Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 - GeoVax Labs, Inc. | ex23-1.htm |
| EXCEL - IDEA: XBRL DOCUMENT - GeoVax Labs, Inc. | Financial_Report.xls |
| EX-5.1 - EXHIBIT 5.1 - GeoVax Labs, Inc. | ex5-1.htm |
As Filed with the Securities and Exchange Commission on March 20, 2015.
Registration No. 333-______
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933
GEOVAX LABS, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
2834 |
87-0455038 |
|
(State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
1900 Lake Park Drive, Suite 380, Smyrna, Georgia 30080, (678) 384-7220
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
|
Robert T. McNally, Ph.D. President & Chief Executive Officer GeoVax Labs, Inc. 1900 Lake Park Drive, Suite 380 Fax: (678) 384-7283 |
With a copy to: T. Clark Fitzgerald III 271 17th Street NW, Suite 2400 Tel: (404) 879-2455 Fax: (404) 870-4869 |
|
(Name, address, including zip code, and telephone number, including area code, of agent for service) | |
Approximate date of commencement of proposed sale to the public: From time to time after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box. ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer ☐ |
Accelerated filer ☐ |
Non-accelerated filer ☐ |
Smaller reporting company ☒ |
Calculation of Registration Fee
|
Title of Each Class Of Securities To Be Registered |
Amount To Be Registered (1) |
Proposed Maximum Offering Price Per Unit |
Proposed Maximum Aggregate Offering Price |
Amount of Registration Fee |
|
Common stock, $0.001 par value per share underlying Series C convertible preferred stock, $0.001 par value per share |
16,666,666 (2) |
$0.16 (3) |
$2,666,667 (3) |
$309.87 |
|
Common stock underlying Outstanding Series D and Series F Warrants, as well as the Maxim Warrant |
34,666,665 (4) |
0.22 (5) |
7,626,666 (5) |
886.22 |
|
Common stock underlying outstanding Series E Warrants |
16,666,666 (4) |
$0.18 (5) |
3,000,000 (5) |
348.60 |
|
TOTAL |
67,999,997 |
|
$13,626,666 |
$1,544.69 (6) |
|
(1) |
In accordance with Rule 416(a), the Registrant is also registering hereunder an indeterminate number of shares that may be issued and resold resulting from stock splits, stock dividends or similar transactions. |
|
(2) |
Represents shares of the Registrant’s common stock underlying shares of Series C convertible preferred stock being registered for resale that have been issued to the selling stockholders named in this registration statement. |
|
(3) |
Estimated pursuant to Rule 457(c) of the Securities Act of 1933 solely for the purpose of computing the amount of the registration fee based on the average of the bid and asked prices reported on the over-the-counter bulletin board on March 16, 2015, which was $0.16 per share. |
|
(4) |
Represents shares of common stock issuable upon exercise of warrants to purchase shares of common stock held by selling stockholders named in this registration statement. |
|
(5) |
Calculated in accordance with Rule 457(g) based upon the aggregate exercise price of the warrants held by selling stockholders named in this registration statement. |
|
(6) |
The Registrant previously filed Form S-1 (333-165828) on March 31, 2010, and paid a filing fee of $2,852. The Registrant did not sell any securities pursuant to that Form S-1, and it was withdrawn in December, 2011. Subsequently, the Registrant applied $1,257.24 of the previously paid filing fee against amounts due for Form S-1 (333-180535) filed on April 3, 2012. Pursuant to Rule 457(p), the Registrant hereby applies $1,544.69 of the remaining previously paid filing fee against amounts due herewith. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. The selling stockholders may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting offers to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED MARCH 20, 2015
PROSPECTUS
GEOVAX LABS, INC.
Up to 67,999,997 Shares of Common Stock
This prospectus relates to up to 67,999,997 shares of common stock, $0.001 par value, of GeoVax Labs, Inc., or the “Company,” that may be sold from time to time by the selling stockholders named in this prospectus, which includes up to:
|
|
● |
16,666,666 shares of common stock underlying Series C Convertible Preferred Stock, par value $0.01 per share, which we refer to as “Series C Preferred Stock; and |
|
|
● |
Approximately 51,333,331 shares of common stock issuable to the selling stockholders upon the exercise of Series D, E, and F Warrants and one other warrant (the “Maxim Warrant”), which we refer to collectively as the “2015 Warrants”. |
The prices at which the selling stockholders may sell the shares will be determined by the prevailing market price for the shares or in negotiated transactions. The shares included in this prospectus may be reoffered and sold directly by the selling stockholders in accordance with one or more of the methods described in the plan of distribution, which begins on page 53 of this prospectus.
We will not receive any proceeds from the sales of outstanding shares of common stock by the selling stockholders, but we will receive funds from the exercise of the 2015 Warrants held by the selling stockholders to the extent they are exercised for cash.
Our common stock is registered under Section 12(g) of the Securities Exchange Act of 1934 and quoted on the over-the-counter market under the symbol “GOVX.” On March 16, 2015, the last reported sale price for our common stock as reported on the over-the-counter market was $0.16 per share.
This prospectus may only be used where it is legal to offer and sell the shares covered by this prospectus. We have not taken any action to register or obtain permission for this offering or the distribution of this prospectus in any country other than the United States.
___________
Investing in the common stock involves a high degree of risk. See “Risk Factors” beginning on page 3 for a discussion of these risks.
___________
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this Prospectus is ______, 2015
TABLE OF CONTENTS
|
PROSPECTUS SUMMARY |
1 |
|
COMPANY OVERVIEW |
1 |
|
RISK FACTORS |
3 |
|
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS |
11 |
|
USE OF PROCEEDS |
12 |
|
MARKET FOR OUR COMMON STOCK AND RELATED STOCKHOLDER MATTERS |
12 |
|
BUSINESS |
13 |
|
AVAILABLE INFORMATION |
28 |
|
SELECTED FINANCIAL DATA |
28 |
|
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
28 |
|
SECURITY OWNERSHIP OF PRINCIPAL STOCKHOLDERS, DIRECTORS AND OFFICERS |
35 |
|
DIRECTORS AND EXECUTIVE OFFICERS |
36 |
|
EXECUTIVE COMPENSATION |
38 |
|
DIRECTOR COMPENSATION |
42 |
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS |
43 |
|
SELLING STOCKHOLDERS |
45 |
|
DESCRIPTION OF SECURITIES |
47 |
|
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION FOR SECURITIES ACT LIABILITIES |
52 |
|
PLAN OF DISTRIBUTION |
53 |
|
LEGAL MATTERS |
54 |
|
EXPERTS |
54 |
|
WHERE YOU CAN FIND MORE INFORMATION |
54 |
|
INDEX TO FINANCIAL STATEMENTS |
F-1 |
|
PART II – INFORMATION NOT REQUIRED IN PROSPECTUS |
II-1 |
___________
You should rely only on the information contained in this prospectus and any free-writing prospectus prepared by or on behalf of us or to which we have referred you. We have not authorized anyone to provide you with additional or different information. The information contained in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or of any sale of our securities. Unless the context otherwise requires, references to “we,” “our,” “us,” or the “Company” mean GeoVax Labs, Inc.
We obtained industry and market data used throughout this prospectus through our research, surveys and studies conducted by third parties and industry and general publications. We have not independently verified market and industry data from third-party sources.
PROSPECTUS SUMMARY
The following is only a summary. We urge you to read the entire prospectus, including the more detailed consolidated financial statements, notes to the consolidated financial statements and other information included. Investing in our securities involves risks. Therefore, please carefully consider the information provided under the heading “Risk Factors” starting on page 3. You should not invest unless you can afford to lose your entire investment.
COMPANY OVERVIEW
GeoVax Labs, Inc. (“GeoVax” or the “Company”) is a clinical-stage biotechnology company developing human vaccines against infectious diseases using our novel vaccine platform. Our platform supports production of non-infectious virus-like particles (VLPs) from the cells of the person receiving the vaccine. Producing non-infectious virus-like particles in the person being vaccinated circumvents the need to purify virus-like particles for inoculation. The production of virus-like particles in the person being vaccinated mimics a natural infection, stimulating both the humoral and cellular arms of the immune system to recognize, prevent and control the target infection should it appear.
Our current development programs are focused on vaccines against Ebola and Marburg viruses, and a vaccine against Human Immunodeficiency Virus (HIV). We believe our technology and vaccine development expertise is well-suited for a wide variety of human infectious diseases for which there is an unmet medical need, and we intend to pursue expansion of our product pipeline as resources permit.
Our Ebola/Marburg vaccine program was initiated during 2014 with the goal of developing monovalent vaccines capable of controlling existing outbreaks as well as a multivalent vaccine for preventing future outbreaks. We plan to conduct preclinical animal immunogenicity and challenge studies during 2015 for both vaccines with human clinical testing to begin in late 2016.
Our most advanced HIV vaccine program is focused on the clade B subtype of HIV prevalent in the Americas and Western Europe. Our preventive clade B HIV vaccine has successfully completed Phase 2a human clinical testing and is targeted to enter a follow-on clinical trial in 2015. It has shown outstanding safety and excellent and highly reproducible immunogenicity (Journal of Infectious Diseases volume 203, pg 610 and volume 210 pg 99). We also are investigating our HIV vaccines for their potential to contribute to combination therapies for therapeutic treatment leading to a cure for HIV infections. We are also extending our HIV vaccine effort to the most common virus subtype affecting the developing world, clade C. For clade C, we have jointly developed and licensed via Emory University one vaccine from the National Institutes of Health (NIH), completed lead discovery for a second vaccine, and initiated early preclinical research using both approaches. Each of our vaccine development programs is discussed in greater detail in the sections that follow below.
Our vaccine development activities have been, and continue to be, financially supported by the U.S. government. This support has been both in the form of research grants awarded directly to us, as well as indirect support for the conduct of our human clinical trials. This is discussed further under “Support from the United States Government” below.
Our HIV vaccine technology was developed in collaboration with researchers at Emory University, the NIH, and the Centers for Disease Control and Prevention (CDC). The technology developed by the collaboration is exclusively licensed to us from Emory University. We also have nonexclusive licenses to certain patents owned by the NIH. Our Ebola/Marburg vaccines have been developed with technology licensed from, and in collaboration with, the NIH.
We are incorporated in Delaware, and our offices and laboratory facilities are located in Smyrna, Georgia (metropolitan Atlanta).
Company Background
We are incorporated under the laws of the State of Delaware. Our principal corporate offices are located at 1900 Lake Park Drive, Suite 380, Smyrna, Georgia 30080 (metropolitan Atlanta). Our telephone number is (678) 384-7220. The address of our web site is www.geovax.com. Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and all amendments to those reports, are available to you free of charge through the “Investors” section of our web site as soon as reasonably practicable after such materials have been electronically filed with, or furnished to, the Securities and Exchange Commission. Information contained on our web site does not form a part of this prospectus.
The Offering
|
|
Common stock offered by selling stockholders |
|
Up to 67,999,997 shares including 16,666,666 shares of common stock underlying Series C Preferred Stock owned by selling stockholders and approximately 51,333,331 shares of common stock issuable upon the exercise of 2015 Warrants held by the selling stockholders. This number represents approximately 68% of our current outstanding common stock, on a fully diluted basis. |
|
|
|
|
|
|
|
Common stock outstanding before the offering |
|
31,950,813 shares (1) |
|
|
|
|
|
|
|
Common stock outstanding after the offering, assuming all the shares of Series C Preferred Stock are converted into common stock and the 2015 Warrants are exercised for cash. |
|
99,950,810 shares (1) |
|
|
|
|
|
|
|
Proceeds to us |
|
We will not receive any proceeds from the sale of common stock covered by this prospectus. We will, however, receive approximately $10.6 million from the exercise of the 2015 Warrants held by the selling stockholders, if they are exercised in full for cash. |
|
|
|
|
|
|
|
Trading Symbol |
|
GOVX |
|
|
|
|
|
|
|
Risk Factors |
|
There are significant risks involved in investing in our Company, including our history of operating losses and our need for continued funding. For a discussion of these and other risk factors you should consider before buying our common stock. See “Risk Factors” beginning on page 3. |
__________________
(1) The number of shares of our common stock to be outstanding after this offering is based on 31,950,813 shares outstanding as of March 16, 2015 and excludes:
|
|
● |
1,197,529 shares of common stock reserved for future issuance under our equity incentive plans. As of March 16, 2015, there were options to purchase 1, 177,500 shares of our common stock outstanding under our equity incentive plans with a weighted average exercise price of $3.49 per share; |
|
|
● |
5,108,826 shares of common stock issuable upon exercise of currently outstanding warrants (but not including the Series D, E and F Warrants and the Maxim Warrant) as of March 16, 2015, with a weighted average exercise price of $0.57 per share; and |
|
● |
285,714 shares of common stock issuable upon conversion of our outstanding Series B Convertible Preferred Stock, which we refer to as our “Series B Preferred Stock.” |
RISK FACTORS
Investing in our securities involves a high degree of risk. You should carefully review and consider the risks, uncertainties and other factors described below before you decide whether to purchase our securities. Any of these factors could materially and adversely affect our business, financial condition, operating results and prospects and could negatively impact the market price of our common stock, and you may lose some or all of your investment. The risks and uncertainties described below are not the only ones facing our Company. Additional risks and uncertainties that we are unaware of, or that we currently deem immaterial, may also impair our business operations. You should also refer to the information contained in this prospectus, including our financial statements and the related notes.
Risks Related to Our Business
We have a history of operating losses, and we expect losses to continue for the foreseeable future.
We have had no product revenue to date and there can be no assurance that we will ever generate any product revenue. We have experienced operating losses since we began operations in 2001. As of December 31, 2014, we had an accumulated deficit of approximately $29.8 million. We expect to incur additional operating losses and expect cumulative losses to increase as our research and development, pre-clinical, clinical, manufacturing and marketing efforts expand. Our ability to generate revenue and achieve profitability depends on our ability to successfully complete the development of our product candidates, conduct pre-clinical tests and clinical trials, obtain the necessary regulatory approvals, and manufacture and market the resulting products. Unless we are able to successfully meet these challenges, we will not be profitable and may not remain in business.
Our business will require continued funding. If we do not receive adequate funding, we will not be able to continue our operations.
To date, we have financed our operations principally through the private placement of our equity securities and through NIH grants. We will require substantial additional financing at various intervals for our operations, including clinical trials, operating expenses, intellectual property protection and enforcement, for pursuit of regulatory approvals, and for establishing or contracting out manufacturing, marketing and sales functions. There is no assurance that such additional funding will be available on terms acceptable to us or at all. If we are not able to secure the significant funding that is required to maintain and continue our operations at current levels, or at levels that may be required in the future, we may be required to delay clinical studies or clinical trials, curtail operations, or obtain funds through collaborative arrangements that may require us to relinquish rights to some of our products or potential markets.
The costs of conducting all of our human clinical trials to date for our preventive HIV vaccine have been borne by the HVTN, funded by the NIH, and we expect NIH support for additional clinical trials. GeoVax incurs costs associated with manufacturing the clinical vaccine supplies and other study support. We cannot predict the level of support we will receive from the HVTN or the NIH for any additional clinical trials of our HIV vaccines.
Our operations are also partially supported by the NIH grants awarded to us to support our HIV/AIDS vaccine program. As of December 31, 2014, there is approximately $229,000 of unused grant funds remaining and available for use during 2015. We are pursuing additional grants from the federal government for both our HIV and Ebola/Marburg vaccine programs. However, as we progress to the later stages of our vaccine development activities, government financial support may be more difficult to obtain, or may not be available at all. Furthermore, there is some risk that actual funding for grants could be delayed, cut back, or eliminated due to government budget constraints. Therefore, it will be necessary for us to look to other sources of funding in order to finance our development activities.
We expect that our current working capital, combined with proceeds from the grants awarded to us from the NIH will be sufficient to support our planned level of operations through the first quarter of 2016. We will need to raise additional funds to significantly advance our vaccine development programs and to continue our operations beyond the first quarter of 2016. In order to meet our operating cash flow requirements we plan to seek sources of non-dilutive capital through government grant programs and clinical trial support. We may also plan additional offerings of our equity securities, debt, or convertible debt instruments. Should the financing we require to sustain our working capital needs be unavailable or prohibitively expensive when we require it, the consequences could have a material adverse effect on our business, operating results, financial condition and prospects.
Risks Related to Development and Commercialization of Product Candidates and Dependence on Third Parties
Our products are still being developed and are unproven. These products may not be successful
To become profitable, we must generate revenue through sales of our products. However, our products are in varying stages of development and testing. Our products have not been proven in human clinical trials and have not been approved by any government agency for sale. If we cannot successfully develop and prove our products and processes, or if we do not develop other sources of revenue, we will not become profitable and at some point we would discontinue operations.
Whether we are successful will be dependent, in part, upon the leadership provided by our management. If we were to lose the services of any of these individuals, our business and operations may be adversely affected. Further, we may not carry key man life insurance on certain of our executive officers or directors.
Whether our business will be successful will be dependent, in part, upon the leadership provided by our officers, particularly our President and Chief Executive Officer and our Chief Scientific Officer. The loss of the services of these individuals may have an adverse effect on our operations. Although we carry some key man life insurance on Dr. Harriet L. Robinson, the amount of such coverage may not be sufficient to offset any adverse economic effects on our operations and we do not carry key man insurance on any of our other executive officers or directors. Further, our employees, including our executive officers and directors, are not subject to any covenants not to compete against the Company, and our business could be adversely affected if any of our employees or directors engaged in an enterprise competitive with the Company.
Regulatory and legal uncertainties could result in significant costs or otherwise harm our business.
To manufacture and sell our products, we must comply with extensive domestic and international regulation. In order to sell our products in the United States, approval from the FDA is required. Satisfaction of regulatory requirements, including FDA requirements, typically takes many years, and if approval is obtained at all, it is dependent upon the type, complexity and novelty of the product, and requires the expenditure of substantial resources. We cannot predict whether our products will be approved by the FDA. Even if they are approved, we cannot predict the time frame for approval. Foreign regulatory requirements differ from jurisdiction to jurisdiction and may, in some cases, be more stringent or difficult to meet than FDA requirements. As with the FDA, we cannot predict if or when we may obtain these regulatory approvals. If we cannot demonstrate that our products can be used safely and successfully in a broad segment of the patient population on a long-term basis, our products would likely be denied approval by the FDA and the regulatory agencies of foreign governments.
We will face intense competition and rapid technological change that could result in products that are superior to the products we will be commercializing or developing.
The market for vaccines that protect against or treat human infectious diseases is intensely competitive and is subject to rapid and significant technological change. We will have numerous competitors in the United States and abroad, including, among others, large companies with substantially greater resources than us. These competitors may develop technologies and products that are more effective or less costly than any of our future technology or products or that could render our technology or products obsolete or noncompetitive. If our technology or products are not competitive, we may not be able to remain in business.
Our product candidates are based on new medical technology and, consequently, are inherently risky. Concerns about the safety and efficacy of our products could limit our future success.
We are subject to the risks of failure inherent in the development of product candidates based on new medical technologies. These risks include the possibility that the products we create will not be effective, that our product candidates will be unsafe or otherwise fail to receive the necessary regulatory approvals, and that our product candidates will be hard to manufacture on a large scale or will be uneconomical to market.
Many pharmaceutical products cause multiple potential complications and side effects, not all of which can be predicted with accuracy and many of which may vary from patient to patient. Long term follow-up data may reveal additional complications associated with our products. The responses of potential physicians and others to information about complications could materially affect the market acceptance of our products, which in turn would materially harm our business.
We may experience delays in our clinical trials that could adversely affect our financial results and our commercial prospects.
We do not know whether planned clinical trials will begin on time or whether we will complete any of our clinical trials on schedule, if at all. Product development costs will increase if we have delays in testing or approvals or if we need to perform more or larger clinical trials than planned. Significant delays may adversely affect our financial results and the commercial prospects for our products, and delay our ability to become profitable.
We rely heavily on the HVTN, independent clinical investigators, and other third party service providers for successful execution of our clinical trials, but do not control many aspects of their activities. We are responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as Good Clinical Practices, for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. Third parties may not complete activities on schedule, or may not conduct our clinical trials in accordance with regulatory requirements or our stated protocols. The failure of these third parties to carry out their obligations could delay or prevent the development, approval and commercialization of our product candidates. There is also a risk of changes in clinical trial strategy and timelines due to the HVTN and the NIH altering their trial strategy.
Failure to obtain timely regulatory approvals required to exploit the commercial potential of our products could increase our future development costs or impair our future sales.
None of our vaccines are approved by the FDA for sale in the United States or by other regulatory authorities for sale in foreign countries. To exploit the commercial potential of our technologies, we are conducting and planning to conduct additional pre-clinical studies and clinical trials. This process is expensive and can require a significant amount of time. Failure can occur at any stage of testing, even if the results are favorable. Failure to adequately demonstrate safety and efficacy in clinical trials could delay or preclude regulatory approval and restrict our ability to commercialize our technology or products. Any such failure may severely harm our business. In addition, any approvals we obtain may not cover all of the clinical indications for which approval is sought, or may contain significant limitations in the form of narrow indications, warnings, precautions or contraindications with respect to conditions of use, or in the form of onerous risk management plans, restrictions on distribution, or post-approval study requirements.
State pharmaceutical marketing compliance and reporting requirements may expose us to regulatory and legal action by state governments or other government authorities.
In recent years, several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs and file periodic reports on sales, marketing, pricing and other activities. Similar legislation is being considered in other states. Many of these requirements are new and uncertain, and available guidance is limited. Unless we are in full compliance with these laws, we could face enforcement action, fines, and other penalties and could receive adverse publicity, all of which could harm our business.
We may be subject to new federal and state legislation to submit information on our open and completed clinical trials to public registries and databases.
In 1997, a public registry of open clinical trials involving drugs intended to treat serious or life-threatening diseases or conditions was established under the FDA Modernization Act, or the FDMA, to promote public awareness of and access to these clinical trials. Under the FDMA, pharmaceutical manufacturers and other trial sponsors are required to post the general purpose of these trials, as well as the eligibility criteria, location and contact information of the trials. Since the establishment of this registry, there has been significant public debate focused on broadening the types of trials included in this or other registries, as well as providing for public access to clinical trial results. A voluntary coalition of medical journal editors has adopted a resolution to publish results only from those trials that have been registered with a no-cost, publicly accessible database, such as www.clinicaltrials.gov. Federal legislation was introduced in the fall of 2004 to expand www.clinicaltrials.gov and to require the inclusion of trial results in this registry. The Pharmaceutical Research and Manufacturers of America also issued voluntary principles for its members to make results from certain clinical trials publicly available and established a website for this purpose. Other groups have adopted or are considering similar proposals for clinical trial registration and the posting of clinical trial results. Failure to comply with any clinical trial posting requirements could expose us to negative publicity, fines and other penalties, all of which could materially harm our business.
Recently enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our drug candidates and affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our drug candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any drug candidates for which we obtain marketing approval.
Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the PPACA, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
Among the provisions of the PPACA of importance to our potential drug candidates are:
|
● |
an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
|
● |
an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for branded and generic drugs, respectively; |
|
● |
expansion of healthcare fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers and enhanced penalties for non-compliance; |
|
● |
a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D; |
|
● |
extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
|
● |
expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the federal poverty level beginning in 2014, thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
|
● |
expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
|
● |
the new requirements under the federal Open Payments program and its implementing regulations; |
|
● |
a new requirement to annually report drug samples that manufacturers and distributors provide to physicians; and |
|
● |
a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. These changes include aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On March 1, 2013, the President signed an executive order implementing the 2% Medicare payment reductions, and on April 1, 2013, these reductions went into effect. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on customers for our drugs, if approved, and, accordingly, our financial operations.
We expect that the PPACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved drug. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our drugs.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for drugs. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our drug candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
We may not be successful in establishing collaborations for product candidates we may seek to commercialize, which could adversely affect our ability to discover, develop, and commercialize products.
We expect to seek collaborations for the development and commercialization of product candidates in the future. The timing and terms of any collaboration will depend on the evaluation by prospective collaborators of the clinical trial results and other aspects of our vaccine’s safety and efficacy profile. If we are unable to reach agreements with suitable collaborators for any product candidate, we will be forced to fund the entire development and commercialization of such product candidates, ourselves, and we may not have the resources to do so. If resource constraints require us to enter into a collaboration agreement early in the development of a product candidate, we may be forced to accept a more limited share of any revenues this product may eventually generate. We face significant competition in seeking appropriate collaborators. Moreover, these collaboration arrangements are complex and time-consuming to negotiate and document. We may not be successful in our efforts to establish collaborations or other alternative arrangements for any product candidate. Even if we are successful in establishing collaborations, we may not be able to ensure fulfillment by collaborators of their obligations or our expectations.
We do not have manufacturing, sales or marketing experience.
We do not have experience in manufacturing, selling, or marketing vaccines. To obtain the expertise necessary to successfully manufacture, market, and sell our vaccines, we will require the development of our own commercial infrastructure and/or collaborative commercial arrangements and partnerships. Our ability to execute our current operating plan is dependent on numerous factors, including, the performance of third party collaborators with whom we may contract.
Our vaccines under development may not gain market acceptance.
Our vaccines may not gain market acceptance among physicians, patients, healthcare payers and the medical community. Significant factors in determining whether we will be able to compete successfully include:
|
● |
the efficacy and safety of our vaccines; |
|
● |
the time and scope of regulatory approval; |
|
● |
reimbursement coverage from insurance companies and others; |
|
● |
the price and cost-effectiveness of our products; and |
|
● |
the ability to maintain patent protection. |
We may be required to defend lawsuits or pay damages for product liability claims.
Product liability is a major risk in testing and marketing biotechnology and pharmaceutical products. We may face substantial product liability exposure in human clinical trials and for products that we sell after regulatory approval. We carry product liability insurance and we expect to continue such policies. However, product liability claims, regardless of their merits, could exceed policy limits, divert management’s attention, and adversely affect our reputation and the demand for our products.
Risks Related to Our Intellectual Property
We could lose our license rights to our important intellectual property if we do not fulfill our contractual obligations to our licensors.
Our rights to significant parts of the technology we use in our vaccines are licensed from third parties and are subject to termination if we do not fulfill our contractual obligations to our licensors. Termination of intellectual property rights under any of our license agreements could adversely impact our ability to produce or protect our vaccines. Our obligations under our license agreements include requirements that we make milestone payments to our licensors upon the achievement of clinical development and regulatory approval milestones, royalties as we sell commercial products, and reimbursement of patent filing and maintenance expenses. Should we become bankrupt or otherwise unable to fulfill our contractual obligations, our licensors could terminate our rights to critical technology that we rely upon.
Other parties may claim that we infringe their intellectual property or proprietary rights, which could cause us to incur significant expenses or prevent us from selling products.
Our success will depend in part on our ability to operate without infringing the patents and proprietary rights of third parties. The manufacture, use and sale of new products have been subject to substantial patent rights litigation in the pharmaceutical industry. These lawsuits generally relate to the validity and infringement of patents or proprietary rights of third parties. Infringement litigation is prevalent with respect to generic versions of products for which the patent covering the brand name product is expiring, particularly since many companies that market generic products focus their development efforts on products with expiring patents. Pharmaceutical companies, biotechnology companies, universities, research institutions or other third parties may have filed patent applications or may have been granted patents that cover aspects of our products or our licensors’ products, product candidates or other technologies.
Future or existing patents issued to third parties may contain patent claims that conflict with our products. We expect to be subject to infringement claims from time to time in the ordinary course of business, and third parties could assert infringement claims against us in the future with respect to our current products or with respect to products that we may develop or license. Litigation or interference proceedings could force us to:
|
● |
stop or delay selling, manufacturing or using products that incorporate, or are made using the challenged intellectual property; |
|
● |
pay damages; or |
|
● |
enter into licensing or royalty agreements that may not be available on acceptable terms, if at all. |
Any litigation or interference proceedings, regardless of their outcome, would likely delay the regulatory approval process, be costly and require significant time and attention of our key management and technical personnel.
Any inability to protect intellectual property rights in the United States and foreign countries could limit our ability to manufacture or sell products.
We will rely on trade secrets, unpatented proprietary know-how, continuing technological innovation and, in some cases, patent protection to preserve our competitive position. Our patents and licensed patent rights may be challenged, invalidated, infringed or circumvented, and the rights granted in those patents may not provide proprietary protection or competitive advantages to us. We and our licensors may not be able to develop patentable products. Even if patent claims are allowed, the claims may not issue, or in the event of issuance, may not be sufficient to protect the technology owned by or licensed to us. If patents containing competitive or conflicting claims are issued to third parties, we may be prevented from commercializing the products covered by such patents, or may be required to obtain or develop alternate technology. In addition, other parties may duplicate, design around or independently develop similar or alternative technologies.
We may not be able to prevent third parties from infringing or using our intellectual property, and the parties from whom we may license intellectual property may not be able to prevent third parties from infringing or using the licensed intellectual property. We generally will attempt to control and limit access to, and the distribution of, our product documentation and other proprietary information. Despite efforts to protect this proprietary information, unauthorized parties may obtain and use information that we may regard as proprietary. Other parties may independently develop similar know-how or may even obtain access to these technologies.
The laws of some foreign countries do not protect proprietary information to the same extent as the laws of the United States, and many companies have encountered significant problems and costs in protecting their proprietary information in these foreign countries.
Neither the U.S. Patent and Trademark Office nor the courts have established a consistent policy regarding the breadth of claims allowed in pharmaceutical patents. The allowance of broader claims may increase the incidence and cost of patent interference proceedings and the risk of infringement litigation. On the other hand, the allowance of narrower claims may limit the value of our proprietary rights.
Risks Related To This Offering and Our Securities
The market price of our common stock is highly volatile.
The market price of our common stock has been, and is expected to continue to be, highly volatile. Certain factors, including announcements of new developments by us or other companies, regulatory matters, new or existing medicines or procedures, concerns about our financial position, operating results, litigation, government regulation, developments or disputes relating to agreements, patents or proprietary rights, may have a significant impact on the market price of our stock. In addition, potential dilutive effects of future sales of shares of common stock by us, and subsequent sales of common stock by the holders of warrants and options could have an adverse effect on the market price of our shares.
Our common stock does not have a vigorous trading market and investors may not be able to sell their securities when desired.
We have a limited active public market for our common shares. A more active public market, allowing investors to buy and sell large quantities of our common stock, may never develop. Consequently, investors may not be able to liquidate their investments in the event of an emergency or for any other reason.
We have never paid dividends and have no plans to do so.
Holders of shares of our common stock are entitled to receive such dividends as may be declared by our Board of Directors. To date, we have paid no cash dividends on our shares of common stock and we do not expect to pay cash dividends on our common stock in the foreseeable future. We intend to retain future earnings, if any, to provide funds for operations of our business. Therefore, any potential return investors may have in our common stock will be in the form of appreciation, if any, in the market value of their shares of common stock.
If we fail to maintain an effective system of internal controls, we may not be able to accurately report our financial results or prevent fraud.
We are subject to reporting obligations under the United States securities laws. The Securities and Exchange Commission, or the SEC, as required by the Sarbanes-Oxley Act of 2002, adopted rules requiring every public company to include a management report on such company’s internal controls over financial reporting in its annual report. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent fraud. As a result, our failure to achieve and maintain effective internal controls over financial reporting could result in the loss of investor confidence in the reliability of our financial statements, which in turn could negatively impact the trading price of our stock.
If we fail to remain current in our reporting requirements, our securities could be removed from the OTC Market, which would limit the ability of broker-dealers to sell our securities and the ability of stockholders to sell their securities in the secondary market.
United States companies trading on the OTC Market must be reporting issuers under Section 12 of the Exchange Act, and must be current in their reports under Section 13. If we fail to remain current on our reporting requirements, we could be removed from the OTC Market. As a result, the market liquidity for our securities could be severely adversely affected by limiting the ability of broker-dealers to sell our securities and the ability of stockholders to sell their securities in the secondary market.
We expect to need additional capital, and the sale of additional shares or other equity securities could result in additional dilution to our stockholders.
We believe that our current cash and cash equivalents, combined with anticipated cash flow from our NIH grants, and without consideration given to any potential proceeds from the exercise of the Series A or C Warrants, will be sufficient to meet our anticipated cash needs into the first quarter of 2016. In order to meet our operating cash flow requirements we plan additional offerings of our equity securities, debt, or convertible debt instruments. The sale of additional equity securities could result in additional dilution to our stockholders. Certain equity securities, such as convertible preferred stock, or warrants, may contain anti-dilution provisions which could result in the issuance of additional shares at lower prices if we sell other shares below specified prices. The incurrence of indebtedness would result in debt service obligations and could result in operating and financing covenants that would restrict our operations. We cannot assure investors that financing will be available in amounts or on terms acceptable to us, if at all.
Our directors and executive officers beneficially own a significant amount of our common stock and will be able to exercise significant influence on matters requiring stockholder approval.
As of March 9, 2015, our directors and executive officers collectively beneficially own approximately 8.0% of our common and Emory University beneficially owns 14.5%. If our directors and executive officers move to act in concert with Emory University, they may be able to exert significant influence over the election of directors and the outcome of most corporate actions requiring stockholder approval and our business, which may have the effect of delaying or precluding a third party from acquiring control of us.
The exercise of options or warrants or conversion of our Series B or Series C Preferred Stock may depress our stock price and may result in significant dilution to our common stockholders.
There are a significant number of outstanding warrants and options to purchase our stock and we have issued Series B and Series C Convertible Preferred Stock that is convertible into our Common Stock. If the market price of our Common Stock exceeds the exercise price of outstanding warrants and options or the conversion prices of the Series B or Series C Convertible Preferred Stock, holders of those securities may be likely to exercise their warrants and options or convert their preferred shares and sell the Common Stock acquired upon exercise or conversion of such securities, as applicable, in the open market. Sales of a substantial number of shares of our Common Stock in the public market by holders of warrants, options, or preferred shares may depress the prevailing market price for our Common Stock and could impair our ability to raise capital through the future sale of our equity securities. Additionally, if the holders of outstanding options, warrants, or preferred shares exercise those options or warrants or convert those preferred shares, as applicable, our common stockholders will incur dilution in their relative percentage ownership. The prospect of this possible dilution may also impact the price of our Common Stock.
Our outstanding options and warrants include warrants to purchase up to 2,690,666 shares of our Common Stock that were originally issued in March 2012, as well as the 2015 Warrants we issued in February 2015. Of these, warrants to purchase up to 34,666,665 shares have an exercise price of $0.22 per share, and warrants to purchase up to 19,357,332 shares have an exercise price of $0.18 per share. These warrants contain anti-dilution provisions, which may, under certain circumstances, reduce the exercise price (but have no effect on the number of shares subject to the warrants) to match if we sell or grant options to purchase, including rights to reprice, our common stock or common stock equivalents at a price lower than the exercise price of the warrants, or if we announce plans to do so. This potential reduction in exercise price could reduce the funds the Company receives upon exercise of the warrants, and increase the likelihood that a dilutive issuance will occur.
Our common stock is and likely will remain subject to the SEC’s “penny stock” rules, which make it more difficult to sell.
Our common stock is currently and may remain classified as a “penny stock.” The SEC rules regarding penny stocks may have the effect of reducing trading activity in our shares, making it more difficult for investors to sell. Under these rules, broker-dealers who recommend such securities to persons other than institutional accredited investors must:
|
• |
make a special written suitability determination for the purchaser; |
|
• |
receive the purchaser’s written agreement to a transaction prior to sale; |
|
• |
provide the purchaser with risk disclosure documents which identify certain risks associated with investing in “penny stocks” and which describe the market for these “penny stocks” as well as a purchaser’s legal remedies; |
|
• |
obtain a signed and dated acknowledgment from the purchaser demonstrating that the purchaser has received the required risk disclosure document before a transaction in a “penny stock” can be completed; and |
|
• |
give bid and offer quotations and broker and salesperson compensation information to the customer orally or in writing before or with the confirmation. |
These rules make it more difficult for broker-dealers to effectuate customer transactions and trading activity in our securities and may result in a lower trading volume of our common stock and lower trading prices.
Certain provisions of our certificate of incorporation which authorize the issuance of additional shares of preferred stock may make it more difficult for a third party to effect a change in control.
Our certificate of incorporation authorizes our Board of Directors to issue up to 10,000,000 shares of preferred stock. We have issued 100 shares of Series B Convertible Preferred Stock and 3,000 shares of our Series C Convertible Preferred Stock. We believe the terms of these preferred shares would not have a substantial impact on the ability of a third party to effect a change in control. The remaining shares of preferred stock may be issued in one or more series, the terms of which may be determined at the time of issuance by our Board of Directors without further action by the stockholders. These terms may include voting rights including the right to vote as a series on particular matters, preferences as to dividends and liquidation, conversion rights, redemption rights and sinking fund provisions. The issuance of any preferred stock could diminish the rights of holders of our common stock, and therefore could reduce the value of our common stock. In addition, specific rights granted to future holders of preferred stock could be used to restrict our ability to merge with, or sell assets to, a third party. The ability of our Board of Directors to issue preferred stock could make it more difficult, delay, discourage, prevent or make it more costly to acquire or effect a change-in-control, which in turn could prevent the stockholders from recognizing a gain in the event that a favorable offer is extended and could materially and negatively affect the market price of our common stock.
Certain provisions of the Series A and C warrants we issued in March 2012, as well as the 2015 Warrants we issued in February 2015, may make it more difficult for a third party to effect a change in control.
The Series A, C, D, E and F Warrants and the Maxim Warrant contain provisions which permit the holders to require the payment to them of an amount of cash equal to the value (based on a Black-Scholes computation) of the remaining unexercised portion of the warrants on the date of the consummation of a fundamental transaction (as defined, but generally a change in control of the Company) that is (i) an all cash transaction, (ii) a “going private” transaction, or (ii) a transaction involving a person or entity not traded on a national securities exchange. The prospect of making such payments may discourage a potential third party acquirer.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements. Forward-looking statements relate to future events or our future financial performance. We generally identify forward-looking statements by terminology such as “may,” “will,” “should,” “expects,” “plans,” “anticipates,” “could,” “intends,” “target,” “projects,” “contemplates,” “believes,” “estimates,” “predicts,” “potential” or “continue” or the negative of these terms or other similar words, although not all forward-looking statements contain these words. Forward-looking statements include, but are not limited to, statements regarding our or our management’s expectations, hopes, beliefs, intentions or strategies regarding the future, such as our estimates regarding anticipated operating losses, future performance, future revenues and projected expenses; our liquidity and our expectations regarding our needs for and ability to raise additional capital; our ability to manage our expenses effectively and raise the funds needed to continue our business; our ability to retain the services of our current executive officers, directors and principal consultants; our ability to obtain and maintain regulatory approval of our existing products and any future products we may develop; the initiation, timing, progress and results of our preclinical and clinical trials, research and development programs; regulatory and legislative developments in the United States and foreign countries; the timing, costs and other limitations involved in obtaining regulatory approval for any product; the further preclinical or clinical development and commercialization of our product candidates; the potential benefits of our product candidates over other therapies; our ability to enter into any collaboration with respect to product candidates; the performance of our third-party manufacturers; our ability to obtain and maintain intellectual property protection for our products and operate our business without infringing upon the intellectual property rights of others; the successful development of our sales and marketing capabilities; the size and growth of the potential markets for our products and our ability to serve those markets; the rate and degree of market acceptance of any future products; our reliance on key scientific management or personnel; the payment and reimbursement methods used by private or governmental third-party payers; and other factors discussed elsewhere in this prospectus or any document incorporated by reference herein or therein.
The words “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “expect,” “plan” and similar expressions may identify forward-looking statements, but the absence of these words does not mean that a statement is not forward-looking. The forward-looking statements contained in this prospectus are based on our current expectations and beliefs concerning future developments and their potential effects on us. There can be no assurance that future developments affecting us will be those that we have anticipated. These forward-looking statements involve a number of risks, uncertainties (some of which are beyond our control) or other assumptions that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. These risks and uncertainties include, but are not limited to, those factors described in the section titled “Risk Factors.” Should one or more of these risks or uncertainties materialize, or should any of our assumptions prove incorrect, actual results may vary from those projected in these forward-looking statements. We undertake no obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws. “Risk Factors” and “Business,” as well as other sections in this prospectus or incorporated by reference into this prospectus, discuss some of the factors that could contribute to these differences.
The forward-looking statements made in this prospectus relate only to events as of the date on which the statements are made. We undertake no obligation to update any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events. Other factors besides those described in this prospectus could also affect our actual results.
This prospectus also contains market data related to our business and industry. These market data include projections that are based on a number of assumptions. While we believe these assumptions to be reasonable and sound as of the date of this prospectus, if these assumptions turn out to be incorrect, actual results may differ from the projections based on these assumptions. As a result, our markets may not grow at the rates projected by these data, or at all. The failure of these markets to grow at these projected rates may have a material adverse effect on our business, results of operations, financial condition and the market price of our common stock.
USE OF PROCEEDS
We will not receive proceeds from the sales by the selling stockholders. If the 2015 Warrants are exercised for cash, then we will receive the proceeds payable by the selling stockholders upon exercise of those warrants. If all of the 2015 Warrants are exercised in full for cash, we will receive approximately $10.6 million. We will use these proceeds, if received, for general working capital purposes.
MARKET FOR OUR COMMON STOCK AND RELATED STOCKHOLDER MATTERS
Market Information
Our common stock is currently traded on the OTCQB Market under the symbol “GOVX”. The following table sets forth the high and low bid prices for our common stock for the periods indicated. The prices represent quotations between dealers and do not include retail mark-up, markdown, or commission, and do not necessarily represent actual transactions. On March 16, 2015, the last reported sale price for our common stock as reported in the OTCQB Market was $0.16 per share.
|
High |
Low |
||||||||
|
2015 |
|||||||||
|
First Quarter (through March 16, 2015) |
$ | 0.24 | $ | 0.14 | |||||
|
2014 |
|||||||||
|
Fourth Quarter |
$ | 0.51 | $ | 0.13 | |||||
|
Third Quarter |
$ | 0.26 | $ | 0.19 | |||||
|
Second Quarter |
$ | 0.37 | $ | 0.21 | |||||
|
First Quarter |
$ | 0.60 | $ | 0.34 | |||||
|
2013 |
|||||||||
|
Fourth Quarter |
$ | 0.97 | $ | 0.36 | |||||
|
Third Quarter |
$ | 0.51 | $ | 0.36 | |||||
|
Second Quarter |
$ | 0.63 | $ | 0.43 | |||||
|
First Quarter |
$ | 0.85 | $ | 0.55 | |||||
Holders
On March 16, 2015, there were approximately 700 holders of record of our common stock. Because many of our shares of common stock are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
Dividends
We have not paid any dividends since our inception and do not contemplate paying dividends in the foreseeable future. Any future determination as to the declaration and payment of dividends, if any, will be at the discretion of our Board of Directors and will depend on then existing conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects and other factors our Board of Directors may deem relevant.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table sets forth certain information as of December 31, 2014 with respect to compensation plans under which our equity securities are authorized for issuance.
|
Plan Category |
Number of securities to be issued upon exercise of outstanding options, warrants and rights |
Weighted-average exercise price of outstanding options, warrants and rights |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
|
Equity compensation plans approved by stockholders (1) |
720,000 |
$5.51 |
-0- |
|
Equity compensation plans not approved by stockholders (2) |
463,100 |
$0.38 |
16,900 |
|
(1) |
Represents shares to be issued pursuant to the GeoVax Labs, Inc. 2006 Equity Incentive Plan (the “Stock Option Plan”), originally approved by our stockholders effective September 30, 2006. A description of the Stock Option Plan and other information concerning the Stock Option Plan can be found in footnote 9 to our 2014 consolidated financial statements contained in our Annual Report on Form 10-K for the year ended December 31, 2014. |
|
(2) |
Represents increases to the shares available pursuant to the Stock Option Plan approved by our Board of Directors. |
BUSINESS
Overview
GeoVax Labs, Inc. (“GeoVax” or the “Company”) is a clinical-stage biotechnology company developing human vaccines against infectious diseases using our novel vaccine platform. Our platform supports production of non-infectious virus-like particles (VLPs) from the cells of the person receiving the vaccine. Producing non-infectious virus-like particles in the person being vaccinated circumvents the need to purify virus-like particles for inoculation. The production of virus-like particles in the person being vaccinated mimics a natural infection, stimulating both the humoral and cellular arms of the immune system to recognize, prevent and control the target infection should it appear.
Our current development programs are focused on vaccines against Ebola and Marburg viruses, and a vaccine against Human Immunodeficiency Virus (HIV). We believe our technology and vaccine development expertise is well-suited for a wide variety of human infectious diseases for which there is an unmet medical need, and we intend to pursue expansion of our product pipeline as resources permit.
Our Ebola/Marburg vaccine program was initiated during 2014 with the goal of developing monovalent vaccines capable of controlling existing outbreaks as well as a multivalent vaccine for preventing future outbreaks. We plan to conduct preclinical animal immunogenicity and challenge studies during 2015 for both vaccines with human clinical testing to begin in late 2016.
Our most advanced HIV vaccine program is focused on the clade B subtype of HIV prevalent in the Americas and Western Europe. Our preventive clade B HIV vaccine has successfully completed Phase 2a human clinical testing and is targeted to enter a follow-on clinical trial in 2015. It has shown outstanding safety and excellent and highly reproducible immunogenicity (Journal of Infectious Diseases volume 203, pg 610 and volume 210 pg 99). We also are investigating our HIV vaccines for their potential to contribute to combination therapies for therapeutic treatment leading to a cure for HIV infections. We are also extending our HIV vaccine effort to the most common virus subtype affecting the developing world, clade C. For clade C, we have jointly developed and licensed via Emory University one vaccine from the National Institutes of Health (NIH), completed lead discovery for a second vaccine, and initiated early preclinical research using both approaches. Each of our vaccine development programs is discussed in greater detail in the sections that follow below.
Our vaccine development activities have been, and continue to be, financially supported by the U.S. government. This support has been both in the form of research grants awarded directly to us, as well as indirect support for the conduct of our human clinical trials. This is discussed further under “Support from the United States Government” below.
Our HIV vaccine technology was developed in collaboration with researchers at Emory University, the NIH, and the Centers for Disease Control and Prevention (CDC). The technology developed by the collaboration is exclusively licensed to us from Emory University. We also have nonexclusive licenses to certain patents owned by the NIH. Our Ebola/Marburg vaccines have been developed with technology licensed from, and in collaboration with, the NIH.
We are incorporated in Delaware, and our offices and laboratory facilities are located in Smyrna, Georgia (metropolitan Atlanta).
Our Technology
Vaccines typically contain agents (antigens) that resemble disease-causing microorganisms. Traditional vaccines are often made from weakened or killed forms of the virus or from its surface proteins. Many newer vaccines use recombinant DNA (deoxyribonucleic acid) technology to generate vaccine antigens in bacteria or cultured cells from specific portions of the DNA sequence of the target pathogen. The generated antigens are then purified and formulated for use in a vaccine. The most successful of these purified antigens have been non-infectious virus-like particles (VLPs) as exemplified by vaccines for hepatitis B (Merck’s Recombivax® and GSK’s Engerix®) and Papilloma viruses (GSK’s Cervarix®, and Merck’s Gardasil®). Our approach uses recombinant DNA or recombinant viruses to produce VLPs in the person being vaccinated. In human clinical trials of our HIV vaccines, we have demonstrated that our VLPs, expressed in the cells of the person being vaccinated, are safe, yet elicit both strong and durable humoral and cellular immune response.
All of our vaccines are designed to produce self-assembling non-infectious VLPs in the cells of the person being vaccinated. VLPs train the body’s immune system to recognize and kill the authentic virus should it appear. VLPs also train the immune system to recognize and kill infected cells to control infection and reduce the length and severity of disease. One of the biggest challenges with VLP-based vaccines is to design the vaccines in such a way that the VLPs will be recognized by the immune system in the same way as the authentic virus would be. When VLPs for enveloped viruses like HIV, Ebola, and Marburg are produced in vivo, they include not only the protein antigens, but also an envelope consisting of membranes from the vaccinated individual’s cells. In this way, they are highly similar to the virus generated in a person’s body during a natural infection. VLPs produced externally, by contrast, have no envelope; or, envelopes from the cultured cells (typically hamster or insect cells) used to produce them. We believe our technology provides distinct advantages by producing VLPs that more closely resemble the authentic virus, which in turn, allows the body’s immune system to more readily recognize the authentic virus. By producing VLPs in vivo, we avoid potential purification issues associated with in vitro production of VLPs.
DNA and MVA as Vaccine Vectors. Our HIV vaccines incorporate two delivery components (or vectors): a recombinant plasmid DNA vaccine, and a recombinant MVA (modified vaccinia Ankara) vaccine. Our Ebola and Marburg vaccines use only the MVA vector. Both our DNA and MVA vaccines express sufficient vaccine genes to support the production of non-infectious VLPs. The VLPs cannot cause disease because they contain mutated or deleted enzymatic functions that are essential for virus replication. The virus-like particles display trimeric membrane bound forms of the viral envelope glycoprotein (Env for HIV or GP for Ebola or Marburg). This is important because the natural form of the envelope glycoprotein elicits multi-target antibody capable of recognizing incoming virus and blocking infections. Expression of multiple proteins by the vaccines is essential for the formation of VLPs. The multiple proteins also provide more targets for immune responses such as cytotoxic T-cells. Elicitation of multi-target humoral and cellular responses limits immune escape, just as multi-drug therapies limit drug escape.
|
|
|
|
Ebola VLPs |
HIV VLPs |
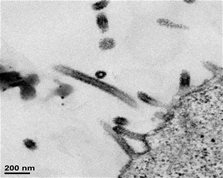
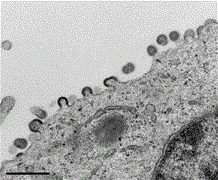
Figure 1. Electron micrographs showing the virus-like particles (VLPs) elicited by GeoVax vaccines from human cells. Note that the Ebola VLPs on the left self-assemble into the rod-like shape of the authentic Ebola virus, while the HIV VLPs shown on the right take on the spherical shape of the authentic HIV virus. While below the resolution of these micrographs, both types of VLPs display what we believe to be the native form of their respective viral envelope glycoproteins which we believe is key to generating an effective immune humoral response.
We selected MVA for use as the live viral component of our vaccines because of its well-established safety record and because of the ability of this vector to carry sufficient viral proteins to produce virus-like particles. MVA was originally developed as a safer smallpox vaccine for use in immune compromised humans. It was developed by attenuating the standard smallpox vaccine by making over 500 passages of the virus in chicken embryos or chick embryo fibroblasts, which resulted in a virus with limited ability to replicate in human cells but did not compromise the ability of MVA to grow on avian cells, which are used for manufacturing the virus. The deletions also resulted in the loss of immune evasion genes which assist the spread of wild type smallpox infections, even in the presence of human immune responses. MVA was safely administered to over 120,000 people in the 1970s as a smallpox vaccine.
Induction of T-cell and Antibody Immune Responses. In both preclinical and clinical trials, our HIV vaccines have been shown to induce both humoral (antibody) and cellular (T-cell) responses against HIV. The induction of both antibodies and T-cells is beneficial because these immune responses work through different mechanisms. Antibodies prevent infection by blocking viruses from infecting cells. In preclinical simian vaccine studies using repeated rectal challenges with moderate doses of virus, the avidity, or tightness, of antibody binding to the surface envelope glycoprotein of HIV correlates with the prevention of infection (The Journal of Infectious Diseases, 204:164 (2011)). In high dose challenges that infect all animals at the first exposure, the avidity of the antibody for envelope glycoprotein correlates with reduced levels of virus replication (Journal of Virology, 83:4102 (2009)). Similarly, antibody responses are believed to be critical for vaccine-elicited protection against Ebola and Marburg infection (Expert Review of Vaccines, 10:63 (2011)). These results likely reflect the tightly binding antibody both blocking infection as well as tagging virus and infected cells for destruction, by white blood cells such as macrophages, neutrophils and natural killer cells. Our vaccines elicit CD8+ T-cells, a type of T-cell that can recognize and kill cells that become infected by virus (without antibody tagging). For HIV, CD8+ T-cells are important for the control of the virus that has established an infection. For Ebola and Marburg, antibodies can stop or slow the progress of infection, but T cells are important for clearing the infection by killing remaining infected cells.
Background – Viruses and Vaccines
What are Viruses? Viruses are microscopic organisms consisting of genetic material comprised of DNA (deoxyribonucleic acid) or RNA (ribonucleic acid), surrounded by a protein, lipid (fat), or glycoprotein coat. Viruses invade healthy, living host cells in order to replicate and spread. In many cases, the body’s immune system can recognize and effectively combat an infection caused by a virus. However, with certain viral infections, the body’s immune system is unable to fully destroy or inhibit the replication of the virus, which results in persistent and ongoing viral replication resulting in disease.
Infections caused by viruses can be chronic or acute. Chronic infections, such as those caused by HIV, do not typically self-resolve with time and can cause chronic disease. Acute infections associated with viruses, such as influenza, generally last for a relatively short period of time, and self-resolve in most immunocompetent individuals. However, certain acute infections, such as those caused by Ebola and Marburg, can overwhelm the immune system, resulting in serious disease and death.
Viruses can also be characterized as either active or latent. An active virus can cause a persistent infection or disease over an extended period of time. A latent virus will remain in the body for very long periods of time after the initial infection and generally will only cause disease when the body’s immune system weakens, fails or is suppressed, allowing the virus to once again replicate. Vaccines have been widely used to prevent active viral infections from occurring. Latent infections are more difficult to address with vaccines. A latent virus does not replicate actively and can “fly below the radar” of the immune system in that it does not provide the immune system with targets for antibody and T-cell responses.
Viruses that develop resistance to antiviral drugs are increasingly becoming a challenge in the treatment of viral infections, particularly those that are chronic in nature. The ability of viruses to mutate spontaneously during replication allows drug-resistant strains to emerge when patients are using drugs that are not potent enough to quickly and completely inhibit viral replication. Drug-resistant mutant viruses, while initially low in number, eventually become the predominant strain in an infected patient as those strains that remain susceptible to the drug are inhibited from replicating. Once this occurs, the treatment benefit of that particular antiviral drug diminishes, resulting in treatment failure and the need for an alternate therapy with different or possibly new drugs, or classes of drugs. In general, viruses that cause chronic infections, such as HIV, are more likely to develop drug resistance due to the long-term and persistent exposure of the virus to the antiviral therapy.
What are Vaccines? Vaccines represent an approach to broaden the ability to prevent serious infectious diseases caused by both viruses and bacteria. A vaccine is a substance introduced into the human body that teaches the immune system to detect and destroy a pathogen (a virus or other pathogen that causes disease). All vaccines contain some harmless form or part of the pathogen they target or of a highly similar pathogen. They exert their effects through the adaptive immune response, an arm of the immune system that learns to recognize and control specific pathogens.
There are several types of vaccines:
|
● |
Whole-killed/Whole-inactivated vaccines: The active ingredient in these vaccines is an intact virus or bacterium that has been killed or otherwise stripped of its ability to infect humans. Examples include the cholera and injectable polio vaccines. This approach has not been applied to the development of vaccines against HIV due to lack of success in animal experiments and the difficulty of developing an inactivation method capable of ensuring that the product will be entirely free of active virus. Similarly, inactivated Ebola vaccines have not shown great promise in animal models, and any production process starting with live Ebola or Marburg virus would require such extreme containment measures that it would be difficult to operate at industrial scale. |
|
● |
Live attenuated vaccines: These vaccines use a form of the targeted pathogen that is highly unlikely to be harmful—one capable, say, of multiplying, but not causing disease. Examples include the measles vaccine and the oral vaccine against polio, which has been widely deployed in global eradication efforts. Such vaccines can be very effective because they closely mimic the behavior of the targeted pathogen, giving the immune system a truer picture of what it would be up against. Due to the risk that attenuated HIV, Ebola, or Marburg might revert to its disease-causing form, this approach has not been applied to the development of HIV, Ebola, or Marburg vaccines. |
|
● |
Subunit vaccines: Vaccines of this variety are composed of purified pieces of the pathogen (known as antigens) that generate a vigorous, protective immune response. Common subunit vaccines include the seasonal flu and hepatitis B vaccines. This approach was employed to devise the first AIDS vaccine candidate tested in humans, which failed to induce protection from HIV infection. To date, subunit vaccines have failed to protect nonhuman primates against Ebola infection (Human Vaccines, 6:439 (2010)). |
|
● |
Purified VLP vaccines: Purified VLP vaccines consist only of virus-like particles, which are composed of certain viral proteins but do not contain the genetic material of the virus. Unlike subunit vaccines, VLPs typically provide viral antigens in their native form. Due to their structural similarity to actual viruses, VLPs are excellent immunogens capable of raising potent antibody and cellular immune responses. Purified VLPs need to be manufactured and purified in large quantities. They also are difficult to make for relatively fragile viruses with lipid membrane envelopes such as HIV, Ebola, or Marburg vaccines. Examples of successful vaccines using purified VLPs include vaccines for hepatitis B (Merck’s Recombivax® and GSK’s Engerix®) and Papilloma viruses (GSK’s Cervarix®, and Merck’s Gardasil®). |
|
● |
Expressed VLP vaccines: These vaccines are designed to produce self-assembling non-infectious VLPs in the cells of the person being vaccinated. When VLPs for enveloped viruses like HIV, Ebola, and Marburg are produced in vivo, they include not only the protein antigens, but also an envelope consisting of membranes from the vaccinated individual’s cells. In this way, they are highly similar to the virus generated in a person’s body during a natural infection. Purified VLPs produced externally, by contrast, have no envelope; or, envelopes from the cultured cells (typically hamster or insect cells) used to produce them. By producing VLPs in vivo, potential purification issues associated with in vitro production of VLPs are avoided. GeoVax employs this approach in our vaccine design. |
|
● |
DNA vaccines: These vaccine candidates are also designed to train the immune system to recognize a piece of the targeted bacterium or virus. The difference is that the active ingredients are not the purified antigens themselves but circles of DNA, called plasmids, which carry genes encoding those antigens. Human cells passively take up these plasmids and produce the antigens which, in turn, train the immune system to recognize the targeted pathogen. |
|
● |
Recombinant viral vaccines: These vaccines, like DNA vaccines, introduce genes for targeted antigens into the body. But the genes are inserted into a virus that actively infects human cells. The viruses chosen as vectors are safe to use because they do not ordinarily cause disease in humans and/or have been stripped of their ability to proliferate. |
Our Ebola & Marburg Vaccine Program
About Ebola and Marburg. Ebola Hemorrhagic Fever (EHF) and the related disease Marburg Virus Disease (MVD) are highly contagious, extremely deadly diseases that, if not contained by quarantine, are capable of threatening populations worldwide. Since 1976, when Ebola was first discovered, at least 28 outbreaks have occurred. The recent Ebola outbreak in West Africa is significantly larger than any previous epidemic, the first to reach urban areas and the first to lead to person-to-person transmission in the United States. As of February 2015, the current epidemic has resulted in over 22,500 infections with over 9,000 deaths (40% fatality rate). No approved preventive or therapeutic products exist for EHF or MVD.
Ebola and Marburg naturally infect animals including bats, creating reservoirs of Ebola and Marburg that, like rabies, cannot be completely eradicated. The rapid urbanization of many areas of Sub-Saharan Africa and the ease of modern air travel create conditions that facilitate the epidemic spread of EHF and MVD, which previously had been limited to localized outbreaks in villages. EHF is caused by ebolaviruses (Ebola), and MVD is caused by marburgviruses (Marburg). Ebola and Marburg are members of the family Filoviridae. Ebolaviruses are more diverse than marburgviruses and are divided into five subtypes: Zaire, Sudan, Bundibugyo, Tai Forest, and Reston. Zaire is the most lethal of the strains and is responsible for the current epidemic. Sudan and Bundibugyo are also lethal but have caused fewer and less severe outbreaks.
A challenge in Ebola and Marburg vaccine development is the need to create products that are effective both in containing an epidemic (in which rapid responses are critical) and in routine immunization (in which the duration of immunity is important). Ideal countermeasures to Ebola and Marburg would include a single-shot strain-specific epidemic vaccine capable of rapidly producing protective antibodies and T cells, and a routine vaccine capable of eliciting durable immunity to the lethal strains of Ebola (Zaire, Sudan and Bundibugyo) as well as Marburg. An effective vaccine against Ebola and/or Marburg would dramatically reduce the epidemic spread of infections as well as the transmission of Ebola and/or Marburg from natural animal hosts to humans.
Research on Ebola vaccines is progressing rapidly amongst a number of different pharmaceutical companies, with recombinant chimpanzee adenovirus (ChAd3), rare-serotype adenovirus (Ad26) and vesicular stomatitis virus (VSV) candidates already in clinical trials and several other vaccines scheduled to begin clinical trials. However, none of these vaccines has an ideal design, nor are any of them well suited for use in proactive immunization of populations to prevent future epidemics. The adenovirus vaccines require boosting with MVA to raise protective immune responses, and the two-product regimen (adenovirus and MVA) dramatically raises manufacturing costs and the complexity of vaccination. The replication competent VSV recombinants have already shown risk signals in the current trial, necessitating a temporary halt to the trial followed by resumption at a lower vaccine dose. The potential dose-limiting toxicity of the VSV vaccines raises safety concerns for large-scale vaccinations and also could pose threats to immunocompromised people, such as those infected with HIV. None of the competitors’ vaccines produce virus-like particles, a desirable characteristic, which is discussed in detail elsewhere in this document. To the best of our knowledge, no non-GeoVax vaccine candidates share this characteristic. One or more of the current candidates may well show success in stemming the current epidemic. However, the world must be prepared with the optimal vaccine for the next epidemic when it occurs. All of the vaccines currently in clinical trials are designed to protect against one, or at most two, strains of Ebola. To be successful, an optimal vaccine should be safe, effective, and long lasting, all at a reasonable cost. Our analysis suggests that the GeoVax designs are well suited to achieve this aim.
Our Ebola/Marburg Vaccines. To address the unmet need for a product to prevent EHF and MVD, we are developing a series of Ebola and Marburg vaccines, which combine our proven MVA technology with advanced vaccine design. We are developing individual vaccines (monovalent) that will address each of the lethal strains of Ebola virus (Zaire, Sudan and Bundibugyo), as well as Marburg virus. We also plan to develop a multivalent vaccine, which will incorporate multiple monovalent vaccines to protect against the three strains of Ebola and Marburg with a single product.
For testing purposes, our first focus will be the monovalent vaccine for the Zaire strain of Ebola, which we plan to test in the widely used guinea pig challenge model during 2015. This would be followed by testing in the more rigorous non-human primate model, with an expected start date in late 2015. We are planning to begin preclinical testing of our multivalent vaccine several months after the monovalent Zaire vaccine. An initial proof-of-concept study is planned which will allow us to identify which strains and how many strains are needed in the multivalent vaccine; this may be followed (if necessary) by another study to provide additional data on the vaccine, especially with regard to durability of immune responses. We are also planning to initiate IND-enabling toxicology studies in late 2015, with the goal of starting Phase 1 human clinical trials, for both our monovalent and multivalent vaccines, in late 2016.
We are self-funding the early development work on our vaccines, including the guinea pig challenge studies, but the later-stage testing and clinical trials will be dependent upon the availability of sufficient financial resources. We intend to seek funding from U.S. government agencies and/or world health organizations to assist us in this regard.
We believe our Ebola/Marburg vaccines will demonstrate a unique combination of advantages that set them apart from any other products in development for prevention of EHF.
|
● |
VLP immunogens. Our GEO-EM01 vector (the active component of the GOVX-E301 product) has been demonstrated to express noninfectious Ebola VLPs in human cells. VLPs mimic the structure of ebolavirus particles and display the vaccine antigens in conformations that are highly similar to those present in live virions. Our prior experience with VLP-expressing HIV vaccines suggests that VLPs expressed by MVA raise highly durable antibody responses, the best durability seen in the field of HIV vaccines. |
|
● |
Expression of VLPs by a live vector. Unlike purified VLP vaccines, the GeoVax vaccines are intended to produce VLPs in the cells of the vaccinated person. This strategy carries several advantages. The live, VLP-expressing vector provides antigens in three different forms: as VLPs, as proteins on the surface of MVA-infected cells, and as proteins expressed within MVA-infected cells. Each type of antigen is recognized differently by the immune system, contributing to the breadth and potency of the immune response. Also, unlike VLPs produced in cell culture, the VLPs expressed by the GeoVax vaccines bud from the cells of the vaccinated person, just as infectious Ebola or Marburg would do if the person were exposed to the virus rather than the vaccine. In this way, the VLPs produced in the cells of the vaccinated person are structurally more similar to actual Ebola or Marburg virions. This structural similarity focuses the immune response on the actual antigens of interest rather than eliciting responses against antigens in non-native forms or irrelevant proteins from the membranes of cultured cells. |
|
● |
The excellent safety of MVA. Our vaccines use the MVA vector, which is highly attenuated. Originally developed as a safer alternative to vaccinia, MVA has shown excellent safety in over 120,000 human subjects. It is widely recognized as a safe vector for recombinant vaccines and has been shown to be safe in immunocompromised individuals and in SIV (the primate version of HIV) infected macaques. The attenuation of MVA allows it to be used in high doses, potentially enabling a protective single-dose regimen in an epidemic situation. Though two other MVA vectors do not express VLPs and are components of other vaccines in clinical development, these other MVA vectors are used in combination with novel adenovirus vectors, which have only limited safety data in humans. |
|
● |
The ability of MVA to raise antibody and T-cell responses. The field of Ebola immunology is developing rapidly, and researchers have not yet reached a solid consensus on a correlate of protection. Recent studies, including anecdotal results from passive antibody therapy of infected patients, point toward neutralizing antibody as the most important immune response. However, certain animal challenge studies have suggested that binding (rather than neutralizing) antibody correlates best with protection, and other studies have indicated T-cell responses are critical for clearing infections. MVA-vectored vaccines are very efficient at raising both antibody and T-cell responses. |
|
● |
Antigens against the current epidemic. A vaccine will be most effective if it provides antigens as similar as possible to those in circulating strains of the pathogen. For this reason, we have designed our Zaire ebolavirus vaccines against a genetic sequence from the current epidemic. In this way, our product maximizes the probability of delivering a vaccine antigen that is as close as possible to the circulating pathogen. |
|
● |
Rapid induction of responses. The MVA vector is highly effective at raising protective responses quickly. Vaccinia, the parental vector for MVA, was used successfully in immunization of people who had come in contact with smallpox-infected individuals. This fact and results from GeoVax’s HIV trials suggest that the GeoVax Ebola and Marburg vaccines should be well-suited to epidemic situations in which a protective response must be raised quickly. |
|
● |
Homologous prime-boost regimen. Published data indicate that, while a single immunization may be sufficient to provide short-term protection in an epidemic situation, a multiple-dose strategy is often superior for raising the durable responses that are required in routine preventive vaccination campaigns. Our MVAs are designed to be used in homologous prime-boost regimens, in which multiple doses of the same vaccine are given. The homologous prime-boost strategy is simpler and more economical than heterologous prime-boost products such as the adenovirus-MVA combinations currently being tested. Relative to a product that requires a heterologous prime-boost regimen, our MVAs are simpler and less expensive to manufacture, test, distribute, and use. |
|
● |
Experience with the use of MVA in prime-boost regimens. MVAs are highly effective at boosting immune responses, as demonstrated in previous work on Ebola as well as preclinical and clinical trials of HIV vaccines. Our results with MVA prime-boost regimens in HIV trials suggest that MVA alone is highly effective (more effective than DNA and MVA combined) at raising antibody responses. For this reason, we believe that the MVA-MVA prime-boost strategy will be ideal for routine vaccination of populations with our GOVX-E301 product. Also, though we have no current plans to develop our MVAs as boosts for other vaccines, we recognize that any of our MVAs could potentially be used as a heterologous boost to a different (for example, adenovirus) priming vaccine if future data indicate that a heterologous regimen is desirable. |
|
● |
The excellent thermal stability of MVA. To be appropriate for use in remote regions of the world, a vaccine must be stable enough to remain potent despite suboptimal cold chain logistics. In addition, to be suitable for storage in national stockpiles, Ebola vaccines must remain stable over several years of storage. MVA vaccines are highly stable in both liquid and lyophilized dosage forms. An ongoing stability study of our MVA vaccine against HIV has shown excellent stability over more than six years of storage. |
|
● |
Manufacturability of MVA-vectored vaccines. If designed with genetically stable inserts, MVA-vectored vaccines can reliably be manufactured in large quantities. In addition to the established Chick Embryo Fibroblast (CEF) cell substrate, we have also investigated novel continuous cell lines for manufacture of our vaccines against HIV, and believe they could potentially be used for manufacture of our MVA vaccines against Ebola. Continuous cell lines offer virtually unlimited scalability as well as greater process consistency and efficiency. |
Our HIV/AIDS Vaccine Program
About HIV/AIDS. HIV is a retrovirus that carries its genetic code in the form of RNA. Retroviruses use RNA and the reverse transcriptase enzyme to create DNA from the RNA template. The HIV-1 virus enters human cells and copies its viral RNA to produce complementary DNA (cDNA) that is subsequently inserted into the chromosomes, which are the genetic material of a cell. HIV preferentially infects and replicates in T-cells, which are a type of white blood cell. Infection of T-cells alters them from immunity mediating cells to cells that produce and release HIV. This process results in the destruction of the immune defenses of infected individuals and ultimately, the development of AIDS.
There are several AIDS-causing HIV virus subtypes, or clades, that are found in different regions of the world. These clades are identified as clade A, clade B and so on. The predominant clade found in Europe, North America, parts of South America, Japan and Australia is clade B, whereas the predominant clades in Africa are clades A and C. In India, the predominant clade is clade C. Each clade differs by at least 20% with respect to its genetic sequence from other clades. These differences may mean that vaccines or treatments developed against HIV of one clade may only be partially effective or ineffective against HIV of other clades. Thus, there is often a geographical focus to designing and developing HIV vaccines.
HIV, even within clades, has a high rate of mutation that supports a significant level of genetic variation. In drug treatment programs, virus mutation can result in the development of drug resistance, referred to as virus drug escape, thereby rendering drug therapy ineffective. Hence, we believe that multi-drug therapy is very important. If several drugs are active against virus replication, the virus must undergo multiple simultaneous mutations to escape, which is less likely. The same is true for immune responses. HIV can escape single targeted immune responses. However, our scientists believe if an immune response is directed against multiple targets, which are referred to as epitopes, virus escape is much less frequent. Vaccination against more than one of the proteins found in HIV increases the number of targets for the immune response as well as the chance that HIV will not escape the vaccine-stimulated immune response, thus resulting in protection against infection or the development of clinical AIDS if infection occurs.
HIV infects and gradually destroys T-cells and macrophages, which are white blood cells that play key roles in protecting humans against infectious disease caused by viruses, bacteria, fungi and other micro-organisms. Opportunistic infections by organisms, normally posing no problem for control by a healthy immune system, can ravage persons with immune systems damaged by HIV infections. Destruction of the immune system occurs over years. The average onset of the clinical disease recognized as AIDS occurs after three to ten years of HIV infection if the virus is not treated effectively with drugs, but the time to developing AIDS is highly variable.
AIDS is considered by many in the scientific and medical community to be the most lethal infectious disease in the world. According to the report published by UNAIDS/WHO, at the end of 2012, an estimated 36 million people were living with HIV worldwide, with approximately 2.5 million newly infected in 2012 alone. Approximately 25 million people infected with HIV have died since the 1981 start of the HIV pandemic. The United States currently has an estimated 1.1 million HIV-infected individuals, with approximately 55,000 new infections per year.
At present, the standard approach to treating HIV infection is to inhibit viral replication through the use of combinations of drugs. Available drugs include reverse transcriptase inhibitors, protease inhibitors, integration inhibitors and inhibitors of cell entry to block multiple essential steps in virus replication. However, HIV is prone to genetic changes that can produce strains that are resistant to currently approved drugs. When HIV acquires resistance to one drug within a class, it can often become resistant to the entire class, meaning that it may be impossible to re-establish control of a genetically altered strain by substituting different drugs in the same class. Furthermore, these treatments continue to have significant limitations which include toxicity, patient non-adherence to the treatment regimens and cost. As a result, over time, viruses acquire drug-resistant mutations, and many patients develop intolerance to the medications or simply give up taking the medications due to the side effects.
According to the International AIDS Vaccine Initiative (IAVI), the cost and complexity of new treatment advances for AIDS puts them out of reach for most people in the countries where treatment is most needed. As noted above, in industrialized nations, where drugs are more readily available, side effects and increased rates of viral resistance have raised concerns about their long term use. AIDS vaccines, therefore, are seen by many as the most promising way to end the HIV/AIDS pandemic. It is expected that vaccines for HIV/AIDS, once developed, will be used universally and administered worldwide by organizations that provide health care services, including hospitals, medical clinics, the military, prisons and schools.
Our Preventive HIV Vaccine Program
Prevention of HIV infection remains a worldwide unmet medical need, even in the United States and other first world countries where effective antiretroviral therapies are available. Current antiretroviral therapies do not eliminate HIV infection, requiring individuals to remain on antiretroviral drugs for their entire lives. In the United States, it is estimated that of the 1.1 million infected individuals, for various reasons (lack of diagnosis, linkage to care, patient compliance, etc.) only 25% ultimately remain in HIV care with their viral load sufficiently suppressed to prevent spread of HIV. As a result, the annual incidence of new HIV infections has remained virtually unchanged for the past 20 years. Furthermore, the annual financial burden to the U.S. taxpayer for HIV education, prevention, and treatment costs borne through Medicaid, Medicare, and the Ryan White Act is more than $16 billion annually, and the estimated lifetime medical costs for an individual infected with HIV is $500,000.
Work on our HIV vaccines began during the 1990s at Emory University in Atlanta, Georgia, under the direction of Dr. Harriet L. Robinson, who is now our Chief Scientific Officer. The vaccine technology was developed in collaboration with researchers at the NIH and the CDC.
Our most clinically advanced vaccine development program is a DNA/MVA vaccine regimen designed to protect against the clade B subtype of the HIV virus. Clade B is prevalent in the Americas and Western Europe. An estimated 3.3 million people are infected with clade B HIV virus worldwide, with 187,000 new infections in 2012.
We have two HIV vaccine components under development: a recombinant DNA vaccine, and a recombinant MVA vaccine. Both the DNA and MVA vaccines contain sufficient HIV genes to support the production of non-infectious virus-like particles. These VLPs display the native trimeric membrane-bound form of the HIV envelope glycoprotein (Env) that mediates entry into cells and is the target for protective antibody. When used together, the recombinant DNA component primes immune responses, which are boosted by administration of the recombinant MVA component. This prime-boost strategy elicits high avidity antibodies (tightly binding antibodies) and cytotoxic T cells. The antibodies can block infections and initiate the killing of virus and infected cells by bound antibody signaling destruction by virion capture, antibody-dependent cellular cytotoxicity, phagocytosis and complement mediated lysis. We may also pursue development of our MVA vaccine component as a standalone HIV vaccine, or in combination with other vaccine components.
Clinical trials of our preventive HIV vaccine have been conducted by the HIV Vaccine Trials Network (HVTN). The HVTN is the largest worldwide clinical trials network dedicated to the development and testing of HIV/AIDS vaccines. Support for the HVTN comes from the National Institute of Allergy and Infectious Diseases (NIAID), part of the NIH. The HVTN’s HIV Vaccine Trial Units are located at leading research institutions in 27 cities on four continents.
We have completed multiple Phase 1 trials and a Phase 2a trial (HVTN 205) of various dosing regimens and formulations of our vaccines. These vaccines have been evaluated in nearly 500 humans. All of the clinical trials of our preventive vaccines have been conducted by the HVTN, and fully funded by the NIH.
We are actively engaged in discussions with the HVTN and NIAID regarding the design of our next clinical study and various trial designs are being considered. Our vaccine is currently the only vaccine being contemplated for efficacy trials for prevention of clade B HIV infection. However, the HVTN believes the best path forward will be to test our vaccines in combination with a protein boost. Protein boosts may augment antibody responses that can block virus infections (neutralizing antibody) and cause antibody dependent cellular cytotoxicity (ADCC antibody). Proteins added to HIV vaccines have shown some success in other trials. The HVTN believes this “dual-action” approach will be a prudent and cost-effective path forward for supporting large clinical trials. Our current expectation is that the next clinical trial will begin in late 2015 and will be a follow-on study to the HVTN 205 trial, in which those trial participants are given a protein boost to evaluate their immune responses. Information from this trial would then inform the design of future, larger clinical trials.
The HVTN is continuing to consider future efficacy studies, and members are working to develop collaborative clinical development plans, as well as initiating regulatory planning. The plans for large-scale clinical trials may change as researchers continue to gather information from our earlier studies and are influenced by results from other vaccine trials. Trial start dates are dependent on many factors and are likely to change.
Preventive HIV Vaccine Program – Clade C. Subject to the availability of funding support from governmental or nongovernmental organizations, we also plan to develop vaccines designed for use to combat the subtypes of HIV that predominate in the developing countries. We have licensed from the U.S. National Institutes of Health (NIH) the modified vaccine Ankara (MVA) construct for the clade C subtype of HIV prevalent in South Africa and India, and we have completed lead discovery using a novel approach to vaccination against clade C. We have performed initial process development studies for the NIH-developed vaccine and initiated early development work on the other, newer clade C vaccine. Depending on the results of animal studies and the focus of government support, we may advance either or both of the clade C vaccines into the clinic.
Preclinical Studies. We conducted preclinical efficacy trials of our preventive HIV vaccines by vaccinating non-human primates with simian immunodeficiency virus prototypes of our HIV vaccines and then testing them for resistance to simian immunodeficiency virus infection. The experimental data produced by these trials documented the ability of the simian prototypes of our vaccines to induce immune responses that can prevent infection as well as reduce the levels of viral replication in those animals that become infected.
Completed Human Clinical Trials -- Preventive HIV Vaccine
Phase 1 Human Clinical Trials. All of our preventive vaccination trials in humans have been conducted by the HVTN, a network that is funded and supported by the NIH. The HVTN is the largest worldwide clinical trials network focused on the development and testing of HIV/AIDS vaccines. The results of a two group, 30 participant, Phase 1 trial (designated HVTN 045) are published in AIDS RESEARCH AND HUMAN RETROVIRUSES 22:678 (2006) and of a four group 120 participant trial (HVTN 065) in The Journal of Infectious Diseases 203:610 (2011). Our Phase 1 trials have tested both safety and dosing regimens.
In our first Phase 1 clinical trial, HVTN 045, our DNA vaccine was tested without MVA boosting to document the safety of the DNA. Our second Phase 1 clinical trial, HVTN 065, was designed to test the combined use of DNA and MVA and consisted of a dose escalation as well as regimen studies. The low dose consisted of 0.3 mg of DNA and 1x107 tissue culture infectious doses (TCID50) of MVA. Once safety was demonstrated for the low dose in 10 participants, the full dose (3 mg of DNA and 1x108 TCID50 of MVA) was administered to 30 participants. A single dose of DNA at time 0 followed by MVA at weeks 8 and 24, a DMM regimen, and three doses of MVA administered at weeks 0, 8 and 24, an MMM regimen, were also tested in 30 participants each. Participants were followed for 12 months to assess vaccine safety and to measure vaccine-induced immune responses.
Data from the HVTN 065 trial again documented the safety of the vaccine products but also showed that the DDMM and MMM regimens induced different patterns of immune responses. The full dose DDMM regimen induced higher response rates of CD4++ T-cells (77%) and CD8++ T-cells (42%) compared to the MMM regimen (43% CD4+ and 17% CD8+ response rates). In contrast, the highest response rates and highest titers of antibodies to the HIV Env protein were induced in the group that received only the MVA using the MMM regimen. Antibody response rates were documented to be higher for the MMM group using three different assays designed to measure total binding antibody levels for an immune dominant portion of the Env protein (27% for DDMM and 75% for MMM), binding of antibodies to the gp120 subunit of the envelope glycoprotein (81% for DDMM and 86% for MMM) and neutralizing antibodies (7% for DDMM and 30% for MMM). The 1/10th dose DDMM regimen induced overall similar T-cell responses but reduced antibody responses while the response rates were intermediate in the DMM group.
The HVTN also sponsored and conducted a Phase 1 clinical trial in humans (HVTN 094) of the adjuvanted form of our vaccine that co-expresses GM-CSF in the DNA priming vaccine. We have designated the GM-CSF-adjuvanted version of our DNA/MVA vaccine regimen as GOVX-B21, and the unadjuvanted version as GOVX-B11. During December 2013, we reviewed preliminary results from HVTN 094. Based on excellent preclinical non-human primate data, this trial was originally initiated with the expectation that GOVX-B21 would be carried forward into Phase 2 testing by the HVTN, with support by the NIH. However, comparison of data between HVTN 094 and the Phase 2a trial, HVTN 205 (see below) did not show a significant benefit from adding the adjuvant to the vaccine for preventive use; therefore GOVX-B21 was not advanced in further clinical testing (results to be published).
Phase 2 Human Clinical Trials. Based on the safety and the immunogenicity results in the HVTN 045 and HVTN 065 trials, the full dose DNA/MVA and MVA-only regimens were selected for testing by the HVTN in a Phase 2a trial (designated HVTN 205) which was completed in 2012 and the subject of an oral presentation at the AIDS Vaccine 2012 Conference in September 2012, with further analysis presented at the AIDS Vaccine Meeting in Barcelona, Spain, in October 2013 and a publication in the Journal of Infectious Diseases (volume 210, pg 99) in 2014. HVTN 205 was designed to evaluate the safety and immunogenicity of our vaccines in healthy, HIV-uninfected adults. In HVTN 205, 299 participants were randomly assigned to three study arms: 149 participants received two injections of our DNA vaccine followed by two injections of our MVA vaccine (DDMM arm), 75 participants received three MVA injections and one placebo injection (MMPM arm), and 75 participants received four injections of placebo. After the final vaccination, antibody responses against the HIV Envelope protein (Env), the target for protective antibody, were detected in 93.2% of the DDMM arm (the vaccination regimen selected for further clinical study). At six months after final vaccination (the latest time point tested), gp140 IgG antibody response titers in the DDMM arm had declined by less than 3-fold, with response rates only declining from 100% to 84%, indicating significant durability of the antibody response. Additionally, HVTN 205 also showed that the antibody responses after vaccination had high affinity binding, a characteristic which has been associated with prevention of HIV infection in preclinical models. The study also showed low response rates for serum IgA, a desirable characteristic because serum IgA competed with serum IgG for reducing the risk of infection in the one partially protective (31%) AIDS vaccine trial in Thailand. Response rates for serum IgG3, an isotype associated with activating innate methods of protection such as complement (C’)-mediated lysis and antibody-dependent cellular cytotoxicity were excellent (91%).
HIV Immunotherapy Program
Current antiretroviral therapies, though highly effective at suppressing HIV viral load, are unable to eliminate HIV infection entirely. A major challenge in the development of HIV therapeutics is the ability of HIV to persist in host cells in a latent proviral form, invisible to the immune system and inaccessible to antiretroviral drugs. In response to this problem, the NIH and other leaders in the HIV field have developed a new concept: the “shock and kill” strategy, in which patients remain on standard-of-care anti-retroviral drug therapy while a second drug (“shock agent”) is used to activate latent HIV and a third drug (“kill agent”) is used to recognize and eliminate cells that harbor the latent HIV reservoir. A shock and kill therapy could potentially contribute to a cure for HIV.
Observations from a pilot Phase 1 clinical trial of our HIV vaccines (GV-TH-01 – discussed below) have led us to postulate that our DNA vaccines may be effective as a shock agent and that a subsequent, precisely timed MVA inoculation may reduce viral reservoirs. The Company is currently considering the best course of action for advancing its HIV immunotherapy program. Future therapeutic studies of GeoVax’s vaccine may investigate vaccine’s ability to act as a “shock agent” in a shock and kill therapy in combination with standard of care antiretroviral drug therapy to seek a cure. The timetable and specific clinical plans will be dependent upon the Company’s ability to secure external funding for the program, and on the nature of any potential collaborations GeoVax may establish.
Preclinical Studies – Therapeutic Vaccine. In 2007-2008, data were generated in three pilot studies on therapeutic vaccination in simian immunodeficiency virus-infected non-human primates. The vaccine used in these pilot studies was specific for simian immunodeficiency virus but with the design features of our HIV/AIDS vaccine. In these pilot studies, conducted at Yerkes National Primate Research Center of Emory University, non-human primates were infected, drug-treated, vaccinated and then drug-interrupted. Following treatment interruption, median levels of virus in blood, measured as viral RNA, were 10 to 1000-times lower than those measured prior to drug and vaccine treatment. The therapeutic reductions in virus levels were best for animals placed on drugs within 12 weeks of infection with lower levels of protection being achieved in animals that were placed on drugs at 3 months or later after infection.
Phase 1 Trial (Treatment Interruption). In early 2014, we completed a Phase 1 clinical trial (GV-TH-01) investigating the therapeutic use of our vaccines in HIV-infected patients. GV-TH-01 is an open label Phase 1 treatment interruption trial investigating the safety and immunogenicity of our DNA/MVA vaccine regimen in 9 HIV-infected patients who initiated drug treatment within 18 months of seroconversion and had stably controlled virus for at least 6 months. Patients were vaccinated with two DNA inoculations followed by two MVA inoculations at intervals of two months. Eight weeks following the last inoculation, patients suspended drug therapy for a 12-week period. Vaccinated patients’ ability to control the time and temporal height of re-emergent virus in the absence of drugs was then observed. Drug treatment was re-instituted after 12 weeks, and trial participants were observed for an additional 6 months. The primary endpoint of this study was to evaluate the safety of our vaccine in HIV-positive patients with well-controlled infections who are being treated with oral HIV medications. An exploratory objective of the study was to evaluate the ability of the vaccinated patient to control re-emergent virus during the drug treatment interruption period.
Analysis of GV-TH-01 data indicates that, during the vaccination phase of the trial, enhanced CD8++ T cells were elicited in 8 of 9 participants and enhanced CD4++ T cell in 5 of 9 participants. Antibody responses were boosted in 4 of 9 participants. Analyses during the treatment interruption phase of the trial suggested that individuals with the best immune responses had lower levels of re-emergent virus. These levels however were not sufficiently low to prevent immune escape and the reinstitution of progression towards AIDS. Excellent safety was observed throughout the trial, with none of the participants needing to reinstate antiretroviral drugs during the treatment interruption phase of the trial (data being compiled for publication).
Support from the United States Government
With the exception of the GV-TH-01 Phase 1 therapeutic trial (treatment interruption protocol), all of our human clinical trials to date have been conducted by the HVTN and funded by NIH. This financial support has been provided by the NIH directly to the HVTN, so has not been recognized in our financial statements. Our responsibility for these clinical trials has been to provide sufficient supplies of vaccine materials and technical expertise when necessary.
In addition to clinical trial support from the NIH, our operations are partially funded by NIH research grants. In September 2007, the NIH awarded us an Integrated Preclinical/Clinical AIDS Vaccine Development (IPCAVD) grant to support our HIV/AIDS vaccine program. We utilized this funding to further our HIV/AIDS vaccine development, optimization and production. The aggregate award (including subsequent amendments) totaled approximately $20.4 million, and there was approximately $75,000 remaining and available for use as of December 31, 2014. In September 2012, the NIH awarded us an additional grant of approximately $1.9 million to support development of versions of our HIV/AIDS vaccines to address the clade C subtype of the HIV virus prevalent in the developing world. All funding pursuant to this grant has been utilized. In July 2013, the NIH awarded us a Small Business Innovative Research (SBIR) grant for approximately $277,000 to support preclinical studies evaluating the ability of protein boosts to augment antibody responses. The initial grant award was approximately $277,000 for the first year of a two-year project period beginning August 1, 2013. In July 2014, the NIH awarded us approximately $290,000 for the second year of the project period. We recorded grant revenues of $258,267, $154,563, and $-0- for the years ended December 31, 2014, 2013 and 2012, respectively, related to this grant, and there was approximately $154,000 of unrecognized grant funds remaining and available for use pursuant to this grant as of December 31, 2014.
Please refer to our Financial Statements beginning on page F-1 of this Form 10-K, and to “Management's Discussion and Analysis of Financial Condition and Results of Operations”, for additional information regarding revenue and funds availability.
Regulations
Regulation by governmental authorities in the United States and other countries is a significant factor in our ongoing research and development activities and in the manufacture of our products under development. Complying with these regulations involves considerable time and expense.
In the United States, drugs are subject to rigorous federal and state regulation. Our products are regulated under the Federal Food, Drug and Cosmetic Act, as amended (FD&C Act), and the regulations promulgated thereunder, and other federal and state statutes and regulations. These laws govern, among other things, the testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of medications and medical devices. Product development and approval within this regulatory framework is difficult to predict, takes a number of years and involves great expense. The steps required before a human vaccine may be marketed in the United States include:
|
● |
pre-clinical laboratory tests, in vivo pre-clinical studies and formulation studies; |
|
● |
manufacturing and testing of the product under strict compliance with current Good Manufacturing Practice (cGMP) regulations; |
|
● |
the submission to the FDA of an Investigational New Drug (IND) application for human clinical testing which must become effective before human clinical trials can commence; |
|
● |
adequate and well-controlled human clinical trials to establish the safety and efficacy of the product; |
|
● |
the submission of a Biologics License Application to the FDA, along with the required user fees; |
|
● |
FDA approval of the Biologics License Application prior to any commercial sale or shipment of the product; and |
|
● |
postmarketing requirements imposed by FDA. |
Each of these steps is described further below. Before marketing any drug or biologic for human use, the product sponsor must obtain FDA approval. In addition, each manufacturing establishment must be registered with the FDA and must pass a Pre-Approval Inspection (PAI) before introducing any new drug or biological product into commercial distribution. Because GeoVax does not manufacture vaccines for human use within our own facilities, we must ensure compliance both in our own operations and in the outsourced manufacturing operations. All FDA-regulated manufacturing establishments (both domestic establishments and foreign establishments that export products to the United States) are subject to inspections by the FDA and must comply with the FDA’s Good Manufacturing Practices for products, drugs and devices.
FDA determines compliance with applicable statutes and regulations through documentation review, investigations, and inspections. Several enforcement mechanisms are available to FDA, ranging from a simple demand to correct a minor deficiency to mandatory recalls, closure of facilities, and even criminal charges for the most serious violations.
Preclinical Testing. Preclinical testing includes laboratory evaluation of chemistry and formulation, as well as cell culture and animal studies to assess the safety and potential efficacy of the product. Preclinical safety tests and certain other pivotal preclinical studies must be conducted by laboratories that comply with the FDA’s Good Laboratory Practices, or GLP. The results of pre-clinical testing are submitted to the FDA as part of the IND application and are reviewed by the FDA prior to the commencement of human clinical trials. Unless the FDA objects to an IND, the IND becomes effective 30 days following its receipt by the FDA.
cGMP-Compliant Manufacturing and Testing. FDA has issued, and frequently updates, extensive regulations on current Good Manufacturing Practice (cGMP). Any drug, biologic, or device for human use, whether commercial or investigational, must be manufactured under these regulations. cGMP regulations include a wide variety of requirements covering personnel, documentation, facilities, equipment, testing procedures, and many other aspects of manufacturing and testing.
Clinical Trials. Clinical trials involve the administration of investigational drugs to volunteers or to patients under the supervision of a qualified, medically trained clinical investigator. Clinical trials are conducted in accordance with Good Clinical Practices under protocols that detail the objectives of the trial, the parameters to be used to monitor safety and the efficacy criteria to be evaluated. Each protocol and the qualifications of the investigators who plan to carry it out must be submitted to the FDA as part of the IND. Further, each clinical trial must be conducted under the auspices of an independent institutional review board at the institution where the trial will be conducted. The institutional review board will consider, among other things, ethical factors, the safety of human subjects and the possible liability of the institution.
Clinical trials are typically conducted in three sequential phases, but the phases may overlap. In the Phase 1 clinical trial, the initial introduction of the product into healthy human subjects, the vaccine is tested for safety (including adverse side effects) and dosage tolerance. The Phase 2 clinical trial is the proof of principal stage and involves trials in a limited patient population to determine whether the product induces the desired effect (for our vaccines this means immune responses) and to better determine optimal dosage. The continued identification of possible safety risks is also a focus. When there is evidence that the product may be effective and has an acceptable safety profile in Phase 2 clinical trials, Phase 3 clinical trials are undertaken to evaluate clinical efficacy and to test for safety within an expanded patient population. Phase 3 trials are completed using multiple clinical study sites which are geographically dispersed. The manufacturer or the FDA may suspend clinical trials at any time if either believes that the individuals participating in the trials are being exposed to unacceptable health risks.
Biologics License Application and FDA Approval Process. The results and details of the pre-clinical studies and clinical trials are submitted to the FDA in the form of a Biologics License Application (BLA), which is equivalent to the New Drug Application (NDA) submitted by companies seeking to market new drugs. If the BLA is approved, the manufacturer may market the product in the United States. Under the Prescription Drug User Fee Act (PDUFA), FDA charges user fees to applicants to offset the costs of its operations. The PDUFA user fee for a new vaccine is over $2 million, unless the applicant obtains a waiver or reduction through certain programs designed to encourage development of certain types of products.
Postmarketing Requirements. FDA frequently imposes postmarketing requirements as a condition of NDA or BLA approval. Common postmarketing requirements include additional clinical trials (Phase 4 trials) or observational studies. Postmarketing requirements are especially relevant to our Ebola and Marburg vaccines. We intend to pursue approval of these vaccines using the accelerated approval process, in which FDA grants approval based on performance against a criterion other than actual protection against the disease but requires the manufacturer to monitor and submit data on efficacy of the approved product. Unlike pathogens such as human papillomavirus, Ebola and Marburg are not constantly in circulation; instead, they occur in sporadic but extremely deadly outbreaks. For this reason, it would be impractical and potentially unethical to attempt to perform a traditional Phase 3 trial in which vaccinated participants are compared against unvaccinated participants to determine the efficacy of the vaccine in preventing infection with Ebola or Marburg. The accelerated approval process allows FDA to approve a new medicine based on its performance against a surrogate endpoint (in the case of Ebola or Marburg, its performance in raising immune responses). We anticipate that, as a condition of receiving accelerated approval, GeoVax would agree to monitor the real-world performance of our Ebola and Marburg vaccines.
International Approval. Whether or not the FDA has approved the drug, approval of a product by regulatory authorities in foreign countries must be obtained prior to the commencement of commercial sales of the drug in such countries. The requirements governing the conduct of clinical trials and drug approvals vary widely from country to country, and the time required for approval may be longer or shorter than that required for FDA approval.
Other Regulations. In addition to FDA regulations, our business activities may also be regulated by the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other present and potential future federal, state or local regulations. Violations of regulatory requirements at any stage may result in various adverse consequences, including regulatory delay in approving or refusal to approve a product, enforcement actions, including withdrawal of approval, labeling restrictions, seizure of products, fines, injunctions and/or civil or criminal penalties. Any product that we develop must receive all relevant regulatory approvals or clearances before it may be marketed.
Manufacturing
We do not have the facilities or expertise to manufacture any of the clinical or commercial supplies of any of our products. To be successful, our products must be manufactured in commercial quantities in compliance with regulatory requirements and at an acceptable cost. To date, we have not commercialized any products, nor have we demonstrated that we can manufacture commercial quantities of our product candidates in accordance with regulatory requirements. If we cannot manufacture products in suitable quantities and in accordance with regulatory standards, either on our own or through contracts with third parties, it may delay clinical trials, regulatory approvals and marketing efforts for such products. Such delays could adversely affect our competitive position and our chances of achieving profitability. We cannot be sure that we can manufacture, either on our own or through contracts with third parties, such products at a cost or in quantities that are commercially viable.
We currently rely and intend to continue to rely on third-party contract manufacturers to produce vaccines needed for research and clinical trials. We have entered into arrangements with third party manufacturers for the supply of our DNA and MVA vaccines for use in our planned clinical trials. These suppliers operate under the FDA’s Good Manufacturing Practices and (in the case of European manufacturers) similar regulations of the European Medicines Agency. We anticipate that these suppliers will be able to provide sufficient vaccine supplies to complete our currently planned clinical trials. Various contractors are generally available in the United States and Europe for manufacture of vaccines for clinical trial evaluation, however, it may be difficult to replace existing contractors for certain manufacturing and testing activities and costs for contracted services may increase substantially if we switch to other contractors.
Development of Improved Manufacturing Techniques for MVA – The MVA component of our vaccine is currently manufactured in cells that are cultured from embryonated chicken eggs, which is a reliable method to manufacture large quantities of vaccine. In an attempt to find a means to reduce costs for large-scale manufacturing, we have explored a number of approaches to producing MVA in continuous cell lines that can be grown in bioreactors. In this process we have identified a duck stem-cell-derived line (termed EB66), that is proprietary to Valneva S.E., France. We are currently working with Valneva on the use of EB66 cells for the growth of our MVA vaccines. We are hopeful that upon completion of process development we will be producing vaccine at significantly higher titers in a much more advanced and scalable process, allowing for quality improvements over the current process as well as meaningful cost reductions.
Competition
The biopharmaceutical industry and the vaccine market is competitive and subject to rapid and substantial technological change. Developments by others may render our proposed vaccination technologies noncompetitive or obsolete, or we may be unable to keep pace with technological developments or other market factors. Technological competition in the industry from pharmaceutical and biotechnology companies, universities, governmental entities and others diversifying into the field is intense and is expected to increase. Many of the pharmaceutical companies that compete with us have significantly greater research and development capabilities than we have, as well as substantially more marketing, manufacturing, and financial resources. In addition, acquisitions of, or investments in, small pharmaceutical or biotechnology companies by such large corporations could increase their research, financial, marketing, manufacturing and other resources. Competitive technologies may ultimately prove to be safer, more effective or less costly than any vaccine that we develop.
There are currently no FDA licensed and commercialized Ebola vaccines, Marburg vaccines, or HIV vaccines available in the world market. We are aware of several development-stage and established enterprises, including major pharmaceutical and biotechnology firms, which are actively engaged in vaccine research and development in these areas. For Ebola, these include Johnson & Johnson, GlaxoSmithKline, and Merck. For HIV, these include Novartis, Sanofi-Aventis and GlaxoSmithKline. Other HIV vaccines are in varying stages of research, testing and clinical trials including those supported by the NIH Vaccine Research Center, the U.S. Military, IAVI, the European Vaccine Initiative, and the South African AIDS Vaccine Initiative. We may also experience competition from companies that have acquired or may acquire technologies from companies, universities and other research institutions. As these companies develop their technologies, they may develop proprietary technologies which may materially and adversely affect our business.
If any of our competitors develop products with efficacy or safety profiles significantly better than our products, we may not be able to commercialize our products, and sales of any of our commercialized products could be harmed. Some of our competitors and potential competitors have substantially greater product development capabilities and financial, scientific, marketing and human resources than we do. Competitors may develop products earlier, obtain FDA approvals for products more rapidly, or develop products that are more effective than those under development by us. We will seek to expand our technological capabilities to remain competitive; however, research and development by others may render our technologies or products obsolete or noncompetitive, or result in treatments or cures superior to ours.
Our competitive position will be affected by the disease indications addressed by our product candidates and those of our competitors, the timing of market introduction for these products and the stage of development of other technologies to address these disease indications. For us and our competitors, proprietary technologies, the ability to complete clinical trials on a timely basis and with the desired results, and the ability to obtain timely regulatory approvals to market these product candidates are likely to be significant competitive factors. Other important competitive factors will include the efficacy, safety, ease of use, reliability, availability and price of products and the ability to fund operations during the period between technological conception and commercial sales.
Our Intellectual Property
We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary rights are described by valid and enforceable patents or are effectively maintained as trade secrets. Accordingly, we are pursuing and will continue to pursue patent protection for our proprietary technologies developed through our collaborations with Emory University, the NIH, and the CDC, or developed by us alone. Our patent portfolio, described more fully below, includes claims directed to DNA and MVA based HIV vaccines, their genetic inserts expressing multiple HIV protein components, composition, structure, claim of immunization against multiple subtypes of HIV, routes of administration, safety and other related factors and methods of therapeutic and prophylactic use thereof including administration regimes. Also included are claims directed to preventive vaccines against Ebola and Marburg viruses and use thereof. As of January 1, 2015, we are the licensee of at least eight issued or allowed U.S. patents and at least 12 issued or allowed non-U.S. patents. We are actively pursuing one U.S. provisional application and one international patent application as the owner of record, in addition to at least six U.S. patent applications and at least 15 non-U.S. patent applications in six jurisdictions under license.
We are the exclusive, worldwide licensee of a number of patents and patent applications, which we refer to as the Emory Technology, owned, licensed or otherwise controlled by Emory University for HIV or smallpox vaccines pursuant to a License Agreement originally entered into on August 23, 2002 and restated on June 23, 2004, which we refer to as the Emory License. Through the Emory License we are also a non-exclusive licensee of four issued United States patents owned by the NIH related to the ability of our MVA vector vaccine to operate as a vehicle to deliver HIV virus antigens, and also to induce an immune response in humans. The four issued United States patents owned by the NIH expire in 2023. All of our obligations with respect to the HIV NIH-owned MVA patents are covered by the Emory License. The Emory License expires on the expiration date of the last to expire of the patents licensed thereunder including those that are issued on patents currently pending. We will not know the final termination date of the Emory License until such patents are issued. The Company may terminate the Emory University License upon 90 days’ written notice. The Emory License also contains standard provisions allowing Emory University to terminate upon breach of contract by the Company or upon the Company’s bankruptcy.
The Emory License, among other contractual obligations, requires payments based on the following:
|
● |
Milestone Payments. An aggregate of $3,450,000 is potentially due to Emory University in the future upon the achievement of clinical development and regulatory approval milestones as defined in the Emory License. To date, we have paid a nominal milestone fee upon entering Phase 2 clinical trials for our preventive HIV/AIDS vaccine. |
|
● |
Maintenance Fees. The Company has achieved the specified milestones and met its obligations with regard to the related payments, and no maintenance fees are (or will be) owed to Emory University. |
|
● |
Royalties. Upon commercialization of products covered by the Emory License, we will owe royalties to Emory University of between 5% and 7.5%, depending on annual sales volume, of net sales made directly by GeoVax. The Emory License also requires minimum annual royalty payments of $3 million in the third year following product launch, increasing annually to $12 million in the sixth year. |
|
● |
Sublicense Royalties. In the event that we sublicense a covered product to a third party, we will owe royalties to Emory University based on all payments, cash or noncash, that we receive from our sublicensees. Those royalties will be 19% of all sublicensing consideration we receive prior to the first commercial sale of a related product. Commencing with the first commercial sale, the royalty owed to Emory University will be 27.5% of all sublicensing consideration we receive. |
|
● |
Patent Reimbursements. During the term of the Emory License, we are obligated to reimburse Emory University for ongoing third party costs in connection with the filing, prosecution and maintenance of patent applications subject to the Emory License. The expense associated with these ongoing patent cost reimbursements to Emory University amounted to $179,958, $98,042, and $89,885 for the years ended December 31, 2014, 2013 and 2012, respectively. |
We may only use the Emory Technology for therapeutic or prophylactic HIV or smallpox vaccines. Emory University also reserved the right to use the Emory Technology for research, educational and non-commercial clinical purposes. Due to the use of federal funds in the development of the Emory Technology, the U.S. Government has the irrevocable, royalty-free, paid-up right to practice and have practiced certain patents throughout the world, should it choose to exercise such rights.
We are not a party to any litigation, opposition, interference, or other potentially adverse proceeding with regard to our patent positions. However, if we become involved in litigation, interference proceedings, oppositions or other intellectual property proceedings, for example as a result of an alleged infringement or a third-party alleging an earlier date of invention, we may have to spend significant amounts of money and time and, in the event of an adverse ruling, we could be subject to liability for damages, invalidation of our intellectual property and injunctive relief that could prevent us from using technologies or developing products, any of which could have a significant adverse effect on our business, financial conditions or results of operations. In addition, any claims relating to the infringement of third-party proprietary rights, or earlier date of invention, even if not meritorious, could result in costly litigation, lengthy governmental proceedings, divert management’s attention and resources and require us to enter royalty or license agreements which are not advantageous if available at all.
In addition to patent protection, we also attempt to protect our proprietary products, processes and other information by relying on trade secrets and non-disclosure agreements with our employees, consultants and certain other persons who have access to such products, processes and information. Under these agreements, all inventions conceived by employees are our exclusive property. Nevertheless, there can be no assurance that these agreements will afford significant protection against misappropriation or unauthorized disclosure of our trade secrets and confidential information.
We cannot be certain that any of the current pending patent applications we have licensed, or any new patent applications we may file or license, will ever be issued in the United States or any other country. Even if issued, there can be no assurance that those patents will be sufficiently broad to prevent others from using our products or processes. Furthermore, our patents, as well as those we have licensed or may license in the future, may be held invalid or unenforceable by a court, or third parties could obtain patents that we would need to either license or to design around, which we may be unable to do. Current and future competitors may have licensed or filed patent applications or received patents, and may acquire additional patents or proprietary rights relating to products or processes competitive to ours. In addition, any claims relating to the infringement of third-party proprietary rights, or earlier date of invention, even if not meritorious, could result in costly litigation, lengthy governmental proceedings, divert management’s attention and resources and require us to enter royalty or license agreements which are not advantageous to us, if available at all.
Research and Development
Our expenditures for research and development activities were $1,812,969, $2,914,878, and $3,043,522 during the years ended December 31, 2014, 2013 and 2012, respectively. As our vaccines continue to go through the process to obtain regulatory approval, we expect our research and development costs to continue to increase as human clinical trials proceed. We have not yet formulated any plans for marketing and sales of any vaccine candidate we may successfully develop. Compliance with environmental protection laws and regulations has not had a material effect on our capital expenditures, earnings or competitive position to date.
Properties and Employees
We lease approximately 8,400 square feet of office and laboratory space located at 1900 Lake Park Drive, Suite 380, Smyrna, Georgia under a lease agreement which began November 1, 2009, with an original expiration date of December 31, 2014. We have renewed the lease for an additional 12 months, with two successive 12-month renewal options. We believe this space is adequate for our current needs. As of March 15, 2015, we had five full-time and two part-time employees. None of our employees are covered by collective bargaining agreements and we believe that our employee relations are good.
Corporate Background
Our primary business is conducted by our subsidiary, GeoVax, Inc., which was incorporated under the laws of Georgia in June 2001. The predecessor of our parent company, GeoVax Labs, Inc. (the reporting entity) was originally incorporated in June 1988 under the laws of Illinois as Dauphin Technology, Inc. (“Dauphin”). In September 2006, Dauphin completed a merger with GeoVax, Inc. As a result of the merger, GeoVax, Inc. became a wholly-owned subsidiary of Dauphin, and Dauphin changed its name to GeoVax Labs, Inc. In June 2008, the Company was reincorporated under the laws of Delaware. We currently do not conduct any business other than GeoVax, Inc.’s business of developing new products for the treatment or prevention of human diseases. Our principal offices are located in Smyrna, Georgia (metropolitan Atlanta).
AVAILABLE INFORMATION
Our website address is www.geovax.com. We make available on this website under “Investors – SEC Reports,” free of charge, our proxy statements, annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports as soon as reasonably practicable after we electronically file or furnish such materials to the SEC. We also make available our Code of Ethics on this website under the heading “Investors – Corporate Governance”. Information contained on our website is not incorporated into this prospectus.
SELECTED FINANCIAL DATA
The following selected financial data are derived from our audited consolidated financial statements. The historical results presented below are not necessarily indicative of the results to be expected for any future period. The information set forth below should be read in conjunction with the information contained in “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, and our consolidated financial statements and the related notes, beginning on page F-1 of this prospectus.
|
Years Ended December 31, |
||||||||||||||||||||
|
2014 |
2013 |
2012 |
2011 |
2010 |
||||||||||||||||
|
Statement of Operations Data: |
||||||||||||||||||||
|
Total revenues (grant income) |
$ | 882,956 | $ | 2,417,550 | $ | 2,657,327 | $ | 4,899,885 | $ | 5,185,257 | ||||||||||
|
Net loss |
(2,733,555 | ) | (2,284,943 | ) | (2,135,140 | ) | (2,346,826 | ) | (2,474,328 | ) | ||||||||||
|
Basic and diluted net loss per common share |
(0.10 | ) | (0.11 | ) | (0.12 | ) | (0.15 | ) | (0.18 | ) | ||||||||||
|
As of December 31, |
||||||||||||||||||||
|
2014 |
2013 |
2012 |
2011 |
2010 |
||||||||||||||||
|
Balance Sheet Data: |
||||||||||||||||||||
|
Total assets |
1,333,198 | 2,839,576 | 1,477,970 | 1,645,142 | 2,357,834 | |||||||||||||||
|
Total stockholders’ equity |
1,146,175 | 2,527,227 | 1,150,935 | 703,607 | 1,836,226 | |||||||||||||||
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read together with “Selected Financial Data” and our consolidated financial statements and the related notes, beginning on page F-1 of this prospectus. This discussion contains forward-looking statements that involve risks and uncertainties because they are based on current expectations and relate to future events and our future financial performance. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many important factors, including those set forth under “Risk Factors” and elsewhere in this prospectus.
Overview
GeoVax is a clinical-stage biotechnology company developing innovative human vaccines using our novel DNA/MVA platform technology. Our lead development programs are focused on Ebola, Marburg, and HIV. Our HIV vaccine technology was developed in collaboration with researchers at Emory University, the NIH, and the CDC, and is exclusively licensed to us from Emory University. We also have nonexclusive licenses to certain patents owned by the NIH. Our Ebola/Marburg vaccines have been developed with technology licensed to us from the NIH.
Our most advanced HIV vaccines under development address the clade B subtype of the HIV virus that is most prevalent in North America and Western Europe. Our preventive clade B HIV vaccine has successfully completed Phase 2a clinical trials and we are currently planning the next stage of human clinical testing. We also are investigating our HIV vaccines for their potential to contribute to combination therapies for therapeutic treatment leading to a cure for HIV infections. We have begun earlier preclinical studies to develop HIV vaccine candidates for the clade C subtype of HIV prevalent in the developing world. Our Ebola and Marburg vaccine development efforts have recently been initiated and we expect to begin preclinical animal studies during 2015, with the goal of beginning human clinical testing in 2016.
We have neither received regulatory approval for any of our vaccine candidates, nor do we have any commercialization capabilities; therefore, it is possible that we may never successfully derive significant product revenues from any of our existing or future development programs or product candidates.
We expect for the foreseeable future our operations will result in a net loss on a quarterly and annual basis. As of December 31, 2014, we had an accumulated deficit of $29.8 million.
Critical Accounting Policies and Estimates
This discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires management to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and related disclosure of contingent assets and liabilities. On an ongoing basis, management evaluates its estimates and adjusts the estimates as necessary. We base our estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ materially from these estimates under different assumptions or conditions.
Our significant accounting policies are summarized in Note 2 to our consolidated financial statements for the year ended December 31, 2014. We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our consolidated financial statements:
Revenue Recognition
We recognize revenue in accordance with the SEC’s Staff Accounting Bulletin No. 101, Revenue Recognition in Financial Statements, as amended by Staff Accounting Bulletin No. 104, Revenue Recognition, (“SAB 104”). SAB 104 provides guidance in applying U.S. generally accepted accounting principles (“GAAP”) to revenue recognition issues, and specifically addresses revenue recognition for upfront, nonrefundable fees received in connection with research collaboration agreements. During 2014, 2013 and 2012, our revenue consisted of grant funding received from the NIH. Revenue from these arrangements is approximately equal to the costs incurred and is recorded as income as the related costs are incurred.
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”), which creates a new Topic, Accounting Standards Codification Topic 606. The standard is principle-based and provides a five-step model to determine when and how revenue is recognized. The core principle is that an entity should recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 is effective for the Company beginning in 2017 and allows for either full retrospective adoption or modified retrospective adoption. We are currently evaluating the impact of the adoption of ASU 2014-09 on our financial statements.
Stock-Based Compensation
We account for stock-based transactions in which the Company receives services from employees, directors or others in exchange for equity instruments based on the fair value of the award at the grant date. Compensation cost for awards of common stock is estimated based on the price of the underlying common stock on the date of issuance. Compensation cost for stock options or warrants is estimated at the grant date based on each instrument’s fair value as calculated by the Black-Scholes option pricing model. We recognize stock-based compensation cost as expense ratably on a straight-line basis over the requisite service period for the award.
Liquidity and Capital Resources
At December 31, 2014, we had cash and cash equivalents of $1,101,651 and total assets of $1,333,198, as compared to $2,513,861 and $2,839,576, respectively, at December 31, 2013. Working capital totaled $1,038,472 at December 31, 2014, compared to $2,385,990 at December 31, 2013.
Sources and Uses of Cash
We have funded our activities to date primarily from government grants and clinical trial assistance, and from sales of our equity securities. Due to our significant research and development expenditures, we have not been profitable and have generated operating losses since our inception in 2001. We will continue to require substantial funds to continue these activities. Our primary sources of cash are from sales of our equity securities and from government grant funding. We believe that our existing cash resources, combined with the proceeds from the NIH grants discussed below will be sufficient to fund our planned operations through the first quarter of 2016. We will require additional funds to continue our planned operations beyond that date. We are currently seeking sources of non-dilutive capital through government grant programs and clinical trial support, and we may also conduct additional offerings of our equity securities. However, additional funding may not be available on favorable terms or at all and if we fail to obtain additional capital when needed, we may be required to delay, scale back, or eliminate some or all of our research and development programs as well as reduce our general and administrative expenses.
Cash Flows from Operating Activities
Net cash used in operating activities was $2,250,107, $1,694,592, and $2,441,247 for the years ended December 31, 2014, 2013 and 2012, respectively. Generally, the differences between periods are due to fluctuations in our net losses, offset by non-cash charges such as depreciation and stock-based compensation expense, and by net changes in our assets and liabilities. Our net losses generally fluctuate based on expenditures for our research activities, offset by government grant revenues.
The NIH has funded the costs of conducting all of our human clinical trials (Phase 1 and Phase 2a) to date for our preventive HIV vaccines, with GeoVax incurring costs associated with manufacturing the clinical vaccine supplies and other study support. We are actively engaged in discussions with the HVTN and NIAID regarding the design of our next clinical study and various trial designs are being considered. Our vaccine is currently the only vaccine being contemplated for efficacy trials for prevention of clade B HIV infection. However, the HVTN believes the best path forward will be to test our vaccines in combination with a protein boost. Protein boosts may augment antibody responses that can block virus infections (neutralizing antibody) and cause antibody dependent cellular cytotoxicity (ADCC antibody). Protein added to HIV vaccines have shown some success in other trials. The HVTN believes this “dual-action” approach will be a prudent and cost-effective path forward for supporting large clinical trials. Our current expectation is that the next clinical trial will begin in late 2015 and will be a follow-on study to the HVTN 205 trial, in which those trial participants are given a protein boost to evaluate their immune responses. Information from this trial would then inform the design of future, larger clinical trials.
The HVTN and NIH are continuing to consider future efficacy studies, and members are working to develop collaborative clinical development plans, as well as initiating regulatory planning. The plans for large-scale clinical trials may change as researchers continue to gather information from our earlier studies and are influenced by results from other vaccine trials. Trial start dates are dependent on many factors and are likely to change.
During 2014, we completed a Phase 1 clinical trial (GV-TH-01) investigating the therapeutic use of our GOVX-B11 vaccine in HIV-infected patients. We received no federal assistance in conducting this study.
Observations from a pilot Phase 1 clinical trial of our HIV vaccines (GV-TH-01 – discussed below) have led us to postulate that our DNA vaccines may be effective as a shock agent and that a subsequent, precisely timed MVA inoculation may reduce viral reservoirs. The Company is currently considering the best course of action for advancing its HIV immunotherapy program. Future therapeutic studies of GeoVax’s vaccine may investigate vaccine’s ability to act as a “shock agent” in a shock and kill therapy in combination with standard of care antiretroviral drug therapy to seek a cure. The timetable and specific clinical plans will be dependent upon the Company’s ability to secure external funding for the program, and on the nature of any potential collaborations GeoVax may establish.
In addition to clinical trial support from the NIH for our preventive HIV vaccines, our operations have been partially funded by NIH research grants. We record the funding we receive pursuant to these grants as revenue at the time the related expenditures are incurred. As of December 31, 2014, there was an aggregate of approximately $229,000 of unused grant funds available for use during 2015. We intend to pursue additional grants from the federal government but cannot be assured of success. As we progress to the later stages of our vaccine development activities, government financial support may be more difficult to obtain, or may not be available at all. Therefore, it will be necessary for us to look to other sources of funding in order to finance our clinical trials and other vaccine development activities.
Cash Flows from Investing Activities
Our investing activities have consisted predominantly of capital expenditures. Capital expenditures for the years ended December 31, 2014, 2013 and 2012, were $35,503, $86,603, and $-0-, respectively.
Cash Flows from Financing Activities
Net cash provided by financing activities was $873,400, $3,259,131, and $2,309,192 for the years ended December 31, 2014, 2013 and 2012, respectively.
The cash generated by our financing activities during 2011 relates to the sale of our common stock to individual accredited investors in a private placement offering initiated during December 2011. During January 2012, we received an additional $310,160 from stock sales pursuant to this offering (including $36,800 received in payment of a stock subscription receivable from December 2011).
In March 2012, we sold 2,200 shares of our Series A Convertible Preferred Stock, as well as accompanying warrants to purchase 8,799,999 shares of common stock, to a group of institutional investors for an aggregate purchase price of $2.2 million. Net proceeds to the Company, after deduction of placement agent fees and other expenses, were approximately $2.0 million. The cash generated by our financing activities during 2012 also includes $310,160 received in January 2012 related to the sale of our common stock to individual accredited investors in a private placement offering which was initiated during December 2011.
In January 2013, we reduced the exercise price of 2,933,333 of certain stock purchase warrants from $0.75 to $0.60 per share. In consideration for the reduction of the exercise price, the holders of the warrants immediately exercised 1,766,667 of the warrants for cash, resulting in total proceeds to the Company of $1,060,000. We also extended the expiration date of the 1,166,666 unexercised warrants from March 21, 2013 to May 21, 2013. In May 2013, we reduced the exercise price of the 1,166,666 remaining warrants from $0.60 to $0.50 per share. In consideration for the reduction of the exercise price, the holders of the warrants immediately exercised all of the remaining warrants for cash, resulting in total proceeds to the Company of $583,333.
In December 2013, we sold 1,650 shares of our Series B Convertible Preferred Stock to a group of institutional investors for an aggregate purchase price of $1.65 million. Net proceeds to the Company, after deduction of transaction expenses, were approximately $1.6 million. No warrants were issued in connection with the transaction.
In October 2014, we entered into an agreement with certain warrant holders to purchase shares of our common stock with respect to the payment to them of a warrant exercise fee of $0.075 per share for each share purchased upon exercise of warrants held by them. In exchange for the fee, they immediately exercised warrants for an aggregate of 3,176,000 shares of our common stock, resulting in proceeds to us of $873,400 (net of the exercise fee).
Our capital requirements, particularly as they relate to our research and development activities, have been and will continue to be significant. We anticipate incurring additional losses for several years as we expand our clinical programs and proceed into higher cost human clinical trials. Conducting clinical trials for our vaccine candidates in development is a lengthy, time-consuming and expensive process. We will not generate revenues from the sale of our technology or products for at least several years, if at all. For the foreseeable future, we will be dependent on obtaining financing from third parties in order to maintain our operations, including our clinical program. Such capital may not be available on terms acceptable to the Company or at all. If we fail to obtain additional funding when needed, we would be forced to scale back or terminate our operations, or to seek to merge with or to be acquired by another company.
In February 2015, we sold shares of Series C convertible preferred stock to certain institutional investors for an aggregate purchase price of $3.0 million, and five-year Series D warrants to purchase an aggregate of 16,666,666 shares of our common stock at $0.22 per share. Net proceeds to the Company, after deduction of placement agent fees and other expenses, were approximately $2.7 million. The preferred stock is convertible at any time into shares of our common stock at $0.18 per share, subject to adjustment as provided in the certificate of designation. We also granted to the investors an additional purchase right, evidenced in the form of one-year Series E warrants to purchase up to 16,666,666 of our common stock with an exercise price of $0.18 per share, and five-year Series F warrants to purchase up to 16,666,666 shares of our common stock at $0.22 per share. The Series E warrants are immediately exercisable. The Series F warrants only become exercisable at the time, and to the extent, that the Series E warrants are exercised.
We expect that our current working capital (including the net proceeds from the February 2015 financing event discussed above) combined with the remaining available funds from the NIH grants will be sufficient to support our planned level of operations through the first quarter of 2016. We will require additional funds to continue our planned operations beyond that date. We are currently seeking sources of non-dilutive capital through government grant programs and clinical trial support, and we may also conduct additional offerings of our equity securities, although there can be no assurance that we will be able to do so. While we believe that we will be successful in obtaining the necessary financing to fund our operations through government grants and clinical trial support, exercise of stock purchase warrants, or other sources, there can be no assurances that such additional funding will be available to us on reasonable terms or at all. Should the financing we require to sustain our working capital needs be unavailable or prohibitively expensive when we require it, the consequences could have a material adverse effect on our business, operating results, financial condition and prospects.
We have no off-balance sheet arrangements that are likely or reasonably likely to have a material effect on our financial condition or results of operations.
Contractual Obligations
Contractual obligations represent future cash commitments and liabilities under agreements with third parties, and exclude contingent liabilities for which we cannot reasonably predict future payment. Additionally, the expected timing of payment of the obligations presented below is estimated based on current information. Timing of payments and actual amounts paid may be different depending on the timing of receipt of goods or services or changes to agreed-upon terms or amounts for some obligations.
The following table represents our contractual obligations as of December 31, 2014, aggregated by type (in thousands):
|
Payments Due by Period |
||||||||||||||||||||
|
Contractual Obligations |
Total |
Less than 1 Year |
1-3 Years |
4-5 Years |
More than 5 years |
|||||||||||||||
|
Operating Lease Obligations (1) |
$ | 146 | $ | 146 | $ | -- | $ | -- | $ | -- | ||||||||||
|
Firm Purchase Commitments (2) |
151 | 151 | -- | -- | -- | |||||||||||||||
|
Emory University – License Agreement (3) |
-- | -- | -- | -- | -- | |||||||||||||||
|
Total |
$ | 297 | $ | 297 | $ | -- | $ | -- | $ | -- | ||||||||||
|
(1) |
Our operating lease obligations relate to the facility lease for our 8,430 square foot facility in Smyrna, Georgia, which houses our laboratory operations and our administrative offices. The lease (as amended), expires on December 31, 2015, with two successive 12-month renewal options. |
|
(2) |
Firm purchase commitments relate to contracts for research activities related to NIH grants. |
|
(3) |
Pursuant to the Emory License, we have committed to make potential future milestone and royalty payments which are contingent upon the occurrence of future events. Such events include development milestones, regulatory approvals and product sales. Because the achievement of these milestones is currently neither probable nor reasonably estimable, the contingent payments have not been included in the table above or recorded on our Consolidated Balance Sheets. The aggregate total of all potential milestone payments included in the Emory License (excluding royalties on net sales) is approximately $3.5 million. |
As of December 31, 2014, except as disclosed in the table above, we had no other material firm purchase obligations or commitments for capital expenditures and no committed lines of credit or other committed funding or long-term debt. We have employment agreements with our executive officers and a consulting agreement with a member of our Board of Directors, each of which may be terminated with no more than 90 days advance written notice.
Net Operating Loss Carryforwards
At December 31, 2014, we had consolidated net operating loss carryforwards for income tax purposes of $64.6 million, which will expire in 2019 through 2034 if not utilized. Approximately $42.6 million of our net operating loss carryforwards relate to the operations of our predecessor, Dauphin Technology, Inc. prior to the 2006 merger between Dauphin Technology, Inc. and GeoVax, Inc. We also have research and development tax credits of approximately $826,000 available to reduce income taxes, if any, which will expire in 2022 through 2034 if not utilized. The amount of net operating loss carryforwards and research tax credits available to reduce income taxes in any particular year may be limited in certain circumstances. Based on an assessment of all available evidence including, but not limited to, our limited operating history in our core business and lack of profitability, uncertainties of the commercial viability of our technology, the impact of government regulation and healthcare reform initiatives, and other risks normally associated with biotechnology companies, we have concluded that it is more likely than not that these net operating loss carryforwards and credits will not be realized and, as a result, a 100% deferred tax valuation allowance has been recorded against these assets.
Results of Operations
Net Loss
We recorded net losses of $2,733,555, $2,284,943, and $2,135,140 for the years ended December 31, 2014, 2013 and 2012, respectively. Our operating results typically fluctuate due to the timing of activities and related costs associated with our vaccine research and development activities and our general and administrative costs, as described in more detail below.
Grant Revenue
We recorded grant revenues of $882,956, $2,417,550, and $2,657,327 for the years ended December 31, 2014, 2013 and 2012, respectively. Grant revenues relate to grants from the NIH in support of our HIV vaccine development activities. We record revenue associated with these grants as the related costs and expenses are incurred. The difference in our grant revenues from period to period is directly related to our expenditures for activities supported by the grants, and can fluctuate significantly based on the timing of the related expenditures. There is an aggregate of approximately $229,000 in approved grant funds remaining and available for use as of December 31, 2014, which we anticipate recognizing as revenue during 2015. Additional detail concerning our grant revenues is discussed below.
In September 2007, the NIH awarded us a grant entitled “GM-CSF-Adjuvanted Clade C DNA/MVA and MVA/MVA Vaccines”. The aggregate award (including subsequent amendments) totaled approximately $20.4 million. We recorded grant revenues of $624,689, $833,390, and $2,227,924 for the years ended December 31, 2014, 2013 and 2012, respectively, related to this grant, and there is $75,464 of unrecognized grant funds remaining and available for use pursuant to this grant as of December 31, 2014.
In September 2012, the NIH awarded us a grant entitled “Immunogens and Manufacturing” to support our HIV/AIDS vaccine development program. The grant award was for approximately $1.9 million. We recorded grant revenues of $-0-, $1,429,597, and $429,403 for the years ended December 31, 2014, 2013 and 2012, respectively, related to this grant, and all funding pursuant to this grant has been utilized as of December 31, 2014.
In July 2013, the NIH awarded us a Small Business Innovative Research (SBIR) grant entitled “Enhancing Protective Antibody Responses for a GM-CSF Adjuvanted HIV Vaccine.” The initial grant award was approximately $277,000 for the first year of a two year project period beginning August 1, 2013. In July 2014, the NIH awarded us approximately $290,000 for the second year of the project period. We recorded grant revenues of $258,267, $154,563, and $-0- for the years ended December 31, 2014, 2013 and 2012, respectively, related to this grant, and there is $153,501 of unrecognized grant funds remaining and available for use pursuant to this grant as of December 31, 2014.
Research and Development
Our research and development expenses were $1,812,969, $2,914,878, and $3,043,522 for the years ended December 31, 2014, 2013 and 2012, respectively. Research and development expense for these periods includes stock-based compensation expense of $32,134, $41,539, and $78,140 for 2014, 2013 and 2012, respectively (see discussion under “Stock-Based Compensation Expense” below). Since our inception, all of our research and development efforts have been focused on development of human vaccines – initially with a focus on HIV/AIDS vaccines, and with a recent expansion to vaccines for Ebola and Marburg. Our research activities conducted pursuant to our NIH grants are also focused solely on the development of human vaccines.
Our research and development expenses can fluctuate considerably on a period-to-period basis, depending on our need for vaccine manufacturing by third parties, the timing of expenditures related to our grants from the NIH, the timing of costs associated with clinical trials being funding directly by us, and other factors. The overall decrease in research and development expense from 2013 to 2014 can mostly be attributed to lower expenditures related to the activities supported by our grants from the NIH, and lower expenditures associated with a Phase 1 trial of our therapeutic HIV vaccine, which was completed during the first quarter of 2014. We have not received any government support for clinical trials of our therapeutic vaccine. Our research and development costs do not include costs incurred by the HVTN in conducting clinical trials of our preventive HIV vaccines; those costs are funded directly to the HVTN by the NIH.
We cannot predict the level of support we may receive from the HVTN, NIH, or other federal agencies (or divisions thereof) for our future clinical trials. We expect that our research and development costs will increase in the future as we progress into the later stage human clinical trials for our HIV vaccines and as we expand our Ebola and Marburg vaccine development program.
Our vaccine candidates still require significant, time-consuming and costly research and development, testing and regulatory clearances. Completion of clinical development will take several years or more, but the length of time generally varies substantially according to the type, complexity, novelty and intended use of a product candidate. The NIH has funded the costs of conducting all of our completed and ongoing human clinical trials to date for our preventive HIV vaccine, with GeoVax incurring costs associated with manufacturing the clinical vaccine supplies and other study support. We are having discussions with the HVTN and NIH with regard to the conduct of an additional trial of our preventive vaccine, and we expect the NIH will provide support for this trial as well. We intend to seek government and/or third party support for future clinical human trials, but there can be no assurance that we will be successful.
The duration and the cost of future clinical trials may vary significantly over the life of the project as a result of differences arising during development of the human clinical trial protocols, including, among others:
|
● |
the number of patients that ultimately participate in the clinical trial; |
|
● |
the duration of patient follow-up that seems appropriate in view of the results; |
|
● |
the number of clinical sites included in the clinical trials; and |
|
● |
the length of time required to enroll suitable patient subjects. |
Due to the uncertainty regarding the timing and regulatory approval of clinical trials and pre-clinical studies, our future expenditures are likely to be highly volatile in future periods depending on the outcomes of the trials and studies. From time to time, we will make determinations as to how much funding to direct to these programs in response to their scientific, clinical and regulatory success, anticipated market opportunity and the availability of capital to fund our programs.
In developing our product candidates, we are subject to a number of risks that are inherent in the development of products based on innovative technologies. For example, it is possible that our vaccines may be ineffective or toxic, or will otherwise fail to receive the necessary regulatory clearances, causing us to delay, extend or terminate our product development efforts. Any failure by us to obtain, or any delay in obtaining, regulatory approvals could cause our research and development expenditures to increase which, in turn, could have a material adverse effect on our results of operations and cash flows. Because of the uncertainties of clinical trials, estimating the completion dates or cost to complete our research and development programs is highly speculative and subjective. As a result of these factors, we are unable to accurately estimate the nature, timing and future costs necessary to complete the development of our product candidates. In addition, we are unable to reasonably estimate the period when material net cash inflows could commence from the sale, licensing or commercialization of such product candidates, if ever.
General and Administrative Expense
Our general and administrative expenses were $1,807,605, $1,792,160, and $1,752,765 for the years ended December 31, 2014, 2013 and 2012, respectively. General and administrative costs include officers’ salaries, legal and accounting costs, patent costs, amortization expense associated with intangible assets, and other general corporate expenses. General and administrative expense includes stock-based compensation expense of $446,969, $360,565, and $231,936 for 2014, 2013 and 2012, respectively (see discussion under “Stock-Based Compensation Expense” below). We expect that our general and administrative costs may increase in the future in support of expanded research and development activities and other general corporate activities.
Stock-Based Compensation Expense
We recorded total stock-based compensation expense of $479,103, $402,104, and $310,076 during the years ended December 31, 2014, 2013 and 2012, respectively, which was allocated to research and development expense or general and administrative expense according to the classification of cash compensation paid to the employee, consultant or director to whom the stock compensation was granted. In addition to amounts related to the issuance of stock options to employees, the figures include amounts related to common stock and stock purchase warrants issued to consultants and non-employee directors. For the three years ended December 31, 2014, stock-based compensation expense was allocated as follows:
|
2014 |
2013 |
2012 |
||||||||||
|
General and administrative expense |
$ | 446,969 | $ | 360,565 | $ | 231,936 | ||||||
|
Research and development expense |
32,134 | 41,539 | 78,140 | |||||||||
|
Total stock option expense |
$ | 479,103 | $ | 402,104 | $ | 310,076 | ||||||
Other Income
Interest income was $ 4,063, $4,545, and $3,820 for the years ended December 31, 2014, 2013 and 2012, respectively. The variances between years are primarily attributable to the cash available for investment and to interest rate fluctuations.
Impact of Inflation
For the three-year period ended December 31, 2014, we do not believe that inflation and changing prices had a material impact on our operations or on our financial results.
Off-Balance Sheet Arrangements
We have not entered into off-balance sheet financing arrangements, other than operating leases.
SECURITY OWNERSHIP OF
PRINCIPAL STOCKHOLDERS, DIRECTORS AND OFFICERS
Based solely upon information made available to us, the following table sets forth information with respect to the beneficial ownership of our common stock as of March 9, 2015 by (1) each director; (2) each of our Named Executive Officers; (3) all executive officers and directors as a group; and (4) each additional person who is known by us to beneficially own more than 5% of our common stock. Except as otherwise indicated, the holders listed below have sole voting and investment power with respect to all shares of common stock beneficially owned by them.
|
Amount and Nature |
||||||||
|
of Beneficial |
Percent |
|||||||
|
Name of Beneficial Owner (1) |
Ownership |
of Class (2) |
||||||
|
Directors and Executive Officers: |
||||||||
|
Randal Chase |
- | - | ||||||
|
David A. Dodd (3) |
382,124 | 1.2 |
% | |||||
|
Dean G. Kollintzas (4) |
153,712 | * | ||||||
|
Robert T. McNally (5) |
261,380 | * | ||||||
|
Mark W. Reynolds (6) |
208,666 | * | ||||||
|
Harriet L. Robinson (7) |
1,479,273 | 4.6 |
% | |||||
|
John N. Spencer, Jr. (8) |
167,412 | * | ||||||
|
All executive officers and directors as a group (6 persons) (9) |
2,652,567 | 8.0 |
% | |||||
|
Other 5% Stockholders: |
||||||||
|
Emory University (10) |
4,621,405 | 14.5 |
% | |||||
|
Sabby Healthcare Master Fund, Ltd (11) |
3,428,400 | 9.99 |
% | |||||
|
Sabby Volatility Warrant Master Fund, Ltd (12) |
3,431,500 | 9.99 |
% | |||||
|
Welch & Forbes LLC (13) |
1,703,464 | 5.3 |
% | |||||
|
___________ |
||||||||
| * Less than 1% | |
|
(1) |
Except as otherwise indicated, the business address of each director and executive officer listed is c/o GeoVax Labs, Inc., 1900 Lake Park Drive, Suite 380, Smyrna, Georgia 30080. |
|
(2) |
This table is based upon information supplied by officers and directors, and with respect to principal stockholders, Schedules 13D and 13G filed with the SEC. Beneficial ownership is determined in accordance with the rules of the SEC. Applicable percentage ownership is based on 31,950,813 shares of common stock outstanding as of March 9, 2015. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, shares of common stock subject to options or warrants currently exercisable, or exercisable within 60 days of March 9, 2015, as well as shares of preferred stock which may be converted at any time at the option of the holder, are deemed outstanding. |
|
(3) |
Includes options and warrants to purchase 254,399 shares of common stock exercisable within 60 days of March 9, 2015. |
|
(4) |
Includes options and warrants to purchase 138,787 shares of common stock exercisable within 60 days of March 9, 2015. |
|
(5) |
Includes options and warrants to purchase 219,175 shares of common stock exercisable within 60 days of March 9, 2015. |
|
(6) |
Includes options and warrants to purchase 172,266 shares of common stock exercisable within 60 days of March 9, 2015. |
|
(7) |
Dr. Robinson shares voting and investment power over 1,024,472 shares with Welch & Forbes LLC, whose ownership is described below. Includes options and warrants to purchase 305,547 shares of common stock exercisable within 60 days of March 9, 2015. |
|
(8) |
Includes options and warrants to purchase 138,787 shares of common stock exercisable within 60 days of March 9, 2015. Mr. Spencer shares voting and investment power with his spouse with respect to 28,625 shares and a warrant for 22,388 shares which are owned jointly by them |
|
(9) |
Includes options and warrants to purchase 1,229,361 shares of common stock exercisable within 60 days of March 9, 2015. Unless otherwise noted, none of our Directors or Executive Officers have pledged any of their beneficially-owned shares as security for any obligation. |
|
(10) |
The address for this stockholder is Administration Building, 201 Dowman Drive, Atlanta, Georgia 30322. |
|
(11) |
The address for this stockholder is c/o Ogier Fiduciary Services (Cayman) Limited, 89 Nexus Way, Camana Bay, Grand Cayman KY1-9007, Cayman Islands. Includes 1,062,508 shares of common stock, 8,333,333 shares of common stock issuable upon conversion of Series C Preferred Stock, and warrants to purchase 17,270,332 shares of common stock exercisable within 60 days of March 9, 2015. The Series C Preferred Stock, and the warrants owned by this stockholder contain exercise and conversion limitations providing that a holder thereof may not convert or exercise (as the case may be) to the extent (but only to the extent) that, if after giving effect to such conversion or exercise (as the case may be), the holder or any of its affiliates would beneficially own in excess of either 4.99% (for conversion of the Series C Preferred Stock) or 9.99% for exercise of warrants (the “Maximum Percentage”) of the outstanding shares of common stock immediately after giving effect to such conversion or exercise (as the case may be). To the extent the above limitation applies, the determination of whether a share of preferred stock or warrant shall be exercisable or convertible (vis-à-vis other convertible, exercisable or exchangeable securities owned by the holder) shall, subject to such Maximum Percentage limitation, be determined on the basis of the first submission to the Company for conversion, exercise or exchange (as the case may be). Sabby Management, LLC shares voting and investment power with respect to these shares on behalf of this stockholder. As manager of Sabby Management, LLC, Hal Mintz also shares voting and investment power on behalf of this stockholder. Each of Sabby Management, LLC and Hal Mintz disclaim beneficial ownership over the securities listed except to the extent of their pecuniary interest therein. Except as described above, none of the holders has had, within the past three years, any position, office or other material relationship with the Company or any of our predecessors or affiliates. |
|
(12) |
The address for this stockholder is c/o Ogier Fiduciary Services (Cayman) Limited, 89 Nexus Way, Camana Bay, Grand Cayman KY1-9007, Cayman Islands. Includes 1,032,426 shares of common stock, 8,333,333 shares of common stock issuable upon conversion of Series C Preferred Stock, and warrants to purchase 17,270,332 shares of common stock exercisable within 60 days of March 9, 2015. The Series C Preferred Stock, and the warrants owned by this stockholder contain exercise and conversion limitations providing that a holder thereof may not convert or exercise (as the case may be) to the extent (but only to the extent) that, if after giving effect to such conversion or exercise (as the case may be), the holder or any of its affiliates would beneficially own in excess of 4.99% (for conversion of the Series C Preferred Stock) or 9.99% (for exercise of warrants) (the “Maximum Percentage”) of the outstanding shares of common stock immediately after giving effect to such conversion or exercise (as the case may be). To the extent the above limitation applies, the determination of whether a share of preferred stock or warrant shall be exercisable or convertible (vis-à-vis other convertible, exercisable or exchangeable securities owned by the holder) shall, subject to such Maximum Percentage limitation, be determined on the basis of the first submission to the Company for conversion, exercise or exchange (as the case may be). Sabby Management, LLC shares voting and investment power with respect to these shares on behalf of this stockholder. As manager of Sabby Management, LLC, Hal Mintz also shares voting and investment power on behalf of this stockholder. Each of Sabby Management, LLC and Hal Mintz disclaim beneficial ownership over the securities listed except to the extent of their pecuniary interest therein. Except as described above, none of the holders has had, within the past three years, any position, office or other material relationship with the Company or any of our predecessors or affiliates. |
|
(13) |
The address for this stockholder is 45 School Street, Boston, Massachusetts 02108. Includes 1,024,472 shares held by Dr. Robinson as to which the stockholder shares voting and dispositive power. |
DIRECTORS AND EXECUTIVE OFFICERS
The following table sets forth certain information with respect to our directors and executive officers:
|
Name |
Age |
Current Position |
|
David A. Dodd (1)(2)(3) |
65 |
Chairman of the Board of Directors |
|
Robert T. McNally, Ph.D. |
67 |
President and Chief Executive Officer, Director |
|
Mark W. Reynolds, CPA |
53 |
Chief Financial Officer and Corporate Secretary |
|
Harriet L. Robinson, Ph.D. |
77 |
Chief Scientific Officer, Director |
|
Randal D. Chase, Ph.D. |
65 |
Independent Director |
|
Dean G. Kollintzas (1)(2)(3) |
41 |
Independent Director |
|
John N. Spencer, Jr. (1)(2)(3) |
74 |
Independent Director |
______________________
|
(1) |
Member of the Compensation Committee of the Board of Directors. |
|
(2) |
Member of the Nominating and Governance Committee of the Board of Directors. |
|
(3) |
Member of the Audit Committee of the Board of Directors. |
David A. Dodd. Mr. Dodd joined the Board of Directors in March 2010 and became Chairman of our Board of Directors on January 1, 2011. Since April 2013, he has served as President and Chief Executive Officer, and as a member of the Board of Directors, of Aeterna Zentaris Inc., an oncology and endocrinology drug development company. He is also the Chief Executive Officer of RiversEdge BioVentures, an investment and advisory firm focused on the life sciences and pharmaceuticals industries, which he founded in 2009. He has more than 35 years of executive experience in the healthcare industry. From December 2007 to June 2009, Mr. Dodd was President, Chief Executive officer and Chairman of BioReliance Corporation, an organization that provided biological safety testing, viral clearance testing, genetic and mammalian technology testing and laboratory animal diagnostic services testing. From October 2006 to April 2009, he served as non-executive chairman of Stem Cell Sciences Plc. Before that, Mr. Dodd served as President, Chief Executive Officer and Director of Serologicals Corporation before it was sold to Millipore Corporation in July 2006 for $1.5 billion. For five years prior to his employment by Serologicals Corporation, Mr. Dodd served as President and Chief Executive Officer of Solvay Pharmaceuticals, Inc. and Chairman of its subsidiary Unimed Pharmaceuticals, Inc. The Board of Directors has concluded that Mr. Dodd should serve on the Board of Directors due to his experience in the pharmaceutical industry, as well as his background in general management, business transformation, corporate partnering, and mergers and acquisitions.
Robert T. McNally, Ph.D. Dr. McNally joined the Board of Directors in December 2006 and was appointed as our President and Chief Executive Officer effective April 1, 2008. From 2000 to March 2008, Dr. McNally served as Chief Executive Officer of Cell Dynamics LLC, a cGMP laboratory services company. Previously, Dr. McNally was a co-founder and Senior Vice President of Clinical Research for CryoLife, Inc., a pioneering company in transplantable human tissues. He has over 34 years of experience in academic and corporate clinical investigations, management, research, business, quality and regulatory affairs Dr. McNally is a Fellow of the American Institute for Medical and Biological Engineering, serves on the advisory boards of the Petit Institute for Bioengineering and Dupree College of Management at the Georgia Institute of Technology, and is a former Chairman of Georgia Bio, a trade association. Dr. McNally graduated with a Ph.D. in biomedical engineering from the University of Pennsylvania. The Board of Directors has concluded that Dr. McNally should serve on its Board of Directors by virtue of his prior business and scientific experience, including his experience as Chief Executive Officer of Cell Dynamics, LLC and as Senior Vice President of Clinical Research for CryoLife, Inc., and due to his intimate involvement with the Company’s ongoing operations as its President and Chief Executive Officer.
Mark W. Reynolds, CPA Mr. Reynolds joined the Company on a part-time basis in October 2006 as Chief Financial Officer and Corporate Secretary, becoming a full-time employee in January 2010. From 2003 to 2006, before being named Chief Financial Officer of GeoVax Labs, Inc., Mr. Reynolds provided financial and accounting services to GeoVax, Inc. as an independent contractor. From 2004 to 2008, Mr. Reynolds served as Chief Financial Officer for HealthWatchSystems, Inc. a privately-held company in the consumer healthcare industry. From 2004 to 2006, he served as Chief Financial Officer for Duska Therapeutics, Inc., a publicly-held biotechnology company. From 1988 to 2002, Mr. Reynolds worked for CytRx Corporation, a publicly-held biopharmaceutical company, where he first served as Controller and then as Chief Financial Officer. Mr. Reynolds began his career as an auditor with Arthur Andersen & Co. from 1985 to 1988. He is a certified public accountant and earned a master’s of accountancy degree from the University of Georgia.
Harriet L. Robinson, Ph.D. Dr. Robinson joined the Company as Senior Vice President, Research and Development on a part-time basis in November 2007 and on a full-time basis in February 2008, and was elected to the Board of Directors in June 2008. She is a co-founder of GeoVax, Inc. and has served as chief of its scientific advisory board since formation of the company in 2001. From 1999 to February 2008, Dr. Robinson served as the Asa Griggs Candler Professor of Microbiology and Immunology at Emory University in Atlanta, Georgia, and from 1998 to February 2008 as Chief, Division of Microbiology and Immunology, Yerkes National Primate Center and Professor at the Emory University School of Medicine. She was Professor, Department of Microbiology & Immunology, at the University of Massachusetts Medical Center from 1988 to 1997 and Staff, then Senior, then Principal Scientist at the University of Massachusetts Worcester Foundation for Experimental Biology from 1977 to 1987. Dr. Robinson received a bachelor of arts degree from Swarthmore College and M.S. and Ph.D. degrees from the Massachusetts Institute of Technology. The Board of Directors has concluded that Dr. Robinson should serve on its Board of Directors by virtue of her extensive knowledge of the Company’s technology as its scientific founder.
Randal D. Chase, Ph.D. Dr. Chase joined the Board of Directors in March 2015. Since 2011, Dr. Chase has served as a business advisor and consultant to companies in the life science sector. From 2006 to 2011, he served as President and Chief Executive Officer of Immunovaccine, Inc., a clinical-stage biotechnology company developing vaccines against cancer and infectious diseases. Dr. Chase is also a former president of Shire Biologics, North American Vaccine, Pasteur Merieux Connaught, and Quadra Logic Technologies, Inc. His early career was at Bristol Myers and Glaxo Pharmaceuticals. Dr. Chase has also served as a member of the board of directors for numerous companies, and recently served as Chairman of the Board for Medicago, Inc. until its sale to Mitsubishi Tanabe Pharma Corporation in 2013. He currently serves as Chairman of the Board for Medimabs, Inc., a privately-held antibody company. Dr. Chase attended the Senior Executive Program of the London Business School in the United Kingdom, holds a bachelor of sciences degree in biochemistry from Bishop’s University and a Ph.D. in biochemistry from the University of British Columbia. Dr. Chase completed a post doctoral fellowship at the McArdle Cancer Institute of the University of Wisconsin. The Board of Directors has concluded that Dr. Chase should serve on the Board of Directors due to his extensive leadership experience in the pharmaceutical industry, and the vaccine industry in particular.
Dean G. Kollintzas. Mr. Kollintzas joined the Board of Directors upon consummation of the merger with GeoVax, Inc. in September 2006. Since 2001 Mr. Kollintzas has been an intellectual property attorney specializing in biotechnology and pharmaceutical licensing, FDA regulation, and corporate/international transactions. Mr. Kollintzas received a microbiology degree from the University of Illinois and a J.D. from Franklin Pierce Law Center. He is a member of the Wisconsin and American Bar Associations. Since 2004, Mr. Kollintzas has been in private practice. In 2014, he founded Procare Clinical, LLC, a clinical trial management company headquartered in Naperville, IL. The Board of Directors has concluded that Mr. Kollintzas should serve on the Board of Directors by virtue of his experience with intellectual property matters, biotechnology and pharmaceutical licensing, and FDA regulation.
John N. (Jack) Spencer, Jr., CPA Mr. Spencer joined the Board of Directors upon consummation of the merger with GeoVax, Inc. in September 2006. Mr. Spencer is a certified public accountant and was a partner of Ernst & Young LLP where he spent more than 38 years until he retired in 2000. Mr. Spencer also serves as a director of MRI Interventions, Inc. (Nasdaq: MRIC), a medical device company, where he also chairs the audit committee and serves on the compensation committee. He served as the Temporary Chief Financial Officer of Applied Genetic Technologies Corporation from November 2013 until February 2014 while that company prepared its initial public offering. He also serves as a consultant to various companies primarily relating to financial accounting and reporting matters. Mr. Spencer received a bachelor of science degree from Syracuse University, and he earned an M.B.A. degree from Babson College. He also attended the Harvard Business School Advanced Management Program. The Board of Directors has concluded that Mr. Spencer should serve on the Board of Directors by virtue of his experience at Ernst & Young LLP where he was the partner in charge of that firm’s life sciences practice for the southeastern United States, and his clients included a large number of publicly-owned and privately-held medical technology companies, together with his continuing expertise as a director of, and a consultant to, other publicly owned and privately held companies.
EXECUTIVE COMPENSATION
The tables and disclosures that follow set forth the compensation and certain other information with respect to our “Named Executive Officers”. The Named Executive Officers for 2014 include our chief executive officer and the two other most highly compensated individuals who were serving as executive officers as of December 31, 2014. Our Named Executive Officers for 2014 were:
● Robert T. McNally, Ph.D., President and Chief Executive Officer
● Mark W. Reynolds, Chief Financial Officer
● Harriet L. Robinson, Ph.D., Chief Scientific Officer
Employment Agreements
Robert T. McNally. On March 20, 2008, GeoVax entered into an employment agreement with Robert T. McNally, Ph.D. to become our President and Chief Executive Officer effective April 1, 2008. The employment agreement has no specified term. The employment agreement provided for an initial annual salary of $200,000 to Dr. McNally, subject to periodic increases as determined by the Compensation Committee. The Board of Directors may also approve the payment of a discretionary bonus annually. Dr. McNally is eligible for grants of awards from our 2006 Equity Incentive Plan (the “Plan”) and is entitled to participate in any and all benefits in effect from time-to-time for employees generally. We may terminate the employment agreement, with or without cause. If we terminate the employment agreement without cause, we will be required to provide Dr. McNally at least 30 days prior notice of the termination and one week of severance pay for each full year of service as President and Chief Executive Officer ($19,038 as of December 31, 2014, paid as salary continuance). Dr. McNally may terminate the employment agreement at any time by giving us 60 days notice. In that event, he would not receive severance. In October 2013, our Board of Directors approved an amendment to the employment agreement with Dr. McNally. The 2013 amendment includes severance provisions in the event of a change in control (as defined in the amendment) and a qualifying termination of employment. See the discussion under “Potential Payments Upon Change-in-Control” below. In February 2014, Dr. McNally reduced his time commitment to the company from 100% to 60%, and his base salary was adjusted proportionately from $275,000 to $165,000.
Mark W. Reynolds. On January 1, 2010, GeoVax entered into an amended and restated employment agreement with Mark W. Reynolds, our Chief Financial Officer. The employment agreement has no specified term. The employment agreement provides for an initial annual salary of $212,600 to Mr. Reynolds. The Board of Directors may also approve the payment of a discretionary bonus annually. Mr. Reynolds is eligible for grants of awards from our Plan and is entitled to participate in any and all benefits in effect from time-to-time for employees generally. We may terminate the employment agreement, with or without cause. If we terminate the employment agreement without cause, we will be required to provide Mr. Reynolds at least 30 days prior notice of the termination and one week of severance pay for each full year of service as Chief Financial Officer ($32,708 as of December 31, 2014, paid as salary continuance). Mr. Reynolds may terminate the employment agreement at any time by giving us 60 days notice. In that event, he would not receive severance. In October 2013, our Board of Directors approved an amendment to the employment agreement with Mr. Reynolds. The 2013 amendment includes severance provisions in the event of a change in control (as defined in the amendment) and a qualifying termination of employment. See the discussion under “Potential Payments Upon Change-in-Control” below. In December 2014, the Compensation Committee awarded Mr. Reynolds a bonus of $2,000 and approved an increase to his base salary from $212,600 to $223,230, effective January 1, 2015.
Harriet L. Robinson. On November 19, 2007, GeoVax entered into an employment agreement with Harriet L. Robinson, our Chief Scientific Officer. The employment agreement has no specified term. The employment agreement provided for an initial base salary of $250,000 to Dr. Robinson, subject to periodic increases as determined by the Compensation Committee. Dr. Robinson initially worked part-time for the Company, and became a full-time employee in February 2008. In April 2013, Dr. Robinson reduced her time commitment to the Company from 100% to 80%, and her base salary was adjusted proportionately from $250,000 to $212,600. The Board of Directors may also approve the payment of a discretionary bonus annually. Dr. Robinson is eligible for grants of awards from our Plan and is entitled to participate in any and all benefits in effect from time-to-time for employees generally. We may terminate the employment agreement, with or without cause. If we terminate the employment agreement without cause, we will be required to provide Dr. Robinson at least 30 days prior notice of the termination and one week of severance pay for each full year of service ($28,619 as of December 31, 2014, paid as salary continuance). Dr. Robinson may terminate the employment agreement at any time by giving us 60 days notice. In that event, she would not receive severance. In April 2013, Dr. Robinson reduced her time commitment to the company to 80% in conjunction with a prorata reduction of her then annualized salary of $265,750 to $212,600. In October 2013, our Board of Directors approved an amendment to the employment agreement with Dr. Robinson. The 2013 amendment includes severance provisions in the event of a change in control (as defined in the amendment) and a qualifying termination of employment. See the discussion under “Potential Payments Upon Change-in-Control” below.
In October 2006 GeoVax Labs, Inc. and our subsidiary, GeoVax, Inc. entered into indemnification agreements with Messrs. McNally, Reynolds, Kollintzas and Spencer. Pursuant to these agreements, we have agreed to indemnify them to the full extent permitted by Illinois and Georgia law against certain liabilities incurred by these individuals in connection with specified proceedings if they acted in a manner they believed in good faith to be in or not opposed to the best interests of the Company and, with respect to any criminal proceeding, had no reasonable cause to believe that such conduct was unlawful. The agreements also provide for the advancement of expenses to these individuals subject to specified conditions.
Potential Payments Upon a Change-in-Control
Our 2006 Equity Incentive Plan contains provisions that could lead to an accelerated vesting of options or other awards. In the event of certain change-in-control transactions described in the Plan, (i) outstanding options or other awards under the Plan may be assumed, converted or replaced; (ii) the successor corporation may substitute equivalent options or other awards or provide substantially similar consideration to Plan participants as were provided to stockholders (after taking into account the existing provisions of the options or other awards); or (iii) the successor corporation may replace options or awards with substantially similar shares or other property.
In the event the successor corporation (if any) refuses to assume or substitute options or other awards as described (i) the vesting of any or all options or awards granted pursuant to the Plan will accelerate upon the change-in-control transaction, and (ii) any or all options granted pursuant to the Plan will become exercisable in full prior to the consummation of the change-in-control transaction at such time and on such conditions as the Compensation Committee determines. If the options are not exercised prior to the consummation of the change-in-control transaction, they shall terminate at such time as determined by the Compensation Committee. Subject to any greater rights granted to Plan participants under the Plan, in the event of the occurrence of a change-in-control transaction any outstanding options or other awards will be treated as provided in the applicable agreement or plan of merger, consolidation, dissolution, liquidation, or sale of assets.
If the Company experienced a change-in-control transaction described in the Plan on December 31, 2014, the value of accelerated options for each Named Executive Officer, based on the difference between the closing price of our common stock on the OTC Market on December 31, 2014, and, if lower, the exercise price per share of each option for which vesting would be accelerated for each Named Executive Officer, would be $0.
Our employment agreements with each Named Executive Officer provide for payment to each Named Executive Officer if we terminate such Named Executive Officer’s employment without cause. If each Named Executive Officer was terminated without cause on December 31, 2014, the following amounts, which represent one week of pay for each full year of service to the Company, would be payable to each Named Executive Officer as salary continuance under the terms of such Named Executive Officer’s employment agreement: Dr. McNally - $19,038; Mr. Reynolds - $32,708; and Dr. Robinson - $28,619.
In October 2013, our Board of Directors approved amendments to the employment agreements with each Named Executive Officer These 2013 amendments include severance provisions in the event of a change in control and a qualifying termination of employment. Specifically, if a Named Executive Officer is terminated at any time during the three month period which immediately precedes a change in control (as defined in the amendment) or during the one year period following a change in control, then the Company would pay an amount in cash equal to (a) a multiple of the Named Executive Officer’s then base salary and target annual bonus (3x for Dr. McNally, 2x for Mr. Reynolds, and 2x for Dr. Robinson), (b) a multiple of the cost to provide 401(k) or other deferred compensation or health and welfare benefits to the Named Executive Officer (3x for Dr. McNally, 2x for Mr. Reynolds, and 2x for Dr. Robinson), and (c) a tax gross-up payment (if an excise tax is imposed by § 4999 of the Internal Revenue Code or any related interest or penalties are incurred by the officer) pursuant to the amendment. The amendments also provide for full and complete vesting of all stock option grants held by the Named Executive Officers.
Summary Compensation Table
The following narrative, table, and footnotes set forth information concerning the total compensation earned during the fiscal years ended December 31, 2014 and 2013 by our Named Executive Officers. The individual components of the total compensation reflected in the table are broken out as follows:
Salary. Base salary earned during 2014 and 2013. The terms of the Employment Agreements governed the base salaries for Dr. McNally, Mr. Reynolds, and Dr. Robinson.
Bonus. The amount of cash bonuses paid during 2014 and 2013.
Option Awards. The awards disclosed under the heading “Option Awards” consist of the aggregate grant date fair value of the stock option grants during 2014 and 2013 computed in accordance with Financial Accounting Standards Board Accounting Standards Codification Topic 718, Compensation – Stock Compensation (“FASB ASC Topic 718”). For a discussion of the various assumptions made and methods used for determining such amounts, see footnotes 2 and 9 to our 2014 consolidated financial statements contained in our Annual Report on Form 10-K for the year ended December 31, 2014.
All Other Compensation. The amounts include under “All Other Compensation” are described in the footnotes to the table.
|
Name and Principal Position |
Year |
Salary ($) |
Bonus ($) |
Option Awards ($) |
All Other Compensation ($)(3) |
Total ($) |
|||||||||||||||
|
Robert T. McNally |
2014 |
$ | 174,167 | $ | - | $ | 4,080 | (1) | $ | 6,967 | $ | 185,214 | |||||||||
| President and Chief Executive Officer | 2013 | 275,000 | - | 12,930 | (2) | 10,200 | 298,130 | ||||||||||||||
|
Mark W. Reynolds |
2014 |
212,600 | 2,000 | 4,080 | (1) | 8,546 | 227,226 | ||||||||||||||
| Chief Financial Officer | 2013 | 212,600 | - | 12,930 | (2) | 8,504 | 234,034 | ||||||||||||||
|
Harriet L. Robinson |
2014 |
212,600 | - | 4,080 | (1) | 8,504 | 225,184 | ||||||||||||||
| Chief Scientific Officer | 2013 | 225,887 | - | 12,930 | (2) | 9,035 | 247,852 | ||||||||||||||
|
(1) |
Grant date fair value of stock option grant on December 9, 2014 for 30,000 shares with an exercise price of $0.17 per share, vesting over a three-year period. As of December 31, 2014, none of these shares have vested and are exercisable |
|
(2) |
Grant date fair value of stock option grant on December 18, 2013 for 30,000 shares with an exercise price of $0.53 per share, vesting over a three-year period. As of December 31, 2014, 10,000 of these shares have vested and are exercisable |
|
(3) |
Amounts shown in the “All Other Compensation” column represent employer contributions to the Company’s 401(k) retirement plan. |
Outstanding Equity Awards at Fiscal Year-End
GeoVax has awarded stock options to its senior management and other employees. The terms of these awards typically provide for vesting over a defined period of time, generally three years. The options expire if not exercised within ten years from the date of grant. The Company does not have a formula for determining stock option awards. Awards are generally based on the subjective judgment of the President and Chief Executive Officer and on the Compensation Committee’s subjective judgment.
The following table sets forth certain information with respect to unexercised options previously awarded to our Named Executive Officers that were outstanding as of December 31, 2014.
|
Option Awards | ||||||||
|
Number of Securities Underlying Unexercised Options |
||||||||
|
Name |
(#) Exercisable |
(#) Unexercisable |
Option Exercise Price ($) |
Option Expiration Date | ||||
|
Robert McNally
|
- 10,000 20,000 30,000 10,000 10,000 10,000 48,000 10,000 26,400 |
30,000 20,000 10,000 - - - - - - - |
(1) (3) |
$ 0.17 0.53 0.66 0.91 1.98 7.00 5.50 8.50 8.05 17.75 |
12/9/24 12/18/23 12/11/22 12/30/21 12/10/20 12/2/19 12/11/18 6/17/18 12/5/17 3/14/17 | |||
|
Mark Reynolds |
- 10,000 16,666 25,000 10,000 10,000 10,000 10,000 36,000 |
30,000 20,000 8,334 - - - - - - |
(1) (2) (3) |
0.17 0.53 0.66 0.91 1.98 7.00 5.50 8.05 17.75 |
12/9/24 12/18/23 12/11/22 12/30/21 12/10/20 12/2/19 12/11/18 12/5/17 3/14/17 | |||
|
Harriet Robinson |
- 10,000 16,666 25,000 10,000 10,000 10,000 |
30,000 20,000 8,334 - - - - |
(1) (2) (3) |
0.17 0.53 0.66 0.91 1.98 7.00 5.50 |
12/9/24 12/18/23 12/11/22 12/30/21 12/10/20 12/2/19 12/11/18 | |||
|
(1) |
These stock options vest and become exercisable in three equal installments on December 9, 2015, 2016 and 2017. |
|
(2) |
These stock options vest and become exercisable in two equal installments on December 18, 2015 and 2016. |
|
(3) |
These stock options vest and become exercisable on December 11, 2015. |
Other Benefits Provided to Executive Officers
Mr. Reynolds and Dr. Robinson are eligible for health insurance and 401(k) benefits at the same level and subject to the same conditions as provided to all other employees. Dr. McNally is eligible for 401(k) benefits at the same level and subject to the same conditions as provided to all other employees, but he is currently ineligible for health insurance due to his time commitment to the Company being less that the required 30 hours per week. GeoVax participates in a multi-employer defined contribution retirement plan (the “401k Plan”) administered by a third party service provider; and the Company contributes to the 401k Plan on behalf of all its eligible employees based upon the same matching formula. The amounts shown in the Summary Compensation Table under the heading “Other Compensation” represent the value of the Company’s matching contributions to the 401(k) accounts of these executive officers. Executive officers did not receive any other perquisites or other personal benefits or property from the Company or any other source.
DIRECTOR COMPENSATION
The following table sets forth information concerning the compensation earned for service on our Board of Directors during the fiscal year ending December 31, 2014 by each individual who served as a director at any time during the fiscal year.
|
Name |
Fees Earned or Paid in Cash ($) |
Stock Awards ($) |
(2)(3) Option Awards ($) |
Non-Equity Incentive Plan Compensation ($) |
Non-qualified Deferred Compensation Earnings ($) |
All Other Compensation ($) |
Total ($) |
|||||||||||||||||||||
|
David A. Dodd |
48,550 | - | 3,400 | - | - | - | 51,950 | |||||||||||||||||||||
|
Dean G. Kollintzas |
28,500 | - | 3,400 | - | - | - | 31,900 | |||||||||||||||||||||
|
Robert T. McNally (1) |
- | - | - | - | - | - | - | |||||||||||||||||||||
|
Harriet L. Robinson (1) |
- | - | - | - | - | - | - | |||||||||||||||||||||
|
John N. Spencer, Jr. |
37,450 | - | 3,400 | - | - | - | 40,850 | |||||||||||||||||||||
|
(1) |
Dr. McNally and Dr. Robinson, who were employees of the Company during the fiscal year ended December 31, 2014, received no compensation for their service as directors. All amounts related to their compensation as Named Executive Officers during the fiscal year ended December 31, 2014 and prior years are included in the “Summary Compensation Table”. |
|
(2) |
Amounts shown in the “Option Awards” column represent the aggregate grant date fair value of awards computed in accordance with FASB ASC Topic 718. For a discussion of the various assumptions made and methods used for determining such amounts, see footnotes 2 and 9 to our 2014 consolidated financial statements contained in our Annual Report on Form 10-K for the year ended December 31, 2014. On December 9, 2014, Messrs. Dodd, Kollintzas and Spencer were each granted options to purchase 25,000 shares of our common stock with an exercise price of $0.17 per share. |
|
(3) |
The table below shows the aggregate numbers of option awards outstanding for each non-employee director as of December 31, 2014. |
|
Name |
Aggregate Option Awards Outstanding as of December 31, 2014 (#) |
|
David A. Dodd |
136,400 |
|
Dean G. Kollintzas |
166,400 |
|
John N. Spencer, Jr. |
166,400 |
Director Compensation Plan
In March 2007, the Board of Directors approved a recommendation from the Compensation Committee for director compensation, which we refer to as the “Director Compensation Plan.” It was subsequently amended in March 2008, December 2009, and in December 2010. The Director Compensation Plan applies only to non-employee directors. Directors who are employees of the Company receive no compensation for their service as directors or as members of committees.
Cash Fees
For 2014, each non-employee director received an annual retainer (paid quarterly) of $5,000 for service as a member of the Audit Committee and $3,300 for service as a member of the Compensation Committee or the Nominating and Corporate Governance Committee. The Chairman of the Audit Committee received an annual retainer of $9,000, and the Chairman of each of the Compensation Committee and the Nominating and Corporate Governance Committee received an annual retainer of $6,000. These retainers were also paid quarterly. Non-employee directors also received fees for each Board of Directors or Committee meeting attended as follows: $3,000 for in person Board of Directors meetings ($1,500 for telephonic meetings), $1,000 for in person Committee meeting chaired ($750 for telephonic meetings), and $500 for in person Committee meeting attended as a non-chair member ($400 for telephonic meetings). Mr. Dodd, the non-employee Chairman of the Board during 2013, received an annual retainer of $30,000 (paid quarterly) and was not entitled to additional fees for Board meetings attended, but did receive additional fees for committees on which he serves.
Stock Option Grants
Each of our current non-employee directors received a grant of options to purchase 26,400 shares of common stock on the date that such non-employee director was first elected or appointed. We currently do not have a formula for determining annual stock option grants to directors (upon their re-election to the Board of Directors, or otherwise). Such option grants are currently determined by the Board of Directors, upon recommendation by the Compensation Committee based on the Compensation Committee’s annual deliberations and review of the director compensation structure of similar companies. At its meeting in December 2014, upon a recommendation of the Compensation Committee, the Board of Directors approved an annual stock option grant of 25,000 shares to its non-employee members.
Expense Reimbursement
All directors are reimbursed for expenses incurred in connection with attending meetings of the Board of Directors and committees.
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
Policies and Procedures for Approval of Related Party Transactions
Our Audit Committee is responsible for reviewing and approving all transactions or arrangements between the Company and any of our directors, officers, principal stockholders or any of their respective affiliates, associates or related parties, other than transactions with officers which are covered by the duties of the Compensation Committee. In determining whether to approve or ratify a related party transaction, the Audit Committee will discuss the transaction with management and will consider all relevant facts and circumstances available to it including:
|
● |
whether the terms of the transaction are fair to the Company and at least as favorable to the Company as would apply if the transaction did not involve a related party. |
|
● |
whether there are demonstrable business reasons for the Company to enter into the transaction. |
|
● |
whether the transaction would impair the independence of a non-employee director; and |
|
● |
whether the transaction would present an improper conflict of interest for any director or executive officer, taking into account the size of the transaction, the direct or indirect nature of the related party’s interest in the transaction and the ongoing nature of any proposed relationship, and any other factors the Audit Committee deems relevant. |
These policies are in writing and included in the Company’s minute book.
Our Board of Directors has made the following findings and adopted the following policies (in writing) regarding related party transactions:
|
● |
The Company has not made and will not make loans or loan guarantees on behalf of any director, officer, beneficially owner of more than 5% of our common stock, or other person constituting a Promoter, as such term is defined in the NASAA Statement of Policy Regarding Corporate Securities Definitions. |
|
● |
The Company has not engaged and will not engage in material transactions with any director, officer, beneficial owner of more than 5% of our common stock, or other person constituting a Promoter, as such term is defined in the NASAA Statement of Policy Regarding Corporate Securities Definitions, except as described below or as otherwise approved by our Audit Committee consistent with the policies and procedures described below. |
|
● |
The Company will make any future material affiliated transactions on terms that are no less favorable to the Company than those that can be obtained from unaffiliated third parties. |
|
● |
A majority of the Company’s Audit Committee will approve all future material transactions. |
|
● |
The Company’s officers, directors, and counsel will: |
|
o |
consider their due diligence and assure that there is a reasonable basis for these representations, and |
|
o |
consider whether to embody the representations in the issuer’s charter or bylaws. |
Transactions with Related Parties
Emory University is a significant stockholder of the Company, and our primary product candidates are based on technology rights subject to a license agreement with Emory University, which we refer to as the Emory License. The Emory License, among other contractual obligations, requires payments based on milestone achievements, royalties on sales by the Company or on payments to the Company by our sublicensees, and payment of maintenance fees in the event certain milestones are not met within the time periods specified in the Emory License. We may terminate the Emory License upon 90 days prior written notice. In any event, the Emory License expires on the date of the latest expiration date of the underlying patents. We are also obligated to reimburse Emory University for certain ongoing costs in connection with the filing, prosecution and maintenance of patent applications subject to the Emory License. The expense associated with these ongoing patent reimbursements to Emory University amounted to $179,958 for the year ended December 31, 2014.
On October 14, 2014, we entered into a letter agreement with Sabby Healthcare Master Fund, Ltd. and Sabby Volatility Warrant Master Fund, Ltd. with respect to the payment to them of a warrant exercise fee of $0.075 per share for each share purchased upon exercise of Series A or Series C Common Stock Purchase Warrants (“Warrants”) held by them. Each of these parties at that time held Warrants to acquire an aggregate of 2,666,666 shares of our common stock. They agreed to exercise Warrants equal to 9.98% of the outstanding shares of GeoVax (3,176,000 shares in the aggregate) upon execution of the letter, and we paid the exercise fee of $238,200 subsequent to our receipt of the exercise price.
On February 25, 2015, we entered into a Securities Purchase Agreement (the “Securities Purchase Agreement”) with Sabby Healthcare Master Fund, Ltd. and Sabby Volatility Warrant Master Fund, Ltd. (collectively, the “Purchasers”) providing for the issuance and sale to the Purchasers of an aggregate of 3,000 shares of our Series C Convertible Preferred Stock (the “Preferred Shares”) and related warrants for gross proceeds to the Company of $3.0 million. Each Preferred Share is initially convertible into approximately 5,555.55 shares of our Common Stock for an aggregate total of 16,666,666 shares of our Common Stock (the “Conversion Shares”). The terms of the Preferred Shares include anti-dilution provisions. Pursuant to the Certificate of Designation which authorized the Series C Convertible Preferred Stock, the Preferred Shares may be converted at any time at the option of the Purchasers into shares of our Common Stock at a conversion price of $0.18 per share (the “Conversion Price”). The Certificate of Designation contains price adjustment provisions, which may, under certain circumstances, (i) reduce the Conversion Price on several future dates, including the effective date of the registration statement to be filed to cover resale of the Conversion Shares, according to a formula based on the then-current market price for our common stock. We closed this transaction on February 27, 2015.
Pursuant to the Securities Purchase Agreement, each Purchaser was also issued a Series D Warrant, a Series E Warrant and a Series F Warrant (collectively, the “Warrants”), each to purchase up to a number of shares of the Company’s Common Stock equal to 100% of the Conversion Shares underlying the Preferred Shares issued to such Purchaser pursuant to the Securities Purchase Agreement (up to 16,666,666 shares in the aggregate for each of the three series of warrants, or approximately 50,000,000 shares in total) (the “Warrant Shares”). The Series D Warrants have an exercise price of $0.22 per share, are exercisable immediately, and have a term of exercise equal to five years from the date of issuance. The Series E Warrants have an exercise price of $0.18 per share, are exercisable immediately, and have a term of exercise equal to one year from the date of issuance. The Series F Warrants have an exercise price of $0.22 per share and have a term of exercise equal to five years from the date of issuance, but only vest and become exercisable upon, and in proportion to, the exercise of the one-year Series E Warrants held by each Purchaser (or its assigns). The Warrants contain anti-dilution and price adjustment provisions, which may, under certain circumstances, (i) reduce the exercise price on several future dates, including the effective date of the registration statement to be filed to cover resale of the shares subject to the Warrants, according to a formula based on the then-current market price for our common stock and (ii) reduce the exercise price to match if we sell or grant options to purchase, including rights to reprice, our common stock or common stock equivalents at a price lower than the exercise price of the Warrants, or if we announce plans to do so. The number of shares subject to warrants will not increase due to such reductions in exercise price. We also issued the Maxim Warrant to our placement agent to acquire 1,333,333 shares of our common stock at $0.22 per share on substantially the same terms and conditions of the Series D warrants.
The Purchasers also have the right to participate in certain future financings, subject to certain exceptions, and may invest up to 75% of the aggregate amount invested at that time. If we do not hold a shareholders’ meeting on or before June 30, 2015 for the purpose of obtaining shareholder approval of an increase in our authorized common stock sufficient to cover the shares to be issued upon exercise of all the Warrants issued in connection with the Securities Purchase Agreement (including shares to be issued to Maxim Partners LLC, an affiliate of our placement agent, Maxim Group, LLC, upon exercise of the Maxim Warrant), we must pay penalties until approval is obtained. The Preferred Shares do not have voting rights except as required by law and are not entitled to a dividend. When issued, the Conversion Shares will have the voting rights afforded to all shares of Common Stock. The Preferred Shares have a liquidation preference equal to the initial purchase price.
On February 25, 2015, in connection with the closing of the private placement, we entered into a Registration Rights Agreement (the “Registration Rights Agreement”) with the Purchasers. Under the Registration Rights Agreement, we are required to file a registration statement within 30 calendar days after signing the Registration Rights Agreement. Our failure to meet the filing deadlines and other requirements set forth in the Registration Rights Agreement may subject us to monetary penalties.
SELLING STOCKHOLDERS
This prospectus relates to the resale by the selling stockholders named below from time to time of up to a total of 67,999,997 shares that are owned by or issuable to the selling stockholders. The number of shares is subject to adjustment as described at “Description of Securities.” The common stock offered by this prospectus is being offered by the selling stockholders for their own accounts.
Private Placement Transaction
On February 27, 2015, we completed a private placement transaction and issued to a total of 3,000 shares of Series C Preferred Stock, with a stated value of $1,000 per share and a conversion price of $0.18, to two accredited investors. Each share of Series C Preferred Stock is convertible into 5,555.55 shares of our common stock. Each purchaser of a share of Series C Preferred Stock also acquired a Series D, a Series E, and a Series F Warrant to purchase a share of our common stock. See “Description of Securities” for details of the terms of these warrants. We also issued a warrant to Maxim Partners LLC to purchase 1,333,333 shares of our stock on substantially the same terms and conditions as the Series D warrants. The Series C Convertible Preferred Stock and the 2015 warrants were issued in reliance upon exemptions provided by Section 4(2) of the Securities Act for the offer and sale of securities not involving a public offering and Regulation D promulgated thereunder.
Selling Stockholders
The table below, which was prepared based on information supplied to us by the selling stockholders, sets forth information regarding the beneficial ownership of outstanding shares of our common stock owned by the selling stockholders and the shares that they may sell or otherwise dispose of from time to time under this prospectus. Each of the selling stockholders, or their respective transferees, donees or their successors, may resell, from time to time, all, some or none of the shares of our common stock covered by this prospectus, as provided in this prospectus under the section entitled “Plan of Distribution” and in any applicable prospectus supplement. However, we do not know when, in what amount, or at what specific prices the selling stockholders may offer their shares for sale under this prospectus, if any.
The number of shares disclosed in the table below as “beneficially owned” are those beneficially owned as determined under the rules of the SEC. Such information is not necessarily indicative of ownership for any other purpose. Under the rules of the SEC, a person is deemed to be a “beneficial owner” of a security if that person has or shares “voting power,” which includes the power to vote or to direct the voting of such security, or “investment power,” which includes the power to dispose of or to direct the disposition of such security. In computing the number of shares beneficially owned by a selling stockholder and the percentage of ownership of that selling stockholder, shares of common stock underlying shares of Series C Preferred Stock, options or warrants held by that selling stockholder that are convertible or exercisable, as the case may be, within 60 days of March 9, 2015 are included, subject to the ”Maximum Percentage” limitation described in footnote 1 to the table below. Those shares, however, are not deemed outstanding for the purpose of computing the percentage ownership of any other selling stockholder. Each selling stockholder’s percentage of ownership in the following table is based upon 31,950,813 shares of our common stock outstanding as of March 9, 2015. Please note that the column titled “Total Shares that may be Offered and Sold Hereby” includes all shares that may be issued.
Unless otherwise indicated and subject to community property laws where applicable, the selling stockholders named in the following table have, to our knowledge, sole voting and investment power with respect to the shares beneficially owned by them. In addition, none of the selling stockholders has any family relationships with our officers, directors or controlling stockholders. Furthermore, no selling stockholder is a registered broker-dealer or an affiliate of a registered broker-dealer, except that Maxim Partners LLC is an affiliate of Maxim Group, LLC, a broker-dealer registered with the Securities and Exchange Commission.
Information concerning any of the selling stockholders may change from time to time, and any changed information will be presented in a prospectus supplement as necessary. Please carefully read the footnotes located below the table in conjunction with the information presented in the table.
|
Selling Stockholder Name |
Beneficial Ownership Prior to this Offering (1) |
Shares that may be Offered and Sold Hereby |
Beneficial Ownership After this Offering (2) |
% Holding After Completion of this Offering |
||||||||||||
|
Sabby Volatility Warrant Master Fund, Ltd. (3) |
3,431,500 | (4) | 33,333,332 | 603,666 | 0.6 | % | ||||||||||
|
Sabby Healthcare Master Fund, Ltd. (3) |
3,428,400 | (5) | 33,333,332 | 603,666 | 0.6 | % | ||||||||||
|
Maxim Partners LLC |
1,333,333 | (6) | 1,333,333 | 0 | 0 | % | ||||||||||
______________________
* Less than 1.0%
|
(1) |
Includes shares of common stock beneficially owned by the selling stockholders as of March 9, 2015, subject, in the case of the two Sabby entities, to the highest Maximum Percentage of 9.99% as discussed at footnote 3. |
|
(2) |
These shares have been separately registered for resale pursuant to the Securities Act and may also be sold. |
|
(3) |
The number of shares in the “Shares that may be Offered and Sold Hereby” column above reflect 100% of the shares issuable upon conversion of the Series C Preferred Stock and upon exercise of the 2015 Warrants. The Series C Preferred Stock and the 2015 Warrants contain exercise and conversion limitations providing that a holder thereof may not convert or exercise (as the case may be) to the extent (but only to the extent) that, if after giving effect to such conversion or exercise (as the case may be), the holder or any of its affiliates would beneficially own in excess of 4.99% (with respect to the Series C Preferred Stock) or 9.99% (with respect to the 2015 Warrants), as applicable (the “Maximum Percentage”) of the outstanding shares of common stock immediately after giving effect to such conversion or exercise (as the case may be). To the extent the above limitation applies, the determination of whether a share of preferred stock or warrant shall be exercisable or convertible (vis-à-vis other convertible, exercisable or exchangeable securities owned by the holder) shall, subject to such Maximum Percentage limitation, be determined on the basis of the first submission to the Company for conversion, exercise or exchange (as the case may be). Accordingly, the number of shares of common stock set forth in the table as being registered for a selling stockholder may exceed the number of shares of common stock that the selling stockholder could own beneficially at any given time through its ownership of the Series C Preferred Stock and the 2015 Warrants. |
|
(4) |
Includes 1,032,426 shares of common stock, 8,333,333 shares issuable upon conversion of Series C Preferred Stock, 24,999,999 shares issuable upon exercise of the Series D, E, and F Warrants, and 603,666 shares issuable upon exercise of Series A Warrants. Sabby Management, LLC shares voting and investment power with respect to these shares on behalf of this stockholder as well as Sabby Healthcare Master Fund, Ltd. As manager of Sabby Management, LLC, Hal Mintz also shares voting and investment power on behalf of this stockholder. Each of Sabby Management, LLC and Hal Mintz disclaim beneficial ownership over the securities covered by this prospectus except to the extent of their pecuniary interest therein. |
|
(5) |
Includes 1,062,508 shares of common stock, 8,333,333 shares issuable upon conversion of Series C Preferred Stock, 24,999,999 shares issuable upon exercise of the Series D, E, and F Warrants, and 603,666 shares issuable upon exercise of Series A Warrants. Sabby Management, LLC shares voting and investment power with respect to these shares on behalf of this stockholder as well as Sabby Volatility Warrant Master Fund, Ltd. As manager of Sabby Management, LLC, Hal Mintz also shares voting and investment power on behalf of this stockholder. Each of Sabby Management, LLC and Hal Mintz disclaim beneficial ownership over the securities covered by this prospectus except to the extent of their pecuniary interest therein. |
|
(6) |
Includes approximately 1,333,333 shares of common stock issuable upon exercise of the Maxim Warrant. |
DESCRIPTION OF SECURITIES
Capital Stock
The following description of our capital stock is summarized from, and qualified in its entirety by reference to, our certificate of incorporation, as amended, including the certificates of designation, as amended, setting forth the terms of our Series B Preferred Stock and Series C Preferred Stock, all of which have been previously filed with the SEC and are incorporated herein by reference. This summary is not intended to give full effect to provisions of statutory or common law. We urge you to review the following documents because they, and not this summary, define the rights of a holder of shares of common stock, Series B and Series C Preferred Stock:
|
|
• |
the General Corporation Law of the State of Delaware, or the “DGCL”, as it may be amended from time to time; |
|
|
• |
our certificate of incorporation, as it may be amended or restated from time to time, and |
|
|
• |
our bylaws, as they may be amended or restated from time to time. |
General
Our authorized capital stock currently consists of 85,000,000 shares, which are divided into two classes consisting of 75,000,000 shares of common stock, par value $0.001 per share, and 10,000,000 shares of preferred stock, par value $0.01 per share. We currently plan to ask our stockholders to authorize an increase in our authorized shares of common stock to 150,000,000, which we believe will enable us to issue all of the shares of common stock that we are required to issue upon conversion of our 2015 Warrants. There can be no assurance this will be proposed by us or approved by our stockholders
Common Stock
As of March 16, 2015, there were issued and outstanding 31,950,813 shares of common stock, options to purchase 1,177,500 shares of common stock and warrants to purchase 56,422,157 shares of common stock, including the 54,023,997 shares in the aggregate subject to the unexercised Series A, C, D, E and F Warrants. In addition, 285,714 shares of our common stock are reserved for issuance upon conversion of the outstanding Series B Preferred Stock , and16,666,666 shares of our common stock are reserved for issuance upon conversion of the outstanding Series C Preferred Stock.
Holders of our common stock are entitled to one vote for each share held in the election of directors and in all other matters to be voted on by the stockholders. There is no cumulative voting in the election of directors. Holders of common stock are entitled to receive dividends as may be declared from time to time by our Board of Directors out of funds legally available therefor, and subject to the rights of holders of our Series B and Series C Preferred Stock. In the event of liquidation, dissolution or winding up of the Company, holders of common stock are to share in all assets remaining after the payment of liabilities, and satisfaction of the liquidation preference of our outstanding Series B and Series C Preferred Stock. Holders of common stock have no pre-emptive or conversion rights and are not subject to further calls or assessments. There are no redemption or sinking fund provisions applicable to the common stock. The rights of the holders of the common stock are subject to any rights that may be fixed for holders of preferred stock, such as the Series B and Series C Preferred Stock. All of the outstanding shares of common stock are fully paid and non-assessable.
Series A Convertible Preferred Stock
We are authorized to issue up to 2,200 shares of our Series A convertible preferred stock, which we refer to as the “Series A Preferred Stock.” No shares of our Series A Preferred Stock, $0.01 par value are outstanding.
Series B Convertible Preferred Stock
We are authorized to issue up to 1,650 shares of our Series B Preferred Stock, which we refer to as the “Series B Preferred Stock.” As of March 16, 2015, 100 shares of our Series B Preferred Stock, $0.01 par value, were outstanding.
The Series B Preferred Stock is convertible at the option of the holder at any time into shares of common stock at a conversion ratio determined by dividing the $1,000 stated value of the Series A convertible preferred stock by a conversion price of $0.35 per share. As of March 16, 2015, an aggregate of 285,714 shares of our common stock are issuable upon conversion of the 100 outstanding shares of Series B Preferred Stock. The conversion price of the Series B Preferred Stock is subject to adjustment in the case of stock splits, stock dividends, combinations of shares, similar recapitalization transactions and certain pro-rata distributions to common stockholders.
Subject to limited exceptions, a holder of the Series B Preferred Stock will not have the right to convert any portion of its Series B Preferred Stock if the holder, together with its affiliates, would beneficially own in excess of 9.99% of the number of shares of our common stock outstanding immediately after giving effect to its conversion.
The holders of Series B Preferred Stock will be entitled to receive any securities or rights to acquire securities or property granted or issued by us pro rata to the holders of our common stock to the same extent as if such holders had converted all of their shares of Series B Preferred Stock. No distribution may be made on the common stock so long as any dividend due on the Series B Preferred Stock remains unpaid. In the event of a fundamental transaction, such as a merger, consolidation, sale of substantially all assets and similar reorganizations or recapitalizations, the holders of Series B Preferred Stock will be entitled to receive, upon conversion of their shares, any securities or other consideration received by the holders of our common stock pursuant to the fundamental transaction.
Except as required by law, holders of the Series B Preferred Stock are not entitled to voting rights; provided, however, that the affirmative vote of the holders of a majority of the outstanding shares of Series B Preferred Stock is required to take certain actions that may alter or change adversely the rights or preferences of the holders of Series B Preferred Stock, increase the number of shares of Series B Preferred Stock, or authorize a new class ranking senior or pari passu to the Series B Preferred Stock. The Series B Preferred Stock has a liquidation preference equal to $1,000 per share.
The securities purchase agreement and related registration rights agreement, as well as the certificate of designation authorizing the Series B Preferred Stock include certain other agreements and covenants for the benefit of the holders of the Series B Preferred Stock, including several restrictions that have now expired, and a requirement to use our best efforts to maintain the listing or trading of our common stock on one or more specified United States securities exchanges or regulated quotation services.
Series C Convertible Preferred Stock
We are authorized to issue up to 3,000 shares of our Series C Convertible Preferred Stock, which we refer to as the “Series C Preferred Stock.” As of March 16, 2015, 3,000 shares of our Series C Preferred Stock, par value $0.01 per share, were outstanding.
The Series C Preferred Stock is convertible at the option of the holder at any time into shares of common stock at a conversion ratio determined by dividing the $1,000 stated value of the Series C Preferred Stock by a conversion price of $0.18 per share. As of March 16, 2015, an aggregate of 16,666,666 shares of our common stock are issuable upon conversion of the outstanding shares of Series C Preferred Stock. The conversion price of the Series C Preferred Stock is subject to adjustment in the case of stock splits, stock dividends, combinations of shares, similar recapitalization transactions and certain pro-rata distributions to common stockholders. The conversion price is also subject to additional price adjustment provisions, which may, under certain circumstances, reduce the conversion price on several future dates, including the effective date of the SEC registration statement to be filed to cover resale of the shares issuable upon conversion of the Series C Preferred Stock, according to formulas based on the then-current market price for our common stock
Subject to limited exceptions, a holder of the Series C Preferred Stock will not have the right to convert any portion of its Series C Preferred Stock if the holder, together with its affiliates, would beneficially own in excess of 4.99% of the number of shares of our common stock outstanding immediately after giving effect to its conversion. Upon 61 days’ prior notice from a holder, the 4.99% limitation may be increased to 9.99% for that holder and its affiliates.
The holders of Series C Preferred Stock will be entitled to receive any securities or rights to acquire securities or property granted or issued by us pro rata to the holders of our common stock to the same extent as if such holders had converted all of their shares of Series A convertible preferred stock. No distribution may be made on the common stock so long as any dividend due on the Series C Preferred Stock remains unpaid. In the event of a fundamental transaction, such as a merger, consolidation, sale of substantially all assets and similar reorganizations or recapitalizations, the holders of Series C Preferred Stock will be entitled to receive, upon conversion of their shares, any securities or other consideration received by the holders of our common stock pursuant to the fundamental transaction.
Except as required by law, holders of the Series C Preferred Stock are not entitled to voting rights; provided, however, that the affirmative vote of the holders of a majority of the outstanding shares of Series C Preferred Stock is required to take certain actions that may alter or change adversely the rights or preferences of the holders of Series C Preferred Stock, increase the number of shares of Series C Preferred Stock, or authorize a new class ranking senior or pari passu to the Series C Preferred Stock. The Series C Preferred Stock has a liquidation preference equal to $1,000 per share.
The securities purchase agreement and related registration rights agreement, as well as the certificate of designation authorizing the Series C Preferred Stock include certain other agreements and covenants for the benefit of the holders of the Series C Preferred Stock, including restrictions on our ability to issue additional equity securities for a period of 90 days after the effective date of this prospectus (and until the shareholders approve an increase in the authorized shares of common stock), and to issue additional debt or equity securities with a variable conversion or exercise price until no Series C Preferred Stock remains outstanding. We also agreed to provide the holders with the right to participate in future financings, up to 75% of the financing amount, for 18 months from February 25, 2015, and undertook to use our best efforts to maintain the listing or trading of our common stock on one or more specified United States securities exchanges or regulated quotation services.
Undesignated Preferred Stock
Subject to the restrictions set forth in the certificate of designation for our Series B and Series C Preferred Stock, our Board of Directors has the authority to issue up to 9,996,900 additional shares of preferred stock in one or more series and fix the number of shares constituting any such series, the voting powers, designations, preferences and relative, participating, optional or other special rights and qualifications, limitations or restrictions thereof, including the dividend rights, dividend rate, terms of redemption (including sinking fund provisions), redemption price or prices, conversion rights and liquidation preferences of the shares constituting any series, without any further vote or action by the stockholders. For example, the Board of Directors is authorized to issue a stockholder of preferred stock that would have the right to vote, separately or with any other stockholder of preferred stock, on any proposed amendment to our certificate of incorporation, or on any other proposed corporate action, including business combinations and other transactions.
We will not offer preferred stock unless the offering is approved by a majority of our independent directors. The independent directors will have access, at our expense, to our counsel or independent counsel.
The Series A, B and C Warrants
Pursuant to the terms of the securities purchase agreement for the sale of the Series A Preferred Stock, each purchaser was also issued a Series A Warrant, a Series B Warrant and a Series C Warrant, each to purchase up to a number of shares of the Company’s common stock equal to 100% of the common stock underlying the preferred shares issued to such purchaser pursuant to the securities purchase agreement (up to 2,933,333 shares in the aggregate for each series of warrants, or 8,799,999 shares in total). The Series A Warrants have a current exercise price of $0.18 per share, are fully exercisable, and have a term of exercise equal to five years from the date of issuance. There are currently no Series B Warrants outstanding. The Series C Warrants have a current exercise price of $0.18 per share and have a term of exercise equal to five years from the date of issuance.
The exercise price of the warrants and, in some cases, the number of shares issuable upon exercise, are subject to adjustment in the case of stock splits, stock dividends, combinations of shares, similar recapitalization transactions and certain pro-rata distributions to common stockholders. The exercise price, but not the number of shares of common stock issuable upon exercise, will also be adjusted to match the lower price if we sell or grant (or announce a sale or grant) of any shares of common stock or securities convertible into, or rights to acquire, common stock at an effective price per share that is lower than the then exercise price, except in the event of certain exempt issuances. In addition, upon exercise of the warrants the warrant holders will be entitled to receive any securities or rights to acquire securities or property granted or issued by us pro rata to the holders of our common stock to the same extent as if such holders had then exercised the warrants. In the event of a fundamental transaction, such as a merger, consolidation, sale of substantially all assets and similar reorganizations or recapitalizations, the warrant holders will be entitled to receive, upon exercise of their warrants, any securities or other consideration received by the holders of common stock pursuant to the fundamental transaction. Under certain circumstance, after a fundamental transaction, holders may be entitled to receive a cash payment equal to the value of the Series A or C Warrants, computed as provided in those warrants. Any successor to us or surviving entity shall assume the obligations under the warrants.
The warrant holders must surrender payment in cash of the aggregate exercise price of the shares being acquired upon exercise of the warrants. If there is no effective registration statement registering, or no current prospectus available for the resale of the shares issuable upon exercise of the warrants, then the warrants may be exercised on a “net” or “cashless” basis. No fractional shares of common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price.
Subject to limited exceptions, a holder of the Series A or C Warrants will not have the right to exercise the warrant if the holder, together with its affiliates, would beneficially own in excess of 4.99% of the number of shares of our common stock outstanding immediately after giving effect to its conversion. Upon 61 days’ prior notice from a holder, the 4.99% limitation may be increased to 9.99% for that holder. The Series A and Series C Warrants held by two investors have been amended to increase the beneficial ownership limitation to 9.99%.
The Series D, E and F Warrants
Pursuant to the terms of the securities purchase agreement for the sale of the Series C Preferred Stock, each purchaser was also issued a Series D Warrant, a Series E Warrant and a Series F Warrant, each such warrant representing the right to purchase up to a number of shares of the Company’s Common Stock equal to 100% of the Common Stock underlying the preferred shares issued to such purchaser pursuant to the securities purchase agreement (up to16,666,666 shares in the aggregate for each series of warrants, or approximately 50,000,000 shares in total). The Series D Warrants have an exercise price of $0.22 per share, are exercisable immediately, and have a term of exercise equal to five years from the date of issuance. The Series E Warrants have an exercise price of $0.18 per share, are exercisable immediately, and have a term of exercise equal to one year from the date of issuance. The Series F Warrants have an exercise price of $0.22 per share and have a term of exercise equal to five years from the date of issuance, but only vest and become exercisable upon, and in proportion to, the exercise of the one-year Series E Warrants held by each Purchaser (or its assigns). The Warrants contain anti-dilution and price adjustment provisions, which may, under certain circumstances, (i) reduce the exercise price on several future dates, including the effective date of the registration statement to be filed to cover resale of the shares subject to the Warrants, according to a formula based on the then-current market price for our common stock and (ii) reduce the exercise price to match if we sell or grant options to purchase, including rights to reprice, our common stock or common stock equivalents at a price lower than the exercise price of the Warrants, or if we announce plans to do so. The number of shares subject to warrants will not increase due to such reductions in exercise price. In addition, upon exercise of the warrants the warrant holders will be entitled to receive any securities or rights to acquire securities or property granted or issued by us pro rata to the holders of our common stock to the same extent as if such holders had then exercised the warrants. In the event of a fundamental transaction, such as a merger, consolidation, sale of substantially all assets and similar reorganizations or recapitalizations, the warrant holders will be entitled to receive, upon exercise of their warrants, any securities or other consideration received by the holders of common stock pursuant to the fundamental transaction. Under certain circumstance, after a fundamental transaction, holders may be entitled to receive a cash payment equal to the value of the Series D, E or F Warrants, computed as provided in those warrants. Any successor to us or surviving entity shall assume the obligations under the warrants.
The warrant holders must surrender payment in cash of the aggregate exercise price of the shares being acquired upon exercise of the warrants. If there is no effective registration statement registering, there are insufficient authorized shares of our common stock available, or there is no current prospectus available for the resale of the shares issuable upon exercise of the warrants, then the warrants may be exercised on a “net” or “cashless” basis. No fractional shares of common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price.
Subject to limited exceptions, a holder of the Series D, E or F Warrants will not have the right to exercise the warrant if the holder, together with its affiliates, would beneficially own in excess of 9.99% of the number of shares of our common stock outstanding immediately after giving effect to its conversion.
The Maxim Warrant
As noted, the Company also issued the Maxim Warrant to its placement agent in February 2015. The Maxim Warrant grants the right to acquire 1,333,333 shares of our common stock at $0.22 per share on substantially the same terms and conditions of the Series D Warrants.
Delaware Anti-Takeover Law
We have elected not to be subject to certain provisions of Delaware law that could make it more difficult to acquire us by means of a tender offer, a proxy contest, open market purchases, removal of incumbent directors and otherwise. These provisions, summarized below, are expected to discourage types of coercive takeover practices and inadequate takeover bids and to encourage persons seeking to acquire control of us to first negotiate with our Board of Directors.
In general, Section 203 of the DGCL prohibits a publicly held Delaware corporation from engaging in various “business combination” transactions with any interested stockholder for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
|
|
• |
the transaction is approved by the corporation’s board of directors prior to the date the interested stockholder obtained interested stockholder status; |
|
|
• |
upon consummation of the transaction that resulted in the stockholder’s becoming an interested stockholder, the stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding those shares owned by (a) persons who are directors and also officers and (b) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
|
|
• |
on or subsequent to the date the business combination is approved by the corporation’s board of directors and authorized at an annual or special meeting of stockholders by the affirmative vote of at least 66 2 / 3 % of the outstanding voting stock that is not owned by the interested stockholder. |
A “business combination” is defined to include mergers, asset sales and other transactions resulting in financial benefit to a stockholder. In general, an “interested stockholder” is a person who, together with affiliates and associates, owns or within three years, did own, 15% or more of a corporation’s voting stock.
Section 203 applies to Delaware corporations that have a class of voting stock that is listed on a national securities exchange or held of record by more than 2,000 stockholders; provided, however, the restrictions of this statute will not apply to a corporation if:
|
|
• |
the corporation’s original charter contains a provision expressly electing not to be governed by the statute; |
|
|
• |
the corporation’s board of directors adopts an amendment to the corporation’s bylaws within 90 days of the effective date of the statute expressly electing not to be governed by it; |
|
|
• |
the stockholders of the corporation adopt an amendment to its charter or bylaws expressly electing not to be governed by the statute (so long as such amendment is approved by the affirmative vote of a majority of the shares entitled to vote); |
|
|
• |
a stockholder becomes an interested stockholder inadvertently and as soon as practicable divests himself of ownership of a sufficient number of shares so that he ceases to be an interested stockholder, and during the three year period immediately prior to a business combination, would not have been an interested stockholder but for the inadvertent acquisition; |
|
|
• |
the business combination is proposed prior to the consummation or abandonment of a merger or consolidation, a sale, lease, exchange, mortgage, pledge, transfer or other disposition of assets of the corporation or a proposed tender or exchange offer for 50% or more of the outstanding voting shares of the corporation; or |
|
|
• |
the business combination is with an interested stockholder who became an interested stockholder at a time when the restrictions contained in the statutes did not apply. |
Our certificate of incorporation includes a provision electing not to be governed by Section 203 of the DCGL. Accordingly, our board of directors does not have the power to reject certain business combinations with interested stockholders based on Section 203 of the DCGL.
Indemnification
Section 145 of the Delaware General Corporation Law, or DGCL, provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to an action, suit or proceeding (other than an action by or in the right of the corporation) by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the corporation’s request in such a capacity for another entity against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with the action, suit or proceeding. The power to indemnify applies (i) if such person is successful on the merits or otherwise in defense of any action, suit or proceeding or (ii) if such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful. The power to indemnify applies to actions brought by or in the right of the corporation as well, but only to the extent of defense expenses (including attorneys’ fees but excluding amounts paid in settlement), actually and reasonably incurred and not to any satisfaction of judgment or settlement of the claim itself, and with the further limitation that in such actions no indemnification shall be made in the event of any adjudication of negligence or misconduct in the performance of his duties to the corporation, unless a court believes that in light of all the circumstances indemnification should apply.
Our bylaws provide that we may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the Company) by reason of the fact that the person is or was a director, officer, employee or agent of the Company, or is or was serving at the request of the Company as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with such action, suit or proceeding if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the Company, and, with respect to any criminal action or proceeding, had no reasonable cause to believe the person’s conduct was unlawful. Our bylaws also provide that we may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the Company to procure a judgment in its favor by reason of the fact that the person is or was a director, officer, employee or agent of the Company, or is or was serving at the request of the Company as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses (including attorneys’ fees) actually and reasonably incurred by the person in connection with the defense or settlement of such action or suit if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the Company and except that no indemnification shall be made in respect of any claim, issue or matter as to which such person shall have been adjudged to be liable to the Company unless and only to the extent that the Delaware Court of Chancery or the court in which such action or suit was brought shall determine upon application that, despite the adjudication of liability but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Delaware Court of Chancery or such other court shall deem proper.
Under our bylaws, expenses (including attorneys’ fees) incurred by an officer or director in defending any civil, criminal, administrative or investigative action, suit or proceeding may be paid by the Company in advance of the final disposition of such action, suit or proceeding upon receipt of an undertaking by or on behalf of such director or officer to repay such amount if it shall ultimately be determined that such person is not entitled to be indemnified by the Company. Such expenses (including attorneys’ fees) incurred by former directors and officers or other employees and agents may be so paid upon such terms and conditions, if any, as we deem appropriate.
The indemnification and advancement of expenses provided by our bylaws is not exclusive, both as to action in such person’s official capacity and as to action in another capacity while holding such office.
Our bylaws also provide that we may purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of the Company, or is or was serving at the request of the Company as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against any liability asserted against such person and incurred by such person in any such capacity, or arising out of such person’s status as such, whether or not the Company would have the power to indemnify such person against such liability under our bylaws. The Company maintains an insurance policy providing for indemnification of its officers, directors and certain other persons against liabilities and expenses incurred by any of them in certain stated proceedings and under certain stated conditions.
In October 2006, GeoVax and our subsidiary, GeoVax, Inc. entered into indemnification agreements with Messrs. McNally, Reynolds, Kollintzas and Spencer. Pursuant to these agreements, we have agreed to hold harmless and indemnify these directors and officers to the full extent authorized or permitted by applicable Illinois and Georgia law against certain expenses and other liabilities actually and reasonably incurred by these individuals in connection with certain proceedings if they acted in a manner they believed in good faith to be in or not opposed to the best interests of the Company and, with respect to any criminal proceeding, had no reasonable cause to believe that such conduct was unlawful. The agreements also provide for the advancement of expenses to these individuals subject to specified conditions. Under these agreements, we will not indemnify these individuals for expenses or other amounts for which applicable Illinois and Georgia law prohibit indemnification. The obligations under these agreements continue during the period in which these individuals are our directors or officers and continue thereafter so long as these individuals shall be subject to any proceeding by reason of their service to the Company, whether or not they are serving in any such capacity at the time the liability or expense incurred for which indemnification can be provided under the agreements.
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION
FOR SECURITIES ACT LIABILITIES
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling the registrant pursuant to the foregoing provisions, the registrant has been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
In the event that a claims for indemnification against such liabilities (other than our payment of expenses incurred or paid by a director, officer or controlling person in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by us is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
PLAN OF DISTRIBUTION
Each selling stockholder and any of their pledgees, assignees and successors-in-interest may, from time to time, sell any or all of their securities covered hereby on the OTC Market or any other stock exchange, market or trading facility on which the securities are traded or in private transactions. These sales may be at fixed or negotiated prices. A selling stockholder may use any one or more of the following methods when selling securities:
|
|
● |
ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
|
|
● |
block trades in which the broker-dealer will attempt to sell the securities as agent but may position and resell a portion of the block as principal to facilitate the transaction; |
|
|
● |
purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
|
|
● |
an exchange distribution in accordance with the rules of the applicable exchange; |
|
|
● |
privately negotiated transactions; |
|
|
● |
settlement of short sales entered into after the effective date of the registration statement of which this prospectus is a part; |
|
|
● |
in transactions through broker-dealers that agree with the selling stockholder to sell a specified number of such securities at a stipulated price per security; |
|
|
● |
through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
|
|
● |
a combination of any such methods of sale; or |
|
|
● |
any other method permitted pursuant to applicable law. |
The selling stockholders may also sell securities under Rule 144 under the Securities Act of 1933, as amended (the “Securities Act”), if available, rather than under this prospectus.
Broker-dealers engaged by the selling stockholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the selling stockholders (or, if any broker-dealer acts as agent for the purchaser of securities, from the purchaser) in amounts to be negotiated, but, except as set forth in a supplement to this Prospectus, in the case of an agency transaction not in excess of a customary brokerage commission in compliance with FINRA Rule 2440; and in the case of a principal transaction a markup or markdown in compliance with Financial Industry Regulation Authority (FINRA) IM-2440.
In connection with the sale of the securities or interests therein, the selling stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the securities in the course of hedging the positions they assume. The selling stockholders may also sell securities short and deliver these securities to close out their short positions, or loan or pledge the securities to broker-dealers that in turn may sell these securities. The selling stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or create one or more derivative securities which require the delivery to such broker-dealer or other financial institution of securities offered by this prospectus, which securities such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The selling stockholders and any broker-dealers or agents that are involved in selling the securities may be deemed to be “underwriters” within the meaning of the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the securities purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Each selling stockholder has informed the Company that it does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the securities. In no event shall any broker-dealer receive fees, commissions and markups which, in the aggregate, would exceed eight percent (8%).
The Company is required to pay certain fees and expenses incurred by the Company incident to the registration of the securities. The Company has agreed to indemnify the selling stockholders against certain losses, claims, damages and liabilities, including liabilities under the Securities Act.
Because selling stockholders may be deemed to be “underwriters” within the meaning of the Securities Act, they will be subject to the prospectus delivery requirements of the Securities Act including Rule 172 thereunder. In addition, any securities covered by this prospectus which qualify for sale pursuant to Rule 144 under the Securities Act may be sold under Rule 144 rather than under this prospectus. The selling stockholders have advised us that there is no underwriter or coordinating broker acting in connection with the proposed sale of the resale securities by the selling stockholders.
We agreed to keep this prospectus effective until the earlier of (i) the date on which the securities may be resold by the selling stockholders without registration and without regard to any volume or manner-of-sale limitations by reason of Rule 144 (assuming a cashless exercise of each Series A, B, and C Warrant), without the requirement for the Company to be in compliance with the current public information under Rule 144 under the Securities Act or any other rule of similar effect or (ii) all of the securities have been sold pursuant to this prospectus or Rule 144 under the Securities Act or any other rule of similar effect. The resale securities will be sold only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, the resale securities covered hereby may not be sold unless they have been registered or qualified for sale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
Under applicable rules and regulations under the Exchange Act, any person engaged in the distribution of the resale securities may not simultaneously engage in market making activities with respect to the common stock for the applicable restricted period, as defined in Regulation M under the Securities Act, prior to the commencement of the distribution. In addition, the selling stockholders will be subject to applicable provisions of the Exchange Act and the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and sales of the common stock by the selling stockholders or any other person. We will make copies of this prospectus available to the selling stockholders and have informed them of the need to deliver a copy of this prospectus to each purchaser at or prior to the time of the sale (including by compliance with Rule 172 under the Securities Act).
The holder of the Maxim Warrant, Maxim Partners LLC, is an affiliate of the placement agent. The Placement Agent Agreement provides that we will indemnify the placement agent, its affiliates and persons who control the placement agent, against certain liabilities caused by, arising out of, or in connection with the placement agent’s acting on our behalf, and if we are unable to provide this indemnification to contribute to payments that the placement agent may be required to make in respect of those liabilities.
Affiliations
The placement agent and certain of its affiliates are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. The placement agent and certain of its affiliates have, from time to time, performed, and may in the future perform, various financial advisory and investment banking services for us, for which they received or will receive customary fees and expenses. We are not under any contractual obligation to engage the placement agent to provide investment banking, lending, asset management and financial advisory services to us in the future. If the placement agent provides such services to us after this offering, we may pay the placement agent fair and reasonable fees that would be determined at that time in an arm’s length negotiation, and pursuant to applicable FINRA rules.
In the ordinary course of their various business activities, the placement agent and certain of its affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for its own account and for the accounts of its customers, and such investment and securities activities may involve securities or instruments of us. The placement agent and certain of its affiliates may also make investment recommendations or publish or express independent research views in respect of such securities or instruments and may at any time hold, or recommend to clients that they acquire, long or short positions in such securities and instruments.
LEGAL MATTERS
The validity of the securities offered by this prospectus will be passed upon by Womble Carlyle Sandridge & Rice LLP, Atlanta, Georgia.
EXPERTS
The consolidated financial statements of GeoVax, Labs, Inc. and subsidiary as of December 31, 2014 and 2013 appearing in this Prospectus and Registration Statement have been audited by Porter Keadle Moore, LLC, an independent registered public accounting firm, as stated in their report appearing elsewhere herein, and are included in reliance upon such report and upon the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement, several supplements, and one post-effective amendment to the registration statement on Form S-1 under the Securities Act with respect to the securities offered by this prospectus. This prospectus does not contain all of the information included in the registration statement as so supplemented and amended. For further information pertaining to us and our securities, you should refer to the registration statement and the exhibits and schedules filed with the registration statement. Whenever we make reference in this prospectus to any of our contracts, agreements or other documents, the references are not necessarily complete, and you should refer to the exhibits attached to the registration statement for copies of the actual contract, agreement or other document.
We are subject to the informational requirements of the Securities Exchange Act and file annual, quarterly and current reports, proxy statements and other information with the SEC. You can read our SEC filings, including the registration statement, over the Internet at the SEC’s website at www.sec.gov. You may also read and copy any document we file with the SEC at its Public Reference Room at 100 F Street, N.E., Washington, D.C., 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330.
GEOVAX LABS, INC.
INDEX TO 2014 CONSOLIDATED FINANCIAL STATEMENTS
|
Report of Independent Registered Public Accounting Firm |
F-2 |
|
Consolidated Balance Sheets as of December 31, 2014 and 2013 |
F-3 |
|
Consolidated Statements of Operations for the years ended December 31, 2014, 2013 and 2012 |
F-4 |
|
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2014, 2013 and 2012 |
F-5 |
|
Consolidated Statements of Cash Flows for the years ended December 31, 2014, 2013 and 2012 |
F-6 |
|
Notes to Consolidated Financial Statements |
F-7 |
|
Financial Statement Schedule: |
|
|
Schedule II – Valuation and Qualifying Accounts for the years ended |
|
|
December 31, 2014, 2013 and 2012 |
F-18 |

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors
GeoVax Labs, Inc.
Atlanta, Georgia
We have audited the accompanying consolidated balance sheets of GeoVax Labs, Inc. and subsidiary (the “Company”) as of December 31, 2014 and 2013, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014. Our audits also included the financial statement schedule of the Company listed in Item 15(a). These financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion of the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of GeoVax Labs, Inc. and subsidiary as of December 31, 2014 and 2013, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
/s/ Porter Keadle Moore, LLC
Atlanta, Georgia
March 20, 2015
235 Peachtree Street NE | Suite 1800 | Atlanta, Georgia 30303 | Phone 404.588.4200 | Fax 404.588.4222
GEOVAX LABS, INC.
CONSOLIDATED BALANCE SHEETS
|
December 31, |
||||||||
|
2014 |
2013 |
|||||||
|
ASSETS |
||||||||
|
Current assets: |
||||||||
|
Cash and cash equivalents |
$ | 1,101,651 | $ | 2,513,861 | ||||
|
Grant funds receivable |
79,341 | 140,909 | ||||||
|
Prepaid expenses and other current assets |
44,503 | 43,569 | ||||||
|
Total current assets |
1,225,495 | 2,698,339 | ||||||
|
Property and equipment, net |
96,693 | 120,227 | ||||||
|
Other assets |
11,010 | 21,010 | ||||||
|
Total assets |
$ | 1,333,198 | $ | 2,839,576 | ||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
|
Current liabilities: |
||||||||
|
Accounts payable |
$ | 55,616 | $ | 155,943 | ||||
|
Accrued expenses |
52,490 | 96,406 | ||||||
|
Amounts payable to Emory University (a related party) (Note 12) |
78,917 | 60,000 | ||||||
|
Total current liabilities |
187,023 | 312,349 | ||||||
|
Commitments (Note 6) |
||||||||
|
Stockholders’ equity: |
||||||||
|
Preferred stock, $.01 par value: |
||||||||
|
Authorized shares – 10,000,000 |
||||||||
|
Series A convertible preferred stock, $1,000 stated value; -0- and 71 shares issued and outstanding at December 31, 2014 and 2013, respectively |
- | 60,586 | ||||||
|
Series B convertible preferred stock, $1,000 stated value; 100 and 1,650 shares issued and outstanding at December 31, 2014 and 2013, respectively |
76,095 | 1,255,569 | ||||||
|
Common stock, $.001 par value: |
||||||||
|
Authorized shares – 75,000,000 |
||||||||
|
Issued and outstanding shares – 31,950,813 and 23,765,180 at December 31, 2014 and 2013, respectively |
31,951 | 23,765 | ||||||
|
Additional paid-in capital |
30,823,769 | 28,239,392 | ||||||
|
Accumulated deficit |
(29,785,640 | ) | (27,052,085 | ) | ||||
|
Total stockholders’ equity |
1,146,175 | 2,527,227 | ||||||
|
Total liabilities and stockholders’ equity |
$ | 1,333,198 | $ | 2,839,576 | ||||
See accompanying notes to consolidated financial statements.
GEOVAX LABS. INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
|
Years Ended December 31, |
||||||||||||
|
2014 |
2013 |
2012 |
||||||||||
|
Grant revenue |
$ | 882,956 | $ | 2,417,550 | $ | 2,657,327 | ||||||
|
Operating expenses: |
||||||||||||
|
Research and development |
1,812,969 | 2,914,878 | 3,043,522 | |||||||||
|
General and administrative |
1,807,605 | 1,792,160 | 1,752,765 | |||||||||
|
Total operating expenses |
3,620,574 | 4,707,038 | 4,796,287 | |||||||||
|
Loss from operations |
(2,737,618 | ) | (2,289,488 | ) | (2,138,960 | ) | ||||||
|
Other income: |
||||||||||||
|
Interest income |
4,063 | 4,545 | 3,820 | |||||||||
|
Total other income |
4,063 | 4,545 | 3,820 | |||||||||
|
Net loss |
$ | (2,733,555 | ) | $ | (2,284,943 | ) | $ | (2,135,140 | ) | |||
|
Basic and diluted: |
||||||||||||
|
Loss per common share |
$ | (0.10 | ) | $ | (0.11 | ) | $ | (0.12 | ) | |||
|
Weighted average shares outstanding |
26,645,140 | 21,212,327 | 18,315,669 | |||||||||
See accompanying notes to consolidated financial statements.
GEOVAX LABS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
|
Series A Convertible |
Series B Convertible |
Additional |
Total |
|||||||||||||||||||||||||||||||||
|
Preferred Stock |
Preferred Stock |
Common Stock |
Paid-in |
Accumulated |
Stockholders’ |
|||||||||||||||||||||||||||||||
|
Shares |
Amount |
Shares |
Amount |
Shares |
Amount |
Capital |
Deficit |
Equity |
||||||||||||||||||||||||||||
|
Balance at December 31, 2011 |
- | $ | - | - | $ | - | 16,442,611 | $ | 16,443 | $ | 23,319,166 | $ | (22,632,002 | ) | $ | 703,607 | ||||||||||||||||||||
|
Sale of common stock for cash |
- | - | - | - | 407,999 | 408 | 272,952 | - | 273,360 | |||||||||||||||||||||||||||
|
Sale of convertible preferred stock and warrants for cash |
2,200 | 871,614 | - | - | - | - | 1,127,418 | - | 1,999,032 | |||||||||||||||||||||||||||
|
Conversion of preferred stock to common stock |
(1,412 | ) | (559,418 | ) | - | - | 1,882,667 | 1,882 | 557,536 | - | - | |||||||||||||||||||||||||
|
Stock-based compensation expense |
- | - | - | - | - | - | 310,076 | - | 310,076 | |||||||||||||||||||||||||||
|
Net loss for the year ended December 31, 2012 |
- | - | - | - | - | - | - | (2,135,140 | ) | (2,135,140 | ) | |||||||||||||||||||||||||
|
Balance at December 31, 2012 |
788 | 312,196 | - | - | 18,733,277 | 18,733 | 25,587,148 | (24,767,142 | ) | 1,150,935 | ||||||||||||||||||||||||||
|
Sale of common stock for cash upon warrant exercise |
- | - | - | - | 2,933,333 | 2,933 | 1,640,400 | - | 1,643,333 | |||||||||||||||||||||||||||
|
Issuance of common stock for services |
- | - | - | - | 50,000 | 50 | 20,450 | - | 20,500 | |||||||||||||||||||||||||||
|
Sale of convertible preferred stock for cash |
- | 360,229 | 1,650 | 1,255,569 | - | - | - | - | 1,615,798 | |||||||||||||||||||||||||||
|
Conversion of preferred stock to common stock |
(717 | ) | (611,839 | ) | - | - | 2,048,570 | 2,049 | 609,790 | - | - | |||||||||||||||||||||||||
|
Stock-based compensation expense |
- | - | - | - | - | - | 381,604 | - | 381,604 | |||||||||||||||||||||||||||
|
Net loss for the year ended December 31, 2013 |
- | - | - | - | - | - | - | (2,284,943 | ) | (2,284,943 | ) | |||||||||||||||||||||||||
|
Balance at December 31, 2013 |
71 | 60,586 | 1,650 | 1,255,569 | 23,765,180 | 23,765 | 28,239,392 | (27,052,085 | ) | 2,527,227 | ||||||||||||||||||||||||||
|
Sale of common stock for cash upon warrant exercise |
- | - | - | - | 3,176,000 | 3,176 | 870,224 | - | 873,400 | |||||||||||||||||||||||||||
|
Issuance of common stock for services |
- | - | - | - | 378,205 | 378 | 99,622 | - | 100,000 | |||||||||||||||||||||||||||
|
Conversion of preferred stock to common stock |
(71 | ) | (60,586 | ) | (1,550 | ) | (1,179,474 | ) | 4,631,428 | 4,632 | 1,235,428 | - | - | |||||||||||||||||||||||
|
Stock-based compensation expense |
- | - | - | - | - | - | 379,103 | - | 379,103 | |||||||||||||||||||||||||||
|
Net loss for the year ended December 31, 2014 |
- | - | - | - | - | - | - | (2,733,555 | ) | (2,733,555 | ) | |||||||||||||||||||||||||
|
Balance at December 31, 2014 |
- | $ | - | 100 | $ | 76,095 | 31,950,813 | $ | 31,951 | $ | 30,823,769 | $ | (29,785,640 | ) | $ | 1,146,175 | ||||||||||||||||||||
See accompanying notes to consolidated financial statements.
GEOVAX LABS. INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
Years Ended December 31, |
||||||||||||
|
2014 |
2013 |
2012 |
||||||||||
|
Cash flows from operating activities: |
||||||||||||
|
Net loss |
$ | (2,733,555 | ) | $ | (2,284,943 | ) | $ | (2,135,140 | ) | |||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
|
Depreciation and amortization |
69,037 | 78,862 | 93,643 | |||||||||
|
Stock-based compensation expense, including common stock issued for services |
479,103 | 402,104 | 310,076 | |||||||||
|
Changes in assets and liabilities: |
||||||||||||
|
Grant funds receivable |
61,568 | 125,339 | (82,733 | ) | ||||||||
|
Prepaid expenses and other current assets |
(934 | ) | (1,268 | ) | (12,593 | ) | ||||||
|
Accounts payable and accrued expenses |
(125,326 | ) | (14,686 | ) | (614,500 | ) | ||||||
|
Total adjustments |
483,448 | (590,351 | ) | (306,107 | ) | |||||||
|
Net cash used in operating activities |
(2,250,107 | ) | (1,694,592 | ) | (2,441,247 | ) | ||||||
|
Cash flows from investing activities: |
||||||||||||
|
Purchase of property and equipment |
(35,503 | ) | (86,603 | ) | - | |||||||
|
Net cash used in investing activities |
(35,503 | ) | (86,603 | ) | - | |||||||
|
Cash flows from financing activities: |
||||||||||||
|
Proceeds from sale of common stock |
873,400 | 1,643,333 | 310,160 | |||||||||
|
Proceeds from sale of preferred stock |
- | 1,615,798 | 1,999,032 | |||||||||
|
Net cash provided in financing activities |
873,400 | 3,259,131 | 2,309,192 | |||||||||
|
Net increase (decrease) in cash and cash equivalents |
(1,412,210 | ) | 1,477,936 | (132,055 | ) | |||||||
|
Cash and cash equivalents at beginning of period |
2,513,861 | 1,035,925 | 1,167,980 | |||||||||
|
Cash and cash equivalents at end of period |
$ | 1,101,651 | $ | 2,513,861 | $ | 1,035,925 | ||||||
Supplemental disclosure of non-cash financing activities:
As discussed in Note 7, during the year ended December 31, 2014, 71 shares of Series A Convertible Preferred Stock were converted into 202,857 shares of common stock and 1,550 shares of Series B Convertible Preferred Stock were converted into 4,428,571 shares of common stock. During the year ended December 31, 2013, 717 shares of Series A Convertible Preferred Stock were converted into 2,048,570 shares of common stock. During the year ended December 31, 2012, 1,412 shares of Series A Convertible Preferred Stock were converted into 1,882,667 shares of common stock.
See accompanying notes to consolidated financial statements.
GEOVAX LABS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years Ended December 31, 2014, 2013 and 2012
|
|
|
1. |
Description of Business |
GeoVax Labs, Inc. (“GeoVax” or the “Company”), is a clinical-stage biotechnology company developing innovative human vaccines using our novel DNA/MVA platform technology. Our lead programs are focused on Ebola virus and Human Immunodeficiency Virus (HIV). Our vaccine delivery technology generates virus-like particles (VLPs) that are effective at eliciting safe and effective immune responses. All of the clinical trials for our preventive HIV vaccine have been conducted by the HIV Vaccine Trials Network (HVTN) with funding from the National Institutes of Health (NIH). Our proprietary Ebola vaccine technology has been developed internally, while our HIV vaccine technology was developed in collaboration with Emory University, the NIH, and the Centers for Disease Control and Prevention (CDC) and is exclusively licensed to us.
Our most advanced vaccines under development address the clade B subtype of the HIV virus that is most prevalent in the developed world – primarily North America and Western Europe. Our preventive clade B HIV vaccine has successfully completed Phase 2a clinical trials and we are currently planning the next stage of human clinical testing. We are also planning clinical trials to evaluate our clade B HIV vaccine as an immunotherapy agent for individuals already infected with HIV. We have begun early-stage preclinical studies to develop HIV vaccine candidates for the clade C subtype of HIV prevalent in the developing world. Our Ebola vaccine development efforts were initiated in 2014 and we expect to conduct preclinical animal studies during 2015, with the goal of beginning human clinical testing in 2016.
We operate in a highly regulated and competitive environment. The manufacturing and marketing of pharmaceutical products require approval from, and are subject to, ongoing oversight by the Food and Drug Administration (FDA) in the United States, by the European Medicines Agency (EMA) in the European Union, and by comparable agencies in other countries. Obtaining approval for a new pharmaceutical product is never certain and may take many years and may involve expenditure of substantial resources. Our goal is to build a profitable company by generating income from products we develop and commercialize, either alone or with one or more potential strategic partners.
GeoVax is incorporated under the laws of the State of Delaware and our principal offices are located in Smyrna, Georgia (metropolitan Atlanta area).
|
|
|
2. |
Summary of Significant Accounting Policies |
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of GeoVax Labs, Inc. together with those of our wholly-owned subsidiary, GeoVax, Inc. All intercompany transactions have been eliminated in consolidation.
Basis of Presentation
The accompanying consolidated financial statements have been prepared assuming that we will continue as a going concern, which contemplates realization of assets and the satisfaction of liabilities in the normal course of business for the twelve-month period following the date of these consolidated financial statements. We are devoting substantially all of our present efforts to research and development. We have funded our activities to date from government grants and clinical trial assistance, and from sales of our equity securities. We will continue to require substantial funds to continue these activities. We believe that our existing cash resources, combined with the proceeds from the NIH grants discussed in Note 5 and the net proceeds of the financing transaction discussed in Note 14, will be sufficient to fund our planned operations through the first quarter of 2016.
We expect we will need to raise additional funds to significantly advance our vaccine development programs and we are currently exploring sources of non-dilutive capital through government grant programs and clinical trial support. However, additional funding may not be available on favorable terms or at all. If we fail to obtain additional capital when needed, we may be required to delay, scale back, or eliminate some or all of our research and development programs as well as reduce our general and administrative expenses.
In August 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update 2014-15, Presentation of Financial Statements – Going Concern (“ASU 2014-15”), which requires management of all entities to evaluate whether there are conditions and events that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the financial statements are issued, and to make certain disclosures if it concludes that substantial doubt exists or when its plans alleviate substantial doubt about the entity’s ability to continue as a going concern. ASU 2014-15 is effective for the Company for annual reporting periods beginning in 2016 and for interim reporting periods starting in the first quarter of 2017. We are currently evaluating the impact of the adoption of ASU 2014-15 on our financial statements.
Development-Stage Enterprise
In June 2014, the FASB issued Accounting Standards Update 2014-10, Development Stage Entities (Topic 915) ("ASU 2014-10"). The amendments in ASU 2014-10 remove the definition of a development stage entity from Topic 915, thereby removing the distinction between development stage entities and other reporting entities from U.S. generally accepted accounting principles (“GAAP”). In addition, the amendments eliminate the requirements for development stage entities to (1) present inception-to-date information on the statements of income, cash flows, and shareholder’s equity, (2) label the financial statements as those of a development stage entity, (3) disclose a description of the development stage activities in which the entity is engaged, and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. For public business entities, those amendments are effective for annual reporting periods beginning after December 15, 2014, and interim periods therein. Early adoption is permitted. We have evaluated this accounting standard and determined it to have a material impact on our financial statements. We adopted ASU-2014-10 effective June 30, 2014 and the effects of the adoption are reflected in our consolidated financial statements and footnotes contained herein.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results may differ from those estimates.
Cash and Cash Equivalents
We consider all highly liquid investments with a maturity of three months or less when purchased to be cash equivalents. Our cash and cash equivalents consist primarily of bank deposits and money market accounts. The recorded values approximate fair market values due to the short maturities.
Fair Value of Financial Instruments and Concentration of Credit Risk
Financial instruments that subject us to concentration of credit risk consist primarily of cash and cash equivalents, which are maintained by a high credit quality financial institution. The carrying values reported in the balance sheets for cash and cash equivalents approximate fair values.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation and amortization. Expenditures for maintenance and repairs are charged to operations as incurred, while additions and improvements are capitalized. We calculate depreciation using the straight-line method over the estimated useful lives of the assets which range from three to five years. We amortize leasehold improvements using the straight-line method over the term of the related lease.
Impairment of Long-Lived Assets
We review long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of the assets to the future net cash flows expected to be generated by such assets. If we consider such assets to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceeds the expected future net cash flows from the assets.
Accrued Liabilities
As part of the process of preparing our financial statements, we estimate expenses that we believe we have incurred, but have not yet been billed by our third party vendors. This process involves identifying services and activities that have been performed by such vendors on our behalf and estimating the level to which they have been performed and the associated cost incurred for such service as of each balance sheet date.
Net Loss Per Share
Basic and diluted loss per common share are computed based on the weighted average number of common shares outstanding. Common share equivalents consist of common shares issuable upon conversion of convertible preferred stock, and upon exercise of stock options and stock purchase warrants. All common share equivalents are excluded from the computation of diluted loss per share since the effect would be anti-dilutive. Common share equivalents which could potentially dilute basic earnings per share in the future, and which were excluded from the computation of diluted loss per share, totaled approximately 6.6 million, 14.4 million, and 13.3 million at December 31, 2014, 2013 and 2012, respectively.
Revenue Recognition
We recognize revenue in accordance with U.S. Securities and Exchange Commission (“SEC”) Staff Accounting Bulletin No. 101, Revenue Recognition in Financial Statements, as amended by Staff Accounting Bulletin No. 104, Revenue Recognition, (“SAB 104”). SAB 104 provides guidance in applying GAAP to revenue recognition issues, and specifically addresses revenue recognition for upfront, nonrefundable fees received in connection with research collaboration agreements. During 2014, 2013 and 2012, our revenue consisted of grant funding received from the NIH (see Note 5). Revenue from these arrangements is approximately equal to the costs incurred and is recorded as income as the related costs are incurred.
In May 2014, the FASB issued Accounting Standards Update 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”), which creates a new Topic, Accounting Standards Codification Topic 606. The standard is principle-based and provides a five-step model to determine when and how revenue is recognized. The core principle is that an entity should recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 is effective for the Company beginning in 2017 and allows for either full retrospective adoption or modified retrospective adoption. We are currently evaluating the impact of the adoption of ASU 2014-09 on our financial statements.
Research and Development Expense
Research and development expense primarily consists of costs incurred in the discovery, development, testing and manufacturing of our product candidates. These expenses consist primarily of (i) fees paid to third-party service providers to perform, monitor and accumulate data related to our preclinical studies and clinical trials, (ii) costs related to sponsored research agreements, (iii) the costs to procure and manufacture materials used in clinical trials, (iv) laboratory supplies and facility-related expenses to conduct development, and (v) salaries, benefits, and stock-based compensation for personnel. These costs are charged to expense as incurred.
Patent Costs
Our expenditures relating to obtaining and protecting patents are charged to expense when incurred, and are included in general and administrative expense.
Period to Period Comparisons
Our operating results are expected to fluctuate for the foreseeable future. Therefore, period-to-period comparisons should not be relied upon as predictive of the results for future periods.
Income Taxes
We account for income taxes using the liability method. Under this method, deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted rates in effect for the year in which temporary differences are expected to be recovered or settled. Deferred tax assets are reduced by a valuation allowance unless, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will be realized.
Stock-Based Compensation
We account for stock-based transactions in which the Company receives services from employees, directors or others in exchange for equity instruments based on the fair value of the award at the grant date. Compensation cost for awards of common stock is estimated based on the price of the underlying common stock on the date of issuance. Compensation cost for stock options or warrants is estimated at the grant date based on each instrument’s fair value as calculated by the Black-Scholes option pricing model. We recognize stock-based compensation cost as expense ratably on a straight-line basis over the requisite service period for the award. See Note 9 for additional stock-based compensation information.
Recent Accounting Pronouncements
Except as discussed above, there have been no recent accounting pronouncements or changes in accounting pronouncements which we expect to have a material impact on our financial statements, nor do we believe that any recently issued, but not yet effective, accounting standards if currently adopted would have a material effect on our financial statements.
|
|
|
3. |
Property and Equipment |
Property and equipment as shown on the accompanying Consolidated Balance Sheets is composed of the following as of December 31, 2014 and 2013:
|
2014 |
2013 |
|||||||
|
Laboratory equipment |
$ | 510,106 | $ | 474,602 | ||||
|
Leasehold improvements |
115,605 | 115,605 | ||||||
|
Other furniture, fixtures & equipment |
28,685 | 28,685 | ||||||
|
Total property and equipment |
654,396 | 618,892 | ||||||
|
Accumulated depreciation and amortization |
(557,703 | ) | (498,665 | ) | ||||
|
Property and equipment, net |
$ | 96,693 | $ | 120,227 | ||||
Depreciation and amortization expense was $59,037, $68,862, and $73,720 during the years ended December 31, 2014, 2013 and 2012, respectively.
|
|
|
4. |
Other Assets |
Other assets as shown on the accompanying Consolidated Balance Sheets include the following as of December 31, 2014 and 2013:
|
2014 |
2013 |
|||||||
|
Technology licenses |
$ | 248,855 | $ | 248,855 | ||||
|
Deposits |
11,010 | 11,010 | ||||||
|
Accumulated amortization – technology licenses |
(248,855 | ) | (238,855 | ) | ||||
|
Total other assets |
$ | 11,010 | $ | 21,010 | ||||
Amortization expense related to technology licenses was $10,000, $10,000, and $19,923 during the years ended December 31, 2014, 2013 and 2012, respectively.
|
|
|
5. |
Government Grants |
We record revenue associated with government grants as the related costs and expenses are incurred and such revenue is reported as a separate line item in our statements of operations. Grant revenues relate to grants from the NIH in support of our HIV vaccine development activities. During 2014, 2013, and 2012, we recorded $882,956, $2,417,550, and $2,657,327, respectively, of revenue associated with these grants. Additional detail concerning our grant revenues is discussed below.
In September 2007, the NIH awarded us a grant entitled “GM-CSF-Adjuvanted Clade C DNA/MVA and MVA/MVA Vaccines”. The aggregate award (including subsequent amendments) totaled approximately $20.4 million. We recorded grant revenues of $624,689, $833,390, and $2,227,924 for the years ended December 31, 2014, 2013 and 2012, respectively, related to this grant, and there is $75,464 of unrecognized grant funds remaining and available for use pursuant to this grant as of December 31, 2014.
In September 2012, the NIH awarded us a grant entitled “Immunogens and Manufacturing” to support our HIV/AIDS vaccine development program. The grant award was for approximately $1.9 million. We recorded grant revenues of $-0-, $1,429,597, and $429,403 for the years ended December 31, 2014, 2013 and 2012, respectively, related to this grant, and all funding pursuant to this grant has been utilized as of December 31, 2014.
In July 2013, the NIH awarded us a Small Business Innovative Research (SBIR) grant entitled “Enhancing Protective Antibody Responses for a GM-CSF Adjuvanted HIV Vaccine.” The initial grant award was $276,690 for the first year of a two year project period beginning August 1, 2013. In July 2014, the NIH awarded us $289,641 for the second year of the project period. We recorded grant revenues of $258,267, $154,563, and $-0- for the years ended December 31, 2014, 2013 and 2012, respectively, related to this grant, and there is $153,501 of unrecognized grant funds remaining and available for use pursuant to this grant as of December 31, 2014.
|
|
|
6. |
Commitments |
Lease Agreements
We lease approximately 8,400 square feet of office and laboratory space located in Smyrna, Georgia (metropolitan Atlanta). Rent expense for the years ended December 31, 2014, 2013 and 2012 was $117,084, $117,879, and $118,801, respectively. The original 62-month lease term expired on December 31, 2014 and we have renewed the lease for an additional 12 months, with two successive 12-month renewal options. Future minimum lease payments total $146,092 in 2015.
Other Commitments
In the normal course of business, we may enter into various firm purchase commitments related to production and testing of our vaccine material, conduct of clinical trials, and other research-related activities. As of December 31, 2014, we had approximately $151,439 of unrecorded outstanding purchase commitments to our vendors and subcontractors, all of which we expect will be due in 2015.
|
7. |
Preferred Stock |
Series A Convertible Preferred Stock
In March 2012, we issued 2,200 shares of our Series A Convertible Preferred Stock, $1,000 stated value (“Series A Preferred Stock”), which were originally convertible into 2,933,333 shares of our common stock, and warrants to purchase up to 8,799,999 shares of our common stock for gross proceeds of $2.2 million. Net proceeds, after deduction of placement agent fees and other expenses, were approximately $2.0 million.
Each share of Series A Preferred Stock was entitled to a liquidation preference equal to the initial purchase price, had no voting rights, and was not entitled to a dividend. The Series A Preferred Stock was convertible at any time at the option of the holders into shares of our common stock. The initial conversion price was $0.75 and during 2012, 1,412 of the Series A Preferred Shares were converted at this price into an aggregate of 1,882,667 shares of our common stock. Effective December 11, 2013, we amended the designation of the Series A Preferred Stock in connection with the issuance of our Series B Convertible Preferred Stock (see discussion below). The amendment had the effect of reducing the conversion price of the then-outstanding Series A Preferred Stock to $0.35 and during the remainder of 2013, 717 shares of the Series A Preferred Stock were converted at this price into an aggregate of 2,048,570 shares of our common stock. The remaining 71 shares of Series A Preferred Stock were converted into 202,857 shares of our common stock in January 2014, and there are no shares of Series A Preferred Stock outstanding at December 31, 2014.
We assessed the Series A Preferred Stock and the related warrants under ASC Topic 480, “Distinguishing Liabilities from Equity” (“ASC 480”), ASC Topic 815, “Derivatives and Hedging” (“ASC 815”), and ASC Topic 470, “Debt” (“ASC 470”). The preferred stock contains an embedded feature allowing an optional conversion by the holder into common stock which meets the definition of a derivative. However, we determined that the preferred stock is an “equity host” (as described by ASC 815) for purposes of assessing the embedded derivative for potential bifurcation and that the optional conversion feature is clearly and closely associated to the preferred stock host; therefore the embedded derivative does not require bifurcation and separate recognition under ASC 815. We determined there to be a beneficial conversion feature (“BCF”) requiring recognition at its intrinsic value. Since the conversion option of the preferred stock was immediately exercisable, the amount allocated to the BCF was immediately accreted to preferred dividends, resulting in an increase in the carrying value of the preferred stock. We also assessed the warrants issued in connection with the financing under ASC 815 and determined that they did not initially meet the definition of a derivative, but will require evaluation on an on-going basis. As of December 31, 2014, we determined that the warrants still did not meet the definition of a derivative.
The following is a summary of the allocation of net proceeds and reconciliation to the carrying value of the Series A Preferred Stock at December 31, 2014:
|
Net proceeds |
$ | 1,999,032 | ||
|
Fair value of warrants (recorded to Additional Paid-in Capital) |
(1,127,418 | ) | ||
|
Beneficial conversion feature (recorded to Additional Paid-in Capital) |
(762,667 | ) | ||
|
Net proceeds allocated to preferred stock |
108,947 | |||
|
Accretion of beneficial conversion feature (deemed dividend) |
762,667 | |||
|
Initial carrying value of preferred stock |
871,614 | |||
|
Accretion of beneficial conversion feature (deemed dividend) related to issuance of Series B Convertible Preferred Stock |
360,229 | |||
|
Conversions to common stock |
(1,231,843 | ) | ||
|
Carrying value at December 31, 2014 |
$ | -0 | - |
Series B Convertible Preferred Stock
In December 2013, we issued 1,650 shares of our Series B Convertible Preferred Stock, $1,000 stated value (“Series B Preferred Stock”), which was originally convertible into 4,714,286 shares of our common stock, for gross proceeds of $1.65 million. Net proceeds, after deduction of transaction expenses, were approximately $1.6 million. No warrants were issued in connection with the transaction.
Each share of Series B Preferred Stock has a liquidation preference equal to the initial purchase price, has no voting rights, and is not entitled to a dividend. The Series B Preferred Stock may be converted at any time at the option of the holders into shares of our common stock at a conversion price of $0.35. During 2014, 1,550 shares of the Series B Preferred Stock were converted into 4,428,571 shares of our common stock. As of December 31, 2014, there were 100 shares of Series B Preferred Stock outstanding, convertible into 285,714 shares of our common stock.
In conjunction with the issuance of the Series B Preferred Stock, we entered into an agreement with the holders of the Series A Preferred Stock to amend the designation of the Series A Preferred Stock. The amendment had the effect of reducing the conversion price of the then-outstanding 788 Series A Preferred Shares from $0.75 to $0.35.
We assessed the Series B Preferred Stock using the same methodology as for the Series A Preferred Stock (see discussion above), and resulting in the same determinations. The following is a summary of the allocation of net proceeds and reconciliation to the carrying value of the Series B Preferred Stock at December 31, 2014:
|
Net proceeds |
$ | 1,615,798 | ||
|
Beneficial conversion feature – Series A Preferred Stock (recorded to Additional Paid-in Capital) |
(360,229 | ) | ||
|
Beneficial conversion feature – Series B Preferred Stock (recorded to Additional Paid-in Capital) |
(754,286 | ) | ||
|
Net proceeds allocated to preferred stock |
501,283 | |||
|
Accretion of beneficial conversion feature (deemed dividend) |
754,286 | |||
|
Conversions to common stock |
(1,179,474 | ) | ||
|
Carrying value at December 31, 2014 |
$ | 76,095 |
|
|
|
8. |
Common Stock |
Common Stock Transactions
During December 2011 and January 2012, we sold an aggregate of 1,066,519 of our common stock to a group of individual accredited investors (including members of our board of directors and management --see Note 12) for an aggregate purchase price of $714,570. We also issued to the investors five-year warrants to purchase an aggregate of 1,599,784 shares of common stock at a price of $1.00 per share.
During January and May 2013, we issued an aggregate of 2,933,333 shares of our common stock pursuant to the exercise of certain stock purchase warrants, resulting in total net proceeds of $1,643,333 (see “Stock Purchase Warrants” below).
During October 2013, we issued 50,000 shares of our common stock to a consultant in exchange for services and recorded general and administrative expense of $20,500 related to the issuance (see Note 9 – “Other Non-Employee Stock-Based Compensation Expense”).
During July and November 2014, we issued an aggregate of 378,205 shares of our common stock to a consultant in exchange for services and recorded aggregate general and administrative expense of $100,000 related to the issuances (see see Note 9 – “Other Non-Employee Stock-Based Compensation Expense”).
During October 2014, we issued an aggregate of 3,176,000 shares of our common stock pursuant to the exercise of certain stock purchase warrants, resulting in total net proceeds of $873,400 (see “Stock Purchase Warrants” below).
During 2014, 2013 and 2012, we issued shares of our common stock related to conversions of our Series A and Series B Preferred Stock (see Note 7) as follows:
|
2014 |
2013 |
2012 |
||||||||||
|
Conversion of Series A Preferred Shares |
202,857 | 2,048,570 | 1,882,667 | |||||||||
|
Conversion of Series B Preferred Shares |
4,428,571 | -0- | -0- | |||||||||
Stock Purchase Warrants
As of December 31, 2014, we have the following stock purchase warrants outstanding:
|
Expiration Date |
Number of Shares |
Weighted Average Exercise Price |
||||||
|
December 31, 2016 |
1,806,159 | $ | 1.00 | |||||
|
January 16, 2017 |
45,000 | 1.00 | ||||||
|
January 31, 2017 |
567,001 | 1.00 | ||||||
|
March 21, 2017 |
2,690,666 | 0.35 | ||||||
|
Total Outstanding at December 31, 2014 |
5,108,826 | $ | 0.66 | |||||
During January 2013, we reduced the exercise price of warrants to purchase 2,933,333 shares of our common stock from $0.75 to $0.60 per share. In consideration for the reduction of the exercise price, the holders of the warrants immediately exercised 1,766,667 of the warrants for cash, resulting in total proceeds to the Company of $1,060,000. We also extended the expiration date of the 1,166,666 unexercised warrants from March 21, 2013 to May 21, 2013. We recorded general and administrative expense of $218,551 associated with these warrant modifications. During May 2013, we reduced the exercise price of the 1,166,666 remaining warrants from $0.60 to $0.50 per share. In consideration for the reduction of the exercise price, the holders of the warrants immediately exercised all of the remaining warrants for cash, resulting in total proceeds to the Company of $583,333. We recorded general and administrative expense of $19,617 associated with this warrant modification.
During September 2014, we reduced the exercise price of warrants to purchase 818,376 shares of our common stock from $16.50 to $1.00 per share, and extended the expiration dates from December 31, 2014 to December 31, 2016. We recorded general and administrative expense of $39,711 associated with these modifications.
During October 2014, we entered into an agreement with certain holders of warrants to purchase shares of our common stock with respect to the payment to them of a warrant exercise fee of $0.075 per share for each share purchased upon exercise of warrants held by them. In exchange for the fee, they immediately exercised warrants for an aggregate of 3,176,000 shares of our common stock, resulting in proceeds to us of $873,400 (net of the exercise fee).
Common Stock Reserved
A summary of common stock reserved for future issuance as of December 31, 2014 is as follows:
|
Stock Purchase Warrants |
5,108,826 | |||
|
Stock Option Plan |
1,197,529 | |||
|
Series B Convertible Preferred Stock |
285,715 | |||
|
Total |
6,592,070 |
|
9. |
Stock-Based Compensation |
Stock Option Plan
In 2006, we adopted the GeoVax Labs, Inc. 2006 Equity Incentive Plan (the “Stock Option Plan”) for the granting of qualified incentive stock options (“ISO’s”), nonqualified stock options, restricted stock awards or restricted stock bonuses to employees, officers, directors, consultants and advisors of the Company. The exercise price for any option granted may not be less than fair value (110% of fair value for ISO’s granted to certain employees). Options granted under the Stock Option Plan have a maximum ten-year term and generally vest over three years. The Company has reserved 1,200,000 shares of its common stock for issuance under the Stock Option Plan.
Certain information concerning the Stock Option Plan as of December 31, 2014, and a summary of activity during the year then ended is presented below:
|
Number of Shares |
Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term (yrs) |
Aggregate Intrinsic Value |
|||||||||||||
|
Outstanding at December 31, 2013 |
1,197,044 | $ | 3.79 | |||||||||||||
|
Granted |
217,500 | 0.17 | ||||||||||||||
|
Exercised |
- | - | ||||||||||||||
|
Forfeited or expired |
(231,444 | ) | 1.89 | |||||||||||||
|
Outstanding at December 31, 2014 |
1,183,100 | $ | 3.50 | 6.6 | $ | -0- | ||||||||||
|
Exercisable at December 31, 2014 |
785,594 | $ | 5.09 | 5.2 | $ | -0- | ||||||||||
Additional information concerning our stock options for the years ended December 31, 2014, 2013 and 2012 is as follows:
|
2014 |
2013 |
2012 |
||||||||||
|
Weighted average fair value of options granted |
$ | 0.14 | $ | 0.43 | $ | 0.59 | ||||||
|
Intrinsic value of options exercised |
-0- | -0- | -0- | |||||||||
|
Total fair value of options vested |
97,707 | 165,490 | 319,920 | |||||||||
We use the Black-Scholes model for determining the grant date fair value of our stock option grants. This model utilizes certain information, such as the interest rate on a risk-free security with a term generally equivalent to the expected life of the option being valued and requires certain other assumptions, such as the expected amount of time an option will be outstanding until it is exercised or expired, to calculate the fair value of stock options granted. The significant assumptions we used in our fair value calculations were as follows:
|
2014 |
2013 |
2012 |
||||||||||
|
Weighted average risk-free interest rates |
1.98 | % | 2.3 | % | 1.1 | % | ||||||
|
Expected dividend yield |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
|
Expected life of option (years) |
7.0 | 7.0 | 6.7 | |||||||||
|
Expected volatility |
94.88 | % | 96.60 | % | 105.2 | % | ||||||
Stock-based compensation expense related to the Stock Option Plan was $101,191, $143,435, and $310,076 during the years ended December 31, 2014, 2013 and 2012, respectively. Stock option expense is allocated to research and development expense or to general and administrative expense based on the nature of the services provided by the related individuals. For the three years ended December 31, 2014, stock option expense was allocated as follows:
|
2014 |
2013 |
2012 |
||||||||||
|
General and administrative expense |
$ | 69,057 | $ | 101,896 | $ | 231,936 | ||||||
|
Research and development expense |
32,134 | 41,539 | 78,140 | |||||||||
|
Total stock option expense |
$ | 101,191 | $ | 143,435 | $ | 310,076 | ||||||
As of December 31, 2014, there was $110,235 of unrecognized compensation expense related to stock-based compensation arrangements pursuant to the Stock Option Plan. The unrecognized compensation expense is expected to be recognized over a weighted average remaining period of 1.9 years.
Other Non-Employee Stock-Based Compensation
We recorded general and administrative expense of $100,000, $20,500, and $-0- during the years ended December 31, 2014, 2013 and 2012, respectively, related to the issuance of our common stock in exchange for services rendered by non-employees (See Note 8 – “Common Stock Transactions”).
We recorded general and administrative expense of $39,712, $238,168, and $-0- during the years ended December 31, 2014, 2013 and 2012, respectively, related to modifications made to certain stock purchase warrants (see Note 8 – “Stock Purchase Warrants”).
During 2014, we recorded general and administrative expense of $238,200 related to a warrant exercise fee paid to certain holders of our stock purchase warrants as an incentive for the holders to immediately certain warrants (see Note 8 – “Stock Purchase Warrants”).
As of December 31, 2014, there was no unrecognized expense related to any non-employee stock-based compensation arrangements.
|
|
|
10. |
Retirement Plan |
We participate in a multi-employer defined contribution retirement plan (the “401k Plan”) administered by a third party service provider; and the Company contributes to the 401k Plan on behalf of its employees based upon a matching formula. During the years ended December 31, 2014, 2013 and 2012 our contributions to the 401k Plan were $35,567, $43,132, and $50,500, respectively.
|
11. |
Income Taxes |
At December 31, 2014, we have a consolidated federal net operating loss (“NOL”) carryforward of approximately $64.6 million, available to offset against future taxable income which expires in varying amounts in 2019 through 2034. Additionally, we have approximately $826,000 in research and development (“R&D”) tax credits that expire in 2022 through 2034 unless utilized earlier. No income taxes have been paid to date.
Section 382 of the Internal Revenue Code contains provisions that may limit our utilization of our NOL and R&D tax credit carryforwards in any given year as a result of significant changes in ownership interests that have occurred in past periods or may occur in future periods.
Deferred income taxes reflect the net effect of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets and liabilities included the following at December 31, 2014 and 2013:
|
2014 |
2013 |
|||||||
|
Deferred tax assets: |
||||||||
|
Net operating loss carryforward |
$ | 22,831,626 | $ | 21,983,109 | ||||
|
Research and development tax credit carryforward |
825,896 | 799,248 | ||||||
|
Stock-based compensation expense |
2,396,805 | 2,233,909 | ||||||
|
Total deferred tax assets |
26,054,327 | 25,016,266 | ||||||
|
Deferred tax liabilities |
||||||||
|
Depreciation |
(7,149 | ) | (13,386 | ) | ||||
|
Total deferred tax liabilities |
(7,149 | ) | (13,386 | ) | ||||
|
Net deferred tax assets |
26,047,178 | 25,002,881 | ||||||
|
Valuation allowance |
(26,047,178 | ) | (25,002,881 | ) | ||||
| $ | -0- | $ | -0- | |||||
We have established a full valuation allowance equal to the amount of our net deferred tax assets due to uncertainties with respect to our ability to generate sufficient taxable income to realize these assets in the future. A reconciliation of the income tax benefit on losses at the U.S. federal statutory rate to the reported income tax expense is as follows:
|
2014 |
2013 |
2012 |
||||||||||
|
U.S. federal statutory rate applied to pretax loss |
$ | (929,409 | ) | $ | (776,881 | ) | $ | (725,948 | ) | |||
|
Permanent differences |
1,734 | 3,138 | 2,674 | |||||||||
|
Research and development credits |
26,648 | 14,047 | 21,236 | |||||||||
|
Change in valuation allowance |
901,027 | 759,696 | 702,038 | |||||||||
|
Reported income tax expense |
$ | -0- | $ | -0- | $ | -0- | ||||||
|
|
|
12. |
Related Party Transactions |
We are obligated to reimburse Emory University (a significant stockholder of the Company) for ongoing costs in connection with the filing, prosecution and maintenance of patent applications subject to a license agreement for technology associated with the vaccines we are developing. The expense associated with these ongoing patent cost reimbursements to Emory amounted to $179,958, $98,042, and $89,885 for the years ended December 31, 2014, 2013, and 2012, respectively.
In connection with our IPCAVD grant from the NIH (see Note 5), we entered into subcontracts with Emory for the purpose of conducting research and development activities related to the grant. During 2014, 2013, and 2012, we recorded $-0-, $252,478 and $552,403, respectively, of expense associated with these subcontracts. All amounts paid to Emory under these subcontracts are reimbursable to us pursuant to the NIH grant.
In December 2011 and January 2012, members of our management and Board of Directors participated in a private placement offering of our common stock and warrants (see Note 8), whereby they purchased an aggregate of 380,954 shares of our common stock for a total purchase price of $255,239 and received five-year warrants to purchase an additional 571,432 shares of our common stock exercisable at $1.00 per share.
|
|
|
13. |
Selected Quarterly Financial Data (unaudited) |
A summary of selected quarterly financial data for 2014 and 2013 is as follows:
|
2014 Quarter Ended |
||||||||||||||||
|
March 31 |
June 30 |
September 30 |
December 31 |
|||||||||||||
|
Revenue from grants |
$ | 157,340 | $ | 180,441 | $ | 322,086 | $ | 223,089 | ||||||||
|
Net loss |
(615,918 | ) | (679,537 | ) | (514,515 | ) | (923,585 | ) | ||||||||
|
Net loss per share |
(0.02 | ) | (0.03 | ) | (0.02 | ) | (0.03 | ) | ||||||||
|
2013 Quarter Ended |
||||||||||||||||
|
March 31 |
June 30 |
September 30 |
December 31 |
|||||||||||||
|
Revenue from grants |
$ | 797,040 | $ | 441,561 | $ | 1,004,211 | $ | 174,738 | ||||||||
|
Net loss |
(696,797 | ) | (526,284 | ) | (190,148 | ) | (871,714 | ) | ||||||||
|
Net loss per share |
(0.03 | ) | (0.02 | ) | (0.01 | ) | (0.04 | ) | ||||||||
|
14. |
Subsequent Event |
On February 27, 2015, we sold shares of our Series C convertible preferred stock to certain institutional investors for an aggregate purchase price of $3.0 million. The preferred stock is convertible at any time into shares of our common stock at $0.18 per share (16,666,666 shares in the aggregate), subject to possible adjustment as provided in the certificate of designation.
Pursuant to the terms of the securities purchase agreement, the investors also received five-year Series D warrants to purchase an aggregate of 16,666,666 shares of our common stock at $0.22 per share. The Series D warrants are immediately exercisable. We also granted to the investors a one-year additional purchase right, evidenced in the form of Series E warrants, to purchase up to 16,666,666 of our common stock for one year with an exercise price of $0.18 per share, and five-year Series F warrants to purchase up to 16,666,666 shares of our common stock at $0.22 per share. The Series D warrants are immediately exercisable. The Series F warrants only vest and become exercisable at the time, and to the extent, that the Series E warrants are exercised. The placement agent for the offering was granted a Series D warrants (exercisable immediately) to purchase 1,333,333 shares of our common stock at $0.22 per share.
GEOVAX LABS, INC.
SCHEDULE II – VALUATION AND QUALIFYING ACCOUNTS
For the Years Ended December 31, 2014, 2013 and 2012
|
Additions |
||||||||||||||||||||
|
Description |
Balance at Beginning Of Period |
Charged to Costs and Expenses |
Charged to Other Accounts |
(1) Deductions |
Balance at End Of Period |
|||||||||||||||
|
Reserve Deducted in the Balance Sheet From the Asset to Which it Applies: |
||||||||||||||||||||
|
Allowance for Deferred Tax Assets |
||||||||||||||||||||
|
Year ended December 31, 2014 |
$ | 25,002,881 | $ | 1,044,297 | $ | -0- | $ | -0- | $ | 26,047,178 | ||||||||||
|
Year ended December 31, 2013 |
27,295,741 | 862,736 | -0- | $ | (3,155,596 | ) | 25,002,881 | |||||||||||||
|
Year ended December 31, 2012 |
27,591,230 | 817,472 | -0- | (1,112,961 | ) | 27,295,741 | ||||||||||||||
|
(1) |
Deductions represent the effect of expiring NOL carryforwards from prior year. |
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 13. Other Expenses of Issuance and Distribution.
The following table sets forth the estimated costs and expenses of the Registrant in connection with the offering described in the registration statement.
|
SEC registration fee |
$ | 1,545 | ||
|
Legal fees and expenses |
76,000 | |||
|
Accounting fees and expenses |
5,000 | |||
|
Miscellaneous |
3,600 | |||
|
TOTAL |
$ | 86,145 |
ITEM 14. Indemnification of Directors and Officers.
Section 145 of the Delaware General Corporation Law (the “DGCL”), provides, among other things, that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding (other than an action by or in the right of the corporation) by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the corporation’s request as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with the action, suit or proceeding. The power to indemnify applies (i) if such person is successful on the merits or otherwise in defense of any action, suit or proceeding or (ii) if such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful. The power to indemnify applies to actions brought by or in the right of the corporation as well, but only to the extent of defense expenses (including attorneys’ fees but excluding amounts paid in settlement), actually and reasonably incurred and not to any satisfaction of judgment or settlement of the claim itself, and with the further limitation that in such actions no indemnification shall be made in the event of any adjudication of negligence or misconduct in the performance of his duties to the corporation, unless a court believes that in light of all the circumstances indemnification should apply.
Our bylaws provide that we may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the Company) by reason of the fact that the person is or was a director, officer, employee or agent of the Company, or is or was serving at the request of the Company as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with such action, suit or proceeding if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the Company, and, with respect to any criminal action or proceeding, had no reasonable cause to believe the person’s conduct was unlawful. Our bylaws also provide that we may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the Company to procure a judgment in its favor by reason of the fact that the person is or was a director, officer, employee or agent of the Company, or is or was serving at the request of the Company as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses (including attorneys’ fees) actually and reasonably incurred by the person in connection with the defense or settlement of such action or suit if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the Company and except that no indemnification shall be made in respect of any claim, issue or matter as to which such person shall have been adjudged to be liable to the Company unless and only to the extent that the Delaware Court of Chancery or the court in which such action or suit was brought shall determine upon application that, despite the adjudication of liability but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Delaware Court of Chancery or such other court shall deem proper.
Under our bylaws, expenses (including attorneys’ fees) incurred by an officer or director in defending any civil, criminal, administrative or investigative action, suit or proceeding may be paid by the Company in advance of the final disposition of such action, suit or proceeding upon receipt of an undertaking by or on behalf of such director or officer to repay such amount if it shall ultimately be determined that such person is not entitled to be indemnified by the Company. Such expenses (including attorneys’ fees) incurred by former directors and officers or other employees and agents may be so paid upon such terms and conditions, if any, as we deem appropriate.
The indemnification and advancement of expenses provided by our bylaws is not exclusive, both as to action in such person’s official capacity and as to action in another capacity while holding such office.
Our bylaws also provide that we may purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of the Company, or is or was serving at the request of the Company as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against any liability asserted against such person and incurred by such person in any such capacity, or arising out of such person’s status as such, whether or not the Company would have the power to indemnify such person against such liability under our bylaws. The Company maintains an insurance policy providing for indemnification of its officers, directors and certain other persons against liabilities and expenses incurred by any of them in certain stated proceedings and under certain stated conditions.
In October 2006, GeoVax and our subsidiary, GeoVax, Inc. entered into indemnification agreements with Messrs. McNally, Reynolds, Kollintzas and Spencer. Pursuant to these agreements, we have agreed to hold harmless and indemnify these directors and officers to the full extent authorized or permitted by applicable Illinois and Georgia law against certain expenses and other liabilities actually and reasonably incurred by these individuals in connection with certain proceedings if they acted in a manner they believed in good faith to be in or not opposed to the best interests of the Company and, with respect to any criminal proceeding, had no reasonable cause to believe that such conduct was unlawful. The agreements also provide for the advancement of expenses to these individuals subject to specified conditions. Under these agreements, we will not indemnify these individuals for expenses or other amounts for which applicable Illinois and Georgia law prohibit indemnification. The obligations under these agreements continue during the period in which these individuals are our directors or officers and continue thereafter so long as these individuals shall be subject to any proceeding by reason of their service to the Company, whether or not they are serving in any such capacity at the time the liability or expense incurred for which indemnification can be provided under the agreements.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling the registrant pursuant to the foregoing provisions, the registrant has been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
ITEM 15. Recent Sales of Unregistered Securities
On October 22, 2013, we issued 50,000 shares of our common stock to ProActive Capital Resources Group, LLC for services rendered pursuant to a consulting agreement with the Company. For this transaction the Company relied upon Section 4(2) of the Securities Act and Rule 506 promulgated thereunder to issue the common stock. The shares were offered to a single accredited investor who acquired the shares for investment in a transaction that did not involve a general solicitation.
On December 11, 2013, we entered into a Securities Purchase Agreement with three accredited investors identified therein providing for the issuance and sale to the purchasers of an aggregate of 1,650 shares of our Series B convertible preferred stock for gross proceeds to the Company of $1,650,000, as described in our Form 8-K filed December 17, 2013. The Company relied on an exemption from the registration requirements of the Securities Act afforded by Section 4(a)(2) thereof and Rule 506 of Regulation D. For information regarding the terms of the Series B convertible preferred stock, see “Description of Securities” in the prospectus accompanying this registration statement. The accredited investors acquired the shares for investment for their own accounts in a transaction that did not involve a general solicitation.
On February 25, 2015, we entered into a Securities Purchase Agreement (the “Securities Purchase Agreement”) with two accredited investors providing for the issuance and sale to the investors of an aggregate of 3,000 shares of our Series C Convertible Preferred Stock (the “Series C Preferred Stock”) and related warrants for gross proceeds to the Company of $3.0 million. Each share of Series C Preferred Stock is initially convertible into approximately 5,555.55 shares of our Common Stock for an aggregate total of 16,666,666 shares of our Common Stock (the “Conversion Shares”). The terms of the Preferred Shares include antidilution provisions. We closed this transaction on February 27, 2015. Pursuant to the Securities Purchase Agreement, each investor was also issued a Series D Warrant, a Series E Warrant and a Series F Warrant (collectively, the “Warrants”), each to purchase up to a number of shares of the Company’s Common Stock equal to 100% of the Conversion Shares underlying the Preferred Shares issued to such Purchaser pursuant to the Securities Purchase Agreement (up to 16,666,666 shares in the aggregate for each of the three series of warrants, or approximately 50,000,000 shares in total). Additional detail is set forth in our Form 8-K filed March 2, 2015. The Company also issued a warrant to its placement agent to acquire 1,333,333 shares of our common stock at $0.22 per share on substantially the same terms and conditions of the Series D warrants. The Company relied on an exemption from the registration requirements of the Securities Act afforded by Section 4(a)(2) thereof and Rule 506 of Regulation D. For information regarding the terms of the Series C Preferred Stock, see “Description of Securities” in the prospectus accompanying this registration statement. The accredited investors acquired the shares for investment for their own accounts in a transaction that did not involve a general solicitation.
ITEM 16. Exhibits and Financial Statement Schedules
|
(a) |
The exhibits filed with this registration statement are set forth on the exhibit index following the signature page and are incorporated by reference in their entirety into this item. |
|
(b) |
Financial Statement Schedules: |
Schedule II—Valuation and Qualifying Accounts for the years ended December 31, 2014, 2013 and 2012 is included in the Financial Statements at page F-18.
All other financial statement schedules have been omitted because they are not applicable or not required or because the information is included elsewhere in the Consolidated Financial Statements or the Notes thereto.
ITEM 17. Undertakings.
|
(a) |
The undersigned registrant hereby undertakes: |
|
|
1. |
To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: | |
|
|
i. |
To include any prospectus required by section 10(a) (3) of the Securities Act of 1933; | |
|
|
ii. |
To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and | |
|
|
iii. |
To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement. | |
|
|
2. |
That, for the purpose of determining any liability under the Securities Act of 1933, as amended, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
|
|
3. |
To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
|
|
4. |
That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities: The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
|
|
i. |
Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; |
|
|
ii. |
Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
|
|
iii. |
The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
|
|
iv. |
Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
|
(b) |
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by us of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933 and will be governed by the final adjudication of such issue. |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Smyrna, State of Georgia, on the 20th day of March, 2015.
|
|
GEOVAX LABS, INC. |
| |
|
|
|
|
|
|
|
|
|
|
|
|
By: |
/s/ Robert T. McNally |
|
|
|
|
Robert T. McNally Ph.D. |
|
|
|
|
President and Chief Executive Officer |
|
POWERS OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Robert T. McNally his or her true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign and file any and all amendments (including post-effective amendments) to this Registration Statement, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
|
Name |
|
Title |
Date |
|
|
|
|
|
|
/s/ Robert T. McNally |
|
Director |
March 20, 2015 |
|
Robert T. McNally |
|
President & Chief Executive Officer |
|
|
|
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
/s/ Mark W. Reynolds |
|
Chief Financial Officer |
March 20, 2015 |
|
Mark W. Reynolds |
|
(Principal Financial and Accounting Officer) |
|
|
|
|
|
|
|
/s/ Randal D. Chase |
|
Director |
March 20, 2015 |
|
Randal D. Chase |
|||
|
/s/David A Dodd |
|
Director |
March 20, 2015 |
|
David A. Dodd |
|
|
|
|
|
|
|
|
|
/s/Dean G. Kollintzas |
|
Director |
March 20, 2015 |
|
Dean G. Kollintzas |
|
|
|
|
|
|
|
|
|
/s/ Harriet L. Robinson |
|
Director |
March 20, 2015 |
|
Harriet L. Robinson |
|
|
|
|
|
|
|
|
|
/s/ John N. Spencer, Jr. |
|
Director |
March 20, 2015 |
|
John N. Spencer, Jr. |
|
|
|
EXHIBIT INDEX
Exhibit
| Number | Description |
|
3.1 |
Certificate of Incorporation (4) |
|
3.1.1 |
Certificate of Amendment to the Certificate of Incorporation of GeoVax Labs, Inc. filed April 13, 2010 (8) |
|
3.1.2 |
Certificate of Amendment to the Certificate of Incorporation of GeoVax Labs, Inc. filed April 27, 2010 (9) |
|
3.1.3 |
Certificate of Amendment to the Certificate of Incorporation of GeoVax Labs, Inc. filed August 2, 2013 (15) |
|
3.2 |
Bylaws (4) |
|
4.1.1 |
Form of Certificate of Designation of Preferences, Rights and Limitations of Series A Convertible Preferred Stock filed March 20, 2012 (12) |
|
4.1.2 |
Amendment to Certificate of Designation of Series A Convertible Preferred Stock filed December 13, 2013 (17) |
|
4.1.3 |
Form of Stock Certificate for the Series A Convertible Preferred Stock (11) |
|
4.2.1 |
Form of Certificate of Designation of Preferences, Rights and Limitations of Series B Convertible Preferred Stock filed December 13, 2013 (17) |
|
4.2.2 |
Form of Stock Certificate for the Series B Convertible Preferred Stock (17) |
|
4.3.1 |
Form of Certificate of Designation of Preferences, Rights and Limitations of Series C Convertible Preferred Stock filed February 27, 2015 (19) |
|
4.3.2 |
Form of Stock Certificate for the Series C Convertible Preferred Stock (19) |
|
5.1 * |
Opinion of Womble Carlyle Sandridge & Rice, LLP |
|
10.1 ** |
Employment Agreement between GeoVax Labs, Inc. and Robert T. McNally effective as of April 1, 2008 (5) |
|
10.2 ** |
Employment Agreement between GeoVax, Inc. and Mark W. Reynolds Amended and Restated effective as of January 1, 2010 (7) |
|
10.3 ** |
Employment Agreement between GeoVax, Inc. and Harriet Robinson effective as of November 19, 2007 (7) |
|
10.4 ** |
Amendment No. 1 to Employment Agreement between GeoVax Labs, Inc. and Robert T. McNally dated October 22, 2013 (16) |
|
10.5 ** |
Amendment No. 1 to Employment Agreement between GeoVax Labs, Inc. and Harriet Robinson dated October 22, 2013 (16) |
|
10.6 ** |
Amendment No. 1 to Employment Agreement between GeoVax Labs, Inc. and Mark W. Reynolds dated October 22, 2013 (16) |
|
10.7 ** |
GeoVax Labs, Inc. 2006 Equity Incentive Plan (2) |
|
10.8 |
License Agreement (as amended and restated) between GeoVax, Inc. and Emory University, dated August 23, 2002 (1) |
|
10.9 |
Office and Laboratory Lease between UCB, Inc. and GeoVax, Inc. (6) |
|
10.9.1 |
Amendment to Lease Agreement between UCB, Inc. and GeoVax, Inc. (20) |
|
10.10 |
Summary of the GeoVax Labs, Inc. Director Compensation Plan (7) |
|
10.11 |
Form of Warrant dated December 30, 2011 (10) |
|
10.12 |
Form of Common Stock Purchase Warrants (11) |
|
10.13 |
Form of Securities Purchase Agreement dated March 16, 2012 (12) |
|
10.14 |
Form of Registration Rights Agreement dated March 16, 2012 (12) |
|
10.15 |
Form of Series A Warrant dated March 16, 2012 (12) |
|
10.16 |
Form of Series B Warrant dated March 16, 2012 (12) |
|
10.17 |
Form of Series C Warrant dated March 16, 2012 (12) |
|
10.18 |
Warrant Reset Offer Agreements dated January 17, 2013 (15) |
|
10.19 |
Warrant Reset Offer Agreements dated May 14, 2013 (14) |
|
10.20 |
Securities Purchase Agreement dated December 11, 2013 with Form of Registration Rights Agreement (17) |
|
10.21 |
Amendment Agreement and Consent of Holders of Series A Convertible Preferred Stock dated December 11, 2013 (17) |
|
10.22 |
Form of Letter Agreement dated October 14, 2014 providing for payment of warrant exercise fee (18) |
|
10.23 |
Form of Securities Purchase Agreement dated February 25, 2015 (19) |
|
10.24 |
Form of Registration Rights Agreement dated February 25, 2015 (19) |
|
10.25 |
Form of Series D Warrant dated February 27, 2015 (19) |
|
10.26 |
Form of Series E Warrant dated February 27, 2015 (19) |
|
10.27 |
Form of Series F Warrant dated February 27, 2015 (19) |
|
10.28 |
Form of Maxim warrant dated February 27, 2015 (19) |
|
14.1 |
Code of Ethics (3) |
|
21.1 |
Subsidiaries of the Registrant (3) |
|
23.1 * |
Consent of Porter Keadle Moore, LLC |
|
23.2 * |
Consent of Womble Carlyle Sandridge & Rice, LLP (contained in the opinion filed as Exhibit 5.1 hereof) |
|
101 *,*** |
The following financial information from GeoVax Labs, Inc. Annual Report on Form 10-K for the year ended December 31, 2014, formatted in Extensible Business Reporting Langue (XBRL): (i) Consolidated Balance Sheets as of December 31, 2014 and December 31, 2013, (ii) Consolidated Statements of Operations for the years ended December 31, 2014, 2013 and 2012, (iii) Consolidated Statements of Stockholders' Equity for the ended December 31, 2014, 2013 and 2012, (iv) Consolidated Statements of Cash Flows for the years ended December 31, 2014, 2013 and 2012, and (v) Notes to Condensed Consolidated Financial Statements. |
___________________
| * | Filed herewith. |
| ** | Indicates a management contract or compensatory plan or arrangement. |
|
*** |
Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files in Exhibit 101 hereto are deemed not filed or part of a registration statement or prospectus for purposes of Section 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended and otherwise are not subject to liability under those sections. |
|
(1) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed October 4, 2006. |
|
(2) |
Incorporated by reference from the registrant’s definitive Information Statement (Schedule 14C) filed August 18, 2006. |
|
(3) |
Incorporated by reference from the registrant’s Annual Report on Form 10-K filed March 28, 2007. |
|
(4) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed June 23, 2008. |
|
(5) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed March 24, 2008. |
|
(6) |
Incorporated by reference from the registrant’s Quarterly Report on Form 10-Q filed November 6, 2009. |
|
(7) |
Incorporated by reference from the registrant’s Annual Report on Form 10-K filed March 8, 2010. |
|
(8) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed April 14, 2010. |
|
(9) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed April 28, 2010. |
|
(10) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed January 5, 2012. |
|
(11) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed February 6, 2012 |
|
(12) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed March 22, 2012. |
|
(13) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed January 17, 2013. |
|
(14) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed May 15, 2013 |
|
(15) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed August 2, 2013. |
|
(16) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed October 23, 2013. |
|
(17) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed December 17, 2013. |
|
(18) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed October 15, 2014. |
|
(19) |
Incorporated by reference from the registrant’s Current Report on Form 8-K filed March 2, 2015. |
|
(20) |
Incorporated by reference from the registrant’s Annual Report on Form 10-K filed March 20, 2015. |
II-7