Attached files
| file | filename |
|---|---|
| EX-23.1 - INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRMS CONSENT - AutoGenomics, Inc. | exhibit231.htm |
As filed with the Securities and Exchange Commission on October 30, 2014
Registration No. 333-199197 .
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
______________________
Amendment No. 1 to
Form S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
_____________________
AutoGenomics, Inc.
(Exact name of registrant as specified in its charter)
______________________
Delaware | 3826 | 80-0252299 |
(State or other jurisdiction of incorporation or organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification No.) |
2980 Scott Street
Vista, California 92081
(760) 477-2248
(Address, including zip code and telephone number, including area code, of registrant’s principal executive offices)
______________________
Fareed Kureshy
President and Chief Executive Officer
AutoGenomics, Inc.
2980 Scott Street
Vista, California 92081
(760) 477-2248
(Name, address, including zip code and telephone number, including area code, of agent for service)
Todd A. Hentges Bingham McCutchen LLP 600 Anton Boulevard, 18th Floor Costa Mesa, California 92626-7653 (714) 830-0600 | copy to: | Charles S. Kim Sean M. Clayton David Peinsipp Cooley LLP 4401 Eastgate Mall San Diego, California 92121-1909 (858) 550-6000 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer | ¨ | Accelerated filer | ¨ | |
Non-accelerated filer | x | (Do not check if a smaller reporting company) | Smaller reporting company | ¨ |
THE REGISTRANT HEREBY AMENDS THIS REGISTRATION STATEMENT ON SUCH DATE OR DATES AS MAY BE NECESSARY TO DELAY ITS EFFECTIVE DATE UNTIL THE REGISTRANT SHALL FILE A FURTHER AMENDMENT WHICH SPECIFICALLY STATES THAT THIS REGISTRATION STATEMENT SHALL THEREAFTER BECOME EFFECTIVE IN ACCORDANCE WITH SECTION 8(a) OF THE SECURITIES ACT OF 1933, OR UNTIL THE REGISTRATION STATEMENT SHALL BECOME EFFECTIVE ON SUCH DATE AS THE COMMISSION, ACTING PURSUANT TO SAID SECTION 8(a), MAY DETERMINE.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any jurisdiction where such offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED , 2014
PRELIMINARY PROSPECTUS

Shares
Common Stock
$ per share
AutoGenomics, Inc. is offering shares of its common stock. This is our initial public offering, and no public market currently exists for our common stock. We expect the initial public offering price to be between $ and $ per share. We have applied to list our common stock on the NASDAQ Global Market under the symbol "AGMX."
We are an "emerging growth company," as defined under federal securities laws, and have elected to comply with certain reduced public company reporting requirements available to such companies.
Investing in our common stock involves a high degree of risk. Please read "Risk Factors" beginning on page 13.
Per Share | Total | |
Initial public offering price | $ | $ |
Underwriting discounts and commissions (1) | $ | $ |
Proceeds, before expenses, to us | $ | $ |
(1) We refer you to "Underwriting" beginning on page 128 of this prospectus for additional information regarding total underwriting compensation.
Delivery of the shares of common stock is expected to be made on or about , 2014. We have granted the underwriters an option for a period of 30 days to purchase, on the same terms and conditions set forth above, up to an additional shares of our common stock to cover over-allotments. If the underwriters exercise the option in full, the total underwriting discount payable by us will be $ and the total proceeds to us, before expenses, will be $ million.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Stifel | Canaccord Genuity | Cantor Fitzgerald & Co. |
The date of this prospectus is , 2014.

Not all products have received all necessary domestic or international regulatory approvals or clearances for commercial sale. The vast majority of products sold by AutoGenomics are offered for sale to allow for the collection of research data, and may only be used for clinical purposes by laboratories certified under the Clinical Laboratory Improvements Amendments of 1988 and that have incorporated the products into laboratory-developed tests pursuant to guidelines issued by the College of American Pathologists. The U.S. Food and Drug Administration has not adopted these guidelines and AutoGenomics is not permitted to represent these products as in vitro diagnostic products.
TABLE OF CONTENTS
____________________________
Through and including , 2014 (25 days after the date of this prospectus), all dealers that effect transactions in our common stock, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealer's obligation to deliver a prospectus when acting as an underwriter and with respect to their unsold allotments or subscriptions.
You should rely only on the information contained or incorporated by reference in this prospectus. Neither we nor the underwriters have authorized anyone to provide you with information different from, or in addition to, the information contained in this prospectus. We are offering to sell shares of our common stock and seeking offers to buy shares of common stock only in jurisdictions where offers and sales are permitted. The information contained in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or of any sale of our common stock.
In this prospectus "we," "us," "our," "Company" and "AutoGenomics" refer to AutoGenomics, Inc. Unless otherwise indicated, all information in this prospectus assumes no exercise of the underwriters' over-allotment option.
PROSPECTUS SUMMARY
This summary highlights certain information contained in this prospectus. Because this is only a summary, it does not contain all of the information that may be important to you. For a more complete understanding of the information that you may consider important in making your investment decision, we encourage you to read this entire prospectus. Among the other information in this prospectus, you should carefully consider the information set forth under the heading "Risk Factors" and our financial statements and accompanying notes included elsewhere in this prospectus. Unless the context requires otherwise, the words "we," "us," "our," "Company" and "AutoGenomics" refer to AutoGenomics, Inc.
Our Company
We are a commercial stage molecular diagnostics company offering an innovative and proprietary technology platform to clinical reference laboratories, specialty clinics and hospital laboratories. Our platform consists of a family of multiplexing INFINITI analyzers, an extensive and expanding menu of genetic test panels, and proprietary microarrays, reagent modules and other related consumables. Our INFINITI analyzers, which include the INFINITI, the INFINITI PLUS and the recently introduced INFINITI HIGH THROUGHPUT SYSTEM, or INFINITI HTS, are easy to use and automate a number of the discrete processes of genetic analysis with minimal manual intervention, which reduces our customers' need for multiple, specialized instruments, and offer a variety of throughput capabilities together with a demonstrated high level of accuracy and reproducibility. Our genetic test panels are focused on large and growing markets primarily in the areas of personalized medicine, women's health, infectious diseases and genetic disorders. Genetic tests are performed on our INFINITI analyzers using our proprietary BioFilmChip microarrays, related Intellipac Reagent Management Modules and other related consumables.
Our genetic tests provide tools to physicians, clinicians and other health care providers to improve detection, treatment and monitoring of a broad spectrum of diseases and conditions. We currently offer 62 test panels for use on our analyzers and have 16 additional test panels in development. In the area of personalized medicine, we offer test panels in pain management, cardiovascular health assessment, oncology and mental health. These test panels offer customers the ability to identify a patient's genetic information, which can assist in improving drug efficacy, identifying non-responders and ultra-, normal- and poor-metabolizers and reducing adverse patient response. In the area of women's health, we offer test panels in cervical cancer, sexually transmitted diseases, or STDs, and vaginal infections. These test panels identify a wide spectrum of specific organisms simultaneously, which can eliminate the need for laboratories to perform multiple tests. The depth and breadth of our menu of test panels, all of which are designed to run on any of our INFINITI analyzers, allows laboratories to utilize laboratory space, labor and capital investment efficiently and conduct genetic tests in less time, thereby improving laboratory economics.
The proprietary design of our platform also allows us to introduce new and enhanced test panels to our menu of genetic tests without modifying our INFINITI analyzers. Based on our own research and customer demand, we intend to continue to increase the number of test panels offered in each of our target market segments, which we believe will further increase the utility of our platform to our customers. For example, we are developing several new test panels to guide the dosing and selection of statin drug therapies, to aid healthcare providers in the treatment of drug addiction and to assess risk and help guide therapy in the treatment of age-related macular degeneration. These test panels are currently in alpha trials and we expect to commercialize these test panels in 2015.
We launched our INFINITI HTS during February 2014 to meet the demand for high test-volume capabilities from our customers, particularly in the areas of personalized medicine and women's health. Our INFINITI HTS is a scalable, automated system comprised of three separate operating modules that provides flexible configuration to maximize workflow efficiency, and has the capacity to process up to 6,912 patient results per day with a single laboratory technician utilizing the same consumables as our other INFINITI analyzers. We believe our INFINITI HTS has the highest processing and throughput capacity of any molecular diagnostics system available in our target markets today. Additionally, for customers who seek a
1
fully automated and integrated solution, and have lower throughput requirements, we offer INFINITI and INFINITI PLUS analyzers designed to operate on a "load and go" basis, in which a technician only needs to load prepared samples along with the test-specific consumables into the analyzer to generate test results.
The following table illustrates the test capabilities of our primary INFINITI analyzers:
Instrument | Capacity per run (1) | Patient results (1) |
INFINITI HTS | 384 samples | up to 6,912 per day |
INFINITI PLUS | 48 samples | up to 192 per day |
INFINITI | 24 samples | up to 96 per day |
(1) Figures assume the use of our four-patient multiple patient array test panel over a 24-hour period.
In late 2013, as we prepared to launch our INFINITI HTS, we began operating our CLIA-certified laboratory to complement and support our efforts to attract customers entering the molecular diagnostics market, and potential INFINITI HTS customers in particular. Our laboratory services allow our customers to more quickly offer new or additional tests during the period when they are awaiting delivery, installation, training and validation of an INFINITI analyzer. Our CLIA-certified laboratory also provides new and existing customers with an alternative source of capacity to assist with their overflow testing needs. In addition, our laboratory enhances our product development efforts and allows us to offer hands-on training opportunities to our customers. Since the launch of our INFINITI HTS, many of these customers have utilized the services of our laboratory to supplement and facilitate sales of medium- and high-volume test products to their physicians, clinicians and healthcare providers.
We commenced sales of our INFINITI HTS in February 2014 through a small direct sales force in the United States. Our net revenue was $18.7 million for the nine months ended September 30, 2014, as compared to $13.3 million for the nine months ended September 30, 2013. Excluding net revenue from one historically large customer, Natural Molecular Testing Corporation, or NMTC, our net revenue for the nine months ended September 30, 2013 was $8.2 million. We had no revenue from NMTC in the nine months ended September 30, 2014. Excluding net revenue from NMTC, our net revenue grew 130% for the nine months ended September 30, 2014 as compared to the same period in 2013. Our net loss was $1.0 million for the nine months ended September 30, 2014, as compared to $6.8 million for the nine months ended September 30, 2013. Our accumulated deficit as of September 30, 2014 was $104.2 million.
For a comparison of our net revenue, including net revenue from NMTC and excluding net revenue from NMTC, by period for the nine months ended September 30, 2014 and 2013, and the years ended December 31, 2013 and 2012, please see "Management's Discussion and Analysis of Financial Condition and Results of Operations - Results of Operations - As Adjusted Financial Information for the Nine Months Ended September 30, 2014 and 2013 and Years Ended December 31, 2013 and 2012 (unaudited)."
Our Market Opportunity
Molecular diagnostics, or MDx, refers to the detection of DNA and RNA and their variations and mutations, and the use of such information to determine a patient’s susceptibility to disease, diagnose disease and infection, evaluate response to therapy and drug efficacy, identify non-responders and ultra-, normal- and poor-metabolizers and reduce adverse events. Frost & Sullivan, a market research company, estimated in 2012 that the MDx market will reach $6.2 billion in the United States during 2014 and forecasted a compound annual growth rate for the market in excess of 11%. The Centers for Medicare and Medicaid Services of the Department of Health and Human Services, or CMS, estimated in June 2014 that there were more than 5,900 independent clinical reference laboratories and specialty clinics and more than 8,900 hospital-based laboratories in the United States. We believe that less than 10% of these laboratories are currently performing MDx testing, and that we have an opportunity to expand our customer base by enabling more laboratories to participate in this growing market.
2
Our Target Markets
Personalized medicine
The optimal matching of treatment options to a patient's specific genetic profile has emerged as an important trend in medicine. More targeted and effective pharmacogenomic-based treatments have the potential to improve healthcare outcomes and lower healthcare costs, which we believe will lead to increased use of genetic testing. Many patients do not currently achieve the best possible outcome with the first drug that they are prescribed in treatment. Studies have linked the variation in efficacy of a patient's response to a prescribed drug treatment to patient-specific differences in the genes that code for drug-metabolizing enzymes, drug transporters or drug targets, which can be identified through the use of genetic testing. Our target markets in personalized medicine include:
Pain management. More than 116 million adult Americans suffer from acute or chronic pain each year. Studies have shown that only 58% of patients taking prescription medication reported pain relief. Additionally, approximately 80% of post-operative patients experience adverse events from pain medications. As of October 24, 2014, we offered 11 test panels in the area of pain management, which detect multiple mutations in specific drug-metabolizing genes and can enable physicians to select optimal therapies for pain management.
Cardiovascular health assessment. According to a report from the World Health Organization, or WHO, cardiovascular diseases were responsible for 30% of global deaths in 2008. The WHO estimates that by 2030 23.6 million people will die annually from some form of cardiovascular disease. As of October 24, 2014, we offered 20 test panels in the area of cardiovascular health assessment, five of which have received U.S. Food and Drug Administration, or FDA, 510(k) clearance, including our Plavix responder and Warfarin sensitivity test panels.
Mental health. Drugs associated with mental health, including drugs focused on central nervous system disorders, and treatment of depression and anti-psychotic therapy, represent a major component of overall pharmaceutical sales. According to the Centers for Disease Control and Prevention, or CDC, as much as 11% of the U.S. population is taking antidepressants at a given time. Despite this prevalence, approximately 30 to 40% of patients do not respond to the first medication prescribed. The resulting trial-and-error process delays effective treatment for patients and increases healthcare costs. Genetic testing provides the information to allow physicians to select the most appropriate drug or combination of drugs specific to the patient's genetic makeup. As of October 24, 2014, we offered 12 test panels in the area of mental health, which identify mutations in drug-metabolizing genes that can enable healthcare providers to select optimal therapies.
Women's health
According to the CDC, approximately 79 million Americans are infected with Human papillomavirus, or HPV, and approximately 14 million become infected each year. Current regulations from the U.S. Preventive Services Task Force recommend that MDx HPV tests for women be utilized in conjunction with cytology (Pap test) in standard five year intervals. The CDC estimates that there are nearly 20 million new cases of STDs each year, costing the nation approximately $16 billion in healthcare costs annually. As of October 24, 2014, we offered 19 test panels in the area of women's health. Key tests in this segment include four test panels for HPV testing and nine test panels designed to identify various microorganisms related to STDs and vaginal infections. We believe our women's health test panels are the most comprehensive menu of test panels in the market currently.
Other markets
We also target the markets of: (i) oncology to help manage chemotherapy treatments for breast, colorectal, lung, melanoma and thyroid cancers; (ii) infectious diseases for detection of influenza, nontuberculous mycobacteria, respiratory viruses and tuberculosis; and (iii) genetic disorders including, but not limited to, tests for identification of carriers of gene mutations associated with Bloom disease, Canavan disease, cystic fibrosis and Familial Mediterranean fever.
3
Limitations of Traditional Testing Methods
Traditional testing methods have a number of drawbacks and limitations, including:
• | Throughput limitations. Traditional systems in the market have throughput constraints and typically process a limited number of patient samples simultaneously. |
• | High operating cost per reportable result. Many existing systems require specialized personnel and training, involve time-consuming protocols, require supplementary, discrete instrumentation and result in high labor costs. |
• | Limited testing menu. Many existing MDx systems have limited test menus, leading to a need for a laboratory to purchase many different systems, all which require their own training, use and maintenance, as well as their own laboratory bench space and inventory. |
• | Inability to multiplex. Many existing systems are only able to examine one biomarker at a time, and, in order to make a diagnosis, the laboratory must perform repeated tests on a sample. Serial testing is expensive, time-consuming and requires higher sample volumes. |
• | Limited automation. Many existing systems automate only certain steps in the MDx testing process and do not enable testing of multiple patient samples in a single microarray. |
• | Need for specialized labor. Many existing systems require specialized laboratory technicians and in some cases specialized training, adding to labor costs. |
Our Solution
Our platform has been designed to enable a broad range of clinical reference laboratories, specialty clinics and hospital laboratories to start performing, or to more cost-effectively perform, genetic testing across a wide range of throughput levels, which we believe will drive adoption and use of our platform as well as expand the potential of the MDx testing market. Our platform has a number of key advantages, including:
• | Significantly higher throughput and better workflow. Our broad offering of INFINITI analyzers is designed to address our target customers' varied throughput and workflow requirements. Using one INFINITI HTS, laboratories are capable of producing up to 6,912 patient results per day. |
• | Improved laboratory economics. Our INFINITI and INFINITI PLUS eliminate the need for complex protocols and manual intervention once a test is initiated, which is intended to improve the laboratory's economics by simplifying workflow and reducing the need for highly skilled technicians. Our INFINITI HTS allows individual components to remain active during the workflow process, which can significantly improve throughput time and reduce costs. |
• | Broad menu of tests. We currently offer 62 test panels as part of our platform and have an additional 16 test panels in development. We believe that this represents the broadest available test menu on a single system in the market today. |
• | Multiple patient array technology. Our proprietary multiple patient array, or MPA, technology is designed to test up to eight distinct patient samples on a single microarray. |
• | Ability to multiplex. Many diseases and patient responses to therapy are caused by multiple genetic mutations that necessitate testing for multiple biomarkers to diagnose those diseases or to predict or monitor therapy response. |
• | Increased accuracy of results. By reducing the risk of human error and contamination, we believe that our platform can provide more accurate and more reproducible test results compared to other less automated systems. |
Our Products and Technology
Our platform is comprised of five key technological innovations: (1) our INFINITI analyzers; (2) our multiplexing test format; (3) our BioFilmChip microarrays; (4) our multiple patient array technology; and (5) our Intellipac Reagent Management Modules.
4
Our INFINITI analyzers
Our platform includes a family of analyzers, each of which is designed to address customer-specific needs based on the customer's workflow and throughput requirements, and automate the discrete processes in genetic analysis. Our INFINITI HTS is a scalable, multiplexing, random access, microarray-based system comprised of three independent operating modules. Our INFINITI and INFINITI PLUS analyzers integrate and automate the discrete processes of sample handling, reagent management, hybridization, detection, results analysis and reporting in a self-contained system. All of our INFINITI analyzers use similar underlying proprietary technology and the same consumables to generate consistent patient results presented in an identical format.
Our multiplexing test methods and multiple patient microarray
Our multiplexing technology allows our INFINITI analyzers to detect multiple biomarkers across multiple genes at the same time on a single microarray, eliminating the need to process multiple tests separately. For example, our pain management panel can detect multiple mutations associated with six different genes to determine the efficacy, toxicity and dosing of specific drugs. In addition, our test panel for HPV detects and genotypes all 14 high-risk types of HPV simultaneously on a single microarray from a single sample. Our proprietary MPA technology enables our INFINITI analyzers to process multiple patient samples on a single microarray, reducing our costs, increasing our test production capacity, and improving our customers' laboratory economics while increasing their throughput. For example, our MTBC OCTA (drug-resistant tuberculosis test panel) is designed to test up to eight patient samples on a single microarray, increasing throughput for our laboratory customers by up to 700%, while reducing their operating cost per reportable result by up to 87%, as compared to our single patient microarrays.
Our BioFilmChip microarrays and Intellipac Reagent Management Modules
Our proprietary BioFilmChip microarrays utilize our proprietary and patented manufacturing processes, and can be printed with up to 1,024 individual features of biochemical sensors, or biomarkers, depending on the requirements of the test. Our assay-specific Intellipac Reagent Management Modules provide the proprietary reagent used to complete our genetic tests. The reagent module is designed to communicate all relevant information about a test to the INFINITI analyzer without any intervention from the operator, saving time by automatically recording all pertinent test details, and reducing the possibility for human errors which could occur in manual systems by eliminating contamination risk.
Sales and Marketing
Our goal is to achieve broad adoption of our INFINITI analyzers in the market, and drive utilization to generate sales of our high-margin testing consumables. Our current sales and marketing strategy is focused on the broad introduction of our INFINITI HTS to customers in high-volume testing market segments, particularly personalized medicine and women's health, and emphasizes the high throughput testing capacity, ease of use, enhanced workflow, increased efficiency, low cost, high quality and consistent results of our platform as well as the potential versatility afforded by our broad menu of genetic test panels.
In the United States, we offer our family of INFINITI analyzers either through direct sales, monthly rental plans or our Reagent Access Plan, or RAP. Under the RAP, an INFINITI analyzer is placed at the customer’s location at no initial cost to the customer, and is subject to contractual minimum consumables purchasing thresholds. We have established a small direct sales force that is regionally deployed and performs market development as well as account management. We plan to significantly expand our sales force within the next 12 months.
Internationally, we market and sell our platform through our network of distributors. We have established 12 distributor relationships to market our platform in 23 countries outside the United States. We plan to expand our distributor network outside the United States to enter new markets and capitalize on growing international demand.
5
Manufacturing and Research and Development
We manufacture our family of INFINITI analyzers, BioFilmChips and Intellipac Reagent Management Modules at our facility in Vista, California. Our facility is registered with the FDA as a Medical Device Manufacturing Establishment. We have established a quality system that is in compliance with the FDA's Quality Systems Regulations and we have obtained ISO 13485:2003 certification for our facility. Our research and development efforts focus on developing additional genetic tests and INFINITI analyzers, improving or enhancing current tests and INFINITI analyzers, and supporting clinical trials for certain genetic tests.
Our Strategy
Our objective is to become a leading provider of genetic tests to clinical reference laboratories, specialty clinics and hospital laboratories in multiple growing market segments. One key element of our strategy is to focus on high-volume customers, particularly in the areas of personalized medicine and women’s health. We plan to continue to expand into other market segments through the development of additional test panels.
To achieve our objectives, we intend to:
• | Increase placements of our recently launched INFINITI HTS analyzer and expand penetration in the highest-volume testing market segments. |
• | Target molecular diagnostics laboratories with high potential utilization of our INFINITI and INFINITI PLUS analyzers. |
• | Develop and launch test panels in new areas and enhance our current test panels. |
• | Significantly expand our domestic sales force, increase marketing expenditures and expand international distribution. |
• | Pursue additional regulatory clearances, approvals and certifications for products and facilities, as necessary. |
• | Utilize our CLIA-certified laboratory to enhance and complement our product offerings. |
Government Regulation
We have received FDA 510(k) clearance for our INFINITI and INFINITI Plus analyzers and five of our genetic test panels: CYP450 2C19, Factor II, Factor V, Factor II/V and Warfarin. Products for which we have received 510(k) clearance accounted for 35%, 18% and 14% of our net revenue for the nine months ended September 30, 2014, and the years ended December 31, 2013 and 2012, respectively. We are currently in clinical trials collecting data to support a submission for 510(k) clearance for our INFINITI HTS, both of our CYP450 2C19 Plus test panels and all three of our CYP450 2D6 test panels. We intend to use a portion of the proceeds of this offering to conduct additional clinical trials for the collection of data, and to submit for 510(k) clearance, for an additional seven of our test panels that, together with our existing and in-process 510(k) cleared test panels, made up approximately 80% of consumables revenue for the nine months ended September 30, 2014. We expect to conduct additional clinical trials for the collection of data, and to submit for 510(k) clearance, for test panels based on a variety of factors, including:
• | the regulatory environment for the use of genetic tests, in particular the FDA's requirements and limitations on marketing RUO tests, which may not be marketed as in vitro diagnostic products; |
• | the demand by our existing and target customers for particular genetic tests that have received regulatory approvals or clearances; |
• | the competitive environment for the use of genetic tests that have received regulatory approvals or clearances versus similar tests that have not; and |
• | the size of the available market for the particular test, given the significant expense and time required to obtain regulatory approvals or clearances. |
6
Internationally, we have obtained a Conformité Européenne, or CE, mark for our INFINITI and INFINITI PLUS analyzers and a total of 24 of our tests. This designation is supported by completed clinical and validation studies that demonstrate the analytical performance of each CE marked test. The CE mark facilitates the marketing and sale of our CE marked analyzers and tests in the European Union and the European Economic Area as well as certain other international markets.
Our test panels that are not 510(k) cleared are offered for sale in the United States under the RUO designation. These RUO tests are labeled "For Research Use Only. Not for use in diagnostic procedures." as required by FDA regulations. Sales of these test panels represented 65%, 82% and 86% of our net revenue for the nine months ended September 30, 2014, and the years ended December 31, 2013 and 2012, respectively.
RUO test panels may only be used in the United States for clinical purposes by laboratories and other facilities certified under CLIA that have incorporated these products into their laboratory developed tests, or LDTs, pursuant to guidelines issued by the College of American Pathologists. We believe that nearly all of our RUO product sales are incorporated into LDTs. In order to develop an LDT utilizing our products, these certified laboratories and other facilities must develop and validate a test protocol that includes specimen collection, DNA extraction, polymerase chain reaction, or PCR, amplification, hybridization and detection, and data analysis, interpretation and reporting. Our products provide components that can be used by these certified laboratories and other facilities for the PCR amplification, hybridization and detection portions of these LDTs. We sell each of these components individually, as ordered by the customer in its discretion, and not as a kit or system. The validation process engaged in by these certified laboratories and other facilities can involve validation of the sample collection and extraction process, establishing limits of detection and analytical sensitivity, testing for specificity and cross-reactivity, including interfering substances, validation for assay accuracy, precision and reproducibility, and establishing reportable ranges of test results for the test system and reference values that will be measured against as controls. This validation process also requires verifying the result from the LDT against known standard samples or the results of a high-standard laboratory testing method such as sequencing, and can involve the testing of a large number of patient samples. This process may take from several weeks to several months or more to complete, and thus requires a significant investment by the customer.
We believe that all sales of our RUO products in the United States are to customers that are either certified in the manner described above and have incorporated our products into their LDTs, or that use such products for research only. We are not permitted to represent our RUO products as in vitro diagnostic products. We therefore train our personnel to only market these products to laboratories for research or investigational use in the collection of research data, and to not promote any off-label uses of our products.
We believe that we are in compliance with existing FDA rules and regulations governing our business, including those governing the marketing and sale of RUO tests; however, a significant change in existing laws, or their enforcement, may require us to change our business model or our business practices to maintain compliance with these laws. For instance, in June 2011, the FDA issued a Draft Guidance entitled "Commercially Distributed In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only: Frequently Asked Questions," and in November 2013 issued a guidance document entitled "Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only," or the RUO Guidance, which highlights the FDA's interpretation that distribution of RUO products with any labeling, advertising or promotion that suggests that clinical laboratories can validate the test through their own procedures and subsequently offer it for clinical diagnostic use as an LDT is in conflict with RUO status. The RUO Guidance further articulates the FDA's position that any assistance offered in performing clinical validation or verification, or similar specialized technical support, to clinical laboratories, is in conflict with RUO status. The FDA has generally exercised its enforcement discretion to not enforce applicable regulations with respect to LDTs. However, the FDA has repeatedly indicated since 2010 that it intends to reconsider its policy regarding enforcement and to begin drafting an oversight framework for such tests, and most recently, on July 31, 2014, the FDA notified the U.S. Congress of the FDA's intent to modify its enforcement discretion approach to LDTs in a risk-based manner. If the RUO Guidance were to be enforced, it would limit our marketing of RUO test panels to general discovery laboratories and require us to seek FDA clearance for
7
our RUO test panels, which may require significant time and investment on our part and may reduce our revenues or increase our costs and adversely affect our business, prospects, results of operations or financial condition.
Risks Affecting Us
Our business is subject to numerous risks, as more fully described in the section entitled "Risk Factors" elsewhere in this prospectus, including the following:
• | Financial risks. We have a history of net losses and negative cash flows with an accumulated deficit of $104.2 million as of September 30, 2014, and may not be able to achieve or maintain profitability in the current fiscal year or in future fiscal periods. |
• | Business risks. There is limited information available to evaluate our business, particularly with respect to our recently launched INFINITI HTS, and we face significant competition. |
• | Product and customer risks. Our financial results depend on commercial acceptance of our platform and tests and the development of additional tests. |
• | Regulatory risks. The majority of our net revenue is derived from the sale of products designated for research use only, or RUO; the FDA announced July 31, 2014 its intention to modify its enforcement discretion approach to laboratory developed tests, which may adversely affect our ability to sell our RUOs; violations of these regulations by us could significantly limit our ability to sell our products to our target customers, or otherwise require us to obtain regulatory approvals for our products at considerable time and expense. |
• | Legal risks. Our success depends in part on our ability to operate without infringing or misappropriating the proprietary rights of others, our ability to own or license patents that are adequate to reduce competition and our ability to license intellectual property from third parties for certain tests and manufacturing processes needed for our business. |
Company Information
We were incorporated as Neuron Technologies, Incorporated in California in April 1999, and changed our name to AutoGenomics, Incorporated in August 2000. We subsequently changed our name to AutoGenomics, Inc. in October 2002. We reincorporated in Delaware in November 2008. Our principal executive offices are located at 2980 Scott Street, Vista, California 92081. Our telephone number is (760) 477-2248. Our website address is www.autogenomics.com. Information contained in or that can be accessed through our website is not incorporated by reference into this prospectus and should not be considered to be part of this prospectus.
INFINITI, BioFilmChip, Intellipac and QMatic are our trademarks. All other service marks, trademarks and trade names referred to in this prospectus are the property of their respective owners. Some of our trademarks are referred to in this prospectus from time to time, solely for convenience, without their associated ® and ™ symbols. We retain, however, our rights to these trademarks notwithstanding such presentation, and we will assert, to the fullest extent under applicable law, our rights to our trademarks.
8
Implications of Being an Emerging Growth Company
We are an "emerging growth company," as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. As an emerging growth company, we are eligible to comply with less stringent disclosure requirements than those applicable to larger, more established companies. We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) following the fifth anniversary of the completion of this offering, (b) in which we have total annual gross revenue of at least $1.0 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
So long as we qualify as an emerging growth company, we will, among other things, be exempted from (i) the auditor attestation requirement in the assessment of internal controls over financial reporting of Section 404(b) of the Sarbanes-Oxley Act; (ii) various existing and forthcoming executive compensation related disclosures, for example: "say-on-pay," "pay-for-performance" and "CEO pay ratio;" (iii) any rules that might be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotation or supplemental auditor discussion and analysis reporting; and (iv) having to solicit advisory say-on-pay, say-on-frequency and say-on golden-parachute shareholder votes on executive compensation under Section 14A of the Securities Exchange Act of 1934, as amended, and will be permitted to comply with the SEC's detailed executive compensation disclosure requirements on the same basis as a smaller reporting company.
In addition, under Section 107(b) of the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
9
The Offering
Common stock to be offered by us | shares |
Common shares to be outstanding immediately after this offering | shares |
Over-allotment option | We have granted the underwriters an option for 30 days from the date of this prospectus to purchase up to additional shares of our common stock at the initial public offering price to cover over-allotments. |
Use of proceeds | We anticipate that we will use the net proceeds from this offering as follows: (i) to fund capital expenditures for expansion of manufacturing capabilities, (ii) to expand our sales and marketing efforts, (iii) to fund research and development, including regulatory efforts, (iv) to satisfy all of our outstanding notes payable and certain of our accounts payable and (v) the balance for working capital and general corporate purposes. |
NASDAQ Global Market listing | We have applied to list our common stock on the NASDAQ Global Market under the symbol "AGMX." |
Risk factors | Investing in our common stock involves a high degree of risk. You should carefully read and consider the information set forth under the heading "Risk Factors" and all other information set forth in this prospectus before deciding to invest in our common stock. |
The number of shares of common stock outstanding after this offering is based on 11,722,251 shares of common stock outstanding as of October 24, 2014, after giving effect to the conversion of our convertible preferred stock into 7,974,932 shares of common stock immediately prior to the completion of this offering and excludes the following:
• | 1,469,055 shares of common stock issuable upon exercise of options outstanding at a weighted average exercise price of $6.25 per share; |
• | 282,221 and 82,500 shares of common stock reserved for future issuance under our 2008 Equity Incentive Award Plan and 2008 Employee Stock Purchase Plan, respectively; and |
• | warrants to purchase 3,892,249 shares of common stock at a weighted average exercise price of $6.04 per share. |
Unless otherwise indicated, all information in this prospectus assumes an initial public offering price of $ per share. The conversion into common stock of our Series C, Series E and Series NC Convertible Preferred Stock is predicated on the offering referred to in this prospectus resulting in net proceeds to us of $25 million or more.
Except as otherwise indicated, all information in this prospectus assumes:
• | no issuance of any options under our 2008 Equity Incentive Award Plan and no exercise of any outstanding warrants or options, in each case after the respective dates as of which information is presented; and |
• | no exercise by the underwriters of their over-allotment option. |
10
Summary Financial Data
The following tables summarize our financial data as of the periods indicated below. We have derived the following summary statement of operations data for the years ended December 31, 2013 and 2012 from our audited financial statements included elsewhere in this prospectus, which have been prepared in accordance with U.S. generally accepted accounting principles. The statements of operations data for the nine months ended September 30, 2014 and 2013, and the balance sheet data as of September 30, 2014, are derived from our unaudited financial statements included elsewhere in this prospectus. We have prepared unaudited financial information on the same basis as the audited financial statements and have included, in our opinion, all adjustments, consisting only of normal recurring adjustments, that we consider necessary for a fair presentation of the financial information set forth in those statements.
Our historical results are not necessarily indicative of the results that may be expected in the future, and our interim results are not necessarily indicative of the results expected for the full fiscal year. The summary historical financial data should be read together with "Management's Discussion and Analysis of Financial Condition and Results of Operations" and the financial statements included elsewhere in this prospectus.
Nine Months Ended September 30, | Years Ended December 31, | ||||||||||||||
2014 | 2013 | 2013 | 2012 | ||||||||||||
(in thousands, except share and per share data) | |||||||||||||||
(unaudited) | |||||||||||||||
Statements of Operations Data: | |||||||||||||||
Net revenue | $ | 18,725 | $ | 13,273 | $ | 18,083 | $ | 18,409 | |||||||
Cost of sales and services | 6,645 | 5,799 | 7,758 | 7,147 | |||||||||||
Gross profit | 12,080 | 7,474 | 10,325 | 11,262 | |||||||||||
Operating expenses: | |||||||||||||||
Research and development | 1,540 | 1,715 | 2,306 | 2,355 | |||||||||||
General and administrative | 3,308 | 7,210 | 8,302 | 4,715 | |||||||||||
Sales and marketing | 5,334 | 1,898 | 2,998 | 2,269 | |||||||||||
Total operating expenses | 10,182 | 10,823 | 13,606 | 9,339 | |||||||||||
Income/(loss) from operations | 1,898 | (3,349 | ) | (3,281 | ) | 1,923 | |||||||||
Interest expense, net | (1,361 | ) | (1,186 | ) | (1,820 | ) | (2,602 | ) | |||||||
Gains/(loss) on extinguishment of debt | (1,872 | ) | (140 | ) | (140 | ) | (1,996 | ) | |||||||
Other income/(expense), net | (5 | ) | (2,095 | ) | (1,877 | ) | 5 | ||||||||
Change in the fair value of stock warrant liabilities | 375 | 5 | 46 | 163 | |||||||||||
Net loss | $ | (965 | ) | $ | (6,765 | ) | $ | (7,072 | ) | $ | (2,507 | ) | |||
Net loss from continuing operations per common share, basic and diluted | $ | (0.26 | ) | $ | (2.22 | ) | $ | (2.21 | ) | $ | (0.95 | ) | |||
Weighted average shares used in per share amounts | 3,743,084 | 3,042,029 | 3,200,286 | 2,652,275 | |||||||||||
11
As of September 30, 2014 | ||||||||||
Actual | Pro forma (1) | Pro forma as adjusted (2) | ||||||||
(unaudited and in thousands) | ||||||||||
Balance Sheet Data: | ||||||||||
Cash and cash equivalents | $ | 556 | $ | 556 | ||||||
Current assets | 12,719 | 12,719 | ||||||||
Total assets | 15,372 | 15,372 | ||||||||
Total debt (3) | 18,491 | 18,491 | — | |||||||
Convertible preferred stock (4) | 53,165 | — | — | |||||||
Total stockholders' equity/(deficit) | (75,995 | ) | (22,830 | ) | ||||||
(1) | On a pro forma basis after giving effect to the conversion of all outstanding shares of convertible preferred stock into common stock, which will occur immediately prior to the completion of this offering. |
(2) | On a pro forma as adjusted basis after giving effect to (i) the conversion of all outstanding shares of convertible preferred stock into common stock, which will occur immediately prior to the completion of this offering, (ii) the sale of shares of our common stock in this offering at an assumed initial offering price of $ per share, the midpoint of the range on the cover page of this prospectus, and after deducting estimated underwriting discounts and commissions and our estimated offering expenses and (iii) the application of $21.2 million of the net proceeds of this offering to repay the principal and accrued interest as of September 30, 2014 under our outstanding promissory notes. A $1.00 increase or decrease in the assumed initial public offering price of $ per share would increase or decrease, as applicable, the net proceeds to us from this offering by approximately $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and our estimated offering expenses. |
(3) | Amounts include principal only of promissory notes payable as of September 30, 2014. |
(4) | Our convertible preferred stock has been classified as temporary equity on our balance sheets instead of in stockholders' equity/(deficit) due to the possibility of the occurrence of certain change in control events that are outside of our control, including our sale or transfer of control, which trigger the rights of holders of the convertible preferred stock to force redemption. Accordingly, these shares are considered contingently redeemable. We have adjusted the carrying values of the convertible preferred stock to their liquidation values at each period end. |
As Adjusted Financial Information for the Nine Months Ended September 30, 2014 and 2013 and Years Ended December 31, 2013 and 2012 (unaudited)
The unaudited as adjusted financial data below sets forth the net revenue for the nine-month periods ended September 30, 2014 and 2013 and the years ended December 31, 2013 and 2012, excluding all revenue attributable to NMTC (in thousands).
Nine Months Ended September 30, | Years Ended December 31, | ||||||||||||||||||||||
2014 | 2013 | Change | 2013 | 2012 | Change | ||||||||||||||||||
Net revenue (as reported) | $ | 18,725 | $ | 13,273 | $ | 5,452 | $ | 18,083 | $ | 18,409 | $ | (326 | ) | ||||||||||
Net revenue from NMTC | — | 5,116 | (5,116 | ) | 5,116 | 7,749 | (2,633 | ) | |||||||||||||||
As adjusted net revenue | $ | 18,725 | $ | 8,157 | $ | 10,568 | $ | 12,967 | $ | 10,660 | $ | 2,307 | |||||||||||
12
RISK FACTORS
Before deciding to invest in our common stock, you should carefully consider each of the following risk factors and all of the other information set forth in this prospectus. The following risks and the risks described elsewhere in this prospectus, including in the section entitled "Management's Discussion and Analysis of Financial Condition and Results of Operations," could materially harm our business, financial condition, future results and cash flow. If that occurs, the trading price of our common stock could decline, and you could lose all or part of your investment. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently believe to be immaterial may also adversely affect our business.
Financial Risks
We have a history of operating losses and negative cash flows and may not be able to achieve or maintain profitability.
We have incurred substantial costs to develop our technology since our inception in 1999. As of September 30, 2014, we had an accumulated deficit of $104.2 million. We expect to continue to spend substantial financial and other resources on introducing new products, expanding our sales and marketing activities, further developing our technology and manufacturing capabilities, engaging in laboratory testing, manufacturing new products and seeking regulatory approvals. As a result, we will need to continue to generate additional revenue in order to achieve and maintain profitability. Our ability to generate additional revenue will depend on our ability to successfully implement our business strategies and address the risks and uncertainties facing us, and we cannot assure you that we will be successful in these efforts. Even if we do address these risks successfully and implement our business strategies, we may not generate sufficient revenue to maintain profitability. We cannot assure you that we would be able to increase profitability on a quarterly or annual basis in the future, or at all.
We may not be able to meet our cash requirements without obtaining additional capital from external sources, and if we are unable to do so, we may have to curtail or cease operations.
We anticipate that our current cash and cash equivalents and cash provided by this offering and our operating activities will be sufficient to meet our currently estimated cash requirements for at least the next 12 months; however, we expect capital outlays and operating expenditures to increase over the next several years as we expand our infrastructure, test panel commercialization, training and support, manufacturing and research and development activities. We operate in a market that makes our prospects difficult to evaluate, and we may need additional financing to execute on our current or future business strategies. The amount of additional capital we may need to raise depends on many factors, including:
• | the level of research and development investment required to maintain and improve our technology, including efforts to expand our menu of test panels, invest in the development of new products and to seek regulatory approval for new products; |
• | the amount of future cash provided by or used in operating activities, including expansion of our sales force and marketing activities; |
• | our need or decision to acquire or license complementary technologies or acquire complementary businesses; |
• | changes in regulatory policies or laws that affect our operations; and |
• | the costs of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights. |
We cannot be certain that additional capital will be available when and as needed or that our actual cash requirements will not be greater than anticipated. If we require additional capital at a time when investment in diagnostics companies, or in the marketplace in general, is limited due to the then-prevailing market or other conditions, we may not be able to raise such funds at the time that we desire or any time thereafter. If we are unable to raise additional capital, we may be required to curtail some or all of our operations, including commercialization and research and development efforts, and forced to forego otherwise valuable business opportunities. Any failure to raise additional capital when needed could have an adverse effect on us. In addition, if we raise additional funds through the issuance of common stock, preferred stock or convertible securities, the percentage ownership of our stockholders could be significantly diluted, and any preferred stock or convertible securities may have rights, preferences or privileges senior to those of common stockholders. If we obtain additional debt financing, a substantial portion of our operating cash flow or other cash resources may be dedicated to the payment of principal
13
and interest on such indebtedness, and the terms of the debt securities issued could impose significant restrictions on our operations. If we raise additional funds through collaborations and licensing arrangements, we may be required to relinquish significant rights to our technologies or products, or grant licenses on terms that are not favorable to us.
Our ability to use our net operating loss and research and development credit carryforwards may be subject to limitations.
As of September 30, 2014, we had approximately $72.4 million of net operating loss carryforwards and $1.2 million of research and development credit carryforwards for U.S. federal tax purposes. Realization of any tax benefit from our carryforwards is dependent on our ability to generate future taxable income and the absence of certain "ownership changes" of our common stock. An "ownership change," as defined in the applicable federal income tax rules, could place significant limitations, on an annual basis, on the amount of our future taxable income that may be offset by our carryforwards. Such limitations, in conjunction with the net operating loss expiration provisions, could effectively eliminate our ability to utilize a substantial portion of our carryforwards.
Although we have not conducted a study to make this determination, it is possible that we have incurred one or more "ownership changes" in the past, in which case our ability to use our carryforwards may be limited. In addition, the issuance of shares of our common stock (including due to this offering) could cause an "ownership change" which could also limit our ability to use our carryforwards. Other issuances of shares of our common stock which could cause an "ownership change" include the issuance of shares of common stock upon future conversion or exercise of outstanding options and warrants.
Business Risks
There is limited information available to evaluate our business, products and strategy.
We are a commercial stage company in the rapidly evolving market for molecular diagnostics, or MDx, and we face numerous risks and uncertainties. We recently launched our INFINITI HTS and we cannot predict the challenges, expenses, difficulties, complications and delays frequently encountered in the launch of a new product. Further, our operations are subject to many of the risks inherent in the growth of a business. We cannot assure you that we will be successful in continuing to place our INFINITI analyzers and targeting high-volume users of genetic tests, achieving anticipated revenue growth or achieving and maintaining profitability. Our failure to meet any of these goals could have an adverse effect on us and may force us to reduce or cease our operations.
Our operating results may be variable and unpredictable.
Due to the nature of the molecular diagnostics testing market and facets of our business, our revenue and operating results may be difficult to predict and may vary significantly from period to period. The sales cycles for our analyzers is generally between three and six months, which makes it difficult for us to accurately forecast revenue in a given period. Specifically, initial sales of our consumables are often dependent on completing customer validation processes that may be time-consuming and unique to each customer. In addition to its length, the sales cycle associated with our products is subject to a number of significant risks, including the budgetary constraints of our customers, their inventory management practices and possibly internal acceptance reviews and the timing of U.S. Food and Drug Administration, or FDA, approval and review, all of which are beyond our control. Sales of our products also involve the purchasing decisions of clinical reference laboratories, specialty clinics and hospital laboratories, which can require many levels of pre-approvals, further lengthening sales time. These purchasing decisions are subject to a number of significant risk factors beyond their and our control and are difficult for us to predict. For example, clinical reference laboratories, specialty clinics and hospital laboratories may purchase fewer of our consumables due to a decline in the volumes of tests needed at these facilities. As a result, we may expend considerable resources on unsuccessful sales efforts or we may not be able to complete sales as anticipated. Our manufacturing, installation and training cycles for our INFINITI analyzers also typically involve a significant investment of working capital by us before we begin to receive revenue from our customers, and can be dependent in part on our inventory levels and the availability of our working capital. As a result, we may incur inventory-carrying and other working capital expenses that may vary over time and that may make our operating results difficult to forecast.
Our revenue and operating results are also variable and difficult to predict due to differences in revenue recognition when our customers purchase, versus rent or lease, our INFINITI analyzers. Changes in any of these
14
relative mixes can have an impact on the timing of our recognition of revenue, as instrument sales result in revenue to us upon the delivery of the instrument and instrument leases or rentals involve the recapture of instrument costs through the sale of consumables, or on our gross margin, as consumable sales typically have significantly higher margins than those of instrument sales.
Further, our revenue and operating results are variable and difficult to predict because many of our test panels have been launched relatively recently. As a result, we do not have sufficient history with these new test panels to reliably predict revenue for those tests. Any period-to-period variations in our revenue and operating results may cause our stock price to fluctuate significantly in the future.
Our inability to pay our indebtedness and trade payables could adversely affect our reputation and business prospects.
In the past, we have been in default on indebtedness owed to various persons due to our inability to repay our debts by their applicable maturity dates. We have also periodically been unable to timely pay various trade payables, including amounts owed to our suppliers, service providers and landlord. We currently are behind on making timely payments to some of our suppliers and service providers. These events have had a negative impact on our reputation and perceived creditworthiness, and may make it more difficult for us to maintain existing relationships and enter into new relationships with suppliers and service providers, or to obtain future financing, particularly debt financing. In addition, concerns about our long-term ability to continue operations may deter potential customers from investing the time and resources necessary to install and train on our analyzers and use our platform to conduct genetic testing. Our reputation has been harmed from our past failure to timely pay our debt and trade payables, and it may take us some time to repair our reputation. In the meantime, we may receive less favorable terms from our suppliers and service providers, find it more difficult to obtain financing, and face challenges convincing customers to invest in the long-term use of our platform.
We face significant competition from large competitors that are well capitalized and from alternative technologies and products.
We face, and will continue to face, significant competition from organizations such as large in vitro diagnostics companies and large reference laboratories that compete directly or indirectly with us in the general molecular diagnostics market. We compete in an industry characterized by: (i) rapid technological change, (ii) evolving industry standards and regulatory requirements, (iii) emerging competition and (iv) new product introductions. Our competitors may develop and commercialize products and technologies that may mitigate any advantages our products may currently have and that may compete successfully with our products and technologies. Since several potentially competing companies and institutions have greater financial resources, more established brand names and larger existing customer bases than we do, they may be able to: (a) provide broader services and product lines, (b) make greater investments in research and development, (c) undertake more extensive sales efforts and marketing campaigns and (d) adopt more aggressive pricing policies than we are able. Some of our competitors are also our suppliers, and there can be no assurance that these suppliers will continue to provide us products at competitive prices or at all. Many of our competitors have also been able to enter into long-term, exclusive agreements with major potential customers, often by offering favorable pricing and other terms. Until these agreements expire, our ability to place our analyzers with and sell our testing consumables to these customers will be limited. Even after exclusive agreements expire, we may not be able to compete with the terms offered by our competitors in their efforts to extend exclusive relationships with these major potential customers. In addition, we compete against companies on the basis of the relative regulatory status of their and our products. Some of our competitors may have already received regulatory approval and conducted extensive clinical trials for tests we seek to sell. Any tests that we offer on a research use only, or RUO, basis may be at a competitive disadvantage to similar tests offered by others that have received FDA clearances or approvals, particularly as the FDA has announced its intention to modify its enforcement discretion approach to laboratory developed tests, or LDTs, which may have an adverse effect on sales of our RUO products even in advance of final regulations taking effect.
We also expect to continue to face competition from enhanced or alternative technologies and products. One or more of our competitors could develop a product that is superior to a product we offer or intend to offer or our technology and products may be rendered obsolete or uneconomical by advances in existing technologies. At least
15
one of our competitors currently offer on a commercial basis self-contained, integrated systems for molecular diagnostics testing that are similar in many ways to our products. If we are unable to keep pace with technological advances in the molecular diagnostics market, our business, financial condition and results of operations may suffer.
Our business may be adversely affected if we fail to capture a share of the rapidly growing molecular diagnostics testing market.
There are barriers to the adoption of widespread genetic testing, including the need to increase physician education on molecular research use and diagnostic testing and to keep physicians up-to-date on new and emerging technologies, as well as overcoming coverage denials from insurance companies. We cannot assure you that we will be successful in capturing a significant share of the MDx market by expanding the sale of our products at the prices we currently project. In the event that the market in general, or our market share in particular, does not grow as we expect, our business may be adversely affected.
Growth in our business could strain our managerial, operational, manufacturing, customer support, sales, financial and information systems resources.
The anticipated future growth necessary to expand our operations will place a significant strain on our managerial, operational, manufacturing, sales, financial and information systems resources. These increased demands could cause us to operate our business less effectively, which in turn could cause deterioration in the financial performance of our business. To increase revenue and achieve growth, we will need to expand our sales efforts and continue to improve and develop our products. The future growth of our business will also require us to expand our customer support resources, including adding additional personnel for customer training and technical product support. If our customer support infrastructure does not keep pace with future growth of our customer base, we may lose customers and our reputation and future sales potential may be harmed. In addition, we will need to improve our financial and managerial controls, reporting systems and procedures, and will also need to expand, train and manage our workforce. We may be unable to hire, train and retain a sufficient number of qualified personnel or successfully manage our growth. This growth may also place increased burdens on our international distributors, increase the complexities we face related to international sales and increase our inventory-related risk. Future growth will also make it difficult for us to adequately predict the expenditures we will need to make in the future. If we do not make the necessary capital or other expenditures to accommodate our anticipated growth, or if we are unable to manage our growth effectively, our business, financial condition and results of operations will suffer. We cannot anticipate all of the demands that our expanding operations will impose on our business, controls and procedures, personnel and systems, and our failure to appropriately address such demands could have an adverse effect on us.
The loss of the services of one or more of our key personnel or failure to attract and retain other highly qualified personnel in the future could adversely affect operations and result in a loss of revenue.
We are dependent on the continued services and performance of senior management and educated, technically-trained personnel. We currently do not have employment or severance agreements with any of these persons other than Fareed Kureshy, our President and Chief Executive Officer. Certain of the members of our senior management, including certain of our named executive officers, are at, close to or past traditional retirement age in the United States. Our business depends and will depend in the future on the ability to identify, attract, hire, train, retain and motivate senior management and other highly skilled technical, managerial, marketing and customer service personnel. Competition for such personnel is intense, and we cannot assure you that we will be able to successfully attract and retain sufficiently qualified personnel in the future. While we do not currently expect any of our senior management or other technically-trained personnel to retire or otherwise cease employment with us in the near-term, we will need to effectively plan for management succession to maintain effective leadership and continuity in our business. The failure to attract and retain necessary senior management and technically-trained personnel and to successfully plan for management succession could adversely affect our business and result in a loss of revenue.
16
We rely on the innovation and resources of larger industry participants and public programs to advance genetic research, establish the medical relevance of biomarkers and educate physicians and clinicians on molecular diagnostics.
The link between the genetic variations that our products detect and the underlying disease states, or responses to medications, is not always fully medically validated, and our current and future products may involve biomarkers whose medical relevance is unproven. Additionally, the availability of validated biomarkers is dependent on significant investment in genetic research, often funded through public programs for which there are no assurances of ongoing support. The adoption of molecular diagnostics is dependent to a great extent on the education and training of physicians and clinicians. We do not have the resources to undertake such training, and we are relying on larger industry participants and professional medical colleges to establish, communicate and educate physicians and clinicians regarding the use and application of molecular diagnostics. The genetic test results produced by our platform (including as used by our in-house laboratory) are reported to our customers in a format that also indicates potential options for treatment and dosing based on the specific test result. These presented options are based on publicly available information and open-source data repositories. We do not independently verify this information. If this information were to be incorrect, or if in the future similar information for genetic tests that we develop were not publicly available, we would be required to develop or obtain such information through our own or private resources, which could be expensive and time-consuming and which would in turn adversely affect our business and results of operations.
We may be unsuccessful in our long-term goal of expanding our product offerings outside the United States.
For the nine months ended September 30, 2014, and the years ended December 31, 2013 and 2012, 14%, 14% and 9%, respectively, of our revenue was from customers in countries outside the United States, including countries in Europe, Asia, the Middle East and South America. We are dependent on third-party distributor relationships in these countries to market, sell and provide customer and technical support for our products. Distributors may not commit the necessary resources to market, sell and support our products to the level of our expectations. If distributors do not perform adequately, or we are unable to locate distributors in particular geographic areas, our ability to realize long-term international revenue growth would be adversely affected. Furthermore, there are risks inherent in doing business internationally, including:
• | imposition of governmental controls and changes in laws, regulations or policies; |
• | political and economic instability; |
• | changes in U.S. and other national government trade policies affecting the markets for our products; |
• | changes in regulatory practices, tariffs and taxes; and |
• | currency exchange rate fluctuations, devaluations and other conversion restrictions. |
Any of these factors could have an adverse effect on our business, results of operations or financial condition.
Manufacturing risks, shortages and inefficiencies may adversely affect our production ability.
We must manufacture or engage third parties to manufacture components of our products in sufficient quantities and on a timely basis, while maintaining product quality and acceptable manufacturing costs and complying with regulatory requirements. In determining the required quantities of our products and the manufacturing schedule, we must make significant judgments and estimates based on historical experience, inventory levels, current market trends and other related factors. Because of the inherent nature of estimates, there could be significant differences between our estimates and the actual amounts of products we require. We generally manufacture our INFINITI analyzers only when ordered by our customers, and a significant influx of instrument orders could result in significant backlogs. In such a situation our ability to manufacture our INFINITI analyzers could be adversely impacted if we do not have sufficient working capital or labor on hand to support the purchasing of components, and the assembly of the same, necessary for the manufacture of our analyzers. For example, from February 2014 through September 30, 2014, we received a higher number of orders for our INFINITI HTS than anticipated, and our ability to fulfill all of these orders has been delayed due to manufacturing lead time, influenced by parts availability and supplier availability. In the future, our inability to timely fulfill analyzer orders could have an adverse impact on our revenue and results of operations.
17
We may also experience unforeseen technical complications in the processes we use to develop, manufacture, customize or receive orders for our products. Such complications could materially delay or limit the use of products we attempt to commercialize, substantially increase the anticipated cost of our products or prevent us from implementing our processes at appropriate quality and scale levels, thereby causing our business to suffer.
We have relationships with suppliers who are sources of raw materials and components for our product offerings. We do not have long-term agreements with our suppliers and have not arranged for alternate suppliers. It may be difficult to find alternate suppliers in a timely manner or on terms acceptable to us. Additionally, the availability of some components of the INFINITI analyzers may be limited as only a few outside vendors produce them. In the event that we become subject to a shortage of components or raw materials, our business, financial condition and results of operations could be adversely affected.
We will incur increased costs as a result of being a public company, and our management will be required to devote substantial time to new compliance matters.
We will incur significant legal, accounting, and other expenses as a public company, including costs resulting from public company reporting obligations under the Securities Exchange Act of 1934, as amended, and the rules and regulations regarding corporate governance practices, including those under the Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act, and the listing requirements of the NASDAQ Global Market.
Our management and other personnel will need to devote a substantial amount of time to ensure that we comply with all of these requirements. Moreover, despite recent reforms made possible by the JOBS Act, the reporting requirements, rules, and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly, particularly after we are no longer an "emerging growth company." Any changes that we make to comply with these obligations may not be sufficient to allow us to satisfy our obligations as a public company on a timely basis, or at all.
We also expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. These factors could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, particularly to serve on our audit and compensation committees, or as executive officers.
Our business and future operating results may be adversely affected by catastrophic or other events outside of our control.
We develop and manufacture our platform in our facility located in Vista, California. Any damage to our facilities or the manufacturing equipment we use would be costly and could require substantial lead-time to repair or replace. Our business and operating results may be harmed due to interruption of our manufacturing by events outside of our control, including earthquakes and fires. Other possible disruptions may include power loss and telecommunications failures. In the event of a disruption, we may lose customers and we may be unable to regain those customers thereafter. Our insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
Product Risks
Our financial results depend on commercial acceptance of our platform, its menu of tests and the development of additional tests and other products.
Our future depends on the success of our platform, which depends primarily on its acceptance by our targets customers as a reliable, accurate and cost-effective replacement for traditional MDx testing methods. Many clinical reference laboratories, specialty clinics and hospital laboratories already use molecular diagnostics testing instruments in which they have made substantial investments, and may be reluctant to change their current procedures for performing such analyses. In addition, many laboratories and clinics currently rely on LDTs that are redundant to LDTs that they would create and validate using our RUO products. These laboratories and clinics may be reluctant to discontinue using their LDTs due to the time and expense already invested in developing and validating these tests and their familiarity with these tests compared to our platform. If we are unable to displace
18
molecular diagnostics testing instruments and LDTs currently in use, our market opportunity will be limited and we may not be able to achieve significant sales of our products.
We continue to seek to develop additional tests for our platform as well as additional products to respond to what we perceive to be the needs of clinical reference laboratories, specialty clinics and hospital laboratories; however, we cannot guarantee that we will be able to develop enough additional tests or other products in a timely manner or in a manner that is cost-effective or at all. The development of new or enhanced tests and other products is a complex and uncertain process requiring the accurate anticipation of technological and market trends, as well as precise technological execution. We are currently not able to estimate when or if we will be able to develop, obtain licenses to third-party intellectual property on commercially acceptable terms for, obtain regulatory approval or clearance for, commercialize or sell, additional tests or enhance existing products. If we are unable to increase sales of our platform or to successfully develop, obtain licenses to third-party intellectual property on commercially acceptable terms for, obtain regulatory approval or clearance for, and commercialize, additional tests or other products, our revenue, prospects and ability to achieve or maintain profitability would be impaired.
We are subject to risks relating to the placement of INFINITI analyzers under our Reagent Access Plans.
In the United States, we offer the INFINITI analyzers through direct sale, monthly rental plans and our Reagent Access Plan, or RAP. As of September 30, 2014 and December 31, 2013, we had 58 and 48, respectively, INFINITI analyzers placed with customers under RAPs, representing 24.8% and 23.3%, respectively, of our total analyzer placements. Under our RAP, an INFINITI analyzer is placed at the customer's location at no direct cost to the customer with the intent of generating recurring demand for our high-margin testing consumables, including our test-specific BioFilmChips and Intellipac Reagent Management Modules. The customers under our RAPs are required by the terms of the plans to purchase minimum amounts of consumables; however, in some cases, customers have not met this requirement. We have the right to reclaim an INFINITI analyzer placed with a customer pursuant to a RAP if the customer does not purchase the minimum amount of consumables. During 2014 and 2013, we reclaimed two and 14 INFINITI analyzers, respectively, either because the respective customer failed to purchase the minimum amount of consumables or otherwise elected not to retain the INFINITI analyzer at the end of the term of the applicable RAP. We bear the costs of producing these INFINITI analyzers and rely on the revenue from the sale of consumables to recoup our costs. As a result, we could expend significant resources to produce INFINITI analyzers for our RAPs and not generate enough revenue from the sale of consumables to recoup our costs, in which case our business could be adversely affected.
Our products are subject to recalls even after receiving FDA clearance or approval, or after affixing the CE marking, which would harm our reputation and business.
We are subject to medical device reporting regulations (e.g., MDR, vigilance reporting) that require us to report to the FDA or governmental authorities in other countries if our products cause or contribute to a death or serious injury, or malfunction in a way that would be reasonably likely to contribute to death or serious injury if the malfunction were to recur. The FDA and similar governmental authorities in other countries have the authority to require the recall of our products in the event of material deficiencies or defects in design or manufacturing. To date, we have not been required to make any reports under the MDR or vigilance reporting regulations and we have not conducted any product recalls. A government mandated, or voluntary, recall by us could occur as a result of component failures, manufacturing errors or design defects, including defects in labeling. Any recall would divert managerial and financial resources and could harm our reputation with customers. There can be no assurance that there will not be product recalls in the future or that such recalls would not have an adverse effect on our business.
If our products contain undetected defects or errors or do not operate as expected, we could incur significant unexpected expenses, experience product returns and lost sales, suffer damage to our brand and reputation and be subject to product liability or other claims and product recalls and other field or regulatory actions.
Our products are complex and may contain undetected defects, errors or failures, particularly when first introduced or when new versions are released. Some errors and defects may be discovered only after a product has been installed and used by the customer. To the extent our products incorporate technologies of third parties, we will be dependent on the reliability of these technologies. If our products contain undetected defects or errors or do not operate as expected, we could experience decreased sales and increased product returns, loss of customers and market share and increased service, warranty and insurance costs. In addition, our reputation and brand could be
19
damaged, and we could face potential legal claims regarding our products and be subject to product recalls or corrections and other field or regulatory actions. A successful product liability or other claim or product recall could result in negative publicity and further harm our reputation, result in unexpected expenses and adversely impact our business, financial condition and results of operations. In addition, we may be subject to claims resulting from false identification of genetic variations or other misdiagnoses made in connection with our tests. Litigating any such claims could be costly. We could expend significant funds during any litigation proceeding brought against us. Further, if a court were to require us to pay damages to a plaintiff or if we agree to pay damages in settlement of a claim, the amount of such damages could adversely affect our business, financial condition and results of operations.
We have limited experience with the specialized film coating process needed to produce our BioFilmChip microarrays, and there are limited alternative sources for this aspect of our manufacturing process.
Our BioFilmChip microarrays are formed by coating polyester film with several layers of emulsion. To date, we have outsourced the special film coating process needed to produce our BioFilmChip microarrays. In connection with the establishment of manufacturing operations at our Vista, California facility, we purchased the film coating equipment required to conduct this process ourselves. We have not yet installed this equipment and have no experience as a company conducting the film coating process. If we are unable to successfully operate and maintain this equipment and produce the coated film necessary for our BioFilmChip microarrays, we will have to continue to outsource this operation to a third party. Film coating capacity is limited and is becoming less available in the marketplace as film vendors move away from analog formats and focus increasingly on digital formats. Although we believe we have adequate coated film for the foreseeable future, we cannot guarantee that we would be able to obtain an alternative supply if we are unable to manufacture the coated film ourselves.
We will need to expand manufacturing capacity by ourselves or with third parties.
We will need either to continue to build internal manufacturing capacity or contract with one or more manufacturing partners, or both. We currently rely solely on internal manufacturing of our end products. Due to the complexity of our manufacturing process and the advanced technologies we employ, we may encounter difficulties in manufacturing our end products. We may not be able to build manufacturing capacity internally or find one or more suitable manufacturing partners, or both, to meet the volume, regulatory and quality requirements necessary to be successful in the market. If our products do not consistently meet our customers' performance expectations or the requirements of the FDA and authorities in other countries, we may be unable to generate sufficient revenue to become profitable. Significant additional resources, implementation of additional manufacturing equipment and changes in our manufacturing processes and organization may be required for the scale-up of each new product prior to commercialization or to meet increasing customer demand once commercialization begins, and this work may not be successfully or efficiently completed. Any delay in establishing or inability to expand our manufacturing capacity could delay our ability to develop or sell our products, which would result in lost revenue and adversely harm our business, financial condition and results of operations.
Customer Risks
We have in the past derived a significant portion of our revenue from a small number of customers and the loss of one or more of these customers, a significant decline in sales to one or more of these customers, or a delay in collection of payments from one or more of these customers could have an adverse effect on our financial condition and results of operations.
We have in the past derived a significant portion of our revenue from a few major customers. For fiscal years 2013 and 2012, we had two customers that together comprised 43% and 59%, respectively, of our total revenue, although during the nine months ended September 30, 2014 no customer accounted for more than 10% of our revenue. With the recent adoption of our INFINITI HTS, we expect that a significant and increasing portion of our revenue will be derived from those of our customers that conduct high-volume testing. This could lead to a significant percentage of our revenue being concentrated in a small number of customers, unless and until we are able to increase the number of placements of our INFINITI HTS. Our continued business relationship with these major customers, and the amount of purchases and timing of payment by these major customers, may be impacted by several factors beyond our control, including product offerings by our competitors, pricing pressures or the financial health of these customers. The loss of any of these major customers, or a significant decline in sales to or a delay in collection of payments from any of these customers, would adversely affect our business, financial
20
condition and results of operations. For example, in the fourth quarter of 2012 one of our key customers, who was then utilizing a beta-trial version of our INFINITI HTS, significantly decreased its ordering volumes during the quarter, which resulted in substantially lower net revenue for that quarter compared to the prior quarter. This customer subsequently filed for U.S. bankruptcy protection during 2013, and as a result we recorded $4.5 million in bad debt expense in 2013.
Our business may suffer if we have difficulty acquiring and retaining customers.
A large part of our business strategy depends on our ability to place our INFINITI analyzers with customers that are high-volume users of genetic tests in our areas of focus to drive sales of our consumable products. We may not succeed in attracting and retaining a sufficiently large customer base for our platform in order to execute on our strategy. The FDA may restrict the ability of laboratories certified under the Clinical Laboratories Improvements Act of 1988, or CLIA, to offer LDTs, which would have an adverse impact on our sale of RUO consumable products to those laboratories. If we are unable to establish a sufficient installed base of INFINITI analyzers, in particular our INFINITI HTS analyzers, or if customers with which we place INFINITI analyzers do not use them, we may not be able to generate revenue or successfully implement our business strategy. In addition, if we are unable to bring our products to market as quickly as anticipated or to acquire and develop our customer base as quickly as we have projected, our ability to generate revenue could be adversely impacted.
Failure by our customers or distributors to pay for products we have sold to them, or failure or third-party payors to cover, or adequately reimburse our customers for the use of our tests, could impact our ability to continue sales and have an adverse effect on our financial condition and results of operations.
In certain cases, we may not be able to recognize revenue on sales even though we may have delivered products to our customers or distributors. For example, we have in the past sold, and may continue to sell, our INFINITI analyzers to certain of our international distributors where our ability to collect payment was not reasonably assured. Although our customers and distributors are required to pay us for the products that we sell to them, from time to time certain of our customers and distributors have not paid us or have delayed payment. In particular, some of our distributors experience long delays in receiving reimbursement from foreign government payors and healthcare providers, which in turn causes them to withhold payment to us. As of September 30, 2014 and December 31, 2013, approximately $7.4 million and $6.3 million, respectively, of our net accounts receivable were 90 days or more past due. This included, as of September 30, 2014 and December 31, 2013, approximately $4.5 million in net accounts receivable from one of our former major customers that filed for U.S. bankruptcy protection. We recorded $4.5 million in bad debt expense in 2013 related to this customer.
In the past we have deferred, and in the future we may be required to defer, the recognition of revenue where the collection of payment is not reasonably assured until such payment is received. Deferring the recognition of revenue could negatively impact our results of operations for any period in which the related sales occurred. Although we have reserved for or deferred the recognition of revenue related to the amount of all late payments on our balance sheets, we also may incur unexpected costs or losses resulting from these sales in the event of any delays or failures to make payment for the related products. If we are unable to collect payment from our customers or distributors for products we have sold to them, we may never be able to recover our costs to manufacture and sell such products, which could have an adverse effect on our financial condition and results of operations.
We sell our products primarily to clinical reference laboratories, specialty clinics and hospital laboratories, substantially all of which receive reimbursement for the healthcare services they provide to their customers from third-party payors, such as Medicare and Medicaid, private insurance plans and managed care programs. These third-party payors may deny reimbursement if they determine that a medical product was not used in accordance with cost-effective treatment methods, as determined by the third-party payor, or was used for an unapproved indication. Third-party payors also may refuse to reimburse for procedures and devices deemed to be experimental or medically unnecessary. In the United States, the American Medical Association assigns specific Current Procedural Terminology, or CPT, codes, which are necessary for reimbursement of diagnostic tests. Once the CPT code is established, the Centers for Medicare and Medicaid Services of the Department of Health and Human Services establishes reimbursement payment levels and coverage rules under Medicaid and Medicare, and private payors independently establish rates and coverage rules. Although there are previously assigned CPT codes for the tests that we offer and most of our tests are approved for reimbursement by Medicare and Medicaid as well as most third-party payors, certain of our future products may not be approved for reimbursement, and past coverage of our
21
current tests is not a guarantee for future coverage. Third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for medical products and services. Increasingly, Medicare, Medicaid and other third-party payors are challenging the prices charged for medical services, including diagnostic tests. Levels of reimbursement may decrease in the future, and future legislation, regulation or reimbursement policies of third-party payors may adversely affect the demand for and price levels of our products. If our customers are not reimbursed for our products, they may reduce or discontinue purchases of our products or fail to make payment on amounts owed to us. For example, in 2013 our then-largest customer quit purchasing our products and sought protection under U.S. bankruptcy laws as a result of being unable to obtain reimbursement for medical services being performed by such customer.
If our customers are unwilling or unable to validate our platform or follow required protocols, our business could be harmed.
In most cases, we are dependent on our customers to validate our INFINITI analyzers and the associated tests we offer. These test validation processes are done on a test-by-test basis and can take anywhere from several weeks to several months or more to complete, and we do not generate revenue from these customers for sales of the tests being validated during these processes. Individual laboratories have their own validation procedures and protocols that our system and tests must pass and we cannot predict how long each validation process will take or whether laboratories will be willing or able to validate our INFINITI analyzer. To the extent that any laboratories are unwilling or unable to validate our INFINITI analyzer, we will not be able to generate meaningful revenue from sales to these laboratories.
Proper use of our INFINITI analyzers also requires our customers to follow certain sample collection and other protocols. Unlike our INFINITI and INFINITI PLUS analyzers, which are embodied in a single instrument box, our INFINITI HTS is a modular system with three instrument boxes, and so requires the observation by our customers of additional and more detailed protocols. If we are unable to develop or properly train our customers on these protocols or if, despite our efforts, customers do not follow the required protocols, the applicable INFINITI analyzer may not generate a result or a correct result. With respect to international customers, we are generally dependent on our distributors to provide support services. Failure by our customers to follow required protocols may lead to a perception, even if unfounded, that the applicable INFINITI analyzer does not function properly. Such perceptions could damage our reputation and brand and reduce the frequency that our customers use our INFINITI analyzers and purchase our consumables.
The spending policies of our customers have a significant effect on the demand for our products.
Our targeted customers include clinical reference laboratories, specialty clinics and hospital laboratories, and their respective spending policies can have a significant effect on the demand for our products. These policies are based on a wide variety of factors, including governmental regulation or price controls, reimbursement policies, the resources available for purchasing diagnostic tests, the spending priorities among various types of diagnostic tests and the policies regarding capital expenditures during recessionary periods. Any decrease in spending by clinical reference laboratories, specialty clinics and hospital laboratories could cause our revenue to decline. We have no control over our customers' spending policies. As a result, we are subject to significant volatility in revenue, which could result in an adverse effect on our operating results.
Regulatory Risks
We conduct business in a heavily regulated industry, and changes in regulations or the FDA's enforcement discretion, or violations of regulations or guidance by us, could adversely affect our business, prospects, results of operations or financial condition.
The clinical laboratory testing industry is highly regulated, and we cannot assure you that the regulatory environment in which we operate will not change significantly and adversely in the near future. In particular, the laws and regulations governing the marketing of molecular research use or diagnostic products for use as laboratory-developed tests are extremely complex and in many instances there are no significant regulatory or judicial interpretations of these laws and regulations. While we believe that we are currently in material compliance with applicable laws and regulations as historically enforced by the FDA, we cannot assure you that the FDA or other regulatory agencies would agree with our determination, and a determination that we have violated these laws, or a public announcement that we are being investigated for possible violations of these laws, could adversely affect our
22
business, prospects, results of operations or financial condition. Additionally, in November 2013 the FDA issued a Guidance document entitled "Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only," or the RUO Guidance, which highlights the FDA's interpretation that distribution of RUO products with any labeling, advertising or promotion that suggests that clinical laboratories can validate the test through their own procedures and subsequently offer it for clinical diagnostic use as an LDT is in conflict with RUO status. The RUO Guidance further articulates the FDA's position that any assistance offered in performing clinical validation or verification, or similar specialized technical support, to clinical laboratories, is in conflict with RUO status. More recently, on July 31, 2014, the FDA notified the United States Congress of its intent to modify its enforcement discretion approach to LDTs in a risk-based manner, and this may require us to change our business model in order to maintain compliance with these laws. If we engage in any activities that are in conflict with the RUO status held by the majority of tests that we sell, we may be subject to immediate, severe and broad FDA enforcement action that would adversely affect our ability to continue operations. Accordingly, if the FDA finds that we are distributing our RUO products in a manner that is inconsistent with its guidance, we may be forced to stop distribution of our RUO tests until we are in compliance, which, would reduce our revenue, increase our costs and adversely affect our business, prospects, results of operations and financial condition.
Our failure to comply with regulatory requirements or receive any required regulatory clearance or approval for our products or operations in the United States or abroad would adversely affect our revenue and potential for future growth.
As a manufacturer of molecular research use and diagnostics products, our products are medical devices that are subject to extensive regulation in the United States by the FDA, and by respective authorities in foreign countries where we do business. The FDA regulates virtually all aspects of a medical device's design and testing, manufacture, safety, labeling, storage, recordkeeping, reporting, clearance and approval, promotion and distribution. The FDA also regulates the export of medical devices to foreign countries. Failure to comply with the regulatory requirements of the FDA and other applicable U.S. regulatory requirements may subject a company to administrative or judicially imposed sanctions ranging from warning letters to criminal penalties or product withdrawal or recall. Unless an exemption applies, a medical device must receive either clearance or premarket approval from the FDA before it can be marketed in the United States. The FDA's 510(k) clearance process for Class II devices usually takes from three to 12 months, but may take significantly longer. The premarket approval process for Class III devices generally takes from one to three years from the time the application is filed with the FDA, but also can be significantly longer. Premarket approval typically requires extensive clinical data and can be significantly longer, more expensive and more uncertain than the 510(k) clearance process. Despite the time, effort and expense expended, there can be no assurance that a particular device ultimately will be cleared or approved by the FDA through either the 510(k) clearance process or the premarket approval process.
Medical devices may only be marketed for the indications for which they are approved or cleared. We may be required to obtain additional new premarket approvals, premarket approval supplements or 510(k) clearances to market additional products or for new indications for or modifications to our existing products. We cannot be certain that we would obtain additional premarket approvals or 510(k) clearances in a timely manner or at all. We have modified various aspects of our devices in the past and we determined that new approvals, supplements or clearances were not required. The FDA may not agree with our decisions not to seek approvals, supplements or clearances for particular device modifications. If the FDA requires us to obtain premarket approvals, supplement approvals, or 510(k) clearances for any modification to a previously cleared or approved device, we may be required to cease manufacturing and marketing the modified device or to recall such modified device until we obtain FDA clearance or approval and we may be subject to significant regulatory fines or penalties. For example, we have not yet submitted a 510(k) notification for our INFINITI HTS analyzer and are presently selling it on an RUO basis only. Our ability to obtain 510(k) clearance for the INFINITI HTS may be adversely impacted by our existing sale of the device as RUO equipment. In addition, we cannot assure you that the FDA will clear or approve such submissions in a timely manner, if at all.
A significant portion of our revenue is derived from genetic test panels sold as research use only and that are not intended for diagnostic purposes. Sales of our RUO products accounted for 65%, 82% and 86% of our net revenue for the nine months ended September 30, 2014, and years ended December 31, 2013 and 2012, respectively. Under the FDA's current enforcement discretion approach to LDTs, laboratories regulated under the Clinical Laboratory Improvement Amendments of 1988, or CLIA, may validate the use of LDTs, specifically for use in their laboratory
23
using any labeled products, which may include our RUO-labeled products. While the FDA does not regulate LDTs, it has issued guidance on acceptable distribution practices for in vitro diagnostic products labeled for Research Use Only and on responding to unsolicited requests for off-label information. We have procedures designed to comply with these requirements and we believe that the sale of our products complies with the FDA's laws and regulations. If the FDA disagrees with the marketing of RUO products, or in its intended modification of LDT enforcement discretion imposes substantial changes to the regulation or enforcement of LDTs that are marketed by us, the broad use of RUO tests may be limited and a significant reduction in the sale of these products could occur. A decline in the sales of our RUO products would reduce our revenue, increase our operating expenses and adversely affect our business and results of operations.
On December 4, 2009, the FDA issued a letter to us indicating that certain of our promotional materials for our RUO tests may include diagnostic/clinical claims which may cause these products to be in vitro diagnostic tests for clinical use for which FDA clearance or approval is required. In addition, the FDA noted that the allegations in its letter may not constitute an exhaustive list of potential violations and noted that we are required to comply with the RUO requirements for all of our products that have not received FDA clearance or approval. We responded to the FDA's allegations in a letter dated December 14, 2009 and believe that we have taken appropriate action to modify and revise the promotional materials in light of the FDA's concerns. However, the FDA has not responded to our submission and has not confirmed its agreement with our approach. If the FDA does not agree with our approach, it could require that we take additional steps, initiate enforcement action or require that we recall or withdraw certain or all of our RUO products from the market. In addition, we could be subject to enforcement through other government agencies or private litigants, based on allegations similar to those contained in the FDA's December 14, 2009 letter, including but not limited to actions seeking fines, injunctions, consent decrees, civil penalties and criminal prosecution, any of which could material adversely impact our business. Our failure to comply with the FDA's regulatory requirements, obtain FDA clearance or approvals of new or existing products or product modifications or enhancements that we develop in the future, any limitations imposed by the FDA on such products' development or use, or the costs of obtaining FDA clearance or approvals, could have an adverse effect on our business, particularly given the proportion of our business that is reliant upon RUO products.
In many of the foreign countries in which we market our products, including those countries in the European Union, we are subject to extensive regulations similar to those of the FDA. As discussed under "Business — Government Regulation," there are additional regulatory requirements outside of the United States, including certifications, registrations or approvals that may require third-party certification of compliance with ISO 13485:2003, a quality management system standard developed by the International Organization for Standardization, or ISO. This certification must be satisfied in order for us to market our products in those jurisdictions. If we fail to satisfy such requirements or obtain the requisite certifications, registrations or approvals in these countries, it could have an adverse effect on our international operations.
Government payors, such as Medicare and Medicaid, and non-government payors, such as health plans, continue to implement controls and limits on the utilization and reimbursement of healthcare services, including in the area of genetic testing.
Government and non-government payors have in the past sought, and continue to seek, to reduce and limit utilization and reimbursement of healthcare services, including in the area of genetic testing. The Medicare Clinical Laboratory Fee Schedule is currently under review, and amounts paid to providers under such schedule is expected to change in upcoming years. Analyte-specific billing codes for genetic testing are also under review and are undergoing changes over time, and CMS has in the past expressed the opinion that certain analyte-specific billing codes should not be covered under Medicare. We expect that government and non-government payors will continue over time to expend efforts to reduce utilization of genetic testing services, adopt increasingly more stringent cost controls and otherwise reduce reimbursements. These efforts, if successful, may have an adverse impact on the volume of our sales of genetic testing products and in-house laboratory services or on the profitability of our genetic testing products and in-house laboratory services, or both.
Federal budgetary limitations and changes in healthcare policy, such as the creation of broad limits for diagnostic products or requirements that Medicare patients pay for portions of clinical laboratory tests or services received, could substantially diminish the sale of, or inhibit the utilization of diagnostic tests, increase costs, divert management’s attention and adversely affect our ability to generate revenue and achieve profitability. For example,
24
on several occasions, the U.S. Congress has considered imposing a 20% patient co-insurance amount for clinical laboratory services, which would require beneficiaries to pay a portion of the cost of their clinical laboratory testing. Although that requirement has not been enacted at this time, such an obligation could be imposed at some point in the future, which would make it more difficult for us to collect adequate reimbursement for our products, and thus might adversely affect our business and product sales.
If the FDA or another regulatory agency determines that we have promoted off-label use of our products, or are not in compliance with restrictions on the marketing of products labeled for research use only, we may be subject to various penalties, including civil or criminal penalties.
The FDA and other regulatory agencies actively enforce regulations prohibiting the promotion of a medical device for a use that has not been cleared or approved by the FDA. Use of a device outside its cleared or approved indications is known as "off-label" use. If the FDA or another regulatory agency determines that our promotional materials or training constitutes promotion of an off-label use, it could require us to modify our training or promotional materials or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalties.
As required by the FDA, we have a policy in place requiring that our labeling and marketing practices comply with FDA regulations for products labeled for research use only. We also have a policy in place to refrain from making statements that could be considered off-label promotion of our products. Although our policy is to refrain from making statements that could be considered off-label promotion of our products, the FDA or another regulatory agency could conclude that we have engaged in off-label promotion or marketing of products labeled for research use only. If the FDA disagrees with the marketing of these products, we may be subject to a variety of enforcement actions ranging from public warning letters, fines, injunctions, consent decrees and civil penalties to suspension or delayed issuance of approvals, seizure or recall of our products, total or partial shutdown of production, withdrawal of approvals or clearances already granted, and criminal prosecution. In addition, the off-label use of our products may increase the risk of injury to patients, and, in turn, the risk of product liability claims. Product liability claims are expensive to defend and could divert our management’s attention and result in substantial damage awards against us. In addition, violations of the Federal Food, Drug, and Cosmetic Act may lead to investigations alleging violations of federal and state health care fraud and abuse laws, as well as state consumer protection laws, which may lead to costly penalties and may adversely impact our business.
If we fail to comply with the FDA's Quality System regulation and similar requirements in other jurisdictions, our manufacturing could be delayed, and our product sales and profitability could suffer.
Our manufacturing processes are required to comply with the FDA's Quality System regulation, which covers the procedures concerning (and documentation of) the design, testing, production processes, controls, quality assurance, labeling, packaging, storage and shipping of our devices. We also are subject to similar requirements in other jurisdictions and to state requirements and licenses applicable to manufacturers of medical devices. In addition, we must engage in extensive recordkeeping and reporting and must make available our manufacturing facilities and records for periodic and unscheduled inspections by governmental agencies, including the FDA, state authorities and comparable agencies in other countries. Moreover, failure to pass a Quality System regulation inspection or to comply with these and other applicable regulatory requirements could result in disruption of our operations and manufacturing delays. Failure to take adequate corrective action could result in, among other things, significant fines, suspension of approvals, seizures or recalls of products, operating restrictions and criminal prosecutions. Any failure to comply with applicable requirements could adversely affect our product sales and profitability.
Failure to comply with the Clinical Laboratory Improvement Amendments of 1988 and state laws governing clinical laboratories would negatively affect our business, and we may be required to expend significant amounts of resources to comply with these requirements.
We are subject to the CLIA a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. CLIA, which is administered by CMS, is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, participation in proficiency testing, patient test management, quality control, quality assurance and inspections. The failure to
25
comply with CLIA requirements can result in enforcement action, including the revocation, suspension, or limitation of our CLIA certificate, as well as a directed plan of correction, state on-site monitoring, civil money penalties, civil injunctive suit or criminal penalties. We must maintain CLIA compliance and certification to be eligible to bill for tests provided to Medicare beneficiaries. If we were to be found out of compliance with CLIA program requirements and subjected to sanction, our business and reputation could be harmed. Even if it were possible for us to bring our laboratory back into compliance, we could incur significant expenses and potentially lose revenue in doing so.
We are also required to maintain a license to conduct testing in California. California laws establish standards for day-to-day operation of our clinical laboratory, including the training and skills required of personnel and quality control. Several other states require that we hold licenses to test specimens from patients residing in those states, and we currently hold licenses in all these states. Other states may adopt similar requirements in the future. Finally, we may in the future become subject to regulation in foreign jurisdictions if we engage in international sales of our genetic testing laboratory services. If we are not in compliance with a particular state’s or foreign jurisdiction’s licensing requirements, we may be unable to accept test specimens from patients residing in that state or foreign jurisdiction, as applicable, which could adversely affect our business.
We have a current certificate of accreditation under CLIA to perform testing. To renew our CLIA certificate, we are subject to survey and inspection every two years to assess compliance with program standards. Moreover, CLIA inspectors may make random inspections of our laboratory. The standards applicable to the testing that we perform may change over time. New interpretations of current regulations or future changes in regulations under CLIA or other governmental regulatory bodies, may make it difficult or impossible for us to comply with CLIA and other applicable regulations, which would significantly harm our business. We can offer no assurance that we will be able to operate profitably should regulatory compliance requirements become substantially more costly in the future.
The imposition of additional healthcare legislation could significantly impact our earnings and negatively impact our results of operations and cash flows.
In March 2010, significant reforms to the healthcare system in the form of the Patient Protection and Affordable Care Act and the Health Care and Education Reconciliation Act of 2010 were adopted as law in the United States. This legislation included provisions that, among other things, reduce or limit Medicare reimbursement (including in respect of clinical laboratory fees such as those collected by our in-house laboratory) and impose an excise tax of 2.3% on U.S. sales of most medical devices, including some of our products. Other elements of this legislation, such as comparative effectiveness research, new requirements to report certain financial arrangements with physicians and teaching hospitals, an independent payment advisory board, payment system reforms, including shared savings pilots, and other provisions, could meaningfully change the way health care is developed and delivered, and may materially impact numerous aspects of our business. The long-term impact of this law on us is uncertain. However, this new legislation, or any future legislation, could reduce medical procedure volumes, lower reimbursement for our products, and impact the demand for our products or the prices at which we sell our products. In addition, various healthcare reform proposals have also emerged at the state level. We cannot predict with certainty what healthcare initiatives, if any, will be implemented at the state level, or what the ultimate effect of federal healthcare reform or any future legislation or regulation may have on us or on our customers’ purchasing decisions regarding our products and services.
We are subject, directly or indirectly, to federal and state healthcare fraud and abuse and other laws and regulations and could face substantial penalties if we are unable to fully comply with such laws.
As a provider of clinical laboratory testing services, we are subject to extensive and frequently changing federal, state and local laws and regulations governing various aspects of our business. For example, we are subject to healthcare fraud and abuse regulation and enforcement by both the federal government and the states in which we conduct our business. These healthcare laws and regulations include, for example:
• | the federal Anti-Kickback Law, which constrains our marketing practices, educational programs, pricing policies, and relationships with healthcare providers or other entities, by prohibiting, among other things, persons or entities from soliciting, receiving, offering or providing remuneration, directly or indirectly, in return for or to induce either the referral of an individual for, or the purchase, lease order or recommendation of, any good, facility, item or services for which payment may be made, in whole or in part, under a federal healthcare program such as the Medicare and Medicaid programs; |
26
• | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent, and which may apply to entities like us to the extent that our interactions with customers may affect their billing or coding practices; |
• | the federal Health Insurance Portability and Accountability Act of 1996, as amended, or HIPAA, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information, and also established federal crimes for knowingly and willfully executing a scheme to defraud any healthcare benefit program or making false statements in connection with the delivery of or payment for healthcare benefits, items or services, and which imposed certain requirements relating to privacy, security, and transmission of individually identifiable health information; |
• | the federal physician self-referral law, commonly known as the Stark Law, which prohibits a physician from making a referral to an entity for certain designated health services reimbursed by Medicare or Medicaid if the physician (or a member of the physician’s family) has a financial relationship with the entity, and which also prohibits the submission of any claims for reimbursement for designated health services furnished pursuant to a prohibited referral; |
• | the federal Physician Payment Sunshine Act, and its implementing regulations, which requires manufacturers of certain drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children's Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and Human Services information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; |
• | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
• | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payor, including commercial insurers and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, with differing effects. |
In addition, our in-house laboratory is also subject to certification and licensing requirements, laws relating to the health and safety of laboratory workers and the handling, transportation and disposal of medical specimens, infectious and hazardous waste and radioactive materials, and laws, rules and regulations of government and non-government payors relating to billing and reimbursement of genetic tests. Without limiting the above, the clinical laboratory industry is subject to particular scrutiny regarding:
• | test ordering and billing practices; |
• | marketing, sales and pricing practices; |
• | insurance; |
• | "anti-markup" legislation; |
• | health information privacy and security; and |
• | consumer protection and unfair trade practices. |
We are unable to predict what additional federal or state legislation or regulatory initiatives may be enacted in the future regarding our business or the healthcare industry in general, or what effect such legislation or regulations may have on us. Federal or state governments may impose additional restrictions or adopt interpretations of existing laws that could have an adverse effect on us.
We incur significant costs in complying with these laws and regulations. Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations, or our sales techniques or product placement strategies, are found to be in violation of, or to encourage or assist the violation by third parties of, any of the laws described above or any other governmental regulations that apply to us, or if we fail to maintain, renew or obtain necessary permits, licenses and approvals related to our in-house laboratory, we may be subject to
27
penalties, including civil and criminal penalties, damages, fines, exclusion from the Medicare and Medicaid programs, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, suspension or revocation of certifications or licenses that are required to operate our business, injunctions and other associated remedies, the curtailment or restructuring of our operations, denial or withdrawal of product clearances, or private "qui tam" actions brought by individual whistleblowers in the name of the government, any of which could have an adverse effect on our business. We currently rely on our general Code of Ethics and Professional Standards policy, and do not have detailed and sophisticated compliance programs in place, in regard to our relationships with healthcare providers, and we will need to implement a detailed and sophisticated compliance program as our operations grow. Any penalties, damages, fines, exclusions, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. The risk of our being found in violation of these laws is increased by the fact that many of these laws are broad and their provisions are open to a variety of interpretations. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
We use hazardous chemicals and biological materials in our business. Any claims relating to improper handling, storage or disposal of these materials could be time consuming and costly.
Our research and development and manufacturing processes, as well as the operations of our CLIA-certified in-house laboratory, involve the controlled use of hazardous materials, including chemicals, and biological materials, and our operations produce hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from these materials. We may be sued for any injury or contamination that results from our use or the use by third parties of these materials, and our liability may exceed our insurance coverage and our total assets. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous materials and specified waste products, as well as the discharge of pollutants into the environment and human health and safety matters. Compliance with environmental laws and regulations may be expensive, and may impair our research, development and production efforts. If we fail to comply with these requirements, we could incur substantial costs, including civil or criminal fines and penalties, cleanup costs, or capital expenditures for control equipment or operational changes necessary to achieve and maintain compliance. In addition, we cannot predict the impact on our business of new or amended environmental laws or regulations, or any changes in the way existing and future laws and regulations are interpreted and enforced.
As a public company, we will be subject to the requirements related to Section 404 of the Sarbanes-Oxley Act. If we are unable to comply with these requirements in a timely manner, it may affect the reliability of our internal control over financial reporting and negatively impact our business and stock price.
Beginning with our annual report on Form 10-K for the year ending December 31, 2015, we will be required to furnish a report by management on the effectiveness of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act. In addition, our independent registered public accounting firm will be required to attest to the effectiveness of our internal controls over financial reporting beginning with our annual report on Form 10-K following the later of the year following our first annual report required by the Securities and Exchange Commission, and the date on which we are no longer an "emerging growth company." We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) following the fifth anniversary of the completion of this offering, (b) in which we have total annual gross revenue of at least $1.0 billion or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
The process of designing and implementing effective internal controls and procedures is a continuous effort that requires us to anticipate and react to changes in our business and economic and regulatory environments. We cannot assure you that our efforts will be adequate to satisfy our reporting obligations as a public company or that we will be able to successfully complete the procedures and certification and attestation requirements related to Section 404 of the Sarbanes-Oxley Act or that we or our independent registered public accounting firm will not identify additional material weaknesses or significant deficiencies in our internal control over financial reporting. If we identify any reason that we cannot certify as to the effectiveness of our internal control over financial reporting, we could incur additional costs remedying the cause of our failure to certify as to the effectiveness of our internal
28
control over financial reporting, and our reputation, investor perceptions of us and the price of our common stock could be adversely affected and you may not be able to rely on our financial statements.
Failure to timely or accurately bill for our in-house laboratory services in compliance with applicable rules and regulations could have an adverse effect on our business.
Our in-house laboratory bills, and is reimbursed by, various healthcare payors from time to time, including Medicare, Medicaid and insurance companies. The rules and regulations surrounding this billing and reimbursement process are complicated, non-uniform and constantly changing. Changes in these rules and regulations, or the manner in which they are implemented by the applicable payors, may result in additional costs, time and expense to us, which may in turn affect our profitability and our results of operations. Our failure to timely and correctly bill our applicable healthcare payors, and their failure or reluctance to timely reimburse us, could result in an increase in the age of our in-house laboratory accounts receivable and may have an adverse effect on our cash flows and as a result, our results of operations.
In addition, our in-house laboratory may from time-to-time perform tests in advance of payment and without certainty as to the outcome of the billing process. In cases where we do receive a fixed fee per test, we may still have disputes over pricing and billing. We receive payment from individual patients and from a variety of payors, such as commercial insurance carriers and governmental programs. Each payor typically has different billing requirements. Among the factors complicating our billing of third-party payors are:
• | disputes among payors regarding which party is responsible for payment; |
• | disparity in coverage among various payors; |
• | different process, information and billing requirements among payors; and |
• | incorrect or missing billing information, which is required to be provided by the prescribing clinician. |
Additionally, from time to time, payors change processes that may affect timely payment. These changes may result in uneven cash flow or impact the timing of revenue recognized with these payors. These billing complexities, and the related uncertainty in obtaining payment for our products, could negatively affect our revenue, cash flow and profitability.
Our in-house laboratory operations and reputation may be impaired if we do not comply with applicable privacy and information securities laws, regulations and policies.
Our in-house laboratory may from time to time receive, produce and/or maintain sensitive patient data and other personal patient information, including legally protected health information, and personally identifiable information about our customers, payors and patients. The secure processing, storage, maintenance and transmission of this critical information is vital to our operations and business strategy. If we do not adequately safeguard that data and information and it were to become available to persons or entities that should not have access to it, our business could be impaired, our reputation could suffer and we could be subject to fines, penalties and litigation. Unauthorized access, loss or dissemination could also disrupt our operations, including our ability to process tests, provide test results, bill payors or patients, process claims and appeals, provide customer assistance services, conduct research and development activities, collect, process and prepare company financial information, provide information about our tests and other patient and physician education and outreach efforts through our website, and manage the administrative aspects of our business and damage our reputation, any of which could adversely affect our business. Any such breach could also result in the compromise of our trade secrets and other proprietary information, which could adversely affect our competitive position. In addition, the interpretation and application of consumer, health-related, privacy and data protection laws in the United States, Europe and elsewhere are often uncertain, contradictory and in flux. It is possible that these laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. Complying with these various laws could cause us to incur substantial costs or require us to change our business practices and compliance procedures in a manner adverse to our business.
29
If we seek FDA approvals of one or more of our genetic test panels, we may fail to receive favorable clinical results from such test panels that require clinical trials, and even if we receive favorable clinical results, we may still fail to receive the necessary clearances or approvals to market these genetic test panels.
We continually invest in the research and development of new products to expand the menu of testing options for our platform. To commercialize our products, we may be required to undertake time-consuming and costly development activities, sometimes including clinical trials, for which the outcome is uncertain. We do not currently intend to seek PMA approval from the FDA for any of our current genetic test panels. However, if we do determine to seek such approval, we will not know whether clinical trials of our genetic test panels or any other pre-clinical or clinical trials will begin on time or be completed on schedule, if at all. The commencement and completion of clinical trials can be delayed for a number of reasons, including delays related to:
• | obtaining sufficient funding for the clinical trial; |
• | satisfying regulatory requirements to commence a clinical trial; |
• | reaching agreement on acceptable terms with prospective clinical research organizations, or CROs, clinical investigators and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs, clinical investigators and clinical trial sites; |
• | obtaining institutional review board approval to initiate and conduct a clinical trial at a prospective site; |
• | identifying, recruiting and training suitable clinical investigators; and |
• | identifying, recruiting and enrolling subjects to participate in clinical trials. |
Clinical trials may also be delayed, halted or repeated as a result of ambiguous or negative interim results or unforeseen complications in testing. In addition, a clinical trial may be suspended or terminated by us, the FDA, the institutional review board overseeing the clinical trial, any of our clinical trial sites, or other regulatory authorities due to a number of factors, including:
• | failure by us, our employees, our CROs or their employees to conduct the clinical trial in accordance with all applicable FDA or other regulatory requirements or our clinical protocols; |
• | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
• | lack of adequate funding to continue the clinical trial, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional trials and studies and increased expenses associated with the services of our CROs and other third parties; |
• | lack of effectiveness of any product candidate during clinical trials; |
• | slower than expected rates of subject recruitment and enrollment rates in clinical trials; |
• | failure of our CROs or other third-party contractors to comply with all contractual requirements or to perform their services in a timely or acceptable manner; or |
• | unfavorable results from ongoing clinical trials and pre-clinical studies. |
Clinical investigations of in vitro diagnostic tests, including our products and product candidates, typically are exempt from the investigational device exemption, or IDE, requirements. Thus, we do not need the FDA's prior approval to conduct clinical trials of our products, provided the testing is non-invasive, does not require an invasive sampling procedure that presents a significant risk, does not intentionally introduce energy into the subject and is not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established diagnostic device or procedure. The FDA requires each sponsor of a clinical trial to make this determination in the first instance, but the FDA can review any decision. If the FDA disagrees with our decision not to submit an IDE for the clinical trials we conduct, the agency may retroactively require us to submit an IDE and halt the clinical trial until the IDE is approved.
Products that appear promising during early development and preclinical studies may, nonetheless, fail to demonstrate the results needed to support regulatory approval or commercial viability. Even if we receive favorable clinical results, we may still fail to obtain the necessary FDA clearance and approvals. Any such failure, or any material delay in obtaining the necessary clearance or approvals, could adversely affect our business, financial condition and results of operations.
30
Our international operations are subject to trade and anti-corruption laws and regulations.
Due to the international scope of our operations, we are subject to a complex system of import- and export-related laws and regulations, including U.S. regulations issued by the Bureau of Industry and Security and the Office of Foreign Assets Control, as well as the counterparts of these agencies in other countries. Any alleged or actual violations of these U.S. and foreign regulations may subject us to government scrutiny, investigation and civil and criminal penalties, and may limit our ability to import or export our products or to provide services outside the United States. We cannot predict the nature, scope or effect of future regulatory requirements to which our operations might be subject or the manner in which existing laws might be administered or interpreted.
In addition, the U.S. Foreign Corrupt Practices Act and similar foreign anti-corruption laws generally prohibit companies and their intermediaries from making improper payments or providing anything of value to improperly influence foreign government officials for the purpose of obtaining or retaining business, or obtaining an unfair advantage. Recent years have seen a substantial increase in the global enforcement of anti-corruption laws. Our international distributors are domiciled in areas of the world with laws, rules and business practices that differ from those in the United States. As a result, we face the reputational and legal risk that our international distributors will violate applicable laws, rules and business practices. If we or our international distributors are determined to be in violation of these laws, rules and business practices, it may result in severe criminal or civil sanctions for us or our distributors, could disrupt our business, and could result in an adverse effect on our reputation, business and results of operations or financial condition.
Legal Risks
We may not be able to compete effectively if we are unable to protect our proprietary rights.
Our success depends in part on our ability to protect our intellectual property rights, which involve complex legal, scientific and factual questions and uncertainties. We rely upon patent, trade secret, copyright and contract laws to protect proprietary technology and trademark law to protect brand identities. We cannot be certain that we have taken adequate steps to prevent misappropriation of our technology or that our competitors will not independently develop technologies that are substantially equivalent or superior to ours. We also cannot be certain that we have taken adequate steps to protect our inventions through patents or our brand identities through trademarks. To date we have not sought trademark protection for our brand identities in countries outside of the United States, and this may adversely affect our ability to prevent unauthorized use of these brand identities after we, or our distributors, have expended resources to increase their recognition. Our success depends in part on our ability to obtain and maintain patent protection for our products and their use, maintain trade secret protection and operate without infringing upon the proprietary rights of others. Our policy is to protect our proprietary technology through patents, where appropriate, and in other cases, through trade secrets. In addition, we rely on the licenses of patents of third parties. We cannot assure you that any patent applications filed by, assigned to, or licensed to us will be granted, that any patents issued to or licensed by us will provide us with any competitive advantages or adequate protection for inventions, that any patents issued to or licensed by us will not be challenged, invalidated, held to be unenforceable, or circumvented by others or that issued patents, or patents that may be issued, will provide protection against competitive products or otherwise be commercially valuable.
If we fail to obtain and/or maintain sufficient patent protection for our products, our ability to realize a competitive advantage for these products could be materially affected, which may have an adverse effect on our results of operations. In particular, if we fail to obtain and/or maintain patent protection, competitors may be able to produce products that would be directly competitive with our products using similar or identical processes with impunity. Moreover, should any government or court limit or alter patent rights relating to specific genetic information or diagnostic methods, our business could be materially and adversely affected.
Patent law relating to the scope of claims in the fields of healthcare and biosciences generally, and molecular diagnostics in particular, is evolving. Our patent rights and those of others are subject to this uncertainty. For example, our assays utilize specific primer sequences to target genes correlated with various diseases and physiological states. In Mayo Collaborative Services v. Prometheus Laboratories, Inc., 132 S. Ct. 1289 (U.S. 2012), the U.S. Supreme Court invalidated a set of claims that were based on determining a correlation between the concentration of a marker molecule and a disease state. The following year, the Court invalidated claims directed towards specific gene sequences as involving naturally occurring DNA in Association for Molecular Pathology v.
31
Myriad Genetics, Inc., 133 S. Ct. 2107 (U.S. 2013). How these and related issues are eventually resolved by the courts could adversely impact us with respect to the scope of our patent protection, as well as validity of patents held by others.
Our patent rights on our products might conflict with the patent rights of others, whether existing now or in the future. We cannot assure you that the inventors of the patents and applications that we own and license were the first to invent or the first to file on the claimed inventions, and if others were determined to be the first inventors, such determination could materially reduce or even eliminate the value of such patents. We also cannot assure you that a third party does not have or will not obtain patents that dominate the patents we own or license now or in the future. Consequently, we could be subject to charges of patent infringement. The defense and prosecution of patent claims are both costly and time-consuming even if the outcome is ultimately in our favor. An adverse outcome could subject us to significant liabilities to third parties, require disputed rights to be licensed from third parties or require us to cease selling the affected products. As our products are distributed in the marketplace, we expect to face opposition to our intellectual property by competitors. If we face opposition to our intellectual property, we likely will incur substantial costs defending our patents.
Additionally, we rely on trade secrets and proprietary know-how, which we seek to protect, in part, by confidentiality agreements with our collaborators, employees and consultants. We cannot assure you that these agreements will not be breached, that we will have adequate remedies for any such breach or that our trade secrets will not otherwise become known or be independently developed by competitors.
Our success will depend partly on our ability to operate without infringing or misappropriating the proprietary rights of others.
The in vitro diagnostics industry has a history of patent litigation and likely will continue to experience patent litigation suits. A third party could claim that we are infringing a patent, which could prevent us from selling our INFINITI analyzers and tests, or from developing new technologies we expect to utilize or products we expect to sell. We may be sued for infringing or misappropriating the proprietary rights of others. We may have to pay substantial damages, including treble damages and the opposing party's attorney fees, for past infringement if it is determined that our products or their use infringe on a third party's proprietary rights. In addition, in the future we may need to obtain one or more licenses to the intellectual property of others, and the failure to obtain such licenses could prevent us from manufacturing or selling the affected products. Our platform utilizes a wide variety of technologies and we cannot assure you that we have identified or can identify all inventions and patents that may be infringed by the development, manufacture, sale and use of our platform, including its various components. If relevant patents are upheld as valid and enforceable and we are found to infringe, we could be prevented from selling our system unless we can obtain a license to use technology or ideas covered by such patents or are able to redesign our platform to avoid infringement. A license may not be available at all or on commercially reasonable terms, and we may not be able to redesign our platform to avoid infringement.
The landscape for intellectual property rights in the field of genetics is complex. Although a significant amount of intellectual property in the field of genetics is already in the public domain, some of our existing and future tests for our platform could require modification, or the acquisition of certain rights from third parties related to those tests which we have not yet obtained, to avoid infringement. We may not be able to make such modifications or obtain such rights at all or on terms that are commercially acceptable. If we sell an otherwise infringing test without such modifications or such rights, a third party patent holder may sue us for patent infringement. If a court finds that we infringed the third party's patent and that the infringement was willful, we could be ordered to pay treble damages and the patent holder’s attorneys' fees, and we may be enjoined from further selling our tests, which may adversely impact our business, financial condition or results of operations.
We also enter into collaborative agreements with research and academic institutions for the development of additional or enhanced tests to be run on our system. Under these agreements, the institution typically retains the intellectual property rights to any inventions or discoveries made solely by the institution, while we receive a right to negotiate a license to these intellectual property rights. If these institutions independently develop intellectual property that is necessary for us to sell new tests that arise from the collaboration, there can be no assurance that we will be able to obtain a license through our negotiation rights. Acquisition of any necessary intellectual property rights from third parties related to any of our existing or future tests, including licenses to patents, could also be
32
time-consuming and expensive, and could require us to pay significant royalties to third parties. And likewise any such rights may not be available on commercially reasonable terms or at all.
We cannot assure you that any third party intellectual property rights to use technology or biomarkers that we determine are necessary to our business and the development of new tests for our platform can be acquired on commercially reasonable terms or at all. Resulting limitations on our ability to offer certain tests or expand our menu of test panels could have an adverse effect on our business, financial condition and results of operations and negatively impact a key basis upon which we compete.
We have hired and will continue to hire individuals who have experience in molecular diagnostics and these individuals may have confidential trade secret or proprietary information of third parties. We cannot assure you that these individuals will not use this third-party information in connection with performing services for us or otherwise reveal this third-party information to us. Thus, we could be sued for misappropriation of proprietary information and trade secrets. Such claims are expensive to defend and could divert our attention and result in substantial damage awards and injunctions that could have an adverse effect on our business, financial condition or results of operations.
We may need to initiate lawsuits to protect or enforce our patents or other proprietary rights, which would be expensive and, if unsuccessful, may cause us to lose some of our intellectual property rights.
To protect or enforce our patent rights, it may be necessary for us to initiate patent litigation proceedings against third parties, such as infringement suits or interference proceedings. These lawsuits would be expensive, take significant time and would divert management's attention from other business concerns. In the event that we seek to enforce any of our patents against an infringing party, it is likely that the party defending the claim will seek to invalidate the patents we assert, which could put our patents at risk of being invalidated, held unenforceable, or interpreted narrowly, and our patent applications at risk of not being issued. Further, these lawsuits may provoke the defendants to assert claims against us. The patent position of in vitro diagnostics firms is highly uncertain, involves complex legal and factual questions and recently has been the subject of much litigation. We cannot assure you that we will prevail in any of such suits or proceedings or that the damages or other remedies awarded to us, if any, will be commercially valuable.
In the normal course of business, we are subject to litigation, which, if adversely determined, could cause us to incur substantial losses.
We are, from time to time, during the normal course of operating our businesses, subject to various litigation claims and legal disputes. Some of the litigation claims may not be covered under our insurance policies or our insurance carriers may seek to deny coverage. As a result, we might be required to incur significant legal fees, which may have an adverse impact on our financial position. In addition, because we cannot predict the outcome of any action, it is possible that, as a result of current or future litigation, we will be subject to adverse judgments or settlements that could significantly reduce our earnings or result in losses. See "Business — Legal Proceedings."
Risks Relating to this Offering and our Common Stock
There has been no prior market for our common stock, and an active trading market may not develop.
Prior to this offering, there has been no public market for our common stock. An active trading market may not develop following the closing of this offering or, if developed, may not be sustained. The lack of an active market may impair your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. The lack of an active market may also reduce the fair market value and increase the volatility of your shares. An inactive market may also impair our ability to raise capital by selling shares and may impair our ability to acquire other companies or technologies by using our shares as consideration.
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for investors purchasing shares in this offering.
The initial public offering price for the shares of our common stock sold in this offering will be determined by negotiation between the representatives of the underwriters and us. This price may not reflect the market price of our common stock following this offering. In addition, the market price of our common stock is likely to be highly volatile and may fluctuate substantially due to many factors, including:
• | actual or anticipated fluctuations in our results of operations; |
33
• | variance in our financial performance from the expectations of market analysts; |
• | conditions and trends in the markets we serve; |
• | changes in our pricing policies or the pricing policies of our competitors; |
• | legislation or regulatory policies, practices or actions; |
• | the announcements of significant new products, contracts, acquisitions or strategic alliances by us or our competitors; |
• | the commencement or outcome of litigation; |
• | our ability to repay remaining indebtedness after completion of this offering; |
• | our sale of common stock or other securities in the future or sales of our common stock by our significant stockholders; |
• | changes in market valuation or earnings of our competitors; |
• | the trading volume of our common stock; |
• | the loss of key personnel; |
• | changes in the estimated future size and growth rate of our markets; |
• | general economic conditions; and |
• | general volatility of the stock market. |
These broad market and industry factors may materially harm the market price of our common stock, regardless of our operating performance. In the past, following periods of volatility in the market price of a company's securities, securities class action litigation has often been instituted against that company. Such litigation, if instituted against us, could result in substantial costs and a diversion of management’s attention and resources, which could adversely harm our financial condition and results of operations.
If equity research analysts do not publish research or reports about our business or if they issue unfavorable commentary or downgrade our common stock, the price of our common stock could decline.
The trading market for our common stock will rely in part on the research and reports that equity research analysts publish about our business and us. The price of our stock could decline if one or more equity research analysts downgrade our stock or if those analysts issue other unfavorable commentary or cease publishing reports about our business or us.
We currently do not intend to pay dividends on our common stock and, consequently, your only opportunity to achieve a return on your investment is if the price of our common stock appreciates.
We currently do not plan to declare dividends on shares of our common stock for the foreseeable future. See "Dividend Policy" for more information. Consequently, your only opportunity to achieve a return on your investment in our company will be if the market price of our common stock appreciates and you sell your shares at a profit. There is no guarantee that the price of our common stock that will prevail in the market after this offering will ever exceed the price that you pay. In addition, the lack of dividends may limit the number of potential buyers of our common stock.
Future sales of our common stock may depress our share price.
After this offering, we will have shares of common stock outstanding, based upon our outstanding shares as of October 24, 2014 after giving effect to the conversion of our convertible preferred stock into common stock. Sales of substantial amounts of our common stock in the public market following this offering, or the perception that these sales may occur, could cause the market price of our common stock to decline. After the lock-up agreements pertaining to this offering expire, additional stockholders will be able to sell their shares in the public market, subject to legal restrictions on transfer. As described in "Shares Eligible for Future Sale — Registration Rights," we also have granted certain of our security holders registration rights which, subject to certain limitations, will permit them to cause us to file a registration statement with respect to the registrable securities held by them. As soon as practicable upon completion of this offering, we also intend to file a registration statement covering shares of our common stock issuable upon exercise of outstanding stock options or reserved for future issuance under our equity incentive and stock purchase plans. We may also sell additional shares of common stock in subsequent public
34
offerings, which may adversely affect market prices for our common stock. See "Shares Eligible for Future Sale" for more information.
We have broad discretion in the use of the net proceeds from this offering, and our investment of these proceeds may not yield a favorable return.
Our management will have broad discretion in the application of the net proceeds from this offering, including for any of the purposes described in "Use of Proceeds." Accordingly, you will have to rely upon the judgment of our management with respect to the use of the proceeds. Our management may spend a portion or all of the net proceeds from this offering in ways that our stockholders may not desire or that may not yield a significant return or any return at all. Of the net proceeds from this offering, we intend to use approximately $ million to satisfy outstanding accounts payable to advisers and service providers incurred in connection with our prior capital raising activities conducted from 2008 through 2013, and approximately $ million to retire existing debt obligations. The payment of these amounts will not enhance our business operations or increase our revenue. The failure by our management to effectively apply the remaining net proceeds from this offering could harm our business. Pending their use, we may also invest the net proceeds from this offering in a manner that does not produce income or that loses value.
As a new investor, you will experience immediate and substantial dilution as a result of this offering.
The initial public offering price of our common stock will be considerably more than the net tangible book value per share of our outstanding common stock. Accordingly, investors purchasing shares of common stock in this offering will:
• | pay a price per share that substantially exceeds the value of our assets after subtracting liabilities; and |
• | contribute % of the total amount invested to fund our company, and will own % of the shares of common stock outstanding after this offering and the use of proceeds therefrom. |
To the extent that outstanding stock options and warrants are exercised, there will be further dilution to new investors. See "Dilution" for more information.
Certain provisions of our corporate governing documents and Delaware law could make an acquisition of our company more difficult.
Our certificate of incorporation and bylaws contain provisions that may make the acquisition of our company more difficult without the approval of our board of directors. For example, our certificate of incorporation and bylaws:
• | authorize the issuance of preferred stock that can be created and issued by our board of directors without prior stockholder approval, commonly referred to as "blank check" preferred stock, with rights senior to those of our common stock; |
• | limit the persons who can call special stockholder meetings; |
• | provide that a supermajority vote of our stockholders is required to amend our bylaws and some portions of our certificate of incorporation; |
• | establish advance notice requirements to nominate persons for election to our board of directors or to propose matters that can be acted on by stockholders at stockholder meetings; |
• | do not provide for cumulative voting in the election of directors; and |
• | provide for the filling of vacancies on our board of directors between annual meetings by action of a majority of the directors and not by the stockholders. |
These and other provisions in our organizational documents could allow our board of directors to affect your rights as a stockholder in a number of ways, including making it more difficult for stockholders to replace members of the board of directors. Because our board of directors is responsible for approving the appointment of members of our management team, these provisions could in turn affect any attempt to replace the current management team. These provisions could also limit the price that investors would be willing to pay in the future for shares of our common stock.
35
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which, subject to certain exceptions, impose certain restrictions on mergers and other business combinations between any holder of 15% or more of our common stock and us. See "Description of Capital Stock — Provisions of Our Certificate of Incorporation and Bylaws and Delaware Law That May Have an Anti-Takeover Effect."
36
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements that reflect our current views with respect to, among other things, future events or our future financial performance. All statements in this prospectus other than statements of historical fact, including statements regarding future events, our future financial performance, business strategy and plans, predictions about our markets, and objectives of management for future operations, are forward-looking statements.
In some cases, you can identify forward-looking statements by terminology including "anticipates," "believes," "can," "continue," "could," "estimates," "expects," "intends," "may," "might," "plans," "potential," "predicts," "should," "will" or "would," or the negative of these terms or other comparable terminology. Forward-looking statements used in this prospectus address topics such as, among other things:
• | our business strategy; |
• | our development of new products; |
• | our anticipated growth strategies; |
• | future demand for our products; |
• | anticipated trends in our business, trends in the market for our products and trends in the adoption of new technologies; |
• | competition and competitive factors or trends in our market; |
• | uncertainty regarding our future operating results, including our projected sales, net sales, financial results, cash flows, pricing and similar items; |
• | our intended use of proceeds from this offering; |
• | our plans for additional clinical trials and seeking regulatory clearances for our INFINITI HTS and additional test panels; |
• | our ability to continue to control costs and maintain quality; |
• | anticipated changes in our expenses over future periods; |
• | the impact of U.S. health care reform legislation and other health care policy changes; |
• | the protection or acquisition of, investment in or legal proceedings regarding our intellectual property; |
• | our ability to obtain FDA or other regulatory clearances or approvals; |
• | the regulatory environment for our products and our business, and our ability to comply with and adapt to such environment; and |
• | the plans, objectives, expectations and intentions expressed in this prospectus that are not historical facts. |
These statements are only predictions. Actual events or results may differ materially. These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in "Risk Factors." We cannot guarantee future results, levels of activity, performance or achievements. We undertake no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, or to conform these statements to actual results or changes in our expectations.
Moreover, we operate in a competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this prospectus may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements.
37
MARKET, INDUSTRY AND OTHER DATA
Unless otherwise indicated, information contained in this prospectus concerning our industry and the market segments in which we operate, including our general expectations and market position, market opportunity and market size, is based on information from various sources, on assumptions that we have made that are based on that information and other similar sources and on our knowledge of the markets for our products. This information involves a number of assumptions and limitations, and you are cautioned not to give undue weight to or reliance on such estimates. In addition, projections, assumptions and estimates of our future performance and the future performance of the industry in which we operate is necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in "Risk Factors" and elsewhere in this prospectus. These and other factors could cause results to differ materially from those expressed in the estimates made by the independent parties and by us.
38
USE OF PROCEEDS
We estimate that our net proceeds from this offering will be approximately $ million, based upon an assumed initial public offering price of $ per share, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us, or approximately $ million if the underwriters' option to purchase additional shares is exercised in full. A $1.00 increase or decrease in the assumed initial public offering price of $ per share would increase or decrease, as applicable, the net proceeds to us from this offering by approximately $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, an increase or decrease in the number of shares we sell in the offering will increase or decrease, as applicable, our net proceeds by an amount equal to such number of shares multiplied by the public offering price, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
We intend to use the net proceeds of this offering for the following purposes:
• | $ million to fund capital expenditures for expansion of manufacturing capabilities; |
• | $ million to expand our sales and marketing efforts; |
• | $ million to fund research and development, including $ million to fund our regulatory efforts related to seeking 510(k) clearance for the following seven test panels: CYP450-2B6 QUAD, CYP450-3A4, CYP450-3A5, CYP450-3A4/3A5, CYP450-3A4/3A5 duplex, bacterial vaginosis and CT-NG QUAD; |
• | $ million to satisfy all of our outstanding notes payable and certain of our accounts payable; and |
• | the balance for working capital and general corporate purposes. |
The expected use of the net proceeds of this offering represents our current intentions based upon our present plans and business condition. The amounts and timing of our actual expenditures may vary significantly and will depend upon numerous factors, such as the level of research and development investment required by our business (including for clinical studies and trials), cash flows from operations, the anticipated growth of our business and other factors. Pending the allocation of the net proceeds of this offering, we intend to invest the net proceeds in short-term, interest-bearing obligations, investment grade instruments, and certificates of deposit or guaranteed obligations of the U.S. government.
We are required by the terms of our outstanding subordinated promissory notes to use 50% of the net cash proceeds of this offering to repay outstanding senior and subordinated indebtedness. As of September 30, 2014, we had outstanding total indebtedness and accrued interest under these promissory notes of approximately $21.2 million, and our promissory notes have a weighted-average fixed annual interest rate of 8.6%. These notes mature in December 2014, June 2015 and November 2015.
DIVIDEND POLICY
We do not anticipate paying any cash dividends after this offering and for the foreseeable future. Instead, we anticipate that all of our earnings on our common stock will be used to provide working capital, to support our operations and to finance the growth and development of our business. Any future determination relating to dividend policy will be made at the discretion of our board of directors and will depend on our future earnings, capital requirements, financial condition, future prospects, financing arrangements, applicable Delaware law, which provides that dividends are only payable out of surplus or current net profits, and other factors our board of directors deems relevant.
39
CAPITALIZATION
The following table sets forth our capitalization as of September 30, 2014:
• | on an actual basis; |
• | on a pro forma basis to reflect the assumed conversion of all outstanding shares of our Series A, Series B, Series C, Series D, Series E and Series NC Convertible Preferred Stock into 7,974,932 shares of our common stock. All preferred stock will convert automatically to common stock immediately prior to the closing of this offering, except for Series C, Series E and Series NC Convertible Preferred Stock, each of which converts automatically only if this offering results in net proceeds to us of at least $25.0 million after deducting underwriting discounts and commissions, which we expect to occur in connection with the completion of this offering; and |
• | on a pro forma as adjusted basis to additionally reflect the sale of shares of our common stock in this offering at an assumed initial offering price of $ per share, and after deducting estimated underwriting discounts and commissions and our estimated offering expenses, and the application of approximately $21.2 million of the net proceeds of this offering to repay principal and accrued interest as of September 30, 2014 under all of our outstanding promissory notes, including notes to related parties. |
40
The pro forma and pro forma as adjusted information below is illustrative only and our capitalization following the completion of this offering will be adjusted based on the actual initial public offering price and other terms of this offering determined at pricing. You should read this table together with "Use of Proceeds," "Selected Financial Data," "Management's Discussion and Analysis of Financial Condition and Results of Operations," "Description of Capital Stock" and our financial statements and the related notes appearing elsewhere in this prospectus.
As of September 30, 2014 | |||||||||||
Actual | Pro forma | Pro forma as adjusted (1) | |||||||||
(unaudited and in thousands, except share and per share data) | |||||||||||
Senior and subordinated promissory notes | $ | 21,174 | $ | 21,174 | $ | — | |||||
Convertible preferred stock, $0.01 par value, 30,000,000 authorized, actual, pro forma and pro forma as adjusted (2) | |||||||||||
Series A Convertible Preferred Stock, $0.01 par value, 1,589,600 authorized, 1,042,290 issued and outstanding, actual, 0 issued and outstanding, pro forma and pro forma as adjusted | 4,990 | — | — | ||||||||
Series B Convertible Preferred Stock, $0.01 par value, 4,515,858 authorized, 1,626,519 issued and outstanding, actual, 0 issued and outstanding, pro forma and pro forma as adjusted | 12,288 | — | — | ||||||||
Series C Convertible Preferred Stock, $0.01 par value, 6,850,000 authorized, 2,336,083 issued and outstanding, actual, 0 issued and outstanding, pro forma and pro forma as adjusted | 17,647 | — | — | ||||||||
Series D Convertible Preferred Stock, $0.01 par value, 3,423,258 authorized, 1,288,651 issued and outstanding, actual, 0 issued and outstanding, pro forma and pro forma as adjusted | 11,126 | — | — | ||||||||
Series E Convertible Preferred Stock, $0.01 par value, 5,500,000 authorized, 266,655 issued and outstanding, actual, 0 issued and outstanding, pro forma and pro forma as adjusted | 1,381 | — | — | ||||||||
Series NC Convertible Preferred Stock, $0.01 par value, 4,287,074 authorized, 1,414,734 issued and outstanding, actual, 0 issued and outstanding pro forma and pro forma as adjusted | 5,733 | — | — | ||||||||
Stockholders’ equity/(deficit): | |||||||||||
Common stock, $0.01 par value, 220,000,000 authorized, actual, pro forma and pro forma as adjusted, 3,747,319 issued and outstanding, actual, 11,722,251 issued and outstanding, pro forma and issued and outstanding, pro forma as adjusted | 114 | 114 | |||||||||
Additional paid-in capital | 28,053 | 81,218 | |||||||||
Accumulated deficit | (104,162 | ) | (104,162 | ) | |||||||
Total stockholders' equity/(deficit) | (75,995 | ) | (22,830 | ) | |||||||
Total capitalization | $ | (1,656 | ) | $ | (1,656 | ) | |||||
(1) | A $1.00 increase or decrease in the assumed initial public offering price of $ per share in this offering would increase or decrease, as applicable, each of additional paid-in capital, total stockholders' equity/(deficit) and total capitalization by $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, an increase or decrease in the number of shares we sell in the offering will increase or decrease, as applicable, our net proceeds by an amount equal to such increased or decreased number of shares multiplied by the public offering price, less estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
(2) | Our convertible preferred stock has been classified as temporary equity on our balance sheets instead of stockholders’ deficit due to the possibility of the occurrence of certain change in control events that are outside of our control, including our sale or transfer of control, which trigger the rights of holders of the convertible preferred stock to cause its redemption. Accordingly, these shares are considered contingently redeemable. |
41
DILUTION
As of September 30, 2014, we had a net tangible book deficit of $76.0 million, or approximately $20.28 per share of common stock, not taking into account the conversion of all outstanding series of convertible preferred stock into common stock. Net tangible book deficit per share represents the amount of our total tangible assets less total liabilities, divided by the number of common shares outstanding.
Included in our total liabilities as of September 30, 2014 is approximately $21.2 million of indebtedness and accrued interest under our outstanding promissory notes.
After giving effect to the conversion of all outstanding series of convertible preferred stock into common stock, as of September 30, 2014 we would have had a net tangible book deficit of $22.8 million, or $1.95 per share of common stock. Pro forma net tangible book deficit per share represents the amount of our total tangible assets less our total liabilities, divided by the number of shares of common stock outstanding as of September 30, 2014 after giving effect to the conversion of all outstanding series of convertible preferred stock into shares of common stock, which will occur immediately prior to the completion of this offering.
After giving effect to the sale by us of shares of common stock in this offering at an assumed initial public offering price of $ per share, and after deducting underwriting discounts and commissions and the estimated offering expenses payable by us, our pro forma net tangible book value as of September 30, 2014 would have been approximately $ million, or approximately $ per share. This amount represents an immediate increase in pro forma net tangible book value of $ per share to our existing stockholders and an immediate dilution in pro forma net tangible book value of approximately $ per share to new investors purchasing shares of common stock in this offering at the assumed initial public offering price. We determine dilution to new investors participating in this offering by subtracting the pro forma net tangible book value per share after this offering from the amount of cash that a new investor will pay for a share of common stock in this offering.
Assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting underwriting discounts and commissions, a $1.00 increase or decrease in the assumed initial public offering price of $ per share would increase or decrease, as applicable, the pro forma net tangible book value by $ per share to our existing stockholders, and the dilution in pro forma net tangible book value by $ per share to new investors participating in this offering.
The following table illustrates this dilution on a per share basis:
Actual net tangible book deficit per share as of September 30, 2014 | $ | (20.28 | ) | |
Assumed initial public offering price per share | ||||
Increase in pro forma net tangible book value upon conversion of convertible stock | $ | 18.33 | ||
Pro forma net tangible book deficit per share as of September 30, 2014 | $ | (1.95 | ) | |
Increase in pro forma as adjusted net tangible book value per share attributable to this offering | ||||
Pro forma as adjusted net tangible book value per share after this offering | ||||
Dilution in pro forma as adjusted net tangible book value per share to new stockholders | ||||
The discussion above assumes no exercise of the underwriters’ over-allotment option. If the underwriters' over-allotment option is exercised in full our adjusted pro forma net tangible book value at September 30, 2014 would have been $ million, or $ per share of common stock, representing an immediate increase in pro forma net tangible book value of $ per share of common stock to our existing stockholders and an immediate dilution of $ per share to investors participating in this offering.
In addition, the above discussion and table assume no exercise of stock options or warrants to purchase common stock or preferred stock. As of September 30, 2014, we had outstanding options to purchase a total of 1,469,055 shares of common stock and warrants to purchase a total of 3,892,249 shares of common stock, at weighted average exercise prices of $6.25 per share and $6.04 per share, respectively.
If all of the outstanding options and warrants as of September 30, 2014 with exercise prices at or below $ per share, had been exercised (and the convertible preferred stock issuable upon the exercise of the preferred stock
42
purchase warrants was converted into common stock), our pro forma net tangible book deficit would have been $ per share of common stock, representing an immediate increase in pro forma net tangible book value of $ per share of common stock and dilution in pro forma net tangible book value to investors in this offering would be $ per share of common stock.
43
SELECTED FINANCIAL DATA
The selected financial data set forth below is derived in part from and should be read in conjunction with our financial statements, the related notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" included elsewhere in this prospectus. The statement of operations data for each of the years ended December 31, 2013 and 2012, and the balance sheet data as of December 31, 2013 and 2012 were derived from our audited financial statements appearing elsewhere in this prospectus. The statement of operations data for the nine months ended September 30, 2014 and 2013 and the balance sheet data as of September 30, 2014 were derived from our unaudited financial statements appearing elsewhere in this prospectus. The unaudited financial data, in management’s opinion, has been prepared on the same basis as the audited financial statements and related notes included elsewhere in this prospectus, and include all adjustments, consisting only of normal recurring adjustments, that our management considers necessary for a fair presentation of the information for the periods presented. The results of operations for the years ended December 2013 and 2012 and for the nine months ended September 30, 2014 and 2013 are not necessarily indicative of the results that may be expected for the year ended December 31, 2014 or any other period.
Nine Months Ended September 30, | Years Ended December 31, | ||||||||||||||
2014 | 2013 | 2013 | 2012 | ||||||||||||
(in thousands, except share and per share data) | |||||||||||||||
(unaudited) | |||||||||||||||
Statement of Operations Data: | |||||||||||||||
Product revenue, net | $ | 9,022 | $ | 13,044 | $ | 16,938 | $ | 18,307 | |||||||
Service revenue, net | 9,703 | 229 | 1,145 | 102 | |||||||||||
Net revenue | $ | 18,725 | $ | 13,273 | $ | 18,083 | $ | 18,409 | |||||||
Cost of sales | 3,968 | 4,269 | 5,726 | 7,147 | |||||||||||
Cost of services | 2,677 | 1,530 | 2,032 | — | |||||||||||
Gross profit | 12,080 | 7,474 | 10,325 | 11,262 | |||||||||||
Operating expenses: | |||||||||||||||
Research and development | 1,540 | 1,715 | 2,306 | 2,355 | |||||||||||
General and administrative | 3,308 | 7,210 | 8,302 | 4,715 | |||||||||||
Sales and marketing | 5,334 | 1,898 | 2,998 | 2,269 | |||||||||||
Total operating expenses | 10,182 | 10,823 | 13,606 | 9,339 | |||||||||||
Income/(loss) from operations | 1,898 | (3,349 | ) | (3,281 | ) | 1,923 | |||||||||
Interest expense, net | (1,361 | ) | (1,186 | ) | (1,820 | ) | (2,602 | ) | |||||||
Gains/(loss) on extinguishment of debt | (1,872 | ) | (140 | ) | (140 | ) | (1,996 | ) | |||||||
Other income/(expense), net | (5 | ) | (2,095 | ) | (1,877 | ) | 5 | ||||||||
Change in the fair value of stock warrant liabilities | 375 | 5 | 46 | 163 | |||||||||||
Net loss | $ | (965 | ) | $ | (6,765 | ) | $ | (7,072 | ) | $ | (2,507 | ) | |||
Net loss from continuing operations per common share, basic and diluted | $ | (0.26 | ) | $ | (2.22 | ) | $ | (2.21 | ) | $ | (0.95 | ) | |||
Weighted average common shares used in per share amounts | 3,743,084 | 3,042,029 | 3,200,286 | 2,652,275 | |||||||||||
44
As of September 30, | As of December 31, | ||||||||||
2014 | 2013 | 2012 | |||||||||
(in thousands) | |||||||||||
(unaudited) | |||||||||||
Balance Sheet Data: | |||||||||||
Cash and cash equivalents | $ | 556 | $ | 2,053 | $ | 332 | |||||
Current Assets | 12,719 | 8,267 | 6,250 | ||||||||
Total assets | 15,372 | 10,392 | 10,047 | ||||||||
Total debt | 18,491 | 18,995 | 17,529 | ||||||||
Convertible preferred stock | 53,165 | 53,165 | 53,165 | ||||||||
Total stockholders' equity/(deficit) | (75,995 | ) | (77,387 | ) | (76,206 | ) | |||||
45
MANAGEMENT'S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with "Selected Financial Data" and our financial statements and the related notes appearing elsewhere in this prospectus. This discussion may contain forward-looking statements that are based upon current expectations that involve risks and uncertainties. Our actual results and the timing of many events could differ materially from those anticipated in forward-looking statements as a result of many factors, including those set forth under "Risk Factors" and elsewhere in this prospectus. You should carefully read the "Risk Factors" section of this prospectus to gain an understanding of the important factors that could cause actual results to differ materially from our forward-looking statements. Please also see the section titled "Special Note Regarding Forward-Looking Statements."
Overview
We are a commercial stage molecular diagnostics company offering an innovative and proprietary technology platform to clinical reference laboratories, specialty clinics and hospital laboratories. Our platform consists of a family of multiplexing INFINITI analyzers, an extensive and expanding menu of genetic test panels, and proprietary microarrays, reagent modules and other related consumables. Our INFINITI analyzers, which include the INFINITI, the INFINITI PLUS and the recently introduced INFINITI HIGH THROUGHPUT SYSTEM, or INFINITI HTS, are easy to use and automate a number of the discrete processes of genetic analysis with minimal manual intervention, which reduces our customers' need for multiple, specialized instruments, and offer a variety of throughput capabilities together with a demonstrated high level of accuracy and reproducibility. Our genetic test panels are focused on large and growing markets primarily in the areas of personalized medicine, women's health, infectious diseases and genetic disorders. Genetic tests are performed on our INFINITI analyzers using our proprietary BioFilmChip microarrays, related Intellipac Reagent Management Modules and other related consumables.
In the United States, we offer our family of INFINITI analyzers to our customers either through direct sales, monthly rental plans and our Reagent Access Plan, or RAP. Under the RAP, an INFINITI analyzer is placed at the customer’s location at no initial cost to the customer. We then seek to recover the cost of the analyzer over time by generating significant recurring demand for our testing consumables, including our test-specific BioFilmChips, which are our proprietary microarrays, and Intellipac Reagent Management Modules, which provide the proprietary reagent used to complete our genetic tests. We have established a nine-person domestic direct sales force that is regionally deployed and performs market development as well as account management. We intend to significantly expand the size of our sales force within the next 12 months.
Internationally, we offer our INFINITI analyzers solely through our network of distributors that sell to our customers. We have established 12 distributor relationships to market our platform in 23 countries outside the United States. Our distributor agreements allow the purchase of our products at a discounted price. The distributors then resell our products to customers in the territories covered by their respective agreements. We plan to expand our distributor network outside the United States. During the nine months ended September 30, 2014 and 2013, and the years ended December 31, 2013 and 2012, sales outside the United States accounted for 14%, 10%, 14% and 9% of product sales, respectively. We expect sales growth outside of the United States to keep pace generally with our anticipated growth of domestic sales. We primarily sell our BioFilmChips, Intellipac Reagent Management Modules and other consumables pursuant to standard purchase orders.
As a commercial stage company in the rapidly evolving market for molecular diagnostics, we face numerous risks and uncertainties. The sales cycles for our analyzers is typically lengthy, which makes it difficult for us to accurately forecast revenue for any given period. The manufacturing, installation and training cycles for our INFINITI analyzers also typically involve a significant investment of working capital by us before we begin to receive revenue from our customers, particularly under our RAP, and our ability to fulfill orders for our analyzers is primarily dependent on manufacturing lead time. Our manufacturing lead time can be affected by ordering and delivery delays, parts availability, supplier availability as well as market demand, because many of the components for our analyzers demand spans across multiple segments of manufacturing and industry. As a result, we may incur inventory-carrying and other working capital expenses that may vary over time and may make our operating results in any given period difficult for us to forecast. Our revenue and operating results in a given period may also vary due to the changing mix among any of the following factors:
46
• | whether our customers purchase, rent or lease our INFINITI analyzers, each of which impact the way in which we recognize revenue; |
• | the mix of sales of our INFINITI analyzers as compared to consumables; and |
• | the introduction of new or enhanced tests. |
Changes in any of these relative mixes can have an impact on the timing of our recognition of revenue, as analyzer sales result in revenue to us upon the delivery of the analyzer and analyzer leases or rentals involve the recapture of analyzer costs through the sale of consumables, or on our gross margin, as consumables sales typically have significantly higher margins than those of analyzer sales. Further, our revenue and operating results are difficult to predict because many of our tests have been launched relatively recently, and we have a number of tests in development. As a result, we do not have sufficient history with these new and in-development tests to reliably forecast revenue for those tests. As of September 30, 2014, we had cash and cash equivalents of $0.6 million. We have historically been exposed to a concentration of credit risk with a small number of large customers, but we have seen a decrease in that concentration with the expansion of our customer base, which we expect to continue. For the nine months ended September 30, 2014, no customer accounted for 10% or more of our revenue, and for the nine months ended September 30, 2013 one customer, Natural Molecular Testing Corporation, or NMTC, accounted for 40% of our revenue. For the years ended December 31, 2013 and 2012, our two largest customers each period, NMTC and AI Biotech, accounted for 31% and 12%, and 43% and 16%, respectively, of our revenue.
Key Factors Affecting Performance
Sales of test panels
We generate substantially all of our revenue through the sale of our genetic tests directly to customers and through our CLIA-certified laboratory, and we expect this trend to continue for the foreseeable future. Average revenue per microarray and average tests per microarray are key factors we use to measure our business. For the nine months ended September 30, 2014 and 2013, and for the years ended December 31, 2013 and 2012, our average revenue per microarray was $91.39, $80.29, $66.73 and $81.53, respectively, and our average tests per microarray was 1.98, 1.64, 1.66 and 1.73, respectively. For purposes of calculating average revenue per microarray, we consider a "microarray" to consist of a single BioFilmChip and the portion of the associated Intellipac Reagent Management Module and related amplification mixture required to perform the genetic test. We calculate our average tests per microarray based on the average number of tests available to run on each microarray that is included in the foregoing calculation. Our wide variety of genetic test panels are sold at various prices and, accordingly, our average revenue per microarray is impacted by the mix of tests sold during each period. However, we expect our average test per microarray to increase as our multiple patient arrays, or MPAs, further penetrate the market and duplex and quad test panels replace single and duplex test panels.
Due to the wide variety in throughput capability for, and the levels of customer utilization of, our analyzers, we believe that the total number of analyzers sold, rented and placed with customers does not provide a direct correlation to our future revenue growth, and thus we do not view it as a key metric for our business. Additionally, many of our customers utilize more than one model of our analyzers, and because the same consumables can be used on all of our analyzers, we are not able to accurately calculate our consumables sold by each type of analyzer or revenue dollars earned by each type of analyzer.
Since its introduction in February 2014, we have received 20 orders from customers for INFINITI HTS analyzers. As of September 30, 2014, we have delivered 17 HTS analyzers and we expect to deliver the remaining HTS orders by the end of the year. Customer demand has been outpacing our manufacturing lead time, but we do not expect this trend to continue. Of these remaining orders, two analyzers are sales where we expect to recognize revenue upon shipping, and one is a RAP. Under the RAP, an INFINITI analyzer is placed at the customer's location at no initial cost to the customer, subject to contractual minimum consumables sales thresholds. If these consumables sales thresholds are not met, we have the right to reclaim our INFINITI analyzer from the customer. During the nine months ended September 30, 2014 no analyzers were reclaimed, and for the year ended December 31, 2013, we reclaimed 14 INFINITI analyzers either because the respective customer failed to purchase the minimum amount of consumables or otherwise elected not to retain the INFINITI analyzer at the end of the term of the applicable RAP. We primarily sell our consumables pursuant to standard purchase orders that may be issued and filled or canceled on relatively short notice. Accordingly, we do not have a material backlog of consumables.
47
Investment in infrastructure and growth
Our ability to increase our sales of genetic tests and to further penetrate our target markets are dependent in large part on our ability to invest in our infrastructure and in our sales and marketing efforts. In order to drive future growth, we plan to hire additional sales personnel and invest in marketing our products to our target customers both in the United States and internationally. This will lead to corresponding increases in our operating expenses, and thus may negatively impact our operating results in the short term, although we believe that these investments will grow and improve our business and financial condition over the long term. Marketing and selling to our target customers involves a substantial amount of time and effort, as it generally involves physical site visits and an investment in educating potential customers regarding the capabilities and benefits of molecular diagnostics testing in general and of our INFINITI analyzers specifically. There can be a significant amount of time invested in marketing to a customer before that customer makes a purchasing decision, followed by what can be a material amount of time invested by the customer in validating the applicable INFINITI analyzer. As a result, it will be difficult for us to determine whether and when our investments in sales and infrastructure will be recovered through sales to new customers or additional sales to existing customers. In the past it has taken approximately six months to complete a sales cycle for our INFINITI analyzer. Recent demand for our product has reduced this sales cycle to closer to three months, although we expect our sales cycle trend to return to its historical levels in the foreseeable future.
We will also incur substantial operating costs in connection with our transition to operating as a public company.
Utilization of our products
We believe there is significant opportunity to increase the number and type of genetic test panels we sell as awareness of opportunities in molecular diagnostics testing becomes more apparent to our targeted customer base. We have invested significant time and effort to develop detailed educational materials to be used by, and to inform, our customers’ sales and marketing networks. We believe that this will assist our customers in taking advantage of meaningful growth opportunities in molecular diagnostics testing in our target market segments, which may result in increased demand for our products. We believe that as more of our present and future customers realize the potential of molecular diagnostics testing, our sales will continue to grow and diversify.
For a discussion of additional factors affecting our performance, see "Risk Factors."
Financial Operations Overview
Revenue
We generate revenue from the sale of our products and services. Our products include our INFINITI analyzers and our high-margin consumables used to run tests with our analyzers. Our consumables include our BioFilmChips, Intellipac Reagent Management Modules and various other disposable products consumed in the testing process. Generally, the pricing of our consumables for customers that participate in our RAP is higher than for our customers who own, or rent monthly, an INFINITI analyzer; however, we may discount the price of consumables depending on the volume and nature of the products purchased by our customers. Revenue generated by our CLIA-certified laboratory is classified as services revenue in our financial statements. We expect that increases in our revenue will be driven largely by the growth of the overall molecular diagnostics market, increased utilization of our products by our existing customers, the expansion of our domestic sales force and international distribution network, the placement of additional INFINITI analyzers with new and with existing customers, and our development of additional and enhanced tests for sale in connection with our platform.
In late 2013, as we prepared to launch our INFINITI HTS, we began operations of a CLIA-certified laboratory to complement and support our efforts to attract customers entering the molecular diagnostics market, in particular potential INFINITI HTS customers. This laboratory is located at our headquarters in Vista, California and is licensed in the State of California as well as a number of other applicable states. Our laboratory provides molecular testing services to other reference laboratories in the areas of personalized medicine, women’s health, oncology, infectious diseases and genetic disorders. We expect that growth in revenue generated from our laboratory will decline as a percentage of overall growth in revenue as we manufacture and sell our INFINITI HTS analyzers, because substantially all of our laboratory revenue is currently generated from sales to INFINITI HTS customers. As new customers complete validation of their INFINITI HTS analyzers and begin conducting their own genetic testing, we
48
expect that our laboratory will provide fewer test services to them, and that the services revenue generated from sales to these customers will transition to product revenue. We anticipate that our overall laboratory revenue will stabilize and remain relatively flat for the foreseeable future as new INFINITI HTS customers utilize the services of our CLIA-certified laboratory during the time that sales to these customers are consummated, their INFINITI HTS analyzers are manufactured and delivered, and our customers validate their own laboratory developed tests, or LDTs, on our INFINITI HTS analyzers.
The variability of our revenue from period to period depends in part on our product sales mix of INFINITI analyzers and consumables as well as tests received from our HTS customers for genetic testing. We anticipate that our revenue from the sale of consumables will become a larger portion of our total revenue over time as we expand our customer base and our customers increase their utilization of our INFINITI analyzers. In the United States, we believe that for the foreseeable future our RAPs will continue to account for more placements of INFINITI analyzers than direct sales.
Cost of sales and services
Cost of sales and cost of services includes manufacturing and laboratory expenses related to product costs, including materials, labor, general manufacturing overhead and other costs, and royalty obligations. Cost of sales also includes estimated product warranty costs. Our general manufacturing overhead and other costs include indirect labor, facilities costs and depreciation of capitalized manufacturing equipment. Cost of sales also includes depreciation (over a three year period) of capitalized INFINITI analyzers placed under our RAPs, since we place the analyzers at the customers’ facility at no direct cost to the customer. We expect cost of sales to decline slightly as a percentage of revenue, as our consumables sales mix shifts to higher average tests per microarray.
Research and development
Research and development expenses include (i) the cost of personnel and consultants related to our activities in developing and improving our INFINITI analyzers and the development and expansion of our menu of test panels and (ii) expenditures related to clinical studies and trials undertaken to validate and obtain FDA and other regulatory clearances for our genetic tests. We expense all research and development costs in the period in which they are incurred. Continued development and improvement of our INFINITI analyzers and expansion of our menu of test panels are key aspects of our business strategy and we expect that our research and development expenditures will increase for the foreseeable future. We expect research and development costs to remain generally flat as a percentage of revenue.
General and administrative
General and administrative expenses include the costs of personnel and consultants charged to administration, finance and accounting, human resources and regulatory affairs. General and administrative expenses also include corporate, administration, accounting, patent, facility and other costs and depreciation. We expect general and administrative expenses to increase for the foreseeable future as a result of the need to hire additional personnel to support the anticipated growth of our business and as a result of additional legal and accounting expenses related to compliance with operating as a public company, including Securities and Exchange Commission and securities exchange listing requirements, directors’ and officers’ insurance premiums and investor relations expenses. We expect general and administrative costs to decline slightly as a percentage of revenue over time.
Sales and marketing
Sales and marketing expenses include the commissions owed to our customers for laboratory test referrals, costs of personnel related to selling and marketing our INFINITI analyzers, customer support, advertising, trade show participation and other expenses. We expect our sales and marketing expenses to increase significantly for the foreseeable future, as we incur significant commission expense to our customers when we collect payments from our laboratory testing of patient samples they provide. Additionally, we plan to expand our sales and marketing organization to drive the growth in sales of our INFINITI analyzers and consumables products, and as we increase our technical customer support personnel to service this additional growth. We expect sales and marketing costs to be generally flat or slightly increase as a percentage of revenue over time.
49
Interest income
Interest income includes interest earned on our cash balances. These balances are invested primarily in money market accounts.
Interest expense
Interest expense relates to our outstanding promissory notes, which are more fully described in this prospectus under the heading "Use of Proceeds" and in "Note 3. Promissory Notes" of our financial statements included elsewhere in this prospectus. We intend to satisfy all of our outstanding promissory note indebtedness in connection with the completion of this offering, and as a result do not expect this expense to recur during the foreseeable future.
Promissory notes and warrants
We have issued promissory notes in connection with a number of private placement financings since 1999. In certain of these financings, the promissory notes were issued with separate common stock or preferred stock warrants to purchase shares of our common stock. The fair value of these warrants is allocated as a debt discount using the relative fair value allocation method determined using the Black-Scholes valuation model. The discount is amortized using the effective interest method over the term of the related promissory notes.
Change in the fair value of warrant liabilities
One of our outstanding warrants to purchase shares of our common stock provides for adjustment of the exercise price based upon subsequent issuances at prices per share that are below the exercise price of the warrant, and is classified as a liability and recorded at fair value regardless of the timing of the redemption feature or the redemption price or the likelihood of redemption. Our outstanding warrants to purchase shares of our convertible preferred stock are also classified as a liability and recorded at fair value regardless of the timing of the redemption feature or the redemption price or the likelihood of redemption. These warrants are subject to re-measurement at each balance sheet date and any change in fair value is recognized as a component of other interest income (expense), net. We will continue to adjust the liability for changes in fair value until the earlier of the conversion, exercise or expiration of the warrants. The last of these preferred stock warrants expired in August 2014.
All of our other outstanding warrants to purchase shares of our common stock that do not provide for exercise price adjustments were determined to be equity instruments and thus included in stockholders deficit upon issuance and are not subject to re-measurement.
Critical Accounting Policies, Significant Judgments and Estimates
Our financial statements and related notes included elsewhere in this prospectus have been prepared in accordance with U.S. generally accepted accounting policies. The preparation of these statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue, costs and expenses and related disclosures. We base such estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Changes in accounting estimates are reasonably likely to occur from period to period. Accordingly, actual results could differ significantly from the estimates made by company management. We evaluate our estimates and assumptions on an ongoing basis. To the extent that there are material differences between these estimates and actual results, our future financial statement preparation, financial condition, results of operations and cash flows will be affected.
The following accounting policies involve a greater degree of judgment and complexity than our other accounting policies. Accordingly, we believe these policies are the most critical to understanding and evaluating our financial condition and results of operations.
Revenue recognition
Our revenue is primarily derived from sales of our INFINITI analyzers, and the BioFilmChips, Intellipac Reagent Management Modules and various disposable items consumed by our diagnostic testing process (collectively, our consumables) sold directly to our customers or as service revenue through our CLIA-certified laboratory. We recognize revenue when persuasive evidence of an arrangement exists, the price to the buyer is fixed or determinable, collectability is reasonably assured and risk of loss transfers to the customer, normally upon shipment. Revenue from our CLIA-certified laboratory also requires the ordered test to be completed and the test
50
results to be accessible by the ordering physician prior to revenue recognition. For sales that include customer-specific acceptance criteria, revenue is recognized when the acceptance criteria have been met. We extend credit to customers based upon the evaluation of the customer's financial condition. Collectability is assessed based on a number of factors, including payment history and the creditworthiness of a customer. If we determine that collection is not reasonably assured, we do not recognize revenue until collection becomes reasonably assured, which is generally upon receipt of cash payment. Our service revenue is derived primarily from the CLIA-certified laboratory's genetic testing services, as well as warranty service on INFINITI analyzers and technical and customer services provided to purchased analyzers. Our laboratory's testing services are substantially all performed for our INFINITI HTS analyzer customers as they await delivery, installation and validation of the analyzer. Our laboratory service revenues are reported net of a contractual rate allowance based on published rate tables and historical experience. In addition, we incur sales and marketing fees to our contractual customers based on a fixed percentage of net service revenue collected from third-party insurers or responsible parties. This fee is included in sales and marketing costs in our accompanying statement of operations.
In instances where final acceptance of the product or analyzer is required, we defer revenue until all the acceptance criteria have been met. The rights granted in the RAP in exchange for a minimum annual purchase commitment constitutes a leasing arrangement. When a customer enters into a RAP agreement, the lease revenue is dependent on purchases of consumables, which is not measurable at the inception of the lease, and thus is accounted for as contingent rentals in their entirety. Accordingly, lease revenues are excluded from minimum lease payments but included in revenue as the consumables are purchased. The cost of the leased equipment is depreciated over its useful life and the expense included in cost of sales.
Arrangements to sell products to customers frequently include multiple deliverables, including the sale of the INFINITI analyzer combined with the sale of consumables. We sell internationally through a network of distributors. Sales to these foreign distributors include training of their field agents who are responsible for installation, training and maintenance for their customers internationally. We do not provide any price protection, rights of return, or extended payment terms to our distributors. We follow authoritative guidance on multiple-element arrangements, and we allocate arrangement consideration in multiple-element revenue arrangements at the inception of an arrangement to all deliverables or those packages in which all components of the package are delivered at the same time, based on the relative selling price method in accordance with the selling price hierarchy, which includes: (i) vendor-specific objective evidence, or VSOE, if available; (ii) third-party evidence if VSOE is not available; and (iii) best estimate of selling price, if neither VSOE nor third-party evidence is available. We establish selling price using management's best estimate, considering internal factors such as historical selling prices, pricing practices and controls and customer segment pricing strategies.
Contractual rate allowance
We are exposed to a certain level of uncertainty regarding the revenues earned from our laboratory services. We have not entered into contractual or capitation agreements for Medicare or private insurance, and estimate revenues based on published price lists and payment history. The difference between the amount we expect to collect and the amount reimbursed is reserved against using a contractual rate allowance; revenue is presented net of the contractual rate allowance.
Allowance for uncollectable accounts
We regularly monitor customer payments and specifically identify outstanding amounts to be allowed for, as well as record general accruals based on the age, history and experience of outstanding receivables with our customers and with third-party payors.
Warranty reserve
We provide a one-year warranty on domestic sales of our INFINITI analyzers. Accruals are provided for the estimated warranty expense at the time the associated revenue is recognized. The estimates of warranty expense are based on actual expenses incurred historically to service our INFINITI analyzers. We also provide a warranty for consumables products, estimated on a variety of inputs including historical performance, historical costs, projected reliability and service costs. Warranty expense accruals inherently require the use of management judgment. If we were to experience an increase in warranty claims or if our cost of servicing these warranties increased, our gross
51
margins would be adversely affected. We periodically review our actual warranty expense to ensure that it is not materially different than our estimates.
Deferred tax valuation allowance
We account for income taxes utilizing the asset and liability method. Under this method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and for tax loss carryforwards. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income or expense in the period that includes the enactment date. Future tax benefits are recognized to the extent that realization of such benefit is more likely than not.
At December 31, 2013, we had federal tax net operating loss carryforwards of $72.4 million and state tax net operating loss carryforwards of $41.2 million. The federal tax net operating loss carryforwards will begin to expire in 2020. The state tax net operating loss carryforwards began to expire in 2012. Utilization of net operating loss carryforwards, credit carryforwards and certain deductions may be subject to substantial annual limitation as a result of ownership change limitations provided by the Internal Revenue Code of 1986, as amended, or the Internal Revenue Code, and similar state provisions. The tax benefits related to future utilization of federal and state net operating loss carryforwards, credit carryforwards and other deferred tax assets may be limited or lost if cumulative changes in ownership exceed 50% within any three-year period.
Convertible preferred stock warrant liabilities
We use the Black-Scholes valuation model to calculate the fair value of our preferred stock warrants. The inputs utilized in this valuation model are discussed below. Each of these inputs is subjective and generally requires significant judgment to determine. The last of our preferred stock warrants expired in August 2014.
• | Fair value of our preferred stock. Because our stock is not publicly traded, we must estimate the fair value of our preferred stock, as discussed below. |
• | Expected term. The expected term is the remaining contractual life of the applicable preferred stock warrant. |
• | Volatility. As we do not have a trading history for our preferred stock, the expected stock price volatility for our preferred stock is estimated by taking the average historic price volatility for industry peers based on daily price observations over a period equivalent to the expected term of the preferred stock warrants. Industry peers consist of several public companies similar in size, stage of life cycle and financial leverage. We do not rely on implied volatilities of our industry peers' publicly traded common stock because the volume of such trading activity has been relatively low. The volatility for all periods presented was calculated within the range of 52% and 57%. |
• | Risk-free interest rate. The risk-free interest rate is based on the yields of U.S. Treasury securities with maturities similar to the contractual term of the preferred stock warrants. |
• | Dividend yield. We have not historically paid cash dividends or distributions and do not intend to pay cash dividends or distributions in the foreseeable future. Consequently, we used an expected dividend yield of zero. |
The fair value of the preferred stock underlying our outstanding preferred stock warrants has historically been determined by our board of directors. Because there has been no public market for our capital stock, our board of directors has determined the fair value of the preferred stock at the time of issuance by considering a number of objective and subjective factors including valuations performed by unrelated third-party specialists, which included valuations of comparable companies, operating and financial performance, lack of liquidity of capital stock and general and industry-specific economic outlook. For each period in which the fair value of preferred stock warrant liabilities was assessed, our business enterprise value was first determined. This enterprise value then was allocated across our capital structure using a Probability Weighted Expected Return Method, or PWERM, model as the primary approach and an Option Pricing Model, or OPM, analysis as a confirming approach. Under the PWERM model, various future potential liquidity scenarios were forecasted and the allocable value to each class of security in our capital structure in each respective scenario was determined. The probability that each potential liquidity event would occur was then assessed. The future proceeds were then discounted to determine the present value of proceeds in each scenario for each security class. The probability weighted present value of each security class was then determined to conclude the value of the preferred stock. Under the confirming OPM analysis, the Black-Scholes
52
option pricing framework was used to determine the proceeds allocable to each security class. A series of option tranches was established whereby each tranche represented the value allocable to various security classes between computed breakpoints. Break points are the value inflection points, or indifference points, whereby the allocation to securities change based on the underlying economic rights of each security class. The breakpoints served as the strike prices in the Black-Scholes option calculations and tranche values were determined based on the difference between option values in each tranche. The proceeds of the value of each option tranche were then allocated to each respective security class based on the economic rights to the security class between breakpoints. The value allocable to each respective security class was then totaled across all option tranches to conclude the fair value of each security class.
We have classified our convertible preferred stock warrants as a liability and have re-measured the liability to estimated fair value at December 31, 2013 and 2012 using the Black-Scholes valuation model. These warrants expired during August 2014 and have no fair value at September 30, 2014.
Stock-based compensation
Compensation expense related to stock-based transactions, including employee stock options, is measured and recognized in the financial statements based on fair value. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service period and classified in the statement of operations based on the department to which the related employee reports. Determining the fair value of stock-based awards at the grant date requires judgment. We use the Black-Scholes valuation model to determine the fair value of grants. The determination of the grant date fair value of grants using a valuation model is affected by assumptions regarding a number of complex and subjective variables. These variables include the fair value of our common stock, the expected term of our options, our expected stock price volatility over the expected term of the options, stock option exercise and cancellation behaviors, risk-free interest rates, and expected dividends, which are estimated as follows:
• | Fair value of our common stock. Because our stock is not publicly traded, we must estimate the fair value of our common stock, as discussed in "Common stock valuations" below. |
• | Expected term. We estimate the expected term of our options using the simplified method allowed under SEC guidance. |
• | Volatility. As we do not have a trading history for our common stock, the expected stock price volatility for our common stock is estimated by taking the average historic price volatility for industry peers based on daily price observations over a period equivalent to the expected term of the stock options. Industry peers consist of several public companies similar in size, stage of life cycle and financial leverage. We do not rely on implied volatilities of traded options in our industry peers' common stock because the volume of such trading activity has been relatively low. We intend to continue to consistently apply this process using the same or similar public companies until a sufficient amount of historical information regarding the volatility of our own common stock price becomes available, or until circumstances change such that the identified companies are no longer similar to us, in which case we would utilize more suitable companies whose share prices are publicly available in the calculation. The volatility for all periods presented was calculated within the range of 52% and 57%. |
• | Risk-free interest rate. The risk-free interest rate is based on the yields of U.S. Treasury securities with maturities similar to the expected term of the options for each option group. |
• | Dividend yield. We have not historically paid cash dividends or distributions and do not intend to pay cash dividends or distributions in the foreseeable future. Consequently, we used an expected dividend yield of zero. |
Common stock valuations
We are required to estimate the fair value of the common stock underlying our stock-based awards when performing the fair value calculations with the Black-Scholes valuation model. The fair value of the common stock underlying our stock-based awards was determined on each grant date by our board of directors, with input from management. This fair value has been calculated on an as-converted basis for each period, assuming for such purposes that all outstanding convertible preferred stock has been converted into common stock without taking into account any dividend or liquidation preferences. All options to purchase shares of our common stock granted under our incentive stock option plans are intended to be granted with an exercise price per share no less than the fair value per share of our common stock underlying those options on the date of grant, based on the information known to us
53
on the date of grant. In the absence of a public trading market for our common stock, on each grant date, we develop an estimate of the fair value of our common stock in order to determine an exercise price for the option grants. We have determined the fair value of our common stock using methodologies, approaches and assumptions consistent with the American Institute of Certified Public Accountants, or AICPA, Audit and Accounting Practice Aid Series: Valuation of Privately Held Company Equity Securities Issued as Compensation, or the AICPA Practice Guide. In addition, our board of directors considered various objective and subjective factors, along with input from management to determine the fair value of our common stock, including:
• | valuations performed by third-party specialists; |
• | the prices at which we sold shares of common and preferred stock to third parties; |
• | the superior rights and preferences of the preferred stock relative to our common stock at the time of each grant; |
• | our results of operations and financial position; |
• | our stage of development, status of our research and development efforts and business strategy; |
• | the lack of an active public market for our common and our preferred stock; and |
• | the likelihood of achieving a liquidity event such as an initial public offering, or IPO, or sale of our company in light of prevailing market conditions. |
The determination of fair value of our common stock also involves complex and subjective judgments including estimates of revenue, earnings, assumed market growth rates and estimated costs, as well as appropriate discount rates. At the time of each of the below-referenced valuations, the estimates used in the discounted cash flow approach included our best estimates of our revenue and revenue growth rates for several years into the future. Although each time we prepared such forecasts we did so based on assumptions that we believed to be reasonable and appropriate at that time, there can be no assurance that any such estimates for earlier periods or for future periods will prove to be accurate.
Our board of directors utilizes the methodologies described above to calculate our implied enterprise value as of each stock option grant date, taking into account our ability or inability to achieve certain milestones, principally among which was the completion of an IPO. A PWERM analysis is then used to allocate a portion of this implied enterprise value based on six possible scenarios: (i) an IPO of our common stock in the immediate future at a price per share of our common stock based on an implied enterprise value equal to a multiple of our management's estimate of our then current-year revenue; (ii) an IPO of our common stock at the end of the subsequent fiscal year at a price per share of our common stock based on an implied enterprise value equal to a multiple of our management's estimate of our next fiscal year's revenue; (iii) a merger or sale of our company at a date that is approximately two and one-half years in the future at a price per share of our common stock based on an implied enterprise value equal to a multiple of our management's estimate of that future fiscal year's revenue; (iv) a merger or sale of our company at the end of the second future fiscal year ending after the then-current fiscal year at a price per share of our common stock based on an implied enterprise value equal to a multiple of our management's estimate of that future fiscal year's revenue; (v) a dissolution of our company with no value available to be distributed to the holders of our common stock; and (vi) remaining a private company. For the IPO scenarios, the value assigned to our common stock was determined using the number of outstanding shares of our common stock assuming the conversion of all outstanding shares of our preferred stock into shares of our common stock. For the merger or sale scenarios, a break point analysis was used to determine the various enterprise values at which holders of each series of our outstanding shares of preferred stock would elect to convert their shares of our preferred stock into shares of our common stock and the points at which the holders of outstanding options and warrants to acquire shares of our common stock would exercise their options or warrants. The values allocated to our common stock as of a future date under each scenario by the PWERM analysis are then discounted using a weighted-average cost of capital to calculate an implied present value of our common stock under the PWERM analysis.
There is inherent uncertainty in these estimates and if we had made different assumptions than those used, the amount of our stock-based compensation expense, net loss and net loss per share amounts could have been significantly different. Following the closing of this offering, the fair value per share of our common stock for purposes of determining stock-based compensation expense will be the closing price of our common stock as reported on The NASDAQ Global Market on the applicable grant date.
54
The following table illustrates our stock option grant information since January 1, 2013, including the estimated fair value of our common stock on the date of grant:
Grant date | Number of shares subject to options granted | Option exercise price | Estimated value of common stock per share at date of grant | |||
October 15, 2013 | 423,529 | $6.67 | $6.67 | |||
December 23, 2013 | 119,390 | $6.67 | $6.67 | |||
Our board of directors determined that the fair value of our common stock at October 15, 2013 and December 23, 2013 was equal to the price per share paid during a private placement equity transaction during September 2013. Additionally, a retrospective analysis as at December 31, 2013 finalized during May 2014 supported such valuation. These values could materially differ from our common stock's share value upon completion of this offering for many reasons, included but not limited to the uncertainties surrounding our ability to complete this offering, cashflow constraints, the potential price volatility of shares of our common stock, the impact of our large accounts payable obligations, and our not operating on a profitable basis.
Useful lives of property and equipment
Property and equipment is recorded at cost and is depreciated on a straight-line basis over the following estimated useful lives:
Reagent access plan assets | 3 or 5 years |
Equipment | 5-7 years |
Computer equipment and software | 3 years |
Leasehold improvements | various |
Furniture | 5 years |
Management must use judgment in determining the estimated useful lives of its assets. Changes in circumstances, such as technological advances, changes to our business model, changes in our business strategy, or changes in the planned use of property and equipment, could result in the actual useful lives differing from our current estimates.
Allowance for excess and obsolete inventory
We record our inventories at cost and regularly perform analysis and market value adjustments. The value that we will realize through sales on our inventory cannot be known with exact certainty, we rely on past sales experience and future sales forecasts to project it. In analyzing our inventory levels, we classify certain inventory as either excess or obsolete. These classifications are maintained for all classes of inventory.
Recently Issued Accounting Standards
Under Section 107(b) of the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. Management has reviewed all recent accounting standard’s updates issued by the Financial Accounting Standards Board and anticipates no material impact from any of their adoption.
55
Results of Operations
The financial data set forth below is derived in part from and should be read in conjunction with our financial statements and the related notes included elsewhere in this prospectus. The following tables set forth selected items in our statements of operations for the periods presented, together with the difference in amounts over such periods in dollars (in thousands) and in percentages.
Nine Months Ended September 30, | Years Ended December 31, | ||||||||||||||||||||||
2014 | 2013 | Change | 2013 | 2012 | Change | ||||||||||||||||||
(unaudited) | |||||||||||||||||||||||
Product revenue, net | $ | 9,022 | $ | 13,044 | $ | (4,022 | ) | $ | 16,938 | $ | 18,307 | $ | (1,369 | ) | |||||||||
Service revenue, net | 9,703 | 229 | 9,474 | 1,145 | 102 | 1,043 | |||||||||||||||||
Net revenue | 18,725 | 13,273 | 5,452 | 18,083 | 18,409 | (326 | ) | ||||||||||||||||
Cost of sales | 3,968 | 4,269 | (301 | ) | 5,726 | 7,147 | (1,421 | ) | |||||||||||||||
Cost of services | 2,677 | 1,530 | 1,147 | 2,032 | — | 2,032 | |||||||||||||||||
Gross profit | 12,080 | 7,474 | 4,606 | 10,325 | 11,262 | (937 | ) | ||||||||||||||||
Operating expenses: | |||||||||||||||||||||||
Research and development | 1,540 | 1,715 | (175 | ) | 2,306 | 2,355 | (49 | ) | |||||||||||||||
General and administrative | 3,308 | 7,210 | (1) | (3,902 | ) | 8,302 | (1) | 4,715 | 3,587 | ||||||||||||||
Sales and marketing | 5,334 | 1,898 | 3,436 | 2,998 | 2,269 | 729 | |||||||||||||||||
Total operating expenses | 10,182 | 10,823 | (641 | ) | 13,606 | 9,339 | 4,267 | ||||||||||||||||
Income/(loss) from operations | 1,898 | (3,349 | ) | 5,247 | (3,281 | ) | 1,923 | (5,204 | ) | ||||||||||||||
Interest expense, net | (1,361 | ) | (1,186 | ) | (175 | ) | (1,820 | ) | (2,602 | ) | 782 | ||||||||||||
Gains/(loss) on extinguishment of debt | (1,872 | ) | (140 | ) | (1,732 | ) | (140 | ) | (1,996 | ) | 1,856 | ||||||||||||
Other income/(expense), net | (5 | ) | (2,095 | ) | 2,090 | (1,877 | ) | 5 | (1,882 | ) | |||||||||||||
Change in the fair value of stock warrant liabilities | 375 | 5 | 370 | 46 | 163 | (117 | ) | ||||||||||||||||
Net income/(loss) | $ | (965 | ) | $ | (6,765 | ) | $ | 5,800 | $ | (7,072 | ) | $ | (2,507 | ) | $ | (4,565 | ) | ||||||
(1) | Includes $4.5 million in bad debt expense relating to NMTC as described below. |
Comparison of the Nine Months Ended September 30, 2014 and 2013 (unaudited)
Income/(loss) from operations and net loss. The improvement in income/(loss) from operations and net loss was primarily a result of lower general and administrative expenses in the nine months ended September 30, 2014 compared to the same period in 2013 due to $4.5 million bad debt expense in 2013, and a $5.4 million increase in net revenue during 2014 as compared to 2013. The improvement was also partially offset by an increase in sales and marketing expenses resulting from commissions resulting from the introduction of our INFINITI HTS analyzer and the increased volume of operations in our CLIA-certified laboratory. The net loss during the nine months ended September 30, 2014 improved further due to other expenses in the same period in 2013 resulting from terminated financing transactions.
Our largest customer up to and during 2013 was NMTC. During January 2013, the Centers for Medicare and Medicaid Services, or CMS, temporarily suspended reimbursement for certain diagnostic tests while it determined reimbursement rates and implemented new molecular diagnostic CPT codes. This suspension adversely affected the cash flows of several of our customers, including NMTC, who became delinquent on its 2013 receivables during the first quarter of 2013, and filed for Chapter 11 protections during the fourth quarter of 2013. Accordingly, we recorded a total of $4.5 million in bad debt expense in 2013 associated with NMTC's delinquency. In September 2013, CMS issued "gap-fill" pricing decisions for molecular diagnostic CPT codes and such guidance has been consistent with reimbursements we have received.
56
Net revenue. The increase in net revenue during the nine months ended September 30, 2014 is due primarily to the $9.5 million increase in service revenue from the sales of our genetic test panels by our CLIA-certified laboratory to our INFINITI HTS customers. This increase was partially offset by a $4.0 million decrease in product revenues due to the loss of a significant customer, NMTC, during 2013 due to bankruptcy. Excluding revenue from NMTC recorded during the nine months ended September 30, 2013, net revenue increased by $10.6 million, or 130%, during the nine months ended September 30, 2014 as compared to the same period in 2013. Net revenue was comprised of $9.0 million product revenue and $9.7 million service revenue for the nine months ended September 30, 2014, and $13.0 million and $0.2 million during the same period in 2013, respectively. The increase in service revenue during the first nine months of 2014 as compared to the same period in 2013 was due to the recent introduction of our INFINITI HTS and the resulting sale of services by our CLIA-certified laboratory to new INFINITI HTS customers during the delivery, installation, training and validation period. We expect that in future periods a significant portion of services revenue will transition to product revenue as new INFINITI HTS customers complete this installation, training and validation period, and for this transitioning services revenue to be replaced by revenue generated from new INFINITI HTS customers.
Cost of sales and services. Total cost of sales and services increased during the nine months ended September 30, 2014 compared to the same period in 2013, due to an increase in CLIA laboratory services. Gross profit margin was 64.6% for the nine months ended September 30, 2014 as compared to 56.3% for the nine months ended September 30, 2013. We expect gross margins to not vary significantly between cost of sales and cost of services. Cost of sales was $4.0 million and $4.3 million, and cost of services was $2.7 million and $1.5 million, for the nine months ended September 30, 2014 and 2013, respectively. The increase in cost of services during the first nine months of 2014 as compared to the same period in 2013 was due to the recent introduction of our INFINITI HTS and the resulting sale of services by our CLIA-certified laboratory to new INFINITI HTS customers.
Operating expenses. Research and development expenses consists of personnel costs and costs to develop our diagnostic testing products and future generations of testing platforms. These costs remained relatively unchanged between the nine month periods ended September 30, 2014 and September 30, 2013.
General and administrative expenses decreased during the nine month period ended September 30, 2014 primarily due to bad debt expense of $4.5 million recorded in the nine month period ended September 30, 2013. Without this bad debt expense, general and administrative expenses increased $0.6 million due to an increase in employee headcount.
Sales and marketing expenses increased during the nine months ended September 30, 2014 primarily due to the introduction of the INFINITI HTS and commencement of operations of our CLIA-certified laboratory. Substantially all of the patient samples provided to our laboratory for testing are from current INFINITI HTS analyzer customers, and we are obligated to pay sales and marketing fees based on a fixed percentage of lab testing revenues collected from third-party insurers.
Non-operating expenses. Net interest expense remained relatively unchanged between the two periods.
Other expenses decreased during the nine months ended September 30, 2014 compared to the same period in 2013 as a result of the terminated IPO efforts during the first quarter of 2013 and the expensing of previously deferred related costs during the year.
The change in the fair value of warrant liabilities increased during the nine months ended September 30, 2014 due to the expiration of preferred stock warrants.
Gains/(losses) on extinguishment of debt. During the nine months ended September 30, 2014, certain of our notes payable were refinanced at maturity and such refinances were considered extinguishment of debt as the terms of the new debt were substantially different than the refinanced debt. Such extinguishment resulted in non-cash expenses of $1.9 million for the nine months ended September 30, 2014, representing the fair value of the warrants granted in conjunction with such refinances, and had no impact on our liquidity.
Comparison of the Years Ended December 31, 2013 and 2012
Income/(loss) from operations and net loss. The increase in loss from operations and net loss during 2013 was primarily driven by the increase in general and administrative expenses due to (i) $4.5 million in bad debt expense
57
resulting from allowing for of all accounts receivables of NMTC during 2013 with no such expense in 2012, (ii) a $0.4 million increase in stock-based compensation and (iii) a $0.3 million decrease in net revenue.
Net revenue. Net revenue for the years ended December 31, 2013 and 2012 decreased very slightly between the two periods, as the loss of one large customer was replaced with a larger number of smaller customers. As a percentage of net revenue, markets outside the United States were 14% and 7% for the years ended December 31, 2013 and 2012, respectively. Net revenue was comprised of $16.9 million product revenue and $1.1 million service revenue during 2013, and $18.3 million and $0.1 million during 2012, respectively.
Cost of sales and services. The decrease in cost of sales is due to the loss of a major customer, NMTC, in early 2013 and the resulting reduction in product sales. The increase in cost of services is primarily due to expenses resulting from the commencement of operations of our CLIA-certified laboratory during 2013 with no such expense in 2012.
Operating expenses. Research and development expenses consisting of personnel costs and costs to develop our diagnostic testing products and future generations of testing platforms. These costs remained relatively unchanged between the two periods.
General and administrative expenses increased primarily due to $4.5 million in bad debt expense resulting from the reserve for outstanding accounts receivable of NMTC during 2013. The increase was partially offset by a decrease in legal expenses resulting from 2012 financing activities that did not reoccur during 2013.
Sales and marketing expenses increased primarily due to increased expenses from the commencement of operations of our CLIA-certified laboratory, in addition to stock-based compensation expense and salary and wage increases resulting from additional headcount during 2013 as compared to 2012.
Non-operating expenses. Net interest expense decreased primarily as a result of refinancing of promissory notes during 2012 and associated common stock warrants recorded as a debt premium and amortized as a credit interest expense, with no such refinancings occurring during 2013.
Other expenses increased during 2013 as a result of the expenses relating to terminated financing transactions during 2013.
The change in the fair value of warrant liabilities is attributable to a decrease in the fair value of our common stock and in the contractual life over which the associated preferred stock warrant liabilities are marked to market.
Gains/(losses) on extinguishment of debt. During the year ended December 31, 2012, certain of our notes payable were refinanced at maturity and such refinances were considered extinguishment of debt as the terms of the new debt were substantially different than the refinanced debt. Such extinguishment resulted in non-cash expenses $2.0 million for the year ended December 31, 2012, representing the fair value of the warrants granted in conjunction with such refinances, and had no impact on our liquidity.
As Adjusted Financial Information for the Nine Months Ended September 30, 2014 and 2013 and Years Ended December 31, 2013 and 2012 (unaudited)
The unaudited as adjusted financial data below sets forth the net revenue for the nine month periods ended September 30, 2014 and 2013 and the years ended December 31, 2013 and 2012 excluding all revenue attributable to NMTC (in thousands).
Nine Months Ended September 30, | Years Ended December 31, | ||||||||||||||||||||||
2014 | 2013 | Change | 2013 | 2012 | Change | ||||||||||||||||||
Net revenue (as reported) | $ | 18,725 | $ | 13,273 | $ | 5,452 | $ | 18,083 | $ | 18,409 | $ | (326 | ) | ||||||||||
Net revenue from NMTC | — | 5,116 | (5,116 | ) | 5,116 | 7,749 | (2,633 | ) | |||||||||||||||
As adjusted net revenue | $ | 18,725 | $ | 8,157 | $ | 10,568 | $ | 12,967 | $ | 10,660 | $ | 2,307 | |||||||||||
At each of September 30, 2014 (unaudited) and December 31, 2013, $4.5 million in net accounts receivable due from NMTC remains unpaid. Because of the uncertainty regarding collectability of this receivable, we allowed for 100% of this receivable during 2013. We believe these 130% and 22% increases in as adjusted net revenue for the
58
nine months ended September 30, 2014 and year ended December 31, 2013 compared to the same periods in prior years, respectively, are important metrics of our continuing business, by presenting only revenues which we expect to continue. We monitor our revenue concentration as a measure of our market penetration and as a factor in determining our technology's adoption.
Liquidity and Capital Resources
Historical cash flows
From our inception in April 1999 through September 30, 2014, we have financed our operations primarily through sales of privately placed shares of convertible preferred stock, promissory notes and associated warrants and net cash provided by operating activities.
Our primary uses of cash are to fund operating expenses, inventory purchases, service our debt and the acquisition of machinery and equipment. Cash used to fund operating expenses excludes the impact of non-cash items such as the provision for excess and obsolete inventory, equipment depreciation, stock-based compensation and non-cash interest expense and is impacted by the timing of when we pay these expenses as reflected in the change in our outstanding accounts payable and accrued expenses. Acquisitions of machinery and equipment primarily consist of our cost to manufacture our INFINITI analyzers utilized in our RAPs, purchases of laboratory equipment, computer hardware and software and the cost of facility improvements.
As of September 30, 2014 (unaudited), we had cash and cash equivalents of $0.6 million compared to $2.1 million and $0.3 million as of December 31, 2013 and 2012, respectively. Our long-term liquidity could be negatively impacted by our indebtedness related to promissory notes with maturity dates in the last quarter of 2015. However, we are required to use the proceeds of the offering referred to in this prospectus to repay these notes, and so the impact of these notes is not considered significant. See "Use of Proceeds."
The following table summarizes our cash flows for each of the periods indicated (in thousands):
Nine Months Ended September 30, | Years Ended December 31, | ||||||||||||||
2014 | 2013 | 2013 | 2012 | ||||||||||||
(unaudited) | |||||||||||||||
Net cash provided by/(used in) operating activities | $ | (790 | ) | $ | (54 | ) | $ | (1,774 | ) | $ | 654 | ||||
Net cash provided by/(used in) investing activities | $ | (708 | ) | $ | (519 | ) | (809 | ) | (355 | ) | |||||
Net cash provided by/(used in) financing activities | 1 | 4,274 | 4,304 | (28 | ) | ||||||||||
Net increase/(decrease) in cash | $ | (1,497 | ) | $ | 3,701 | $ | 1,721 | $ | 271 | ||||||
Operating activities. Net cash used in operating activities for the nine months ended September 30, 2014 compared to 2013 was primarily driven by increases in operating expenses from the growth in our CLIA-laboratory.
Net cash provided by operating activities for the year ended December 31, 2013 compared to 2012 was primarily driven by a reduction in cash collections from accounts receivable during 2013, due to NMTC, with no such cash collection shortfalls in 2012.
Investing activities. Net cash used in investing activities for all periods noted above consisted primarily of invested capital and facility investment to manufacture the INFINITI analyzers utilized in our RAPs and purchases of machinery and equipment, including furniture, computer equipment and software, in support of all functional areas of the business.
Financing activities. Net cash provided by financing activities for the nine months ended September 30, 2014 compared to 2013 was primarily driven by $4.2 million in proceeds from the issuance of common stock during 2013, with no such issuances in 2014.
Net cash provided by financing activities for the year ended December 31, 2013 compared to 2012 was primarily driven by $4.2 million in proceeds from the issuance of common stock, with no such issuances in 2012.
Capital resources. We anticipate that our current cash and cash equivalents, together with cash provided by this offering and our operating activities will be sufficient to meet our currently estimated cash requirements for at least
59
the next 12 months. We have discussed the expected use of the net proceeds from this offering elsewhere in this prospectus.
We expect capital outlays and operating expenditures to increase over the next several years as we expand our infrastructure, commercialization, manufacturing and research and development activities, and as a result we may need additional capital financing. The amount of additional capital we may need to raise depends on many factors, including:
• | the level of research and development investment required to maintain and improve our technology, including efforts to expand our menu of test panels, to fund clinical studies and trials of our tests and to invest in the development of new analyzers; |
• | the amount of future cash provided by or used in operating activities; |
• | the costs of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights; |
• | our need or decision to acquire or license complementary technologies or acquire complementary businesses; and |
• | changes in regulatory policies or laws that affect our operations. |
We cannot be certain that additional capital will be available when and as needed or that our actual cash requirements will not be greater than anticipated. If we require additional capital at a time when investment in diagnostics companies or in the marketplace in general is limited by the then prevailing market or other conditions, we may not be able to raise such funds at the time that we desire or any time thereafter. In addition, if we raise additional funds through the issuance of common stock or securities convertible into shares of common stock, the percentage ownership of our stockholders could be significantly diluted, and these newly issued securities may have rights, preferences or privileges senior to those of existing stockholders. If we obtain additional debt financing, a substantial portion of our operating cash flow may be dedicated to the payment of principal and interest on such indebtedness, and the terms of the debt securities issued could impose significant restrictions on our operations. If we raise additional funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our technologies or products, or grant licenses on terms that are not favorable to us.
Promissory notes. As of September 30, 2014 (unaudited), we had $2.3 million in principal amount of outstanding promissory notes with an interest rate of 8.5% due December 2014, $1.0 million in principal amount of outstanding promissory notes with an interest rate of 10.0% due June 2015, $14.5 million in principal amount of outstanding promissory notes with an interest rate of 8.5% due November 2015, and $0.1 million in principal amount of outstanding promissory notes with an interest rate of 8.5% due November 2015. During the nine months ended September 30, 2014 and the year ended December 31, 2012, certain of our notes payable were refinanced at maturity and such refinances were considered extinguishment of debt as the terms of the new debt were substantially different than the refinanced debt. Such extinguishment resulted in non-cash expenses of $1.9 million and $2.0 million for the nine months ended September 30, 2014 and the year ended December 31, 2012, respectively, representing the fair value of the warrants granted in conjunction with such refinances, and had no impact on our liquidity.
We granted the holders of our $14.5 million in principal amount of outstanding promissory notes with an interest rate of 8.5% due November 2015 a security interest in our accounts receivable and proceeds therefrom. We are required to use 50% of the proceeds of this offering to repay substantially all of our outstanding promissory notes, and we intend to use a portion of the proceeds of this offering to repay all of our outstanding indebtedness.
60
Contractual Obligations
As of September 30, 2014, the annual amounts of future minimum payments under certain of our contractual obligations were:
Payments due by period | |||||||||||||||||||
Total | Less than 1 year | 1-3 years | 3-5 years | More than 5 years | |||||||||||||||
(unaudited and in thousands) | |||||||||||||||||||
Contractual obligations: | |||||||||||||||||||
Promissory notes (1) | 21,225 | $ | 4,396 | $ | 16,829 | $ | — | $ | — | ||||||||||
Operating lease (2) | 38,655 | 1,834 | 3,967 | 4,249 | 28,605 | ||||||||||||||
Total | $ | 59,880 | $ | 6,230 | $ | 20,796 | $ | 4,249 | $ | 28,605 | |||||||||
(1) | From September 2008 through March 2013, we issued $17.5 million aggregate principal amount of promissory notes in private placements to certain accredited investors of which $2.5 million in aggregate principal amount was issued to directors, executive officers, and holders of more than 5% of our common stock. The notes bear a weighted-average annual interest rate of 8.6%, with the principal and accrued interest (shown above accrued through September 30, 2014), with substantially all maturing during November 2015. We can prepay our outstanding promissory notes at any time without penalty. We are required to use 50% of our cash proceeds from this offering to repay our senior and subordinated promissory notes. See "Use of Proceeds." |
(2) | Our lease for our corporate offices in Vista, California commenced on February 1, 2009 and will expire in December 2029. This facility houses our research and development, manufacturing and warehousing operations and our administrative offices. |
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements or any special-purpose entities.
Qualitative and Quantitative Disclosures about Market Risk
Our exposure to market risk is currently confined to our cash and cash equivalents. We have not used derivative financial instruments for speculation or trading purposes. In addition, we have not invested in auction rate securities. Our promissory notes have a weighted-average fixed annual interest rate of 8.6%. These notes will be retired in connection with the completion of this offering. See "Use of Proceeds."
The primary objective of our investment activities is to preserve our capital for the purpose of funding operations while at the same time maximizing the income we receive from our investments without significantly increasing risk. To achieve these objectives, we invest our cash primarily in liquid money market funds. As a result, we believe we have minimal interest rate risk. Due to our limited funds invested in interest bearing accounts, a one percentage point change in the average interest rate on invested funds would have had a minimal effect on interest income for the nine months ended September 30, 2014 and 2013 and years ended December 31, 2013 and 2012.
Our international sales are all denominated and paid in U.S. dollars and as a result we believe we are not exposed to foreign exchange currency risk.
61
BUSINESS
Overview
We are a commercial stage molecular diagnostics company offering an innovative and proprietary technology platform to clinical reference laboratories, specialty clinics and hospital laboratories. Our platform consists of a family of multiplexing INFINITI analyzers, an extensive and expanding menu of genetic test panels, and proprietary microarrays, reagent modules and other related consumables. Our INFINITI analyzers, which include the INFINITI, the INFINITI PLUS and the recently introduced INFINITI HIGH THROUGHPUT SYSTEM, or INFINITI HTS, are easy to use and automate a number of the discrete processes of genetic analysis with minimal manual intervention, which reduces our customers' need for multiple, specialized instruments, and offer a variety of throughput capabilities together with a demonstrated high level of accuracy and reproducibility. Our genetic tests are focused on large and growing markets primarily in the areas of personalized medicine, women's health, infectious diseases and genetic disorders. Genetic tests are performed on our INFINITI analyzers using our proprietary BioFilmChip microarrays, related Intellipac Reagent Management Modules and other related consumables.
We believe genetic testing is improving the way patients are diagnosed, monitored and managed, by providing actionable information that can lead to enhanced patient care. Our genetic tests provide tools to physicians, clinicians and other health care providers to improve detection, treatment and monitoring of a broad spectrum of diseases and conditions. Frost & Sullivan estimated in 2012 that the molecular diagnostics market will reach $6.2 billion in the United States during 2014 and forecasted a compound annual growth rate for the market in excess of 11%. The Centers for Medicare and Medicaid Services, or CMS, of the Department of Health and Human Services estimated in June 2014 that there were more than 5,900 independent clinical reference laboratories and specialty clinics, and more than 8,900 hospital-based laboratories, in the United States. We believe that less than 10% of these laboratories are currently performing molecular diagnostics testing, and that we have an opportunity to expand our customer base as more laboratories seek to participate in this growing market.
We currently offer 62 test panels for use on our analyzers and have 16 additional test panels in development. In the area of personalized medicine, we offer test panels in pain management, cardiovascular health assessment, oncology and mental health. These test panels offer our customers the ability to identify a patient's genetic information, which can assist in improving drug efficacy, identifying non-responders and ultra-, normal- and poor-metabolizers, and reducing adverse patient response. In the area of women's health, we offer test panels in cervical cancer, sexually transmitted diseases, or STDs, and vaginal infections. These test panels identify a wide spectrum of specific organisms simultaneously, which can eliminate the need for laboratories to perform multiple tests. The depth and breadth of our menu of genetic test panels, all of which are designed to run on any of our INFINITI analyzers, allows laboratories to utilize space, labor and capital investment efficiently and to conduct genetic tests in less time, thereby improving laboratory economics. The proprietary design of our platform also allows us to introduce new and enhanced test panels to our menu of genetic tests without modifying our INFINITI analyzers. Based on our own research and customer demand, we intend to continue to increase the number of tests offered in each of our target market segments, which we believe will further increase the utility of our platform to our customers.
We launched our INFINITI HTS during February 2014 to meet the demand for high test-volume capabilities from our customers, particularly in the areas of personalized medicine and women's health. Our INFINITI HTS is a scalable, automated system comprised of three separate operating modules that provides flexible configuration to maximize workflow efficiency, and has the capacity to process up to 6,912 patient results per day with a single laboratory technician utilizing the same consumables as our other INFINITI analyzers. We believe our INFINITI HTS has the highest processing and throughput capacities of any molecular diagnostics system available in our target markets today. Additionally, for customers who seek a fully automated and integrated solution, and have lower throughput requirements, we offer INFINITI and INFINITI PLUS analyzers designed to operate on a "load and go" basis, in which a technician only needs to load prepared samples along with the test-specific consumables into the analyzer to generate test results.
We believe that compared to traditional genetic testing methods, our platform can significantly improve workflow, throughput and testing accuracy, while reducing our customers' operating cost per reportable result, thereby improving laboratory economics. We believe that these and other attributes of our platform can decrease the cost and complexity of genetic testing and reduce the need for specialized laboratory personnel, training, equipment
62
and facilities.
In late 2013, as we prepared to launch our INFINITI HTS, we began operations of a CLIA-certified laboratory to complement and support our efforts to attract customers entering the molecular diagnostics market, and potential INFINITI HTS customers in particular. Our laboratory services allow our customers to more quickly offer new or additional tests during the period when they are awaiting delivery, installation, training and validation of an INFINITI analyzer. Our CLIA-certified laboratory also provides new and existing customers with an alternative source of capacity to assist with their overflow testing needs. In addition, our laboratory enhances our product development efforts and allows us to offer hands-on training opportunities for our customers. Since the launch of our INFINITI HTS, many of these customers have utilized the services of our laboratory's INFINITI HTS capabilities to supplement and facilitate sales of medium- and high-volume test products to physicians, clinicians and healthcare providers.
Since the introduction of our INFINITI HTS and commencement of operations in our CLIA-certified laboratory, we have focused our sales and marketing efforts primarily on clinical reference laboratories, specialty clinics and hospital laboratories with high-volume genetic testing opportunities in the personalized medicine and women’s health market segments. We believe that our platform, together with our CLIA-certified laboratory and our training capabilities, will continue to attract additional customers, and enable existing laboratories and clinics to start performing genetic testing on a cost-effective basis in a high-volume setting.
We have received FDA 510(k) clearance for our INFINITI and INFINITI Plus analyzers and five of our genetic test panels: CYP450 2C19, Factor II, Factor V, Factor II/V and Warfarin. Products for which we have received 510(k) clearance accounted for 35%, 18% and 14% of our net revenue for the nine months ended September 30, 2014, and the years ended December 31, 2013 and 2012, respectively. We are currently in clinical trials collecting data to support a submission for 510(k) clearance for our INFINITI HTS, both of our CYP450 2C19 Plus test panels and all three of our CYP450 2D6 test panels. We intend to use a portion of the proceeds of this offering to conduct additional clinical trials for the collection of data, and to submit for 510(k) clearance, for an additional seven of our test panels that, together with our existing and in-process 510(k) cleared test panels, made up approximately 80% of consumables revenue for the nine months ended September 30, 2014. We expect to conduct additional clinical trials for the collection of data, and to submit for 510(k) clearance, for test panels based on a variety of factors, including:
• | the regulatory environment for the use of genetic tests, in particular the FDA's requirements and limitations on marketing RUO tests, which may not be marketed as in vitro diagnostic products; |
• | the demand by our existing and target customers for particular genetic tests that have received regulatory approvals or clearances; |
• | the competitive environment for the use of genetic tests that have received regulatory approvals or clearances versus similar tests that have not; and |
• | the size of the available market for the particular test, given the significant expense and time required to obtain regulatory approvals or clearances. |
Internationally, we have obtained a Conformité Européenne, or CE, mark for our INFINITI and INFINITI PLUS analyzers and a total of 24 of our tests. This designation is supported by completed clinical and validation studies that demonstrate the analytical performance of each CE marked test. The CE mark facilitates the marketing and sale of our CE marked analyzers and tests in the European Union and the European Economic Area as well as certain other international markets.
Our test panels that are not 510(k) cleared are offered for sale in the United States under the RUO designation. These RUO tests are labeled "For Research Use Only. Not for use in diagnostic procedures." as required by FDA regulations. Sales of these test panels represented 65%, 82% and 86% of our net revenue for the nine months ended September 30, 2014, and the years ended December 31, 2013 and 2012, respectively.
RUO test panels may only be used in the United States for clinical purposes by laboratories and other facilities certified under CLIA that have incorporated these products into their LDTs pursuant to guidelines issued by the College of American Pathologists. We believe that nearly all of our RUO product sales are incorporated into laboratory developed tests, or LDTs. In order to develop an LDT utilizing our products, these certified laboratories and other facilities must develop and validate a test protocol that includes specimen collection, DNA extraction,
63
polymerase chain reaction, or PCR, amplification, hybridization and detection, and data analysis, interpretation and reporting. Our products provide components that can be used by these certified laboratories and other facilities for the PCR amplification, hybridization and detection portions of these LDTs. We sell each of these components individually, as ordered by the customer in its discretion, and not as a kit or system. The validation process engaged in by these certified laboratories and other facilities can involve validation of the sample collection and extraction process, establishing limits of detection and analytical sensitivity, testing for specificity and cross-reactivity, including interfering substances, validation for assay accuracy, precision and reproducibility, and establishing reportable ranges of test results for the test system and reference values that will be measured against as controls. This validation process also requires verifying the result from the LDT against known standard samples or the results of a high-standard laboratory testing method such as sequencing, and can involve the testing of a large number of patient samples. This process may take from several weeks to several months or more to complete, and thus requires a significant investment by the customer.
We believe that all sales of our RUO products in the United States are to customers that are either certified in the manner described above and have incorporated our products into their LDTs, or that use such products for research only. We are not permitted to represent our RUO products as in vitro diagnostic products. We therefore train our personnel to only market these products to laboratories for research or investigational use in the collection of research data, and to not promote any off-label uses of our products.
We believe that we are in compliance with existing FDA rules and regulations governing our business, including those governing the marketing and sale of RUO tests; however, a significant change in existing laws, or their enforcement, may require us to change our business model or our business practices to maintain compliance with these laws. For instance, in June 2011, the FDA issued a Draft Guidance entitled "Commercially Distributed In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only: Frequently Asked Questions," and in November 2013 issued a guidance document entitled "Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only," or the RUO Guidance, which highlights the FDA's interpretation that distribution of RUO products with any labeling, advertising or promotion that suggests that clinical laboratories can validate the test through their own procedures and subsequently offer it for clinical diagnostic use as an LDT is in conflict with RUO status. The RUO Guidance further articulates the FDA's position that any assistance offered in performing clinical validation or verification, or similar specialized technical support, to clinical laboratories, is in conflict with RUO status. The FDA has generally exercised its enforcement discretion to not enforce applicable regulations with respect to LDTs. However, the FDA has repeatedly indicated since 2010 that it intends to reconsider its policy regarding enforcement and to begin drafting an oversight framework for such tests, and most recently, on July 31, 2014, the FDA notified the U.S. Congress of the FDA's intent to modify its enforcement discretion approach to LDTs in a risk-based manner. If the RUO Guidance were to be enforced, it would limit our marketing of RUO test panels to general discovery laboratories and require us to seek FDA clearance for our RUO test panels, which may require significant time and investment on our part and may reduce our revenues or increase our costs and adversely affect our business, prospects, results of operations or financial condition.
Our Strategy
Our objective is to become a leading provider of genetic tests to clinical reference laboratories, specialty clinics and hospital laboratories in multiple growing market segments. Our INFINITI analyzers can address a wide range of customer throughput requirements and, combined with our large and expanding menu of test panels, we believe gives us a significant competitive advantage within the molecular diagnostics market. We believe that our ability to develop new tests to meet customer demand will enable us to take advantage of emerging opportunities in genetic testing and drive additional consumables sales. One key element of our strategy is to focus on high-volume customers, particularly in the areas of personalized medicine and women's health. We plan to continue to expand into other market segments through the development of additional test panels.
To achieve our objectives, we intend to:
• | Increase placements of our recently launched INFINITI HTS and expand penetration in the highest volume testing market segments. Our INFINITI HTS and menu of test panels provide an easy to use, automated solution with greater breadth of diagnostic information at a lower operating cost per reported result as compared to traditional testing methods. We believe that our INFINITI HTS's process automation, ability to multiplex and broad menu of test panels in the area of high-volume testing are compelling features that are attractive to |
64
customers in our target market segments. We believe that these factors will lead to increased INFINITI HTS market penetration.
• | Target molecular diagnostics laboratories with high potential utilization of our INFINITI and INFINITI PLUS analyzers. We believe that our INFINITI and INFINITI PLUS analyzers' automation, "load and go" design and broad menu of test panels designed to address various throughput requirements, will generate demand from both medium to larger clinical reference laboratories seeking a more flexible and efficient molecular diagnostics platform and from smaller clinical reference laboratories, specialty clinics and hospital laboratories for whom it has not previously been cost-effective to develop their own tests. |
• | Develop and launch test panels in new areas and enhance our current test panels. We plan to add test panels for cholesterol-lowering drugs (statin drug therapy), susceptibility to drug addiction and risk of age-related macular degeneration during 2015. In addition, we continue to create test panels that combine multiple genetic tests on a single microarray to enable our customers to increase throughput capability and significantly decrease the operating cost per reportable result and improve laboratory economics. We believe that continuing to expand and enhance our broad menu of genetic tests will drive additional placements of our INFINITI analyzers and capture additional consumable sales. |
• | Significantly expand our domestic sales force, increase marketing expenditures and expand international distribution of our products. We believe there is a meaningful opportunity to further penetrate existing markets and customers by expanding our direct U.S. sales and marketing organization. In the future, we plan to strengthen and enhance specific target market opportunities by hiring application specialists with in-depth knowledge about those markets, such as transfusion centers, newborn screening and drug addiction and rehabilitation centers. We also plan to expand our existing distributor network outside the United States to enter new markets and capitalize on growing international demand. |
• | Expand FDA approvals. We intend to pursue additional regulatory clearances, approvals and certifications for products and facilities, as necessary. We have received U.S. Food and Drug Administration, or FDA, 510(k) clearance for our INFINITI and INFINITI PLUS analyzers and five of our genetic tests, and we have submitted an additional notification to the FDA for 510(k) clearance of our UGT1A1 test. |
• | Utilize our CLIA-certified laboratory to enhance and complement our product offerings. Our CLIA-certified laboratory enables us to provide a broad range of services to assist clinical reference laboratories and specialty clinics as they enter the molecular diagnostics market using our platform. We expect to attract a number of customers to our platform by providing an enhanced level of customer support, including services offered by our CLIA-certified laboratory. |
Our Market Opportunity
Molecular diagnostics, or MDx, refers to the detection of genetic material, including DNA and RNA and their variations and mutations, and the use of such information to diagnose disease, determine a patient's susceptibility to disease, evaluate response to drug therapy or establish a patient's prognosis. MDx is one of the largest and fastest growing segments in the $50 billion in vitro diagnostics industry. Frost & Sullivan estimated in 2012 that the MDx market will reach $6.2 billion in the United States during 2014 and forecasted a compound annual growth rate for the market in excess of 11%. CMS has estimated in June 2014 there were more than 5,900 independent clinical reference laboratories and specialty clinics and more than 8,900 hospital-based laboratories in the United States. We believe that less than 10% of these laboratories are currently performing MDx testing, and that we have an opportunity to expand our customer base by enabling more laboratories to participate in this growing market.
A fundamental goal of personalized medicine is to deliver the right treatment to the right patient at the right time, and we believe MDx testing is helping to achieve this objective. Increasing knowledge of genetic biomarkers and continued genetic biomarker discoveries in the areas of drug metabolism, drug dosing, use of genetic information to increase the efficacy of chemotherapy agents, identification of infectious diseases and use of genetic biomarkers in monitoring the progression of diseases are driving the growth of MDx testing companies. There is a growing awareness that personalized, predictive and preventative medicine initiatives can improve healthcare outcomes and lower costs. This can be achieved through the earlier diagnosis of disease, improved monitoring of disease progression and more personalized treatment based on a patient's distinct genetic profile. We believe the genetic information generated by our platform can enable a physician to differentiate patient-specific characteristics, design personalized treatment approaches and ultimately improve patient outcomes.
65
Genetic testing is increasingly used as a primary tool to provide actionable information to the healthcare provider for improved patient care. Information provided by genetic testing is playing a key role in guiding therapy, assessing disease risk, improving drug efficacy, identifying non-responders and ultra-, normal- and poor-metabolizers and reducing adverse events. For example, in personalized medicine, the identification of genetic biomarkers that affect drug-metabolizing enzymes has improved dosing of drugs for conditions as wide-ranging as pain management, cardiac assessment and mental health, and has benefited patients by helping them avoid adverse events, adverse drug interactions or ineffective treatments.
Our Target Markets
Personalized medicine
More targeted and effective pharmacogenomic-based treatments have the potential to improve healthcare outcomes and lower healthcare costs, which we believe will lead to increased use of genetic testing. Healthcare providers are able to better treat patients by enhancing efficacy while minimizing possible adverse events by conducting genetic tests prior to prescribing drugs. Many pharmaceutical companies, researchers and pharmacy benefit companies are screening drugs for differences in efficacy and toxicity among individuals with varied genetic profiles. Regulatory agencies have revised drug labels to improve safety and efficacy based on information provided by genetic testing.
Many patients do not currently achieve the best possible outcome with the first drug that they are offered in treatment. For example, according to The Case For Personalized Medicine, 4th edition (2014), published by the Personalized Medicine Coalition, a large number of patients, including 38% of those being treated for depression, 40% of those being treated for asthma, 43% of those being treated for diabetes and 50% of those being treated for arthritis will not respond to initial drug treatments. Studies have linked the variation in efficacy of a patient's response to a prescribed drug treatment to differences in the genes that code for drug-metabolizing enzymes, drug transporters or drug targets, which can be identified through the use of genetic testing. Genetic and other molecular diagnostic testing can assist a physician in selecting an optimal drug therapy in the first instance, and eliminate the otherwise commonly used trial-and-error method of identifying optimal drug therapy for an individual patient. Key tests in our personalized medicine market segment include test panels to assess drug metabolism in pain management, cardiac assessment and mental health. Our leading test panels in this area are our CYP450 2C19, CYP450 2D6I and CYP450 3A4/CYP450 3A5, which are used to determine gene variants that affect the metabolism and efficacy of drugs such as platelet therapy, those for treatment of psychiatric disorders and those prescribed for pain management.
Our target markets in personalized medicine include the following segments:
Pain management. More than 116 million adult Americans suffer from acute or chronic pain each year, and drugs are the "first line" of treatment for most forms of pain. The goal of successful pain management is to effectively control patient pain without causing excess side effects from the medication prescribed. However, studies have shown that only 58% of patients taking prescription medication reported pain relief. Additionally, approximately 80% of post-operative patients experience adverse events from pain medications. In particular, opioids such as hydrocodone, codeine, oxycodone and morphine are frequently prescribed for the relief of chronic and moderate to severe post-surgical pain. According to the IMS Health National Prescription Audit, hydrocodone alone accounted for more than 135.3 million prescriptions in 2012, and many of these patients can experience adverse events from these medications. Opioid-related adverse events include nausea, constipation and respiratory depression, which can be severe. Additionally, opioids are highly addictive and induce drug-resistance and tolerance. As of October 24, 2014, we offered 11 test panels in the area of pain management, which detect multiple mutations associated with six different drug-metabolizing genes: CYP450 2B6, CYP450 2C9, CYP450 2C19, CYP450 2D6, CYP450 3A4 and CYP450 3A5. Identification of these mutations can assist physicians in improving selection and dosing of drugs and better manage patient treatment by reducing adverse events and improving efficacy.
Cardiovascular health assessment. According to a report from the World Health Organization, or WHO, cardiovascular diseases were responsible for 30% of global deaths in 2008. The WHO estimates that by 2030, 23.3 million people will die annually from some form of cardiovascular disease. With the increasing prevalence of cardiovascular diseases, information generated by genetic testing is becoming increasingly important for drug efficacy, managing cardiovascular condition and overall treatment of disease. For example, it is now widely
66
accepted that the efficacy of one of the most commonly used cardiovascular drugs, Plavix (clopidogrel), to reduce the risk of heart attack, unstable angina, stroke prevention of stent thrombosis and cardiovascular death following acute coronary syndrome, is heavily influenced by a patient's genetics. Plavix has received a "black box" warning label from the FDA that identifies the chance of limited effectiveness for patients who are poor metabolizers of the drug, and that informs physicians of the availability of genetic tests to identify such patients. Plavix is metabolized into its active form by CYP450 2C19. By utilizing genetic testing, including the test panels that we offer, the cardiologist can determine whether the patient is a poor-, normal- or ultra-metabolizer or a non-responder before the drug therapy is administered. As of October 24, 2014, we offered 20 test panels in the area of cardiovascular health assessment, five of which have received FDA 510(k) clearance, including our Plavix responder and Warfarin sensitivity test panels.
Mental health. Drugs associated with mental health, including drugs focused on central nervous system disorders, and treatment of depression and anti-psychotic therapy, represent a major component of overall pharmaceutical sales. According to the Centers for Disease Control and Prevention, or CDC, as much as 11% of the U.S. population is taking antidepressants at a given time. Despite this prevalence, approximately 30 to 40% of patients do not respond to the first medication prescribed. The resulting trial and error process delays effective treatment for patients and increases healthcare costs. Genetic testing can provide information to allow physicians to select a more appropriate drug or combination of drugs specific to the patient's genetic makeup and improve patient outcomes. Additionally, many doctors are taking greater precautions in choosing medications for young patients, because of potential developmental issues which could result from psychiatric drug treatment for minors, such as for attention deficit hyperactivity disorder. As of October 24, 2014, we offered 12 test panels in the area of mental health. Our test panels identify mutations in the drug metabolizing genes ApoE, CYP450 2D6, 2C19, 2B6 and 2C9, and can enable the healthcare provider to select optimal combinations of anti-convulsive, anti-depression and anti-psychotic drugs or stimulants for an individual patient.
Women's health
The women's health MDx testing market represents a substantial and growing market opportunity. According to the CDC, approximately 79 million Americans are infected with Human papillomavirus, or HPV, and approximately 14 million become infected each year. Current regulations from the U.S. Preventive Services Task Force recommend that MDx HPV tests for women be utilized in conjunction with cytology (Pap test) in standard five year intervals. Additionally, the CDC estimates that there are nearly 20 million new cases of STDs each year, costing the nation approximately $16 billion in healthcare costs annually. MDx testing in this market is currently a minority of the tests available, but is expanding significantly due to increased applications, better performance and improved pathogen identification capabilities. As of October 24, 2014, we offered 19 test panels in the area of women's health.
Key tests in this segment include four test panels for HPV testing and nine test panels designed to identify various microorganisms related to STDs and vaginal infections. Our HPV test panels are designed for screening and genotyping all 14 high-risk types of HPV simultaneously. Our STD panels are designed to identify the presence of six pathogens in one test panel: Chlamydia trachomatis, Neisseria gonorrhoeae, Ureaplasma urealyticum, Mycoplasma genitalium, Mycoplasma hominis and Trichomonas vaginalis. In addition, we have panels to test for Bacterial vaginosis (BV) and Candida vaginitis. Our Resolve-QUAD detects Gardnerella vaginalis, Mobiluncus spp., Candida albicans, Candida glabrata and Trichomonas vaginalis. Resolve-QUAD is able to detect incidences of BV, candidiasis and trichomoniasis. Our panels are designed to screen for multiple microorganisms simultaneously on a single sample. We believe our women's health test panels are the most comprehensive menu of test panels in the market currently.
Other markets
We also target the markets of: (i) oncology to help manage chemotherapy treatments for breast, colorectal, lung, melanoma and thyroid cancers; (ii) infectious diseases for detection of influenza, nontuberculous mycobacteria, respiratory viruses and tuberculosis; and (iii) genetic disorders including, but not limited to, tests for identification of carriers of gene mutations associated with Bloom disease, Canavan disease, cystic fibrosis and Familial Mediterranean fever.
67
New test panels in development
In addition to our current menu of 62 test panels, we are developing new test panels for the following markets:
Statin drug therapy. Statin drugs are some of the most prescribed medications in the world and are used to reduce the level of low-density lipoprotein cholesterol, or LDL, in the blood. According to the CDC, there are currently more than 71 million American adults (over 20 years of age) with high LDL. The Clinical Research Institute at Duke University estimates that over 43 million Americans aged 40 to 75 are currently prescribed the statin class of cholesterol lowering drugs, which will increase to 56 million Americans, half the population of this age group. However, statin LDL-lowering drugs can cause significant adverse events, including muscle aches, cramps and muscle weakness, and if not controlled, can lead to more serious muscular injury in certain patients. Our statin drug management test panel and decision support system is designed to identify individuals who are genetically susceptible to elevated liver enzymes or liver damage, or muscle pain (myalgia), while on certain statins and who may develop muscular injury (myopathy) while on high dose statin therapy. We are collaborating with Genomas, Inc., a biomedical company and genetic reference laboratory affiliated with Hartford Hospital, which is the teaching hospital for the University of Connecticut Medical School, on development of the test panel and decision support system. We expect this test panel will provide a reliable, cost-effective platform for clinical laboratories to provide results and guidance to physicians to optimize treatment strategies for LDL disorders. Our statin drug management panel will identify 34 mutations and will include a decision software application to develop risk scores to help guide a physician in the selection and dosing of statin therapies. This test panel is currently in alpha trials and we expect to launch commercially in 2015.
Drug addiction. Scientific advances have improved our understanding of drug abuse and addiction. Addiction is now recognized as a chronic relapsing brain disease expressed in the form of compulsive behaviors, and this understanding has improved healthcare providers' ability to both prevent and treat addiction. We believe that the use of genetic information for diagnosis, treatment, and prevention strategies can lead to reduced relapse and enhanced quality of life for many patients who are addicted or at risk for addiction. Drugs that have the ability to activate the mesolimbic brain reward pathway and increase dopamine levels have the potential to cause addiction in certain patients, suggesting that individual differences in a marker of dopamine function can influence a person's susceptibility to drug abuse. Our test panel identifies markers associated with multiple genes and 16 variants to aid healthcare providers in addiction and rehabilitation centers to use these genetic biomarkers to better understand why certain therapies are ineffective and when to change a certain drug regimen. This test panel is currently in alpha trials and we expect to launch commercially in 2015.
Age-related macular degeneration. Age-related macular degeneration, or AMD, is the leading cause of central vision impairment in persons over 50 years of age in developed countries. Both genetic and environmental factors play major roles in AMD etiology, and multiple gene variants and lifestyle factors such as smoking have been associated with the disease. We have developed a test panel to identify 21 genetic mutations for the potential of improved risk prediction and therapeutic intervention in AMD patients. Our AMD panel identifies the following genes associated with AMD: C3, C2, CBF, ABCA1, VEGFA and TIPM3. We believe this test can provide essential information to assess risk and help guide therapy for wet and dry forms of AMD. This test panel is currently in alpha trials and we expect to launch commercially in 2015.
Limitations of Traditional Testing Methods
A variety of traditional genetic testing methods have been developed, including DNA sequencing, gene expression and genotyping, to detect genetic biomarkers. These traditional testing methods have a number of drawbacks and limitations, including:
• | Throughput limitations. There is increasing demand for high throughput systems that can perform large test volumes in a single day, and deliver multiple multiplexed results per patient. Traditional systems in the market have throughput constraints and typically process a limited number of patient samples simultaneously. |
• | High operating cost per reportable result. Because many existing systems require specialized personnel and training to complete complex and extensive protocols, the tests can be time-consuming and result in high labor costs. These processes may also use a significant amount of reagent, which can be costly. The use of supplementary, discrete instrumentation to perform semi-manual tests also increases costs, as compared to a system that automates all the discrete processes of genetic testing. |
68
• | Limited testing menu. Many existing MDx systems have limited test menus. As a result, a laboratory may need to purchase additional systems with specific testing capability to satisfy its needs. This requires separate training of operators on the use and maintenance of each system and may require a significant amount of laboratory bench space and inventory. The combination of these factors often makes in-house MDx testing impractical, particularly for small and mid-size laboratories. |
• | Inability to multiplex. In many cases, the predisposition to a genetic disorder, or the presence of a particular disease, condition or genetic variance affecting therapy, is caused by multiple genetic mutations that necessitate testing for multiple biomarkers to diagnose those diseases, conditions or variances. Many traditional technologies are only able to examine one biomarker at a time, and, in order to make a diagnosis, the laboratory must perform repeated tests on a sample. Without multiplexing capabilities, serial testing is required which is expensive, time consuming and requires higher sample volumes. |
• | Limited automation. While a number of companies offer systems that can perform MDx tests, these systems tend to automate only certain steps in the testing process. The manual handling of samples can lead to an increased risk of sample contamination, and the lack of automation results in multiple, complex steps to achieve patient results and can increase the probability of operator error if any step is omitted or performed out of sequence. In addition, many of these traditional systems do not enable the testing of multiple patient samples in a single microarray, which reduces the cost-benefit of higher throughput. |
• | Need for specialized labor. Specialized laboratory technicians and in some cases specialized training are required to properly perform and evaluate the quality and accuracy of the results of most traditional MDx technologies. |
Our Solution
Our platform has been designed to enable a broad range of clinical reference laboratories, specialty clinics and hospital laboratories to more cost-effectively perform, or enter the market to perform genetic testing at variable levels of throughput, which we believe will drive adoption and use of our platform as well as expand the potential of the MDx testing market. We believe that many medium-sized clinical reference laboratories, specialty clinics and hospital laboratories are seeking to add or expand MDx capabilities to treat patients efficiently and provide a comprehensive offering. We believe that this trend is being facilitated in part by new technologies like ours.
Our platform has a number of key advantages, including:
• | Significantly higher throughput and better workflow. Our broad offering of INFINITI analyzers is designed to address our target customers' varied throughput and workflow requirements. Using one INFINITI HTS, laboratories are capable of producing up to 6,912 patient results per day. We believe that we can substantially increase a laboratory's workflow by enabling them to perform their tests on our highly integrated and automated system, which reduces or eliminates the need for laboratories to run tests in batches. |
• | Improved laboratory economics. Our INFINITI and INFINITI PLUS eliminate the need for complex protocols and manual intervention once a test is initiated, which is intended to reduce the laboratory's operating cost per reportable result by simplifying workflow and reducing the need for highly skilled technicians. Our INFINITI HTS allows individual components to remain active during the workflow process, which can significantly improve throughput time and reduce costs. |
• | Broad menu of test panels. We currently offer 62 test panels, which we believe represents the broadest available menu of test panels on a single system in the market today. The depth and breadth of our menu of test panels, all of which are designed to run on any of our INFINITI analyzers, allows laboratories to utilize space, labor and capital investment more efficiently and to conduct more genetic tests in less time at a lower operating cost per sample. In addition, we intend to increase the number of test panels available for use on our platform, including in the areas of statin drug therapy, drug addiction and AMD, and believe that laboratories using our system will be able to broaden their MDx offerings without significant additional capital investment or operator training. |
• | Multiple patient array technology. Our proprietary multiple patient array, or MPA, technology is designed to test up to eight distinct patient samples on a single microarray. This enhances throughput by up to 700% while reducing operating cost per sample by up to 87% as compared to our single patient microarrays. Our MPA technology is particularly well suited for addressing high-volume test markets such as HPV and tuberculosis. |
69
• | Ability to multiplex. The analysis of multiple genes from a single patient sample is commonly referred to as multiplexing. Many diseases and patient responses to therapy are caused by multiple genetic mutations that necessitate testing for multiple biomarkers to diagnose those diseases or to predict or monitor therapy response. Our platform is able to detect up to 1,024 individual features of biochemical sensors, or biomarkers, within a single microarray, which reduces the amount of sample needed, the time required to run the test and the need for multiple test panels. |
• | Increased accuracy of results. Manual handling of samples is the most common cause of contamination in existing technologies. By reducing the risk of human error and contamination, we believe that our INFINITI analyzers can provide more accurate and reproducible test results compared to other less automated systems. In addition, where certain systems only use target or signal amplification (e.g., PCR amplification), we believe that our combined target and signal amplification technologies can increase the sensitivity and specificity over these widely used stand-alone amplification methods. |
Our Products and Technology
Our platform is comprised of five key technological innovations: (1) our INFINITI analyzers; (2) our multiplexing test format; (3) our BioFilmChip microarrays; (4) our multiple patient array technology; and (5) our Intellipac Reagent Management Modules.
Our INFINITI analyzers
Our platform includes a family of analyzers, each of which is designed to address customer-specific needs based on the customer's workflow and throughput requirements. Our platform includes our INFINITI, INFINITI PLUS and INFINITI HTS analyzers. Our INFINITI analyzers automate the discrete processes in genetic analysis and offer a wide range of sample throughput. All of our INFINITI analyzers use the same proprietary technology and consumables to generate consistent patient results presented in an identical format. The following table illustrates the test capabilities of our primary INFINITI analyzers:
Instrument | Capacity per run (1) | Patient results (1) |
INFINITI HTS | 384 samples | up to 6,912 per day |
INFINITI PLUS | 48 samples | up to 192 per day |
INFINITI | 24 samples | up to 96 per day |
(1) Figures assume the use of our four-patient multiple patient array test panel over a 24-hour period.
Our newest analyzer, the INFINITI HTS, was commercially launched in February 2014 following a three-year design and development process. Our INFINITI HTS is a scalable, multiplexing, random access, microarray-based system comprised of three independent operating modules. We believe that the INFINITI HTS has the highest processing capacities of any molecular diagnostics system available in the market today. Our INFINITI HTS was developed in response to the demand from clinical reference laboratories and specialty clinics that perform MDx testing in personalized medicine and women's health. This platform features a hybridization chamber (INFINITI Incubator), a multi-channel microarray processing system (INFINITI Processor) and a built-in high resolution imaging system (INFINITI Ace). The modularity of the system is the key to its scalability. With rapid processing of samples on the INFINITI Incubator and scanning time of less than ten minutes for 96 multiplexed microarrays, the system significantly enhances test throughput capabilities for our laboratory customers.

The INFINITI HTS is designed to operate in a random and continuous access mode, allowing simultaneous processing of multiple tests within the same run, and is capable of producing up to 6,912 patient results per day with a single technician. Once testing is completed, the INFINITI HTS transmits test results to the laboratory's
70
information system for electronic transmission to the ordering physician. The INFINITI HTS automates the discrete processes of genetic analysis and is a scalable system which provides flexible configuration options to maximize high-volume work efficiency.
Our INFINITI and INFINITI PLUS analyzers are automated, multiplexing, continuous flow, random access microarray instruments that are designed to integrate all the discrete processes of genetic testing, including sample handling, reagent management and detection for the analyses of DNA sequences, into a self-contained system. These INFINITI analyzers feature a built-in confocal microscope and temperature cycler for target and signal amplification. They operate on a "load and go" basis. This means that to run a test, an operator need only load prepared samples into the applicable INFINITI analyzer, along with the test-specific BioFilmChips and Intellipac Reagent Management Modules, and the testing process will run to completion without any intervention or supervision from the operator. |  |
INFINITI Analyzer | |
From the perspective of the technician, the test protocols are identical for each of our genetic tests, which eliminates the need for additional training of operators when additional tests are added. After the test is completed, the INFINITI analyzer generates an electronic report that can be transmitted directly to a laboratory information system.
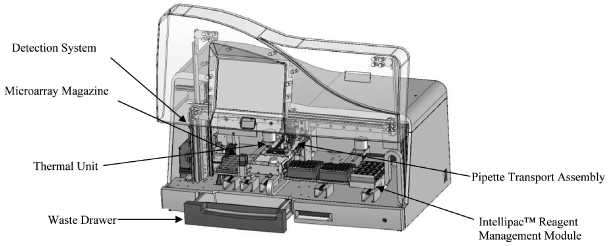
The hardware in each of our INFINITI analyzers is controlled by our proprietary QMatic scheduling software, which is embedded within the onboard computer. The QMatic software has a schedule manager that is designed to control all operations of the analyzer, including test protocol, fluid handling, robotics, optical detection and results analysis. Key features include: (1) processing of multiple samples simultaneously; (2) running multiple samples and multiple tests at the same time; (3) test protocol monitoring; and (4) multiplexing different samples on the single microarray for increased throughput and reduced cost.
Tests are processed automatically and read by the built-in confocal microscope. The optics module in our INFINITI analyzers is a lightproof assembly comprised of a camera, a laser and a photomultiplier tube. Using an excitation wavelength from the laser light source, the camera takes micron-level pictures of reference fluorescent dye spots on the BioFilmChip. The integrated software uses that data to calculate the location of the spots on the specific microarray, and the optics module scans those calculated locations, and analyzes the microspots on the microarray.
Our multiplexing test methods
Our multiplexing technology allows our INFINITI analyzers to detect multiple biomarkers across multiple genes at the same time on a single microarray, eliminating the need to process multiple tests separately. For example, our pain management panel can detect multiple mutations associated with six different genes (CYP450 2B6, CYP450 2C9, CYP450 2C19, CYP450 2D6, CYP450 3A4 and CYP450 3A5) to determine the efficacy, toxicity and
71
dosing of specific drugs. In addition, our genetic test for HPV detects and genotypes all 14 high-risk types of HPV simultaneously on a single microarray from a single sample.
Our BioFilmChip microarrays
Our proprietary BioFilmChip microarrays consist of multiple layers of a hydro-gel matrix coated on polyester film sandwiched between a plastic base and a reaction body to form a microarray. Utilizing our proprietary and patented manufacturing processes, we coat polyester film with four different layers of emulsions and then cut the film into sections for each BioFilmChip. Each of the emulsion layers has a unique formulation designed to address a specific function necessary for the film to act as the base of the microarray. The microarrays are printed with up to 1,024 individual features of biochemical sensors depending on the requirements of the test. These spotted microarrays are then packaged into specifically designed magazines and are used on all INFINITI analyzers. We believe this process provides us a manufacturing, scale and operating cost advantage.
The BioFilmChip has a set of unique universal immobilized capture probes. A target gene-specific detection primer is hybridized (the reaction of complementary DNA sequences) to the PCR amplicon of the sample. PCR is an enzymatic reaction that makes millions of copies of DNA. The PCR-copied products are known as PCR amplicons. The hybridized detection primer is designed to extend in the presence of a fluorescence precursor to produce a fluorescent extension product that hybridizes to the microarray (the process increases the signal so that the detection system can read it). The detection primer has two complementary parts: one target-specific and the other immobilized capture probe-specific. After hybridization and washing of the excess unbound probes, the chip is scanned for fluorescence and the data is analyzed. Where certain systems only use target or signal amplification, we believe our combined target and signal amplification technologies can increase the sensitivity and specificity over these widely-used stand-alone amplification methods. For example, we have conducted internal studies that have demonstrated that our system has achieved sensitivity to the level of detection of ten copies of mycobacterium tuberculosis and five copies of HPV, which we believe is more sensitive than many other available tests.


BioFilm cross-sectional view Microarray top view



Microarray side view Microarray magazine Intellipac reagent module
Multiple patient microarray
Our proprietary MPA technology enables our INFINITI analyzers to process multiple patient samples on a single microarray. This differentiating technology substantially reduces our per test product costs, reduces our inventory costs and increases our test production capacity. It also can create increased efficiency for our customers by lowering their operating costs and increasing their throughput, which leads to improved laboratory capacity, equipment utilization and improved economics. For example, our MTBC OCTA (drug-resistant tuberculosis test panel) is designed to test up to eight patient samples on a single microarray, increasing throughput for our laboratory
72
customers by up to 700% while reducing their operating cost per sample by up to 87%, as compared to our single patient microarrays. We are also developing an application to produce an MPA that permits samples from up to ten patients for certain tests to be processed simultaneously on a single microarray. Our MPA technology first processes the samples individually, where each sample is assigned a separate set of detection primer tags. The detection primer tags correspond to one of eight separate zones on the microarray (one zone for each sample). After the dye-labeled probes bind to the specific site on the microarray that corresponds to its detection primer tag, the INFINITI analyzers scan for the presence of the dye-labeled probes at the appropriate sites to determine whether the genetic variant being tested for is present in each of the samples. We can produce up to 1,024 microspots on a single BioFilmChip. Results are analyzed and presented in numerical format, and the electronic results report is transmitted directly to a laboratory information system.
 |  |
Images of low- to medium-density spotted microarrays
Our Intellipac Reagent Management Modules
Our assay specific Intellipac Reagent Management Modules provide the proprietary reagent used to complete our genetic tests. Our platform will only operate with our proprietary reagent modules. The reagent module is designed to communicate all relevant information about a test to the INFINITI analyzer without any intervention from the laboratory technician, saving time by automatically recording all pertinent test details, and reducing the possibility for human errors which could occur in manual systems by eliminating contamination risk. A read-write memory chip embedded in the reagent module saves test-specific information, including reagent identification, expiration dates, lot number, amount of reagent remaining for future tests, specific instructions for test processing, the time last used and the serial number for the instrument. The INFINITI analyzer opens the reagent module and breaks the seal, reads all the needed information and prompts the laboratory technician for further action if needed.
Limitations of microarray test methods
In general, microarray test methods are subject to the possibility of false negative test results due to unexpected gene sequence variations. Nearly all genetic tests are designed to detect a sequence in the given region on the targeted genome. Because genetic test methods are premised upon identifying these specific gene sequences, the phenomenon of sequence variance in a gene away from the expected, mapped sequence being measured will result in the sample not being detected and the result being reported as a negative test. This is common to all genetic tests,
73
and as a result laboratory technicians and molecular biologists are generally aware of this possibility. Although unexpected gene sequence variations tend to be rare occurrences, when we or other industry participants identify that unexpected gene sequence variations are recurring, and that there is a market demand to test for this sequence variance, we may be inclined to modify our tests or develop new tests, to the extent commercially feasible, to capture the newly identified variations. These test modifications or new test developments go through our standard development process, and can involve significant time and expense.
Our Current Menu of Test Panels
We currently offer 62 test panels for use with our family of INFINITI analyzers. In the area of personalized medicine, we offer test panels in pain management, mental health, cardiovascular health assessment and oncology. In the area of women's health, we offer test panels in cervical cancer, STDs and vaginal infections. We also offer test panels in infectious diseases (including tests for tuberculosis and respiratory viruses), genetic disorders and newborn screening.
74
Product (1) | Application | Market Segments | ||||||||
Pain management (11 tests) | Mental health (12 tests) | Cardiovascular risk assessment (20 tests) | Other personalized medicine (21 tests) | Women’s health (19 tests) | Oncology (23 tests) | Infectious disease (20 tests) | Genetic disorders (6 tests) | Newborn screening (3 tests) | ||
5-FU (Fluorouracil) | Metabolism of leading cancer drug/toxicity assessment | s | s | |||||||
ApoE QUAD | Drug metabolism | s | s | s | ||||||
Ashkenazi Jewish Panel | Diseases more prevalent in the Ashkenazi Jewish population | s | s | |||||||
Bacterial Vaginosis QUAD (3) | Bacterial infection - Sexually transmitted disease | s | s | |||||||
BRAF (3) | KRAS and BRAF amino acid changes - Metastatic Colorectal Cancer | s | s | |||||||
Breast Cancer Panel – AJ QUAD | Breast cancer risk | s | s | s | ||||||
Candida Plus QUAD | Fungal infection – sexually transmitted disease | s | s | |||||||
Candida Vaginitis QUAD (3) | Fungal infection - Sexually transmitted disease | s | s | |||||||
CFTR-15 | Cystic fibrosis gene mutation | s | s | |||||||
CFTR-31 (3) | Cystic fibrosis gene mutation | s | s | |||||||
CHEK-2 | Familial breast cancer risk | s | s | s | ||||||
CT-NG QUAD | Gonorrhea, chlamydia | s | s | |||||||
CYP450 2B6 QUAD | Drug metabolism | s | s | |||||||
CYP450 2C19 (2) (3) | Drug metabolism | s | s | s | s | |||||
CYP450 2C19 Plus (3) | Drug metabolism | s | s | s | s | |||||
CYP450 2C19 Plus Duplex | Drug metabolism | s | s | s | s | |||||
CYP450 2C9 - VKORC1 (3) | Sensitivity to Warfarin, drug metabolism | s | s | s | s | |||||
CYP450 2D6-BC | Drug metabolism | s | s | s | s | |||||
CYP450 2D6I (3) | Drug metabolism | s | s | s | s | |||||
CYP450 2D6T | Efficacy of leading breast cancer drug | s | s | s | ||||||
CYP450 3A4 | Drug metabolism | s | s | s | s | |||||
CYP450 3A5 | Drug metabolism | s | s | s | s | |||||
CYP450 3A4-3A5 | Drug metabolism | s | s | s | s | |||||
CYP450 3A4-3A5 Duplex | Drug metabolism | s | s | s | s | |||||
EGFR | Lung cancer efficacy | s | s | |||||||
Factor II (2) (3) | Increased risk of blood clots | s | ||||||||
Factor II QUAD | Increased risk of blood clots | s | ||||||||
Factor II Plus | Increased risk of blood clots | s | ||||||||
Factor II Plus - V Panel | Increased risk of blood clots | s | ||||||||
Factor II/V (2) (3) | Increased risk of blood clots | s | ||||||||
Factor II/V QUAD | Increased risk of blood clots | s | ||||||||
75
Factor V (2) (3) | Increased risk of blood clots | s | ||||||||
Factor V QUAD | Increased risk of blood clots | s | ||||||||
Factors Plus QUAD | Increased risk of blood clots | s | ||||||||
FII - FV - MTHFR Panel (3) | Increased risk of blood clots | s | ||||||||
FII - FV - MTHFR QUAD | Increased risk of blood clots | s | ||||||||
FII Plus-FV MTHFR Panel | Increased risk of blood clots | s | ||||||||
Flu A-sH1N1 (3) | Flu A-sH1N1 | s | ||||||||
FMF Panel (3) | MEFV gene mutation | s | ||||||||
HPV - HR HEX | Cervical cancer risk | s | s | s | ||||||
HPV - HR QUAD (3) | Cervical cancer risk | s | s | s | ||||||
HPV Genotyping (3) | Cervical cancer risk | s | s | s | ||||||
HPV Quad (3) | Cervical cancer risk | s | s | s | ||||||
KRAS (3) | KRAS amino acid changes | s | s | |||||||
KRAS-BRAF (3) | KRAS and BRAF amino acid changes - Metastatic Colorectal Cancer | s | s | |||||||
Leuko QUAD | Gonorrhea, chlamydia, trichomonas vaginalis | s | s | |||||||
MDR-1 | Drug metabolism | s | s | |||||||
MDR-TB | Drug resistance to leading treatments for tuberculosis | s | ||||||||
Mobi QUAD | Mobiluncus infection | s | s | |||||||
MTBC OCTA | Drug resistance to leading treatments for tuberculosis | s | ||||||||
MTHFR (3) | Increased risk of blood clots | s | s | s | ||||||
MTHFR QUAD | Increased risk of blood clots | s | s | s | ||||||
NAT-2 | Bladder and other types of cancer risk | s | s | |||||||
NTM | Nontuberculous mycobacterium infection | s | ||||||||
Resolve QUAD | Trichomonas, Candida, Gardnerella, Mobiluncus | s | s | |||||||
RVP 10 | Respiratory virus infection | s | ||||||||
RVP Plus (3) | Respiratory virus infection | s | ||||||||
STD-6 QUAD | Gonorrhea, chlamydia, trichomonas and others | s | s | |||||||
UGT1A1 (3) | Initial dosing of leading cancer drug | s | s | |||||||
UroGen QUAD (3) | Ureaplasma urealyticum, mycoplasma genitalium, mycoplasma hominis | s | s | |||||||
UroGen Plus QUAD | Ureaplasma urealyticum, mycoplasma genitalium, mycoplasma hominis | s | s | |||||||
Warfarin Assay (2) (3) | Sensitivity to Warfarin | s | s | |||||||
(1) | Unless otherwise indicated as having received 510(k) clearance, the tests listed in this table have not been cleared or approved for diagnostic use by the FDA and are sold on an RUO basis in the United States. |
(2) | We have received 510(k) clearance for our Warfarin Assay, CYP450 2C19, Factor II, Factor II/V and Factor V test panels. |
(3) | These tests are CE-marked for the international market. |
76
Descriptions of our test panels
The following is a description of our test panels that are available for sale.
5-FU (Fluorouracil). Our 5-FU, test is designed to detect and identify point mutations in the Dihydropyrimidine Dehyrogenase, MTHFR and Thymidylate Synthase genes that affect the toxicity and efficacy of 5-FU, a leading chemotherapy drug. Fluorouracil is given as a treatment for several types of cancer, including colon, rectal, breast, stomach and pancreatic cancers.
ApoE QUAD. Our ApoE QUAD test is designed to identify two mutations on the apolipoprotein E gene that affects the function of ApoE in four samples simultaneously on a single BioFilmChip.
Ashkenazi Jewish Panel. Our Ashkenazi Jewish Panel test is designed to detect 31 genetic mutations associated with susceptibility to eight genetic diseases that occur more frequently in the Ashkenazi Jewish population, including Tay-Sachs disease, Canavan disease, Familial Dysautonomia, Gaucher’s disease, Fanconi Anemia, Niemann-Pick disease and Bloom Syndrome.
Bacterial Vaginosis QUAD. Our Bacterial Vaginosis QUAD test is designed to detect the presence of the organisms that can cause six sexually transmitted bacterial infections: Bacteroides fragilis, Gardnerella vaginalis, Mobiluncus mulieris, Mobiluncus curtisii, Atopobium vaginae and Prevotella bivia in four samples simultaneously on a single BioFilmChip.
BRAF Panel. Our BRAF Panel is designed to identify select variants in regions important for BRAF activity, which we believe could assist in developing proper therapeutic treatment of certain cancers.
Breast Cancer Panel-AJ QUAD. Our Breast Cancer Panel-AJ test is designed to identify three genetic mutations common in the Ashkenazi Jewish population in the breast cancer 1, or BRCA1, and breast cancer 2, or BRCA2, genes that are associated with an increased risk of developing breast cancer in four samples simultaneously on a single BioFilmChip.
Candida Vaginitis QUAD. Our Candida Vaginitis QUAD test is designed to detect the presence of the organisms that can cause five sexually transmitted fungal infections: Candida albicans, Candida parapsilosis, Candida tropicalis, Candida glabrata and Candida krusei in four samples simultaneously on a single BioFilmChip.
CFTR-15. Our CFTR-15 test is designed to detect the presence of 15 mutations in the cystic fibrosis transmembrane conductance regulator, or CFTR, gene, which are associated with an increased risk of developing cystic fibrosis. This panel of variants are targeted for the international market.
CFTR-31. Our CFTR-31 test is designed to detect the presence of 31 mutations in the CFTR gene, which are associated with an increased risk of developing cystic fibrosis.
CHEK-2. Our CHEK-2 test is designed to identify select variants of the CHEK-2 gene that are associated with an increased risk of developing breast cancer. Mutations in the CHEK-2 gene also have been associated with other hereditary and somatic (not inherited) cancers, including prostate, lung, colon, kidney, thyroid and ovarian cancers.
CT-NG QUAD. Our CT-NG QUAD test is designed to detect the presence of Chlamydia trachomatis and Neisseria gonorrhea in four different samples simultaneously on a single BioFilmChip.
CYP2C19 (single and quad formats). Our CYP2C19 test aids clinicians in determining therapeutic strategy for drugs such as proton pump inhibitors and anti-epileptics which are metabolized CYP450 2C19. This test received 510(k) clearance from the FDA in October 2010.
CYP450 2B6 QUAD. Our CYP450 2B6 test is designed to detect seven mutations in the CYP450 2B6 gene that affect the efficacy of many pain management drugs, including Methadone and HIV drugs like Efavirenz, in four samples simultaneously on a single BioFilmChip.
CYP450 2C19 (single and duplex formats). Our single test panel and two test panel CYP450 2C19 Plus tests are designed to identify select variants of the CYP450 2C19 gene that affect the efficacy of many drugs, including the anticoagulant Plavix (clopidogrel), diazepam (an antiepileptic drug) and pantoprazole (a proton pump inhibitor).
77
CYP450 2C9-VKORC1. Our CYP450 2C9-VKORC1 test is designed to identify select variants of the CYP450 2C9 and VKORC1 genes that affect the metabolism of warfarin. The test is designed to detect expanded numbers of CYP450 2C9/VKORC1 variants found in certain ethnic groups as compared to our Warfarin test.
CYP450 2D6BC. Our CYP450 2D6BC test is a modified version of the CYP450 2D6I assay, specifically optimized for use with various types of buccal samples.
CYP450 2D6I. Our CYP450 2D6I test is designed to identify select variants of the CYP450 2D6 gene that affect the efficacy of many drugs, including the antidepressants Paroxetine and Fluoxetine and the cancer drug Tamoxifen.
CYP450 2D6T. Our CYP450 2D6T test is designed to identify select variants of the CYP450 2D6 gene that affect the efficacy of Tamoxifen, a breast cancer drug.
CYP450 3A4 / CYP450 3A5/CYP450 3A4-3A5. Our CYP450 3A4, CYP450 3A5 and CYP450 3A4-3A5 tests are designed to identify select variants of the CYP450 3A4 and CYP450 3A5 genes that affect the metabolism of many drugs, including most calcium channel blockers, most benzodiazepines and human immunodeficiency virus, or HIV, protease inhibitors. The presence of these variants may be used to determine the cause of amplified side effects to a drug and unresponsiveness to the therapeutic effects of a drug.
EGFR. Our EGFR test is designed to identify receptors that may indicate whether cancer patients will respond to drugs that inhibit EGFR expression, such as Tarceva and Gefitinib.
Factor II / Factor V / FII/V Panel. Our Factor II, Factor V and FII/V Panel tests are designed to identify select variants of the Factor II and Factor V genes associated with an increased risk of developing blood clots. Each of these tests received 510(k) clearance from the FDA in February 2007.
Factor II Plus / Factor II Plus–V Panel / FII-FV-MTHFR Panel / FII Plus-FV-MTHFR Panel. Our Factor II Plus, Factor II Plus-V Panel, FII-FV-MTHFR Panel, and FII Plus-FV-MTHFR Panel single test panel and four test panel thrombophilia tests are designed to identify select variants of the Factor II, Factor V and Methylenetetrahydrofolate reductase, or MTHFR, genes associated with an increased risk of developing blood clots.
Factor II QUAD / Factor II-V QUAD / Factor V QUAD / Factors Plus QUAD / FII-FV-MTHFR QUAD. Our Factor II QUAD, Factor II-V QUAD, Factor V QUAD, Factors Plus QUAD and FII-FV-MTHFR QUAD are four test-panel thrombophilia tests designed to identify select variants of the Factor II, Factor V and MTHFR genes associated with an increased risk of developing blood clots.
Flu A-sH1N1. Our Flu A-sH1N1 test is designed to detect the presence of Flu A and 2009 H1N1, common respiratory viruses found in the human respiratory tract.
FMF Panel. Our FMF panel is designed to detect 13 genetic mutations in the MEFV gene, with intent to cover the most prominent disease-causing mutations in all afflicted ethnicities.
HPV-HR HEX. Our HPV-HR HEX test is designed to screen six different samples for 14 high-risk types of HPV simultaneously on a single BioFilmChip.
HPV-HR QUAD. Our HPV–HR QUAD test is designed to genotype 14 high-risk types of HPV in four different samples simultaneously on a single BioFilmChip.
HPV Genotyping. Our HPV Genotyping test is designed to identify 26 high- and low-risk HPV types and help identify women at high risk for the development of cervical disease and cervical cancer.
HPV QUAD. Our HPV QUAD test is designed to screen four different samples for 13 high-risk and two low-risk types of HPV simultaneously on a single BioFilmChip.
KRAS. Our KRAS test is designed to identify select mutations of the KRAS gene in colorectal cancer patients, who may not benefit from anti-EGFR therapies, such as Cetuximab and Panitumumab.
KRAS-BRAF. Our KRAS-BRAF test is designed to identify select mutations of the BRAF gene and the KRAS gene in metastatic colorectal cancer patients who have been found to not respond to certain anti-EGFR therapies, such as Cetuximab and Panitumumab.
78
Leuko QUAD. Our Leuko QUAD test is designed to detect Chlamydia trachomatis, Neisseria gonorrhea and Trichomonas vaginalis in four samples simultaneously on a single BioFilmChip.
MDR-1. Our MDR-1 test is designed to identify select variants of the MDR-1 gene, which is helpful in predicting how certain drugs will penetrate through different pharmacological barriers in the body and how new anti-cancer or anti-parasite agents will interact with MDR-1 and potential liver toxicity.
MDR-TB. Our multidrug-resistant tuberculosis, or MDR-TB, test is designed to detect the presence of tuberculosis and assess drug resistance to the tuberculosis treatments rifampin, isoniazid and pyrazinamide.
Mobi QUAD. Our Mobi QUAD test is designed to detect the presence of the organisms that can cause two sexually transmitted bacterial infections, Mobiluncus mulieris and Mobiluncus curtisii, in four samples simultaneously on a single BioFilmChip.
MTBC OCTA. Our MTBC OCTA test is designed to detect the presence of tuberculosis for eight different samples simultaneously on a single BioFilmChip.
MTHFR/ MTHFR QUAD. Our MTHFR Panel thrombophilia single test panel and four test panel is designed to identify select variants of the MTHFR gene associated with an increased risk of developing blood clots.
NAT-2. Our N-acetyltransferase 2, or NAT-2, test is designed to identify select variants of the NAT-2 gene which are associated with an increased risk of developing bladder and other types of cancer.
NTM. Our nontuberculous mycobacterium, or NTM, test is designed to detect the presence of 12 of the most common pathogenic NTMs from a single sample.
Resolve-QUAD. Our Resolve-QUAD uses the principle of the Nugent System in an advanced molecular-based platform to detect Gardnerella vaginalis, Mobiluncus spp., Candida albicans, Candida glabrata and Trichomonas vaginalis at the same time in four samples simultaneously on a single BioFilmChip. The Resolve-QUAD is also able to detect incidences of BV, candidiasis and trichomoniasis in a single test, and can differentiate episodes of uncomplicated Candidiasis caused by C.albicans from complicated candidiasis caused by C.glabrata and thus help physicians to render appropriate treatment.
Respiratory Virus 10 Panel. Our Respiratory Virus Panel test is designed to detect the presence of ten common respiratory viruses found in the human respiratory tract.
Respiratory Virus Panel. Our Respiratory Virus Panel test is designed to detect the presence of 24 common respiratory viruses found in the human respiratory tract.
STD-6 QUAD. Our STD-6 QUAD test is designed to detect the presence of six common microorganisms that cause sexually transmitted microorganisms (Chlamydia trachomatis, Neisseria gonorrhea, Trichomonas vaginalis, Ureaplasma urealyticum, Mycoplasma hominis and Mycoplasma genitalium) in four different samples simultaneously on a single BioFilmChip.
UGT1A1. Our UGT1A1 test is designed to identify a select variant of the UGT1A1 gene that affects the toxicity of Irinotecan, a leading cancer drug.
Urogen QUAD. Our Urogen QUAD test is designed to detect Ureaplasma urealyticum, Mycoplasma hominis and Mycoplasma genitalium in four samples simultaneously on a single BioFilmChip.
Urogen Plus QUAD. Our Urogen Plus QUAD test is designed to detect Ureaplasma urealyticum, Ureaplasma parvum, Mycoplasma hominis and Mycoplasma genitalium in four samples simultaneously on a single BioFilmChip.
Warfarin Assay. Our Warfarin test is designed to identify 14 gene variants of the CYP450 2C9 and Vitamin K epoxide reductase, or VKORC1, in individuals at risk for sensitivity to Warfarin, an oral anticoagulant drug, by identifying certain gene variants associated with responsiveness. Warfarin decreases the clotting ability of blood preventing thrombosis. It is used for indication of atrial fibrillation, venous thrombosis, heart valve replacement, and acute myocardiac infarction. This test received 510(k) clearance from the FDA in October 2010.
79
Test panels in development
We have a demonstrated track record of successfully developing genetic tests to address evolving customer demands. We currently have 16 additional test panels in development for use on our INFINITI analyzers. The proprietary design of our platform allows us to introduce new and enhanced tests to our menu of test panels without modifying the INFINITI analyzers. Based on our own research and customer demand, we intend to increase the number of tests offered in each of our target market segments, which we believe will further increase the utility of our platform to our customers. Our test panels in development include the following:
Product in Development | Application | Market Segments | ||||||||
Pain management | Mental health | Cardiovascular risk assessment | Other personalized medicine | Women's health | Oncology | Infectious disease | Genetic disorders | Newborn screening | ||
Alpha-1-antitrypsin | Alpha-1-antitrypsin deficiency | s | s | |||||||
AMD | Age-related macular degeneration | s | ||||||||
Blood Banking Panel | Transfusion medicins | |||||||||
C. diff | Clostridium difficile, which can cause intestinal disease | s | ||||||||
Carbamazepine | Management of anti-epilepsy drug | s | s | s | ||||||
Drug Addiction Panel | Addiction to alcohol, tobacco, and opioid drugs | s | ||||||||
HSV 1/2 | Detects Herpes Simplex Virus | s | s | |||||||
IL-28B | HCV anti-viral response | s | s | |||||||
MRSA-HAI | Hospital acquired infection | s | ||||||||
PIK3CA | PIK3CA gene, which is linked to ovarian cancer | s | ||||||||
RVP—10 | Respiratory virus infection | s | ||||||||
Statin Panel | Management of statin drugs | s | ||||||||
TB-MDR-8 | Drug resistance tuberculosis; 8 patient format | s | ||||||||
Thalassemia | Genetic Blood Disorder | s | ||||||||
TPMT | Thiopurine metabolism | s | s | |||||||
Warfarin Resistance Panel | Warfarin Resistance Panel | s | s | |||||||
Descriptions of our test panels in development
Alpha-1-antitrypsin. We are developing our alpha-1-antitrypsin, or AAT, test to identify two mutations on the SERPINA1 gene that confer AAT deficiency, causing COPD and liver disease.
AMD. We are developing our Age-Related Macular Degeneration, or AMD, panel which will test for 22 genetic mutations and help improve risk prediction and therapeutic intervention for AMD patients.
80
Blood Banking. We are developing our Blood Banking panel to genotype various blood groups to serve as an aide in transfusion medicine.
C. difficile. We are developing our C. difficile test to detect the presence of Clostridium difficile, or C. difficile, a bacterium that can cause symptoms ranging from diarrhea to life-threatening inflammation of the colon.
Carbamazepine. We are developing our carbamazepine test to identify a select variant of the human leukocyte antigen, or HLA, gene that is associated with an increased risk of serious dermal toxicities to carbamazepine in individuals with Asian ancestry. Carbamazepine is a drug used to treat epilepsy, bipolar disorder and neuropathic pain.
Drug Addiction Panel. We are developing our drug addiction panel to test for addiction to substances such as alcohol, tobacco and opioid drugs.
HSV1/2. We are developing our HSV1/2 test to detect and differentiate the herpes simplex virus 1 and 2, which cause cold sores and genital ulcers, respectively.
IL-28B. We are developing our IL-28B test to identify a mutation in the interleukin-28 gene, which can predict HCV anti-viral response.
MRSA-HAI. We are developing our MRSA-HAI test to detect the presence of certain microorganisms that may be associated with hospital acquired infections, or HAIs, including MRSA.
PIK3CA. We are developing our PIK3CA test to identify a select variant of the PIK3CA gene which is linked to gynecological malignancy, including ovarian cancer.
RVP-10. We are developing our RVP-10 test to detect the presence of ten common respiratory viruses found in the human respiratory tract.
Statin drug therapy. We are developing our statin (cholesterol lowering drugs) test that will identify individuals who are genetically susceptible to muscle pain (myalgia) and who may develop muscular injury (myopathy) when taking these drugs.
TB-MDR-8. We are developing our TB-MDR-8 test to detect the presence of tuberculosis and assess drug resistance to the tuberculosis treatments rifampin, isoniazid and pyrazinamide in eight patient samples simultaneously on a single BioFilmChip.
Thalassemia. We are developing our Thalassemia test to identify select variants of the beta globin genes associated with beta thalassemia. Beta thalassemia is a group of blood disorders that affect the production of normal hemoglobin and causes different forms of anemia.
TPMT. We are developing our TPMT test to identify mutations in the TPMT gene that affect the metabolism of thiopurine drugs.
Warfarin Resistance Panel. We are developing our Warfarin Resistance Panel to identify with a single test select variants of the CYP450 2C9, VKORC1, Factor II and Factor V genes that play a role in resistance to warfarin.
Our decision to seek FDA approvals or clearances domestically, and CE marking internationally, for our genetic tests is made on a test-by-test basis, and is based on a variety of factors, including the regulatory environment for the use of genetic tests, the demand by our existing and target customers for particular genetic tests that have received regulatory approvals or clearances of particular genetic tests, the competitive environment for the use of genetic tests that have received regulatory approvals or clearances versus similar tests that have not, and the expense and time required to obtain regulatory approvals or clearances for particular genetic tests.
81
Sales and Marketing
Our current sales and marketing strategy is focused on the broad introduction of our INFINITI HTS to customers in high-volume testing market segments, particularly personalized medicine and women's health. Our target customers are clinical reference laboratories, specialty clinics and hospital laboratories who are seeking a more flexible and efficient MDx platform or who are seeking to enter the MDx market. Our marketing efforts emphasize the high throughput testing capacity, ease of use, enhanced workflow, increased efficiency, low cost, high quality and consistent results of our system as well as the potential versatility afforded by our broad menu of test panels. Our goal is to achieve broad adoption of our INFINITI analyzers in the market, and drive utilization to generate sales of our high-margin testing consumables.
In the United States, we offer our family of INFINITI analyzers either through direct sales, monthly rental plans or our Reagent Access Plan, or RAP. Under the RAP, an INFINITI analyzer is placed at the customer's location at no initial cost to the customer, and is subject to contractual minimum consumables purchasing thresholds. If these purchasing thresholds are not met, we have the right to reclaim our INFINITI analyzer from the customer.
To execute our domestic marketing strategy, we have established a nine-person direct sales force that is regionally deployed and performs market development as well as account management. We plan to significantly expand our sales force within the next 12 months. To supplement our sales and marketing effort, we maintain a customer support team comprised of training specialists, hotline staff, field service engineers and product managers. Our technical services team are experienced molecular biologists and engineers with expertise on the operation of our platform who provide prompt assistance to our customers when necessary. Our product managers focus on the major MDx market segments of personalized medicine, women's health, oncology, infectious diseases and genetic disorders to develop product-specific strategies and to identify market opportunities to maximize market penetration.
We market and sell our platform internationally through our network of distributors. We have established 12 distributor relationships for the marketing of our platform in 23 countries outside the United States. We plan to expand our existing distributor network outside the United States to enter new markets and capitalize on growing international demand. Our distributor agreements allow the purchase of our analyzers and consumables at a discounted price. The distributors then resell our products to customers in the territories covered by their respective agreements. Under these agreements, the distributors generally are responsible for all customer sales, support and service activities relating to our products in the applicable countries. These agreements generally also provide the distributors with the exclusive right to sell products in the applicable country or countries, so long as they meet minimum purchase requirements. For those agreements that do not include minimum purchase requirements, the distributor is required to use best efforts to meet specified sales objectives.
In addition to our selling activities, we align with key opinion leaders at leading institutions and clinical research laboratories to help increase scientific and commercial awareness of our system, demonstrate its benefits relative to existing technologies and accelerate its adoption in the MDx market. We also seek to increase awareness of our products through participation at trade shows, academic conferences, online webinars and hospital-based grand rounds.
In late 2013, as we prepared to launch our INFINITI HTS, we began operating our CLIA-certified laboratory to complement and support our efforts to attract customers entering the molecular diagnostics market, and potential INFINITI HTS customers in particular. This laboratory is located at our headquarters in Vista, California and is licensed in the State of California as well as a number of other applicable states. Our laboratory provides molecular testing services to other reference laboratories in the areas of personalized medicine, women's health, oncology, infectious diseases and genetic disorders. Our laboratory utilizes our INFINITI PLUS and INFINITI HTS systems and proprietary consumables, and is committed to providing reliable and accurate results by means of proper sample identification and timely reporting of results. Policies and procedures utilized by our laboratory are designed to meet applicable federal and state license and accreditation requirements for clinical diagnostic laboratories, including in respect of applicable CLIA, OSHA, EPA and FDA rules and regulations. We believe that our laboratory supports and enhances our strategy to become a leading provider of genetic tests to a broad array of customers within our targeted market segments. In addition to capitalizing on the capabilities of the INFINITI HTS and INFINITI PLUS analyzers, our laboratory provides our customers with an alternative source for genetic testing services when we have placed INFINITI analyzers at their location.
82
When a laboratory acquires a new analyzer and other critical laboratory equipment, there is a period of time after delivery and installation of the equipment for the laboratory to train their personnel and complete validation of the equipment and the selected tests. Traditionally, a laboratory would not market its services or obtain patient samples during the training and validation period. Our laboratory provides testing services for patient samples originated by our customers during this period and offers training on our platform for their laboratory personnel. This allows our laboratory customers to begin marketing and selling their services and generate revenue during the validation and training process.
Manufacturing
We operate within an approximately 125,000 square foot building in Vista, California, that houses our research and development, manufacturing and warehousing operations, our in-house laboratory and our administrative offices, under a lease that expires in December 2029.
We manufacture our family of INFINITI analyzers, BioFilmChips and Intellipac Reagent Management Modules at our facility. We utilize a fully integrated enterprise resource planning software system developed specifically for medical manufacturing companies. We have relationships with vendors who are sources of raw materials and components for our product offerings, including reagents, oligonucleotides used for microarray spotting and film coating for our microarrays.
Our facility is registered with the FDA as a Medical Device Manufacturing Establishment. We have established a quality system that is in compliance with the FDA's Quality Systems Regulations and we have obtained ISO 13485:2003 certification for our facility. Our quality system covers all phases of product design and development, the manufacturing operations from purchasing to assembly to shipping, and product distribution, installation and servicing.
Research and Development
Our research and development efforts focus on developing additional genetic tests and INFINITI analyzers, improving or enhancing current tests and INFINITI analyzers, and supporting clinical trials for certain test panels. We spent $1.5 million in the nine months ended September 30, 2014, and $2.3 million and $2.4 million on research and development in the years ended December 31, 2013 and 2012, respectively. We believe that continuing to develop our broad menu of test panels will increase the value of our system, drive additional placements and increase consumables sales.
We continue to focus our research and development efforts on enhancing existing tests as new genetic research becomes available. We also have conducted, and intend to conduct, clinical trials to support our FDA and other regulatory clearances or approvals, as necessary, and to explore opportunities to enhance our platform. We have entered into, and expect to continue to enter into, collaborative relationships with leading research and academic institutions for the development of additional or enhanced test panels to further increase the depth and breadth of our menu of test panels. Where appropriate, we enter into royalty agreements with our collaborative partners to license intellectual property for use in our test panels.
Competition
We face competition in the molecular testing markets primarily from laboratory-developed tests, or LDTs, and products developed by companies such as Abbott Laboratories Inc., Becton, Dickinson and Company, Cepheid, GenMark Diagnostics, Inc., Hologic, Inc., Luminex Corporation, Qiagen N.V., Roche Holding Ltd. and Thermo Fisher Scientific Inc. Due to the breadth of our testing menu and the throughput capabilities of our INFINITI analyzers, we believe we do not compete with any of these companies directly across all of our market segments. We believe that we compete with products offered by these companies on the basis of test menu, cost-effectiveness, ease of use, accuracy of results, throughput and multiplexing capability, laboratory services and support and, with respect to some of our tests, FDA clearance.
Many of our competitors have significantly greater financial, technical, research and other resources and larger, more established marketing, sales and distribution services organizations than we do. Many of our competitors also offer broader product lines outside of the molecular research use or diagnostic testing market, and many have greater brand recognition than we do. Moreover, our competitors may make rapid technological developments that may result in our technologies and products becoming obsolete before we recover the expenses incurred to develop them
83
or before they generate significant revenue. Our success will depend, in part, on our ability to establish successful marketing, sales and distribution efforts.
Intellectual Property, Licenses and Collaborations
As of October 24, 2014, we had eight issued patents and one pending patent application in the United States. Our issued patents expire between 2018 and 2026, and the pending patent application, if issued, is expected to expire in 2023, absent any adjustments or extensions. Our patents are directed to certain of the technologies relating to the method and design of the BioFilmChip and its film technology, the design of our INFINITI analyzers, the Intellipac Reagent Management Module and the system's chemistry methods, system robotics and signal enhancements.
In addition to our own genetic test development efforts, we are currently using, and intend to use in the future, certain tests and biomarkers in our platform that have been developed by third parties or by us in collaboration with third parties. While a significant amount of intellectual property in the field of genetics is already in the public domain, one of our tests requires, and some of the future tests developed by us, or by third parties on our behalf for use in our system, may require, that we license the right to use certain intellectual property from third parties and pay customary royalties or make one time payments.
We currently license certain patents used in our UGT1A1 test from the Mayo Foundation for Medical Education and Research, or Mayo Laboratories. This license requires that we pay a royalty of six percent of net sales from our genetic test panels that incorporate the patented technology. The patents covered by this license expire between July 5, 2015 and May 28, 2024. The license expires when the last patent covered by the license expires. This license is terminable by us upon 90 days notice or by the licensor upon certain bankruptcy or liquidation events involving us, or 30 days after licensor's written notice of our failure to observe a material obligation of the license. We have granted a license to one of our patents relating to oligonucleotides of CYP450 1A1, CYP450 3A4, CYP450 2D6 and NAT-2 to Mayo Laboratories. The license requires the licensee to pay, among other amounts, a royalty equal to six percent of net sales of any products that incorporate the patented technology. The license is terminable by the licensee upon 30 days notice or by us upon the licensee's failure to cure a payment default within 45 days or its failure to cure a material breach of the license other than a payment default within 90 days.
In June 2014, we entered into an agreement with Genomas, Inc., a biomedical company and genetic reference laboratory affiliated with Hartford Hospital, which is the teaching hospital for the University of Connecticut Medical School, focused in personalized medicine to jointly develop genetic tests and DNA-guided diagnostic systems for optimal selection of statin (cholesterol-lowering) drugs and for improved delivery of statin therapy for the treatment of cardiovascular disease, obesity and diabetes. The objective of the collaboration is to provide clinicians and physicians with newly developed genetic tests and a decision support system and software package that assigns risk based on genetic results, which will allow healthcare providers to select statins, prescribe and dose these drugs on a DNA-guided, personalized basis to more effectively guide the therapy for each patient.
Government Regulation
The healthcare industry, and thus our business, is subject to extensive federal, state, local and foreign regulation. Both federal and state governmental agencies continue to subject the healthcare industry to intense regulatory scrutiny, including heightened civil and criminal enforcement efforts. We believe that we have structured our business operations and relationships with our customers to comply with applicable legal requirements. However, it is possible that governmental entities or other third parties could interpret these laws differently and assert otherwise. We discuss below the statutes and regulations that are most relevant to our business and most frequently cited in enforcement actions.
U.S. Food and Drug Administration regulation
The Federal Food, Drug and Cosmetic Act, or FFDCA, includes within the definition of a medical device any instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory, intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment or prevention of disease, in man or other animals. Our in vitro diagnostic products are considered by the FDA to be medical devices. Among other things, pursuant to the FFDCA and its implementing regulations, the FDA regulates the research, testing, manufacturing, safety, labeling, storage, recordkeeping, pre-market activities, marketing and promotion, and sales and distribution of medical devices in the United States to
84
ensure that medical products distributed domestically are safe and effective for their intended uses. In addition, the FDA regulates the export of medical devices manufactured in the United States to international markets.
FDA's pre-market clearance and approval requirements
Unless an exemption applies, each medical device that we wish to market in the United States must first receive either clearance of a 510(k) pre-market notification or approval of a pre-market application, or PMA, from the FDA. The FDA's 510(k) clearance process usually takes from three to 12 months, but it can take significantly longer and clearance is never guaranteed. The process of obtaining PMA approval is much more costly, lengthy and uncertain. It generally takes from one to three years or even longer and approval is not guaranteed. Our Factor II, Factor V, and Factor II-V Panel tests received 510(k) clearances in February 2007. Our Warfarin test received 510(k) clearance in January 2008 and our CYP2C19 test received 510(k) clearance in October 2010. All of these tests are currently cleared for use with the INFINITI analyzers.
Whether a device must undergo either the 510(k) clearance or PMA approval process is based upon statutory criteria. These criteria include the level of risk that the agency determines is associated with the device and a determination whether the product is a type of device that is similar to devices that are already legally marketed. Devices are classified into Class I, Class II or Class III.
Class I devices are those for which safety and effectiveness can be assured by adherence to the FDA's general regulatory controls for medical devices, which include compliance with the applicable portions of the FDA's Quality System Regulations, facility registration and product listing, reporting of adverse medical events, and appropriate, truthful and non-misleading labeling, advertising and promotional materials, or General Controls. Most Class I devices are exempt from pre-market regulation; some may require FDA clearance through the 510(k) notification process.
Class II devices are subject to the FDA's General Controls, and any other special controls as deemed necessary by the FDA to establish the safety and effectiveness of the device. Pre-market review and clearance by the FDA for Class II devices is generally accomplished through the 510(k) pre-market notification process which most often requires analytical and clinical data to demonstrate that the proposed device is "substantially equivalent" in intended use and in safety and effectiveness to a legally marketed device (known as "predicate device"). 510(k) pre-market notification submissions are subject to user fees, unless a specific exemption applies. We believe that most of our tests are Class II devices. Devices deemed to pose relatively less risk are placed in either Class I or II, which requires the manufacturer to submit a pre-market notification requesting 510(k) clearance, unless an exemption applies. The pre-market notification must demonstrate that the proposed device is "substantially equivalent" in intended use and in safety and effectiveness to a legally marketed "predicate device" that is either in Class I or Class II or is a Class III preamendment device which is a device that was in commercial distribution before May 28, 1976, for which the FDA has not yet called for submission of a PMA application. We believe that most of our tests are Class II devices for which the 510(k) clearance process is available, and we believe our HPV-HR Quad test is a Class III device for which the more stringent PMA requirements are applicable.
Class III devices are those devices which are deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, have a new intended use, or use advanced technology that is not substantially equivalent to that of a "predicate device." These devices are required to undergo the PMA process of scientific and regulatory review to evaluate the safety and effectiveness of a Class III device. These devices almost always require formal clinical trials to demonstrate safety and effectiveness. A PMA application must provide extensive preclinical and clinical trial data and also information about the device and its components regarding, among other things, device design, manufacturing and labeling. Pre-market approval applications (and PMA supplements) are subject to significantly higher user fees than are 510(k) pre-market notifications. We believe our HPV-HR Quad test is a Class III device for which the more stringent PMA requirements are applicable.
Medical devices can be marketed only for the indications for which they are cleared or approved. Any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require a PMA approval. We have made modifications to our 510(k) cleared system and tests, but have determined that, in our view, based on FDA guidance as to when to submit a 510(k) notification for changes to a cleared device, new 510(k) clearances or PMA approvals are not required. Should the FDA disagree with our determination, we may need to submit a 50(k) notification or a PMA application,
85
we also may be required to cease marketing and/or recall the modified device until we obtain a new 510(k) clearance or PMA approval.
A clinical trial may be required in support of a 510(k) submission and generally is required for a PMA application. Clinical trials generally require an Investigational Device Exemption, or IDE, from the FDA for a specified number of patients, unless the product is exempt from IDE requirements or deemed a non significant risk device eligible for more abbreviated IDE requirements. The IDE application must be supported by appropriate data, such as animal and laboratory testing results. Clinical trials may begin 30 days after the submission of the IDE application unless the FDA or the appropriate institutional review boards at the clinical trial sites place the trial on clinical hold. In the future, we expect to submit additional 510(k) notifications or PMA applications in order to market new claims, uses or products. Clinical investigations of in vitro diagnostic tests, including our products and product candidates, typically are exempt from the IDE requirements. Thus, we do not need FDA's prior approval to conduct clinical trials on our products, provided the testing is non-invasive, does not require an invasive sampling procedure that presents a significant risk, does not intentionally introduce energy into the subject and is not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established diagnostic device or procedure.
A significant portion of our revenue is derived from genetic test panels sold as research use only, or RUO, and are not intended for diagnostic purposes. Laboratories regulated under the Clinical Laboratory Improvement Amendments of 1988, or CLIA, may currently validate the use of LDTs specifically for use in their laboratory using any labeled products, which may include our RUO-labeled products. On July 31, 2014, the FDA notified Congress of its intent to publish, on or after September 30, 2014, draft guidance for a new framework for regulation of LDTs. The FDA stated its intention to modify its approach to enforcement discretion of LDTs in a risk-based manner, which will require certain LDTs to obtain 510(k) clearance or PMA approval, as appropriate, in order to continue to be marketed. The FDA has also has issued guidance on acceptable distribution practices for in vitro diagnostic products labeled for Research Use Only and on responding to unsolicited requests for off-label information. We have procedures designed to comply with these requirements and we believe that the sale of our products complies with FDA's laws and regulations. If the FDA disagrees with the marketing of RUO products, or imposes substantial changes to the regulation or enforcement of LDTs, the broad use of RUO tests may be limited and a significant reduction in the sale of these products could occur. A decline in the sales of our RUO products could reduce our revenue, increase our operating expenses and adversely affect our business and results of operations.
Fraud and abuse laws
As a clinical laboratory and diagnostic test manufacturer, we are subject to a number of laws in the United States whose purpose is to eliminate fraud and abuse in federal healthcare programs, as well as within the states which include without limitation the following:
Anti-Kickback statutes, Federal False Claims Act and similar laws
The federal healthcare program's Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, overtly or covertly, in cash or in kind, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. The definition of "remuneration" has been broadly interpreted to include anything of value, including, for example, gifts, discounts, the furnishing of free supplies, equipment or services, credit arrangements, payments of cash and waivers of payments. Several courts have interpreted the statutes intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. Penalties for violations include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal healthcare programs. In addition, some kickback allegations have been claimed to violate the Federal False Claims Act, discussed in more detail below. Additionally, the intent standard under the Anti-Kickback Statute was amended by the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, collectively the Affordable Care Act, to a stricter standard such that a person or entity need not have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the Affordable Care Act codified case law that a
86
claim including items or services resulting from a violation of the Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the Federal False Claims Act.
The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements, Congress authorized the U.S. Office of Inspector General, or OIG, to issue a series of regulations, known as "safe harbors." These safe harbors set forth requirements that, if met in their entirety, will assure healthcare providers and other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal, or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny, and potential enforcement, by government enforcement authorities, such as the OIG.
Many states have adopted laws similar to the Anti-Kickback Statute. Some of these state prohibitions apply to referral of patients for healthcare items or services reimbursed by any payor, not only the Medicare and Medicaid programs, and do not contain identical safe harbors.
Government officials have focused their enforcement efforts on marketing of healthcare services and products, among other activities, and have brought cases against numerous pharmaceutical and medical device companies, and certain sales and marketing personnel for allegedly offering unlawful inducements to potential or existing customers in an attempt to procure their business. Another development affecting the healthcare industry is the increased use of the federal civil False Claims Act and, in particular, actions brought pursuant to the False Claims Act's "whistleblower" or "qui tam" provisions. The False Claims Act imposes liability on any person or entity who, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in any monetary recovery. In recent years, the number of suits brought by private individuals has increased dramatically. Similarly, the federal health care program civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. In addition, various states have enacted false claim laws analogous to the False Claims Act. Many of these state laws apply where a claim is submitted to any third-party payor and not merely a federal healthcare program.
When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties of $5,500 to $11,000 for each separate false claim. There are many potential bases for liability under the False Claims Act. Liability arises, primarily, when an entity knowingly submits, or causes another to submit, a false claim for reimbursement to the federal government. The False Claims Act has been used to assert liability on the basis of inadequate care, kickbacks and other improper referrals, improper use of Medicare numbers when detailing the provider of services, and allegations as to misrepresentations with respect to the services rendered. Our future activities relating to the reporting of discount and rebate information and other information affecting federal, state and third party reimbursement of our products, and the sale and marketing of our products, may be subject to scrutiny under these laws. We are unable to predict whether we would be subject to actions under the False Claims Act or a similar state law, or the impact of such actions. However, the costs of defending such claims, as well as any sanctions imposed, could significantly adversely affect our financial performance.
Federal and state physician self-referral prohibitions
We are subject to the federal physician self-referral prohibitions, commonly known as the Stark Law, and to similar state law restrictions, such as California's Physician Ownership and Referral Act, or PORA. Together these restrictions generally prohibit us from billing a patient or any governmental or private payor for certain designated health services, including diagnostic services, when the physician ordering the service, or any member of such physician's immediate family, has a financial interest, such as an ownership or investment interest in or compensation arrangement with us, unless the arrangement meets an exception to the prohibition. Both the Stark Law and PORA, as well as many other state law equivalents, contain an exception for compensation paid to a physician for personal services rendered by the physician. If we enter into these types of arrangements in a manner
87
that does not comply with Stark, PORA or similar state laws, we would be required to refund any payments we receive pursuant to a referral prohibited by these laws to the patient, the payor or the Medicare program, as applicable. In addition, sanctions for a violation of the Stark Law include the following:
• | denial of payment for the services provided in violation of the prohibition; |
• | refunds of amounts collected by an entity in violation of the Stark Law; |
• | a civil penalty of up to $15,000 for each bill or claim for service arising out of the prohibited referral; |
• | the imposition of up to three times the amounts for each item or service wrongfully claimed; |
• | possible exclusion from federal healthcare programs, including Medicare and Medicaid; and |
• | a civil penalty of up to $100,000 for each arrangement or scheme that the parties know (or should know) has a principal purpose of circumventing the Stark Law's prohibition. |
These prohibitions apply regardless of the reasons for the financial relationship and the referral. No finding of intent to violate the Stark Law is required for a violation. In addition, knowing violations of the Stark Law may also serve as the basis for liability under the Federal False Claims Act.
Further, a violation of PORA is a misdemeanor and could result in civil penalties and criminal fines. Finally, other states have self-referral restrictions with which we have to comply, some of which differ from those imposed by federal and California law. While we have attempted to comply with the Stark Law, PORA and similar laws of other states, it is possible that some of our financial arrangements with physicians could be subject to regulatory scrutiny at some point in the future, and we cannot provide assurance that we will be found to be in compliance with these laws following any such regulatory review.
HIPAA and other fraud and privacy regulations
Among other things, the Health Insurance Portability and Accountability Act of 1996, as amended, or HIPAA, created two federal crimes: healthcare fraud and false statements relating to healthcare matters. HIPAA prohibits knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private payors, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines, imprisonment and/or exclusion from government-sponsored programs.
In addition to creating two federal healthcare crimes, regulations implementing HIPAA also establish uniform standards governing the conduct of certain electronic healthcare transactions and protecting the security and privacy of individually identifiable health information maintained or transmitted by healthcare providers, health plans and healthcare clearinghouses, which are referred to as "covered entities." Three standards have been promulgated under HIPAA's regulations: the Standards for Privacy of Individually Identifiable Health Information, which restrict the use and disclosure of certain individually identifiable health information; the Standards for Electronic Transactions, which establish standards for common healthcare transactions, such as claims information, plan eligibility, payment information and the use of electronic signatures; and the Security Standards, which require covered entities to implement and maintain certain security measures to safeguard certain electronic health information. As a covered entity, and also in our capacity as a business associate to certain of our customers, we are subject to these standards. While the government intended this legislation to reduce administrative expenses and burdens for the healthcare industry, our compliance with certain provisions of these standards entails significant costs for us, and our failure to comply could lead to enforcement action that could have an adverse effect on our business. If we or our operations are found to be in violation of HIPAA or its implementing regulations, we may be subject to potentially significant penalties, including civil and criminal penalties, damages and fines.
In addition to federal regulations issued under HIPAA, many states and foreign jurisdictions have enacted privacy and security statutes or regulations that, in some cases, are more stringent than those issued under HIPAA. In those cases, it may be necessary to modify our planned operations and procedures to comply with the more stringent laws. If we fail to comply with applicable state laws and regulations, we could be subject to additional sanctions.
88
Physician payment "sunshine" laws
There has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Affordable Care Act imposed, among other things, new annual reporting requirements for covered manufacturers (including manufacturers of certain laboratory tests, including some of our products) for certain payments and "transfers of value" provided to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit timely, accurately and completely the required information for all payments, transfers of value and ownership or investment interests may result in civil monetary penalties of up to an aggregate of $150,000 per year and up to an aggregate of $1 million per year for "knowing failures." Covered manufacturers were required to report detailed payment data for the first reporting period (August 1, 2013 to December 31, 2013) under this law and submit legal attestation to the completeness and accuracy of such data by June 30, 2014. Thereafter, covered manufacturers must submit reports by the 90th day of each subsequent calendar year. In addition, certain states require implementation of commercial compliance programs and compliance with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, impose restrictions on marketing practices, and/or tracking and reporting of gifts, compensation and other remuneration or items of value provided to physicians and other healthcare professionals and entities
If our operations are found to be in violation of any of the health regulatory laws described above or any other laws that apply to us, we may be subject to penalties, including potentially significant criminal, civil and/or administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings and the curtailment or restructuring of our operations.
Healthcare reform
A primary trend in the U.S. healthcare industry is cost containment. The federal government and state legislatures have attempted to control healthcare costs in part by limiting coverage and the amount of reimbursement for particular drug products, including implementing price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. By way of example, the Affordable Care Act contains provisions that may reduce the profitability of drug products. The Affordable Care Act, among other things, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care plans, imposed mandatory discounts for certain Medicare Part D beneficiaries and subjected manufacturers to new annual fees based on pharmaceutical companies' share of sales to federal healthcare programs.
Other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. On August 2, 2011, the President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not recommend and Congress did not enact legislation to reduce the deficit by at least $1.2 trillion for the years 2013 through 2021, triggering the legislation's automatic reduction to several government programs. This includes reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and will remain in effect through 2024 unless additional Congressional action is taken. On January 2, 2013, the President signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products or add additional pricing pressures.
CLIA laboratory certification
The conduct and provision of our tests are regulated under the Clinical Laboratory Improvement Amendments of 1988, or CLIA. CLIA requires us to maintain federal certification. CLIA imposes requirements relating to test processes, personnel qualifications, facilities and equipment, recordkeeping, quality assurance and participation in proficiency testing. CLIA compliance and certification are also a condition for participation by clinical laboratories in the Medicare Program and for eligibility to bill for services provided to governmental healthcare program beneficiaries. As a condition of CLIA certification, our laboratory is subject to survey and inspection every other
89
year, in addition to being subject to additional random inspections. The biennial survey is conducted by CMS, a CMS agent (typically a state agency), or, if the laboratory is accredited, a CMS-approved accreditation organization. Sanctions for failure to meet these certification, accreditation and licensure requirements include suspension, revocation or limitation of a laboratory's CLIA certification, accreditation or license, which is necessary to conduct business, cancellation or suspension of the laboratory's ability to receive Medicare or Medicaid reimbursement, as well as imposition of plans to correct deficiencies, injunctive actions and civil monetary and criminal penalties. The loss or suspension of a CLIA certification, imposition of a fine or other penalties, or future changes in the CLIA law or regulations (or interpretation of the law or regulations) could harm our business.
California laboratory licensing
In addition to federal certification requirements of laboratories under CLIA, licensure is required and maintained for our clinical laboratory under California law. Such laws establish standards for the day-to-day operation of a clinical laboratory, including the training and skills required of personnel and quality control. In addition, California laws mandate proficiency testing, which requires the laboratory to verify at least twice annually the accuracy of any test or procedure performed by that laboratory. We maintain a current license in good standing with the California Department of Public Health. If our clinical laboratory is out of compliance with California standards, the California Department of Public Health may suspend, restrict or revoke our license to operate our clinical laboratory, assess substantial civil money penalties or impose specific corrective action plans. Any such actions could harm our business.
Other states' laboratory testing license requirements
In addition to California, certain states such as New York, Florida, Pennsylvania, Maryland and Rhode Island require out-of-state laboratories to be licensed if the laboratories accept specimens from those states. Potential sanctions for violation of state statutes and regulations include significant fines, the disapproval of licensure applications and the suspension or loss of various licenses, certificates and authorizations, which could harm our business. CLIA does not preempt state laws that have established laboratory quality standards that are at least as stringent as federal law.
Third party coverage and reimbursement
Our primary customers are clinical laboratories that bill many different payor groups. The majority of reimbursement dollars for traditional laboratory services are provided by health maintenance organizations, or HMOs, and other managed care plans, as well as government healthcare programs, such as Medicare and Medicaid. HMOs and other managed care plans typically contract with a limited number of clinical laboratories and then designate the laboratory or laboratories to be used for tests ordered by participating physicians. We are currently an out-of-network provider with payors which means we do not have a contract with payors to pay a specific rate for our tests. We are subject to applicable state laws regarding who should be billed, how they should be billed, how business should be conducted, and how patient obligations regarding cost sharing should be handled. In addition, if we become an "in-network" provider for certain payors in the future, we will also be subject to the terms of contracts (which could include reduced reimbursement rates) and may be subject to discipline, breach of contract actions, non-renewal or other contractually provided remedies for non-compliance with the contract's requirements and/or applicable laws.
We generally bill third-party payors and individual patients for testing services on a test-by-test basis. Third-party payors include Medicare, Medicaid and private insurance companies, each of which has different billing requirements. Medicare and Medicaid reimbursement programs are complex and ambiguous, and are continuously being evaluated and modified by the CMS. Our ability to receive timely reimbursements from third-party payors is dependent on our ability to correct and complete missing and incorrect billing information. Missing and incorrect information on reimbursement submissions slows down the billing process and increases the aging of accounts receivable. We must bill Medicare directly for tests performed for Medicare patients and must accept Medicare's fee schedule for the covered tests as payment in full. State Medicaid programs are generally prohibited from paying more than the Medicare fee schedule. Our lab has contracted with a healthcare billing services management company to manage its third-party billing.
There are a number of factors that influence coverage and reimbursement for diagnostic tests. In the United States, the American Medical Association assigns specific CPT codes, which are necessary for reimbursement of
90
diagnostic tests. Once the CPT code is established, the Centers for Medicare and Medicaid Services establish reimbursement payment levels and coverage rules under Medicaid and Medicare, and private payors establish rates and coverage rules independently. However, the availability of a CPT Code is not a guarantee of coverage, nor of adequate reimbursement levels, and the revenues generated from our tests will depend, in part, on the extent to which third-party payors provide coverage and establish adequate reimbursement levels.
United States and other government regulations governing coverage and reimbursement for diagnostic testing may affect directly or indirectly the design of our products and the potential market for their use. The availability of third-party reimbursement for our products and services may be limited or uncertain. Third-party payors may deny coverage if they determine that the prescribed product or service has not received appropriate FDA or other government regulatory clearances, is not used in accordance with cost-effective treatment methods as determined by the payor, or is deemed by the third-party payor to be experimental, unnecessary or inappropriate. Furthermore, third-party payors, including federal and state healthcare programs, government authorities, private managed care providers, private health insurers and other organizations, are increasingly challenging the prices, examining the medical necessity and reviewing the cost-effectiveness of healthcare products and services, including laboratory tests. Such payors may limit coverage of our tests to specific, limited circumstances, may not provide coverage at all, or may not provide adequate reimbursement rates, if covered. Further, one payor's determination to provide coverage does not assure that other payors will also provide coverage for the test. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to maintain our revenue and growth. Coverage policies and third-party reimbursement rates may change at any time.
Foreign regulation
In order for us to market our products in other countries, we must comply with extensive safety and quality regulations and obtain regulatory approvals or registrations for these countries. ISO 13485 Medical devices – Quality management system is an International Organization for Standardization, or ISO, standard, that represents the requirements for a comprehensive quality management system for the design and manufacture of medical devices. Compliance to this ISO standard is required by most, if not all, foreign countries. We are certified to ISO 13485:2003, the current version of the standard. Our current certification is valid until February 21, 2016. Continued certification to the ISO standard requires periodic audits by a recognized certification body such a European Notified Body. The requirements for approvals or registrations and the time required for regulatory review, if required, vary from country to country.
Many foreign countries in which we market or may market our products have regulatory bodies and restrictions similar to those of the FDA. International sales are subject to foreign government regulation, the requirements of which vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA approval and the requirements may differ.
European countries. In the European Economic Area, or EEA, a medical device can only be placed on the market if it is in conformity with the essential requirements set out in the European Medical Devices Directives (Directive 93/42/EEC on Medical Devices, Directive 90/385/EEC on Active Implantable Medical Devices and Directive 98/79/EC on in vitro diagnostic medical devices). In vitro diagnostic medical devices that comply with the requirements of the IVDD Directive are entitled to bear the CE conformity mark, indicating that the device meets minimum standards of performance, safety and quality (i.e., the essential requirements), and can be commercially distributed throughout the EEA and other countries outside the EEA that have accepted the CE marking as an acceptable certification of efficacy and safety of medical devices. For certain classes of devices, certification by a recognized European Notified Body may be required to permit the manufacturer to affix the CE marking on its products and commercially distribute those products throughout the EEA.
The CE marking of conformity was affixed to our INFINITI analyzer in accordance with the IVDD Directive in June 2008 and to our INFINITI PLUS analyzer in September 2012. We also have CE marked 24 of our tests. The CE marking allows us to market these analyzers and tests in the EEA. Our tests that are not CE marked are offered to European laboratories on an RUO basis to conduct research projects and other pre-market activities. We are working with our international distributors to prioritize and fulfill requirements for registration, licensing and approvals required by foreign regulatory agencies.
91
Facilities
Our corporate offices are located at 2980 Scott Street, Vista, California, in approximately 125,000 square feet occupied under a lease that commenced on February 1, 2009 and will expire on December 31, 2029. This facility houses our research and development, manufacturing and warehousing operations and our administrative offices.
Employees
As of October 24, 2014, we had 114 full-time employees. None of our employees are represented by a labor union and we consider our employee relations to be good.
Legal Proceedings
We are from time to time subject to various claims and legal actions in the ordinary course of business. We believe that there are currently no claims or legal actions against us that, in management's judgment based on information currently available, are likely to have an adverse effect on our results of operations or financial condition. However, regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
92
MANAGEMENT
The following table sets forth information about our directors and executive officers. The biographies for Mr. Kureshy and our other directors, below, include information on the directors' experience, qualifications, attributes and skills that have led our board to conclude that they should serve on our board.
Name | Age | Position | ||
Fareed Kureshy | 71 | Founder, Chairman, President and Chief Executive Officer | ||
John R. Zacamy | 65 | Chief Financial Officer and Secretary | ||
Ramanath Vairavan | 66 | Co-Founder, Senior Vice President, Lab Services | ||
Shailendra Singh, Ph.D. | 75 | Co-Founder, Vice President, System Development | ||
Jim Canfield | 55 | Vice President, Sales and Marketing | ||
Stephen Allison | 69 | Director | ||
Charles Birmingham | 63 | Director | ||
Joseph S. Coyne, Dr.P.H., MPH. | 66 | Director | ||
Laurence M. Demers, Ph.D. | 76 | Director | ||
Thomas V. Hennessey, Jr. | 66 | Director | ||
Adam Levinson | 44 | Director | ||
Peter Wilding, Ph.D. | 79 | Director | ||
Executive Officers
Fareed Kureshy. Mr. Kureshy has been our Chairman, President and Chief Executive Officer since our founding in April 1999. Mr. Kureshy has over 30 years of entrepreneurial leadership experience in the healthcare industry. Mr. Kureshy served as President and Chief Executive Officer of Sequenom, Inc. from 1996 to 1997, and as President of Behring Diagnostics, Inc. and PB Diagnostics Systems, Inc. from 1992 to 1996, where he set up the infrastructure to design, develop, manufacture and market platform technologies in the areas of chemistry, immunoassays and DNA analysis. Mr. Kureshy also spent 12 years at Abbott Laboratories from 1976 to 1987 where he held various management positions. Mr. Kureshy holds undergraduate degrees in physics, chemistry and mathematics from Karachi University, an undergraduate degree in engineering from Northrop University and has completed graduate engineering studies. He was awarded an M.B.A. from Southern Methodist University. Our board of directors has determined that Mr. Kureshy brings to the board knowledge of our business and his historical understanding of our operations combined with his extensive entrepreneurial leadership, operations, strategic planning and marketing experience in the healthcare industry in which we operate.
John R. Zacamy. Mr. Zacamy joined us as our Chief Financial Officer in September 2014, and prior to that served as a strategic advisor to the Company beginning in December 2009. Mr. Zacamy founded Zacamy & Co. LLC, a strategic advisory firm with a focus on healthcare and technology, in 2009, and served as its Managing Director and President until September 2014. From 2005 to 2009, Mr. Zacamy was the Managing Director and President of Cross Lake Capital LLC, a private equity and investment firm. Mr. Zacamy held senior positions at Morgan Stanley & Co. Incorporated for 15 years, and at Deutsche Bank Securities Inc. (and its predecessor companies) for 15 years, specializing in corporate finance, mergers and acquisitions and capital markets transactions. Mr. Zacamy holds an A.B. from Colby College and an M.B.A. from Tuck School of Business at Dartmouth College.
Ramanath Vairavan. Mr. Vairavan has been Senior Vice President, Lab Services since January 2014. From January 2007 to January 2014 he served as Senior Vice President, Sales and Marketing and he has been an executive officer of AutoGenomics since its founding in April 1999. Mr. Vairavan has over 30 years of experience in the healthcare industry in the areas of research, product development, manufacturing, sales, marketing and business development. He began his career with Hoechst AG / Behring Diagnostics, Inc. where he held various management positions in research, development, operations, international sales and marketing in the United States, Singapore and Germany. Mr. Vairavan received a B. Tech. in chemical engineering from University of Madras, India, an M.S. in biomedical engineering from Washington University and an M.B.A. from Fairleigh Dickinson University.
93
Shailendra Singh, Ph.D. Dr. Singh has been Vice President, System Development of AutoGenomics since its founding in April 1999. Dr. Singh has 34 years of experience in software engineering, systems development and integration, the last 29 of which have been spent in the healthcare industry and participating in the invention and development of eight medical instruments. Dr. Singh has held the position of Vice President of Systems Development at Sequenom, Inc., PB Diagnostics Systems, Inc. and Behring Diagnostics, Inc. Dr. Singh received a B.S. in electrical engineering, an M.E. in electrical engineering and a Ph.D. in electrical engineering from Southern Methodist University.
Jim Canfield. Mr. Canfield joined AutoGenomics during 2013 as Vice President, Sales & Marketing. From 2011 through 2013, Mr. Canfield tended to his personal investments. Mr. Canfield served as Director of Sales for Netline from October 2011 to June 2012, and Chief Sales Officer for Preparis, Inc from April 2010 to September 2011 and has more than 25 years experience leading sales and marketing teams in both the Clinical Diagnostic and Interactive Media Industries. He is an expert at marketing cutting edge technology. Notable companies at which he has held leadership positions include Abbott Diagnostics, Idexx Labs, Quest Diagnostics, Headhunter.net (CareerBuilder), and Knowledgestorm (Tech Target). During his career, Mr. Canfield has worked extensively with the venture capital community and has raised both public and private funds. Mr. Canfield is a graduate of Southern Illinois University and holds a B.S. degree in Industrial Technology.
Directors
Stephen Allison. Mr. Allison joined our board of directors in April 2011. Mr. Allison has over 30 years of experience advising and working for organizations in the healthcare and technology sectors and is currently an independent business consultant. From 2006 to 2007, Mr. Allison was Vice President, Chief Financial Officer of Gensym Corporation, a leading provider of software and services for mission-critical solutions in real time. Prior to Gensym, he was CFO for Webhire, Inc. from 2000 to 2003, PRI Automation from 1997 to 2000 and Helix Technology from 1995 to 1997. From 1991 until 1995, Mr. Allison was Vice President, Finance for Behring Diagnostics, Inc. an international company engaged in the development, manufacture and sale of immunoassay systems. Mr. Allison holds a B.A. degree in Economics from Columbia University and an M.B.A. from the Amos Tuck School of Business Administration at Dartmouth College. Our board of directors has determined that Mr. Allison brings to our board his knowledge of public accounting, his public company management experience, having served as chief financial officer of several public companies, and knowledge of the healthcare sectors in which we operate.
Charles Birmingham. Mr. Birmingham joined our board of directors in August 2012. He has over 30 years of experience in growth-oriented health care technology and service companies. Most recently, he was President and COO of Intelligent Healthcare from December 2012 to May 2014, a company that provides analytics to improve the quality of health care services at some of the leading, clinically integrated health systems; the company was acquired by ZirMed, Inc., of Louisville, Kentucky in May 2014. Before joining Intelligent Healthcare, Mr. Birmingham was Vice President of Corporate Development of CareMore Health Plan, recently acquired by WellPoint, Inc., where he was responsible for the national expansion of health plans and new product development. There, he also served as General Manager of CareMore Touch focusing on clinical and business operations, development and distribution of the Medicare Special Needs Plan. Mr. Birmingham has also been a Board member and COO of a health information exchange company that offered a platform for data exchange and "Cloud" computing linking reference labs to physicians. This company was ultimately acquired by IBM. Mr. Birmingham is a nationally recognized expert in the integration of Medicare and Medicaid services for seniors and in Medicaid behavioral managed care. He led the development of the $200 million Massachusetts Mental Health and Substance Abuse Managed Care and ran the Medicare special needs plans for low income seniors at Secure Horizons. He also led the sales team of a Medicare Advantage plan for chronically ill seniors that grew from $10 million in annual revenue to $1 billion in 12 months. Mr. Birmingham is a Board member of National Healthcare Services, a venture capital fund specializing in health care technology. Mr. Birmingham earned an MBA at Vanderbilt University and a B.S. in Psychology from St. Joseph’s University in Philadelphia, PA. Our board of directors has determined that Mr. Birmingham brings to our board an extensive knowledge of the healthcare industry in general and the reimbursement environment in which our customers operate.
Joseph S. Coyne, Dr.P.H., MPH. Dr. Coyne joined our board of directors in July 2013. He has 30 years of healthcare experience in international health finance, health information technology and mergers/acquisitions/joint
94
ventures. He has served as a financial advisor to Kaiser Health Plan, Adventist Health System, Chase Bank, Stanford Medical Center, Rush Medical Center and Beijing Association of Teaching Hospitals. From 1999 to present, he has served as a healthcare finance faculty member at Washington State University, in the Department of Health Policy and Administration, and from 2004 to present as the Director of the Center for International Health Services Research & Policy. From 1990 to 1999, he served as co-founder and Senior Partner with Healthcare DataBank, Inc. From 1989 to 1990, he was Chief Operating Officer with Heals Health Plan. Dr. Coyne is a founding member of both the National Healthcare Services and National Healthcare Reinsurance Company Board of Directors. Dr. Coyne has a B.A. in Business Administration, cum laude, at the University of Notre Dame, and earned DrPH and MPH degrees from the University of California, Berkeley. Our board of directors has determined that Dr. Coyne brings to our board his strategic financial growth analysis expertise from a global perspective.
Laurence M. Demers, Ph.D., DABCC, FACB. Dr. Laurence M. Demers has served as a director of AutoGenomics since March 2008. He is Distinguished Professor Emeritus of Pathology and Medicine at the M. S. Hershey Medical Center of The Pennsylvania State University, Hershey, PA. Dr. Demers is a diplomat of the American Board of Clinical Chemistry, a fellow of the National Academy of Clinical Biochemistry, past president of the AACC as well as past president of the National Academy of Clinical Biochemistry, and the founding director of Clinical Chemistry at the Hershey Medical Center where he has practiced for over 35 years. His primary research interests have been in the area of biochemical endocrinology, laboratory automation, thyroid disease, metabolic bone disease, molecular diagnostics and breast cancer. Dr. Demers has had extensive experience as principal investigator of clinical trials for both the pharmaceutical and diagnostic industries. Dr. Demers received an A.B. degree in Biochemistry from Merrimack College, a Ph.D degree from the State University of New York, Upstate Medical University, Syracuse, NY and his post-doctoral fellowship training was obtained at Harvard Medical School, Boston, MA from 1970 to 1973. He was the recipient of an NIH Fogarty Fellowship award for a visiting Professorship at the University of Oxford, John Radcliffe Hospital, UK from 1981 to 1982 in the Nuffield Department of Obstetrics and Gynecology. He has published over 750 manuscripts, chapters and abstracts from his research experience. Our board of directors has determined that Dr. Demers brings to our board his scientific knowledge and understanding of our product offerings and knowledge of the pharmaceutical industry and the diagnostic industry in which we operate.
Thomas V. Hennessey, Jr. Mr. Hennessey has served as a director of AutoGenomics since 2001. He joined AutoGenomics in January 2008 as Chief Operating Officer and Chief Financial Officer and served in those capacities until August 2013. Mr. Hennessey served as AutoGenomics’ Chief Financial Officer as an independent contractor in December 2007 and as a non-employee director from January 2007 through November 2007. Mr. Hennessey has 35 years of experience in the medical and technology industries. Mr. Hennessey was an independent consultant to AutoGenomics from January 2003 until December 2007. From 2001 until 2003, he was Chief Operating Officer and Chief Financial Officer of Photovac, Inc. He has also served as Chief Operating Officer and Chief Financial Officer for several public and private start-up companies, including Behring Diagnostics, Inc., AutoImmune, Inc., Medical Diagnostics, Inc., Cambridge Heart, Inc. and Sonamed Corporation. Mr. Hennessey received a S.B. and a S.M. degree in mechanical engineering from Massachusetts Institute of Technology and an M.B.A. from Harvard Business School. Our board of directors had determined that Mr. Hennessey brings to the board his knowledge of our operations as well as management and financial experience, having served as Chief Operating Officer and Chief Financial Officer for several public and private start-up companies in the medical and technology industries in which we operate.
Adam Levinson. Mr. Levinson joined our board of directors in October 2013 and is the Chief Investment Officer and Founder of the Fortress Asia Macro Fund, Co-Chief Investment Officer of the Fortress Macro Funds and Chief Executive Officer of Fortress Investment Group (Singapore) Pte. Ltd. Mr. Levinson is also a member of the Management Committee of Fortress Investment Group. Prior to joining Fortress in June 2002, Mr. Levinson was a fund manager at Tudor Investment Corporation and a proprietary trader for ten years at Goldman, Sachs & Co. Additionally, Mr. Levinson was the Head of Foreign Exchange at Goldman Sachs Japan from 1998 to 1999. Mr. Levinson is also the Founder and Managing Partner of Graticule Asset Management Asia (GAMA), which is expected to begin operations in late 2014. Additionally, Mr. Levinson is a member of the Board of Directors at Fortress Investment Group as an Independent Director. Mr. Levinson received a B.A. in Government, magna cum laude, Phi Beta Kappa from Cornell University. Our board of directors has determined that Mr. Levinson brings to our board a sophisticated knowledge of financial markets and business acumen.
95
Peter Wilding, Ph.D. Dr. Wilding joined our board of directors in October 2012. He is professor emeritus in the department of pathology and laboratory medicine at the University of Pennsylvania Medical Center in Philadelphia, a position he has held since 2003, previously he was director of clinical chemistry from 1986 to 2003. For the past 20 years, he has concentrated on the creation of new technology involving microfabricated devices now known as microchips for clinical analyses, resulting in 20 patents. Dr. Wilding has held many offices in organizations associated with laboratory medicine, including that of president of the American Association of Clinical Chemistry and has served as an adviser or consultant to the UK Department of Health, World Health Organization, International Federation of Clinical Chemistry and Laboratory Medicine, National Institutes of Health, and National Academy of Sciences. In 1988, he co-founded the ChemCore Corp. which merged to form Caliper Technologies Corp. that had a successful IPO in 1996. He has served on Technology Advisory Committees or the Board of Directors of several bio-technology companies. Dr. Wilding is a Fellow of the Royal College of Pathologists (United Kingdom), the Royal Society of Chemistry (United Kingdom), and the National Academy of Clinical Biochemistry. He was certified by the American Board of Bioanalysis as a High Complexity Clinical Laboratory Director. Dr. Wilding received his B.Sc. with Honors in medical biochemistry in 1961 and his Ph.D. in clinical chemistry in 1965, both from the University of Birmingham, United Kingdom. Dr. Wilding was elected to our board of directors beginning in October 2012 pursuant to the right granted to the holders of our Series C Convertible Preferred Stock in our certificate of incorporation, acting by majority vote, to elect one director to our board of directors.
Corporate Governance and Board Composition
Our board of directors is composed of eight directors. All directors hold office until the next annual meeting of stockholders and until a successor has been duly elected and qualified. A resolution of the board of directors may change the authorized number of directors; however, our certificate of incorporation and bylaws currently limit the number to not more than 15 nor fewer than five directors. Our board of directors has determined that each of our non-employee directors is independent within the meaning of NASDAQ rules.
Board Structure and Committees Composition
Our board of directors directs the management of our business and affairs, as provided by Delaware law, and conducts its business through meetings of the board of directors and standing committees. Our board of directors currently has an audit committee, compensation committee and nominating committee. Our board of directors may establish other committees to facilitate the management of our business. The membership and function of each of the committees are described below.
Audit committee. The audit committee of our board of directors consists of Stephen Allison (Chairman), Charles Birmingham and Laurence M. Demers, Ph.D. The audit committee, which is composed solely of independent directors, has sole authority for hiring the company's independent auditors, oversees the results and scope of the audit and other services provided by our independent auditors, oversees our audit and control functions and reviews all related persons transactions. Our board of directors has determined that Mr. Allison qualifies as an "audit committee financial expert" as defined by the rules under the Exchange Act. The background and experience of each of our audit committee members are set forth above.
Compensation committee. The compensation committee of our board of directors consists of Laurence M. Demers, Ph.D. (Chairman), Adam Levinson and Stephen Allison. The compensation committee, comprised solely of independent directors, oversees our compensation plans and is responsible for setting the compensation of our chief executive officer. Such oversight includes reviewing and, as it deems appropriate, recommending to our board of directors policies, practices and procedures relating to the compensation of our directors, officers and other managerial employees and exercising authority under our equity incentive plans.
Nominating and corporate governance committee. The nominating and corporate governance committee of our board of directors consists of Peter Wilding, Ph.D. (Chairman), Joseph S. Coyne, Dr.P.H., MPH and Fareed Kureshy. The nominating and corporate governance committee is responsible for recruiting and retaining qualified persons to serve on our board of directors, including proposing such individuals to the board of directors for nomination for election as directors, evaluating the performance, size and composition of the board of directors and overseeing our corporate governance programs and compliance activities. The nominating and corporate governance committee considers written suggestions from stockholders, including potential nominees for election as directors.
96
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics that applies to all of our employees, officers and directors, including those officers responsible for financial reporting. The code of business conduct and ethics is available on our website at www.autogenomics.com. Information contained in or that can be accessed through our website is not incorporated by reference into this prospectus and should not be considered to be part of this prospectus. Any amendments to the code, or any waivers of its requirements with respect to our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions will be promptly disclosed on our website.
Compensation Committee Interlocks and Insider Participation
None of the members of our compensation committee has at any time during our last completed fiscal year been one of our officers or employees. None of our executive officers currently serves, or during our last completed fiscal year has served, as a member of the board of directors or compensation committee of any entity that has one or more executive officers on our board of directors or compensation committee.
Director Compensation
The non-employee members of our board of directors are reimbursed for travel, lodging and other reasonable expenses incurred in attending board of directors or committee meetings. We do not currently provide any cash compensation to our non-employee directors. From time to time, we have granted stock options to our non-employee directors as compensation for their services, although there has been no formal policy in place with respect to such awards. During 2013, we granted stock options to our directors pursuant to which the optionee may purchase up to an aggregate of 87,450 shares of our common stock at a price of $6.67 per share, and had vesting periods of zero or 12 months, expiring in 2023.
Following the completion of this offering, we will provide cash compensation in the form of an annual retainer of $35,000 for each non-employee director. We will also pay an additional annual retainer of $10,000 to the lead non-employee director, $10,000 to the chairman of our audit committee, $7,500 to each other non-employee member of our audit committee, $7,000 to the chairman of our compensation committee, $5,000 to each other non-employee member of our compensation committee, $5,000 to the chairman of our nominating and corporate governance committee and $3,000 to each other non-employee member of our nominating and corporate governance committee. In lieu of receiving this cash compensation, the directors will be able to elect to receive restricted stock units with an equal value on their date of grant. We will continue to reimburse our non-employee directors for their reasonable expenses incurred in attending meetings of our board of directors and committees of the board of directors.
Following the completion of this offering, any non-employee director who is first elected to the board of directors will be eligible to receive a restricted stock unit covering 3,300 shares of our common stock in connection with his or her initial election to the board of directors. In addition, on the date of each annual meeting of our stockholders following this offering, each non-employee director will be eligible to receive a restricted stock unit covering 1,650 shares of our common stock.
The restricted stock units granted to non-employee directors described above will vest one year after they were granted, subject to the director's continuing service on our board of directors on that date. The terms of these restricted stock units are described in more detail under "Executive Compensation — Employee Plans — 2008 Equity Incentive Award Plan."
97
The following table summarizes the aggregate number of outstanding stock options at, and compensation received by our non-employee directors during the year ended, December 31, 2013.
Name | Outstanding options | Option awards ($) (1) | |
Stephen Allison | 47,100 | 104,895 | |
Charles Birmingham | 10,500 | 69,930 | |
Joseph S. Coyne, Dr.P.H., MPH. | 10,500 | 69,930 | |
Laurence M. Demers, Ph.D. | 57,000 | 104,895 | |
Thomas V. Hennessey, Jr. (2) | 162,314 | 46,620 | |
Adam Levinson | 9,000 | 59,940 | |
Peter Wilding, Ph.D. | 18,950 | 126,207 | |
(1) | Represents the full grant date fair value of the stock option grants awarded during fiscal year 2013, as computed in accordance with FASB Accounting Standards Codification Topic 718. For a discussion of the valuation assumptions, see Note 8 to our financial statements included elsewhere in this prospectus. |
(2) | Mr. Hennessey served as Chief Operating Officer and Chief Financial Officer until August 2013; his compensation from employment and director services is reflected in the summary compensation table below. |
98
EXECUTIVE COMPENSATION
Summary Compensation Table
The following table summarizes the compensation that we paid to our named executive officers during the year ended December 31, 2013.
Name and principal position | Year | Salary ($) | Option awards ($) (1) | Total ($) | |||||||
Fareed Kureshy | 2013 | 296,154 | 330,165 | 626,319 | |||||||
President and Chief Executive Officer | 2012 | 255,769 | 86,500 | 342,269 | |||||||
Thomas V. Hennessey, Jr. (2) | 2013 | 107,692 | 23,345 | 131,037 | |||||||
Former Chief Operating Officer, Chief Financial Officer and Secretary | 2012 | 138,461 | 173,000 | 311,461 | |||||||
Shailendra Singh, Ph.D. | 2013 | 186,153 | — | 186,153 | |||||||
Vice President, System Development | 2012 | 165,769 | — | 165,769 | |||||||
Ramanath Vairavan | 2013 | 175,572 | — | 175,572 | |||||||
Senior Vice President, Lab Services | 2012 | 183,076 | — | 183,076 | |||||||
(1) | Represents the grant date fair value for 2013 of stock options granted to our named executive officers, computed in accordance with FASB ASC Topic 718, Stock Compensation. For information regarding assumptions made in connection with this valuation, please see Note 1 to our financial statements included elsewhere in this prospectus. |
(2) | Mr. Hennessey was an executive officer until August 31, 2013. He continues to serve on our board of directors. Option awards in 2013 in the amount of $23,345 were related to non-employee service on our board of directors. |
Base salary and bonuses
Our compensation committee of our board of directors reviews and recommends to our board of directors the base salary and bonus compensation of our named executive officers. Our board of directors, without our named executive officers present, ratifies and approves all compensation matters for our named executive officers. Compensation for our named executive officers has historically been highly individualized, resulted from arm's length negotiations and been based on a variety of informal factors including our financial condition and available resources, our need for that particular position to be filled and the compensation levels of our other executive officers, each as of the time of the applicable compensation decision. We have historically operated with limited cash resources. Consequently, our board of directors and compensation committee have been limited in the ability to consider significant increases in cash compensation for our named executive officers.
Our board of directors and our compensation committee has not undertaken a formal review of relevant market compensation data in setting named executive officer compensation. Instead, our board of directors and our compensation committee rely primarily upon the judgment of their members in making compensation decisions, after reviewing our performance and financial condition and carefully evaluating a named executive officer's performance during the year against established goals, leadership qualities, operational performance, business responsibilities, career with us, current compensation arrangements and long-term potential to enhance stockholder value. To date, base salary and bonuses for our named executive officers have been determined and paid on a discretionary basis in those instances where the compensation committee and the board of directors desire to reward outstanding performance during the fiscal year by our named executive officers. Bonuses are not based upon the achievement of specified performance goals, and due to continuing cash constraints, no bonuses were paid for services performed in 2012 or 2013.
In 2012 and 2013, base salary for our named executive officers was limited due to continuing economic constraints on our business. We expect base salary and cash bonuses for our named executive officers to increase, in some cases significantly, following the completion of this offering, as we align our named executive officer
99
compensation with general market practices and if our financial and operating performance increases in future periods. For example, on January 3, 2013, we signed an employment agreement with Fareed Kureshy, our President, Chief Executive Officer and Chairman of our board of directors, that provides for an annual base salary of $450,000 for Mr. Kureshy following the completion of this offering, with annual increases of at least five percent, which represents a substantial increase to his 2013 base salary and reflects the fact that Mr. Kureshy has in recent past periods, including during 2010 through 2013, accepted significantly reduced pay due to significant financial constraints. See "— Executive employment agreements" below. Decisions regarding salary increases in future periods may take into account the executive officer's current salary, equity ownership and the amounts paid to an executive officer's peers inside our company by conducting an internal analysis, which compares the pay of each executive officer to other members of the management team. Other than for Mr. Kureshy as described above, no formulaic base salary increases are provided to our named executive officers. This strategy is consistent with our intent of offering compensation that is both cost-effective, competitive and consistent with our financial resources.
Long-term equity incentives
The goals of our long-term, equity-based incentive awards are to align the interests of our named executive officers with the interests of our stockholders. Because vesting is based on continued employment, our equity-based incentives also encourage the retention of our named executive officers through the vesting period of the awards. In determining the size of the long-term equity incentives to be awarded to our named executive officers, we take into account a number of internal factors, such as the relative job scope, the value of existing long-term incentive awards, individual performance history, prior contributions to us, the size of prior grants, competitive considerations and our limited cash resources. Our compensation committee does not refer to formal competitive market data in determining long-term equity incentive awards. Based upon these factors, the compensation committee determines the size of the long-term equity incentives at levels it considers appropriate to create a meaningful opportunity for reward predicated on the creation of long-term stockholder value.
To reward and retain our named executive officers in a manner that best aligns employees' interests with stockholders' interests, we use stock options as the primary incentive vehicles for long-term compensation. We believe that stock options are an effective tool for meeting our compensation goal of increasing long-term stockholder value by tying the value of the stock options to our future performance. Because employees are able to profit from stock options only if our stock price increases relative to the stock option's exercise price, we believe stock options provide meaningful incentives to employees to achieve increases in the value of our stock over time.
We use stock options to compensate our named executive officers both in the form of initial grants in connection with the commencement of employment and additional or "refresher" grants. To date there has been no set program for the award of additional or "refresher" grants, and the compensation committee retains discretion to make stock option awards to employees at any time, including in connection with the promotion of an employee, to reward an employee, for retention purposes or for other circumstances recommended by management or the compensation committee.
The exercise price of each stock option granted under our incentive stock option plans is the fair market value of our common stock on the grant date, as determined by our board of directors. Stock option awards to our named executive officers typically vest over a four-year period as follows: 25% of the shares underlying the option vest on each of the first four anniversaries of the vesting commencement date established at the time of grant. For new hire awards, the vesting commencement date is typically the new employee's date of hire. For awards to existing employees or service providers the vesting commencement date is typically the same as the grant date. Some of the option awards granted to our named executive officers in the past were fully vested on the date of grant. The board of directors granted these awards in consideration of the officer's past service to our company. We do not have any security ownership requirements for our named executive officers.
In 2012, we granted Mr. Kureshy and Mr. Hennessey options to purchase 16,500 and 33,000, respectively, shares of our common stock at an exercise price per share of $5.24. These grants were made pursuant to our 2008 Equity Incentive Award Plan, as further described below under "— Employee Plans — 2008 Equity Incentive Award Plan." The compensation committee granted these awards based on its subjective review of each executive's performance during the previous year, its review and consideration of the named executive officers' respective unvested equity holdings, its desire to reward the executives for service to the company, its recognition of the executives having received significantly reduced base salary during 2010 through 2013, and its determination that
100
such awards were advisable in order to continue to retain and incentivize such officers. In January 2013, pursuant to the Employment Agreement with Mr. Kureshy described below under "— Executive employment agreements," we granted Mr. Kureshy options to purchase 99,000 shares of our common stock at an exercise price of $9.85 per share. The total shares subject to this grant will vest in two equal annual installments.
As a privately owned company, there has been no active market for our common stock. Accordingly, we have had no program, plan or practice pertaining to the timing of stock option grants to named executive officers coinciding with the release of material non-public information.
Retirement savings
Our employees, including our named executive officers, are eligible to participate in our 401(k) plan. Pursuant to our 401(k) plan, employees may elect to reduce their eligible compensation by up to the statutorily prescribed annual limit of $17,500 in 2014 (additional elective deferrals not to exceed $5,500 are available to those employees 50 years of age or older) and to have the amount of this reduction contributed to our 401(k) plan. We do not make matching or profit sharing contributions under our 401(k) plan.
Perquisites, health and welfare benefits and other compensation
The establishment of competitive benefit packages for our employees is an important factor in attracting and retaining highly qualified personnel.
Health and welfare benefits
Our named executive officers are eligible to participate in all of our employee benefit plans, including our medical, dental, vision, group life and disability insurance plans, in each case on the same basis as other employees. We believe that these health and welfare benefits help ensure that we have a productive and focused workforce through reliable and competitive health and other benefits.
Perquisites
We do not provide significant perquisites or personal benefits to our named executive officers.
Post-termination and change in control benefits
Except for the employment agreement described below under "— Executive employment agreements," we do not have any employment or severance agreements with our named executive officers.
As described above, from time to time we grant our named executive officers stock options under our equity incentive plans. For a description of the change in control provisions in such equity incentive plans applicable to these stock options, see "— Employee Plans — 2008 Equity Incentive Award Plan" and "— Employee Plans — 2000 Equity Incentive Plan" below.
Executive employment agreements
We entered into an employment agreement with Mr. Kureshy on January 3, 2013. The employment agreement has an initial term of three years, ending January 3, 2016, which can be renewed for additional one year terms upon the mutual consent of us and Mr. Kureshy. Mr. Kureshy will serve as our President and Chief Executive Officer and the Chairman of our board of directors. Under the terms of the employment agreement, Mr. Kureshy will be paid an annual base salary of $450,000 per year following the completion of this offering. Beginning on the first anniversary of the employment agreement, Mr. Kureshy's then current base salary will be increased by five percent per year. Our board of directors may authorize an increase greater than five percent based on the board's evaluation of Mr. Kureshy's annual performance, our financial condition and our ability to pay Mr. Kureshy an increased base salary. Mr. Kureshy is also entitled to an annual bonus in the target amount of at least fifty percent of his then current base salary; provided, however, that any bonus is determined in the discretion of the compensation committee of our board of directors and is based on our performance and Mr. Kureshy's achievement of the quantitative and qualitative goals to be set by the compensation committee.
Upon a change in control (as defined in the employment agreement) of the Company, Mr. Kureshy will earn a cash bonus in an amount equal to one percent of the net proceeds of the transaction constituting the change of control, which will be paid at the same time and in the same form as the proceeds of the transaction that are payable
101
to us or our stockholders in connection with the consummation of the transaction. We have agreed to take all actions necessary to ensure that immediately prior to any change in control of the Company any stock options, restricted stock awards or other rights to acquire any of our capital stock granted to Mr. Kureshy, which are subject to vesting or a right of repurchase, will immediately vest in full and all rights to repurchase will lapse in their entirety, and the stock options will become exercisable for all the shares of capital stock subject to the option.
Pursuant to the employment agreement, we granted an option to purchase 99,000 shares of our common stock to Mr. Kureshy on January 14, 2013 with an exercise price of $9.85 per share. The shares of common stock subject to this grant will vest and become exercisable in two equal installments on each anniversary of the date of grant.
Under the terms of the employment agreement, (i) in no event will Mr. Kureshy accrue less than three weeks of vacation each calendar year and (ii) Mr. Kureshy is entitled to accrue up to the greater of (A) 16 weeks of vacation time or (B) the aggregate amount of vacation allowed to be accrued under our vacation benefit policy. Mr. Kureshy is eligible to participate in all benefit plans that are generally available to other employees of similar employment positions, subject to meeting the applicable eligibility requirements.
Mr. Kureshy's employment agreement grants him rights in the event of certain terminations of his employment:
• | If we terminate his employment for cause (as defined in the employment agreement), or Mr. Kureshy terminates his employment without good reason (as defined in the employment agreement), Mr. Kureshy will be entitled only to (A) the salary and other benefits accrued under the employment agreement through the effective date of his termination and (B) the reimbursement of certain expenses incurred in performing services under the employment agreement. |
• | If we terminate his employment without cause, or Mr. Kureshy terminates his employment for good reason, Mr. Kureshy will be entitled to receive: (A) all base salary and other benefits accrued under the employment agreement through the date of such separation from service (as defined in the employment agreement); (B) reimbursement of certain expenses incurred in performing services under the employment agreement; and (C) a lump sum severance benefit (as defined in the employment agreement) equal to two and one-half times his base salary as in effect immediately prior to the date of such separation from service. Mr. Kureshy will only be entitled to the severance benefit if he executes and delivers to us a general release within 50 days after the date of such separation from service and he does not revoke the release. Additionally, all stock options, restricted stock awards and other rights to acquire any of our capital stock granted to Mr. Kureshy that are subject to vesting or any right of repurchase will immediately vest in full and all of our rights to repurchase such shares will lapse in their entirety. |
• | In the event of his termination due to death or disability (as defined in the employment agreement), Mr. Kureshy (or his estate in the event of his death) is entitled to receive: (A) all base salary and other benefits accrued under the employment agreement through the date of termination; (B) reimbursement for certain expenses incurred in performing services under the employment agreement; and (C) a lump sum severance amount equal to two and one-half times his base salary as in effect immediately prior to the date of such termination. Additionally, all stock options, restricted stock awards and other rights to acquire any of our capital stock granted to Mr. Kureshy that are subject to vesting or any right of repurchase will immediately vest in full and all of our rights to repurchase such shares will lapse in their entirety. |
We have agreed to ensure that, with respect to any stock options granted to Mr. Kureshy after January 3, 2013, that Mr. Kureshy, or his estate or other people entitled to exercise such stock options following Mr. Kureshy's death, will be entitled to exercise such stock option for not less than three years after the date of termination (but not after the expiration of the original term of the stock option). We have also agreed to reimburse Mr. Kureshy in the event certain excise taxes are imposed on any portion of the compensation or benefits payable to Mr. Kureshy under the employment agreement. We do not have employment agreements with any of our other named executive officers.
Accounting for stock-based compensation
We account for stock-based payments in accordance with the requirements of stock-based compensation guidance.
102
Outstanding Stock Options at Fiscal Year End
The following table shows grants of stock options outstanding on December 31, 2013 to each of our named executive officers.
Name | Number of securities underlying option awards (exercisable) (1) | Number of securities underlying option awards (unexercisable) | Option exercise price ($) | Option expiration date | |||||
Fareed Kureshy | 99,000 | — | (2) | 2.152 | 1/11/2018 | ||||
99,000 | — | (2) | 8.303 | 2/26/2020 | |||||
8,646 | — | (3) | 6.091 | 9/29/2020 | |||||
17,325 | — | (4) | 5.242 | 8/3/2021 | |||||
8,250 | 8,250 | (5) | 5.242 | 5/16/2022 | |||||
49,500 | 49,500 | (8) | 6.670 | 1/14/2023 | |||||
Thomas V. Hennessey, Jr. | 51,150 | — | 8.333 | 6/13/2018 | |||||
11,550 | — | 7.970 | 5/27/2019 | ||||||
13,200 | — | 8.303 | 3/30/2020 | ||||||
15,477 | — | (3) | 6.091 | 9/29/2020 | |||||
30,937 | — | (4) | 5.242 | 8/3/2021 | |||||
16,500 | 16,500 | (6) | 5.242 | 5/16/2022 | |||||
0 | 7,000 | (7) | 6.670 | 11/1/2023 | |||||
Shailendra Singh, Ph.D. | 5,940 | — | 7.970 | 5/27/2019 | |||||
3,300 | — | 8.303 | 3/30/2020 | ||||||
5,445 | — | (3) | 6.091 | 9/29/2020 | |||||
10,890 | — | (4) | 5.242 | 8/3/2021 | |||||
Ramanath Vairavan | 11,550 | — | 7.970 | 5/27/2019 | |||||
3,300 | — | 8.303 | 3/30/2020 | ||||||
6,270 | — | (3) | 6.091 | 9/29/2020 | |||||
12,375 | — | (4) | 5.242 | 8/3/2021 | |||||
(1) | Except as described below, each option grant has a ten year term from the date of grant and vests as follows: 25% of the total shares subject to the option vest on each of the first four anniversaries of the date of grant subject to the named executive officers continued service. |
(2) | This option was not granted under our 2000 Equity Incentive Plan or our 2008 Equity Incentive Award Plan. The terms of these options are substantially the same as those for our 2000 Equity Incentive Plan. |
(3) | All of the shares subject to this grant were vested on December 31, 2010. |
(4) | All of the shares subject to this grant were vested on September 30, 2011. |
(5) | 50% of the total shares subject to this grant vest on each of the first two anniversaries of the date of grant subject to the named executive officers continued service. All of the shares subject to this grant vested and became exercisable on May 16, 2014. |
(6) | 50% of the total shares subject to this grant vest on each of the first two anniversaries of the date of grant subject to the named executive officers continued service. |
(7) | All of the shares subject to this grant will vest on November 1, 2014. |
(8) | 50% of the total shares subject to this grant vest on each of the first two anniversaries of January 3, 2013, subject to the named executive officer's continued service. |
103
Employee Plans
2008 Equity Incentive Award Plan
Our board of directors has adopted, and our stockholders have approved, a 2008 Equity Incentive Award Plan, or the 2008 Plan. The 2008 Plan became effective upon its approval by our stockholders on November 2, 2008. The purpose of the 2008 Plan is to promote the success and enhance the value of our company by continuing to link the personal interests of participants to those of our stockholders and by providing participants with an incentive for outstanding performance.
A summary of the principal provisions of the 2008 Plan is set forth below. This summary is qualified in its entirety by reference to the 2008 Plan itself. The form of the 2008 Plan is filed as an exhibit to the registration statement of which this prospectus is a part.
Shares available for awards. We reserved initially 660,000 shares of our common stock for issuance under the 2008 Plan. The number of shares reserved under the 2008 Plan will be increased by (1) the number of shares of common stock available for issuance and not subject to awards granted under our 2000 Equity Incentive Plan as of the effective date of the 2008 Plan and (2) the number of shares of common stock related to awards granted under our 2000 Equity Incentive Plan on or before the effective date of the 2008 Plan that are repurchased, forfeited, expired or are canceled on or after the effective date of the 2008 Plan. As of October 24, 2014, the aggregate number of shares available for issuance under the 2008 Plan as a result of grants that were repurchased, forfeited, expired or canceled in the 2000 Plan was 581,605 shares. The maximum number of shares of common stock that may be added to the share reserve under the 2008 Plan pursuant to the preceding two sentences will not exceed 2,178,000 shares. The 2008 Plan contains an "evergreen provision" that mandates an annual increase in the number of shares available for issuance under the 2008 Plan on January 1 of each year, beginning on the first January 1 following the completion of this offering. The annual increase in the number of shares shall be equal to the lesser of:
• | 3.5% of our outstanding common stock on the applicable January 1st of the applicable year; |
• | 495,000 shares; and |
• | a lesser number of shares as determined by our board of directors. |
As of October 24, 2014, options to purchase 1,318,410 shares of our common stock had been issued under the 2008 Plan, 8,745 of the options granted had been exercised and 371,268 of the options granted had been forfeited, resulting in 938,397 options outstanding. The 2008 Plan also provides for an aggregate limit on the number of shares of common stock that may be issued under the 2008 Plan over the course of its ten-year term of 7,788,000 shares, subject to changes in our capital structure as set forth in the 2008 Plan. To the extent that an award terminates, expires, lapses for any reason or is settled in cash, any shares of common stock subject to the award will again be available for the grant of an award pursuant to the 2008 Plan. Any shares of common stock tendered or withheld to satisfy the grant or exercise price or tax withholding obligation with respect to any award and any shares forfeited by a participant or repurchased by us will be available for subsequent grant under the 2008 Plan.
Administration. The compensation committee of our board of directors administers the 2008 Plan (except with respect to any award granted to non-employee directors, which is administered by our full board of directors). Following the completion of this offering, to administer the 2008 Plan, our compensation committee must consist of at least two members of our board of directors, each of whom is a "non-employee director" for purposes of Rule 16b-3 under the Exchange Act, and, with respect to awards that are intended to be performance-based awards, an "outside director" for purposes of Section 162(m) of the Internal Revenue Code. Subject to the terms and conditions of the 2008 Plan, our compensation committee has the authority to select the persons to whom awards are to be made, to determine the type or types of awards to be granted to each person, the number of shares to be subject to such awards and the terms and conditions of such awards, and to make all other determinations and decisions and to take all other actions necessary or advisable for the administration of the 2008 Plan. Our compensation committee is also authorized to establish, adopt or revise rules relating to administration of the 2008 Plan, subject to certain restrictions. Our board of directors may at any time revert to itself the authority to administer the 2008 Plan. The board of directors and any committee exercising discretion under the 2008 Plan from time to time are referred to in this section as the plan administrator.
104
Eligibility. Options, stock appreciation rights, or SARs, restricted stock and other awards under the 2008 Plan may be granted to individuals who are our officers or employees or are the officers or employees of our parent or subsidiary companies, if any, on the date of grant. Such awards may also be granted to our non-employee directors and consultants but only employees may be granted incentive stock options, or ISOs. As of October 24, 2014, there were seven non-employee directors and approximately 114 employees who are eligible for awards under the 2008 Plan. Certain individuals who are consultants covered by active consulting agreements are also eligible for awards.
Awards. The 2008 Plan provides that the plan administrator may grant or issue stock options, SARs, restricted stock, restricted stock units, dividend equivalents, stock payments and performance bonus awards, or any combination thereof. The plan administrator will consider each award grant subjectively, considering factors such as the individual performance of the recipient and the anticipated contribution of the recipient to the attainment of our long-term goals.
At such time after the offering date when the Company is subject to the requirements of Section 162(m) of the Internal Revenue Code, the maximum number of shares of our common stock that may be subject to awards granted to any one participant during any calendar year is 660,000 shares and the maximum amount that may be paid to a participant in cash during any calendar year with respect to one or more awards payable in cash is $2,000,000.
Awards under the 2008 Plan will be evidenced by a written award agreement that sets forth the terms, conditions and limitations for each award, as determined by the plan administrator.
• | Stock options. Stock options, including incentive stock options, as defined under Section 422 of the Internal Revenue Code, and nonqualified stock options, may be granted pursuant to the 2008 Plan. The option exercise price of all incentive stock options granted pursuant to the plan will not be less than 100% of the fair market value of our common stock on the date of grant. No incentive stock option may be granted to a grantee who owns more than 10% of the total combined voting power of all classes of our capital stock on the date of grant unless the exercise price is at least 110% of the fair market value at the time of grant. |
Notwithstanding whether an option is designated as an incentive stock option, to the extent that the aggregate fair market value of the shares with respect to which such option (and other options designated as incentive stock options) is exercisable for the first time by any optionee during any calendar year exceeds $100,000, such excess will be treated as a nonqualified stock option.
The plan administrator will determine the methods of payment of the exercise price of an option, including, without limitation, cash, shares of our common stock with a fair market value on the date of delivery equal to the exercise price of the option or exercised portion thereof (including shares issuable upon exercise of the option) or other property acceptable to the plan administrator (including the delivery of a notice that the participant has placed a market sell order with a broker with respect to shares then issuable upon exercise of the option, and that the broker has been directed to pay a sufficient portion of the net proceeds of the sale to us in satisfaction of the option exercise price, provided that payment of such proceeds is then made to us not later than settlement of such sale). However, no participant who is a director or an executive officer of our company within the meaning of Section 13(k) of the Exchange Act will be permitted to pay the exercise price of an option in any method that would violate Section 13(k) of the Exchange Act.
Stock options shall vest and may be exercised as determined by the plan administrator, but in no event may incentive stock options be exercised after the tenth anniversary of the date of grant. However, in the case of an incentive stock option granted to a person who owns more than 10% of the total combined voting power of all classes of our capital stock on the date of grant, such term will not exceed five years.
• | Restricted stock. Eligible employees, consultants and directors may be issued restricted stock in such amounts and on such terms and conditions as determined by the plan administrator. Restricted stock will be evidenced by a written restricted stock agreement, which will contain restrictions on transferability and other such restrictions as the plan administrator may determine, including, without limitation, limitations on the right to vote restricted stock or the right to receive dividends on the restricted stock. These restrictions may lapse separately or in combination at such times, pursuant to such circumstances, in such installments, or otherwise, as the plan administrator determines at the time of grant of the award or thereafter. Typically, restricted stock may be repurchased by us at the original purchase price or forfeited for no consideration if the conditions or restrictions are not met. |
105
• | Stock appreciation rights. A SAR is the right to receive payment of an amount equal to the excess of the fair market value of a share of our common stock on the date of exercise of the SAR over the per share exercise price of the SAR. The plan administrator may issue SARs in such amounts and on such terms and conditions as it may determine, consistent with the terms of the 2008 Plan. SARs shall vest and may be exercised as determined by the plan administrator. The plan administrator may elect to pay SARs in cash, in our common stock or in a combination of cash and our common stock. |
• | Restricted stock units. Restricted stock units may be granted to any eligible employee, consultant or director in such amounts and subject to such terms and conditions as are determined by the plan administrator. At the time of grant, the plan administrator will specify the date or dates on which the restricted stock units will become fully vested and nonforfeitable, and may specify such conditions to vesting as it deems appropriate. At the time of grant, the plan administrator will specify the distribution date or event applicable to each grant of restricted stock units which will be no earlier than the vesting date or dates of the award. On the distribution date or event, we will transfer to the participant one unrestricted, fully transferable share of our common stock for each restricted stock unit scheduled to be paid out on such date and not previously forfeited. The plan administrator will specify the purchase price, if any, to be paid by the participant to us for such shares of our common stock. |
• | Dividend equivalents. Dividend equivalents are rights to receive the equivalent value (in cash or our common stock) of dividends paid on our common stock. They represent the value of the dividends per share paid by us, calculated with reference to the number of shares that are subject to any award held by the participant, other than options or SARs. |
• | Stock payments. Stock payments include payments in the form of our common stock or options or other rights to purchase our common stock, in each case made in lieu of all or any portion of the compensation that would otherwise be paid to the participant. The number of shares will be determined by the plan administrator and may be based upon specific performance criteria determined appropriate by the plan administrator. |
• | Performance bonus awards. Any eligible employee, consultant or director selected by the plan administrator may be granted one or more performance-based awards in the form of a cash bonus payable upon the attainment of performance goals that are established by the plan administrator and relate to any one or more performance criteria determined appropriate by the plan administrator on a specified date or dates or over any period or periods determined by the plan administrator. Any such cash bonus paid to a "covered employee" within the meaning of Section 162(m) of the Internal Revenue Code may be, but need not be, qualified performance-based compensation as described below. |
Qualified performance-based compensation. The plan administrator may grant awards to employees who are or may be "covered employees," as defined in Section 162(m) of the Internal Revenue Code, that are intended to be "qualified performance-based compensation" within the meaning of Section 162(m) of the Internal Revenue Code in order to preserve the deductibility of these awards for federal income tax purposes. Participants are only entitled to receive payment for "qualified performance-based compensation" for any given performance period to the extent that pre-established performance goals set by the plan administrator for the period are satisfied. These pre-established performance goals must be based on one or more of the following performance criteria: net earnings (either before or after interest, taxes, depreciation and amortization), sales or revenue, net income (either before or after taxes), operating earnings, cash flow (including, but not limited to, operating cash flow and free cash flow), return on net assets, return on stockholders' equity, return on assets, return on capital, return on sales, gross or net profit margin, working capital, earnings per share and price per share. These performance criteria may be measured in absolute terms or as compared to performance in an earlier period or as compared to any incremental increase or as compared to results of a peer group. With regard to a particular performance period, the plan administrator will have the discretion to select the length of the performance period, the type of performance-based awards to be granted, and the goals that will be used to measure the performance for the period. In determining the actual size of an individual performance-based award for a performance period, the plan administrator may reduce or eliminate (but not increase) the award to take into account additional factors the plan administrator deems relevant to the performance for the period. Generally, a participant will have to be employed by us or a parent or subsidiary company of ours, if any, throughout the performance period to be eligible for a performance-based award for any period.
Adjustments. If there is any stock dividend, stock split, combination or exchange of shares, merger, consolidation or other distribution (other than normal cash dividends) of our assets to stockholders, or any other
106
change affecting the shares of our common stock or the share price of our common stock other than an equity restructuring (as defined in the 2008 Plan), the plan administrator will make such equitable adjustments, if any, as the plan administrator in its discretion may deem appropriate to reflect such change with respect to (1) the aggregate number and type of shares that may be issued under the 2008 Plan (including, but not limited to, adjustments of the number of shares available under the plan and the maximum number of shares which may be subject to one or more awards to a participant pursuant to the plan during any calendar year), (2) the terms and conditions of any outstanding awards (including, without limitation, any applicable performance targets or criteria with respect thereto) and (3) the grant or exercise price per share for any outstanding awards under the plan. If there is any equity restructuring, (1) the number and type of securities subject to each outstanding award and the grant or exercise price per share for each outstanding award, if applicable, will be proportionately adjusted and (2) the plan administrator will make proportionate adjustments to reflect such equity restructuring with respect to the aggregate number and type of shares that may be issued under the 2008 Plan (including, but not limited to, adjustments of the number of shares available under the plan and the maximum number of shares which may be subject to one or more awards to a participant pursuant to the plan during any calendar year). Adjustments in the event of an equity restructuring will not be discretionary. Any adjustment affecting an award intended as "qualified performance-based compensation" will be made consistent with the requirements of Section 162(m) of the Internal Revenue Code. The plan administrator also has the authority under the 2008 Plan to take certain other actions with respect to outstanding awards in the event of a corporate transaction, including provision for the cash-out, termination, assumption or substitution of such awards.
Repricing. The plan administrator may, without stockholder approval, (1) amend any award to reduce the per share exercise price of such an award below the per share exercise price as of the date the award is granted and (2) grant an award in exchange for, or in connection with, the cancellation or surrender of an award having a higher per share exercise price.
Corporate transactions. In the event of a change of control where the acquirer does not continue, convert, assume or replace awards granted under the 2008 Plan, awards issued under the 2008 Plan will be subject to accelerated vesting such that 100% of the awards will become vested and exercisable or payable, as applicable, immediately prior to a change of control. Under the 2008 Plan, a change of control is generally defined as:
• | a transaction or series of related transactions (other than an offering of our stock to the general public through a registration statement filed with the SEC) whereby any person or entity or related group of persons or entities (other than us, our subsidiaries, if any, an employee benefit plan maintained by us or our subsidiaries, if any, or a person or entity that, prior to such transaction, directly or indirectly controls, is controlled by, or is under common control with, us) directly or indirectly acquires beneficial ownership (within the meaning of Rule 13d-3 under the Exchange Act) of more than 50% of the total combined voting power of our securities outstanding immediately after such acquisition; |
• | during any two-year period, individuals who, at the beginning of such period, constitute our board of directors together with any new director(s) whose election by our board of directors or nomination for election by our stockholders was approved by a vote of at least two-thirds of the directors then still in office who either were directors at the beginning of the two-year period or whose election or nomination for election was previously so approved, cease for any reason to constitute a majority of our board of directors; |
• | our consummation (whether we are directly or indirectly involved through one or more intermediaries) of (x) a merger, consolidation, reorganization or business combination, (y) the sale or other disposition of all or substantially all of our assets in any single transaction or series of transactions or (z) the acquisition of assets or stock of another entity, in each case other than a transaction: |
• | which results in our voting securities outstanding immediately before such transaction continuing to represent (either by remaining outstanding or by being converted into voting securities of the company or the person that, as a result of the transaction, controls, directly or indirectly, the company or owns, directly or indirectly, all or substantially all of the company's assets or otherwise succeeds to our business (the "successor entity")), directly or indirectly, at least a majority of the combined voting power of the successor entity’s outstanding voting securities immediately after the transaction; and |
• | after which no person or group beneficially owns voting securities representing 50% or more of the combined voting power of the successor entity (but no person or group will be treated as beneficially |
107
owning 50% or more of combined voting power of the successor entity solely as a result of the voting power held in the company prior to the transaction).
Amendment and termination of the 2008 Plan. Our board of directors may terminate, amend or modify the 2008 Plan. However, stockholder approval of any amendment to the 2008 Plan will be obtained to the extent necessary and desirable to comply with any applicable law, regulation or stock exchange rule, or for any amendment to the 2008 Plan that increases the number of shares available under the 2008 Plan. If not terminated earlier by the board of directors, the 2008 Plan will terminate in 2018.
2000 Equity Incentive Plan
Our board of directors initially adopted our 2000 Equity Incentive Plan, or the 2000 Plan, on December 13, 2000. We terminated the 2000 Plan in connection with the approval of the 2008 Plan, and, therefore, we will not make any further awards of options or stock purchase rights under the 2000 Plan. Outstanding stock options granted under the 2000 Plan will continue to be governed by the 2000 Plan until they are exercised, expire or otherwise terminate.
The purpose of the 2000 Plan was to further our growth, development and financial success by providing incentives to certain key persons by assisting them in acquiring shares of common stock so that they may benefit directly from our growth, development and financial success.
A summary of the principal provisions of the 2000 Plan is set forth below. This summary is qualified in its entirety by reference to the 2000 Plan itself. The 2000 Plan has been filed as an exhibit to the registration statement of which this prospectus is a part.
Shares available for awards. Subject to certain adjustments set forth in the 2000 Plan, the maximum number of shares of our common stock that could have been issued or awarded under the 2000 Plan was 2,178,000 shares. As of October 24, 2014, options to purchase 253,048 shares of our common stock were outstanding and 105,600 shares of our common stock had been issued pursuant to stock purchase rights under the 2000 Plan.
To the extent that an award expires, terminates or is not exercised, any shares of common stock subject to such award that have not been issued will again be available for the grant of an award pursuant to the 2008 Plan. Any shares of our common stock acquired by a participant pursuant the 2000 Plan that are reacquired by us will also again be available for the grant of an award pursuant to the 2008 Plan.
Administration. Either the board of directors or a committee of the board of directors will administer the 2000 Plan. The board of directors and any committee exercising discretion under the 2000 Plan from time to time are referred to in this section as the plan administrator. Subject to the terms of the 2000 Plan, the plan administrator had express authority to determine the eligible persons who will receive awards, the number of shares of common stock to be covered by each award and the terms and conditions of awards. The plan administrator has broad discretion to prescribe, amend and rescind rules relating to the 2000 Plan and its administration, to interpret the 2000 Plan and the terms of all award agreements, to determine the rights and obligations of participants of the 2000 Plan, to authorize the amendment of the terms of any outstanding awards under the plan.
Eligibility. Stock options and stock purchase rights under the 2000 Plan were granted to individuals who were then our employees, consultants, advisors and members of our board of directors or who were employees, consultants, advisors or members of the board of directors of our subsidiaries, if any. Only employees may be granted ISOs.
Awards. The 2000 Plan provides for the grant of stock options and stock purchase rights. The options and stock purchase rights are evidenced by option agreements and restricted stock purchase agreements, respectively, which set forth the terms, conditions and limitations for each award, as determined by the plan administrator. Such terms and conditions include certain rights of repurchase, rights of first offer, rights of first refusal and call rights in our favor.
Subject to certain adjustments set forth in the plan, the maximum number of shares of common stock that could be subject to awards granted to any one participant pursuant to the 2000 Plan was 264,000 shares.
• | Stock options. Stock options, including incentive stock options, as defined under Section 422 of the Internal Revenue Code, and nonqualified stock options, could be granted pursuant to the 2000 Plan. The option exercise |
108
price of all stock options granted pursuant to the plan could not be less than 85% of the fair market value of our common stock on the date of grant. No stock option could be granted to a grantee who owned more than 10% of the total combined voting power of all classes of our capital stock on the date of grant unless the exercise price was at least 110% of the fair market value at the time of grant.
Notwithstanding whether an option is designated as an incentive stock option, to the extent that the aggregate fair market value of the shares with respect to which such option is exercisable for the first time by any optionee during any calendar year exceeds $100,000, such excess will be treated as a nonqualified stock option. The plan administrator will determine the methods of payment of the exercise price of an option, including, without limitation, cash or other consideration acceptable to the plan administrator.
Stock options may be exercised as determined by the plan administrator, but no options were granted that could be exercised after the tenth anniversary of the date of grant. However, in accordance with tax rules governing incentive tax options, no incentive stock options were granted to a person who owned more than 10% of the total combined voting power of all classes of our capital stock on the date of grant with a term exceeding five years.
• | Stock purchase rights. Stock purchase rights could have been granted pursuant to the 2000 Plan. The exercise price of a stock purchase right granted pursuant to the plan could not have been less than 85% of the fair market value of our common stock on the date of grant or at the time the participant purchases shares of common stock pursuant thereto, and the exercise price of any stock purchase right granted pursuant to the plan to any person who owns stock possessing more than 10% of the total combined voting power of all classes of our stock must have been not less than 100% of the fair market value on the date of grant or at the time the participant purchases shares of common stock pursuant thereto. |
Adjustments. If the outstanding shares of our common stock are increased, decreased or exchanged for different securities through a stock split, stock dividend, recapitalization, reorganization or similar capital adjustment other than in connection with a merger or consolidation (as described in the 2000 Plan), appropriate adjustments will be made in (1) the aggregate number of shares of common stock covered by the plan and/or the kind of shares subject to the plan, (2) the number and kind of shares of stock which may be purchased pursuant to the exercise of outstanding awards granted under the plan and (3) the exercise price or purchase price of outstanding awards granted under the plan.
Corporate transactions. Except as otherwise provided in any option agreement between the participant and us, in the event of a corporate transaction (as defined in the 2000 Plan) involving our company in which options are not assumed or substituted with an equivalent option by the surviving corporation or its parent or subsidiary, if any, such options will become exercisable in full immediately prior to the consummation of such corporate transaction and will terminate upon the consummation of such corporate transaction to the extent not exercised prior to such consummation. At least ten days prior to the consummation of any corporate transaction, we will give each participant written notice that states whether (1) the outstanding options will be expressly assumed or substituted with equivalent options or (2) such participant will have the right to exercise such awards immediately prior to such consummation of the corporate transaction. Any shares of our common stock issued pursuant to any stock purchase right pursuant to the 2000 Plan shall, except as otherwise provided in the applicable restricted stock purchase agreement, become fully vested upon the consummation of a corporate transaction (as defined in the 2000 Plan).
The 2000 Plan defines a corporate transaction as any of the following stockholder approved transactions to which our company is a party:
• | a merger or consolidation in which our company is not the surviving entity; |
• | the sale, transfer or other disposition of all or substantially all of the assets of our company (including the capital stock of our company's subsidiary corporations, if any) in connection with the complete liquidation or dissolution of our company; |
• | any merger in which our company is the surviving entity but in which the shares of our company's capital stock outstanding immediately prior to such transaction are converted into the right to receive cash, debt securities and/or equity securities of another corporation; or |
• | the sale by the holders of more than 90% of the outstanding shares of our company's capital stock in a single transaction or a series of related transactions. |
109
Amendment and termination of the 2000 Plan. Our board of directors may amend or modify the 2000 Plan. The 2000 Plan was terminated in connection with our adoption of the 2008 Plan and as of the effective date of the 2008 Plan, no further awards will be granted under the 2000 Plan.
Options issued outside of the 2000 Plan and the 2008 Plan
We have issued options to purchase our common stock to certain of our directors, executive officers and key employees that are not subject to either the 2008 Plan or the 2000 Plan. The terms of these options are substantially the same as those issued under the 2000 Plan. As of October 24, 2014, there were outstanding options to purchase 324,000 shares of our common stock that were not issued under either the 2008 Plan or the 2000 Plan. The issuance of these options was exempt from registration under Section 4(2) of the Securities Act.
2008 Employee Stock Purchase Plan
Our board of directors adopted, and our stockholders have approved, our 2008 Employee Stock Purchase Plan, or the Purchase Plan. The compensation committee of the board of directors administers the Purchase Plan. The Purchase Plan is designed to allow our eligible employees and the eligible employees of our designated subsidiaries, if any, to purchase shares of common stock with accumulated payroll deductions. The Purchase Plan will become effective on the business day immediately prior to the date on which the registration statement filed with respect to the Purchase Plan becomes effective, but we expect that the first offering period under the Purchase Plan will not commence until the first May 15 or November 15 following the date on which the registration statement filed by us with respect the Purchase Plan becomes effective. We have initially reserved a total of 82,500 shares of our common stock for issuance under the Purchase Plan. The Purchase Plan contains an "evergreen provision" that mandates an annual increase in the number of shares available for issuance under the Purchase Plan on January 1 of each year, beginning on the first January 1 following the completion of this offering and continuing through, and including, January 1, 2018. The annual increase shall be equal to the lesser of:
• | 1% of our outstanding common stock on January 1st of the applicable year; |
• | 66,000 shares; and |
• | a lesser number of shares determined by our board of directors. |
The Purchase Plan also provides for an aggregate limit on the number of shares of common stock that may be issued under the Purchase Plan over the course of its ten-year term of 742,500 shares. The material terms of the Purchase Plan are summarized below. The Purchase Plan has been filed as an exhibit to the registration statement of which this prospectus is a part.
The Purchase Plan will have consecutive six-month offering periods. Under the Purchase Plan, purchases will be made on the last day of each offering period. We expect the first offering period under the Purchase Plan will commence on the first May 15 or November 15 following the date on which the registration statement filed by us with respect to the Purchase Plan becomes effective. A new six-month offering period will commence on each May 15 and November 15 thereafter during the term of the Purchase Plan. Our compensation committee or board of directors may change the frequency and duration of offering periods under the Purchase Plan.
Individuals scheduled to work more than 20 hours per week for greater than five calendar months per year, may join an offering period on the first day of the offering period to the extent such individual does not, immediately after any rights under the Purchase Plan are granted, own (directly or through attribution) stock possessing 5% or more of the total combined voting power or value of all classes of our common or other stock of ours or a parent or subsidiary of ours, if any. As of October 24, 2014, substantially all of our 114 full-time employees would have been eligible to participate in the Purchase Plan if it were in effect.
Participants may contribute up to 20% of their cash earnings through payroll deductions, and the accumulated deductions will be applied to the purchase of shares on each purchase date. Unless otherwise designated by the compensation committee, the purchase price per share will be equal to 85% of the fair market value per share on the first day of the offering period or, if lower, 85% of the fair market value per share on the purchase date. In each calendar year, no employee is permitted to purchase shares under the Purchase Plan at a rate in excess of $25,000 worth of shares, based on the fair market value per share determined as of the first day of the offering period, for each year such a purchase right is outstanding.
110
In the event there is a specified type of change in our capital structure, such as a stock dividend or stock split, the compensation committee may make appropriate adjustments to (a) the number and type of shares that may be issued under the Purchase Plan, including the maximum number of shares by which the share reserve may increase automatically each year, (b) the class(es) and number of shares and price per share of stock subject to outstanding rights and (c) the purchase price with respect to any outstanding rights.
In the event of certain significant corporate transactions and other events or a change in control (as defined in the Purchase Plan), the compensation committee may provide for (a) the replacement or termination of outstanding rights in exchange for cash or other rights or property, (b) the assumption or substitution of outstanding rights by the successor or survivor corporation or parent or subsidiary thereof, if any, (c) the adjustment in the number and type of shares of stock subject to outstanding rights or in the terms and conditions of outstanding rights and rights which may be granted in the future, (d) the use of participants’ accumulated payroll deductions to purchase stock on a new purchase date prior to the next purchase date and termination of any rights under ongoing offering periods or (e) the termination of all outstanding rights.
Under the Purchase Plan, a change in control has the same definition as given to such term in the 2008 Plan.
The compensation committee may amend, suspend or terminate the Purchase Plan. However, stockholder approval of any amendment to the Purchase Plan will be obtained for any amendment which changes the aggregate number of shares that may be sold pursuant to rights under the Purchase Plan, changes the corporations or classes of corporations whose employees are eligible to participate in the Purchase Plan or changes the Purchase Plan in any manner that would cause the Purchase Plan to no longer be an "employee stock purchase plan" within the meaning of Section 423(b) of the Internal Revenue Code. The Purchase Plan will terminate no later than the tenth anniversary of the Purchase Plan’s initial adoption by our board of directors.
401(k) Plan
We provide a basic savings plan, or 401(k) plan, which is intended to qualify under Section 401(a) of the Internal Revenue Code. Pre-tax contributions by our employees or any contributions by us to the 401(k) plan, and the investment earnings thereon, are not taxable to employees until withdrawn from our 401(k) plan. Roth contributions by our employees to the 401(k) plan are made on an after-tax basis and neither the investment earnings thereon, nor the withdrawals, subject to certain holding requirements, are taxable to employees. If our 401(a) plan qualifies under Section 401(k) of the Internal Revenue Code, contributions by us, if any, will be deductible by us when made.
Our full-time employees are eligible to participate in our 401(k) plan. Pursuant to our 401(k) plan, employees may elect to reduce their eligible compensation by up to the statutorily prescribed annual limit of $17,500 in 2014 and to have the amount of this reduction contributed to our 401(k) plan. Participants who are 50 years or older also may make "catch-up" contributions, which in calendar year 2014 may be up to an additional $5,500 above the statutory limit. Under the 401(k) plan, each participant is fully vested in his or her elective deferred contributions when contributed. Participant contributions are held and invested, pursuant to the participant's instructions, by the plan's trustee. We have the ability to make matching or profit sharing contributions to our 401(k) plan in our discretion. We have not historically made such contributions, and do not currently have any plans to do so in the immediate future. The 401(k) plan does not offer the ability to invest in our securities.
Indemnification of Directors and Officers and Limitation of Liability
We are subject to the Delaware General Corporate Law, or DGCL. Section 145 of the DGCL authorizes a corporation's board of directors to grant indemnity to directors and officers in terms sufficiently broad to permit such indemnification under certain circumstances for liabilities, including reimbursement for expenses incurred, arising under the Securities Act.
Certificate of incorporation and bylaws
Our certificate of incorporation and bylaws require us to indemnify our directors, officers and other persons serving at our request as a director, officer or agent of another entity to the fullest extent permitted by the DGCL. We are required to advance expenses, as incurred, to the covered persons in connection with defending a legal proceeding if we have received an undertaking by that person to repay all such amounts if it is determined that he or she is not entitled to be indemnified by us.
111
Our certificate of incorporation eliminates the personal liability of our directors for monetary damages for breach of fiduciary duty as a director, except for liability for:
• | any breach of the director's duty of loyalty to the corporation or its stockholders; |
• | acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; |
• | unlawful payments of dividends or unlawful stock repurchases, redemptions or other distributions, as provided in Section 174 of the DGCL; or |
• | any transaction from which the director derived an improper personal benefit. |
Indemnification agreements
We have entered into indemnification agreements with each of our directors and executive officers. These agreements provide indemnification to our directors and executive officers under certain circumstances for acts or omissions which may not be covered by directors' and officers' liability insurance, and may, in some cases, be broader than the specific indemnification provisions contained under the DGCL.
Indemnification for Securities Act liability
Insofar as indemnification for liabilities arising under the Securities Act may be permitted for directors, officers or persons controlling us pursuant to the foregoing, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
Insurance policies
We maintain an insurance policy covering our directors and officers with respect to certain liabilities, including liabilities arising under the Securities Act or otherwise.
112
PRINCIPAL STOCKHOLDERS
The following table sets forth information with respect to the beneficial ownership of our common stock as of October 24, 2014:
• | each person or group of affiliated persons known by us to own beneficially more than 5% of our outstanding common stock; |
• | each of our named executive officers; |
• | each of our directors; and |
• | all of our executive officers and directors as a group. |
We have determined beneficial ownership in accordance with the rules of the SEC. Except as otherwise indicated in the footnotes below, all of the shares reflected in the table are shares of common stock and we believe, based on the information furnished to us, that all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws.
Percentage ownership calculations are based on 11,722,251 shares outstanding as of October 24, 2014, and assumes the conversion of all outstanding series of convertible preferred stock into common stock. In computing the number of shares of common stock beneficially owned by a person and the percentage ownership of that person, we have deemed outstanding shares of common stock subject to options or warrants held by that person that are exercisable within 60 days after October 24, 2014. We have not deemed these shares outstanding for the purpose of computing the percentage ownership of any other person. Beneficial ownership representing less than one percent is denoted with an asterisk "*."
Unless otherwise indicated below, the address for each of the beneficial owners in the table below is c/o AutoGenomics, Inc., 2980 Scott Street, Vista, California 92081.
Shares beneficially owned | |||||
Name and address of beneficial owner | Number | Percent | |||
5% stockholders (other than directors and named executive officers) | |||||
Dennis A. Repp (1) | 1,707,935 | 15 | % | ||
Named executive officers and directors | |||||
Fareed Kureshy (2) | 1,425,529 | 12 | % | ||
Thomas V. Hennessey, Jr. (3) | 248,336 | 2 | % | ||
Shailendra Singh, Ph.D. (4) | 642,015 | 5 | % | ||
Ramanath Vairavan (5) | 257,153 | 2 | % | ||
Stephen Allison (6) | 70,200 | * | |||
Charles Birmingham (7) | 20,428 | * | |||
Joseph S. Coyne, Dr.P.H., MPH. (8) | 10,500 | * | |||
Laurence M. Demers, Ph.D. (9) | 144,339 | 1 | % | ||
Adam Levinson (10) | 642,888 | 6 | % | ||
Peter Wilding, Ph.D. (11) | 18,950 | * | |||
All executive officers and directors as a group (12 persons) | 3,712,700 | 32 | % | ||
* | Represents less than 1% of the outstanding shares of common stock. |
(1) | Includes (i) warrants exercisable for 36,300 shares of common stock, (ii) 891,280 shares of Series B Convertible Preferred Stock convertible into 324,432 shares of common stock, 527,273 shares of Series C Convertible Preferred Stock convertible into 205,168 shares of common stock, 61,539 shares of Series D Convertible Preferred Stock convertible into 23,166 shares of common stock, 1,503,011 shares of Series NC Convertible Preferred Stock convertible into 495,994 shares of common stock and warrants exercisable for 622,876 shares of common stock held by A R Properties, of which Mr. Repp is the General Partner. The business address of Mr. Repp is 3419 Via Lido #262, Newport Beach, California 92663. |
113
(2) | Includes options to purchase 339,471 shares of common stock and 4,915 shares of Series A Convertible Preferred Stock convertible into 3,244 shares of common stock. The shares of Series A Convertible Preferred Stock are held jointly by Fareed Kureshy and Dawn Beth Kureshy. |
(3) | Includes (i) warrants to purchase 3,300 shares of common stock, options to purchase 162,314 shares of common stock, and 12,658 shares of Series NC Convertible Preferred Stock convertible into 4,177 shares of common stock, and 39,600 shares of common stock and (ii) 44,668 shares of Series A Convertible Preferred Stock convertible into 29,481 shares of common stock and 26,000 shares of Series C Convertible Preferred Stock convertible into 9,464 shares of common stock. |
(4) | Consists of 616,440 shares of common stock and options to purchase 25,575 shares of common stock. |
(5) | Consists of 223,658 shares of common stock and options to purchase 33,495 shares of common stock. |
(6) | Consists of 19,800 shares of common stock, warrants exercisable for 3,300 shares of common stock and options to purchase 47,100 shares of common stock. |
(7) | Consists of 27,274 shares of Series B Convertible Preferred Stock convertible into 9,928 shares of common stock held by ABHV Partnership, of which Mr. Birmingham is a partner. Mr. Birmingham disclaims beneficial ownership of the shares held by ABHV Partners. Also includes options to purchase 10,500 shares of common stock. |
(8) | Consists of options to purchase 10,500 shares of common stock. |
(9) | Consists of 58,763 shares of Series E Convertible Preferred Stock convertible into 21,390 shares of common stock, options to purchase 57,000 shares of common stock, warrants exercisable for 62,649 shares of common stock and 3,300 share of common stock. |
(10) | Consists of 573,888 shares of common stock, warrants exercisable for 60,000 shares of common stock and options to purchase 9,000 shares of common stock. |
(11) | Consists of options to purchase 18,950 shares of common stock. |
114
CERTAIN RELATIONSHIPS AND RELATED PERSONS TRANSACTIONS
In addition to the director and executive compensation arrangements discussed above in "Management" and "Executive Compensation," we have been a party to the following transactions since January 1, 2011, in which any director, executive officer or holder of more than 5% of any class of our voting stock, or any member of the immediate family of or entities affiliated with any of them, had or will have a material interest.
Warrant Issuances
As of October 24, 2014, we had outstanding warrants to purchase an aggregate of 3,892,249 shares of our common stock. These warrants were issued in connection with equity and promissory note financings and in connection with services rendered to us. The participants in these warrant issuances included the following directors, executive officers and holders of more than 5% of our capital stock. The following table presents the number of shares issued to these related parties in these financings:
Participants (1) | Warrants to purchase common stock |
5% Stockholders (other than directors and executive officers) | |
Dennis A. Repp | 659,176 (2) |
Directors and Executive Officers | |
Laurence M. Demers, Ph.D, Director | 62,649 (3) |
Adam Levinson, Director | 60,000 (4) |
Donald E. Pogorzelski, Former Director | 219,151 (5) |
John R. Zacamy, Chief Financial Officer and Secretary | 23,265 (6) |
(1) | Additional detail regarding these stockholders and their equity holdings is provided in "Principal Stockholders" appearing elsewhere in this prospectus. |
(2) | In January 2011 and May 2011, we issued warrants to A R Properties, of which Mr. Repp is the General Partner, in connection with our promissory notes financings to purchase 9,900 shares of our common stock and 19,800 shares of our common stock, each at an exercise price of $9.09 per share, respectively. In August 2011, we issued a warrant to purchase 3,300 shares of our common stock at an exercise price of $5.24 per share as consideration for services provided to us. In November 2011 and March 2012, we issued warrants to A R Properties in connection with our promissory notes financings to purchase 20,625 shares of our common stock and 24,750 shares of our common stock, each at an exercise price of $5.24 per share, respectively. In November 2012, in connection with an exchange of notes payable aggregating $2.6 million for 1,498,637 shares of Series NC convertible preferred stock, we cancelled warrants to purchase 8,903, 21,300, 9,372, 12,375, 93,720, 9,900, 19,800, 20,625, and 24,750 shares of our common stock at exercise prices of $21.21, $21.21, $12.12, $12.12, $15.15, $15.15, $9.09, $9.09 and $5.24, respectively, and issued an aggregate of 222,876 warrants to purchase shares of our common stock at an exercise price of $4.55. In February 2014, we issued warrants to A R Properties to purchase 400,000 shares of our common stock at an exercise price of $5.31 per share in connection with the refinancing of a promissory note issued by us to A R Properties. See "— Equity and Promissory Notes Financings" below. |
(3) | In August 2011, we issued a warrant to purchase 3,300 shares of our common stock at an exercise price of $5.24 per share to Dr. Demers in connection with his service on our board of directors. In November 2011, in connection with our sale of Series E Convertible Preferred Stock, we issued to Dr. Demers a warrant to purchase 29,088 shares of our common stock at an exercise price of $8.33 per share. In December 2011, we issued a warrant to purchase 8,250 shares of our common stock at an exercise price of $5.24 per share to Dr. Demers in connection with our promissory notes financings. In May 2012, we issued a warrant to Dr. Demers to purchase 3,300 shares of common stock at an exercise price of $5.24 in connection with his service as chairman of the compensation committee of our board of directors. In November 2012, in connection with a refinancing of a note payable in the amount of $55,000 including accrued interest into a note payable of $55,000, we canceled a warrant to purchase 8,250 shares of our common stock at an exercise price of $5.24 |
115
and issued a warrant to purchase 12,375 shares of our common stock at an exercise price of $5.30. See "— Equity and Promissory Notes Financings" below.
(4) | In October 2013, we issued to Mr. Levinson a warrant to purchase 60,000 shares of our common stock at an exercise price of $6.67 in connection with the purchase by Mr. Levinson of 573,888 shares of common stock at a price of $6.97 in an equity fundraising. See "— Equity and Promissory Notes Financings" below. |
(5) | Mr. Pogorzelski left our board of directors in February 2013. In November 2011, in connection with our sale of Series E Convertible Preferred Stock, we issued to Mr. Pogorzelski a warrant to purchase 36,000 shares of our common stock at an exercise price of $8.33 per share. In January and March 2012, we issued warrants to purchase 41,250 and 89,100 shares of our common stock, respectively, each at an exercise price of $5.24 per share to Mr. Pogorzelski in connection with our promissory note financings. In November 2012, in connection with an exchange of notes payable aggregating $1.2 million for 137,928 shares of Series NC convertible preferred stock and a new note in the amount of $1.0 million, we cancelled warrants to purchase 41,250 and 89,100 shares of our common stock each at an exercise price of $5.24 and issued a warrant to purchase 24,750 shares of our common stock at an exercise price of $4.55 and warrants to purchase an aggregate of 158,400 shares of our common stock at and exercise price of $5.30. See "— Promissory Notes Financings" below. |
(6) | John R. Zacamy was the Managing Director and President of Zacamy & Co. LLC until September 2014. In November 2012, in connection with our sale of Series E Convertible Preferred Stock, we issued a warrant to Zacamy & Co. LLC to purchase 23,265 shares of our common stock at an exercise price of $8.33 per share. |
Equity and Promissory Notes Financings
As of September 30, 2014, we had an aggregate of $21.2 million in principal and accrued interest outstanding under promissory notes that were issued as private placements. These promissory notes had a weighted average annual interest rate of 8.6%, with the principal and accrued interest balances generally due three years after the date of issuance. We may elect to prepay the notes at any time without penalty. In the private placements, we also issued warrants to purchase an aggregate of 2,314,402 shares of our common stock with a weighted average exercise price of $7.09 per share. The warrants were exercisable upon issuance and have terms of five years. Dennis A. Repp, Laurence M. Demers and Donald E. Pogorzelski participated in the financings and purchased $2,245,000, $200,000 and $1,250,000 aggregate principal amount of notes and received warrants to purchase 222,876, 22,836 and 130,350 shares of our common stock, respectively. As of September 30, 2014, there was an aggregate of $340,000, $13,000 and $0 of accrued interest outstanding under the notes issued to these persons, respectively. In April 2010, we extended to April 2012 the maturity dates of notes with aggregate principal amounts of $1,000,000 and $50,000 held by Mr. Repp and Dr. Demers, respectively. In connection with those extensions, we canceled warrants and issued new warrants to purchase 93,720 and 4,686 shares of our common stock to each of these individuals, respectively. Each of the new warrants had an exercise price of $15.15 per share and expired six and one-half years after the issuance date of the original warrants. In November 2012, Mr. Repp and Mr. Pogorzelski surrendered outstanding promissory notes payable of $2,245,000 and $150,000 aggregate principal and received 1,498,637 and 134,545 shares, respectively, of Series NC Convertible Preferred Stock. Also in November 2012, Mr. Pogorzelski and Dr. Demers exchanged outstanding promissory notes payable of $1,000,000 and $50,000 aggregate principal and received new subordinated promissory notes bearing rates of interest of 8.5% and maturity dates in November 2015 aggregating $1,000,000 and $55,000, respectively. Our directors, executive officers and holders of more than 5% of our common stock that participated in the private placements and the extensions, cancellations and reissuances described above received warrants to purchase a number of shares of our common stock in the same proportion to their respective note investments as all other investors participating in the same private placement, extension, cancellation or reissuance. In January 2013, Mr. Repp purchased a $2.4 million promissory note from us in a private placement. This note has an annual interest rate of 8.5%, an original maturity date of March 31, 2013, which was refinanced to December 31, 2014, and is prepayable at any time without premium or penalty. During November 2011 through August 2012, we issued and sold shares of our Series E Convertible Preferred Stock to investors in a private placement. This private placement included the sale of 72,727, 58,763 and 47,000 shares of our Series E Convertible Preferred Stock to Mr. Pogorzelski, Dr. Demers, and Zacamy & Co. LLC, of which John R. Zacamy was the Managing Director and President until September 2014, respectively, for $200,000, $161,598, and $129,250, respectively. In September 2013, we issued and sold in a private placement 537,888 shares of our common stock to Genomic Generation Partners LLC, of which Adam Levinson is a partner, for $4,000,000.
116
Employment Relationships
Saeed Kureshy, our Vice President of Operations, is the brother of Fareed Kureshy, our Founder, President, Chief Executive Officer and Chairman of the board. Mr. Saeed Kureshy received employment compensation in the amounts of $181,003 and $132,769 for the fiscal years 2013 and 2012, respectively, which included stock-based compensation in the amount of $14,355 for fiscal year 2013 associated with the grant of options to purchase 3,300 shares of our common stock at a weighted average exercise price of $6.67 per share.
On February 27, 2013, we entered into an agreement with Zacamy & Co. LLC, or Zacamy & Co., pursuant to which Zacamy & Co. provides consulting services to the Company. Christina Zacamy, the President and Managing Director of Zacamy & Co., is the wife of John Zacamy, our Chief Financial Officer. Under the terms of the agreement, we pay Zacamy & Co. a fee of $10,000 per month, in addition to reimbursement for all out-of-pocket expenses. In addition, the agreement provides that we pay Zacamy & Co. a mutually agreed upon completion fee upon the closing of any transaction for which Zacamy & Co. serves as advisor. No fee will be payable to Zacamy & Co. from the completion of this transaction.
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and executive officers, as described in "Executive Compensation — Indemnification of Directors and Officers and Limitation of Liability."
Registration Rights Agreements
We have entered into a registration rights agreement with an investor beneficially owned by one of the members of the board of directors, as well as with the holders of our Series C Convertible Preferred Stock and warrants to purchase our Series C Convertible Preferred Stock, including certain of our directors and beneficial owners of more than 5% of our outstanding common stock. See "Shares Eligible for Future Sale — Registration Rights."
Review, Approval or Ratification of Transactions with Related Persons
Pursuant to its written charter, our audit committee must review and approve all related person transactions, which includes any transactions with an executive officer, director, beneficial owner of more than 5% of our outstanding common stock, or any of such persons' immediate family members in which the amount involved exceeds $120,000, and in which any such persons had or will have a direct or indirect material interest. For a discussion of the composition and responsibilities of our audit committee see "Management — Board Structure and Committees Composition — Audit committee." In determining whether to approve a related person transaction, our audit committee will consider such matters as it deems appropriate under the circumstances. After considering these factors, our audit committee will decide whether the related person transaction is in our best interests and will approve or reject the transaction accordingly.
117
DESCRIPTION OF CAPITAL STOCK
The following is a description of our capital stock, after giving effect to the automatic conversion of our outstanding preferred stock in connection with the completion of this offering, and the material provisions of our certificate of incorporation and bylaws. The following is only a summary and is qualified by the provisions of our certificate of incorporation and bylaws, copies of which are available as set forth under "Where You Can Find More Information."
Our authorized capital stock consists of 220 million shares of common stock, par value $0.01 per share, and 30 million shares of preferred stock, par value $0.01 per share.
Common Stock
As of October 24, 2014, there were 3,747,319 shares of common stock outstanding, and we had 400 stockholders (including holders of our convertible preferred stock). The number of shares of common stock outstanding as of October 24, 2014 does not include (i) 7,974,932 shares of common stock issuable upon the conversion of outstanding shares of convertible preferred stock, (ii) 3,892,249 shares of common stock issuable upon the exercise of outstanding warrants to purchase common stock, and (iii) 1,469,055 shares of common stock issuable upon the exercise of outstanding options to purchase common stock. The holders of our common stock are entitled to the following rights:
Voting rights
Each share of our common stock entitles its holder to one vote per share on all matters to be voted upon by the stockholders. There is no cumulative voting, which means that a holder or group of holders of more than 50% of the shares of our common stock can elect all of our directors.
Dividend rights
The holders of our common stock are entitled to receive dividends when and as declared by our board of directors from legally available sources, subject to any restrictions in our certificate of incorporation or prior rights of the holders of our preferred stock. See "Dividend Policy."
Liquidation rights
In the event of our liquidation or dissolution, the holders of our common stock are entitled to share ratably in the assets available for distribution after the payment of all of our debts and other liabilities, subject to the prior rights of the holders of our preferred stock.
Other matters
The holders of our common stock have no subscription, redemption or conversion privileges. There are no sinking fund provisions applicable to our common stock. Our common stock does not entitle its holder to preemptive rights. All of the outstanding shares of our common stock are fully paid and nonassessable. The rights, preferences and privileges of the holders of our common stock are subject to the rights of the holders of shares of any series of preferred stock that we may issue in the future.
Preferred Stock
Effective immediately prior to the completion of this offering, each outstanding share of our Series A Convertible Preferred Stock will convert into 0.6600 shares of our common stock, each outstanding share of our Series B, Series C and Series E Convertible Preferred Stock will convert into 0.3640 shares of our common stock, each outstanding share of our Series D Convertible Preferred Stock will convert into 0.3764 shares of our common stock and each outstanding share of our Series NC Convertible Preferred Stock will convert into 0.3300 shares of our common stock. Effective upon the completion of this offering, each warrant exercisable for preferred stock will become exercisable for a number of shares of common stock based on the same conversion ratios.
Following completion of this offering our board of directors, without further vote or action by our stockholders, may fix the designations, powers, preferences and rights, and the qualifications, limitations or restrictions thereof including dividend rights, dividend rates, conversion rights, voting rights, terms of redemption, redemption prices, liquidation preferences and the number of shares constituting any class or series, of up to an aggregate of 9.1 million
118
shares of our preferred stock, and may authorize the issuance of the same. The issuance of our preferred stock may have the effect of delaying, deferring, or preventing a change in control of our company without further action by the stockholders and may adversely affect the voting and other rights, including the right to dividends and to payments upon liquidation, of the holders of our common stock. Upon the completion of this offering, no shares of our preferred stock will be outstanding, and we have no present plans to issue any shares of preferred stock.
Provisions of Our Certificate of Incorporation and Bylaws and Delaware Law That May Have an Anti-Takeover Effect
Certificate of incorporation and bylaws
Certain provisions of Delaware law, our certificate of incorporation and our bylaws may be deemed to have an anti-takeover effect and may delay, deter or prevent a tender offer or takeover attempt that a stockholder might consider to be in its best interests, including attempts that might result in a premium being paid over the market price for the shares held by stockholders.
Our certificate of incorporation and bylaws contain provisions that could have the effect of discouraging potential acquisition proposals or tender offers or delaying or preventing a change of control of our company. In particular, our certificate of incorporation and bylaws, as applicable, among other things:
• | provide that special meetings of the stockholders may be called only by the chairman of our board, chief executive officer or a majority of the members of our board of directors; |
• | provide that any action required or permitted to be taken by the stockholders of our company must be effected at a duly called annual or special meeting of the stockholders and the taking of action by written consent of the stockholders in lieu of a meeting of the stockholders is specifically denied; |
• | establish procedures with respect to stockholder proposals and stockholder nominations, including requiring that advance written notice of a stockholder proposal or director nomination generally must be received at our principal executive offices not less than 90 nor more than 120 days prior to the first anniversary date of the previous year’s annual meeting of stockholders; |
• | do not include any provision for cumulative voting in the election of directors or for any other matters. Under cumulative voting, a minority stockholder holding a sufficient number of shares may be able to ensure the election of one or more directors. The absence of cumulative voting may have the effect of limiting the ability of minority stockholders to effect changes in the board of directors and, as a result, may have the effect of deterring a hostile takeover or delaying or preventing changes in control or management of our company; |
• | provide that the exact number of directors on our board may be fixed from time to time exclusively by resolution of our board of directors within a range of five to 15 directors and that newly-created directorships and vacancies on our board of directors may be filled by a majority of directors in office, although less than a quorum; |
• | provide that the board of directors may determine the number of committee members on each board committee at any time, and may remove any individual board committee member at any time for any reason. Any vacancy or newly-created committee memberships may be filled by our board of directors; |
• | provide that a director may be removed from office by the affirmative vote of at least 66-2/3% of the total voting power of the outstanding shares entitled to vote generally in the election of directors, voting together as a single class; |
• | provide that our board of directors has the power to alter, amend or repeal the bylaws without stockholder approval; |
• | require that the stockholders may alter, amend or repeal our bylaws only with the vote of holders of 66-2/3% of the voting power of the outstanding shares entitled to vote generally in the election of directors; and |
• | require that the vote of holders of 66-2/3% of the voting power of the outstanding shares entitled to vote generally in the election of directors is required to amend various provisions of our certificate of incorporation, including provisions relating to: |
• | the number of directors on our board of directors; the election, qualification and term of office of our directors; |
•filling vacancies on our board of directors;
119
•removal of members of our board of directors; and
•amendments to our certificate of incorporation.
Our certificate of incorporation authorizes our board of directors, without further vote or action by the stockholders, to issue, after giving effect to the completion of this offering and the automatic conversion of all of our shares of outstanding Series A, Series B, Series C, Series D, Series E and Series NC Convertible Preferred Stock into shares of common stock, up to an additional 9.1 million shares of preferred stock, par value $0.01 per share, in one or more classes or series, and to fix or alter:
• | the number of shares constituting any class or series; |
• | the designations, powers and preferences of each class or series; |
• | the relative, participating, optional and other special rights of each class or series; and |
• | any qualifications, limitations or restrictions on each class or series. |
The above provisions are intended to promote continuity and stability in the composition of our board of directors and in the policies formulated by the board and to discourage certain types of transactions that may involve an actual or threatened change of control. These provisions are expected to reduce our vulnerability to unsolicited acquisition attempts as well as discourage certain tactics that may be used in proxy fights. Such provisions, however, could discourage others from making tender offers for our shares and, as a consequence, may inhibit fluctuations in the market price of our common stock that could result from actual or rumored takeover attempts. These provisions also could operate to prevent changes in our management.
Delaware takeover statute
We are subject to the provisions of Section 203 of the DGCL. Subject to certain exceptions, Section 203 prohibits a Delaware corporation from engaging, under certain circumstances, in a "business combination" with an "interested stockholder" for a period of three years following the date that the stockholder became an interested stockholder, unless:
• | prior to the date of the business combination, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; |
• | on consummation of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock of the interested stockholder) those shares owned: |
• by persons who are directors and also officers, and
• | by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
• | at or subsequent to such time, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66-2/3% of the outstanding voting stock that is not owned by the interested stockholder. |
A "business combination" includes:
• | any merger or consolidation involving the corporation and the interested stockholder; |
• | any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; |
• | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; |
• | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; or |
• | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
Subject to various exceptions, an "interested stockholder" is an entity or person who, together with affiliates and associates, owns (or within three years from the date of determination, did own) 15% or more of the corporation's
120
outstanding voting stock. This statute could delay, defer or prohibit a merger or other takeover or a change of control of our company. We also anticipate that this statute may discourage business combinations or other attempts that might result in a premium over the market price for the shares of common stock held by our stockholders.
NASDAQ
We have applied to list our common stock on the NASDAQ Global Market under the symbol "AGMX."
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is American Stock Transfer & Trust Company, LLC.
121
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been no public market for our common stock, and a significant public market for our common stock may not develop or be sustained after this offering. Future sales of significant amounts of our common stock, including shares of our outstanding common stock and shares of our common stock issued upon exercise of outstanding options, in the public market after this offering could adversely affect the prevailing market price of our common stock and could impair our future ability to raise capital through the sale of our equity securities.
Sale of Restricted Shares and Lock-Up Agreements
Upon the closing of this offering, shares of common stock will be outstanding assuming no exercise of the underwriters' over-allotment option and no exercise of currently outstanding options or warrants. Of these shares, the shares of common stock sold in this offering, plus any additional shares sold upon exercise of the underwriters' over-allotment option, will be freely tradable without restriction under the Securities Act, unless they are held by our affiliates, as that term is defined in Rule 144 under the Securities Act and the rules and regulations promulgated thereunder.
The remaining shares of common stock held by existing stockholders are restricted securities as that term is defined in Rule 144 under the Securities Act or are subject to the contractual restrictions described below. Of these remaining securities:
• | shares may be sold as of the date of this prospectus; |
• | additional shares may be sold beginning 90 days after the date of this prospectus; and |
• | additional shares may be sold upon expiration of the lock-up period described below, although a portion of those shares will be subject to volume limitations pursuant to Rule 144. |
Restricted securities may be sold in the public market only if registered or if they qualify for an exemption from registration under Rule 144 or 701 under the Securities Act, which rules are summarized below.
Lock-Up Agreements
All of our officers and directors, together with other stockholders, owning approximately % of our common stock on an as-converted basis have agreed, subject to certain exceptions, not to offer, pledge, announce the intention to sell, sell, contract to sell, transfer or dispose of, directly or indirectly, any shares of our common stock, or any securities convertible into or exercisable or exchangeable for shares of our common stock, for a period of 180 days after the date of this prospectus, without the prior written consent of Stifel, Nicolaus & Company, Incorporated and Canaccord Genuity Inc. Stifel, Nicolaus & Company, Incorporated and Canaccord Genuity Inc. currently do not anticipate shortening or waiving any of the lock-up agreements or other contractual obligations restricting the sale of securities and do not have any pre-established conditions for such modifications or waivers; however, Stifel, Nicolaus & Company, Incorporated and Canaccord Genuity Inc. may, in their sole discretion, at any time, and without notice, release for sale in the public market all or any portion of the shares subject to the lock-up agreement. See "Underwriting — No Sales of Similar Securities."
Rule 144
In general, under Rule 144 as currently in effect, once we have been subject to public company reporting requirements for at least 90 days, a person who is not one of our affiliates and who is not deemed to have been one of our affiliates for purposes of the Securities Act at any time during the 90 days preceding a sale and who has beneficially owned the shares proposed to be sold for at least six months, including the holding period of any prior owner other than an affiliate, is entitled to sell those shares without complying with the manner of sale, volume limitation or notice provisions of Rule 144, subject to our compliance with the public information requirements of Rule 144. If such a person has beneficially owned the shares proposed to be sold for at least one year, including the holding period of any prior owner other than our affiliates, then that person is entitled to sell those shares without complying with any of the requirements of Rule 144.
122
In general, under Rule 144 as currently in effect, our affiliates or persons selling shares on behalf of our affiliates are entitled to sell upon expiration of the lock-up agreements described above, within any three-month period, a number of shares that does not exceed the greater of:
• | 1% of the number of shares of common stock then outstanding, which will equal approximately shares immediately after this offering; or |
• | the average weekly trading volume of the common stock during the four calendar weeks preceding the filing of a notice on Form 144 with respect to that sale. |
Such sales by affiliates may commence beginning 90 days after the date of this prospectus, subject to continued availability of current public information about us. Sales under Rule 144 by our affiliates or persons selling shares on behalf of our affiliates are also subject to certain manner of sale provisions and notice requirements.
Rule 701
Rule 701 generally allows a stockholder who purchased shares of our common stock pursuant to a written compensatory plan or contract and who is not deemed to have been an affiliate of our company during the immediately preceding 90 days to sell these shares in reliance upon Rule 144, but without being required to comply with the public information or holding period provisions of Rule 144. Rule 701 also permits affiliates of our company to sell their Rule 701 shares under Rule 144 without complying with the holding period requirements of Rule 144. All holders of Rule 701 shares, however, are required to wait until 90 days after the date of this prospectus before selling such shares pursuant to Rule 701.
Options
In addition to the shares of common stock outstanding immediately after this offering, as of October 24, 2014, there were outstanding options to purchase 1,469,055 shares of our common stock. As soon as practicable upon completion of this offering, we intend to file a registration statement on Form S-8 under the Securities Act covering shares of our common stock issued or reserved for issuance under our stock plans and other eligible shares of our common stock. Accordingly, shares of our common stock registered under such registration statement will be available for sale in the open market upon exercise of related options by the holders, subject to vesting restrictions with us, contractual lock-up restrictions, and/or market stand-off provisions applicable to each option agreement that prohibit the sale or other disposition of the shares of common stock underlying the options for a period of 180 days after the date of this prospectus without the prior written consent from us or our underwriters.
Registration Rights
We have entered into a registration rights agreement with an investor beneficially owned by one of the members of our board of directors and with the holders of our Series C Convertible Preferred Stock and warrants to purchase our Series C Convertible Preferred Stock. Pursuant to these agreements, these holders are entitled to cause us to register the shares of common stock or other securities issuable upon conversion or exercise of such preferred stock and warrants (referred to as "registrable securities"), subject to the terms and conditions of the agreements. Pursuant to the lock-up agreements described above, holders of a majority of the registrable securities have agreed, on behalf of all such holders, not to exercise their registration rights until 180 days after the completion of this offering. However, following this 180-day period, subject to certain limitations, these holders will be entitled to cause us to include their registrable securities in certain registrations by us of our common stock under the Securities Act. If, in connection with any underwritten registration of our common stock, the managing underwriter in its judgment imposes a limitation on the number of shares to be registered, the number of a holder's registrable securities to be included in the registration may be limited on a pro rata basis. In addition, at any time after we become eligible to register securities for resale on Form S-3, subject to certain limitations, the holders will be entitled to cause us to prepare and file a Form S-3 for their registrable securities. In the event that a holder causes us to file a Form S-3, we will be obligated to maintain its effectiveness for a period of 180 days, subject to certain exceptions. However, notwithstanding the provisions described above, we will not be required to effect a registration to the extent that all of a holder's registrable securities may be sold under Rule 144 during any 90-day period.
123
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS
The following is a summary of the material U.S. federal income tax consequences to non-U.S. holders (as defined below) of the acquisition, ownership and disposition of our common stock issued for cash pursuant to this offering. This summary is not a complete analysis of all of the potential U.S. federal income tax consequences relating to the acquisition, ownership and disposition of our common stock, nor does it address any U.S. federal estate or gift tax consequences or any tax consequences arising under any state, local or foreign tax laws, or under the 3.8% Medicare contribution tax or under any other U.S. federal tax laws. This summary is based on the Internal Revenue Code of 1986, as amended, or the Code, Treasury Regulations promulgated thereunder, judicial decisions, and published rulings and administrative procedures of the Internal Revenue Service, or IRS, all as in effect as of the date hereof. These authorities are subject to change, and to differing interpretations, possibly retroactively, resulting in U.S. federal income tax consequences different from those summarized below. No ruling has been or will be sought from the IRS with respect to the matters summarized below, and there can be no assurance that the IRS will not take a contrary position regarding the tax consequences of the acquisition, ownership or disposition of our common stock, or that any such contrary position would not be sustained by a court.
This summary is limited to non-U.S. holders who purchase our common stock issued pursuant to this offering for cash and who hold our common stock as a "capital asset" within the meaning of the Code (generally, property held for investment). This summary does not address all of the U.S. federal income tax consequences that may be relevant to a particular holder in light of the holder's particular circumstances. Special rules under the U.S. federal income tax laws, which are not addressed in this summary, may apply to certain non-U.S. holders, including foreign governments or entities they control; U.S. expatriates or former long-term residents of the United States; partnerships, other pass-through entities, or beneficial owners of interests in those entities; "controlled foreign corporations" and their shareholders; "passive foreign investment companies" and their shareholders; corporations that accumulate earnings to avoid U.S. federal income tax; banks; financial institutions; insurance companies; brokers, dealers or traders in securities, commodities or currencies; tax-exempt pension funds or other tax-exempt organizations; tax-qualified retirement plans; persons subject to the alternative minimum tax; persons that own, or have owned, actually or constructively, more than 5% of our common stock; and persons holding our common stock as part of a hedging or conversion transaction, straddle, constructive sale, or other risk reduction strategy.
PROSPECTIVE INVESTORS SHOULD CONSULT THEIR TAX ADVISORS REGARDING THE PARTICULAR U.S. FEDERAL INCOME TAX CONSEQUENCES TO THEM OF ACQUIRING, OWNING AND DISPOSING OF OUR COMMON STOCK, AS WELL AS ANY TAX CONSEQUENCES ARISING UNDER ANY STATE, LOCAL OR FOREIGN TAX LAWS AND ANY OTHER U.S. FEDERAL TAX LAWS. PROSPECTIVE INVESTORS SHOULD ALSO CONSULT THEIR TAX ADVISORS REGARDING THE POTENTIAL IMPACT OF ANY APPLICABLE INCOME TAX TREATY.
Definition of Non-U.S. Holder
For purposes of this summary, a "non-U.S. holder" means a beneficial owner of our common stock that is, for U.S. federal income tax purposes:
• | a nonresident alien individual; |
• | a foreign corporation (or an entity treated as a foreign corporation for U.S. federal income tax purposes); |
• | a foreign estate; or |
• | a foreign trust. |
In the case of a holder that is classified as a partnership for U.S. federal income tax purposes, the tax treatment of a partner in that partnership generally will depend upon the status of the partner and the activities of the partner and the partnership.
Distributions on Our Common Stock
If we make cash or other property distributions (other than certain stock distributions) on our common stock, or effect certain redemptions that are treated as distributions on our common stock, those distributions or redemptions will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Amounts not treated as dividends for U.S. federal income tax purposes will be allocated ratably among the shareholder's shares of common stock with
124
respect to which the distribution is paid, will constitute a return of capital, and will first be applied against and reduce a shareholder's adjusted tax basis in those shares of common stock, but not below zero. Distributions in excess of our current and accumulated earnings and profits and in excess of the shareholder's tax basis in its shares will constitute capital gain realized on the sale or other disposition of the common stock and will be treated as described under "— Gain on Disposition of Our Common Stock" below. A non-U.S. holder's adjusted tax basis in a share of common stock is generally the purchase price of the share, reduced by the amount of any distributions constituting a return of capital with respect to that share.
Any dividends paid to a non-U.S. holder of our common stock generally will be subject to U.S. federal withholding tax at a rate of 30% of the gross amount of the dividends, or such lower rate as is specified by an applicable income tax treaty. To receive the benefit of a reduced treaty rate, a non-U.S. holder must furnish to us or our paying agent a valid IRS Form W-8BEN, Form W-8BEN-E or other applicable form certifying under penalties of perjury the holder's qualification for the reduced rate. This certification must be provided to us or our paying agent prior to the payment of dividends and must be updated periodically. Special certification requirements apply to non-U.S. holders that hold common stock through certain foreign intermediaries. Non-U.S. holders that do not timely provide us or our paying agent with the required certification, but that qualify for a reduced treaty rate, may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. If we are not able to determine whether or not a distribution will exceed current and accumulated earnings and profits at the time the distribution is made, we may withhold tax on the entire amount of any distribution at the same rate as we would withhold on a dividend. However, a non-U.S. holder may obtain a refund of amounts that we withhold to the extent attributable to the portion of the distribution in excess of our current and accumulated earnings and profits.
If a non-U.S. holder holds our common stock in connection with the conduct of a trade or business in the United States, and dividends paid on the common stock are effectively connected with the holder's U.S. trade or business (and, if required by an applicable income tax treaty, are attributable to a permanent establishment or fixed base maintained by the non-U.S. holder in the United States, as defined under the applicable treaty), the non-U.S. holder will be exempt from U.S. federal withholding tax on the dividends. To claim the exemption, the non-U.S. holder must furnish to us or our paying agent a properly executed IRS Form W-8ECI (or other applicable form) prior to the payment of the dividends.
Any dividends paid on our common stock that are effectively connected with a non-U.S. holder's U.S. trade or business (and if required by an applicable income tax treaty, are attributable to a permanent establishment or fixed base maintained by the non-U.S. holder in the United States, as defined under the applicable treaty) generally will be subject to U.S. federal income tax on a net income basis at the regular graduated U.S. federal income tax rates generally applicable to U.S. persons or at such lower rate as may be specified by an applicable income tax treaty. A non-U.S. holder that is a foreign corporation also may be subject to an additional branch profits tax equal to 30% (or such lower rate as is specified by an applicable income tax treaty) of a portion of its effectively connected earnings and profits for the taxable year, as adjusted for certain items.
Gain on Disposition of Our Common Stock
Subject to the discussion below regarding backup withholding and FATCA, a non-U.S. holder generally will not be subject to U.S. federal income tax on any gain realized upon the sale, exchange or other taxable disposition of our common stock, unless:
• | the gain is effectively connected with the non-U.S. holder's conduct of a trade or business in the United States, and, if required by an applicable income tax treaty, is attributable to a permanent establishment or fixed base, as defined under the applicable treaty, maintained by the non-U.S. holder in the United States, in which case the non-U.S. holder will be required to pay tax on the net gain derived from the sale, exchange, or other taxable disposition (net of certain deductions or credits) under regular graduated U.S. federal income tax rates generally applicable to U.S. persons or at such lower rate as may be specified by an applicable income tax treaty and in the case of a non-U.S. holder that is treated as corporation for U.S. federal income tax purposes, such non-U.S. holder may be subject to a branch profits tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty; |
• | the non-U.S. holder is a nonresident alien individual present in the United States for 183 days or more during the taxable year of the sale, exchange or other taxable disposition, and certain other requirements are met in |
125
which case the non-U.S. holder will be subject to U.S. federal income tax at a flat 30% rate (or such lower rate as is specified by an applicable income tax treaty) on the gain derived from the sale, exchange, or other taxable disposition, which gain may be offset by U.S. source capital losses (even though the non-U.S. holder is not considered a resident of the U.S.) provided that the non-U.S. holder has timely filed U.S. federal income tax returns reporting those losses; or
• | our common stock constitutes a "United States real property interest" by reason of (i) our having a status as a "United States real property holding corporation," or USRPHC, for U.S. federal income tax purposes at any time within the shorter of the five-year period preceding the disposition or the non-U.S. holder's holding period for our common stock, and, (ii) in the case where shares of our common stock are regularly traded on an established securities market, the non-U.S. holder having owned, directly or indirectly, more than 5% of our common stock at any time within the shorter of the five-year period preceding the disposition or the non-U.S. holder's holding period for our common stock. In that case, the non-U.S. holder will be required to pay tax on the net gain derived from the sale, exchange, or other taxable disposition (net of certain deductions or credits) under regular graduated U.S. federal income tax rates generally applicable to U.S. persons or at such lower rate as may be specified by an applicable income tax treaty, and the gross proceeds from the applicable transaction may be subject to a 10% withholding tax, which the non-U.S. holder may claim as a credit against the non-U.S. holder's U.S. federal income tax liability. There can be no assurance that our common stock will be treated as regularly traded on an established securities market for this purpose. We believe that we are not currently and will not become a USRPHC. However, because the determination of whether we are a USRPHC at any time depends on the fair market value of our U.S. real property interests relative to the fair market value of our other business assets, there can be no assurance that we will not become a USRPHC in the future. Generally, we would be a USRPHC if the fair market value of our U.S. real property interests were to equal or exceed fifty percent of the sum of the fair market values of our worldwide real property interests and other assets used or held for use in a trade or business, all as determined for U.S. federal income tax purposes. |
Information Reporting and Backup Withholding
We must report annually to the IRS and to each non-U.S. holder the amount of dividends and other distributions paid to the non-U.S. holder and the amount of tax, if any, withheld with respect to those distributions. The IRS may make this information available to the tax authorities in the country in which the non-U.S. holder is resident.
In addition, a non-U.S. holder may be subject to information reporting requirements and backup withholding with respect to dividends paid on, and the proceeds of disposition of, shares of our common stock, unless, generally, the non-U.S. holder certifies under penalties of perjury (on IRS Form W-8BEN, Form W-8BEN-E or other applicable forms) that the non-U.S. holder is not a U.S. person or otherwise establishes an exemption. The backup withholding rate is 28%. Additional rules relating to information reporting requirements and backup withholding with respect to payments of the proceeds from the disposition of shares of our common stock are as follows:
• | If the proceeds are paid to or through the U.S. office of a broker, the proceeds generally will be subject to backup withholding and information reporting, unless the non-U.S. holder certifies under penalties of perjury (on IRS Form W-8BEN, Form W-8BEN-E or other applicable forms) that the non-U.S. holder is not a U.S. person or otherwise establishes an exemption. |
• | If the proceeds are paid to or through a non-U.S. office of a broker that is not a U.S. person and is not a foreign person with certain specified U.S. connections, which we refer to below as a "U.S.-related person," information reporting and backup withholding generally will not apply. |
• | If the proceeds are paid to or through a non-U.S. office of a broker that is a U.S. person or a U.S.-related person, the proceeds generally will be subject to information reporting (but not to backup withholding), unless the non-U.S. holder certifies under penalties of perjury (on IRS Form W-8BEN, Form W-8BEN-E or other applicable forms) that the non-U.S. holder is not a U.S. person. |
Backup withholding is not a tax. Any amounts withheld from a non-U.S. holder under the backup withholding rules may be allowed as a refund or a credit against the non-U.S. holder's U.S. federal income tax liability, provided that the non-U.S. holder timely furnishes the required information to the IRS.
126
FATCA
FATCA imposes a 30% U.S. withholding tax on dividends on our common stock and the gross proceeds from a disposition of our common stock paid to:
• | a "foreign financial institution" (as specifically defined for purposes of FATCA) unless the institution enters into an agreement with the U.S. Treasury to collect and disclose information regarding the institution's U.S. account holders (including certain account holders that are foreign entities with U.S. owners) and to withhold on certain payments, or unless it otherwise qualifies for an exemption, and |
• | a "non-financial foreign entity" (also specifically defined for purposes of FATCA) unless the entity provides the payor with a certification that it does not have any substantial direct or indirect U.S. owners or provides information identifying the substantial U.S. owners of the entity (which generally include any U.S. person who directly or indirectly owns more than 10% of the entity) or unless the entity agrees to report that information to the IRS or otherwise qualifies for an exemption. |
FATCA withholding applies to dividends paid after June 30, 2014 (or, in certain circumstances, after later dates) and to gross proceeds from sales or other dispositions of our common stock after December 31, 2016. Where applicable, intergovernmental agreements between the Unites States and other countries with respect to the implementation of FATCA and non-U.S. laws, regulations and other authorities enacted or issued with respect to those intergovernmental agreements may modify the requirements under FATCA described above. You are encouraged to consult with your own tax advisor regarding the possible implications of this legislation on your investment in our common stock.
The summary of material U.S. federal income tax consequences above is included for general information purposes only. Potential purchasers of our common stock are urged to consult their own tax advisors to determine the U.S. federal, state, local and non-U.S. tax considerations of purchasing, owning and disposing of our common stock.
127
UNDERWRITING
Stifel, Nicolaus & Company, Incorporated and Canaccord Genuity Inc. are acting as representatives of the underwriters named below. Subject to the terms and conditions set forth in an underwriting agreement dated the date of this prospectus, each of the underwriters named below has severally agreed to purchase from us the aggregate number of shares of common stock set forth opposite their respective names below:
Name | Number of Shares |
Stifel, Nicolaus & Company, Incorporated | |
Canaccord Genuity Inc. | |
Cantor Fitzgerald & Co. | |
Total | |
The underwriting agreement provides that the obligations of the several underwriters are subject to various conditions, including approval of legal matters by counsel. The nature of the underwriters' obligations commits them to purchase and pay for all of the shares of common stock listed above if any are purchased. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
The underwriters expect to deliver the shares of common stock to purchasers on or about , 2014.
Over-Allotment Option
We have granted a 30-day over-allotment option to the underwriters to purchase up to a total of additional shares of our common stock from us at the initial public offering price, less the underwriting discount payable by us, as set forth on the cover page of this prospectus. If the underwriters exercise this option in whole or in part, then each of the underwriters will be separately committed, subject to the conditions described in the underwriting agreement, to purchase the additional shares of our common stock in proportion to their respective commitments set forth in the table above.
Determination of Offering Price
Prior to this offering, there has been no public market for our common stock. The initial public offering price will be determined through negotiations between us and the representatives. In addition to currently prevailing general conditions in the equity securities markets, including current market valuations of publicly traded companies considered comparable to our company, the factors to be considered in determining the initial public offering price will include:
• | our results of operations; |
• | our current financial condition; |
• | our future prospects; |
• | our management; |
• | our markets; |
• | the economic conditions in and future prospects for the industry in which we compete; and |
• | other factors we deem relevant. |
We cannot assure you that an active or orderly trading market will develop for our common stock or that our common stock will trade in the public markets subsequent to this offering at or above the initial public offering price.
Commissions and Discounts
The underwriters propose to offer the shares of common stock directly to the public at the initial public offering price set forth on the cover page of this prospectus, and at this price less a concession not in excess of $ per share of common stock to other dealers specified in a master agreement among underwriters who are members of
128
the Financial Industry Regulatory Authority, Inc. After this offering, the offering price, concessions and other selling terms may be changed by the underwriters. Our common stock is offered subject to receipt and acceptance by the underwriters and to the other conditions, including the right to reject orders in whole or in part.
The following table summarizes the compensation to be paid to the underwriters by us and the proceeds, before expenses, payable to us:
Total | |||||
Without | With | ||||
Per Share | Over-Allotment | Over-Allotment | |||
Public offering price | $ | $ | $ | ||
Underwriting discount | $ | $ | $ | ||
Proceeds, before expenses, to us | $ | $ | $ | ||
We estimate that the total expenses of this offering, excluding underwriting discounts and commissions, will be $ million, all of which will be paid by us. We have also agreed to reimburse the underwriters for certain of their expenses totaling approximately $ as set forth in the underwriting agreement.
Indemnification of Underwriters
We will indemnify the underwriters against some civil liabilities, including liabilities under the Securities Act and liabilities arising from breaches of our representations and warranties contained in the underwriting agreement. If we are unable to provide this indemnification, we will contribute to payments the underwriters may be required to make in respect of those liabilities.
No Sales of Similar Securities
The underwriters will require all of our directors and officers and substantially all of our stockholders to agree not to offer, sell, agree to sell, directly or indirectly, or otherwise dispose of any shares of common stock or any securities convertible into or exchangeable for shares of common stock without the prior written consent of Stifel, Nicolaus & Company, Incorporated and Canaccord Genuity Inc. for a period of 180 days after the date of this prospectus, subject to specified limited exceptions. Stifel, Nicolaus & Company, Incorporated and Canaccord Genuity Inc., in their sole discretion, may release any of the securities subject to these agreements at any time, which, in the case of officers and directors, shall be with notice.
We have agreed that for a period of 180 days after the date of this prospectus, we will not, without the prior written consent of Stifel, Nicolaus & Company, Incorporated and Canaccord Genuity Inc., offer, sell or otherwise dispose of any shares of common stock, except for the shares of common stock offered in this offering, the shares of common stock issuable upon exercise of options, warrants and convertible securities outstanding on the date of this prospectus and other specified limited exceptions.
NASDAQ Global Market Listing
We have applied to list our common stock on the NASDAQ Global Market under the symbol "AGMX."
Short Sales, Stabilizing Transactions and Penalty Bids
In order to facilitate this offering, persons participating in this offering may engage in transactions that stabilize, maintain, or otherwise affect the price of our common stock during and after this offering. Specifically, the underwriters may engage in the following activities in accordance with the rules of the Securities and Exchange Commission.
Short sales
Short sales involve the sales by the underwriters of a greater number of shares than they are required to purchase in the offering. Covered short sales are short sales made in an amount not greater than the underwriters' over-allotment option to purchase additional shares from us in this offering. The underwriters may close out any covered short position by either exercising their over-allotment option to purchase shares or purchasing shares in the open market. In determining the source of shares to close out the covered short position, the underwriters will
129
consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the over-allotment option. Naked short sales are any short sales in excess of such over-allotment option. The underwriters must close out any naked short position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of the common stock in the open market after pricing that could adversely affect investors who purchase in this offering.
Stabilizing transactions
The underwriters may make bids for or purchases of the shares for the purpose of pegging, fixing, or maintaining the price of the shares, so long as stabilizing bids do not exceed a specified maximum.
Penalty bids
If the underwriters purchase shares in the open market in a stabilizing transaction or syndicate covering transaction, they may reclaim a selling concession from the underwriters and selling group members who sold those shares as part of this offering. Stabilization and syndicate covering transactions may cause the price of the shares to be higher than it would be in the absence of these transactions. The imposition of a penalty bid might also have an effect on the price of the shares if it discourages presales of the shares.
The transactions above may occur on the NASDAQ Global Market or otherwise. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of the shares. If these transactions are commenced, they may be discontinued without notice at any time.
Discretionary Sales
The underwriters have informed us that they do not expect to confirm sales of common stock offered by this prospectus to accounts over which they exercise discretionary authority without obtaining the specific approval of the account holder.
Electronic Distribution
A prospectus in electronic format may be made available on the internet sites or through other online services maintained by one or more of the underwriters participating in this offering, or by their affiliates. Other than the prospectus in electronic format, the information on any underwriter's website and any information contained in any other website maintained by an underwriter is not part of the prospectus or the registration statement of which this prospectus forms a part, has not been approved or endorsed by us or any underwriter in its capacity as underwriter and should not be relied upon by investors.
Relationships
The underwriters and their respective affiliates are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, principal investment, hedging, financing and brokerage activities. Certain of the underwriters and their affiliates have in the past provided, and may in the future from time to time provide, investment banking and other financing and banking services to us, for which they have in the past received, and may in the future receive, customary fees and reimbursement for their expenses. In the ordinary course of their various business activities, the underwriters and their respective affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments including bank loans for their own account and for the accounts of their customers and may at any time hold long and short positions in such securities and instruments. Such investment and securities activities may involve our securities and instruments.
Notice to Residents of the European Economic Area
In relation to each member state of the European Economic Area that has implemented the Prospectus Directive (each, a relevant member state), with effect from and including the date on which the Prospectus Directive is implemented in that relevant member state (the relevant implementation date), an offer of securities described in this prospectus may not be made to the public in that relevant member state other than:
• | to any legal entity that is authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities; |
130
• | to any legal entity that has two or more of (i) an average of at least 250 employees during the last financial year; (ii) a total balance sheet of more than €43,000,000 and (iii) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts; |
• | to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive) subject to obtaining the prior consent of the representatives; or |
• | in any other circumstances that do not require the publication of a prospectus pursuant to Article 3 of the Prospectus Directive, |
provided that no such offer of securities shall require us or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Directive. For purposes of this provision, the expression an "offer of securities to the public" in any relevant member state means the communication in any form and by any means of sufficient information on the terms of the offer and the securities to be offered so as to enable an investor to decide to purchase or subscribe the securities, as the expression may be varied in that member state by any measure implementing the Prospectus Directive in that member state, and the expression "Prospectus Directive" means Directive 2003/71/EC and includes any relevant implementing measure in each relevant member state.
We have not authorized and do not authorize the making of any offer of securities through any financial intermediary on their behalf, other than offers made by the underwriters with a view to the final placement of the securities as contemplated in this prospectus. Accordingly, no purchaser of the securities, other than the underwriters, is authorized to make any further offer of the securities on behalf of us or the underwriters.
Notice to Residents of the United Kingdom
This prospectus is only being distributed to, and is only directed at, persons in the United Kingdom that are qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive (Qualified Investors) that are also (i) investment professionals falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005 (the Order) or (ii) high net worth entities, and other persons to whom it may lawfully be communicated, falling within Article 49(2)(a) to (d) of the Order (all such persons together being referred to as relevant persons). This prospectus and its contents are confidential and should not be distributed, published or reproduced (in whole or in part) or disclosed by recipients to any other persons in the United Kingdom. Any person in the United Kingdom that is not a relevant person should not act or rely on this document or any of its contents.
Notice to Residents of France
This prospectus has not been prepared in the context of a public offering of financial securities in France within the meaning of Article L.411-1 of the French Code Monétaire et Financier and Title I of Book II of the Reglement Général of the Autorité des marchés financiers (the AMF) and therefore has not been and will not be filed with the AMF for prior approval or submitted for clearance to the AMF. Consequently, the shares of our common stock may not be, directly or indirectly, offered or sold to the public in France and offers and sales of the shares of our common stock may only be made in France to qualified investors (investisseurs qualifiés) acting for their own account, as defined in and in accordance with Articles L.411-2 and D.411-1 to D.411-4, D.734-1, D.744-1, D.754-1 and D.764-1 of the French Code Monétaire et Financier. Neither this prospectus nor any other offering material may be released, issued or distributed to the public in France or used in connection with any offer for subscription on sale of the shares of our common stock to the public in France. The subsequent direct or indirect retransfer of the shares of our common stock to the public in France may only be made in compliance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 through L.621-8-3 of the French Code Monétaire et Financier.
Notice to Residents of Germany
Each person who is in possession of this prospectus is aware of the fact that no German securities prospectus (wertpapierprospekt) within the meaning of the securities prospectus act (wertpapier-prospektgesetz, the act) of the federal republic of Germany has been or will be published with respect to the shares of our common stock. In particular, each underwriter has represented that it has not engaged and has agreed that it will not engage in a public offering in the federal republic of Germany (ôffertliches angebot) within the meaning of the act with respect to any of the shares of our common stock otherwise than in accordance with the act and all other applicable legal and regulatory requirements.
131
Notice to Residents of Switzerland
The securities which are the subject of the offering contemplated by this prospectus may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange, or SIX, or on any other stock exchange or regulated trading facility in Switzerland. This prospectus has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. None of this prospectus or any other offering or marketing material relating to the securities or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
None of this prospectus or any other offering or marketing material relating to the offering, us or the securities have been or will be filed with or approved by any Swiss regulatory authority. In particular, this prospectus will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA and the offer of securities has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes, or CISA. The investor protection afforded to acquirers of interests in collective investment schemes under the CISA does not extend to acquirers of the securities.
Notice to Residents of the Netherlands
The offering of the shares of our common stock is not a public offering in The Netherlands. The shares of our common stock may not be offered or sold to individuals or legal entities in The Netherlands unless (i) a prospectus relating to the offer is available to the public, which has been approved by the Dutch Authority for the Financial Markets (Autoriteit Financiële Markten) or by the competent supervisory authority of another state that is a member of the European Union or party to the Agreement on the European Economic Area, as amended or (ii) an exception or exemption applies to the offer pursuant to Article 5:3 of The Netherlands Financial Supervision Act (Wet op het financieel toezicht) or Article 53 paragraph 2 or 3 of the Exemption Regulation of the Financial Supervision Act, for instance due to the offer targeting exclusively "qualified investors" (gekwalificeerde beleggers) within the meaning of Article 1:1 of The Netherlands Financial Supervision Act.
Notice to Residents of Japan
The underwriters will not offer or sell any of the shares of our common stock directly or indirectly in Japan or to, or for the benefit of, any Japanese person or to others, for re-offering or re-sale directly or indirectly in Japan or to any Japanese person, except in each case pursuant to an exemption from the registration requirements of, and otherwise in compliance with, the Financial Instruments and Exchange Law of Japan and any other applicable laws and regulations of Japan. For purposes of this paragraph, "Japanese person" means any person resident in Japan, including any corporation or other entity organized under the laws of Japan.
Notice to Residents of Hong Kong
The underwriters and each of their affiliates have not (i) offered or sold, and will not offer or sell, in Hong Kong, by means of any document, any shares of our common stock other than (a) to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571) of Hong Kong and any rules made under that Ordinance or (b) in other circumstances which do not result in the document being a "prospectus" as defined in the Companies Ordinance (Cap. 32) of Hong Kong or which do not constitute an offer to the public within the meaning of that Ordinance; and (ii) issued or had in its possession for the purposes of issue, and will not issue or have in its possession for the purposes of issue, whether in Hong Kong or elsewhere any advertisement, invitation or document relating to the shares of our common stock which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the securities laws of Hong Kong) other than with respect to the shares of our common stock which are or are intended to be disposed of only to persons outside Hong Kong or only to "professional investors" within the meaning of the Securities and Futures Ordinance and any rules made under that Ordinance. The contents of this document have not been reviewed by any regulatory authority in Hong Kong. You are advised to exercise caution in relation to the offer. If you are in any doubt about any of the contents of this document, you should obtain independent professional advice.
132
Notice to Residents of Singapore
This document has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this document and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of shares of our common stock may not be circulated or distributed, nor may shares of our common stock be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore (the Securities and Futures Act), (ii) to a relevant person, or any person pursuant to Section 275(1A), and in accordance with the conditions, specified in Section 275 of the Securities and Futures Act or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the Securities and Futures Act.
Where shares of our common stock are subscribed or purchased under Section 275 by a relevant person, which is:
(a) | a corporation (which is not an accredited investor) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or |
(b) | a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary is an accredited investor, shares, debentures and units of shares and debentures of that corporation or the beneficiaries’ rights and interest in that trust shall not be transferable for six months after that corporation or that trust has acquired the shares of our common stock under Section 275 except: |
(1) to an institutional investor or to a relevant person, or to any person pursuant to an offer that is made on terms that such rights or interest are acquired at a consideration of not less than $200,000 (or its equivalent in a foreign currency) for each transaction, whether such amount is to be paid for in cash or by exchange of securities or other assets;
(2) where no consideration is given for the transfer; or
(3) by operation of law.
133
LEGAL MATTERS
The validity of the shares of common stock offered by us under this prospectus will be passed upon for us by Bingham McCutchen LLP, Costa Mesa, California. As of the date of this prospectus, an investment partnership that includes certain Bingham McCutchen LLP attorneys holds 87,754 shares of our preferred stock that will be converted into an aggregate of 40,903 shares of our common stock upon the completion of this offering. This investment partnership also holds warrants for the purchase of 4,686 shares of our common stock. The underwriters are represented by Cooley LLP, San Diego, California.
EXPERTS
Marcum LLP, independent registered public accounting firm, has audited our financial statements at and for the years ending December 31, 2013 and 2012, as set forth in their report. We have included our financial statements in the prospectus and elsewhere in the registration statement in reliance on Marcum LLP's report, given their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 (including the exhibits, schedules and amendments to the registration statement) under the Securities Act with respect to the shares of common stock offered by this prospectus. This prospectus does not contain all of the information set forth in the registration statement. For further information about us and the new shares of common stock to be sold in this offering, we refer you to the registration statement. Statements contained in this prospectus as to the contents of any contract, agreement or other document to which we make reference are not necessarily complete. In each instance, we refer you to the copy of such contract, agreement or other document filed as an exhibit to the registration statement, each such statement being qualified in all respects by the more complete description of the matter involved.
Upon completion of this offering, we will become subject to the reporting and information requirements of the Exchange Act, and, as a result, will file periodic and current reports, proxy statements, and other information with the SEC. You may read and copy this information at the Public Reference Room of the SEC located at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the Public Reference Room. Copies of all or any part of the registration statement may be obtained from the SEC's offices upon payment of fees prescribed by the SEC. The SEC maintains an Internet site that contains periodic and current reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of the SEC’s website is http://www.sec.gov.
134
AutoGenomics, Inc.
INDEX TO FINANCIAL STATEMENTS
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Audit Committee of the
Board of Directors and Stockholders
of AutoGenomics, Inc.
We have audited the accompanying balance sheets of AutoGenomics, Inc. (the “Company”) as of December 31, 2012 and 2013, and the related statements of operations, changes in convertible preferred stock and stockholders’ deficit and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of AutoGenomics, Inc. as of December 31, 2012 and 2013, and the results of its operations and its cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America.
/s/ Marcum LLP
Irvine, CA
August 8, 2014
F-2
AutoGenomics, Inc.
Balance Sheets
(in thousands, except share and per share data)
September 30, | December 31, | ||||||||||
2014 | 2013 | 2012 | |||||||||
Assets | (unaudited) | ||||||||||
Current assets: | |||||||||||
Cash and cash equivalents | $ | 556 | $ | 2,053 | $ | 332 | |||||
Accounts receivable (net of allowances of $6,334, $6,150 and $136) | 8,210 | 2,975 | 2,962 | ||||||||
Inventory, net | 3,041 | 2,926 | 2,721 | ||||||||
Unbilled accounts receivable, net | 683 | — | — | ||||||||
Prepaid and other current assets | 229 | 313 | 235 | ||||||||
Total current assets | 12,719 | 8,267 | 6,250 | ||||||||
Property, equipment and improvements, net | 1,762 | 1,474 | 1,234 | ||||||||
Deferred costs | 694 | 454 | 2,366 | ||||||||
Other long-term assets | 197 | 197 | 197 | ||||||||
Total assets | $ | 15,372 | $ | 10,392 | $ | 10,047 | |||||
Liabilities and stockholders’ deficit | |||||||||||
Current liabilities: | |||||||||||
Accounts payable | $ | 9,961 | $ | 10,056 | $ | 10,110 | |||||
Accrued expenses | 3,322 | 1,410 | 1,931 | ||||||||
Related party promissory notes, current portion, net | 2,642 | 2,602 | — | ||||||||
Senior promissory notes, current portion, net | 1,047 | — | 2,447 | ||||||||
Subordinated promissory notes, current portion, net | 706 | 676 | 624 | ||||||||
Preferred stock warrant liabilities | — | 385 | 438 | ||||||||
Common stock warrant liabilities | 42 | 32 | 25 | ||||||||
Other current liabilities | 248 | 477 | 380 | ||||||||
Total current liabilities | 17,968 | 15,638 | 15,955 | ||||||||
Deferred rent and other long-term liabilities | 3,455 | 3,259 | 2,675 | ||||||||
Related party promissory notes, long-term portion, net | 177 | 60 | 1,064 | ||||||||
Subordinated promissory notes, long-term portion, net | 16,602 | 15,657 | 13,394 | ||||||||
Commitments and contingencies | |||||||||||
Convertible preferred stock, $0.01 par value, 30,000,000 shares authorized: | |||||||||||
Series A Convertible Preferred Stock, 1,589,600 shares authorized; 1,579,227 shares issued and outstanding at September 30, 2014, December 31, 2013 and 2012; liquidation preference in the aggregate of $4,990 at September 30, 2014, December 31, 2013 and December 31, 2012 | 4,990 | 4,990 | 4,990 | ||||||||
Series B Convertible Preferred Stock, 4,515,858 shares authorized; 4,468,369 shares issued and outstanding at September 30, 2014, December 31, 2013 and 2012; liquidation preference in the aggregate of $12,288 at September 30, 2014, December 31, 2013 and December 31, 2012 | 12,288 | 12,288 | 12,288 | ||||||||
Series C Convertible Preferred Stock, 6,850,000 shares authorized; 6,417,680 shares issued and outstanding at September 30, 2014, December 31, 2013 and 2012; liquidation preference in the aggregate of $17,647 at September 30, 2014, December 31, 2013 and December 31, 2012 | 17,647 | 17,647 | 17,647 | ||||||||
Series D Convertible Preferred Stock, 3,423,258 shares authorized; 3,423,258 shares issued and outstanding at September 30, 2014, December 31, 2013 and 2012; liquidation preference in the aggregate of $11,126 at September 30, 2014, December 31, 2013 and December 31, 2012 | 11,126 | 11,126 | 11,126 | ||||||||
Series E Convertible Preferred Stock, 5,500,000 shares authorized; 732,555 shares issued and outstanding at September 30, 2014, December 31, 2013 and 2012; liquidation preference in the aggregate of $1,381 at September 30, 2014, December 31, 2013 and December 31, 2012 | 1,381 | 1,381 | 1,381 | ||||||||
Series NC Convertible Preferred Stock, 4,287,074 shares authorized; 4,287,074 shares issued and outstanding at September 30, 2014, December 31, 2013 and 2012; liquidation preference in the aggregate $5,733 at September 30, 2014, December 31, 2013 and December 31, 2012 | 5,733 | 5,733 | 5,733 | ||||||||
Stockholders’ deficit: | |||||||||||
Common stock, $0.01 par value, 220,000,000 shares authorized; 3,747,319, 3,742,369 and 2,856,083 issued and outstanding at September 30, 2014, December 31, 2013 and 2012, respectively | 114 | 113 | 87 | ||||||||
Additional paid-in-capital | 28,053 | 25,697 | 19,832 | ||||||||
Accumulated deficit | (104,162 | ) | (103,197 | ) | (96,125 | ) | |||||
Total stockholders’ deficit | (75,995 | ) | (77,387 | ) | (76,206 | ) | |||||
Total liabilities and stockholders’ deficit | $ | 15,372 | $ | 10,392 | $ | 10,047 | |||||
See accompanying notes to financial statements
F-3
AutoGenomics, Inc.
Statements of Operations
(in thousands, except share and per share data)
Nine Months Ended September 30, | Years Ended December 31, | ||||||||||||||
2014 | 2013 | 2013 | 2012 | ||||||||||||
(unaudited) | |||||||||||||||
Product revenue, net | $ | 9,022 | $ | 13,044 | $ | 16,938 | $ | 18,307 | |||||||
Service revenue, net | 9,703 | 229 | 1,145 | 102 | |||||||||||
Net revenue | 18,725 | 13,273 | 18,083 | 18,409 | |||||||||||
Cost of sales | 3,968 | 4,269 | 5,726 | 7,147 | |||||||||||
Cost of services | 2,677 | 1,530 | 2,032 | — | |||||||||||
Gross profit | 12,080 | 7,474 | 10,325 | 11,262 | |||||||||||
Operating expenses: | |||||||||||||||
Research and development | 1,540 | 1,715 | 2,306 | 2,355 | |||||||||||
General and administrative | 3,308 | 7,210 | 8,302 | 4,715 | |||||||||||
Sales and marketing | 5,334 | 1,898 | 2,998 | 2,269 | |||||||||||
Total operating expenses | 10,182 | 10,823 | 13,606 | 9,339 | |||||||||||
Income/(loss) from operations | 1,898 | (3,349 | ) | (3,281 | ) | 1,923 | |||||||||
Interest expense, net | (1,361 | ) | (1,186 | ) | (1,820 | ) | (2,602 | ) | |||||||
Loss on extinguishment of debt | (1,872 | ) | (140 | ) | (140 | ) | (1,996 | ) | |||||||
Other income/(expense), net | (5 | ) | (2,095 | ) | (1,877 | ) | 5 | ||||||||
Change in the fair value of stock warrant liabilities | 375 | 5 | 46 | 163 | |||||||||||
Net income/(loss) | $ | (965 | ) | $ | (6,765 | ) | $ | (7,072 | ) | $ | (2,507 | ) | |||
Net income/(loss) per share attributable to common stockholders | |||||||||||||||
Basic and diluted | $ | (0.26 | ) | $ | (2.22 | ) | $ | (2.21 | ) | $ | (0.95 | ) | |||
Shares to compute net income/(loss) per share attributable to common stockholders | |||||||||||||||
Basic and diluted | 3,743,084 | 3,042,029 | 3,200,286 | 2,652,275 | |||||||||||
See accompanying notes to financial statements
F-4
AutoGenomics, Inc.
Statements of Convertible Preferred Stock and Stockholders’ Deficit
(in thousands, except share and per share data)
Series A | Series B | Series C | Series D | Series E | Series NC | |||||||||||||||||||||||||||||||||||||||
Convertible | Convertible | Convertible | Convertible | Convertible | Convertible | Common | ||||||||||||||||||||||||||||||||||||||
Preferred Stock | Preferred Stock | Preferred Stock | Preferred Stock | Preferred Stock | Preferred Stock | Stock | Additional | Total | ||||||||||||||||||||||||||||||||||||
Issued | Issued | Issued | Issued | Issued | Issued | Issued | paid-in | Accumulated | Stockholders’ | |||||||||||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | capital | Deficit | Deficit | ||||||||||||||||||||||||||||
Balance at December 31, 2011 | 1,579,227 | $ | 4,990 | 4,468,369 | $ | 12,288 | 6,417,680 | $ | 17,647 | 3,423,258 | $ | 11,126 | 685,555 | $ | 1,292 | — | $ | — | 2,633,833 | $ | 81 | $ | 14,288 | $ | (93,618 | ) | $ | (79,249 | ) | |||||||||||||||
Issuance of common stock for exercise of stock options | — | — | — | — | — | — | — | — | — | — | — | — | 192,250 | 5 | 107 | — | 112 | |||||||||||||||||||||||||||
Stock-based compensation related to issuance of stock options to employees | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 737 | — | 737 | |||||||||||||||||||||||||||
Issuance of Series E convertible preferred stock at $2.75 in conjunction with conversion of debt and related accrued interest, net of issuance costs of $12,186 | — | — | — | — | — | — | — | — | 47,000 | 89 | — | — | — | — | 40 | — | 40 | |||||||||||||||||||||||||||
Issuance of Series NC convertible preferred stock at $1.75 and common stock warrants in conjunction with conversion of debt and related accrued interest | — | — | — | — | — | — | — | — | — | — | 4,287,074 | 5,733 | — | — | 1,769 | — | 1,769 | |||||||||||||||||||||||||||
Issuance of stock in connection with premises lease negotiations | — | — | — | — | — | — | — | — | — | — | — | — | 30,000 | 1 | 165 | — | 166 | |||||||||||||||||||||||||||
Issuance of common stock warrants in connection with subordinated notes | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 316 | — | 316 | |||||||||||||||||||||||||||
Issuance of common stock warrants in connection with financing consulting | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 103 | — | 103 | |||||||||||||||||||||||||||
Modification of warrants | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 2,307 | 2,307 | ||||||||||||||||||||||||||||
Net loss and comprehensive loss | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | (2,507 | ) | (2,507 | ) | |||||||||||||||||||||||||
Balance at December 31, 2012 | 1,579,227 | $ | 4,990 | 4,468,369 | $ | 12,288 | 6,417,680 | $ | 17,647 | 3,423,258 | $ | 11,126 | 732,555 | $ | 1,381 | 4,287,074 | $ | 5,733 | 2,856,083 | $ | 87 | $ | 19,832 | $ | (96,125 | ) | $ | (76,206 | ) | |||||||||||||||
Issuance of common stock | — | — | — | — | — | — | — | — | — | — | — | — | 573,888 | 16 | 3,865 | — | 3,881 | |||||||||||||||||||||||||||
Issuance of common stock for exercise of stock options | — | — | — | — | — | — | — | — | — | — | — | — | 249,698 | 8 | 211 | — | 219 | |||||||||||||||||||||||||||
Stock-based compensation related to issuance of stock options to employees | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 1,233 | — | 1,233 | |||||||||||||||||||||||||||
Issuance of common stock to consultants | — | — | — | — | — | — | — | — | — | — | — | — | 62,700 | 2 | 416 | — | 418 | |||||||||||||||||||||||||||
Modification of warrants | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 140 | — | 140 | |||||||||||||||||||||||||||
Net loss and comprehensive loss | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | (7,072 | ) | (7,072 | ) | |||||||||||||||||||||||||
Balance at December 31, 2013 | 1,579,227 | $ | 4,990 | 4,468,369 | $ | 12,288 | 6,417,680 | $ | 17,647 | 3,423,258 | $ | 11,126 | 732,555 | $ | 1,381 | 4,287,074 | $ | 5,733 | 3,742,369 | $ | 113 | $ | 25,697 | $ | (103,197 | ) | $ | (77,387 | ) | |||||||||||||||
Issuance of common stock | — | — | — | — | — | — | — | — | — | — | — | — | 4,950 | 1 | 3 | — | 4 | |||||||||||||||||||||||||||
Stock-based compensation related to issuance of stock options to employees | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 481 | — | 481 | |||||||||||||||||||||||||||
Issuance of common stock warrants in connection with subordinated notes | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 1,872 | — | 1,872 | |||||||||||||||||||||||||||
Net loss and comprehensive loss | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | (965 | ) | (965 | ) | |||||||||||||||||||||||||
Balance at September 30, 2014 (unaudited) | 1,579,227 | $ | 4,990 | 4,468,369 | $ | 12,288 | 6,417,680 | $ | 17,647 | 3,423,258 | $ | 11,126 | 732,555 | $ | 1,381 | 4,287,074 | $ | 5,733 | 3,747,319 | $ | 114 | $ | 28,053 | $ | (104,162 | ) | $ | (75,995 | ) | |||||||||||||||
F-5
AutoGenomics, Inc.
Statements of Cash Flows
(in thousands)
Nine months ended September 30, | Years ended December 31, | ||||||||||||||
2014 | 2013 | 2013 | 2012 | ||||||||||||
(unaudited) | |||||||||||||||
Operating Activities | |||||||||||||||
Net income/(loss) | $ | (965 | ) | $ | (6,765 | ) | $ | (7,072 | ) | $ | (2,507 | ) | |||
Adjustments to reconcile net loss to net cash and cash equivalents provided by/(used in) operating activities: | |||||||||||||||
Depreciation and amortization | 420 | 423 | 569 | 600 | |||||||||||
Amortization of debt discount/premium | 30 | (7 | ) | 31 | 522 | ||||||||||
Amortization of deferred financing costs | 178 | 185 | 257 | 60 | |||||||||||
Expense of deferred IPO costs | — | 1,873 | — | — | |||||||||||
Allowance for doubtful accounts | 184 | 4,444 | 6,014 | 84 | |||||||||||
Allowance for excess and obsolete inventory | (3 | ) | (52 | ) | (456 | ) | 219 | ||||||||
Employee stock-based compensation | 481 | 704 | 1,233 | 737 | |||||||||||
Nonemployee stock-based compensation | — | — | 418 | 103 | |||||||||||
Change in fair value of stock warrant liabilities | (375 | ) | (5 | ) | (46 | ) | (164 | ) | |||||||
Non cash interest expense | 1,162 | 725 | 1,502 | 2,020 | |||||||||||
Loss on extinguishment of debt | 1,872 | 140 | 140 | 1,996 | |||||||||||
Deferred rent | 195 | 433 | 620 | 455 | |||||||||||
Interest paid on promissory notes | (10 | ) | (267 | ) | (287 | ) | (176 | ) | |||||||
Changes in operating assets/liabilities | |||||||||||||||
Accounts receivable | (6,101 | ) | (3,997 | ) | (6,028 | ) | (1,181 | ) | |||||||
Inventory | (112 | ) | 145 | 251 | (1,245 | ) | |||||||||
Prepaid and other current assets | (333 | ) | (268 | ) | 1,577 | (2,546 | ) | ||||||||
Accounts payable | 853 | 1,808 | — | 927 | |||||||||||
Other current liabilities and accrued expenses | 1,734 | 427 | (497 | ) | 750 | ||||||||||
Cash provided by/(used in) operating activities | (790 | ) | (54 | ) | (1,774 | ) | 654 | ||||||||
Investing activities | |||||||||||||||
Purchase of property and equipment | (708 | ) | (519 | ) | (809 | ) | (355 | ) | |||||||
Cash used in investing activities | (708 | ) | (519 | ) | (809 | ) | (355 | ) | |||||||
Financing activities | |||||||||||||||
Proceeds from the issuance of common stock | 1 | 4,176 | 4,099 | 113 | |||||||||||
Proceeds from the issuance of promissory notes | — | 2,750 | 3,000 | 1,769 | |||||||||||
Payment of promissory notes principal | — | (2,652 | ) | (2,795 | ) | (1,910 | ) | ||||||||
Net cash provided by/(used in) financing activities | 1 | 4,274 | 4,304 | (28 | ) | ||||||||||
Increase/(decrease) in cash and cash equivalents | (1,497 | ) | 3,701 | 1,721 | 271 | ||||||||||
Cash and cash equivalents at the beginning of the period | 2,053 | 332 | 332 | 61 | |||||||||||
Cash and cash equivalents at the end of the period | $ | 556 | $ | 4,033 | $ | 2,053 | $ | 332 | |||||||
Nine months ended September 30, | Years ended December 31, | ||||||||||||||
2014 | 2013 | 2013 | 2012 | ||||||||||||
(unaudited) | |||||||||||||||
Supplemental disclosure of cash flow information: | |||||||||||||||
Interest paid | $ | 10 | $ | 268 | $ | 287 | $ | 6 | |||||||
Income taxes paid | $ | 23 | $ | 16 | $ | 44 | $ | 4 | |||||||
Non-cash investing and financing activities | |||||||||||||||
Change in classification of reagent access plan analyzers to/from inventory and property and equipment | $ | 366 | $ | 189 | $ | 334 | $ | 47 | |||||||
Modifications to warrants and notes payable | $ | — | $ | 140 | $ | 140 | $ | 2,307 | |||||||
Issuance of common stock warrants in connection with notes payable financing | $ | 1,872 | $ | — | $ | — | $ | 316 | |||||||
Issuance of common stock warrants in connection with the conversion of notes payable to Series NC convertible preferred stock | $ | — | $ | — | $ | — | $ | 1,769 | |||||||
Surrender of notes payable and related interest in exchange for Series NC convertible preferred stock | $ | — | $ | — | $ | — | $ | 5,733 | |||||||
Surrender of accounts payable in exchange for Series E convertible preferred stock and common stock warrants | $ | — | $ | — | $ | — | $ | 129 | |||||||
Issuance of common stock in exchange for rent expense | $ | — | $ | — | $ | — | $ | 166 | |||||||
Issuance of common stock in exchange for consulting expense | $ | — | $ | — | $ | 418 | $ | — | |||||||
Issuance of note payable in exchange for past due rent expense | $ | 997 | $ | — | $ | — | $ | — | |||||||
Refinance of existing note payable | $ | 2,404 | $ | — | $ | — | $ | — | |||||||
See accompanying notes to financial statements
F-6
AutoGenomics, Inc.
Notes to Financial Statements
1. Organization, liquidity and management’s plan and summary of significant accounting policies
Organization and business
AutoGenomics, Inc., or the Company, incorporated in California and commenced its operations in 1999, and in October 2008, the Company reincorporated in Delaware. The Company designs, develops, manufactures and markets a fully integrated molecular diagnostics platform. The Company's platform consists of a family of multiplexing INFINITI analyzers, an extensive and expanding menu of genetic tests, and proprietary microarrays, reagent modules and other related consumables. The Company's menu of genetic tests is focused primarily on the growing markets of personalized medicine, women's health, infectious diseases and genetic disorders. The Company's genetic tests are performed on our INFINITI analyzers using our proprietary BioFilmChip microarrays, related Intellipac Reagent Management Modules and other related consumables.
Liquidity and management’s plan
The Company did not achieve positive cash flow from operations in the nine months ended September 30, 2014 or the 12 months ended December 31, 2013 and the Company’s current liabilities were greater than current assets as of those periods and December 31, 2012, and there can be no guarantee that the Company will generate positive cash flow in the future. As a result, the Company is continuing to explore alternatives for additional sources of cash flows and financing to fund the Company’s operations. However, there can be no assurance that such cash flows will continue or that financing will be available to the Company if and when needed or that such financing would be available under favorable terms. If the Company is unable to obtain sufficient funding or generate sufficient cash from operations, it may be required to significantly curtail its planned operations, which may have a material, adverse impact on the Company.
Basis of presentation
The accompanying financial statements have been prepared in accordance with accounting principles generally
accepted in the United States of America (“GAAP”).
The financial statements as of September 30, 2014 and for the nine months ended September 30, 2014 and 2013 are unaudited. The unaudited financial statements have been prepared on the same basis as the audited financial statements and, in the opinion of management, include all adjustments, consisting of normal recurring adjustments, considered necessary to fairly state the financial information set forth therein, in accordance with GAAP.
The results of operations for the audited years ended December 31, 2013 and 2012 and the unaudited interim financial statements for the nine months ended September 30, 2014 and 2013 are not necessarily indicative of the results which may be reported for any other interim period or for the year ended December 31, 2014.
Certain reclassifications have been made to prior years’ financial statements to conform to the 2014 presentation, which did not affect previously reported results of operations.
Use of estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and disclosures made to the accompanying notes to the financial statements. The Company’s critical accounting policies that involve significant judgment and estimates include revenue recognition, contractual rate allowance, allowance for uncollectable accounts, warranty reserve, deferred tax valuation allowance, convertible preferred stock warrant liabilities, stock-based compensation, common stock valuations, useful lives of property and equipment and allowance for excess and obsolete inventory. Actual results could differ from those estimates and assumptions.
F-7
Cash and cash equivalents
The Company considers all highly liquid investments with an original maturity of three months or less when purchased to be cash equivalents.
Concentration of credit risk
Financial instruments, which potentially subject the Company to concentrations of credit risk, consist primarily of cash and cash equivalents and accounts receivable. The Company limits its exposure to credit loss by placing its cash with high credit-quality financial institutions. At times, such deposits may be in excess of insured limits. The Company has not experienced any losses on its deposits of cash and cash equivalents. For the nine months ended September 30, 2014, no customer accounted for more than 10% of revenue, and for the nine months ended September 30, 2013 one customer, Natural Molecular Testing Corporation, or NMTC, accounted for 40% of revenue. For the years ended December 31, 2013 and 2012, the two largest customers each period, NMTC and AI Biotech, or AIB, accounted for 31% and 12%, and 43% and 16% of revenue, respectively.
For the nine months ended September 30, 2014, and year ended December 31, 2013, NMTC accounts receivable balance was 33% and 48% of outstanding accounts receivable, respectively, but was fully allowed for. For the year ended December 31, 2012, the two largest customers each period, AIB and NMTC, accounted for 41% and 30%, respectively, of outstanding accounts receivable balances.
During the nine months ended September 30, 2014 and 2013, and the years ended December 31, 2013 and 2012, sales outside the United States accounted for 14%, 10%, 14% and 9% of revenue, respectively.
Fair value of financial instruments
The carrying amounts shown for the Company’s cash and cash equivalents, accounts receivable, prepaid expenses, accounts payable and accrued expenses approximate their fair value due to the short-term nature of these items. The carrying amount of common stock and convertible preferred stock warrant liabilities represents their respective fair values (see note 5 Common stock and convertible preferred stock warrant liabilities).
The carrying values of notes payable approximate fair values principally because the Company's credit worthiness has not changed significantly since issuing such note. Fair value is defined as the exchange price that would be received for an asset or an exit price paid to transfer a liability in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The current accounting guidance for fair value measurements defines a three-level valuation hierarchy for disclosures as follows:
Level I — Unadjusted quoted prices in active markets for identical assets or liabilities
Level II — Inputs other than quoted prices included within Level I that are observable, unadjusted quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data
Level III — Unobservable inputs that are supported by little or no market activity requiring the Company to develop its own assumptions
The categorization of a financial instrument within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement. The Company’s financial instruments consist of Level I assets and Level III liabilities. Level I assets consist of money market funds. Level III liabilities consist of the convertible preferred stock and common stock warrant liabilities, the fair values of which are measured using the Black-Scholes valuation model. The Company determined that using alternative valuation models, such as a Monte Carlo simulation model, would result in de minimus differences.
The Company may elect to use fair value to measure accounts and loans receivable, available-for-sale and held-to-maturity securities, equity method investments, accounts payable, guarantees and issued debt. Other eligible items
F-8
include firm commitments for financial instruments that otherwise would not be recognized at inception and non-cash warranty obligations where a warrantor is permitted to pay a third party to provide the warranty goods or services. If the use of fair value is elected, any upfront costs and fees related to the item such as debt issuance costs must be recognized in earnings and cannot be deferred. The fair value election is irrevocable and generally made on an instrument-by-instrument basis, even if a company has similar instruments that it elects not to measure based on fair value. Unrealized gains and losses on existing items for which fair value has been elected are reported as a cumulative adjustment to beginning retained earnings and any changes in fair value are recognized in earnings. The Company has elected not to apply the fair value option to its financial assets and liabilities.
Segment
The Company’s business is considered as operating in one segment, molecular diagnostic testing, based upon the Company’s organizational structure, the way in which the operations are managed and evaluated by the chief operating decision maker and the manner in which the Company records its financial information. The Company has two primary revenue streams, product revenues and laboratory service revenues. Service revenues are generated principally from current or identified future product customers, and the laboratory services compliment the sale of the Company's products.
Accounts receivable and allowance for doubtful accounts
The Company performs ongoing credit evaluations of its customers. Accounts receivable are recorded at invoiced amounts, net of the Company’s estimated allowances for doubtful accounts. The allowance for doubtful accounts is based on an assessment of the Company’s ability to collect on customer accounts receivable. The Company regularly reviews the allowance by considering certain factors such as historical experience, industry data, credit quality, age of accounts receivable balances and current economic conditions that may affect a customer’s ability to pay. In cases where the Company is aware of circumstances that may impair a customer’s ability to meet their financial obligations, the Company records a specific allowance against amounts due from the customer and thereby reduces the net recognized receivable to the amount the Company reasonably believes will be collected. The Company writes-off accounts receivable against the allowance when it determines the balance is uncollectible and no longer actively pursues collection of the receivable.
The following table summarizes (in thousands) the activity in the allowance for doubtful accounts receivable during the nine months ended September 30, 2014 and the years ended December 31, 2013 and 2012. Increases to the allowance are reflected as "Charged to costs and expenses." Accounts receivable that have been recovered or written off against the allowance are reflected as "Allowance reductions/write-offs."
For the period ended | Balance at beginning of period | Charged to costs and expenses | Allowance reductions/ write-offs | Balance at end of period | |||||||||||
September 30, 2014 (unaudited) | $ | 6,150 | $ | 211 | $ | (27 | ) | $ | 6,334 | ||||||
December 31, 2013 | $ | 136 | $ | 4,491 | $ | 1,523 | $ | 6,150 | |||||||
December 31, 2012 | $ | 52 | $ | 350 | $ | (266 | ) | $ | 136 | ||||||
In January 2013, the Centers for Medicare & Medicaid Services, or CMS, temporarily suspended reimbursement for certain diagnostic tests while it determined applicable reimbursement rates. This suspension negatively affected the cash flows of several of the Company’s customers including a significant customer of the Company that accounted for 31% and 43% of sales revenue in 2013 and 2012, respectively. In 2013, the customer paid in full all 2012 receivables, but became significantly delinquent on paying 2013 invoices during the first half of 2013. The Company provided an allowance for all remaining receivables from this customer during March and April 2013, recording a combined expense of $4.5 million. In October 2013, this customer filed for Chapter 11 protection.
F-9
Inventory, net
The Company reports its inventories net of its reserve for slow-moving and obsolete parts. Inventories are valued at the lower of cost or market value using the first-in first-out method and include direct labor, materials, and manufacturing overhead. In the event inventories are written down, the new value becomes the cost basis, and cannot be subsequently marked up even with changes in underlying facts or circumstances. The Company periodically reviews inventory for evidence of slow-moving or obsolete parts, and the estimated reserve is based on management’s reviews of inventories on hand, compared to estimates of future usage and sales, and assumptions about the likelihood of obsolescence.
Unbilled accounts receivable
The Company recognizes revenue from laboratory services upon completion of the genetic testing service and when the ordering physician can access the testing results. Services performed but not billed are recorded as revenue and are accounted for as unbilled accounts receivable, net of contractual rate allowances.
Property, equipment and improvements
Property, equipment and improvements includes manufacturing, laboratory, office and computer hardware and software, and is stated at cost and depreciated over the estimated useful lives of the assets (three to seven years) using the straight-line method. Leasehold improvements are stated at cost and amortized over the lesser of the expected remaining lease term or the remaining useful life of the asset.
Equipment placed with customers under the Company’s Reagent Access Plan, or RAP, is recorded at the Company’s cost to manufacture and the related depreciation expense is charged to cost of sales over the equipment’s expected life. INFINITI and INFINITI PLUS analyzers have an expected service life of three years, while INFINITI HTS analyzers have an expected service life of five years.
Deferred costs
Deferred costs are comprised of offering costs related to financing activities, such as private debt placements or initial public offering, or IPO costs, and are amortized over the life of the financing or expensed upon the completion of an event. The company has deferred offering costs related to a planned IPO of $0.7 million for the nine months ended September 30, 2014.
Other long-term assets
Other long-term assets consist of funds on deposit with our vendors of $0.2 million for all periods presented.
Warranty reserves
The Company provides a standard twelve-month warranty on all INFINITI analyzers sold domestically. The cost of warranties is recognized in cost of sales at the time revenue is earned based on the estimated cost to repair over its warranty period. Cost of sales reflects not only estimated warranty expense for equipment sold in the current period, but also any changes in estimated warranty expense for the portion of the aggregate installed base that is under warranty. The Company also estimates a warranty reserve for consumable products. Estimated warranty expense is based on a variety of inputs, including historical performance in the customers’ environment, historical costs incurred in servicing equipment and projected equipment reliability and service costs. Should actual service rates or costs differ from estimates, which are based on historical data and projections of future costs, revisions to the estimated warranty liability would be required. The Company reviews these inputs, at a minimum, on an annual basis. The liability for warranties is included in other current liabilities in the accompanying balance sheets.
Promissory notes
The Company has issued promissory notes pursuant to a number of private placements since 1999. At times these promissory notes were issued with separate warrants to purchase the Company’s common stock. These warrants are treated as equity instruments. The fair value of such warrants was determined using the Black-Scholes valuation
F-10
model and was allocated as a debt discount using the relative fair value allocation method. The discount has been amortized using the effective interest method over the term of the related notes payable.
Stock split
During January 2013 the Company completed a 1-for-0.33 reverse stock split for shares of its common stock. All per share amounts reflect this reverse stock split at September 30, 2014 and December 31, 2013, and retrospectively at December 31, 2012.
Common stock and convertible preferred stock warrant liabilities
The Company has issued freestanding warrants to purchase shares of its common stock that include an anti-dilution provision which provides for the adjustment of the exercise price upon the occurrence of certain events. The Company has classified the fair value of this warrant as a liability on the balance sheet. The Company will continue to adjust the liability for changes in fair value until the earlier of the expiration of this warrant or its exercise, at which time the liability will be reclassified into stockholders’ deficit. The Company records any change in fair value as a component of other income or expense.
The Company has issued freestanding warrants to purchase shares of its convertible preferred stock. The Company has classified the fair value of these warrants as liabilities on the balance sheet as they correspond to the treatment of the preferred stock as temporary equity. The Company will continue to adjust the liability for changes in fair value until the earlier of the expiration of the warrants or their exercise, at which time the liability will be reclassified into stockholders’ deficit. The Company records any change in fair value as a component of other income or expense.
Revenue recognition
The Company’s product revenue is derived primarily from the sales of its genetic testing technology through its INFINITI analyzers and the BioFilmChip, Intellipac Reagent Management Module, and various disposable items consumed by the diagnostic testing process, collectively referred to as consumables. The INFINITI analyzers and all consumables are offered through direct sale in domestic markets and through distributors in international markets. The INFINITI analyzers also are available to domestic customers through the Company’s RAP. In such arrangements, customers receive an analyzer at no direct cost in return for a commitment to purchase minimum quantities of consumables based on an agreed to pricing schedule over the contract period, which is usually two years, and which may be extended at the option of the customer or the Company. The Company reserves the option to cancel the arrangement and repossess the analyzer if minimum quantities of consumables are not purchased. These analyzers are recorded in the Company’s financial records as capitalized assets and are depreciated over an estimated useful life of, typically, three years. When the analyzer is returned to the Company it is placed back into inventory at its then net book value. Analyzers that are returned are subject to a review and refurbishment process designed to enable the return of the analyzer to the field as soon as practicable. Any costs associated with the refurbishment of an analyzer are capitalized and added to the Company’s cost basis in the analyzer. The new basis, including refurbishment costs, either is amortized to cost of sales over the useful life of the analyzer once the refurbished unit is placed with a new customer under the RAP, or is charged to cost of sales upon the sale of the refurbished analyzer. Throughout the term of the placement agreement, the Company retains title to the analyzer.
The Company's service revenue is derived primarily from the CLIA-certified laboratory's genetic testing services, as well as warranty service on INFINITI analyzers and technical and customer services provided to purchased analyzers. The laboratory's testing services are substantially all performed for INFINITI HTS analyzer customers as they await delivery, installation and validation of the analyzer. Laboratory service revenues are reported net of a contractual rate allowance based on published rate tables and historical experience. In addition, the Company incurs sales and marketing fees to its contractual customers based on a fixed percentage of net service revenues collected from third-party insurers or responsible parties. This fee is included in sales and marketing costs in the accompanying statement of operations.
Revenue is recognized when persuasive evidence of an arrangement exists, the price to the buyer is fixed or determinable, collectability is reasonably assured and risk of loss transfers, which normally is upon shipment of product or delivery of service. For sales that include customer specific acceptance criteria, revenue is recognized
F-11
when the acceptance criteria have been met. Credit is extended based upon the evaluation of the customer’s financial condition. Collectability is assessed based on a number of factors, including payment history and the creditworthiness of a customer. If it is determined that collection is not reasonably assured, revenue is not recognized until collection becomes reasonably assured, which is generally upon receipt of cash. The Company does not request collateral from its customers. The Company recognizes revenue when the products have been shipped and the title and risk of loss have been transferred. For laboratory services revenue is recognized when the completed test results are accessible by the ordering physician.
In instances where final acceptance of the product or system is required, revenue is deferred until all the acceptance criteria have been met. The rights granted in the RAP in exchange for a minimum annual purchase commitment constitutes a leasing arrangement. When a customer enters into a RAP agreement, the lease revenue is dependent on purchases of consumables, which is not measurable at the inception of the lease, and thus is accounted for as contingent rentals in their entirety. Accordingly, lease revenues are excluded from minimum lease payments but included in revenue as the consumables are purchased. The cost of the leased equipment is depreciated over its useful life and the expense included in cost of sales.
Arrangements to sell products to customers frequently include multiple deliverables, including the sale of the INFINITI analyzers combined with the sale of consumables. Sales to foreign distributors include training of their field agents who will be responsible for installation, training, and maintenance for their customers internationally. The Company does not provide any price protection, rights of return, or extended payment terms to the distributors. Prior to January 1, 2011 revenue was recognized upon completion of training, as the Company could not establish vendor-specific objective evidence, or VSOE, of the training. Beginning on January 1, 2011, the Company adopted new authoritative guidance on multiple-element arrangements, using the prospective method for all arrangements entered into or materially modified from the date of adoption. Under this new guidance, the Company allocates arrangement consideration in multiple-element revenue arrangements at the inception of an arrangement to all deliverables or those packages in which all components of the package are delivered at the same time, based on the relative selling price method in accordance with the selling price hierarchy, which includes: (i) VSOE if available; (ii) third-party evidence if VSOE is not available, and (iii) best estimate of selling price if neither VSOE nor third-party evidence is available. In 2011, the Company established selling prices using management’s best estimate, considering internal factors such as historical selling prices, pricing practices and controls, and customer segment pricing strategies.
Implementation of this new authoritative guidance has had an insignificant impact on reported revenue as compared to revenue under previous guidance.
Shipping and handling fees and costs
The Company records shipping and handling amounts in cost of sales. All shipping and handling fees and costs are charged to expense as incurred. No significant expense was incurred for the nine months ended September 30, 2014 and 2013 or for the years ended December 31, 2013 and 2012.
Research and development costs
Research and development costs consist primarily of salaries and related personnel costs, stock-based compensation, fees paid to outside parties, facilities costs, travel costs, depreciation and materials used in research and development. All research and development costs are charged to expense as incurred.
Advertising costs
All advertising costs are charged to expense as incurred. No significant expense was incurred for the nine months ended September 30, 2014 and 2013 or for the years ended December 31, 2013 and 2012.
Income taxes
The Company has adopted the provisions of ASC 740, Income Taxes. When tax returns are filed, it is highly certain that some positions taken would be sustained upon examination by the taxing authorities, while others are subject to uncertainty about the merits of the position taken or the amount of the position that would be ultimately
F-12
sustained. In accordance with the accounting guidance, the benefit of a tax position is recognized in the financial statements in the period during which management believes it is more likely than not that the position will be sustained upon examination, based on all available evidence, including the resolution of appeals or litigation processes, if any. Tax positions taken are not offset or aggregated with other positions. Tax positions that meet the more-likely-than-not recognition threshold are measured as the largest amount of tax benefit that is more than fifty percent likely of being realized upon settlement with the applicable taxing authority. The portion of the benefits associated with tax positions taken that exceeds the amount measured as described above should be reflected as a liability for unrecognized tax benefits in the accompanying balance sheets along with any associated interest and penalties that would be expensed through income tax expense and payable to the taxing authorities upon examination.
Stock-based compensation
The Company recognizes compensation expense related to stock-based transactions, including employee stock options, based on their fair value. The Company recognizes compensation expense over the vesting period using the straight-line method and classifies these amounts in the statement of operations based on the department to which the related employee reports. The Company uses the Black-Scholes valuation model to calculate the fair value of stock options.
The Company accounts for stock options issued to non-employees based on the fair value of the awards determined using the Black-Scholes valuation model. The fair value of stock options is recognized over the vesting period. Grants are made to non-employees based on services provided by the non-employee.
Comprehensive income/(loss)
The Company does not have any components of comprehensive income/(loss) other than net loss for the periods presented.
Recently issued accounting standards
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board, or the FASB, or other standard setting bodies that the Company adopts as of the specified effective date. The impact of recently issued standards that are not yet effective will not have a material impact on the Company's financial position or results of operations upon adoption.
In July 2013, the FASB issued ASU 2013-11, "Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists." ASU 2013-11 provides guidance on the presentation of unrecognized tax benefits related to any disallowed portion of net operating loss carryforwards, similar tax losses, or tax credit carryforwards, if they exist. ASU 2013-11 is effective for fiscal years beginning after December 15, 2013. The adoption of ASU 2013-11 did not have a material impact on the Company’s consolidated financial statements.
In August 2014, the FASB issued ASU 2014-15, “Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern”. ASU 2014-15 provides guidance on presentation of management’s plans, when conditions or events, considered in the aggregate, raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued, which are intended to mitigate those conditions or events that will alleviate the substantial doubt. ASU 2014-15 is effective for fiscal years ending after December 15, 2016. The adoption of ASU 2014-15 is not expected to have a material impact on the Company’s financial statements.
F-13
2. Balance sheet details
Inventory, net, consisted of the following (in thousands):
September 30, | December 31, | ||||||||||
2014 | 2013 | 2012 | |||||||||
(unaudited) | |||||||||||
Raw materials | $ | 2,474 | $ | 2,051 | $ | 1,719 | |||||
Work in process | 86 | 468 | 643 | ||||||||
Finished goods | 481 | 407 | 359 | ||||||||
Total | $ | 3,041 | $ | 2,926 | $ | 2,721 | |||||
The following table summarizes (in thousands) the activity in the allowance for excess and obsolete inventory during the nine months ended September 30, 2014 and the years ended December 31, 2013 and 2012. Increases to the allowance are reflected as "Charged to costs and expenses." "Allowance reductions/write-offs" is comprised primarily of inventory that has been scrapped.
For the period ended | Balance at beginning of period | Charged to costs and expenses | Allowance reductions/ write-offs | Balance at end of period | |||||||||||
September 30, 2014 (unaudited) | $ | 442 | $ | 376 | $ | (379 | ) | $ | 439 | ||||||
December 31, 2013 | $ | 898 | $ | 0 | $ | (456 | ) | $ | 442 | ||||||
December 31, 2012 | $ | 679 | $ | 504 | $ | (285 | ) | $ | 898 | ||||||
Property, equipment and improvements consisted of the following (in thousands):
September 30, | December 31, | ||||||||||||
Estimated Life | 2014 | 2013 | 2012 | ||||||||||
(unaudited) | |||||||||||||
Reagent access plan assets | 3 or 5 years | $ | 2,140 | $ | 1,774 | $ | 1,516 | ||||||
Equipment | 5-7 years | 2,495 | 2,391 | 2,122 | |||||||||
Computer equipment and software | 3 years | 242 | 239 | 232 | |||||||||
Leasehold improvements | various | 199 | 175 | 159 | |||||||||
Furniture | 5 years | 156 | 156 | 156 | |||||||||
$ | 5,232 | $ | 4,735 | $ | 4,185 | ||||||||
Less accumulated depreciation and amortization | 3,470 | 3,261 | 2,951 | ||||||||||
Total | $ | 1,762 | $ | 1,474 | $ | 1,234 | |||||||
Depreciation and amortization of property, equipment and improvements, including capitalized INFINITI analyzers in RAPs, was $0.4 million, $0.4 million, $0.6 million and $0.6 million for the nine months ended September 30, 2014 and 2013 (unaudited), and the years ended December 31, 2013 and 2012, respectively.
F-14
Deferred costs consisted of the following (in thousands):
September 30, | December 31, | ||||||||||
2014 | 2013 | 2012 | |||||||||
(unaudited) | |||||||||||
Deferred private placement costs | $ | 277 | $ | 454 | $ | 712 | |||||
Deferred IPO costs | 417 | — | 1,654 | ||||||||
$ | 694 | $ | 454 | $ | 2,366 | ||||||
Private placement costs pertain to the refinancing of promissory notes during November 2012, and are amortized over the life of the new promissory notes maturing in November 2015. The amortization during the nine months ended September 30, 2014 and the year ended December 31, 2013, was $0.2 million and $0.2 million, with no material amortization during 2012.
Accrued expenses consisted of the following (in thousands):
September 30, | December 31, | ||||||||||
2014 | 2013 | 2012 | |||||||||
(unaudited) | |||||||||||
Payroll and related expenses | $ | 1,056 | $ | 694 | $ | 692 | |||||
Professional fees | 1,399 | 318 | 416 | ||||||||
Initial public offering – terminated offering | — | — | 524 | ||||||||
Initial public offering | — | — | — | ||||||||
Other | 867 | 398 | 299 | ||||||||
$ | 3,322 | $ | 1,410 | $ | 1,931 | ||||||
The following table summarizes the activity in accrued warranty costs included in Other Current Liabilities (in thousands):
For the period ended | Balance at beginning of period | Charged to costs and expenses | Costs incurred | Balance at end of period | |||||||||||
September 30, 2014 (unaudited) | $ | 88 | $ | 205 | $ | (87 | ) | $ | 206 | ||||||
December 31, 2013 | $ | 83 | $ | 62 | $ | (57 | ) | $ | 88 | ||||||
December 31, 2012 | $ | 75 | $ | 82 | $ | (74 | ) | $ | 83 | ||||||
3. Promissory notes
During the year ended December 31, 2012, the Company issued $1.7 million in short term notes payable bearing interest at rates between 10% and 12% per annum with maturity dates up to six months after issuance. In connection with these notes, the Company issued freestanding warrants to purchase 201,667 shares of the Company’s common stock at an exercise price of $5.19 per share. The warrants were exercisable immediately and expire five years after the date of issuance. The fair value of the warrants was determined using the Black-Scholes valuation model and was used to calculate the debt discount of $0.3 million. This amount is being amortized to interest expense using the effective interest method over the term of the notes.
In November 2012, the holders of $7.5 million in aggregate principal and accrued interest on the Company’s outstanding subordinated promissory notes with rates of interest ranging from 6.0% to 12.0% that were then past due or scheduled to come due in the immediate future surrendered their right to payment of those promissory notes in
F-15
exchange for an aggregate of 1,414,734 shares of the Company’s Series NC Convertible Preferred Stock, as converted to common. In connection with this surrender and issuance, these noteholders surrendered warrants for the purchase of an aggregate of 644,255 shares of the Company’s common stock with exercise prices ranging from $5.00 per share to $5.19 per share in exchange for warrants of substantially like tenor for the purchase of an aggregate of 644,255 shares of the Company’s common stock with exercise price of $4.50 per share.
Also in November 2012, the holders of $14.4 million in aggregate principal amount and accrued interest on the Company’s outstanding subordinated promissory notes with rates of interest ranging from 6.5% to 13.5% that were then past due or scheduled to come due in the immediate future surrendered their right to payment of those promissory notes in exchange for new promissory notes with an aggregate principal amount of $14.4 million bearing interest at 8.5% per annum due in November 2015. In connection with this surrender and issuance, these noteholders surrendered warrants for the purchase of an aggregate of 1,167,690 shares of the Company’s common stock with exercise prices ranging from $5.19 per share to $15.00 per share in exchange for warrants of substantially like tenor for the purchase of an aggregate of 1,751,535 shares of the Company’s common stock with exercise prices of $5.25 per share that are exercisable through November 2017.
On December 28, 2012, Tregale Group Ltd, the holder of approximately $2.2 million in principal amount under promissory notes that had been in default beginning in the first quarter of 2012, filed with the Supreme Court of the State of New York a request for judicial intervention and motion for summary judgment in lieu of complaint, demanding payment of the principal and interest outstanding under its promissory notes as well as reimbursement of certain legal and other expenses. In January 2013, the Company issued and sold a subordinated promissory note in the amount of $2.4 million to one of its existing investors, who is a holder of more than 5% of the Company’s capital stock. This promissory note had a maturity date of March 31, 2013, an annual interest rate of 8.5%, and may be prepaid at any time without premium or penalty. The Company used the proceeds of this sale to retire the outstanding principal and interest owed to Tregale Group Ltd under its outstanding promissory notes but did not settle the legal and other expenses. In May 2013, the Court granted the motion for summary judgment in lieu of complaint and ordered the Company to reimburse Tregale Group Ltd its legal and other expenses. The Company had $250,000 in accrued expenses related to this matter as of December 31, 2013. During February 2014, the $2.4 million in notes payable with maturity dates in 2013 were refinanced to December 2014 and November 2015. Included in this refinance were the issuance of 400,000 Common Stock Warrants with a strike price of $5.31 and expiring in seven years.
In April, 2013, the Company issued a subordinated note in the amount of $0.6 million with a maturity date of March 31, 2014 to an existing noteholder in exchange for an outstanding subordinated note in the same amount with a maturity date of March 31, 2013. The new note has an annual interest rate of 8.5%. In return for the extension, the Company also issued a warrant to the noteholder to purchase 90,273 shares of common stock with a term of 5 years at an exercise price of $5.25 per share. In September 2014, the lender waived a payment default and the maturity date of this note was extended to December 31, 2014.
In February 2014, the Company issued a senior note in the amount of $2.3 million with a maturity date of December 15, 2014 and a subordinated note in the amount of $0.1 million with a maturity date of November 12, 2015 to an existing noteholder in exchange for outstanding subordinated notes in the same amount with maturity dates of March 31, 2014. The new notes have an annual interest rate of 8.5%. In return for the extension, the Company also issued a warrant to the noteholder to purchase 400,000 shares of common stock with a term of 7 years at an exercise price of $5.31 per share.
During April 2014, $1.0 million due to the Company's landlord, all of which was payable at December 31, 2013, was converted into a note payable due June 30, 2015 and carrying 10.0% interest. The note contains an accelerated timeline provision for payment upon the completion of a capital event.
Refinanced promissory notes where the present value of the future cash flows, under the modified notes, did not exceed the present value of the future cash flows under the original notes by more than 10%, were treated as a modification and the increase in the fair value of the warrants resulting from the modification was recorded as a discount to the notes and is being amortized over the new term of the notes using the effective interest method. For
F-16
notes where the present value of future cash flows changed by more than 10% under modified terms as compared to original terms, the Company accounted for the modification as a debt extinguishment, and accordingly recorded the notes at fair value as calculated using future discounted cash flows. The gain or loss on extinguishment is recorded in the statement of operations in the period in which the extinguishment occurred. The gain or loss on extinguishment was determined by calculating the difference between the net carrying amount of the extinguished debt (which includes principal, accrued interest, original fair value of the associated warrants originally recorded in equity, unamortized discount and unamortized deferred financing cost) and the fair value of the modified debt (which includes fair value of modified debt, fair value of modified warrants and any amendment related fees). The fair value of the debt was determined using Level III input as described in (note 9 Fair value measurements). The refinancing of the subordinated promissory notes is summarized as follows.
February 2012 refinancing to modify the terms of the applicable subordinated promissory notes and related warrants:
Modification | Extinguishment | ||||||
Original notes and warrants | |||||||
Note principal amount | $ | 250,000 | $ | 400,000 | |||
Original maturity date(s) | April 2012 | February 2012 | |||||
Common stock warrant shares | 71,000 | 113,600 | |||||
Warrant exercise price | $ | 7.00 | $ | 4.00 | |||
Warrant expiration date | 5 years from original issuance | 5 years from original issuance | |||||
Modified notes and warrants | |||||||
Note principal amount | $ | 250,000 | $ | 400,000 | |||
Extended maturity date(s) | June 2012 | April 2012 | |||||
Common stock warrant shares | 71,000 | 113,600 | |||||
Warrant exercise price | $ | 3.00 | $ | 3.00 | |||
Warrant expiration date | 6.5 years from original issuance | 6.5 years from original issuance | |||||
Increase in fair value of warrants | $ | 36,000 | $ | 47,000 | |||
Gain on extinguishment | N/A | $ | 78,000 | ||||
The gain on extinguishment is comprised of a reduction in the fair value of the debt partially offset by the increase in fair value of warrants and write off of deferred financing costs.
The warrant cancellations and reissuances, summarized above, include the cancellation of original warrants and reissuance of new warrants to purchase 145,735 shares of the Company’s common stock beneficially held by members of the board of directors of the Company, officers of the Company, and a significant stockholder.
F-17
November 2012 refinancing to exchange promissory notes for Series NC Preferred Stock and related warrants:
Modification | Extinguishment | ||||
Original notes and warrants | |||||
Note principal amount (including accumulated interest) | N/A | $ | 7,502,000 | ||
Original maturity date(s) | March 2012 – December 2012 | ||||
Common stock warrant shares | 644,255 | ||||
Warrant exercise price | $5.00 to $5.19 | ||||
Warrant expiration date | 5 years from original issuance | ||||
Extinguished note and modified warrants | |||||
Note principal amount (including accumulated interest) | N/A | $ | 0 | ||
Common stock warrant shares | 644,255 | ||||
Warrant exercise price | $ | 4.50 | |||
Warrant expiration date | November 2017 | ||||
Gain/loss on extinguishment | $ | — | |||
November 2012 refinancing to exchange promissory notes and related warrants:
Modification | Extinguishment | ||||
Original notes and warrants | |||||
Note principal amount and accrued interest | N/A | $ | 14,454,000 | ||
Original maturity date(s) | March 2012 – December 2012 | ||||
Common stock warrant shares | 3,503,070 | ||||
Warrant exercise price | $1.73 to $5.00 | ||||
Warrant expiration date | 5 years from original issuance | ||||
Modified notes and warrants | |||||
Note principal amount (including accumulated interest) | N/A | $ | 14,454,000 | ||
Extended maturity date(s) | November 2015 | ||||
Common stock warrant shares | 1,751,535 | ||||
Warrant exercise price | $ | 5.25 | |||
Warrant expiration date | November 2017 | ||||
Increase in fair value of warrants | $ | 4,947,000 | |||
Loss on extinguishment | $ | 2,074,000 | |||
The loss on extinguishment is comprised of an increase in the fair value of the warrants partially offset by the decrease in the fair value of the debt.
F-18
March 2013 refinancing to modify the terms of the applicable promissory notes and related warrants:
Modification | Extinguishment | ||||
Original notes and warrants | |||||
Note principal amount | N/A | $ | 619,000 | ||
Original maturity date(s) | March 2013 | ||||
Modified notes and warrants | |||||
Note principal amount (including accumulated interest) | N/A | $ | 636,000 | ||
Extended maturity date(s) | March 2014 | ||||
Common stock warrant shares | 90,273 | ||||
Warrant exercise price | $ | 5.25 | |||
Warrant expiration date | November 2017 | ||||
Increase in fair value of warrants | $ | 140,000 | |||
Loss on extinguishment | $ | 140,000 | |||
The loss on extinguishment is comprised of an increase in the fair value of the warrants partially offset by the decrease in the fair value of the debt.
February 2014 refinancing to modify the terms of the applicable promissory notes and related warrants:
Modification | Extinguishment | ||||
Original notes and warrants | |||||
Note principal amount | N/A | $ | 2,404,000 | ||
Original maturity date(s) | March 2014 | ||||
Modified notes and warrants | |||||
Note principal amount (including accumulated interest) | N/A | $ | 2,404,000 | ||
Extended maturity date(s) | December 2014 and November 2015 | ||||
Common stock warrant shares | 400,000 | ||||
Warrant exercise price | $ | 5.31 | |||
Warrant expiration date | February 2021 | ||||
Increase in fair value of warrants | $ | 1,872,000 | |||
Loss on extinguishment | $ | 1,872,000 | |||
The loss on extinguishment is comprised of an increase in the fair value of the warrants.
F-19
The components of the Company’s promissory notes payable consisted of (in thousands):
September 30, 2014 | December 31, | ||||||||||
2013 | 2012 | ||||||||||
(unaudited) | |||||||||||
Senior promissory note, 13.0% interest, due February 2012 | $ | — | $ | — | $ | 2,200 | |||||
Senior promissory note, 8.5% interest, due March 2013 | — | 2,298 | — | ||||||||
Senior promissory note, 8.5% interest, due December 2014 | 2,298 | — | — | ||||||||
Subordinated promissory notes, 8.0% interest, due March 2013 | — | — | 500 | ||||||||
Subordinated short term notes, 8.5% interest, due March 2014 | — | 106 | — | ||||||||
Subordinated promissory notes, 8.5% interest, due March 2014, extended to December 31, 2014 | 636 | 636 | — | ||||||||
Senior promissory note, 10.0% interest, due June 2015 | 997 | — | — | ||||||||
Subordinated promissory notes, 8.5% interest, due November 2015 | 14,560 | 14,452 | 14,454 | ||||||||
Total | 18,491 | 17,492 | 17,154 | ||||||||
Unamortized premium/(discount) | (50 | ) | (79 | ) | (111 | ) | |||||
Accrued interest | 2,734 | 1,582 | 486 | ||||||||
Outstanding balance | $ | 21,175 | $ | 18,995 | $ | 17,529 | |||||
The balance of notes payable is as follows as September 30, 2014 (in thousands):
Payments due by period | |||||||||||
Total | 2014 | 2015 | |||||||||
(unaudited and in thousands) | |||||||||||
Subordinated promissory notes | 18,491 | $ | 2,934 | $ | 15,557 | ||||||
4. Commitments and contingencies
Operating lease
The Company leases its corporate headquarters and manufacturing facility under a non-cancelable operating lease agreement expiring in December 2029. Rent expense under the lease was $1.5 million, $1.9 million, $2.5 million and $2.5 million for the nine months ended September 30, 2014 and 2013 (unaudited), and years ended December 31, 2013 and 2012, respectively. The terms of the facility lease provide for rental payments on a monthly basis with periodic rent escalations over the term of the lease as well as penalties of 11% of the monthly lease payment for late payment.
The amounts of future minimum lease commitments under the operating lease at September 30, 2014, are as follows (in thousands):
Payments due by period | ||||||||||||||||||||
Total | 2014 | 2015 | 2016 | 2017 | 2018 | 2019 & Beyond | ||||||||||||||
(unaudited) | ||||||||||||||||||||
Operating lease | 38,655 | 1,834 | 1,924 | 2,043 | 2,095 | 2,154 | 28,605 | |||||||||||||
F-20
The Company recognizes rent expense on a straight-line basis over the lease period and has accrued for rent expense incurred but not paid. Certain incentives, including discounted rental payments and tenant improvement allowances, were granted when the original lease was executed.
Accumulated discounted rental payments recorded to deferred rent totaled $3.3 million, $3.1 million and $2.5 million as of September 30, 2014, December 31, 2013 and 2012, respectively, and the balance of the tenant improvement allowance stayed consistent at $0.2 million. The Company amortizes the allowance on a straight-line basis over the term of the lease.
The Company at times has been unable to remit timely lease payments and the remediation of these defaults has resulted in amendments to the original lease that included rent escalations and extensions of the original leasehold expiration date.
License agreements
The Company has license agreements with third parties entitling the Company to receive royalty payments and obligating the Company to make royalty payments. These license agreements expire at various times through May 2024. The Company records royalty payments as earned; however, such royalty payments have historically been immaterial in amount. The Company did not receive any royalty payments in fiscal years 2012 and 2013 or for the first nine months of 2014. The Company records royalty expense concurrently with the recording of the revenue giving rise to the royalty payment obligation under the terms of the applicable license agreements; however, such royalty expense has historically been immaterial in amount.
Litigation
The Company is from time to time subject to various claims and legal actions in the ordinary course of business. The Company believes that there are currently no claims or legal actions against the Company that, in management’s judgment based on information currently available, are likely to have a material adverse effect on the results of operations or financial condition. However, regardless of outcome, litigation can have an adverse impact on the Company because of defense and settlement costs, diversion of management resources and other factors.
5. Common and convertible preferred stock warrant liabilities
Common stock warrant liabilities
During 2010 and 2011 the Company issued two common stock warrants to investors that provided to the investors anti-dilution protection in respect of their warrants. The 2010 warrant was surrendered when the note was refinanced in November 2012 as discussed in Note 3. The conversion features of these warrants include anti-dilution protection and accordingly are accounted for as derivative liabilities on a stand-alone basis from other common stock warrants.
The Company uses the Black-Scholes valuation model to calculate the fair value of its common stock warrant liability. The Company determined that using alternative valuation models, such as a Monte Carlo simulation model, would result in de minimus differences. The fair value of the common stock warrant liability was estimated at the balance sheet dates using the following assumptions:
September 30, | December 31, | ||||||||||
2014 | 2013 | 2012 | |||||||||
(unaudited) | |||||||||||
Fair value | $ | 7.95 | $ | 6.60 | $ | 5.31 | |||||
Exercise price | $ | 4.50 | $ | 4.50 | $ | 4.50 | |||||
Contractual term (yrs) | 1.6 | 2.3 | 3.3 | ||||||||
Stock price volatility | 53 | % | 53 | % | 54 | % | |||||
Risk-free interest rate | 0.43 | % | 0.43 | % | 0.35 | % | |||||
F-21
Outstanding common stock warrants consisted of:
Issue date | Term | Exercise price per warrant share | Number of shares of underlying stock | ||||
April 2011 | 5 years | $ | 4.50 | 10,621 | |||
The fair value at September 30, 2014 (unaudited), December 31, 2013 and 2012 was $42,000, $32,000 and $25,000, respectively.
In November 2012, the Company and certain note holders agreed to the conversion into shares of Series NC Convertible Preferred Stock of notes payable that had matured and that were delinquent or that would mature by December 2012. In connection with that conversion, existing warrants to purchase shares of common stock at exercise prices between $5.19 and $15.00 per share were cancelled and replaced with new warrants to purchase common stock with an exercise price of $4.50 per share. Pursuant to the terms of the warrant to purchase shares of common stock issued in April 2011 and described above, the Company cancelled a warrant to purchase 10,621 shares of common stock at an exercise price of $9.00 and replaced it with a new warrant to purchase 10,621 shares of common stock with like terms and a strike price of $4.50 per share.
The Company has issued additional common stock warrants which were not accounted for as liabilities. See Note 7 for further details.
Convertible preferred stock warrant liabilities
The Company uses the Black-Scholes valuation model to calculate the fair value of its preferred stock warrants. The Company determined that using alternative valuation models, such as a Monte Carlo simulation model, would result in de minimus differences. The inputs utilized in this valuation model are discussed below. Each of these inputs is subjective and generally requires significant judgment to determine.
• | Fair value of preferred stock – Because the Company’s stock is not traded publicly, the Company must estimate the fair value of its preferred stock as discussed below. |
• | Expected term – The expected term is the remaining contractual life of the applicable preferred stock warrant. |
• | Volatility – As the Company does not have a trading history for its preferred stock, the expected stock price volatility for its preferred stock is estimated by taking the average historic price volatility for industry peers. The historic price volatility is determined by averaging daily price observations over a period equivalent to the expected term of the preferred stock warrant. The industry peers consist of several publicly traded companies similar in size, stage of life cycle, and financial leverage. The Company does not rely on implied volatilities of the companies in the industry peer group because the volume of activity has been relatively low. |
• | Risk-free interest rate – The risk-free interest rate is based on the yields of U.S. Treasury securities with maturities similar to the contractual term of the preferred stock warrants. |
• | Dividend yield – The Company has not paid cash dividends or distributions historically and does not intend to pay cash dividends or distributions in the foreseeable future. Consequently, the Company used an expected dividend yield of zero. |
F-22
The fair value of the preferred stock warrants was estimated at the balance sheet dates using the following assumptions (fair value and exercise price are presented on a common stock equivalents basis):
September 30, | December 31, | ||||||||||
2014 | 2013 | 2012 | |||||||||
(unaudited) | |||||||||||
Fair value | $ | 3.96 | $ | 4.49 | $ | 5.31 | |||||
Exercise price | $3.75-$4.30 | $3.75-$4.30 | $3.50-$4.05 | ||||||||
Contractual term (yrs) | 0 | 0.6 | 0.6 | ||||||||
Stock price volatility | 52 | % | 52 | % | 51 | % | |||||
Risk-free interest rate | 0.13 | % | 0.13 | % | 0.16 | % | |||||
These warrants expired in August 2014. Because there has been no public market for the Company’s stock, the board of directors historically has determined the fair value of the preferred stock at the time of issuance by considering a number of objective and subjective factors including valuations performed by unrelated third-party specialists, which included valuations of comparable companies, operating and financial performance, lack of liquidity of capital stock and the general and industry-specific economic outlook, among other factors. For each period in which the fair values of preferred stock warrant liabilities was assessed, the business enterprise value of the Company was first determined. This enterprise value then was allocated across the Company’s capital structure using a Probability Weighted Expected Return Method (PWERM) model as the primary approach and an Option Pricing Model (OPM) analysis as a confirming approach. Under the PWERM model, various future potential liquidity scenarios were forecast and the allocable value to each class of security in the Company’s capital structure in each respective scenario was determined. The probability that each potential liquidity event would occur then was assessed. The future proceeds then were discounted to determine the present value of proceeds in each scenario for each security class. The probability weighted present value of each security class then was determined to conclude the value of the preferred stock. Under the confirming OPM analysis, the Black Scholes option-pricing framework was used to determine the proceeds allocable to each security class. A series of option tranches was established whereby each tranche represented the value allocable to various security classes between computed breakpoints. Breakpoints are the value inflection points, or indifference points, whereby the allocations to securities change based on the underlying economic rights of each security class. The breakpoints served as the strike prices in the Black Scholes option calculations and tranche values were determined based on the difference between option values in each tranche. The proceeds of the value of each option tranche then were allocated to each respective security class based on the economic rights to the security class between breakpoints. The value allocable to each respective security class then was totaled across all option tranches to conclude the fair value of each security class.
The Company’s convertible preferred stock historically has been issued by the Company at a purchase price that reflects a premium to the fair market value of the Company’s common stock. These premiums have been negotiated at arms’-length between the Company and the various participating investors in the offering and sale of such convertible preferred stock, and generally result from the preferential position that the Company’s convertible preferred stock maintains in relation to the Company’s common stock with respect to matters such as preferences upon dividends and preferences upon liquidation (See note 6 Convertible preferred stock).
6. Convertible preferred stock
Upon the occurrence of certain change in control events that are outside of the control of the Company, including sale or transfer of control of the Company, holders of the convertible preferred stock can force the Company to redeem these shares for cash. Accordingly, these shares are considered contingently redeemable and are classified as temporary equity on the accompanying balance sheets.
The rights, preferences and privileges of the convertible preferred stock are as follows:
Dividends
F-23
The holders of Series A, Series B, Series C, Series D and Series E Convertible Preferred Stock are entitled to receive noncumulative dividends, in preference and in priority to any dividends on common stock, at a rate of $0.19, $0.17, $0.17, $0.20 and $0.165 per share, respectively, when and if declared by the board of directors. As of September 30, 2014, the board of directors had not declared any dividends.
Conversion
All preferred stock will convert automatically to common stock immediately prior to the closing of an underwritten public offering of common stock under the Securities Act of 1933, as amended, except for Series C Convertible Preferred Stock, Series E Convertible Preferred Stock and Series NC Convertible Preferred Stock, each of which converts automatically only if such public offering results in proceeds of $25,000,000 or more, which the Company expects to occur in connection with the completion of this offering. In addition, each individual series of convertible preferred stock will convert automatically to common stock upon the vote of a majority of the outstanding shares of each series. The conversion price of each series of the Company’s convertible preferred stock is adjusted on a broad-based weighted average basis if the Company issues certain types of securities at a price per share below the then current conversion price of the applicable series of convertible preferred stock. This conversion feature was evaluated and determined not to be subject to derivative accounting.
Outstanding shares as of September 30, 2014 (unaudited) are as follows:
Number of Shares | |||||||
Convertible | Shares | Conversion | As Converted | Liquidation | |||
Preferred Stock | Issued | Factor | To Common | Preference | |||
Series A | 1,579,227 | 0.6600 | 1,042,290 | $3.16 | |||
Series B | 4,468,369 | 0.3640 | 1,626,519 | $2.75 | |||
Series C | 6,417,680 | 0.3640 | 2,336,083 | $2.75* | |||
Series D | 3,423,258 | 0.3764 | 1,288,651 | $3.25 | |||
Series E | 732,555 | 0.3640 | 266,655 | $2.75* | |||
Series NC | 4,287,074 | 0.3300 | 1,414,734 | $1.75 | |||
Total | 7,974,932 | ||||||
* Indicates liquidation preference priority,
Liquidation preference
Upon a liquidation, dissolution or winding up of the Company, the holders of the Series E Convertible Preferred Stock are entitled to receive liquidation preferences (subject to appropriate adjustments for stock splits, stock dividends and other similar reclassifications), plus any declared but unpaid dividends, in priority over any payments to the holders of Series A, Series B, Series C, Series D and Series NC Convertible Preferred Stock and the holders of common stock. Upon completion of the required distributions to the holders of Series E Convertible Preferred Stock, the holders of Series C Convertible Preferred Stock are entitled to receive liquidation preferences (subject to appropriate adjustments for stock splits, stock dividends and other similar reclassifications), plus any declared but unpaid dividends, in priority over any payments to the holders of Series A, Series B, Series D and Series NC Convertible Preferred Stock and the holders of common stock. Upon completion of the required distributions to the holders of Series E and Series C Convertible Preferred Stock, the holders of Series A, Series B, Series D and Series NC Convertible Preferred Stock are entitled to receive liquidation preferences (subject to appropriate adjustments for stock splits, stock dividends and other similar reclassifications), plus any declared but unpaid dividends, in priority over any payments to the holders of common stock.
Voting
The holders of convertible preferred stock (other than Series NC convertible Preferred Stock) vote on an equivalent basis with the holders of common on an as-converted basis.
F-24
7. Common stock warrants
Included in the Company’s outstanding warrants as of September 30, 2014 are warrants exercisable for 2,271,237, 644,255, 366,278, 85,000 and 40,000 shares of its common stock in connection with the issuance of promissory notes, the conversion of promissory notes into Series NC Convertible Preferred Stock, the sale of Series E Convertible Preferred Stock, the provision of services rendered to the Company by non-employee consultants as compensation for those services, and compensation to lead non-employee members of the board of directors, respectively.
In November 2012, the Company and the applicable note holders of $14.4 million in notes payable and accrued interest agreed to the exchange of promissory notes maturing in December 2012 for new notes maturing in November 2015. In connection with this agreement, the Company cancelled the warrant to purchase 1,167,690 shares of its common stock with exercise prices ranging from $5.19 per share to $15.00 per share and issued a new warrant to purchase 1,751,535 shares of its common stock at an exercise price of $5.25 per share, which will expire in November 2017.
Also in November 2012, the Company and the applicable note holders of $7.5 million in notes payable and accrued interest agreed to the exchange of promissory notes maturing in December 2012 for Series NC Convertible Preferred Stock. In connection with this agreement, the Company cancelled warrants to purchase 644,255 shares of its common stock with an exercise price between $5.00 and $5.19 per share and issued new warrants to purchase 644,255 shares of its common stock at an exercise price of $4.50 per share, which will expire in November 2017.
In April, 2013, the Company issued a subordinated note in the amount of $636,000 with a maturity date of March 31, 2014 to an existing noteholder in exchange for an outstanding subordinated note in the same amount with a maturity date of March 31, 2013. The new note has an annual interest rate of 8.5%. In return for the extension, the Company also issued a warrant to the noteholder to purchase 90,273 shares of common stock with a term of 5 years at an exercise price of $5.25 per share. In September 2014, the lender waived a payment default and the maturity date of this note was extended to December 31, 2014.
In September 2013, the Company raised $4.0 million in a private placement pursuant to the issuance of 573,888 shares of the Company’s common stock at $6.97 per share. The Company also issued warrants to the investor to purchase an additional 60,000 shares of the Company’s common stock with a term of 4 years at an exercise price of $6.97 per share.
F-25
These warrants do not have any provisions for the adjustment of their strike prices, and have therefore been accounted for as equity instruments. The following summarizes transactions related to common stock warrants:
Number of Shares | Weighted- average exercise price per share | Weighted- average contractual life (years) | |||||||
Outstanding and exercisable – January 1, 2012 | 2,620,761 | $ | 11.06 | 2.64 | |||||
Issued | 2,604,647 | 5.15 | 4.80 | ||||||
Cancelled | (1,793,825 | ) | 12.06 | — | |||||
Exercised | — | — | — | ||||||
Expired | (58,872 | ) | $ | 4.64 | — | ||||
Outstanding and exercisable – December 31, 2012 | 3,372,711 | 6.09 | 4.46 | ||||||
Issued | 180,273 | 1.75 | — | ||||||
Cancelled | (30,000 | ) | 1.75 | — | |||||
Exercised | — | — | — | ||||||
Expired | (16,500 | ) | $ | 1.82 | — | ||||
Outstanding and exercisable – December 31, 2013 | 3,506,484 | 6.09 | 3.52 | ||||||
Issued | 400,000 | 5.31 | 4.37 | ||||||
Expired | (14,235 | ) | 0.76 | 0.10 | |||||
Outstanding and exercisable – September 30, 2014 (unaudited) | 3,892,249 | $ | 6.04 | 3.62 | |||||
The Company had 13,492,909, 13,294,168 and 13,648,010 shares of common stock reserved for future issuance at September 30, 2014 (unaudited), December 31, 2013 and 2012, respectively, upon the conversion or exercise, as applicable, of its outstanding common stock options, common stock and preferred stock warrants (assuming conversion of the preferred stock warrants into common stock), and convertible preferred stock.
8. Stock-based compensation
In December 2000, the Company adopted the 2000 Equity Incentive Plan, or the 2000 Plan, which now has expired. The 2000 Plan allowed for the grant of stock options and the right to acquire restricted stock to employees, directors and consultants of the Company. The terms and conditions of specific awards were set at the discretion of the Company’s board of directors although generally options under the 2000 Plan vest in four annual installments of 25% and are generally immediately exercisable. The exercise price of incentive stock options was required to be not less than 100% of the fair market value of the Company’s common stock on the date of grant and the exercise price of any option granted to a 10% stockholder was required to be not less than 110% of the fair market value of the Company’s common stock on the date of grant. Options granted under the 2000 Plan expire no later than 10 years from the date of grant. Unvested common shares obtained upon early exercise of options are subject to repurchase by the Company at the original issue price. As of September 30, 2014, there were no unvested common shares outstanding that originally had been issued upon early exercise of options. The 2000 Plan expired in 2010 and no shares remain available for grant under the 2000 Plan.
In October 2008, the Company adopted the 2008 Equity Incentive Award Plan, or the 2008 Plan. The 2008 Plan allows for the grant of stock options and the right to acquire restricted stock to employees, directors and consultants of the Company. The terms and conditions of specific awards are set at the discretion of the Company’s board of directors although generally options vest in four annual installments of 25%. The exercise price of incentive stock options shall not be less than 100% of the fair market value of the Company’s common stock on the date of grant and the exercise price of any option granted to a 10% stockholder may be no less than 110% of the fair market value of the Company’s common stock on the date of grant. Options granted under the 2008 Plan expire no later than 10 years from the date of grant. Unvested common shares obtained upon early exercise of options are subject to
F-26
repurchase by the Company at the original issue price. The 2008 Plan reserved 2,000,000 shares of common stock pursuant to awards under the 2008 Plan. In addition, shares available for issuance and not subject to awards and shares forfeited under the 2000 Plan, aggregating 1,573,392 shares, were transferred to and made available for option grants pursuant to the 2008 Plan. In aggregate, 2,655,389 shares remained available for grant under the 2008 Plan at September 30, 2014.
The Company has issued options to purchase shares of the Company’s common stock to certain of the Company’s directors, executive officers and key employees that are not subject to either the 2008 Plan or the 2000 Plan. The terms of these options are substantially the same as those issued under the 2000 Plan.
A summary of outstanding options at the periods indicated is as follows:
September 30, | December 31, | |||||||
2014 | 2013 | 2012 | ||||||
Outstanding Options | (unaudited) | |||||||
2000 plan | ||||||||
Vested | 248,098 | 288,292 | 470,826 | |||||
Unvested | — | — | — | |||||
Subtotal | 248,098 | 288,292 | 470,826 | |||||
2008 plan | ||||||||
Vested | 685,483 | 472,095 | 521,611 | |||||
Unvested | 211,474 | 490,667 | 197,408 | |||||
Subtotal | 896,957 | 962,762 | 719,019 | |||||
Non-plan | ||||||||
Vested | 264,000 | 165,000 | 323,000 | |||||
Unvested | 60,000 | 269,000 | 50,000 | |||||
Subtotal | 324,000 | 434,000 | 373,000 | |||||
Total Outstanding Options | 1,469,055 | 1,685,054 | 1,562,845 | |||||
F-27
The following table summarizes stock option transactions from January 1, 2012 through September 30, 2014:
Options | Weighted-average exercise price per share | Weighted-average remaining contractual terms (years) | Intrinsic Value | ||||||||||
(in thousands) | |||||||||||||
Outstanding – January 1, 2012 | 1,858,740 | $ | 4.70 | ||||||||||
Granted | 90,750 | $ | 5.24 | ||||||||||
Exercised | (190,328 | ) | $ | 0.61 | $ | 872 | |||||||
Forfeited | (211,615 | ) | $ | 5.12 | |||||||||
Outstanding – December 31, 2012 | 1,547,547 | $ | 4.70 | ||||||||||
Granted | 542,919 | $ | 6.67 | ||||||||||
Exercised | (249,698 | ) | $ | 0.88 | $ | 1,444 | |||||||
Forfeited | (155,714 | ) | $ | 7.20 | |||||||||
Outstanding – December 31, 2013 | 1,685,054 | $ | 6.18 | ||||||||||
Exercised | (4,950 | ) | $ | 0.75 | $ | 33 | |||||||
Forfeited | (211,049 | ) | $ | 6.67 | |||||||||
Outstanding – September 30, 2014 (unaudited) | 1,469,055 | $ | 6.20 | ||||||||||
Vested and exercisable at September 30, 2014 (unaudited) | 1,197,581 | $ | 6.19 | 5.0 | $ | — | |||||||
Unvested at September 30, 2014 (unaudited) | 271,474 | $ | 6.54 | 8.2 | $ | — | |||||||
Determining the fair value of common stock option grants
The Company and its Board of Directors use the Black-Scholes valuation model to calculate the fair value of its common stock option grants. The inputs utilized in this valuation model are discussed below. Each of these inputs is subjective and generally requires significant judgment to determine.
• | Expected term - The expected life of employee and non-employee director stock options represents the average of the contractual term of the options and the weighted-average vesting period, as permitted under the simplified method. The Company has elected to use the simplified method, as the Company does not have enough historical exercise experience to provide a reasonable basis upon which to estimate the expected term and the stock option grants are considered "plain vanilla" options. |
• | Expected volatility - The expected volatility is derived from the historical stock volatilities of several comparable publicly listed peer companies over a period approximately equal to the expected term of the options because the Company has limited information on the volatility of its common stock since the Company has no trading history. When making the selections of the comparable industry peers to be used in the volatility calculation, the Company considered the size, operational and economic similarities to its principle business operations. |
• | Fair value of common stock - Because there has been no public market for the Company’s common stock, the board of directors has determined the fair value of the common stock at the time of the option grant by considering a number of objective and subjective factors including valuations performed by unrelated third-party specialists, which included valuations of comparable companies, operating and financial performance, lack of liquidity of capital stock and general and industry-specific economic outlook, amongst other factors. This fair value has been calculated on an as-converted basis for each period, assuming for such purposes that all outstanding convertible preferred stock has been converted into common stock without taking into account any dividend or liquidation preferences. Once the Company’s common stock is |
F-28
listed on an established stock exchange or national market system, the fair value will be based on the publicly traded share price.
• | Risk-free interest rate - The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant for zero coupon U.S. Treasury notes with maturities approximately equal to the expected term of the options. |
• | Expected dividend - The expected dividend has been zero as the Company has never paid dividends and has no expectations to do so. |
• | Forfeiture rate - The Company estimated its forfeiture rate at 4% based on an analysis of its actual forfeitures and will continue to evaluate the adequacy of the forfeiture rate based on actual forfeiture experience, analysis of employee turnover behavior, and other factors. The impact from a forfeiture rate adjustment will be recognized in full in the period of adjustment, and if the actual number of future forfeitures differs from that estimated, the Company may be required to record adjustments to stock-based compensation expense in future periods. |
The fair value of employee stock options was estimated at the grant date using the following assumptions (No 2014 grants, unaudited):
December 31, | |||||||
2013 | 2012 | ||||||
Fair value | $ | 6.67 | $ | 5.19 | |||
Risk-free interest rate | 1.37% - 1.76% | 0.90 | % | ||||
Dividend yield | — | — | |||||
Expected life of options (years) | 5.00 – 6.25 | 5.50 | |||||
Stock price volatility | 68.00% - 72.00% | 69.89 | % | ||||
The recognized employee stock-based compensation in the statement of operations is as follows (in thousands):
Nine months ended September 30, | Years ended December 31, | ||||||||||||||
2014 | 2013 | 2013 | 2012 | ||||||||||||
(unaudited) | |||||||||||||||
Cost of sales | $ | 64 | $ | 74 | $ | 108 | $ | 111 | |||||||
Research and development | 51 | 65 | 82 | 153 | |||||||||||
General and administrative | 130 | 364 | 741 | 407 | |||||||||||
Sales and marketing | 236 | 201 | 302 | 66 | |||||||||||
Total | $ | 481 | $ | 704 | $ | 1,233 | $ | 737 | |||||||
No income tax benefit has been recognized in the statement of operations for the periods presented. The total compensation cost related to unvested stock option grants not yet recognized as of September 30, 2014 (unaudited) was $2.2 million, and the weighted average period over which these grants was expected to vest is 5.5 years.
Stock based compensation expense related to non-employee consultants totaled $0.1 million for the year ended December 31, 2013, and there was no such expense for the nine months ended September 30, 2014 (unaudited) and 2013 and for the year ended December 31, 2012, respectively.
The Company has adopted an Employee Stock Purchase Plan which contains an "evergreen provision" that allows for an annual increase in the number of shares available for issuance. This plan provides for an aggregate limit on the number of shares of common stock that may be issued over the course of its ten-year term of 2,250,000 shares. The Company has not issued any shares or awards under this plan, as it is not effective.
F-29
9. Fair value measurements
Assets and Liabilities Measured at Fair Value on a Recurring Basis
Fair values based on quoted market prices for identical assets are considered Level I, while those using significant unobservable inputs are Level III. Fair value inputs used to value the common stock warrant liabilities and preferred stock warrant liabilities and the required Level III disclosures are included in note 5 Common and convertible preferred stock warrant liabilities. The following table summarizes the Company’s assets and liabilities that require fair value measurements on a recurring basis and their respective input levels based on the fair value hierarchy (in thousands):
Fair Value Measurements at | |||||||||||
September 30, 2014 | December 31, | ||||||||||
2013 | 2012 | ||||||||||
Level I | (unaudited) | ||||||||||
Money Market Funds | $ | 4 | $ | 378 | $ | 2 | |||||
Level III | |||||||||||
Common Stock Warrant Liabilities | 42 | 32 | 25 | ||||||||
Preferred Stock Warrant Liabilities | — | 385 | 438 | ||||||||
$ | 46 | $ | 795 | $ | 465 | ||||||
Preferred and common stock warrant liabilities
The following table summarizes the activity in the common stock and preferred stock warrant liabilities measured at fair value on a recurring basis using significant unobservable inputs (Level III):
Preferred stock warrant liabilities | Common stock warrant liabilities | ||||||
Balance at January 1, 2012 | $ | 412 | $ | 215 | |||
Changes in warrant liability fair value | 26 | (190 | ) | ||||
Balance at December 31, 2012 | 438 | 25 | |||||
Changes in warrant liability fair value | (53 | ) | 7 | ||||
Balance at December 31, 2013 | 385 | 32 | |||||
Changes in warrant liability fair value | (385 | ) | 10 | ||||
Balance at September 30, 2014 (unaudited) | $ | — | $ | 42 | |||
Certain assets and liabilities are measured at fair value on a nonrecurring basis and therefore are not included in the tables above. Modified notes payable determined to be extinguished were measured and recognized at fair value on the extinguishment date. The fair values of these notes were calculated using discounted cash flow models that utilized certain unobservable inputs (Level III).
F-30
10. Net income/(loss) per share
Basic net loss per share is calculated by dividing net loss applicable to common stockholders by the weighted-average number of shares outstanding during the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by adjusting the weighted-average number of shares outstanding for the dilutive effect of common stock equivalents outstanding for the period, determined using the treasury-stock, or the if-converted, method. For purposes of the diluted net loss per share calculation, redeemable convertible preferred stock, warrants to purchase redeemable convertible preferred stock, stock options and unvested restricted stock are considered to be common stock equivalents, but have been excluded from the calculation of diluted net loss per share, as their effect, including the related impact to the numerator of the fair value adjustment of the warrant and the impact to the denominator of the warrant shares, would be anti-dilutive for all periods presented. Therefore, basic and diluted net loss per share applicable to common stockholders were the same for all periods presented.
Nine months ended September 30, | Years ended December 31, | ||||||||||||||
2014 | 2013 | 2013 | 2012 | ||||||||||||
(in thousands except share and per share data) | |||||||||||||||
(unaudited) | |||||||||||||||
Numerator: | |||||||||||||||
Net loss applicable to common shareholders | $ | (965 | ) | $ | (6,765 | ) | $ | (7,072 | ) | $ | (2,507 | ) | |||
Denominator: | |||||||||||||||
Weighted-average number of common shares used in computing net loss per share applicable to common stockholders - basic and diluted | 3,743,084 | 3,042,029 | 3,200,286 | 2,652,275 | |||||||||||
Net loss per share applicable to common stockholders - basic and diluted | $ | (0.26 | ) | $ | (2.22 | ) | $ | (2.21 | ) | $ | (0.95 | ) | |||
Historical outstanding anti-dilutive securities not included in diluted net loss per share calculation | |||||||||||||||
Preferred stock | 7,974,932 | 7,933,578 | 7,933,578 | 7,933,578 | |||||||||||
Preferred stock warrants | — | 122,552 | 122,552 | 245,103 | |||||||||||
Common stock warrants | 3,892,249 | 3,506,484 | 3,552,984 | 3,906,484 | |||||||||||
Common stock options | 1,469,055 | 1,788,923 | 1,685,054 | 1,562,845 | |||||||||||
Total | 13,336,236 | 13,351,537 | 13,294,168 | 13,648,010 | |||||||||||
F-31
11. Income taxes
The Company reported net losses for the nine months ended September 30, 2014 (unaudited) and 2013 and for all years through December 31, 2013. Accordingly, no provision for income taxes has been recorded for the nine months ended September 30, 2014 (unaudited) and 2013 or for the years ended December 31, 2013 and 2012.
The effective tax rate on income taxes is reconciled to the statutory federal income tax rate as follows:
December 31, 2013 | December 31, 2012 | ||||
Tax computed at the federal statutory rate | (34) | % | (34) | % | |
State income tax, net of federal benefit | (3) | % | (4) | % | |
Permanent differences and other | 0 | % | (2) | % | |
State tax adjustments | (4) | % | 96 | % | |
Change in tax rate | 0 | % | 63 | % | |
Stock option compensation | 6 | % | 9 | % | |
Changes in valuation allowance | 35 | % | (129) | % | |
Actual income tax rate | 0 | % | 0 | % | |
Deferred tax assets and liabilities are determined based on differences between financial reporting and tax bases of assets and liabilities, and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. Significant components of the deferred tax assets are as follows:
December 31, 2013 | December 31, 2012 | ||||||
Deferred tax assets: | |||||||
Net operating losses | $ | 27,843 | $ | 26,643 | |||
Research tax credits | 2,062 | 1,905 | |||||
Accrued expenses | 3,184 | 1,358 | |||||
Intangible assets | 1,946 | 2,638 | |||||
Stock-based compensation | 941 | 952 | |||||
Other | 205 | 361 | |||||
36,181 | 33,857 | ||||||
Valuation allowance | (36,002 | ) | (33,789 | ) | |||
Total deferred tax assets | 179 | 68 | |||||
Deferred tax liabilities: | |||||||
Fixed assets | (179 | ) | (68 | ) | |||
Total deferred tax liabilities | (179 | ) | (68 | ) | |||
Total deferred tax assets, net of valuation allowance | $ | — | $ | — | |||
A valuation allowance of approximately $36.0 million, $36.0 million and $33.8 million at September 30, 2014 (unaudited), December 31, 2013 and December 31, 2012, respectively, has been recorded to offset net deferred tax assets, as the Company is unable to conclude that it is more likely than not that such deferred tax assets will be realized.
At December 31, 2013, the Company had federal tax net operating loss carryforwards of $72.4 million, California tax net operating loss carryforwards of $41.2. The federal net operating loss carryforwards will begin to expire in
F-32
2020. A portion of the state net operating loss carryforwards expired in 2012 and the remaining state net operating losses will continue to expire between 2014 and 2033 if not previously utilized.
At December 31, 2013, the Company had federal research and development credit carryforwards of $1.2 million and state research credit carryforwards of $1.4 million. The federal research and development credit carryforwards begins to expire in 2022. The state research credit carryforwards do not expire.
The Company is subject to U.S. federal and state income taxes. The Company is no longer subject to U.S. federal or state income tax examinations for years ended before December 31, 2010 and December 31, 2009, respectively; however, to the extent allowed by law, the tax authorities may have the right to examine prior periods where net operating losses or tax credits were granted.
The tax benefits related to future utilization of federal and state net operating loss carryforwards, credit carryforwards, and other deferred tax assets may be limited or lost if cumulative changes in ownership exceeds 50% within any three-year period. The Company has not completed a study to assess whether an ownership change has occurred or whether there have been multiple ownership changes since the Company’s formation due to the complexity and cost associated with such a study, and the fact that there may be additional such ownership changes in the future. Such a limitation may occur as a result of a change in ownership upon the planned initial public offering. If the Company has experienced an ownership change at any time since its formation, utilization of the NOL or credit carryforwards to offset future taxable income and taxes, respectively, would be subject to an annual limitation under Section 382 of the Code, which is determined by first multiplying the value of the Company’s stock at the time of the ownership change by the applicable long-term, tax-exempt rate, and then could be subject to additional adjustments, as required. Any limitation may result in expiration of all or a portion of the NOL or credit carryforwards before utilization. Until a study is completed and any limitation known, no amounts of domestic NOL or credit carryforwards are being considered as an uncertain tax position or disclosed as unrecognized tax benefits under FIN 48 since no benefits have been realized to date.
The research and development tax credit expired as of December 31, 2013. During May 2014 H.R. 4438 passed to reinstate and make permanent the research and development tax credit. Management believes it is uncertain whether the credit will be revived or not, and if it were in future the extent to which retroactive credits will be allowed is also uncertain. If the tax impact of the research and development credit was recognized, the Company does not anticipate any federal income tax benefit due to the existence of deferred tax assets offset by a valuation allowance.
12. Employee savings plan
The Company maintains a defined contribution 401(k) plan available to eligible employees. Employee contributions are voluntary and are determined on an individual basis, limited to the maximum amount allowable under federal tax regulations. The Company, at its discretion, may make certain contributions to the 401(k) plan. No contributions were approved or funded by the Company through September 30, 2014 (unaudited).
13. Subsequent events
The Company has evaluated subsequent events through October 30, 2014, the date of the issuance of these financial statements, and except as disclosed in Note 3 and 7, no events require adjustment to, or disclosure in, the accompanying financial statements.
F-33
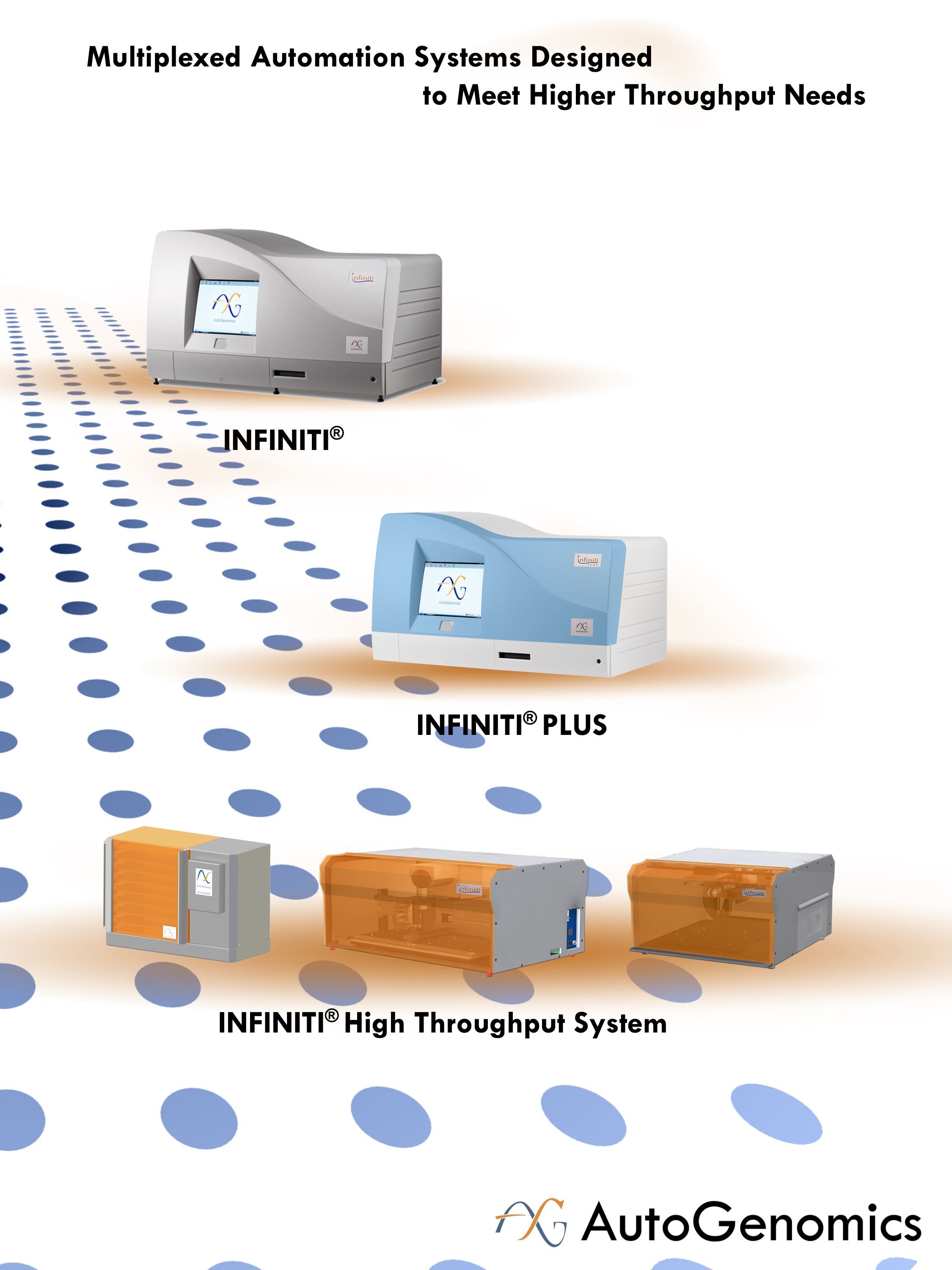

Shares
Common Stock
__________________
PROSPECTUS
, 2014
__________________
Stifel
Canaccord Genuity
Cantor Fitzgerald & Co.
Neither we nor any of the underwriters have authorized anyone to provide information difference from that contained in this prospectus. When you make a decision about whether to invest in our common stock, you should not rely upon any information other than the information in this prospectus. Neither the delivery of this prospectus nor the sale of our common stock means that information contained in this prospectus is correct after the date of this prospectus. This prospectus is not an offer to sell or solicitation of an offer to buy these shares of common stock in any circumstances under which the offer or solicitation is unlawful.
Part II
Information Not Required in Prospectus
Item 13. Other expenses of issuance and distribution.
The table below lists various expenses, other than underwriting discounts and commissions, we expect to incur in connection with the sale and distribution of the securities being registered hereby. All the expenses are estimates, except the Securities and Exchange Commission registration fee, the FINRA filing fee and the NASDAQ listing fee.
Type | Amount | |||
Securities and Exchange Commission Registration Fee | $ | 7,728 | ||
FINRA Filing Fee | 9,500 | |||
NASDAQ Fee | 125,000 | |||
Legal Fees and Expenses | 1,500,000 | |||
Accounting Fees and Expenses | 200,000 | |||
Printing and Engraving Expenses | 50,000 | |||
Transfer Agent and Registrar Fees | 20,000 | |||
Miscellaneous Expenses | 87,772 | |||
Total | $2,000,000 | |||
* To be filed by amendment.
Item 14. Indemnification of Directors and Officers.
Section 102 of the Delaware General Corporation Law, or DGCL, as amended, allows a corporation to eliminate the personal liability of directors of a corporation to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, except where the director breached the duty of loyalty, failed to act in good faith, engaged in intentional misconduct or knowingly violated a law, authorized the payment of a dividend or approved a stock repurchase in violation of Delaware corporate law or obtained an improper personal benefit. Our certificate of incorporation eliminates this liability.
Section 145 of the DGCL provides for the indemnification of officers, directors and other corporate agents in terms sufficiently broad to indemnify such persons under circumstances for liabilities (including reimbursement for expenses incurred) arising under the Securities Act. Our certificate of incorporation and bylaws provide for indemnification of our officers, directors, employees and agents to the extent and under the circumstances permitted under the DGCL.
The Underwriting Agreement (Exhibit 1.1) provides for indemnification by the underwriters of us, our directors and officers, and by us of the underwriters, for some liabilities arising under the Securities Act, and affords some rights of contributions with respect thereto.
We have executed indemnification agreements with each of our directors and each of our executive officers. These agreements provide indemnification to our directors and executive officers under certain circumstances for acts or omissions that may be sufficiently broad to indemnify such persons for liabilities arising under the Securities Act.
II-1
Item 15. Recent Sales of Unregistered Securities.
Since January 1, 2011, we have issued the following securities that were not registered under the Securities Act of 1933, as amended:
(a) Stock Option Grants
From January 1, 2011 through October 24, 2014, we granted stock options to our employees, directors and consultants pursuant to which the optionees may purchase up to an aggregate of 827,442 shares of our common stock at a weighted average exercise price of $6.25 per share. Of the options we granted during this period, options to purchase a total of 282,221 shares have been forfeited or canceled. The sale and issuance of the stock options were exempt from registration under Section 4(2) of the Securities Act or Rule 701 under the Securities Act.
(b) Issuances of Common Stock and related Warrants
From September 2011 through October 2013, we issued and sold 440,751 shares of our common stock to our employees, directors and consultants upon the exercise of stock options at a weighted average exercise price of $0.76 per share for an aggregate exercise price equal to approximately $0.3 million. These underlying stock options were granted between January 2002 and May 2011 pursuant to our equity incentive compensation plans and agreements.
In December 2012, we issued 29,700 shares of our common stock to the former landlord for our Vista, California facility in connection with an amendment and forbearance agreement that we entered into with the landlord. This amendment and forbearance agreement provided for an extension of the period of time in which we had to satisfy our past due amounts owed to the landlord, which then totaled $1.1 million. The past due amount has since been satisfied by the issuance of a promissory note to the landlord in the principal amount of $997,000 with an interest at a rate of 10% per annum and maturity date of June 30, 2015.
In September 2013, we issued and sold 573,888 shares of our common stock to an investor at $6.97 per share, for an aggregate price of $4.0 million. In connection with this issuance, we issued this investor warrants for the purchase of 60,000 shares of our common stock at an exercise price of $6.97 per share and an expiration date of September 2018. One of the beneficial owners of this investor was subsequently elected to our board of directors at our 2013 annual stockholder meeting.
In December 2013, we issued 17,700 shares of our common stock to consultants for professional fees performed during 2013, in a non-cash exchange for services valued at approximately $0.1 million.
In December 2013, we issued 45,000 shares of common stock to a consultant in exchange for the surrender and cancellation of warrants to purchase 24,750 and 30,000 shares of common stock originally issued in October 2011 and March 2013 at exercise prices of $0.76 and $0.75, respectively.
No underwriters were involved in the foregoing sales of securities. The sale and issuance of the securities described in this Item 15(b) above were exempt from registration under Section 4(2) of the Securities Act or Rule 701 under the Securities Act.
(c) Issuances of Preferred Stock and related Warrants
From April 2011 through July 2012, we issued 2,991 shares of our Series C Convertible Preferred Stock to three existing holders at a weighted average exercise price of $3.30 per share for an aggregate exercise price of $9,870, pursuant to the exercise by such holders of then outstanding warrants to purchase such shares that were originally issued in connection with the sale and issuance of our Series C Convertible Preferred Stock in 2007.
In May, July and August 2011, warrants to purchase 59,072 shares of our Series C Convertible Preferred Stock at an exercise price of $3.30 per share were due to expire. These warrants were originally issued in connection with the sale and issuance of our Series C Convertible Preferred Stock in 2007. We offered to extend the expiration date of these warrants to May 2012, July 2012 and August 2012, respectively, in exchange for an increase in the exercise price of such warrants from $3.30 per share to $3.80 per share. Holders of warrants for the purchase of 51,800 of these warrants accepted the offer. The remaining warrants, for the purchase of 7,272 shares of our Series C Convertible Preferred Stock, expired in accordance with their respective terms.
In July and August 2011, warrants to purchase 348,910 shares of our Series C Convertible Preferred Stock at an exercise price of $2.75 per share were due to expire. These warrants were originally issued in connection with the
II-2
sale and issuance of our Series C Convertible Preferred Stock in 2007. We offered to extend the expiration date of these warrants to July 2012 and August 2012, respectively, in exchange for an increase in the exercise price of such warrants from $2.75 per share to $3.25 per share. Holders of warrants for the purchase of 334,364 shares of our Series C Convertible Preferred Stock accepted the offer. The remaining warrants, for the purchase of 14,546 shares of our Series C Convertible Preferred Stock, expired in accordance with their respective terms.
In November 2011, we issued and sold an aggregate of 685,555 shares of our Series E Convertible Preferred Stock to eight investors at a purchase price of $2.75 per share, for an aggregate purchase price of $1.9 million. As part of this offering, we also issued to these investors five-year warrants for the purchase of 339,351 shares of our common stock at an exercise price of $8.33 per share.
In July and August 2012, warrants to purchase 42,300 shares of our Series C Convertible Preferred Stock at an exercise price of $3.80 per share were due to expire. These warrants were originally issued in connection with the sale and issuance of our Series C Convertible Preferred Stock in 2007 (and, as described above, were amended in 2011). We offered to extend the expiration date of these warrants to July 2013 and August 2013, respectively, in exchange for an increase in the exercise price of such warrants from $3.80 per share to $4.05 per share. Holders of warrants for the purchase of 34,064 of these warrants accepted the offer. The remaining warrants, for the purchase of 8,236 shares of our Series C Convertible Preferred Stock, expired in accordance with their respective terms.
In July and August 2012, warrants to purchase 342,000 shares of our Series C Convertible Preferred Stock at an exercise price of $3.25 per share were due to expire. These warrants were originally issued in connection with the sale and issuance of our Series C Convertible Preferred Stock in 2007 (and, as described above, were amended in 2011). We offered to extend the expiration date of these warrants to July 2013 and August 2013, respectively, in exchange for an increase in the exercise price of such warrants from $3.25 per share to $3.50 per share. Holders of warrants for the purchase of 308,727 of these warrants accepted the offer. The remaining warrants, for the purchase of 33,273 shares of our Series C Convertible Preferred Stock, expired in accordance with their respective terms.
In August 2012, we issued and sold 47,000 shares of our Series E Convertible Preferred Stock at a purchase price of $2.75 per share, for an aggregate purchase price in the form of surrender of accounts payable of $129,250. In connection with this sale, we also issued to this investor a five year warrant for the purchase of 23,265 shares of our common stock at an exercise price of $8.33 per share.
In November 2012, the applicable holders of $7.5 million in aggregate principal amount of and accrued interest on our outstanding subordinated promissory notes with rates of interest ranging from six percent to 12% that were then past due or scheduled to come due in the immediate future surrendered their right to payment of those promissory notes in exchange for an aggregate of 4,290,136 shares of our Series NC Convertible Preferred Stock. In connection with this surrender and issuance, these note holders surrendered warrants for the purchase of an aggregate of 637,813 shares of our common stock with exercise prices ranging from $5.24 per share to $15.15 per share in exchange for warrants of a like tenor for the purchase of an aggregate of 637,813 shares of our common stock with exercise prices of $4.55 per share that are exercisable through November 2017.
In July and August 2013, warrants to purchase 35,700 shares of our Series C Convertible Preferred Stock at an exercise price of $4.05 were due to expire. These warrants were originally issued in connection with the sale and issuance of our Series C Convertible Preferred Stock in 2007 (and, as described above, were amended in 2012). Prior to their expiration, we extended the expiration date of all of these warrants to July 2014 and August 2014, respectively, in exchange for an increase in the exercise price of such warrants from $4.05 per share to $4.1325 per share.
In July and August 2013, warrants to purchase 318,727 shares of our Series C Convertible Preferred Stock at an exercise price of $3.50 were due to expire. These warrants were originally issued in connection with the sale and issuance of our Series C Convertible Preferred Stock in 2007 (and, as described above, were amended in 2012). Prior to their expiration, we extended the expiration date of all of these warrants to July 2014 and August 2014, respectively, in exchange for an increase in the exercise price of such warrants from $3.50 per share to $3.5825 per share.
No underwriters were involved in the foregoing sales of securities. The securities described above were sold and issued to accredited investors in reliance upon the exemption from the registration requirements of the Securities
II-3
Act, as set forth in Section 3(a)(9) and Section 4(2) of the Securities Act and Rule 506 of Regulation D under the Securities Act.
(d) Issuances of Subordinated Promissory Notes and related Warrants
In August 2011, we and the applicable note holder exchanged promissory notes with an aggregate principal amount of $3,071,000 that were originally set to mature in July 2011 for promissory notes with an aggregate principal amount of $3,900,000, set to expire December 2011 and February 2012. In connection with this exchange, we canceled warrants to purchase 281,160 shares of our common stock at an exercise price of $12.12 per share held by the note holder and reissued a warrant to purchase 429,000 shares of our common stock at an exercise price of $9.09 to the note holder which will expire in August 2016.
In October and December 2011, we and the applicable note holders amended promissory notes with an aggregate principal amount of $595,000 that were set to expire in November and December 2011, by extending the maturity dates to dates in 2012. In connection with this amendment, we amended warrants to purchase 55,764 shares of our common stock by reducing the exercise price from $12.12 per share to $9.09 per share.
From November 2011 through January 2012, we issued $695,000 aggregate principal amount of promissory notes to certain accredited investors with an interest rate of 12.0% per annum and maturity date in April 2012. In connection with this offering, we issued warrants to purchase 114,675 shares of our common stock at an exercise price of $5.24 per share to the note holders which will expire five years from their date of issue.
In February 2012, we and the applicable note holder amended a certain promissory note with a principal amount of $250,000 set to mature in April 2012, by extending the maturity date to June 2012. In connection with this amendment, we amended a warrant to purchase 23,430 shares of our common stock at an exercise price of $15.15 per share held by the note holder, by reducing the exercise price to $9.09 per share and extending the term of the warrant until June 2017.
In February 2012, we and the applicable note holders amended certain promissory notes with an aggregate principal amount of $400,000 set to mature in February 2012, by extending the maturity dates to April 2012. In connection with these amendments, we amended warrants to purchase 37,488 shares of our common stock at an exercise price of $12.12 per share held by the note holder, by reducing the exercise price to $9.09 per share and extending the term of the warrant until April 2017.
In March 2012, we issued $1,150,000 aggregate principal amount of promissory notes to certain accredited investors with an interest rate of 10.0% per annum, and with the principal and accrued interest balances generally due in June 2012. In connection with the issuance of these notes, we issued warrants to purchase 113,850 shares of our common stock with an exercise price of $5.24 per share which will expire five years from their date of issue.
In November 2012, the applicable holders of $14.4 million in aggregate principal amount of and accrued interest on our outstanding subordinated promissory notes with rates of interest ranging from six percent to 13% that were then past due or scheduled to come due in the immediate future surrendered their right to payment of those promissory notes in exchange for new promissory notes in aggregate principal amount of $14.4 million with eight and one-half percent rates of interest and November 2015 maturity dates. In connection with this surrender and issuance, these note holders surrendered warrants for the purchase of an aggregate of 1,156,013 shares of our common stock with exercise prices ranging from $5.24 per share to $15.15 per share in exchange for warrants of a like tenor for the purchase of an aggregate of 1,734,020 shares of our common stock with exercise prices of $5.30 per share that are exercisable through November 2017.
In April 2013, warrants for 90,273 shares of our common stock were issued as part of the refinance of $0.6 million of subordinated notes with an expiring maturity date of March 31, 2013 and a new maturity date of March 31, 2014. The warrants had an exercise price of $5.25 per share and an expiration date of April 2018.
In February 2014, we and the applicable note holder amended a certain promissory note with a principal amount of $2.3 million set to mature in March 2013, by extending the maturity date to December 2014. In connection with this amendment and issuance, we issued a warrant to this note holder for 400,000 shares of our common stock with an exercise price of $5.31 per share, which will expire seven years from its date of issue. In March 2013, we issued a promissory note to this note holder in the principal amount of $350,000.00 with an interest rate of 8.5% and a November 2015 maturity date. In July 2013, we issued a promissory note to this note holder in the principal amount of $250,000 with an interest rate of 8.5% and a December 2013 maturity date.
II-4
No underwriters were involved in the foregoing sales of securities. The sale and issuance of the securities described in this Item 15(d) above were exempt from registration under Section 3(a)(9) and Section 4(2) of the Securities Act or Rule 506 of Regulation D under the Securities Act.
(e) Issuances of Other Warrants
In August 2011, we issued to certain non-employee members of our board of directors warrants to purchase an aggregate of 33,000 shares of our common stock at an exercise price of $5.24 per share. These warrants will expire five years from their date of issue.
In May 2012, we issued to certain non-employee members of our board of directors warrants to purchase an aggregate of 9,900 shares of our common stock at an exercise price of $5.24 per share. These warrants will expire five years from their date of issue.
No underwriters were involved in the foregoing sales of securities. The securities described in this Item 15(c) above were sold and issued to accredited investors in reliance upon the exemption from the registration requirements of the Securities Act, as set forth in Section 4(2) of the Securities Act or Rule 506 of Regulation D under the Securities Act.
Item 16. Exhibits and Financial Statement Schedules.
See "Exhibit Index." All schedules have been omitted because they are not required or because the required information is given in the financial statements or notes to those statements.
Item 17. Undertakings.
The undersigned registrant hereby undertakes to provide to the underwriter at the closing specified in the underwriting agreement, certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended, may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933, as amended, and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933, as amended, and will be governed by the final adjudication of such issue.
The registrant hereby undertakes that:
(a) | For purposes of determining any liability under the Securities Act of 1933, as amended, the information omitted from a form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in the form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act of 1933, as amended, shall be deemed to be part of this registration statement as of the time it was declared effective. |
(b) | For the purpose of determining any liability under the Securities Act of 1933, as amended, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-5
Signatures
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Vista, State of California, on this day of October 30, 2014.
AUTOGENOMICS, INC. | ||
By: | /s/ Fareed Kureshy | |
Fareed Kureshy | ||
Chairman, President and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Act of 1933, as amended, this Registration Statement has been signed by the following persons in the capacities indicated below on the dates indicated below.
Name | Title | Date | ||
/s/ Fareed Kureshy | Chairman, President, Chief Executive Officer and Director (principal executive officer) | October 30, 2014 | ||
Fareed Kureshy | ||||
/s/ John R. Zacamy | Chief Financial Officer and Secretary (principal financial officer and principal accounting officer) | October 30, 2014 | ||
John R. Zacamy | ||||
/s/ Stephen Allison* | Director | October 30, 2014 | ||
Stephen Allison | ||||
/s/ Charles Birmingham* | Director | October 30, 2014 | ||
Charles Birmingham | ||||
/s/ Joseph S. Coyne, Dr.P.H., MPH* | Director | October 30, 2014 | ||
Joseph S. Coyne, Dr.P.H., MPH | ||||
/s/ Laurence M. Demers, Ph.D.* | Director | October 30, 2014 | ||
Laurence M. Demers, Ph.D. | ||||
/s/ Thomas V. Hennessey, Jr.* | Director | October 30, 2014 | ||
Thomas V. Hennessey, Jr. | ||||
/s/ Adam Levinson* | Director | October 30, 2014 | ||
Adam Levinson | ||||
/s/ Peter Wilding, Ph.D.* | Director | October 30, 2014 | ||
Peter Wilding, Ph.D. | ||||
*By: | /s/ Fareed Kureshy | |
Fareed Kureshy | ||
Attorney-in-fact | ||
Exhibit Index
Exhibit Number | Exhibit Description | ||
1.1 | * | Form of Underwriting Agreement | |
3.1 | ** | Certificate of Incorporation of the Registrant, as amended | |
3.2 | ** | Amended and Restated Bylaws of the Registrant | |
4.1 | * | Form of Common Stock Certificate | |
4.5 | ** | Agreement to Furnish Debt Instruments | |
5.1 | * | Opinion of Bingham McCutchen LLP, related to the shares of common stock being sold in the initial public offering | |
10.1 | ** | Indemnification Agreement dated April 28, 2010, by and between the Registrant and Ramanath Vairavan (all other Indemnification Agreements, which are substantially identical in all material respects, except as to the parties thereto and the dates of execution, are omitted pursuant to Instruction 2 to Item 601 of Regulation S-K) | |
10.2 | #** | 2000 Equity Incentive Plan and forms of agreements relating thereto | |
10.3 | #** | Form of Nonstatutory Stock Option Agreement used for certain grants outside of the 2000 Equity Incentive Plan | |
10.4 | #** | 2008 Equity Incentive Award Plan and form of stock option agreement relating thereto | |
10.5 | #** | 2008 Employee Stock Purchase Plan | |
10.6 | #** | Non-Employee Director Compensation Policy | |
10.7 | ** | Registration Rights Agreement dated July 19, 2006, by and among the Registrant, MESA Development Inc. of Nevada and the purchasers of the Registrant’s Series C Convertible Preferred Stock | |
10.8 | ** | Registration Rights Agreement dated September 27, 2013, by and between the Registrant and Genomic Generation Partners, LLC | |
10.9 | ** | Non-Exclusive Sub-License Agreement dated April 14, 2006, between the Registrant and Mayo Foundation for Medical Education and Research | |
10.10 | ** | Nonexclusive Patent License Agreement dated April 14, 2006, between the Registrant and the Mayo Foundation for Medical Education and Research | |
10.11 | ** | Amended and Restated Air Commercial Real Estate Standard Industrial/Commercial Single Tenant Lease-Net, dated July 1, 2013, between Garrison Vista LLC, as successor-in-interest to PCCP DJ Ortho, LLC, and the Registrant | |
10.12 | ** | Promissory Note (No. 201401), issued by the Registrant to PCCP DJ Ortho, LLC in the principal amount of $997,000 | |
10.13 | ** | Warrant to Purchase Common Stock (No. CS-07), dated January 15, 2010, issued to Dennis Repp (all other Warrants to Purchase Common Stock that are substantially identical in all material respects to this Warrant to Purchase Common Stock, except as to the parties thereto, number of warrant shares, exercise price and expiration date, are omitted pursuant to Instruction 2 of Item 601 of Regulation S-K) | |
10.14 | ** | Warrant to Purchase Common Stock (No. E-01), dated November 29, 2011, issued to Roland F. Smith (all other Warrants to Purchase Common Stock that are substantially identical in all material respects to this Warrant to Purchase Common Stock, except as to the parties thereto, number of warrant shares, are omitted pursuant to Instruction 2 of Item 601 of Regulation S-K) | |
10.15 | ** | 8-1/2% Subordinated Note (No. 201211-001), issued by the Registrant to Argus Reinsurance, Ltd in the principal amount of $312,671.23 (all other 8-1/2% Subordinated Notes which are substantially identical in all material respects to this 8-1/2% Subordinated Note, except as to the parties thereto and principal amounts, are omitted pursuant to Instruction 2 of Item 601 of Regulation S-K) | |
10.16 | ** | Security Agreement by and between the Registrant and Citibank, N.A., acting through its Citibank Agency & Trust Division, as collateral agent for the holders of the New Notes (as defined therein) | |
10.17 | ** | Warrant to Purchase Common Stock (No. 201211-050) issued to AR Properties (all other Warrants to Purchase Common Stock which are substantially identical in all material respects to this Warrant to Purchase Common Stock, except as to the parties thereto, number of warrant shares and exercise prices, are omitted pursuant to Instruction 2 of Item 601 of Regulation S-K) | |
10.18 | ** | Promissory Note (No. 201301-001), issued by the Registrant to A R Properties in the principal amount of $2,400,000 | |
10.19 | ** | 8-1/2% Subordinated Note (No. 201304-001), issued by the Registrant to MemorialCare Innovation Fund, LP, in the principal amount of $635,725, and letter agreement dated September 25, 2014 from Registrant to MemorialCare Innovation Fund, LP. | |
10.20 | #** | Employment Agreement dated January 3, 2013, by and between the Registrant and Fareed Kureshy | |
23.1 | Consent of Marcum LLP | ||
23.2 | * | Consent of Bingham McCutchen LLP (included in Exhibit 5.1) | |
24.1 | ** | Power of Attorney (filed with the SEC on October 7, 2014) | |
# | Indicates a management contract or compensatory plan or arrangement. |
* | To be filed by amendment. |
** Previously filed.