Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - PROVECTUS BIOPHARMACEUTICALS, INC. | Financial_Report.xls |
| EX-23 - EX-23 - PROVECTUS BIOPHARMACEUTICALS, INC. | d862762dex23.htm |
| EX-3.2 - EX-3.2 - PROVECTUS BIOPHARMACEUTICALS, INC. | d862762dex32.htm |
| EX-31.2 - EX-31.2 - PROVECTUS BIOPHARMACEUTICALS, INC. | d862762dex312.htm |
| EX-31.1 - EX-31.1 - PROVECTUS BIOPHARMACEUTICALS, INC. | d862762dex311.htm |
| EX-32 - EX-32 - PROVECTUS BIOPHARMACEUTICALS, INC. | d862762dex321.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2014
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 001-36457
PROVECTUS BIOPHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 90-0031917 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
7327 Oak Ridge Highway, Suite A, Knoxville, Tennessee 37931
(Address of principal executive offices) (Zip Code)
866-594-5999
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Common Stock, par value $.001 per share
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, par value $.001 per share
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ¨ Yes x No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ¨ Yes x No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. x Yes ¨ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). x Yes ¨ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | x | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). ¨ Yes x No
The aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold as of June 30, 2014 was $142,203,263 (computed on the basis of $0.86 per share).
The number of shares outstanding of the registrant’s common stock, par value $.001 per share, as of March 2, 2015 was 185,171,159.
DOCUMENTS INCORPORATED BY REFERENCE
The information required by Part III is incorporated by reference to portions of the definitive proxy statement to be filed within 120 days after December 31, 2014, pursuant to Regulation 14A under the Securities Exchange Act of 1934 in connection with the annual meeting of stockholders to be held on June 19, 2015.
Table of Contents
Table of Contents
CAUTIONARY NOTE REGARDING FORWARD LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements regarding, among other things, our anticipated financial and operating results. Forward-looking statements reflect our management’s current assumptions, beliefs, and expectations. Words such as “anticipate,” “believe,” “estimate,” “seek,” “expect,” “intend,” “plan,” and similar expressions are intended to identify forward-looking statements. While we believe that the expectations reflected in our forward-looking statements are reasonable, we can give no assurance that such expectations will prove correct. Forward-looking statements are subject to risks and uncertainties that could cause our actual results to differ materially from the future results, performance, or achievements expressed in or implied by any forward-looking statement we make. Some of the relevant risks and uncertainties that could cause our actual performance to differ materially from the forward-looking statements contained in this report are discussed below under the heading “Risk Factors” and elsewhere in this Annual Report on Form 10-K. We caution investors that these discussions of important risks and uncertainties are not exclusive, and our business may be subject to other risks and uncertainties which are not detailed there. Investors are cautioned not to place undue reliance on our forward-looking statements. We make forward-looking statements as of the date on which this Annual Report on Form 10-K is filed with the Securities and Exchange Commission (“SEC”), and we assume no obligation to update the forward-looking statements after the date hereof whether as a result of new information or events, changed circumstances, or otherwise, except as required by law.
1
Table of Contents
| ITEM 1. | BUSINESS. |
General
Provectus Biopharmaceuticals, Inc., a Delaware corporation formed in 2002, together with its six wholly owned subsidiaries and one majority owned subsidiary managed on a consolidated basis, referred to herein as “we,” “us,” and “our,” is a development-stage biopharmaceutical company that is primarily engaged in developing ethical pharmaceuticals for oncology and dermatology indications. Our goal is to develop alternative treatments that are safer, more effective, less invasive and more economical than conventional therapies. We develop and intend to license or market and sell our two prescription drug candidates, PV-10 and PH-10. We also hold patents and other intellectual property which we believe may be used in over-the-counter products, which we refer to as OTC products, and various other non-core technologies. We have transferred all our intellectual property related to OTC products and non-core technologies to our subsidiaries and have designated such subsidiaries as non-core to our primary business of developing our oncology and dermatology prescription drug candidates.
Prescription Drugs
We focus on developing our prescription drug candidates PV-10 and PH-10. We are developing PV-10 for treatment of several life threatening cancers including metastatic melanoma, liver cancer, and breast cancer. We are developing PH-10 to provide minimally invasive treatment of chronic severe skin afflictions such as psoriasis and atopic dermatitis, a type of eczema. We believe that our prescription drug candidates will be safer and more specific than currently existing products. All of our prescription drug candidates are in either the pre-clinical or clinical trial stage.
The table below sets forth our two prescription drug candidates and our progress in developing those candidates for the indications shown:
| PV-10 Melanoma |
• | Protocol for Phase 3 study for treatment of locally advanced cutaneous melanoma submitted to FDA in 2014 for study in 2015 | ||
| • |
Phase 1b/2 combination study of PV-10 + immune checkpoint blockade is being designed 2014 into 2015 | |||
| • | Type C FDA meeting December 2013, and January 2015 | |||
| • | Prepare for Breakthrough Therapy Designation request 2013 into 2014 | |||
| • | Finalized Phase 2 data October 2012 and September 2013 | |||
| • | End-of-Phase 2 FDA meeting April 2010, March 2011, and October 2011 | |||
| • | Phase 2 study completed May 2010 | |||
| • | Phase 2 treatments completed September 2009 | |||
| • | Phase 2 recruitment completed May 2009 | |||
| • | Phase 2 study initiated September 2007 | |||
| • | Orphan drug status January 2007 | |||
| PH-10 Psoriasis |
• | Full Phase 2c study report submitted to FDA February 2014 | ||
| • | Toxicity study research and development for advanced studies 2012, 2013, 2014 and into 2015 | |||
| • | Phase 2c randomized study final data collection February 2012 | |||
| • | Phase 2c randomized study initiated December 2010 and completed August 2011 | |||
| • | Phase 2 study completed April 2010 | |||
| • | Phase 2 recruitment completed October 2009 | |||
| • | Replacement Phase 2 initiated July 2009 due to dose regimen change | |||
| • | Phase 2 study initiated November 2007 | |||
| PH-10 Atopic Dermatitis |
• | Toxicity study research and development for advanced studies 2012, 2013, 2014 and into 2015 | ||
| • | Phase 2 study completed September 2009 | |||
| • | Phase 2 recruitment completed June 2009 | |||
| • | Phase 2 study initiated June 2008 | |||
| PV-10 Breast Cancer |
• |
Assessing further development 2013, 2014 and 2015 in conjunction with Moffitt Cancer Center research | ||
2
Table of Contents
| • | Phase 1 study completed July 2008 | |||
| • | Phase 1 initial cohort treatment completed April 2006 | |||
| • | Phase 1 study initiated October 2005 | |||
| PV-10 Liver Metastasis |
• | Phase 1b/2 study being planned 2014 into 2015 | ||
| • | Phase 1 protocol expansion September 2012 through 2014 into 2015 | |||
| • | Orphan drug status April 2011 | |||
| • | Phase 1 patient accrual and treatment completed January 2011 | |||
| • | Phase 1 study initiated October 2009 | |||
| PV-10 Mechanism of Action |
• |
Moffitt Cancer Center initiates Phase 1 feasibility study to detect immune cell infiltration into melanomas treated by PV-10 in January 2013 into 2014 and 2015 | ||
| PH-10 Mechanism of Action |
• |
Phase 2 study initiated January 2015 | ||
In addition to clinical trials, patients enrolled in the expanded access or compassionate use program for PV-10 are also receiving PV-10 treatments for cutaneous and subcutaneous cancer indications.
Oncology (PV-10)
We believe our prescription drug candidate PV-10, a novel investigational drug, may afford competitive advantage compared to currently available options for the treatment of certain types of cancer; particularly solid tumors. We are developing PV-10, a sterile injectable form of rose bengal disodium (Rose Bengal), for direct injection into tumors. It is an ablative immunotherapy or immuno-chemoablative agent that when injected intralesionally is tantamount to an “in situ” vaccination following acute and durable necrosis of diseased tissue. Because PV-10 is retained in diseased or damaged tissue but quickly dissipates from healthy tissue, we believe we can develop therapies that confine treatment to cancerous tissue and reduce collateral impact on healthy tissue. We have conducted phase 1 and phase 2 studies of PV-10 for the treatment of recurrent and metastatic melanoma, and phase 1 studies of PV-10 for the treatment of liver and breast cancers, each of which are described in more detail below. Furthermore, we expect to commence a phase 3 study of PV-10 to treat locally advanced cutaneous melanoma in the first quarter of 2015, which is described in more detail below.
Recurrent or Locally Advanced Cutaneous Melanoma and Widely Metastatic [Melanoma] Disease
A Type C meeting was held with the U.S. Food and Drug Administration’s (the “FDA” or the “Agency”) Division of Oncology Products 2 on December 16, 2013. The purpose of the meeting was to determine which of the available paths that our novel investigational oncology drug PV-10 will take in pursuit of initial FDA approval and commercialization. As a result of this meeting, we submitted data from our phase 2 study in a formal breakthrough therapy designation (BTD) request.
We believe that this meeting with the FDA is another significant step forward in streamlining the pathway to initial U.S. approval of PV-10 as the first local agent for recurrent locoregionally advanced melanoma. These patients suffer with troublesome, disfiguring disease that can persist for many years before presenting at distant sites. Our meeting with the FDA established the parameters for submission of a BTD request tailored to addressing the pressing needs of these patients.
The meeting and official meeting minutes provided valuable guidance on a number of issues surrounding the approval path of PV-10:
| • | The FDA agreed with us that treatment of cutaneous and subcutaneous tumors in patients with locally advanced cutaneous melanoma (i.e., recurrent, in-transit or satellite melanoma that has not yet spread from the skin to distant sites) could provide clinical benefit to such patients, particularly if the measured objective responses in patients’ disease correlated to a demonstrated treatment effect on one or more symptoms of their disease (e.g., pain, infection or significant bleeding). |
| • | The FDA agreed to work with us to quantify symptom control in this patient population. |
| • | In reference to discussions on the potential for breakthrough therapy designation, “FDA advised Provectus to provide objective response rates with adequate information to evaluate the symptomatic treatment effects (e.g. pain, infection, bleeding) in patients presenting with locally advanced cutaneous melanoma who received PV-10 to all lesions.” |
3
Table of Contents
The phase 2 study of PV-10 showed:
| • | Among all 80 intent-to-treat melanoma patients, 26% achieved a complete response and another 25% achieved a partial response (shrinkage by at least 30%) of their injected study tumors (51% objective response rate, confidence interval 40-63%). |
| • | In the subgroup of melanoma patients that received PV-10 injection into all known disease (28 of the 80 ITT patients), 50% achieved a complete response (71% ORR, CI 51-87%). |
| • | In the subgroup of melanoma patients with locally advanced cutaneous melanoma that received PV-10 injection into all known disease or only had 1 or 2 designated bystander tumors untreated (54 of the 80 ITT patients), a complete response was achieved in 232 of 363 injected tumors (64% of lesions) with the vast majority of these tumors requiring only 1 or 2 injections. |
We believe these data show that if a tumor is accessible to PV-10 injection, the drug is likely to destroy that tumor. If approved, PV-10 would be the first tissue-sparing local therapy for recurrent or locally advanced cutaneous melanoma.
Non-specific local treatments that temporarily reduce tumor burden, such as surgery and radiation, are the most commonly used cancer therapies today. Furthermore, we believe our clinical and immunologic mechanism data show that it may be possible to delay or prevent melanoma metastasis to distant sites. Measurement of tumor shrinkage via objective response criteria has been considered direct clinical benefit in drug approvals for other skin cancers, and we believe a similar case can be made for PV-10 in locally advanced cutaneous melanoma. As advised by the FDA, we submitted data from the 28 patients in our phase 2 study who had all existing disease treated in a formal BTD request.
In the BTD request, Provectus presented an analysis of the sub-group of 54 patients in phase 2 study PV-10-MM-02 having melanoma confined to cutaneous and subcutaneous sites and where all disease was followed; any new clinically relevant disease constituted progression with no further assessment for response. Complete response of all injected lesions was observed in 37% of these patients. In 28 of these patients having all of their baseline disease injected, complete response was observed in 50% of patients (Confidence Interval: 31-69%). These 28 patients had as many as 20 lesions confined to the skin, and experienced a mean PFS of 9.8 months.
On May 23, 2014, we announced that we received notification from the FDA that the data upon which the Company based its request for designation of PV-10 as a Breakthrough Therapy for the treatment of patients with locally advanced cutaneous melanoma was insufficient to demonstrate substantial improvement over existing therapies. As a result, the FDA declined to designate PV-10 as a Breakthrough Therapy at that time. Further data may cause the Agency to revisit this decision at a later date.
In the notification letter the FDA stated, “We have reviewed your request and while we have determined that treatment of ‘locally advanced cutaneous melanoma’ meets the criteria for a serious or life-threatening disease or condition, the preliminary clinical evidence you submitted does not indicate that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. Therefore, designation as a Breakthrough Therapy cannot be granted at this time.”
On June 2, 2014, there were two presentations in the Poster Highlights Session, Melanoma/Skin Cancers, during the American Society of Clinical Oncology (ASCO) annual meeting in Chicago, Illinois. The first highlighted abstract, presented by Sanjiv S. Agarwala, MD, of the St. Luke’s Cancer Center, Bethlehem, PA, entitled “Efficacy of intralesional rose bengal in patients receiving injection in all existing melanoma in phase II study PV-10-MM-02” (abstract 9027), may be viewed at: [http://abstracts.asco.org/144/AbstView_144_132320.html]. The second highlighted abstract, presented by Amod A. Sarnaik, MD, of Moffitt Cancer Center (“Moffitt”), Tampa, FL, entitled “Assessment of immune and clinical efficacy after intralesional PV-10 in injected and uninjected metastatic melanoma lesions” (abstract 9028), may be viewed at: [http://abstracts.asco.org/144/AbstView_144_132288.html].
4
Table of Contents
In the phase 2 PV-10 trial, when all existing lesions were injected with PV-10, tumors were no longer detectable (complete response) in 50% of the patients (Confidence Interval: 31-69%). This subgroup analysis supports the potential of PV-10 as a single agent and provides a rationale for a PV-10 phase 3 randomized controlled trial in locally advanced melanoma patients.
This phase 3 randomized controlled trial of PV-10 in patients with unresectable locally advanced cutaneous melanoma will assess response to PV-10 versus that of systemic chemotherapy in patients who have disease limited to cutaneous and subcutaneous sites and who have failed or are ineligible for systemic immunotherapy. Progression-free survival and complete response rate will be assessed using standard criteria (RECIST 1.1). Overall survival and exploratory assessment of patient reported outcomes related to lesion pain and other melanoma symptoms will also be assessed. The study is expected to commence in the first quarter of 2015, and will allow for interim assessment when 50% of the required events have occurred (i.e., disease progressions).
The Moffitt abstract provided interim results of a pilot clinical trial designed to investigate the local and immunologic effects of tumor ablation with PV-10. Lead author, Dr. Sarnaik, noted “In the peripheral blood of patients after PV-10 injection, we saw a significant increase in circulating T-cells, including CD3+ and cytotoxic CD8+ cells. This suggests an immunologic-mediated antitumor response is engendered by PV-10. We are hoping to undertake combination trials that combine PV-10 with the promising systemic immunotherapies being developed by our medical oncology colleagues.”
In the Moffitt poster at ASCO, Dr. Sarnaik and co-authors reported interim results of this pilot clinical trial designed to investigate the immunologic basis of this bystander or systemic immunologic response. In this single institution translational study, a target lesion and a bystander were biopsied prior to treatment of the target lesion with PV-10. Both lesions were then resected within 7-14 days of target lesion injection and compared to pre-treatment biopsies. Peripheral blood was also collected pre-treatment, at the time of resection and at day 28. The researchers note “treatment with IL PV-10 led to pCR (pathologic complete response) in the post-treatment biopsies of both PV-10 injected and uninjected study lesions in 4 of the 8 patients, and all 8 exhibited at least partial regression of the injected lesion.” The abstract continues, “six of the 8 patients had metastatic disease refractory to previous ipilimumab, anti-PD-1 and/or vemurafenib therapy.” Based on T cells isolated from the peripheral blood of the patients, the authors conclude that, “IL PV-10 treatment can lead to systemic anti-melanoma immunity and pCR in injected and uninjected lesions including treatment-refractory tumors.”
We observed that these results delineate two development paths to generate data sufficient for a new drug application (NDA) for PV-10 in melanoma. Our focus in 2014 and the first quarter of 2015 has been initiation of the phase 3 randomized controlled trial. We also expect to begin the more exploratory combinatorial work that potentially addresses the needs of patients with more advanced metastatic disease.
We are not alone in advocating for an intralesional approach in the treatment of cancer. For melanoma patients with recurrent or in-transit disease confined to their skin this approach has been used to treat patients for many years, as evidenced by guidelines published by the National Comprehensive Cancer Network (NCCN Guidelines®) defining the standard of care for cancer treatment in the United States. Intralesional injection with BCG and certain immunomodulatory agents, local ablation, topical therapy for superficial lesions and regional radiotherapy are consensus interventions for these patients, while systemic therapy remains an option and participation in a clinical trial is the preferred option. We believe that, in this context, PV-10 is well positioned to show superiority in phase 3 testing as a single agent.
For those patients who do not have all disease accessible to injection, medical oncologists have stated that using an agent like PV-10 to prime the immune system could be synergistic in combination with a systemic agent. Our patent application on this strategy was published in 2012 and we have been vigorously pursuing this approach. We believe the nonclinical research we first presented at the Society for Immunotherapy of Cancer (SITC) annual meeting that year, together with ongoing translational clinical research on PV-10’s mechanism of action we are sponsoring at Moffitt and our own phase 2 data, provide a rationale for combination testing of PV-10. This development track, separate from the phase 3 study, using PV-10 in combination with checkpoint protein inhibitors could present a path forward for patients with significant disease burden not amenable to intralesional injection.
On October 28, 2014, we announced that data on PV-10, obtained in clinical trial PV-10-MM-02 [ClinicalTrials.gov Identifier NCT00521053], has been published by the Annals of Surgical Oncology (ASO). The peer-reviewed article, entitled “Phase 2 Study of Intralesional PV-10 in Refractory Metastatic Melanoma”, is available as an Epub ahead of print, and may be accessed at [http://dx.doi.org/10.1245/s10434-014-4169-5]. The Annals of Surgical Oncology is the official journal of the Society of Surgical Oncology (SSO) and the American Society of Breast Surgeons. Annals is published monthly by Springer.
5
Table of Contents
On November 13, 2014, we announced that the protocol for our phase 3 study of PV-10 as a treatment for melanoma is now available on: [http://clinicaltrials.gov/ct2/show/study/NCT02288897].
The protocol states that the study is “an international multicenter, open-label, randomized controlled trial (RCT) of single-agent intralesional PV-10 versus systemic chemotherapy with dacarbazine (DTIC) or temozolomide (TMZ) to assess treatment of locally advanced cutaneous melanoma in patients who are BRAF V600E wild-type and have failed or are not otherwise candidates for ipilimumab or another immune checkpoint inhibitor. Subjects in the comparator arm will receive the Investigator’s choice of dacarbazine or temozolomide as determined by Investigator preference and/or local availability of the agent. Effectiveness will be assessed by comparison of progression-free survival (PFS) between all intent-to-treat (ITT) subjects in the two study treatment arms.”
The Primary Outcome Measure is progression-free survival (PFS) to be assessed every 12 weeks up to 18 months. The Secondary Outcome Measures include complete response rate (CRR) and its duration (to be assessed every 12 weeks up to 18 months); the change in total symptom score from baseline using the patient reported Skindex-16 instrument (to be assessed 12 weeks after Day 1); Overall survival (OS) to be assessed every 12 weeks up to 18 months; and number of participants with adverse events assessed every 4 weeks until 28 days after last treatment. Safety and tolerability will be assessed by monitoring the frequency, duration, severity and attribution of adverse events and evaluating changes in laboratory values and vital signs.
On December 22, 2014, we announced that we would be meeting with the FDA to review certain operational aspects of the protocol for our planned phase 3 clinical trial of intralesional PV-10, as a treatment for melanoma.
On February 9, 2015, we announced that we have held a Type C meeting with the FDA to review certain operational aspects of the protocol for our planned phase 3 clinical trial of intralesional PV-10 as a treatment for melanoma. The meeting was held by teleconference on January 29, 2015.
As noted in our press release of December 22, 2014, when we submitted the protocol to the Agency in November 2014, we included a brief list of questions about certain operational aspects of the protocol. The FDA subsequently indicated that a formal meeting was appropriate to assure that these questions were addressed in a timely and comprehensive manner. As is typical for such meetings, we provided a more extensive list of questions in the formal meeting package. This led to a very thorough and helpful review of the protocol as a result of the meeting. Topics formally reviewed included subject eligibility requirements, primary and secondary study end points, and study lesion definitions and conventions for defining disease progression.
The outcome of the FDA’s review does not affect the fundamental design of the study nor the patient population, but does affect certain details concerning some secondary end points and statistical analysis matters, such as the treatment of missing data. We are making a number of small changes to the protocol in light of this review, and intend to issue a final version and start enrolling the phase 3 in the first quarter of 2015. No further review is required by the FDA in order for us to proceed.
We have eight sites, four in the U.S. and four in Australia, in our expanded access program currently using PV-10 for melanoma and other cutaneous malignancies. We expect that they will provide a path to starting enrollment quickly upon completion of the changes to the protocol in the first quarter of 2015. In addition, we have been qualifying additional sites that will join the study pending action by their respective Institutional Review Boards.
While we believe the rapid ablative effect immediately evident in patients treated with PV-10 highlights our path to initial approval, the bystander effect, or secondary immunomodulatory benefit of PV-10 as a result of direct ablation, continues to be of scientific interest and studies to quantify systemic tumor-specific immune response in cancer patients are ongoing. This is why we term the overall function of PV-10 as ablative immunotherapy. This emerging understanding of the secondary effect of tumor ablation with PV-10 is an important foundation for future studies to assess the long-term impact of PV-10 on distant metastasis and possible combination strategies for use of PV-10 in the treatment of cancer patients with more advanced disease. PV-10 is therefore becoming known as an ablative immunotherapy, and we believe it is therefore a next generation ablative immunological treatment.
Ongoing immunologic mechanism of action studies at Moffitt have been conducted from 2011 through 2014, and thus far in 2015, to characterize the systemic benefit of PV-10. A feasibility study to detect immune cell infiltration into melanomas treated by PV-10 commenced in January 2013.
6
Table of Contents
In August, 2013, Moffitt stated that a single injection (PV-10) may revolutionize melanoma treatment. In their initial study, researchers injected a single dose of PV-10 into mice with melanoma. The result was a significant reduction in the skin cancer lesions, as well as a sizable reduction in melanoma tumors that had spread to the lungs. The researchers said the dye solution appeared to produce a robust anti-tumor immune response and may be safer than existing immunological agents.
Moffitt is currently in the middle of their first human clinical trial of PV-10 for advanced melanoma patients. In addition to monitoring the response of injected melanoma tumors, Moffitt is also measuring the boost in the anti-tumor immune cells of patients after injection. The initial study appears in PLOS ONE, an open-access, peer-reviewed online journal.
On April 7, 2014, we announced that a poster presentation detailing significant decrease in melanoma cells in patients’ injected tumors 7-14 days after intralesional PV-10 treatment that was accompanied by similar decrease in uninjected bystander tumors was presented April 6 by researchers from Moffitt at the American Association for Cancer Research Annual Meeting in San Diego, CA. These clinical and pathologic changes were accompanied by increases in important immune cell populations detected in the patients’ peripheral blood.
The poster presentation, based upon abstract #630, entitled “Induction of anti-melanoma immunity after intralesional ablative therapy,” was authored by Hao Liu, Krithika Kodumudi, Amy Weber, Amod A. Sarnaik and Shari Pilon-Thomas of Moffitt. The mechanism of regression of uninjected lesions is under investigation at Moffitt (NCT01760499). Further information can be accessed at the following NIH Registry link: [http://www.clinicaltrials.gov/ct2/show/NCT01760499].
Moffitt researchers presented clinical data on 8 melanoma patients that demonstrated significant decreases in melanoma cells in injected tumors and uninjected bystander tumors 7-14 days after PV-10 injection as evidenced by pathologic evaluation confirmed with immunohistochemical staining of biopsy specimens for melA (a marker of melanoma). The researchers showed that these changes in tumors were accompanied by increased populations of CD3+, CD4+ and CD8+ T cells along with NKT cells in peripheral blood. T cells from one patient were purified and exhibited increased interferon-gamma expression when exposed to the patient’s pre-treatment melanoma cells.
In addition, Moffitt’s team found that PV-10 was cytotoxic to B16 mouse melanoma cells with minimal cytotoxicity to normal skin cells (fibroblasts). This cytotoxicity occurred via necrosis with minimal evidence of apoptosis. The PV-10 treatment of B16 tumors in mice led to release of HMGB1, a soluble Damage Associated Molecule Pattern (DAMP) that is important in activation of dendritic cells; such dendritic cells from these mice were selectively active against B16 tumor cells. PV-10 treatment of B16 tumors in mice also led to infiltration of dendritic cells into the lymph nodes draining the treated tumors; no infiltration was observed in non-draining nodes.
Dr. Pilon-Thomas of Moffitt stated, “These data are exciting and illustrate successful translation of our pre-clinical work in mice to clinical results in melanoma patients. With only 8 patients we’ve been able to clearly observe statistically significant increases in beneficial T cell populations in peripheral blood. Ironically, the original aim of the trial to assess tumor-infiltrating lymphocytes was thwarted when biopsies of patient tumors collected just 7-14 days after PV-10 injection no longer contained viable tumor tissue. We are following up both the human data and continuing to design more experiments in mice to better explain the systemic immune effects elicited by PV-10 ablation.”
Abstract #630 can be accessed at the following link: [http://www.abstractsonline.com/Plan/ViewAbstract.aspx?mID=3404&sKey=9a90b661-024e-4702-894a-d3f419f9925e&cKey=3ee0b61c-784a-4e56-8ec7-c9b3d868a8b6&mKey=6ffe1446-a164-476a-92e7-c26446874d93].
On November 10, 2014, we announced that data on PV-10 for intralesional (IL) treatment of cancer was featured in a poster presentation at the Society for Immunotherapy of Cancer [SITC] 29th Annual Meeting on Saturday, November 8, 2014. The presentation, titled “Efficacy of Intralesional Injection with PV-10 in Combination with Co-Inhibitory Blockade in a Murine Model of Melanoma,” is available at http://www.pvct.com/publications/SITCposter2014.pdf.
The poster, presented by Dr Shari Pilon-Thomas of Moffitt, concludes that the new data “support combination therapy with IL PV-10 and co-inhibitory blockade.” In clinical trials, IL PV-10 has induced regression of both injected lesions and uninjected bystander lesions in patients with melanoma, and tumor ablation with PV-10 has been shown to increase certain T-cell populations in patients’ peripheral blood. In the study reported at SITC, the team from Moffitt measured whether IL PV-10 and co-inhibitory blockade could improve anti-tumor immunity and regression of melanoma in mice.
7
Table of Contents
The testing assessed response of injected and uninjected B16 melanoma tumors in mice receiving PV-10 alone or in combination with one of three agents designed for co-inhibitory blockade. The tested agents targeted either CLTA-4, PD-1 or PD-L1, the three most common clinical targets for co-inhibitory blockade. In each case, combination of PV-10 with co-inhibitory blockade led to improved tumor response and enhanced anti-tumor immunity of T-cells. Further testing with the anti-PD-L1 agent showed that these improvements could apply to both injected and uninjected tumors.
We believe this important work further validates use of an intralesional therapy with a systemic immunotherapy, and solidifies our plans for a promising second path for development of PV-10. In addition to use as a single-agent therapy for cutaneous melanoma (the focus of our phase 3 study), we believe these findings support commencement of clinical testing of PV-10 in combination anti-CLTA-4, anti-PD-1 or anti-PD-L1 agents. We are assessing strategies to allow this work to commence in a timely and cost-effective manner so that we can begin translating these model test results into human clinical data.
We also report ongoing progress with our Compassionate Use Program for PV-10 for non-visceral cancers. With well over 100 patients enrolled in eight centers across the U.S. and Australia, the protocol enables subjects to undergo more frequent and extensive treatments of PV-10 over a longer period of time than was allowed under the protocol used for the phase 2 trials. Its dosage has been very helpful with planning for the phase 3 melanoma study as well as treating other types of cutaneous and subcutaneous cancers, and we are gratified we can provide PV-10 now for patients that request it who have no other available option.
We are continuing to assess how much additional work we should do by ourselves, and when to partner with a larger company to further co-develop PV-10, as well as potential paths to accelerated and expedited approval in the U.S. and abroad, including in China and India.
On August 18, 2014, we announced that we entered into a Memorandum of Understanding (“MOU”) with Sinopharm-China State Institute of Pharmaceutical Industry (“Sinopharm-CSIPI”), the leader among all pharmaceutical research institutes in China, and Sinopharm A-THINK Pharmaceutical Co., Ltd. (“Sinopharm A-THINK”), the only injectable anti-tumor drug research and development, manufacture and distribution integrated platform within Sinopharm Group. The MOU term, as extended pursuant to an amendment entered into on November 13, 2014, continues to May 16, 2015.
The key component of the MOU provides that “Sinopharm-CSIPI and Sinopharm A-THINK desire to obtain an exclusive license to commercialize PV-10 within [the People’s Republic of] China territory, and [Provectus] is willing to grant such license to Sinopharm.” Prior to May 16, 2015, the parties will seek to enter into a definitive licensing contract, subject to additional negotiation, due diligence, and any required regulatory and corporate approvals. If such an agreement is reached, we intend to manufacture PV-10 in the USA and to have Sinopharm A-THINK distribute PV-10 in China.
The MOU contains customary provisions regarding confidential information, publicity, and intellectual property, and is non-binding upon the parties (except for certain non-material provisions). The MOU shall continue in effect until the earliest of the replacement of the MOU with a definitive agreement, one month prior written notice by either the Company or Sinopharm, or the expiration of the MOU.
We have signed agreements with two manufacturers to supply us with clinical-quality PV-10, and we now have sufficient quantities of PV-10 available to commence the phase 3 trial and to undertake our other development activities. To assure smooth execution of the study we have lined up specialty contract research organizations (CROs) and other service providers with expertise in clinical operations and integrated data management. As is standard in our industry, this includes a full-service, international CRO who will coordinate the global efforts of this team of specialists.
We have worked with this team to establish an independent Clinical Trial Data Monitoring Committee (DMC). The FDA states “A clinical trial DMC is a group of individuals with pertinent expertise that reviews on a regular basis accumulating data from one or more ongoing clinical trials. The DMC advises the sponsor regarding the continuing safety of trial subjects and those yet to be recruited to the trial, as well as the continuing validity and scientific merit of the trial.” The DMC will ensure that our study provides patients with maximum possible safety while protecting the scientific validity and integrity of the data we gather.
8
Table of Contents
Liver Cancer
According to Global Cancer Facts & Figures, 2nd Edition, liver cancer is the fifth leading cause of deaths related to cancer in the world in men and seventh in women. Approximately 750,000 people are newly diagnosed annually with primary liver cancer, also known as Hepatocellular carcinoma (HCC), with China alone accounting for about 55% of the cases diagnosed each year. The world market for liver cancer drugs is projected to exceed $2.0 billion by 2015 and does not include the full impact of the China market potential.
Early detection is difficult and as a result, most cases reach an advanced metastatic stage and are unresectable. If the cancer cannot be completely removed, the disease is usually deadly within three to six months. Malignant lesions in the liver arising from HCC or metastases from a wide range of cancers represent an ongoing treatment challenge for oncologists. HCC is one of the most common malignancies worldwide, and its incidence is rapidly increasing in the United States. The liver is a common site of metastases from solid tumors, particularly those arising in the gastrointestinal tract. Other tumors, such as lung and breast cancer and melanoma, also readily spread to the liver.
In 2009, we began a phase 1 study of PV-10 to assess the safety, tolerability and pharmacokinetics of single intralesional injections of PV-10 with subjects with either recurrent hepatocellular carcinoma or cancer metastatic to the liver. In January 2011, we completed patient accrual of all subjects in the phase 1 study. The primary outcome measure was safety, including systemic and locoregional adverse events. The secondary outcome measures were (i) lesion distribution and retention of PV-10 following injection, (ii) ORR of target and measurable bystander lesions (if present) by modified RECIST, (iii) changes in markers of hepatic function, including ALP, ALT, AST, total bilirubin and GGT, and (iv) pharmacokinetics of PV-10 in the bloodstream following intralesional injection.
Final results for PV-10 as a treatment for liver cancer are very encouraging as they show the treatment was generally well-tolerated, with substantial evidence of efficacy. We believe PV-10’s ability to selectively target and destroy cancer cells without harming surrounding healthy tissue make it a potentially attractive therapy for cancers of the liver, which can be very serious and difficult to treat if they cannot be fully removed through surgery. Based upon the initial results of our PV-10 phase 1 trial for liver cancer, and the growing confidence we have in PV-10 as a viable treatment for non-resectable liver cancer, we are currently designing a phase 1b/ 2 study with the potential for expedited approval.
In April 2011, we received orphan drug designation by the FDA for Rose Bengal, the active ingredient in PV-10, for the treatment of HCC, the most common form of primary liver cancer.
In September 2012, we commenced an expansion of the phase 1 study, which we continued in 2013, 2014, and thus far in 2015. Drug-drug metabolic interaction nonclinical studies of PV-10 and sorafenib provided the data to support additional work within the regulatory framework for this important indication.
We collaborated with XenoTech, a preclinical CRO and pioneer in collaborative research surrounding in vitro drug metabolism and pharmacokinetics (DMPK) services, in writing an article describing a study to determine the potential of rose bengal disodium to cause drug-drug interactions which has been published by Xenobiotica, a peer-reviewed scientific journal that publishes comprehensive research papers on pharmacokinetics (the study of distribution, metabolism, disposition and excretion of drugs). The published research indicated that the risk of PV-10 causing clinically relevant drug-drug interactions is likely minimal.
The study was undertaken prior to initiation of the now ongoing testing of PV-10 plus sorafenib (cohort 2) in a clinical trial of PV-10 intralesional injection in hepatocellular carcinoma patients taking a stable dose of sorafenib. Sorafenib is a competitive inhibitor of cytochrome P450 (CYP) drug metabolism enzymes and is reliant on the UDP-glucuronosyltransferase (UGT) pathway for efficient clearance. CYP and UGT enzymes help to biotransform small lipophilic drugs like sorafenib into water-soluble excretable metabolites.
Provectus researchers collaborated with XenoTech’s experts to design the appropriate in vitro experiments necessary to assess the risk for potential liability when rose bengal is co-administered with other drugs in humans. Rose Bengal, known for inducing singlet oxygen on exposure to light, can cause erroneous results in conventional in vitro test systems. These assay artifacts were shown to be test system dependent in DMPK studies. XenoTech scientists successfully tailored experiments to ascertain CYP and UGT inhibition potential in more appropriate model systems.
We have recently expanded our exploratory phase 1 study of cancers of the liver to three centers (St. Luke’s University Health Network, Bethlehem, Pennsylvania, and The Southeastern Center for Digestive Disorders & Pancreatic Cancer, Tampa, Florida, in addition to Sharp Memorial Hospital, San Diego, California), and we are evaluating the addition of several more centers to further advance this initial effort. We are working with our investigators to report results from long-term
9
Table of Contents
follow-up of our initial patients in the coming months. We are assessing strategies to accelerate transition to phase 2 testing in a randomized controlled trial, either alone or in combination with systemic therapy. Any combination studies in the liver are likely to follow similar development strategies to those outlined above for melanoma and rely on much of the same foundational science.
The current phase 1 study, initially designed solely to establish safety of percutaneous injection of PV-10 into liver tumors (that is, injection through the skin), is providing valuable data crucial for planning such phase 2 development. This trial is open to patients with hepatocellular carcinoma or other cancers metastatic to the liver who have at least one tumor that has either originated in or spread to the liver and are not candidates for surgery or transplant. All patients enrolled in this open-label study receive the same treatment: an interventional radiologist injects PV-10 percutaneously into a single liver tumor. Patients with multiple injectable tumors may later receive further PV-10 to their other tumors. We have received numerous inquiries about this study from researchers as well as patients and their doctors, and refer these to our investigators through the contact information available on the clinicaltrials.gov website. We plan to commence the phase 1b/2 liver study in early 2015. This study has potential for generating sufficient data to support expedited approval under one or more FDA programs.
Breast Cancer
In 2005, we began a phase 1 study of PV-10 to assess the safety and tolerability of injections of PV-10 into recurrent breast carcinoma. We completed the phase 1 study in 2008. The primary outcome measure was systemic and locoregional adverse experience. The secondary outcome measures were (i) histopathologic response of PV-10 injected lesions and (ii) wound healing of PV-10 injected lesions.
The goals of the phase 1 clinical trial were to determine the safety of the treatment and the appropriate dosage. We have also wanted to show that PV-10 has multi-indication potential. We continued to demonstrate this objective in 2011 through 2014, and expect to do so in 2015. We are now in a position for a phase 2 study in recurrent breast carcinoma with our lead oncology drug product candidate PV-10. We are evaluating potential for further development of PV-10 to treat recurrent breast cancer based on the published data provided by Moffitt as well as interest to address this important indication.
Other Indications
The compassionate use program for PV-10 is only available for cancer indications that do not involve treatment of visceral organs and are not subject to enrollment in ongoing clinical trials. These indications include certain breast cancers, basal cell carcinoma, squamous cell carcinoma, certain head and neck cancers and melanoma. Compassionate use programs provide patients with access to experimental therapeutics prior to FDA approval.
The protocol for the compassionate use program enables subjects to undergo more frequent and extensive treatments of PV-10 over a longer period of time than was allowed under the protocol used for the phase 2 trial of PV-10. Based on the success of the compassionate use program, its dose regimen served as the blueprint for the phase 3 study for melanoma. The majority of patients enrolled in the program have been treated for melanoma, with other patients for other indications such as recurrent squamous cell carcinoma and refractory scalp sarcoma.
Additionally, we are considering a clinical study of PV-10 for pancreatic cancer as well as other solid tumor indications.
Dermatology (PH-10)
Our prescription drug candidate PH-10 is an aqueous hydrogel formulation of Rose Bengal for topical administration to the skin. It is a novel nonsteroidal anti-inflammatory agent that interacts with ambient and other light sources. We are developing PH-10 for the treatment of cutaneous skin disorders, specifically psoriasis and atopic dermatitis, and we believe that PH-10 may be successful in treating other skin diseases. We believe that PH-10 offers a superior treatment for psoriasis and atopic dermatitis because it selectively treats diseased tissue with negligible potential for side effects in healthy tissue.
We have been actively discussing licensing transactions with a number of potential out licensing partners for PH-10. We believe that our phase 2c trial of PH-10 for psoriasis will further solidify the commercial viability of PH-10 in these discussions. In August 2011, we completed follow-up of all phase 2c patients and communicated data of the study to both prospective partners as well as the public market in early 2012. In January 2015, we commenced a mechanism of action study of PH-10 to better characterize the unique immunologic signaling aspects along with PH-10 safety and efficacy.
10
Table of Contents
Psoriasis
Psoriasis is a common chronic disorder of the skin characterized by dry scaling patches, called “plaques,” for which current treatments are few and those that are available have potentially serious side effects. There is no known cure for the disease at this time. According to the National Institutes of Health, as many as 7.5 million Americans, or approximately 2.2 percent of the U.S. population, have psoriasis. The National Psoriasis Foundation reports that approximately 125 million people worldwide, 2 to 3 percent of the total population, have psoriasis. It also reports that total direct and indirect health care costs of psoriasis for patients exceed $11 billion annually.
According to the National Psoriasis Foundation, the majority of psoriasis sufferers, those with mild to moderate cases, are treated with topical steroids that can have unpleasant side effects. None of the other treatments for moderate cases of psoriasis have proven completely effective. The 25-30% of psoriasis patients who suffer from more severe cases generally are treated with more intensive drug therapies or PUVA, a light-based therapy that combines the drug Psoralen with exposure to ultraviolet A light. While PUVA is one of the more effective treatments, it increases a patient’s risk of skin cancer.
Our phase 1 study for PH-10 was initiated in April 2001 to evaluate the safety of three different doses of PH-10 in separate patient segment groups. Subjects in the study each received a single dose of PH-10 followed by administration of green light on psoriatic plaques. Subjects were examined post-treatment, with a final follow-up examination at 90 days.
Our phase 2 study of PH-10 for treatment of psoriasis was initiated in 2009 and completed in April 2010. There were 30 subjects treated in the completed phase 2 study, and an additional six subjects were treated in an earlier study that was terminated in favor of an increased dosing frequency. Consistent with the preliminary data that we announced in December 2009, 70% of the 30 subjects enrolled in the phase 2 clinical trial of PH-10 for psoriasis demonstrated improvement in their Psoriasis Severity Index (PSI) scores at the end of four weeks of daily treatment with PH-10. In addition, 86% of subjects reported no or only mild pruritus (itching) by week four of the trial, and no significant safety issues were noted. At the four week interval substantial improvement was observed across all standard disease assessment scores.
During 2010, we initiated a phase 2c clinical trial of PH-10 for psoriasis. This multicenter, randomized controlled phase 2c study enrolled 99 subjects at four different sites, which began in December 2010. The subjects were randomized sequentially by center to one of four treatment cohorts, and assessed efficacy and safety of topical PH-10 applied once daily to areas of mild to moderate plaque psoriasis. The primary efficacy endpoint was “treatment success,” a static endpoint assessed at day 29 after initial PH-10 treatment and defined as 0 or 1 on all Psoriasis Severity Index (PSI) components and 0 or 1 on the Plaque Response scale. The primary safety endpoint was incidence of adverse experiences, including pain and dermatologic/skin toxicity (incidence, severity, frequency, duration and causality). The secondary outcome measures were (i) Psoriasis Severity Index (PSI) score changes at each visit from day 1 pre-treatment, (ii) Plaque Response score changes at each visit from day 1 pre-treatment, and (iii) Pruritus Self-Assessment score changes at each visit from day 1 pre-treatment.
The phase 2c trial was conducted at four sites in the U.S. including the Mount Sinai School of Medicine in New York City, Wake Research Associates in Raleigh, North Carolina, Dermatology Specialists in Oceanside, California, and International Dermatology Research in Miami, Florida. With over 90 subjects, this trial is the largest dermatological trial that we have conducted to date.
The results of this study helped define the parameters necessary for the design of a pivotal phase 3 trial, and it was an important milestone on the regulatory pathway leading towards commercialization. In addition, we have held discussions with a number of potential out licensing partners, and we believe this phase 2c trial has further solidified the commercial viability of PH-10 in these discussions. We have also continued important toxicity study research and development in 2012 through 2014 and into 2015 to prepare for a phase 3 study and to support a New Drug Approval filing.
On December 23, 2014, we announced that the protocol for our phase 2 study of the mechanism of action of PH-10 in psoriasis is now available on ClinicalTrials.gov, Identifier NCT02322086: [https://www.clinicaltrials.gov/ct2/show/NCT02322086]. The purpose of the trial is to study the safety and efficacy of PH-10, a 0.005% preparation of Rose Bengal, in the treatment of psoriasis.
Officially titled, “A Phase 2 Study of Cellular and Immunologic Changes in the Skin of Subjects Receiving PH-10 Aqueous Hydrogel to Plaque Psoriasis,” total enrollment is expected to consist of 30 patients. Subjects will apply PH-10 vehicle daily for 28 consecutive days followed by active PH-10 daily for 28 consecutive days to their plaque psoriasis areas on the trunk or extremities (excluding palms, soles, scalp, facial and intertriginous sites). Biopsies of one target plaque will be collected at baseline (at least 7 days prior to first study treatment on Day 1) and at Days 29 and 64, with a 7-day interval between biopsy at Day 29 at the end of vehicle application and commencement of application of active PH-10 on Day 36. Study data from each subject will serve as an internal control (i.e., with assessment at baseline and at the end of application of PH-10 vehicle) for evaluation of clinical and cellular response to active investigational agent.
11
Table of Contents
The protocol states that the multicenter study is designed to assess treated psoriatic plaque for “changes in immunologic, structural and hyperproliferative state and for any evidence of cellular atypia” when treated with PH-10 and to “correlate observed changes in the skin with clinical response to treatment.” These assessments are expected to advance the understanding of the mechanism of action of PH-10 in psoriasis and other inflammatory dermatoses, such as atopic dermatitis, and further substantiate the safety profile of the agent. Biopsy specimens will be assessed for changes in epidermal hyperplasia (i.e., disordered condition of the skin creating thickening and scaling); infiltration with immune cells; and molecular markers of inflammation. Correlation of clinical response to these cellular and molecular changes will be performed at the plaque level using Psoriasis Severity Index (PSI) assessment data. Safety will be assessed by monitoring the frequency, duration, severity and attribution of clinical adverse events; evaluating changes in laboratory values and vital signs; and by correlation of clinical adverse events with observed histopathologic and immunohistopathologic changes in the skin.
By capturing data at the clinical and cellular level, we expect this study to allow us to establish how PH-10 affects psoriatic plaque and other similar inflammatory diseases of the skin, and to relate the safety profile from earlier studies to such effects. We believe that understanding these effects with this level of detail will allow us to properly position PH-10 within the competitive landscape and should provide crucial safety data to support extended dosing. We expect this effort to provide a comparable level of understanding of the effects of PH-10 in diseased skin to the keen insight we have gained through our clinical and nonclinical mechanism studies of PV-10, our novel investigational cancer drug, in melanoma and other cancers. Because there are no good model systems for psoriasis, we believe this study affords a critical opportunity to link the clinical effects we have observed to changes in well-established immunologic drivers of the disease. The study will be performed at three centers in the United States.
On January 29, 2015, we announced that we have opened recruitment for our PH-10 mechanism study. PH-10 has already been testing phase 1 and 2 studies and a total of 226 patients. In this study, we are looking at possible changes in the immunologic, structural and hyperproliferative state of the skin in the target plaque and evidence of cellular atypia following PH-10 application. We will use this data to aid in further development of PH-10 with our objective to co-develop or license PH-10 with dermatological partner as we continue to prepare to advance PH-10 for approval as topical anti-inflammatory non-steroidal agent for treating psoriasis and other inflammatory dermatoses. According to clinicaltrials.gov, the estimated completion date of the study is January 2016.
Atopic Dermatitis
Atopic Dermatitis, the most severe and common type of eczema, is a long-term skin disease that causes dry and itchy skin, rashes on the face, inside the elbows, behind the knees, and on the hands and feet. Scratching of the afflicted skin can cause redness, swelling, cracking, weeping clear fluid, crusting, thick skin, and scaling. According to the National Eczema Association, physicians estimate that 65% of eczema patients are diagnosed in the first year of life and 90% of patients experience it before age five. Often the symptoms fade during childhood, though most will have atopic dermatitis for life. The National Eczema Association estimates that atopic dermatitis affects over 30 million Americans.
In 2008, we initiated a phase 2 study of PH-10 for the treatment of atopic dermatitis. This phase 2 study assessed whether topical PH-10 applied once daily to mild, moderate or severe atopic dermatitis may ameliorate inflammation of the skin when activated by ambient light. The subjects applied PH-10 daily for 28 days to skin areas affected by atopic dermatitis. The subjects were assessed weekly during the treatment period and for four weeks following the treatment period. The primary outcome measures were (i) treatment success, defined as a score of 0 to 1 at day 28, the end of the study treatment period, by the Investigator’s Global Assessment (IGA) scoring system for atopic dermatitis status, and (ii) adverse experience, including pain and dermatologic/skin toxicity (incidence, severity, frequency, duration and causality) during the eight weeks following treatment.
Data from the subjects indicated that a substantial majority of subjects had improvement in the Eczema Area Severity Index (EASI) during four weeks of treatment. The treatments were generally well tolerated with no significant safety issues identified. At the four week interval substantial improvement was observed across all standard disease assessment scores. We have also continued important toxicity study research and development in 2012, 2013, 2014 and thus far in 2015 to prepare for continued development in this indication and to support a New Drug Approval filing.
Other Indications
We have investigated the use of PH-10 for treatment of actinic keratosis (also called solar keratosis or senile keratosis), which is the most common pre-cancerous skin lesion among fair-skinned people and is estimated to occur in over 50% of elderly fair-skinned persons living in sunny climates. We have previously conducted a phase I clinical trial of PH-10 for
12
Table of Contents
actinic keratosis to examine the safety profile of a single treatment using topical PH-10 with green light photoactivation. No significant safety concerns were identified in the study. We have decided to prioritize further clinical development of PH-10 for treatment of psoriasis and atopic dermatitis rather than actinic keratosis at this time since the market is much larger for psoriasis and atopic dermatitis.
We have also conducted pre-clinical studies of PH-10 for use in treating severe acne vulgaris. Moderate to severe forms of the disease have proven responsive to several photodynamic regimens, and we anticipate that PH-10 can be used as an advanced treatment for this disease. Our pre-clinical studies show that the active ingredient in PH-10 readily kills bacteria associated with acne. This finding, coupled with our clinical experience in psoriasis, atopic dermatitis, and actinic keratosis, suggests that therapy with PH-10 will exhibit no significant side effects and will afford improved performance relative to other therapeutic alternatives. If correct, this would be a major advance over currently available products for severe acne.
The active ingredient in PH-10 is photoactive in that it reacts to light of certain wavelengths thereby potentially increasing its therapeutic effects. We believe that photodynamic treatment regimens can deliver a higher therapeutic effect at lower dosages of active ingredient, thus minimizing potential side effects including damage to nearby healthy tissues. PH-10 is especially responsive to green light, which is strongly absorbed by the skin and thus only penetrates the body to a depth of about three to five millimeters. For this reason, in the past we have investigated PH-10 combined with green-light activation, for topical use in surface applications where serious damage could result if medicinal effects were to occur in deeper tissues.
Over-the-Counter Pharmaceuticals
We have designated our subsidiary that holds our OTC products, GloveAid and Pure-ific, Pure-Stick, Pure N Clear as non-core. The potential further development and licensure of our OTC products would likely be facilitated by selling a majority stake of the underlying assets of the non-core subsidiary holding the OTC products. This transaction would likely be accomplished through a non-core spin-out process, which would enable the non-core subsidiary to become a separate publicly held company. The new public entity could then raise funds without diluting the ownership of the then current stockholders of the Company, although there can be no assurance that this process will occur.
GloveAid
Personnel in many occupations and industries now use disposable gloves daily in the performance of their jobs, including airport security personnel, food handling and preparation personnel, health care workers such as hospital and blood bank personnel, laboratory researchers, police, fire and emergency response personnel, postal and package delivery handlers and sorters, and sanitation workers.
Accompanying the increased use of disposable gloves is a mounting incidence of chronic skin irritation. To address this market, we have developed GloveAid, a hand cream with both antiperspirant and antibacterial properties, to increase the comfort of users’ hands during and after the wearing of disposable gloves. During 2003, we ran a pilot scale run at the manufacturer of GloveAid.
Pure-ific
Our Pure-ific line of products includes two quick-drying sprays, Pure-ific and Pure-ific Kids, that immediately kill up to 99.9% of germs on skin and prevent regrowth for six hours. We have determined the effectiveness of Pure-ific based on our internal testing and testing performed by Paratus Laboratories H.B., an independent research lab. Pure-ific products help prevent the spread of germs and thus complement our other OTC products designed to treat irritated skin or skin conditions such as acne, eczema, dandruff and fungal infections. Our Pure-ific sprays have been designed with convenience in mind and are targeted towards mothers, travelers, and anyone concerned about the spread of sickness-causing germs. During 2003 and 2004, we identified and engaged sales and brokerage forces for Pure-ific. We emphasized getting sales in independent pharmacies and mass (chain stores) markets. The supply chain for Pure-ific was established with the ability to support large-scale sales and a starting inventory was manufactured and stored in a contract warehouse/fulfillment center. In addition, a website for Pure-ific was developed with the ability for supporting online sales of the antibacterial hand spray. During 2005 and 2006, most of our sales were generated from customers accessing our website for Pure-ific and making purchases online. We discontinued our proof-of-concept program in November 2006 and have, therefore, ceased selling our Pure-ific product. We now intend to license the Pure-ific product, a strategy we have been discussing with interested groups. Additionally, we also intend to sell a majority stake in the underlying assets via a non-core spin-out transaction, as discussed below.
On December 15, 2011, we sold Units to accredited investors which included shares of common stock in Pure-ific and a warrant to purchase 3/4 of a share of the Company’s common stock. A total of 666,666 Units were sold for gross proceeds of $500,000 resulting in the sale of a 33% non-controlling interest in Pure-ific. At the time of the sale and as of December 31,
13
Table of Contents
2011, the carrying value of the net assets in Pure-ific was $0. The sale also resulted in the issuance of warrants to purchase 500,000 shares of the Company’s common stock at an exercise price of $1.25 per share with a five-year term. We intend to use the proceeds, after deducting offering expenses of approximately $56,500, to spin-off Pure-ific as a new publicly-traded company, a process we have initiated but have not yet completed. Network 1 Financial Securities, Inc., served as placement agent for the offering.
Acne
Our acne products Pure-Stick and Pure N Clear work by decreasing the production of fats, oils and sweat that create an environment conducive to unchecked growth of bacteria. Secondly, the products also act to reduce the number of bacteria already present. Pure-Stick and Pure N Clear represent new formulations of proven, safe ingredients that achieve both steps required to successfully treat acne. Since Pure-Stick and Pure N Clear are applied topically to affected areas there are no safety concerns with healthy skin. The unique combinations have allowed the Company to secure patent protection for these products.
Medical Devices
We have non-core medical device technologies that we believe may address two major markets:
| • | cosmetic treatments, such as reduction of wrinkles and elimination of spider veins and other cosmetic blemishes; and |
| • | therapeutic uses, including photoactivation of PH-10, other prescription drugs and non-surgical destruction of certain skin cancers. |
We expect to further develop our non-core medical devices through partnerships with, or selling our assets to, third-party device manufacturers or, if appropriate opportunities arise, through acquisition of one or more device manufacturers. Additionally, the Company also intends to sell a majority stake in the underlying assets via a non-core spin-out transaction.
Photoactivation
Our clinical tests of PH-10 for dermatology have in the past utilized a number of commercially available lasers for activation of the drug. This approach has several advantages, including the leveraging of an extensive base of installed devices present throughout the pool of potential physician-adopters for PH-10. Access to such a base could play an integral role in early market capture. However, since the use of such lasers, which were designed for occasional use in other types of dermatological treatment, is potentially too cumbersome and costly for routine treatment of the large population of patients with psoriasis, we have begun investigating potential use of other types of photoactivation hardware, such as light booths. The use of such booths is consistent with current care standards in the dermatology field, and may provide a cost-effective means for addressing the needs of patients and physicians alike. We anticipate that such photoactivation hardware would be developed, manufactured, and supported in conjunction with one or more third-party device manufacturers.
Laser-Based Treatment of Melanoma
We have conducted extensive research on ocular melanoma at the Massachusetts Eye and Ear Infirmary (a teaching affiliate of Harvard Medical School) using a new laser treatment that may offer significant advantage over current treatment options. A single quick non-invasive treatment of ocular melanoma tumors in a rabbit model resulted in elimination of over 90% of tumors, and may afford significant advantage over invasive alternatives, such as surgical excision, enucleation, or radiotherapy implantation. Ocular melanoma is rare, with approximately 2,000 new cases annually in the U.S. However, we believe that our extremely successful results could be extrapolated to treatment of primary melanomas of the skin, which have an incidence of over 60,000 new cases annually in the U.S. and a 6% five-year survival rate after metastasis of the tumor. We have performed similar laser treatments on large (averaging approximately 3 millimeters thick) cutaneous melanoma tumors implanted in mice, and have been able to eradicate over 90% of these pigmented skin tumors with a single treatment. Moreover, we have shown that this treatment stimulates an anti-tumor immune response that may lead to improved outcome at both the treatment site and at sites of distant metastasis. From these results, we believe that a device for laser treatment of primary melanomas of the skin and eye is nearly ready for human studies. We anticipate partnering with, or selling our assets to, a medical device manufacturer to bring it to market in reliance on a 510(k) notification. For more information about the 510(k) notification process, see “Federal Regulation of Therapeutic Products” below.
14
Table of Contents
Research and Development
We continue to actively develop projects that are product-directed and are attempting to conserve available capital and achieve full capitalization of our company through equity and convertible debt offerings, generation of product revenues, and other means. All ongoing research and development activities are directed toward maximizing shareholder value and advancing our corporate objectives in conjunction with our OTC product licensure, our current product development and maintaining our intellectual property portfolio.
Research and development costs totaling $5,137,927 for 2014 included payroll of $1,395,321, consulting and contract labor of $2,355,780, lab supplies and pharmaceutical preparations of $790,653, legal of $384,061, insurance of $115,957, rent and utilities of $87,623, and depreciation expense of $8,532. Research and development costs totaling $3,595,555 for 2013 included payroll of $1,459,057, consulting and contract labor of $1,317,472, lab supplies and pharmaceutical preparations of $310,160, legal of $262,720, insurance of $161,268, rent and utilities of $78,512, and depreciation expense of $6,366. Research and development costs totaling $5,005,459 for 2012 included payroll of $2,536,818, consulting and contract labor of $2,008,270, lab supplies and pharmaceutical preparations of $47,808, legal of $231,430, insurance of $97,728, rent and utilities of $77,238, and depreciation expense of $6,167.
Production
We have determined that the most efficient use of our capital in further developing our OTC products is to license the products. The Company has been discussing this strategy with interested groups. Additionally, the Company also intends to sell a majority stake in the underlying assets via a non-core spin-out transaction.
Sales
We have not had any significant sales of any of our OTC products, though we commenced limited sales of Pure-ific, our antibacterial hand spray in 2004 through 2006, in a proof-of-concept program. We discontinued our proof-of-concept program in 2006 and have, therefore, ceased selling our OTC products. We will continue to seek additional markets for our products through existing distributorships that market and distribute medical products, ethical pharmaceuticals, and OTC products for the professional and consumer marketplaces through licensure, partnership and asset sale arrangements, and through potential merger and acquisition candidates.
In addition to developing and selling products ourselves on a limited basis, we are negotiating actively with a number of potential licensees for several of our intellectual properties, including patents and related technologies. To date, we have not yet entered into any licensing agreements; however, we anticipate consummating one or more such licenses in the future.
15
Table of Contents
Intellectual Property
Patents
We hold a number of U.S. patents covering the technologies we have developed and are continuing to develop for the production of prescription drugs, non-core technologies and OTC pharmaceuticals. All patents material to an understanding of the Company are included and a cross reference to a discussion that explains the patent technologies and products that are identified for each patent in the following table:
| U.S. Patent No |
Title and Cross Reference |
Issue Date |
Expiration Date | |||
| 5,829,448 | Method for improved selectivity in activation of molecular agents; see discussion under Medical Devices in Description of Business | November 3, 1998 | October 30, 2016 | |||
| 5,832,931 | Method for improved selectivity in photo-activation and detection of diagnostic agents; see discussion under Medical Devices in Description of Business | November 10, 1998 | October 30, 2016 | |||
| 5,998,597 | Method for improved selectivity in activation of molecular agents; see discussion under Medical Devices in Description of Business | December 7, 1999 | October 30, 2016 | |||
| 6,042,603 | Method for improved selectivity in photo-activation of molecular agents; see discussion under Medical Devices in Description of Business | March 28, 2000 | October 30, 2016 | |||
| 6,331,286 | Methods for high energy phototherapeutics; see discussion under Oncology in Description of Business | December 18, 2001 | December 21, 2018 | |||
| 6,451,597 | Method for enhanced protein stabilization and for production of cell lines useful production of such stabilized proteins; see discussion under Material Transfer Agreement in Description of Intellectual Property | September 17, 2002 | April 6, 2020 | |||
| 6,468,777 | Method for enhanced protein stabilization and for production of cell lines useful production of such stabilized proteins; see discussion under Material Transfer Agreement in Description of Intellectual Property | October 22, 2002 | April 6, 2020 | |||
| 6,493,570 | Method for improved imaging and photodynamic therapy; see discussion under Oncology in Description of Business | December 10, 2002 | December 10, 2019 | |||
| 6,495,360 | Method for enhanced protein stabilization for production of cell lines useful production of such stabilized proteins; see discussion under Material Transfer Agreement in Description of Intellectual Property | December 17, 2002 | April 6, 2020 | |||
| 6,519,076 | Methods and apparatus for optical imaging; see discussion under Medical Devices in Description of Business | February 11, 2003 | October 30, 2016 | |||
| 6,525,862 | Methods and apparatus for optical imaging; see discussion under Medical Devices in Description of Business | February 25, 2003 | October 30, 2016 | |||
| 6,541,223 | Method for enhanced protein stabilization and for production of cell lines useful production of such stabilized proteins; see discussion under Material Transfer Agreement in Description of Intellectual Property | April 1, 2003 | April 6, 2020 | |||
| 6,986,740 | Ultrasound contrast using halogenated xanthenes; see discussion under Oncology in Description of Business | January 17, 2006 | September 9, 2023 | |||
| 6,991,776 | Improved intracorporeal medicaments for high energy phototherapeutic treatment of disease; see discussion under Oncology in Description of Business | January 31, 2006 | March 5, 2023 | |||
16
Table of Contents
| U.S. Patent No |
Title and Cross Reference |
Issue Date |
Expiration Date | |||
| 7,036,516 | Treatment of pigmented tissues using optical energy; see discussion under Medical Devices in Description of Business | May 2, 2006 | January 28, 2020 | |||
| 7,201,914 | Combination antiperspirant and antimicrobial compositions; see discussion under Over-the-Counter Pharmaceuticals in Description of Business | April 10, 2007 | May 15, 2024 | |||
| 7,338,652 | Diagnostic Agents for Positron Emission Imaging; see discussion under Oncology in Description of Business | March 4, 2008 | September 25, 2025 | |||
| 7,346,387 | Improved Selectivity in Photo-Activation and Detection of Molecular Diagnostic Agents; see discussion under Medical Devices in Description of Business | March 18, 2008 | October 30, 2016 | |||
| 7,353,829 | Improved Methods and Apparatus For Multi-Photon Photo-Activation of Therapeutic Agents; see discussion under Medical Devices in Description of Business | April 8, 2008 | April 23, 2020 | |||
| 7,384,623 | A Radiosensitizer Agent comprising Tetrabromoerythrosin; see discussion under Oncology in Description of Business | June 10, 2008 | August 25, 2019 | |||
| 7,390,668 | Intracorporeal photodynamic medicaments for photodynamic treatment containing a halogenated xanthene or derivative; see discussion under Dermatology in Description of Business | June 24, 2008 | March 6, 2021 | |||
| 7,402,299 | Intracorporeal photodynamic medicaments for photodynamic treatment containing a halogenated xanthene or derivative; see discussion under Dermatology in Description of Business | July 22, 2008 | October 2, 2025 | |||
| 7,427,389 | Diagnostic Agents for Positron Emission Imaging; see discussion under Oncology in Description of Business | September 23, 2008 | July 7, 2026 | |||
| 7,648,695 | Improved Medicaments for chemotherapeutic treatment of disease; see discussion under Oncology in Description of Business | January 19, 2010 | July 6, 2021 | |||
| 7,863,047 | Improved intracorporeal medicaments for photodynamic treatment of disease; see discussion under Dermatology in Description of Business | January 4, 2011 | October 30, 2016 | |||
| 8,470,296 | Improved intracorporeal medicaments for high energy photodynamic treatment of disease; see discussion under Dermatology in Description of Business | June 25, 2013 | June 28, 2022 | |||
| 8,530,675 | Process for the synthesis rose bengal and related xanthenes; see discussion under Oncology in Description of Business | September 10, 2013 | July 13, 2031 | |||
| 8,557,298 | Chemotherapeutic agents for cancer; see discussion under Oncology in Description of Business | October 15, 2013 | June 23, 2020 | |||
We continue to pursue patent applications on numerous other developments we believe to be patentable. We consider our issued patents, our pending and patent applications, and any patentable inventions which we may develop to be extremely valuable assets of our business.
Material Transfer Agreement
We have entered into a “Material Transfer Agreement” dated as of July 31, 2003 with Schering-Plough Animal Health Corporation, which we refer to as “SPAH”, the animal-health subsidiary of Schering-Plough Corporation, a major international pharmaceutical company which is still in effect. Under the Material Transfer Agreement, we will provide SPAH with access to some of our patented technologies to permit SPAH to evaluate those technologies for use in animal-health
17
Table of Contents
applications. If SPAH determines that it can commercialize our technologies, then the Material Transfer Agreement obligates us and SPAH to enter into a license agreement providing for us to license those technologies to SPAH in exchange for progress payments upon the achievement of goals.
The Material Transfer Agreement covers four U.S. patents that cover biological material manufacturing technologies (i.e., biotech related). The Material Transfer Agreement continues indefinitely, unless SPAH terminates it by giving us notice or determines that it does not wish to secure from us a license for our technologies. The Material Transfer Agreement can also be terminated by either of us in the event the other party breaches the agreement and does not cure the breach within 30 days of notice from the other party. We cannot assure you that SPAH will determine that it can commercialize our technologies or that the goals required for us to obtain progress payments from SPAH will be achieved.
The Company has received no “progress payments” in relation to its Material Transfer Agreement with SPAH. Progress payments could potentially total $50,000 for the first cell line for which SPAH uses our technology and $25,000 for each use of the same technology thereafter. We do not know how many cell lines SPAH may have and we currently have no indication from SPAH that it intends to use any of our technologies in the foreseeable future.
Additionally, the Company also intends to sell a majority stake in these underlying assets via a non-core spin-out transaction.
Competition
In general, the pharmaceutical and biotechnology industries are intensely competitive, characterized by rapid advances in products and technology. A number of companies have developed and continue to develop products that address the areas we have targeted. Some of these companies are major pharmaceutical companies and biotechnology companies that are international in scope and very large in size, while others are niche players that may be less familiar but have been successful in one or more areas we are targeting. Existing or future pharmaceutical, device, or other competitors may develop products that accomplish similar functions to our technologies in ways that are less expensive, receive faster regulatory approval, or receive greater market acceptance than our products. Many of our competitors have been in existence for considerably longer than we have, have greater capital resources, broader internal structure for research, development, manufacturing and marketing, and are in many ways further along in their respective product cycles.
While it is possible that eventually we may compete directly with major pharmaceutical companies, we believe it is more likely that we will enter into joint development, marketing, or other licensure arrangements with such competitors. Eventually, we believe that we will be acquired.
We also have a number of market areas in common with traditional skincare cosmetics companies, but in contrast to these companies, our products are based on unique, proprietary formulations and approaches. For example, we are unaware of any products in our targeted OTC skincare markets that are similar to our Pure-ific product. Further, proprietary protection of our products may help limit or prevent market erosion until our patents expire.
Federal Regulation of Therapeutic Products
All of the prescription drugs we currently contemplate developing will require approval by the FDA prior to sales within the United States and by comparable foreign agencies prior to sales outside the United States. The FDA and comparable regulatory agencies impose substantial requirements on the manufacturing and marketing of pharmaceutical products and medical devices. These agencies and other entities extensively regulate, among other things, research and development activities and the testing, manufacturing, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion of our proposed products. While we attempt to minimize and avoid significant regulatory bars when formulating our products, some degree of regulation from these regulatory agencies is unavoidable. Some of the things we do to attempt to minimize and avoid significant regulatory bars include the following:
| • | Using chemicals and combinations already allowed by the FDA; |
| • | Using drugs that have been previously approved by the FDA and that have a long history of safe use; and |
| • | Using chemical compounds with known safety profiles |
The regulatory process required by the FDA, through which our drug or device products must pass successfully before they may be marketed in the U.S., generally involves the following:
| • | Preclinical laboratory and animal testing; |
| • | Submission of an application that must become effective before clinical trials may begin; |
18
Table of Contents
| • | Adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended indication; and |
| • | FDA approval to market a given product for a given indication after the appropriate application has been filed. |
For pharmaceutical products, preclinical tests include laboratory evaluation of the product, its chemistry, formulation and stability, as well as animal studies to assess the potential safety and efficacy of the product. Where appropriate (for example, for human disease indications for which there exist inadequate animal models), we will attempt to obtain preliminary data concerning safety and efficacy of proposed products using carefully designed human pilot studies. We will require sponsored work to be conducted in compliance with pertinent local and international regulatory requirements, including those providing for Institutional Review Board approval, national governing agency approval and patient informed consent, using protocols consistent with ethical principles stated in the Declaration of Helsinki and other internationally recognized standards. We expect any pilot studies to be conducted outside the United States; but if any are conducted in the United States, they will comply with applicable FDA regulations. Data obtained through pilot studies will allow us to make more informed decisions concerning possible expansion into traditional FDA-regulated clinical trials.
If the FDA is satisfied with the results and data from preclinical tests, it will authorize human clinical trials. Human clinical trials typically are conducted in three sequential phases which may overlap. Each of the three phases involves testing and study of specific aspects of the effects of the pharmaceutical on human subjects, including testing for safety, dosage tolerance, side effects, absorption, metabolism, distribution, excretion and clinical efficacy.
Phase 1 clinical trials include the initial introduction of an investigational new drug into humans. These studies are closely monitored and may be conducted in patients, but are usually conducted in healthy volunteer subjects. These studies are designed to determine the metabolic and pharmacologic actions of the drug in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. While the FDA can cause us to end clinical trials at any phase due to safety concerns, phase 1 clinical trials are primarily concerned with safety issues. We also attempt to obtain sufficient information about the drug’s pharmacokinetics and pharmacological effects during phase 1 clinical trial to permit the design of well-controlled, scientifically valid, phase 2 studies.
Phase 1 studies also evaluate drug metabolism, structure-activity relationships, and the mechanism of action in humans. These studies also determine which investigational drugs are used as research tools to explore biological phenomena or disease processes. The total number of subjects included in phase 1 studies varies with the drug, but is generally in the range of 20 to 80.
Phase 2 clinical trials include the early controlled clinical studies conducted to obtain some preliminary data on the effectiveness of the drug for a particular indication or indications in patients with the disease or condition. This phase of testing also helps determine the common short-term side effects and risks associated with the drug. Phase 2 studies are typically well-controlled, closely monitored, and conducted in a relatively small number of patients, usually involving several hundred people.
Phase 3 studies are expanded controlled and uncontrolled trials. They are performed after preliminary evidence suggesting effectiveness of the drug has been obtained in phase 2, and are intended to gather the additional information about effectiveness and safety that is needed to evaluate the overall benefit-risk relationship of the drug. Phase 3 studies also provide an adequate basis for extrapolating the results to the general population and transmitting that information in the physician labeling. Phase 3 studies usually include several hundred to several thousand people.
Applicable medical devices can be cleared for commercial distribution through a notification to the FDA under Section 510(k) of the applicable statute. The 510(k) notification must demonstrate to the FDA that the device is as safe and effective and substantially equivalent to a legally marketed or classified device that is currently in interstate commerce. Such devices may not require detailed testing. Certain high-risk devices that sustain human life, are of substantial importance in preventing impairment of human health, or that present a potential unreasonable risk of illness or injury, are subject to a more comprehensive FDA approval process initiated by filing a premarket approval, also known as a “PMA,” application (for devices) or accelerated approval (for drugs).
We have established a core clinical development team and have been working with outside FDA consultants to assist us in developing product-specific development and approval strategies, preparing the required submittals, guiding us through the regulatory process, and providing input to the design and site selection of human clinical studies. Historically, obtaining FDA approval for photodynamic therapies has been a challenge. Wherever possible, we intend to utilize lasers or other activating systems that have been previously approved by the FDA to mitigate the risk that our therapies will not be approved by the FDA. The FDA has considerable experience with lasers by virtue of having reviewed and acted upon many 510(k) and premarket approval filings submitted to it for various photodynamic and non-photodynamic therapy laser applications, including a large number of cosmetic laser treatment systems used by dermatologists.
19
Table of Contents
The testing and approval process requires substantial time, effort, and financial resources, and we may not obtain FDA approval on a timely basis, if at all. Success in preclinical or early-stage clinical trials does not assure success in later-stage clinical trials. The FDA or the research institution sponsoring the trials may suspend clinical trials or may not permit trials to advance from one phase to another at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Once issued, the FDA may withdraw a product approval if we do not comply with pertinent regulatory requirements and standards or if problems occur after the product reaches the market. If the FDA grants approval of a product, the approval may impose limitations, including limits on the indicated uses for which we may market a product. In addition, the FDA may require additional testing and surveillance programs to monitor the safety and/or effectiveness of approved products that have been commercialized, and the agency has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs. Further, later discovery of previously unknown problems with a product may result in restrictions on the product, including its withdrawal from the market.
Marketing our products abroad will require similar regulatory approvals by equivalent national authorities and is subject to similar risks. To expedite development, we may pursue some or all of our initial clinical testing and approval activities outside the United States, and in particular in those nations where our products may have substantial medical and commercial relevance. In some such cases, any resulting products may be brought to the U.S. after substantial offshore experience is gained. Accordingly, we intend to pursue any such development in a manner consistent with U.S. standards so that the resultant development data is maximally applicable for potential FDA approval.
OTC products are subject to regulation by the FDA and similar regulatory agencies, but the regulations relating to these products are much less stringent than those relating to prescription drugs and medical devices. The types of OTC products developed and previously sold by us only require that we follow cosmetic rules relating to labeling and the claims that we make about our product. The process for obtaining approval of prescription drugs with the FDA does not apply to the OTC products, which we have sold. The FDA can, however, require us to stop selling our product if we fail to comply with the rules applicable to our OTC products.
Employees
We currently employ four persons, all of whom are full-time employees. We currently engage four full-time consultants, including a lab technician, a contract research associate, an analytical chemist, and an information technology consultant. We also work with various vendors and disclose on our corporate website that we currently have human resources focused on our activities that equate to fifty-five (55) full-time equivalents, including our eight full-time employees and consultants.
Our executive officers and directors are:
H. Craig Dees, Ph.D., 63, has served as our Chief Executive Officer and as a member of our board of directors since we acquired PPI, a privately held Tennessee corporation on April 23, 2002. Before joining us, from 1997 to 2002 he served as senior member of the management team of Photogen Technologies, Inc., including serving as a member of the board of directors of Photogen from 1997 to 2000. Prior to joining Photogen, Dr. Dees served as a Group Leader at the Oak Ridge National Laboratory and as a senior member of the management teams of LipoGen Inc., a medical diagnostic company which used genetic engineering technologies to manufacture and distribute diagnostic assay kits for auto-immune diseases, and TechAmerica Group Inc., now a part of Boehringer Ingelheim Vetmedica, Inc., the U.S. animal health subsidiary of Boehringer Ingelheim GmbH, an international chemical and pharmaceutical company headquartered in Germany. He earned a Ph.D. in Molecular Virology from the University of Wisconsin–Madison in 1984.
Timothy C. Scott, Ph.D., 56, has served as our President and as a member of our board of directors since we acquired PPI on April 23, 2002. Prior to joining us, Dr. Scott was a senior member of the Photogen management team from 1997 to 2002, including serving as Photogen’s Chief Operating Officer from 1999 to 2002, as a director of Photogen from 1997 to 2000, and as interim CEO for a period in 2000. Before joining Photogen, he served as senior management of Genase LLC, a developer of enzymes for fabric treatment and held senior research and management positions at Oak Ridge National Laboratory. Dr. Scott earned a Ph.D. in Chemical Engineering from the University of Wisconsin–Madison in 1985.
Eric A. Wachter, Ph.D., 52, currently serves as our Chief Technology Officer since May 14, 2012 and prior to that served as Executive Vice President – Pharmaceuticals and as a member of our board of directors since we acquired PPI on April 23, 2002 until May 14, 2012. Prior to joining us, from 1997 to 2002 he was a senior member of the management team of Photogen, including serving as Secretary and a director of Photogen since 1997 and as Vice President and Secretary and a director of Photogen since 1999. Prior to joining Photogen, Dr. Wachter served as a senior research staff member with Oak Ridge National Laboratory. He earned a Ph.D. in Chemistry from the University of Wisconsin–Madison in 1988.
20
Table of Contents
Peter R. Culpepper, 55, was appointed to serve as our Chief Financial Officer in February 2004 and is also our Chief Operating Officer. Previously, Mr. Culpepper served as Chief Financial Officer for Felix Culpepper International, Inc. from 2001 to 2004; was a Registered Representative with AXA Advisors, LLC from 2002 to 2003; has served as Chief Accounting Officer and Corporate Controller for Neptec, Inc. from 2000 to 2001; has served in various Senior Director positions with Metromedia Affiliated Companies from 1998 to 2000; has served in various Senior Director and other financial positions with Paging Network, Inc. from 1993 to 1998; and has served in a variety of financial roles in public accounting and industry from 1982 to 1993. He earned a Masters in Business Administration in Finance from the University of Maryland–College Park in 1992. He earned an AAS in Accounting from the Northern Virginia Community College–Annandale, Virginia in 1985. He earned a B.A. in Philosophy from the College of William and Mary–Williamsburg, Virginia in 1982. He is a licensed Certified Public Accountant in both Tennessee and Maryland.
Equity Issuances and Financing During 2014
During the three months ended March 31, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $137,500. During the three months ended March 31, 2014, the Company issued 733,000 warrants to consultants in exchange for services. Consulting costs charged to operations were $900,317.
During the three months ended June 30, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $140,250. During the three months ended June 30, 2014, the Company issued 202,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $450,002. During the three months ended June 30, 2014, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $5,000,000. The Company accepted subscriptions, in the aggregate, for 2,000,000 shares of common stock and five year warrants to purchase 2,000,000 shares of common stock. Investors received five year fully vested warrants to purchase up to 100% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $3.00 per share. The purchase price for each share of common stock together with the warrants was $2.50. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $650,000 and issued five year fully vested warrants to purchase 300,000 shares of common stock with an exercise price of $2.50 to Network 1 Financial Securities, Inc., which represents 15% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc.
During the three months ended September 30, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $68,500. During the three months ended September 30, 2014, the Company issued 6,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $4,189. During the three months ended September 30, 2014, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $3,586,300. The Company accepted subscriptions, in the aggregate, for 3,586,300 shares of common stock and five year warrants to purchase 1,793,150 shares of common stock. Investors received five year fully vested warrants to purchase up to 50% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants was $1.00. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $466,219 and issued five year fully vested warrants to purchase 358,630 shares of common stock with an exercise price of $1.25 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc.
During the three months ended December 31, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $72,000. During the three months ended December 31, 2014, the Company issued 1,503,913 warrants to consultants in exchange for services. Consulting costs charged to operations were $966,819. During the three months ended December 31, 2014, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $4,198,300. The Company accepted subscriptions, in the aggregate, for 4,198,300 shares of common stock and five year warrants to purchase 2,099,150 shares of common stock. Investors received five year fully vested warrants to purchase up to 50% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants was $1.00. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $545,779 and issued five year fully vested warrants to purchase 419,830 shares of common stock with an exercise price of $1.25 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc.
21
Table of Contents
The issuances of the securities were exempt from the registration requirements of the Securities Act of 1933 by virtue of Section 4(a)(2) and Rule 506 promulgated under Regulation D thereunder as transactions not involving a public offering.
Available Information
Our website is located at www.pvct.com. We make available free of charge through this website our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed with or furnished to the SEC pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. Reference to our website does not constitute incorporation by reference of the information contained on the site and should not be considered part of this document.
All filings made by us with the SEC may be copied or read at the SEC’s Public Reference Room at 100 F Street NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room may be obtained by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC as we do. The website is http://www.sec.gov.
| ITEM 1A. | RISK FACTORS. |
Our business and its future performance may be affected by various factors, the most significant of which are discussed below.
We are a development stage company, have no prescription drug products approved for commercial sale, have incurred substantial losses, and expect to incur substantial losses and negative operating cash flow for the foreseeable future.
Our company is a development stage company that has no prescription drug products approved for commercial sale. We have never generated any substantial revenues and may never achieve substantial revenues or profitability. As of December 31, 2014, we have incurred net losses of $156 million in the aggregate since inception in January 2002. We expect to incur substantial losses and negative operating cash flow for the foreseeable future. We may never achieve or maintain profitability, even if we succeed in developing and commercializing one or more of our prescription drug candidates, OTC products, or non-core technologies. We also expect to continue to incur significant operating expenditures and anticipate that our operating and capital expenses may increase substantially in the foreseeable future as we:
| • | continue to develop and seek regulatory approval for our prescription drug candidates PV-10 and PH-10; |
| • | seek licensure of PV-10, PH-10, our OTC products, and our other non-core technologies; |
| • | further develop our non-core technologies; |
| • | implement additional internal systems and infrastructure; and |
| • | hire additional personnel. |
We also expect to experience negative operating cash flow for the foreseeable future as we fund our operating losses and any future capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability could negatively impact the value of our common stock.
All of our existing prescription drug candidates are in early stages of development. It may be several years, if ever, until we have a prescription drug product available for commercial resale. If we do not successfully develop and license or commercialize our prescription drug candidates, or sell or license our OTC products or non-core technologies, we will not achieve revenues or profitability in the foreseeable future, if at all. If we are unable to generate revenues or achieve profitability, we may be unable to continue our operations.
We may need additional capital to conduct our operations and commercialize and/or further develop our prescription drug candidates in 2016 and beyond, and our ability to obtain the necessary funding is uncertain.
We estimate that our existing capital resources will be sufficient to fund our current and planned operations until 2016. However, we may need additional capital in 2016 and beyond as we continue to develop and seek commercialization of our prescription drug candidates. We intend to proceed as rapidly as possible with licensure of PH-10 on the basis of our expanding phase 2 atopic dermatitis and psoriasis results, which continue to be developed. We potentially may license PV-10
22
Table of Contents
depending on the timing for the optimal deal structure for our stockholders. We are also focusing on PV-10 geographic licensing and partnering opportunities in such countries as China and India. We are also focusing on potential co-development partnering opportunities with combination of PV-10 and immune checkpoint blockade or systemic immunotherapy agents. We intend to also proceed as rapidly as possible with the sale or licensure of our OTC products and other non-core technologies. Although we believe that there is a reasonable basis for our expectation that we will become profitable due to both the licensure of PH-10 and PV-10, and the sale or licensure of our OTC products and non-core technologies, we cannot assure you that we will be able to achieve, or maintain, a level of profitability sufficient to meet our operating expenses.
We have based our estimate of capital needs on assumptions that may prove to be wrong, and we cannot assure you that estimates and assumptions will remain unchanged. For example, we are currently assuming that we will continue to operate without any significant staff or other resources expansion. We intend to acquire additional funding through public or private equity or debt financings or other financing sources that may be available. Additional financing may not be available on acceptable terms, or at all. As discussed in more detail below, additional equity financing could result in significant dilution to stockholders. Further, in the event that additional funds are obtained through licensing or other arrangements, these arrangements may require us to relinquish rights to some of our products, product candidates, and technologies that we would otherwise seek to develop and commercialize ourselves. If sufficient capital is not available, we may be required to delay, reduce the scope of, or eliminate one or more of our programs, any of which could have a material adverse effect on our business and may impair the value of our patents and other intangible assets.
Our prescription drug candidates are at an intermediary stage of development and may never obtain U.S. or international regulatory approvals required for us to commercialize our prescription drug candidates.
We will need approval of the FDA to commercialize our prescription drug candidates in the U.S. and approvals from the FDA equivalent regulatory authorities in foreign jurisdictions to commercialize our prescription drug candidates in those jurisdictions.
We are continuing to pursue clinical development of our most advanced prescription drug candidates, PV-10 and PH-10, for use as treatments for specific conditions. The continued and further development of these prescription drug candidates will require significant additional research, formulation and manufacture development, and pre-clinical and extensive clinical testing prior to their regulatory approval and commercialization. Pre-clinical and clinical studies of our prescription drug candidates may not demonstrate the safety and efficacy necessary to obtain regulatory approvals. Pharmaceutical and biotechnology companies have suffered significant setbacks in advanced clinical trials, even after experiencing promising results in earlier trials. Pharmaceutical drug and medical device products that appear to be promising at early stages of development may not reach the market or be marketed successfully for a number of reasons, including the following:
| • | a product may be found to be ineffective or have harmful side effects during subsequent pre-clinical testing or clinical trials, |
| • | a product may fail to receive necessary regulatory clearance, |
| • | a product may be too difficult to manufacture on a large scale, |
| • | a product may be too expensive to manufacture or market, |
| • | a product may not achieve broad market acceptance, |
| • | others may hold proprietary rights that will prevent a product from being marketed, and |
| • | others may market equivalent or superior products. |
Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our research and clinical approaches will result in drugs that the FDA considers safe for humans and effective for indicated uses. The FDA has substantial discretion in the drug approval process and may require us to conduct additional nonclinical and clinical testing or to perform post-marketing studies. The approval process may also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals may:
| • | delay commercialization of, and our ability to derive product revenues from, our product candidates; |
| • | impose costly procedures on us; and |
| • | diminish any competitive advantages that we may otherwise enjoy. |
23
Table of Contents
We do not expect any prescription drug and other product candidates that we are developing to be commercially available without a partner. Our research and product development efforts may not be successfully completed and may not result in any successfully commercialized products. Further, after commercial introduction of a new product, discovery of problems through adverse event reporting could result in restrictions on the product, including withdrawal from the market and, in certain cases, civil or criminal penalties.
Even if we comply with all FDA requests, we cannot be sure that we will ever obtain regulatory clearance for any of our prescription drug or other product candidates. Failure to obtain FDA approval of any of our product candidates will severely undermine our business by reducing our number of salable products and, therefore, corresponding product revenues.
In foreign jurisdictions, we must receive approval from the appropriate regulatory authorities before we can commercialize our drugs. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval procedures described above.
We are subject to securities class action lawsuits that could adversely affect our business. This litigation, and potential similar or related litigation, could result in substantial damages and may divert management’s time and attention from our business.
Beginning on May 27, 2014, three putative securities class action lawsuits (the “Federal Class Actions”) were commenced in the United States District Court for the Middle District of Tennessee against us, and certain of our officers and directors, alleging violations by the defendants of Sections 10(b) and 20(a) of the Exchange Act and Rule 10b-5 promulgated thereunder. The Federal Class Actions allege, among other things, that the defendants made false and materially misleading statements and failed to disclose material information regarding our application to the FDA for BTD of PV-10.
On July 9, 2014, the Company and the Federal Class Action plaintiffs filed joint motions to consolidate the cases and transfer them to United States District Court for the Eastern District of Tennessee. By order dated July 16, 2014, the United States District Court for the Middle District of Tennessee consolidated the Federal Class Actions and transferred them to the United States District Court for the Eastern District of Tennessee. On November 26, 2014, the United States District Court for the Eastern District of Tennessee (the “Court”) entered an order appointing Fawwaz Hamati as the Lead Plaintiff in the Securities Litigation, with the Law Firm of Glancy Binkow & Goldberg, LLP as counsel to Lead Plaintiff. On February 3, 2015, the Court entered an order compelling the Lead Plaintiff to file a consolidated amended complaint within 60 days of entry of the order. As of March 4, 2015, the Lead Plaintiff has yet to file a consolidated amended complaint.
In addition, on June 4, 2014, a shareholder derivative lawsuit captioned Hurtado v. Provectus Biopharmaceuticals, Inc., et al. was filed derivatively on behalf of the Company against H. Craig Dees, Timothy C. Scott, Jan E. Koe, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, the “Individual Defendants”), and against the Company as a nominal defendant in the United States District Court for the Middle District of Tennessee (the “Hurtado Shareholder Derivative Lawsuit”). The Hurtado Shareholder Derivative Lawsuit alleges (i) breach of fiduciary duties, and (ii) abuse of control, both claims based on the Plaintiff’s allegations that the Individual Defendants recklessly permitted the Company to disclose false and misleading information and failed to implement adequate controls and procedures to ensure the accuracy of the Company’s disclosures.
On July 25, 2014, the United States District Court for the Middle District of Tennessee entered an order transferring the Hurtado Shareholder Derivative Lawsuit to the United States District Court for the Eastern District of Tennessee and, in light of the pending Federal Class Actions, relieving the Individual Defendants from responding to the complaint in the Hurtado Shareholder Derivative Lawsuit pending further order from the United States District Court for the Eastern District of Tennessee.
On October 24, 2014, Paul Montiminy brought a shareholder derivative complaint on behalf of the Company in the United States District Court for the Eastern District of Tennessee (the “Montiminy Shareholder Derivative Lawsuit”) against the Individual Defendants. Like the Hurtado Shareholder Derivative Lawsuit, the Montiminy Shareholder Derivative Lawsuit alleges (i) breach of fiduciary duties and (ii) gross mismanagement of the assets and business of the Company, both claims based on Mr. Montiminy’s allegations that the Individual Defendants recklessly permitted the Company to make certain false and misleading disclosures regarding the likelihood that PV-10 would qualify for BTD.
On December 29, 2014, the United States District Court for the Eastern District of Tennessee (the “Court”) entered an order consolidating the Hurtado Shareholder Derivative Lawsuit and the Montiminy Derivative Lawsuit. On February 25, 2015, the parties submitted a proposed agreed order staying the Hurtado and Montiminy Shareholder Derivative Lawsuits until the Court issues a ruling on the anticipated motion to dismiss the amended consolidated complaint to be filed in the Securities Litigation. As of March 4, 2015, the Court has not yet entered the proposed agreed order staying the Hurtado and Montiminy Shareholder Derivative Lawsuits.
24
Table of Contents
Finally, on October 28, 2014, Chris Foley, derivatively on behalf of the Company, filed a shareholder derivative complaint in the Chancery Court of Knox County, Tennessee against the Individual Defendants and against the Company as a nominal defendant (the “Foley Shareholder Derivative Lawsuit”). The Foley Shareholder Derivative Lawsuit asserts the exact same facts and legal claims as the Montiminy Shareholder Derivative Lawsuit. Since the filing of the Foley Shareholder Derivative Lawsuit, the parties have submitted a proposed agreed order staying the Foley Derivative Lawsuit until the United States District Court for the Eastern District of Tennessee issues a ruling on the anticipated motion to dismiss the amended consolidated complaint to be filed in the Securities Litigation.
In each of the three Shareholder Derivative Lawsuits, the Company is a nominal defendant only. As such, the plaintiffs seek relief from the Individual Defendants, but not the Company itself.
We intend to defend these actions vigorously. We are currently unable to estimate a range of payments, if any, that we may be required to pay or may agree to pay with respect to the Federal Class Actions, the Hurtado Shareholder Derivative Lawsuit, the Montiminy Shareholder Derivative Lawsuit, and the Foley Shareholder Derivative Lawsuit. We believe that the resolution of these suits will not result in a material adverse effect to our consolidated financial statements. However, due to the inherent uncertainties that accompany litigation of this nature, there can be no assurance that we will be successful, and an adverse resolution of any of the lawsuits could have a material adverse effect on our consolidated financial statements. Furthermore, these actions may divert management’s time and attention from our business, and we could be forced to expend significant resources and pay significant costs and expenses, including legal fees, in connection with defending the lawsuits.
We did not obtain and may not obtain or maintain the benefits associated with breakthrough therapy designation.
On March 21, 2014, we submitted a request for breakthrough therapy designation (BTD) to the FDA for PV-10 in the treatment of metastatic melanoma in the United States. The FDA denied the request in May 2014, but stated that a new request may be submitted if we obtain new clinical evidence that supports BTD. Accordingly, we are not entitled to the benefits of BTD, including expedited development and review of PV-10 in the treatment of melanoma.
If we resubmit such request for BTD, we may not be granted BTD, or even if granted, we may not receive the benefits associated with BTD. This may result from a failure to maintain breakthrough therapy status if PV-10 is no longer considered to be a breakthrough therapy. For example, a drug’s development program may be granted BTD using early clinical testing that shows a much higher response rate than available therapies. However, subsequent interim data derived from a larger study may show a response that is substantially smaller than the response seen in early clinical testing. Another example is where BTD is granted to two drugs that are being developed for the same use. If one of the two drugs gains traditional approval, the other would not retain its designation unless its sponsor provided evidence that the drug may demonstrate substantial improvement over the recently approved drug. When BTD is no longer supported by emerging data or the designated drug development program is no longer being pursued, the FDA may choose to send a letter notifying the sponsor that the program is no longer designated as a BTD program.
We depend on the successful completion of clinical trials for our product candidates, including PV-10. The positive clinical results obtained for our product candidates in prior clinical studies may not be repeated in future clinical studies.
Before obtaining regulatory approval for the sale of our product candidates, including PV-10, we must conduct additional clinical trials to demonstrate the safety and efficacy of our product candidates. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more of our clinical trials can occur at any stage of testing. The outcome of pre-clinical testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, pre-clinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in pre-clinical studies and clinical trials have nonetheless failed to obtain marketing approval for their products.
In October 2012, we presented final top-line data from the phase 2 trial of PV-10 for metastatic melanoma, and in March 2014, applied for BTD with the FDA, which was subsequently denied pending new clinical evidence that supports BTD. We (i) are conducting an expanded phase 1 trial for PV-10 for metastatic liver cancer, which is expected to be completed in early 2015; (ii) have completed a phase 1 clinical study for PV-10 for recurrent breast cancer; (iii) are conducting a phase 1 trial for
25
Table of Contents
PV-10 in an investigator initial study to ascertain the feasibility of detecting immune cell infiltrates in injected melanoma tumors which is expected to be completed in early 2015; (iv) are conducting a phase 2 clinical trial for mechanism of action of PH-10 for psoriasis; (v) have completed multiple phase 2 clinical trials for PH-10 for psoriasis and atopic dermatitis; and (vi) expect to commence a phase 3 clinical trial to assess response to intralesional PV-10 versus that of systemic chemotherapy in patients with disease confined to cutaneous and subcutaneous sites. Meetings with scientific advisors, investigators and advocates in the field have led us to expect a starting date for the phase 3 clinical study sometime in the first quarter of 2015. However, we have never conducted a phase 3 clinical trial. The positive results we have seen to date in our phase 2 clinical trials of PV-10 for metastatic melanoma do not ensure that later clinical trials will demonstrate similar results. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy characteristics despite having progressed satisfactorily through preclinical studies and initial clinical testing. A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience, have suffered significant setbacks in phase 3 clinical development; even after seeing promising results in earlier clinical trials.
We may experience a number of unforeseen events during clinical trials for our product candidates, including PV-10, that could delay or prevent the commencement and/or completion of our clinical trials, including the following:
| • | regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
| • | the clinical study protocol may require one or more amendments delaying study completion; |
| • | clinical trials of our product candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs; |
| • | the number of subjects required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be insufficient or slower than we anticipate or subjects may drop out of these clinical trials at a higher rate than we anticipate; |
| • | clinical investigators or study subjects fail to comply with clinical study protocols; |
| • | trial conduct and data analysis errors may occur, including, but not limited to, data entry and/or labeling errors; |
| • | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; |
| • | we might have to suspend or terminate clinical trials of our product candidates for various reasons, including a finding that the subjects are being exposed to unacceptable health risks; |
| • | regulators or institutional review boards may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements; |
| • | the cost of clinical trials of our product candidates may be greater than we anticipate; |
| • | the supply or quality of our clinical trial materials or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; and |
| • | our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators to suspend or terminate the trials. |
We expect our research and development expenses to increase in connection with our ongoing activities, particularly if we commence a phase 3 clinical trial with respect to PV-10 as planned, and undertake additional clinical trials of our other product candidates. Because successful development of our product candidates is uncertain, we are unable to estimate the actual funds required to complete research and development and commercialize our products under development; however, we believe we have sufficient cash on hand to fund the planned phase 3 clinical trial with respect to PV-10.
Negative or inconclusive results of our future clinical trials of PV-10, or any other clinical trial we conduct, could cause the FDA to require that we repeat or conduct additional clinical studies. Despite the results reported in earlier clinical trials for PV-10, we do not know whether any clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market our product candidates, including PV-10. If later stage clinical trials do not produce favorable results, our ability to obtain regulatory approval for our product candidates, including PV-10, may be adversely impacted.
26
Table of Contents
Delays in clinical trials are common and have many causes, and any delay could result in increased costs to us and jeopardize or delay our ability to obtain regulatory approval.
Clinical testing is expensive, difficult to design and implement, can take many years to complete, and is uncertain as to outcome. We may experience delays in clinical trials at any stage of development and testing of our product candidates. Our planned clinical trials may not begin on time, have an effective design, enroll a sufficient number of subjects, or be completed on schedule, if at all.
Events which may result in delays or unsuccessful completion of clinical trials, including our future clinical trials for PV-10, include the following:
| • | inability to raise funding, if necessary, to initiate or continue a trial; |
| • | delays in obtaining regulatory approval to commence a trial; |
| • | delays in reaching agreement with the FDA on final trial design; |
| • | imposition of a clinical hold following an inspection of our clinical trial operations or trial sites by the FDA or other regulatory authorities; |
| • | delays in reaching agreement on acceptable terms with prospective contract research organizations (CROs) and clinical trial sites; |
| • | delays in obtaining required institutional review board (IRB) approval at each site; |
| • | delays in recruiting suitable patients to participate in a trial; |
| • | delays in having subjects complete participation in a trial or return for post-treatment follow-up; |
| • | delays caused by subjects dropping out of a trial due to side effects or otherwise; |
| • | delays caused by clinical sites dropping out of a trial; |
| • | time required to add new clinical sites; and |
| • | delays by our contract manufacturers to produce and deliver sufficient supply of clinical trial materials. |
If initiation or completion of any of our clinical trials for our product candidates, including PV-10, are delayed for any of the above reasons, our development costs may increase, the approval process could be delayed, any periods during which we may have the exclusive right to commercialize our product candidates may be reduced and our competitors may bring products to market before us. Any of these events could impair our ability to generate revenues from product sales and impair our ability to generate regulatory and commercialization milestones and royalties, all of which could have a material adverse effect on our business.
Clinical trials are very expensive, time consuming and difficult to design and implement.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time consuming. We estimate that current or future clinical trials of our prescription drug candidates will take additional years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by several factors, including:
| • | unforeseen safety issues; |
| • | determination of dosing issues; |
| • | lack of effectiveness during clinical trials; |
| • | slower than expected rates of patient recruitment; |
| • | inability to monitor patients adequately during or after treatment; and |
| • | inability or unwillingness of medical investigators to follow our clinical protocols. |
In addition, we or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our submissions or the conduct of these trials.
27
Table of Contents
The results of our clinical trials may not support our claims concerning our prescription drug candidates.
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our claims concerning our prescription drug candidates. Success in nonclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and nonclinical testing. The clinical trial process may fail to demonstrate that our product candidates are safe for humans or effective for indicated uses. This failure would cause us to abandon a product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay our ability to commercialize our product candidates and generate product revenues. In addition, we anticipate that our clinical trials will involve only a small patient population. Accordingly, the results of such trials may not be indicative of future results over a larger patient population.
Physicians and patients may not accept and use our prescription drug candidates.
Even if the FDA approves our prescription drug candidates, physicians and patients may not accept and use them. Acceptance and use of our prescription drug products will depend upon a number of factors including:
| • | perceptions by members of the healthcare community, including physicians, about the safety and effectiveness of our prescription drug products; |
| • | cost-effectiveness of our prescription drug products relative to competing products; |
| • | availability of reimbursement for our prescription drug products from government or other healthcare payers; and |
| • | effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any. |
Because we expect sales or licensure of our prescription drug candidates, if approved, to generate substantially all of our revenues for the foreseeable future, the failure of any of these drugs to find market acceptance would harm our business and could require us to seek additional financing.
We have no sales, marketing or distribution capabilities for our prescription drug candidates or our OTC products and non-core technologies.
We currently have no sales, marketing or distribution capabilities. We do not anticipate having the resources in the foreseeable future to allocate to the sales and marketing of our prescription drug candidates or our OTC products and non-core technologies. Our future success depends, in part, on our ability to enter into and maintain such collaborative relationships, the collaborator’s strategic interest in the products under development and such collaborator’s ability to successfully market and sell any such products. We intend to proceed as rapidly as possible with licensure of PH-10 on the basis of our Phase 2 atopic dermatitis and psoriasis results, which are in process of being further developed. We have determined that the most efficient use of our capital in further developing our OTC products is to license the products. There can be no assurance that we will be able to establish or maintain relationships with third party collaborators or develop in-house sales and distribution capabilities. To the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties, and there can be no assurance that such efforts will be successful. In addition, there can also be no assurance that we will be able to market and sell our product in the United States or overseas.
We cannot be sure that our OTC products or non-core technologies will be licensed or sold in the marketplace.
In order for our OTC products to become commercially successful and our non-core technologies to be further developed, we must license or sell those products and technologies. We have been discussing this strategy with interested groups, though we cannot be sure that we will be successful in licensing or selling such products or technologies.
Competition in the prescription pharmaceutical and biotechnology industries is intense, and we may be unable to succeed if our competitors have more funding or better marketing.
The pharmaceutical and biotechnology industries are intensely competitive. Other pharmaceutical and biotechnology companies and research organizations currently engage in or have in the past engaged in research efforts related to treatment of dermatological conditions or cancers of the skin, liver and breast, which could lead to the development of products or therapies that could compete directly with the prescription drug and other product candidates, and OTC products that we are seeking to develop and market.
28
Table of Contents
Many companies are also developing alternative therapies to treat cancer and dermatological conditions and, in this regard, are our competitors. Many of the pharmaceutical companies developing and marketing these competing products have significantly greater financial resources and expertise than we do in:
| • | research and development; |
| • | manufacturing; |
| • | preclinical and clinical testing; |
| • | obtaining regulatory approvals; and |
| • | marketing. |
Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Academic institutions, government agencies, and other public and private research organizations may also conduct research, seek patent protection, and establish collaborative arrangements for research, clinical development, and marketing of products similar to ours. These companies and institutions compete with us in recruiting and retaining qualified scientific and management personnel as well as in acquiring technologies complementary to our programs.
In addition to the above factors, we expect to face competition in the following areas:
| • | product efficacy and safety; |
| • | the timing and scope of regulatory consents; |
| • | availability of resources; |
| • | reimbursement coverage; |
| • | price; and |
| • | patent position, including potentially dominant patent positions of others. |
Since our prescription candidates PV-10 and PH-10 have not yet been approved by the FDA or introduced to the marketplace, we cannot estimate what competition these products might face when they are finally introduced, if at all. We cannot assure you that these products will not face significant competition for other prescription drugs and generic equivalents.
If we are unable to secure or enforce patent rights, trademarks, trade secrets or other intellectual property our business could be harmed.
We may not be successful in securing or maintaining proprietary patent protection for our products and technologies we develop or license. In addition, our competitors may develop products similar to ours using methods and technologies that are beyond the scope of our intellectual property protection, which could reduce our anticipated sales. While some of our products have proprietary patent protection, a challenge to these patents can subject us to expensive litigation. Litigation concerning patents, other forms of intellectual property, and proprietary technology is becoming more widespread and can be protracted and expensive and can distract management and other personnel from performing their duties.
We also rely upon trade secrets, unpatented proprietary know-how, and continuing technological innovation to develop a competitive position. We cannot assure you that others will not independently develop substantially equivalent proprietary technology and techniques or otherwise gain access to our trade secrets and technology, or that we can adequately protect our trade secrets and technology.
If we are unable to secure or enforce patent rights, trademarks, trade secrets, or other intellectual property, our business, financial condition, results of operations and cash flows could be materially adversely affected. If we infringe on the intellectual property of others, our business could be harmed.
We could be sued for infringing patents or other intellectual property that purportedly cover products and/or methods of using such products held by persons other than us. Litigation arising from an alleged infringement could result in removal from the market, or a substantial delay in, or prevention of, the introduction of our products, any of which could have a material adverse effect on our business, financial condition, results of operations, and cash flows.
If we do not update and enhance our technologies, they will become obsolete.
The pharmaceutical market is characterized by rapid technological change, and our future success will depend on our ability to conduct successful research in our fields of expertise, to discover new technologies as a result of that research, to develop
29
Table of Contents
products based on our technologies, and to commercialize those products. While we believe that our current technology is adequate for our present needs, if we fail to stay at the forefront of technological development, we will be unable to compete effectively. Our competitors are using substantial resources to develop new pharmaceutical technologies and to commercialize products based on those technologies. Accordingly, our technologies may be rendered obsolete by advances in existing technologies or the development of different technologies by one or more of our current or future competitors.
If we lose any of our key personnel, we may be unable to successfully execute our business plan.
Our business is presently managed by four key employees:
| • | H. Craig Dees, Ph.D., our Chief Executive Officer; |
| • | Timothy C. Scott, Ph.D., our President; |
| • | Eric A. Wachter, Ph.D. our Chief Technology Officer; and |
| • | Peter R. Culpepper, CPA, MBA, our Chief Financial Officer and Chief Operating Officer. |
In addition to their responsibilities for management of our overall business strategy, Drs. Dees, Scott and Wachter are our chief researchers in the fields in which we are developing and planning to develop our prescription drug and other product candidates, and our OTC products. The loss of any of these key employees could have a material adverse effect on our operations, and our ability to execute our business plan might be negatively impacted. Any of these key employees may leave their employment with us if they choose to do so, and we cannot assure you that we would be able to hire similarly qualified employees if any of our key employees should choose to leave.
Because we have only four employees in total, our management may be unable to successfully manage our business.
In order to successfully execute our business plan, our management must succeed in all of the following critical areas:
| • | Researching diseases and possible therapies in the areas of dermatology and skin care, oncology, and biotechnology; |
| • | Developing our prescription drug and other product candidates, and OTC products based on our research; |
| • | Marketing and selling developed products; |
| • | Obtaining additional capital to finance research, development, production, and marketing of our products; and |
| • | Managing our business as it grows. |
As discussed above, we currently have only four employees, all of whom are full-time employees. The greatest burden of succeeding in the above areas, therefore, falls on Drs. Dees, Scott, Wachter, and Mr. Culpepper. Focusing on any one of these areas may divert their attention from our other areas of concern and could affect our ability to manage other aspects of our business. We cannot assure you that our management will be able to succeed in all of these areas or, even if we do so succeed, that our business will be successful as a result. We have added, including our employees, a total of fifty-five (55) human resources on a full-time equivalent basis. While we have not historically had difficulty in attracting employees, our small size and limited operating history may make it difficult for us to attract and retain employees in the future, which could further divert management’s attention from the operation of our business.
The market price of our common stock has been highly volatile due to several factors that will continue to affect the price of our common stock.
Our common stock has traded as low as $0.30 per share and as high as $6.03 per share during the period beginning on January 1, 2013 and ending on December 31, 2014. We believe that our common stock is subject to wide price fluctuations because of several factors, including:
| • | absence of meaningful earnings and ongoing need for external financing; |
| • | a relatively thin trading market for our common stock, which causes trades of small blocks of stock to have a significant impact on our stock price; |
| • | general volatility of the stock market and the market prices of other publicly-traded companies; and |
| • | investor sentiment regarding equity markets generally, including public perception of corporate ethics and governance and the accuracy and transparency of financial reporting. |
Financings that may be available to us under current market conditions frequently involve sales at prices below the prices at which our common stock trades on the NYSE MKT, as well as the issuance of warrants or convertible equity or debt that
30
Table of Contents
require exercise or conversion prices that are calculated in the future at a discount to the then market price of our common stock. The current economic downturn has made the financings available to development-stage companies like us more dilutive in nature than they would otherwise be.
Any agreement to sell, or convert debt or equity securities into, our common stock at a future date and at a price based on the then current market price will provide an incentive to the investor or third parties to sell our common stock short to decrease the price and increase the number of shares they may receive in a future purchase, whether directly from us or in the market.
Our stock price is below $5.00 per share and is treated as a “penny stock”, which places restrictions on broker-dealers recommending the stock for purchase.
Our common stock is defined as “penny stock” under the Exchange Act and its rules. The SEC has adopted regulations that define “penny stock” to include common stock that has a market price of less than $5.00 per share, subject to certain exceptions. These rules include the following requirements:
| • | broker-dealers must deliver, prior to the transaction, a disclosure schedule prepared by the SEC relating to the penny stock market; |
| • | broker-dealers must disclose the commissions payable to the broker-dealer and its registered representative; |
| • | broker-dealers must disclose current quotations for the securities; and |
| • | a broker-dealer must furnish its customers with monthly statements disclosing recent price information for all penny stocks held in the customer’s account and information on the limited market in penny stocks. |
Additional sales practice requirements are imposed on broker-dealers who sell penny stocks to persons other than established customers and accredited investors. For these types of transactions, the broker-dealer must make a special suitability determination for the purchaser and must have received the purchaser’s written consent to the transaction prior to sale. If our common stock remains subject to these penny stock rules these disclosure requirements may have the effect of reducing the level of trading activity in the secondary market for our common stock. As a result, fewer broker-dealers may be willing to make a market in our stock, which could affect a shareholder’s ability to sell their shares.
Future sales by our stockholders may adversely affect our stock price and our ability to raise funds in new stock offerings.
Sales of our common stock in the public market following any prospective offering could lower the market price of our common stock. Sales may also make it more difficult for us to sell equity securities or equity-related securities in the future at a time and price that our management deems acceptable. The recent economic downturn has made the financings available to development-stage companies like us more dilutive in nature than they would otherwise be.
We currently intend to retain all of our future earnings rather than pay a cash dividend.
We have never declared or paid cash dividends on our common stock. We currently intend to retain all of our future earnings, if any, for use in our business and therefore do not anticipate paying any cash dividends on our common stock in the foreseeable future.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS. |
None.
| ITEM 2. | PROPERTIES. |
We currently lease approximately 6,000 square feet of space outside of Knoxville, Tennessee for our corporate office and operations. Our monthly rental charge for these offices is approximately $5,000 per month, and the lease is on an annual basis, renewable for one year at our option. We have a lease commitment of $0 as of December 31, 2014. We believe that these offices generally are adequate for our needs currently and in the immediate future.
| ITEM 3. | LEGAL PROCEEDINGS. |
Except as described below, we are not involved in any legal proceedings nor are we party to any pending claims that we believe could reasonably be expected to have a material adverse effect on our business, financial condition, or results of operations.
31
Table of Contents
Kleba Shareholder Derivative Lawsuit
On January 2, 2013, Glenn Kleba, derivatively on behalf of the Company, filed a shareholder derivative complaint in the Circuit Court for the State of Tennessee, Knox County (the “Court”), against H. Craig Dees, Timothy C. Scott, Eric A. Wachter, and Peter R. Culpepper (collectively, the “Executives”), Stuart Fuchs, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, together with the Executives, the “Individual Defendants”), and against the Company as a nominal defendant (the “Shareholder Derivative Lawsuit”). The Shareholder Derivative Lawsuit alleged (i) breach of fiduciary duties, (ii) waste of corporate assets, and (iii) unjust enrichment, all three claims based on Mr. Kleba’s allegations that the defendants authorized and/or accepted stock option awards in violation of the terms of the Company’s 2002 Stock Plan (the “Plan”) by issuing stock options in excess of the amounts authorized under the Plan and delegated to defendant H. Craig Dees the sole authority to grant himself and the other Executives cash bonuses that Mr. Kleba alleges to be excessive.
In April 2013, the Company’s Board of Directors appointed a special litigation committee to investigate the allegations of the Shareholder Derivative Complaint and make a determination as to how the matter should be resolved. The special litigation committee conducted its investigation, and proceedings in the case were stayed pending the conclusion of the committee’s investigation. The Company has established a reserve of $100,000 for potential liabilities because such is the amount of the self-insured retention of its insurance policy. On February 21, 2014, an Amended Shareholder Derivative Complaint was filed which added Don B. Dale (“Mr. Dale”) as a plaintiff.
On March 6, 2014, the Company filed a Joint Notice of Settlement (the “Notice of Settlement”) in the Shareholder Derivative Lawsuit. In addition to the Company, the parties to the Notice of Settlement are Mr. Kleba, Mr. Dale and the Individual Defendants.
On June 6, 2014, the Company, in its capacity as a nominal defendant, entered into a Stipulated Settlement Agreement and Mutual Release (the “Settlement”) in the Shareholder Derivative Lawsuit. In addition to the Company and the Individual Defendants, Plaintiffs Glenn Kleba and Don B. Dale are parties to the Settlement.
By entering into the Settlement, the settling parties have resolved the derivative claims to their mutual satisfaction. The Individual Defendants have not admitted the validity of any claims or allegations and the settling plaintiffs have not admitted that any claims or allegations lack merit or foundation. Under the terms of the Settlement, (i) the Executives each agreed (A) to re-pay to the Company $2.24 Million of the cash bonuses they each received in 2010 and 2011, which amount equals 70% of such bonuses or an estimate of the after-tax net proceeds to each Executive; provided, however, that subject to certain terms and conditions set forth in the Settlement, the Executives are entitled to a 2:1 credit such that total actual repayment may be $1.12 Million each; (B) to reimburse the Company for 25% of the actual costs, net of recovery from any other source, incurred by the Company as a result of the Shareholder Derivative Lawsuit; and (C) to grant to the Company a first priority security interest in 1,000,000 shares of the Company’s common stock owned by each such Executive to serve as collateral for the amounts due to the Company under the Settlement; (ii) Drs. Dees and Scott and Mr. Culpepper agreed to retain incentive stock options for 100,000 shares but shall forfeit 50% of the nonqualified stock options granted to each such Executive in both 2010 and 2011. The Settlement also requires that each of the Executives enter into new employment agreements with the Company, which were entered into on April 28, 2014, and that the Company adhere to certain corporate governance principles and processes in the future. Under the Settlement, Messrs. Fuchs and Smith and Dr. McMasters have each agreed to pay the Company $25,000 in cash, subject to reduction by such amount that the Company’s insurance carrier pays to the Company on behalf of such defendant pursuant to such defendant’s directors and officers liability insurance policy. The Settlement also provides for an award to plaintiffs’ counsel of attorneys’ fees and reimbursement of expenses in connection with their role in this litigation, subject to Court approval.
On July 24, 2014, the Court approved the terms of the proposed Settlement and awarded $911,000 to plaintiffs’ counsel for attorneys’ fees and reimbursement of expenses in connection with their role in the Shareholder Derivative Lawsuit. The payment to plaintiff’s counsel was made by the Company during October 2014 and is recorded as other current assets at December 31, 2014. The Company is seeking reimbursement of the full amount from insurance and if the full amount is not received from insurance, the amount remaining will be reimbursed to the Company from the Individual Defendants.
On October 3, 2014, the Settlement was effective and stock options for Drs. Dees and Scott and Mr. Culpepper were rescinded, totaling 2,800,000. At December 31, 2014, a Gain on Settlement of $4,178,345, net of discount, was recorded for the total due from the Executives. A Short-term Receivable was recorded for $733,333 and a Long-term Receivable was recorded for $3,378,345. A discount for implied interest of $301,655 was recorded as an offset to the Gain on Settlement in the consolidated statements of operations. $66,667 was repaid by the Executives as of December 31, 2014. The cash settlement amounts will be repaid to the Company over a period of five years with the first payment due in October 2015 and the final payment is expected to be received by October 3, 2019.
32
Table of Contents
Class Action Lawsuits
On May 27, 2014, Cary Farrah and James H. Harrison, Jr., individually and on behalf of all others similarly situated (the “Farrah Case”), and on May 29, 2014, each of Paul Jason Chaney, individually and on behalf of all others similarly situated (the “Chaney Case”), and Jayson Dauphinee, individually and on behalf of all others similarly situated (the “Dauphinee Case”) (the plaintiffs in the Farrah Case, the Chaney Case and the Dauphinee Case collectively referred to as the “Plaintiffs”), each filed a class action lawsuit in the United States District Court for the Middle District of Tennessee against the Company, H. Craig Dees, Timothy C. Scott and Peter R. Culpepper (the “Defendants”) alleging violations by the Defendants of Sections 10(b) and 20(a) of the Exchange Act and Rule 10b-5 promulgated thereunder. Specifically, the Plaintiffs in each of the Farrah Case, the Chaney Case and the Dauphinee Case allege that the Defendants are liable for making false statements and failing to disclose adverse facts known to them about the Company, in connection with the Company’s application to the FDA for Breakthrough Therapy Designation (“BTD”) of the Company’s melanoma drug, PV-10, in the Spring of 2014, and the FDA’s subsequent denial of the Company’s application for BTD. The Company intends to defend vigorously against all claims in these complaints. However, in view of the inherent uncertainties of litigation and the early stage of this litigation, the outcome of these cases cannot be predicted at this time. Likewise, the amount of any potential loss cannot be reasonably estimated. No amounts have been recorded in the consolidated financial statements as the outcome of these cases cannot be predicted and the amount of any potential loss is not estimable at this time.
On July 9, 2014, the Plaintiffs and the Defendants filed joint motions in the Farrah Case, the Chaney Case and the Dauphinee Case to consolidate the cases and transfer them to United States District Court for the Eastern District of Tennessee. By order dated July 16, 2014, the United States District Court for the Middle District of Tennessee entered an order consolidating the Farrah Case, the Chaney Case and the Dauphinee Case (collectively and, as consolidated, the “Securities Litigation”) and transferred the Securities Litigation to the United States District Court for the Eastern District of Tennessee.
On November 26, 2014, the United States District Court for the Eastern District of Tennessee (the “Court”) entered an order appointing Fawwaz Hamati as the Lead Plaintiff in the Securities Litigation, with the Law Firm of Glancy Binkow & Goldberg, LLP as counsel to Lead Plaintiff. On February 3, 2015, the Court entered an order compelling the Lead Plaintiff to file a consolidated amended complaint within 60 days of entry of the order. As of March 4, 2015, the Lead Plaintiff has yet to file a consolidated amended complaint.
Hurtado Shareholder Derivative Lawsuit
On June 4, 2014, Karla Hurtado, derivatively on behalf of the Company, filed a shareholder derivative complaint in the United States District Court for the Middle District of Tennessee against H. Craig Dees, Timothy C. Scott, Jan E. Koe, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, the “Individual Defendants”), and against the Company as a nominal defendant (the “Hurtado Shareholder Derivative Lawsuit”). The Hurtado Shareholder Derivative Lawsuit alleges (i) breach of fiduciary duties and (ii) abuse of control, both claims based on Ms. Hurtado’s allegations that the Individual Defendants (a) recklessly permitted the Company to make false and misleading disclosures and (b) failed to implement adequate controls and procedures to ensure the accuracy of the Company’s disclosures.
On July 25, 2014, the United States District Court for the Middle District of Tennessee entered an order transferring the case to the United States District Court for the Eastern District of Tennessee and, in light of the pending Securities Litigation, relieving the Individual Defendants from responding to the complaint in the Hurtado Shareholder Derivative Lawsuit pending further order from the United States District Court for the Eastern District of Tennessee.
As a nominal defendant, no relief is sought against the Company itself in the Hurtado Shareholder Derivative Lawsuit.
Montiminy Shareholder Derivative Lawsuit
On October 24, 2014, Paul Montiminy brought a shareholder derivative complaint on behalf of the Company in the United States District Court for the Eastern District of Tennessee (the “Montiminy Shareholder Derivative Lawsuit”) against H. Craig Dees, Timothy C. Scott, Jan E. Koe, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, the “Individual Defendants”). Like the Hurtado Shareholder Derivative Lawsuit, the Montiminy Shareholder Derivative Lawsuit alleges (i) breach of fiduciary duties and (ii) gross mismanagement of the assets and business of the Company, both claims based on Mr. Montiminy’s allegations that the Individual Defendants recklessly permitted the Company to make certain false and
33
Table of Contents
misleading disclosures regarding the likelihood that the Company’s melanoma drug, PV-10, would qualify for BTD. As a practical matter, the factual allegations and requested relief in the Montiminy Shareholder Derivative Lawsuit are substantively the same as those in the Hurtado Shareholder Derivative Lawsuit.
On December 29, 2014, the United States District Court for the Eastern District of Tennessee (the “Court”) entered an order consolidating the Hurtado Shareholder Derivative Lawsuit and the Montiminy Derivative Lawsuit. On February 25, 2015, the parties submitted a proposed agreed order staying the Hurtado and Montiminy Shareholder Derivative Lawsuits until the Court issues a ruling on the anticipated motion to dismiss the amended consolidated complaint to be filed in the Securities Litigation. As of March 4, 2015, the Court has not yet entered the proposed agreed order staying the Hurtado and Montiminy Shareholder Derivative Lawsuits.
Again, as in the Hurtado Shareholder Derivative Lawsuit, no relief is sought against the Company itself; the action is against the Individual Defendants only.
Foley Shareholder Derivative Complaint
On October 28, 2014, Chris Foley, derivatively on behalf of the Company, filed a shareholder derivative complaint in the Chancery Court of Knox County, Tennessee against H. Craig Dees, Timothy C. Scott, Jan E. Koe, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, the “Individual Defendants”), and against the Company as a nominal defendant (the “Foley Shareholder Derivative Lawsuit”). The Foley Shareholder Derivative Lawsuit was brought by the same attorney as the Montiminy Shareholder Derivative Lawsuit, Paul Kent Bramlett of Bramlett Law Offices. Other than the difference in the named plaintiff, the complaints in the Foley Shareholder Derivative Lawsuit and the Montiminy Shareholder Derivative Lawsuit are identical. Since the filing of the Foley Shareholder Derivative Lawsuit, the parties have submitted a proposed agreed order staying the Foley Derivative Lawsuit until the United States District Court for the Eastern District of Tennessee issues a ruling on the anticipated motion to dismiss the amended consolidated complaint to be filed in the Securities Litigation.
| ITEM 4. | MINE SAFETY DISCLOSURES. |
Not applicable.
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES. |
Market Information and Holders
On May 16, 2014, our common stock ceased to be traded on the OTCQB Marketplace operated by OTC Markets Group and is now trading on the NYSE MKT. Our trading symbol remains “PVCT.” The following table sets forth the range of high and low sale prices of our common stock for the periods indicated since January 1, 2013:
| High | Low | |||||||
| 2014 |
||||||||
| First Quarter (January 1 to March 31) |
$ | 6.03 | $ | 1.16 | ||||
| Second Quarter (April 1 to June 30) |
$ | 3.75 | $ | 0.30 | ||||
| Third Quarter (July 1 to September 30) |
$ | 1.20 | $ | 0.81 | ||||
| Fourth Quarter (October 1 to December 31) |
$ | 1.10 | $ | 0.75 | ||||
| 2013 |
||||||||
| First Quarter (January 1 to March 31) |
$ | 0.88 | $ | 0.55 | ||||
| Second Quarter (April 1 to June 30) |
$ | 0.80 | $ | 0.58 | ||||
| Third Quarter (July 1 to September 30) |
$ | 1.14 | $ | 0.58 | ||||
| Fourth Quarter (October 1 to December 31) |
$ | 2.59 | $ | 0.75 | ||||
The closing price for our common stock on March 2, 2015 was $0.88. High and low sale price information was obtained from data provided by Yahoo! Inc.
As of March 2, 2015, we had 1,224 active shareholders of record of our common stock.
34
Table of Contents
Dividend Policy
We have never declared or paid any cash dividends on our capital stock. We currently plan to retain future earnings, if any, to finance the growth and development of our business and do not anticipate paying any cash dividends in the foreseeable future. We may incur indebtedness in the future which may prohibit or effectively restrict the payment of dividends, although we have no current plans to do so. Any future determination to pay cash dividends will be at the discretion of our board of directors.
Stock Performance Graph
The following graph shows the changes, over the past five-year period, in the value of $100 invested in Provectus common stock, the NASDAQ Composite Total Return Index and a Peer group of companies composed of development stage, biopharmaceutical companies that have a focus on developing oncology compounds. The graph assumes that all dividends are reinvested.
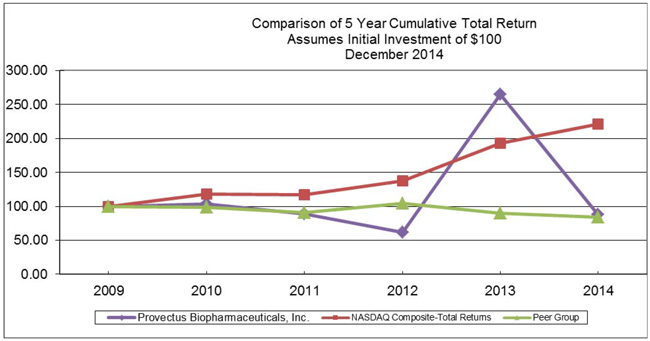
| 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | |||||||||||||||||||
| Provectus Biopharmaceuticals, Inc. |
$ | 100.00 | $ | 103.30 | $ | 89.01 | $ | 61.54 | $ | 264.84 | $ | 87.91 | ||||||||||||
| NASDAQ Composite-Total Returns |
$ | 100.00 | $ | 118.02 | $ | 117.04 | $ | 137.47 | $ | 192.62 | $ | 221.02 | ||||||||||||
| Peer Group |
$ | 100.00 | $ | 98.43 | $ | 91.12 | $ | 104.52 | $ | 90.20 | $ | 83.78 | ||||||||||||
Recent Issuances of Unregistered Securities
During the three months ended March 31, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $137,500. During the three months ended March 31, 2014, the Company issued 733,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $900,317.
During the three months ended June 30, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $140,250. During the three months ended June 30, 2014, the Company issued 202,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $450,002. During the three months ended June 30, 2014, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $5,000,000. The Company accepted subscriptions, in the aggregate, for 2,000,000 shares of common stock and five year warrants to purchase 2,000,000 shares of common stock. Investors received five year fully vested warrants to purchase up to 100% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $3.00 per share. The purchase price for each share of common stock together with the warrants was $2.50. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $650,000 and issued five year fully vested warrants to purchase 300,000 shares of common stock with an exercise price of $2.50 to Network 1 Financial Securities, Inc., which represents 15% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc.
35
Table of Contents
During the three months ended September 30, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $68,500. During the three months ended September 30, 2014, the Company issued 6,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $4,189. During the three months ended September 30, 2014, the Company commenced a private offering of up to $15 million of common stock and five-year warrants to accredited investors. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants is $1.00. The Company plans to use the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. is serving as placement agent for the offering. During the three months ended September 30, 2014, the Company received subscriptions, in the aggregate, for 3,586,300 shares of common stock and five year warrants to purchase 1,793,150 shares of common stock for an aggregate of $3,586,300. Investors will receive five year fully vested warrants to purchase up to 50% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants is $1.00. The Company plans to use the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. is serving as placement agent for the offering. In connection with the offering, the Company paid $466,219 and issued five year fully vested warrants to purchase 358,630 shares of common stock with an exercise price of $1.25 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock subscribed for by investors solicited by Network 1 Financial Securities, Inc.
During the three months ended December 31, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $72,000. During the three months ended December 31, 2014, the Company issued 1,503,913 warrants to consultants in exchange for services. Consulting costs charged to operations were $966,819. During the three months ended December 31, 2014, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $4,198,300. The Company accepted subscriptions, in the aggregate, for 4,198,300 shares of common stock and five year warrants to purchase 2,099,150 shares of common stock. Investors received five year fully vested warrants to purchase up to 50% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants was $1.00. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $545,779 and issued five year fully vested warrants to purchase 419,830 shares of common stock with an exercise price of $1.25 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc.
The issuances of the securities were exempt from the registration requirements of the Securities Act of 1933 by virtue of Section 4(a)(2) and Rule 506 promulgated under Regulation D thereunder as transactions not involving a public offering.
For the issuance of securities to executives, see table labeled “Equity Compensation Plan Information” to be contained in the definitive Proxy Statement for our Annual Meeting of Stockholders to be held on June 19, 2015, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act, incorporated by reference in Part III, Item 12 of this Annual Report on Form 10-K.
36
Table of Contents
| ITEM 6. | SELECTED FINANCIAL DATA. |
The following table sets forth our selected consolidated financial data and has been derived from our audited consolidated financial statements. Consolidated balance sheets as of December 31, 2014 and 2013, as well as consolidated statements of operations for the years ended December 31, 2014, 2013, and 2012, and the report thereon are included elsewhere in this Annual Report on Form 10-K. The information below should be read in conjunction with our audited consolidated financial statements (and notes thereon) and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” included below in Item 7.
| Years ended December 31, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| (all amounts in thousands except per share data) | ||||||||||||||||||||
| Consolidated Statement of Operations Data: |
||||||||||||||||||||
| Gain on settlement – net of discount |
$ | 4,178 | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Operating expenses |
||||||||||||||||||||
| Research and development |
5,138 | 3,596 | 5,006 | 8,808 | 8,417 | |||||||||||||||
| General and administrative |
11,002 | 8,761 | 8,661 | 11,962 | 11,605 | |||||||||||||||
| Amortization |
671 | 671 | 671 | 671 | 671 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating loss |
(12,633 | ) | (13,028 | ) | (14,338 | ) | (21,441 | ) | (20,693 | ) | ||||||||||
| Other income, net |
2,390 | (14,670 | ) | 1,769 | 2,006 | 2,141 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
(10,243 | ) | (27,698 | ) | (12,569 | ) | (19,435 | ) | (18,552 | ) | ||||||||||
| Dividends on preferred stock |
— | (1,188 | ) | (183 | ) | (247 | ) | (10,408 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss applicable to common stockholders |
$ | (10,243 | ) | $ | (28,886 | ) | $ | (12,752 | ) | $ | (19,682 | ) | $ | (28,960 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted loss per common share |
$ | (0.06 | ) | $ | (0.22 | ) | $ | (0.11 | ) | $ | (0.19 | ) | $ | (0.37 | ) | |||||
| Weighted average number of common shares outstanding – basic and diluted |
175,828 | 132,001 | 112,987 | 105,725 | 78,818 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and marketable securities |
$ | 17,392 | $ | 15,696 | $ | 1,222 | $ | 7,705 | $ | 8,087 | ||||||||||
| Patents, net |
3,584 | 4,255 | 4,926 | 5,598 | 6,268 | |||||||||||||||
| Other assets |
5,208 | 57 | 56 | 47 | 48 | |||||||||||||||
| Total assets |
26,184 | 20,008 | 6,204 | 13,350 | 14,403 | |||||||||||||||
| Current liabilities |
847 | 513 | 511 | 263 | 1,350 | |||||||||||||||
| Warrant liability |
147 | 12,866 | 1,300 | 3,067 | 2,353 | |||||||||||||||
| Preferred stock |
— | — | 2 | 4 | 5 | |||||||||||||||
| Common stock |
185 | 160 | 118 | 110 | 91 | |||||||||||||||
| Additional paid-in capital |
181,299 | 152,520 | 122,626 | 115,690 | 96,953 | |||||||||||||||
| Accumulated deficit |
(156,294 | ) | (146,051 | ) | (118,353 | ) | (105,784 | ) | (86,350 | ) | ||||||||||
| Total stockholders’ equity |
25,190 | 6,629 | 4,393 | 10,020 | 10,699 | |||||||||||||||
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS. |
The following discussion is intended to assist in the understanding and assessment of significant changes and trends related to our results of operations and our financial condition together with our consolidated subsidiaries. This discussion and analysis should be read in conjunction with the consolidated financial statements and notes thereto included in this Annual Report on Form 10-K. Historical results and percentage relationships set forth in the statement of operations, including trends which might appear, are not necessarily indicative of future operations.
Critical Accounting Policies
Long-Lived Assets
We review the carrying values of our long-lived assets for possible impairment whenever an event or change in circumstances indicates that the carrying amount of the assets may not be recoverable. Any long-lived assets held for disposal are reported at the lower of their carrying amounts or fair value less cost to sell. Management has determined there to be no impairment.
Patent Costs
Internal patent costs are expensed in the period incurred. Patents purchased are capitalized and amortized over their remaining lives, which range from 2-7 years. Annual amortization of the patents is expected to approximate $671,000 for each of the next two years, $659,000 in 2017 and 2018, and $547,000 in 2019.
Stock-Based Compensation
The compensation cost relating to share-based payment transactions is measured based on the fair value of the equity or liability instruments issued and is expensed on a straight-line basis. For purposes of estimating the fair value of each stock
37
Table of Contents
option, on the date of grant, we utilize the Black-Scholes option-pricing model. The Black-Scholes option valuation model was developed for use in estimating the fair value of traded options which have no vesting restrictions and are fully transferable. In addition, option valuation models require the input of highly subjective assumptions including the expected volatility factor of the market price of the company’s common stock (as determined by reviewing its historical public market closing prices).
Warrants to non-employees are generally vested and nonforfeitable upon the date of the grant. Accordingly, fair value is determined on the grant date.
Research and Development
Research and development costs are charged to expense when incurred. An allocation of payroll expenses to research and development is made based on a percentage estimate of time spent. The research and development costs include the following: payroll, consulting and contract labor, lab supplies and pharmaceutical preparations, legal, insurance, rent and utilities, and depreciation.
Derivative Instruments
The warrants issued in conjunction with convertible preferred stock in March and April 2010 private placements include a reset provision if the Company issues additional warrants, in certain circumstances as defined in the agreement, below the exercise price of $1.00. Effective January 1, 2009, the reset provision of these warrants preclude equity accounting treatment under ASC 815. Accordingly the Company is required to record the warrants as liabilities at their fair value upon issuance and remeasure the fair value at each period end with the change in fair value recorded in the statement of operations. When the warrants are exercised or cancelled, they are reclassified to equity. The Company uses the Monte-Carlo Simulation model to estimate the fair value of the warrants. Significant assumptions used at December 31, 2014 include a weighted average term of 0.2 years, a 5% probability that the warrant exercise price would be reset, a volatility of 63.7% and a risk free interest rate that ranges between 0.03% and 0.04%. Significant assumptions used at December 31, 2013 include a weighted average term of 1.2 years, a 5% probability that the warrant exercise price would be reset, a volatility range between 64.7% and 69.5% and a risk free interest rate range between 0.13% and 0.38%.
Additionally, the Series A and Series C Warrants issued in conjunction with the January 2011 registered direct public offering include a reset provision if the Company issues additional warrants, in certain circumstances as defined in the agreement, below the exercise price of $1.12. During 2012, the warrant exercise price was reset to $0.675. Significant assumptions used at December 31, 2014 include a weighted average term of 1.0 years, a 5% probability that the warrant exercise price would be further reset, a volatility of 159.2% and a risk free interest rate of 0.25%. Significant assumptions used at December 31, 2013 include a weighted average term of 2.0 years, a 5% probability that the warrant exercise price would be further reset, a volatility of 64.7% and a risk free interest rate that ranges between 0.38% and 0.78%.
On February 22, 2013, the Company entered into a Securities Purchase Agreement with certain accredited investors for the issuance and sale in a private placement of an aggregate of $2,550,000 of Units at a purchase price of $0.75 per Unit. Each Unit consists of one share of Series A 8% Convertible Preferred Stock, par value $.001 per share, and a warrant to purchase one and one-quarter shares of the Company’s common stock, par value $.001 per share (subject to adjustment) at an exercise price of $1.00 per whole share (subject to adjustment). The total Series A 8% Convertible Preferred Stock issued was 3,400,001 shares, and the total warrants were 4,250,000. The Company used the net proceeds of the private placement for working capital, FDA trials, securing licensing partnerships, and general corporate purposes.
The Company determined that the warrants issued in February, 2013 with the Series A 8% Convertible Preferred Stock should be classified as liabilities in accordance with ASC 815 because the warrants in question contain exercise price reset features that require the exercise price of the warrants be adjusted if the Company issues certain other equity related instruments at a lower price per share. The preferred stock was determined to have characteristics more akin to equity than debt. As a result, the conversion option was determined to be clearly and closely related to the preferred stock and therefore does not need to be bifurcated and classified as a liability. At June 30, 2014, there are no remaining 2013 warrants and therefore no associated warrant liability. Significant assumptions used at December 31, 2013 include a weighted average term of 4.1 years, a 5% probability that the warrant exercise price would be reset, volatility of 67.2% and a risk free interest rate range between 0.78% and 1.78%.
38
Table of Contents
Fair Value of Financial Instruments
The carrying amounts reported in the consolidated balance sheets for cash and cash equivalents, short-term settlement receivable, and accounts payable approximate their fair value because of the short-term nature of these items. Cash equivalents are measured on a recurring basis within the fair value hierarchy using Level 1 inputs.
The fair value of derivative instruments is determined by management with the assistance of an independent third party valuation specialist. Certain derivatives with limited market activity are valued using externally developed models that consider unobservable market parameters.
Contractual Obligations—Leases
We lease office and laboratory space in Knoxville, Tennessee, on an annual basis, renewable for one year at our option.
Capital Resources and Liquidity
Our ability to continue as a going concern is reasonably assured due to our financing completed during 2014 and thus far from option exercises in 2015 and the partial repayment of bonuses and costs associated with the settlement of the Shareholder Derivative Lawsuit (see Note 9 to the financial statements). Given our current rate of expenditures and our ability to curtail or defer certain controllable expenditures, we do not need to raise additional capital to further develop PV-10 on our own to treat melanoma, HCC and cancers of the liver, recurrent breast carcinoma, and other indications because we plan to license PH-10 for psoriasis and other related indications described as inflammatory dermatoses, strategically monetize PV-10, and also complete the spin-out of Pure-ific Corporation and the other non-core subsidiaries. Additionally, our existing funds are sufficient to meet minimal necessary expenses until well into 2016.
We believe our continued development of PV-10 with existing funds will yield proof-of-concept evidence to support expected best-in-class clinical benefit to treat a wide range of solid tumor recurrences due to its unique ablative immunotherapy or immuno-chemoablation mechanism of action. Likewise, we believe our development of PH-10 with existing funds will yield proof-of-concept evidence to support expected best-in-class clinical benefit to treat a wide range of inflammatory dermatoses due to its unique non-steroidal anti-inflammatory mechanism of action.
Our cash and cash equivalents were $17,391,601 at December 31, 2014, compared with $15,696,243 at December 31, 2013. The increase of approximately $1.7 million was due primarily to an increase of sales of common stock and warrants as well as exercises of warrants and stock options offset partially by approximately $4 million more cash that was used in operating activities. Additionally, thus far in 2015, the Company received approximately $0.3 million in cash due to stock options exercises and $0.2 million from the repayment of bonuses and costs associated with the settlement of the Shareholder Derivative Lawsuit.
By managing variable cash expenses due to minimal fixed costs, we believe our cash and cash equivalents on hand at December 31, 2014, together with approximately $0.5 million received thus far in 2015 due to exercises of stock options and the repayment of bonuses and costs associated with the settlement of the Shareholder Derivative Lawsuit will be sufficient to meet our current and planned operating needs until well into 2016 without consideration being given to additional cash inflows that might occur from the exercise of existing warrants or future sales of equity securities, although we may, in our sole discretion, direct Alpha Capital Anstalt (“Investor”) to purchase up to $30 million of our common stock per an existing agreement with Investor. In addition, on April 30, 2014, the Company entered into a Controlled Equity OfferingSM Sales Agreement with Cantor Fitzgerald & Co., as sales agent (“Cantor”), under which the Company may issue and sell shares of its common stock having an aggregate offering price of up to $50,000,000 from time to time through Cantor, acting as sales agent.
We are seeking to improve our cash flow through both the global licensure of PH-10 on the basis of our Phase 2 atopic dermatitis and psoriasis results, and the geographic licensure of PV-10 on the basis of our Phase 2 metastatic melanoma and Phase 1 liver results in certain areas of the world, as well as pursuing a strategic investment strategy, including equity sales to potential pharmaceutical and or biotech partners. In addition, the data now available and forthcoming from Moffitt in Tampa, Florida has been and is expected to be particularly helpful in supporting our development plans with both the FDA and prospective partners. The geographic areas of interest for PV-10 principally include China, India, Russia, Brazil, Japan and Middle East and North Africa (MENA). We are encouraged by the interest in both PV-10 and PH-10 on a geographic basis and are continuing discussions with potential partners.
We are also considering the global licensure of PV-10 as well since it has come to our attention that this is of interest to potential partners. We have provided data on a confidential basis to both potential global and geographic partners for both PV-10 and PH-10 via a secure electronic data room that is monitored 24 hours a day, seven days a week and houses formal data submissions to the FDA as well as various corporate governance related documents.
39
Table of Contents
We also expect to continue with the majority stake asset sale and licensure of our non-core assets. However, the primary objective of the Company is to strategically monetize the core value of PV-10 and PH-10 through various transactions, leveraging value creation up to and including an appropriate merger and acquisition transaction that includes upfront cash and acquirer stock in exchange for Company ownership as well as a contingency value right (CVR) to facilitate potential upside post-acquisition. We believe regulatory clarity, including one or more breakthrough therapy designations, is determined by specifying the expected approval pathways of both PV-10 and PH-10. This may include the potential for expedited approval for PV-10 to treat locally advanced recurrent melanoma as we commence phase 3 the first quarter of 2015 with PV-10 to treat this indication. Such clarity may help facilitate transactions with potential partners. Additionally, the existing and forthcoming mechanism of action related clinical and nonclinical data for both PV-10 and PH-10 will further aid in both regulatory clarity and transactions with potential partners.
However, we cannot assure you that we will be successful in either licensing of PH-10 or PV-10, any equity transaction, any merger or acquisition transaction or selling a majority stake of the OTC and other non-core assets via a spin-out transaction and licensing our existing non-core products. Moreover, even if we are successful in improving our current cash flow position, we nonetheless plan to seek additional funds to meet our long-term requirements in 2015 and beyond. We anticipate that these funds will otherwise come from the proceeds of private placements, the exercise of existing warrants outstanding, or public offerings of debt or equity securities. While we believe that we have a reasonable basis for our expectation that we will be able to raise additional funds, we cannot assure you that we will be able to complete additional financing in a timely manner. In addition, any such financing may result in significant dilution to stockholders.
We believe that our financial position and corporate governance are such that we will continue to meet the relevant listing requirements of NYSE MKT, although there can be no assurance that we will continue to be listed on NYSE MKT. We expect that the existing and forthcoming clinical and nonclinical mechanism of action data for both PV-10 and PH-10 will aid in both regulatory clarity and transactions with potential partners. The Company’s current cash position is sufficient to meet our obligations. In addition, management is returning $8.96 million to the Company as a result of the previously announced settlement of the Shareholder Derivative Lawsuit (subject to a 2:1 credit to the executives, such that total actual repayment by the executives may be $1.12 million per executive which would total $4.48 million), and we further enhanced our strength by management’s recent exercise of options. In total, we believe we have adequate funds to operate without a further injection of capital through well into 2016. We believe the existing cash position of the Company is sufficient to fund our operations through obtaining interim data from the planned phase 3 melanoma study as well as other planned programs including generating key liver data, and clinical mechanism of action data for both PV-10 and PH-10.
We have provided data on a confidential basis to both potential global and geographic partners for both PV-10 for oncology, and PH-10 for dermatology, via a secure electronic data room. We are encouraged by the number of companies doing due diligence on our technologies. For instance, we recently so far in 2015 had a team in India meeting with potential partners and have two teams focused in China working with potential partners there. We also have begun to consider co-development transactions with one or more pharmaceutical or biotech companies to combine PV-10 with immunology agents such as those referred to as checkpoint protein inhibitors. Whenever we obtain a Memorandum of Understanding (MOU), definitive agreement or similar indication of interest from a potential partner, we will issue a press release and file a Form 8-K with the SEC to notify the market. Furthermore, our strategy for the benefit of stockholders is a series of partnerships followed by an acquisition of the Company along the lines of Celgene/Abraxis, although there can be no assurance that such partnerships or acquisition will occur. The Company is not in discussions regarding the sale of its business and there can be no assurance that the Company will be able to monetize PV-10 or PH-10 in the manner described herein.
On August 18, 2014, we announced that we entered into a Memorandum of Understanding (“MOU”) with Sinopharm-China State Institute of Pharmaceutical Industry (“Sinopharm-CSIPI”), the leader among all pharmaceutical research institutes in China, and Sinopharm A-THINK Pharmaceutical Co., Ltd. (“Sinopharm A-THINK”), the only injectable anti-tumor drug research and development, manufacture and distribution integrated platform within Sinopharm Group. The MOU term, as extended pursuant to an amendment entered into on November 13, 2014, continues to May 16, 2015. This agreement is intended to enhance our reach into China and will bolster our efforts in developing partnering opportunities in various countries in Asia including China, India and Japan, where we have held numerous detailed discussions with pharmaceutical companies over the last year. We are already seeing the results of efforts to enter into partnerships from the activity in our electronic data room. The Company is not in discussions regarding the sale of its business and there can be no assurance that the Company will be able to monetize PV-10 or PH-10 in the manner described herein.
40
Table of Contents
Plan of Operation
We have implemented our integrated business plan, including execution of the current and next phases in clinical development of our pharmaceutical products and continued execution of research programs for new research initiatives.
Our current plans include continuing to operate with our four employees during the immediate future, as well as four primary consultants and various vendor relationships totaling fifty-five (55) full-time equivalents, and anticipate adding additional personnel or contract research organizations if necessary in the next 12 months. Our current plans also include minimal purchases of new property, plant and equipment, and increased research and development for additional clinical trials.
We believe that our investigational drugs PV-10 and PH-10 provide us with two products in multiple indications, which have been shown in clinical trials to be safe to treat serious cancers and diseases of the skin, and important immunologic data has been corroborated and characterized by institutions such as Moffitt in Tampa, Florida, and another leading research facility. We continue to develop clinical trials for these products to show their safety and efficacy, which we believe will continue to be shown based on data in previous studies, and which result in one or more license transactions with pharmaceutical and or biotech companies. Together with our non-core technologies, which we intend to sell or license in the future, we believe this combination represents the foundation for maximizing shareholder value this year and beyond.
Comparison of the Years Ended December 31, 2014 and 2013
Gain on Settlement
The gain on settlement, net of discount, of $4,178,345 occurred in 2014 as a result from accounting for the settlement of the Shareholder Derivative Lawsuit described in Note 9 to the financial statements. The settlement is a one-time event.
Research and development
Research and development costs totaling $5,137,927 for 2014 included payroll of $1,395,321, consulting and contract labor of $2,355,780, lab supplies and pharmaceutical preparations of $790,653, legal of $384,061, insurance of $115,957, rent and utilities of $87,623, and depreciation expense of $8,532. Research and development costs totaling $3,595,555 for 2013 included payroll of $1,459,057, consulting and contract labor of $1,317,472, lab supplies and pharmaceutical preparations of $310,160, legal of $262,720, insurance of $161,268, rent and utilities of $78,512, and depreciation expense of $6,366.
The increase in consulting and contract labor of approximately $1.0 million in 2014 over 2013 is primarily the result of the preparation of phase 3 PV-10 for locally advanced cutaneous melanoma and further development in other PV-10 and PH-10 programs. The increase in lab supplies and pharmaceutical preparations of approximately $0.5 million in 2014 over 2013 is primarily the result of the preparation of additional phase 3 PV-10 drug supply, as well as for other PV-10 programs, along with phase 2 PH-10 mechanism of action drug supply. The increase in both consulting and contract labor, and lab supplies and pharmaceutical preparations represents virtually all of the increase in research and development expenses in 2014 versus 2013.
General and administrative
General and administrative expenses increased by $2,241,062 for 2014 to $11,002,326 from $8,761,264 in 2013. General and administrative expenses were very similar for both periods; however, almost $600,000 in increased expense is due to the higher stock price of our common stock during the three months ended March 31, 2014 versus the three months ended March 31, 2013, which resulted in higher noncash expenses charged to operations for the value of both common stock and warrants issued for services. Additionally, legal expense increased by about $500,000 primarily due to our NYSE MKT listing and the Controlled Equity OfferingSM Sales Agreement with Cantor and investor relations and related travel expenses increased approximately $1,100,000 in 2014 over 2013.
Investment income
Investment income is immaterial for all periods presented.
Change in fair value of warrant liability
Change in fair value of warrant liability increased by $17,055,523 to a gain of $2,384,393 in 2014 from a loss of $14,671,130 in 2013. This activity results from accounting for the warrant liability described in Notes 3(c), 3(d), 3(e) and 8 to the financial statements which is primarily attributed to a decrease in our common stock price, warrant exercises and a reduction in the remaining life of warrants outstanding.
41
Table of Contents
Cash Flow
Our cash and cash equivalents were $17,391,601 at December 31, 2014, compared with $15,696,243 at December 31, 2013. The increase of approximately $1.7 million was due primarily to an increase of sales of common stock and warrants as well as exercises of warrants and stock options offset partially by approximately $4 million more cash that was used in operating activities in 2014 versus 2013. Additionally thus far in 2015, the Company received approximately $0.3 million in cash due to stock options exercises and $0.2 million from the repayment of bonuses and costs associated with the settlement of the Shareholder Derivative Lawsuit. At our current cash expenditure rate, our cash and cash equivalents will be sufficient to meet our current and planned needs until well into 2016 without additional cash inflows from the exercise of existing warrants, stock options, or sales of equity securities.
Comparison of the Years Ended December 31, 2013 and 2012
Research and development
Research and development costs totaling $3,595,555 for 2013 included payroll of $1,459,057, consulting and contract labor of $1,317,472, lab supplies and pharmaceutical preparations of $310,160, legal of $262,720, insurance of $161,268, rent and utilities of $78,512, and depreciation expense of $6,366. Research and development costs totaling $5,005,459 for 2012 included payroll of $2,536,818, consulting and contract labor of $2,008,270, lab supplies and pharmaceutical preparations of $47,808, legal of $231,430, insurance of $97,728, rent and utilities of $77,238, and depreciation expense of $6,167.
The decrease in payroll in 2013 over 2012 is primarily the result of the termination of bonuses and reduced stock-based compensation expense from stock options. The reduction in payroll represents most of the decrease in research and development expenses in 2013 versus 2012. Additionally, consulting and contract labor decreased in 2013 over 2012 due to reduction in warrants for services in 2013 versus 2012.
General and administrative
General and administrative expenses increased by $100,224 for 2013 to $8,761,264 from $8,661,040 in 2012. The increase is primarily due to an increase in investor relations expense offset by the termination of bonuses and reduced stock-based compensation expense from stock options.
Investment income
Investment income is immaterial for all periods presented.
Change in fair value of warrant liability
Change in fair value of warrant liability increased by $16,439,048 to a loss of $14,671,130 in 2013 from a gain of $1,767,918 in 2012. This activity results from accounting for the warrant liability described in Notes 3(c), 3(d), 3(e) and 8 to the financial statements which is primarily attributed to a significant increase in our common stock price.
Cash Flow
Our cash and cash equivalents were $15,696,243 at December 31, 2013, compared with $1,221,701 at December 31, 2012.
Liquidity and Capital Resources
As noted above, our present cash and cash equivalents are currently sufficient to meet our short-term operating needs. Excess cash will be used to finance any additional phases in clinical development of our pharmaceutical products that we may decide to undertake ourselves versus with a partner. We anticipate that any required funds for our operating and development needs in 2015 and beyond may come from a partnership agreement or from the proceeds of public or private sales of equity or debt securities or the exercise of existing warrants and stock options outstanding. While we believe that we have a reasonable basis for our expectation that we will be able to raise additional funds if necessary, we cannot assure you that we will be able to complete additional financing in a timely manner. In addition, any such financing may result in significant dilution to stockholders.
42
Table of Contents
Recent Accounting Pronouncements
In May 2014, the FASB issued Accounting Standards Update No. 2014-09, Revenue from Contracts with Customers (ASU 2014-09), which supersedes nearly all existing revenue recognition guidance under U.S. GAAP. The core principle of ASU 2014-09 is to recognize revenues when promised goods or services are transferred to customers in an amount that reflects the consideration to which an entity expects to be entitled for those goods or services. ASU 2014-09 defines a five step process to achieve this core principle and, in doing so, more judgment and estimates may be required within the revenue recognition process than are required under existing U.S. GAAP.
The standard is effective for annual periods beginning after December 15, 2016, and interim periods therein, using either of the following transition methods: (i) a full retrospective approach reflecting the application of the standard in each prior reporting period with the option to elect certain practical expedients, or (ii) a retrospective approach with the cumulative effect of initially adopting ASU 2014-09 recognized at the date of adoption (which includes additional footnote disclosures). We are currently evaluating the impact of our pending adoption of ASU 2014-09 on our consolidated financial statements and have not yet determined the method by which we will adopt the standard in 2017. The Company currently does not have revenues but will consider any related impact going forward.
In June 2014, the FASB issued Accounting Standards Update 2014-10, Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation (ASU 2014-10), which eliminates the concept of a development stage entity (DSE) from U.S. GAAP. This change rescinds certain financial reporting requirements that have historically applied to DSEs and is intended to result in cost-savings for affected entities, such as certain start-up or research and development entities. The new standard also changes one related aspect of the variable interest entity (VIE) consolidation guidance in Topic 810.
ASU 2014-10 is effective for public entities for annual reporting periods beginning after December 15, 2014 and interim periods therein. Early adoption is permitted. We early adopted ASU 2014-10 in our consolidated financial statements as of the third quarter of fiscal 2014.
In August 2014, the FASB issued Accounting Standards Update 2014-15, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern (ASU 2014-15), which addresses when and how to disclose going-concern uncertainties in the financial statements. ASU 2014-15 requires management to perform interim and annual assessments of an entity’s ability to continue as a going concern within one year after the date the financial statements are issued. An entity must provide certain disclosures if conditions or events raise substantial doubt about the entity’s ability to continue as a going concern. ASU 2014-15 applies to all entities and is effective for annual periods ending after December 15, 2016, and interim periods thereafter, with early adoption permitted. The amended guidance is not expected to have a material impact on the Company’s consolidated financial statements.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK. |
We had no holdings of financial or commodity instruments as of December 31, 2014, other than cash and cash equivalents, short-term deposits, money market funds and interest bearing investments in U.S. governmental debt securities. We have accounted for certain warrants issued in March and April 2010, January 2011 and February 2013 as liabilities at their fair value upon issuance, which are remeasured at each period end with the change in fair value recorded in the statement of operations. See Note 3 of the consolidated financial statements contained in this Annual Report on Form 10-K.
All of our business is transacted in U.S. dollars and, accordingly, foreign exchange rate fluctuations have not had an impact on us, and they are not expected to have an impact on us in the foreseeable future.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
The financial statements required by this Item are included as a separate section of this report commencing on page F-1.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
Not applicable.
| ITEM 9A. | CONTROLS AND PROCEDURES. |
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, has concluded that our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended (the Act)) were effective as of December 31, 2014, based on the evaluation of these controls and procedures required by Rule 13a-15(b) or 15d-15(b) of the Act.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control system was designed to provide reasonable assurance regarding the preparation and fair presentation of
43
Table of Contents
published financial statements in accordance with generally accepted accounting principles. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation in accordance with generally accepted accounting principles. Management conducted an assessment of our internal control over financial reporting as of December 31, 2014 using the framework specified in Internal Control – Integrated Framework (2013), published by the Committee of Sponsoring Organizations of the Treadway Commission. Based on its assessment, the Chief Executive Officer and Chief Financial Officer concluded that our internal control over financial reporting at December 31, 2014 was effective.
Our independent registered public accounting firm, BDO USA, LLP, assessed the effectiveness of the Company’s internal control over financial reporting. BDO USA, LLP has issued an attestation report on our internal control over financial reporting as of December 31, 2014, which is set forth below.
Changes in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting that occurred during the fourth quarter of 2014 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
44
Table of Contents
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Provectus Biopharmaceuticals, Inc.
Knoxville, Tennessee
We have audited Provectus Biopharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Provectus Biopharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying “Item 9A, Management’s Report on Internal Control Over Financial Reporting”. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Provectus Biopharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Provectus Biopharmaceuticals, Inc., as of December 31, 2014 and 2013, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014 and our report dated March 12, 2015 expressed an unqualified opinion thereon.
/s/ BDO USA, LLP
Chicago, Illinois
March 12, 2015
45
Table of Contents
| ITEM 9B. | OTHER INFORMATION. |
None.
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE. |
The information called for by this item is incorporated herein by reference to the definitive Proxy Statement for our Annual Meeting of Stockholders to be held on June 19, 2015, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act.
| ITEM 11. | EXECUTIVE COMPENSATION. |
The information called for by this item is incorporated herein by reference to the definitive Proxy Statement for our Annual Meeting of Stockholders to be held on June 19, 2015, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
The information called for by this item is incorporated herein by reference to the definitive Proxy Statement for our Annual Meeting of Stockholders to be held on June 19, 2015, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE. |
The information called for by this item is incorporated herein by reference to the definitive Proxy Statement for our Annual Meeting of Stockholders to be held on June 19, 2015, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act.
| ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES. |
The information called for by this item is incorporated herein by reference to the definitive Proxy Statement for our Annual Meeting of Stockholders to be held on June 19, 2015, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act.
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES. |
Financial Statements
See Index to Consolidated Financial Statements in “Financial and Supplementary Data.”
Financial Statement Schedules
None
Exhibits
Exhibits required by Item 601 of Regulation S-K are incorporated herein by reference and are listed on the attached Exhibit Index, which appears immediately after the Consolidated Financial Statements of our Annual Report on Form 10-K.
46
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
March 12, 2015
| PROVECTUS BIOPHARMACEUTICALS, INC. | ||
| By: | /s/ H. Craig Dees | |
| H. Craig Dees, Ph.D. | ||
| Chief Executive Officer and Chairman of the Board | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacity and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ H. Craig Dees |
Chief Executive Officer (principal executive officer) and Chairman of the Board |
March 12, 2015 | ||
| H. Craig Dees, Ph.D. | ||||
| /s/ Peter R. Culpepper |
Chief Financial Officer (principal financial officer), Chief Operating Officer and Chief Accounting Officer |
March 12, 2015 | ||
| Peter R. Culpepper | ||||
| /s/ Timothy C. Scott |
President and Director | March 12, 2015 | ||
| Timothy C. Scott | ||||
| /s/ Jan Koe |
Director | March 12, 2015 | ||
| Jan Koe | ||||
| /s/ Kelly M. McMasters |
Director | March 12, 2015 | ||
| Kelly M. McMasters, M.D., Ph.D. | ||||
| /s/ Alfred E. Smith, IV |
Director | March 12, 2015 | ||
| Alfred E. Smith, IV | ||||
47
Table of Contents
The following financial statements are included in Part II, Item 8:
Table of Contents
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Provectus Biopharmaceuticals, Inc.
Knoxville, Tennessee
We have audited the accompanying consolidated balance sheets of Provectus Biopharmaceuticals, Inc., as of December 31, 2014 and 2013 and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Provectus Biopharmaceuticals, Inc. at December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Provectus Biopharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and our report dated March 12, 2015 expressed an unqualified opinion thereon.
/s/ BDO USA, LLP
Chicago, Illinois
March 12, 2015
F-1
Table of Contents
PROVECTUS BIOPHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
| December 31, 2014 |
December 31, 2013 |
|||||||
| Assets |
||||||||
| Current Assets |
||||||||
| Cash and cash equivalents |
$ | 17,391,601 | $ | 15,696,243 | ||||
| Short-term receivable - settlement |
733,333 | — | ||||||
| Other current assets |
978,000 | — | ||||||
|
|
|
|
|
|||||
| Total Current Assets |
19,102,934 | 15,696,243 | ||||||
|
|
|
|
|
|||||
| Equipment and furnishings, less accumulated depreciation of $437,863 and $429,331, respectively |
92,171 | 30,113 | ||||||
| Patents, net of amortization of $8,131,737 and $7,460,617, respectively |
3,583,708 | 4,254,828 | ||||||
| Long-term receivable – settlement, net of discount |
3,378,345 | — | ||||||
| Other assets |
27,000 | 27,000 | ||||||
|
|
|
|
|
|||||
| $ | 26,184,158 | $ | 20,008,184 | |||||
|
|
|
|
|
|||||
| Liabilities and Stockholders’ Equity |
||||||||
| Current Liabilities |
||||||||
| Accounts payable – trade |
$ | 440,702 | $ | 348,869 | ||||
| Accrued consulting expense |
91,282 | 61,282 | ||||||
| Other accrued expenses |
315,738 | 102,795 | ||||||
|
|
|
|
|
|||||
| Total Current Liabilities |
847,722 | 512,946 | ||||||
|
|
|
|
|
|||||
| Long-Term Liability |
||||||||
| Warrant liability |
146,560 | 12,866,572 | ||||||
|
|
|
|
|
|||||
| Total Liabilities |
994,282 | 13,379,518 | ||||||
|
|
|
|
|
|||||
| Stockholders’ Equity |
||||||||
| Preferred stock; par value $.001 per share; 25,000,000 shares authorized; Series A 8% convertible preferred stock, 0 and 33,334 shares issued and outstanding, respectively, liquidation preference $0.75 (for 2013 in aggregate $25,001) |
— | 33 | ||||||
| Common stock; par value $.001 per share; 300,000,000 shares authorized; 184,796,275 and 159,751,724 shares issued and outstanding, respectively |
184,796 | 159,752 | ||||||
| Paid-in capital |
181,298,890 | 152,519,701 | ||||||
| Accumulated deficit |
(156,293,810 | ) | (146,050,820 | ) | ||||
|
|
|
|
|
|||||
| Total Stockholders’ Equity |
25,189,876 | 6,628,666 | ||||||
|
|
|
|
|
|||||
| $ | 26,184,158 | $ | 20,008,184 | |||||
|
|
|
|
|
|||||
See accompanying notes to consolidated financial statements.
F-2
Table of Contents
PROVECTUS BIOPHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| Year Ended December 31, 2014 |
Year Ended December 31, 2013 |
Year Ended December 31, 2012 |
||||||||||
| Gain on settlement – net of discount |
$ | 4,178,345 | $ | — | $ | — | ||||||
| Operating expenses |
||||||||||||
| Research and development |
5,137,927 | 3,595,555 | 5,005,459 | |||||||||
| General and administrative |
11,002,326 | 8,761,264 | 8,661,040 | |||||||||
| Amortization |
671,120 | 671,120 | 671,120 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating loss |
(12,633,028 | ) | (13,027,939 | ) | (14,337,619 | ) | ||||||
| Investment income |
5,645 | 1,325 | 1,347 | |||||||||
| Gain (loss) on change in fair value of warrant liability |
2,384,393 | (14,671,130 | ) | 1,767,918 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (10,242,990 | ) | $ | (27,697,744 | ) | $ | (12,568,354 | ) | |||
| Dividends on preferred stock |
— | (1,188,648 | ) | (183,187 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net loss applicable to common shareholders |
$ | (10,242,990 | ) | $ | (28,886,392 | ) | $ | (12,751,541 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted loss per common share |
$ | (0.06 | ) | $ | (0.22 | ) | $ | (0.11 | ) | |||
| Weighted average number of common shares outstanding – basic and diluted |
175,828,004 | 132,000,796 | 112,986,636 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
F-3
Table of Contents
PROVECTUS BIOPHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
| Preferred Stock | Common Stock | |||||||||||||||||||||||||||
| Number of Shares |
Par Value | Number of Shares |
Par Value | Paid in capital |
Accumulated Deficit |
Total | ||||||||||||||||||||||
| Balance, at January 1, 2012 |
3,531,665 | $ | 3,531 | 110,596,798 | $ | 110,597 | $ | 115,690,334 | $ | (105,784,722 | ) | $ | 10,019,740 | |||||||||||||||
| Issuance of stock for services |
— | — | 550,000 | 550 | 455,950 | — | 456,500 | |||||||||||||||||||||
| Issuance of warrants for services |
— | — | — | — | 1,512,026 | — | 1,512,026 | |||||||||||||||||||||
| Issuance of common stock and warrants pursuant to Regulation D |
— | — | 6,227,647 | 6,228 | 4,784,316 | — | 4,790,544 | |||||||||||||||||||||
| Preferred stock conversions into common stock |
(1,053,480 | ) | (1,053 | ) | 1,053,480 | 1,053 | — | — | — | |||||||||||||||||||
| Employee compensation from stock options |
— | — | — | — | 183,028 | — | 183,028 | |||||||||||||||||||||
| Net loss for the year ended 2012 |
— | — | — | — | — | (12,568,354 | ) | (12,568,354 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance, at December 31, 2012 |
2,478,185 | $ | 2,478 | 118,427,925 | $ | 118,428 | $ | 122,625,654 | $ | (118,353,076 | ) | $ | 4,393,484 | |||||||||||||||
| Issuance of stock for services |
— | — | 750,000 | 750 | 525,250 | — | 526,000 | |||||||||||||||||||||
| Issuance of warrants for services |
— | — | — | — | 1,786,824 | — | 1,786,824 | |||||||||||||||||||||
| Reclassification of warrant liability |
— | — | — | — | 4,402,078 | — | 4,402,078 | |||||||||||||||||||||
| Exercise of warrants and stock options |
— | — | 6,319,594 | 6,320 | 3,427,072 | — | 3,433,392 | |||||||||||||||||||||
| Issuance of common stock and warrants pursuant to Regulation D |
— | — | 28,409,353 | 28,409 | 18,390,926 | — | 18,419,335 | |||||||||||||||||||||
| Issuance of preferred stock and warrants pursuant to Regulation D |
3,400,001 | 3,400 | — | — | 1,248,650 | — | 1,252,050 | |||||||||||||||||||||
| Preferred stock conversions into common stock |
(5,844,852 | ) | (5,845 | ) | 5,844,852 | 5,845 | — | — | — | |||||||||||||||||||
| Dividends on preferred stock |
— | — | — | — | (29,063 | ) | — | (29,063 | ) | |||||||||||||||||||
| Employee compensation from stock options |
— | — | — | — | 142,310 | — | 142,310 | |||||||||||||||||||||
| Net loss for the year ended 2013 |
— | — | — | — | — | (27,697,744 | ) | (27,697,744 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance, at December 31, 2013 |
33,334 | $ | 33 | 159,751,724 | $ | 159,752 | $ | 152,519,701 | $ | (146,050,820 | ) | $ | 6,628,666 | |||||||||||||||
| Issuance of stock for services |
— | — | 300,000 | 300 | 417,950 | — | 418,250 | |||||||||||||||||||||
| Issuance of warrants for services |
— | — | — | — | 2,321,327 | — | 2,321,327 | |||||||||||||||||||||
| Reclassification of warrant liability |
— | — | — | — | 10,335,619 | — | 10,335,619 | |||||||||||||||||||||
| Cash proceeds from exercise of warrants and stock options |
— | — | 14,926,617 | 14,926 | 4,475,831 | — | 4,490,757 | |||||||||||||||||||||
| Issuance of common stock and warrants pursuant to Regulation D |
— | — | 9,784,600 | 9,785 | 11,112,817 | — | 11,122,602 | |||||||||||||||||||||
| Preferred stock conversions into common stock |
(33,334 | ) | (33 | ) | 33,334 | 33 | — | — | — | |||||||||||||||||||
| Employee compensation from stock options |
— | — | — | — | 115,645 | — | 115,645 | |||||||||||||||||||||
| Net loss for the year ended 2014 |
— | — | — | — | — | (10,242,990 | ) | (10,242,990 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance, at December 31, 2014 |
— | $ | — | 184,796,275 | $ | 184,796 | $ | 181,298,890 | $ | (156,293,810 | ) | $ | 25,189,876 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
See accompanying notes to consolidated financial statements.
F-4
Table of Contents
PROVECTUS BIOPHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Year Ended December 31, 2014 |
Year Ended December 31, 2013 |
Year Ended December 31, 2012 |
||||||||||
| Cash Flows From Operating Activities |
||||||||||||
| Net loss |
$ | (10,242,990 | ) | $ | (27,697,744 | ) | $ | (12,568,354 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities |
||||||||||||
| Depreciation |
8,532 | 6,366 | 6,167 | |||||||||
| Amortization of patents |
671,120 | 671,120 | 671,120 | |||||||||
| Compensation through issuance of stock options |
115,645 | 142,310 | 183,028 | |||||||||
| Issuance of stock for services |
418,250 | 526,000 | 456,500 | |||||||||
| Issuance of warrants for services |
2,321,327 | 1,786,824 | 1,512,026 | |||||||||
| (Gain) loss on change in fair value of warrant liability |
(2,384,393 | ) | 14,671,130 | (1,767,918 | ) | |||||||
| Gain on settlement |
(4,178,345 | ) | — | — | ||||||||
| (Increase) decrease in assets |
||||||||||||
| Settlement receivable |
66,667 | — | — | |||||||||
| Other current assets |
(978,000 | ) | — | — | ||||||||
| Increase (decrease) in liabilities |
||||||||||||
| Accounts payable |
91,833 | 105,434 | 142,333 | |||||||||
| Accrued expenses |
242,943 | (103,912 | ) | 106,367 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(13,847,411 | ) | (9,892,472 | ) | (11,258,731 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash Flows From Investing Activities |
||||||||||||
| Capital expenditures |
(70,590 | ) | (6,650 | ) | (15,885 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in investing activities |
(70,590 | ) | (6,650 | ) | (15,885 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash Flows From Financing Activities |
||||||||||||
| Net proceeds from sales of preferred stock and warrants |
— | 2,550,000 | — | |||||||||
| Net proceeds from sales of common stock and warrants |
11,122,602 | 18,419,335 | 4,790,544 | |||||||||
| Proceeds from exercises of warrants and stock options |
4,490,757 | 3,433,392 | — | |||||||||
| Cash paid for preferred dividends |
— | (29,063 | ) | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
15,613,359 | 24,373,664 | 4,790,544 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net change in cash and cash equivalents |
$ | 1,695,358 | $ | 14,474,542 | $ | (6,484,072 | ) | |||||
| Cash and cash equivalents, at beginning of period |
$ | 15,696,243 | $ | 1,221,701 | $ | 7,705,773 | ||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents, at end of period |
$ | 17,391,601 | $ | 15,696,243 | $ | 1,221,701 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental Disclosure of Noncash Investing and Financing Activities
|
||||||||||||
| Year Ended December 31, 2014 |
Year Ended December 31, 2013 |
Year Ended December 31, 2012 |
||||||||||
| Reclassification of warrant liability to equity due to exercise of warrants |
$ | 10,335,619 | $ | 4,402,078 | $ | — | ||||||
See accompanying notes to consolidated financial statements.
F-5
Table of Contents
PROVECTUS BIOPHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Significant Accounting Policies
Nature of Operations
Provectus Biopharmaceuticals, Inc. (together with its subsidiaries, the “Company”) is a biopharmaceutical company that is focusing on developing minimally invasive products for the treatment of psoriasis and other topical diseases, and certain forms of cancer including melanoma, breast cancer, and cancers of the liver. To date, the Company has no revenues from planned principal operations. The Company’s activities are subject to significant risks and uncertainties, including failing to successfully develop and license or commercialize the Company’s prescription drug candidates, or sell or license the Company’s OTC products or non-core technologies.
Principles of Consolidation
Intercompany balances and transactions have been eliminated in consolidation.
Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments with a maturity of three months or less when purchased to be cash equivalents.
Cash Concentrations
Cash and cash equivalents are maintained at financial institutions and, at times, balances may exceed federally insured limits of $250,000 although the Company seeks to minimize this through treasury management. We have never experienced any losses related to these balances.
Equipment and Furnishings
Equipment and furnishings are stated at cost. Depreciation of equipment is provided for using the straight-line method over the estimated useful lives of the assets. Computers and laboratory equipment are being depreciated over five years; furniture and fixtures are being depreciated over seven years.
Long-Lived Assets
The Company reviews the carrying values of its long-lived assets for possible impairment whenever an event or change in circumstances indicates that the carrying amount of the assets may not be recoverable. Any long-lived assets held for disposal are reported at the lower of their carrying amounts or fair value less cost to sell. Management has determined there to be no impairment.
Patent Costs
Internal patent costs are expensed in the period incurred. Patents purchased are capitalized and amortized over the remaining life of the patent.
Patents at December 31, 2014 were acquired as a result of the merger with Valley Pharmaceuticals, Inc. (“Valley”) on November 19, 2002. The majority stockholders of Provectus also owned all of the shares of Valley and therefore the assets acquired from Valley were recorded at their carry-over basis. The patents are being amortized over the remaining lives of the patents, which range from 2-7 years at December 31, 2014. Annual amortization of the patents is expected to approximate $671,000 for each of the next two years, $659,000 in 2017 and 2018, and $547,000 in 2019.
F-6
Table of Contents
Research and Development
Research and development costs are charged to expense when incurred. An allocation of payroll expenses to research and development is made based on a percentage estimate of time spent. The research and development costs include the following: payroll, consulting and contract labor, lab supplies and pharmaceutical preparations, legal, insurance, rent and utilities, and depreciation.
Income Taxes
The Company accounts for income taxes under the liability method in accordance with Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 740 “Income Taxes”. Under this method, deferred income tax assets and liabilities are determined based on differences between financial reporting and tax basis of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is established if it is more likely than not that all, or some portion, of deferred income tax assets will not be realized. The Company has recorded a full valuation allowance to reduce its net deferred income tax assets to zero. In the event the Company were to determine that it would be able to realize some or all its deferred income tax assets in the future, an adjustment to the deferred income tax asset would increase income in the period such determination was made.
The Company recognizes the effect of income tax positions only if those positions are more likely than not of being sustained upon an examination. Any recognized income tax positions would be measured at the largest amount that is greater than 50% likely of being realized. Changes in recognition or measurement would be reflected in the period in which the change in judgment occurs. The Company would recognize any corresponding interest and penalties associated with its income tax positions in income tax expense. There were no income taxes, interest or penalties incurred in 2014, 2013 or 2012. Tax years going back to 2011 remain open for examination by the IRS.
Basic and Diluted Loss Per Common Share
Basic and diluted loss per common share is computed based on the weighted average number of common shares outstanding. Loss per share excludes the impact of outstanding options and warrants and convertible preferred stock as they are antidilutive. Potential common shares excluded from the calculation for the years ended December 31, 2014, 2013 and 2012, respectively, are 63,235,956, 73,037,416 and 30,038,017 from warrants, 10,845,098, 15,322,206 and 15,140,956 from options, and 0, 33,334 and 2,478,185 from convertible preferred shares.
Derivative Instruments
The warrants issued in conjunction with convertible preferred stock in March and April 2010 private placements include a reset provision if the Company issues additional warrants, in certain circumstances as defined in the agreement, below the exercise price of $1.00. Effective January 1, 2009, the reset provision of these warrants preclude equity accounting treatment under ASC 815. Accordingly, the Company is required to record the warrants as liabilities at their fair value upon issuance and remeasure the fair value at each period end with the change in fair value recorded in the statement of operations. When the warrants are exercised or cancelled, they are reclassified to equity. The Company uses the Monte-Carlo Simulation model to estimate the fair value of the warrants. Significant assumptions used at December 31, 2014 include a weighted average term of 0.2 years, a 5% probability that the warrant exercise price would be reset, a volatility of 63.7% and a risk free interest rate that ranges between 0.03% and 0.04%. Significant assumptions used at December 31, 2013 include a weighted average term of 1.2 years, a 5% probability that the warrant exercise price would be reset, a volatility range between 64.7% and 69.5% and a risk free interest rate range between 0.13% and 0.38%.
Additionally, the Series A and Series C Warrants issued in conjunction with the January 2011 registered direct public offering include a reset provision if the Company issues additional warrants, in certain circumstances as defined in the agreement, below the exercise price of $1.12. During 2012, the warrant exercise price was reset to $0.675. Significant assumptions used at December 31, 2014 include a weighted average term of 1.0 years, a 5% probability that the warrant exercise price would be further reset, a volatility of 159.2% and a risk free interest rate range of 0.25%. Significant assumptions used at December 31, 2013 include a weighted average term of 2.0 years, a 5% probability that the warrant exercise price would be further reset, a volatility of 64.7% and a risk free interest rate that ranges between 0.38% and 0.78%.
On February 22, 2013, the Company entered into a Securities Purchase Agreement with certain accredited investors for the issuance and sale in a private placement of an aggregate of $2,550,000 of Units at a purchase price of $0.75 per Unit. Each Unit consists of one share of Series A 8% Convertible Preferred Stock, par value $.001 per share, and a warrant to purchase one and one-quarter shares of the Company’s common stock, par value $.001 per share (subject to adjustment) at an exercise price of $1.00 per whole share (subject to adjustment). The total Series A 8% Convertible Preferred Stock issued was 3,400,001 shares, and the total warrants were 4,250,000. The Company used the net proceeds of the private placement for working capital, FDA trials, securing licensing partnerships, and general corporate purposes.
F-7
Table of Contents
The Company determined that warrants issued in February 2013 with the Series A 8% Convertible Preferred Stock should be classified as liabilities in accordance with ASC 815 because the warrants in question contain exercise price reset features that require the exercise price of the warrants be adjusted if the Company issues certain other equity related instruments at a lower price per share. The preferred stock was determined to have characteristics more akin to equity than debt. As a result, the conversion option was determined to be clearly and closely related to the preferred stock and therefore does not need to be bifurcated and classified as a liability. At June 30, 2014, there are no remaining 2013 warrants and therefore no associated warrant liability. Significant assumptions used at December 31, 2013 include a weighted average term of 4.1 years, a 5% probability that the warrant exercise price would be reset, volatility of 67.2% and a risk free interest rate range between 0.78% and 1.78%.
Reclassification of Prior Period Balances
Certain reclassifications have been made to prior period amounts to conform to current-year presentation.
Fair Value of Financial Instruments
The carrying amounts reported in the consolidated balance sheets for cash and cash equivalents, short-term settlement receivable, other current assets and accounts payable approximate their fair value because of the short-term nature of these items. Cash equivalents are measured on a recurring basis within the fair value hierarchy using Level 1 inputs.
The fair value of derivative instruments is determined by management with the assistance of an independent third party valuation specialist. Certain derivatives with limited market activity are valued using externally developed models that consider unobservable market parameters.
Stock-Based Compensation
The compensation cost relating to share-based payment transactions is measured based on the fair value of the equity or liability instruments at date of issuance and is expensed on a straight-line basis. The Company utilizes the Black-Scholes option-pricing model for purposes of estimating the fair value of each stock option on the date of grant. The Black-Scholes option-pricing model was developed for use in estimating the fair value of traded options which have no vesting restrictions and are fully transferable. In addition, option valuation models require the input of highly subjective assumptions including the expected volatility factor of the market price of the Company’s common stock (as determined by reviewing its historical public market closing prices).
Warrants to non-employees are generally vested and nonforfeitable upon the date of the grant. Accordingly fair value is determined on the grant date.
Recent Accounting Pronouncements
In May 2014, the FASB issued Accounting Standards Update No. 2014-09, Revenue from Contracts with Customers (ASU 2014-09), which supersedes nearly all existing revenue recognition guidance under U.S. GAAP. The core principle of ASU 2014-09 is to recognize revenues when promised goods or services are transferred to customers in an amount that reflects the consideration to which an entity expects to be entitled for those goods or services. ASU 2014-09 defines a five step process to achieve this core principle and, in doing so, more judgment and estimates may be required within the revenue recognition process than are required under existing U.S. GAAP.
The standard is effective for annual periods beginning after December 15, 2016, and interim periods therein, using either of the following transition methods: (i) a full retrospective approach reflecting the application of the standard in each prior reporting period with the option to elect certain practical expedients, or (ii) a retrospective approach with the cumulative effect of initially adopting ASU 2014-09 recognized at the date of adoption (which includes additional footnote disclosures). We are currently evaluating the impact of our pending adoption of ASU 2014-09 on our consolidated financial statements and have not yet determined the method by which we will adopt the standard in 2017. The Company currently does not have revenues but will consider any related impact going forward.
In June 2014, the FASB issued Accounting Standards Update 2014-10, Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation (ASU 2014-10), which eliminates the concept of a development stage entity (DSE) from U.S. GAAP. This change rescinds certain financial reporting requirements that have historically applied to DSEs and is intended to result in cost-savings for affected entities, such as certain start-up or research and development entities. The new standard also changes one related aspect of the variable interest entity (VIE) consolidation guidance in Topic 810.
F-8
Table of Contents
ASU 2014-10 is effective for public entities for annual reporting periods beginning after December 15, 2014 and interim periods therein. Early adoption is permitted. We early adopted ASU 2014-10 in our consolidated financial statements as of the third quarter of fiscal 2014.
In August 2014, the FASB issued Accounting Standards Update 2014-15, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern (ASU 2014-15), which addresses when and how to disclose going-concern uncertainties in the financial statements. ASU 2014-15 requires management to perform interim and annual assessments of an entity’s ability to continue as a going concern within one year after the date the financial statements are issued. An entity must provide certain disclosures if conditions or events raise substantial doubt about the entity’s ability to continue as a going concern. ASU 2014-15 applies to all entities and is effective for annual periods ending after December 15, 2016, and interim periods thereafter, with early adoption permitted. The amended guidance is not expected to have a material impact on the Company’s consolidated financial statements.
2. Commitments
Leases
The Company leases office and laboratory space in Knoxville, Tennessee on an annual basis, renewable for one year at our option. Rent expense was $60,000, $55,379 and $55,378 for the years ended December 31, 2014, 2013 and 2012, respectively.
Employee Agreements
On April 28, 2014, the Company entered into amended and restated executive employment agreements (the “Employment Agreements”) with each of the following executive officers of the Company: H. Craig Dees, Ph.D. to serve as its Chief Executive Officer, Timothy C. Scott, Ph.D. to serve as its President, Eric A. Wachter, Ph.D. to serve as its Chief Technology Officer, and Peter R. Culpepper to serve as its Chief Financial Officer and Chief Operating Officer (collectively, the “executives”).
Each Employment Agreement provides that such executive will be employed for an initial term of five years, subject to automatic renewal for successive one-year periods, unless the executive or the Company (i) terminates the Employment Agreement and the executive’s employment thereunder as provided in the Employment Agreement or (ii) provides notice of his or its intent not to renew. Each executive’s initial base salary is $500,000 per year, and any increases to such executive’s base salary shall be determined by the Compensation Committee of the Company’s Board of Directors in its sole discretion (the “Compensation Committee”). The executives are also eligible for annual bonuses and annual equity incentive awards as determined by the Compensation Committee in its sole discretion.
Each of the Employment Agreements generally provides that in the event that the executive’s employment is terminated (i) voluntarily by the executive without Good Reason (as defined in the Employment Agreement), or (ii) by the Company for Cause (as defined in the Employment Agreement), the Company shall pay the executive’s compensation only through the last day of the employment period and, except as may otherwise be expressly provided, the Company shall have no further obligation to the executive. In the event that the executive’s employment is terminated by the Company other than for Cause (including death or disability), or if the executive voluntarily resigns for Good Reason, for so long as the executive is not in breach of his continuing obligations under the non-competition, non-solicitation and confidentiality restrictions contained in the Employment Agreement, the Company shall continue to pay the executive (or his estate) an amount equal to his base salary in effect immediately prior to the termination of his employment for a period of 24 months, to be paid in accordance with the Company’s regular payroll practices through the end of the fiscal year in which termination occurs and then in one lump sum payable to the executive in the first month of the calendar year following termination, as well as any prorated bonuses determined by the Compensation Committee, plus benefits on a substantially equivalent basis to those which would have been provided to the executive.
During the term of each executive’s employment by the Company, and for a period of twenty-four (24) months following termination of employment, in the event that such executive voluntarily terminates his employment with the Company other than for Good Reason or such executive is terminated for Cause, then neither the executive nor any other person or entity with executive’s assistance shall (i) participate in any business that is directly competitive with the Company’s business or (ii) directly or indirectly, solicit any employee of the Company to quit or terminate their employment with the Company or employ as an employee, independent contractor, consultant, or in any other position, any person who was an employee of the Company or the Company’s affiliates within the preceding six months, subject to certain exceptions. In addition, without the express written consent of the Company, each executive shall not at any time (either during or after the termination of executive’s employment) use (other than for the benefit of the Company) or disclose to any other business entity proprietary or confidential information concerning the Company, any of their affiliates, or any of its officers. Neither shall such executive disclose any of the Company’s or the Company’s affiliates’ trade secrets or inventions of which he gained knowledge during his employment with the Company (subject to certain exceptions).
F-9
Table of Contents
3. Equity Transactions
Common Stock Issued for Services
(a) During the three months ended March 31, 2012, the Company issued 175,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $160,000. During the three months ended June 30, 2012, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $64,500. During the three months ended September 30, 2012, the Company issued 225,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $184,750. During the three months ended December 31, 2012, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $47,250. As the fair market value of these services was not readily determinable, these services were valued based on the fair market value of stock at grant date.
During the three months ended March 31, 2013, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $48,750. During the three months ended June 30, 2013, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $49,500. During the three months ended September 30, 2013, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $51,250. During the three months ended December 31, 2013, the Company issued 275,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $214,000. As the fair market value of these services was not readily determinable, these services were valued based on the fair market value of stock at grant date.
During the three months ended March 31, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $137,500. During the three months ended June 30, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $140,250. During the three months ended September 30, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $68,500. During the three months ended December 31, 2014, the Company issued 75,000 shares of common stock to consultants in exchange for services. Consulting costs charged to operations were $72,000. As the fair market value of these services was not readily determinable, these services were valued based on the fair market value of stock at grant date.
Warrants Issued for Services
(b) During the three months ended March 31, 2012, the Company issued 1,003,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $475,668. During the three months ended March 31, 2012, 1,500 warrants expired. During the three months ended June 30, 2012, the Company issued 454,500 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $183,908. During the three months ended June 30, 2012, 4,368,644 warrants expired. During the three months ended September 30, 2012, the Company issued 1,732,135 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $721,753. During the three months ended September 30, 2012, 122,833 warrants expired. During the three months ended December 31, 2012, the Company issued 452,500 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $130,697. During the three months ended December 31, 2012, 987,667 warrants expired. As the fair market value of these services was not readily determinable, these services were valued based on the fair market value, determined using the Black-Scholes option-pricing model. The fair market value for the warrants issued in 2012 ranged from $0.24 to $0.47 per warrant.
During the three months ended March 31, 2013, the Company issued 1,924,973 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $409,640. During the three months ended March 31, 2013, 859,833 expired warrants were forfeited. During the three months ended June 30, 2013, the Company issued 2,605,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $931,655. During the three months ended June 30, 2013, 1,051,500 expired warrants were forfeited. During the three months ended September 30, 2013, the Company issued 442,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $186,223. During the three months ended September 30, 2013, 136,500 expired warrants were forfeited. During the three months ended December 31, 2013, the Company issued 209,473 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $259,306. During the three months ended December 31, 2013, 247,973 expired warrants were forfeited. During the three months ended December 31, 2013, 4,480,005 warrants were exercised on a cashless basis resulting in 2,386,004 shares being issued. During the three months ended December 31, 2013, 3,899,840 warrants were exercised for $3,412,392 resulting in 3,899,840 common shares issued. As the fair market value of these services was not readily determinable, these services were valued based on the fair market value of the warrants, determined using the Black-Scholes option-pricing model. The fair market value for the warrants issued in 2013 ranged from $0.10 to $1.97 per warrant.
F-10
Table of Contents
During the three months ended March 31, 2014, the Company issued 733,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $900,317. During the three months ended March 31, 2014, 121,500 expired warrants were forfeited. During the three months ended March 31, 2014, 12,522,198 warrants were exercised on a cashless basis resulting in 9,100,824 common shares being issued. During the three months ended March 31, 2014, 3,036,218 warrants were exercised for $2,672,364 resulting in 3,036,218 common shares issued. During the three months ended June 30, 2014, the Company issued 202,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $450,002. During the three months ended June 30, 2014, 315,000 expired warrants were forfeited. During the three months ended June 30, 2014, 1,594,082 warrants were exercised on a cashless basis resulting in 915,467 common shares being issued. During the three months ended June 30, 2014, 372,000 warrants were exercised for $372,000 resulting in 372,000 common shares issued. During the three months ended September 30, 2014, the Company issued 6,000 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $4,189. During the three months ended September 30, 2014, 228,500 expired warrants were forfeited. During the three months ended December 31, 2014, the Company issued 1,503,913 fully vested warrants to consultants in exchange for services. Consulting costs charged to operations were $966,819. During the three months ended December 31, 2014, 1,027,635 expired warrants were forfeited. As the fair market value of these services was not readily determinable, these services were valued based on the fair market value of the warrants, determined using the Black-Scholes option-pricing model. The fair market value for the warrants issued in 2014 ranged from $0.55 to $2.56 per warrant.
There are no provisions or obligations that would require the Company to cash settle any of its outstanding warrants. The equity classification of certain of the Company’s warrants is appropriate considering that these warrants provide the counterparties the right to purchase a fixed number of shares at a fixed price and the terms are not subject to any potential adjustment.
Private Offerings of Common Stock and Warrants
(c) The Company determined that warrants issued January 13, 2011 and referred to as Series A Warrants and Series C Warrants should be classified as liabilities in accordance with ASC 815 because the warrants in question contain exercise price reset features that require the exercise price of the warrants be adjusted if the Company issues certain other equity related instruments at a lower price per share. The value of the warrant liability was determined based on the Monte-Carlo Simulation model at the date the warrants were issued. The warrant liability is then revalued at each subsequent quarter. At December 31, 2012, the Series A Warrants and the Series C Warrants exercise price of $1.12 per share was reduced to $0.675 per share due to a new issuance price, net of commissions, from a private offering of common stock and warrants to accredited investors during the three months ended December 31, 2012 and pursuant to their exercise price reset provision. For the year ended December 31, 2012 there was a gain recognized from the revaluation of the warrant liability of $495,338. During the three months ended December 31, 2013, 1,269,520 of the Series A Warrants were exercised. During the three months ended December 31, 2013, 748,663 of the Series C Warrants were exercised. The Company determined the fair value of the Series A and Series C Warrants exercised on the date of exercise and adjusted the related warrant liability accordingly. The adjusted fair value of the Series A and Series C Warrants exercised of $1,620,081 was reclassified into additional paid-in capital. For the year ended December 31, 2013 there was a loss recognized from the revaluation of the warrant liability of $3,873,187. During the three months ended March 31, 2014, 858,825 of the Series A Warrants were exercised. During the three months ended March 31, 2014, 697,092 of the Series C Warrants were exercised. The Company determined the fair value of the Series A and Series C Warrants exercised on the date of exercise and adjusted the related warrant liability accordingly. The adjusted fair value of the Series A and Series C Warrants exercised in 2014 of $3,911,370 was reclassified into additional paid-in capital. For the year ended December 31, 2014 there was a loss recognized from the revaluation of the warrant liability of $959,320.
During the three months ended June 30, 2012 the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $2,077,796. The Company accepted subscriptions, in the aggregate, for 1,855,176 shares of common stock, and five year warrants to purchase 1,855,176 shares of common stock. Investors received five year fully vested warrants to purchase up to 100% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants was $1.12. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company issued five year fully vested warrants to purchase 371,035 shares of common stock with an exercise price of $1.12 to Network 1 Financial Securities, Inc., which represents 20% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc. During the three months ended December 31, 2012 the Company completed a private offering of
F-11
Table of Contents
common stock and warrants to accredited investors for gross proceeds of $2,379,365. The Company accepted subscriptions, in the aggregate, for 3,172,486 shares of common stock, and five year warrants to purchase 3,172,486 shares of common stock. Investors received five year fully vested warrants to purchase up to 100% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.00 per share. The purchase price for each share of common stock together with the warrants was $0.75. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $279,317 and issued five year fully vested warrants to purchase 317,249 shares of common stock with an exercise price of $1.00 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc. During the three months ended December 31, 2012, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $710,000. The Company accepted subscriptions, in the aggregate, for 946,666 shares of common stock, and five year warrants to purchase 946,666 shares of common stock. Investors received five year fully vested warrants to purchase up to 100% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.00 per share. The purchase price for each share of common stock together with the warrants was $0.75. The Company used the proceeds for working capital and other general corporate purposes. Maxim Group LLC served as placement agent for the offering. In connection with the offering, the Company paid $97,300 and issued five year fully vested warrants to purchase 94,667 shares of common stock with an exercise price of $1.00 to Maxim Group LLC, which represents 10% of the total number of shares of common stock sold to investors solicited by Maxim Group LLC.
During the three months ended March 31, 2013, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $4,045,510. The Company accepted subscriptions, in the aggregate, for 5,394,013 shares of common stock, and five year warrants to purchase 7,277,264 shares of common stock. Investors received five year fully vested warrants to purchase up to 100% to 150% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.00 per share. The purchase price for each share of common stock together with the warrants was $0.75. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $522,640 and issued five year fully vested warrants to purchase 539,401 shares of common stock with an exercise price of $1.00 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc. During the three months ended June 30, 2013 the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $2,641,501. The Company accepted subscriptions, in the aggregate, for 3,522,001 shares of common stock, and five year warrants to purchase 5,283,003 shares of common stock. Investors received five year fully vested warrants to purchase up to 150% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.00 per share. The purchase price for each share of common stock together with the warrants was $0.75. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $314,173, accrued $32,500 at June 30, 2013 which was paid in July 2013 and issued five year fully vested warrants to purchase 352,200 shares of common stock with an exercise price of $1.00 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc.
During the three months ended September 30, 2013, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $4,613,037. The Company accepted subscriptions, in the aggregate, for 6,150,718 shares of common stock and five year warrants to purchase 9,226,077 shares of common stock. Investors received five year fully vested warrants to purchase up to 150% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.00 per share. The purchase price for each share of common stock together with the warrants was $0.75. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $564,686 and issued five year fully vested warrants to purchase 615,072 shares of common stock with an exercise price of $1.00 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc. During the three months ended September 30, 2013, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $2,687,500. The Company accepted subscriptions, in the aggregate, for 3,583,333 shares of common stock and five year warrants to purchase 5,375,000 shares of common stock. Investors received five year fully vested warrants to purchase up to 150% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.00 per share. The purchase price for each share of common stock together with the warrants was $0.75. The Company used the proceeds for working capital and other general corporate purposes. Maxim Group LLC served as placement agent for the offering. In connection with the offering, the Company paid $349,375 and issued five year fully vested warrants to purchase 358,333 shares of common stock with an exercise price of $1.00 to Maxim Group LLC, which represents 10% of the total number of shares of
F-12
Table of Contents
common stock sold to investors solicited by Maxim Group LLC. During the three months ended December 31, 2013, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $5,820,588. The Company accepted subscriptions, in the aggregate, for 7,760,784 shares of common stock and five year warrants to purchase 11,641,176 shares of common stock. Investors received five year fully vested warrants to purchase up to 150% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.00 per share. The purchase price for each share of common stock together with the warrants was $0.75. The Company plans to use the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $747,302 and issued five year fully vested warrants to purchase 776,078 shares of common stock with an exercise price of $1.00 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc. During the three months ended December 31, 2013, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $1,312,500. The Company accepted subscriptions, in the aggregate, for 1,750,000 shares of common stock and five year warrants to purchase 2,625,000 shares of common stock. Investors received five year fully vested warrants to purchase up to 150% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.00 per share. The purchase price for each share of common stock together with the warrants was $0.75. The Company used the proceeds for working capital and other general corporate purposes. Maxim Group LLC served as placement agent for the offering. In connection with the offering, the Company paid $170,625 and issued five year fully vested warrants to purchase 175,000 shares of common stock with an exercise price of $1.00 to Maxim Group LLC, which represents 10% of the total number of shares of common stock sold to investors solicited by Maxim Group LLC.
During the three months ended June 30, 2014, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $5,000,000. The Company accepted subscriptions, in the aggregate, for 2,000,000 shares of common stock and five year warrants to purchase 2,000,000 shares of common stock. Investors received five year fully vested warrants to purchase up to 100% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $3.00 per share. The purchase price for each share of common stock together with the warrants was $2.50. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $650,000 and issued five year fully vested warrants to purchase 300,000 shares of common stock with an exercise price of $2.50 to Network 1 Financial Securities, Inc., which represents 15% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc. During the three months ended September 30, 2014, the Company commenced a private offering of up to $15 million of common stock and five-year warrants to accredited investors. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants is $1.00. The Company plans to use the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. is serving as placement agent for the offering. During the three months ended September 30, 2014, the Company received subscriptions, in the aggregate, for 3,586,300 shares of common stock and five year warrants to purchase 1,793,150 shares of common stock for an aggregate of $3,586,300. Investors will receive five year fully vested warrants to purchase up to 50% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants is $1.00. The Company plans to use the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. is serving as placement agent for the offering. In connection with the offering, the Company paid $466,219 and issued five year fully vested warrants to purchase 358,630 shares of common stock with an exercise price of $1.25 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock subscribed for by investors solicited by Network 1 Financial Securities, Inc. During the three months ended December 31, 2014, the Company completed a private offering of common stock and warrants to accredited investors for gross proceeds of $4,198,300. The Company accepted subscriptions, in the aggregate, for 4,198,300 shares of common stock and five year warrants to purchase 2,099,150 shares of common stock. Investors received five year fully vested warrants to purchase up to 50% of the number of shares purchased by the investors in the offering. The warrants have an exercise price of $1.25 per share. The purchase price for each share of common stock together with the warrants was $1.00. The Company used the proceeds for working capital and other general corporate purposes. Network 1 Financial Securities, Inc. served as placement agent for the offering. In connection with the offering, the Company paid $545,779 and issued five year fully vested warrants to purchase 419,830 shares of common stock with an exercise price of $1.25 to Network 1 Financial Securities, Inc., which represents 10% of the total number of shares of common stock sold to investors solicited by Network 1 Financial Securities, Inc.
Private Offering of Convertible Preferred Stock with Warrants
(d) In March and April 2010, the Company issued 8% Convertible Preferred Stock with warrants. The Company determined that warrants issued with the 8% Convertible Preferred Stock should be classified as liabilities in accordance with ASC 815 because the warrants in question contain exercise price reset features that require the exercise price of the warrants be adjusted if the Company issues certain other equity related instruments at a lower price per share. The value of the warrant
F-13
Table of Contents
liability was determined based on the Monte-Carlo Simulation model at the date the warrants were issued. The warrant liability is then revalued at each subsequent quarter. For the year ended December 31, 2012, there was a gain recognized from the revaluation of the warrant liability of $1,272,580. During the three months ended December 31, 2013, 1,146,662 of the warrants included in the warrant liability were exercised. The Company determined the fair value of the warrants exercised on the date of exercise and adjusted the related warrant liability accordingly. The adjusted fair value of the warrants exercised of $765,997 was reclassified into additional paid-in capital. For the year ended December 31, 2013, there was a loss recognized from the revaluation of the warrant liability of $6,911,583. During the three months ended March 31, 2014, 1,756,665 of the warrants included in the warrant liability were exercised. During the three months ended June 30, 2014, 133,232 of the warrants included in the warrant liability were exercised. The Company determined the fair value of the warrants exercised on the date of exercise and adjusted the related warrant liability accordingly. The adjusted fair value of the warrants exercised in 2014 of $2,377,133 was reclassified into additional paid-in capital. For the year ended December 31, 2014, there was a gain recognized from the revaluation of the warrant liability of $4,222,519.
Dividends on the 8% Convertible Preferred Stock accrued at an annual rate of 8% of the original issue price and are payable in either cash or common stock. If the dividend is paid in common stock, the number of shares of common stock will equal the quotient of the amount of cash dividends divided by the market price of the stock on the dividend payment date. The dividends are payable quarterly on the 15th day after the quarter-end. The Company has a deficit and, as a result, the dividends will be recorded against additional paid-in capital. In January 2012, the Company issued 64,183 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of January 15, 2012. At March 31, 2012, the Company recognized dividends of $50,631 which are included in dividends on preferred stock on the consolidated statement of operations. In April 2012, the Company issued 58,490 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of April 16, 2012. At June 30, 2012, the Company recognized dividends of $51,194 which are included in dividends on preferred stock on the consolidated statement of operations. In July 2012, the Company issued 61,424 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of July 16, 2012. At September 30, 2012, the Company recognized dividends of $43,884 which are included in dividends on preferred stock on the consolidated statement of operations. In October 2012, the Company issued 69,222 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of October 15, 2012. At December 31, 2012, the Company recognized dividends of $37,478 which are included in dividends on preferred stock on the consolidated statement of operations. In January 2013, the Company issued 61,022 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of January 15, 2013. At March 31, 2013, the Company recognized dividends of $21,921 which are included in dividends on preferred stock on the consolidated statement of operations. In April 2013, the Company issued 29,384 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of April 15, 2013. At June 30, 2013, the Company recognized dividends of $22,164 which are included in dividends on preferred stock on the consolidated statement of operations. In July 2013, the Company issued 34,598 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of July 15, 2013. At September 30, 2013, the Company recognized dividends of $10,586 which are included in dividends on preferred stock on the consolidated statement of operations. In October 2013, the Company issued 12,066 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of October 15, 2013. At December 31, 2013, the Company recognized no dividends due because of the full conversion of preferred stock to common stock as of December 31, 2013.
During the three months ended March 31, 2012 there were 100,000 shares of the Company’s redeemable preferred stock that converted into 100,000 shares of the Company’s common stock. During the three months ended September 30, 2012 there were 490,000 shares of the Company’s redeemable preferred stock that converted into 490,000 shares of the Company’s common stock. During the three months ended December 31, 2012, there were 463,480 shares of the Company’s redeemable preferred stock that converted into 463,480 shares of the Company’s common stock. During the three months ended March 31, 2013, there were 593,000 shares of the Company’s redeemable preferred stock that converted into 593,000 shares of the Company’s common stock. During the three months ended June 30, 2013 there were 403,520 shares of the Company’s redeemable preferred stock that converted into 403,520 shares of the Company’s common stock. During the three months ended September 30, 2013, there were 734,999 shares of the Company’s redeemable preferred stock that converted into 734,999 shares of the Company’s common stock. During the three months ended December 31, 2013, there were 746,666 shares of the Company’s redeemable preferred stock that converted into 746,666 shares of the Company’s common stock. At December 31, 2013 there was no 8% Convertible Preferred Stock outstanding.
(e) On February 22, 2013, the Company entered into a Securities Purchase Agreement with certain accredited investors for the issuance and sale in a private placement of an aggregate of $2,550,000 of Units at a purchase price of $0.75 per Unit. Each Unit consists of one share of Series A 8% Convertible Preferred Stock, par value $.001 per share, and a warrant to purchase one and one-quarter shares of the Company’s common stock, par value $.001 per share (subject to adjustment) at an exercise price of $1.00 per whole share (subject to adjustment). The total Series A 8% Convertible Preferred Stock issued was 3,400,001 shares, and the total warrants were 4,250,000. The Company used the net proceeds of the private placement for working capital, FDA trials, securing licensing partnerships, and general corporate purposes.
F-14
Table of Contents
The Company determined that warrants issued in February, 2013 with the Series A 8% Convertible Preferred Stock should be classified as liabilities in accordance with ASC 815 because the warrants in question contain exercise price reset features that require the exercise price of the warrants be adjusted if the Company issues certain other equity related instruments at a lower price per share.
The preferred stock was determined to have characteristics more akin to equity than debt. As a result, the conversion option was determined to be clearly and closely related to the preferred stock and therefore does not need to be bifurcated and classified as a liability. The proceeds received from the issuance of the preferred stock were first allocated to the fair value of the warrants with the remainder allocated to the preferred stock. The fair value of the preferred stock if converted on the date of issuance was greater than the value allocated to the preferred stock. As a result, a beneficial conversion amount was recorded upon issuance. The fair value of the warrants recorded from the February 2013 issuance was $1,297,950 resulting in a beneficial conversion amount of $1,025,950. The beneficial conversion has been recorded as a deemed dividend as of March 31, 2013 and is included in dividends on preferred stock on the consolidated statements of operations.
The value of the warrant liability was determined based on the Monte-Carlo Simulation model at the date the warrants were issued. The warrant liability is then revalued at each subsequent quarter. During the three months ended December 31, 2013, 2,400,000 of the warrants included in the warrant liability were exercised, resulting in 2,400,000 common shares being issued. The Company determined the fair value of the warrants exercised on the date of exercise and adjusted the related warrant liability accordingly. The adjusted fair value of the warrants exercised of $2,016,000 was reclassified into additional paid-in capital. For the year ended December 31, 2013, there was a loss recognized from the revaluation of the warrant liability of $3,886,360. During the three months ended March 31, 2014, 1,650,000 of the warrants included in the warrant liability were exercised. During the three months ended June 30, 2014, 200,000 of the warrants included in the warrant liability were exercised, which is the remainder of the 2013 warrants. The Company determined the fair value of the warrants exercised on the date of exercise and adjusted the related warrant liability accordingly. The adjusted fair value of the warrants exercised in 2014 of $4,047,116 was reclassified into additional paid-in capital. For the year ended December 31, 2014, there was a loss recognized from the revaluation of the warrant liability of $878,806.
Dividends on the Series A 8% Convertible Preferred Stock accrued at an annual rate of 8% of the original issue price and are payable in either cash or common stock. If the dividend is paid in common stock, the number of shares of common stock will equal the quotient of the amount of cash dividends divided by the market price of the stock on the dividend payment date. The dividends are payable quarterly on the 15th day after the quarter-end. The Company paid the dividends in common stock although was required to pay the initial dividends due in cash. The Company has a deficit and, as a result, the dividends are recorded against additional paid-in capital. At March 31, 2013, the Company recognized dividends of $29,063 which are included in dividends on preferred stock on the consolidated statement of operations and were paid in April 2013. At June 30, 2013, the Company recognized dividends of $50,860 which are included in dividends on preferred stock on the consolidated statement of operations. In July 2013, the Company issued 79,401 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of July 15, 2013. At September 30, 2013, the Company recognized dividends of $28,104 which are included in dividends on preferred stock on the consolidated statement of operations. In October 2013, the Company issued 32,033 shares of common stock in dividends on preferred stock in lieu of cash dividends due as of October 15, 2013. At December 31, 2013, the Company recognized no dividends due because of the full conversion of preferred stock to common stock as of January 15, 2014. In 2014, the Company recognized no dividends because of the conversion of all outstanding preferred stock to common stock as of January 15, 2014.
During the three months ended September 30, 2013, there were 441,667 shares of the Company’s Series A 8% Convertible Preferred Stock that converted into 441,667 shares of the Company’s common stock. During the three months ended December 31, 2013, there were 2,925,000 shares of the Company’s Series A 8% Convertible Preferred Stock that converted into 2,925,000 shares of the Company’s common stock. In January 2014, there were 33,334 shares of the Company’s Series A 8% Convertible Preferred Stock that converted into 33,334 shares of the Company’s common stock. As of January 15, 2014, there were no shares of Series A 8% Convertible Preferred Stock outstanding.
Common Stock Purchase Agreements
(f) In December 2010, we entered into a purchase agreement with Lincoln Park Capital Fund, LLC, pursuant to which the Company could, in our sole discretion, direct Lincoln Park to purchase up to an additional $30,000,000 of our common stock over the 30-month term of the purchase agreement at no less than $0.75 per share. On June 23, 2013, our agreement with Lincoln Park Capital Fund, LLC expired.
F-15
Table of Contents
On July 22, 2013 the Company entered into a Purchase Agreement with Alpha Capital Anstalt pursuant to which the Company may, in the Company’s sole discretion, direct the purchase up to $30,000,000 of the Company’s common stock over the 30-month term of the Purchase Agreement. From time to time during the term of the Purchase Agreement, the Company may, in its sole discretion direct the purchase up to 100,000 shares of the Company’s common stock at a per share purchase price equal to the lesser of (i) the lowest sale price of the Company’s common stock reported on the OTCQB or NYSE MKT on the purchase date and (ii) the arithmetic average of the three lowest closing sale prices for the Company’s common stock during the 12 consecutive business days ending on the business day immediately preceding the purchase date. The Company may, under certain circumstances, at its discretion, increase the amount of common stock that it sells on each purchase date. The committed obligation under any single regular purchase shall not exceed $250,000, unless the parties mutually agree to increase the dollar amount of any regular purchase. In no event may Alpha Capital Anstalt purchase shares of the Company’s common stock for less than $0.75 per share. In consideration of entering into the Purchase Agreement and making the commitment to purchase the Purchase Shares, the Company issued 250,000 shares of the Company’s common stock to Alpha Capital Anstalt. Costs charged to operations for this commitment fee were $162,500. The Purchase Agreement may be terminated by the Company at any time, at its discretion, without cost to the Company. As of December 31, 2014, the Company had the full amount of the Purchase Agreement available for use.
4. Stock Incentive Plan and Warrants
Options
The Company maintained two long-term incentive compensation plans which have been terminated; namely, the Provectus Pharmaceuticals, Inc. 2002 Stock Plan, which provided for the issuance of 18,450,000 shares of common stock pursuant to stock options, and the 2012 Stock Plan, which provided for the issuance of up to 20,000,000 shares of common stock pursuant to stock options. Currently, the Provectus Biopharmaceuticals, Inc. 2014 Equity Compensation Plan provides for the issuance of up to 20,000,000 shares of common stock pursuant to stock options for the benefit of eligible employees and directors of the Company.
Options granted under the 2002, 2012 and 2014 Stock Plans were either “incentive stock options” within the meaning of Section 422 of the Internal Revenue Code or options which were not incentive stock options. The stock options are exercisable over a period determined by the Board of Directors (through its Compensation Committee), but generally no longer than 10 years after the date they are granted.
For stock options granted to employees during 2014, 2013 and 2012, the Company has estimated the fair value of each option granted using the Black-Scholes option pricing model with the following assumptions:
| 2014 | 2013 | 2012 | ||||
| Weighted average fair value per options granted |
$0.77 | $0.57 | $0.73 | |||
| Significant assumptions (weighted average) risk-free interest rate at grant date |
0.25% | 0.25% | 0.25% | |||
| Expected stock price volatility |
85% – 92% | 83% – 85% | 83% – 87% | |||
| Expected option life (years) |
10 | 10 | 10 |
On May 14, 2012, the Company issued 50,000 stock options to a newly appointed member of the board. On June 28, 2012, the Company issued 200,000 stock options to its re-elected members of the board. All of the stock options issued in 2012 vest on the date of grant and have an exercise price equal to the fair market price on the date of issuance.
One employee of the Company exercised 18,750 options at an exercise price of $0.32 per share of common stock for $6,000 and 25,000 options at an exercise price of $0.60 per share of common stock for $15,000 during the three months ended June 30, 2013. One former non-employee member of the board forfeited 25,000 stock options on May 29, 2013. On August 19, 2013, the Company issued 250,000 stock options to its re-elected members of the board. All of the stock options issued in 2013 vest on the date of grant and have an exercise price equal to the fair market price on the date of issuance.
One employee of the Company exercised 25,000 options at an exercise price of $0.95 per share of common stock for $23,750, 14,248 options at an exercise price of $0.75 per share of common stock for $10,686 and 600,000 options at an exercise price of $0.93 per share of common stock for $558,000 during the three months ended March 31, 2014. Another employee of the Company exercised 300,000 options at an exercise price of $1.10 per share of common stock for $330,000
F-16
Table of Contents
during the three months ended March 31, 2014. Another employee of the Company exercised 189,624 options at an exercise price of $1.10 per share of common stock for $208,586 during the three months ended March 31, 2014. One employee of the Company forfeited 300,000 stock options on February 26, 2014. One employee of the Company exercised 25,000 options at an exercise price of $0.95 per share of common stock for $23,750 during the three months ended June 30, 2014. Another employee of the Company exercised 100,000 options at an exercise price of $1.25 per share of common stock for $125,000 during the three months ended June 30, 2014. A former non-employee member of the board of directors exercised 25,000 options at an exercise price of $0.95 per share of common stock for $23,750 during the three months ended June 30, 2014. One employee of the Company forfeited 25,000 stock options on May 27, 2014. On July 29, 2014, the Company issued a total of 150,000 stock options to its three re-elected non-employee members of the board of directors. All of the stock options issued in 2014 vested on the date of grant and have an exercise price equal to the fair market price on the date of issuance. One employee of the Company exercised 96,875 options at an exercise price of $0.64 per share of common stock for $62,000, and 126,361 options at an exercise price of $0.64 per share of common stock for $80,871 during the three months ended December 31, 2014. Three employees of the Company had fully vested options rescinded during the three months ended December 31, 2014 due to the terms of the settlement discussed in Note 9.
The compensation cost relating to share-based payment transactions is measured based on the fair value of the equity or liability instruments issued. For purposes of estimating the fair value of each stock option on the date of grant, the Company utilized the Black-Scholes option-pricing model. The Black-Scholes option-pricing model was developed for use in estimating the fair value of traded options, which have no vesting restrictions and are fully transferable. In addition, option-pricing models require the input of highly subjective assumptions including the expected volatility factor of the market price of the Company’s common stock (as determined by reviewing its historical public market closing prices). Included in the results for the year ended December 31, 2014, is $115,645 of stock-based compensation expense which relates to the fair value of stock options vested in 2014. Included in the results for the year ended December 31, 2013, is $142,310 of stock-based compensation expense which relates to the fair value of stock options vested in 2013. Included in the results for the year ended December 31, 2012, is $183,028 of stock-based compensation expense which relates to the fair value of stock options vested in 2012.
The following table summarizes the options granted, exercised, outstanding and exercisable as of December 31, 2012, 2013 and 2014:
| Shares | Exercise Price Per Share |
Weighted Average Exercise Price |
||||||||||
| Outstanding at January 1, 2012 |
14,890,956 | $ | 0.32 – 1.50 | $ | 0.98 | |||||||
| Granted |
250,000 | $ | 0.84 – 0.93 | $ | 0.86 | |||||||
| Exercised |
— | — | — | |||||||||
| Forfeited |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Outstanding and exercisable at December 31, 2012 |
15,140,956 | $ | 0.32 – 1.50 | $ | 0.97 | |||||||
|
|
|
|
|
|
|
|||||||
| Outstanding at January 1, 2013 |
15,140,956 | $ | 0.32 – 1.50 | $ | 0.97 | |||||||
| Granted |
250,000 | $ | 0.67 | $ | 0.67 | |||||||
| Exercised |
(43,750 | ) | $ | 0.32 – 0.60 | $ | 0.48 | ||||||
| Forfeited |
(25,000 | ) | $ | 0.60 | $ | 0.60 | ||||||
|
|
|
|
|
|
|
|||||||
| Outstanding and exercisable at December 31, 2013 |
15,322,206 | $ | 0.62 – 1.50 | $ | 0.97 | |||||||
|
|
|
|
|
|
|
|||||||
| Outstanding at January 1, 2014 |
15,322,206 | $ | 0.62 – 1.50 | $ | 0.97 | |||||||
| Granted |
150,000 | $ | 0.88 | $ | 0.88 | |||||||
| Settlement |
(2,800,000 | ) | $ | 0.93 – 1.00 | $ | 0.97 | ||||||
| Exercised |
(1,502,108 | ) | $ | 0.64 – 1.25 | $ | 0.96 | ||||||
| Forfeited |
(325,000 | ) | $ | 0.95 – 1.10 | $ | 1.09 | ||||||
|
|
|
|
|
|
|
|||||||
| Outstanding and exercisable at December 31, 2014 |
10,845,098 | $ | 0.64 – 1.50 | $ | 0.97 | |||||||
|
|
|
|
|
|
|
|||||||
F-17
Table of Contents
The following table summarizes information about stock options outstanding at December 31, 2014 in order of issuance from oldest to newest.
| Exercise Price |
Number Outstanding at December 31, 2014 |
Weighted Average Remaining contractual Life |
Outstanding Weighted Average Exercise price |
Number Exercisable at December 31, 2014 |
Exercisable Weighted Average Exercise Price |
|||||||||||||||
| $0.64 |
376,764 | 0.00 years | $ | 0.64 | 376,764 | $ | 0.64 | |||||||||||||
| $0.75 |
708,334 | 0.42 years | $ | 0.75 | 708,334 | $ | 0.75 | |||||||||||||
| $0.94 |
575,000 | 0.92 years | $ | 0.94 | 575,000 | $ | 0.94 | |||||||||||||
| $1.02 |
4,135,000 | 1.50 years | $ | 1.02 | 4,135,000 | $ | 1.02 | |||||||||||||
| $1.50 |
200,000 | 2.50 years | $ | 1.50 | 200,000 | $ | 1.50 | |||||||||||||
| $1.16 |
50,000 | 3.42 years | $ | 1.16 | 50,000 | $ | 1.16 | |||||||||||||
| $1.00 |
150,000 | 3.50 years | $ | 1.00 | 150,000 | $ | 1.00 | |||||||||||||
| $1.04 |
250,000 | 4.50 years | $ | 1.04 | 250,000 | $ | 1.04 | |||||||||||||
| $1.16 |
250,000 | 5.50 years | $ | 1.16 | 250,000 | $ | 1.16 | |||||||||||||
| $1.00 |
1,600,000 | 5.50 years | $ | 1.00 | 1,600,000 | $ | 1.00 | |||||||||||||
| $1.04 |
250,000 | 6.50 years | $ | 1.04 | 250,000 | $ | 1.04 | |||||||||||||
| $0.99 |
50,000 | 6.50 years | $ | 0.99 | 50,000 | $ | 0.99 | |||||||||||||
| $0.93 |
1,600,000 | 6.67 years | $ | 0.93 | 1,600,000 | $ | 0.93 | |||||||||||||
| $0.93 |
50,000 | 7.38 years | $ | 0.93 | 50,000 | $ | 0.93 | |||||||||||||
| $0.84 |
200,000 | 7.50 years | $ | 0.84 | 200,000 | $ | 0.84 | |||||||||||||
| $0.67 |
250,000 | 8.71 years | $ | 0.67 | 250,000 | $ | 0.67 | |||||||||||||
| $0.88 |
150,000 | 9.67 years | $ | 0.88 | 150,000 | $ | 0.88 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| 10,845,098 | 3.85 years | $ | 0.97 | 10,845,098 | $ | 0.97 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
The total intrinsic value of options exercised during the year ended December 31, 2014 which were in the money was $1,327,300.
The total intrinsic value of options exercised during the year ended December 31, 2013 which were in the money was $7,000.
There were no options exercised during the year ended December 31, 2012.
The following is a summary of nonvested stock option activity for the year ended December 31, 2014:
| Number of Shares | Weighted Average Grant-Date Fair Value |
|||||||
| Nonvested at December 31, 2013 |
— | $ | — | |||||
| Granted |
150,000 | $ | 0.77 | |||||
| Vested |
(150,000 | ) | $ | 0.77 | ||||
| Canceled |
— | — | ||||||
|
|
|
|
|
|||||
| Nonvested at December 31, 2014 |
— | $ | — | |||||
|
|
|
|
|
|||||
As of December 31, 2014, there was no unrecognized compensation cost related to nonvested share-based compensation arrangements granted under the Plan.
F-18
Table of Contents
The following is a summary of the aggregate intrinsic value of shares outstanding and exercisable at December 31, 2014. The aggregate intrinsic value of stock options outstanding and exercisable is defined as the difference between the market value of the Company’s stock as of the end of the period and the exercise price of the stock options which are in the money.
| Number of Shares | Aggregate Intrinsic Value |
|||||||
| Outstanding and Exercisable at December 31, 2014 |
10,845,098 | $ | 128,199 | |||||
|
|
|
|
|
|||||
Warrants
The following table summarizes the warrants granted, exercised, outstanding and exercisable as of December 31, 2012, 2013 and 2014.
| Warrants | Exercise Price Per Warrant |
Weighted Average Exercise Price |
||||||||||
| Outstanding at January 1, 2012 |
25,119,247 | $ | 0.91 – 2.00 | $ | 1.15 | |||||||
| Granted |
10,399,414 | $ | 0.68 – 1.50 | $ | 1.00 | |||||||
| Exercised |
— | — | — | |||||||||
| Forfeited |
(5,480,644 | ) | $ | 0.91 – 1.50 | $ | 1.29 | ||||||
|
|
|
|
|
|
|
|||||||
| Outstanding and exercisable at December 31, 2012 |
30,038,017 | $ | 0.68 – 2.00 | $ | 1.05 | |||||||
|
|
|
|
|
|
|
|||||||
| Outstanding at January 1, 2013 |
30,038,017 | $ | 0.68 – 2.00 | $ | 1.05 | |||||||
| Granted |
53,675,050 | $ | 0.68 – 1.12 | $ | 1.00 | |||||||
| Exercised |
(8,379,845 | ) | $ | 0.68 – 1.25 | $ | 0.93 | ||||||
| Forfeited |
(2,295,806 | ) | $ | 0.68 – 1.12 | $ | 0.87 | ||||||
|
|
|
|
|
|
|
|||||||
| Outstanding and exercisable at December 31, 2013 |
73,037,416 | $ | 0.68 – 2.00 | $ | 1.03 | |||||||
|
|
|
|
|
|
|
|||||||
| Outstanding at January 1, 2014 |
73,037,416 | $ | 0.68 – 2.00 | $ | 1.03 | |||||||
| Granted |
9,415,673 | $ | 1.00 – 3.00 | $ | 1.61 | |||||||
| Exercised |
(17,524,498 | ) | $ | 0.68 – 1.50 | $ | 1.01 | ||||||
| Forfeited |
(1,692,635 | ) | $ | 0.95 – 1.25 | $ | 1.07 | ||||||
|
|
|
|
|
|
|
|||||||
| Outstanding and exercisable at December 31, 2014 |
63,235,956 | $ | 0.68 – 3.00 | $ | 1.12 | |||||||
|
|
|
|
|
|
|
|||||||
The following table summarizes information about warrants outstanding at December 31, 2014.
| Exercise Price |
Number Outstanding and Exercisable at December 31, 2014 |
Weighted Average Remaining Contractual Life in Years |
Weighted Average Exercise Price |
|||||||||
| $0.68 |
134,994 | 1.00 | $ | 0.68 | ||||||||
| $0.95 |
187,467 | 0.00 | $ | 0.95 | ||||||||
| $1.00 |
48,844,755 | 3.30 | $ | 1.00 | ||||||||
| $1.12 |
1,793,036 | 1.42 | $ | 1.12 | ||||||||
| $1.25 |
8,782,703 | 2.98 | $ | 1.25 | ||||||||
| $1.50 |
670,001 | 1.30 | $ | 1.50 | ||||||||
| $1.75 |
200,000 | 1.00 | $ | 1.75 | ||||||||
| $2.00 |
323,000 | 2.25 | $ | 2.00 | ||||||||
| $2.50 |
300,000 | 4.33 | $ | 2.50 | ||||||||
| $3.00 |
2,000,000 | 4.33 | $ | 3.00 | ||||||||
|
|
|
|
|
|
|
|||||||
| 63,235,956 | 3.19 | $ | 1.12 | |||||||||
|
|
|
|
|
|
|
|||||||
F-19
Table of Contents
5. Related Party Transactions
The Company paid one non-employee member of the board $54,000 for consulting services performed as of December 31, 2012. The Company paid another non-employee member of the board $75,000 for consulting services performed as of December 31, 2012 and issued 100,000 fully vested warrants in exchange for services. Consulting costs charged to operations were $47,520 for the services for which these warrants were issued. As the fair market value of these services was not readily determinable, these services were valued based on the fair market value, determined using the Black-Scholes option-pricing model. The Company paid a third non-employee member of the board $75,000 for consulting services performed as of December 31, 2012.
The Company paid one non-employee member of the board $54,000 for consulting services performed as of December 31, 2013. The Company paid another non-employee member of the board $75,000 for consulting services performed as of December 31, 2013. The Company paid a third non-employee member of the board $75,000 for consulting services performed as of December 31, 2013.
The Company paid one of the Company’s directors $6,000 as of March 31, 2014, all of which was paid as part of his overall compensation of an aggregate of $85,000 for board and committee service.
6. Income Taxes
Reconciliations between the statutory federal income tax rate and the Company’s effective tax rate follow:
| 2014 | 2013 | 2012 | ||||||||||||||||||||||
| Years Ended December 31, | Amount | % | Amount | % | Amount | % | ||||||||||||||||||
| Federal statutory rate |
$ | (3,483,000 | ) | (34.0 | ) | $ | (9,417,000 | ) | (34.0 | ) | $ | (4,273,000 | ) | (34.0 | ) | |||||||||
| State taxes |
(461,000 | ) | (4.5 | ) | (1,246,000 | ) | (4.5 | ) | (566,000 | ) | (4.5 | ) | ||||||||||||
| Adjustment to valuation allowance |
4,862,000 | 47.7 | 5,015,000 | 18.1 | 4,596,000 | 36.5 | ||||||||||||||||||
| Non-deductible compensation |
— | — | — | — | 924,000 | 7.0 | ||||||||||||||||||
| (Gain) loss on warrant liability |
(918,000 | ) | (9.2 | ) | 5,648,000 | 20.4 | (681,000 | ) | (5.0 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Actual tax benefit |
$ | — | — | $ | — | — | $ | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
The components of the Company’s deferred income taxes are summarized below:
| December 31, | 2014 | 2013 | ||||||
| Deferred tax assets |
||||||||
| Net operating loss carry-forwards |
$ | 34,046,000 | $ | 30,995,000 | ||||
| Stock-based compensation |
6,344,000 | 6,299,000 | ||||||
| Warrants for services |
5,421,000 | 4,527,000 | ||||||
|
|
|
|
|
|||||
| Deferred tax asset |
45,811,000 | 41,821,000 | ||||||
| Deferred tax liabilities |
||||||||
| Patent amortization |
(1,380,000 | ) | (1,638,000 | ) | ||||
| Valuation allowance |
(44,431,000 | ) | (40,183,000 | ) | ||||
|
|
|
|
|
|||||
| Net deferred taxes |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
A valuation allowance against deferred tax assets is required if, based on the weight of available evidence, it is more likely than not that some or all of the deferred tax assets may not be realized. The Company is in the development stage and realization of the deferred tax assets is not considered more likely than not. As a result, the Company has recorded a full valuation allowance for the net deferred tax assets.
Since inception of the Company on January 17, 2002, the Company has generated tax net operating losses of approximately $99 million, expiring in 2022 through 2034. The tax loss carry-forwards of the Company may be subject to limitation by Section 382 of the Internal Revenue Code with respect to the amount utilizable each year. This limitation reduces the Company’s ability to utilize net operating loss carry-forwards. The amount of the limitation has been quantified by the Company. The Company completed a Section 382 study for the period from inception through the year ended December 31, 2014 and recorded a limitation of $3.2 million of their net operating loss carry-forward.
F-20
Table of Contents
The Company has determined that there are no uncertain tax positions as of December 31, 2014 or 2013 and does not expect any significant change within the next year.
7. 401(K) Profit Sharing Plan
Contributions made by the Company in 2012 totaled approximately $132,000 and were included in other accrued expenses. Contributions made by the Company in 2013 totaled approximately $226,000. Contributions made by the Company in 2014 totaled approximately $320,000.
8. Fair Value of Financial Instruments
The FASB’s authoritative guidance on fair value measurements establishes a framework for measuring fair value, and expands disclosure about fair value measurements. This guidance enables the reader of the financial statements to assess the inputs used to develop those measurements by establishing a hierarchy for ranking the quality and reliability of the information used to determine fair values. Under this guidance, assets and liabilities carried at fair value must be classified and disclosed in one of the following three categories:
Level 1: Quoted market prices in active markets for identical assets or liabilities.
Level 2: Observable market based inputs or unobservable inputs that are corroborated by market data.
Level 3: Unobservable inputs that are not corroborated by market data.
In determining the appropriate levels, the Company performs a detailed analysis of the assets and liabilities that are measured and reported on a fair value basis. At each reporting period, all assets and liabilities for which the fair value measurement is based on significant unobservable inputs are classified as Level 3. The fair value of certain of the Company’s financial instruments, including Cash and cash equivalents, short-term receivable and Accounts payable, approximates the carrying value due to the relatively short maturity of such instruments. The fair value of derivative instruments is determined by management with the assistance of an independent third party valuation specialist. The warrant liability is a derivative instrument and is classified as Level 3. The Company used the Monte-Carlo Simulation model to estimate the fair value of the warrants. Significant assumptions used are as follows:
| December 31, 2014 | December 31, 2013 | December 31, 2012 | ||||
| 2010 Warrants: |
||||||
| Weighted average term |
0.2 years | 1.2 years | 2.2 years | |||
| Probability the warrant exercise price would be reset |
5% | 5% | 5% | |||
| Volatility |
63.7% | 66.5% to 69.5% | 58.9% to 63.4% | |||
| Risk free interest rate |
0.03% to 0.04% | 0.13% to 0.38% | 0.25% to 0.36% | |||
| 2011 Warrants: |
||||||
| Weighted average term |
1.0 years | 2.0 years | 3.0 years | |||
| Probability the warrant exercise price would be reset |
5% | 5% | 5% | |||
| Volatility |
159.2% | 64.7% | 58.9% to 63.4% | |||
| Risk free interest rate |
0.25% | 0.38% to 0.78% | 0.25% to 0.36% | |||
| 2013 Warrants: |
||||||
| Weighted average term |
N/A | 4.1 years | N/A | |||
| Probability the warrant exercise price would be reset |
N/A | 5% | N/A | |||
| Volatility |
N/A | 67.2% | N/A | |||
| Risk free interest rate |
N/A | 0.78% to 1.78% | N/A |
At December 31, 2014, there are no remaining 2013 warrants and therefore no associated warrant liability.
The warrant liability measured at fair value on a recurring basis is as follows:
| Total | Level 1 | Level 2 | Level 3 | |||||||||||||
| Derivative instruments: |
||||||||||||||||
| Warrant liability at December 31, 2014 |
$ | 146,560 | $ | — | $ | — | $ | 146,560 | ||||||||
| Warrant liability at December 31, 2013 |
$ | 12,866,572 | $ | — | $ | — | $ | 12,866,572 | ||||||||
F-21
Table of Contents
A reconciliation of the warranty liability measured at fair value on a recurring basis with the use of significant unobservable inputs (Level 3) from January 1, 2013 to December 31, 2014 is as follows:
| Balance at January 1, 2013 |
$ | 1,299,570 | ||
| Issuance of warrants |
1,297,950 | |||
| Net loss included in earnings |
14,671,130 | |||
| Exercise of warrants |
(4,402,078 | ) | ||
|
|
|
|||
| Balance at December 31, 2013 |
$ | 12,866,572 | ||
|
|
|
|||
| Balance at January 1, 2014 |
$ | 12,866,572 | ||
| Issuance of warrants |
— | |||
| Net gain included in earnings |
(2,384,393 | ) | ||
| Exercise of warrants |
(10,335,619 | ) | ||
|
|
|
|||
| Balance at December 31, 2014 |
$ | 146,560 | ||
|
|
|
9. Litigation
Kleba Shareholder Derivative Lawsuit
On January 2, 2013, Glenn Kleba, derivatively on behalf of the Company, filed a shareholder derivative complaint in the Circuit Court for the State of Tennessee, Knox County (the “Court”), against H. Craig Dees, Timothy C. Scott, Eric A. Wachter, and Peter R. Culpepper (collectively, the “Executives”), Stuart Fuchs, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, together with the Executives, the “Individual Defendants”), and against the Company as a nominal defendant (the “Shareholder Derivative Lawsuit”). The Shareholder Derivative Lawsuit alleged (i) breach of fiduciary duties, (ii) waste of corporate assets, and (iii) unjust enrichment, all three claims based on Mr. Kleba’s allegations that the defendants authorized and/or accepted stock option awards in violation of the terms of the Company’s 2002 Stock Plan (the “Plan”) by issuing stock options in excess of the amounts authorized under the Plan and delegated to defendant H. Craig Dees the sole authority to grant himself and the other Executives cash bonuses that Mr. Kleba alleges to be excessive.
In April 2013, the Company’s Board of Directors appointed a special litigation committee to investigate the allegations of the Shareholder Derivative Complaint and make a determination as to how the matter should be resolved. The special litigation committee conducted its investigation, and proceedings in the case were stayed pending the conclusion of the committee’s investigation. The Company has established a reserve of $100,000 for potential liabilities because such is the amount of the self-insured retention of its insurance policy. On February 21, 2014, an Amended Shareholder Derivative Complaint was filed which added Don B. Dale (“Mr. Dale”) as a plaintiff.
On March 6, 2014, the Company filed a Joint Notice of Settlement (the “Notice of Settlement”) in the Shareholder Derivative Lawsuit. In addition to the Company, the parties to the Notice of Settlement are Mr. Kleba, Mr. Dale and the Individual Defendants.
On June 6, 2014, the Company, in its capacity as a nominal defendant, entered into a Stipulated Settlement Agreement and Mutual Release (the “Settlement”) in the Shareholder Derivative Lawsuit. In addition to the Company and the Individual Defendants, Plaintiffs Glenn Kleba and Don B. Dale are parties to the Settlement.
By entering into the Settlement, the settling parties have resolved the derivative claims to their mutual satisfaction. The Individual Defendants have not admitted the validity of any claims or allegations and the settling plaintiffs have not admitted that any claims or allegations lack merit or foundation. Under the terms of the Settlement, (i) the Executives each agreed (A) to re-pay to the Company $2.24 Million of the cash bonuses they each received in 2010 and 2011, which amount equals 70% of such bonuses or an estimate of the after-tax net proceeds to each Executive; provided, however, that subject to certain terms and conditions set forth in the Settlement, the Executives are entitled to a 2:1 credit such that total actual repayment may be $1.12 Million each; (B) to reimburse the Company for 25% of the actual costs, net of recovery from any other source, incurred by the Company as a result of the Shareholder Derivative Lawsuit; and (C) to grant to the Company a first priority security interest in 1,000,000 shares of the Company’s common stock owned by each such Executive to serve as collateral for
F-22
Table of Contents
the amounts due to the Company under the Settlement; (ii) Drs. Dees and Scott and Mr. Culpepper agreed to retain incentive stock options for 100,000 shares but shall forfeit 50% of the nonqualified stock options granted to each such Executive in both 2010 and 2011. The Settlement also requires that each of the Executives enter into new employment agreements with the Company, which were entered into on April 28, 2014, and that the Company adhere to certain corporate governance principles and processes in the future. Under the Settlement, Messrs. Fuchs and Smith and Dr. McMasters have each agreed to pay the Company $25,000 in cash, subject to reduction by such amount that the Company’s insurance carrier pays to the Company on behalf of such defendant pursuant to such defendant’s directors and officers liability insurance policy. The Settlement also provides for an award to plaintiffs’ counsel of attorneys’ fees and reimbursement of expenses in connection with their role in this litigation, subject to Court approval.
On July 24, 2014, the Court approved the terms of the proposed Settlement and awarded $911,000 to plaintiffs’ counsel for attorneys’ fees and reimbursement of expenses in connection with their role in the Shareholder Derivative Lawsuit. The payment to plaintiff’s counsel was made by the Company during October 2014 and is recorded as other current assets at December 31, 2014. The Company is seeking reimbursement of the full amount from insurance and if the full amount is not received from insurance, the amount remaining will be reimbursed to the Company from the Individual Defendants.
On October 3, 2014, the Settlement was effective and stock options for Drs. Dees and Scott and Mr. Culpepper were rescinded, totaling 2,800,000. At December 31, 2014, a Gain on Settlement of $4,178,345, net of discount, was recorded for the total due from the Executives. A Short-term Receivable was recorded for $733,333 and a Long-term Receivable was recorded for $3,378,345. A discount for implied interest of $301,655 was recorded as an offset to the Gain on Settlement in the consolidated statements of operations. $66,667 was repaid by the Executives as of December 31, 2014. The cash settlement amounts will be repaid to the Company over a period of five years with the first payment due in October 2015 and the final payment is expected to be received by October 3, 2019.
Class Action Lawsuits
On May 27, 2014, Cary Farrah and James H. Harrison, Jr., individually and on behalf of all others similarly situated (the “Farrah Case”), and on May 29, 2014, each of Paul Jason Chaney, individually and on behalf of all others similarly situated (the “Chaney Case”), and Jayson Dauphinee, individually and on behalf of all others similarly situated (the “Dauphinee Case”) (the plaintiffs in the Farrah Case, the Chaney Case and the Dauphinee Case collectively referred to as the “Plaintiffs”), each filed a class action lawsuit in the United States District Court for the Middle District of Tennessee against the Company, H. Craig Dees, Timothy C. Scott and Peter R. Culpepper (the “Defendants”) alleging violations by the Defendants of Sections 10(b) and 20(a) of the Exchange Act and Rule 10b-5 promulgated thereunder. Specifically, the Plaintiffs in each of the Farrah Case, the Chaney Case and the Dauphinee Case allege that the Defendants are liable for making false statements and failing to disclose adverse facts known to them about the Company, in connection with the Company’s application to the FDA for Breakthrough Therapy Designation (“BTD”) of the Company’s melanoma drug, PV-10, in the Spring of 2014, and the FDA’s subsequent denial of the Company’s application for BTD. The Company intends to defend vigorously against all claims in these complaints. However, in view of the inherent uncertainties of litigation and the early stage of this litigation, the outcome of these cases cannot be predicted at this time. Likewise, the amount of any potential loss cannot be reasonably estimated. No amounts have been recorded in the consolidated financial statements as the outcome of these cases cannot be predicted and the amount of any potential loss is not estimable at this time.
On July 9, 2014, the Plaintiffs and the Defendants filed joint motions in the Farrah Case, the Chaney Case and the Dauphinee Case to consolidate the cases and transfer them to United States District Court for the Eastern District of Tennessee. By order dated July 16, 2014, the United States District Court for the Middle District of Tennessee entered an order consolidating the Farrah Case, the Chaney Case and the Dauphinee Case (collectively and, as consolidated, the “Securities Litigation”) and transferred the Securities Litigation to the United States District Court for the Eastern District of Tennessee.
On November 26, 2014, the United States District Court for the Eastern District of Tennessee (the “Court”) entered an order appointing Fawwaz Hamati as the Lead Plaintiff in the Securities Litigation, with the Law Firm of Glancy Binkow & Goldberg, LLP as counsel to Lead Plaintiff. On February 3, 2015, the Court entered an order compelling the Lead Plaintiff to file a consolidated amended complaint within 60 days of entry of the order. As of March 4, 2015, the Lead Plaintiff has yet to file a consolidated amended complaint.
F-23
Table of Contents
Hurtado Shareholder Derivative Lawsuit
On June 4, 2014, Karla Hurtado, derivatively on behalf of the Company, filed a shareholder derivative complaint in the United States District Court for the Middle District of Tennessee against H. Craig Dees, Timothy C. Scott, Jan E. Koe, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, the “Individual Defendants”), and against the Company as a nominal defendant (the “Hurtado Shareholder Derivative Lawsuit”). The Hurtado Shareholder Derivative Lawsuit alleges (i) breach of fiduciary duties and (ii) abuse of control, both claims based on Ms. Hurtado’s allegations that the Individual Defendants (a) recklessly permitted the Company to make false and misleading disclosures and (b) failed to implement adequate controls and procedures to ensure the accuracy of the Company’s disclosures.
On July 25, 2014, the United States District Court for the Middle District of Tennessee entered an order transferring the case to the United States District Court for the Eastern District of Tennessee and, in light of the pending Securities Litigation, relieving the Individual Defendants from responding to the complaint in the Hurtado Shareholder Derivative Lawsuit pending further order from the United States District Court for the Eastern District of Tennessee.
As a nominal defendant, no relief is sought against the Company itself in the Hurtado Shareholder Derivative Lawsuit.
Montiminy Shareholder Derivative Lawsuit
On October 24, 2014, Paul Montiminy brought a shareholder derivative complaint on behalf of the Company in the United States District Court for the Eastern District of Tennessee (the “Montiminy Shareholder Derivative Lawsuit”) against H. Craig Dees, Timothy C. Scott, Jan E. Koe, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, the “Individual Defendants”). Like the Hurtado Shareholder Derivative Lawsuit, the Montiminy Shareholder Derivative Lawsuit alleges (i) breach of fiduciary duties and (ii) gross mismanagement of the assets and business of the Company, both claims based on Mr. Montiminy’s allegations that the Individual Defendants recklessly permitted the Company to make certain false and misleading disclosures regarding the likelihood that the Company’s melanoma drug, PV-10, would qualify for BTD. As a practical matter, the factual allegations and requested relief in the Montiminy Shareholder Derivative Lawsuit are substantively the same as those in the Hurtado Shareholder Derivative Lawsuit.
On December 29, 2014, the United States District Court for the Eastern District of Tennessee (the “Court”) entered an order consolidating the Hurtado Shareholder Derivative Lawsuit and the Montiminy Derivative Lawsuit. On February 25, 2015, the parties submitted a proposed agreed order staying the Hurtado and Montiminy Shareholder Derivative Lawsuits until the Court issues a ruling on the anticipated motion to dismiss the amended consolidated complaint to be filed in the Securities Litigation. As of March 4, 2015, the Court has not yet entered the proposed agreed order staying the Hurtado and Montiminy Shareholder Derivative Lawsuits.
Again, as in the Hurtado Shareholder Derivative Lawsuit, no relief is sought against the Company itself; the action is against the Individual Defendants only.
Foley Shareholder Derivative Complaint
On October 28, 2014, Chris Foley, derivatively on behalf of the Company, filed a shareholder derivative complaint in the Chancery Court of Knox County, Tennessee against H. Craig Dees, Timothy C. Scott, Jan E. Koe, Kelly M. McMasters, and Alfred E. Smith, IV (collectively, the “Individual Defendants”), and against the Company as a nominal defendant (the “Foley Shareholder Derivative Lawsuit”). The Foley Shareholder Derivative Lawsuit was brought by the same attorney as the Montiminy Shareholder Derivative Lawsuit, Paul Kent Bramlett of Bramlett Law Offices. Other than the difference in the named plaintiff, the complaints in the Foley Shareholder Derivative Lawsuit and the Montiminy Shareholder Derivative Lawsuit are identical. Since the filing of the Foley Shareholder Derivative Lawsuit, the parties have submitted a proposed agreed order staying the Foley Derivative Lawsuit until the United States District Court for the Eastern District of Tennessee issues a ruling on the anticipated motion to dismiss the amended consolidated complaint to be filed in the Securities Litigation.
10. Subsequent Events
The Company has evaluated subsequent events through the date of the filing of these financial statements.
F-24
Table of Contents
11. Selected Quarterly Financial Data (Unaudited)
The following tables present a summary of quarterly results of operations for 2014 and 2013:
| Three Months Ended | ||||||||||||||||
| March 31, 2014 |
June 30, 2014 |
September 30, 2014 |
December 31, 2014 |
|||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Consolidated Statement of Operations Data: |
||||||||||||||||
| Gain on settlement – net of discount |
$ | — | $ | — | $ | — | $ | 4,178 | ||||||||
| Total operating loss not including gain on settlement |
(4,382 | ) | (4,160 | ) | (3,826 | ) | (4,443 | ) | ||||||||
| Other income (expense), net |
(2,285 | ) | 3,517 | 77 | 1,081 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net income (loss) |
(6,667 | ) | (643 | ) | (3,749 | ) | 816 | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net income (loss) applicable to common stockholders |
$ | (6,667 | ) | $ | (643 | ) | $ | (3,749 | ) | $ | 816 | |||||
|
|
|
|
|
|
|
|
|
|||||||||
| Basic and diluted income (loss) per common share |
$ | (0.04 | ) | $ | (0.00 | ) | $ | (0.02 | ) | $ | 0.00 | |||||
| Weighted average number of common shares outstanding – basic and diluted |
168,860 | 175,554 | 179,089 | 182,057 | ||||||||||||
| Three Months Ended | ||||||||||||||||
| March 31, 2013 |
June 30, 2013 |
September 30, 2013 |
December 31, 2013 |
|||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Consolidated Statement of Operations Data: |
||||||||||||||||
| Total operating loss |
$ | (3,247 | ) | $ | (3,287 | ) | $ | (3,650 | ) | $ | (2,844 | ) | ||||
| Other income (expense), net |
(923 | ) | 909 | (903 | ) | (13,754 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
(4,170 | ) | (2,378 | ) | (4,553 | ) | (16,598 | ) | ||||||||
| Dividends on preferred stock |
(1,077 | ) | (72 | ) | (38 | ) | — | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss applicable to common stockholders |
$ | (5,247 | ) | $ | (2,450 | ) | $ | (4,591 | ) | $ | (16,598 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Basic and diluted loss per common share |
$ | (0.04 | ) | $ | (0.02 | ) | $ | (0.03 | ) | $ | (0.11 | ) | ||||
| Weighted average number of common shares outstanding – basic and diluted |
120,702 | 127,115 | 131,574 | 148,314 | ||||||||||||
F-25
Table of Contents
| Exhibit No. |
Description | |
| 3.1 | Certificate of Incorporation of Provectus Biopharmaceuticals, Inc. (the “Company”) (incorporated by reference to Exhibit 3.1 of the Company’s annual report on Form 10-K filed with the SEC on March 13, 2014) | |
| 3.2† | Certificate of Amendment to Certificate of Incorporation of Provectus Biopharmaceuticals, Inc. | |
| 3.3 | Certificate of Designation for the Company’s 8% Convertible Preferred Stock (incorporated by reference to Exhibit 3.2 of the Company’s annual report on Form 10-K filed with the SEC on March 13, 2014) | |
| 3.4 | Certificate of Designation for the Company’s Series A 8% Convertible Preferred Stock (incorporated by reference to Exhibit 3.3 of the Company’s annual report on Form 10-K filed with the SEC on March 13, 2014) | |
| 3.5 | Bylaws of the Company (incorporated by reference to Exhibit 3.4 of the Company’s annual report on Form 10-K filed with the SEC on March 13, 2014) | |
| 4.1 | Specimen certificate for the Company’s common shares, $.001 par value per share (incorporated by reference to Exhibit 4.1 of the Company’s annual report on Form 10-KSB filed with the SEC on April 15, 2003). | |
| 4.2 | Form of Series A Warrant issued to each of the purchasers identified on the signature pages of the Securities Purchase Agreement dated as of January 13, 2011 (incorporated by reference to Exhibit 4.1 of the Company’s current report on Form 8-K filed with the SEC on January 13, 2011). | |
| 4.3 | Form of Series B Warrant issued to each of the purchasers identified on the signature pages of the Securities Purchase Agreement dated as of January 13, 2011 (incorporated by reference to Exhibit 4.2 of the Company’s current report on Form 8-K filed with the SEC on January 13, 2011). | |
| 4.4 | Form of Series C Warrant issued to each of the purchasers identified on the signature pages of the Securities Purchase Agreement dated as of January 13, 2011 (incorporated by reference to Exhibit 4.3 of the Company’s current report on Form 8-K filed with the SEC on January 13, 2011). | |
| 4.5 | Form of Warrant issued to Lincoln Park Capital, LLC (incorporated by reference to Exhibit 4.1 of the Company’s current report on Form 8-K filed with the SEC on December 23, 2010). | |
| 4.6 | Form of Warrant issued to investors in connection with the offering of the Company’s 8% Convertible Preferred Stock (incorporated by reference to Exhibit 10.2 of the Company’s current report on Form 8-K filed on March 12, 2010). | |
| 4.7 | Form of Warrant issued to investors in connection with the offering of the Company’s Series A 8% Convertible Preferred Stock (incorporated by reference to Exhibit 10.2 of the Company’s current report on Form 8-K filed on February 28, 2013). | |
| 10.1* | Amended and Restated 2012 Stock Plan (incorporated herein by reference to Appendix A of the Company’s definitive proxy statement filed on April 30, 2012). | |
| 10.2* | Confidentiality, Inventions and Non-competition Agreement dated as of November 26, 2002 between the Company and H. Craig Dees (incorporated by reference to Exhibit 10.8 of the Company’s annual report on Form 10-KSB filed on April 15, 2003). | |
| 10.3* | Confidentiality, Inventions and Non-competition Agreement dated as of November 26, 2002 between the Company and Timothy C. Scott (incorporated by reference to Exhibit 10.9 of the Company’s annual report on Form 10-KSB filed on April 15, 2003). | |
| 10.4* | Confidentiality, Inventions and Non-competition Agreement dated as of November 26, 2002, between the Company and Eric A. Wachter (incorporated by reference to Exhibit 10.10 of the Company’s annual report on Form 10-KSB filed on April 15, 2003). | |
| 10.5 | Material Transfer Agreement dated as of July 31, 2003 between Schering-Plough Animal Health Corporation and the Company (incorporated by reference to Exhibit 10.15 of the Company’s quarterly report on Form 10-QSB filed on August 14, 2003). | |
| 10.6 | Securities Purchase Agreement dated as of January 13, 2011, by and between the Company and the purchasers identified on the signature pages thereto (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K filed on January 13, 2011). | |
Table of Contents
| Exhibit No. |
Description | |
| 10.7 | Purchase Agreement dated as of December 22, 2010, by and between the Company and Lincoln Park Capital Fund, LLC (incorporated by reference to Exhibit 10.2 of the Company’s current report on Form 8-K filed on December 23, 2010). | |
| 10.8 | Registration Rights Agreement dated as of December 22, 2010, by and between the Company and Lincoln Park Capital Fund, LLC (incorporated by reference to Exhibit 10.2 of the Company’s current report on Form 8-K filed on December 23, 2010). | |
| 10.9 | Form of Securities Purchase Agreement by an among the Company and the investors set forth on the signature pages affixed thereto used in connection with the offering of the 8% Convertible Preferred Stock and related warrants (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K filed on March 12, 2010). | |
| 10.10 | Form of Registration Rights Agreement by and among the Company and the stockholders set forth on the signature pages affixed thereto used in connection with the offering of the 8% Convertible Preferred Stock and related warrants (incorporated by reference to Exhibit 10.3 of the Company’s current report on Form 8-K filed on March 12, 2010). | |
| 10.11 | Form of Securities Purchase Agreement by and among the Company and the investors set forth on the signature pages affixed thereto used in connection with the offering of the Series A 8% Convertible Preferred Stock and related warrants (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K filed on February 28, 2013). | |
| 10.12 | Form of Registration Rights Agreement by and among the Company and the stockholders set forth on the signature pages affixed thereto used in connection with the offering of the Series A 8% Convertible Preferred Stock and related warrants (incorporated by reference to Exhibit 10.3 of the Company’s current report on Form 8-K filed on February 28, 2013). | |
| 10.13* | Executive Employment Agreement by and between the Company and H. Craig Dees, Ph.D., dated April 28, 2014 (incorporated by reference to Exhibit 10.13 of the Company’s annual report on Form 10-K filed with the SEC on April 30, 2014). | |
| 10.14* | Executive Employment Agreement by and between the Company and Eric Wachter, Ph.D., dated April 28, 2014 (incorporated by reference to Exhibit 10.14 of the Company’s annual report on Form 10-K filed with the SEC on April 30, 2014). | |
| 10.15* | Executive Employment Agreement by and between the Company and Timothy C. Scott, Ph.D., dated April 28, 2014 (incorporated by reference to Exhibit 10.15 of the Company’s annual report on Form 10-K filed with the SEC on April 30, 2014). | |
| 10.16* | Executive Employment Agreement by and between the Company and Peter Culpepper dated April 28, 2014 (incorporated by reference to Exhibit 10.16 of the Company’s annual report on Form 10-K filed with the SEC on April 30, 2014). | |
| 10.17 | 2014 Equity Compensation Plan (incorporated herein by reference to Appendix A of the Company’s definitive proxy statement filed on April 30, 2014). | |
| 10.18 | Controlled Equity OfferingSM Sales Agreement, dated April 30, 2014, by and between Provectus Biopharmaceuticals, Inc. and Cantor Fitzgerald & Co. (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K filed on April 30, 2014). | |
| 10.19 | Stipulated Settlement Agreement and Mutual Release, dated June 6, 2004, by and among the Company as nominal defendant, H. Craig Dees, Timothy C. Scott, Eric A. Wachter, Peter R. Culpepper, Stuart Fuchs, Kelly M. McMasters, and Alfred E. Smith, IV, as defendants, and Glenn Kleba and Don B. Dale, as plaintiffs (Exhibits Omitted) (incorporated by reference to Exhibit 10.6 of the Company’s quarterly report on Form 10-Q filed on August 7, 2014). | |
| 14 | Code of Ethics (incorporated by reference to Exhibit 14 of the Company’s annual report on Form 10-K filed on March 16, 2011). | |
| 21 | Subsidiaries of the Company (incorporated by reference to Exhibit 21 of the Company’s annual report on Form 10-K filed on March 16, 2011). | |
Table of Contents
| Exhibit No. |
Description | |
| 23† | Consent of Independent Registered Public Accounting Firm | |
| 31.1† | Certification of CEO pursuant to Rules 13a-14(a) of the Securities Exchange Act of 1934. | |
| 31.2† | Certification of CFO pursuant to Rules 13a-14(a) of the Securities Exchange Act of 1934. | |
| 32† | Certification Pursuant to 18 U.S.C. Section 1350. | |
| 101 | The following financial information from Provectus Biopharmaceuticals, Inc.’s Annual Report on Form 10-K for the period ended December 31, 2014, filed with the SEC on March 12, 2015, formatted in Extensible Business Reporting Language (XBRL): (i) the Consolidated Balance Sheet as of December 31, 2014 and December 31, 2013; (ii) the Consolidated Statements of Operations for the years ended December 31, 2014, 2013 and 2012; (iii) the Consolidated Statements of Equity for the years ended December 31, 2014, 2013 and 2012; (iv) the Consolidated Statements of Cash Flows for the years ended December 31, 2014, 2013 and 2012; and (v) Notes to Consolidated Financial Statements.** | |
| † | Filed herewith. |
| * | Indicates a management contract or compensatory plan or arrangement. |
| ** | Pursuant to Rule 406T of Regulation S-T, the XBRL related information in Exhibit 101 to this Annual Report on Form 10-K shall not be deemed to be “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be deemed part of a registration statement, prospectus or other document filed under the Securities Act or the Exchange Act, except as shall be expressly set forth by specific reference in such filings. |