Attached files
| file | filename |
|---|---|
| EX-3.1 - EX-3.1 - XBiotech Inc. | d833047dex31.htm |
| EX-3.3 - EX-3.3 - XBiotech Inc. | d833047dex33.htm |
| EX-3.2 - EX-3.2 - XBiotech Inc. | d833047dex32.htm |
| EX-10.4 - EX-10.4 - XBiotech Inc. | d833047dex104.htm |
| EX-10.6 - EX-10.6 - XBiotech Inc. | d833047dex106.htm |
| EX-10.8 - EX-10.8 - XBiotech Inc. | d833047dex108.htm |
| EX-21.1 - EX-21.1 - XBiotech Inc. | d833047dex211.htm |
| EX-14.1 - EX-14.1 - XBiotech Inc. | d833047dex141.htm |
| EX-23.1 - EX-23.1 - XBiotech Inc. | d833047dex231.htm |
| EX-10.1 - EX-10.1 - XBiotech Inc. | d833047dex101.htm |
| EX-10.2 - EX-10.2 - XBiotech Inc. | d833047dex102.htm |
| EX-10.5 - EX-10.5 - XBiotech Inc. | d833047dex105.htm |
| EX-10.7 - EX-10.7 - XBiotech Inc. | d833047dex107.htm |
| EX-10.3 - EX-10.3 - XBiotech Inc. | d833047dex103.htm |
Table of Contents
As filed with the Securities and Exchange Commission on February 2, 2015
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
XBIOTECH INC.
(Exact name of registrant as specified in its charter)
| Canada | 2834 | N/A | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
8201 E. Riverside Drive
Building 4, Suite 100
Austin, TX 78744
(512) 386-2900
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
John Simard, Chief Executive Officer
XBiotech Inc.
8201 E. Riverside Drive
Building 4, Suite 100
Austin, TX 78744
(512) 386-2900
(Name, address, including zip code, and telephone number, including area code, of agent for service)
With copies to:
| John F. Anderson, Esq. Stikeman Elliott LLP 666 Burrard Street, Suite 1700 Vancouver, BC Canada V6C 2X8 (604) 631-1307 |
Laura M. Holm, Esq. Quarles & Brady LLP 1395 Panther Lane, Suite 300 Naples, FL 34109 (239) 434-4905 |
James R. Tanenbaum Esq. Morrison & Foerster LLP 250 West 55th Street New York, NY 10019-9601 (212) 468-8163 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this Registration Statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | x (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Calculation of Registration Fee
|
| ||||
| Title of Each Class of Securities to be Registered |
Proposed Maximum Aggregate |
Amount of Registration Fee(1) | ||
| Common stock, no par value per share |
$5,810.00 | |||
| Total Registration Fee |
$5,810.00 | |||
|
| ||||
|
| ||||
| (1) | Calculated pursuant to Rule 457(o) of the rules and regulations under the Securities Act of 1933. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this prospectus is not complete and may be changed. We may not sell these securities until the Securities and Exchange Commission declares our registration statement effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED FEBRUARY 2, 2015.

|
XBiotech Inc. | |||||||
| Shares of Common Stock
| ||||||||
| This is our initial public offering and no public market currently exists for our shares. We are selling shares of our common stock. We expect that the initial public offering price will be between $ and $ per share.
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act, of the JOBS Act, and, as such, may elect to comply with certain reduced reporting requirements for this prospectus and future filings after this offering.
|
Best Efforts Offering: The method of distribution being used by the underwriters in this offering differs somewhat from that traditionally employed in underwritten public offerings. The underwriters have agreed to use their best efforts to procure potential purchasers for the shares of common stock offered pursuant to this Prospectus.
The shares are being offered on an all or none basis. All investor funds received from the date of this Prospectus to the closing date will be deposited into escrow with an escrow agent until closing. If, on the closing date, investor funds are not received for the full amount of shares to be sold in this offering, the offering will terminate and any funds received will be returned promptly.
A more detailed description of this process is included in “Underwriting” beginning on page [—]. | |||||||
| THE OFFERING | PER SHARE | TOTAL MINIMUM OFFERING |
||||||
| Initial Public Offering Price | $ | $ | ||||||
| Underwriting Discounts and Commissions | $ | $ | ||||||
| Proceeds to Us | $ | $ | ||||||
| The underwriters expect to deliver the shares of our common stock on , 2015. | ||||||||
| Proposed NASDAQ Symbol: XBIT | ||||||||
Investing in our common stock involves a high degree of risk. See “Risk Factors” beginning on page 8.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed on the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.

The date of this prospectus is , 2015.
Table of Contents
| Page No. |
||||
| 1 | ||||
| 8 | ||||
| 29 | ||||
| 31 | ||||
| 32 | ||||
| 32 | ||||
| 33 | ||||
| 34 | ||||
| MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
34 | |||
| 43 | ||||
| 67 | ||||
| 75 | ||||
| 78 | ||||
| 79 | ||||
| 80 | ||||
| 85 | ||||
| 88 | ||||
| 92 | ||||
| 101 | ||||
| 103 | ||||
| 103 | ||||
| 103 | ||||
You should rely only on the information contained in this Prospectus. We have not authorized anyone to provide you with different or additional information. If anyone provides you with different or inconsistent information, you should not rely on it. The information contained in this Prospectus is accurate only as of the date of this Prospectus, regardless of the time of delivery of this Prospectus or any sale of securities described in this Prospectus. This Prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted. You should assume that the information appearing in this Prospectus, as well as information we have previously filed with the Securities and Exchange Commission (“SEC”) is accurate as of the date on the front of those documents only. Our business, financial condition, results of operations and prospects may have changed since those dates.
i
Table of Contents
This summary highlights certain information about us, this offering and information appearing elsewhere in this Prospectus. This summary is not complete and does not contain all of the information that you should consider before investing in our securities. To fully understand this offering and its consequences to you, you should carefully read this entire Prospectus and any free writing Prospectus we distribute, including the information contained under the heading “Risk Factors” in this Prospectus beginning on page 9 and the financial statements and related notes thereto. Unless the context requires otherwise, in this Prospectus, the term “XBiotech,” “we,” “us,” “our,” the “Company” and “our company” refer to XBiotech, Inc. and its subsidiaries.
Our Business
XBiotech is a clinical-stage biopharmaceutical company engaged in discovering and developing “True Human™” monoclonal antibodies for treating a variety of diseases. True Human™ monoclonal antibodies are those which occur naturally in human beings—as opposed to being derived from animal immunization or otherwise engineered. We believe that naturally occurring monoclonal antibodies have the potential to be safer and more effective than their non-naturally occurring counterparts. While focused on bringing our lead product candidate to market, we also have developed a “True Human™” pipeline and manufacturing system.
The majority of our efforts to date have concentrated on developing MABp1 (also known as Xilonix™, CA-18C3, CV-18C3, RA-18C3, and T2-18C3), a therapeutic antibody which specifically neutralizes interleukin-1 alpha (IL-1a). IL-1a is a pro-inflammatory protein produced by leukocytes and other cells, where it plays a key role in inflammation. When unchecked, inflammation can contribute to the development and progression of a variety of different diseases, such as cancer, vascular disease, inflammatory skin disease, and diabetes. Our clinical studies have shown that blocking IL-1a with MABp1 may have a beneficial effect in several diseases.
We completed a Phase I/II clinical trial for MABp1 (Xilonix™) as a treatment for cancer at MD Anderson Cancer Center. The results of this study, published in Lancet Oncology in April 2014, found that in the 52 patients with metastatic cancer (18 tumor types) who participated, MABp1 was well tolerated, with no dose-limiting toxicities or immunogenicity. Moreover, within eight weeks of starting therapy many patients began to improve with respect to constitutional symptoms. An imaging method, known as dual energy X-ray absorptiometry (DEXA), revealed that many of the patients improved physically, in terms of gaining lean body mass; and patient reported outcomes documented that many were recovering from pain, fatigue and appetite loss. Finally, we found that in the patients with colorectal cancer, DEXA-measured recovery was associated with significant improvement in survival.
We received a fast track designation from the FDA in October 2012 to develop Xilonix™ as a treatment in the setting of metastatic colorectal cancer. The purpose of the fast track designation is to aid in the development, and expedite the review, of drugs that have the potential to treat a serious or life-threatening disease. Currently we have two Phase III studies underway — one launched in the US for advanced refractory colorectal cancer and another in Europe for symptomatic colorectal cancer. If these trials are successful, we will seek marketing approvals for MABp1 (Xilonix™) in Europe and/or in the United States. Assuming such marketing approvals are obtained, we would distribute and sell this product through our own direct sales force or with a commercial partner.
We are also investigating MABp1 in clinical trials for other indications including vascular disease, type II diabetes, acne and psoriasis. In a randomized Phase II study involving 43 patients, we evaluated MABp1 for its ability to reduce adverse events after balloon angioplasty, atherectomy or stent placement in patients undergoing revascularization procedures for blockage of the superficial femoral artery (SFA), a major artery in the leg. While the study did not involve a large enough patient population to provide a statistically significant outcome, results from this study showed an important trend towards the reduction of restenosis and reduced incidence of
1
Table of Contents
Major Adverse Cardiovascular Events (“MACE”) in treated patients compared to the control group. In 2012, we obtained a fast track designation to develop MABp1 as a therapy to reduce the need for re-intervention after treatment of peripheral vascular disease with angioplasty or other endovascular methods of treatment.
In a Phase II pilot study completed in 2012, we tested MABp1 in patients with Type 2 diabetes. A treatment-related decline in HbA1c, and increased serum levels of pro-insulin and C-peptide (indicators of improved glucose control and pancreas function, respectively) were observed. We also conducted two phase II pilot studies in skin disease, evaluating the potential benefit of MABp1 in subjects with (1) moderate to severe plaque psoriasis and (2) moderate to severe acne vulgaris. The psoriasis study revealed rapid improvements in the Psoriasis Area and Severity Index (PASI), with patients having a median of 43% improvement within 35 days. In the acne study, treated patients exhibited a continual improvement in lesions over the course of therapy, with up to 42% reduction in eight weeks; and interestingly, these patients had a statistically significant improvement in anxiety, as measured by the Hospital Anxiety and Depression Scale (HADS). We continue to analyze our clinical results, and prioritize further clinical initiatives for MABp1 in oncology, SFA, diabetes, psoriasis and acne.
We recently filed an Investigational New Drug Application (IND) for a Phase I/II randomized clinical study to assess our True Human Antibody therapy for the treatment of serious infections due to Staphylococcus aureus. This product candidate was identified from an individual that harbored a natural antibody capable of neutralizing S. aureus, including drug-resistant strains of the bacteria. The study is currently on clinical hold while, at the request of the FDA, we complete an animal toxicology study. We expect to complete the animal study during the first quarter of 2015.
More recently, we have begun using our True Human™ antibody technology to begin developing a therapy for Ebola virus infection. We recently received a blood donation from an Ebola-recovered patient which we have now confirmed contains high levels of anti-Ebola antibodies. A product candidate derived from this blood is expected to go into animal studies in 2015.
To produce our product candidates, we have developed a manufacturing system that employs simple disposable bioreactor technology. Our manufacturing operation is currently located within our forty-six thousand square foot facility in Austin, Tx. To accommodate larger-scale commercial manufacturing needs, we purchased 48 acres of industrial-zoned property located five miles from Austin’s central business district. In September 2014, we commenced ground-breaking on a new manufacturing facility on this property. Construction is estimated to be completed by late 2015, and we expect to begin operating in the new facility in early 2016. The new facility will be capable of producing several hundred thousand doses of antibody annually.
2
Table of Contents
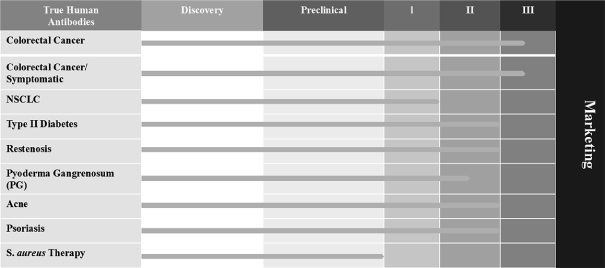
Our product development status for the first quarter of 2015 is as follows:
Our Strategy
Our objective is to fundamentally change the way therapeutic antibodies are developed and commercialized, and become a leading biopharmaceutical company focused on the discovery, development and commercialization of therapeutic True HumanTM antibodies. The key goals of our business strategy are to:
| • | Obtain regulatory approval to market and sell XilonixTM in the United States, Europe and other markets, and begin commercial sale of XilonixTM; |
| • | Continue our research and clinical work on infectious diseases, including S. aureus; |
| • | Review our clinical results for SFA, diabetes, psoriasis and acne and determine additional research or clinical studies which we may conduct in the future; |
| • | Discover other True HumanTM antibody therapies using our proprietary platform; and |
| • | Leverage our manufacturing technology. |
Risks Associated with Our Business
An investment in our common stock involves a high degree or risk. We are subject to a number of risks of which you should be aware of before you decide to buy our common stock. These risks are discussed more fully in “Risk Factors.” The following highlights a few of the most significant risks which we face:
| • | We have a history of losses. As of September 30, 2014, we had an accumulated deficit of $87 million. We expect to continue to incur losses for the foreseeable future, and we have never achieved or sustained profitability. |
| • | We will likely need to obtain additional capital to continue operations. |
| • | Our success depends on the regulatory approval and commercialization of Xilonix™ and any future product candidates. |
| • | We are subject to regulatory approval processes that are lengthy, time consuming and unpredictable; we may not obtain approval for Xilonix™ or any of our future product candidates from the FDA or foreign regulatory authorities. |
3
Table of Contents
| • | It is difficult and costly to protect our intellectual property rights. |
Implications of Being an Emerging Growth Company
As a company with less than $1.0 billion in revenues during our last fiscal year, we qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. An emerging growth company may take advantage of specified reduced reporting requirements that are otherwise applicable generally to public companies. These provisions include:
| • | A requirement to have only two years of audited financial statements and only two years of related management’s discussion and analysis; |
| • | An exemption from compliance with the auditor attestation requirement on the effectiveness of our internal controls over financial reporting; |
| • | An exemption from compliance with any requirement that the Public Company Accounting Oversight Board may adopt regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
| • | Reduced disclosure about our executive compensation arrangements; and |
| • | Exemptions from the requirements to obtain a non-binding advisory vote on executive compensation or a shareholder approval of any golden parachute arrangements. |
Under the JOBS Act, we will remain an “emerging growth company” until the earliest of: (a) the last day of the fiscal year during which we have total annual gross revenue of $1.0 billion or more; (b) the last day of the fiscal year following the fifth anniversary of the effective date of the registration statement of which this Prospectus forms a part; (c) the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt; or (d) the date on which we are deemed to be a “large accelerated filer” under the Securities Exchange Act of 1934, as amended, or the Exchange Act (we will qualify as a large accelerated filer as of the first day of the first fiscal year after we have (i) more than $700 million in outstanding common equity held by our non-affiliates and (ii) been public for at least 12 months; the value of our outstanding common equity will be measured each year on the last day of our second fiscal quarter).
We may choose to take advantage of some of the available benefits under the JOBS Act, and have taken advantage of some reduced reporting requirements in this Prospectus. Accordingly, the information contained herein may be different from the information contained in Prospectuses from other US public companies.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This provision allows an emerging growth company to delay the adoption of some accounting standards until those standards would otherwise apply to private companies. However, we are choosing to “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
Corporate Information
We were incorporated under the Canada Business Corporations Act on March 22, 2005 and were continued under the British Columbia Business Corporations Act on September 23, 2005. We have four wholly-owned subsidiaries: XBiotech USA, Inc., formed under the laws of Delaware, XBiotech Switzerland AG formed under the laws of Switzerland; XBiotech Japan K.K,. formed under the laws of Japan and XBiotech Germany GmbH formed under the laws of Germany.
4
Table of Contents
Unless the context otherwise requires, any reference to “XBiotech”, “we”, “our” and “us” in this Prospectus refers to XBiotech, Inc. and its subsidiaries. Our principal place of business is at 8201 E. Riverside Side, Building 4, Suite 100, Austin, TX 78744. Our telephone number is (512) 386-2900 and our facsimile number is (512) 386-5505. We also maintain a web site at www.xbiotech.com. The information contained in, or that can be accessed through our website is not a part of this Prospectus.
5
Table of Contents
The Offering
| Common Stock to be offered: |
shares of common stock |
| Common stock to be outstanding after the offering |
shares(1) |
| Trading symbol: |
We will apply to list our common stock on the NASDAQ Capital Market under the trading symbol “XBIT.” |
| Use of proceeds: |
We estimate that the net proceeds from this offering will be approximately $ million, based upon an assumed initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this Prospectus), after deducting the estimated underwriting discounts and commissions, and estimated offering expenses payable by us. We currently expect to use the net proceeds from this offering: to fund clinical trials for Xilonix™ and our other products, and for working capital and general corporate purposes. See “Use of Proceeds” for more information. |
| Risk factors: |
This offering involves a high degree of risk. You should not consider a purchase of the shares unless you can afford to lose your entire investment. See “Risk Factors,” as well as other cautionary statements throughout this Prospectus, before investing in shares of our common stock. |
| (1) | The number of shares of common stock outstanding after this offering is based on 27,546,632 shares of common stock outstanding at January 22, 2015 and excludes as of that date: (a) 493,000 shares of common stock issuable upon exercise of warrants outstanding at a weighted-average exercise price of $15.00 per share; (b) 4,929,165 shares issuable upon exercise of stock options at a weighted-average exercise price of $7.10 per share and (c) 1,070,835 shares of common stock available for grant under our 2005 Incentive Stock Option Plan. |
6
Table of Contents
Summary Financial Data
The following selected consolidated statements of operations data for the years ended December 31, 2012 and 2013 are derived from our audited financial statements included elsewhere in this Prospectus. The selected statements of operations data for the nine months ended September 30, 2013 and 2014, and balance sheet data at September 30, 2014, are derived from our unaudited financial statements, included elsewhere in this Prospectus, which have been prepared on a basis consistent with our audited financial statements and, in the opinion of management, include all adjustments, consisting of normal recurring adjustments, necessary for a fair presentation of our financial position and results of operations. The results of operations for any interim period are not necessarily indicative of results to be expected for the entire year. The following data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and the related notes included elsewhere in this Prospectus.
| Year Ended December 31, | Nine Months Ended September 30, |
|||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||
| Statement of Operations Data |
||||||||||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
$ | 13,334 | $ | 7,935 | $ | 5,610 | $ | 9,424 | ||||||||
| General and administrative |
1,829 | 1,990 | 1,828 | 6,435 | ||||||||||||
| Total operating expenses |
15,163 | 9,925 | 7,438 | 15,859 | ||||||||||||
| Loss from operations |
(15,163 | ) | (9,925 | ) | (7,438 | ) | (15,859 | ) | ||||||||
| Other income (loss): |
||||||||||||||||
| Interest income |
3 | 1 | 1 | — | ||||||||||||
| Foreign exchange gain (loss) |
— | (3 | ) | (3 | ) | 24 | ||||||||||
| Total other income (loss): |
3 | (2 | ) | (2 | ) | 24 | ||||||||||
| Net loss |
$ | (15,160 | ) | $ | (9,927 | ) | $ | (7,440 | ) | $ | (15,835 | ) | ||||
| Net loss per common share—basic and diluted |
$ | (0.70 | ) | $ | (0.44 | ) | $ | (0.33 | ) | $ | (0.66 | ) | ||||
| Weighted average number of common shares—basic and diluted |
21,594,369 | 22,520,416 | 22,341,240 | 24,173,485 | ||||||||||||
| At September 30, 2014 | ||||||
| Actual | Pro forma As adjusted | |||||
| (in thousands) | ||||||
| Balance sheet data |
||||||
| Cash and cash equivalents |
$ | 15,497 | ||||
| Working capital |
13,428 | |||||
| Total assets |
19,661 | |||||
| Total shareholders’ equity |
17,218 | |||||
7
Table of Contents
Investing in our common stock involves a number of risks. You should not invest unless you are able to bear the complete loss of your investment. In addition to the risks and investment considerations discussed elsewhere in this Prospectus, the following factors should be carefully considered by anyone purchasing the securities offered by this Prospectus. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations. If any of the following risks actually occur, our business could be harmed. In such case, the trading price of our common stock could decline and investors could lose all or a part of the money paid to buy our common stock.
Risks Related to our Financial Condition and Capital Requirements
We have incurred significant losses every quarter since our inception and anticipate that we will continue to incur significant losses in the future.
We are a clinical-stage pharmaceutical company with no revenue and a limited operating history. Investment in pharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate efficacy or an acceptable safety profile, gain regulatory approval or become commercially viable. We do not have any products approved by regulatory authorities for marketing or commercial sale and have not generated any revenue from product sales, or otherwise, to date, and we continue to incur significant research, development and other expenses related to our ongoing operations. As a result, we are not profitable and have incurred losses in every reporting period since our inception in 2005. For the years ended December 31, 2012 and December 31, 2013, we reported a net loss of $15.2 million and $9.9 million, respectively. For the nine months ended September 30, 2014, we reported a net loss of $15.8 million. As of September 30, 2014, we had an accumulated deficit since inception of $87.3 million.
We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses will increase as we continue the research and development of, and seek regulatory approvals for, Xilonix™ and any of our other product candidates, and potentially begin to commercialize any products that may achieve regulatory approval. We may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our financial condition. The amount of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues. Our prior losses and expected future losses have had, and will continue to have, an adverse effect on our financial condition. If Xilonix™ or any other product candidate fails in clinical trials or does not gain regulatory approval, or if approved, fails to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
Even if this offering is successful, we will need to raise significant additional funding, which may not be available on acceptable terms, if at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate our product development efforts or other operations.
Since inception, we have dedicated a majority of our resources to the discovery and development of our proprietary preclinical and clinical product candidates, and we expect to continue to expend substantial resources doing so for the foreseeable future. These expenditures will include costs associated with conducting research and development, manufacturing product candidates and products approved for sale, conducting preclinical experiments and clinical trials and obtaining and maintaining regulatory approvals, as well as commercializing any products later approved for sale. During the nine months ended September 30, 2014, we recognized approximately $9.4 million in expenses associated with research and development and clinical trials.
Our anticipated net proceeds from this offering are not expected to be sufficient to complete clinical development of any of our product candidates nor prepare to commercializing any product candidate which
8
Table of Contents
receives regulatory approval. Accordingly, we will likely require substantial additional capital beyond this offering to continue our clinical development and potential commercialization activities. Our future capital requirements depend on many factors, including but not limited to:
| • | the number and characteristics of the future product candidates we pursue; |
| • | the scope, progress, results and costs of independently researching and developing any of our future product candidates, and conducting preclinical research and clinical trials; |
| • | the timing of, and the costs involved in, obtaining regulatory approvals for any future product candidates we develop independently; |
| • | the cost of future commercialization activities for Xilonix™ and the cost of commercializing independently any future products approved for sale; |
| • | the cost of manufacturing our future products; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patents, including litigation costs and the outcome of any such litigation. |
We are unable to estimate the funds we will actually require to complete research and development of our product candidates or the funds required to commercialize any resulting product in the future.
Upon the completion of this offering, based upon our anticipated operating expenditures, we expect that the net proceeds from this offering, with our cash and cash equivalents of approximately $57.3 million as of December 31, 2014, will be sufficient to fund our current operations for at least the next 36 months. However, our operating plan may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. Raising funds in the future may present additional challenges and future financing may not be available in sufficient amounts or on terms acceptable to us, if at all.
Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
The terms of any financing arrangements we enter into may adversely affect the holdings or the rights of our shareholders and the issuance of additional securities, by us, or the possibility of such issuance, may cause the market price of our shares to decline. The sale of additional equity or convertible securities would dilute all of our shareholders. The incurrence of indebtedness would result in increased fixed payment obligations and, potentially, the imposition of restrictive covenants. Those covenants may include limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborators or otherwise at an earlier stage than otherwise would be desirable resulting in the loss of rights to some of our product candidates or other unfavorable terms, any of which may have a material adverse effect on our business, operating results and prospects. In addition, additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our products.
Risks Related to Our Business
We currently have no source of product revenue and may never become profitable.
To date, we have not generated any revenues from commercial product sales, or otherwise. Our ability to generate revenue from product sales and achieve profitability will depend upon our ability, alone or with any future collaborators, to commercialize products successfully, including Xilonix™ or any future product
9
Table of Contents
candidates that we may develop, in-license or acquire in the future. Even if we are able to achieve regulatory approval successfully for Xilonix™ or any future product candidates, we do not know when any of these products will generate revenue from product sales, if at all. Our ability to generate revenue from product sales from Xilonix™ or any of our other product candidates also depends on a number of additional factors, including our ability to:
| • | complete development activities, including the necessary clinical trials; |
| • | complete and submit new drug applications, or NDAs, to the US Food and Drug Administration, or FDA, and obtain regulatory approval for indications for which there is a commercial market; |
| • | complete and submit applications to, and obtain regulatory approval from, foreign regulatory authorities; |
| • | establish our manufacturing operations; |
| • | develop a commercial organization capable of sales, marketing and distribution for Xilonix™ and any products for which we obtain marketing approval and intend to sell ourselves in the markets in which we choose to commercialize on our own; |
| • | find suitable distribution partners to help us market, sell and distribute our approved products in other markets; |
| • | obtain coverage and adequate reimbursement from third-party payors, including government and private payors; |
| • | achieve market acceptance for our products, if any; |
| • | establish, maintain and protect our intellectual property rights; and |
| • | attract, hire and retain qualified personnel. |
In addition, because of the numerous risks and uncertainties associated with pharmaceutical product development, including that Xilonix™ or any other product candidates may not advance through development or achieve the endpoints of applicable clinical trials, we are unable to predict the timing or amount of increased expenses, or when or if we will be able to achieve or maintain profitability. In addition, our expenses could increase beyond expectations if we decide to or are required by the FDA, or foreign regulatory authorities, to perform studies or trials in addition to those that we currently anticipate. Even if we are able to complete the development and regulatory process for Xilonix™ or any other product candidates, we anticipate incurring significant costs associated with commercializing these products.
Even if we are able to generate revenues from the sale of Xilonix™ or any other product candidates that may be approved, we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce our operations.
Our future success is dependent on the regulatory approval and commercialization of Xilonix™ and any of our other product candidates.
We do not have any products that have gained regulatory approval. Our lead product, Xilonix™, is currently in 2 Phase III clinical trials in the United States and in Europe, respectively. As a result, our near-term prospects, including our ability to finance our operations and generate revenue, are substantially dependent on our ability to obtain regulatory approval for, and, if approved, to successfully commercialize Xilonix™ in a timely manner. We cannot commercialize Xilonix™ or our other product candidates in the United States without first obtaining regulatory approval for each product from the FDA; similarly, we cannot commercialize Xilonix™ or our other product candidates outside of the United States without obtaining regulatory approval from comparable foreign regulatory authorities. The FDA review process typically takes years to complete and approval is never
10
Table of Contents
guaranteed. Before obtaining regulatory approvals for the commercial sale of any Xilonix™ or our other product candidates for a target indication, we must demonstrate with substantial evidence gathered in preclinical and well-controlled clinical studies, generally including two well-controlled Phase 3 trials, and, with respect to approval in the United States, to the satisfaction of the FDA, that the product candidate is safe and effective for use for that target indication and that the manufacturing facilities, processes and controls are adequate. Obtaining regulatory approval for marketing of Xilonix™ or our future product candidates in one country does not ensure we will be able to obtain regulatory approval in other countries but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in other countries.
Even if Xilonix™ or any of our other product candidates were to successfully obtain approval from the FDA and comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, or may be subject to burdensome post-approval study or risk management requirements. If we are unable to obtain regulatory approval for Xilonix™ in one or more jurisdictions, or any approval contains significant limitations, we may not be able to obtain sufficient funding or generate sufficient revenue to continue the development of any of our other product candidates that we are developing or may discover, in-license, develop or acquire in the future. Also, any regulatory approval of any of Xilonix™ or our other product candidates, once obtained, may be withdrawn. Furthermore, even if we obtain regulatory approval for Xilonix™, the commercial success of Xilonix™ will depend on a number of factors, including the following:
| • | development of a commercial organization or establishment of a commercial collaboration with a commercial infrastructure; |
| • | establishment of commercially viable pricing and obtaining approval for adequate reimbursement from third-party and government payors; |
| • | our ability to manufacture quantities of Xilonix™ using commercially sufficient processes and at a scale sufficient to meet anticipated demand and enable us to reduce our cost of manufacturing; |
| • | our success in educating physicians and patients about the benefits, administration and use of Xilonix™; |
| • | the availability, perceived advantages, relative cost, relative safety and relative efficacy of alternative and competing treatments; |
| • | the effectiveness of our own or our potential strategic collaborators’ marketing, sales and distribution strategy and operations; |
| • | acceptance of Xilonix™ as safe and effective by patients and the medical community; and |
| • | a continued acceptable safety profile of Xilonix™ following approval. |
Many of these factors are beyond our control. If we are unable to successfully commercialize Xilonix™, we may not be able to earn sufficient revenues to continue our business.
Because the results of earlier clinical trials are not necessarily predictive of future results, Xilonix™ which is currently in Phase III clinical trials, or any other product candidate we advance into clinical trials, may not have favorable results in later clinical trials or receive regulatory approval.
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will generate adequate data to demonstrate the efficacy and safety of an investigational drug. A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience, have suffered significant setbacks in clinical trials, even after seeing promising results in earlier clinical trials. Despite the results reported in earlier clinical trials for Xilonix™, we do not know whether the clinical trials we are conducting, or may conduct, will demonstrate adequate efficacy and safety to result in regulatory approval to market Xilonix™ or any of our other product candidates in any particular jurisdiction. Even if we believe that we
11
Table of Contents
have adequate data to support an application for regulatory approval to market our product candidates, the FDA or other applicable foreign regulatory authorities may not agree and may require us to conduct additional clinical trials. If later-stage clinical trials do not produce favorable results, our ability to achieve regulatory approval for any of our product candidates may be adversely impacted.
If we are unable to enroll subjects in clinical trials, we will be unable to complete these trials on a timely basis.
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of subjects to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, ability to obtain and maintain patient consents, risk that enrolled subjects will drop out before completion, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. Furthermore, we rely on Clinical Research Organizations (“CR’s”) and clinical trial sites to ensure the proper and timely conduct of our clinical trials, and while we have agreements governing their committed activities, we have limited influence over their actual performance.
If we experience delays in the completion or termination of, any clinical trial of Xilonix™ or any future product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, and jeopardize our ability to commence product sales, which would impair our ability to generate revenues and may harm our business, results of operations, financial condition and cash flows and future prospects. In addition, many of the factors that could cause a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of Xilonix™ or our other product candidates.
The regulatory approval processes of the FDA and comparable foreign regulatory authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for Xilonix™ or our other product candidates, our business will fail.
The time required to obtain approval by the FDA and comparable foreign regulatory authorities is unpredictable, but typically takes many years following the commencement of preclinical studies and clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate, and it is possible that neither Xilonix™ nor any other product candidates we are developing or may discover, in-license or acquire and seek to develop in the future will ever obtain regulatory approval.
Our product candidates could fail to receive marketing approval from the FDA or a comparable foreign regulatory authority for many reasons, including:
| • | disagreement over the design or implementation of our clinical trials; |
| • | failure to demonstrate that a product candidate is safe; |
| • | failure of clinical trials to meet the level of statistical significance required for approval; |
| • | failure to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| • | disagreement over our interpretation of data from preclinical studies or clinical trials; |
| • | disagreement over whether to accept efficacy results from clinical trial sites outside the United States where the standard of care is potentially different from that in the United States; |
12
Table of Contents
| • | the insufficiency of data collected from clinical trials of Xilonix™ or our other product candidates to support the submission and filing of an NDA or other submission or to obtain regulatory approval; |
| • | irreparable or critical compliance issues relating to our manufacturing process; or |
| • | changes in the approval policies or regulations that render our preclinical and clinical data insufficient for approval. |
The FDA or a comparable foreign regulatory authority may require more information, including additional preclinical or clinical data to support approval, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program altogether. Even if we do obtain regulatory approval, Xilonix™ or our other product candidates may be approved for fewer or more limited indications than we request, approval contingent on the performance of costly post-marketing clinical trials, or approval with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. In addition, if Xilonix™ or our other product candidate produces undesirable side effects or safety issues, the FDA may require the establishment of Risk Evaluation Mitigation Strategies, or REMS, or a comparable foreign regulatory authority may require the establishment of a similar strategy, that may, restrict distribution of our products and impose burdensome implementation requirements on us. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
Even if we believe our current or planned clinical trials are successful, the FDA may not agree that our completed clinical trials provide adequate data on the safety or efficacy of Xilonix™ or our other product candidates to permit us to proceed to additional clinical trials. Approval by comparable foreign regulatory authorities does not ensure approval by the FDA and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other countries or by the FDA. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. We may not be able to file for regulatory approvals and even if we file we may not receive the necessary approvals to commercialize our products in any market.
Xilonix™ or our other product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following any marketing approval.
Undesirable side effects caused by Xilonix™ or our other product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authority. If toxicities occur in our current or future clinical trials they could cause delay or even discontinuance of further development of Xilonix™ or other product candidates, which would impair our ability to generate revenues and would have a material adverse effect our business, results of operations, financial condition and cash flows and future prospects. To date, the majority of adverse events observed in clinical trials of Xilonix™ have been mild and have not resulted in discontinuation of therapy. There have been no serious side effects observed that appear to be related to administration of Xilonix in our clinical trials. There can be no assurance that side-effects from Xilonix™ in future clinical trials will continue to be mild or that side-effects in general will not prompt the discontinued development of Xilonix™ or other product candidates. If serious side effects or other safety or toxicity issues are experienced in our clinical trials in the future, we may not receive approval to market Xilonix™ or any other product candidates, which could prevent us from ever generating revenues or achieving profitability. Results of our trials could reveal an unacceptably high severity and prevalence of side effects. In such an event, our trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled subjects to complete the trial or result in potential product liability claims. Any of these occurrences may have a material adverse effect on our business, results of operations, financial condition and cash flows and future prospects.
13
Table of Contents
Additionally, if Xilonix™ or any of our other product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such product, a number of potentially significant negative consequences could result, including:
| • | we may be forced to suspend marketing of such product; |
| • | regulatory authorities may withdraw their approvals of such product; |
| • | regulatory authorities may require additional warnings on the label that could diminish the usage or otherwise limit the commercial success of such product; |
| • | the FDA or other regulatory bodies may issue safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warnings about such product; |
| • | the FDA may require the establishment or modification of REMS or a comparable foreign regulatory authority may require the establishment or modification of a similar strategy that may, for instance, restrict distribution of our product and impose burdensome implementation requirements on us; |
| • | we may be required to change the way the product is administered or conduct additional clinical trials; |
| • | we could be sued and held liable for harm caused to subjects or patients; |
| • | we may be subject to litigation or product liability claims; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved.
Even if Xilonix™ or our other product candidates receive regulatory approval, they may still face future development and regulatory difficulties.
Even if we obtain regulatory approval for Xilonix™ or another product candidate, it would be subject to ongoing requirements by the FDA and comparable foreign regulatory authorities governing the manufacture, quality control, further development, labeling, packaging, storage, distribution, safety surveillance, import, export, advertising, promotion, recordkeeping and reporting of safety and other post-market information. The safety profile of any product will continue to be closely monitored by the FDA and comparable foreign regulatory authorities after approval. If the FDA or comparable foreign regulatory authorities become aware of new safety information after approval of Xilonix™ or any other product candidate, they may require labeling changes or establishment of a REMS or similar strategy, impose significant restrictions on a product’s indicated uses or marketing, or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. For example, the label ultimately approved for Xilonix™, if it achieves marketing approval, may include restrictions on use.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current good manufacturing practices, or cGMP, and other regulations. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, our manufacturing facility, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. If we, our product candidates or our manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
| • | issue warning letters or untitled letters; |
| • | impose restrictions on the marketing or manufacturing of the product candidates; |
14
Table of Contents
| • | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; |
| • | require us or any future collaborator to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
| • | seek an injunction or impose civil or criminal penalties or monetary fines; |
| • | suspend or withdraw regulatory approval; |
| • | suspend any ongoing clinical trials; |
| • | refuse to approve pending applications or supplements to applications filed by us; |
| • | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
| • | seize or detain products, refuse to permit the import or export of products, or require us to initiate a product recall. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize Xilonix™ or any other product candidates and generate revenue.
The FDA strictly regulates the advertising and promotion of drug products, and drug products may only be marketed or promoted for their FDA approved uses, consistent with the product’s approved labeling. Advertising and promotion of any product candidate that obtains approval in the United States will be heavily scrutinized by the FDA, the Department of Justice, or the DOJ, the Office of Inspector General of the Department of Health and Human Services, or HHS, state attorneys general, members of Congress and the public. Violations, including promotion of our products for unapproved or off-label uses, are subject to enforcement letters, inquiries and investigations, and civil, criminal and/or administrative sanctions by the FDA. Additionally, advertising and promotion of, any product candidate that obtains approval outside of the United States will be heavily scrutinized by comparable foreign regulatory authorities.
In the United States, engaging in impermissible promotion of our future products for off-label uses can also subject us to false claims litigation under federal and state statutes, which can lead to civil, criminal and/or administrative penalties and fines and agreements that materially restrict the manner in which we promote or distribute our drug products. These false claims statutes include the federal False Claims Act, which allows any individual to bring a lawsuit against a pharmaceutical company on behalf of the federal government alleging submission of false or fraudulent claims, or causing to present such false or fraudulent claims, for payment by a federal program such as Medicare or Medicaid. If the government prevails in the lawsuit, the individual may share in any fines or settlement funds. Since 2004, these False Claims Act lawsuits against pharmaceutical companies have increased significantly in volume and breadth, leading to several substantial civil and criminal settlements based on certain sales practices promoting off-label drug uses. This growth in litigation has increased the risk that a pharmaceutical company will have to defend a false claim action, pay settlement fines or restitution, agree to comply with burdensome reporting and compliance obligations, and be excluded from the Medicare, Medicaid and other federal and state healthcare programs. If we do not lawfully promote our approved products, we may become subject to such litigation and, if we are not successful in defending against such actions, those actions could have a material adverse effect our business, results of operations, financial condition and cash flows and future prospects.
Existing government regulations may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of Xilonix™ or any other product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and/or be subject to fines or enhanced government oversight and reporting obligations, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
15
Table of Contents
Failure to obtain regulatory approval in foreign jurisdictions would prevent Xilonix™ or any other product candidates from being marketed in those jurisdictions.
In order to market and sell our products in the European Union and many other jurisdictions, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the United States generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain countries. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. A failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory approval process in others. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market. If we are unable to obtain approval of Xilonix™ for any of our other product candidates by regulatory authorities in the European Union or another jurisdiction, the commercial prospects of that product candidate may be significantly diminished and our business prospects could decline.
Even if we are able to commercialize Xilonix™ or our other product candidates, the products may not receive coverage and adequate reimbursement from third-party payors, which could harm our business.
Our ability to commercialize any products successfully will depend, in part, on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government authorities, private health insurers, health maintenance organizations and third-party payors. Patients who are prescribed medications for the treatment of their conditions generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. Coverage and adequate reimbursement from government healthcare programs, such as Medicare and Medicaid, and private health insurers are critical to new product acceptance. Patients are unlikely to use Xilonix™ or our other product candidates unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our product candidates. A primary trend in the US healthcare industry and elsewhere is cost containment. As a result, government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. Third-party payors may also seek additional clinical evidence, beyond the data required to obtain marketing approval, demonstrating clinical benefits and value in specific patient populations before covering our products for those patients. We cannot be sure that coverage and adequate reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If coverage and reimbursement are not available or are available only at limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, obtaining coverage does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sales and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may only be temporary. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used. Reimbursement rates may also be based in part on existing reimbursement amounts for lower cost drugs or may be bundled into the payments for other services. Net prices for drugs may be reduced by
16
Table of Contents
mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage and reimbursement determination process is often a time-consuming and costly process with no assurance that coverage and adequate reimbursement will be obtained or applied consistently. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own coverage and reimbursement policies. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
We have never marketed a drug before, and if we are unable to establish an effective sales force and marketing infrastructure, or enter into acceptable third-party sales and marketing or licensing arrangements, we may be unable to generate any revenue.
We do not currently have an infrastructure for the sales, marketing and distribution of pharmaceutical drug products and the cost of establishing and maintaining such an infrastructure may exceed the cost-effectiveness of doing so. In order to market any products that may be approved by the FDA and comparable foreign regulatory authorities, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not become profitable. We will be competing with many companies that currently have extensive and well-funded sales and marketing operations. Without an internal commercial organization or the support of a third party to perform sales and marketing functions, we may be unable to compete successfully against these more established companies.
Xilonix™ and our other product candidates, if approved, may not achieve adequate market acceptance among physicians, patients, and healthcare payors and others in the medical community necessary for commercial success.
Even if we obtain regulatory approval for Xilonix™ or any of our other product candidates, such product(s) may not gain market acceptance among physicians, healthcare payors, patients or the medical community. Our commercial success also depends on coverage and adequate reimbursement of our product candidates by third-party payors, including government payors, generally, which may be difficult or time-consuming to obtain, may be limited in scope and may not be obtained in all jurisdictions in which we may seek to market our products. Market acceptance of any of our product candidates for which we receive approval depends on a number of factors, including:
| • | the efficacy and safety of such product candidates as demonstrated in clinical trials; |
| • | the clinical indications for which the product candidate is approved; |
| • | acceptance by physicians and patients of the product candidate as a safe and effective treatment; |
| • | the potential and perceived advantages of product candidates over alternative treatments; |
| • | the safety of product candidates seen in a broader patient group, including a product candidate’s use outside the approved indications; |
| • | the prevalence and severity of any side effects; |
| • | product labeling or product insert requirements of the FDA or other regulatory authorities; |
| • | the timing of market introduction of our products as well as competitive products; |
| • | the cost of treatment in relation to alternative treatments; |
17
Table of Contents
| • | the availability of coverage and adequate reimbursement and pricing by third-party payors and government authorities; |
| • | relative convenience and ease of administration; |
| • | the effectiveness of our sales and marketing efforts and those of our collaborators; and |
| • | unfavorable publicity relating to the product candidate. |
If any of our product candidates are approved but fail to achieve market acceptance among physicians, patients, or healthcare payors, we will not be able to generate significant revenues, which would compromise our ability to become profitable.
Our Ebola Research May Not Succeed.
Although we recently received a blood sample from a recovered Ebola victim in the hope that we will be able to identify Ebola-resistant antibodies that might used to produce a drug to help fight that deadly disease, there is no assurance that we will identify appropriate antibodies and be able to isolate them for possible drug production. Even if we succeed in isolating appropriate antibodies, the regulatory process to determine their safety and effectiveness would likely take years to complete at substantial expense, with no assurance that a safe and effective drug will ever be produced.
Even an Effective Ebola Drug Might Not Be Commercially Successful.
Even if we ultimately succeed in creating a safe and effective drug based on human antibodies that resist Ebola, there is no assurance it would be commercially successful. Competitive products might become available faster or with lower costs or adverse risks to patients, resulting in few sales of any product developed by XBiotech. Occurrences of Ebola might become sufficiently rare, or victims of Ebola might be sufficiently impoverished, that commercial production is uneconomic. XBiotech’s promises regarding donation of five percent of any gross revenues from an Ebola drug developed using the blood sample it recently received may adversely impact XBiotech and its shareholders.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The development and commercialization of new drug products is highly competitive. We face competition with respect to our current lead product candidate, Xilonix™ for the treatment of colorectal cancer from Vectibix ® by Amgen; Erbitux by Bristol Myers Squibb; Cyramza ® by Eli Lilly and Company and Avastin by Genentech/Roche. Our competitors in the other therapeutic categories that we are addressing which are non-small cell lung cancer, restenosis in peripheral vascular disease, diabetes and psoriasis are Humira from AbbVie, Remicade and Stellara from J&J, Enbrel from Amgen and Necitmumab from Eli Lilly and Company. In the infectious disease area, there are no currently approved monoclonal antibody products, although many have been tried. The leading small-molecule antibiotics are Vancomycin, originally from Eli Lilly and Company now in a generic form from Baxter, Sandoz, Akorn and Hospira; Cubicin (Daptomycin) from Cubist and Dalvance from Durata.
There are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of products for the treatment of the disease indications for which we are developing our future product candidates. Some of these competitive products and therapies are based on scientific approaches that are the same as or similar to our approach, and others are based on entirely different approaches. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
18
Table of Contents
More established companies may have a competitive advantage over us due to their greater size, cash flows and institutional experience. Compared to us, many of our competitors may have significantly greater financial, technical and human resources. As a result of these factors, our competitors may obtain regulatory approval of their products before we do, which will limit our ability to develop or commercialize Xilonix™ or any of our other product candidates. In addition, many companies are developing new therapeutics, and we cannot predict what the standard of care will be as our product candidates progress through clinical development.
Our failure to identify, acquire, develop and commercialize successfully additional product candidates or approved products other than Xilonix™ could impair our ability to grow.
Although a substantial amount of our efforts will focus on the continued clinical testing and potential approval of our most advanced product candidate, Xilonix™, a key element of our growth strategy is to acquire, develop and/or market additional products and product candidates. All of these potential product candidates remain in the discovery and clinical study stages. Research programs to identify product candidates require substantial technical, financial and human resources, whether or not any product candidates are ultimately identified. Because our internal research capabilities are limited, we may be dependent upon pharmaceutical and biotechnology companies, academic scientists and other researchers to sell or license products or technology to us. The success of this strategy depends partly upon our ability to identify, select and acquire promising pharmaceutical product candidates and products. The process of proposing, negotiating and implementing a license or acquisition of a product candidate or approved product is lengthy and complex. Other companies, including some with substantially greater financial, marketing and sales resources, may compete with us for the license or acquisition of product candidates and approved products. We have limited resources to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and integrate them into our current infrastructure. Moreover, we may devote resources to potential acquisitions or in-licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts. Any product candidate that we acquire may require additional development efforts prior to commercial sale, including extensive clinical testing and approval by the FDA and applicable foreign regulatory authorities. All product candidates are prone to risks of failure typical of pharmaceutical product development, including the possibility that a product candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot provide assurance that any products that we develop or approved products that we acquire will be manufactured profitably or achieve market acceptance.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of Xilonix™ and any other product candidates in clinical trials and will face an even greater risk if we commercially sell any products that we may develop. Product liability claim may be brought against us by subjects enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our products. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for any product candidates or products that we may develop; |
| • | termination of clinical trial sites or entire trial programs; |
| • | injury to our reputation and significant negative media attention; |
| • | withdrawal of clinical trial participants; |
| • | significant costs to defend the related litigation; |
| • | substantial monetary awards to trial subjects or patients; |
| • | loss of revenue; |
19
Table of Contents
| • | diversion of management and scientific resources from our business operations; and |
| • | the inability to commercialize our product candidates. |
We intend to obtain insurance coverage for products to include the sale of commercial products if we obtain marketing approval for Xilonix™ or our other product candidates, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
We will need to expand our operations and grow the size of our organization, and we may experience difficulties in managing this growth.
As of January 22, 2015, we had 55 employees. As our development and commercialization plans and strategies develop, or as a result of any future acquisitions, we will need additional managerial, operational, sales, marketing, scientific, and financial headcount and other resources. Our management, personnel and systems currently in place may not be adequate to support this future growth. Future growth would impose significant added responsibilities on members of management, including:
| • | managing our clinical trials effectively, which we anticipate being conducted at numerous clinical sites; |
| • | identifying, recruiting, maintaining, motivating and integrating additional employees with the expertise and experience we will require; |
| • | managing our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors and other third parties; |
| • | managing additional relationships with various strategic partners, suppliers and other third parties; |
| • | improving our managerial, development, operational and finance reporting systems and procedures; and |
| • | expanding our facilities. |
Our failure to accomplish any of these tasks could prevent us from successfully growing our Company.
We are highly dependent on our Chief Executive Officer.
Our future success depends in significant part on the continued service of our Chief Executive Officer, John Simard. Mr. Simard is critical to the strategic direction and overall management of our company as well as our research and development process. Although we have an employment agreement with Mr. Simard, it has no specific duration. The loss of Mr. Simard could adversely affect our business, financial condition and operating results.
We depend on key personnel to operate our business, and many members of our current management team are new. If we are unable to retain, attract and integrate qualified personnel, our ability to develop and successfully grow our business could be harmed.
In addition to the continued services of Mr. Simard, we believe that our future success is highly dependent on the contributions of our significant employees, as well as our ability to attract and retain highly skilled and experienced sales, research and development and other personnel in the United States and abroad. Some of our significant employees, include our Medical Director, our Vice President of Manufacturing, our Vice President of Quality, our Director of Research, our Director of Quality Control and our Vice President of Finance and Human Resources. Changes in our management team may be disruptive to our business.
20
Table of Contents
All of our employees, including our Chief Executive Officer, are free to terminate their employment relationship with us at any time, subject to any applicable notice requirements, and their knowledge of our business and industry may be difficult to replace. If one or more of our executive officers or significant employees leaves, we may not be able to fully integrate new personnel or replicate the prior working relationships, and our operations could suffer. Qualified individuals with the breadth of skills and experience in the pharmaceutical industry that we require are in high demand, and we may incur significant costs to attract them. Many of the other pharmaceutical companies that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Competition for qualified personnel is particularly intense in the Austin area, where our headquarters are located. Our failure to retain key personnel could impede the achievement of our research, development and commercialization objectives.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations in the United States and elsewhere, including, as a result of our leased laboratory space, those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes.
We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological or hazardous materials. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Business disruptions could seriously harm our future revenues and financial condition and increase our costs and expenses.
Our operations could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or manmade disasters or business interruptions, for which we are predominantly self-insured. We do not carry insurance for all categories of risk that our business may encounter. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on third-parties to supply various items which are critical for producing our product candidates. Our ability to produce clinical supplies of product candidates could be disrupted, if the operations of these suppliers are affected by a man-made or natural disaster or other business interruption. The ultimate impact on us, our significant suppliers and our general infrastructure of being consolidated in certain geographical areas is unknown, but our operations and financial condition could suffer in the event of a major earthquake, fire or other natural disaster. Further, any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our business, results of operations, financial condition and cash flows from future prospects.
21
Table of Contents
Risks Related to Intellectual Property
If we are unable to obtain or protect intellectual property rights, our competitive position could be harmed.
We depend on our ability to protect our proprietary technology. We rely on trade secret, patent, copyright and trademark laws, and confidentiality, licensing and other agreements with employees and third parties, all of which offer only limited protection. Our commercial success will depend in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our proprietary technology and products. Where we deem appropriate, we seek to protect our proprietary position by filing patent applications in the United States and abroad related to our novel technologies and products that are important to our business. The patent positions of biotechnology and pharmaceutical companies generally are highly uncertain, involve complex legal and factual questions and have in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patents, including those patent rights licensed to us by third parties, are highly uncertain.
The steps we have taken to protect our proprietary rights may not be adequate to preclude misappropriation of our proprietary information or infringement of our intellectual property rights, both inside and outside the United States. The rights already granted under any of our currently issued patents and those that may be granted under future issued patents may not provide us with the proprietary protection or competitive advantages we are seeking. If we are unable to obtain and maintain patent protection for our technology and products, or if the scope of the patent protection obtained is not sufficient, our competitors could develop and commercialize technology and products similar or superior to ours, and our ability to successfully commercialize our technology and products may be adversely affected.
With respect to patent rights, we do not know whether any of existing patents or pending patent applications for any of our technologies or product candidates will result in the issuance of patents that protect such technologies or products candidates, or if any of our issued patents will effectively prevent others from commercializing competitive technologies and products. Our pending patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications. Further, the examination process may require us to narrow the claims for our pending patent applications, which may limit the scope of patent protection that may be obtained if these applications issue. Because the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, issued patents that we own or have licensed from third parties may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in the loss of patent protection, the narrowing of claims in such patents or the invalidity or unenforceability of such patents, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection for our technology and products. Protecting against the unauthorized use of our patented technology, trademarks and other intellectual property rights is expensive, difficult and, in some cases, may not be possible. In some cases, it may be difficult or impossible to detect third-party infringement or misappropriation of our intellectual property rights, even in relation to issued patent claims, and proving any such infringement may be even more difficult.
Intellectual property rights do not necessarily address all potential threats to any competitive advantage we may have.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| • | Others may be able to make compounds that are the same as or similar to MABp1 or our future product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed. |
| • | We might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed. |
22
Table of Contents
| • | We or any of our licensors or strategic partners might not have been the first to file patent applications covering certain of our inventions. |
| • | Others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights. |
| • | It is possible that our pending patent applications will not lead to issued patents. |
| • | Issued patents that we own or have exclusively licensed may not provide us with any competitive advantages, or may be held invalid or unenforceable, as a result of legal challenges by our competitors. |
| • | Our competitors might conduct research and development activities in the United States and other countries that provide a safe harbor from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets. |
| • | We may not develop additional proprietary technologies that are patentable. |
| • | The patents of others may have an adverse effect on our business. |
Our technology may be found to infringe third-party intellectual property rights.
Third parties may in the future assert claims or initiate litigation related to their patent, copyright, trademark and other intellectual property rights in technology that is important to us. The asserted claims and/or litigation could include claims against us, our licensors or our suppliers alleging infringement of intellectual property rights with respect to our products or components of those products. Regardless of the merit of the claims, they could be time consuming, result in costly litigation and diversion of technical and management personnel, or require us to develop a non-infringing technology or enter into license agreements. We cannot assure you that licenses will be available on acceptable terms, if at all. Furthermore, because of the potential for significant damage awards, which are not necessarily predictable, it is not unusual to find even arguably unmeritorious claims resulting in large settlements. If any infringement or other intellectual property claim made against us by any third party is successful, or if we fail to develop non-infringing technology or license the proprietary rights on commercially reasonable terms and conditions, our business, operating results and financial condition could be materially adversely affected.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to:
| • | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| • | abandon an infringing drug or therapy candidate; |
| • | redesign our products or processes to avoid infringement; |
| • | stop using the subject matter claimed in the patents held by others; |
| • | pay damages; or |
| • | defend litigation or administrative proceedings which may be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources. |
Risks Related to This Offering and Owning Shares of our Common Stock
Purchasers in this offering will suffer immediate dilution.
If you purchase common stock in this offering, the value of your shares based on our actual book value will immediately be less than the offering price you paid. This reduction in the value of your equity is known as dilution. Based upon the pro forma net tangible book value of our common stock at September 30, 2014, your
23
Table of Contents
shares may be worth less per share than the price you paid in the offering. If the options and warrants we previously granted are exercised, additional dilution will occur. As of January 22, 2015, options to purchase 4,929,165 shares of common stock at a weighted-average exercise price of $7.10 per share were outstanding, and warrants to purchase 493,000 shares of common stock at a weighted-average exercise price of $15.00 per share were outstanding. Furthermore, if we raise additional funding by issuing additional equity securities, the newly-issued shares will further dilute your percentage ownership of our shares and may also reduce the value of your investment.
Our share price may be volatile, which could subject us to securities class action litigation and prevent you from being able to sell your shares at or above the offering price.
Our stock could be subject to wide fluctuations in response to many risk factors listed in this section, and others beyond our control, including:
| • | results of our clinical trials; |
| • | results of clinical trials of our competitors’ products; |
| • | regulatory actions with respect to our products or our competitors’ products; |
| • | actual or anticipated fluctuations in our financial condition and operating results; |
| • | actual or anticipated changes in our growth rate relative to our competitors; |
| • | actual or anticipated fluctuations in our competitors’ operating results or changes in their growth rate; |
| • | competition from existing products or new products that may emerge; |
| • | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments; |
| • | issuance of new or updated research or reports by securities analysts; |
| • | fluctuations in the valuation of companies perceived by investors to be comparable to us; |
| • | share price and volume fluctuations attributable to inconsistent trading volume levels of our shares; |
| • | additions or departures of key management or scientific personnel; |
| • | disputes or other developments related to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| • | announcement or expectation of additional financing efforts; |
| • | sales of our common stock by us, our insiders or our other shareholders; |
| • | market conditions for biopharmaceutical stocks in general; and |
| • | general economic and market conditions. |
Furthermore, the stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies. These fluctuations often have been unrelated or disproportionate to the operating performance of those companies. These broad market and industry fluctuations, as well as general economic, political and market conditions such as recessions, interest rate changes or international currency fluctuations, may negatively impact the market price of shares of our common stock. In addition, such fluctuations could subject us to securities class action litigation, which could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business. If the market price of shares of our common stock after this offering does not exceed the initial public offering price, you may not realize any return on your investment in us and may lose some or all of your investment.
24
Table of Contents
Insiders will continue to have substantial control over our company after this offering and could delay or prevent a change in corporate control.
After this offering, our directors, executive officers and principal shareholders, together with their affiliates, will beneficially own, in the aggregate, at least shares or approximately % of our outstanding common stock, and could own up to shares or % of our outstanding common stock if they fully exercise their outstanding stock options or shares. As a result, these shareholders, if acting together, will have the ability to determine the outcome of matters submitted to our shareholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our assets. In addition, these persons, acting together, will have the ability to control the management and affairs of our company. Accordingly, this concentration of ownership may harm the market price of our common stock by:
| • | delaying, deferring or preventing a change in control of our company; |
| • | impeding a merger, consolidation, takeover or other business combination involving our company; or |
| • | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of our company. |
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
We currently intend to allocate the net proceeds that we will receive from this offering as described below in the “Use of Proceeds” section of this Prospectus. However, our management will have broad discretion in the actual application of the net proceeds, and we may elect to allocate proceeds differently from that described in “Use of Proceeds” if we believe it would be in our best interests to do so. Our shareholders may not agree with the manner in which our management chooses to allocate and spend the net proceeds. The failure by our management to apply these funds effectively could have a material adverse effect on our business. Pending their use, we may invest the net proceeds from this offering in a manner that does not produce income or that loses value.
Provisions in our charter documents and under Canadian law could make an acquisition of us, which may be beneficial to our shareholders, more difficult and may prevent attempts by our shareholders to replace or remove our current management.
Our authorized preferred capital stock is available for issuance from time to time at the discretion of our board of directors, without shareholder approval. Our articles grant our board of directors the authority, subject to the corporate law of British Columbia, to determine or alter the special rights and restrictions granted to or imposed on any wholly unissued series of preferred shares, and such rights may be superior to those of our common shares.
Limitations on the ability to acquire and hold our common shares may be imposed by the Competition Act (Canada). This legislation permits the Commissioner of Competition of Canada to review any acquisition of a significant interest in us. This legislation grants the Commissioner jurisdiction to challenge such an acquisition before the Canadian Competition Tribunal if the Commissioner believes that it would, or would be likely to, result in a substantial lessening or prevention of competition in any market in Canada. The Investment Canada Act (Canada) subjects an acquisition of control of a company by a non-Canadian to government review if the value of our assets as calculated pursuant to the legislation exceeds a threshold amount. A reviewable acquisition may not proceed unless the relevant minister is satisfied that the investment is likely to be a net benefit to Canada.
Any of the foregoing could prevent or delay a change of control and may deprive or limit strategic opportunities for our shareholders to sell their shares.
25
Table of Contents
We may be a passive foreign investment company for US tax purposes which may negatively affect US investors.
For US federal income taxation purposes, we will be a passive foreign investment company, or PFIC, if in any taxable year either: (a) 75% or more of our gross income consists of passive income; or (b) 50% or more of the value of our assets is attributable to assets that produce, or are held for the production of, passive income. If we meet either test, our shares held by a US person in that year will be PFIC shares for that year and all subsequent years in which they are held by that person. Because in the past our gross income consisted mostly of interest, we have been a PFIC in prior taxable years. We may also be a PFIC in future taxable years. Gain realized by a US investor from the sale of PFIC shares is taxed as ordinary income, as opposed to capital gain, and subject to an interest charge unless the US person has timely made one of the tax elections described in the section titled “Material United States and Canadian Tax Considerations—US Material Federal Income Tax Consequences.”
We are governed by the corporate laws in British Columbia, Canada which in some cases have a different effect on shareholders than the corporate laws in Delaware, United States.
The material differences between the BCBCA as compared to the Delaware General Corporation Law, or the DGCL, which may be of most interest to shareholders include the following: (i) for material corporate transactions (such as mergers and amalgamations, other extraordinary corporate transactions, amendments to our articles) the BCBCA generally requires two-thirds majority vote by shareholders, whereas DGCL generally only requires a majority vote of shareholders for similar material corporate transactions; (ii) the quorum for shareholders meetings is not prescribed under the BCBCA and is only two persons representing 20% of the issued shares under our articles, whereas under DGCL, quorum requires a minimum of one-third of the shares entitled to vote to be present and companies’ certificates of incorporation frequently require a higher percentage to be present; (iii) under the BCBCA a holder of 5% or more of our common shares can requisition a special meeting at which any matters that can be voted on at our annual meeting can be considered, whereas the DGCL does not give this right; (iv) our articles require two-thirds majority vote by shareholders to pass a resolution for one or more directors to be removed, whereas DGCL only requires the affirmative vote of a majority of the shareholders; however, many public company charters limit removal of directors to a removal for cause; and (v) our articles may be amended by resolution of our directors to alter our authorized share structure, including to consolidate or subdivide any of our shares, whereas under DGCL, a majority vote by shareholders is generally required to amend a corporation’s certificate of incorporation and a separate class vote may be required to authorize alterations to a corporation’s authorized share structure. We cannot predict if investors will find our common shares less attractive because of these material differences. If some investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and our share price may be more volatile.
Future sales, or the possibility of future sales, of a substantial number of our common shares could adversely affect the price of the shares and dilute shareholders.
Future sales of a substantial number of our common shares, or the perception that such sales will occur, could cause a decline in the market price of our common shares. Following the completion of this offering and based on the midpoint of the price range stated on the front cover of this Prospectus, we will have common shares outstanding. This includes the common shares sold in this offering, which may be resold in the public market immediately without restriction, unless purchased by our affiliates. Approximately % of the common shares outstanding after this offering is expected to be held by existing shareholders. Our CEO and [list others] will be subject to the lock-up agreements described in the “Shares Available for Future Sale” section of this Prospectus. If, after the end of such lock-up agreements, these shareholders sell substantial amounts of common shares in the public market, or the market perceives that such sales may occur, the market price of our common shares and our ability to raise capital through an issue of equity securities in the future could be adversely affected.
26
Table of Contents
In addition, in the future, we may issue additional common shares or other equity or debt securities convertible into common shares in connection with a financing, acquisition, litigation settlement, employee arrangements or otherwise. Any such issuance could result in substantial dilution to our existing shareholders and could cause our common share price to decline.
We are an “emerging growth company” as that term is used in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we intend to take advantage of reduced disclosure and governance requirements applicable to emerging growth companies, which could result in our common stock being less attractive to investors and adversely affect the market price of our common stock or make it more difficult to raise capital as and when we need it.
We are an “emerging growth company” as that term is used in the JOBS Act, and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved and exemptions from any rules that the Public Company Accounting Oversight Board may adopt requiring mandatory audit firm rotation or a supplement to the auditor’s report on the financial statements. We currently intend to take advantage of some, but not all, of the reduced regulatory and reporting requirements that will be available to us under the JOBS Act, so long as we qualify as an “emerging growth company.” For example, so long as we qualify as an “emerging growth company,” we may elect not to provide you with certain information, including certain financial information and certain information regarding compensation of our executive officers, that we would otherwise have been required to provide in filings we make with the Securities and Exchange Commission, or SEC, which may make it more difficult for investors and securities analysts to evaluate our company.
We cannot predict if investors will find our common stock less attractive because we will rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company, which in certain circumstances could be for up to five years. See “Prospectus Summary—Implications of Being an Emerging Growth Company.”
Because of the exemptions from various reporting requirements provided to us as an “emerging growth company” we may be less attractive to investors and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our financial accounting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our business, results of operations, financial condition and cash flows and future prospects may be materially and adversely affected.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, shareholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our common shares.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404 or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require
27
Table of Contents
prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common shares.
We will be required to disclose changes made in our internal controls and procedures on a quarterly basis and our management will be required to assess the effectiveness of these controls annually. However, for as long as we are an “emerging growth company” under the JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. We could be an “emerging growth company” for up to five years. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. In addition, our management and independent registered public accounting firm did not perform an evaluation of our internal control over financial reporting as of December 31, 2013 or December 31, 2012, in accordance with the provisions of the Sarbanes-Oxley Act. Had we and our independent registered public accounting firm performed such an evaluation, control deficiencies may have been identified by management or our independent registered public accounting firm, and those control deficiencies could have also represented one or more material weaknesses. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
28
Table of Contents
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This Prospectus and the documents incorporated by reference herein contain forward-looking statements that involve substantial risks and uncertainties. All statements, other than statements of historical facts, included in this Prospectus, including, without limitation, statements regarding the assumptions we make about our business and economic model, our dividend policy, business strategy and other plans and objectives for our future operations, are forward-looking statements.
These forward-looking statements include declarations regarding our management’s beliefs and current expectations. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “would,” “could,” “expects,” “plans,” “contemplate,” “anticipates,” “believes,” “estimates,” “predicts,” “projects,” “intend” or “continue” or the negative of such terms or other comparable terminology, although not all forward-looking statements contain these identifying words. Forward-looking statements are subject to inherent risks and uncertainties in predicting future results and conditions that could cause the actual results to differ materially from those projected in these forward-looking statements. Some, but not all, of the forward-looking statements contained in this Prospectus and the documents incorporated by reference herein include, among other things, statements about the following:
| • | our ability to obtain regulatory approval to market and sell Xilonix™ in the United States, Europe and elsewhere; |
| • | the initiation, timing, cost, progress and success of our research and development programs, preclinical studies and clinical trials for Xilonix™ and other product candidates; |
| • | our ability to advance product candidates into, and successfully complete, clinical trials; |
| • | our ability to successfully commercialize the sale of Xilonix™ in the United States, Europe and elsewhere; |
| • | our ability to recruit sufficient numbers of patients for our future clinical trials for our pharmaceutical products; |
| • | our ability to achieve profitability; |
| • | our ability to obtain funding for our operations, including research funding; |
| • | our ability to identify additional new products using our True Human™ antibody discovery platform; |
| • | the implementation of our business model and strategic plans; |
| • | our ability to develop and commercialize product candidates for orphan and niche indications independently; |
| • | our commercialization, marketing and manufacturing capabilities and strategy; |
| • | our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others; |
| • | our expectations regarding federal, state and foreign regulatory requirements; |
| • | the therapeutic benefits, effectiveness and safety of our product candidates; |
| • | the accuracy of our estimates of the size and characteristics of the markets that may be addressed by our products and product candidates; |
| • | the rate and degree of market acceptance and clinical utility of Xilonix™ and future products, if any; |
| • | the timing of and our and our collaborators’ ability to obtain and maintain regulatory approvals for our product candidates; |
| • | our ability to maintain and establish collaborations; |
29
Table of Contents
| • | our use of proceeds from this offering; |
| • | our expectations regarding market risk, including interest rate changes and foreign currency fluctuations; |
| • | our belief in the sufficiency of our cash flows to meet our needs for at least the next 12 to 24 months; |
| • | our expectations regarding the timing during which we will be an emerging growth company under the JOBS Act; |
| • | our ability to engage and retain the employees required to grow our business; |
| • | our future financial performance and projected expenditures; |
| • | developments relating to our competitors and our industry, including the success of competing therapies that are or become available; and |
| • | estimates of our expenses, future revenue, capital requirements and our needs for additional financing. |
You should also read the matters described in “Risk Factors” and the other cautionary statements made in this Prospectus as being applicable to all related forward-looking statements wherever they appear in this Prospectus. We cannot assure you that the forward-looking statements in this Prospectus will prove to be accurate and therefore you are encouraged not to place undue reliance on forward-looking statements. You should read this Prospectus completely.
30
Table of Contents
We estimate that the net proceeds to us from the sale of the common shares in this offering will be approximately $ million, based upon an assumed initial public offering price of $ per common share (the midpoint of the price range set forth on the cover page of this Prospectus) and after deducting estimated underwriting discounts and commissions, estimated fees payable in connection with the concurrent private placement and estimated offering expenses payable by us. Each $1.00 increase (decrease) in the assumed initial public offering price of $ per common share would increase (decrease) the net proceeds to us from this offering by approximately $ million, assuming the number of common shares offered by us, as set forth on the cover page of this Prospectus, remains the same. We may also increase or decrease the number of common shares we are offering. Each increase (decrease) of 500,000 common shares in the number of shares offered by us would increase (decrease) the net proceeds to us from this offering by approximately $ million, assuming that the assumed initial public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. We do not expect that a change in the initial public offering price or the number of common shares by these amounts would have a material effect on our use of the proceeds from this offering, although it may accelerate the time at which we will need to seek additional capital.
We currently expect to use the net proceeds from this offering as follows:
| • | approximately $30 million to complete our Phase III trials for Xilonix™ in the United States and in Europe; |
| • | approximately $12 million on plant and equipment infrastructure to complete the manufacturing and R&D facilities under construction, and; |
| • | approximately $2 million to complete our Phase II PG study; |
| • | approximately $4 million to complete a Phase I/II study for a treatment for S. aureus infections; |
| • | the remainder for working capital and general corporate purposes. |
This expected use of the net proceeds of this offering represents our intentions based on our current plans and business conditions, which could change in the future as our plans and business conditions evolve. As of the date of this Prospectus, we cannot predict with certainty all of the particular uses for the net proceeds to be received upon the closing of this offering or the amounts that we will actually spend on the uses set forth above. The amounts, allocation and timing of our actual expenditures will depend upon numerous factors, including:
| • | the focus and results of our research, drug discovery and preclinical development activities; |
| • | the type, number, costs and results of any clinical trials for our product candidates; |
| • | regulatory actions relating to our product candidates; |
| • | competitive and technological developments; and |
| • | the rate of growth, if any, of our business. |
31
Table of Contents
To date, we have not declared or paid cash dividends on our shares of common stock. We do not anticipate paying cash dividends to the holders of common stock at any time in the foreseeable future. As a result, you will benefit only from your investment in our common stock through any stock appreciation, and should not expect to receive any dividend payments.
If you invest in our common stock, your ownership interest will be diluted to the extent of the difference between the offering price per share of our common stock and the net tangible book value per share of our common stock immediately after completion of this offering. Net tangible book value per share represents total tangible assets less total liabilities, divided by the number of shares of common stock outstanding. As of September 30, 2014, the net tangible book value of our common stock was approximately $17.2 million, or approximately $0.71 per share based upon 24,339,767 shares outstanding.
After giving effect to our sale of common stock in this offering at the offering price of $ per share and the receipt and application of the estimated net proceeds, our pro forma net tangible book value as of September 30, 2014 would be $ or $ per share. This represents an immediate increase $ in net tangible book value of $ per share to existing shareholders and an immediate dilution in net tangible book value of $ per share to purchasers of securities in this offering. The following table illustrates this pro forma per share dilution:
| Assumed public offering price per share |
||
| Net tangible book value per share as of September 30, 2014 |
||
| Pro forma increase per share attributable to existing investors |
||
|
| ||
| Pro forma net book value per share after this offering |
||
|
| ||
| Dilution per share to new investors |
||
|
|
32
Table of Contents
The following table sets forth our cash and cash equivalents and capitalization as of September 30, 2014:
| • | on an actual basis; and |
| • | on a pro forma as further adjusted basis to give effect to our sale of shares of common stock in this offering at an offering price of $ per share (the mid-point of our offering range on the cover of this Prospectus), after deducting the estimated underwriters commissions and estimated offering expenses payable by us. |
| You should read this table in conjunction with “Use of Proceeds” above as well as our “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and financial statements and the related notes appearing elsewhere in this Prospectus. |
Actual, as of, September 30, 2014 |
Pro forma, as further adjusted for this offering | ||||
| (in thousands) | ||||||
| Shareholders’ equity: |
||||||
| Common stock, no par value, unlimited shares authorized, 24,339,767 and shares outstanding at September 30, 2014 and after this offering |
$ | 104,619 | ||||
| Accumulated deficit |
(87,279 | ) | ||||
| Accumulated other comprehensive loss |
(122 | ) | ||||
|
|
|
| ||||
| Total Shareholders’ Equity |
17,218 | |||||
|
|
|
| ||||
| Total Capitalization |
$ | 17,218 | ||||
|
|
|
| ||||
33
Table of Contents
The following selected consolidated statements of operations data for the years ended December 31, 2012 and 2013 are derived from our audited financial statements included elsewhere in this Prospectus. The selected statements of operations data for the nine months ended September 30, 2013 and 2014, and the selected balance sheet data at September 30, 2014, are derived from our unaudited financial statements, included elsewhere in this Prospectus, which have been prepared on a basis consistent with our audited financial statements and, in the opinion of management, include all adjustments, consisting of normal recurring adjustments, necessary for a fair presentation of our financial position and results of operations. The results of operations for any interim period are not necessarily indicative of results to be expected for the entire year. The following data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and the related notes included elsewhere in this Prospectus.
Below is a table, which is a summary of our financial statements. It should be noted that from September 30, 2014 to December 31, 2014, we issued 3,206,864 shares of common stock for approximately $48.0 million in cash, increasing our cash on hand to $57.3 million.
| Year Ended December 31, | Nine Months Ended September 30, |
|||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||
| Statement of Operations Data |
||||||||||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
$ | 13,334 | $ | 7,935 | $ | 5,610 | $ | 9,424 | ||||||||
| General and administrative |
1,829 | 1,990 | 1,828 | 6,435 | ||||||||||||
| Total operating expenses |
15,163 | 9,925 | 7,438 | 15,859 | ||||||||||||
| Loss from operations |
(15,163 | ) | (9,925 | ) | (7,438 | ) | (15,859 | ) | ||||||||
| Other income (loss): |
||||||||||||||||
| Interest income |
3 | 1 | 1 | — | ||||||||||||
| Foreign exchange gain (loss) |
— | (3 | ) | (3 | ) | 24 | ||||||||||
| Total other income (loss): |
3 | (2 | ) | (2 | ) | 24 | ||||||||||
| Net loss |
$ | (15,160 | ) | $ | (9,927 | ) | $ | (7,440 | ) | $ | (15,835 | ) | ||||
| Net loss per common share—basic and diluted |
$ | (0.70 | ) | $ | (0.44 | ) | $ | (0.33 | ) | $ | (0.66 | ) | ||||
| Weighted average number of common shares—basic and diluted |
21,594,369 | 22,520,416 | 22,341,240 | 24,173,485 | ||||||||||||
| As of September 30, 2014 | ||||||
| Actual | Pro forma As adjusted | |||||
| (in thousands) | ||||||
| Balance sheet data |
||||||
| Cash and cash equivalents |
$ | 15,497 | ||||
| Working capital |
13,428 | |||||
| Total assets |
19,661 | |||||
| Total shareholders’ equity |
17,218 | |||||
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes appearing at the end of this Prospectus. Some of the information contained in this discussion and analysis is set forth elsewhere in this Prospectus and includes forward-looking statements that involve risks and uncertainties. You should review the “Risk Factors” section of
34
Table of Contents
this Prospectus for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
XBiotech is a clinical-stage biopharmaceutical company engaged in discovering and developing “True Human™” monoclonal antibodies for treating a variety of different diseases. True Human™ monoclonal antibodies are those which occur naturally in human beings—as opposed to being derived from animal immunization technologies or otherwise engineered. We believe that naturally occurring monoclonal antibodies have the potential to be safer and more effective than their non-naturally occurring counterparts. While primarily focused on bringing our lead product candidate to market, we have also developed a proprietary “True Human™” monoclonal antibody discovery platform and manufacturing system.
We have never been profitable and, as of September 30, 2014, we had an accumulated deficit of $87.3 million. We had net losses of $9.9 million and $15.2 million for the year ended December 31, 2013 and 2012, respectively, and a net loss of $15.8 million for the nine months ended September 30, 2014. We expect to incur significant and increasing operating losses for the foreseeable future as we advance our drug candidates from discovery through preclinical testing and clinical trials and seek regulatory approval and eventual commercialization. In addition to these increasing research and development expenses, we expect general and administrative costs to increase as we add personnel and begin to operate as a public company. We will need to generate significant revenues to achieve profitability, and we may never do so.
Revenues
To date, we have not generated any revenue. Our ability to generate revenue and become profitable depends on our ability to successfully commercialize our lead product candidate, Xilonix™, or any other product candidate we may advance in the future.
Research and Development Expenses
Research and development expense consists of expenses incurred in connection with identifying and developing our drug candidates. These expenses consist primarily of salaries and related expenses, stock-based compensation, the purchase of equipment, laboratory and manufacturing supplies, facility costs, costs for preclinical and clinical research, development of quality control systems, quality assurance programs and manufacturing processes. We charge all research and development expenses to operations as incurred.
Clinical development timelines, likelihood of success and total costs vary widely. We do not currently track our internal research and development costs or our personnel and related costs on an individual drug candidate basis. We use our research and development resources, including employees and our drug discovery technology, across multiple drug development programs. As a result, we cannot state precisely the costs incurred for each of our research and development programs or our clinical and preclinical drug candidates. From inception through September 30, 2014, we have recorded total research and development expenses, including stock-based compensation, of $65.0 million. Our total research and development expenses for the years ended December 31, 2012 and 2013 were $13.3 million and $7.9 million, respectively, and $9.4 million for the nine months ended September 30, 2014. Stock-based compensation accounted for $2.2 million and $0.6 million for the years ended December 31, 2012 and 2013, respectively, and $0.8 million for the nine months ended September 30, 2014. In 2014, we estimate that our total cash spend on research and development will be approximately $13 million, consisting of $2.4 million of clinical trial expenses for the European trial of Xilonix™, $2.3 million for the US clinical trial for Xilonix™, $4.5 million for all expenses related to drug manufacturing and process developments, $1 million for purchasing scientific equipment and $2.8 million on other research and development expenses not attributable to any specified discovery and development program.
35
Table of Contents
Research and development expenses, as a percentage of total operating expenses for the years ended December 31, 2012 and 2013 were 88% and 80%, respectively, and 59% for the nine months ended September 30, 2014. The percentages, excluding stock-based compensation, for the years ended December 31, 2012 and 2013, and the nine months ended September 30, 2014, were 90%, 80% and 79%, respectively.
We expect our clinical costs to be substantial and to increase as we advance Xilonix™ through Phase III clinical trials in Europe and the US and research and development costs to increase as we move other drug candidates into preclinical testing and clinical trials. Based on the results of our preclinical studies, we expect to selectively advance some drug candidates into clinical trials. We anticipate that we will select drug candidates and research projects for further development on an ongoing basis in response to their preclinical and clinical success and commercial potential.
During 2014 and throughout 2015, we intend to continue our Phase II study in Pyoderma Gangrenosum, or PG. Given that we continue to see positive results in the Phase II study, we intend to seek breakthrough designation from the FDA and launch a pivotal Phase III study in this indication. After the Phase II clinical trial for PG is completed, we intend to conduct an end of Phase II meeting with the FDA and propose a pivotal, randomized controlled study design that will prove efficacy of the MABp1 antibody for the treatment of PG. To our knowledge, there is currently no established regulatory approval path for this condition, and the endpoint the Company intends to propose is a composite of changing wound characteristics. A response rate will be measured at a set time point for the treated arm and the placebo controlled arm, and the difference between the responses will need to reach statistical significance for the trial to be successful. As this is an orphan condition, with a relatively small incidence (~3000 US patients), the Company has anticipated the need to conduct this pivotal study in both the US and Europe.
A clinical study to test our antibody therapy for S. aureus infections is planned based on FDA feedback on the IND filing that we made in December 2014.
Due to the fact that our drug candidates are in the early stage of development, we cannot estimate anticipated completion dates and when we might receive material net cash inflows from our research and development projects.
General and Administrative Expenses
General and administrative expense consists primarily of salaries and related expenses for personnel in administrative, finance, business development and human resource functions, as well as the legal costs of pursuing patent protection of our intellectual property and patent filing and maintenance expenses, stock–based compensation, and professional fees for legal services. Our total general and administration expenses for the years ended December 31, 2012 and 2013 were $1.8 million and $2.0 million, respectively, and $6.4 million for the nine months ended September 30, 2014. Stock-based compensation accounted for $0.6 million and $0.2 million for years the ended December 31, 2012 and 2013, respectively, and $4.2 million for the nine months ended September 30, 2014. We estimate that a total of $2.8 million excluding stock-based compensation will be spent on general and administrative expense for the twelve months ended December 31, 2014.
After this offering, we anticipate increases in general and administrative expense relating to operating as a public company. These increases will likely include legal fees, accounting fees and directors’ and officers’ insurance premiums as well as fees for investor relations services.
Critical Accounting Policies
Our Management’s Discussion and Analysis of our Financial Condition and Results of Operations is based on our financial statements, which have been prepared in conformity with generally accepted accounting principles in the US, or US GAAP. The preparation of our financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and expenses incurred during the reported periods.
36
Table of Contents
We base estimates on our historical experience, known trends and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in the notes to our financial statements appearing in this Prospectus, we believe that the following accounting policies are the most critical to understanding and evaluating our reported financial results.
Stock-Based Compensation
Stock-based awards are measured at fair value at each grant date. We recognize stock-based compensation expenses ratably over the requisite service period of the option award.
Determination of the Fair Value of Stock-Based Compensation Grants
The determination of the fair value of stock-based compensation arrangements is affected by a number of variables, including estimates of the fair value of our common stock, expected stock price volatility, risk-free interest rate and the expected life of the award. We value stock options using the Black-Scholes option-pricing model, which was developed for use in estimating the fair value of traded options that are fully transferable and have no vesting restrictions. Black-Scholes and other option valuation models require the input of highly subjective assumptions, including the expected stock price volatility. If we made different assumptions, our stock-based compensation expenses, net loss, and net loss per common share could be significantly different. During all of the periods presented, we issued common stock for cash consideration to new investors. We believe that such transactions represent the best evidence of fair value of our common stock. Therefore, we used the sales price of our common stock during these periods as the fair value of our common stock.
The following summarizes the assumptions used for estimating the fair value of stock options granted during the periods indicated:
| Year Ended December 31, | Nine Months Ended September 30, | |||||
| 2012 | 2013 | 2014 | ||||
| (unaudited) | ||||||
| Weighted-average grant date fair value per share |
$10.09 | $11.37 | $7.43 | |||
| Expected volatility |
79% | 73% | 70% | |||
| Risk-free interest rate |
1.57%-1.93% | 2.04%-3.84% | 0.69%-2.73% | |||
| Expected life (in years) |
6.25-10 | 6.25-10 | 3-10 | |||
| Dividend yield |
— | — | — | |||
We have assumed no dividend yield because we do not expect to pay dividends in the foreseeable future, which is consistent with our past practice. The risk-free interest rate assumption is based on observed interest rates for US Treasury securities with maturities consistent with the expected life of our stock options. The expected life represents the period of time the stock options are expected to be outstanding and is based on the simplified method when the stock option includes “plain vanilla” terms. Under the simplified method, the expected life of an option is presumed to be the midpoint between the vesting date and the end of the agreement term. We used the simplified method due to the lack of sufficient historical exercise data to provide a reasonable basis upon which to otherwise estimate the expected life of the stock options. For stock options that did not include “plain vanilla” terms we used the contractual life of the stock option as the expected life. Such stock options consisted primarily of options issued to our board of directors that were immediately vested at issuance. Expected volatility is based on historical volatilities for publicly traded stock of comparable companies over the estimated expected life of the stock options.
37
Table of Contents
We based our estimate of pre-vesting forfeitures, or forfeiture rate, on historical forfeiture rates. We apply the estimated forfeiture rate to the total estimated fair value of the awards, as derived from the Black-Scholes model, to compute the stock-based compensation expenses, net of pre-vesting forfeitures, to be recognized in our consolidated statements of operations.
Determination of the Fair Value of Common Stock on Grant Dates
Prior to this offering, we have been a private company with no active public market for our common stock. Our board of directors periodically determined for financial reporting purposes the estimated per share fair value of our common stock at various dates. The board of directors considered all objective and subjective factors that they believed to be relevant including most notably the recent sales activity of our common stock. Significant factors considered were:
| • | the recent sales of our common stock to new investors for cash consideration; |
| • | the fact that we are a privately-held biotechnology company and our common stock is illiquid; |
| • | the nature and history of our business; |
| • | current and forecasted economic conditions, both generally and specific to our industry; |
| • | the state of the initial public offering market for similarly situated privately-held technology companies. |
The following table summarizes by issuance date the number of shares of our common stock sold to investors, including new unrelated investors, from January 1, 2012 through the date of this Prospectus.
| Issuance Date |
Number of Shares | Price Per Share | ||||||
| May to September 2012 |
476,548 | $ | 15.00 | |||||
| August 2013 |
1,204,510 | $ | 10.00 | |||||
| January to June 2014 |
1,195,000 | $ | 10.00 | |||||
| July to December 2014 |
3,584,530 | $ | 15.00 | |||||
Pursuant to the AICPA Audit and Accounting Practice Aid, Valuation of Privately-Held Company Equity Securities Issued as Compensation, privately held enterprises may sometimes engage in arm’s-length cash transactions with unrelated parties for issuances of their equity securities, and the cash exchanged in such a transaction is, under certain conditions, an observable price that serves the same purpose as a quoted market price. Our board of directors believes the sale of common stock by the Company to new unrelated investors represents an observable price since the equity securities in the transaction are the same securities as those for which the fair value determination is being made, and the transaction is a current transaction between willing parties, that is, other than in a forced or liquidation sale and other than under terms or conditions arising from a previous transaction.
We recognized $2,816,000 and $739,000 of stock-based compensation expenses in the years ended December 31, 2012 and 2013; $570,000 and $4,972,000 of stock-based compensation expenses for the nine months ended September 30, 2013 and 2014.
Results of Operations
Nine Months Ended September 30, 2014 and 2013
Revenue. We did not record any revenue during the nine months ended September 30, 2014 or 2013.
Research and Development. Research and development expenses increased by $3.81 million, or 68%, to $9.42 million for the nine months ended September 30, 2014, compared to $5.61 million for the nine months
38
Table of Contents
ended September 30, 2013. Research and development expense consists of direct costs which include salaries and related costs of research and development personnel, clinical trial management personnel, the costs of consultants, outsourcing, materials and supplies associated with research and development projects and clinical trial expenses related to the trials, the data management as well as monitoring expenses. Indirect research and development costs include stock option expenses related to R&D and clinical trial personnel, facilities, depreciation, patents and other indirect overhead costs. The increase was due to a $2.74 million increase in clinical trials expenses; $380,000 increase in stock option expense and a $554,000 increase in compensation expense.
General and Administrative. General and administrative expense increased by $4.6 million, or 252%, to $6.4 million for the nine months ended September 30, 2014 compared to $1.8 million for the nine months ended September 30, 2013. The increase was primarily related to a $4 million charge to expense the fair value of stock options granted to board members for board service and to non-employees for consulting services.
Years Ended December 31, 2013 and 2012
Revenue. We did not record any revenue during the years ended December 31, 2013 or 2012.
Research and Development. Research and development expenses decreased by $5.4 million to $7.9 million for the year ended December 31, 2013, compared to $13.33 million for the year ended December 31, 2012. This decrease was due to a $1.67 million decrease in stock-based compensation and a $1.7 million decrease in salaries due to a reduction in employees in September 2012. In addition the cost for clinical trials and research and development consumables decreased by almost $1 million each.
General and Administrative. General and administrative expense increased to $1.99 million for the year ended December 31, 2013 compared to $1.82 million for the year ended 2012. The increase was primarily related to increases in expenses related to professional fees.
Liquidity and Capital Resources
Our cash requirements could change materially as a result of the progress of our research and development and clinical programs, licensing activities, acquisitions, divestitures or other corporate developments.
Since our inception on March 22, 2005, we have funded our operations principally through the private placement of equity securities, which have provided aggregate cash proceeds of approximately $131.61 million.
In evaluating alternative sources of financing we consider, among other things, the dilutive impact, if any, on our shareholders, the ability to leverage shareholder returns through debt financing, the particular terms and conditions of each alternative financing arrangement and our ability to service our obligations under such financing arrangements.
As of December 31, 2014, we had cash on hand of approximately $57.3 million.
Sources and Uses of Cash
Net cash used in operating activities was $11.6 million in 2012, $8.9 million in 2013, and $8.76 million for the nine months ended September 30, 2014. Net cash used in operating activities for these periods consisted primarily of our net loss, partially offset by depreciation and share based compensation.
In 2012 and 2013, net cash of $0.6 million and $0.1 million, respectively, was used in investing activities to purchase property and equipment. $0.8 million was used in investing activities for the nine months ended September 30, 2014.
39
Table of Contents
Net cash provided by financing activities was $7.3 million and $12.1 million in fiscal 2012 and 2013, respectively; substantially all of which was from the issuance of shares of our common stock to investors. For the nine months ended September 30, 2014, net cash provided by financing activities was $17.8 million; substantially all of which was from the issuance of shares of our common stock to investors.
Capital Expenditure Requirements
We expect to continue to incur substantial operating losses in the future. We will not receive any product revenue until a drug candidate has been approved by the FDA or similar regulatory agencies in other countries and successfully commercialized. We expect to expend between $30 million and $50 million over the next twelve months to fund our current operations. We currently anticipate that our cash, cash equivalents of approximately $57.3 million as of December 31, 2014, together with the proceeds from this offering and cash flow, will be sufficient to fund our operations at least through the next 36 months. However, we will need to raise substantial additional funds to continue our operations and bring future products to market. We cannot be certain that any of our programs will be successful or that we will be able to raise sufficient funds to complete the development and commercialization of any of our drug candidates currently in development, should they succeed. Additionally, we plan to continue to evaluate in-licensing and acquisition opportunities to gain access to new drugs or drug targets that would fit with our strategy. Any such transaction would likely increase our funding needs in the future.
Our future funding requirements will depend on many factors, including but not limited to:
| • | the size and complexity of our research and development programs; |
| • | the scope and results of our preclinical testing and clinical trials; |
| • | continued scientific progress in our research and development programs; |
| • | the time and expense involved in seeking regulatory approvals; |
| • | competing technological and market developments; |
| • | the acquisition, licensing and protection of intellectual property rights; and |
| • | the cost of establishing manufacturing capabilities and conducting commercialization activities. |
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never do, we expect to finance future cash needs primarily through public or private equity offerings, debt financings or strategic collaborations. We do not know whether additional funding will be available on acceptable terms, or at all. Moreover, without the proceeds from this offering, or if we are unable to obtain alternative sources of funding, we do not expect to be able to continue our operations.
Contractual Obligations and Commitments
On January 12, 2008, the Company entered a lease agreement to lease its facility in Austin, Texas, USA. On September 15, 2010, the Company entered into a second lease agreement to lease additional space in Austin, TX, USA. On March 20, 2013, the company extended the lease for another 21 months with the same terms and rental rates as the current lease. Rent expense was $552,646 and $484,558 for the years ended December 31, 2013 and 2012, respectively. The future minimum lease payments are as follows as of December 31, 2013 (in thousands):
| Contractual Obligations |
Total | Less than 1 Year |
1 - 3 Years | 3 - 5 Years | More than 5 Years |
|||||||||||||||
| Operating facility leases |
$ | 470,000 | $ | 403,000 | $ | 67,000 | $ | $ | — | |||||||||||
| Total contractual obligations |
$ | 470,000 | $ | 403,000 | $ | 67,000 | $ | $ | — | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
40
Table of Contents
Income Taxes
We have established four wholly-owned subsidiaries, XBiotech USA, Inc., a United States based company incorporated in Delaware, which employs all of our employees, XBiotech Switzerland AG, XBiotech Japan K.K. and XBiotech Germany GmbH. We are required to file separate income tax returns for all the subsidiaries. Canadian income tax rules require us to treat the United States based operations as an arm’s length company and require the services provided by the United States subsidiary to be charged to us at fair market value. All profit and losses are eliminated upon consolidation.
All Operations
We have incurred net operating losses on a consolidated basis for the years ended December 31, 2012 and 2013, accordingly, we did not pay or record any Canadian or United States federal taxes. As of December 31, 2013, we had non-capital loss carry forwards of $52.8 million (approximately $44.5 million in Canada and approximately $8.3 million in the US), which expire over various periods beginning in 2025.
A full valuation allowance is provided to offset our deferred tax assets because the realization of the benefit does not meet the more likely than not criteria. In the event that we determine that we will be able to utilize our deferred tax assets in the future, an adjustment to the valuation allowance would increase net income in the period such a determination is made.
Inflation
We do not believe that inflation has had a material impact on our business and operating results during the periods presented.
Foreign Currency Fluctuations
For a description of the effect on us of foreign currency fluctuations, see “Quantitative and Qualitative Disclosure of Market Risks”.
Related Party Transactions
For a description of our related party transactions, see “Certain Relationships and Related Party Transactions”.
Off-Balance Sheet Arrangements
Since inception, we have not engaged in any off-balance sheet activities, including the use of structured finance, special purpose entities or variable interest entities.
Quantitative and Qualitative Disclosure of Market Risks
Our concentration of credit risk consists principally of cash and cash equivalents. Our exposure to market risk is limited primarily to interest income sensitivity, which is affected by changes in the general level of Canadian and United States interest rates, particularly because the majority of our investments are in money market accounts.
Our investment policy restricts investments to high-quality investments and limits the amounts invested with any one issuer, industry, or geographic area. The goals of our investment policy are as follows: preservation of capital; assurance of liquidity needs; best available return on invested capital; and minimization of capital taxation and a reduction of impact on Scientific Research and Experimental Development refundable tax credits
41
Table of Contents
under the Income Tax Act (Canada). Some of the securities in which we invest may be subject to market risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with an interest rate fixed at the then-prevailing rate and the prevailing interest rate later rises, the principal amount of our investment will probably decline. To minimize this risk, in accordance with our investment policy, we maintain our portfolio of cash equivalents, short-term marketable securities and restricted cash in a variety of securities, including money market mutual funds, T-bills, GICs, and commercial papers. The risk associated with fluctuating interest rates is limited to our investment portfolio. Due to the short term nature of our investment portfolio we believe we have minimal interest rate risk arising from our investments.
We are also exposed to market risk related to change in foreign currency exchange rates. We contract with CROs that are located in Europe, which are denominated in foreign currencies. We are subject to fluctuations in foreign currency rates in connection with these agreements. We do not currently hedge our foreign currency exchange rate risk. As of December 31, 2013 and September 30, 2014, we had minimal or no liabilities denominated in foreign currencies.
Recent Accounting Pronouncements
The Jumpstart Our Business Startups Act of 2012, or JOBS Act, provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we are choosing to “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
In June 2014 the Financial Accounting Standards Board (FASB) issued ASU 2014-10, Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810 Consolidation. These updates remove the definition of a development stage entity from the Master Glossary of the Accounting Standards Codification, thereby removing the financial reporting distinction between development stage entities and other reporting entities from US GAAP. In addition, the amendments eliminate the requirements for development stage entities to (1) present inception-to-date information in the statements of income, cash flows, and shareholder equity, (2) label the financial statements as those of a development stage entity, (3) disclose a description of the development stage activities in which the entity is engaged, and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. This standard is effective for annual reporting periods beginning after December 15, 2014. The Company has early adopted this standard in the presentation of its 2013 financial statements.
42
Table of Contents
Overview
XBiotech is a clinical-stage biopharmaceutical company engaged in discovering and developing “True Human™” monoclonal antibodies for treating a variety of diseases. True Human™ monoclonal antibodies are those which occur naturally in human beings—as opposed to being derived from animal immunization or otherwise engineered. We believe that naturally occurring monoclonal antibodies have the potential to be safer and more effective than their non-naturally occurring counterparts. While focused on bringing our lead product candidate to market, we also have developed a “True Human™” pipeline and manufacturing system.
The majority of our efforts to date have concentrated on developing MABp1 (also known as Xilonix™, CA-18C3, CV-18C3, RA-18C3, and T2-18C3), a therapeutic antibody which specifically neutralizes interleukin-1 alpha (IL-1a). IL-1a is a pro-inflammatory protein produced by leukocytes and other cells, where it plays a key role in inflammation. When unchecked, inflammation can contribute to the development and progression of a variety of different diseases such as cancer, vascular disease, inflammatory skin disease, and diabetes. Our clinical studies have shown that blocking IL-1a with MABp1 may have a beneficial effect in several diseases.
We completed a Phase I/II clinical trial for MABp1 (Xilonix™) as a treatment for cancer at MD Anderson Cancer Center. The results of this study, published in Lancet Oncology in April 2014, found that in the 52 patients with metastatic cancer (18 tumor types) who participated, MABp1 was well tolerated, with no dose-limiting toxicities or immunogenicity. Moreover, within eight weeks of starting therapy many patients began to improve with respect to constitutional symptoms. An imaging method, known as dual energy X-ray absorptiometry (DEXA), revealed that many of the patients improved physically, in terms of gaining lean body mass; and patient reported outcomes documented that many were recovering from pain, fatigue and appetite loss. Finally, we found that in the patients with colorectal cancer, DEXA-measured recovery was associated with significant improvement in survival.
We received a fast track designation from the FDA in October 2012 to develop Xilonix™ as a treatment in the setting of metastatic colorectal cancer. The purpose of the fast track designation is to aid in the development, and expedite the review, of drugs that have the potential to treat a serious or life-threatening disease. Currently we have two Phase III studies underway — one launched in the US for advanced refractory colorectal cancer and another in Europe for symptomatic colorectal cancer. If these trials are successful, we will seek marketing approvals for MABp1 (Xilonix™) in Europe and/or in the United States. Assuming such marketing approvals are obtained, we would distribute and sell this product through our own direct sales force or with a commercial partner.
We are also investigating MABp1 in clinical trials for other indications including vascular disease, type II diabetes, acne and psoriasis. In a randomized Phase II study involving 43 patients, we evaluated MABp1 for its ability to reduce adverse events after balloon angioplasty, atherectomy or stent placement in patients undergoing revascularization procedures for blockage of the superficial femoral artery (SFA), a major artery in the leg. While the study did not involve a large enough patient population to provide a statistically significant outcome, results from this study showed an important trend towards the reduction of restenosis and reduced incidence of Major Adverse Cardiovascular Events (“MACE”) in treated patients compared to the control group. In 2012, we obtained a fast track designation to develop MABp1 as a therapy to reduce the need for re-intervention after treatment of peripheral vascular disease with angioplasty or other endovascular methods of treatment.
In a Phase II pilot study completed in 2012, we tested MABp1 in patients with Type 2 diabetes. A treatment-related decline in HbA1c, and increased serum levels of pro-insulin and C-peptide (indicators of improved glucose control and pancreas function, respectively) were observed. We also conducted two phase II pilot studies in skin disease, evaluating the potential benefit of MABp1 in subjects with (1) moderate to severe plaque psoriasis and (2) moderate to severe acne vulgaris. The psoriasis study revealed rapid improvements in the Psoriasis Area and Severity Index (PASI), with patients having a median of 43% improvement within
43
Table of Contents
35 days. In the acne study, treated patients exhibited a continual improvement in lesions over the course of therapy, with up to 42% reduction in eight weeks; and interestingly, these patients had a statistically significant improvement in anxiety, as measured by the Hospital Anxiety and Depression Scale (HADS). We continue to analyze our clinical results, and prioritize further clinical initiatives for MABp1 in oncology, SFA, diabetes, psoriasis and acne.
We recently filed an Investigational New Drug Application (IND) for a True Human™ Antibody therapy we are developing to treat infections due to Staphylococcus aureus. This product candidate was identified from an individual that harbored a natural antibody capable of neutralizing drug-resistant strains of S. aureus. This study is currently on clinical hold while, on the request of the FDA, we complete an animal toxicology study.
More recently, we have begun using our True Human™ antibody technology to begin developing a therapy for Ebola virus infection. We recently received a blood donation from an Ebola-recovered patient which we have now confirmed contains high levels of anti-Ebola antibodies. A product candidate derived from this blood is expected to go into animal studies in 2015.
To produce our product candidates, we have developed a manufacturing system that employs simple disposable bioreactor technology. Our manufacturing operation is currently located within our forty-six thousand square foot facility in Austin, Tx. To accommodate larger-scale commercial manufacturing needs, we purchased 48 acres of industrial-zoned property located 5 miles from Austin’s central business district. In September 2014, we commenced ground-breaking on a new manufacturing facility on this property. Construction is estimated to be completed by late 2015, and we expect to begin operating in the new facility in early 2016. The new facility will be capable of producing several hundred thousand doses of antibody annually.
A Background on Therapeutic Antibodies
A century ago scientists and physicians envisioned being able to custom design therapeutic agents that were highly specific for a single biological target. By selectively attacking disease while sparing healthy tissue, these “magic bullets” were thought to be ideal therapeutic agents. It was not until the early 1970’s, however, that this vision was realized when Kohler and Milstein developed a ground-breaking method for making target-specific monoclonal antibodies—a Nobel prize-winning endeavor. Using this new approach, numerous monoclonal antibody-based research, diagnostic, and therapeutic products have been developed.
Kohler and Milstein’s discovery was based on their knowledge that the immune system of higher animals produces antibodies as a method of protecting them from various potentially damaging agents such as viruses, bacteria, and diseased cells. White blood cells known as B cells produce billions of different types of antibodies, each with a unique potential to selectively attach to and neutralize different disease targets. The vast array of possible treatments based on antibodies lead to the development of what is now a major industry around the use of therapeutic antibodies.
True Human™ Antibodies
White blood cells in the human body secrete billions of different antibodies that circulate through the blood to react and protect us from toxins, infectious agents or even other unwanted substances produced by our body. True human™ antibodies, as the name implies, are simply those that are derived from a natural antibody identified from the blood of an individual.
To develop a True Human™ antibody therapy, donors are screened to find an individual that has a specific antibody that matches the desired characteristics needed to obtain the intended medical benefit. White blood cells from that individual are obtained, the unique gene that produced the antibody is cloned, and the genetic information is used to produce an exact replica of the antibody sequence. A True Human™ antibody is therefore not to be confused with other marketed antibodies, such as so-called fully human antibodies—where antibody reactivity is developed through gene sequence engineering in the laboratory.
44
Table of Contents
Fundamental Science of True Human Antibodies
To appreciate the background safety and tolerability of True Human™ antibodies, it is important to consider the fundamental biology of natural antibody production.
Billions of different white blood cells secrete billions of unique antibodies every day into the circulation. The vast number of different antibodies (and cells that produce them) are essential to enable adequate molecular diversity to ward off all potential infectious or toxic threats. In other words, since antibodies act to bind and thereby neutralize unwanted agents, any given circulating antibody must be able to react with a potentially limitless number of existing or evolving disease entities.
The staggering number of different antibodies needed to achieve this level of preparedness, however, is a daunting concept from a genetics point of view. If an individual antibody gene was needed to encode each of a billion different antibodies, there would be 20,000-times as many genes needed just for antibodies as there would be needed to encode the rest of the entire human genome! Individual cells would need to be gigantic, and monumental resources would be required to make, copy and maintain all of the DNA. Clearly, the system of antibodies could not have evolved to protect us, had not an elegant solution emerged to deal with this genetic conundrum.
Thus a hallmark of the immune physiology of all vertebrates (all have antibodies) is the ability to recombine and selectively mutate a relatively small number of gene segments to create a phenomenal and effectively unlimited number of antibody genes. By rearranging, recombining and mutating the genetic code, specialized white blood cells, or B lymphocytes, are able to create an unlimited array of antibody genes. The consequence of this genetic engineering, however, is that each antibody gene is unique to the individual B lymphocyte that created it—and no copy of the gene exists in the human germline. The only place to find a unique antibody gene is in the individual cells that created it.
The extraordinary process of gene rearrangement and mutation results in a multitude of unique B lymphocytes and consequently an incredibly diverse repertoire of antibodies in any given individual.
Elucidating the mechanisms behind the production of unique antibody genes must be considered one of the major achievements of medical research in the 20th century. Yet unfolding this mystery created another problem to solve: If antibodies were not produced from genes encoded in the human genome and the products of these genes were new to the body, why were these antibody molecules not recognized by the immune system as foreign substances—like any other foreign substance that they were intended to eradicate? How could the body distinguish the apparently “foreign” antibody molecules from the bona fide infectious intruders?
Unraveling the genetics of antibody production led to another major advance in medicine: the discovery of how an endless array of antibody proteins could be made in a way that individual molecules were always tolerated by the body.
In the early 1990s research began to demonstrate that the production of antibodies was not an unregulated process. Rather, it was learned that the antibodies produced by each and every B lymphocyte were subject to intense scrutiny. Studies showed that B lymphocytes which produced acceptable antibodies were stimulated to grow while those that produced “autoreactive” antibodies were not. B lymphocytes that produced “good” antibodies were stimulated to proliferate, and enabled to produce copious amounts of antibody in the event it was needed to ward off a harmful agent. B lymphocytes that rearranged genes to produce antibodies that were ineffective or were autoreactive were given signals that instructed them to engage in a process of programmed cell death. Thus B lymphocytes producing harmful or useless antibodies are simply killed off. This mechanism for creating antibody diversity on the one hand, while protecting the individual from a mass of unwanted or intolerable antibody molecules on the other, was as elegant as it was fundamental to the success of vertebrate immune physiology.
45
Table of Contents
This process of “selection” has been elucidated in great detail. There can be no more important feature of immune physiology than the process of selection. Selection is a fundamental step to enable the body to produce an extremely diverse set of antibody molecules without, in the process, producing an array of novel molecules that cause harm.
Industry Context
Until now each and every therapeutic antibody on the market has been derived from animals and/or through gene sequence modification in the laboratory to produce a desired antibody reactivity. Marketed antibodies to date, described as “fully human”, are not derived from human gene sequences that have undergone the crucial process of selection in a human.
Without exception, all marketed products to date that are described as “fully human”, are in fact engineered and are not selected based on natural tolerance in the human body. The use of the term fully human to describe these products has thus created considerable confusion. To our knowledge, there are at present no True Human™ antibodies currently marketed. If successful in clinical development, our lead product Xilonix™ is expected to be the first True Human™ therapeutic antibody to be commercialized.
Platform Technology
There are significant technical challenges in identifying and cloning genes for True Human™ antibodies. A key problem to overcome can be to first identify individuals with the desired antibody reactivity. This can involve screening hundreds of donors to enable the identification of a single, clinically relevant antibody—discovered from literally trillions of irrelevant background antibody molecules in the blood of donors. We screen human donors to find an individual who has in his or her blood a specific antibody that we believe will be protective against a certain disease. White blood cells from that individual can then be isolated, and the unique gene that produced the antibody obtained.
Novel cloning technologies developed at XBiotech have enabled us to clone the crucial antibody gene sequences from these donors in order to reproduce a True Human™ antibody for use in clinical therapy. A True Human™ monoclonal antibody should therefore not be confused with other marketed therapeutic monoclonal antibodies, such as those currently referred to as fully human antibodies.
Market Opportunity
We have a number of indications in various stages of clinical or pre-clinical development with significant market opportunities. These include oncology, diabetes, dermatology and infectious disease indications. At present, our therapy for colorectal cancer, in which we are conducting two Phase III clinical studies, is the most advanced.
Cancer Business and the Market for Colorectal Therapies
The development of new therapies for the treatment of cancer continues to be a fundamental area of focus and growth in the pharma and biopharmaceutical industry. Detailed and thorough analysis of the oncology business, including that for colorectal cancer, is available from a number of research specialists. In brief, over the past decade expenditures in R&D and revenues derived from oncology related sales have continued to grow largely as anticipated. According to the independent research group IMS Health, the market for cancer drugs grew at an annual rate of about 5% between 2008 and 2013, which is slower than previous years, but still makes it by far the largest sector of the pharmaceutical industry. In 2017, they predict between $74 and $84 billion in sales, double that of the second place area of sales, which is diabetes.
46
Table of Contents
The three major regional markets for cancer drugs are the United States, Europe and Japan, with US sales representing about 40% of the global market share. The largest revenue earners in the oncology space are the therapeutic antibodies Avastin, Herceptin and Rituximab, with combined sales reported to be over $16 billion in 2010.
The market for colorectal cancer (CRC) drugs is expected to grow, according to the independent research group RnRMarketResearch.com, at a compound annual growth rate of 1.8%, to reach $9.4 billion in annual sales over the next 5 years. The US market for CRC therapeutics commands about 44% of the global business, more than double that of the next biggest market, Japan.
According to RnRMarketResearch.com, however, growth for the CRC drug business is expected to be tempered by increasing availability of biosimilar competitors, particularly those challenging leading products such as Avastin and Herceptin. There is also a substantial pipeline for CRC therapeutics across the industry in various stages of commercialization that are expected to bring new drugs to patients and competition to the market place.
An intense industry focus on developing new therapies for CRC is not surprising since this a highly prevalent form of cancer. According the American Cancer Society, Surveillance Research 2015, nearly 130,000 men and women in the United States will be diagnosed with CRC in 2015. This represents almost 20% of all forms of cancer that will be diagnosed in 2015. In terms of mortality, as many as 50,000 people are expected to die from the disease in 2015 and many more will be refractory to existing therapy.
Our therapeutic antibody Xilonix™ is being evaluated as a monotherapy to treat advanced stages of colorectal cancer. If Xilonix™ therapy proves successful in both its Phase III clinical programs, it will have demonstrated the ability to not only increase survival of patients, but also to facilitate recovery and reduce debilitating symptoms of the disease.
We are aware of no other cancer therapy either marketed or in development that achieves overall survival benefit and in the course of treatment improves quality of life, and facilitates physical recovery of advanced cancer patients. While the market for CRC drugs is competitive and dynamic, in the event marketing approval is established for Xilonix™, we expect this therapy to be a highly valued and unique therapy in oncology, providing for significant market share in advanced CRC.
Our Strategy
Our objective is to fundamentally change the way therapeutic antibodies are developed and commercialized, and become a leading biopharmaceutical company focused on the discovery, development and commercialization of therapeutic True Human™ antibodies. The key goals of our business strategy are to:
| • | Obtain regulatory approval to market and sell Xilonix™ in the United States, Europe and other markets, and begin commercial sale of Xilonix™; |
| • | Continue our research and clinical work on infectious diseases, including S. aureus; |
| • | Review our clinical results for SFA, diabetes, psoriasis and acne and determine additional research or clinical studies which we may conduct in the future; |
| • | Discover other True Human™ antibody therapies using our proprietary platform; and |
| • | Leverage our manufacturing technology. |
47
Table of Contents
Product Pipeline
Our product development status for the first quarter of 2015 is as follows:
Competition
There continues to be a highly active commercial pipeline of therapeutic antibodies globally, involving a complex array of development cycles as products reach the end of their patent life and as new candidate products proceed into pivotal studies and approach registration. While there are numerous independent reviews on the subject in both trade journals and academic press, XBiotech has analyzed and attempted to synthesize these publicly available data in order to understand, at least at the broadest level, the competitive landscape for its products.
While we believe True Human™ therapeutic antibodies are an important positive differentiating factor from other monoclonal antibodies currently marketed, we also believe the unique activity of our anti-cancer therapeutic Xilonix™, which is being tested for its ability to both improve well being and extend life, if proven in our phase III clinical programs, will be highly differentiated in the market place for colorectal cancer therapeutics. However, regardless of the potential advantages or uniqueness of Xilonix™ in the market, we do nevertheless expect these products to compete head-to-head with the numerous existing candidate antibody products in development, including emerging biosimilar therapeutic antibodies.
48
Table of Contents
As reported in the National Review of Drug Discovery (October 2010), more than 140 “fully human” antibodies had entered clinical studies and from 2004-2013 nineteen antibody therapeutics were approved in the US for marketing, with some antibodies receiving marketing approval in multiple indications (Reichert JM, Antibodies to Watch in 2014. mAbs. 6:4, 799-802). Thus the competitive landscape is both broad and complex (See below Table).
| Drug |
Company |
Generic Name |
Status of Drug |
Indication | ||||
| Cancer Treatments |
||||||||
| Yervoy ® |
Bristol-Myers Squibb | Ipilmumab | FDA Approved | Melanoma | ||||
| Vectibix ® |
Amgen | Panitumumab | FDA Approved | Colorectal Cancer | ||||
| Arzerra® |
GlaxoSmithKline | Ofatumumab | FDA Approved | Leukemia | ||||
| Cyramza™ |
Lilly | Ramucirumab | FDA Approved | Non Small-Cell Lung Cancer | ||||
| Opdivo™ |
Bristol-Myers Squibb | Nivolumab | FDA Approved | Melanoma | ||||
| Cyramza™ |
Lilly | Ramucirumab | FDA Approved | Gastric and Colorectal Cancer | ||||
| Sylvant® |
Janssen | Siltuximab | FDA Approved | Myeloma (Multicentric Castleman’s Disease) | ||||
| Entyvio® |
Takeda Pharmaceuticals | Vedolizumab | FDA Approved | Ulcerative Colitis and Crohn’s disease | ||||
| Daratumumab |
Janssen | Daratumumab | Multiple Myeloma | |||||
| Necitumumab |
Eli Lilly | Necitumumab | Pending FDA | Non Small-Cell Lung Cancer | ||||
| Patritumab |
Daiichi Sanko | Patritumab | Phase 3 | Non Small-Cell Lung Cancer; Head and Neck Cancer | ||||
| MEDI-4736 |
AstraZeneca | MEDI-4736 | Phase 3 | Non Small-Cell Lung Cancer | ||||
| RG7446 |
Roche/Genentech | RG7446 | Phase 3 | Non Small-Cell Lung Cancer | ||||
| Cardiovascular Disease Treatments |
Novartis | Canakinumab | Phase 3 | Recurrent CV events | ||||
| Pyoderma Gangrenosum Treatments |
Xoma/Servier | Gevokizumab | Phase 3 | PG | ||||
| Acne or Psoriasis Treatments |
||||||||
| Secukinumab |
Novartis | Pending FDA | Psoriasis | |||||
| Brodalumab |
Amgen | Phase 3 | Psoriasis | |||||
| Guselkumab |
Janssen | Phase 3 | Psoriasis | |||||
Recent Developments
In 2014, at least four antibody drugs—ramucirumab, siltuximab, vedolizumab and pembrolizumab—for the treatment of gastric cancer, Castleman disease, Crohn’s disease and melanoma achieved regulatory approval for marketing authorization in the United States. While this was the first marketing approval for each of these molecules, we can assume more approvals will be forthcoming for other indications for these agents in the future.
Ramucirumab is a classical anti-angiogenic, targeting the VEGF receptor system similar to that of the highly successful Avastin™ product. Such an anti-angiogenic does have overlap with Xilonix™, since the latter is expected, albeit not proven, to have anti-angiogenic properties through its inhibition of several downstream pathways related to IL-1 signaling. However, there are numerous anti-VEGF antibodies, in addition to several
49
Table of Contents
small molecule inhibitors of the pathway, and we expect that some of these may be seen as “me too” therapies and may suffer from difficulties with differentiation in the market place. Moreover, ramucirumab is indicated for treatment of advanced gastric cancer or gastro-esophageal adenocarcinoma and may not find utility for the treatment of colorectal cancer or other cancers relevant to Xilonix™. Also the approved dosage of 8 mg/kg is quite high, and considering the conventional production processes involved in manufacturing this product, pricing of the drug will be highly constrained at the upper end of the spectrum compared to what would be possible for Xilonix™. Ramucirumab is reported to be involved in a Phase 3 clinical study for non-small cell lung cancer (NCT01168973) and colorectal cancer (NCT01183780), but there is no indication of efficacy at this time.
The approval for the anti-IL-6 antibody siltuximab does indicate that it has potential anti-cancer applications, although Castleman’s disease is a rare and poorly defined malignancy and therapeutic activity for the drug in other cancer indications is uncertain. This approval is of interest to XBiotech, since we have both a True Human™ anti-IL-6 antibody in the pre-clinical pipeline and have seen responsiveness to Xilonix™ in Castleman’s disease. In our Phase I/II clinical study conducted at MD Anderson Cancer Center with Xilonix™, a single patient with Castleman’s who had previously received and failed anti-IL-6 therapy had a durable response to Xilonix™ therapy, including dramatic reduction in IL-6 levels in the blood. In the same study, in patients with other cancer types treated with Xilonix™, the therapy was found to have an effect on reducing IL-6 levels. This link between Xilonix™ therapy and reduction in IL-6 levels was anticipated, since Xilonix™ neutralizes a target that mediates signals to induce IL-6 production in the body. Our True Human™ anti-IL-6 antibody is not currently scheduled for clinical development nor is there any immediate plans to launch a clinical program with the antibody. While we have considered launching a Castleman’s clinical study with Xilonix™, the extremely small size of the patient population led us to focus Xilonix™ development in other areas where there is much greater unmet patient need.
The FDA granted marketing approval for Vedolizumab to treat ulcerative colitis and Crohn’s disease (CD). MABp1 is considered to have the potential to treat gastro-intestinal ailments with important inflammatory features. Excellent preclinical reports in animal models support the use of MABp1 in these indications, but no clinical programs have been initiated or are in the immediate scope at this time. It would be some time before XBiotech could launch a clinical study for these indications and vedolizumab would be expected to be well established in the market before we could complete clinical development and receive approval for such a therapy. Vedolizumab is a humanized murine antibody that is unlikely to achieve good tolerability or long term efficacy due to immunogenicity. Therefore, providing comparable efficacy, we expect that an opportunity to effectively compete with this product, even with its foothold in the market, would be available given that MABp1 is a True Human™ antibody.
Pembrolizumab is an anticancer agent that was approved late in 2014 for the treatment of melanoma. This antibody works by activating the immune system, such that it may stimulate an immune-mediated anti-tumor cytotoxicity. There are a number of antibodies that are in development for, or have reached the oncology market, such as ipilimumab, based on this general mechanism of action. While approvals have been forthcoming with these therapies, these agents share in common severe side effects without achieving high rates of durable remission. In advanced malignant disease, these agents have and will likely continue to achieve success in the market place. Both pembrolizumab and ipilimumab are approved for the treatment of melanoma, a disease for which we currently have no clinical development plans for Xilonix™. However, with respect to the overall competitiveness of these approaches vis-a-is Xilonix™, our Phase III clinical programs are intended to demonstrate overall survival benefit that is generally consistent with those observed with cytotoxic therapies in refractory disease. Additionally, in the case of Xilonix™, we are also attempting to demonstrate improved life quality and physical recovery. We expect that Xilonix™ will therefore compete favorably with this new and emerging class of anti-cancer antibodies.
50
Table of Contents
CURRENT CLINICAL INVESTIGATION ACTIVITY
Current Clinical Activity
European Registration Study Oncology
Currently, we have a double-blinded, placebo-controlled Phase III registration study underway in Europe. Clinical sites are located in a number of different EU member states, and the addition of sites in Russia is expected soon. The study aims to evaluate MABp1, or Xilonix™, as an anticancer therapy in patients with symptomatic colorectal cancer.
The primary objective of this study is to assess the efficacy of Xilonix™ in reversing symptoms in patients with symptomatic colorectal cancer. By blocking a substance that helps tumors grow and spread, Xilonix™ therapy may not only slow tumor growth, but also may improve symptoms of muscle loss, fatigue, appetite loss, and pain in patients with colorectal cancer.
The efficacy of the therapy will be measured by assessing the change in these symptoms for patients treated with Xilonix™ versus those treated with placebo. Reversal of muscle loss will be assessed with a type of X-ray called a DEXA scanner. Improvement in pain, appetite loss, and fatigue will be measured with a questionnaire that is completed by patients enrolled in the trial.
The study, which started in July 2014, will enroll at least 276 patients and is expected to be completed by mid-2015. As of January 30, 2015 about 98 patients had been enrolled. If the study endpoints are satisfactorily achieved, we expect to submit a registration package to the EMA and possible other foreign regulatory agencies.
US Registration Study Oncology
A Phase III randomized study using Xilonix™ was started in March 2013 (IND #114,759). The study, which was recruiting patients at over sixty cancer centers in the US, was halted by us in September 2014 to propose changes in inclusion criteria to the FDA to enable faster patient recruitment. Amendments were proposed and agreed to by the FDA in order to correct what we believed to be unnecessary enrollment barriers.
Changes to the study protocol included eliminating the 5% weight loss requirement for patients and terminating the use of megesterol acetate (megace) in the control arm. The new agreed upon protocol will enable recruitment of all advanced, refractory colorectal patients regardless of weight loss, and use a 2:1 double-blinded randomization against placebo rather than a 1:1 randomization against megace. We expect to begin treating patients under the new protocol in February 2015. Enrollment under the revised protocol is expected in to be completed in 2016.
While we regretted the disruption to the Phase III study and impact on patients and caregivers, the protocol revision allowed an important analysis to be performed on the intended primary and secondary endpoints of the study. This analysis provided important insights into the activity of Xilonix™ in the patient population. Forty patients had entered the study with approximately equal numbers in each arm. The findings were not statistically significant due to the relatively small number of patients (the statistical model was designed for 656 patients), but the trends observed were encouraging and suggest continuation of the study.
Phase II study Pyoderma Gangrenosum (“PG”)
A phase II open-label exploratory study is underway to evaluate MABp1 for treatment of the rare skin disorder PG (IND# 112,459). PG is a chronic condition characterized by inflamed, non-healing skin ulcerations. The study is evaluating safety and efficacy of the therapy to facilitate wound healing and is being conducted at 5 sites in the United States. Primary endpoints of study involve clinicians’ and patients’ global assessment at day 28 from baseline. Patients who are found to be responding to therapy, but who have not yet experienced complete resolution of their lesion(s) after 28 days of therapy may participate in up to 3 additional 28 day cycles.
51
Table of Contents
Ten patients are scheduled to be enrolled. We plan to evaluate patient responses in the first quarter of 2015 and based on these results may elect to seek breakthrough designation with the FDA.
Phase I/II Study for Staphylococcus Aureus
We recently filed an Investigational New Drug Application (IND) for a Phase I/II randomized clinical study to assess our True Human Antibody therapy for the treatment of serious infections due to Staphylococcus aureus. This product candidate was identified from an individual that harbored a natural antibody capable of neutralizing S. aureus, including drug-resistant strains of the bacteria. The study is currently on clinical hold while, at the request of the FDA, we complete an animal toxicology study. We expect to complete the animal study in the first quarter of 2015.
52
Table of Contents
SUMMARY OF CLINICAL FINDINGS TO DATE
Safety
Our lead product under development, MABp1, is derived from a natural human immune response. We expected that this would facilitate better tolerability when used as a therapeutic compared to humanized or “fully human” monoclonal antibodies. Antibody therapies are known to be associated with significant risk for infusion reactions, including serious anaphylactic reactions. We believe that these reactions are the result of using antibodies that were not derived from natural human immunity but rather had engineered specificities. Based on scientific principles of antibody physiology, a fundamentally important premise was that our True HumanTM antibody therapy should be safer and result in less infusion-related complications than engineered human antibodies when used in clinical studies.
As illustrated in the table below, therapeutic monoclonal antibodies, even those so-called “fully human,” have been associated with infusion reactions (see table below). MABp1 has been administered over 700 times to 170 patients in seven different clinical trials. As of January 30, 2015, there has not been an incident of an infusion reaction with MABp1, nor has there been a single reported incidence of “probably” or “definitely” drug-related serious adverse events (SAEs) of any kind across all the studies.
Reports of Infusions Reactions from Leading marketed “Fully Human” Antibody Products Versus MABp1 (Table below).
| Fully Human Antibodies | Target | Incidence of Infusion Reactions* | ||
| Trastuzumab |
HER-2 | 40% | ||
| Alemtuzumab |
CD52 | 10-35% had Grade ³3 | ||
| Natalizumab |
a4-integrin | 11-24% | ||
| Tocilizumab |
IL-6 | 8%, Fatal Anaphylaxis | ||
| Bevacizumab |
VEGF | 3% | ||
| True HumanTM | ||||
| Xilonix |
IL-1a | 0% | ||
| * | Source of data: Herceptin® [trastuzumab]. Package Insert; San Francisco, CA: Genentech, Inc; June 2014. Campath® [alemtuzumab]. Package Insert; Cambridge, MA: Genzyme Corporation; August 2009. Tysabri® [natalizumab]. Package Insert; Cambridge, MA: Biogen Idec Inc.; December 2013. Actemra® [tocilizumab]. Package Insert; San Francisco, CA: Genentech, Inc.; November 2014. Avastin® [bevacizumab]. Package Insert; San Francisco, CA: Genentech, Inc.; November 2014. |
Interpretation of Results from a Previous Phase I/II Oncology Study with Xilonix
Non-Small-Cell Lung Cancer (“NSCLC”)
Sixteen evaluable NSCLC patients were treated using MABp1 monotherapy (Xilonix™) as part of the Phase 1/II clinical trial for MABp1 (“Xilonix”) at MD Anderson Cancer Study in Houston, Texas. The study design was single arm, and examined radiographic tumor response, change in lean body mass as measured by DEXA, change in quality of life, and overall survival. NSCLC Patients with both pulmonary and non-pulmonary or only non-pulmonary metastases have been reported to have median time to death from date of disease progression of 3.2 months1 which was reported in Changes in the Natural History of Non-small Cell Lung Cancer (NSCLC))—Comparison of Outcomes and Characteristics in Patients with Advanced NSCLC Entered in Eastern Cooperative Oncology Group Trials Before and After 1990 (Heather Wakelee and et al, 2006 American Cancer Society).
The NSCLC patients treated with MABp1 monotherapy all entered the study with progressive refractory disease and all had pulmonary and/or non-pulmonary metastasis.
53
Table of Contents
Overall survival for the NSCLC patients treated in this study was 7.6 months, which is notably greater than 3.2 months. Stratification based on prior anti-EGFR therapy revealed a median survival of 9.4 months (IQR 7.6-12.5) for those pretreated with Tarceva® (N=10) versus a survival of 4.8 months (IQR 4.3-5.7) for those without (N=6, logrank p=0.187).
The first figure below compares patient overall survival after treatment with MABp1 with that observed in another study with Tarceva®, a drug recently approved for treating NSCLC. The median survival for patients treated with Xilonix™ was 7.6 months. It should be noted that 63% of the Xilonix™ patients had taken Tacerva® and failed. This compares with median survival for a similar patient population treated with Tarceva®, which is a historical control group, where survival was only 6.7 months. Overall survival in the control population in the Tarceva® study was 4.7 months. The comparison between overall survival observed with Xilonix™ and that of the Tarceva® study should be viewed with caution, since the patient populations or supportive care or other factors may have been different between the two studies making direct comparison difficult.
The second figure compares overall survival of MABp1 treated patients based on whether or not they received pre-treatment with Tarceva®. These findings suggest a remarkable interaction between Tarceva® pre-treatment and MABp1. Survival in the Tarceva® pre-treated group was nearly double compared to those who had not received Tarceva® previously.
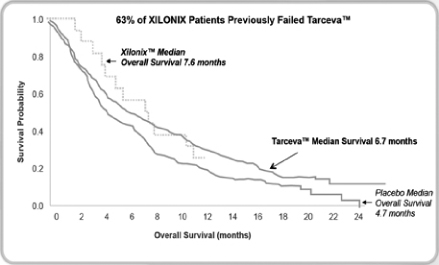
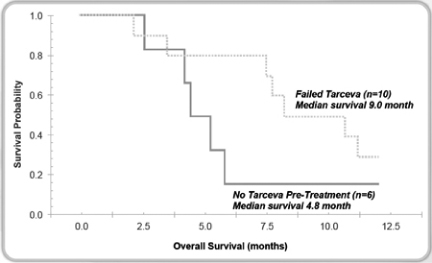
54
Table of Contents
When overall survival was analyzed according to pre-treatment status, it was found that having received, but failed Tarceva®, correlated with significantly increased survival. The Kaplan-Meier curves compare survival of patients who had either received and failed (green line) or had not received (gray line) treatment of Tarceva® prior to receiving MABp1 treatment. This subset analysis reveals that patients who had received Tarceva® treatment prior to receiving MABp1 live longer on average than patients who did not receive Tarceva®. This type of analysis may help in designing future trials of MABp1.
Colorectal Cancer
Fourteen colorectal carcinoma patients were treated using MABp1 monotherapy, as part of a trial of the Phase I/II clinical trials at MD Anderson Cancer Center in Houston, Texas. The study design was single arm, and examined radiographic tumor response, change in lean body mass as measured by DEXA, change in quality of life, and overall survival. Patients with colorectal cancer (n=14) were assessed for overall survival and the median overall survival for these patients was 8.7 months (IQR 6.4–22.1). Survival outcomes in patients with colorectal cancer who had increases in lean body mass showed numerically longer survival than those who lost lean body mass during the study (median 19.3 months vs 6.6 months; log-rank p=0.098). Overall survival for colorectal cancer patients was determined to be significantly greater than what is expected for this patient population irrespective of treatment strategies. The advanced, refractory colorectal cancer patients treated with MABp1 had a median survival of 8.7 months. Based on large clinical studies where similar patient populations were involved, median survival for this patient group receiving only placebo is expected to be about 4.7 months (Jonker D., et al. Cetuximab for the Treatment of Colorectal Cancer. N Engl J Med 2007;357:2040-8.). A recently approved drug, Regorafinib™, improved survival in this patient population to a median of 6.0 months.
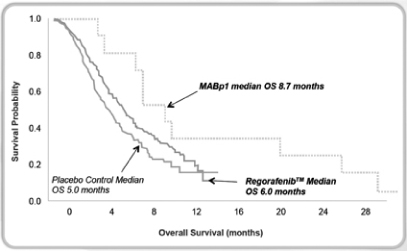
The Kaplan-Meier curves show overall survival of colorectal cancer patients treated with MABp1 (green line) compared to overall survival observed for the drug Regorafenib™, which was used in a similar patient population, a historical control group, a study that recently resulted in marketing approval for the drug. The median overall survival (“OS”) of the cohort of refractory colorectal cancer patients treated with MABp1 was 8.7 months. Patients treated in the Regorafenib™ pivotal trial had a median OS of 6.0 months compared with 5.0 months for the placebo controls. The comparison between overall survival observed with Xilonix and that of the historical control group Regorafenib study should be viewed with caution, since the patient populations or supportive care or other factors may have been different between the two studies making direct comparison difficult.
55
Table of Contents
Anorexia/Cachexia
A subset of thirty advanced cancer patients that were treated as part of the Phase I/II clinical trial at MD Anderson Cancer Center in Houston, Texas were assessed for anorexia/cachexia using a variety of means, including dual energy X-ray absorptiometry (DEXA) scans at screening and after an 8-week follow-up period. Cachexia is an irreversible loss of muscle mass observed in the setting of a chronic disease. All patients had very advanced stage disease, having failed a median of five previous treatment regimens, including chemo/radiotherapy. Seventy-seven percent (23 of 30) of the patients evaluated had demonstrable weight loss as formal evidence of cachexia, while otherwise the advanced disease status also suggested cachexia.
Cachexia in advanced cancer is well known to correlate with poor survival outcomes. Yet in the absence of therapeutic agents to reverse this cachexia process, it has never been formally established that a measure of LBM, on its own, might offer a means to predict survival outcomes in cancer patients. XBiotech believes that the observed increases in LBM, and improved survival outcomes seen with increased LBM, were due to a combination of anti-cachexia, anti-tumor and anti-metastatic effects of the antibody therapy, which resulted in blocking tumor-associated inflammatory processes.
A measure of LBM is a means of assessing weight gain that is attributable to tissue other than fat. Gain of fat is not considered to be a crucial indicator of recovery from cachexia, since it is wasting of muscle (i.e. diaphragmatic or pulmonary) that can result in death. Most cachexia patients (21/30 or 70%) showed an average increase in LBM of 1.9 kg after three infusions of antibody therapy (p<.001). Patients were evaluated with DEXA no more than 2 weeks prior to the start of treatment. Antibody infusions were given at day 0, day 21 and day 42 and DEXA scans were performed at day 57.
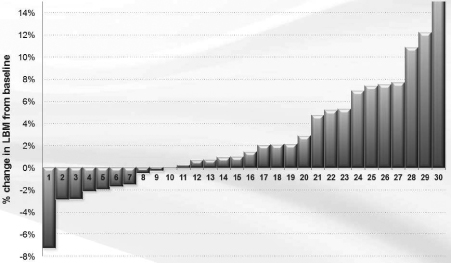
56
Table of Contents
Each vertical bar on the chart above represents the change in lean body mass (LBM) for a single patient during 8 weeks of therapy. LBM change is expressed as a percentage of total body weight from baseline. The statistical significance of the result was was determined by comparing the LBM change for responders vs. non-responders 3.0±1.4 kg (p=0.002).
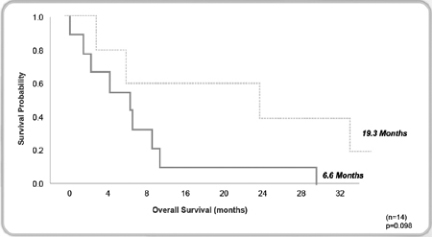
Overall survival was analyzed in the above Kaplan-Meyer graph for patients with advanced colorectal cancer treated with Xilonix according to whether or not they had gained LBM. An increase in LBM was measured in 5 of 14 (36%) patients. The median survival time for LBM gainers was 19.3 months compared to 6.6 months in those with no evidence of gain (p= 0.098).
Results from a Phase II Randomized Study in Cardiovascular Disease
XBiotech completed a multi-center Phase II clinical study in cardiovascular medicine with 43 patients in 2013 (IND# 110,908). The study was conducted at nine investigative sites in the United States. This Phase II randomized, controlled clinical study evaluated the therapeutic antibody MABp1 for its ability to reduce adverse events after balloon angioplasty, atherectomy or stent placement in patients undergoing revascularization procedures for blockage of a major artery (superficial femoral artery or SFA) in the leg. Interim data from this study was submitted to the FDA and resulted in fast track designation for this drug development program in the fall of 2012.
While the study was exploratory in nature and not powered with patient numbers to provide a statistically significant outcome, clinical results to date have shown an important trend towards the reduction of restenosis, and reduced incidence of Major Adverse Cardiovascular Events (MACE) in treated patients compared to controls. Patients were monitored for restenosis, or MACE, including heart attack or stroke.
All of the subjects included in this trial had symptomatic peripheral vascular disease, characterized by claudication, rest pain, or limited gangrene. All patients had hemodynamically significant occlusion of the femoral artery. Subjects eligible for enrollment had to be undergoing endovascular intervention as a part of standard of care treatment. Enrolled subjects were randomized to receive (i) study drug plus standard of care or (ii) standard of care following surgery.
Data analyzed from the study suggests a beneficial treatment effect at 15 weeks. The patients received intravenous infusions of MABp1 at day 0 peri-operative, and at days 14, 28 and 42 post-operative. At 15 weeks no patients (0/22) in the treatment arm had experienced re-occlusion (restenosis) of the treated artery, whereas
57
Table of Contents
3 patients (3/21) had restenosis in the control arm. Three patients (3/22) had MACE in the treatment arm compared to 5 (5/21) in the control group.
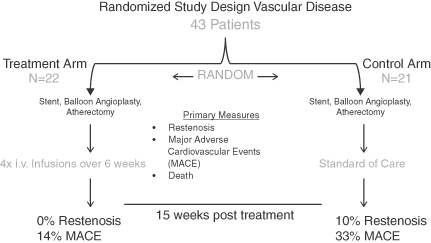
These data, together with the FDA fast track designation, have supported advancement of the clinical trial program for the treatment of vascular injury and disease.
Dermatology
Inflammatory skin conditions encompass a wide range of diagnoses from common conditions such as acne, eczema, and psoriasis, to more rare conditions such as pyoderma gangrenosum. One common factor unifying the pathophysiology of these conditions is IL-1a, a pro-inflammatory cytokine present in keratinocytes and inflammatory cells present in skin lesions. We believe that blockade of IL-1a will prove to be a safe and effective treatment for numerous dermatologic conditions.
Psoriasis
XBiotech completed a multicenter, single arm Phase II study of MABp1 in eight patients with moderate to severe plaque psoriasis (IND# 112,459). This clinical trial was launched after a dramatic response was observed in a psoriasis patient who was treated with MABp1 on a compassionate basis. The patient seen was a 48 year-old male with Type I psoriasis vulgaris. After a single treatment, the patient showed almost complete resolution of psoriasis lesions within 10 days. The Phase II exploratory study involved providing psoriasis patients three subcutaneous injections of the antibody to evaluate safety, pharmacokinetics and preliminary efficacy of the treatment. Numerous efficacy assessments were made, including the Psoriasis Area and Severity Index (PASI) and performance measures, assessed with the use of the Dermatology Life Quality Index (DLQI) Questionnaire and Physicians Global Assessment (PGA). Findings revealed rapid improvement in patients treated with MABp1, with a median response of 43% improvement in PASI score in just 35 days.
Acne
A Phase II exploratory study launched in 2012 and completed in 2013 evaluated MABp1 therapy in moderate to severe acne vulgaris (IND # 112,459). The acne study was a single-arm, multicenter study conducted in the United States under. This study examined changes in the number of inflammatory acne lesions, as well as patient reported changes in psychiatric symptoms. Eleven patients were administered open-label, subcutaneous injections of MABpl over a six-week period (ClinicalTrials.gov NCT01474798). Objectives were assessment of safety, change in inflammatory lesion count and change in psychosocial functioning using two validated questionnaires.
58
Table of Contents
Patients showed significant improvement in the number of facial inflammatory lesions after treatment with MABp1. Median inflammatory lesion counts decreased 36% (IQR -44% to 1%). Anxiety scores improved (from median 6 to 1) as well as self-image assessment (2.3±0.9 to 2.1±0.1) as measured by the Hospital Anxiety and Depression Scale and the modified Body Image Disturbance Questionnaire, respectively. There were no serious adverse events, or adverse events greater than grade I.
Diabetes (Type 2)
XBiotech conducted a Phase II pilot study to test MABp1 in seven patients with Type 2 diabetes at the University Hospital in Basel, Switzerland. The study was headed by an endocrinologist and expert on the role of inflammatory disease in diabetes, Dr. Marc Donath, Head of Endocrinology, Diabetes and Metabolism at University Hospital of Basel. This study was conducted with appropriate due diligence and authorization equivalent to a US FDA IND, under the Swiss regulatory authority SwissMedic.
The clinical study assessed safety and pharmacokinetics of MABp1 in the diabetic patient population. The study also examined patients to determine if their diabetes improved, including assessing pancreas function and glucose control.
Patients were given a low dose of MABp1 intravenously every two weeks for a total of four doses (Days 0, 14, 28, and 42). To be eligible for treatment, patients needed to have been diagnosed with Type 2 diabetes according to American Diabetes Association diagnostic criteria at least three months prior to the study.
To examine the trend of glycated hemoglobin (HbA1c) levels along the study time points, a trend analysis was performed on patients who completed all visits. Compared to baseline, after the 60 days period of treatment HbAlc was reduced by 0.14±0.21% (p=0.15), fasting C-peptide was increased by 88% (p=0.03), pro-insulin by 48% (p=0.03) and insulin by 74% (P=0.ll). Systolic blood pressure decreased by 11 mmHg (p=0.2). Both HbAlc and blood pressure rebounded to baseline levels thirty days after the end of MABp1 application. Treatment with MABpl was well tolerated and no adverse events occurred during the study. This increase in HbA1c after removal of the drug, further suggested the activity for antibody therapy in these type 2 diabetic patients.
Intellectual property
XBiotech has developed a large international intellectual property (IP) portfolio to protect important aspects of its technology, services and products, including patents, trademarks and trade secrets. To date, XBiotech’s patent portfolio consists of 16 patent families, and includes 39 issued/allowed patents and approximately 100 pending patent applications in various countries around the world. XBiotech’s IP portfolio is designed to protect XBiotech’s drug products, therapies and to some extent, its discovery technology. It includes patents and applications that protect MABp1 as a composition of matter and methods of using anti-IL-1a antibodies for the treatment of various diseases including cancer, vascular disorders, inflammatory skin diseases, diabetes, and arthritis. XBiotech’s IP portfolio also includes patents and applications directed to some aspects of our proprietary antibody discovery platform, as well as treating Staphylococcus aureus (S. aureus) infections.
With respect to its 39 issued/allowed patents, XBiotech owns the rights to the patent families as described in more detail below.
A. Interleukin-1 Alpha Antibodies and Methods of Use. This patent family relates to the development of specific True HumanTM monoclonal antibodies, including MABp1, that include (i) an antigen-binding variable region that exhibits very high binding affinity for human IL-1a and (ii) a constant region that is effective at both activating the complement system though C1q binding and binding to several different Fc receptors. XBiotech has been granted 25 patents in this family for interleukin-1 alpha antibodies and methods of use; including ten in the U.S. (two allowed, but not issued), four in Australia, one in China, one in Hong Kong, two in Israel, one in Japan, one in Mexico, one in New Zealand, one in the Philippines, one in Russia and two in South Africa. Patents in this family have a term at least through 2029.
59
Table of Contents
B. Treatment of Cancer with Anti-IL-1a Antibodies. This patent family relates to the use of anti-IL-1a antibodies to inhibit the metastatic potential of tumors by interrupting the physiological role tumor-derived IL-1a plays in tumor metastasis. XBiotech has been granted three patents for this family; including one in Australia, one in Canada and one in Europe. Patents in this family have a term at least through 2027.
C. Treatment of Neoplastic Diseases. This patent family relates to the administration of anti-IL-1a antibodies to treat various tumor-associated diseases and the administration of a monoclonal antibody that specifically binds IL-1a to reduce the size of tumors in human patients suffering from cancer. We have been issued one patent in New Zealand. Patents in this family have a term at least through 2027.
D. Diagnosis, Treatment, and Prevention of Vascular Disorders. This patent family relates to methods of diagnosing, treating and preventing a variety of vascular disorder using IL-1a autoantibody. We have been issued six patents in this family, including one in the U.S., two in Australia, two in Europe and one in Japan. Patents in this family have a term at least through 2027.
E. Compositions and Methods for Treating S. Aureus Infections. This patent family relates to new antibodies for treating S. aureus infections. XBiotech acquired use of these patents pursuant to its exclusive license agreement with STROX Biopharmaceuticals, LLC. This patent family includes three patents in the U.S. and one patent in Australia. Patents in this family have a term at least through 2027.
Because the patent positions of pharmaceutical, biotechnology, and diagnostics companies are highly uncertain and involve complex legal and factual questions, the patents owned and licensed by us, or any future patents, may not prevent other companies from developing similar or therapeutically equivalent products or ensure that others will not be issued patents that may prevent the sale of our products or require licensing and the payment of significant fees or royalties. Furthermore, to the extent that any of our future products or methods are not patentable, that such products or methods infringe upon the patents of third parties, or that our patents or future patents fail to give us an exclusive position in the subject matter claimed by those patents, we will be adversely affected. We may be unable to avoid infringement of third party patents and may have to obtain a license, defend an infringement action, or challenge the validity of the patents in court. A license may be unavailable on terms and conditions acceptable to us, if at all. Patent litigation is costly and time consuming, and we may be unable to prevail in any such patent litigation or devote sufficient resources to even pursue such litigation.
Other Commercial Agreements
We have licensed certain proprietary technology from a Swiss company related to manufacturing antibodies on a clinical and commercial scale pursuant to a license agreement dated as of January 16, 2015. Our license for this proprietary technology is world-wide, non-exclusive and continues until the expiration of the last valid claim of a patent has expired. We also have the right to sublicense this proprietary technology. We are required to make royalty payments once we commence commercial sales of our products, which use these antibodies. We also are required to make royalty payments when we or a strategic partner are manufacturing a product which uses these antibodies, and have initiated Phase II clinical trials for the product. We have the right to terminate the license upon sixty days written notice.
Government Regulation
The development, manufacture, distribution, marketing and advertising of drug products are subject to extensive regulation by federal, state and local governmental authorities in the United States, including the FDA, and by similar agencies in other countries. Any product that we develop must receive all relevant regulatory approvals or clearances before it may be marketed in a particular country. Gaining regulatory approval of a drug product candidate requires the expenditure of substantial resources over an extended period of time. As a result, larger companies with greater financial resources will likely have a competitive advantage over us.
60
Table of Contents
Development Activities
To gain regulatory approval of our products, we must demonstrate, through experiments, preclinical studies and clinical trials that each of our drug product candidates meets the safety and efficacy standards established by the FDA and other international regulatory authorities. In addition, we must demonstrate that all development-related laboratory, clinical and manufacturing practices comply with regulations of the FDA, other international regulators and local regulators.
Regulations establish standards for such things as drug substances and materials; drug manufacturing operations and facilities and analytical laboratories and medical development laboratories processes and environments; in each instance, in connection with research, development, testing, manufacture, quality control, labeling, storage, record keeping, approval, advertising and promotion, and distribution of product candidates, on a product-by-product basis.
Pre-clinical Studies and Clinical Trials
Development testing generally begins with laboratory testing and experiments, as well as research studies using animal models to obtain preliminary information on a product’s efficacy and to identify any safety issues. The results of these studies are compiled along with other information in an IND application, which is filed with the FDA. After resolving any questions raised by the FDA, which may involve additional testing and animal studies, clinical trials may begin. Regulatory agencies in other countries generally require a Clinical Trial Application (CTA) to be submitted and approved before each trial can commence in each country.
Clinical trials normally are conducted in three sequential phases and may take a number of years to complete. Phase 1 consists of testing the drug product in a small number of humans, normally healthy volunteers, to determine preliminary safety and tolerable dose range. Phase 2 usually involves studies in a limited patient population to evaluate the effectiveness of the drug product in humans having the disease or medical condition for which the product is indicated, determine dosage tolerance and optimal dosage and identify possible common adverse effects and safety risks. Phase 3 consists of additional controlled testing at multiple clinical sites to establish clinical safety and effectiveness in an expanded patient population of geographically dispersed test sites to evaluate the overall benefit-risk relationship for administering the product and to provide an adequate basis for product labeling. Phase 4 clinical trials may be conducted after approval to gain additional experience from the treatment of patients in the intended therapeutic indication. Similarly, we may not be able to obtain trademark protection for our current nomenclature with some of our products and processes.
The conduct of clinical trials is subject to stringent medical and regulatory requirements. The time and expense required to establish clinical sites, provide training and materials, establish communications channels and monitor a trial over a long period of time is substantial. The conduct of clinical trials at institutions located around the world is subject to foreign regulatory requirements governing human clinical trials, which vary widely from country to country. Delays or terminations of clinical trials could result from a number of factors, including stringent enrollment criteria, slow rate of enrollment, size of patient population, having to compete with other clinical trials for eligible patients, geographical considerations and others. Clinical trials are monitored by the regulatory agencies as well as medical advisory and standards boards, which could determine at any time to reevaluate, alter, suspend, or terminate a trial based upon accumulated data, including data concerning the occurrence of adverse health events during or related to the treatment of patients enrolled in the trial, and the regulator’s or monitor’s risk/benefit assessment with respect to patients enrolled in the trial. If they occur, such delays or suspensions could have a material impact on our bile salt development programs.
Regulatory Review
The results of preclinical and clinical trials are submitted to the FDA in an NDA, with comparable filings submitted to other international regulators. After the initial submission, the FDA has a period of time in which it
61
Table of Contents
must determine if the NDA is complete. After an NDA is submitted, although the statutory period provided for the FDA’s review is less than one year, dealing with questions or concerns of the agency and, taking into account the statutory timelines governing such communications, may result in review periods that can take several years. If an NDA is accepted for filing, following the FDA’s review, the FDA may grant marketing approval, request additional information, or deny the application if it determines that the application does not provide an adequate basis for approval. If the FDA grants approval, the approval may be conditioned upon the conduct of post-marketing clinical trials or other studies to confirm the product’s safety and efficacy for its intended use. Until the FDA has issued its approval, no marketing activities can be conducted in the United States. Similar regulations apply in other countries.
Fast Track and Breakthrough Designations
The FDA has various programs, including fast track and breakthrough therapy designations, which are intended to expedite the process for reviewing drugs. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification. Generally, drugs that are eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that offer meaningful benefits over existing treatments.
Fast track designation is intended to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need. Designation may be granted on the basis of preclinical data. A sponsor of a drug that receives fast track designation will typically have more frequent interactions with FDA during drug development. In addition, products that have been designated as fast track can obtain rolling review.
Breakthrough therapy designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. The criteria for breakthrough therapy designation require preliminary clinical evidence that demonstrates the drug may have substantial improvement on at least one clinically significant endpoint over available therapy. A breakthrough therapy designation conveys all of the fast track program features, more intensive FDA guidance on an efficient drug development program, an organizational commitment involving senior managers, and eligibility for rolling review and priority review.
A key difference between fast track designation and breakthrough designation is what needs to be demonstrated to qualify for the programs. A breakthrough therapy designation is for a drug that treats a serious or life-threatening condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies. In contrast, a fast track designation is for a drug that treats a serious or life-threatening condition, and nonclinical or clinical data demonstrate the potential to address unmet medical needs for the serious condition.
Although FDA has granted fast track designations for Xilonix™ to treat colorectal cancer and for MABp1 to reduce the need for re-intervention after SFA, such designations may not result in a faster development or review time, do not increase the odds of approval, and may be rescinded at any time if these drug candidates do not continue to meet the qualifications for these programs.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the US, or more than 200,000 individuals in the US and for which there is no reasonable expectation that the cost of developing and making a drug product available in the US for this type of disease or condition will be recovered from sales of the product. Because of the small population of sufferers and severity of PG, we expect that PG will be classified as an orphan indication by the FDA. Orphan product designation must be requested before submitting an NDA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
62
Table of Contents
If a product that has orphan designation subsequently receives the first FDA approval for such drug for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Competitors, however, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity. Orphan product exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same product as defined by the FDA or if our product candidate is determined to be contained within the competitor’s product for the same indication or disease. If a drug designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity. Orphan drug status in the EU has similar, but not identical, benefits, including up to ten years of exclusivity.
Manufacturing Standards
The FDA and other international regulators establish standards and routinely inspect facilities and equipment, analytical and quality laboratories and processes used in the manufacturing and monitoring of products. Prior to granting approval of a drug product, the agency will conduct a pre-approval inspection of the manufacturing facilities, and the facilities of suppliers, to determine that the drug product is manufactured in accordance with current good manufacturing practices (“cGMP”) regulations and product specifications. Following approval, the FDA will conduct periodic inspections. If, in connection with a facility inspection, the FDA determines that a manufacturer does not comply with cGMP regulations and product specifications, the FDA will issue an inspection report citing the potential violations and may seek a range of remedies, from administrative sanctions, including the suspension of our manufacturing operations, to seeking civil or criminal penalties.
International Approvals
If we succeed in gaining regulatory approval to market our products in the United States, we will still need to apply for approval with other international regulators. Regulatory requirements and approval processes are similar in approach to that of the United States. With certain exceptions, although the approval of the FDA carries considerable weight, international regulators are not bound by the findings of the FDA and there is a risk that foreign regulators will not accept a clinical trial design or may require additional data or other information not requested by the FDA. In Europe, there is a centralized procedure available under which the EMEA will conduct the application review and recommend marketing approval to the European Commission, or not, for the sale of drug products in the EU countries.
Post-approval Regulation
Following the grant of marketing approval, the FDA regulates the marketing and promotion of drug products. Promotional claims are generally limited to the information provided in the product package insert for each drug product, which is negotiated with the FDA during the NDA review process. In addition, the FDA enforces regulations designed to guard against conflicts of interest, misleading advertising and improper compensation of prescribing physicians. The FDA will review, among other things, direct-to-consumer advertising, prescriber-directed advertising and promotional materials, sales representative communications to healthcare professionals, promotional programming and promotional activities on the Internet. The FDA will also monitor scientific and educational activities. If the FDA determines that a company has promoted a product for an unapproved use (“off-label”), or engaged in other violations, it may issue a regulatory letter and may require corrective advertising or other corrective communications to healthcare professionals. Enforcement actions may also potentially include product seizures, injunctions and civil or criminal penalties. The consequences of such an action and the related adverse publicity could have a material adverse effect on a developer’s ability to market its drug and its business as a whole.
63
Table of Contents
Following approval, the FDA and other international regulators will continue to monitor data to assess the safety and efficacy of an approved drug. A post-approval discovery of previously unknown problems or failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or a recall or withdrawal of the product from the market, as well as possible civil or criminal sanctions. Similar oversight is provided by regulators in jurisdictions outside the US.
None of our products under development has been approved for marketing in the United States or elsewhere. We may not be able to obtain regulatory approval for any of our products under development. If we do not obtain the requisite governmental approvals or if we fail to obtain approvals of the scope we request, we or our licensees or strategic alliance or marketing partners may be delayed or precluded entirely from marketing our products, or the commercial use of our products may be limited. Such events would have a material adverse effect on our business, financial condition and results of operations.
Other Healthcare Laws and Regulations
If we obtain regulatory approval for any of our product candidates, we may also be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we conduct our business. These laws may impact, among other things, our proposed sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
| • | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent; |
| • | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| • | the federal Physician Payment Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the Centers for Medicare & Medicaid Services information related to payments and other transfers of value to physicians, other healthcare providers, and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members; |
| • | HIPAA, as amended by HITECH, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; and |
| • | state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that apply to us, we may be subject to penalties, including civil and criminal penalties,
64
Table of Contents
damages, fines, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and impact our financial results.
Manufacturing
Our drug product candidates, including Xilonix™, are manufactured at our corporate headquarters in Austin, Texas. Currently, manufacturing scale is largely suitable for supplying clinical trial demands. If and when we receive regulatory approval from the FDA and other regulatory agencies, we will need to manufacture these pharmaceutical products on a commercial scale.
Our manufacturing program involves a disposable bioreactor, in-house designed bioreactor platforms, and a mostly disposable downstream process. The disposable “bioreactor” is a single use container, essentially a large plastic bag, which is used to contain growth media. Cells suspended in the growth media proliferate and secrete antibody, which can then be harvested and purified for use in preparation of a drug product.
The plastic bags used as bioreactors come pre-sterilized and are simply discarded after use. Other components of the manufacturing process are also disposable. These include many “downstream” systems used in the antibody purification. This simple bioreactor system and other disposable components translate into minimal plant infrastructure and dramatically less capital costs and staffing. We believe that it results in a more reliable production process with less risk of contamination.
Simple, disposable technology, together with in-house engineering, dramatically reduces capital requirements and go-forward infrastructure and operational complexities, compared to conventional processes centered on clean-in-place, stainless steel bioreactor technology. The 1,000 liter bioreactor systems we are using permit seamless and continuous production scale-up, from clinical study programs to market. The flexibility, scalability and low infrastructure requirements allows us to move as quickly and efficiently as possible to transition from clinical programs, to commercial launch of products.
Sales and Marketing
We intend to build the commercial infrastructure in the United States and Europe necessary to effectively support the commercialization of all of our product candidates, if and when we believe a regulatory approval of the first of such product candidates in a particular geographic market appears imminent. The commercial infrastructure for Xilonix™, our oncology product, typically consists of a targeted, specialty sales force that calls on a limited and focused group of physicians supported by sales management, medical liaisons, internal sales support, an internal marketing group, and distribution support. To develop the appropriate commercial infrastructure, we will have to invest significant amounts of financial and management resources, some of which will be committed prior to any confirmation that any of our product candidates will be approved.
Outside of the United States and Europe, where appropriate, we may elect in the future to utilize strategic partners, distributors, or contract sales forces to assist in the commercialization of our products. In certain instances we may consider building our own commercial infrastructure.
Legal Proceedings
From time to time, we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, in the opinion of our management, would reasonably be expected to have a material adverse effect on our business, financial condition, operating results or cash flows if determined adversely to us. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
65
Table of Contents
Environmental Matters
Our operations require the use of hazardous materials (including biological materials) which subject us to a variety of federal, provincial and local environmental and safety laws and regulations. Some of the regulations under the current regulatory structure provide for strict liability, holding a party potentially liable without regard to fault or negligence. We could be held liable for damages and fines as a result of our, or others’, business operations should contamination of the environment or individual exposure to hazardous substances occur. We cannot predict how changes in laws or development of new regulations will affect our business operations or the cost of compliance.
Employees
As of January 22, 2015, we had 55 employees, 14 of whom hold a Ph.D. or M.D. (or equivalent) degree. None of our employees are represented by a labor union. We have not experienced any work stoppages, and we consider our relations with our employees to be good.
Facilities
We operate 46,000 square foot facility in Austin, Texas, which includes our corporate headquarters, research and development, clinical and manufacturing activities. We are in the process of constructing a new manufacturing and research facility on a 48 acre site in Austin, Texas. We began construction at our new site in September 2014 and expect to be completed by mid-2015.
66
Table of Contents
The following table set forth information with respect to our executive officer, significant employees and directors as of January 22, 2015:
| NAME |
AGE | POSITION(S) | ||||
| Executive Officer |
||||||
| John Simard |
53 | Founder, President, Chief Executive Officer & Chairman of the Board | ||||
| Significant Employees |
||||||
| Dr. David J. Combs, Ph.D. |
36 | Vice President, Manufacturing | ||||
| Norma I. Gonzalez |
58 | Vice President, Quality | ||||
| Queena Han, C.P.A. |
48 | Vice President, Finance & Human Resources | ||||
| Dr. Sushma Shivaswamy, Ph.D. |
37 | Vice President Research and Development | ||||
| Dr. Michael Stecher, M.D. |
38 | Medical Director | ||||
| Non-Employee Directors |
||||||
| Fabrizio Bonanni |
68 | Director | ||||
| W. Thorpe McKenzie |
67 | Director | ||||
| Daniel Vasella, M.D. |
61 | Director | ||||
Executive Officer
John Simard, our founder, has served as our President and Chief Executive Officer and a member of our board of Directors since March 2005. Prior to XBiotech, he was founder and Chief Executive Officer of CTL ImmunoTherapies Corp., a developer of therapeutic vaccines to treat cancer and chronic infectious disease; he also founded and was initial Chief Executive Officer of AlleCure Corp., of Valencia, California, a developer of allergy treatments and immune-modulating therapies. In 2001, AlleCure and CTL ImmunoTherapies merged to form MannKind Corp., where Mr. Simard served as Corporate Vice President and Member of the Board until his departure in 2002. Mr. Simard invented the core technologies which form the basis for each of the commercialization programs. Operationally, he has championed integrated operations approach, undertaking R&D, manufacturing and clinical regulatory programs under one roof. Mr. Simard holds a degree in Biochemistry from the University of Saskatchewan and attended graduate studies in Medical Biophysics/Immunology at the University of Toronto. He has dozens of issued patents related to cancer therapy, therapeutic vaccines and therapeutic antibodies, as well as substantial peer-reviewed scientific publications and the textbook “Immune Response Genes.”
Our board of directors believes that Mr. Simard possesses specific attributes that qualify him to serve as a director, including his extensive executive leadership experience, his role as founder of the company, his many years of service on our board of directors and as our Chief Executive Officer, and extensive knowledge of our company and industry.
Significant Employees
Dr. David J. Combs, Ph.D. has been employed by XBiotech since April 2010, initially as manufacturing process development scientist and now as Vice President Manufacturing. Dr. Combs oversees a team that manages all aspects of the manufacturing program—from initiation of cell culture process to fill and finish of drug product. Dr. Combs also oversees a team of research scientists working on both upstream and downstream process development, including media development and product purification technology. In addition, he heads up efforts to develop a custom bioreactor system that will reduce XBiotech’s capital requirements for manufacturing expansion. Dr. Combs previously worked for The Cancer Research Institute at Scott and White Memorial Hospital where he served as Senior Scientist, in charge of pre-clinical development, process development, manufacturing and formulation of immunotoxins used for clinical trials. While at Scott and White, Dr. Combs was involved in the design and set up of a cGMP facility and has extensive experience designing and running
67
Table of Contents
bioreactors used for the production of therapeutics. Dr. Combs has a B.Sc. in Biology from Tarleton State University and a Ph.D. in Cellular and Molecular Biology from The University of Texas at Austin.
Norma I. Gonzalez has served as our Vice President of Quality since February 2008. Ms. Gonzalez was one of the first employees to join XBiotech’s operation in Austin, Texas in 2008. She played a crucial role in launching XBiotech’s manufacturing program and establishing the Company’s bioreactor system for the production of clinical trial drug product. Ms. Gonzalez transitioned to developing a full quality program, to enable cGMP compliance and growth of the Company’s manufacturing program. Ms. Gonzalez continues to prepare the Company to enable pivotal study and commercial scale production. Before joining XBiotech, Ms. Gonzalez was Director of Quality at a world-leading manufacturer of heart valves, Carbomedics. During her tenure at Carbomedics, Ms. Gonzalez held various roles including Director of R&D, Director of Manufacturing Mechanical Heart Valve, and Director of Tissue valve and Quality. Prior to Carbomedics, Ms. Gonzalez worked for Shiley Caribbean as a Quality Control & Manufacturing Manager as well as Baxter-Travenol as a Microbiology/QC Supervisor. Ms. Gonzalez holds a B.Sc. in Biology from University of Puerto Rico.
Queena Han has been employed by XBiotech since April 2008 beginning as our controller, and now as our Vice President of Finance and Human Resources. Ms. Han is responsible for reporting, auditing, taxation, internal control, insurance, legal and regulatory compliance regarding all financial functions, and maintenance of cash flow position. She serves as a liaison among directors, investors and the Company. She is also responsible for overseeing Human Resources within the Company. Ms. Han has over 20 years experience in accounting and finance, and has held several executive positions. Prior to joining XBiotech, she served as Chief Financial Officer (CFO) for a public company with a nation-wide pay phone hardware and service business. Ms. Han has a B.A in accounting and holds the Chartered Professional Accountants designation in Canada. She is a professional member of SHRM and holds the Human Resource Management Certification from the University of Texas at Austin.
Dr. Sushma Shivaswamy, Ph.D. has been with XBiotech since May 2009 when she joined as a senior research scientist and quickly advanced to lead the R&D group as Director of Research. The multidisciplinary R&D group has pioneered development of the Company’s technology platform for discovery of True Human™ antibodies. Dr. Shivaswamy has lead the group in the development of new approaches for screening human blood for novel True Human™ antibodies based on super-high-stringency mining technology. She has also led the group to establish the Company’s new molecular cloning and proteomics strategies for identifying the True Human™ therapeutics. Dr. Shivaswamy’s program has also included development of the high-output manufacturing cell lines, for producing antibodies in mammalian cells for large-scale commercial production. Dr. Shivaswamy has an academic background in regulation of eukaryotic gene expression and molecular genetics. Prior to joining XBiotech, Dr. Shivaswamy was a postdoctoral researcher at the Center for Systems and Synthetic Biology at the University of Texas at Austin. She has a Ph.D. degree in Molecular Biology from the Center for Cellular and Molecular Biology, India.
Dr. Michael Stecher, M.D. has served as our Medical Director since June 2010. Dr. Stecher has pioneered the development of therapeutic antibody therapy to treat chronic inflammatory diseases through targeting the interleukin-1 system. He has worked to develop novel clinical approaches and strategies for multiple disease indications and has launched and managed numerous clinical programs in oncology, vascular disease, endocrinology and dermatology. Dr. Stecher has collaborated with an acclaimed group of international physician researchers and scientists to help produce breakthrough clinical data on diseases characterized by chronic inflammatory processes. Dr. Stecher is board certified in family medicine and a graduate from the University of Kansas School of Medicine. He completed his work in family practice medicine at Austin Brackenridge Hospital where he served as chief resident of Family Medicine.
Non-Employee Directors
Fabrizio Bonanni has served as a director since August 2013. For over a decade, Dr. Bonanni headed up the manufacturing program for biological drugs at Amgen, Inc. In one capacity or another, Dr. Bonanni was the
68
Table of Contents
senior operating officer responsible for Amgen Inc.’s biological drug production from 1999-2012. His titles included Executive Vice President Operations (2007-2012), Senior Vice President, Manufacturing, Senior Vice President, Quality and Compliance. Earlier, Dr. Bonanni held various management positions at Baxter International, Inc. from 1974 to 1999, including positions as Corporate Vice President, Regulatory and Clinical Affairs and Corporate Vice President, Quality Systems.
Dr. Bonnani was selected to serve on our board of directors based on his extensive experience with biopharmaceutical companies and their operations.
W. Thorpe McKenzie has served as a director since February 2009. Mr. McKenzie is Managing Director of Pointer Management Company, Chattanooga, Tennessee, which he co-founded in 1990 to invest in hedge funds and similar types of partnerships utilizing a fund of funds approach. From 1982 until 1990, he was a private investor in New York City, and a director of several public and private companies. From 1980 until 1982, he was founding general partner of TIGER, a global hedge fund. From 1971 until 1980, he was a Vice President of Kidder, Peabody & Co., Inc. in New York. Mr. McKenzie is a graduate of the University of North Carolina in Chapel Hill, and the Wharton Graduate division of the University of Pennsylvania in Philadelphia.
Mr. McKenzie was selected to serve on our board of directors based on his experience with corporate financings and his role as an investor in XBiotech.
Dr. Daniel Vasella has served as a director since November 2014. Dr. Vasella is the Honorary Chairman and Former Chairman and Chief Executive Officer, Novartis AG, a company that engages in the research, development, manufacture and marketing of health care products worldwide. Dr. Vasella served as Chairman of Novartis from 1999 to February 2013 and as Chief Executive Officer from 1996 to January 2010. From 1992 to 1996, Dr. Vasella held the positions of Chief Executive Officer, Chief Operating Officer, Senior Vice President and Head of Worldwide Development and Head of Corporate Marketing at Sandoz Pharma Ltd. Dr. Vasella is a director of American Express, Inc., PepsiCo, Inc., a member of the International Business Leaders Advisory Council for the Mayor of Shanghai, a foreign honorary member of the Academy of Arts and Sciences, a trustee of the Carnegie Endowment for International Peace and a member of several industry associations and educational institutions.
Dr. Vasella was selected to serve on our Board based on has extensive senior management, operating and leadership experience through his business career at Novartis and elsewhere. Dr. Vasella brings to the board his core business and leadership skills, his global marketing experience, and his experience leading a highly regulated, global business in rapidly changing markets, as well as his public company director experience.
Board Composition
Our board of directors is currently composed of four members. Three of our directors are independent within the meaning of the independent director guidelines of The NASDAQ Capital Market, or NASDAQ. Our articles and by-laws provide that the number of directors shall be at least one up to a maximum of ten and will be fixed from time to time by resolution of the board of directors. Each of our directors is subject to election at each annual meeting of our shareholders. There are no family relationships among any of the directors or executive officers.
Director Independence
Upon the completion of this offering, we anticipate that our common shares will be listed on NASDAQ. Rule 5605 of the NASDAQ Marketplace Rules, or the NASDAQ Listing Rules, requires that independent directors compose a majority of a listed company’s board of directors within one year of listing. In addition, the NASDAQ Listing Rules require that, subject to specified exceptions, each member of a listed company’s audit, compensation, and nominating and corporate governance committees be independent and that audit committee
69
Table of Contents
members also satisfy independence criteria set forth in Rule 10A-3 under the Exchange Act of 1934. Under NASDAQ Listing Rule 5605(a)(2), a director will only qualify as an “independent director” if, in the opinion of our board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. In order to be considered independent for purposes of Rule 10A-3 under the Exchange Act, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee: (1) accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries; or (2) be an affiliated person of the listed company or any of its subsidiaries. In addition to satisfying general independence requirements under the NASDAQ Listing Rules, members of the compensation committee must also satisfy additional independence requirements set forth in NASDAQ Listing Rule 5605(d)(2). In order to be considered independent for purposes of NASDAQ Listing Rule 5605(d)(2), a member of a compensation committee of a listed company may not, other than in his or her capacity as a member of the compensation committee, the board of directors, or any other board committee, accept, directly or indirectly any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries. Additionally, the board of directors of the listed company must consider whether the compensation committee member is an affiliated person of the listed company or any of its subsidiaries and if so, must determine whether such affiliation would impair the director’s judgment as a member of the compensation committee.
In January 2015, our board of directors undertook a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our board of directors determined that Dr. Bonanni and Dr. Vasella do not have any relationships that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that he is considered an “independent” director as that term is defined under the applicable rules and regulations of the SEC and the NASDAQ Listing Rules. The Board also determined that W. Thorpe McKenzie does not have any relationships that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each is considered an “independent” director as that term is defined under the applicable rules and regulations of the NASDAQ Listing Rules. In making those determinations, our board of directors considered the current and prior relationships that each non-employee director has with our company and all other facts and circumstances our board of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director.
Lead Independent Director
Our corporate governance guidelines provide that one of our independent directors shall serve as a lead independent director at any time when an independent director is not serving as the Chairman of the board of directors. Our board of directors has appointed Dr. Vasella to serve as our lead independent director. As lead independent director, Dr. Vasella will preside over periodic meetings of our independent directors, coordinate activities of the independent directors and perform such additional duties as our board of directors may otherwise determine and delegate.
Role of the Board in Risk Oversight
We face a number of risks, including those described in the section of this Prospectus captioned “Risk Factors.” Our board of directors believes that risk management is an important part of establishing, updating and executing on our business strategy. Our board of directors, as a whole and at the committee level, has oversight responsibility relating to risks that could affect the corporate strategy, business objectives, compliance, operations, and the financial condition and our performance. Our board of directors focuses its oversight on our most significant risks and on our processes to identify, prioritize, assess, manage and mitigate those risks. Our board of directors and its committees receive regular reports from members of our senior management on areas of material risk to the company, including strategic, operational, financial, legal and regulatory risks. While our
70
Table of Contents
board of directors has an oversight role, management is principally tasked with direct responsibility for management and assessment of risks and the implementation of processes and controls to mitigate their effects on us.
The audit committee, as part of its responsibilities, oversees the management of financial risks, including accounting matters, liquidity and credit risks, corporate tax positions, insurance coverage, and cash investment strategy and results. The audit committee is also responsible for overseeing the management of risks relating to the performance of our internal audit function, if required, and its independent registered public accounting firm, as well as our systems of internal controls and disclosure controls and procedures. The compensation committee is responsible for overseeing the management of risks relating to our executive compensation and overall compensation and benefit strategies, plans, arrangements, practices and policies. The nominating and corporate governance committee oversees the management of risks associated with our overall compliance and corporate governance practices, and the independence and composition of our board of directors. These committees provide regular reports, on at least a quarterly basis, to the full board of directors.
Board Committees
Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee. Our board of directors may establish other committees to facilitate the management of our business. The composition and functions of each committee are described below. Members serve on these committees until their resignation or until otherwise determined by our board of directors.
Audit Committee
Our audit committee consists of Dr. Bonanni and Dr. Vasella. Our board of directors determined that Dr. Bonanni and Dr. Vasella are independent under the NASDAQ Listing Rules and Rule 10A-3(b)(1) of the Exchange Act. The chair of our audit committee has not yet been assigned. Our board of directors will undertake to determine who on the board is suitable as an “audit committee financial expert” within the meaning of the SEC regulations and appoint such member in a timely manner. Our board of directors also determined that each member of our audit committee can read and understand fundamental financial statements in accordance with applicable requirements. In arriving at these determinations, the board of directors examined each audit committee member’s scope of experience and the nature of their employment in the corporate finance sector. The functions of this committee include:
| • | direct responsibility for the appointment, compensation, retention (including termination) and oversight of our independent auditors (our independent auditors report directly the audit committee); |
| • | helping to ensure the independence and performance of the independent registered public accounting firm; |
| • | discussing the scope and results of the audit with the independent registered public accounting firm, and reviewing, with management and the independent accountants, our interim and year-end operating results; |
| • | developing procedures for employees to submit concerns anonymously about questionable accounting or audit matters; |
| • | reviewing our policies on risk assessment and risk management; |
| • | reviewing related party transactions; |
| • | preparation of the audit committee report that the SEC requires to be included in our annual proxy statement; |
| • | obtaining and reviewing a report by the independent registered public accounting firm at least annually, that describes our internal quality-control procedures, any material issues with such procedures, and any steps taken to deal with such issues when required by applicable law; and |
71
Table of Contents
| • | approving (or, as permitted, pre-approving) all audit and all permissible non-audit services, other than de minimis non-audit services, to be performed by the independent registered public accounting firm. |
Compensation Committee
Our compensation committee consists of Dr. Bonanni, and Dr. Vasella. Our board of directors determined that Dr. Bonanni, and Dr. Vasella are independent under the NASDAQ Listing Rules, are “non-employee directors” as defined in Rule 16b-3 promulgated under the Exchange Act and are “outside directors” as that term is defined in Section 162(m) of the Internal Revenue Code of 1986, as amended, or Section 162(m). The chair of our compensation committee is Dr. Vasella. The functions of this committee include:
| • | reviewing and approving, or recommending that our board of directors approve, the compensation of our chief executive officer and other executive officers including in all cases base salary, bonus, benefits and other perquisites; |
| • | reviewing and recommending to our board of directors the compensation of our directors; |
| • | reviewing and approving, or recommending that our board of directors approve, the terms of compensatory arrangements with our executive officers; |
| • | administering our stock and equity incentive plans; |
| • | selecting independent compensation consultants and assessing conflict of interest compensation advisers; |
| • | reviewing and approving, or recommending that our board of directors approve, incentive compensation and equity plans; and |
| • | reviewing and establishing general policies relating to compensation and benefits of our employees and reviewing our overall compensation philosophy and objectives. |
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Mr. McKenzie and Dr. Bonnani. Our board of directors determined that Mr. McKenzie and Dr. Bonanni is independent under the NASDAQ Listing Rules. The chair of our nominating and corporate governance committee is Mr. McKenzie. The functions of this committee include:
| • | identifying, evaluating and selecting, or recommending that our board of directors approve, nominees for election to our board of directors and its committees; |
| • | evaluating the performance of our board of directors and of individual directors; |
| • | considering and making recommendations to our board of directors regarding the composition and structure of our board of directors and its committees; |
| • | reviewing developments in corporate governance practices; |
| • | evaluating the adequacy of our corporate governance practices and reporting; |
| • | reviewing management succession plans; |
| • | developing and making recommendations to our board of directors regarding corporate governance guidelines and matters; and |
| • | overseeing an annual evaluation of the board of directors’ performance. |
72
Table of Contents
Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics that applies to all of our employees, officers, including our principal executive officer, principal financial officer and principal accounting officer or controller, or persons performing similar functions and agents and representatives, including directors, officers and consultants responsible for financial reporting. The full text of our Code of Business Conduct and Ethics will be posted on our website at www.xbiotech.com. We intend to disclose future amendments to certain provisions of our Code of Business Conduct and Ethics, or waivers of such provisions applicable to any principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions, and our directors, on our website identified above.
Compensation Committee Interlocks and Insider Participation
In fiscal 2013, Dr. Bonanni and Mr. McKenzie served on our Compensation Committee. None of the members of the compensation committee is currently or has been at any time an officer or an employee of our company. None of our executive officers currently serves, or has served during the last three years, as a member of the board of directors or compensation committee of any entity that has one or more executive officers serving as a member of our board of directors or compensation committee.
Non-Employee Director Compensation
Non-employee directors of the Company do not receive cash compensation for their services as directors or as members of committees of the Board, but are reimbursed for their reasonable expenses incurred for attending meetings. At the discretion of the Board, non-employee directors are eligible to receive stock options under our 2005 Plan as compensation for serving as directors. We did not make any option grants to non-employee directors in 2013. When determining the amount and vesting schedules for option grants, the Board takes into account a variety of factors, including previous option grants made to the director.
The following table sets forth, as of and for the year ended December 31, 2013, all options held by our Dr. Bonanni and Mr. McKenzie, two non-employee directors and a former director, Mr. MacKay-Dunn.
| Name |
Aggregate Option Awards at December 31, 2013 |
|||
| Hector MacKay-Dunn, former director |
75,000 | |||
| W. Thorpe McKenzie |
1,250,000 | |||
| Fabrizio Bonanni |
0 | |||
On January 1, 2014, the Company granted to Mr. McKeznie options to acquire up to 90,000 shares of our common stock at an exercise price of $15.00 per share and a term expiring 10 years following the effective date of grant.
On January 1st, 2014, the Company granted to Hector MacKay-Dunn, a former director, options to acquire up to 75,000 shares of our common stock at an exercise price of $10.00 per share and a term expiring 10 years following the effective date of grant.
On March 1st, 2014, the Company granted to Dr. Bonanni options to acquire up to 33,333 shares of our common stock at an exercise price of $10.00 per share and a term expiring 10 years following the effective date of grant.
On March 31st, 2014, the Company granted to John Simard options to acquire up to 500,000 shares of our common stock at an exercise price of $10.00 per share and a term expiring 10 years following the effective date of grant. As of January 22, 2015, 100,000 options were vested.
73
Table of Contents
We entered into a Board Member Agreement with Dr. Vasella on November 4, 2014, when he joined our Board of Directors. This agreement provides that Dr. Vasella would receive an initial option grant to purchase 125,000 shares of our common stock upon joining the Board and during each year of service on the Board will receive an annual option grant to purchase an additional 125,000 shares of our common stock. These options will be fully vested upon the grant date and the expiration date is ten years after the grant date. The exercise price of the options is equal to closing price of the Company’s common stock on the most recent day of trading prior to grant, if the Company is public, or the price in the most recent equity financing. The Company also agreed to provide indemnification to Dr. Vasella and to reimburse him for all ordinary business expenses incurred in connection with his service on the Board, including first class airfare for all international travel on behalf of the Company
Future Director Compensation
Following the completion of this offering, we intend to implement a formal policy pursuant to which our non-employee directors will be eligible to receive annual cash retainers and stock awards as compensation for service on our board of directors and committees of our board of directors.
74
Table of Contents
Our named executive officer, or CEO consisted of John Simard, our Chief Executive Officer for fiscal 2012 and 2013.
Summary Compensation Table
| Name and Principal Position |
Year | Salary ($) | Bonus ($)(1) | All other Compensation(2) ($) |
Total ($) | |||||||||||||||
| John Simard |
2012 | $ | 240,000 | $ | 84,000 | $ | 406.00 | $ | 324,406 | |||||||||||
| President, Chief Executive Officer |
2013 | $ | 240,000 | $ | 84,000 | $ | 316.00 | $ | 324,316 | |||||||||||
| (1) | Amounts represent annual discretionary bonuses earned pursuant to the CEO’s employment agreement. |
| (2) | Amounts represent premiums for health insurance. |
Employment Agreement
We entered into an employment agreement and change of control agreement with John Simard, our Chief Executive Officer and President on March 22, 2005. The employment agreement is for an indefinite term. Mr. Simard’s current annual base salary is $550,000 per year and he is eligible for an annual incentive payment of up to 35% of his base salary, subject to the achievement or performance metrics set by the board. The employment agreement contains customary non-competition and non-solicitation provisions which apply for a period of six (6) months after Mr. Simard’s employment is terminated for any reason. In addition, Mr. Simard agrees that all intellectual property developed by him during the term of his employment agreement shall be our property. If Mr. Simard is terminated without cause, if he resigns for good reason or if there is a change in control, he is entitled to certain severance benefits. For details regarding our current obligations under such circumstances, please see “Termination Benefits.”
Potential Payments and Benefits upon Termination or Change in Control
Mr. Simard may voluntarily resign for any reason by providing us with three months prior notice. We may elect to waive all or a portion of such notice by paying to Mr. Simard his base salary that he would have earned if he had remained employed by us for the full duration of such notice period.
If Mr. Simard terminates his employment within 12 months after a “change of control” for “good reason” (as such terms are defined in his change of control agreement) or if he is terminated without cause, we will make a lump sum payment to him equal to twelve month of his base salary, plus other sum owed to him for arrears of salary, vacation pay and, if awarded, his performance bonus, subject to his prior resignation as a director. Additionally, if Mr. Simard terminates his employment within 12 months after a change of control or for good reason, all unvested stock options held by him will immediately vest on such termination and will survive and be exercisable by Mr. Simard, along with his vested options, in accordance with the terms of the option agreements. To the extent permitted by applicable law, we will provide health, medical, dental and other insurance benefits to Mr. Simard for a period of one year after his termination date.
75
Table of Contents
Outstanding Equity Awards at Fiscal Year-End December 31, 2013
The following table contains information regarding outstanding equity awards held at December 31, 2013, by our Chief Executive Officer.
| Option Awards | ||||||||||||||
| Name |
Number of Securities Underlying Unexercised Options (#) Exercisable |
Number of Securities Underlying Unexercised Options (#) Unexercisable |
Option Exercise Price ($) |
Option Expiration Date | ||||||||||
| John Simard |
240,000 | — | $ | 1.17 | 4/16/17 | |||||||||
| 50,000 | — | $ | 2.50 | 12/31/17 | ||||||||||
| 50,000 | — | $ | 2.50 | 1/1/19 | ||||||||||
| 100,000 | 400,000 | $ | 7.50 | 4/11/21 | ||||||||||
Option Exercises and Stock Vested 2013
Our Chief Executive Officer did not exercise any options or receive any stock awards in fiscal 2013. The Company did not grant any stock options or stock awards to the Chief Executive Officer in fiscal 2013.
Securities Authorized for Issuance under Incentive Compensation Plans
The following table sets forth certain information regarding grants under the Company’s equity compensation plans as of December 31, 2013:
Equity Compensation Plans as of December 31, 2013
| Plan category |
Number of securities to be issued upon exercise of outstanding options (a) |
Weighted-average exercise price of outstanding options (b) |
Number of securities remaining available for future issuance under Incentive Compensation Plans (excluding securities reflected in column (a)) (c) |
|||||||||
| Equity compensation plans approved by shareholders(1) |
2,165,000 | $ | 4.92 | 2,606,501 | ||||||||
|
|
|
|
|
|
|
|||||||
| Total |
2,165,000 | $ | 4.92 | 2,606,501 | ||||||||
|
|
|
|
|
|
|
|||||||
All options or shares relate to XBiotech’s 2005 Equity Incentive Plan, which was approved by its shareholders in 2005.
2005 Plan
We have reserved an aggregate of 6,000,000 shares of common stock for issuance under our 2005 Plan which provides for the grants of stock options to directors, officers, employees and consultants. Our Compensation Committee administers the 2005 Plan including, without limitation, the selection of recipients of stock options under the 2005 Plan, the granting of stock options, the determination of the terms and conditions of any such options, the interpretation of the 2005 Plan and any other action they deem appropriate in connection with the administration of the 2005 Plan.
76
Table of Contents
The exercise price of any options granted under our 2005 Plan must at least be equal to the fair market value of our common shares on the date of grant, express in terms of money, as determined by the Compensation Committee, in its sole discretion, provided that such price may not be less than the lowest price permitted under the applicable rules and regulations of all regulatory authorities to which the Company is subject, including the stock exchange on which the Company’s shares are listed.
The term of the options is at the discretion of the Compensation Committee, but may not exceed 10 years from the grant date. The options expire on the earlier of the expiration date or the date three months following the day on which the participant ceases to be a director, officer or employee of, or consultant to, the Company, of in the event of the termination of the participant with cause, the date of such terminating. All options are nontransferable and may be exercised only by the participant, or in the event of the death of the participant, a legal representative until the earlier of the options’ expiry date of the first anniversary of the participants’ death or such other date as may be specified by the Compensation Committee. As of December 31, 2013, we had granted an aggregate of 3,393,499 options under the 2005 Plan.
77
Table of Contents
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth information relating to the beneficial ownership of our common shares as of January 22, 2015 as adjusted to reflect the sale of common shares offered by us in this offering, for:
| • | John Simard, our Chief Executive Officer; |
| • | each of our directors; |
| • | our Chief Executive Officer and directors as a group; and |
| • | each person, or group of affiliated persons, known by us to beneficially own more than 5% of our outstanding common shares. |
We have determined beneficial ownership in accordance with the rules of the SEC, and thus it represents sole or shared voting or investment power with respect to our securities. Unless otherwise indicated below, to our knowledge, the persons and entities named in the table have sole voting and sole investment power with respect to all shares that they beneficially owned, subject to community property laws where applicable. The information does not necessarily indicate beneficial ownership for any other purpose, including for purposes of Sections 13(d) and 13(g) of the Securities Act.
We have based our calculation of the percentage of beneficial ownership prior to this offering on shares of our common stock outstanding as of January 22, 2015. We have based our calculation of the percentage of beneficial ownership after this offering on shares of our common stock outstanding immediately after the completion of this offering. We have deemed shares of our common stock subject to stock options that are currently exercisable or exercisable within 60 days of January 22, 2015 to be outstanding and to be beneficially owned by the person holding the stock option for the purpose of computing the percentage ownership of that person. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Except as otherwise noted below, the address for each person or entity listed in the table is c/o 8201 E Riverside Drive, Bldg 4,Ste.100, Austin, TX 78744.
| PERCENTAGE OF SHARES BENEFICIALLY OWNED | ||||||||||
| NAME OF BENEFICIAL OWNER | NUMBER OF SHARES BENEFICIALLY OWNED |
BEFORE OFFERING |
AFTER OFFERING | |||||||
| Chief Executive Officer and Directors(1) |
||||||||||
| John Simard |
7,593,267 | 23.37 | % | |||||||
| Fabrizio Bonanni |
66,666 | * | ||||||||
| W. Thorpe McKenzie(2) |
5,540,996 | 14.49 | % | |||||||
| Daniel Vasella |
155,000 | * | ||||||||
| All Directors and Chief Executive Officers as a Group(3) |
13,355,929 | 36.21 | % | |||||||
| 5% or Greater Shareholders |
||||||||||
| Joseph Karl Gut |
2,461,000 | 8.94 | % | |||||||
| * | Less than 1% |
| (1) | These figures include shares of common stock underlying stock options held by our Chief Executive Officer and directors that are immediately exercisable or schedule to become immediately exercisable within 60 days of January 22, 2015. Underlying stock options include the following amounts:—Dr. Bonanni—66,666; Mr. McKenzie—1,355,000; Mr. Simard—940,000 and Dr. Vasella—125,000. |
| (2) | Includes 610,996 shares held by McBiotech, LLC, a corporation controlled by Mr. McKenzie, 100,000 shares held by the McKenzie Charity and 100,000 shares held by Mr. McKenzie’s spouse. |
| (3) | Includes 2,486,666 shares of common stock underlying stock options held by our Chief Executive Officer and directors (4 persons in total) that are immediately exercisable or are scheduled to become exercisable within 60 days of January 22, 2015, 2014. |
78
Table of Contents
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Related Transactions
The following is a description of transactions since January 1, 2013 to which we have been a party, in which the amount involved exceeds $120,000, or that we have chosen to voluntarily disclose in which any of our directors, executive officers, or holders of more than 5% of our shares, or an affiliate or immediate family member thereof, had or will have a direct or indirect material interest, other than compensation arrangements which are described under the sections of this Prospectus captioned “Management—Director Compensation” and “Executive Compensation.”
Hector MacKay-Dunn, a former director of the Company, was a partner of Farris, Vaughan, Wills & Murphy, LLP (“Farris Law Firm”) a Canadian law firm which rendered legal services to the Company. The Company paid legal fees to the Farris Law Firm for services rendered in the amount of approximately $40,585 and $34,590 in 2012 and 2013, respectively. We believe that the fees charged for services provided by the Farris Law Firm are on terms at least as favorable as those that we could secure from a non-affiliated law firm.
Related Person Transaction Policy
We have adopted a formal, written policy, which will become effective as of the effective date of the registration statement of which this Prospectus forms a part, that our executive officers, directors (including director nominees), holders of more than 5% of any class of our voting securities, and any member of the immediate family of or any entities affiliated with any of the foregoing persons, are not permitted to enter into a related party transaction with us without the prior approval or, in the case of pending or ongoing related party transactions, ratification of our audit committee. For purposes of our policy, a related party transaction is a transaction, arrangement or relationship where we were, are or will be involved and in which a related party had, has or will have a direct or indirect material interest, other than transactions available to all of our employees.
Indemnification Agreements and Directors’ and Officers’ Liability Insurance
We will enter into indemnification agreements with our directors in addition to the indemnification provided for under the BCBCA and in our articles. These agreements, among other things, require us to indemnify our directors for certain expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by a director in any action or proceeding arising out of their services as one of our directors or any other company or enterprise to which the person provides services at our request. We believe that these indemnification agreements are necessary to attract and retain qualified persons as directors.
Requirements under the British Columbia Business Corporations Act
Pursuant to the British Columbia Business Corporations Act, or BCBCA, directors and officers are required to act honestly and in good faith with a view to the best interests of the company. Under the BCBCA, subject to certain limited exceptions, a director who holds a disclosable interest in a material contract or transaction is not entitled to vote on any director’s resolution approving such contract or transaction. A director or senior officer, with certain exceptions, has a disclosable interest in a contract or transaction if:
| (a) | the contract or transaction is material to the company; |
| (b) | the company has entered, or proposes to enter, into the contract or transaction; |
| (c) | either of the following applies to the director or senior officer: |
| (i) | the director or senior officer has a material interest in the contract or transaction; |
| (ii) | the director or senior officer is a director or senior officer of, or has a material interest in, a person who has a material interest in the contract or transaction. |
79
Table of Contents
Authorized and Outstanding Stock
The Company’s authorized share capital as described in its Articles of Incorporation consists of an unlimited number of common shares and preferred shares without par value.
As of September 30, 2014, the Company had outstanding 24,339,767 common shares and no preferred shares.
Voting Rights
Holders of common shares are entitled to one vote in respect of each common share held at any meeting of the Company. Except as otherwise provided with respect to any particular series of preferred shares and except as otherwise required by law, the registered holders of preferred shares shall not be entitled as a class to receive notice of or to attend to vote at any meetings of the Company.
Under our articles that will be in effect upon the closing of this offering, the holders of our common shares will be entitled to one vote for each common share held on all matters submitted to a vote of the shareholders, including the election of directors. Our articles to be in effect upon the completion of this offering do not provide for cumulative voting rights. Because of this, the holders of a plurality of the common shares entitled to vote in any election of directors can elect all of the directors standing for election, if they should so choose.
Dividends
Subject to the BCBCA, and subject to the prior rights of any holders of preferred shares, the holders of the common shares in the absolute discretion of the directors, shall be entitled to receive, and the Company shall pay thereon, out of moneys of the Company properly applicable to the payment of dividends, when declared by the directors, only such dividends as may be declared from time to time in respect of the common shares. The preferred shares are entitled to preference over the common shares with respect to the payment of dividends.
Liquidation Rights
Subject to the prior payment to the holders of the preferred shares described below, in the event of the liquidation, dissolution or winding-up of the Company or other distribution of the assets of the Company among its shareholders, the holders of the common shares shall be entitled to share pro rata in the distribution of the balance of the assets. The preferred shares shall be entitled to a preference over the common shares with respect to the distribution of assets of the Company, whether voluntary or involuntary, or in the event of any other distribution of assets of the Company among its shareholders for the purpose of winding up its affairs; and the preferred shares may be given such other preference not inconsistent with the Articles of Incorporation.
Other Rights
The following Drag Along Rights and Tag Along Rights will not apply to the Company once the Company’s shares are registered under the 1934 Exchange Act.
Drag Along Rights: If selling shareholders have agreed to transfer to a purchaser, or group of purchasers (the “Purchaser”), equity securities of the Company which represent more than 60% of the common shares of the Company on a fully diluted basis, and the Purchaser offers to each of the other shareholders to purchase the remaining equity securities on equivalent terms and conditions, the other shareholders will be required to sell all of their remaining equity securities.
80
Table of Contents
Tag Along Rights: If any shareholder becomes entitled to transfer its equity securities to a Purchaser who controls, or afterwards would control, a majority of the voting power of the Company on a fully diluted basis, each other shareholder may elect to sell its equity securities of the Company to the Purchaser for a price as determined by the Articles of Incorporation, but generally not less than the price offered by the Purchaser to the original shareholder. If the Purchaser refuses to purchase the equity securities of the electing other shareholders, the originally proposed sale shall not be made.
Corporate Governance
Under the BCBCA, we are required to hold a general meeting of our shareholders at least once every year, provided that the meeting must not be held later than 15 months after the preceding annual general meeting. Under the Company’s Articles, the location of the shareholders meeting shall be anywhere in North America, as determined by the directors.
Subject to limited exceptions under the BCBCA, a notice specifying the date, time and location of a shareholders meeting must be sent to each shareholder entitled to attend the meeting and to each director. Upon the Company’s shares becoming registered under the 1934 Exchange Act, the notice must be sent not less than 21 days prior to the meeting and not more than 2 months before the meeting.
Under the Company’s Articles, all business transacted at a special meeting of shareholders that is not an annual general meeting, except business relating to the conduct of or voting at the meeting, is deemed to be special business. At an annual general meeting, all business is special business except for the following: (a) business relating to the conduct of or voting at the meeting; (b) consideration of any financial statements of the Company presented to the meeting; (c) consideration of any reports of the directors or auditor; (d) the setting or changing of the number of directors; (e) the election or appointment of directors; (f) the appointment of an auditor; (g) the setting of the remuneration of an auditor; (h) business arising out of a report of the directors not requiring the passing of a special resolution or an exceptional resolution; and (i) any other business which, under the Company’s Articles or the BCBCA, may be transacted at a meeting of shareholders without prior notice of the business being given to the shareholders.
Notice of a meeting of shareholders at which special business is to be transacted must:
| (a) | state the general nature of the special business; and |
| (b) | if the special business includes considering, approving, ratifying, adopting or authorizing any document or the signing of or giving of effect to any document, have attached to it a copy of the document or state that a copy of the document will be available for inspection by shareholders: |
| (i) | at the meeting; or |
| (ii) | at the Company’s records office, or at such other reasonably accessible location in British Columbia as is specified in the notice, during statutory business hours on any one or more specified days before the day set for the holding of the meeting. |
Under the Company’s Articles, our board of directors has the power at any time to call a meeting of our shareholders. In addition, subject to the requirements of the BCBCA, the holders of not less than 5% of our shares that carry the right to vote at a meeting sought to be held can also requisition our board of directors to call a meeting of our shareholders for the purposes stated in the requisition. If our board of directors does not call the meeting within 21 days after receiving the requisition, our shareholders can call the meeting and the expenses reasonably incurred by such shareholders in requisitioning, calling and holding the meeting must be reimbursed by us.
Those entitled to vote at a meeting are entitled to attend meetings of our shareholders. Every shareholder entitled to vote may appoint a proxyholder to attend the meeting in the manner and to the extent authorized and with the authority conferred by the proxy. Directors, auditors, legal counsels, secretary (if any), and any other
81
Table of Contents
persons invited by the chair of the meeting or with the consent of those at the meeting are entitled to attend any meeting of our shareholders but will not be counted in quorum or be entitled to vote at the meeting unless he or she or it is a shareholder or proxyholder entitled to vote at the meeting.
Certain Takeover Bid Requirements
Unless such offer constitutes an exempt transaction, an offer made by a person, an “offeror”, to acquire outstanding shares of a Canadian entity that, when aggregated with the offeror’s holdings (and those of persons or companies acting jointly with the offeror), would constitute 20% or more of the outstanding shares in a class, would be subject to the take-over provisions of Canadian securities laws. The foregoing is a limited and general summary of certain aspects of applicable securities law in the provinces and territories of Canada, all in effect as of the date hereof.
In addition to those takeover bid requirements noted above, the acquisition of our shares may trigger the application of statutory regimes including among others, the Investment Canada Act (Canada) and the Competition Act (Canada).
Limitations on the ability to acquire and hold our common stock may be imposed by the Competition Act (Canada). This legislation permits the Commissioner of Competition, or the Commissioner, to review any acquisition of control over or of a significant interest in us. This legislation grants the Commissioner jurisdiction, for up to one year, to challenge this type of acquisition before the Canadian Competition Tribunal on the basis that it would, or would be likely to, substantially prevent or lessen competition in any market in Canada.
This legislation also requires any person who intends to acquire our common stock to file a notification with the Canadian Competition Bureau if certain financial thresholds are exceeded and if that person (and their affiliates) would hold more than 20% of our common stock. If a person already owns 20% or more of our common stock, a notification must be filed when the acquisition of additional shares would bring that person’s holdings to over 50%. Where a notification is required, the legislation prohibits completion of the acquisition until the expiration of a statutory waiting period, unless the Commissioner provides written notice that she does not intend to challenge the acquisition.
There is no limitation imposed by Canadian law or our articles on the right of non-residents to hold or vote our common stock, other than those imposed by the Investment Canada Act. The Investment Canada Act requires any person that is a “non-Canadian” (as defined in the Investment Canada Act) who acquires control of an existing Canadian business, where the acquisition of control is not a reviewable transaction, to file a notification with Industry Canada. The Investment Canada Act generally prohibits the implementation of a reviewable transaction unless, after review, the relevant minister is satisfied that the investment is likely to be of net benefit to Canada. Under the Investment Canada Act, the acquisition of control of us (either through the acquisition of our common stock or all or substantially all our assets) by a non-Canadian who is a World Trade Organization member country investor, including a US investor, would be reviewable only if the value of our assets was equal to or greater than a specified amount. The specified amount for 2014 is CAD$354.0 million. The threshold amount is subject to an annual adjustment on the basis of a prescribed formula in the Investment Canada Act to reflect changes in Canadian gross domestic product.
As a result of recent amendments to the Investment Canada Act substantial changes to the review threshold are pending. If and when these amendments come into force, the review threshold will increase to CAD$600.0 million (and eventually to CAD$1.0 billion) and will no longer be calculated on the basis of the book value of the Canadian business assets, but rather its “enterprise value”.
The acquisition of a majority of the voting interests of an entity is deemed to be acquisition of control of that entity. The acquisition of less than a majority but one-third or more of the voting shares of a corporation or an equivalent undivided ownership interest in the voting shares of a corporation is presumed to be an acquisition of
82
Table of Contents
control of that corporation unless it can be established that, on the acquisition, the corporation is not controlled in fact by the acquirer through the ownership of voting shares. The acquisition of less than one-third of the voting shares of a corporation is deemed not to be an acquisition of control of that corporation. Certain transactions in relation to our common shares would be exempt from review by the Investment Canada Act including:
| • | the acquisition of our common shares by a person in the ordinary course of that person’s business as a trader or dealer in securities; |
| • | the acquisition of control of us in connection with the realization of security granted for a loan or other financial assistance and not for any purpose related to the provisions of the Investment Canada Act; and |
| • | the acquisition of control of us by reason of an amalgamation, merger, consolidation or corporate reorganization following which ultimate direct or indirect control in fact of us, through the ownership of our voting shares, remains unchanged. |
Under the new national security regime in the Investment Canada Act, review on a discretionary basis may also be undertaken by the federal government in respect of a much broader range of investments by a non-Canadian to “acquire, in whole or in part, or to establish an entity carrying on all or any part of its operations in Canada.” The relevant test is whether such an investment by a non-Canadian could be “injurious to national security.” The Minister of Industry has broad discretion to determine whether an investor is a non-Canadian and may be subject to national security review. Review on national security grounds is at the discretion of the federal government and may occur on a pre- or post-closing basis.
There is no law, governmental decree or regulation in Canada that restricts the export or import of capital or which would affect the remittance of dividends or other payments by us to non-Canadian holders of our common shares or preferred shares, other than withholding tax requirements.
Neither our articles to be in effect upon the completion of this offering nor by-laws to be in effect upon the completion of this offering contain any change of control limitations with respect to a merger, acquisition or corporate restructuring that involves us.
This summary is not a comprehensive description of relevant or applicable considerations regarding such requirements and, accordingly, is not intended to be, and should not be interpreted as, legal advice to any prospective purchaser and no representation with respect to such requirements to any prospective purchaser is made. Prospective investors should consult their own Canadian legal advisors with respect to any questions regarding securities law in the provinces and territories of Canada.
Actions Requiring a Special Majority
Under the BCBCA and the Company’s Articles, certain corporate actions require the approval of a special majority of shareholders, meaning holders of shares representing not less than 66 2⁄3% of those votes cast in respect of a shareholder vote addressing such matter. Subject to the BCBCA, those items requiring the approval of a special majority generally relate to fundamental changes with respect to our business, and include among others, resolutions: (i) to change all or any of the Company’s unissued, or fully paid issued, shares with par value into shares without par value or any of its unissued shares without par value into shares with par value; (ii) alter the identifying name of any of the Company’s shares; (iii) create special rights or restrictions for, and attach those special rights or restrictions to, the shares of any class or series of shares, whether or not any or all of those shares have been issued; (iv) vary or delete any special rights or restrictions attached to the shares of any class or series of shares, whether or not any or all of those shares have been issued; (v) remove a director before the expiry of his or her term; and (vi) providing for a sale, lease or exchange of all or substantially all of the Company’s property.
83
Table of Contents
Advance Notice Procedures and Shareholder Proposals
Under the BCBCA, shareholders may make proposals for matters to be considered at the annual general meeting of shareholders. Such proposals must be sent to us in advance of any proposed meeting by delivering a timely written notice in proper form to our registered office in accordance with the requirements of the BCBCA. The notice must include information on the business the shareholder intends to bring before the meeting.
Transfer Agent and Registrar
The Transfer Agent and Registrar for shares of our common stock is . Our Transfer Agent and Registrar’s telephone number is .
84
Table of Contents
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been no public market for our common stock, and although we expect that our common stock will be approved for listing on The NASDAQ Capital Market, we cannot assure investors that there will be an active public market for our common stock following this offering. We cannot predict what effect, if any, sales of our shares in the public market or the availability of shares for sale will have on the market price of our common stock. Future sales of substantial amounts of common stock in the public market, including shares issued upon exercise of outstanding options, or the perception that such sales may occur, however, could adversely affect the market price of our common stock and also could adversely affect our future ability to raise capital through the sale of our common stock or other equity-related securities of ours at times and prices we believe appropriate.
Upon completion of this offering, we will have shares of common stock issued and outstanding. All of the common stock expected to be sold in this offering will be freely tradable without restriction or further registration under the Securities Act of 1933, as amended, or the Securities Act, unless held by our “affiliates,” as that term is defined in Rule 144 under the Securities Act. The remaining outstanding shares of common stock will be deemed “restricted securities” as that term is defined under Rule 144. Restricted securities may be sold in the public market only if their offer and sale is registered under the Securities Act or if the offer and sale of those securities qualify for an exemption from registration, including exemptions provided by Rules 144 and 701 under the Securities Act, which are summarized below.
As a result of the lock-up agreements and market stand-off provisions described below and the provisions of Rules 144 or 701, the common stock that will be deemed “restricted securities” will be available for sale in the public market following the completion of this offering as follows:
| • | shares will be eligible for sale on the date of this Prospectus; and |
| • | shares will be eligible for sale upon expiration of the lock-up agreements and market stand-off provisions described below, beginning more than 180 days after the date of this Prospectus. |
We may issue common stock from time to time for a variety of corporate purposes, including in capital-raising activities through future public offerings or private placements, in connection with exercise of stock options, vesting of restricted stock units and other issuances relating to our employee benefit plans and as consideration for future acquisitions, investments or other purposes. The number of shares of common stock that we may issue may be significant, depending on the events surrounding such issuances. In some cases, the shares we issue may be freely tradable without restriction or further registration under the Securities Act; in other cases, we may grant registration rights covering the shares issued in connection with these issuances, in which case the holders of the common stock will have the right, under certain circumstances, to cause us to register any resale of such shares to the public.
Lock-Up
We, our directors and officers and substantially all of the holders of our equity securities have agreed, subject to certain exceptions, not to offer, sell or transfer any of our shares of common stock or securities convertible into or exchangeable or exercisable for our common stock, for 180 days after the date of this Prospectus without first obtaining the written consent of WR Hambrecht on behalf of the underwriters, after the date of this Prospectus. These agreements are described in the section of this Prospectus captioned “Underwriting.”
Our underwriters have advised us that they have no present intent or arrangement to release any shares of common stock subject to a lock-up, and will consider the release of any lock-up on a case-by-case basis. There are no existing agreements between the underwriters and any of our shareholders who have or will execute a lock-up agreement, providing consent to the sale of common stock prior to the expiration of the lock-up period.
85
Table of Contents
Following the lock-up periods set forth in the agreements described above, and assuming that the representatives of the underwriters do not release any parties from these agreements and that there is no extension of the lock-up period, all of the shares of common shares that are restricted securities or are held by our affiliates as of the date of this Prospectus will be eligible for sale in the public market in compliance with Rule 144 under the Securities Act.
In addition to the restrictions contained in the lock-up agreements described above, our amended and restated investor rights agreement, as amended, contains market stand-off provisions imposing restrictions on the ability of certain of our shareholders to offer, sell or transfer our equity securities for a period of 180 days following the effective date of this registration statement.
Rule 144
In general, under Rule 144, as currently in effect, once we have been subject to the public company reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act, for at least 90 days, a person (or persons whose common shares are required to be aggregated) who is not deemed to have been one of our “affiliates” for purposes of Rule 144 at any time during the three months preceding a sale, and who has beneficially owned restricted securities within the meaning of Rule 144 for at least six months, including the holding period of any prior owner other than one of our “affiliates,” is entitled to sell those shares in the public market (subject to the lock-up agreement referred to above, if applicable) without complying with the manner of sale, volume limitations or notice provisions of Rule 144, but subject to compliance with the public information requirements of Rule 144. If such a person has beneficially owned the shares proposed to be sold for at least one year, including the holding period of any prior owner other than “affiliates,” then such person is entitled to sell such shares in the public market without complying with any of the requirements of Rule 144 (subject to the lock-up agreement referred to above, if applicable). In general, under Rule 144, as currently in effect, once we have been subject to the public company reporting requirements of the Exchange Act for at least 90 days, our “affiliates,” as defined in Rule 144, who have beneficially owned the common shares proposed to be sold for at least six months are entitled to sell in the public market, upon expiration of any applicable lock-up agreements and within any three-month period, a number of those common shares that does not exceed the greater of:
| • | 1% of the number of shares of common stock then outstanding, which will equal approximately shares of common stock immediately after this offering (calculated on the basis of the assumptions described above and assuming no exercise of outstanding options or warrants); or |
| • | the average weekly trading volume of our common shares on The NASDAQ Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to such sale. |
Such sales under Rule 144 by our “affiliates” or persons selling shares on behalf of our “affiliates” are also subject to certain manner of sale provisions, notice requirements and to the availability of current public information about us. Notwithstanding the availability of Rule 144, the holders of substantially all of our restricted securities have entered into lock-up agreements as referenced above and their restricted securities will become eligible for sale (subject to the above limitations under Rule 144) upon the expiration of the restrictions set forth in those agreements.
Rule 701
In general, under Rule 701 as currently in effect, any of our employees, directors, officers, consultants or advisors who acquired common shares from us in connection with a written compensatory stock or option plan or other written agreement in compliance with Rule 701 under the Securities Act before the effective date of the registration statement of which this Prospectus is a part (to the extent such common shares are not subject to a lock-up agreement) is entitled to rely on Rule 701 to resell such common shares beginning 90 days after we become subject to the public company reporting requirements of the Exchange Act in reliance on Rule 144, but without compliance with the holding period requirements contained in Rule 144. Accordingly, subject to any
86
Table of Contents
applicable lock-up agreements, beginning 90 days after we become subject to the public company reporting requirements of the Exchange Act, under Rule 701 persons who are not our “affiliates,” as defined in Rule 144, may resell those common shares without complying with the minimum holding period or public information requirements of Rule 144, and persons who are our “affiliates” may resell those common shares without compliance with Rule 144’s minimum holding period requirements (subject to the terms of the lock-up agreement referred to above, if applicable).
Equity Incentive Plan
We intend to file with the SEC a registration statement under the Securities Act covering the common stock that we may issue upon exercise of outstanding options under our 2005 Plan and the common stock that we may issue pursuant to future awards under our 2005 Plan. Such registration statement is expected to be filed and become effective as soon as practicable after the completion of this offering. Accordingly, common stock registered under such registration statement will be available for sale in the open market following its effective date, subject to Rule 144 volume limitations and the lock-up agreements described above, if applicable.
87
Table of Contents
MATERIAL CANADIAN TAX CONSIDERATIONS
In the opinion of Stikeman Elliott LLP, Canadian counsel to us, the following is, as of the date hereof, a general summary of the principal Canadian federal income tax considerations under the Tax Act generally applicable to purchasers who acquire common shares pursuant to this offering and who, for the purposes of the Tax Act and at all relevant times, hold such common shares as capital property and deal at arm’s length and are not affiliated with us and the underwriter (each a “Holder”). Common shares will generally be considered to be capital property to a Holder unless such common shares are held by such Holder in the course of carrying on a business, or were acquired by such Holder in a transaction or transactions considered to be an adventure in the nature of trade.
This summary does not apply to a purchaser of common shares (i) that is a “financial institution”, as defined in the Tax Act for purposes of the mark-to-market rules; (ii) an interest in which is or would constitute a “tax shelter investment” as defined in the Tax Act; (iii) that is a “specified financial institution” as defined in the Tax Act; (iv) that reports its Canadian tax results in a currency other than the Canadian currency; or (v) that has or will enter into a “synthetic disposition arrangement” or a “derivative forward agreement”, as those terms are defined in the Tax Act, in respect of common shares pursuant to this offering. All such purchasers should consult their own tax advisors with respect to an investment in common shares. Additional considerations, not discussed herein, may be applicable to a Holder that is a corporation resident in Canada, and is, or becomes as part of a transaction or event or series of transactions or events that includes the acquisition of the common shares, controlled by a non-resident corporation for purposes of the “foreign affiliate dumping” rules in section 212.3 of the Tax Act. Such Holders should consult their tax advisors with respect to the consequences of acquiring common shares. This summary is based on the current provisions of the Tax Act and the regulations thereunder, the Convention Between Canada and the United States of America with Respect to Taxes on Income and on Capital, signed September 26, 1980, as amended (the “Canada-U.S. Tax Treaty”), counsel’s understanding of the current published administrative practices and assessing policies of the Canada Revenue Agency (the “CRA”), and all specific proposals to amend the Tax Act and the regulations thereunder announced by the Minister of Finance (Canada) prior to the date hereof (“Tax Proposals”). This summary assumes that the Tax Proposals will be enacted in their current form and does not otherwise take into account or anticipate any changes in the law or in the administrative practices and assessing policies of the CRA, whether by judicial, governmental or legislative decisions or action, and whether prospective or retroactive in effect, nor does it take into account tax legislation or considerations of any province or territory of Canada or any jurisdiction other than Canada.
The summary is of a general nature only, is not exhaustive of all income tax considerations, and is not intended to be, and should not be construed to be, legal or tax advice to any particular Holder of the common shares and no representation with respect to the Canadian tax consequences to any particular Holder is made. This summary is not exhaustive of all Canadian federal income tax considerations. The relevant tax considerations applicable to the acquiring, holding and disposing of common shares pursuant to this offering may vary according to the status of the purchaser, the jurisdiction in which the purchaser resides or carries on business and the purchaser’s own particular circumstances. Accordingly, holders should consult with their own tax advisors with respect to the income tax consequences to them of acquiring, holding or disposing of the common shares.
Certain Canadian Federal Income Tax Considerations for Canadian Holders
The following portion of the summary is applicable to a Holder who at all relevant times is resident or deemed to be resident in Canada for the purposes of the Tax Act and any applicable tax treaty or convention (a “Canadian Holder”). Certain Canadian Holders to whom common shares might not constitute capital property may make the irrevocable election provided by subsection 39(4) of the Tax Act, in qualifying circumstances, to have the common shares and every other “Canadian security” (as defined in the Tax Act) owned by such Canadian Holder in the taxation year of the election and in all subsequent taxation years deemed to be capital property to the Holder. Canadian Holders should consult their own tax advisors for advice as to whether an election under subsection 39(4) of the Tax Act is available and/or advisable in their particular circumstances.
88
Table of Contents
Dividends
A Canadian Holder will be required to include in computing such Canadian Holder’s income for a taxation year the amount of any taxable dividends (including deemed dividends) received on common shares. In the case of a Canadian Holder who is an individual (other than certain trusts) such dividends will be subject to the gross-up and dividend tax credit rules applicable to taxable dividends received by an individual from taxable Canadian corporations, including the enhanced gross-up and dividend tax credit for “eligible dividends” properly designated as such by us. There may be restrictions on the ability of the Company to so designate any dividend as an eligible dividend, and the Company has made no commitments in this regard. Taxable dividends received by such Canadian Holder may give rise to alternative minimum tax under the Tax Act.
In the case of a Canadian Holder that is a corporation, the amount of any taxable dividends (including deemed dividends) received on common shares that is included in its income will generally be deductible in computing such Canadian Holder’s taxable income for that taxation year. A Canadian Holder that is a “private corporation” (as defined in the Tax Act) or any other corporation resident in Canada and controlled, whether by reason of a beneficial interest in one or more trusts or otherwise, by or for the benefit of an individual (other than a trust) or a related group of individuals (other than trusts), may be liable to pay a 33 1⁄3% refundable tax under Part IV of the Tax Act on dividends received on the common shares to the extent that such dividends are deductible in computing the Canadian Holder’s taxable income for the taxation year. A Canadian Holder that is, throughout the relevant taxation year, a “Canadian-controlled private corporation” (as defined in the Tax Act) may be liable to pay a refundable tax of 6 2⁄3% on its “aggregate investment income” for the taxation year, which is defined to include any dividends received or deemed to have been received on the common shares to the extent that such dividends are not deductible in computing such Canadian Holder’s taxable income.
Disposition of Common Shares
A Canadian Holder who disposes of or is deemed to have disposed of a common share (except to the Company) will generally realize a capital gain (or capital loss) equal to the amount by which such Canadian Holder’s proceeds of disposition in respect of the common share exceeds (or is exceeded by) the aggregate of the adjusted cost base of such common share to the Canadian Holder and any reasonable expenses associated with the disposition. The cost to a Canadian Holder of a common share acquired pursuant to this offering generally will be averaged with the adjusted cost base of any other common shares owned by such Canadian Holder as capital property for the purposes of determining the adjusted cost base of each such common share to such Canadian Holder.
A Canadian Holder will generally be required to include in computing such Canadian Holder’s income for a taxation year of a disposition, one-half of the amount of any capital gain (a “taxable capital gain”) realized in such taxation year, and subject to and in accordance with the provisions of the Tax Act, will generally be required to deduct one-half of the amount of any capital loss incurred by a Canadian Holder (an “allowable capital loss”) against taxable capital gains realized by the Canadian Holder in the taxation year. Allowable capital losses in excess of taxable capital gains realized in a taxation year may generally be deducted by the Canadian Holder against taxable capital gains realized in any of the three preceding taxation years or any subsequent taxation year, subject to detailed rules contained in the Tax Act in this regard. Capital gains realized by a Holder who is an individual (other than certain trusts) may be subject to alternative minimum tax.
The amount of any capital loss realized on the disposition or deemed disposition of a common share by a Canadian Holder that is a corporation may, in certain circumstances, be reduced by the amount of dividends previously received or deemed to have been received by the Canadian Holder on such common share to the extent and in the circumstances prescribed by the Tax Act. Similar rules may apply to a corporation that is a member of a partnership or beneficiary of a trust that owns common shares or that is itself a member of a partnership or a beneficiary of a trust that owns common shares.
89
Table of Contents
A Canadian Holder that is, throughout the relevant taxation year, a “Canadian-controlled private corporation” (as defined in the Tax Act) may be liable to pay an additional refundable tax of 6 2⁄3% on its “aggregate investment income” for the taxation year, which is defined to include an amount in respect of taxable capital gains.
Certain Canadian Federal Income Tax Considerations for Non-Canadian Holders
The following portion of the summary is applicable to a Holder that, at all relevant times for the purposes of the Tax Act and any applicable tax treaty: (i) is not (and is not deemed to be) a resident in Canada, (ii) does not use or hold (and will not use or hold) and is not deemed to use or hold the common shares in, or in the course of, carrying on a business in Canada, and (iii) does not carry on an insurance business in Canada and elsewhere and is not an “authorized foreign bank” as defined in the Tax Act (a “Non-Canadian Holder”).
The 2014 Canadian Federal Budget released on February 11, 2014 contained proposed rules for consultation with respect to treaty shopping. These rules are not discussed herein. Non-Canadian Holders should consult their own tax advisors with respect to the potential application of these rules to their particular circumstance.
Dividends
Dividends paid or credited (or deemed to be paid or credited) on the common shares to a Non-Canadian Holder will generally be subject to withholding tax under the Tax Act at a rate of 25%, subject to a reduction under the provisions of an applicable tax treaty. For Non-Canadian Holders who are resident in the United States for purposes of and entitled to the benefits of the Canada-U.S. Tax Treaty, and are the beneficial owner of such dividends on the common shares (a “U.S. Holder”), the Canadian withholding tax will generally be reduced to the rate of 15%. This rate is further reduced to 5% in the case of such U.S. Holder that is a company for purposes of the Canada-U.S. Treaty that owns at least 10% of our issued and outstanding voting shares at the time the dividend is paid or deemed to be paid. In addition, under the Canada-U.S. Treaty, dividends may be exempt from Canadian withholding tax if paid to certain U.S. Holders that are qualifying religious, scientific, literary, educational or charitable tax-exempt organizations and qualifying trusts, companies, organizations or other arrangements operated exclusively to administer or provide pension, retirement or employee benefits that are exempt from tax in the U.S. and that have complied with specific administrative procedures.
Disposition of Common Shares
A Non-Canadian Holder will not be subject to tax under the Tax Act in respect of a capital gain realized upon the disposition of common shares unless the common shares are “taxable Canadian property” (as defined in the Tax Act) to the Non-Canadian Holder, and the gain is not otherwise exempt from tax in Canada pursuant to the terms of an applicable tax treaty. Provided the common shares are listed on a designated stock exchange (which currently includes the TSX and NASDAQ) at the time of disposition, the common shares generally will not constitute taxable Canadian property to a Non- Canadian Holder unless at any time during the 60 months immediately preceding the disposition, (i) (a) the Non-Canadian Holder, (b) persons with whom the Non-Canadian Holder does not deal at arm’s length, and (c) pursuant to certain Tax Proposals, partnerships in which the Non-Canadian Holder or persons referred to in (b) hold a membership interest directly or indirectly through one or more partnerships, individually or collectively owned at least 25% of the issued shares of any class or series of our capital stock and (ii) more than 50% of the fair market value of the shares of the Company was derived directly or indirectly from one or any combination of real or immoveable property situated in Canada, “Canadian resource properties” (as defined in the Tax Act), “timber resource properties” (as defined in the Tax Act) or an option, interest or right in such property, whether or not such property exists. For a U.S. Holder, even if the common shares are taxable Canadian property, no Canadian taxes will generally be payable on a capital gain realized on the disposition of the common shares unless the value of the common shares is derived principally from real property situated in Canada.
90
Table of Contents
In the event the common shares are (or are deemed to be) taxable Canadian property to a Non-Canadian Holder and a capital gain realized on the disposition of such common shares is not exempt from tax under the Tax Act by virtue of the terms of an applicable tax treaty, such Non-Resident Holder will realize a capital gain (or capital loss) generally in the circumstances and computed in the manner described above under “Certain Canadian Federal Income Tax Considerations for Canadian Holders—Disposition of Common Shares”. A Non-Canadian Holder whose common shares are taxable Canadian property may be required to file a Canadian income tax return reporting the disposition of such common shares. Non-Canadian Holders whose common shares are taxable Canadian property should consult their own tax advisors for advice having regard to their particular circumstances.
Eligibility for Investment
In the opinion of Stikeman Elliott LLP, Canadian counsel to the Company, based on the current provisions of the Tax Act and the regulations (the “Regulations”) thereunder, provided that the common shares are listed on a “designated stock exchange”, as defined in the Tax Act (which currently includes the TSX and NASDAQ), a common share acquired under this prospectus will be a “qualified investment” under the Tax Act and the Regulations for a trust governed by a “registered retirement savings plan” (“RRSP”), a “registered retirement income fund” (“RRIF”), a “tax-free savings account” (“TFSA”), a “registered education savings plan”, a “deferred profit sharing plan” or a “registered disability savings plan” (as those terms are defined in the Tax Act).
Notwithstanding that a common share may be a qualified investment for a TFSA, RRSP or RRIF (a “Registered Plan”), if the common share is a “prohibited investment” within the meaning of the Tax Act for a Registered Plan, the holder or annuitant of the Registered Plan, as the case may be, will be subject to penalty taxes as set out in the Tax Act. A common share will generally not be a “prohibited investment” for a Registered Plan if the holder or annuitant, as the case may be, (i) deals at arm’s length with the Company for the purposes of the Tax Act, and (ii) does not have a “significant interest” (as defined in the Tax Act) in the Company. In addition, a common share will not be a “prohibited investment” if the common share is “excluded property” as defined in the Tax Act for a Registered Plan.
Purchasers of the common shares should consult their own tax advisers with respect to whether common shares would be prohibited investments having regard to their particular circumstances.
91
Table of Contents
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES
The following is a summary of certain material U.S. federal income tax consequences to a U.S. Holder (as defined below) arising from and relating to the acquisition, ownership, and disposition of our common shares. Except where noted, this summary deals only with common stock that is held as a capital asset by a U.S. Holder.
This summary is for general information purposes only and does not purport to be a complete analysis or listing of all potential U.S. federal income tax consequences that may apply to a U.S. Holder as a result of the acquisition, ownership, and disposition of our common shares. In addition, this summary does not take into account the individual facts and circumstances of any particular U.S. Holder that may affect the U.S. federal income tax consequences of the acquisition, ownership, and disposition of common shares. In addition, taxes other than federal income taxes, such as foreign (in addition to Canadian as discussed above in “Material Canadian Income Tax Consequences”), state and local taxes, and federal estate and gift taxes, may affect U.S. Holder’s acquisition, ownership and disposition of our common shares.
This summary is not intended to be, and should not be construed as, legal or U.S. federal income tax advice with respect to any U.S. Holder. Each U.S. Holder should consult, and must rely upon, its own tax advisor regarding the U.S. federal income, U.S. state and local, and foreign tax consequences of the acquisition, ownership, and disposition of our common shares with specific reference to its own tax situation.
SCOPE OF THIS SUMMARY
Authorities
This summary is based on the Internal Revenue Code of 1986, as amended (the “Code”), Treasury regulations (“Regulations”) (whether final, temporary, or proposed), published rulings of the Internal Revenue Service (the “IRS”), published administrative positions of the IRS, the Treaty and U.S. court decisions that are applicable and, in each case, as in effect and available, as of the date of this prospectus. Any of the authorities on which this summary is based could be changed in a material and adverse manner at any time, and any such change could be applied on a retroactive basis. This summary does not discuss the potential effects, whether adverse or beneficial, of any proposed legislation that, if enacted, could be applied on a retroactive basis. The authorities on which this summary is based are subject to various interpretations. No rulings have been or will be sought from the IRS with respect to the transactions described herein. Accordingly, there can be no assurance that the IRS will not challenge the views expressed herein or that a court will not sustain such a challenge.
For purposes of this summary, a “U.S. Holder” is a beneficial owner of our common shares that, for U.S. federal income tax purposes, is (a) an individual who is a citizen or resident of the U.S., (b) a corporation, or any other entity classified as a corporation for U.S. federal income tax purposes, that is created or organized in or under the laws of the U.S., any state in the U.S., or the District of Columbia, (c) an estate if the income of such estate is subject to U.S. federal income tax regardless of the source of such income, or (d) a trust if (i) such trust has validly elected to be treated as a U.S. person for U.S. federal income tax purposes or (ii) a U.S. court is able to exercise primary supervision over the administration of such trust and one or more U.S. persons have the authority to control all substantial decisions of such trust.
Non-U.S. Holders
For purposes of this summary, a “non-U.S. Holder” is a beneficial owner of common shares other than a U.S. Holder. This summary does not address the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares by non-U.S. Holders. Accordingly, a non-U.S. Holder should consult, and must rely upon, its own tax advisor regarding the U.S. federal income, U.S. state and local, and foreign tax consequences (including the potential application of and operation of any income tax treaties) of the acquisition, ownership, and disposition of our common shares.
92
Table of Contents
U.S. Holders Subject to Special U.S. Federal Income Tax Rules Not Addressed
This summary is general and does not address the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares by U.S. Holders that are subject to special provisions under the Code, including, but not limited to:
| • | Tax consequences to holders of common shares that are tax-exempt organizations, or qualified retirement plans, individual retirement accounts or other tax-deferred accounts; |
| • | Tax consequences to holders of common shares that are dealers in securities or currencies or holders that are traders in securities that elect to apply a mark-to-market accounting method, financial institutions, insurance companies, real estate investment trusts, or regulated investment companies; |
| • | Tax consequences to holders of common shares that have a “functional currency” other than the U.S. dollar; |
| • | Tax consequences to holders of common shares that are liable for the alternative minimum tax under the Code; |
| • | Tax consequences to persons holding the common shares as part of a straddle, hedging transaction, conversion transaction, constructive sale, or other arrangement involving more than one position; |
| • | Holders that acquired our common shares in connection with the exercise of employee stock options or otherwise as compensation for services; |
| • | Tax consequences to holders of common shares that are held other than as a capital asset within the meaning of Section 1221 of the Code; or |
| • | Holders of common shares that own (directly, indirectly, or constructively) 10 percent or more of the total combined voting power of all classes of our shares entitled to vote. |
U.S. Holders that are subject to special provisions under the Code, including U.S. Holders described immediately above, should consult, and must rely upon, their own tax advisors regarding the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares.
If an entity that is classified as a partnership for U.S. federal income tax purposes holds our common shares, the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares to such partnership and the partners of such partnership generally will depend on the activities of the partnership and the status of such partners. Partners of entities that are classified as partnerships for U.S. federal income tax purposes should consult, and must rely upon, their own tax advisors regarding the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares.
Tax Consequences Other than U.S. Federal Income Tax Consequences Not Addressed
This summary does not address the U.S. state and local, U.S. federal estate and gift, or foreign tax consequences to U.S. Holders of the acquisition, ownership, and disposition of our common shares. Each U.S. Holder should consult, and must rely upon, its own tax advisor regarding the U.S. state and local, U.S. federal estate and gift, and foreign tax consequences of the acquisition, ownership, and disposition of our common shares. (See, however, “Material Canadian Income Tax Consequences).
If you are considering the purchase of the common shares, you should consult, and must rely upon, your own tax advisors concerning the particular U.S. federal income tax consequences to you of the purchase, ownership and disposition of the common shares, as well as the consequences to you arising under the laws of any other taxing jurisdiction.
93
Table of Contents
U.S. Federal Income Tax Consequences of the Acquisition, Ownership, and Disposition of Common Shares
Distributions on Common Shares
General Taxation of Distributions
Subject to the “passive foreign investment company”, or PFIC, rules discussed below, a U.S. Holder that receives a distribution, including a constructive distribution, with respect to our common shares will be required to include the amount of such distribution in gross income as a dividend (without reduction for any Canadian income tax withheld from such distribution) to the extent of our current or accumulated “earnings and profits.” To the extent that a distribution exceeds our current and accumulated “earnings and profits,” such distribution will be treated (a) first, as a tax-free return of capital to the extent of a U.S. Holder’s tax basis in the common shares and, (b) thereafter, as gain from the sale or exchange of such common shares. (See “Disposition of common shares” below). Dividends received on the common shares generally will not be eligible for the “dividends received deduction”.
Reduced Tax Rates for Certain Dividends
A dividend paid by us generally will be taxed at the preferential tax rates applicable to long-term capital gains if (a) we are a “qualified foreign corporation” (as defined below), (b) the U.S. Holder receiving such dividend is an individual, estate, or trust, and (c) such dividend is paid on common shares that have been held by such U.S. Holder for at least 61 days during the 121-day period beginning 60 days before the “ex-dividend date.”
We generally will be a “qualified foreign corporation” under Section 1(h)(11) of the Code (a QFC) if (a) we were incorporated in a possession of the U.S., (b) we are eligible for the benefits of the Treaty, or (c) the common shares are readily tradable on an established securities market in the U.S. However, even if we satisfy one or more of such requirements, we will not be treated as a QFC if we are a PFIC for the taxable year during which we pay a dividend or for the preceding taxable year.
As discussed herein, we believe that we were a PFIC for previous taxable years, expect that we will be a PFIC for the current taxable year and may be a PFIC in subsequent taxable years. (See “Passive Foreign Investment Company” below). Accordingly, we do not expect to be a QFC for the current taxable year and may not be a QFC in subsequent taxable years.
Distributions Paid in Foreign Currency
The amount of a distribution received on the common shares in foreign currency generally will be equal to the U.S. dollar value of such distribution based on the exchange rate applicable on the date of receipt. A U.S. Holder that does not convert foreign currency received as a distribution into U.S. dollars on the date of receipt generally will have a tax basis in such foreign currency equal to the U.S. dollar value of such foreign currency on the date of receipt. Such a U.S. Holder generally will recognize ordinary income or loss on the subsequent sale or other taxable disposition of such foreign currency (including an exchange for U.S. dollars).
Disposition of Common Shares
A U.S. Holder will recognize gain or loss on the sale or other taxable disposition of our common shares in an amount equal to the difference, if any, between (a) the amount of cash plus the fair market value of any property received and (b) such U.S. Holder’s adjusted tax basis in the common shares sold or otherwise disposed of. Subject to the PFIC rules discussed below, any such gain or loss generally will be capital gain or loss, which will be long-term capital gain or loss if the common shares are held for more than one year.
Preferential tax rates apply to long-term capital gains of a U.S. Holder that is an individual, estate, or trust. Deductions for capital losses are subject to significant limitations under the Code.
94
Table of Contents
Foreign Tax Deduction or Credit
A U.S. Holder that pays (whether directly or through withholding) Canadian income tax with respect to dividends received on our common shares generally will be entitled, at the election of such U.S. Holder, to receive either a deduction or a credit for such Canadian income tax paid. Generally, a credit will reduce a U.S. Holder’s U.S. federal income tax liability on a dollar-for-dollar basis, whereas a deduction will reduce a U.S. Holder’s income subject to U.S. federal income tax. This election is made on a year-by-year basis and applies to all foreign taxes paid (whether directly or through withholding) by a U.S. Holder during a taxable year.
Complex limitations apply to the foreign tax credit, including the general limitation that the credit cannot exceed the proportionate share of a U.S. Holder’s U.S. federal income tax liability that such U.S. Holder’s “foreign source” taxable income bears to such U.S. Holder’s worldwide taxable income. In applying this limitation, a U.S. Holder’s various items of income and deduction must be classified, under complex rules, as either “foreign source” or “U.S. source.” In addition, this limitation is calculated separately with respect to specific categories of income (including “passive income,” “general income,” and certain other categories of income). Gain or loss recognized by a U.S. Holder on the sale or other taxable disposition of common shares and foreign currency gains generally will be treated as “U.S. source” for purposes of applying the foreign tax credit rules.
Dividends received on the common shares generally will be treated as “foreign source” and generally will be categorized as “passive income” or, in the case of certain U.S. Holders, “general income” for purposes of applying the foreign tax credit rules. The foreign tax credit rules are complex, and each U.S. Holder should consult, and must rely upon, its own tax advisor regarding the foreign tax credit rules.
Information Reporting; Backup Withholding Tax
Generally, information reporting requirements will apply to all payments made within the United States or by a U.S. payor or U.S. middleman, of dividends on, or proceeds arising from the sale or other taxable disposition of our common shares, unless you are an exempt recipient, such as a corporation. Additionally, if you fail to provide your taxpayer identification number, or in the case of dividend payments, fail either to report in full dividend income or to make certain certifications, you may be subject to backup withholding at the rate of 28 percent.
Any amounts withheld under the backup withholding rules will be allowed as a refund or a credit against your U.S. federal income tax liability provided the required information is timely furnished to the IRS. Each U.S. Holder should consult with and rely upon its own tax advisor regarding the information reporting and backup withholding tax rules.
Medicare Tax
For taxable years beginning after December 31, 2012, a U.S. Holder that is an individual or estate, or a trust that does not fall into a special class of trusts that is exempt from such tax, will be subject to a 3.8% tax (the “Medicare Tax”) on the lesser of (a) the U.S. Holder’s “net investment income” in the case of individuals, and the “undistributed net investment income” in the case of estates and trusts, for the relevant taxable year, and (b) the excess of the U.S. Holder’s modified adjusted gross income for the taxable year over a certain threshold (which in the case of individuals will be between $125,000 and $250,000, depending on the individual’s circumstances). A U.S. Holder’s net investment income will generally include its income from dividends, interest, rents, royalties and annuities and its net gains from the disposition of the common stock, unless such interest income or net gains are derived in the ordinary course of the conduct of a trade or business (other than a trade or business that consists of certain passive or trading activities). See the discussion below under “Passive Foreign Investment Company” regarding application of Medicare Tax to PFICs. If you are a U.S. Holder that is an individual, estate or trust, you should consult with, and must rely upon, your tax advisors regarding the applicability of the Medicare tax to your income and gains in respect of your investment in our common stock.
95
Table of Contents
Foreign Asset Reporting
Certain U.S. Holders who are individuals (and under proposed regulations, certain entities) may be required to report information relating to an interest in our ordinary shares, subject to certain exceptions (including an exception for shares held in accounts maintained by U.S. financial institutions). U.S. Holders are urged to consult with, and must rely upon, their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of our ordinary shares.
Passive Foreign Investment Company
We generally will be a PFIC under Section 1297(a) of the Code if, for a taxable year, (a) 75 percent or more of our gross income for such taxable year is passive income, or (b) on average, 50 percent or more of the assets held by us either produce passive income or are held for the production of passive income, based on the fair market value of such assets (or on the adjusted tax basis of such assets, if we are not publicly traded and make an election). “Passive income” includes, for example, dividends, interest, certain rents and royalties, certain gains from the sale of stock and securities, excess of foreign currency gains over foreign currency losses and certain gains from commodities transactions.
For purposes of the PFIC income test and asset test described above, if we own, directly or indirectly, 25 percent or more of the total value of the outstanding shares of another foreign corporation, we will be treated as if we (a) held a proportionate share of the assets of such other foreign corporation, and (b) received directly a proportionate share of the income of such other foreign corporation. In addition, for purposes of the PFIC income test and asset test described above, “passive income” does not include any interest, dividends, rents, or royalties that are received or accrued by us from a “related person” (as defined in Section 954(d)(3) of the Code), to the extent such items are properly allocable to the income of such related person that is not passive income. Finally, if a foreign corporation that subject to the accumulated earnings tax and would otherwise be a PFIC owns 25% of more of the stock, by value, of a U.S. corporation, in determining whether such a foreign corporation is a PFIC, the stock of the U.S. subsidiary is not treated as a passive asset, and any income received with respect to that stock is not treated as passive income.
Because we are a clinical-stage biopharmaceutical company which has not yet recognized significant operating income and our gross income consists mostly of interest, we have been a PFIC for previous taxable years. We may also be a PFIC in the current taxable year as well as future taxable years until we generate significant operating income. A U.S. Holder can avoid the adverse U.S. federal income tax consequences of holding shares in a PFIC by making a QEF Election (see “QEF Election”, below). Under a QEF Election, generally, an electing U.S. Holder will be required each taxable year in which we are a PFIC to recognize, as ordinary income, a pro rata share of our earnings, and to recognize, as capital gain, a pro rata share of our net capital gain. Accordingly, because we expect that we only will be a PFIC in taxable years in which we do not generate any net income, an electing U.S. Holder would not have any income inclusions as a result of the QEF Election. Furthermore, in any taxable year in which we generate significant operating income, we may cease to be a PFIC and the QEF Election will not be applicable.
The determination of whether we were, or will be, a PFIC for a taxable year depends, in part, on the application of complex U.S. federal income tax rules, which are subject to various interpretations. In addition, whether we will be a PFIC for the current taxable year and each subsequent taxable year depends on our assets and income over the course of each such taxable year and, as a result, cannot be predicted with certainty as of the date of this prospectus. Accordingly, there can be no assurance that the IRS will not challenge the determination made by us concerning our PFIC status or that we were not, or will not be, a PFIC for any taxable year.
Default PFIC Rules Under Section 1291 of the Code
If we are a PFIC, the U.S. federal income tax consequences to a U.S. Holder of the acquisition, ownership, and disposition of common shares will depend on whether such U.S. Holder makes an election to treat us as a
96
Table of Contents
“qualified electing fund” or “QEF” under Section 1295 of the Code (a QEF Election) or a mark-to-market election under Section 1296 of the Code (a Mark-to-Market Election). A U.S. Holder that does not make either a QEF Election or a Mark-to-Market Election will be referred to in this summary as a “Non-Electing U.S. Holder.”
A Non-Electing U.S. Holder will be subject to the rules of Section 1291 of the Code with respect to (a) any gain recognized on the sale or other taxable disposition of common shares, and (b) any excess distribution received on the common shares. A distribution generally will be an “excess distribution” to the extent that such distribution (together with all other distributions received in the current taxable year) exceeds 125 percent of the average of actual distributions received during the three preceding taxable years (or during a U.S. Holder’s holding period for the common shares, if shorter).
Under Section 1291 of the Code, any gain recognized on the sale or other taxable disposition of our common shares, and any excess distribution received on our common shares, must be ratably allocated to each day in a Non-Electing U.S. Holder’s holding period for such common shares. The amount of any such gain or excess distribution allocated to prior years of such Non-Electing U.S. Holder’s holding period for the common shares (other than years prior to our first taxable year beginning after December 31, 1986 for which we were not a PFIC) will be subject to U.S. federal income tax at the highest tax rate applicable to ordinary income in each such prior year. A Non-Electing U.S. Holder will be required to pay interest on the resulting tax liability for each such prior year, calculated as if such tax liability had been due in each such prior year. Such a Non-Electing U.S. Holder that is not a corporation must treat any such interest paid as “personal interest,” which is not deductible. The amount of any such gain or excess distribution allocated to the current year of such Non-Electing U.S. Holder’s holding period for the common shares will be treated as ordinary income in the current year, and no interest charge will be incurred with respect to the resulting tax liability for the current year. In addition, dividend distributions made to a U.S. Holder will not qualify for preferential rates of taxation, as discussed above under “Reduced Tax Rates for Certain Dividends.”
If we are a PFIC for any taxable year during which a Non-Electing U.S. Holder holds our common shares, we will continue to be treated as a PFIC with respect to such Non-Electing U.S. Holder, regardless of whether we cease to be a PFIC in one or more subsequent taxable years (“Once a PFIC, Always a PFIC Rule”). A Non-Electing U.S. Holder may terminate this deemed PFIC status by electing to recognize gain (which will be taxed under the rules of Section 1291 of the Code discussed above) as if such common shares were sold for their fair market value on the last day of the last taxable year for which we were a PFIC (“Deemed Sale Election”). If we continue to be a PFIC following a Deemed Sale Election, in order to make the Deemed Sale Election, the Non-Electing Shareholder must also make a QEF Election (discussed below).
Finally, if we are a PFIC and own shares of another foreign corporation that also is a PFIC, under certain indirect ownership rules, a disposition by us of the shares of such other foreign corporation or a distribution received by us from such other foreign corporation generally will be treated as an indirect disposition by a U.S. Holder or an indirect distribution received by a U.S. Holder, subject to the rules of Section 1291 of the Code discussed above. To the extent that gain recognized on the actual disposition by a U.S. Holder of our common shares or income recognized by a U.S. Holder on an actual distribution received on our common shares was previously subject to U.S. federal income tax under these indirect ownership rules, such amount generally should not be subject to U.S. federal income tax.
QEF Election
A U.S. Holder that makes a timely QEF Election generally will not be subject to the rules of Section 1291 of the Code as discussed above. A U.S. Holder that makes a timely QEF Election generally will recognize capital gain or loss on the sale or other taxable disposition of our common shares. Furthermore, for each taxable year in which we are a PFIC, an electing U.S. Holder will recognize, for U.S. federal income tax purposes, such U.S. Holder’s pro rata share of (a) our net capital gain, which will be taxed as long-term capital gain to such U.S. Holder, and (b) and our ordinary earnings, which will be taxed as ordinary income to such U.S. Holder.
97
Table of Contents
Accordingly, an electing U.S. Holder would not have any income inclusions as a result of the QEF Election so long as we do not generate any net income. Generally, “net capital gain” is the excess of (a) net long-term capital gain over (b) net short-term capital loss, and “ordinary earnings” are the excess of (a) “earnings and profits” over (b) net capital gain. A U.S. Holder that makes a QEF Election will include such U.S. Holder’s pro rata share of our net capital gain and ordinary earnings for each taxable year in which we are a PFIC, even though such amounts may not be distributed to such U.S. Holder by us. A U.S. Holder that makes a QEF Election may, subject to certain limitations, elect to defer payment of current U.S. federal income tax on such amounts, subject to an interest charge. If such U.S. Holder is not a corporation, any such interest paid will be treated as “personal interest,” which is not deductible.
A U.S. Holder that makes a timely QEF Election generally (a) may receive a tax-free distribution from us to the extent that such distribution represents our “earnings and profits” that were previously included in income by the U.S. Holder because of such QEF Election, and (b) will adjust such U.S. Holder’s tax basis in our common shares to reflect the amount included in income or allowed as a tax-free distribution because of such QEF Election. A QEF Election generally will be “timely” if it is made for the first year in a U.S. Holder’s holding period for our common shares in which we are a PFIC. In this case, a U.S. Holder may make a timely QEF Election by filing the appropriate QEF Election documents with such U.S. Holder’s U.S. federal income tax return for such first year.
If we were a PFIC in a prior year in a U.S. Holder’s holding period for the common shares and we continue to be a PFIC, then in order to purge the taint of the Once a PFIC, Always a PFIC Rule, such U.S. Holder must make a QEF Election and a Deemed Sale Election (discussed above). A Deemed Sale Election in this circumstance will require the U.S. Holder to recognize gain (which will be taxed under the rules of Section 1291 of the Code discussed above) as if the common shares were sold on the qualification date for an amount equal to the fair market value of the common shares on the qualification date. The “qualification date” is the first day of the first taxable year in which we were a QEF with respect to such U.S. Holder. Technically, a QEF Election can be made for any taxable year, regardless of the prior PFIC status of a foreign corporation. However, if the QEF Election is not made for first year of the U.S. Holder’s holding period, and no Deemed Sale Election is made, both the QEF rules and the excess distribution rules of Section 1291 (discussed above) apply simultaneously because the PFIC taint remains. Thus, if we cease to be a PFC, QEF inclusions continue to be required, earnings from post-PFIC years continue to be tainted, distributions during post-PFIC years are subject to the excess distribution rules (to the extent not previously taxed under the QEF rules), and gain on disposition is treated as an excess distribution. Finally, under very limited circumstances, a U.S. Holder may make a retroactive QEF Election if such U.S. Holder failed to file the QEF Election documents in a timely manner.
A QEF Election will apply to the taxable year for which such QEF Election is made and to all subsequent taxable years, unless such QEF Election is invalidated or terminated or the IRS consents to revocation of such QEF Election. If a U.S. Holder makes a QEF Election and, in a subsequent taxable year, we cease to be a PFIC, the QEF Election will remain in effect (although it will not be applicable) during those taxable years in which we are not a PFIC. Accordingly, if we become a PFIC in another subsequent taxable year, the QEF Election will be effective and the U.S. Holder will be subject to the QEF rules described above during any such subsequent taxable year in which we qualify as a PFIC. In addition, the QEF Election will remain in effect (although it will not be applicable) with respect to a U.S. Holder even after such U.S. Holder disposes of all of such U.S. Holder’s direct and indirect interest in our common shares. Accordingly, if such U.S. Holder reacquires an interest in our common stock, such U.S. Holder will be subject to the QEF rules described above for each taxable year in which we were a PFIC.
In the event we are a PFIC, we will satisfy record keeping requirements that apply to a QEF and supply U.S. Holders with information that such U.S. Holders require to report under the QEF rules. Each U.S. Holder should consult its own tax advisor regarding the availability of, and procedure for making, a QEF Election.
98
Table of Contents
Mark-to-Market Election
A U.S. Holder may make a Mark-to-Market Election only if our common shares are marketable stock. Our common shares generally will be “marketable stock” if the common shares are regularly traded on a qualified exchange or other market. For this purpose, a “qualified exchange or other market” includes (a) a national securities exchange that is registered with the U.S. Securities and Exchange Commission, (b) the national market system established pursuant to section 11A of the Securities and Exchange Act of 1934, or (c) a foreign securities exchange that is regulated or supervised by a governmental authority of the country in which the market is located, provided that (i) such foreign exchange has trading volume, listing, financial disclosure, surveillance, and other requirements designed to prevent fraudulent and manipulative acts and practices, remove impediments to and perfect the mechanism of a free, open, fair, and orderly market, and protect investors (and the laws of the country in which the foreign exchange is located and the rules of the foreign exchange ensure that such requirements are actually enforced), and (ii) the rules of such foreign exchange effectively promote active trading of listed stocks. If the common shares are traded on such a qualified exchange or other market, the common shares generally will be “regularly traded” for any calendar year during which the common shares are traded, other than in de minimis quantities, on at least 15 days during each calendar quarter.
A Mark-to-Market Election applies to the taxable year in which such Mark-to-Market Election is made and to each subsequent taxable year, unless our common shares cease to be “marketable stock” or the IRS consents to revocation of such election. Each U.S. Holder should consult, and must rely upon, its own tax advisor regarding the availability of, and procedure for making, a Mark-to-Market Election.
A U.S. Holder that makes a Mark-to-Market Election generally will not be subject to the rules of Section 1291 of the Code discussed above. However, if a U.S. Holder makes a Mark-to-Market Election after the beginning of such U.S. Holder’s holding period for the common shares and such U.S. Holder has not made a timely QEF Election, the rules of Section 1291 of the Code discussed above will apply to certain dispositions of, and distributions on, our common shares. A U.S. Holder that makes a Mark-to-Market Election will include in ordinary income, for each taxable year in which we are a PFIC, an amount equal to the excess, if any, of (a) the fair market value of the common shares as of the close of such taxable year over (b) such U.S. Holder’s adjusted tax basis in such common shares. A U.S. Holder that makes a Mark-to-Market Election will be allowed a deduction in an amount equal to the lesser of (a) the excess, if any, of (i) such U.S. Holder’s adjusted tax basis in the common shares over (ii) the fair market value of such common shares as of the close of such taxable year or (b) the excess, if any, of (i) the amount included in ordinary income because of such Mark-to-Market Election for prior taxable years over (ii) the amount allowed as a deduction because of such Mark-to-Market Election for prior taxable years.
A U.S. Holder that makes a Mark-to-Market Election generally will adjust such U.S. Holder’s tax basis in the common shares to reflect the amount included in gross income or allowed as a deduction because of such Mark-to-Market Election. In addition, upon a sale or other taxable disposition of our common shares, a U.S. Holder that makes a Mark-to-Market Election will recognize ordinary income or loss (not to exceed the excess, if any, of (a) the amount included in ordinary income because of such Mark-to-Market Election for prior taxable years over (b) the amount allowed as a deduction because of such Mark-to-Market Election for prior taxable years).
U.S. Tax Return Filing Requirements
In addition, all U.S. Holders (including certain deemed U.S. Holders) may be required to file annual tax returns (including IRS Form 8621) containing such information as the U.S. Treasury may require. For example, if a U.S. Holder owns ordinary shares during any year in which we are classified as a PFIC and the U.S. Holder recognizes gain on a disposition of our ordinary shares or receives distributions with respect to our ordinary shares, the U.S. Holder generally will be required to file an IRS Form 8621 with respect to the Company, generally with the U.S. Holder’s federal income tax return for that year. The failure to file this form when required could result in substantial penalties.
99
Table of Contents
Medicare Tax
Regulations issued under Section 1411 of the Code address the application of the Medicare Tax in the PFIC context. An inclusion from a PFIC as to which a QEF Election is in effect will not be taken into account as net investment income for purposes of the Medicare Tax. Instead, actual distributions out of previously taxed income will be treated as net investment income and taxable to U.S. Holders for purposes of the Medicare Tax, even though such distributions are excluded from gross income for regular income tax purposes. A U.S. Holder that is an individual, a trust, or an estate can elect to reflect the same inclusions in net investment income for Medicare Tax purposes as the inclusions taken into account for income tax purposes. The election is generally irrevocable. To the extent that an excess distribution under Section 1291 of the Code is allocated to prior years in a U.S. Holder’s holding period during which we are a PFIC, the applicability of the Medicare Tax to such distributions is unclear. Each U.S. Holder should consult, and must rely upon, its own tax advisor regarding the application of the Medicare Tax in the PFIC context.
The PFIC rules are complex, and each U.S. HOLDER should consult its own tax advisor regarding the PFIC rules and how the PFIC rules may affect the U.S. federal income tax consequences of the acquisition, ownership, and disposition of common shares.
100
Table of Contents
We and the underwriters will enter into an underwriting agreement with respect to the shares being offered. Subject to certain conditions, each underwriter has severally agreed to use its best efforts to procure potential purchasers for the shares of our common stock offered hereby. This offering is being undertaken on a best efforts only basis. The underwriters are not required to take or pay for any specific number or dollar amount of our common stock. WR Hambrecht + Co is the representative of the underwriters.
| Underwriters |
Number of Shares | |
| WR Hambrecht + Co |
||
| Total |
The shares are being offered on an all or none basis. Investor funds will be deposited into an escrow account for the benefit of investors set up with [Bank]. The offering will not be completed unless we sell the number of shares specified on the cover page of this prospectus. All investor funds received prior to the closing will be deposited into escrow with the escrow agent until closing. The escrow account will be opened on the date of this prospectus and will remain open until the closing date. The escrow agent will invest all funds it receives after the pricing of the offering in a non-interest bearing account in accordance with Rule 15c2-4 under the Exchange Act. The escrow agent will not accept any investor funds until the date of this prospectus. On the closing date, the escrow agent will notify the underwriters whether the full amount necessary to purchase the shares to be sold in this offering has been received. If, on the closing date, investor funds are not received in respect of the full amount of shares to be sold in this offering, then all investor funds that were deposited into the escrow account will be returned promptly to investors, and the offering will terminate.
The following table shows the per share and total underwriting discount to be paid to the underwriters.
Shares sold by the underwriters to the public will initially be offered at the initial public offering price set forth on the cover of this prospectus. Any shares sold by the underwriters to securities dealers may be sold at a discount of up to $ per share from the initial public offering price. After the initial offering of the shares, the representative may change the offering price and the other selling terms. The offering of the shares by the underwriters is subject to receipt and acceptance and subject to the underwriters’ right to reject any order in whole or in part.
We and our officers, directors, and holders of substantially all of our common stock have agreed, or will agree, with the underwriters, subject to certain exceptions, not to dispose of or hedge any of their common stock or securities convertible into or exchangeable for shares of common stock during the period from the date of this prospectus continuing through the date 180 days after the date of this prospectus, except with the prior written consent of the representative. This agreement does not apply, in our case, to securities issued pursuant to existing employee benefit plans or securities issued upon exercise of options and other exceptions, and in the case of our officers, directors and other holders of our securities, exercise of options issued pursuant to a stock option or similar plans, and other exceptions. See “Shares Eligible for Future Sale” for a discussion of certain transfer restrictions.
Prior to the offering, there has been no public market for the shares. We intend to apply to have our shares of common stock listed on The NASDAQ Capital Market under the symbol “XBIT” In order to meet one of the requirements for listing the common stock on The NASDAQ Capital Market, the underwriters have undertaken to sell lots of 100 or more shares to a minimum of 400 beneficial holders.
101
Table of Contents
Any underwriter who is a qualified market maker on The NASDAQ Capital Market may engage in passive market making transactions on The NASDAQ Capital Market in accordance with Rule 103 of Regulation M, during the business day prior to the pricing of the offering, before the commencement of offers or sales. Passive market makers must comply with applicable volume and price limitations and must be identified as passive market makers. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for such security; if all independent bids are lowered below the passive market maker’s bid, however, the passive market maker’s bid must then be lowered when certain purchase limits are exceeded.
We have agreed to indemnify the several underwriters against certain liabilities, including liabilities under the Securities Act.
The underwriters and their respective affiliates are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. Certain of the underwriters and their respective affiliates have, from time to time, performed, and may in the future perform, various financial advisory and investment banking services for us, for which they received or will receive customary fees and expenses.
In the ordinary course of their various business activities, the underwriters and their respective affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers, and such investment and securities activities may involve securities and/or instruments of the issuer. The underwriters and their respective affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or instruments, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
102
Table of Contents
Legal matters relating to Canadian law, the offering and the validity of the common shares offered in this offering are being passed upon for us by Stikeman Elliott LLP, Vancouver, British Columbia. Legal matters relating to U.S. law and the offering are being passed upon for us by Quarles & Brady LLP, Naples, Florida. The underwriters have been advised by Morrison & Foerster LLP, New York, New York, with respect to certain matters involving US law.
The consolidated financial statements of XBiotech, Inc. at December 31, 2013 and 2012 and for the years then ended appearing in this Prospectus and Registration Statement have been audited by Ernst & Young LLP, independent registered public accounting firm, as set forth in their report appearing elsewhere herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
We have filed with the SEC a registration statement on Form S-1 (File Number 333- ) under the Securities Act with respect to the securities we are offering. This Prospectus, which constitutes part of the registration statement, does not contain all of the information included in the registration statement and exhibits. For further information pertaining to us and our common stock, you should refer to the registration statement and to its exhibits. Statements contained in this Prospectus about the contents of any contract or any other document are not necessarily complete and, in each instance, we refer you to the copy of the contract or other documents filed as an exhibit to the registration statement. Each of these statements is qualified in all respects by this reference.
You may obtain copies of this information by mail from the Public Reference Section of the SEC, 100 F Street, N.E., Room 1580, Washington, D.C. 20549, at prescribed rates. You may obtain information on the operation of the public reference rooms by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports, proxy statements and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov.
As a result of this offering, we will become subject to the information and reporting requirements of the Exchange Act and, in accordance with this law, will file periodic reports, proxy statements and other information with the SEC. These periodic reports, proxy statements and other information will be available for inspection and copying at the SEC’s public reference facilities and the website of the SEC referred to above. We also maintain a website at www.xbiotech.com. Upon completion of this offering, you may access these materials free of charge as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. Information contained on our website is not a part of this prospectus.
103
Table of Contents
XBiotech, Inc.
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
| F-8 |
F-1
Table of Contents
Report of Registered Independent Public Accountant
The Board of Directors and Stockholders of
XBiotech, Inc.
We have audited the accompanying consolidated balance sheets of XBiotech, Inc. (the Company) as of December 31, 2013 and 2012, and the related statements of operations, comprehensive loss, shareholders’ equity and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purposes of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of XBiotech, Inc. at December 31, 2013 and 2012, and the results of its operations and its cash flows for the years then ended in conformity with US generally accepted accounting principles.
/s/ Ernst & Young LLP
Austin, Texas
December 9, 2014, except as to Note 11, which is as of January 22, 2015
F-2
Table of Contents
XBiotech, Inc.
(in thousands, except share data)
| December 31 | September 30 2014 |
|||||||||||
| 2012 | 2013 | |||||||||||
| (unaudited) | ||||||||||||
| Assets |
||||||||||||
| Current assets: |
||||||||||||
| Cash |
$ | 4,167 | $ | 7,244 | $ | 15,497 | ||||||
| Prepaid expenses and other current assets |
197 | 449 | 374 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total current assets |
4,364 | 7,693 | 15,871 | |||||||||
| Property and equipment, net |
4,105 | 3,380 | 3,790 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total assets |
$ | 8,469 | $ | 11,073 | $ | 19,661 | ||||||
|
|
|
|
|
|
|
|||||||
| Liabilities and shareholders’ equity |
||||||||||||
| Current liabilities: |
||||||||||||
| Accounts payable |
$ | 457 | $ | 561 | $ | 1,218 | ||||||
| Accrued expenses |
610 | 284 | 1,225 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total current liabilities |
1,067 | 845 | 2,443 | |||||||||
| Long-term liabilities: |
||||||||||||
| Deferred rent |
29 | — | — | |||||||||
| Capital lease |
1 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total liabilities |
1,097 | 845 | 2,443 | |||||||||
| Shareholders’ equity: |
||||||||||||
| Preferred stock, no par value, unlimited shares authorized, no shares outstanding |
— | — | — | |||||||||
| Common stock, no par value, unlimited shares authorized, 21,547,591, 22,752,101 and 24,339,767 (unaudited) shares outstanding at December 31, 2012 and 2013 and September 30, 2014, respectively |
69,023 | 81,807 | 104,619 | |||||||||
| Accumulated other comprehensive loss |
(134 | ) | (135 | ) | (122 | ) | ||||||
| Accumulated deficit |
(61,517 | ) | (71,444 | ) | (87,279 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Total shareholders’ equity |
7,372 | 10,228 | 17,218 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total liabilities and shareholders’ equity |
$ | 8,469 | $ | 11,073 | $ | 19,661 | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes.
F-3
Table of Contents
XBiotech, Inc.
Consolidated Statements of Operations
(in thousands, except share and per share data)
| Year Ended December 31, | Nine Months Ended September 30, |
|||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| (unaudited) | (unaudited) | |||||||||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
$ | 13,334 | $ | 7,935 | $ | 5,610 | $ | 9,424 | ||||||||
| General and administrative |
1,829 | 1,990 | 1,828 | 6,435 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
15,163 | 9,925 | 7,438 | 15,859 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(15,163 | ) | (9,925 | ) | (7,438 | ) | (15,859 | ) | ||||||||
| Other income (loss): |
||||||||||||||||
| Interest income |
3 | 1 | 1 | — | ||||||||||||
| Foreign exchange gain (loss) |
— | (3 | ) | (3 | ) | 24 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total other income (loss) |
3 | (2 | ) | (2 | ) | 24 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (15,160 | ) | $ | (9,927 | ) | $ | (7,440 | ) | $ | (15,835 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss per share—basic and diluted |
$ | (0.70 | ) | $ | (0.44 | ) | $ | (0.33 | ) | $ | (0.66 | ) | ||||
| Shares used to compute basic and diluted net loss per share |
21,594,369 | 22,520,416 | 22,341,240 | 24,173,485 | ||||||||||||
See accompanying notes.
F-4
Table of Contents
XBiotech, Inc.
Consolidated Statements of Comprehensive Loss
(in thousands)
| Year Ended December 31, | Nine Months Ended September 30, |
|||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| (unaudited) | (unaudited) | |||||||||||||||
| Net loss |
$ | (15,160 | ) | $ | (9,927 | ) | $ | (7,440 | ) | $ | (15,835 | ) | ||||
| Foreign currency translation adjustment |
— | (1 | ) | — | 13 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Comprehensive loss |
$ | (15,160 | ) | $ | (9,928 | ) | $ | (7,440 | ) | $ | (15,822 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
See accompanying notes.
F-5
Table of Contents
XBiotech, Inc.
Consolidated Statements of Shareholders’ Equity
(in thousands, except per share data)
| Common Stock | Accumulated Other Comprehensive Loss |
Accumulated Deficit |
Total | |||||||||||||||||
| Number of Shares |
Amount | |||||||||||||||||||
| Balance at January 1, 2012 |
21,344,043 | $ | 58,924 | $ | (134 | ) | $ | (46,357 | ) | $ | 12,433 | |||||||||
| Net loss |
— | — | — | (15,160 | ) | (15,160 | ) | |||||||||||||
| Issuance of common stock |
476,548 | 7,148 | — | — | 7,148 | |||||||||||||||
| Cancellation of shares |
(300,000 | ) | — | — | — | — | ||||||||||||||
| Issuance of common stock under stock option plan |
27,000 | 135 | — | — | 135 | |||||||||||||||
| Share-based compensation expense |
— | 2,816 | — | — | 2,816 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2012 |
21,547,591 | 69,023 | (134 | ) | (61,517 | ) | 7,372 | |||||||||||||
| Net loss |
— | — | — | (9,927 | ) | (9,927 | ) | |||||||||||||
| Foreign currency translation adjustment |
— | — | (1 | ) | — | (1 | ) | |||||||||||||
| Issuance of common stock and warrants |
1,204,510 | 12,045 | — | — | 12,045 | |||||||||||||||
| Share-based compensation expense |
— | 739 | — | — | 739 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2013 |
22,752,101 | 81,807 | (135 | ) | (71,444 | ) | 10,228 | |||||||||||||
| Net loss (unaudited) |
— | — | — | (15,835 | ) | (15,835 | ) | |||||||||||||
| Foreign currency translation adjustment (unaudited) |
— | — | 13 | — | 13 | |||||||||||||||
| Issuance of common stock (unaudited) |
1,587,666 | 17,840 | — | — | 17,840 | |||||||||||||||
| Share-based compensation expense (unaudited) |
— | 4,972 | — | — | 4,972 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at September 30, 2014 (unaudited) |
24,339,767 | $ | 104,619 | $ | (122 | ) | $ | (87,279 | ) | $ | 17,218 | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
See accompanying notes.
F-6
Table of Contents
XBiotech, Inc.
Consolidated Statements of Cash Flows
(In Thousands)
| Year Ended December 31, | Nine Months Ended September 30, |
|||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| (unaudited) | (unaudited) | |||||||||||||||
| Operating activities |
||||||||||||||||
| Net loss |
$ | (15,160 | ) | $ | (9,927 | ) | $ | (7,440 | ) | $ | (15,835 | ) | ||||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||||||
| Depreciation |
946 | 784 | 626 | 433 | ||||||||||||
| Share-based compensation expense |
2,816 | 739 | 569 | 4,972 | ||||||||||||
| Changes in operating assets and liabilities: |
||||||||||||||||
| Prepaid expenses and other current assets |
(49 | ) | (252 | ) | (124 | ) | 76 | |||||||||
| Accounts payable |
(199 | ) | 103 | 50 | 657 | |||||||||||
| Accrued expenses |
80 | (326 | ) | (235 | ) | 940 | ||||||||||
| Deferred rent |
(68 | ) | (29 | ) | (29 | ) | — | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net cash used in operating activities |
(11,634 | ) | (8,908 | ) | (6,583 | ) | (8,757 | ) | ||||||||
| Investing activities |
||||||||||||||||
| Purchase of property and equipment |
(550 | ) | (59 | ) | (8 | ) | (843 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net cash used in investing activities |
(550 | ) | (59 | ) | (8 | ) | (843 | ) | ||||||||
| Financing activities |
||||||||||||||||
| Issuance of common stock and warrants |
7,148 | 12,045 | 12,045 | 17,840 | ||||||||||||
| Issuance of common stock under stock option plan |
135 | — | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net cash provided by financing activities |
7,283 | 12,045 | 12,045 | 17,840 | ||||||||||||
| Effect of foreign exchange rate on cash |
— | (1 | ) | — | 13 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net change in cash |
(4,901 | ) | 3,077 | 5,454 | 8,253 | |||||||||||
| Cash, beginning of period |
9,068 | 4,167 | 4,167 | 7,244 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Cash, end of period |
$ | 4,167 | $ | 7,244 | $ | 9,621 | $ | 15,497 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
See accompanying notes.
F-7
Table of Contents
XBiotech, Inc.
Notes to Consolidated Financial Statements
1. Organization
XBiotech, Inc. (XBiotech or the Company) was incorporated in Canada on March 22, 2005. XBiotech USA Inc., a wholly-owned subsidiary of the Company, was incorporated in Delaware, United States in November 2007. XBiotech Schweiz AG, a wholly-owned subsidiary of the Company, was incorporated in Zug, Switzerland in August 2010. XBiotech Japan KK, a wholly-owned subsidiary of the Company, was incorporated in Tokyo, Japan in March 2013. XBiotech GmbH, a wholly-owned subsidiary of the Company, was incorporated in Germany in January 2014.
Since its inception, XBiotech has focused on advancing technology to rapidly identify and clone antibodies from individuals that have resistance to disease. At the heart of the Company is a proprietary technical knowhow to translate natural human immunity into therapeutic product candidates.
In 2005, the Company began to develop a new framework for commercial manufacturing, using technology that required less capital, fewer operators and provided greater flexibility than standard industry practices.
With the manufacturing capability to produce its True HumanTM antibody therapy, in 2010 the Company began a clinical trial program. The first clinical trial program at MD Anderson Cancer Center began treating the sickest cancer patients irrespective of tumor type. Soon thereafter, the Company used the same antibody therapy in various clinical studies at treatment centers around the US and abroad to investigate the antibody effect in patients that had vascular disease, leukemia, type 2 diabetes, psoriasis or acne.
The Company’s headquarters are located in Austin, Texas.
The Company continues to be subject to a number of risks common to companies in similar stages of development. Principal among these risks are the uncertainties of technological innovations, dependence on key individuals, development of the same or similar technological innovations by the Company’s competitors and protection of proprietary technology. The Company’s ability to fund its planned clinical operations, including completion of its planned trials, is expected to depend on the amount and timing of cash receipts from future collaboration or product sales and/or financing transactions. The Company believes that its cash and cash equivalents of $7.2 million at December 31, 2013 and the additional cash and cash equivalents it has raised through additional common stock issuances through December 2014 will enable the Company to maintain its current and planned operations for the foreseeable future.
2. Significant Accounting Policies
Basis of Presentation
These consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States, or US GAAP.
Unaudited Interim Financial Information
The accompanying interim consolidated balance sheet at September 30, 2014, the consolidated statements of operations, comprehensive loss and cash flows for the nine months ended September 30, 2013 and 2014, and the consolidated statement of shareholders’ equity for the nine months ended September 30, 2014 are unaudited. These unaudited interim consolidated financial statements have been prepared in accordance with US GAAP. In the opinion of management of the Company, the unaudited interim consolidated financial statements have been prepared on the same basis as the audited consolidated financial statements and include all adjustments necessary for a fair presentation. The results of operations for the nine months ended September 30, 2014 are not necessarily indicative of the results to be expected for the year ended December 31, 2014 or for any other period.
F-8
Table of Contents
Basis of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All significant intercompany transactions have been eliminated upon consolidation.
Use of Estimates
The preparation of financial statements in accordance with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported values of amounts in the financial statements and accompanying notes. Actual results could differ from those estimates.
The Company utilizes significant estimates and assumptions in determining the fair value of its common stock. The board of directors determined the estimated fair value of the Company’s common stock based on a number of objective and subjective factors, including the prices at which the Company sold shares of its common stock to third parties and external market conditions affecting the biotechnology industry sector.
Research and Development Costs
All research and development costs are charged to expense as incurred. Research and development costs include salaries and personnel-related costs, consulting fees, fees paid for contract research services, the costs of laboratory equipment and facilities, license fees and other external costs. Costs incurred to acquire licenses for intellectual property to be used in research and development activities with no alternative future use are expensed as incurred as research and development.
Nonrefundable advance payments for goods or services to be received in the future for use in research and development activities are deferred and capitalized. The capitalized amounts are expensed as the related goods are delivered or the services are performed.
Income Taxes
The Company accounts for income taxes in accordance with ASC 740, Accounting for Income Taxes (“ASC 740”), which provides for deferred taxes using an asset and liability approach. The Company recognizes deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. The Company determines its deferred tax assets and liabilities based on differences between financial reporting and tax bases of assets and liabilities, which are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. Valuation allowances are provided if, based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
The Company accounts for uncertain tax positions in accordance with the provisions of ASC 740. When the uncertain tax positions exist, the Company recognizes the tax benefit of tax positions to the extent that the benefit will more likely than not be realized. The determination as to whether the tax benefit will more than not be realized is based upon the technical merits of the tax position as well as consideration of the available facts and circumstances. As of December 31, 2012 and 2013, the Company does not have any significant uncertain tax positions.
Stock-Based Compensation
The Company accounts for its stock-based compensation awards in accordance with ASC Topic 718, Compensation-Stock Compensation (“ASC 718”). ASC 718 requires all stock-based payments to employees, including grants of employee stock options, to be recognized in the statements of operations based on their grant date fair values. For stock options granted to employees and to members of the board of directors for their services on the board of directors, the Company estimates the grant date fair value of each option award using the
F-9
Table of Contents
Black-Scholes option-pricing model. The use of the Black-Scholes option-pricing model requires management to make assumptions with respect to the expected term of the option, the expected volatility of the common stock consistent with the expected life of the option, risk-free interest rates and expected dividend yields of the common stock. For awards subject to service-based vesting conditions, the Company recognizes stock-based compensation expense, net of estimated forfeitures, equal to the grant date fair value of stock options on a straight-line basis over the requisite service period.
Stock-based compensation expense recognized in the years ended December 31, 2012 and 2013, and for the nine month periods ended September 30, 2013 and 2014 was included in the following line items on the Consolidated Statement of Operations.
| Year Ended December 31, |
Nine Months Ended to September 30, |
|||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| (unaudited) | (unaudited) | |||||||||||||||
| Research and development |
$ | 2,226 | $ | 551 | $ | 416 | $ | 796 | ||||||||
| General and administrative |
590 | 188 | 153 | 4,176 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total stock-based compensation expense |
$ | 2,816 | $ | 739 | $ | 569 | $ | 4,972 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The fair value of each option is estimated on the date of grant using the Black-Scholes method with the following assumptions:
| Year Ended December 31, | Nine Months Ended September 30, | |||||
| 2012 | 2013 | 2014 | ||||
| (unaudited) | ||||||
| Dividend yield |
— | — | — | |||
| Expected volatility |
79% | 73% | 70% | |||
| Risk-free interest rate |
1.57%–1.93% | 2.04%–3.84% | 0.69%–2.73% | |||
| Expected life (in years) |
6.25–10 | 6.25–10 | 3-10 | |||
| Weighted-average grant date fair value per share |
$10.09 | $11.37 | $7.43 | |||
No related tax benefits were recognized for the years ended December 31, 2012 and 2013.
Cash and Cash Equivalents
The Company considers highly liquid investments with a maturity of three months or less when purchased to be cash equivalents. Cash and cash equivalents, which consist primarily of cash on deposit in US, Swiss, and Canadian banks. Cash and cash equivalents are stated at cost which approximates fair value.
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to credit risk consist primarily of cash and cash equivalents. The Company holds these investments in highly-rated financial institutions, and limits the amounts of credit exposure to any one financial institution. These amounts at times may exceed federally insured limits. The Company has not experienced any credit losses in such accounts and does not believe it is exposed to any significant credit risk on these funds. The Company has no off-balance sheet concentrations of credit risk, such as foreign currency exchange contracts, option contracts or other hedging arrangements.
Fair Value Measurements
The Company follows ASC Topic 820, Fair Value Measurements and Disclosures, which establishes a fair value hierarchy for those instruments measured at fair value that distinguishes between assumptions based on market
F-10
Table of Contents
date (observable inputs) and the Company’s own assumptions (unobservable inputs). The hierarchy consists of three levels:
| • | Level 1—Unadjusted quoted prices in active markets for identical assets or liabilities. |
| • | Level 2—Quoted prices for similar assets and liabilities in active markets, quoted prices in markets that are not active, or inputs which are observable, either directly or indirectly, for substantially the full term of the asset or liability. |
| • | Level 3—Unobservable inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability in which there is little, if any, market activity for the asset or liability at the measurement date. |
At December 31, 2012 and 2013 and September 30, 2014 (unaudited) the Company did not have any assets or liabilities that were remeasured at fair value on a recurring basis. The carrying amounts reflected in the balance sheets for cash, prepaid expenses and other current assets, accounts payable, and accrued expenses approximate their fair values at December 31, 2012 and 2013 and September 30, 2014 (unaudited), due to their short-term nature.
Property and Equipment
Property and equipment, which consists of land, construction in process, furniture and fixtures, computers and office equipment, scientific equipment, leasehold improvements and vehicles are stated at cost and depreciated over the estimated useful lives of the assets, with the exception of land and construction in process which is not depreciated, using the straight line method:
| • Furniture and fixtures |
7 years | |
| • Office equipment |
5 years | |
| • Leasehold improvements |
Shorter of asset’s useful life or remaining lease term | |
| • Scientific equipment |
5 years | |
| • Vehicles |
5 years | |
Costs of major additions and betterments are capitalized; maintenance and repairs, which do not improve or extend the life of the respective assets, are charged to expense as incurred. Upon retirement or sale, the cost of the disposed asset and the related accumulated depreciation are removed from the accounts and the resulting gain or loss is recognized.
Impairment of Long-Lived Assets
The Company periodically evaluates its long-lived assets for potential impairment in accordance with ASC Topic 360, Property, Plant and Equipment. Potential impairment is assessed when there is evidence that events or changes in circumstances indicate that the carrying amount of an asset may not be recovered. Recoverability of these assets is assessed based on undiscounted expected future cash flows from the assets, considering a number of factors, including past operating results, budgets and economic projections, market trends and product development cycles. If impairments are identified, assets are written down to their estimated fair value. The Company has not recognized any impairment through December 31, 2013.
Deferred Offering Costs
Deferred offering costs, which consist of direct incremental legal, accounting and other professional service fees relating to this proposed offering, are capitalized. The deferred offering costs will be offset against the proceeds
F-11
Table of Contents
from this proposed offering upon the consummation of the offering. In the event the offering is terminated, deferred offering costs will be expensed.
Foreign Currency Transactions
Certain transactions are denominated in a currency other than the Company’s functional currency of the US dollar, and the Company generates assets and liabilities that are fixed in terms of the amount of foreign currency that will be received or paid. At each balance sheet, the Company adjusts the assets and liabilities to reflect the current exchange rate, resulting in a translation gain or loss. Transaction gains and losses are also realized upon a settlement of a foreign currency transaction in determining net loss for the period in which the transaction is settled.
Comprehensive Income (Loss)
ASC 220, Comprehensive Income, requires that all components of comprehensive income (loss), including net income (loss), be reported in the financial statements in the period in which they are recognized. Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources, including unrealized gains and losses on investments and foreign currency translation adjustments.
Segment and Geographic Information
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision making group, in making decisions on how to allocate resources and assess performance. The Company’s chief operating decision maker is the chief executive officer. The Company and the chief operating decision maker view the Company’s operations and manage its business as one operating segment. Substantially all of the Company’s operations are in the US geographic segment.
Net Loss Per Share
Net loss per share (“EPS”) is computed by dividing net loss by the weighted average number of common shares outstanding during each period. Diluted EPS is computed by dividing net loss by the weighted average number of common shares and common share equivalents outstanding (if dilutive) during each period. The number of common share equivalents, which include stock options, is computed using the treasury stock method.
Subsequent Events
The Company considered events or transactions occurring after the balance sheet date but prior to the date the consolidated financial statements are available to be issued for potential recognition or disclosure in its consolidated financial statements. Subsequent events have been evaluated through the date the financial statements were available to be issued.
Recent Accounting Pronouncements
In June 2014 the Financial Accounting Standards Board (FASB) issued ASU 2014-10, Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810 Consolidation. These updates remove the definition of a development stage entity from the Master Glossary of the Accounting Standards Codification, thereby removing the financial reporting distinction between development stage entities and other reporting entities from US GAAP. In addition, the amendments eliminate the requirements for development stage entities to (1) present inception-to-date information in the statements of income, cash flows, and shareholder equity, (2) label the financial statements as those of a development stage entity, (3) disclose a
F-12
Table of Contents
description of the development stage activities in which the entity is engaged, and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. This standard is effective for annual reporting periods beginning after December 15, 2014. The Company has early adopted this standard in the presentation of its 2013 financial statements.
3. Property and Equipment
Property and equipment consisted of the following at December 31 (in thousands):
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| Computer and office equipment |
$ | 249 | $ | 252 | ||||
| Furniture and fixtures |
126 | 126 | ||||||
| Land |
1,418 | 1,418 | ||||||
| Leasehold improvements |
731 | 731 | ||||||
| Scientific equipment |
3,573 | 3,629 | ||||||
| Vehicle |
30 | 30 | ||||||
| Construction in process |
738 | 738 | ||||||
| Accumulated depreciation |
(2,760 | ) | (3,544 | ) | ||||
|
|
|
|
|
|||||
| $ | 4,105 | $ | 3,380 | |||||
|
|
|
|
|
|||||
Depreciation expense related to property and equipment amounted to approximately $946,000 and $784,000 for the years ended December 31, 2012 and 2013, respectively.
4. Accrued Expenses
Accrued expenses consist of the following at December 31, 2012 and 2013 (in thousands):
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| Accrued compensation and related |
$ | 161 | $ | 104 | ||||
| Accrued professional fees |
39 | 75 | ||||||
| Other |
410 | 105 | ||||||
|
|
|
|
|
|||||
| $ | 610 | $ | 284 | |||||
|
|
|
|
|
|||||
5. Common Stock
Pursuant to its Articles, the Company has an unlimited number of common shares available for issuance with no par value.
In 2012, the Company issued approximately 477,000 shares of common stock at a price of $15.00 per share for total proceeds of approximately $7.1 million.
In 2012, the Company cancelled 300,000 shares of previously existing restricted common stock, such common stock had been previously issued to a consultant in return for specified services, Such services were never received by the Company and, accordingly, the Company chose to cancel the relationship. The Company had not recognized any share-based compensation related to such common stock as no services had been provided and vesting had not occurred. Accordingly, the cancellation of such common stock did not result in the recapture of any previously recognized share-based compensation.
In August 2013, the Company issued approximately 1.2 million shares of common stock at a price of $10.00 per share for total proceeds of approximately $12.0 million. Each share had one warrant attached, which would be exercisable for 180 days into a single common share in the Company at a price of $10.00 per share.
F-13
Table of Contents
In February 2014, the Company issued approximately 1.2 million shares of common stock for total proceeds of approximately $12 million from the exercise of warrants by common stock shareholders. In July through September of 2014, the Company issued approximately 392,666 shares of common stock at a price of $15.00 per share for total proceeds of approximately $5.9 million. Each share had one warrant attached, which would be exercisable for 180 days into a single common share in the Company at a price of $15.00 per share. As of September 30, 2014, the Company had total warrants outstanding for the purchase of 392,666 shares of common stock at a price of $15.00 per share.
6. Common Stock Options
On November 11, 2005, the Board of Directors of the Company adopted a stock option plan (the Plan) pursuant to which the Company may grant incentive stock options to directors, officers, employees or consultants of the Company or an affiliate or other persons as the Compensation Committee may approve.
In February and August 2011, the Company granted stock options to a non-employee consultant that vest over 3 and 4 years, respectively. The fair value of these awards is remeasured at each reporting date until the date the consultant’s services are completed.
All options will be non-transferable and may be exercised only by the participant, or in the event of the death of the participant, a legal representative until the earlier of the options’ expiry date or the first anniversary of the participant’s death, or such other date as may be specified by the Compensation Committee.
The term of the options is at the discretion of the Compensation Committee, but may not exceed 10 years from the grant date. The options expire on the earlier of the expiration date or the date three months following the day on which the participant ceases to be a director, officer or employee of or consultant to the company, or in the event of the termination of the participant with cause, the date of such termination.
The number of common shares reserved for issuance to any one person pursuant to this Plan shall not, in aggregate, exceed 5% of the total number of outstanding common shares. The exercise price per common share under each option will be the fair market value of such shares at the time of the grant. Upon share option exercise, the Company issues new shares of common stock.
A summary of changes in common stock options issued under the Plan is as follows:
| Options | Exercise Price | Weighted- Average Exercise Price |
||||||||
| Options outstanding at January 1, 2012 |
3,884,999 | $0.65–10.00 | $ | 4.53 | ||||||
| Granted |
302,167 | 7.50–15.00 | 14.89 | |||||||
| Exercised |
(27,000 | ) | 5.00 | 5.00 | ||||||
| Forfeitures |
(490,500 | ) | 1.25–7.50 | 4.74 | ||||||
| Options outstanding at December 31, 2012 |
3,669,666 | 0.65–15.00 | 7.01 | |||||||
| Granted |
40,000 | 15.00 | 15.00 | |||||||
| Exercised |
— | |||||||||
| Forfeitures |
(316,167 | ) | 2.50–15.00 | 8.39 | ||||||
| Options outstanding at December 31, 2013 |
3,393,499 | 0.65–15.00 | 6.02 | |||||||
| Granted (unaudited) |
1,022,833 | 2.5-15.00 | 9.75 | |||||||
| Exercised (unaudited) |
— | — | — | |||||||
| Forfeitures (unaudited) |
(58,000 | ) | 2.50–15.00 | 10.63 | ||||||
|
|
|
|||||||||
| Options outstanding at September 30, 2014 (unaudited) |
4,358,332 | 0.65-15.00 | 6.17 | |||||||
|
|
|
|||||||||
F-14
Table of Contents
The weighted average fair value of the options issued to directors, employees and consultants during the years ended December 31, 2013 and 2012 and the nine months ended September 30, 2014 was $11.37, $10.09 and $7.43 (unaudited), respectively. Options with an intrinsic value of $2.51 and $4.22 became vested during 2012 and 2013, respectively. The total intrinsic value of options exercisable and total options outstanding at December 31, 2013 was $16,345,000 and $16,425,000, respectively. The total fair value of options vested during the years ended December 31, 2012 and 2013 was $2,955,000 and $632,500, respectively.
As of December 31, 2013, there was approximately $668,000 of unrecognized compensation cost, related to stock options granted under the Plan which will be amortized to stock compensation expense over the next three years. As of September 30, 2014, there was approximately $3,238,000 (unaudited) of unrecognized compensation cost, related to stock options granted under the Plan which will be amortized to stock compensation expense over the next 2.03 years.
The following table summarizes information concerning outstanding options under the Plan as of December 31, 2013:
| Options outstanding | Options Vested and Outstanding |
|||||||||||||||||||||
| Exercise Price |
Number | Weighted- Average Remaining Contractual Life |
Weighted- Average Exercise Price |
Number | Weighted- Average Exercise Price |
|||||||||||||||||
| $ | 0.65 | 60,000 | $ | 1.72 | 0.65 | 60,000 | $ | 0.65 | ||||||||||||||
| 0.94 | 50,000 | 2.95 | 0.94 | 50,000 | 0.94 | |||||||||||||||||
| 1.17 | 270,000 | 3.29 | 1.17 | 270,000 | 1.17 | |||||||||||||||||
| 2.50 | 693,500 | 4.71 | 2.50 | 693,500 | 2.50 | |||||||||||||||||
| 3.75 | 1,000,000 | 6.12 | 3.75 | 1,000,000 | 3.75 | |||||||||||||||||
| 5.00 | 137,000 | 6.36 | 5.00 | 126,924 | 5.00 | |||||||||||||||||
| 7.50 | 734,499 | 7.14 | 7.50 | 668,679 | 7.50 | |||||||||||||||||
| 10.00 | 250,000 | 8.00 | 10.00 | 250,000 | 10.00 | |||||||||||||||||
| 15.00 | 198,500 | 8.95 | 15.00 | 162,200 | 15.00 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| 3,393,499 | 6.02 | 5.16 | 3,281,303 | 5.02 | ||||||||||||||||||
7. Net Loss Per Share
The following summarizes the computation of basic and diluted net loss per share for the years ended December 31, 2012 and 2013 and the nine months ended September 30, 2013 and 2014 (in thousands, except share and per share data):
| Year Ended December 31, | Nine Months Ended September 30, |
|||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| (unaudited) | (unaudited) | |||||||||||||||
| Net loss |
$ | (15,160 | ) | $ | (9,927 | ) | $ | (7,440 | ) | $ | (15,835 | ) | ||||
| Weighted-average number of common shares—basic and diluted |
21,594,369 | 22,520,416 | 22,341,240 | 24,173,485 | ||||||||||||
| Net loss per share—basic and diluted |
$ | (0.70 | ) | $ | (0.44 | ) | $ | (0.33 | ) | $ | (0.66 | ) | ||||
F-15
Table of Contents
The following potentially dilutive securities outstanding, prior to the use of the treasury stock method or if-converted method, have been excluded from the computation of diluted weighted-average common shares outstanding, because including them would have had an anti-dilutive effect due to the losses reported.
| Year Ended December 31, | Nine Months Ended September 30, |
|||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| (unaudited) | (unaudited) | |||||||||||||||
| Stock options |
3,669,666 | 3,393,499 | 3,393,499 | 4,358,332 | ||||||||||||
| Warrants to purchase common stock |
— | 1,204,510 | 1,204,510 | 392,666 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
3,669,666 | 4,598,009 | 4,598,009 | 4,750,998 | ||||||||||||
8. Income Taxes
The Company recorded no provision for income taxes for the years ended December 31, 2012 and 2013 due to the reported net losses in each year.
A reconciliation of the Company’s Canadian federal statutory income tax rate to the Company’s effective income tax rate is as follows for the years ended December 31, 2012 and 2013:
| 2012 | 2013 | |||||||
| Income tax benefit computed at federal tax rate |
13.5 | % | 13.5 | % | ||||
| Change in valuation allowance |
(8.4 | ) | (10.0 | ) | ||||
| Stock compensation and other |
(5.1 | ) | (3.5 | ) | ||||
|
|
|
|
|
|||||
| Total |
— | % | — | % | ||||
|
|
|
|
|
|||||
During the years ended December 31, 2012 and 2013, the Company had no interest and penalties related to income taxes.
As of December 31, 2013, the Company has unused net operating losses of approximately $52.8 million (approximately $44.5 million in Canada and approximately $8.3 million in the US) available to reduce taxable income of future years. The net operating losses expire as follows (in thousands):
| 2025 |
$ | 508 | ||
| 2026 |
722 | |||
| 2027 |
689 | |||
| 2028 |
2,850 | |||
| 2029 |
5,362 | |||
| 2030 |
7,863 | |||
| 2031 |
13,458 | |||
| 2032 |
11,009 | |||
| 2033 |
10,336 |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company has established a valuation allowance due to uncertainties regarding the realization of deferred tax assets based upon
F-16
Table of Contents
the Company’s lack of earnings history. Significant components of the Company’s deferred tax assets and liabilities as of December 31, 2012 and 2013 are as follows (in thousands):
| 2012 | 2013 | |||||||
| Deferred tax assets: |
||||||||
| Noncapital losses |
$ | 7,201 | $ | 8,820 | ||||
| Qualifying research and development credits |
729 | 878 | ||||||
| Depreciation |
— | 68 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
7,930 | 9,766 | ||||||
| Deferred tax liabilities: |
||||||||
| Depreciation |
316 | — | ||||||
| Accrued liabilities |
100 | 74 | ||||||
| Share issuance costs |
430 | 431 | ||||||
|
|
|
|
|
|||||
| Total deferred tax liabilities |
846 | 505 | ||||||
|
|
|
|
|
|||||
| Net deferred tax asset |
7,084 | 9,261 | ||||||
| Valuation allowance for deferred tax assets |
(7,084 | ) | (9,261 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax asset including valuation allowance |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
The valuation allowance increased by approximately $2.2 million during the year ended December 31, 2013.
The Company applies the accounting guidance in ASC 740 related to accounting for uncertainty in income taxes. The Company’s reserves related to taxes are based on a determination of whether, and how much of, a tax benefit taken by the Company in its tax filings or positions is more likely than not to be realized following resolution of any potential contingencies present related to the tax benefit. As of December 31, 2012 and 2013, the Company had no unrecognized tax benefits.
The Company files federal income tax returns in Canada, US, Switzerland, Germany, and Japan. The Company also files income tax returns in Texas in the US The statute of limitations for assessment by local taxing authorities is open for tax years ended December 2013, 2012, 2011 and 2010. There are currently no federal or state income tax audits in progress.
9. Related-Party Transactions
In both 2012 and 2013 approximately $240,000 in salaries and wages were paid to a shareholder/director and in 2013 approximately $168,000 in bonuses were paid to the same shareholder/director. Also, legal fees of approximately $41,000 and $35,000 were paid to a law firm for legal services rendered in which a director of the Company is a senior partner in 2012 and 2013, respectively.
10. Commitments
On January 12, 2008, the Company entered a lease agreement to lease its facility in Austin, Texas, USA. On September 15, 2010, the Company entered into a second lease agreement to lease additional space in Austin, TX, USA. Both leases expire in 2013. On March 20, 2013, the company extended the lease for another 21 months with the same terms and rental rates as the current lease. Rent expense was approximately $552,000 and $484,000 for the years ended December 31, 2013 and 2012, respectively. The future minimum lease payments are as follows as of December 31, 2013 (in thousands):
| 2014 |
$ | 403 | ||
| 2015 |
67 |
F-17
Table of Contents
11. Subsequent Events
In March 2014, the Company entered into a licensing agreement with STROX Biopharmaceuticals, LLC (“STROX) pursuant to which STROX granted to the Company an exclusive worldwide license in all fields to the patent portfolio owned by STROX relating to non-protein A-binding antibodies for treating bacterial infections. The Company paid STROX $30,000 and issued 50,000 fully vested options for the purchase of shares of the Company’s common stock with an exercise price of $10.00 per share. The Company will be required to pay a royalty of 10% of net sales related to such patent portfolio. The $30,000 of cash paid and the fair value of the 50,000 options issued ($292,000) were expensed immediately as research and development costs as the license acquired is to be used for a specific research and development project and has no current commercially viable alternative future use. At such time when the Company records revenues that are subject to royalties owed to STROX, the Company will record such royalties as cost of revenues in the period in which the related revenue is recorded.
From October 1, 2014 to January 22, 2015, the company received subscription agreements from investors to purchase a total of $3,083,865 shares of common stock at $15.00 per share, or $46 million. As of January 22, 2015, the Company had received the full amount from these investors pursuant to the respective subscription agreements.
In December 2014, approximately 108,000 warrants were exercised for $15.00 per share for a total of $1.62 million.
In December 2014, 15,000 stock options were exercised at $10 per share for a total of $150,000.
F-18
Table of Contents
Until [ ], 2015 ( days after the date of this Prospectus), all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a Prospectus. This is in addition to the dealers’ obligation to deliver a Prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
XBIOTECH, INC.
Shares of Common Stock
Prospectus
, 2015
Table of Contents
PART II
INFORMATION NOT REQUIRED IN THE PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution. |
The following table sets forth the estimated costs and expenses of XBiotech, Inc. (“we,” “us” or the “Company”) in connection with the offering described in the registration statement.
| SEC Registration Fee |
$ | |||
| FINRA Filing Fee |
[ | ] | ||
| Legal Fees and Expenses |
[ | ] | ||
| Accounting Fees and Expenses |
[ | ] | ||
| Transfer Agent Fees |
[ | ] | ||
| Escrow Agent Fees |
[ | ] | ||
| Printing Expenses |
[ | ] | ||
| Reimbursed Placement Agent Expenses |
[ | ] | ||
| Other Expenses |
[ | ] | ||
|
|
|
|||
| Total Expenses |
$ | [ | ] | |
|
|
|
| Item 14. | Indemnification of Directors and Officers. |
XBiotech Inc. (“we,” “us” or “our company”) is subject to the provisions of Part 5, Division 5 of the Business Corporations Act (British Columbia) (the “BCBCA”).
Under Section 160 of the BCBCA, we may, subject to Section 163 of the BCBCA:
| (1) | indemnify an individual who: |
| • | is or was a director or officer of our company; |
| • | is or was a director or officer of another corporation (i) at a time when such corporation is or was an affiliate of our company; or (ii) at our request, or |
| • | at our request, is or was, or holds or held a position equivalent to that of, a director or officer of a partnership, trust, joint venture or other unincorporated entity, and including, subject to certain limited exceptions, the heirs and personal or other legal representatives of that individual (collectively, an “eligible party”), against all eligible penalties to which the eligible party is or may be liable; and |
| (2) | after final disposition of an eligible proceeding, pay the expenses actually and reasonably incurred by an eligible party in respect of that proceeding, where: |
“eligible penalty” means a judgment, penalty or fine awarded or imposed in, or an amount paid in settlement of, and eligible proceeding.
“eligible proceeding” means a proceeding in which an eligible party or any of the heirs and personal or other legal representatives of the eligible party, by reason of the eligible party being or having been a director or officer of, or holding or having held a position equivalent to that of a director or officer of, our company or an associated corporation (i) is or may be joined as a party, or (ii) is or may be liable for or in respect of a judgment, penalty or fine in, or expenses related to, the proceeding.
“proceeding” includes any legal proceeding or investigative action, whether current, threatened , pending or completed.
Under Section 161 of the BCBCA, and subject to Section 163 of the BCBCA, we must, after the final disposition of an eligible proceeding, pay the expenses actually and reasonably incurred by an eligible party in
II-1
Table of Contents
respect of that proceeding if the eligible party (i) has not been reimbursed for those expenses, and (ii) is wholly successful, on the merits or otherwise, in the outcome of the proceeding or is substantially successful on the merits in the outcome of the proceeding.
Under Section 162 of the BCBCA, and subject to Section 163 of the BCBCA, we may pay, as they are incurred in advance of the final disposition of an eligible proceeding, the expenses actually and reasonably incurred by an eligible party in respect of the proceeding, provided that we must not make such payments unless we first receive from the eligible party a written undertaking that, if it is ultimately determined that the payment of expenses is prohibited under Section 163 of the BCBCA, the eligible party will repay the amounts advanced.
Under Section 163 of the BCBCA, we must not indemnify an eligible party against eligible penalties to which the eligible party is or may be liable or pay the expenses of an eligible party in respect of that proceeding under Sections 160, 161 or 162 of the BCBCA, as the case may be, if any of the following circumstances apply:
| • | if the indemnity or payment is made under an earlier agreement to indemnify or pay expenses and, at the time that the agreement to indemnify or pay expenses was made, we were prohibited from giving the indemnity or paying the expenses by our memorandum or articles; |
| • | if the indemnity or payment is made otherwise than under an earlier agreement to indemnify or pay expenses and, at the time that the indemnity or payment is made, we are prohibited from giving the indemnity or paying the expenses by our memorandum or articles; |
| • | if, in relation to the subject matter of the eligible proceeding, the eligible party did not act honestly and in good faith with a view to the best interests of our company or the associated corporation, as the case may be; or |
| • | in the case of an eligible proceeding other than a civil proceeding, if the eligible party did not have reasonable grounds for believing that the eligible party’s conduct in respect of which the proceeding was brought was lawful. |
If an eligible proceeding is brought against an eligible party by or on behalf of our company or by or on behalf of an associated corporation, we must not either indemnify the eligible party against eligible penalties to which the eligible party is or may be liable, or pay the expenses of the eligible party under Sections 160, 161 or 162 of the BCBCA, as the case may be, in respect of the proceeding.
Under Section 164 of the BCBCA, and despite any other provision of Part 5, Division 5 of the BCBCA and whether or not payment of expenses or indemnification has been sought, authorized or declined under Part 5, Division 5 of the BCBCA, on application of our company or an eligible party, the Supreme Court of British Columbia may do one or more of the following:
| • | order us to indemnify an eligible party against any liability incurred by the eligible party in respect of an eligible proceeding; |
| • | order us to pay some or all of the expenses incurred by an eligible party in respect of an eligible proceeding; |
| • | order the enforcement of, or payment under, an agreement of indemnification entered into by us; |
| • | order us to pay some or all of the expenses actually and reasonably incurred by any person in obtaining an order under Section 164 of the BCBCA; or |
| • | make any other order the court considers appropriate. |
Section 165 of the BCBCA provides that we may purchase and maintain insurance for the benefit of an eligible party or the heirs and personal or other legal representatives of the eligible party against any liability that may be incurred by reason of the eligible party being or having been a director or officer of, or holding or having held a position equivalent to that of a director or officer of, our company or an associated corporation.
II-2
Table of Contents
Under our articles, and subject to the BCBCA, we must indemnify an eligible party and his or her heirs and legal personal representatives against all eligible penalties to which such person is or may be liable, and we must, after the final disposition of an eligible proceeding, pay the expenses actually and reasonably incurred by such person in respect of that proceeding. Each eligible party is deemed to have contracted with our company on the terms of the indemnity contained in our articles.
Under our articles, and subject to the BCBCA, we may agree to indemnify and may indemnify any person (including an eligible party) against eligible penalties and pay expenses incurred in connection with the performance of services by that person for us.
Under our articles, and subject to the BCBCA, we may advance expenses to an eligible party.
Pursuant to our articles, the failure of an eligible party to comply with the BCBCA or our articles does not, of itself, invalidate any indemnity to which he or she is entitled under our articles.
Under our articles, we may purchase and maintain insurance for the benefit of an eligible person (or his or her heirs or legal personal representatives) against any liability incurred by him or her as a director, officer or person who holds or held such equivalent position.
| Item 15. | Recent Sales of Unregistered Securities |
The following list sets forth information regarding all unregistered securities sold by us by us since January 1, 2012. No underwriters were involved in the sales.
2012
During the month of May to September 2012, we raised approximately $7,148,220 from the sale of 476,548 shares our common stock to 30 investors at price of $15 per share.
During the year 2012, we granted options to seven new employees, under our 2005 Plan to purchase an aggregate of 122,167 shares of common shares at exercise prices ranging from $7.50 per share to $15 per common share. In December 12, 2012, we granted all employees 5,000 stock options each to purchase an aggregate of 180,000 shares of common stock at an exercise price of $15.00 per share.
On December 21, 2012, one consultant exercised options to purchase 27,000 shares at an exercise price of $5.00 per share.
2013
On February 1st, 2013, we granted options to one employee under our 2005 Plan to purchase an aggregate of 40,000 shares of common shares at exercise prices of $15 per common share. In year 2013, we granted options to eight new employees 4,500 each under our 2005 Plan to purchase an aggregate of 36,000 shares of common shares at exercise prices ranging from $7.50 per share to $15 per common share.
In August 2013, we raised approximately $12 million by selling 1.2 million units to 10 investors at a purchase price of $10.00 per share. We issued approximately 1,204,510 shares of our common stock and each share had an attached warrant which allowed them to purchase shares within 180 days at an exercise price of $10.00 per share.
2014
In February 2014, 7 investors exercised warrants to acquire 1,195,000 shares of our common stock at an exercise price of $10.00 per share.
II-3
Table of Contents
In December 2014, 15,000 stock options were exercised at $10 per share for a total of $150,000.
From July to January 22, 2015, we raised approximately $53.8 million by selling 3,584,530 shares of our common stock to 59 investors at a purchase price of $ 15 per share.
In 2014, we granted options to employees under our 2005 Plan to purchase an aggregate of 415,500 shares of common shares at exercise prices ranging from $10.00 to 15.00 per common share. From January 1, 2014 to January 22, 2015, we granted options to certain of our directors and officers under our 2005 Plan to purchase an aggregate of 886,666 shares of common shares at exercise prices ranging from $10.00 to $15.00 per common share. We also granted options to five consultants to purchase an aggregate of 170,500 shares of common shares at exercise prices ranging from $10.00 to $15.00 per common share.
All of these issuances were deemed to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act, as transactions by an issuer not involving a public offering. The purchasers of securities in each such transaction represented their intention to acquire the securities for investment only and not with a view to offer or sell, in connection with any distribution of the securities, and appropriate legends were affixed to the share certificates and instruments issued in such transactions.
| Item 16. | Exhibits and Financial Statement Schedules. |
A list of exhibits filed herewith is included on the Exhibit Index which immediately follows the signature page of this registration statement and is incorporated herein by reference.
The following exhibits are filed as part of this Registration Statement:
| Exhibit |
Description | |
| 1.1 | Form of underwriting agreement+ | |
| 3.1 | Certificate of Continuation dated September 23, 2005, issued by the Registrar of Companies, Province of British Columbia, Canada | |
| 3.2 | Notice of Articles, dated December 8, 2005, issued by the Registrar of Companies, Province of British Columbia, Canada | |
| 3.3 | Articles of XBiotech, Inc. | |
| 5.1 | Legal opinion of Stikeman Elliott LLP + | |
| 10.1 | Executive Employment Agreement dated as of March 22, 2005 between XBiotech and John Simard* | |
| 10.2 | Change in Control Agreement dated as of March 22, 2005 between XBiotech and John Simard* | |
| 10.3 | Confidentiality and Assignment of Inventions Agreement dated as of March 22, 2005 between XBiotech and John Simard | |
| 10.4 | XBiotech 2005 Incentive Stock Option Plan* | |
| 10.5 | Form of indemnification agreement between XBiotech and each director of XBiotech* | |
| 10.6 | Agreement of Lease by and between NNN Met Center 4-9, LP and XBiotech dated January 14, 2008 and the First Amendment dated January 17, 2008, the Second Amendment dated August 2010 and the Third Amendment dated March 2013 | |
| 10.7 | Agreement of Lease by and between NNN Met Center 4-9, LLP and XBiotech, Inc. for Suite 600 dated August 16, 2010 and First Amendment dated March 2013 | |
II-4
Table of Contents
| Exhibit |
Description | |
| 10.8 | Board Member Agreement dated November 4, 2014 between XBiotech, Inc. and Daniel Vasella * | |
| 14.1 | Code of conduct for directors, officers and employees | |
| 21.1 | List of subsidiaries | |
| 23.1 | Consent of Ernst & Young LLP | |
| 23.2 | Consent of Stikeman Elliott LLP (included in Exhibit 5.1) + | |
| 24.1 | Power of attorney (included on signature page) | |
| + | to be filed by amendment |
| * | Indicates management contract or compensatory plan |
| Item 17. | Undertakings |
| (a) | The undersigned registrant hereby undertakes: |
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | To include any Prospectus required by Section 10(a)(3) of the Securities Act of 1933; |
| (ii) | To reflect in the Prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of Prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement. |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement; |
| (2) | That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (4) | That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities: |
The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
| (i) | Any preliminary Prospectus or Prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; |
II-5
Table of Contents
| (ii) | Any free writing Prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
| (iii) | The portion of any other free writing Prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
| (iv) | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
| (b) | Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue. |
| (c) | The undersigned hereby undertakes that: |
| (1) | For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of Prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of Prospectus filed by the registrant pursuant to Rule 424(b) (1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (2) | For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of Prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-6
Table of Contents
SIGNATURES
Pursuant to the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized in the City of Austin, State of Texas, on February 2, 2015.
| XBIOTECH INC., | ||
| /S/ JOHN SIMARD | ||
| Name: | John Simard | |
| Title: | President and Chief Executive Officer | |
| (Principal Executive Officer) | ||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints John Simard, as his true and lawful attorney-in-fact and agent with full power of substitution, for him in any and all capacities, to sign any and all amendments to this registration statement (including post-effective amendments or any abbreviated registration statement and any amendments thereto filed pursuant to Rule 462(b) under the Securities Act of 1933 increasing the number of securities for which registration is sought), and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact, proxy and agent full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully for all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact, proxy and agent, or his substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement on Form S-1 has been signed by the following persons in the capacities and on the dates indicated.
| Signature and Title |
Date | |
| /S/ JOHN SIMARD John Simard, Chief Executive Officer (Principal Executive Officer) and Director |
February 2, 2015 | |
| /S/ QUEENA HAN Queena Han, Vice President of Finance & Human Resources (Principal Financial Officer and Principal Accounting Officer) |
February 2, 2015 | |
| /S/ FABRIZIO BONANNI Fabrizio Bonanni, Director |
February 2, 2015 | |
| /S/ W. THORPE MCKENZIE W. Thorpe McKenzie, Director |
February 2, 2015 | |
| /S/ DANIEL VASELLA Daniel Vasella, Director |
February 2, 2015 | |
II-7
Table of Contents
EXHIBIT INDEX
| Exhibit |
Description | |
| 1.1 | Form of underwriting agreement+ | |
| 3.1 | Certificate of Continuation dated September 23, 2005, issued by the Registrar of Companies, Province of British Columbia, Canada | |
| 3.2 | Notice of Articles, dated December 8, 2005, issued by the Registrar of Companies, Province of British Columbia, Canada | |
| 3.3 | Articles of XBiotech, Inc. | |
| 5.1 | Legal opinion of Stikeman Elliott LLP+ | |
| 10.1 | Executive Employment Agreement dated as of March 22, 2005 between XBiotech and John Simard* | |
| 10.2 | Change in Control Agreement dated as of March 22, 2005 between XBiotech and John Simard* | |
| 10.3 | Confidentiality and Assignment of Inventions Agreement dated as of March 22, 2005 between XBiotech and John Simard | |
| 10.4 | XBiotech 2005 Incentive Stock Option Plan* | |
| 10.5 | Form of indemnification agreement between XBiotech and each director of XBiotech* | |
| 10.6 | Agreement of Lease by and between NNN Met Center 4-9, LP and XBiotech dated January 14, 2008 and the First Amendment dated January 17, 2008, the Second Amendment dated August 2010 and the Third Amendment dated March 2013 | |
| 10.7 | Agreement of Lease by and between NNN Met Center 4-9, LLP and XBiotech, Inc. for Suite 600 dated August 16, 2010 and First Amendment dated March 2013 | |
| 10.8 | Board Member Agreement dated November 4, 2014 between XBiotech, Inc. and Daniel Vasella* | |
| 14.1 | Code of conduct for directors, officers and employees | |
| 21.1 | List of subsidiaries | |
| 23.1 | Consent of Ernst & Young LLP | |
| 23.2 | Consent of Stikeman Elliott LLP (included in Exhibit 5.1)+ | |
| 24.1 | Power of attorney (included on signature page) | |
| * | Indicates management contract or compensatory plan |
| + | To be filed by amendment |