Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Medisun Precision Medicine Ltd. | Financial_Report.xls |
| EX-32.2 - EXHIBIT 32.2 - Medisun Precision Medicine Ltd. | ex32_2apg.htm |
| EX-32.1 - EXHIBIT 32.1 - Medisun Precision Medicine Ltd. | ex32_1apg.htm |
| EX-31.2 - EXHIBIT 31.2 - Medisun Precision Medicine Ltd. | ex31_2apg.htm |
| EX-31.1 - EXHIBIT 31.1 - Medisun Precision Medicine Ltd. | ex31_1apg.htm |
| EX-10.13 - EXHIBIT 10.13 - Medisun Precision Medicine Ltd. | ex10_13apg.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
[X] ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal years ended December 31, 2014
or
[ ] TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from ______to ______
Commission File Number: 000-54907
ACCUREXA INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
47-2999657 |
|
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
|
|
|
|
113 Barksdale Newark, DE 19711 (Address of principal executive offices, including Zip Code)
| |
|
(415) 494-7850 (Registrant’s telephone number, including area code) | |
Securities registered under Section 12 (b) of the Exchange Act: None
Securities registered under Section 12 (g) of the Exchange Act: Common stock, $0.0001 par value (the “Common Stock”)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
[ ] Yes [X] No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
[ ] Yes [X] No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
[X] Yes [ ] No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
[X] Yes [ ] No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.
[X] Yes [ ] No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer [ ] Accelerated filer [ ] Non-accelerated filer [ ] (Do not check if a smaller reporting company) Smaller reporting company [X] |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
[ ] Yes [X] No
There were a total of 5,993,816 shares of the registrant’s common stock outstanding as of March 25, 2015.
DOCUMENTS INCORPORATED BY REFERENCE
None.
2
Table of Contents
|
|
|
Page | ||||
|
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS |
|
5 | ||||
|
|
|
|
|
| ||
|
PART I |
|
|
|
6 | ||
|
|
|
|
|
|
|
|
|
|
|
Item 1. |
|
Business |
|
6 |
|
|
|
|
|
|
|
|
|
|
|
Item 1A. |
|
Risk Factors |
|
13 |
|
|
|
|
|
|
|
|
|
|
|
Item 1B. |
|
Unresolved Staff comments |
|
28 |
|
|
|
|
|
|
|
|
|
|
|
Item 2. |
|
Properties |
|
28 |
|
|
|
|
|
|
|
|
|
|
|
Item 3. |
|
Legal Proceedings |
|
28 |
|
|
|
|
|
|
|
|
|
|
|
Item 4. |
|
Mine Safety Disclosures |
|
28 |
|
|
|
|
|
|
|
|
|
PART II |
|
|
|
29 | ||
|
|
|
|
|
|
|
|
|
|
|
Item 5. |
|
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
|
29 |
|
|
|
|
|
|
|
|
|
|
|
Item 6. |
|
Selected Financial Data |
|
31 |
|
|
|
|
|
|
|
|
|
|
|
Item 7. |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
32 |
|
|
|
|
|
|
|
|
|
|
|
Item 7A. |
|
Quantitative and Qualitative Disclosures About Market Risk |
|
35 |
|
|
|
|
|
|
|
|
|
|
|
Item 8. |
|
Financial Statements and Supplementary Data |
|
36 |
|
|
|
|
|
|
|
|
|
|
|
Item 9. |
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
|
37 |
|
|
|
|
|
|
|
|
|
|
|
Item 9A. |
|
Controls and Procedures |
|
37 |
|
|
|
|
|
|
|
|
|
|
|
Item 9B. |
|
Other Information |
|
39 |
|
|
|
|
|
|
|
|
|
PART III |
|
|
|
39 | ||
|
|
|
|
|
|
|
|
|
|
|
Item 10. |
|
Directors, Executive Officers of the Registrant |
|
39 |
|
|
|
|
|
|
|
|
|
|
|
Item 11. |
|
Executive Compensation |
|
41 |
|
|
|
|
|
|
|
|
|
|
|
Item 12. |
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
42 |
|
|
|
|
|
|
|
|
|
|
|
Item 13. |
|
Certain Relationships and Related Transactions, and Director Independence |
|
43 |
|
|
|
|
|
|
|
|
|
|
|
Item 14. |
|
Principal Accountant Fees and Services |
|
43 |
|
|
|
|
|
|
|
|
|
PART IV |
|
|
|
44 | ||
|
|
|
|
|
|
|
|
|
|
|
Item 15. |
|
Exhibits and Financial Statement Schedules |
|
44 |
|
|
|
|
|
|
|
|
|
SIGNATURES |
|
|
|
45 | ||
3
CAUTIONARY NOTE REGARDING FORWARD LOOKING STATEMENTS
This annual report on Form 10-K and other reports that we file with the SEC contain statements that are considered forward-looking statements. These forward-looking statements are not historical facts and can be identified by use of terminology such as believe, hope, may, anticipate, should, intend, plan, will, expect, estimate, project, positioned, strategy and similar expressions. You should be aware that these forward-looking statements are subject to risks and uncertainties that are beyond our control. For a discussion of these risks, you should read this entire annual report on Form 10-K carefully, especially the risks discussed under Risk Factors. The assumptions underlying the forward looking statements included in this annual report on Form 10-K do not guarantee our future performance, and actual results could differ from those contemplated by these forward looking statements. The assumptions used for purposes of the forward-looking statements specified in the following information represent estimates of future events and are subject to uncertainty as to possible changes in economic, legislative, industry, and other circumstances. As a result, the identification and interpretation of data and other information and their use in developing and selecting assumptions from and among alternatives require the exercise of judgment. To the extent that the assumed events do not occur, the outcome may vary substantially from anticipated or projected results. In the light of these risks and uncertainties, there can be no assurance that the results and events contemplated by the forward-looking statements contained in this annual report on Form 10-K will in fact transpire. You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of their dates. We do not undertake any obligation to update or revise any forward-looking statements.
We are an "emerging growth company" under applicable Securities and Exchange Commission rules and are subject to reduced public company reporting requirements. See "Implications of Being an Emerging Growth Company" and "Risk Factors".
4
PART I
ITEM 1. BUSINESS
Introduction
We are a development stage company. We were incorporated in Delaware on August 29, 2012. We are focused on developing and commercializing novel neurological therapies based on our proprietary BranchPoint device delivering therapeutics directly into specific regions of the brain. The BranchPoint device can deliver therapeutics through the radial deployment of a flexible delivery catheter to large and anatomically complex brain targets through a single initial brain penetration. The BranchPoint device was developed at the University of California, San Francisco (UCSF) with $1.8 million in funding from the California Institute for Regenerative Medicine (CIRM). It is based on a neurosurgical delivery platform that we have exclusively licensed from UCSF. It may enable new approaches to neurological therapy and be modified for the delivery of a broad range of novel therapeutics, such as stem cells to treat neurodegenerative diseases, chemotherapeutics to brain tumors and gene therapy vectors.
Furthermore, we have licensed a photoacoustic technology platform from the University of Arkansas for Medical Sciences (UAMS) that may allow the detection, capturing and targeted destruction of metastatic circulating tumor cells (CTCs). We have completed a proof-of-concept clinical trial and are seeking a strategic partner for further clinical development
We cannot assure you that we will be successful with our development activities. We have an office at 113 Barksdale, Newark, Delaware 19711 and our telephone number is (415) 494-7850.
Implications of Being an Emerging Growth Company
As a company with less than $1.0 billion in revenue, we qualify as an "emerging growth company" as defined in the Jumpstart our Business Startups Act of 2012, or the JOBS Act. An emerging growth company may take advantage of specified reduced reporting and other burdens that are otherwise applicable generally to public companies. These provisions include:
·
exemption from the auditor attestation requirement on the effectiveness of our internal controls over financial reporting;
·
reduced disclosure about the company's executive compensation arrangements; and
·
no requirements for non-binding advisory votes on executive compensation or golden parachute arrangements.
We may take advantage of these provisions until December 31, 2017 or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we have more than $1.0 billion in annual revenues, have more than $700 million in market value of our capital stock held by non-affiliates, or issue more than $1.0 billion of non-convertible debt over a three-year period. We may choose to take advantage of some but not all of these reduced burdens. To the extent that we take advantage of these reduced burdens, the information that we provide stockholders may be different than you might get from other public companies in which you hold equity interests.
Section 107(b) of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other companies.
Our BranchPoint Device
Our BranchPoint device can deliver therapeutics through the radial deployment of a flexible delivery catheter to large and anatomically complex brain targets through a single initial brain penetration. Clinicians can "tailor" therapeutic delivery to individual patient anatomy and specific disease targets, which may enhance the efficacy of therapies. The BranchPoint device also gives surgeons more precise control of the volume of therapeutics delivered and may ensure that the therapeutics delivered into the brain stay in the brain, avoiding the problem of reflux out to the brain surface. This modern and "easy to use" platform technology allows "real-time" monitoring of therapeutic delivery under interventional MRI guidance (iMRI), which can improve the accuracy of delivery, reduce the risk of complications, and increase patient safety. Currently available neurosurgical devices can deliver a straight, rigid needle to single brain locations. However, this basic strategy limits the size and "shape" of the brain treatment area. In order to deliver to larger and more complex brain targets, the surgeon needs to penetrate the brain multiple
5
times with such straight needles, which may increase the risk of bleeding and stroke, and may reduce the efficacy of therapeutics due to reflux, whereby therapeutics injected using straight needles can flow back out to the brain surface along the needle's path. The BranchPoint device was originally developed at UCSF with $1.8 million in funding from the California Institute for Regenerative Medicine (CIRM). It is based on a neurosurgical delivery platform that can enable new approaches to neurological therapy and be modified for the delivery of a broad range of novel therapeutics, such as stem cells to treat neurodegenerative diseases, chemotherapeutics to brain tumors and gene therapy vectors.
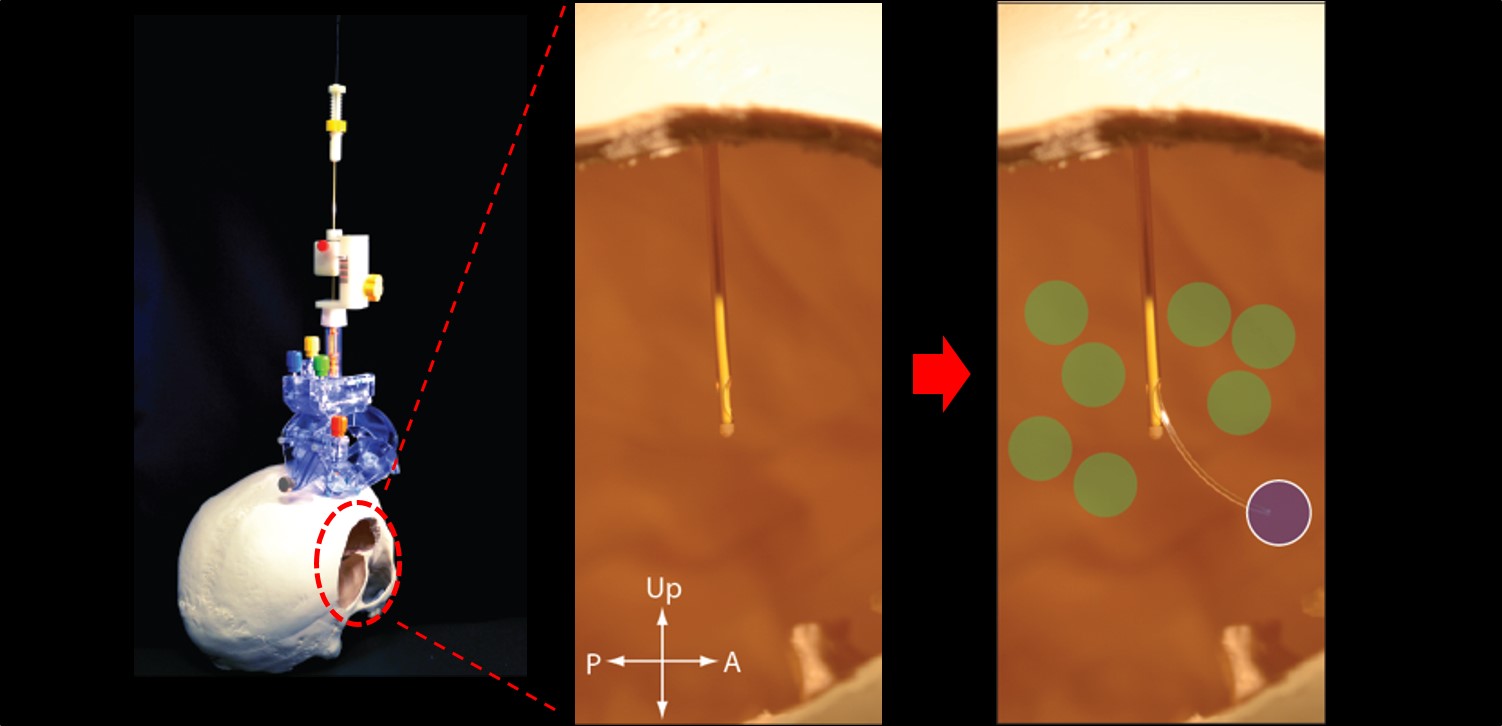
Fig. 1: The BranchPoint device delivers to large, complex targets through single penetration
Development Pipeline
Effective and safe delivery of therapeutics to the brain is often limited by the blood-brain barrier and lack of accurate, anatomic targeting of the intended site in the brain. Our BranchPoint device is a single platform that allows the delivery of multiple novel therapeutics under "real-time" MRI guidance (Fig. 2).
We are currently developing two programs in our pipeline.
1.
BranchPoint device for the delivery of therapeutics, such as stem cells or gene therapy vectors:
·
We plan to file a 510(k) submission of our BranchPoint device to the FDA (US Food and Drug Administration) in 2015.
·
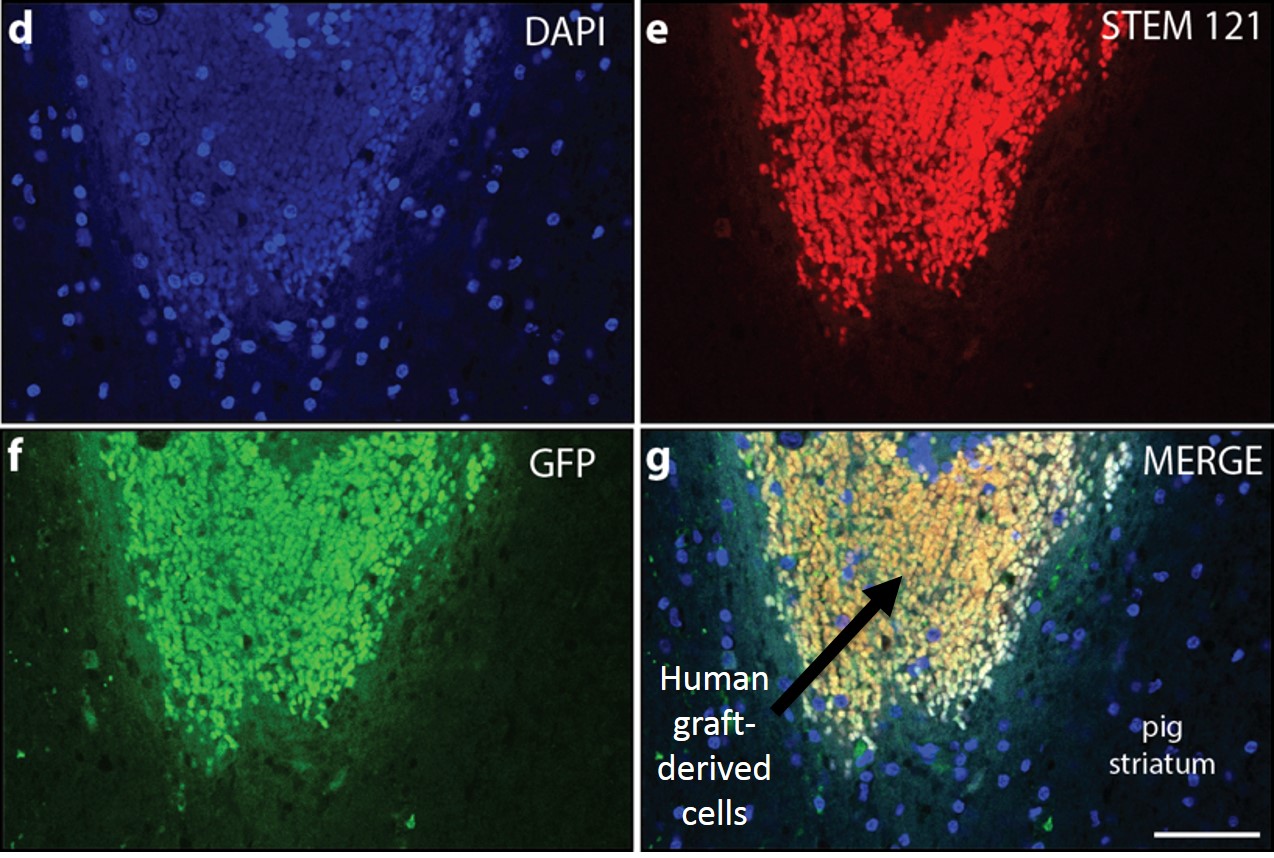
The feasibility of delivering human dopaminergic neural stem cells for the treatment of Parkinson's Disease has been demonstrated in animal studies (Fig. 3).
2.
ACX-31:
·
ACX-31 could deliver temozolomide, a chemotherapy drug, directly to the tumor site with our BranchPoint device. ACX-31 is contingent on BranchPoint's 510(k) approval.
·
temozolomide is a generic, approved chemotherapy drug that is indicated for the treatment of adult patients with newly diagnosed glioblastoma multiforme concomitantly with radiotherapy and then as maintenance treatment. Before temozolomide became generic, it generated US sales of $420 million and global sales of $910 million under its brand name Temodar in 2012.
·
Glioblastoma multiforme (GBM) is the most common and most aggressive malignant primary brain tumor in humans. Approximately 16,000 new patients are diagnosed with GBM in the US each year. Median survival without treatment is 4.5 months. With treatment, concomitant and adjuvant temozolomide chemotherapy with radiation significantly improves, from 12.1 months to 14.6 months, progression free survival and overall survival in GBM patients, as demonstrated in recent randomized clinical trials.
6

Fig. 2: The BranchPoint device may allow delivery of multiple therapeutics
|
Fig. 3: Immunohistochemistry analysis of dopaminergic neural stem cells grafted to the pig striatum: (d) 4'-6-diamidino-2-phenylindole nuclear stain (e) STEM 121-positive cells at the graft site (f) Green Fluorescent Protein-positive cells at the graft site (g) Merged images showing the border between graft-derived cells and pig striatum |
7
UCSF License Agreement
On September 16, 2014 (“Effective Date”), we entered into an exclusive license agreement (“UCSF License”) with the Regents of the University of California acting through its Office of Innovation, Technology, and Alliances, University of California San Francisco (“UCSF”) in regards to the exclusive licensing of a medical stereotactic device for the delivery of therapeutics to the human brain, characterized as “Microinjection Brain Catheter”, a.k.a. BranchPoint device ("Invention"). The invention was made in the course of research at UCSF by Drs. Daniel A. Lim, Matthew Silvestrini, and Tejal A. Desai, (collectively, the “Inventors”) and claimed in U.S. Patent Application No. PCT/US2013/052301, Microinjection Catheter; UC Case No. SF2012-063 (“Patent Rights”). The Invention was developed under funding from the California Institute for Regenerative Medicine ("CIRM") and sponsored in part by the National Institutes of Health. The UCSF License remains in effect from the Effective Date until expiration or abandonment of the Patent Rights.
Under the UCSF License, we are obligated to develop, manufacture, market and sell the invention, and have been granted the right to sublicense to third parties. Specifically, the UCSF License requires us to: (i) market the Invention for research use within three (3) months from the Effective Date; (ii) sell the Invention for research use within 12 months of the Effective Date; (iii) file and finalize any necessary regulatory documentation for FDA 510(k) approval within 6 months after the 510(k) application has been filed; (iv) market the Invention for clinical use within 6 months of receiving market approval from FDA or equivalent foreign regulatory agency; (v) sell the Invention for clinical use within 12 months of receiving market approval from FDA or equivalent foreign regulatory agency; (vi) within 1 year of the Effective Date, raise at least $750,000 in funding or revenue; (vii) market the Invention in the United States within 6 months of receiving approval from the FDA; and (viii) use commercially reasonable efforts to fill the market demand for the Invention following commencement of marketing. We are also required to pay UCSF 35% of our net sales or any sublicense royalty as defined therein, and a non-refundable license issue fee of $50,000. For a full description of further payment obligations and the UCSF License itself, the license agreement was filed as exhibit 10.1 to the Form 8-K as of September 17, 2014.
UCSF Patent
Our UCSF License includes U.S. Patent Application No. PCT/US2013/052301, Microinjection Catheter; UC Case No. SF2012-063 which is summarized as follows. The patent application can be accessed under the following link: http://www.google.com/patents/WO2014018871A1?cl=en
Background of the Invention:
Cell therapy has shown great promise in the treatment of a wide range of neurological diseases, including Parkinson's disease (PD), Huntington's disease, and stroke. To date, cell therapies have been delivered to the human brain with a stereotactically inserted straight cannula. While effective for small animal experimental models, straight cannula transplantation strategies present significant challenges when scaled-up for human therapy. The human brain is 800 to 2300 times larger than that of rodents used for preclinical research. With a straight cannula, cell delivery to the larger target volumes of human brain requires several independent brain penetrations. Some patients with PD have received up to 16 separate penetrations for transplantation to the putamen. Every transcortical brain penetration injures normal brain tissue and threatens hemorrhagic stroke.
In one approach to translational scale -up, very large numbers of cells can be delivered to a single location or along a short segment of the cannula tract. Unfortunately, the implantation of a large mass of cells within a confined location can severely impair graft viability, resulting in necrosis at the center of the transplant.
Another approach has been to insert a large host catheter, which is comprised of a number of internal passages, or lumens for the advancement of micro-catheters. These internal passages within the host catheter exit at specified distal orifice locations around the distal end to allow the delivery of a media to a desired target area. Using this approach, the host catheter is inserted into the center of the desired target, or delivery area in the patient. Then, the micro-catheters are inserted into the various lumens, where multiple doses can be delivered to each of the distal orifice locations along the elongate member. This method allows a larger target area to be covered without the need for multiple cranial penetrations. The introduction of a relatively large host catheter displaces a larger amount of tissue and the use of multiple micro catheters makes the ability to deliver a metered injection more difficult due to their variable lengths. A problem with at least some prior systems is that the delivered media may not stay at the desired delivery site. In a phenomenon called reflux, a portion of the media may flow back up the penetration shaft, significantly reducing the amount of media that remains at the treatment site. Larger injection volumes worsen the reflux of infused materials along the penetration tract making cell dosing unpredictable in terms of numbers as well as final graft location.
8
In most clinical trials, a syringe is used to deliver cells through the inserted cannula. Unless the syringe is kept in constant motion, the cells naturally sediment to the most dependent location, usually the end attached to cannula. Thus, the first partial injection volume from a syringe may contain far more cells than those dispensed later, further contributing to unpredictable variability of cell dosing. The use of a syringe having a larger diameter than the catheter main lumen may make it difficult to control the volume of each dose, and may subject the cells to shear and other mechanical forces that result in the decrease of cell viability for cell transplantation.
Brief Summary of the Invention:
An insertion device for delivering media inside a patient includes an outer guide tube having a closed end configured for insertion into patient tissue. The outer guide tube defines a side port in a wall of the guide tube near the closed end. The insertion device also includes an inner guide tube nested within the outer guide tube and movable axially within the outer guide tube. The inner guide tube includes a deflector at an end within the outer guide tube. The device also includes a catheter nested within the inner guide tube and movable axially within the inner guide tube. The catheter has a dispensing end that defines one more dispensing holes for dispensing the media. The deflector of the inner guide tube is positionable in relation to the opening of the outer guide tube such that it deflects the dispensing end of the catheter outward through the opening in the outer guide tube when the catheter is advanced axially within the inner guide tube.
UAMS License
On December 15, 2012 (“Effective Date”), we executed an Exclusive License Agreement (“UAMS License”) with the Board of Trustees of the University of Arkansas (“UofA”) acting on behalf of the University of Arkansas for Medical Sciences (“UAMS”) to commercially develop certain patents and patent applications owned by UAMS throughout the world for the life of the patents. Our UAMS License includes three patents (i) U.S. Patent Application Serial No. 12/334,217, Device and Method for In Vivo Flow Cytometry Using the Detection of Photoacoustic Waves; UAMS ID No. 2008-16; (ii) U.S. Patent Application Ser. No.: 12/945,576 Device and Method for In Vivo Noninvasive Magnetic Manipulation of Circulating Objects in BioFlows; UAMS ID No. 2008-16 CIP; (iii) U.S. Patent Application Ser. No.: 13/253,767 Device and Method for In Vivo Detection of Clots Within Circulatory Vessels; UAMS ID No. 2008-16 CIP2. Our three licensed patents cover a method for in vivo flow cytometry using the detection of photoacoustic waves by which targeted objects moving in the bloodstream absorb the energy level of a pulsed laser, and then either emit an acoustic wave that can be detected by an ultrasound transducer, or can be destroyed if a sufficient laser energy level is absorbed. The license agreement was filed as exhibit 10.2 to the Form 10 as of February 27, 2013.
As previously disclosed in our filing on Form 8-K on February 24, 2014, in the normal course of events, we confirmed, on or about February 3, 2014, that a portion of one of the three licensed patent applications, U.S. Patent Application Serial No. 12/334,217, Device and Method for In Vivo Flow Cytometry Using the Detection of Photoacoustic Waves; UAMS ID No. 2008-16, had been rejected by the United States Patent and Trademark Office and U.S. Patent Application Serial No. 12/334,217 was then abandoned prior to the date of our UAMS License. The basis for the rejection was primarily that the rejected portion of the application had previously been published in a scientific journal by the inventor Prof. Vladimir Zharov, an employee of the UAMS, prior to the filing of the application and therefore was not patentable.
We completed a proof-of-concept clinical trial, titled “In vivo real-time detection of circulating melanoma cells” which was conducted at UAMS under the direction of Prof. Laura Hutchins, Director, Division of Hematology/Oncology as the principal investigator and Prof. Vladimir Zharov, Director of the Philips Classic Laser and Nanomedicine Laboratories as sub-investigator, both of whom are employees of UAMS. The clinical trial examined 10 healthy control subjects in order to characterize the photoacoustic baseline signals and validated the sensitivity of CTC (circulating tumor cells) detection in 10 patients who had melanoma. We are currently seeking a strategic partner for further clinical development.
Device Approval Process
Our contemplated products will be regulated as medical devices and will be subject to extensive regulation by the FDA and other regulatory authorities in the US. The Food, Drug, and Cosmetic Act (“FD&C Act”) and other federal and state statutes and regulations govern the research, design, development, preclinical and clinical testing, manufacturing, safety, approval or clearance, labeling, packaging, storage, record keeping, servicing, promotion, import and export, and distribution of medical devices.
Unless an exemption applies, each medical device we wish to commercially distribute in the U.S. will require either prior premarket notification, or 510(k) clearance, or premarket approval (PMA) from the FDA. The FDA classifies medical devices
9
into one of three classes. Devices requiring fewer controls because they are deemed to pose lower risk are placed in Class I or II. Class I devices are subject to general controls such as labeling, premarket notification, and adherence to the FDA’s Quality System Regulation (a set of current good manufacturing practice requirements put forth by the FDA which govern the methods used in, and the facilities and controls used for, the design, manufacture, packaging, labeling, storage, installation and servicing of finished devices) (“QSR”). Class II devices are subject to special controls such as performance standards, post-market surveillance, FDA guidelines, as well as general controls. Some Class I and Class II devices are exempted by regulation from the premarket notification, or 510(k), clearance requirement or the requirement of compliance with certain provisions of the QSR. Devices are placed in Class III, which requires approval of a PMA application, if insufficient information exists to determine that the application of general controls or special controls are sufficient to provide reasonable assurance of safety and effectiveness, or they are life-sustaining, life-supporting or implantable devices, or the FDA deems these devices to be “not substantially equivalent” either to a previously 510(k) cleared device or to a “pre-amendment” Class III device in commercial distribution before May 28, 1976, for which PMA applications have not been required. A PMA application must be supported by valid scientific evidence, which typically requires extensive data, including technical, pre-clinical, clinical, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device. A PMA application must include, among other things, a complete description of the device and its components, a detailed description of the methods, facilities and controls used to manufacture the device, and proposed labeling. A PMA application also must be accompanied by a user fee, unless exempt. For example, the FDA does not require the submission of a user fee for a small business’s first PMA. After a PMA application is submitted and found to be sufficiently complete, the FDA begins an in-depth review of the submitted information. During this review period, the FDA may request additional information, or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA maybe convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA generally will conduct a pre-approval inspection of the manufacturing facility to ensure compliance with the QSR, which requires manufacturers to follow design, testing, control, documentation, and other quality assurance procedures.
The FDA can delay, limit, or deny approval of a PMA application for many reasons, including:
·
the product may not be safe or effective to the FDA’s satisfaction;
·
the data from our pre-clinical studies and clinical trials may be insufficient to support approval;
·
the manufacturing process or facilities we use may not meet applicable requirements; and
·
changes in FDA approval policies or adoption of new regulations may require additional data.
If the FDA evaluations of both the PMA application and the manufacturing facilities are favorable, the FDA will either issue an approval letter, or approvable letter, which usually contains a number of conditions which must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of the device for certain indications. If the FDA’s evaluation of the PMA or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA may also determine that additional clinical trials are necessary, in which case the PMA approval may be delayed while the trials are conducted and the data acquired is submitted in an amendment to the PMA. Even with additional trials, the FDA may not approve the PMA application. The PMA process can be expensive, uncertain, and lengthy and a number of devices for which FDA approval has been sought by other companies have never been approved for marketing.
New PMA applications or PMA supplements may be required for modifications to the manufacturing process, labeling and device specifications, materials or design of a device that is approved through the PMA process. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA application, and may not require as extensive clinical data or the convening of an advisory panel.
Clinical trials are almost always required to support a PMA application, and are sometimes required for a 510(k) clearance. These trials generally require submission of an application for an Investigational Device Exemption (“IDE”) to the FDA. If a trial is considered a “Non-Significant Risk” (“NSR”) study subject to abbreviated IDE regulations, a formal IDE submission is not required by the FDA. An IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and eligible for more abbreviated IDE requirements. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent form are approved by appropriate institutional review boards (“IRBs”) at the clinical trial sites. The FDA’s approval of an IDE allows clinical testing to go forward, but does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and effectiveness, even if the
10
trial meets its intended success criteria. All clinical trials must be conducted in accordance with the FDA’s IDE regulations that govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. . Required records and reports are subject to inspection by the FDA. The results of clinical testing may be unfavorable or, even if the intended safety and effectiveness success criteria are achieved, may not be considered sufficient for the FDA to grant approval or clearance of a product.
Although we believe our clinical trials will provide favorable data to support our PMA application, upon evaluation the FDA may conclude differently. Delays in receipt of or failure to receive FDA approval, the withdrawal of previously received approvals, or failure to comply with existing or future regulatory requirements would have a material adverse effect on our business, financial condition, and results of operations. Even if granted, the approvals may include significant limitations on the intended use and indications for use for which our products may be marketed.
After a device is approved or cleared and placed in commercial distribution, numerous regulatory requirements apply. These include:
·
establishing registration and device listing;
·
implementing QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures;
·
labeling regulations, which prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling;
·
medical device reporting regulations, which require that manufacturers report to the FDA if a device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; and
·
corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FD&C Act that may present a risk to health.
Also, the FDA may require us to conduct post market studies or order us to establish and maintain a system for tracking our products through the chain of distribution to the patient level. The FDA enforces regulatory requirements by conducting periodic, unannounced inspections and market surveillance.
Failure to comply with applicable regulatory requirements, including those applicable to the conduct of our clinical trials, can result in enforcement action by the FDA, which may lead to any of the following sanctions:
·
warning letters;
·
fines and civil penalties;
·
unanticipated expenditures;
·
delays in approving or refusal to approve our applications, including supplements;
·
withdrawal of FDA approval;
·
product recall or seizure;
·
interruption of production;
·
operating restrictions;
·
injunctions; and
·
criminal prosecution.
Our products will need to be manufactured in compliance with current Good Manufacturing Practices (“cGMP”) requirements set forth in the QSR. The QSR requires a quality system for the design, manufacture, packaging, labeling, storage, installation and servicing of marketed devices, and includes extensive requirements with respect to quality management and organization, device design, equipment, purchase and handling of components, production and process controls, packaging and labeling controls, device evaluation, distribution, installation, complaint handling, servicing, and record keeping. The FDA enforces the QSR through periodic unannounced inspections If the FDA believes that we are not in compliance with QSR, it can shut down the manufacturing operations, require recall of our products, refuse to approve new marketing applications, institute legal proceedings to detain or seize products, enjoin future violations, or assess civil and criminal penalties against us or our officers or other employees. Any such action by the FDA would have a material adverse effect on our business. We cannot assure you that we will be able to comply with all applicable FDA regulations.
11
Non-FDA Government Regulation
The advertising of our products will be subject to both FDA and Federal Trade Commission regulations. In addition, the sale and marketing of our products will be subject to a complex system of federal and state laws and regulations intended to deter, detect, and respond to fraud and abuse in the healthcare system. These laws and regulations restrict and may prohibit pricing, discounting, commissions and other commercial practices that may be typical outside of the healthcare business. In particular, anti-kickback and self-referral laws and regulations will limit our flexibility in crafting promotional programs and other financial arrangements in connection with the sale of our products and related services, especially with respect to physicians seeking reimbursement through Medicare or Medicaid. These federal laws include, by way of example, the following:
·
the anti-kickback statute prohibits certain business practices and relationships that might affect the provision and cost of healthcare services reimbursable under Medicare, Medicaid and other federal healthcare programs, including the payment or receipt of remuneration for the referral of patients whose care will be paid by Medicare or other federal healthcare programs;
·
the physician self-referral prohibition, commonly referred to as the Stark Law, which prohibits referrals by physicians of Medicare or Medicaid patients to providers of a broad range of designated healthcare services in which the physicians or their immediate family members have ownership interests or with which they have certain other financial arrangements;
·
the anti-inducement law, which prohibits providers from offering anything to a Medicare or Medicaid beneficiary to induce that beneficiary to use items or services covered by either program;
·
the Civil False Claims Act, which prohibits any person from knowingly presenting or causing to be presented false or fraudulent claims for payment by the federal government, including the Medicare and Medicaid programs; and
·
the Civil Monetary Penalties Law, which authorizes the US Department of Health and Human Services (“HHS”) to impose civil penalties administratively for fraudulent or abusive acts.
Sanctions for violating these federal laws include criminal and civil penalties that range from punitive sanctions, damage assessments, money penalties, imprisonment, denial of Medicare and Medicaid payments, or exclusion from the Medicare and Medicaid programs, or both. These laws also impose an affirmative duty on those receiving Medicare or Medicaid funding to ensure that they do not employ or contract with persons excluded from the Medicare and other government programs.
Many states have adopted or are considering legislative proposals similar to the federal fraud and abuse laws, some of which extend beyond the Medicare and Medicaid programs to prohibit the payment or receipt of remuneration for the referral of patients and physician self-referrals regardless of whether the service was reimbursed by Medicare or Medicaid. Many states have also adopted or are considering legislative proposals to increase patient protections, such as limiting the use and disclosure of patient-specific health information. These state laws typically impose criminal and civil penalties similar to the federal laws.
In the ordinary course of their business, medical device manufacturers and suppliers have been and are subject regularly to inquiries, investigations and audits by federal and state agencies that oversee these laws and regulations. Recent federal and state legislation has greatly increased funding for investigations and enforcement actions, which have increased dramatically over the past several years. This trend is expected to continue. Private enforcement of healthcare fraud also has increased, due in large part to amendments to the Civil False Claims Act in 1986 that were designed to encourage private persons to sue on behalf of the government. These whistleblower suits by private persons, known as qui tam relaters, may be filed by almost anyone, including physicians and their employees and patients, our employees, and even competitors. The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), in addition to its privacy provisions, created a series of new healthcare-related crimes.
Competition
The business for delivery of therapeutics to the human brain is highly competitive. Our products must gain acceptance by the medical community and show clinically meaningful advantages in performance. There are several groups and companies working on competitive technologies although in various stages. Ivar Mendez, a neurosurgeon at Dalhousie University in Halifax, Canada, has created the Halifax Injector, which delivers cells by tiny computer-controlled motors. Researchers at Vanderbilt University, headed up by Prof. Robert J. Webster III and Asst. Prof. of Neurological Surgery Kyle Weaver, are developing a robotic steerable needle, which they call the 'Active Cannula' system. This is focused primarily on removing blood clots and not for stem cell deployment although future uses could include it. Renishaw's neuroinspire software system is a software base guidance system that builds a map of the brain via MRI scans and produces a 3D image for deciding on a path for needle based biopsies. The Renishaw system incorporates the use of traditional straight needles and used for biopsies. Many researchers working on regenerative medicine and stem cell deployment to the brain will continue to use straight needles.
12
We believe that our BranchPoint device is unique. Additionally, the projected low cost of our BranchPoint device compared to robotic or steerable needles utilizing micro-motors could be a significant advantage and create a more cost-effective solution for the delivery of therapeutics to the brain.
Backlog
We are in a development stage and do not have any backlog.
Proposed Sales, Marketing and Advertising
We will seek commercial partners to market the products upon approval by the FDA.
Environmental Matters
Laws and regulations relating to protection of the environment have not had and should not have a material impact on our business.
Proprietary Rights
In addition to our patent application we have entered into an employment agreement with our CEO and intend to enter into similar agreements with other key employees that require them to keep all of our proprietary information confidential. We cannot assure that such protections will prove adequate should they be challenged in litigation.
Research and Development
We anticipate that our expenses in 2015 will include product development, trial and contractor expenses, and professional fees.
Employees
Our CEO is employed by us on a part time basis. We believe that our business can be carried forward by consultants and independent contractors in 2015.
Seasonality
We do not anticipate that our business will be seasonal to any material extent.
ITEM 1A. RISK FACTORS
Risks Relating To Our Business
You should carefully consider the risks described below before investing in our publicly traded securities. The risks described below are not the only ones facing us. Our business is also subject to the risks that affect many other companies, such as competition, technological obsolescence, labor relations, general economic conditions, geopolitical events, climate change and international operations. Additional risks not currently known to us or that we currently believe are immaterial also may impair our business operations and our liquidity.
We are in an early development stage and have limited resources and are dependent on conducting successful clinical trials and raising additional capital.
To date our activities have involved obtaining and executing license agreements with UAMS and UCSF, and raising sufficient funds for a clinical trial and product development. As of December 31, 2014, we had $555,506 cash and cash equivalents on hand. This is not sufficient to complete our product development as a standalone company, which we estimate will cost approximately $5 to $7 million over the next four to five years. We do not have any commitments for the additional capital we require and management believes that our ability to raise additional capital is highly dependent on successful product development. We will be able to fund the current stage of product development with cash on hand, but if any stage of our product
13
development produces ambiguous or unfavorable results, it is unlikely that we will be able to raise additional funds for subsequent stages and our business will fail and our stock will become virtually worthless.
Our auditors have qualified their opinion based on our ability to continue as a going concern.
Our auditors qualified their report that we will continue as a going concern because we have no revenues, have incurred recurring losses and recurring negative cash flow from operating activities, and have an accumulated deficit. If we are unable to raise additional funds and continue as a going concern, investors in our stock will lose their money.
Even if our product development is successful and we raise additional capital, our shareholders may suffer substantial dilution.
We will require $5 to $7 million in additional capital to complete our clinical trials as a standalone company. We do not have any commitments for those funds, but are dependent on conducting successful product development and seeking additional investors. The terms of investment of any additional investors may result in substantial dilution to the holders of our common stock.
Our limited ability to protect our intellectual property, and the possibility that our technology could inadvertently infringe technology owned by others, may adversely affect our ability to compete.
We rely on a patent application obtained from UCSF under our UCSF License to protect our intellectual property rights. A successful challenge to the ownership of our technology could materially damage our business prospects. Our competitors may assert that our technologies or products infringe on their patents or proprietary rights. We may be required to obtain from others licenses that may not be available on commercially reasonable terms, if at all. Problems with intellectual property rights could increase the cost of our proposed products or delay or preclude our new product development and commercialization. If infringement claims against us are deemed valid, we may not be able to obtain appropriate licenses on acceptable terms or at all. Litigation could be costly and time-consuming but may be necessary to protect our technology license positions or to defend against infringement claims. UCSF has applied for a United States patent which is the subject of our UCSF License. No assurance can be given that this patent will be granted, that, if granted, this patent will provide us with meaningful protection from infringement by others or that any patent that we may be granted will not be held by a court to infringe on the rights of others. The loss of patent protection could materially adversely affect our business.
The patent applications that we licensed from the University of California, San Francisco (UCSF) under our UCSF License could become not patentable and impact the viability of our UCSF License.
A portion of one of the three licensed patent applications, U.S. Patent Application Serial No. 12/334,217, Device and Method for In Vivo Flow Cytometry Using the Detection of Photoacoustic Waves; UAMS ID No. 2008-16, had been rejected by the United States Patent and Trademark Office and U.S. Patent Application Serial No. 12/334,217 was then abandoned prior to the date of our UAMS License. The basis for the rejection was primarily that the rejected portion of the application had previously been published in a scientific journal by the inventor Prof. Vladimir Zharov, an employee of the UAMS, prior to the filing of the application and therefore was not patentable. U.S. Patent Application No. PCT/US2013/052301, Microinjection Catheter; UC Case No. SF2012-063 which we licensed under our UCSF License could also be rejected by the Unites States Patent and Trademark Office and become not patentable.
The Federal government has “March-in Rights” to grant additional licenses to the patents that we licensed from UCSF under our UCSF License.
The work resulting in the invention of the device described in the patent applications that we licensed was generated with the assistance of Federal grant funding. Therefore, the Federal government has the rights established and described in 35 U.S.C. §§ 200-212. Of particular relevance is 35 U.S.C. § 202(c)(4), which in respect to any invention in which we elect rights, provides the Federal agency that gave the grant funding a nonexclusive, nontransferrable, irrevocable, paid-up license to practice or have practiced for or on behalf of the United States any subject invention throughout the world: Provided, that the funding agreement may provide for such additional rights, including the right to assign or have assigned foreign patent rights in the subject invention, as are determined by the agency as necessary for meeting the obligations of the United States under any treaty, international agreement, arrangement of cooperation, memorandum of understanding, or similar arrangement, including military agreement relating to weapons development and production. Also of particular relevance is 35 U.S.C. § 203, which establishes the “March-in Rights” granted to the government. In summary, the statute provides the power for the Federal agency that gave the grant funding to bestow on a third-party applicant an additional license to the patents if that Federal agency determines that (1) we have not
14
taken sufficient steps to achieve practical application of the technology; (2) action is necessary to alleviate health or safety needs not currently being addressed by us; (3) public use requirements specified by Federal regulations are not being met by us; or (4) the technology is not being manufactured substantially in the United States. To date, despite multiple applications requesting such action, a Federal agency such as the NIH (National Institutes of Health) has never exercised the powers provided for in 35 U.S.C. § 203.
Our BranchPoint device may not be able to deliver therapeutics to brain targets, improve accuracy, reduce risk of complications or increase patient safety.
We demonstrated the ability of our BranchPoint device to deliver therapeutics to brain targets and improve accuracy of therapeutic delivery under interventional MRI guidance (iMRI) in animal and human cadaver studies. The ability of our BranchPoint device to reduce risk of complications or increase patient safety, however, needs to be demonstrated in either pre-approval or post-approval clinical trials which could be requested by the FDA upon 510(k) submission. If we conduct a clinical trial, such a trial could fail to demonstrate the ability of our BranchPoint device to perform as expected.
We are subject to the periodic reporting requirements of the Securities Exchange Act of 1934 that requires us to incur audit fees and legal fees in connection with the preparation of such reports. These additional costs could reduce or eliminate our ability to earn a profit.
We are required to file periodic reports with the Securities and Exchange Commission pursuant to the Securities Exchange Act of 1934 and the rules and regulations promulgated thereunder. In order to comply with these requirements, our independent registered public accounting firm has to review our financial statements on a quarterly basis and audit our financial statements on an annual basis. Moreover, our legal counsel has to review and assist in the preparation of such reports. The costs charged by these professionals for such services cannot be accurately predicted because factors such as the number and type of transactions that we engage in and the complexity of our reports cannot be determined at this time and have a major effect on the amount of time to be spent by our auditors and attorneys. However, our incurring these costs obviously is an expense to our operations and thus has a negative effect on our ability to meet our overhead requirements and earn a profit. We may be exposed to potential risks resulting from new requirements under Section 404 of the Sarbanes-Oxley Act of 2002. If we cannot provide reliable financial reports or prevent fraud, our business and operating results could be harmed, investors could lose confidence in our reported financial information, and the trading price of our common stock, if a market ever develops, could drop significantly.
Since approximately a quarter of our current assets are in overseas accounts, it may prove difficult for U.S. investors to reach those assets to satisfy any claims.
We maintain approximately a quarter of our current assets in overseas accounts for strategic reasons that management believes are beneficial to the Company and its shareholders. However, in the event U.S. investors obtained a judgment or award against the Company for claims under federal or state securities laws, enforcement of such judgment or award would become more difficult as to those assets that are overseas.
We currently do not have, and may never develop, any commercialized products.
We are a development stage company and currently do not have any commercialized products or any significant source of revenue. We have invested substantially all of our time and resources since inception in developing our products. Our development products may require additional development and clinical evaluation and they will require regulatory approval, significant marketing efforts and substantial additional investment before they can provide us with any revenue. Commercialization of each of our products remains subject to certain significant risks. Our efforts may not lead to commercially successful products for a number of reasons, including:
·
we may not be able to obtain regulatory approvals for our development products, or the approved indication may be narrower than we seek;
·
any of our development products may not prove to be safe and effective in clinical trials to the FDA’s satisfaction;
·
physicians may not receive any reimbursement from third-party payers, or the level of reimbursement may be insufficient to support widespread adoption of our development products;
·
we may experience delays in our continuing development program;
15
·
any products that are approved by regulators may not be accepted in the marketplace by physicians or patients;
·
we may not have adequate financial or other resources to complete the continued development or to commence the commercialization of our products and we may not have adequate financial or other resources to achieve significant commercialization of our products;
·
we may not be able to manufacture our products in commercial quantities or at an acceptable cost; and
·
rapid technological change may make our technology and products obsolete.
If we are unable to obtain regulatory approval for or successfully commercialize our products, we will be unable to generate revenue.
We have not received, and may never receive, FDA approval to market any of our products.
We do not have the necessary regulatory approvals to market any of our products in the U.S. or in any foreign market. We plan initially to launch our products, once approved, in the U.S. The regulatory approval may process involve, among other things, successfully completing clinical trials. The FDA may requires us to prove the safety and effectiveness of our products to the FDA’s satisfaction. This process can be expensive and uncertain, and requires detailed and comprehensive scientific and human clinical data. FDA review may take years after an application is filed. The FDA may never grant approval. The FDA can delay, limit, or deny approval of an application for many reasons, including:
·
any of our products may not be safe or effective to the FDA’s satisfaction;
·
the data we may obtain from our pre-clinical studies and clinical trials may be insufficient to support approval;
·
the manufacturing process or facilities we use may not meet applicable requirements; and
·
changes in FDA approval policies or adoption of new regulations may require additional data.
The FDA may not consider the data we may gather in clinical trials sufficient to support approval of our products. The FDA may determine that additional clinical trials or data are necessary, in which case regulatory approval may be delayed for several months or even years while the trials are conducted and the data acquired are submitted in an amendment to initial application. The occurrence of unexpected findings in connection with any clinical trial may prevent or delay obtaining regulatory approval, and may adversely affect coverage or reimbursement determinations. If we are unable to complete our clinical trials necessary to successfully support our intended applications, our ability to commercialize our products, and our business, financial condition, and results of operations would be materially adversely affected, thereby threatening our ability to continue operations.
If any of our products are approved by the FDA, they may be approved only for narrow indications.
Even if approved, our products may not be approved for the indications that are necessary or desirable for successful commercialization. If the use of our products is restricted, then the size of the market for our products and the rate of acceptance of our products by physicians may be adversely affected.
If we wish to modify any of our products after receiving FDA approval, including changes in indications or other modifications that could affect safety and effectiveness, additional approvals could be required from the FDA, we may be required to submit extensive pre-clinical and clinical data, depending on the nature of the changes. Any request by the FDA for additional data, or any requirement by the FDA that we conduct additional clinical studies, could delay the commercialization of our devices and require us to make substantial additional research, development and other expenditures. We may not obtain the necessary regulatory approvals to market our products in the U.S. or anywhere else. Any delay in, or failure to receive or maintain, approval for our products could prevent us from generating revenue or achieving profitability, and our business, financial condition, and results of operations would be materially adversely affected.
Any of our products may not be commercially viable if we fail to obtain an adequate level of reimbursement by Medicare and other third party payers. The markets for our products may also be limited by the indications for which its use may be reimbursed.
16
The availability of medical insurance coverage and reimbursement for newly approved products is uncertain. In the U.S., physicians and other healthcare providers are generally reimbursed for all or part of the cost of patient treatment by Medicare, Medicaid, or other third-party payers.
The commercial success of our products in both domestic and international markets will significantly depend on whether third-party coverage and reimbursement are available for services involving our products. Medicare, Medicaid, health maintenance organizations and other third-party payers are increasingly attempting to contain healthcare costs by limiting both the scope of coverage and the level of reimbursement of new products, and as a result, they may not cover or provide adequate payment for the use of our products. In order to obtain satisfactory reimbursement arrangements, we may have to agree to a fee or sales price lower than the fee or sales price we might otherwise charge. Even if Medicare and other third-party payers decide to cover procedures involving our products, we cannot be certain that the reimbursement levels will be adequate. Accordingly, even if our products or future products we develop are approved for commercial sale, unless government and other third-party payers provide adequate coverage and reimbursement for our products, some physicians may be discouraged from using them, and our sales would suffer.
Medicare reimburses for use of medical products in a variety of ways, depending on where and how a product is used. However, Medicare only provides reimbursement if the Centers for Medicare and Medicaid Services (“CMS”) determines that a certain product should be covered and that the use of the product is consistent with the coverage criteria. A coverage determination can be made at the local level by the Medicare administrative contractor (formerly called carriers and fiscal intermediaries), a private contractor that processes and pays claims on behalf of CMS for the geographic area where the services were rendered, or at the national level by CMS through a national coverage determination. There are new statutory provisions intended to facilitate coverage determinations for new products, but it is unclear how these new provisions will be implemented. Coverage presupposes that the product has been cleared or approved by the FDA and further, that the coverage will be no broader than the indication as approved or cleared by the FDA, but coverage can be narrower. A coverage determination may be so limited that relatively few patients will qualify for a covered use of a product. Should a very narrow coverage determination be made for our products, it may undermine the commercial viability of our products.
Obtaining a coverage determination, whether local or national, is a time-consuming, expensive and highly uncertain proposition, especially for new products, and inconsistent local determinations are possible. On average, according to an industry report, Medicare coverage determinations for medical products lag 15 months to five years or more behind FDA approval for a product. The Medicare statutory framework is also subject to administrative rulings, interpretations and discretion that affect the amount and timing of reimbursement made under Medicare. Medicaid coverage determinations and reimbursement levels are determined on a state by state basis, because Medicaid, unlike Medicare, is administered by the states under a state plan filed with the Secretary of the U.S. Department of Health and Human Services (“HHS”). Medicaid generally reimburses at lower levels than Medicare. Moreover, Medicaid programs and private insurers are frequently influenced by Medicare coverage determinations.
The FDA may require additional clinical trials and any adverse results in such clinical trials, or difficulties in conducting such clinical trials, could have a material adverse effect on our business.
The FDA may require us to conduct additional clinical studies upon evaluation of our regulatory submission. The occurrence of unexpected findings in connection with any initial or subsequent clinical trial required by the FDA may prevent or delay obtaining regulatory approval. In addition subsequent clinical studies would require the expenditure of additional company resources and could be a long and expensive process subject to unexpected delays. Any adverse results in such clinical trials, or difficulties in conducting such clinical trials, could have a material adverse effect on our business.
We expect to operate in a highly competitive market, we may face competition from large, well-established pharmaceutical or medical device companies with significant resources, and we may not be able to compete effectively.
Our products must gain acceptance by the medical community and show clinically meaningful advantages in performance. However, our present and future competitors may enjoy
·
significantly greater name recognition;
·
established relations with healthcare professionals, customers and third-party payers;
·
established distribution networks;
17
·
additional lines of products, and the ability to offer rebates, higher discounts or incentives to gain a competitive advantage;
·
greater experience in conducting research and development, manufacturing, clinical trials, obtaining regulatory approval for products, and marketing approved products; and
·
greater financial and human resources for product development, sales and marketing, and patent litigation.
As a result, we may not be able to compete effectively against these companies or their products.
Technological breakthroughs could render our products obsolete.
The medical field is subject to rapid technological change and product innovation. Our products are based on our proprietary technology, but a number of companies and medical researchers are pursuing new technologies. Companies in the medical field with significantly greater financial, technical, research, marketing, sales and distribution and other resources have expertise and interest in the delivery of therapeutics to the human brain targets. Some of these companies are working on potentially competing products or therapies.
In addition, the National Institutes of Health and other supporters of medical research are presumptively seeking ways to improve patient diagnosis and treatment by sponsoring corporate and academic research. There can be no assurance that one or more of these companies will not succeed in developing or marketing technologies and products or services that demonstrate better safety or effectiveness, superior clinical results, greater ease of use or lower cost than our products, or that such competitors will not succeed in obtaining regulatory approval for introducing or commercializing any such products or services prior to us.
FDA approval of a commercially viable alternative to our products produced by a competitor could significantly reduce market acceptance of our products. Any of the above competitive developments could have a material adverse effect on our business, financial condition, and results of operations. There is no assurance that products, services, or technologies introduced prior to or subsequent to the commercialization of our products will not render our products less marketable or obsolete.
For initial or additional clinical trials required for our products by the FDA or with respect to clinical trials relating to the development of our technology for other applications, we depend on clinical investigators and clinical sites and other third parties to manage the trials and to perform related data collection and analysis, and, as a result, we may face costs and delays that are outside of our control.
With respect to any additional clinical studies for our products which may required by the FDA or with respect to clinical trials relating to the development of our core technology for other applications, we rely on clinical investigators and clinical sites, some of which are private practices, and some of which are research university- or government-affiliated, to enroll patients in our clinical trials. We may rely on: pathologists and pathology laboratories; a contract research organization to assist in monitoring, collection of data, and ensuring FDA Good Clinical Practices (“GCP”) are observed at our sites; a consultant biostatistician; and other third parties to manage the trial and to perform related data collection and analysis.
However, we may not be able to control the amount and timing of resources that clinical sites and other third parties may devote to our clinical trials. If these clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials, or if the clinical sites fail to comply adequately with the clinical protocols, we will be unable to complete these trials, which could prevent us from obtaining regulatory approvals for our products or other products developed from our technology. Our agreements with clinical investigators and clinical sites for clinical testing place substantial responsibilities on these parties and, if these parties fail to perform as expected, our trials could be delayed or terminated.
If these clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain are compromised due to their failure to adhere to our clinical protocols or for other reasons, our clinical trials may be extended, delayed or terminated, and we may be unable to obtain regulatory approval for, or successfully commercialize, our products or other products developed from our technology.
18
In addition to the foregoing, any initial or additional clinical studies for any of our products which may required by the FDA and any clinical trials relating to the development of our technology for other applications may be delayed or halted, or be inadequate to support regulatory approval, for numerous other reasons, including, but not limited to, the following:
·
the FDA, an Institutional Review Board (“IRB”) or other regulatory authorities place our clinical trial on hold;
·
patients do not enroll in clinical trials at the rate we expect;
·
patient follow-up is not at the rate we expect;
·
IRBs and third-party clinical investigators delay or reject our trial protocol;
·
third-party organizations do not perform data collection and analysis in a timely or accurate manner;
·
regulatory inspections of our clinical trials or manufacturing facilities, among other things, require us to undertake corrective action or suspend or terminate our clinical trials, or invalidate our clinical trials;
·
changes in governmental regulations or administrative actions; and
·
the interim or final results of the clinical trial are inconclusive or unfavorable as to safety or effectiveness.
If our products are approved for reimbursement, we anticipate experiencing significant pressures on pricing.
Even if Medicare covers a product for certain uses, that does not mean that the level of reimbursement will be sufficient for commercial success.
We expect to experience pricing pressures in connection with the commercialization of our products and our future products due to efforts by private and government-funded payers to reduce or limit the growth of healthcare costs, the increasing influence of health maintenance organizations, and additional legislative proposals to reduce or limit increases in public funding for healthcare services. Private payers, including managed care payers, increasingly are demanding discounted fee structures and the assumption by healthcare providers of all or a portion of the financial risk. Efforts to impose greater discounts and more stringent cost controls upon healthcare providers by private and public payers are expected to continue. Payers frequently review their coverage policies for existing and new diagnostic tools and can, sometimes without advance notice, deny or change their coverage policies. Significant limits on the scope of services covered or on reimbursement rates and fees on those services that are covered could have a material adverse effect on our ability to commercialize our products and therefore, on our liquidity and our business, financial condition, and results of operations.
Our products may never achieve market acceptance even if we obtain regulatory approvals.
Even if we obtain regulatory approval, patients and physicians may not endorse our products. Physicians tend to be slow to change their diagnostic and medical treatment practices because of perceived liability risks arising from the use of new products and the uncertainty of third party reimbursement. Physicians may not utilize any of our products until there is long-term clinical evidence to convince them to alter their existing methods of diagnosing or evaluating suspicious lesions or other conditions addressed by our products and there are recommendations from prominent physicians that our products are effective. We cannot predict the speed at which physicians may adopt the use of any of our products. If our products receive the appropriate regulatory approvals but do not achieve an adequate level of acceptance by patients, physicians and healthcare payers, we may not generate significant product revenue and we may not become profitable. The degree of market acceptance of our products will depend on a number of factors, including:
·
perceived effectiveness of our products;
·
convenience of use;
·
cost of use of our products;
·
availability and adequacy of third-party coverage or reimbursement;
19
·
approved indications and product labeling;
·
publicity concerning our products or competitive products;
·
potential advantages over alternative diagnostic methodologies;
·
introduction and acceptance of competing products or technologies; and
·
extent and success of our sales, marketing and distribution efforts.
The success of our products will depend upon the acceptance by physicians and hospitals. We will be subject to intense scrutiny before physicians will be comfortable incorporating our products in their treatment approaches. We believe that recommendations by respected physicians and marketing by established licensees will be essential for the development and successful marketing of our products; however, there can be no assurance that any such recommendations will be obtained. To date, the medical community has had little exposure to us and our products.
Because the medical community is often skeptical of new companies and new technologies, we may be unable to gain access to potential customers in order to demonstrate the operational effectiveness of our products. Even if we gain access to potential customers, no assurance can be given that physicians will perceive a need for or accept our products, even after we receive approval from the FDA for marketing the product.
We intend to contract with third parties in order to commercialize our products.
To the extent that we enter into arrangements with third parties to perform marketing and distribution services in the U.S., our product revenue could be lower and our costs higher than if we directly marketed our products. Furthermore, to the extent that we enter into co-promotion or other marketing and sales arrangements with other companies, any revenue received will depend on the skills and efforts of others, and we do not know whether these efforts will be successful. If we are unable to establish and maintain adequate sales, marketing and distribution capabilities, independently or with others, we will not be able to generate product revenue, and may not become profitable.
We have no in-house manufacturing capabilities and manufacturing personnel, and intend to rely on third parties for manufacturing. Accordingly, our manufacturing operations will be dependent upon third-party suppliers, making us vulnerable to supply problems and price fluctuations, which could harm our business.
If our products are approved, we intend to rely on third party licensees for their manufacture. Our success will therefore be dependent on those third parties’ ability to manufacture our products that meet all FDA requirements and are cost effective and reliable. Any failures on the part of those third parties could negatively affect our results. Our products are complex and may contain undetected design defects and errors when first introduced, or errors that may be introduced when enhancements are released. Such defects and errors may occur despite our testing, and may not be discovered until after our devices have been shipped to and used by our customers. The existence of these defects and errors could result in costly repairs, returns of devices, diversion of development resources and damage to our reputation in the marketplace. Any of these conditions could have a material adverse impact on our business, financial condition, and results of operations. In addition, when we contract with third-party manufacturers for the production of our products, these manufacturers may inadvertently produce devices that vary from devices we have produced in unpredictable ways that cause adverse consequences.
We intend to enter into a contract for commercial production of our devices once commercial specifications for the devices have been finalized, but we may not be able to enter such an agreement on mutually acceptable terms. Failure to enter into such an agreement would require us to expand our own manufacturing facilities or obtain such services elsewhere. Our planned reliance upon an outside provider for assembly and production services subjects us to the risk of adverse consequences from delays and defects caused by the failure of such outside supplier to meet its contractual obligations. The failure by us or our supplier to produce a sufficient number of devices that can operate according to our specifications could delay the commercial sale of our products, and would adversely affect both our ability to successfully commercialize any such product and our business, financial condition and results of operations.
We will not be able to sell our products unless and until its design is verified and validated in accordance with current good manufacturing practices as set forth in the U.S. medical device Quality System Regulation.
20
We have not yet successfully completed all the steps necessary to verify and validate the design of our products that are required to be performed prior to commercialization. If we are delayed or unable to complete verification and validation successfully, we will not be able to sell our products, and we will not be able to meet our plans for the commercialization of our products. Later discovery of previously unknown problems with our products, including manufacturing problems, or failure to comply with regulatory requirements such as the FDA QSR, may result in restrictions on our products or its manufacturing processes, withdrawal of our products from the market, patient or physician notification, voluntary or mandatory recalls, fines, withdrawal of regulatory approvals, refusal to approve pending applications or supplements to approved applications, refusal to permit the import or export of our products, product seizures, injunctions or the imposition of civil or criminal penalties. Should any of these enforcement actions occur, our business, financial condition and results of operations could be materially and adversely affected.
Assuming that our products are approved by regulatory authorities, if we or our suppliers fail to comply with ongoing regulatory requirements, or if we experience unanticipated problems with our products, they could be subject to restrictions or withdrawal from the market.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data and promotional activities for such product, will be subject to continuous review and periodic inspections by the FDA and other regulatory bodies. In particular, we and our suppliers are required to comply with the QSR and other regulations which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage, promotion, distribution, and shipping of our products, and with record keeping practices.
We also will be subject to ongoing FDA requirements, including required submissions of safety and other post-market information and reports and registration and listing requirements. To the extent that we contract with third parties to manufacture some of our products, our manufacturers will be required to adhere to cGMP requirements enforced by the FDA as part of QSR, or similar regulations required by regulatory agencies in other countries. The manufacturing facilities of our contract manufacturers must be inspected or must have been inspected, and must be in full compliance with cGMP requirements before approval for marketing. The FDA enforces the QSR and other regulatory requirements through unannounced inspections. We have not yet been inspected by the FDA for any of our products and will have to complete such an inspection successfully before we ship any products.
We are involved in a heavily regulated sector, and our ability to remain viable will depend on favorable government decisions at various points by various agencies.
From time to time, legislation is introduced in the U.S. Congress that could significantly change the statutory provisions governing the approval, manufacture, and marketing of a medical product. Additionally, healthcare is heavily regulated by the federal government, and by state and local governments. The federal laws and regulations affecting healthcare change constantly, thereby increasing the uncertainty and risk associated with any healthcare related venture, including our business and our products. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance, or interpretations changed, and what the impact of such changes, if any, may be.
The federal government regulates healthcare through various agencies, including but not limited to the following: (i) the FDA, which administers the Food, Drug, and Cosmetic Act, as well as other relevant laws; (ii) CMS, which administers the Medicare and Medicaid programs; (iii) the Office of Inspector General (“OIG”) which enforces various laws aimed at curtailing fraudulent or abusive practices, including by way of example, the Anti-Kickback Law, the Anti-Physician Referral Law, commonly referred to as Stark, the Anti-Inducement Law, the Civil Money Penalty Law, and the laws that authorize the OIG to exclude healthcare providers and others from participating in federal healthcare programs; and (iv) the Office of Civil Rights, which administers the privacy aspects of the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”). All of the aforementioned are agencies within HHS. Healthcare is also provided or regulated, as the case may be, by the Department of Defense through its TriCare program, the Public Health Service within HHS under the Public Health Service Act, the Department of Justice through the Federal False Claims Act and various criminal statutes, and state governments under Medicaid and other state sponsored or funded programs and their internal laws regulating all healthcare activities.
In addition to regulation by the FDA as a medical device manufacturer, we are subject to general healthcare industry regulations. The healthcare industry is subject to extensive federal, state and local laws and regulations relating to:
·
billing for services;
21
·
quality of medical equipment and services;
·
confidentiality, maintenance and security issues associated with medical records and individually identifiable health information;
·
false claims; and
·
labeling products.
These laws and regulations are extremely complex and, in some cases, still evolving. In many instances, the industry does not have the benefit of significant regulatory or judicial interpretation of these laws and regulations. If our operations are found to be in violation of any of the federal, state or local laws and regulations that govern our activities, we may be subject to the applicable penalty associated with the violation, including civil and criminal penalties, damages, fines or curtailment of our operations. The risk of being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Any action against us for violation of these laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s time and attention from the operation of our business.
The application of the privacy provisions of HIPAA is uncertain.
HIPAA, among other things, protects the privacy and security of individually identifiable health information by limiting its use and disclosure. HIPAA directly regulates “covered entities” (insurers, clearinghouses, and most healthcare providers) and indirectly regulates “business associates” with respect to the privacy of patients’ medical information. Certain entities that receive and process protected health information are required to adopt certain procedures to safeguard the security of that information.
It is uncertain whether we would be deemed to be a covered entity under HIPAA, and it is unlikely that based on our current business model, we would be a business associate. Nevertheless, we will likely be contractually required to physically safeguard the integrity and security of the patient information that we or our physician customers receive, store, create or transmit. If we fail to adhere to our contractual commitments, then our physician customers may be subject to civil monetary penalties, and this could adversely affect our ability to market our products. We also may be liable under state laws governing the privacy of health information.
We may become subject to claims of infringement or misappropriation of the intellectual property rights of others, which could prohibit us from shipping affected products, require us to obtain licenses from third parties or to develop non-infringing alternatives, and subject us to substantial monetary damages and injunctive relief. Our patents may also be subject to challenge on validity grounds, and our patent applications may be rejected.
Third parties could, in the future, assert infringement or misappropriation claims against us with respect to our current or future products. Whether a product infringes a patent involves complex legal and factual issues, the determination of which is often uncertain. Therefore, we cannot be certain that we have not infringed the intellectual property rights of such third parties. Our potential competitors may assert that some aspect of the intellectual property utilized in our products infringes their patents. Because patent applications may take years to issue, there also may be applications now pending of which we are unaware that may later result in issued patents on which our owned or licensed intellectual property infringes. There also may be existing patents of which we are unaware that one or more components of our products’ systems may inadvertently infringe.
Any infringement or misappropriation claim could cause us to incur significant costs, could place significant strain on our financial resources, divert management’s attention from our business and harm our reputation. If the relevant patents were upheld as valid and enforceable and we were found to infringe, we could be prohibited from selling our product that is found to infringe unless we could obtain licenses to use the technology covered by the patent or are able to design around the patent. We may be unable to obtain a license on terms acceptable to us, if at all, and we may not be able to redesign our products to avoid infringement. A court could also order us to pay compensatory damages for such infringement, plus prejudgment interest and could, in addition, treble the compensatory damages and award attorney fees. These damages could be substantial and could harm our reputation, business, financial condition, and operating results. A court also could enter orders that temporarily, preliminarily or permanently enjoin us and our customers from making, using, selling, offering to sell or importing our products, and/or could enter an order mandating that we undertake certain remedial activities. Depending on the nature of the relief ordered by the court, we could become liable for additional damages to third parties.
22
We also may rely on our patents, patent applications and other intellectual property rights to give us a competitive advantage. Whether a patent is valid, or whether a patent application should be granted, is a complex matter of science and law, and therefore we cannot be certain that, if challenged, our patents, patent applications and/or other intellectual property rights would be upheld. If one or more of those patents, patent applications and other intellectual property rights are invalidated, rejected or found unenforceable, that could reduce or eliminate any competitive advantage we might otherwise have had.
New product development in the medical industry is both costly and labor intensive with very low success rates for successful commercialization; if we cannot successfully develop or obtain future products, our growth would be delayed.
Our long-term success is dependent, in large part, on the design, development and commercialization of our products and other new products and services in the medical industry. The product development process is time-consuming, unpredictable and costly. There can be no assurance that we will be able to develop or acquire new products, successfully complete clinical trials, obtain the necessary regulatory clearances or approvals required from the FDA on a timely basis, or at all, manufacture our potential products in compliance with regulatory requirements or in commercial volumes, or that our products or other potential products will achieve market acceptance.
In addition, changes in regulatory policy for product approval during the period of product development, and regulatory agency review of each submitted new application, may cause delays or rejections. It may be necessary for us to enter into licensing arrangements in order to market effectively any new products or new indications for existing products. There can be no assurance that we will be successful in entering into such licensing arrangements on terms favorable to us or at all. Failure to develop, obtain necessary regulatory clearances or approvals for, or successfully market potential new products could have a material adverse effect on our business, financial condition, and results of operations.
We face the risk of product liability claims and may not be able to obtain or maintain adequate insurance.
Our business exposes us to the risk of product liability claims that is inherent in the testing, manufacturing and marketing of medical devices, including those that may arise from the misuse or malfunction of, or design flaws in, our products. We may be subject to product liability claims if any of our products causes, or merely appears to have caused, an injury or if a patient alleges that any of our products failed to provide appropriate treatment. Claims may be made by patients, healthcare providers or others involved with our products.
Each of our products will require regulatory approval prior to commercialization in the U.S. The clinical studies of our products may be considered by the FDA as “Non-Significant Risk.” Consequently, the trials may be conducted under the auspices of an abbreviated Investigational Device Exemption. We therefore may only maintain limited domestic clinical trial liability insurance, as may be required by clinical sites. We intend to obtain clinical trial liability insurance in certain European countries where required by statute or clinical site policy. Although we intend to obtain general liability insurance that we believe will be appropriate, and anticipate obtaining adequate product liability insurance before commercialization of any of our products, this insurance is and will be subject to deductibles and coverage limitations.
Our anticipated product liability insurance may not be available to us in amounts and on acceptable terms, if at all, and if available, the coverages may not be adequate to protect us against any future product liability claims. If we are unable to obtain insurance at an acceptable cost or on acceptable terms with adequate coverage, or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which may harm our business. A product liability claim, recall or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could result in significant costs and significant harm to our business.
We may be subject to claims against us even if the apparent injury is due to the actions of others. For example, we rely on the expertise of physicians, nurses and other associated medical personnel to operate our devices. If these medical personnel are not properly trained or are negligent, we may be subjected to liability. These liabilities could prevent or interfere with our product commercialization efforts. Defending a suit, regardless of merit, could be costly, could divert management attention and might result in adverse publicity, which could result in the withdrawal of, or inability to recruit, clinical trial volunteers, or result in reduced acceptance of any of our devices in the market.
Insurance and surety companies have reassessed many aspects of their business and, as a result, may take actions that could negatively affect our business. These actions could include increasing insurance premiums, requiring higher self-insured retentions and deductibles, reducing limits, restricting coverages, imposing exclusions, and refusing to underwrite certain risks
23
and classes of business. Any of these actions may adversely affect our ability to obtain appropriate insurance coverage at reasonable costs, which could have a material adverse effect on our business, financial condition and results of operations.
Failure to obtain and maintain regulatory approval in foreign jurisdictions will prevent us from marketing abroad.
We intend to seek partners to develop and market our products internationally. Outside the U.S., we can market a product only if we receive a marketing authorization and, in some cases, pricing approval, from the appropriate regulatory authorities. The approval procedure varies among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval.
The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval, in addition to other risks. Foreign regulatory bodies have established varying regulations governing product standards, packaging requirements, labeling requirements, import restrictions, tariff regulations, duties and tax requirements. We may not obtain foreign regulatory approvals on a timely basis, if at all. Foreign regulatory agencies, as well as the FDA, periodically inspect manufacturing facilities both in the U.S. and abroad. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We have not taken any significant actions to obtain foreign regulatory approvals. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize any of our products in any market on a timely basis, or at all. Our inability or failure to comply with varying foreign regulation, or the imposition of new regulations, could restrict our sale of products internationally.
We may not be able to find a strategic partner to continue further clinical development of our photoacoustic device that we licensed under our UAMS License.
On December 15, 2012 (“Effective Date”), we executed an Exclusive License Agreement (“UAMS License”) with the Board of Trustees of the University of Arkansas (“UofA”) acting on behalf of the University of Arkansas for Medical Sciences (“UAMS”) to commercially develop certain patents and patent applications owned by UAMS throughout the world for the life of the patents. Our UAMS License includes three patents (i) U.S. Patent Application Serial No. 12/334,217, Device and Method for In Vivo Flow Cytometry Using the Detection of Photoacoustic Waves; UAMS ID No. 2008-16; (ii) U.S. Patent Application Ser. No.: 12/945,576 Device and Method for In Vivo Noninvasive Magnetic Manipulation of Circulating Objects in BioFlows; UAMS ID No. 2008-16 CIP; (iii) U.S. Patent Application Ser. No.: 13/253,767 Device and Method for In Vivo Detection of Clots Within Circulatory Vessels; UAMS ID No. 2008-16 CIP2.
As previously disclosed in our filing on Form 8-K on February 24, 2014, in the normal course of events, we confirmed, on or about February 3, 2014, that a portion of one of the three licensed patent applications, U.S. Patent Application Serial No. 12/334,217, Device and Method for In Vivo Flow Cytometry Using the Detection of Photoacoustic Waves; UAMS ID No. 2008-16, had been rejected by the United States Patent and Trademark Office and U.S. Patent Application Serial No. 12/334,217 was then abandoned prior to the date of our UAMS License. The basis for the rejection was primarily that the rejected portion of the application had previously been published in a scientific journal by the inventor Prof. Vladimir Zharov, an employee of the UAMS, prior to the filing of the application and therefore was not patentable. Thus, we may not be able to find a strategic partner to continue further clinical development, despite completing a proof-of-concept clinical trial.
Risks Relating to our Common Stock
Currently, there is no active, liquid public market for our securities, and there can be no assurances that any liquid public market will ever develop or that our common stock will be quoted for trading and, even if quoted, it is likely to be subject to significant price fluctuations.
Prior to the date of this filing on Form 10-K, there has not been any established active, liquid trading market for our common stock. We cannot predict the extent to which investor interest in us will lead to the development of an active, liquid trading market. Active trading markets generally result in lower price volatility and more efficient execution of buy and sell orders for investors.
In addition, our common stock is unlikely to be followed by any market analysts, and there may be few institutions acting as market makers for the common stock. Either of these factors could adversely affect the liquidity and trading price of our common stock. Until our common stock is fully distributed and an orderly market develops in our common stock, if ever, the price at which it trades is likely to fluctuate significantly. Prices for our common stock will be determined in the marketplace and may be
24
influenced by many factors, including the depth and liquidity of the market for shares of our common stock, developments affecting our business, including the impact of the factors referred to elsewhere in these Risk Factors, investor perception, and general economic and market conditions. No assurances can be given that an orderly or liquid market will ever develop for the shares of our common stock. Because of the anticipated low price of the securities, many brokerage firms may not be willing to effect transactions in these securities.
Any market that develops in shares of our common stock will be subject to the penny stock restrictions which will create a lack of liquidity and make trading difficult or impossible.
SEC Rule 15g-9 (as most recently amended and effective on September 12, 2005) establishes the definition of a "penny stock," for purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to a limited number of exceptions. It is likely that our shares will be considered to be penny stocks for the immediately foreseeable future. This classification severely and adversely affects the market liquidity for our common stock. For any transaction involving a penny stock, unless exempt, the penny stock rules require that a broker-dealer approve a person's account for transactions in penny stocks and the broker-dealer receive from the investor a written agreement to the transaction setting forth the identity and quantity of the penny stock to be purchased.
In order to approve a person's account for transactions in penny stocks, the broker-dealer must obtain financial information and investment experience and objectives of the person and make a reasonable determination that the transactions in penny stocks are suitable for that person and that person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks.
The broker-dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prepared by the SEC relating to the penny stock market, which, in highlight form, sets forth:
·
the basis on which the broker-dealer made the suitability determination, and
·
that the broker-dealer received a signed, written agreement from the investor prior to the transaction.
Disclosure also has to be made about the risks of investing in penny stock in both public offerings and in secondary trading and commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks.
Because of these regulations, broker-dealers may not wish to engage in the above-referenced necessary paperwork and disclosures and/or may encounter difficulties in their attempt to sell shares of our common stock, which may affect the ability of selling shareholders or other holders to sell their shares in the secondary market and have the effect of reducing the level of trading activity in the secondary market. These additional sales practice and disclosure requirements could impede the sale of our securities, if and when our securities become publicly traded. In addition, the liquidity for our securities may decrease, with a corresponding decrease in the price of our securities. Our shares in all probability will be subject to such penny stock rules for the foreseeable future and our shareholders will, in all likelihood, find it difficult to sell their securities.
We are an “emerging growth company” as defined in the JOBS Act and we cannot be certain whether the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
Management intends to take full advantage of the regulatory relief afforded by the JOBS Act. We are an “emerging growth company,” as defined in the JOBS Act, and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding an annual non-binding advisory vote on executive compensation and nonbinding stockholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we will rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may become more volatile.
25
In addition, Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we are choosing to “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. We will be required, under Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a control deficiency, or combination of control deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company as defined in the JOBS Act, we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the independent registered public accounting firm attestation requirement.
Our compliance with Section 404 will require that we incur substantial accounting expense and expend significant management efforts. We currently do not have an internal audit group, and we will need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge, and compile the system and process documentation necessary to perform the evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting once that firm begin its Section 404 reviews, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by the NASDAQ, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
We are incurring increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we are incurring significant legal, accounting and other expenses that we did not incur as a private company, and these expenses may increase even more after we are no longer an "emerging growth company." We will be subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Protection Act, as well as rules adopted, and to be adopted, by the SEC and the NASDAQ Stock Market. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, we expect these rules and regulations to substantially increase our legal and financial compliance costs and to make some activities more time-consuming and costly. We cannot predict or estimate the amount or timing of additional costs we may incur to respond to these requirements.
Our Board of Directors is authorized to issue shares of preferred stock, which may have rights and preferences detrimental to the rights of the holders of our common shares.
26
We are authorized to issue up to 2,000,000 shares of preferred stock, $0.0001 par value. As of the date of this filing on Form 10-K, we have not issued any shares of preferred stock. Our preferred stock may bear such rights and preferences, including dividend and liquidation preferences, as the Board of Directors may fix and determine from time to time. Any such preferences may operate to the detriment of the rights of the holders of the common stock being offered hereby.
Our Articles of Incorporation provide for indemnification of officers and directors at our expense and limit their liability which may result in a major cost to us and hurt the interests of our shareholders because corporate resources may be expended for the benefit of officers and/or directors.
Our Articles of Incorporation and applicable Delaware law provide for the indemnification of our directors, officers, employees, and agents, under certain circumstances, against attorney's fees and other expenses incurred by them in any litigation to which they become a party arising from their association with or activities on our behalf. We will also bear the expenses of such litigation for any of our directors, officers, employees, or agents, upon such person's promise to repay us, therefore if it is ultimately determined that any such person shall not have been entitled to indemnification. This indemnification policy could result in substantial expenditures by us, which we will be unable to recoup. In addition, limitations on indemnification which we are allowed to offer may discourage qualified persons from serving as our officers or directors.
We anticipate our stock being quoted on the OTCQB which may result in limited liquidity and the inability of our stockholders to maintain accurate price quotations of their stock.
Until our shares of common stock qualify for inclusion in the NASDAQ system, if ever, the trading of our securities, if any, will be in the over-the-counter market which is commonly referred to as the OTCQB as operated by OTC Markets Inc. As a result, an investor may find it difficult to dispose of, or to obtain accurate quotations as to the price of our securities.
It is anticipated that our stock price will be volatile and the value of your shares may be subject to sudden decreases.
Our stock price is likely to be volatile. The stock market in general and the market for medical technology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. The following factors, in addition to other risk factors described in this section and general market and economic conditions, may have a significant impact on the market price of our common stock:
·
results of our research and development efforts and our clinical trials;
·
the timing of regulatory approval for our products;
·
failure of any of our products, if approved, to achieve commercial success;
·
the announcement of new products or product enhancements by us or our competitors;
·
regulatory developments in the US and foreign countries;
·
ability to manufacture our products to commercial standards;
·
developments concerning our clinical collaborators, suppliers or marketing partners;
·
changes in financial estimates or recommendations by securities analysts;
·
public concern over our products;
·
developments or disputes concerning patents or other intellectual property rights;
·
product liability claims and litigation against us or our competitors;
·
the departure of key personnel;
·
changes in the structure of and third-party reimbursement in the US and other countries;
27
·
general economic, industry and market conditions; and
·
future sales of our common stock by us to fund our operations.
A decline in the market price of our common stock could cause you to lose some or all of your investment and may adversely impact our ability to attract and retain employees and raise capital. In addition, stockholders may initiate securities class action lawsuits if the market price of our stock drops significantly. Whether or not meritorious, litigation brought against us could result in substantial costs and could divert the time and attention of our management. Our insurance to cover claims of this sort may not be adequate.
Your stock ownership will be diluted by our issuance of additional securities, including in connection with subsequent rounds of financing.
We will need further financing for the continued growth of our business to achieve our planned growth. No assurance can be given as to the availability of additional financing or, if available, the terms upon which it may be obtained. Raising additional financing may result in our issuing additional securities, which could result in the dilution of your ownership percentage of us. We may also decide to issue shares in exchange for the acquisition of other companies in order to expand business operations. The issuance of any such shares will have the effect of further diluting your ownership percentage.
There may be restrictions on your ability to resell shares of Common Stock under Rule 144.
Currently, Rule 144 under the Securities Act permits the public resale of securities under certain conditions after a six or twelve month holding period by the seller, including requirements with respect to the manner of sale, sales volume restrictions, filing requirements and a requirement that certain information about the issuer is publicly available (the “Rule 144 resale conditions”). At the time that stockholders intend to resell their shares under Rule 144, there can be no assurances that we will be subject to the reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”) or, if so, current in our reporting requirements under the Exchange Act, in order for stockholders to be eligible to rely on Rule 144 at such time. In addition to the foregoing requirements of Rule 144 under the federal securities laws, the various state securities laws may impose further restrictions on the ability of a holder to sell or transfer the shares of Common Stock.
We do not anticipate paying dividends.
We never have paid any dividends on our common stock and we do not intend to pay any dividends in the foreseeable future.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES.
Our present operations do not require a permanent staffed office. We have entered a “Virtual Offices Services Agreement with an unaffiliated party which allows us to receive mail and book rooms on an “as needed” basis for an hourly fee ranging from $30 to $150 per hour depending on the size of the room. We pay $79 per month for these services and believe that they are sufficient for our foreseeable needs. The agreement is on a month to month basis and management believes that if required, the Company could obtain similar services at a similar cost.
ITEM 3. LEGAL PROCEEDINGS.
We are not currently a defendant in any legal proceeding or governmental proceeding nor are we currently aware of any pending legal proceeding or governmental proceeding proposed to be initiated against us. There are no proceedings in which any of our current directors, executive officers or affiliates, or any registered or beneficial shareholder, is an adverse party or has a material interest adverse to us.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
28
PART II
ITEM 5. MARKET FOR COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Common Stock
We are authorized to issue 20,000,000 shares of common stock. There are 5,993,816 shares of our common stock issued and outstanding as of the date of this prospectus which shares are held by approximately 60 shareholders. The holders of our common stock:
|
|
• |
Have equal ratable rights to dividends from funds legally available for payment of dividends when, as and if declared by the Board of Directors; |
|
|
• |
are entitled to share ratably in all of the assets available for distribution to holders of common stock upon liquidation, dissolution or winding up of our affairs; |
|
|
• |
do not have preemptive, subscription or conversion rights, or redemption; and |
|
|
• |
are entitled to one non-cumulative vote per share on all matters submitted to stockholders for a vote at any meeting of stockholders. |
Any market that develops in shares of our common stock will be subject to the penny stock restrictions which will make trading difficult or impossible regarding negative implications of being classified as a "Penny Stock."
Preferred Stock
We are authorized to issue 2,000,000 shares of preferred stock with designations, rights and preferences determined from time to time by our Board of Directors. No shares of preferred stock have been designated, issued or outstanding. Accordingly, our Board of Directors is empowered, without stockholder approval, to issue up to 2,000,000 shares of preferred stock with voting, liquidation, conversion, or other rights that could adversely affect the rights of the holders of the common stock. Although we have no present intention to issue any shares of preferred stock, there can be no assurance that we will not do so in the future.
Among other rights, our Board of Directors may determine, without further vote or action by our stockholders:
|
|
• |
the number of shares and the designation of the series; |
|
|
• |
whether to pay dividends on the series and, if so, the dividend rate, whether dividends will be cumulative and, if so, from which date or dates, and the relative rights of priority of payment of dividends on shares of the series; |
|
|
• |
whether the series will have voting rights in addition to the voting rights provided by law and, if so, the terms of the voting rights; |
|
|
• |
whether the series will be convertible into or exchangeable for shares of any other class or series of stock and, if so, the terms and conditions of conversion or exchange; |
|
|
• |
whether or not the shares of the series will be redeemable and, if so, the dates, terms and conditions of redemption and whether there will be a sinking fund for the redemption of that series and, if so, the terms and amount of the sinking fund; and |
|
|
• |
the rights of the shares of the series in the event of our voluntary or involuntary liquidation, dissolution or winding up and the relative rights or priority, if any, of payment of shares of the series. |
We presently do not have plans to issue any shares of preferred stock. However, preferred stock could be used to dilute a potential hostile acquirer. Accordingly, any future issuance of preferred stock or any rights to purchase preferred shares may have the effect of making it more difficult for a third party to acquire control of us. This may delay, defer or prevent a change of control in our Company or an unsolicited acquisition proposal. The issuance of preferred stock also could decrease the amount of earnings attributable to, and assets available for distribution to, the holders of our common stock and could adversely affect the rights and powers, including voting rights, of the holders of our common stock.
Authorized but Un-issued Capital Stock
Delaware law does not require stockholder approval for any issuance of authorized shares. However, the marketplace rules of the NASDAQ, which would apply only if our common stock were listed on the NASDAQ, require stockholder approval of certain
29
issuances of common stock equal to or exceeding 20% of the then-outstanding voting power or then-outstanding number of shares of common stock, including in connection with a change of control of the Company, the acquisition of the stock or assets of another company or the sale or issuance of common stock below the book or market value price of such stock. These additional shares may be used for a variety of corporate purposes, including future public offerings to raise additional capital or to facilitate corporate acquisitions.
One of the effects of the existence of un-issued and unreserved common stock may be to enable our Board of Directors to issue shares to persons friendly to current management, which issuance could render more difficult or discourage an attempt to obtain control of our board by means of a merger, tender offer, proxy contest or otherwise, and thereby protect the continuity of our management and possibly deprive the stockholders of opportunities to sell their shares of our common stock at prices higher than prevailing market prices.
Shareholder Matters
Certain provisions of Delaware law create rights that might be deemed material to our shareholders. Other provisions might delay or make more difficult acquisitions of our stock or changes in our control or might also have the effect of preventing changes in our management or might make it more difficult to accomplish transactions that some of our shareholders may believe to be in their best interests.
Transfer Agent
The transfer agent for our common stock is Nevada Agency and Transfer Company whose address is 50 West Liberty Street, Suite 880, Reno NV 89501. Its telephone number is 775-322-0626.
Market Information
Our Common Stock currently only trades on the OTCQB operated by OTC Markets Inc. under the symbol “ACXA”. Our Common Stock commenced trading under the symbol on June 16, 2014.
The following historical quotations obtained online at www.yahoo.com reflects the high and low bids for our Common Stock based on inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions:
|
Quarter Ended |
High ($) |
Low ($) |
|
December 31, 2014 |
$5.40 |
$3.20 |
|
September 30, 2014 |
$6.00 |
$5.35 |
|
June 30, 2014 |
$4.40 |
$4.00 |
As of March 17, 2015, our Common Stock closed at a price of $2.50.
Shareholders
As of March 17, 2015, we had 5,993,816 shares of common stock issued and outstanding and approximately 60 stockholders of record of our common stock.
Dividends
We have never declared or paid any cash dividends on our common stock. The payment by us of dividends, if any, in the future rests within the discretion of our board of directors and will depend, among other things, upon our earnings, capital requirements, debt covenants and financial condition, as well as other relevant factors. We do not intend to pay any cash dividends in the foreseeable future, but intend to retain all earnings, if any, for use in our business. There are no restrictions in our articles of incorporation or bylaws that prevent us from declaring dividends. The Delaware General Corporation Law, however, does prohibit us from declaring dividends where, after giving effect to the distribution of the dividend, we would not be able to pay our debts as they become due in the usual course of business, or our total assets would be less than the sum of our total liabilities plus the amount that would be needed to satisfy the rights of shareholders who have preferential rights superior to those receiving the distribution. Management is not aware of any such rights.
30
Securities Approved For Issuance Under Equity Compensation Plans
As of the end of our fiscal year ended December 31, 2014, we had no outstanding equity award and no equity compensation plan in effect under which any shares of our common stock were authorized for issuance. We do not have any compensation plan or individual compensation arrangement under which our common stock or other equity securities are authorized for issuance to employees or non-employees in exchange for consideration in the form of goods or service as described in FAS 123.
Recent Sales of Unregistered Securities
The following is a summary of all transactions during our fiscal year ended December 31, 2014. Shares issued for cash consideration paid to us are valued at the purchase price per share; all other shares are valued as stated. All shares issued were issued as “restricted” shares of our common stock except as otherwise expressly stated.
On January 10, 2014, we sold 5,000 shares for $10,000 to an investor in an isolated transaction. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On January 16, 2014, we sold 4,500 shares for $3,500 to an investor in an isolated transaction. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On July 15, 2014, we sold 15,000 shares for $30,000 to an investor in an isolated transaction. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On August 14, 2014, we issued 450,000 shares of our Company’s common stock to a consultant to provide development services over 36 months per our consulting agreement. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On September 16, 2014, in connection with entering into the UCSF License, we purchased a prototype from a contract manufacturer in exchange for 2,150,000 shares of our Company’s common stock. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On February 4, 2015, we issued 57,000 shares of our Company’s common stock to a consultant to provide investor relations services over 3 months per our consulting agreement. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On February 10, 2015, we issued 150,000 shares of our Company’s common stock to a consultant to provide media services over 6 months per our consulting agreement. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
There were no underwriters employed in connection with any of the transactions described above.
Purchase of Equity Securities by the Company and Affiliated Purchasers
Not Applicable.
ITEM 6. SELECTED FINANCIAL DATA
The Company, as a “smaller reporting company” (as defined by §229.10(f)(1)), is not required to provide the information required by this Item.
31
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following plan of operation together with our financial statements and related notes appearing elsewhere in this annual report. This plan of operation contains forward-looking statements that involve risks, uncertainties, and assumptions. The actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors.
Overview
We are a development stage company. We were incorporated in Delaware on August 29, 2012. We are focused on developing and commercializing novel neurological therapies based on our proprietary BranchPoint device delivering therapeutics directly into specific regions of the brain. The BranchPoint device can deliver therapeutics through the radial deployment of a flexible delivery catheter to large and anatomically complex brain targets through a single initial brain penetration. The BranchPoint device was developed at the University of California, San Francisco (UCSF) with $1.8 million in funding from the California Institute for Regenerative Medicine (CIRM). It is based on a neurosurgical delivery platform that we have exclusively licensed from UCSF. It can enable new approaches to neurological therapy and be modified for the delivery of a broad range of novel therapeutics, such as stem cells to treat neurodegenerative diseases, chemotherapeutics to brain tumors and gene therapy vectors.
Furthermore, we have licensed a photoacoustic technology platform from the University of Arkansas for Medical Sciences (UAMS) that allows the detection, capturing and targeted destruction of metastatic circulating tumor cells (CTCs). We have completed a proof-of-concept clinical trial and are seeking a strategic partner for further clinical development
We cannot assure you that we will be successful with our development activities.
Recent Developments
On September 16, 2014 (“Effective Date”), we entered into an exclusive license agreement (“UCSF License”) with the Regents of the University of California acting through its Office of Innovation, Technology, and Alliances, University of California San Francisco (“UCSF”) in regards to the exclusive licensing of a medical stereotactic device for the delivery of therapeutics to the human brain, characterized as “Microinjection Brain Catheter”, a.k.a. BranchPoint device ("Invention"). The invention was made in the course of research at UCSF by Drs. Daniel A. Lim, Matthew Silvestrini, and Tejal A. Desai, (collectively, the “Inventors”) and claimed in U.S. Patent Application No. PCT/US2013/052301, Microinjection Catheter; UC Case No. SF2012-063 (“Patent Rights”). The Invention was developed under funding from the California Institute for Regenerative Medicine ("CIRM") and sponsored in part by the National Institutes of Health. The UCSF License remains in effect from the Effective Date until expiration or abandonment of the Patent Rights.
Under the UCSF License, we are obligated to develop, manufacture, market and sell the invention, and have been granted the right to sublicense to third parties. Specifically, the UCSF License requires us to: (i) market the Invention for research use within three (3) months from the Effective Date; (ii) sell the Invention for research use within 12 months of the Effective Date; (iii) file and finalize any necessary regulatory documentation for FDA 510(k) approval within 6 months after the 510(k) application has been filed; (iv) market the Invention for clinical use within 6 months of receiving market approval from FDA or equivalent foreign regulatory agency; (v) sell the Invention for clinical use within 12 months of receiving market approval from FDA or equivalent foreign regulatory agency; (vi) within 1 year of the Effective Date, raise at least $750,000 in funding or revenue; (vii) market the Invention in the United States within 6 months of receiving approval from the FDA; and (viii) use commercially reasonable efforts to fill the market demand for the Invention following commencement of marketing. We are also required to pay UCSF 35% of our net sales or any sublicense royalty as defined therein, and a non-refundable license issue fee of $50,000. For a full description of further payment obligations and the UCSF License itself, the license agreement was filed as exhibit 10.1 to the Form 8-K as of September 17, 2014.
Plan of Operations
Our current plan is to continue the development of two programs in our pipeline under FDA’s Quality System Regulation 21 CFR.
1.
We are preparing the submission of a 510(k) application of our BranchPoint device to the FDA in 2015. The testing of our BranchPoint device and design history files have been finalized. Our contract manufacturer is finalizing the
32
necessary validation work before our regulatory consultant can draft and submit our 510(k) application. Once our BranchPoint device obtained regulatory approval from the FDA, our device may be used for the delivery of therapeutics such as stem cells or gene therapy vectors. We may then commercialize our BranchPoint device on our own or through a commercial collaboration with a strategic partner.
2.
We are developing our ACX-31 program to deliver temozolomide directly to a tumor site with our BranchPoint device. temozolomide is a generic, approved chemotherapy drug that is indicated for the treatment of adult patients with newly diagnosed glioblastoma multiforme concomitantly with radiotherapy and then as maintenance treatment. ACX-31 is contingent on BranchPoint's 510(k) approval. Local delivery of temozolomide has been demonstrated to be superior to oral administration in an animal model. In the scientific publication Brem S, Tyler BM, Li K, Pradilla G, Legnani F, Caplan J, et al. Local delivery of temozolomide by biodegradable polymers is superior to oral administration in a rodent glioma model. Cancer Chemother Pharmacol 2007;60:643-50., it was demonstrated that that intracranial concentrations of temozolomide increased threefold compared with orally delivered temozolomide. In a rodent glioma model, animals treated with a single temozolomide polymer (50% w/w) had a median survival of 28 days (P < 0.001 vs. controls, P < 0.001 vs. oral treatment), whereas animals treated with oral temozolomide had a median survival of 22 days compared to control animals (median survival of 13 days). Animals treated with two temozolomide polymers (50% w/w) had a median survival of 92 days (P < 0.001 vs. controls, P < 0.001 vs. oral treatment). The percentage of long-term survivors (LTS) for groups receiving intracranial temozolomide ranged from 25 to 37.5%; there were no LTS with oral temozolomide treatment. Animals treated with radiation therapy (XRT) and intracranial temozolomide (median survival not reached, LTS = 87.5%) demonstrated improved survival compared to those with intracranial temozolomide alone (median survival, 41 days; LTS = 37.5%), or oral temozolomide and XRT (median survival, 43 days, LTS = 38.9%). We are anticipating to conduct studies to demonstrate the ability of our BranchPoint device to deliver temozolomide directly to the tumor site.
The development of our products is estimated to cost approximately $5-7 million over 4-5 years as a standalone company. However, the Company may seek to pursue a strategic collaboration partnership which may accelerate the development timeline, share expenses, and also provide access to ex-US markets. The development of our products may also cost substantially more than $5-7 million and may require a substantially longer development timeline than 4-5 years. Higher development expenses and delays may be caused by but are not limited to sub-optimal product performance and continued product optimization cycles, adverse clinical results and repeat of clinical trials, or delays of an FDA approval. In such a scenario, we may not be able to secure sufficient funding or a strategic collaboration partnership and could cease operations under such circumstances.
By using our own capital resources and accessing external funding sources, we expect to operate as a technology development company with limited capital expenditures focused on commercial applications by pursuing (i) product development; (ii) capturing and protecting intellectual property; and (iii) expanding our commercial partner relationships. By employing this business model, we believe that we can effectively deploy capital and maximize shareholder return.
As we continue the development of our pipeline programs, we are seeking strategic partnerships with established healthcare companies to pursue further development, regulatory approval and commercialization of our programs. We do not expect to manufacture finished products in-house, nor conduct direct or indirect sales of products which may allow the Company to avoid significant capital investment in production facilities and sales and marketing teams. It is difficult to predict whether we will be able to enter into beneficial commercial partner relationships with recognized healthcare companies and our ultimate success remains uncertain.
Need for Additional Capital
To become profitable and competitive, and execute strategic acquisitions, we may have to raise additional capital. If we are unable to raise additional equity capital to develop our business and continue earning revenues, we might have to suspend or cease operations and our investors may lose their investment.
We have no assurance that future financings will be available to us on acceptable terms. If financing is not available on satisfactory terms, we may be unable to continue, develop, or expand our operations. Equity financing could result in additional dilution to existing shareholders.
33
Liquidity and Capital Resources
The reader is referred to our financial statements included elsewhere herein. As of December 31, 2014, we had $555,506 in cash and cash equivalents. We are actively seeking additional capital investment as the total course of our product development over the next four to five years is estimated by management to be from $5 million to $7 million as a standalone company. We cannot assure you that we will have access to the funding required for our product development or that if available it will be available on terms that are not dilutive to our present shareholders. If the funding is not available, we may have to severely curtail or cease operations. We presently intend to raise a minimum of $500,000 and a maximum of $2,500,000 in the next twelve months, but these plans are subject to change based on many factors, including, but not limited to, the availability of funds to us. We believe that our OTCQB listing will assist us in our funding efforts. Most of the funds we raise will be applied directly towards continued development of our pipeline products.
Results of Operations for the Twelve Months ended December 31, 2014
Revenues
We had no revenues as a development stage company for the twelve months ended December 31, 2014 and 2013, respectively.
Operating Expenses
Depreciation and Amortization Expenses: Depreciation and Amortization expenses were $94,298 and $11,817 for the twelve months ended December 31, 2014 and 2013, respectively. The increase was due to the amortization of prepaid assets in connection with prototype optimization.
Impairment Loss: Impairment losses were $4,304,927 and $95,940 for the twelve months ended December 31, 2014 and 2013, respectively. The increase was caused by the write-off of the licensing fees, which were incurred in connection with the UAMS License, and the write-off of the fixed assets, which were purchased in connection with the UCSF License, because further development is uncertain and contingent on the availability of sufficient funding.
Research and Development: Research and development expenses were $177,414 and $529,874 for the twelve months ended December 31, 2014 and 2013, respectively. The decrease was due to the completion of the proof-of-concept clinical trial that was conducted in connection with the UAMS License in 2013.
General and Administrative: General and administrative expenses were $123,127 and $122,100 for the twelve months ended December 31, 2014 and 2013, respectively. They consisted of employee salaries, legal and audit fees, and other administrative expenses.
Net Loss
We had a net loss of $4,737,670 and $770,709 for the twelve months ended December 31, 2014 and 2013, respectively. The increase was due to higher impairment losses caused by write-offs.
Off-Balance Sheet Arrangements
We do not have any off balance sheet arrangements that are reasonably likely to have a current or future effect on our financial condition, revenues, and results of operations, liquidity or capital expenditures.
CRITICAL ACCOUNTING POLICIES AND USE OF ESTIMATES
We prepare our financial statements in accordance with generally accepted accounting principles in the United States of America. The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and related disclosures of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Significant accounting policies and methods used in preparation of the financial statements are described in Note 1 to our financial statements included in this Form 10-k. We evaluate our estimates and assumptions on a regular basis, based on historical experience and other relevant factors. Actual results could differ materially from these estimates and assumptions. The following critical accounting policies are impacted by judgments, assumptions and estimates used in preparation of our financial statements included in this registration statement.
34
Patent Rights
We capitalized as an asset the licensing fees incurred in connection with the exclusive license agreement (“UAMS License”) executed between the Company and the University of Arkansas for Medical Sciences (“UAMS”) on December 15, 2012 (“Effective Date”). The capitalized licensing fees of $87,499 net of an accumulated amortization expense of $115,072 were written off as an impairment loss as of December 31, 2014 because, as previously disclosed in a filing on Form 8-K on February 24, 2014, the Company confirmed on or about February 3, 2014, that a portion of one of the three licensed patent applications, U.S. Patent Application Serial No. 12/334,217, Device and Method for In Vivo Flow Cytometry Using the Detection of Photoacoustic Waves; UAMS ID No. 2008-16, had been rejected by the United States Patent and Trademark Office and U.S. Patent Application Serial No. 12/334,217 was then abandoned prior to the date of our UAMS License. The basis for the rejection was primarily that the rejected portion of the application had previously been published in a scientific journal by the inventor Prof. Vladimir Zharov, an employee of the UAMS, prior to the filing of the application and therefore was not patentable.
Prepaid Assets
On August 14, 2014, we engaged a consultant to provide development services over 36 months in exchange for 450,000 shares of the Company’s common stock. The 450,000 issued shares of common stock are valued at $2 per share, equal to the price per share paid by an investor in the most recent sale of the Company’s shares of common stock on July 15, 2014, and are capitalized in the amount of $900,000 as prepaid assets on the Company’s balance sheet. These prepaid assets are amortized on a straight-line basis over the 36-month term of the consulting agreement.
Fixed Assets
On September 16, 2014, in connection with entering into the UCSF License, we purchased a prototype from a contract manufacturer in exchange for 2,150,000 shares of the Company’s common stock. The 2,150,000 issued shares of common stock are valued at $2 per share, equal to the price per share paid by an investor in the most recent sale of the Company’s shares of common stock on July 15, 2014, and are capitalized in the amount of $4,300,000 as fixed assets on the Company’s balance sheet. These fixed assets were amortized on a straight-line basis over the remaining 15 years of estimated patent life of the patent rights underlying the UCSF License, until they were written off as of December 31, 2014 because further development of the prototype is uncertain and contingent on the availability of sufficient funding.
On October 13, 2014 we purchased equipment to support the development of our prototype device which are capitalized in the amount of $162,294 as fixed assets on the Company’s balance sheet. The equipment is amortized on a straight-line basis over 5 years.
Research and Development Expenses
We expense all of our research and development expenses in the period in which they are incurred. At such time as our products are determined to be commercially available, we will capitalize those development expenditures that are related to the maintenance of the commercial products, and amortize these capitalized expenditures over the estimated life of the commercial product. The estimated life of the commercial product will be based on management’s estimates, including estimates of current and future industry conditions. A significant change to these assumptions could impact the estimated useful life of our commercial products resulting in a change to amortization expense and impairment charges.
Impact of New Accounting Standards
The Company does not expect the adoption of recently issued accounting pronouncements to have a significant impact on the Company's results of operations, financial position, or cash flow.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable
35
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Set forth below is a list of audited financial statements for the years ended December 31, 2014 and 2013.
INDEX TO FINANCIAL STATEMENTS
|
Report of Independent Registered Public Accounting Firm Seale & Beers, CPAs |
F-1 | |||||||
|
|
|
|
|
|
|
|
|
|
|
Balance Sheets at December 31, 2014 and 2013 |
F-2 | |||||||
|
|
|
|
|
|
|
|
|
|
|
Statements of Operations for the year ended December 31, 2014 and 2013 |
F-3 | |||||||
|
|
|
|
|
|
|
|
|
|
|
Statements of Cash Flow for the year ended December 31, 2014 and 2013 |
F-4 | |||||||
|
|
|
|
|
|
|
|
|
|
|
Statements of Stockholders' Deficit for the year ended December 31, 2014 and 2013 |
F-5 | |||||||
|
|
|
|
|
|
|
|
|
|
|
Notes to Financial Statements |
F-6 | |||||||
36
SEALE AND BEERS, CPAs
PCAOB & CPAB REGISTERED AUDITORS
www.sealebeers.com
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Accurexa, Inc.
We have audited the accompanying balance sheets of Accurexa, Inc. as of December 31, 2014, and the related statements of income, stockholders’ equity (deficit), and cash flows for the period ended December 31, 2014. Accurexa, Inc.’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Accurexa, Inc. as of December 31, 2014, and the related statements of income, stockholders’ equity (deficit), and cash flows for the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has no revenues, has incurred recurring losses and recurring negative cash flow from operating activities, and has an accumulated deficit which raises substantial doubt about its ability to continue as a going concern. Management’s plans concerning these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Seale and Beers, CPAs
Seale and Beers, CPAs
Las Vegas, Nevada
March 25, 2015
F-1
|
ACCUREXA INC. | |||||||
|
Balance Sheets | |||||||
|
(Audited) | |||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2014 |
|
December 31, 2013 |
|
ASSETS |
|
|
| ||||
|
|
|
|
|
|
|
|
|
|
Current assets |
|
|
|
|
| ||
|
|
Cash and cash equivalents |
|
$ |
555,506 |
$ |
818,070 | |
|
|
Total current assets |
|
|
555,506 |
|
818,070 | |
|
|
|
|
|
|
|
|
|
|
Patent rights, net |
|
|
- |
|
94,251 | ||
|
Prepaid Assets |
|
|
786,685 |
|
- | ||
|
Fixed Assets, net |
|
|
157,320 |
|
- | ||
|
|
|
|
|
|
|
|
|
|
|
Total assets |
|
$ |
1,499,511 |
$ |
912,321 | |
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS' EQUITY |
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
CURRENT LIABILITIES |
|
|
|
|
| ||
|
|
Accounts payable and other accruals, including related party liabilities of $105,000 as of December 31, 2014 and $45,000 as of December 31, 2013 |
|
$ |
266,026 |
$ |
209,679 | |
|
|
Total current liabilities |
|
|
266,026 |
|
209,679 | |
|
|
|
|
|
|
|
|
|
|
|
Convertible Notes, net of unamortized debt discount of $16,940 |
|
|
353,060 |
|
350,000 | |
|
|
|
|
|
|
|
|
|
|
|
Total liabilities |
|
|
619,085 |
|
559,679 | |
|
|
|
|
|
|
|
|
|
|
Commitments and contingencies |
|
|
|
|
| ||
|
STOCKHOLDERS' EQUITY |
|
|
|
|
| ||
|
|
Common stock |
|
|
|
|
| |
|
|
|
Authorized 20,000,000 shares at par value of $ 0.0001 each Issued and outstanding 5,786,816 shares as of December 31, 2014 and 3,162,316 shares as of December 31, 2013 |
|
|
579 |
|
316 |
|
|
Common Stock Issuable |
|
|
2,000 |
|
- | |
|
|
Additional paid-in capital |
|
|
6,425,151 |
|
1,161,960 | |
|
|
Accumulated deficit |
|
|
(5,547,305) |
|
(809,634) | |
|
|
|
Total stockholders' deficit |
|
|
880,425 |
|
352,642 |
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders' deficit |
|
$ |
1,499,511 |
$ |
912,321 | |
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these financial statements. | |||||||
F-2
|
ACCUREXA INC. | |||||||
|
Statements of Operations | |||||||
|
(Audited) | |||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2014 |
|
Year Ended December 31, 2013 |
|
|
|
|
|
|
|
|
|
|
Revenue |
|
$ |
- |
$ |
- | ||
|
|
|
|
|
|
|
|
|
|
Operating Expenses |
|
|
|
|
| ||
|
|
Depreciation and Amortization Expense |
|
|
94,298 |
|
11,817 | |
|
|
Impairment Loss |
|
|
4,304,927 |
|
95,940 | |
|
|
Research & Development |
|
|
177,414 |
|
529,874 | |
|
|
General and Administrative |
|
|
123,127 |
|
122,100 | |
|
Total Operating Expenses |
|
|
4,699,766 |
|
759,730 | ||
|
|
|
|
|
|
|
|
|
|
Loss from Operations |
|
|
(4,699,766) |
|
(759,730) | ||
|
|
|
|
|
|
|
|
|
|
Interest Income (Expense) |
|
|
(37,904) |
|
(8,364) | ||
|
Realized Income (Loss) |
|
|
- |
|
(2,615) | ||
|
Loss before provisions for income taxes |
|
|
(4,737,670) |
|
(770,709) | ||
|
|
|
|
|
|
|
|
|
|
Provision for income taxes |
|
|
- |
|
- | ||
|
Net Loss |
|
$ |
(4,737,670) |
$ |
(770,709) | ||
|
|
|
|
|
|
|
|
|
|
Loss per common share - basic and fully diluted: |
|
$ |
(1.19) |
$ |
(0.24) | ||
|
|
|
|
|
|
|
|
|
|
Weighted average number of basic and fully diluted common shares outstanding |
|
|
3,976,122 |
|
3,149,541 | ||
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these financial statements. | |||||||
F-3
|
ACCUREXA INC. | ||||||
|
Statements of Cash Flows | ||||||
|
(Audited) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2014 |
|
Year Ended December 31, 2013 |
|
|
|
|
|
|
|
|
|
Cash flows from operations: |
|
|
|
| ||
|
|
Loss from continuing operations |
$ |
(4,737,670) |
$ |
(770,709) | |
|
|
Adjustment to reconcile net loss to net cash used in operating activities: |
|
| |||
|
|
|
Depreciation and Amortization |
|
94,298 |
|
11,817 |
|
|
|
Impairment Loss |
|
4,304,927 |
|
95,940 |
|
|
|
Amortization of non-cash R&D expenses |
|
113,315 |
|
- |
|
|
|
Amortization of discount on convertible notes |
|
3,060 |
|
- |
|
|
|
Realized Gain/Loss on marketable securities |
|
- |
|
2,615 |
|
|
Changes in operating assets and liabilities: |
|
|
|
| |
|
|
|
Accounts payable and other accruals |
|
58,346 |
|
146,624 |
|
Net cash used in operations |
|
(163,724) |
|
(513,713) | ||
|
|
|
|
|
|
|
|
|
Investment activities: |
|
|
|
| ||
|
|
Investment in fixed assets |
|
(162,294) |
|
- | |
|
|
Investment in patent rights |
|
- |
|
- | |
|
|
Investment in marketable securities |
|
- |
|
(2,615) | |
|
Net cash used in investment activities |
|
(162,294) |
|
(2,615) | ||
|
|
|
|
|
|
|
|
|
Financing activities: |
|
|
|
| ||
|
|
Convertible notes received |
|
20,000 |
|
350,000 | |
|
|
Share subscriptions received |
|
43,454 |
|
35,953 | |
|
Net cash provided by financing activities |
|
63,454 |
|
385,953 | ||
|
|
|
|
|
|
|
|
|
Net (decrease) / increase in cash |
|
(262,564) |
|
(130,375) | ||
|
|
|
|
|
|
|
|
|
Cash, beginning of period |
|
818,070 |
|
948,445 | ||
|
Cash, end of period |
$ |
555,506 |
$ |
818,070 | ||
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash flow information: |
|
|
|
| ||
|
|
Cash paid for interest |
$ |
35,000 |
$ |
- | |
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these financial statements. | ||||||
F-4
|
ACCUREXA INC. | ||||||
|
Statement of Stockholders' Equity | ||||||
|
(Audited) | ||||||
|
|
|
|
|
|
|
|
|
|
Common Stock |
Amount |
Common Stock Issuable |
Additional paid-in capital |
Accumulated (Deficit) |
Stockholders' Equity |
|
|
|
|
|
|
|
|
|
Balance December 31, 2012 |
3,126,316 |
$ 313 |
$ - |
$ 1,126,010 |
$ (38,925) |
$ 1,087,398 |
|
|
|
|
|
|
|
|
|
Shares issued pursuant to private placement on February 6, 2013 |
10,000 |
1 |
- |
9,952 |
- |
9,953 |
|
Shares issued pursuant to Bill Callahan consulting agreement on June 15, 2013 |
26,000 |
3 |
- |
25,997 |
- |
26,000 |
|
Net (loss) for the period |
- |
- |
- |
- |
(770,709) |
(770,709) |
|
|
|
|
|
|
|
|
|
Balance December 31, 2013 |
3,162,316 |
$ 316 |
$ - |
$ 1,161,960 |
$ (809,634) |
$ 352,642 |
|
|
|
|
|
|
|
|
|
Shares issued pursuant to private placements |
24,500 |
2 |
- |
43,452 |
- |
43,454 |
|
Common Stock Issuable to Mr. Callahan |
- |
- |
2,000 |
- |
- |
2,000 |
|
Shares issued pursuant to consulting agreement |
450,000 |
45 |
- |
899,955 |
- |
900,000 |
|
Shares issued pursuant to asset purchase agreement |
2,150,000 |
215 |
- |
4,299,785 |
- |
4,300,000 |
|
Discount on Convertible Note |
- |
- |
- |
20,000 |
- |
20,000 |
|
Net (loss) for the period |
- |
- |
- |
- |
(4,737,670) |
(4,737,670) |
|
|
|
|
|
|
|
|
|
Balance December 31, 2014 |
5,786,816 |
$ 579 |
$ 2,000 |
$ 6,425,151 |
$ (5,547,305) |
$ 880,425 |
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these financial statements. | ||||||
F-5
ACCUREXA INC.
NOTES TO THE FINANCIAL STATEMENTS
December 31, 2014
1 - SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Description of Business
Accurexa, Inc. (the “Company”) was incorporated under the laws of the state of Delaware on August 29, 2012. The Company elected its fiscal year ending on December 31. The Company is a development stage company as defined in the Accounting Standards Codification 915, Development Stage Entities. All activities of the Company to date relate to its organization, initial funding and share issuances, and research & development.
The Company is focused on developing and commercializing novel neurological therapies based on its proprietary BranchPoint device delivering therapeutics directly into specific regions of the brain. The BranchPoint device can deliver therapeutics through the radial deployment of a flexible delivery catheter to large and anatomically complex brain targets through a single initial brain penetration. The BranchPoint device was developed at the University of California, San Francisco (UCSF) with $1.8 million in funding from the California Institute for Regenerative Medicine (CIRM). It is based on a neurosurgical delivery platform that the Company has exclusively licensed from UCSF. It may enable new approaches to neurological therapy and be modified for the delivery of a broad range of novel therapeutics, such as stem cells to treat neurodegenerative diseases, chemotherapeutics to brain tumors and gene therapy vectors.
Furthermore, the Company has licensed a photoacoustic technology platform from the University of Arkansas for Medical Sciences (UAMS) that allows the detection, capturing and targeted destruction of metastatic circulating tumor cells (CTCs). The Company has completed a proof-of-concept clinical trial and is seeking a strategic partner for further clinical development.
Accounting Basis
These financial statements are prepared on the accrual basis of accounting in conformity with accounting principles generally accepted in the United States of America.
Use of Estimates
Management uses estimates and assumptions in preparing financial statements. Those estimates and assumptions affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities, and the reported revenues and expenses. Actual results could differ from these estimates.
Cash and Cash Equivalents
Cash equivalents comprise certain highly liquid instruments with a maturity of three months or less when purchased.
Concentration of Credit Risk and Financial Instruments
Financial instruments which potentially subject the Company to concentration of credit risk consist principally of cash balances maintained at creditworthy financial institutions. The Company maintained cash balances in bank checking and savings accounts which, at times, either may exceed insured limits set or are not insured by the Federal Deposit Insurance Corporation (FDIC). As of December 31, 2014, $118,301 of the Company’s cash balances at an offshore bank were not insured by the FDIC. The Company has not experienced any losses in such accounts and believes it is not exposed to any significant credit risk on its cash and cash equivalents.
Patent Rights
Patent rights are carried at cost net of accumulated amortization on a straight-line basis over their estimated remaining patent lives. Estimated patent life is 20 years from the date on which the application for a patent was filed.
F-6
ACCUREXA INC.
NOTES TO THE FINANCIAL STATEMENTS
December 31, 2014
Fair Value of Financial Instruments
The Company’s financial instruments consist of cash and accounts payable. The carrying values of financial instruments reflected in these financial statements approximate their fair values due to the short-term maturity of the instruments.
Earnings (Loss) per Share
The basic earnings (loss) per share are calculated by dividing the Company’s net income available to common shareholders by the weighted average number of common shares outstanding during the reported period. The diluted earnings (loss) per share are calculated by dividing the Company’s net income (loss) available to common shareholders by the diluted weighted average number of shares outstanding during the year. The diluted weighted average number of shares outstanding is the basic weighted number of shares adjusted as of the first of the period for any potentially dilutive debt or equity. There are no diluted shares outstanding.
Revenue Recognition
The Company has no current source of revenue; therefore the Company has not yet adopted any policy regarding the recognition of revenue.
Start-up Cost
The Company accounts for start-up costs pursuant to the provisions of the Accounting Standard Codification 720-15. Accounting for start-up costs require all costs incurred in connection with the start-up and organization of the Company be expensed as incurred.
Research and Development Expenses
We expense all of our research and development expenses in the period in which they are incurred. At such time as our products are determined to be commercially available, we will capitalize those development expenditures that are related to the maintenance of the commercial products, and amortize these capitalized expenditures over the estimated life of the commercial product. The estimated life of the commercial product will be based on management’s estimates, including estimates of current and future industry conditions. A significant change to these assumptions could impact the estimated useful life of our commercial products resulting in a change to amortization expense and impairment charges.
Income Taxes
The Company accounts for income taxes pursuant to the provisions of the Accounting Standards Codification 740, Accounting for Income Taxes, which requires an asset and liability approach to calculate deferred income taxes. The asset and liability approach requires the recognition of deferred tax liabilities and assets for the expected future tax consequences of temporary difference between the carrying amounts and the tax basis of assets and liabilities. As a result of the initial period’s incurred loss the deferred tax asset has been fully reserved.
Recent Accounting Pronouncements
On June 10, 2014, the FASB issued ASU 2014-10, Elimination of Certain Financial Reporting Requirements, including Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation. The content resulting from the issuance of ASU 2014-10 eliminates inception-to-date presentation and other disclosure requirements in ASC Topic 915 for entities previously considered development stage entities. Early adoption is permitted, and the Company has elected to make an early adoption of ASU 2014-10.
The Company's management has evaluated all other recently issued accounting pronouncements through the filing date of these financial statements and does not believe that any of these pronouncements will have a material impact on the Company's financial position and results of operations.
F-7
ACCUREXA INC.
NOTES TO THE FINANCIAL STATEMENTS
December 31, 2014
2 - GOING CONCERN
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As shown in the accompanying financial statements, the Company has incurred net losses and negative cash flows from operating activities for the year ended December 31, 2014, and has an accumulated deficit of $5,547,305 as of December 31, 2014. The Company has relied upon raising capital through private placements to fund its ongoing operations to date, and expects to continue to do so, as it has yet to generate cash from its operating activities. These factors raise substantial doubt about the Company’s ability to continue as a going concern. The Company intends to raise additional capital through equity and/or debt financings as well as through entering into sub-license agreements with strategic partners in order to ensure the Company continues operations. The financial statements do not include any adjustments that might be necessary if the Company is unable to continue as a going concern.
3 – PATENT RIGHTS
The Company capitalized as an asset the licensing fees incurred in connection with the exclusive license agreement (“UAMS License”) executed between the Company and the University of Arkansas for Medical Sciences (“UAMS”) on December 15, 2012 (“Effective Date”). The capitalized licensing fees of $87,499 net of an accumulated amortization expense of $115,072 were written off as an impairment loss as of December 31, 2014 because, as previously disclosed in a filing on Form 8-K on February 24, 2014, the Company confirmed on or about February 3, 2014, that a portion of one of the three licensed patent applications, U.S. Patent Application Serial No. 12/334,217, Device and Method for In Vivo Flow Cytometry Using the Detection of Photoacoustic Waves; UAMS ID No. 2008-16, had been rejected by the United States Patent and Trademark Office and U.S. Patent Application Serial No. 12/334,217 was then abandoned prior to the date of our UAMS License. The basis for the rejection was primarily that the rejected portion of the application had previously been published in a scientific journal by the inventor Prof. Vladimir Zharov, an employee of the UAMS, prior to the filing of the application and therefore was not patentable.
Gross carrying amount was $0 and $94,252 as of December 31, 2014 and 2013, respectively. Accumulated amortization was $115,072 and $108,319 as of December 31, 2014 and 2013, respectively. Amortization expense was $94,251, of which $87,499 was recorded as impairment loss as of December 31, 2014. Amortization expense was $107,757, of which $95,940 was recorded as impairment loss as of December 31, 2013.
4 – PREPAID ASSETS
On August 14, 2014, the Company engaged a consultant to provide development services over 36 months in exchange for 450,000 shares of the Company’s common stock. The 450,000 issued shares of common stock are valued at $2 per share, equal to the price per share paid by an investor in the most recent sale of the Company’s shares of common stock on July 15, 2014, and are capitalized in the amount of $900,000 as prepaid assets on the Company’s balance sheet. These prepaid assets are amortized on a straight-line basis over the 36-month term of the consulting agreement.
5 – FIXED ASSETS
On September 16, 2014, in connection with entering into the UCSF License, the Company purchased a prototype from a contract manufacturer in exchange for 2,150,000 shares of the Company’s common stock. The 2,150,000 issued shares of common stock are valued at $2 per share, equal to the price per share paid by an investor in the most recent sale of the Company’s shares of common stock on July 15, 2014, and are capitalized in the amount of $4,300,000 as fixed assets on the Company’s balance sheet. These fixed assets were amortized on a straight-line basis over the remaining 15 years of estimated patent life of the patent rights underlying the UCSF License, until they were written off as of December 31, 2014 because further development of the prototype is uncertain and contingent on the availability of sufficient funding.
On October 13, 2014 the Company purchased equipment to support the development of its prototype device which are capitalized in the amount of $162,294 as fixed assets on the Company’s balance sheet. The equipment is amortized on a straight-line basis over 5 years.
F-8
ACCUREXA INC.
NOTES TO THE FINANCIAL STATEMENTS
December 31, 2014
Gross carrying amount was $157,320 and $0 as of December 31, 2014 and 2013, respectively. Accumulated amortization was $82,572 and $0 as of December 31, 2014 and 2013, respectively. Depreciation expense was $4,304,974, of which $4,217,428 was recorded as impairment loss as of December 31, 2014. There was no depreciation expense and impairment loss as of December 31, 2013.
6 – INCOME TAXES
The Company provides for income taxes under ASC Topic 740 which requires the use of an asset and liability approach in accounting for income taxes. Deferred tax assets and liabilities are recorded based on the differences between the financial statement and tax bases of assets and liabilities and the tax rates in effect currently.
ASC 740 requires the reduction of deferred tax assets by a valuation allowance if, based on the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. In the Company’s opinion, it is uncertain whether they will generate sufficient taxable income in the future to fully utilize the net deferred tax asset. Accordingly, a valuation allowance equal to the deferred tax asset has been recorded. The total deferred tax asset is $1,610,808 which is calculated by multiplying a 34% estimated tax rate by the cumulative NOL of $4,737,670. The total valuation allowance is a comparable $1,610,808. Details are as follows:
|
|
Twelve Months Ended December 31, 2014 |
|
Deferred Tax Asset |
1,610,808 |
|
Valuation Allowance |
(1,610,808) |
|
Current Taxes Payable |
0.00 |
|
Income Tax Expense |
$ 0.00 |
Below is a chart showing the estimated corporate federal net operating loss (NOL) and the year in which it will expire.
|
Year |
Amount |
Expiration |
|
2012 |
$ 8,564 |
2032 |
|
2013 |
$ 262,041 |
2033 |
|
2014 |
$ 1,610,808 |
2034 |
|
Total NOL |
$ 1,881,413 |
|
7 – RELATED PARTY TRANSACTIONS
Accounts payable includes $105,000 of accrued salary due to the Company’s Chief Executive Officer as of December 31, 2014.
8 – STOCKHOLDERS’ EQUITY
The Company has authorized 20,000,000 Common Shares and 2,000,000 Preferred Shares at a par value of $0.0001 per share.
On January 10, 2014, we sold 5,000 shares for $10,000 to an investor in an isolated transaction. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On January 16, 2014, we sold 4,500 shares for $3,500 to an investor in an isolated transaction. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
F-9
ACCUREXA INC.
NOTES TO THE FINANCIAL STATEMENTS
December 31, 2014
On July 15, 2014, we sold 15,000 shares for $30,000 to an investor in an isolated transaction. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On August 14, 2014, we issued 450,000 shares of our Company’s common stock to a consultant to provide development services over 36 months per our consulting agreement. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On September 16, 2014, in connection with entering into the UCSF License, we purchased a prototype from a contract manufacturer in exchange for 2,150,000 shares of our Company’s common stock. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On February 4, 2015, we issued 57,000 shares of our Company’s common stock to a consultant to provide investor relations services over 3 months per our consulting agreement. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
On February 10, 2015, we issued 150,000 shares of our Company’s common stock to a consultant to provide media services over 6 months per our consulting agreement. These shares were issued pursuant to Regulation D under the Securities Act of 1933, as amended, were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and bear an appropriate restrictive legend.
There were no underwriters employed in connection with any of the transactions described above.
9 - CONVERTIBLE NOTES
On July 25, 2013, the Company entered into a secured convertible note (“Note”) under which the Company received $350,000 from the convertible note holder (“Holder”) and is obligated to pay to the Holder the full principal amount after 36 months from the date of the Note, plus an interest at the rate of 10.0% per year payable at the end of each year from the date of the Note. The Holder has the right to convert the Note, in whole or in part, into shares of common stock of the Company (“Common Stock”) at a fixed rate of $2.00 per share (the “Conversion Price”) at any time. The number of shares issuable on full conversion was 175,000 and their fair market value exceeded the principal amount of the Note by $210,000 as of December 31, 2014. The Company may prepay the Note in whole or in part at any time for cash on 15 business days’ prior written notice, subject to the right of the Holder to convert into shares of Common Stock of the Company prior to any prepayment. The Note agreement was filed on August 14, 2013 as an exhibit to Form 10-Q for the three months ended September 30, 2013.
On July 15, 2014, the Company entered into a secured convertible note (“Note”) under which the Company received $20,000 from the convertible note holder (“Holder”) and is obligated to pay to the Holder the full principal amount after 36 months from the date of the Note. The Holder has the right to convert the Note, in whole or in part, into shares of common stock of the Company (“Common Stock”) at a "Variable Conversion Price" of 50% multiplied by the Market Price (representing a discount rate of 50%). “Market Price” means the average of the Closing Trading Prices for the Common Stock during the ten (10) Trading Day period ending on the latest complete Trading Day prior to the Conversion Date. The Company may prepay the Note in whole or in part at any time for cash on 15 business days’ prior written notice, subject to the right of the Holder to convert into shares of Common Stock of the Company prior to any prepayment. The number of shares issuable on full conversion was 12,500 and their fair market value exceeded the principal amount of the Note by $20,000 as of December 31, 2014. The Company has determined the value associated with the beneficial conversion feature in connection with the notes to be $20,000. The aggregate beneficial conversion feature was accreted and charged to interest expense in the amount of $3,060 as of December 31, 2014, and will be amortized until July 15, 2017.
F-10
ACCUREXA INC.
NOTES TO THE FINANCIAL STATEMENTS
December 31, 2014
10 – SUBSEQUENT EVENTS
On January 12, 2015, the Company and the Lim Development Group (“Consultant”) entered into a consulting agreement (“Agreement”) under which the Consultant provides scientific advisory to the Company in the development, partnering and commercialization of the “Microinjection Brain Catheter” (a.k.a. BranchPoint device) that the Company exclusively licensed from the University of California San Francisco (“UCSF”) on September 16, 2014. The Consultant will also serve as a member of the Company’s newly formed Scientific Advisory Board. The term of the Agreement shall be two years and may be extended by mutual agreement of both parties. As consideration for provided services during the term of the Agreement, the Consultant shall receive either a project fee or an hourly rate, either of which will be determined and agreed upon by both parties prior to commencement of any work or project. Under the Agreement, the Company also issued a promissory note (“Note”) in the amount of $200,000 and at an interest rate of 5.00% per annum to the Consultant. Under the Note, the Consultant shall have the right, exercisable at any time at the Consultant’s sole discretion, to convert all or a portion of the outstanding principal amount of and all accrued interest under the Note, in whole or in part, into shares of common stock of the Company (the “Shares”) at a conversion price of $0.20 per share. The Shares are issuable pursuant to Regulation D under the Securities Act of 1933, as amended, are to be exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”), and are to bear an appropriate restrictive legend. A full description of the Agreement was filed as exhibit 10.1 and of the Note was filed as exhibit 10.2 to the Form 8-K as of January 13, 2015.
On February 18, 2015, the Company entered into a consulting agreement (“Agreement”) with the Capital Communications Group (“Consultant”) under which the Consultant assists the Company in its efforts to gain greater recognition and awareness among relevant investors in the public capital markets on a non-exclusive basis. In connection with the Agreement, the Company issued a four-year Warrant (“Warrant”) to the Consultant under which the Consultant is entitled to purchase from the Company up to 200,000 shares of the Company’s Common Stock (“Warrant Shares”) at an exercise price of $0.50 per Share (“Exercise Price”). The Warrant is exercisable, in whole or in part, during the term commencing on the issuance date of the Warrant on February 18, 2015 and ending on February 18, 2019 (the “Exercise Period”). A full description of the Agreement is filed herewith as exhibit 10.13.
F-11
ITEM 9. CHANGES AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURES
None
ITEM 9A. CONTROLS AND PROCEDURES
Regulations under the Securities Exchange Act of 1934 (the “Exchange Act”) require public companies to maintain “disclosure controls and procedures,” which are defined as controls and other procedures that are designed to ensure that information required to be disclosed by the issuer in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by an issuer in the reports that it files or submits under the Exchange Act is accumulated and communicated to the issuer's management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
We conducted an evaluation, with the participation of our Chief Executive Officer who is also our Principal Financial Officer, of the effectiveness of our disclosure controls and procedures as of December 31, 2014. Based on that evaluation, our Chief Executive Officer and Principal Financial Officer has concluded that as of December 31, 2014, our disclosure controls and procedures were not effective at the reasonable assurance level due to the material weaknesses described below.
In light of the material weaknesses described below, we performed additional analysis and other post-closing procedures to ensure our financial statements were prepared in accordance with generally accepted accounting principles. Accordingly, we believe that the financial statements included in this report fairly present, in all material respects, our financial condition, results of operations and cash flows for the periods presented.
A material weakness is a control deficiency (within the meaning of the Public Company Accounting Oversight Board (PCAOB) Auditing Standard No. 2) or combination of control deficiencies that result in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected. Management has identified the following two material weaknesses which have caused management to conclude that, as of December 31, 2014, our disclosure controls and procedures were not effective at the reasonable assurance level:
1. We do not have written documentation of our internal control policies and procedures. Written documentation of key internal controls over financial reporting is a requirement of Section 404 of the Sarbanes-Oxley Act which is applicable to us for the year ending December 31, 2014. Management evaluated the impact of our failure to have written documentation of our internal controls and procedures on our assessment of our disclosure controls and procedures and has concluded that the control deficiency that resulted represented a material weakness.
2. We do not have sufficient segregation of duties within accounting functions, which is a basic internal control. Due to our size and nature, segregation of all conflicting duties may not always be possible and may not be economically feasible. However, to the extent possible, the initiation of transactions, the custody of assets and the recording of transactions should be performed by separate individuals. Management evaluated the impact of our failure to have segregation of duties on our assessment of our disclosure controls and procedures and has concluded that the control deficiency that resulted represented a material weakness.
To address these material weaknesses, management performed additional analyses and other procedures to ensure that the financial statements included herein fairly present, in all material respects, our financial position, results of operations and cash flows for the periods presented.
Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, the issuer’s principal executive and principal financial officers and effected by the issuer’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America and includes those policies and procedures that:
37
|
|
1. |
Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the issuer; |
|
|
|
|
|
|
2. |
Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the issuer; and |
|
|
|
|
|
|
3. |
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the issuer’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Because of the inherent limitations of internal control, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
As of the end of our most recent quarter, management assessed the effectiveness of our internal control over financial reporting based on the criteria for effective internal control over financial reporting established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO") and SEC guidance on conducting such assessments. Based on that evaluation, they concluded that, as of December 31, 2014, such internal control over financial reporting was not effective. This was due to deficiencies that existed in the design or operation of our internal control over financial reporting that adversely affected our internal controls and that may be considered to be material weaknesses.
The matters involving internal control over financial reporting that our management considered to be material weaknesses under the standards of the Public Company Accounting Oversight Board were: (1) lack of a functioning audit committee due to a lack of a majority of independent members and a lack of a majority of outside directors on our board of directors, resulting in ineffective oversight in the establishment and monitoring of required internal controls and procedures; and (2) inadequate segregation of duties consistent with control objectives of having segregation of the initiation of transactions, the recording of transactions and the custody of assets. The aforementioned material weaknesses were identified by our Chief Executive Officer in connection with the review of our financial statements as of December 31, 2014.
Management believes that the material weaknesses set forth in items (1) and (2) above did not have an effect on our financial results. However, management believes that the lack of a functioning audit committee and the lack of a majority of outside directors on our board of directors results in ineffective oversight in the establishment and monitoring of required internal controls and procedures, which could result in a material misstatement in our financial statements in future periods.
This annual report does not include an attestation report of the Company's registered public accounting firm regarding internal control over financial reporting. Management's report was not subject to attestation by the Company's registered public accounting firm pursuant to temporary rules of the SEC that permit the Company to provide only the management's report in this annual report.
Management's Remediation Initiatives
In an effort to remediate the identified material weaknesses and other deficiencies and enhance our internal controls, we have initiated, or plan to initiate, the following series of measures:
We will increase our personnel resources and technical accounting expertise within the accounting function when funds are available to us. First, we will create a position to segregate duties consistent with control objectives of having separate individuals perform (i) the initiation of transactions, (ii) the recording of transactions and (iii) the custody of assets. Second, we will create a senior position to focus on financial reporting and standardizing and documenting our accounting procedures with the goal of increasing the effectiveness of the internal controls in preventing and detecting misstatements of accounting information. Third, we plan to appoint one or more outside directors to our board of directors who shall be appointed to an audit committee resulting
38
in a fully functioning audit committee who will undertake the oversight in the establishment and monitoring of required internal controls and procedures such as reviewing and approving estimates and assumptions made by management when funds are available to us.
Management believes that the appointment of one or more outside directors, who shall be appointed to a fully functioning audit committee, will remedy the lack of a functioning audit committee and a lack of a majority of outside directors on our Board. Due to our small size and limited resources we could experience delays in implementation.
ITEM 9B. OTHER INFORMATION
On August 4, 2014, we filed a Certificate of Amendment to our Certificate of Incorporation with the Delaware Secretary of State to change the name of our Company from “Cyto Wave Technologies Inc.” to “Accurexa Inc.”. The new CUSIP number for the Company’s common stock is 00439W 100. A copy of the Certificate of Amendment was filed as Appendix A to the Schedule 14C Information Statement as of August 4, 2014.
PART III
ITEM 10. DIRECTORS AND EXECUTIVE OFFICERS
Our directors and executive officers, and their respective ages, positions and offices, are as follows:
|
Name Age Position(s) George Yu 42 President & CEO and Director Anchie Kuo 55 Chairman of the Board and Director Luc Meyer 72 Director |
Our Chief Executive Officer is also serving as our principal financial officer for the Company.
George Yu
(age 42) has been the President and Chief Executive Officer of the Company since inception. He has over 15 years of experience in start-up companies, finance, operations, corporate development and the medical field. His well-rounded background allowed him to start the Company as a founder and raise a common stock funding round. He has the experience to manage device development, clinical trials and to raise additional funding that is critical for the success of the Company. He served, from September 2009 to August 2010, as the Managing Partner, Bay2Peak S.A., a financial advisory and investment management firm, and from September 2005 to August 2009, as the Managing Partner of Bay2Peak Strategies, Ltd., a financial advisory and investment management firm, where he was responsible in both instances for the sourcing and execution of transactions, including financings, mergers and acquisitions, and investor relations. Prior to that, Mr. Yu served in various operational management and consulting roles and has worked at the management consulting firm Bain & Company, with small-cap hedge funds and in investment banking at Lehman Brothers. Mr. Yu holds a Medical Doctor Degree from the University of Tuebingen, Germany, and a Master in Business Administration in Finance and Economics from Columbia Business School.
Anchie Kuo
(age 55) is a physician, venture capitalist and entrepreneur with over 25 years of experience in the US healthcare industry, serving on the board of over 15 companies. In addition, he has significant experience in the healthcare industry in Asia as the owner and CEO of the first FDA registered suture manufacturer in China. He has an extensive network of international relationships that could the assist the Company in exploring strategic collaboration partnerships and fundraising. He could also assist the Company in identifying assets or other small companies that would allow the Company to accelerate its growth through licensing or M&A transactions. His background as the CEO of an FDA registered suture manufacturer could allow him to provide valuable regulatory insights to the Company. Dr. Kuo is currently the CEO of Landbridge, based in San Francisco, since January 2012, a company dedicated to helping companies in the US and China with licensing, business development, and financing opportunities.
39
From January 2005 to August 2011, Dr. Kuo was the CEO of Arc Medical Supplies Co., Ltd., a medical products manufacturer that distributed products in China, Europe, and the US. From February 2009 to July 2010, he was the President & CEO of Sinobiomed Inc., a developer of genetically engineered recombinant protein drugs and vaccines, based in Shanghai, China. Sinobiomed Inc. sold its protein drug and vaccine business in December 2010 and changed its company name to Sitoa Global Inc. in August 2011 after licensing an e-commerce technology in June 2011. From 2000 to 2004, Dr. Kuo was a Managing Partner and co-Founder of Genaissance Capital Group, a top quartile healthcare focused hedge fund. From 1994 to 2000, Dr. Kuo co-founded and became a Managing Director of BankAmerica Ventures (Scale Ventures Partners), a venture group that managed approximately $1.2 billion of assets. From 1990 to 1994, Dr. Kuo began in business development at Pfizer Hospital Products Group, a subsidiary of Pfizer Inc., where he was a manger responsible for acquisitions and venture investments. Dr. Kuo received his Bachelors or Arts (Economics) from Dartmouth College in Hanover, New Hampshire. Then Dr. Kuo received his M.D. from Dartmouth Medical School in Hanover, New Hampshire.
Luc H. Meyer
(age 72) has been a retired chef since 2006 and is currently involved in a number of commercial real estate and restaurant investments, in philanthropy including an art gallery, publishing of a cookbook and extensive travel across the world. He opened the Left Bank Restaurant with his wife Liz Meyer in Vail, Colorado in 1970, and was later involved in 9 different restaurants as chef and owner. He was born in France and had worked as a chef for over 50 years in France, Switzerland, Canada, Bahamas, Virgin Islands and Vail, Colorado where he settled.
Scientific Advisory Board
Daniel A. Lim, MD PhD, is Assistant Professor of Neurological Surgery at the University of California, San Francisco (UCSF) and the lead inventor of the BranchPoint technology. He has over 18 years of research experience in the neural stem cell field and is also a board-qualified neurosurgeon at the University of California, San Francisco and the San Francisco Veterans Affairs Medical Center. Dr. Lim was the principal investigator of a $1.8 million California Institute for Regenerative Medicine (CIRM) project that funded the development of the BranchPoint technology. In 2009, he edited a book describing the use of interventional MRI (iMRI) in neurosurgery. From 2010 to 2011, he served as co-surgeon in a Phase 1 clinical trial of neural cell transplantation to the brain of patients with Pelizaeus-Merzbacher disease (PMD). In 2012, he served as the local chairman of the biannual conference of the American Association of Stereotactic and Functional Neurosurgeons (ASSFN), and there he presented his work on the development of the BranchPoint technology, which was published in 2013 as an article featured on the cover of Stereotactic and Functional Neurosurgery. Ongoing work on iMRI for RBD-based cell delivery was reported in a news article online at Nature (http://www.nature.com/news/devices-aim-to-deliver-on-stem-cell-therapies-1.12532). The iMRI-compatible version of BranchPoint was published in Molecular Therapy in 2014. His basic science research on neural stem cells has resulted in over 50 peer-reviewed articles and book chapters. Dr. Lim serves as a member of the advisor board of the Rosenman Institute (http://qb3.org/rosenman), a non-profit organization whose mission is to drive medical device innovation for improvements in patient care by helping drive research concepts into innovation. For his basic science research, Dr. Lim received an NIH Director's New Innovator Award (2009-2014) and in 2010, he was a Kavli Fellow of the U.S. National Academy of Sciences.
Family Relationships
There are no family relationships, or other arrangements or understandings between or among any of the directors, executive officers or other person pursuant to which such person was selected to serve as a director or officer.
Involvement in certain legal proceedings
Our directors, executive officers and control persons have not been involved in any of the following events during the past five years:
·
any bankruptcy petition filed by or against any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time;
·
any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses);
·
being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities or banking activities; or
40
·
being found by a court of competent jurisdiction (in a civil action), the Securities and Exchange Commission or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated.
Indemnification of Directors and Officers
Our certificate of incorporation provides to the fullest extent permitted by Delaware law, that our directors or officers shall not be personally liable to us or our stockholders for damages for breach of such director’s or officer’s fiduciary duty. The effect of this provision of our certificate of incorporation is to eliminate the right of us and our stockholders (through stockholders’ derivative suits on behalf of our company) to recover damages against a director or officer for breach of the fiduciary duty of care as a director or officer (including breaches resulting from negligent or grossly negligent behavior, except under certain situations defined by statute). We believe that the indemnification provisions in our certificate of incorporation are necessary to attract and retain qualified persons as directors and officers.
In addition to the indemnification provided by our certificate of incorporation permitted by Delaware law, the Company agrees in the employment agreement with our Chief Executive Officer (“Executive”) to indemnify the Executive from and against any and all losses, claims, damages and liabilities, joint and several (collectively, “Losses”), to which the Executive may become subject under any applicable federal or state law, arising from or related to the Executive’s positions, conducts, activities, duties, or omissions at the Company; provided that the Company will not be liable to the extent that any Loss is found in a final judgment in a court to have resulted primarily from the Executive’s intentional violation of the law. The Company will reimburse the Executive for all expenses (including reasonable counsel fees and expenses) as such may be incurred in connection with the investigation of or preparation for or defense of any pending or threatened claim or any action or proceeding arising thereof, whether or not such the Company is a party. The indemnification provided for in the employment agreement shall be in addition to any rights that the Executive may have at common law or otherwise.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 (the Securities Act) may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the Securities and Exchange commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suite or proceeding) is asserted by such director officer or controlling person in connection with the securities being registered, we will submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
ITEM 11. EXECUTIVE COMPENSATION
Summary Compensation Table
The following table sets forth information concerning all cash and non-cash compensation awarded to, earned by or paid to the named persons for services rendered in all capacities during the noted periods. No other executive officer received total annual salary and bonus compensation in excess of $100,000:
|
Name and Principal Position |
Year |
Salary ($) |
Bonus ($) |
Stock Awards ($) |
Option Awards ($) |
Non-Equity Incentive Plan Compensation ($) |
Nonqualified Deferred Compensation Earnings ($) |
All Other Compensation ($) |
Total ($) |
|
George Yu President & CEO |
2014 2013 |
60,000 (#) 60,000 (##) |
- - |
- - |
- - |
- - |
- - |
- - |
60,000 60,000 |
|
Note: (#) This salary has accrued and will be paid upon us having adequate resources without interest and penalty as determined by the Company’s Board of Directors. (##) $35,000 were paid out as salary while $25,000 has accrued and will be paid upon us having adequate resources without interest and penalty as determined by the Company’s Board of Directors.
| |||||||||
41
Narrative Disclosure to Summary Compensation Table
On September 1, 2012, we entered into an employment agreement with George Yu, our president and CEO, which expired August 31, 2014, but renews for subsequent one year terms unless terminated by either party at least 60 days before the expiration of a term. Mr. Yu’s salary under his agreement is $5,000 per month and contains typical clauses with respect to non-disclosure, confidentiality and non-disparagement. Mr. Yu’s salary has been accrued in 2014.
Retirement Benefits and Change of Control
Not Applicable.
Director Compensation
The following table discloses the compensation of the directors of the Company for the Company’s fiscal year ended December 31, 2014 and 2013, respectively:
|
Name |
Fees Earned or Paid in Cash ($) |
Stock Awards ($) |
Option Awards ($) |
Non-Equity Incentive Plan Compensation ($) |
Nonqualified Deferred Compensation Earnings ($) |
All Other Compensation ($) |
Total ($) |
|
George Yu |
2014 2013 |
- - |
- - |
- - |
- - |
- - |
- - |
|
Anchie Kuo |
2014 2013 |
- - |
- - |
- - |
- - |
- - |
- - |
|
Luc H. Meyer |
2014 2013 |
- - |
- - |
- - |
- - |
- - |
- - |
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT.
The following table sets forth certain information as of December 31, 2014, with respect to our officers and directors and any person (including any “group” as that term is used in Section 13(d)(3) of the Exchange Act) who is known to us to be the beneficial owner of more than five percent of our common stock, being our only class of voting securities, under the current rules of the Securities and Exchange Commission regarding beneficial ownership:
|
Name of beneficial owner |
Position |
Number of shares owned |
Percent of Class (3) |
|
George Yu |
President, CEO and Director |
200,000 |
3.3% |
|
Anchie Kuo |
Director |
60,000 |
1.0% |
|
Elizabeth A. & Luc H. Meyer |
Director |
350,000 |
5.8% |
|
Abazu Ltd. |
Shareholder |
1,150,000 (1) |
19.2% |
|
Cercade Ltd. |
Shareholder |
1,000,000 (2) |
16.7% |
|
Luneel Corp. |
Shareholder |
450,000 |
7.5% |
|
All officers and directors as a group (3 persons) |
|
610,000 |
10.1% |
|
Notes: (1) These shares are deemed to be indirectly controlled and owned by George Yu in his capacity as a director of Abazu Ltd. (2) These shares are deemed to be indirectly controlled and owned by George Yu in his capacity as a director of Cercade Ltd. (3) Applicable percentage ownership is based on 5,993,816 shares of our common stock outstanding as of March 25, 2015. Beneficial ownership is determined in accordance with the rules of the Securities and Exchange Commission and generally includes voting or investment power with respect to securities.
| |||
42
Changes in control
There are no arrangements, including any pledge by any person of our securities, known to us the operation of which may at a subsequent date result in a change in control of our company.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
Upon our incorporation we issued 2,000,000 shares to our 34 founders. George Yu received 200,000 founder shares and Anchie Kuo received 60,000 shares. All of such transactions with the Company’s founders were exempt from registration by reason of Section 4(2) of the Securities Act of 1933, as amended (the “Act”). All of the shares issued in such transactions bear an appropriate restrictive legend.
We engaged in a private offering of our common stock and Elizabeth A. and Luc H. Meyer purchased 350,000 shares for $350,000 (the same price per share as all other purchasers in our private offering). A total of 965,000 shares were sold in the private offering to 10 investors. These shares were issued in a private offering pursuant to Regulation D under the Securities Act of 1933, as amended, and each of the investors therein represented in writing that such investor was an accredited investor as that term is defined in Regulation D and that the investor was acquiring the shares for his/her own account and for investment. The private offering was, accordingly, also exempt by reason of Section 4(6) of the Act.
Under his retainer agreement with the Company in October 2012, Mr. Hariton, our counsel, was issued 10,000 shares in a transaction exempt under Section 4(2) of the Securities Act. The certificate for these shares bears a restrictive legend. The shares were expensed by us at $10,000, the price in the private offering.
No underwriter participated in the foregoing transactions, and no underwriting discounts or commissions were paid, nor was any general solicitation or general advertising conducted. The securities bear a restrictive legend and stop transfer instructions are noted on our stock transfer records.
As disclosed under Item 6. Executive Compensation, Mr. Yu has an employment agreement with the Company.
Director Independence
We believe that the following directors of our company are considered “independent” under Rule 400(a)(15) of the National Association of Securities Dealers listing standards: Anchie Kuo and Luc H. Meyer.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The following table discloses the fees billed by our auditor in connection with the audit of the Company’s annual financial statements for the year ended December 31, 2014 and 2013.
43
PART IV
ITEM 15. FINANCIAL STATEMENTS AND EXHIBITS
(a) Financial Statements. See Index to Financial Statements on page F-1.
(b) Exhibits:
|
Exhibit No. |
|
Description |
|
|
|
|
|
3.1 |
|
Certificate of Incorporation, August 29, 2012 (incorporated by reference to Form 10 of the Company filed on February 27, 2013) |
|
3.2 |
|
Amended and Restated Certificate of Incorporation, dated December 12, 2012 (incorporated by reference to Form 10 of the Company filed on February 27, 2013) |
|
3.3 |
|
By –Laws (incorporated by reference to Form 10, Amendment No. 2 of the Company filed on April 23, 2013) |
|
4.1 |
|
Form of Stock Certificate (incorporated by reference to Form 10, Amendment No. 2 of the Company filed on April 23, 2013) |
|
10.1 |
|
Virtual Office Lease (incorporated by reference to Form 10 of the Company filed on February 27, 2013) |
|
10.2 |
|
UAMS License (incorporated by reference to Form 10 of the Company filed on February 27, 2013) |
|
10.3 |
|
Employment Agreement with George Yu (incorporated by reference to Form 10 of the Company filed on February 27, 2013) |
|
10.4 |
|
Form of Subscription Agreement (incorporated by reference to Form 10 of the Company filed on February 27, 2013) |
|
10.5 |
|
U.S. Patent Application 12/334,217 – (incorporated by reference to Form 10, Amendment No. 4 of the Company filed on June 5, 2013) |
|
10.6 |
|
U.S. Patent Application 12/945,576 – (incorporated by reference to Form 10, Amendment No. 4 of the Company filed on June 5, 2013) |
|
10.7 |
|
U.S. Patent Application 13/253,767 – (incorporated by reference to Form 10, Amendment No. 4 of the Company filed on June 5, 2013) |
|
10.8 |
|
Convertible Note Agreement – (incorporated by reference to Form 10-Q of the Company filed on August 14, 2013) |
|
10.9 |
|
Rejection and abandonment of U.S. Patent Application Serial No. 12/334,217 by the United States Patent and Trademark Office prior to date of UAMS License – (incorporated by reference to Form 8-K of the Company filed on February 24, 2014) |
|
10.10 |
|
Certificate of Amendment to Certificate of Incorporation – (incorporated by reference to Appendix A filed to Schedule 14C Information Statement of the Company as of August 4, 2014) |
|
10.11 |
|
UCSF License Agreement with the Regents of the University of California, dated as of September 16, 2014 – (incorporated by reference to Form 8-K of the Company filed on September 17, 2014) |
|
10.12 |
|
Consulting Agreement between the Company and the Lim Development Group, dated as of January 12, 2015, and Promissory Note issued to the Lim Development Group, dated as of January 12, 2015 – (incorporated by reference to Form 8-K of the Company filed on January 13, 2015) |
|
10.13 |
|
Consulting and Warrant Agreement between the Company and Capital Group Communications, Inc., dated as of February 18, 2015 – Filed herewith |
|
22.1 |
|
Subsidiaries: None |
|
23.1 |
|
Consent of Auditors (incorporated by reference to Form 10 of the Company filed on February 27, 2013) |
|
31.1 |
|
Certifications of Principal Executive Officer filed pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
31.2 |
|
Certifications of Principal Financial Officer filed pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
32.1 |
|
Certifications of Principal Executive Officer furnished pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
32.2 |
|
Certifications of Principal Financial Officer furnished pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
44
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Accurexa, Inc.
(Registrant)
Date:
March 25, 2015
By:
/s/ George Yu
Name:
George Yu
Title:
President and Chief Executive Officer
45