Attached files
| file | filename |
|---|---|
| 8-K - 8-K - TREVENA INC | a2222386z8-k.htm |
Exhibit 99.1
BUSINESS
Overview
We are a clinical stage biopharmaceutical company that discovers, develops and intends to commercialize therapeutics that use a novel approach to target G protein coupled receptors, or GPCRs. Using our proprietary product platform, we have identified and advanced three differentiated product candidates into the clinic as follows:
· TRV130: We recently announced top-line data from our Phase 2a/b clinical trial of TRV130 in postoperative pain. At doses of 2 mg and 3 mg of TRV130 administered every three hours, the trial achieved its primary endpoint of statistically greater pain reduction than placebo for 48 hours, which we believe demonstrates proof of concept for TRV130. The 3 mg dose of TRV130 also showed statistically superior analgesic efficacy over the 48-hour trial period compared to 4 mg of morphine administered every four hours. Additionally, in the first three hours of dosing, when pain was most severe, the 1 mg, 2 mg and 3 mg doses of TRV130 demonstrated superior analgesic efficacy in the trial compared to placebo, and the 2 mg and 3 mg doses of TRV130 demonstrated superior analgesic efficacy compared to 4 mg of morphine. There were no serious adverse events reported in the trial, which we believe suggests that these levels of pain relief can be achieved safely. Over the 48-hour trial period, the tolerability of TRV130 at doses of 2 mg and 3 mg administered every three hours was similar to that of 4 mg of morphine administered every four hours. Based on these data, we plan to move into Phase 3 preparations, which we expect to occur in parallel with a second Phase 2 trial for TRV130 that we plan to commence in December 2014. We also anticipate that we will initiate a Phase 3 clinical trial for TRV130 in the first quarter of 2016. These data complement the data generated in our Phase 1b trial, in which TRV130 showed superior efficacy with an improved tolerability profile following a single dose of TRV130 relative to a 10 mg dose of morphine in a human evoked-pain model. We hold a U.S. patent covering the composition of matter and methods of use for TRV130. We have retained all worldwide development and commercialization rights to TRV130, and plan to commercialize it in acute care markets such as hospitals and ambulatory surgery centers if it receives regulatory approval.
· TRV734: We have completed a first Phase 1 single ascending dose clinical trial for TRV734, an oral follow-on to TRV130 for the treatment of moderate to severe acute and chronic pain. We have completed enrollment in a second Phase 1 multiple ascending dose clinical trial and expect to report data from this trial early in the first quarter of 2015. We have retained all worldwide development and commercialization rights to TRV734.
· TRV027: We have completed a Phase 2a clinical trial and in early 2014 we initiated a Phase 2b clinical trial of TRV027 for acute heart failure, or AHF. Enrollment in this trial is ongoing, with over 250 patients recruited out of planned enrollment of approximately 500 patients. More than 65 sites in 12 countries are now open and recruiting, and we expect patient enrollment will conclude in the third quarter of 2015. We expect to report top-line data from this trial in the fourth quarter of 2015. Actavis plc, or Actavis, has the exclusive option to license TRV027 from us. We plan for TRV027 to be commercialized in the acute care hospital market if it receives regulatory approval.
We also have identified a new product candidate, TRV250, from our preclinical δ-opioid receptor program focused on central nervous system, or CNS, indications and plan to advance TRV250 to preclinical studies in 2015 that would support our submission of an investigational new drug application, or IND, to the U.S. Food and Drug Administration, or FDA.
Our Pipeline

Our Platform
GPCRs are a large family of cell surface receptors that trigger two signaling pathways, G protein and β-arrestin, and are implicated in cellular function and disease processes. More than 30% of all currently marketed therapeutics target GPCRs. Currently available therapeutics that target GPCRs, or GPCR ligands, are typically not signal specific, and therefore either inhibit both the G protein and β-arrestin pathways (an antagonist ligand) or activate both pathways (an agonist ligand). This lack of signal specificity often results in a suboptimal therapeutic profile for these drugs because in many cases one of the pathways is associated with a beneficial therapeutic effect and the other is associated with limiting that benefit or with an undesirable side effect (see Figure 1). We use our proprietary Advanced Biased Ligand Explorer, or ABLE, product platform to identify “biased” ligands, which are compounds that activate one of the two signaling pathways of the GPCR while inhibiting the other (see Figure 2). This signaling specificity is the basis for our drug discovery and development approach, which is to identify selective GPCR biased ligands and develop them into differentiated clinical products. While some GPCRs trigger other signaling pathways in addition to G protein and β-arrestin, most GPCRs trigger those two pathways.
Our ABLE product platform is a collection of proprietary biological information, in vitro assays, know-how and expertise that we use to identify unique GPCR-targeted biased ligands with attractive pharmaceutical properties. In vitro assays are laboratory tests performed outside of a living organism. Our in vitro assays use cells that have the receptor of interest on the cell surface, where G protein and β-arrestin signaling from that receptor can be measured to determine if a particular ligand is biased, and if so whether it is a G protein or β- arrestin biased ligand. Our assays can also measure different cellular responses resulting from signaling through β-arrestin and can thereby help us to associate pharmacological responses with molecular signaling. Most components of our ABLE product platform are maintained as trade secrets, but the output of the product platform is reflected in the product candidates that we have advanced into clinical testing and the research we have published in numerous peer- reviewed journals. We believe that our ABLE product platform provides us with an important competitive advantage in identifying further opportunities for efficient and high-impact biased ligand drug discovery, development and commercialization.
We were founded in late 2007 to discover and develop product candidates based on biased ligands, a concept discovered by our scientific founder, Dr. Robert Lefkowitz, who was awarded the 2012 Nobel Prize in Chemistry in part for his elucidation of the multiple pathways that a GPCR engages. We believe that we are the first company to progress a GPCR biased ligand into clinical trials. The
members of our executive management team have held senior positions at leading pharmaceutical and biotechnology companies and possess substantial experience across the spectrum of drug discovery, development and commercialization.
Figure 1: Mechanism of current GPCR-targeted drugs

Figure 2: Mechanism of our biased ligands—the next generation of GPCR-targeted drugs

Our Strategy
Our goal is to build a leading biopharmaceutical company leveraging our expertise in biased ligands to develop and commercialize innovative, best- in-class drugs targeting established GPCRs. Key elements of our business strategy to achieve this goal are to:
Rapidly advance clinical development of our three lead product candidates to commercialization.
We plan to develop and commercialize TRV130 for the treatment of moderate to severe acute postoperative pain and other indications where intravenous, or IV, therapy is preferred. Specific uses could include the treatment of pain related to surgery as well as nonsurgical settings like severe burn or end-of-life care facilities. The efficacy of drugs targeting the µ-opioid receptor is well-established. We have announced top-line data from our Phase 2a/b trial of TRV130 in postoperative pain in which we observed statistically significant analgesic efficacy compared to placebo and evidence for beneficial
differentiation from a benchmark dose of morphine. We also plan to initiate a second Phase 2 clinical trial for TRV130 in December 2014 in a soft tissue surgery model to further inform Phase 3 development and to further evaluate the potential for an improved therapeutic profile compared to morphine.
We plan to develop TRV734 for oral use in moderate to severe acute and chronic pain. We intend to seek a collaborator with experience in developing and commercializing controlled-substance therapeutics in acute and chronic care pain markets while retaining rights to commercialize TRV734 in hospital surgical settings in the United States. We completed our first Phase 1 clinical trial, in which we observed bioavailability and CNS activity of TRV734 after oral dosing, and we have completed enrollment in a second Phase 1 trial to support Phase 2 development. We expect to report data for the second trial early in the first quarter of 2015.
We plan to complete our Phase 2b clinical trial for TRV027 for the treatment of AHF by the end of 2015. If this trial is successful and Actavis exercises its option, Actavis will be responsible for all costs associated with further development and commercialization of TRV027. If the option is exercised, we will be entitled to an upfront option exercise fee and certain contingent milestone payments and royalties, which we intend to use to further develop and potentially commercialize our proprietary portfolio.
Establish commercialization and marketing capabilities in the United States, initially in acute care markets, for any of our product candidates that are approved or that we anticipate may be approved.
If any of our products beyond TRV027 receive or are anticipated to receive regulatory approval, we intend to build a focused sales force and establish marketing capabilities to commercialize those products to specialists in the United States, initially in acute care settings such as hospitals and ambulatory surgery centers.
We intend to retain full commercialization rights in the United States for TRV130. After the availability of Phase 2 clinical data for TRV130, we may seek collaborators for commercializing TRV130 outside the United States to offset risk and preserve capital.
For TRV734, we intend to seek a collaborator with experience in developing and commercializing controlled-substance therapeutics in acute and chronic care pain markets, thereby leveraging their expertise while retaining rights to commercialize TRV734 in hospital surgical settings and other settings where we may commercialize TRV130 in the United States.
If Actavis exercises its option to license TRV027, Actavis will be responsible for commercialization of TRV027 worldwide. We have the option to negotiate with Actavis for co-promotion rights in the United States, although Actavis has no obligation to grant us any co-promotion rights. We expect that TRV027, if approved, would be used primarily in the acute care setting, thereby providing an opportunity to leverage the commercial infrastructure we plan to implement to market our other product candidates, if any of them are approved.
Expand our CNS product portfolio by advancing TRV250, our δ -opioid receptor product candidate.
We aim to develop TRV250, which we believe will be the first selective δ-opioid receptor-targeted therapeutic for the treatment of CNS disorders. Based on the initial profile of TRV250, we anticipate focusing our initial development efforts on treatment-refractory migraine headaches. According to Decision Resources, a healthcare consulting company, the acute episodic migraine market encompassed approximately 12 million drug-treated patients in 2013 in the United States, representing approximately $2.2 billion of sales. We estimate that approximately 20% to 30% of these patients either do not respond to or cannot tolerate the market-leading triptan drug class and an additional 30% would benefit from improved efficacy compared to these drugs.
We believe TRV250 also may have utility in other CNS areas such as depression, Parkinson’s disease or neuropathic pain. We intend to conduct preclinical work beginning in 2015 designed to support the filing of an IND for TRV250. We also intend to seek a collaborator for TRV250 with CNS development and worldwide commercialization expertise, while potentially retaining commercialization rights in the United States.
Leverage our ABLE product platform to continue to discover and develop a pipeline of innovative biased ligand therapeutics and expand our product platform’s impact through external collaborations.
We have used our ABLE product platform to identify four potential therapeutics targeting GPCRs and have also identified additional high-value GPCR targets. As part of our longer term strategy, we plan to initiate internal drug discovery efforts in indications characterized by significant unmet medical need. We also intend to selectively collaborate on discovery and development programs to leverage the potential of our ABLE product platform.
CNS Portfolio
TRV130
TRV130 is a small molecule G protein biased ligand at the µ-opioid receptor, which we are developing as a first-line treatment for patients experiencing moderate to severe acute pain where IV administration is preferred. TRV130 activates the µ-opioid receptor G protein pathway, which in preclinical studies was associated with analgesia, and inhibits the β-arrestin pathway at the same receptor, which in preclinical studies was associated with limiting opioid analgesia and with promoting opioid-induced respiratory depression and constipation. Given its pharmacokinetic, tolerability and efficacy profile in our Phase 1 and Phase 2a/b trials, we believe that both the inpatient and outpatient settings could be appropriate for TRV130 use. The current focus of our clinical trials is on surgical patients. We believe offering superior analgesia or reducing the adverse side effects typically associated with the activation of the µ-opioid receptor will position TRV130, if approved, to more effectively treat moderate to severe acute pain than currently available µ-opioid therapies. We have an issued U.S. patent that covers TRV130, compositions comprising TRV130 and methods of using TRV130 and this patent is expected to expire no earlier than 2032.
Disease
According to data from IMS Health, in 2013 there were approximately 47 million hospital inpatient stays and outpatient visits during which reimbursement claims for injectable opioids were made, 20 million of which involved a surgical procedure. In terms of the total potential market opportunity, the World Health Organization estimates that over 230 million major surgical procedures are performed each year worldwide. According to Life Science Intelligence, a market research firm, over 30 million inpatient surgical procedures and 42 million outpatient or ambulatory surgical procedures were performed in the United States in 2013. According to the European Commission, about 30 million hospital inpatient surgeries are performed collectively in France, Germany, the United Kingdom, Italy and Spain each year. Accordingly, we believe that there is a large potential commercial opportunity for TRV130 in the treatment of surgical pain, if approved.
Treatment options for moderate to severe, acute postoperative pain
The typical treatment paradigm in developed markets for management of moderate to severe, acute postoperative pain is to initiate injectable or IV pain medication in the preoperative or immediate postoperative period to provide rapid and effective pain relief. As soon as it is safe and practical, a transition is typically made to oral pain medication, allowing patients to take medication home with them.
Opioid analgesics like morphine, fentanyl and hydromorphone are mainstays of pain treatment in the immediate postoperative period. Despite the development and adoption of guidelines for the management of postoperative pain and the extensive use of current treatments, significant unmet need remains. In a 2012 survey of 300 surgical patients in the United States, over 80% of patients reported postoperative pain after the first analgesic medication had been administered, and 40% of this pain was reported to be moderate or severe. The effectiveness of currently available μ-opioid agonists is limited in part because their doses are limited by severe side effects such as respiratory depression, nausea and vomiting, constipation and postoperative ileus, or POI, which is a condition that most commonly occurs after surgery involving interruption of movement of the intestines in which the bowel enters spasm and stops passing food and waste.
A recent survey we conducted in a sample of 72 U.S. surgeons and anesthesiologists suggests that the most important attribute driving physicians’ choice of an IV opioid is analgesic efficacy. In the same survey, respondents stated that injectable non-opioid analgesics are currently used to supplement IV opioids for post-surgical pain management in about 60% of hospital inpatient cases. These drugs, such as IV non-steroidal anti- inflammatory drugs, or NSAIDs, IV acetaminophen or local anesthetics such as bupivacaine, have their own potential side effects in the cardiovascular and gastrointestinal, or GI, systems as well as the liver. We estimate that recently introduced branded versions of these drugs can add $50 to $300 per patient per day to the cost of managing patients with moderate to severe postoperative pain in the United States.
None of these non-opioid analgesic approaches has displaced the use of opioid analgesics as the cornerstone of IV therapy for acute moderate to severe pain. However, the level of analgesic efficacy achievable with opioid medicines is limited as a result of dose-limiting side effects. Opioid-related side effects, including respiratory depression, nausea and vomiting, and constipation, can limit opioid dosing and may contribute to inadequate pain relief reported by patients and physicians with currently prescribed opioids:
· Morphine, fentanyl and hydromorphone are all associated with reduced respiratory rate and reduced tidal volume, which is the amount of air inhaled or exhaled in a single breath. Although serious complications or deaths from opioid-induced respiratory depression are rare, with our estimate being about 80,000 cases per year in U.S. hospitals, fear of respiratory depression represents a major barrier to the effective use of opioids in the management of postoperative pain because physicians are cautious about increasing dose. We believe this contributes to the limited effectiveness of pain relief with current IV opioids.
· In several published surveys, patients faced with surgery list the avoidance of postoperative nausea and vomiting, or PONV, as a leading concern. PONV occurs in approximately one third of surgical patients following treatment with IV opioids. We believe that there are over 5 million cases of opioid- induced PONV annually in U.S. hospitals for inpatients alone. The constipating effects of opioid drugs are also problematic and costly for surgical patients, who are typically not considered ready for discharge until they have had a meal or a bowel movement. POI is a condition in which the bowel enters spasm and stops passing food and waste, which most commonly occurs after surgery involving interruption of movement of the intestines. POI is exacerbated by anesthetics and opioid analgesics, and occurs in at least 10% of patients following invasive abdominal procedures. We believe that opioid-induced PONV, opioid-induced constipation, and POI together add approximately $5 billion to the cost of hospital inpatient post-surgical recovery in the United States annually.
Key differentiating attributes of TRV130
We believe that TRV130 may offer several potential advantages over existing opioid treatments for postoperative pain, any of which may contribute to higher levels of pain relief for TRV130 compared to these drugs. These potential advantages are as follows:
· Efficacy
· Highly effective. Based on top-line data from our Phase 2a/b clinical trial of TRV130 for treatment of postoperative pain following bunionectomy, a 3mg dose of TRV130 administered every three hours showed statistically superior analgesic efficacy over the 48-hour trial period compared to 4 mg of morphine administered every four hours Additionally, 2mg and 3mg doses of TRV130 demonstrated statistically superior analgesic efficacy compared to 4 mg of morphine in the first three hours of dosing, when pain was most severe. There were no serious adverse events reported in the trial, which we believe suggests that these levels of pain relief can be achieved safely. Over the 48-hour trial period, the tolerability of TRV130 at doses of 2 mg and 3 mg administered every three hours was similar to that of 4 mg of morphine administered every four hours. At the highest dose tested, TRV130 was associated with a mean change from baseline pain score of approximately seven (severe pain) to approximately one (mild pain) by the first data collection point five minutes after dosing. By contrast, a standard dose of morphine was associated with a change from the same baseline to a score of approximately five (moderate pain). These data are complementary to data from our Phase 1b trial in healthy subjects using an evoked-pain model, in which 3 mg of TRV130 showed superior analgesia compared to a 10 mg dose of morphine and produced less respiratory depression, less severe nausea and less frequent vomiting compared to morphine. If future pivotal trials of TRV130 continue to provide evidence of an improved therapeutic profile with respect to key safety and tolerability concerns, we believe that TRV130, if approved, may represent an improvement over unbiased µ-opioid agonists, which are the current standard of care.
· Fast acting. In preclinical studies, TRV130 delivered maximal efficacy at only five minutes after dosing. In our Phase 1 clinical trial, we also observed full pharmacodynamic response in the form of pupil constriction in humans at ten minutes after dosing. Pupil constriction is a well-established biomarker for the analgesic efficacy of opioid drugs. We also observed full analgesic effect in the Phase 1b evoked-pain model at the first practical data collection point of 10 minutes after dosing, and in our Phase 2a/b postoperative pain trial we observed maximum analgesia five to 15 minutes after dosing. If our pivotal clinical trials confirm this rapid time to peak effect, we believe the market potential of TRV130, if approved, could be broadened into the peri-operative pain market where fentanyl is commonly used today.
· Drug safety and tolerability
· Reduced respiratory depression risk. In a Phase 1b clinical trial in healthy subjects using an evoked-pain model, TRV130 showed less respiratory depression compared to a10 mg dose of morphine and delivered superior analgesia. In a preclinical proof of concept study, TRV130 showed less respiratory depression at equivalent analgesic doses compared to morphine.
· Reduced PONV. In our Phase 1b clinical trial in healthy subjects using an evoked-pain model, subjects treated with TRV130 showed less severe nausea and less frequent vomiting at a dose eliciting greater analgesia compared to a high dose of morphine. This was consistent with our Phase 1 data in which TRV130 showed no nausea or vomiting at doses eliciting equivalent or greater pupil constriction compared to high doses of morphine or fentanyl that would be expected to result in a 20% to 30% incidence of nausea and vomiting. A reduction in PONV, if supported by future clinical trials, would be a meaningful advantage for physicians, patients and payors.
· Reduced POI and constipation. If we are able to demonstrate its safety and efficacy in clinical trials, in the absence of negative GI side effects, we believe TRV130, if approved, would be an attractive treatment option for patients. In preclinical studies, TRV130 caused
significantly less constipation compared to morphine at doses delivering equivalent analgesia. If these potential benefits translate to the clinical setting, and TRV130 is approved, we believe it could offer the possibility of meaningful cost savings to hospitals.
Clinical experience
We have had an active IND for TRV130 for moderate to severe acute pain with the FDA since January 2012. Since then, we have completed our Phase 2 a/b clinical trial of TRV130 in postoperative pain in 333 patients, four other clinical trials in 121 healthy subjects and one part of a two-part multiple- ascending dose trial in healthy volunteers, the second part of which is ongoing. These trials include:
Phase 2a/b trial of TRV130 in acute postoperative pain following bunionectomy.
The aim of our Phase 2a/b clinical trial was to evaluate TRV130’s efficacy and tolerability in the management of postoperative pain using morphine as a benchmark. The trial was a multicenter, randomized, double-blind, placebo- and active-controlled, multiple dose, adaptive trial in 333 women and men undergoing a primary unilateral first-metatarsal bunionectomy surgery at four sites in the United States. Patients were randomized after surgery to receive TRV130, morphine or placebo to manage their pain. Pain intensity was measured using validated numeric rating scales ranging from ten (most severe pain) to zero (no pain) at multiple time points up to 48 hours. Based on these scales, analgesic efficacy was assessed with a time-weighted average change in pain score over 48 hours—a well established measure of changes in the intensity of pain over time and an FDA-recommended endpoint for pain studies. The trial was conducted in two parts, with the goal of providing information on efficacy and dose- and interval-ranging and furthering the differentiation of TRV130 compared to morphine. In the first part, a pilot phase, patients were randomized to receive one of four doses of TRV130 (1 mg, 2 mg, 3 mg or 4 mg), morphine or placebo, all given at four hour intervals. In the second part of the trial, an adaptive phase, eight cohorts of approximately 25 patients each were randomized successively to one of two adaptive doses of TRV130 given every three hours, morphine given every four hours, and placebo given every three or four hours hours in a double-blind, double-dummy fashion. In this adaptive phase, doses of 0.5 mg, 1 mg, 2 mg and 3 mg of TRV130 were evaluated. Rescue medication consisting of acetaminophen or ketorolac was used in all groups. In total, 141 patients were treated in the pilot phase and 192 patients were treated in the adaptive phase. The second part of the trial was originally planned to include ten cohorts of 25 patients each, but after progressing through the pilot phase and eight of the ten planned cohorts in the second phase, we elected to close enrollment in the trial following a pre- specified interim analysis because the trial had met its objectives.
We recently announced top-line data from this trial. At doses of 2 mg and 3 mg of TRV130 administered every three hours, the trial achieved its primary endpoint of statistically greater pain reduction than placebo for 48 hours, which we believe demonstrates proof of concept for TRV130. Over the 48-hour trial period, the 3mg dose of TRV130 administered every three hours also showed statistically superior analgesic efficacy compared to the 4mg dose of morphine administered every four hours. Additionally, in the first three hours of dosing, when pain was most severe, the 1 mg, 2 mg and 3 mg doses of TRV130 demonstrated superior analgesic efficacy in the trial compared to placebo, and the 2 mg and 3 mg doses of TRV130 demonstrated superior analgesic efficacy compared to the 4 mg dose of morphine.
There were no serious adverse events reported in the trial. Both the 2mg and 3 mg doses of TRV130 showed overall tolerability over the 48-hour trial period similar to the 4 mg dose of morphine administered every four hours. Adverse events attributable to TRV130 were largely opioid-related, with the most frequently reported events being dizziness, headache, somnolence, nausea, vomiting, flushing and itching. Adverse effects were generally dose-related.
These top-line results of the adaptive phase of the trial are summarized in more detail below:
· Primary endpoint—TRV130 compared to placebo over 48 hours. Over 48 hours, doses of 2 mg and 3 mg of TRV130 administered at three hour intervals achieved statistically more reduction in pain intensity compared to placebo administered at three or four hour intervals. The 2 mg dose of TRV130 reduced the time-weighted average pain score by 1.4 points more than placebo and the 3 mg dose of TRV130 reduced the time-weighted average pain score by 2.4 points more than placebo . These results were statistically significant, with one-sided p-values of 0.0024 and less than 0.0001, respectively, for the 2 mg and 3 mg doses of TRV130. P-value is a conventional statistical method for measuring the statistical significance of clinical results. A p-value of 0.05 or less represents statistical significance, meaning that there is a 1-in-20 or less likelihood that the observed results occurred by chance. The mean baseline pain rating was approximately seven out of ten, a pain level considered severe.
· TRV130 compared to morphine over 48 hours. Over 48 hours, 3 mg of TRV130 administered at three hour intervals achieved statistically more reduction in pain intensity compared to 4 mg of morphine administered at four hour intervals, reducing the time-weighted average pain score compared to placebo by 1.0 point more than morphine. This result was statistically significant, with a one-sided p-value of 0.014.
Figure 3 summarizes these results from the adaptive phase of the trial, comparing the least squares mean time-weighted average pain intensity difference over the 48-hour trial period for the four doses of TRV130 and morphine, each compared to placebo.
Figure 3: Pain relief from TRV130 and morphine compared to placebo over 48 hours

· TRV130 compared to placebo and morphine over three hours. When study pain was most severe, during the first three hours after the initial dose, TRV130 at 1 mg, 2 mg and 3 mg showed statistically more reduction in pain intensity compared to placebo, reducing the time-weighted average pain score by 1.0 point, 2.4 points and 3.0 points, respectively, more than placebo and with one-sided p-values of 0.021, less than 0.0001 and less than 0.0001, respectively. Likewise, TRV130 at 2 mg and 3 mg showed statistically more reduction in pain intensity compared to 4 mg of morphine, reducing the time-weighted average pain score by 1.2 points and 1.8 points, respectively, more than morphine, with one-sided p-values of 0.0029 and less than 0.0001, respectively. The 3 mg dose of TRV130 achieved a reduction in least squares mean pain intensity of approximately six points, with notable efficacy at five minutes, the first pain intensity assessment after dosing.
Figure 4 summarizes these results from the adaptive phase of the trial, comparing the mean pain score from one to ten at various measurement points over the first three hours after the initial dose of placebo, 4 mg of morphine or one of the four doses of TRV130.
Figure 4: Pain intensity for TRV130, morphine and placebo over first three study hours

· Patient-reported peak pain relief after first dose. Consistent with these findings, more patients reported statistically greater peak pain relief during the first three-hour dosing period for 2 mg and 3 mg doses TRV130 compared to 4 mg of morphine, with p-values of 0.005 and less than 0.0001, respectively. Of patients receiving 1 mg, 2 mg or 3 mg of TRV130, 13%, 31% and 52%, respectively, reported complete peak pain relief during this period compared to 0% and 8% of patients receiving placebo and 4 mg morphine, respectively.
Figure 5 summarizes these results from the adaptive phase of the trial, indicating the percentage of responding patients taking placebo, morphine or one of the four doses of TRV130 reporting various levels of peak pain relief, from no or little relief to complete relief, during the first three-hour dosing period.
Figure 5: Level of peak pain relief from TRV130, morphine and placebo over first three study hours

The efficacy demonstrated by TRV130 in the first three hours was reproduced upon repeat dosing, leading to sustained differences from placebo at both the 2 mg and 3 mg doses (Figure 6). During the final 24 hours of the trial, pain intensity decreased in patients receiving placebo, which we believe reduced the amount of efficacy that could be measured for TRV130.
Figure 6: Pain intensity for TRV130, morphine and placebo over 48 hours

As noted above, there were no serious adverse events reported in the trial. Adverse events were generally dose-related. Adverse events attributable to TRV130 were largely opioid-related, with the
most frequently reported events, representing a greater than 10% incidence in any group, being dizziness, headache, somnolence, nausea, vomiting, flushing and itching, as reflected below in Figure 7.
Figure 7: Spontaneously reported opioid-related adverse events

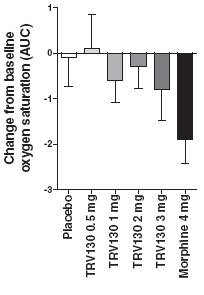
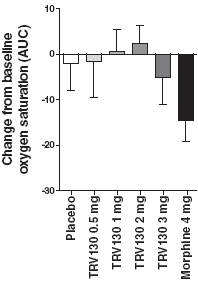
In addition, all four doses of TRV130 showed trends of less respiratory depression than morphine, as measured by oxygen saturation, after three hours following the first dose (hours 0-3) and after 24 hours following the first dose, the last time of oxygen saturation measurement (hours 0-24), as shown in Figure 8.
Figure 8: Change from baseline in oxygen desaturation levels from 0-3 and 0-24 hours
|
hours 0-3 |
|
hours 0-24 |
|
|
|
|
|
|
|
|
Phase 1b proof of concept exploratory trial in healthy subjects using an evoked-pain model
The aims of this trial were to characterize the analgesic efficacy and safety and tolerability of a single dose of TRV130 as compared to a single 10 mg dose of morphine. We employed a double-blind, five-period crossover design with 30 healthy male subjects each randomized to receive a 2-minute infusion of three dose levels of TRV130 (1.5 mg, 3.0 mg and 4.5 mg), 10 mg of morphine, and placebo in random order. We used an evoked-pain model, the cold pain test, to evaluate the analgesic effects of TRV130. The cold pain test is an established model to evaluate opioid effectiveness. We measured time to hand removal, or latency, from a temperature-controlled cold water bath. We used visual analog scale measurements of nausea and measured respiratory depression through ventilatory response to hypercapnia, another well-known experimental model.
At both the 3.0 mg and 4.5 mg doses, TRV130 showed superior efficacy as compared to a 10 mg morphine dose that was statistically significant with a p-value of less than 0.05 at the ten and 30 minute time points after dosing. The durability of the analgesic effect was similar to morphine as shown in Figure 9. In addition, the time to peak effect was more rapid than morphine.
Overall, TRV130 was well tolerated in the trial. Subjects receiving TRV130 showed less severe nausea and less frequent vomiting at the 1.5 mg and 3.0 mg doses as compared to a 10 mg dose of morphine. TRV130 also showed less respiratory depression compared to morphine, measured as minute volume, or MV, area under the curve over 4 hours as shown in Figure 10. MV is a product of respiratory rate and tidal volume, or the amount of air exhaled in a single breath, and thereby captures the body’s ability to expel carbon dioxide. The reduction in respiratory depression was statistically significant as compared to a 10 mg morphine dose with a p-value of less than 0.05 at all TRV130 doses. The 3.0 mg dose of TRV130 therefore demonstrated superior efficacy, less severe nausea, less vomiting and less respiratory depression in this trial as compared to 10 mg morphine, suggesting that TRV130, if approved, may have a better analgesic profile compared to existing unbiased µ-opioid agonists.
Figure 9: Analgesic effect of TRV130 as compared to
morphine in an evoked-pain model
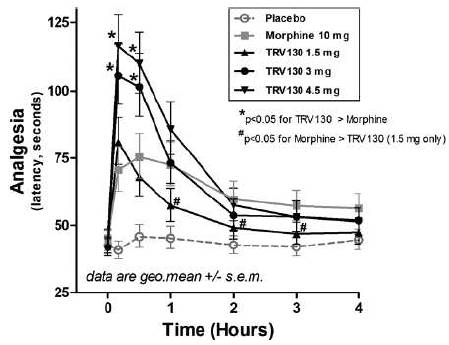
Figure 10: Less respiratory depression with TRV130 as compared to morphine

Three part phase 1 clinical trial in healthy subjects
The primary objectives of this trial were to evaluate the pharmacokinetics and tolerability of TRV130. We also obtained pharmacodynamic data by measuring pupil constriction. At historically efficacious doses, morphine and fentanyl cause approximately 1 to 2 mm of pupil constriction.
Based on the pharmacokinetics data from these trials, we expect TRV130, if approved, could be administered by IV bolus, or continuous infusion, including by way of patient-controlled analgesic device, making it potentially convenient and easy to use for postoperative pain. Specific pharmacokinetic data obtained from these trials is highlighted below:
· TRV130 showed a dose-dependent increase in exposure.
· In vitro data suggest that TRV130 is metabolized by at least two liver enzymes: CYP2D6 and CYP3A4. Approximately 2% to 21% of the population has low levels of CYP2D6 activity. In Part B of the trial, we evaluated TRV130 in a group of these poor metabolizers in order to understand whether dose adjustments will be required in this group. The maximum TRV130 plasma concentration in this group was in the upper range of that observed in non- poor metabolizers, suggesting that the poor metabolizers should exhibit similar tolerability to non-poor metabolizers. There was a reduction in clearance by approximately 50% in the poor metabolizers suggesting that a lower frequency of dosing may be required to offer effective pain relief.
· Reducing infusion time when administering TRV130 as a bolus in Part C of the trial did not significantly alter the exposure, suggesting that TRV130 could be administered as an intermittent bolus infusion without compromising drug exposure.
Overall, TRV130 was well tolerated. In Part A of the Phase 1 clinical trial, when TRV130 was administered as a one-hour infusion, there was no nausea or vomiting reported at doses up to 4 mg/hr that produced a reduction in pupil diameter of approximately 2.5 mm. When the dose was increased to 7 mg/hr, four subjects receiving TRV130 experienced nausea and four experienced vomiting, thus establishing the non-tolerated dose.
In Part A of this Phase 1 clinical trial in healthy subjects, one subject who received 0.25 mg/hr TRV130 experienced a severe episode of vasovagal syncope during which he fainted and his pulse stopped, which were classified as serious adverse events. He recovered without medical intervention and experienced no known adverse consequences from this event. Certain potential triggers of vasovagal syncope were removed from the trial protocol, and dose escalation proceeded up to 7 mg/hr (28-fold higher than the 0.25 mg/hr dose at which the syncope occurred). No additional vasovagal syncope events were reported in the trial or in any other TRV130 trial.
In Part C of the trial, TRV130 was administered to six subjects with each subject receiving on successive days a 1.5 mg dose with an infusion time of 30 minutes, 15 minutes, five minutes and one minute. TRV130 was well tolerated with pupil constriction of approximately 1 mm. We used these data to design a further intravenous bolus trial as described below to evaluate higher bolus doses.
Phase 1 IV bolus trial
In a follow-up trial with bolus doses of 2.0, 3.0 or 3.5 mg administered over two minutes, TRV130 was well tolerated up to 3.5 mg (the highest dose in the trial). One subject experienced mild nausea when 3.5 mg TRV130 was given. No nausea was reported at the lower doses. When 3.5 mg of TRV130 was administered, pupil diameter decreased by approximately 2 mm from baseline, in line with high-dose morphine or fentanyl.
Phase 1 drug-drug interaction trial
To further explore TRV130’s metabolic profile in the clinic, a single dose of TRV130 was administered to healthy subjects in conjunction with ketoconazole, a CYP3A4 inhibitor. TRV130 was safe and generally well-tolerated in the presence of ketoconazole and there was no clinically meaningful change in TRV130 exposure.
Phase 1 two-part multiple ascending dose trial
The first part of this trial evaluated the maximum tolerated dose and pharmacokinetics of TRV130 when multiple doses were given, and also measured pharmacodynamic effects of TRV130, including pupil constriction and cold pain test analgesia. The results of this part of the trial were consistent with earlier trials, showing reproducible pharmacokinetics and pharmacodynamic effects of TRV130. Safety and tolerability of TRV130 were also consistent with earlier trials, and no unexpected adverse effects were observed. The second part of the trial, which is testing the effects of subjects’ metabolic capacity on potential duration of action of TRV130, is in progress and we expect to release data from this part of the trial by the end of 2014.
Preclinical studies
In preclinical models, TRV130 showed analgesic efficacy comparable to morphine but reached peak effect more quickly than morphine. Time to peak effect occurred within five minutes for TRV130 compared to 30 minutes for morphine. TRV130 had a significantly improved therapeutic index, compared to morphine, of analgesia to respiratory depression, measured as blood carbon dioxide, or pCO2, and analgesia to constipation, measured using two GI motility assays. This was consistent with basic research studies in which morphine given to β-arrestin2 knockout mice showed increased analgesia, less respiratory depression and less constipation than morphine given to wild-type mice.
Clinical development strategy
We believe that the early clinical and preclinical data generated suggest that TRV130 may have superior analgesia with fewer dose-limiting safety and tolerability disadvantages compared to existing opioid analgesics. If confirmed in further trials, we believe that this profile will justify TRV130, if approved, as a preferred opioid analgesic for the intravenous treatment of moderate to severe acute pain.
We are also conducting a Phase 1 clinical trial to explore the safety, tolerability, pharmacokinetics and pharmacodynamics of multiple ascending doses of TRV130 in healthy volunteers. This trial consists of two parts. We have completed the first part of this trial, in which we observed safety, tolerability, pharmacokinetics and pharmacodynamics after repeat-dosing consistent with our expectations from earlier single-dose trials. The second part of the trial, which is testing the effects of subjects’ CYP2D6 metabolic capacity on TRV130 potential duration of action, is ongoing.
Separately, we are conducting an additional Phase 1 trial to evaluate the absorption, metabolism and excretion of TRV130 in healthy subjects. This additional Phase 1 trial is ongoing and we expect it to conclude in the first half of 2015.
We expect to initiate a second Phase 2 clinical trial of TRV130 in December 2014 with the goal of evaluating analgesic efficacy following a soft tissue surgery and exploring TRV130’s safety and tolerability profile compared to morphine. We expect to report top-line data from this trial in mid-2015. This trial will employ as-needed dosing to broaden dosing information beyond the fixed-interval dosing used in the bunionectomy trial. In this trial, TRV130, morphine or placebo will be administered as an initial loading dose followed by delivery of on-demand doses via a patient-controlled analgesia device. Approximately 200 patients who have undergone uncomplicated, elective abdominoplasty surgery will be enrolled in the trial, with approximately 40 receiving placebo, 80 receiving TRV130 and 80 receiving morphine. The primary endpoint of the trial will be the efficacy of TRV130 compared to placebo over 24 hours, which may serve as a registration endpoint in Phase 3 development. In parallel with the Phase 2 abdominoplasty clinical trial, we intend to commence Phase 3 preparations for TRV130, with the goal of initiating our first of two Phase 3 clinical trials in the first quarter of 2016. We expect that the Phase 2 abdominoplasty trial, if the data are promising, along with data from the Phase 2a/b clinical trial of treatment of postoperative pain following bunionectomy, would support Phase 3 development in soft and hard tissue pain, which we believe would be required by the FDA for approval of TRV130 for broad use in moderate to severe pain. In addition, we plan to complete other clinical trials that would support Phase 3 clinical development. Core pivotal studies in the Phase 3 program could closely resemble the Phase 2 trials, with additional trials exploring the therapeutic potential more broadly. This approach may enable an NDA for a broad acute moderate to severe pain label and may also guide commercial positioning.
We plan to initially target TRV130 for the treatment of moderate to severe, acute postoperative pain where IV administration is preferred. If our trials for this indication are successful, we believe there may be additional opportunities to expand the target indications in subsequent trials. Other potential patient populations for the eventual use of TRV130 include perioperative use (including sustained dosing for the most painful surgery types); non-surgical hospitalized patients such as burn victims (including debridement); end-of-life palliative care; emergency service trauma care; renal stones; sickle cell crises and military applications. We may also explore other dosage forms, such as transmucosal or transdermal administration for breakthrough or chronic pain, respectively, in additional separate trials.
Commercialization
We plan to develop and commercialize TRV130 for IV administration ourselves, if approved. We intend to build acute care commercial capabilities, initially in the United States, and to retain full U.S. rights. In the United States, sales of injectable analgesics have increased by more than 70% between
2011 and 2013 to approximately $660 million, according to IMS health. We may seek collaborators for commercializing TRV130 outside the United States after the availability of full Phase 2 data to offset risk and preserve capital.
Manufacturing
We have carried out TRV130 drug substance synthesis, performed by a third party, at a scale up to 2 kg per batch. Phase 3 synthetic process development and regulatory compliance studies are in progress. Currently we manufacture drug substance and drug product, both with third parties, at single sites, but we plan to qualify additional sites in connection with any Phase 3 trials.
Competition
If TRV130 is approved for IV treatment of moderate to severe acute pain, it will compete with widely used, currently marketed opioid analgesics, such as morphine, hydromorphone and fentanyl. The effectiveness of these agents is limited by well-known adverse side effects, such as respiratory depression, nausea and vomiting, constipation and POI. TRV130 may also compete against Ofirmev, marketed by Mallinckrodt plc, and Exparel, marketed by Pacira Pharmaceuticals, Inc., which are reformulations of existing products and are typically used in combination with opioids.
We are aware of a number of products in development that are aimed at improving the treatment of moderate to severe, acute postoperative pain while reducing undesirable side effects. The most advanced product candidates are reformulations of existing opioids, such as a fentanyl ionophoresis patch, in development by The Medicines Company, and sufentanil nanotab, in development by AcelRx. In addition, Cara Therapeutics Inc. is developing an IV and oral peripherally restricted κ-opioid receptor agonist, which will likely be used in combination with opioids.
Intellectual property
Our TRV130 patent portfolio is wholly owned by us. The portfolio includes one issued U.S. Patent, which claims among other things, TRV130, compositions comprising TRV130 and methods of using TRV130. The portfolio also includes one pending U.S. patent application claiming TRV130, other compounds and/or methods of making or using the same. If issued, the pending U.S. application is predicted to expire no earlier than 2032, subject to any disclaimers or extensions. A related Patent Cooperation Treaty, or PCT, application was filed and national patent applications have been filed in South Korea, the European Patent Office, the Eurasian Patent Office, Australia, Brazil, Canada, Israel, India, Japan, China, and New Zealand. Any patents resulting from these national patent applications, if issued, are expected to expire no earlier than 2032, subject to any disclaimers or extensions.
TRV734
TRV734 is a small molecule G protein biased ligand at the µ-opioid receptor, which we are developing as a first-line, orally administered compound for the treatment of moderate to severe acute and chronic pain. Like TRV130, TRV734 takes advantage of a well-established mechanism of pain relief by targeting the µ-opioid receptor, but does so with enhanced selectivity for the G protein signaling pathway, which in preclinical studies was linked to analgesia, as opposed to the β-arrestin signaling pathway, which in preclinical studies was associated with side effects. Subject to successful preclinical and clinical development and regulatory approval, we believe TRV734 may have an improved efficacy and side effect profile as compared to current commonly prescribed oral analgesics, such as oxycodone. We have filed patent applications covering TRV734 and methods of using TRV734.
Data from IMS Health show that opioid drug sales across the United States, Europe and Japan were approximately $11 billion in 2013. Despite widespread use, there are significant limitations to existing therapies with respect to efficacy, constipation, nausea and vomiting and respiratory depression. Dose-limiting side-effects may translate into inadequate pain control. The constipating effects of chronic opioids are particularly problematic because they do not lessen over time, while efficacy does
tend to reduce over time for a particular dose level. Numerous approaches have been attempted to mitigate constipation. Laxatives, peripherally restricted opioid antagonists, such as naloxegol, methylnaltrexone and alvimopan, and multimodal analgesia, such as the opioid/SNRI tapentadol, are only partially effective and can raise problematic new side effects in an attempt to mitigate the adverse effects of opioid analgesics. Based on the very large market and substantial limitations confronting current analgesics, we believe a new opioid with a more precisely targeted mechanism of action and an improved therapeutic profile could provide a significant product opportunity in the acute and chronic pain markets.
Clinical experience
We have had an active IND for TRV734 since January 2014. In 2014, we completed our first Phase 1 trial of TRV734, which tested single ascending doses and the relative bioavailability of oral TRV734 in healthy subjects. In this trial, we observed that TRV734 was pharmacologically active at a range of safe and well-tolerated doses. We believe that the data from this trial suggest that TRV734 provides dose-related exposure, speed of onset, and duration of action suitable for treating moderate to severe acute pain. TRV734 elicited dose-related increases in plasma concentrations, with peak plasma concentrations reached approximately one hour after dosing and a terminal half-life consistent with use for treating acute pain. Pupil constriction indicative of analgesia was observed at doses of 80 mg and higher, and mild-to-moderate adverse effects were reported at the maximum explored dose of 250 mg. We believe this suggests that the analgesic efficacy of TRV734 may be separable from opioid-related adverse effects. No clinically significant changes in vital signs, laboratory values or ECG parameters, and no severe or serious adverse events, were reported.
Preclinical data
TRV734 has shown a similar profile to TRV130 in in vitro and in vivo studies. It is highly selective for the µ-opioid receptor, where, like the most powerful opioid analgesics, it is a strong agonist of G protein coupling. TRV734 is distinct from those analgesics in its very weak recruitment of β-arrestins to the µ-opioid receptor. In our preclinical studies, TRV734 showed analgesic effects in preclinical pain models similar to oxycodone and morphine. In the same studies, TRV734 caused less constipation compared to equivalently analgesic doses of oxycodone and morphine. TRV734 is active after oral administration in mice and rats, has high oral bioavailability and has been well tolerated in non-human primates.
Based on these data and data for TRV130, we believe that TRV734 may offer an improved efficacy profile as compared to current opioid therapies or equivalent efficacy with an improved GI tolerability and respiratory safety profile.
Clinical development strategy
We intend to seek a collaborator with experience in developing and commercializing controlled-substance therapeutics in chronic care pain markets thereby leveraging their expertise while still retaining rights to commercialize TRV734 in acute care settings, including hospitals, in the United States.
We have completed enrollment in a second Phase 1 clinical trial, which is a multiple ascending dose trial evaluating the safety, tolerability, pharmacodynamics and pharmacokinetics of TRV734 given as a single dose and as multiple ascending doses in healthy volunteers. The trial is designed to enable Phase 2 development, and is being conducted in two parts with approximately 70 healthy volunteers randomized to participate in the trial. The first part will assess the safety, tolerability, pharmacodynamics and pharmacokinetics of single 125 mg doses of TRV734 in an open-label, randomized, three-period crossover trial in which subjects are fasted, fed a standard meal or fed a high-fat meal. This portion of the trial is designed to explore how changes in absorption may modify the performance of TRV734 and to identify the best administration paradigm for the second part of the
trial. The second part of the trial will assess the safety, tolerability, pharmacodynamics and pharmacokinetics of multiple ascending doses of TRV734 in a double-blind, double-dummy, randomized, active- and placebo-controlled adaptive trial. Oxycodone immediate release 10 mg is used as a benchmark for a variety of pharmacodynamic measures intended to evaluate the analgesic and adverse effect profile of TRV734. We expect to release top line data for both parts of the trial early in the first quarter of 2015. We plan to continue development of TRV734 by conducting activities to support Phase 2 clinical trials. We also plan to seek a collaboration with a third party to support later-stage development and commercialization efforts.
Manufacturing
We have carried out TRV734 drug substance synthesis, performed by a third party, at a scale up to 2 kg per batch. A formulated tablet is being developed for Phase 2 clinical trials.
Intellectual property
Our TRV734 patent portfolio, which is wholly owned by us, includes one pending U.S. patent application claiming TRV734, other compounds and/or methods of making or using the same. If issued, we expect the pending U.S. application will expire no earlier than 2032, subject to any disclaimers or extensions. A related PCT application was filed and national patent applications have been filed in South Korea, the European Patent Office, the Eurasian Patent Office, Australia, Brazil, Canada, Israel, India, Japan, China, and New Zealand. Any patents resulting from these national patent applications, if issued, are predicted to expire no earlier than 2032, subject to any disclaimers or extensions.
TRV250
In November 2014, we identified a new product candidate, TRV250, a small molecule G protein biased ligand of the δ-opioid receptor. Based on the initial profile of TRV250, we anticipate focusing our initial development efforts on the treatment of treatment-refractory migraine headaches. According to Decision Resources, a healthcare consulting company, the acute episodic migraine market encompassed approximately 12 million drug-treated patients in 2013 in the United States, representing approximately $2.2 billion of sales. We estimate that approximately 20% to 30% of these patients either do not respond to or cannot tolerate the market-leading triptan drug class, and an additional 30% would benefit from improved efficacy compared to these drugs.
We believe our preclinical data support targeting the δ-opioid receptor for the treatment of CNS disorders. Prior approaches to modulate this receptor have been limited by a significant risk of seizure associated with this target. By contrast, TRV250 is a potent δ-opioid receptor ligand that displayed strong efficacy in animal models of migraine and other CNS disorders with reduced seizure liability through selectively activating G protein coupling without engaging β-arrestin. These in vivo data are further supported by data for δ-agonists in β-arrestin knockout mice suggesting that β-arrestin plays a role in seizures. We intend to advance TRV250 into preclinical studies in 2015 designed to support our submission of an IND to the FDA. We also intend to seek a collaborator for TRV250 with CNS development and worldwide commercialization expertise, while potentially retaining commercialization rights in the United States. Phase 1 clinical trials could include electroencephalogram studies to specifically assess seizure liability.
We have two provisional patent applications directed to compounds that modulate the δ-opioid receptor. One of the applications is solely owned by us and the other is co-owned by us and Ligand Pharmaceuticals Incorporated. We have an exclusive worldwide, paid up, royalty-free license to any compound or method of use in the field of pharmaceuticals disclosed in the Ligand co-owned application. We expect that any compound that modulates the δ-opioid receptor we choose to pursue under our development program would be covered by the application solely owned by us. These applications are eligible for worldwide filing and may be used to establish non-provisional applications that, if issued, are predicted to expire no earlier than 2035.
Cardiovascular Program
TRV027
TRV027 is a peptide β-arrestin biased ligand that targets the AT1R, inhibiting G protein signaling and activating β-arrestin signaling. We are developing TRV027 for the treatment of AHF in combination with standard diuretic therapy. In our Phase 2a clinical trial, TRV027 rapidly reduced blood pressure and preserved renal, or kidney, function, while preserving cardiac performance. We currently are enrolling patients in a Phase 2b clinical trial to evaluate the safety and efficacy of TRV027 in AHF. If our clinical development of TRV027 is successful and the product ultimately is approved by regulatory authorities, we believe TRV027 would be used as a first-line in-hospital AHF treatment. We also believe TRV027 could improve AHF symptoms, shorten length of hospital stay and potentially lower readmission rates and mortality rates after hospital discharge. U.S. patents covering the composition of matter and method of use of TRV027 have issued and are expected to expire no earlier than 2031 and 2029, respectively.
Disease
Heart failure is the inability of the heart to supply adequate blood flow, and therefore oxygen, to peripheral tissues and organs. When the heart is failing, mechanisms are triggered by the body to maintain blood pressure and tissue perfusion. One such mechanism is the activation of the renin-angiotensin system, or RAS, of which angiotensin II is a key mediator. Through angiotensin II, RAS increases blood pressure and stimulates the kidneys to retain both sodium and water. These mechanisms maintain cardiac performance in the short term, but in the longer term, the heart must pump against higher pressure, referred to as afterload, and is overstretched when filled, referred to as preload. These effects make the failing heart pump less efficiently and lead to progressive damage to the muscular tissue of the heart.
There are over 20 million people living with heart failure in the United States and Europe, according to the American Heart Association and the European Society of Cardiology. AHF, also sometimes referred to as acute decompensated heart failure, is heart failure requiring hospitalization. AHF patients present with fluid overload and severe dyspnea, a serious shortness of breath sometimes described as “air hunger,” leading to an inability to perform simple functions such as standing and walking short distances. AHF can also lead to organ dysfunction, including in the kidneys and heart. Most patients experiencing an AHF event have a worsening of existing chronic heart failure, although an estimated 25% of AHF hospitalizations represent new diagnoses of heart failure.
According to National Hospital Discharge Survey data, in the United States there were over 5 million hospital discharges in 2010 where heart failure was listed as a component of the diagnosis, over 1 million of which listed heart failure as the primary diagnosis. Based on national hospital discharge statistics from 25 countries in Europe, we estimate that there were a total of 1.6 million hospitalizations with a primary heart failure diagnosis in 2010 in those countries. Despite long hospital stays, up to approximately 50% of AHF patients remain symptomatic on discharge according to data from ADHERE, a national U.S. registry of over 100,000 patients admitted to the hospital with AHF between 2000 and 2005. In addition, the risk of readmission is 25% after 30 days and the one-year mortality rate is approximately 30%. Combined, these poor outcomes result in a substantial burden to the healthcare system. In 2012, the American Heart Association estimated the annual direct medical cost of treating heart failure in the United States to be almost $21 billion.
Current treatment options for AHF
We believe there is a significant unmet medical need for improved treatments for AHF. The current approach to treating patients with AHF involves facilitating the excretion of accumulated fluid with loop diuretics like furosemide; improving hemodynamics by reducing preload and afterload blood
pressure with vasodilators like nitroglycerin; and directly stimulating the heart to contract more forcefully with inotropes like dobutamine. None of these approaches has been robustly shown to improve patient outcomes in AHF, and each therapy has specific adverse effects that limit its clinical utility.
The mainstay of therapy for AHF is loop diuretics, such as furosemide. In AHF patients, fluid removal is important to relieve symptoms and to improve tissue oxygenation. Furosemide facilitates excretion of excess fluid, but aggressive diuresis can lead to renal dysfunction. Worsening renal function in AHF patients is associated with higher mortality and increased risk of hospital readmission. Diuretic therapy has also been shown to precipitate activation of RAS, further exacerbating the vicious cycle of heart failure.
After diuretics, IV vasodilators, such as nitroglycerin, nitroprusside and nesiritide, are the most common medications used for the treatment of AHF. These vasodilators effectively reduce blood pressure, but each is associated with undesirable side effects and other limitations. Hypotension, or low blood pressure, is the most common serious side effect of vasodilating agents. Nitroglycerin raises RAS, and its use is also hampered by rapid development of tolerance, such that the medication becomes less effective the longer that it is used. Nitroprusside is associated with possible cyanide toxicity and cannot be used without intensive monitoring, so its use is limited. Nesiritide is infrequently used, which we believe is due to uncertainties about its efficacy and safety.
In severe cases, and those characterized by very low cardiac output, physicians sometimes resort to the use of inotropes, which work by increasing cardiac contractility by mobilizing calcium but at the expense of increased oxygen consumption and risk of arrhythmia. These agents can improve symptoms in the short term but have been shown to increase mortality. In addition, these drugs are only used in patients who have AHF associated with low ejection fraction. This sub-group of AHF patients represents approximately half of all patients who present for urgent AHF treatment.
There remains an unmet need for better therapeutic approaches to treat AHF that can improve blood circulation through vasodilation, facilitate fluid excretion by the kidneys and enhance cardiac function through a novel mechanism not requiring calcium mobilization. Based on our preclinical studies and our clinical trials conducted to date, we believe TRV027 has the potential to meet this unmet need, and may prove to be more effective than currently available treatment options, reducing hospital readmission rates, mortality rates and length of hospital stay, while improving symptoms more rapidly and more completely.
Key differentiating attributes of TRV027
We believe that TRV027, when used with current standard of care, particularly loop diuretics like furosemide,will have the following potential advantages:
· Efficacy
· Targets RAS, a mechanism that is central to the disease. RAS blockade has been shown to have morbidity and mortality benefits in chronic heart failure. We believe that TRV027, if approved, could be the first therapy to bring modulation of RAS to the acute hospital setting, allowing the physician to improve blood circulation while protecting the heart and kidneys.
· Benefits the three key organ systems affected by AHF. In our preclinical studies and Phase 1b and 2a clinical trials, TRV027 has shown beneficial effects on the blood vessels, heart and kidneys. TRV027 could improve patient symptoms and outcomes by rapidly lowering afterload and preload blood pressure, sustaining cardiac output, and preserving kidney performance as a result of the lower blood pressure.
· Enhances furosemide’s effects on pulmonary capillary wedge pressure. Pulmonary capillary wedge pressure is a pharmacodynamic marker of dyspnea, a main symptom of AHF. Loop diuretics, like furosemide, facilitate excretion of excess fluid and are frequently used to manage AHF patients. Loop diuretics also activate RAS, which may compromise their ability to fully resolve symptoms, and may contribute to the estimated 50% of AHF patients who are still symptomatic at the time of discharge from the hospital. We believe that administering TRV027 in combination with furosemide may improve dyspnea directly by decreasing pressure on the heart and in the lungs and indirectly by allowing furosemide to work more effectively without the negative consequences of RAS activation.
· Drug safety and tolerability
· Favorable drug safety profile. TRV027 is a small peptide that is highly specific for the angiotensin receptor, so we believe that off-target adverse effects would not be expected. In clinical trials to date, TRV027 has been well-tolerated in healthy subjects and in patients with advanced chronic congestive heart failure, in each case at doses up to 20-fold higher than the expected efficacious dose. In preclinical toxicology studies, TRV027 had a favorable profile at doses up to 500 times the expected therapeutic dose.
· Self-limiting blood pressure effect. In our Phase 2a clinical trial, there was a dose-dependent decrease in blood pressure up to doses of 1 µg/kg/min. No further reduction in blood pressure was seen at doses up to 3 µg/kg/min. We believe that this characteristic would offer a safety advantage over current vasodilators, which can cause dangerous hypotension.
· Rapidly reversible effects on blood pressure. In our three completed clinical trials, TRV027 had a very short half-life and its effects were rapidly reversible. In the acute care setting, we believe this should allow the physician to alter the dose and avoid prolonged hypotension.
· Action specific to target pathophysiology. In our three completed clinical trials, TRV027 lowered blood pressure only in subjects with elevated measures of RAS activity, the target pathophysiology. This is important for any drug that is used in emergency rooms when the initial diagnosis may be uncertain.
Clinical experience
We have had an active IND, for TRV027 for AHF with the FDA since February 2010. Since then, we have completed three clinical trials of TRV027:
· A Phase 2a clinical trial in medically fragile subjects with advanced stable heart failure, low ejection fraction and a clinical indication for right-heart catheterization. Ejection fraction is a measure of the volume of blood pumped by the heart. Right-heart catheterization is a procedure that allows measurement of intracardiac and intravascular pressures on the side of the heart leading to the lungs. This procedure is not commonly used for the treatment of AHF patients, so this trial enabled us to profile the hemodynamic effects of TRV027 in a comparatively stable chronic heart failure population that could be considered an AHF forerunner population.
· A Phase 1b clinical trial in subjects with moderate heart failure and concomitant renal dysfunction. Selecting a stable population allowed us to directly measure renal plasma flow, or RPF, and glomerular filtration rate, or GFR, two common measures used to evaluate renal safety.
· A Phase 1 clinical trial in healthy subjects to evaluate pharmacokinetics and tolerability prior to moving into chronic stable heart failure subjects.
Phase 2a hemodynamics trial in advanced stable heart failure subjects
The primary objectives of this trial were to characterize the safety and tolerability of TRV027 in subjects with advanced stable heart failure and to measure its effects on blood circulation, also known as hemodynamics. Due to the wide dose-range available following the Phase 1 clinical trial, we elected to employ a step-wise dose titration over five hours with the dose increased to a target dose 10-fold higher than the starting dose. This highest dose was continued for nine hours as a steady state infusion, for a total infusion time of 14 hours, to evaluate the stability of TRV027’s hemodynamic effects. Reversibility of TRV027’s effects was then studied for four hours after the infusion was discontinued. Three dosing regimens were evaluated in 24 subjects: 0.1 µg/kg/min titrated up to 1 µg/kg/min; 0.3 µg/kg/min titrated up to 3 µg/kg/min; and 1 µg/kg/min titrated up to 10 µg/kg/min. In total, 14 different doses were studied across the three different dosing regimens. Nine additional subjects received placebo in a double blind manner. Based on the preclinical and Phase 1 data, we were expecting the hemodynamic effects of TRV027 to depend on elevation of RAS activity. The data were therefore analyzed based on plasma renin activity, or PRA, elevation, with high PRA subjects defined as those with PRA levels greater than 5.82 ng/ml/hr, which is the upper limit of lab normal range. PRA is an enzyme in the RAS cascade and measures RAS activity. Eleven of the 24 treated subjects had high PRA. We believe that these high PRA subjects represent a sicker population more relevant to AHF, and we anticipate that most AHF patients will have high PRA.
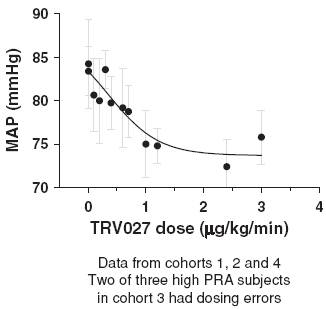
In this trial TRV027 produced a dose-related decrease in mean arterial pressure, or MAP, in subjects with elevated PRA, as shown in Figure 11. The reduction in MAP was sustained during the steady state infusion period and reversed during the washout period following the end of the infusion. This reversal of effect was statistically significant compared to both placebo and normal PRA subjects with p-values of less than 0.01 and 0.001, respectively. The decrease in MAP in the high PRA subjects compared to subjects receiving placebo in the maintenance phase was also statistically significant, with a p-value of less than 0.05.
Figure 11: Effect of TRV027 on mean arterial pressure in advanced stable
heart failure subjects with elevated PRA
We also observed evidence of pharmacologic effects on PCWP in the subjects with elevated PRA. PCWP dropped in subjects with high PRA during the titration phase and this was sustained during the
maintenance phase and reversed during the wash-out phase. The interpretation of the results in the titration and maintenance phases was complicated by a baseline drift in PCWP in the placebo group, however, the increase in PCWP when the TRV027 infusion was stopped was clear and statistically significant in high PRA compared to normal PRA subjects, with a p-value of less than 0.01, as shown in Figure 12.
Figure 12: Reversal of effect of TRV027 on pulmonary capillary wedge pressure
in advanced stable heart failure subjects

In this trial, there was no apparent change in cardiac index or heart rate observed in subjects with normal or high PRA following administration of TRV027. Cardiac index is a well accepted measurement of how well the heart is functioning as a pump by directly correlating the volume of blood pumped by the heart with an individual’s body surface area. This contrasts with the response of heart failure subjects to acute administration of the angiotensin receptor blocker, or ARB, losartan, which has been shown to decrease cardiac index in some studies.
TRV027 was well tolerated in this medically fragile population. Despite the substantial reduction in MAP in TRV027-treated high-PRA subjects, there was no apparent increase in heart rate or in levels of cystatin-C or creatinine, which are biomarkers of renal function. This suggests that the blood pressure reduction was accompanied by preservation of kidney function. This result was consistent with our observations in preclinical studies. One subject in the lowest-dose cohort in this trial experienced hypotension necessitating dose reduction and then discontinuation of the TRV027 infusion. No other TRV027-related clinically significant adverse events were reported.
Phase 1b renal safety trial in stable chronic heart failure subjects
The primary objective of this trial was to explore the pharmacokinetics and renal safety of TRV027, co-administered with furosemide, in 17 subjects with a history of heart failure and concomitant renal dysfunction. Two cohorts of six subjects and one cohort of five subjects were enrolled in this two-period crossover trial. All of the subjects had moderate heart failure and concomitant renal dysfunction.
TRV027 was administered using a standard dosing paradigm, with doses of 1.25 mg/hr, 6.25 mg/hr and 31.25 mg/hr (equivalent to 0.35 µg/kg/min, 1.74 µg/kg/min and 8.68 µg/kg/min, respectively, for a 60 kg person), without weight correction. The plasma concentrations obtained were similar to those
obtained when TRV027 was administered on a per-kg basis to subjects with normal kidney function, suggesting that a standard dosing approach with no adjustment for weight or renal impairment is appropriate, which would facilitate use in the emergency room where patients are not routinely weighed.
TRV027 was well tolerated in these renally impaired subjects. There were no TRV027-related clinically significant or serious adverse events reported. Previously published research has shown that oral furosemide administration produces a reduction in GFR that can be inhibited by blocking the effects of elevated angiotensin II. In our trial, however, there was no effect of the single dose of furosemide on GFR or RPF; therefore, it was not possible to show a renal protective effect of TRV027. The trial did, however, show that TRV027 itself preserved GFR and RPF, before and after furosemide administration. In this trial, co-administration of TRV027 did not impair furosemide’s effect on diuresis or urinary sodium excretion.
Taken together, we believe the Phase 2a and Phase 1b clinical trials in stable chronic heart failure subjects provide evidence suggesting that TRV027 may have a beneficial effect on the heart, the blood vessels and kidney function in patients suffering from AHF, consistent with the data we had obtained in preclinical studies.
Phase 1 clinical trial
The Phase 1 clinical trial was a single center, crossover trial evaluating four-hour infusions of TRV027 in 20 healthy subjects at doses ranging from 0.01 to 20 µg/kg/min. The primary objective of the trial was to evaluate the tolerability and pharmacokinetics of TRV027. TRV027 was well tolerated with no serious adverse events or clinically significant adverse events reported even at doses up to 20 times higher than the expected therapeutic dose. There was a linear increase in exposure with dose and TRV027 was rapidly cleared when the infusion was stopped, suggesting that it will potentially be easy to reverse any unexpected hypotensive effects. There was no urinary excretion of TRV027 so we do not expect any dose adjustments to be required for renal insufficiency. We believe this characteristic may make TRV027 easy to use in the emergency room.
Preclinical studies
In a paced dog animal model of heart failure, TRV027 decreased MAP and PCWP. TRV027 also increased renal blood flow and moderately increased cardiac output. In another paced dog model study, TRV027 was studied in combination with furosemide and showed additive effects on reducing PCWP, which would be consistent with beneficial effects on dyspnea in the clinic. In addition, combining the data in normal dogs, paced dogs and paced dogs treated with furosemide, we observed meaningful blood pressure decreases only in animals with elevated RAS, which is consistent with the data seen in the clinical trials and we believe provides further evidence supporting the premise that TRV027 only works in patients with the target pathophysiology. Furthermore, the dose response observed in paced dogs was consistent with that observed in subjects in the Phase 2a clinical trial.
To examine the direct effects of TRV027 on cardiac contractility, we studied the hemodynamic effects of TRV027 compared to the unbiased ARB telmisartan in normal rats using a micromanometer conductance catheter. TRV027 treatment increased cardiac contractility independent of its effects on blood pressure, as measured by end systolic pressure volume relationship, or ESPVR, a common measure of cardiac output independent of blood pressure, and it also decreased MAP. This compared to telmisartan, which similarly decreased MAP but also decreased ESPVR (see Figure 13). Telmisartan is an unbiased ARB that inhibits both the G protein and β-arrestin AT1R pathways. In addition, in in vitro studies, TRV027 stimulated cardiomyocyte contractility through a β-arrestin dependent mechanism and selectively activated a subset of downstream signaling pathways seen with the full agonist, angiotensin II.
Figure 13: Effect of TRV027 on MAP and cardiac contractility in normal rats

The mechanism by which TRV027 increased cardiac contractility in in vivo studies does not appear to involve calcium mobilization seen in currently marketed inotropes. Calcium mobilization is linked to pro- arrhythmic effects. In a study we conducted in rats, a β-arrestin biased AT1R ligand closely related to TRV027 increased contractility through a myofilament calcium sensitization mechanism, a novel mechanism of cardiac contractility that does not involve calcium mobilization. In in vivo studies, this related ligand prevented hypertrophy and prevented cardiac apoptosis, suggesting a potential cardioprotective effect. Furthermore, cardiac stress in mice induces AT1R, β-arrestin-dependent cardioprotective signaling, suggesting that AT1R β-arrestin biased ligands could be potentially cardioprotective.
Clinical development strategy
We are enrolling patients in a Phase 2b clinical trial to evaluate the safety and efficacy of TRV027 in AHF. This is a randomized double-blind, placebo controlled trial comparing TRV027 plus standard of care to standard of care alone. The primary objective of this trial is to evaluate the effects of three doses of TRV027, 1.0 mg/hr, 5.0 mg/hr and 25 mg/hr, on a composite of clinically important outcomes: mortality, worsening heart failure, hospital readmission rate, dyspnea and length of hospital stay. Our trial design contemplates that approximately 500 patients will be enrolled and randomized. We are targeting early administration of TRV027, ideally within six hours of arrival at the hospital. TRV027 will then continue to be administered for a minimum of 48 hours and up to 96 hours. We believe administration of TRV027 soon after hospital admission will improve in-hospital mortality rates and shorten length of hospital stay. We are enrolling patients with both low ejection fraction and preserved ejection fraction since RAS elevation is a key component of both conditions. This trial has enrolled over 250 patients towards the objective of 500 patients. More than 65 sites in 12 countries are now open and recruiting, and patient enrollment is expected to conclude in the third quarter of 2015. We plan to conduct an interim analysis and, depending on the outcome of that analysis, enrollment into one or more of the active dose groups may be discontinued. Since the initiation of this trial, the data safety monitoring board for the trial has reviewed safety data from the trial on two separate occasions and has recommended each time that the trial continue administering all three doses under investigation. We expect to release top-line data from this trial in the fourth quarter of 2015.
We believe that an endpoint measuring dyspnea or worsening of heart failure during hospitalization in Phase 3 clinical trials could form the basis for FDA approval of TRV027. However, we believe the FDA may be open to other well-defined benefit parameters, such as a hospitalization benefit or a patient and caregiver quality of life benefit. The composite endpoint tested in Phase 2b will facilitate our evaluation of potential alternative proposals to be discussed with the FDA at an end-of-Phase 2 meeting.
Option and License Agreements with Actavis
On May 3, 2013, we entered into an option agreement and a license agreement with Actavis plc (formerly Forest Laboratories Holdings Limited), under which we granted to Actavis an exclusive option to license TRV027. If Actavis exercises this option, the license agreement will become effective and Actavis will have an exclusive worldwide license to develop and commercialize TRV027 and specified related compounds. At our request, Actavis will consider in good faith whether to grant us the right to co-promote the licensed products in the United States under terms to be agreed upon by the parties, but it has no obligation to provide co-promotion rights to us. Actavis will be responsible for subsequent development, regulatory approval and commercialization of TRV027 at Actavis’ expense.
Under the option agreement, we will conduct, at our expense, a Phase 2b clinical trial of TRV027 in acute heart failure. The Phase 2b clinical trial will be conducted pursuant to a mutually agreed upon development plan and under the oversight of a joint development committee, which has an equal number of representatives from us and from Actavis, with operational authority during the option period retained by us, subject to Actavis’ right to assume control in certain circumstances if we fail to conduct the development activities adequately.
Actavis may exercise its option during the pendency of the Phase 2b clinical trial or during a specified time period after we deliver the data from the Phase 2b clinical trial to Actavis. During the option period, we are not permitted to negotiate for or enter into any agreement with a third party for the development and commercialization of TRV027 and its related compounds. Under specified circumstances linked to adverse changes in the market or related to the results from the Phase 2b trial of TRV027, Actavis has the right to renegotiate the terms of the license agreement. If Actavis exercises such right, we will be obligated to negotiate in good faith with Actavis for a period of time the terms of any new arrangement. If we and Actavis are unable to agree on the terms of any new arrangement, then the option agreement will terminate and for a specified period of time thereafter we may not offer a license to any third party on terms better than those last proposed either by us or by Actavis during the negotiations. If Actavis does not exercise the option during the specified period, its option will expire and the license agreement will not become effective. In that case, we would be free to enter into a collaboration arrangement with another party for the development and commercialization of TRV027 or to pursue development and commercialization on our own.
We received no consideration upon the grant of the option to Actavis. If Actavis exercises the option, we would receive a $65 million option exercise fee and could potentially receive up to $365 million in additional payments depending upon the achievement of future development and commercial milestones. We also could receive tiered royalties between 10% and 20% on net sales of licensed products worldwide, with the royalty rates on net sales of licensed products in the United States being somewhat higher than the royalty rates on net sales of licensed products outside the United States. The term of the royalty on sales of TRV027 for a given country would extend until the latest to occur of (i) ten years from first commercial sale of TRV027 in that country, (ii) the expiration of the last to expire patent claiming TRV027 that is sufficient to block the entrance of a generic version of the product, or (iii) the expiration of any period of exclusivity granted by applicable law or any regulatory authority in such country that confers exclusive marketing rights on the product.
If the license agreement becomes effective, Actavis has the right to grant sublicenses under the license agreement to affiliates and third parties. Any sublicensing does not act to relieve Actavis of any of its obligations under the license agreement, including Actavis’ obligation to make milestone payments to us with respect to TRV027 or pay royalties to us on sales of TRV027 by such sublicensee. Under the license, both Actavis and we have the right to terminate the agreement in the event of an uncured material breach or insolvency of the other party. In addition, Actavis is permitted to terminate the license agreement without cause at any time upon prior written notice or immediately for product safety reasons. Following a termination of the license agreement, all licenses granted to Actavis would
terminate, and Actavis would grant to us an exclusive royalty bearing license under specified patents and know-how to develop and commercialize reverted licensed products. If not terminated, the license agreement would remain in effect until the expiration of the last royalty term for the last licensed product.
Manufacturing
TRV027 drug substance has been made by a third party at a scale up to 2 kg per batch. We are exploring potential process improvements, which we will implement as appropriate as development progresses. Currently drug substance and drug product are each manufactured at single sites, but additional sites are planned for qualification in connection with any Phase 3 clinical trials.
Commercialization
If Actavis exercises its option to license TRV027, Actavis will have the exclusive rights to commercialize TRV027 and will be responsible for all commercialization activities at Actavis’s expense. At our request, Actavis will consider in good faith whether to grant us the right to co-promote TRV027 in the United States under terms to be agreed upon by the parties, but it has no obligation to provide co-promotion rights to us. If Actavis does not exercise its option to license TRV027 and we are successful in obtaining necessary regulatory approval, we might pursue commercialization on our own or seek to collaborate with a third party for commercialization, particularly outside the United States.
Competition
If TRV027 is approved for the indication of AHF, it will be used with standard loop diuretic therapy and may result in reduced need for vasodilators and/or inotropes. We also are aware of three product candidates in mid- to late-stage clinical development for AHF, specifically serelaxin, which is being developed by Novartis and currently is in Phase 3 clinical trials in patients with AHF; omecamtiv mecarbil, which is being developed by Amgen in collaboration with Cytokinetics Incorporated and currently is in Phase 2b clinical trials in patients with AHF and chronic heart failure; and ularitide, which is being developed by Cardiorentis and currently is in Phase 3 clinical trials for AHF. In addition, several product candidates are in mid- to late-stage clinical development for treating chronic heart failure which may, if approved, reduce the incidence of acute heart failure. These product candidates include LCZ-696 from Novartis, and Mydicar from Celladon.
Intellectual Property
Our TRV027 patent portfolio is wholly owned by us. The portfolio includes three issued U.S. patents that claim, among other things, TRV027, compositions comprising TRV027 and methods of using TRV027, and issued patents in New Zealand and China. The issued U.S. patents covering the composition of matter and methods of using TRV027 are expected to expire no earlier than 2031 and 2029, respectively, subject to any disclaimers or extensions available under the Hatch-Waxman Act. The TRV027 patent portfolio also includes two pending U.S. patent applications, which claim a genus of compounds that would encompass TRV027 and methods of using such compounds. If the two pending U.S. patent applications were to issue, they would be expected to expire no earlier than 2029, subject to any disclaimers or extensions. Outside of the United States, we have pending patent applications in Australia, Canada, the European Patent Office, Hong Kong, India, and Japan that are directed to TRV027. The patents from these applications, if issued, are predicted to expire in 2029, subject to any disclaimers or extensions.
Additionally, the TRV027 patent portfolio includes two U.S. provisional applications directed to, among other things, synthesis of TRV027, crystalline and amorphous forms of TRV027, and methods of preparing crystalline and amorphous forms of TRV027. Any patents resulting from these patent applications, if issued are expected to expire no earlier than 2035. The TRV027 patent portfolio is subject to Actavis’ option for an exclusive license.
Intellectual Property
We strive to protect the proprietary technologies that we believe are important to our business, including seeking and maintaining patent protection intended to cover the composition of matter of our product candidates, their methods of use, related technology and other inventions that are important to our business. We also rely on trade secrets and careful monitoring of our proprietary information to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
Our success will depend significantly on our ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to our business, defend and enforce our patents, maintain our licenses to use intellectual property owned by third parties, preserve the confidentiality of our trade secrets and operate without infringing valid and enforceable patents and other proprietary rights of third parties. We also rely on know-how, and continuing technological innovation to develop, strengthen and maintain our proprietary position in the field of modulating G protein coupled receptors with biased ligands.
One or more third parties may hold intellectual property, including patent rights, that is important or necessary to the development of our products. It may be necessary for us to use the patented or proprietary technology of third parties to commercialize our products, in which case we would be required to obtain a license from these third parties on commercially reasonable terms, or our business could be harmed, possibly materially. If we were not able to obtain a license, or were not able to obtain a license on commercially reasonable terms, our business could be harmed, possibly materially.
We plan to continue to expand our intellectual property estate by filing patent applications directed to dosage forms, methods of treatment and additional biased modulators of G protein coupled receptors. We anticipate seeking patent protection in the United States and internationally for compositions of matter covering the compounds, the chemistries and processes for manufacturing these compounds and the use of these compounds in a variety of therapies.
The patent positions of biopharmaceutical companies like us are generally uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and the patent’s scope can be modified after issuance. Consequently, we do not know whether any of our product candidates will be protectable or remain protected by enforceable patents. We cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient proprietary protection from competitors. Any patents that we hold may be challenged, circumvented or invalidated by third parties.
Because many patent applications in the United States and certain other jurisdictions are maintained in secrecy for 18 months, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain that we will be able to obtain patent protection for the inventions disclosed and/or claimed in our pending patent applications. Moreover, we may have to participate in interference proceedings declared by the United States Patent and Trademark Office or a foreign patent office to determine priority of invention or in post-grant challenge proceedings, such as oppositions, inter-partes review, post grant review or a derivation proceeding, that challenge our entitlement to an invention or the patentability of one or more claims in our patent applications or issued patents. Such proceedings could result in substantial cost, even if the eventual outcome is favorable to us.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a PCT application or a non-provisional patent application, subject to any disclaimers or extensions. The term of a patent in the United States can be adjusted and extended due
to the failure of the United States Patent and Trademark Office following certain statutory and regulation deadlines for issuing a patent.
In the United States, the patent term of a patent that covers an FDA- approved drug may also be eligible for patent term extension, which permits patent term restoration as compensation for a portion of the patent term lost during clinical development and the FDA regulatory review process. The Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under clinical development and regulatory review. Patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other non-United States jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our pharmaceutical products receive FDA approval, we expect to apply for patent term extensions on patents covering those products. Although, we intend to seek patent term extensions to any of our issued patents in any jurisdiction where these are available there is no guarantee that the applicable authorities, including the United States Patent and Trademark Office, will agree with our assessment of whether such extensions should be granted, and even if granted, the length of such extensions.
We also rely on trade secret protection for our confidential and proprietary information. Although we take steps to protect our proprietary information and trade secrets, including through contractual means with our employees and consultants, third parties may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose our technology. Thus, we may not be able to meaningfully protect our trade secrets. It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information concerning our business or financial affairs developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual, and which are related to our current or planned business or research and development or made during normal working hours, on our premises or using our equipment or proprietary information, are our exclusive property.
Manufacturing
We do not have any manufacturing facilities. We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates for preclinical and clinical testing, as well as for commercial manufacture if our product candidates receive marketing approval. At this time, none of our contract manufacturing agreements limit where, or with whom we can contract for commercial manufacture or distribution. It is our intention that by the time of any regulatory approvals for commercialization, we will have negotiated long-term commitments with at least one primary and one secondary supplier for each manufacturing and distribution function.
Commercialization
We have not yet established a sales, marketing or product distribution infrastructure. Subject to successfully completing product development and receiving marketing approvals, we expect to commence commercialization activities for our products other than TRV027 by building a focused sales and marketing organization in the United States, initially in the acute care area. We believe that such an organization will be able to address the community of physicians who are the key specialists in treating the patient populations for which our product candidates are being developed. We further believe that this sales organization could be adapted and expanded to provide support for TRV027 in
the acute care setting if Actavis does not exercise its option to license TRV027. Outside the United States, we expect to enter into distribution and other marketing arrangements with third parties for any of our product candidates that obtain marketing approval. We also intend to license out commercial rights for products that require a substantial primary care presence.
We plan to build a marketing and sales management organization to create and implement marketing strategies for any products that we market through our own sales organization and oversee and support our sales force. In parallel with building this organization, we plan to develop educational initiatives with respect to approved products and relationships with thought leaders in relevant fields of medicine.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. While we believe that our technology, knowledge, experience and scientific resources provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. Products in development by other companies may provide efficacy, safety, convenience and other benefits that are not provided by currently marketed therapies. As a result, they may provide significant competition for any of our product candidates for which we obtain marketing approval.
Some of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
The key competitive factors affecting the success of all of our therapeutic product candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third party payors.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected in many cases by insurers or other third party payors seeking to encourage the use of generic products. Generic products that broadly address these indications are currently on the market for the indications that we are pursuing, and additional products are expected to become available on a generic basis over the coming years. If our product candidates achieve marketing approval, we expect that they will be priced at a significant premium over competitive generic products.
Government Regulation and Product Approval
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, import and export of pharmaceutical products such as those we are developing. The processes for obtaining regulatory approvals in the United States and in foreign countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
FDA Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implemented regulations. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending new drug applications, or NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
· completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practice, or GLP, regulations;
· submission to the FDA of an IND which must become effective before human clinical trials may begin;
· approval by an independent institutional review board, or IRB, at each clinical site before each trial may be initiated;
· performance of human clinical trials, including adequate and well-controlled clinical trials, in accordance with good clinical practices, or GCP, to establish the safety and efficacy of the proposed drug product for each indication;
· submission to the FDA of an NDA;
· completion of an FDA advisory committee review, if applicable;
· satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practices, or cGMP, and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity, as well as satisfactory completion of an FDA inspection of selected clinical sites to determine GCP compliance;
· FDA review and approval of the NDA; and
· Some of our potential products are anticipated to require DEA review and scheduling activities prior to launch.
Preclinical Studies
Preclinical studies include laboratory evaluation of drug substance chemistry, toxicity and drug product formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Manufacture of drug substance, drug product and the labeling and distribution of clinical supplies must all comply with cGMP standards. Some preclinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must continue to oversee the clinical trial while it is being conducted. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their ClinicalTrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined. In Phase 1, the drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an initial indication of its effectiveness. In Phase 2, the drug typically is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. In Phase 3, the drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product and to provide adequate information for the labeling of the product.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture,
controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has agreed to certain performance goals regarding the timing of its review of an application.
In addition, under the Pediatric Research Equity Act an NDA or supplement to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, to mitigate any identified or suspected serious risks and ensure safe use of the drug. The REMS plan could include medication guides, physician communication plans, assessment plans and elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools. We expect that the µ-opioid agonist products will be subject to a REMS, since currently marketed opioid products are subject to this requirement.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
The FDA typically refers a question regarding a novel drug to an external advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured, referred to as a Pre-Approval Inspection, or PAI. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical trial sites to assure compliance with GCP.
The testing and approval process for an NDA requires substantial time, effort and financial resources, and each may take several years to complete. Data obtained from preclinical and clinical testing are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval of an NDA on a timely basis, or at all.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure
final approval of the NDA and may require additional clinical or preclinical testing in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. For some products, an additional step of DEA review and scheduling is required.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, including a boxed warning, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization or impose other conditions, including distribution restrictions or other risk management mechanisms under a REMS which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market.
Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition
of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
· restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
· fines, warning letters or holds on post-approval clinical trials;
· refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals;
· product seizure or detention, or refusal to permit the import or export of products; or
· injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Although physicians, in the practice of medicine, may prescribe approved drugs for unapproved indications, pharmaceutical companies generally are required to promote their drug products only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
DEA Regulation
Both TRV130 and TRV734 will be regulated as a “controlled substance” as defined in the Controlled Substances Act of 1970, or CSA, which establishes registration, security, recordkeeping, reporting, storage, distribution and other requirements administered by the DEA. The DEA is concerned with the control of handlers of controlled substances, and with the equipment and raw materials used in their manufacture and packaging, in order to prevent loss and diversion into illicit channels of commerce.
The DEA regulates controlled substances as Schedule I, II, III, IV or V substances. Schedule I substances by definition have no established medicinal use, and may not be marketed or sold in the United States. A pharmaceutical product may be listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest risk of abuse and Schedule V substances the lowest relative risk of abuse among such substances. TRV130 and TRV734, if approved, are expected to be listed by the DEA as Schedule II controlled substances under the CSA. Consequently, their manufacture, shipment, storage, sale and use will be subject to a high degree of regulation.
Annual registration is required for any facility that manufactures, distributes, dispenses, imports or exports any controlled substance. The registration is specific to the particular location, activity and controlled substance schedule. For example, separate registrations are needed for import and manufacturing, and each registration will specify which schedules of controlled substances are authorized.
The DEA typically inspects a facility to review its security measures prior to issuing a registration. Security requirements vary by controlled substance schedule, with the most stringent requirements applying to Schedule I and Schedule II substances. Required security measures include background checks on employees and physical control of inventory through measures such as cages, surveillance
cameras and inventory reconciliations. Records must be maintained for the handling of all controlled substances, and periodic reports made to the DEA, for example distribution reports for Schedule I and II controlled substances, Schedule III substances that are narcotics, and other designated substances. Reports must also be made for thefts or losses of any controlled substance, and to obtain authorization to destroy any controlled substance. In addition, special authorization and notification requirements apply to imports and exports.
In addition, a DEA quota system controls and limits the availability and production of controlled substances in Schedule I or II. Distributions of any Schedule I or II controlled substance must also be accompanied by special order forms, with copies provided to the DEA. The DEA may adjust aggregate production quotas and individual production and procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments. Our, or our contract manufacturers’, quota of an active ingredient may not be sufficient to meet commercial demand or complete clinical trials. Any delay or refusal by the DEA in establishing our, or our contract manufacturers’, quota for controlled substances could delay or stop our clinical trials or product launches.
To meet its responsibilities, the DEA conducts periodic inspections of registered establishments that handle controlled substances. Individual states also regulate controlled substances, and we and our contract manufacturers will be subject to state regulation with respect to the distribution of these products.
Federal and State Fraud and Abuse and Data Privacy and Security Laws and Regulations
In addition to FDA restrictions on marketing of pharmaceutical products, federal and state fraud and abuse laws restrict business practices in the biopharmaceutical industry. These laws include anti-kickback and false claims laws and regulations as well as data privacy and security laws and regulations.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any item or service reimbursable under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting some common activities from prosecution, the exemptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated.
The reach of the Anti-Kickback Statute was also broadened by the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively PPACA, which, among other things, amended the intent requirement of the federal Anti-Kickback Statute such that a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act or the civil monetary penalties statute, which imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. PPACA also created new federal requirements for reporting, by applicable manufacturers of covered drugs of payments and other transfers of value to physicians and teaching hospitals.
The federal False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. Several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of products for unapproved, and thus non-reimbursable, uses.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created new federal criminal statutes that prohibit knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Also, many states have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations, including the final omnibus rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of the federal or state laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including criminal and significant civil monetary penalties, damages, fines, imprisonment, exclusion from participation in government healthcare programs, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
Coverage and Reimbursement
The commercial success of our product candidates and our ability to commercialize any approved product candidates successfully will depend in part on the extent to which governmental payor programs at the federal and state levels, including Medicare and Medicaid, private health insurers and other third-party payors provide coverage for and establish adequate reimbursement levels for our
product candidates. Government health administration authorities, private health insurers and other organizations generally decide which drugs they will pay for and establish reimbursement levels for healthcare. In particular, in the United States, private health insurers and other third-party payors often provide reimbursement for products and services based on the level at which the government provides reimbursement through the Medicare or Medicaid programs for such treatments. In the United States, the European Union and other potentially significant markets for our product candidates, government authorities and third-party payors are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which often has resulted in average selling prices lower than they would otherwise be. Further, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European Union will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical coverage and reimbursement policies and pricing in general.
Third-party payors are increasingly imposing additional requirements and restrictions on coverage and limiting reimbursement levels for medical products. For example, federal and state governments reimburse covered prescription drugs at varying rates generally below average wholesale price. These restrictions and limitations influence the purchase of healthcare services and products. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drug products for a particular indication. Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Our product candidates may not be considered medically necessary or cost-effective. A payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in drug development. Legislative proposals to reform healthcare or reduce costs under government insurance programs may result in lower reimbursement for our products and product candidates or exclusion of our products and product candidates from coverage. The cost containment measures that healthcare payors and providers are instituting and any healthcare reform could significantly reduce our revenue from the sale of any approved product candidates. We cannot provide any assurances that we will be able to obtain and maintain third-party coverage or adequate reimbursement for our product candidates in whole or in part.
Impact of Healthcare Reform on Coverage, Reimbursement and Pricing
The United States and some foreign jurisdictions are considering enacting or have enacted a number of additional legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. For example, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Part D plans include both standalone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay
for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, any negotiated prices for our future products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from Medicare Part D may result in a similar reduction in payments from non-governmental payors.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes of Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. If third-party payors do not consider our product candidates to be cost-effective compared to other available therapies, they may not cover our product candidates, once approved, as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
PPACA became law in March 2010 and substantially changes the way healthcare is financed by both governmental and private insurers. Among other cost containment measures, the PPACA establishes an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents; a new Medicare Part D coverage gap discount program; and a new formula that increases the rebates a manufacturer must pay under the Medicaid Drug Rebate Program. In the future, there may continue to be additional proposals relating to the reform of the U.S. healthcare system, some of which could further limit the prices we are able to charge for our product candidates, once approved, or the amounts of reimbursement available for our product candidates once they are approved.
In addition, other legislative changes have been proposed and adopted since PPACA was enacted. In August 2011, the President signed into law the Budget Control Act of 2011, as amended, which, among other things, created the Joint Select Committee on Deficit Reduction to propose spending reductions to Congress. The Joint Select Committee on Deficit Reduction did not achieve its targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reductions to several government programs. These reductions include aggregate reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013, and will remain in effect through 2024 unless additional Congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These and other healthcare reform initiatives may result in additional reductions in Medicare and other healthcare funding.
Exclusivity and Approval of Competing Products
Hatch-Waxman Patent Exclusivity
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s product or a method of using the product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an abbreviated new drug application, or ANDA, or 505(b)(2) NDA. Generally, an ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths, dosage form and route of administration as the listed drug and has been shown to be bioequivalent through in vitro or in vivo testing or otherwise to the listed drug. ANDA applicants are not required to conduct or submit results of preclinical or clinical tests to prove the safety or effectiveness of their drug product, other than the requirement for bioequivalence testing. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug. 505(b)(2) NDAs generally are submitted for changes to a previously approved drug product, such as a new dosage form or indication.
The ANDA or 505(b)(2) NDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book, except for patents covering methods of use for which the ANDA applicant is not seeking approval. Specifically, the applicant must certify with respect to each patent that:
· the required patent information has not been filed;
· the listed patent has expired;
· the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or
· the listed patent is invalid, unenforceable or will not be infringed by the new product.
Generally, the ANDA or 505(b)(2) NDA cannot be approved until all listed patents have expired, except when the ANDA or 505(b)(2) NDA applicant challenges a listed drug. A certification that the proposed product will not infringe the already approved product’s listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the applicant does not challenge the listed patents or indicate that it is not seeking approval of a patented method of use, the ANDA or 505(b)(2) NDA application will not be approved until all the listed patents claiming the referenced product have expired.
If the ANDA or 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the application has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days after the receipt of notice of the Paragraph IV certification automatically prevents the FDA from approving the ANDA or 505(b)(2) NDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
Hatch-Waxman Non-Patent Exclusivity
Market and data exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications for competing products. The FDCA provides a five-year period of non-patent data exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any
other new drug containing the same active moiety, which is the molecule or ion responsible for the activity of the drug substance. During the exclusivity period, the FDA may not accept for review an ANDA or a 505(b)(2) NDA submitted by another company that contains the previously approved active moiety. However, an ANDA or 505(b)(2) NDA may be submitted after four years if it contains a certification of patent invalidity or noninfringement.
The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA, or supplement to an existing NDA or 505(b)(2) NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant, are deemed by the FDA to be essential to the approval of the application or supplement. Three-year exclusivity may be awarded for changes to a previously approved drug product, such as new indications, dosages, strengths or dosage forms of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and, as a general matter, does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for generic versions of the original, unmodified drug product. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric Exclusivity
Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the non-patent exclusivity periods described above. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or Orange Book listed patent protection cover the drug are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot approve an ANDA or 505(b)(2) application owing to regulatory exclusivity or listed patents. When any of our products is approved, we anticipate seeking pediatric exclusivity when it is appropriate.
Foreign Regulation
In order to market any product outside of the United States, we would need to comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of our products. For example, in the European Union, we must obtain authorization of a clinical trial application, or CTA, in each member state in which we intend to conduct a clinical trial. Whether or not we obtain FDA approval for a product, we would need to obtain the necessary approvals by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others.
Employees
As of September 30, 2014, we had 39 employees, all of whom are located in the United States. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be good.