares, stock units, including restricted stock units and stock appreciation rights may be granted to employees, consultants, and outside directors of the Company. Options granted may be either ISOs or NSOs.
Stock options are governed by stock option agreements between the Company and recipients of stock options. ISOs and NSOs may be granted under the 2013 Plan at an exercise price of not less than 100% of the fair market value of the common stock on the date of grant, determined by the Compensation Committee of the Board of Directors. Options become exercisable and expire as determined by the Compensation Committee, provided that the term of ISOs may not exceed ten years from the date of grant. Stock option agreements may provide for accelerated exercisability in the event of an optionee's death, disability, or retirement or other events.
Any outside director who was not previously an employee and who first joins the Company's Board of Directors on or after the effective date of the 2013 Plan will be automatically granted an initial NSO to purchase 35,000 shares of common stock upon first becoming a member of the Board of Directors. Twenty-five percent of the shares subject to the initial option will vest and become exercisable on the first anniversary of the date of grant. The balance (i.e. the remaining 75%) will vest and become exercisable over three years in equal monthly installments. On the first business day after each regularly scheduled annual meeting of stockholders, each outside director who was not elected to the Board of Directors for the first time at such meeting and who will continue serving as a member of the Board of Directors thereafter will be automatically granted an option to purchase 10,000 shares of common stock, provided that the outside director has served on the Board of Directors for at least six months. Each annual option will vest and become exercisable on the first anniversary of the date of grant, or immediately prior to the next regular annual meeting of the Company's stockholders following the date of grant if the meeting occurs prior to the first anniversary date. The options granted to outside directors will have a per share exercise price equal to 100% of the fair market value of the underlying shares on the date of grant and will become fully vested in the event of a change of control. In addition, such options will terminate on the earlier of (i) the day before the 10th anniversary of the date of grant or (ii) the date 12 months after the termination of the outside director's service for any reason.
110
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
12. Stock Incentive Plans (Continued)
The following table summarizes activity under the Company's stock option plans (aggregate intrinsic value in thousands):
| |
Shares Available for Grant |
Stock Options Outstanding |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (Years) |
Aggregate Intrinsic Value |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Balance—December 31, 2011 |
474,961 | 1,429,737 | $ | 1.55 | 8.22 | $ | 1,221 | |||||||||
Additional options authorized |
743,100 | — | ||||||||||||||
Granted |
(931,944 | ) | 931,944 | 2.78 | ||||||||||||
Canceled |
61,269 | (61,269 | ) | 1.97 | ||||||||||||
Exercised |
— | (72,743 | ) | 1.05 | ||||||||||||
| | | | | | | | | | | | | | | | | |
Balance—December 31, 2012 |
347,386 | 2,227,669 | $ | 2.06 | 8.17 | $ | 4,311 | |||||||||
Additional options authorized |
1,950,000 | — | ||||||||||||||
Granted |
(695,029 | ) | 695,029 | 5.32 | ||||||||||||
Canceled |
185,445 | (185,445 | ) | 2.69 | ||||||||||||
Exercised |
— | (377,966 | ) | 1.46 | ||||||||||||
| | | | | | | | | | | | | | | | | |
Balance—December 31, 2013 |
1,787,802 | 2,359,287 | $ | 3.07 | 7.84 | $ | 26,964 | |||||||||
Additional options authorized |
845,732 | — | ||||||||||||||
Granted |
(1,488,492 | ) | 1,488,492 | 13.38 | ||||||||||||
Canceled |
196,210 | (196,210 | ) | 9.37 | ||||||||||||
Exercised |
— | (402,100 | ) | 1.68 | ||||||||||||
| | | | | | | | | | | | | | | | | |
Balance—December 31, 2014 |
1,341,252 | 3,249,469 | $ | 7.59 | 7.88 | $ | 12,400 | |||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Options vested and exercisable—December 31, 2014 |
1,370,944 | $ | 2.92 | 6.44 | $ | 9,287 | ||||||||||
Options vested and expected to vest—December 31, 2014 |
3,092,476 | $ | 7.32 | 7.81 | $ | 12,295 | ||||||||||
The aggregate intrinsic value was calculated as the difference between the exercise price of the options to purchase common stock and the fair market value of the Company's common stock, which was $9.66 per share as of December 31, 2014 and $14.50 per share as of December 31, 2013.
The weighted average fair value of options to purchase common stock granted was $9.08, $4.19 and $1.95 for the years ended December 31, 2014, 2013 and 2012, respectively.
The weighted average fair value of stock options vested was $3.07, $2.12 and $1.40 per share for the years ended December 31, 2014, 2013 and 2012, respectively. The aggregate estimated grant date fair value of employee options to purchase common stock vested during the years ended December 31, 2014 and 2013 was $1.6 million and $1.3 million, respectively.
The weighted-average fair value of stock options exercised was $1.18 and $0.97 for the years ended December 31, 2014 and 2013, respectively. The intrinsic value of stock options exercised was $3.2 million, $4.9 million and $0.2 million for the years ended December 31, 2014, 2013 and 2012, respectively.
111
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
12. Stock Incentive Plans (Continued)
Stock-based Compensation
The following table summarizes stock-based compensation expense related to stock options for the years ended December 31, 2014, 2013 and 2012, and are included in the consolidated statements of operations and comprehensive loss as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Cost of revenue |
$ | 51 | $ | 34 | $ | 26 | ||||
Research and development |
790 | 250 | 131 | |||||||
Selling and marketing |
707 | 169 | 111 | |||||||
General and administrative |
2,000 | 794 | 407 | |||||||
| | | | | | | | | | | |
Total stock-based compensation expense |
$ | 3,548 | $ | 1,247 | $ | 675 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
As of December 31, 2014, the Company had $9.9 million of unrecognized compensation expense related to unvested stock options, which is expected to be recognized over an estimated weighted-average period of 3.0 years.
The estimated grant-date fair value of employee stock options was calculated using the Black-Scholes option-pricing model, based on the following assumptions:
| |
Year Ended December 31, | |||||
|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||
Weighted-average volatility |
70.19 - 78.54% | 80.42 - 81.41% | 82.07 - 84.33% | |||
Weighted-average expected term (years) |
5.50 - 6.08 | 5.00 - 6.08 | 5.00 - 6.08 | |||
Risk-free interest rate |
1.66 - 2.04% | 0.88 - 2.11% | 0.65 - 1.19% | |||
Expected dividend yield |
— | — | — | |||
The estimated grant-date fair value of non-employee stock options was calculated using the Black-Scholes option-pricing model, based on the following assumptions:
| |
Year Ended December 31, | |||||
|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||
Weighted-average volatility |
73.20 - 74.48% | 77.86 - 78.14% | 81.14 - 82.11% | |||
Weighted-average expected term (years) |
8.75 - 10.00 | 7.72 - 9.75 | 8.23 - 9.93 | |||
Risk-free interest rate |
2.09 - 2.20% | 2.59 - 2.99% | 1.43 - 1.77% | |||
Expected dividend yield |
— | — | — | |||
Equity-based Compensation
In February 2013, the Company's Board of Directors authorized the grant of 100,498 fully vested stock options at a fair value of $2.59, determined using the Black-Scholes option-pricing valuation model, resulting in a $259,000 expense in the year ended December 31, 2012. Upon issuance of the options, the
112
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
12. Stock Incentive Plans (Continued)
accrued liability was reclassified into additional paid-in capital. For the years ended December 31, 2014 and 2013, the Company paid executive bonuses only in the form of cash.
13. Genzyme Co-Promotion Agreement
In January 2012, the Company and Genzyme Corporation ("Genzyme") executed a co-promotion agreement for the co-exclusive rights and license to promote and market the Company's Afirma thyroid diagnostic solution in the United States and in 40 named countries. In exchange, the Company received a $10.0 million upfront co-promotion fee from Genzyme in February 2012. Under the terms of the agreement, Genzyme will receive a percentage of U.S. cash receipts that the Company has received related to Afirma as co-promotion fees. The percentage was 50% in 2012, 40% from January 2013 through February 2014, and 32% beginning in February 2014. Genzyme's obligation to also spend up to $500,000 for qualifying clinical development activities in countries that require additional testing for approval expired in July 2014.
On August 12, 2014, the Company signed a binding Letter of Agreement with Genzyme to amend terms of the co-promotion agreement. On November 7, 2014, the Company signed an Amended and Restated U.S. Co-Promotion Agreement ("Amended Agreement") with Genzyme. Under the Amended Agreement, the co-promotion fees Genzyme will receive as a percentage of U.S. cash receipts were reduced from 32% to 15% beginning January 1, 2015. Through August 11, 2014, the Company amortized the $10.0 million upfront co-promotion fee over a four-year period, which was management's best estimate of the life of the agreement, in part because after that period either party could have terminated the agreement without penalty. Effective August 12, 2014, the Company extended the amortization period from January 2016 to June 2016, the modified earliest period either party could terminate the agreement without penalty. The Company accounted for the change in accounting estimate prospectively. Either party may terminate the agreement with six months prior notice, however, under the Amended Agreement, neither party can terminate the agreement for convenience prior to June 30, 2016. The agreement with Genzyme expires in 2027.
On November 7, 2014, the Company signed a binding Letter of Agreement (the "LOA") with Genzyme to negotiate a potential co-promotion agreement to promote Afirma GEC in countries other than the United States. The LOA provided an exclusive negotiation period to attempt to negotiate in good faith until December 12, 2014 a mutually agreeable co-promotion agreement ("Ex-U.S. Co-Promotion Agreement") pursuant to which the companies could co-promote the test in six initial countries. During such exclusive negotiation period, both parties were prohibited from negotiating with any third party with respect to those six countries. On December 12, 2014 the parties amended the LOA to extend the exclusive negotiation period from December 12, 2014 to January 31, 2015. During the extended period, the terms of the original co-promotion agreement with respect to countries outside the U.S. would remain in effect, including the payment to Genzyme of 32% of net revenues received by the Company on the test in countries outside the U.S. On January 30, 2015 the parties further amended the LOA to extend the exclusive negotiation period to February 14, 2015.
On February 13, 2015, the Company signed an Ex-U.S. Co-Promotion Agreement with Genzyme for the promotion of the Afirma GEC test with exclusivity in five countries outside the United States initially and in other countries agreed to from time to time. See Note 18, Subsequent Event.
The Company incurred $12.0 million, $8.6 million and $5.5 million in co-promotion expense in the years ended December 31, 2014, 2013 and 2012, respectively, which is included in selling and marketing
113
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
13. Genzyme Co-Promotion Agreement (Continued)
expenses in the consolidated statements of operations and comprehensive loss. The Company's outstanding obligation to Genzyme totaled $6.0 million and $6.7 million at December 31, 2014 and December 31, 2013, respectively. Of the $6.0 million obligation at December 31, 2014, $2.7 million is included in accounts payable and $3.3 million is included in accrued liabilities in the Company's consolidated balance sheets. Of the $6.7 million obligation at December 31, 2013, $1.8 million is included in accounts payable and $4.9 million is included in accrued liabilities in the Company's consolidated balance sheets.
The Company amortized $2.3 million, $2.5 million and $2.4 million of the $10.0 million up-front co-promotion fee in the years ended December 31, 2014, 2013 and 2012, respectively, which is reflected as a reduction to selling and marketing expenses in the consolidated statements of operations and comprehensive loss.
14. Thyroid Cytopathology Partners
In 2010, the Company entered into an arrangement with Pathology Resource Consultants, P.A. ("PRC") to set up and manage a specialized pathology practice to provide testing services to the Company. There is no direct monetary compensation from the Company to PRC as a result of this arrangement. The Company's service agreement is with the specialized pathology practice, Thyroid Cytopathology Partners, ("TCP"), and is effective through December 31, 2015, and thereafter automatically renews every year unless either party provides notice of intent not to renew at least 12 months prior to the end of the then-current term. Under the service agreement, the Company pays TCP based on a fixed price per test schedule, which is reviewed periodically for changes in market pricing. Subsequent to December 2012, an amendment to the service agreement allows TCP to use a portion of the Company's facility in Austin, Texas. The Company does not have an ownership interest in or provide any form of financial or other support to TCP.
The Company has concluded that TCP represents a variable interest entity and that the Company is not the primary beneficiary as it does not have the ability to direct the activities that most significantly impact TCP's economic performance. Therefore, the Company does not consolidate TCP. All amounts paid to TCP under the service agreement are expensed as incurred and included in cost of revenue in the consolidated statements of operations and comprehensive loss. The Company incurred $4.0 million, $3.2 million and $1.8 million in the years ended December 31, 2014, 2013 and 2012, respectively, in cytopathology testing and evaluation services expenses with TCP. The Company's outstanding obligations to TCP for cytopathology testing services were $1.1 million and $0.6 million as of December 31, 2014 and 2013, respectively, and are included in accounts payable in the Company's consolidated balance sheets.
TCP reimburses the Company for a proportionate share of the Company's rent and related operating expense costs for the leased facility. TCP's portion of rent and related operating expense costs for the shared space at the Austin, Texas facility was $86,000 and $49,000 for the years ended December 31, 2014 and 2013. TCP reimbursed the Company $59,000 in 2013 and the resulting excess payment of $10,000 was included in accounts payable in the Company's consolidated balance sheets at December 31, 2013.
15. Income Taxes
The Company generated a pretax loss of $29.4 million, $25.6 million and $18.6 million in the United States for the years ended December 31, 2014, 2013 and 2012, respectively. Since inception, the Company has not generated any pretax income or loss outside of the United States. The Company did not record a provision or benefit for income taxes during the years ended December 31, 2014, 2013 and 2012.
114
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
15. Income Taxes (Continued)
The Company follows FASB ASC No. 740, Income Taxes for the Computation and Presentation of its Tax Provision. The following table presents a reconciliation of the tax expense computed at the statutory federal rate and the Company's tax expense for the period presented (in thousands):
| |
Year Ended December, 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
U.S. federal taxes at statutory rate |
$ | (9,987 | ) | $ | (8,697 | ) | $ | (6,341 | ) | |
State tax (net of federal benefit) |
5 | 11 | (1,074 | ) | ||||||
Permanent differences |
64 | 790 | (23 | ) | ||||||
Incentive stock options |
672 | 355 | 284 | |||||||
Tax credits |
(461 | ) | (502 | ) | (113 | ) | ||||
Change in valuation allowance |
9,707 | 8,043 | 7,267 | |||||||
| | | | | | | | | | | |
Total |
$ | — | $ | — | $ | — | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax
115
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
15. Income Taxes (Continued)
purposes. Significant components of the Company's deferred tax assets and liabilities are as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Current deferred tax assets: |
||||||||||
Genzyme co-promotion agreement |
$ | 793 | $ | 1,005 | $ | 1,001 | ||||
Accruals and deferred rent |
1,819 | 599 | 148 | |||||||
| | | | | | | | | | | |
Gross deferred tax assets |
2,612 | 1,604 | 1,149 | |||||||
Valuation allowance |
(2,312 | ) | (1,603 | ) | (1,145 | ) | ||||
| | | | | | | | | | | |
Net deferred tax assets |
300 | 1 | 4 | |||||||
| | | | | | | | | | | |
Non-current deferred tax assets: |
||||||||||
Net operating loss carryforwards |
41,971 | 28,569 | 20,536 | |||||||
Research and development credits |
1,916 | 1,455 | 954 | |||||||
Stock-based compensation |
826 | 313 | 154 | |||||||
Genzyme co-promotion agreement |
202 | 787 | 2,048 | |||||||
Accruals, deferred rent and other |
1,562 | 106 | 9 | |||||||
| | | | | | | | | | | |
Gross deferred tax assets |
46,477 | 31,230 | 23,701 | |||||||
Valuation allowance |
(41,127 | ) | (31,216 | ) | (23,622 | ) | ||||
| | | | | | | | | | | |
Net deferred tax assets |
5,350 | 14 | 79 | |||||||
| | | | | | | | | | | |
Deferred tax liabilities: |
||||||||||
Property and equipment |
(60 | ) | (15 | ) | (83 | ) | ||||
In-process research and development |
(5,590 | ) | — | — | ||||||
| | | | | | | | | | | |
Gross deferred tax liabilities |
(5,650 | ) | (15 | ) | (83 | ) | ||||
| | | | | | | | | | | |
Net non-current deferred tax liabilities |
(300 | ) | (1 | ) | (4 | ) | ||||
| | | | | | | | | | | |
Total deferred tax assets |
$ | — | $ | — | $ | — | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The Company has established a full valuation allowance against its net deferred tax assets due to the uncertainty surrounding realization of such assets. The valuation allowance increased $10.6 million and $8.1 million during the years ended December 31, 2014 and 2013, respectively.
As of December 31, 2014, the Company had net operating loss carryforwards of approximately $113.6 million and $60.6 million available to reduce future taxable income, if any, for federal and state income tax purposes, respectively. Of these amounts, $0.2 million represent federal and state deductions from stock-based compensation, which will be recorded as an adjustment to additional paid-in capital when they reduce tax payable. The U.S. federal net operating loss carryforwards will begin to expire in 2026 while for state purposes, the net operating losses will begin to expire in 2018.
As of December 31, 2014, the Company had net credit carryforwards of approximately $2.2 million and $1.6 million available to reduce future taxable income, if any, for federal and state income tax purposes, respectively. The federal credit carryforwards begin to expire in 2028. California credits have no expiration date.
116
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
15. Income Taxes (Continued)
The Internal Revenue Code of 1986, as amended, imposes restrictions on the utilization of net operating losses and tax credits in the event of an "ownership change" of a corporation. Accordingly, a company's ability to use net operating losses and tax credits may be limited as prescribed under Internal Revenue Code Section 382 and 383 ("IRC Section 382"). Events which may cause limitations in the amount of the net operating losses or tax credits that the Company may use in any one year include, but are not limited to, a cumulative ownership change of more than 50% over a three-year period. Utilization of the federal and state net operating losses may be subject to substantial annual limitation due to the ownership change limitations provided by the IRC Section 382 rules and similar state provisions. In the event the Company has any changes in ownership, net operating losses and research and development credit carryovers could be limited and may expire unutilized.
Uncertain Tax Positions
As of December 31, 2014, the Company had unrecognized tax benefits of $1.6 million, none of which would currently affect the Company's effective tax rate if recognized due to the Company's deferred tax assets being fully offset by a valuation allowance. The Company does not anticipate that the amount of unrecognized tax benefits relating to tax positions existing at December 31, 2014 will significantly increase or decrease within the next 12 months.
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows (in thousands):
| |
Year Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | 2012 | |||||||
Unrecognized tax benefits, beginning of period |
$ | 727 | $ | 481 | $ | 341 | ||||
Gross increases—tax position in prior period |
548 | 68 | 67 | |||||||
Gross increases—current period tax positions |
296 | 178 | 73 | |||||||
| | | | | | | | | | | |
Unrecognized tax benefits, end of period |
$ | 1,571 | $ | 727 | $ | 481 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
It is the Company's policy to include penalties and interest expense related to income taxes as a component of other income (expense), net, and interest expense, respectively, as necessary. There was no interest expense or penalties related to unrecognized tax benefits recorded through December 31, 2014.
The Company's major tax jurisdictions are the United States and California. All of the Company's tax years will remain open for examination by the Federal and state tax authorities for three and four years, respectively, from the date of utilization of the net operating loss or research and development credit. The Company does not have any tax audits pending.
16. 401(k) Plan
The Company sponsors a 401(k) defined contribution plan covering all employees. There were no employer contributions to the plan in the years ended December 31, 2014 and 2013.
117
VERACYTE, INC.
Notes to Consolidated Financial Statements (Continued)
17. Selected Quarterly Financial Data (Unaudited)
The following table presents selected unaudited consolidated financial data for each of the eight quarters in the two-year period ended December 31, 2014. The Company believes this information reflects all recurring adjustments necessary to fairly present this information when read in conjunction with the Company's financial statements and the related notes. Net loss per common share, basic and diluted, for the four quarters of each fiscal year may not sum to the total for the fiscal year because of the different number of shares outstanding during each period. The results of operations for any quarter are not necessarily indicative of the results to be expected for any future period.
Quarter Ended
|
March 31 | June 30 | September 30 | December 31 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands, except share and per share data) |
||||||||||||
2014: |
|||||||||||||
Total revenues |
$ | 7,476 | $ | 8,677 | $ | 9,838 | $ | 12,199 | |||||
Net loss |
(6,674 | ) | (6,655 | ) | (7,902 | ) | (8,142 | ) | |||||
Net loss per common share, basic and diluted |
(0.32 | ) | (0.31 | ) | (0.37 | ) | (0.36 | ) | |||||
Shares used to compute net loss per common share, basic and diluted |
21,148,342 | 21,237,196 | 21,648,660 | 22,508,250 | |||||||||
2013: |
|||||||||||||
Total revenues |
$ | 4,384 | $ | 5,068 | $ | 5,594 | $ | 6,838 | |||||
Net loss |
(6,895 | ) | (6,490 | ) | (6,303 | ) | (5,892 | ) | |||||
Net loss per common share, basic and diluted |
(9.04 | ) | (7.53 | ) | (6.59 | ) | (0.42 | ) | |||||
Shares used to compute net loss per common share, basic and diluted |
763,021 | 861,839 | 955,890 | 13,944,239 | |||||||||
18. Subsequent Event (Unaudited)
On February 13, 2015, the Company signed an Ex-U.S. Co-Promotion Agreement with Genzyme for the promotion of the Afirma GEC test with exclusivity in five countries outside the United States initially and in other countries agreed to from time to time. The term of the agreement is January 1, 2015 and continuing until December 31, 2019 with extension of the agreement possible upon agreement of the parties. Country specific terms have been established under this agreement for Brazil and Singapore and a right of first negotiation has been established for Canada, the Netherlands and Italy. The Company will pay Genzyme 25% of net revenue in Brazil and Singapore over a five-year period commencing January 1, 2015. Beginning in the fourth year of the agreement, if the Company terminates the agreement for convenience, the Company may be required to pay a termination fee contingent on the number of GEC billable results generated.
118
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
On June 4, 2014, our Audit Committee of the Board of Directors approved the dismissal of PricewaterhouseCoopers LLP, or PwC, as our independent registered public accounting firm.
The reports of PwC on the financial statements for the fiscal years ended December 31, 2013 and 2012 did not contain an adverse opinion or a disclaimer of opinion, nor were such reports qualified or modified as to uncertainty, audit scope, or accounting principles, except that the PwC report on our financial statements as of December 31, 2011 and 2012 and for each of the two years in the period ended December 31, 2012, included in our registration statement on Form S-1 (File No. 333-191282) and related prospectus dated October 29, 2013, contained an emphasis of matter paragraph relating to our experience of recurring operating losses and negative cash flows from operations as described in Note 2 to such financial statements.
During the fiscal years ended December 31, 2013 and 2012, and through June 4, 2014, we did not have any disagreements with PwC on any matter of accounting principles or practices, financial statement disclosure or auditing scope, or procedure, which disagreements, if not resolved to the satisfaction of PwC, would have caused it to make reference to the subject matter of the disagreements in connection with its reports on the financial statements for such periods.
During the fiscal years ended December 31, 2013 and 2012, and through June 4, 2014, there were no "reportable events" as defined in Item 304(a)(1)(v) of Regulation S-K.
On June 4, 2014, the Audit Committee approved the appointment of Ernst & Young LLP, or EY, as our independent registered public accounting firm for the fiscal year ending December 31, 2014. During the fiscal years ended December 31, 2013 and 2012 and in the subsequent interim period through June 4, 2014, neither we nor anyone acting on our behalf has consulted with EY on any of the matters or events set forth in Item 304(a)(2) of Regulation S-K.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We maintain "disclosure controls and procedures," as such term is defined in Rule 13a-15(e) under the Securities Exchange Act of 1934, or Exchange Act, that are designed to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in Securities and Exchange Commission rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating our disclosure controls and procedures, management recognized that disclosure controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the disclosure controls and procedures are met. Our disclosure controls and procedures have been designed to meet reasonable assurance standards. Additionally, in designing disclosure controls and procedures, our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible disclosure controls and procedures. The design of any disclosure controls and procedures also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions.
Based on their evaluation as of the end of the period covered by this Annual Report on Form 10-K, our Chief Executive Officer and Chief Financial Officer have concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
119
Management's Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) under the Exchange Act. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of the effectiveness of internal control to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2014 using the criteria established in Internal Control—Integrated Framework ("1992 Framework") issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO").
Based on our evaluation using those criteria, our management has concluded that, as of December 31, 2014, our internal control over financial reporting was effective to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
This Annual Report on Form 10-K does not include an attestation report of our registered public accounting firm on our internal control over financial reporting due to an exemption established by the JOBS Act for "emerging growth companies."
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting (as such term is defined in Rule 13a-15(f) under the Exchange Act) identified in connection with the evaluation identified above that occurred during the quarter ended December 31, 2014 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
120
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item with respect to directors is incorporated by reference from the information under the caption "Election of Directors," contained in our Proxy Statement to be filed with the Securities and Exchange Commission no later than 120 days from the end of the Company's last fiscal year ended December 31, 2014 in connection with the solicitation of proxies for our 2015 Annual Meeting of Stockholders to be held on May 18, 2015, or Proxy Statement. Certain information required by this item concerning executive officers is set forth in Part I of this Report under the caption "Executive Officers of the Registrant" and is incorporated herein by reference.
There have been no material changes to the procedures by which stockholders may recommend nominees to our Board of Directors.
Item 405 of Regulation S-K calls for disclosure of any known late filing or failure by an insider to file a report required by Section 16(a) of the Exchange Act. This disclosure is contained in the section entitled "Section 16(a) Beneficial Ownership Reporting Compliance" in the Proxy Statement and is incorporated herein by reference.
We have adopted a Code of Business Conduct and Ethics that applies to all of our officers and employees, including our President and Chief Executive Officer, our Chief Financial Officer and other employees who perform financial or accounting functions. The Code of Business Conduct and Ethics sets forth the basic principles that guide the business conduct of our employees. We have also adopted a Senior Financial Officers' Code of Ethics that specifically applies to our President and Chief Executive Officer, our Chief Financial Officer, and key management employees. Stockholders may request a free copy of our Code of Business Conduct and Ethics and our Senior Financial Officers' Code of Ethics by contacting Veracyte, Inc., Attention: Chief Financial Officer, 7000 Shoreline Court, Suite 250, South San Francisco, California 94080.
To date, there have been no waivers under our Code of Business Conduct and Ethics or Senior Financial Officers' Code of Ethics. We intend to disclose future amendments to certain provisions of our Code of Business Conduct and Ethics or Senior Financial Officers' Code of Ethics or waivers of such Codes granted to executive officers and directors on our website at http://www.veracyte.com within four business days following the date of such amendment or waiver.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated by reference from the information under the captions "Election of Directors—Director Compensation" and "Executive Compensation" contained in the Proxy Statement for the 2015 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2014.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated by reference to the disclosure appearing under the headings "Security Ownership of Certain Beneficial Owners and Management" and "Executive Compensation—Equity Compensation Plan Information" contained in the Proxy Statement for the 2015 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2014.
121
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is incorporated by reference from the information under the caption "Election of Directors—Certain Relationships and Related Transactions" and "—Director Independence" contained in the Proxy Statement for the 2015 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2014.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item is incorporated by reference from the information under the caption "Ratification of the Appointment of Independent Registered Public Accounting Firm—Principal Accountant Fees and Services" contained in the Proxy Statement for the 2015 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2014.
122
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
- (a)
- Documents filed as part of this report
1. Financial Statements:
Reference is made to the Index to Financial Statements of Veracyte, Inc. included in Item 8 of Part II hereof.
2. Financial Statement Schedules
All schedules have been omitted because they are not required, not applicable, or the required information is included in the financial statements or notes thereto.
3. Exhibits
See Item 15(b) below. Each management contract or compensating plan or arrangement required to be filed has been identified.
- (b)
- Exhibits
| Exhibit Number |
Description | ||
|---|---|---|---|
| 2.1 | Agreement and Plan of Merger, dated September 4, 2014, by and among Veracyte, Inc., Full Moon Acquisition, Inc., Allegro Diagnostics Corp., Andrey Zarur, as the Stockholders' Agent, Kodiak Venture Partners III, L.P., Kodiak III Entrepreneurs Fund, L.P. and Catalyst Health Ventures L.P. (incorporated by reference to Exhibit 2.1 to the Registrant's Form 10-Q for the quarterly period ended September 30, 2014 filed November 13, 2014). | ||
| 3.1 | Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 to the Registrant's Current Report on Form 8-K filed November 8, 2013). | ||
| 3.2 | Amended and Restated Bylaws of the Registrant (incorporated by reference to Exhibit 3.2 to the Registrant's Current Report on Form 8-K filed November 8, 2013). | ||
| 4.1 | Form of Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 4.2 | Second Amended and Restated Investors Rights Agreement, dated November 6, 2012, between the Registrant and certain investors (incorporated by reference to Exhibit 4.2 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 4.3 | Amendment to Second Amended and Restated Investors Rights Agreement, dated June 14, 2013, between the Registrant and certain investors (incorporated by reference to Exhibit 4.3 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 10.1 | # | Form of Indemnification Agreement between the Registrant and its officers and directors (incorporated by reference to Exhibit 10.1 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
123
| Exhibit Number |
Description | ||
|---|---|---|---|
| 10.2 | # | 2008 Stock Plan and forms of agreements thereunder (incorporated by reference to Exhibit 10.2 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.3 | # | 2013 Stock Incentive Plan and forms of agreements thereunder (incorporated by reference to Exhibit 10.3 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.4 | Lease Agreement dated as of February 10, 2010 between ARE-San Francisco No 17, LLC and the Registrant (incorporated by reference to Exhibit 10.4 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 10.5 | First Amendment to Lease Agreement entered into as of July 11, 2012 between ARE-San Francisco No 17, LLC and the Registrant (incorporated by reference to Exhibit 10.5 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 10.6 | Lease Agreement between Riata Holdings, L.P., as landlord, and the Registrant, as tenant, dated November 28, 2012 (incorporated by reference to Exhibit 10.6 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 10.7 | First Amendment to Lease Agreement dated as of January 7, 2014 by and between Riata Holdings, L.P. and the Registrant (incorporated by reference to Exhibit 10.7 to the Registrant's Annual Report on Form 10-K for the year ended December 31, 2013 filed March 20, 2014). | ||
| 10.8 | † | Co-promotion Agreement dated as of January 18, 2012 between Genzyme Corporation and the Registrant (incorporated by reference to Exhibit 10.7 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.9 | Amendment to Co-promotion Agreement, effective April 9, 2013, between Genzyme Corporation and the Registrant (incorporated by reference to Exhibit 10.8 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 10.10 | Loan and Security Agreement dated as of June 26, 2013 between Silicon Valley Bank and the Registrant (incorporated by reference to Exhibit 10.9 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 10.11 | # | Employment Agreement, dated as of February 15, 2008, between Bonnie Anderson and the Registrant (incorporated by reference to Exhibit 10.10 to the Registrant's Registration Statement on Form S-1 (File No. 333- 191282), as amended, declared effective on October 29, 2013). | |
| 10.12 | # | Amendment to Bonnie Anderson Employment Agreement, dated as of December 22, 2008, between Bonnie Anderson and the Registrant (incorporated by reference to Exhibit 10.11 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
124
| Exhibit Number |
Description | ||
|---|---|---|---|
| 10.13 | # | Amendment No. 2 to Bonnie Anderson Employment Agreement, effective as of March 11, 2009, between Bonnie Anderson and the Registrant (incorporated by reference to Exhibit 10.12 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.14 | # | Change of Control and Severance Agreement, effective as of August 24, 2012, between Bonnie Anderson and the Registrant (incorporated by reference to Exhibit 10.13 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.15 | # | Change of Control and Severance Agreement, effective as of August 24, 2012, between Christopher Hall and the Registrant (incorporated by reference to Exhibit 10.14 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.16 | # | Change of Control and Severance Agreement, effective as of April 8, 2013, between Shelly Guyer and the Registrant (incorporated by reference to Exhibit 10.15 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.17 | Reserved. | ||
| 10.18 | # | Offer Letter dated as of April 8, 2013 with Shelly D. Guyer (incorporated by reference to Exhibit 10.17 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.19 | # | Offer Letter dated as of January 28, 2010 with Christopher M. Hall (incorporated by reference to Exhibit 10.18 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013. | |
| 10.20 | † | Pathology Services Agreement dated as of November 12, 2010 between Brazos Valley Pathology, P.A. D/B/A Reitpath and the Registrant (incorporated by reference to Exhibit 10.19 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | |
| 10.21 | Approval of the Registrant to the Assignment of the Pathology Services Agreement with Brazos Valley Pathology to Thyroid Cytopathology Partners, P.A. as of May 18, 2011 (incorporated by reference to Exhibit 10.20 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 10.22 | First Amendment to Pathology Services Agreement dated as of December 19, 2012 between Thyroid Cytopathology Partners, P.A. and the Registrant (incorporated by reference to Exhibit 10.21 to the Registrant's Registration Statement on Form S-1 (File No. 333-191282), as amended, declared effective on October 29, 2013). | ||
| 10.23 | Second Amendment to Pathology Services Agreement dated as of January 1, 2014 by and between the Registrant and Thyroid Cytopathology Partners, P.A. (incorporated by reference to Exhibit 10.23 to the Registrant's Annual Report on Form 10-K for the year ended December 31, 2013 filed March 20, 2014). | ||
| 10.24 | Consent and First Amendment to Loan and Security Agreement dated as of December 18, 2014 between Silicon Valley Bank and the Registrant (incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed December 18, 2014). | ||
125
| Exhibit Number |
Description | ||
|---|---|---|---|
| 10.25 | † | Amended and Restated U.S. Co-Promotion Agreement between Veracyte, Inc. and Genzyme Corporation (incorporated by reference to Exhibit 10.1 to the Registrant's Form 10-Q for the quarterly period ended September 30, 2014 filed November 13, 2014). | |
| 10.26 | *†† | Ex-U.S. Co-Promotion Agreement between Veracyte, Inc. and Genzyme Corporation. | |
| 23.1 | * | Consent of Ernst & Young LLP, Independent Registered Public Accounting Firm. | |
| 23.2 | * | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm. | |
| 24.1 | * | Power of Attorney (see the signature page of this Annual Report on Form 10-K). | |
| 31.1 | * | Principal Executive Officer's Certifications Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | * | Principal Financial Officer's Certifications Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | ** | Certification Pursuant to 18 U.S.C. § 1350 (Section 906 of Sarbanes-Oxley Act of 2002). | |
| 32.2 | ** | Certification Pursuant to 18 U.S.C. § 1350 (Section 906 of Sarbanes-Oxley Act of 2002). | |
| 101.INS | XBRL Instance Document | ||
| 101.SCH | XBRL Taxonomy Extension Schema | ||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase | ||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase | ||
| 101.LAB | XBRL Taxonomy Extension Label Linkbase | ||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase | ||
- †
- Confidential
treatment has been granted with respect to certain portions of this Exhibit.
- ††
- Confidential
treatment has been requested with respect to certain portions of this Exhibit.
- #
- Indicates
management contract or compensatory plan or arrangement.
- *
- Filed
herewith.
- **
- In accordance with Item 601(b)(32)(ii) of Regulation S-K and SEC Release No. 34-47986, the certifications furnished in Exhibits 32.1 and 32.2 hereto are deemed to accompany this Form 10-K and will not be deemed "filed" for purposes of Section 18 of the Securities Exchange Act of 1934 (the "Exchange Act") or deemed to be incorporated by reference into any filing under the Exchange Act or the Securities Act of 1933 except to the extent that the registrant specifically incorporates it by reference.
Copies of the above exhibits not contained herein are available to any stockholder, upon payment of a reasonable per page fee, upon written request to: Chief Financial Officer, Veracyte, Inc., 7000 Shoreline Court, Suite 250, South San Francisco, California 94080.
- (c)
- Financial Statement Schedules
Reference is made to Item 15(a) 2 above.
126
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| VERACYTE, INC. | ||||
By: |
/s/ BONNIE H. ANDERSON Bonnie H. Anderson President and Chief Executive Officer |
|||
Date: March 24, 2015
KNOW ALL PERSONS BY THESE PRESENT, that each person whose signature appears below constitutes and appoints Bonnie H. Anderson and Shelly D. Guyer, and each of them, his true and lawful attorneys-in-fact, each with full power of substitution, for him or her in any and all capacities, to sign any amendments to this annual report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact or their substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons, on behalf of the registrant on the dates and the capacities indicated.
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ BONNIE H. ANDERSON Bonnie H. Anderson |
President, Chief Executive Officer (Principal Executive Officer) and Director | March 24, 2015 | ||
/s/ SHELLY D. GUYER Shelly D. Guyer |
Chief Financial Officer (Principal Financial Officer) |
March 24, 2015 |
||
/s/ DUNCAN T. POWELL Duncan T. Powell |
Senior Vice President (Principal Accounting Officer) |
March 24, 2015 |
||
/s/ BRIAN G. ATWOOD Brian G. Atwood |
Chairman of Board of Directors |
March 24, 2015 |
||
/s/ JOHN L. BISHOP John L. Bishop |
Director |
March 24, 2015 |
127
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ BROOK H. BYERS Brook H. Byers |
Director | March 24, 2015 | ||
/s/ FRED E. COHEN, M.D., D.PHIL. Fred E. Cohen, M.D., D.Phil. |
Director |
March 24, 2015 |
||
/s/ KARIN EASTHAM Karin Eastham |
Director |
March 24, 2015 |
||
/s/ ROBERT S. EPSTEIN Robert S. Epstein |
Director |
March 24, 2015 |
||
/s/ EVAN JONES Evan Jones |
Director |
March 24, 2015 |
||
/s/ JESSE I. TREU, PH.D. Jesse I. Treu, Ph.D. |
Director |
March 24, 2015 |
128