Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Synthetic Biologics, Inc. | Financial_Report.xls |
| EX-31.1 - EXHIBIT 31.1 - Synthetic Biologics, Inc. | v393218_ex31-1.htm |
| EX-32.1 - EXHIBIT 32.1 - Synthetic Biologics, Inc. | v393218_ex32-1.htm |
| EX-32.2 - EXHIBIT 32.2 - Synthetic Biologics, Inc. | v393218_ex32-2.htm |
| EX-31.2 - EXHIBIT 31.2 - Synthetic Biologics, Inc. | v393218_ex31-2.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-Q
(Mark One)
| x | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the quarterly period ended September 30, 2014 |
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES ACT OF 1934 |
| For the transition period from ____________ to ____________ |
Commission File Number: 1-12584
SYNTHETIC BIOLOGICS, INC.
(Name of small business issuer in its charter)
| Nevada | 13-3808303 |
| (State or other jurisdiction of incorporation or organization) | (IRS Employer Identification Number) |
| 155 Gibbs Street, Suite 412 | |
| Rockville, MD | 20850 |
| (Address of principal executive offices) | (Zip Code) |
| 617 Detroit Street, Suite 100 | |
| Ann Arbor, MI | 48104 |
| (Mailing Address) | (Zip Code) |
Registrant’s telephone number, including area code:
(734) 332-7800
Securities registered pursuant to Section 12(b) of the Act:
Common Stock, $0.001 par value per share
Securities registered pursuant to Section 12(g) of the Act:
None.
(Title of Class)
Indicate by check mark whether the issuer: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated file, a non-accelerated file, or a smaller reporting company. See the definitions of “large accelerated filer, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ |
| Non-Accelerated filer | ¨ | Smaller reporting company | x |
(Do not check if a smaller reporting company)
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
As of November 10, 2014 the registrant had 72,513,144 shares of common stock outstanding.
|
|
SYNTHETIC BIOLOGICS, INC.
FORM 10-Q
TABLE OF CONTENTS
| 2 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Balance Sheets
(In thousands except share and per share amounts)
| September 30, 2014 | December 31, 2013 | |||||||
| (Unaudited) | (Audited) | |||||||
| Assets | ||||||||
| Current Assets | ||||||||
| Cash and cash equivalents | $ | 3,286 | $ | 14,625 | ||||
| Prepaid expenses and other current assets | 1,382 | 1,591 | ||||||
| Total Current Assets | 4,668 | 16,216 | ||||||
| Property and equipment, net | 67 | 37 | ||||||
| Deposits and other assets | 6 | 4 | ||||||
| Total Assets | $ | 4,741 | $ | 16,257 | ||||
| Liabilities and Stockholders' Equity | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 873 | $ | 142 | ||||
| Accrued liabilities | 111 | 885 | ||||||
| Total Current Liabilities | 984 | 1,027 | ||||||
| Total Liabilities | 984 | 1,027 | ||||||
| Stockholders' Equity | ||||||||
| Preferred stock, $0.001 par value; 10,000,000 shares authorized, | ||||||||
| none issued and outstanding | - | - | ||||||
| Common stock, $0.001 par value; 100,000,000 shares authorized, | ||||||||
| 58,535,010 issued and 58,453,528 outstanding and | ||||||||
| 58,295,808 issued and 58,214,326 outstanding | 58 | 58 | ||||||
| Additional paid-in capital | 98,261 | 96,430 | ||||||
| Accumulated deficit | (94,562 | ) | (81,258 | ) | ||||
| Total Synthetic Biologics, Inc. and Subsidiaries Equity | 3,757 | 15,230 | ||||||
| Non-controlling interest | - | - | ||||||
| Total Equity | 3,757 | 15,230 | ||||||
| Total Liabilities and Equity | $ | 4,741 | $ | 16,257 | ||||
See accompanying notes to unaudited consolidated financial statements.
| 3 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Statements of Operations
(In thousands except share and per share amounts)
(Unaudited)
| For the three months ended September 30, | For the nine months ended September 30, | |||||||||||||||
| 2014 | 2013 | 2014 | 2013 | |||||||||||||
| Operating Costs and Expenses: | ||||||||||||||||
| General and administrative | $ | 1,218 | $ | 1,890 | $ | 4,154 | $ | 4,270 | ||||||||
| Research and development | 3,693 | 1,475 | 9,247 | 3,796 | ||||||||||||
| Total Operating Costs and Expenses | 4,911 | 3,365 | 13,401 | 8,066 | ||||||||||||
| Loss from Operations | (4,911 | ) | (3,365 | ) | (13,401 | ) | (8,066 | ) | ||||||||
| Other Income (Expense): | ||||||||||||||||
| Interest income | 1 | 11 | 2 | 32 | ||||||||||||
| Other income (expense) | - | (14 | ) | 95 | (59 | ) | ||||||||||
| Total Other Income (Expense) | 1 | (3 | ) | 97 | (27 | ) | ||||||||||
| Net Loss | (4,910 | ) | (3,368 | ) | (13,304 | ) | (8,093 | ) | ||||||||
| Net Loss Attributable to Non-controlling Interest | - | - | - | - | ||||||||||||
| Net Loss Attributable to Synthetic Biologics, Inc. and Subsidiaries | $ | (4,910 | ) | $ | (3,368 | ) | $ | (13,304 | ) | $ | (8,093 | ) | ||||
| Net Loss Per Share - Basic and Dilutive | $ | (0.08 | ) | $ | (0.08 | ) | $ | (0.23 | ) | $ | (0.18 | ) | ||||
| Net Loss Per Share Attributable to Synthetic Biologics, Inc. and Subsidiaries | $ | (0.08 | ) | $ | (0.08 | ) | $ | (0.23 | ) | $ | (0.18 | ) | ||||
| Weighted average number of shares outstanding during the period - Basic and Dilutive | 58,453,528 | 44,654,414 | 58,356,025 | 44,636,935 | ||||||||||||
See accompanying notes to unaudited consolidated financial statements.
| 4 |
Synthetic Biologics, Inc. and Subsidiaries
Consolidated Statements of Cash Flows
(In thousands)
(Unaudited)
| For the nine months ended September 30, | ||||||||
| 2014 | 2013 | |||||||
| Cash Flows From Operating Activities: | ||||||||
| Net Loss | $ | (13,304 | ) | $ | (8,093 | ) | ||
| Adjustments to reconcile net loss to net cash | ||||||||
| used in operating activities: | ||||||||
| Stock-based compensation | 1,827 | 1,202 | ||||||
| Depreciation | 13 | 33 | ||||||
| Provision for uncollectible note and interest receivables | - | 763 | ||||||
| Loss on sale of equipment | - | 58 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | 209 | 800 | ||||||
| Deposits and other assets | (2 | ) | 33 | |||||
| Accounts payable | 731 | 189 | ||||||
| Accrued liabilities | (774 | ) | - | |||||
| Net Cash Used In Operating Activities | (11,300 | ) | (5,015 | ) | ||||
| Cash Flows From Investing Activities: | ||||||||
| Purchases of property and equipment | (43 | ) | (25 | ) | ||||
| Net Cash Used In Investing Activities | (43 | ) | (25 | ) | ||||
| Cash Flows From Financing Activities: | ||||||||
| Proceeds from issuance of common stock for stock option exercises | 4 | 231 | ||||||
| Net Cash Provided By Financing Activities | 4 | 231 | ||||||
| Net decrease in cash | (11,339 | ) | (4,809 | ) | ||||
| Cash at beginning of period | 14,625 | 9,954 | ||||||
| Cash at end of period | $ | 3,286 | $ | 5,145 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for interest | $ | - | $ | - | ||||
| Cash paid for taxes | $ | - | $ | - | ||||
See accompanying notes to unaudited consolidated financial statements.
| 5 |
Synthetic Biologics, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
(Unaudited)
| 1. | Organization and Nature of Operations and Basis of Presentation |
Description of Business
Synthetic Biologics, Inc. (the “Company” or “Synthetic Biologics”) is a clinical-stage biotechnology company developing pathogen-specific therapies for serious infections and diseases, with a focus on protecting the microbiome. The Company is developing an oral biologic to protect the gastrointestinal (GI) microflora from the effects of intravenous (IV) antibiotics for the prevention of C. difficile infection, an oral treatment to reduce the impact of methane producing organisms on constipation-predominant irritable bowel syndrome (C-IBS) and a monoclonal antibody combination for the treatment of Pertussis. In addition, the Company is developing a Phase 2 oral estriol drug for the treatment of relapsing-remitting multiple sclerosis (MS) and cognitive dysfunction in MS.
| Therapeutic Area | Product Candidate | Status | ||
| Relapsing-remitting MS |
Trimesta (oral estriol) |
Patient follow-up is complete in the Phase 2 trial and the lead principal investigator from University of California, Los Angeles (UCLA) presented additional Phase 2 clinical outcome data, including more detailed results on improvements in cognitive and disability measures at the ACTRIMS-ECTRIMS Joint Meeting in September 2014 | ||
| Cognitive dysfunction in MS |
Trimesta (oral estriol) |
Patient enrollment underway in Phase 2 clinical trial | ||
| C. difficile infection prevention |
SYN-004 (oral enzyme) |
Intend to initiate Phase 1a and 1b SYN-004 clinical trials during 4th quarter 2014 | ||
| Constipation-predominant irritable bowel syndrome (C-IBS) |
SYN-010 (oral compound) |
Intend to initiate Phase 2 clinical trial during 1st half of 2015; Collaboration with Cedars-Sinai Medical Center | ||
| Pertussis |
SYN-005 (monoclonal antibodies) |
Positive preclinical research findings reported in 2014; Collaborations with Intrexon and The University of Texas at Austin | ||
| Acinetobacter infection |
SYN-001 (monoclonal antibody) |
Discovery stage; Collaboration with Intrexon | ||
| IBS |
SYN-007 (biologic) |
Discovery stage; We are still evaluating an IP option; Collaboration with Intrexon |
Basis of Presentation
The accompanying consolidated financial statements have been prepared pursuant to the rules and regulations of Securities and Exchange Commission (“SEC”) for interim financial information. Accordingly, they do not include all of the information and notes required by U.S. Generally Accepted Accounting Principles (“GAAP”) for complete financial statements. The accompanying consolidated financial statements include all adjustments, comprised of normal recurring adjustments, considered necessary by management to fairly state our results of operations, financial position and cash flows. The operating results for the interim periods are not necessarily indicative of results that may be expected for any other interim period or for the full year. These consolidated financial statements should be read in conjunction with the consolidated financial statements and notes thereto included in our Annual Report on Form 10-K for the year ended December 31, 2013 (“2013 Form 10-K”) as filed with the SEC. The interim results for the three and nine months ended September 30, 2014, are not necessarily indicative of results for the full year.
| 6 |
The consolidated financial statements are prepared in conformity with U.S. GAAP, which requires the use of estimates, judgments and assumptions that affect the amounts of assets and liabilities at the reporting date and the amounts of revenue and expenses in the periods presented. We believe that the accounting estimates employed are appropriate and the resulting balances are reasonable; however, due to the inherent uncertainties in making estimates actual results could differ from the original estimates, requiring adjustments to these balances in future periods.
Change in Filing Status
On June 30, 2014, the Company exceeded the $75.0 million public float threshold to trigger accelerated filer status with the SEC. Consequently the Company will no longer be a Smaller Reporting Company (SRC) and the Company’s auditors must provide an auditor attestation on our internal controls to be included in the Company’s Form 10-K for the year ending December 31, 2014. Such Form 10-K will continue to provide scaled SRC-level disclosures; the larger reporting company disclosures will commence in the Company’s Form 10-Q for the three months ended March 31, 2015.
| 2. | Management’s Plan |
The Company has incurred an accumulated deficit of $94.6 million through September 30, 2014. With the exception of the quarter ended June 30, 2010, the Company has incurred negative cash flow from operations since it started the business. The Company has spent, and expects to continue to spend, substantial amounts in connection with implementing its business strategy, including the planned product development efforts, clinical trials, and research and discovery efforts.
The actual amount of funds the Company will need to operate is subject to many factors, some of which are beyond our control. These factors include the following:
| · | the progress of research activities; | |
| · | the number and scope of research programs; | |
| · | the progress of preclinical and clinical development activities; | |
| · | the progress of the development efforts of parties with whom the Company has entered into research and development agreements; | |
| · | costs associated with additional clinical trials of product candidates; | |
| · | the ability to maintain current research and development licensing arrangements and to establish new research and development, and licensing arrangements; | |
| · | the ability to achieve milestones under licensing arrangements; | |
| · | the costs involved in prosecuting and enforcing patent claims and other intellectual property rights; and | |
| · | the costs and timing of regulatory approvals. |
The Company has based its estimate on assumptions that may prove to be wrong. The Company may need to obtain additional funds sooner or in greater amounts than it currently anticipates. Potential sources of financing include strategic relationships, public or private sales of the Company’s shares or debt and other sources.
The Company may seek to access the public or private equity markets when conditions are favorable due to long-term capital requirements. The Company does not have any committed sources of financing at this time, and it is uncertain whether additional funding will be available when needed on terms that will be acceptable to it, or at all. If the Company raises funds by selling additional shares of common stock or other securities convertible into common stock, the ownership interest of the existing stockholders will be diluted. If the Company is not able to obtain financing when needed, it may be unable to carry out the business plan. As a result, the Company may have to significantly limit its operations and its business, financial condition and results of operations would be materially harmed.
| 3. | Fair Value of Financial Instruments |
The fair value accounting standards define fair value as the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is determined based upon assumptions that market participants would use in pricing an asset or liability. Fair value measurements are rated on a three-tier hierarchy as follows:
| · | Level 1 inputs: Quoted prices (unadjusted) for identical assets or liabilities in active markets; |
| · | Level 2 inputs: Inputs, other than quoted prices included in Level 1, that are observable either directly or indirectly; and |
| · | Level 3 inputs: Unobservable inputs for which there is little or no market data, which require the reporting entity to develop its own assumptions. |
If the inputs used to measure fair value fall in different levels of the fair value hierarchy, the hierarchy level is based upon the lowest level of input that is significant to the fair value measurement.
| 7 |
Cash and cash equivalents include money market accounts of $3.2 million and $11.0 million as of September 30, 2014 and December 31, 2013, respectively, that are measured using Level 1 inputs.
| 4. | Selected Balance Sheet Information |
Prepaid expenses and other current assets (in thousands):
| September 30, 2014 | December 31, 2013 | |||||||
| Intrexon prepaid research and development expenses | $ | 1,073 | $ | 1,361 | ||||
| Prepaid insurance | 51 | 177 | ||||||
| Prepaid credit cards | 65 | - | ||||||
| Prepaid expenses | 193 | 53 | ||||||
| Total | $ | 1,382 | $ | 1,591 | ||||
The anticipated Intrexon research and development expenses for the next twelve months are classified as a current asset. The Company may terminate the arrangement at any time and receive a cash refund of the remaining balance minus any amounts owed to Intrexon.
Property and equipment (in thousands):
| September 30, 2014 | December 31, 2013 | |||||||
| Computer and office equipment | $ | 88 | $ | 45 | ||||
| Software | 11 | 11 | ||||||
| 99 | 56 | |||||||
| Less accumulated depreciation | (32 | ) | (19 | ) | ||||
| Total | $ | 67 | $ | 37 | ||||
| 5. | Stock-Based Compensation |
Stock Incentive Plan
During 2001, the Company’s Board of Directors and stockholders approved the 2001 Stock Incentive Plan (the “2001 Stock Plan”). The total number of shares of stock with respect to which stock options and stock appreciation rights may be granted to any one employee of the Company or a subsidiary during any one-year period under the 2001 Stock Plan shall not exceed 250,000. All awards pursuant to the 2001 Stock Plan shall terminate upon the termination of the grantee’s employment for any reason. Awards include options, restricted shares, stock appreciation rights, performance shares and cash-based awards (the “Awards”). The 2001 Stock Plan contains certain anti-dilution provisions in the event of a stock split, stock dividend or other capital adjustment, as defined in the plan. The 2001 Stock Plan provides for a Committee of the Board to grant Awards and to determine the exercise price, vesting term, expiration date and all other terms and conditions of the Awards, including acceleration of the vesting of an Award at any time. As of September 30, 2014, there were 682,449 options issued and outstanding under the 2001 Stock Plan.
On March 20, 2007, the Company’s Board of Directors approved the 2007 Stock Incentive Plan (the “2007 Stock Plan”) for the issuance of up to 2,500,000 shares of common stock to be granted through incentive stock options, nonqualified stock options, stock appreciation rights, dividend equivalent rights, restricted stock, restricted stock units and other stock-based awards to officers, other employees, directors and consultants of the Company and its subsidiaries. This plan was approved by stockholders on November 2, 2007. The exercise price of stock options under the 2007 Stock Plan is determined by the compensation committee of the Board of Directors, and may be equal to or greater than the fair market value of the Company’s common stock on the date the option is granted. The total number of shares of stock with respect to which stock options and stock appreciation rights may be granted to any one employee of the Company or a subsidiary during any one-year period under the 2007 plan shall not exceed 250,000. Options become exercisable over various periods from the date of grant, and generally expire ten years after the grant date. As of September 30, 2014, there were 428,657 options issued and outstanding under the 2007 Stock Plan.
On November 2, 2010, the Company’s Board of Directors and stockholders approved the 2010 Stock Incentive Plan (“2010 Stock Plan”) for the issuance of up to 3,000,000 shares of common stock to be granted through incentive stock options, nonqualified stock options, stock appreciation rights, dividend equivalent rights, restricted stock, restricted stock units and other stock-based awards to officers, other employees, directors and consultants of the Company and its subsidiaries. On October 22, 2013, the stockholders approved and adopted an amendment to the Company’s 2010 Incentive Stock Plan to increase the number of shares of the Company’s common stock reserved for issuance under the Plan from 3,000,000 to 6,000,000. The exercise price of stock options under the 2010 Stock Plan is determined by the compensation committee of the Board of Directors, and may be equal to or greater than the fair market value of the Company’s common stock on the date the option is granted. Options become exercisable over various periods from the date of grant, and generally expire ten years after the grant date. As of September 30, 2014, there were 4,675,000 options issued and outstanding under the 2010 Stock Plan.
| 8 |
In the event of an employee’s termination, the Company will cease to recognize compensation expense for that employee. There is no deferred compensation recorded upon initial grant date, instead, the fair value of the stock-based payment is recognized ratably over the stated vesting period.
The Company has applied fair value accounting for all share based payment awards since inception. The fair value of each option or warrant granted is estimated on the date of grant using the Black-Scholes option pricing model. The Black-Scholes assumptions used in the three and nine months ended September 30, 2014 and 2013 are as follows:
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||
| 2014 | 2013 | 2014 | 2013 | |||||||||||||
| Exercise price | $1.71 - $1.78 | - | $1.71 - $2.91 | $ | 1.74 | |||||||||||
| Expected dividends | 0% | - | 0% | 0 | % | |||||||||||
| Expected volatility | 101% - 107% | - | 101% - 150% | 148 | % | |||||||||||
| Risk free interest rate | 1.62% - 1.78% | - | 1.62% - 1.78% | 0.77 | % | |||||||||||
| Expected life of option | 5 years | - | 5 years - 10 years | 5 years | ||||||||||||
| Expected forfeitures | 0% | - | 0% | 0 | % | |||||||||||
The Company records stock-based compensation based upon the stated vested provisions in the related agreements. The vesting provisions for these agreements have various terms as follows:
| · immediate vesting, | ||
| · half vesting immediately and remaining over three years, | ||
| · quarterly over three years, | ||
| · annually over three years, | ||
| · one-third immediate vesting and remaining annually over two years, | ||
| · one half immediate vesting and remaining over nine months, | ||
| · one quarter immediate vesting and remaining over three years, | ||
| · one quarter immediate vesting and remaining over 33 months; and | ||
| · monthly over three years. |
During the nine months ended September 30, 2014, the Company granted 2,187,500 options to employees, Board members and consultants having an approximate fair value of $4.7 million based upon the Black-Scholes option pricing model. During the same period in 2013, the Company granted 117,500 options to employees and consultants having an approximate fair value of $185,000 based upon the Black-Scholes option pricing model.
A summary of stock option activities as of September 30, 2014, and for the year ended December 31, 2013, is as follows:
| Weighted | ||||||||||||||||||
| Average | ||||||||||||||||||
| Weighted | Remaining | Aggregate | ||||||||||||||||
| Average Exercise | Contractual | Intrinsic | ||||||||||||||||
| Options | Price | Life | Value | |||||||||||||||
| Balance - December 31, 2012 | 4,453,746 | $ | 1.78 | 6.43 years | $ | 1,308,000 | ||||||||||||
| Granted | 222,500 | $ | 1.69 | |||||||||||||||
| Exercised | (291,666 | ) | $ | 0.79 | ||||||||||||||
| Forfeited | (475,000 | ) | $ | 2.30 | ||||||||||||||
| Balance - December 31, 2013 | 3,909,580 | $ | 1.78 | 5.59 years | $ | 785,000 | ||||||||||||
| Granted | 2,187,500 | $ | 2.44 | |||||||||||||||
| Exercised | (6,583 | ) | $ | 0.58 | ||||||||||||||
| Forfeited | (304,391 | ) | $ | 1.93 | ||||||||||||||
| Balance - September 30, 2014 - outstanding | 5,786,106 | $ | 2.03 | 5.92 years | $ | 1,027,000 | ||||||||||||
| Balance - September 30, 2014 - exercisable | 3,597,772 | $ | 1.81 | 5.26 years | $ | 996,000 | ||||||||||||
| Grant date fair value of options granted - September 30, 2014 | $ | 4,679,000 | ||||||||||||||||
| Weighted average grant date fair value - September 30, 2014 | $ | 2.14 | ||||||||||||||||
| Grant date fair value of options granted - December 31, 2013 | $ | 350,000 | ||||||||||||||||
| Weighted average grant date fair value - December 31, 2013 | $ | 1.57 | ||||||||||||||||
| 9 |
Stock-based compensation expense included in general and administrative expenses and research and development expenses relating to stock options issued to employees and consultants for the three months ended September 30, 2014 and 2013 was $610,000 and $338,000, respectively, and $1.8 million and $1.2 million for the nine month periods ended September 30, 2014 and 2013, respectively.
As of September 30, 2014, total unrecognized stock-based compensation expense related to stock options was $4.4 million, which is expected to be expensed through September 2017.
| 6. | Stock Purchase Warrants |
On October 25, 2012, the Company entered into a Common Stock Purchase Agreement with certain accredited investors. As part of this agreement, the Company issued warrants to purchase 635,855 shares of common stock to the placement agent, or its permitted assigns. The warrants have an exercise price of $1.60 and a life of five years. The warrants vested immediately and expire October 25, 2017. Since these warrants were granted as part of an equity raise, the Company has treated them as a direct offering cost. The result of the transaction has no affect to equity. Warrants outstanding as of September 30, 2014 were 316,522.
On March 15, 2012, the Company entered into a consulting agreement for a financial communications program, for a period of twelve months that began on February 20, 2012. As compensation for such program, the consultant is paid a monthly fee and will be issued a performance warrant exercisable for 250,000 shares of the Company’s common stock based on achievement of certain stock price milestones. Upon initiation of the program, 50,000 of the performance warrants vested. The performance warrant is exercisable for a period of two years from the date of issuance for an exercise price equal to the price ($2.20 per share) of the Company’s common stock on the date of execution (March 15, 2012). In March 2013, the performance warrants’ vesting period was extended to March 14, 2014. All other provisions of the performance warrants remain unchanged. These warrants expired unexercised on March 14, 2014.
On December 20, 2011, the Company entered into a consulting agreement for financial advisory services, for a period of twelve months. As compensation for such services, the consultant was paid a monthly fee and on February 2, 2012, was issued warrants exercisable for 100,000 shares of the Company’s common stock. The warrant is exercisable upon issuance for a period of five years from the date of issue at an exercise price equal to the price ($1.14 per share) of the Company’s common stock on the date of issue. As of September 30, 2014, all of these warrants have been exercised.
A summary of warrant activity for the Company for the nine months ended September 30, 2014 and for the year ended December 31, 2013 is as follows:
| Weighted Average | ||||||||
| Number of Warrants | Exercise Price | |||||||
| Balance at December 31, 2012 | 1,632,501 | $ | 1.99 | |||||
| Granted | - | - | ||||||
| Exercised | - | - | ||||||
| Forfeited | - | - | ||||||
| Balance at December 31, 2013 | 1,632,501 | 1.99 | ||||||
| Granted | - | - | ||||||
| Exercised | (232,619 | ) | 1.47 | |||||
| Forfeited | (454,896 | ) | 1.88 | |||||
| Balance at September 30, 2014 | 944,986 | $ | 2.16 | |||||
Stock-based compensation expense included in general and administrative expenses relating to warrants issued to consultants was $0 for the three months ended September 30, 2014 and 2013 and $0 and $22,000 for the nine months ended September 30, 2014 and 2013, respectively.
| 10 |
A summary of all outstanding and exercisable warrants as of September 30, 2014 is as follows:
| Weighted Average | ||||||||||||||||
| Exercise | Warrants | Warrants | Remaining | Aggregate | ||||||||||||
| Price | Outstanding | Exercisable | Contractual Life | Intrinsic Value | ||||||||||||
| $ | 1.60 | 316,522 | 316,522 | 3.07 years | $ | 57,000 | ||||||||||
| $ | 2.22 | 517,257 | 517,257 | 2.16 years | $ | - | ||||||||||
| $ | 3.30 | 61,207 | 61,207 | 0.66 years | $ | - | ||||||||||
| $ | 3.75 | 50,000 | 50,000 | 1.38 years | $ | - | ||||||||||
| $ | 2.58 | 944,986 | 944,986 | 2.33 years | $ | 57,000 | ||||||||||
| 7. | Stockholders’ Equity |
During the nine months ended September 30, 2014, the Company issued 6,583 shares of common stock, in connection with the exercise of stock options, for proceeds of approximately $4,000. The Company also issued 232,619 shares of common stock, in connection with cashless warrant exercises for the nine months ended September 30, 2014.
| 8. | Net Loss per Share |
Net loss per share is computed by dividing net loss by the weighted average number of common shares outstanding. Diluted loss per share is computed by dividing net loss by the weighted average number of common shares outstanding including the effect of common share equivalents. All common equivalent shares were anti-dilutive at September 30, 2014 and 2013, as such there is no separate computation for diluted loss per share. The number of options and warrants for the purchase of common stock, that were excluded from the computations of net loss per common share for the nine months ended September 30, 2014 were 5,786,106 and 944,986, respectively, and for the nine months ended September 30, 2013 were 3,804,580 and 1,632,501, respectively.
| 9. | Recent Accounting Pronouncements and Developments |
In May 2014, the Financial Accounting Standards Board issued a comprehensive new standard which amends revenue recognition principles and provides a single set of criteria for revenue recognition among all industries. The new standard provides a five step framework whereby revenue is recognized when promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosure pertaining to revenue recognition in both the interim and annual periods. The standard is effective for interim and annual periods beginning after December 15, 2016 and allows for adoption using a full retrospective method, or a modified retrospective method. The Company is currently assessing the method of adoption and the expected impact the new standard has on its financial position and results of operations.
In August 2014, the Financial Accounting Standards Board ("FASB") issued new guidance for presentation of financial statements for going-concern, which addresses when and how to disclose going-concern uncertainties in the financial statements. The new standard requires management to perform interim and annual assessments of an entity’s ability to continue as a going concern within one year after the date the financial statements are issued. An entity must provide certain disclosures if conditions or events raise substantial doubt about the entity’s ability to continue as a going concern. The new guidance applies to all entities and is effective for annual periods ending after December 15, 2016, and interim periods thereafter, with early adoption permitted. The amended guidance is not expected to have a material impact on the Company’s consolidated financial statements.
| 10. | Subsequent Event |
On October 10, 2014, the Company sold a total of 14,059,616 units at a purchase price of $1.47, with each unit consisting of one share of the Company’s common stock, par value of $0.001 per share and a warrant to purchase 0.5 shares of common stock in a registered direct offering for gross proceeds of $20.7 million and net proceeds of $18.9 million.
Each warrant has an exercise price of $1.75 per share, is exercisable immediately after the date of issuance and will expire five years from the date on which it is initially exercisable. The common stock and the warrants are immediately separable and were issued separately.
| 11 |
ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL INFORMATION AND RESULTS OF OPERATIONS
The following discussion should be read in conjunction with the attached unaudited consolidated financial statements and notes thereto, and with our audited consolidated financial statements and notes thereto for the fiscal year ended December 31, 2013, found in our Annual Report on Form 10-K for the year ended December 31, 2013. In addition to historical information, the following discussion contains forward-looking statements that involve risks, uncertainties and assumptions. Where possible, we have tried to identify these forward looking statements by using words such as “anticipate,” “believe,” “intends,” or similar expressions. Our actual results could differ materially from those anticipated by the forward-looking statements due to important factors and risks including, but not limited to, those set forth under “Risk Factors” in this 10-Q and as applicable in Part I, Item 1A of our Annual Report on Form 10-K for the year ended December 31, 2013.
Overview
We are a clinical-stage biotechnology company developing pathogen-specific therapies for serious infections and diseases, with a focus on protecting the microbiome. We are developing an oral biologic to protect the gastrointestinal (GI) microflora from the effects of intravenous (IV) antibiotics for the prevention of C. difficile infection, an oral treatment to reduce the impact of methane producing organisms on constipation-predominant irritable bowel syndrome (C-IBS) and a monoclonal antibody combination for the treatment of Pertussis. In addition, we are developing a Phase 2 oral estriol drug for the treatment of relapsing-remitting multiple sclerosis (MS) and cognitive dysfunction in MS.
Product Pipeline:
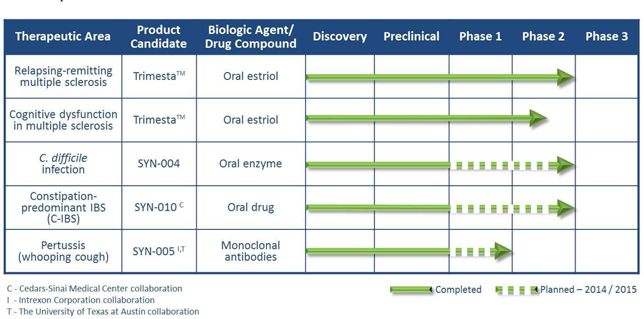
Summary of Pathogen-Specific Therapy Programs:
| · | C. difficile infections (CDI): We are in preclinical development of a novel second-generation oral enzyme candidate, SYN-004, for co-administration with commonly used IV beta-lactam antibiotics intended to prevent the development of and severe effects from C. difficile infections (CDI). CDIs are a leading type of hospital acquired infections (HAIs) that generally occur secondary to treatment with IV antibiotics. Designed to be given orally to protect the gut while certain IV beta-lactam antibiotics (penicillins and cephalosporins) fight the primary infection, SYN-004 is believed to have a similar profile to its first-generation predecessor, which demonstrated protection of the gut flora (microbiome) during treatment with certain penicillins, with the potential to act against a broader spectrum of IV beta-lactam antibiotics. Beta-lactam antibiotics are a mainstay in hospital infection management and include the commonly used penicillin and cephalosporin classes of antibiotics. Approximately 14.4 million patients are administered "SYN-004 target" IV beta-lactam antibiotics annually representing an estimated target market for SYN-004 of 117.6 million beta-lactam doses purchased by U.S. hospitals. The addressable market for SYN-004 is significant. Currently there are no approved treatments designed to protect the microbiome from the damaging effects of IV antibiotics. This worldwide opportunity could represent a multi-billion dollar market.* In October 2014, the U.S. Patent and Trademark Office issued a Notice of Allowance for a composition of matter patent application that has claims to compositions of matter and pharmaceutical compositions of beta-lactamases, including SYN-004, and carries a patent term to at least 2031. We remain on schedule to initiate Phase 1a and 1b clinical studies in the fourth quarter of 2014. Preliminary Phase 1 topline data is expected by year-end 2014, and a Phase 2a trial of SYN-004 is planned to begin in the first half of 2015, followed by the initiation of a Phase 2b clinical trial in the second half of 2015. Clinical manufacturing of SYN-004 in accordance with GMP guidelines has commenced, and completion of manufacturing for Phase 1 and 2 trials is expected during the second half of 2014. |
| * | This information is an estimate derived from the use of information under license from the following IMS Health Incorporated information service: CDM Hospital database for full year 2012. IMS expressly reserves all rights, including rights of copying, distribution and republication. |
| 12 |
| · | C-IBS: In December 2013, through our majority-owned subsidiary, Synthetic Biomics, Inc., we entered into a worldwide exclusive license agreement with Cedars-Sinai Medical Center (CSMC) for the right to develop products for therapeutic and prophylactic treatments for acute and chronic diseases, including the development of SYN-010 to target C-IBS. An investigational team led by Mark Pimentel, M.D., at CSMC has discovered that these products may reduce the production of methane gas by certain gastrointestinal (GI) microorganisms. Methane produced by these organisms is perceived as an underlying cause of bloating, pain and constipation associated with C-IBS, and may contribute to the pathology of other diseases. In September 2014, we announced that our candidate, SYN-010, is a modified release formulation of a statin being designed to reduce the impact of methane producing organisms on C-IBS. A 505(b)(2) regulatory pathway is anticipated for the development of SYN-010. An extensive intellectual property portfolio including granted use patents and pending patent applications for SYN-010 has been licensed to us by CSMC. Additional worldwide patent filings having composition of matter claims, which were recently filed by CSMC and licensed to us, could extend patent protection of SYN-010 to 2035. Based on guidance from the members on our IBS clinical advisory board, we plan to complete a Phase 1 pharmacokinetics/pharmacodynamics trial of SYN-010 in the first half of 2015. We also expect to file an Investigational New Drug (IND) application with the U.S. FDA in the first quarter of 2015, followed by the initiation of a Phase 2 clinical trial, also in the first half of 2015, with Phase 2 data anticipated during the first half of 2015. |
| · | Pertussis: In December 2012, in collaboration with Intrexon Corporation (NYSE: XON) (Intrexon), we initiated development of a monoclonal antibody (mAb) therapy for the treatment of Pertussis infections, more commonly known as whooping cough. We are developing a therapy comprising a combination of two mAbs, SYN-005, designed to target and neutralize pertussis toxin as a prophylaxis for high-risk newborns and in order to reduce the mortality rate in infected infants. To further the development of this potential therapy for Pertussis, we entered into an agreement with The University of Texas at Austin (UT) to license the rights to certain research and pending patents related to pertussis antibodies. We have patents pending on compositions and uses of SYN-005 and we have an issued U.S. patent on other pertussis mAbs from UT. According to the World Health Organization, each year, B. pertussis infection is estimated to cause up to 300,000 deaths worldwide, primarily among unvaccinated infants. Positive preclinical research findings for SYN-005 were reported in April 2014, and again in September 2014, for our proprietary mAb combination therapy for treating Pertussis, in non-human primate studies. We intend to seek non-dilutive funding to support the clinical development of SYN-005 for the prophylaxis and treatment of Pertussis. Based on positive non-human primate and murine model findings, we intend to file an IND application in 2015 to support a Phase 1 clinical trial expected to initiate during the second half of 2015, with topline Phase 1 data expected during 2015. This is expected to be followed by the initiation of a Phase 2/3 trial during 2016. In addition, in September 2014 we received a U.S. Orphan Drug designation for SYN-005 for the treatment of Pertussis. |
| · | Acinetobacter infections: In September 2012, in collaboration with Intrexon, we initiated efforts to develop a mAb therapy for the treatment of Acinetobacter infections. Many strains of Acinetobacter are multidrug-resistant and pose an increasing global threat to hospitalized patients, wounded military personnel and those affected by natural disasters. A treatment for Acinetobacter infections represents a billion dollar market opportunity. This program is in the discovery stage and the generation of a panel of antibodies is ongoing. |
| · | IBS: In December 2013, in collaboration with Intrexon, and partially utilizing the intellectual property optioned from CSMC, we announced an intent to develop biologic approaches targeted at the prevention, and acute and chronic treatment of a subset of IBS pathologies specifically caused by auto-antibodies. This program is in the early discovery stage, and we are still evaluating the option. |
Summary of Multiple Sclerosis Program:
| · | Relapsing-Remitting MS: Patient follow-up is complete in the Phase 2, investigator-initiated, randomized (n=158), double-blinded, placebo-controlled trial which evaluated our drug candidate, Trimesta, in women with relapsing-remitting MS at 16 sites across the U.S. Positive Phase 2 topline efficacy and safety results were presented in April 2014 by lead principal investigator, Dr. Rhonda Voskuhl of the University of California, Los Angeles (UCLA) David Geffen School of Medicine at the 66th American Academy of Neurology Annual Meeting. Dr. Voskuhl presented additional Phase 2 clinical outcome data, including more detailed results on improvements in cognitive and disability measures, at the 2014 Joint Americas and European Committees for Treatment and Research in Multiple Sclerosis Meeting (ACTRIMS-ECTRIMS) in Boston in September 2014. The data as reported by Dr. Voskuhl for the UCLA-led Phase 2 study demonstrated the potential of Trimesta to have a novel dual mechanism of action for both the anti-inflammatory effects that improve relapse rate, and a neuroprotective effect that improves standard measures of disability and cognition. Further analyses of the MRI data are ongoing, with topline data expected during the first quarter of 2015. This investigator-initiated clinical trial is supported by grants exceeding $8 million, awarded primarily by the National Multiple Sclerosis Society (NMSS) in partnership with the NMSS’s Southern California chapter, and the National Institutes of Health. Annual worldwide sales of MS therapies are forecasted to be $17.8 billion in 2019. We have licensed from UCLA issued method of treatment patents in the U.S. for MS therapy with estriol and estriol combination therapies (including estriol with Copaxone®), and four new provisional patent applications have been filed based on the recent Phase 2 clinical results. We are engaging with the neurology community and potential strategic partners, as we determine next steps for Trimesta. |
| 13 |
| · | Cognitive Dysfunction in MS: Trimesta is also being developed for the treatment of cognitive dysfunction in female MS patients. This 12-month randomized, double-blind, placebo-controlled Phase 2 clinical trial is being conducted at four sites in the United States, including UCLA. The primary endpoint is the effect on cognitive function as assessed by Paced Auditory Serial Addition Test (PASAT). Patient enrollment is ongoing. The majority of the costs of this trial are being funded by grants from foundations and charitable organizations and we have pledged approximately $500,000 to UCLA to partially fund this trial, payable over three years. An estimated 50-65% of MS patients are expected to develop disabilities due to cognitive dysfunction and there is currently no approved treatment for this indication. |
Since our inception in January 2001, our efforts and resources have been focused primarily on acquiring and developing our product candidates, our clinical trials, raising capital, manufacturing and recruiting personnel. As of June 30, 2010, we emerged from the development stage after entering into a sublicense agreement with Meda AB and receiving an up-front payment of $2.5 million. We consider this sublicense agreement to be an indication that we commenced our principal operations. Meda AB informed us that due to the decision of the European Medicines Agency (EMA) to limit the use of flupirtine for long-term pill and systemic use, it has decided to postpone the planned fibromyalgia clinical trials in the U.S.
To date, we have financed our operations primarily through public and private sales of our common stock, and we expect to continue to seek to obtain the required capital in a similar manner. We have incurred an accumulated deficit of $94.6 million through September 30, 2014. We cannot provide any assurance that we will be able to achieve profitability on a sustained basis, if at all, obtain the required funding, obtain the required regulatory approvals, or complete additional corporate partnering or acquisition transactions.
Recent Developments
On October 10, 2014, we sold a total of 14,059,616 units at a purchase price of $1.47, with each unit consisting of one share of our common stock, and a warrant to purchase 0.5 shares of common stock in a registered direct offering for gross proceeds of $20.7 million and net proceeds of $18.9 million.
Pipeline Programs and Therapeutic Areas
Pathogen-Specific Therapy Programs
We are developing pathogen-specific therapies for serious infections and diseases, with a focus on protecting the microbiome. Infectious disease outbreaks are increasing while intervention options are declining due to widespread multidrug-resistant bacteria, increasing numbers of immuno-compromised patients (e.g., the elderly and cancer patients), and the isolation of new pathogens. We are developing an oral biologic to protect the gastrointestinal microflora from the effects of certain IV beta-lactam antibiotics for the prevention of CDI, an oral treatment to reduce the impact of methane producing organisms on C-IBS and a monoclonal antibody combination for the treatment of Pertussis. We have discovery stage programs targeting Acinetobacter infections and the prevention and treatment of a root cause of a subset of IBS.
Microbiome-Focused Therapies:
Our C. difficile and C-IBS programs are focused on protecting the microbiome, or our gut flora, which is home to millions of bacteria and composed of a natural balance of both “good” beneficial bacteria and “bad” pathogenic bacteria. When that natural balance of all of these bacteria is disrupted, a person’s health is compromised.
C. difficile:
According to the Agency for Healthcare Research and Quality, aggregate costs associated with CDI related stays in the hospital were $8.2 billion in the U.S. during 2009. CDI is a rising global HAI problem in which the toxins produced by C. difficile bacteria result in diarrhea antibiotic-associated diarrhea (AAD), and in the most serious cases, pseudomembranous colitis (erosion of the lower GI tract) that can lead to death. The Centers for Disease Control and Prevention (CDC) identified C. diff as an “urgent public health threat,” particularly given its resistance to many drugs used to treat other infections. CDI is a major, unintended risk associated with the prophylactic or therapeutic use of IV antibiotics, which may alter the natural balance of microflora that normally protect the GI tract, leading to C. difficile overgrowth and infection. Other risk factors for CDI include hospitalization, prolonged length of stay, underlying illness, immune-compromising conditions including the administration of chemotherapy, and advanced age.
CDI is a widespread and often drug resistant infectious disease, and it is estimated that 1.1 million patients are infected with C. diff annually in the U.S.*, and it has been reported that 30,000 patients die with a C. diff infection each year. CDI has surpassed methicillin-resistant staphylococcus aureus (MRSA) as the most frequent infection acquired in the hospital. Controlling the spread of CDI has proven challenging, as the C. difficile spores are easily transferred to patients via normal contact with healthcare personnel and other inanimate objects. There is currently no vaccine or approved product for the prevention of C. diff infection.
* This information is an estimated derived from the use of information under license from the following IMS Health Incorporated information service: CDM Hospital database for full year 2012. IMS expressly reserves all rights, including rights of copying, distribution and republication.
| 14 |
C. difficile: Acquisition of Clinical-Stage Program
In November 2012, we acquired a series of oral beta-lactamase enzymes (P1A, P2A and P3A) and related assets targeting the prevention of CDI, the leading HAI that generally occurs secondary to treatment with IV antibiotics. The acquired assets include a pre-IND package for P3A (now referred to as SYN-004), Phase 1 and Phase 2 clinical data for P1A, manufacturing processes and data, and a portfolio of issued and pending U.S. and international patents intended to support an IND and Biologics License Application (BLA) with the FDA. Utilizing this portfolio of assets, we intend to develop a proprietary oral beta-lactamase enzyme product candidate, SYN-004. When co-administered with certain IV beta-lactam antibiotics, it is expected that SYN-004 can degrade the antibiotic that is excreted in the GI tract, thus preserving the natural balance of the patient's microflora, and preventing opportunistic infections including CDI. Beta-lactam antibiotics are a mainstay in hospital infection management and include the commonly used penicillin and cephalosporin classes of antibiotics. Approximately 14.4 million patients are administered "SYN-004 target" IV beta-lactam antibiotics annually, representing an estimated target market for SYN-004 of 117.6 million beta-lactam doses purchased by U.S. hospitals. The addressable market is significant and currently there are no approved treatments designed to protect the microbiome from the damaging effects of IV antibiotics. This worldwide opportunity could represent a multi-billion dollar market.*
* This information is an estimated derived from the use of information under license from the following IMS Health Incorporated information service: CDM Hospital database for full year 2012. IMS expressly reserves all rights, including rights of copying, distribution and republication.
C. difficile: Oral Enzyme Background
Beta-lactamase enzymes have the ability to degrade beta-lactam antibiotics that may be excreted into the GI tract. P1A (the first generation candidate) showed acceptable safety and tolerability in a Phase 1 study. In addition, two Phase 2 clinical studies demonstrated that P1A had the ability to preserve GI microflora in hospitalized patients treated with intravenous ampicillin or the combination of piperacillin and tazobactam.
C. difficile: Preclinical and Clinical Development
Compared to the first generation oral enzyme candidate, P1A, we believe that the second generation candidate, SYN-004 (formerly P3A), will have activity against a broader spectrum of beta-lactam antibiotics, including both penicillins and certain cephalosporins. Due to the structural similarities between P1A and SYN-004, and based on previous discussions with the FDA, it is anticipated that certain preclinical data collected on P1A may be used in support of an IND for our new product candidate, SYN-004.
In June 2014, we formed a Clinical Advisory Board (CAB) to support development of SYN-004. The CAB is comprised of industry leaders Mark Wilcox, M.D., (Chairman), Curtis Donskey, M.D., Ciarán Kelly, M.D. and Tom Louie, M.D., all of whom are expected to provide expertise and guidance on each aspect of the C. diff clinical program.
In August 2014, we announced an agreement with Evonik for GMP manufacturing of our proprietary oral beta-lactamase enzyme, SYN-004, for use in the planned clinical trials. Evonik plans to formulate and encapsulate enterically coated SYN-004 for oral delivery using material generated by our API manufacturer FUJIFILM Diosynth Biotechnologies UK Limited.
We remain on schedule to initiate Phase 1a and 1b clinical studies in the fourth quarter of 2014. Preliminary Phase 1 topline data is expected by year-end 2014, and a Phase 2a trial of SYN-004 is planned to begin in the first half of 2015, followed by the initiation of a Phase 2b clinical trial in the second half of 2015. Clinical manufacturing of SYN-004 in accordance with GMP guidelines has commenced, and completion of manufacturing for Phase 1 and 2 trials is expected during the second half of 2014.
C. difficile: Intellectual Property
In October 2014, the U.S. Patent and Trademark Office issued a Notice of Allowance for a composition of matter patent application that has claims to compositions of matter and pharmaceutical compositions of beta-lactamases, including SYN-004, and carries a patent term to at least 2031. In addition to the newly allowed patent, we have numerous related granted and pending U.S. and international patent applications that are central to our intellectual property estate.
C-IBS:
Irritable Bowel Syndrome (IBS) is a functional GI disorder characterized by gas, abdominal pain, bloating and diarrhea or constipation, or alternating episodes of both. According to reports published by The International Foundation for Functional Gastrointestinal Disorders (IFFGD), IBS affects an estimated 10 to 15 percent of the population, or as many as 40 million Americans. The illness affects both men and women; two-thirds of diagnosed sufferers are women. The onset of IBS can begin anytime from adolescence to adulthood. Four bowel patterns may be seen with IBS, including: C-IBS (constipation predominant), D-IBS (diarrhea predominant), M-IBS (mixed diarrhea and constipation) and A-IBS (alternating diarrhea and constipation). We intend to focus on the development of SYN-010, an oral treatment to reduce the impact of methane producing organisms on C-IBS.
It has been reported that one-third of all IBS patients have C-IBS. Current FDA-approved therapies for the treatment of C-IBS include AMITIZA® (lubiprostone) and LINZESS® (linaclotide). Prescription and over-the-counter laxatives are also used by C-IBS patients for symptomatic relief. According to GlobalData, sales of approved drugs to treat C-IBS in seven major markets are projected to reach $1.3 billion by 2018.
| 15 |
C-IBS: Acquisition of Clinical-Stage Program
In December 2013, we entered into a worldwide exclusive license agreement with CSMC for the right to develop products for therapeutic and prophylactic treatments for acute and chronic diseases. We licensed and optioned from CSMC a portfolio of intellectual property comprised of several U.S. and international patents and pending patent applications for various fields of use, including C-IBS, obesity and diabetes. An investigational team led by Mark Pimentel, M.D. at CSMC has discovered that these products may reduce the production of methane gas by certain GI microorganisms. Methane produced by these organisms is perceived as the underlying cause of bloating, pain and constipation associated with C-IBS, and may contribute to the pathology of other diseases. Initially we will focus on the development of SYN-010, an oral treatment being designed to reduce the impact of methane producing organisms on C-IBS.
IBS: Gas Producing Organisms Background
In the 1990’s, research showed that IBS patients (over a given time) produced five times more gas than did people without IBS. Since the only source of those gases was bacterial, the initial presumption was that IBS patients had excessive bacteria in the colon. Subsequent studies showed that IBS patients had excessive quantities of gas in the small bowel; these data were the catalyst for studying small bowel bacteria in IBS. Normally the small intestine contains a very small quantity of bacteria. In published studies, indirect measures of small bowel bacteria suggest that 84% of IBS sufferers have excessive quantities of bacteria typically found in the colon. The CSMC investigational team led by Dr. Pimentel is researching a recent theory that defines IBS as a bacterial disease. Gut microflora that should normally be confined to the large intestine inappropriately colonize the small intestine. This process is referred to as small intestine bacterial overgrowth (SIBO), which results in gas, bloating, abdominal pain and altered stool habits characterized by IBS.
C-IBS: Methane Producing Organisms Background
Further research by the CSMC investigational team led by Dr. Pimentel is focused on the C-IBS patient population. Extensive studies conducted by Dr. Pimentel and collaborators have shown that overproduction of methane gas is directly associated with bloating, pain and constipation in C-IBS patients. CSMC investigators have discovered that inhibiting intestinal methane production may reverse constipation associated with C-IBS, and can be beneficial in other major diseases such as obesity and type 2 diabetes.
C-IBS: Preclinical and Clinical Development
Ongoing efforts led by Dr. Pimentel include formulating and testing non-antibiotic FDA-approved oral drug candidates for ultimate product registration via potential expedited pathways. Such candidates are intended for the reduction or elimination of methane gas production within the intestines, with the goal of having little or no unintended impact on a patient's normal intestinal microflora. Initially we will focus on the development of an oral treatment to reduce the impact of methane producing organisms on C-IBS.
In April 2014, we formed a CAB to support development of SYN-010, and also announced that gastroenterologist and lead investigator for the C-IBS program, Dr. Mark Pimentel, will Chair the CAB. In October 2014, we announced the expansion of the C-IBS CAB to include William Chey, M.D., Gail M. Comer, M.D., Anthony J. Lembo, M.D., and, Philip Schoenfeld, M.D., MSEd, MSc.
In September 2014, we announced that our candidate, SYN-010, is a modified release formulation of a statin being designed to reduce the impact of methane producing organisms on C-IBS. A 505(b)(2) regulatory pathway is anticipated for the development of SYN-010.
Based on guidance from the members on our IBS CAB, we plan to complete a Phase 1 pharmacokinetics/pharmacodynamics trial of SYN-010 in the first half of 2015. We also expect to file an IND with the U.S. FDA in the first quarter of 2015, followed by the initiation of a Phase 2 clinical trial, also in the first half of 2015, with Phase 2 data anticipated during the first half of 2015.
C-IBS: Intellectual Property
An extensive intellectual property portfolio including granted use patents and pending patent applications for SYN-010 has been licensed to us by CSMC. Additional worldwide patent filings having composition of matter claims, which were recently filed by CSMC and licensed to us, could extend patent protection of SYN-010 to 2035.
Monoclonal Antibodies:
Monoclonal Antibodies for Infectious Diseases
Acting as the body's army, antibodies are proteins, generally found in the bloodstream, that provide immunity in detecting and destroying pathogens, such as viruses and bacteria and their associated toxins. MAbs can also be designed and produced as therapeutic agents, utilizing protein engineering and recombinant production technologies. The mAbs being developed under our collaboration with Intrexon are intended to supplement a patient's own immune system by providing the means to specifically and rapidly neutralize and/or clear specific pathogens and toxins of interest in a process known as “passive immunity”. Many pathogens that cause infectious diseases are innately resistant to, or over time have developed increased resistance to, antibiotics and other drugs.
| 16 |
Intrexon Collaboration: Monoclonal Antibodies for Infectious Diseases
In August 2012, we entered into a worldwide exclusive channel collaboration (“Second ECC”) with Intrexon through which we intend to develop a series of mAb therapies for the treatment of certain infectious diseases not adequately addressed by existing therapies. Utilizing Intrexon’s comprehensive suite of proprietary technologies, including the mAbLogix TM platform for rapid discovery of fully human mAbs and the LEAP TM cell processing station, our initial efforts will target three infectious disease indications.*** We also have the option to target an additional five infectious disease indications under this collaboration. To date, we have initiated development of a mAb therapy for the treatment of Pertussis and Acinetobacter infections.
***mAbLogixTM and LEAPTM are registered trademarks of Intrexon Corporation.
Pertussis:
Bordetella pertussis (B. pertussis) is a gram-negative bacterium that infects the upper respiratory tract, causing uncontrollable, and violent coughing. Antibiotic treatment does not have a major effect on the course of Pertussis, because while it can eliminate the B. pertussis bacteria from the respiratory tract, it does not neutralize the pertussis toxin. Infants with Pertussis often require hospitalization in pediatric intensive care units, frequently requiring mechanical ventilation. Pertussis in adults generally leads to a chronic cough referred to as the "cough of 100 days." The incidence of Pertussis is increasing due to a less effective acellular vaccine introduced in the 1990s, exposure of unvaccinated and under-vaccinated individuals including infants who are not yet fully vaccinated, exposure of individuals whose immunity has diminished over time, as well as asymptomatic carriers.
According to the World Health Organization there are 50 million cases of whooping cough and B. pertussis infection that are estimated to cause up to 300,000 deaths each year worldwide, primarily among unvaccinated infants. Recent news reports throughout the U.S. indicate that the pertussis vaccine introduced in the 1990s does not provide long-term protection and, as a result, whooping cough cases have increased to a 60-year high.
Pertussis: Intrexon Collaboration and The University of Texas at Austin Agreement
In December 2012, we initiated mAb development for the treatment of Pertussis focusing on toxin neutralization pursuant to our August 2012 collaboration with Intrexon. Unlike antibiotics, we are developing a therapy comprising a combination of two mAbs, SYN-005, to target and neutralize the pertussis toxin as a prophylaxis for high-risk newborns and in order to reduce the mortality rate in infected infants.
To further the development of this potential therapy for pertussis, we have entered into an agreement with The University of Texas at Austin to license the rights to certain research and pending patents related to pertussis antibodies. These research efforts are being conducted at the Cockrell School of Engineering in the laboratory of Assistant Professor, Jennifer A. Maynard, Ph.D., the Laurence E. McMakin, Jr. Centennial Faculty Fellow in the McKetta Department of Chemical Engineering. Dr. Maynard brings to the project her expertise in defining the key neutralizing epitopes of pertussis toxin to optimize the potential efficacy of antibody therapeutics.
Pertussis: Preclinical and Clinical Development
Working with our collaborator, Intrexon, and our academic collaborator, The University of Texas at Austin, we established a combination of two humanized antibodies designed to neutralize pertussis toxin, a major cause of pertussis-mediated infant morbidity and mortality. Benchtop studies demonstrated high affinity binding to the toxin, as well as potent neutralization of the toxin. In addition, the antibodies were highly efficacious in a murine model of pertussis in which they completely mitigated elevations of the white blood cell count that is characteristic of the illness.
In April 2014, and again in September 2014, we received positive preclinical research findings for SYN-005, our proprietary mAb combination therapy for treating Pertussis (whooping cough), in two non-human primate studies (n=15). In the second pertussis study in particular, SYN-005 was associated with favorable decreases in white blood cell counts within two days and the achievement of nearly normal levels within one week of treatment with SYN-005.
We intend to seek non-dilutive funding to support the clinical development of SYN-005 for the prophylaxis and treatment of Pertussis. Based on positive non-human primate and murine model findings, we intend to file an IND application in 2015 to support a Phase 1 clinical trial expected to initiate during the second half of 2015, with topline Phase 1 data expected during 2015. This is expected to be followed by the initiation of a Phase 2/3 trial during 2016. In addition, in September 2014 we received a U.S. Orphan Drug designation for SYN-005 for the treatment of Pertussis.
Pertussis: Intellectual Property
We have patents pending on compositions and uses of SYN-005 and we have an issued U.S. patent on other pertussis mAbs from UT.
| 17 |
Acinetobacter Infections:
Acinetobacter baumanii is a difficult to treat pathogen due to its rapid and well-established development of resistance to most antibiotics, making it a multidrug-resistant pathogen. In addition, as a biofilm-forming pathogen, Acinetobacter baumanii has the ability to survive up to twice as long as non-biofilm-forming pathogens. In the U.S., Acinetobacter baumanii has been reported to be the cause of up to 2.6% of hospital acquired infections, 1.3% of bloodstream infections and 7.0% of ICU respiratory tract infections, and more than half of the Acinetobacter baumanii isolates are multidrug-resistant. According to published articles, mortality rates associated with Acinetobacter infections as high as 43.0% are reported in hospitals and ICU settings. While Acinetobacter baumanii is a well-documented pathogen in the hospital setting, this pathogen also poses an increasing danger to wounded servicemen and women in military treatment centers and to those treated in trauma centers following natural disasters.
A treatment for Acinetobacter infections represents a billion dollar market opportunity.
Acinetobacter: Intrexon Collaboration
In September 2012, we initiated a mAb discovery and development program for Acinetobacter infections pursuant to our August 2012 collaboration with Intrexon. This program is in the discovery stage and the generation of a panel of antibodies is ongoing.
IBS:
Existing IBS therapies, which are primarily focused on supportive care, are unlikely to address the treatment needs of the patient population with auto-antibodies, an underlying immune-specific pathology. Through our collaboration with Intrexon, we intend to address the unmet medical need in these patients with personalized medicine and target the root causes of a subset of IBS-associated pathologies.
IBS: Intrexon Collaboration
In December 2013, in collaboration with Intrexon, and partially utilizing the intellectual property optioned from CSMC, we announced an intent to develop biologic approaches targeted at the prevention, and acute and chronic treatment of a subset of IBS pathologies specifically caused by auto-antibodies.
We intend to utilize intellectual property optioned from CSMC. According to an increasing body of recent work conducted by CSMC, a subset of IBS cases appear to be causally initiated by one or more encounters with acute infectious gastroenteritis, such as the foodborne illness, Campylobacter jejuni . CSMC has identified a novel autoimmune target for this subset of IBS cases because of the development of cross-reacting antibodies between a bacterial toxin and a protein important for controlling GI motility. This program is in the early discovery stage, and we are still evaluating the option.
Multiple Sclerosis Program
Relapsing-Remitting MS:
MS is a progressive neurological disease in which the body loses the ability to transmit messages along the central nervous system, leading to pain, loss of muscle control, paralysis, cognitive impairment and in some cases death. According to the National Multiple Sclerosis Society (NMSS), more than 2.3 million people worldwide (approximately 400,000 patients in the U.S. of which approximately 65% are women) have been diagnosed with MS. The diagnosis is typically made in young adults, ages 20 to 50. According to the NMSS, approximately 85% of MS patients are initially diagnosed with the relapsing-remitting form, and 10-15% with other progressive forms.
There are nine FDA-approved therapies for the treatment of relapsing-remitting MS: Betaseron®, Rebif®, Avonex®, Copaxone®, Tysabri ®, Gilenya ®, Extavia ®, Aubagio ® and Tecfidera ®. Many of these therapies provide only a modest benefit for patients with relapsing-remitting MS. All of these drugs except Gilenya®, Aubagio® and Tecfidera® require frequent (daily, weekly & monthly) injections (or infusions) on an ongoing basis and can be associated with unpleasant side effects (such as flu-like symptoms) and high rates of non-compliance among users. Despite the availability of therapies for the treatment of relapsing-remitting MS, the disease is highly underserved and exacts a heavy personal and economic toll. Annual worldwide sales of MS therapies were estimated at $14.1 billion in 2012.
Relapsing-Remitting MS: Background
Research has shown that pregnant women with MS tend to experience a spontaneous reduction of disease symptoms during pregnancy, particularly in the third trimester. The PRIMS (Pregnancy In MS) study published in 1998, a landmark observational clinical study published in the New England Journal of Medicine followed 254 women with MS during 269 pregnancies and for up to one year after delivery. The PRIMS study demonstrated that relapse rates were significantly reduced by 71% (p < 0.001) through the third trimester of pregnancy compared to pre-pregnancy-rates, and that relapse rates increased by 120% (p < 0.001) during the first three months after birth (post-partum) and then return to pre-pregnancy rates. It has been hypothesized that the female hormone, estriol, produced by the placenta during pregnancy, plays a role in “fetal immune privilege”, a process that prevents a mother’s immune system from attacking and rejecting the fetus. The maternal levels of estriol increase linearly through the third trimester of pregnancy until birth, whereupon it abruptly returns to low circulating levels. The anti-autoimmune effects of estriol are thought to be responsible for the therapeutic effects of pregnancy on MS.
| 18 |
Rhonda Voskuhl, M.D., Director, UCLA MS program, UCLA Department of Neurology, has found that plasma levels of estriol achieved during pregnancy have potent immunomodulatory effects. Dr. Voskuhl further postulated and tested in a pilot clinical study that oral doses of estriol may have a therapeutic benefit when administered to non-pregnant female MS patients by, in essence, mimicking the spontaneous reduction in relapse rates seen in MS patients during pregnancy.
Estriol has been approved and marketed for over 40 years throughout Europe and Asia for the oral treatment of post-menopausal symptoms. It has never been approved by the U.S. FDA for any indication.
Relapsing-Remitting MS: Clinical Development
Trimesta (oral estriol) is being developed as an adjunctive oral once-daily treatment for relapsing-remitting MS in women. An investigator-initiated, 10-patient, 22-month, single-agent, crossover clinical trial to study the therapeutic effects of 8 mg. of oral Trimesta taken daily in non-pregnant female relapsing-remitting MS patients was completed in the U.S. The total volume and number of gadolinium-enhancing lesions were measured by brain magnetic resonance imaging (an established neuroimaging measure of disease activity in MS). Over the next three months of treatment with Trimesta, the median total enhancing lesion volumes decreased by 79% (p = 0.02) and the number of lesions decreased by 82% (p = 0.09). They remained decreased during the next 3 months of treatment, with lesion volumes decreased by 82% (p = 0.01), and numbers decreased by 82% (p = 0.02). Following a six-month drug holiday during which the patients were not on any drug therapies, median lesion volumes and numbers returned to near baseline pretreatment levels. Trimesta therapy was reinitiated during a four-month retreatment phase of this clinical trial. The relapsing-remitting MS patients again demonstrated a decrease in enhancing lesion volumes of 88% (p = 0.008) and a decrease in the number of lesions by 48% (p = 0.04) compared with original baseline scores.
Patient follow-up is complete in the Phase 2, investigator-initiated, randomized (n=158), double-blinded, placebo-controlled trial which evaluated our drug candidate, Trimesta, in women with relapsing-remitting MS at 16 sites across the U.S. Positive Phase 2 topline efficacy and safety results were presented in April 2014 by lead principal investigator, Dr. Rhonda Voskuhl of UCLA at the 66th American Academy of Neurology Annual Meeting. Dr. Voskuhl presented additional Phase 2 clinical outcome data, including more detailed results on improvements in cognitive and disability measures, at the 2014 Joint ACTRIMS-ECTRIMS in Boston in September 2014. The data as reported by Dr. Voskuhl for the UCLA-led Phase 2 study demonstrated the potential of Trimesta to have a novel dual mechanism of action for both the anti-inflammatory effects that improve relapse rate, and a neuroprotective effect that improves standard measures of disability and cognition. Specifically, Dr. Voskuhl reported the following results:
(i) Annualized relapse rate: a 47% reduction in annualized relapse rate in the Trimesta+Copaxone® arm as compared to the placebo+Copaxone® arm (active control arm) at 12 months of therapy (p= 0.02), meeting the primary outcome of the trial. These improvements in annualized relapse rate were sustained during the 24 months of therapy. When compared to the placebo+Copaxone® arm at 24 months, the Trimesta+Copaxone ® arm demonstrated a 32% lower relapse rate (p= 0.11).
(ii) Cognitive disability: Patients in the Trimesta+Copaxone® arm who had Paced Auditory Serial Addition Test (PASAT) scores lower than 55 before treatment experienced an approximately 12%, or 6 point, improvement in cognitive scores within 12 months of treatment (p<0.05). This improvement from baseline was sustained throughout the 24 month study. In addition, a significantly larger proportion of patients in the Trimesta+Copaxone® arm demonstrated sustained improvement in cognition during the entire 24 month period, as approximately 33% of the patients showed sustained improvement of at least 3 points during this time period, compared to only about 21% in the placebo+ Copaxone® arm (p<0.05).
(iii) Physical disability: Expanded Disability Status Scale (EDSS) scores in the Trimesta+Copaxone® arm significantly improved during 24 month follow-up by at least 0.5 point (p=0.03) compared to the placebo+Copaxone® arm which experienced no change in EDSS scores. The between group difference showed a positive trend (p=0.25). The 25 foot walk test showed a significant difference, while the patients in the Trimesta+Copaxone® arm were stable during the study, those in the active control arm did worse. The between group difference (p=0.02).
In addition, adjunctive oral Trimesta plus injectable standard of care Copaxone® demonstrated a strong safety profile and was well tolerated by women in the study. Further analyses of the MRI data are ongoing, with topline data expected during the first quarter of 2015. We are engaging with the neurology community and potential strategic partners, as we determine next steps for Trimesta. Annual worldwide sales of MS therapies are forecasted to be $17.8 billion in 2019.
This investigator-initiated clinical trial is supported by grants exceeding $8 million, awarded primarily by the National Multiple Sclerosis Society (NMSS) in partnership with the NMSS’s Southern California chapter, and the National Institutes of Health.
Relapsing-Remitting MS: Intellectual Property
| 19 |
In March 2014, we announced that the U.S. Patent & Trademark Office issued U.S. Patent No. 8,658,627 entitled, Pregnancy Hormone Combination for Treatment of Autoimmune Diseases, to the Regents of the University of California. The patent includes claims to the use of our drug candidate, Trimesta (oral estriol), in conjunction with a gestagen for the treatment of multiple sclerosis (MS) and other autoimmune diseases. The patent also includes a claim for the administration of Trimesta, a gestagen and a third standard of care MS agent, such as glatiramer acetate injection (Copaxone®), interferon beta-1a (Avonex®, Rebif®), interferon beta-1b (Betaseron®, Extavia®) or sphingosine-1-phosphate receptor modulator (Gilenya®).
In April 2013, we announced that the U.S. Patent & Trademark Office issued U.S. Patent No. 8,372,826 entitled, Estriol Therapy for Multiple Sclerosis and Other Autoimmune Diseases, to the Regents of the University of California which includes claims to the use of our drug candidate, Trimesta (oral estriol), in combination with glatiramer acetate injection (Copaxone®). According to Teva Pharmaceutical Industries Ltd.’s Form 20-F for the year ended December 31, 2013, filed with the SEC on February 10, 2014. Copaxone ® is the number one selling drug for multiple sclerosis with approximately $4.3 billion in global annual sales. Currently marketed exclusively by Teva Pharmaceutical Industries Ltd., Copaxone® is expected to face generic competition in the U.S., as certain patent terms began to expire in May 2014.
Through our wholly owned subsidiary, we hold the exclusive worldwide license to issued U.S. Patents 8,658,627, 8,372,826 and 6,936,599 and pending patents for multiple sclerosis and other autoimmune diseases covering the uses of our drug candidate, Trimesta. Four new provisional patent applications have been filed based on the recent Phase 2 clinical results.
Cognitive Dysfunction in MS:
According to the NMSS and the Multiple Sclerosis Society of Canada publication, Hold that Thought! Cognition and MS, it is fairly common for people with MS to complain of cognitive difficulties, such as remembering things, finding the right words and the ability to concentrate. Among MS patients, 50-65% have some degree of cognitive dysfunction.
The major areas of cognition that may be affected include complex attention and executive functions. Complex attention involves multitasking, the speed with which information can be processed, learning and memory, and perceptual skills; executive functions include problem solving, organizational skills, the ability to plan, and word finding. Just as the nature, frequency, and severity of MS-related physical problems can widely vary, not all people with MS will have cognitive dysfunction, and no two people will experience exactly the same type or severity.
Cognitive Dysfunction in MS: Background
In the investigator-initiated, 10-patient, 22-month, single-agent, crossover clinical trial conducted by Dr. Rhonda Voskuhl, a statistically significant 14% improvement from baseline in the PASAT cognitive testing scores (p = 0.04) was observed in relapsing-remitting MS patients after six months of Trimesta therapy. PASAT is a routine cognitive test performed in patients with a wide variety of neuropsychological disorders such as MS. The PASAT scores are expressed as a mean percent change from baseline.
Cognitive Dysfunction in MS: Clinical Development
Our Trimesta (oral estriol) drug candidate is also being developed for the treatment of cognitive dysfunction in female MS patients. This randomized, double-blind, placebo-controlled Phase 2 clinical trial to evaluate Trimesta’s potential neuroprotective and therapeutic effect on cognitive dysfunction in female MS patients is currently enrolling relapsing-remitting or secondary-progressive female MS patients at four clinical sites in the United States, including UCLA. Up to 64 patients between the ages of 18 and 50 will be randomized 1:1 into the treatment and placebo groups. Dr. Voskuhl will administer either oral Trimesta or a matching placebo, in addition to an FDA-approved MS treatment, including Copaxone ®, Avonex®, Betaseron®, Extavia®, Rebif®, Gilenya®, Aubagio® and Tecfidera®. Each patient will be dosed and monitored for one year after being enrolled. The primary endpoint in this clinical trial being run under an investigator-initiated IND application is expected to be improvement in PASAT cognitive testing scores versus matching placebo. We and a private foundation have pledged to equally support this new clinical trial, and we will also provide Trimesta drug supply. The trial also received contributions from several other supporters. Patient recruitment and enrollment into this trial is ongoing.
Critical Accounting Policies
The consolidated financial statements are prepared in conformity with U.S. GAAP, which require the use of estimates, judgments and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses in the periods presented. We believe that the accounting estimates employed are appropriate and resulting balances are reasonable; however, due to inherent uncertainties in making estimates, actual results could differ from the original estimates, requiring adjustments to these balances in future periods. The critical accounting estimates that affect the consolidated financial statements and the judgments and assumptions used are consistent with those described in the MD&A section in our Annual Report on Form 10-K for the year ended December 31, 2013.
| 20 |
Recent Accounting Pronouncements and Developments
In May 2014, the Financial Accounting Standards Board issued a comprehensive new standard which amends revenue recognition principles and provides a single set of criteria for revenue recognition among all industries. The new standard provides a five step framework whereby revenue is recognized when promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosure pertaining to revenue recognition in both the interim and annual periods. The standard is effective for interim and annual periods beginning after December 15, 2016 and allows for adoption using a full retrospective method, or a modified retrospective method. We are currently assessing the method of adoption and the expected impact the new standard has on its financial position and results of operations.
In August 2014, the Financial Accounting Standards Board ("FASB") issued new guidance for presentation of financial statements for going-concern, which addresses when and how to disclose going-concern uncertainties in the financial statements. The new standard requires management to perform interim and annual assessments of an entity’s ability to continue as a going concern within one year after the date the financial statements are issued. An entity must provide certain disclosures if conditions or events raise substantial doubt about the entity’s ability to continue as a going concern. The new guidance applies to all entities and is effective for annual periods ending after December 15, 2016, and interim periods thereafter, with early adoption permitted. The amended guidance is not expected to have a material impact on our consolidated financial statements.
Results of Operations
Three Months Ended September 30, 2014 and 2013
General and Administrative Expenses
General and administrative expenses decreased by 36% to $1.2 million for the three months ended September 30, 2014, from $1.9 million for the three months ended September 30, 2013. This decrease is primarily the result of bad debt expense of $763,000 associated with the determination that the note receivable and interest receivable from the sale of Adeona Clinical Laboratory was uncollectible for the quarter ended September 30, 2013. The charge relating to stock-based compensation expense was $377,000 for the three months ended September 30, 2014, compared to $264,000 for the three months ended September 30, 2013.
Research and Development Expenses
Research and development expenses increased by 150% to $3.7 million for the three months ended September 30, 2014, from $1.5 million for the three months ended September 30, 2013. This increase is primarily the result of increased program costs associated with expanded research, development and manufacturing activities in our pathogen-specific pipeline, including our C. diff, C-IBS and Pertussis programs. Research and development expenses also include a charge relating to non-cash stock-based compensation expense of $232,000 for the three months ended September 30, 2014, compared to $74,000 for the three months ended September 30, 2013.
Other Income (Expense)
Other income was $1,000 for the three months ended September 30, 2014, compared to other expense of $3,000 for the three months ended September 30, 2013.
Net Loss
Our net loss was $4.9 million, or $0.08 per common share for the three months ended September 30, 2014, compared to a net loss of $3.4 million, or $0.08 per common share for the three months ended September 30, 2013.
Nine Months Ended September 30, 2014 and 2013
General and Administrative Expenses
General and administrative expenses decreased to $4.2 million for the nine months ended September 30, 2014, from $4.3 million for the nine months ended September 30, 2013. This decrease of 3% is primarily the result of bad debt expense of $763,000 associated with the determination that the note receivable and interest receivable from the sale of Adeona Clinical Laboratory was uncollectible for the quarter ended September 30, 2013, offset by supplemental compensation granted by our Board of Directors to our executive officers and increased stock-based compensation expense in 2014. The charge relating to stock-based compensation expense was $1.3 million for the nine months ended September 30, 2014, compared to $916,000 for the nine months ended September 30, 2013.
Research and Development Expenses
Research and development expenses increased to $9.2 million for the nine months ended September 30, 2014, from $3.8 million for the nine months ended September 30, 2013. This increase of 144% is primarily the result of increased program costs associated with expanded research, development and manufacturing activities in our pathogen-specific pipeline, including our C. diff, C-IBS and Pertussis programs. Research and development expenses also include a charge relating to non-cash stock-based compensation expense of $550,000 for the nine months ended September 30, 2014, compared to $286,000 for the nine months ended September 30, 2013.
Other Income (Expense)
Other income was $97,000 for the nine months ended September 30, 2014, compared to other expense of $27,000 for the nine months ended September 30, 2013.
Net Loss
Our net loss was $13.3 million, or $0.23 per common share for the nine months ended September 30, 2014, compared to a net loss of $8.1 million, or $0.18 per common share for the nine months ended September 30, 2013.
Liquidity and Capital Resources
We have financed our operations since inception primarily through proceeds from equity financings, corporate partnering license fees, laboratory revenues and miscellaneous equipment sales.
| 21 |
Our cash totaled $3.3 million as of September 30, 2014, a decrease of $11.3 million from December 31, 2013. During the nine months ended September 30, 2014, the primary use of cash was for working capital requirements and operating activities which resulted in a net loss of $13.3 million for the nine months ended September 30, 2014.
On October 10, 2014, we sold a total of 14,059,616 units at a purchase price of $1.47, with each unit consisting of one share of our common stock, and a warrant to purchase 0.5 shares of common stock in a registered direct offering for gross proceeds of $20.7 million and net proceeds of $18.9 million.
Our continued operations will primarily depend on our ability to raise additional capital from various sources including equity and debt financings, as well as, license fees from potential corporate partners, joint ventures and grant funding. Such additional funds may not become available on acceptable terms and there can be no assurance that any additional funding that we do obtain will be sufficient to meet our needs in the long term. We will continue to fund operations from cash on hand and through the similar sources of capital previously described. We can give no assurance that any additional capital that we are able to obtain will be sufficient to meet our needs.
Current and Future Financing Needs
We have incurred an accumulated deficit of $94.6 million through September 30, 2014. With the exception of the quarter ended September 30, 2010, we have incurred negative cash flow from operations since we started our business. We have spent, and expect to continue to spend, substantial amounts in connection with implementing our business strategy, including our planned product development efforts, our clinical trials, and our research and discovery efforts.
However, the actual amount of funds we will need to operate is subject to many factors, some of which are beyond our control. These factors include the following:
| · the progress of our research activities; | ||
| · the number and scope of our research programs; | ||
| · the progress of our preclinical and clinical development activities; | ||
| · the progress of the development efforts of parties with whom we have entered into research and development agreements; | ||
| · our ability to maintain current research and development licensing arrangements and to establish new research and development and licensing arrangements; | ||
| · our ability to achieve our milestones under licensing arrangements; | ||
| · the costs involved in prosecuting and enforcing patent claims and other intellectual property rights; and | ||
| · the costs and timing of regulatory approvals. |
We have based our estimate on assumptions that may prove to be wrong. We may need to obtain additional funds sooner or in greater amounts than we currently anticipate. Potential sources of financing include strategic relationships, public or private sales of our shares or debt and other sources. We may seek to access the public or private equity markets when conditions are favorable due to our long-term capital requirements. We do not have any committed sources of financing at this time, and it is uncertain whether additional funding will be available when we need it on terms that will be acceptable to us, or at all. If we raise funds by selling additional shares of common stock or other securities convertible into common stock, the ownership interest of our existing stockholders will be diluted. If we are not able to obtain financing when needed, we may be unable to carry out our business plan. As a result, we may have to significantly limit our operations and our business, financial condition and results of operations would be materially harmed.
| 22 |
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Synthetic Biologics, Inc. is a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and is not required to provide the information required under this item.
ITEM 4. CONTROLS AND PROCEDURES
| (a) | Evaluation of disclosure controls and procedures |
Pursuant to Rule 13a-15(b) under the Securities Exchange Act of 1934 (“Exchange Act”), the Company carried out an evaluation, with the participation of the Company’s management, including the Company’s Chief Executive Officer (“CEO”) and Chief Financial Officer (“CFO”), of the effectiveness of the Company’s disclosure controls and procedures (as defined under Rule 13a-15(e) under the Exchange Act) as of the end of the period covered by this report. Based upon that evaluation, the Company’s CEO and CFO concluded that the Company’s disclosure controls and procedures are effective as of September 30, 2014 to ensure that information required to be disclosed by the Company in the reports that the Company files or submits under the Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to the Company’s management, including the Company’s CEO and CFO, as appropriate, to allow timely decisions regarding required disclosure.
| (b) | Changes in Internal Control over Financial Reporting |
There has been no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act) that occurred during our fiscal quarter ended September 30, 2014, that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
| 23 |
None.
The following information updates, and should be read in conjunction with, the information disclosed in Part 1, Item 1A, “Risk Factors,” of our Annual Report on Form 10-K for the fiscal year ended December 31, 2013, which was filed with the Securities and Exchange Commission on March 31, 2014. There have been no material changes from the risk factors disclosed in our Form 10-K for the year ended December 31, 2013, other than as set forth below.
RISKS RELATING TO OUR BUSINESS
We will need to raise additional capital to operate our business.
With the exception of the three months ended June 30, 2010, we have experienced significant losses since inception and have a significant accumulated deficit. We expect to incur additional operating losses in the future and therefore expect our cumulative losses to increase. With the exception of the quarter ended June 30, 2010, and limited laboratory revenues from Adeona Clinical Laboratory, which we sold in March 2012, we have generated very minimal revenues. We do not expect to derive revenue from any source in the near future until we or our potential partners successfully commercialize our products. As of September 30, 2014, our accumulated deficit totaled approximately $94.6 million on a consolidated basis. Until such time as we receive approval from the FDA and other regulatory authorities for our product candidates, we will not be permitted to sell our products and therefore will not have product revenues from the sale of products. For the foreseeable future we will have to fund all of our operations and capital expenditures from equity and debt offerings, cash on hand, licensing fees and grants. If our current cash, cash equivalents and short-term investments are not sufficient to sustain our operations, we will need to seek additional sources of financing and such additional financing may not be available on favorable terms, if at all. If we do not succeed in raising additional funds on acceptable terms, we may be unable to complete planned preclinical and clinical trials or obtain approval of our product candidates from the FDA and other regulatory authorities. In addition, we could be forced to delay, discontinue or curtail product development, forego sales and marketing efforts, and forego licensing in attractive business opportunities. Any additional sources of financing will likely involve the issuance of our equity or debt securities, which will have a dilutive effect on our stockholders.
We cannot determine if or when we may generate any additional revenue from our sublicense with Meda AB due to recent developments in Europe.
On May 6, 2010, we entered into a sublicense agreement with Meda AB whereby we were given the right to receive certain milestone payments totaling $17.5 million (including an upfront payment of $2.5 million that was received in 2010), plus certain royalties on our flupirtine program. The successful achievement of the various milestones set forth in the sublicense agreement is not within our control and we are dependent upon Meda AB for achievement of such milestones. Meda AB has recently informed us that due to the decision of the European Medicines Agency (EMA) to limit the use of flupirtine for long-term pill and systemic use, it has postponed its planned fibromyalgia clinical trials in the U.S. Therefore, there can be no assurance given that the various milestones set forth in the sublicense agreement will be achieved by Meda AB, or that Meda AB will develop flupirtine for fibromyalgia in the U.S., Canada or Japan so that we could receive any additional milestone payments or royalties on sales in connection with the sublicense agreement. If the clinical trials do not recommence, we will not receive any additional milestone or royalty payments from the sublicense agreement with Meda AB.
RISKS RELATING TO OUR STOCK
Our principal stockholder has the ability to influence the vote on matters submitted to our stockholders and subsequent sales by such stockholder could adversely affect the market for our stock.
Through Intrexon Corporation and NRM VII Holdings I, LLC, Randal J. Kirk indirectly, beneficially owns approximately 12.3 million shares of our common stock. As a result, he will be able to exert influence over issues submitted to our stockholders, including the election of our Board of Directors and the vote on issues. The sale of a number of shares by our principal stockholder could have an adverse effect on the market for our stock and our share price.
Holders of our warrants issued in our October 2014 offering have no rights as common stockholders until they exercise their warrants and acquire our common stock and limited liquidity for the warrants.
Until the holders of the warrants we issued in our October 2014 offering acquire shares of our common stock by exercising their warrants, the holders have no rights as a stockholder with respect to the shares of common stock underlying their warrants. Upon exercise of the warrants, the holders will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date.
Because there is no established public trading market for the warrants we issued, the liquidity of the warrants is limited. We do no expect a market to develop, nor do we intend to apply to list the warrants on any securities exchange.
| 24 |
ITEM 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
None.
ITEM 3. DEFAULTS UPON SENIOR SECURITIES
None.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
None.
| 25 |
| 31.1 | Certification of Principal Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) * |
| 31.2 | Certification of Principal Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) * |
| 32.1 | Certification of Principal Executive Officer pursuant to Section 1350 of the Sarbanes-Oxley Act of 2002 * |
| 32.2 | Certification of Principal Financial Officer pursuant to Section 1350 of the Sarbanes-Oxley Act of 2002 * |
| EX-101.INS | XBRL Instance Document * | |
| EX-101.SCH | XBRL Taxonomy Extension Schema * | |
| EX-101.CAL | XBRL Taxonomy Extension Calculation Linkbase * | |
| EX-101.DEF | XBRL Taxonomy Extension Definition Linkbase * | |
| EX-101.LAB | XBRL Taxonomy Extension Label Linkbase * | |
| EX-101.PRE | XBRL Taxonomy Extension Presentation Linkbase * |
*Filed herewith.
| 26 |
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned.
| SYNTHETIC BIOLOGICS, INC. | ||||
| By: | /s/ Jeffrey Riley | |||
| Jeffrey Riley | ||||
| President and Chief Executive Officer | ||||
| (Principal Executive Officer) | ||||
| Date: November 14, 2014 | ||||
| By: | /s/ C. Evan Ballantyne | |||
| C. Evan Ballantyne | ||||
| Chief Financial Officer | ||||
| (Principal Financial Officer) | ||||
| Date: November 14, 2014 | ||||
| 27 |
| Term | Definition | |
| Adverse Event | Any adverse change in health or “side-effect” that occurs in a person participating in a clinical trial, from the time they consent to joining the trial until a pre-specified period of time after their treatment has been completed. | |
| BLA - Biologics License Application | An application in the U.S. through which biologic sponsors formally propose that the FDA approve a new biologic for sale and marketing. | |
| Clinical Study/Trial | A research study that is conducted to find out if a treatment or procedure is safe and/or effective in humans. | |
| Controlled Clinical Trial | A clinical study that compares patients receiving a specific treatment to patients receiving an alternate treatment for the condition of interest. The alternate treatment may be another active treatment, standard of care for the condition and/or a placebo (inactive) treatment. | |
| Double-blinded Study/Trial | Both the participant and the researcher are unaware of who is receiving the active treatment or the placebo. | |
| FDA - Food & Drug Administration | The U.S. government agency that ensures that medicines, medical devices, prescription medical foods and radiation-emitting consumer products are safe and effective. Authorized by Congress to enforce the Federal Food, Drug, and Cosmetic Act and several other public health laws, the agency monitors the manufacture, import, transport, storage, and sale of $1 trillion worth of goods annually. | |
| GMP - Good Manufacturing Practice | Regulations that require that manufacturers, processors, and packagers of drugs, medical devices, some food, and blood take proactive steps to ensure that their products are consistently produced, pure, and stable. GMP regulations require a quality approach to manufacturing, enabling companies to minimize or eliminate instances of contamination, mix-ups, and errors. | |
| Monoclonal Antibodies (mAbs) | Acting as the body's army, antibodies are proteins, generally found in the bloodstream, that provide immunity in detecting and destroying pathogens, such as viruses and bacteria and their associated toxins. | |
| IND - Investigational New Drug | An application in the U.S. submitted to the FDA for a new drug or biologic that, if allowed, will be used in a clinical trial. | |
| IRB - Institutional Review Board | A committee designated to formally approve, monitor, and review biomedical research at an institution involving human studies. Institutional Review Boards aim to protect the rights and welfare of the research subjects. | |
| NDA - New Drug Application | An application in the U.S. through which drug sponsors formally propose that the FDA approve a new pharmaceutical for sale and marketing. | |
| Open-label Clinical Study/Trial | A trial in which both the treating physician and the patient know they are receiving the experimental treatment. | |
| Phase 1 Clinical Trial | A Phase 1 trial represents an initial study in a small group of patients to primarily test for safety. |
| Phase 2 Clinical Trial | A Phase 2 trial represents a study in a larger number of patients to assess the safety and efficacy of a product. | |
| Phase 3 Clinical Trial | Phase 3 trials are initiated to establish safety and efficacy in an expanded patient population and at multiple clinical trial sites and are generally larger than trials in earlier phases of development. |
| 28 |
| Placebo | An inactive pill or liquid. Many studies compare an active drug to a placebo to determine whether any changes seen during the study can be attributed to the active drug. | |
| Principal Investigator | This is the study director who is ultimately responsible for the conduct of the study. | |
| Prospective Clinical Study/Trial | A clinical study/trial in which participants are identified and then followed throughout the study going forward in time. | |
| Protocol | A clinical study/trial’s plan - includes the schedule of tests, requirements for participation, procedures, and medications. | |
| Randomized Study/Trial | Participants in a study are assigned by chance to either one or more of the active treatment group(s) or the placebo group. | |
| Single-blinded Study/Trial | One party, either the participant or the researcher, does not know if the participant is taking the active treatment or the placebo. | |
| Study/Trial Coordinator | Staff member who is often the primary contact for research participants and coordinates their care and evaluations throughout the study. |
| 29 |