Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 - Viatar CTC Solutions Inc. | s100531_ex23-1.htm |
| EX-4.1 - EXHIBIT 4.1 - Viatar CTC Solutions Inc. | s100531_ex4-1.htm |
| EXCEL - IDEA: XBRL DOCUMENT - Viatar CTC Solutions Inc. | Financial_Report.xls |
As filed with the Securities and Exchange Commission on November 26, 2014
Registration No. 333- 199619
| UNITED STATES |
| SECURITIES AND EXCHANGE COMMISSION |
| Washington, D.C. 20549 |
| FORM S-1/A (Amendment No.1) |
| REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933 |
| Viatar CTC Solutions Inc. |
| (Exact name of Registrant as specified in its charter) |
| Delaware | 3841 | 26-1581305 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
| 116 John Street, Suite 10, Lowell, Massachusetts 01852 |
| (617) 299-6590 |
| (Address, including zip code, and telephone number, |
| including area code, of Registrant’s principal executive offices) |
| Ilan Reich |
| Chief Executive Officer |
| Viatar CTC Solutions Inc. |
| 116 John Street, Suite 10 |
| Lowell, Massachusetts 01852 |
| (617) 299-6590 |
| (Name, address, including zip code, and telephone number, including area code, of agent for service) |
Copies to:
Gregory Sichenzia, Esq.
Darrin M. Ocasio, Esq.
Sichenzia Ross Friedman Ference LLP
61 Broadway, 32nd Floor
New York, NY 10006
(212) 930-9700
Approximate date of commencement of proposed sale to the public: As soon as practicable after this registration statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box: x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ | |||
| Non-accelerated filer ¨ (Do not check if a smaller reporting company) | Smaller reporting company x |
| CALCULATION OF REGISTRATION FEE |
| Title of Each Class of Securities to Be Registered |
Amount to Be Registered |
Proposed Maximum Offering Price Per Unit (1) |
Proposed Maximum Aggregate Offering Price |
Amount of Registration Fee |
||||||||||||
| Common Stock (2) | 1,164,571 | $ | 3.50 | $ | 4,075,999 | $ | 473.63 | |||||||||
| Common Stock underlying Warrants (2) | 37,500 | $ | 3.50 | $ | 131,250 | $ | 15.25 | |||||||||
| Total | 1,202,071 | $ | 488.88 | (3) | ||||||||||||
| (1) | Pursuant to Rule 416 under the Securities Act of 1933, this Registration Statement also covers any additional securities that may be offered or issued in connection with any stock split, stock dividend or similar transaction. |
| (2) | Estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(a) under the Securities Act of 1933, as amended. |
| (3) | Previously paid. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the Securities and Exchange Commission (the “SEC”) declares our registration statement effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to Completion, dated November 26, 2014
Prospectus
Viatar CTC Solutions Inc.
1,202,071 Shares of Common Stock
The selling stockholders identified in this prospectus are offering 1,202,071 shares of common stock, par value $0.001 per share (“Common Stock”) of Viatar CTC Solutions Inc.(the “Company,” “we,” “us,” or “our”)including 1,164,571 shares of Common Stock and 37,500 shares of Common Stock issuable upon exercise of certain warrants of the Company.
These securities will be offered for sale by the selling stockholders identified in this prospectus in accordance with the methods and terms described in the section of this prospectus titled “Plan of Distribution.” We will not receive any of the proceeds from the sale of these shares. The selling stockholders will offer and sell the shares at $3.50 per share until the price of our Common Stock is quoted on the OTC Bulletin Board and thereafter at prevailing market prices or privately negotiated prices. The selling stockholders may be deemed “underwriters” within the meaning of the Securities Act of 1933, as amended, in connection with the sale of their Common Stock under this prospectus. We will pay all the expenses incurred in connection with the offering described in this prospectus, with the exception of brokerage expenses, fees, discounts and commissions, which will all be paid by the selling stockholders. Our Common Stock is more fully described in the section of this prospectus titled “Description of Securities.”
No public market currently exists for our Common Stock. Although we intend to request a registered broker-dealer apply to have our Common Stock quoted on the OTC Bulletin Board, public trading of our Common Stock may never materialize or even if materialized, be sustained. If our Common Stock is quoted on the on the OTC Bulletin Board, then the sale price to the public will vary according to prevailing market prices or privately negotiated prices by the selling stockholders.
We are an “emerging growth company” under the federal securities laws and are subject to reduced public company reporting requirements. Investing in our Common Stock involves a high degree of risk. You may lose your entire investment. See “Risk Factors” beginning on page 3. You should read the entire prospectus before making an investment decision.
Neither the SEC nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is ________________, 2014
TABLE OF CONTENTS
This summary highlights information contained in greater detail elsewhere in this prospectus. You should read the entire prospectus carefully before making an investment in our Common Stock. You should carefully consider, among other things, our consolidated financial statements and the related notes and the sections entitled “Risk Factors,” “Business” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus. Unless the context otherwise requires, the terms “Viatar,” “the Company,” “we,” “us” and “our” in this prospectus refer to Viatar CTC Solutions Inc., and its subsidiaries.
Overview
We were a limited liability company organized in the State of Delaware on December 18, 2007 under the name “Vizio Medical Devices LLC” and converted into a Delaware corporation on February 25, 2014 upon filing a certificate of conversion and a certificate of incorporation under the name “Viatar CTC Solutions Inc.”
We are a medical technology company focused on cancer molecular diagnostics and cancer therapy, both based on the principle of removing blood-borne circulating tumor cells. We are a development stage company which is conducting research and planning to commercialize two products that utilize our proprietary blood-borne circulating tumor cells removal technology:
| · | The ViatarTM Collection System for Molecular Analysis is designed to collect and purify a statistically significant quantity of blood-borne circulating tumor cells from up to 50 mL of blood and these circulating tumor cells collected in our device are then used in subsequent, downstream applications, like DNA sequencing and other genetic analysis technologies used primarily for research. |
| · | The ViatarTM Therapeutic Oncopheresis System is designed to remove blood-borne circulating tumor cells from a patient’s blood as a new cancer therapy for metastatic disease, which occurs when cancers spreads from one organ to another not directly connected with it. |
We anticipate launching the ViatarTM Collection System for Molecular Analysis during 2016: initially with a CE Mark in Europe and Canada, which is the foreign equivalent of compliance with United States Food and Drug Administration clearance to commercially market a medical device, followed by a 510(k) designation in the United States, which means that the United States Food and Drug Administration has given permission to market the new device. We are currently engaged in research collaborations with several blood-borne circulating tumor cells technology platform companies with the goal of marketing and selling the ViatarTM Collection System for Molecular Analysis through private label and distribution arrangements. We anticipate launching the ViatarTM Therapeutic Oncopheresis System during 2016 with a CE Mark in Europe and Canada. Introduction in the United States will take several years, based on the anticipated Premarket Approval classification by the United States Food and Drug Administration, which requires us to submit an exhaustive application to the United States Food and Drug Administration and receive approval prior to beginning commercial marketing of the device. The Premarket Approval application includes information on how the device was designed and manufactured, as well as preclinical and clinical studies, demonstrating that it is safe and effective for its intended use. We anticipate marketing and selling the ViatarTM Therapeutic Oncopheresis System on a direct basis through our own dedicated sales force in key large markets (such as the United States, Germany, France, Italy, UK and Japan), and through distributors in other markets.
Both of our oncology products are in the development stage, with significant design, materials validation, manufacturing scale-up and preclinical testing necessary before they can be used in human clinical trials. None of the capabilities of our two oncology products have been demonstrated yet in statistically significant clinical trials.
An investment in our Common Stock involves a high degree of risk. You should carefully consider the risks summarized below. The risks are discussed more fully in the “Risk Factors” section of this prospectus on page 3. These risks include, but are not limited to, the following:
| • | we are an early-stage company with total net losses of $18.02 million from inception (December 18, 2007) to September 30, 2014. Before 2011, we were also pursuing a business plan relating to ambulatory removal of excess fluids for kidney failure and congestive heart failure patients. We discontinued that program due to technical difficulties and the scarcity of funds. The portion of our accumulated deficit that relates to those activities for the period from inception through December 30, 2010 is approximately $9 million. |
| • | we may never achieve sustained profitability; |
| • | our business depends upon our ability to complete product development activities and achieve sales of our two oncology applications; |
| • | our current cash resources are insufficient to fund our operations for an extended period of time; |
| • | our business depends on executing our business strategy, developing collaboration relationships with other life science companies and major cancer research centers, and gaining acceptance of our products in the market; |
| 1 |
| • | our business depends on satisfying any applicable United States (including Food and Drug Administration) and international regulatory requirements with respect to new medical devices including validation studies of the materials and manufacturing processes used in our devices and pre-clinical testing to demonstrate safety in animals, as well human clinical studies to demonstrate efficacy for specific disease conditions; | |
| • | our business depends on our ability to effectively compete with other therapies, technologies and services that now exist or may hereafter be developed; | |
| • | we depend on our chief executive officer; | |
| • | we depend on our ability to attract and retain scientists, clinicians and sales personnel with extensive experience in oncology, who are in short supply; | |
| • | we need to obtain or maintain patents or other appropriate protection for the intellectual property utilized in our current and planned products; and | |
| • | Our largest stockholder, chief executive officer and chairman of the board, Mr. Ilan Reich, has the ability to control our business direction and could delay or prevent a change in corporate control. |
Company Information
We maintain our principal executive offices at 116 John Street, Suite 10, Lowell, Massachusetts 01852. Our telephone number is (617) 299-6590 and our website address is www.viatarctcsolutions.com. The information contained in, or that can be accessed through, our website is not incorporated into and is not part of this prospectus. We were incorporated in Delaware on December 18, 2007 as a limited liability company and converted to a corporation on February 25, 2014.
Implications of Being an Emerging Growth Company
As a company with less than $1 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act, or JOBS Act, enacted in April 2012, and may take advantage of reduced reporting requirements that are otherwise applicable to public companies. These provisions include, but are not limited to:
| • | being permitted to present only two years of audited financial statements and only two years of related Management’s Discussion and Analysis of Financial Condition and Results of Operations in our SEC filings; |
| • | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act; |
| • | reduced disclosure obligations regarding executive compensation in periodic reports, proxy statements and registration statements; and |
| • | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We may take advantage of these provisions until the last day of our fiscal year following the fifth anniversary of the date of the first sale of our common equity securities pursuant to an effective registration statement under the Securities Act. However, if certain events occur before the end of such five-year period, including if we become a “large accelerated filer,” our annual gross revenues exceed $1 billion or we issue more than $1 billion of non-convertible debt in any three-year period, we will cease to be an emerging growth company before the end of such five-year period.
In addition, Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. We have elected to take advantage of the extended transition period for complying with new or revised accounting standards.
Going Concern
As described in auditor’s report on our financial statements, our auditors have included a “going concern” provision in their opinion on our financial statements, expressing substantial doubt that we can continue as an ongoing business for the next twelve months.
The Offering
| Common Stock offered by the selling stockholders | 1,202,071 shares of Common Stock, including 1,164,571 shares of Common Stock and 37,500 shares of Common Stock issuable upon exercise of certain warrants of the Company | |
| Common Stock Outstanding before this offering | 17,118,622 shares | |
| Common stock to be outstanding after this offering assuming exercise of all the warrants being offered herein) | 17,156,122 shares | |
| 2 |
| Use of proceeds | We will not receive any of the proceeds from the sale of shares to be offered by the selling stockholders. See “Use of Proceeds.” | |
| Risk factors | You should carefully consider the information set forth in this prospectus and, in particular, the specific factors set forth in the “Risk Factors” section beginning on page 3 of this prospectus before deciding whether or not to invest in shares of our Common Stock. | |
Investing in our Common Stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below, together with all of the other information in this prospectus, including our consolidated financial statements and related notes, before investing in our Common Stock. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that affect us. If any of the following risks occur, our business, financial condition, operating results, and prospects could be materially harmed. In that event, you could lose part or all of your investment.
Risks Relating to Our Financial Condition and Capital Requirements
We are an early stage company with a history of net losses; we expect to incur net losses in the future, and we may never achieve sustained profitability.
We have historically incurred substantial net losses, including net losses of $4.39 million in 2012 and $1.06 million in 2013, and we have never been profitable. At September 30, 2014, our total net loss since inception was approximately $18.02 million. Before 2011, we were pursuing a business plan relating to an ambulatory device for removal of excess fluids from kidney failure and congestive heart failure patients, which was unrelated to oncology. The portion of our accumulated deficit that relates to those activities during the period from inception through December 31, 2010 is approximately $9 million.
We expect our losses to continue as a result of costs relating to our research and development, clinical and regulatory activities, as well as sales and marketing costs. These losses have had, and will continue to have, an adverse effect on our working capital, total assets and stockholders’ equity. Because of the numerous risks and uncertainties associated with our commercialization efforts, we are unable to predict when we will become profitable, and we may never become profitable. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our inability to achieve and then maintain profitability would negatively affect our business, financial condition, results of operations and cash flows.
Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
As described in auditor’s report on our financial statements, our auditors have included a “going concern” provision in their opinion on our financial statements, expressing substantial doubt that we can continue as an ongoing business for the next twelve months. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. If we cannot secure the financing needed to continue as a viable business, our stockholders may lose some or all of their investment in us.
| 3 |
We will need to raise additional capital.
We believe our current cash resources and committed borrowing capacity are insufficient to satisfy our liquidity requirements at our current level of operations. We expect that we will need to raise additional financing later in the third quarter of 2014. Such financing may not be available to us on favorable terms, if at all. Without that new financing, we would need to scale back our general and administrative activities and certain of our research and development activities. Our forecast pertaining to the adequacy of our current financial resources supporting our current and anticipated level of operations is a forward-looking statement and involves risks and uncertainties.
We will also need to raise additional capital to expand our business to meet our long-term business objectives. Additional financing, which is not in place at this time, may be from the sale of equity or convertible or other debt securities in a public or private offering, from an additional credit facility or strategic partnership coupled with an investment in us or a combination of both. We may be unable to raise sufficient additional financing on terms that are acceptable to us, if at all. Failure to raise additional capital in sufficient amounts would significantly impact our ability to expand our business. For further discussion of our liquidity requirements as they relate to our long-term plans, see the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources.”
Risks Relating to Our Business and Strategy
If we are unable to generate sufficient sales for our two oncology products once commercialized, our revenues will be insufficient for us to achieve profitability.
We are in varying stages of research and development for our two oncology products and have not commenced any sales of our products. If we are unable to generate sufficient sales for those products once they are commercialized, we will not produce sufficient revenues to become profitable.
If we are unable to execute our sales and marketing strategy for our two oncology products and are unable to gain acceptance in the market, we may be unable to generate sufficient revenue to sustain our business.
Although we believe that our two oncology products represent a promising commercial opportunity, our products may never gain significant acceptance in the marketplace and therefore may never generate substantial revenue or profits for us. We will need to establish a market and build that market through collaborations with other companies, physician education, awareness programs and the publication of clinical trial results. Gaining acceptance in medical communities requires publication in leading peer-reviewed journals of results from studies using our planned products. The process of publication in leading medical journals is subject to a peer review process and peer reviewers may not consider the results of our studies sufficiently novel or worthy of publication. Failure to have our studies published in peer-reviewed journals would limit the adoption of our two oncology products.
Our ability to successfully market our two oncology products will depend on numerous factors, including:
| • | conducting clinical studies with our two medical devices in collaboration with key thought leaders to demonstrate their use and value in important medical decisions; |
| • | whether our potential collaboration partners vigorously support our offerings; |
| • | the success of the sales force which we intend to hire; |
| • | whether healthcare providers believe our two oncology products provide clinical or therapeutic utility; |
| • | whether the medical community accepts that such products are sufficiently meaningful in patient care and treatment decisions; and |
| • | whether health insurers, government health programs and other third-party payors will cover and pay for such products and, if so, whether they will adequately reimburse us. |
Failure to achieve widespread market acceptance of our two oncology products would materially harm our business, financial condition and results of operations.
| 4 |
If we cannot develop new products to keep pace with rapid advances in technology, medicine and science, our operating results and competitive position could be harmed.
In recent years, there have been numerous advances in technologies relating to the diagnosis and treatment of cancer. Several new cancer drugs have been approved, and a number of new drugs in clinical development may increase patient survival time. There have also been advances in methods used to identify patients likely to benefit from these drugs based on analysis of biomarkers and DNA sequencing. We must continuously develop new oncology products and enhance any existing products to keep pace with evolving standards of care. Our products could become obsolete unless we continually innovate and expand them to demonstrate benefit in the diagnosis and treatment of patients with cancer, which would have a material adverse effect on our business, financial condition and results of operations.
If our two oncology products do not continue to perform as expected, our operating results, reputation and business will suffer.
Our success depends on the market’s confidence that we can continue to provide reliable, high-quality products. We believe that our customers are likely to be particularly sensitive to safety issues, defects and errors. As a result, the failure of our two oncology products to perform as expected would significantly impair our reputation and public image, and we may be subject to legal claims arising from any safety issues, defects or errors.
If our sole facility becomes damaged or inoperable, or we are required to vacate the facility, our ability to sell and provide our two oncology products and pursue our research and development efforts may be jeopardized.
We currently conduct virtually all of our business from our facility in Lowell, Massachusetts. We do not have any other facilities. Our facility and equipment could be harmed or rendered inoperable by natural or man-made disasters, including fire, storm, earthquake, flooding and power outages, which may render it difficult or impossible for us to run our business. This may result in the loss of customers or harm to our reputation or relationships with scientific or clinical collaborators, and we may be unable to regain those customers or repair our reputation in the future. Furthermore, our facility and the equipment we use to perform our research and development work could be costly and time-consuming to repair or replace. We carry insurance for damage to our property and the disruption of our business, but this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, if at all.
If we cannot compete successfully with our competitors, we may be unable to increase or sustain our revenues or achieve and sustain profitability.
Our principal competition comes from mainstream diagnostic methods, used by pathologists and oncologists for many years, which focus on tumor tissue analysis. It may be difficult to change the methods or behavior of oncologists to incorporate our CTC molecular diagnostic system in their practices in conjunction with or instead of tissue biopsies and analysis. In addition, companies offering capital equipment and kits or reagents to local pathology laboratories represent another source of potential competition. These kits are used directly by the pathologist, which can facilitate adoption.
While virtually all of the companies which offer molecular diagnostic tests are potential customers and/or collaboration partners for our molecular diagnostic system, CTC analysis is a new area of science and we cannot predict what tests or technologies others will develop that may compete with or provide results similar or superior to the results we are able to achieve with our product. Currently, among the category of companies which we regard as potential customers and/or collaboration partners are Janssen Diagnostics, LLC (a unit of Johnson & Johnson), which markets its CellSearch® test; Atossa Genetics markets its ArgusCYTE® test, as well as public companies such as Alere (Adnagen) and Illumina as well as many private companies, including Apocell, EPIC Sciences, Clearbridge Biomedics, Cynvenio Biosystems, Fluxion Biosciences, RareCells, ScreenCell and Silicon Biosystems. Many of these groups, in addition to operating research and development laboratories, are establishing CLIA-certified testing laboratories while others are focused on selling equipment and reagents.
CTC removal via therapeutic oncopheresis is a new field and we cannot predict whether oncologists will accept it as a valid treatment methodology for metastatic cancers; or whether pharmaceutical and biopharmaceutical companies will develop targeted drugs or vaccines that achieve similar or better outcomes. Mechanical separation of one species of cells from the blood is a long-established technology, and several research groups are investigating methods of using leukapheresis (either alone or in conjunction with a separation medium such as beads) to remove CTCs from a cancer patient’s blood. Although we believe that those methods will be too cumbersome, expensive and have adverse side effects, there can be no assurance that other technologies will be developed which compete effectively against our therapeutic oncopheresis system.
| 5 |
We expect that pharmaceutical and biopharmaceutical companies will increasingly focus attention and resources on the personalized cancer diagnostic sector as the potential and prevalence of molecularly targeted oncology therapies approved by the FDA along with companion diagnostics increases. For example, the FDA has recently approved three such agents—Xalkori® from Pfizer Inc. along with its companion anaplastic lymphoma kinase FISH test from Abbott Laboratories, Inc., Zelboraf® from Daiichi-Sankyo/Genentech/Roche along with its companion B-raf kinase V600 mutation test from Roche Molecular Systems, Inc. and Tafinlar® from GlaxoSmithKline along with its companion B-raf kinase V600 mutation test from bioMerieux. These recent FDA approvals are only the second, third and fourth instances of simultaneous approvals of a drug and companion diagnostic, the first being the 1998 approval of Genentech’s Herceptin® for HER2 positive breast cancer along with the HercepTest from partner Dako A/S.
There are a number of companies which are focused on the oncology diagnostic market, such as Biodesix, Caris, Clarient, Foundation Medicine, Neogenomics, Response Genetics, Agendia, Genomic Health, and Genoptix, who while not currently offering CTC tests are selling to the medical oncologists and pathologists and could develop or offer CTC tests. Large laboratory services companies, such as Sonic USA, Quest and LabCorp, provide more generalized cancer diagnostic testing.
Additionally, projects related to cancer diagnostics and particularly genomics have received increased government funding, both in the United States and internationally. As more information regarding cancer genomics becomes available to the public, we anticipate that more products aimed at identifying targeted treatment options will be developed and that these products may compete with ours. In addition, competitors may develop their own versions of our current or planned products in countries where we did not apply for patents or where our patents have not issued and compete with us in those countries, including encouraging the use of their test by physicians or patients in other countries.
Some of our present and potential competitors have widespread brand recognition and substantially greater financial and technical resources and development, production and marketing capabilities than we do. Others may develop lower-priced CTC-based tests that payors, pathologists and oncologists could view as functionally equivalent to the benefits that can be obtained using our molecular diagnostic system and DNA sequencing, which could force us to lower the list price of our medical device and impact our operating margins and our ability to achieve and maintain profitability. In addition, technological innovations that result in the creation of enhanced diagnostic tools that are more sensitive or specific than DNA sequencing may enable other clinical laboratories, hospitals, physicians or medical providers to provide specialized diagnostic tests in a more patient-friendly, efficient or cost-effective manner than is currently possible. If we cannot compete successfully against current or future competitors, we may be unable to increase or create market acceptance and sales of our current or planned medical devices, which could prevent us from increasing or sustaining our revenues or achieving or sustaining profitability.
We expect to continue to incur significant expenses to develop and market oncology products, which could make it difficult for us to achieve and sustain profitability.
In recent years, we have incurred significant costs in connection with the development of our oncology products. For the year ended December 31, 2012, our research and development expenses were $4.23 million. For the year ended December 31, 2013, our research and development expenses were $0.68 million. We expect our expenses to increase from that level for the foreseeable future as we conduct studies of our two oncology tests, establish a sales and marketing organization, drive adoption of and reimbursement for those products and develop new oncology products. As a result, we need to generate significant revenues in order to achieve sustained profitability.
| 6 |
Clinical studies are important in demonstrating to both customers and payors a medical device’s clinical relevance and value. If we are unable to identify collaborators willing to work with us to conduct clinical studies, or the results of those studies do not demonstrate that a medical device provides clinically meaningful health outcomes and value, commercial adoption of such device may be slow, which would negatively impact our business.
Clinical studies show when and how to use a given medical device, and describe the particular clinical situations or settings in which it can be applied and the expected results. Clinical studies also show the impact of the results on patient care and management. Clinical studies are typically performed with collaborating oncologists at medical centers and hospitals, and generally result in peer-reviewed publications. Sales and marketing representatives use these publications to demonstrate to customers how to use a medical device, as well as why they should use it. These publications are also used with payors to obtain coverage for a procedure or test utilizing a medical device, helping to assure there is appropriate reimbursement.
We will need to conduct clinical studies for our two oncology products to drive adoption in the marketplace and reimbursement. Should we not be able to perform these studies, or should their results not provide clinically meaningful data and value for oncologists, adoption of our tests could be impaired and we may not be able to obtain reimbursement for them.
The loss of key members of our executive management team could adversely affect our business.
Our success in implementing our business strategy depends largely on the skills, experience and performance of key members of our executive management team, including Ilan Reich, our Chief Executive Officer. The collective efforts of each of these persons and others working with them as a team are critical to us as we continue to develop our technologies, tests and research and development and sales programs. As a result of the difficulty in locating qualified new management, the loss or incapacity of existing members of our executive management team could adversely affect our operations.. We do not maintain “key person” life insurance on any of our employees.
In addition, we rely on collaborators, consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our collaborators, consultants and advisors are generally employed by employers other than us and may have commitments under agreements with other entities that may limit their availability to us.
The loss of a key member of our management team, the failure of a key member of our management team to perform in his or her current position or our inability to attract and retain skilled employees could result in our inability to continue to grow our business or to implement our business strategy.
There is a scarcity of experienced professionals in our industry. If we are not able to retain and recruit personnel with the requisite technical skills, we may be unable to successfully execute our business strategy.
The specialized nature of our industry results in an inherent scarcity of experienced personnel in the field. Our future success depends upon our ability to attract and retain highly skilled personnel, including scientific, technical, commercial, business, regulatory and administrative personnel, necessary to support our anticipated growth, develop our business and perform certain contractual obligations. Given the scarcity of professionals with the scientific knowledge that we require and the competition for qualified personnel among life science businesses, we may not succeed in attracting or retaining the personnel we require to continue and grow our operations.
| 7 |
Our inability to attract, hire and retain a sufficient number of qualified sales professionals in key large markets, and engage qualified distributors in small markets, would hamper our sales for our molecular diagnostic system and our therapeutic oncopheresis system, as well as our ability to expand geographically and to successfully commercialize any other products or services we may develop.
To succeed in selling our two oncology products, we must build a sales force in key large markets and engage qualified distributors in small markets. These personnel and entities need to have extensive experience in oncology and close relationships with medical oncologists, nurses, pathologists and other hospital personnel. To achieve our marketing and sales goals, we will need to build our sales and commercial infrastructure, with which to date we have had little experience. Sales professionals with the necessary technical and business qualifications are in high demand, and there is a risk that we may be unable to attract, hire and retain the number of sales professionals with the right qualifications, scientific backgrounds and relationships with decision-makers at potential customers needed to achieve our sales goals. We expect to face competition from other companies in our industry, some of whom are much larger than us and who can pay greater compensation and benefits than we can, in seeking to attract and retain qualified sales and marketing employees. If we are unable to hire and retain qualified sales and marketing personnel, and engage qualified distributors, our business will suffer.
Our dependence on commercialization partners for sales of tests could limit our success in realizing revenue growth.
We intend to grow our business through the use of commercialization partners for the sales and marketing of our two oncology products, and to do so we must enter into agreements with these partners to sell, market or commercialize our medical devices. These agreements may contain exclusivity provisions and generally cannot be terminated without cause during the term of the agreement. We may need to attract additional partners to expand the markets in which we sell our medical devices. These partners may not commit the necessary resources to market and sell our two oncology products to the level of our expectations, and we may be unable to locate suitable alternatives should we terminate our agreement with such partners or if such partners terminate their agreement with us. Any relationships we form with commercialization partners are subject to change over time. If we cannot replace any diminution in revenues from a given distributor, our results will be weakened.
If current or future commercialization partners do not perform adequately, or we are unable to locate commercialization partners, we may not realize revenue growth.
We depend on third parties for the supply of blood samples and other biological materials that we use in our research and development efforts. If the costs of such samples and materials increase or our third party suppliers terminate their relationship with us, our business may be materially harmed.
We have relationships with suppliers and institutions that provide us with blood samples and other biological materials that we use in developing and validating our current test and our planned future tests. If one or more suppliers terminate their relationship with us or are unable to meet our requirements for samples, we will need to identify other third parties to provide us with blood samples and biological materials, which could result in a delay in our research and development activities and negatively affect our business. In addition, as we grow, our research and academic institution collaborators may seek additional financial contributions from us, which may negatively affect our results of operations.
We currently rely on third-party suppliers for critical materials needed to build our two oncology products, and any problems experienced by them could result in a delay or interruption of their supply to us.
We currently purchase raw materials for the single-use filter circuits in both of our oncology products under purchase orders and do not have long-term contracts with the supplier of these materials. If suppliers were to delay or stop producing our materials, or if the prices they charge us were to increase significantly, or if they elected not to sell to us, we would need to identify other suppliers. We could experience delays in manufacturing critical components while finding another acceptable supplier, which could impact our results of operations. The changes could also result in increased costs associated with qualifying the new materials and in increased operating costs. Further, any prolonged disruption in a supplier’s operations could have a significant negative impact on our ability to manufacture those filter circuits in a timely manner.
Some of the components used in our current or planned products are currently sole-source, and substitutes for these components might not be able to be obtained easily or may require substantial design or manufacturing modifications. Any significant problem experienced by one of our sole source suppliers may result in a delay or interruption in the supply of components to us until that supplier cures the problem or an alternative source of the component is located and qualified. Any delay or interruption would likely lead to a delay or interruption in our manufacturing operations. The inclusion of substitute components must meet our product specifications and could require us to qualify the new supplier with the appropriate government regulatory authorities.
| 8 |
If we were sued for product liability or professional liability, we could face substantial liabilities that exceed our resources.
The marketing, sale and use of our two oncology products and any future products could lead to the filing of product liability claims against us if someone alleges that our medical devices failed to perform as designed. We may also be subject to liability for errors in the results we provide directly or indirectly to physicians or for a misunderstanding of, or inappropriate reliance upon, the information we provide. A product liability or professional liability claim could result in substantial damages and be costly and time-consuming for us to defend.
Although we believe that our existing product and professional liability insurance is adequate, our insurance may not fully protect us from the financial impact of defending against product liability or professional liability claims. Any product liability or professional liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any product liability lawsuit could damage our reputation; result in the recall of medical devices; or cause current partners to terminate existing agreements and potential partners to seek other partners. Any of these outcomes could impact our results of operations.
We may acquire other businesses or form joint ventures or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, increase our debt or cause us to incur significant expense.
As part of our business strategy, we may pursue acquisitions of businesses and assets. We also may pursue strategic alliances and joint ventures that leverage our core technology and industry experience to expand our offerings or distribution. We have no experience with acquiring other companies and limited experience with forming strategic alliances and joint ventures. We may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could have a material adverse effect on our financial condition, results of operations and cash flows. Integration of an acquired company also may disrupt ongoing operations and require management resources that would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could have a material negative effect on our results of operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance or joint venture.
To finance any acquisitions or joint ventures, we may choose to issue shares of our Common Stock or preferred stock as consideration, which would dilute the ownership of our stockholders. If the price of our Common Stock is low or volatile, if such a market for our Common Stock exists, we may not be able to acquire other companies or fund a joint venture project using our stock as consideration. Alternatively, it may be necessary for us to raise additional funds for acquisitions through public or private financings. Additional funds may not be available on terms that are favorable to us, or at all.
If we cannot support demand for our two oncology products once they achieve commercialization, including successfully managing the evolution of our technology and manufacturing platforms, our business could suffer.
As our volume of medical devices and single-use fluid circuits grows, we will need to increase our manufacturing capacity, implement automation, increase our scale and related processing, customer service, billing, collection and systems process improvements and expand our internal quality assurance program and technology to support those functions on a larger scale. Any increases in scale, related improvements and quality assurance may not be successfully implemented and appropriate personnel may not be available. As additional medical devices are commercialized, we may need to bring new equipment on line, implement new systems, technology, controls and procedures and hire personnel with different qualifications. Failure to implement necessary procedures or to hire the necessary personnel could result in a higher cost of processing or an inability to meet market demand. We cannot assure you that we will be able to manufacture our products once they achieve commercialization on a timely basis at a level consistent with demand, that our efforts to scale our commercial operations will not negatively affect the quality of our products or that we will respond successfully to the growing complexity of our business as it grows. If we encounter difficulty meeting market demand or quality standards for our two oncology products once they achieve commercialization, our reputation could be harmed and our future prospects and business could suffer, which may have a material adverse effect on our financial condition, results of operations and cash flows.
| 9 |
We may encounter manufacturing problems or delays that could result in lost revenue.
We currently manufacture our medical devices on a small scale basis at our Lowell facility and intend to continue to do so. We believe we currently have adequate manufacturing capacity for the first two years after commercialization. If demand for our medical devices increases significantly, we will need to either expand our manufacturing capabilities or outsource to other manufacturers. If we or third party manufacturers engaged by us fail to manufacture and deliver our medical devices in a timely manner, our relationships with our customers could be seriously harmed. We cannot assure you that manufacturing or quality control problems will not arise as we attempt to increase the production or that we can increase our manufacturing capabilities and maintain quality control in a timely manner or at commercially reasonable costs. If we cannot manufacture our medical devices consistently on a timely basis because of these or other factors, it could have a significant negative impact on our ability to perform tests and generate revenues.
International expansion of our business would expose us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the United States.
Our business strategy contemplates international expansion, including partnering with cancer researchers and companies outside the United States, in particular Europe and Canada. Doing business internationally involves a number of risks, including:
| • | multiple, conflicting and changing laws and regulations such as tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses; |
| • | failure by us or our distributors to obtain regulatory approvals for the sale or use of our current test and our planned future tests in various countries; |
| • | difficulties in managing foreign operations; |
| • | complexities associated with managing government payor systems, multiple payor-reimbursement regimes or self-pay systems; |
| • | logistics and regulations associated with shipping products manufactured in one country to another, including infrastructure conditions and transportation delays; |
| • | limits on our ability to penetrate international markets; |
| • | financial risks, such as longer payment cycles, difficulty enforcing contracts and collecting accounts receivable and exposure to foreign currency exchange rate fluctuations; |
| • | reduced protection for intellectual property rights, or lack of them in certain jurisdictions, forcing more reliance on our trade secrets, if available; |
| • | natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; and |
| • | failure to comply with the Foreign Corrupt Practices Act, including its books and records provisions and its anti-bribery provisions, by maintaining accurate information and control over sales activities and distributors’ activities. |
Any of these risks, if encountered, could significantly harm our future international expansion and operations and, consequently, have a material adverse effect on our financial condition, results of operations and cash flows.
Declining general economic or business conditions may have a negative impact on our business.
Continuing concerns over United States health care reform legislation and energy costs, geopolitical issues, the availability and cost of credit and government stimulus programs in the United States and other countries have contributed to increased volatility and diminished expectations for the global economy. These factors, combined with low business and consumer confidence and high unemployment, precipitated an economic slowdown and recession. If the economic climate does not improve, or it deteriorates, our business, including our access to patient samples and the addressable market for diagnostic tests that we may successfully develop, as well as the financial condition of our suppliers and our third-party payors, could be adversely affected, resulting in a negative impact on our business, financial condition and results of operations.
| 10 |
Intrusions into our computer systems could result in compromise of confidential information.
Despite the implementation of security measures, our technology or systems that we interface with, including the Internet and related systems, may be vulnerable to physical break-ins, hackers, improper employee or contractor access, computer viruses, programming errors, or similar problems. Any of these might result in confidential medical, business or other information of other persons or of ourselves being revealed to unauthorized persons.
There are a number of state, federal and international laws protecting the privacy and security of health information and personal data. As part of the American Recovery and Investment Act 2009, or ARRA, Congress amended the privacy and security provisions of the Health Insurance Portability and Accountability Act, or HIPAA. HIPAA imposes limitations on the use and disclosure of an individual’s healthcare information by healthcare providers, healthcare clearinghouses, and health insurance plans, collectively referred to as covered entities. The HIPAA amendments also impose compliance obligations and corresponding penalties for non-compliance on individuals and entities that provide services to healthcare providers and other covered entities, collectively referred to as business associates. ARRA also made significant increases in the penalties for improper use or disclosure of an individual’s health information under HIPAA and extended enforcement authority to state attorneys general. The amendments also create notification requirements for individuals whose health information has been inappropriately accessed or disclosed: notification requirements to federal regulators and in some cases, notification to local and national media. Notification is not required under HIPAA if the health information that is improperly used or disclosed is deemed secured in accordance with encryption or other standards developed by the U.S. Department of Health and Human Services, or HHS. Most states have laws requiring notification of affected individuals and state regulators in the event of a breach of personal information, which is a broader class of information than the health information protected by HIPAA. Many state laws impose significant data security requirements, such as encryption or mandatory contractual terms to ensure ongoing protection of personal information. Activities outside of the United States implicate local and national data protection standards, impose additional compliance requirements and generate additional risks of enforcement for non-compliance. We may be required to expend significant capital and other resources to ensure ongoing compliance with applicable privacy and data security laws, to protect against security breaches and hackers or to alleviate problems caused by such breaches.
We depend on our information technology and telecommunications systems, and any failure of these systems could harm our business.
We depend on information technology and telecommunications systems for significant aspects of our operations. In addition, third-party vendors we may utilize for billing and collections may depend upon telecommunications and data systems provided by outside vendors and information we provide on a regular basis. These information technology and telecommunications systems support a variety of functions, including manufacturing, product tracking, quality control, customer service and support, billing and reimbursement, research and development activities, sales and marketing activities, regulatory and clinical activities, and our general and administrative activities. Information technology and telecommunications systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our information technology and telecommunications systems, failures or significant downtime of our information technology or telecommunications systems or those used by our third-party service providers could prevent us from manufacturing our products or managing our business operations. Any disruption or loss of information technology or telecommunications systems on which critical aspects of our operations depend could have an adverse effect on our business.
| 11 |
Regulatory Risks Relating to Our Business
Healthcare policy changes, including recently enacted legislation reforming the U.S. health care system, may have a material adverse effect on our financial condition, results of operations and cash flows.
The 2010 Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the ACA, makes a number of substantial changes in the way health care is financed by both governmental and private insurers. Among other things, the ACA:
| • | Mandates a reduction in payments for clinical laboratory services paid under the Medicare Clinical Laboratory Fee Schedule annual Consumer Price Index update of 1.75% for the years 2011 through 2015. In addition, a permanent productivity adjustment is made to the fee schedule payment amount, which could range from 1.1% to 1.4% each year over the next 10 years. These changes in payments may apply to some or all of the tests based on DNA sequencing which utilize our molecular diagnostic system which is furnished to Medicare beneficiaries. |
| • | Establishes an Independent Payment Advisory Board to reduce the per capita rate of growth in Medicare spending if spending exceeds a target growth rate. The Independent Payment Advisory Board has broad discretion to propose policies, which may have a negative impact on payment rates for services, including clinical laboratory services and therapeutic treatments such as our therapeutic oncopheresis system, beginning in 2016, and for hospital services beginning in 2020. |
| • | Requires each medical device manufacturer to pay an excise tax equal to 2.3% of the price for which such manufacturer sells its medical devices, beginning in 2013. We believe that at this time this tax does not apply to our current test or to our products that are in development; nevertheless, this could change in the future if either the FDA or the Internal Revenue Service, which regulates the payment of this excise tax, changes its position. |
Although some of these provisions may negatively impact payment rates for clinical laboratory tests and therapeutic treatments, the ACA also extends coverage to over 30 million previously uninsured people, which may result in an increase in the demand for our two oncology products. The mandatory purchase of insurance has been strenuously opposed by a number of state governors, resulting in lawsuits challenging the constitutionality of certain provisions of the ACA. In 2012, the Supreme Court upheld the constitutionality of the ACA, with the exception of certain provisions dealing with the expansion of Medicaid coverage under the law. Therefore, most of the law’s provisions are going into effect in 2013 and 2014. Congress has also proposed a number of legislative initiatives, including possible repeal of the ACA. At this time, it remains unclear whether there will be any changes made to the ACA, whether in part or in its entirety.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. The Budget Control Act of 2011, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers and suppliers of up to 2% per fiscal year, starting in 2013. The full impact on our business of the ACA and the sequester law is uncertain. In addition, the Middle Class Tax Relief and Job Creation Act of 2012, or MCTRJCA, mandated an additional change in Medicare reimbursement for clinical laboratory tests and therapeutic treatments. This legislation requires a rebasing of the Medicare Clinical Laboratory Fee Schedule to effect a 2% reduction in payment rates otherwise determined for 2013. This will serve as a base for 2014 and subsequent years. In January 2013, as a result of the changes mandated by the ACA and MCTRJCA, the Centers for Medicare & Medicaid Services, or CMS, reduced its reimbursement for laboratory tests for 2013 by approximately 3%.
Some of the tests and treatments that might utilize our two oncology products are subject to the Medicare Physician Fee Schedule and, under the current statutory formula, the rates for these services are updated annually. For the past several years, the application of the statutory formula would have resulted in substantial payment reductions if Congress failed to intervene. In the past, Congress passed interim legislation to prevent the decreases. On November 1, 2013, the CMS issued its 2014 Physician Fee Schedule Final Rule, or the 2014 Final Rule. In the 2014 Final Rule, CMS called for a reduction of approximately 23.7% in the 2014 conversion factor that is used to calculate physician reimbursement. If in future years Congress does not adopt interim legislation to block or offset, and/or CMS does not moderate, any substantial CMS-proposed reimbursement reductions, the resulting decrease in payments from Medicare could adversely impact our revenues and results of operations.
| 12 |
In addition, many of the Current Procedure Terminology, or CPT, codes that healthcare providers would use to bill for tests and treatments using our two oncology products once they are commercialized are subject to periodic revision. We cannot predict whether future health care initiatives will be implemented at the federal or state level, or how any future legislation or regulation may affect us. The expansion of government’s role in the U.S. health care industry as a result of the ACA’s implementation as well as changes to the reimbursement amounts paid by Medicare or other payors for our current test and our planned future cancer diagnostic tests may reduce our profits, if any and have a materially adverse effect on our business, financial condition, results of operations and cash flows.
Our commercial success could be compromised if third-party payors, including managed care organizations and Medicare, do not provide coverage and reimbursement, breach, rescind or modify their contracts or reimbursement policies or delay payments for our two oncology products once they are commercialized.
Oncologists may not utilize our two oncology products once they are commercialized unless third-party payors, such as managed care organizations and government payors such as Medicare and Medicaid, pay a substantial portion of the price. Coverage and reimbursement by a third-party payor may depend on a number of factors, including a payor’s determination that tests or treatments using our technologies are:
| • | not experimental or investigational; |
| • | medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; |
| • | supported by peer-reviewed publications; and |
| • | included in clinical practice guidelines. |
Uncertainty surrounds third-party payor reimbursement of any test or treatment incorporating new technology, including our two oncology products. Technology assessments of new medical devices conducted by research centers and other entities may be disseminated to interested parties for informational purposes. Third-party payors and health care providers may use such technology assessments as grounds to deny coverage for a test or procedure which uses those medical devices. Technology assessments can include evaluation of clinical studies, which define how a medical device is used in a particular clinical setting or situation.
Because each payor generally determines for its own enrollees or insured patients whether to cover or otherwise establish a policy to reimburse our two oncology products once they are commercialized, seeking payor approvals is a time-consuming and costly process. We cannot be certain that coverage for our two oncology products will be provided in the future. If we cannot obtain coverage and reimbursement from private and governmental payors such as Medicare and Medicaid for our two oncology products once they are commercialized, our ability to generate revenues could be limited, which may have a material adverse effect on our financial condition, results of operations and cash flow.
We will indirectly depend on Medicare and a limited number of private payors for a significant portion of our revenues and if these or other payors stop providing reimbursement or decrease the amount of reimbursement for our two oncology products once they are commercialized, our revenues could suffer.
We believe that our two oncology products once they are commercialized will be used by a significant number of patients with Medicare coverage. While we will sell our medical devices to healthcare providers or clinical testing services, who in turn will bill Medicare for tests or treatments that utilize our medical devices, we cannot provide you with assurance that Medicare and other third-party payors will not change their coverage policies or cancel future contracts with such healthcare providers at any time, review and adjust the rate of reimbursement or stop paying for our medical devices altogether, which would reduce our total revenues.
| 13 |
Payors have increased their efforts to control the cost, utilization and delivery of health care services. In the past, measures have been undertaken to reduce payment rates for and decrease utilization of the clinical laboratory testing and therapeutic treatments generally. Because of the cost-trimming trends, third-party payors that may cover and provide reimbursement for our two oncology products may suspend, revoke or discontinue coverage at any time, or may reduce the reimbursement rates payable to healthcare providers who might utilize our medical devices. Any such action could have a negative impact on our revenues, which may have a material adverse effect on our financial condition, results of operations and cash flows.
Long payment cycles of Medicare, Medicaid and/or other third-party payors, or other payment delays, could hurt our cash flows and increase our need for working capital.
Medicare and Medicaid have complex billing and documentation requirements that the healthcare providers and clinical testing services who might utilize our medical devices must satisfy in order to receive payment, and the programs can be expected to carefully audit and monitor their compliance with these requirements. Those healthcare providers must also comply with numerous other laws applicable to billing and payment for healthcare services, including privacy laws. Failure to comply with these requirements may result in non-payment, refunds, exclusion from government healthcare programs, and civil or criminal liabilities, any of which may have a material adverse effect on our revenues and earnings. In addition, failure by third-party payors to properly process payment claims to those healthcare providers or clinical testing services in a timely manner could delay our receipt of payment for our products, which may have a material adverse effect on our cash flows.
If we do not receive regulatory approvals, we may not be able to commercialize our technology.
We will need FDA approval to market our products in the United States and approvals from foreign regulatory authorities to market products outside the United States including validation studies of the materials and manufacturing processes used in our devices and pre-clinical testing to demonstrate safety in animals, as well human clinical studies to demonstrate efficacy for specific disease conditions. We have not yet filed an application with the FDA to obtain approval to market any of our proposed products. If we fail to obtain regulatory approval for the marketing of products, we will be unable to sell such products and will not be able to sustain operations.
If we seek to market our two oncology products in Europe, we need to receive a CE Mark. If we do not obtain a CE Mark for our products, we will be unable to sell these products in Europe and countries that recognize the CE Mark.
The regulatory review and approval process, which may include evaluation of preclinical studies and clinical trials of products, as well as the evaluation of manufacturing processes, is lengthy, expensive and uncertain. Securing regulatory approval for our oncology products may require the submission of extensive preclinical and clinical data and supporting information to regulatory authorities to establish such products’ safety and effectiveness for each indication.
Regulatory authorities generally have substantial discretion in the approval process and may either refuse to accept an application, or may decide after review of an application that the data submitted is insufficient to allow approval of the product. If regulatory authorities do not accept or approve our applications, they may require that we conduct additional clinical, preclinical or manufacturing studies and submit that data before regulatory authorities will reconsider such application. We may need to expend substantial resources to conduct further studies to obtain data that regulatory authorities believe is sufficient. Depending on the extent of these studies, approval of applications may be delayed by several years, or may require us to expend more resources than we may have available. It is also possible that additional studies may not suffice to make applications approvable. If any of these outcomes occur, we may be forced to abandon our applications for approval, which might cause us to cease operations.
We are subject to federal and state healthcare fraud and abuse laws and regulations and could face substantial penalties if we are unable to fully comply with such laws.
We are subject to health care fraud and abuse regulation and enforcement by both the federal government and the states in which we conduct our business. These health care laws and regulations include, for example:
| 14 |
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from soliciting, receiving, offering or providing remuneration, directly or indirectly, in return for or to induce either the referral of an individual for, or the purchase order or recommendation of, any item or services for which payment may be made under a federal health care program such as the Medicare and Medicaid programs; |
| • | the federal physician self-referral prohibition, commonly known as the Stark Law, which prohibits physicians from referring Medicare or Medicaid patients to providers of “designated health services” with whom the physician or a member of the physician’s immediate family has an ownership interest or compensation arrangement, unless a statutory or regulatory exception applies; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which established federal crimes for knowingly and willfully executing a scheme to defraud any health care benefit program or making false statements in connection with the delivery of or payment for health care benefits, items or services; |
| • | federal false claims laws, which, prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, false or fraudulent claims for payment to the federal government; and |
| • | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payor, including commercial insurers. |
The risk of our being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Further, the ACA, among other things, amends the intent requirement of the federal Anti-Kickback Statute and certain criminal health care fraud statutes. Where the intent requirement has been lowered, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, because of amendments enacted in 2009 as part of the Fraud Enforcement and Recovery Act, the government may now assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes. Any action brought against us for violation of these laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If our operations are found to be in violation of any of these laws and regulations, we may be subject to any applicable penalty associated with the violation, including civil and criminal penalties, damages and fines, and/or exclusion from participation in Medicare or state or federal health care programs, we could be required to refund payments received by us, and we could be required to curtail or cease our operations. Any of the foregoing consequences could seriously harm our business and our financial results.
Clinical research is heavily regulated and failure to comply with human subject protection regulations may disrupt our research program leading to significant expense, regulatory enforcement, private lawsuits and reputational damage.
Clinical research is subject to federal, state and, for studies conducted outside of the United States, international regulation. At the federal level, the FDA imposes regulations for the protection of human subjects and requirements such as initial and ongoing institutional board review; informed consent requirements, adverse event reporting and other protections to minimize the risk and maximize the benefit to research participants. Many states impose human subject protection laws that mirror or in some cases exceed federal requirements. HIPAA also regulates the use and disclosure of Protected Health Information in connection with research activities. Research conducted overseas is subject to a variety of national protections such as mandatory ethics committee review, as well as laws regulating the use, disclosure and cross-border transfer of personal data. The costs of compliance with these laws may be significant and compliance with regulatory requirements may result in delay. Noncompliance may disrupt our research and result in data that is unacceptable to regulatory authorities, data lock or other sanctions that may significantly disrupt our operations.
| 15 |
Intellectual Property Risks Related to Our Business
Our collaborators may assert ownership or commercial rights to inventions we develop from our use of the biological materials which they provide to us, or otherwise arising from the collaboration.
We collaborate with various institutions, physicians, researchers and companies in scientific matters. We may not always have written agreements with certain of such collaborators, or the written agreements we have do not cover intellectual property rights. Also, we rely on numerous third parties to provide us with blood samples and biological materials that we use to develop tests. If we cannot successfully negotiate sufficient ownership and commercial rights to any inventions that result from our use of a third party collaborator’s materials, or if disputes arise with respect to the intellectual property developed with the use of a collaborator’s samples, or data developed in a collaborator’s study, we may be limited in our ability to capitalize on the market potential of these inventions or developments.
If we are unable to maintain intellectual property protection, our competitive position could be harmed.
Our ability to protect our proprietary discoveries and technologies affects our ability to compete and to achieve sustained profitability. Currently, we rely on a combination of U.S. and foreign patent applications, copyrights, trademarks and trademark applications, confidentiality or non-disclosure agreements, material transfer agreements, consulting agreements, work-for-hire agreements and invention assignment agreements to protect our intellectual property rights. We also maintain certain company know-how, trade secrets and technological innovations designed to provide us with a competitive advantage in the market place as trade secrets. Currently, we own over ten U.S. and foreign patent applications (all of which are in the review stage and have not been issued), and we do not license any patent. While we intend to pursue additional patent applications, it is possible that our pending patent applications and any future applications may not result in issued patents. Even if patents are issued, third parties may independently develop similar or competing technology that avoids our patents. Further, we cannot be certain that the steps we have taken will prevent the misappropriation of our trade secrets and other confidential information as well as the misuse of our patents and other intellectual property, particularly in foreign countries where we have not filed for patent protection.
From time to time the U.S. Supreme Court, other federal courts, the U.S. Congress or the U.S. Patent and Trademark Office, or USPTO, may change the standards of patentability and any such changes could have a negative impact on our business.
We may face intellectual property infringement claims that could be time-consuming and costly to defend, and could result in our loss of significant rights and the assessment of treble damages.
From time to time we may face intellectual property infringement or misappropriation claims from third parties. Some of these claims may lead to litigation. The outcome of any such litigation can never be guaranteed, and an adverse outcome could affect us negatively. For example, were a third-party to succeed on an infringement claim against us, we may be required to pay substantial damages, including treble damages if such infringement were found to be willful. In addition, we could face an injunction, barring us from conducting the allegedly infringing activity. The outcome of the litigation could require us to enter into a license agreement which may not be pursuant to acceptable or commercially reasonable or practical terms or which may not be available at all.
It is also possible that an adverse finding of infringement against us may require us to dedicate substantial resources and time in developing non-infringing alternatives, which may or may not be possible. In the case of diagnostic tests, we would also need to include non-infringing technologies which would require us to re-validate the test. Any such re-validation, in addition to being costly and time consuming, may be unsuccessful.
Finally, we may initiate claims to assert or defend our own intellectual property against third parties. Any intellectual property litigation, irrespective of whether we are the plaintiff or the defendant, and regardless of the outcome, is expensive and time-consuming, and could divert our management’s attention from our business and negatively affect our operating results or financial condition.
| 16 |
Risks Relating to Our Common Stock
Our Common Stock is considered a “penny stock” and will likely continue to be a penny stock in the public market. The application of the “penny stock” rules to our Common Stock could limit the trading and liquidity of our Common Stock, adversely affect the market price of our Common Stock and increase the transaction costs to sell those shares.
We anticipate applying for quotation of our Common Stock on the OTCBB upon the effectiveness of the registration statement of which this prospectus forms a part. Our Common Stock is a “low-priced” security or “penny stock” under rules promulgated under the Securities Exchange Act of 1934, as amended, and, assuming that a public market for our Common Stock develops, it will likely continue to be a penny stock. In accordance with these rules, broker-dealers participating in transactions in low-priced securities must first deliver a risk disclosure document which describes the risks associated with such stocks, the broker-dealer’s duties in selling the stock, the customer’s rights and remedies and certain market and other information. Furthermore, the broker-dealer must make a suitability determination approving the customer for low-priced stock transactions based on the customer’s financial situation, investment experience and objectives. Broker-dealers must also disclose these restrictions in writing to the customer, obtain specific written consent from the customer, and provide monthly account statements to the customer. The effect of these restrictions will likely decrease the willingness of broker-dealers to make a market in our Common Stock, will decrease liquidity of our Common Stock and will increase transaction costs for sales and purchases of our Common Stock as compared to other securities.
The stock market in general has experienced volatility that often has been unrelated to the operating performance of companies. These broad fluctuations may be the result of unscrupulous practices that may adversely affect the price of our stock, regardless of our operating performance.
Investors should be aware that, according to SEC Release No. 34-29093 dated April 17, 1991, the market for penny stocks has suffered from patterns of fraud and abuse. Such patterns include (1) control of the market for the security by one or a few broker-dealers that are often related to the promoter or issuer; (2) manipulation of prices through prearranged matching of purchases and sales and false and misleading press releases; (3) boiler room practices involving high-pressure sales tactics and unrealistic price projections by inexperienced sales persons; (4) excessive and undisclosed bid-ask differential and markups by selling broker-dealers; and (5) the wholesale dumping of the same securities by promoters and broker-dealers after prices have been manipulated to a desired level, along with the resulting inevitable collapse of those prices and with consequent investor losses. The occurrence of these patterns or practices could increase the volatility of our share price.
Future issuances of additional shares of our Common Stock will dilute stockholders.
In the future, we may issue our authorized but previously unissued equity securities, resulting in the dilution of the ownership interests of our present stockholders. We may also issue additional shares of our Common Stock or other securities that are convertible into or exercisable for Common Stock in connection with hiring or retaining employees or consultants, future acquisitions, future sales of our securities for capital raising purposes, or for other business purposes, including at a price (or exercise prices) below the price at which shares of our Common Stock are currently offered. The future issuance of any such additional shares of our Common Stock or other securities may create downward pressure on the trading price of our Common Stock.
There is no assurance that shares of our Common Stock will trade on a public market or will ever trade on a recognized exchange. Therefore you may be unable to liquidate your investment in our stock.
There is no established public trading market for our Common Stock. Our shares have not been listed or quoted on any exchange or quotation system. There can be no assurance that a market maker will agree to file the necessary documents with FINRA, which operates the OTCBB, nor can there be any assurance that such an application for quotation on the OTCBB or any other exchange or OTC Market will be approved or that a regular trading market will develop or that if developed, will be sustained. In the absence of a trading market, an investor may be unable to liquidate his investment. There can be no assurance that volume, liquidity or a regular trading market for our Common Stock will develop or that, if developed, will be sustained.
| 17 |
Our largest stockholder, Chief Executive Officer and Chairman of the Board, Mr. Ilan Reich, has the ability to control our business direction and could delay or prevent a change in corporate control.
Our largest stockholder, Chief Executive Officer and Chairman of the Board, Mr. Ilan Reich, has the ability to control our business by virtue of his ownership of approximately 14.8% of issued and outstanding shares of our Common Stock and 100% of our issued and outstanding Series A Participating Preferred Stock (“Series A Preferred Stock”). As specified in the certificate of designation for Series A Preferred Stock, any matter submitted to a vote of stockholders shall be approved by the holders of a majority of the outstanding shares of Series A Preferred Stock, voting as a separate class in addition to approval of holders of Common Stock or any other junior securities voting together with Common Stock. In addition, pursuant our bylaws, only holders of Series A Preferred Stock can remove any or all directors for cause or without cause. As such, Mr. Reich is likely in a position to control the Board and the officers and will also have controlling influence over the outcome of all corporate transactions or other matters, including mergers, consolidations and the sale of all or substantially all of our assets, and also the power to prevent or cause a change in control. While we have no current plans with regard to any merger, consolidation or sale of substantially all of our assets, the interests of Mr. Reich may still differ from the interests of the other stockholders and this concentration of ownership might harm the value of our Company.
If we are unable to favorably assess the effectiveness of our internal control over financial reporting, investors may lose confidence in our financial reporting and our stock price could be materially adversely affected if there exists a market for our Common Stock.
We are subject to the requirements of Section 404 of the Sarbanes-Oxley Act of 2002 and are required to document and test our internal control over financial reporting. If we fail to remediate any significant deficiencies or material weaknesses in internal controls that may be identified in the future, we may be unable to conclude that our internal control over financial reporting is effective. Under the JOBS Act, issuers that qualify as “emerging growth companies” will not be required to provide an auditor’s attestation report on internal controls for so long as the issuer qualifies as an emerging growth company. We currently qualify as an emerging growth company under the JOBS Act and we may choose not to provide an auditor’s attestation report on internal controls. However, if we cannot favorably assess the effectiveness of our internal control over financial reporting, or if we require an attestation report from our independent registered public accounting firm in the future and that firm is unable to provide an unqualified attestation report on the effectiveness of our internal controls over financial reporting, investor confidence and, in turn, our stock price could be materially adversely affected if there exists such a market for our Common Stock.
We qualify as an “emerging growth company,” and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our Common Stock less attractive to investors.
We qualify as an emerging growth company, as defined in the JOBS Act. For as long as we continue to be qualified as an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements and exemptions from the requirements of holding nonbinding advisory votes on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the market value of our Common Stock held by non-affiliates exceeds $700 million as of any June 30 before that time or if we have total annual gross revenue of $1.0 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three year period before that time, we would cease to be an emerging growth company immediately. Even after we no longer qualify as an emerging growth company, we may still qualify as a “smaller reporting company” which would allow us to take advantage of many of the same exemptions from disclosure requirements including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in registration statements, periodic reports and proxy statements. We cannot predict if investors will find our Common Stock less attractive because we may rely on these exemptions. If some investors find our Common Stock less attractive as a result, there may be a less active trading market for our Common Stock and our stock price may be more volatile if such a market for our Common Stock exists.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have elected to take advantage of the benefits of this extended transition period. Our financial statements may therefore not be comparable to those of companies that comply with such new or revised accounting standards.
| 18 |
Anti-takeover provisions of our certificate of incorporation, our bylaws and Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove the current members of our board and management.
Certain provisions of our certificate of incorporation and bylaws could discourage, delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. Our certificate of incorporation and bylaws include provisions that:
| • | Require that any matter submitted to stockholder vote shall be approved by the holders of Series A Preferred Stock, voting as a separate class in addition to approval of holders of Common Stock or any other junior securities voting together with Common Stock; |
| • | Authorize our board of directors to issue up to 20,000,000 shares of preferred stock in one or more series and to fix the powers, preferences and rights of each series without stockholder approval; |
| • | Provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in a timely manner, and also specify requirements as to the form and content of such notice; |
| • | Provide that any or all of the directors may be only removed for cause or without cause by the vote of a majority of the shares of the Series A Preferred Stock; |
| • | Permit corporate actions to be approved by written consent of stockholders without a meeting, without prior notice and without a vote; |
| • | Authorize the board of directors to determine the number of directors and to fill vacancies on the board of directors; |
| • | Specify that only the board of directors or officers instructed by the board may call a special meeting of the stockholders; and |
| • | Provide that our certificate of incorporation can only be altered, amended or repealed by the affirmative vote of the holders of a majority of all of the then outstanding shares of Series A Preferred Stock, voting as a single class, provided that there are at least 700,000 shares of Series A Preferred Stock outstanding. |
These provisions, alone or together, could delay or prevent hostile takeovers and changes in control. These provisions may also frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, or DGCL, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our Common Stock, from merging or combining with us for a prescribed period of time.
Holders of our Series A Preferred Stock have voting rights that adversely affect voting rights of common stockholders.
Under our certificate of incorporation, bylaws and certificate of designation for Series A Preferred Stock, holders of our Series A Preferred Stock have certain rights, preferences and privileges that adversely affect the rights of the holders of our Common Stock, including but not limited to the following:
| • | our certificate of incorporation can only be altered, amended or repealed by the affirmative vote of the holders of a majority of all of the then outstanding shares of Series A Preferred Stock, voting as a single class, provided that there are at least 700,000 shares of Series A Preferred Stock outstanding; |
| • | any or all of the directors may be only removed for cause or without cause by the vote of a majority of the shares of the Series A Preferred Stock; and |
| • | the amendment, repeal or adoption of new bylaws of the Company is subject to the approval of holders of Series A Preferred Stock. |
| 19 |
These rights shift the ability to control the Company to the holders of Series A Preferred Stock and could negatively affect the market for our Common Stock, if any.
We do not expect to pay cash dividends for the foreseeable future.
We do not intend to pay cash dividends on shares of our Common Stock for the foreseeable future. Any determination to pay dividends in the future will be at the discretion of our board of directors and will depend upon results of operations, financial performance, contractual restrictions, restrictions imposed by applicable law and other factors our board of directors deems relevant. Accordingly, you will have to rely on capital appreciation, if any, to earn a return on your investment in our Common Stock. Investors seeking cash dividends in the foreseeable future should not purchase our Common Stock.
We may raise capital by issuing securities that may result in dilution to our existing stockholders or contain rights, preferences or privileges senior to our Common Stock. Issuing any such securities may adversely affect the value of your investment.
If we raise additional funds through the issuance of equity or convertible debt securities, the percentage ownership of the Company held by existing stockholders will be reduced and our stockholders may experience significant dilution. In addition, new securities may contain rights, preferences or privileges that are senior to those of our Common Stock. Issuing a significant number of shares of Common Stock or securities that are senior to our Common Stock could have a material adverse effect on the market price of our Common Stock and result in a decline in the value of your investment.
Future sales by our stockholders may adversely affect our stock price and our ability to raise funds.
1,202,071 shares of our Common Stock including 37,500 shares of Common Stock issuable upon exercise of certain warrants could be sold in the public market if the registration statement of which this prospectus is a part is declared effective by the SEC. In addition, 15,086,159 shares of Common Stock (including 347,500 shares of Common Stock issuable upon exercise of certain warrants) will be eligible under Rule 144 for sale in the public market from time to time after the effective date of the registration statement, subject to the volume limitation, public notice, manner-of-sale and current public information requirements of Rule 144. Sales of a significant number of shares of our Common Stock in the public market could lower market price for our Common Stock. Sales may also make it more difficult for us to sell equity securities or equity-related securities in the future at a time and price that management deems acceptable or at all.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus, including the sections entitled “Prospectus Summary,” “Risk Factors,” “Use of Proceeds,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and “Business,” contains forward-looking statements. The words “believe,” “may,” “will,” “potentially,” “estimate,” “continue,” “anticipate,” “intend,” “could,” “would,” “project,” “plan” “expect,” and similar expressions that convey uncertainty of future events or outcomes are intended to identify forward-looking statements.
These forward-looking statements include, but are not limited to, statements concerning the following:
| · | our ability to maintain an adequate rate of revenue growth; |
| · | our business plan and our ability to effectively manage our growth; |
| · | costs associated with defending intellectual property infringement and other claims ; |
| · | our ability to attract and retain end-customers; |
| · | our ability to further penetrate our existing customer base; |
| · | our ability to displace existing products in established markets; |
| · |
| · | our ability to timely and effectively scale and adapt our existing technology; |
| · | our ability to innovate new products and bring them to market in a timely manner; |
| · | our ability to expand internationally; |
| · | the effects of increased competition in our market and our ability to compete effectively; |
| 20 |
| · | the effects of seasonal trends on our results of operations; |
| · | our expectations concerning relationships with third parties; |
| · | the attraction and retention of qualified employees and key personnel; |
| · | our ability to maintain, protect, and enhance our brand and intellectual property; and |
| · | future acquisitions of or investments in complementary companies, products, services or technologies. |
These forward-looking statements are subject to a number of risks, uncertainties, and assumptions, including those described in “Risk Factors” and elsewhere in this prospectus. Moreover, we operate in a very competitive and rapidly changing environment, and new risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties, and assumptions, the forward-looking events and circumstances discussed in this prospectus may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements.
You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. We undertake no obligation to update publicly any forward-looking statements for any reason after the date of this prospectus to conform these statements to actual results or to changes in our expectations, except as required by law.
You should read this prospectus and the documents that we reference in this prospectus and have filed with the SEC as exhibits to the registration statement of which this prospectus is a part with the understanding that our actual future results, levels of activity, performance, and events and circumstances may be materially different from what we expect.
MARKET AND INDUSTRY DATA
The industry and market data presented in this prospectus are inherently estimates and are based upon third-party data, information made public by online market research, brokerage analyst reports, internet companies and our own internal estimates. While we believe that this data is reasonable, in some cases this data is based on our own and others’ estimates. Accordingly, you are cautioned not to place undue reliance on the industry and market data included in this prospectus.
The net proceeds from any disposition of the shares covered hereby would be received by the selling stockholders. We will not receive any of the proceeds from any such sale of the Common Stock offered by this prospectus.
MARKET FOR COMMON EQUITY AND RELATED SHAREHOLDER MATTERS
There is currently no established public trading market for our Common Stock. We anticipate applying for quotation of our Common Stock on the OTCBB upon the effectiveness of the registration statement of which this prospectus forms a part. To have our securities quoted on the OTCBB we must be a company that (1) reports its current financial information to the SEC, banking regulators or insurance regulators; and (2) has at least one market maker who completes and files a Form 211 with FINRA. The OTCBB differ substantially from national and regional stock exchanges because they: (a) operates through communication of bids, offers and confirmations between broker-dealers, rather than one centralized market or exchange; and (b) securities admitted to quotation are offered by one or more broker-dealers rather than “specialists” which operate in stock exchanges. We recently engaged a market maker to assist us to apply for quotation on the OTCBB but we are not able to determine the length of time that such application process will take. Such time frame is dependent on comments we receive, if any, from FINRA regarding our Form 211 application.
| 21 |
We have approximately 106 record holders of our Common Stock as of November 25, 2014. The transfer agent and registrar for our Common Stock is VStock Transfer, LLC, 77 Spruce Street, Suite 201, Cedarhurst, NY 11516.
We have never declared or paid any cash dividends on our Common Stock. We currently intend to retain any future earnings and do not expect to pay any cash dividends on our Common Stock for the foreseeable future. Any determination to pay dividends in the future will be at the discretion of our board of directors and will be dependent on a number of factors, including our earnings, capital requirements and overall financial conditions.
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes included elsewhere in this prospectus.
Overview
We are a biotechnology company focused on developing and marketing cancer molecular diagnostics and cancer therapy products, both of which are based on the principle of removing blood-borne circulating tumor cells (CTCs). We are seeking to commercialize two products which utilize our proprietary CTC removal technology:
| · | The ViatarTM Collection System for Molecular Analysis is designed to collect and purify a statistically significant quantity of CTCs from up to 50 mL of blood as the “front end” for DNA sequencing and other genetic analysis technologies used primarily for research. |
| · | The ViatarTM Therapeutic Oncopheresis System is designed to remove CTCs from a patient’s blood as a new cancer therapy for metastatic disease. |
We anticipate launching the ViatarTM Collection System for Molecular Analysis during 2016 with a CE Mark in Europe and Canada, followed by a 510K designation in the United States. We are currently engaged in research collaborations with several CTC platform technology companies with the goal of marketing and selling the ViatarTM Collection System for Molecular Analysis through private label and distribution arrangements.
We anticipate launching the ViatarTM Therapeutic Oncopheresis System during 2016 with a CE Mark in Europe and Canada. Introduction in the United States will take several years, based on the anticipated PMA classification by the FDA. We anticipate marketing and selling the ViatarTM Therapeutic Oncopheresis System on a direct basis through our own dedicated sales force in key large markets (such as the United States, Germany, France, Italy, UK and Japan), and through distributors in other markets.
Key Factors Affecting our Results of Operations and Financial Condition
Our overall long-term growth plan depends on our ability to develop and commercialize our two oncology products. We are working with several CTC-based diagnostic companies to optimize our ViatarTM Collection System for Molecular Analysis to their process flow, and we intend to enter into additional such collaborations in the coming year. These activities will help facilitate market adoption of that product, and we anticipate having to complete various studies with clinical samples to demonstrate its efficacy. We will also seek to publish the results of those studies in peer-reviewed scientific journals. Our ability to complete such clinical studies, as well as to perform clinical studies for the ViatarTM Therapeutic Oncopheresis System is dependent on our ability to foster strong collaborative relationships with cancer researchers, other CTC technology platform companies, and companies which make DNA sequencing equipment or perform such services; to conduct the appropriate clinical studies; and to obtain favorable clinical data.
We did not generate any revenue in 2012 or 2013 from commercial sales. Assuming we are successful in completing development activities for the ViatarTM Collection System for Molecular Analysis, we expect to generate revenues during 2016 from sales of that product. However, significant revenues and growth will depend on market adoption of that product. Similarly, assuming we are successful in completing development activities for the ViatarTM Therapeutic Oncopheresis System, we expect to generate revenues in 2016; however, significant revenues and growth will depend on clinical studies showing its efficacy in mitigating the spread and growth of metastatic tumors and regulatory approval in the US.
| 22 |
Our operating expenses consist of general and administrative; research and development; clinical and regulatory; and business development. These expenses principally consist of personnel costs, outside services (such as cost of supporting clinical studies and regulatory consultants), laboratory equipment and consumables, rent and overhead, and legal and accounting fees. All of these expense categories will increase in the near-term, principally as a result of hiring additional personnel and contracting with outside engineering firms to complete the development of our two products; expanding the range and scope of clinical trials (both with cancer researchers as well as companies we collaborate with); hiring additional personnel to handle business development, sales and marketing activities; and hiring additional personnel or outside services to handle administrative functions. In addition, there is approximately $347,000 of past due invoices to service providers that need to be scheduled for payment as of September 30, 2014.
We expect there to be an element of seasonality to our business, as research activities and patient treatment or monitoring with CTC-based tests is likely to decline during vacation and holiday seasons. We expect this trend of seasonality to continue for the foreseeable future.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of our financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expenses and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates based on historical experience and make various assumptions, which management believes to be reasonable under the circumstances, which form the basis for judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
The notes to our financial statements, which are included elsewhere in this prospectus, contain a summary of our significant accounting policies. We consider the accounting policies discussed below as being critical to the understanding of our current and future results of our operations.
Revenue Recognition
We plan to recognize revenue in accordance with ASC 605, Revenue Recognition, and ASC 954-605, Health Care Entities, Revenue Recognition which requires that four basic criteria must be met before revenue can be recognized: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred and title and the risks and rewards of ownership have been transferred to the client or services have been rendered; (3) the price is fixed or determinable; and (4) collectability is reasonably assured. For contract partners, revenue is recorded based upon the contractually agreed upon fee schedule. When assessing collectability, we will consider whether we have sufficient payment history to reliably estimate a payor’s individual payment patterns.
Accounts Receivable and Bad Debts
We will carry accounts receivable at original invoice amounts, less an estimate for doubtful receivables, based on a review of all outstanding amounts on a periodic basis. The estimate for doubtful receivables will be determined from an analysis of the accounts receivable on a quarterly basis, and be recorded as bad debt expense. Since we will only recognize revenue to the extent we expect to collect such amounts, bad debt expense related to receivables from product revenue will be recorded in general and administrative expense in the statements of operations. Accounts receivable will be written off when deemed uncollectible. Recoveries of accounts receivable previously written off will be recorded when received.
| 23 |
Stock-Based Compensation Expense
In December 2004, the FASB issued ASC 718, Compensation – Stock Compensation, or ASC 718. Under ASC 718, companies are required to measure the compensation costs of share-based compensation arrangements based on the grant-date fair value and recognize the costs in the financial statements over the period during which employees are required to provide services. Share-based compensation arrangements include options, restricted stock, restricted stock units, performance-based awards, share appreciation rights, and employee share purchase plans. As such, compensation cost is measured on the date of grant at fair value. The Company amortizes such compensation amounts, if any, over the respective vesting periods of the award. The Company uses the Black-Scholes-Merton option pricing model, or the Black-Scholes Model, an acceptable model in accordance with ASC 718, that requires the input of highly complex and subjective variables, including the expected life of the award and the expected stock price volatility over a period equal to or greater than the expected life of the award.
Equity instruments (“instruments”) issued to anyone other than employees are recorded on the basis of the fair value of the instruments, as required by ASC 718. ASC 505, Equity Based Payments to Non-Employees, or ASC 505, defines the measurement date and recognition period for such instruments. In general, the measurement date is when either (a) a performance commitment, as defined, is reached or (b) the earlier of (i) the non-employee performance is complete or (ii) the instruments are vested. The measured value related to the instruments is recognized over a period based on the facts and circumstances of each particular grant as defined in ASC 505.
The Company recognizes the compensation expense for all share-based compensation granted based on the grant date fair value estimated in accordance with ASC 718. The Company generally recognizes the compensation expense on a straight-line basis over the employee’s requisite service period.
Results of Operations
Quarters Ended September 30, 2014 and 2013
The following table sets forth certain information concerning our results of operations for the periods shown:
| Quarters Ended September 30, | Change | |||||||||||||||
| 2014 | 2013 | Dollars | Percent | |||||||||||||
| Revenue | ||||||||||||||||
| Sales | $ | - | $ | - | $ | - | 0 | % | ||||||||
| Expenses | ||||||||||||||||
| Research and Development | ||||||||||||||||
| Research and Development | 548,157 | 168,082 | 380,075 | 226 | % | |||||||||||
| Clinical & Regulatory | 4,164 | 33,800 | (29,636 | ) | (88 | )% | ||||||||||
| Total R&D | 552,321 | 201,882 | 350,439 | 174 | % | |||||||||||
| General and Administrative | ||||||||||||||||
| General and Administrative | 133,491 | 84,781 | 48,710 | 57 | % | |||||||||||
| Business Development | 2,540 | 13,627 | (11,087 | ) | (81 | )% | ||||||||||
| Total G&A | 136,031 | 98,408 | 37,623 | 38 | % | |||||||||||
| Total Expenses | $ | 688,352 | $ | 300,290 | $ | 388,062 | 129 | % | ||||||||
Revenue
We did not have any Commercial sales for the quarter ended September 30, 2014. We do not expect to achieve significant sales until we obtain product certification.
Expenses
Research and Development Expenses
Research and development (“R&D”) expenses consist of R&D and clinical trial and regulatory expenses.
| 24 |
R&D Expenses
Research and development expenses were $548,157 for the quarter ended September 30, 2014, an increase of $380,075 or 226%, compared with $168,082 for the quarter ended September 30, 2013. The increase was due to higher staffing levels and more supplies to support the oncology program, including use of an on-site biological testing laboratory. In addition, the Company awarded restricted stock to employees issued pursuant to the 2014 Equity Incentive Plan which resulted in an immediate expense of $346,379 recorded to research and development.
Clinical Trial and Regulatory Expenses
Clinical trial and regulatory expenses were $4,164 for the quarter ended September 30, 2014, a decrease of $29,636, or 88%, compared with $33,800 for the quarter ended September 30, 2013. The decrease was due to fewer clinical trials and regulatory requirements during the quarter ended September 30, 2014 as compared to the corresponding quarter in the prior year.
General and Administrative Expenses
General and administrative (“G&A”) expenses consist of G&A and business development expenses.
G&A Expenses
G&A expenses were $133,491 for the quarter ended September 30, 2014, an increase of $48,710 or 57%, compared with $84,781 for the quarter ended September 30, 2013. The increase was due to financial advisory, legal and accounting expenses incurred in conjunction with the preparation of a private placement memorandum for fundraising activities and a draft registration statement for a public listing.
Business Development Expenses
Business development expenses were $2,540 for the quarter ended September 30, 2014, a decrease of $11,087, or 81%, compared with $13,627 for the quarter ended September 30, 2013. The decrease was due to participation in fewer meetings and events to attract new investors.
Other Income
Change in Value of Derivative Liability
The Company recorded a gain on the change in fair value of the Company’s embedded derivative liability of $0 during the quarter ended September 30, 2014 as compared to a gain of $5,400 during the quarter ended September 30, 2013. The change is mainly attributable to the elimination of the derivative liability.
Net Loss
Net loss was $706,015 for the quarter ended September 30, 2014, an increase of $404,942 or 135%, compared with $301,073 for the quarter ended September 30, 2013. The increase was primarily due to higher expenses, particularly, the stock based compensation to employees.
| 25 |
Nine Months Ended September 30, 2014 and 2013
The following table sets forth certain information concerning our results of operations for the periods shown:
| Nine Months Ended September 30, | Change | |||||||||||||||
| 2014 | 2013 | Dollars | Percent | |||||||||||||
| Revenue | ||||||||||||||||
| Sales | $ | 9,000 | $ | - | $ | 9,000 | 100 | % | ||||||||
| Expenses | ||||||||||||||||
| Research and Development | ||||||||||||||||
| Research and Development | 907,152 | 532,178 | 374,974 | 70 | % | |||||||||||
| Clinical & Regulatory | 75,298 | 73,800 | 1,498 | 2 | % | |||||||||||
| Total R&D | 982,450 | 605,978 | 376,472 | 62 | % | |||||||||||
| General and Administrative | ||||||||||||||||
| General and Administrative | 772,159 | 194,225 | 577,934 | 298 | % | |||||||||||
| Business Development | 34,017 | 38,579 | (4,562 | ) | (12 | )% | ||||||||||
| Total G&A | 806,176 | 232,804 | 573,372 | 246 | % | |||||||||||
| Total Expenses | $ | 1,788,626 | $ | 838,782 | $ | 949,844 | 113 | % | ||||||||
Revenue
Commercial sales of $9,000 for the nine months ended September 30, 2014 arose in connection with evaluation testing of our filters being conducted by a diagnostic CTC company. We do not expect to achieve significant sales until we obtain product certification.
Expenses
R&D Expenses
R&D expenses consist of R&D and clinical trial and regulatory expenses.
R&D Expenses
R&D expenses were $907,152 for the nine months ended September 30, 2014, an increase of $374,974 or 70%, compared with $532,178 for the nine months ended September 30, 2013. The spending level in this category was virtually unchanged, due to higher staffing levels and more supplies to support the oncology program, including use of an on-site biological testing laboratory during the quarter ended September 30, 2014, notwithstanding the discontinuance in 2013 of the dialysis and congestive heart failure programs and the Company’s decision to focus solely on the oncology program. In addition, during the nine months ended September 30, 2014, the Company awarded restricted stock to employees issued pursuant to the 2014 Equity Incentive Plan which resulted in an immediate expense of $346,379 recorded to research and development.
Clinical Trial and Regulatory Expenses
Clinical trial and regulatory expenses were $75,298 for the nine months ended September 30, 2014, an increase of $1,498, or 2%, compared with $73,800 for the nine months ended September 30, 2013. The increase was due to a new clinical study for the oncology project and regulatory consulting activities in conjunction with obtaining and maintaining our ISO 13485 certification. This certification is a prerequisite for being a medical device manufacturer in Europe and Canada, and serves as the foundation for FDA approval in the US.
G&A Expenses
G&A expenses consist of G&A and business development expenses.
G&A Expenses
G&A expenses were $772,159 for the nine months ended September 30, 2014, an increase of $577,934 or 298%, compared with $194,225 for the nine months ended September 30, 2013. The increase was due to financial advisory, legal and accounting expenses incurred in conjunction with the preparation of a private placement memorandum for fundraising activities and a draft registration statement for a public listing.
| 26 |
Business Development Expenses
Business development expenses were $34,017 for the nine months ended September 30, 2014, a decrease of $4,562, or 12%, compared with $38,579 for the nine months ended September 30, 2013. The decrease was due to participation in fewer meetings and events to attract new investors.
Other Income
Change in Value of Derivative Liability
The Company recorded a gain on the change in fair value of the Company’s embedded derivative liability of $1,800 during the nine months ended September 30, 2014 as compared to a gain of $10,800 during the nine months ended September 30, 2013. The change is mainly attributable to the fluctuation in the assumed market value of the Company’s stock price.
Net Loss
Net loss was $1,795,489 for the nine months ended September 30, 2014, an increase of $961,503 or 115%, compared with $833,986 for the nine months ended September 30, 2013. The increase was primarily due to higher expenses.
Years Ended December 31, 2013 and 2012
The following table sets forth certain information concerning our results of operations for the periods shown:
| Year Ended December 31, | Change | |||||||||||||||
| 2013 | 2012 | Dollars | Percent | |||||||||||||
| Revenue | ||||||||||||||||
| Sales | $ | - | $ | - | $ | - | - | % | ||||||||
| Expenses | ||||||||||||||||
| Research and Development | ||||||||||||||||
| Research and Development | 580,019 | 4,076,977 | (3,496,958 | ) | (86 | )% | ||||||||||
| Clinical & Regulatory | 102,130 | 150,781 | (48,651 | ) | (32 | )% | ||||||||||
| Total R&D | 682,149 | 4,227,758 | (3,545,609 | ) | (84 | )% | ||||||||||
| General and Administrative | ||||||||||||||||
| General and Administrative | 440,967 | 320,946 | 120,021 | 37 | % | |||||||||||
| Business Development | 104,260 | 117,586 | (13,326 | ) | (11 | )% | ||||||||||
| Total G&A | 545,227 | 438,532 | 106,695 | 24 | % | |||||||||||
| Total Expenses | $ | 1,227,376 | $ | 4,666,290 | $ | (3,438,914 | ) | (74 | )% | |||||||
Revenue
Commercial sales have not commenced as of December 31, 2013.
Operating Expenses
Research and Development Expenses
R&D expenses consist of R&D, clinical trial and regulatory expenses. The R&D component of this category was $580,019 for the year ended December 31, 2013, a decrease of $3,496,958 or 86%, compared with $4,076,977 for the year ended December 31, 2012. The decrease was due to the decision in the first quarter of 2013 to cease funding the dialysis and congestive heart failure projects due to a dearth of financing and technical issues; and, instead, to focus our resources on the oncology program. Clinical trial and regulatory expenses were $102,130 for the year ended December 31, 2013, a decrease of $48,651, or 32%, compared with $150,781 for the year ended December 31, 2012. The decrease was due to the discontinuation of animal studies for the dialysis project, offset by a new clinical study for the oncology project and regulatory consultant activities in conjunction with obtaining ISO 13485 certification. This certification is a prerequisite for being a medical device manufacturer in Europe and Canada, and serves as the foundation for FDA approval in the US.
General and Administrative Expenses
G&A expenses consist of G&A and business development expenses. The G&A component of this category was $440,967 for the year ended December 31, 2013, an increase of $120,021 or 37%, compared with $320,946 for the year ended December 31, 2012. The increase was due to higher legal expenses for patents and litigation, and higher rent expense because of a full year of occupancy. Business development expenses were $104,260 for the year ended December 31, 2013, a decrease of $13,326 or 11%, compared with $117,586 for the year ended December 31, 2012. The decrease was due to participation in fewer meetings and events to attract new investors.
Other Income
Change in Value of Derivative Liability
The Company recorded a gain on the change in fair value of the Company’s embedded derivative liability of $16,200 during the year ended December 31, 2013 as compared to a gain of $32,400 during the year ended December 31, 2012. The change is mainly attributable to the fluctuation in the assumed value of the Company’s stock price .
Income Tax Benefit
Income tax benefit was $147,997 for the year ended December 31, 2013, a decrease of $94,019 or 39%, compared with $242,016 for the year ended December 31, 2012. These amounts were received from a New York City biotech tax credit program; the decrease was due to the lower amount of eligible research and development expenses in 2013. The Company will not be eligible to participate in that program after 2013.
The Company is currently under audit by New York City tax authorities for the 2011, 2012, and 2013 tax years. The Company applied for New York City biotechnology tax credits and received certificates of eligibility and funds from the NYC Department of Finance in the amounts of $147,997, $242,016, and $236,874 for tax years 2013, 2012, and 2011, respectively. These credits have been recognized as income tax benefits on the consolidated statements of operations in the year of application. Audit outcomes and the timing of the audit settlements are subject to uncertainty. If the New York City biotechnology tax credits are disallowed, the Company will have to repay the tax credits received, in aggregate of $626,887, plus interest and penalties. The Company determined it was more-likely-than-not the benefit would be realized when the benefits were recognized.
| 27 |
Net Loss
Net loss was $1,063,179 for the year ended December 31, 2013, a decrease of $3,328,607 or 76%, compared with $4,391,786 for the year ended December 31, 2012. The decrease was primarily due to lower research and development expenses arising from the decision to discontinue work on the dialysis and congestive heart failure device; offset by higher general and administrative expenses and lower income tax benefit.
Liquidity and Capital Resources
We are actively working to improve our financial position and enable the growth of our business by raising new capital. During 2013 and 2012 we raised approximately $445,000 and $540,000, respectively, through equity financing, $160,000 and $0, respectively, through debt financing, and we received $242,000 and $237,000, respectively, of income tax refunds.
During the nine months ended September 30, 2014 and 2013, we received gross proceeds of approximately $830,000 and $122,996, respectively, through equity financing, $0 and $160,000, respectively, through debt financing, and we received $147,997 and $242,016, respectively, of income tax refunds.
We have yet to generate significant revenues. As a result, we have incurred cumulative net losses of $18,022,269 and negative cash flows from operations of $13,682,281 since our inception. Our ability to continue as a going concern relies on the continued availability of funds from equity and debt financing and other sources. Our consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Cash Flows
Cash Used in Operating Activities
Net cash used in operating activities was $600,876 for the year ended December 31, 2013, compared to net cash used in operating activities of $2,668,139 for the year ended December 31, 2012.
Net cash used in operating activities was $812,933 for the nine months ended September 30, 2014, compared to net cash used in operating activities of $291,449 for the nine months ended September 30, 2013. The change in cash used in operating activities was a result of the overall increase in expenses.
In all periods, the primary use of cash in operating activities was to fund operations.
Cash Used in Investing Activities
Inasmuch as we expense all purchased assets used in research and development activities, there was no cash used in investing activities in 2013 or 2012.
There was no cash used in investing activities in the nine months ended September 30, 2014 and 2013.
Cash Provided by Financing Activities
Net cash provided by financing activities was $605,000 for the year ended December 31, 2013, compared to net cash provided by financing activities of $540,000 for the year ended December 31, 2012. Our primary source of financing in all periods consisted of equity capital contributed by current and new investors. Our ability to continue as a going concern relies on the continued availability of financing from these and other sources.
Net cash provided by financing activities was $880,000 for the nine months ended September 30, 2014, compared to net cash provided by financing activities of $282,996 for the nine months ended September 30, 2013. Our primary source of financing in all periods consisted of equity capital contributed by current and new investors. Our ability to continue as a going concern relies on the continued availability of financing from these and other sources.
| 28 |
Contingencies
We are currently under audit by New York City authorities for the 2011 and 2012 tax years. The Company applied for New York City biotechnology tax credits and received certificates of eligibility and funds from the NYC Department of Finance in the amounts of $147,997, $242,016, and $236,874 for tax years 2013, 2012, and 2011, respectively. Audit outcomes and the timing of the audit settlements are subject to uncertainty. If the New York City biotechnology tax credits are disallowed, we will have to repay the tax credits received, in aggregate of $626,887, plus interest and penalties.
Subsequent Events
During October 2014, the Company received $50,000 for the payoff of notes originally issued to purchase common stock receivable.
On October 8, 2014, pursuant to the 2014 Equity Incentive Plan, the holders of 595,000 shares of vested Preferred Stock exchanged the preferred shares for 595,000 shares of common stock.
During October 2014, the Company paid $5,000 as the final payment of the Bridgemedica settlement.
During November 2014, the Company granted 25,000 restricted shares to an employee pursuant to the Equity Incentive Plan.
During November 2014, the Company received an additional $50,000 for the payoff of notes originally issued to purchase common stock receivable.
Capital Resources and Expenditure Requirements
Without further financing, we believe our current cash resources are insufficient to satisfy our liquidity requirements at our current level of operations. We expect that we will need to raise additional financing by mid-January 2015, which might not be available on favorable terms, if at all. We can provide no assurances that any sources of a sufficient amount of financing will be available to us on favorable terms, if at all. If we are unable to raise a sufficient amount of financing in a timely manner, we would likely need to scale back our general and administrative activities and certain of our research and development activities. Our forecast pertaining to our current financial resources and the costs to support our general and administrative and research and development activities are forward-looking statements and involve risks and uncertainties. Actual results could vary materially and negatively as a result of a number of factors, including: our ability to secure financing and the amount thereof; the costs of operating our research, development and clinical programs, including with collaborators; the costs of expanding our business development activities; our ability to manage the costs for manufacturing our products; the costs of maintaining, expanding and protecting our intellectual property portfolio, including potential litigation costs and liabilities; the costs of additional general and administrative personnel, including accounting and finance, legal and human resources, as a result of ultimately becoming a public company; our ability to collect revenues; and other risks discussed in the section entitled “Risk Factors”.
Our auditor’s report on our financial statements includes an explanatory paragraph expressing substantial doubt that we can continue as a going concern for the next twelve months. During the first three quarters of 2014, once we chose to focus solely on the oncology program, we experienced net cash outflows of approximately $271,000 per quarter, as compared to approximately $97,000 per quarter in the first nine months of 2013 when we were undergoing that transition. Assuming that we are successful in raising additional funds in 2014 and consequently expand our operations, we would expect our net cash outflow to increase to $350,000 or more per quarter.
Furthermore, we may need to raise additional capital to expand our business to meet our long-term business objectives. We expect that our operating expenses and capital expenditures will increase in the future as we expand our business. Until we can generate a sufficient amount of revenues to finance our cash requirements, which we may never do, we may need to continue to raise additional capital to fund our operations.
We may raise additional capital to fund our current operations and to fund expansion of our business to meet our long-term business objectives through public or private equity offerings, debt financings, borrowings or strategic partnerships coupled with an investment in our company or a combination thereof. If we raise additional funds through the issuance of convertible debt securities, or other debt securities, these securities could be secured and could have rights senior to those of our Common Stock. In addition, any new debt incurred by us could impose covenants that restrict our operations. The issuance of any new equity securities will also dilute the interest of our current stockholders. Given the risks associated with our business, including our unprofitable operating history and our ability or inability to commercialize our oncology products, additional capital may not be available when needed on acceptable terms, or at all. If adequate funds are not available, we will need to curb our expansion plans or limit our research and development activities, which would have a material adverse impact on our business prospects and results of operations.
| 29 |
Off-Balance Sheet Arrangements
We have not engaged in any off-balance sheet arrangements as defined in Item 303(a)(4) of Regulation S-K.
Overview
We were a limited liability company organized in the State of Delaware on December 18, 2007 under the name “Vizio Medical Devices LLC” and converted into a Delaware corporation on February 25, 2014 upon filing a certificate of conversion and a certificate of incorporation under the name “Viatar CTC Solutions Inc.”
Before 2011, we were also pursuing a business plan relating to ambulatory removal of excess fluids for kidney failure and congestive heart failure patients. We discontinued those programs during the first quarter of 2013 due to technical difficulties and the scarcity of funds. In connection with the discontinuation of our dialysis and congestive heart failure programs, we disposed of our assets for those development programs through termination of all of our license and research agreements with Columbia University and others. As a result we no longer have any assets or rights to the numerous patents and patent applications which were previously associated with those development program.s.We are not subject to any penalty or other payments due to the termination of those development programs.
We are a medical technology company focused on developing and marketing products to collect cancer cells for molecular diagnostics by others and as cancer therapy, both of which are based on the principle of removing blood-borne circulating tumor cells (CTCs). These CTCs detach from solid tumors and enter the bloodstream carrying the DNA of the tumor and seeding new tumors. CTCs are rare and difficult to distinguish from other blood cells. However, we believe our technology is substantially more effective, both in a sensitivity and specificity context, at removing CTCs when compared to other available CTC capture technologies. We are seeking to commercialize two products which utilize our proprietary CTC removal technology:
| · | The ViatarTM Collection System for Molecular Analysis is designed to collect and purify a statistically significant quantity of CTCs from up to 50 mL of blood as the “front end” for DNA sequencing, curing and other genetic analysis technologies used primarily for research. Due to the rapidly declining cost of DNA sequencing, it is gaining favor as the technology of choice for research and new drug development related to the many variants of mutations in cancer cells. This analysis is critical in understanding the complex biology of cancer as the disease progresses, developing new drugs and crafting a personalized approach to treatment. | |
| · | The ViatarTM Therapeutic Oncopheresis System is designed to remove CTCs from a patient’s blood as a new cancer therapy for metastatic disease. The initial focus will be gastric, lung, breast, colon or prostate cancer patients who have secondary tumors in the lungs, brain and bones. Secondary tumors are responsible for 90% of cancer deaths, yet there are no effective or low-cost treatments. Unlike chemotherapy drugs and immunotherapy vaccines, which seek to treat metastatic cancer by targeting specific cells or pathways, the ViatarTM Therapeutic Oncopheresis System seeks to reduce the tumor burden by the wholesale removal of CTCs via size-based filtration (similar to removal of toxins by dialysis). We believe this can augment the body’s immune system and slow the disease’s progression, thereby making some types of metastatic cancer a chronic rather than fatal condition. |
We anticipate launching the ViatarTM Collection System for Molecular Analysis during 2016: initially with a CE Mark in Europe and Canada, followed by a 510K designation in the United States. This product consists of a small, bench top base unit (pumps, hardware and control system) and a single use fluid circuit. It is designed for use in a laboratory setting at a hospital, research lab or outpatient oncology clinic. The output from our product will be a tube of CTCs suitable for DNA amplification and sequencing, or analysis by virtually all other companies’ CTC technology platforms. We are currently engaged in research collaborations with several CTC technology platform companies with the goal of marketing and selling the ViatarTM Collection System for Molecular Analysis through private label and distribution arrangements.
| 30 |
We anticipate launching the ViatarTM Therapeutic Oncopheresis System during 2016 with a CE Mark in Europe and Canada. Introduction in the United States will take several years, based on the anticipated PMA classification by the FDA. Our strategy is to conduct clinical trials outside the United States and then use the results of those trials to support approval of an investigational device exemption by the FDA. The ViatarTM Therapeutic Oncopheresis System will consist of a small base unit (pumps, hardware and control system) and a single use extracorporeal fluid circuit that is connected to a cancer patient’s central venous catheter. A treatment session will take approximately four hours, during which time the patient’s blood is circulated through our proprietary size-based filter. Most normal blood components—plasma, red blood cells and platelets—will be safely returned to the patient, while some white blood cells (WBCs) and numerous CTCs will be removed and discarded.
The ViatarTM Therapeutic Oncopheresis System is designed for use in a chemotherapy, dialysis or apheresis clinic in a hospital or outpatient setting by nurses or technicians who are trained to administer infusion, dialysis or apheresis treatments. We anticipate marketing and selling the ViatarTM Therapeutic Oncopheresis System on a direct basis through our own dedicated sales force in key large markets (such as the United States, Germany, France, Italy, UK and Japan), and through distributors in other markets.
Our headquarters facility is located at 116 John Street, Suite 10, Lowell, Massachusetts 01852, where we conduct research, development and manufacturing activities.
Industry Overview
Cancer Market Overview
Cancer is a group of diseases characterized by excessive, uncontrolled growth of cells at various sites in the body. Cancer is a difficult to treat disease, and currently represents one of the top ten causes of death in all regions around the world.
The 2011 Pulitzer Prize book for general nonfiction, entitled The Emperor of All Maladies: A Biography of Cancer, astutely noted: “Cancer, as we now know, is a disease caused by the uncontrolled growth of a single cell. This growth is unleashed by mutations—changes in DNA that specifically affect genes that incite unlimited cell growth…The secret to battling cancer, then, is to find means to prevent these mutations from occurring in susceptible cells, or to find means to eliminate the mutated cells without compromising normal growth. The conciseness of that statement belies the enormity of the task.”
Nowhere is this dilemma more evident than in finding the means to understand and mitigate the metastatic process, which accounts for over 90% of cancer-related mortality. While considerable progress has been made in the decades-long war on cancer, there is a striking paucity of good news in battling the metastatic process in solid tumor cancers. According to an article published by Mayo Clinic Proceedings, the handful of recently approved drugs which show some efficacy in targeting metastasized cancers cost upwards of $100,000 per patient and yet provide only a scant few months of longevity over legacy chemotherapy drugs.
According to the World Health Organization (“WHO”), the global incidence of new cancer cases in 2012 (the latest data available at the global level) was estimated at approximately 14.1 million. Among genders, men had a higher incidence of all types of cancer than women –7.43 million as compared to 6.66 million – as well as a higher overall mortality from cancer. In the American, European and Western Pacific regions, cancer is the second leading cause of death after cardiovascular disease.
Lung, breast, colorectal, stomach and prostate cancers account for the majority of cancer deaths worldwide. Lung cancer is also the leading cause of cancer deaths among men in high-income countries, while breast cancer causes the majority of cancer deaths among women in these countries. In the US, cancer is the second leading cause of death, with one in four deaths attributed to it. Lung, colon, prostate and breast cancer account for the majority of cancer-related deaths in the US.
The following tables present the top five most common types of cancers worldwide and their associated mortality in 2012, based on WHO data:
| Type of Cancer | Incidence | Mortality | ||
| Lung | 1.8 million | 1.6 million | ||
| Breast | 1.7 million | 0.5 million | ||
| Colorectal | 1.4 million | 0.7 million | ||
| Stomach | 1.0 million | 0.7 million | ||
| Prostate | 1.1 million | 0.3 million | ||
| All types | 14.1 million | 8.2 million |
| 31 |
According to the GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012 published by the WHO International Agency for Research on Cancer, the incidence of cancer rises with increasing incomes, as the incidence rate of all cancers combined in high-income countries was more than double that of low-income countries. In upper-middle-income and high-income countries, prostate and breast cancers are the most commonly diagnosed in males and females respectively, while lung and colorectal cancers are the next most common types in both sexes. In the case of breast cancer, women in the European region had the highest rates followed by the Americas; however, breast cancer was also among the most commonly diagnosed type of cancer in low income countries as well. Men in the region of the Americas had the highest incidence rates of prostate cancer, followed by the European region; the lowest rate of prostate cancer occurred in the South-East Asia Region.
In the next few decades, the WHO predicts that cancer will be an increasingly important cause of morbidity and mortality worldwide, regardless of the region’s development level. As the global population ages and its longevity increases, annual cancer deaths are projected to increase to approximately 14.6 million in 2035.
The following table shows the projected growth in the incidence of cancer cases globally, based on geographic region using WHO data:
| Region | 2012 Incidence | 2015 Projected Incidence | ||
| Africa | 0.6 million | 0.7 million | ||
| Americas | 2.9 million | 3.1 million | ||
| Europe | 3.7 million | 3.9 million | ||
| Eastern Mediterranean | 0.6 million | 0.6 million | ||
| Southeast Asia | 1.7 million | 1.8 million | ||
| Western Pacific | 4.5 million | 5.0 million | ||
| Global | 14.1 million | 15.2 million |
Overview of the Disease of Cancer
Cancer constitutes a heterogeneous class of diseases, characterized by uncontrolled cell growth that results from a combination of both environmental and hereditary risk factors. Many different tissue types can become malignant, such as breast, lung, liver, and skin, and even within a particular tumor there is heterogeneity, with certain cancer cells in a patient bearing mutations which are lacking in other cells nearby. Only in recent years have DNA sequencing and other genetic analysis techniques progressed enough to enable researchers to understand many cancers at a cellular and molecular level, attribute specific cancers to associated genetic changes, and determine the extent to which these changes are seen in a patient’s tumor.
Cancer cells contain genetic alterations compared to normal human cells. Common genetic abnormalities correlated to cancer include gains or losses of genetic material on specific chromosomal regions, or loci, or changes in specific genes, or mutations, which ultimately result in detrimental cellular changes followed by cancerous or pre-cancerous conditions. In addition, mutations within gene sequences, or single nucleotide variations, can give rise to aberrant proteins that do not perform their functions correctly, leading to uncontrolled cell growth. Such genetic alterations can be a result of multiple factors, including genetic predisposition, environmental or lifestyle factors, or viral infections. Importantly, these genetic changes can be identified using DNA sequencing to help guide appropriate treatment.
Primary tumors consist of two major types of cells: the malignant cells and the stroma cells. While stroma cells are normal cells that support and nourish the tumor, the malignant cells are a heterogeneous group of cancerous cells that also have the potential to give rise to disease metastasis via dissemination of CTCs. CTCs are recognized as cancer cells that detached from primary or secondary tumors and enter the bloodstream, traveling nearby or to distant organs and forming new tumors.
| 32 |
Primary tumors are rarely the determining survival factor among different types of cancers, since primary tumors can typically be removed effectively by localized surgical resection and/or radiation. Even if cancer is diagnosed at an early stage and treated by one of these methods, cancer is a major cause of mortality worldwide because of the lethal effect of tumors which metastasize. Metastasis is more difficult to treat than a primary tumor due to its systemic nature. CTCs that leave the primary tumor are able to colonize distant organs in the body and initiate the process of metastasis. The majority of CTCs die in the bloodstream, due to the harsh environment; very few of them are capable of metastasizing, and even fewer can form distant tumors of lethal size. Nevertheless, this small percentage of CTCs — estimated at 1-2% — is sufficient to initiate and continue the blood-borne metastasis process, which leads to the progression of the disease and is responsible for 90% of cancer-related deaths.
CTC Research and Applications
Just after the Civil War (1869), an autopsy discovered the existence of cancer cells in the blood of a patient who had died from a metastasized tumor. From the 1940s through the 1970s, researchers learned that: a gram of a metastasizing tumor produces roughly one million CTCs each day; CTCs are generated by virtually all 27 types of solid tumors; and CTCs are responsible for seeding metastatic secondary tumors at distant sites throughout a patient’s body, most often through the circulatory system. In other words, CTCs are both the messengers for propagating the disease and carry within their DNA the blueprint for a given cancer variant. These revelations led to multiple, but failed attempts to develop technologies to collect, process and analyze CTCs.
The field of CTC technology platforms is now experiencing a renaissance, beginning with FDA approval in 2004 of the Johnson & Johnson CellSearch device as a diagnostic tool with prognostic significance for patients with prostate, colon and breast cancer. The early promise of CellSearch spurred numerous researchers and companies in the US, Asia and Europe to develop some forty-five new diagnostic CTC technology platforms: most capture CTCs either by identifying proteins expressed by those cells (immunogenic-based) and/or by separating them based on size relative to other blood components (size-based filtration). The goal of this research has been to isolate a sufficiently large number of CTCs with high purity and in a viable, intact state.
CTCs have tremendous utility in cancer research, aiding scientists in deciphering the complex biology of cancer as it progresses from a localized, primary tumor to metastasis. An enhanced scientific understanding of the characteristics of CTCs, as well as the factors that determine or influence their migration from tumors into circulation and to distant organs, provides valuable insights into the mechanism of metastasis and the development of new therapeutic approaches. Additionally, the detection and analysis of CTCs has significant potential in the diagnosis and management of cancer, because current tools — such as tumor tissue biopsy, imaging technologies and biomarker development — have numerous limitations.
Limitations of Traditional Cancer Diagnostic and Profiling Approaches
Cancer is difficult to diagnose and manage due to its heterogeneity at morphologic, genetic and clinical levels. Traditional methods of diagnosis for solid tumors, routinely used as the initial step in cancer detection, involve a tissue biopsy followed by a pathologist examining a thin slice of potentially cancerous tissue under a microscope. A recently obtained tissue sample is used in combination with chemical staining techniques to enable analysis of the biopsy. After staining, the pathologist determines through visual inspection whether the biopsy contains normal or cancerous cells, with those that are deemed cancerous being graded on a level of aggressiveness. Often an analysis of biomarkers relevant to that tumor type is also performed on the tissue, ranging from immunohistochemistry to mutation analysis by various means such as microarrays and DNA sequencing. After the diagnosis, a clinical workup is performed according to established guidelines for the specific cancer type. From there, the physician determines the stage of progression of the cancer based on a series of clinical measures, such as size, grade, metastasis rates, symptoms and patient history, and decides on a treatment plan that may include surgery, watchful waiting, radiation, chemotherapy, or stem cell transplant.
| 33 |
This type of analysis is dependent on the availability of a recently obtained tissue biopsy for the pathologist to analyze. Such a biopsy is often not available. A tumor may not be readily accessible for biopsy, a patient’s condition may be such that a biopsy is not advised, and for routine periodic patient monitoring to evaluate potential progression or recurrence, a biopsy is a fairly invasive procedure and not typically performed. As the length of time between when the original biopsy, diagnosis or surgery is conducted to the current evaluation of the patient increases, the likelihood that an original biopsy specimen is truly representative of the current disease condition declines, as does the usefulness of the original biopsy for making treatment decisions. This risk intensifies in situations where a drug therapy is being administered, because the drug can put selective pressure on the tumor cells to adapt and change.
Similarly, the heterogeneity referred to above means that different parts or areas of the same tumor can have different molecular features or properties. In evaluating a biopsy specimen, the pathologist will take a few thin slices of the tumor for microscopic review rather than exhaustively analyzing the whole tumor mass. The pathologist can only report on the tumor sections analyzed and if other parts of the tumor have different features, such as biomarkers corresponding to specific treatments, they can be missed. A more representative analysis of the entire tumor, as well as any metastases if they are present, is very helpful.
CTC Technology Applications
The accelerated pace of CTC research in the past few years has been driven by the pressing need to overcome the limitations of traditional cancer diagnostic and profiling approaches. The following table shows the current and potential applications for CTC removal and analysis technologies, both in research and clinical practice:
| Application | Description | Benefits | ||
| Research & drug development | Understand the biology of cancer at the molecular and cellular level
Develop biomarkers and understanding of mutated DNA that aid in the discovery of new drug targets
Selection of patients likely to respond to therapy for clinical trials |
Develop and confirm theoretical models for cancer progression
Accelerate the drug development process
Increase the probability of success and regulatory approval
Reduce clinical trial costs | ||
| Early diagnosis | Liquid biopsy to diagnose cancer types based on presence, number and characteristics of CTCs | Non-invasive test, compared to traditional biopsy
Less expensive, more frequent diagnostic test | ||
| Survival prognosis | Assess the progression-free and overall survival for cancer patients based on number and characteristics of CTCs in peripheral blood | Non-invasive test
Less expensive, more frequent disease monitoring | ||
| Therapy monitoring | Enumeration and molecular analysis of CTCs to predict patients’ response to therapy | Select and adjust therapy for each patient based on predicted response
Less expensive, more frequent disease monitoring
Determine risk of disease recurrence | ||
| Therapeutic oncopheresis | Mechanical separation of CTCs from cancer patients’ blood, using central venous access | Reduce tumor burden and augment the body’s immune system
Slow the disease’s progression, thereby making metastatic cancer a chronic rather than fatal condition |
| 34 |
Mechanism of Metastatic Spread
In order to understand the technological challenges in capturing CTCs for diagnostics or therapy, it is important to appreciate the fact that CTCs are highly heterogeneous: both within a cancer type and depending on the stage of the disease.
CTCs detach from primary or secondary tumors - via mechanisms such as necroptosis, apoptosis or secretion - and enter the bloodstream (intravasation), traveling to distant organs (blood circulation) and forming new tumors (extravasation). CTCs can leave the primary tumor earlier in its formation process or after the tumor reaches a certain critical mass of cells. See diagram below.
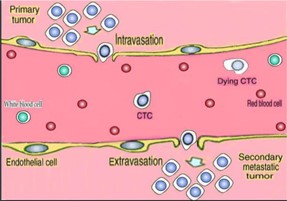
The mechanism of intravasation - the process by which CTCs enter the bloodstream - is believed to be facilitated by the epithelial-mesenchymal transition (EMT). This transition appears to occur in principal tumor cells that have stem cell-like properties and are therefore the most invasive ones. During EMT, tumor cells lose their epithelial characteristics and acquire migratory properties that enhance their invasiveness and permit them to enter the blood circulation. Cell markers associated with EMT include increased expression levels of the TWIST-1, ZEB1, mesenchymal intermediate filament vimentin – a marker also found in certain subpopulations of WBCs – and reduced levels of epithelial cytokeratins.
While in the bloodstream, CTCs are believed to possess a semi-mesenchymal phenotype. However, the vast majority of CTCs will die within 24 hours in that harsh environment due to attacks by WBCs and the high shear rates in the heart; or persist in a dormant, inactive state for years. Very few CTCs are capable of metastasizing, and even fewer can form distant tumors. Nevertheless, this small percentage of CTCs – estimated at 1-2% – have been called the decathlon athletes of CTCs and are sufficient to initiate and continue the blood-borne metastatic process, which leads to the progression of the disease and is ultimately responsible for the overwhelming majority of cancer-related deaths.
In order to reach distant organs and form new tumors, CTCs need to extravasate – exit the blood vessels - and survive in the new environment; such cells may potentially acquire more aggressive phenotypes that initiate the tumor formation process, or may re-enter the blood circulation to travel to other organs or back to the initial tumor.
After CTCs extravasate in new locations, they are believed to undergo a reverse EMT process – the mesenchymal-to-epithelial transition (MET) – and assimilate in the new environment. While some CTCs have genes that might make them more benign or less likely to survive in other locations in the body, others have different patterns of gene expression that enable them to lodge themselves in new locations, establishing a new tumor. Such cells initiate micrometastases (fewer than 100 cells) that may eventually develop into overt metastases that are finally visible using current MRI, CT scan or ultrasound imaging techniques.
In theory, CTCs can reach the majority of organs in the body and initiate metastasis. However, research indicates that different types of cancer give rise to metastasis at different sites; for example, prostate cancer metastasizes preferentially in the bone, while breast cancer cells give rise to new tumors in the liver, lungs, or bone. This preferential behavior is due to the heterogeneity of tumors and their CTCs, and the consequently different ability of cells to extravasate and survive in new locations.
| 35 |
Characteristics of CTCs
The normal cellular components of blood principally consist of red blood cells (RBCs), leukocytes (white blood cells, or WBCs) and platelets. CTCs are abnormal blood cell components shed from tumors, and are invariably collected from a patient’s peripheral blood even though they are present throughout the circulatory system. Since all blood cells must pass through extremely narrow capillaries as they circulate through the body, CTCs in peripheral blood are found in extremely low numbers because they must circulate and survive through an obstacle course of harsh conditions, such as the heart, lungs and capillaries before they can appear in the peripheral blood.
The following table shows the typical number and sizes of blood cells in typical humans:
| Type of Cell | Average number of cells per milliliter of blood |
Approximate cell diameter, in microns | ||
| RBC | 5 billion | 8 | ||
| Platelets | 300 million | 2 | ||
| WBC | 7 million | 10-15 | ||
| CTC | <1,000 | 15-20 |
Aside from size and number, another important characteristic of blood cells is their deformability: the ability to change shape in order to traverse through the narrow capillaries which separate arterial blood (blood pumped out by the heart) from venous blood (blood returning to the heart after passing through a capillary). RBCs are known to be highly deformable, effectively and very quickly changing shape from a donut to a sausage as they pass through capillaries. Platelets and WBCs are less deformable than RBCs; but they nevertheless must and do traverse through capillaries, albeit at a slower rate. CTCs are the least deformable type of blood cells by several orders of magnitude, and it is that characteristic –in addition to size differences — which our technology exploits in a manner which is unique and different from all other CTC technologies.
In the early era of CTC research, CTCs were described as nucleated cells of epithelial origin that are typically significantly larger in diameter than normal blood cells. However, as scientific research revealed the heterogeneity of tumors, it became evident that the morphology of CTCs varies by disease type, its stage at the time of diagnosis, and its pre- or post-treatment state. Currently a common, definite set of criteria that define all CTCs has not been established. Additionally, due to the heterogeneity of tumor cells, scientists have not identified yet a universal set of markers that distinguishes CTCs from other types of blood cells. Recent studies also suggest that CTC markers differ from those of the primary tumor cells, and they may also change during the course of a given cancer therapy. As a result of these challenges, the molecular characterization of CTCs has been technically difficult. With the ever-declining cost of DNA sequencing, however, it is now possible to more completely characterize and differentiate CTCs once they are captured.
CTC Collection and Analysis Technologies
CTC collection and analysis technologies rely on one of several categories of methods, each of which exploits various physical or biological properties that distinguish these cells from RBCs, platelets and WBCs. The methods which are most commonly used for CTC detection are: size, density, electrical properties, and cell surface antigens, as well as automated scanning of all non-RBCs.
| 36 |
The following table provides an overview of each method, some of the companies with CTC platform technologies, as well as their advantages and limitations.
| Method | Technology/Company | Sample
Size & Description |
Advantages | Disadvantages | ||||
| Size-based | ScreenCell/ScreenCell ClearCell/Clearbridge Biomedix GEN/Parsortix |
≤ 10 mL Size based filtration through a polymer filter |
Inexpensive Permits molecular analysis |
Low specificity and sensitivity: Contaminated with large WBCs Low number of CTCs recovered | ||||
| Density-based | OncoQuick/Greiner Bio-One RosetteSep/StemCell Technology |
≤ 10 mL Density gradient centrifugation |
Rapid and simple
|
Low specificity and sensitivity: Contaminated with large WBCs Low number of CTCs recovered | ||||
| Dielectro- phoresis | ApoStream/ApoCell DEPArray/Silicon Biosystems |
≤ 10 mL Electrical fields used to separate CTCs based on slower elution times |
Detects broader range of non-EpCam CTCs | Low specificity and sensitivity: Contaminated with large WBCs Low number of CTCs recovered | ||||
| Immuno-magnetic capture | CellSearch/Johnson & Johnson AdnaTest/AdnaTest MACS/Milteny Biotec
|
≤ 10 mL Positive selection of CTCs by using antibodies to epithelial markers |
Regulatory approval Useful in detecting tumor-specific markers |
Low specificity and sensitivity: Contaminated with large WBCs Low number of CTCs recovered | ||||
| Microfluidic immune-magnetic | CellSearch 2nd generation/J&J CEE/Biocept IsoFlux/Fluxion Biosystems |
≤ 10 mL Positive and negative selection of CTCs by using antibodies to epithelial markers |
High sensitivity High throughput Detects a broader range of CTCs |
Low specificity Low number of CTCs recovered | ||||
| Automated scanning | HD-CTC Chip/Epic Sciences
|
≤ 10 mL Negative enrichment via RBC lysis; digital microscopy |
Detects broader range of CTCs Allows visual inspection |
Lack of enrichment causes unspecific immunofluorescent staining Low sensitivity |
Our Technology
The common disadvantages of alternative technology platforms are the rarity of CTCs which are present in a standard blood sample of 10 mL or less (which hinders the sensitivity of any analysis); and the overwhelming number of WBCs as compared to CTCs (which reduces the specificity and purity of CTCs). The result is a wide variance in results using the same patient’s blood on different technology platforms, due to the difficulty in obtaining a statistically significant number of CTCs from such a small-sized sample. In other words, even if a given CTC technology platform can recover all of the CTCs which might be present in a single milliliter of blood (say, 1,000 cells), unless they represent 5-10% of the total cells in the sample being analyzed by DNA sequencing or other genetic analysis techniques, the mutations present in those CTCs will be lost in the background as noise in relation to the normal DNA sequence represented by the WBCs. So the problem to be solved is both quantity and purity of CTCs removed.
| · | Our molecular diagnostic CTC technology platform—the ViatarTM Collection System for Molecular Analysis—is unique because it utilizes a larger blood sample to collect a far greater absolute quantity of CTCs from circulating blood without clogging (up to 50 mL drawn from a patient, instead of just 10mL) than alternative technology platforms, while also enhancing purity by reducing the number of WBCs. The output from our device can then be used as the raw material for processing and analysis by all other diagnostic CTC technology platforms, as well as directly for DNA sequencing. As such, it is a “front-end” rather than a competitor to other companies’ CTC technology platforms for research and development, early diagnosis, survival prognosis and therapy monitoring. |
| 37 |
| · | Our therapeutic CTC technology platform—the ViatarTM Therapeutic Oncopheresis System—will utilize wholesale removal of a significant quantity of CTCs as a cancer therapy by treating a patient’s entire blood volume (much like dialysis removes toxins). |
Our perspective is that the existing CTC technology platforms are driven by the quest to solve a scarcity problem: how to find an extremely low number of CTCs out of the 5.4 billion cells (mostly RBCs) which inhabit a given milliliter of blood—an infinitesimal percentage indeed. Compounding the tenacity of that problem is their universal reliance on a standard blood draw sample of only 1 to 10 mL as the pool (blood) in which to go searching for the target species (CTCs). In essence, conventional wisdom defines CTCs as being rare because everyone is seeking to find and collect them from an extraordinarily small sample size. Yet for over forty years researchers have known with a good degree of precision that tumors generate millions of CTCs per day: hardly a rare or scarce amount.
We believe that rarity is in the eye of the beholder. Our proprietary CTC technology overcomes the conventional 10 mL limitation on sample size by capturing CTCs from up to 50 mLs of blood (for the diagnostic application), or up to 5 liters of blood (for the therapeutic application). This represents a significantly larger sample size, which inherently improves the sensitivity of other companies’ diagnostic CTC technology platforms or DNA sequencing; and enables a low-cost therapeutic application.
Two key features of our technology differentiate it from other CTC technology platforms:
| · | Our cross-flow filtration technology exploits the differential size and deformability properties between CTCs and normal blood components. Small but numerous cells—RBCs (2 x 8 microns) and platelets (2 microns in diameter)—are separated using a filter with small pores (e.g., 8 microns). See diagram below. The larger component in normal blood—WBCs—are typically 10-15 microns in diameter; while CTCs are typically 15-20 microns in diameter and much less deformable. These are retained for further processing and analysis by other companies’ CTC technology platform or DNA sequencing (in the case of diagnostic applications), or removed and discarded (in the case of the therapeutic application). |
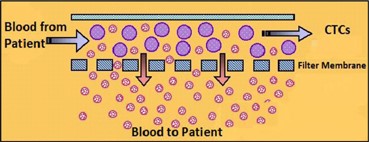
| · | Our filter is exceptionally thin compared to others’ filters. It is comprised of a micro-machined polymer that allows the passage of ~50 million RBCs, platelets and WBCs per second, while retaining viable CTCs for downstream analysis. Design algorithms balance blood flow across and through the filter, while maintaining low trans-membrane pressure. Since the force necessary to drive cells through a pore is a cube relationship to the depth of the pore, other companies’ thicker filters are significantly less efficient and can only process a small volume of blood before their pores become clogged with normal blood components. This is the defining technical feature that enables our technology to process a large volume of blood, as compared to other technologies, that are structurally limited to a very small sample size because they deploy dead-end filtration through a thicker membrane material. |
The efficacy of our technology for molecular diagnostics has been demonstrated through in vitro and in vivo tests using both animal and human blood, including blood from cancer patients. We are currently conducting research collaborations with other companies’ CTC technology platforms to validate these results.
| 38 |
The efficacy of the concept of mechanically removing CTCs as a cancer therapy to slow the progression of metastatic cancer was demonstrated in a study published by others in 2011. Using magnetic filtration of the ascites of mice that were infected with human ovarian carcinoma, the researchers removed CTCs and showed that the median time to death increased by 32.4% with only one filtration session. Similarly, due to the reduction in malignant cell burden due to that one filtration session, tumor progression slowed to 10.77 times the rate of the control group, which received no treatment.
The efficacy of a cancer therapy based on the concept of obstructing the metastatic process has been demonstrated by three cancer immunotherapy drugs approved since 2010 by the FDA (Provenge, Yervoy, and Keytruda) which prolong the lives of patients with metastatic disease. The mode of action for each of them is to sensitize white blood cells to target and attack cell signal pathways in the tumor microenvironment which flourishes once CTCs leave the primary tumor and begin extravasation at distant sites. These immunotherapies are the most significant advance in oncology treatment in decades, and offer the first glimmer of hope in reducing the 90% fatality rate once a patient’s cancer has metastasized and reaches Stage IV. .
On a parallel track, numerous clinical studies published by others since the 2004 introduction of CellSearch demonstrate that a reduction in the absolute number of CTCs in a patient’s blood correlates with favorable overall survival. This proposition holds true for all major types of cancer over a range of different drug regimens. In other words, if a patient’s CTC count drops from a “danger zone” of 10 CTCs to a “safe zone” of 5 or fewer CTCs—due to any therapy—they will live longer.
The ViatarTM Therapeutic Oncopheresis System is derived from those two paradigms. It utilizes mechanical filtration of blood as the therapy to remove CTCs, and a dosage regimen that will be determined through human clinical trials to maintain the absolute number of CTCs in the “safe zone” which delimits the probability of favorable survival.
Both of our oncology products are in the development stage, with significant design, materials validation, manufacturing scale-up and preclinical testing necessary before they can be used in human clinical trials. None of the capabilities of our two oncology products have been demonstrated yet in statistically significant clinical trials.
Commercialization Strategy
We anticipate launching the ViatarTM Collection System for Molecular Analysis during 2016: initially with a CE Mark in Europe and Canada, and later with a 510K designation in the United States. This product consists of a small, bench top base unit (pumps, hardware and control system) and a single use fluid circuit. It is intended for use primarily for research in a laboratory setting at a hospital, research lab or outpatient oncology clinic. The output from our product is a tube of CTCs suitable for DNA amplification and sequencing, or analysis by virtually all other companies’ CTC technology platforms. We are currently engaged in research collaborations with several companies with the goal of marketing and selling the ViatarTM Collection System for Molecular Analysis through private label and distribution arrangements. We believe that these prospective diagnostic partners will value the ability to augment existing instrumentation without causing a significant retool from either a manufacturing or regulatory standpoint.
Our research collaborations are with ScreenCell, Fluxion and Memorial Sloan-Kettering Cancer Center (“MSKCC”). We entered into the collaboration agreement with ScreenCell on December 2, 2013. The collaboration is one year and extends for further one year periods unless terminated upon advanced notice. Our collaboration with Fluxion is one year, ending on December 12, 2014. Under these collaboration agreements, each party is responsible for its costs and expenses (except that ScreenCell has purchased custom-designed filters from us). We own all of the intellectual property related to the filtration of CTCs under the collaboration agreement with MSKCC while we share equally with ScreenCell or Fluxion the intellectual property rights arising from those collaborations with them. We previously had a research agreement with Vivonics (related to the discontinued dialysis project), but it was terminated in 2013.
We anticipate launching the ViatarTM Therapeutic Oncopheresis System during 2016 with a CE Mark in Europe and Canada. Introduction in the United States will take several years, based on the anticipated PMA classification by the FDA. Our strategy is to conduct clinical trials outside the United States and then use the results of those trials to support approval of an investigational device exemption by the FDA. The ViatarTM Therapeutic Oncopheresis System will consist of a small base unit (pumps, hardware and control system) and a single use extracorporeal fluid circuit that is connected to a cancer patient’s central venous catheter. A treatment session will take approximately four hours, during which time the patient’s blood is circulated through our proprietary size-based filter. Virtually all normal blood components—plasma, red blood cells, white blood cells and platelets—will be safely returned to the patient, while some white blood cells and numerous CTCs will be removed and discarded.
The ViatarTM Therapeutic Oncopheresis System is intended for use in a chemotherapy, dialysis or apheresis clinic in a hospital or outpatient setting by nurses or technicians who are trained to administer infusion, dialysis or apheresis treatments. We anticipate marketing and selling the ViatarTM Therapeutic Oncopheresis System on a direct basis through our own dedicated sales force in key large markets (such as the United States, Germany, France, Italy, UK and Japan), and through distributors in other markets.
Our Business Strategy
We plan to provide the cancer research community and clinicians with the raw material – CTCs – needed for a wide range of molecular diagnostic applications; and to provide a therapy for treating metastatic cancers.
CTC-based tests. In the case of molecular diagnostics using CTC-based tests, the value added by our technology is improved sensitivity and specificity beyond what is available from other companies’ CTC technology platforms. But we do not intend to develop our own analytic techniques or clinical tests for cancer: that is an arena in which billions of dollars are spent each year and it is well beyond the scope of our expertise or resources.
Instead, our business strategy is to offer our CTC technology platform for molecular diagnostics as a “front end” component to (i) other companies’ CTC technology platforms (see the table at page 37 above), and (ii) direct DNA sequencing of CTCs using established tools and protocols (marketed by companies such as Illumina and Thermo Fisher). In this manner, our technology will enhance the breadth and utility of those applications and avoid trapping us in a situation of having to pick a winner from among the many analytic techniques which the cancer researcher community and clinicians have at their disposal. Given the rapid pace of technology change in cancer research, we believe that this will prove to be a durable strategy, especially as the cost of DNA sequencing continues to decline and it becomes the technology of choice for cancer research, new drug development and the treatment of cancers using a personalized approach.
| 39 |
The global market for CTC technology platforms is estimated at $910 million in 2012 according to a marketing intelligence report, “Circulating Tumor Cells and Cancer Stem Cells,” published by Kalorama Information (“Kalorama Information”). The key customers are researchers at major cancer medical centers, biotech and pharmaceutical companies who utilize them to identify and develop new cancer therapies based on the knowledge derived from CTC-driven research. Accordingly, that research segment is estimated to account for 97% of the global market ($885 million); with the clinical segment accounting for the remainder ($25 million) due to a limited number of clinically-validated and regulatory-approved CTC-based tests. Within that dominant research segment, approximately 73% is estimated to be research that is focused on breast and prostate cancers, with lung and colon cancers accounting for most of the remainder (22%). The geographic breakdown for CTC technology platforms in 2012 is estimated as 56% in the US, 31% in Europe and 13% in the rest of the world.
The global market for CTC technology platforms is projected to reach $3.6 billion in 2017, according to Kalorama Information. Most of this growth is projected to arise in the research segment (e.g., major cancer medical centers, biotech and pharmaceutical companies), given the long lead time for clinical adoption of new diagnostic tests (especially those which are used as companion diagnostics to new drugs). Other growth drivers are increasing cancer incidence, diagnosis and treatment rates. See the following table (annual data shown in millions of dollars):
| Market Segment | 2011 | 2012 | 2013 | 2017 | Cumulative
Annual Growth Rate (2012-2017) |
|||||||||||||||
| Diagnostics | 22 | 25 | 31 | 75 | 24.6 | % | ||||||||||||||
| Research | 830 | 885 | 960 | 3,480 | 31.5 | % | ||||||||||||||
| Total | 852 | 910 | 991 | 3,555 | 31.3 | % | ||||||||||||||
In the long term we anticipate that the clinical adoption of CTC-based diagnostics will become a major segment of the overall market for CTC technology platforms, and it will supplant the use of traditional imaging techniques (e.g., MRI, CT scans and ultrasound) for early diagnosis and tracking the progression of various cancers. But until that shift occurs, over the next five years our marketing strategy will be guided by the dominance of the research segment in the overall market for CTC technology platforms.
Current CTC-based tests used in the research segment range in price from approximately $500 to $5,000, depending on the CTC technology platform or DNA amplification and sequencing tools being used. While these prices for specific tests will undoubtedly decrease over time given the inexorable march of innovation, the overall market is expected to continue to expand. It is worth noting that these prices are based on list prices set by the manufacturers and negotiated discounts for major research customers; and do not entail reimbursement by third party payors such as Medicare, private health plans or foreign healthcare systems.
Our strategy is to negotiate pricing for the ViatarTM Collection System for Molecular Analysis with the objective that it constitutes 25% of the total end-user price of the CTC-based test regimen or DNA sequencing analysis used in the research segment. We believe that this is an appropriate metric for measuring the value attributable to our product in improving the results currently obtainable from CTC technology platforms when weighed against the overall pricing pressures and cost constraints that exist in the research segment. The clinical segment of CTC-based tests is anticipated to remain an immaterial portion of the global market for the next five years, and there is no clear viewpoint on the reimbursement terms that third party payors (such as Medicare, private insurance plans and foreign healthcare systems) will allow for CTC-based tests. Therefore, it is not possible for us to determine a revenue goal for the clinical segment of CTC-based tests.
CTC removal as a cancer therapy. The second leg of our business strategy is to deploy our unique CTC removal technology as an extracorporeal device for the wholesale removal of CTCs from metastatic cancer patients as a therapy to mitigate the growth and spread of secondary tumors: the ViatarTM Therapeutic Oncopheresis System.
| 40 |
The practice of separating or taking away (apheresis) a component or species from blood and returning the remainder to the circulation has been a mainstream medical therapy for over five decades. Examples include dialysis (removal of metabolic toxins from patients with end stage renal disease); plasmapheresis (collection of plasma and blood components such as immune globulin, rare WBC and RBC antibodies); LDL apheresis (removal of low density in patients with familial hypercholesterolemia, a rare genetic condition; stem cell harvesting (collection of circulating bone marrow cells for use in bone marrow transplantation); and leukapheresis (removal of various WBC sub-groups for transfusion into patients who need them).
We previously conducted animal testing in 2012 on a prototype extracorporeal CTC removal device, but subsequently changed the type of filter being used. Our current strategy is to build an apheresis system specifically targeted to remove CTCs as a cancer therapy (therapeutic oncopheresis) based on filtration of 20 mL or more per minute. This would translate into treating a cancer patient’s entire blood volume during a four hour session (which is equivalent to a single dialysis session). We anticipate launching the ViatarTM Therapeutic Oncopheresis System during 2016 with a CE Mark in Europe and Canada. Introduction in the United States will take several years, based on the anticipated PMA classification by the FDA.
Our strategy is to conduct clinical trials outside the United States and then use the results of those trials to support approval of an investigational device exemption by the FDA. These clinical trials will serve as the basis for regulatory approval and reimbursement. While it is not possible to predict what reimbursement schedule third party payors might set for the ViatarTM Therapeutic Oncopheresis System, existing apheresis applications (except for dialysis) are reimbursed at approximately $1,500 to $1,800 per treatment session. Given the fact that the number of patients with metastatic cancer is significantly greater than all current apheresis treatments combined (including dialysis), we assume that third party payors will need to assess the therapeutic benefits of our technology versus other therapeutic modalities (including drugs which cost approximately $90,000 or more annually). Our strategic goal is to lower the cost of cancer care so that it can be affordable by healthcare systems and a broader range of patients. Toward that end, we plan to periodically revise our pricing schedule downward so that the ViatarTM Therapeutic Oncopheresis System remains a low-cost therapy.
Competition
Our principal competition comes from mainstream diagnostic and therapeutic methods, used by pathologists and oncologists for many years. These include on tumor tissue analysis (in the case of molecular diagnostics) and chemotherapy drugs (in the case of therapeutic oncopheresis). It may be difficult to change the methods or behavior of researchers and oncologists to incorporate our products into their practices.
We also face competition from companies that offer products or are conducting research to develop products for CTC-based testing in various cancers, including companies such as Johnson & Johnsons, Atossa Genetics, Alere, Illumina, Apocell, EPIC Sciences, Clearbridge Biomedics, Cynvenio Biosystems, Fluxion Biosciences, RareCells, ScreenCell and Silicon Biosystems.
We expect that pharmaceutical and biopharmaceutical companies will increasingly focus attention and resources on the personalized cancer diagnostic and therapy sector as the potential and prevalence increases of molecularly targeted oncology therapies approved by the FDA along with companion diagnostics. For example, the FDA has recently approved three such agents—Xalkori® from Pfizer Inc. along with its companion anaplastic lymphoma kinase FISH test from Abbott Laboratories, Inc., Zelboraf® from Daiichi-Sankyo/Genentech/Roche along with its companion B-raf kinase V600 mutation test from Roche Molecular Systems, Inc. and Tafinlar® from GlaxoSmithKline along with its companion B-raf kinase V600 mutation test from bioMerieux. These recent FDA approvals are only the second, third and fourth instances of simultaneous approvals of a drug and companion diagnostic. The first approval was the 1998 approval of Genentech’s Herceptin® for HER2 positive breast cancer along with the HercepTest from partner Dako A/S. Our competitors may invent and commercialize CTC technology platforms or tests that compete with ours.
There are a number of companies which are focused on the oncology diagnostic market, such as Biodesix, Caris, Clarient, Foundation Medicine, Response Genetics, Neogenomics, Agendia, Genomic Health, and Genoptix, and which are selling to the medical oncologists and pathologists. Large laboratory services companies, such as Sonic USA, Quest and LabCorp, provide more generalized cancer diagnostic testing.
| 41 |
Additionally, projects related to cancer diagnostics and genomics have received increased government funding, both in the United States and internationally. As more information regarding cancer genomics becomes available to the public, we anticipate that more products aimed at identifying targeted treatment options will be developed and that these products may compete with ours. In addition, competitors may develop their own versions of our current and planned tests in countries where we did not apply for patents or where our patents have not issued and compete with us in those countries, including encouraging the use of their test by physicians or patients in other countries.
Third-Party Suppliers and Manufacturers
Some of the components used in our current or planned products are currently sole-source, and substitutes for these components might not be able to be obtained easily or may require substantial design or manufacturing modifications. Any significant problem experienced by one of our sole source suppliers may result in a delay or interruption in the supply of components to us until that supplier cures the problem or an alternative source of the component is located and qualified. Any delay or interruption would likely lead to a delay or interruption in our manufacturing operations. The inclusion of substitute components must meet our product specifications and could require us to qualify the new supplier with the appropriate government regulatory authorities.
We anticipate manufacturing our two oncology products at our facility in Lowell, MA. Additional clean room space, production equipment and quality control equipment will be needed. We will source sub-components and sub-assemblies from third party manufacturers, depending on cost, quality and volume requirements. We anticipate renting additional space for our manufacturing and warehouse needs; most likely at or near our current location. There is sufficient commercial space at reasonable prices in the vicinity of our current location.
Intellectual Property
Our business is dependent on a combination of patent applications, trademarks, trademark applications, trade secrets and industry know-how, in order to protect the proprietary aspects of our technology and assure that we can manufacture and sell our products.
The Company’s patent portfolio applicable to our oncology products consists of ten patent applications (three in the United States and seven international), with anticipated expiration dates of 2030 to 2034 (assuming they are issued). These are now either in pre-publication phase or in various stages of examination. The table below sets forth the information regarding our these patent applications:
| 42 |
| Description | Jurisdiction | Filed Date | Status | |||
| Methods, systems, and devices for separating tumor cells | United Arab Emirates | 3/31/2011 | Pending | |||
| Methods, systems, and devices for separating tumor cells | Australia | 3/31/2011 | Pending | |||
| Methods, systems, and devices for separating tumor cells | Canada | 3/31/2011 | Pending | |||
| Methods, systems, and devices for separating tumor cells | European Community | 3/31/2011 | Pending | |||
| Methods, systems, and devices for separating tumor cells | Israel | 3/31/2011 | Pending | |||
| Methods, systems, and devices for separating tumor cells | Mexico | 3/31/2011 | Pending | |||
| Methods, systems, and devices for separating tumor cells | US | 3/31/2011 | Pending | |||
| Methods, systems, and devices for separating tumor cells | US | 1/24/2014 | Pending | |||
| Extracorporeal fluidic device for collecting circulating tumor cells and method of use thereof | US | 8/6/2013 | Pending | |||
| Methods, systems and devices for separating tumor cells | South Africa | 3/31/2011 | Pending |
| 43 |
In connection with the discontinuation of our dialysis and congestive heart failure programs, we terminated all of our license and research agreements with Columbia University and others. There were no penalty or other payments made, and we no longer have any rights to the numerous patents and patent applications previously associated with those development programs.
We own the trade name Viatar in the United States, Canada, the 28 member countries of the European Community, Japan, Mexico and Russia. We also have trade name applications for Viatar pending in six other foreign countries. We learned that another CTC technology company, named Viator Technologies Inc. (“Viator”), was causing confusion with our Viatar trade name, so during 2014, we sent a cease-and-desist letter to Viator with a demand it stop all use of the Viator Technologies name in association with its CTC detection system. In the third quarter of 2014 they agreed to do so.
Government Regulation
Our products are classified as medical devices under the United States Food, Drug, and Cosmetic Act (FDCA). The FDCA requires these products, when sold in the United States, to be safe and effective for their intended use and to comply with the regulations administered by the United States Food and Drug Administration (FDA). Our medical device products are also regulated by comparable agencies in non-U.S. countries where our products may be sold.
The FDA’s regulatory requirements include:
| · | Establishment Registration. We must register with the FDA each facility where regulated products are developed or manufactured. The FDA periodically inspects these facilities. |
| · | Marketing Authorization . We must obtain FDA authorization to begin marketing a regulated, non-exempted product in the United States. For our ViatarTM Collection System for Molecular Analysis, this authorization will be obtained by the submission and approval of a 510(k) pre-market notification, which generally provides data on the performance of the product to allow the FDA to determine substantial equivalence to a product already in commercial distribution in the United States. Our ViatarTM Therapeutic Oncopheresis System will probably go through a much stricter and time-consuming formal pre-market approval process which includes the review of non-clinical laboratory studies and clinical investigations, as well as an inspection by the FDA prior to market approval. |
| · | Quality Management System. We are required to establish a quality management system that includes procedures for ensuring regulated products are developed, manufactured and distributed in accordance with specified standards. We also must establish procedures for investigating and responding to customer complaints regarding the performance of regulated products. In December 2013 we obtained an ISO 13485 certification, which utilizes many of the same standards for an FDA-approved quality management system. |
| · | Labeling. The labeling for our products must contain specified information. In some cases, the FDA must review and approve the labeling and any quality assurance protocols specified in the labeling. |
| · | Imports and Exports. The FDCA establishes requirements for importing products into and exporting products from the United States. In general, any limitations on importing and exporting products apply only to products that have not received marketing authorization. |
| · | Post-Market Reporting. After regulated products have been distributed to customers, we may receive product complaints requiring us to investigate and report to the FDA certain events involving the products. We also must notify the FDA when we conduct recalls involving our products. |
If the FDA finds that a manufacturer has failed to comply with FDA laws and regulations or that a medical device is ineffective or poses an unreasonable health risk, it can institute or seek a wide variety of enforcement actions and remedies, ranging from a public warning letter to more severe actions such as:
| · | fines, injunctions and civil penalties; |
| · | recall or seizure of products; |
| · | operating restrictions, partial suspension or total shutdown of production; |
| · | refusing requests for 510(k) clearance or PMA approval of new products; |
| · | withdrawing 510(k) clearance or PMA approvals already granted; and |
| · | criminal prosecution. |
The FDA also has the authority to require repair, replacement or refund of the cost of any medical device.
In the European Union, a single medical device regulatory approval process exists. Regulated products must meet minimum standards of performance, safety, and quality (known as the “essential requirements”), and then, according to their classification, comply with one or more of a selection of conformity assessment routes. Unlike United States regulations, which require most devices to undergo some level of premarket review by the FDA, the European Union regulations allow manufacturers to bring many devices to market using a process in which the manufacturer certifies that the device conforms to the essential requirements for that device. Certain products must go through a more formal pre-market review process. We do not anticipate that either of our products will require that more formal pre-market review process.
| 44 |
We are also required to report device failures and injuries potentially related to product use in a timely manner to the competent authorities of the European Union countries. A number of other countries, including Australia, Brazil, Canada, China and Japan, have also adopted or are in the process of adopting standards for medical devices sold in those countries.
We will also be subject to various healthcare related laws regulating fraud and abuse, pricing and sales and marketing practices and the privacy and security of health information, including the United States federal regulations described below. Many states, foreign countries and supranational bodies have also adopted laws and regulations similar to, and in some cases more stringent than, the federal regulations discussed above and below.
| · | The Federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal health care program, such as Medicare or Medicaid. |
| · | The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) prohibits knowingly and willfully (1) executing a scheme to defraud any health care benefit program, including private payors, or (2) falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services. In addition, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, also restricts the use and disclosure of personal health information, mandates the adoption of standards relating to the privacy and security of individually identifiable health information and requires us to report certain breaches of unsecured, individually identifiable health information. |
| · | The Physician Payments Sunshine Act requires manufacturers of medical devices covered under Medicare and Medicaid to record transfers of value to physicians and teaching hospitals and to report this data beginning in 2013 to the Centers for Medicare and Medicaid Services for subsequent public disclosure. Similar reporting requirements have also been enacted on the state level, and an increasing number of countries worldwide either have adopted or are considering similar laws requiring transparency of interactions with health care professionals. |
| · | The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal health care program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in any monetary recovery. |
If we or our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil, criminal and administrative penalties, damages, fines, disgorgement, exclusion from participating in government healthcare programs, contractual damages, reputational harm and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could materially adversely affect our ability to operate our business and our financial results.
Segment and Geographical Information
We operate in one reportable business segment and our current operations are solely in the United States.
Employees
As of November 25, 2014, we had a total of seven full-time employees. In addition, we use consultants for research and development, clinical, regulatory, legal and administrative activities. We plan to hire additional staff as we expand research, production, business development, and sales and marketing programs. None of our employees is represented by a labor union.
| 45 |
We have a lease for approximately 3,000 square feet of space in Lowell, Massachusetts for use as our corporate headquarters, including manufacturing and research laboratories. This lease expires in 2015, and there is ample space available in the same building or nearby for expansion.
In the normal course of business, we may be involved in legal proceedings or threatened legal proceedings. We are not party to any legal proceedings or aware of any threatened legal proceedings which are expected to have a material adverse effect on our financial condition, results of operations or liquidity.
In 2012, we sued Bridgemedica, an engineering firm, in New York State Supreme Court for $1.5 million of damages arising from breach of contract and other claims relating to non-performance under various engineering development contracts. Bridgemedica counterclaimed for $179,000 for delinquent payments. In April 2014, we settled the lawsuit with Bridgemedica. Pursuant to the settlement agreement, we issued 150,000 shares of Common Stock valued at $1.00 per share and agree to pay four cash installments of $5,000 each to Bridgemedica LLC. At June 30, 2014, we had accrued $5,000 of expenses payable to Bridgemedica. We have incurred expenses in connection with this lawsuit in the amount of approximately $25,000. During October, 2014, the Company made its third payment of $5,000 to Bridgemedica. As of the date of this registration statement, $5,000 remained unpaid.
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Executive Officers and Directors
The following set forth information regarding the executive officers and directors of the Company. We anticipate adding several additional independent directors within the next few months with operational experience in finance and life sciences companies, as well as medical researchers and practitioners who specialize in oncology.
| Executive Officers & Directors | Position/Title | Age | Position Held Since | |||
| Ilan Reich | Chairman, Chief Executive Officer and Chief Financial Officer | 60 | 2008 | |||
| Charles Andrew McCartney | Director | 58 | * | |||
| Francisco J. Esteva, MD, PhD | Director | 50 | * |
* The directors will be appointed prior to the effectiveness of this registration statement.
Ilan Reich has more than 15 years of CEO experience in medical device and instrumentation companies and has served as our Chairman and CEO since he founded the Company in 2008. From 2001 until 2007, Mr. Reich served as a corporate advisor on a variety of projects, including for RainDance Technologies, Warburg Pincus, and Gabelli Partners. From 1998-2001, he served as Executive Vice President (between January 22, 1998 and December 22, 1998) a director (between January 22, 1998 and January 4, 2001), President (from December 22, 1998 to January 4, 2001) and Co-CEO (between March 8, 2000 and January 4, 2001) of Inamed Corporation, a medical device manufacturer of implants for plastic surgery, aesthetics and obesity.
Earlier, Mr. Reich served as a Vice President of Corporate and Business Development for a private equity investor. Mr. Reich began his career as a corporate and securities lawyer and spent approximately 8 years advising on more than 100 public company M&A transactions. Mr. Reich received his J.D. from Columbia University School of Law in 1979 (where he was an editor of the Columbia Law Review) and B.A. in political science from Columbia College in 1976. Mr. Reich brings to the Board executive leadership experience, extensive knowledge of the business and operation of the Company and industry expertise.
In 1986, Mr. Reich pleaded guilty to two counts of mail and securities fraud arising from his tipping confidential information to a third party at two intervals during 1980 to 1984 regarding merger and acquisition transactions in which Mr. Reich’s law firm was involved. Mr. Reich did not receive any compensation for providing that information. He paid a fine to the US Treasury, served a prison term, was disbarred as an attorney in New York, and consented to an injunction on future violations. In 1995, Mr. Reich was readmitted to the New York bar. Mr. Reich's career was the subject of a profile in Fortune Magazine in June 2007.
| 46 |
Charles Andrew McCartney, has been affiliated with Bel Health Investment Partners, a private equity firm, since 2011, where he currently serves as a member of the Operating Committee and was CEO of Skila, its first portfolio company. Prior to that, from 2008 he was affiliated with DW Healthcare Partners Equity, a healthcare focused private equity firm since February 2008, where he worked with general partners to identify and acquire growth phase healthcare companies. Prior to that, Mr. McCartney was the Interim Chief Executive Officer of Iapyx Medical from February to September 2007; and held various management positions in life science companies, including President, Chief Operating Officer and Chief Executive Officer of First Circle Medical, Inc., a privately held emerging medical company focusing on the treatment of chronic Hepatitis C infection, from 1999 to 2008. Mr. McCartney has many years of experience in the medical and biotechnology industries across multiple disciplines including financing, research and development, operations and business development. Mr. McCartney received his B.A in Government and Foreign Affairs from University of Virginia in 1979. Mr. McCartney’s board management expertise in the medical and biotechnology industries provide relevant experience in a number of strategic and operating areas and led the Board to conclude that he should serve as a director of the Company.
Francisco J. Esteva, MD, PhD, is Professor of Medicine, Director of Breast Medical Oncology, Co-Director of Developmental Therapeutics, and Associate Director of Clinical Investigation at New York University Langone Medical Center. From 1997 to 2013, he was at the University of Texas MD Anderson Cancer Center, most recently as a Professor. Dr. Esteva is board certified in medical oncology, an active member of the American Society of Clinical Oncology and the Eastern Cooperative Oncology Group, a Fellow of the American College of Physicians and an elected member of the American Society for Clinical Investigation. He received his MD and PhD degrees from the University of Zaragoza School of Medicine in Spain. Dr. Esteva is an author of more than 200 publications in the area of breast cancer research and treatment. He is dedicated to the clinical development of novel therapies for breast cancer, with a passion for improving the survival and quality of life of cancer patients through innovative research and compassionate patient care. Dr. Esteva’s experience and training as a practicing physician, a scientist and a leader in cancer research and treatment enable him to bring valuable insights to the Board, including through his understanding of the scientific nature of our business and the ability to assist us with product development.
Board Committees and Independence
We are not required to have any independent members of the Board of Directors. Our Board of Directors has determined that all of our directors except Mr. Ilan Reich are independent under applicable SEC rules. As we do not have any board committees, the Board as a whole carries out the functions of audit, nominating and compensation committees.
Family Relationships
There are no family relationships between any of our executive officers or directors.
Involvement in Certain Legal Proceedings
Our directors and executive officers have not been involved in any of the following events during the past ten years:
| 1. | any bankruptcy petition filed by or against such person or any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time; |
| 2. | any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); |
| 3. | being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining him from or otherwise limiting his involvement in any type of business, securities or banking activities or to be associated with any person practicing in banking or securities activities; |
| 4. | being found by a court of competent jurisdiction in a civil action, the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; |
| 5. | being subject of, or a party to, any federal or state judicial or administrative order, judgment decree, or finding, not subsequently reversed, suspended or vacated, relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| 47 |
| 6. | being subject of or party to any sanction or order, not subsequently reversed, suspended, or vacated, of any self-regulatory organization, any registered entity or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
Code of Ethics
We have a code of ethics that applies to all employees, including the Company’s executive officers and directors.
Compensation Committee Interlocks and Insider Participation
None of the members of our Board except Mr. Reich is or has been one of our officers or employees. None of our executive officers currently serves, or in the past year has served, as a member of the compensation committee or director (or other board committee performing equivalent functions or, in the absence of any such committee, the entire board of directors) of any entity that has one or more executive officers serving on our Board.
The following table provides certain summary information concerning compensation awarded to, earned by or paid to our Chief Executive Officer for the fiscal years 2013 and 2012.
| Name and principal position |
Year | Salary | Bonus | Stock awards |
Option awards |
Nonequity incentive plan compensation |
Nonqualified deferred compensation earnings |
All other compensation |
Total | |||||||||||||||||||||||||||
| Ilan Reich Chief Executive |
2013 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | |||||||||||||||||||
| Officer and Chairman | 2012 | $ | 252,000 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 252,000 | |||||||||||||||||||
Employment Agreements
None.
Equity Compensation Plan Information
2014 Equity Incentive Plan
On March 26, 2014, our Board of Directors approved the adoption of the 2014 Equity Incentive Plan (the “2014 Plan”), which provides for the issuance of 3,000,000 shares of Common Stock (in the form of grants or options) to employees, consultants and directors. Unless prematurely terminated by the Board, the 2014 Plan terminates on March 26, 2024. As of November 25, 2014, an aggregate of 920,000 shares of Common Stock have been granted under the 2014 Plan (including the 595,000 shares described below).
The 2014 Plan also provides for the exchange of an aggregate of 595,000 shares of Series A Preferred Stock (originally issued by Vizio Medical Devices LLC to seven people under the 2010 Equity Incentive Plan as Class B member units) for an aggregate of 595,000 shares of Common Stock and the return of those exchanged shares to Mr. Reich, who was the managing member (all as contemplated by the 2010 Equity Incentive Plan). This exchange occurred on October 8, 2014.
Outstanding Equity Awards at Fiscal Year-End
None.
| 48 |
Director Compensation
The following table provides information regarding compensation during the year ended December 31, 2013 earned by directors of Vizio Medical Devices LLC who are not executive officers. None of those individuals currently serves on the Board of Directors.
| Name | Fees earned or paid in cash ($) |
Stock awards (1) ($) |
Option awards ($) |
Non-equity incentive plan compensation ($) |
Nonqualified deferred compensation earnings ($) |
All other compensation ($) |
Total ($) |
|||||||||||||||||||||
| Yaron Reich | 0 | 50,000 | 0 | 0 | 0 | 0 | 50,000 | |||||||||||||||||||||
| Sumit Rai | 0 | 50,000 | 0 | 0 | 0 | 0 | 50,000 | |||||||||||||||||||||
(1) Represents 50,000 shares of Common Stock valued at $1.00 per share. Please see note 3 to the accompanying consolidated financial statements.
For the fiscal year ending December 31, 2014, our directors will receive an annual retainer of $10,000 for attending up to six board and committee meetings either in person or telephonically with $250 per hour for additional board or committee meetings. In addition, they will receive 75,000 shares of Common Stock each year for services on the Board of Directors.
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
Except as disclosed below, there have been no transactions, or proposed transactions, which have materially affected or will materially affect us in which any director, executive officer or beneficial holder of more than 5% of the outstanding Common Stock, or any of their respective relatives, spouses, associates or affiliates, has had or will have any direct or material indirect interest in the past two fiscal years. We have no policy regarding entering into transactions with affiliated parties.
In January 2013, we borrowed an aggregate of $60,000 in exchange for a demand note from Mr. Yaron Reich, a former member of the board of managers of Vizio Medical Devices LLC and the brother of Mr. Ilan Reich, our Chairman and Chief Executive Officer. The loan was interest free. In February 2014 this note was converted into 60,000 shares of Common Stock and a warrant to purchase 60,000 shares of Common Stock at $1 per share, expiring in 2019.
Policies and Procedures for Related Party Transactions
We do not have a formal written policy for the review and approval of transactions with related parties. Our Board is responsible for reviewing and approving or ratifying related-persons transactions.
Director Independence
We are not currently listed on any national securities exchange that has a requirement that the board of directors be independent. However, our Board has determined that each of Messrs. McCartney and Esteva to be an independent director as defined by the applicable rules of the SEC.
The following table sets forth information with respect to our Common Stock known to us to be beneficially owned by the selling stockholders as of November 25, 2014. No selling stockholder has, or within the past three years has had, any position, office or other material relationship with us or any of our predecessors or affiliates other than as a result of the ownership of our securities.
Up to 1,202,071 shares of common stock are being offered by the selling stockholders under this prospectus and include the following:
On February 4, 2014, the Company sold to Amir Zada 50,000 shares of Common Stock and option to purchase 50,000 shares of Common Stock at $1.0 per share, expiring on June 30, 2014. On May 28, 2014, Mr. Zada acquired 50,000 shares of Common Stock upon exercise of this option.
On June 18, 2014, the Company sold an aggregate of 25,000 units (the “Units”) in a private placement of our securities to Marc Lesnick at a purchase price of $2.00 per Unit pursuant to a confidential private placement memorandum for an aggregate purchase price of $50,000. Each Unit consists of (i) one share of the Company’s Common Stock and (ii) a five year warrant (the “Warrant”) to purchase one share of Common Stock at an exercise price of $2.00 per share. The Warrant may be exercised on a cashless basis. On June 24, 2014, the Company sold additional 12,500 Units to Stephen & Judith Wertheimer for gross proceeds of $25,000. On July 21, 2014, the Company sold 12,500 shares of Common Stock and a five year warrant to purchase 12,500 shares of Common Stock at $2.00 per share to Robert Anello for gross proceeds of $25,000.
From February 26, 2008 through August 4, 2010, the Company sold an aggregate of 2,475,000 Class A Units, which were subsequently converted into 2,475,000 shares of Common Stock, to I. Reich Family LP for total consideration of $2,475,000 in cash, of which shares the selling stockholder is offering 618,750 shares for sale under this registration statement.
On August 27, 2014, the Company issued 250,000 and 75,000 shares of Common Stock under the 2014 Equity Incentive Plan to two engineering employees, Stephen Keaney and Kate Soojian, respectively, as compensation for services.
On September 14, 2014, the Company issued 45,871 shares of Common Stock to Columbia Technology Ventures for cancellation of $102,228 accounts payable.
| 49 |
| Common | Beneficial Ownership of Common |
|||||||||||||||||||
| Beneficial Ownership of | Stock | Stock | ||||||||||||||||||
| Common Stock Prior | Saleable | After the | ||||||||||||||||||
| to the Offering (1) | Pursuant | Offering (1) | ||||||||||||||||||
| Number of | Percent of | to This | Number of | Percent of | ||||||||||||||||
| Name of Selling Stockholder | Shares | Class (2) | Prospectus | Shares | Class (2) | |||||||||||||||
| Amir Zada | 100,000 | * | 100,000 | 0 | 0 | |||||||||||||||
| Marc Lesnick | 50,000 | (3) | * | 50,000 | (3) | 0 | 0 | |||||||||||||
| Stephen & Judith Wertheimer | 25,000 | (4) | * | 25,000 | (4) | 0 | 0 | |||||||||||||
| Robert Anello | 37,500 | * | 37,500 | 0 | 0 | |||||||||||||||
| I. Reich Family LP (5) | 2,475,000 | 14.5 | % | 618,750 | 1,856,250 | 10.8 | % | |||||||||||||
| Columbia Technology Ventures (6) | 366,878 | 2.1 | % | 45,821 | 321,057 | 1.9 | % | |||||||||||||
| Stephen Keaney (7) | 275,000 | 1.6 | % | 250,000 | 25,000 | * | ||||||||||||||
| Kate Soojian (7) | 75,000 | * | 75,000 | 0 | 0 | |||||||||||||||
| Total | 1,202,071 | |||||||||||||||||||
| 50 |
* represents less than 1%
| (1) | Assumes that all of the shares held by the selling stockholder covered by this prospectus are sold and that the selling stockholder acquires no additional shares of common stock before the completion of this offering. However, as the selling stockholder can offer all, some, or none of its common stock, no definitive estimate can be given as to the number of shares that the selling stockholder will ultimately offer or sell under this prospectus. |
| (2) | Calculated based on 17,118,622 shares of Common Stock outstanding as of November 24, 2014. |
| (3) | Includes 25,000 shares of Common Stock issuable upon exercise of warrants. | |
| (4) | Includes 12,500 shares of Common Stock issuable upon exercise of warrants. | |
| (5) | Ilan Reich has the voting and investment control over the securities held by I. Reich Family LP. | |
| (6) | The Trustees of Columbia University has the voting and investment control over the securities held by Columbia Technology Ventures. The Company entered into licensing agreements with the selling stockholder for patents and patent applications pertaining to cardiology, nephrology and other potential medical and commercial applications. These licensing agreements were terminated in September 2014. | |
| (7) | Engineering employees of the Company. |
We are registering the shares of Common Stock on behalf of the selling stockholders. Sales of shares may be made by selling stockholders, including their respective donees, transferees, pledgees or other successors-in-interest directly to purchasers or to or through underwriters, broker-dealers or through agents. Sales may be made from time to time on the OTC Bulletin Board or any exchange upon which our shares may trade in the future, in the over-the-counter market or otherwise, at market prices prevailing at the time of sale, at prices related to market prices, or at negotiated or fixed prices. The shares may be sold by one or more of, or a combination of, the following:
| 51 |
| · | a block trade in which the broker-dealer so engaged will attempt to sell the shares as agent but may position and resell a portion of the block as principal to facilitate the transaction (including crosses in which the same broker acts as agent for both sides of the transaction); |
| · | purchases by a broker-dealer as principal and resale by such broker-dealer, including resales for its account, pursuant to this prospectus; |
| · | ordinary brokerage transactions and transactions in which the broker solicits purchases; |
| · | through options, swaps or derivatives; |
| · | in privately negotiated transactions; |
| · | in making short sales or in transactions to cover short sales; |
| · | put or call option transactions relating to the shares; and |
| · | any other method permitted under applicable law. |
The selling stockholders may effect these transactions by selling shares directly to purchasers or to or through broker-dealers, which may act as agents or principals. These broker-dealers may receive compensation in the form of discounts, concessions or commissions from the selling stockholders and/or the purchasers of shares for whom such broker-dealers may act as agents or to whom they sell as principals, or both (which compensation as to a particular broker-dealer might be in excess of customary commissions). The selling stockholders have advised us that they have not entered into any agreements, understandings or arrangements with any underwriters or broker-dealers regarding the sale of their securities.
The selling stockholders may enter into hedging transactions with broker-dealers or other financial institutions. In connection with those transactions, the broker-dealers or other financial institutions may engage in short sales of the shares or of securities convertible into or exchangeable for the shares in the course of hedging positions they assume with the selling stockholders. The selling stockholders may also enter into options or other transactions with broker-dealers or other financial institutions which require the delivery of shares offered by this prospectus to those broker-dealers or other financial institutions. The broker-dealer or other financial institution may then resell the shares pursuant to this prospectus (as amended or supplemented, if required by applicable law, to reflect those transactions).
The selling stockholders and any broker-dealers that act in connection with the sale of shares may be deemed to be “underwriters” within the meaning of Section 2(11) of the Securities Act of 1933, and any commissions received by broker-dealers or any profit on the resale of the shares sold by them while acting as principals may be deemed to be underwriting discounts or commissions under the Securities Act. The selling stockholders may agree to indemnify any agent, dealer or broker-dealer that participates in transactions involving sales of the shares against liabilities, including liabilities arising under the Securities Act. We have agreed to indemnify each of the selling stockholders and each selling shareholder has agreed, severally and not jointly, to indemnify us against some liabilities in connection with the offering of the shares, including liabilities arising under the Securities Act.
The selling stockholders will be subject to the prospectus delivery requirements of the Securities Act. We have informed the selling stockholders that the anti-manipulative provisions of Regulation M promulgated under the Securities Exchange Act of 1934 may apply to their sales in the market.
Selling stockholders also may resell all or a portion of the shares in open market transactions in reliance upon Rule 144 under the Securities Act, provided they meet the criteria and conform to the requirements of Rule 144.
Upon being notified by a selling shareholder that a material arrangement has been entered into with a broker-dealer for the sale of shares through a block trade, special offering, exchange distribution or secondary distribution or a purchase by a broker or dealer, we will file a post-effective amendment to this registration statement, disclosing:
| · | the name of each such selling shareholder and of the participating broker-dealer(s); |
| · | the number of shares involved; |
| · | the initial price at which the shares were sold; |
| · | the commissions paid or discounts or concessions allowed to the broker-dealer(s), where applicable; |
| 52 |
| · | that such broker-dealer(s) did not conduct any investigation to verify the information set out or incorporated by reference in this prospectus; and |
| · | other facts material to the transactions. |
We are paying all expenses and fees in connection with the registration of the shares. The selling stockholders will bear all brokerage or underwriting discounts or commissions paid to broker-dealers in connection with the sale of the shares.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth the beneficial ownership of our Common Stock as of November 25, 2014 by:
| • | each person, or group of affiliated persons, whom we know to beneficially own more than 5% of our Common Stock; | ||
| • | each of our executive officers and our directors; and | ||
| • | all of our executive officers and directors as a group. |
| Title of Class | Name and Address of Beneficial Owner (3) | Amount
and Nature of Ownership (1) |
Percent
Of Class (1) (2) |
||||||||
| Principal Stockholders | |||||||||||
| Common Stock | I. Reich Family LP (4) | 2,475,000 | 14.5 | % | |||||||
| Common Stock | Winchester Fund of Funds (5) | 1,750,000 | 10.2 | % | |||||||
| Common Stock | Yaron Reich | 1,030,000 | (6) | 6.0 | % | ||||||
| All Principal Stockholders as a Group | 5,255,000 | 30.6 | % | ||||||||
| % | |||||||||||
| Executive Officers and Directors | |||||||||||
| Common Stock | Ilan Reich | 2,485,000 | (7) | 14.5 | % | ||||||
| Common Stock | Charles Andrew McCartney | 0 | 0 | ||||||||
| Common Stock | Francisco J. Esteva | 0 | 0 | ||||||||
| All Executive Officers and Directors as a Group (three persons) | 2,485,000 | 14.5 | % | ||||||||
| 53 |
Less than 1% of the outstanding shares of our Common Stock.
| (1) | Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. Shares of Common Stock subject to options or warrants currently exercisable or convertible, or exercisable or convertible within 60 days of August 12, 2014 are deemed outstanding for computing the percentage of the person holding such option or warrant but are not deemed outstanding for computing the percentage of any other person. |
| (2) | Percentage based on 17,118,622 shares of Common Stock outstanding as of November 25, 2014. |
| (3) | Unless otherwise noted, the address is c/o Viatar CTC Solutions Inc., 116 John Street, Suite 10, Lowell, Massachusetts 01852. |
| (4) | Mr. Ilan Reich has sole voting and investment control over shares owned by the I. Reich Family LP. The address of this entity is 29 Clive Hills Rd., Short Hills, NJ 07078. |
| (5) | Mr. Ali Taj has voting and investment control over shares owned by Winchester Fund of Funds. The address of this entity is 1301 Dove Street, Suite 890, Newport Beach, CA 92660. |
| (6) | Includes 970,000 shares of Common Stock, and warrants to purchase 60,000 shares of Common Stock exercisable within 60 days. Mr. Yaron Reich is the brother of Mr. Ilan Reich. |
| (7) | Includes (i) 10,000 shares of Common Stock held directly by Mr. Ilan Reich, and (ii) 2,475,000 shares of Common Stock held by I. Reich Family LP, over which Mr. Ilan Reich holds voting and dispositive power through the corporate general partner. |
Mr. Ilan Reich also owns 4,000,000 shares of our Series A Preferred Stock, representing 100% of the issued and outstanding shares of Series A Preferred Stock. Each share of Series A Preferred Stock is entitled to one vote on all matters submitted to stockholders for vote. For further information regarding Series A Preferred Stock, please see below “Description of Securities.”
General
Our certificate of incorporation, which became effective on February 25, 2014 when we converted from a Delaware limited liability company to a Delaware corporation and changed our name from “Vizio Medical Devices LLC” to “Viatar CTC Solutions Inc.”, authorizes us to issue up to 100,000,000 shares of Common Stock, par value $0.001 per share, and 20,000,000 shares of preferred stock, par value $0.001 per share.
As of November 25, 2014, there were 17,118,622 shares of Common Stock outstanding, held of record by approximately 105 stockholders. The number of shares of Common Stock outstanding as of that date does not include (i) 435,000 shares of Common Stock issuable upon the exercise of our outstanding warrants, and (ii) 3,000,000 shares of Common Stock reserved for future issuance under our 2014 Equity Incentive Plan of which 920,000 have already been issued.
The following descriptions of our capital stock and provisions of our certificate of incorporation and bylaws are summaries and are qualified by reference to the certificate of incorporation and the bylaws and applicable law.
Common Stock
The holders of our Common Stock are entitled to the following rights:
| 54 |
Voting Rights
Holders of our Common Stock are entitled to one vote per share in the election of directors and on all other matters on which stockholders are entitled or permitted to vote. All matters presented to stockholders for a vote require the approval of both Common Stock and the Series A Preferred Stock (which votes as a separate class) except that only the approval of holders of Series A Preferred Stock is required to alter, amend or repeal the Company’s Certificate of Incorporation and to remove any or all directors for cause or without cause, provided there are at least 700,000 shares of Series A Preferred Stock outstanding. Holders of our Common Stock are not entitled to cumulative voting rights.
Dividend Rights
Subject to the terms of any outstanding series of preferred stock, the holders of our Common Stock are entitled to dividends in the amounts and at times as may be declared by the board of directors out of funds legally available therefor.
Liquidation Rights
Upon liquidation or dissolution or winding up of the Company, holders of Common Stock are entitled to receive cash or property with a fair market value of $13,470,000 (the “Common Stock Preference”) from the net assets available for distribution to stockholders after we have paid, or provided for payment of all of our debts and liabilities (the “Debt”). In addition to the Common Stock Preference, holders of our Common Stock are also entitled to share ratably in all net assets available for distribution to stockholders after we have paid, or provided for payment of, the Debt, the Common Stock Preference and any liquidation preferences to holders of our preferred stock.
Other Matters
Holders of our Common Stock have no redemption, conversion or preemptive rights. There are no sinking fund provisions applicable to our Common Stock. The rights, preferences and privileges of the holders of our Common Stock are subject to the rights of the holders of shares of any series of preferred stock that we may issue in the future.
Preferred Stock
Our certificate of incorporation authorizes 20,000,000 shares of preferred stock. Our board of directors has the authority to issue preferred stock in one or more classes or series and to fix the designations, powers, preferences and rights, and the qualifications, limitations or restrictions thereof, including dividend rights, conversion right, voting rights, terms of redemption, liquidation preferences and the number of shares constituting any class or series, without further vote or action by the stockholders. Although we have no present plans to issue any shares of preferred stock beyond the shares of Series A Participating Preferred Stock currently outstanding, the issuance of shares of preferred stock, or the issuance of rights to purchase such shares, could decrease the amount of earnings and assets available for distribution to the holders of Common Stock, could adversely affect the rights and powers, including voting rights, of the Common Stock, and could have the effect of delaying, deterring or preventing a change of control of us or an unsolicited acquisition proposal.
Series A Participating Preferred Stock
Our board of directors has designated 4,000,000 shares of our preferred stock as Series A Participating Preferred Stock, par value $0.001 per share (the “Series A Preferred Stock”). As of November 25, 2014, there were 4,000,000 shares of our Series A Preferred Stock outstanding, all of which are owned by Ilan Reich. The holders of our Series A Preferred Stock are entitled to the following rights:
Rank
Except otherwise provided in the certificate of incorporation, the Series A Preferred Stock shall rank senior to all Common Stock or each other class of capital stock of the Company or series of preferred stock of the Company existing or hereafter created (collectively referred to, together with all classes of Common Stock, as “Junior Securities”) with respect to dividend distributions and distribution upon liquidation, winding up or dissolution of the Company.
| 55 |
Voting Rights
Holders of our Series A Preferred Stock are entitled to one vote per share, voting as a separate class, on any matter submitted to a vote of holders of Junior Securities, provided that there are at least 700,000 shares of Series A Preferred Stock outstanding. All matters presented to stockholders for a vote require the approval of both Common Stock (and any other securities which vote together with Common Stock and the Series A Preferred Stock (which votes as a separate class) except that only the approval of holders of Series A Preferred Stock is required to alter, amend or repeal the Company’s Certificate of Incorporation and to remove any or all directors for cause or without cause, provided there are at least 700,000 shares of Series A Preferred Stock outstanding. In addition, holders of Series A Preferred Stock are also entitled to vote on the amendment, repeal or adoption of any new Bylaws of the Company. Holders of our Series A Preferred Stock are not entitled to cumulative voting rights.
Dividend Rights
The Series A Preferred Stock is entitled to dividends in the amounts and at times as may be declared by the board of directors out of funds legally available therefor provided that our board of directors shall not authorize the payment of any dividend solely on Series A Preferred Stock. Series A Preferred Stock shall participate in all dividends declared on shares of Junior Securities and shall be entitled to receive their ratable share of 20% of the dividends to be paid to Junior Securities. The Company may not declare or pay any dividend for Junior Securities unless Series A Preferred Stock is paid dividend first.
Liquidation and Distribution Rights
Upon liquidation or dissolution or winding up of the Company, holders of Series A Preferred Stock are entitled to share ratably 20% of the net assets available for distribution to stockholders after we have paid, or provided for payment of, (i) the Debt, and (ii) the Common Stock Preference. The net assets remaining after payment of the Debt and the Common Stock Preference shall be distributed 20% to the holders of our Series A Preferred Stock and 80% to the holders of our Junior Securities.
Purchase Rights
If we issue or sell any options, convertible securities or rights to purchase stock, warrants, securities or other property pro rata to all the holders of any class of Junior Securities (the “Purchase Rights”), then holders of Series A Preferred Stock will be entitled to 20% of the Purchase Rights with respect to Junior Securities.
Other Matters
Holders of our Series A Preferred Stock have no redemption or preemptive rights. There are no sinking fund provisions applicable to our Series A Preferred Stock. The Company may not take any action which would materially affect any of the rights, preferences and privileges of our Series A Preferred Stock without the approval of at least 75% of the then outstanding shares of Series A Preferred Stock.
Warrants
As of November 25, 2014, there were warrants to purchase an aggregate of 435,000 shares of our Common Stock issued and outstanding, with exercise prices ranging from $1.00 to $2.00 per share, subject to adjustment and expiration dates ranging from 2014 to 2019. The warrants will expire five years from the date of issuance and may be exercised on a cashless basis if there is no effective registration statement covering the shares of Common Stock issuable upon exercise of the warrants. Upon satisfaction of certain criteria, the Company may redeem all the warrants at $0.001 per share.
Convertible Note
In April 2013, we borrowed $100,000 in exchange for a 4% interest-only, convertible promissory note due April 30, 2015 and a five year warrant to purchase 50,000 shares of Common Stock at $2.00 each. The note is convertible at any time prior to maturity at the option of the holder into 50,000 shares of Common Stock.
| 56 |
We have agreed to register an aggregate of 1,202,071 shares of Common Stock (including 37,500 shares of Common Stock issuable upon exercise of certain warrants). An aggregate of 15,086,159 shares of Common Stock (including 347,500 shares of Common Stock issuable upon exercise of certain warrants) will become eligible under Rule 144 for sale in the public market from time to time after the effective date of the registration statement relating to this prospectus upon expiration of their applicable holding periods, subject to the volume limitation, public notice, manner-of-sale and current public information requirements of Rule 144. On February 29, 2009, January 22, 2011, and December 19, 2012, a total of 1, 225, 000 shares, 800,000 shares and 1,000,000 shares of our Common Stock, respectively, became eligible for sale under Rule 144, subject to volume limitation, public notice, manner-of-sale and current public information requirements of Rule 144.
Anti-Takeover Effects of Delaware Law and Our Certificate of Incorporation and Bylaws
The provisions of Delaware law, our certificate of incorporation and our bylaws described below may have the effect of delaying, deferring or discouraging another party from acquiring control of us.
Delaware Law
We are a Delaware corporation and consequently are also subject to certain anti-takeover provisions of the Delaware General Corporation Law (the “DCGL”). Under Section 203 of the DCGL, a Delaware corporation may not engage in any business combination with any interested stockholder for a period of three years following the date such stockholder became an interested stockholder, unless:
| • | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; |
| • | upon completion of the transaction that resulted in the interested stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding (a) shares owned by persons who are directors and also officers, and (b) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
| • | on or subsequent to the date of the transaction, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder. |
Generally, a business combination includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. An interested stockholder is any person who, together with such person’s affiliates and associates (i) owns 15% or more of a corporation’s voting securities or (ii) is an affiliate or associate of a corporation and was the owner of 15% or more of the corporation’s voting securities at any time within the three-year period immediately preceding a business combination of the corporation governed by Section 203. We expect the existence of this provision to have an anti-takeover effect with respect to transactions our board of directors does not approve in advance. We also anticipate that Section 203 may discourage takeover attempts that might result in a premium over the market price, once a market exists, for the shares of Common Stock held by our stockholders.
Our Certificate of Incorporation and Bylaws
The provisions of our certificate of incorporation and bylaws may have an anti-takeover effect and may delay, defer or prevent a tender offer or takeover attempt that a stockholder might consider in his or her best interest, including those attempts that might result in a premium over the market price for the Common Stock. Among other things, our certificate of incorporation and bylaws:
| · | provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in a timely manner, and also specify requirements as to the form and content of such notice; |
| · | provide that any or all of the directors may be only removed for cause or without cause by the vote of a majority of the shares of the Series A Preferred Stock; |
| · | Permit corporate actions to be approved by written consent of stockholders without a meeting, without prior notice and without a vote; |
| · | authorize the board of directors to determine the number of directors and to fill vacancies on the board; |
| · | provide that only the board of directors or officers instructed by the board may call a special meeting of the stockholders; |
| · | provide that our certificate of incorporation can only be altered, amended or repealed by the affirmative vote of the holders of a majority of all of the then outstanding shares of Series A Preferred Stock, voting as a single class, provided that there are at least 700,000 shares of Series A Preferred Stock outstanding; |
| · | provide for the issuance of “blank check” preferred stock; and |
| 57 |
| · | provide that any matter submitted to a vote of holders of Junior Securities shall be approved by the holders of Series A Preferred Stock in addition to approval of holders of Common Stock or any other junior securities voting together with Common Stock, provided that there are at least 700,000 shares of Series A Preferred Stock outstanding |
DISCLOSURE OF COMMISSION POSITION OF INDEMNIFICATION FOR
SECURITIES ACT LIABILITIES
Section 145 of the Delaware General Corporation Law (the “DGCL”) provides that a Delaware corporation may indemnify directors, officers, employees and agents against expenses (including attorney’s fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with any threatened, pending or completed action, suit or proceeding in which such person is made a party by reason of such person being or having been a director, officer, employee or agent to the corporation, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was unlawful.
The DGCL provides that Section 145 is not exclusive of other rights to which those seeking indemnification may be entitled under any bylaw, agreement, vote of stockholders, or disinterested directors or otherwise.
Section 102(b)(7) of the DGCL permits a corporation to provide in its certificate of incorporation or an amendment thereto, to eliminate or limit the personal liability of a director to the corporation and its stockholders for monetary damages arising out of certain breaches of their fiduciary duty.
The Company’s Certificate of Incorporation provides for the elimination of a director’s liability to the Company and its stockholders for monetary damages for breach of fiduciary duty, except for liability (i) for any breach of the director’s duty of loyalty to the Registrant or its stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of the law, (iii) under Section 174 of the DGCL, or (iv) for any transaction from which the director derived an improper personal benefit.
The Company’s Certificate of Incorporation and Bylaws provide that the Company shall indemnify each of its directors and executive officers to the fullest extent not prohibited by the DGCL and may indemnify certain other persons as set forth in the DGCL. The Company may advance expenses to its directors and officers in connection with defending a proceeding.
The rights conferred in the Certificate of Incorporation and Bylaws are not exclusive, and the Company is authorized to enter into indemnification agreements with its directors, officers, employees, and agents and to obtain insurance to indemnify such persons.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. No pending material litigation or proceeding involving our directors, executive officers, employees or other agents as to which indemnification is being sought exists, and we are not aware of any pending or threatened material litigation that may result in claims for indemnification by any of our directors or executive officers.
| 58 |
The validity of the shares of Common Stock offered hereby will be passed upon for us by Sichenzia Ross Friedman Ference LLP. Sichenzia Ross Friedman Ference LLP is the beneficial owner of 75,000 shares of our Common Stock.
Our consolidated financial statements as of December 31, 2012 and 2013, and for the two years then ended, included in this Prospectus and Registration Statement, have been audited by Rosenberg, Rich Baker Berman & Company, an independent registered public accounting firm, as stated in their report appearing herein. Such consolidated financial statements have been so included in reliance upon the report of such firm given upon their authority as experts in accounting and auditing.
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the shares of Common Stock being offered by this prospectus. This prospectus, which constitutes a part of the registration statement, does not contain all of the information set forth in the registration statement, some of which is contained in exhibits to the registration statement as permitted by the rules and regulations of the SEC. For further information with respect to us and our Common Stock, we refer you to the registration statement, including the exhibits filed as a part of the registration statement. Statements contained in this prospectus concerning the contents of any contract or any other document is not necessarily complete. If a contract or document has been filed as an exhibit to the registration statement, please see the copy of the contract or document that has been filed. Each statement is this prospectus relating to a contract or document filed as an exhibit is qualified in all respects by the filed exhibit. You may obtain copies of this information by mail from the Public Reference Section of the SEC, 100 F Street, N.E., Room 1580, Washington, D.C. 20549, at prescribed rates. You may obtain information on the operation of the public reference rooms by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports, proxy statements, and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov.
As a result of this offering, we will become subject to the information and reporting requirements of the Securities Exchange Act of 1934 and, in accordance with this law, will file periodic reports and other information with the SEC. These periodic reports and other information will be available for inspection and copying at the SEC’s public reference facilities and the website of the SEC referred to above. We also maintain a website at www.viatarctcsolutions.com. Upon completion of this offering, you may access these materials free of charge as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. Information contained on our website is not a part of this prospectus and the inclusion of our website address in this prospectus is an inactive textual reference only.
CTC : Cells that have shed into the blood stream from a primary tumor and circulate in it (CTCs). They are the seeds for subsequent growth of additional tumors (metastasis) in distant organs, triggering a mechanism that is responsible for the vast majority of cancer deaths.
DNA Sequencing : The process of determining the precise order of nucleotides within a DNA molecule. Nucleotides are the four bases—adenine, guanine, cytosine and thymine—in a strand of DNA. There are over 3 billion such base pairs in a DNA molecule. DNA sequencing is used to determine the complete genome, or blueprint, for a species; as well as variations (mutations) for a given disease condition.
DNA Amplification : The process of producing two identical replicas from one original DNA molecule. This process occurs in all living organisms and is the basis for biological inheritance and genetics. It is also used in DNA sequencing to enhance the quantity of molecules being analyzed.
510(k) Pre-Market Notification : A filing by a medical device manufacturer with the Food and Drug Administration (FDA) at least 90 days in advance of its intent to market a device. This allows the FDA to determine whether the proposed device is substantially equivalent to a device already on the market in 1976, or whether it is new and therefore requires further regulatory materials and review.
Mutation : A change of the nucleotide sequence in the DNA molecule of an organism; these can arise from replication errors during DNA amplification, or the insertion or deletion of segments of DNA. Mutations can either have no effect, alter the functioning of a gene, or prevent a gene from operating properly.
CE Mark : A CE logo and four digit identification number of the notified body which assessed the conformity of a product to the European directives, or regulatory system, which is applicable to a given medical device (and other products) in the European Economic Area. It is a symbol of free marketability in that region, and is recognizable worldwide as the European equivalent of compliance with FDA regulatory requirements. One key distinction, however, is that the CE system focuses primarily on safety, whereas the FDA focuses on both safety and efficacy of a given product.
PMA Classification : The most stringent type of medical device marketing application required by the FDA (one category above 510(k) pre-market notification). Unlike the 510(k) pathway, the manufacturer must submit an exhaustive application to the FDA and must receive approval prior to beginning commercial marketing of a device. The PMA application includes information on how the device was designed and manufactured, as well as preclinical and clinical studies, demonstrating that it is safe and effective for its intended use.
| 59 |
Dialysis: A process for removing waste and excess water from the blood, which is used as an artificial replacement for lost kidney function in people with renal failure.
Apheresis : A medical technology in which the blood of a patient or donor is passed through an instrument that separates out one particular constituent and safely returns the remainder to the circulation. Dialysis is the most widely practiced and well-known form of apheresis.
Stroma cells : Connective tissue cells of any organ, which support the functional cells of that organ. Stroma provides an extracellular matrix on which tumors can grow.
Malignant cells : Cells which are not self-limited in growth, are capable of invading into adjacent tissues, and may be capable of spreading to distant organs. These cells typically have multiple mutations and reduced expression of DNA repair enzymes.
Metastasis : The spread of a cancer from one organ to another not directly connected with it. Cancer occurs after one single cell in a tissue is progressively genetically damaged to produce cells with uncontrolled proliferation. As these cells transform, they are able to invade into the blood circulation and invade a secondary site.
Oncopheresis : An apheresis process in which circulating cancer tumor cells are separated from the blood by means of an extracorporeal circuit, with the normal blood components being safely returned to the body.
EMT : Epithelial-mesenchymal transition, is a process by which epithelial cells lose their cell polarity and cell-cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells; these are stromal cells that can differentiate into a variety of cell types. EMT is implicated in the initiation of metastasis for cancer progression.
ZEB1 : Zinc finger E-box-binding homeobox, is a protein in humans that is encoded by the ZEB1 gene. Mutations in this gene have a contributing role in lung cancer invasiveness and metastatic development.
TWIST1 : Twist-related protein 1, is protein in humans that is encoded by the TWIST1 gene. Mutations in this gene have a contributing role in breast cancer.
Mesenchymal intermediate filament vimentin : A protein in humans that is encoded by the VIM gene, which is expressed in mesenchymal cells. It is a marker for cells undergoing EMT during both normal development and metastatic progression.
FDA : Food and Drug Administration, is an agency of the US federal government which regulates the sale of medical devices and drugs.
Leukapheresis : An apheresis process in which white blood cells are separated from blood. It may be used to decrease a very high white blood cell count, to obtain a patient’s own blood cells for later transplant, or to obtain cells for research purposes. It may also be used for oncopheresis, although it is expensive, cannot be repeated on a frequent basis, and results in the loss of vital, normal blood cells.
Necroptosis : A programmed form of passive cell death, which is regulated by certain proteins.
Apotosis : The process of programmed cell death. While 50 to 70 billion human cells die each day due to the normal cell growth/death cycle, defective apoptosis has been implicated in the progression of cancer.
Plasmapheresis : An apheresis process by which certain blood cells and plasma are separated from blood, with the plasma collected and frozen for use in the manufacture of a variety of medications; it is also used as a therapy to treat a variety of disorders, including lupus and myasthenia gravis.
| 60 |
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Audited Financial Statements for the Years ended December 31, 2013 and 2012
Condensed Consolidated Financial Statements for the Nine and Three Months Ended September 30, 2014 and 2013
| 61 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Members of
Vizio Medical Devices, LLC and Subsidiary
We have audited the accompanying consolidated balance sheets of Vizio Medical Devices, LLC and Subsidiary (a development stage company) as of December 31, 2013 and 2012, and the related consolidated statements of operations, members’ deficit, and cash flows for the two years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We did not audit the financial statements of Vizio Medical Devices, LLC and Subsidiary for the period December 18, 2007 (date of inception) to December 31, 2011. Those statements were audited by other auditors whose reports have been furnished to us and our opinion, insofar as it relates to amounts for the period from inception to December 31, 2011, included in the cumulative totals, is based solely upon the report of the other auditors.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Vizio Medical Devices, LLC and Subsidiary as of December 31, 2013 and 2012, and the results of its operations and its cash flows for the two years then ended, in conformity with U.S. generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has not generated any revenue to date, has incurred net losses and negative cash flows from operations since inception. These conditions raise substantial doubt about its ability to continue as a going concern. Management’s plans regarding those matters also are described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
As discussed in Note 10, the accompanying consolidated financial statements of Vizio Medical Devices LLC and Subsidiary have been restated as of December 31, 2013 and 2012, and for the years then ended to reflect to the correction of an error in the Company's accounting for the interest of the promissory notes relating to the common stock subscription agreements.
/s/ Rosenberg Rich Baker Berman & Company
Somerset, New Jersey
March 21, 2014
Except for Notes 10 and 11 as to which are October 24, 2014
| 62 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Consolidated Balance Sheets
December 31, 2013 and 2012
| 2013 | 2012 | |||||||
| (As restated) | (As restated) | |||||||
| ASSETS | ||||||||
| Cash and cash equivalents | $ | 66,769 | $ | 62,645 | ||||
| Tax refund receivable | 147,997 | 242,016 | ||||||
| TOTAL ASSETS | $ | 214,766 | $ | 304,661 | ||||
| LIABILITIES AND MEMBERS' DEFICIT | ||||||||
| Accrued expenses | $ | 469,700 | $ | 374,476 | ||||
| Demand note payable | 60,000 | - | ||||||
| Derivative liability | 10,800 | 27,000 | ||||||
| Total current liabilities | 540,500 | 401,476 | ||||||
| Convertible note payable, net | 73,980 | - | ||||||
| TOTAL LIABILITIES | 614,480 | 401,476 | ||||||
| Commitments and contingencies | ||||||||
| EQUITY (DEFICIT) | ||||||||
| Members' deficit | (390,045 | ) | (36,469 | ) | ||||
| Noncontrolling interest | (9,669 | ) | (60,346 | ) | ||||
| TOTAL DEFICIT | (399,714 | ) | (96,815 | ) | ||||
| TOTAL LIABILITIES AND DEFICIT | $ | 214,766 | $ | 304,661 | ||||
See notes to consolidated financial statements.
| 63 |
Viatar CTC Solutions Inc. and Subsidiary
(A Development Stage Company)
Consolidated Statements of Operations (Restated)
Years Ended December 31, 2013 and 2012 and the Period
from December 18, 2007 (Inception) through December 31, 2013
| The Period from | ||||||||||||
| December 18, 2007 | ||||||||||||
| Year Ended December 31, | (Date of Inception) to | |||||||||||
| 2013 | 2012 | December 31, 2013 | ||||||||||
| (As restated) | (As restated) | (As restated) | ||||||||||
| REVENUE | $ | - | $ | - | $ | - | ||||||
| EXPENSES | ||||||||||||
| Research and development | 682,149 | 4,227,758 | 14,896,299 | |||||||||
| General and administrative | 545,227 | 438,532 | 2,047,962 | |||||||||
| TOTAL EXPENSES | 1,227,376 | 4,666,290 | 16,944,261 | |||||||||
| OTHER INCOME | ||||||||||||
| Change in value of derivative liability | 16,200 | 32,400 | 71,800 | |||||||||
| Interest and dividends | - | 88 | 18,794 | |||||||||
| Income tax benefit | 147,997 | 242,016 | 626,887 | |||||||||
| TOTAL OTHER INCOME | 164,197 | 274,504 | 717,481 | |||||||||
| NET LOSS | (1,063,179 | ) | (4,391,786 | ) | (16,226,780 | ) | ||||||
| Net loss attributable to noncontrolling interest in consolidated subsidiary | (198,318 | ) | (1,466,815 | ) | (2,274,310 | ) | ||||||
| NET LOSS ATTRIBUTABLE TO MEMBERS | $ | (864,861 | ) | $ | (2,924,971 | ) | $ | (13,952,470 | ) | |||
See notes consolidated financial statements.
| 64 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Consolidated Statements of Members' Deficit (Restated)
| Vizio Member Units | Noncontrolling | |||||||||||||||||||
| Class A Units | Class B Units | Vizio Member | Interest | Total | ||||||||||||||||
| Members' equity - December 18, 2007 | - | - | $ | - | - | $ | - | |||||||||||||
| Issuance of Class A member units | 1,945,000 | - | 1,847,750 | - | 1,847,750 | |||||||||||||||
| Issuance of Class B member units | - | 4,000,000 | - | - | - | |||||||||||||||
| Issuance of member warrants | - | - | 97,250 | 97,250 | ||||||||||||||||
| Exercise of member warrants | 350,000 | - | 350,000 | 350,000 | ||||||||||||||||
| Net loss | - | - | (1,734,485 | ) | (1,734,485 | ) | ||||||||||||||
| Equity - December 31, 2008 | 2,295,000 | 4,000,000 | 560,515 | - | 560,515 | |||||||||||||||
| Issuance of Class A member units | 300,000 | - | 575,000 | - | 575,000 | |||||||||||||||
| Exercise of member warrants | 415,000 | - | 415,000 | - | 415,000 | |||||||||||||||
| Net loss | - | - | (1,539,261 | ) | - | (1,539,261 | ) | |||||||||||||
| Equity - December 31, 2009 | 3,010,000 | 4,000,000 | 11,254 | - | 11,254 | |||||||||||||||
| Issuance of Class A member units | 4,765,000 | - | 4,678,000 | - | 4,678,000 | |||||||||||||||
| Issuance of Class A member units for license | 155,500 | - | 155,500 | - | 155,500 | |||||||||||||||
| Issuance of member warrants | - | - | 87,000 | - | 87,000 | |||||||||||||||
| Contribution of services from noncontrolling interest | - | - | - | 200,000 | 200,000 | |||||||||||||||
| Net loss | - | - | (3,833,603 | ) | (198,112 | ) | (4,031,715 | ) | ||||||||||||
| Equity - December 31, 2010 | 7,930,500 | 4,000,000 | 1,098,151 | 1,888 | 1,100,039 | |||||||||||||||
| Issuance of Class A member units | 3,845,000 | - | 3,845,000 | - | 3,845,000 | |||||||||||||||
| Issuance of Class A member units for patent expenses | 269,245 | - | 269,245 | - | 269,245 | |||||||||||||||
| Issuance of Class A member units for equity raised | 300,000 | - | - | - | - | |||||||||||||||
| Contribution of services from noncontrolling interest | - | - | - | 367,000 | 367,000 | |||||||||||||||
| Net loss | - | - | (3,055,289 | ) | (411,065 | ) | (3,466,354 | ) | ||||||||||||
| Equity (deficit) - December 31, 2011 | 12,344,745 | 4,000,000 | 2,157,107 | (42,177 | ) | 2,114,930 | ||||||||||||||
| Issuance of Class A member units | 745,500 | - | 983,647 | - | 983,647 | |||||||||||||||
| Issuance of member warrants | - | - | 123,353 | - | 123,353 | |||||||||||||||
| Receivable from issuance of Class A member units | - | - | (567,000 | ) | - | (567,000 | ) | |||||||||||||
| Issuance of Class A member units for license | 91,395 | - | 91,395 | - | 91,395 | |||||||||||||||
| Issuance of Class A member units for equity raised | 446,500 | - | - | - | - | |||||||||||||||
| Issuance of Class A member units for services | 100,000 | - | 100,000 | - | 100,000 | |||||||||||||||
| Contribution of services from noncontrolling interest | - | - | - | 1,448,646 | 1,448,646 | |||||||||||||||
| Net loss | - | - | (2,924,971 | ) | (1,466,815 | ) | (4,391,786 | ) | ||||||||||||
| Deficit - December 31, 2012 | 13,728,140 | 4,000,000 | (36,469 | ) | (60,346 | ) | (96,815 | ) | ||||||||||||
| Issuance of Class A member units | 485,000 | - | 453,195 | - | 453,195 | |||||||||||||||
| Issuance of member warrants | - | - | 134,330 | - | 134,330 | |||||||||||||||
| Receivable from issuance of Class A member units | - | - | (110,000 | ) | - | (110,000 | ) | |||||||||||||
| Issuance of Class A member units for license | 14,605 | - | 14,605 | - | 14,605 | |||||||||||||||
| Issuance of Class A member units for patent expenses | 51,812 | - | 51,812 | - | 51,812 | |||||||||||||||
| Issuance of Class A member units for equity raised | 117,004 | - | - | - | - | |||||||||||||||
| Issuance of Class A member units for services | 100,000 | - | 100,000 | - | 100,000 | |||||||||||||||
| Acquisition of noncontrolling interest | - | - | (132,657 | ) | 132,657 | - | ||||||||||||||
| Contribution of services from noncontrolling interest | - | - | - | 116,338 | 116,338 | |||||||||||||||
| Net loss | - | - | (864,861 | ) | (198,318 | ) | (1,063,179 | ) | ||||||||||||
| Members' deficit - December 31, 2013 | 14,496,561 | 4,000,000 | (390,045 | ) | (9,669 | ) | (399,714 | ) | ||||||||||||
See notes to consolidated financial statements.
| 65 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Consolidated Statements of Cash Flows (Restated)
Years Ended December 31, 2013 and 2012 and the Period
from December 18, 2007 (Inception) through December 31, 2013
| The Period from | ||||||||||||
| December 18, 2007 | ||||||||||||
| Year Ended December 31, | (Date of Inception) to | |||||||||||
| 2013 | 2012 | December 31, 2013 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||||||
| Net loss | $ | (1,063,179 | ) | $ | (4,391,786 | ) | $ | (16,226,780 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Issuance of member units for patent expenses | 51,812 | - | 321,057 | |||||||||
| Issuance of member units for services | 100,000 | 100,000 | 200,000 | |||||||||
| Contribution on noncontrolling interest | 130,943 | 1,540,041 | 2,393,484 | |||||||||
| Amortization of discount on debt | 6,505 | - | 6,505 | |||||||||
| Deferred costs | - | - | 103,883 | |||||||||
| Change in derivative liability | (16,200 | ) | (32,400 | ) | 10,800 | |||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Tax refund receivable | 94,019 | (5,142 | ) | (147,997 | ) | |||||||
| Accrued expenses | 95,224 | 121,148 | 469,700 | |||||||||
| Net cash used in operating activities | (600,876 | ) | (2,668,139 | ) | (12,869,348 | ) | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||||||
| Proceeds from demand note payable | 60,000 | - | 60,000 | |||||||||
| Proceeds from convertible note payable | 100,000 | - | 100,000 | |||||||||
| Issuance of member units | 445,000 | 540,000 | 12,115,000 | |||||||||
| Exercise of member warrants | - | - | 765,000 | |||||||||
| Payment of deferred costs | - | - | (103,883 | ) | ||||||||
| Net cash provided by financing activities | 605,000 | 540,000 | 12,936,117 | |||||||||
| Net increase (decrease) in cash and cash equivalents | 4,124 | (2,128,139 | ) | 66,769 | ||||||||
| Cash and cash equivalents - beginning of the period | 62,645 | 2,190,784 | - | |||||||||
| Cash and cash equivalents - end of the period | $ | 66,769 | $ | 62,645 | $ | 66,769 | ||||||
| Supplemental disclosure of cash flow information: | ||||||||||||
| Cash paid for interest | $ | 2,000 | $ | - | $ | 2,000 | ||||||
| Supplemental disclosure of non-cash financing activities: | ||||||||||||
| Promissory notes received from issuance of member units | $ | 110,000 | $ | 567,000 | $ | 677,000 | ||||||
| Discount on debt issued with warrants | $ | 32,525 | $ | - | $ | 32,525 | ||||||
See notes to consolidated financial statements.
| 66 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
| 1. | Development Stage operations AND GOING CONCERN |
Vizio Medical Devices LLC and Subsidiary designs and manufactures medical devices for oncology applications which remove circulating tumor cells for diagnostic and therapeutic purposes.
Vizio Medical Devices LLC (“Vizio”) is a Delaware limited liability company with perpetual duration that was formed on December 18, 2007. On February 25, 2014 Vizio converted to a Delaware C corporation and changed its name to Viatar CTC Solutions Inc. (“Viatar CTC Solutions”) (see Note 7).
Vizio conducts all of its operations through its subsidiary, Viatar LLC (“Viatar”). Viatar was formed on September 23, 2010 as a Delaware limited liability company with perpetual duration.
Viatar CTC Solutions, its predecessor Vizio, and Viatar are together referred to as the “Company.”
The members of a limited liability company are not liable for the debts, obligations or liabilities of the limited liability company. Vizio is managed by a board of managers which shall consist of one to five managers. The original Class B member of Vizio can appoint one manager and the Class A members who own 250,000 or more units can appoint additional managers. As of December 31, 2013, there were three managers on the board of managers of Vizio. Viatar is managed by a board of managers which shall consist of one or more managers. A member who owns 250 or more units can appoint additional managers. As of December 31, 2013, there was one manager on the board of managers of Viatar.
The Company is in the development stage and has not generated any revenues. As a result, the Company has incurred net losses of $16,226,780 and negative cash flows from operations of $12,869,348 since its inception. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. The Company’s continued existence is dependent upon several factors, including (a) obtaining funding, whether in the form of equity or, debt investments, licensing revenues, strategic collaboration payments or grants from government or other sources, (b) success in achieving its research and development goals, (c) obtaining regulatory approvals, and (d) gaining market acceptance and/or distribution or strategic partners for its products. The Company has raised $13.5 million of equity capital from non-institutional investors since its inception, and management is continually pursuing additional funding sources.
| 67 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 2. | Summary of Significant Accounting Policies |
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of Vizio, Viatar and amounts related to a noncontrolling interest in Viatar. The noncontrolling interest represents a 0.03% and 50% ownership in Viatar at December 31, 2013 and 2012, respectively. All significant intercompany accounts and transactions have been eliminated in consolidation.
Basis of Accounting
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America.
Cash and Cash Equivalents
The Company considers all highly liquid debt instruments purchased with original maturities of three months or less to be cash equivalents.
Cash and cash equivalents are held in a money market fund that holds US government and agency bonds, and highly-rated securities issued by finance companies. This fund is not federally insured.
Syndication Costs
Investment banking fees and related costs associated with the issuance of member units are charged to members’ equity. Investment banking fees and related costs that are paid in anticipation of raising capital are recorded as deferred costs until such capital is raised or subsequently expensed if such capital is not raised.
Research and Development
Research and development costs consist of ongoing testing and research and are expensed as incurred. Government grants received by the Company for research and development are recorded as reductions of the related expenses in the period in which they are reported.
| 68 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 2. | Summary of Significant Accounting Policies (continued) |
Income Taxes
No provision or benefit for federal or state income taxes has been included in the accompanying financial statements because such taxable income or loss passes through to, and is reportable by, the members. No tax liability has been incurred since inception as the Company has incurred losses.
For the year ended December 31, 2013, the Company applied for biotechnology tax credits from New York City and recorded a receivable of $147,997. For the year ended December 31, 2012, the Company received biotechnology tax credits from New York City of $242,016. These credits have been recorded as income tax benefits in the consolidated statements of operations using the flow-through method as discussed in ASC Topic 740, “Income Taxes.”
On April 30, 2014, the Company received the tax refund receivable of $147,997.
The Company files income tax returns with the U.S. federal government and various state and local jurisdictions. The Company is no longer subject to tax examinations by tax authorities for years prior to 2010.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of income and expenses during the reporting period. Significant estimates subject to such estimates and assumptions include the valuation of debt and equity instruments issued for services and in connection with agreements and contracts entered in to by the Company. Actual results could differ from those estimates.
Derivatives
The Company evaluated its options, warrants or other contracts to determine if those contracts or embedded components of those contracts qualify as derivatives to be separately accounted for under ASC Topic 815, “Derivatives and Hedging.” The result of this accounting treatment is that the fair value of the derivative is marked-to-market each balance sheet date and recorded as a liability. In the event that the fair value is recorded as a liability, the change in fair value is recorded in the statements of operations as other income (expense). Upon conversion or exercise of a derivative instrument, the instrument is marked to fair value at the conversion date and then that fair value is reclassed to equity. Equity instruments that are initially classified as equity that become subject to reclassification under ASC Topic 815 are reclassified to liability at the fair value of the instrument on the reclassification date.
| 69 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 2. | Summary of Significant Accounting Policies (continued) |
Reclassifications
Certain amounts from the prior year financial statements have been reclassified to conform to the current year presentation.
| 3. | MEMBERS’ deficit |
Vizio’s capital structure consists of the authorization to issue 16,000,000 Class A member units and 4,000,000 Class B member units. Vizio is also authorized to issue warrants. Each issued Class A member unit has one vote. The Class B member units and the warrants do not have voting rights. The Class B member units were originally issued to the managing member of Vizio and represent a non-dilutable 20% economic interest in its profits. The profits and losses of Vizio, as defined in the operating agreement, are allocated to the members in accordance with their respective ownership percentage interests, provided that such percentage interest is adjusted to reflect the 20% profits interest that is allocable to the holders of the Class B member units. The Class B member units are not allocated any losses; and before the Class B member units are allocated any profits, the Class A member units are to first be allocated an aggregate amount of profits equal to the aggregate amount of losses previously allocated to them.
As of December 31, 2013, 14,496,561 Class A member units and 4,000,000 Class B member units were issued and outstanding. As of December 31, 2012, 13,728,140 Class A member units and 4,000,000 Class B member units were issued and outstanding.
During 2013 and 2012, Vizio issued an aggregate of 140,000 and 378,000 Class A member units, respectively, in exchange for $210,000 and $567,000, respectively, principal amount of a 5% secured promissory note due December 31, 2014. As of December 31, 2013 and 2012, the outstanding note balance was $721,782, and $574,927 respectively, including $44,782 and $7,927, respectively of accrued interest receivable.
As of December 31, 2013 and 2012, Vizio issued an aggregate of 150,000 and 75,000 warrants, respectively, in connection with the sale of Class A member units. The warrants are not transferable without prior written consent of Vizio. The warrants are exercisable at prices ranging from $1 to $2 each and expire in 2017 and 2018. The fair value of the warrants issued in 2013 and 2012 were $101,805 and $123,353, respectively, based on the Black-Scholes option pricing model.
| 70 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 3. | MEMBERS’ deficit (CONTINUED) |
In 2013 and 2012, Vizio issued 14,605 and accrued 13,063 and issued 91,395 Class A member units, respectively, to the noncontrolling interest pursuant to an anti-dilution adjustment originating in 2010 in consideration for the grant of the license of intellectual property. It provides for the issuance of additional Class A member units as is necessary to maintain a 2% ownership interest in Vizio. This annual anti-dilution adjustment is in effect until the first to occur of (x) the first approval for a product, or (y) $10 million of capital being raised at a valuation that is greater than $1 per Class A member unit. The anti-dilution adjustments are in lieu of cash royalty payments by Vizio or its sublicensees. The derivative liability associated with the anti-dilution adjustment was $10,800 and $27,000 at December 31, 2013 and 2012, respectively. The Company estimated the fair value of the derivative liability using a probability weighted average approach that considered the possible outcomes based on a assumptions related to the timing and probability of expected capital raises and product approval.
In 2013, Vizio issued 51,812 Class A member units to a licensor in lieu of cash reimbursement for $51,812 of accrued patent expenses. In 2013 and 2012, Vizio issued an aggregate of 117,004 and 446,500 Class A member units, respectively, for the cost of raising equity and 100,000 each year for annual service by the non-officer board of managers. In 2011, Vizio issued 300,000 units for equity raised.
In 2010, Vizio and its managing member, who was then the owner of all of the Class B member units, jointly implemented an Equity Incentive Plan (the “Incentive Plan”) to provide employees and consultants with incentive compensation awards. The managing member contributed 1,000,000 of the 4,000,000 Class B member units discussed above to the Plan. The units are not transferable without prior written consent of Vizio. As of December 31, 2013, the participants in the Plan hold 595,000 vested and 345,000 unvested Class B member units; an additional 60,000 Class B member units are reserved for future issuance under the Plan.
Viatar’s capital structure consists of the authorization to issue 1,000,000 member units. Each issued member unit has one vote. The profits and losses of Viatar, as defined in the operating agreement, are allocated to the members in accordance with their respective ownership percentage interests. During 2013, Vizioi acquired 420 units from the other member of Viatar in exchange for an exclusive license to appropriate patents owned by Viatar. Viatar accounted for this as an equity transaction whereby the fair value of the noncontrolling interest was adjusted to reflect the change in ownership of Viatar, with no corresponding gain or loss recognized in the consolidated statements of operations. As of December 31, 2013 and 2012, 304,000 and 1,000 member units, respectively, were issued and outstanding, of which Vizio held 303,920 and 500, respectively, and another entity held 80 and 500, respectively.
| 71 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 4. | CONVERTIBLE NOTE PAYABLE |
| In April 2013, Vizio borrowed $100,000 in exchange for a 4% interest-only, convertible promissory note due April 30, 2015 and a five year warrant to purchase 50,000 Class A member units at $2 each. The note is convertible at any time prior to maturity at the option of the holder into 50,000 Class A member units. The fair value of the warrant issued was $32,525 based on the Black-Scholes option pricing model. The fair value of the warrant was recorded as a discount and is being amortized over two years based on the straight line method, which approximates the effective interest method. The unamortized discount at December 31, 2013 was $26,020. The note requires interest payments semi-annually. |
| 5. | OPERATING LEASE |
The Company has entered into an operating lease for its office space for $3,103 per month through June 2015. The following are the future minimum rental payments to be paid under the non-cancelable operating lease in effect at December 31, 2013:
| Year Ending December 31, | Amount | |||
| 2014 | $ | 37,240 | ||
| 2015 | $ | 18,620 | ||
| 6. | Research and development and grant |
During 2013 and 2012, Viatar conducted its research and development activities on the oncology projects through its own staff in Lowell, MA, as well as consultants and component manufacturers in the United States with expertise in specific technical and regulatory areas. During 2012, Viatar also engaged Mount Sinai School of Medicine in New York City.
During 2012, Vizio conducted research activities on medical devices for dialysis and congestive heart failure patients through its own staff in Walpole and Lowell, MA; it also engaged Columbia University (“Columbia”) and Bridgemedica LLC (“Bridgemedica”), as well as consultants and component manufacturers in the United States and Europe with expertise in specific technical areas. In the first quarter of 2013, Vizio discontinued support for those activities due to financial constraints and unresolved technical hurdles.
The Company owns or controls all of the intellectual property generated by employees and third parties, either via license agreements or contractual provisions. The Company’s contracts for research and development activities are generally for one year or at-will.
| 72 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 6. | Research and development and grant (CONTINUED) |
In 2011, the National Institutes of Health (“NIH”) issued a grant for an aggregate of $1.95 million for research and development of a medical device for dialysis patients, as a Phase II award under the U.S. government’s Small Business Technology Transfer program. The grant expires in 2014. Vivonics, Inc. (an engineering firm) is the official grant recipient, Columbia is the academic partner, and Vizio is the commercialization partner. In 2013, Vivonics paid $25,984 to Vizio under the NIH grant for research services.
| 7. | COMMITMENTS AND CONTINGENCIES |
The Company’s patent portfolio consists of a total of over 30 licensed and owned patents and patent applications.
There are 3 core patent groups with a broad range of claims applicable to Viatar’s oncology products; these have been filed as 14 applications: three in the United States and 11 in foreign jurisdictions, with anticipated expiration dates of 2030 to 2034 (assuming they are issued). These are now either in pre-publication phase or in various stages of examination. All of the 14 patents and applications relevant to Viatar’s oncology products are owned by Viatar.
The Company’s patent portfolio also includes 8 issued patents and 8 patent applications that pertain to cardiology, nephrology and other potential medical or commercial applications. These are licensed from Columbia University under customary terms for milestones, royalties and other payments; but the Company will not incur any costs for them in conducting its oncology products business. In addition, the Company has licensing agreements with other companies for coating and filter manufacturing techniques which are not relevant to Viatar’s oncology products and will lapse over time.
The licensed patents expire at various times from 2018 to 2029, while the last-dated pending patent applications (if issued) would expire in 2031. The Company’s license agreements have broad, exclusive “field of use” clauses, which cover all currently planned product applications on a worldwide basis. These agreements also contain customary provisions regarding royalty rates, minimum annual payments, milestone payments, sublicense payments, and payments in the event of a change of control.
| 73 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 7. | COMMITMENTS AND CONTINGENCIES (CONTINUED) |
The total royalties payable under the Company’s license agreements (none of which pertain to Viatar’s oncology products) once commercial sales commence will range from 2.75% to 10% of the net sales price for a given product category or component, depending on product mix, expiration of certain patents, and the level of sales. The total minimum annual royalty payments for the first year after regulatory approval is obtained will be $200,000 or zero depending on the product category; thereafter minimum annual payments will be zero or in the range of $400,000 to $1,000,000, depending primarily on whether the Company obtains regulatory approval for additional product categories. In the event the Company achieves $50 million of annual sales, either cumulatively or for certain product categories in a single year, additional payments of $500,000 would be due. Aggregate milestone payments of $300,000 are due pursuant to one license agreement during the period from 2012 until various regulatory approvals are first obtained, but the licensor has waived that provision for 2012 and 2013. In addition, the license agreement with Columbia provides that the Company will be required to pay a one-time fee equal to 5% of the valuation of the Company, as defined in the license agreement, in connection with certain “Trigger Events,” as defined in the license agreement, such as a sale, merger or initial public offering, among other events. This arrangement does not pertain to Viatar’s oncology products, since they do not utilize any of the intellectual property covered by the license agreement with Columbia.
The license agreement with Columbia provides for an annual cap of $200,000 in 2012 on the Company’s patent expense reimbursement obligation. The annual cap for subsequent years has not been negotiated, and the cap will expire in its entirety once the Company obtains regulatory approval to sell a licensed product or raises an aggregate of $15 million of financing.
In 2012, Vizio sued Bridgemedica in New York State Supreme Court for $1.5 million of damages arising from breach of contract and other claims relating to non-performance under various engineering development contracts. Bridgemedica counterclaimed for $179,000 for delinquent payments.
| 74 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 8. | FAIR VALUE OF FINANCIAL INSTRUMENTS |
The Company follows the provisions of ASC 820 for fair value measurements of its financial assets and liabilities. The accounting standard for fair value measurements provides a framework for measuring fair value and requires expanded disclosures regarding fair value measurements. Fair value is defined as the price that would be received for an asset or the exit price that would be paid to transfer a liability in the principal or most advantageous market in an orderly transaction between market participants on the measurement date. The accounting standard established a fair value hierarchy which requires an entity to maximize the use of observable inputs, where available. This hierarchy prioritizes the inputs into three broad levels, as follows: Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Level 3 inputs are unobservable inputs based on the Company's own assumptions used to measure assets and liabilities at fair value. An asset or liability's classification within the hierarchy is determined based on the lowest level input that is significant to the fair value measurement. The Company’s financial instruments measured at fair value on a recurring basis consisted of the following at December 31, 2013 and 2012:
| Total Carrying Value at December 31, 2013 | ||||||||||||
| (Level 1) | (Level 2) | (Level 3) | ||||||||||
| Derivative Liability | $ | - | $ | - | $ | 10,800 | ||||||
| Total Carrying Value at December 31, 2012 | ||||||||||||
| (Level 1) | (Level 2) | (Level 3) | ||||||||||
| Derivative Liability | $ | - | $ | - | $ | 27,000 | ||||||
The following is a summary of activity of Level 3 liabilities for the years ended December 31, 2013 and 2012:
| Balance at December 31, 2011 | $ | (59,400 | ) | |
| Change in fair value 2012 | 32,400 | |||
| Balance at December 31, 2012 | (27,000 | ) | ||
| Change in fair value 2013 | 16,200 | |||
| Balance at December 31, 2013 | $ | (10,800 | ) |
| 75 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 8. | FAIR VALUE OF FINANCIAL INSTRUMENTS (CONTINUED) |
The Company estimates the fair value of the convertible note and warrants liability utilizing the Black-Scholes option pricing model, which is dependent upon several variables such as the expected note term, expected volatility of the stock price over the expected note term, expected risk-free interest rate over the expected note term, and the expected dividend yield rate over the expected note term. The Company believes this valuation methodology is appropriate for estimating the fair value of the liability. The following table summarizes the assumptions the Company utilized to estimate the fair value of the convertible note and warrants liability at December 31, 2013:
| Assumptions | December 31, 2013 | |
| Expected term (years) | 5 | |
| Expected volatility | 100% | |
| Risk-free interest rate | 0.725% - 1.748% | |
| Dividend yield | 0.00% |
The expected warrant term is based on the remaining contractual term. The expected volatility is based on historical-volatility of peer entities whose stock prices were publicly available. The risk-free interest rate is based on the U.S. Treasury yields with terms equivalent to the expected term of the related warrant at the valuation date. Dividend yield is based on historical trends.
The carrying value of the Company’s other financial assets and liabilities approximates fair value at December 31, 2013 and 2012.
| 9. | RELATED PARTY TRANSACTION |
In January 2013, Vizio borrowed an aggregate of $60,000 in exchange for a demand note from a member of the board of managers. The loan is interest free.
| 10. | RESTATEMENT OF PREVIOUSLY ISSUED FINANCIAL STATEMENTS |
In October 2014, management concluded that the interest receivable recognized in relation to the issuance of certain promissory notes related to a receivable from the issuance of Class A member units was improperly recorded in the Company’s financial statements. The interest recognized on the promissory notes should have been recorded in Members’ Deficit.
| 76 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 10. | RESTATEMENT OF PREVIOUSLY ISSUED FINANCIAL STATEMENTS (CONTINUED) |
The above non-cash restatements for the correction of an error affect the financial statements as follows:
Consolidated Balance Sheets
| December 31, 2013 | ||||||||
| As Previously | As | |||||||
| Reported | Restated | |||||||
| Total assets | $ | 259,548 | $ | 214,766 | ||||
| Total liabilities | 614,480 | 614,480 | ||||||
| Total Vizio Medical Devices members' deficit | (354,786 | ) | (390,045 | ) | ||||
| Noncontrolling interest | (146 | ) | (9,669 | ) | ||||
| December 31, 2012 | ||||||||
| As Previously | As | |||||||
| Reported | Restated | |||||||
| Total assets | $ | 312,588 | $ | 304,661 | ||||
| Total liabilities | 401,476 | 401,476 | ||||||
| Total Vizio Medical Devices members' deficit | (31,190 | ) | (36,469 | ) | ||||
| Noncontrolling interest | (57,698 | ) | (60,346 | ) | ||||
Consolidated Statements of Operations
| For the Year Ended December 31, 2013 | ||||||||
| As Previously | As | |||||||
| Reported | Restated | |||||||
| Net loss | $ | (1,026,324 | ) | $ | (1,063,179 | ) | ||
| Net loss attributable to noncontrolling interest | (191,443 | ) | (198,318 | ) | ||||
| Net loss attributable to members | (834,881 | ) | (864,861 | ) | ||||
| For the Year Ended December 31, 2012 | ||||||||
| As Previously | As | |||||||
| Reported | Restated | |||||||
| Net loss | $ | (4,383,859 | ) | $ | (4,391,786 | ) | ||
| Net loss attributable to noncontrolling interest | (1,464,167 | ) | (1,466,815 | ) | ||||
| Net loss attributable to members | (2,919,692 | ) | (2,924,971 | ) | ||||
| 77 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 11. | SUBSEQUENT EVENTS |
During January and February 2014 Vizio issued 686,668 Class A member units, consisting of 487,500 Class A member units, 16,668 Class A member units issued in connection with an anti-dilution adjustment under a license agreement, 82,500 Class A member units for the cost of raising equity and 100,000 Class A member units issued to the non-officer members of the board of managers. These issuances are included in the number of shares of common stock of Viatar CTC Solutions issued and outstanding just after the conversion of Vizio from an LLC into a C corporation.
On February 25, 2014 Vizio converted from a limited liability company to a C corporation pursuant to Delaware law, and changed its name to Viatar CTC Solutions Inc. Each Class A member unit was converted into one share of common stock and each Class B member unit was converted into one share of Series A Preferred Stock (the “Preferred Stock”). Each warrant to purchase Class A member units was converted into one warrant to purchase one share of common stock.
Viatar CTC Solutions’ capital structure consists of authorization to issue 100,000,000 shares of common stock and 20,000,000 shares of preferred stock, which may be issued in series as designated by the Board of Directors. Each series of Preferred Stock shall have such voting and other rights as designated at the time of establishment by the Board of Directors.
Each issued share of common stock and Preferred Stock has one vote; and the approval of a majority of the Preferred Stock, voting as a separate class, is required to approve any corporate action. The Preferred Stock is entitled to participate in dividends, rights issuances and property distributions on the same terms that the Class B member units had since the formation of the Company; namely, 20% of the dividends, rights and property distributed to common stockholders. Upon liquidation of the Company, the Preferred Stock is entitled to 20% of the liquidation value, subject to a priority $13,470,000 allocation to the common shares. This formulation preserves the right of the former Class B member units to participate in profits once the holders of Class A member units recouped their aggregate capital contributions as of February 25, 2014.
As of February 25, 2014, immediately following the conversion of the Company from a limited liability company to a C corporation, 15,196,292 shares of common stock and 4,000,000 shares of Preferred Stock are issued and outstanding. In addition, 575,259 shares of common stock are due to professional advisory firms for services related to the conversion of which 344,196 shares are restricted and held in escrow for 12 months. Also as of that date, 410,000 shares of common stock are reserved for issuance pursuant to warrants at prices ranging from $1 to $2 per share and expiring in 2017 through 2019, and 3,000,000 shares of common stock to be reserved for future issuance to employees, directors and consultants under the Plan.
| 78 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Notes to Consolidated Financial Statements (Restated)
December 31, 2013 and 2012
(Continued)
| 11. | SUBSEQUENT EVENTS (CONTINUED) |
In February 1, 2014, the demand note was converted into 60,000 shares of common stock and a warrant to purchase 60,000 shares of common stock at $1 per share, expiring in 2019.
In February 25, 2014, the Company issued 231,063 shares for $1 per share in connection with services performed for an equity raise.
In March 2014, Viatar CTC Solutions issued 75,000 shares of common stock for $2 per share.
During the second quarter 2014, the Company issued 73,750 shares of common shares for proceeds of $125,000 which included 11,250 shares of common stock for issuance costs.
During the second quarter 2014, the Company received proceeds of $50,000 for the exercise of 50,000 previously issued warrants.
On April 8, 2014, the Company settled with Bridgemedica. The settlement requires the Company to issue 150,000 shares of common stock and pay four installments totaling $20,000 cash to Bridgemedica. The shares were issued during the second quarter 2014 at $1 per share. As of the date of this report, $5,000 remains unpaid.
On August 21, 2014, the consulting and license agreement between Aquamarijn and the Company was terminated, which in effect ends the 2% anti-dilution adjustment.
On August 27, 2014, the Company issued 325,000 shares of common stock to various employees, of which 150,000 vested immediately and 175,000 will vest in 2015 and 2016.
On September 14, 2014, Viatar CTC Solutions issued 45,821 shares of Common Stock to Columbia University in connection with the termination of the license and research agreements.
During October 2014, the Company received $50,000 for the payoff of notes originally issued to purchase common stock receivable.
On October 8, 2014, pursuant to the 2014 Equity Incentive Plan, the holders of 595,000 shares of vested Preferred Stock exchanged the preferred shares for 595,000 shares of common stock.
| 79 |
UNAUDITED PRO FORMA CONSOLIDATED FINANCIAL INFORMATION
The following unaudited pro forma consolidated balance sheet of Vizio Medical Devices LLC & Subsidiaries at December 31, 2013 gives effect to our conversion to a corporation. The following unaudited pro forma consolidated statement of operations for the year ended December 31, 2013 gives effect to the conversion as if it had occurred on the first day of the year.
The pro forma adjustments are preliminary and have been made solely for purposes of developing the pro forma financial information. As a result, the pro forma information does not purport to be indicative of what the financial condition or results of operations would have been had the transaction been completed on the applicable dates of this pro forma financial information. The pro forma financial information is based upon the historical financial statements of Vizio Medical Devices LLC & Subsidiaries.
The following unaudited pro forma consolidated financial information is derived from the historical financial statements of Vizio Medical Devices LLC & Subsidiaries and has been prepared to illustrate the effects of the conversion to a corporation of Vizio Medical Devices LLC & Subsidiaries. The pro forma consolidated financial information should be read in conjunction with the historical consolidated financial statements and the accompanying notes of Vizio Medical Devices LLC & Subsidiaries.
| 80 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Unaudited Pro Forma Consolidated Balance Sheet
December 31, 2013
| 2013 (As | Pro Forma | 2013 (As if | ||||||||||
| reported) | Adjustment | Corporation) | ||||||||||
| ASSETS | ||||||||||||
| CURRENT ASSETS | ||||||||||||
| Cash and cash equivalents | $ | 66,769 | $ | - | $ | 66,769 | ||||||
| Tax refund receivable | 147,997 | - | 147,997 | |||||||||
| TOTAL ASSETS | $ | 214,766 | $ | - | $ | 214,766 | ||||||
| LIABILITIES AND DEFICIT | ||||||||||||
| CURRENT LIABILITIES | ||||||||||||
| Accrued expenses | $ | 469,700 | $ | - | $ | 469,700 | ||||||
| Demand note payable | 60,000 | - | 60,000 | |||||||||
| Derivative liability | 10,800 | - | 10,800 | |||||||||
| Total current liabilities | 540,500 | - | 540,500 | |||||||||
| Convertible note payable, net | 73,980 | - | 73,980 | |||||||||
| TOTAL LIABILITIES | 614,480 | - | 614,480 | |||||||||
| Commitments and contingencies | ||||||||||||
| EQUITY (DEFICIT) | ||||||||||||
| Members' deficit | (390,045 | ) | 390,045 | - | ||||||||
| Series A preferred stock, $.001 par value, 20,000,000 shares authorized, 4,000,000 shares issued and outstanding | 4,000 | 4,000 | ||||||||||
| Common stock, $.001 par value, 100,000,000 shares authorized, 14,496,561 shares issued and outstanding. | 14,497 | 14,497 | ||||||||||
| Common stock subscription and interest receivable | (721,782 | ) | (721,782 | ) | ||||||||
| Additional paid-in capital | 14,265,710 | 14,265,710 | ||||||||||
| Deficit accumulated during development stage | (13,952,470 | ) | (13,952,470 | ) | ||||||||
| (390,045 | ) | - | (390,045 | ) | ||||||||
| Noncontrolling interest | (9,669 | ) | - | (9,669 | ) | |||||||
| TOTAL DEFICIT | (399,714 | ) | - | (399,714 | ) | |||||||
| TOTAL LIABILITIES AND DEFICIT | $ | 214,766 | $ | - | $ | 214,766 | ||||||
| 81 |
Vizio Medical Devices LLC and Subsidiary
(A Development Stage Company)
Unaudited Pro Forma Consolidated Statement of Operations
Year Ended December 31, 2013
| 2013 (As | Pro Forma | 2013 (As if | ||||||||||
| reported) | Adjustment | Corporation) | ||||||||||
| REVENUE | $ | - | $ | - | $ | - | ||||||
| EXPENSES | ||||||||||||
| Research and development | 682,149 | - | 682,149 | |||||||||
| General and administrative | 545,227 | - | 545,227 | |||||||||
| TOTAL EXPENSES | 1,227,376 | - | 1,227,376 | |||||||||
| OTHER INCOME (EXPENSE) | ||||||||||||
| Change in value of derivative liability | 16,200 | - | 16,200 | |||||||||
| Income tax benefit | 147,997 | - | 147,997 | |||||||||
| TOTAL OTHER INCOME (EXPENSE) | 164,197 | - | 164,197 | |||||||||
| NET LOSS | (1,063,179 | ) | - | (1,063,179 | ) | |||||||
| Net loss attributable to noncontrolling interest in consolidated subsidiary | (198,318 | ) | - | (198,318 | ) | |||||||
| NET LOSS ATTRIBUTABLE TO MEMBERS | $ | (864,861 | ) | $ | 864,861 | $ | - | |||||
| NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS | $ | - | $ | (864,861 | ) | $ | (864,861 | ) | ||||
| Weighted-average common shares outstanding | ||||||||||||
| Basic and diluted | - | 14,184,778 | 14,184,778 | |||||||||
| Net loss per common share | ||||||||||||
| Basic and diluted | $ | - | $ | (0.06 | ) | $ | (0.06 | ) | ||||
| 82 |
Viatar CTC Solutions Inc. and Subsidiary
Condensed Consolidated Financial Statements
For the Nine and Three Months Ended September 30, 2014 and 2013
| 83 |
Viatar CTC Solutions Inc. and Subsidiary
Table of Contents
For the Nine and Three Months Ended September 30, 2014 and 2013
| 84 |
| Viatar CTC Solutions Inc. and Subsidiary |
| Condensed Consolidated Balance Sheets |
| September 30, 2014 | December 31, 2013 | |||||||
| (Unaudited) | ||||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash and cash equivalents | $ | 133,836 | $ | 66,769 | ||||
| Accounts receivable | 4,530 | - | ||||||
| Tax refund receivable | - | 147,997 | ||||||
| TOTAL ASSETS | $ | 138,366 | $ | 214,766 | ||||
| LIABILITIES AND STOCKHOLDERS' DEFICIT | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accrued expenses | $ | 346,987 | $ | 469,700 | ||||
| Demand note payable | - | 60,000 | ||||||
| Derivative liability | - | 10,800 | ||||||
| Total current liabilities | 346,987 | 540,500 | ||||||
| Convertible note payable, net | 88,616 | 73,980 | ||||||
| TOTAL LIABILITIES | 435,603 | 614,480 | ||||||
| Commitments and contingencies | ||||||||
| STOCKHOLDERS' DEFICIT | ||||||||
| Series A preferred stock, $.001 par value, 20,000,000 shares authorized, 4,000,000 shares issued and outstanding at September 30, 2014 and December 31, 2013 | 4,000 | 4,000 | ||||||
| Common stock, $.001 par value, 100,000,000 shares authorized, 16,179,426 and 14,496,561 shares issued and outstanding at September 30, 2014 and December 31, 2013, respectively | 16,180 | 14,497 | ||||||
| Common stock subscription and interest receivable | (743,900 | ) | (721,782 | ) | ||||
| Additional paid-in capital | 16,183,960 | 14,265,710 | ||||||
| Accumulated deficit | (15,747,679 | ) | (13,952,470 | ) | ||||
| Total stockholders' deficit | (287,439 | ) | (390,045 | ) | ||||
| Noncontrolling interest | (9,798 | ) | (9,669 | ) | ||||
| TOTAL STOCKHOLDERS' DEFICIT | (297,237 | ) | (399,714 | ) | ||||
| TOTAL LIABILITIES AND STOCKHOLDERS' DEFICIT | $ | 138,366 | $ | 214,766 | ||||
See notes to condensed consolidated financial statements.
| 85 |
| Viatar CTC Solutions Inc. and Subsidiary |
| Condensed Consolidated Statements of Operations |
| (Unaudited) |
| For the Nine Months Ended September 30, | For the Three Months Ended September 30, | |||||||||||||||
| 2014 | 2013 | 2014 | 2013 | |||||||||||||
| REVENUE | ||||||||||||||||
| Sales | $ | 9,000 | $ | - | $ | - | $ | - | ||||||||
| EXPENSES | ||||||||||||||||
| Research and development | 982,450 | 605,978 | 552,321 | 201,882 | ||||||||||||
| General and administrative | 806,176 | 232,804 | 136,031 | 98,408 | ||||||||||||
| TOTAL EXPENSES | 1,788,626 | 838,782 | 688,352 | 300,290 | ||||||||||||
| OTHER INCOME (EXPENSE) | ||||||||||||||||
| Change in value of derivative liability | 1,800 | 10,800 | - | 5,400 | ||||||||||||
| Interest and dividends | - | - | - | (179 | ) | |||||||||||
| Interest expense | (17,663 | ) | (6,004 | ) | (17,663 | ) | (6,004 | ) | ||||||||
| Income tax benefit | - | - | - | - | ||||||||||||
| TOTAL OTHER INCOME (EXPENSE) | (15,863 | ) | 4,796 | (17,663 | ) | (783 | ) | |||||||||
| NET LOSS | (1,795,489 | ) | (833,986 | ) | (706,015 | ) | (301,073 | ) | ||||||||
| Net (loss) income attributable to noncontrolling interest in consolidated subsidiary | (280 | ) | (191,410 | ) | (194 | ) | 6,158 | |||||||||
| NET LOSS ATTRIBUTABLE TO STOCKHOLDERS | $ | (1,795,209 | ) | $ | (642,576 | ) | $ | (705,821 | ) | $ | (307,231 | ) | ||||
| LOSS PER COMMON SHARE - BASIC AND DILUTED: | ||||||||||||||||
| Net Loss Attributable to Common Stockholders | $ | (0.12 | ) | $ | (0.05 | ) | $ | (0.04 | ) | $ | (0.02 | ) | ||||
| Weighted Average Shares | 15,556,312 | 14,092,717 | 15,918,177 | 14,252,232 | ||||||||||||
See notes to condensed consolidated financial statements.
| 86 |
| Viatar CTC Solutions Inc. and Subsidiary |
| Condensed Consolidated Statements of Stockholders' Deficit |
| For the Nine Months Ended September 30, 2014 and the Year Ended December 31, 2013 |
| (Unaudited) |
| Common Stock | Total | |||||||||||||||||||||||||||||||||||||||
| Subscription | Additional | Deficit Accumulated | Stockholders' | |||||||||||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | and Interest | Paid-in | During Development | Equity | Noncontrolling | Total | |||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Receivable | Capital | Stage | (Deficit) | Interest | Deficit | |||||||||||||||||||||||||||||||
| Balance, January 1, 2013 | 4,000,000 | $ | 4,000 | 13,728,140 | $ | 13,728 | $ | (574,927 | ) | $ | 13,608,339 | $ | (13,087,609 | ) | $ | (36,469 | ) | $ | (60,346 | ) | $ | (96,815 | ) | |||||||||||||||||
| Issuance of stock and warrants, net of offering costs of $117,004 | - | - | 462,004 | 462 | - | 587,063 | - | 587,525 | - | 587,525 | ||||||||||||||||||||||||||||||
| Receivable from issuance of stock | - | - | 140,000 | 140 | (210,000 | ) | (140 | ) | - | (210,000 | ) | - | (210,000 | ) | ||||||||||||||||||||||||||
| Receipt of promissory note | - | - | - | - | 100,000 | - | - | 100,000 | - | 100,000 | ||||||||||||||||||||||||||||||
| Issuance of stock for license | - | - | 14,605 | 15 | - | 14,590 | - | 14,605 | - | 14,605 | ||||||||||||||||||||||||||||||
| Issuance of stock for patent expenses | - | - | 51,812 | 52 | - | 51,760 | - | 51,812 | - | 51,812 | ||||||||||||||||||||||||||||||
| Issuance of stock for services | - | - | 100,000 | 100 | - | 99,900 | - | 100,000 | - | 100,000 | ||||||||||||||||||||||||||||||
| Interest on common stock receivable | - | - | - | - | (36,855 | ) | 36,855 | - | - | - | - | |||||||||||||||||||||||||||||
| Acquisition of non-controlling interest | - | - | - | - | - | (132,657 | ) | - | (132,657 | ) | 132,657 | - | ||||||||||||||||||||||||||||
| Contribution of services from noncontrolling interest | - | - | - | - | - | - | - | - | 116,338 | 116,338 | ||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (864,861 | ) | (864,861 | ) | (198,318 | ) | (1,063,179 | ) | ||||||||||||||||||||||||||
| Balance, December 31, 2013 | 4,000,000 | 4,000 | 14,496,561 | 14,497 | (721,782 | ) | 14,265,710 | (13,952,470 | ) | (390,045 | ) | (9,669 | ) | (399,714 | ) | |||||||||||||||||||||||||
| Issuance of stock and warrants, net of offering costs of $93,750 | - | - | 671,250 | 671 | - | 829,329 | - | 830,000 | - | 830,000 | ||||||||||||||||||||||||||||||
| Issuance of stock upon exercise of warrants | - | - | 50,000 | 50 | - | 49,950 | - | 50,000 | - | 50,000 | ||||||||||||||||||||||||||||||
| Issuance of stock upon conversion of demand note | - | - | 60,000 | 60 | - | 59,940 | - | 60,000 | - | 60,000 | ||||||||||||||||||||||||||||||
| Issuance of stock for license | - | - | 29,731 | 30 | - | 29,701 | - | 29,731 | - | 29,731 | ||||||||||||||||||||||||||||||
| Issuance of stock for services | - | - | 481,063 | 481 | - | 480,582 | - | 481,063 | - | 481,063 | ||||||||||||||||||||||||||||||
| Issuance of stock to settle liability | - | - | 45,821 | 46 | - | 91,596 | - | 91,642 | - | 91,642 | ||||||||||||||||||||||||||||||
| Issuance of stock per incentive plan | - | - | 345,000 | 345 | - | 346,034 | - | 346,379 | - | 346,379 | ||||||||||||||||||||||||||||||
| Transfer from derivative liability classification to equity classification | - | - | - | - | - | 9,000 | 9,000 | 9,000 | ||||||||||||||||||||||||||||||||
| Interest on common stock receivable | - | - | - | - | (22,118 | ) | 22,118 | - | - | - | ||||||||||||||||||||||||||||||
| Contribution of services from noncontrolling interest | - | - | - | - | - | - | - | - | 151 | 151 | ||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (1,795,209 | ) | (1,795,209 | ) | (280 | ) | (1,795,489 | ) | ||||||||||||||||||||||||||
| Balance, September 30, 2014 | 4,000,000 | $ | 4,000 | 16,179,426 | $ | 16,180 | $ | (743,900 | ) | $ | 16,183,960 | $ | (15,747,679 | ) | $ | (287,439 | ) | $ | (9,798 | ) | $ | (297,237 | ) | |||||||||||||||||
See notes to condensed consolidated financial statements.
| 87 |
| Viatar CTC Solutions Inc. and Subsidiary |
| Condensed Consolidated Statements of Cash Flows |
| For the Nine Months Ended September 30, 2014 and 2013 |
| (Unaudited) |
| 2014 | 2013 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (1,795,489 | ) | $ | (833,986 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Issuance of stock for patent expenses | - | 51,812 | ||||||
| Issuance of stock for services | 676,441 | 125,000 | ||||||
| Contribution of services from noncontrolling interest | 29,882 | 130,961 | ||||||
| Amortization of discount on debt | 14,636 | 4,337 | ||||||
| Deferred costs | - | - | ||||||
| Change in derivative liability | (1,800 | ) | (10,800 | ) | ||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | (4,530 | ) | - | |||||
| Tax refund receivable | 147,997 | 242,016 | ||||||
| Accrued expenses | 119,930 | (789 | ) | |||||
| Net cash used in operating activities | (812,933 | ) | (291,449 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from demand note payable | - | 60,000 | ||||||
| Proceeds from convertible note payable | - | 100,000 | ||||||
| Issuance of stock and warrants | 830,000 | 122,996 | ||||||
| Exercise of warrants | 50,000 | - | ||||||
| Net cash provided by financing activities | 880,000 | 282,996 | ||||||
| Net increase in cash and cash equivalents | 67,067 | (8,453 | ) | |||||
| Cash and cash equivalents - beginning of the period | 66,769 | 62,645 | ||||||
| Cash and cash equivalents - end of the period | $ | 133,836 | $ | 54,192 | ||||
| Supplemental disclosure of cash flow information: | ||||||||
| Cash paid for interest | $ | 2,000 | $ | - | ||||
| Supplemental disclosure of non-cash financing activities: | ||||||||
| Conversion of demand note payable to common stock | $ | 60,000 | $ | - | ||||
| Transfer from derivative liability classification to equity classification | $ | 9,000 | $ | - | ||||
| Issuance of stock for settlement of payable | $ | 242,643 | $ | - | ||||
| Promissory notes received from issuance of stock | $ | - | $ | 150,000 | ||||
| Discount on debt issued with warrants | $ | - | $ | 32,525 | ||||
See notes to condensed consolidated financial statements.
| 88 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
| 1. | NATURE OF OPERATIONS AND GOING CONCERN |
Viatar CTC Solutions Inc., formerly known as Vizio Medical Devices LLC (“Vizio”), and Subsidiary is a development stage company focused on medical devices for oncology applications which remove circulating tumor cells for diagnostic and therapeutic purposes.
Vizio was a Delaware limited liability company with perpetual duration that was formed on December 18, 2007. On February 25, 2014, Vizio converted to a Delaware C corporation and changed its name to Viatar CTC Solutions Inc. (“Viatar CTC Solutions”).
Viatar CTC Solutions conducts all of its operations through its subsidiary, Viatar LLC (“Viatar”). Viatar was formed on September 23, 2010 as a Delaware limited liability company with perpetual duration.
Viatar CTC Solutions and Viatar are together referred to as the “Company”.
Since inception, the Company has not generated significant revenues. As a result, the Company has incurred net losses of $18,022,269 and negative cash flows from operations of $13,682,281 since its inception. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. The Company’s continued existence is dependent upon several factors, including (a) obtaining funding, whether in the form of equity, or debt investments, licensing revenues, strategic collaboration payments or grants from government or other sources, (b) success in achieving its research and development goals, (c) obtaining regulatory approvals, and (d) gaining market acceptance and/or distribution or strategic partners for its products. The Company has raised approximately $13.9 million of equity capital from non-institutional investors since its inception, and management is continually pursuing additional funding sources.
| 2. | Summary of Significant Accounting Policies |
Basis of Accounting
The accompanying unaudited condensed consolidated financial statements have been prepared on the accrual basis of accounting in accordance with U.S. generally accepted accounting principles (“GAAP”). The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
| 89 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
(Continued)
| 2. | Summary of Significant Accounting Policies (continued) |
Basis of Accounting (Continued)
The financial statements contained herein have been prepared by the Company in accordance with GAAP for interim financial information. Accordingly, they do not include all information and footnotes required by GAAP for complete financial statements. The balance sheet at December 31, 2013 was derived from audited financial statements but does not include all disclosures required by accounting principles generally accepted in the United States of America. In the opinion of management, the interim financial statements include all adjustments, consisting of normal recurring adjustments, necessary for a fair statement of the results of the periods presented. The results of operations for the nine and three months ended September 30, 2014 are not necessarily indicative of the results that may be expected for the full year. These unaudited condensed consolidated financial statements should be read in conjunction with the Company's audited consolidated financial statements and notes thereto for the year ended December 31, 2013.
Principles of Consolidation
The accompanying condensed consolidated financial statements include the accounts of Viatar CTC Solutions, Viatar and amounts related to a noncontrolling interest in Viatar. The noncontrolling interest represents a 0.01% and 0.03% ownership interest in Viatar at September 30, 2014 and December 31, 2013, respectively. All significant intercompany accounts and transactions have been eliminated in consolidation.
Income Taxes
In the ordinary course of business, there is inherent uncertainty in quantifying income tax positions. The Company assesses its income tax positions and records tax assets or liabilities for all years subject to examination based upon management’s evaluation of the facts and circumstances and information available at the reporting dates. For those tax positions where it is more-likely-than-not that a tax benefit will be sustained, we have recorded the largest amount of tax benefit with a greater than 50% likelihood of being realized upon ultimate settlement with a taxing authority that has full knowledge of all relevant information. For those income tax positions where it is not more-likely-than-not that a tax benefit will be sustained, no tax benefit has been recognized in the financial statements. The Company records interest and penalties related to uncertain tax positions as a component of income tax expense or benefit.
The Company files income tax returns with the U.S. federal government and various state and local jurisdictions. The Company is no longer subject to tax examinations by tax authorities for years prior to 2011.
Stock-Based Compensation
In December 2004, the FASB issued ASC 718, Compensation – Stock Compensation, or ASC 718. Under ASC 718, companies are required to measure the compensation costs of share-based compensation arrangements based on the grant-date fair value and recognize the costs in the financial statements over the period during which employees are required to provide services. Share-based compensation arrangements include options, restricted stock, restricted stock units, performance-based awards, share appreciation rights, and employee share purchase plans. As such, compensation cost is measured on the date of grant at fair value. The Company amortizes such compensation amounts, if any, over the respective vesting periods of the award. The Company uses the Black-Scholes-Merton option pricing model, or the Black-Scholes Model, an acceptable model in accordance with ASC 718, that requires the input of highly complex and subjective variables, including the expected life of the award and the expected stock price volatility over a period equal to or greater than the expected life of the award.
| 90 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
(Continued)
| 2. | Summary of Significant Accounting Policies (continued) |
Equity instruments (“instruments”) issued to anyone other than employees are recorded on the basis of the fair value of the instruments, as required by ASC 718. ASC 505, Equity Based Payments to Non-Employees, or ASC 505, defines the measurement date and recognition period for such instruments. In general, the measurement date is when either (a) a performance commitment, as defined, is reached or (b) the earlier of (i) the non-employee performance is complete or (ii) the instruments are vested. The measured value related to the instruments is recognized over a period based on the facts and circumstances of each particular grant as defined in ASC 505.
The Company recognizes the compensation expense for all share-based compensation granted based on the grant date fair value estimated in accordance with ASC 718. The Company generally recognizes the compensation expense on a straight-line basis over the employee’s requisite service period.
Recent Accounting Pronouncements
In May 2014, the FASB issued Accounting Standards Update (“ASU”) 2014-09, Revenue from Contracts with Customers (Topic 606). The new standard supersedes a majority of existing revenue recognition guidance under US GAAP, and requires companies to recognize revenue when it transfers goods or services to a customer in an amount that reflects the consideration to which a company expects to be entitled. Companies may need to use more judgment and make more estimates while recognizing revenue, which could result in additional disclosures to the financial statements. Topic 606 allows for either a “full retrospective” adoption or a “modified retrospective” adoption. The standard is effective for the Company beginning January 1, 2017. The Company is currently evaluating which method it will use and the revenue recognition impact this guidance will have once implemented.
In June 2014, the FASB issued ASU 2014-10, Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation. The guidance in ASU 2014-10 removes all incremental financial reporting requirements from GAAP for development-stage entities, including the removal of Topic 915 from the FASB Accounting Standards Codification. In addition, the update adds an example disclosure in Risks and Uncertainties (Topic 275) to illustrate one way that an entity that has not begun planned principal operations could provide information about the risks and uncertainties related to the company’s current activities.
The accounting standards update also removes an exception provided to development stage entities in Consolidations (Topic 810) for determining whether an entity is a variable interest entity—which may change the consolidation analysis, consolidation decision, and disclosure requirements for a company that has an interest in a company in the development stage. ASU 2014-10, effective the first annual period beginning after December 15, 2014, will no longer require the presentation and disclosure requirements in Topic 915. The revised consolidation standards are effective one year later, in annual periods beginning after December 15, 2015. The Company adopted this ASU during the quarter ended September 30, 2014.
In June 2014, the Financial Accounting Standards Board issued Accounting Standards Update 2014-12, Compensation- Stock Compensation. The amendments in this update apply to reporting entities that grant their employees share-based payments in which the terms of the award provide that a performance target can be achieved after the requisite service period. The amendments in this Update are effective for annual periods and interim periods within those annual periods beginning after December 15, 2015, and early adoption is permitted. The Company is currently evaluating the effects of ASU 2014-12 on the consolidated financial statements.
In August 2014, the Financial Accounting Standards Board issued Accounting Standards Update 2014-15, Presentation of Financial Statements-Going Concern. The Update provides U.S. GAAP guidance on management’s responsibility in evaluating whether there is substantial doubt about a company’s ability to continue as a going concern and about related footnote disclosures. For each reporting period, management will be required to evaluate whether there are conditions or events that raise substantial doubt about a company’s ability to continue as a going concern within one year from the date the financial statements are issued. The amendments in this Update are effective for the annual period ending after December 15, 2016, and for annual periods and interim periods thereafter. The Company is currently evaluating the effects of ASU 2014-15 on the consolidated financial statements.
| 91 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
(Continued)
| 3. | Stockholders’ EQUITY (deficit) |
On February 25, 2014, Vizio converted from a limited liability company to a C corporation pursuant to Delaware law, and changed its name to Viatar CTC Solutions Inc. Each Class A member unit was converted into one share of common stock and each Class B member unit was converted into one share of Series A Preferred Stock (the “Preferred Stock”). Each warrant to purchase Class A member units was converted into one warrant to purchase one share of common stock. The 2010 Incentive Plan was amended to provide for the issuance of common stock instead of Class B member units, and 3,000,000 shares of common stock are reserved for future issuance.
Viatar CTC Solutions’ capital structure consists of the authorization to issue 100,000,000 shares of common stock and 20,000,000 shares of Preferred Stock, which may be issued in series as designated by the Board of Directors. Each series of Preferred Stock shall have such voting and other rights as designated at the time of establishment by the Board of Directors.
Each issued share of common stock and Preferred Stock has one vote and the approval of a majority of the Preferred Stock, voting as a separate class, is required to approve any corporate action. The Preferred Stock is entitled to participate in dividends, rights issuances and property distributions on the same terms that the Class B member units had since the formation of the Company; namely, 20% of the dividends, rights and property distributed to common stockholders. Upon liquidation of the Company, the Preferred Stock is entitled to 20% of the liquidation value, subject to a priority $13,470,000 allocation to the common shares. This formulation preserves the right of the former Class B member units to participate in profits once the holders of Class A member units recouped their aggregate capital contributions.
As of February 25, 2014, immediately following the conversion of the Company from a limited liability company to a C corporation, 15,196,292 shares of common stock and 4,000,000 shares of Preferred Stock were issued and outstanding. Also as of that date, 410,000 shares of common stock were reserved for issuance pursuant to warrants at prices ranging from $1 to $2 per share and expiring in 2014 through 2019.
As of September 30, 2014, 16,179,426 shares of common stock and 4,000,000 shares of Preferred Stock were issued and outstanding.
| 92 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
(Continued)
| 3. | Stockholders’ EQUITY (deficit) (Continued) |
During the nine months ended September 30, 2014 and 2013, Viatar CTC Solutions issued an aggregate of 0 and 140,000 shares of common stock, respectively, in exchange for $0 and $210,000, respectively, of secured promissory notes in principal amounts at 5% due December 31, 2014. As of September 30, 2014 and December 31, 2013, the outstanding note balance was $743,900 and $721,782, respectively.
During the nine months ended September 30, 2014 and 2013, Viatar CTC Solutions issued 29,731 and 14,605 and accrued 0 and 13,063 shares of common stock, respectively, to Aquamarijn Microfiltration BV (“Aquamarijn”), the holder of the noncontrolling interest pursuant to an anti-dilution adjustment originating in 2010 in consideration for the grant of the license of intellectual property. It provides for the issuance of additional common stock as is necessary to maintain a 2% ownership interest in Viatar CTC Solutions. This annual anti-dilution adjustment is in effect until the first to occur of (x) the first approval for a product, or (y) $10 million of capital being raised at a valuation that is greater than $1 per share of common stock. The anti-dilution adjustments are in lieu of cash royalty payments by Viatar CTC Solutions or its sub-licensees. The derivative liability associated with the anti-dilution adjustment was $0 and $10,800 at September 30, 2014 and December 31, 2013, respectively. The Company estimated the fair value of the derivative liability using a probability weighted approach that considered the possible outcomes based on assumptions related to the timing and probability of expected capital raises and product approval. The agreement providing for the anti-dilution adjustment was terminated as of August 21, 2014 and the derivative ceased to exist and was therefore reclassified to equity.
During the nine months ended September 30, 2014, Viatar CTC Solutions raised $830,000 through the issuance of 671,250 shares of common stock, of which 93,750 shares were issued as offering costs.
During the nine months ended September 30, 2014 and 2013, Viatar CTC Solutions issued an aggregate of 210,000 and 50,000 warrants, respectively, in connection with the sale of common stock. The warrants are not transferable without prior written consent of Viatar CTC Solutions. The warrants are exercisable at prices ranging from $1 to $2 each and expire between 2014 and 2019. The fair value of the warrants issued in the nine months ended September 30, 2014 and 2013 were $125,121 and $32,525, respectively, based on the Black-Scholes option pricing model. During the nine months ended September 30, 2014, 50,000 warrants were exercised for $50,000 and 50,000 shares of common stock.
| 93 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
(Continued)
| 3. | Stockholders’ EQUITY (deficit) (Continued) |
During the nine months ended September 30, 2014, professional advisory firms were paid $35,000, issued 231,063 shares, and an additional 344,196 shares are restricted and held in escrow for 12 months for financial and legal services related to the conversion to a C corporation and a planned equity offering. The shares issued to these firms were valued at $1 per share totaling $231,063 which have been expensed to general and administrative on the condensed consolidated statements of operations as no equity was raised from the offering, which closed in June 2014. No shares were issued in the nine months ended September 30, 2013 for the cost of raising equity.
For each of the nine month periods ended September 30, 2014 and 2013, 100,000 shares of common stock were issued for annual service by the non-officer board of managers. The shares were valued at the grant date value of $1.00 per share based upon the most recent issuance price at the time of issuance and immediately recorded as stock-based compensation to research and development expenses. As of September 30, 2014 and 2013, $100,000 in stock-based compensation has been expensed on the condensed consolidated balance sheets. In April 2014, an additional 150,000 shares were issued to Bridgemedica as a result of a settlement in lieu of cash for prior services performed (see Note 7).
On August 27, 2014, the Company issued 345,000 shares of restricted common stock to various employees, of which 150,000 vested immediately and 195,000 will vest in 2015 and 2016. The shares were valued at the grant date value of $2.00 per share based upon the most recent issuance price at the time of issuance and the Company expensed an aggregate $346,379 as stock based compensation to research and development expenses. . All shares of restricted stock that were issued and outstanding are being amortized over their respective vesting periods. The unrecognized compensation expense at September 30, 2014 was $343,426.
| 4. | CONVERTIBLE NOTE PAYABLE |
In April 2013, Viatar CTC Solutions borrowed $100,000 in exchange for a 4% interest-only, convertible promissory note due April 30, 2015 and a five-year warrant to purchase 50,000 shares of common stock at $2 each. The note is convertible at any time prior to maturity at the option of the holder into 50,000 shares of common stock. The fair value of the warrant issued was $32,525 based on the Black-Scholes option pricing model. The fair value of the warrant was recorded as a discount and is being amortized over two years based on the straight line method, which approximates the effective interest method. The unamortized discount at September 30, 2014 and December 31, 2013 was $11,384 and $26,020. The note requires interest payments semi-annually.
| 94 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
(Continued)
| 5. | Income Taxes |
The Company is currently under audit by New York City tax authorities for the 2011, 2012, and 2013 tax years. The Company applied for New York City biotechnology tax credits and received certificates of eligibility from the NYC Department of Finance in the amounts of $147,997, $242,016, and $236,874 for tax years 2013, 2012, and 2011, respectively. These credits have been recognized as income tax benefits on the condensed consolidated statements of operations in the year of application. As of September 30, 2014, the 2011, 2012, and 2013 tax refunds have been received for these amounts. Audit outcomes and the timing of the audit settlements are subject to uncertainty. If the New York City biotechnology tax credits are disallowed, the Company will have to repay the tax credits received, in aggregate of $626,887, plus interest and penalties. The Company determined it was more-likely-than-not the benefit would be realized when the benefits were recognized. As of September 30, 2014, management’s judgment has not changed.
| 6. | Research and development COSTS and grant |
In 2011, the National Institutes of Health (“NIH”) issued a grant for an aggregate of $1.95 million for research and development of a medical device for dialysis patients, as a Phase II award under the U.S. government’s Small Business Technology Transfer program. The grant expires in September 2014. Vivonics, Inc. (an engineering firm) is the official grant recipient, Columbia is the academic partner, and Viatar CTC Solutions is the commercialization partner. During the nine months ended September 30, 2014 and 2013, Vivonics paid $0 and $39,000, respectively, to Viatar CTC Solutions under the NIH grant for research services.
| 7. | COMMITMENTS AND CONTINGENCIES |
In 2012, Viatar CTC Solutions sued Bridgemedica in New York State Supreme Court for $1.5 million of damages arising from breach of contract and other claims relating to non-performance under various engineering development contracts. Bridgemedica counterclaimed for $179,000 for delinquent payments.
On April 8, 2014, the Company settled with Bridgemedica. The Company issued 150,000 shares of common stock and will pay four installments totaling $20,000 cash to Bridgemedica. At December 31, 2013, accrued expenses on the condensed consolidated balance sheets included $168,593 payable to Bridgemedica. At September 30, 2014 and December 31, 2013, accrued expenses on the condensed consolidated balance sheets included $5,000 and $168,593 payable to Bridgemedica, respectively. The outstanding balance due to Bridgemedica was repaid in October 2014.
| 8. | FAIR VALUE OF FINANCIAL INSTRUMENTS |
The Company follows the provisions of ASC 820 for fair value measurements of its financial assets and liabilities. The accounting standard for fair value measurements provides a framework for measuring fair value and requires expanded disclosures regarding fair value measurements. Fair value is defined as the price that would be received for an asset or the exit price that would be paid to transfer a liability in the principal or most advantageous market in an orderly transaction between market participants on the measurement date. The accounting standard established a fair value hierarchy which requires an entity to maximize the use of observable inputs, where available. This hierarchy prioritizes the inputs into three broad levels, as follows: Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Level 3 inputs are unobservable inputs based on the Company's own assumptions used to measure assets and liabilities at fair value. An asset or liability's classification within the hierarchy is determined based on the lowest level input that is significant to the fair value measurement. The Company’s financial instruments measured at fair value on a recurring basis consisted of the following at September 30, 2014 and December 31, 2013:
| 95 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
(Continued)
| Total Carrying Value at September 30, 2014 | ||||||||||||
| (Level 1) | (Level 2) | (Level 3) | ||||||||||
| Derivative Liability | $ | - | $ | - | $ | - | ||||||
| Total Carrying Value at December 31, 2013 | ||||||||||||
| (Level 1) | (Level 2) | (Level 3) | ||||||||||
| Derivative Liability | $ | - | $ | - | $ | 10,800 | ||||||
The following is a summary of activity of Level 3 liabilities for the nine months ended September 30, 2014:
| Balance at December 31, 2013 | $ | (10,800 | ) | |
| Reclassified to equity when liability ceased to exist | 9,000 | |||
| Change in fair value during the nine months ended September 30, 2014 | 1,800 | |||
| Balance at September 30, 2014 | $ | - |
| 96 |
Viatar CTC Solutions Inc. and Subsidiary
Notes to the Condensed Consolidated Financial Statements
September 30, 2014
(Unaudited)
(Continued)
| 8. | FAIR VALUE OF FINANCIAL INSTRUMENTS (CONTINUED) |
The Company estimates the fair value of the convertible note and warrants liability utilizing the Black-Scholes option pricing model, which is dependent upon several variables such as the expected note term, expected volatility of its stock price over the expected note term, expected risk-free interest rate over the expected note term, and the expected dividend yield rate over the expected note term. The Company believes this valuation methodology is appropriate for estimating the fair value of the liability. The following table summarizes the assumptions the Company utilized to estimate the fair value of the convertible note and warrants liability at September 30, 2014:
| Assumptions | September 30, 2014 | |
| Expected term (years) | 5.00 | |
| Expected volatility | 100% | |
| Risk-free interest rate | 0.725% - 1.748% | |
| Dividend yield | 0.00% |
The expected warrant term is based on the remaining contractual term. The expected volatility is based on historical-volatility of peer entities whose stock prices were publicly available. The risk-free interest rate is based on the U.S. Treasury yields with terms equivalent to the expected term of the related warrant at the valuation date. Dividend yield is based on historical trends.
The carrying value of the Company’s other financial assets and liabilities approximates fair value at September 30, 2014 and December 31, 2013.
| 9. | RELATED PARTY TRANSACTION |
In January 2013, Vizio borrowed an aggregate of $60,000 in exchange for a demand note from a member of the board of managers. In February 2014, the demand note was converted into 60,000 shares of common stock and a warrant to purchase 60,000 shares of common stock at $1 per share, expiring in 2019. The loan was interest free.
| 10. | SUBSEQUENT EVENTS |
Subsequent to September 30, 2014 and in addition to disclosures elsewhere in the unaudited condensed consolidated financial statements:
During October 2014, the Company received $50,000 for the payoff of notes originally issued to purchase common stock receivable.
On October 8, 2014, pursuant to the 2014 Equity Incentive Plan, the holders of 595,000 shares of vested Preferred Stock exchanged the preferred shares for 595,000 shares of common stock.
During October 2014, the Company paid $5,000 as the final payment of the Bridgemedica settlement.
During November 2014, the Company granted 25,000 restricted shares to an employee pursuant to the Equity Incentive Plan.
During November 2014, the Company received $50,000 for the payoff of notes originally issued to purchase common stock receivable.
| 97 |
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 13. OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION.
The following table sets forth all expenses to be paid by the Registrant, other than underwriting discounts and commissions, upon completion of this offering. All amounts shown are estimates except for the SEC registration fee and the FINRA filing fee.
| SEC registration fee | 488.88 | * | ||
| Printing and engraving | 100 | * | ||
| Legal fees and expenses | 100,000 | * | ||
| Accounting fees and expenses | 5,000 | * | ||
| Blue sky fees and expenses (including legal fees) | 7,000 | * | ||
| Transfer agent and registrar fees | 5,000 | * | ||
| Miscellaneous | 10,000 | * | ||
| Total | $ | 127,588.88 | * |
| * | Estimated. |
ITEM 14. INDEMNIFICATION OF DIRECTORS AND OFFICERS.
Section 145 of the Delaware General Corporation Law (the “DGCL”) provides that a Delaware corporation may indemnify directors, officers, employees and agents against expenses (including attorney’s fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with any threatened, pending or completed action, suit or proceeding in which such person is made a party by reason of such person being or having been a director, officer, employee or agent to the corporation, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was unlawful.
The DGCL provides that Section 145 is not exclusive of other rights to which those seeking indemnification may be entitled under any bylaw, agreement, vote of stockholders, or disinterested directors or otherwise.
Section 102(b)(7) of the DGCL permits a corporation to provide in its certificate of incorporation or an amendment thereto, to eliminate or limit the personal liability of a director to the corporation and its stockholders for monetary damages arising out of certain breaches of their fiduciary duty.
The Company’s Certificate of Incorporation provides for the elimination of a director’s liability to the Company and its stockholders for monetary damages for breach of fiduciary duty, except for liability (i) for any breach of the director’s duty of loyalty to the Registrant or its stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of the law, (iii) under Section 174 of the DGCL, or (iv) for any transaction from which the director derived an improper personal benefit.
The Company’s Certificate of Incorporation and Bylaws provide that the Company shall indemnify each of its directors and executive officers to the fullest extent not prohibited by the DGCL and may indemnify certain other persons as set forth in the DGCL. The Company may advance expenses to its directors and officers in connection with defending a proceeding.
The rights conferred in the Certificate of Incorporation and Bylaws are not exclusive, and the Company is authorized to enter into indemnification agreements with its directors, officers, employees, and agents and to obtain insurance to indemnify such persons.
| 98 |
ITEM 15. RECENT SALES OF UNREGISTERED SECURITIES.
During the past three years, we have sold the following securities which were not registered under the Securities Act:
Prior to our conversion into a corporation on February 25, 2014
From November 7, 2011 through August 10, 2012, we issued an aggregate of 1,495,000 Class A Member Units to a total of 11 accredited investors at $1.00 per unit for aggregate proceeds of $2,095,395.
On January 3, 2012, we issued 91,395 Class A Member Units to a consultant for compensation under certain consulting agreement.
On January 4, 2012, we issued a total of 100,000 Class A Member Units to two directors (and their designees) as compensation for their service.
From January 4, 2012 through June 21, 2012, we issued a total of 446, 500 Class A Member Units to four consultants as compensation for financing service.
On January 2, 2013, we issued 14,605 Class A Member Units to one consultant as compensation for services. On the same day, we also issued a total of 100,000 Class A Member Units to two directors (and their designees) as compensation for their service.
On June 3, 2013, we issued 51,812 Class A Member Units to one consultant as payment in lieu of patent legal expenses under certain license agreement.
On September 12, 2013, we issued an aggregate of 117,004 Class A Member Units to three consultants as compensation for financing service.
On January 2, 2014, we issued a consultant 29,731 Class A Member Units to one consultant as license payment in lieu of royalties.
On January 14, 2014, we issued an aggregate of 100,000 Class A Member Units to two directors (and their designees) as compensation for service.
On February 14, 2014, we issued an aggregate of 442, 441 Class A Member Units to four consultants as fee for financing.
From September 2, 2012 through December 7, 2012, we sold a total of 97,500 Class A Member Units to an aggregate of eight accredited investors at $2.00 per unit for total proceeds of $195,000.
From June 21, 2013 through January 16, 2014, we sold a total of 180,000 Class A Member Units to an aggregate of five accredited investors at $1.00 per unit for total proceeds of $180,000.
From July 23, 2012 to July 12, 2013, we sold 518,000 Class A Units at $1.50 per unit to six accredited investors, in exchange for $777,000 of promissory notes.
| 99 |
From January 22, 2014 to February 14, 2014, we sold a total of 205,000 Class A Member Units to an aggregate of eight accredited investors at $1.00 per unit for total proceeds of $205,000.
On February 1, 2014, we issued a member 60,000 Class A Member Units and a warrant to purchase 60,000 Class A Member Units at $1.00 per share with a term expiring on February 28, 2019 for the conversion of a demand note of $60,000.
From January 22, 2014 to February 6, 2014, we sold a total of 77,500 Class A Member Units to an aggregate of eight accredited investors at $2.00 per unit for total proceeds of $155,000.
From October 5, 2012 through January 7, 2014, we sold a total of 100,000 Class A Member Units and warrants to purchase a total of 100,000 Class A Member Units for gross proceeds of $200,000. The warrants have an exercise price of $1.00 with expiration dates ranging from October 31, 2017 through January 31, 2019.
From February 8, 2013 through February 4, 2014, we sold to four investors a total of 335,000 Class A Member Units and five year warrants to purchase a total of 335,000 Class A Member Units at various exercise prices ranging from $1.00 to $2.00 per unit for a total cash consideration of $285,000.
The above securities were issued pursuant to the exemption from registration set forth in Section 4(a)(2) of the Securities Act of 1933, as amended, and Rule 506 of Regulation D promulgated thereunder. The investors represented to us that they are accredited investors. We believe that the investors had adequate information about us as well as the opportunity to ask questions and receive responses from our management.
Since our conversion into a corporation on February 25, 2014, we issued the following unregistered securities:
Upon the conversion into a corporation, we issued an aggregate of 15,189,979 shares of our Common Stock to former holders of Class A member units of our predecessor, Vizio Medical Devices LLC, in connection with our conversion from a limited liability company into a corporation. We also issued an aggregate of 4,000,000 shares of our Series A Preferred Stock to holders of Class B member units, and warrants to purchase a total of 410,000 shares of our Common Stock to holders of warrants to purchase Class A member units of our predecessor. The issuance of the shares of Common Stock and Series A Preferred Stock and the warrants were exempt from registration under the Securities Act of 1933, as amended, in reliance on Section 3(a)(9) thereof.
On February 25, 2014, the Company issued 231,063 shares of Common Stock to its counsel and consultants as consideration for services in connection with an equity raise.
On March 19, 2014, we sold 75,000 shares of Common Stock to one investor at $2.00 per share for a total purchase price of $150,000. On May 28, 2014, we issued 50,000 shares of Common Stock to one investor upon exercise of certain warrants at $1.00 per share.
On April 8, 2014, the Company issued a total of 11,250 shares of Common Stock to three consultants as fees for financing services. On the same day, the Company issued 150,000 shares Common Stock to Bridgemedica pursuant to the settlement agreement with Bridgemedica.
On May 11, 2014, we sold to one investor 25,000 shares of Common Stock and warrants to purchase 25,000 shares of Common Stock an exercise price of $2.00 for a total purchase price of $50,000. The warrants expire on May 31, 2019.
On June 18, 2014, we sold an aggregate of 25,000 units (the “Units”) in a private placement of our securities to one investor (the “Investor”) at a purchase price of $2.00 per Unit pursuant to a confidential private placement memorandum (the “PPM”) for an aggregate purchase price of $50,000. On June 24, 2014, we sold additional 12,500 Units to two investors for gross proceeds of $25,000. Each Unit consists of (i) one share of the Company’s common stock and (ii) a five year warrant (the “Warrant”) to purchase one share of Common Stock at an exercise price of $2.00 per share. The Warrant may be exercised on a cashless basis. The Company is prohibited from effecting the exercise of a Warrant to the extent that, as a result of such exercise, the holder would beneficially own more than 4.99%, in the aggregate, of the issued and outstanding shares of the Company’s Common Stock calculated immediately after giving effect to the issuance of shares of common stock upon the exercise of the Warrant, which limitation may be increased to 9.99% upon not less than 61 days’ prior notice to the Company.
On July 21, 2014, we sold 12,500 shares of Common Stock and 12,500 Warrants to one investor for gross proceeds of $25,000.
On August 27, 2014, the Company issued a total of 325,000 shares of Common Stock to various employees under the 2014 Incentive Plan as compensation for services.
On September 14, 2014, the Company issued 45,821 shares of Common Stock to Columbia University in connection with the termination of the license and research agreements.
| 100 |
On October 8, 2014, we issued an aggregate of 595,000 shares of Common Stock under the 2014 Incentive Plan to seven persons who received these shares in exchange for their 595,000 shares of Series A Preferred Stock.
None of the foregoing transactions involved any underwriters, underwriting discounts or commissions, or any public offering. The Registrant believes the offers, sales and issuances of the above securities were exempt from registration under the Securities Act by virtue of Rule 506 of Regulation D promulgated thereunder and Section 4(a)(2) of the Securities Act because the issuance of securities to the recipients did not involve a public offering. The recipients of the securities in each of these transactions represented their intentions to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof, and appropriate legends were placed upon the stock certificates issued in these transactions. All recipients had adequate access, through their relationships with the Company, to information about the Company. The sales of these securities were made without any general solicitation or advertising.
ITEM 16. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES.
(a) Exhibits.
We have filed the exhibits listed on the accompanying Exhibit Index of this Registration Statement.
(b) Financial Statement Schedules.
All financial statement schedules are omitted because the information called for is not required or is shown either in the consolidated financial statements or in the notes thereto.
ITEM 17. UNDERTAKINGS.
The undersigned registrant hereby undertakes:
1. To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
i. To include any prospectus required by section 10(a)(3) of the Securities Act of 1933;
ii. To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement.
| 101 |
iii. To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
2. That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
3. To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
4. That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser:
A. Each prospectus filed by the registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and
B. Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5), or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii), or (x) for the purpose of providing the information required by section 10(a) of the Securities Act of 1933 shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which that prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date; or
C. Each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
5. Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
6. That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities:
The registrant undertakes that in a primary offering of its securities pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the registrant or used or referred to by the registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the registrant or its securities provided by or on behalf of the registrant; and
(iv) Any other communication that is an offer in the offering made by the registrant to the purchaser.
| 102 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the Registrant has duly caused this registration statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in Lowell, Massachusetts, on the 26th day of November, 2014.
| VIATAR CTC SOLUTIONS INC. | ||
| By: | /s/Ilan Reich | |
| Ilan Reich | ||
| Chief Executive Officer and Chief Financial Officer | ||
Pursuant to the requirements of the Securities Act of 1933, this registration statement on Form S-1 has been signed by the following persons in the capacities and on the dates indicated.
Signature |
Title |
Date | ||
| /s/ Ilan Reich | Chief Executive Officer, Chief Financial Officer and Chairman (Principal Executive Officer and Principal Financial and Accounting Officer) | November 26, 2014 | ||
| Ilan Reich | ||||
| 103 |
EXHIBIT INDEX
| Exhibit Number |
Description | |
| 3.1 | Certificate of Conversion (1) | |
| 3.2 | Certificate of Incorporation (1) | |
| 3.3 | Certificate of Amendment (1) | |
| 3.4 | Bylaws (1) | |
| 4.1* | Specimen common stock certificate. | |
| 4.2 | Form of warrant (1) | |
| 5.1** | Opinion of Sichenzia Ross Friedman Ference LLP. | |
| 10.1+ | 2014 Equity Incentive Plan and forms of agreements thereunder.(1) | |
| 10.2 | Director Consent Letter - Charles Andrew McCartney (1) | |
| 10.3 | Director Consent Letter - Francisco J. Esteva (1) | |
| 21.1 | List of subsidiaries.(1) | |
| 23.1* | Consent of Rosenberg, Rich Baker Berman & Company | |
| 23.2** | Consent of Sichenzia Ross Friedman Ference LLP (included in Exhibit 5.1). | |
| 101.INS | XBRL Instance Document *** | |
| 101.SCH | XBRL Taxonomy Extension Schema Document *** | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document *** | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document *** | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document *** | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document *** | |
(1) Incorporated herein by reference to the exhibits to the Company’s Registration Statement on Form S-1 filed with the SEC on October 27, 2014.
| * | Filed herewith. |
| ** | To be filed by amendment. |
| + | Indicates a management contract or compensatory plan. |
| *** | Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files on Exhibit 101 hereto are deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections. |
| 104 |