Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - GALECTIN THERAPEUTICS INC | Financial_Report.xls |
| EX-31.1 - EX-31.1 - GALECTIN THERAPEUTICS INC | d826567dex311.htm |
| EX-23.1 - EX-23.1 - GALECTIN THERAPEUTICS INC | d826567dex231.htm |
| EX-31.2 - EX-31.2 - GALECTIN THERAPEUTICS INC | d826567dex312.htm |
| EX-32.2 - EX-32.2 - GALECTIN THERAPEUTICS INC | d826567dex322.htm |
| EX-32.1 - EX-32.1 - GALECTIN THERAPEUTICS INC | d826567dex321.htm |
| EX-21.1 - EX-21.1 - GALECTIN THERAPEUTICS INC | d826567dex211.htm |
Table of Contents
Index to Financial Statements
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | Annual report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the fiscal year ended December 31, 2014
| ¨ | Transition report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the transition period from to
Commission File No. 001-31791
GALECTIN THERAPEUTICS INC.
| Nevada | 04-3562325 | |
| (State or other jurisdiction of incorporation) |
(I.R.S. Employer Identification No.) | |
| 4960 Peachtree Industrial Blvd., Suite 240, Norcross, GA | 30071 | |
| (Address of Principal Executive Offices) | (Zip Code) | |
(678) 620-3186
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, $0.001 Par Value Per Share | The NASDAQ Capital Market | |
| Units, each consisting of two shares of Common Stock and one Warrant to purchase one share of Common Stock | The NASDAQ Capital Market | |
| Common Stock Purchase Warrants | The NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ¨ NO x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES x NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES x NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ Accelerated filer x Non-accelerated filer ¨ Smaller reporting company ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YES ¨ NO x
The aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was sold, or the average bid and asked price of such common equity, as of June 30, 2014 was $245.7 million.
The number of shares outstanding of the registrant’s common stock as of March 6, 2015 was 23,546,315.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive Proxy Statement for the 2015 Annual Meeting of Stockholders are incorporated by reference into Part III of this Report.
Table of Contents
Index to Financial Statements
FOR THE YEAR ENDED DECEMBER 31, 2014
Table of Contents
Index to Financial Statements
PART I
| Item 1. | Business |
Overview
We are a clinical stage biopharmaceutical company engaged in drug research and development to create new therapies for fibrotic disease and cancer. Our drug candidates are based on our method of targeting galectin proteins, which are key mediators of biologic and pathologic functions. We use naturally occurring, readily-available plant materials as starting material in manufacturing processes to create proprietary complex carbohydrates with specific molecular weights and other pharmaceutical properties. These complex carbohydrate molecules are appropriately formulated into acceptable pharmaceutical formulations. Using these unique carbohydrate-based candidate compounds that largely bind and inhibit galectin proteins, particularly galectin-3, we are undertaking the focused pursuit of therapies for indications where galectins have a demonstrated role in the pathogenesis of a given disease. We focus on diseases with serious, life-threatening consequences to patients and those where current treatment options are limited. Our strategy is to establish and implement clinical development programs that add value to our business in the shortest period of time possible and to seek strategic partners when a program becomes advanced and requires additional resources.
We endeavor to leverage our scientific and product development expertise as well as established relationships with outside sources to achieve cost-effective and efficient development. These outside sources, amongst others, provide us with expertise in preclinical models, pharmaceutical development, toxicology, clinical development, pharmaceutical manufacturing, sophisticated physical and chemical characterization, and commercial development. We also have established several collaborative scientific discovery programs with leading experts in carbohydrate chemistry and characterization. These discovery programs are generally aimed at the targeted development of new carbohydrate molecules which bind galectin proteins and offer alternative options to larger market segments in our primary disease indications. We also have established a discovery program aimed at the targeted development of small molecules (non-carbohydrate) which bind galectin proteins and may afford options for alternative means of drug delivery (e.g., oral) and as a result expand the potential uses of our compounds. Another discovery program seeks to identify the molecular interactions of molecules with the galectin-receptor. We are pursuing a development pathway to clinical enhancement and commercialization for our lead compounds in immune enhancement for cancer therapy as well as in both liver fibrosis and fatty liver disease. All of our proposed products are presently in development, including pre-clinical and clinical trials.
We were founded in July 2000 as Pro-Pharmaceuticals, Inc., a Massachusetts corporation. On April 25, 2001, DTR-Med Pharma Corp. (“DTR”), which was incorporated in Nevada on January 26, 2001, entered into a stock exchange agreement with Pro-Pharmaceuticals, Inc., whereby DTR acquired all of the outstanding shares of common stock of Pro-Pharmaceuticals, Inc. On May 10, 2001, DTR changed its name to “Pro- Pharmaceuticals, Inc.” and on June 7, 2001, the Massachusetts corporation was merged into the Nevada corporation. On May 26, 2011, Pro-Pharmaceuticals, Inc. changed its name to “Galectin Therapeutics Inc.” In October, 2012, we moved our headquarters to a suburb of Atlanta, GA to be closer to a center of discovery collaboration while maintaining a contract laboratory operation in the Boston area.
Our Drug Development Programs
Galectins are a class of proteins that are made by many cells in the body. As a group, these proteins are able to bind to sugar molecules that are part of other proteins in and on the cells of our body. Galectin proteins act as a kind of molecular glue, bringing together molecules that have sugars on them. Galectin proteins, in particular galectin-3, are known to be markedly increased in a number of important diseases including scarring of organs (e.g. liver, lung, kidney, and heart) and cancers of many kinds. The increase in galectin protein promotes the disease and is detrimental to the patient. Published data show that mice lacking the galectin-3 gene, and thus unable to produce galectin-3, are incapable of developing liver fibrosis in response to toxic insult to the liver and in fatty liver disease.
We have two compounds in development, GR-MD-02 and GM-CT-01, both of which have shown promise in preclinical studies in treatment of fibrosis and in cancer therapy. However, we are currently focusing on
1
Table of Contents
Index to Financial Statements
development of GR-MD-02 intended to be used in the treatment of liver fibrosis associated with fatty liver disease (NASH) and in cancer therapy in combination with immune-system modifying agent(s). Both of our proprietary, patented compounds are derived from completely different, natural, readily available, starting materials, which, following chemical processing, both exhibit the property of binding to and inhibiting galectin proteins.
Our product pipeline is shown below:
| Indication |
Drug | Status | ||
| Fibrosis |
||||
| NASH with Advanced Fibrosis |
GR-MD-02 | IND submitted January 2013, FDA indicated on March 1, 2013 that we could proceed with a Phase 1 US clinical trial. Phase 1 clinical trial started Q2-2013. Results from the three cohorts of the Phase 1 clinical trial were reported in 2014, with final results reported in January 2015. End of Phase 1 meeting held with FDA in 2014 and Phase 2 clinical program expected to begin in Q2 2015. | ||
| Lung Fibrosis |
GR-MD-02 | In pre-clinical development | ||
| Kidney Fibrosis |
GR-MD-02 | In pre-clinical development | ||
| Cardiac Fibrosis |
GR-MD-02 and GM-CT-01 |
In pre-clinical development | ||
| Cancer Immunotherapy |
||||
| Melanoma |
GR-MD-02 | Investigator IND filed in December 2013. Phase 1B study in process. |
Fibrosis. GR-MD-02 is our lead product candidate for treatment of fibrotic disease. Our preclinical data show that GR-MD-02 has a powerful therapeutic effect on liver fibrosis as shown in several relevant animal models. Therefore, we chose GR-MD-02 as the lead candidate in a development program targeted initially at fibrotic liver disease associated with non-alcoholic steatohepatitis (NASH, or fatty liver disease). In January 2013, an Investigational New Drug (“IND”) was submitted to the FDA with the goal of initiating a Phase 1 study in patients with NASH and advanced liver fibrosis to evaluate the human safety of GR-MD-02 and pharmacodynamics biomarkers of disease. On March 1, 2013, the FDA indicated we could proceed with a US Phase 1 clinical trial for GR-MD-02 with a development program aimed at obtaining support for a proposed indication of GR-MD-02 for treatment of NASH with advanced fibrosis. The Phase 1 trial was completed and demonstrated that GR-MD-02 up to 8 mg/Kg, i.v. was safe and well tolerated and the human pharmacokinetic data defined a drug dose for use in the planned Phase 2 trials. An “End of Phase 1 Meeting” was held with FDA which, amongst other items, provided guidance on the primary endpoint for the Phase 2 clinical trial.
Our drug candidate provides a promising new approach for the therapy of fibrotic diseases, and liver fibrosis in particular. Fibrosis is the formation of excess connective tissue (collagen and other proteins plus cellular elements such as myofibroblasts) in response to damage, inflammation or repair. When the fibrotic tissue becomes confluent, it obliterates the cellular architecture, leading to scarring and dysfunction of the underlying organ.
Cancer Immunotherapy. We believe there is potential for galectin inhibition to play a key role in the burgeoning area of cancer immunotherapy. For example, there have been several recent approvals of drugs that enhance a patient’s immune system to fight cancer. With many additional vaccines and immune stimulatory agents in development, industry analysts forecast that this market could generate over $35 billion in sales over the next 10 years. It is our goal to use a galectin inhibitor to enhance the immune system function to fight cancer in a way that complements other approaches to this type of therapy. Our drug candidates provide a promising new therapeutic approach to enhance the activity of the immune system against cancer cells. Preclinical studies have indicated that GR-MD-02 enhances the immune response to and more specifically increased tumor
2
Table of Contents
Index to Financial Statements
shrinkage and enhanced survival in immune competent mice with prostate, breast, melanoma and sarcoma cancers when combined with one of the immune checkpoint inhibitors, anti-CTLA-4 or anti-PD-1. These preclinical data have led to the filing of an Investigator-sponsored IND and the initiation of a study of GR-MD-02 in combination with Yervoy® (ipilimumab) in a Phase 1B study of patients with metastatic melanoma. This study is being conducted under the sponsorship of Providence Portland Medical Center’s Earle A. Chiles Research Institute (EACRI).
We believe the mechanism of action for GM-CT-01 and GR-MD-02 is based upon interaction with, and inhibition of, galectin proteins, particularly galectin-3, which are expressed at high levels in certain pathological states including inflammation, fibrosis and cancer. While GM-CT-01 and GR-MD-02 are capable of binding to multiple galectin proteins, we believe that they have the greatest affinity for galectin-3, the most prominent galectin implicated in pathological processes. Blocking galectin in cancer and liver fibrosis has specific salutary effects on the disease process, as discussed below.
Liver Fibrosis: New Approach for a Significant Unmet Medical Need
When an internal organ is exposed to chronic disease one of the responses is that scar tissue is laid down in the organ (this process is called fibrosis). The longer the disease affects the organ, the more fibrous tissue is deposited and this ultimately results in the failure of the organ. This chronic fibrosis of organs may occur in the liver, lung, kidney, and heart, as well as others and, as a result, fibrosis of organs has been estimated to account for as much as 45% of all mortality. Scientific findings during the last few years indicate that the galectin-3 protein is critically important in this fibrotic process in multiple organs.
In the liver, fibrosis is the end result of multiple inflammatory conditions and infections. Progressive liver fibrosis leads to scarring (cirrhosis), which results in reduction of liver function, multiple medical complications and ultimately death. It is estimated that 1-2 million patients have cirrhosis in the United States with close to 50,000 losing their lives yearly. Only a fraction of patients’ lives, approximately 6,200 per year, are saved by liver transplantation at a cost of approximately $350,000 per transplantation. One condition in particular that frequently leads to cirrhosis is non-alcoholic steatohepatitis, or NASH, a liver disease characterized by the accumulation of fat in the liver with associated inflammation and fibrosis, which can lead to end-stage cirrhosis requiring liver transplantation. The National Institute of Health estimates that 9 to 15 million Americans are affected by NASH, and other sources suggest it may be as many as 28 million people have NASH, and forecasts that the number of Americans affected by this disease is growing due to obesity and diabetes, with the potential to become the leading cause of liver cirrhosis and liver transplantation in the future. Liver transplantation is currently the only therapeutic approach to NASH or other forms of liver fibrosis as, to the best of our knowledge, there are no drug therapies on the market. Organ transplantation is a difficult, risky and costly procedure as organ availability is scarce and there is the risk of developing cirrhosis in the transplanted liver from the same disease that damaged the patient’s original liver and therefore, there is a great need for other therapeutic options. All diseases that affect the liver (viral hepatitis, alcoholic liver disease, and fatty liver as examples) lead to the development of scarring of the liver.
The primary focus of the company is to use galectin inhibitors to block galectin-3 and treat organ scarring or fibrosis in the liver. There are no approved therapies for treatment of liver fibrosis. We believe that our drug candidates have the potential to treat NASH and other forms of liver fibrosis. Scientific evidence suggests that galectin-3 is essential for the development of liver fibrosis in animals. Published data show that mice lacking the galectin-3 gene, and thus unable to produce galectin-3, are incapable of developing liver fibrosis in response to toxic insult to the liver and in fatty liver disease. Moreover, mice that do not have the galectin-3 gene are resistant to lung and kidney fibrosis. These published data show that galectin-3 is a critical protein for the development of organ fibrosis. Our drugs, based on experiments in well characterized animal models, are also potentially useful in scarring or fibrosis of other organs such as lung and kidney which expands the possibilities for therapeutic indications.
3
Table of Contents
Index to Financial Statements
We have evaluated the ability of GR-MD-02 to block galectin-3 in animal models of liver fibrosis, the conclusions of which yielded positive results. Our pre-clinical data show that GR-MD-02 may have a therapeutic effect on liver fibrosis as shown in several relevant animal models. Therefore, we chose GR-MD-02 as the lead candidate in a development program targeted initially at fibrotic liver disease associated with NASH.
We evaluated GR-MD-02 in pre-clinical toxicology and pharmacology studies during 2013 and filed an IND with the FDA in January 2013 for initiating human studies in patients with NASH. In February 2013 we entered into an agreement with CTI Clinical Trial Services to assist with the design, development and conduct of one or more clinical research studies, specifically for services with respect to our Phase 1 clinical trials to evaluate safety of GR-MD-02 in patients with NASH. The FDA notified us in March 2013 that we may proceed with a Phase 1 clinical trial for patients with NASH and we began enrolling patients in the Phase 1 clinical trial in the third quarter of 2013. In August 2013, GR-MD-02 was granted Fast Track designation by the FDA for NASH with hepatic fibrosis, commonly known as fatty liver disease with advanced fibrosis. In January 2014, we completed the enrollment of the first cohort of patients in the Phase 1 trial with no serious adverse events being reported. We reported initial safety and tolerability results from the first cohort of patients on June 30, 2014. The second cohort of this Phase 1 trial began and enrollment was completed in April 2014. In July 2014, we reported the results from the second cohort of patients. Enrollment of the third cohort of Phase 1 began in July 2014 with interim results presented in November 2014 with the final report on cohort 3 presented in January 2015. The results of the Phase 1 study demonstrate that (i) GR-MD-02 was safe and well tolerated by patients with advanced NASH liver fibrosis after IV administration of four doses of 2 mg/kg, 4 mg/kg and 8mg/kg lean body weight, (ii) Pharmacokinetics revealed drug exposure in humans at the 8 mg/kg dose that was equivalent to the upper range of the targeted therapeutic dose determined from effective doses in NASH animal models, (iii) Disease Serum Marker Effect showed there was a statistically significant, dose-dependent reduction in FibroTest® scores due to a statistically significant reduction in alpha-2 macroglobulin (A2M) serum levels, and (iv) Liver Stiffness Effect, as measured by FibroScan® showed that there was a signal of reduced liver stiffness in patients receiving GR-MD-02. The reduction seen in A2M does not necessarily mean fibrosis got better in this short study, but does suggest changes in the fibrogenic process that might lead to an improvement in fibrosis with longer-term therapy. These Phase 1 results in NASH patients with advanced fibrosis provide a firm foundation for entry into a Phase 2 development program.
The company held an “End of Phase 1 meeting” with FDA and, amongst other things, received clear guidance on the primary endpoint for a Phase 2 trial. Our Phase 2 clinical trial design targets a patient population with cirrhosis due to NASH. The study endpoints will include those that are closely associated with outcomes in patients with cirrhosis Primary endpoint: Hepatic venous pressure gradient (HVPG). Planned secondary endpoints include: morphometric analysis of collagen on liver biopsies and other secondary endpoints will include non-invasive tests to evaluate for correlation with HVPG and liver collagen. We have awarded the contract for the primary Phase 2 study to a well-known CRO with experience in NASH trials and expect to initiate a Phase 2 clinical trial in the first half of 2015 to assess the efficacy of GR-MD-02 in patients with NASH and advanced liver fibrosis. The timing of initial results from the Phase 2 trial are dependent upon the trial design, and, amongst other factors, the rate of patient enrollment; and the trial design is being finalized. Our Phase 2 clinical program currently includes one additional clinical trial in patients with NASH with advanced fibrosis to fully characterize human response to GR-MD-02 and to better position the Company for a successful Phase 3 clinical trial program.
GR-MD-02 is a proprietary, patented galactoarabino-rhamnogalacturonan polysaccharide polymer that is comprised predominantly of galacturonic acid, galactose, arabinose, rhamnose, and smaller amounts of other sugars. Structural studies have shown that GR-MD-02 binds to galectin-1 and to galectin-3 with binding affinity to galectin-3 being significantly greater than binding to galectin-1. With respect to GR-MD-02, we currently have a number of issued US patents including one composition of matter patent, one method of manufacture patent, one method of use patient in patients with NASH, one method of use patent in patients with liver fibrosis, and one method of use patent in patients with diabetic kidney disease. Additional patent applications are pending with respect to, amongst other uses, cancer immunotherapy, lung fibrotic disease, and inflammatory disease
4
Table of Contents
Index to Financial Statements
associated with increase in inducible nitric oxide synthase. Patents have been granted with respect to liver fibrosis, NASH, and liver fibrosis in combination with other therapeutic agents. Compounds for subcutaneous administration and oral delivery are currently under pre-clinical development.
Galectin Inhibition in Cancer Therapy
We believe the potential exists for galectin inhibition to play an important role in cancer therapy. Galectin proteins, particularly galectin-1 and galectin-3, have been shown to be highly expressed in the majority of cancers and have multiple roles in promoting cancer progression, including tumor cell invasion, metastasis, angiogenesis, and tumor evasion of the immune system.
The role of galectins in cancer immunotherapy can be understood through the “Galectin Effect”, a recent discovery of how tumors avoid the body’s own immune system, i.e., the tumors secrete galectin proteins that block the body’s efforts to fight tumors. Our current program to block the “Galectin Effect” is based on the research of Dr. Pierre van der Bruggen (of the Ludwig Institute of Cancer Research in Brussels, Belgium), demonstrating that galectin-3, which is produced by the vast majority of human cancers, binds to and blocks the actions of tumor-infiltrating T-lymphocytes, the major immune cell in the body’s defense against cancers. In addition, Dr. Will Redmond of the Earl Chiles Cancer Research Institute in Portland Oregon has shown that our galectin inhibitors can enhance the anti-tumor immunogenic effect of other immunotherapies based on targeting lymphocyte checkpoints such as CTLA4. Based on these results, we believe that the body’s immune cells may be unable to attack and kill tumor cells in the presence of galectins. Using this approach, the mechanism of action for our drugs seeks to block galectins and, in turn, restore the ability of the T-lymphocytes to kill tumor cells.
In May 2012, we initiated a Phase 1/2 clinical trial of GM-CT-01 in Belgium in combination with a tumor vaccine in patients with advanced melanoma, a deadly skin cancer. The Belgian Federal Agency of Medicine and Health Products, or FAMHP, granted approval for this clinical trial, which was being conducted at three centers in Belgium and one in Luxembourg. The only trial site that was initiated was at the Ludwig Cancer Institute in Belgium. In January 2014, the Cancer Centre at the Cliniques universitaires Saint-Luc and the Ludwig Institute for Cancer Research (LICR), in agreement with Galectin Therapeutics, voluntarily placed on hold its Phase 1/2 trial evaluating the safety and efficacy, GM-CT-01, in combination with an experimental peptide vaccine for the treatment of advanced metastatic melanoma. The trial was placed on hold as the investigators were unable to enroll sufficient patients with advanced stage melanoma due to the high selection criteria of patient candidates for the peptide vaccine and the recent availability of Yervoy in Europe as a treatment increasing the overall survival of metastatic melanoma patients. At the time when the trial was halted, three patients had completed the protocol with no serious adverse events. This trial was stopped because of patient enrollment issue and not due to the perceived effects of the drugs on the disease or on safety issues.
The company supported preclinical studies led by tumor immunology expert William L. Redmond, Ph.D., of the Providence Portland Medical Center’s Earle A. Chiles Research Institute (EACRI). The preclinical study found that GR-MD-02 increased tumor shrinkage and enhanced survival in immune competent mice with prostate and breast cancers when combined with one of the immune checkpoint inhibitors, anti-CTLA-4 or anti-PD-1. These findings suggest a role for GR-MD-02 in cancer immunotherapy. These preclinical observations by Dr Redmond provided scientific rationale for proceeding and lead to the filing by Providence Portland Medical Center of an Investigator-sponsored IND to conduct a Phase 1B study to determine if GR-MD-02 enhances the probability of melanoma response with ipilimumab by inducing proliferation, activation and memory function of CD8+ T cells in human patients. The company has licensed the underlying invention from Providence Portland Medical Center. This study represents a novel approach for patients with metastatic melanoma. The IND was approved by FDA in February 2014. This study is being conducted under the sponsorship of Providence Portland Medical Center’s Earle A. Chiles Research Institute (EACRI) and is being supported by the Company.
The study employs a dose escalation of GR-MD-02 in conjunction with the standard therapeutic dose of ipilimumab in patients with advanced melanoma for whom ipilimumab would be considered standard of care. In
5
Table of Contents
Index to Financial Statements
addition to monitoring for toxicity and clinical response by irRECIST criteria on imaging tests, blood samples will be obtained to assess immunologic measures relevant to galectin biology and ipilimumab T-cell check-point inhibition. Galectin Therapeutics will provide its proprietary compound GR-MD-02 to EACRI researchers, as well as supply researchers with supporting analysis of the pharmacokinetics of GR-MD-02 and the right to reference the Company’s open IND on GR-MD-02. To date three patients were treated in the first dosing group without serious adverse events. The second dosing group of GR-MD-02 2 mg/kg is now enrolling.
Patents and Proprietary Rights
Our development and commercial viability, and ultimately our competitiveness, depend on our ability to develop and maintain the proprietary aspects of our technology and operate without infringing on the proprietary rights of others. We rely on a combination of patent, trademark, trade secret and copyright law and contract restrictions to protect the proprietary aspects of our technologies. We seek to limit disclosure of our intellectual property by requiring employees, consultants, and any third parties with access to our proprietary information to execute confidentiality agreements and by restricting access to that information.
In August 2014, we received a notice of allowance from the U.S. Patent and Trademark Office for patent application number 13/573,442 titled “Composition of Novel Carbohydrate Drug for Treatment of Human Diseases.” The patent covers composition and chemical structural claims for compounds that includes the Company’s lead galectin inhibitor compound GR-MD-02 and will expire in December 2031. Claims include multiple routes of administration, including intravenous, subcutaneous and oral. The application also covers therapeutic formulations for use in the treatment of NASH (fatty liver disease), cancer and fibrotic, inflammatory and autoimmune disorders in which galectin proteins are involved, at least in part, in the pathogenesis. Additional specific claims encompass liver fibrosis, kidney fibrosis, lung fibrosis or heart fibrosis. The patent, assigned U.S. Patent No. 8,871,925, was issued October 28, 2014.
In May 2014, we received notice of allowance from the U.S. Patent and Trademark Office for patent application number 13/998,197 titled “Galactose-Pronged Carbohydrate Compounds for the Treatment of Diabetic Nephropathy and Associated Disorders.” The patent covers both composition claim for and uses of the Company’s carbohydrate-based galectin inhibitor compound GR-MD-02 in patients with diabetic nephropathy, a type of progressive kidney disease that occurs in individuals with diabetes. Diabetic nephropathy is the major cause for chronic renal failure in the United States. The patent, assigned U.S. Patent No. 8,828,971, was issued September 9, 2014.
In February 2014, we received notice of issuance that the U.S. Patent and Trademark Office issued patent number 8,658,787 to the Company for its application titled “Galacto-rhamnogalacturonate compositions for the treatment of non-alcoholic steatohepatitis and non-alcoholic fatty liver disease.” The patent covers the Company’s carbohydrate-based galectin inhibitor compound GR-MD-02 for use in patients with fatty liver disease with or without fibrosis or cirrhosis, providing patent protection through 2031. The major claims are for methods of obtaining galectin inhibitor compounds, obtaining a composition for parenteral or enteral administration in an acceptable pharmaceutical carrier and administering to a subject having at least one of the following: fatty liver, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, non-alcoholic hepatitis with liver fibrosis, non-alcoholic steatohepatitis with cirrhosis, or non-alcoholic steatohepatitis with cirrhosis and hepatocellular carcinoma. The use covers reversing or slowing the progression of disease activity or medical consequences of the disease. Applications are pending in multiple countries to extend patent protection globally.
In January 2014, we received a notice of allowance from the U.S. Patent and Trademark Office for Patent Application Number 13/550,962 titled “Galactose-Pronged Polysaccharides in a Formulation for Anti-fibrotic Therapies.” The patent covers both composition claim for and uses of the Company’s carbohydrate-based galectin inhibitor compound GR-MD-02 for use in patients with liver fibrosis in combination with other potential therapeutic agents. The patent covers use of GR-MD-02 with agents directed at multiple targets, some of which are currently in clinical development for fibrotic disorders including monoclonal antibodies to connective tissue growth factor, integrins, and TGF-ß1. The patent, assigned U.S. Patent No. 8,722,645, was issued May 13, 2014.
6
Table of Contents
Index to Financial Statements
In July 2012, we received a notice of issuance from the U.S. Patent and Trademark Office for the U.S. Patent number 8,236,780 issued on August 7, 2013 titled “Galactose-prolonged polysaccharides in a formulation for antifibrotic therapies”. This methods patent covers key methods of derivation and use for our carbohydrate-based galectin inhibitor compound for use in patients with chronic liver disease associated with the development of fibrosis, established liver fibrosis or end-stage scarring, or cirrhosis. The major claim is for a method of obtaining a galacto-rhamnogalacturan compound from an apple pectin, obtaining a composition for parenteral administration the galacto-rhamnogalacturonan compound in an acceptable pharmaceutical carrier and administering to a subject having at least one of the following: chronic liver disease associated with the development of fibrosis, established liver fibrosis or cirrhosis. The use covers inhibiting or slowing the progression of fibrosis. GR-MD-02 is covered by this patent and it provides opportunities for development of additional compounds in the class.
As of January 31, 2015, we held 12 granted U.S. patents, 14 foreign granted patents (Japan, E.U., New Zealand), 39 international patent applications, and 6 U.S. patent applications. Many of our patents and patent applications cover composition of matter for complex carbohydrate drugs and methods of use for reducing toxicity and enhancing chemotherapeutic drugs by co-administering a polysaccharide with a chemotherapeutic agent or for use in treatment of fibrosis. The scheduled expiration dates of our United States patents span from 2020 to 2033. We have corresponding patent applications pending in Europe, Israel, and Brazil. Additionally, we have patent applications in other areas to utilize our carbohydrate-based compounds to treat disease other than cancer. See “Risk Factors — Risks Related to Our Intellectual Property”. Our competitive position, in part, is contingent upon protection of our intellectual property.
Research
Our primary focus is on the design and testing of agents which target galectins in various in vitro and in vivo systems and which demonstrate efficacy in treatment of experimentally induced fibrosis or enhance immune system responsiveness in various tissues and in live animal models. We contract with independent laboratories and other facilities to conduct our research, which is designed, evaluated and managed by our scientists. While we conduct in house research related to our compounds at SBH laboratories in Massachusetts, we do not anticipate building additional in-house research or development facilities or hiring staff other than for purposes of designing and managing our out-sourced research.
As we develop products eligible for clinical trials, we contract with independent parties to assist in the design of the clinical trial protocols, arrange for and monitor the clinical trials, collect data and analyze data. In addition, certain clinical trials for our products may be conducted by government-sponsored agencies and will be dependent on governmental participation and funding. Our dependence on independent parties and clinical sites involves risks including reduced control over the timing and other aspects of our clinical trials.
In February 2013, the Company established a collaborative drug discovery program with Dr. Geert-Jan Boons’ (“Dr. Boons”) laboratory located in the Complex Carbohydrate Research Center at the University of Georgia. This on-going program is focused on the discovery of new carbohydrate molecules that can be used in the therapy of diseases where galectin proteins play a major role, including cancer, and inflammatory and fibrotic disorders. The aim of this program is to develop a pipeline of drugs that can target galectins. This is an important goal as follow-on compounds for our drugs currently in development and to extend the potential indications and routes of administration. The Complex Carbohydrate Research Center is a world-class program and Dr. Boons is a world renowned and pre-eminent carbohydrate chemist.
In September, 2014, the Company established a collaborative research program with Dr. William Redmonds’ laboratory located at the Providence Portland Medical Center, Portland, Oregon. This program focuses on combination immunotherapy plus galectin inhibition to augment tumor immunogenicity.
7
Table of Contents
Index to Financial Statements
During the years ended December 31, 2014 2013 and 2012, our expenditures for research and development were $8.4 million, $5.7 million and $4.5 million, respectively. We expense all research and development costs as they are incurred.
In January, 2014 we created, with SBH Sciences, Inc. (Natick, Ma), Galectin Sciences, LLC, a collaborative joint venture to research and develop small organic molecule inhibitors of galectin-3 for oral administration.
Using computer molecular modeling techniques coupled with in vitro screening of a variety of compound libraries, SBH Sciences had identified several small organic molecules with promising galectin-3 inhibitory activity in vitro. Galectin Sciences LLC will further develop these unique organic molecule inhibitors of galectin-3 as drug candidates as well as develop additional candidates. Galectin Sciences LLC will build on the scientific body of knowledge amassed by SBH Sciences, coupled with Galectin Therapeutics’ knowledge and expertise of galectins’ pathological role and mechanism of action in inflammation, fibrosis and many cancers. The long-term goal of this effort is to identify and develop drug candidates that are highly specific galectin inhibitors which may be formulated for oral administration. The intermediate term goal is the development of small molecule inhibitors of galectin-3 which exhibit activity in in vivo preclinical disease models of fibrosis and cancer in which galectins play a key role.
Because, increased levels of galectin proteins have been implicated in a very large number of inflammatory, fibrotic and neoplastic diseases; the discovery and development of orally active galectin inhibitors would be a major step towards expanded treatment approaches for these disorders. This early drug discovery effort may lead to drugs that would expand our pipeline as follow on compounds to our first in class galectin inhibitors, GR-MD-02 and GM-CT-01.
Manufacturing and Marketing
We are a development stage company at this time and do not intend to establish internal facilities for the manufacture of our products for clinical or commercial production. To have our products manufactured, we have developed and will continue to develop relationships with third-parties that have established pharmaceutical manufacturing capabilities and expertise. We are not a party to any long-term agreement with any of our suppliers and, accordingly, we have our products manufactured on a purchase-order basis from one of two primary well-known and established pharmaceutical suppliers that meeting FDA requirements.
Because our products are in the development stage, we have not created a sales and marketing staff to commercialize pharmaceutical products. If we develop products eligible for commercial sale, we will need to develop a sales and marketing capability or rely on third parties such as licensees, collaborators, joint venture partners or independent distributors to market and sell those products. Our dependence on third-party manufacturers and marketers will involve risks relating to our reduced control, and other risks including those discussed in “Risk Factors — Risks Related to our Company — There are risks associated with reliance on third parties for manufacturing, marketing, sales, managed care and distribution infrastructure channels.”
Competition
Many biotechnology and pharmaceutical companies are developing new technologies for the treatment of cancer and other diseases. Technologies such as monoclonal antibodies could be competitive with our galectin therapeutic platforms. Other companies are trying to improve the therapeutic profile of widely used protein-based drugs. While these companies may broaden the market for our products they may also provide competitive alternatives to our products. We expect increased competition in the area of galectins will be fueled by a nearly exponential increase in the publication rate of research papers on galectins.
See “Risk Factors — Risks Related to Our Company — We face intense competition in the biotechnology and pharmaceutical industries” for additional discussion related to our current and potential competition.
8
Table of Contents
Index to Financial Statements
Government Regulation
The research, development, testing, manufacture, labeling, promotion, advertising, distribution, and marketing, among other things, of our products are extensively regulated by governmental authorities in the United States and other countries. The FDA regulates drugs under the federal Food, Drug, and Cosmetic Act and its implementing regulations. Failure to comply with the applicable U.S. requirements may subject us to administrative or judicial sanctions, such as FDA refusal to approve pending New Drug Applications (“NDAs”), warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, and/or criminal prosecution.
Drug Approval Process
Drugs may not be marketed in the U.S. until the FDA has approved them. The steps required before a drug may be marketed in the U.S. include:
| 1. | Pre-clinical laboratory tests, animal studies, and formulation studies, |
| 2. | Submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin, |
| 3. | Adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each indication, |
| 4. | Submission to the FDA of a NDA, |
| 5. | Satisfactory completion of an FDA inspection of the manufacturing facility or facilities, at which the drug is produced to assess compliance with current good manufacturing procedures (“cGMP”) established by the FDA, |
| 6. | FDA review and approval of the NDA, and |
| 7. | FDA review and approval of a trademark used in connection with a pharmaceutical. |
Pre-clinical tests include laboratory evaluation of product chemistry, toxicity, and formulation, as well as numerous in vitro and in vivo animal studies. The results of the pre-clinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may begin and the Company must resolve any outstanding FDA concerns or questions before clinical trials can proceed. There is no certainty that submission of an IND will result in the FDA allowing clinical trials to begin.
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators and constant oversight by the FDA or foreign regulatory authorities. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined. Each trial must be reviewed and approved by an independent Institutional Review Board (“IRB”), before it can begin. Study subjects must sign an informed consent form before participating in a clinical trial. Phase 1 usually involves the initial introduction of the investigational drug into patients to evaluate its safety, dosage tolerance, pharmacodynamics, and, if possible, to gain an early indication of its effectiveness. Phase 2 usually involves trials in a limited patient population to (i) evaluate dosage tolerance and appropriate dosage; (ii) identify possible adverse effects and safety risks; and (iii) evaluate preliminarily the efficacy of the drug for specific indications. Phase 3 trials usually further evaluate clinical efficacy and test further for safety by using the drug in its final form in an expanded patient population. There is no assurance that these trials will be completed within a specified period of time, if at all.
9
Table of Contents
Index to Financial Statements
Assuming successful completion of the required clinical testing, the results of the pre-clinical studies and of the clinical studies, together with other detailed information, including information on the manufacture and composition of the drug, are submitted to the FDA in an NDA requesting approval to market the product for one or more indications. Before approving an NDA, the FDA usually will inspect the facilities at which the drug is manufactured, and will not approve the product unless compliance with cGMP is satisfactory. If the FDA evaluates the NDA and the manufacturing facilities as acceptable, the FDA will generally issue an approval letter. If the FDA evaluates the NDA submission or the manufacturing facilities as not acceptable, the FDA will generally outline the deficiencies in the submission and often will request additional testing or information. Even if an applicant submits the requested additional information, the FDA ultimately may decide that the NDA does not satisfy the regulatory criteria for approval. The testing and approval process requires substantial time, effort, and financial resources, and there is no assurance that any approval will be granted on a timely basis, if at all. After approval, certain changes to the approved product, such as adding new indications, manufacturing changes, or additional labeling claims are subject to further FDA review and approval.
See “Risk Factors — Risks Related to the Regulation of Our Products — We will need regulatory approvals to commercialize our products” for additional discussion of regulatory risks related to our drug development program.
FDA Priority Review
FDA procedures provide for priority review of an NDA submitted for drugs that, compared to currently marketed products, offer a significant improvement in the treatment, diagnosis, or prevention of a disease. NDAs that are granted priority review are acted upon more quickly than NDAs given standard review. If we were to seek priority review, there can be no guarantee that the FDA will grant priority review status, that priority review status will affect the time of review, or that the FDA will approve the NDA submitted for any of our product candidates, whether or not priority review status is granted.
Post-Approval Requirements
If FDA approval of one or more of our products is obtained, we will be required to comply with a number of post-approval requirements. For example, holders of an approved NDA are required to report certain adverse reactions to the FDA and to comply with certain requirements concerning advertising and promotional labeling for their products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. In addition, discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved NDA, including withdrawal of the product from the market. Also, new government requirements may be established that could delay or prevent regulatory approval of our products under development.
Regulation Outside the United States
Before our products can be marketed outside of the United States, they are subject to regulatory approval similar to that required in the United States, although the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary widely from country to country. No action can be taken to market any product in a country until an appropriate application has been approved by the regulatory authorities in that country. The current approval process varies from country to country, and the time spent in gaining approval varies from that required for FDA approval. In certain countries, the sales price of a product must also be approved. The pricing review period often begins after market approval is granted. No assurance can be given that even if a product is approved by a regulatory authority, satisfactory prices will be approved for such product.
10
Table of Contents
Index to Financial Statements
Environmental Regulation
Pharmaceutical research and development involves the controlled use of hazardous materials. Biotechnology and pharmaceutical companies must comply with laws and regulations governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain materials, biological specimens and wastes. We do not anticipate building in-house research, development or manufacturing facilities, and, accordingly, do not expect to have to comply directly with environmental regulation. However, our contractors and others conducting research, development or manufacturing activities for us may be required to incur significant compliance cost, and this could in turn could increase our expense or delay our completion of research or manufacturing programs.
Employees
We currently have seven full-time employees, three of whom are involved primarily in management of our pre-clinical research and development and clinical trials and four who were involved primarily in management and administration of our Company. We also utilize contractors who provide product development, manufacture, analytical testing and clinical trial support.
| Item 1A. | Risk Factors |
An investment in our common stock involves a high degree of risk. You should carefully consider the risks described below and the other information before deciding to invest in our common stock. The risks described below are not the only ones facing our Company. Additional risks not presently known to us or that we currently consider immaterial may also adversely affect our business. We have attempted to identify below the major factors that could cause differences between actual and planned or expected results, but we cannot assure you that we have identified all of those factors.
If any of the following risks actually happen, our business, financial condition and operating results could be materially adversely affected. In this case, the trading price of our common stock could decline, and you could lose all or part of your investment.
Risks Related to Our Company
We have incurred net losses to date and must raise additional capital in order to continue to operate after September 30, 2016.
We have incurred net losses in each year of operation since our inception in July 2000. Our accumulated deficit as of December 31, 2014 was $119 million. We had $29.1 million of unrestricted cash as of December 31, 2014. Additionally, in January and February 2015, the Company received approximately $4.1 million in net proceeds from the issuance of common stock at then-current market prices through its at the market (“ATM”) financing arrangement. The Company currently believes there is sufficient cash to fund currently planned operations through the third quarter of 2016. We will require more cash to fund our operations after the third quarter of 2016. However, there can be no assurance that we will be successful in obtaining such new financing or, if available, that such financing will be obtainable on terms favorable to us. If our current clinical trials are unsuccessful or do not produce positive results, it may be particularly difficult for us to raise additional capital. If we do not raise additional cash for operations after the third quarter of 2016, we may not be able to continue operations and may be forced to seek bankruptcy protection.
We may raise capital through public or private equity financings, partnerships, debt financings, bank borrowings, or other sources. Additional funding may not be available on favorable terms or at all. If adequate funds are not otherwise available, we may need to significantly curtail operations. To obtain additional funding, we may need to enter into arrangements that require us to relinquish rights to certain technologies, products and/or potential markets. To the extent that additional capital is raised through the sale of equity, or securities convertible into equity, our equity holders may experience dilution of their proportionate ownership of the Company.
11
Table of Contents
Index to Financial Statements
We are a development stage company and have not yet generated any revenue.
We are a development stage company and have not generated any revenues to date. There is no assurance that we will obtain FDA approval of GR-MD-02, GM-CT-01, or any other of our products in development and, even if we do so, that we will generate revenue sufficient to become profitable. Our failure to generate revenue and profit would likely lead to loss of your investment.
Our ability to generate revenue from product sales and achieve profitability will depend upon our ability to successfully commercialize products, including any of our current product candidates, or other product candidates that we may in-license or acquire in the future. Even if we are able to successfully achieve regulatory approval for these product candidates, we do not know when any of these products will generate revenue from product sales for us, if at all. Our ability to generate revenue from product sales from our current or future product candidates also depends on a number of additional factors, including our ability to:
| • | successfully complete development activities, including the necessary clinical trials; |
| • | complete and submit new drug applications, or NDAs, to the U.S. Food and Drug Administration, or FDA, and obtain regulatory approval for indications for which there is a commercial market; |
| • | complete and submit applications to, and obtain regulatory approval from, foreign regulatory authorities; |
| • | successfully complete all required regulatory agency inspections; |
| • | set a commercially viable price for our products; |
| • | obtain commercial quantities of our products at acceptable cost levels; |
| • | find suitable distribution partners to help us market, sell and distribute our approved products in other markets; and |
| • | obtain coverage and adequate reimbursement from third parties, including government and private payers. |
In addition, because of the numerous risks and uncertainties associated with product development, including that our product candidates may not advance through development or achieve the endpoints of applicable clinical trials, we are unable to predict the timing or amount of increased expenses, or when or if we will be able to achieve or maintain profitability. Even if we are able to complete the development and regulatory process for any product candidates, we anticipate incurring significant costs associated with commercializing these products.
Even if we are able to generate revenues from the sale of our products, we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce our operations
We are largely dependent on the success of our two lead product candidates, GR-MD-02 and GM-CT-01 and we cannot be certain that these product candidates will receive regulatory approval or be successfully commercialized.
We currently have no products for sale and we cannot guarantee that we will ever have any drug products approved for sale. We and our product candidates are subject to extensive regulation by the FDA and comparable regulatory authorities in other countries governing, among other things, research, testing, clinical trials, manufacturing, labeling, promotion, selling, adverse event reporting and recordkeeping. We are not permitted to market any of our product candidates in or outside the United States until we receive approval of a new drug application for a product candidate from the FDA or the equivalent approval from a foreign regulatory authority. Obtaining FDA approval is a lengthy, expensive and uncertain process.
12
Table of Contents
Index to Financial Statements
Before obtaining regulatory approval for the sale of any drug candidate, we must conduct extensive pre-clinical studies and clinical trials to demonstrate the safety and efficacy of our product candidates in humans.
GR-MD-02 our lead product candidate for fibrosis has completed Phase 1 of the human clinical trial phase of drug development in the US and is entering Phase 2 clinical trials in North America. GR-MD-02 is also currently in an investigator sponsored, human Phase 1B clinical trial being conducted by Providence Portland Medical Center in combination with Yervoy® (ipilimumab) in patients with metastatic melanoma. We cannot assure you that these trials will yield successful results, that they will lead to the generation of revenue, or that we will obtain regulatory approval in other countries.
There are currently no FDA clinical trials ongoing for GM-CT-01.
We filed for an IND with the FDA for GR-MD-02 in January 2013 for initiating human clinical trials in patients with NASH, and the FDA notified us in March 2013 that we may proceed with a Phase 1 clinical trial. Our Phase 1 clinical trial began in July 2013 and was completed in 2014. Pre-clinical studies and clinical trials are expensive, time-consuming and ultimately may not be successful. The results of pre-clinical and initial clinical testing of these products may not necessarily indicate the results that will be obtained from later or more extensive testing. Also, it is possible to suffer significant setbacks in advanced clinical trials, even after obtaining promising results in earlier trials. For example, even though GM-CT-01 progressed successfully through Phase 1 and was progressing successfully through Phase 2 human trials (which were only partially completed due to financing issues in 2010), it may fail in Phase 3 trials or in later stages of development. We will engage others to conduct our clinical trials, including clinical research organizations and, possibly, government-sponsored agencies. Pre-clinical studies and clinical trials may not start or be completed as we forecast and may not achieve the desired results. The time required to obtain FDA and other approvals is unpredictable but often can take years following the commencement of clinical trials, depending upon the complexity of the drug candidate.
Even if we receive regulatory approval, we may be unable to commercialize our product candidates.
Even if GR-MD-02, GM-CT-01 and other future product candidates achieve positive results in clinical trials, we may be unable to commercialize them. The availability of government and third party payor reimbursement, and pricing, especially compared to competitor products, could affect our ability to commercialize our product candidates. Our general inability to obtain necessary regulatory approvals and, if obtained, to commercialize our products would substantially impair our viability.
There are risks associated with our reliance on third parties to design trial protocols, arrange for and monitor the clinical trials, and collect and analyze data.
As we develop products eligible for clinical trials, we will contract with independent parties to assist us in the design of the trial protocols, arrange for and monitor the clinical trials, collect data and analyze data. For instance, in February 2013, we entered into an agreement with CTI Clinical Trial Services, Inc. and CTI Clinical Consulting Services, Inc. for the purpose of assisting us in the design, development and conduct of one or more clinical research studies from time to time. In accordance with this agreement, CTI is conducting the Phase 1 clinical trial for GR-MD-02 to evaluate the drug’s safety in subjects with NASH with advanced hepatic fibrosis. In addition, certain clinical trials for our products may be conducted by government-sponsored agencies and will be dependent on governmental participation and funding. Additionally, GR-MD-02 is being studied by Providence Portland Medical Center in an Investigator-sponsored IND to conduct a Phase 1B study to determine if GR-MD-02 enhances the probability of melanoma response with ipilimumab by inducing proliferation, activation and memory function of CD8+ T cells in human patients. This study represents a novel approach for patients with metastatic melanoma. The IND was approved by FDA in February 2014.
Our dependence on independent parties and clinical sites involves risks including reduced control over the timing and other aspects of our clinical trials.
13
Table of Contents
Index to Financial Statements
There are risks associated with our reliance on third parties for manufacturing, marketing, sales, managed care and distribution infrastructure and channels.
We do not have, and do not now intend to develop, facilities for the manufacture of any of our products for clinical or commercial production. At this time, we are not a party to any long-term agreement with any of our suppliers, and accordingly, we have our products manufactured on a purchase-order basis from one of two primary suppliers. We are developing relationships with manufacturers and will enter into collaborative arrangements with licensees or have others manufacture our products on a contract basis. We expect to depend on such collaborators to supply us with products manufactured in compliance with standards imposed by the FDA and foreign regulators.
We have limited experience in marketing, sales or distribution, and we do not intend to develop a sales and marketing infrastructure to commercialize our pharmaceutical products. If we develop commercial products, we will need to rely on licensees, collaborators, joint venture partners or independent distributors to market and sell those products. Thus, we expect that we will be required to enter into agreements with commercial partners to engage in sales, marketing and distribution efforts around our products in development. We may be unable to establish or maintain third-party relationships on a commercially reasonable basis, if at all. In addition, these third parties may have similar or more established relationships with our competitors. If we do not enter into relationships with third parties for the sales and marketing of our proposed products, we will need to develop our own sales and marketing capabilities.
Even if engaged, these distributors may:
| • | fail to satisfy financial or contractual obligations to us; |
| • | fail to adequately market our products; |
| • | cease operations with little or no notice to us; or |
| • | offer, design, manufacture or promote competing formulations or products. |
If we fail to develop sales, managed care, marketing and distribution channels, we would experience delays in generating sales and incur increased costs, which would harm our financial results.
We are exposed to product liability, pre-clinical and clinical liability risks, which could place a financial burden upon us, should we be sued, because we do not currently have product liability insurance beyond our general insurance coverage.
Our business exposes us to potential product liability and other liability risks that are inherent in the testing, manufacturing and marketing of pharmaceutical formulations and products; accordingly, claims may be asserted against us. In addition, the use in our clinical trials of pharmaceutical formulations and products that our potential collaborators may develop and the subsequent sale of such formulations or products by us or our potential collaborators may cause us to assume a portion of or all of the product liability risks. A successful liability claim or series of claims brought against us could have a material adverse effect on our business, financial condition and results of operations.
Because we do not currently have any FDA-approved products or formulations, we do not currently have any product liability insurance covering commercialized products. We may not be able to obtain or maintain adequate product liability insurance on acceptable terms, if at all, or such insurance may not provide adequate coverage against our potential liabilities. Furthermore, our current and potential partners with whom we have collaborative agreements or our future licensees may not be willing to indemnify us against these types of liabilities and may not, themselves, be sufficiently insured or have sufficient liquidity to satisfy any product liability claims. Claims or losses in excess of any product liability insurance coverage that may be obtained by us could have a material adverse effect on our business, financial condition and results of operations.
14
Table of Contents
Index to Financial Statements
We face intense competition in the biotechnology and pharmaceutical industries.
The biotechnology and pharmaceutical industries are intensely competitive. We face direct competition from U.S. and foreign companies focusing on pharmaceutical products, which are rapidly evolving. Our competitors include major multinational pharmaceutical and chemical companies, specialized biotechnology firms and universities and other research institutions. Many of these competitors possess greater financial and other resources, larger research and development staffs and more effective marketing and manufacturing organizations than we possess. In addition, academic and government institutions are increasingly likely to enter into exclusive licensing agreements with commercial enterprises, including our competitors, to market commercial products based on technology developed at such institutions. Our competitors may succeed in developing or licensing technologies and products that are more effective, or succeed in obtaining FDA or other regulatory approvals for product candidates before we do. Acquisitions of, or investments in, competing pharmaceutical or biotechnology companies by large corporations could increase such competitors’ financial, marketing, manufacturing and other resources.
The market for our proposed products is rapidly changing and competitive, and new drugs and new treatments which may be developed by others could impair our ability to maintain and grow our business and remain competitive.
The pharmaceutical and biotechnology industries are subject to rapid and substantial technological change. Developments by others may render our proposed products noncompetitive or obsolete, or we may be unable to keep pace with technological developments or other market factors. Technological competition from pharmaceutical and biotechnology companies, universities, governmental entities and others diversifying into the field is intense and is expected to increase.
As a pre-revenue company engaged in the development of drug technologies, our resources are limited and we may experience technical challenges inherent in such technologies. Competitors have developed or are in the process of developing technologies that are, or in the future may be, the basis for competition. Some of these technologies may have an entirely different approach or means of accomplishing similar therapeutic effects compared to our proposed products. Our competitors may develop drugs that are safer, more effective and less costly than our proposed products and, therefore, present a serious competitive threat to us.
The potential widespread acceptance of therapies that are alternatives to ours may limit market acceptance of our proposed products, even if commercialized. Many of our targeted diseases and conditions can also be treated by other medications. These treatments may be widely accepted in medical communities and have a longer history of use. The established use of these competitive drugs may limit the potential for our technologies, formulations and products to receive widespread acceptance even if commercialized.
Our lack of operating experience may cause us difficulty in managing our growth.
We have limited experience in manufacturing or procuring products in commercial quantities, conducting other later-stage phases of the regulatory approval process, selling pharmaceutical products, or negotiating, establishing and maintaining strategic relationships. Although we have engaged a number of consultants to assist us, any additional growth may require us to expand our management, operational and financial systems and controls. If we are unable to do so, our business and financial condition would be materially harmed. If rapid growth occurs, it may strain our managerial, operational and financial resources.
We depend on key individuals to develop our products and core technologies and pursue collaborative relationships.
We are highly dependent on Peter G. Traber, M.D. Dr. Traber is our Chief Executive Officer and our Chief Medical Officer who, among other things, designs and leads our pre-clinical and clinical studies, as well as our U.S. and European regulatory processes. The loss of Dr. Traber or failure to attract or retain other key personnel could prevent us from developing our products and core technologies and pursuing collaborative relationships.
15
Table of Contents
Index to Financial Statements
We may fail to comply with our reporting and other requirements under federal securities laws.
As a publicly traded company, we are subject to the reporting requirements of the Exchange Act. The Exchange Act requires that we file annual, quarterly and current reports. Our failure to prepare and disclose this information in a timely manner could subject us to penalties under federal securities laws, expose us to lawsuits and restrict our ability to access financing. We may be required to implement additional and expensive finance and accounting systems, procedures and controls as we grow our business and organization to satisfy new reporting requirements, which will increase our costs and require additional management resources.
Risks Related to the Regulation of our Products
We will need regulatory approvals to commercialize our products.
We are required to obtain approval (i) from the FDA in order to sell our products in the U.S. and (ii) from foreign regulatory authorities in order to sell our products in other countries. The FDA’s review and approval process is lengthy, expensive and uncertain. Extensive pre-clinical and clinical data and supporting information must be submitted to the FDA for each indication for each product candidate in order to secure FDA approval. Before receiving FDA clearance to market our proposed products, we will have to demonstrate that our products are safe on the patient population and effective for the diseases that are to be treated. Clinical trials, manufacturing and marketing of drugs are subject to the rigorous testing and approval process of the FDA and equivalent foreign regulatory authorities. The Federal Food, Drug and Cosmetic Act and other federal, state and foreign statutes and regulations govern and influence the testing, manufacture, labeling, advertising, distribution and promotion of drugs and medical devices. As a result, regulatory approvals can take several years to acquire and may further require the expenditure of substantial financial, managerial and other resources. The FDA could reject an application or, in the alternative, require us to conduct additional clinical or other studies as part of the regulatory review process. Delays in obtaining or failure to obtain FDA approvals would delay or prevent the commercialization of our product candidates, which would prevent, defer or decrease our receipt of revenues. In addition, should we receive initial regulatory approval, our product candidates will be subject to extensive and rigorous ongoing domestic and foreign government regulation.
Even if we obtain regulatory approvals, our marketed drugs will be subject to ongoing regulatory review. If we fail to comply with ongoing regulatory requirements, we could lose our approvals to market drugs, in which case our business would be materially adversely affected.
Following regulatory approval in the United States of any drugs we may develop, we will remain subject to continuing regulatory review, including the review of adverse drug experiences and clinical results that are reported after our drug products are made available to patients. This would include results from any post marketing tests or vigilance required as a condition of approval. The manufacturer and manufacturing facilities we use to make any of our drug products will also be subject to periodic review and inspection by the FDA. The discovery of any new or previously unknown problems with the product, manufacturer or facility may result in restrictions on the drug or manufacturer or facility, including withdrawal of the drug from the market. We would continue to be subject to the FDA requirements governing the labeling, packaging, storage, advertising, promotion, recordkeeping, and submission of safety and other post-market information for all of our product candidates, even those that the FDA had approved. If we fail to comply with applicable continuing regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and other adverse consequences.
The drug development process to obtain FDA approval is very costly and time consuming and if we cannot complete our clinical trials in a cost-effective manner, our results of operations may be adversely affected.
Costs and timing of clinical trials may vary significantly over the life of a project owing to the following non-exclusive reasons:
| • | the duration of the clinical trial; |
16
Table of Contents
Index to Financial Statements
| • | the number of sites included in the trials; |
| • | the countries in which the trial is conducted; |
| • | the length of time required and ability to enroll eligible patients; |
| • | the number of patients that participate in the trials; |
| • | the number of doses that patients receive; |
| • | the drop-out or discontinuation rates of patients; |
| • | per patient trial costs; |
| • | third party contractors failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
| • | our drug product candidates having different chemical and pharmacological properties in humans than in lab testing; |
| • | the need to suspend or terminate our clinical trials; |
| • | insufficient or inadequate supply or quality of drug product candidates or other necessary materials to conduct our trials; |
| • | potential additional safety monitoring, or other conditions required by FDA or comparable foreign regulatory authorities regarding the scope or design of our clinical trials, or other studies requested by regulatory agencies; |
| • | problems engaging IRBs to oversee trials or in obtaining and maintaining IRB approval of studies; |
| • | the duration of patient follow-up; |
| • | the efficacy and safety profile of the product candidate; |
| • | the costs and timing of obtaining regulatory approvals; and |
| • | the costs involved in enforcing or defending patent claims or other intellectual property rights. |
Each of the above factors and other unanticipated factors beyond our control could prevent us from gaining approval for our drugs in a cost-effective and timely manner, which could have a material adverse impact on our business.
If users of our proposed products are unable to obtain adequate reimbursement from third-party payers, market acceptance of our proposed products may be limited and we may not achieve revenues or profits.
The continuing efforts of governments, insurance companies, health maintenance organizations and other payers of healthcare costs to contain or reduce costs of health care may affect our future revenues and profitability as well as the future revenues and profitability of our potential customers, suppliers and collaborative partners in addition to the availability of capital. In other words, our ability to commercialize our proposed products will depend in large part on the extent to which appropriate reimbursement levels for the cost of our proposed formulations, products and related treatments are obtained by the health care providers of these products and treatments. At this time we cannot predict the precise impact of the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Affordability Act of 2010, the comprehensive health care reform legislation passed by Congress in March 2010. It is possible that the adoption of this legislation could harm our business, financial condition and results of operations.
Data obtained from clinical trials are not necessarily predictive of future results, may be negative or inconclusive, and are susceptible to varying interpretations, which could delay, limit or prevent regulatory clearances.
Data already obtained, or in the future obtained, from pre-clinical studies and clinical trials do not necessarily predict the results that will be obtained from later pre-clinical studies and clinical trials. Moreover,
17
Table of Contents
Index to Financial Statements
pre-clinical and clinical data may be negative or inconclusive. In addition, data is susceptible to varying interpretations. Negative or inconclusive data, or data interpreted in various ways, could delay, limit or prevent regulatory approval. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after having obtained promising results in earlier trials. Despite the results reported in earlier clinical trials for GR-MD-02, our clinical trials may not demonstrate sufficient levels of safety and efficacy necessary to obtain the requisite regulatory approvals for our drugs, and thus, our proposed drugs may not be approved for marketing. If later-stage clinical trials do not produce favorable results, our ability to achieve regulatory approval for any of our product candidates may be adversely impacted. The failure to adequately demonstrate the safety and effectiveness of a proposed formulation or product under development could delay or prevent regulatory clearance of the potential drug. The resulting delays in commercialization could materially harm our business.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following any marketing approval.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authority. Although we are not currently aware of any undesirable side effects caused by our product candidates, it is possible that they may be identified in the clinical trial process.
As a result of undesirable side effects or safety or toxicity issues that we may experience in our clinical trials, we may not receive approval to market any product candidates, which could prevent us from ever generating revenues or achieving profitability. Results of our trials could reveal an unacceptably high severity and prevalence of side effects. In such an event, our trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order us to cease further development or deny approval of our product candidates for any or all targeted indications. These side effects could affect patient recruitment or the ability of enrolled subjects to complete the trial or result in potential product liability claims.
Additionally, if any of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such product, a number of potentially significant negative consequences could result, including:
| • | we may be forced to suspend marketing of such product; |
| • | regulatory authorities may withdraw their approvals of such product; |
| • | regulatory authorities may require additional warnings on the label that could diminish the usage or otherwise limit the commercial success of such products; |
| • | we may be required to conduct post-market studies; |
| • | we could be sued and held liable for harm caused to subjects or patients; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved.
We will need to obtain FDA approval of any proposed product brand names, and any failure or delay associated with such approval may adversely impact our business.
A pharmaceutical product cannot be marketed in the U.S. or other countries until it has completed rigorous and extensive regulatory review processes, including approval of a brand name. Any brand names we intend to use for our product candidates will require approval from the FDA regardless of whether we have secured a formal trademark registration from the U.S. Patent and Trademark Office, or the PTO. The FDA typically
18
Table of Contents
Index to Financial Statements
conducts a review of proposed product brand names, including an evaluation of potential for confusion with other product names. The FDA may also object to a product brand name if it believes the name inappropriately implies medical claims. If the FDA objects to any of our proposed product brand names, we may be required to adopt an alternative brand name for our product candidates. If we adopt an alternative brand name, we would lose the benefit of our existing trademark applications for such product candidate and may be required to expend significant additional resources in an effort to identify a suitable product brand name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA. We may be unable to build a successful brand identity for a new trademark in a timely manner or at all, which would limit our ability to commercialize our product candidates.
Failure to obtain regulatory approval in international jurisdictions would prevent our product candidates from being marketed abroad.
In order to market and sell our products in the European Union and many other jurisdictions, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the United States generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market. If we are unable to obtain approval of any of our product candidates by regulatory authorities in the European Union or other countries, the commercial prospects of that product candidate may be significantly diminished and our business prospects could decline.
Risks Related to Our Intellectual Property
Our competitive position is contingent upon the protection of our intellectual property.
Development and protection of our intellectual property are critical to our business. All of our intellectual property, patented or otherwise, has been invented and/or developed by employees or former employees of the Company. Our success depends, in part, on our ability to obtain patent protection for our products or processes in the U.S. and other countries, protect trade secrets and prevent others from infringing on our proprietary rights. We will only be able to protect our product candidates from unauthorized making, using, selling, offering to sell or importation by third parties to the extent that we have rights under valid and enforceable patents or trade secrets that cover these activities. If we do not adequately protect our intellectual property, competitors may be able to practice our technologies.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date in the United States. The biotechnology patent situation outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed in our pending patent applications or enforced in our issued patents or in third-party patents.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| • | others may be able to make compounds that are competitive with our product candidates but are not covered by the claims of our patents; |
19
Table of Contents
Index to Financial Statements
| • | we might not have been the first to make the inventions covered by our pending patent applications; |
| • | we might not have been the first to file patent applications for these inventions; |
| • | it is possible that our pending patent applications will not result in issued patents; |
| • | we may not develop additional proprietary technologies that are patentable; or |
| • | the patents of others may have an adverse effect on our business. |
We also may rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Although we require our scientific and technical employees and consultants to enter into broad assignment of inventions agreements, and all of our employees, consultants and corporate partners with access to proprietary information to enter into confidentiality agreements, these agreements may not be honored. Enforcing a claim that a third party illegally obtained, and is using, our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to protect our rights to, or use of, our technology.
Some or all of our patent applications may not issue as patents, or the claims of any issued patents may not afford meaningful protection for our technologies or products. In addition, patents issued to us or our licensors, if any, may be challenged and subsequently narrowed, invalidated or circumvented. Patent litigation is widespread in the biotechnology industry and could harm our business. Litigation might be necessary to protect our patent position or to determine the scope and validity of third-party proprietary rights.
If we choose to go to court to stop someone else from using the inventions claimed in our patents, that individual or company would have the right to ask the court to rule that such patents are invalid and/or should not be enforced against that third party. These lawsuits are expensive and we may not have the required resources to pursue such litigation or to protect our patent rights. In addition, there is a risk that the court will decide that these patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of these patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe our rights in these patents.
Furthermore, a third party may claim that we are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in our normal operations and activities, including making or selling our product candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and technical personnel. There is a risk that a court would decide that we are infringing the third party’s patents and would order us to stop the activities covered by the patents. In addition, there is a risk that a court will order us to pay the other party treble damages for having violated the other party’s patents. The biotechnology industry has produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our products or methods of use either do not infringe the claims of the relevant patent and/or that the patent claims are invalid, and we may not be able to do this. Proving invalidity in the U.S., in particular, is difficult since it requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
Because some patent applications in the United States may be maintained in secrecy until the patents are issued, patent applications in the United States and many foreign jurisdictions are typically not published until eighteen months after filing, and publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our issued patents or
20
Table of Contents
Index to Financial Statements
our pending applications or that we were the first to invent the technology. Our competitors may have filed, and may in the future file, patent applications covering technology similar to ours. Any such patent application may have priority over our patent applications and could further require us to obtain rights to issued patents covering such technologies. If another party has filed a United States patent application on inventions similar to ours, we may have to participate in an interference or other proceeding in the PTO or a court to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, resulting in a loss of our United States patent position with respect to such inventions.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
Obtaining and maintaining our patent protection depends upon compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The PTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
Our failure to secure trademark registration could adversely affect our ability to market our product candidates and our business.
Our trademark applications in the United States, when filed, and any other jurisdictions where we may file may not be allowed for registration, and our registered trademarks may not be maintained or enforced. During trademark registration proceedings, we may receive rejections. Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the PTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our applications and/or registrations, and our applications and/or registrations may not survive such proceedings. Failure to secure such trademark registrations in the United States and in foreign jurisdictions could adversely affect our ability to market our product candidates and our business.
Confidentiality agreements with employees and others may not adequately prevent disclosure of our trade secrets and other proprietary information and may not adequately protect our intellectual property, which could impede our ability to compete.
Because we operate in the highly technical field of biotechnology and pharmaceutical development, we rely in part on trade secret protection in order to protect our proprietary trade secrets and unpatented know-how. However, trade secrets are difficult to protect, and we cannot be certain that others will not develop the same or similar technologies on their own. We have taken steps, including entering into confidentiality agreements with all of our employees, consultants and corporate partners to protect our trade secrets and unpatented know-how. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. We also typically obtain agreements from these parties which provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Enforcing a claim that a party illegally obtained and is using our trade secrets or know-how is difficult, expensive
21
Table of Contents
Index to Financial Statements
and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets or know-how. The failure to obtain or maintain trade secret protection could adversely affect our competitive position.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to Our Common Stock
The market price of our common stock may be volatile and adversely affected by several factors. This could subject us to securities class action litigation and our stockholders could incur substantial losses.
The market price of our common stock could fluctuate significantly in response to various factors and events, including but not limited to:
| • | the results of our pre-clinical studies and clinical trials, including interim results, as well as those of our competitors; |
| • | regulatory actions with respect to our products or our competitors’ products; |
| • | our ability to integrate operations, technology, products and services; |
| • | our ability to execute our business plan; |
| • | operating results below expectations; |
| • | our issuance of additional securities, including debt or equity or a combination thereof, which may be necessary to fund our operating expenses; |
| • | announcements of technological innovations or new products by us or our competitors; |
| • | the success of competitive products; |
| • | loss of any strategic relationship; |
| • | industry developments, including, without limitation, changes in healthcare policies or practices or third-party reimbursement policies; |
| • | regulatory or legal developments in the United States and other countries; |
| • | the level of expenses related to any of our product candidates or clinical development programs; |
| • | disputes or other developments related to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies; |
| • | economic and other external factors; |
| • | period-to-period fluctuations in our financial results; |
| • | sales of our common stock by us, our insiders or our other stockholders; and |
| • | whether an active trading market in our common stock develops and is maintained. |
In addition, the market price for securities of pharmaceutical and biotechnology companies historically has been highly volatile, and the securities markets have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These broad market fluctuations may cause the market price of our common stock to decline substantially.
22
Table of Contents
Index to Financial Statements
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant stock price volatility in recent years. As described below, we are currently defending a consolidated federal securities class action lawsuit and a consolidated shareholder derivative action and we may become involved in additional instances of this type of litigation in the future. Litigation often is expensive and diverts management’s attention and resources, which could materially and adversely affect our business.
Additionally, fluctuations in the trading price or liquidity of our common stock may materially and adversely affect, among other things, the interest of investors to purchase our common stock on the open market and, generally, our ability to raise capital.
We are a defendant in a consolidated class action and in a consolidated shareholder derivative action, and these lawsuits and any future such lawsuits may adversely affect our business, financial condition, results of operations and cash flows
We and certain of our officers and directors are defendants in a consolidated federal securities class action lawsuit and a consolidated shareholder derivative action. These lawsuits are described in Part I, Item 3 “Legal Proceedings” in this Form 10-K. These lawsuits may divert our attention from our ordinary business operations, and we may incur significant expenses associated with their defense (including, without limitation, substantial attorneys’ fees and other fees of professional advisors and potential obligations to indemnify current and former officers and directors who are or may become parties to such actions). Depending on the outcome of the class action lawsuit, we may be required to pay material damages and fines, consent to injunctions on future conduct and/or suffer other penalties, remedies or sanctions. Accordingly, the ultimate resolution of these matters could have a material adverse effect on our business, results of operations, financial condition, liquidity and ability to meet our debt obligations and, consequently, could negatively impact the trading price of our common stock. In addition, there is the potential for additional shareholder litigation and for governmental investigations and/or enforcement actions. Any existing or future shareholder lawsuits and any future governmental investigations and/or enforcement actions could adversely impact our reputation, our relationships with our customers and our ability to generate revenue
Our board of directors has the power to designate, without stockholder approval, additional series of preferred stock, the shares of which could be senior to our common stock and be entitled to conversion or voting rights that adversely affect the holders of our common stock.
Our articles of incorporation authorize the issuance of capital stock including 20,000,000 authorized undesignated shares (8,001,000 designated as of December 31, 2014), and empowers our board of directors to prescribe, by resolution and without stockholder approval, a class or series of undesignated shares, including the number of shares in the class or series and the voting powers, designations, rights, preferences, restrictions and the relative rights in each such class or series. Accordingly, we may designate and issue additional shares or series of preferred stock that would rank senior to the shares of common stock as to dividend rights or rights upon our liquidation, winding-up, or dissolution.
Nevada law and our charter documents could make it more difficult for a third party to acquire us and discourage a takeover, which could depress the trading price of our common stock.
Nevada corporate law and our articles of incorporation and bylaws contain provisions that could discourage, delay, or prevent a change in control of our Company or changes in our management that our stockholders may deem advantageous. For example, holders of our common stock do not have cumulative voting rights in the election of directors, meaning that stockholders owning a majority of our outstanding shares of common stock will be able to elect all of our directors. In addition, because we have more than 200 stockholders of record, we are subject to the “business combinations” provisions of the Nevada Revised Statutes, or NRS. These provisions
23
Table of Contents
Index to Financial Statements
could prohibit or delay a merger or other takeover or change in control attempt and, accordingly, may discourage attempts to acquire our company even though such a transaction may be in our stockholders’ best interest and offer our stockholders the opportunity to sell their stock at a price above the prevailing market price.
One investor and certain directors, by virtue of ownership of our securities and related rights, may be able to control the Company.
The 10X Fund owns all of our issued and outstanding Series B Preferred Stock, which are convertible into 2,000,000 shares of our common stock. The 10X Fund owns related warrants exercisable to purchase an aggregate of 4,000,000 shares of our common stock. As of December 31, 2014, we have issued 1,233,256 shares of our common stock as dividends on the Series B Preferred Stock and 2,000,000 shares of our common stock on the exercise of warrants by 10X Fund. In addition, (i) James C. Czirr, a managing partner of the 10X Fund and Executive Chairman of our board of directors, owns or controls approximately 817,000 shares of our common stock, including shares of Series A on an as converted basis, and has the right to acquire approximately 811,000 additional shares of our common stock upon the exercise of outstanding stock options (approximately 631,000 of which are exercisable as of December 31, 2014); and (ii) Rod D. Martin, a managing partner of the 10X Fund and Vice Chairman of our board of directors, owns or controls approximately 175,000 shares of our common stock and has the right to acquire approximately 41,000 additional shares of our common stock upon the exercise of outstanding stock options (approximately 34,000 of which are exercisable as of December 31, 2014). As of December 31, 2014, on a fully diluted basis, assuming conversion of all Series B Preferred Stock and exercise of all outstanding warrants, the 10X Fund would own approximately 31% of our then outstanding shares of common stock, which, together with the shares of our common stock that would be owned by Mr. Czirr and Mr. Martin (assuming exercise of all vested options at that date), would constitute approximately 35% of the then outstanding shares.
As holder of Series B Preferred Stock, the 10X Fund is entitled to elect three directors in a separate class vote, nominate three directors for election by all shares entitled to vote, and provide or withhold consent to a range of fundamental corporate actions we may wish to undertake, such as recapitalization, sale of our company, and other matters. Such concentration of stock ownership and related rights could have the effect of delaying, deterring or preventing corporate events that our other security holders may desire or consider beneficial to the company.
We may issue additional common stock, which might dilute the net tangible book value per share of our common stock.
Our board of directors has the authority, without action or vote of our stockholders, to issue all or a part of our authorized but unissued shares. Such stock issuances could be made at a price that reflects a discount to, or a premium from, the then-current market price of our common stock. In addition, in order to raise capital, we may need to issue securities that are convertible into or exchangeable for a significant amount of our common stock. We are currently contemplating additional capital raising transactions within the next twelve months, which would likely result in issuances of additional shares which would be dilutive to current shareholders. These issuances would dilute the percentage ownership interest, which would have the effect of reducing your influence on matters on which our stockholders vote, and might dilute the net tangible book value per share of our common stock. You may incur additional dilution if holders of stock options, whether currently outstanding or subsequently granted, exercise their options, or if warrant holders exercise their warrants to purchase shares of our common stock.
A sale of a substantial number of shares of the common stock may cause the price of our common stock to decline.
Our common stock is currently traded on The NASDAQ Capital Market and, despite certain increases of trading volume from time to time, there have been periods when it could be considered “thinly-traded,” meaning
24
Table of Contents
Index to Financial Statements
that the number of persons interested in purchasing our common stock at or near bid prices at any given time may be relatively small or non-existent. Finance transactions resulting in a large amount of newly issued shares that become readily tradable, or other events that cause current stockholders to sell shares, could place downward pressure on the trading price of our stock. Some of our shareholders have registration rights to facilitate sales of large blocks of our common stock. We have filed a shelf registration statement to allow registered sales of up to 9.7 million shares by these shareholders. We may consider additional capital raising transactions within the next twelve months, which would likely result in issuances of additional shares which would be dilutive to current shareholders. In addition, the lack of a robust resale market may require a stockholder who desires to sell a large number of shares of common stock to sell the shares in increments over time to mitigate any adverse impact of the sales on the market price of our stock.
If our stockholders sell, or the market perceives that our stockholders intend to sell for various reasons, including the ending of restriction on resale or the expiration of lock-up agreements such as those entered into in connection with this offering, substantial amounts of our common stock in the public market, including shares issued upon the exercise of outstanding options or warrants, the market price of our common stock could fall. Sales of a substantial number of shares of our common stock may make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate. We may become involved in securities class action litigation that could divert management’s attention and harm our business.
We have not paid cash dividends in the past and do not expect to pay cash dividends in the foreseeable future.
We have never paid cash dividends on our capital stock and do not anticipate paying cash dividends on our capital stock in the foreseeable future. The payment of dividends on our capital stock will depend on our earnings, financial condition and other business and economic factors affecting us at such time as the board of directors may consider relevant. If we do not pay dividends, our common stock may be less valuable because a return on your investment will only occur if the market price of our common stock price appreciates.
At times, our shares of common stock and warrants have been thinly traded, so you may be unable to sell at or near ask prices or even at all if you need to sell your shares or warrants to raise money or otherwise desire to liquidate your shares or warrants.
We cannot predict the extent to which an active public market for our common stock and warrants will develop or be sustained. Our common stock is currently traded on The NASDAQ Capital Market and experiences periods when it could be considered “thinly-traded.” This situation may be attributable to a number of factors, including the fact that we are a small company which is relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume, and that even if we came to the attention of such persons, they tend to be risk averse and would be reluctant to follow an unproven company such as ours or purchase or recommend the purchase of our shares until such time as we became more seasoned and viable. As a consequence, there may be periods of several days, weeks or months when trading activity in our shares is minimal, as compared to a seasoned issuer which has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. We cannot give you any assurance that a broader or more active public trading market for our common stock will be sustained, or that current trading levels will be sustained or not diminish.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
We lease 3,610 square feet for our executive offices located at 4960 Peachtree Industrial Blvd., Norcross, GA. We also lease approximately 300 square feet in Natick, MA, for use by research and development
25
Table of Contents
Index to Financial Statements
consultants and which is collocated with one of our research and development service vendors. We believe these spaces are suitable for our present operations.
| Item 3. | Legal Proceedings |
From time to time, the Company is exposed to litigation relating to its operations. The Company is not currently engaged in any legal proceedings that are expected, individually or in the aggregate, to have a material, adverse effect on its financial condition or results of operations, except as noted below:
Separation Agreement
In February 2009, the Company entered into a Separation Agreement in connection with the resignation of David Platt, Ph.D., the Company’s former Chief Executive Officer and Chairman of the Board of Directors. The Separation Agreement provides for the deferral of a $1.0 million separation payment due to Dr. Platt upon the earlier occurrence of any of the following milestone events: (i) the approval by the Food and Drug Administration for a new drug application (“NDA”) for any drug candidate or drug delivery candidate based on the Company’s GM-CT-01 technology (whether or not such technology is patented), in which case Dr. Platt is also entitled to a fully vested 10-year cashless-exercise stock option to purchase at least 83,334 shares of common stock at an exercise price not less than the fair market value of the common stock determined as of the date of grant; (ii) consummation of a transaction with a pharmaceutical company expected to result in at least $10.0 million of equity investment or $50 million of royalty revenue to the Company, in which case Dr. Platt is also entitled to stock options on the same terms to purchase at least 50,000 shares of common stock; or (iii) the renewed listing of the Company’s securities on a national securities exchange and the achievement of a market capitalization of $100 million. Payment upon the events (i) and (iii) may be deferred up to six months, and if the Company has insufficient cash at the time of any of such events, it may issue Dr. Platt a secured promissory note for such amount. If the Company files a voluntary or involuntary petition for bankruptcy, whether or not a milestone event has occurred, such event shall trigger the obligation to pay the $1.0 million with the result that Dr. Platt may assert a claim for such obligation against the bankruptcy estate. During 2011, when it became probable that the Company could be relisted on a national securities exchange and eventually reach a market capitalization of $100 million, the Company recognized the $1.0 million severance payment due to Dr. Platt which was included in accrued expenses at December 31, 2013.
On October 12, 2012, Dr. Platt commenced a lawsuit under the Massachusetts Wage Act against Dr. Traber and Mr. McGauley who in their capacities as the Company’s Chief Executive Officer and the Company’s former Chief Financial Officer, respectively, can be held individually liable under the Wage Act for non-payment of wages. The lawsuit is based on the facts and issues raised in the arbitration regarding the payment of the $1.0 million separation payment under the Separation Agreement, and other unspecified “wages”. The statute provides that a successful claimant may be entitled to multiple damages, interest and attorney’s fees. On April 29, 2013, the Superior Court allowed Dr. Traber’s and Mr. McGauley’s motion to dismiss. On May 28, 2013, Dr. Platt filed a Notice of Appeal to appeal the Superior Court’s order allowing the defendants’ motion to dismiss. On April 14, 2014, the Appeals Court denied Dr. Platt’s appeal of the dismissal in full.
On March 29, 2013, the Company instituted arbitration before the American Arbitration Association, seeking to rescind or reform the Separation Agreement discussed above. The Company claimed that Dr. Platt fraudulently induced the Company to enter into the Separation Agreement, breached his fiduciary duty to the Company, and was unduly enriched from his conduct. Along with removal of the $1.0 million milestone payment under the Separation Agreement, the Company sought repayment of all separation benefits paid to Dr. Platt to date.
On August 1, 2013, the market capitalization of the Company’s common stock exceeded $100 million and the Company received a letter dated October 1, 2013, demanding payment of the $1 million. As described in the preceding paragraph, the Company had previously instituted an arbitration proceeding against Dr. Platt seeking
26
Table of Contents
Index to Financial Statements
to rescind the Separation Agreement, including the milestone payment provision, and the Company delayed payment pending the outcome of this arbitration. In June 2014, the arbitrator issued a judgment in favor of Dr. Platt. In July 2014, the Company paid the $1 million severance obligation.
Shareholder Class Actions and Derivative Lawsuits
Between July 30, 2014, and August 6, 2014, three putative class action complaints were filed in the United States District Court for the District of Nevada (the “Nevada District Court”) against the Company and certain of its officers and directors on behalf of all persons who purchased or otherwise acquired the Company’s stock between January 6, 2014 and July 28, 2014. The complaints allege that the defendants made false or misleading statements in certain press releases and other public statements in violation of the federal securities laws and seek class certification, unspecified monetary damages, costs, and attorneys’ fees. The Company disputes the allegations in the complaints and intends to vigorously defend against the claims. On August 22, 2014, the Nevada District Court entered an order consolidating the three cases, relieving the defendants of any obligation to respond to the complaints currently on file, and providing that defendants may respond to a consolidated amended complaint after it is filed by a lead plaintiff(s) to be appointed pursuant to the Private Securities Litigation Reform Act of 1995. On January 5, 2015, the Nevada District Court granted Defendants’ motion to transfer the consolidated putative securities class action to the United States District Court for the Northern District of Georgia. The court has not yet appointed a lead plaintiff or plaintiffs, and no consolidated amended complaint has been filed.
On August 1 and 25, 2014, persons claiming to be Galectin shareholders filed putative shareholder derivative complaints in the Nevada District Court, seeking recovery on behalf of the Company against certain of the Company’s directors and officers. On September 10, 2014, the Nevada District Court entered an order consolidating the two cases, relieving the defendants of any obligation to respond to the initial complaints, and providing that defendants may respond to a consolidated amended complaint to be filed by the plaintiffs. On January 5, 2015, the Nevada District Court granted Defendants’ motion to transfer the consolidated putative derivative litigation to the United States District Court for the Northern District of Georgia. The plaintiffs filed a consolidated amended complaint on February 27, 2015. The consolidated amended complaint alleges that the defendants breached their fiduciary duties to the Company’s shareholders by causing or permitting the Company to make allegedly false and misleading public statements concerning the Company’s financial and business prospects. The consolidated amended complaint also alleges that the defendants violated the federal securities laws by allegedly making false or misleading statements of material fact in the Company’s proxy filings, committed “waste” of corporate assets, were unjustly enriched, aided and abetted breaches of fiduciary duties, and that certain defendants breached their fiduciary duties through allegedly improper sales of Galectin stock. The complaints seek unspecified monetary damages on behalf of the Company, corporate governance reforms, disgorgement of profits, benefits and compensation by the defendants, costs, and attorneys’ and experts’ fees. Defendants’ response to the consolidated amended complaint is currently due to be filed on March 30, 2015.
On August 29, 2014, another alleged Galectin shareholder filed a putative shareholder derivative complaint in state court in Las Vegas, Nevada, seeking recovery on behalf of the Company against the same directors and officers who are named as defendants in the derivative litigation pending in the United States District Court for the District of Nevada. The state court derivative plaintiff filed an amended complaint on December 1, 2014, which alleges claims for breach of fiduciary duties, unjust enrichment, and waste of corporate assets, based on allegations that are substantially similar to those in the derivative complaints now pending in the United States District Court for the Northern District of Georgia, and seeks unspecified monetary damages on behalf of the Company, corporate governance reforms, disgorgement of profits, benefits and compensation by the defendants, costs, and attorneys’ and experts’ fees. Defendant filed motion to dismiss the amended complaint on February 26, 2015.
27
Table of Contents
Index to Financial Statements
Estimating an amount or range of possible losses resulting from litigation proceedings is inherently difficult and requires an extensive degree of judgment, particularly where the matters involve indeterminate claims for monetary damages, are in the early stages of the proceedings, and are subject to appeal. In addition, because most legal proceedings are resolved over extended periods of time, potential losses are subject to change due to, among other things, new developments, changes in legal strategy, the outcome of intermediate procedural and substantive rulings and other parties’ settlement posture and their evaluation of the strength or weakness of their case against us. For these reasons, we are currently unable to predict the ultimate timing or outcome of, or reasonably estimate the possible losses or a range of possible losses resulting from, the matters described above. Based on information currently available, the Company does not believe that any reasonably possible losses arising from currently pending legal matters will be material to the Company’s results of operations or financial condition. However, in light of the inherent uncertainties involved in such matters, an adverse outcome in one or more of these matters could materially and adversely affect the Company’s financial condition, results of operations or cash flows in any particular reporting period.
| Item 4. | Mine Safety Disclosures |
Not applicable.
28
Table of Contents
Index to Financial Statements
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Price Range of Common Stock
Our common stock began trading on The NASDAQ Capital Market under the symbol GALT effective March 23, 2012. The high and low sale prices for our common stock as reported on the NASDAQ Capital Market, for the periods indicated as shown below. All share prices reflect the one-for-six reverse split, which was effective March 23, 2012.
| High | Low | |||||||
| Fiscal Year Ended December 31, 2014 |
||||||||
| First Quarter |
$ | 19.11 | $ | 7.90 | ||||
| Second Quarter |
$ | 16.02 | $ | 9.80 | ||||
| Third Quarter |
$ | 16.55 | $ | 4.28 | ||||
| Fourth Quarter |
$ | 5.88 | $ | 3.00 | ||||
| Fiscal Year Ended December 31, 2013 |
||||||||
| First Quarter |
$ | 4.55 | $ | 1.96 | ||||
| Second Quarter |
$ | 5.22 | $ | 3.19 | ||||
| Third Quarter |
$ | 13.21 | $ | 3.96 | ||||
| Fourth Quarter |
$ | 12.75 | $ | 5.35 | ||||
29
Table of Contents
Index to Financial Statements
Performance graph
The following graph shows the value of an investment of $100 on December 31, 2009, in each of Galectin Therapeutics common stock, the Nasdaq Biotech Index, the Nasdaq Composite Index. All values assume reinvestment of the pretax value of dividends and are calculated as of December 31 of each year. The historical stock price performance of the Company’s common stock shown in the performance graph is not necessarily indicative of future stock price performance.
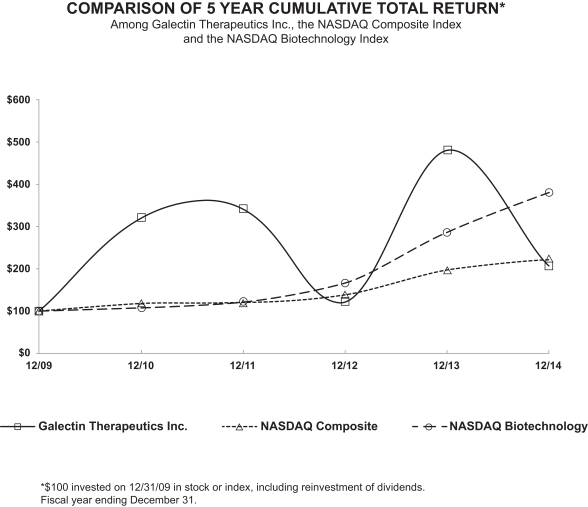
The material in this performance graph is not soliciting material, is not deemed filed with the SEC, and is not incorporated by reference in any filing of the Company under the Securities Act or the Exchange Act, whether made on, before or after the date of this filing and irrespective of any general incorporation language in such filing.
Holders of Common Stock
As of February 27, 2015, there were 124 shareholders of record of our common stock. Because shares of our common stock are held by depositaries, brokers and other nominees, the number of beneficial holders of our shares is substantially larger than the number of record holders. Based on information available to us, we believe there are approximately 10,889 non-objecting beneficial owners of our shares of our common stock in addition to the record holders.
30
Table of Contents
Index to Financial Statements
Dividends
There have been no cash dividends declared on our common stock since our company was formed. Dividends are declared at the sole discretion of our Board of Directors. Our intention is not to declare cash dividends and retain all cash for our operations.
| Item 6. | Selected Financial Data |
| Years ended December 31, | ||||||||||||||||||||
| Consolidated Statement of Income Data: |
2014 | 2013 | 2012 | 2011 | 2010 | |||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
$ | 8,425 | $ | 5,688 | $ | 4,527 | $ | 3,552 | $ | 1,066 | ||||||||||
| General and administrative |
7,005 | 6,416 | 5,372 | 6,857 | 3,817 | |||||||||||||||
| Other income (expense) |
(358 | ) | 16 | 224 | (506 | ) | (746 | ) | ||||||||||||
| Net loss |
(15,788 | ) | (12,088 | ) | (9,675 | ) | (10,915 | ) | (5,629 | ) | ||||||||||
| Preferred stock dividends |
(943 | ) | (867 | ) | (976 | ) | (1,568 | ) | (902 | ) | ||||||||||
| Preferred stock accretion |
(229 | ) | (229 | ) | (230 | ) | (230 | ) | (2,178 | ) | ||||||||||
| Warrant modification |
— | (8,763 | ) | — | — | — | ||||||||||||||
| Net loss applicable to common stockholders |
(16,960 | ) | (21,947 | ) | (10,881 | ) | (12,713 | ) | (8,709 | ) | ||||||||||
| Basic and diluted earnings per share |
$ | (0.78 | ) | $ | (1.30 | ) | $ | (0.72 | ) | $ | (1.06 | ) | $ | (0.93 | ) | |||||
| Dividends paid per share |
— | — | — | — | — | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| Consolidated Balance Sheet Data: |
2014 | 2013 | 2012 | 2011 | 2010 | |||||||||||||||
| (in millions) | ||||||||||||||||||||
| Total assets |
$ | 29,677 | $ | 10,713 | $ | 9,561 | $ | 6,612 | $ | 6,300 | ||||||||||
| Total debt |
— | — | — | — | — | |||||||||||||||
| Total stockholders’ equity (deficit) |
21,195 | 1,481 | 1,165 | (2,125 | ) | (1,694 | ) | |||||||||||||
See Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations and the Consolidated Financial Statements and accompanying notes and previously filed Annual Reports on Form 10-K for further information regarding our consolidated results of operations and financial position for periods reported therein and for known factors that will impact comparability of future results.
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
Overview
We are a clinical stage company engaged in drug research and development to create new therapies for fibrotic disease and cancer. Our drug candidates are based on our method of targeting galectin proteins, which are key mediators of biologic and pathologic functions. We use naturally occurring, readily-available plant materials as starting material in manufacturing processes to create proprietary complex carbohydrates with specific molecular weights and other pharmaceutical properties. These complex carbohydrate molecules are appropriately formulated into acceptable pharmaceutical formulations. Using these unique carbohydrate-based candidate compounds that bind and inhibit galectin proteins, we are undertaking the focused pursuit of therapies for indications where galectins have a demonstrated role in the pathogenesis of a given disease. We focus on diseases with serious, life-threatening consequences to patients and those where current treatment options are limited. Our strategy is to establish and implement clinical development programs that add value to our business in the shortest period of time possible and to seek strategic partners when a program becomes advanced and requires additional resources.
We endeavor to leverage our scientific and product development expertise as well as established relationships with outside sources to achieve cost-effective and efficient development. These outside sources, amongst others,
31
Table of Contents
Index to Financial Statements
provide us with expertise in preclinical models, pharmaceutical development, toxicology, clinical development, pharmaceutical manufacturing, sophisticated physical and chemical characterization, and commercial development. We also have established a collaborative scientific discovery program with leading experts in carbohydrate chemistry and characterization. This discovery program is aimed at the targeted development of new molecules which bind galectin proteins and offer alternative options to larger market segments in our primary disease targets. We are pursuing a development pathway to clinical enhancement and commercialization for our lead compounds in liver fibrosis and fatty liver disease as well as in immune enhancement for cancer therapy. All of our proposed products are presently in development, including pre-clinical and clinical trials.
2012 Common Stock and Warrant Offering with Reverse Split and 2013 and 2014 At Market Issuance Agreements
On March 22, 2012, in anticipation of completing a public offering of securities, we effected a one-for-six reverse stock split of our common stock. All common share and per unit amounts in this report, including the financial statements, have been adjusted to reflect the reverse split. Our common stock began trading on The NASDAQ Capital Market under the symbol GALT on March 23, 2012, and the units and warrants that we sold in the offering began trading on that exchange under the symbols GALTU and GALTW, respectively, on March 28, 2012.
On March 28, 2012, we completed the public offering in which we issued 2,666,722 shares of common stock and related warrants exercisable until March 28, 2017, at $5.63 per share to purchase 1,333,361 shares of common stock for gross proceeds of $12.0 million (net cash proceeds of 10.4 million).
On October 25, 2013, the Company entered into an At Market Issuance Sales Agreement (the “2013 At Market Agreement”) with a sales agent under which the Company may issue and sell shares of its common stock having an aggregate offering price of up to $30.0 million from time to time through the sales agent. Sales of the Company’s common stock through the sales agent, if any, will be made by any method that is deemed an “at the market” offering as defined by the U.S. Securities and Exchange Commission. The Company will pay to the sales agent a commission rate equal to 3.0% of the gross proceeds from the sale of any shares of common stock sold through the sales agent under the 2013 At Market Agreement. As of December 31, 2013, the Company had issued 99,942 shares of its common stock through its 2013 At Market Agreement at an average price of $9.02 per share resulting in gross proceeds of approximately $944,000. The Company incurred one time, initial legal and accounting costs of approximately $82,000 and commissions of $29,000 resulting in net proceeds of $833,000 as of December 31, 2013. In January and February 2014, the Company issued 2,663,647 shares of common stock for net proceeds of approximately $28,178,000 which completed the 2013 At Market Agreement.
On March 30, 2014, the Company entered into an At Market Issuance Sales Agreement (the “2014 At Market Agreement”) with a sales agent under which the Company may issue and sell shares of its common stock having an aggregate offering price of up to $30.0 million from time to time through the sales agent. Sales of the Company’s common stock through the sales agent, if any, will be made by any method that is deemed an “at the market” offering as defined by the U.S. Securities and Exchange Commission. The Company will pay to the sales agent a commission rate equal to 3.0% of the gross proceeds from the sale of any shares of common stock sold through the sales agent under the 2014 At Market Agreement. As of December 31, 2014, the Company had issued 217,622 shares of its common stock through its 2014 At Market Agreement at an average price of $5.49 per share resulting in gross proceeds of approximately $1,196,000. The Company incurred commissions of approximately $36,000 resulting in net proceeds of approximately $1,159,000 as of December 31, 2014. In January and February 2015, the Company issued 1,173,458 shares of common stock for net proceeds of approximately $4,127,000 under the 2014 At Market Agreement.
Our Drug Development Programs
Galectins are a class of proteins that are made by many cells in the body. As a group, these proteins are able to bind to sugar molecules that are part of other proteins in and on the cells of our body. Galectin proteins act as a
32
Table of Contents
Index to Financial Statements
kind of glue, bringing together molecules that have sugars on them. Galectin proteins are known to be markedly increased in a number of important diseases including scaring of organs (e.g. liver, lung, kidney, and heart) and cancers of many kinds. The increase in galectin protein promotes the disease and is detrimental to the patient.
We have two compounds in development that are intended to be used in the treatment of liver fibrosis and fatty liver disease and in cancer therapy. These two compounds are produced from completely different, natural, readily available, starting materials, which, following chemical processing, both exhibit the property of binding to and inhibiting galectin proteins. GR-MD-02, our lead product for treatment of liver fibrosis and fatty liver disease with inflammation and fibrosis and in cancer therapy, is a proprietary complex polysaccharide polymer possessing both linear and globular structures, which is derived from a plant source. GM-CT-01 is a proprietary linear polysaccharide polymer comprised of mannose and galactose that has a precisely defined chemical structure and which is also derived from a plant source.
We believe the mechanism of action for GR-MD-02 and GM-CT-01 is based upon interaction with, and inhibition of, galectin proteins, which are expressed at high levels in certain pathological states including inflammation, fibrosis and cancer. While GR-MD-02 and GM-CT-01 are capable of binding to multiple galectin proteins, we believe that they have the greatest affinity for galectin-3, the most prominent galectin implicated in pathological processes. Blocking galectin in cancer and liver fibrosis has specific salutary effects on the disease process, as discussed below.
Liver Fibrosis
The main initiative in our development strategy is the application of galectin inhibition in connection with liver fibrosis, a condition that leads to cirrhosis. We believe that GR-MD-02 has the potential to treat nonalcoholic steatohepatitis (NASH) and other forms of liver fibrosis. The driving factor for our commitment to galectin inhibition for fibrosis is scientific evidence that strongly suggests that galectin-3 is essential for the development of liver fibrosis in animals. Published data show that mice lacking the galectin-3 gene are incapable of developing liver fibrosis in response to toxin insult to the liver and in fatty liver disease. Moreover, mice that do not have the galectin-3 gene are resistant to lung and kidney fibrosis.
We have evaluated the ability of GR-MD-02 to block galectin-3 in animal models of liver fibrosis, the conclusions of which yielded positive results. Our pre-clinical data show that GR-MD-02 may have a therapeutic effect on liver fibrosis as shown in several relevant animal models. Therefore, we chose GR-MD-02 as the lead candidate in a development program targeted initially at fibrotic liver disease associated with NASH. In January 2013, an Investigational New Drug (“IND”) was submitted to the FDA with the goal of initiating a Phase 1 study in patients with NASH and advanced liver fibrosis to primarily evaluate the human safety of GR-MD-02 and pharmacodynamics biomarkers of disease are also included in the trial design. On March 1, 2013, the FDA indicated we could proceed with a U.S. Phase 1 clinical trial for GR-MD-02 with a development program aimed at obtaining support for a proposed indication of GR-MD-02 for treatment of NASH with advanced fibrosis. In February 2013 we entered into an agreement with Clinical Trial Services Inc. (“CTI”) to conduct a Phase 1 clinical trial of GR-MD-02 to assess safety and preliminary evidence of efficacy in humans. In June 2013, we submitted a Fast Track application to the FDA to help expedite its clinical development program of GR-MD-02 in the treatment of NASH with advanced fibrosis. FDA grants Fast Track designation to help expedite review and approval of drugs in development that treat serious or life threatening diseases and fill an unmet medical need. On August 7, 2013, FDA concluded that the development program for GR-MD-02 meets the criteria for Fast Track designation, and FDA has designated the investigation of GR-MD-02 for non-alcoholic steatohepatitis with hepatic fibrosis as a Fast Track development program. In January 2014, we completed the enrollment of the first cohort of patients in the Phase 1 trial with no serious adverse events being reported. We reported initial safety and tolerability results from the first cohort of patients on June 30, 2014. The second cohort of this Phase 1 trial began and enrollment was completed in April 2014. In July 2014, we reported the results from the second cohort of patients. Enrollment of the third cohort of Phase 1 began in July 2014 with interim results presented in November 2014 with the final report on cohort 3 reported in January 2015. The results of the Phase 1 study
33
Table of Contents
Index to Financial Statements
demonstrate that (i) GR-MD-02 was safe and well tolerated by patients with advanced NASH liver fibrosis after IV administration of four doses of 2 mg/kg, 4 mg/kg and 8mg/kg lean body weight, (ii) Pharmacokinetics revealed drug exposure in humans at the 8 mg/kg dose that was equivalent to the upper range of the targeted therapeutic dose determined from effective doses in NASH animal models, (iii) Disease Serum Marker Effect showed there was a statistically significant, dose-dependent reduction in FibroTest® scores due to a statistically significant reduction in alpha-2 macroglobulin serum levels, and (iv) Liver Stiffness Effect, as measured by FibroScan® showed that there was a signal of reduced liver stiffness in patients receiving GR-MD-02. The reduction seen in A2M does not necessarily mean fibrosis got better in this short study, but does suggest changes in the fibrogenic process that might lead to an improvement in fibrosis with longer-term therapy. These Phase 1 results in NASH patients with advanced fibrosis provide a firm foundation for entry into a Phase 2 development program.
The Company held an “End of Phase 1 meeting” with FDA and, amongst other things, received clear guidance on the primary endpoint for a Phase 2 trial. Preliminary Phase 2 clinical trial design targets a patient population with cirrhosis due to NASH. The study endpoints will include those that are closely associated with outcomes in patients with cirrhosis Primary endpoint: Hepatic venous pressure gradient (HVPG). Planned secondary endpoints include: morphometric analysis of collagen on liver biopsies and other secondary endpoints will include non-invasive tests to evaluate for correlation with HVPG and liver collagen. We have awarded the contract for the primary Phase 2 study to a CRO and expect to initiate a Phase 2 clinical trial in the first half of 2015 to assess the efficacy of GR-MD-02 in patients with NASH and advanced liver fibrosis. The timing of initial results from the Phase 2 trial are dependent upon the trial design, and, amongst other factors, the rate of patient enrollment. Our Phase 2 clinical program is likely to include additional clinical trials to fully characterize human response to GR-MD-02 and to better position the Company for a successful Phase 3 clinical trial program.
Galectin Inhibition in Cancer Therapy
We believe the potential exists for galectin inhibition to play an important role in cancer therapy. Galectin proteins, particularly galectin-1 and galectin-3, have been shown to be highly expressed in the majority of cancers and have multiple roles in promoting cancer progression, including tumor cell invasion, metastasis, angiogenesis, and tumor evasion of the immune system.
We believe there is potential for galectin inhibition to play a key role in the burgeoning area of cancer immunotherapy. For example, there have been two recent approvals of drugs that enhance a patient’s immune system to fight cancer. With many additional vaccines and immune stimulatory agents in development, industry analysts forecast that this market could grow to over $7 billion by 2015. It is our goal to use a galectin inhibitor to enhance the immune system function to fight cancer and, most important, that complements other approaches to this type of therapy. Our drug candidates provide a promising new therapeutic approach to enhance the activity of the immune system against cancer cells. Preclinical studies have indicated that GR-MD-02 and GM-CT-01 enhance the immune response to and more specifically increased tumor shrinkage and enhanced survival in immune competent mice with prostate and breast cancers when combined with one of the immune checkpoint inhibitors, anti-CTLA-4 or anti-PD-1. These preclinical data have led to the filing of an Investigator-sponsored IND and the initiation of a study of GR-MD-02 in combination with Yervoy® (ipilimumab) in a Phase 1B study of patients with metastatic melanoma. This study is being conducted under the sponsorship of Providence Portland Medical Center’s Earle A. Chiles Research Institute (EACRI).
We previously attempted to gain regulatory approval of GM-CT-01 for use in combination with 5-FU (5-Fluorouracil, an anti-cancer chemotherapy drug) containing chemotherapy regiments for metastatic colorectal cancer in Colombia. This approach had been recommended to the Company by key oncology opinion leaders in Colombia and by PROCAPS S.A. (“PROCAPS”), a Colombia-based pharmaceutical company. There has been no approval of GM-CT-01 in a major region such as the U.S. or Europe and it was determined that approval from the regulatory authority in Columbia (INVIMA) would require additional clinical trial data. Although the Company worked with PROCAPS to design a Phase 3 clinical trial, a satisfactory plan could not be agreed upon and we terminated the Agreement with PROCAPS (as described below), effective September 29, 2012, and have
34
Table of Contents
Index to Financial Statements
no current plans to continue attempts to gain approval of GM-CT-01 in Columbia. We had not taken into account projections for any potential revenues from this agreement in our financing plans.
Agreement with PROCAPS S.A.
On March 25, 2010, we granted PROCAPS S.A. (in the form of a definitive term sheet) exclusive rights to market and sell GM-CT-01 to treat cancer in Colombia, South America. PROCAPS is an international, privately held pharmaceutical company based in Barranquilla, Colombia. In October 2010, we received a payment of $200,000 and shipped GM-CT-01 to PROCAPS to be used by PROCAPS to undertake initial steps contemplated by the term sheet. We recorded the $200,000 payment from PROCAPS as deferred revenue on the consolidated balance sheet as of December 31, 2011, to be recognized when the remaining deliverables of the agreement were completed.
On October 18, 2011, we entered into a Collaboration, Supply, Marketing and Distribution Agreement (the “Agreement”) with PROCAPS. The Agreement granted PROCAPS first negotiation rights to enter into similar agreements in other Central and South American countries. We were to be the sole manufacturer and supplier of GM-CT-01 to PROCAPS. The Agreement obligated PROCAPS to procure regulatory approvals necessary for the marketing and sale of GM-CT-01 naming us as the owner of such approvals to the extent permitted by law, or alternatively hold the approvals for our benefit. PROCAPS was required to pay us a stated fee for each dose it purchases and royalties at an incremental rate determined by annual net sales of GM-CT-01. We retained all intellectual property rights to GM-CT-01 and related products and PROCAPS may not produce, modify, reverse engineer, or otherwise interfere with the GM-CT-01 compound. PROCAPS may not manufacture or sell products that compete with GM-CT-01 during the term of the Agreement and for five years thereafter.
PROCAPS had not obtained approval to sell GM-CT-01 in Columbia as required by the Agreement and, as they were in material breach of the Agreement, we terminated the Agreement, effective September 29, 2012. With no further obligations under the Agreement, we recognized the $200,000 payment as Other Income in the Statement of Operations during the year ended December 31, 2012.
Results of Operations from the Years Ended December 31, 2014 and 2013
Research and Development Expense
| Year ended December 31, |
2014 as Compared to 2013 | |||||||||||||||
| 2014 | 2013 | $ Change | % Change | |||||||||||||
| (in thousands, except %) | ||||||||||||||||
| Research and development |
$ | 8,425 | $ | 5,688 | $ | 2,737 | 48 | % | ||||||||
We generally categorize research and development expenses as either direct external expenses, comprised of amounts paid to third party vendors for services, or all other research and development expenses, comprised of employee payroll and general overhead allocable to research and development. We consider a clinical program to have begun upon acceptance by the FDA, or similar agency outside of the United States, to commence a clinical trial in humans, at which time we begin tracking expenditures by the product candidate. Clinical program expenses comprise payments to vendors related to preparation for, and conduct of, all phases of the clinical trial, including costs for drug manufacture, patient dosing and monitoring, data collection and management, oversight of the trials and reports of results. Pre-clinical expenses comprise all research and development amounts incurred before human trials begin, including payments to vendors for services related to product experiments and discovery, toxicology, pharmacology, metabolism and efficacy studies, as well as manufacturing process development for a drug candidate.
We have two product candidates, GR-MD-02 and GM-CT-01; however only GR-MD-02 is in active development. We filed for an IND for GR-MD-02 in January 2013 and in February 2013 we entered into an
35
Table of Contents
Index to Financial Statements
agreement with CTI to conduct a Phase 1 clinical trial of GR-MD-02. In March 2013, the FDA indicated we could proceed with a Phase 1 human clinical trial of GR-MD-02, and we began enrolling patients in the third quarter of 2013. In January 2014, we completed the enrollment of the first cohort of patients in the Phase 1 trial with no serious adverse events being reported. We reported initial safety and tolerability results from the first cohort of patients on June 30, 2014. The second cohort of this Phase 1 trial began and enrollment was completed in April 2014. In July 2014, we reported the results from the second cohort of patients. Enrollment of the third cohort of Phase 1 began in July 2014 with interim results presented in November 2014 with the final report on cohort 3 presented in January 2015. The results of the Phase 1 study demonstrate that (i) GR-MD-02 was safe and well tolerated by patients with advanced NASH liver fibrosis after IV administration of four doses of 2 mg/kg, 4 mg/kg and 8mg/kg lean body weight, (ii) Pharmacokinetics revealed drug exposure in humans at the 8 mg/kg dose that was equivalent to the upper range of the targeted therapeutic dose determined from effective doses in NASH animal models, (iii) Disease Serum Marker Effect showed there was a statistically significant, dose-dependent reduction in FibroTest® scores due to a statistically significant reduction in alpha-2 macroglobulin serum levels, and (iv) Liver Stiffness Effect, as measured by FibroScan® showed that there was a signal of reduced liver stiffness in patients receiving GR-MD-02. The reduction seen in A2M does not necessarily mean fibrosis got better in this short study, but does suggest changes in the fibrogenic process that might lead to an improvement in fibrosis with longer-term therapy. These Phase 1 results in NASH patients with advanced fibrosis provide a firm foundation for entry into a Phase 2 development program.
The company held an “End of Phase 1 meeting” with FDA and, amongst other things, received clear guidance on the primary endpoint for a Phase 2 trial. Preliminary Phase 2 clinical trial design targets a patient population with cirrhosis due to NASH. The study endpoints will include those that are closely associated with outcomes in patients with cirrhosis Primary endpoint: Hepatic venous pressure gradient (HVPG). Planned secondary endpoints include: morphometric analysis of collagen on liver biopsies and other secondary endpoints will include non-invasive tests to evaluate for correlation with HVPG and liver collagen. We have awarded the contract for the primary Phase 2 study to a CRO and expect to initiate a Phase 2 clinical trial in the first half of 2015 to assess the efficacy of GR-MD-02 in patients with NASH and advanced liver fibrosis. The timing of initial results from the Phase 2 trial are dependent upon the trial design, and, amongst other factors, the rate of patient enrollment. Our Phase 2 clinical program is likely to include additional clinical trials to fully characterize human response to GR-MD-02 and to better position the Company for a successful Phase 3 clinical trial program.
Our research and development expenses were as follows:
| Year Ended December 31, |
||||||||
| 2014 | 2013 | |||||||
| (in thousands) | ||||||||
| Direct external expenses: |
||||||||
| Clinical programs and pre-clinical activities |
$ | 6,071 | $ | 3,936 | ||||
| Other research and development expenses: |
||||||||
| Payroll and other |
1,053 | 761 | ||||||
| Stock based compensation |
1,301 | 991 | ||||||
|
|
|
|
|
|||||
| $ | 8,425 | $ | 5,688 | |||||
|
|
|
|
|
|||||
Clinical programs and pre-clinical expenses cost increases for the year ended December 31, 2014, compared to the same period in 2013, were due to increases in costs related to our Phase 1 clinical trial of $367,000, pre-clinical activities in support of planned Phase 2 program of $798,000 and drug manufacturing costs of $970,000. We have completed our Phase 1 trial for GR-MD-02 and are preparing for our Phase 2 program, and expect our clinical and pre-clinical program costs will increase substantially.
Both the time required and costs we may incur in order to commercialize a drug candidate that would result in material net cash inflow are subject to numerous variables, and therefore we are unable at this stage of our
36
Table of Contents
Index to Financial Statements
development to forecast useful estimates. Variables that make estimates difficult include the number of clinical trials we may undertake, the number of patients needed to participate in the clinical trial, patient recruitment uncertainties, trial results as to the safety and efficacy of our product, and uncertainties as to the regulatory agency response to our trial data prior to receipt of marketing approval. Moreover, the FDA or other regulatory agencies may suspend clinical trials if we or an agency believes patients in the trial are subject to unacceptable risks, or find deficiencies in the conduct of the clinical trial. Delays or rejections may also occur if governmental regulation or policy changes during our clinical trials or in the course of review of our clinical data. Due to these uncertainties, accurate and meaningful estimates of the ultimate cost to bring a product to market, the timing of costs and completion of our program and the period during which material net cash inflows will commence are unavailable at this time. However, we expect to continue to have substantial research and development expenses for the foreseeable future as we continue to develop our products.
General and Administrative Expense
| Year ended December 31, |
2014 as Compared to 2013 | |||||||||||||||
| 2014 | 2013 | $ Change | % Change | |||||||||||||
| (in thousands, except %) | ||||||||||||||||
| General and administrative |
$ | 7,005 | $ | 6,416 | $ | 589 | 9 | % | ||||||||
General and administrative expenses consist primarily of salaries including stock based compensation, legal and accounting fees, insurance, investor relations, business development and other office related expenses. The primary reasons for the increase for the year ended December 31, 2014 as compared to the same period for 2013 are due to, increased legal expenses of $407,000 related to our arbitration with Dr. Platt which was settled in 2014 and includes the $150,000 retention of legal fees we paid in connection with the shareholder suits filed in 2014 and increased insurance expense of $115,000.
Other Income and Expense
During the year ended December 31, 2014, other income and expense consisted primarily of the $400,000 loss on equity method investment in Galectin Sciences LLC.
Results of Operations from the Years Ended December 31, 2013 and 2012
Research and Development Expense
| Year ended December 31, |
2013 as Compared to 2012 | |||||||||||||||
| 2013 | 2012 | $ Change | % Change | |||||||||||||
| (in thousands, except %) | ||||||||||||||||
| Research and development |
$ | 5,688 | $ | 4,527 | $ | 1,161 | 26 | % | ||||||||
Our research and development expenses were as follows:
| Year Ended December 31, |
||||||||
| 2013 | 2012 | |||||||
| (in thousands) | ||||||||
| Direct external expenses: |
||||||||
| Clinical programs and pre-clinical activities |
$ | 3,936 | $ | 2,999 | ||||
| Other research and development expenses: |
||||||||
| Payroll and other |
761 | 606 | ||||||
| Stock based compensation |
991 | 922 | ||||||
|
|
|
|
|
|||||
| $ | 5,688 | $ | 4,527 | |||||
|
|
|
|
|
|||||
37
Table of Contents
Index to Financial Statements
Clinical programs and pre-clinical expenses for the year ended December 31, 2013, increased compared to the same period in 2012, due primarily to costs of $1,892,000 related our Phase 1 clinical trial which began in 2013 offset by decreases in expenses for pre-clinical and drug manufacturing totaling $956,000. We expect as we continue our Phase 1 trial for GR-MD-02 in 2014 and prepare for our Phase 2 program, our clinical and pre-clinical program costs will increase substantially.
General and Administrative Expense
| Year ended December 31, |
2013 as Compared to 2012 | |||||||||||||||
| 2013 | 2012 | $ Change | % Change | |||||||||||||
| (in thousands, except %) | ||||||||||||||||
| General and administrative |
$ | 6,416 | $ | 5,372 | $ | 1,044 | 19 | % | ||||||||
General and administrative expenses consist primarily of salaries including stock based compensation, legal and accounting fees, insurance, investor relations, business development and other office related expenses. The primary reasons for the increase for the year ended December 31, 2013 as compared to the same period for 2012 are due to increased stock-based compensation of $928,000, increased legal expenses of $49,000 related to our ongoing litigation with Dr. Platt, increased insurance expense of $92,000 and increased investor relations expense of $66,000, offset by decreased rent expense of $177,000. The primary reason for the increase in stock-based compensation for the year ended December 31, 2013 was due to a modification in September 2013 of certain vested options held by a former board member to extend the contractual exercise period through the original expiration dates as opposed to 90 days after service on the board ended.
Other Income and Expense
During the year ended December 31, 2012, other income and expense consisted primarily of the $200,000 payment from PROCAPS which was previously accounted for as deferred income and recognized upon the termination of the PROCAPS Agreement, as previously described.
Liquidity and Capital Resources
As described above in the Overview and elsewhere in this Annual Report on Form 10-K, we are in the development stage and have not generated any revenues to date. Since our inception on July 10, 2000, we have financed our operations from proceeds of public and private offerings of debt and equity. As of December 31, 2014, we raised a net total of $108.9 million from these offerings. At December 31, 2014, the Company had $29.1 of unrestricted cash and cash equivalents available to fund future operations. Additionally, in January and February 2015, the Company received $4,127,000 in net proceeds from the issuance of common stock at then-current market prices through its at the market (“ATM”) financing arrangement. The Company currently believes there is sufficient cash to fund currently planned operations through September 30, 2016. We will require more cash to fund our operations after September 30, 2016 and believe we will be able to obtain additional financing. However, there can be no assurance that we will be successful in obtaining such new financing or, if available, that such financing will be on terms favorable to us. If we are unsuccessful in raising additional capital to fund operations after September 30, 2016, we may be required to cease operations or seek bankruptcy protection.
2014 compared to 2013
Net cash used in operations increased by $4,942,000 to $12,426,000 for 2014, as compared to $7,484,000 for 2013. Cash operating expenses increased principally due to increased research and development activities primarily related to our fibrosis development and Phase 1 clinical trial for GR-MD-02 begun in 2013.
There were no equipment purchases or other investing activities in 2014.
38
Table of Contents
Index to Financial Statements
Net cash provided by financing activities was $31,465,000 during 2014 as compared to $8,609,000 during 2013, due primarily to the transactions described below.
In 2014, we received $2,128,000 from the exercise of stock options and warrants. Additionally, in 2014, we received $29,337,000 from sales of our common stock through At the Market issuances. In 2013, we received $4,776,000 from the exercise of stock options and warrants. Additionally, in 2013, we received $3,000,000 from a private placement of unregistered common stock and received $833,000 in net proceeds from our at the market stock issuance program.
2013 compared to 2012
Net cash used in operations decreased by $16,000 to $7,484,000 for 2013, as compared to $7,500,000 for 2012. Cash operating expenses increased principally due to increased research and development activities primarily related to our fibrosis development and Phase 1 clinical trial for GR-MD-02 begun in 2013, offset by an increase in accounts payable and accrued expenses at December 31, 2013 over December 31, 2012.
There were no equipment purchases or other investing activities in 2013. Cash provided by investing activities during 2012 consisted of a decrease in restricted cash by $64,000 as our $59,000 secured letter of credit for office space and $10,000 of secured credit cards were released, offset by equipment purchases of $5,000.
Net cash provided by financing activities was $8,609,000 during 2013 as compared to $10,403,000 during 2012, due primarily to the transaction described below.
In 2013, we received $4,776,000 from the exercise of stock options and warrants. Additionally, in 2013, we received $3,000,000 from a private placement of unregistered common stock and received $833,000 in net proceeds from our at the market stock issuance program. On March 28, 2012, we issued 2,666,722 shares of common stock and related $5.63 warrants to purchase 1,333,361 shares of common stock, resulting in gross proceeds of $12,000,000 (net proceeds of $10,403,000).
Operating leases.
In September 2012, the Company entered into an operating lease for office space in Norcross, GA for a term of twenty-six months, beginning on October 1, 2012 and ending November 30, 2014 at a rate of approximately $3,000 per month. In June 2014, the Company signed an amendment to the lease extending the term through November 30, 2017 with a base monthly rental of approximately $3,300 through the extended term. The original lease provided for free rent for the first two months of the lease and required a security deposit of $6,000. In addition to base rental payments included in the contractual obligations table above, the Company is responsible for our pro-rata share of the operating expenses for the building.
In October 2012, the Company entered into an operating lease for office space collocated with lab space for research and development activities. The lease is for a period of one year, beginning on October 1, 2012, for a rate of $15,000 for the term, payable in equal monthly increments. This lease was continued on a month to month basis from October 1, 2013.
Other. We have engaged outside vendors for certain services associated with our clinical trials. These services are generally available from several providers and, accordingly, our arrangements are typically cancellable on 30 days notice.
Off-Balance Sheet Arrangements
We have not created, and are not a party to, any special-purpose or off-balance sheet entities for the purpose of raising capital, incurring debt or operating parts of our business that are not consolidated into our financial
39
Table of Contents
Index to Financial Statements
statements. We do not have any arrangements or relationships with entities that are not consolidated into our financial statements that are reasonably likely to materially affect our liquidity or the availability of capital resources.
Contractual Obligations and Commitments
The following table summarizes contractual obligations and commitments as of December 31, 2014:
| Contractual Obligations |
Payments due by period (in thousands) | |||||||||||||||
| Total | Less than 1 year |
1-3 years |
3-5 years |
More than 5 years | ||||||||||||
| Operating Leases |
$ | 119 | $ | 38 | $ | 81 | ||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total |
$ | 119 | $ | 38 | $ | 81 | ||||||||||
|
|
|
|
|
|
|
|||||||||||
Critical Accounting Policies and Estimates
Our significant accounting policies are more fully described in Note 2 to our consolidated financial statements included elsewhere in this annual report on Form 10-K. Certain of our accounting policies, however, are critical to the portrayal of our financial position and results of operations and require the application of significant judgment by our management, which subjects them to an inherent degree of uncertainty. In applying our accounting policies, our management uses its best judgment to determine the appropriate assumptions to be used in the determination of certain estimates. Our more significant estimates include stock option and warrant liability valuations and performance vesting features of certain of these instruments, useful lives and potential impairment of property and equipment and intangible assets, accrued liabilities, deferred income taxes and cash flow. These estimates are based on our historical experience, terms of existing contracts, our observance of trends in the industry, information available from other outside sources, and on various other factors that we believe to be appropriate under the circumstances. We believe that the critical accounting policies discussed below involve more complex management judgment due to the sensitivity of the methods, assumptions and estimates necessary in determining the related asset, liability, revenue and expense amounts.
Accrued Expenses. As part of the process of preparing our consolidated financial statements, we are required to estimate accrued expenses. This process involves identifying services that third parties have performed on our behalf and estimating the level of service performed and the associated cost incurred on these services as of each balance sheet date in our consolidated financial statements. Examples of estimated accrued expenses include contract service fees in conjunction with pre-clinical and clinical trials, professional service fees, such as those arising from the services of attorneys and accountants and accrued payroll expenses. In connection with these service fees, our estimates are most affected by our understanding of the status and timing of services provided relative to the actual services incurred by the service providers. In the event that we do not identify certain costs that have been incurred or we under- or over-estimate the level of services or costs of such services, our reported expenses for a reporting period could be understated or overstated. The date on which certain services commence, the level of services performed on or before a given date, and the cost of services are often subject to our judgment. We make these judgments based upon the facts and circumstances known to us in accordance with accounting principles generally accepted in the U.S.
Research and Development Expenses. Costs associated with research and development are expensed as incurred. Research and development expenses include, among other costs, salaries and other personnel-related costs, and costs incurred by outside laboratories and other accredited facilities in connection with clinical trials and preclinical studies.
Stock-Based Compensation. Stock-based compensation cost is measured at the grant date based on the fair value of the award and is recognized as expense over the service period, which generally represents the vesting
40
Table of Contents
Index to Financial Statements
period. For awards that have performance based vesting conditions the Company recognizes the expense over the estimated period that the awards are expected to be earned. The Company generally uses the Black-Scholes option-pricing model to calculate the grant date fair value of stock options. For options that only vest upon the achievement of market conditions, the Company values the options using a Monte Carlo model to calculate the grant date fair value of the stock options. The expense related to options that vest based on market conditions is not reversed should those options not ultimately vest. The expense recognized over the service period is required to include an estimate of the awards that will be forfeited. Stock options issued to non-employees are accounted for in accordance with the provisions of ASC Subtopic 505-50, Equity-Based Payments to Non-employees, which requires valuing the stock options using an option pricing model (the Company uses Black-Scholes) and measuring such stock options to their current fair value when they vest.
Forward-Looking Statements
Certain statements made herein that look forward in time or express management’s expectations or beliefs with respect to the occurrence of future events are forward-looking statements as defined under Section 21E of the Securities Exchange Act of 1934, as amended, and are subject to the safe harbor created therein for forward-looking statements. Such statements include, but are not limited to, statements concerning our anticipated operating results, research and development, clinical trials, regulatory proceedings, and financial resources, and can be identified by use of words such as, for example, “anticipate,” “estimate,” “expect,” “project,” “intend,” “plan,” “believe” and “would,” “should,” “could” or “may.” All statements, other than statements of historical facts, included herein that address activities, events, or developments that the Company expects or anticipates will or may occur in the future, are forward-looking statements, including statements regarding: plans and expectations regarding clinical trials; plans and expectations regarding regulatory approvals; our strategy and expectations for clinical development and commercialization of our products; potential strategic partnerships; expectations regarding the effectiveness of our products; plans for research and development and related costs; statements about accounting assumptions and estimates; expectations regarding liquidity and the sufficiency of cash to fund operations through 2015; our commitments and contingencies; and our market risk exposure. Forward-looking statements are based on current expectations, estimates and projections about the industry and markets in which Galectin Therapeutics operates, and management’s beliefs and assumptions. These statements are not guarantees of future performance and involve certain known and unknown risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Such risks and uncertainties are related to and include, without limitation,
| • | our early stage of development, |
| • | we have incurred significant operating losses since our inception and cannot assure you that we will generate revenue or profit, |
| • | our dependence on outside capital, |
| • | we may be unable to enter into strategic partnerships for the development, commercialization, manufacturing and distribution of our proposed product candidates, |
| • | uncertainties related to our technology and clinical trials, |
| • | uncertainties related to any litigation, including shareholder class actions and derivative lawsuits filed, |
| • | we may be unable to demonstrate the efficacy and safety of our developmental product candidates in human trials, intellectual property protection, and we may be unable to improve upon, protect and/or enforce our intellectual property, |
| • | we are subject to extensive and costly regulation by the U.S. Food and Drug Administration (FDA) and by foreign regulatory authorities, which must approve our product candidates in development and could restrict the sales and marketing and pricing of such products, |
| • | competition and stock price volatility in the biotechnology industry, |
41
Table of Contents
Index to Financial Statements
| • | limited trading volume for our stock, concentration of ownership of our stock, and other risks detailed herein and from time to time in our SEC reports. |
We caution investors that actual results or business conditions may differ materially from those projected or suggested in forward-looking statements as a result of various factors including, but not limited to, those described above and in the Risk Factors section of this annual report on Form 10-K. We cannot assure you that we have identified all the factors that create uncertainties. Moreover, new risks emerge from time to time and it is not possible for our management to predict all risks, nor can we assess the impact of all risks on our business or the extent to which any risk, or combination of risks, may cause actual results to differ from those contained in any forward-looking statements. Readers should not place undue reliance on forward-looking statements. We undertake no obligation to publicly release the result of any revision of these forward-looking statements to reflect events or circumstances after the date they are made or to reflect the occurrence of unanticipated events.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Due to the nature of our operations, assets and absence of debt, we are not exposed to any significant market risks at December 31, 2014 and 2013.
| Item 8. | Financial Statements and Supplementary Data |
The financial statements required by this item are attached to this Annual Report on Form 10-K beginning on Page F-1.
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
(a) Evaluation of Disclosure Controls and Procedures
As required by Rule 13a-15 under the Securities Exchange Act of 1934, (the “Exchange Act”) as of the end of the period covered by this Annual Report, we carried out an evaluation, under the supervision and with the participation of our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of our disclosure controls and procedures as of December 31, 2014. Our management has concluded, based on their evaluation, that our disclosure controls and procedures were effective as of December 31, 2014 to ensure that information required to be disclosed by us in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms.
(b) Management’s Annual Report on Internal Control Over Financial Reporting
Management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting. As defined in Rule 13a-15(f) under the Exchange Act, internal control over financial reporting is a process designed by, or under the supervision of, a company’s principal executive and principal financial officers and effected by a company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. It includes those policies and procedures that:
a) Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of a company;
42
Table of Contents
Index to Financial Statements
b) Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of a company are being made only in accordance with authorizations of management and the board of directors of the company; and
c) Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of a company’s assets that could have a material effect on its financial statements.
Because of the inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management has used the criteria established in “Internal Control-Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework), or COSO, to evaluate the effectiveness of the Company’s internal control over financial reporting. Management has selected the COSO 1992 framework for its evaluation as it is a control framework recognized by the SEC and the Public Company Accounting Oversight Board, that is free from bias, permits reasonably consistent qualitative and quantitative measurement of the Company’s internal controls, is sufficiently complete so that relevant controls are not omitted, and is relevant to an evaluation of internal controls over financial reporting.
Management conducted an evaluation of internal controls based on the COSO 1992 framework. The evaluation included a full scale, documented risk assessment, based on the principles described in the framework, and included identification of key controls. Management completed documentation of its testing to verify the effectiveness of the key controls. Based on the evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2014.
The effectiveness of the Company’s internal control over financial reporting has been audited by McGladrey LLP, an independent registered public accounting firm, as stated in their attestation report appearing below, which expresses an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting as of December 31, 2014.
(c) Changes in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting that occurred during the fourth quarter of 2014 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information |
None.
43
Table of Contents
Index to Financial Statements
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
The information required by this Item will be contained in our definitive Proxy Statement to be filed with the Securities and Exchange Commission, or SEC, in connection with our Annual Meeting of Stockholders which is scheduled to be held on May 21, 2015 (the “2015 Proxy Statement”) under the captions “Election of Directors,” “Board of Directors Meetings and Committees of the Board,” “Executive Officers” and “Section 16(a) Beneficial Ownership Reporting Compliance” and is incorporated herein by reference.
We have adopted a Code of Ethics that applies to all our directors, officers and employees. The Code of Ethics is publicly available on our website at www.galectintherapeutics.com. Amendments to the Code of Ethics and any grant of a waiver from a provision of the Code of Ethics requiring disclosure under applicable SEC rules will be disclosed on our website.
| Item 11. | Executive Compensation |
The information required by this Item will be incorporated by reference from the information under the caption “Compensation of Named Executive Officers” contained in our 2015 Proxy Statement.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this item will be incorporated by reference from the information under the caption “Security Ownership of Certain Beneficial Owners and Management” contained in our 2015 Proxy Statement.
| Item 13. | Certain Relationships, Related Transactions and Director Independence |
The information required by this item will be incorporated by reference from the information under the caption “Certain Relationships and Related Transactions” contained in our 2015 Proxy Statement.
| Item 14. | Principal Accountant Fees and Services |
The information required by this item will be incorporated by reference from the information under the captions “Audit Fees”, “Audit-Related Fees,” “Tax Fees,” “All Other Fees” and “Pre-Approval Policies and Procedures” contained in our 2015 Proxy Statement.
44
Table of Contents
Index to Financial Statements
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
| (a) 1. | Consolidated Financial Statement Schedules |
The Consolidated Financial Statements are filed as part of this report.
| 2. | Consolidated Financial Statement Schedules |
| All schedules are omitted because of the absence of conditions under which they are required or because the required information is included in the Consolidated Financial Statements or notes thereto. |
| 3. | Exhibits |
| Exhibit |
Description of Document | |
| 3.1 | Amended and Restated Articles of Incorporation of Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on May 30, 2012.) | |
| 3.2 | Amended and Restated Bylaws of Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on May 30, 2012.) | |
| 3.3 | Certificate of Designation of Preferences, Rights and Limitations of Series A 12% Convertible Preferred Stock of Pro Pharmaceuticals, Inc., as filed with the Secretary of State of the State of Nevada on October 5, 2007. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on October 9, 2007.) | |
| 3.4 | Certificate of Designation of Preferences, Rights and Limitations of Series B-1 Convertible Preferred Stock and Series B-2 Convertible Preferred Stock of Pro Pharmaceuticals, Inc., as filed with the Secretary of State of the State of Nevada on February 11, 2009. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 3.5 | Certificate of Amendment to the Certificate of Designation of Preferences, Rights and Limitations of Series B-1 Convertible Preferred Stock and Series B-2 Convertible Preferred Stock of Pro-Pharmaceuticals, Inc., as filed with the secretary of State of the State of Nevada on August 12, 2009. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q for the period ended June 30, 2009 as filed with the Commission on August 14, 2009.) | |
| 3.6 | Certificate of Amendment No. 2 to the Certificate of Designation of Preferences, Rights and Limitations of Series B-1 Convertible Preferred Stock and Series B-2 Convertible Preferred Stock, as filed with the State of Nevada, on February 17, 2010. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on February 17, 2010.) | |
| 3.7 | Certificate of Amendment with respect to the Amended and Restated Certificate of Designation of Preferences, Rights and Limitation of Series B-1 Convertible Preferred Stock and Series B-2 Convertible Preferred Stock of Pro-Pharmaceuticals, Inc., as filed with the Secretary of State of the State of Nevada on January 26, 2011. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on January 27, 2011.) | |
| 3.8 | Certificate of Designation of Preferences, Rights and Limitation of Series C Super Dividend Convertible Preferred Stock of Pro-Pharmaceuticals, Inc., as filed with the Secretary of State of Nevada on December 30, 2010. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on January 6, 2011.) | |
| 3.9 | Certificate of Change as filed with the Nevada Secretary of State on March 1, 2012. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on March 23, 2012.) | |
45
Table of Contents
Index to Financial Statements
| Exhibit |
Description of Document | |
| 4.1 | Form of Class A-1 Common Stock Purchase Warrant (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 4.2 | Form of Class A-2 Common Stock Purchase Warrant (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 4.3 | Form of Class B Common Stock Purchase Warrant (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 4.4 | Amended Form of Class A-1 Common Stock Purchase Warrant (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on January 27, 2011.) | |
| 4.5 | Amended Form of Class A-2 Common Stock Purchase Warrant (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on January 27, 2011.) | |
| 4.6 | Amended Form of Class B Common Stock Purchase Warrant (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on January 27, 2011.) | |
| 4.7 | Form of Warrant Agreement between Galectin Therapeutics Inc. and Continental Stock Transfer and Trust Company, as warrant agent (including form of warrant certificate) (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on March 23, 2012.) | |
| 10.1† | Pro-Pharmaceuticals, Inc. 2001 Stock Incentive Plan. (Incorporated by reference to the Company’s Quarterly Report on Form 10-QSB for the quarter ended September 30, 2001 filed with the Commission on November 14, 2001.) | |
| 10.2† | Pro-Pharmaceuticals, Inc. 2003 Non-employee Director Stock Incentive Plan. (Incorporated by reference to the Company’s Registration Statement on Form S-8, as filed with the Commission on October 22, 2003.) | |
| 10.3† | Employment Agreement, effective January 2, 2004, between Pro Pharmaceuticals, Inc. and David Platt. (Incorporated by reference to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2003, as filed with the Commission on March 30, 2004.) | |
| 10.4† | Form of Incentive Stock Option Agreement (under the 2001 Stock Incentive Plan). (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q for the period ended September 30, 2004 as filed with the Commission on November 19, 2004.) | |
| 10.5† | Form of Non-Qualified Stock Option Agreement (under the 2001 Stock Incentive Plan). (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q for the period ended September 30, 2004 as filed with the Commission on November 19, 2004.) | |
| 10.6† | Form of Non-Qualified Stock Option Agreement (under the 2003 Non-Employee Director Stock Incentive Plan). (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q for the period ended September 30, 2004 as filed with the Commission on November 19, 2004.) | |
| 10.7 | Form of Common Stock Purchase Warrant. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on February 15, 2008.) | |
| 10.8 | Promissory Note dated February 12, 2009 issued by Pro Pharmaceuticals, Inc. in favor of 10X Fund, L.P. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 10.9 | Security Agreement dated February 12, 2009 between Pro Pharmaceuticals, Inc. and 10X Fund, L.P. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
46
Table of Contents
Index to Financial Statements
| Exhibit |
Description of Document | |
| 10.10 | Escrow Agreement dated February 12, 2009 among Pro Pharmaceuticals, Inc., 10X Fund, L.P. and Investment Law Group of Gillett, Mottern & Walker, LLP, as Escrow Agent. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 10.11 | Registration Rights Agreement dated February 12, 2009 between Pro Pharmaceuticals, Inc. and 10X Fund, L.P. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 10.12† | Separation Agreement dated February 12, 2009 between Pro Pharmaceuticals, Inc. and David Platt, Ph.D. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 10.13† | Pro-Pharmaceuticals, Inc. 2009 Incentive Compensation Plan. (Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Commission on February 18, 2009.) | |
| 10.14† | Form of Restricted Stock Grant Agreement (under the 2009 Incentive Compensation Plan). (Incorporated by reference to the Company’s Annual Report on Form 10-K as filed with the Commission on March 30, 2009.) | |
| 10.15† | Form of Non-Qualified Stock Option Grant Agreement (under the 2009 Incentive Compensation Plan). (Incorporated by reference to the Company’s Annual Report on Form 10-K as filed with the Commission on March 30, 2009.) | |
| 10.16† | Form of Incentive Stock Option Grant Agreement (under the 2009 Incentive Compensation Plan). (Incorporated by reference to the Company’s Annual Report on Form 10-K as filed with the Commission on March 30, 2009.) | |
| 10.17 | Agreement with the 10X Fund L.P., dated February 11, 2010. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on February 17, 2010.) | |
| 10.18† | Common Stock Purchase Warrant dated August 3, 2010 issued to Peter Traber. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on August 13, 2010.) | |
| 10.19 | Letter Agreement Between 10X Fund, L.P. and Pro-Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on August 13, 2010.) | |
| 10.20 | Form of Securities Purchase Agreement for Series C Super Dividend Convertible Preferred Stock (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on January 6, 2011.) | |
| 10.21 | Agreement dated January 21, 2011, between Pro-Pharmaceuticals, Inc. and 10X Fund L.P. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on January 27, 2011.) | |
| 10.22† | Non-Qualified Stock Option Agreement dated March 7, 2011 (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on March 9, 2011.) | |
| 10.23† | Amended Employment Agreement dated March 8, 2011 between Anthony D. Squeglia, and Pro-Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on March 14, 2011.) | |
| 10.24† | Amended Employment Agreement dated March 8, 2011 between Maureen Foley, and Pro-Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on March 14, 2011.) | |
47
Table of Contents
Index to Financial Statements
| Exhibit |
Description of Document | |
| 10.25† | Amended Employment Agreement dated March 31, 2011 between Anatole Klyosov, and Pro-Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on April 6, 2011.) | |
| 10.26† | Employment Agreement dated March 31, 2011 between Eli Zomer and Pro-Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on April 6, 2011.) | |
| 10.27† | Separation Agreement dated March 31, 2011 between Pro-Pharmaceuticals, Inc. and Theodore D. Zucconi (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on April 6, 2011.) | |
| 10.28 | Agreement dated April 22, 2011, between Pro-Pharmaceuticals, Inc. and Sigma-Aldrich, Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on April 28, 2011.) | |
| 10.29† | Employment Agreement dated March 31, 2011 between Peter Traber, and Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on June 2, 2011.) | |
| 10.30† | Employment Agreement dated June 28, 2011 between James C. Czirr, and Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on July 5, 2011.) | |
| 10.31† | Non-Qualified Stock Option Agreement for Peter G. Traber, M.D. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on August 15, 2011.) | |
| 10.32† | Non-Qualified Stock Option Agreement for James C. Czirr (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on August 15, 2011. ) | |
| 10.33† | Consulting Agreement, dated March 2, 2012 between Galectin Therapeutics Inc. and Thomas A. McGauley (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on May 11, 2012.) | |
| 10.34† | Independent Consulting Agreement dated April 30, 2012, between Scott L. Friedman, M.D. and Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on November 9, 2012.) | |
| 10.35† | Amended Employment Agreement dated July 19, 2012 between Maureen Foley and Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on November 9, 2012.) | |
| 10.36† | Amended and Restated Employment Agreement dated December 11, 2014 between Harold H. Shlevin and Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on December 12, 2014.) | |
| 10.37† | Independent Consulting Agreement dated September 19, 2012 between Thomas A. McGauley and Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on November 9, 2012.) | |
| 10.38 | Amended and Restated Master Services Agreement dated February 1, 2013 between Galectin Therapeutics Inc. and CTI Clinical Trial Services, Inc. and CTI Clinical Consulting Services Inc. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on May 10, 2013.) | |
| 10.39 | Amended Form of Class A-2 Common Stock Purchase Warrant (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on August 14, 2013.) | |
48
Table of Contents
Index to Financial Statements
| Exhibit |
Description of Document | |
| 10.40 | Amended Form of Class B Common Stock Purchase Warrant (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on August 14, 2013.) | |
| 10.41† | Employment Agreement dated June 20, 2013 between Jack W. Callicutt and Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on August 14, 2013.) | |
| 10.42† | Amendment to Independent Consulting Agreement dated June 19, 2013 between Thomas A. McGauley and Galectin Therapeutics Inc. (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on August 14, 2013.) | |
| 10.43† | Stock Option Agreement with Thomas A. McGauley dated June 19, 2013 (Incorporated by reference to the Company’s Quarterly Report on Form 10-Q as filed with the Commission on August 14, 2013.) | |
| 10.44 | At Market Issuance Sales Agreement, dated October 25, 2013, by and between Galectin Therapeutics Inc. and MLV & Co. LLC (Incorporated by reference to the Company’s Current Report on Form 8-K as filed with the Commission on October 25, 2013.) | |
| 10.45 | Amendment No. 1 to At Market Issuance Sales Agreement, dated March 21, 2014, by and between Galectin Therapeutics Inc. and MLV & Co. LLC (Incorporated by reference to the Company’s Registration Statement on Form S-3 as filed with the Commission on March 21, 2014.) | |
| 21.1* | Subsidiaries of Galectin Therapeutics Inc. | |
| 23.1* | Consent of McGladrey LLP, an independent registered public accounting firm. | |
| 31.1* | Certification Pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. | |
| 31.2* | Certification Pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. | |
| 32.1*# | Certification Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2*# | Certification Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 101.INS*** | XBRL Instance document. | |
| 101.SCH*** | XBRL Taxonomy Extension Schema Document. | |
| 101.CAL*** | XBRL Taxonomy Calculation Linkbase Document. | |
| 101.DEF*** | XBRL Taxonomy Definition Linkbase Document. | |
| 101.LAB*** | XBRL Taxonomy Label Linkbase Document. | |
| 101.PRE*** | XBRL Taxonomy Presentation Linkbase Document. | |
| * | Filed herewith. |
| # | Furnished herewith and not “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. |
| *** | Submitted electronically herewith. |
| † | Executive Compensation Arrangement pursuant to 601(b)(10)(iii)(A) of Regulation S-K |
49
Table of Contents
Index to Financial Statements
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on March 18, 2015.
| GALECTIN THERAPEUTICS INC. | ||
| By: | /s/ PETER G. TRABER | |
| Name: Peter G. Traber, M.D. | ||
| Title: Chief Executive Officer and President | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ PETER G. TRABER Peter G. Traber, M.D. |
Chief Executive Officer, President and Director (principal executive officer) |
March 18, 2015 | ||
| /s/ JACK W. CALLICUTT Jack W. Callicutt |
Chief Financial Officer (principal financial and accounting officer) |
March 18, 2015 | ||
| /s/ JAMES C. CZIRR James C. Czirr |
Executive Chairman and Director | March 18, 2015 | ||
| /s/ ROD D. MARTIN Rod D. Martin |
Vice-Chairman and Director | March 18, 2015 | ||
| /s/ GILBERT F. AMELIO Gilbert F. Amelio |
Director | March 18, 2015 | ||
| /s/ ARTHUR R. GREENBERG Arthur R. Greenberg |
Director | March 18, 2015 | ||
| /s/ KEVIN D. FREEMAN Kevin D. Freeman |
Director | March 18, 2015 | ||
| /s/ JOHN MAULDIN John Mauldin |
Director | March 18, 2015 | ||
| /s/ GILBERT S. OMENN Gilbert S. Omenn, M.D, Ph.D. |
Director | March 18, 2015 | ||
| /s/ STEVEN PRELACK Steven Prelack |
Director | March 18, 2015 | ||
| /s/ H. PAUL PRESSLER H. Paul Pressler |
Director | March 18, 2015 | ||
| /s/ MARC RUBIN Marc Rubin, M.D. |
Director | March 18, 2015 | ||
50
Table of Contents
Index to Financial Statements
Galectin Therapeutics Inc.
Table of Contents
Index to Financial Statements
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Galectin Therapeutics, Inc.
We have audited the accompanying consolidated balance sheets of Galectin Therapeutics, Inc. and subsidiaries as of December 31, 2014 and 2013, and the related consolidated statements of operations, changes in redeemable convertible preferred stock and stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Galectin Therapeutics, Inc. as of December 31, 2014 and 2013, and the results of its operations and cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Galectin Therapeutics, Inc. and subsidiaries’ internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission in 1992, and our report dated March 18, 2015 expressed an unqualified opinion on the effectiveness of Galectin Therapeutics, Inc.’s internal control over financial reporting
/s/ McGladrey LLP
Charlotte, North Carolina
March 18, 2015
F-1
Table of Contents
Index to Financial Statements
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Galectin Therapeutics, Inc.
We have audited Galectin Therapeutics, Inc. and subsidiaries’ internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission in 1992. Galectin Therapeutics Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (a) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (b) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (c) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Galectin Therapeutics, Inc. and subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission in 1992.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Galectin Therapeutics, Inc. as of December 31, 2014 and 2013, and the related consolidated statements of operations, changes in redeemable convertible preferred stock and stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014 and our report dated March 18, 2015 expressed an unqualified opinion.
/s/ McGladrey LLP
Charlotte, North Carolina
March 18, 2015
F-2
Table of Contents
Index to Financial Statements
GALECTIN THERAPEUTICS INC.
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| (in thousands) | ||||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 29,128 | $ | 10,489 | ||||
| Prepaid expenses and other current assets |
533 | 198 | ||||||
|
|
|
|
|
|||||
| Total current assets |
29,661 | 10,687 | ||||||
|
|
|
|
|
|||||
| Property and equipment, net |
1 | 3 | ||||||
| Intangible assets, net |
15 | 23 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 29,677 | $ | 10,713 | ||||
|
|
|
|
|
|||||
| LIABILITIES, REDEEMABLE CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 906 | $ | 762 | ||||
| Accrued expenses |
729 | 1,651 | ||||||
| Accrued dividends payable |
68 | 73 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
1,703 | 2,486 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
1,703 | 2,486 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies (Note 9) |
||||||||
| Series B-1 12% redeemable convertible preferred stock; 900,000 shares authorized, issued and outstanding at December 31, 2014 and 2013, redemption value and liquidation value: $1,800,000, at December 31, 2014 |
1,731 | 1,715 | ||||||
| Series B-2 12% redeemable convertible preferred stock; 2,100,000 shares authorized, issued and outstanding at December 31, 2014 and 2013, redemption value and liquidation value: $4,200,000, at December 31, 2014 |
3,325 | 3,112 | ||||||
| Series C super dividend convertible preferred stock; 1,000 shares authorized, 176 and 196 issued and outstanding at December 31, 2014 and 2013, respectively, redemption value: $4,835,000, liquidation value: $1,786,000 at December 31, 2014 |
1,723 | 1,919 | ||||||
| Stockholders’ equity: |
||||||||
| Undesignated stock, $0.01 par value; 20,000,000 shares authorized at December 31, 2014 and 2013, 8,001,000 shares designated at December 31, 2014 and 2013 |
||||||||
| Series A 12% convertible preferred stock; 5,000,000 shares authorized, 1,402,500 and 1,452,500 issued and outstanding at December 31, 2014 and 2013, liquidation value $1,445,000 at December 31, 2014, |
567 | 587 | ||||||
| Common stock, $0.001 par value; 50,000,000 shares authorized at December 31, 2014 and 2013, 22,277,283 and 18,386,900 issued and outstanding at December 31, 2014 and 2013, respectively |
22 | 18 | ||||||
| Additional paid-in capital |
139,531 | 102,841 | ||||||
| Retained deficit |
(118,925 | ) | (101,965 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
21,195 | 1,481 | ||||||
|
|
|
|
|
|||||
| Total liabilities, redeemable convertible preferred stock and stockholders’ equity |
$ | 29,677 | $ | 10,713 | ||||
|
|
|
|
|
|||||
See notes to consolidated financial statements.
F-3
Table of Contents
Index to Financial Statements
GALECTIN THERAPEUTICS INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (in thousands, except per share amounts) |
||||||||||||
| Operating expenses: |
||||||||||||
| Research and development |
$ | 8,425 | $ | 5,688 | $ | 4,527 | ||||||
| General and administrative |
7,005 | 6,416 | 5,372 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
15,430 | 12,104 | 9,899 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating loss |
(15,430 | ) | (12,104 | ) | (9,899 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Other income (expense): |
||||||||||||
| Interest income |
42 | 16 | 24 | |||||||||
| Loss from equity method investment in Galectin Sciences, LLC |
(400 | ) | — | — | ||||||||
| Other income |
— | — | 200 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total other income (expense) |
(358 | ) | 16 | 224 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (15,788 | ) | $ | (12,088 | ) | $ | (9,675 | ) | |||
|
|
|
|
|
|
|
|||||||
| Preferred stock dividends |
(943 | ) | (867 | ) | (976 | ) | ||||||
| Preferred stock accretion |
(229 | ) | (229 | ) | (230 | ) | ||||||
| Warrant modification |
— | (8,763 | ) | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss applicable to common stockholders |
$ | (16,960 | ) | $ | (21,947 | ) | $ | (10,881 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (0.78 | ) | $ | (1.30 | ) | $ | (0.72 | ) | |||
| Shares used in computing basic and diluted net loss per share |
21,849 | 16,874 | 15,131 | |||||||||
See notes to consolidated financial statements.
F-4
Table of Contents
Index to Financial Statements
GALECTIN THERAPEUTICS INC.
CONSOLIDATED STATEMENT OF CHANGES IN REDEEMABLE CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY
For the Years Ended December 31, 2014, 2013 and 2012
(amounts in thousands except share data)
| Stockholders’ Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-1 12% Redeemable Convertible Preferred Stock |
Series B-2 12% Redeemable Convertible Preferred Stock |
Series C Super Dividend Convertible Preferred Stock |
Series A 12% Convertible Preferred Stock |
Common Stock | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Amount | Number of Shares |
Amount | Number of Shares |
Amount | Number of Shares |
Amount | Numbe of Shares |
Amount | Additional Paid-In Capital |
Retained Deficit |
Total Stockholders’ Equity |
||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2011 |
900,000 | $ | 1,681 | 2,100,000 | $ | 2,687 | 220 | $ | 2,154 | 1,562,500 | $ | 632 | 12,919,538 | $ | 13 | $ | 66,367 | $ | (69,137 | ) | $ | (2,125 | ) | |||||||||||||||||||||||||||||||
| Accretion of Series B redeemable convertible preferred stock |
17 | 157 | (174 | ) | (174 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||
| Accretion of beneficial conversion feature for Series B-2 |
56 | (56 | ) | (56 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock and warrants, net of issuance costs of $1,597,000 |
2,666,722 | 3 | 10,400 | 10,403 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of shares related to reverse split of common stock |
3,324 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Series A 12% convertible preferred stock dividend |
31,250 | 103 | (103 | ) | — | |||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-1 12% redeemable convertible preferred stock dividend |
95,584 | 224 | (224 | ) | — | |||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-2 12% redeemable convertible preferred stock dividend |
223,027 | 522 | (522 | ) | — | |||||||||||||||||||||||||||||||||||||||||||||||||
| Series C super dividend convertible preferred stock dividend |
46,053 | 127 | (127 | ) | — | |||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to consultants |
11,348 | 26 | 26 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of warrants |
12,177 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of options |
51,830 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation expense |
2,766 | 2,766 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Net loss |
(9,675 | ) | (9,675 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
| Balance at December 31, 2012 |
900,000 | $ | 1,698 | 2,100,000 | $ | 2,900 | 220 | $ | 2,154 | 1,562,500 | $ | 632 | 16,060,853 | $ | 16 | $ | 80,535 | $ | (80,018 | ) | $ | 1,165 | ||||||||||||||||||||||||||||||||
5
Table of Contents
Index to Financial Statements
GALECTIN THERAPEUTICS INC.
CONSOLIDATED STATEMENT OF CHANGES IN REDEEMABLE CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY — (Continued)
For the Years Ended December 31, 2014, 2013 and 2012
(amounts in thousands except share data)
| Stockholders’ Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-1 12% Redeemable Convertible Preferred Stock |
Series B-2 12% Redeemable Convertible Preferred Stock |
Series C Super Dividend Convertible Preferred Stock |
Series A 12% Convertible Preferred Stock |
Common Stock |
|
|
||||||||||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Amount | Number of Shares |
Amount | Number of Shares |
Amount | Number of Shares |
Amount | Numbe of Shares |
Amount | Additional Paid-In Capital |
Retained Deficit |
Total Stockholders’ Deficit |
||||||||||||||||||||||||||||||||||||||||||
| Accretion of Series B redeemable convertible preferred stock |
17 | 158 | (175 | ) | (175 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||
| Accretion of beneficial conversion feature for Series B-2 |
54 | (54 | ) | (54 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Series A 12% convertible preferred stock dividend |
25,062 | 148 | (148 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-1 12% redeemable convertible preferred stock dividend |
36,106 | 178 | (178 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-2 12% redeemable convertible preferred stock dividend |
84,553 | 418 | (418 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series C super dividend convertible preferred stock dividend |
23,848 | 123 | (123 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of warrants |
1.284.948 | 1 | 4,504 | 4,505 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of options |
213,008 | 271 | 271 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock, net of issuance costs of $111 |
599,942 | 1 | 3,832 | 3,833 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series A to common stock |
(110,000 | ) | (45 | ) | 18,387 | 45 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series C super dividend convertible preferred stock to common stock |
(24 | ) | (235 | ) | 40,193 | 235 | 235 | |||||||||||||||||||||||||||||||||||||||||||||||
| Modification of warrants |
8,763 | (8,763 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation expense |
3,789 | 3,789 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Net loss |
(12,088 | ) | (12,088 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
| Balance at December 31, 2013 |
900,000 | $ | 1,715 | 2,100,000 | $ | 3,112 | 196 | $ | 1,919 | 1,452,500 | $ | 587 | 18,386,900 | $ | 18 | $ | 102,841 | $ | (101,965 | ) | $ | 1,481 | ||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
See notes to consolidated financial statements.
6
Table of Contents
Index to Financial Statements
GALECTIN THERAPEUTICS INC.
CONSOLIDATED STATEMENT OF CHANGES IN REDEEMABLE CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY — (Continued)
For the Years Ended December 31, 2014, 2013 and 2012
(amounts in thousands except share data)
| Stockholders’ Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-1 12% Redeemable Convertible Preferred Stock |
Series B-2 12% Redeemable Convertible Preferred Stock |
Series C Super Dividend Convertible Preferred Stock |
Series A 12% Convertible Preferred Stock |
Common Stock | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Amount | Number of Shares |
Amount | Number of Shares |
Amount | Number of Shares |
Amount | Numbe of Shares |
Amount | Additional Paid-In Capital |
Retained Deficit |
Total Stockholders’ Equity |
||||||||||||||||||||||||||||||||||||||||||
| Accretion of Series B redeemable convertible preferred stock |
16 | 157 | (173 | ) | (173 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||
| Accretion of beneficial conversion feature for Series B-2 |
56 | (56 | ) | (56 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Series A 12% convertible preferred stock dividend |
19,490 | 154 | (154 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-1 12% redeemable convertible preferred stock dividend |
32,043 | 206 | (206 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B-2 12% redeemable convertible preferred stock dividend |
74,764 | 480 | (480 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series C super dividend convertible preferred stock dividend |
13,152 | 103 | (103 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of warrants |
572,148 | 1 | 1,675 | 1,676 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of options |
246,445 | 452 | 452 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock |
2,881,269 | 3 | 29,334 | 29,337 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series A to common stock |
(50,000 | ) | (20 | ) | 8,350 | 20 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series C super dividend convertible preferred stock to common stock |
(20 | ) | (196 | ) | 33,756 | 196 | 196 | |||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to consultants |
8,966 | 100 | 100 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation expense |
3,970 | 3,970 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Net loss |
(15,788 | ) | (15,788 | ) | ||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
| Balance at December 31, 2014 |
900,000 | $ | 1,731 | 2,100,000 | $ | 3,325 | 176 | $ | 1,723 | 1,402,500 | $ | 567 | 22,277,283 | $ | 22 | $ | 139,531 | $ | (118,925 | ) | $ | 21,195 | ||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
See notes to consolidated financial statements.
7
Table of Contents
Index to Financial Statements
GALECTIN THERAPEUTICS INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (in thousands) | ||||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: |
||||||||||||
| Net loss |
$ | (15,788 | ) | $ | (12,088 | ) | $ | (9,675 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
10 | 12 | 9 | |||||||||
| Stock-based compensation expense |
4,070 | 3,789 | 2,792 | |||||||||
| Loss from equity method investment in Galectin Sciences LLC |
400 | — | — | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Prepaid expenses and other assets |
(335 | ) | (39 | ) | (55 | ) | ||||||
| Accounts payable and accrued expenses |
(783 | ) | 848 | (577 | ) | |||||||
| Other long-term liabilities |
— | (6 | ) | 6 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(12,426 | ) | (7,484 | ) | (7,500 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: |
||||||||||||
| Equity method investment in Galectin Sciences LLC |
(400 | ) | — | — | ||||||||
| Purchases of property and equipment |
— | — | (5 | ) | ||||||||
| Change in restricted cash and security deposit |
— | — | 69 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
(400 | ) | — | 64 | ||||||||
|
|
|
|
|
|
|
|||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: |
||||||||||||
| Net proceeds from issuance of common stock and warrants |
29,337 | 3,833 | 10,403 | |||||||||
| Net proceeds from exercise of common stock warrants and options |
2,128 | 4,776 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
31,465 | 8,609 | 10,403 | |||||||||
|
|
|
|
|
|
|
|||||||
| NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS |
18,639 | 1,125 | 2,967 | |||||||||
| CASH AND CASH EQUIVALENTS, BEGINNING OF PERIOD |
10,489 | 9,364 | 6,397 | |||||||||
|
|
|
|
|
|
|
|||||||
| CASH AND CASH EQUIVALENTS, END OF PERIOD |
$ | 29,128 | $ | 10,489 | $ | 9,364 | ||||||
|
|
|
|
|
|
|
|||||||
| NONCASH FINANCING ACTIVITIES: |
||||||||||||
| Payment of preferred stock dividends in common stock |
$ | 943 | $ | 867 | $ | 976 | ||||||
See notes to consolidated financial statements.
F-8
Table of Contents
Index to Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| 1. | Nature of Business and Basis of Presentation |
Galectin Therapeutics Inc. (the “Company”) is a clinical stage biopharmaceutical company that is applying its leadership in galectin science and drug development to create new therapies for fibrotic disease and cancer. These candidates are based on the Company’s targeting of galectin proteins which are key mediators of biologic and pathologic function. These compounds also may have application for drugs to treat other diseases and chronic health conditions.
On March 23, 2012, the Company effected a one-for-six reverse stock split. All common share and per share amounts in these financial statements have been adjusted to reflect the effect of the reverse split.
The Company has operated at a loss since its inception and has had no revenues. The Company anticipates that losses will continue for the foreseeable future. At December 31, 2014, the Company had $29,128,000 of unrestricted cash and cash equivalents available to fund future operations. Additionally, in January and February 2015, the Company received $4,127,000 in net proceeds from the issuance of common stock at market through its at the market (“ATM”) financing arrangement.
The Company was founded in July 2000, was incorporated in the State of Nevada in January 2001 under the name “Pro-Pharmaceuticals, Inc.,” and changed its name to “Galectin Therapeutics Inc.” on May 26, 2011. On March 23, 2012, the Company began trading on The NASDAQ Capital Market under the symbol GALT. Prior to March 23, 2012, the Company was traded on the Over-the Counter Bulletin Board (“OTCBB”) under the symbol GALT.OB (previously PRWP.OB) from January 21, 2009 to March 22, 2012 after the Company was delisted from the NYSE Alternext US (“Exchange”), formerly the American Stock Exchange, due to non-compliance with the Exchange minimum shareholders’ equity requirements on January 9, 2009.
The Company is subject to a number of risks similar to those of clinical stage companies, including dependence on key individuals, uncertainty of product development and generation of revenues, dependence on outside sources of capital, risks associated with clinical trials of products, dependence on third-party collaborators for research operations, need for regulatory approval of products, risks associated with protection of intellectual property, and competition with larger, better-capitalized companies. Successful completion of the Company’s development program and, ultimately, the attainment of profitable operations is dependent upon future events, including obtaining adequate financing to fulfill its development activities and achieving a level of revenues adequate to support the Company’s cost structure. There are no assurances that the Company will be able to obtain additional financing on favorable terms, or at all, or successfully market its products.
| 2. | Summary of Significant Accounting Policies |
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States (“GAAP”).
Basis of Consolidation. The consolidated financial statements include the accounts of the Company and Galectin Therapeutics Security Corp., its wholly-owned subsidiary, which was incorporated in Delaware on December 23, 2003 and Galectin Sciences LLC (see Note 10). All intercompany transactions have been eliminated.
F-9
Table of Contents
Index to Financial Statements
Use of Estimates. The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and judgments that may affect the reported amounts of assets, liabilities, equity, revenue, expenses and related disclosure of contingent assets and liabilities. Management’s estimates and judgments include assumptions used in stock option and warrant liability valuations, useful lives of property and equipment and intangible assets, accrued liabilities, deferred income taxes and various other assumptions that are believed to be reasonable under the circumstances. Actual results may differ from those estimates under different assumptions or conditions.
Fair Value Measurements. The Company has certain financial assets and liabilities recorded at fair value. Fair values determined by Level 1 inputs utilize observable data such as quoted prices in active markets. Fair values determined by Level 2 inputs utilize data points other than quoted prices in active markets that are observable either directly or indirectly. Fair values determined by Level 3 inputs utilize unobservable data points in which there is little or no market data, which require the reporting entity to develop its own assumptions. The estimated value of accounts payable and accrued expenses approximates their carrying value due to their short-term nature.
Cash and Cash Equivalents. The Company considers all highly-liquid investments with original maturities of 90 days or less at the time of acquisition to be cash equivalents. The Company had no cash equivalents at December 31, 2014 or 2013.
Prepaid Expenses and Other Current Assets. Prepaid expenses and other assets consist principally of prepaid insurance and prepaid rent on the Company’s leased executive office space.
Property and Equipment. Property and equipment, including leasehold improvements, are stated at cost, net of accumulated depreciation and amortization, and are depreciated or amortized using the straight-line method over the estimated useful lives of the related assets of generally three years for computers and office equipment, five years for furniture and fixtures and the shorter of the useful life or life of the lease for leasehold improvements.
Restricted Cash and Security Deposit. At December 31, 2014 and 2013, the Company had a security deposit of $6,000 for leased office space. The security deposit was included in Prepaid Expenses and Other Current Assets at December 31, 2014.
Intangible Assets. Intangible assets include patent costs, consisting primarily of related capitalized legal fees, which are amortized over an estimated useful life of five years from issuance. Amortization expense in 2014 and 2013 was $8,000 and $7,000, respectively. Gross intangible assets at December 31, 2014 and 2013 totaled $78,000 each year, and accumulated amortization at December 31, 2014 and 2013 totaled $63,000 and $55,000, respectively.
Long-Lived Assets. The Company reviews all long-lived assets for impairment whenever events or circumstances indicate the carrying amount of such assets may not be recoverable. Recoverability of assets to be held or used is measured by comparison of the carrying value of the asset to the future undiscounted net cash flows expected to be generated by the asset. If such asset is considered to be impaired, the impairment recognized is measured by the amount by which the carrying value of the asset exceeds the discounted future cash flows expected to be generated by the asset.
Accrued Expenses. As part of the process of preparing our consolidated financial statements, we are required to estimate accrued expenses. This process involves identifying services that third parties have performed on our behalf and estimating the level of service performed and the associated cost incurred on these services as of each balance sheet date in our consolidated financial statements. Examples of estimated accrued expenses include contract service fees in conjunction with pre-clinical and clinical trials, professional service fees, such as those arising from the services of attorneys and accountants and accrued
F-10
Table of Contents
Index to Financial Statements
payroll expenses. In connection with these service fees, our estimates are most affected by our understanding of the status and timing of services provided relative to the actual services incurred by the service providers. In the event that we do not identify certain costs that have been incurred or we under- or over-estimate the level of services or costs of such services, our reported expenses for a reporting period could be understated or overstated. The date on which certain services commence, the level of services performed on or before a given date, and the cost of services are often subject to our judgment. We make these judgments based upon the facts and circumstances known to us in accordance with accounting principles generally accepted in the U.S.
Warrants. The Company has issued common stock warrants in connection with the execution of certain equity and debt financings. Certain warrants were accounted for as derivative liabilities at fair value. Such warrants did not meet the accounting criteria that a contract should not be considered a derivative instrument if it is (1) indexed to its own stock and (2) classified in stockholders’ equity. Changes in fair value of derivative liabilities are recorded in the consolidated statement of operations under the caption “Change in fair value of warrant liabilities.” Warrants that are not considered derivative liabilities are accounted for at fair value at the date of issuance in additional paid-in capital. The fair value of warrants was determined using the Black-Scholes option-pricing model using assumptions regarding volatility of our common share price, remaining life of the warrant, and risk-free interest rates at each period end. There were no warrant liabilities as of December 31, 2014 or 2013.
Revenue Recognition. The Company records revenue provided that there is persuasive evidence that an arrangement exists, the price is fixed and determinable, services were rendered and collectability is reasonably assured.
Research and Development Expenses. Costs associated with research and development are expensed as incurred. Research and development expenses include, among other costs, salaries and other personnel-related costs, and costs incurred by outside laboratories and other accredited facilities in connection with clinical trials and preclinical studies.
Income Taxes. The Company accounts for income taxes in accordance with the accounting rules that requires an asset and liability approach to accounting for income taxes based upon the future expected values of the related assets and liabilities. Deferred income tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities and for tax loss and credit carry forwards, and are measured using the expected tax rates estimated to be in effect when such basis differences reverse. Valuation allowances are established, if necessary, to reduce the deferred tax asset to the amount that will, more likely than not, be realized.
Comprehensive Income (Loss). Comprehensive income (loss) is defined as the change in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources. The Company does not have any items of comprehensive income (loss) other than net losses as reported.
Concentration of Credit Risk. Financial instruments that subject the Company to credit risk consist of cash and cash equivalents and certificates of deposit. The Company maintains cash and cash equivalents and certificates of deposit with well-capitalized financial institutions. At times, those amounts may exceed federally insured limits. The Company has no significant concentrations of credit risk.
Stock-Based Compensation. Stock-based compensation cost is measured at the grant date based on the fair value of the award and is recognized as expense over the service period, which generally represents the vesting period. For awards that have performance based vesting conditions the Company recognizes the expense over the estimated period that the awards are expected to be earned. The Company generally uses the Black-Scholes option-pricing model to calculate the grant date fair value of stock options. For options
F-11
Table of Contents
Index to Financial Statements
that only vest upon the achievement of market conditions, the Company values the options using a Monte Carlo model to calculate the grant date fair value of the stock options. The expense related to options that vest based on market conditions is not reversed should those options not ultimately vest. The expense recognized over the service period is required to include an estimate of the awards that will be forfeited. Stock options issued to non-employees are accounted for in accordance with the provisions of ASC Subtopic 505-50, Equity-Based Payments to Non-employees, which requires valuing the stock options using an option pricing model (the Company uses Black-Scholes) and measuring such stock options to their current fair value when they vest.
New Accounting Pronouncements. The Company adopted Financial Accounting Standards Board (FASB), Accounting Standards Update No. 2014-10 “Development Stage Entities (Topic 915)” as of June 30, 2014. This new standard modifies financial statement presentation to eliminate the requirement to include inception-to-date information in the statements of operations and cash flows, among other provisions.
In August 2014, the FASB issued Accounting Standard Update No. 2014-15, Disclosure of Uncertainties About an Entity’s Ability to Continue as a Going Concern. The amendments require management to perform interim and annual assessments of an entity’s ability to continue as a going concern and provides guidance on determining when and how to disclose going concern uncertainties in the financial statements. The standard applies to all entities and is effective for annual and interim reporting periods ending after December 15, 2016, with early adoption permitted. The Company is currently evaluating the impact that this new guidance will have on its financial statements.
In July 2013, the FASB issued amended guidance on the financial statement presentation of an unrecognized tax benefit when a net operating loss carryforward, similar tax loss, or tax credit carryforward exists. The guidance requires an unrecognized tax benefit, or a portion of an unrecognized tax benefit, to be presented as a reduction of a deferred tax asset when a net operating loss carryforward, similar tax loss, or tax credit carryforward exists, with certain exceptions. This accounting guidance was effective for annual and interim periods beginning after December 15, 2013. The Company adopted this new guidance beginning with its interim financial statements for the three months ended March 31, 2014. The adoption of this standard did not have a material impact on the Company’s financial statements.
| 3. | Property and Equipment |
Property and equipment consists of the following at December 31:
| 2014 | 2013 | |||||||
| (in thousands) | ||||||||
| Leasehold improvements |
$ | 2 | $ | 2 | ||||
| Computer and office equipment |
13 | 13 | ||||||
| Furniture and fixtures |
59 | 59 | ||||||
|
|
|
|
|
|||||
| Total |
74 | 74 | ||||||
| Less accumulated depreciation and amortization |
(73 | ) | (71 | ) | ||||
|
|
|
|
|
|||||
| Property and equipment—net |
$ | 1 | $ | 3 | ||||
|
|
|
|
|
|||||
Depreciation and amortization expense for the years ended December 31, 2014 and 2013 was $2,000 and $3,000, respectively.
F-12
Table of Contents
Index to Financial Statements
| 4. | Accrued Expenses |
Accrued expenses consist of the following at December 31:
| 2014 | 2013 | |||||||
| (in thousands) | ||||||||
| Legal and accounting fees |
$ | 118 | $ | 103 | ||||
| Accrued compensation |
604 | 526 | ||||||
| Severance agreement (Note 11) |
— | 1,000 | ||||||
| Other |
7 | 22 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 729 | $ | 1,651 | ||||
|
|
|
|
|
|||||
| 5. | Stockholders’ Equity |
At December 31, 2014, the Company had 50,000,000 shares of common stock and 20,000,000 undesignated shares authorized. As of December 31, 2014, 5,000,000 shares have been designated for Series A 12% Convertible Preferred Stock, 900,000 shares have been designated for Series B-1 Convertible Preferred Stock, 2,100,000 shares have been designated for Series B-2 Convertible Preferred Stock, 1,000 shares have been designated for Series C Super Dividend Convertible Preferred Stock and 11,999,000 remain undesignated.
2013 Private Placement of Common Stock
On August 16, 2013, the Company issued 500,000 unregistered shares of its common stock for proceeds of $3,000,000 to a single investor pursuant to a private placement. There were no warrants or placement fees associated with this transaction.
2013 At Market Issuance of Common Stock
On October 25, 2013, the Company entered into an At Market Issuance Sales Agreement (the “2013 At Market Agreement”) with a sales agent under which the Company may issue and sell shares of its common stock having an aggregate offering price of up to $30.0 million from time to time through the sales agent. Sales of the Company’s common stock through the sales agent, if any, will be made by any method that is deemed an “at the market” offering as defined by the U.S. Securities and Exchange Commission. The Company will pay to the sales agent a commission rate equal to 3.0% of the gross proceeds from the sale of any shares of common stock sold through the sales agent under the 2013 At Market Agreement. As of December 31, 2013, the Company had issued 99,942 shares of its common stock through its 2013 At Market Agreement at an average price of $9.02 per share resulting in gross proceeds of approximately $944,000. The Company incurred one time, initial legal and accounting costs of approximately $82,000 and commissions of $29,000 resulting in net proceeds of $833,000 as of December 31, 2013. In January and February 2014, the Company issued 2,663,647 shares of common stock for net proceeds of approximately $28,178,000 which completed the 2013 At Market Agreement.
2014 At Market Issuance of Common Stock
On March 30, 2014, the Company entered into an At Market Issuance Sales Agreement (the “2014 At Market Agreement”) with a sales agent under which the Company may issue and sell shares of its common stock having an aggregate offering price of up to $30.0 million from time to time through the sales agent. Sales of the Company’s common stock through the sales agent, if any, will be made by any method that is deemed an “at the market” offering as defined by the U.S. Securities and Exchange Commission. The Company will pay to the sales agent a commission rate equal to 3.0% of the gross proceeds from the sale of any shares of common stock sold through the sales agent under the 2014 At Market Agreement. As of
F-13
Table of Contents
Index to Financial Statements
December 31, 2014, the Company had issued 217,622 shares of its common stock through its 2014 At Market Agreement at an average price of $5.49 per share resulting in gross proceeds of approximately $1,196,000. The Company incurred commissions of approximately $36,000 resulting in net proceeds of approximately $1,159,000 as of December 31, 2014. In January and February 2015, the Company issued 1,173,458 shares of common stock for net proceeds of approximately $4,127,000 under the 2014 At Market Agreement.
Series A 12% Convertible Preferred Stock — February 4, 2008 Private Placement
On February 4, 2008, the Company closed a private placement begun in October 2007 of its Series A 12% Convertible Preferred Stock (“Series A”) and related warrants. In this transaction, the Company sold units of securities at $6.00 per unit, each unit comprised of (i) one share of Series A Preferred, (ii) a warrant to purchase one share of common stock for $9.00, and (iii) a warrant to purchase one share of common stock for $12.00. Each share of the Series A is entitled to dividends at the rate of 12% per annum payable at the Company’s option in cash or shares of common stock valued at the higher of $6.00 per share or 100% of the value weighted average price of the Company’s share price for the 20 consecutive trading days prior to the applicable dividend payment date. Dividends are payable semi-annually on March 30 and September 30. The dividend paid on the initial dividend payment date is calculated from the date the Company deposited each subscription advance.
The shares of Series A are entitled to vote as a class with the Company’s common stock and each share of Series A is convertible at any time to one-sixth of a share of common stock, subject to adjustment in the event of a stock dividend, stock split or combination, reclassification or similar event. The Company has the right to require conversion if the closing price of the common stock exceeds $18.00 for 15 consecutive trading days and a registration statement covering the resale of the shares of common stock issuable upon conversion of the Series A is then in effect. Each warrant is exercisable solely for cash beginning August 3, 2008 and expired on February 4, 2012. The exercise price of each warrant is adjustable in the event of a stock split or stock combination, capital reorganization, merger or similar event.
As of December 31, 2007, the Company had received subscription advances of $1,667,500 for Series A. In 2008, the Company received additional subscription advances of $75,000 resulting in total gross proceeds of $1,742,500. On February 4, 2008 the Company closed the private placement. The Company incurred $52,000 of cash transaction costs resulting in net cash proceeds of $1,691,000. In addition, the Company incurred $3,000 of costs for 1,400 warrants exercisable at $9.00 issued to placement agents. Proceeds of $984,000 were allocated to investor warrants using the Black-Scholes method with the following assumptions as of February 4, 2008: risk free interest rate 2.51%, volatility 95%, fair market value of the company’s common stock on February 4, 2008, and the share price on the closing date of the transaction of $3.54. The warrants were originally accounted for as freestanding derivative instruments in the consolidated balance sheet formerly under the caption “Warrant Liabilities”. These warrants were originally classified as a liability because the February 2006 warrants contain an anti-dilution provision in the event of a subsequent dilutive issuance and the potential number of shares issuable exceeded the Company’s authorized shares. Changes in fair value were recognized as either a gain or loss in the consolidated statement of operations under the caption “Change in fair value of warrant liabilities”. In the second quarter of 2008, the warrants were reclassified to equity as a result of an amendment to the Company’s articles of incorporation approved at the May 21, 2008 annual meeting of shareholders increasing the Company’s authorized common. Through May 21, 2008, these warrants were marked to market resulting in a reduction in warrant liabilities in the balance sheet and an offsetting credit to change in fair value of warrant liabilities in the statement of operations in the amount of $100,000. The remaining fair value of $502,000 was credited to additional paid-in capital in the balance sheet.
In 2014 and 2013, 50,000 and 110,000 shares of Series A were converted into 8,334 and 18,387 shares of common stock, respectively. Prior to 2013, a total of 180,000 shares of Series A had been converted into 30,000 shares of common stock.
F-14
Table of Contents
Index to Financial Statements
Series B Redeemable Convertible Preferred Stock
On February 12, 2009, the Company entered into a securities purchase agreement (the “10X Agreement”) pursuant to which it agreed to issue and sell to 10X Fund LP, at two or more closings, up to: (i) 3,000,000 shares its Series B convertible preferred stock (“Series B redeemable convertible preferred stock” or “Series B”) with an aggregate stated value of $6.0 million and convertible into 2,000,000 shares of common stock at December 31, 2011 and (ii) warrants to purchase 6,000,000 shares of common stock.
Through a series of closings from February 2009 through May 2010, the Company issued and sold, pursuant to the 10X Agreement, a total of (i) 900,000 shares of Series B-1 convertible preferred stock (“Series B-1 redeemable convertible preferred stock” or “Series B-1”) and related common stock warrants for 1,800,000 shares of common stock and (ii) 2,100,000 shares of Series B-2 convertible preferred stock (“Series B-2 redeemable convertible preferred stock” or “Series B-2”) and related warrants for 4,200,000 shares of common stock for total net proceeds of $5,483,000.
The terms of the Series B are as follows:
Dividends. Holders of the Series B will be entitled to receive cumulative dividends at the rate of 12% per share per annum (compounding monthly) payable quarterly which may, at the Company’s option, be paid in cash or common stock. Pursuant to an agreement with the holder of all shares of Series B, on January 26, 2011, the Company amended and restated the Certificate of Designation of Preferences, Rights and Limitations for the Series B-1 and Series B-2, to provide that dividends are payable in cash or shares of Common Stock valued at 100% of the volume weighted average price of the Common Stock for the 20 consecutive trading days prior to the dividend payment date on and after September 30, 2011. If the Company does not pay any dividend on the Series B, dividends will accrue at the rate of 15% per annum (compounding monthly).
Conversion Rights. Each share of Series B is convertible into two-thirds (approximately 0.667) shares of common stock at the conversion price of $3.00 per share at the option of (i) the holder, at any time and (ii) the Company, at any time after February 12, 2010 (and upon 10 days notice) if the common stock is quoted at or above $9.00 for 15 consecutive trading days and an effective registration statement regarding the underlying shares of common stock is in effect (subject to certain monthly volume limits). Pursuant to an agreement with the holder of all shares of Series B, on January 26, 2011, the Company amended and restated the Certificate of Designation of Preferences, Rights and Limitations for the Series B-1 and Series B-2, to remove the Company’s right to compel conversion of the Series B Preferred Stock to shares of its Common Stock
Redemption Rights. Pursuant to an agreement with the holder of all shares of Series B, on January 26, 2011, the Company amended and restated the Certificate of Designation of Preferences, Rights and Limitations for the Series B-1 and Series B-2, to provide that, upon notice of not less than 30 trading days, a holder of Series B may require the Company to redeem, in whole or in part at any time on or after the earlier of (a) February 12, 2019 or (b) the date of issuance of a promissory note to David Platt (see Note 11) in connection with the achievement of certain milestones under his separation agreement.
The redemption price will be equal to the sum of the stated value of the Series B, plus all accrued but unpaid dividends thereon, as of the redemption date. If the Company fails to pay the redemption price in cash on the redemption date, then the holders of the Series B requesting redemption may, at their sole option, automatically convert their shares of Series B into a promissory note bearing interest at the rate of 15% per year and secured by a lien on all of the Company’s assets. So long as any shares of the Series B remain outstanding, the Company is also subject to restrictions limiting, among other things, amendments to the Company’s organizational documents; the purchase or redemption of the Company’s capital stock; mergers,
F-15
Table of Contents
Index to Financial Statements
consolidations, liquidations and dissolutions; sales of assets; dividends and other restricted payments; investments and acquisitions; joint ventures, licensing agreements, exclusive marketing and other distribution agreements; issuances of securities; incurrence of indebtedness; incurrence of liens and other encumbrances and issuances of any common stock equivalents.
Liquidation Rights. In the event of any liquidation, dissolution or winding up of the Company, either voluntarily or involuntarily, the holders of Series B will receive $2 per share plus accrued and unpaid dividends, payable prior and in preference to any distributions to the holders of Common Stock but pari passu with the holders of the Series A 12% Convertible Preferred Stock.
Voting Rights. Except as noted below, the holder of each share of Series B shall be entitled to the number of votes equal to the number of shares of Common Stock into which such share of Series B would be convertible, and shall otherwise have voting rights and powers equal to the voting rights and powers of the Common Stock. With respect to the election of directors, the holders of the Series B shall vote together as a separate class to elect two (2) members of the Board of Directors (the “Series B Directors”), and the Company shall take all reasonably necessary or desirable actions within its control (including, without limitation, calling special meetings of the Board of Directors, nominating such persons designated by the holders of the Series B as directors on the applicable proxy statements and recommending their election) to permit the holders of the Series B to appoint two additional (2) members of the Board of Directors (the “Series B Nominees”), who shall be subject to election by all shares of voting stock of the Company voting together as a single group, until such time as all authorized shares of Series B have has been issued and sold, after which the number of Series B Nominees shall be three (3), and shall remain three (3) until there are no longer any shares of Series B outstanding. The holders of Series B shall vote together with the holders of Common Stock and other voting capital stock of the Company to elect all other members of the Board of Directors.
Other Restrictions. So long as any shares of the Series B remain outstanding, the Company may not, without the approval of the holders of a majority of the shares of Series B outstanding, among other things, (i) change the size of the Company’s Board of Directors; (ii) amend or repeal the Company’s Articles of Incorporation or Bylaws or file any articles of amendment designating the preferences, limitations and relative rights of any series of preferred stock; (iii) create or increase the authorized amount of any additional class or series of shares of stock that is equal to or senior to Series B; (iv) increase or decrease the authorized number of shares of the Series B; (v) purchase, redeem or otherwise acquire for value any shares of any class of capital stock; (vi) merge or consolidate the Company into or with any other corporation or sell, assign, lease, pledge, encumber or otherwise dispose of all or substantially all of the Company’s assets or those of any subsidiary; (vii) voluntarily or involuntarily liquidate, dissolve or wind up the Company or the Company’s business; (viii) pay or declare dividends on any capital stock other than the Preferred Stock, unless the Series B share ratably in such dividend and all accrued dividends payable with respect to the Series B have been paid prior to the payment or declaration of such dividend; (ix) acquire an equitable interest in, or the assets or business of any other entity in any form of transaction; (x) create or commit us to enter into a joint venture, licensing agreement or exclusive marketing or other distribution agreement with respect to the Company’s products, other than in the ordinary course of business; (xi) permit the Company or any subsidiary to sell or issue any security of such subsidiary to any person or entity other than the Company; (xii) enter into, create, incur, assume or guarantee any indebtedness for borrowed money of any kind (other than indebtedness existing on the initial closing date and approved by Series B shareholders); (xiii) enter into, create, incur or assume any liens of any kind (other than certain permitted liens); (xiv) issue any common stock equivalents; (xv) increase the number of shares of the Company’s common stock that may be issued pursuant to options, warrants or rights to employees, directors, officers, consultants or advisors above 250,000.
Warrants. Each Class A-1 warrant, Class A-2 warrant and Class B warrant is exercisable at $3.00 per share of common stock at any time on or after the date of issuance until the fifth anniversary of the respective issue date. The Company may, upon 30 days notice and so long as an effective registration
F-16
Table of Contents
Index to Financial Statements
statement regarding the underlying shares of common stock is in effect, issue a termination notice with respect to (i) each Class A-1 warrant on any trading day on which the market value of the common stock for each of the 15 previous trading days exceeded $7.50 per share and (ii) each Class A-2 warrant on any trading day on which the market value of the common stock for each of the 15 previous trading days exceeded $10.50 per share. All Class A-1 warrants were exercised for cash proceeds of $3,000,000 in 2011 and 500,000 of the Class A-2 warrants were exercised for cash proceeds of $1,500,000 in 2013. Subsequently, in January 2014, the remaining 500,000 Class A-2 warrants were exercised for cash proceeds of $1,500,000.
The fair value of the warrants issued in connection with the Series B-1 was $1,296,000 at the date of issuance based on the following assumptions: an expected life of 5 years, volatility of 118%, risk free interest rate of 1.79% and zero dividends. The Company allocated the gross proceeds based on the relative fair value of the Series B-1 and the related warrants, resulting in $1,105,000 of the proceeds being allocated to additional paid-in capital. The Company analyzed the Series B-1, post-allocation of the gross proceeds, and determined that there was no beneficial conversion feature at the date of issuance. The issuance costs of the Series B-1 and the amounts allocated to warrants were recorded as a reduction to the carrying value of the Series B-1 when issued, and are accreted to the redemption value of the Series B-1 through the earliest redemption date. Due to the redemption feature, the Company has presented the Series B-1 outside of permanent equity, in the mezzanine of the consolidated balance sheet at December 31, 2014 and 2013.
The fair value of the warrants issued during the year ended December 31, 2010 in connection with the Series B-2 was $4,148,000 at the dates of issuance based on the following assumptions: an expected life of 5 years, volatility of 126% to 129%, risk free interest rates of 2.27% to 2.43% and zero dividends. The fair value of the warrants issued during the year ended December 31, 2009 in connection with the Series B-2 was $5,333,000 at the dates of issuance based on the following assumptions: an expected life of 5 years, volatility of 124% to 127%, risk free interest rates of 1.98% to 2.70% and zero dividends. The Company allocated the gross proceeds based on the relative fair value of the Series B-2 and the related warrants, resulting in $1,028,000 and $1,732,000 of the proceeds being allocated to additional paid-in capital for the years ended December 31, 2010 and 2009, respectively. The issuance costs of the Series B-2 and the amounts allocated to warrants were recorded as a reduction to the carrying value of the Series B-2 when issued, and are accreted to the redemption value of the Series B-2 through the earliest redemption dates. Due to the redemption feature, the Company has presented the Series B-2 outside of permanent equity, in the mezzanine of the consolidated balance sheet at December 31, 2014 and 2013.
The Company analyzed the Series B-2, post-allocation of the gross proceeds, and determined that there was a beneficial conversion feature at the dates of issuance. Because the closing price of the common stock on the closing date was greater than the effective conversion price, $388,000 and $628,000 of the proceeds (limited to the allocation of the proceeds) during the years ended December 31, 2010 and 2009, respectively, were allocated to an embedded beneficial conversion feature of the Series B-2. The amount allocated to the beneficial conversion feature was recorded as a discount to the Series B-2 is being accreted, with such accretion being charged through the earliest redemption dates.
Series C 6% Super Dividend Convertible Preferred Stock
On December 29, 2010, the Company designated and authorized the sale and issuance of up to 1,000 shares of Series C Super Dividend Convertible Preferred Stock (“Series C”) with a par value of $0.01 and a stated value equal to $10,000 (the “Stated Value”).
On December 30, 2010, the Company sold and issued 212 shares of Series C at a price of $10,000 per share for gross proceeds of $2,120,000. The Company incurred $47,000 of cash transaction costs resulting in net cash proceeds of $2,073,000. In addition, the Company issued 500 warrants exercisable at $7.20 to a
F-17
Table of Contents
Index to Financial Statements
placement agent which had a de minimis value. Additionally, in January 2011, the Company sold and issued 13 shares of Series C at a price of $10,000 per share for gross proceeds of $130,000.
The terms of the Series C are as follows:
Conversion Rights. Each holder of Series C may convert all, but not less than all, of his Series C shares plus accrued and unpaid dividends into Common Stock at the price of $6.00 per share of Common Stock (“Conversion Price”), such that approximately 1,667 shares of Common Stock will be issued per each converted share of Series C (accrued and unpaid dividends will be issued as additional shares). At December 31, 2014, the 176 outstanding shares of Series C were convertible into a total of approximately 293,340 shares of Common Stock.
Subject to the continuing obligation to pay post conversion dividends, the Company may convert all, but not less than all, of the Series C (plus all accrued and unpaid dividends) into Common Stock, at the Conversion Price, upon such time that the closing price of the Common Stock is no less than $18.00 per share for 15 consecutive trading days.
Dividends. Holders of Series C shall be entitled to receive cumulative non-compounding dividends at the rate per share of Series C equal to the greater of (i) 6% per annum of the Stated Value (also defined as the “Floor”) or (ii) 2.5% of net sales until the total dividends paid is equal to the initial investment and 1.25% of net sales thereafter. The maximum amount each Series C shareholder will receive in dividend payments is equal to $100,000 (the “Maximum Payout”). For purposes of this dividend calculation, net sales shall mean gross revenues actually received by the Company, from the sale or licensing of the product DAVANAT® (GM-CT-01), less chargebacks, returns, expenses attributable to product recalls, duties, customs, sales tax, freight, insurance, shipping expenses, allowances and other customary deductions.
The dividend shall be payable in arrears semiannually on March 31 and September 30, beginning with the first such date after the original issue date; provided, however, that all dividends and all other distributions shall cease, and no further dividends or other distributions shall be paid, in respect of each share of Series C from and after such time that the Maximum Payout has been paid in respect of such share of Series C. Such dividends shall be payable at the Company’s option either in cash or in duly authorized, fully paid and non-assessable shares of Common Stock valued at the higher of (i) $3.00 per share or (ii) the average of the Common Stock trading price for the ten (10) consecutive trading days ending on the trading day that is immediately prior to the dividend payment date.
Series C Post Conversion Dividend Right. In the event that any share of Series C is converted into Common Stock before the Maximum Payout is paid in respect of such converted share of Series C, then the holder shall have the right to continue to receive dividends in respect of such converted share of Series C equal to the remaining payout (the “Series C Preferred Stock Post Conversion Dividend Right”) which shall be equal to the Maximum Payout less the cumulative dividends received through the conversion date. One share of Series C Preferred Stock Post Conversion Dividend Right shall be issued for each such converted share of Series C. The holder of each Series C Preferred Stock Post Conversion Dividend Right shall receive the remaining payout on an equal basis and in conjunction with the then outstanding shares of Series C and all the other then outstanding Series C Post Conversion Dividend Rights, in the same manner and subject to the same terms and conditions as applicable to the payment of dividends on each share of Series C, except that for purposes of calculating the dividend the Floor shall not apply. The Series C Preferred Stock Post Conversion Dividend Right shall have no stated value, liquidation preference or right to any dividends or distributions other than the remaining payout. The Series C Preferred Stock Post Conversion Right is subject to redemption in the same manner as outstanding Series C shares.
At the date of issuance, the Series C have an embedded dividend right to continue to receive dividend payments after conversion to common stock (the Series C Post Conversion Dividend Right) which requires
F-18
Table of Contents
Index to Financial Statements
bifurcation. The value of this post conversion dividend right on the date of issuance was determined to be de minimis due to the fact that the payment of a dividend stream other than the 6% dividend and conversion of Series C prior to the Company achieving sales of GM-CT-01 was deemed improbable at that time. Upon a conversion of the Series C, the Company will be required to record a liability and the related expense during the period of conversion.
In July 2011, 5 shares of Series C were converted into 8,334 shares of common stock and 5 Series C Post Conversion Dividend Rights (Dividend Rights) were issued. In 2013, 24 shares of Series C were converted into 40,193 shares of common stock and 24 Dividend Rights were issued. In 2014, 20 shares of Series C were converted into 33,756 shares of common stock and 20 Dividend Rights were issued. Per the terms of the Series C, these Dividend Rights shall continue to participate in dividends, however the Floor shall not apply. At December 31, 2014 and 2013, these Dividend Rights were determined to have a de minimis value, as the payment of a dividend is considered improbable at this time. The Company will continue to evaluate and assess the Series C Post Conversion Dividend Right for each reporting period.
Liquidation Rights. In the event of any liquidation, dissolution or winding up of the Company, either voluntarily or involuntarily, the holders of Series C will receive $10,000 per share plus accrued and unpaid dividends, payable prior and in preference to any distributions to the holders of Common Stock but after and subordinate to the Series A 12% Convertible Preferred Stock (“Series A”), Series B-1 and Series B-2, subject to the Maximum Payout.
Redemption. Upon a sale of the Company, the Company shall redeem all of the then outstanding shares of Series C and Series C Preferred Stock Post Conversion Rights within thirty (30) days after the transaction constituting the sale of the Company is closed and such closing is fully funded. The price to redeem a share of Series C and each redeemed Series C Preferred Stock Post Conversion Redemption Right shall be equal to (i) (A) the applicable return on investment (“ROI”) percentage, multiplied by (B) $10,000, minus (ii) the cumulative dividends received through the redemption date. The redemption price shall be payable at the Company’s option either in cash or in shares of common stock valued at the higher of (i) $3.00 per share or (ii) the average market price for the ten consecutive trading days ending immediately prior to the date of redemption. The ROI Percentage shall mean the percentage that applies as of the redemption date, as follows:
| ROI Percentage |
||
| 200% |
before the second anniversary of the date of issuance; | |
| 250% |
on or after the second anniversary of the date of issuance, but before the third anniversary of the date of issuance; | |
| 300% |
on or after the third anniversary of the date of issuance, but before the fourth anniversary of the date of issuance; | |
| 350% |
on or after the fourth anniversary of the date of issuance, but before the fifth anniversary of the date of issuance; | |
| 400% |
on or after the fifth anniversary of the date of issuance, but before the sixth anniversary of the date of issuance; | |
| 450% |
on or after the sixth anniversary of the date of issuance, but before the seventh anniversary of the date of issuance; | |
| 500% |
on or after the seventh anniversary of the date of issuance, but before the eighth anniversary of the date of issuance; and | |
| 550% |
on or after the eighth anniversary of the date of issuance, but before the ninth anniversary of the date of issuance. | |
Due to the redemption feature, the Company has presented the Series C outside of permanent equity, in the mezzanine of the consolidated balance sheets at December 31, 2014 and 2013. At December 31, 2014, the Series C redemption value was $4,835,000.
Voting Rights. The Series C shares have no voting rights.
F-19
Table of Contents
Index to Financial Statements
| 6. | Warrants and Warrant Liabilities |
Warrants
Warrant activity is summarized as follows:
| Outstanding at December 31, 2012 |
7,424,241 | |||
| Issued |
5,000 | |||
| Cancelled |
(1,344,012 | ) | ||
| Exercised |
(50,000 | ) | ||
|
|
|
|||
| Outstanding at December 31, 2013 |
6,035,229 | |||
|
|
|
|||
| Issued |
20,000 | |||
| Cancelled |
(7,500 | ) | ||
| Exercised |
(576,734 | ) | ||
|
|
|
|||
| Outstanding at December 31, 2014 |
5,470,995 | |||
|
|
|
The following table summarizes information with regard to outstanding warrants issued in connection with equity and debt financings and consultants as of December 31, 2014.
| Issued in Connection With |
Number Issued |
Exercise Price |
Exercisable Date | Expiration Date | ||||||||||||
| February 12, 2009 Series B-1 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
1,200,000 | $ | 3.00 | February 12, 2009 | February 12, 2019 | |||||||||||
| May 13, 2009 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
600,000 | $ | 3.00 | May 13, 2009 | May 13, 2019 | |||||||||||
| June 30, 2009 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
333,333 | $ | 3.00 | June 30, 2009 | June 30, 2019 | |||||||||||
| August 12, 2009 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
200,000 | $ | 3.00 | August 12, 2009 | August 12, 2019 | |||||||||||
| September 30, 2009 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
216,666 | $ | 3.00 | September 30, 2009 | September 30, 2019 | |||||||||||
| November 4, 2009 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
206,666 | $ | 3.00 | November 4, 2009 | November 4, 2019 | |||||||||||
| December 8, 2009 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
216,667 | $ | 3.00 | December 8, 2009 | December 8, 2019 | |||||||||||
| January 29, 2010 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
216,667 | $ | 3.00 | January 29, 2010 | January 29, 2020 | |||||||||||
| March 8, 2010 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
223,334 | $ | 3.00 | March 8, 2010 | March 8, 2020 | |||||||||||
| April 30, 2010 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
206,667 | $ | 3.00 | April 30, 2010 | April 30, 2020 | |||||||||||
| May 10, 2010 Series B-2 Transaction |
||||||||||||||||
| $0.50 Investor Warrants—Class B |
380,000 | $ | 3.00 | May 10, 2010 | May 10, 2020 | |||||||||||
| June 15, 2010 Consultant Warrants |
100,000 | $ | 4.26 | June 15, 2010 | June 15, 2015 | |||||||||||
| December 9, 2010 Consultant Warrants |
33,334 | $ | 3.90 | December 9, 2010 | December 9, 2015 | |||||||||||
| December 30, 2010 Placement Agent Warrants |
500 | $ | 7.20 | December 30, 2010 | December 30, 2015 | |||||||||||
| March 28, 2012 Offering Warrants |
1,317,161 | $ | 5.63 | March 28, 2012 | March 28, 2017 | |||||||||||
| October 30, 2014 Consultant Warrants |
20,000 | $ | 5.45 | October 30, 2014 | October 30, 2017 | |||||||||||
|
|
|
|||||||||||||||
| Total outstanding warrants |
5,470,995 | |||||||||||||||
|
|
|
|||||||||||||||
Consultant Warrants
In October 2014, the Company granted warrants to a consultant for the purchase of 20,000 shares of common stock at an exercise price of $5.45 per share. The warrants were valued at $76,000 on issuance based on the following assumptions: an expected life of 3 years, volatility of 117%, risk free interest rate of 0.91% and zero dividends. The warrants vested immediately and the company recognized an expense of
F-20
Table of Contents
Index to Financial Statements
$76000 related to these warrants during the year ended December 31, 2014. These warrants are outstanding at December 31, 2014.
Offering Warrants
On March 28, 2012, the Company sold and issued 1,333,361 Units (2,666,722 shares of common stock and related $5.63 warrants to purchase 1,333,361 shares of common stock) for gross proceeds of $12.0 million (net cash proceeds of $10,403,000 after the underwriting discount and offering costs). The warrants were valued at $4,445,000 as of the issuance date of March 28, 2012, using the closing price of $4.20, a life of 5 years, a volatility of 119% and a risk free interest rate of 1.05%. Based upon the Company’s analysis of the criteria contained in ASC Topic 815-40, “Derivatives and Hedging — Contracts in Entity’s Own Equity” the Company has determined that warrants issued in connection with this financing transaction were not derivative liabilities and therefore, were recorded as additional paid-in capital. At December 31, 2014, 1,317,161 of these warrants remain outstanding.
Warrants Modification
On May 6, 2013, the Company modified the terms of the Class A-2 and Class B warrants that were originally issued to the 10X Fund with the Series B Preferred Stock offering. The Class B warrants were modified to allow for the cashless exercise of all 4,000,000 outstanding Class B warrants. Previously, only half of the Class B warrants allowed for cashless exercise. The Class A-2 warrants for the purchase of 1,000,000 shares of common and all of the Class B warrants had their exercisable life extended by an additional five years. In exchange for these modifications, the 10X Fund agreed to a future amendment of the Company’s Series B certificate of designation to remove the redemption provision such that the Series B Preferred Stock will no longer be redeemable, if and when the Company will no longer be required to issue Dr. Platt a promissory note as may currently be required under the separation agreement (see Note 11). Should the Company amend their Series B certificate of designation in the future as described above, the Company will be required at that time to evaluate whether such amendment is to be accounted for as a modification or an extinguishment of the Company’s Series B Preferred Stock. The Company has accounted for the modified terms of the Class A-2 and Class B warrants pursuant to ASC 718, Stock Compensation, whereby the Company has recognized a charge for the change in fair value of the warrants immediately before and immediately after the modification. In the second quarter of 2013, the Company recognized a one-time charge of $8,763,000 related to the extension of the 5,000,000 warrants. The following assumptions were used to value the extension of the warrants immediately before and immediately after the modification: a) immediately before the modification — an expected life range of 0.77 to 2.01 years, volatility range of 77% to 96%, risk free interest rate range of 0.11% to 0.22% and zero dividends and; b) immediately following the modification — an expected life range of 5.78 to 7.02 years, volatility range of 113% to 122%, risk free interest rate range of 0.74% to 1.19% and zero dividends.
| 7. | Stock-Based Compensation |
Summary of Stock-Based Compensation Plans
At December 31, 2014, the Company had three stock-based compensation plans where the Company’s common stock has been made available for equity-based incentive grants as part of the Company’s compensation programs (the “Incentive Plans”) as follows:
2001 Stock Incentive Plan. In October 2001, the Company’s Board of Directors adopted the Pro-Pharmaceuticals, Inc. 2001 Stock Incentive Plan (the “Incentive Plan”), which permits awards of incentive and nonqualified stock options and other forms of incentive compensation to employees and non-employees such as directors and consultants. Originally, there were 833,334 shares of common stock for issuance upon exercise of grants made under the Incentive Plan. Options granted under the Incentive Plan vest either
F-21
Table of Contents
Index to Financial Statements
immediately or over a period of up to three years, and expire 3 years to 10 years from the grant date. At December 31, 2014, there were no shares were available for future grant under the Incentive Plan as the terms of the Incentive Plan did not allow for grants to be made after 10 years; however, there were 166,167 options outstanding under the Incentive Plan.
2003 Non-Employee Director Stock Option Plan. In 2003, the stockholders approved the Pro-Pharmaceuticals, Inc. 2003 Non-Employee Director Stock Option Plan (the “Director Plan”), which permits awards of stock options to non-employee directors. The stockholders originally reserved 166,667 shares of common stock for issuance upon exercise of grants made under the Director Plan. At December 31, 2014, there were no shares were available for future grant under the Director Plan as the terms of the Director Plan did not allow for grants to be made after 10 years, and there were no options outstanding under the Director Plan.
2009 Incentive Compensation Plan. In February 2009, the Company adopted the 2009 Incentive Compensation Plan (the “2009 Plan”) which originally provided for the issuance of up to 3,333,334, which was subsequently increased to 4,733,334 in May 2014, shares of the Company’s common stock in the form of options, stock appreciation rights, restricted stock and other stock-based awards to employees, officers, directors, consultants and other eligible persons. At December 31, 2014, 1,851,114 shares were available for future grant under the 2009 Plan.
In addition, the Company has awarded 1,477,379 non-plan stock option grants to employees and non-employees. These non-plan grants have vesting periods and expiration dates similar to those options granted under the Incentive Plans. At December 31, 2014, 1,416,669 non-plan grants were outstanding.
Stock-Based Compensation
Following is the stock-based compensation expense related to common stock options, restricted common stock and common stock warrants:
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Research and development |
$ | 1,302 | $ | 991 | $ | 922 | ||||||
| General and administrative |
2,768 | 2,798 | 1,870 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation expense |
$ | 4,070 | $ | 3,789 | $ | 2,792 | ||||||
|
|
|
|
|
|
|
|||||||
The fair value of the options granted is determined using the Black-Scholes option-pricing model. The following weighted average assumptions were used:
| 2014 | 2013 | 2012 | ||||||||||
| Risk-free interest rate |
1.58 | % | 1.17 | % | 0.90 | % | ||||||
| Expected life of the options |
6 years | 5.29 years | 5.81 years | |||||||||
| Expected volatility of the underlying stock |
114 | % | 115 | % | 116 | % | ||||||
| Expected dividend rate |
0 | % | 0 | % | 0 | % | ||||||
As noted above, the fair value of stock options is determined by using the Black-Scholes option pricing model. For all options granted since January 1, 2006 the Company has generally used option terms of between 5 to 10 years, generally with 5 to 6 years representing the estimated life of options granted to employees. The volatility of the common stock is estimated using historical volatility over a period equal to the expected life at the date of grant. The risk-free interest rate used in the Black-Scholes option pricing model is determined by reference to historical U.S. Treasury constant maturity rates with terms equal to the expected terms of the awards. An expected dividend yield of zero is used in the option valuation model, because the Company does not expect to pay any cash dividends in the foreseeable future. At December 31, 2014, the Company does not anticipate any option awards will be forfeited in the calculation of compensation expense due to the limited number of employees that receive stock option grants and the Company’s historical employee turnover.
F-22
Table of Contents
Index to Financial Statements
The following table summarizes the stock option activity in the stock based compensation plans:
| Number of Shares |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (in years) |
Aggregate Intrinsic Value (in thousands) |
|||||||||||||
| Outstanding, December 31, 2011 |
3,091,474 | $ | 6.83 | |||||||||||||
| Granted |
800,000 | 2.13 | ||||||||||||||
| Forfeited/Cancelled |
(299,683 | ) | 8.93 | |||||||||||||
| Exercised |
(51,830 | ) | 2.31 | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Outstanding, December 31, 2012 |
3,539,961 | $ | 5.66 | |||||||||||||
| Granted |
425,426 | 3.89 | ||||||||||||||
| Forfeited/Cancelled |
(403,674 | ) | 14.19 | |||||||||||||
| Exercised |
(213,008 | ) | 2.18 | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Outstanding, December 31, 2013 |
3,348,705 | $ | 4.70 | |||||||||||||
| Granted |
354,823 | 12.72 | ||||||||||||||
| Forfeited/Cancelled |
(124,466 | ) | 3.80 | |||||||||||||
| Exercised |
(246,445 | ) | 1.97 | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Outstanding, December 31, 2014 |
3,332,617 | $ | 5.79 | 6.51 | $ | 1,383 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Exercisable, December 31, 2014 |
2,444,477 | $ | 5.24 | 6.22 | $ | 1,272 | ||||||||||
The aggregate intrinsic value in the table above represents the total pre-tax amount, net of exercise price, which would have been received by option holders if all option holders had exercised all options with an exercise price lower than the market price on December 31, 2014, based on the closing price of the Company’s common stock of $3.47 on that date.
The weighted-average grant-date fair values of options granted during 2014, 2013 and 2012 were $10.75, $3.17 and $1.77, respectively. As of December 31, 2014 and December 31, 2013, there were unvested options to purchase 888,140 and 1,256,735 shares of common stock, respectively. Total expected unrecognized compensation cost related to such unvested options is $4,308,000 at December 31, 2014, which is expected to be recognized over a weighted-average period of 1.61 years.
During the years ended December 31, 2014, 2013 and 2012, the Company issued shares totaling 246,445, 213,008 and 51,830, respectively, upon the exercise of options valued at $411,000 and $378,000, respectively. During the years ended December 31, 2014, 2013 and 2012, the Company received $394,000, $271,000 and 0, respectively, for the exercise of stock options. During 2014, 2013 and 2012, 35,734, 173,669 and 90,253 options were exercised on a cashless basis resulting in the issuance of 26,109, 81,591 and 51,830 shares, respectively. The intrinsic value of options exercised during the years ended December 31, 2014, 2013 and 2012 was $2,677,000, $1,498,000 and 162,000, respectively.
During the years ended December 31, 2014, 2013 and 2012, 676,335, 614,041 and 595,420 options became vested, respectively. The total grant date fair value of options vested during the years ended December 31, 2014, 2013 and 2012 was $3,711,000, $2,406,000 and $2,447,000 respectively.
F-23
Table of Contents
Index to Financial Statements
The following table summarizes additional information regarding outstanding and exercisable options under our stock based compensation plans at December 31, 2014:
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Exercise Price (Range) |
Number of Shares |
Weighted Average Remaining Contractual Life |
Weighted Average Exercise Price |
Number of Shares |
Weighted Average Exercise Price |
|||||||||||||||
| (in years) | ||||||||||||||||||||
| $1.80 – 1.83 |
276,669 | 2.4 | $ | 1.80 | 276,669 | $ | 1.80 | |||||||||||||
| $2.08 – 2.88 |
761,667 | 6.7 | 2.26 | 669,917 | 2.26 | |||||||||||||||
| $3.59 – 4.41 |
354,954 | 8.4 | 4.08 | 213,242 | 3.92 | |||||||||||||||
| $5.00 – 7.56 |
1,568,382 | 6.2 | 6.94 | 1,083,746 | 6.93 | |||||||||||||||
| $8.10 – 11.40 |
44,445 | 6.4 | 8.10 | 44,445 | 8.10 | |||||||||||||||
| $13.38 |
326,500 | 9.1 | 13.38 | 156,458 | 13.38 | |||||||||||||||
|
|
|
|
|
|||||||||||||||||
| 3,332,617 | 6.5 | $ | 5.79 | 2,444,477 | $ | 5.24 | ||||||||||||||
|
|
|
|
|
|||||||||||||||||
Other Stock Based Compensation Transactions
In September 2013, the Company modified certain vested stock options held by a former member of the Company’s board of directors. The individual left the board on May 23, 2013. The modification extended the contractual period of exercise of 103,158 stock options until the end of their original terms instead of such options expiring 3 months after service on the board ended. As a result, the Company recorded a one-time, non-cash charge of $930,000 in general and administrative expenses related to the modification in for the year ended December 31, 2013.
In June 2013, the Company issued 25,000 options to a consultant for consulting services, which vested in August 2013. The options are exercisable at $3.97 per share. These options were valued using the Black-Scholes option-pricing model based on a grant date fair value of the Company’s common stock ranging from $3.97 per share upon grant to $7.25 per share at completion of vesting. The Company recorded a $173,000 charge to stock compensation expense over the vesting period of the options.
| 8. | Loss Per Share |
Basic net loss per common share is computed by dividing the net loss available to common stockholders by the weighted average number of common shares outstanding during the period. Diluted net loss per common share is computed by dividing the net loss available to common stockholders by the weighted average number of common shares and other potential common shares then outstanding. Potential common shares consist of common shares issuable upon the assumed exercise of in-the-money stock options and warrants and potential common shares related to the conversion of the preferred stock. The computation of diluted net loss per share does not assume the issuance of common shares that have an anti-dilutive effect on net loss per share.
F-24
Table of Contents
Index to Financial Statements
| Year Ended December 31, | ||||||||||||
| (in thousands, except share and per share amounts) |
||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Net loss |
$ | (15,788 | ) | $ | (12,088 | ) | $ | (9,675 | ) | |||
| Preferred stock dividends |
(943 | ) | (867 | ) | (976 | ) | ||||||
| Preferred stock accretion |
(229 | ) | (229 | ) | (230 | ) | ||||||
| Warrant modification |
— | (8,763 | ) | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss applicable to common stockholders |
$ | (16,960 | ) | $ | (21,947 | ) | $ | (10,881 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (0.78 | ) | $ | (1.30 | ) | $ | (0.72 | ) | |||
| Shares used in computing basic and diluted net loss per share |
21,849 | 16,874 | 15,131 | |||||||||
Dilutive shares which could exist pursuant to the exercise of outstanding stock instruments and which were not included in the calculation because their affect would have been anti-dilutive are as follows:
| Year Ended December 31, |
||||||||||||
| 2014 (Shares) |
2013 (Shares) |
2012 (Shares) |
||||||||||
| Warrants to purchase shares of common stock |
5,470,995 | 6,035,229 | 7,424,241 | |||||||||
| Options to purchase shares of common stock |
3,332,617 | 3,348,705 | 3,539,961 | |||||||||
| Shares of common stock issuable upon conversion preferred stock |
2,527,103 | 2,568,771 | 2,627,110 | |||||||||
|
|
|
|
|
|
|
|||||||
| 11,330,715 | 11,952,705 | 13,591,312 | ||||||||||
|
|
|
|
|
|
|
|||||||
| 9. | Commitments and Contingencies |
Lease Commitments
In September 2012, the Company entered into an operating lease for office space in Norcross, GA for a term of twenty-six months, beginning on October 1, 2012 and ending November 30, 2014 at a rate of approximately $3,000 per month. In June 2014, the Company signed an amendment to the lease extending the term through November 30, 2017 with a base monthly rental of approximately $3,300 through the extended term. The original lease provided for free rent for the first two months of the lease and required a security deposit of $6,000. In addition to base rental payments included in the contractual obligations table above, the Company is responsible for our pro-rata share of the operating expenses for the building.
In October 2012, the Company entered into an operating lease for office space collocated with lab space for research and development activities. The lease is for a period of one year, beginning on October 1, 2012, for a rate of $15,000 for the term, payable in equal monthly increments. This lease was continued on a month to month basis from October 1, 2013.
Rent expense under the above operating leases was $44,000 and $39,000 for the years ended December 31, 2014 and 2013, respectively.
Future minimum payments under this lease as of December 31, 2014 are as follows (in thousands):
| Year ended December 31, |
||||
| 2015 |
$ | 38 | ||
| 2016 |
40 | |||
| 2017 |
41 | |||
|
|
|
|||
| Total |
$ | 119 | ||
|
|
|
|||
F-25
Table of Contents
Index to Financial Statements
Separation Agreement
In February 2009, the Company entered into a Separation Agreement in connection with the resignation of David Platt, Ph.D., the Company’s former Chief Executive Officer and Chairman of the Board of Directors. The Separation Agreement provides for the deferral of a $1.0 million separation payment due to Dr. Platt upon the earlier occurrence of any of the following milestone events: (i) the approval by the Food and Drug Administration for a new drug application (“NDA”) for any drug candidate or drug delivery candidate based on the Company’s GM-CT-01 technology (whether or not such technology is patented), in which case Dr. Platt is also entitled to a fully vested 10-year cashless-exercise stock option to purchase at least 83,334 shares of common stock at an exercise price not less than the fair market value of the common stock determined as of the date of grant; (ii) consummation of a transaction with a pharmaceutical company expected to result in at least $10.0 million of equity investment or $50 million of royalty revenue to the Company, in which case Dr. Platt is also entitled to stock options on the same terms to purchase at least 50,000 shares of common stock; or (iii) the renewed listing of the Company’s securities on a national securities exchange and the achievement of a market capitalization of $100 million. Payment upon the events (i) and (iii) may be deferred up to six months, and if the Company has insufficient cash at the time of any of such events, it may issue Dr. Platt a secured promissory note for such amount. If the Company files a voluntary or involuntary petition for bankruptcy, whether or not a milestone event has occurred, such event shall trigger the obligation to pay the $1.0 million with the result that Dr. Platt may assert a claim for such obligation against the bankruptcy estate. During 2011, when it became probable that the Company could be relisted on a national securities exchange and eventually reach a market capitalization of $100 million, the Company recognized the $1.0 million severance payment due to Dr. Platt which was included in accrued expenses at December 31, 2013.
On October 12, 2012, Dr. Platt commenced a lawsuit under the Massachusetts Wage Act against Dr. Traber and Mr. McGauley who in their capacities as the Company’s Chief Executive Officer and the Company’s former Chief Financial Officer, respectively, can be held individually liable under the Wage Act for non-payment of wages. The lawsuit is based on the facts and issues raised in the arbitration regarding the payment of the $1.0 million separation payment under the Separation Agreement, and other unspecified “wages”. The statute provides that a successful claimant may be entitled to multiple damages, interest and attorney’s fees. On April 29, 2013, the Superior Court allowed Dr. Traber’s and Mr. McGauley’s motion to dismiss. On May 28, 2013, Dr. Platt filed a Notice of Appeal to appeal the Superior Court’s order allowing the defendants’ motion to dismiss. On April 14, 2014, the Appeals Court denied Dr. Platt’s appeal of the dismissal in full.
On March 29, 2013, the Company instituted arbitration before the American Arbitration Association, seeking to rescind or reform the Separation Agreement discussed above. The Company claimed that Dr. Platt fraudulently induced the Company to enter into the Separation Agreement, breached his fiduciary duty to the Company, and was unduly enriched from his conduct. Along with removal of the $1.0 million milestone payment under the Separation Agreement, the Company sought repayment of all separation benefits paid to Dr. Platt to date.
On August 1, 2013, the market capitalization of the Company’s common stock exceeded $100 million and the Company received a letter dated October 1, 2013, demanding payment of the $1 million. As described in the preceding paragraph, the Company had previously instituted an arbitration proceeding against Dr. Platt seeking to rescind the Separation Agreement, including the milestone payment provision, and the Company delayed payment pending the outcome of this arbitration. In June 2014, the arbitrator issued a judgment in favor of Dr. Platt. In July 2014, the Company paid the $1 million severance obligation.
Shareholder Class Actions and Derivative Lawsuits
Between July 30, 2014, and August 6, 2014, three putative class action complaints were filed in the United States District Court for the District of Nevada (the “Nevada District Court”) against the Company and certain of its officers and directors on behalf of all persons who purchased or otherwise acquired the Company’s stock
F-26
Table of Contents
Index to Financial Statements
between January 6, 2014 and July 28, 2014. The complaints allege that the defendants made false or misleading statements in certain press releases and other public statements in violation of the federal securities laws and seek class certification, unspecified monetary damages, costs, and attorneys’ fees. The Company disputes the allegations in the complaints and intends to vigorously defend against the claims. On August 22, 2014, the Nevada District Court entered an order consolidating the three cases, relieving the defendants of any obligation to respond to the complaints currently on file, and providing that defendants may respond to a consolidated amended complaint after it is filed by a lead plaintiff(s) to be appointed pursuant to the Private Securities Litigation Reform Act of 1995. On January 5, 2015, the Nevada District Court granted Defendants’ motion to transfer the consolidated putative securities class action to the United States District Court for the Northern District of Georgia. The court has not yet appointed a lead plaintiff or plaintiffs, and no consolidated amended complaint has been filed.
On August 1 and 25, 2014, persons claiming to be Galectin shareholders filed putative shareholder derivative complaints in the Nevada District Court, seeking recovery on behalf of the Company against certain of the Company’s directors and officers. On September 10, 2014, the Nevada District Court entered an order consolidating the two cases, relieving the defendants of any obligation to respond to the initial complaints, and providing that defendants may respond to a consolidated amended complaint to be filed by the plaintiffs. On January 5, 2015, the Nevada District Court granted Defendants’ motion to transfer the consolidated putative derivative litigation to the United States District Court for the Northern District of Georgia. The plaintiffs filed a consolidated amended complaint on February 27, 2015. The consolidated amended complaint alleges that the defendants breached their fiduciary duties to the Company’s shareholders by causing or permitting the Company to make allegedly false and misleading public statements concerning the Company’s financial and business prospects. The consolidated amended complaint also alleges that the defendants violated the federal securities laws by allegedly making false or misleading statements of material fact in the Company’s proxy filings, committed “waste” of corporate assets, were unjustly enriched, aided and abetted breaches of fiduciary duties, and that certain defendants breached their fiduciary duties through allegedly improper sales of Galectin stock. The complaints seek unspecified monetary damages on behalf of the Company, corporate governance reforms, disgorgement of profits, benefits and compensation by the defendants, costs, and attorneys’ and experts’ fees. Defendants’ response to the consolidated amended complaint is currently due to be filed on March 30, 2015.
On August 29, 2014, another alleged Galectin shareholder filed a putative shareholder derivative complaint in state court in Las Vegas, Nevada, seeking recovery on behalf of the Company against the same directors and officers who are named as defendants in the derivative litigation pending in the United States District Court for the District of Nevada. The state court derivative plaintiff filed an amended complaint on December 1, 2014, which alleges claims for breach of fiduciary duties, unjust enrichment, and waste of corporate assets, based on allegations that are substantially similar to those in the derivative complaints now pending in the United States District Court for the Northern District of Georgia, and seeks unspecified monetary damages on behalf of the Company, corporate governance reforms, disgorgement of profits, benefits and compensation by the defendants, costs, and attorneys’ and experts’ fees. Defendant filed motion to dismiss the amended complaint on February 26, 2015.
Estimating an amount or range of possible losses resulting from litigation proceedings is inherently difficult and requires an extensive degree of judgment, particularly where the matters involve indeterminate claims for monetary damages, are in the early stages of the proceedings, and are subject to appeal. In addition, because most legal proceedings are resolved over extended periods of time, potential losses are subject to change due to, among other things, new developments, changes in legal strategy, the outcome of intermediate procedural and substantive rulings and other parties’ settlement posture and their evaluation of the strength or weakness of their case against us. For these reasons, we are currently unable to predict the ultimate timing or outcome of, or reasonably estimate the possible losses or a range of possible losses resulting from, the matters described above. Based on information currently available, the Company does not believe that any reasonably possible losses arising from currently pending legal matters will be material to the Company’s results of operations or financial
F-27
Table of Contents
Index to Financial Statements
condition. However, in light of the inherent uncertainties involved in such matters, an adverse outcome in one or more of these matters could materially and adversely affect the Company’s financial condition, results of operations or cash flows in any particular reporting period.
Other Legal Proceedings
The Company records accruals for such contingencies to the extent that the Company concludes that their occurrence is probable and the related damages are estimable. There are no other pending legal proceedings except as noted above.
| 10. | Galectin Sciences LLC |
In January 2014, we created Galectin Sciences, LLC (the “LLC” or “Investee”), a collaborative joint venture co-owned by SBH Sciences, Inc. (“SBH”), to research and develop small organic molecule inhibitors of galectin-3 for oral administration. The LLC was initially capitalized with a $400,000 cash investment to fund future research and development activities, which was provided by the Company, and specific in-process research and development (“IPR&D”) contributed by SBH. The estimated fair value of the IPR&D contributed by SBH, on the date of contribution, was $400,000. Initially, the Company and SBH have a 50% equity ownership interest in the LLC, with neither party having control over the LLC. Accordingly from inception through the fourth quarter of 2014, the Company accounted for its investment in the LLC using the equity method of accounting. Under the equity method of accounting, the Company’s investment was initially recorded at cost with subsequent adjustments to the carrying value to recognize additional investments in or distributions from the Investee, as wells the Company’s share of the Investee’s earnings, losses and/or changes in capital. The estimated fair value of the IPR&D contributed to the LLC was immediately expensed upon contribution as there was no alternative future use available at the point of contribution. The operating agreement provides that if either party does not desire to contribute its equal share of funding required after the initial capitalization, then the other party, providing all of the funding, will have its ownership share increased in proportion to the total amount contributed from inception. In the fourth quarter of 2014, after the LLC had expended the $400,000 in cash, SBH decided not to contribute its share of the funding required. As a result, the Company contributed the $73,000 needed for the fourth quarter of 2014 expenses of the LLC. As a result, the Company’s ownership percentage in the LLC is 54.2% at December 31, 2014. The Company accounts for the interest in the LLC as a consolidated, less than wholly owned subsidiary. The Company’s portion of the LLC’s net loss for 2014, prior to the change in accounting discussed previously, was $400,000, which includes the Company’s proportionate share of the non-cash charge associated with the contributed IPR&D of $200,000.
| 11. | Income Taxes |
The components of the net deferred tax assets are as follows at December 31:
| 2014 | 2013 | |||||||
| (in thousands) | ||||||||
| Operating loss carryforwards |
$ | 30,578 | $ | 25,881 | ||||
| Tax credit carryforwards |
645 | 400 | ||||||
| Other temporary differences |
4,689 | 4,302 | ||||||
|
|
|
|
|
|||||
| 35,912 | 30,583 | |||||||
| Less valuation allowance |
(35,912 | ) | (30,583 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax asset |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
F-28
Table of Contents
Index to Financial Statements
The primary factors affecting the Company’s income tax rates were as follows:
| 2014 | 2013 | 2012 | ||||||||||
| Tax benefit at U.S. statutory rates |
(34 | %) | (34 | %) | (34 | %) | ||||||
| State tax benefit |
(5.3 | %) | (5.3 | %) | (5.3 | %) | ||||||
| Credit |
0 | % | 0 | % | (0.2 | %) | ||||||
| Permanent differences |
5.4 | % | 0.9 | % | 1.4 | % | ||||||
| Expiring state NOL’s |
1.4 | % | 1.8 | % | 3.3 | % | ||||||
| Changes in valuation allowance |
32.5 | % | 36.6 | % | 34.8 | % | ||||||
|
|
|
|
|
|
|
|||||||
| 0 | %% | 0 | % | 0 | % | |||||||
|
|
|
|
|
|
|
|||||||
As of December 31, 2014, the Company has federal and state net operating loss carryforwards totaling $82,150,000 and $42,394,000 respectively, which expire through 2033. The net operating losses include Federal and State excess benefits related to stock options of $707,000 that will be charged to additional paid-in capital when utilized. In addition, the Company has federal and state research and development credits of $468,000 and $176,000, respectively, which expire through 2033. Ownership changes, as defined by Section 382 of the Internal Revenue Code, may have limited the amount of net operating loss carryforwards that can be utilized annually to offset future taxable income. Subsequent ownership changes could further affect the limitation in future years. Because of the Company’s limited operating history and its recorded losses, management has provided, in each of the last two years, a 100% valuation allowance against the Company’s net deferred tax assets.
The Company is subject to taxation in the U.S. and various states. Based on the history of net operating losses all jurisdictions and tax years are open for examination until the operating losses are utilized or the statute of limitations expires. As of December 31, 2014 and 2013, the Company does not have any significant uncertain tax positions.
| 12. | Subsequent Events |
The Company evaluated all events and transactions occurring after December 31, 2014 through the date of which the financial statements were issued and noted no additional items requiring recognition or disclosure.
| 13. | Quarterly financial data (unaudited) |
| 2014 Quarters ended | ||||||||||||||||
| (In thousands except per share data) |
December 31 | September 30 | June 30 | March 31 | ||||||||||||
| Net loss |
$ | (3,731 | ) | $ | (3,518 | ) | $ | (3,429 | ) | $ | (5,110 | ) | ||||
| Net loss applicable to common stockholders |
(3,968 | ) | (3,853 | ) | (3,731 | ) | (5,408 | ) | ||||||||
| Basic and diluted net loss per share |
$ | (0.17 | ) | $ | (0.17 | ) | $ | (0.17 | ) | $ | (0.27 | ) | ||||
| 2013 Quarters ended | ||||||||||||||||
| (In thousands except per share data) |
December 31 | September 30 | June 30 | March 31 | ||||||||||||
| Net loss |
$ | (2,799 | ) | $ | (3,542 | ) | $ | (2,544 | ) | $ | (3,203 | ) | ||||
| Net loss applicable to common stockholders |
(3,111 | ) | (3,723 | ) | (11,641 | ) | (3,472 | ) | ||||||||
| Basic and diluted net loss per share |
$ | (0.14 | ) | $ | (0.22 | ) | $ | (0.72 | ) | $ | (0.22 | ) | ||||
F-29