Attached files
| file | filename |
|---|---|
| EX-31.1 - EXHIBIT 31.1 - SECOND SIGHT MEDICAL PRODUCTS INC | s100869_ex31-1.htm |
| EX-31.2 - EXHIBIT 31.2 - SECOND SIGHT MEDICAL PRODUCTS INC | s100869_ex31-2.htm |
| EX-32.1 - EXHIBIT 32.1 - SECOND SIGHT MEDICAL PRODUCTS INC | s100869_ex32-1.htm |
| EX-21.1 - EXHIBIT 21.1 - SECOND SIGHT MEDICAL PRODUCTS INC | s100869_ex21-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
_____________________________________
FORM 10-K
_____________________________________
(Mark One)
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended December 31, 2014
OR
£ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from ________ to ________
Commission File Number 333-198073
_____________________________________
Second Sight Medical Products,
Inc.
(Exact name of
Registrant as specified in its charter)
_____________________________________
|
California |
02-0692322 |
|
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
12744
San Fernando Road, Building 3, Sylmar, CA
91342
(Address of
principal executive offices, including zip code)
Registrant’s telephone number, including area code: (818) 833-5000
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class |
|
Name of Each Exchange on Which Registered |
|
Common Stock, without par value |
|
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes £ No x
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes £ No x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No £
Indicate by check mark whether the Registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the Registrant was required to submit and post such files). Yes x No £
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer £ |
Accelerated filer £ |
Non-accelerated filer x |
Smaller reporting company £ |
|
(Do not check if a smaller reporting company) |
|||
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes £ No x
As of June 30, 2014, the last business day of the registrant’s most recently completed second quarter, the registrant’s common shares were not publicly traded. The aggregate market value of the common shares held by non-affiliates of the registrant (treating all executive officers and directors of the registrant and holders of 10% or more of the common shares outstanding, for this purpose, as if they may be affiliates of the registrant) was approximately $131,172,000 on December 31, 2014, based on a closing price of $10.26 per common share as reported on the NASDAQ Exchange on such date.
As of March 13, 2015, the number of shares of the Registrant’s common stock outstanding was 35,339,994.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant's Proxy Statement for the 2015 Annual Meeting of Stockholders are incorporated herein by reference in Part III of this Annual Report on Form 10-K to the extent stated herein. Such proxy statement will be filed with the Securities and Exchange Commission within 120 days of the registrant's fiscal year ended December 31, 2014.
SECOND SIGHT MEDICAL PRODUCTS INC.
FORM 10-K
TABLE OF CONTENTS
|
|
|
Page |
|
|
|
|
|
|
|
|
|
Item 1. |
1 |
|
|
Item 1A. |
21 |
|
|
Item 1B. |
37 |
|
|
Item 2. |
37 |
|
|
Item 3. |
37 |
|
|
Item 4. |
37 |
|
|
|
|
|
|
PART II |
||
|
|
|
|
|
Item 5. |
37 |
|
|
Item 6. |
38 |
|
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
39 |
|
Item 7A. |
49 |
|
|
Item 8. |
50 |
|
|
Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
50 |
|
Item 9A. |
50 |
|
|
Item 9B. |
51 |
|
|
|
|
|
|
|
|
|
|
Item 10. |
51 |
|
|
Item 11. |
51 |
|
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters |
51 |
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
51 |
|
Item 14. |
52 |
|
|
|
|
|
|
|
|
|
|
Item 15. |
52 |
|
|
|
|
|
|
53 |
||
i
Overview
Second Sight Medical Products, Inc. is a medical device company that develops, manufactures and markets implantable visual prosthetics to restore some functional vision to blind patients. Our current product, the Argus® II System, treats outer retinal degenerations, such as retinitis pigmentosa, which we refer to as RP in this Form 10-K. RP is a hereditary disease, affecting an estimated 1.5 million people worldwide including about 100,000 people in the United States, that causes a progressive degeneration of the light-sensitive cells of the retina, leading to significant visual impairment and ultimately blindness. The Argus II System is the only retinal prosthesis approved in the United States by the Food and Drug Administration, or FDA, and the first approved retinal prosthesis in the world. By restoring some functional vision in patients who otherwise have total sight loss, the Argus II System can provide benefits which include:
• improving patients’ orientation and mobility, such as locating doors and windows, avoiding obstacles, and seeing the lines of a crosswalk,
• allowing patients to feel more connected with people in their surroundings, such as seeing when someone is approaching or moving away,
• providing patients with enjoyment from being “visual” again, such as locating the moon, tracking groups of players as they move around a field, and watching the moving streams of lights from fireworks, and
• improving patients’ well-being and ability to perform activities of daily living.
The Argus II System provides an artificial form of vision that differs from the vision that normally sighted people have. It does not restore normal vision and it does not slow or reverse the progression of the disease. Results vary among patients. While the majority of patients receive a benefit from the Argus II, some patients report receiving little or no benefit.
Our Company
We were founded in 1998 with the mission to develop, manufacture, and market implantable prosthetic devices that can restore sight to the blind. In 2002, we began a clinical trial of our proof-of-concept device, the Argus I retinal prosthesis, at the University of Southern California. Six human subjects were implanted with the Argus I retinal prosthesis in a study that was designed to demonstrate the feasibility and safety of long-term electrical stimulation of the retina and its ability to restore some functional vision. By 2006, we developed a second generation device, the Argus II Retinal Prosthesis System that, among other attributes, is smaller, has more stimulating electrodes, and is easier to install surgically than the Argus I retinal prosthesis. In that year, we conducted a small pilot study in Mexico, and we utilized data from this pilot study to obtain FDA approval to begin an Investigational Device Exemption (IDE) clinical trial at six hospitals in the US during 2007. In 2008 we expanded the trial to include sites in three European countries. We completed enrollment for this study in August 2009. Based on the long-term results of this study, which demonstrated the benefits of Argus II System, we obtained CE Mark approval in EU in February 2011, and FDA marketing approval, under a Humanitarian Device Exemption, in February 2013. To our knowledge the Argus II System currently is the only retinal prosthesis to be commercialized anywhere in the world and currently is the only such product to obtain FDA marketing approval in the US.
Currently, after more than 15 years of research and development, more than $130 million of investment and over $29 million of direct federal grants received in support of our technology development, we employ over 100 people in the development (engineering and clinical), manufacture, and commercialization of the Argus II System and future products.
Our Markets
Second Sight is the global leader in vision restoration to the blind. We believe that our competitive advantage and ability to maintain market share in the future will be bolstered by the following:
• We have extensive IP, or intellectual property, protection that covers every major aspect of the technology we have developed. We have over 300 granted patents and over 170 patent applications on a worldwide basis. We believe that
1
our IP and our technical approach, which does not rely on light getting to the implant, will result in a device that can deliver cortical stimulation to the brain. Subject to additional research and development, this IP and technology may result in a device that can treat nearly all forms of blindness.
• We have regulatory leadership in that, to our knowledge, we currently possess the only device that is both FDA approved and CE-marked to restore some functional vision to individuals who are or will become blind as a result of RP.
• We continue to achieve meaningful reimbursement levels for the Argus II System in the US and some European countries. We are currently working to expand the number of countries that reimburse us for the Argus II System.
• We plan to offer periodic software upgrades to enhance our customers’ experience, which arise from our strong engineering, research and clinical programs. We plan to offer the next upgrade in late 2016.
• We expect to expand the numbers of eligible blind persons who will benefit from the Argus II System through additional clinical trials, to treat patients blinded age-related macular degeneration (or AMD). Recruitment for this study began in December 2014 and we plan to begin implanting in the study in the first half of 2015.
• We intend to develop a new device, the Orion I visual prosthesis, over approximately the next 24 months that we expect may favorably address almost all other forms of blindness.
During clinical studies the Argus II System generally demonstrated clear and significant improvement in visual function both in the clinic and in patient’s daily lives. Based on these data, the Argus II Retinal Prosthesis System has been approved for marketing in Europe, the US, Canada, Turkey, and at one medical center in Saudi Arabia. We have submitted an application for full regulatory approval in Saudi Arabia. Our current approval is limited to one hospital, and no assurance can be given that we will receive full regulatory approval.
The Argus II System is intended to restore some useful vision to patients who are blind and have lost most or all of their vision due to retinitis pigmentosa (which is the approved US indication for use) or due to outer retinal degeneration (which is the broader CE mark indication for use). While there are several diseases and syndromes that comprise outer retinal degeneration, the two most prominent of these are RP and AMD. We believe that future product development of the Orion I visual prosthesis will expand the market for our products to include nearly all forms of blindness.
Retinitis Pigmentosa (RP)
RP is a group of inherited disorders that affect the retina. The retina is a layer of nerve cells at the back of the eye. RP is a disease that gradually robs relatively young people of their vision over time. Onset of RP is often noted in the teen years or early twenties, typically as night blindness. This is followed by a period of peripheral vision loss, until the patient is left with a tunnel of vision and then no remaining sight. Although there are various genetic causes (over 100) and thus variability in the disease progression, many people with advanced RP have lost all functional vision by their 40s or 50s. The Argus II System works by bypassing rods and cones which are defunct in these patients and sending electrical signals directly to the retina’s remaining healthy cells.
Although there are reported trials for other treatments underway, to our knowledge the Argus II System remains the only approved therapeutic option for end-stage RP in the US, and to our knowledge it is the only treatment option currently commercialized anywhere in the world.
Worldwide, an estimated 1.5 million people suffer from RP,1 which includes about 100,000 in the US.2 Pan-European data is not readily available, but we believe it is reasonable to estimate that the average prevalence throughout Europe is similar to the average prevalence within the US, and so the ratio of populations could be used to estimate the number of Europeans affected as
__________
1 Weleber, R.G. and Gregory-Evans, K. (2001) ‘Retinitis Pigmentosa and allied disorders.’ In Ryan, S.J. (ed.), Retina. Mosby, St. Louis, pp, 362-470.
2 Foundation Fighting Blindness estimates that about 100,000 Americans are affected by RP or similar diseases. (http://www.ffb.ca/documents/File/rp_guide/Guide_to_RP_and_Other_Related_Diseases.pdf)
2
167,000 in the 28 EU countries.3,4 Approximately 25% of people with RP in the US have vision that is 20/200 or worse (legally blind).5 Since the bare light perception or worse vision criterion for the US indication is worse than 20/200, we believe that the subset of patients that can be treated by the Argus II System is less than 25,000 in the US. In Europe, the indicated vision loss is severe to profound which, while better than bare light perception, remains somewhat worse than 20/200. We estimate that the subset of RP patients that can be treated in Europe to be somewhat smaller than 42,000. Worldwide, we estimate that 375,000 people are legally blind due to RP, and that a portion of these would be candidates for the Argus II System.
Age Related Macular Degeneration (AMD)
AMD is a relatively common eye condition and the leading cause of vision loss among people age 65 and older.6 The macula is a small spot near the center of the retina and its damage results in loss of central vision. AMD can start as a blurred area near the center of vision and over time it can grow larger until loss of central vision occurs. Central vision is extremely important for everyday tasks such as reading, writing, and face recognition.
There are three stages of AMD defined in part by the size of drusen (yellow deposits) under the retina. Early and intermediate stage AMD has few symptoms or vision loss. These earlier stages of the disease are usually left untreated or dealt with using diet supplementation. People with advanced AMD have vision loss from damage to the macula. There are two types of late stage AMD:
• Dry AMD or geographic atrophy: There is a breakdown of light sensitive cells in the macula that send visual information to the brain, and the supporting tissue beneath the macula. This damage causes vision loss.
• Wet AMD or neovascular AMD: Blood vessels grow underneath the retina, these vessels might leak blood which may lead to swelling and damage of the macula. This damage may be severe and can progress quickly.
Treatments for AMD:
• The Implantable Miniature Telescope (VisionCare Ophthalmic Technologies, Inc.), a magnifying device that is implanted in the eye, is approved for use in patients with severe to profound vision impairment (best corrected visual acuity of 20/160 to 20/800) due to dry AMD. Some patients who are candidates for the Argus II device may also be candidates for the implantable telescope.
• There are currently no other treatments for AMD after the disease has caused severe to profound vision loss.
• There are currently no established treatments that delay or reverse the progression of Dry AMD other than supplements.
• Therapies exist for Wet AMD that delay the progression of visual impairment or slightly improve the vision, rather than completely curing or reversing its course. These therapies are approved in many regions throughout the world, including the US and EU.
Worldwide, between 20 and 25 million people suffer from vision loss due to AMD,7 and of these about 2 million have vision that is considered legally blind, or worse.8 In the US, just over two million people experience vision loss due to AMD according to a 2010 study by the National Eye Institute. Of the 1.3 million legally blind Americans,9 we estimate that 42.5% (or 552,500) are due to AMD.10 Applying this percent of legally blind due to AMD (42.5%) to the total number of legally blind people in
__________
3 Eurostat. Retrieved 1 January 2013.
4 Haim M. Epidemiology of Retinitis Pigmentosa in Denmark. Acta Ophthalmol Scand Suppl 2002; 1-34.
5 Grover et al., ‘Visual Acuity Impairment in Patients with Retinitis Pigmentosa at Age 45 Years or Older’, Ophthalmology. 1999 Sept; 106(9):1780-5.
6 The Eye Diseases Prevalence Research Group, 2004a; CDC, 2009.
7 Choptar, A., Chakravarthy, U., and Verma, D. ‘Age Related Macular Degeneration’. BJM 2003;326:485.
8 Global Data on Visual Impairments 2010, World Health Organization.
9 National Eye Institute ( http://www.nei.nih.gov/eyedata/blind.asp)
10 Congdon N, O’Colmain B, Klaver CC, et al. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. Apr 2004;122(4):477-485. This percent amount was derived from the rates of different causes of blindness by different races and racial demographic data from 2010 US Census data.
3
Europe (2.55 million),11 we estimate the population of legally blind individuals from AMD to be about 1.08 million individuals in Europe. We believe the Argus II System may be able to help a subset of these legally blind AMD patients who have severe to profound vision loss.
To date, we have not yet implanted any AMD patients with the Argus II device. We are planning to conduct a five subject feasibility study of the use of the Argus II System in patients with dry AMD. This feasibility study will be conducted in the United Kingdom and approval to conduct the study has been obtained from the UK Competent Authority, the Medicines and Healthcare Products Regulatory Agency or MHRA. Patient recruitment in this study began in December 2014. We expect to perform these implants in the first half of 2015. After positive follow-up data have been collected from these five subjects, we intend to conduct a larger pivotal study, of up to 30 subjects, in the US and Europe to collect safety and efficacy data to support market approval for this expanded indication for use for the Argus II System.
We intend to seek FDA approval in the US for use of the Argus II System for AMD. We also intend to explicitly add AMD to our CE label for the use of the Argus II System in patients with AMD in Europe. Our approach will be to implant the electrode array in the central vision area where patients have vision loss and leave any peripheral vision largely unchanged.
Other diseases resulting in blindness that may be treated by Orion I visual prosthesis
Many diseases outside of RP and AMD can also cause blindness. Many of the largest causes of visual impairment (i.e. refractive error and cataracts) are avoidable or curable, and their prolonged or untreated impact on vision is largely observed in developing nations. Some other causes of blindness, such as brain trauma, may also not be suitable for treatment by a cortical stimulator. However, the remaining causes of severe vision loss which include glaucoma, diabetic retinopathy, eye trauma, retinopathy of prematurity and many others can result in severe visual impairment that may prove to be treatable by an Orion I visual prosthesis.
According to the World Health Organization (WHO),12 285 million people suffer from vision loss worldwide. Of these, 39 million people are considered legally blind. The WHO further estimates that 80% of legal blindness is avoidable, leaving 7.8 million legally blind individuals, including those blind due to AMD and RP, or 5.8 million excluding AMD and RP. In the US, 1.3 million people are legally blind,9 of which we estimate 44.3%, or 575,900, are legally blind due to causes other than preventable/treatable conditions, RP or AMD.10
The potentially addressable market for the Orion I visual prosthesis is a subset of the legally blind population cited here, or less than 5.8 million worldwide, approximately 575,900 in the US, and about 1.13 million in Europe.
We intend to use a portion of the proceeds we received from our IPO, completed in November 2014, to support the research and development of the Orion I visual prosthesis. We plan to complete the pre-clinical development of the Orion I device by mid-2016 and currently anticipate conducting our first-in-human study by the end of 2016.
Our Technology
We developed the Argus II System primarily in-house following its clinical conception in the early 1990s by a handful of leading retinal doctors, vision scientists, and engineers, and the subsequent formation of the company in 1998. During this development period we created long term safety, reliability and clinical benefit as we encountered, solved and frequently patented solutions to, a number of significant clinical and engineering challenges. These include:
• Development of an electrode array that can rest on and interface with the delicate retina for multiple years without causing damage to the underlying neurons;
• Miniaturization of the implantable micro-electronics package under the constraints of requiring it to be water tight, durable, biocompatible, and biostable, while featuring over 60 electrical connections;
• Development of a flexible polymer based electrode array that does not break down and leak in-vivo for a period of up to decades as demonstrated by accelerated life tests and over five years of continuous use in patients;
__________
11 Global Data on Visual Impairments 2010, World Health Organization.
12 WHO Fact Sheet number 282, updated October 2013.
4
• Development of a biocompatible and stable connection to join the polymer array to the micro-electronics package;
• Development of an electrode material that can withstand higher charge densities than the known best neurostimulation industry standard (platinum) - thereby enabling the use of smaller (and hence more in a given area) stimulating electrodes;
• Development of a wireless power and data link that meets international standards and produces stable device function with a moving eye;
• Development of stimulation and rehabilitation methods that improve patients’ outcomes; and
• MRI conditional status, that is safe for the patients to undergo MRI under specified conditions.
The Argus II Retinal Prosthesis System consists of an implant, a small portable computer and a pair of glasses with a miniature video camera.
Implant
Our implant is an epiretinal (that is, the retinal surface is the site of stimulation) prosthesis that includes a receiver coil (antenna), electronics, and an electrode array. It is implanted in and around the eye. The array has 60 platinum gray electrodes arranged in a 6x10 grid. Each electrode is 200 µm (0.008”) in diameter. The array covers about 20° of visual field (diagonally). The flexible polymer thin-film electrode array, which follows the curvature of the retina, is attached to the retina over the macula with a retinal tack. The extra-ocular portion of the Argus II Implant is secured to the eye by means of a scleral band and sutures.
 |
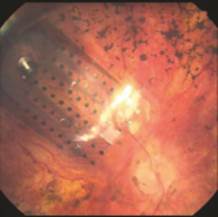 |
| Figure 1: Schematic of Argus II implant as implanted (surgical implantation is typically performed in 2 to 4 hours) | Figure 2: Electrode array. Current version contains 60 platinum gray electrodes |
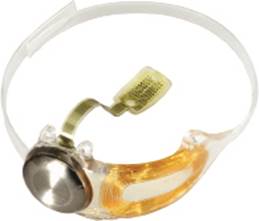
Figure 3: Argus II implant.
5
Externals
The external equipment consists of a pair of glasses and a video processing unit or VPU. The glasses include a miniature video camera and a transmitter coil. The Argus II Clinician Programming Kit is used to program the Argus II System stimulation parameters and video processing strategies for each patient. The software provides modules for electrode control, permitting the clinicians to program the amplitude, pulse-with, and frequency of the stimulation waveform of each electrode.

Figure 4: External Components of the Argus II System
How it works
In a healthy eye, the photoreceptors (rods and cones) on the retina convert light into tiny electrochemical impulses that are sent through the optic nerve and to the brain, where they are decoded into images. If the photoreceptors no longer function correctly (as in RP and AMD), the first step in this process is disrupted and the visual system cannot transform light into images, causing blindness. The Argus II System is designed to bypass damaged photoreceptors altogether and provides real-time visual information to blind patients. The miniature video camera captures a scene and the video is sent to the small VPU where it is processed and transformed into instructions that are sent back to the glasses. These instructions are transmitted wirelessly to the receiver coil in the implant. The signals are then sent to the electrode array, which emits small pulses of electricity. These pulses bypass the damaged photoreceptors and stimulate the retina’s remaining cells, which transmit the visual information along the optic nerve to the brain. This process is intended to create the perception of patterns of light which patients can learn to interpret as real-time visual patterns.

Figure 5: The patient perceives patterns of light created by electrical stimulation.
Long-Term Reliability
The Argus II System has been extensively tested at the component, sub-assembly, and system levels for long term reliability. The hermetic electronics case has been demonstrated to prevent moisture accumulation inside the device for many years. The Argus II implant is specified to last a minimum of five years, however, in vitro tests and actual clinical data suggest the device should last much longer. Production implants have reached more than ten years of lifetime use in accelerated in vitro testing and more than seven years use in real time in patients under active stimulation and normal use conditions.
6
Our Research and Development
Our research and development staff is focused on improving the level of vision that the Argus II System can provide to blind patients and adapting the technology to help a broader audience of blind individuals. A portion of the proceeds from our IPO will go toward supporting the research and development efforts described below.
Increasing Resolution
We believe that increasing the resolution of the system should enhance the user experience, which would increase the value and benefits of the technology to the patient. We believe that we will be able to increase the system resolution by:
• Developing enhanced image processing: Through enhanced image processing, including contrast enhancement and electronic ‘zoom’, one patient so far tested has achieved 20/200 level vision as measured by a grating acuity test.13
• Creating multiple virtual electrodes: we believe we can use software to electronically create a number of virtual electrodes between the physical ones in the Argus II electrode array. This development could potentially enhance the resolution of existing devices by more than one order of magnitude. Although similar approaches have been successful in other neural stimulators, this approach has not yet been tested clinically on the retina.
We expect to spend a portion of the proceeds received from our IPO over the next 24 months on increasing resolution of and other performance enhancing improvements to the Argus II system.
Cortical Stimulator – Orion I
Developing a cortical stimulator is central to our strategy of maintaining our world leadership in restoring sight to those who are blind. There are different diseases that damage the optic nerve or impair the total functioning of the retina. We believe that a cortical stimulator will permit us to bypass the eye and the optic nerve, thereby allowing treatment for a wider variety of disease-related blindness.
Research described in 1968 reported that it was possible for a blind subject to experience light though phosphenes (appearance of light) when the visual cortical region (surface) of the brain was electronically stimulated – just as the Argus II System does in the eye.14 Functional vision corresponding to visual acuities up to 20/1200 was reported in the early 2000s, and two subjects were reported to have a prototype of a functional prosthesis implanted for more than 20 years without infections or other severe medical reactions.15Though these human experiments demonstrated proof of principle, no reliable implantable neurostimulator with a large number of electrodes was available before we developed and introduced the Argus II System.
By implementing relatively minor modifications to the Argus II technology, we believe that the Orion I visual prosthesis can be implanted directly on the surface of the brain in the visual cortex and may be able successfully to restore some functional vision in almost all cases of disease related blindness. Our small electronics case will be implanted under the scalp and the electronic array placed in the visual cortex region of the brain. A transmitter coil similar to the one in current production will send power and signals to the implanted device. We plan to place our electrode array against the medial surface of the visual cortex.
We anticipate that many of the challenges that we encountered and solved in the process of developing the Argus II System are largely the same challenges in developing a product intended for enabling some functional vision through directly stimulating the brain. For example, a robust implant with a large number of electrodes is required for a cortical or retinal visual prosthesis. We believe the knowledge and technology gained in the development of the Argus II System will contribute to accelerating the development of a cortical stimulator directed at treating blindness.
We can also leverage public information learned from other electrical stimulation implants that are FDA-approved for use in the brain such as Medtronic’s Activa deep brain stimulator for Parkinson’s, Essential Tremor and Dystonia and more recently
__________
13 Sahel JA, Mohand-Said S, Stanga PE, Caspi A, Greenberg RJ. Acuboost™: Enhancing the maximum acuity of the Argus II Retinal Prosthesis System. IOVS 2013 May; 1389. ARVO E-Abstract.
14 Brindley, G.S. and W.S. Lewin, The sensations produced by electrical stimulation of the visual cortex. J Physiology 1968. 196(2): p. 479-93.
15 Dobelle, W.H. Artificial vision for the blind by connecting a television camera to the visual cortex. ASAIO J 2000;46:3-9.
7
NeuroPace’s RNS® brain stimulator for epilepsy. Furthermore, we believe that our specific experience obtaining regulatory approval for these types of devices in the United States and other regions will prove to be helpful in our effort to expand and get new products, such as Orion I visual prosthesis, approved throughout the world.
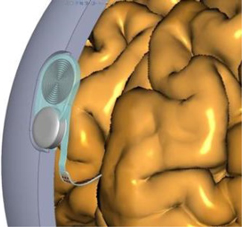
A: Placement of array against the medial surface of the visual cortex. Array is in blue, the electronics capsule in dark gray and the receiver coil in light gray on the outer surface of the skull.
Clinical Trials
Second Sight completed a pre-market clinical trial of the Argus II System and data from this trial supported both the FDA (US) and CE Mark (EU) approval of the Argus II device. Second Sight is currently conducting post-market studies of the Argus II System to continue to collect data regarding the long-term safety and benefit of the Argus II System in patients with severe to profound RP/outer retinal degeneration. We began recruitment for a study to support expanding the indications for use of the Argus II System to Age-Related Macular Degeneration (AMD) in the fourth quarter 2014, and we plan to begin implants in the first half of 2015. We are planning to begin a feasibility study of the Orion I visual cortical prosthesis in the fourth quarter of 2016.
Pre-market Clinical Trial of the Argus II System for Retinitis Pigmentosa/Outer Retinal Degeneration
The Argus II System, indicated for patients with severe to profound outer retinal degeneration (limited to RP in the United States), was studied in a clinical trial of 30 subjects in the U.S and EU. The study is registered at www.clinicaltrials.gov under study ID NCT00407602. The study began in 2007 and as of January 2015, there were over 180 subject-years in the clinical trial. As part of post-market surveillance, this study is continuing and we intend to follow subjects for a total of ten years each.
Data collected in this trial demonstrated that the Argus II System has a reasonable safety profile for an ophthalmic device that requires vitreoretinal surgery to implant. There were no unexpected adverse events. The most common, serious adverse events were conjunctival erosion/dehiscence, hypotony (low eye pressure), endophthalmitis (infection in the eye), retinal tear or detachment, and re-tacking. It was also demonstrated that the device can be safely removed: one implant, including the retinal tack, was safely explanted to resolve an adverse event, and three retinal tacks were safely removed during elective revision surgeries to reposition arrays. All adverse events were treatable with standard practices utilized by ophthalmologists. In general, these events did not adversely affect performance with the Argus II System.16 Furthermore, since approval, we have observed a decrease in the rate of adverse events, most likely in our view due to increased surgical experience with the technology.17
The Argus II System provides visual information that can range, depending on the patient, from light detection to form detection. A sub-study of Argus II System clinical trial patients demonstrated that 72% of patients could identify closed set letters, and a subgroup of six patients was able to consistently read letters of reduced size, the smallest measuring 0.9 cm (1.7°)
__________
16 Sponsor Executive Summary, FDA Ophthalmic Devices Advisory Panel, September 28, 2012. (http://www.fda.gov/AdvisoryCommittees/Calendar/ucm312582.htm).
17 Humayun, M.S., da Cruz L., Dagnelie G., Stanga PE, Ho AC, Greenberg RJ, Birch DG, Duncan JL, Sahel JA. An update on the Argus II epiretinal implant. IOVS 2014 May; 5968. ARVO E-Abstract.
8
at 30 cm and four patients correctly identified unrehearsed two-, three- and four-letter words.18 Patients are able to use this visual information to perform functional tasks (such as, locating windows and doors, following lines in a cross walk), to allow them to feel more connected with others (for example, seeing when a person approaches them or when someone walks away), and to simply enjoy visual perception again (such as, seeing the changing light levels on a TV, tracking groups of players as they move around the field at an athletic event and being able to locate the moon). For people with bare or no light perception, even limited restoration of vision can make a significant difference in their lives. 22
In the clinical trial, the Argus II System provided all 30 subjects with benefit as measured by high-contrast visual function tests. The degree of benefit varied from subject to subject. The Argus II System was also able to provide subjects with clinical benefit as measured by objectively-scored functional vision tests. Subjects performed better with the Argus II system on vs. off on orientation and mobility tests (finding a door and following a line) and on functional vision tasks (sorting white, black and grey socks; following an outdoor sidewalk; and determining the direction of a person walking by).22
An assessment of Argus II System subjects’ functional vision in and around their home by independent, certified low‑vision rehabilitation specialists was also performed. The assessment was called the Functional Low-vision Observer Rated Assessment, or FLORA®. In no cases, did the low vision specialists report that the Argus II System had a negative impact on subjects. In 77% of cases, low vision specialists determined that the subject was receiving (or had received at one time) functional vision and/or well-being benefit from the Argus II System.22 The results from this clinical trial demonstrated that the Argus II System provided benefits for these blind subjects in terms of visual function (how the eye works), functional vision (performance in vision related activities), and well-being. The study also demonstrated that the Argus II System does not pose an unacceptable risk to blind patients with severe to profound RP with bare or no light perception in both eyes. In 2012, after an in depth review of the clinical trial data, a 22 person (19 voting) FDA-convened panel of experts voted unanimously that the benefits of the Argus II System outweighed the risks.19
Post-Market Clinical Trials
Following CE Mark and FDA approval for the Argus II System, Second Sight is conducting two post-market studies of the device (one in EU and one in the US) to collect additional long-term data on the use of the Argus II System in patients with severe to profound outer retinal degeneration (or RP in the United States). Post-market studies are typically conditions of market approval for medical devices.
In the United States, the study is designed to enroll 53 subjects and will follow each subject for five years. Adverse events, visual function, and functional vision data are being collected for all study participants. Enrollment began in February 2014 and seven subjects have been enrolled as of January 31, 2015. The study is registered at www.clinicaltrials.gov under study ID NCT01860092.
In Europe, Second Sight is conducting a post-market study that is designed to enroll 45 subjects and will follow each subject for three years. Adverse events, visual function, and functional vision data are being collected for all study participants. Enrollment began in December 2011 and 36 subjects have been enrolled as of January 31, 2015. The study is registered at www.clinicaltrials.gov under study ID NCT01490827.
In France, Second Sight was selected to receive the first “Forfait Innovation” (Innovation Bundle Payment) from the Ministry of Health, which is a special funding for breakthrough procedures to be introduced into clinical practice. As part of this program Second Sight is conducting a post market study in France which will enroll 18 subjects and follow them for two years. Enrollment began in November 2014 and 2 subjects have been enrolled as of January 31, 2015. The study is registered at www.clinicaltrials.gov under study ID NCT02303288.
Pre-market Clinical Trial of the Argus II System for Age-Related Macular Degeneration
We have begun recruitment in a pilot study of the Argus II System for use in patients with age-related macular degeneration, or AMD. In this study, the Argus II System is being used in its current RP configuration, without any significant modifications. We will be enrolling five subjects in the pilot study who have central vision loss due to dry AMD; the subjects will be followed for three years. The study
__________
18 da Cruz L, Coley BF, Dorn J, et al. The Argus II epiretinal prosthesis system allows letter and word reading and long-term function in patients with profound vision loss. Br J Ophthalmology 2013;97:632-6.
19 Sponsor Executive Summary, FDA Ophthalmic Devices Advisory Panel, September 28, 2012.
9
will be conducted at a single center in the UK. The study is registered at www.clinicaltrials.gov under study ID NCT02227498. Recruitment for this study began in December 2014 and we plan to begin implanting in the study in the first half of 2015.
Assuming the early results from this pilot study are positive, we intend to apply for approval to conduct a larger study of AMD (both wet and dry) in the United States and EU in late 2015. This larger study, which we anticipate will begin in early 2016, will be used to support efforts to obtain regulatory approval to expand the label for the Argus II System to include AMD in its indications for use. The study would also be used to support efforts to obtain reimbursement in the United States and EU for this expanded indication for use. However, there can be no assurance that this pilot study will be successful, and further research and development may be needed.
Future Clinical Trials
Cortical Prosthesis
Following completion of the development effort associated with the Orion I, including verification and validation of the design, we intend to conduct a feasibility clinical trial to assess the safety and benefit of the device in blind individuals. We expect that this feasibility study will begin in 2016.
Our Commercialization Plan
We launched the Argus II System in Italy and Germany in late 2011. We have, since early in 2012, also launched the Argus II System in France, the UK, the Netherlands, Spain and Saudi Arabia. In 2013, the Argus II System received FDA approval, and the product was launched in the US in January 2014, after receiving a required FCC Grant of Equipment Authorization late in 2013. Also in 2014, an investigator-sponsored study in Toronto, Canada has resulted in two unit sales to that Toronto center. We obtained approval for the Argus II System from Health Canada at the end of 2014 and we intend to begin commercializing the device within Canada during 2015. In this early stage of commercialization, we focused on a controlled launch to ensure adequate service to the centers and to integrate new knowledge gained so as to make necessary adjustments to our products and services in the following larger commercial launch. We currently are poised for a broader launch phase, where the treatment will be made available to a larger population of eligible patients. We expect to use up to $4 million of the proceeds from our IPO over the next 18 months to expand the commercial roll out of the Argus II System.
Our successful commercialization of this technology and therapy is dependent on implementing our sales and marketing strategy, and obtaining reimbursement of the Argus II System by payers.
Sales Strategy
During our commercial launch, we are employing a Centers of Excellence sales strategy and deploying the Argus II System at prominent and reputable eye clinics. We believe this strategy represents an efficient use of our capital after giving consideration to the following factors:
• The size of the RP patient population.
• The complexity of the technology, surgery, and treatment paradigm.
• The cost of selecting, qualifying, training and supporting new centers.
When selecting new sites, we focus on high quality health providers utilizing the following considerations:
• Geographic desirability,
• Facility and Surgeon skill and reputation,
• Access to patients,
• Regulatory pathway, and
• Reimbursement environment from government agencies or contractors and third party insurers.
10
In the United States and Canada, as of January 31, 2015, we have 11 centers that have implanted the Argus II retinal prosthesis. Additionally, we have 18 other centers that have been selected as potential implanting centers and that are somewhere in the process of becoming active centers in 2015. We anticipate opening other new centers in subsequent years. We believe that we will be able to serve the domestic RP market by having about 70-80 implanting centers across the US.
In Europe and the Middle East, we currently have 12 centers that actively are implanting and/or recruiting patients to schedule their Argus II retinal prosthesis surgeries (six in Germany, three in France, one in Saudi Arabia, and two in Italy). Additionally, we have 15 other centers that are either preparing to implant patients or are in the qualification process. We intend to continue recruiting additional centers in 2015 to yield further active centers in 2016. We anticipate that annual new center recruitment, in subsequent years will prove to be an important driver of our implant and revenue growth in foreign markets. We believe that we will be able to serve the European markets for RP by having about 100-120 centers across Europe.
To date, we have employed direct sales to service our initial markets during our controlled launch phase. We believe we can more efficiently support centers that are located at distances from our US and European headquarters by securing distributors in several key markets in order to expand our reach of client marketing and support. To date, we have appointed distributors in Spain and Turkey. We expect that our distributors will commit to providing support services that include marketing, market access, reimbursement, sales and service and also commit to annual minimum quantities and volume targets depending on their territories.
To date, we have not faced traditional sales challenges in any of our markets, largely due to the currently unmet clinical need and the lack of any other approved device or competitive treatment for RP caused blindness. Due to pending reimbursement approvals in the United States, many doctors and facilities have expressed interest in providing the Argus II System for their patients but have been unable to do so. We have over 1,700 potential patients with verified contact information in a database as a result of media coverage and news of awards that we have received. Our patient pipeline has over 60 US patients deemed currently eligible by implanting physicians and awaiting reimbursement authorization to be implanted. No assurance can be provided that all these reimbursement authorizations will be received or when these patients might be implanted. Please see ‘Reimbursement’ below.
Due to the high cost of the system, government reimbursement (coding, coverage, and payment) is paramount to being able to provide this device to our patients. Please see ‘Reimbursement’ below. We anticipate that our primary challenges in pursuing and implementing sales efforts will be to maintain a growing patient pipeline while expanding reimbursement coverage.
Marketing Strategy
To date, and for the foreseeable future, our marketing efforts have been primarily focused on promoting both our brand (for both the product and the company) and on raising awareness amongst and educating certain target groups which include the following:
• Potential patients,
• Potential implanting physicians and medical centers, and
• Potential referring physicians (general practitioners, ophthalmologists, optometrists, and low-vision specialists).
To achieve these objectives we have employed a mix of marketing plans and approaches which includes media relations, trade and professional show attendance, exhibition, and podium presence, sponsoring medical symposia, conducting regional education sessions, partnering with patient advocacy groups focused on blindness and retinal degeneration, and a limited amount of advertising. We employ two in-house marketing professionals currently and a number of specialized consultants.
Reimbursement
Reimbursement, which is third party coverage and payment for health care services rendered to patients by government and private insurance providers, varies significantly in form and function across countries.
United States
In the US, hospitals or ambulatory surgery centers, known as ASCs, are the primary purchasers of the Argus II System. Hospitals, ASCs and physicians bill third-party payers, including Medicare, Medicare Advantage and private payers for costs
11
associated with providing the services and acquiring the Argus II System. Regardless of age, Medicare provides coverage to the blind simply on the basis of their disability. While the majority of patients for the Argus II System are insured by Medicare or Medicare Advantage plans, Medicare does not currently universally reimburse for the Argus II System
In order to have adequate reimbursement for devices and services, we are required to obtain coding, coverage, and payment. Additionally, the required codes and payment vary based on the site of service such as in-patient hospital, out-patient hospital, ASC, or physician’s office. Most Argus II System procedures are performed on an out-patient basis, while a small number may be performed in an in-patient setting. We have obtained required codes and payment (for both the procedure and device) from the national office of Medicare in both settings of care.
Reimbursement in the U.S. consists of three basic components: “coding, payment and coverage.”
Coding
Providers use systems of codes to communicate with the payer the patient conditions and diagnoses, services provided, procedures performed, and devices used to treat the patient. These codes include American Medical Association Current Procedural Terminology (CPT) codes, Healthcare Common Procedure Coding System (HCPCS) codes, and International Classification of Diseases-9th Revision (ICD-9) codes.
Second Sight has obtained the required coding from Centers for Medicare and Medicaid Services (CMS) and CPT, including:
• CPT code for the retinal implantation procedure (CPT 0100T), which is utilized by physicians, hospital outpatient departments and ambulatory surgery centers;
• HCPCS Code specific to the Argus II retinal prosthesis implant device (and external components) (HCPCS code C1841) which is utilized by hospital outpatient departments and ambulatory surgery centers; and
• ICD-9 procedure code for the retinal implantation procedure (ICD-9 code 14.81), which is utilized by hospital inpatient departments.
Payment
CMS has established specific Medicare payment rates for the implantation procedure in the hospital inpatient setting, hospital outpatient setting and ambulatory surgery center setting. In addition, Second Sight has been successful in obtaining additional transitional pass-through payment for the Argus II retinal prosthesis device (and external components) which is reimbursed to the hospital outpatient department or ambulatory surgery center. While the majority of these procedures are likely to be performed in a hospital outpatient setting, Second Sight has also obtained new technology add-on payment which provides additional reimbursement if the procedure is performed on an inpatient basis. We expect that specific physician payment rates will be established when CPT 0100T is formally valued by the American Medical Association. Until that time, reimbursement to physicians is based on charges submitted to the payer.
Coverage
Coverage is the process, criteria, and policy used by payers (insurers) to determine whether to pay for a medical procedure or product. Payers make decisions on coverage for a procedure or service based on a number of factors, including medical necessity, effectiveness, and outcomes. We anticipate that the majority of patients receiving the Argus II retinal prosthesis will be insured by Medicare due to age or the nature of their disability. Medicare insures over 47 million beneficiaries for items and services that are “reasonable and necessary for the diagnosis or treatment of an illness or to improve the functioning of a malformed body member”.20 These beneficiaries include people age 65 or older, people under age 65 with certain disabilities and people of all ages with end-stage renal disease.
Currently one regional Medicare Administrative Contractor, or MAC, in the Mid-Atlantic region provides coverage for the Argus II System. We are actively working with seven other MACs and Medicare Advantage Plans to obtain favorable coverage. Several commercial payers and Medicare Advantage Plans cover the Argus II System either through formal policy or
__________
20 Social Security Act §1862 (a)
12
on a case-by-case basis, including Health Net, Independence Blue Cross, AmeriHealth, Priority Health Medicare Advantage, BCBSS of Arkansas, AV Med, Medica, among others. Second Sight is actively engaging the dissenting plans to obtain favorable coverage for the Argus II System through either a formal coverage policy or on a case-by-case basis. Although we expect an increasing number of payers to agree to cover the Argus II System, there can be no assurance that all other MACs, Medicare Advantage Plans or private payers will cover and reimburse our product and the procedures to implant them in whole or in part in the future or that payment rates will be adequate.
Europe
After obtaining the regulatory approval (CE mark) in Europe, innovative medical devices go through a fragmented public reimbursement system across Europe. Many European countries use a Diagnosis Related Group, DRG, or similar type reimbursement coding system for the lump sum payment of in-patient medical procedures. Medical devices are generally included in the lump sum DRG payment. To apply for the creation of a new reimbursement code a new medical device first needs to be in widespread use as part of a hospital medical procedure. Typically it may require two or more years from application to obtain a new DRG code. A common way for these medical devices to be made available in the public healthcare setting, is through hospital research/ innovation budgets/other routes. However, these budgets are small in size, limited to a few hospitals, and quite difficult to access, thereby limiting patient access to new treatments. To address this problem, many countries in Europe have created temporary reimbursement programs for innovative medical products to bridge the time to collect convincing clinical and economic evidence to get a new DRG code. While these programs offer a reasonable opportunity for new medical products to get faster reimbursement, they have stringent requirements of clinical and economic evidence that may vary from country to country, leading to slow adoption rates of reimbursement approvals. Such short-term reimbursement programs, such as “NUB” in Germany, “Forfait Innovation” in France, Local/Regional funding in Italy and “Coverage through Evaluation” in England, can be generally grouped as “coverage with evidence” programs.
Second Sight achieved reimbursement for the Argus II System in Germany in 2011 with a process dedicated to innovative procedures referred to as NUB (Neue Untersuchung und Behandlungsmethoden). This reimbursement under NUB is valid for one year in a specific number of hospitals. Under the NUB program, each hospital negotiates the payment for the procedure with the insurance companies for “additional costs associated with the innovative treatment,” and in 2011 two hospitals managed to get a sustainable funding to start the first procedures. Since 2011, the funding was renewed five times and the number of hospitals obtaining funding increased from two to seven. Over the next few years we expect our Argus II therapy to be covered under the standard payment system which would mean the device would be reimbursed at all centers and the annual negotiation would no longer be necessary. In January 2015 we received renewal of our NUB status for an additional one year term.
In France, we were selected to receive the first “Forfait Innovation” (Innovation Bundle Payment) from the Ministry of Health, which is a special funding for breakthrough procedures to be introduced into clinical practice. France commissioned this program for the first time in 2014, and to our knowledge the Argus II System is one of only two products that were selected for this funding after rigorous healthcare technology assessment by “haute authorité de santé” (French Health Technology Assessment authority). In the longer term, we expect Argus II therapy to be covered under the standard payment system (GHM‑LPP system).
In Italy the Argus II System has been available in the Tuscany region since 2011 under a hospital/regional funding program. Beginning in 2015, it is also available in the Veneto region. We expect that the Argus II System will be available in several other regions as well under similar funding programs. Patients across Italy will then have a possibility of treatment in these centers with prior government approvals required in some of these regions. Within the next several years, we expect the Argus II System to be accessible across Italy with funding from national or regional governments.
The Argus II System is going through a review process under National Specialized Services program in England, as well as under other funding programs in other markets across the EU and Middle East. We expect the Argus II System to become eligible for reimbursement at the national or regional level for eligible patients during the next several years in some of these markets.
A multi-market economic evaluation (Anil Vaidya, Elio Borgonovi, Rod S Taylor, José-Alain Sahel, Stanislao Rizzo, Paulo Eduardo Stanga, Amit Kukreja, Peter Walter; BMC Ophthalmology 2014; 14: 49. Published online 2014 April 14. doi: 10.1186/1471-2415-14-49) conducted on the Argus II System affirms that the Argus II System is a cost-effective intervention compared to the usual care of RP patients. The lifetime analysis ICER (Incremental Cost Effectiveness Ratio) for
13
the Argus II System falls below the published societal willingness to pay in EU. According to this assessment, the Argus II System treatment has a cost that we believe is reasonably priced for a technology addressing a rare population such as retinitis pigmentosa.
Warranty
We generally provide a standard limited warranty for the Argus II System covering replacement over the following periods after implant:
• three years on implanted epiretinal prosthesis
• two years on external components other than batteries and chargers, and
• three months on batteries and chargers.
Based on our experience to date, the Argus II System has proven to be a reliable device generally performing as intended. We have accrued warranty expense of $555,972 as of December 31, 2014, which we believe to be adequate.
Our Competition
The US life sciences industry is highly competitive and well-positioned for future growth. The treatment of blindness varies based on the cause; generally there are seven categories of treatments in development for the treatment of blindness from retinal disease:
• Retinal Prostheses: aimed at giving more visual ability to a blind patient via implanting a device in the eye to stimulate remaining retina cells. Electrical neurostimulation technology has seen growing use in recent years for numerous applications– such as chronic pain, Parkinson’s, Essential tremor, Epilepsy, and others.
• Transplants: transplanting retinal tissue to stimulate remaining retina cells.
• Genetics and Gene Therapy: involves identifying a specific gene that is causing retinal problems (there are over 120 for retinitis pigmentosa alone) resulting in visual impairments and blindness; and inserting healthy genes into an individual’s cells using a virus to treat the diseases.
• Stem Cells: generally involves implanting immature retinal support cells aimed at slowing retinal degeneration.
• Optogenetics Therapy: aimed at slowing down, reversing, and/or eliminating the process by which photoreceptors in the eye are compromised. This therapy also requires infecting the patient’s cells with a virus. However, instead of fixing a gene defect, this approach would cause cells within the eye to become light sensitive. Animal work has shown that these cells are not sensitive enough to respond to ambient light, so this approach currently also requires a light amplifier outside the body to increase light delivered to the retina.
• Nutritional Therapy: involves diets or supplements that are thought to prevent or slow the progress of vision loss.
• Implantable Telescope: VisionCare, Ophthalmic Technologies, Inc. offers an FDA approved implantable minature telescope for AMD, a magnifying device that is implanted in the eye. The VisonCare telescope is approved for use in patients with severe to profound vision impairment (best corrected visual acuity of 20/160 to 20/800) due to dry AMD.
Projects in these six areas are still undergoing either animal or early clinical trials; some, like gene therapy, stem cell therapy and optogenetics remain highly speculative for most conditions. We believe that it is currently unlikely that gene therapy and stem cell therapy will prove effective in end stage RP patients in the near term.
In the area of retinal prosthetics, there are a number of competing efforts underway. We believe that most, if not all of these efforts, are not as advanced as the Argus II System in terms of commercialization, especially in the United States.
Commercial efforts by others include:
• Retina Implant AG: A German company that is developing the Alpha IMS, a wireless sub-retinal implant using the image from the eye’s own optical system Retina Implant AG has a CE mark and to our knowledge expects to seek commercialization of its product during 2015 in EU. To our knowledge, Retina Implant has not applied for or obtained FDA approval to begin a clinical trial in the US as of the date of this report.
14
• Pixium Vision S.A.: A French company that is developing the IRIS (Intelligent Retinal Implant System) that is surgically placed into the eye and attached to the surface of the retina. Similar to our Argus II technology, its system uses a camera and a wireless transmitter. Pixium is reported to be in clinical studies with IRIS and we believe plans to submit a CE mark application in 2015.21 To our knowledge, Pixium Vision has not applied for or obtained FDA approval to begin a clinical trial in the US as of the date of this report.
• NanoRetina Inc., a company based in Israel, and several other early stage companies are reported to have developed intellectual property or technology that may improve retinal prostheses in the future, but to our knowledge none of these efforts has resulted in a completed system that has been tested clinically in patients anywhere.
Academic entities are also working on vision restoring implants. To our knowledge these include Bionic Vision Australia (an early prototype device has been developed and to our knowledge implanted in three human subjects), Boston Retinal Implant project (preclinical phase) and Stanford University (preclinical). Of these projects, we believe most have not yet demonstrated a working implant, only one has begun long-term clinical work in humans, and to our knowledge none has received FDA approval to begin clinical trials in the US.
No other device to our knowledge has been successful in long-term human trials, currently making the Argus II System the sole implant being marketed for treating RP in the US, EU, and Saudi Arabia. We anticipate that our identified competitors are unlikely to obtain significant commercial traction in EU (even should they obtain CE Marks) until they have developed in depth clinical data showing the reliability, effectiveness, and safety of their devices. Based on current FDA guidance for retinal prostheses, we estimate any other competitor is at a minimum five years away from obtaining FDA approval in the US.
Government Regulations
The Argus II System is regulated within the category of medical devices. Medical device products are subject to rigorous FDA and other governmental agency regulations in the United States and as well as in foreign countries. Noncompliance with applicable requirements can result in import detentions, fines, civil penalties, injunctions, suspensions or losses of regulatory approvals or clearances, recall or seizure of products, operating restrictions, denial of export applications, governmental prohibitions on entering into supply contracts, and criminal prosecution. Failure to obtain regulatory approvals or the restriction, suspension or revocation of regulatory approvals or clearances, as well as any other failure to comply with regulatory requirements, would have a material adverse effect on our business, financial condition and results of operations.
The FDA regulates, among other things, the clinical testing, design, manufacture, labeling, packaging, marketing, distribution, post-market surveillance, and record-keeping for these products to ensure that medical products distributed in the United States are safe and effective for their intended uses.
In the United States, the Argus II System is classified as a Class III device, which is reserved for life-sustaining, life‑supporting and implantable devices. The most common path to market approval for Class III devices in the US is the Pre-Market Approval (PMA) process. To obtain PMA approval, the manufacturer must demonstrate that a device is safe and effective for its intended use. Class III devices intended for rare patient populations may also be approved under an alternative regulatory pathway, called the Humanitarian Device Exemption (HDE). To utilize the HDE approval process, a device must be designated a Humanitarian Use Device (HUD). To qualify as a humanitarian use device, the device must be used to treat or diagnose a disease or condition that manifests itself in fewer than 4,000 individuals per year in the United States, and there must be no alternative treatments available in the United States. To obtain HDE approval, the manufacturer is required to demonstrate that a device is safe and provides a probable benefit for its intended use.
Significant changes to existing products and new products, such as the cortical stimulation to be utilized by the Orion I visual prosthesis, must be approved by the FDA prior to distribution. Modifications or enhancements that could significantly affect the safety or effectiveness of the device or that constitute a major change to the intended use of the device will require new PMA or HDE application and approval. Other changes may require a supplement or other change notification that must be reviewed and approved by the FDA. Modified devices for which a new PMA or HDE application, supplement or notification is required cannot be distributed until the application is approved by the FDA. An adverse determination or a request for additional information could delay the market introduction of new products, such as Orion I visual prosthesis, which could have
__________
21 http://www.lerevenu.com/bourse/biotechs-et-medtechs/l-actualite-du-secteur/201405285385f194c6854/pixium-vision-interview-du-pdg-bernard-gilly
15
a material adverse effect on our business, financial condition and results of operations. We may not be able to obtain PMA or HDE approval in a timely manner, if at all, for Orion I visual prosthesis or any future devices or modifications to Orion I visual prosthesis or such devices for which we may submit a PMA or HDE application.
PMA and HDE applications must contain valid scientific evidence to support the safety and effectiveness (or probable benefit for an HDE) of the device, which includes the results of clinical trials, all relevant bench tests, and laboratory and animal studies. The application must also contain a complete description of the device and its components, as well as a detailed description of the methods, facilities and controls used for its manufacture, including, where appropriate, the method of sterilization and its assurance. In addition, the application must include proposed labeling, advertising literature and any required training methods.
If human clinical trials of a device are required in connection with an application and the device presents a significant risk, the sponsor of the trial is required to file an application for an Investigational Device Exemption (IDE) before beginning human clinical trials. Usually, the manufacturer or distributor of the device is the sponsor of the trial. The IDE application must be supported by data, typically including the results of animal and laboratory testing, and a description of how the device will be manufactured. If the application is reviewed and approved by the FDA and one or more appropriate Institutional Review Boards (IRBs), human clinical trials may begin at a specified number of investigational sites with a specified number of patients. If the device presents a non-significant risk to the patient, a sponsor may begin clinical trials after obtaining approval for the study by one or more appropriate institutional review boards, but FDA approval for the commencement of the study is not required. Sponsors of clinical trials are permitted to sell those devices distributed in the course of the study if the compensation received does not exceed the costs of manufacture, research, development and handling. A supplement for an Investigational Device Exemption must be submitted to and approved by the FDA before a sponsor or an investigator may make a significant change to the investigational plan that may affect the plan’s scientific soundness or the rights, safety or welfare of human subjects.
Upon receipt of a PMA or HDE application, the FDA makes a threshold determination as to whether the application is sufficiently complete to permit a substantive review. If the FDA makes this determination, it will accept the application for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the application. An FDA review of a PMA application generally takes one to two years from the date the application is accepted for filing; review of an HDE application can be shorter than a PMA. However, this review period is often significantly extended by requests for more information or clarification of information already provided in the submission. During the review period, the submission may be sent to an FDA-selected scientific advisory panel composed of physicians and scientists with expertise in the particular field. The FDA scientific advisory panel issues a recommendation to the FDA that may include conditions for approval. The FDA is not bound by the recommendations of the advisory panel. Toward the end of the PMA or HDE application review process, the FDA will conduct an inspection of the manufacturer’s facilities to ensure that the facilities are in compliance with applicable good manufacturing practice. If the FDA evaluations of both the PMA/HDE application and the manufacturing facilities are favorable, the FDA will issue a letter. This letter usually contains a number of conditions, which must be met in order to secure final approval of the application. When those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue an approval letter authorizing commercial marketing of the device for specified indications and intended uses.
The PMA/HDE application review process can be expensive, uncertain and lengthy. A number of devices for which PMA/HDE approval has been sought have never been approved for marketing. The FDA may also determine that additional clinical trials are necessary, in which case the approval may be significantly delayed while trials are conducted and data is submitted in an amendment to the PMA/HDE application. Modifications to the design, labeling or manufacturing process of a device that has received PMA/HDE approval may require the FDA to approve supplements or new applications. Supplements to a PMA/HDE application often require the submission of additional information of the same type required for an initial premarket approval, to support the proposed change from the product covered by the original application. The FDA generally does not call for an advisory panel review for PMA/HDE supplements, though applicants may request one. If any PMAs/HDEs are required for our products, we may not be able to meet the FDA’s requirements or we may not receive any necessary approvals. Failure to comply with regulatory requirements or to receive any necessary approvals would have a material adverse effect on our business, financial condition and results of operations.
Regulatory approvals, if granted, may include significant labeling limitations and limitations on the indicated uses for which the product may be marketed. Conditions of approval for a PMA/HDE application also often include the requirement to conduct a post-market study or studies. In addition, to obtain regulatory approvals and clearances, the FDA and some foreign regulatory authorities impose numerous other requirements with which medical device manufacturers must comply. FDA enforcement policy strictly prohibits the marketing of approved medical devices for unapproved uses. Any products we manufacture or distribute under FDA clearances or approvals are subject to pervasive and continuing regulation by the FDA.
16
The FDA also requires us to provide it with information on death and serious injuries alleged to have been associated with the use of our products, as well as any malfunctions that would likely cause or contribute to death or serious injury.
The FDA requires us to register as a medical device manufacturer and list our products. We are also subject to inspections by the FDA to confirm compliance with good manufacturing practice. These regulations require that we manufacture our products and maintain documents in a prescribed manner with respect to manufacturing, testing, quality assurance and quality control activities.
We are also subject to a variety of other controls that affect our business. Labeling and promotional activities are subject to scrutiny by the FDA and, in some instances, by the Federal Trade Commission. The FDA actively enforces regulations prohibiting marketing of products for unapproved users. We are also subject, as are our products, to a variety of state and local laws and regulations in those states and localities where our products are or will be marketed. Any applicable state or local regulations may hinder our ability to market our products in those regions. Manufacturers are also subject to numerous federal, state and local laws relating to matters such as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. We may be required to incur significant costs to comply with these laws and regulations now or in the future. These laws or regulations may have a material adverse effect on our ability to do business.
International sales of our products are subject to the regulatory requirements of each country in which we market our products. The regulatory review process varies from country to country. In EU, the European Union has promulgated rules that require medical products to affix the CE mark, an international symbol of adherence to quality assurance standards and compliance with applicable European medical directives. Once the CE mark has been duly applied to a device, the manufacturer may commercially distribute the product in all countries that are members of the European Union, and in several other countries that recognize the CE Mark, such as Switzerland and Turkey. Similar to the US, once the device has received the CE mark, companies are required to report certain serious adverse events, are required to conduct post-market surveillance, and in some countries are required to register or list the products.
To obtain the CE Mark for the Argus II System, we were required to demonstrate compliance with several European directives and standards, including the Active Implantable Medical Device Directive (AIMDD), and ISO 13485:2003 (“Medical devices, Quality management systems, and Requirements for regulatory purposes”). Second Sight contracts with a European Notified Body, an organization that reviews design documentation for our device and audits us annually to ensure compliance to the AIMDD directive and the ISO 13485 standard. In addition, significant changes to our device design, and new devices or new indications for existing products, would need to be reviewed and approved by the Notified Body, prior to allowing us to apply the CE mark to the new product. Losing the right to affix the CE mark to our Argus II device or any future products could have a material adverse effect on our business, financial condition and results of operations.
In the EU, replacement of the existing Medical Device Directive and Active Implantable Medical Device Directive with new regulations have been proposed and are under review by governing bodies in the EU. Two of the proposed changes that could have a significant impact on the review of Second Sight’s products by European Union regulators include: (1) requiring review of applications for certain high risk devices by an outside committee, which is in addition to review by the Notified Body; and (2) increasing the requirements for clinical data that are used to support an application. If the proposed changes are adopted into law, this could increase the cost and time required to obtain approval for our products in EU.
We will be responsible for obtaining and maintaining regulatory approvals for our products. The inability or failure to comply with the varying regulations or the imposition of new regulations would materially adversely affect our business, financial condition and results of operations.
Our Intellectual Property
Our success depends, partially, on our ability to protect our core technology and intellectual property. We rely on a combination of patents, patent applications, trademarks, trade secrets, including know-how, license agreements, confidentiality procedures, non-disclosure agreements with third parties, employee disclosure and invention assignment agreements, and other contractual rights, to protect our proprietary rights.
As of March 1, 2015, we have:
• 96 pending US patent applications and provisional patent applications
• 220 US granted patents
17
• 76 pending foreign patent applications
• 88 foreign granted patents
Our issued patents expire between March 2018 and October 2033. We cannot assure that any of our patent applications will result in the issuance of a patent or whether the examination process will require us to narrow our claims. In addition, any patents may be contested, circumvented, found unenforceable or invalid, and we may not be able to prevent third parties from infringing them. As we intend to expand our international operations, our patent portfolio, copyright, trademark and trade secret protection may not be available or may be limited in foreign countries.
Our international patents as of March 1, 2015 include:
• 50 Australia;
• 24 France, UK, and Germany;
• 1 France, UK, Germany and Switzerland;
• 1 UK, Germany and Switzerland;
• 9 Japan; and
• 3 Canada.
We have focused on obtaining patents primarily in the US and EU, as we have identified these jurisdictions to be our primary markets. We believe that the significant development and regulatory costs and expense of commercializing a product such as the Argus II System will be a material impediment to any competitor who attempts to market a visual prosthesis if excluded from these markets by not having access to our intellectual property.
We actively seek to identify and protect our intellectual property. We have a dual strategy of filing for and obtaining patents to block potential competitors, and filing patents where we believe our technology would be useful in other products. Our patent portfolio covers many aspects of our implant device and its supporting equipment. We have also patented alternative intellectual property paths that do not cover our device, but could present a possible alternative implant solution to a competitor. However, there can be no assurance that our pending patent applications or any future patent applications will be approved or will not be challenged successfully by third parties, that any issued patents will protect our technology or will not be challenged by third parties, or that the patents of others will not have an adverse effect on our ability to conduct our operations. No assurance can be given that others will not independently develop a similar or competing technology or design around any patents that have been or may be issued to us.
Our Licenses and Agreements
We have exclusive world-wide royalty-bearing licenses on intellectual property related to the Argus II System from Johns Hopkins University and Duke University which we entered into in October 2000, and from the Doheny Eye Institute (DEI) which we entered into in April 2002. Total royalties we pay under these licenses will not exceed 3.25% of our net revenue.
Johns Hopkins University and Duke University
Our license from Johns Hopkins covers two patents and one patent application. The two patents include a patent covering a system of wireless communication between the external part and implanted part of an implanted medical device, and a patent covering a stimulation pattern to preferentially stimulate deeper retinal cells. The patent application covers a system for fitting a visual prosthesis using visually evoked potentials. While the Johns Hopkins patents licensed to us may present a significant impediment to any competitors selling in the US or European markets, they expire in 2018. If the patent application does not issue as a patent, the license agreement will expire in 2018 with the expiration of the last patent covered by the license. If a patent issues on the patent application, the agreement will terminate with the expiration of any patent issuing on that application, which likely will be in 2030. The license provides a maximum royalty of 3% of net sales. The royalty rate is reduced by other royalties paid on the same product, and is also reduced by our meeting certain volume targets. The license can be terminated by Second Sight for any reason. It may be terminated by Johns Hopkins University for breach not corrected within 60 days of written notice.
18
Doheny Eye Institute
The DEI license agreement includes 67 patents and applications for patents which are all co-owned by Second Sight and DEI. The patents cover a wide range of technologies, most of which relate to the Argus II System. As a co-owner of the patents and patent applications, we have the non-exclusive right to use the technology with or without the license agreement. The license agreement pertains to DEI’s share in the patents and patent applications which gives Second Sight an exclusive license. The agreement continues until the last of the patents expire and involves ongoing joint research. We expect that the agreement will continue until the end of 2031 which is the current expiration date of the last patent based on a patent application filed under the agreement. The agreement provides for a 0.5% royalty of net sales. The license may be terminated by breach not corrected within 30 days of written notice, or on insolvency of the licensee.
Grants
We and our partners have been successful at securing a number of grants from the US federal government. These grants support our research and development, are non-dilutive to our equity and do not need to be repaid. The government, however, retains ‘March-In’ rights in connection with these grants - a non-exclusive right to practice inventions developed from grant funding. Grants received by us to-date include:
• R24 EY012893 – Development/Testing of Artificial Retinas for the Blind, in the amount of $13,197,584,
• R01 EY012893 – Research/Development of Artificial Retinas for the Blind, in the amount of $12,917,718,
• RC3 EY020778 – Development and Testing of Low Vision Assessment Tools for Retinal Prosthesis, Robert Greenberg in the amount of $2,988,224, and
•
R41 NS058244 – Hermetic Nanowire
Interconnects for Neural Prostheses, in the amount of $459,172.
Source: http://projectreporter.nih.gov/
In September 2014, we entered into a Joint Research and Development Agreement or JRDA with The Johns Hopkins University Applied Physics Laboratory or APL. The JRDA awarded us a subcontract to conduct applied research under a grant received by APL from the Mann Fund. Under the JRDA, we have agreed to perform applied research regarding integration of APL research into a visual prosthesis system. In October 2014 APL paid us $4.075 million in one lump sum to conduct our portion of the research. The JRDA includes a field limited exclusive license from APL to us for the life of any patents resulting from APL’s portion of the research. Under the JRDA we have agreed to collaborate with APL over a 36 month period to develop an improved video processing system that will enhance a next generation visual prosthesis. The APL portion of the research includes image processing hardware and software for a visual prosthesis. In exchange for the license, we issued 1,000 shares of our common stock to APL, have agreed to pay APL its patent prosecution costs, and to pay APL a royalty of 0.25% of net sales of licensed products. The Mann Fund was established and largely funded more than 15 years ago by Alfred E. Mann, our Chairman and largest shareholder. No assurance can be given that the outcome of this research and development will prove successful.
We also intend to apply for future grants to help offset research and development costs.
Our Manufacturing and Quality Assurance
We have a single manufacturing facility, located at our principal office in Sylmar, California. The manufacturing areas at this location are housed in a single building, and include approximately 10,000 square feet of controlled environment rooms (CERs) suitable for implant manufacturing. At present less than half of this space is being used for Argus II implant production. At the same site are spaces for assembling the external (non-implantable) components of our system and for the labeling, receiving and shipping, and stockroom functions. Finished goods are held at this location and at our contracted fulfillment partner in Europe.
We rely on many suppliers to provide materials and services necessary to produce and test our products. Many of these materials or services are currently provided by sole source suppliers. In a number of instances we maintain sole source suppliers because our current purchasing volumes do not warrant developing more than one supplier. We expect to secure additional providers as our production volumes increase. If we experience a loss of a sole supplier before confirming an alternative, we risk possible disruptions in our operations. We attempt to mitigate the sole source risk, by among other things, increasing parts
19
inventory as a partial hedge against interruptions in parts supply and by actively seeking to develop alternative suppler sources before experiencing any such disruptions.
As of December 31, 2014 our manufacturing department had 42 employees and the quality assurance department had an additional nine members. We operate a day shift and smaller swing shift, and at this staffing level we can manufacture up to 10 devices per month. To support future growth of the sales of the Argus II System, we believe that the space available at the current facility when fully utilized and operating at two full shifts will prove sufficient to build and assemble approximately 100 devices per month.
Employees
As of December 31, 2014, we had 121 employees, including approximately 51 in operations; 17 in selling, marketing and distribution; 37 in clinical and regulatory and research and development; and 16 in administration. Of these employees, we employed 102 in the United States and 19 in Europe. We believe that the continued success of our business will depend, in part, on our ability to attract and retain qualified personnel, and we are committed to developing our people and providing them with opportunities to contribute to our growth and success. None of these employees is covered by a collective bargaining agreement, and we believe our relationship with our employees is good to excellent.
Available Information
Our website address is www.secondsight.com. We make available free of charge through a link provided at such website our Forms 10-K, 10-Q and 8-K as well as any amendments thereto. Such reports are available as soon as reasonably practicable after they are filed with the Securities and Exchange Commission.
20
The statements that are not historical facts contained in this Form 10-K are forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. These statements reflect the current belief, expectations or intent of our management and are subject to and involve certain risks and uncertainties. Many of these risks and uncertainties are outside of our control and are difficult for us to forecast or mitigate. An investment in our common stock is speculative and involves a high degree of risk. In addition to the risks described elsewhere in this Form 10-K and in certain of our other filings with the US Securities and Exchange Commission, the following important factors, among others, could cause our actual results to differ materially from those expressed or implied by us in any forward-looking statements contained herein or made elsewhere by or on behalf of us. The risks described below are not the only risks we face. If any of the events described in the following risk factors actually occurs, or if additional risks and uncertainties later materialize, that are not presently known to us or that we currently deem immaterial, then our business, prospects, results of operations and financial condition could be materially adversely affected. In that event, the trading price of our common stock could decline, and you may lose all or part of your investment in our shares.
Risks Related to Our Dependence on the Argus II System
We depend on the success of our first commercial product, the Argus II System, which received European market clearance (CE Mark) in February 2011 and FDA approval in February 2013, in the United States for RP; and on the regulatory approval of our current product and a new device under development, the Orion I visual prosthesis (a modified version of the Argus II System), to treat other diseases causing blindness, in the US and other countries, which may never occur.
Our future success depends upon building a commercial operation in the US and expanding growth in Europe as well as entering additional markets to commercialize our Argus II System for both RP and AMD. We believe our expanded growth will depend on the further development, regulatory approval and commercialization of the Orion I product, which we anticipate can be used by nearly all profoundly blind individuals. If we fail to expand the use of the Argus II System in a timely manner for other forms of retinal degeneration in addition to RP, or to develop the Orion I product and penetrate the available markets which those applications are intended to serve, we may not be able to expand our markets or to grow our revenue, our stock values could decline and investors may lose money.
Our revenue from sales of Argus II System is dependent upon the pricing and reimbursement guidelines adopted in each country and if pricing and reimbursement levels are inadequate to achieve profitability our operations will suffer.
Our financial success is dependent on our ability to price our products in a manner acceptable to government and private payers while still maintaining our profit margins. Numerous factors that may be beyond our control may ultimately impact our pricing of Argus II System and determine whether we are able to obtain reimbursement or reimbursement at adequate levels from governmental programs and private insurance. If we are unable to obtain reimbursement or our product is not adequately reimbursed, we will experience reduced sales, our revenues likely will be adversely affected, and we may not become profitable.
Obtaining reimbursement approvals is time consuming, requires substantial management attention, and is expensive. Our business will be materially adversely affected if we do not receive approval for reimbursement of the Argus II System under government programs and from private insurers on a timely or satisfactory basis. Limitations on coverage could also be imposed at the local Medicare Administrative Contractor level or by fiscal intermediaries in the US and by regional, or national funding agencies in Europe. Our business could be materially adversely affected if the Medicare program, local Medicare Administrative Contractors or fiscal intermediaries were to make such a determination and deny, restrict or limit the reimbursement of Argus II System. Similarly in Europe these governmental and other agencies could deny, restrict or limit the reimbursement of Argus II System at the hospital, regional or national level. Our business also could be adversely affected if retinal specialists and the facilities within which they operate are not adequately reimbursed by Medicare and other funding agencies for the cost of the procedure in which they implant the Argus II System on a basis satisfactory to the administering retinal specialists and their facilities. If the local contractors that administer the Medicare program and other funding agencies are slow to reimburse retinal specialists or provider facilities for the Argus II System, the retinal specialists may delay their payments to us, which would adversely affect our working capital requirements. If the funding agencies delay reimbursement payments to the hospitals, any increase to their working capital requirements could reduce their willingness to treat blind patients who wish to have our devices implanted. If reimbursement for our products is unavailable, limited in scope or amount, or if pricing is set at unsatisfactory levels, our business will be materially harmed.
Our commercial and financial success depends on the Argus II System being accepted in the market, and if not achieved will result in our not being able to generate revenues to support our operations.
Even if we are able to obtain favorable reimbursement within the markets that we serve, commercial success of our products will depend, among other things, on their acceptance by retinal specialists, ophthalmologists, general practitioners,
21
low vision therapists and mobility experts, hospital purchasing and controlling departments, patients, and other members of the medical community. The degree of market acceptance of any of our product candidates will depend on factors that include:
• cost of treatment,
• pricing and availability of future alternative products,
• the extent of available third-party coverage or reimbursement,
• perceived efficacy of the Argus II System relative to other future products and medical solutions, and
• prevalence and severity of adverse side effects associated with treatment.
The activities of competitive medical device companies, or others, may limit the Argus II System’s revenue.
Our commercial opportunities for the Argus II System may be reduced if our competitors develop or market products that are more effective, are better tolerated, receive better reimbursement terms, are more accepted by physicians, have better distribution channels, or are less costly.
Currently, to our knowledge, no other medical devices comparable to the Argus II System have been approved by regulatory agencies, both in the US and Europe, to restore some functional vision in persons who have become blind due to RP. Other visual prosthesis companies such as Retina Implant AG and Pixium Vision S.A., both based in Europe, are developing retinal implant technologies to partially restore some vision in blind patients. Retina Implant has obtained a CE mark for its Alpha IMS product but has not yet sold it to our knowledge, and to our knowledge neither Retina Implant nor Pixium has filed for market approval with the FDA, nor to our knowledge has either company obtained an Investigational Device Exemption to begin the required clinical trials in the US. These competitive therapies if or when developed or brought to market may result in pricing and market access pressure even if Argus II System is otherwise viewed as a preferable therapy.
Many privately and publicly funded universities and other organizations are engaged in research and development of potentially competitive products and therapies, such as stem cell and gene therapies, some of which may target RP and other indications as our product candidates. These organizations include pharmaceutical companies, biotechnology companies, public and private universities, hospital centers, government agencies and research organizations. Our competitors include large and small medical device and biotechnology companies that may have significant access to capital resources, competitive product pipelines, substantial research and development staffs and facilities, and substantial experience in medical device development.
We may face substantial competition in the future and may not be able to keep pace with the rapid technological changes which may result from others discovering, developing or commercializing products before or more successfully than we do.
In general the development and commercialization of new medical devices is highly competitive and is characterized by extensive research and development and rapid technological change. Our customers consider many factors including product reliability, clinical outcomes, product availability, inventory consignment, price and product services provided by the manufacturer. Market share can shift as a result of technological innovation and other business factors. We believe these risk factors are partially mitigated by the Argus II System being the sole product that is currently available for commercial implantation in the US and Europe. Major shifts in industry market share have occurred in connection with product problems, physician advisories and safety alerts, reflecting the importance of product quality in the medical device industry, and any quality problems with our processes, goods and services could harm our reputation for producing high-quality products and would erode our competitive advantage, sales and market share. Our competitors may develop products or other novel technologies that are more effective, safer or less costly than any that we are developing and if those products gain market acceptance our revenue and financial results could be adversely affected.
If we fail to develop new products or enhance existing products, our leadership in the markets we serve could erode, and our business, financial condition and results of operations may be adversely affected.
22
Risks Related to Our Business and Industry
We have incurred operating losses since inception and may continue to incur losses for the foreseeable future.
We have had a history of operating losses and we expect that operating losses will continue into the near term. Although we have had sales of the Argus II product, these limited sales have not been sufficient to cover our operating expenses. Our ability to generate positive cash flow will also hinge on our ability to correctly price our product to our markets, expand the use of the Argus II System, develop the Orion I visual prosthesis and obtain government and private insurance reimbursement. As of December 31, 2014 we have total stockholders’ equity of $34,618,247 and an accumulated deficit of $(152,663,968). We cannot assure you that we will be profitable even if we successfully commercialize our products. Failure to become and remain profitable may adversely affect the market price of our common stock and our ability to raise capital and continue operations.
Our business is subject to international economic, political and other risks that could negatively affect our results of operations or financial position.
We derive a significant portion of our revenues from Europe, and we anticipate that revenue from Europe and other countries outside the US will increase. Accordingly, our operations are subject to risks associated with doing business internationally, including
• currency exchange variations,
• extended collection timelines for accounts receivable,
• greater working capital requirements,
• multiple legal frameworks and unexpected changes in legal and regulatory requirements,
• the need to ensure compliance with the numerous regulatory and legal requirements applicable to our business in each of these jurisdictions and to maintain an effective compliance program to ensure compliance with these requirements,
• political changes in the foreign governments impacting health policy and trade,
• tariffs, export restrictions, trade barriers and other regulatory or contractual limitations that could impact our ability to sell or develop our products in certain foreign markets,
• trade laws and business practices favoring local competition,
• adverse economic conditions, including the stability and solvency of business financial markets, financial institutions and sovereign nations and the healthcare expenditure of domestic or foreign nations.
The realization of any of these or other risks associated with operating in Europe or other non-U.S. countries could have a material adverse effect on our business, results of operations or financial condition.
We are subject to stringent domestic and foreign medical device regulation and any unfavorable regulatory action may materially and adversely affect our financial condition and business operations.
Our products, development activities and manufacturing processes are subject to extensive and rigorous regulation by numerous government agencies, including the FDA and comparable foreign agencies. To varying degrees, each of these agencies monitors and enforces our compliance with laws and regulations governing the development, testing, manufacturing, labeling, marketing, distribution, and the safety and effectiveness of our medical devices. The process of obtaining marketing approval or clearance from the FDA and comparable foreign bodies for new products, or for enhancements, expansion of the indications or modifications to existing products, could:
• take a significant, indeterminate amount of time,
• result in product shortages due to regulatory delays,
• require the expenditure of substantial resources,
23
• involve rigorous pre-clinical and clinical testing, and possibly post-market surveillance,
• involve modifications, repairs or replacements of our products,
• require design changes of our products,
• result in limitations on the indicated uses of our products, and
• result in our never being granted the regulatory approval we seek.
Any of these occurrences that we might experience will cause our operations to suffer, harm our competitive standing and result in further losses that adversely affect our financial condition.
We have ongoing responsibilities under FDA and international regulations, both before and after a product is commercially released. For example, we are required to comply with the FDA’s Quality System Regulation (QSR), which mandates that manufacturers of medical devices adhere to certain quality assurance requirements pertaining among other things to validation of manufacturing processes, controls for purchasing product components, and documentation practices. As another example, the Medical Device Reporting regulation requires us to provide information to the FDA whenever there is evidence that reasonably suggests that a device may have caused or contributed to a death or serious injury or, that a malfunction occurred which would be likely to cause or contribute to a death or serious injury upon recurrence. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA. If the FDA were to conclude that we are not in compliance with applicable laws or regulations, or that any of our medical devices are ineffective or pose an unreasonable health risk, the FDA could ban such medical devices, detain or seize such medical devices, order a recall, repair, replacement, or refund of such devices, or require us to notify health professionals and others that the devices present unreasonable risks of substantial harm to the public health. The FDA has been increasing its scrutiny of the medical device industry and the government is expected to continue to scrutinize the industry closely with inspections and possibly enforcement actions by the FDA or other agencies. Additionally, the FDA may restrict manufacturing and impose other operating restrictions, enjoin and restrain certain violations of applicable law pertaining to medical devices and assess civil or criminal penalties against our officers, employees, or us. Any adverse regulatory action, depending on its magnitude, may restrict us from effectively manufacturing, marketing and selling our products. In addition, negative publicity and product liability claims resulting from any adverse regulatory action could have a material adverse effect on our financial condition and results of operations.
The number of preclinical and clinical tests that will be required for regulatory approval varies depending on the disease or condition to be treated, the jurisdiction in which we are seeking approval and the regulations applicable to that particular medical device. Regulatory agencies, including those in the US, Canada, Europe and other countries where medical devices are regulated, can delay, limit or deny approval of a product for many reasons. For example,
• a medical device may not be safe or effective,
• regulatory agencies may interpret data from preclinical and clinical testing differently than we do,
• regulatory agencies may not approve our manufacturing processes,
• regulatory agencies may conclude that our device does not meet quality standards for durability, long-term reliability, biocompatibility, electromagnetic compatibility, electrical safety, and
• regulatory agencies may change their approval policies or adopt new regulations.
The FDA may make requests or suggestions regarding conduct of our clinical trials, resulting in an increased risk of difficulties or delays in obtaining regulatory approval in the US. Any of these occurrences could prove materially harmful to our operations and business.
We are also subject to stringent government regulation in European and other foreign countries, which could delay or prevent our ability to sell our products in those jurisdictions.
We intend to pursue market authorizations for the Argus II System and other product candidates in additional jurisdictions. For us to market our products in Europe and some other international jurisdictions, we and our distributors and agents must
24
obtain required regulatory registrations or approvals. The approval procedure varies among countries and jurisdictions and can involve additional testing and the time and costs required to obtain approval may differ from that required to obtain an approval by the FDA. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or jurisdictions or by the FDA. Violations of foreign laws governing use of medical devices may lead to actions against us by the FDA as well as by foreign authorities. We must also comply with extensive regulations regarding safety, efficacy and quality in those jurisdictions. We may not be able to obtain all the required regulatory registrations or approvals, or we may be required to incur significant costs in obtaining or maintaining any regulatory registrations or approvals we receive. Delays in obtaining any registrations or approvals required for marketing our products, failure to receive these registrations or approvals, or future loss of previously obtained registrations or approvals would limit our ability to sell our products internationally. For example, international regulatory bodies have adopted various regulations governing product standards, packaging requirements, labeling requirements, import restrictions, tariff regulations, duties and tax requirements. These regulations vary from country to country. In order to sell our products in Europe, we must maintain our ISO 13485:2003 certification and CE mark certification, which is an international symbol of quality and compliance with applicable European medical device directives. Failure to maintain the ISO 13485:2003 certification or CE mark certification or other international regulatory approvals would prevent us from selling in some countries in Europe and elsewhere. The failure to obtain these approvals could harm our business materially.
Even if we obtain clearance or approval to sell our products, we are subject to ongoing requirements and inspections that could lead to the restriction, suspension or revocation of our clearance.
We, as well as any potential collaborative partners such as distributors, will be required to adhere to applicable FDA regulations regarding good manufacturing practice, which include testing, control, and documentation requirements. We are subject to similar regulations in foreign countries. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Ongoing compliance with good manufacturing practice and other applicable regulatory requirements is strictly enforced in the United States through periodic inspections by state and federal agencies, including the FDA, and in international jurisdictions by comparable agencies. Failure to comply with these regulatory requirements could result in, among other things, warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure to obtain premarket clearance or premarket approval for devices, withdrawal of approvals previously obtained, and criminal prosecution. The restriction, suspension or revocation of regulatory approvals or any other failure to comply with regulatory requirements would limit our ability to operate and could increase our costs.
The CE marking regulations are subject to a significant effort to strengthen the regulatory regime for medical devices which, if adopted, will make clearance process more time consuming and costly for us to obtain access to and continue to market within the European markets.
We are subject to an annual audit of compliance with the rules necessary to support our CE Mark. In 2012 the European Commission proposed a new regulatory scheme which is likely to come into effect in 2015 or 2016. It is anticipated that that the proposals which are currently being discussed by the Council of the European Union, will impose significant additional obligations on medical device companies. The Council of the European Union expects that these proposals will be adopted by the end of 2014 or early 2015. and if so the new regulations on medical devices would likely become effective by 2016. Devices with a current CE marking may have to comply with additional, more challenging regulatory obligations, the details of which are not yet clarified. We expect changes being made to regulations will include stricter requirements for clinical evidence and pre-market assessment of safety and performance, new classifications to indicate risk levels, requirements for third party testing by government accredited groups for some types of medical devices, and tightened and streamlined quality management system assessment procedures. Additionally we anticipate that the new regulations will require clinical evidence as well as analytical performance levels, the details of which are yet to be provided. If the additional provisions proposed by the European Parliament are adopted, this could lead to the involvement of the European Medicines Agency (EMA) in regulation of some types of medical devices, in the qualification and monitoring of notified bodies (NBs), and enhancing the roles of other bodies, including a new Medical Devices Coordination Group (MDCG). The European Parliament’s proposed revisions would impose enhanced competence requirements for NBs and “special notified bodies” (SNBs) for specific categories of devices, such as implantable devices. This could result in stricter conformity assessment procedures. Although the extent of the new regulations is currently uncertain the medical device industry anticipates that there will be significant changes under these initiatives to the regulation of medical devices which will increase the time and costs for obtaining CE marking.
25
We have no large scale manufacturing experience, which could limit our growth.
Our limited manufacturing experience may not enable us to make products in the volumes that would be necessary for us to achieve a significant amount of commercial sales. Our product involves new and technologically complex materials and processes and we currently experience low yields on our manufacturing process. As we move from making small quantities of our product for clinical trials to larger quantities for commercial distribution, we must develop new manufacturing techniques and processes that allow us to scale production. We may not be able to establish and maintain reliable, efficient, full scale manufacturing at commercially reasonable costs in a timely fashion. Difficulties we encounter in manufacturing scale-up, or our failure to implement and maintain our manufacturing facilities in accordance with good manufacturing practice regulations, international quality standards or other regulatory requirements, could result in a delay or termination of production. To date, our manufacturing activities have largely been to provide units for clinical testing and limited initial sales of the Argus II System. We may face substantial difficulties in establishing and maintaining manufacturing for our products at a larger commercial scale and those difficulties may impact the quality of our products and adversely affect our ability to increase sales.
Materials necessary to manufacture Argus II may not be available on commercially reasonable terms, or at all, which may delay development, manufacturing and commercialization of our products.
We rely on numerous suppliers to provide materials, components and services necessary to produce the Argus II System and next generation product candidates. Certain suppliers are currently sole source because of our low manufacturing volumes and our need for specialty technical or other engineering expertise. Our suppliers may be unable or unwilling to deliver these materials and services to us timely as needed or on commercially reasonable terms. Should this occur, we would seek to qualify alternative suppliers or develop in-house manufacturing capability, but may be unable to do so. Substantial design or manufacturing process modifications and regulatory approval might be required to facilitate or qualify an alternate supplier. Even where we could qualify alternative suppliers the substitution of suppliers may be at a higher cost and cause time delays including delays associated with additional possible FDA review, that impede the commercial production of the Argus II System, reduce gross profit margins and impact our abilities to deliver our products as may be timely required to meet demand.
Any failure or delay in completing clinical trials or studies for new product candidates or next generation of the Argus II System and the expense of those trials could adversely affect our business.
Preclinical studies and clinical trials required to demonstrate the safety and efficacy of incremental changes and obtain indication expansion for the next generation of the Argus II System and for new product candidates are time consuming and expensive. If we are required to conduct additional clinical trials or other studies with respect to any of our product candidates beyond those that we have contemplated, if we are unable to successfully complete our clinical trials or other studies or if the results of these trials or studies are not positive or are only modestly positive, we may be delayed in obtaining marketing approval for those product candidates, we may not be able to obtain marketing approval or we may obtain approval for indications that are not as broad as intended. Our product development costs also will increase if we experience delays in testing or approvals.
The completion of clinical trials for our product candidates could be delayed because of our inability to manufacture or obtain from third-parties materials sufficient for use in preclinical studies and clinical trials; delays in patient enrollment and variability in the number and types of patients available for clinical trials; difficulty in maintaining contact with patients after treatment, resulting in incomplete data; poor effectiveness of product candidates during clinical trials; unforeseen safety issues or side effects; and governmental or regulatory delays and changes in regulatory requirements and guidelines.
If we incur significant delays in our clinical trials, our competitors may be able to bring their products to market before we do which could result in harming our ability to commercialize our products or potential products. If we experience any of these occurrences our business will be materially harmed.
To establish our sales and marketing infrastructure, we will need to grow the size of our organization, and we may experience delays or other difficulties in managing this growth.
As our development and commercialization plans and strategies evolve, we will need to expand the size of our employee base for managerial, operational, sales, marketing, financial and other resources. Future growth would impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, motivate and integrate additional employees. Our management team may have to use a substantial amount of time to manage these growth activities. Our future
26
financial performance and our ability to commercialize the Argus II System and our other product candidates and compete effectively will depend, in part, on our ability timely and effectively to manage any future growth and related costs. We may not be able to effectively manage a rapid pace of growth and timely implement improvements to our management infrastructure and control systems.
We may acquire additional businesses or form strategic alliances in the future, and we may not realize the benefits of such acquisitions or alliances.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third-parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may have difficulty in developing, manufacturing and marketing the products of a newly acquired company that enhances the performance of our combined businesses or product lines to realize value from expected synergies. We cannot assure that, following an acquisition, we will achieve the revenues or specific net income that justifies the acquisition.
If we lose key management personnel, or if we fail to recruit additional highly skilled personnel, our ability to identify, develop and commercialize new or next generation product candidates will be impaired, could result in loss of markets or market share and could make us less competitive.
We are highly dependent upon Robert J. Greenberg M.D., Ph.D., our President and Chief Executive Officer, and are also dependent on other members of our senior management. Our executives have significant ophthalmic, regulatory industry, sales and marketing, operational, and/or corporate finance experience. The loss of any management executive or any other principal member of our management team could impair our ability to identify, develop and market new products or effectively deal with regulatory and reimbursement matters.
Our ability to utilize and benefit from our net operating loss carryforwards and certain other tax attributes may be limited.
As of December 31, 2014, we had federal and state of California income tax net operating loss carryforwards, which may be applied to future taxable income, of approximately $107,346,000 and $101,807,000, respectively. To the extent that we continue to generate taxable losses, unused losses will carry forward to offset future taxable income, if any, until these unused losses expire. However, we may be unable to use these losses to offset taxable income before our unused losses expire at various dates that range from 2023 through 2034 for federal net operating losses and from 2015 through 2034 for state net operating losses. Under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, if a corporation undergoes an “ownership change,” generally defined as a greater than 50 percentage point change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating loss, or NOL, carryforwards to offset its post‑change taxable income may be limited. Limitations may also apply to the utilization of other pre-change tax attributes as a result of an ownership change. We have experienced ownership changes in the past. We may experience additional ownership changes in connection with our recent IPO and in the future as a result of shifts in our stock ownership, including shifts in our stock ownership that are outside of our control. As a result, our ability to use our pre-change NOL carryforwards to offset taxable income may be subject to limitations. In addition, there may be periods during which the use of NOL carryforwards is suspended or otherwise limited under state tax law. For these reasons, we may not be able to utilize and benefit from a material portion of our NOL carryforwards and other tax attributes.
We could be adversely affected by violations of the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws.
The U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business. We have adopted policies for compliance with these anti-bribery laws, which often carry substantial penalties. We cannot assure you that our internal control policies and procedures always will protect us from reckless or other inappropriate acts committed by our affiliates, employees or agents. Violations of these laws, or allegations of such violations, could have a material adverse effect on our business, financial position and results of operations and could cause the market value of our common stock to decline.
27
Risks Related to Intellectual Property and Other Legal Matters
If we or our licensors are unable to protect our/their intellectual property, then our financial condition, results of operations and the value of our technology and products could be adversely affected.
Patents and other proprietary rights are essential to our business and our ability to compete effectively with other companies is dependent upon the proprietary nature of our technologies. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop, maintain and strengthen our competitive position. We seek to protect these, in part, through confidentiality agreements with certain employees, consultants and other parties. Our success will depend in part on the ability of our licensors to obtain, maintain (including making periodic filings and payments) and enforce patent protection for their intellectual property, in particular, those patents to which we have secured exclusive rights. Our licensors may not successfully prosecute or continue to prosecute the patent applications which we have licensed. Even if patents are issued in respect of these patent applications, we or our licensors may fail to maintain these patents, may determine not to pursue litigation against entities that are infringing upon these patents, or may pursue such enforcement less aggressively than we ordinarily would. Without adequate protection for the intellectual property that we own or license, other companies might be able to offer substantially identical products for sale, which could unfavorably affect our competitive business position and harm our business prospects.
Even if issued, patents may be challenged, invalidated, or circumvented, which could limit our ability to stop competitors from marketing similar products or limit the length of term of patent protection that we may have for our products.
Litigation or third-party claims of intellectual property infringement or challenges to the validity of our patents would require us to use resources to protect our technology and may prevent or delay our development, regulatory approval or commercialization of improvements in the Argus II System or new product candidates. Further, the validity of some of our patents has been challenged.
Pixium Vision has filed oppositions in the European Patent Office (EPO) challenging the validity of nine European patents owned or exclusively licensed by Second Sight. Retina Implant has joined in one of the nine oppositions. Two of the patents are owned by Johns Hopkins University (JHU) and exclusively licensed to Second Sight. Seven of the patents are owned by Second Sight. Second Sight was successful in the Opposition Division in the two JHU cases. However, in the Appellate Division one of the JHU patents was preserved and one of JHU patents was invalidated. In the third proceeding Pixium was successful in the Opposition Division, and we have appealed. In the fourth case we were successful in the Opposition Division. The fifth case is scheduled for hearing in the Opposition Division on April 23, 2015. The sixth and seventh of these opposition cases have not reached a hearing in the opposition division. We have opposed one Pixium patent. The patent was successfully opposed and significantly narrowed. The eighth and ninth cases were filed in recent months and await initial action. These challenges to our patent portfolio, if successful, may affect our ability to block competitors from utilizing this particular intellectual property, but in our view have no material effect on our ability to make and sell the Argus II System or otherwise have any material effect upon us. Of the nine patents contested, two have reached final resolution with no further appeal available within in the EPO. Of the seven remaining patents, none apply to our current product. To our current knowledge, none apply to any competitive product. These patents represent possible improvements that we, or a competitor, may wish to use in the future. Remaining at issue are seven out of over 300 patents we have to protect our technology. These EPO proceedings involving us and Pixium include:
• EP 1061874 Visual Prosthesis — upheld by the opposition and appellate divisions. No further appeal is available in the EPO.
• EP 1061996 Apparatus for Preferential Outer Retinal Stimulation — upheld by the opposition division, lost in the appellate division. No further appeal is available in the EPO.
• EP 1171188 Retinal Color Prosthesis for Color Sight Restoration — successfully opposed in the opposition division, pending before the Appellate Division.
• EP2219728 Electrode Array for Even Neural Pressure Having Multiple Attachment Points — successfully upheld in the Opposition Division.
• EP1937352 Sub-threshold Stimulation to Precondition Neurons for Supra-threshold Stimulation — successfully opposed in the Opposition Division, pending appeal.
28
• EP2192949 — Return Electrode for a Flexible Circuit Electrode Array - opposition and response filed, pending hearing April 23, 2015 in Munich Germany.
• EP1949437 — Implantable Microelectronic Device and Method of Manufacture — opposition filed.
• EP1945835 — Platinum Electrode Surface Coating and Method for Manufacturing the Same — opposition filed.
• EP2061549 — Package for an Implantable Neural Stimulation Device — opposition filed.
• EP1986733 (Pixium) — Device with Flexible Multilayer System for Contacting or Electro-stimulation of Living Tissue Cells or Nerves — successfully opposed and significantly narrowed.
If we are the target of claims by third parties asserting that our products or intellectual property infringe upon the rights of others we may be forced to incur substantial expenses or divert substantial employee resources from our business and, if successful, those claims could result in our having to pay substantial damages or prevent us from developing one or more product candidates. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
If we experience patent infringement claims, or if we elect to avoid potential claims others may be able to assert, we or our collaborators may choose to seek, or be required to seek, a license from the third-party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. This could harm our business significantly. The cost to us of any litigation or other proceeding, regardless of its merit, even if resolved in our favor, could be substantial. Some of our competitors may be able to bear the costs of such litigation or proceedings more effectively than we can because of their having greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Intellectual property litigation and other proceedings may, regardless of their merit, also absorb significant management time and employee resources.
If we fail to comply with our obligations in the agreements under which we license development or commercialization rights to products or technology from third-parties, we could lose license rights that are important to our business.
We hold exclusive licenses from Johns Hopkins University, Duke University, and the Doheny Eye Institute to intellectual property relating to the Argus II visual prosthesis. These licenses impose various commercialization, milestone payment, profit sharing, insurance and other obligations on us. If we fail to comply with any material obligations, the licensor will have the right to terminate the applicable license, which covers part of the system of the eye implant and thus will be a barrier to manufacture the Argus II System and impair our ability to sell the Argus II. The existing or future patents to which we have rights based on our agreements with Johns Hopkins University, Duke University and the Doheny Eye Institute may be too narrow to prevent third-parties from developing or designing around these patents. Additionally, we may lose our rights to the patents and patent applications we license in the event of a breach or termination of the license agreement. Each license expires with the expiration of the last of the licensed patents. In the case of JHU, the license will expire March 13, 2018, unless extended by additional patents. While the JHU agreement includes a patent which is a significant obstacle to our competitors, it is one of many other patents which in our view present material obstacles to our competitors. The DEI license includes ongoing research, making the expiration date indeterminate, but in any event the expiration date is no earlier than August 8, 2033. The total aggregate royalty on both agreements does not exceed 3.25% of Argus II System net sales. All of the patents in the DEI agreement are co-owned with the Doheny Eye Institute. We license the Doheny Eye Institute’s interest in the patents to maintain our exclusive use on that intellectual property. Should the license terminate we retain the right to utilize the intellectual property, but may not be able to prevent others from doing so, in which case we may lose a competitive advantage.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patented technology, we rely upon, among other things, unpatented proprietary technology, processes, trade secrets and know-how. Any involuntary disclosure to or misappropriation by third-parties of our confidential or proprietary
29
information could enable competitors to duplicate or surpass our technological achievements, potentially eroding our competitive position in our market. We seek to protect confidential or proprietary information in part by confidentiality agreements with our employees, consultants and third-parties. While we require all of our employees, consultants, advisors and any third-parties who have access to our proprietary know-how, information and technology to enter into confidentiality agreements, we cannot be certain that this know-how, information and technology will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. These agreements may be terminated or breached, and we may not have adequate remedies for any such termination or breach. Furthermore, these agreements may not provide meaningful protection for our trade secrets and know-how in the event of unauthorized use or disclosure. To the extent that any of our staff was previously employed by other pharmaceutical, medical technology or biotechnology companies, those employers may allege violations of trade secrets and other similar claims in relation to their medical device development activities for us.
If we are unable to protect the intellectual property used in our products, others may be able to copy our innovations which may impair our ability to compete effectively in our markets.
The strength of our patents involves complex legal and scientific questions and can be uncertain. As of March 1, 2015 we have 308 issued patents and 172 pending patent applications on a worldwide basis. Our patent applications may be challenged or fail to result in issued patents and our existing or future patents may be too narrow to prevent third-parties from developing or designing around our intellectual property and in that event we may lose competitive advantage and our business may suffer.
Further, the patent applications that we license or have filed may fail to result in issued patents. The claims may need to be amended. Even after amendment, a patent may not issue and in that event we may not obtain the exclusive use of the intellectual property that we seek and may lose competitive advantage which could result in harm to our business.
Third-party claims of intellectual property infringement may prevent or delay our commercialization efforts for Argus II and our development and commercialization activities for other product candidates.
Although we are not currently aware of any litigation or other proceedings or third-party claims of intellectual property infringement related to the Argus II System, the medical device industry is characterized by many litigation cases regarding patents and other intellectual property rights. Other parties may in the future allege that our activities infringe their patents or that we are employing their proprietary technology without authorization. We may not have identified all the patents, patent applications or published literature that affect our business either by blocking our ability to commercialize our product, by preventing the patentability of one or more aspects of our products or those of our licensors or by covering the same or similar technologies that may affect our ability to market our product.
In addition, even in the absence of litigation, we may need to obtain licenses from third-parties to advance our research or allow commercialization of our product candidates, and we have done so from time to time. We may fail to obtain future licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be unable to further develop and commercialize one or more of our product candidates, which could harm our business significantly.
We may become involved in future lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may file infringement claims, which can be expensive and time consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours or of our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
The US Patent and Trademark Office may initiate interference proceedings to determine the priority of inventions described in or otherwise affecting our patents and patent applications or those of our collaborators or licensors. An unfavorable outcome could require us to cease using the technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if a prevailing party does not offer us a license on terms that are acceptable to us. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distraction of our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our proprietary rights, particularly in countries where the laws may not protect those rights as fully as in the US.
30
Product liability lawsuits could divert our resources, result in substantial liabilities and reduce the commercial potential of our products.
We face a risk of product liability claims arising from the prosthesis being inserted into the eye, and it is possible that we may be held liable for eye injuries of patients who receive our product. These lawsuits may divert our management from pursuing our business strategy and may be costly to defend. In addition, if we are held liable in any of these lawsuits, we may incur substantial liabilities and may be forced to limit or forego further commercialization of one or more of our products. We maintain product liability insurance that covers our clinical trials and commercial sales, our aggregate coverage limit under these insurance policies for an amount of $5,000,000, and while we believe this amount of insurance currently is sufficient to cover our product liability exposure, these limits may not prove adequate to fully cover potential liabilities. In addition, we may not be able to obtain or maintain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims, which could prevent or inhibit the commercial production and sale of our products. If the use of our products harm or are alleged to harm people, we may be subject to costly and damaging product liability claims that exceed our policy limits and cause us significant losses that could seriously harm our financial condition or reputation.
CE Marking does not absolve us from strict conformity with all applicable European Union legislation and member state regulation where the product is offered and if we do not adhere to these directive and regulations we may incur fines and other penalties that will prevent or delay market penetration of our products.
The CE (European Conformity) marking is a symbol that manufacturers affix to products to indicate that a product conforms to all relevant EU rules and regulations and that the manufacturer has performed all necessary evaluation procedures. Although the CE mark allows manufacturers to place products on the market and permits free movement of goods, it is not a mark of approval by the EU. The manufacturer and its authorized representative in EU are responsible for all aspects of the product assessment, testing, documentation, declaration of conformity and CE marking, even where a formal processing agent, the notified body, is required, as in the case of non-European based manufacturers. In all cases the manufacturer and representative assume the full responsibility and liability even when using the services of a consultant or test laboratory. Liability is not transferrable to third parties, including the notified body which is required for processing the certification. Generally, there is strict liability applied to medical devices subject to the CE marking by directive 85/374/EEC, and testing and reporting does not change or reduce this liability.
Legislative or regulatory reform of the health care system in the US and foreign jurisdictions may adversely impact our business, operations or financial results.
Our industry is highly regulated and changes in law may adversely impact our business, operations or financial results. In March 2010, the Patient Protection and Affordable Care Act, or PPACA, and a related reconciliation bill were signed into law. This legislation changes the current system of healthcare insurance and benefits intended to broaden coverage and control costs. The law also contains provisions that will affect companies in the medical device industry and other healthcare related industries by imposing additional costs and changes to business practices.
Moreover, in some foreign countries, including countries in Europe and Canada, the pricing of approved medical devices is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take 12 months or longer after the receipt of regulatory approval and product launch. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. Our business could be materially harmed if reimbursement of our products is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels.
We cannot predict what healthcare reform initiatives may be adopted in the future. Further federal and state legislative and regulatory developments are likely, and we expect ongoing initiatives in the U.S and Europe. These reforms could have an adverse effect on our ability to obtain timely regulatory approval for new products and on anticipated revenues from the Argus II System and other product candidates, both of which may affect our overall financial condition.
We may incur significant increased costs as a result of operating as a public company, and our management will be required to devote substantial time to compliance requirements.
As a public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. The Sarbanes-Oxley Act of 2002, as well as rules subsequently implemented by the SEC and NASDAQ, have imposed various requirements on public companies, including requiring establishment and maintenance of effective disclosure
31
and financial controls and changes in corporate governance practices. Our management and other personnel will be required to devote a substantial amount of time to these new compliance requirements. Moreover, these rules and regulations will substantially increase our legal and financial compliance costs and will make some activities more time consuming and costly. These rules and regulations will make it more difficult and more expensive for us to maintain our existing director and officer liability insurance or to obtain similar coverage from an alternative provider.
We are an “emerging growth company,” and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
For so long as we remain an “emerging growth company” as defined in the JOBS Act, we may take advantage of certain exemptions from various requirements that are applicable to public companies that are not “emerging growth companies,” including not being required to comply with the independent auditor attestation requirements of Section 404 of the Sarbanes‑Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We may take advantage of these exemptions for so long as we are an “emerging growth company,” which could be as long as five years following the completion of our IPO. Investors may find our common stock less attractive because we rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock, and our stock price may be more volatile or may decline.
In addition, Section 107 of the JOBS Act also provides that an “emerging growth company” can take advantage of an extended transition period for complying with new or revised accounting standards. However, we chose to “opt out” of this extended transition period, and as a result, we intend to comply with new or revised accounting standards on the relevant dates that adoption of those standards may be required for non-emerging growth companies. Our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
We will be required to evaluate our internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act of 2002, and any adverse results from such evaluation could result in a loss of investor confidence in our financial reports and have an adverse effect on our stock price.
Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, we will be required to furnish a report by our management on our internal control over financial reporting the year following our first annual report required to be filed with the SEC. The report will contain, among other matters, an assessment of the effectiveness of our internal control over financial reporting as of the end of our fiscal year, including a statement as to whether or not our internal control over financial reporting is effective. This assessment must include disclosure of any material weaknesses in our internal control over financial reporting identified by management. If we are unable to assert that our internal control over financial reporting is effective, we could lose investor confidence in the accuracy and completeness of our financial reports, which could have an adverse effect on our stock price.
Risks Relating to Our Financial Results and Need for Additional Financing
Fluctuations in our quarterly operating results and cash flows could adversely affect the price of our common stock.
The revenues we generate and our operating results will be affected by numerous factors such as:
• the commercial success of the Argus II System,
• our ability to obtain regulatory approval of the Argus II System in additional jurisdictions,
• our ability to obtain regulatory approval of the Argus II System for treatment of AMD,
• the emergence of products that compete with our product candidates,
• the status of our preclinical and clinical development programs,
• variations in the level of expenses related to our existing product candidates or preclinical and clinical development programs,
• execution of collaborative, licensing or other arrangements, and the timing of payments received or made under those arrangements,
32
• any intellectual property infringement lawsuits to which we may become a party,
• regulatory developments affecting our product candidates or those of our competitors, and
• our ability to obtain reimbursement from government or private payers.
If our quarterly operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Any quarterly fluctuations in our operating results and cash flows may cause the price of our stock to fluctuate substantially. We believe that, in the near term, quarterly comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
We will need additional capital to support our growth. Additional capital, may be difficult to obtain restricting our operations and resulting in additional dilution to our stockholders.
We raised net proceeds of approximately $34.2 million from the IPO we completed in November 2014. Our business will require additional capital for implementation of our long term business plan. We believe our cash, cash equivalents and other investments will be sufficient to fund our operations over approximately the next 18 to 24 months. However, the actual amount of funds that we will need for our business development will be determined by many factors, some of which are beyond our control, and we may need funds sooner than currently anticipated. These factors include:
• the amount of our future operating losses,
• third party expenses relating to the commercialization of the Argus II System,
• the need and cost of conducting additional clinical trials of the Argus II System for other applications,
• the amount of our research and development, including research and development for Orion I visual prosthesis, marketing and general and administrative expenses, and
• regulatory changes and technological developments in our markets.
As we require additional funds, we may seek to fund our operations through the sale of equity securities, additional debt financing and strategic collaboration agreements. We cannot be sure that additional financing from any of these sources will be available when needed or that, if available, the additional financing will be obtained on terms favorable to us or our stockholders. If we raise additional funds by selling shares of our capital stock, the ownership interest of our current stockholders will be diluted. If we are unable to obtain additional funds on a timely basis or on terms favorable to us, we may be required to cease or reduce further commercialization of the Argus II System, to cease or reduce certain research and development projects, to sell some or all of our technology or assets or business units or to merge all or a portion of our business with another entity.
Risks Related to the Securities Market, and Ownership of Our Common Stock
The price of our common stock may be volatile and the value of your investment could decline.
Medical technology stocks have historically experienced high levels of volatility. The trading price of our common stock may fluctuate substantially. The market price of our common stock may be higher or lower than the price you pay, depending on many factors, some of which are beyond our control and may not be related to our operating performance. These fluctuations could cause you to lose substantially all or part of your investment in our common stock. Factors that could cause fluctuations in the trading price of our common stock include:
• announcements of new offerings, products, services, therapies, treatments or technologies, commercial relationships, acquisitions or other events by us or our competitors,
• challenges to our patents and the patents underlying the patents and intellectual property that we license,
• United States and European approvals or denials of our products,
• price and volume fluctuations in the overall stock market from time to time,
33
• significant volatility in the market price and trading volume of technology companies in general,
• fluctuations in the trading volume of our shares or the size of our public float,
• actual or anticipated changes or fluctuations in our results of operations,
• whether our results of operations meet the expectations of securities analysts or investors,
• actual or anticipated changes in the expectations of investors or securities analysts,
• litigation involving us, our industry, or both,
• regulatory developments in the United States, foreign countries, or both,
• general economic conditions and trends,
• major catastrophic events,
• lockup releases, sales of large blocks of our common stock,
• departures of key employees, or
• an adverse impact on the company from any of the other risks cited herein.
In addition, if the market for medical technology stocks or the stock market, in general, experiences a loss of investor confidence, the trading price of our common stock could decline for reasons unrelated to our business, results of operations or financial condition. The trading price of our common stock might also decline in reaction to events that affect other companies in our industry even if these events do not directly affect us. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been brought against that company. If our stock price is volatile, we may become the target of securities litigation. Securities litigation could result in substantial costs and divert our management’s attention and resources from our business. This could have a material adverse effect on our business, results of operations and financial condition.
Sales of substantial amounts of our common stock in the public markets, including sales after the “lock-up” period, or the perception that sales might occur, could reduce the price of our common stock and may dilute your voting power and ownership interest in us.
Sales of a substantial number of shares of our common stock in the public market, or the perception that these sales could occur, could adversely affect the market price of our common stock and may make it more difficult for you to sell your common stock at a time and price that you deem appropriate.
Subject to certain exceptions, our directors, officers and our stockholders beneficially owning 10% or more of our common stock and certain of our consultants have agreed not to offer, sell or agree to sell, directly or indirectly, any shares of common stock without the permission of MDB Capital Group, our IPO underwriter, for a period of 12 months ending November 18, 2015. Certain of our executives, other employees and consultants have agreed to similar lock-up agreements for a period of six months from the date of the IPO. When these lockup periods expire, the locked-up security holders will be able to sell shares in the public market. In addition, the underwriter may, in its sole discretion, release all or some portion of the shares subject to lock-up agreements prior to the expiration of the applicable lock-up period. Our prospectus dated November 18, 2014 disclosed that our chief executive officer may sell up to 100,000 shares of common stock from February 15 through April 15, 2015 to cover his personal tax liability and we currently expect he will do so after March 16, 2015 and prior to April 15, 2015. Sales of a substantial number of such shares by directors, officers or employees, or the perception that such sales may occur, or early release of the lock-up, could cause our share price to fall or make it more difficult for you to sell your common stock at a time and price that you deem appropriate.
Holders of up to approximately 22,898,690 shares of our common stock, and holders of up to 805,000 shares of common stock underlying the underwriter’s warrant, will have rights, subject to some conditions, to require us to file a registration statement covering the sale of their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. We also intend to register the offer and sale of all shares of common stock that we may issue under our equity compensation plans.
34
Certain of our stockholders have the ability to control the outcome of matters submitted for stockholder approval and may have interests that differ from those of our other stockholders.
As of December 31, 2014 our executive officers, key employees, directors and their affiliates beneficially own in the aggregate approximately 65.8% of the outstanding shares of our common stock. As a result, these stockholders, if acting together, may be able to exercise significant influence over all matters requiring stockholder approval, including the election of directors and the approval of significant corporate transactions. They may also have interests that differ from yours and may vote in a manner that is adverse to your interests. This concentration of voting power may have the effect of deterring, delaying or impeding actions that could be beneficial to you, including actions that may be supported by our board of directors, and deprive our shareholders of an opportunity to receive a premium for their common stock as part of sale of our company and might ultimately affect the market price of our common stock.
We have broad discretion in the use of proceeds we obtained in November 2014 from our IPO and may allocate the net proceeds from the IPO in ways with which you may not agree, and if we do not use those proceeds effectively your investment could be harmed.
We obtained net proceeds of approximately $34.2 million from the IPO we completed in November 2014. We intend to use the proceeds of the IPO to expand our sales and marketing efforts, enhance our current product, gain new marketing approvals, and continue research into next generation technology including the Orion I, as well as for working capital and general corporate purposes. Our management will have broad discretion over the specific use of the net proceeds that we received in the offering. Our stockholders may not have an opportunity to evaluate the economic, financial or other information on which we base our decisions on how to use our proceeds and will need to rely upon the judgment of our management with respect to the use of proceeds. As a result, our stockholders may not agree with our decisions. If we do not use the net proceeds that we received in the offering effectively, our business, results of operations and financial condition could be harmed.
Holders of common stock who purchased shares in the IPO, but who do not register and continuously hold shares in their name for two years will lose the Long Term Investor Right and opportunity to receive additional shares from us if those Long Term Investor Rights are triggered.
We granted each purchaser of shares in the IPO the Long Term Investor Right to receive for no additional investment or payment up to one additional share for each share purchased in the offering if the requirements have been met including:
• the purchaser registers those shares in its name, either in certificate or book entry form, within 90 days following the closing date of the IPO,
• the purchaser continuously holds those shares in its name until the second anniversary date of the closing date of the IPO, and
• the price per share of our common stock does not trade at $18.00, which is 200% of the offering price, or greater for any five consecutive trading days during the two year period after the closing date of this offering.
If the holder of the shares has not timely registered the shares purchased in the IPO and does not hold the shares continuously for the two years after the closing date of the offering, or if the price per share of our common stock trades at 200% of the offering price or greater for any five consecutive trading days during the two year period after the closing date of the offering, the Long Term Investor Right will terminate and the holder will lose the opportunity to benefit from receipt of shares if this Long Term Investor Right is triggered.
Holders who have perfected their LTIRs and are keeping those shares registered in their names, may experience delays in their ability to dispose of those shares which could cause partial or full loss of their investment in the event of a rapid decline in our share price.
Our shares are susceptible to financial market volatility and other financial and business related risks that can cause the value of our shares to decline drastically within a short period of time. Holders who have perfected their LTIRs and maintain those shares registered in their names may experience delays in their ability timely to dispose of those shares which can result in partial or full loss of their investment.
35
If the Long Term Investor Right is triggered we will become obligated to deliver additional shares according to a formula limited to no more than one share for each share acquired in this offering which may still lead to partial or substantial loss of investment for those holders who have perfected their LTIRs.
If the Long Term Investor Right is triggered for the delivery of up to one share for each share held by a holder who has perfected and retained his LTIR on the two years anniversary of the closing date of our IPO, holders cannot be assured that they will recover their investment or avoid incurring a loss when the additional shares under the Long Term Investor Right are delivered to you. In the event the Long Term Investor Right is triggered our other stockholders will incur dilution and may experience a decline in the value of their shares.
We do not intend to pay dividends for the foreseeable future and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our common stock.
We have never declared or paid any dividends on our common stock. We intend to retain any earnings to finance the operation and expansion of our business, and we do not anticipate paying any cash dividends in the future. As a result, you may only receive a return on your investment in our common stock if the market price of our common stock increases.
Future sales and issuances of our equity securities or rights to purchase our equity securities, including pursuant to our equity incentive plans, would result in dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
To the extent we raise additional capital by issuing equity securities; our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to existing stockholders.
The public market for our common stock has been volatile since completion of our initial public offering. This volatility may affect the ability of our investors to sell their shares as well as the price at which they sell their shares.
We completed our initial public offering in November 2014. Since that time, our shares have traded from $8.05 per share to $24.02 per share and day-to-day trading often has been volatile. This volatility may continue or increase in the future. The market price for the shares may be significantly affected by factors such as progress in the development of our technology, progress in our pre-clinical and clinical trials, agreements with research facilities or co-development partners, commercialization of our technology, coverage by third party payers, variations in quarterly and yearly operating results, general trends in the medical device industry, and changes in FDA and foreign regulations affecting us and our industry. Furthermore, in recent years the stock market has experienced extreme price and volume fluctuations that are unrelated or disproportionate to the operating performance of the affected companies. Those broad market fluctuations may adversely affect the market price of our common stock.
Substantial future sales of shares of our common stock in the public market could cause our stock price to fall.
If our common stockholders (including those persons who may become common stockholders upon exercise of our options or warrants) sell substantial amounts of our common stock, or the public market perceives that stockholders might sell substantial amounts of our common stock, the market price of our common stock could decline significantly. Such sales also might make it more difficult for us to sell equity or equity-related securities in the future at a time and price that our management deems appropriate.
We have the right to issue shares of preferred stock. If we were to issue preferred stock, it is likely to have rights, preferences and privileges that may adversely affect the common stock.
We are authorized to issue 10,000,000 shares of “blank check” preferred stock, with such rights, preferences and privileges as may be determined from time-to-time by our board of directors. Our board of directors is empowered, without stockholder approval, to issue preferred stock in one or more series, and to fix for any series the dividend rights, dissolution or liquidation preferences, redemption prices, conversion rights, voting rights, and other rights, preferences and privileges for the preferred stock. No shares of preferred stock are presently issued and outstanding and we have no immediate plans to issue shares of preferred stock. The issuance of shares of preferred stock, depending on the rights, preferences and privileges attributable to the preferred stock, could adversely reduce the voting rights and powers of the common stock and the portion of our assets allocated for distribution to common stockholders in a liquidation event, and could also result in dilution in the book value per share of our common stock. The preferred stock could also be utilized, under certain circumstances, as a method for raising additional capital or discouraging, delaying or preventing a change in control of our company, to the detriment of the holders of our common stock. We cannot assure you that we will not, under certain circumstances, issue shares of our preferred stock.
36
Item 1B. Unresolved Staff Comments
Not applicable.
Our principal office and facilities are located at 12744 San Fernando Road, Building 3, Sylmar, California 91342, and consists of approximately 45,351 rentable square feet at a base rent of $30,883 per month. Our lease expires in February 2022 and grants us an option to extend the lease term for an additional 60 months period. We originally rented these premises from Mann Biomedical Park LLC, an entity affiliated with our Chairman of the Board, Alfred E. Mann. We believe that the terms of this lease are at least as favorable as those that may have been obtained from a non-affiliated third party. We believe that these premises are adequate for our foreseeable needs. In November 2014, the industrial center in which these premises are located was sold to an independent third party.
Our European office is located on the Innovation Park at EPFL, Rue Jean Daniel Colladon, CH 1015 Lausanne. The lease consists of 180 square meters at a base rent of 7,079 CHF per month, or currently about $7,100 per month. Our lease is currently monthly with a six month notice required for termination, with the Foundation for the Innovation Park at EPFL.
We are not a party to any pending legal proceedings other than those involving Pixium Vision described in “Risk Factors—Risks Related to Intellectual Property and Other Legal Matters”.
Item 5. Market for Registrant’s Common Equity, Related Shareholder Matters and Issuer Purchases of Equity Securities
(a) Market Price, Dividends and Related Matters
Second Sight’s common stock is traded on the NASDAQ Capital Market under the symbol “EYES.” The following table sets forth the high and low closing sales prices of our common stock as reported on the NASDAQ Capital Market for the following time periods.
|
|
|
High |
|
|
Low |
|
||
|
2014 |
|
|
|
|
|
|
|
|
|
|
$ |
23.60 |
|
|
$ |
10.15 |
|
|
On March 13, 2015, the closing sales price reported for our common stock was $14.28 per share, and as of that date there were approximately 239 shareholders of record.
We have never declared or paid cash dividends on our common stock and do not anticipate paying any dividends in the foreseeable future.
This chart compares the cumulative total stockholder return on our common stock with that of the NASDAQ Composite index and the NASDAQ Medical Equipment index. The chart assumes $100 was invested at the close of the market on November 19, 2014 (the date our common stock first commenced trading on NASDAQ) in our common stock, the NASDAQ Composite index and the NASDAQ Medical Equipment index. The comparison assumes reinvestment of dividends. The comparisons shown in the graph below are based upon historical data. We caution that the stock price performance shown in the graph below is not necessarily indicative of, nor is it intended to forecast, the potential future performance of our common stock.
37

|
|
11/19/14 |
|
|
11/26/14 |
|
|
12/3/14 |
|
|
12/10/14 |
|
|
12/17/14 |
|
|
12/24/14 |
|
|
12/31/14 |
|
|||||||
Second Sight Medical Products, Inc. |
|
$ |
100.00 |
|
|
$ |
86.63 |
|
|
$ |
73.66 |
|
|
$ |
74.81 |
|
|
$ |
66.15 |
|
|
$ |
55.13 |
|
|
$ |
51.38 |
|
NASDAQ Composite Index |
|
$ |
100.00 |
|
|
$ |
102.42 |
|
|
$ |
102.15 |
|
|
$ |
100.23 |
|
|
$ |
99.39 |
|
|
$ |
102.18 |
|
|
$ |
101.39 |
|
NASDAQ Medical Equipment Index |
|
$ |
100.00 |
|
|
$ |
102.53 |
|
|
$ |
103.59 |
|
|
$ |
101.85 |
|
|
$ |
101.77 |
|
|
$ |
104.03 |
|
|
$ |
102.89 |
|
Use of Proceeds from Initial Public Offering
On November 18, 2014, we sold 4,025,000 shares of common stock in our IPO, including 525,000 shares sold upon exercise of the underwriter's over-allotment option pursuant to a registration statement (File No. 333-198073) that we initially filed with the Securities and Exchange Commission in August 2014. Our net proceeds totaled approximately $34.2 million, after offering costs of approximately $5.0 million, including approximately $2.9 million in fair value of warrants and shares of common stock issued in connection with the underwriting and other services rendered for the IPO. In addition to funding our ongoing business operations, we expect to invest the proceeds of the IPO in our business to expand sales and marketing efforts, enhance our current Argus II product, gain regulatory approvals for additional indications, and continue research and development into next generation technology. Through December 31, 2014, the net proceeds from the offering were used as follows: approximately $30.5 million was deposited into various cash and money market funds, and approximately $3.7 million was used to fund ongoing business operations. None of the proceeds were used for construction of plant, building and facilities, the purchase of real estate, or the acquisition of any business.
Item 6. Selected Financial Data
The following selected consolidated financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the notes to those consolidated financial statements. The consolidated statement of operations data set forth below for the years ended December 31, 2014, 2013and 2012 and the consolidated balance sheet data as of December 31, 2014 and 2013 are derived from, and are qualified in their entirety by reference to, the Company’s audited consolidated financial statements included elsewhere in this Form 10-K. The consolidated balance sheet data as of December 31, 2012 is derived from the audited consolidated financial statements not included herein, but which were previously filed with the Securities and Exchange Commission.
38
|
|
|
Fiscal Years Ended December 31, |
|
|||||||||
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
Net sales |
|
$ |
3,397,852 |
|
|
$ |
1,564,933 |
|
|
$ |
1,367,224 |
|
|
Cost of sales |
|
|
3,558,135 |
|
|
|
5,629,320 |
|
|
|
4,396,746 |
|
|
Gross loss |
|
|
(160,283 |
) |
|
|
(4,064,387 |
) |
|
|
(3,029,522 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5,040,969 |
|
|
|
3,248,466 |
|
|
|
3,045,157 |
|
|
|
|
|
2,621,919 |
|
|
|
3,215,290 |
|
|
|
3,726,556 |
|
|
|
|
|
6,844,825 |
|
|
|
3,301,452 |
|
|
|
2,194,590 |
|
|
|
|
|
6,565,464 |
|
|
|
4,167,934 |
|
|
|
4,025,558 |
|
|
|
Total operating expenses |
|
|
21,073,177 |
|
|
|
13,933,142 |
|
|
|
12,991,861 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations |
|
|
(21,233,460 |
) |
|
|
(17,997,529 |
) |
|
|
(16,021,383 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
9,108 |
|
|
|
7,454 |
|
|
|
7,512 |
|
|
Other income, net |
|
|
11,805 |
|
|
|
34,768 |
|
|
|
1,775 |
|
|
Interest expense on convertible promissory notes and loan payable |
|
|
(1,956,555 |
) |
|
|
(1,588,687 |
) |
|
|
(138,934 |
) |
|
Amortization of discount on convertible promissory notes |
|
|
(5,077,535 |
) |
|
|
( 3,424,931 |
) |
|
|
(128,097 |
) |
|
Write-off of unamortized discount on conversion of convertible promissory notes |
|
|
(6,954,610 |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(35,201,247 |
) |
|
$ |
(22,968,925 |
) |
|
$ |
(16,279,127 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share – Basic and diluted |
|
$ |
(1.41 |
) |
|
$ |
(1.02 |
) |
|
$ |
(0.74 |
) |
|
Weighted average shares outstanding – Basic and diluted |
|
|
25,052,806 |
|
|
|
22,521,432 |
|
|
|
21,945,580 |
|
|
|
|
As of December 31, |
|
|||||||||
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
Cash |
|
$ |
619,411 |
|
|
$ |
62,565 |
|
|
$ |
144,754 |
|
|
Money market funds |
|
$ |
33,999,563 |
|
|
$ |
8,611,614 |
|
|
$ |
4,310,038 |
|
|
Working capital |
|
$ |
33,524,991 |
|
|
$ |
9,104,436 |
|
|
$ |
4,275,975 |
|
|
Total assets |
|
$ |
43,069,444 |
|
|
$ |
12,673,421 |
|
|
$ |
7,992,575 |
|
|
Convertible promissory notes payable |
|
$ |
— |
|
|
$ |
19,211,112 |
|
|
$ |
8,273,356 |
|
|
Stockholders’ equity (deficiency) |
|
$ |
34,618,247 |
|
|
$ |
(9,221,071 |
) |
|
$ |
(3,043,823 |
) |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of many factors. The consolidated results of operations for the years ended December 31, 2014, 2013 and 2012 are not necessarily indicative of the results that may be expected for any future period. The following discussion should be read in conjunction with the consolidated financial statements and the notes thereto included in Part IV, Item 15 of this Form 10-K and in conjunction with the “Risk Factors” included in Part I, Item 1A of this Form 10-K.
Business Overview
We were founded in 1998 with a mission to develop, manufacture, and market prosthetic devices that restore vision to the blind. Our principal offices are located in Sylmar, California, approximately 25 miles northwest of downtown Los Angeles. We also have an office in Lausanne, Switzerland, that manages our commercial and clinical operations in Europe and the Middle East.
39
Our first commercial product, the Argus II System, is a retinal prosthesis that can provide some functional vision to individuals blinded by retinitis pigmentosa (RP). The Argus II System is an implantable neurostimulation device that uses electrical stimulation of the retina (based on a wireless video camera feed) to replace the function of the defunct photo-receptors in RP patients.
Our major corporate, clinical and regulatory milestones include:
• In 1998, we were founded.
• In 2002, we commenced clinical trials for our prototype product, the Argus I retinal prosthesis.
• In 2006, we commenced clinical trials for the Argus II System, which later became our first commercial product.
• In 2011, we received marketing approval in Europe (CE Mark) for the Argus II System.
• In 2013, we received marketing approval in the United States (FDA) for the Argus II System.
• In 2014, we completed our initial public offering and began trading on NASDAQ under the symbol “EYES.”
We began selling the Argus II System in Europe at the end of 2011, in Saudi Arabia in 2012, and in the United States and Canada in 2014. We have limited regulatory approval in Canada and Saudi Arabia, and we are currently applying for full approval. To date, all of our sales have been made by our direct sales force, but we plan to add partners and distributors to enhance our coverage of existing and future markets. In 2014, we entered into our first distribution agreements, that cover the countries of Spain and Turkey, and we are at various stages of negotiations with a number of other distributors for countries in Europe and the Middle East.
We have achieved certain insurance reimbursement milestones in the United States (Medicare Transitional Pass Through Payment, New Technology Add-on Payment, and coverage by a number of insurers/payers), but reimbursement hurdles remain as not every payer is covering this technology. In Europe, we have achieved government reimbursement in Germany and have received a positive reimbursement decision in France, and additional reimbursement is being sought in a number of other countries. Obtaining reimbursement from governmental or private insurance companies is critical to our future commercial success. Due to the cost of the Argus II System, our sales will be limited without the availability of third party reimbursement.
Plan of Operation
We currently market and sell our products in the United States, Europe and Saudi Arabia. Over the next two years, we intend to use approximately $2.0 to $4.0 million of the proceeds from our IPO to expand our sales and marketing organizations in these existing markets to increase sales coverage, market penetration and revenue in these markets. Over the next 12 to 18 months, we intend to introduce the Argus II System in additional countries through our direct sales force or by working with partners and distributors.
Over the next two years, we intend to use approximately $4.0 million of the proceeds from our IPO on development and clinical efforts to enhance the external hardware and software of our Argus II System, which could improve the resolution and other performance characteristics of the system. Increasing the resolution of the system may enhance the user experience and increase our potential market size. Image resolution may be achieved by enhanced image processing, including contrast enhancement and electronic zooming. In addition, we believe that, through software enhancements, we may be able to create a number of virtual electrodes between the physical electrodes on the current retinal implant. This could potentially enhance the resolution of existing devices by ten-fold or more.
Currently, our Argus II System is approved for persons suffering from RP. We believe we can expand the market for the Argus II System beyond RP to patients with severe to profound vision loss due to age-related macular degeneration or AMD. We intend to use approximately $2.0 million of the proceeds from our IPO to conduct a pilot study, of about five patients to evaluate the safety and benefit of the Argus II System for use in persons suffering from AMD. Recruitment for this study began in December 2014 and we plan to begin implanting in this study in the first half of 2015. If results from this study are promising, we anticipate beginning a larger scale efficacy trial in 2016 that could lead to marketing approval for the Argus II system for AMD patients in 2019. We estimate that the cost to complete this additional trial would be approximately $4.5 million. If the Argus II System is successfully developed and approved for sale to treat AMD, as to which there can be no assurances, we believe that the potential addressable market opportunity for that device will significantly exceed our existing RP markets for the Argus II System.
40
We also plan to also use approximately $5.0 million of the proceeds from our IPO to conduct preclinical development of a product for cortical stimulation that we refer to as the Orion I visual cortical prosthesis, which we expect will be able to provide some vision restoration to individuals with almost all unpreventable forms of blindness. Our objective in designing and developing the Orion I visual cortical prosthesis is to bypass the retina and optic nerve and to directly stimulate the visual cortex region of the brain. Human clinical testing is likely to take the form of a feasibility study followed by a premarket approval pivotal trial. The details of these trials will be determined collaboratively with the FDA at that time. We cannot accurately estimate the timing or exact cost of these trials at this time. If the Orion I visual cortical prosthesis is successfully developed and approved for sale, as to which there can be no assurances, we believe that the potential addressable market opportunity for that device will greatly exceed our existing RP markets for the Argus II System.
The amounts that we actually spend for any specific purpose may vary significantly and will depend on a number of factors, including, but not limited to, the pace of progress of our commercialization and development efforts, actual needs with respect to research and development, clinical testing, regulatory approval, market conditions, insurance reimbursement, and changes in or revisions to our product, sales and marketing strategies. Investors will be relying on the judgment of our management regarding the application of the proceeds from the sale of our common stock.
Recent Accounting Pronouncements
In August 2014, the FASB issued Accounting Standards Update No. 2014-15 (ASU 2014-15), Presentation of Financial Statements — Going Concern (Subtopic 205-10). ASU 2014-15 provides guidance as to management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. In connection with preparing financial statements for each annual and interim reporting period, an entity’s management should evaluate whether there are conditions or events, considered in the aggregate, that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued (or within one year after the date that the financial statements are available to be issued when applicable). Management’s evaluation should be based on relevant conditions and events that are known and reasonably knowable at the date that the financial statements are issued (or at the date that the financial statements are available to be issued when applicable). Substantial doubt about an entity’s ability to continue as a going concern exists when relevant conditions and events, considered in the aggregate, indicate that it is probable that the entity will be unable to meet its obligations as they become due within one year after the date that the financial statements are issued (or available to be issued). ASU 2014-15 is effective for the annual period ending after December 15, 2016, and for annual periods and interim periods thereafter. Early application is permitted. The Company is currently evaluating the impact the adoption of ASU 2014-15 on the Company’s financial statement presentation and disclosures.
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2014-09 (ASU 2014-09), Revenue from Contracts with Customers. ASU 2014-09 will eliminate transaction- and industry-specific revenue recognition guidance under current U.S. GAAP and replace it with a principle based approach for determining revenue recognition. ASU 2014-09 will require that companies recognize revenue based on the value of transferred goods or services as they occur in the contract. The ASU also will require additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. ASU 2014-09 is effective for reporting periods beginning after December 15, 2016, and early adoption is not permitted. Entities can transition to the standard either retrospectively or as a cumulative-effect adjustment as of the date of adoption. Management is currently assessing the impact the adoption of ASU 2014-09 and has not determined the effect of the standard on the Company’s ongoing financial reporting.
In April 2014, the FASB issued Accounting Standards Update No. 2014-08 (ASU 2014-08), Presentation of Financial Statements (Topic 205) and Property, Plant and Equipment (Topic 360). ASU 2014-08 amends the requirements for reporting discontinued operations and requires additional disclosures about discontinued operations. Under the new guidance, only disposals representing a strategic shift in operations or that have a major effect on the Company’s operations and financial results should be presented as discontinued operations. This new accounting guidance is effective for annual periods beginning after December 15, 2014. The Company is currently evaluating the impact of adopting ASU 2014-08 on the Company’s results of operations or financial condition.
Management does not believe that any other recently issued, but not yet effective, authoritative guidance, if currently adopted, would have a material impact on the Company’s financial statement presentation or disclosures.
41
Critical Accounting Policies and Estimates
The following discussion and analysis of financial condition and results of operations is based upon our consolidated financial statements, which have been prepared in conformity with accounting principles generally accepted in the United States of America. Certain accounting policies and estimates are particularly important to the understanding of our financial position and results of operations and require the application of significant judgment by our management or can be materially affected by changes from period to period in economic factors or conditions that are outside of our control. As a result, they are subject to an inherent degree of uncertainty. In applying these policies, our management uses their judgment to determine the appropriate assumptions to be used in the determination of certain estimates. Those estimates are based on our historical operations, our future business plans and projected financial results, the terms of existing contracts, our observance of trends in the industry, information provided by our customers and information available from other outside sources, as appropriate. See Note 2 to our consolidated financial statements for a more complete description of our significant accounting policies.
Revenue Recognition. Our revenue is derived primarily from the sale of our Argus II System, which is implanted during a surgery, and intended to provide some functional vision to patients blinded by retinitis pigmentosa (RP). We sell to university hospitals, teaching hospitals, large medical centers, and ambulatory surgical centers. We recognize revenue when four basic criteria are met: (1) persuasive evidence of an arrangement exists; (2) surgical implantation has occurred; (3) the price is fixed or determinable; and (4) collectability is reasonably assured. We generally use customer purchase orders or purchase agreements to determine the existence of an arrangement. Sales transactions are based on prices that are determinable at the time we accept the customer’s purchase order. In order to determine whether collection is reasonably assured, we assess a number of factors, including creditworthiness of the customer and medical insurance coverage. If we determine that collection is not reasonably assured, we will defer the recognition of revenue until collection becomes reasonably assured, which is generally upon receipt of payment. We may periodically grant special terms, such as extended payment terms. We defer revenues when these special terms are granted until a final price is fixed and collection becomes reasonably assured. Due to the nature of our revenue recognition policy of recording revenue only after surgical implantation, we have had no returns related to Argus II System recorded as revenue.
Stock-Based Compensation. Pursuant to Financial Accounting Standards Board (“FASB”) ASC 718 Share-Based Payment (“ASC 718”), the Company records stock-based compensation expense for all stock-based awards.
Under ASC 718, the Company estimates the fair value of stock options granted using the Black-Scholes option pricing model. The fair value for awards that are expected to vest is then amortized on a straight-line basis over the requisite service period of the award, which is generally the option vesting term.
• The grant price of the issuances, with certain exceptions, is determined based on the estimated fair value of the shares at the date of grant.
• The risk free interest rate for periods within the contractual life of the option is based on the U.S. treasury yield in effect at the time of grant.
• As permitted by SAB 107, due to the Company’s insufficient history of option activity, management utilizes the simplified approach to estimate the options expected term, which represents the period of time that options granted are expected to be outstanding.
• Volatility is determined based on average historical volatilities of comparable companies in similar industry.
• Expected dividend yield is based on current yield at the grant date or the average dividend yield over the historical period. The Company has never declared or paid dividends and has no plans to do so in the foreseeable future.
Patent Costs. The Company has over 300 domestic and foreign patents. Due to the uncertainty associated with the successful development of one or more commercially viable products based on Company’s research efforts and any related patent applications, all patent costs, including patent-related legal, filing fees and other costs, including internally generated costs, are expensed as incurred. Patent costs are included in general and administrative expenses in the consolidated statements of operations.
Convertible Promissory Notes and Warrants. The warrants and embedded beneficial conversion feature of convertible promissory notes are classified as equity under FASB ASC Topic 815-40 “Derivatives and Hedging — Contracts in Entity’s
42
Own Equity”. The Company allocates the proceeds of the convertible promissory notes between convertible promissory notes and the financial instruments related to warrants associated with convertible promissory notes based on their relative fair values at the commitment date. The fair value of the financial instruments related to warrants associated with convertible promissory notes is determined utilizing the Black-Scholes option pricing model and the respective allocated proceeds to the warrants is recorded in additional paid-in capital. The Company utilized the Black-Scholes option valuation model using the same valuation assumptions as described herein for Stock Based Compensation. The embedded beneficial conversion feature associated with convertible promissory notes is recognized and measured by allocating a portion of the proceeds equal to the intrinsic value of that feature to additional paid-in capital in accordance with ASC Topic 470-20 “Debt — Debt with Conversion and Other Options.” The portion of debt discount resulting from the allocation of proceeds to the financial instruments related to warrants associated with convertible promissory notes is being amortized over the life of the convertible promissory notes. For the portion of debt discount resulting from the allocation of proceeds to the beneficial conversion feature, it is amortized over the term of the notes from the respective dates of issuance.
Long Term Investor Right. Each beneficial owner (“IPO Shareholder”) of the Company’s common stock, who purchased shares directly in the offering (“IPO Shares”), may qualify to receive up to one additional share of common stock from the Company for each share purchased in the offering (“IPO Supplemental Shares”) pursuant to the Long Term Investor Right that was included with each IPO Share. To qualify for receipt of IPO Supplemental Shares, an IPO Shareholder was required to take action to become the direct registered owner of its IPO Shares within 90 days following the closing date of the offering, or by February 22, 2015. Furthermore, IPO Shareholders are required to hold their IPO Shares in their own name and not place them in “street name” or trade them at any time during the 24 month period immediately following the IPO closing date. This Long Term Investors Right is non-detachable and transferable only in limited circumstances.
The Company will issue IPO Supplemental Shares to IPO Shareholders who have not otherwise forfeited their Long Term Investor Right if, during the two-year period immediately following the IPO closing date, the Company’s common stock does not trade at or above $18.00 per share (200% of the IPO price per share) for any five consecutive day period. If the Company’s common stock trades on its principal exchange at 200% of the IPO price per share or greater on five consecutive trading days during the two years after the IPO closing date, the Long Term Investor Right will terminate.
The formula to determine the number of IPO Supplemental Shares to be issued on a trigger of the Long Term Investor Right will be: (i) $18.00 minus (ii) the average of the highest consecutive closing prices in any 90 day trading period on the principal exchange during the two years after the Closing Date (the “Measurement Average”) divided by the Measurement Average. Fractional shares issuable to a qualifying IPO Shareholder resulting from the calculation will be rounded up to the next whole share of common stock, taking into account the aggregate number of Long Term Investor Rights of a holder. As an illustrative example, if the highest average of consecutive closing prices over any 90 calendar day period is $10.00 per share, each Long‑Term Investor Right will be entitled to 0.80 additional shares of common stock, which is calculated as: ($18.00 - $10.00)/$10.00.
The IPO offering price for purposes of the calculation of the amount of common stock to be issued on a Long Term Investor Right will be subject to adjustment in the event of a reorganization, recapitalization or split-up of the Company’s shares, the issuance of a stock dividend or any similar event. The amount of IPO Supplemental Shares, if any, to be issued will be computed by an independent public accountant as soon as practicable following the second anniversary of the Closing Date. The determination by such independent public accountant will be final and binding on the Company and on all qualifying IPO Shareholders and the Company will within about 15 days after receipt of written determination deliver to shareholders certificates evidencing the additional shares.
The Company has identified and will track IPO Investors who have perfected their Rights on a quarterly basis. At the end of each reporting period, the Company will disclose the potential dilutive effect of the Right, including the number of common shares that would be issuable on such date, based on the actual share price movements since the IPO.
The Right is an equity instrument that is accounted for as a component of the actual price per common share paid by the investor in the IPO. For basic earnings per share, the common shares associated with the Right are treated as contingently issuable shares and will not be included in basic earnings per share until the actual number of shares can be calculated and the shares have been issued.
Results of Operations
Net sales. Our net sales are derived primarily from the sale of our Argus II System. We began selling our products in Europe in 2011, Saudi Arabia in 2012, and the United States and Canada in 2014. Our objective is to increase our product
43
revenue over the next several years as we pursue commercialization of our product, as our product becomes more well-known and accepted in the market, and as insurance coverage becomes more widespread.
Cost of sales. Cost of sales includes the salaries, benefits, material, overhead, third party costs, warranty, charges for excess and obsolete inventory, and other costs required to make our Argus II System at our Sylmar, California facility. Historically, our cost of sales has been greater than our revenues, which has resulted in gross losses. However, in the third and fourth quarters of fiscal 2014, due to higher revenues and increased manufacturing output and efficiencies, we generated a positive gross margin for the first time in our operating history. Our product involves new and technologically complex materials and processes. As we move from making small quantities of our product for clinical trials to larger quantities for commercial distribution, we are developing new manufacturing techniques and processes that we expect to allow us to scale production. We are currently experiencing low yields on our manufacturing process, but we expect that over the next few years we will be able to refine our processes and improve our manufacturing yields. Accordingly, as we scale our production over the next few years, we expect that our cost per unit will decrease and we will generate a positive gross profit.
Operating Expenses. We generally recognize our operating expenses as we incur them in four general operational categories: research and development, clinical and regulatory, sales and marketing, and general and administrative. Our operating expenses also include a non-cash component related to the amortization of deferred stock-based compensation allocated to research and development, clinical and regulatory, sales and marketing and general and administrative personnel. From time to time we have received grants from institutions or agencies, such as the National Institutes of Health, to help fund the some of the cost of our development efforts. We have recorded these grants as offsets to the costs as they are incurred to complete the related work.
• Research and development expenses consist primarily of employee compensation and consulting costs related to the design, development, and enhancements of our current and potential future products, offset by grant revenue received in support of specific research projects. We expense our research and development costs as they are incurred. We expect research and development expenses to increase in the future as we pursue further enhancements of our existing product and develop technology for our potential future products, such as the Orion I visual cortical prosthesis. We also expect to receive additional grants in the future that will be offset primarily against research and development costs.
• Clinical and regulatory expenses consist primarily of salaries, travel and related expenses for personnel engaged in clinical and regulatory functions, as well as internal and external costs associated with conducting clinical trials and maintaining relationships with regulatory agencies. We expect clinical and regulatory expenses to increase substantially as we assess the safety and efficacy of enhancements to our current Argus II System, seek to expand the indications for the Argus II System, such as AMD, and prepare to initiate clinical studies of potential future products, such as the Orion I visual cortical prosthesis.
• Sales and marketing expenses consist primarily of salaries, commissions, travel and related expenses for personnel engaged in sales, marketing and business development functions, as well as costs associated with promotional and other marketing activities. We expect sales and marketing expenses to increase substantially as we hire additional sales personnel, initiate additional marketing programs, develop relationships with new distributors, and expand the number of doctors and medical centers that buy and implant our Argus II System and any future products.
• General and administrative expenses consist primarily of salaries and related expenses for executive, legal, finance, human resources, information technology and administrative personnel, as well as recruiting and professional fees, patent filing costs, insurance costs and other general corporate expenses, including rent. We expect general and administrative expenses to increase as we add personnel and incur additional costs related to the growth of our business and operation as a public company.
Interest expense on convertible promissory notes. Interest expense is a non-cash expense associated with the Company’s convertible promissory notes. Simple interest is accrued at 7.5% per annum based on the face value of the convertible promissory notes outstanding during the year. The accrued interest is added to the amount of outstanding debt, but does not earn additional interest. The terms of the convertible promissory notes provided for automatic conversion of principal and accrued interest into equity on our IPO, at $5.00 per share. Accordingly, subsequent to our IPO in the fourth quarter of 2014, the Company no longer incurred interest expense on the convertible promissory notes.
Amortization of discount on convertible promissory notes. As discussed more fully above, our convertible promissory notes issued during 2012 and 2013 were issued with detachable warrants and an embedded beneficial conversion feature,
44
which were recorded as an issuance discount with an offsetting credit to additional paid-in capital. This issuance discount is amortized as a non-cash charge over the term of the convertible promissory note. The terms of the convertible promissory notes provided for conversion into equity on an IPO, at $5.00 per share. At December 31, 2013, the unamortized issuance cost related to our convertible promissory notes was $12,032,146. As a result of our IPO in November 2014, $6,954,610 of unamortized issuance costs were charged to income due to the automatic conversion of all outstanding convertible promissory notes into common stock. Accordingly, subsequent to our IPO in the fourth quarter of 2014, the Company no longer amortized the issuance discount on the convertible promissory notes.
Comparison of the Years Ended December 31, 2014 and 2013
Net Sales. Our net sales increased from $1,564,933 in 2013 to $3,397,852 in 2014, an increase of $1,832,919, or 117%. This increase in product revenue was due to selling more implants in 2014 and as well as attaining a higher average selling price for implanted devices. We sold 29 Argus II Systems that were implanted in 2014, compared to 22 in the prior year. In 2013, all implants were in Europe and the Middle East, whereas in 2014, there were 16 implants in the United States and Canada and 13 in Europe and the Middle East. The decrease in implants in Europe and the Middle East during 2014, as compared to 2013, is primarily due to sites in Italy and Saudi Arabia, which together accounted for 10 implants in 2013, performing no implants in the first three quarters of fiscal 2014, before resuming activity with a combined 4 implants in the fourth quarter of 2014. The increase in average selling price in 2014 was primarily due to establishing a higher selling price on the introduction of the Argus II system in the United States and Canada, combined with a lower level of discounting and free goods in Europe in comparison to 2013.
Cost of sales. Cost of sales decreased from $5,629,320 in 2013 to $3,558,135 in 2014, a decrease of $2,071,185, or 37%. This decrease is primarily due to increasing our production volume and yields in 2014 relative to 2013, resulting in more finished goods and sub-assemblies being accepted into inventory and a lower level of scrapped product being expenses. As we manufacture more products, our manufacturing overhead is spread over more units and our cost per unit produced decreases. Also, as our yields improve and we accept more units into inventory, the amount of scrapped product that is written off to cost of sales decreases. We will continue to invest in improving our manufacturing processes, and we expect that manufacturing yields will improve and cost of sales will decrease relative to our revenues over the next few years, although we expect significant fluctuations on a quarter to quarter basis.
Research and development expense. Research and development expense increased from $3,248,466 in 2013 to $5,040,969 in 2014, an increase of $1,792,503, or 55%. This increase in expense is primarily related to work developing our next generation externals, which includes new eyewear and a new video processing unit. In 2014, we spent $936,222 more on salaries and other compensation costs than in 2013, and we spent $305,255 more on materials, consulting services and other supplies to make and evaluate prototypes. We expect research and development costs to increase in the future as we pursue further enhancements of our existing product and develop technology for our potential future cortical implant product.
Clinical and regulatory expense. Clinical and regulatory expense decreased from $3,215,290 in 2013 to $2,621,919 in 2014, a decrease of $593,371, or 18%. This decrease is primarily attributable to lower levels of staffing in 2014 compared to 2013. We expect clinical and regulatory costs to increase in the future as we conduct clinical trials to assess possible enhancements to our existing product, and to assess the safety and efficacy of our current product for treating blindness due to age related macular degeneration.
Selling and marketing expense. Selling and marketing expense increased from $3,301,452 in 2013 to $6,844,825 in 2014, an increase of $3,543,373, or 107%. This increase in costs is attributable to an increase in personnel, as well as higher costs for marketing and customer awareness programs, as we increased our efforts to commercialize the Argus II System as, beginning in 2014, we began selling our product in the United States, Canada and Spain. While we expect these costs to increase in the future as we increase our selling and marketing resources to accelerate the commercialization of our product, we expect selling and marketing expense to decrease over time when expressed as a percentage of product revenue.
General and administrative expense. General and administrative expense increased from $4,167,934 in 2013 to $6,565,464 in 2014, an increase of $2,397,530, or 58%. This increase is primarily attributable to $848,531 of higher stock‑based compensation charges in 2014, $823,161 in higher compensation costs, which includes $422,643 due to forgiveness of a loan receivable to an officer to finance stock options, $175,000 of expense related to a stock award to the Company’s Chairman, as well as higher spending on patent and audit related fees. The stock-based compensation charge in 2014 includes $637,694 related to option grants to our chief executive officer. After we become a public company, we expect our general and administrative costs
45
to increase as we incur the additional costs of being a public company, including higher legal, accounting, insurance, exchange listing, and other costs.
Interest expense on the convertible promissory notes. Interest expense on the convertible promissory notes increased from $1,588,687 in 2013 to $1,956,555 in 2014, an increase of $367,868, or 23%. This increase is due to the higher average level of debt outstanding during 2014 compared to 2013, although the debt was only outstanding for ten and one-half months in 2014. As a result of the Company’s IPO, effective November 18, 2014, all of the Company’s convertible promissory notes were converted into common stock. After the IPO, the Company will no longer incur interest expense on the convertible promissory notes.
Amortization of issuance discount on convertible promissory notes. Amortization of issuance discount on convertible promissory notes increased from $3,424,931 in 2013 to $5,077,535 in 2014, an increase of $1,652,604, or 48%. This increase is due to the higher average level of debt outstanding during 2014 compared to 2013, although the debt was only outstanding for ten and one-half months in 2014, and to higher value attributed to the beneficial conversion feature associated with promissory notes issued during 2013, but which was only outstanding for a part of 2013. As a result of the Company’s IPO, effective November 18, 2014, all of the Company’s convertible promissory notes were converted into common stock. After the IPO, the Company will no longer incur the amortization of issuance discount on convertible promissory notes.
Write-off of unamortized discount on conversion of convertible promissory notes. The original terms of the Company’s convertible promissory notes specified that the notes automatically converted into common stock of the Company in the event, among other things, of an IPO. Accordingly, as of the IPO date, the Company wrote off $6,954,610 of deferred issuance costs related to the convertible promissory notes that converted into common stock.
Net loss. The net loss was $22,968,925 in 2013, as compared to $35,201,247 in 2014.
Comparison of the Years Ended December 31, 2013 and 2012
Overview. During 2012 the Company completed clinical trials that led to the February 2013 FDA marketing approval for the Argus II System. In early 2013, the Company shifted resources from product development and clinical testing to increase investment in production capabilities and commercialization efforts. This shift in spending was accomplished by decreasing staffing levels in the research and development and clinical and regulatory areas during the first quarter of 2013 while increasing staffing levels in operations and sales and marketing throughout the year. For the 2013 year, total employee count decreased from 108 at January 1 to 104 at December 31, but the mix of employees shifted towards production and commercialization.
Net Sales. Our net sales increased from $1,367,224 in 2012 to $1,564,933 in 2013, an increase of $197,709, or 14%. This increase in product revenue was due to implanting 22 Argus II systems in 2013, as compared to 15 implants in the prior year, offset by a lower average selling price in 2013. Our average selling prices in 2013 declined from 2012, mainly due to reduced pricing in Europe. In some instances, we discounted our prices to introduce our product into new hospital centers, and in other situations, due to the lack of insurance reimbursement or other funding, we gave price discounts to maintain momentum at certain implant centers. We believe that as the market for our product becomes more established, and as insurance reimbursements for this new technology become more standard, pricing in Europe will stabilize. In 2012, our revenues came from Europe; in 2013, revenue came from Europe and the Middle East. Product sales did not commence in the United States and Canada until 2014. We expect smaller price variations, followed by price stabilization, as we enter new markets.
Cost of sales. Cost of sales increased from $4,396,746 in 2012 to $5,629,320 in 2013, an increase of $1,232,574, or 28%. The increase in cost of sales is primarily due to increasing our production capacity, including the addition of direct and indirect personnel to the operations staff, while still experiencing low yields and incurring higher charges related to our allowance for excess and obsolete inventory.
During the year ended December 31, 2013, we increased our allowance for excess and obsolete inventory by $1,042,621, or 188%, to $1,595,792. During 2013, we also increased our manufacturing activity substantially over 2012, which resulted in a $1,336,083, or 104%, increase in work in process inventory at year-end as compared to a year earlier. The large increase in the allowance during 2013 is due to increased production activity and costs during the year without a similar increase in production of goods that conformed to our manufacturing standards. As we increased the amount of work in process inventory and manufacturing activity during the year, we experienced a high level of limited use or unusable subassemblies, as a result of which our ending work in process inventory contained a significant amount of goods that will be discarded. We also implemented
46
design changes during the year, the effect of which was to make obsolete certain older designs, which for the most part consisted of sub-assemblies included in work in process inventory.
We anticipate that this increased allowance will have minimal impact on future operations. As these items are discarded in future periods, they will be charged against the reserve and we expect that there will be nominal impact on the Company’s operations.
We will continue to invest in improving our manufacturing processes. We expect manufacturing yields to improve and production costs to decrease over the next several years, although significant fluctuations may occur on a quarter to quarter basis.
Research and development expense. Research and development expense increased from $3,045,157 in 2012 to $3,248,466 in 2013, an increase of $203,309, or 7%. This increase in expense is primarily due to $426,690 of lower grant revenue offsets in 2013 compared to 2012, and to a higher level of stock-based compensation in 2013. Offsetting these increases, compensation costs were lower in 2013 due to a lower level of staffing. We expect research and development costs to increase in the future as we pursue further enhancements of our existing product and develop technology for our potential future cortical implant product.
Clinical and regulatory expense. Clinical and regulatory expense decreased from $3,726,556 in 2012 to $3,215,290 in 2013, a decrease of $511,266, or 14%. This decrease from 2012 to 2013 is primarily attributable to a lower level of staffing during 2013 compared to 2012, and a lower level of clinical trial activity after the Company received FDA marketing approval for the Argus II System. We expect clinical and regulatory costs will increase in the future as we conduct clinical trials to assess possible enhancements to our existing product, and assess safety and the efficacy of our current product for treating blindness due to age related macular degeneration, or AMD.
Selling and marketing expense. Selling and marketing expense increased from $2,194,590 in 2012 to $3,301,452 in 2013, an increase of $1,106,862, or 50%. This increase in costs is attributable to an increase in selling and marketing personnel, resulting in higher compensation costs, as well as higher marketing and market research related costs. While we expect these costs to increase in the future as we increase our selling and marketing resources to accelerate the commercialization of our product, we expect selling and marketing expense to decrease over time when expressed as a percentage of revenue.
General and administrative expense. General and administrative expense increased from $4,025,558 in 2012 to $4,167,934 in 2013, an increase of $142,376, or 4%. After we become a public Company, we expect these costs to increase as we incur the additional costs of being a public Company, including higher legal, accounting, insurance, exchange listing, and other costs.
Interest expense on the convertible promissory notes. Interest expense on the convertible promissory notes increased from $138,934 in 2012 to $1,588,687 in 2013, an increase of $1,449,753, or 1,043%. This increase is due to the higher average level of debt outstanding during 2013 compared to 2012.
Amortization of issuance discount on convertible promissory notes. Amortization of issuance discount on convertible promissory notes increased from $128,097 in 2012 to $3,424,931 in 2013, an increase of $3,296,834, or 2,574%. This increase is due to the higher average level of debt outstanding during 2013 compared to 2012, and to higher value attributed to the beneficial conversion feature associated with promissory notes issued in 2013. As of December 31, 2013, the unamortized issuance discount on the convertible promissory notes was $12,032,146.
Net loss. Net loss was $16,279,127 for the year ended December 31, 2012, as compared to $22,968,925 for the year ended December 31, 2013.
Liquidity and Capital Resources
On November 18, 2014, we sold 4,025,000 shares of common stock in an IPO, including 525,000 shares sold upon exercise of the underwriter’s over-allotment option, at a price of $9.00 per share. Our net proceeds totaled $34,195,637, after cash offering costs of $2,029,363, and excluding non-cash costs of $2,941,109 for the fair value of warrants and common stock issued in connections with services rendered. We expect to invest the proceeds of the IPO in our business principally to expand sales and marketing efforts, enhance current product, gain regulatory approvals for additional indications, and continue research and development into next generation technology.
In accordance with the original terms of the Company’s convertible promissory notes, the notes converted into the Company’s common stock upon the Company’s IPO. In November 2014, convertible promissory notes with a face value of $29,519,162, plus accrued interest of $3,676,377, converted into 6,639,137 shares of common stock.
47
Working capital was $33,524,991 at December 31, 2014, as compared to $9,104,436 at December 31, 2013, an increase of $24,420,555 or 268%. Working capital was $9,104,436 at December 31, 2013, as compared to $4,275,975 at December 31, 2012, an increase of $4,828,461 or 113%. We use our cash, money market funds and working capital to fund our operating activities.
Cash Flows from Operating Activities
During 2014, we used $17,092,342 of cash in operating activities, consisting primarily of a net loss of $35,201,247, offset by a non-cash charge of $6,954,610 for the write off of unamortized issuance costs related to the automatic conversion of convertible debt triggered by out IPO, and other non-cash charges of $9,556,534 for amortization of discount on convertible notes payable, non-cash interest accrued on convertible notes payable, depreciation and amortization of property and equipment, stock-based compensation, a stock grant to a related party, common stock issued for services, and common stock issuable and decreased by a net change in operating assets and liabilities of $1,597,761. This compares to 2013, when we used $17,426,862 of cash in operating activities, consisting of a net loss of $22,968,925, reduced by non-cash charges of $6,099,284 for depreciation and amortization of property and equipment, stock-based compensation, amortization of discount on convertible notes payable, and non-cash interest accrued on convertible notes payable, and increased by a net change in operating assets and liabilities of $557,221.
During 2013, we used $17,426,862 of cash in operating activities, consisting of a net loss of $22,968,925, reduced by non-cash charges of $6,099,284 for depreciation and amortization of property and equipment, stock-based compensation, amortization of discount on convertible notes payable, and non-cash interest accrued on convertible notes payable, and increased by a net change in operating assets and liabilities of $557,221. This compares to 2012, when we used $15,321,214 of cash in operating activities, consisting of a net loss of $16,279,127, reduced by non-cash charges of $1,499,605 for depreciation and amortization of property and equipment, stock-based compensation, amortization of discount on convertible notes payable, and non-cash interest accrued on convertible notes payable, and increased by a net change in operating assets and liabilities of $541,692.
Cash Flows from Investing Activities
Investing activities in 2014 used $25,948,300 of cash, reflecting $25,387,949 in purchases of money market investments and $560,351 for the purchase of equipment.
Investing activities in 2013 and 2012 used $4,547,580 and $2,847,259 of cash, respectively. Of these totals, $4,301,576 related to investments in money market funds in 2013, compared to $2,651,176 in 2012. We also used $246,004 to purchase property and equipment in 2013, compared to $196,083 in 2012.
Cash Flows from Financing Activities
Financing activities provided $43,803,962 of cash in 2014, including of $34,195,637 net proceeds from our IPO, $9,098,971 from the issuance of 1,299,853 shares of common stock at $7.00 per share in a private placement, and $509,354 from stock option and warrant exercises. Financing activities provided $21,974,617 of cash in 2013, including $19,519,162 from the issuance of convertible promissory notes primarily to existing investors and $2,400,685 from the issuance of 342,955 shares of common stock to new investors at $7.00 per share to new investors and $108,436 from stock option exercises, offset by a convertible note repayment of $53,666.
Financing activities provided $21,974,617 of cash in 2013 compared to $17,984,016 in 2012. In 2013, financing activities included $19,519,162 from the issuance of convertible promissory notes primarily to existing investors and $2,400,685 from the issuance of 342,955 shares of common stock in a private placement to new investors at $7.00 per share and $108,436 from stock option exercises, offset by a convertible note repayment of $53,666. In 2012, financing activities included $10,000,000 from the issuance of convertible promissory notes to existing investors and $7,880,080 from the issuance of 1,576,016 shares of common stock primarily to existing investors at $5.00 per share. Cash provided by stock option exercises was $103,936 in 2012.
Since our inception, we have generated limited revenues from the sale of products and have financed our operations primarily through the issuance of common stock, convertible debt (which has been converted into common stock), and grants from government agencies and other institutions. Our initial public offering (“IPO”) has provided us with sufficient financial resources to fund our operations for a period in excess of the next twelve months. As a result of the IPO, the report by our independent registered public accounting firm on the Company’s 2014 consolidated financial statements does not contain an emphasis paragraph with respect to our ability to continue as a going concern, as did the report by our independent registered public accounting firm on our 2013 consolidated financial statements.
48
Although our objective is to increase revenues from product sales within the next two years, as well as to reduce the related cost of sales, in an amount sufficient to reach operating and cash flow breakeven levels, there can be no assurances that we will be successful in this regard. If we are unsuccessful in being able to fund our operations from internal resources within the next two years, we may consider raising additional debt and/or equity capital. However, there can be no assurances that we will be able to secure any such additional financing on acceptable terms and conditions or at all.
Financial Commitments
Effective August 2012, we entered into a lease agreement (the “Sylmar Lease”) with a Company owned by the major stockholder of the Company for office space for a term of five years that expires on February 28, 2017. The Sylmar Lease included rental of additional space commencing January 1, 2013 and a five year option to renew. The lease requires us to pay real estate taxes, insurance and common area maintenance each year, and is subject to periodic cost of living adjustments. In April 2014, the Sylmar Lease was renegotiated with the term ending on February 28, 2022, and a five year option to renew. The new lease also requires us to pay real estate taxes, insurance and common area maintenance each year and includes automatic increases in base rent each year. In November 2014, the industrial center in which Company’s premises are located was sold to an independent third party.
Our Swiss subsidiary rents office space in Switzerland on a month-to-month basis for CHF 7,079 (approximately $7,100) per month.
Future minimum rental payments required under the operating leases are as follows for the years ended December 31.
|
Years |
|
Amount |
|
|
|
2015 |
|
$ |
778,448 |
|
|
2016 |
|
|
808,068 |
|
|
2017 |
|
|
833,045 |
|
|
2018 |
|
|
858,036 |
|
|
2019 |
|
|
883,777 |
|
|
Thereafter |
|
|
2,004,919 |
|
|
|
|
|
|
|
|
Total |
|
$ |
6,166,293 |
|
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Interest Rate Sensitivity
The primary objective of our investment activities is to maintain the safety of principal and preserve liquidity without incurring significant risk. We invest cash in excess of our current needs in money market funds. In general, money market funds are not considered to be subject to interest rate risk because the interest paid on such funds fluctuates with the prevailing interest rate. As of December 31, 2014, our cash equivalents consisted solely of money market funds.
Exchange Rate Sensitivity
The majority of our product sales and operating expenses are denominated in U.S. dollars. However, since we generate revenue outside of the United States and we have sales, marketing and other operations outside of the United States, we do generate a portion of our revenue and incur a portion of our operating expenses in foreign currencies. Our primary currency for revenues generated outside of the United States is the Euro, and our primary currencies for operating expenses incurred outside of the States are the Swiss Franc and the Euro. If the Euro weakens against the U.S. dollar, our revenue as reported in U.S. dollars will decline. Historically, we have not entered into foreign currency forward contracts to hedge our operating expense exposure to foreign currencies, but we may do so in the future.
49
Item 8. Financial Statements and Supplementary Data
Our financial statements and supplementary data required by this Item are provided in the consolidated financial statements of the Company included in this Form 10-K as listed in Item 15(a) of this Form 10-K.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure of Controls and Procedures
Our management carried out an evaluation, under the supervision and with the participation of our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rule 13a-15(e) of the Exchange Act) as of the end of the period covered by this report. Based on this evaluation, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this report.
The design of any system of control is based upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated objectives under all future events, no matter how remote, or that the degree of compliance with the policies or procedures may not deteriorate. Because of its inherent limitations, disclosure controls and procedures may not prevent or detect all misstatements. Accordingly, even effective disclosure controls and procedures can provide only reasonable assurance of achieving their control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply its judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Management's Report on Internal Control Over Financial Reporting
This Annual Report on Form 10-K does not include a report of management's assessment regarding internal control over financial reporting (as defined in Rule 13a-l5(f) of the Exchange Act) or an attestation report of our independent registered public accounting firm due to a transition period established by the rules of the SEC for newly public companies.
Changes in Internal Control over Financial Reporting
There have been no changes in our internal control over financial reporting that occurred during our most recent fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
50
Certain information required by Part III is omitted from this Annual Report on Form 10-K and is incorporated by reference from our definitive proxy statement relating to our 2015 annual meeting of stockholders, pursuant to Regulation 14A of the Securities Exchange Act of 1934, as amended, also referred to in this Annual Report on Form 10-K as our 2015 Proxy Statement, which we expect to file with the SEC no later than April 30, 2015.
Item 10. Directors, Executive Officers and Corporate Governance
Information regarding our directors, including the audit committee and audit committee financial experts, and executive officers and compliance with Section 16(a) of the Exchange Act will be included in our 2015 Proxy Statement and is incorporated herein by reference.
Item 11. Executive Compensation
The information required by this item regarding executive compensation will be included in our 2015 Proxy Statement and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this item regarding security ownership of certain beneficial owners and management will be included in our 2015 Proxy Statement and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item regarding certain relationships and related transactions and director independence will be included in our 2015 Proxy Statement and is incorporated herein by reference.
51
Item 14. Principal Accounting Fees and Services
The information required by this item regarding principal accounting fees and services will be included in our 2015 Proxy Statement and is incorporated herein by reference.
Item 15. Exhibits, Financial Statement Schedules
(a) The following documents are included in this Annual Report on Form 10-K:
1. The consolidated financial statements listed in the accompanying Index to Consolidated Financial Statements are filed as part of this report.
2. All financial schedules have been omitted because the required information is either presented in the consolidated financial statements or the notes thereto or is not applicable or required.
3. The exhibits required by Item 601 of Regulation S-K and Item 15(b) of this Annual Report on Form 10-K are listed in the Exhibit Index immediately preceding the exhibits and are incorporated herein. We have identified in the Exhibit Index each management contract and compensation plan filed as an exhibit to this Annual Report on Form 10-K in response to Item 15(a) (3) of Form 10-K.
52
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
Dated: March 16, 2015 |
Second Sight Medical Products, Inc. |
|
|
|
|
|
/s/ Robert J. Greenberg |
|
|
Robert J. Greenberg |
|
|
Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
|
Name |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Robert j. Greenberg |
|
Chief Executive Officer and Director |
|
March 16, 2015 |
|
Robert J. Greenberg |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
/s/ Thomas B. Miller |
|
Chief Financial Officer |
|
March 16, 2015 |
|
Thomas B. Miller |
|
(Principal Financial and Accounting Officer) |
|
|
| /s/ Alfred E. Mann |
|
Chairman of the Board |
|
March 16, 2015 |
|
Alfred E. Mann |
|
|
|
|
|
|
|
|
|
|
|
/s/ William J. Link |
|
Director |
|
March 16, 2015 |
|
William J. Link |
|
|
|
|
|
|
|
|
|
|
|
/s/ Aaron Mendelsohn |
|
Director |
|
March 16, 2015 |
|
Aaron Mendelsohn |
|
|
|
|
|
|
|
|
|
|
|
/s/ Gregg Williams |
|
Director |
|
March 10, 2015 |
|
Gregg Williams |
|
|
|
|
53
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
Page |
|
|
|
|
Report of Independent Registered Public Accounting Firm |
F-2 |
|
Consolidated Balance Sheets as of December 31, 2014 and 2013 |
F-3 |
|
Consolidated Statements of Operations for the Years Ended December 31, 2014, 2013 and 2012 |
F-4 |
|
Consolidated Statements of Comprehensive Loss for the Years Ended December 31, 2014, 2013 and 2012 |
F-5 |
|
Consolidated Statements of Stockholders’ Equity (Deficiency) for the Years Ended December 31, 2014, 2013 and 2012 |
F-6 |
|
Consolidated Statements of Cash Flows for the Years Ended December 31, 2014, 2013 and 2012 |
F-8 |
|
Notes to Consolidated Financial Statements for the Years Ended December 31, 2014, 2013 and 2012 |
F-10 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
of Second Sight Medical Products, Inc. and Subsidiary
We have audited the accompanying consolidated balance sheets of Second Sight Medical Products, Inc. and Subsidiary (the “Company”) as of December 31, 2014 and 2013, and the related consolidated statements of operations, comprehensive loss, stockholders’ equity (deficiency), and cash flows for each of the years in the three-year period ended December 31, 2014. The Company’s management is responsible for these consolidated financial statements. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America.
/s/ Gumbiner Savett Inc.
March 16, 2015
Santa Monica, California
F-2
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
|
|
|
December 31, |
|
|||||
|
|
|
2014 |
|
|
2013 |
|
||
|
|
|
|
|
|
|
|
|
|
|
ASSETS |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
|
$ |
619,411 |
|
|
$ |
62,565 |
|
|
|
|
|
33,999,563 |
|
|
|
8,611,614 |
|
|
|
|
|
707,648 |
|
|
|
468,644 |
|
|
|
|
|
5,721,991 |
|
|
|
2,346,770 |
|
|
|
|
|
927,575 |
|
|
|
298,223 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets |
|
|
41,976,188 |
|
|
|
11,787,816 |
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment, net |
|
|
1,004,646 |
|
|
|
723,474 |
|
|
Deposits and other assets |
|
|
88,610 |
|
|
|
162,131 |
|
|
|
|
|
|
|
|
|
|
|
|
Total assets |
|
$ |
43,069,444 |
|
|
$ |
12,673,421 |
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIENCY) |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
|
$ |
513,106 |
|
|
$ |
314,327 |
|
|
|
|
|
1,412,383 |
|
|
|
662,883 |
|
|
|
|
|
1,361,894 |
|
|
|
1,146,028 |
|
|
|
|
|
488,910 |
|
|
|
491,267 |
|
|
|
|
|
599,904 |
|
|
|
68,875 |
|
|
|
|
|
4,075,000 |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities |
|
|
8,451,197 |
|
|
|
2,683,380 |
|
|
|
|
|
|
|
|
|
|
|
|
Convertible promissory notes, including $15,389,215 payable to related parties, including accrued interest of $1,724,096, net of unamortized discount of $12,032,146 |
|
|
— |
|
|
|
19,211,112 |
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities |
|
|
8,451,197 |
|
|
|
21,894,492 |
|
|
|
|
|
|
|
|
|
|
|
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity (deficiency): |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
163,171,005 |
|
|
|
88,311,192 |
|
|
|
|
|
166,250 |
|
|
|
— |
|
|
|
|
|
24,590,368 |
|
|
|
20,785,499 |
|
|
|
|
|
(171,436 |
) |
|
|
(587,543 |
) |
|
|
|
|
(473,972 |
) |
|
|
(267,498 |
) |
|
|
|
|
(152,663,968 |
) |
|
|
(117,462,721 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity (deficiency) |
|
|
34,618,247 |
|
|
|
(9,221,071 |
) |
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity |
|
$ |
43,069,444 |
|
|
$ |
12,673,421 |
|
See accompanying notes to consolidated financial statements.
F-3
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS
|
|
|
Years Ended December 31, |
|
|||||||||
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net sales |
|
$ |
3,397,852 |
|
|
$ |
1,564,933 |
|
|
$ |
1,367,224 |
|
|
Cost of sales |
|
|
3,558,135 |
|
|
|
5,629,320 |
|
|
|
4,396,746 |
|
|
Gross loss |
|
|
(160,283 |
) |
|
|
(4,064,387 |
) |
|
|
(3,029,522 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5,040,969 |
|
|
|
3,248,466 |
|
|
|
3,045,157 |
|
|
|
|
|
2,621,919 |
|
|
|
3,215,290 |
|
|
|
3,726,556 |
|
|
|
|
|
6,844,825 |
|
|
|
3,301,452 |
|
|
|
2,194,590 |
|
|
|
|
|
6,565,464 |
|
|
|
4,167,934 |
|
|
|
4,025,558 |
|
|
|
|
|
21,073,177 |
|
|
|
13,933,142 |
|
|
|
12,991,861 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations |
|
|
(21,233,460 |
) |
|
|
(17,997,529 |
) |
|
|
(16,021,383 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
9,108 |
|
|
|
7,454 |
|
|
|
7,512 |
|
|
Other income, net |
|
|
11,805 |
|
|
|
34,768 |
|
|
|
1,775 |
|
|
Interest expense on convertible promissory notes and loan payable |
|
|
(1,956,555 |
) |
|
|
(1,588,687 |
) |
|
|
(138,934 |
) |
|
Amortization of discount on convertible promissory notes |
|
|
(5,077,535 |
) |
|
|
(3,424,931 |
) |
|
|
(128,097 |
) |
|
Write-off of unamortized discount on conversion of convertible promissory notes |
|
|
(6,954,610 |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(35,201,247 |
) |
|
$ |
(22,968,925 |
) |
|
$ |
(16,279,127 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share – basic and diluted |
|
$ |
(1.41 |
) |
|
$ |
(1.02 |
) |
|
$ |
(0.74 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares outstanding – basic and diluted |
|
|
25,052,806 |
|
|
|
22,521,432 |
|
|
|
21,945,580 |
|
See accompanying notes to consolidated financial statements.
F-4
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
|
|
|
Years Ended December 31, |
|
|||||||||
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(35,201,247 |
) |
|
$ |
(22,968,925 |
) |
|
$ |
(16,279,127 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other comprehensive loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(206,474 |
) |
|
|
(82,364 |
) |
|
|
(43,538 |
) |
|
|
Comprehensive loss |
|
$ |
(35,407,721 |
) |
|
$ |
(23,051,289 |
) |
|
$ |
(16,322,665 |
) |
See accompanying notes to consolidated financial statements.
F-5
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIENCY)
|
|
|
Common Stock |
|
|
Common
Stock |
|
|
Additional Paid-in |
|
|
Notes Receivable for Stock Option |
|
|
Accumulated Other Comprehensive |
|
|
Accumulated |
|
|
Total Stockholders’ Equity |
|
|||||||||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Exercises |
|
|
Loss |
|
|
Deficit |
|
|
(Deficiency) |
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
Balance, December 31, 2011 |
|
|
20,771,801 |
|
|
$ |
77,585,137 |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
3,522,568 |
|
|
$ |
(354,625 |
) |
|
$ |
(141,596 |
) |
|
$ |
(78,214,669 |
) |
|
$ |
2,396,815 |
|
|
Issuance of shares of common stock in connection with private placement |
|
|
1,576,016 |
|
|
|
7,880,080 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
7,880,080 |
|
|
Fair value of warrants issued in connection with convertible promissory notes |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,044,649 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,044,649 |
|
|
Fair value of beneficial conversion feature in connection with convertible promissory notes |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
945,500 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
945,500 |
|
|
Exercise of stock options |
|
|
27,430 |
|
|
|
100,548 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
100,548 |
|
|
Stock-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
907,862 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
907,862 |
|
|
Repayment of notes receivable for stock option exercises, net |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
3,388 |
|
|
|
— |
|
|
|
— |
|
|
|
3,388 |
|
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(16,279,127 |
) |
|
|
(16,279,127 |
) |
|
Foreign currency translation adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(43,538 |
) |
|
|
— |
|
|
|
(43,538 |
) |
|
Comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(43,538 |
) |
|
|
(16,279,127 |
) |
|
|
(16,322,665 |
) |
|
Balance, December 31, 2012 |
|
|
22,375,247 |
|
|
$ |
85,565,765 |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
6,420,579 |
|
|
$ |
(351,237 |
) |
|
$ |
(185,134 |
) |
|
$ |
(94,493,796 |
) |
|
$ |
(3,043,823 |
) |
|
Issuance of shares of common stock in connection with private placement |
|
|
342,955 |
|
|
|
2,400,685 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,400,685 |
|
|
Fair value of warrants issued in connection with convertible promissory notes |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
3,107,379 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
3,107,379 |
|
|
Fair value of beneficial conversion feature in connection with convertible promissory notes |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10,487,645 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10,487,645 |
|
|
Exercise of stock options |
|
|
331,871 |
|
|
|
344,742 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
344,742 |
|
|
Stock-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
769,896 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
769,896 |
|
|
Notes receivable, including amount due from officer of $100,000 for stock option exercises, net |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(236,306 |
) |
|
|
— |
|
|
|
— |
|
|
|
(236,306 |
) |
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(22,968,925 |
) |
|
|
(22,968,925 |
) |
|
Foreign currency translation adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(82,364 |
) |
|
|
— |
|
|
|
(82,364 |
) |
|
Comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(82,364 |
) |
|
|
(22,968,925 |
) |
|
|
(23,051,289 |
) |
|
Balance, December 31, 2013 |
|
|
23,050,073 |
|
|
$ |
88,311,192 |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
20,785,499 |
|
|
$ |
(587,543 |
) |
|
$ |
(267,498 |
) |
|
$ |
(117,462,721 |
) |
|
$ |
(9,221,071 |
) |
See accompanying notes to consolidated financial statements.
F-6
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIENCY)
(Continued)
|
|
Common Stock |
|
|
Common Stock Issuable |
|
|
Additional Paid-in |
|
|
Notes Receivable for Stock Option |
|
|
Accumulated Other Comprehensive |
|
|
Accumulated |
|
|
Total Stockholders’ Equity |
|
|||||||||||||||
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Exercises |
|
|
Loss |
|
|
Deficit |
|
|
(Deficiency) |
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
Issuance of common stock in connection with initial public offering |
|
|
4,025,000 |
|
|
|
36,225,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
36,225,000 |
|
Issuance costs of initial public offering |
|
|
— |
|
|
|
(4,970,472 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(4,970,472 |
) |
Fair value of warrants issued in connection with initial public offering |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,771,604 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,771,604 |
|
Issuance
of common stock in connection with conversion |
|
|
6,639,137 |
|
|
|
33,195,539 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
33,195,539 |
|
Issuance of common in connection with warrant exercise |
|
|
2,059 |
|
|
|
10,295 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10,295 |
|
Issuance of common stock in connection with private placement |
|
|
1,299,853 |
|
|
|
9,098,971 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
9,098,971 |
|
Finders’ fee paid on private placement |
|
|
64,384 |
|
|
|
450,688 |
|
|
|
— |
|
|
|
— |
|
|
|
(450,688 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Exercise of stock options |
|
|
115,029 |
|
|
|
505,595 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
505,595 |
|
Stock-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,474,645 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,474,645 |
|
Common stock cancelled |
|
|
(1,322 |
) |
|
|
(9,308 |
) |
|
|
— |
|
|
|
— |
|
|
|
9,308 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Stock issued in connection with professional services |
|
|
22,215 |
|
|
|
178,505 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
178,505 |
|
Common stock issuable for services |
|
|
— |
|
|
|
— |
|
|
|
16,204 |
|
|
|
166,250 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
166,250 |
|
Stock grant in connection with services by a director |
|
|
25,000 |
|
|
|
175,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
175,000 |
|
Repayment of notes receivable for stock option exercises, net |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(6,536 |
) |
|
|
— |
|
|
|
— |
|
|
|
(6,536 |
) |
Forgiveness of notes receivable from an officer for stock option exercises |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
422,643 |
|
|
|
— |
|
|
|
— |
|
|
|
422,643 |
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(35,201,247 |
) |
|
|
(35,201,247 |
) |
Foreign currency translation adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(206,474 |
) |
|
|
— |
|
|
|
(206,474 |
) |
Comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(206,474 |
) |
|
|
(35,201,247 |
) |
|
|
(35,407,721 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, December 31, 2014 |
|
|
35,241,428 |
|
|
$ |
163,171,005 |
|
|
|
16,204 |
|
|
$ |
166,250 |
|
|
$ |
24,590,368 |
|
|
$ |
(171,436 |
) |
|
$ |
(473,972 |
) |
|
$ |
(152,663,968 |
) |
|
$ |
34,618,247 |
|
See accompanying notes to consolidated financial statements.
F-7
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
Years Ended December 31, |
|
|||||||||
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(35,201,247 |
) |
|
$ |
(22,968,925 |
) |
|
$ |
(16,279,127 |
) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
279,179 |
|
|
|
315,770 |
|
|
|
324,712 |
|
|
|
|
1,474,645 |
|
|
|
769,896 |
|
|
|
907,862 |
|
|
|
|
175,000 |
|
|
|
— |
|
|
|
— |
|
|
|
|
422,643 |
|
|
|
— |
|
|
|
— |
|
|
|
|
5,077,535 |
|
|
|
3,424,931 |
|
|
|
128,097 |
|
|
|
|
1,952,282 |
|
|
|
1,588,687 |
|
|
|
138,934 |
|
|
|
|
6,954,610 |
|
|
|
— |
|
|
|
— |
|
|
|
|
9,000 |
|
|
|
— |
|
|
|
— |
|
|
|
|
166,250 |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
163,576 |
|
|
|
(163,576 |
) |
|
|
|
(239,004 |
) |
|
|
(148,310 |
) |
|
|
97,433 |
|
|
|
|
— |
|
|
|
47,567 |
|
|
|
68,863 |
|
|
|
|
(3,375,221 |
) |
|
|
(559,887 |
) |
|
|
(595,734 |
) |
|
|
|
(555,831 |
) |
|
|
(34,171 |
) |
|
|
(62,397 |
) |
|
|
|
198,779 |
|
|
|
(417,050 |
) |
|
|
492,483 |
|
|
|
|
749,500 |
|
|
|
176,206 |
|
|
|
(129,913 |
) |
|
|
|
215,866 |
|
|
|
283,785 |
|
|
|
(318,765 |
) |
|
|
|
(2,357 |
) |
|
|
29,522 |
|
|
|
65,260 |
|
|
|
|
531,029 |
|
|
|
(98,459 |
) |
|
|
4,654 |
|
|
|
|
4,075,000 |
|
|
|
— |
|
|
|
— |
|
|
Net cash used in operating activities |
|
|
(17,092,342 |
) |
|
|
(17,426,862 |
) |
|
|
(15,321,214 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(560,351 |
) |
|
|
(246,004 |
) |
|
|
(196,083 |
) |
|
| Investment in money market funds | (25,387,949 | ) | (4,301,576 | ) | (2,651,176 | ) | ||||||
Net cash used in investing activities |
|
|
(25,948,300 |
) |
|
|
(4,547,580 |
) |
|
|
(2,847,259 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
43,294,608 |
|
|
|
2,400,685 |
|
|
|
7,880,080 |
|
|
|
|
499,059 |
|
|
|
108,436 |
|
|
|
103,936 |
|
|
|
|
— |
|
|
|
(53,666 |
) |
|
|
— |
|
|
|
|
— |
|
|
|
19,519,162 |
|
|
|
10,000,000 |
|
|
|
|
10,295 |
|
|
|
— |
|
|
|
— |
|
|
Net cash provided by financing activities |
|
|
43,803,962 |
|
|
|
21,974,617 |
|
|
|
17,984,016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Effect of exchange rate changes on cash |
|
|
(206,474 |
) |
|
|
(82,364 |
) |
|
|
(43,538 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash: |
|
|
|
|
|
|
|
|
|
|
|
|
Net increase (decrease) |
|
|
556,846 |
|
|
|
(82,189 |
) |
|
|
(227,995 |
) |
Balance at beginning of year |
|
|
62,565 |
|
|
|
144,754 |
|
|
|
372,749 |
|
Balance at end of year |
|
$ |
619,411 |
|
|
$ |
62,565 |
|
|
$ |
144,754 |
|
See accompanying notes to consolidated financial statements.
F-8
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Continued)
|
|
|
Years Ended December 31, |
|
|||||||||
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental cash flow information: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-cash financing and investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
— |
|
|
$ |
3,107,379 |
|
|
$ |
1,044,649 |
|
|
|
|
$ |
2,771,604 |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
$ |
— |
|
|
$ |
10,487,645 |
|
|
$ |
945,500 |
|
|
|
|
$ |
— |
|
|
$ |
252,165 |
|
|
$ |
2,511 |
|
|
|
|
$ |
33,195,539 |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
$ |
450,688 |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
$ |
169,505 |
|
|
$ |
— |
|
|
$ |
— |
|
|
See accompanying notes to consolidated financial statements.
F-9
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Business Operations
Second Sight Medical Products, Inc. (“Second Sight” or “the Company”), formerly Second Sight LLC, was founded in 1998 as a limited liability company and was subsequently incorporated in the State of California in 2003. Second Sight develops, manufactures and markets implantable prosthetic devices that can restore some functional vision to patients blinded by outer retinal degenerations, such as Retinitis Pigmentosa.
In 2007, Second Sight formed Second Sight (Switzerland) Sarl, initially to manage clinical trials for its products in Europe, and later to manage sales and marketing in Europe and the Middle East. As the laws of Switzerland require at least two corporate stockholders, Second Sight (Switzerland) Sarl is 99.5% owned directly by the Company and 0.5% owned by an executive of Second Sight, who is acting as a nominee of the Company. Accordingly, Second Sight (Switzerland) Sarl is considered 100% owned for financial statement purposes and is consolidated with Second Sight for all periods presented.
The Company’s current product, the Argus II system, entered clinical trials in 2006, received CE Mark approval for marketing and sales in the European Union (“EU”) in 2011, and approval by the United States Food and Drug Administration (“FDA”) for marketing and sales in the United States in 2013. The Company began selling its product in Europe in 2011, in Saudi Arabia in 2013, and in the United States and Canada in 2014.
Since its inception, the Company has generated limited revenues from the sale of products and has financed its operations primarily through the issuance of common stock, convertible debt (which has been converted into common stock), and grants primarily from government agencies. The Company’s initial public offering (“IPO”) in November 2014 has provided it with sufficient financial resources to fund its operations for a period in excess of the next twelve months. As a result of the IPO, the report by the Company’s independent registered public accounting firm on the Company’s 2014 consolidated financial does not contain an emphasis paragraph with respect to the Company’s ability to continue as a going concern, as did the report by the Company’s independent registered public accounting firm on the Company’s 2013 consolidated financial statements.
Although the Company’s objective is to increase revenues from product sales within the next two years, as well as to reduce the related cost of sales, in an amount sufficient to reach operating and cash flow breakeven levels, there can be no assurances that the Company will be successful in this regard. If the Company is unsuccessful in being able to fund its operations from internal resources within the next two years, the Company may consider raising additional debt and/or equity capital. However, there can be no assurances that the Company will be able to secure any such additional financing on acceptable terms and conditions or at all.
2. Summary of Significant Accounting Policies
Principles of Consolidation
The accompanying consolidated financial statements include the financial statements of Second Sight and Second Sight Switzerland. Intercompany balances and transactions have been eliminated in consolidation.
Accounts receivable
Trade accounts receivable are stated net of an allowance for doubtful accounts. The Company performs ongoing credit evaluations of its customers’ financial condition and generally requires no collateral from its customers or interest on past due amounts. Management estimates the allowance for doubtful accounts based on review and analysis of specific customer balances that may not be collectible and how recently payments have been received. Accounts are considered for write-off when they become past due and when it is determined that the probability of collection is remote. There was no allowance for doubtful accounts at December 31, 2014 and 2013.
Inventories
Inventories are stated at the lower of cost or market, determined by the first-in, first-out method. Inventories consist primarily of raw materials, work in progress and finished goods, which includes all direct material, labor and other overhead costs.
F-10
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies (Cont’d)
The Company establishes a reserve to mark down its inventory for estimated unmarketable inventory equal to the difference between the cost of inventory and the estimated net realizable value based on assumptions about the usability of the inventory, future demand and market conditions. If actual market conditions are less favorable than those projected by management, additional inventory reserve may be required.
Property and Equipment
Property and equipment are recorded at historical cost less accumulated depreciation and amortization. Improvements are capitalized, while expenditures for maintenance and repairs are charged to expense as incurred. Upon disposal of depreciable property, the appropriate property accounts are reduced by the related costs and accumulated depreciation. The resulting gains and losses are reflected in the consolidated statements of operations.
Depreciation is provided for using the straight-line method in amounts sufficient to relate the cost of assets to operations over their estimated service lives. Leasehold improvements are amortized over the shorter of the life of the asset or the related lease term. Estimated useful lives of the principal classes of assets are as follows:
|
Lab equipment |
5 – 7 years |
|
Computer hardware and software |
3 – 7 years |
|
Leasehold improvements |
1 – 5 years or the term of the lease, if shorter |
|
Furniture, fixtures and equipment |
5 – 10 years |
The Company reviews its property and equipment for impairment annually or whenever events or changes in circumstances indicate that the carrying value of such assets may not be recoverable. There were no impairment losses recognized in 2014 and 2013.
Depreciation and amortization of property and equipment amounted to $279,179, $315,770 and $324,712 for the years ended December 31, 2014, 2013 and 2012, respectively.
Research and Development
Research and development costs are charged to operations in the period incurred and amounted to $5,040,969, $3,248,466 and $3,045,157 net of grant revenue, for the years ended December 31, 2014, 2013 and 2012, respectively.
Patent Costs
The Company has over 300 domestic and foreign patents. Due to the uncertainty associated with the successful development of one or more commercially viable products based on Company’s research efforts and any related patent applications, all patent costs, including patent-related legal, filing fees and other costs, including internally generated costs, are expensed as incurred. Patent costs were $665,617, $669,011 and $689,633 for the years ended December 31, 2014, 2013 and 2012, respectively, and are included in general and administrative expenses in the consolidated statements of operations.
Revenue Recognition
The Company’s revenue is derived from the sale of its Argus II retinal implant, which is implanted during retina surgery to provide limited vision to patients blinded by Retinitis Pigmentosa.
The Company sells to university hospitals, teaching hospitals and large medical centers. The Company recognizes revenue when four basic criteria are met: (1) persuasive evidence of an arrangement exists; (2) surgical implantation has occurred; (3) the price is fixed or determinable; and (4) collectability is reasonably assured. The Company generally uses customer purchase orders or contracts to determine the existence of an arrangement. Sales transactions are based on prices that are determinable at the time
F-11
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies (Cont’d)
that the customer’s purchase order is accepted by the Company. In order to determine whether collection is reasonably assured, the Company assesses a number of factors, including creditworthiness of the customer and medical insurance coverage. If the Company determines that collection is not reasonably assured, the Company will defer the recognition of revenue until collection becomes reasonably assured, which is generally upon receipt of payment. The Company may periodically grant special terms, such as extended payment terms. The Company defers revenues when these special terms are granted until a final price is fixed and collection becomes reasonably assured. Due to the nature of the Company’s revenue recognition policy of recording revenue only after surgical implantation, the Company has had no returns related to Argus II System recorded as revenue.
Grant Receipts and Liabilities
From time to time, the Company receives grants that help fund specific development programs. Any amounts received pursuant to grants are offset against the related operating expenses as the costs are incurred. During the years ended December 31, 2014, 2013 and 2012 grants offset against operating expenses were $19,245, $174,565 and $601,255, respectively.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions. These estimates and assumptions affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual amounts could differ materially from those estimates.
Concentration of Risk
Credit Risk
Financial instruments that subject the Company to concentrations of credit risk consist primarily of cash, money market funds, and trade accounts receivable. The Company maintains cash and money market funds with financial institutions that management deems reputable, and at times, cash balances may be in excess of FDIC and SIPC insurance limits. The Company extends differing levels of credit to customers, and typically does not require collateral.
The Company also maintains a cash balance at a bank in Switzerland. Accounts at such bank are insured up to an amount specified by the deposit insurance agency of Switzerland.
Customer Concentration
During the years ended December 31, 2014, 2013 and 2012, the following customers comprised more than 10% of revenues:
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||
|
Customer 1 |
|
|
21 |
% |
|
|
0 |
% |
|
|
0 |
% |
|
Customer 2 |
|
|
10 |
% |
|
|
0 |
% |
|
|
0 |
% |
|
Customer 3 |
|
|
6 |
% |
|
|
13 |
% |
|
|
0 |
% |
|
Customer 4 |
|
|
3 |
% |
|
|
31 |
% |
|
|
1 |
% |
|
Customer 5 |
|
|
3 |
% |
|
|
0 |
% |
|
|
29 |
% |
|
Customer 6 |
|
|
0 |
% |
|
|
13 |
% |
|
|
0 |
% |
|
Customer 7 |
|
|
0 |
% |
|
|
12 |
% |
|
|
21 |
% |
|
Customer 8 |
|
|
0 |
% |
|
|
7 |
% |
|
|
32 |
% |
|
Customer 9 |
|
|
0 |
% |
|
|
6 |
% |
|
|
14 |
% |
F-12
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies (Cont’d)
As of December 31, 2014 and 2013, the following customers comprised more than 10% accounts receivable:
|
|
|
2014 |
|
|
2013 |
|
||
|
|
|
|
|
|
|
|
|
|
|
Customer 1 |
|
|
32 |
% |
|
|
9 |
% |
|
Customer 2 |
|
|
20 |
% |
|
|
45 |
% |
|
Customer 3 |
|
|
13 |
% |
|
|
0 |
% |
|
Customer 4 |
|
|
13 |
% |
|
|
0 |
% |
|
Customer 5 |
|
|
0 |
% |
|
|
21 |
% |
|
Customer 6 |
|
|
0 |
% |
|
|
20 |
% |
Geographic Concentration
During the years ended December 31, 2014, 2013 and 2012, regional revenue, based on customer location, consisted of the following:
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
|
47 |
% |
|
|
0 |
% |
|
|
0 |
% |
|
Germany |
|
|
16 |
% |
|
|
32 |
% |
|
|
75 |
% |
|
Canada |
|
|
10 |
% |
|
|
0 |
% |
|
|
0 |
% |
|
Italy |
|
|
8 |
% |
|
|
18 |
% |
|
|
21 |
% |
|
Spain |
|
|
9 |
% |
|
|
0 |
% |
|
|
0 |
% |
|
Saudi Arabia |
|
|
3 |
% |
|
|
31 |
% |
|
|
1 |
% |
|
France |
|
|
3 |
% |
|
|
7 |
% |
|
|
0 |
% |
|
Netherlands |
|
|
0 |
|
|
|
13 |
% |
|
|
0 |
% |
Sources of Supply
Several of the components, materials and services used in the Company’s current Argus II product are available from only one supplier, and substitutes for these items cannot be obtained easily or would require substantial design or manufacturing modifications. Any significant problem experienced by one of the Company’s sole source suppliers could result in a delay or interruption in the supply of components to the Company until that supplier cures the problem or an alternative source of the component is located and qualified. Even where the Company could qualify alternative suppliers, the substitution of suppliers may be at a higher cost and cause time delays that impede the commercial production of the Argus II, reduce gross profit margins and impact the Company’s abilities to deliver its products as may be timely required to meet demand.
Foreign Operations
The accompanying consolidated financial statements as of December 31, 2014 and 2013 include assets amounting to approximately $2,091,000 and $729,000, respectively, relating to operations of the Company in Switzerland. It is always possible unanticipated events in foreign countries could disrupt the Company’s operations.
Fair Value of Financial Instruments
The authoritative guidance with respect to fair value establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three levels, and requires that assets and liabilities carried at fair value be classified and disclosed in one of three categories, as presented below. Disclosure as to transfers in and out of Levels 1 and 2, and activity in Level 3 fair value measurements, is also required.
F-13
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies (Cont’d)
Level 1. Observable inputs such as quoted prices in active markets for an identical asset or liability that the Company has the ability to access as of the measurement date. Financial assets and liabilities utilizing Level 1 inputs include active-exchange traded securities and exchange-based derivatives.
Level 2. Inputs, other than quoted prices included within Level 1, which are directly observable for the asset or liability or indirectly observable through corroboration with observable market data. Financial assets and liabilities utilizing Level 2 inputs include fixed income securities, non-exchange based derivatives, mutual funds, and fair-value hedges.
Level 3. Unobservable inputs in which there is little or no market data for the asset or liability which requires the reporting entity to develop its own assumptions. Financial assets and liabilities utilizing Level 3 inputs include infrequently-traded non-exchange-based derivatives and commingled investment funds, and are measured using present value pricing models.
The Company determines the level in the fair value hierarchy within which each fair value measurement falls in its entirety, based on the lowest level input that is significant to the fair value measurement in its entirety. In determining the appropriate levels, the Company performs an analysis of the assets and liabilities at each reporting period end.
Money market funds are the only financial instrument that is measured and recorded at fair value on the Company’s balance sheet, and they are considered Level 1 valuation securities in both 2013 and 2014.
Stock-Based Compensation
Pursuant to Financial Accounting Standards Board (“FASB”) ASC 718 Share-Based Payment (“ASC 718”), the Company records stock-based compensation expense for all stock-based awards.
Under ASC 718, the Company estimates the fair value of stock options granted using the Black-Scholes option pricing model. The fair value for awards that are expected to vest is then amortized on a straight-line basis over the requisite service period of the award, which is generally the option vesting term.
The fair value of each stock option award is estimated on the date of grant using the Black-Scholes option valuation model. The assumptions used in the Black-Scholes valuation model are as follows:
• The grant price of the issuances, with certain exceptions, is determined based on the estimated fair value of the shares at the date of grant.
• The risk free interest rate for periods within the contractual life of the option is based on the U.S. treasury yield in effect at the time of grant.
• As permitted by SAB 107, due to the Company’s insufficient history of option activity, management utilizes the simplified approach to estimate the options expected term, which represents the period of time that options granted are expected to be outstanding.
• Volatility is determined based on average historical volatilities of comparable companies in similar industry.
• Expected dividend yield is based on current yield at the grant date or the average dividend yield over the historical period. The Company has never declared or paid dividends and has no plans to do so in the foreseeable future.
Long Term Investor Right
Each beneficial owner (“IPO Shareholder”) of the Company’s common stock, who purchased shares directly in the offering (“IPO Shares”), may qualify to receive up to one additional share of common stock from the Company for each share purchased in the offering (“IPO Supplemental Shares”) pursuant to the Long Term Investor Right that was included with each IPO Share.
F-14
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies (Cont’d)
To receive IPO Supplemental Shares, within 90 days following the closing date of the offering, or by February 22, 2015, an IPO Shareholder was required to take action to become the direct registered owner of its IPO Shares. Furthermore, IPO Shareholders are required to hold their IPO Shares in their own name and not place them in “street name” or trade them at any time during the 24 month period immediately following the IPO closing date. This Long Term Investors Right is non-detachable and transferable only in limited circumstances.
The Company will issue IPO Supplemental Shares to IPO Shareholders who have not otherwise forfeited their Long Term Investor Right if, during the two-year period immediately following the IPO closing date, the Company’s common stock does not trade at or above $18.00 per share (200% of the IPO price per share) for any five consecutive day period. If the Company’s common stock trades on its principal exchange at 200% of the IPO price per share or greater on five consecutive trading days during the two years after the IPO closing date, the Long Term Investor Right will terminate.
The formula to determine the number of IPO Supplemental Shares to be issued on a trigger of the Long Term Investor Right will be: (i) $18.00 minus (ii) the average of the highest consecutive closing prices in any 90 day trading period on the principal exchange during the two years after the Closing Date (the “Measurement Average”) divided by the Measurement Average. Fractional shares issuable to a qualifying IPO Shareholder resulting from the calculation will be rounded up to the next whole share of Common Stock, taking into account the aggregate number of Long Term Investor Rights of a holder. As an illustrative example, if the highest average of consecutive closing prices over any 90 calendar day period is $10.00 per share, each Long‑Term Investor Right will be entitled to 0.80 additional shares of common stock, which is calculated as: ($18.00 - $10.00)/$10.00.
The IPO offering price for purposes of the calculation of the amount of common stock to be issued on a Long Term Investor Right will be subject to adjustment in the event of a reorganization, recapitalization or split-up of the Company’s shares, the issuance of a stock dividend or any similar event. The amount of IPO Supplemental Shares, if any, to be issued will be computed by an independent public accountant as soon as practicable following the second anniversary of the Closing Date. The determination by such independent public accountant will be final and binding on the Company and on all qualifying IPO Shareholders and the Company will within 15 days after receipt of written determination deliver to shareholders certificates evidencing the additional shares.
The Company has identified and will track IPO Investors who have perfected their Rights on a quarterly basis. At the end of each reporting period, the Company will disclose the potential dilutive effect of the Right, including the number of common shares that would be issuable on such date, based on the actual share price movements since the IPO.
The Right is an equity instrument that is accounted for as a component of the actual price per common share paid by the investor in the IPO. For basic earnings per share, the common shares associated with the Right are treated as contingently issuable shares and will not be included in basic earnings per share until the actual number of shares can be calculated and the shares have been issued.
Convertible Promissory Notes and Warrants
The warrants and embedded beneficial conversion feature of convertible promissory notes are classified as equity under FASB ASC Topic 815-40 “Derivatives and Hedging — Contracts in Entity’s Own Equity”. The Company allocates the proceeds of the convertible promissory notes between convertible promissory notes and the financial instruments related to warrants associated with convertible promissory notes based on their relative fair values at the commitment date. The fair value of the financial instruments related to warrants associated with convertible promissory notes is determined utilizing the Black‑Scholes option pricing model and the respective allocated proceeds to the warrants is recorded in additional paid-in capital. The Company utilized the Black-Scholes option valuation model using the same valuation assumptions as described herein for Stock Based Compensation. The embedded beneficial conversion feature associated with convertible promissory notes is recognized and measured by allocating a portion of the proceeds equal to the intrinsic value of that feature to additional paid-in capital in
F-15
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies (Cont’d)
accordance with ASC Topic 470-20 “Debt — Debt with Conversion and Other Options.” The portion of debt discount resulting from the allocation of proceeds to the financial instruments related to warrants associated with convertible promissory notes is being amortized over the life of the convertible promissory notes. For the portion of debt discount resulting from the allocation of proceeds to the beneficial conversion feature, it is amortized over the term of the notes from the respective dates of issuance.
Comprehensive Income or Loss
The Company complies with provisions of FASB ASC 220, Comprehensive Income, which requires companies to report all changes in equity during a period, except those resulting from investment by owners and distributions to owners, for the period in which they are recognized. Comprehensive income is defined as the change in equity during a period from transactions and other events from non-owner sources.
Comprehensive and other comprehensive income (loss) is reported on the face of the financial statements. For the years ended December 31, 2014, 2013 and 2012 comprehensive income (loss) is the total of net income (loss) and other comprehensive income (loss) which, for the Company, consists entirely of foreign currency translation adjustments and there were no material reclassifications from other comprehensive loss to net loss during the years ended December 31, 2014, 2013 and 2012.
Foreign Currency Translation and Transactions
The financial statements and transactions of the subsidiary’s operations are reported in the local (functional) currency of Swiss francs (CHF) and translated into US dollars in accordance with U.S. GAAP. Assets and liabilities of those operations are translated at exchange rates in effect at the balance sheet date. The resulting gains and losses from translating foreign currency financial statements are recorded as other comprehensive income (loss). Revenues and expenses are translated at the average exchange rate for the reporting period. Foreign currency translation gains (losses) resulting from exchange rate fluctuations on transactions denominated in a currency other than the foreign operations’ functional currencies are included in expenses in the consolidated statements of operations.
Income Taxes
The Company accounts for income taxes under an asset and liability approach for financial accounting and reporting for income taxes. Accordingly, the Company recognizes deferred tax assets and liabilities for the expected impact of differences between the financial statements and the tax basis of assets and liabilities.
The Company records a valuation allowance to reduce its deferred tax assets to the amount that is more likely than not to be realized. In the event the Company was to determine that it would be able to realize its deferred tax assets in the future in excess of its recorded amount, an adjustment to the deferred tax assets would be credited to operations in the period such determination was made. Likewise, should the Company determine that it would not be able to realize all or part of its deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to operations in the period such determination was made. The Company has incurred losses for tax purposes since inception and has significant tax losses and tax credit carryforwards. These amounts are subject to valuation allowances as it is not likely that they will be realized in the next few years.
Product Warranties
The Company’s policy is to warrant all shipped products against defects in materials and workmanship for two years by replacing failed parts. The Company also provides a three-year manufacturer’s warranty covering implant failure by providing a functionally-equivalent replacement implant. Accruals for product warranties are estimated based on historical warranty experience and current product performance trends, and are recorded at the time revenue is recognized as a component of cost of sales. The warranty liabilities are reduced by material and labor costs used to replace parts over the warranty period in the
F-16
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies (Cont’d)
periods in which the costs are incurred. The Company periodically assesses the adequacy of its recorded warranty liabilities and adjusts the amounts as necessary. The warranty liabilities are included in accrued expenses in the consolidated balance sheets.
Presentation of sales and value added taxes
The Company collects value added tax on its sales in Europe and certain states in the United Sates impose a sales tax on the Company’s sales to nonexempt customers. The Company collects that valued added and sales tax from customers and remits the entire amount to the respective authorities. The Company’s accounting policy is to exclude the tax collected and remitted to the authorities from revenues and cost of revenues.
Net Loss per Share
The Company’s computation of earnings per share (“EPS”) includes basic and diluted EPS. Basic EPS is measured as the income (loss) available to common shareholders divided by the weighted average number of common shares outstanding for the period. Diluted EPS is similar to basic EPS but presents the dilutive effect on a per share basis of potential common shares (e.g., convertible notes payable, convertible preferred stock, preferred stock warrants and common stock options) as if they had been converted at the beginning of the periods presented, or issuance date, if later. Potential common shares that have an anti-dilutive effect (i.e., those that increase income per share or decrease loss per share) are excluded from the calculation of diluted EPS.
Loss per common share is computed by dividing net loss by the weighted average number of shares of common stock outstanding during the respective periods. Basic and diluted loss per common share is the same for all periods presented because all convertible notes payable, common stock warrants and common stock options outstanding were anti-dilutive.
At December 31, 2014, 2013 and 2012, the Company excluded the outstanding securities summarized below, which entitle the holders thereof to ultimately acquire shares of common stock, from its calculation of earnings per share, as their effect would have been anti-dilutive.
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Long Term Investor Rights |
|
|
1,021,021 |
|
|
|
— |
|
|
|
— |
|
|
Underwriter’s warrants |
|
|
805,000 |
|
|
|
— |
|
|
|
— |
|
|
Convertible notes payable |
|
|
— |
|
|
|
6,248,652 |
|
|
|
2,027,082 |
|
|
Warrants associated with convertible debt |
|
|
1,178,707 |
|
|
|
1,180,766 |
|
|
|
400,000 |
|
|
Common stock options |
|
|
3,251,627 |
|
|
|
2,240,568 |
|
|
|
2,727,503 |
|
|
Total |
|
|
6,256,355 |
|
|
|
9,669,986 |
|
|
|
5,154,585 |
|
Recent Accounting Pronouncements
In August 2014, the FASB issued Accounting Standards Update No. 2014-15 (ASU 2014-15), Presentation of Financial Statements — Going Concern (Subtopic 205-10). ASU 2014-15 provides guidance as to management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. In connection with preparing financial statements for each annual and interim reporting period, an entity’s management should evaluate whether there are conditions or events, considered in the aggregate, that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued (or within one year after the date that the financial statements are available to be issued when applicable). Management’s evaluation should be based on relevant conditions and events that are known and reasonably knowable at the date that the financial statements are issued (or at the date that the financial statements are available to be issued when applicable). Substantial doubt about an entity’s ability to continue as a going concern exists when relevant conditions and events, considered in the
F-17
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies (Cont’d)
aggregate, indicate that it is probable that the entity will be unable to meet its obligations as they become due within one year after the date that the financial statements are issued (or available to be issued). ASU 2014-15 is effective for the annual period ending after December 15, 2016, and for annual periods and interim periods thereafter. Early application is permitted. The Company is currently evaluating the impact the adoption of ASU 2014-15 on the Company’s financial statement presentation and disclosures.
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2014-09 (ASU 2014-09), Revenue from Contracts with Customers. ASU 2014-09 will eliminate transaction- and industry-specific revenue recognition guidance under current U.S. GAAP and replace it with a principle based approach for determining revenue recognition. ASU 2014-09 will require that companies recognize revenue based on the value of transferred goods or services as they occur in the contract. The ASU also will require additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. ASU 2014-09 is effective for reporting periods beginning after December 15, 2016, and early adoption is not permitted. Entities can transition to the standard either retrospectively or as a cumulative-effect adjustment as of the date of adoption. Management is currently assessing the impact the adoption of ASU 2014-09 and has not determined the effect of the standard on the Company’s ongoing financial reporting.
In April 2014, the FASB issued Accounting Standards Update No. 2014-08 (ASU 2014-08), Presentation of Financial Statements (Topic 205) and Property, Plant and Equipment (Topic 360). ASU 2014-08 amends the requirements for reporting discontinued operations and requires additional disclosures about discontinued operations. Under the new guidance, only disposals representing a strategic shift in operations or that have a major effect on the Company’s operations and financial results should be presented as discontinued operations. This new accounting guidance is effective for annual periods beginning after December 15, 2014. The Company is currently evaluating the impact of adopting ASU 2014-08 on the Company’s results of operations or financial condition.
Management does not believe that any other recently issued, but not yet effective, authoritative guidance, if currently adopted, would have a material impact on the Company’s financial statement presentation or disclosures.
3. Money Market Funds
Money market funds at December 31, 2014 totaled $33,999,563 and consisted of $268,464 in the City National Rochdale Government Fund Class S, $1,024,101 in a Preferred Deposit, $6,427 in the BBIF Money Fund Class 4, $32,644,788 in the FFI Institutional Fund, and $55,783 held in a deposit account in Switzerland as security for the performance of a contract. Money market funds at December 31, 2013 totaled $8,611,614 and consisted of $768,368 in the City National Rochdale Government Fund Class S, $3,550,845 in a Preferred Deposit, $3,351,104 in the BBIF Money Fund Class 3, and $941,297 in the FFI Institutional Fund.
The investment objective of the City National Rochdale Government Money Market Fund is to preserve principal and maintain a high degree of liquidity while providing current income through a portfolio of liquid, high quality, short-term U.S. Government bonds and notes, at least 80% of which is in U.S. Government securities. The City National Rochdale Government Money Market Fund is managed by City National Rochdale, LLC. The Preferred Business Deposit Fund is managed by Merrill Lynch and is designed to provide liquidity, safety and competitive yields. The investment objective of the BBIF Money Fund is to seek current income, preservation of capital and liquidity through a diversified portfolio of U.S. dollar-denominated short‑term securities with maturities of not more than 397 days (13 months). The BBIF Money Fund is managed by BlackRock Advisors, LLC. The investment objective of the FFI Institutional Fund is to seek maximum current income consistent with liquidity and the maintenance of a portfolio of high-quality, short-term money market securities. The FFI Institutional Fund is managed by BlackRock Advisors, LLC.
F-18
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
3. Money Market Funds (Cont’d)
The following table presents money market funds at their level within the fair value hierarchy at December 31, 2014 and 2013.
|
|
|
Total |
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2014: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds |
|
$ |
33,999,563 |
|
|
$ |
33,999,563 |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2013: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds |
|
$ |
8,611,614 |
|
|
$ |
8,611,614 |
|
|
$ |
— |
|
|
$ |
— |
|
4. Selected Balance Sheet Detail
Inventories, net
Inventories consisted of the following at December 31, 2014 and 2013:
|
|
|
2014 |
|
|
2013 |
|
||
|
|
|
|
|
|
|
|
|
|
|
Raw materials |
|
$ |
610,434 |
|
|
$ |
510,802 |
|
|
Work in process |
|
|
4,729,235 |
|
|
|
2,617,502 |
|
|
Finished goods |
|
|
1,748,966 |
|
|
|
814,258 |
|
|
|
|
|
7,088,635 |
|
|
|
3,942,562 |
|
|
Allowance for excess and obsolescence |
|
|
(1,366,644 |
) |
|
|
(1,595,792 |
) |
|
|
|
$ |
5,721,991 |
|
|
$ |
2,346,770 |
|
Property and equipment, net of accumulated depreciation
Property and equipment consisted of the following at December 31, 2014 and 2013:
|
|
|
2014 |
|
|
2013 |
|
||
|
Laboratory equipment |
|
$ |
3,285,842 |
|
|
$ |
2,986,770 |
|
|
Computer hardware and software |
|
|
1,700,612 |
|
|
|
1,448,640 |
|
|
Leasehold improvements |
|
|
362,408 |
|
|
|
359,173 |
|
|
Furniture, fixtures and equipment |
|
|
135,303 |
|
|
|
129,231 |
|
|
|
|
|
5,484,165 |
|
|
|
4,923,814 |
|
|
Accumulated depreciation and amortization |
|
|
(4,479,519 |
) |
|
|
(4,200,340 |
) |
|
|
|
$ |
1,004,646 |
|
|
$ |
723,474 |
|
5. Related Party Transactions
As of December 31, 2013, three members of the Company’s Board of Directors and certain of their affiliates (collectively, the “Related Party Investors”) held $23,378,808 in face value of the Company’s convertible promissory notes. These convertible notes, which are more-fully described in Note 7, entitled the Related Party Investors to (i) simple interest of 7.5% per annum accrued on the outstanding face value of convertible notes, (ii) warrants to purchase shares of the Company’s common stock at $5.00 per share, and (iii) the right to convert their convertible notes into shares of the Company’s common stock at $5.00 per share upon the occurrence of certain events, one of which was an initial public offering of the Company’s common stock. In June 2014, an entity associated with one of these Related Party Investors assigned $200,000 in face value of these convertible notes payable to unrelated parties. This assignment included all accrued interest and the related 8,000 warrants. As more
F-19
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
5. Related Party Transactions (Cont’d)
fully described in Note 7, all of the Company’s convertible promissory notes were converted into common stock upon the initial public offering of common stock. Accordingly, the Related Party Investors received 5,210,548 shares of the Company’s common stock upon conversion of their convertible promissory notes. As of December 31 2013, the Related Party Investors held convertible promissory notes, including accrued interest, totaling $24,731,240. As of December 31, 2014 and 2013, in connection with the issuance of these convertible notes, the Related Party Investors held warrants to purchase 927,152 and 935,151 shares, respectively, of the Company’s common stock. During the period January 1, 2014 to November 18, 2014 (the date the notes were converted to common stock of the Company), the Company recorded interest expense to the Related Party Investors of $1,533,611. During the year ended December 31, 2013, in connection with these convertible notes, the Company recorded interest expense to the Related Party Investors of $1,239,956. The Related Party Investors purchased these convertible notes on the same terms and conditions as the other investors in the convertible note financings. The Related Party Investors were also stockholders of the Company at the time that they purchased the convertible notes.
The Company’s largest stockholder and chairman is also a substantial contributor to the Alfred E. Mann Foundation for Scientific Research (the “Foundation”). Beginning February 2007, an officer of the Company also became the Chairman of the Board of the Foundation. The Company and the Foundation share certain limited administrative and engineering employees. The shared employees make an allocation of their time between the Company and the Foundation. There are also various other costs shared between the Company and the Foundation. In connection with these shared costs, the Company owed the Foundation $1,004 and $11,887 as of December 31, 2014 and December 31, 2013, respectively.
On May 31, 2011, an officer of the Company entered into a loan agreement with the Company to finance the exercise of stock options to purchase 100,000 shares for $319,000, with a maturity date of May 31, 2016 and interest accruing at 2.26% per annum. On December 11, 2013, the same officer of the Company entered into a second loan agreement with the Company to finance the exercise of stock options to purchase 200,000 shares of common stock for $100,000, with a maturity date of December 31, 2018 and interest accruing at 1.64% per annum. As of December 31, 2013, the balance outstanding pursuant to the two loans, including accrued interest, was $423,217. These loans receivable were recorded in the Company’s financial statements as an offset to stockholders’ equity. In July 2014, the Company’s Board of Directors approved forgiving this note receivable and related accrued interest of $422,643, which amount is included in general and administrative expenses in the Company’s statement of operations for the year ended December 31, 2014.
Prior to November 2014, the Company leased its office and laboratory space in Sylmar, California under an operating lease with Mann Biomedical Park, LLC (formerly Sylmar Biomedical Park, LLC), which is wholly owned by Alfred E. Mann, a stockholder of the Company (see Note 13). In November 2014, the Mann Biomedical Park, LLC was sold to an unrelated third party.
The Company entered into a loan agreement with an entity affiliated with Mr. Mann to lend the Company up to $3.0 million at an annualized interest rate of 1.5% on an unsecured basis. The Company borrowed $2.0 million pursuant to this loan agreement on October 1, 2014, and repaid the loan on November 24, 2014, including $4,274 of interest. As of December 31, 2014, no amounts were due or outstanding under this agreement.
6. Grants
In April 2010, the Company was awarded a development and testing grant of $2,988,224 from the Department of Health and Human Services, National Institutes of Health (NIH). The grant was for three years commencing in May 2010. The grant included managing various subcontracts with designated individuals and their respective institutions. The grant reimburses research costs to develop technology for the prevention, cure and amelioration of the loss of eyesight and other neurologic applications. The Company recorded funding under the grant as an offset to research and development expenses. In 2014, 2013 and 2012, research and development expenses were offset by $19,245, $174,565 and $601,255, respectively.
In August 2010, the Company was awarded a foreign grant of CHF230,000 from the European Union Federal Office for Professional Education and Technology (valued at $251,600 at December 31, 2012) to support training and career development of researchers. In January 2013, the Company had yet to meet the requirements as specified by the grant agreement, and therefore, management decided not to pursue the grant and returned partial advances received pursuant to the grant totaling $163,576.
F-20
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
6. Grants (Cont’d)
In September, 2014, the Company entered into a Joint Research and Development Agreement or JRDA with The Johns Hopkins University Applied Physics Laboratory or APL. The JRDA includes a subcontract to do research under a grant received by APL. Under the JRDA, the Company has agreed to perform research regarding integration of APL research in to a visual prosthesis system. In October, 2014, APL paid the Company $4.075 million in one lump sum to conduct its portion of the research. The JRDA also includes a license from APL to the Company, for the life of any patents resulting from APL’s portion of the research. The APL portion of the research includes image processing enhancements for a visual prosthesis. In exchange for the license, the Company issued 1,000 shares of its common stock to APL, has agreed to pay APL patent prosecution costs, and to pay APL a royalty of .25% of net sales of licensed products. The Company has not commenced any research and development activities under JDRA and as such recorded funds received under this grant as deferred revenue at December 31, 2014.
7. Convertible Promissory Notes and Warrants
During 2010 and 2011, the Company borrowed money in a series of financing rounds by issuing $15,440,511 of convertible notes (the “2010 - 2011 Notes”) primarily to existing stockholders. The notes accrued interest at 7.5% per annum and had a variety of maturity dates. During 2011, all but two of the 2010 and 2011 Notes, with a combined face value $47,001, were converted into 3,195,590 shares of the Company’s common stock at $5.00 per share. In March 2013, the Company redeemed the remaining two notes for $53,666 in cash.
During 2012 and 2013, the Company borrowed money primarily from existing investors in three separate rounds through the issuance of convertible promissory notes (collectively, the “Convertible Notes”) totaling $29,519,162. The first round of Convertible Notes in the amount of $5,000,000 was issued from July through November 2012 (the “July 2012 Notes). The second round of Convertible Notes in the amount of $5,000,000 was issued from October through December 2012 (the “October 2012 Notes”). The third round of Convertible Notes in the amount of $19,519,162 was issued from February through December 2013 (the “February 2013 Notes”). There were no placement fees associated with the Convertible Notes, and other administrative costs were nominal and were expensed as incurred. The July 2012 Notes and the October 2012 Notes had maturity dates of July 31, 2015. The February 2013 Notes had a maturity date of February 28, 2016. The Convertible Notes accrued interest at the rate of 7.5% per annum, which is added to the principal amounts. For the year ended December 31, 2014, the annualized effective interest rate on the July 2012 Notes, the October 2012 Notes and the February 2013 Notes were 18.6%, 19.2%, and 63.3%, respectively. For the year ended December 31, 2013, the annualized effective interest rates on the July 2012 Notes, the October 2012 Notes, and the February 2013 Notes were 14.5%, 14.9% and 33.3%, respectively. For the year ended December 31, 2012, the annualized effective interest rates on the July 2012 Notes and the October 2012 Notes were 14.5% and 14.9%, respectively
The Convertible Notes were due on their respective maturity dates or convertible into the Company’s common stock upon the occurrence of a “capital event,” which is defined as (i) a sale of stock to a third party, excluding existing shareholders, of not less than $15,000,000, (ii) an initial public offering, or (iii) a “qualifying reorganization event” as defined in the Convertible Promissory Note agreement. The Convertible Promissory Note agreement contained a beneficial conversion feature that provided that if the notes were converted due to a capital event then all outstanding principal and interest would be converted into shares of common stock at the lower of the purchase price paid pursuant to the capital event, or at $5.00 per share. If no capital event occurred before the maturity date, the Convertible Promissory Note agreement provided that, at the election of the holder, all outstanding principal and interest could be converted to shares of common stock at $5.00 per share. During 2013, the debt issuance discount recorded in connection with this beneficial conversion feature was $10,487,645.
In connection with the Convertible Notes, the Company issued warrants to purchase 1,180,766 shares of the Company’s common stock. The warrants grant the holder the right to purchase additional shares of common stock of the Company equal to the product of (a) twenty percent, multiplied by (b) the face amount of the convertible note divided by $5.00. The exercise price for each share purchased under the warrant is $5.00. Until their expiration date, the warrants may be exercised at any time, and from time to time, in whole or in part. As originally issued, the warrants expired on the earlier of their expiration dates, upon a change in control event, or within 30 days of prior written notice of a pending IPO. In June 2014, the board of directors amended
F-21
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
7. Convertible Promissory Notes and Warrants (Cont’d)
the warrants to provide that they will not expire on the occurrence of an IPO. The warrants associated with the July 2012 Notes and the October 2012 Notes have an expiration date of July 31, 2017. The warrants associated with the February 2013 Notes have an expiration date of February 28, 2018. During 2013 and 2012, the debt issuance discount recorded in connection with the fair value of warrants issued was $3,107,379 and 1,044,649, respectively.
As of December 31, 2014, there were outstanding warrants associated with the Convertible Notes to purchase 1,178,707 shares of the Company’s common stock, with a weighted average remaining contractual life of 2.97 years.
During the fourth quarter of 2014, because of the successful completion of the Company’s IPO, the Company’s Convertible Notes were automatically converted into 6,639,137 shares of the Company’s common stock, and the unamortized discount on the Convertible Notes of $6,954,610 was written off.
The calculated value of the warrants associated with the Convertible Notes was estimated on the respective dates of grant using the Black-Scholes option-pricing model with the following assumptions:
|
|
|
2013 |
|
|
2012 |
|
||
|
|
|
|
|
|
|
|
|
|
|
Risk-free interest rate |
|
|
0.65% –1.68% |
|
|
|
0.60% – 83% |
|
|
Expected dividend yield |
|
|
0% |
|
|
|
0% |
|
|
Expected volatility |
|
|
57.5% |
|
|
|
63.9% |
|
|
Expected term |
|
|
4.2 – 5.0 years |
|
|
|
4.6 – 5.0 years |
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average grant date calculated fair value |
|
$ |
3.98 |
|
|
$ |
2.61 |
|
A summary for warrant activity the years ended December 31, 2014, 2013, and 2012 is presented below:
|
|
|
Number of Shares |
|
|
Weighted Average Exercise Price |
|
|
Weighted Average Remaining Contractual Life (in Years) |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Warrants outstanding at December 31, 2011 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
|
Granted |
|
|
400,000 |
|
|
|
5.00 |
|
|
|
|
|
|
Exercised |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
Forfeited or expired |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
Warrants outstanding at December 31, 2012 |
|
|
400,000 |
|
|
$ |
5.00 |
|
|
|
|
|
|
Granted |
|
|
780,766 |
|
|
|
5.00 |
|
|
|
|
|
|
Exercised |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
Forfeited or expired |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
Warrants outstanding at December 31, 2013 |
|
|
1,180,766 |
|
|
$ |
5.00 |
|
|
|
|
|
|
Granted |
|
|
805,000 |
|
|
|
11.25 |
|
|
|
|
|
|
Exercised |
|
|
(2,059 |
) |
|
|
5.00 |
|
|
|
|
|
|
Forfeited or expired |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
Warrants outstanding at December 31, 2014 |
|
|
1,983,707 |
|
|
$ |
7.54 |
|
|
|
3.75 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Warrants exercisable at December 31, 2014 |
|
|
1,178,707 |
|
|
$ |
5.00 |
|
|
|
2.97 |
|
F-22
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
7. Convertible Promissory Notes and Warrants (Cont’d)
The estimated aggregate intrinsic value of warrants exercisable at December 31, 2014 and 2013 was approximately $6,200,000 and $2,362,000, respectively.
Convertible promissory notes consisted of the following at December 31, 2013:
|
|
|
July 2012 Notes |
|
|
October 2012 Notes |
|
|
February 2013 Notes |
|
|
Total |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Principal outstanding |
|
$ |
5,000,000 |
|
|
$ |
5,000,000 |
|
|
$ |
19,519,162 |
|
|
$ |
29,519,162 |
|
|
Accrued interest |
|
|
482,331 |
|
|
|
404,072 |
|
|
|
837,693 |
|
|
|
1,724,096 |
|
|
Unamortized discount |
|
|
(554,285 |
) |
|
|
(586,272 |
) |
|
|
(10,891,589 |
) |
|
|
(12,032,146 |
) |
|
|
|
|
4,928,046 |
|
|
|
4,817,800 |
|
|
|
9,465,266 |
|
|
|
19,211,112 |
|
|
Less current portion |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Long-term portion |
|
$ |
4,928,046 |
|
|
$ |
4,817,800 |
|
|
$ |
9,465,266 |
|
|
$ |
19,211,112 |
|
8. Employee Benefit Plans
The Company has a 401(k) Savings Retirement Plan that covers substantially all full-time employees who meet the plan’s eligibility requirements and provides for an employee elective contribution. The Plan provides for employer matching contributions. Employer contributions are discretionary and determined annually by the Board of Directors. For the years ended December 31, 2014, 2013 and 2012, employer contributions to the Plan totaled $126,691, $109,866 and $129,486, respectively.
The Company is required to contribute to a government-sponsored pension plan for the employees of its Switzerland-based subsidiary. For the years ended December 31, 2014, 2013 and 2012, the employer’s portion of the amounts contributed to the subsidiary’s pension plan on behalf of those employees was $100,704, $94,157 and $78,029, respectively.
9. Equity Securities
In June 2014, the Company’s articles of incorporation were amended to increase authorized common shares to 200,000,000, no par value, and to authorize 10,000,000 shares of preferred stock, no par value. The Company’s consolidated financial statements have been retroactively restated to reflect this amendment. The Board of Directors has the authority to establish the rights, preferences, privileges and restrictions granted to and imposed upon the holders of preferred stock and common stock.
November 2014 IPO
On November 18, 2014, the Company sold 4,025,000 shares of common stock in an IPO, including 525,000 shares sold upon exercise of the underwriter’s over-allotment option. Net proceeds to the Company totaled approximately $34.2 million, net of offering costs of approximately $5.0 million, including approximately $2.9 million for the fair value of warrants and common stock issued in connections with services rendered. The proceeds from the IPO are expected to be used by the Company to invest in its business to expand sales and marketing efforts, enhance current product, gain regulatory approvals for additional indications, and continue research and development into next generation technology.
Underwriter’s Warrant
As a component of the IPO underwriting fee, the Company granted the underwriter a warrant to purchase 805,000 shares of the Company’s common stock at an exercise price of $11.25 per share, which was 25 percent above the offering price to the investors. The warrant is exercisable, in whole or in part, for a period commencing 180 days after the effective date of the
F-23
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
9. Equity Securities (Cont’d)
registration statement (November 18, 2014) and ending on the fifth anniversary date of the effective date of the registration statement. The fair value of the warrant issued as part of underwriting fee for the Company’s IPO was estimated to be $2,771,604, using the Black-Scholes option-pricing model with the following assumptions:
|
Risk-free rate of return |
|
|
1.63% |
|
|
Expected dividend yield |
|
|
0% |
|
|
Expected volatility |
|
|
49.92% |
|
|
Expected term |
|
|
5 years |
|
Long Term Investor Right
As of February 22, 2015, the deadline for IPO Shareholders to perfect their Long Term Investor Rights, investors had perfected rights relating to 1,777,799 IPO Shares. For the 90 day period ended on February 22, 2015, the average of the closing prices for the Company’s common stock on NASDAQ was $11.43. Based on this average closing price, an IPO Shareholder who has perfected its Long Term Investor Right would be entitled to additional 0.5743 shares for each share purchased in the offering, rounded up to the next whole share, which represents an aggregate maximum of 1,021,021 shares that are potentially issuable by the Company pursuant to the Long Term Investor Right at such date. The actual number of common shares issuable pursuant to the Long Term Investor Right is dependent on the future stock price of the Company over the two year period subsequent to the November 24, 2014 closing date of the Offering, and could be as low as zero shares.
2014 Private Placement
During 2014, the Company sold 1,299,853 shares of its common stock to new investors at $7.00 per share in a private placement, raising a total of $9,098,971. Related to this stock placement, the Company paid a finder’s fee of 26,785 shares of common stock to Mendelsohn Investment Services, LLC, a firm affiliated with Aaron Mendelsohn, a member of the Company’s Board of Directors. The Company paid an additional finder’s fee of 37,599 shares of common stock to an existing shareholder in connection with this stock placement.
2013 Private Placement
From July 1, 2013 through December 31, 2013, the Company sold 342,955 shares of its common stock to new investors at $7.00 per share in a private placement, raising a total of $2,400,685. No costs were incurred in connection with these issuances.
Common Stock Issuable
Beginning with services rendered in 2014, and with the first payment in June 2015, non-employee members of the Board of Directors will be paid for their services in common stock on June 1 of each year based on the average closing prices for the immediately preceding twenty trading days. For 2014, the Company accrued $166,250 for these services, which equates to 16,204 shares based on the $10.26 closing price for Company’s common stock on December 31, 2014. These shares have not yet been issued and are excluded from the calculation of weighted average common shares outstanding for EPS purposes.
10. Stock-Based Compensation
Under the 2003 Plan, as restated in June 2011, the Company was authorized to issue options covering up to 3,500,000 common stock shares. Effective June 1, 2011, the Company adopted the 2011 Equity Incentive Plan (the “2011 Plan”). The maximum number of shares with respect to which options may be granted under the 2011 Plan is 4,000,000 shares, which is offset and reduced by options previously granted under the 2003 Plan. The option price is determined by the Board of Directors but cannot be less than the fair value of the shares at the grant date. Generally, the options vest ratably over either four or five years and expire ten years from the grant date. Both plans provide for accelerated vesting if there is a change of control, as defined in the plans.
F-24
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
10. Stock-Based Compensation (Cont’d)
No option shall be granted under the 2011 Plan after May 31, 2021. The option price is determined by the Board of Directors. The term of each option will not to exceed ten years and the option exercise is subject to vesting and other conditions.
The Company recognized stock-based compensation cost of $1,474,645, $769,896 and $907,862 during 2014, 2013 and 2012, respectively. The calculated value of each option grant was estimated on the date of grant using the Black-Scholes option‑pricing model with the following assumptions:
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Risk-free interest rate |
|
|
0.3% – 2.2% |
|
|
|
1% |
|
|
|
0.93% – 1.3% |
|
|
Expected dividend yield |
|
|
0% |
|
|
|
0% |
|
|
|
0% |
|
|
Expected volatility |
|
|
50.0% –61.2% |
|
|
|
61.2% |
|
|
|
62.2% |
|
|
Expected term |
|
|
1.5-6.5years |
|
|
|
6.5 years |
|
|
|
6.25-6.5 years |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average grant date calculated fair value |
|
$ |
4.73 |
|
|
$ |
1.58 |
|
|
$ |
2.93 |
|
As the Company has limited stock trading history, the expected volatility is based on the historical volatility of similar companies that have a trading history. The expected term represents the estimated average period of time that the options are expected to remain outstanding. Since the Company does not have sufficient historical data on the exercise of stock options, the expected term is based on the “simplified” method that measures the expected term as the average of the vesting period and the contractual term. The risk free rate of return reflects the grant date interest rate offered for zero coupon U.S. Treasury bonds over the expected term of the options.
A summary of stock option activity for the years ended December 31, 2014, 2013 and 2012 is presented below:
|
|
|
Number of Shares |
|
|
Weighted Average Exercise Price |
|
|
Weighted Average Remaining Contractual Life (in Years) |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options outstanding at December 31, 2011 |
|
|
2,599,053 |
|
|
$ |
4.26 |
|
|
|
|
|
|
Granted |
|
|
190,100 |
|
|
|
5.00 |
|
|
|
|
|
|
Exercised |
|
|
(27,430 |
) |
|
|
3.67 |
|
|
|
|
|
|
Forfeited or expired |
|
|
(34,220 |
) |
|
|
4.67 |
|
|
|
|
|
|
Options outstanding at December 31, 2012 |
|
|
2,727,503 |
|
|
|
4.32 |
|
|
|
|
|
|
Granted |
|
|
500 |
|
|
|
5.00 |
|
|
|
|
|
|
Exercised |
|
|
(331,871 |
) |
|
|
1.05 |
|
|
|
|
|
|
Forfeited or expired |
|
|
(155,564 |
) |
|
|
3.73 |
|
|
|
|
|
|
Options outstanding at December 31, 2013 |
|
|
2,240,568 |
|
|
|
4.84 |
|
|
|
|
|
|
Granted |
|
|
1,377,978 |
|
|
|
7.62 |
|
|
|
|
|
|
Exercised |
|
|
(115,029 |
) |
|
|
4.40 |
|
|
|
|
|
|
Forfeited or expired |
|
|
(251,890 |
) |
|
|
4.44 |
|
|
|
|
|
|
Options outstanding at December 31, 2014 |
|
|
3,251,627 |
|
|
|
6.07 |
|
|
|
5.60 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options exercisable at December 31, 2014 |
|
|
1,870,667 |
|
|
$ |
4.87 |
|
|
|
3.00 |
|
F-25
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
10. Stock-Based Compensation (Cont’d)
The exercise prices of common stock options outstanding and exercisable are as follows at December 31, 2014:
| Exercise Price | Options Outstanding (Shares) | Options Exercisable (Shares) | ||||||||
| $ | 2.50 | 6,000 | 6,000 | |||||||
| $ | 3.75 | 10,000 | 10,000 | |||||||
| $ | 4.25 | 125,000 | 125,000 | |||||||
| $ | 4.75 | 476,159 | 476,159 | |||||||
| $ | 5.00 | 1,592,239 | 1,253,508 | |||||||
| $ | 7.00 | 262,095 | — | |||||||
| $ | 9.00 | 768,134 | — | |||||||
| $ | 14.06 | 12,000 | — | |||||||
| 3,251,627 | 1,870,667 | |||||||||
The estimated aggregate intrinsic value of stock options exercisable at December 31, 2014 and 2013 was approximately $10,080,000 and $3,996,000, respectively. As of December 31, 2014, there was $5,654,532 of total unrecognized compensation cost related to the outstanding stock options that will be recognized over a weighted average period of 3.95 years.
In January 2014, the Company granted a stock option to its chief executive officer to purchase 125,000 shares of common stock at an exercise price of $4.25 per share, exercisable for a period of three years from the date of grant. The stock option grant was fully vested on the date of issuance and was intended to replace an earlier stock option grant with the same exercise price that had expired in January 2014. The stock option was not granted pursuant to the 2011 Plan. The grant date fair value of the stock option, calculated pursuant to the Black-Scholes option-pricing model utilizing a volatility factor of 50% and a dividend rate of 0%, was determined to be $392,737, which was charged to operations as general and administrative expense in the year ended December 31, 2014.
During the year ended December 31, 2014, the Company recorded a charge of $235,347 to extend the exercise period of 232,003 options for four employees who resigned and became consultants for the Company. All unvested options for employees were terminated when they ceased full-time employment with the Company.
The total stock-based compensation recognized for stock-based awards granted in the consolidated statements of operations for the years ended December 31, 2014, 2013 and 2012 is as follows:
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of sales |
|
$ |
191,390 |
|
|
$ |
152,653 |
|
|
$ |
217,887 |
|
|
Research and development |
|
|
292,653 |
|
|
|
229,253 |
|
|
|
136,179 |
|
|
Clinical and regulatory |
|
|
112,999 |
|
|
|
82,686 |
|
|
|
118,022 |
|
|
Selling and marketing |
|
|
141,344 |
|
|
|
101,768 |
|
|
|
145,258 |
|
|
General and administrative |
|
|
736,259 |
|
|
|
203,536 |
|
|
|
290,516 |
|
|
Total |
|
$ |
1,474,645 |
|
|
$ |
769,896 |
|
|
$ |
907,862 |
|
From time to time, the Company has extended full-recourse loans to certain non-officer employees for the purpose of financing stock option exercises. These loans bear interest ranging from 1.27% to 1.91% per annum and are payable over three years in monthly installments of principal and interest. At December 31, 2014 and 2013, the outstanding balance of such loans, including accrued interest, was $108,798 and $24,661, respectively. These loans receivable are recorded in the Company’s consolidated financial statements as an offset to stockholders’ equity. Additionally at December 31, 2014 and 2013, the Company had a receivable in the amount of $10,182 and $12,500, respectively, from a non-officer employee for the exercise of options which has been recorded as an offset to stockholders’ equity in the Company’s consolidated financial statements at December 31, 2013.
F-26
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
10. Stock-Based Compensation (Cont’d)
On December 27, 2013, the Company extended a full-recourse loan totaling $127,165 to a consultant for the purpose of financing the exercise of stock options. The loan bears interest at 1.64% per annum and is repayable in eight equal quarterly installments of $16,192. This loan receivable is recorded in the Company’s consolidated financial statements as an offset to stockholders’ equity. At December 31, 2014 and 2013, the outstanding balance of this loan including accrued interest was $42,373 and $127,165, respectively.
Stock Awards
In July 2014, the Company awarded Alfred E. Mann, the Chairman of the Board of Directors, 25,000 shares of common stock in recognition of services rendered to the Company since inception. These shares were valued at $175,000, or $7.00 per share, and were charged to general and administrative expense in 2014.
In 2014, the Company awarded 21,215 shares to an outside attorney and his staff as part of the fee paid for drafting the Company’s prospectus and S-1 filing. These shares were valued at $169,505, with 10,715 shares valued at $7.00 per shares and the balance valued at $9.00 per share. The cost of these shares is treated as an issuance cost of the Company’s initial public offering and was deducted from the gross proceeds from the offering.
11. Income Taxes
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets as of December 31, 2014 and 2013 are summarized below:
|
|
|
2014 |
|
|
2013 |
|
||
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation |
|
$ |
2,489,000 |
|
|
$ |
1,164,000 |
|
|
Research credits |
|
|
5,436,000 |
|
|
|
5,201,000 |
|
|
Depreciation |
|
|
(13,000 |
) |
|
|
(13,000 |
) |
|
Net operating loss carryforwards |
|
|
43,700,000 |
|
|
|
37,773,000 |
|
|
Inventory reserve |
|
|
544,000 |
|
|
|
684,000 |
|
|
Other |
|
|
492,000 |
|
|
|
241,000 |
|
|
Total deferred tax assets |
|
|
52,648,000 |
|
|
|
45,050,000 |
|
|
Valuation allowance |
|
|
(52,648,000 |
) |
|
|
(45,050,000 |
) |
|
Net deferred tax assets |
|
$ |
— |
|
|
$ |
— |
|
In assessing the potential realization of these deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will be realized. The ultimate realization of deferred tax assets is dependent upon the Company attaining future taxable income during the periods in which those temporary differences become deductible. As of December 31, 2014 and 2013, management was unable to determine if it is more likely than not that the Company’s deferred tax assets will be realized, and has therefore recorded an appropriate valuation allowance against deferred tax assets at such dates.
No federal tax provision has been provided for the years ended December 31, 2014, 2013 and 2012 due to the losses incurred during such periods. The Company’s effective tax rate is different from the federal statutory rate of 34% due primarily to operating losses that receive no tax benefit as a result of a valuation allowance recorded for such losses.
F-27
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
11. Income Taxes (Cont’d)
As of December 31, 2014, the Company had federal and state income tax net operating loss carryforwards, which may be applied to future taxable income, of approximately $107,346,000 and $101,807,000, respectively. The federal net operating loss carryforwards will expire at various dates from 2023 through 2034. The state net operating loss carryforwards will expire at various dates from 2015 through 2034. The Company also has a federal and state research and development tax credit carryforwards totaling approximately $3,209,000 and $3,375,000, respectively. The federal research and development tax credit carryforwards will expire at various dates from 2023 through 2034. The state research and development tax credit carryforwards do not expire.
Pursuant to Internal Revenue Code Sections 382 and 383, use of the Company’s net operating loss and credit carryforwards may be limited if a cumulative change in ownership of more than 50% occurs within any three-year period since the last ownership change. The Company may have had a change in control under these Sections. However, the Company does not anticipate performing a complete analysis of the limitation on the annual use of the net operating loss and tax credit carryforwards until the time that it projects it will be able to utilize these tax attributes.
The Company files income tax returns in the U.S. federal jurisdiction and California and is subject to income tax examinations by federal tax authorities for tax years ended 2011 and later and by California authorities for tax years ended 2010 and later. The Company currently is not under examination by any tax authority. The Company’s policy is to record interest and penalties on uncertain tax positions as income tax expense. As of December 31, 2014, the Company has no accrued interest or penalties related to uncertain tax positions. Second Sight Switzerland, the Company’s foreign subsidiary, has not had any taxable income in the past.
12. Product Warranties
A summary of activity in the Company’s warranty liabilities, which are included in accrued expenses in the accompanying consolidated balance sheets, for the years ended December 31, 2014, 2013 and 2012 is presented below:
|
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, beginning of year |
|
$ |
253,200 |
|
|
$ |
120,440 |
|
|
$ |
35,353 |
|
|
Additional accruals |
|
|
415,276 |
|
|
|
138,490 |
|
|
|
105,125 |
|
|
Payments |
|
|
(112,504 |
) |
|
|
(5,879 |
) |
|
|
(22,574 |
) |
|
Adjustments and other |
|
|
— |
|
|
|
149 |
|
|
|
2,536 |
|
|
Total |
|
$ |
555,972 |
|
|
$ |
253,200 |
|
|
$ |
120,440 |
|
13. Commitments and Contingencies
Lease Commitment
Effective August 2012, the Company entered into a lease agreement (the “Sylmar Lease”) with a company owned by the major stockholder of the Company for office space for a term of five years that expires on February 28, 2017. The Sylmar Lease included rental of additional space commencing January 1, 2013 and a five year option to renew. The lease requires the Company to pay real estate taxes, insurance and common area maintenance each year, and is subject to periodic cost of living adjustments. In April 2014, the Sylmar Lease was renegotiated with the term ending on February 28, 2022, and a five year option to renew. The new lease also requires the Company to pay real estate taxes, insurance and common area maintenance each year and includes automatic increases in base rent each year. In November 2014, the property underlying the Sylmar lease was sold to an unrelated party.
Second Sight Switzerland rents office space in Switzerland on a month-to-month basis for CHF 7,079 (approximately $7,100 at December 31, 2014) per month.
F-28
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
13. Commitments and Contingencies (Cont’d)
Total rent expense was approximately $1,007,000, $766,000 and $458,000 for the years ended December 31, 2014, 2013 and 2012, respectively, and is allocated based on square footage to general and administrative and manufacturing costs in the accompanying consolidated statement of operations.
Future minimum rental payments required under the operating leases are as follows for the years ended December 31.
|
Years |
|
Amount |
|
|
|
|
|
|
|
|
|
2015 |
|
$ |
778,448 |
|
|
2016 |
|
|
808,068 |
|
|
2017 |
|
|
833,045 |
|
|
2018 |
|
|
858,036 |
|
|
2019 |
|
|
883,777 |
|
|
Thereafter |
|
|
2,004,919 |
|
|
|
|
|
|
|
|
Total |
|
$ |
6,166,293 |
|
License Agreements
The Company has exclusive licensing agreements to utilize certain patents. These patents are related to the technology for visual prostheses. There are currently two such agreements that the Company has determined there is a reasonable likelihood of future royalty payments. The Company has agreed to pay the licensors' royalties for licensed products sold or leased by the Company. The royalty rates range from 0.5% to 3.25%, based on related net sales of the patented portion of licensed products, less a credit for royalties paid to others. The 3.25% rate reflects a .25% credit for royalties paid to others. Additional discounts may be possible if we enter additional licenses.
One of the licensing agreements requires the Company to pay the licensors a $5,000 annual maintenance fee for the first seven years and a $10,000 annual maintenance fee each year thereafter for as long as the agreement has not been terminated by the Company. The second of these agreements has no stipulated fees. Pursuant to these agreements, the Company has incurred costs of approximately $45,260, $28,000 and $0 for the years ended December 31, 2014, 2013 and 2012, respectively.
Clinical Trial Agreements
Based upon FDA approval, which was obtained in February 2013, the Company is required to collect follow-up data from subjects enrolled in its pre-approval trial for a period of up to ten years post-implant, which extends this trial through the year 2019. In addition, the Company is conducting two post-market studies to comply with US FDA and European post-market surveillance regulations and requirements. The Company has contracted with various universities, hospitals, and medical practices to provide these services. Payments are based on procedures performed for each subject and are charged to clinical and regulatory expense as incurred. Total amounts charged to expense for the years ended December 31, 2014, 2013 and 2012 were $602,196, $480,950 and $385,556, respectively.
Litigation, Claims and Assessments
Nine oppositions have been filed by a third-party in the European Patent Office, each challenging the validity of a European patent owned or exclusively licensed by the Company. The outcome of the challenges is not certain, however, if successful, they may affect the Company’s ability to block competitors from utilizing its patented technology. Management of the Company does not believe a successful challenge will have a material effect on its ability to manufacture and sell its products, or otherwise have a material effect on its operations.
The Company is party to litigation arising in the ordinary course of business. It is management’s opinion that the outcome of such matters will have not have a material effect on the Company’s financial statements.
F-29
SECOND SIGHT MEDICAL PRODUCTS, INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
14. Subsequent Events
On February 25, 2015, the Board of Directors authorized, subject to shareholder approval, an increase of 2,000,000 shares in the number of shares available for option awards under the 2011 Equity Incentive Plan.
15. Quarterly Financial Summary (unaudited)
|
|
|
Quarters Ended |
|
|||||||||||||
|
|
|
December 31, 2014(1) |
|
|
September 30, 2014 |
|
|
June
30, |
|
|
March
31, |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product sales |
|
$ |
1,520,220 |
|
|
$ |
609,429 |
|
|
$ |
611,477 |
|
|
$ |
656,726 |
|
|
Gross profit (loss) |
|
$ |
99,204 |
|
|
$ |
192,797 |
|
|
$ |
(381,577 |
) |
|
$ |
(70,707 |
) |
|
Operating loss |
|
$ |
(5,565,045 |
) |
|
$ |
(5,649,054 |
) |
|
$ |
(5,562,876 |
) |
|
$ |
(4,456,485 |
) |
|
Net loss |
|
$ |
(13,577,118 |
) |
|
$ |
(7,644,301 |
) |
|
$ |
(7,541,075 |
) |
|
$ |
(6,438,753 |
) |
|
Net loss per share – basic and diluted |
|
$ |
(0.46 |
) |
|
$ |
(0.31 |
) |
|
$ |
(0.32 |
) |
|
$ |
(0.28 |
) |
|
|
|
Quarters Ended |
|
|||||||||||||
|
|
|
December 31, 2013 |
|
|
September 30, 2013 |
|
|
June
30, |
|
|
March
31, |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product sales |
|
$ |
563,631 |
|
|
$ |
235,677 |
|
|
$ |
313,750 |
|
|
$ |
451,875 |
|
|
Gross loss |
|
$ |
(998,347 |
) |
|
$ |
(1,044,756 |
) |
|
$ |
(575,980 |
) |
|
$ |
(1,445,304 |
) |
|
Operating loss |
|
$ |
(4,336,031 |
) |
|
$ |
(4,609,914 |
) |
|
$ |
(4,107,692 |
) |
|
$ |
(4,943,892 |
) |
|
Net loss |
|
$ |
(5,978,001 |
) |
|
$ |
(6,177,009 |
) |
|
$ |
(5,366,571 |
) |
|
$ |
(5,447,344 |
) |
|
Net loss per share – basic and diluted |
|
$ |
(0.27 |
) |
|
$ |
(0.27 |
) |
|
$ |
(0.24 |
) |
|
$ |
(0.24 |
) |
_______________
(1) During the fourth quarter of 2014, the Company wrote-off the unamortized discount on convertible promissory notes of $6,954,610 as a result of the automatic conversion of such notes into common stock upon the closing of the Company’s IPO.
F-30
EXHIBIT INDEX
| Exhibit No. | Exhibit Description | |
| 1.1 | Form of Underwriting Agreement.(1) | |
| 3.1 | Restated Articles of Incorporation of the Registrant(1) | |
| 3.2 | Amended and Restated Bylaws of the Registrant, as currently in effect.(1) | |
| 4.1 | Form of the Registrant’s common stock certificate.(1) | |
| 4.2 | Form of Underwriter’s Warrant.(1) | |
| 10.1 | Form of Indemnification Agreement between Registrant and each of its directors and officers.(1)+ | |
| 10.2 | 2003 Equity Incentive Plan.(1)+ | |
| 10.3 | 2003 Form of Employee Option Agreement.(1)+ | |
| 10.4 | 2011 Equity Incentive Plan.(1)+ | |
| 10.5 | 2011 Form of Employee Option Agreement.(1)+ | |
| 10.6 | 2014 Option Issued to Robert Greenberg – Terms and Conditions.(1)+ | |
| 10.7 | 2014 Executive Officer Option Agreement.(1)+ | |
| 10.8 | Form of Convertible Promissory Note.(1) | |
| 10.9 | Form of Warrant, as amended.(1) | |
| 10.10 | Standard Multi-Tenant Office Lease – Net, dated April 15, 2014, between Registrant and Mann Biomedical Park LLC.(1) | |
| 10.11 | Exclusive License Agreement between Registrant and Johns Hopkins University and Duke University.(1) | |
| 10.12 | Cost Reimbursement Consortium Research Agreement between Registrant and Doheny Eye Institute.(1) | |
| 10.13 | Form of Lock Up Agreement.(1) | |
| 10.14 | Shareholders’ Agreement dated September 5, 2003.(1) | |
| 10.15 | Offer Letter to Thomas Miller dated May 21, 2014.(1)+ | |
| 10.16 | Form of Loan Agreement dated September 30, 2014 between Mann Group LLC and Registrant for $3,000,000, including form of promissory note as Exhibit A thereto.(1) | |
| 10.17 | Joint Research and Development Agreement between Johns Hopkins University Applied Physics Laboratory and Registrant.(1) | |
| 21.1* | List of subsidiaries of the Registrant. | |
| 31.1* | Certification of Principal Executive Officer of Second Sight Medical Products, Inc. pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2* | Certification of Principal Financial and Accounting Officer of Second Sight Medical Products, Inc. pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1* | Certifications of Principal Executive Officer and Principal Financial and Accounting Officer of Second Sight Medical Products, Inc. pursuant to Rule 13a-14(b) under the Exchange Act and 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
_______________
* Included herein.
+ Indicates management contract or compensatory plan
(1) Incorporated by reference to the registrant’s registration statement on Form S-1, file no. 333-198073, originally filed with the Securities and Exchange Commission on August 12, 2014, as amended.
F-31