Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - SEELOS THERAPEUTICS, INC. | apri12312014ex321.htm |
| EX-10.27 - EXHIBIT 10.27 - SEELOS THERAPEUTICS, INC. | apri12312014ex1027.htm |
| EX-31.1 - EXHIBIT 31.1 - SEELOS THERAPEUTICS, INC. | apri12312014ex311.htm |
| EX-21 - EXHIBIT 21 - SEELOS THERAPEUTICS, INC. | apri12312014ex21.htm |
| EX-32.2 - EXHIBIT 32.2 - SEELOS THERAPEUTICS, INC. | apri12312014ex322.htm |
| EX-23.1 - EXHIBIT 23.1 - SEELOS THERAPEUTICS, INC. | apri12312014ex231.htm |
| EX-31.2 - EXHIBIT 31.2 - SEELOS THERAPEUTICS, INC. | apri12312014ex312.htm |
| EX-10.28 - EXHIBIT 10.28 - SEELOS THERAPEUTICS, INC. | apri12312014ex1028.htm |
| EX-10.26 - EXHIBIT 10.26 - SEELOS THERAPEUTICS, INC. | apri12312014ex1026.htm |
| EXCEL - IDEA: XBRL DOCUMENT - SEELOS THERAPEUTICS, INC. | Financial_Report.xls |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
(Mark One)
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2014
OR
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 0-22245
APRICUS BIOSCIENCES, INC.
(Exact Name of Registrant as Specified in Its Charter)
Nevada | 87-0449967 | |
(State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) | |
11975 El Camino Real, Suite 300, San Diego, CA 92130
(Address of Principal Executive Offices) (Zip Code)
(858) 222-8041
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Exchange on Which Registered | |
Common Stock, par value $.001 | The NASDAQ Capital Market | |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ¨ No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ¨ No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K(§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act (check one):
Large accelerated filer | o | Accelerated filer | ý | ||||
Non-accelerated filer | o | (do not check if a smaller reporting company) | Smaller Reporting Company | o | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No ý
As of March 11, 2015, 50,414,481 shares of the common stock, par value $.001, of the registrant were outstanding.
The aggregate market value of the common stock held by non-affiliates, based upon the last sale price of the registrant’s common stock on June 30, 2014, was approximately $85.4 million. Shares of common stock held by each officer and director and by each person who is known to own 10% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates of the Company. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required to be disclosed in Part III of this report is incorporated by reference from the registrant’s Proxy Statement for the 2015 Annual Meeting of Stockholders, which Proxy Statement will be filed no later than 120 days after the end of the fiscal year covered by this report.
Table of Contents
Page | ||
3
PART I.
Cautionary Note Regarding Forward-Looking Statements
This report includes “forward-looking statements” within the meaning of Section 21E of the Exchange Act. Those statements include statements regarding the intent, belief or current expectations of Apricus Biosciences, Inc. and its Subsidiaries (“we,” “us,” “our,” the “Company” or “Apricus”) and our management team. Any such forward-looking statements are not guarantees of future performance and involve risks and uncertainties, and actual results may differ materially from those projected in the forward-looking statements. These risks and uncertainties include but are not limited to those risks and uncertainties set forth in Item 1A of this Report. In light of the significant risks and uncertainties inherent in the forward-looking statements included in this Report, the inclusion of such statements should not be regarded as a representation by us or any other person that our objectives and plans will be achieved. Further, these forward-looking statements reflect our view only as of the date of this report. Except as required by law, we undertake no obligations to update any forward-looking statements and we disclaim any intent to update forward-looking statements after the date of this report to reflect subsequent developments. Accordingly, you should also carefully consider the factors set forth in other reports or documents that we file from time to time with the Securities and Exchange Commission (“SEC”).
ITEM 1. BUSINESS
Overview
We are a Nevada corporation that was initially formed in 1987. We have operated in the pharmaceutical industry since 1995, with a current primary focus on the development and commercialization of products and product candidates in the areas of specialty urology and rheumatology. Our proprietary drug delivery technology is a permeation enhancer called NexACT® and we have one approved drug, Vitaros®, which uses the NexACT® delivery system and is approved for the treatment of erectile dysfunction (“ED”) in Canada and through the European Decentralized Procedure (“DCP”) in Europe. Vitaros® was launched by our licensee partners in certain territories in Europe in the second half of 2014 and we expect that commercial launches will continue to occur throughout 2015. We have a second-generation Vitaros® product candidate (“Room Temperature Vitaros®”) in development, which is a proprietary stabilized dosage formulation that is expected to be stored at room temperature conditions. RayVa™, our product candidate that also utilizes our proprietary permeation enhancer for the treatment of Raynaud's Phenomenon, received clearance in May 2014 from the United States Food and Drug Administration (“FDA”) to begin clinical studies, and our Phase 2a clinical trial began in December 2014.
In October 2014, we entered into an agreement to license the exclusive United States development and commercialization rights for fispemifene, a tissue-specific selective estrogen receptor modulator (“SERM”) designed to treat secondary hypogonadism, chronic prostatitis and lower urinary tract symptoms in men. We expect to commence a Phase 2 clinical trial with fispemifene during the first half of 2015.
We are seeking to out-license our product candidate, Femprox®, for female sexual interest / arousal disorder (“FSIAD”), to one or more partners for future development.
Growth Strategy
To develop and commercialize our proprietary products and product candidates, through these primary initiatives:
Commercialize Vitaros® through partnerships
We currently have commercial partnerships for Vitaros® with the following pharmaceutical companies in the countries indicated:
4
Partner | Licensed Territory |
Abbott Laboratories Limited (“Abbott”) | Canada |
Takeda Pharmaceuticals International GmbH (“Takeda”) | United Kingdom (the “UK”) |
Hexal AG (“Sandoz”) | Germany, Austria, Belgium, Luxemburg, the Netherlands, Denmark, Finland, Iceland, Norway, Sweden and Switzerland, Malaysia, Indonesia, the Philippines, Thailand, Taiwan, Vietnam, Hong Kong and Singapore |
Laboratoires Majorelle (“Majorelle”) | France, Monaco and certain African countries |
Bracco SpA (“Bracco”) | Italy, Vatican City and San Marino |
Recordati Ireland Ltd. (“Recordati”) | Spain, Ireland, Portugal, Greece, Cyprus, the CEE countries (Central and Eastern Europe), Russia and the rest of the CIS countries (former Soviet republics), Ukraine, Georgia, Turkey and certain African countries |
Neopharm Scientific Limited (“Neopharm”) | Israel and the Palestinian National Authority |
Elis Pharmaceuticals Limited (“Elis”) | Gulf States and certain Middle Eastern countries |
Global Harvest Pharmaceutical Corporation (“Global Harvest”) | Australia and New Zealand |
During 2014, we commenced shipment of our product, Vitaros®, to certain commercialization partners. Takeda, our commercialization partner in the UK, became the first to market Vitaros® with its launch in June 2014. In August 2014, Sandoz launched Vitaros® in Sweden and Germany and in November 2014, Sandoz launched Vitaros® in Belgium. We expect additional launches in other approved countries by our commercial partners during 2015.
In 2009, we sold the commercial rights to Vitaros® in the United States to Warner Chilcott Company, Inc. (“Warner Chilcott”), now a subsidiary of Actavis plc. (“Actavis”). We believe there is a significant commercial opportunity for Vitaros® in the United States and are in discussions with Actavis to explore the various options available to move the Vitaros® clinical development program forward in the United States
Develop and commercialize in the United States additional technologies and products based upon proprietary technologies developed in-house or acquired from third-parties
In October 2014, we signed an in-license agreement with Forendo Pharma Ltd., (“Forendo”) under which we were granted certain exclusive rights to develop and commercialize fispemifene, a tissue-specific SERM designed to treat secondary hypogonadism, chronic prostatitis and lower urinary tract symptoms in men, in the United States We intend to initiate a Phase 2 clinical trial in the first half of 2015 and depending on those results and based upon the feedback received from the FDA, initiate a Phase 3 clinical trial in 2016.
In May 2014, we received clearance from the FDA to begin clinical testing of RayVa™, our product candidate for the treatment of Raynaud’s Phenomenon secondary to scleroderma. Raynaud's Phenomenon is characterized by vasoconstriction in the hands, feet or other extremities, resulting in reduced blood flow and the sensation of pain, which can become severe. RayVa™ combines alprostadil, a vasodilator, with our proprietary permeation enhancer DDAIP.HCl, and is applied as an on-demand topical cream to affected extremities. We initiated a Phase 2a clinical trial for RayVa™ in December 2014.
We also plan to continue to evaluate other product candidates and we may also acquire or develop other complementary products leveraging our regulatory and development experience.
Establish new Vitaros® and RayVa™ licensing partnerships with pharmaceutical companies
In the future, we will seek new partnerships to license, develop, and commercialize Vitaros® and RayVa™ in markets not covered by existing partnerships. For Vitaros®, these territories primarily consist of the following countries and regions: (1) Latin America: Mexico, Brazil and other Central and South American countries, (2) Japan and (3) China. For RayVa™, these territories consist of all countries outside of the United States. We expect that any such agreements will provide us with one or more of the following: up-front payments, the right to receive regulatory and sales milestone payments and/or royalty payments.
NexACT® Drug Delivery Technology
The NexACT® drug delivery technology is designed to enhance the delivery of an active drug to the patient. If successful, the combination of our NexACT ® technology with active drugs could improve therapeutic outcomes and reduce systemic side effects that often accompany existing medications.
The NexACT® technology consists of a small molecule permeation enhancer called Dodecyl 2-(N,N dimethylamino)-propionate (“DDAIP”) that enables the rapid absorption of high concentrations of an active pharmaceutical ingredient directly at the target site, which is designed to enhance the delivery of an active drug to the patient.
5
NexACT® was designed to enable multi-route administration of active drugs across numerous therapeutic classes. The NexACT® technology has been tested in human clinical trials by us and our partners as a means of transdermal delivery of drugs (through the skin) and has been shown in pre-clinical animal studies to have the potential to serve as an effective vehicle for the delivery of a wide range of drugs and drug classes, via numerous routes of administration, including transdermal (topical), oral, subcutaneous, rectal and buccal (absorbed in the mouth).
NexACT® is based on proprietary permeation enhancers that are biodegradable, biocompatible, and mimic the composition of human skin. NexACT® has been tested in human clinical trials in over 5,000 patients, including those subjects exposed to Vitaros®, Femprox®, and RayVa™. In these clinical trials, NexACT® demonstrated a favorable safety profile, with minimal serious adverse events that were likely attributed to the active ingredients in the drug candidates.
Product and Product Candidate Portfolio
Vitaros® for Erectile Dysfunction
Vitaros®, our lead product for the treatment of ED, is a topically-applied cream formulation of alprostadil, a vasodilator and NexACT®, which directly increases blood flow to the penis causing an erection. Alprostadil is one of several treatment options for ED, and is a widely accepted alternative to the PDE5 inhibitors, such as Viagra®. Following the approval by the European and Canadian Health Authorities, Vitaros® has been deemed a safe and effective treatment, and has the potential to address a meaningful market opportunity due to its patient-friendly form of administration versus both other alprostadil dosage forms and its non-systemic safety profile.
The current leading ED medications are taken in pill form and work by inhibiting an enzyme called PDE5. We believe there is a need for new, safe and effective treatments, especially for those patients who cannot or prefer not to take or do not respond to oral medications. Vitaros® is a topically-applied, on-demand, non-PDE5 inhibitor that may be appropriate for ED patients who:
1. | Want a fast-acting and on-demand treatment; |
2. | Prefer a locally-acting treatment instead of an oral systemic treatment; |
3. | Have contraindications to PDE5 inhibitors due to medications or concurrent disease (estimated to be approximately 18% of the ED market); |
4. | Are healthy enough to take the PDE5 inhibitors but stop taking them because they are non-responders (estimated to be approximately 21% of the ED market); or |
5. | Drop out because of poor tolerability or side effects from oral PDE5 inhibitors. |
Factors such as these lead to an estimated 31% drop out rate after initial prescription for patients taking sildenafil citrate, which increases to an estimated 48% drop-out rate after three years of taking the drug.
In clinical studies, Vitaros® showed efficacy in patients suffering from ED, including men who did not respond to sildenafil citrate. The side effects reported were localized and transient. According to the European Male Aging Study (“EMAS”) assessing ED prevalence in eight countries, 30% of men in that study reported moderate or severe ED increasing to 64% in men 70 years or older. In Germany and Spain, the prevalence of ED was reported to be between 18-19% for men 40 years or older while in France, the percentage climbs to 32%. With an overall ex-United States ED market affecting nearly 150 million men worldwide and representing approximately $2.0 billion in revenue, we believe that Vitaros® represents a major market opportunity, particularly as a distinct product that addresses a significant underserved population.
Vitaros® is currently manufactured by Therapex, a division of E-Z-EM Canada Inc., a wholly-owned subsidiary of Bracco SpA in Italy (“Therapex”) and by Groupe Parima, Inc. Our third-party manufacturers are subject to numerous regulations, including Good Manufacturing Practices, or cGMPs, FDA regulations governing manufacturing processes and related activities and similar foreign regulations. Both of these manufacturers are located in Canada and are capable of providing commercial product for our partners. We currently have manufacturing and supply agreements in place with many of our commercialization partners; however, in 2015, we expect our partners will begin to work directly with our third-party manufacturers.
The first-generation Vitaros® product (“Cold Chain Vitaros®”) is stored in one chamber of our AccuDose® dispenser. This single-chamber formulation requires that the product be stored by customers in a refrigerator until a short time prior to use. Cold Chain Vitaros® was the product used in the Company’s clinical trials, and was approved in Canada and Europe. In November 2010, Health Canada approved Cold Chain Vitaros® for a current shelf-life of nine months for the 330 micrograms (“mcg”) product and six months for the 220 mcg product. These shelf-life durations are calculated at a temperature of 2°C-8°C. At room temperature conditions, Cold Chain Vitaros® has an approved shelf-life of up to seven days. Therefore, Cold Chain Vitaros® can be conveniently carried by the patient and brought up to room temperature prior to use. In June 2013, through the European DCP, Vitaros® was approved for a current shelf-life of eighteen months for the 300 mcg product and nine months for the 200 mcg product. These shelf-life durations are calculated at a temperature of 2°C-8°C. At room temperature conditions, Cold Chain Vitaros® has an approved shelf-life in Europe of up to three days through the European DCP.
6
It is expected that the product ingredients in our second-generation Vitaros® product candidate (“Room Temperature Vitaros®”), will be stored in two separate chambers. This will allow alprostadil to be segregated from ingredients that cause it to become unstable at room temperature. The contents of each of the two chambers are then mixed in the dispenser immediately prior to use. This mixture is expected to result in the same pharmaceutical formulation as the approved Cold Chain Vitaros®. This proprietary stabilized dosage form is expected to allow the product to be stored at room temperature conditions with a current target shelf-life duration for Room Temperature Vitaros® of twenty-four months. We plan to perform the necessary equivalence and stability studies to market Room Temperature Vitaros® in Canada, Europe and other future-approved territories, and to seek to increase the shelf-life over time. If successfully developed, this second-generation product candidate is expected to be filed for approval in Europe in 2016.
Competition for Vitaros®
There is significant competition and financial incentive to develop, market and sell drugs for the treatment of ED. Leading drugs approved for ED indications are PDE5 inhibitors that target the vascular system, such as sildenafil citrate (sold by Pfizer under the trade name Viagra®), vardenafil (sold by GlaxoSmith-Kline under the trade name Levitra®) , tadalafil (sold by Lilly under the trade name Cialis®) and avanafil (sold in the United States by Endo Pharmaceuticals, Inc. under the trade name Stendra® and sold in Europe and New Zealand by The Menarini Group under the trade name Spedra®). In addition, we are aware of other PDE5 inhibitors under development. As patents for the three major PDE5 inhibitors, sildenafil citrate, tadalafil and vardenafil, are expiring over various dates in each country, we anticipate that generic PDE5 inhibitors will impact the overall market for ED products. Generic PDE5 inhibitors are being sold at lower prices than their brand equivalents. Other drugs approved for ED indications include alprostadil for injection directly into the penis (sold by Pfizer under the trade name Caverject Impulse®, and Edex, sold in the United States by Endo Pharmaceuticals, Inc.), and alprostadil in urethral suppository format (sold by Meda under the trade name MUSE®). In addition, a variety of devices, including vacuum devices and surgical penile implants, have been approved for ED indications. We are aware of a number of companies developing new drugs for ED indications including Futura Medical Inc., which is developing MED 2002, a topical gel applied directly to the penis for the treatment of ED. MED2002 is based on the active compound glyceryl trinitrate within a patented gel delivery system. We are not aware of any company actively developing a topical alprostadil drug for ED.
Commercialization of Vitaros®
United States
In February 2009, we sold the United States rights for Vitaros® and the specific United States patents and trademarks covering Vitaros® for the treatment of ED to Warner Chilcott, now Actavis plc. Under the terms of the agreement, we received gross proceeds of $2.5 million as an up-front payment and we are eligible to receive an additional payment of $2.5 million upon receipt of a New Drug Application (“NDA”) approval from the FDA. The purchase agreement gives us the right to reference their work on Vitaros® in our future filings outside the United States, which may benefit us in international partnering opportunities as any additional data generated may further validate the safety of the product and enhance its potential value. We are not able to provide guidance on the Actavis development and commercialization plans for Vitaros® until further clarity is provided by Actavis.
Canada
Vitaros® was approved in November 2010 in Canada for the treatment of ED. In January 2012, we entered into a license agreement with Abbott, granting Abbott the exclusive rights to market Vitaros® for the treatment of ED in Canada. To date, we have received $2.5 million in up-front payments and are eligible to receive up to an additional $13.2 million in regulatory and sales milestones payments, plus tiered royalty payments based on Abbott’s sales of the product. We understand that Abbott Canada continues to work toward the launch of Vitaros® in Canada.
Europe
In June 2013, we received approval in Europe through the DCP for commercialization of Vitaros®, giving us the right to sell Vitaros® in multiple countries in the European Union. We then entered into the national step where each country makes decisions on region-specific issues such as approval of translations for labeling, and pricing or reimbursement, if applicable. Once this step is completed in any individual country, the product can be marketed in that country. The national step is performed by the Marketing Authorization Holder (“MAH”). In countries where Vitaros® has been licensed to a partner, that partner will be the MAH and make the submissions. Vitaros® is currently approved for marketing in the Netherlands, Germany, the UK, Ireland, Italy, France, Belgium, Luxembourg, Spain and Sweden and is currently launched, or expected to be launched in 2015, in the countries where we have commercial partners.
Italy
In December 2010, we entered into a license agreement with Bracco, granting Bracco the exclusive rights to market Vitaros® for the treatment of ED in Italy. To date, we have received $1.3 million in upfront payments and regulatory milestones. We are eligible to receive up to a total of €4.5 million ($5.5 million as of December 31, 2014) in regulatory and sales milestone payments and
7
payments for certain regulatory filing costs. Additionally, we are entitled to receive tiered double-digit royalties on Bracco’s sales of the product. In November 2013, the Italian Medicines Agency granted national phase approval to Vitaros® indicated for the treatment of ED patients in Italy with ED.
Germany, Benelux, the Nordics and Switzerland
In February 2012, we entered into a license agreement with Sandoz, granting Sandoz the exclusive rights to market Vitaros® in Germany for the treatment of ED. In December 2013, we amended and restated the agreement to include Austria, Belgium, Denmark, Finland, Iceland, Luxembourg, Norway, the Netherlands, Sweden and Switzerland (the “Expanded Territory”). In June 2014, we entered into a Manufacturing and Supply Agreement with Sandoz whereby we or our contract manufacturer will manufacture Vitaros® product and supply the product to Sandoz on a cost plus basis. To date, we have received $4.0 million in upfront payments and launch milestones and are eligible to receive up to an additional €0.2 million ($0.2 million as of December 31, 2014) in regulatory milestones, up to $1.5 million in marketing launch milestones, and up to €41.75 million ($50.7 million as of December 31, 2014) in aggregate sales milestones. In addition, we are entitled to receive tiered double-digit royalties on Sandoz’ sales of the product.
In 2013, Germany's Federal Institute for Drugs and Medical Devices, the Netherlands’ Medicines Evaluations Board and Sweden’s National Board of Health and Welfare each granted national phase approval to Vitaros® indicated for the treatment of patients with ED. In January 2014, Belgium’s Ministry of Social Affairs, Public Health and Environment granted national phase approval to Vitaros® indicated for the treatment of patients with ED. Sandoz launched Vitaros® in Germany and Sweden in August 2014 and in Belgium in November 2014.
We have filed a marketing application in Switzerland with Swissmedic, the Swiss Agency for Therapeutic Products, for Vitaros® as a treatment for patients with ED. The Swiss regulatory comments for the marketing approval of Vitaros® are expected in the second quarter of 2015.
France, Monaco and certain African countries
In November 2013, we entered into a license agreement with Majorelle, granting Majorelle the exclusive rights to market Vitaros® for the treatment of ED in France, Monaco and certain African countries. In September 2014, we entered into a Manufacturing and Supply Agreement with Majorelle whereby we or our contract manufacturer will manufacture Vitaros® product and supply the product to Majorelle on a cost plus basis. In addition, during the first quarter of 2015, Groupe Parima began manufacturing product for Majorelle’s commercial launch anticipated in the first half of 2015.
In December 2013, France's National Agency for Medicines and Health Products Safety granted national phase approval to Vitaros® indicated for the treatment of patients with ED. To date, we have received $2.0 million in up-front payments and launch milestones and are eligible to receive up to an additional $2.0 million in marketing launch milestones and €15.5 million ($18.8 million as of December 31, 2014) in sales milestones, as well as double-digit tiered royalties on Majorelle’s sales of the product. In a related negotiation, Majorelle made severance payments to certain former employees of Scomedica SAS, NexMed Europe SAS and NexMed Pharma SAS (the “French Subsidiaries”) for an aggregate amount of approximately $2.0 million on our behalf.
The United Kingdom
In September 2012, we entered into a license agreement with Takeda, granting Takeda the exclusive rights to market Vitaros® for the treatment of ED in the UK. In September 2013, we entered into a Manufacturing and Supply Agreement with Takeda whereby we or our contract manufacturer will manufacture Vitaros® product and supply the product to Takeda on a cost plus basis. In August 2013, the United Kingdom’s Medicines and Healthcare Products Regulatory Agency granted national phase approval to Vitaros® indicated for the treatment of ED. To date, we have received $1.0 million in up-front payments and are eligible to receive up to a total of €34.65 million ($42.1 million as of December 31, 2014) in regulatory and sales milestone payments and payments for certain regulatory filing costs. Additionally, we are entitled to receive double-digit tiered royalties on Takeda’s sales of the product. Takeda launched Vitaros® in the United Kingdom in June 2014.
Spain, Ireland, Portugal, Greece, Cyprus, the CEE countries, Russia as well as the other CIS countries, Ukraine, Georgia, Turkey and certain African countries
In February 2014, we entered into a license agreement with Recordati, granting Recordati the exclusive rights to market Vitaros® for the treatment of ED in Spain, Ireland, Portugal, Greece, Cyprus, the CEE Countries (Central and Eastern Europe), Russia as well as the other CIS Countries (former Soviet republics), Ukraine, Georgia, Turkey and certain African countries. In June 2014, we entered into a Manufacturing and Supply Agreement with Recordati whereby we or our contract manufacturer will manufacture Vitaros® product and supply the product to Recordati on a cost plus basis. To date, we have received $2.5 million in up-front payments and are entitled to receive up to €1.0 million ($1.2 million as of December 31, 2014) in launch payments and €34.5 million ($41.9 million as of December 31, 2014) in sales milestones. Additionally, we are entitled to receive tiered double-digit royalties based on Recordati’s sales of the product.
8
Rest of World
The Middle East
In January 2011, we entered into a license agreement with Elis, granting Elis the exclusive rights to market Vitaros® for the treatment of ED in the United Arab Emirates, Oman, Bahrain, Qatar, Saudi Arabia, Kuwait, Lebanon, Syria, Jordan, Iraq and Yemen. To date, we have received $0.1 million in upfront payments and are eligible to receive up to $2.1 million in regulatory and sales milestone payments, plus tiered double-digit royalties based on Elis’ sales of the product.
In February 2011, we entered into a license agreement with Neopharm, granting Neopharm the exclusive rights to market Vitaros® for the treatment of ED in Israel and the Palestinian Territories. To date, we have received $0.1 million in upfront payments and are eligible to receive up to $4.35 million in regulatory and sales milestone payments, plus tiered double-digit royalties based on Neopharm’s sales of the product.
Elis and Neopharm are responsible for the registration process in their respective territories.
Asia
In February 2015, we amended our license agreement with Sandoz to grant Sandoz exclusive rights to market Vitaros® for the treatment of ED in the following countries: Malaysia, Indonesia, the Philippines, Thailand, Taiwan, Vietnam, Hong Kong and Singapore (the “Expanded APAC Territory”). To date, we have earned $0.4 million in upfront payments and are eligible to receive an additional regulatory milestone payment of $0.1 million as well as tiered double-digit royalties based on Sandoz’ sales of the product.
We have not yet established development or commercial plans for Japan or China.
Australia and New Zealand
In June 2009, we entered into a license agreement with Global Harvest, granting Global Harvest the exclusive rights to market Vitaros® for the treatment of ED in Australia and New Zealand. We are eligible to receive royalty payments on Global Harvest’s sales of the product. Global Harvest filed for approval with the Therapeutic Goods Administration (“TGA”) in Australia in December 2014 with a preliminary evaluation by the TGA expected mid-2015.
Latin America
We are currently in discussions with potential commercialization partners for the licensing rights to market Vitaros® for the treatment of ED in the Latin American territory.
RayVaTM for the Treatment of Raynaud’s Phenomenon Secondary to Scleroderma
RayVaTM is our product candidate for the treatment of Raynaud’s Phenomenon secondary to scleroderma. Raynaud’s Phenomenon secondary to scleroderma is a disorder of the small blood vessels of the extremities, which affects approximately 100,000 people in the United States RayVa utilizes our NexACT® technology, combining alprostadil and DDAIP in an on-demand topical application to the affected areas. There are currently no approved prescription treatments in the United States for Raynaud’s Phenomenon and we are unaware of any other products currently in development to treat Raynaud’s Phenomenon secondary to scleroderma.
We began patient enrollment in December 2014 for a Phase 2a proof-of-concept clinical trial. Upon successful completion of the Phase 2a proof-of-concept study, we plan to initiate a Phase 2b clinical trial.
Fispemifene for the Treatment of Secondary Hypogonadism
In October 2014, we licensed from Forendo the exclusive United States rights to develop and commercialize fispemifene, a tissue-specific SERM designed to treat secondary hypogonadism, chronic prostatitis and lower urinary tract symptoms in men. Fispemifene acts using the body’s own regulatory mechanisms, through the hypothalamus and pituitary glands, to normalize production of testosterone by the testes whereas testosterone replacement therapies do not. Fispemifene has also been shown to provide other benefits such as reduction of prostate inflammation, improved urodynamics, and preservation of bone density, among others. We plan to initiate a Phase 2 clinical trial during the first half of 2015.
Competition for Fispemifene
There is significant competition and financial incentive to develop, market and sell drugs for the treatment of hypogonadism (a syndrome consisting of “Low-Testosterone” or “Low-T” and related symptoms). Leading drugs approved and on the market for Low-T belong to a class called testosterone replacement therapy (“TRT”). This class consists of branded and generic topical testosterone gels, patches, buccal tabs, implantables, and injectables, such as Androgel (sold by Abbott Labs), Axiron (sold by Eli Lilly), and Testim, Testopel, Striant, Aveed and Fortesta (sold by Endo Pharmaceuticals, Inc.) that increase hormone levels via external testosterone supplementation. In addition, we are aware of other testosterone formulations under development, which include a subcutaneous delivery from Antares and oral formulations from Clarus Therapeutics and Lipocine. As patents expire
9
for the leading topical testosterone formulation, Androgel, we anticipate that generic gel alternatives will impact the overall market for testosterone replacement. There are no other FDA-approved SERMs for hypogonadism, which differ from TRT in that SERMs increase the body’s own production of endogenous testosterone. A future potential competitor to fispemifene is enclomifene citrate (developed by Repros Therapeutics). Enclomifene is the trans-isomer of a compound named clomiphene, which is aSERM approved for the treatment of infertility. We are aware that clomiphene is sometimes used off-label to treat hypogonadism despite not being approved by the FDA for that use. Beyond enclomifene, we are not aware of any other company actively developing a SERM for hypogonadism.
Femprox® for Female Sexual Interest & Arousal Disorder
We have developed Femprox®, an alprostadil-based cream product candidate which also utilizes our NexACT® technology. Femprox® is intended for the treatment of FSIAD. Women with FSIAD have a lack of sexual desire and a persistent or recurring inability to attain or maintain sufficient sexual excitement. The lack of sufficient sexual excitement may be due to decreased subjective responsiveness, lack of genital lubrication and swelling or other somatic responses. This disorder may be related to a medical/physiologic problem such as reduced genital blood flow and not necessarily related to psychological/hormonal factors as in other types of Female Sexual Dysfunction (“FSD”). FSIAD has been recognized as a sexual disorder diagnosis by the United States American Psychiatric Association as described in the Diagnostic and Statistical Manual of Mental Diseases, 5th Edition (“DSM-5”) issued in May 2013.
Femprox® utilizes alprostadil, which induces vasodilation through a direct effect on vascular smooth muscle. This results in genital engorgement and surface lubrication, which are components of genital arousal. NexACT®, as a patented proprietary permeation enhancer, opens the cellular tight junction, allowing drugs to permeate through epidermal barriers, enabling rapid absorption of high concentrations of alprostadil cream directly to the target site. The Femprox® cream will be contained in our proprietary unit-dose dispenser for easy application to the clitoris and distal anterior vaginal wall.
We have completed seven clinical studies to date on Femprox®. Approximately 100 women were exposed to Femprox® in Phase 1 clinical trials, including a hemodynamic assessment. In Phase 2/3 studies, approximately 350 female sexual arousal disorder patients were treated with various dosages of Femprox® to evaluate safety and efficacy.
In a Phase 2/3 study conducted in 2005, we initiated clinical trials on approximately 400 patients. The results of the Phase 2/3 study were that all three dose levels of Femprox® met the primary endpoint. The highest dose level met all secondary endpoints resulting in statistically significant and clinically relevant responses compared to the placebo group. In addition, the Phase 2/3 study showed a favorable safety and tolerability profile with only five patients (1.2%) withdrawing from the study because of local adverse events. No drug-related serious adverse events were reported in the Phase 2/3 study.
We met with the United States FDA in August 2013 and with the European regulatory authorities in the Netherlands and Germany in January 2014, where we received regulatory guidance for the further development of Femprox®. Our strategic goal for this asset is to enter into one or more agreements with third parties for the development and ultimate commercialization of Femprox®, if successfully developed.
Competition for Femprox®
There is limited competition to develop, market and sell drugs for the treatment of FSIAD. At this time, there are no approved drugs for the treatment of FSIAD in the United States We are aware of a non-hormone oral drug, flibanserin, owned by privately-held Sprout Pharmaceuticals, Inc., that has been investigated for treatment of premenopausal women with hypoactive sexual desire disorder, and remains under review with the FDA. In addition, Palatin Technologies, Inc. is currently in Phase 3 development with a melanocortin receptor agonist for FSD. A number of hormonal therapies have been commercialized for other indications, including progestin, androgen and localized estrogen therapies, but none have been approved by the FDA for FSIAD or FSD indications. We are not aware of any company actively developing a topical alprostadil drug for FSD.
Patent Portfolio
We currently own or exclusively license approximately 331 issued patents which will expire from 2017 through 2032, approximately, and 194 patent applications. Should the patent applications issue, they may extend our patent exclusivity on our NexACT® technology, our acquired products and on our other products and technologies throughout the world until approximately 2032, based upon the potential expiration date of the last to expire of those patent applications. Patents covering Vitaros® for ED have been issued in Australia, Canada, Eurasia, Europe, Hong Kong, Israel, Japan, Mexico, New Zealand, Singapore, South Africa, South Korea, Turkey, Taiwan, and the United States. We have licensed our patent rights to Vitaros® to commercial partners in a number of these countries and we are actively seeking commercial partners in other jurisdictions.
In the United States, we hold 16 United States patents in connection with our NexACT® technology and our NexACT®-based products under development. In January 2015, the United States Patent and Trademark Office (“USPTO”) issued to us a United States patent related to methods for treating Raynaud’s Phenomenon, that is secondary to systemic sclerosis.
10
In October 2014, we exclusively licensed United States patents and applications from Forendo. The licensed patents and applications include five United States patents and two United States patent applications related to the SERM, fispemifene, for investigational treatment for urological conditions in men. These patents will expire between 2020 and 2028.
To further strengthen our global patent position on our proprietary products under development and to expand the patent protection to other markets, we have filed foreign patent applications, many of which correspond to our issued United States patents and pending United States patent applications. These foreign filings have resulted in numerous issued patents and currently pending patent applications.
While we have obtained patents and have patent applications pending, the extent of effective patent protection in the United States and other countries is highly uncertain. No consistent policy addresses the breadth of claims allowed in or the degree of protection afforded under patents of medical and pharmaceutical companies. Patents we currently own or may obtain might not be sufficiently broad to protect us against competitors with similar technology. Any of our patents could be invalidated or circumvented.
The holders of competing patents could determine to commence a lawsuit against us and may even prevail in any such lawsuit. Litigation could result in substantial cost to and diversion of effort by us, which may harm our business. In addition, our efforts to protect or defend our proprietary rights may not be successful or, even if successful, may result in substantial cost to us.
Trademark Portfolio
We currently own approximately 113 registered trademarks, 47 pending trademark applications and six allowed pending trademark applications worldwide. We own registered trademarks for Vitaros®, Femprox® and NexACT® in certain countries and territories throughout the world.
While we have obtained registered trademarks, have trademark applications pending and may have common law trademark rights where applicable, the extent of effective trademark protection in the United States and other countries is highly uncertain. Trademarks we currently own or may obtain might not be sufficiently broad to protect us against competitors. Any of our trademarks could be invalidated or circumvented.
Even where we have registered trademarks, competitors could seek to invalidate these registrations. Any such litigation could result in substantial cost to and diversion of effort by us, which may harm our business. In addition, our efforts to protect or defend our proprietary rights may not be successful or, even if successful, may result in substantial cost to us.
Governmental Regulation
Government authorities in the United States (including federal, state and local authorities) and in other countries, extensively regulate, among other things, the manufacturing, research and clinical development, marketing, labeling and packaging, storage, distribution, post-approval monitoring and reporting, advertising and promotion, pricing and export and import of pharmaceutical products, such as our products and product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Moreover, failure to comply with applicable regulatory requirements may result in, among other things, warning letters, clinical holds, civil or criminal penalties, recall or seizure of products, injunction, disbarment, partial or total suspension of production or withdrawal of the product from the market. Any agency or judicial enforcement action could have a material adverse effect on us.
United States Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. The process required by the FDA before product candidates may be marketed in the United States generally involves the following:
• | submission to the FDA of an IND which must become effective before human clinical trials may begin and must be updated annually; |
• | completion of extensive preclinical laboratory tests and preclinical animal studies, all performed in accordance with the FDA’s Good Laboratory Practice, or GLP, regulations; |
• | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate for each proposed indication; |
• | submission to the FDA of an NDA after completion of all pivotal clinical trials; |
• | a determination by the FDA within 60 days of its receipt of an NDA to file the NDA for review; |
• | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities at which the active pharmaceutical ingredient, or API, and finished drug product are produced and tested to assess compliance with cGMP regulations; and |
11
• | FDA review and approval of an NDA prior to any commercial marketing or sale of the drug in the United States. |
An IND is a request for authorization from the FDA to administer an investigational drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human studies. The IND also includes results of animal studies or other human studies, as appropriate, as well as manufacturing information, analytical data and any available clinical data or literature to support the use of the investigational new drug. An IND must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to the proposed clinical trials. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before clinical trials can begin. Accordingly, submission of an IND may or may not result in the FDA allowing clinical trials to commence.
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators in accordance with Good Clinical Practices, or GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety, and the efficacy criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical trial site’s IRB before the trials may be initiated, and the IRB must monitor the study until completed. There are also requirements governing the reporting of ongoing clinical trials and clinical trial results to public registries.
The clinical investigation of a drug is generally divided into three phases. Although the phases are usually conducted sequentially, they may overlap or be combined. The three phases of an investigation are as follows:
• | Phase I. Phase I includes the initial introduction of an investigational new drug into humans. Phase I clinical trials are typically closely monitored and may be conducted in patients with the target disease or condition or in healthy volunteers. These studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the investigational drug in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness. During Phase I clinical trials, sufficient information about the investigational drug’s pharmacokinetics and pharmacological effects may be obtained to permit the design of well-controlled and scientifically valid Phase II clinical trials. The total number of participants included in Phase I clinical trials varies, but is generally in the range of 20 to 80. |
• | Phase II. Phase II includes controlled clinical trials conducted to preliminarily or further evaluate the effectiveness of the investigational drug for a particular indication(s) in patients with the disease or condition under study, to determine dosage tolerance and optimal dosage, and to identify possible adverse side effects and safety risks associated with the drug. Phase II clinical trials are typically well-controlled, closely monitored, and conducted in a limited patient population, usually involving no more than several hundred participants. |
• | Phase III. Phase III clinical trials are generally controlled clinical trials conducted in an expanded patient population generally at geographically dispersed clinical trial sites. They are performed after preliminary evidence suggesting effectiveness of the drug has been obtained, and are intended to further evaluate dosage, clinical effectiveness and safety, to establish the overall benefit-risk relationship of the investigational drug product, and to provide an adequate basis for product approval. Phase III clinical trials usually involve several hundred to several thousand participants. |
A pivotal study is a clinical study which adequately meets regulatory agency requirements for the evaluation of a drug candidate’s efficacy and safety such that it can be used to justify the approval of the product. Generally, pivotal studies are also Phase III studies but may be Phase II studies if the trial design provides a well-controlled and reliable assessment of clinical benefit, particularly in situations where there is an unmet medical need.
The FDA, the IRB or the clinical trial sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move forward at designated check points based on access to certain data from the study. We may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate.
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, detailed investigational drug product information is submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. The application includes all relevant data available from pertinent preclinical and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including studies
12
initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational drug product to the satisfaction of the FDA.
Once the NDA submission has been accepted for filing, the FDA’s goal is to review applications for new molecular entities within ten months of the filing date or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months from the filing date. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it typically follows such recommendations.
After the FDA evaluates the NDA and conducts inspections of manufacturing facilities where the drug product and/or its API will be produced, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may require additional clinical data and/or an additional pivotal Phase III clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. Even if such additional information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA could also approve the NDA with a Risk Evaluation and Mitigation Strategies, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling, development of adequate controls and specifications, or a commitment to conduct one or more post-market studies or clinical trials. Such post-market testing may include Phase IV clinical trials and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. Regulatory approval of oncology products often requires that patients in clinical trials be followed for long periods to determine the overall survival benefit of the drug.
After regulatory approval of a drug product is obtained, we are required to comply with a number of post-approval requirements. The holder of an approved NDA must report, among other things, certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information, and to comply with requirements concerning advertising and promotional labeling for the approved product. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval to ensure and preserve the long term stability of the drug product. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes extensive procedural, substantive and record keeping requirements. In addition, changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates. Future FDA and state inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
Available Special Regulatory Procedures
The Hatch-Waxman Amendments
ANDA Approval Process
The Hatch-Waxman Act, established abbreviated FDA approval procedures for drugs that are shown to be equivalent to proprietary drugs previously approved by the FDA through its NDA process. Approval to market and distribute these drugs is obtained by filing an ANDA with the FDA. An ANDA is a comprehensive submission that contains, among other things, data and information pertaining to the active pharmaceutical ingredient, drug product formulation, specifications and stability of the generic drug, as well as analytical methods, manufacturing process validation data and quality control procedures. Premarket applications for generic drugs are termed abbreviated because they generally do not include preclinical and clinical data to demonstrate safety and effectiveness. Instead, a generic applicant must demonstrate that its product is bioequivalent to the innovator drug.
13
In certain situations, an applicant may obtain ANDA approval of a generic product with a strength or dosage form that differs from a referenced innovator drug pursuant to the filing and approval of an ANDA Suitability Petition. The FDA will approve the generic product as suitable for an ANDA application if it finds that the generic product does not raise new questions of safety and effectiveness as compared to the innovator product. A product is not eligible for ANDA approval if the FDA determines that it is not equivalent to the referenced innovator drug, if it is intended for a different use, or if it is not subject to an approved Suitability Petition. However, such a product might be approved under an NDA, with supportive data from clinical trials.
505(b)(2) NDAs
As an alternative path to FDA approval for modifications to formulations or uses of products previously approved by the FDA, an applicant may submit an NDA under Section 505(b)(2) of the FDCA. Section 505(b)(2) was enacted as part of the Hatch-Waxman Amendments and permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant. If the 505(b)(2) applicant can establish that reliance on FDA’s previous findings of safety and effectiveness is scientifically appropriate, it may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements, including clinical trials, to support the change from the approved branded reference drug. The FDA may then approve the new product candidate for all, or some, of the label indications for which the branded reference drug has been approved, as well as for any new indication sought by the 505(b)(2) applicant.
Orange Book Listing
In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to list with the FDA certain patents whose claims cover the applicant’s product. Upon approval of an NDA, each of the patents listed in the application for the drug is then published in the Orange Book. Any applicant who files an ANDA seeking approval of a generic equivalent version of a drug listed in the Orange Book or a 505(b)(2) NDA referencing a drug listed in the Orange Book must certify to the FDA that (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. This last certification is known as a paragraph IV certification. A notice of the paragraph IV certification must be provided to each owner of the patent that is the subject of the certification and to the holder of the approved NDA to which the ANDA or 505(b)(2) application refers. The applicant may also elect to submit a ‘‘section viii’’ statement certifying that its proposed label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. If the reference NDA holder and patent owners assert a patent challenge directed to one of the Orange Book listed patents within 45 days of the receipt of the paragraph IV certification notice, the FDA is prohibited from approving the application until the earlier of 30 months from the receipt of the paragraph IV certification expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the applicant. The ANDA or 505(b)(2) application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the branded reference drug has expired as described in further detail below.
Non-Patent Exclusivity
In addition to patent exclusivity, the holder of the NDA for the listed drug may be entitled to a period of non-patent exclusivity, during which the FDA cannot approve an ANDA or 505(b)(2) application that relies on the listed drug. For example, a pharmaceutical manufacturer may obtain five years of non-patent exclusivity upon NDA approval of a new chemical entity, or NCE, which is a drug that contains an active moiety that has not been approved by FDA in any other NDA. An ‘‘active moiety’’ is defined as the molecule or ion responsible for the drug substance’s physiological or pharmacologic action. During the five year exclusivity period, the FDA cannot accept for filing any ANDA seeking approval of a generic version of that drug or any 505(b)(2) NDA for the same active moiety and that relies on the FDA’s findings regarding that drug, except that FDA may accept an application for filing after four years if the follow-on applicant makes a paragraph IV certification. A drug, including one approved under Section 505(b)(2), may obtain a three-year period of exclusivity for a particular condition of approval, or change to a marketed product, such as a new formulation for a previously approved product, if one or more new clinical studies (other than bioavailability or bioequivalence studies) was essential to the approval of the application and was conducted/sponsored by the applicant. Should this occur, the FDA would be precluded from approving any ANDA or 505(b)(2) application for the protected modification until after that three-year exclusivity period has run. However, unlike NCE exclusivity, the FDA can accept an application and begin the review process during the exclusivity period.
Europe/Rest of World Government Regulation
In addition to regulations in the United States, we are subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the
14
commencement of human clinical trials. In Europe, for example, a clinical trial application, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
To obtain regulatory approval of an investigational drug under European Union regulatory systems, we must submit a marketing authorization application. The application used to file the NDA in the United States is similar to that required in Europe, with the exception of, among other things, country-specific document requirements.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Authorization Procedures in the European Union
Medicines can be authorized in the European Union by using either the centralized authorization procedure or national authorization procedures.
• | Centralized procedure. The EMA implemented the centralized procedure for the approval of human medicines to facilitate marketing authorizations that are valid throughout the European Union. This procedure results in a single marketing authorization issued by the EMA that is valid across the European Union, as well as Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for human medicines that are: derived from biotechnology processes, such as genetic engineering, contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions, and officially designated orphan medicines. |
• | For medicines that do not fall within these categories, an applicant has the option of submitting an application for a centralized marketing authorization to the EMA, as long as the medicine concerned is a significant therapeutic, scientific or technical innovation, or if its authorization would be in the interest of public health. |
• | National authorization procedures. There are also two other possible routes to authorize medicinal products in several countries, which are available for investigational drug products that fall outside the scope of the centralized procedure: |
• | Decentralized procedure. Using the decentralized procedure, an applicant may apply for simultaneous authorization in more than one European Union country of medicinal products that have not yet been authorized in any European Union country and that do not fall within the mandatory scope of the centralized procedure. |
Mutual recognition procedure. In the mutual recognition procedure, a medicine is first authorized in one European Union Member State, in accordance with the national procedures of that country. Following this, further marketing authorizations can be sought from other European Union countries in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization.
Other Health Care Laws
We may also be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments where we may market our product candidates, if approved. These laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, physician sunshine and privacy and security laws and regulations.
The federal Anti-Kickback Statute prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The Anti-Kickback Statute is subject to evolving interpretations. In the past, the government has enforced the Anti-Kickback Statute to reach large settlements with healthcare companies based on sham consulting and other financial arrangements with physicians. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act. The majority of states also have anti-kickback laws which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers.
15
Additionally, the civil False Claims Act prohibits knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the United States government. Actions under the False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the United States, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers' and manufacturers' compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created new federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
There has also been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the Affordable Care Act, among other things, imposed new reporting requirements on drug manufacturers for payments made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Drug manufacturers were required to begin collecting data on August 1, 2013 and submit reports to the government by March 31, 2014 and June 30, 2014, and the 90th day of each subsequent calendar year. Certain states also mandate implementation of compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians.
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations, including the final omnibus rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts.
Coverage and Reimbursement
Sales of our products and product candidates, once approved, will depend, in part, on the extent to which the costs of our products will be covered by third-party payors, such as government health programs, private health insurers and managed care organizations. Third-party payors generally decide which drugs they will cover and establish certain reimbursement levels for such drugs. In particular, in the United States, private health insurers and other third-party payors often provide reimbursement for products and services based on the level at which the government (through the Medicare or Medicaid programs) provides reimbursement for such treatments. Patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products. Sales of our products and product candidates, if approved, will therefore depend substantially on the extent to which the costs of products and our product candidates will be paid by third-party payors. Additionally, the market for our products and product candidates will depend significantly on access to third-party payors’ formularies without prior authorization, step therapy, or other limitations such as approved lists of treatments for which third-party payors provide coverage and reimbursement. Additionally, coverage and reimbursement for therapeutic products can differ significantly from payor to payor. One third-party payor’s decision to cover a particular medical product or service does not ensure that other payors will also provide coverage for the medical product or service, or will provide coverage at an adequate reimbursement rate. As a result, the coverage determination process will require
16
us to provide scientific and clinical support for the use of our products to each payor separately and will be a time-consuming process.
In addition, the United States government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our future net revenue and results. Decreases in third-party reimbursement for our products and product candidates or a decision by a third-party payor to not cover our products or product candidates could reduce physician usage of our products and product candidates, if approved, and have a material adverse effect on our sales, results of operations and financial condition.
Health Care Reform
In the United States and foreign jurisdictions, there have been a number of legislative and regulatory changes to the healthcare system that could affect our future results of operations. There have been and continue to be a number of initiatives at the United States federal and state levels that seek to reduce healthcare costs.
In particular, in the United States, the Affordable Care Act has had, and is expected to continue to have, a significant impact on the healthcare industry. The Affordable Care Act was designed to expand coverage for the uninsured while at the same time containing overall healthcare costs. The Affordable Care Act, among other things, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, established annual fees and taxes on manufacturers of certain branded prescription drugs, and established a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Substantial new provisions affecting compliance were also enacted, which may require us to modify our business practices with healthcare providers and entities.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. In August 2011, the President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This included reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will stay in effect through 2024 unless additional Congressional action is taken. Additionally, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
Segment and Geographic Area Information
We currently operate in a single segment, through which we develop pharmaceutical products. See note 1 to our consolidated financial statements for further details on our segment and geographic area information. For financial information regarding our business, see “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
Employees
As of March 11, 2015, we had 23 full time employees in the United States None of our employees are represented by a collective bargaining agreement. We believe that we have a good relationship with our employees.
Available Information
We file annual, quarterly and current reports, proxy statements and other information with the SEC, and we have an Internet website address at http://www.apricusbio.com. We make available free of charge on our Internet website address our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Sections 13(a) or 15(d) of the Exchange Act as well as our proxy statements as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. You may also read and copy any document we file at the SEC’s public reference room located at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-732-0330 for further information on the operation of such public reference room. You also can request copies of such documents, upon payment of a duplicating fee, by writing to the SEC at 450 Fifth Street, N.W., Washington, D.C. 20549 or obtain copies of such documents from the SEC’s website at http://www.sec.gov.
17
ITEM 1A. | RISK FACTORS |
Risks Related to the Company
We expect to continue to require external financing to fund our operations, which may not be available.
We expect to require external financing to fund our long-term operations. As of December 31, 2014, we had cash and cash equivalents of approximately $11.4 million. We believe we have sufficient cash reserves and access to cash to fund our on-going operations through 2015, however, we expect to continue to have net cash outflows from operations in 2015 as we further develop Room Temperature Vitaros®, conduct a Phase 2a development program for RayVa™, initiate a Phase 2b development program for fispemifene and incur other operating costs. While we have historically generated modest revenues from our operations, we do not believe that revenues will be sufficient for the foreseeable future to fund our long-term ongoing operations, including the development and commercialization of our product candidates and general and administrative expenses. Given our current lack of profitability and limited capital resources, we may not be able to fully execute all the elements of our strategic plan, including seeking additional market approvals and commercializing Vitaros®, developing and implementing a partnering strategy for Femprox®, and completing our development programs for RayVa™ and fispemifene. If we are unable to accomplish these objectives, our business prospects would be diminished and we will likely be unable to achieve profitability.
We have a history of operating losses and an accumulated deficit, and we may be unable to generate sufficient revenue to achieve profitability in the future.
We only began generating revenues from the commercialization of Vitaros® in the third quarter of 2014, we have never been profitable and we have incurred an accumulated deficit of approximately $289.9 million from our inception through December 31, 2014. We have incurred these losses principally from costs incurred in funding the research, development and clinical testing of our product candidates, from our general and administrative expenses and from our efforts to support commercialization of Vitaros® by our partners. We expect to continue to incur significant operating losses and capital expenditures for the foreseeable future.
Our ability to generate revenues and become profitable depends, among other things, on (1) the successful commercialization of Vitaros® in major markets outside the United States, and (2) the successful development, approval and commercialization of our product candidates including fispemifene, Femprox® and RayVaTM. If we are unable to accomplish these objectives, we may be unable to achieve profitability and would need to raise additional capital to sustain our operations.
Revenues based on Vitaros® represent a substantial portion of our current and expected future revenues.
Our marketing partners are obligated to pay us royalties on their sales of Vitaros®. These payments are expected to be a substantial portion of our ongoing revenues for some time. As a result, any setback that may occur with respect to Vitaros® could significantly impair our operating results and/or reduce the market price of our stock. Setbacks for Vitaros® could include problems with shipping, distribution, manufacturing, product safety, marketing, government regulation or reimbursement, licenses and approvals, intellectual property rights, competition with existing or new products and physician or patient acceptance of the product, as well as higher than expected total rebates, returns or discounts.
In markets where Vitaros® is approved, we are substantially dependent on marketing partners to successfully commercialize Vitaros®.
In markets where Vitaros® has received regulatory approval, we do not have or expect to have any sales or marketing infrastructure. Accordingly, our operating results and long-term success is substantially dependent on the commercialization efforts of our marketing partners in Canada, the UK, Sweden, Germany, the Netherlands, Ireland, Italy, France, Luxembourg, Spain and Belgium. In jurisdictions where we have commercialized our products with partners the amount of revenue we receive from product sales will be lower than if we commercialized directly, as we will be required to share the revenues with our partners. If our partners’ commercialization efforts for Vitaros® are unsuccessful, we may realize little or no revenue from sales in such markets.
In addition, distribution of Vitaros® requires cold-chain distribution, whereby the product must be maintained between specified temperatures. If a difficulty arises in our partner’s cold-chain distribution processes, through our partner’s failure to maintain Vitaros® between specified temperatures, Vitaros® could be damaged or spoiled and rendered unusable. Our marketing partners may also be required to repackage Vitaros® in certain smaller territories where Vitaros® has been approved, or our marketing partners may make claims about applications of Vitaros® beyond uses approved by regulators. Any failure by our partners to comply with packaging, labeling, advertising or promoting requirements in any jurisdiction may result in restrictions on the marketing or manufacturing of Vitaros®, withdrawal of the product from the market or voluntary or mandatory product recalls, which could negatively affect our potential future revenues.
Any failure of our partners to adequately perform their obligations under our license agreements for Vitaros® or any of our other product candidates or the termination of such agreements could have a material and adverse impact on our business.
We and our licensees depend upon third party manufacturers for our products Vitaros®, Fispemifene, RayVa™ and Femprox® and for the raw materials, components, chemical supplies, and dispensers required for our finished products.
18
We do not manufacture any of our products or product candidates. As such, we are dependent on third party manufacturers for the supply of these products and product candidates. The manufacturing process for our products is highly regulated and regulators may refuse to qualify new suppliers and/or terminate manufacturing at existing facilities that they believe do not comply with regulations. Further, our commercial partners may require changes in the product specifications which could cause delays or additional costs to be incurred. The inability of our contract manufacturers to successfully produce commercial quantities of Vitaros® with an acceptable shelf-life could delay or prevent a commercial launch in certain territories, which would negatively affect our potential future revenues.
Our third-party manufacturers and suppliers are subject to numerous regulations, including Good Manufacturing Practices, FDA regulations governing manufacturing processes and related activities and similar foreign regulations. Our third-party manufacturers and suppliers are independent entities who are subject to their own operational and financial risks that are out of our control. If we or any of these third-party manufacturers or suppliers fail to perform as required or fail to comply with the regulations of the FDA and other applicable governmental authorities, our ability to deliver our products on a timely basis or receive royalties or continue our clinical trials would be adversely affected. Also, the manufacturing processes of our manufacturing partners may be found to violate the proprietary rights of others, which could interfere with their ability to manufacture products on a timely and cost effective basis.
In addition, we and our licensees are also dependent on third party manufacturers and suppliers of raw materials, components, chemical supplies for the active drugs in our products and product candidates under development for the formulation and supply of our NexACT® enhancers and finished products. We are dependent on these third-party manufacturers for dispensers that are essential in the production of our products Vitaros®, Femprox® and other products and product candidates. These raw materials, components, chemical supplies, finished products and dispensers must be supplied on a timely basis and at satisfactory quality levels.
If our third party product manufacturers or suppliers of raw materials, components, chemical supplies, finished products and dispensers fail to produce quality products on time and in sufficient quantities or if we are unable to secure adequate alternative sources of supply for such materials, components, chemicals, finished products and dispensers, our results would suffer, as we or our licensees would encounter costs and delays in re-validating new third party suppliers.
Our financial prospects depend in part on the ability of our contract manufacturers and our suppliers to produce and deliver Vitaros® in Canada, Europe and other countries within the approved product specifications. If Vitaros® is not able to be manufactured and provided to customers within the desired specifications and if those specifications cannot be maintained in accordance with approved label requirements, the expected sales by our partners may not be possible and our financial results would be negatively impacted.
We are dependent upon our suppliers and manufacturers of active drug substance, proprietary excipient and other components used in Vitaros® to produce and deliver these materials for Vitaros® manufacturing according to the approved quality specifications filed with the regulatory authorities and according to GMP. If these suppliers or manufacturers are not able to supply these materials in a consistent and timely manner or fail to meet the regulatory requirements to include Vitaros® product specifications, then Vitaros® would not be able to be manufactured.
Similarly, we are dependent upon contract manufacturers to produce Vitaros® dosage form according to the approved specifications for each territory. If the manufacturers are not able to make Vitaros® for any reason, such as an unexpected plant shutdown, failure of certain inspections by regulatory authorities, equipment failure or inability to meet approved regulatory specifications for Vitaros®, then Vitaros® would not be able to be delivered to our partners.
It is possible that our contract manufacturers will not be able to successfully manufacture according to the requirements, and any unforeseen delay, inability to manufacture, or any unforeseen circumstance whereby the approved product label cannot be maintained could significantly impact our financial results.
The product specifications for Vitaros®, and other pharmaceutical products, are governed by the applicable jurisdiction’s regulatory authorities and those specifications may affect the ability of our partners to manufacture a product with a desired product shelf-life, prescribing information or other product characteristics that impact their marketing goals. Such product specifications are specific to each individual jurisdiction’s market-approval directives and are generally not applicable to those product specifications approved by other countries’ regulatory authorities.
The manufacturing specifications for producing Vitaros® in Canada affect the expected shelf-life that can be achieved for the product. Abbott, our marketing partner in Canada, is working with their contract manufacturer to optimize the shelf-life period for the cold-chain product prior to launch. If any of our partners are unable to achieve the desired product shelf life within approved specifications, our financial results could be negatively impacted.
Pre-clinical and clinical trials are inherently unpredictable. If we or our partners do not successfully conduct the clinical trials or gain regulatory approval, we or our partners may be unable to market our product candidates.
19
Through pre-clinical studies and clinical trials, our product candidates, such as fispemifene, RayVa™, and Femprox®, must be demonstrated to be safe and effective for their indicated uses. Results from pre-clinical studies and early clinical trials may not be indicative of, or allow for, prediction of results in later-stage testing. Many of the pre-clinical studies that we have conducted are in animals with “models” of human disease states. Although these tests are widely used as screening mechanisms for drug candidates before being advanced to human clinical studies, results in animal studies are less reliable predictors of safety and efficacy than results of human clinical studies. Future clinical trials may not demonstrate the safety and effectiveness of our product candidates or may not result in regulatory approval to market our product candidates. Commercial sales in any territory cannot begin until approval is received from the applicable regulatory authorities, including the FDA in the United States.
Our business is dependent in part on the success of our product candidates, which will require significant additional clinical testing before we can seek regulatory approval and potentially commercialize products.
Our future success depends in part on our ability to obtain regulatory approval for, and then successfully commercialize our product candidates. Our product candidates will require additional clinical and non-clinical development, regulatory review and approval in multiple jurisdictions, substantial investment, access to sufficient commercial manufacturing capacity and significant marketing efforts before we can generate any revenues from product sales. We are not permitted to market or promote our product candidates in the United States before we receive regulatory approval from the United States FDA and comparable foreign regulatory authorities in overseas jurisdictions, and we may not receive such regulatory approvals on a timely basis, or at all.
Our clinical development plan for RayVa includes a Phase 2a clinical trial, a Phase 2b clinical trial and up to two Phase 3 clinical trials in patients with Raynaud’s Phenomenon secondary to scleroderma. We initiated the Phase 2a clinical trial in December 2014 and will assess those results prior to initiating future clinical studies. Our clinical development plan for fispemifene includes Phase 2 and 3 clinical trials in patients with secondary hypogonadism. We expect to initiate the Phase 2 clinical trial in the second quarter of 2015. There is no guarantee that these clinical trials will commence or be completed on time or at all, and the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials. Even if such regulatory authorities agree with the design and implementation of our clinical trials, we cannot guarantee you that such regulatory authorities will not change their requirements in the future. In addition, even if the clinical trials are successfully completed, we cannot guarantee that the FDA or foreign regulatory authorities will interpret the results as we do, and more trials could be required before we submit our product candidates for approval. To the extent that the results of the clinical trials are not satisfactory to the FDA or foreign regulatory authorities for support of a marketing application, approval of our product candidates may be significantly delayed, or we may be required to expend significant additional resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates.
We cannot anticipate when or if we will seek regulatory review of our product candidates for any indication. An NDA must include extensive pre-clinical and clinical data and supporting information to establish the drug candidate’s safety and effectiveness for each desired indication. The NDA must also include significant information regarding the chemistry, manufacturing and controls for the product. Obtaining approval of an NDA is a lengthy, expensive and uncertain process and may not be obtained on a timely basis, or at all. We have not received marketing approval for any product candidates in the United States, and we cannot be certain that our product candidates will be successful in clinical trials or receive regulatory approval for any indication. If we do not receive regulatory approvals for and successfully commercialize our product candidates on a timely basis or at all, we may not be able to continue our operations. Even if we successfully obtain regulatory approvals to market our product candidates, our revenues will be dependent, in part, on our ability to commercialize our product candidates and on the favorableness of the labeling language granted as well as the size of the markets in the territories for which we gain regulatory approval and have commercial rights. If the markets for the treatment of Raynaud’s Phenomenon secondary to scleroderma or secondary hypogonadism, chronic prostatitis and lower urinary tract symptoms in men are not as significant as we estimate, our business and prospects will be harmed.
If we are unable to adequately establish, maintain and protect our intellectual property rights, we may incur substantial litigation costs and may be unable to generate significant product revenue.
Protection of the intellectual property for our products and product candidates is of material importance to our business in the United States and other countries. We have sought and will continue to seek proprietary protection for our product candidates to attempt to prevent others from commercializing equivalent products. Our success may depend on our ability to (1) obtain effective patent protection within the United States and internationally for our proprietary technologies and products, (2) defend patents we own, (3) preserve our trade secrets and (4) operate without infringing upon the proprietary rights of others. In addition, we have agreed to indemnify certain of our partners for certain liabilities with respect to the defense, protection and/or validity of our patents and would also be required to incur costs or forgo revenue if it is necessary for our partners to acquire third party patent licenses in order for them to exercise the licenses acquired from us.
While we have obtained patents and have many patent applications pending, the extent of effective patent protection in the United States and other countries is highly uncertain and involves complex legal and factual questions. No consistent policy addresses the breadth of claims allowed in, or the degree of protection afforded under, patents of medical and pharmaceutical comp
20
anies. Patents we currently own or may obtain might not be sufficiently broad enough to protect us against competitors with similar technology. Any of our patents could be invalidated or circumvented.
Furthermore, holders of competing patents could allege that our activities infringe on their rights and could potentially prevail in litigation against us. We have also sold certain patents in transactions where we have licensed rights to our drug candidates. In certain of these transactions, we have agreed to indemnify the purchaser from third party patent claims, which could expose us to potentially significant damages for patents that we no longer own. Any litigation could result in substantial cost to us and would divert management’s attention, which may harm our business. In addition, our efforts to protect or defend our proprietary rights may not be successful or, even if successful, may result in substantial cost to us.
We face a high degree of competition.
We are engaged in a highly competitive industry. We and our licensees compete against many companies and research institutions that research, develop and market products in areas similar to those in which we operate. For example, Viagra®(Pfizer), Cialis®(Lilly), Levitra®(Glaxo Smith Kline), Stendra®(Endo Pharmaceuticals, Inc.), and Spedra®(Menarini Group) are currently approved for treatment of ED. Various companies have testosterone replacement therapies on the market for hypogonadism, such as Androgel (Abbott Labs), Axiron (Eli Lilly) and Testim, Testopel, Striant, Aveed and Fortesta (Endo Pharmaceuticals, Inc.).
These and other competitors may have specific expertise and development technologies that are better than ours. Many of these competitors, which include large pharmaceutical companies, have substantially greater financial resources, larger research and development capabilities and substantially greater experience than we do. Accordingly, our competitors may successfully develop competing products. We are also competing with other companies and their products with respect to manufacturing efficiencies and marketing capabilities, areas where we have limited or no direct experience.
A number of other companies have attempted to gain approval in the United States and foreign countries for products similar to Femprox® for indications similar to Female Sexual Interest and Arousal Disorder and have not been successful.
There have been numerous other companies that have tried to gain regulatory approval for a product in the United States or in any other country to treat FSIAD. To date, to our knowledge, no such products have been approved by the FDA or any other regulatory agency and no products are currently on the market for this disorder. A number of companies such as BioSante for its drug LibiGel®, Proctor & Gamble for its drug Intrinsa® and Boehringer Ingelheim for its drug Girosa®, have invested substantial resources in pre-clinical and clinical development on such products and have failed to have them approved by the FDA. There is no guarantee that our product candidate, Femprox®, will be approved by the FDA or any other regulatory agency or that we will realize any revenues from sales of or for the partnering agreements for Femprox®.
Our pharmaceutical expenditures may not result in commercially successful products.
We cannot be sure our business expenditures will result in the successful acquisition, development or launch of products that will prove to be commercially successful or will improve the long-term profitability of our business. If such business expenditures do not result in successful acquisition, development or launch of commercially successful brand products, our results of operations and financial condition could be materially adversely affected.
Business development activity involves numerous risks, including the risks that we may be unable to integrate an acquired business successfully and that we may assume liabilities that could adversely affect us.
In order to augment our product pipeline or otherwise strengthen our business, we may decide to acquire or license additional businesses, products and technologies. Acquisitions could require us to raise significant capital and involve many risks, including, but not limited to, the following:
• | difficulties in achieving identified financial revenue synergies, growth opportunities, operating synergies and cost savings; |
• | difficulties in assimilating the personnel, operations and products of an acquired company, and the potential loss of key employees; |
• | difficulties in consolidating information technology platforms, business applications and corporate infrastructure; |
• | difficulties in integrating our corporate culture with local customs and cultures; |
• | possible overlap between our products or customers and those of an acquired entity that may create conflicts in relationships or other commitments detrimental to the integrated businesses; |
• | our inability to achieve expected revenues and gross margins for any products we may acquire; |
• | the diversion of management’s attention from other business concerns; |
• | risks and challenges of entering or operating in markets in which we have limited or no prior experience, including the unanticipated effects of export controls, exchange rate fluctuations, foreign legal and regulatory requirements, and foreign political and economic conditions; and |
21
• | difficulties in reorganizing, winding-down or liquidating operations if not successful. |
In addition, foreign acquisitions involve numerous risks, including those related to changes in local laws and market conditions and due to the absence of policies and procedures sufficient to assure compliance by a foreign entity with United States regulatory and legal requirements. Business development activities require significant transaction costs, including substantial fees for investment bankers, attorneys, and accountants. Any acquisition could result in our assumption of material unknown and/or unexpected liabilities. We also cannot assure you that we will achieve any cost savings or synergies relating to recent or future acquisitions. Additionally, in any acquisition agreement, the negotiated representations, warranties and agreements of the selling parties may not entirely protect us, and liabilities resulting from any breaches could exceed negotiated indemnity limitations. These factors could impair our growth and ability to compete, divert resources from other potentially more profitable areas, or otherwise cause a material adverse effect on our business, financial position and results of operations.
The financial statements of acquired companies, or those that may be acquired in the future, are prepared by management of such companies and are not independently verified by our management. In addition, any pro forma financial statements prepared by us to give effect to such acquisitions may not accurately reflect the results of operations of such companies that would have been achieved had the acquisition of such entities been completed at the beginning of the applicable periods.
We may be subject to product liability and similar claims, which may lead to a significant financial loss if our insurance coverage is inadequate.
We are exposed to potential product liability risks inherent in the development, testing, manufacturing, marketing and sale of human therapeutic products. Product liability insurance for the pharmaceutical industry is extremely expensive, difficult to obtain and may not be available on acceptable terms, if at all. Although we maintain various types of insurance, we have no guarantee that the coverage limits of such insurance policies will be adequate. If liability claims were made against us, it is possible that our insurance carriers may deny, or attempt to deny, coverage in certain instances. A successful claim against us if we are uninsured, or which is in excess of our insurance coverage, if any, could have a material adverse effect upon us and on our financial condition.
Our business and operations would be adversely impacted in the event of a failure or security breach of our information technology infrastructure.
We rely upon the capacity, reliability and security of our information technology hardware and software infrastructure, including internet-based systems, and our ability to expand and update this infrastructure in response to our changing needs. We are constantly updating our information technology infrastructure. Any failure to manage, expand and update our information technology infrastructure or any failure in the operation of this infrastructure could harm our business.
Despite our implementation of security measures, our systems and those of our business partners may be vulnerable to damages from cyber-attacks, computer viruses, natural disasters, unauthorized access, telecommunication and electrical failures, and other similar disruptions. Our business is also potentially vulnerable to break-ins, sabotage and intentional acts of vandalism by third parties as well as employees. Any system failure, accident or security breach could result in disruptions to our operations, could lead to the loss of trade secrets or other intellectual property, could lead to the public exposure of personal information of our employees, clinical trial participants and others, and could result in a material disruption to our clinical and commercialization activities and business operations. To the extent that any disruption or security breach results in a loss or damage to our data, or inappropriate disclosure of confidential information, it could harm our business and cause us to incur liability. In addition, we may be required to incur significant costs to protect against damage caused by these disruptions or security breaches in the future.
If we fail to attract and retain senior management and key scientific personnel, we may be unable to successfully operate our business.
Our success depends, in part, on our ability to attract, retain and motivate highly qualified management and scientific personnel and on our ability to develop and maintain important relationships with healthcare providers, clinicians and scientists. We are highly dependent upon our senior management and scientific staff. We have incurred attrition at the senior management level in the past, and although we have employment agreements with five of our executives, these agreements are generally terminable at will at any time, and, therefore, we may not be able to retain their services as expected. The loss of services of one or more members of our senior management and scientific staff could delay or prevent us from successfully operating our business. Competition for qualified personnel in the biotechnology and pharmaceuticals field is intense, particularly in the San Diego, California area, where our offices are located. We may need to hire additional personnel to support commercial efforts for Vitaros®. We may not be able to attract and retain qualified personnel on acceptable terms.
Our ability to maintain, expand or renew existing business relationships and to establish new business relationships, particularly in the drug development sector, also depends on our ability to subcontract and retain scientific staff with the skills necessary to keep pace with continuing changes in drug development technologies.
22
From time to time we are subject to various legal proceedings, which could expose us to significant liabilities.
We, as well as certain of our officers and distributors, are subject, from time to time, to a number of legal proceedings, including those legal proceedings described in the notes to the consolidated financial statements included in this Annual Report on Form 10-K. Litigation is inherently unpredictable, and these claims and disputes may result in significant legal fees and expenses regardless of merit and could divert management’s time and other resources. If we are unable to successfully defend or settle any claims asserted against us, we could be liable for damages and be required to alter or cease certain of our business practices or product lines. Any of these outcomes could cause our business, financial performance and cash position to be negatively impacted. There is no guarantee of a successful result in any of these lawsuits regardless of merit, either in defending these claims or in pursuing counterclaims.
Management’s determination that a material weakness exists in our internal controls over financial reporting could have a material adverse impact on our ability to produce timely and accurate financial statements.
The Sarbanes-Oxley Act requires that we report annually on the effectiveness of our internal controls over financial reporting. Among other things, we must perform systems and processes evaluation testing. This includes an assessment of our internal controls to allow management to report on, and our independent public accounting firm to attest to, our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. Management performed an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2014 using criteria established by the Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this assessment, management determined that, as of December 31, 2014, material weaknesses exist in our internal control over financial reporting over the accounting for and disclosures of technical accounting matters in the consolidated financial statements and effective monitoring and oversight over the controls in the financial reporting process. Because of these material weaknesses, management concluded that the Company did not maintain effective internal control over financial reporting as of December 31, 2014, based on the COSO framework.
For information on the progress of the remediation of the material weaknesses, see the material weaknesses and remediation efforts section under Item 9A. Controls and Procedures below. Our future assessment, or the future assessment by our independent registered public accounting firm, may reveal additional material weaknesses in our internal controls. If not remediated, a material weakness, and any future potential material weaknesses identified by management could result in future errors in our financial statements or in documents we file with the SEC.
The terms of our credit facility place restrictions on our operating and financial flexibility.
On October 17, 2014, we entered into a loan and security agreement, or the credit facility, with Oxford Finance LLC, or Oxford, and certain other lenders party thereto from time to time, or the lenders, including Silicon Valley Bank, or SVB, that is secured by substantially all of our assets, excluding intellectual property. The principal balance under the credit facility was $5.0 million at the closing of the loan and security agreement on October 17, 2014.
The credit facility includes affirmative and negative covenants applicable to us and any subsidiaries we create in the future. The affirmative covenants include, among others, covenants requiring us to maintain our legal existence and governmental approvals, deliver certain financial reports and maintain insurance coverage. The negative covenants include, among others, restrictions on our transferring collateral, incurring additional indebtedness, engaging in mergers or acquisitions, paying dividends or making other distributions, making investments, creating liens, selling assets, and suffering a change in control, in each case subject to certain exceptions.
The credit facility also includes events of default, the occurrence and continuation of which provide Oxford, as collateral agent, with the right to exercise remedies against us and the collateral securing the term loans under the credit facility, including foreclosure against our properties securing the credit facilities, including our cash. These events of default include, among other things, our failure to pay any amounts due under the credit facility, a breach of covenants under the credit facility, our insolvency, a material adverse change, the occurrence of any default under certain other indebtedness in an amount greater than $250,000, and a final judgment against us in an amount greater than $250,000.
If we fail to comply with our obligations in our intellectual property licenses and funding arrangements with third parties, we could lose rights that are important to our business.
23
We are party to a license agreement with Forendo Pharma Ltd. that imposes diligence, development and commercialization timelines, milestone payment, royalty, insurance and other obligations on us. Under our existing licensing agreement, we are obligated to pay royalties on net product sales of fispemifene to the extent they are covered by the agreement. If we fail to comply with our obligations, our counterparties may have the right to terminate these agreements, in which event we might not be able to develop, manufacture or market any product that is covered by these agreements or may face other penalties under the agreements. Such an occurrence could materially adversely affect the value of product candidates being developed using rights licensed to us under any such agreement. Termination of these agreements or reduction or elimination of our rights under these agreements may result in our having to negotiate new or reinstated agreements with less favorable terms, or cause us to lose our rights under these agreements, including our rights to important intellectual property or technology.
We may enter into license agreements in the future that could also impose diligence, development and commercialization timelines, milestone payments, royalty, insurance and other obligations.
Industry Risks
Instability and volatility in the financial markets in the global economy are likely to have a negative impact on our ability to raise necessary funds.
During the past several years, there has been substantial volatility in financial markets due in part to the global economic environment. In addition, there has been substantial uncertainty in the capital markets and access to financing is uncertain. These conditions are likely to have an adverse effect on our industry, licensing partners and business, including our financial condition, results of operations and cash flows.
We expect to need to raise capital through equity sales and/or incur indebtedness, if available, to finance operations. However, continued volatility in the capital markets may have an adverse effect on our ability to fund our business strategy through sales of capital stock or through borrowings, in the public or private markets on terms that we believe to be reasonable, if at all.
Changes in trends in the pharmaceutical and biotechnology industries, including difficult market conditions, could adversely affect our operating results.
Industry trends and economic and political factors that affect pharmaceutical, biotechnology and medical device companies also affect our business. In the past, mergers, product withdrawals, liability lawsuits and other factors in the pharmaceutical industry have slowed decision-making by pharmaceutical companies and delayed drug development projects. Continuation or increases in these trends could have an adverse effect on our business.
The biotechnology, pharmaceutical and medical device industries generally, and more specifically drug discovery and development, are subject to increasingly rapid technological changes. Our competitors might develop technologies, services or products that are more effective or commercially attractive than our current or future technologies, services or products, or that render our technologies, services or products less competitive or obsolete. If competitors introduce superior technologies, services or products and we cannot make enhancements to our technologies, services or products to remain competitive, our competitive position, and in turn our business, revenue and financial condition, would be materially and adversely affected.
We and our licensees are subject to numerous and complex government regulations which could result in delay and expense.
Governmental authorities in the United States and other countries heavily regulate the testing, manufacture, labeling, distribution, advertising and marketing of our proposed product candidates. None of our proprietary products under development have been approved for marketing in the United States. Before any products we develop are marketed, FDA and comparable foreign agency approval must be obtained through an extensive clinical study and approval process.
The failure to obtain requisite governmental approvals for our product candidates under development in a timely manner, or at all, would delay or preclude us and our licensees from marketing our product candidates or limit the commercial use of our product candidates, which could adversely affect our business, financial condition and results of operations.
Because we intend that our product candidates will also be sold and marketed outside the United States, we and/or our licensees will be subject to foreign regulatory requirements governing the conduct of clinical trials, product licensing, pricing and reimbursements. These requirements vary widely from country to country. The failure to meet each foreign country’s requirements could delay the introduction of our proposed product candidates in the respective foreign country and limit our revenues from sales of our proposed product candidates in foreign markets.
We face uncertainty related to healthcare reform, pricing and reimbursement, which could reduce our revenue.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could, among other things, prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell Vitaros® or any products for which we obtain marketing approval.
24
For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation Act, collectively the Affordable Care Act, was enacted to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for health care and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Among the provisions of the Affordable Care Act of importance to our potential drug candidates are the following:
• | an annual, nondeductible fee payable by any entity that manufactures or imports specified branded prescription drugs and biologic agents; |
• | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; |
• | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
• | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries under their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
• | extension of manufacturers’ Medicaid rebate liability to individuals enrolled in Medicaid managed care organizations; |
• | expansion of eligibility criteria for Medicaid programs; |
• | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
• | a new requirement to annually report drug samples that manufacturers and distributors provide to physicians; and a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
Although it is too early to determine the full effect of the Affordable Care Act, the law has continued the downward pressure on pharmaceutical pricing, especially under the Medicare program, and increased the industry’s regulatory burdens and operating costs.
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. For example, in August 2011, the President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This included reductions to Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013 and, due to subsequent legislative amendments to the statute, will stay in effect through 2024 unless additional Congressional action is taken. Additionally, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products and product candidates or additional pricing pressures.
If reimbursement for our products is substantially less than we expect in the future, or rebate obligations associated with them are substantially increased, our business could be materially and adversely impacted. Further, numerous foreign governments are also undertaking efforts to control growing healthcare costs through legislation, regulation and voluntary agreements with medical care providers and pharmaceutical companies.
Sales of Vitaros® and other product candidates, if approved, will depend in part on the availability of coverage and reimbursement from third-party payers such as United States and foreign government insurance programs, including Medicare and Medicaid, private health insurers, health maintenance organizations and other health care related organizations. Both the federal and state governments in the United States and foreign governments continue to propose and pass new legislation affecting coverage and reimbursement policies, which are designed to contain or reduce the cost of health care. Further federal and state proposals and healthcare reforms are likely that could limit the prices that can be charged for the product candidates that we develop and may further limit our commercial opportunity. There may be future changes that result in reductions in current coverage and reimbursement levels for our products and we cannot predict the scope of any future changes or the impact that those changes would have on our operations.
Adoption by the medical community of Vitaros® and other product candidates, if approved, may be limited if third-party payers will not offer coverage. Cost control initiatives may decrease coverage and payment levels for drugs, which in turn would negatively affect the price that we will be able to charge. We are unable to predict all changes to the coverage or reimbursement methodologies that will be applied by private or government payers to any drug candidate we have in development. Any denial of private or government payer coverage or inadequate reimbursement for our products could harm our business and reduce our revenue.
25
The FDA regulatory approval process is lengthy and time-consuming, and if we experience significant delays in the clinical development and regulatory approval of our product candidates, our business may be substantially harmed.
We may experience delays in commencing and completing clinical trials of our product candidates. We do not know whether planned clinical trials will begin on time, need to be redesigned, enroll patients on time or be completed on schedule, if at all. Any of our planned clinical trials may be delayed for a variety of reasons, including delays related to:
• | the availability of financial resources for us to commence and complete our planned clinical trials; |
• | reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and clinical trial sites; |
• | obtaining independent institutional review board, or IRB, approval at each clinical trial site; |
• | obtaining regulatory approval to commence clinical trials in each country; |
• | recruiting a sufficient number of eligible patients to participate in a clinical trial; |
• | having patients complete a clinical trial or return for post-treatment follow-up; |
• | clinical trial sites deviating from trial protocol or dropping out of a trial; |
• | adding new clinical trial sites; or |
• | manufacturing sufficient quantities of our product candidate for use in clinical trials. |
Patient enrollment is a significant factor in the timing of clinical trials and is affected by many factors including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the clinical trial, the design of the clinical trial, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages or potential side effects of the drug candidate being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating.
We could encounter delays if physicians encounter unresolved ethical issues associated with enrolling patients in clinical trials of our product candidates in lieu of prescribing existing treatments that have established safety and efficacy profiles. Further, a clinical trial may be suspended or terminated by us, the IRBs in the institutions in which such trials are being conducted, the Data Monitoring Committee for such trial (if included), or by the FDA or other regulatory authorities due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. Furthermore, we rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials. While we have agreements governing the CROs’ services, we have limited influence over their actual performance. If we experience termination of, or delays in the completion of, any clinical trial of our product candidates, the commercial prospects for our product candidates will be harmed, and our ability to generate product revenues will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product development and approval process and jeopardize our ability to commence product sales and generate revenues from our product candidates. Any of these occurrences may harm our business, prospects, financial condition and results of operations. Furthermore, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
If we are unable to obtain regulatory approval of our product candidates, we will not be able to commercialize our product candidates and our business will be adversely impacted.
If we fail to obtain regulatory approval to market our product candidates, we will be unable to sell our product candidates, which will impair our ability to generate additional revenues. To receive approval, we must, among other things, demonstrate with substantial evidence from clinical trials that the product candidate is both safe and effective for each indication for which approval is sought. Failure can occur in any stage of development. Satisfaction of the approval requirements is unpredictable but typically takes several years following the commencement of clinical trials, and the time and money needed to satisfy them may vary substantially, based on the type, complexity and novelty of the pharmaceutical product. We cannot predict if or when our existing and planned clinical trials will generate the data necessary to support an NDA and if, or when, we might receive regulatory approvals for our product candidates.
Our product candidates could fail to receive regulatory approval for many reasons, including the following:
• | the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
26
• | we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that our product candidates are safe and effective for any of the proposed indications; |
• | the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
• | we may be unable to demonstrate that our product candidates’ clinical and other benefits outweigh their safety risks; |
• | the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
• | the data collected from clinical trials of our product candidates may not be sufficient to the satisfaction of the FDA or comparable foreign regulatory authorities to support the submission of an NDA or other comparable submission in foreign jurisdictions or to obtain regulatory approval in the United States or elsewhere; |
• | the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; |
• | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval; and |
• | even after following regulatory guidance or advice, the FDA or comparable foreign regulatory authorities may still reject our ultimate regulatory submissions since their guidance is generally considered non-binding and the regulatory authorities have the authority to revise or adopt new and different guidance at any time. |
This lengthy approval process as well as the unpredictability of future clinical trial results may result in our failure to obtain regulatory approval to market our product candidates, which would significantly harm our business, prospects, financial condition and results of operations. In addition, any approvals that we obtain may not cover all of the clinical indications for which we are seeking approval, or could contain significant limitations in the form of narrow indications, warnings, precautions or contra-indications with respect to conditions of use. In such event, our ability to generate revenues would be greatly reduced and our business would be harmed.
Even if we receive regulatory approval for our product candidates, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates, if approved, could be subject to labeling and other restrictions and market withdrawal and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our product candidates.
Any regulatory approvals that we receive for our product candidates may contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require additional risk management activities and labeling which may limit distribution or patient/prescriber uptake. An example would be the requirement of a risk evaluation and mitigation strategy (“REMS”) in order to approve our product candidates, which could entail requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA or a comparable foreign regulatory authority approves our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for our product candidates will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, and registration. We are also required to maintain continued compliance with cGMP requirements and current good clinical practice, or cGCP, requirements for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with our product candidates or other manufacturers’ products in the same class, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
• | restrictions on the marketing or manufacturing of our product candidates, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
• | fines, warning letters or holds on clinical trials; |
• | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of license approvals; |
• | product seizure or detention, or refusal to permit the import or export of our product candidates; and |
• | injunctions or the imposition of civil or criminal penalties. |
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we
27
are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business, prospects, financial condition and results of operations.
Our relationships with investigators, health care professionals, consultants, third-party payors, and customers are subject to applicable healthcare regulatory laws, which could expose us to penalties.
Our business operations and arrangements with investigators, healthcare professionals, consultants, marketing partners, third-party payors and customers, may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations. These laws may constrain the business or financial arrangements and relationships through which we conduct our operations, including how we research, market, sell and distribute our products and product candidates for which we obtain marketing approval. Such laws include:
• | the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it to have committed a violation; in addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
• | the federal False Claims Act imposes criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
• | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
• | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
• | the federal Physician Payment Sunshine Act, which requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the government information related to payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and requires applicable manufacturers and group purchasing organizations to report annually to the government ownership and investment interests held by the physicians described above and their immediate family members and payments or other “transfers of value” to such physician owners (manufacturers are required to submit reports to the government by the 90th day of each calendar year); and |
• | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third party payors, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws governing the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable healthcare laws. If our operations are found to be in violation of any of these or any other health regulatory laws that may apply to us, we may be subject to significant penalties, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement, individual imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations. Defending against any such actions can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired.
28
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. Results of our trials could reveal a high and unacceptable severity and prevalence of undesirable side effects. In such an event, our trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Additionally if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
• | regulatory authorities may withdraw approvals of such product; |
• | regulatory authorities may require additional warnings on the label; |
• | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
• | we could be sued and held liable for harm caused to patients; and |
• | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
Our employees, independent contractors, principal investigators, CROs, consultants, commercial partners and vendors are subject to a number of regulations and standards.
We are exposed to the risk that employees, independent contractors, principal investigators, CROs, consultant and vendors may engage in fraudulent or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates: (1) the laws of the FDA and other similar foreign regulatory bodies; including those laws that require the reporting of true, complete and accurate information to the FDA and other similar foreign regulatory bodies, (2) manufacturing standards, (3) healthcare fraud and abuse laws in the United States and similar foreign fraudulent misconduct laws, or (4) laws that require the true, complete and accurate reporting of financial information or data. These laws may impact, among other things, our current activities with principal investigators and research subjects, as well as proposed and future sales, marketing and education programs. In particular, the promotion, sales and marketing of healthcare items and services, as well as certain business arrangements in the healthcare industry, are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commission(s), certain customer incentive programs and other business arrangements generally. Activities subject to these laws also involve the improper use of information obtained in the course of patient recruitment for clinical trials. If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our potential exposure under such laws will increase significantly, and our costs associated with compliance with such laws are also likely to increase. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Risks Related to Owning Our Common Stock
We are vulnerable to volatile stock market conditions.
The market prices for securities of biopharmaceutical and biotechnology companies, including ours, have been highly volatile. The market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. In addition, future announcements, such as the results of testing and clinical trials, the status of our relationships with third-party collaborators, technological innovations or new therapeutic products, governmental regulation, developments in patent or other proprietary rights, litigation or public concern as to the safety of products developed by us or others and general market conditions concerning us, our competitors or other biopharmaceutical companies, may have a significant effect on the market price of our common stock. In the past, when the market price of a stock has been volatile, holders of that stock have been more likely to initiate securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management
29
We do not expect to pay dividends on our common stock in the foreseeable future.
Although our stockholders may in the future receive dividends if and when declared by our board of directors, we do not intend to declare dividends on our common stock in the foreseeable future. Therefore, you should not purchase our common stock if you need immediate or future income by way of dividends from your investment.
We may issue additional shares of our capital stock that could dilute the value of your shares of common stock.
We are authorized to issue 85,000,000 shares of our capital stock, consisting of 75,000,000 shares of our common stock and 10,000,000 shares of our preferred stock. Pursuant to the Common Stock Purchase Agreement with Aspire Capital Fund, LLC (“Aspire Capital”) entered into in August 2014, we may, from time to time under certain restrictions (see note 1 to our consolidated financials for further details), sell up to $22.0 million worth of our common stock, of which $18.2 million remained available as of December 31, 2014. In light of our future capital needs, we may also issue additional shares of common stock at below current market prices or convertible securities in addition to the $10.8 million raised as a result of our February 2015 financing. These issuances would dilute the book value of existing stockholders common stock and could depress the value of our common stock.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
We currently lease two properties in San Diego for approximately 19,000 square feet, consisting of corporate offices and a warehouse facility.
ITEM 3. LEGAL PROCEEDINGS
We are a party to certain litigation that is either judged to be not material or that arises in the ordinary course of business from time to time. We intend to vigorously defend our interests in these matters. We expect that the resolution of these matters will not have a material adverse effect on our business, financial condition or results of operations. However, due to the uncertainties inherent in litigation, no assurance can be given as to the outcome of these proceedings.
Versailles Civil Court Summons
In June 2014, consistent with the Global Settlement Agreement (“GSA”) signed in February 2014, the Works Council withdrew its previously submitted €4.1 million claim in the Civil Court, all parties accepted the withdrawal and the Civil Court judge closed the discussions between all parties. The final procedural step occurred in October 2014, when we received a written judgment from the Civil Court acknowledging the dismissal of the claim and the closure of the litigation. Given the existence of the aforementioned ratified GSA, the accepted withdrawal and the closure of the discussions by the Civil Court judge, it was concluded during the second quarter of 2014 that we were relieved of all claims previously asserted by the French Works Council.
Pursuant to the aforementioned license and settlement agreements, Majorelle agreed to make certain severance payments of approximately $2.0 million to the former French Subsidiaries’ employees on behalf of us, a portion of which were made in May 2014. In addition, the Works Council and the Judicial Liquidator and Trustee of our former French Subsidiaries as well as each of the former French Subsidiaries’ employees, waived all claims they had asserted or could have asserted against us related to the liquidation and reorganization of the French Subsidiaries.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II.
30
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our Common Stock is traded on the NASDAQ Capital Market (“NASDAQ”) under the symbol “APRI.”
On March 11, 2015, the last reported sales price for our Common Stock on NASDAQ was $2.19 per share, and we had approximately 127 holders of record of our Common Stock. One of our shareholders is Cede & Co., a nominee for Depository Trust Company, or DTC. Shares of common stock that are held by financial institutions as nominees for beneficial owners are deposited into participant accounts at DTC, and are considered to be held of record by Cede & Co. as one stockholder.
The following table sets forth the range of the high and low sales prices for our Common Stock as reported by NASDAQ for each quarter in 2013 and 2014.
2014 | 2013 | |||||||||||||||
High | Low | High | Low | |||||||||||||
First quarter | $ | 2.63 | $ | 2.06 | $ | 3.34 | $ | 2.04 | ||||||||
Second quarter | $ | 2.40 | $ | 2.01 | $ | 3.41 | $ | 2.28 | ||||||||
Third quarter | $ | 2.22 | $ | 1.51 | $ | 2.41 | $ | 1.91 | ||||||||
Fourth quarter | $ | 1.64 | $ | 0.95 | $ | 2.68 | $ | 1.72 | ||||||||
Dividends
We have never paid cash dividends on our Common Stock and do not have any plans to pay cash dividends in the foreseeable future. Our Board of Directors anticipates that any earnings that might be available to pay dividends will be retained to finance our business.
Equity Compensation Plan
Information about our equity compensation plans is incorporated by reference in Item 12 of Part III of this Annual Report on Form 10-K.
Unregistered Sales of Equity Securities and Use of Proceeds
None.
31
Performance Graph
The following graph shows the cumulative total stockholder return of an investment of $100 in cash on December 31, 2009 through December 31, 2014, for (i) our common stock, (ii) the NASDAQ Composite Index and (iii) the NASDAQ Biotech Index. Pursuant to applicable SEC rules, all values assume reinvestment of the full amount of all dividends; however, no dividends have been declared on our common stock to date. The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
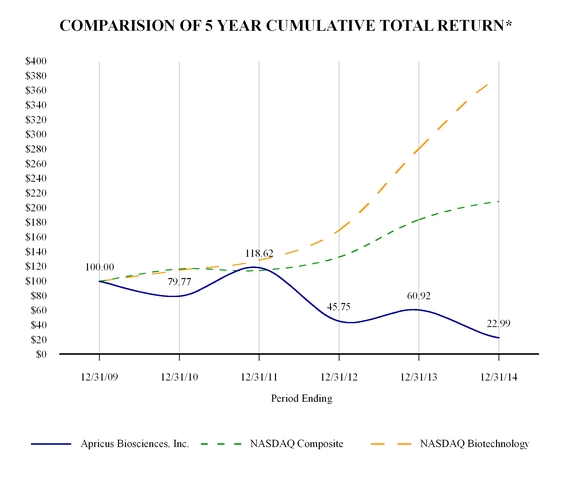
*$100 invested on 12/31/09 in stock or index, including reinvestment of dividends. Fiscal year ending December 31, 2014.
ITEM 6. SELECTED FINANCIAL DATA
The selected consolidated financial data set forth below as of December 31, 2014 and 2013, and for each of the fiscal years ended December 31, 2014, 2013 and 2012, are derived from our audited consolidated financial statements included elsewhere in this report. This information should be read in conjunction with those consolidated financial statements, the notes thereto, and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The selected consolidated financial data set forth below as of December 31, 2012, 2011 and 2010, and for each of the fiscal years ended December 31, 2011 and 2010, are derived from our audited consolidated financial statements that are contained in reports previously filed with the SEC, not included herein.
32
Five-Year Selected Financial Data
For the Years Ended December 31, (1) | ||||||||||||||||||||
2014 | 2013(2)(3)(4) | 2012 (2)(3)(4) | 2011(2)(4) | 2010(2) | ||||||||||||||||
(In thousands, except share and per share data) | ||||||||||||||||||||
Statements of Operations Data: | ||||||||||||||||||||
Total revenue | $ | 9,259 | $ | 2,511 | $ | 7,945 | $ | 3,603 | $ | 4,446 | ||||||||||
Gross profit (loss) | 8,341 | (120 | ) | 3,705 | 1,745 | 889 | ||||||||||||||
Total operating expenses | 30,900 | 17,247 | 28,715 | 20,032 | 22,418 | |||||||||||||||
Loss from continuing operations | (22,477 | ) | (15,870 | ) | (25,676 | ) | (18,225 | ) | (29,636 | ) | ||||||||||
Income (loss) from discontinued operations | 691 | (1,068 | ) | (6,095 | ) | 108 | 128 | |||||||||||||
Net loss | (21,786 | ) | (16,938 | ) | (31,771 | ) | (18,117 | ) | (29,508 | ) | ||||||||||
Basic and diluted loss per common share (5) | ||||||||||||||||||||
Loss from continuing operations | $ | (0.57 | ) | $ | (0.46 | ) | $ | (0.94 | ) | $ | (0.91 | ) | $ | (2.50 | ) | |||||
Income (loss) from discontinued operations | $ | 0.02 | $ | (0.03 | ) | $ | (0.22 | ) | $ | 0.01 | $ | 0.01 | ||||||||
Net loss | $ | (0.55 | ) | $ | (0.49 | ) | $ | (1.16 | ) | $ | (0.90 | ) | $ | (2.49 | ) | |||||
Weighted average shares outstanding, basic and diluted loss per share | 39,540,409 | 34,413,253 | 27,458,184 | 20,023,456 | 11,847,703 | |||||||||||||||
As of December 31, | ||||||||||||||||||||
2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
(In thousands) | ||||||||||||||||||||
Consolidated Balance Sheets Data | ||||||||||||||||||||
Cash & cash equivalents | $ | 11,400 | $ | 21,405 | $ | 15,130 | $ | 7,435 | $ | 9,146 | ||||||||||
Total assets | 14,809 | 23,310 | 23,879 | 16,616 | 18,864 | |||||||||||||||
Long term liabilities | 5,984 | 578 | 6,492 | 1,777 | 4,980 | |||||||||||||||
Accumulated deficit | (289,852 | ) | (268,066 | ) | (251,128 | ) | (219,357 | ) | (201,240 | ) | ||||||||||
(1) | In December 2009, we acquired Bio-Quant, Inc. (“Bio-Quant”) for $13.7 million, which included the issuance of promissory notes for $12.1 million and 0.3 million shares of common stock valued at $1.6 million. The results of Bio-Quant’s operations have been included from the date of acquisition through June 2011, the date that Bio-Quant was sold to an unrelated third party. Costs associated with the merger of $0.6 million were expensed during 2009. In connection with the valuation of the future expected cash flows and the goodwill related to Bio-Quant at December 31, 2010, an impairment charge of $9.1 million was recorded in 2010 representing the then recorded goodwill from this acquisition. A loss on the sale of $2.8 million was recognized during 2011 and a recovery of the loss was recognized during 2012, 2013, and 2014 for earn-out payments received that, at the time of sale, were considered to have no value, in the amount of $0.3 million, $0.3 million and $0.1 million. We amended the agreement in June 2014 and received a one-time cash payment of $0.6 million. We recorded the gain of $0.7 million in 2014 as discontinued operations within our consolidated statement of operations. Historically, the Company reflected the operations and subsequent cash collections associated with the sale of the business as a component of continuing operations, as recovery on sale of subsidiary within the consolidated statements of operations. However, the Company has elected to not correct these prior period amounts which were deemed not material to prior period statements. |
(2) | In June 2013, we determined that the BQ Kits division would be offered for sale to qualified buyers and in July 2013, it was sold to an unrelated third-party. For years 2013 through 2010 presented above, it is presented as discontinued operations. |
(3) | On July 12, 2012, by way of contribution, we accepted 100% percent of the outstanding common shares of Finesco SAS, for an aggregate purchase price, net of cash paid for costs and cash acquired, of $6.7 million, and included the issuance of 2.6 million shares of common stock valued at $8.6 million. The results of Finesco’s operations were included from the date of acceptance. During the fourth quarter of 2012, we recorded a charge in the amount of $8.3 million for the impairment of the goodwill associated with the Finesco acquisition and a related charge recorded as tax expense in the amount $1.3 million partially offset by $0.8 million in tax benefit recorded in 2012 after the acceptance of the Finesco shares to record a valuation allowance on the recoverability of the deferred tax assets acquired as part of the Finesco transaction. Also in the fourth quarter of 2012, we made the decision to cease funding of our former French Subsidiaries and the businesses were deconsolidated in April 2013. |
33
(4) | In December 2011, we acquired TopoTarget USA, Inc., for $3.5 million, which included the issuance of 0.3 million in shares of common stock valued at $1.7 million. In February 2012, we also acquired the co-promotion rights to sell Granisol® in the United States and other territories. In March 2013, following our strategic decision to divest this business, we sold to Biocodex, Inc. (“Biocodex”) all of our rights and certain information, property and inventory related to the Totect® assets for $1.5 million plus the right to receive from Biocodex double-digit, tiered, decreasing royalties. We retained all liabilities related to Totect®. We recorded a net loss of $1.4 million during the first quarter of 2013 related to the sale. The net results of these operations are reported as discontinued operations for the year ended December 31, 2013 and 2012. |
ITEM 7. | MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Disclosures Regarding Forward-Looking Statements
This report includes “forward-looking statements” within the meaning of Section 21E of the Exchange Act. Those statements include statements regarding the intent, belief or current expectations of Apricus Biosciences, Inc. and Subsidiaries (“we,” “us,” “our,” the “Company” or “Apricus”) and our management team. Any such forward-looking statements are not guarantees of future performance and involve risks and uncertainties, and actual results may differ materially from those projected in the forward-looking statements. These risks and uncertainties include but are not limited to those risks and uncertainties set forth in Item 1A of this Report. In light of the significant risks and uncertainties inherent in the forward-looking statements included in this Report, the inclusion of such statements should not be regarded as a representation by us or any other person that our objectives and plans will be achieved. Further, these forward-looking statements reflect our view only as of the date of this report. Except as required by law, we undertake no obligations to update any forward-looking statements and we disclaim any intent to update forward-looking statements after the date of this report to reflect subsequent developments. Accordingly, you should also carefully consider the factors set forth in other reports or documents that we file from time to time with the Securities and Exchange Commission.
Results of Operations
Revenues and gross profit from continuing operations were as follows (in thousands, except percentages):
Year Ended December 31, | 2014 vs 2013 | 2013 vs 2012 | |||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | |||||||||||||||||||
License fee revenue | $ | 8,454 | $ | 941 | $ | 4,276 | $ | 7,513 | 798 | % | $ | (3,335 | ) | (78 | )% | ||||||||||
Royalty revenue | 36 | — | — | 36 | N/M | — | N/M | ||||||||||||||||||
Product sales | 769 | 21 | 23 | 748 | 3,562 | % | (2 | ) | (9 | )% | |||||||||||||||
Contract service revenue | — | 1,549 | 3,646 | (1,549 | ) | (100 | )% | (2,097 | ) | (58 | )% | ||||||||||||||
Total revenue | 9,259 | 2,511 | 7,945 | 6,748 | 269 | % | (5,434 | ) | (68 | )% | |||||||||||||||
Cost of product sales | 918 | 23 | 10 | 895 | 3,891 | % | 13 | 130 | % | ||||||||||||||||
Cost of service revenue | — | 2,608 | 4,230 | (2,608 | ) | (100 | )% | (1,622 | ) | (38 | )% | ||||||||||||||
Gross profit (loss) | $ | 8,341 | $ | (120 | ) | $ | 3,705 | $ | 8,461 | N/M | $ | (3,825 | ) | (103 | )% | ||||||||||
Revenue
The $6.7 million increase in total revenue during the year ended December 31, 2014 as compared to the prior year is primarily due to an increase in license fee revenue from $0.9 million in 2013 to $8.5 million in 2014. Significant components of license fee revenue in 2014 were $4.0 million from Majorelle for the fair value of the license, consisting of an upfront payment of $1.8 million, a $0.2 million payment received for National Phase approval in France, and $2.0 million in connection with certain severance payments made by Majorelle on our behalf; an upfront license payment of approximately $2.5 million from Recordati; and $2.0 million from Sandoz, consisting of $0.5 million for each of the launches of Vitaros® in Sweden and Belgium and $1.0 million that had been previously deferred awaiting the satisfaction of a contractual condition that was met in the fourth quarter of 2014. Comparatively, in 2013, we received $0.6 million and $0.3 million in license fee revenue from Bracco and Sandoz, respectively, as a result of substantive milestones received upon regulatory approvals in Italy and Germany, respectively.
Product sales in 2014 are the result of the commencement of shipping Vitaros® product to our commercialization partners. Contract service revenue resulted from our former French Subsidiaries which were deconsolidated in April 2013.
We expect our cash inflows from operations during 2015 will result from licensing and milestone revenues received from commercial partners as well as product sales for our Vitaros® product. The timing of these revenues are uncertain, as such our revenue may vary significantly between periods.
34
The $5.4 million decrease in total revenue during the year ended December 31, 2013 as compared to the prior year was primarily due to $4.3 million in upfront license fees from our commercial partners Sandoz, Abbott and Takeda, received during the year ended December 31, 2012. This was partially offset by $0.6 million in license fee revenue in 2013 from Bracco and $0.3 million in license fee revenue from Sandoz as a result of substantive milestones received upon regulatory approvals in Italy and Germany, respectively. This decrease was also due to decreased contract service revenue in 2013 as compared to the prior year due to the loss of certain contract service customers in France in 2013 as compared to 2012, following the deconsolidation of our former French Subsidiaries in April 2013.
Cost of Product Sales
Our cost of product sales includes direct material costs associated with the production of inventories. Cost of product sales also includes the cost of manufactured samples provided to our licensee partners free of charge, which contributed to our negative margin. Cost of product sales for the year ended December 31, 2014 of $0.9 million resulted from the commencement of shipping Vitaros® product in 2014.
Cost of Service Revenue
Our cost of service revenue includes compensation, related personnel expenses and contract services to support our contract service revenue. The $2.6 million decrease in cost of service revenue during the year ended December 31, 2014, as compared to the prior year, is due to the absence of contract services related to our former French Subsidiaries, which were deconsolidated in April 2013.
The $1.6 million decrease in cost of service revenue during the year ended December 31, 2013, as compared to the prior year, is also due to contract services related to our former French Subsidiaries, which were included in our statements of operations beginning July 2012 and deconsolidated in April 2013.
Operating Income (Expense)
Operating income (expense) from continuing operations were as follows (in thousands, except percentages):
Year Ended December 31, | 2014 vs 2013 | 2013 vs 2012 | |||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | |||||||||||||||||||
Operating expense (income) | |||||||||||||||||||||||||
Research and development | $ | 21,288 | $ | 5,123 | $ | 5,375 | $ | 16,165 | 316 | % | $ | (252 | ) | (5 | )% | ||||||||||
General and administrative | 11,418 | 13,554 | 15,336 | (2,136 | ) | (16 | )% | (1,782 | ) | (12 | )% | ||||||||||||||
Gain on contract settlement | (910 | ) | (534 | ) | — | (376 | ) | 70 | % | (534 | ) | N/M | |||||||||||||
Recovery on sale of subsidiary | (50 | ) | (255 | ) | (250 | ) | 205 | (80 | )% | (5 | ) | 2 | % | ||||||||||||
Deconsolidation of former French Subsidiaries | (846 | ) | (641 | ) | — | (205 | ) | 32 | % | (641 | ) | N/M | |||||||||||||
Impairment on goodwill and intangible assets | — | — | 8,254 | — | N/M | (8,254 | ) | (100 | )% | ||||||||||||||||
Total operating expense | 30,900 | 17,247 | 28,715 | 13,653 | 79 | % | (11,468 | ) | (40 | )% | |||||||||||||||
Loss from operations | $ | (22,559 | ) | $ | (17,367 | ) | $ | (25,010 | ) | $ | (5,192 | ) | 30 | % | $ | 7,643 | (31 | )% | |||||||
Research and Development Expenses
Research and development costs are expensed as incurred and include the cost of compensation and related expenses, as well as expenses for third parties who conduct research and development on our behalf. The $16.2 million increase in our research and development expenses during the year ended December 31, 2014, as compared to the prior year, results primarily from a charge of $13.6 million as a result of the fispemifene in-license agreement with Forendo in October 2014 as well as other consulting and outside services for the development of Room Temperature Vitaros® and RayVa™. We expect to continue to incur expenses in 2015 related to the further development of Room Temperature Vitaros®, RayVa™ and fispemifene.
The $0.3 million decrease in our research and development expenditures during the year ended December 31, 2013, as compared to the prior year, reflects a decrease in license fee expenses related to our purchase of a PediatRx license in 2012 offset by an increase in consulting services to support our regulatory filings for Vitaros® in Europe.
General and Administrative Expenses
General and administrative expenses include expenses for personnel, finance, legal, business development and investor relations. The $2.1 million decrease in general and administrative expenses during 2014, as compared to the prior year, is primarily due to a decrease in salary-related expenses as a result of the deconsolidation of our former French Subsidiaries in April 2013. In addition,
35
we incurred higher legal expenses in 2013 related to the disposition of certain assets and businesses and certain litigation expenses. Consulting and professional fees also decreased in 2014 as compared to 2013.
The $1.8 million decrease in general and administrative expenses during 2013, as compared to the prior year, is due to compensation charges in 2012 of $0.5 million in severance expenses related to the departure of our former CEO and associated non-cash charges in the amount of $0.7 million related to the acceleration of the former CEO’s unvested options. Additionally, expenses related to our former French Subsidiaries decreased $0.9 million in 2013 as compared to the prior period following the deconsolidation of the former French Subsidiaries in April 2013. This was partially offset by higher legal expenses related to the disposition of certain assets and businesses and certain litigation expenses.
Gain on Contract Settlement
The $0.9 million gain on contract settlement recorded during 2014 represents the fair value of 388,888 escrowed common shares that were returned to us in connection with the settlement with former managers of the French Subsidiaries. These shares were restored as authorized, unissued common stock in March 2014. The $0.5 million gain on contract settlement recorded during 2013 represents the difference between the $1.2 million in common shares issued to TopoTarget in exchange for the extinguishment of $1.7 million of contingent consideration.
Recovery on Sale of Subsidiary
In June 2014, we amended our stock purchase agreement with Biotox and received a one-time cash payment of approximately $0.6 million in exchange for relinquishing our rights to future minimum payments. Prior to the amendment of the agreement, we also received payments of approximately $0.1 million for a total received from BioTox of $0.7 million in 2014. We have rights to certain potential future payments upon a change of control of BioTox within a specified time frame. These potential future payments will be recorded if and when realized.
We recorded a gain of approximately $0.7 million as discontinued operations within our statement of operations in 2014. Historically, we reflected the operations and subsequent cash collections associated with the sale of the business as a component of continuing operations, on the line recovery on sale of subsidiary within our consolidated statements of operations. However, we have elected not to correct these prior period amounts which are deemed immaterial. In both 2013 and 2012, we received $0.3 million and $0.3 million in payments from the buyer of Bio-Quant, which were recognized as a recovery on the sale of subsidiary in the respective periods.
Deconsolidation of Former French Subsidiaries
As a result of our former French Subsidiaries entering into judicial liquidation procedures in April 2013, we deconsolidated the former French Subsidiaries in the second quarter of 2013. This deconsolidation resulted in a non-cash gain of $0.6 million in 2013. At that time, we also recorded a liability of $2.8 million, equal to the net deconsolidated liabilities. In June 2014, consistent with the Global Settlement Agreement (“GSA”) signed in February of that year, the Works Council withdrew its previously submitted €4.1 million claim in the Versailles Civil Court (the “Civil Court”), all parties accepted the withdrawal and the Civil Court judge closed the discussions between all parties. The final procedural step occurred in October 2014, when we received a written judgment from the Civil Court acknowledging the dismissal of the claim and the closure of the litigation. Given the existence of the aforementioned ratified GSA, the accepted withdrawal and the closure of the discussions by the Civil Court judge, it was concluded during the second quarter of 2014 that we were relieved of all claims previously asserted by the French Works Council.
Pursuant to the aforementioned license and settlement agreements, Majorelle agreed to make certain severance payments of approximately $2.0 million to the former French Subsidiaries’ employees on our behalf. In addition, the Works Council and the Judicial Liquidator and Trustee of our former French Subsidiaries as well as each of the former French Subsidiaries’ employees, waived all claims they had asserted or could have asserted against us related to the liquidation and reorganization of the French Subsidiaries. As a result, during the second quarter of 2014, we released the approximate $2.8 million liability previously recorded in connection with the deconsolidation of the former French Subsidiaries and recognized approximately $0.8 million as a gain on deconsolidation as follows:
Release of deconsolidation liability | $ | 2.8 | ||
Less: Payments made by Majorelle on our behalf | (2.0 | ) | ||
Gain on deconsolidation of former French Subsidiaries | $ | 0.8 | ||
36
Impairment on Goodwill and Intangible Assets
In 2012, France implemented changes in reimbursement policy to favor generic drugs. This change in policy resulted in our former French Subsidiaries experiencing a loss and interruption in certain key contract agreements. Accordingly, we determined that the goodwill associated with our former French Subsidiaries was impaired and recorded a charge of $8.3 million to write down the entire balance of goodwill as of December 31, 2012. This impairment is presented in impairment of goodwill and intangible assets in our consolidated statements of operations.
Other Income and Expense
Other income and expense were as follows (in thousands, except percentages):
Year Ended December 31, | 2014 vs 2013 | 2013 vs 2012 | |||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | |||||||||||||||||||
Other income (expense) | |||||||||||||||||||||||||
Interest expense, net | $ | (339 | ) | $ | (727 | ) | $ | (325 | ) | $ | 388 | (53 | )% | $ | (402 | ) | 124 | % | |||||||
Loss on extinguishment of debt | (82 | ) | — | — | (82 | ) | N/M | — | N/M | ||||||||||||||||
Gain on sale of investment | — | 2,600 | — | (2,600 | ) | (100 | )% | 2,600 | N/M | ||||||||||||||||
Other income (expense), net | 503 | (376 | ) | 175 | 879 | (234 | )% | (551 | ) | (315 | )% | ||||||||||||||
Total other income (expense) | $ | 82 | $ | 1,497 | $ | (150 | ) | $ | (1,415 | ) | (95 | )% | $ | 1,647 | N/M | ||||||||||
Interest Expense, Net
Interest expense decreased $0.4 million during 2014 as compared to the prior year primarily due to the repayment of $1.5 million in principal on the 2012 Convertible Notes in April 2014, resulting in a reduction of interest expense. The terms of the 2012 Convertible Notes were amended during the fourth quarter of 2014 and the remainder of the balance was satisfied at that time. We will continue to recognize interest expense in connection with our credit facility with Oxford and SVB (see note 6 to our consolidated financial statements for further details).
Interest expense increased $0.4 million during 2013 as compared to the prior year, primarily as a result of interest expense in connection with the amortization of the discount related to the 2012 Convertible Notes (see note 6 to our consolidated financial statements for further details) as well as non-cash interest expense related to contingent consideration, which has been eliminated following the settlement agreement with TopoTarget during the third quarter of 2013.
Loss on Extinguishment of Debt
In October 2014, we amended the terms of our 2012 Convertible Notes due December 31, 2014 and repaid the remaining aggregate principal balance of $1.2 million with accrued interest. We incurred a loss on extinguishment of debt of approximately $0.l million during the fourth quarter of 2014, which consisted of the fair value of warrants issued to the former note holders in connection with the amendment, payment to the note holders in the amount of interest payments due under the original agreement terms, the remaining debt discount, and legal fees incurred in connection with the amendment.
Gain on Sale of Investment
We previously held an investment in a privately-held biotechnology company, which was valued at zero in our consolidated financial statements as of December 31, 2012. In 2013, we sold our investment in the entity and realized net proceeds of approximately $2.6 million, which was reflected as a gain on sale of investment during the fourth quarter of 2013 in our consolidated statement of operations.
Other Income (Expense), Net
Other income (expense), net, increased $0.9 million during 2014 as compared to the prior year primarily due to the change in the market value of the derivative liability related to the 2012 Convertible Notes. The 2012 Convertible Notes were satisfied during the fourth quarter of 2014 and therefore, the related derivative liability was removed from the balance sheet during the fourth quarter of 2014 (see note 6 to our consolidated financial statements for further details).
Other income (expense), net, decreased $0.6 million during 2013 as compared to the prior year due to $0.3 million of expense associated with the change in the market value of the derivative liability related to the 2012 Convertible Notes (see note 6 to our consolidated financial statements for further details) as well as the absence of rental income in 2013 as compared to $0.4 million in 2012.
37
Liquidity, Capital Resources and Financial Condition
We have experienced net losses and negative cash flows from operations each year since our inception. Through December 31, 2014, we had an accumulated deficit of $289.9 million and recorded a net loss of approximately $21.8 million for the year ended December 31, 2014. We have been principally financed through the sale of our common stock and other equity securities, debt financing and up-front payments received from commercial partners for our products under development. Funds raised in recent periods include approximately $15.8 million from our May 2013 follow-on public offering, approximately $3.7 million during 2014 from the sale of common stock from our committed equity financing facility with Aspire Capital (see note 7 to our consolidated financial statements for further details) and $2.2 million during 2014 from the sale of common stock via our “at-the-market” (“ATM”) stock selling facility, which was terminated in August 2014. These and other cash-generating activities should not necessarily be considered an indication of our ability to raise additional funds in the future.
In February 2015, we entered into subscription agreements with certain purchasers pursuant to which we agreed to sell an aggregate of 6,043,955 shares of our common stock and issued warrants to purchase up to an additional 3,021,977 shares of our common stock. Each share of common stock was priced at $1.82 per unit and included one-half of warrant to purchase a share of common stock. The warrants have an exercise price of $1.82 per share, are exercisable beginning six months and one day after the date of issuance and expire on the seventh anniversary of the date of issuance. The total net proceeds from the offering were $10.8 million after deducting expenses of approximately $0.2 million.
Based upon our current business plan, the access to additional capital under our committed equity financing facility, the potential to borrow an additional amount of up to $5.0 million under our credit facility, and the $10.8 million received from our February 2015 financing, we believe we have sufficient cash to fund our on-going operations through the first quarter of 2016. We expect to have net cash outflows from operations in 2015 as we initiate a Phase 2 clinical trial for fispemifene, continue to develop Room Temperature Vitaros®, further our Phase 2a clinical trial for RayVa™, and incur other operating expenses. As of December 31, 2014, net open purchase orders, less any accruals or invoices charged or amounts paid, totaled approximately $5.3 million.
Based on our recurring losses, negative cash flows from operations and working capital levels, we will need to raise substantial additional funds to finance our operations. If we are unable to maintain sufficient financial resources, including by raising additional funds when needed, our business, financial condition and results of operations will be materially and adversely affected. There can be no assurance that we will be able to obtain the needed financing on reasonable terms or at all. Additionally, equity financings may have a dilutive effect on the holdings of our existing stockholders.
We currently have an effective shelf registration statement on Form S-3 filed with the SEC under which we may offer from time to time any combination of debt securities, common and preferred stock and warrants. We have approximately $89.0 million available under the S-3 shelf registration statement (No. 333-198066) of which $18.2 million is currently reserved under the committed equity financing facility. Our equity financing facility may be terminated by us by giving proper written notice. The rules and regulations of the SEC or any other regulatory agencies may restrict our ability to conduct certain types of financing activities, or may affect the timing of and amounts we can raise by undertaking such activities.
Even if we are successful in obtaining additional cash resources to support further development of our products, we may still encounter additional obstacles such as our development activities not being successful, our products not proving to be safe or effective, clinical development work not being completed in a timely manner or at all, or anticipated products not being commercially viable or successfully marketed. Additionally, our business could require additional financing if we choose to accelerate product development expenditures in advance of receiving up-front payments from development and commercial partners. If our efforts to raise additional capital when needed through equity or debt financings are unsuccessful, we may be required to delay or scale-back our development plans, reduce costs and personnel and cease to operate as a going concern. These financial statements do not include any adjustments that might result from the outcome of this uncertainty.
38
Cash Flow Summary
The following table summarizes selected items in our consolidated statements of cash flows (in thousands):
2014 | 2013 | 2012 | ||||||||||
Net cash used in operating activities from continuing operations | $ | (18,031 | ) | $ | (15,103 | ) | $ | (12,397 | ) | |||
Net cash (used in) provided by investing activities from continuing operations | (530 | ) | 3,059 | 1,368 | ||||||||
Net cash provided by financing activities from continuing operations | 7,865 | 16,631 | 20,766 | |||||||||
Net cash provided by (used in) discontinued operations | 691 | 1,688 | (2,285 | ) | ||||||||
Effect of exchange rate changes on cash | — | — | 243 | |||||||||
Net (decrease) increase in cash and cash equivalents | $ | (10,005 | ) | $ | 6,275 | $ | 7,695 | |||||
Operating Activities from Continuing Operations
Cash used in operating activities increased by $2.9 million in 2014 as compared to 2013 due to an increase in net loss from continuing operations of $6.6 million from 2013 to 2014, adjusted for non-cash items including $5.9 million of research and development (“R&D”) expense paid to Forendo in shares of common stock, stock based compensation expense of $1.7 million, a gain of $0.8 million related to the deconsolidation of our former French Subsidiaries, and a $0.9 million gain on contract settlement. The change in net operating assets resulted mainly from the payment of liabilities associated with our former French Subsidiaries, release of deferred revenue largely related to the recognition of the Majorelle upfront payment which were offset by an increase in accrued expenses related to future consideration due to Forendo (see note 3 to our consolidated financial statements for further details).
Cash used in operating activities increased by $2.7 million in 2013 as compared to 2012 due to a decrease in net loss from continuing operations of $9.8 million from 2012 to 2013, adjusted for non-cash items including stock-based compensation expense of $2.0 million, a gain of $0.6 million related to the deconsolidation of our former French Subsidiaries, and a $0.5 million gain on contract settlement. The change in net operating assets resulted mainly from a decrease in accounts payable and accrued compensation offset by an increase in deferred revenue.
Cash used in operating activities decreased by $2.6 million in 2012 as compared to 2011 due to a decrease in net loss from continuing operations of $7.5 million from 2011 to 2012, adjusted for non-cash items including $8.3 million of impairment charges to goodwill and intangible assets, stock based compensation expense of $2.9 million as well as a $1.3 million deferred tax provision. The change in net operating assets resulted mainly from a decrease in prepaid expenses and other current assets offset by an increase in accounts payable.
Investing Activities from Continuing Operations
Cash used in investing activities totaled $0.5 million in 2014 which included fixed asset purchases of $0.6 million offset by proceeds of $0.1 million from the recovery of loss on sale of subsidiary.
Cash provided by investing activities totaled $3.1 million in 2013. We had proceeds of $3.7 million from the sale of our New Jersey facility, offset by fixed asset purchases of $0.6 million and $0.3 million for the deposit of restricted cash.
Cash provided by investing activities totaled $1.4 million in 2012 primarily as a result of acquiring cash of $2.1 million in the acquisition of our former French Subsidiaries, partially offset by purchases of $0.4 million in fixed assets.
Financing Activities from Continuing Operations
Cash provided by financing activities totaled $7.9 million in 2014. We received proceeds of approximately $5.9 million from the sale of common stock under our ATM stock selling facility and our committed equity financing facility with Aspire Capital. We also had proceeds of $4.7 million from the issuance of notes payable in October 2014. Offsetting these transactions was the repayment of $2.75 million in principal on our 2012 Convertible Notes (see note 6 to our consolidated financial statements for further details on both debt-related activities).
Cash provided by financing activities totaled $16.6 million in 2013. We received proceeds of $16.6 million from the sale of common stock, from the sale of common stock, primarily in connection with a May 2013 financing.
Cash provided by financing activities totaled $20.8 million in 2012. We received proceeds of $20.4 million from the sale of common stock, primarily in connection with a February 2012 financing, offset by the net extinguishment of convertible notes payable.
39
Off-Balance Sheet Arrangements
As of December 31, 2014, we did not have any off-balance sheet arrangements as defined in Item 303(a)(4)(ii) of Regulation S-K.
Contractual Obligations
As of December 31, 2014, future minimum payments due under our contractual obligations are as follows (in thousands):
Payments Due by Period | ||||||||||||||||||||
Total | Less Than 1 Year | 1-3 Years | 3-5 Years | After 5 Years | ||||||||||||||||
Notes payable, including interest | $ | 6,268 | $ | 644 | $ | 3,758 | $ | 1,866 | $ | — | ||||||||||
Operating lease obligations | 931 | 515 | 416 | — | — | |||||||||||||||
Deferred compensation, including interest | 495 | 180 | 315 | — | — | |||||||||||||||
Capital lease obligations | 11 | 6 | 5 | — | — | |||||||||||||||
Total | $ | 7,705 | $ | 1,345 | $ | 4,494 | $ | 1,866 | $ | — | ||||||||||
We also have significant contractual obligations related to our clinical trial expenditures with clinical research organizations (“CROs”). As of December 31, 2014, net open purchase orders which include obligations to our CROs, less any accruals or invoices charged or amounts paid, totaled approximately $5.3 million. These payments are generally cancellable upon notice without penalty and therefore these obligations are not included in the table above.
Certain employees have employment agreements that provide for severance compensation in the event of a termination or a change in control. These agreements generally provide for a severance payment of up to 12 months of the applicable base salary in effect at the time of termination and continued health benefits at our expense for up to 12 months, each of which will be recorded as a liability when and if incurred. No obligation is recorded in the table above relating to the aforementioned agreements.
Recent Accounting Pronouncements
See note 2 to our consolidated financial statements for a discussion of recent accounting pronouncements and their effect, if any, on us.
Critical Accounting Estimates and Policies
The preparation of financial statements in accordance with United States generally accepted accounting principles (“GAAP”) requires management to make estimates and assumptions that affect the amounts reported in our consolidated financial statements and accompanying notes. Management bases its estimates on historical experience, market and other conditions, and various other assumptions it believes to be reasonable. Although these estimates are based on management’s best knowledge of current events and actions that may impact us in the future, the estimation process is, by its nature, uncertain given that estimates depend on events over which we may not have control. If market and other conditions change from those that we anticipate, our consolidated financial statements may be materially affected. In addition, if our assumptions change, we may need to revise our estimates, or take other corrective actions, either of which may also have a material effect in our consolidated financial statements. We review our estimates, judgments, and assumptions used in our accounting practices periodically and reflect the effects of revisions in the period in which they are deemed to be necessary. We believe that these estimates are reasonable; however, our actual results may differ from these estimates.
We believe that the following critical accounting policies and estimates have a higher degree of inherent uncertainty and require our most significant judgments. In addition, changes in the accounting estimates we use are reasonably likely to occur from time to time and had we used estimates different from any of these, our consolidated financial statements could have been materially different from those presented. Members of our senior management have discussed the development and selection of our critical accounting policies and estimates, and our disclosure regarding them, with the Audit Committee of our Board of Directors. Our accounting policies are more fully described in note 1 to the consolidated financial statements in Part II, Item 8 of this Form 10-K.
40
Revenue Recognition
We generate revenues from the licensing of technology rights and the sale of products, and historically, from the performance of pre-clinical testing services and contract sales services. Payments received under commercial arrangements, such as the licensing of technology rights, may include non-refundable fees at the inception of the arrangements, milestone payments for specific achievements designated in the agreements, and royalties on the sale of products.
We recognize revenue when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) Our price to the buyer is fixed or determinable; and (4) collectability is reasonably assured.
License Fee Revenue
Consideration received for our license arrangements may consist of non-refundable upfront license fees, various performance or sales milestones, royalties upon sales of product, and the delivery of product and/or research services to the licensor. We consider a variety of factors in determining the appropriate method of accounting under our license agreements, including whether the various elements can be separated and accounted for individually as separate units of accounting. Deliverables under the arrangement will be separate units of accounting, provided (i) a delivered item has value to the customer on a standalone basis; and (ii) if the arrangement includes a general right of return relative to the delivered item and delivery or performance of the undelivered item is considered probable and substantially in our control.
We account for revenue arrangements with multiple elements by separating and allocating consideration in a multiple-element arrangement according to the relative selling price of each deliverable. If an element can be separated, an amount is allocated based upon the relative selling price of each element. We determine the relative selling price of a separate deliverable using the price we charge other customers when we sell that product or service separately; however, if the product or service is not sold separately and third party pricing evidence is not available, we will use our best estimate of selling price.
We defer recognition of non-refundable upfront fees if we have continuing performance obligations without which the technology, right, product or service conveyed in conjunction with the non-refundable fee has no utility to the licensee that is separate and independent of our performance under the other elements of the arrangement. Non-refundable, up-front fees that are not contingent on any future performance by us and require no consequential continuing involvement on our part are recognized as revenue when the license term commences and the licensed data, technology and/or compound is delivered. The specific methodology for the recognition of the revenue is determined on a case-by-case basis according to the facts and circumstances of the applicable agreement.
We evaluate milestone payments on an individual basis and revenues are recognized upon achievement of the associated milestone, provided that (i) the milestone event is substantive and its achievability was not reasonably assured at the inception of the agreement and (ii) the amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with the milestone event.
Long-Lived Assets
We review our long-lived assets for impairment whenever events or circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of an asset and its eventual disposition are less than its carrying amount. If such asset is considered impaired, the amount of the impairment loss recognized is measured as the amount by which the carrying value of the asset exceeds the fair value of the asset, the fair value of which is determined based upon discounted cash flows or appraised values, depending on the nature of the asset. There were no impairment charges recorded in 2014 related to our long-lived assets.
Stock Based Compensation
In preparation of our consolidated financial statements, we calculate the value of stock options issued to employees, non-employee contractors and the Board of Directors and warrants issued to investors. The fair value of each option and warrant is estimated on the date of grant using the Black-Scholes option pricing model. The Black-Scholes option pricing model is a generally accepted method of estimating the fair value of stock options and warrants.
The Black-Scholes option pricing model requires us to estimate our dividend yield rate, expected volatility and risk free interest rate over the life of the option. The use of estimates on these factors may cause the fair value of the option to be under or overestimated (see note 8 to our consolidated financial statements for the current estimates used in the Black-Scholes option pricing model).
Clinical Trial Accruals
In preparation of our consolidated financial statements, we are required to estimate our expenses resulting from our obligations under contracts with vendors and consultants and clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations which vary from contract to contract, and may result in payment flows that do not match the periods over which materials or services are provided to us under such contracts. Our objective is to reflect the
41
appropriate clinical trial expenses in our financial statements by matching those expenses with the period in which the services and efforts are expended. We account for these expenses according to the progress of the clinical trial as measured by patient progression and the timing of various aspects of the trial. We determine accrual estimates through financial models, taking into account discussion with applicable personnel and outside service providers as to the progress or state of consummation of trials, or the services completed. During the course of a clinical trial, we adjust our rate of clinical expense recognition if actual results differ from our estimates. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on the facts and circumstances known to us at that time. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in our reporting amounts that are too high or too low for any particular period. Through December 31, 2014, there have been no material adjustments to our prior period estimates of expenses for clinical trials.
Income Taxes
We recognize deferred taxes under the asset and liability method of accounting for income taxes by which deferred income taxes are recognized for differences between the financial statement and tax bases of assets and liabilities at enacted statutory tax rates in effect for the years in which the differences are expected to reverse. The effect on deferred taxes of a change in tax rates is recognized in income in the period that includes the enactment date. In addition, valuation allowances are established, when necessary, to reduce deferred tax assets to the amounts expected to be realized.
In consideration of our accumulated losses and lack of historical ability to generate taxable income to utilize our deferred tax assets, we have determined it is not more likely than not we will be able to realize any benefit from our temporary differences and have recorded a full valuation allowance. If we become profitable in the future at levels which cause management to conclude that it is more likely than not that we will realize all or a portion of the net operating loss carry-forward, we would record the estimated net realized value of the deferred tax asset at that time and would then provide for income taxes at a rate equal to our combined federal and state effective rates, which would be approximately 40% under current tax laws. Subsequent revisions to the estimated net realizable value of the deferred tax asset could cause our provision for income taxes to vary significantly from period to period.
Our policy is to recognize interest and penalties related to income tax matters in income tax expense. As the unrecognized tax benefits relate to un-utilized deferred tax assets and because we have generated net operating losses since inception for both federal and state income tax purposes and no tax liabilities, penalties or interest have been recognized for balance sheet or statement of operations purposes as of and for the period ended December 31, 2014, 2013 or 2012.
ITEM 7A. | QUALITATIVE AND QUANTITATIVE DISCLOSURES ABOUT MARKET RISK |
Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of United States interest rates, particularly because the majority of our investments are in short-term marketable securities. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. Some of the securities may be subject to market risk. This means that a change in prevailing interest rates may cause the value of the investment to fluctuate. For example, if we purchase a security that was issued with a fixed interest rate and the prevailing interest rate later rises, the value of our investment will probably decline. To minimize this risk, we typically invest all, or substantially all, of our cash in money market funds that invest primarily in government securities. Our investment policy also permits investments in a variety of securities including commercial paper and government and non-government debt securities. In general, money market funds are not subject to market risk because the interest paid on such funds fluctuates with the prevailing interest rate. As of December 31, 2014 and 2013, we did not have any holdings of derivative financial or commodity instruments. We conduct a portion of our business in currencies other than our United States dollar functional currency. These transactions give rise to monetary assets and liabilities that are denominated in currencies other than the United States dollar. The value of these monetary assets and liabilities are subject to changes in currency exchange rates from the time the transactions are originated until settlement in cash. Our foreign currency exposures are primarily concentrated in the Euro and both realized and unrealized gains or losses on the value of these monetary assets and liabilities are included in the determination of net income.
All of our cash and cash equivalents is in cash accounts and highly liquid. If a 10% change in interest rates were to have occurred on December 31, 2014, this change would not have had a material effect on the fair value of our investment portfolio as of that date nor our net loss for the years then ended. Due to the short holding period of our investments, we have concluded that we do not have a material financial market risk exposure.
42
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
INDEX TO FINANCIAL STATEMENTS
PAGE | |
Reports of Independent Registered Public Accounting Firm | |
Financial Statements: | |
Consolidated Balance Sheets as of December 31, 2014 and 2013 | |
Consolidated Statements of Operations and Other Comprehensive Loss for the years ended December 31, 2014, 2013 and 2012 | |
Consolidated Statements of Cash Flows for the years ended December 31, 2014, 2013 and 2012 | |
Consolidated Statements of Changes in Stockholders’ Equity for the years ended December 31, 2014, 2013 and 2012 | |
Notes to the Consolidated Financial Statements | |
43
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
Apricus Biosciences, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations and other comprehensive loss, of changes in stockholders’ equity and of cash flows present fairly, in all material respects, the financial position of Apricus Biosciences, Inc. and its subsidiaries at December 31, 2014 and 2013, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company did not maintain, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) because material weaknesses in internal control over financial reporting related to the accounting for and disclosures of technical accounting matters in the consolidated financial statements and the monitoring and oversight over the controls in the financial reporting process existed as of that date. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis. The material weaknesses referred to above are described in Management’s Report on Internal Control Over Financial Reporting appearing under Item 9A. We considered these material weaknesses in determining the nature, timing, and extent of audit tests applied in our audit of the December 31, 2014 consolidated financial statements, and our opinion regarding the effectiveness of the Company’s internal control over financial reporting does not affect our opinion on those consolidated financial statements. The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in management's report referred to above. Our responsibility is to express opinions on these financial statements and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
PricewaterhouseCoopers LLP
San Diego, California
March 16, 2015
44
Apricus Biosciences, Inc. and Subsidiaries
Consolidated Balance Sheets
(In thousands, except share and per share data)
December 31, 2014 | December 31, 2013 | ||||||
Assets | |||||||
Current assets | |||||||
Cash and cash equivalents | $ | 11,400 | $ | 21,405 | |||
Accounts receivable | 678 | 59 | |||||
Restricted cash | 290 | 332 | |||||
Inventories | 275 | 336 | |||||
Prepaid expenses and other current assets | 646 | 132 | |||||
Total current assets | 13,289 | 22,264 | |||||
Property and equipment, net | 1,358 | 955 | |||||
Other long term assets | 162 | 91 | |||||
Total assets | $ | 14,809 | $ | 23,310 | |||
Liabilities and stockholders’ equity | |||||||
Current liabilities | |||||||
Notes payable | $ | 153 | $ | — | |||
Convertible notes payable, net | — | 2,600 | |||||
Accounts payable | 860 | 926 | |||||
Accrued expenses | 4,555 | 2,119 | |||||
Accrued compensation | 1,112 | 952 | |||||
Deferred revenue | 226 | 1,800 | |||||
Derivative liability | — | 517 | |||||
Deconsolidation of former French Subsidiaries | — | 2,846 | |||||
Total current liabilities | 6,906 | 11,760 | |||||
Long term liabilities | |||||||
Notes payable, net | 4,626 | — | |||||
Deferred revenue | 1,000 | — | |||||
Other long term liabilities | 358 | 578 | |||||
Total liabilities | 12,890 | 12,338 | |||||
Commitments and contingencies | |||||||
Stockholders’ equity: | |||||||
Preferred stock, $.001 par value, 10,000,000 shares authorized, no shares issued or outstanding as of December 31, 2014 and 2013 | — | — | |||||
Common stock, $.001 par value, 75,000,000 shares authorized, 44,330,006 and 37,541,404 issued and outstanding as of December 31, 2014 and 2013, respectively | 44 | 38 | |||||
Additional paid-in-capital | 291,727 | 279,000 | |||||
Accumulated deficit | (289,852 | ) | (268,066 | ) | |||
Total stockholders’ equity | 1,919 | 10,972 | |||||
Total liabilities and stockholders’ equity | $ | 14,809 | $ | 23,310 | |||
The accompanying notes are an integral part of these consolidated financial statements.
45
Apricus Biosciences, Inc. and Subsidiaries
Consolidated Statements of Operations
And Other Comprehensive Loss
(In thousands, except share and per share data)
For the Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
License fee revenue | $ | 8,454 | $ | 941 | $ | 4,276 | |||||
Royalty revenue | 36 | — | — | ||||||||
Product sales | 769 | 21 | 23 | ||||||||
Contract service revenue | — | 1,549 | 3,646 | ||||||||
Total revenue | 9,259 | 2,511 | 7,945 | ||||||||
Cost of product sales | 918 | 23 | 10 | ||||||||
Cost of service revenue | — | 2,608 | 4,230 | ||||||||
Gross profit (loss) | 8,341 | (120 | ) | 3,705 | |||||||
Operating expense (income) | |||||||||||
Research and development | 21,288 | 5,123 | 5,375 | ||||||||
General and administrative | 11,418 | 13,554 | 15,336 | ||||||||
Gain on contract settlement | (910 | ) | (534 | ) | — | ||||||
Recovery loss on sale of subsidiary | (50 | ) | (255 | ) | (250 | ) | |||||
Deconsolidation of former French Subsidiaries | (846 | ) | (641 | ) | — | ||||||
Impairment on goodwill and intangible assets | — | — | 8,254 | ||||||||
Total operating expense | 30,900 | 17,247 | 28,715 | ||||||||
Loss from continuing operations before other income (expense) | (22,559 | ) | (17,367 | ) | (25,010 | ) | |||||
Other income (expense) | |||||||||||
Interest expense, net | (339 | ) | (727 | ) | (325 | ) | |||||
Loss on extinguishment of debt | (82 | ) | — | — | |||||||
Gain on sale of investment | — | 2,600 | — | ||||||||
Other income (expense), net | 503 | (376 | ) | 175 | |||||||
Total other income (expense) | 82 | 1,497 | (150 | ) | |||||||
Loss from continuing operations before income tax expense | (22,477 | ) | (15,870 | ) | (25,160 | ) | |||||
Income tax expense | — | — | (516 | ) | |||||||
Loss from continuing operations | (22,477 | ) | (15,870 | ) | (25,676 | ) | |||||
Income (loss) from discontinued operations | 691 | (1,068 | ) | (6,095 | ) | ||||||
Net loss | $ | (21,786 | ) | $ | (16,938 | ) | $ | (31,771 | ) | ||
Basic and diluted loss per common share | |||||||||||
Loss per share from continuing operations | $ | (0.57 | ) | $ | (0.46 | ) | $ | (0.94 | ) | ||
Income (loss) per share from discontinued operations | 0.02 | (0.03 | ) | (0.22 | ) | ||||||
Net loss per share | $ | (0.55 | ) | $ | (0.49 | ) | $ | (1.16 | ) | ||
Weighted average common shares outstanding used for basic and diluted loss per share | 39,540,409 | 34,413,253 | 27,458,184 | ||||||||
Net loss | $ | (21,786 | ) | $ | (16,938 | ) | $ | (31,771 | ) | ||
Other comprehensive income | |||||||||||
Foreign currency translation adjustments | — | — | 641 | ||||||||
Comprehensive loss | $ | (21,786 | ) | $ | (16,938 | ) | $ | (31,130 | ) | ||
The accompanying notes are an integral part of these consolidated financial statements.
46
Apricus Biosciences, Inc. and Subsidiaries
Consolidated Statements of Cash Flows
(In thousands, except share data)
For the Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cash flows from operating activities of continuing operations: | |||||||||||
Net loss | $ | (21,786 | ) | $ | (16,938 | ) | $ | (31,771 | ) | ||
Gain (loss) from discontinued operations | 691 | (1,068 | ) | (6,095 | ) | ||||||
Net loss from continuing operations | (22,477 | ) | (15,870 | ) | (25,676 | ) | |||||
Adjustments to reconcile net loss from continuing operations to net cash used in operating activities of continuing operations: | |||||||||||
Shares issued in connection with the fispemifene in-license agreement | 5,904 | — | — | ||||||||
Deconsolidation of former French Subsidiaries | (846 | ) | (641 | ) | — | ||||||
Gain on contract settlement | (910 | ) | (534 | ) | — | ||||||
Depreciation and amortization | 170 | 77 | 203 | ||||||||
Non cash interest expense | 163 | 250 | 37 | ||||||||
Loss on debt extinguishment | 51 | — | — | ||||||||
Stock-based compensation expense | 1,731 | 1,992 | 2,917 | ||||||||
Recovery on loss on sale of subsidiary | (50 | ) | (255 | ) | (250 | ) | |||||
Derivative liability revaluation | (517 | ) | 274 | — | |||||||
Interest on contingent consideration | — | 242 | — | ||||||||
Non-cash deferred compensation | — | — | 640 | ||||||||
Impairment charges on property held for sale | — | — | 656 | ||||||||
Impairment charges on goodwill and intangible assets | — | — | 8,254 | ||||||||
Deferred tax provision | — | — | 1,261 | ||||||||
Other | — | 131 | — | ||||||||
Changes in operating assets and liabilities of continuing operations, net of assets and liabilities acquired and divested: | |||||||||||
Accounts receivable | (620 | ) | 253 | 682 | |||||||
Inventories | 61 | (341 | ) | — | |||||||
Prepaid expenses and other current assets | (464 | ) | 143 | (928 | ) | ||||||
Other assets | 2 | (52 | ) | — | |||||||
Accounts payable | (66 | ) | (920 | ) | 681 | ||||||
Deconsolidation of Former French Subsidiaries | (2,000 | ) | — | — | |||||||
Accrued expenses | 2,465 | (476 | ) | (1,348 | ) | ||||||
Accrued compensation | 160 | (140 | ) | 967 | |||||||
Deferred revenue | (574 | ) | 1,021 | (490 | ) | ||||||
Other liabilities | (214 | ) | (257 | ) | (3 | ) | |||||
Net cash used in operating activities from continuing operations | (18,031 | ) | (15,103 | ) | (12,397 | ) | |||||
Cash flows from investing activities of continuing operations: | |||||||||||
Purchase of fixed assets | (580 | ) | (573 | ) | (436 | ) | |||||
Proceeds from sale of subsidiary | 50 | 255 | 250 | ||||||||
Proceeds from the sale of property and equipment | — | 3,657 | — | ||||||||
Deposit of restricted cash | — | (280 | ) | — | |||||||
Cash acquired from acquisitions | — | — | 2,067 | ||||||||
Cash paid for acquisitions | — | — | (513 | ) | |||||||
Net cash (used in) provided by investing activities from continuing operations | (530 | ) | 3,059 | 1,368 | |||||||
Cash flows from financing activities of continuing operations: | |||||||||||
Issuance of common stock, net of offering costs | 5,913 | 16,612 | 20,410 | ||||||||
Proceeds from issuance of notes payable, net | 4,729 | — | — | ||||||||
Proceeds from exercise of warrants | — | 46 | 40 | ||||||||
Payment under convertible notes | (2,750 | ) | — | (4,000 | ) | ||||||
47
Release of restricted cash, net | 42 | — | — | ||||||||
Repurchase and retirement of stock | (42 | ) | — | — | |||||||
Reissuance of convertible notes payable | — | — | 3,413 | ||||||||
Changes in derivative liability | — | — | 906 | ||||||||
Proceeds from the exercise of stock options | — | — | 10 | ||||||||
Repayment of capital lease obligations | (27 | ) | (27 | ) | (13 | ) | |||||
Net cash provided by financing activities from continuing operations | 7,865 | 16,631 | 20,766 | ||||||||
Cash flows from discontinued operations: | |||||||||||
Net cash provided by (used in) operating activities of discontinued operations | 16 | 38 | (1,985 | ) | |||||||
Net cash provided by (used in) investing activities of discontinued operations | 675 | 1,650 | (300 | ) | |||||||
Net cash provided by (used in) discontinued operations | 691 | 1,688 | (2,285 | ) | |||||||
Effect of exchange rate changes on cash | — | — | 243 | ||||||||
Net (decrease) increase in cash and cash equivalents | (10,005 | ) | 6,275 | 7,695 | |||||||
Cash and cash equivalents, beginning of period | 21,405 | 15,130 | 7,435 | ||||||||
Cash and cash equivalents, end of period | $ | 11,400 | $ | 21,405 | $ | 15,130 | |||||
Supplemental disclosure of cash flow information: | |||||||||||
Cash paid for interest | $ | 193 | $ | 238 | $ | 318 | |||||
Non-cash investing and financing activities: | |||||||||||
Liability incurred in connection with fixed asset purchases | $ | (7 | ) | $ | — | $ | — | ||||
Issuance of 193,798 common warrants to debtholders | $ | 104 | $ | — | $ | — | |||||
Issuance of 486,923 shares of common stock upon conversion of convertible note | $ | — | $ | 1,737 | $ | — | |||||
Issuance of 688,717 shares of common stock to TopoTarget | $ | — | $ | 1,543 | $ | — | |||||
Release of restricted cash | $ | — | $ | (337 | ) | $ | — | ||||
Release of obligations related to short-term loans | $ | — | $ | 270 | $ | — | |||||
Issuance of 373,134 shares of common stock to PediatRx Inc. for co-promote agreement | $ | — | $ | — | $ | 1,000 | |||||
Issuance of 2,592,592 shares of common stock to former Finesco shareholders at date of contribution | $ | — | $ | — | $ | 8,556 | |||||
The accompanying notes are an integral part of these consolidated financial statements.
48
Apricus Biosciences, Inc. and Subsidiaries
Consolidated Statements of Changes in Stockholders’ Equity
(In thousands, except share data)
Common Stock (Shares) | Common Stock (Amount) | Additional Paid-In Capital | Accumulated Other Comprehensive Income | Accumulated Deficit | Total Stockholders’ Equity | |||||||||||||||||
Balance as of December 31, 2011 | 21,347,986 | $ | 21 | $ | 224,154 | $ | — | $ | (219,357 | ) | $ | 4,818 | ||||||||||
Issuance of common stock upon exercise of stock options | 5,000 | — | 10 | — | — | 10 | ||||||||||||||||
Issuance of stock to employees, consultants and Board of Director members | 147,761 | — | — | — | — | — | ||||||||||||||||
Stock-based compensation expense | 2,917 | — | — | 2,917 | ||||||||||||||||||
Issuance of common stock for co-promote agreement | 373,134 | — | 1,000 | — | — | 1,000 | ||||||||||||||||
Issuance of common stock for Finesco transaction | 2,592,592 | 3 | 8,553 | — | — | 8,556 | ||||||||||||||||
Issuance of common stock and warrants, net of offering costs | 5,453,601 | 6 | 20,404 | — | — | 20,410 | ||||||||||||||||
Issuance of common stock upon exercise of warrants | 17,595 | — | 40 | — | — | 40 | ||||||||||||||||
Foreign currency translation adjustment | 641 | 641 | ||||||||||||||||||||
Net loss | — | — | — | — | (31,771 | ) | (31,771 | ) | ||||||||||||||
Balance as of December 31, 2012 | 29,937,669 | 30 | 257,078 | 641 | (251,128 | ) | 6,621 | |||||||||||||||
Issuance of restricted stock to employees and Board of Director members | 95,645 | — | — | — | — | — | ||||||||||||||||
Stock-based compensation expense | 1,992 | — | — | 1,992 | ||||||||||||||||||
Issuance of common stock, net of offering costs | 312,450 | — | 792 | — | — | 792 | ||||||||||||||||
Issuance of common stock and warrants, net of offering costs | 6,000,000 | 6 | 15,814 | — | — | 15,820 | ||||||||||||||||
Issuance of common stock upon exercise of convertible notes | 486,923 | 1 | 1,736 | — | — | 1,737 | ||||||||||||||||
Issuance of common stock to TopoTarget | 688,717 | 1 | 1,542 | — | — | 1,543 | ||||||||||||||||
Issuance of common stock upon exercise of warrants | 20,000 | — | 46 | — | — | 46 | ||||||||||||||||
Elimination of cumulative translation adjustment upon deconsolidation of former French Subsidiaries | — | — | — | (641 | ) | (641 | ) | |||||||||||||||
Net loss | — | — | — | — | (16,938 | ) | (16,938 | ) | ||||||||||||||
Balance as of December 31, 2013 | 37,541,404 | 38 | 279,000 | — | (268,066 | ) | 10,972 | |||||||||||||||
Issuance of restricted stock to employees and Board of Director members | 26,728 | — | — | — | — | — | ||||||||||||||||
Repurchase and retirement of stock | (19,338 | ) | — | (42 | ) | — | — | (42 | ) | |||||||||||||
Stock-based compensation expense | — | — | 1,731 | — | — | 1,731 | ||||||||||||||||
Issuance of common stock and warrants, net of offering costs | 3,570,030 | 3 | 6,047 | — | — | 6,050 | ||||||||||||||||
Issuance of common stock in connection with fispemifene in-license agreement | 3,600,070 | 3 | 5,901 | — | — | 5,904 | ||||||||||||||||
49
Return of common stock in connection with contract settlement | (388,888 | ) | — | (910 | ) | — | — | (910 | ) | |||||||||||||
Net loss | — | — | — | — | (21,786 | ) | (21,786 | ) | ||||||||||||||
Balance as of December 31, 2014 | 44,330,006 | $ | 44 | $ | 291,727 | $ | — | $ | (289,852 | ) | $ | 1,919 | ||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
50
Apricus Biosciences, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Organization
Apricus Biosciences, Inc. and Subsidiaries (“Apricus” or the “Company”) is a Nevada corporation initially formed in 1987. The Company has operated in the pharmaceutical industry since 1995 with a current primary focus on the development and commercialization of products and product candidates in the areas of specialty urology and rheumatology. The Company’s proprietary drug delivery technology is a permeation enhancer called NexACT® and the Company has one approved drug,Vitaros®, which uses the NexACT® delivery system, and is approved for the treatment of erectile dysfunction (“ED”) in Canada and through the European Decentralized Procedure (“DCP”) in Europe. Vitaros® was launched by the Company’s licensee partners in certain territories in Europe in the second half of 2014 and the Company expects that commercial launches will continue to occur throughout 2015. The Company has a second generation Vitaros® product candidate (“Room Temperature Vitaros®”) in development, which is a proprietary stabilized dosage formulation that is expected to be stored at room temperature conditions. RayVa™, the Company’s product candidate which also utilizes its proprietary permeation enhancer for the treatment of Raynaud’s Phenomenon secondary to scleroderma, received clearance from the United States Food and Drug Administration (“FDA”) in May 2014 to begin clinical studies, and the Company’s Phase 2a clinical trial began in December 2014.
In October 2014, the Company entered into an agreement to license the exclusive United States development and commercialization rights for fispemifene, a tissue-specific selective estrogen receptor modulator (“SERM”) designed to treat secondary hypogonadism, chronic prostatitis and lower urinary tract symptoms in men (see note 3 for further details).
Basis of Presentation and Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All significant intercompany accounts and transactions have been eliminated in consolidation. Certain prior year items have been reclassified to conform to the current year presentation.
Use of Estimates
The preparation of these consolidated financial statements in conformity with generally accepted accounting principles (“GAAP”) requires the Company to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses. The Company’s most significant estimates relate to whether revenue recognition criteria have been met, accounting for clinical trials, the valuation of stock based compensation, the impairment of long-lived assets and valuation allowances for the Company’s deferred tax assets. The Company’s actual results may differ from these estimates under different assumptions or conditions.
Liquidity
The accompanying consolidated financial statements have been prepared on a basis that contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company had an accumulated deficit of approximately $289.9 million as of December 31, 2014, recorded a net loss of approximately $21.8 million for the year ended December 31, 2014 and has principally been financed through the sale of its common stock and other equity securities, debt financing and up-front payments received from commercial partners for the Company’s products under development. Funds raised in recent periods from the sale of common stock include approximately 1) $15.8 million from the Company’s May 2013 follow-on public offering, 2) $3.7 million in net proceeds during 2014 from the sale of common stock from its committed equity financing facility with Aspire Capital Fund, LLC (“Aspire Capital”) (see note 7 for further details) and 3) $2.2 million in net proceeds during 2014 from the sale of common stock via its “at-the-market” (“ATM”) stock selling facility, which was terminated in August 2014. These and other cash-generating activities should not necessarily be considered an indication of the Company’s ability to raise additional funds in the future.
In February 2015, the Company entered into subscription agreements with certain purchasers pursuant to which it agreed to sell an aggregate of 6,043,955 shares of its common stock and issued warrants to purchase up to an additional 3,021,977 shares of its common stock. Each share of common stock was priced at $1.82 and included one half of a warrant to purchase a share of common stock. The warrants have an exercise price of $1.82 per share, are exercisable beginning six months and one day after the date of issuance and expire on the seventh anniversary of the date of issuance. The total net proceeds from the offering were $10.8 million after deducting expenses of approximately $0.2 million. The subscription agreements require that the Company obtain permission from certain of the purchasers prior to selling shares under its committed equity financing facility.
Based upon its current operating plan, the access to additional capital under its committed equity financing facility, potential to borrow an additional amount of up to $5.0 million under our credit facility, and the $10.8 million received from the Company’s February 2015 financing, the Company believes it has sufficient cash to fund its on-going operations through the first quarter of 2016.
51
Based on its recurring losses, negative cash flows from operations and working capital levels, the Company will need to raise substantial additional funds to finance its operations. If the Company is unable to maintain sufficient financial resources, including by raising additional funds when needed, its business, financial condition and results of operations will be materially and adversely affected. There can be no assurance that the Company will be able to obtain the needed financing on reasonable terms or at all. Additionally, equity or debt financings may have a dilutive effect on the holdings of the Company’s existing stockholders.
Cash and Cash Equivalents
Cash equivalents represent all highly liquid investments with an original maturity date of three months or less and were not significant as of December 31, 2014 and 2013.
Restricted Cash
Short term restricted cash of $0.3 million is primarily restricted cash held in escrow for environmental remediation services to be performed and for taxes in connection with the sale of our New Jersey facility, both of which are the obligation of the Company. The Company has recorded a liability for the environmental remediation as well as tax liabilities, both of which are included in accrued liabilities. These liabilities represent the best estimate of the fair value of the total obligations and are expected to be satisfied within the current year and are therefore classified as current restricted cash and current liabilities, respectively.
Concentration of Credit Risk
From time to time, the Company maintains cash in bank accounts that exceed the FDIC insured limits. The Company has not experienced any losses on its cash accounts. It performs credit evaluations of its customers, but generally does not require collateral to support accounts receivable. Laboratoires Majorelle, Recordati Ireland Ltd., and Hexal AG accounted for approximately 45%, 27%, and 27%, respectively, of total revenues during the year ended December 31, 2014. In addition, one of these companies comprised 75% of the Company’s accounts receivable as of December 31, 2014.
Inventory Valuation
Inventories are stated at the lower of cost or estimated realizable value. The Company capitalizes inventory costs associated with its products after regulatory approval when, based on management's judgment, future commercialization is considered probable and the future economic benefit is expected to be realized. Otherwise, such costs are expensed as research and development. The Company periodically analyzes its inventory levels to identify inventory that may expire prior to expected sale or has a cost basis in excess of its estimated realizable value, and writes-down such inventories as appropriate. In addition, the Company’s products are subject to strict quality control and monitoring which is performed throughout the manufacturing process, which takes place at its contract manufacturer. If certain batches or units of product no longer meet quality specifications or become obsolete due to expiration, the Company records a charge to cost of sales to write down such inventory to its estimated realizable value.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation. Depreciation is provided on a straight-line basis over the estimated useful lives of the assets. The Company estimates useful lives as follows:
• | Machinery and equipment: three to five years |
• | Furniture and fixtures: ten years |
• | Computer software: five years |
Amortization of leasehold improvements and capital lease equipment is provided on a straight-line basis over the shorter of their estimated useful lives or the lease term. The costs of additions and betterments are capitalized, and repairs and maintenance costs are charged to operations in the periods incurred (see note 5 for further details).
Leases
Leases are reviewed and classified as capital or operating at their inception. The Company records rent expense associated with operating leases on a straight-line basis over the term of the lease. The difference between rent payments and straight-line rent expense is recorded as deferred rent in accrued liabilities.
Impairment of Long-Lived Assets
The Company reviews for impairment of long-lived assets whenever events or circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment loss is recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. If such assets are considered impaired, the amount of the impairment loss recognized is measured as the amount by which the carrying value of the asset exceeds the fair value of the asset, fair value being determined based upon future cash flows or appraised values, depending on the nature of the asset.
52
Debt Issuance Costs
Amounts paid related to debt financing activities are capitalized and amortized over the term of the loan.
Revenue Recognition
The Company generates revenues from the licensing of technology rights and the sale of products, and historically, from the performance of pre-clinical testing services and contract sales services. Payments received under commercial arrangements, such as the licensing of technology rights, may include non-refundable fees at the inception of the arrangements, milestone payments for specific achievements designated in the agreements, and royalties on the sale of products.
The Company recognizes revenue when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) the Company’s price to the buyer is fixed or determinable; and (4) collectability is reasonably assured.
License Fee Revenue
Consideration received for the Company’s license arrangements may consist of non-refundable upfront license fees, various performance or sales milestones, royalties upon sales of product, and the delivery of product and/or research services to the licensor. The Company considers a variety of factors in determining the appropriate method of accounting under its license agreements, including whether the various elements can be separated and accounted for individually as separate units of accounting. Deliverables under the arrangement will be separate units of accounting, provided (i) a delivered item has value to the customer on a standalone basis; and (ii) if the arrangement includes a general right of return relative to the delivered item and delivery or performance of the undelivered item is considered probable and substantially in the Company’s control.
The Company accounts for revenue arrangements with multiple elements by separating and allocating consideration in a multiple-element arrangement according to the relative selling price of each deliverable. If an element can be separated, an amount is allocated based upon the relative selling price of each element. The Company determines the relative selling price of a separate deliverable using the price it charges other customers when it sells that product or service separately; however, if the product or service is not sold separately and third party pricing evidence is not available, the Company will use its best estimate of selling price.
The Company defers recognition of non-refundable upfront fees if it has continuing performance obligations without which the technology, right, product or service conveyed in conjunction with the non-refundable fee has no utility to the licensee that is separate and independent of its performance under the other elements of the arrangement. Non-refundable, up-front fees that are not contingent on any future performance by the Company and require no consequential continuing involvement on its part are recognized as revenue when the license term commences and the licensed data, technology and/or compound is delivered. The specific methodology for the recognition of the revenue is determined on a case-by-case basis according to the facts and circumstances of the applicable agreement.
Product Sales Revenue
The Company has supply and manufacturing agreements with certain of its licensee partners for the manufacture and delivery of Vitaros® product. These agreements do not permit the Company’s licensee partners to return product, unless the product sold to the licensee partner is short-dated as defined in each respective license agreement. In those cases, the Company defers revenue recognition on these shipments until the right of return no longer exists, which is the earlier of: (i) evidence that the product has been sold through to the end customer or (ii) the right of return expires. As such, the Company does not have a sales and returns allowance recorded as of December 31, 2014.
In 2014, the Company commenced shipping of its Vitaros® product to its licensee partners and recognized product sales revenue on certain of these shipments since the criteria for revenue recognition was met.
Royalty Revenue
The Company relies on its commercial partners to sell its product, Vitaros®, in approved markets. The Company receives royalties from licensee partners based upon the amount of sales of licensed Vitaros® product consummated by its commercial partners. Royalty revenues are computed based on sales reported to the Company by its licensee partners on a quarterly basis and agreed upon royalty rates for the respective license agreement.
53
Contract Service Revenue
Revenue from contract sales services resulted primarily from the Company’s former subsidiaries, Scomedica SAS, NexMed Europe SAS and NexMed Pharma SAS (the “French Subsidiaries”). The revenue was based on the number of medical visits plus an incentive based on the sales growth of the targeted pharmaceutical products. Revenue associated with medical visits was recognized in the accounting period in which services were rendered. For research services, the Company determined the period in which the performance obligation occurred and recognized revenue using the proportional performance method when the level of effort to complete its performance obligations under an arrangement was able to be reasonably estimated. The Company does not anticipate future revenues from contract services.
Cost of Product Sales
The Company’s cost of product sales includes direct material and manufacturing overhead associated with the production of inventories. Cost of product sales is also affected by manufacturing efficiencies, allowances for scrap or expired material and additional costs related to initial production quantities of new products.
Deferred Cost of Product Sales
Deferred cost of product sales is stated at the lower of cost or net realizable value and includes product sold where title has transferred, but the criteria for revenue recognition have not been met. The Company’s deferred cost of product sales is included in prepaid expenses and other current assets in the consolidated balance sheets.
Research and Development
Research and development costs are expensed as incurred and include the cost of compensation and related expenses, as well as expenses for third parties who conduct research and development on the Company’s behalf, pursuant to development and consulting agreements in place.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred income taxes are recorded for temporary differences between financial statement carrying amounts and the tax basis of assets and liabilities. Deferred tax assets and liabilities reflect the tax rates expected to be in effect for the years in which the differences are expected to reverse. A valuation allowance is provided if it is more likely than not that some or all of the deferred tax assets will not be realized.
The Company also follows the provisions of accounting for uncertainty in income taxes which prescribes a model for the recognition and measurement of a tax position taken or expected to be taken in a tax return, and provides guidance on derecognition, classification, interest and penalties, disclosure and transition.
Loss per Common Share
Basic and diluted net loss per common share is computed by dividing net loss by the weighted-average number of common shares outstanding for the respective period, without consideration of common stock equivalents as they would have an anti-dilutive effect on per share amounts.
The following securities that could potentially decrease net loss per share in the future are not included in the determination of diluted loss per share as they are anti-dilutive and are as follows:
Year Ended December 31, | |||||||||
2014 | 2013 | 2012 | |||||||
Outstanding stock options | 3,955,548 | 2,351,237 | 2,213,916 | ||||||
Outstanding warrants | 6,859,682 | 6,185,492 | 3,205,492 | ||||||
Unvested restricted stock | — | 26,728 | 112,705 | ||||||
Convertible notes payable | — | 1,065,891 | 1,544,402 | ||||||
10,815,230 | 9,629,348 | 7,076,515 | |||||||
Stock-Based Compensation
The estimated grant date fair value of stock options granted to employees and directors is calculated based upon the closing stock price of the Company’s common stock on the date of the grant and recognized as stock-based compensation expense over the expected service period. The Company estimates the fair value of each option award on the date of grant using the Black-Scholes option pricing model.
Segment Information
The Company operates under one segment which develops pharmaceutical products.
54
Geographic Information
Revenues by geographic area for the Company’s continuing operations are as follows (in thousands):
Year Ended December 31, | ||||||||||||
2014 | 2013 | 2012 | ||||||||||
France | $ | 4,150 | $ | 921 | $ | 2,970 | ||||||
Europe- Other | 5,109 | 1,590 | 2,475 | |||||||||
North America | — | — | 2,500 | |||||||||
$ | 9,259 | $ | 2,511 | $ | 7,945 | |||||||
(1) | Amounts included in Europe-other have not been broken out by country as it is impractical to do so given the nature and structure of the license agreements which cover multiple territories. See note 2 for further details related to these agreements. |
All of the Company’s net long-lived assets were located in the United States in 2014 and 2013.
Recent Accounting Pronouncements
In November 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-16, Derivatives and Hedging (Topic 815): Determining Whether the Host Contract in a Hybrid Financial Instrument Issued in the Form of a Share is More Akin to Debt or to Equity. This update clarifies how current guidance should be interpreted in evaluating the economic characteristics and risks of a host contract in a hybrid financial instrument that is issued in the form of a share. In addition, it clarifies that in evaluating the nature of a host contract, an entity should assess the substance of the relevant terms and features (that is, the relative strength of the debt-like or equity-like terms and features given the facts and circumstances) when considering how to weight those terms and features. The effects of initially adopting the new standard should be applied on a modified retrospective basis to existing hybrid financial instruments issued in a form of a share as of the beginning of the fiscal year for which the amendments are effective. Retrospective application is permitted to all relevant prior periods. The new standard is effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2015. Early adoption is permitted. The Company is currently in the process of evaluating whether the adoption of this update will have a material effect on its consolidated financial statements and related disclosures.
In August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements -Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. The amendments in this update will require management to assess, at each annual and interim reporting period, the entity’s ability to continue as a going concern and, if management identifies conditions or events that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued, to disclose in the notes to the entity’s financial statements the principal conditions or events that raised substantial doubt about the entity’s ability to continue as a going concern, management’s evaluation of their significance, and management’s plans that alleviated or are intended to alleviate substantial doubt about the entity’s ability to continue as a going concern. This new standard is effective for annual periods ending after December 15, 2016 and early application is permitted. The Company is currently in the process of evaluating whether the adoption of this update will have a material effect on its consolidated financial statements and related disclosures.
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers, which requires entities to recognize revenue in the way it expects to be entitled for the transfer of promised goods or services to customers. This pronouncement is effective for annual reporting periods beginning after December 15, 2016, including interim periods within that reporting period and is to be applied retrospectively, with early application not permitted. The Company is currently evaluating the effect that this pronouncement will have on its financial statements and related disclosures.
In April 2014, the FASB issued ASU 2014-08, Reporting Discontinued Operations and Disclosures of Disposals of Components of an Entity. This ASU raises the threshold for a disposal to qualify as discontinued operations and requires new disclosures for individually material disposal transactions that do not meet the definition of a discontinued operation. Under the new standard, companies report discontinued operations when they have a disposal that represents a strategic shift that has or will have a major impact on operations or financial results. This update will be applied prospectively and is effective for annual periods, and interim periods within those years, beginning after December 15, 2014. Early adoption is permitted provided the disposal was not previously disclosed. This update will not have a material impact on the Company's reported results of operations and financial position.
55
2. VITAROS® LICENSING AGREEMENTS
Abbott Laboratories Limited
In January 2012, the Company entered into a license agreement with Abbott Laboratories Limited (“Abbott”), granting Abbott the exclusive rights to commercialize Vitaros® for the treatment of ED in Canada. The product was approved by Health Canada in late 2010. To date, the Company has received $2.5 million in upfront payments and is eligible to receive an additional $13.2 million in aggregate regulatory and sales milestone payments, plus tiered royalty payments based on Abbott’s sales of the product.
Bracco SpA
In December 2010, the Company entered into a license agreement with Bracco SpA (“Bracco”), granting Bracco the exclusive rights to commercialize Vitaros® for the treatment of ED in Italy. Vitaros® was granted national phase approval for the treatment of ED in Italy in November 2013. To date, the Company has received $1.3 million in upfront payments and regulatory milestones and is eligible to receive up to an additional €4.5 million ($5.5 million as of December 31, 2014) in regulatory and sales milestone payments, plus tiered double-digit royalties based on Bracco’s sales of the product.
Hexal AG, an affiliate within the Sandoz Division of the Novartis Group of Companies
In February 2012, the Company entered into a license agreement with Hexal AG, an affiliate within the Sandoz Division of the Novartis Group of Companies (“Sandoz”), granting Sandoz the exclusive rights to commercialize Vitaros® for the treatment of ED in Germany. In December 2013, the Company amended and restated its license agreement with Sandoz to include the following countries as part of the exclusive license agreement: Austria, Belgium, Denmark, Finland, Iceland, Luxemburg, the Netherlands, Norway, Sweden and Switzerland (the “Expanded Territory”). In June 2014, the Company entered into a Manufacturing and Supply Agreement with Sandoz whereby the Company’s or its contract manufacturer will manufacture Vitaros® product and supply the product to Sandoz on a cost plus basis. Vitaros® has been granted national phase approval for the treatment of ED in Germany, the Netherlands, Sweden, Belgium, and Luxembourg. The Company filed a marketing application in Switzerland with Swissmedic, the Swiss Agency for Therapeutic Products, for Vitaros® for the treatment of ED and is awaiting regulatory comments from Swissmedic.
In December 2013, upon amendment, the Company recorded $2.0 million of deferred revenue for the upfront payment received since Sandoz was entitled to a $2.0 million refund if certain regulatory and manufacturing conditions were not met. In December 2014, the Company met the manufacturing requirement and recognized $1.0 million of the upfront payment as license fee revenue during the fourth quarter of 2014. The results of the regulatory condition will be determined by December 2016.
In 2014, Sandoz launched Vitaros® in Germany, Sweden and Belgium. The Company recorded $1.0 million in aggregate license fee revenue in 2014 as a result of the launches in Sweden and Belgium in August 2014 and November 2014, respectively.
To date, the Company has received $4.0 million in upfront payments and launch milestone payments and is eligible to receive up to €0.2 million ($0.2 million as of December 31, 2014) in regulatory milestones, up to $1.5 million in marketing launch milestones, and up to €41.75 million ($50.7 million as of December 31, 2014) in sales milestones. Additionally, the Company is entitled to receive tiered double-digit royalties on Sandoz’ sales of the product.
In February 2015, the Company amended its license agreement with Sandoz to grant exclusive rights to commercialize Vitaros® in the following countries: Malaysia, Indonesia, the Philippines, Thailand, Taiwan, Vietnam, Hong Kong and Singapore (the “Expanded APAC Territory”). Under the amended agreement, the Company earned an upfront payment of $0.4 million and is entitled to receive an additional regulatory milestone payment of $0.1 million upon marketing authorization in the Expanded APAC Territory as well as tiered double-digit royalties based on Sandoz’ sales of the product.
Laboratoires Majorelle
In November 2013, the Company entered into a license agreement with Laboratoires Majorelle (“Majorelle”), granting Majorelle the exclusive rights to market Vitaros® for the treatment of ED in France, Monaco and certain countries in Africa. In December 2013, in a related negotiation, Majorelle agreed to make severance payments to certain former employees of the Company’s former French Subsidiaries for an aggregate amount of approximately $2.0 million on behalf of the Company. In September 2014, the Company entered into a Manufacturing and Supply Agreement with Majorelle whereby the Company’s or its contract manufacturer will manufacture Vitaros® product and supply the product to Majorelle on a cost plus basis. In addition, during the first quarter of 2015, Groupe Parima began manufacturing product for Majorelle under its own manufacturing and supply agreement. Vitaros® was granted national phase approval for the treatment of ED in France in December 2013.
The Company concluded that the fair value of the Vitaros® license granted was equal to $4.0 million or the sum of the $1.8 million upfront payment received, the $0.2 million payment received for National Phase approval in France, and the $2.0 million paid by Majorelle on behalf of the Company. During the second quarter of 2014, upon withdrawal of the Works Council Claim in June 2014 (see note 4 for further details regarding the claim), the Company recognized $3.0 million of the $4.0 million Vitaros® license fair value as license fee revenue in its statement of operations. During the third quarter of 2014, the Company met the remaining
56
contractual condition to deliver a specified amount of Vitaros® and therefore, the remaining $1.0 million of deferred revenue that had previously been deferred was recognized as license fee revenue in the Company’s consolidated statement of operations. To date, the Company has received $2.0 million in upfront payments and regulatory milestones, and is eligible to receive up to $2.0 million in additional regulatory milestone payments and €15.5 million ($18.8 million as of December 31, 2014) in sales milestones, plus tiered double-digit royalties based on Majorelle’s sales of the product.
Recordati Ireland Ltd.
In February 2014, the Company entered into a license agreement with Recordati Ireland Ltd. (“Recordati”), granting Recordati the exclusive rights to market Vitaros® for the treatment of ED in Spain, Ireland, Portugal, Greece, Cyprus, the CEE Countries (Central and Eastern Europe), Russia and the other CIS Countries (former Soviet Republics), Ukraine, Georgia, Turkey and certain countries in Africa. In June 2014, the Company entered into a Manufacturing and Supply Agreement with Recordati whereby the Company or its contract manufacturer will manufacture Vitaros® product and supply the product to Recordati on a cost plus basis. Vitaros® has been granted national phase approval for the treatment of ED in Ireland and Spain.
The Company received $2.5 million in upfront payments in February 2014 which were recorded as deferred revenue. Upon execution of the manufacturing and supply agreement in June 2014, the Company recorded the deferred revenue as license fee revenue of $2.5 million. To date, the Company has received $2.5 million in upfront payments and is eligible to receive up to €1.0 million ($1.2 million as of December 31, 2014) in commercial launch payments and €34.5 million ($41.9 million as of December 31, 2014) in sales milestones. Additionally, the Company is entitled to receive tiered double-digit royalties based on Recordati’s sales of the product.
Takeda Pharmaceuticals International GmbH
In September 2012, the Company entered into a license agreement with Takeda Pharmaceuticals International GmbH (“Takeda”), granting Takeda the exclusive rights to market Vitaros® for the treatment of ED in the United Kingdom. In September 2013, the Company entered into a Manufacturing and Supply Agreement with Takeda whereby the Company or its contract manufacturer will manufacture Vitaros® product and supply the product to Takeda on a cost plus basis. Vitaros® was granted national phase approval in August 2013 for the treatment of ED in the UK. To date, the Company has received $1.0 million in upfront payments and is eligible to receive up to €34.65 million ($42.1 million as of December 31, 2014) in up-front license fees and aggregate milestone payments plus tiered double-digit royalty payments. Takeda launched Vitaros® in the United Kingdom in June 2014.
Actavis plc
Warner Chilcott Company, Inc., now a subsidiary of Actavis plc, acquired the Vitaros® United States commercial rights in 2009. To date, the Company has received $2.5 million in upfront payments and is eligible to receive an additional $2.5 million upon receipt of an NDA approval from the FDA.
3. IN-LICENSING AGREEMENT
On October 17, 2014, the Company entered into a license agreement and stock issuance agreement with Forendo Pharma Ltd. (“Forendo”), under which the Company was granted exclusive rights in the United States to develop and commercialize fispemifene, a tissue-specific SERM designed to treat secondary hypogonadism, chronic prostatitis and lower urinary tract symptoms in men.
In exchange for the license, the Company issued to Forendo approximately 3.6 million shares of common stock with a value of $5.9 million based on the Company’s closing stock price on the date of the agreement and made an upfront cash payment of $5.0 million. Additionally, the Company may be obligated to pay Forendo up to $45.0 million based on completion of certain regulatory milestones, up to $260.0 million in sales milestones, plus tiered double-digit royalties based on its sales of the product in the United States.
As part of the agreement, the Company is obligated to pay Forendo $2.5 million upon the earlier of 1) initiation of a Phase 2b clinical trial or 2) April 1, 2015. Since the agreement is not terminable prior to payment, the payment was considered deferred consideration and was recorded as a liability as of December 31, 2014.
The Company recognized research and development expense of $13.6 million upon the completion of the transaction. The $13.6 million is the sum of the following: $5.0 million upfront payment made in October 2014; $5.9 million in common stock issued to Forendo; the additional $2.5 million cash consideration due no later than April 1, 2015, and transaction costs of $0.2 million.
57
4. CEASED AND DISCONTINUED OPERATIONS
In June 2014, consistent with the Global Settlement Agreement (“GSA”) signed in February 2014, the Works Council withdrew its previously submitted €4.1 million claim in the Versailles Civil Court (the “Civil Court”), all parties accepted the withdrawal and the Civil Court judge closed the discussions between all parties. The final procedural step occurred in October 2014, when the Company received a written judgment from the Civil Court acknowledging the dismissal of the claim and the closure of the litigation. Given the existence of the aforementioned ratified GSA, the accepted withdrawal and the closure of the discussions by the Civil Court judge, it was concluded during the second quarter of 2014 that the Company was relieved of all claims previously asserted by the French Works Council.
Pursuant to the aforementioned license and settlement agreements, Majorelle agreed to make certain severance payments of approximately $2.0 million to the former French Subsidiaries’ employees on behalf of the Company, a portion of which were made in May 2014. In addition, the Works Council and the Judicial Liquidator and Trustee of the Company’s former French Subsidiaries as well as each of the former French Subsidiaries’ employees, waived all claims they had asserted or could have asserted against the Company related to the liquidation and reorganization of the French Subsidiaries. As a result, during the second quarter of 2014, the Company released the approximate $2.8 million liability previously recorded in connection with the deconsolidation of the former French Subsidiaries and recognized approximately $0.8 million as a gain on deconsolidation as follows:
Release of deconsolidation liability | $ | 2.8 | ||
Less: Payments made by Majorelle on the Company’s behalf | (2.0 | ) | ||
Gain on deconsolidation of former French Subsidiaries | $ | 0.8 | ||
Sale of Bio-Quant
In June 2014, the Company and BioTox amended its stock purchase agreement and the Company received a one-time cash payment of approximately $0.6 million in exchange for relinquishing its rights to minimum payments in the future. Prior to the amendment of the agreement, the Company also received payments of approximately $0.1 million for a total received from BioTox of $0.7 million in 2014. The Company has rights to certain potential future payments upon a change of control of BioTox within a specified time frame. These potential future payments will be recorded if and when realized.
The Company has recorded the gain of approximately $0.7 million as discontinued operations within its statement of operations in 2014. Historically, the Company reflected the operations and subsequent cash collections associated with the sale of the business as a component of continuing operations, on the line recovery on sale of subsidiary within the consolidated statements of operations. However, the Company has elected not to correct these prior period amounts which were deemed not material to prior period financial statements.
5. OTHER FINANCIAL INFORMATION
Inventory
Inventory is comprised of the following (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Raw materials | $ | 106 | $ | 209 | |||
Work in process | 169 | 127 | |||||
Inventory | $ | 275 | $ | 336 | |||
58
Property and Equipment
Property and equipment are comprised of the following (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Leasehold improvements | $ | 43 | $ | 20 | |||
Machinery and equipment | 1,385 | 847 | |||||
Capital lease equipment | 76 | 76 | |||||
Computer software | 142 | 134 | |||||
Furniture and fixtures | 37 | 34 | |||||
Total property and equipment | 1,683 | 1,111 | |||||
Less: accumulated depreciation and amortization | (325 | ) | (156 | ) | |||
Property and equipment, net | $ | 1,358 | $ | 955 | |||
Depreciation expense totaled $0.2 million, $0.1 million and $0.2 million for the years ended December 31, 2014, 2013, and 2012, respectively.
Accrued Expenses
Accrued expenses are comprised of the following (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Deferred consideration to Forendo (Note 3) | $ | 2,500 | $ | — | |||
Outside research and development services | 838 | 298 | |||||
Professional fees | 625 | 997 | |||||
Deferred compensation | 176 | 184 | |||||
Environmental remediation | 126 | 168 | |||||
Other | 290 | 472 | |||||
$ | 4,555 | $ | 2,119 | ||||
Other Long Term Liabilities
Other long term liabilities are comprised of the following (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Deferred compensation | $ | 312 | $ | 487 | |||
Deferred rent | 41 | 80 | |||||
Capital lease payable | 5 | 11 | |||||
$ | 358 | $ | 578 | ||||
In 2002, the Company entered into an employment agreement with Y. Joseph Mo, Ph.D., pursuant to which Dr. Mo served as the Company’s Chief Executive Officer and President. Under the employment agreement, Dr. Mo was entitled to severance, payable monthly for 180 months, upon termination of his employment. Dr. Mo’s employment was terminated in December 2005. In addition to the long-term portion above, the Company had a balance of $0.2 million classified as short-term deferred severance compensation as of December 31, 2014 and December 31, 2013.
Gain on Contract Settlement
The $0.9 million gain on contract settlement recorded during 2014 represents the fair value of 388,888 escrowed shares of common stock that were returned to the Company in connection with a settlement with former managers of the French Subsidiaries. These shares were restored as authorized, unissued common stock in March 2014. The $0.5 million gain on contract settlement recorded during 2013 represents the difference between the $1.2 million in common shares issued to TopoTarget in exchange for the extinguishment of $1.7 million of contingent consideration.
59
6. DEBT
Credit Facility
On October 17, 2014 (the “closing date”), the Company entered into a loan and security agreement (the “credit facility”) with Oxford Finance LLC (“Oxford”) and Silicon Valley Bank (“SVB”) (the “lenders”), pursuant to which the lenders agreed to make term loans totaling up to $10.0 million available to the Company. The proceeds from these loans are designated to pay off existing indebtedness and for working capital and general business purposes. The first $5.0 million term loan was funded on the closing date of the credit facility. A second term loan of up to a principal amount of $5.0 million may be funded at the Company’s request prior to April 30, 2015, subject to certain conditions. The lenders have first priority over all other potential creditors. Substantially all of the Company’s current and future assets, other than intellectual property, have been pledged as collateral. The lenders have the right to declare the loan immediately due and payable in an event of default under the credit facility, which includes, among other things, a material adverse change in the Company’s business, operations, or financial condition or a material impairment in the prospect of repayment of the loan. As of December 31, 2014, the Company was in compliance with all covenants under the credit facility.
The first term loan bears interest at an annual rate of 7.95%. The repayment schedule provides for interest-only payments in arrears for the first 12 months followed by consecutive equal monthly payments of principal and interest in arrears through the maturity date, or October 1, 2018. The Company has the option to prepay the outstanding balance of the term loan in full prior to the maturity date, subject to a prepayment fee of up to 3%. Upon repayment of each term loan, the Company is also required to make a final payment to the lenders equal to 6.00% of the original principal amount of each term loan funded. This final payment is being accreted over the life of the credit facility using the effective interest method.
On the closing date of the credit facility, the Company issued to Oxford and SVB warrants to purchase up to 193,798 shares of common stock at an exercise price of $1.29 per share. The warrants expire ten years from the date of issuance. The initial fair value of the warrants was recorded as a discount to the principal balance and is being amortized over the life of the credit facility using the effective interest method.
The Company’s notes payable balance as of December 31, 2014 consisted of the following (in thousands):
Notes payable | $ | 5,000 | ||
Add: accretion of final payment fee | 16 | |||
Less: unamortized debt discount | (237 | ) | ||
4,779 | ||||
Less: current portion of notes payable, net | (153 | ) | ||
$ | 4,626 | |||
The debt issuance costs, accretion of the final payment and amortization of the warrants are included in interest expense in the Company’s consolidated financial statements. The Company recognized interest expense related to the credit facility of $0.1 million during the year ended December 31, 2014.
Convertible Notes Payable
On October 17, 2014, the Company amended the terms of its 7% Convertible Notes (“2012 Convertible Notes”) due December 31, 2014 and repaid the remaining aggregate principal balance of $1.225 million with accrued interest. In addition, the Company issued warrants to the former note holders for the right to purchase up to an aggregate of 480,392 shares of common stock, at an exercise price of $2.55 per share. The warrants are exercisable through December 31, 2015. The Company incurred a loss on extinguishment of debt of approximately $0.1 million during the fourth quarter of 2014, which consisted of the fair value of the warrants, an additional payment to the note holders in the amount of the remaining interest payments prior to the amendment, the write-off of the remaining debt discount, and legal fees incurred as part of the amended terms.
The Company’s convertible notes payable balance as of December 31, 2013 consisted of the following (in thousands):
Convertible notes payable | $ | 4,000 | ||
Less: conversions to common stock | (1,250 | ) | ||
2,750 | ||||
Less: unamortized debt discount | (150 | ) | ||
$ | 2,600 | |||
The Company recognized interest expense related to its convertible notes payable of $0.2 million, $0.5 million and $0.3 million during the years ended December 31, 2014, 2013 and 2012, respectively.
60
7. STOCKHOLDERS' EQUITY
Preferred Stock
The Company is authorized to issue 10.0 million shares of preferred stock, par value $0.001, of which 1.0 million shares are designated as Series A Junior Participating Preferred Stock, 800 are designated as Series B 8% Cumulative Convertible Preferred Stock, 600 are designated as Series C 6% Cumulative Convertible Preferred Stock and 50,000 have been designated as Series D Junior Participating Cumulative Preferred Stock. No shares of preferred stock were outstanding as of December 31, 2014 or 2013.
Common Stock Offerings
Ascendiant Offering Agreement
During 2014, 2013 and 2012, the Company sold an aggregate of 954,922, 312,450, and 515,329 shares of common stock, respectively, under the Ascendiant Offering Agreement resulting in offering proceeds of approximately $2.2 million, $0.8 million, and $2.0 million, respectively. The agreement with Ascendiant was terminated by the Company in August 2014.
Aspire Common Stock Purchase Agreement
In August 2014, the Company and Aspire Capital entered into a Common Stock Purchase Agreement (the “Aspire Purchase Agreement”), which provides that Aspire Capital is committed to purchase, if the Company chooses to sell and at the Company’s discretion (pursuant to obtaining the permission mentioned in the paragraph below), an aggregate of up to $22.0 million of shares of the Company’s common stock over the 24-month term of the Aspire Purchase Agreement. The Aspire Purchase Agreement can be terminated at any time by the Company by delivering notice to Aspire Capital. The shares will be sold at a price equal to the lower of the lowest sales price of the Company’ s common stock on the purchase date or the average of the lowest three closing sales prices for the twelve business days prior to the purchase date. Under the Aspire Purchase Agreement, the Company delivered to Aspire Capital a commitment fee in the form of common stock of 255,161 shares at a value of $0.4 million, in consideration for Aspire Capital's obligation to purchase up to $22.0 million of the Company’s common stock. During 2014, the Company sold approximately 2.4 million additional shares of its common stock to Aspire Capital at a weighted average sales price of $1.62 per share, for aggregate net proceeds of $3.7 million. As of December 31, 2014, the Company had $18.2 million remaining under the terms of the Aspire Purchase Agreement.
February 2015 Financing
In February 2015, the Company entered into subscription agreements with certain purchasers pursuant to which it agreed to sell an aggregate of 6,043,955 shares of its common stock and issued warrants to purchase up to an additional 3,021,977 shares of its common stock. Each share of common stock was priced at $1.82 per unit and included one half of a warrant to purchase a share of common stock. The warrants have an exercise price of $1.82 per share, are exercisable beginning six months and one day after the date of issuance and expire on the seventh anniversary of the date of issuance. The total net proceeds from the offering were $10.8 million after deducting expenses of approximately $0.2 million. The subscription agreements also require that the Company obtain permission from certain of the purchasers of the subscription agreements prior to selling shares under its committed equity financing facility.
Warrants
A summary of warrant activity during the year ended December 31, 2014 is as follows:
Common Shares Issuable upon Exercise | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in years) | ||||||
Outstanding at December 31, 2013 | 6,185,492 | $ | 4.01 | 3.6 | ||||
Issued | 674,190 | 2.19 | ||||||
Exercised | — | — | ||||||
Cancelled | — | — | ||||||
Outstanding at December 31, 2014 | 6,859,682 | 3.83 | 2.7 | |||||
Exercisable at December 31, 2014 | 6,859,682 | $ | 3.83 | 2.7 | ||||
During the years ended December 31, 2013, and 2012, the Company received proceeds of $50,000 and $40,000 from the exercise of 20,000, and 17,595 warrants, respectively.
In connection with the credit facility entered into in October 2014, the Company issued warrants to the lenders to purchase up to 193,798 shares of common stock at an exercise price of $1.29 per share. The warrants expire ten years from each date of issuance. The Company
61
also issued warrants to its former convertible note holders to purchase up to an aggregate of 480,392 shares of common stock at an exercise price of $2.55 per share. The warrants are exercisable through December 31, 2015 (see note 6 for further details regarding the warrants issued in connection with the debt arrangements).
The following table shows the number of outstanding warrants by exercise price and date of expiration as of December 31, 2014:
Shares Issuable Upon Exercise | Exercise Price | Expiration Date | |||||
716,356 | $ | 2.27 | October 2015 | ||||
480,392 | $ | 2.55 | December 2015 | ||||
2,469,136 | $ | 5.25 | February 2017 | ||||
3,000,000 | $ | 3.40 | May 2018 | ||||
193,798 | $ | 1.29 | January 2024 | ||||
6,859,682 | |||||||
8. EQUITY COMPENSATION PLANS
As of December 31, 2014, the Company had two share-based compensation plans: the 2012 Stock Long Term Incentive Plan (the “2012 Plan”) and the NexMed, Inc. 2006 Stock Incentive Plan (“the 2006 Plan”). Both plans provide for the issuance of incentive and non-incentive stock options, restricted and unrestricted stock awards, stock unit awards and stock appreciation rights. Options granted generally vest over a period of one to four years and have a maximum term of 10 years from the date of grant. As of December 31, 2014, an aggregate of 6.8 million shares of common stock are authorized under the Company’s equity compensation plans, of which 1.6 million common shares are available for future grants.
Stock Options
A summary of stock option activity during the year ended December 31, 2014 is as follows:
Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in years) | Total Aggregate Intrinsic Value | |||||||||
Outstanding as of December 31, 2013 | 2,351,237 | $ | 3.10 | 8.6 | $ | 444,012 | ||||||
Granted | 2,173,506 | 1.96 | ||||||||||
Exercised | — | — | ||||||||||
Cancelled | (569,195 | ) | 2.37 | |||||||||
Outstanding as of December 31, 2014 | 3,955,548 | $ | 2.58 | 7.5 | $ | — | ||||||
Vested or expected to vest as of December 31, 2014 | 3,837,625 | $ | 2.61 | 7.4 | $ | — | ||||||
Exercisable as of December 31, 2014 | 1,613,222 | $ | 3.42 | 5.1 | $ | — | ||||||
As of December 31, 2014, 2013, and 2012, there were 1,613,222, 752,815 and 856,868 options exercisable, respectively. The aggregate intrinsic value of options exercised during the years ended December 31, 2012 was approximately $16,000.
Stock Awards
A summary of stock award activity during the year ended December 31, 2014 is as follows:
Number of Shares | Weighted Average Grant Date Fair Value Per Share | |||||
Nonvested as of December 31, 2013 | 26,728 | $ | 4.58 | |||
Granted | — | — | ||||
Vested | (26,728 | ) | 4.58 | |||
Forfeited | — | — | ||||
Nonvested as of December 31, 2014 | — | $ | — | |||
Share-Based Compensation
62
The value of stock grants is calculated based upon the closing stock price of the Company’s common stock on the date of the grant. For stock options granted to employees and directors, the Company recognizes compensation expense based on the grant-date fair value over the requisite service period of the awards, which is the vesting period. The Company estimates the fair value of each option award on the date of grant using the Black-Scholes option pricing model.
The following table presents the weighted average assumptions used by the Company to estimate the fair value of stock option grants using the Black-Scholes option-pricing model, as well as the resulting weighted average fair values:
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Risk-free interest rate | 1.58% - 1.96% | 1.08% - 1.85% | 0.6% - 1.1% | ||||||||
Volatility | 80.75 | % | 70.00 | % | 70.00 | % | |||||
Dividend yield | 0.00 | % | 0.00 | % | 0.00 | % | |||||
Expected term | 5.25- 6.08 years | 5.25-6.25 years | 5.25-6.0 years | ||||||||
Forfeiture rate | 3.60 | % | 2.66 | % | 2.66 | % | |||||
Weighted average fair value | $ | 1.25 | $ | 1.52 | $ | 1.95 | |||||
Expected Volatility. The Company uses analysis of historical volatility to compute the expected volatility of its stock options.
Expected Term. The expected life assumptions are based on the simplified method set forth in SEC’s Staff Accounting Bulletin 14.
Risk-Free Interest Rate. The interest rate used in valuing awards is based on the yield at the time of grant of a United States Treasury security with an equivalent remaining term.
Dividend Yield. The Company has never paid cash dividends, and does not currently intend to pay cash dividends, and thus has assumed a 0% dividend yield.
Pre-Vesting Forfeitures. Estimates of pre-vesting option forfeitures are based on the Company’s experience. The Company adjusts its estimate of forfeitures over the requisite service period based on the extent to which actual forfeitures differ, or are expected to differ, from such estimates. Changes in estimated forfeitures are recognized through a cumulative catch-up adjustment in the period of change and also impact the amount of compensation expense to be recognized in future periods. Adjustments have not been significant to date.
As of December 31, 2014, there was $2.7 million in unrecognized compensation cost related to non-vested stock options expected to be recognized over a weighted average period of 2.9 years.
The value of stock awards is calculated based upon the closing stock price of the Company’s common stock on the date of the grant and is expensed over the vesting period of the award. As of December 31, 2014 there are no non-vested stock awards outstanding and therefore no unrecognized compensation cost related to non-vested restricted stock.
The following table summarizes the total stock-based compensation expense resulting from share-based awards recorded in the Company’s consolidated statements of operations (in thousands):
Year Ended December 31, | ||||||||||||
2014 | 2013 | 2012 | ||||||||||
Research and development | $ | 267 | $ | 225 | $ | 299 | ||||||
General and administrative | 1,464 | 1,767 | 2,618 | |||||||||
$ | 1,731 | $ | 1,992 | $ | 2,917 | |||||||
9. INCOME TAXES
The Company has incurred losses since inception, which have generated United States net operating loss carry forwards (“NOLs”) of approximately $174.7 million for federal income tax purposes. These carry forwards are available to offset future taxable income and expire beginning in 2018 through 2034 for federal income tax purposes.
Utilization of the NOLs may be subject to a substantial annual limitation due to ownership change limitations that may have occurred or that could occur in the future, as required under Internal Revenue Code Section 382 (“Section 382”), as well as similar state and foreign provisions. These ownership changes may limit the amount of NOLs that can be utilized annually to offset future taxable income. In general, an “ownership change” as defined by Section 382 results from a transaction or series of transactions
63
over a three-year period resulting in an ownership change of more than 50 percentage points of the outstanding stock of a company by certain stockholders or public groups. Since the Company’s formation, the Company has raised capital through the issuance of capital stock on several occasions which, combined with the purchasing stockholders’ subsequent disposition of those shares, may have resulted in such an ownership change, or could result in an ownership change in the future upon subsequent disposition.
The Company has not completed a study to assess whether an ownership change has occurred or whether there have been multiple ownership changes since the Company’s formation due to the complexity and cost associated with such a study, and the fact that there may be additional such ownership changes in the future. If the Company has experienced an ownership change at any time since its formation, utilization of the NOLs would be subject to an annual limitation under Section 382, which is determined by first multiplying the value of the Company’s stock at the time of the ownership change by the applicable long-term, tax-exempt rate, and then could be subject to additional adjustments, as required. Any limitation may result in expiration of a portion of the NOL before utilization. Further, until a study is completed and any limitation known, no amounts are being considered as an uncertain tax position or disclosed as an unrecognized tax benefit under authoritative accounting guidance. Any NOLs that will expire prior to utilization as a result of such limitations will be removed from deferred tax assets with a corresponding reduction of the valuation allowance with no net effect on income tax expense or the effective tax rate.
Details of income tax expense are as follows (in thousands):
December 31, | ||||||||||||
2014 | 2013 | 2012 | ||||||||||
Current | ||||||||||||
Federal | $ | — | $ | — | $ | — | ||||||
State | — | — | — | |||||||||
Foreign | — | — | — | |||||||||
Total current | — | — | — | |||||||||
Deferred | ||||||||||||
Federal | — | — | — | |||||||||
State | — | — | — | |||||||||
Foreign | — | — | 516 | |||||||||
Total deferred | — | — | 516 | |||||||||
Total income tax expense | $ | — | $ | — | $ | 516 | ||||||
Deferred tax assets consist of the following:
December 31, | ||||||||
2014 | 2013 | |||||||
Net operating tax loss carryforwards | $ | 60,380 | $ | 59,927 | ||||
Research and development tax credits | 534 | 404 | ||||||
Deferred compensation | 168 | 269 | ||||||
Other accruals and reserves | 670 | 504 | ||||||
Basis of intangible assets | 4,610 | (45 | ) | |||||
Total deferred tax asset | 66,362 | 61,059 | ||||||
Less valuation allowance | (66,362 | ) | (61,059 | ) | ||||
Net deferred tax asset | $ | — | $ | — | ||||
The net operating loss carryforwards and tax credit carryforwards resulted in a noncurrent deferred tax asset as of December 31, 2014 and 2013 of approximately $60.9 million and $60.3 million, respectively. In consideration of the Company’s accumulated losses and the uncertainty of its ability to utilize this deferred tax asset in the future, the Company has recorded a full valuation allowance as of such dates.
The Company follows the provisions of income tax guidance which provides recognition criteria and a related measurement model for uncertain tax positions taken or expected to be taken in income tax returns. The guidance requires that a position taken or expected to be taken in a tax return be recognized in the financial statements when it is more likely than not that the position would be sustained upon examination by tax authorities. Tax positions that meet the more likely than not threshold are then measured using a probability weighted approach recognizing the largest amount of tax benefit that is greater than 50% likely of being realized upon ultimate settlement. The Company’s Federal income tax returns for 2011 to 2014 are still open and subject to audit. In addition, net operating losses arising from prior years are also subject to examination at the time they are utilized in future
64
years. Unrecognized tax benefits, if recognized, would have no effect on the Company’s effective tax rate. The Company’s policy is to recognize interest and penalties related to unrecognized tax benefits in income tax expense. As of December 31, 2014, the Company has not recorded any interest and penalties related to uncertain tax positions. The Company does not foresee any material changes to unrecognized tax benefits within the next twelve months.
A reconciliation of the Company’s unrecognized tax benefits from January 1, 2014 through December 31, 2014 is provided in the following table (in thousands):
Year Ended December 31, | ||||||||
2014 | 2013 | |||||||
Beginning balance | $ | 2,795 | $ | 2,879 | ||||
Increase in current period positions | 34 | 41 | ||||||
Decrease in prior period positions | (7 | ) | (125 | ) | ||||
Ending balance | $ | 2,822 | $ | 2,795 | ||||
The reconciliation of income taxes computed using the statutory United States income tax rate and the provision (benefit) for income taxes for continuing operations for the years ended December 31, 2014, 2013, and 2012, are as follows:
Year Ended December 31, | |||||||||
2014 | 2013 | 2012 | |||||||
Federal statutory tax rate | (34 | )% | (34 | )% | (34 | )% | |||
State taxes, net of federal benefit | (1 | )% | (1 | )% | (3 | )% | |||
Valuation allowance | 22 | % | 37 | % | 20 | % | |||
Prior year true-ups | 17 | % | 1 | % | 6 | % | |||
Foreign rate difference | — | % | — | % | 2 | % | |||
Permanent differences | (3 | )% | (1 | )% | 11 | % | |||
Tax credits | (1 | )% | (2 | )% | — | % | |||
Income tax expense | — | % | — | % | 2 | % | |||
For the years ended December 31, 2014, 2013, and 2012, the Company’s effective tax rate differs from the federal statutory rate principally due to net operating losses and other temporary differences for which no benefit was recorded.
10. COMMITMENTS AND CONTINGENCIES
Operating Leases
In December 2011, the Company entered into a five year lease agreement for its headquarters location in San Diego, California expiring December 31, 2016. The Company has an option to extend the lease an additional five years. The lease term contains a base rent of approximately $24,000 per month with 3% annual escalations, plus a supplemental real estate tax and operating expense charge to be determined annually. The Company received a five month base rent abatement with the lease agreement. This abatement is recoverable by the landlord on a straight line amortized basis over 60 months should the Company terminate the lease early for any reason.
In June 2014, the Company entered into a two and one-half year sublease agreement for additional office space within the same building as its headquarters location in San Diego, California expiring December 31, 2016.
For the years ended December 31, 2014, 2013, and 2012, rent expense for continuing operations totaled $0.4 million for each year.
Future minimum rental payments under operating leases as of December 31, 2014 are as follows (in thousands):
2015 | $ | 515 | ||
2016 | 416 | |||
Total | $ | 931 | ||
The Company has significant contractual obligations related to its clinical trial expenditures with clinical research organizations (“CROs”). As of December 31, 2014, net open purchase orders which include obligations to our CROs, less any accruals or invoices charged or amounts paid, totaled approximately $5.3 million. These payments are generally cancellable upon notice and do not have any penalties upon cancellation and therefore these obligations are not included in the table above.
65
Additionally, certain employees have agreements that provide for severance compensation in the event of termination or a change in control. These agreements can provide for a severance payment of up to 12 months of base salary and bonus in effect at the time of termination and continued health benefits at the Company’s cost for up to 12 months.
Legal Matters
The Company is a party to certain litigation that is either judged to be not material or that arises in the ordinary course of business from time to time. The Company intends to vigorously defend its interests in these matters. The Company expects that the resolution of these matters will not have a material adverse effect on its business, financial condition or results of operations. However, due to the uncertainties inherent in litigation, no assurance can be given as to the outcome of these proceedings.
11. SELECTED QUARTERLY FINANCIAL INFORMATION (Unaudited)
The following table presents the Company’s unaudited quarterly results of operations for the years ended December 31, 2014 and 2013 (in thousands, except per share data):
2014 | ||||||||||||||||
1st Quarter | 2nd Quarter | 3rd Quarter | 4th Quarter | |||||||||||||
Total revenue | $ | — | $ | 5,454 | $ | 1,898 | $ | 1,907 | ||||||||
Gross profit (loss) | — | 5,379 | 1,413 | 1,549 | ||||||||||||
Loss (income) from continuing operations | (3,262 | ) | 1,205 | (3,132 | ) | (17,288 | ) | |||||||||
Income (loss) from discontinued operations | — | 672 | 19 | — | ||||||||||||
Net loss (income) | (3,262 | ) | 1,877 | (3,113 | ) | (17,288 | ) | |||||||||
Basic loss (income) per share | ||||||||||||||||
(Loss) income from continuing operations | (0.09 | ) | 0.03 | (0.08 | ) | (0.40 | ) | |||||||||
(Loss) income from discontinued operations | — | 0.02 | — | — | ||||||||||||
Net loss | (0.09 | ) | 0.05 | (0.08 | ) | (0.40 | ) | |||||||||
Diluted loss (income) per share | ||||||||||||||||
Loss from continuing operations | (0.09 | ) | 0.03 | (0.08 | ) | (0.40 | ) | |||||||||
(Loss) income from discontinued operations | — | 0.02 | — | — | ||||||||||||
Net loss | $ | (0.09 | ) | $ | 0.05 | $ | (0.08 | ) | $ | (0.40 | ) | |||||
2013 (1) | ||||||||||||||||
1st Quarter | 2nd Quarter | 3rd Quarter | 4th Quarter | |||||||||||||
Total revenue | $ | 929 | $ | 1,192 | $ | 28 | $ | 362 | ||||||||
Gross profit (loss) | (922 | ) | 490 | 13 | 299 | |||||||||||
Loss from continuing operations | (6,950 | ) | (4,086 | ) | (3,199 | ) | (1,635 | ) | ||||||||
Loss from discontinued operations | (1,723 | ) | 151 | 214 | 290 | |||||||||||
Net loss | (8,673 | ) | (3,935 | ) | (2,985 | ) | (1,345 | ) | ||||||||
Basic and diluted loss per share | ||||||||||||||||
Loss from continuing operations | (0.23 | ) | (0.12 | ) | (0.09 | ) | (0.04 | ) | ||||||||
Loss from discontinued operations | (0.06 | ) | — | 0.01 | — | |||||||||||
Net loss | $ | (0.29 | ) | $ | (0.12 | ) | $ | (0.08 | ) | $ | (0.04 | ) | ||||
(1) | In June 2013, the Company determined that the BQ Kits division would be offered for sale to qualified buyers and in July 2013, it was sold to an unrelated third-party. For all quarters included above, it is presented as discontinued operations. |
66
ITEM 9. CHANGES IN DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our disclosure controls and procedures are designed to ensure that information required to be disclosed by us in reports that we file or submit under the Securities Exchange Act (“SEC”) of 1934, as amended (“the Exchange Act”), is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to our management, including our principal executive officer and principal financial officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
Under the supervision and with the participation of our management, including the Chief Executive Officer (“CEO”) (principal executive officer) and the VP, finance and Chief Accounting Officer (“CAO”) (principal financial officer), we conducted an evaluation of the effectiveness of our disclosure controls and procedures, as such term is defined under Rules 13a-15(e) and 15d-15(e) under the Exchange Act, as of December 31, 2014. Based on this evaluation, our CEO and our CAO concluded that our disclosure controls and procedures were not effective as of December 31, 2014 because of the material weaknesses in internal control over financial reporting described below.
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control over financial reporting is a process designed, under the supervision and with the participation of our principal executive officer and principal financial officer, overseen by our Board of Directors and implemented by our management and other personnel, to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of our financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes policies and procedures that:
• | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
• | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of our management and directors; and |
• | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements. |
Because of its inherent limitations, our internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management performed an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2014 using criteria established in the Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this assessment, management determined that, as of December 31, 2014, material weaknesses exist in our internal control over financial reporting over the accounting for and disclosures of technical accounting matters in the consolidated financial statements and effective monitoring and oversight over the controls in the financial reporting process. Because of these material weaknesses, management concluded that the Company did not maintain effective internal control over financial reporting as of December 31, 2014, based on the COSO framework. For information on the progress of the remediation of the material weaknesses, see Material Weaknesses and Remediation Efforts below.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
The deficiencies resulted in an audit adjustment with respect to the consolidated financial statements for the interim period ended March 31, 2014 related to the cash flows presentation of certain noncash disclosures. Additionally, the material weaknesses could result in a misstatement of account balances or disclosures that would result in a material misstatement to the annual or interim consolidated financial statements that would not be prevented or detected.
67
The effectiveness of the Company’s internal control over financial reporting as of December 31, 2014 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which appears under Item 8.
Material Weaknesses and Remediation Efforts
The material weaknesses in our internal control over financial reporting described above were disclosed in Item 9A, Controls and Procedures of our annual report on Form 10-K, for the prior year ended December 31, 2013.
Management is responsible for implementing changes and improvements in the internal control over financial reporting and for remediating the control deficiencies that gave rise to these material weaknesses. We are committed to remediating the control deficiencies that constitute these material weaknesses and have implemented changes to our internal control over financial reporting. Although these material weaknesses continue to exist as of December 31, 2014, management believes that progress has been made in that regard during the year ended December 31, 2014. Specifically, the following specific actions were taken:
• | we added more experienced accounting staff with the requisite skills and experience to support our structure and financial reporting requirements during 2014; |
• | during 2014 we utilized qualified outside consulting personnel, where necessary, in support of our complex technical accounting matters; |
• | during 2014 we retained an outside consulting firm to review the design of our internal control over financial reporting to ensure that the processes and intended changes to the processes are addressing the relevant financial statement assertions and presentation and disclosure matters, including the monitoring and oversight of controls in the financial reporting process; and |
• | during the fourth quarter of 2014 we completed our review of the design of our internal controls over financial reporting, including those associated with the analysis of technical matters and the monitoring and oversight controls in the financial reporting process. We have also completed our implementation of changes to the underlying processes and controls to address the relevant financial statement assertions and presentation and disclosure. |
Changes in Internal Control Over Financial Reporting
There were changes in internal control over financial reporting, as discussed above under Material Weaknesses and Remediation Efforts, during the quarter ended December 31, 2014 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. | OTHER INFORMATION |
None.
68
PART III.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Code of Ethics
We have adopted a Code of Ethics, as amended, that applies to our Chief Executive Officer and our Chief Accounting Officer and to all of our other officers, directors and employees. The Code of Ethics is available in the Corporate Governance section of the Investors page on our website at www.apricusbio.com. We will disclose future amendments to, or waivers from, certain provisions of our code of ethics, if any, on the above website within four business days following the date of such amendment or waiver.
The other information required by this item is incorporated herein by reference to the Proxy Statement under the sections “Executive Compensation,” “Directors Compensation,” and “Board of Directors and Committees; Corporate Governance” to be filed with the Securities and Exchange Commission in connection with our 2014 Annual Meeting of Stockholders.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated herein by reference to the Proxy Statement under the sections “Executive Compensation,” “Directors Compensation,” and “Board of Directors and Committees; Corporate Governance.”
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated herein by reference to the Proxy Statement under the section “Security Ownership of Certain Beneficial Owners and Management.”
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is incorporated herein by reference to the Proxy Statement under the sections “Certain Relationships and Related Party Transactions” and “Board of Directors and Committees; Corporate Governance.”
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item is incorporated herein by reference to the Proxy Statement under the section “Fees for Independent Registered Public Accounting Firm.”
PART IV.
ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(a) 1. Financial Statements:
The information required by this item is included in Item 8 of Part II of this Form 10-K.
2. Financial Statement Schedules
The information required by this item is included in Item 8 of Part II of this Form 10-K.
3. Exhibits
The following exhibits are incorporated by reference or filed as part of this report.
69
EXHIBITS NO. | DESCRIPTION | |
2.1† | Stock Purchase Agreement, dated December 15, 2011, by and among Apricus Biosciences Inc., TopoTarget A/S, and TopoTarget USA, Inc. (incorporated herein by reference to Exhibit 2.1 to the Company’s Form 10-K filed with the Securities and Exchange Commission on March 13, 2012). | |
2.2 | Stock Contribution Agreement, dated June 19, 2012, by and among Apricus Biosciences, Inc., Finesco SAS, Scomedica SA and the shareholders of Finesco named therein (incorporated herein by reference to Exhibit 2.1 to the Company’s Current Report form 8-K, filed with the Securities and Exchange Commission on July 13, 2012). | |
2.3† | Asset Purchase Agreement by and between Apricus Pharmaceuticals USA, Inc. and Biocodex, Inc., dated March 26, 2013 (incorporated herein by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on April 1, 2013). | |
2.4 | Amendment to Stock Purchase Agreement, dated June 13, 2014, by and between Apricus Biosciences, Inc. and Samm Solutions, Inc. (doing business as BTS Research and formerly doing business as BioTox Sciences) (incorporated herein by reference to Exhibit 2.1 to the Company’s Form 10-Q filed with Securities and Exchange Commission on August 11, 2014). | |
3.1 | Amended and Restated Articles of Incorporation of Apricus Biosciences, Inc. (incorporated herein by reference to Exhibit 2.1 to the Company’s Registration Statement on Form 10-SB filed with the Securities and Exchange Commission on March 14, 1997). | |
3.2 | Certificate of Amendment to Articles of Incorporation of Apricus Biosciences, Inc., dated June 22, 2000 (incorporated herein by reference to Exhibit 3.2 to the Company’s Form 10-K filed with the Securities and Exchange Commission on March 31, 2003). | |
3.3 | Certificate of Amendment to Articles of Incorporation of Apricus Biosciences, Inc., dated June 14, 2005 (incorporated herein by reference to Exhibit 3.4 to the Company’s Form 10-K filed with the Securities and Exchange Commission on March 16, 2006). | |
3.4 | Certificate of Amendment to Amended and Restated Articles of Incorporation of Apricus Biosciences, Inc., dated March 3, 2010 (incorporated herein by reference to Exhibit 3.6 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2010). | |
3.5 | Certificate of Correction to Certificate of Amendment to Amended and Restated Articles of Incorporation of Apricus Biosciences, Inc., dated March 3, 2010 (incorporated herein by reference to Exhibit 3.7 to the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2010). | |
3.6 | Certificate of Designation for Series D Junior-Participating Cumulative Preferred Stock (incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-A12GK filed with the Securities and Exchange Commission on March 24, 2011). | |
3.7 | Certificate of Change filed with the Nevada Secretary of State (incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the Securities Exchange Commission on June 17, 2010). | |
3.8 | Certificate of Amendment to Amended and Restated Articles of Incorporation of Apricus Biosciences, Inc., dated September 10, 2010 (incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on September 10, 2010). | |
70
3.9 | Fourth Amended and Restated Bylaws, dated December 18, 2012 (incorporated herein by reference to Exhibit 3.9 to the Company’s Form 10-K filed with the Securities and Exchange Commission on March 18, 2013). | |
3.10 | Certificate of Withdrawal of Series D Junior Participating Cumulative Preferred Stock, dated May 15, 2013 (incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on May 16, 2013). | |
4.1 | Form of Warrant, dated September 17, 2010 (incorporated herein by reference to Exhibit 4.6 of Amendment No. 2 to the Company’s Registration Statement on Form S-1 (File No. 333-169132) filed with the Securities and Exchange Commission on September 28, 2010). | |
4.2 | Form of Warrant Certificate (incorporated herein by reference to Exhibit 4.7 of Amendment No. 2 to the Company’s Registration Statement on Form S-1 (File No. 333-169132) filed with the Securities and Exchange Commission on September 28, 2010). | |
4.3 | Form of Common Stock Certificate (incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on March 24, 2011). | |
4.4 | Form of Warrant (incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on February 13, 2012). | |
4.5 | Form of Warrant (incorporated herein by reference to Exhibit 1.1 to the Company’s Current Report on From 8-K filed with the Securities and Exchange Commission on May 24, 2013). | |
4.6 | Form of Warrant issued to the Holders under the Amendment Agreement, dated as of October 17, 2014, by and among Apricus Biosciences, Inc., The Tail Wind Fund Ltd., Solomon Strategic Holdings, Inc., and Tail Wind Advisory & Management Ltd. (incorporated herein by reference to Exhibit 4.1 to the Company’s Form 8-K filed with the Securities and Exchange Commission on October 20, 2014). | |
4.7 | Form of Warrant issued to the lenders under the Loan and Security Agreement, dated as of October 17, 2014, by and among Apricus Biosciences, Inc., NexMed (U.S.A.), Inc., NexMed Holdings, Inc. and Apricus Pharmaceuticals USA, Inc., as borrowers, Oxford Finance LLC, as collateral agent, and the lenders party thereto from time to time including Oxford Finance LLC and Silicon Valley Bank. (incorporated herein by reference to Exhibit 4.2 to the Company’s Form 8-K filed with the Securities and Exchange Commission on October 20, 2014). | |
10.1* | Employment Agreement, dated February 26, 2002, by and between NexMed, Inc. and Dr. Y. Joseph Mo (incorporated herein by reference to Exhibit 10.7 to the Company’s Form 10-K filed with the Securities and Exchange Commission on March 29, 2002). | |
10.2* | Amendment, dated September 26, 2003, to Employment Agreement by and between NexMed, Inc. and Dr. Y. Joseph Mo dated February 26, 2002 (incorporated herein by reference to Exhibit 10.4 to the Company’s Form 10-Q filed with the Securities and Exchange Commission on November 12, 2003). | |
10.3* | NexMed, Inc. 2006 Stock Incentive Plan (incorporated herein by reference to Annex A of the Company’s Definitive Proxy Statement filed with the Securities and Exchange Commission on April 6, 2006). | |
71
10.4* | NexMed, Inc. Amendment to 2006 Stock Incentive Plan (incorporated herein by reference to Appendix A of the Company’s Definitive Proxy Statement filed with the Securities and Exchange Commission on April 18, 2008). | |
10.5 | Asset Purchase Agreement, dated February 3, 2009, by and between Warner Chilcott Company, Inc. and NexMed, Inc. (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on February 5, 2009). | |
10.6 | License Agreement, dated February 3, 2009, by and between NexMed, Inc. and Warner Chilcott Company, Inc. (incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on February 5, 2009). | |
10.7* | Apricus Biosciences, Inc. 2012 Stock Long Term Incentive Plan (incorporated by reference to Exhibit A of the Registrant’s Definitive Proxy Statement filed on April 6, 2012). | |
10.8 | Warrant Agent Agreement, dated September 17, 2010, by and between Apricus Biosciences, Inc. and Wells Fargo Bank, N.A. (incorporated by reference to Exhibit 10.30 of Amendment No. 2 to the Company’s Registration Statement on Form S-1 (File No. 333-169132) filed with the Securities and Exchange Commission on September 28, 2010). | |
10.9 | Employment Agreement by and between Apricus Biosciences, Inc. and Richard W. Pascoe, dated March 18, 2013 (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on March 20, 2013). | |
10.10 | Settlement Agreement and Release, dated as of September 23, 2013, by and between Apricus Biosciences, Inc. and Topotarget A/S (incorporated by reference to Exhibit 10.1 of Amendment No. 1 to the Company's Registration Statement on Form S-3 (File No. 333-191679) filed with the Securities and Exchange Commission on October 31, 2013). | |
10.11 | Form of Stock Option Grant Notice and Stock Option Agreement under the Apricus Biosciences, Inc. 2012 Stock Long Term Incentive Plan (incorporated herein by reference to Exhibit 10.1 to the Company’s Form 10-Q filed with the Securities and Exchange Commission on August 11, 2014). | |
10.12 | Non-Employee Director Compensation Policy (incorporated herein by reference to Exhibit 10.2 to the Company’s Form 10-Q filed with the Securities and Exchange Commission on August 11, 2014). | |
10.13 | Common Stock Purchase Agreement, dated August 12, 2014, by and between the Company and Aspire Capital Fund, LLC (incorporated herein by reference to Exhibit 10.1 to the Company’s Form 8-K filed with the Securities and Exchange Commission on August 12, 2014). | |
10.14 | Registration Rights Agreement, dated August 12, 2014, by and between the Company and Aspire Capital Fund, LLC (incorporated herein by reference to Exhibit 10.2 to the Company’s Form 8-K filed with the Securities and Exchange Commission on August 12, 2014). | |
10.15† | License Agreement by and between NexMed (U.S.A.), Inc. and Forendo Pharma Ltd., dated as of October 17, 2014 (incorporated herein by reference to Exhibit 10.1 to the Company’s Form 8-K filed with the Securities and Exchange Commission on October 20, 2014). | |
10.16 | Stock Issuance Agreement, by and among Apricus Biosciences, Inc., Forendo Pharma Ltd. and Birch & Lake Partners, LLC, dated as of October 17, 2014 (incorporated herein by reference to Exhibit 10.2 to the Company’s Form 8-K filed with the Securities and Exchange Commission on October 20, 2014). | |
72
10.17 | Loan and Security Agreement by and among Apricus Biosciences, Inc., NexMed (U.S.A.), Inc., NexMed Holdings, Inc. and Apricus Pharmaceuticals USA, Inc., as borrowers, Oxford Finance LLC, as collateral agent, and the lenders party thereto from time to time, including Oxford Finance LLC and Silicon Valley Bank, dated as of October 17, 2014 (incorporated herein by reference to Exhibit 10.3 to the Company’s Form 8-K filed with the Securities and Exchange Commission on October 20, 2014). | |
10.18 | Employment Agreement by and between Apricus Biosciences, Inc. and Brian Dorsey, dated December 1, 2014. | |
10.19 | Employment Agreement by and between Apricus Biosciences, Inc. and Dr. Barbara Troupin, dated December 12, 2014. | |
10.2 | Employment Agreement by and between Apricus Biosciences, Inc. and Catherine Bovenizer, dated January 12, 2015. | |
21 | Subsidiaries. | |
23.1 | Consent of PricewaterhouseCoopers LLP, independent registered public accounting firm. | |
31.1 | Chief Executive Officer’s Certificate, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
31.2 | Chief Accounting Officer’s Certificate, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
32.1 | Chief Executive Officer’s Certificate, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. (1) | |
32.2 | Chief Accounting Officer’s Certificate, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. (1) | |
101.INS | XBRL Instance Document. (1) | |
101.SCH | XBRL Taxonomy Extension Schema. (1) | |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase. (1) | |
101.DEF | XBRL Taxonomy Extension Definition Linkbase. (1) | |
101.LAB | XBRL Taxonomy Extension Label Linkbase. (1) | |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase. (1) | |
(1) | Furnished, not filed. |
* | Management compensatory plan or arrangement. |
† | Confidential treatment has been requested for portions of this exhibit. These portions have been omitted and filed separately with the Securities and Exchange Commission. |
73
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
Apricus Biosciences, Inc. | |
Date: March 16, 2015 | /s/ CATHERINE BOVENIZER |
Catherine Bovenizer | |
Vice President, Chief Accounting Officer | |
Signature | Title | Date | ||
/s/ RICHARD W. PASCOE | Chief Executive Officer and Director | March 16, 2015 | ||
Richard W. Pascoe | ||||
/s/ CATHERINE BOVENIZER | Vice President, Chief Accounting Officer | March 16, 2015 | ||
Catherine Bovenizer | ||||
/s/ KLEANTHIS G. XANTHOPOULOS, PH.D. | Chairman of the Board of Directors | March 16, 2015 | ||
Kleanthis G. Xanthopoulos, Ph.D. | ||||
/s/ RUSTY RAY | Director | March 16, 2015 | ||
Rusty Ray | ||||
/s/ DEIRDRE Y. GILLESPIE, M.D. | Director | March 16, 2015 | ||
Deirdre Y. Gillespie, M.D. | ||||
/s/ PAUL V. MAIER | Director | March 16, 2015 | ||
Paul V. Maier | ||||
/s/ WENDELL WIERENGA | Director | March 16, 2015 | ||
Wendell Wierenga, Ph.D. | ||||
/s/ SANDFORD D. SMITH | Director | March 16, 2015 | ||
Sandford D. Smith | ||||
74
EXHIBIT INDEX
10.26 | Employment Agreement by and between Apricus Biosciences, Inc. and Brian Dorsey, dated December 1, 2014. | |
10.27 | Employment Agreement by and between Apricus Biosciences, Inc. and Dr. Barbara Troupin, dated December 12, 2014. | |
10.28 | Employment Agreement by and between Apricus Biosciences, Inc. and Catherine Bovenizer, dated January 12, 2015. | |
21 | Subsidiaries. | |
23.1 | Consent of PricewaterhouseCoopers LLP, independent registered public accounting firm. | |
31.1 | Chief Executive Officer’s Certificate, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
31.2 | Chief Accounting Officer’s Certificate, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
32.1 | Chief Executive Officer’s Certificate, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. (1) | |
32.2 | Chief Accounting Officer’s Certificate, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. (1) | |
101.INS | XBRL Instance Document. (1) | |
101.SCH | XBRL Taxonomy Extension Schema. (1) | |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase. (1) | |
101.LAB | XBRL Taxonomy Extension Labels Linkbase. (1) | |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase. (1) | |
101.DEF | XBRL Taxonomy Extension Definition Linkbase. (1) | |
(1) | Furnished, not filed. |
75