Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Cell Source, Inc. | Financial_Report.xls |
| EX-23.1 - EXHIBIT 23.1 - Cell Source, Inc. | v392018_ex23-1.htm |
As filed with the Securities and Exchange Commission on October 24, 2014
Registration No. 333-197972
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 2 to
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
CELL SOURCE, INC.
(Exact name of registrant as specified in its charter)
| Nevada | 2836 | 32-0379665 | ||
| (State or jurisdiction of | (Primary Standard Industrial | (I.R.S. Employer | ||
| incorporation or organization) | Classification Code Number) | Identification No.) |
65 Yigal Alon Street
Tel Aviv Israel 67433
(Address of principal executive offices) (zip code)
011 972 3 562-1755
(Registrant’s telephone number, including area code)
(Former Name or Former Address, if Changed Since Last Report)
Copies to:
Gregory Sichenzia, Esq.
David Manno, Esq.
Sichenzia Ross Friedman Ference LLP
61 Broadway
New York, New York 10006
Phone: (212) 930-9700
Fax: (212) 930-9725
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement is declared effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box. x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
¨ Large accelerated filer
¨ Accelerated filer
¨ Non-accelerated filer
x Smaller reporting company
CALCULATION OF REGISTRATION FEE
| Title of each class of
securities to be registered | Amount to be
Registered (1) | Proposed Maximum
Offering Price Per Security | Proposed Maximum
Aggregate Offering Price | Amount of
Registration Fee | ||||||||||||
| Common Stock, $0.001 par value per share | 9,618,648 | $ | 0.70 | (2) | $ | 6,733,053.60 | $ | 782.38 | * | |||||||
(1) Securities being registered hereunder include (a) 4,859,324 shares of common stock; (b) 4,759,324 shares of common stock issuable upon exercise of warrants; and (c) pursuant to Rule 416, such indeterminate number of additional shares of common stock as may be issued after the date hereof as a result of stock splits, stock dividends or similar transactions.
(2) Estimated solely for purposes of calculating the registration fee pursuant to Rule 457(c) under the Securities Act of 1933, as amended, using the average of the high and low prices as reported on October 23, 2014
THE REGISTRANT HEREBY AMENDS THIS REGISTRATION STATEMENT ON SUCH DATE OR DATES AS MAY BE NECESSARY TO DELAY ITS EFFECTIVE DATE UNTIL THE REGISTRANT SHALL FILE A FURTHER AMENDMENT WHICH SPECIFICALLY STATES THAT THIS REGISTRATION STATEMENT SHALL THEREAFTER BECOME EFFECTIVE IN ACCORDANCE WITH SECTION 8(A) OF THE SECURITIES ACT OF 1933 OR UNTIL THE REGISTRATION STATEMENT SHALL BECOME EFFECTIVE ON SUCH DATE AS THE SECURITIES AND EXCHANGE COMMISSION, ACTING PURSUANT TO SUCH SECTION 8(A), MAY DETERMINE.
*Previously paid.
| 2 |
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
PRELIMINARY PROSPECTUS, SUBJECT TO COMPLETION, DATED OCTOBER 24, 2014
CELL SOURCE, INC.
9,618,648 SHARES OF COMMON STOCK
This prospectus relates to the sale of up to 9,618,648 shares of common stock of Cell Source, Inc. by the Selling Stockholders, including 4,859,324 outstanding shares and 4,759,324 shares issuable upon the exercise of outstanding warrants.
There are no underwriting arrangements to sell the shares of common stock that are being registered hereunder. The prices at which the selling stockholders may sell shares will be determined by the prevailing market price for the shares or in privately negotiated transactions. We will not receive any proceeds from the sale of these shares by the selling stockholders. All expenses of registration incurred in connection with this offering are being borne by us, but all selling and other expenses incurred by the selling stockholders will be borne by the selling stockholders.
The Company’s common stock is quoted under the symbol “CLCS" on the OTCQB. There has been no active trading of our common stock. On October 23, 2014, the last reported sale price of our common stock as reported on the OTCQB was $0.70 per share.
We are an emerging growth company as that term is used in the Jumpstart Our Business Startups Act of 2012, and, as such, we have elected to take advantage of certain reduced public company reporting requirements for this prospectus and future filings.
Investing in our common stock is highly speculative and involves a high degree of risk. You should carefully consider the risks and uncertainties in the section entitled “Risk Factors” beginning on page 9 of this prospectus before making a decision to purchase our stock.
We may amend or supplement this prospectus from time to time by filing amendments or supplements as required. You should read the entire prospectus and any amendments or supplements carefully before you make your investment decision.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 2014.
| 3 |
CELL SOURCE, INC.
TABLE OF CONTENTS
No person is authorized in connection with this prospectus to give any information or to make any representations about us, the Selling Stockholders, the securities or any matter discussed in this prospectus, other than the information and representations contained in this prospectus. If any other information or representation is given or made, such information or representation may not be relied upon as having been authorized by us or any Selling Stockholder. This prospectus does not constitute an offer to sell, or a solicitation of an offer to buy the securities in any circumstances under which the offer or solicitation is unlawful. Neither the delivery of this prospectus nor any distribution of securities in accordance with this prospectus shall, under any circumstances, imply that there has been no change in our affairs since the date of this prospectus. The prospectus will be updated and updated prospectuses made available for delivery to the extent required by the federal securities laws.
| 4 |
This summary highlights information contained elsewhere in this prospectus. It does not contain all of the information that you should consider before investing in our securities. You should read the entire prospectus carefully, including the section entitled “Risk Factors” and our consolidated financial statements and the related notes. Some of the statements contained in this prospectus, including statements under “Prospectus Summary” and “Risk Factors” as well as those noted in the documents incorporated herein by reference, are forward-looking statements and may involve a number of risks and uncertainties. We note that our actual results and future events may differ significantly based upon a number of factors. The reader should not put undue reliance on the forward-looking statements in this document, which speak only as of the date on the cover of this prospectus.
About Us
Cell Source, Inc. (the "Company") is a Nevada corporation formed on June 6, 2012 under the name Ticket to See, Inc. ("TTSI"). Prior to the Share Exchange (as defined below), we did not have any significant assets or operations.
Cell Source, Inc. is the parent company of Cell Source Ltd. ("Cell Source Israel"). Cell Source Israel was founded in Israel in 2011 in order to commercialize a suite of inventions relating to certain cancer treatments. Our target indications include treatment of lymphoma, multiple myeloma and BCLL (which is a common form of leukemia), facilitating transplantation acceptance (initially bone marrow transplantation and subsequently organ transplantation) and ultimately treating a variety of non-malignant diseases. Our lead prospective product is our patented Veto-Cell immune system management technology, which is an immune tolerance biotechnology that enables the selective blocking of immune responses. Our Veto-Cell immune system management technology is based on technologies patented, owned, and licensed to us by Research and Development Company Limited, Yeda Research and Development Limited, the commercial arm of Weizmann Institute of Science in Rehovot Israel ("Yeda"). We do not currently own any patents or other intellectual property and our total portfolio of intellectual property is limited to our rights to Yeda’s intellectual property pursuant to our license agreement with Yeda. We are an early stage biomedical technology development company and we do not have any revenues and no immediate source of revenue is planned in the near term. We do not produce nor do we sell any products or services. In order to commence commercial activity, we will have to complete human treatment protocols and then have them approved for human clinical trials for each of our product candidates. The development of these protocols can take between 1 to 4 years per product. The various stages of approval in the United States with respect to the FDA can take between five and eight years if not longer. This means that it is quite likely that we not begin selling products in the United States for another eight years or longer. The Company currently has only one product which is in a slightly more advanced stage of development. This product, the megadose drug combination, already has a final human treatment protocol which has been developed. A hospital in Italy, on its own initiative, plans to conduct human clinical trials for this product on a self-directed basis in the near future. The hospital, which applied for approval on May 14th, 2014, recently received final written approval from the Italian Medicine Association (AIFA), which is the Italian equivalent of the United States FDA, to begin a trial with a small number of patients. While this trial is not being conducted by the Company itself, because it involves the same treatment protocol the Company plans to use in the future, successful results in this trial can act as a springboard to a company sponsored study of the same protocol in Italy and potentially elsewhere. There is no assurance that any trials will be successful, and there is no assurance that any products will be safe and/or effective in treating patients. Furthermore, there can be no assurances that we will be granted any regulatory approval or that we will be able to sell any products anywhere in the world.
The Company's corporate headquarters is located at 65 Yigal Alon Street, 23rd Floor, Tel Aviv 67433, Israel, and the telephone number at such address is (972) 3 562-1755. The Company’s U.S. contact information is: 57 W. 57th St., Suite 400 New York, NY 10019 and the telephone number at such address is (646) 416-7896.
References to “we,” “us,” “our” and similar words refer to the Company and its subsidiaries after giving effect to the Share Exchange. References to “TTSI” refer to the Company and its business prior to the Share Exchange.
As of June 30, 2014 we had $889,332 in cash. Our historical burn rate was approximately $150,000 per month. However, we estimate that our current burn rate is approximately $100,000 per month and, as a result, we expect that the cash we currently have available will fund our operations through November 2014.
Recent Developments
Share Increase
On May 7, 2014, the Board of Directors and the majority stockholder of TTSI adopted resolutions approving an amendment (the "Amendment") of the Company's Articles of Incorporation to increase the number of authorized shares. Prior to the Amendment, the authorized shares of the Company consisted of 75,000,000 shares of common stock, $0.001 par value. The Amendment was filed with the Secretary of State of the State of Nevada on May 20, 2014 and increased the number of shares of common stock that the Company is authorized to issue from 75,000,000 shares to 200,000,000 shares. The Company also authorized 10,000,000 shares of preferred stock, par value $0.001, for designation in one or more series, with such designations, preferences and relative, participating, optional, or other special rights and qualifications, limitations, or restrictions thereof, as may, from time to time, be adopted by the Company's Board of Directors.
Name Change
On June 23, 2014, the majority stockholder of TTSI adopted resolutions approving an amendment of the Company's Articles of Incorporation to change the name of the corporation from Ticket to See, Inc. to Cell Source, Inc. The Amendment was filed with the Secretary of State of the State of Nevada on June 23, 2014 and changed the name of the Corporation from Ticket to See, Inc. to Cell Source, Inc., effective June 26, 2014. In connection with the name change, the trading symbol of the Company’s common stock was changed from TTSE to CLCS.
Share Exchange
On June 30, 2014 (the “Closing Date”), TTSI entered into and closed a Share Exchange Agreement (the “Share Exchange Agreement”) with Cell Source Israel and 100% of the shareholders of Cell Source Israel (the “CSL Shareholders”) whereby Cell Source Israel became the wholly-owned subsidiary of TTSI and TTSI changed its name to Cell Source, Inc. (the "Share Exchange"), and whereby certain CSL Shareholders, holding 18,245,923 of the outstanding shares of Cell Source Israel, transferred to the Company an aggregate of 18,245,923 shares of Cell Source Israel’s ordinary shares, each of nominal value of NIS 0.01 (“CSL Ordinary Shares”) in exchange for an aggregate of 18,245,923 newly issued shares of the Company's Common Stock, par value $0.001 per share (the “Company Common Stock” or the "Common Stock"). The aggregate of 18,245,923 shares of newly issued Company Common Stock represents 78.5% of the outstanding shares of Company Common Stock following the Closing Date. In addition, outstanding five (5) year warrants to acquire 4,859,324 CSL Ordinary Shares at an exercise price of $0.75 per share (the "CSL Warrants") were exchanged for newly issued warrants to purchase shares of Company Common Stock (the “Company Warrants”), which Company Warrants contain substantially similar terms as the CSL Warrants. In addition, outstanding warrants to acquire 2,043,835 CSL Ordinary Shares held by Dr. Reisner and Yeda were exchanged for warrants to purchase shares of Company Common Stock (the “Researcher Company Warrants”), which Researcher Company Warrants contain substantially similar terms as their warrants to acquire CSL Ordinary Shares. The aggregate of 6,903,159 Company Warrants and Researcher Company Warrants represents 77.5% of the outstanding warrants to purchase Common Stock of the Company following the Closing Date.
| 5 |
Cell Source Israel's Private Placement
Beginning in November 2013, Cell Source Israel collected and entered into a series of subscription agreements (the “Subscription Agreement”) with certain accredited investors (the “Investors”) in a private placement offering (the “Private Placement”). Cell Source Israel held closings of the Private Placement between December 9, 2013 through April 7, 2014, pursuant to which Cell Source Israel sold an aggregate of 4,759,324 Units (the "Units"), at a purchase price of $0.75 per Unit, for gross proceeds of $3,569,475. Each Unit consists of one (1) share of CSL Ordinary Shares and one (1) CSL Warrant. Each CSL Warrant entitled the holder to purchase one (1) share of CSL Ordinary Shares for a five (5) year period at an exercise price of $0.75 per share. In connection with the Private Placement, Cell Source Israel relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act of 1933, as amended (the “Securities Act”) and Rule 506 as promulgated under the Securities Act for transactions not involving a public offering.
Under the Subscription Agreement, the Investors were granted the following rights for a period of five (5) years commencing on the closing of the Private Offering: (i) in the event any shares of CSL Ordinary Shares or securities convertible, exchangeable or exercisable for CSL Ordinary Shares are issued at a price less than $0.75 per share (“Adjustment Event”), subject to certain adjustments, then additional CSL Ordinary Shares, or equivalents, will be issued to the Investors such that the aggregate holdings of the Investors is equal to the aggregate holding had such Investors initially purchased at the applicable lower price by which securities were issued in the Adjustment Event (except that certain issuances set forth in the Subscription Agreement would not be an Adjustment Event); and (ii) upon any financing by Cell Source whereby CSL Ordinary Shares or securities convertible into CSL Ordinary Shares are issued or sold (a “Subsequent Financing”), Investors have the right to participate in such Subsequent Financing (subject to customary exemptions). The Investors were also granted the right to elect up to two (2) independent board members. On May 29, 2014, the majority of the Investors granted certain groups of shareholders the right to elect, subject to the closing of the Share Exchange Agreement, Yoram Drucker, Itamar Shimrat, David Zolty, Ben Friedman and Dennis Brown to the Board of Directors of the Company. Furthermore, pursuant to the Subscription Agreement, in the event that the Registration Statement is declared effective, the Company is obligated to issue to certain founders of Cell Source Israel (Isaac Braun, Saar Dickman, Itamar Shimrat and Yoram Drucker) warrants to purchase an aggregate of 3,000,000 shares of Company Common Stock at an exercise price of $0.75 per share, subject to the same adjustments and terms as the Company Warrants.
In connection with the Private Placement, Cell Source Israel also entered into a Registration Rights Agreement (the “Registration Rights Agreement”) with the Investors, pursuant to which Cell Source Israel agreed to file a registration statement (the “Registration Statement”), registering for resale (i) all CSL Ordinary Shares, or securities into which they were exchanged, that were included in the Units; and (ii) all CSL Ordinary Shares, or equivalent securities, issuable upon exercise of the Investor Warrants or upon exercise of warrants into which the Investor Warrants were exchanged.
As a result of the Share Exchange, the Company assumed the obligations of Cell Source Israel under the Subscription Agreement and Registration Rights Agreement.
In July and August 2014, the Company, Cell Source Israel and the majority of the Investors entered into Amendment No. 1 (the “RRA Amendment”) to the Registration Rights Agreement in order to amend a definition in the Registration Rights Agreement to more accurately reflect the understanding of the parties. Pursuant to the RRA Amendment, the definition relating to the deadline to file the Registration Statement was corrected such that the Company became obligated to file the Registration Statement on or prior to the 60th day after the closing of the Share Exchange Agreement (the “Registration Filing Date”). The RRA Amendment did not change any other term of the Registration Rights Agreement, including the obligation of the Company to get the Registration Statement declared effective within 120 days of the Registration Filing Date.
The foregoing descriptions of the Private Placement and related agreements and transactions do not purport to be complete and are qualified in their entirety by reference to the complete text of such agreements.
| 6 |
Implications of being an Emerging Growth Company
As a company with less than $1.0 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act, or JOBS Act, enacted in April 2012. An “emerging growth company” may take advantage of reduced reporting requirements that are otherwise applicable to public companies. These provisions include, but are not limited to:
| · | being permitted to present only two years of audited financial statements and only two years of related Management’s Discussion & Analysis of Financial Condition and Results of Operations in this prospectus; |
| · | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act; |
| · | reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and |
| · | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We may take advantage of these provisions until the last day of our fiscal year following the fifth anniversary of the date of the first sale of our common equity securities pursuant to an effective registration statement under the Securities Act of 1933, as amended, or the Securities Act, which such fifth anniversary will occur in 2018. However, if certain events occur prior to the end of such five-year period, including if we become a “large accelerated filer,” our annual gross revenues exceed $1.0 billion or we issue more than $1.0 billion of non-convertible debt in any three-year period, we will cease to be an emerging growth company prior to the end of such five-year period.
We have elected to take advantage of certain of the reduced disclosure obligations and may elect to take advantage of other reduced reporting requirements in future filings. As a result, the information that we provide to our stockholders may be different than you might receive from other public reporting companies in which you hold equity interests.
In addition, Section 107 of the JOBS Act also provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we are choosing to “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that its decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
Notwithstanding the above, we are also currently a “smaller reporting company”, meaning that we are not an investment company, an asset-backed issuer, nor a majority-owned subsidiary of a parent company that is not a smaller reporting company, and has a public float of less than $75 million and annual revenues of less than $50 million during the most recently completed fiscal year. Some of the reduced disclosure and other requirements available to us as a result of the JOBS Act may continue to be available to us after we are no longer considered an “emerging growth company”. Specifically, similar to “emerging growth companies”, “smaller reporting companies” are able to provide simplified executive compensation disclosures in their filings; are exempt from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that independent registered public accounting firms provide an attestation report on the effectiveness of internal control over financial reporting; and have certain other decreased disclosure obligations in their SEC filings, including, among other things, only being required to provide two years of audited financial statements in annual reports. Decreased disclosures in our SEC filings due to our status as an “emerging growth company” or “smaller reporting company” may make it harder for investors to analyze our results of operations and financial prospects.
| 7 |
About this Offering
This prospectus includes 9,618,648 shares of Company Common Stock offered by the Selling Stockholders consisting of (i) 4,759,324 shares of Common Stock issued pursuant to the Private Placement; (ii) 4,759,324 shares of Common Stock issuable upon exercise of Company Warrants issued pursuant to the Private Placement; and (iii) 100,000 shares of Common Stock issued for legal services provided to the Company.
Estimated Use of Proceeds
This prospectus relates to shares of our Common Stock that may be offered and sold from time to time by the Selling Stockholders. We will not receive any of the proceeds resulting from the sale of Common Stock by the Selling Stockholders. However, we may generate proceeds from the cash exercise of the Company Warrants. We intend to use those proceeds for general corporate purposes.
Financial Results
Our planned principal operations are the development and commercialization of new cell therapy products focused on treatment of blood cancers, certain non-malignant disorders and organ transplantations and regeneration. We are currently conducting research and development activities in order to facilitate the transition of the patent technology we license from the laboratory to clinical trials. We have a limited operating history. Therefore, there is limited historical financial information upon which to base an evaluation of our performance. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. We have not generated any revenues to date and Cell Source Israel has generated net losses since it began its operations in 2011, including $1,783,650 for the year ended December 31, 2013 and $1,806,320 for the six months ended June 30, 2014. We expect to incur substantial additional net expenses over the next several years as our research, development, and commercial activities increase. As of June 30, 2014 we had cumulative losses of approximately $5,747,000. The amount of future losses and when, if ever, we will achieve profitability are uncertain. Our ability to generate revenue and achieve profitability will depend on, among other things, successful completion of the preclinical and clinical development of our product candidates; obtaining necessary regulatory approvals from the U.S. Food and Drug Administration (the “FDA”) and international regulatory agencies; successful manufacturing, sales, and marketing arrangements; and raising sufficient funds to finance our activities. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations may be materially adversely affected.
| 8 |
Summary of the Shares Issued Pursuant to this Prospectus
The following is a summary of the shares being offered by the Company:
| Common Stock offered by the Selling Stockholders | 9,618,648 shares of Common Stock of which 4,759,324 shares are issuable upon exercise of outstanding warrants. |
| Common Stock outstanding prior to the offering | 23,579,256 (1) | |
| Common Stock to be outstanding after the offering | 28,338,580, assuming the full exercise of the warrants to purchase 4,759,324 share of Common Stock that are included in this prospectus. |
| Use of proceeds | We will not receive any proceeds from the sale of shares in this offering by the Selling Stockholders. However, we may generate proceeds from the cash exercise of the Company Warrants. We intend to use those proceeds for general corporate purposes. |
(1) Based upon the total number of issued and outstanding shares as of October 23, 2014.
An investment in the Company’s Common Stock involves a high degree of risk. You should carefully consider the risks described below as well as other information provided to you in this prospectus, including information in the section of this document entitled “Information Regarding Forward Looking Statements.” The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us or that we currently believe are immaterial may also impair our business operations. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected, the value of our Common Stock could decline, and you may lose all or part of your investment.
Risks related to our Business and our Industry
We have a limited operating history and a history of operating losses, and expect to incur significant additional operating losses.
Our planned principal operations are the development and commercialization of new cell therapy products focused on treatment of blood cancers, certain non-malignant disorders and organ transplantations and regeneration. We are currently conducting research and development activities in order to facilitate the transition of the patent technology we license from the laboratory to clinical trials. We have a limited operating history. Therefore, there is limited historical financial information upon which to base an evaluation of our performance. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. We have generated net losses since we began operations, including $1,783,650 for year ended December 31, 2013 and $1,806,320 for the six months ended June 30, 2014. We expect to incur substantial additional net expenses over the next several years as our research, development, and commercial activities increase. The amount of future losses and when, if ever, we will achieve profitability are uncertain. Our ability to generate revenue and achieve profitability will depend on, among other things, successful completion of the preclinical and clinical development of our product candidates; obtaining necessary regulatory approvals from the U.S. Food and Drug Administration (the “FDA”) and international regulatory agencies; successful manufacturing, sales, and marketing arrangements; and raising sufficient funds to finance our activities. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations may be materially adversely affected.
We may need to secure additional financing.
We anticipate that we will incur operating losses for the foreseeable future. Our historical cash burn rate was approximately $150,000 per month. However, we estimate that our current burn rate is approximately $100,000 per month and, as a result, we expect that the cash we currently have on hand will fund our operations through November 2014. We may require additional funds for our anticipated operations and if we are not successful in securing additional financing, we may be required to delay significantly, reduce the scope of or eliminate one or more of our research or development programs, downsize our general and administrative infrastructure, or seek alternative measures to avoid insolvency, including arrangements with collaborative partners or others that may require us to relinquish rights to certain of our technologies, product candidates or products.
Our auditors have issued a “going concern” audit opinion.
Our independent auditors have indicated, in their report on our December 31, 2013 financial statements, that there is substantial doubt about our ability to continue as a going concern. A “going concern” opinion indicates that the financial statements have been prepared assuming we will continue as a going concern and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets, or the amounts and classification of liabilities that may result if we do not continue as a going concern. Therefore, you should not rely on our balance sheet as an indication of the amount of proceeds that would be available to satisfy claims of creditors, and potentially be available for distribution to shareholders, in the event of liquidation.
| 9 |
We are an early-stage company with an unproven business strategy and may never achieve commercialization of our candidate products or profitability.
We are at an early stage of development and commercialization of our technologies and product candidates. We have not yet begun to market any products and, accordingly, have not begun to generate revenues from the commercialization of our products. Our products will require significant additional clinical testing and investment prior to commercialization. A commitment of substantial resources by ourselves and, potentially, our partners to conduct time-consuming research and clinical trials will be required if we are to complete the development of our product candidates. There can be no assurance that any of our product candidates will meet applicable regulatory standards, obtain required regulatory approvals, be capable of being produced in commercial quantities at reasonable costs or be successfully marketed. Most of our product candidates are not expected to be commercially available for several years, if at all.
We are dependent on our collaborative partners and service providers the loss of which would hurt our business.
Our strategy is to enter into various arrangements with corporate and academic collaborators, licensors, licensees, service providers and others for the research, development, clinical testing and commercialization of our products. We intend to or have entered into agreements with academic, medical and commercial organizations to research, develop and test our products. In addition, we intend to enter into corporate partnerships to commercialize the Company’s core products. There can be no assurance that such collaborations can be established on favorable terms, if at all.
Should any collaborative partner or service provider fail to appropriately research, develop, test or successfully commercialize any product to which the Company has rights, our business may be adversely affected. Failure of a collaborative partner or service provider to successfully conduct or complete their activities or to remain a viable collaborative partner or commercialize enterprise for any particular program could delay or halt the development or commercialization of any products arising out of such program. While management believes that collaborative partners and service providers will have sufficient economic motivation to continue their activities, there can be no assurance that any of these collaborations or provisions of required services will be continued or result in successfully commercialized products.
Notably, we maintain an exclusive worldwide license to certain intellectual property owned by Yeda pursuant to the Yeda License Agreement, as further discussed in the Intellectual Property section hereinafter. If we should default under the License Agreement, then our rights to Yeda’s intellectual property would extinguish, and we would lose all rights to operate the licenses. In such an event, we would effectively cease to operate unless we re-obtained licensing with Yeda.
In addition, there can be no assurance that the collaborative research or commercialization partners will not pursue alternative technologies or develop alternative products either on their own or in collaboration with others, including our competitors, as a means for developing treatments for the diseases or conditions targeted by our programs.
Our ability and our collaborators' ability to sell therapeutic products will depend to a large extent upon reimbursement from health care insurance companies.
Our success may depend, in part, on the extent to which reimbursement for the costs of therapeutic products and related treatments will be available from third-party payers such as government health administration authorities, private health insurers, managed care programs, and other organizations. Over the past decade, the cost of health care has risen significantly, and there have been numerous proposals by legislators, regulators, and third-party health care payers to curb these costs. Some of these proposals have involved limitations on the amount of reimbursement for certain products. Similar federal or state health care legislation may be adopted in the future and any products that we or our collaborators seek to commercialize may not be considered cost-effective. Adequate third-party insurance coverage may not be available for us or our collaborative partners to establish and maintain price levels that are sufficient for realization of an appropriate return on investment in product development.
We do not own any patents and rely on the patents we license from Yeda Research and Development Limited.
We do not currently own any patents and only have an exclusive worldwide license to certain intellectual property owned by Yeda pursuant to a license agreement between us and Yeda. Under the license agreement with Yeda, Yeda retains ownership of the licensed patents. If we were to default under the license agreement, then our rights to Yeda’s intellectual property would be extinguished and we would lose all rights to operate the license. In such an event, we would effectively cease to operate unless we re-obtained licensing with Yeda.
We are dependent on protecting our proprietary rights.
Our success and competitive position and future overall revenues will depend in part on our ability to obtain and maintain patent protection over the patents that we have an exclusive license to use for our product candidates, methods, process and other technologies to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. Although our patents and related technologies are owned by Yeda, under our exclusive license agreement, we are required to pay all patent related expenses for applications, renewals, etc., as well as any and all legal or other costs associated with the defending and protecting such proprietary rights. However, we cannot predict:
| § | the degree and range of protection any patents will afford us against competitors, including whether third parties will find ways to invalidate or otherwise circumvent the patents that we license; |
| § | whether or not others will obtain patents claiming aspects similar to those covered by the patents that we license or |
| § | whether we will need to initiate litigation or administrative proceedings, which may be costly whether we win or lose. |
For a complete list of the patents that we license from Yeda, please see pages 35 through 41 of this prospectus.
| 10 |
We may be required to obtain licenses from third parties to avoid infringing patents or other proprietary rights. No assurance can be given that any licenses required under any such patents or proprietary rights would be made available, if at all, on terms we find acceptable. If we do not obtain such licenses, we could encounter delays in the introduction of products, or could find that the development, manufacture or sale of products requiring such licenses could be prohibited.
A number of pharmaceutical, biopharmaceutical and biotechnology companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to or affect our business. Some of these technologies, applications or patents may conflict with our technologies or patent applications. Such conflict could limit the scope of the patents, if any, that we may be able to obtain. Such conflict may also result in the denial of our patent applications. In addition, if patents that cover our activities are issued to other companies, there can be no assurance that we would be able to obtain licenses to these patents at a reasonable cost or be able to develop or obtain alternative technology. If we do not obtain such licenses, we could encounter delays in the introduction of products, or could find that the development, manufacture or sale of products requiring such licenses could be prohibited. In addition, we could incur substantial costs in defending ourselves in suits brought against us on patents that our products might infringe or in filing suits against others to have such patents declared invalid.
Patent applications in the U.S. are maintained in secrecy and not published if either: i) the application is a provisional application or, ii) the application is filed and we request no publication, and certify that the invention disclosed “has not and will not” be the subject of a published foreign application. Otherwise, U.S. applications or foreign counterparts, if any, publish 18 months after the priority application has been filed. Since publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be certain that we or any licensor were the first creator of inventions covered by pending patent applications or that we or such licensor was the first to file patent applications for such inventions. Moreover, we might have to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention, which could result in substantial cost to us, even if the eventual outcome were favorable to us. There can be no assurance that our patents, if issued, would be held valid or enforceable by a court or that a competitor’s technology or product would be found to infringe such patents.
Much of our know-how and technology may not be patentable. To protect our rights, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. There can be no assurance, however, that these agreements will provide meaningful protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Further, our business may be adversely affected by competitors who independently develop competing technologies, especially if we obtain no, or only narrow, patent protection.
We are subject to various government regulations.
The manufacture and sale of human therapeutic and diagnostic products in the U.S., Canada and foreign jurisdictions are governed by a variety of statutes and regulations. These laws require approval of manufacturing facilities, controlled research and testing of products and government review and approval of a submission containing manufacturing, preclinical and clinical data in order to obtain marketing approval based on establishing the safety and efficacy of the product for each use sought, including adherence to current Good Manufacturing Practice (or cGMP) during production and storage, and control of marketing activities, including advertising and labeling.
The products we are currently developing will require significant development, preclinical and clinical testing and investment of substantial funds prior to their commercialization. The process of obtaining required approvals can be costly and time-consuming, and there can be no assurance that future products will be successfully developed and will prove to be safe and effective in clinical trials or receive applicable regulatory approvals. Markets other than the U.S. and Canada have similar restrictions. Potential investors and shareholders should be aware of the risks, problems, delays, expenses and difficulties which we may encounter in view of the extensive regulatory environment which controls our business.
We may become subject to increased government regulation.
Increased government regulation could: (i) reduce our revenues; (ii) increase our operating expenses; and (iii) expose us to significant liabilities. We cannot be sure what effect any future material noncompliance by us with any future laws and regulations or any material changes in current laws and regulations could have on our business, operating results and financial condition.
| 11 |
If we are unable to keep up with rapid technological changes in our field or compete effectively, we will be unable to operate profitably.
We are engaged in a rapidly changing field. Other products and therapies that will compete directly with the products that we are seeking to develop and market currently exist or are being developed. Competition from fully integrated pharmaceutical companies and more established biotechnology companies is intense and is expected to increase. Most of these companies have significantly greater financial resources and expertise in discovery and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and marketing than us. Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical and established biopharmaceutical or biotechnology companies. Many of these competitors have significant products that have been approved or are in development and operate large, well-funded discovery and development programs. Academic institutions, governmental agencies and other public and private research organizations also conduct research, seek patent protection and establish collaborative arrangements for therapeutic products and clinical development and marketing. These companies and institutions compete with us in recruiting and retaining highly qualified scientific and management personnel. In addition to the above factors, we will face competition based on product efficacy and safety, the timing and scope of regulatory approvals, availability of supply, marketing and sales capability, reimbursement coverage, price and patent position. There is no assurance that our competitors will not develop more effective or more affordable products, or achieve earlier patent protection or product commercialization, than our own.
Other companies may succeed in developing products earlier than ourselves, obtaining Health Canada, European Medicines Agency (the “EMEA”) and FDA approvals for such products more rapidly than we will, or in developing products that are more effective than products we propose to develop. While we will seek to expand our technological capabilities in order to remain competitive, there can be no assurance that research and development by others will not render our technology or products obsolete or non-competitive or result in treatments or cures superior to any therapy we develop, or that any therapy we develop will be preferred to any existing or newly developed technologies.
Clinical trials for our product candidates are expensive and time consuming, and their outcome is uncertain.
The process of obtaining and maintaining regulatory approvals for new therapeutic products is expensive, lengthy and uncertain. Costs and timing of clinical trials may vary significantly over the life of a project owing to any or all of the following non-exclusive reasons:
| • | the duration of the clinical trial; |
| • | the number of sites included in the trials; |
| • | the countries in which the trial is conducted; |
| • | the length of time required and ability to enroll eligible patients; |
| • | the number of patients that participate in the trials; |
| • | the number of doses that patients receive; |
| • | the drop-out or discontinuation rates of patients; |
| • | per patient trial costs; |
| • | third party contractors failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
| • | our final product candidates having different properties in humans than in laboratory testing; |
| • | the need to suspend or terminate our clinical trials; |
| • | insufficient or inadequate supply of quality of necessary materials to conduct our trials; |
| • | potential additional safety monitoring, or other conditions required by FDA or comparable foreign regulatory authorities regarding the scope or design of our clinical trials, or other studies requested by regulatory agencies; |
| • | problems engaging institutional review boards (“IRB”) to oversee trials or in obtaining and maintaining IRB approval of studies; |
| • | the duration of patient follow-up; |
| • | the efficacy and safety profile of a product candidate; |
| • | the costs and timing of obtaining regulatory approvals; and |
| • | the costs involved in enforcing or defending patent claims or other intellectual property rights. |
Late stage clinical trials are especially expensive, typically requiring tens of millions of dollars, and take years to reach their outcomes. Such outcomes often fail to reproduce the results of earlier trials. It is often necessary to conduct multiple late stage trials, including multiple Phase III trials, in order to obtain sufficient results to support product approval, which further increases the expense. Sometimes trials are further complicated by changes in requirements while the trials are under way (for example, when the standard of care changes for the disease that is being studied in the trial). Accordingly, any of our current or future product candidates could take a significantly longer time to gain regulatory approval than we expect, or may never gain approval, either of which could delay or stop the commercialization of our product candidates.
We may be required to suspend or discontinue clinical trials due to unexpected side effects or other safety risks that could preclude approval of our product candidates.
Our clinical trials may be suspended at any time for a number of reasons. For example, we may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to the clinical trial patients. In addition, the FDA or other regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical trial patients.
| 12 |
Administering any product candidate to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidates for any or all targeted indications. Ultimately, some or all of our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects as a result of participating in our clinical trials.
We may not receive regulatory approvals for our product candidates or there may be a delay in obtaining such approvals.
Our products and our ongoing development activities are subject to regulation by regulatory authorities in the countries in which we or our collaborators and distributors wish to test, manufacture or market our products. For instance, the FDA will regulate the product in the U.S. and equivalent authorities, such as the European Medicines Agency (“EMA”), will regulate in Europe. Regulatory approval by these authorities will be subject to the evaluation of data relating to the quality, efficacy and safety of the product for its proposed use, and there can be no assurance that the regulatory authorities will find our data sufficient to support product approval.
The time required to obtain regulatory approval varies between countries. In the U.S., for products without “Fast Track” status, it can take up to eighteen (18) months after submission of an application for product approval to receive the FDA’s decision. Even with Fast Track status, FDA review and decision can take up to twelve (12) months.
Different regulators may impose their own requirements and may refuse to grant, or may require additional data before granting, an approval, notwithstanding that regulatory approval may have been granted by other regulators. Regulatory approval may be delayed, limited or denied for a number of reasons, including insufficient clinical data, the product not meeting safety or efficacy requirements or any relevant manufacturing processes or facilities not meeting applicable requirements as well as case load at the regulatory agency at the time.
Delays in our clinical trials could result in us not achieving anticipated developmental milestones when expected, increased costs and delay our ability to obtain regulatory approval and commercialize our product candidates.
Delays in our ability to commence or enroll patients for our clinical trials could result in us not meeting anticipated clinical milestones and could materially impact our product development costs and delay regulatory approval of our product candidates. We do not know whether planned clinical trials will be commenced or completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including:
| · | delays in the development of manufacturing capabilities for our product candidates to enable their consistent production at clinical trial scale |
| · | delays in the commencement of clinical trials as a result of clinical trial holds or the need to obtain additional information to complete an Investigational New Drug Application (IND) |
| · | delays in obtaining regulatory approval to commence new trials |
| · | adverse safety events experienced during our clinical trials |
| · | insufficient efficacy during trials leading to withdrawal of product candidate |
| · | delays in obtaining clinical materials |
| · | slower than expected patient recruitment for participation in clinical trials; and |
| · | delays in reaching agreement on acceptable clinical trial agreement terms with prospective sites or obtaining institutional review board approval. |
If we do not successfully commence or complete our clinical trials on schedule, the price of our common stock may decline.
Preclinical studies and Phase 1 or 2 clinical trials of our product candidates may not predict the results of subsequent human clinical trials.
Preclinical studies, including studies of our product candidates in animal models, may not accurately predict the result of human clinical trials of those product candidates. In particular, promising animal studies suggesting the efficacy of our products may not predict the ability of these products to treat humans. Our technology may be found not to be efficacious when studied in human clinical trials.
To satisfy FDA or foreign regulatory approval standards for the commercial sale of our product candidates, we must demonstrate in adequate and controlled clinical trials that our product candidates are safe and effective. Success in early clinical trials, including Phase 2 trials, does not ensure that later clinical trials will be successful. Our initial results from Phase 1/2 clinical trials also may not be confirmed by later analysis or subsequent larger clinical trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier clinical trials.
Our product candidates must undergo rigorous clinical testing, the results of which are uncertain and could substantially delay or prevent us from bringing them to market.
Before we can obtain regulatory approval for a product candidate, we must undertake extensive clinical testing in humans to demonstrate safety and efficacy to the satisfaction of the FDA or other regulatory agencies. Clinical trials of new drug candidates sufficient to obtain regulatory marketing approval are expensive and take years to complete.
| 13 |
We cannot be certain of successfully completing clinical testing within the time frame we have planned, or at all. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent us from receiving regulatory approval or commercializing our product candidates, including the following:
| · | our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing or to abandon programs; |
| · | the results obtained in earlier stage clinical testing may not be indicative of results in future clinical trials; |
| · | clinical trial results may not meet the level of statistical significance required by the FDA or other regulatory agencies; |
| · | enrollment in our clinical trials for our product candidates may be slower than we anticipate, resulting in significant delays and additional expense; |
| · | we, or regulators, may suspend or terminate our clinical trials if the participating patients are being exposed to unacceptable health risks; and |
| · | the effects of our product candidates on patients may not be the desired effects or may include undesirable side effects or other characteristics that may delay or preclude regulatory approval or limit their commercial use, if approved. |
Completion of clinical trials depends, among other things, on our ability to enroll a sufficient number of patients, which is a function of many factors, including:
| · | the therapeutic endpoints chosen for evaluation; |
| · | the eligibility criteria defined in the protocol; |
| · | the perceived benefit of the investigational drug under study; |
| · | the size of the patient population required for analysis of the clinical trial’s therapeutic endpoints; |
| · | our ability to recruit clinical trial investigators and sites with the appropriate competencies and experience; |
| · | our ability to obtain and maintain patient consents; and |
| · | competition for patients by clinical trial programs for other treatments. |
We may experience difficulties in enrolling patients in our clinical trials, which could increase the costs or affect the timing or outcome of these clinical trials. This is particularly true with respect to diseases with relatively small patient populations.
We have conducted and may in the future conduct clinical trials for our products or product candidates outside the United States and the FDA may not accept data from such trials.
We have conducted and may in the future choose to conduct one or more of our clinical trials outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of such study data by the FDA is subject to certain conditions. For example, the study must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The study population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. Generally, the patient population for any clinical studies conducted outside of the United States must be representative of the population for whom we intend to label the product in the United States. In addition, such studies would be subject to the applicable local laws and FDA acceptance of the data would be dependent upon its determination that the studies also complied with all applicable U.S. laws and regulations. There can be no assurance the FDA will accept data from trials conducted outside of the United States. If the FDA does not accept any such data, it would likely result in the need for additional trials, which would be costly and time consuming and delay aspects of our business plan.
We may fail to comply with regulatory requirements.
Our success will be dependent upon our ability, and our collaborative partners’ abilities, to maintain compliance with regulatory requirements, including cGMP, and safety reporting obligations. The failure to comply with applicable regulatory requirements can result in, among other things, fines, injunctions, civil penalties, total or partial suspension of regulatory approvals, refusal to approve pending applications, recalls or seizures of products, operating and production restrictions and criminal prosecutions.
Regulatory approval of our product candidates may be withdrawn at any time.
After regulatory approval has been obtained for medicinal products, the product and the manufacturer are subject to continual review, including the review of adverse experiences and clinical results that are reported after our products are made available to patients, and there can be no assurance that such approval will not be withdrawn or restricted. Regulators may also subject approvals to restrictions or conditions, or impose post-approval obligations on the holders of these approvals, and the regulatory status of such products may be jeopardized if such obligations are not fulfilled. If post-approval studies are required, such studies may involve significant time and expense.
The manufacturer and manufacturing facilities we use to make any of our products will also be subject to periodic review and inspection by the FDA or EMA, as applicable. The discovery of any new or previously unknown problems with the product, manufacturer or facility may result in restrictions on the product or manufacturer or facility, including withdrawal of the product from the market. We will continue to be subject to the FDA or EMA requirements, as applicable, governing the labeling, packaging, storage, advertising, promotion, recordkeeping, and submission of safety and other post-market information for all of our product candidates, even those that the FDA or EMA, as applicable, had approved. If we fail to comply with applicable continuing regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and other adverse consequences.
There may not be a viable market for our products.
We believe that there will be many different applications for our products. We also believe that the anticipated market for our products will continue to expand. These assumptions may prove to be incorrect for a variety of reasons, including competition from other products and the degree of our products’ commercial viability.
We rely on key personnel and, if we are unable to retain or motivate key personnel or hire qualified personnel, we may not be able to grow effectively.
We are dependent on our Chief Executive Officer, Itamar Shimrat, our Executive Chairman, Yoram Drucker, and on scientific and drug development staff and consultants, including Professor Yair Reisner, the loss of services of one or more of whom could materially adversely affect us.
| 14 |
We currently do not have full-time employees, but we retain the services of part-time staff on an independent contractor/consultant and contract-employment basis. Our ability to manage growth effectively will require us to continue to implement and improve our management systems and to recruit and train new employees. Although we have done so in the past and expect to do so in the future, there can be no assurance that we will be able to successfully attract and retain skilled and experienced personnel.
We may be subject to foreign exchange fluctuation.
We maintain our accounts in both U.S. dollars and Israeli shekels. A portion of our expenditures are in foreign currencies, most notably in U.S. dollars, and therefore we are subject to foreign currency fluctuations, which may, from time to time, impact our financial position and results. We may enter into hedging arrangements under specific circumstances, typically through the use of forward or futures currency contracts, to minimize the impact of increases in the value of the U.S. dollar. In order to minimize our exposure to foreign exchange fluctuations we may hold sufficient U.S. dollars to cover our expected U.S. dollar expenditures.
We may be exposed to potential product and clinical trials liability.
Our business exposes us to potential product liability risks, which are inherent in the testing, manufacturing, marketing and sale of therapeutic products. Human therapeutic products involve an inherent risk of product liability claims and associated adverse publicity. While we will continue to take precautions we deem appropriate, there can be no assurance that we will be able to avoid significant product liability exposure. We do not currently maintain liability insurance coverage as such insurance is expensive and difficult to obtain. In the event clinical trials are commenced, we plan to obtain liability insurance coverage in the jurisdictions applicable to such clinical trials. However, when we seek such insurance, it may not be available on acceptable terms, if at all. The inability to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims could prevent or inhibit our ability to conduct clinical trials in certain jurisdiction or the commercialization of our current or potential products. A product liability claim brought against us in a clinical trial or a product withdrawal could have a material adverse effect upon us and our financial condition. Should the insurance coverage be insufficient in amount or scope to address multiple and diverse claims, liabilities not covered by insurance could represent a significant financial liability for Cell Source. Since Yeda does not conduct human trials, there is no need for Cell Source to have insurance for trials there. When Cell Source begins to contract facilities at hospitals to conduct human trials on its behalf, it will ensure that full and proper insurance coverage will be in place with respect to such clinical facilities. Cell Source plans to insure its participation in any and all clinical trials, above and beyond whatever insurance coverage is already held by the institutions and facilities providing services with respect to such clinical trials.
Our directors and officers may be exposed to liability.
We do not currently maintain directors and officers liability insurance, also known as “D&O Insurance.” However, we plan to obtain D&O Insurance for our directors and officers at or around the period immediately following the closing of the Share Exchange Agreement.
We may become subject to liabilities related to risks inherent in working with hazardous materials.
Our discovery and development processes involve the controlled use of hazardous and radioactive materials. We are subject to federal, state, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by such laws and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result and any such liability could exceed our resources. We are not specifically insured with respect to this liability. Although we believe that we are in compliance in all material respects with applicable environmental laws and regulations and currently do not expect to make material capital expenditures for environmental control facilities in the near-term, there can be no assurance that we will not be required to incur significant costs to comply with environmental laws and regulations in the future, or that our operations, business or assets will not be materially adversely affected by current or future environmental laws or regulations.
We identified a material weakness in our internal control over financial reporting and if the remediation procedures we have undertaken are unable to successfully remediate the existing material weakness, then the accuracy and timing of our financial reporting may be adversely affected.
In preparing our financial statements as of and for the year ended December 31, 2013, we identified control deficiencies in the design and operation of our internal control over financial reporting that together constituted a material weakness in our internal control over financial reporting. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis. The material weaknesses identified were that we did not have adequate accounting systems and our accounting staff was inadequate both in terms of the number of personnel and their expertise in U.S. GAAP and SEC rules and regulations. As such, our controls over financial reporting were not designed or operating effectively. We believe we have remediated this material weakness as of the date of this filing. However, sufficient time has not passed to allow us to test the effects of such remedies on the efficacy of our controls over financial reporting.
The material weakness in our internal control over financial reporting was attributable to inadequate accounting systems. In addition, our accounting staff was inadequate both in terms of the number of personnel and their expertise in U.S. GAAP and SEC rules and regulations. In response to this material weakness, we engaged the services of additional personnel with knowledge of U.S. GAAP and public company financial reporting expertise to enhance our financial management and reporting infrastructure, and further develop and document our accounting policies and financial reporting procedures.
| 15 |
Our independent certified public accounting firm is not required to perform an evaluation of our internal control over financial reporting in accordance with the provisions of the Sarbanes-Oxley Act by virtue of our status as an “emerging growth company” as defined in the JOBS Act. In light of the control deficiencies and the resulting material weakness that were identified as a result of the limited procedures we did perform, it is possible that additional material weaknesses and significant control deficiencies may have been identified if we and our independent registered public accounting firm performed an evaluation of our internal control over financial reporting in accordance with the provisions of the Sarbanes-Oxley Act. However, for as long as we remain an “emerging growth company” as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the requirement that our independent registered public accounting firm provide an attestation on the effectiveness of our internal control over financial reporting.
If we failed to remediate the material weakness or if in the future we fail to meet the demands that will be placed upon us as a public company, we may be unable to accurately report our financial results, or report them within the timeframes required by law or stock exchange regulations. Failure to comply with Section 404 of the Sarbanes-Oxley Act could also potentially subject us to sanctions or investigations by the SEC or other regulatory authorities.
Risks Related to Our Common Stock
There is not an active liquid trading market for the Company’s Common Stock.
The Company voluntarily reports under the Exchange Act and its Common Stock is eligible for quotation on the OTC Markets. However, there is no regular active trading market in the Company’s Common Stock, and we cannot give an assurance that an active trading market will develop. If an active market for the Company’s Common Stock develops, there is a significant risk that the Company’s stock price may fluctuate dramatically in the future in response to any of the following factors, some of which are beyond our control:
| • | variations in our quarterly operating results; |
| • | announcements that our revenue or income are below analysts’ expectations; |
| • | general economic slowdowns; |
| • | sales of large blocks of the Company’s Common Stock; and |
| • | announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships, joint ventures or capital commitments. |
Our Common Stock is subject to the “penny stock” rules of the Securities and Exchange Commission, which may make it more difficult for stockholders to sell our Common Stock.
The SEC has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require that a broker or dealer approve a person’s account for transactions in penny stocks, and the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased.
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must obtain financial information and investment experience objectives of the person, and make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks.
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form sets forth the basis on which the broker or dealer made the suitability determination, and that the broker or dealer received a signed, written agreement from the investor prior to the transaction.
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of the Company’s Common Stock if and when such shares are eligible for sale and may cause a decline in the market value of its stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stock.
| 16 |
We may not be able to attract the attention of brokerage firms because we became a public company by means of a reverse acquisition.
Because we became public through a “reverse acquisition," securities analysts of brokerage firms may not provide coverage of us since there is little incentive to brokerage firms to recommend the purchase of our Common Stock. No assurance can be given that brokerage firms will want to conduct any secondary offerings on behalf of the Company in the future.
Applicable regulatory requirements, including those contained in and issued under the Sarbanes-Oxley Act of 2002, may make it difficult for the Company to retain or attract qualified officers and directors, which could adversely affect the management of its business and its ability to obtain or retain listing of its Common Stock.
The Company may be unable to attract and retain those qualified officers, directors and members of board committees required to provide for effective management because of the rules and regulations that govern publicly held companies, including, but not limited to, certifications by principal executive officers. The enactment of the Sarbanes-Oxley Act has resulted in the issuance of a series of related rules and regulations and the strengthening of existing rules and regulations by the SEC, as well as the adoption of new and more stringent rules by the stock exchanges. The perceived increased personal risk associated with these changes may deter qualified individuals from accepting roles as directors and executive officers.
Further, some of these changes heighten the requirements for board or committee membership, particularly with respect to an individual’s independence from the corporation and level of experience in finance and accounting matters. The Company may have difficulty attracting and retaining directors with the requisite qualifications. If the Company is unable to attract and retain qualified officers and directors, the management of its business and its ability to obtain or retain listing of our shares of Common Stock on any stock exchange (assuming the Company elects to seek and are successful in obtaining such listing) could be adversely affected.
If the Company fails to maintain an effective system of internal controls, it may not be able to accurately report its financial results or detect fraud. Consequently, investors could lose confidence in the Company’s financial reporting and this may decrease the trading price of its stock.
The Company must maintain effective internal controls to provide reliable financial reports and detect fraud. The Company has been assessing its internal controls to identify areas that need improvement. It is in the process of implementing changes to internal controls, but has not yet completed implementing these changes. Failure to implement these changes to the Company’s internal controls or any others that it identifies as necessary to maintain an effective system of internal controls could harm its operating results and cause investors to lose confidence in the Company’s reported financial information. Any such loss of confidence would have a negative effect on the trading price of the Company’s stock.
Voting power of our shareholders is highly concentrated by insiders.
Our officers, directors and affiliates currently own approximately 28% of our outstanding common stock. Assuming the selling shareholders sell all of the shares and shares issuable upon exercise of the warrants issued to them, our officers, directors and affiliates (were they also to exercise all of their warrants) will on a pro forma basis own approximately 35% of our outstanding common stock. Such concentrated control of the Company may adversely affect the value of our ordinary shares. If you acquire our ordinary shares, you may have no effective voice in our management. Sales by our insiders or affiliates, along with any other market transactions, could affect the value of our ordinary shares.
We do not intend to pay dividends for the foreseeable future.
We have paid no dividends on our Common Stock to date and it is not anticipated that any dividends will be paid to holders of our Common Stock in the foreseeable future. While our future dividend policy will be based on the operating results and capital needs of the business, it is currently anticipated that any earnings will be retained to finance our future expansion and for the implementation of our business plan. As an investor, you should take note of the fact that a lack of a dividend can further affect the market value of our stock, and could significantly affect the value of any investment in our Company.
Our articles of incorporation allow for our board to create a new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our Common Stock.
Our Board of Directors has the authority to fix and determine the relative rights and preferences of preferred stock. Our Board of Directors have the authority to issue up to 10,000,000 shares of our preferred stock terms of which may be determined by the Board without further stockholder approval. As a result, our Board of Directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of Common Stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our Common Stock. In addition, our Board of Directors could authorize the issuance of a series of preferred stock that has greater voting power than our Common Stock or that is convertible into our Common Stock, which could decrease the relative voting power of our Common Stock or result in dilution to our existing stockholders. Although we have no present intention to issue any additional shares of preferred stock or to create any additional series of preferred stock, we may issue such shares in the future.
| 17 |
If our registration statement becomes effective, there will be a significant number of shares of Common Stock eligible for sale, which could depress the market price of such shares.
We have agreed to file a registration statement with the SEC to register the shares of our Common Stock issued or issuable in connection with the Private Placement. Following the effective date of such registration statement a large number of shares of Common Stock will be available for sale in the public market, which could harm the market price of the stock.
You may experience dilution of your ownership interests because of the future issuance of additional ordinary shares.
In the future, we may issue additional authorized but previously unissued equity securities, resulting in the dilution of the ownership interests of our shareholders. We may also issue additional shares of our securities that are convertible into or exercisable for ordinary shares, as the case may be, in connection with hiring or retaining employees, future acquisitions, future sales of its securities for capital raising purposes, or for other business purposes. The future issuance of any such additional shares may create downward pressure on the value of our securities. There can be no assurance that we will not be required to issue additional shares, warrants or other convertible securities in the future in conjunction with any capital raising efforts, including at a price (or exercise prices) below the price at which our shares may be valued or are trading in a public market.
As an issuer of “penny stock," the protection provided by the federal securities laws relating to forward looking statements does not apply to us.
Although federal securities laws provide a safe harbor for forward-looking statements made by a public company that files reports under the federal securities laws, this safe harbor is not available to issuers of penny stocks. As a result, we will not have the benefit of this safe harbor protection in the event of any legal action based upon a claim that the material provided by us contained a material misstatement of fact or was misleading in any material respect because of our failure to include any statements necessary to make the statements not misleading. Such an action could hurt our financial condition.
Our issuance of Common Stock upon exercise of warrants or options may depress the price of our Common Stock.
As of the date of this prospectus, we have 23,579,256 shares of Common Stock and warrants to purchase 8,503,159 shares of Common Stock. The issuance of shares of Common Stock upon exercise of outstanding warrants or options could result in substantial dilution to our stockholders, which may have a negative effect on the price of our Common Stock.
We will incur increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. We will be subject to the reporting and other requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act, the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and the Dodd-Frank Wall Street Reform and Protection Act. These rules and regulations will require, among other things, that we file annual, quarterly and current reports with respect to our business and financial condition and establish and maintain effective disclosure and financial controls and corporate governance practices. We expect these rules and regulations to substantially increase our legal and financial compliance costs and to make some activities more time-consuming and costly, particularly after we are no longer an "emerging growth company" as defined in the recently enacted Jumpstart Our Business Startups Act of 2012, or the JOBS Act. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. For example, we expect that these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance. We estimate that we will incur between $1 million and $2.5 million annually in expenses in response to these requirements.
.
If we take advantage of specified reduced disclosure requirements applicable to an "emerging growth company" under the JOBS Act, the information that we provide to stockholders may be different than they might receive from other public companies.
As a company with less than $1 billion in revenue during our last fiscal year, we qualify as an "emerging growth company" under the JOBS Act. As an emerging growth company, we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable generally to public companies. These provisions include:
| · | Only two years of audited financial statements in addition to any required unaudited interim financial statements with correspondingly reduced "Management's Discussion and Analysis of Financial Condition and Results of Operations" disclosure; |
| · | Reduced disclosure about our executive compensation arrangements; |
| · | No non-binding advisory votes on executive compensation or golden parachute arrangements; |
| · | Exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting. |
| 18 |
We may take advantage of these exemptions for up to five years or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we have more than $1 billion in annual revenues, we have more than $700 million in market value of our stock held by non-affiliates, or we issue more than $1 billion of non-convertible debt over a three-year period. We may choose to take advantage of some but not all of these reduced burdens. We have not taken advantage of any of these reduced reporting burdens in this prospectus, although we may choose to do so in future filings. If we do, the information that we provide stockholders may be different than you might get from other public companies in which you hold stock.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, stockholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our common stock.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act, or the subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our consolidated financial statements or identify other areas for further attention or improvement. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
We will be required to disclose changes made in our internal controls and procedures on a quarterly basis and our management will be required to assess the effectiveness of these controls annually. However, for as long as we are an “emerging growth company” under the JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. We could be an emerging growth company for up to five years. An independent assessment of the effectiveness of our internal controls could detect problems that our management's assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
Statements contained in this prospectus and the Form S-1 Registration Statement of which this prospectus is a part may be “forward-looking statements.” Forward-looking statements include, but are not limited to, statements that express our intentions, beliefs, expectations, strategies, predictions or any other statements relating to our future activities or other future events or conditions. These statements are based on current expectations, estimates and projections about our business based, in part, on assumptions made by management. These statements are not guarantees of future performance and involve risks, uncertainties and assumptions that are difficult to predict. Therefore, actual outcomes and results may, and are likely to, differ materially from what is expressed or forecasted in the forward-looking statements due to numerous factors, including those described above and those risks discussed from time to time in this prospectus, including the risks described under “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operation” in this prospectus and in other documents which we file with the Securities and Exchange Commission. In addition, such statements could be affected by risks and uncertainties related to:
| • | our ability to raise funds for general corporate purposes and operations, including our clinical trials; |
| • | the commercial feasibility and success of our technology; |
| • | our ability to recruit qualified management and technical personnel; |
| • | the success of our clinical trials; |
| • | our ability to obtain and maintain required regulatory approvals for our products; and |
| • | the other factors discussed in the “Risk Factors” section and elsewhere in this prospectus. |
Any forward-looking statements speak only as of the date on which they are made, and except as may be required under applicable securities laws, we do not undertake any obligation to update any forward-looking statement to reflect events or circumstances after the date of this current report.
| 19 |
The Selling Stockholders will receive all of the proceeds from the sale of the shares offered by them under this prospectus. We will not receive any proceeds from the sale of the shares by the Selling Stockholders covered by this prospectus. However, we will generate proceeds from any cash exercise of the warrants by the Selling Stockholders, if any. There can be no assurance that any of the Selling Stockholders will exercise all or any of their warrants. We intend to use any proceeds received for general corporate purposes.
MARKET FOR OUR COMMON STOCK AND RELATED STOCKHOLDER MATTERS
Market Information
The Company’s common stock is quoted under the symbol “CLCS” on the OTCQB. There has been no active trading of our Company’s common stock.
There was no reported trading in our common stock prior to March 13, 2014. Since March 13, 2014, there has been limited trading in our common stock. The following table sets forth the range of high and low bid prices of our common stock as reported and summarized on the OTCQB for the periods indicated. These prices are based on inter-dealer bid and asked prices, without markup, markdown, commissions, or adjustments and may not represent actual transactions.
2014 Fiscal Year
| High | Low | |||||||
| First Quarter ended March 31, 2014 | $ | 0.10 | *** | $ | 0.10 | *** | ||
| Second Quarter ended June 30, 2014 | $ | 1.55 | $ | 0.10 | ||||
| Third Quarter ended September 30, 2014 | $ | 1.75 | $ | 0.75 | ||||
| Fourth Quarter ended December 30, 2014* | $ | 0.95 | $ | 0.70 | ||||
| * through October 23, 2014 | ||||||||
2013 Fiscal Year
| High | Low | |||||||
| First Quarter ended March 31, 2013 | $ | *** | $ | *** | ||||
| Second Quarter ended June 30, 2013 | $ | *** | $ | *** | ||||
| Third Quarter ended September 30, 2013 | $ | *** | $ | *** | ||||
| Fourth Quarter ended December 31, 2013 | $ | *** | $ | *** | ||||
2012 Fiscal Year
| High | Low | |||||||
| First Quarter ended March 31, 2012 | $ | *** | $ | *** | ||||
| Second Quarter ended June 30, 2012 | $ | *** | $ | *** | ||||
| Third Quarter ended September 30, 2012 | $ | *** | $ | *** | ||||
| Fourth Quarter ended December 31, 2012 | $ | *** | $ | *** | ||||
***trade data is unavailable prior to March 13, 2014.
Transfer Agent
Our transfer agent is Globex Transfer, LLC, 780 Deltona Blvd., Suite 202, Deltona, FL 32725.
Holders
As of October 23, 2014, there were approximately 100 holders of record of the Company’s common stock.
Outstanding
As of October 23, 2014: (i) 8,503,159 shares of common stock are subject to outstanding warrants to purchase, or securities convertible into, common stock; (ii) 0 shares of common stock can be sold pursuant to Rule 144 under the Securities Act; and (iii) 0 shares of common stock are being, or has been publicly proposed to be, publicly offered by the Company.
Dividends
The Company has never declared or paid any cash dividends on its Common Stock. The Company currently intends to retain future earnings, if any, to finance the expansion of its business. As a result, the Company does not anticipate paying any cash dividends in the foreseeable future.
Securities Authorized for Issuance Under Equity Compensation Plans
As of the date of the filing of this prospectus, the Company does not have any equity compensation plan.
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATION
This Management Discussion and Analysis (“MD&A”) contains “forward-looking statements," which represent our projections, estimates, expectations or beliefs concerning among other things, financial items that relate to management’s future plans or objectives or to our future economic and financial performance. In some cases, you can identify these statements by terminology such as “may,” “should,” “plans,” “believe,” “will,” “anticipate,” “estimate,” “expect,” “project” or “intend,” including their opposites or similar phrases or expressions.
You should be aware that these statements are projections or estimates as to future events and are subject to a number of factors that may tend to influence the accuracy of the statements. These forward-looking statements should not be regarded as a representation by the Company or any other person that the events or plans of the Company will be achieved. You should not unduly rely on these forward-looking statements, which speak only as of the date of this MD&A. Except as may be required under applicable securities laws, we undertake no obligation to publicly revise any forward-looking statement to reflect circumstances or events after the date of this MD&A or to reflect the occurrence of unanticipated events. You should, however, review the factors and risks we describe under “Risk Factors” in this prospectus. Actual results may differ materially from any forward looking statement.
| 20 |
Overview
Our wholly-owned subsidiary, Cell Source Israel was founded in 2011 as a privately held company located in Tel Aviv, Israel. Our business is based on over ten (10) years of prominent research at the Weizmann Institute, the commercial arm of Yeda, from whom we license patented technology. Our exclusive, world-wide license provides us with access to certain discoveries, inventions and other intellectual property generated by Dr. Reisner, formerly Head of the Immunology Department at the Weizmann Institute, together with others. Dr. Reisner leads a team at the Weizmann Institute to continue the development of these technologies in order to facilitate the transition of those technologies from the laboratory to clinical trials. We also collaborate with Dr. Herman Einsele and Dr. Franco Aversa, leading figures in bone marrow transplantation for cancer treatment and research, both of whom plan to serve on our Scientific Advisory Board and will oversee our initial clinical trials which, when started, will focus on addressing cancer through cell therapy accompanied by bone marrow transplants.
Our lead prospective product is our patented Veto-Cell immune system management technology, which is an immune tolerance biotechnology that enables the selective blocking of immune responses. The Company’s target indications include: lymphoma, multiple myeloma and BCLL (a form of leukemia treatment), facilitating transplantation acceptance (initially bone -marrow transplantation and subsequently organ transplantation), and ultimately treating a variety of non-malignant diseases.
Prior to commercializing its products, the Company must conduct human clinical trials and obtain FDA approval and/or approvals from comparable foreign regulatory authorities.
Generally speaking, as a preclinical biotechnology firm, Cell Source needs to go through several necessary steps in order to commercialize its products and commence revenue generation. These steps are per product, but can run in parallel for multiple products, which are each in different stages of the development “pipeline”, so that, for example, when a certain product is already in a human clinical trial, another product may still be in preclinical development and a third may be awaiting regulatory approval to commence human trials. These can also take place in parallel, and varied stages, for the same product in different geographic jurisdictions. The typical steps per product (and range of time frame for each) are:
| 1. | Complete development of human treatment protocol (2-5 years) |
| 2. | Apply for and receive approval to commence human trials (9-18 months) |
| 3. | Recruit patients (1-6 months) |
| 4. | Conduct Phase I trials showing safety of product (1-2 years) |
| 5. | Apply for and receive approval to conduct trials showing product efficacy (6-12 months) |
| 6. | Data collecting and analysis (6-12 months) |
| 7. | Conduct Phase II efficacy trials (2-3 years) |
| 8. | Data collecting and analysis (6-12 months) |
| 9. | Apply for and receive approval to conduct trials showing efficacy in larger numbers of patients (6-12 months) |
| 10. | Conduct Phase III efficacy trials with larger numbers of patients (2-4 years) |
| 11. | Data collecting and analysis (6-12 months) |
| 12. | Apply for and receive approval for production scale manufacturing facilities (6-12 months) |
| 13. | Contract third party or establish own production facilities (6-30 months) |
| 14. | Contract third party or establish own distribution platform (6-18 months) |
| 15. | Commence manufacturing and distribution (6-12 months) |
Please note that steps 12-15 can be conducted in parallel with some of the steps above. In the case of Cell Source and other firms that treat terminal patients with either rare diseases or those for which there is currently no effective treatment, or where preclinical studies indicate a reasonable expectation to increase life expectancy and survival rates by a substantive margin, several of these steps can be combined and or shortened, subject to regulatory discretion. For example, Phase I and II (safety and efficacy) can be combined in a single concurrent step; approvals for subsequent steps can be accelerated; in some countries patients can already be treated commercially after the end of Phase II, foregoing the requirement for Phase III data.
Although we have provided estimated timeframes for each step above, no assurances can be made that such timeframes are accurate or that they would not be delayed for one or more reasons. At any stage of human clinical trial, there could be problems with either safety or efficacy of treatment. In these instances the Company could be required to reformulate the treatment and proceed with additional patients, which could involve a delay of months or years, depending on whether we would have to seek approval from the very beginning of the approval process. There can also be a delay of up to 1 to 2 years between phases of human clinical trial, as the regulator may wish to take additional time to review the approval of a subsequent stage. Furthermore, if a significant modification to the treatment is required, the application process begins again from the very first stage. If the treatment is not effective at all or if it’s harmful to patients, even after modifications are made, it is possible that the trials may be halted completely and the product candidates permanently withdrawn. While the timescales presented here are representative of the typical experience, there is no assurance that there will not be significant delays at any stage or step in the process or a complete failure of trials.
The specific detailed next steps the company must take to get the treatments or products to market include the following:
We have not submitted any drug applications to the FDA and do not have anything pending for approval with the FDA. Cell Source itself has not had any contact with any regulator anywhere regarding treatment approvals or clinical trials associated with regulatory approvals. We are aware that a hospital in Italy in May, 2014 independently requested and in September, 2014 received approval to conduct a trial with the same protocol that we plan to use, but we are not mentioned in the application nor in the approval. However, we may indirectly benefit from the outcome of the trial, if successful, although we are not the sponsor of this trial. There are no written or verbal agreements between the hospital and Cell Source regarding the use of the technology. That said, Cell Source is aware and in favor of the hospital plans to use the technology and would of course find a positive initial outcome encouraging. Since the treatment is being done on compassionate grounds as a non-commercial clinical trial, there is no legal requirement for the hospital to obtain approval to use the treatment protocol.
| 21 |
Cell Source then plans to commence a follow-on full Phase I/II study in Parma, Italy that is meant to demonstrate safety and efficacy and is anticipated to last between 2 and 3 years. The local regulator will then determine whether the product will then be made available for sale, or whether it will require a Phase III study. Since the product involves treatments that are already in the clinic but in combination in a new way, and since Cell Source aspires to demonstrate a significant increase in survival rates, this treatment may conceivably be made available commercially on a “compassionate grounds” basis immediately following the conclusion of Phase I/II, which could be in 2019. In the event that a full Phase III study is required, a further 2-3 years may be required.
For the Veto-Cell applications for reducing rejection in Bone Marrow Transplants and for eradicating lymphoma cells, Cell Source expects to commence a Phase I/II human clinical study in Italy, and subsequently in Germany, starting sometime in 2016. Since this technology does not involve elements that are already in clinical use, Cell Source anticipates that Phase I/II studies will last until 2018 until 2019. These would be followed by completion of Phase II and Phase III, which would last another 2-3 years each, so that full approval, if successful, would be expected sometime in 2024. In Germany there is a possibility of approval for commercial use on a “compassionate grounds” basis at the end of Phase II, which could take place by 2022. In the US, Cell Source may or may not be permitted by the FDA to submit European trial results as “supporting data”. If not, Cell Source would have to go through the full FDA approval process, which, commencing in 2015, would last until between 2021 and 2023 for the Megadose Drug Combination. For the Veto-Cell this would commence in 2016 or 2017 and could last until 2024 to 2025. It is possible that Cell Source treatments could qualify for any or all of Fast Track, Breakthrough Therapy, Accelerated Approval and Priority Review designation under the FDA, which would hasten their approval if successful.
The costs for each step of development, in terms of clinical trials, are delineated below:
Cell Source estimates the cost of clinical trials alone to be up to $5 million over the coming two years and another $25-50 million in order to reach commercialization for both the Megadose Drug Combination and the Veto-Cell products. This would mean that Cell Source will need to secure one or more significant capital infusions in order to reach the point that meaningful revenues could be incurred.
Cell Source will require additional financing for any and all of the steps described above.
The exhibit below represents the planned time line for implementation:
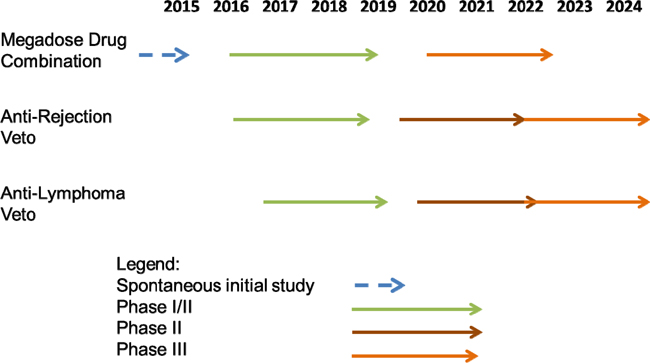
| 22 |
Results of Operations
Three Months Ended June 30, 2014 Compared to Three Months Ended June 30, 2013
The following table presents selected items in our unaudited condensed consolidated statements of operations for the three months ended June 30, 2014 and 2013, respectively:
| For The | ||||||||
| Three Months Ended | ||||||||
| June 30, | ||||||||
| 2014 | 2013 | |||||||
| Revenues | $ | - | $ | - | ||||
| Operating Expenses | ||||||||
| Research and development | 220,995 | 87,436 | ||||||
| Research and development - related party | 398,845 | 329,753 | ||||||
| Selling, general and administrative | 397,338 | 79,100 | ||||||
| Total Operating Expenses | 1,017,178 | 496,289 | ||||||
| Loss From Operations | (1,017,178 | ) | (496,289 | ) | ||||
| Other (Expense) Income | ||||||||
| Interest expense | - | (362,001 | ) | |||||
| Change in fair value of derivative liability | 31,100 | 54,300 | ||||||
| Total Other Income (Expense) | 31,100 | (307,701 | ) | |||||
| Net Loss | $ | (986,078 | ) | $ | (803,990 | ) | ||
Research and Development
Research and development expense was $619,840 and $417,189 for the three months ended June 30, 2014 and 2013, respectively, an increase of $202,651, or 49%, primarily because the proceeds from our recent equity financing permitted us to expand our research and development expenses, including expenses associated with key patents entering the National Phase in a number of countries around the world.
Selling, General and Administrative
Selling, general and administrative expense was $397,338 and $79,100 for the three months ended June 30, 2014 and 2013, respectively, an increase of $318,238, or 402%, primarily as a result of legal and professional fees associated with our Share Exchange transaction which was prepared for and closed in the current period.
Interest Expense
Interest expense for the three months ended June 30, 2014 and 2013, was $0 and $362,001, respectively. Interest expense during the three months ended June 30, 2013 was primarily related to the amortization of debt discount associated with our convertible notes.
Change in Fair Value of Derivative Liability
The change in fair value of derivative liability for the three months ended June 30, 2014 and 2013 was $31,100 and $54,300, respectively, which represents the change in fair value of the warrants and embedded conversion options associated with our convertible notes that were deemed to be derivative liabilities.
| 23 |
Six Months Ended June 30, 2014 Compared to Six Months Ended June 30, 2013
The following table presents selected items in our unaudited condensed consolidated statements of operations for the six months ended June 30, 2014 and 2013, respectively:
| For The | ||||||||
| Six Months Ended | ||||||||
| June 30, | ||||||||
| 2014 | 2013 | |||||||
| Revenues | $ | - | $ | - | ||||
| Operating Expenses | ||||||||
| Research and development | 545,412 | 193,806 | ||||||
| Research and development - related party | 611,203 | 402,629 | ||||||
| Selling, general and administrative | 633,305 | 127,853 | ||||||
| Total Operating Expenses | 1,789,920 | 724,288 | ||||||
| Loss From Operations | (1,789,920 | ) | (724,288 | ) | ||||
| Other (Expense) Income | ||||||||
| Interest expense | - | (396,534 | ) | |||||
| Change in fair value of derivative liability | (16,400 | ) | 64,500 | |||||
| Total Other Income (Expense) | (16,400 | ) | (332,034 | ) | ||||
| Net Loss | $ | (1,806,320 | ) | $ | (1,056,322 | ) | ||
Research and Development
Research and development expense was $1,156,615 and $596,435 for the six months ended June 30, 2014 and 2013, respectively, an increase of $560,180, or 94%, primarily because the proceeds from our recent equity financing permitted us to expand our research and development expenses, including expenses associated with key patents entering the National Phase in a number of countries around the world.
Selling, General and Administrative
Selling, general and administrative expense was $633,305 and $127,853 for the six months ended June 30, 2014 and 2013, respectively, an increase of $505,452, or 395%, primarily as a result of legal and professional fees associated with our Share Exchange transaction, which was prepared for and closed in the current period.
Interest Expense
Interest expense for the six months ended June 30, 2014, and 2013 was $0 and $396,534, respectively. Interest expense during the six months ended June 30, 2013 was primarily related to the amortization of debt discount associated with our convertible notes.
Change in Fair Value of Derivative Liability
The change in fair value of derivative liability for the six months ended June 30, 2014 and 2013 was $(16,400) and $64,500, respectively, which represents the change in fair value of the warrants and embedded conversion options associated with our convertible notes that were deemed to be derivative liabilities.
| 24 |
Yeah Ended December 31, 2013 Compared to Year Ended December 31, 2012
| For the Year Ended December 31 | ||||||||||||||||
| 2013 $ | 2012 $ | Change $ | Change % | |||||||||||||
| Research and Development | (1,135,513 | ) | (1,219,515 | ) | 84,002 | 7 | % | |||||||||
| General and Administrative | (319,857 | ) | (447,264 | ) | 127,407 | 28 | % | |||||||||
| Foreign Exchange (Gain) Loss | 21,339 | 64,565 | 43,226 | 67 | % | |||||||||||
| Interest Expense | (681,780 | ) | (8,633 | ) | (673,147 | ) | -7797 | % | ||||||||
| Net Loss | (1,783,650 | ) | (1,661,912 | ) | (121,738 | ) | -7 | % | ||||||||
Research and Development
Research and development expenses decreased to $1,135,513 for the year ended December 31, 2013 from $1,219,515 for year ended December 31, 2012.
Additionally, contracted research and travel were lower during the year ended December 31, 2013 compared to the year ended December 31, 2012. Contracted research costs were lower in the current period due to the initiation of the agreement with Yeda in the year ended December 31, 2012. Travel has decreased in the current period compared to the prior period as a result of cash shortages.
General and Administrative
General and administrative expenses were $319,857 for year ended December 31, 2013 compared to $447,264 for the year ended December 31, 2012. The principal reasons for the decrease were due to higher professional fees and personnel costs incurred in the prior period. The decrease in professional fees related to costs incurred for the initiation of our first financial statement audit, legal fees related to the updating of our corporate records and for business development fees. Personnel costs increased in the year ended December 31, 2013 compared to the year ended December 31, 2012 due to an increase in salaries paid in the current period compared to the prior period.
Foreign Exchange (Gain) Loss
Our functional currency is the Israeli Shekel, but we report our results in USD. The translation gains and losses are reported in other comprehensive loss/income. Foreign exchange gains and losses are the result of our incurring expenses in USD and then translating those USD expenses into Shekels. We will continue to incur some expenses in USD and as a result will continue to be exposed to foreign exchange gains and losses in part because it is expected that the Shekel will continue to be our functional currency.
We recognized a foreign exchange loss of $21,339 for the year ended December 31, 2013 compared to a loss of $64,565 for the year ended December 31, 2012. The change was due to changes in the exchange rate between the Israeli Shekel and the USD and to varying levels of USD accounts payable.
Interest Expense
Interest expense for the year ended December 31, 2013 was $681,780 as compared to $8,633 for the year ended December 31, 2012. The increase is primarily attributable to an increase in the amortization of debt discount.
Liquidity and Capital Resources
We measure our liquidity in a number of ways, including the following:
| June 30, | December 31, | |||||||
| 2014 | 2013 | |||||||
| (unaudited) | ||||||||
| Cash | $ | 889,332 | $ | 28,878 | ||||
| Working capital deficiency | $ | (1,679,565 | ) | $ | (697,334 | ) | ||
We have not generated any revenues since our inception, we have recurring net losses, we have a working capital deficiency as of June 30, 2014 and December 31, 2013 of $1,679,565 and $697,334, respectively, and we have used cash in operations of $2,149,810 and $340,090 during the six months ended June 30, 2014 and 2013, respectively. These conditions raise substantial doubt about our ability to continue as a going concern. Based upon our working capital deficiency, forecast for continued operating losses, and cash burn rate of approximately $100,000 per month, we expect that the cash we currently have available will fund our operations through November 2014.
Our ability to continue our operations is dependent on management’s plans, which include the raising of capital through debt and/or equity markets with some additional funding from other traditional financing sources, including term notes, until such time that funds provided by operations are sufficient to fund working capital requirements. We may need to incur additional liabilities with certain related parties to sustain our existence. If we were not to continue as a going concern, we would likely not be able to realize our assets at values comparable to the carrying value or the fair value estimates reflected in the balances set out in the preparation of our financial statements.
There can be no assurances that the Company will be successful in generating additional cash from equity or debt financings or other sources to be used for operations. Should the Company not be successful in obtaining the necessary financing to fund its operations, the Company would need to curtail certain or all operational activities and/or contemplate the sale of its assets if necessary.
During the six months ended June 30, 2014 and 2013, our sources and uses of cash were as follows:
Net Cash Used in Operating Activities
We experienced negative cash flows from operating activities for the six months ended June 30, 2014 and 2013 in the amounts of $2,149,810 and $340,090, respectively. The net cash used in operating activities for the six months ended June 30, 2014 was primarily due to cash used to fund a net loss of $1,806,320, adjusted for net non-cash expenses in the aggregate amount of $59,425, and $402,915 of net cash used to fund changes in the levels of operating assets and liabilities, primarily as a result of payments to vendors due to improved cash availability. The net cash used in operating activities for the six months ended June 30, 2013 was primarily due to cash used to fund a net loss of $1,056,322, adjusted for non-cash expenses in the aggregate amount of $409,024 partially offset by $307,208 of net cash provided due to changes in the levels of operating assets and liabilities, primarily as a result of increases in accounts payable and accrued expenses, due to cash constraints during the period.
Net Cash Used in Investing Activities
Net cash used in investing activities was $2,582 for the six months ended June 30, 2014, which was related to purchases of property and equipment.
Net Cash Provided by Financing Activities
Net cash provided by financing activities for the six months ended June 30, 2014 and 2013 was $3,012,846 and $210,000, respectively. The net cash provided by financing activities during the six months ended June 30, 2014 was attributable to $3,012,846 of net proceeds from our IPO (gross proceeds of $3,067,996 less $55,150 of issuance costs). The net cash provided by financing activities during the six months ended June 30, 2013 was attributable $210,000 of proceeds from the issuance of convertible notes.
| 25 |
During the years ended December 31, 2013 and 2012, our sources and uses of cash were as follows:
Operating Activities
Net cash used in operating activities decreased to $893,942 for the year ended December 31, 2013 from $1,535,208 for the year ended December 31, 2012. The principal uses of funds were for services supporting the development of the Company’s business plan and research and development fees as well as the costs of the daily operations of the business.
Financing Activities
We received $551,497 in net proceeds from the issuance of 735,327 shares of common stock during the year ended December 31, 2013 compared to $810,000 in proceeds from the issuance of 2,250,000 shares of common stock during the year ended December 31, 2012. In addition we received $210,000 and $300,000 during the years ended December 31, 2013 and 2012, respectively, from the issuance of convertible notes.
| 26 |
Off Balance Sheet Arrangements
We do not have any off balance sheet arrangements.
Critical Accounting Policies
Our discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe to be reasonable under the circumstances. Actual results may differ from these estimates. We believe the judgments and estimates required by the following accounting policies to be critical in the preparation of our consolidated financial statements.
As a company with less than $1.0 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act, or JOBS Act, enacted in April 2012. An “emerging growth company” may take advantage of reduced reporting requirements that are otherwise applicable to public companies. We have elected to take advantage of certain of the reduced disclosure obligations and may elect to take advantage of other reduced reporting requirements in future filings. As a result, the information that we provide to our stockholders may be different than you might receive from other public reporting companies in which you hold equity interests.
Significant Factors, Assumptions, and Methodologies Used in Estimating Fair Value of Common Stock
We performed valuations to estimate the fair value of our common stock at June 30, 2014, December 31, 2013 and December 31, 2012 (Valuation Dates). To determine the value of our common stock at each Valuation Date, we considered the following three possible valuation methods: (1) the income approach, (2) the market approach and (3) the cost approach to estimate our enterprise value.
The income approach focuses on the income-producing capability of a business by estimating value based on the expectation of future cash flows that a company will generate - such as cash earnings, cost savings, tax deductions, and the proceeds from disposition. These cash flows are discounted to the present using a rate of return that incorporates the risk-free rate for the use of funds, the expected rate of inflation, and risks associated with the particular investment. The selected discount rate is generally based on rates of return available from alternative investments of similar type, quality and risk.
The market approach valuation method measures the value of an asset or business through an analysis of recent sales or offerings of comparable investments or assets. When applied to the valuation of equity interests, consideration is given to the financial condition and operating performance of the entity being appraised relative to those of publicly traded entities operating in the same or similar lines of business, potentially subject to corresponding economic, environmental and political factors and considered to be reasonable investment alternatives.
In addition to the income approach and market approach valuation methods, we also considered the cost approach as a valuation method. This approach measures the value of an asset by the cost to reconstruct or replace it with another of like utility.
The Company selected a Market Approach to estimate the fair value of the Common shares as the Company sold shares of Common Stock to third parties in 2012 and 2013.
| · | During the year ended December 31, 2012, the Company entered into an agreement with a group of investors whereby the investors purchased for $810,000 of cash 2,250,000 shares of common stock, which at $0.36 per share represented a fair value of the common stock. |
| · | During the year ended December 31, 2013, the Company entered into an agreement with a group of investors whereby, the investors purchased 735,327 common units for cash proceeds of $551,497 at $0.75 per unit. Each unit consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. |
| · | During the six months ended June 30, 2014, the Company entered into an agreement with a group of investors whereby the Investors purchased 4,090,661 common units for cash proceeds of $3,067,996. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. |
| 27 |
Using an option pricing method and the relative fair values, the Company derived the implied equity value for the Common Stock based on the sale of the Units described above.
| December 31, 2013 | Six Months End | |||||||||||||||||||||||
| Common stock equivalents | Fair Value | Allocated % | Common stock equivalents | Fair Value | Allocated % | |||||||||||||||||||
| Common stock | 735,327 | $ | 551,497 | 54 | % | 4,090,661 | $ | 3,067,996 | 57 | % | ||||||||||||||
| Warrants | 735,327 | $ | 470,800 | 46 | % | 4,090,661 | $ | 2,320,054 | 43 | % | ||||||||||||||
| Relative fair value of the common stock | $ | 0.40 | Relative fair value of the common stock | $ | 0.43 | |||||||||||||||||||
There is inherent uncertainty in our forecasts and projections, and if we had made different assumptions and estimates than those described previously, the determined fair value of our common stock as of each of the Valuation Dates could have been materially different.
Derivative liability
We account for certain warrants under the authoritative guidance on accounting for derivative financial instruments indexed to, and potentially settled in, a company’s own stock, on the understanding that in compliance with applicable securities laws, the warrants require the issuance of securities upon exercise and do not sufficiently preclude an implied right to net cash settlement. We classify warrants in our balance sheet as a derivative liability which is fair valued at each reporting period subsequent to the initial issuance. We use a probability-weighted Black-Scholes pricing model to value the warrants. Determining the appropriate fair-value model and calculating the fair value of warrants requires considerable judgment. Any change in the estimates (specifically probabilities) used may cause the value to be higher or lower than that reported. The estimated volatility of our common stock at the date of issuance, and at each subsequent reporting period, is based on the historical volatility of similar life sciences companies. The risk-free interest rate is based on rates published by the government for bonds with a maturity similar to the expected remaining life of the warrants at the valuation date. The expected life of the warrants is assumed to be equivalent to their remaining contractual term.
Overview
TTSI Corporate History
Cell Source, Inc., f/k/a Ticket To See Inc., was incorporated in the State of Nevada on June 6, 2012 with a mission to make the buying and selling of advance event tickets easier, more accessible, and cost-effective for clients (venues / artists / promoters) and consumers. This was to be done by providing an online, print-your-own ticketing platform for ticketed events of all kinds, including special events, attractions, and shows and exhibits.
Cell Source Israel Corporate History
Prior to the Share Exchange, Cell Source Israel was a privately held company located in Tel Aviv, Israel. Cell Source Israel was founded in 2011 in order to commercialize a suite of inventions that were the result of over ten (10) years of research at the Weizmann Institute of Science in Rehovot, Israel (“Weizmann Institute”). Pursuant to a Research and License Agreement by and between Cell Source Israel and Yeda Research and Development Company Limited ("Yeda"), dated October 3, 2011, as amended on April 1, 2014 (the “Yeda License Agreement”), Yeda, the commercial arm of the Weizmann Institute, granted Cell Source Israel an exclusive license to certain patents, discoveries, inventions, and other intellectual property generated (together with others) by Yair Reisner, Ph.D. (“Dr. Reisner”), head of the Immunology Department at the Weizmann Institute.
Our Business
We are a cell therapy company focused on effectively treating lymphoma and other blood cell cancers. Our technology seeks to address one of the most fundamental challenges within human immunology: how to tune the immune response such that it tolerates selected desirable foreign cells, but continues to attack all other (undesirable) targets. A number of severe medical conditions are characterized by this need. These include:
| · | Haematological malignancies (leukemias, lymphomas, etc.). One of the most effective treatments for these conditions is bone marrow transplantation. However, this is a risky and difficult procedure primarily because of potential conflicts between host and donor immune systems. |
| 28 |
| · | Non-malignant haematological conditions (such as sickle cell anemia) which could also be largely treated by bone marrow transplantation if the procedure did not pose such threatening conflicts between host and donor immune systems. |
| · | Organ failure and transplantation. A variety of conditions can be treated by the transplantation of vital organs. However, transplantation is limited both by the problem of rejection and an insufficient supply of available donor organs. |
Discussion
Haematological Malignancies
Haematological malignancies (blood cancers) comprise a variety of lymphomas and leukemias. A very important treatment protocol for these malignancies involves the use of hematopoietic stem cell transplantation (“HSCT”). To the best of our knowledge, approximately 50,000 bone marrow transplantations are performed annually worldwide (table below). Our technology will be immediately applicable to, at a minimum, the 47% of worldwide bone marrow transplants that are allogeneic (using cells taken from another individual).
Source: Worldwide Network for Blood and Marrow Transplantations
HSCT has a curative effect when successful. However, it is very risky. HSCT involves destroying the patient’s native immune system with radiation or chemotherapy (myeloablation) before the transplantation, and then suppressing immune response (immunosuppression) with drugs to manage the conflicts between host and donor cells, often for the rest of the patient’s life. Approximately 50% of all transplant patients die within two (2) years of transplantation due to either aggressive pre-transplant immune suppression or post-transplant complications such as infections.
Myeloablation and immunosuppression are dangerous and difficult to tolerate, especially in patients over age 50. Therefore HSCT has been largely off-limits to the older patient population and has traditionally been used only when said older patient is clearly terminal.
This means that:
| a) | Many blood cancer patients are not candidates for the primary treatment (HSCT) that represents a potential cure; |
| b) | there is high mortality among those patients who are candidates for HSCT and do undergo the procedure; and |
| c) | those patients who successfully undergo and survive HSCT take dangerous, expensive, and quality-of-life reducing immunosuppression medications, typically for a prolonged period of time. |
| 29 |
There is widespread awareness of the need for improved immune-system management technologies for HSCT – both to improve outcomes of transplantations that have already taken place and to make transplantation safe enough to become appropriate for older patients and those with earlier-stage diseases.
We aspire to use Veto-Cell technology to dramatically improve the outcomes of the allogeneic transplantations already being performed, and thereby to rapidly penetrate the current market. However, our target population greatly exceeds those patients who currently undergo HSCT, as the firm’s tolerizing technology could potentially make allogeneic transplantation an option for a much larger proportion of the diseased population. The following table shows the prevalence of the specific haematological malignancies on which we will focus:
|
Initial Malignancy Indications (note estimates for North America and EU only (1)) |
Prevalence (Number patients) |
Annual Bone Marrow Transplantations | ||
| Non-Hodgkins Lymphoma | 823,000 | 8,700 | ||
| Multiple Myeloma | 134,000 | 13,500 | ||
| Chronic Lymphocytic Leukemia | 117,000 | 1,500 | ||
| Total | 1,074,000 | 23,700 |
(1) assumes European Union prevalence is approximately same as US
Source: Medtrack, Centers for Disease Control, Journal of Clinical Oncology.
For the purposes of this document, it is assumed that the immediate candidates for Cell Source-enabled HSCT will be the subset of cancer patients that today receive transplantations as part of their cancer treatment (rightmost column in table above). We believe that these patients will benefit from Veto-Cell adjunct therapy, as such therapy aspires to improve the success and reduce the risk and mortality of a procedure that they are having anyway. With time, as Veto-Cell treatment becomes more widespread and data is accumulated, we believe that the percentage of patients that will be referred for Veto-Cell enabled HSCT will increase significantly.
It is also important to note that incidence of these diseases is increasing, with up to a 77 percent increase in the number of newly diagnosed hematologic malignancies among the older population expected to occur over the next 20 years. See Mohamed L. Sorror et al., Long-term Outcomes Among Older Patients Following Nonmyeloablative Conditioning and Allogeneic Hematopoietic Cell Transplantation for Advanced Hematologic Malignancies, J. Am. Med. Ass'n, Nov. 2, 2011, at 1874.
HSCT Market Trends
There are four important market trends affecting the haematological malignancies market:
| (1) | As noted above, increasing incidence of these disorders in the West, largely driven by the aging population. |
| (2) | Improvement and proliferation of HSCT treatments. |
| (3) | A “virtuous circle” of lowered death rate due to better transplantations leading to more aggressive focus on HSCT. |
| (4) | The growing use of “reduced intensity conditioning,” i.e., lower myoablative dosing, which makes the procedure more survivable for older patients. |
| 30 |
The trends are highlighted on the above charts. The incidence (above left) of leukemias and other blood cancers has been rising. At the same time (above right) death rate from these conditions has been falling. The improving death rate is largely due to the proliferation of better HSCT techniques. (Source: Bell Potter Securities.)
These trends have led to, and been driven by, the increasing number of transplantations as shown in the graphic below. Note that the most significant growth since 2000 is in the area of allogeneic transplantations, including transplantations from unrelated donors (light-blue line).
However, despite the above trends, the use of HSCT, especially allogeneic, remains generally limited to younger, healthier patients because of the risks associated with the myeloablative treatments required to reduce the host immune response and Graft versus Host Disease (“GVHD”). This means that the “gold-standard” of treatment is largely unavailable to the age cohort that makes up the majority of sufferers of these diseases.
The Company aspires to addresses this issue in a distinctive manner by significantly reducing the need for myeloablative treatment and avoiding the risk of GVHD, thereby improving the outlook for allogeneic transplantations and enabling their use in a much larger population set.
Relevant Non-Malignant Diseases
While Hematological malignancies represent the Company's initial focus, the Company's selective immune response blocking technology may also be effective in treating certain non-malignant blood and immune system disorders. This would represent an additional growth opportunity for the Company.
The target non-malignant diseases are widespread. The Company's first non-malignant disorder target is expected to be sickle cell anemia. This is a serious and relatively common disease.
Sickle cell anemia can be treated by HSCT which replaces the defective bone marrow cells. However, because of HSCT’s riskiness, the procedure is currently used only in extreme cases. If successful in enabling safer HSCT, the Company will make this treatment available to a broader set of sickle cell anemia sufferers. As the therapy would be introduced in the form of bone marrow transplantation, we assume that only patients with relatively severe forms of the disease will initially be candidates. As such, only a minority of sickle cell anemia patients will be treatment candidates.
A second target within non-malignant disorders is support of organ transplantations (kidney, liver, etc.). Approximately 60,000 such procedures are conducted in North America and the EU each year. As with bone marrow transplantations, organ transplantations require substantial immunosuppression to prevent rejection. This ongoing treatment is dangerous, quality-of-life reducing, and costly. The Company's Veto-Cell technology can potentially be used to selectively reduce immune response to the transplanted organ, thus reducing the need for aggressive immunosuppression post transplantation.
Market Access and Channels
The market for transplantation therapies is relatively concentrated. There are approximately 1,400 transplantation centers worldwide, with the vast majority of them concentrated in the Americas (primarily North America) and Europe as indicated in the chart below.
| 31 |

A relatively small subset of the above centers (often termed “Centers of Excellence”) tends to set the practice standards for the entire transplantation community. Therefore, as discussed in the Strategy section, the Company plans to focus its initial penetration strategy on a relatively small group of influential centers.
Reimbursement issues for our therapies are expected to be relatively straightforward. Once clinical effectiveness and regulatory approval are established, the value-proposition for payors and providers is expected to be clear and compelling. Issues connected with immunosuppression and rejection constitute a major component of bone marrow transplantation costs, and significant improvement in this area is expected to bring substantive cost-savings for payors.
Sector Focus
We are in the general space of cell therapies. This is an emerging field, described by industry analysts as having “Blockbuster potential for regenerative treatments in indications with high level of unmet need." (Datamonitor.)
Within the cell therapy field, our initial focus is on allogeneic therapies (treatments using donor derived—as opposed to patient derived—cells), with a focus on haploidentical transplantations (transplantations that use cells from partially matched—as opposed to fully matched—donors and recipients). While potentially valuable, allogeneic therapies are relatively complex, risky, and expensive. A key driver of this complexity and associated costs is the conflict between host and donor immune systems, as discussed above.
Our technology, which in preclinical studies has shown the ability to enable tolerance of donor cells without affecting other immune processes, is fundamentally enabling. We expect it to significantly increase the safety, reduce the cost, and therefore broaden the scope of indications for such procedures.
Over time, we aspire to apply these technologies to autologous therapies (the processing and re-transplantation of an individual’s own cells) for example for the treatment of B cell malignancies. All of these treatments would take the form of non-invasive cell suspension treatments administered intravenously. The currently planned treatment modality of fully personalized medicine (i.e., using the patient’s own cells or those of a donor provided expressly by that patient) could, in some cases, eventually be supplanted by a more generic “off the shelf” modality offering which would be marketed as a pre-packaged suspension of cells and medium, taken and stored in advance for each cell “type” and then shipped to patients with the same “type” who have never met the donor. This delivery model is a longer term aspiration for us and is beyond the scope of our current market share projections.
| 32 |
Our Value Drivers
Our current positioning in the cell-therapy and cancer therapy value chain is typical of an early clinical stage company: developing, validating and attaining regulatory approvals for the various applications of our technology platforms. Going forward, once the products are commercialized, physician and patient interest in these treatments is expected to drive insurer reimbursement for patients – a key demand lever. The generic value chain for biotechnology development commences with an invention which is formulated, patented and successful in pre-clinical animal trials. We have already passed this stage with our core platforms (Veto-Cell and Organsource) for which we have an exclusive license to use (or exclusive option to license) from Yeda, the owner of these patents. The next steps in development include human trials (testing first safety and then efficacy). Finally, the offering earns regulatory approval and patient treatment, along with the ensuing revenues, can commence. This can be a particularly lengthy process in the United States and therefore some medical treatments are approved in Europe or Asia and generate revenues there prior to commencing U.S. sales. Recently passed “fast track” regulation in the U.S. is aimed at getting critical treatments for life threatening conditions to patients more quickly.
Our successful preclinical validation of the Megadose Drug Combination treatment and the Veto-Cell treatment involved basic laboratory research including both in-vivo (live) animal trials and in-vitro (in a glass dish) human cell trials. This validates the protocol prior to commencing human clinical trials. Human clinical trials fine-tune the treatment protocol and confirm both safety and efficacy in treating patents. In parallel, the patents on the core technology go into the national phase in various countries and are emended with claims associated with exact treatment protocols, bolstering the protection afforded by already issued patents on the base technology.
In some cases, successful biotech companies have been able to capitalize on positive human clinical results (even prior to full approval for patient treatment) by either signing lucrative non-dilutive distribution option deals or by being partially or fully acquired by larger market participants. There is no indication or assurance that we are currently under consideration for any option or acquisition deal.
We are poised to commence human trials for the Megadose Drug Combination treatment and concurrently finalize human treatment protocols and seek approval for the Veto-Cell based treatments.
We have had positive preclinical results for three of our cell therapy treatments and for our organ generation and regeneration treatments. Yeda, the proprietary owners of the patents underlying our technologies from whom we license our patents, has been granted patents for its original Veto-Cell and for organ generation. The revised versions of the veto cell, additional organs for “Organsource,” and the combination of the Megadose treatment with a currently FDA approved drug (as a combined treatment) are the subject of a pending patent that leverages the priority of the already granted parents for organ generation, Veto-Cell and Megadose, respectively. We plan to conduct human clinical trials with terminal patients in remission. If these trials are successful, they will demonstrate both safety (the patients survived and were not harmed) and initial indications of efficacy (there are signs of prolonging the progression free period).
Science and Technology Overview:
Our Technology Portfolio is comprised of two proprietary platforms. All the patents are owned by Yeda, and we license them exclusively on a worldwide basis. The two platforms are: Anti Third Party Veto-Cell and Organsource. Each platform already has been granted patents and has further patents pending. The total relevant patent portfolio consists of 6 inventions, 14 patent families, 10 granted patents and 88 pending patents. The patents for the Veto Cell and Megadose Drug Combination and T-regulatory cells are already licensed. Those for the organ platform are covered under an exclusive option agreement whereby Cell Source has until December 31st, 2015 to decide whether it wants to license those patents. We currently license all of the patents related to our Anti Third Party Veto-Cell technology from Yeda. The key terms of the agreement pursuant to which we license all of Yeda’s patents related to our technology is set forth in the section entitled “Intellectual Property” herein. The license period (per product, per country) is for the full life of the patents, and expires at the later of the patent expiration date in that country or 15 years after the date that the FDA or local equivalent regulatory authority in each country approves that particular product for sale in that country. As long as Cell Source pays either a nominal license fee of $50,000 per year (total for use of all the products) or pays royalties on product sales on at least one product as per the license agreement, the license will remain in effect continuously and expire only with the expiration of the patent or 15 years after regulatory approval (later of the two) per product per country as described above. Cell Source voluntarily sponsors Research at the Weizmann Institute for the sake of developing its products and treatments from initial invention through to finalization of human treatment protocols. Cell Source has recently extended the initial research period, which terminated in October of 2014, for a further four years through October 2018.
Although Yeda has applied for and been granted various patents related to our technology, a granted patent only provides Yeda, and the Company by virtue of its exclusive license, the right to use the underlying invention. However, in order for our cell-therapy and cancer therapy to be legally sold and administered to patients, the FDA or similar regulatory agencies must approve its use. In other words, having a patent provides legal “freedom to operate” for a certain technology, and may provide the ability to prevent others from using the same technology without the patent holder’s permission. However, in order to legally manufacture and distribute products, a company must go through all of the typical approval steps delineated in the Overview section above.
The following sections provide an overview of each platform. Further information on the underlying science is available upon written request and the execution of an appropriate nondisclosure agreement.
Our primary focus is the Veto-Cell platform. When we licensed the Veto-Cell platform at our inception, we also were granted an exclusive option to license from Yeda the Organsource platform.
| 33 |
Our licensed technology portfolio consists of 6 inventions, 14 patent families, 10 granted patents and 88 pending patents. The patents for the organ regeneration platform are supported by Cell Source, but we have until the end of 2015 to determine whether we wish to license them. The following table lists the patents and patent applications that Yeda holds and which we have a license to use or an exclusive option to license for use in each of the below-referenced countries:
Cell Source currently licenses the following patents held by Yeda:
| Name: VETO CELLS EFFECTIVE IN PREVENTING GRAFT REJECTION AND DEVOID OF GRAFT VERSUS HOST POTENTIAL | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA (Basic) | 6,544,506 | 05-Jan-2000 | 05-Jan-2020 | Granted | Yeda Research and Development Co. Ltd. |
| USA (National Phase) | 7,270,810 | 28-Dec-2000 | 1-Dec-2021 | Granted | Yeda Research and Development Co. Ltd. |
| Europe | 1244803 | 28-Dec-2000 | 28-Dec-2020 | Granted | Yeda Research and Development Co. Ltd. |
| Israel | 150440 | 28-Dec-2000 | 28-Dec-2020 | Granted | Yeda Research and Development Co. Ltd. |
| Name: USE OF ANTI THIRD PARTY CENTRAL MEMORY T CELLS FOR ANTI-LEUKEMIA/LYMPHOMA TREATMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 13/821,255 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 2013-527738 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 2,810,632 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| China | 201180053858.9 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 2013-7008892 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 225102 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | BR 11 2013 005756 4 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2013/002668 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Singapore | 201301743-9 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 11773325.3 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Hong Kong | 14100513.2 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| 34 |
| Name: ANTI THIRD PARTY CENTRAL MEMORY T CELLS, METHODS OF PRODUCING SAME AND USE OF SAME IN TRANSPLANTATION AND DISEASE TREATMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 14/343,053 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 12769743.1 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 2014-529143 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 2,848,121 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| China | 201280054739.X | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Australia | 2012305931 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 10-2014-7009267 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| New Zealand | 622749 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| South Africa | 2014/01993 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| India | 577/MUMNP/2014 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 231397 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 2014110897 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | BR 11 2014 005355 3 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2014/002771 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Singapore | 11201400513P | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Name: ANTI THIRD PARTY CENTRAL MEMORY T CELLS, METHODS OF PRODUCING SAME AND USE OF SAME IN TRANSPLANTATION AND DISEASE TREATMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 13/126,472 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 09764302.7 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 212587 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| India | 905/MUMNP/2011 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| China | 200980153053.4 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 2011121630 | 29-Oct-2009 | 29-Oct-2029 | Granted | Yeda Research and Development Co. Ltd. |
| Name: UNIVERSAL DONOR-DERIVED TOLEROGENIC CELLS FOR INDUCING NON-SYNGENEIC TRANSPLANTATION TOLERANCE | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| US | 11/990,628 | 21-Aug-2006 | 21-Aug-2026 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 06796063.3 | 21-Aug-2006 | 21-Aug-2026 | Pending | Yeda Research and Development Co. Ltd. |
| Europe (Divisional) | 12161171.9 | 21-Aug-2006 | 21-Aug-2026 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 189688 | 21-Aug-2006 | 21-Aug-2026 | Granted | Yeda Research and Development Co. Ltd. |
| 35 |
| Name: A COMBINATION THERAPY FOR A STABLE AND LONG TERM ENGRAFTMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| Singapore | 11201403459X | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2014/007647 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | BR 11 2014 015960 2 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 2014128479 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 233303 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| India | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| South Africa | 2014/05071 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| New Zealand | 627272 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Australia | 2012355990 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| China | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 12859036.1 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| USA | 14/367,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Name: A COMBINATION THERAPY FOR A STABLE AND LONG TERM ENGRAFTMENT USING SPECIFIC PROTOCOLS FOR T/B CELL DEPLETION | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| Singapore | 11201403456U | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2014/007648 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | BR 11 2014 015959 9 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 233302 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| India | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| South Africa | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| New Zealand | 627549 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Australia | 2012355989 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| China | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| USA | 14/367,923 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| 36 |
Cell Source currently has an option to license, and in the interim is fully funding the following patents:
| Name: METHODS OF TREATING DISEASE BY TRANSPLANTATION OF DEVELOPING ALLOGENEIC OR XENOGENEIC ORGANS OR TISSUES | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| Mexico | 319957 | 04-Mar-2004 | 04-Mar-2024 | Granted | Yeda Research and Development Co. Ltd. |
| Mexico (Divisional) | MX/a/2014/001950 | 04-Mar-2004 | 04-Mar-2024 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 2216033 | 04-Mar-2004 | 04-Mar-2024 | Granted | Yeda Research and Development Co. Ltd. |
| Name: METHODS OF TREATING DISEASE BY TRANSPLANTATION OF ALLOGENEIC OR XENOGENEIC ORGANS OR TISSUES | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| Europe | 11179593.6 | 04-Mar-2004 | 04-Mar-2024 | Pending | Yeda Research and Development Co. Ltd. |
| Name: THE USE OF DEVELOPING ALLOGENEIC OR XENOGENEIC ORGANS OR TISSUES FOR DISEASE TREATMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| Israel | 170622 | 04-Mar-2004 | 04-Mar-2024 | Pending | Yeda Research and Development Co. Ltd. |
| Name: METHODS OF KIDNEY TRANSPLANTATION UTILIZING DEVELOPING NEPHRIC TISSUE | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| Israel | 218961 | 01-Sep-2002 | 01-Sep-2022 | Pending | Yeda Research and Development Co. Ltd. |
| Name: THERAPEUTIC TRANSPLANTATION USING DEVELOPING, HUMAN OR PORCINE, RENAL OR HEPATIC, GRAFTS | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 7,780,993 | 19-Jan-2005 | 23-Apr-2023 | Granted | Yeda Research and Development Co. Ltd. |
| USA (Divisional) | 12/777,292 | 19-Jan-2005 | 19-Jan-2025 | Pending | Yeda Research and Development Co. Ltd. |
| Name: DISEASE TREATMENT VIA DEVELOPING NON-SYNGENEIC GRAFT TRANSPLANTATION | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 11/664,530 | 02-Oct-2005 | 02-Oct-2023 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 1809734 | 02-Oct-2005 | 02-Oct-2025 | Granted | Yeda Research and Development Co. Ltd. |
| Europe (Divisional) | 11193959.1 | 02-Oct-2005 | 02-Oct-2025 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 182363 | 02-Oct-2005 | 02-Oct-2025 | Pending | Yeda Research and Development Co. Ltd. |
| Name: MAMMALIAN FETAL PULMONARY CELLS AND THERAPEUTIC USE OF SAME | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 14/363,814 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 12813990.4 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 61/568,240 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 61/568,240 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| China | 61/568,240 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Australia | 2012348574 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 10-2014-7018702 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| New Zealand | 627071 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| South Africa | 02014/04958 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| India | 1366/MUMNP/2014 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 233022 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 2014127338 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | 61/568,240 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2014/006756 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Singapore | 11201402902V | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Philippines | 1-2014-501309 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
Platform I – Veto-Cell:
Background
Our Veto-Cell technology is a next generation cell-therapy technology that enables the selective attenuation of the immune system. In other words, pre-clinical studies suggest that the treatment has the ability to reduce the immune response to selective “threats,” with low risk for adverse side effects.
What makes the Veto-Cell approach unique is the degree to which it leverages the inherent specificity of the human immune system. The immune system defends the body by creating a specific stream of T-cell clones for each of millions of individual threats. A given T-cell will attack only its specific target, ignoring all other threats. Our technology might enable the physician to selectively attenuate immune response, thus effectively “switching-off” an individual stream of T-cell clones without affecting any other such streams of T-cell clones dispatched by the immune system to attack unwanted incursions.
The technology is based on the discovery that certain T-cells have the property of attracting and proactively neutralizing immune attacks on them.
| 37 |
The technology has achieved distinctive results in animal live trial models. See, e.g., Thorsten Zenz, Exhausting T cells in CLL, Blood, Feb. 28, 2013, at 1485. If it succeeds in human clinical trials, we believe that it may have meaningful and potentially broad impact on the field of bone marrow transplantation:
| 1. | Significantly improve outcomes of transplantations by reducing host rejection rate of T-cell depleted bone marrow, markedly reducing both the risk of graft-versus-host-disease and the need for using aggressive amounts of immunosuppression medications. This would significantly reduce the bone marrow transplant mortality rate (currently 50%) and therefore lead to broader use of this treatment. |
| 2. | Substantively increase the number of transplantations by enabling lower myeloablative conditioning and therefore making the therapy accessible to older and sicker patients (who today may not survive ablation). |
| 3. | Further increase the number of transplantations by making transplantation appropriate for other indications (for which today transplantation would be considered an inappropriately risky treatment). |
In addition, our Veto-Cell technology may possibly play a role in the treatment of a number of serious and currently poorly treated non-malignant diseases. Furthermore, initial animal trials have shown potential anti-lymphoma activity.
Mechanism
Our Veto-Cell is a CD8 central memory anti-3rd party T-cell that has five critical properties:
| 1. | It has an outer surface coating that triggers attack by specific host T-cells (and only those specific T-cells). |
| 2. | It can annihilate an attacking T-cell without itself being damaged (specifically, it exposes or releases a death-signaling molecule when an attacking T-cell binds to it). |
| 3. | It has been oriented to attack cells of a simulated third party (i.e., neither host nor donor) and thus exhibits markedly reduced risk of GVHD or graft rejection. |
| 4. | It is long-lived and endures in the body for extended periods. |
| 5. | It migrates to the thymus and lymph nodes. |
The outcome is that when a large number of these cells are introduced into the body, they effectively eliminate the T-cell clones that the immune system dispatches to attack the desirable, transplanted bone marrow cells.
Thus, for example, if a population of Veto-Cells is derived from a donor, they will express the same peptide as do the donor’s cells. Therefore, the specific stream of host T-cells that would ordinarily attack the donor stem-cells, are instead directed to “decoy” Veto-Cells and disabled before they reach the transplantation.
Described in a Blood editorial as a “substantial advance in Cell Therapy," a notable characteristic of our Veto-Cell is that this mechanism is quite specific. Only those specific T-cell clones that were generated to attack cells from this specific donor are disabled. The rest of the immune system essentially remains intact.
This is in marked contrast with conventional immunosuppression which degrades the entire immune system and is therefore associated with severe risk of infection and, in the case of bone marrow transplantations, high mortality.
This effect is long-lived. Firstly, the Veto-Cells themselves are long-lived memory cells. Secondly, when infused with bone marrow cells the latter migrate to the thymus where, over time, they create a new “identity” in the host and initiate “chimerism,” where the host and donor cells peacefully co-exist. This chimerism has the effect of "educating" new T-cells being generated by the thymus to tolerate donor cells. This tolerance can become permanent.
Target Indications
Our Veto-Cell technology, an intravenously administered cell suspension, if successful, could initially be used in bone marrow and other transplantations associated with malignant disorders (i.e., cancers). At a later stage, Veto-Cell technology may be applied to selected non-malignant conditions. The following sections provide a brief overview of the use of the Veto-Cell technology in both of these scenarios.
| i. | Bone Marrow Transplantation |
In order to describe the effect of Veto-Cells in transplantation, it is helpful to first briefly review the state of the art:
| 38 |
In a conventional bone marrow transplant, the recipient first receives myeloablative conditioning – powerful chemotherapy and/or radiation therapy intended to destroy his/her own bone marrow cells. This has a threefold purpose:
| 1. | It destroys the host T-cells so they will not attack (reject) the donor bone marrow cells. |
| 2. | It makes space in the host bone marrow for the new donor cells. |
| 3. | It destroys diseased host blood cells so that they do not proliferate and cause relapse following the procedure. |
In practice however, there are two major problems:
| · | Host rejection – the myeloablative conditioning does not destroy all the host T-cells. Those that remain may aggressively attack the donor bone marrow cells before they can engraft. |
| · | “Graft versus Host Disease” (GVHD) –the transplanted cells include donor T-cells which recognize the host's body as foreign and attack it. |
Both rejection and GVHD are potentially life-threatening complications in and of themselves and also lead to the use of dangerous and costly immunosuppression medications. The Megadose technology addresses the foregoing two problems by introducing an extremely large population of selected donor cells into the host. This overwhelms the remaining host immune system, and therefore, reduces the risk of rejection. It also reduces the risk of GVHD, as the donor cells are selected so as to minimize the number of accompanying T-cells.
Megadose is a well-developed technology and is now used in clinical treatments where a “mismatched” bone marrow blood cancer transplantation is in order.
| ii. | Veto-Cell in Transplantation |
The Veto-Cell technology is a next generation of the Megadose concept. In a transplantation scenario, a population of donor Veto-Cells is created to "escort" the bone marrow cells when they are transplanted. This population is created by identifying donor cells with Veto-Cell properties, exposing them to simulated 3rd party cells (i.e., selecting only those that react to a third person and therefore by definition will not react to either host or donor), and expanding their population in the lab.
The Veto-Cells are then introduced into the host along with the transplanted stem-cells. The host mounts its normal immune response to the donor cells by generating a population of T-cell clones that will bind to any cells expressing markers from this specific donor. In a conventional transplantation, these T-cells would bind to and destroy donor stem-cells thus causing rejection of the transplant.
However, when the transplantation is accompanied by large numbers of Veto-Cells, this rejection mechanism is “ambushed." Since the Veto-Cells express the same donor markers as the stem-cells, the host T-cell clones will attempt to bind to the donor-derived Veto-Cells as noted above, which act as decoys by attracting and then counterattacking and killing the clones before they ever reach the bone barrow transplantation.
| iii. | Direct Anti-Cancer Effect |
A further effect of Veto-Cells has been noted in mouse and in-vitro studies: donor Veto-Cells selectively attack host lymphoma malignant cells. This effect has been robust in animals, in fact completely eradicating lymphoma in mouse models (see Development Status section below).
The direct anti-cancer effect has been documented for several human B cell malignant lines, however, preliminary experiments with human anti-3rd party veto cells prepared in a slightly different protocol than that used for the mouse studies, indicate that further optimization and verification are required before killing fresh human B-CLL or myeloma tumor cells could become a feasible option.
If this effect transfers to human patients, it may have significant therapeutic value for the above disorders, which as noted hereafter in the Marketing Strategy section, are among the largest blood cancer markets.
| iv. | In Non-Malignant Diseases |
As discussed above, there are two major categories of non-malignant disorders that the Veto-Cell technology aspires to address: non-malignant hematological disorders and organ transplantations.
In the case of organ transplantations and congenital non-malignant hematological disorders, the goal of the veto cells is to enable transplantation (bone marrow or organ) by reducing host/donor immune system conflicts.
| 39 |
For example, in the case of congenital non-malignant diseases such as sickle cell anemia, the body’s bone marrow produces “flawed” cells. An effective treatment is HSCT which replaces the flawed host bone marrow with healthy donor cells. These cells then produce healthy blood cells, basically curing the anemia. As noted elsewhere however, today HSCT is a risky procedure because of the graft/host immune conflicts. It is therefore used infrequently to treat sickle cell disease. The Veto-Cell tolerizing technology would increase the target population for this treatment by significantly reducing these conflicts and by extension the procedure’s risk. Likewise, if permanent tolerance to donor hematopoietic cells is induced under safe conditions, the new immune status could permit acceptance of a kidney from the same donor, without further requirement for a toxic immune suppression currently used in organ transplantation. This means that patients who today are required to take expensive and sometimes debilitating anti-rejection medication daily for the rest of their lives would no longer have to do so.
Development Status
The Veto-Cell platform has been extensively tested by in vitro studies (on both human and mouse disease) and confirmed in animal trials. The results appear to be consistently effective.
| 1. | Immune-system management: |
The following images show some example data from the Veto-Cell animal studies. Skin of black mice has been grafted onto the backs of white mice. The data show that T-cells from host and donor mice are fully coexisting in the treatment group using the Megadose treatment (“chimerism”). This is done using high levels of immune suppression that are associated with high mortality. Our Megadose drug combination aspires to produce the same results with lower, safer levels of immune suppression.
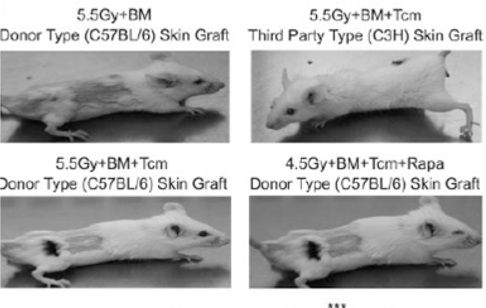
| 2. | Anti-lymphoma tumor cells action: |
The anti- lymphoma tumor effect also appears to be consistently effective. The data below shows mice with non-Hodgkins lymphoma treated with Veto-Cell therapy.
The control group mice (A) have pronounced tumors by day 21, and have all died by day 28. By contrast, the Veto-Cell treatment group (B) show no tumor and all are still healthy by day 100.
| 40 |
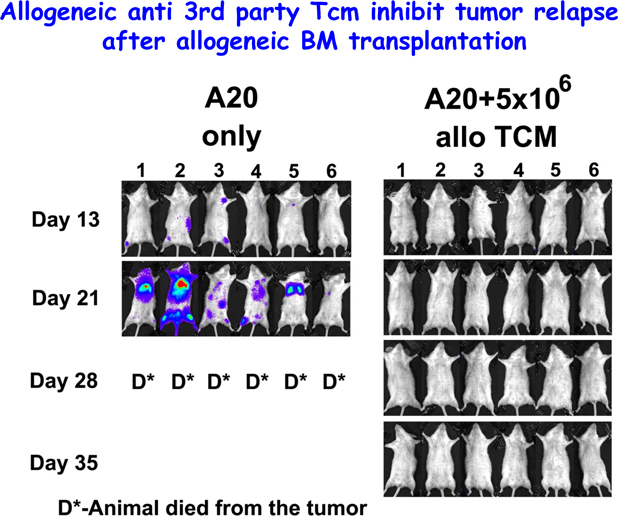
| 41 |

We are less confident about the status of anti-human B-CLL as initial experiments in-vitro were not satisfactory as outlined in our progress report: “While marked progress has been made in developing a protocol for the generation of human Tcm either from normal donors or from B-CLL patients, the Tcm fail to kill autologous B-CLL tumor cells in-vitro. This problem might arise from the secretion of protective cytokines which can accumulate during the culture. This problem which is currently addressed as outlined below, can adversely affect our plans to start a clinical trial with autologous Tcm in patients with B-CLL.”
Administration
We envision that Veto-Cell therapy will be administered in an in-patient setting, typically as part of the existing preparation procedures for bone marrow transplantations. Blood will be taken from the donor. The frozen blood will be sent to a regional Company center where the Veto-Cells will be developed and expanded – a process that lasts up to two weeks. The Veto-Cells will then be sent to the transplantation center where they will be infused to the patient intravenously along with the transplantation.
Patent Status
The original Veto-Cell is protected by three granted patents and multiple additional pending patents in various countries. The patent for the current central memory or Tcm Veto-Cell, which is slated for human clinical trials, is still pending; however, the patent benefits from the priority of the previous Veto-Cell patents because these earlier versions act as “prior art” thereby bolstering the current patent application. The patents provide coverage on the Veto-Cell technology as discussed in the IP section below.
Development Roadmap
The Veto-Cell platform roadmap comprises three main programs as outlined in the table below. The specific clinical trials planned for each are detailed in the Clinical Trials section of this document.
| 42 |
| Offering | Objective | Major Activities | Estimated start date | |||
| Megadose drug combination (a distinct treatment from the Veto platform) | Validate and introduce new commercial treatment to increase engraftment of allogeneic bone marrow transplantations | 1. Regulatory approval and treatment protocols 2. Conduct human clinical trials 3. Develop plan for commercial exploitation |
• Commence a formal company-sponsored Phase I/II clinical trial by the end of 2014 • Interim analysis within 18 months thereafter
| |||
| Anti-lymphoma veto cell | Validate the possibility of introducing commercial Veto-cell treatment for lymphoma, multiple myeloma and B-cell chronic lymphocytic leukemia (BCLL) based on autologous transplantation | 4. Define feasibility of using human veto cells for killing fresh CLL tumor cells ex-vivo or in experimental mouse models 5. Develop large scale production protocol (GMP process) 6. Conduct human clinical trials to validate safety and efficacy |
• Protocol validation and production process development already underway • If preclinical studies are successful , human trials would be the next step
| |||
| Anti-rejection veto cell | Validate and introduce commercial Veto-cell therapy for reducing rejection in allogeneic bone marrow transplantation for blood-cancers | 7. Develop large scale production protocol (GMP process) 8. Conduct human clinical trials to validate safety and efficacy |
• Production process development already underway • Production process for this Investigational New Drug (IND) may be approved in 18-24 months, Human trials may be approved in 24-30 months depending on regulatory approval cycle |
Platform II – Organsource:
Overview
Organsource is Yeda's patented technology, as referenced above. Yeda has been granted patents in the United States and Europe. Organsource addresses the growing shortage of organs for human transplantation. Cell Source has an exclusive option to license this technology, which option has been extended until December 31st, 2015.
The key discovery has two elements:
| · | Embryonic tissue (taken from an animal or human fetus during gestation), which can be identified as organ precursors that will grow into specific organs (e.g., kidney, liver, pancreas), can be harvested at a very specific moment in the gestation period where they have just then become “committed” organ precursors and thus have not yet begun to generate the acute levels of rejection otherwise typical for xenotransplant (i.e., between species) which have been problematic in other earlier studies transplanting porcine tissue into humans. |
| · | These pre-organs can be successfully transplanted into a host, even of another species, and grow into functional organs in the host with only the level of organ rejection associated with an allogeneic organ donor, which can currently be managed through medication. Incidentally, this post transplantation rejection could potentially be further reduced by using Veto-Cells. |
This means that porcine embryonic tissue can potentially become a source for human organ replacement. We intend to exercise our option to license this technology and intellectual property.
Background
The main focus of the Organsource work to date has been demonstrating that organ precursor tissue can be successfully transplanted into both rodents and primates from pigs.
| 43 |
Pigs have long been considered the ideal source of organs for human transplantation for two reasons:
| · | Their organs are similarly sized to humans, and |
| · | They have large litters so can provide extensive supply (unlike for example monkeys). |
However, others’ previous experimental efforts to transplant porcine organs into primates have shown only limited success because a certain marker on pig blood vessels causes a hyper-immune response in primates (which, for example, have immediately killed organ recipients in trials with monkeys).
Mechanism
The Organsource technology avoids the hyper rejection problem by extracting embryonic pig tissue in a highly specific development window. Cells within this momentary window can grow inside the host using blood vessels of the host, not donor, origin. Therefore, they do not trigger the host hyper-immune response. However these embryonic organ precursors have developed sufficient organ differentiation to act as pre-organs in the host, and they grow into functional developed organs, in the case of primates, within a few months.
Specifically, a mouse with Type 1 diabetes received a transplanted porcine pre-pancreas, which grew into a full sized pancreatic organ largely composed of beta cells which secrete insulin, thus effectively treating diabetes in the mouse. Similar results have also been achieved in monkeys.
Target Indications
Organsource could theoretically provide a significant new source of transplantation organs for major human organ needs.
Work so far indicates positive results for growing a pancreas to replace one in which beta cells have been chemically disabled leading to a disease similar to that found in Type 1 Diabetes.
Development Status
Organsource is at an early stage of development relative to the Veto-Cell platform. However, in-vitro results and animal trials have shown positive progress. For example:
| · | Porcine spleen tissue was successfully implanted into a mouse, effectively treating hemophilia. |
| · | Embryonic lung cells have shown effectiveness in repairing injured mouse lungs and are currently being tested on cystic-fibrosis mice. In principal, these could potentially be used to effectively treat several major lung diseases. |
| · | Porcine pancreatic cells were successfully infused into monkeys where they effectively corrected chemically induced diabetes. The chart below shows exogenous insulin requirements of the subject animal (vertical axis) as a function of the number of days following the transplantation (horizontal axis). |
Note that within days of the transplantation, the insulin requirements drop sharply, indicating that the porcine cells are now producing insulin in the monkey, and 10 months after transplantation the monkey is diabetes free. Considering that in these experiments the recipient body weight is small, and a large dose of tissue was used for transplantation, it could be argued that our approach might not be feasible for treating large human adults. In other words, the number of porcine pancreases required for the dosage for treating a large human being may prove to be prohibitive.

| 44 |
Administration
Administration of Organsource is may be less invasive than a typical organ transplantation procedure. In the case of smaller organs such as the pancreas, Organsource transplantation requires only a relatively minor procedure. This is because precursor cells rather than full grown organs are being introduced.
Since the embryonic implants can promptly attract blood vessels they therefore can be placed in sites in the body nearer to the surface of the skin instead of deeper internal sites such as the pancreatic cavity.
Patent status
A U.S. patent has been granted for porcine liver generation and a European patent was recently granted for pancreas generation. Patents for heart, kidney, lung, and lymph glands are pending. There is also a patent pending for repairing existing lung tissue using human embryonic cells.
Development Roadmap
Our Organsource roadmap is to continue animal testing in vivo, with an eventual aspiration to human trials. Organsource’s next animal tests will attempt to regenerate healthy lungs in mice that currently have diseased lungs and to refine the process of and specific tissue doses required for regenerating pancreases in monkeys and to address organ rejection in such pancreatic procedures. Organsource’s first human trials, which we don’t forecast occurring within the next five years, will most likely concern porcine pancreatic tissue to humans or using human embryonic cells to effectively treat diseased lungs.
Products and Services
Currently, we do not have any products, and there is no assurance that we will be able to develop any products.
Our initial products will likely be based on the Veto-Cell platform. We are also about to commence human trials for a new product that combines Dr. Reisner’s existing Megadose technology with an existing generic FDA approved drug. This combination of products has a potential to be an early source of revenues. We expect that spontaneous trials on compassionate grounds by a University using technology which we license from Yeda will commence in 2014 and Company sponsored trials will commence in 2015 or early 2016. Additionally, the Organsource platform may potentially generate products and revenues in the longer term.
The following products are currently planned and represent most of the projected revenues presented in the financial section:
| 1. | “Anti-rejection” veto-cell tolerance therapy for both matched and mismatched allogeneic bone marrow transplantations. This is our flagship (as an initial platform for increasing transplantation success) and is focused on allogeneic bone marrow transplantations. |
Treatment will comprise a course of infusions of Veto-Cells derived from the donor and processed in a Company facility that will be accessible to the transplantation center at the time of transplantation.
| 2. | “Anti-cancer” veto-cell therapy for lymphoma, multiple myeloma and BCLL. This is an intravenous cell-suspension-based cell-therapy focused on lymphocyte cancers. |
This therapy will comprise a course of infusions derived from the patient’s own blood and prepared for autologous transplantation. (In cases of allogeneic transplantation, donor cells will be used.) This treatment exploits the observed effect that Veto-Cells tend to selectively attack lymphoma cells that is described in the Technology section.
| 3. | Veto-Cell tolerance therapy for non-malignant disorders. This is the application of Veto-Cell technology to treatment of non-malignant (i.e., non-cancerous) diseases. As discussed in the Technology section, a custom treatment would be developed for each selected disorder. |
Target indications for Veto-Cell therapy for nonmalignant disorders are likely to be: tolerizing therapy for allogeneic transplantations for sickle cell anemia and aplastic anemia (by using bone marrow transplantations as referenced in no. 2 above) and tolerizing therapy for conventional organ transplantations.
| 45 |
Our Overall Development Status and Future Development Program
Prior to commercializing its products, the Company must conduct human clinical trials and obtain FDA approval and/or approvals from comparable foreign regulatory authorities.
Generally speaking, as a preclinical biotechnology firm, Cell Source needs to go through several necessary steps in order to commercialize its products and commence revenue generation. These steps are per product, but can run in parallel for multiple products, which are each in different stages of the development “pipeline”, so that, for example, when a certain product is already in a human clinical trial, another product may still be in preclinical development and a third may be awaiting regulatory approval to commence human trials. These can also take place in parallel, and varied stages, for the same product in different geographic jurisdictions. The typical steps per product (and range of time frame for each) are:
| 16. | Complete development of human treatment protocol (2-5 years) |
| 17. | Apply for and receive approval to commence human trials (9-18 months) |
| 18. | Recruit patients (1-6 months) |
| 19. | Conduct Phase I trials showing safety of product (1-2 years) |
| 20. | Apply for and receive approval to conduct trials showing product efficacy (6-12 months) |
| 21. | Data collecting and analysis (6-12 months) |
| 22. | Conduct Phase II efficacy trials (2-3 years) |
| 23. | Data collecting and analysis (6-12 months) |
| 24. | Apply for and receive approval to conduct trials showing efficacy in larger numbers of patients (6-12 months) |
| 25. | Conduct Phase III efficacy trials with larger numbers of patients (2-4 years) |
| 26. | Data collecting and analysis (6-12 months) |
| 27. | Apply for and receive approval for production scale manufacturing facilities (6-12 months) |
| 28. | Contract third party or establish own production facilities (6-30 months) |
| 29. | Contract third party or establish own distribution platform (6-18 months) |
| 30. | Commence manufacturing and distribution (6-12 months) |
Notably, steps 12-15 can be conducted in parallel with some of the steps above. In the case of Cell Source and other firms that treat terminal patients with either rare diseases or those for which there is currently no effective treatment, or where preclinical studies indicate a reasonable expectation to increase life expectancy and survival rates by a substantive margin, several of these steps can be combined and or shortened, subject to regulatory discretion. For example, Phase I and II (safety and efficacy) can be combined in a single concurrent step; approvals for subsequent steps can be accelerated; in some countries patients can already be treated commercially after the end of Phase II, foregoing the requirement for Phase III data.
The specific detailed next steps the company must take to get the treatments or products to market include the following:
In the case of the Megadose Drug Combination, the Hematology and Bone Marrow Transplantation Unit of the University of Parma in Italy on May 14, 2014 requested and on October 23, 2014 obtained approval from the Italian Medicine Association (the Italian equivalent of the US FDA) to conduct human clinical trials using the “Megadose + Drug Combination”. While this is a small initial study conducted solely by the University of Parma, the protocol employs the specific technology which is licensed by Cell Source from Yeda Research and Development Limited. The University of Parma on May 14, 2014 filed an investigational new drug application (IND) in Italy, which IND was approved on October 23, 2014. Our final human treatment protocols for Veto cell treatments are still under development. For this reason, Cell Source has yet to file an IND for the Veto Cell treatment. Furthermore, the organ regeneration dosages and final animal protocols are still under development and will therefore require significant further development prior to filing an IND for the organ regeneration products. Furthermore, we have not submitted any drug applications to the FDA and do not have anything pending for approval with the FDA. Cell Source itself has not had any contact with any regulator anywhere regarding treatment approvals or clinical trials associated with regulatory approvals. We are aware that the aforementioned hospital in Italy has independently requested approval to conduct a trial with the same protocol that we plan to use, but we are not mentioned in the application, nor in the approval. However, we may indirectly benefit from the outcome of the trial, if successful, although we are not the sponsor of this trial. There are no written or verbal agreements between the hospital and Cell Source regarding the use of the technology. That said, Cell Source is aware and in favor of the hospital plans to use the technology and would find a positive initial outcome encouraging. Since the treatment is being done on compassionate grounds as a non-commercial clinical trial, there is no legal requirement for the hospital to obtain approval to use the treatment protocol.
Cell Source then plans to commence a follow-on full Phase I/II study in Parma, Italy that is meant to demonstrate safety and efficacy and is anticipated to last between 2 and 3 years. The local regulator will then determine whether the product will then be made available for sale, or whether it will require a Phase III study. Since the product involves treatments that are already in the clinic but in combination in a new way, and since Cell Source aspires to demonstrate a significant increase in survival rates, this treatment may conceivably be made available commercially on a “compassionate grounds” basis immediately following the conclusion of Phase I/II, which could be in 2019. In the event that a full Phase III study is required, a further 2-3 years may be required.
For the Veto-Cell applications for reducing rejection in Bone Marrow Transplants and for eradicating lymphoma cells, Cell Source expects to commence a Phase I/II human clinical study in Italy, and subsequently in Germany, starting sometime in 2016. Since this technology does not involve elements that are already in clinical use, Cell Source anticipates that Phase I/II studies will last until 2018 until 2019. These would be followed by completion of Phase II and Phase III, which would last another 2-3 years each, so that full approval, if successful, would be expected sometime in 2024. In Germany there is a possibility of approval for commercial use on a “compassionate grounds” basis at the end of Phase II, which could take place by 2022. In the US, Cell Source may or may not be permitted by the FDA to submit European trial results as “supporting data”. If not, Cell Source would have to go through the full FDA approval process, which, commencing in 2015, would last until between 2021 and 2023 for the Megadose Drug Combination. For the Veto-Cell this would commence in 2016 or 2017 and could last until 2024 to 2025. It is possible that Cell Source treatments could qualify for any or all of Fast Track, Breakthrough Therapy, Accelerated Approval and Priority Review designation under the FDA, which would hasten their approval if successful.
| 46 |
The costs for each step of development, in terms of clinical trials, are delineated below:
Cell Source estimates the cost of clinical trials alone to be up to $5 million over the coming two years and another $25-50 million in order to reach commercialization for both the Megadose Drug Combination and the Veto-Cell products. This would mean that Cell Source will need to secure one or more significant capital infusions in order to reach the point that meaningful revenues could be incurred.
Cell Source will require additional financing for any and all of the steps described above.
The following table summarizes the development plan for the coming few years:
Competition
The development and commercialization of new cell therapies is highly competitive. Our products are focused on treatment of blood cancers, non-malignant blood disorders and organ transplantations. Various products are currently marketed for the treatment of blood cancers. A number of companies are also developing new treatments. In addition to competition from a variety of other nascent unconventional medical treatments, we also face competition from established pharmaceutical and biotechnology companies, as well as from academic institutions, government agencies and private and public research institutions worldwide. For instance, our competitors include the technology developed by Micromet, Inc., which was since acquired by Amgen Inc. and Avila Therapeutics, a wholly-owned subsidiary of Celgene Corporation.
| 47 |
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Earlier stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. While our commercial opportunities may be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than our own products, we believe that if our human trials show efficacy at the same levels of our animal trials, we would have the potential to develop at least a niche market share.
We expect that our ability to compete effectively will depend upon our capacity to:
| · | successfully and complete adequate and well-controlled clinical trials that demonstrate statistically significant safety and efficacy and to obtain all requisite regulatory approvals in a timely and cost-effective manner; |
| · | effectively use patents and possibly exclusive partnership agreements with important treatment facilities to maintain a stable competitive stance for our Technology; |
| · | attract and retain appropriate clinical and commercial personnel and service providers; and |
| · | establish adequate distribution relationships for our products. |
Failure in efficiently developing and executing these capabilities may have an adverse effect on our business, financial condition or results of operations.
Strategy Overview
Our strategy is based on two underlying drivers: (a) that animal studies show Veto-Cell technology to be consistently effective; and (b) that the lead indications (certain blood cancers) are relatively common, have high mortality and have limited treatment options today.
Based on the foregoing drivers, we have developed a business plan with the objective of obtaining regulatory approvals and subsequently launching product sales with a focus on Europe, Asia and the United States.
Key Strategy Elements
We are pursuing a staged entry strategy. The first several years will be narrowly focused, both in terms of market segments (Lymphoma, 3rd party clinical trials and bone marrow transplantation) and products (Megadose drug combination and then, hopefully, our Veto-Cell platform).
Subsequently, we plan to broaden the segmentation strategy to include additional bone marrow transplantation indications as well as selected genetic non-malignant diseases. The product strategy may also be broadened to include the Organsource platform.
Our strategy can be summarized as follows:
| Strategy Element | Introductory period (years 1 -3 post FDA approval) |
Years 4+ | ||
| Market Segments |
· Lymphomas and BCLL · Other bone marrow transplantation in acute fatal conditions with no current medical treatment |
· Lower criticality/higher volume segments (non-malignant diseases)
| ||
| Product Rollout |
· Autologous Veto-Cell therapy for lymphocyte cancers · Allogeneic Veto-Cell therapy for bone marrow transplantation tolerance |
· Veto-Cell therapy for additional transplantation indications · Veto-Cell therapy for non-malignant disorders · Trials of Organsource platform | ||
| Customer/ Geographic Focus |
· North America · Western Europe · China |
· North America, Western & Eastern Europe, Russia, Brazil, selected Asian markets | ||
| Channels/Go to Market | · Direct relationships with leading transplantation centers |
· Additional relationships with leading oncologists · Sponsored conferences for oncologists and transplantation providers in key regions | ||
| Pricing | · Consistent with other cell therapy offerings currently associated with transplantations | · Potentially higher volume, lower cost for “off the shelf” offerings | ||
| Operations |
· Three operating centers : · US East Coast · US West Coast · Western Europe · Operating centers in lab-space leased from major transplantation centers. |
· Eight operating centers · US East & West Coasts, Western Europe, Russia, Brazil, Japan, China, Australia/New Zealand · Operating centers in company-owned facilities |
| 48 |
Segment Selection
Within the general market for immune therapies, we have selected target market segments (i.e., medical conditions) for initial focus based on two (2) key criteria:
| 1. | Severity of medical need: degree of severity of the indication and the effectiveness of existing treatments. These criteria help determine the proper regulatory pathway. |
| 2. | Technology relevance: relative value of the ability to manage immune response to the treatment of a given indication. |
We will initially focus on indications that score highly with respect to both criteria (e.g., Multiple Myeloma and BCLL). These conditions may qualify for Fast Track status with the FDA, and, due to the cost of current treatment alternatives, could potentially support profitable price points for effective new treatments.
Product Rollout
Cell Source plans to seek approval initially in Europe (Italy and Germany) and, in parallel but with a delayed start, in the US and China and possibly Taiwan. A successful Phase I/II trial in Europe, which could be concluded by 2018, would serve as a strong foundation for trials in other countries. Cell Source plans to approach the FDA (US) and CFDA (China) with interim data from Italian trials by 2017, with a view to commencing trials in both jurisdictions based on that data. Limited sales on a “compassionate grounds” basis may commence as early as 2019 in Europe and Asia, and, depending on qualification for Fast Track or other Accelerated Approval status paths, may be available in the US by 2021. Full approval by the FDA in the US can take as long as 8 years, or 2025.
Future products may include Veto-Cell tolerance inducement therapy for allogeneic bone marrow transplantations and Veto-Cell cell therapy for lymphoma.
Following the initial market penetration and establishment of solid market positioning, we plan to broaden the product offering to address a wider variety of indications which may include custom Veto-Cell developments for specific non-malignant diseases and continued work on Organsource.
Customer/Geographic Focus
Assuming positive clinical trials, we will initially focus our sales efforts of Veto-Cell autologous therapy on centers dealing with Stage 4 Lymphomas. High profile, high volume HSCT facilities can be targeted to market the Megadose drug combination, possibly augmented in the future by Veto-Cell therapy.
Current plans are to introduce the products first in North America and Western Europe, and, perhaps concurrently, in China. Focusing on key transplantation facilities in target geographic markets will allow us to both refine the administration of our products and bolster our reputation in respective markets.
After the introductory period, we plan to expand its activities in its initial markets while simultaneously broadening geographic coverage. In Stage 2, we plan to initiate active marketing efforts in the remaining Western European countries, Japan, Australia, Eastern Europe, Russia and Brazil.
Marketing Strategy
The initial target market is concentrated and networked. It comprises the approximately 40 leading transplantation centers in the target geographies. As discussed in the Market Access and Channels section, these centers are well connected to each other and tend to quickly share innovations and best practices.
The planned penetration strategy is to introduce Veto-Cell into the best-known and most influential centers in North America and Western Europe, and benefit from the exposure and industry leadership provided by these centers.
| 49 |
This initial penetration strategy includes incorporating these centers into the clinical trials so as to expose and involve their medical leadership.
In the longer term, we plan to drive use and awareness within and across the broader oncology community in order to encourage oncologists to refer their patients to centers that already use our products and therapies and to encourage pull-influence on additional centers to adopt our products and therapies.
The broader provider community will be addressed by attending conventions where research and best clinical practices are shared, seminars are conducted, and networking opportunities are provided for the physicians.
Operating Strategy
Veto-Cell doses are to be prepared by our personnel (not outsourcers) in our facilities. This is to both protect trade-secrets and directly control quality during the initial stages.
The graphic below outlines the general operating model in each geographic market.
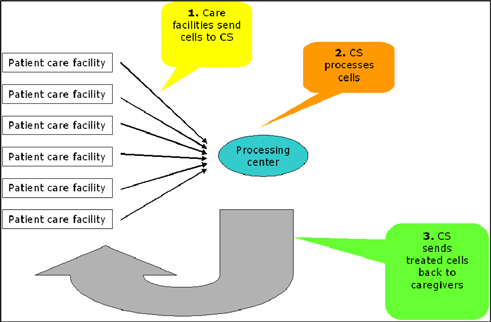
Patient care facilities send frozen cells (donor and/or host depending on application) to a Cell Source processing center. Most likely, the first processing center will consist of lab space leased from a large transplantation center. Such a transplantation center has appropriate equipment and infrastructure, along with available production capacity, and will also represent an immediate market for our offerings for use in their own procedures. The Cell Source processing center processes the cells and sends the treated cells and appropriate protocols back to the caregiver for infusion at time of transplantation.
In the introductory period, we plan on establishing two centers in the U.S. (East and West Coast), one in Western Europe (most likely Germany), and one in China. Specific locations and timing are to be determined. Initially, we plan to outsource production capacity from existing facilities at or adjacent to large hospitals. Subsequently, sales from these centers can justify and fund stand-alone facilities.
The general goal of the initial four centers is to support the FDA process, provide full coverage for the North American and European markets, and provide access to the developing Chinese market. Following the introductory period in each respective market, we may elect to migrate the production facilities from leased space in transplantation center laboratories to company-owned stand-alone facilities.
In general, we assume a capital cost per stand-alone production facility of $8M. This estimate is based, in part, on the projected high costs of GMP “clean rooms,” each of which can cost $1 million to set up. We will need to obtain financing in order to fund the setup of such facilities. There can be no assurance that financing will be available in amounts or on terms acceptable to us, if at all.
Clinical Trials Overview
We will initially focus our clinical trials on certain lymphomas and leukemias, for which our Veto-Cell technology constitutes a potential breakthrough. These two indications have unmet needs, as evidenced by the recent acquisitions of lymphoma/leukemia biotechnology firms Micromet and Avila Therapeutics for $1.16B and over $350M in cash, respectively. These acquisitions are especially notable because their respective lead treatment candidates were then only in Phases 1 and 2 of clinical trials.
| 50 |
We plan to initiate a company-sponsored "Phase 1/2" clinical trials by the end of 2014. These trials combine traditional Phase 1 safety trials with Phase 2 efficacy trials inasmuch as they are safety trials conducted on sick patients, so they are able to both establish safety and show initial indications of efficacy concurrently. The goal is to demonstrate safety and initial efficacy in several indications. Management has structured the trials such that an additional goal of showing initial markers pointing to prolonging progression-free survival may be possible already within Phase 1/2.
The chart below provides an overview of the current trials plan, which can of course vary based on both finalization of human protocols and timing or regulatory approvals:
Trial Plans
Trials will be conducted concurrently in Parma, Italy and Wurzburg, Germany. Multiple trials are planned on at least 16 patients. Patients are expected to be age 55 and older. The conditions chosen are ones which are associated with high mortality in this patient age-group today. This means that we may obtain a limited scope of patient reimbursement from government insurance in Europe on compassionate grounds for the treatment of said age group upon successful completion of Phase 2 trials. The following trials are planned for each center:
| Italy | Germany | |||
| 1 | Megadose + currently FDA approved drug | 1 | Megadose + currently FDA approved drug | |
| 2 | Anti-rejection Veto-Cell | 2 | Anti-rejection Veto-Cell | |
| 3 | Anti-lymphoma veto cell | 3 | Anti-lymphoma Veto-Cell | |
| 4 | Possibly sickle cell anemia or aplastic anemia (allogeneic transplant) | |||
| 5 | Possibly Veto-Cell to obviate need for organ transplant immune suppression treatment | |||
Regulatory Issues Overview
We seek regulatory approval from the U.S. FDA, the EMA in Europe and similar agencies elsewhere to both produce and sell our products.
Key approvals in Europe, where both treatment and limited insurance reimbursement may be possible at the end of Phase 2 trials, are expected to accelerate approval by the U.S. FDA. Given the importance of the U.S. market, we will conduct trials with a view to conforming with FDA guidelines so as to utilize clinical data gathered outside the U.S. in seeking to qualify for FDA approval.
In the longer-term, we may also seek regulatory approval for selected Organsource applications. In addition, we are exploring potential sources of near-term revenue, namely the combination of the broadly used Megadose with already FDA-approved agents.
| 51 |
Regulatory Process and Expectations
We will develop our clinical trial protocols with the support of experienced FDA and EMA consultants.
The clinical trials outlined in the previous section are designed to lead to regulatory approval for Veto-Cell-based therapy in treating blood cancers and bone marrow transplantation applications.
Interim Revenue Opportunities
As noted above, while the clear focus is to conclude Phase 3 approval for cancer treatments, the Company is also exploring complementary “quick win” opportunities for generating revenue before additional FDA approvals are received, namely:
| 1. | Treating European patients after the end of Phase 2 (in some cases possibly with insurance reimbursement available); and |
| 2. | Exploring an interim improved bone marrow transplantation mechanism by combining Megadose with current FDA-approved agents. |
Intellectual Property
Pursuant to the Yeda License Agreement, Yeda granted the Company an exclusive license to certain patents, discoveries, inventions and other intellectual property generated (together with others) by Dr. Reisner as head of the Immunology Department at the Weizmann Institute. Under the Yeda License Agreement, The Company grants Yeda an industry-standard 4% royalty on sales of patented products. Currently, the Company voluntarily funds research (on its own behalf) and the Weizmann Institute for the preclinical development of its products, and plans to do so in the foreseeable future. Should the Company elect to curtail such funding, it would have to pay a $50,000 annual license fee until such times as payment of royalties commences. The Yeda License Agreement also requires the Company to proceed with the development of the technologies on a timely basis.
The license period (per product, per country) is for the full life of the patents, and expires at the later of the patent expiration date in that country or 15 years after the date that the FDA or local equivalent regulatory authority in each country approves that particular product for sale in that country. As long as Cell Source pays either a nominal license fee of $50,000 per year (total for use of all the products) or pays royalties on product sales on at least one product as per the license agreement, the license will remain in effect continuously and expire only with the expiration of the patent or 15 years after regulatory approval (later of the two) per product per country as described above. Cell Source voluntarily sponsors Research at the Weizmann Institute for the sake of developing its products and treatments from initial invention through to finalization of human treatment protocols. Cell Source has recently extended the initial research period, which terminated in October of 2014, for a further four years through October 2018.
Also under the Yeda License Agreement, the Company agreed to fund Yeda’s research until October 3, 2018, with an aggregate annual payment of US$800,000 paid in quarterly US$200,000 installments. However, in the event that the Company and Yeda execute a new research and license agreement, then the Company will annually fund research in the amount of US $900,000 until Oct. 3, 2018. Such a new research and license agreement must be in accordance with the Evaluation and Exclusive Option Agreement by and between the Company and Yeda, dated Oct. 3, 2011, as amended on April 1, 2014 and June 22, 2014 (the “E&O Agreement). Among other things, the E&O Agreement grants Cell Source an option to negotiate a commercial license in the field of organ transplantation with Yeda (the “Option to Negotiate”). The Option to Negotiate requires an initiation fee of $200,000 payable to Yeda, which may be paid on the later of (i) the date on which the Option to Negotiate is granted and (ii) the date on which the Company receives an aggregate investment amount of at least US $10,000,000. Pursuant to an amendment to the license agreement, the Option to Negotiate expires on December 31, 2015.
If the Company fails to achieve any one of the milestones set forth in the Yeda License Agreement, which are listed below, then Yeda will be entitled to (i) modify the related license such that it will become non-exclusive or (ii) terminate the Yeda License Agreement upon thirty (30) days written notice:
| (a) | Within three (3) years of the signature of the Yeda License Agreement to commence Phase I clinical trials with respect to the Megadose Drug Combination; |
| (b) | Within five (5) years of the signature of the Yeda License Agreement to commence Phase II clinical trials with respect to the Megadose Drug Combination, unless the Company shall have invested, during such five (5) year period, above an aggregate amount of at least US$5,000,000 in research and development in respect of the Megadose Drug Combination; |
| (c) | Within either (8) years of the signature of the Yeda License Agreement to receive a FDA, EMEA or CFDA approval in respect of at least one (1) Product; |
| (d) | To achieve commercialization of at least one (1) Product within twelve (12) months of the date of FDA, EMEA or CFDA approval; or |
| (e) | In case of a commercial sale of any Product having commenced, there shall be a period of twelve (12) months or more during which no sales of any Product shall take place (except as a result of force majeure or other factors beyond the control of the Company). |
| 52 |
Additionally, the Yeda License Agreement also provides that:
| · | Funding the Research. Within 60 days of receiving any capital investment in the Company in excess of US $2,000,000 and provided that the Company has not paid Yeda by that date an option initiation fee of $200,000, as set forth in the E&O Agreement, Cell Source will pay Yeda 20% from such excess investment up to the sum of US $200,000 (the “Additional Research Payment”). The Additional Research Payment shall be allocated by Yeda to support research activities of Dr. Reisner. |
| · | Title. All right, title and interest in and to the Licensed Information and the Patents (as those terms are defined in the Yeda License Agreement ) and all right, title and interest in and to any drawings, plans, diagrams, specifications, other documents, models, or any other physical matter in any way containing, representing or embodying any of the foregoing, vest and shall vest in Yeda and subject to the license granted in the Yeda License Agreement. |
| · | Patents. Both Yeda and the Company shall consult with one another on the filing of patent applications for any portion of Licensed Information and/or corresponding to patent application existing at the time the Yeda License Agreement was executed. Yeda shall retain outside patent counsel that will be approved by Cell Source, to prepare, file and prosecute patent applications. All applications will be filed in Yeda’s name. |
| · | Patents; Patent Infringements. Where the Company determines that a third party is infringing one or more of the Patents or is sued, in prosecuting or defending such litigation, the Company must pay any expenses or costs or other liabilities incurred in connection with such litigation (including attorney’s fees, costs and other sums awarded to the counterparty in such action). The Company agreed to indemnify Yeda against any such expenses or costs or other liabilities. |
| · | License. With regard to the expiration of Patents, a Product is deemed to be covered by a Patent so long as such Product is protected by “Orphan Drug” status (or the like). The Company has an exclusive worldwide license under the Licensed Information and the Patents for the development, manufacture and sales of the Products. License remains in force in each country with respect to each Product until the later of (i) the expiration of the last Patent in such country covering such Product or (ii) the expiration of a 15-year period commencing the day FDA New Drug Approval is received for a Product in such country. |
The Company may grant a Sublicense only with the prior written consent, which shall not be withheld unreasonably provided that:
| i. | the proposed Sublicence is for monetary consideration only; |
| ii. | the proposed Sublicence is to be granted in a bona fide armslength commercial transaction; |
| iii. | a copy of the agreement granting the Sublicence and all amendments thereof shall be made available to Yeda, 14 days before their execution and Cell Source shall submit to Yeda copies of all such Sublicenses and all amendments thereof promptly upon execution thereof; and |
| iv. | the proposed Sublicence is made by written agreement, the provisions of which are consistent with the terms of the Licence and contain, inter alia, the following terms and conditions, including: the Sublicense shall expire automatically on the termination of the Licence for any reason. |
However, Yeda’s prior written consent is not needed if the sublicense is limited to China, and the Company grants it to a Chinese affiliated entity of the Company.
| · | Termination. The Yeda License Agreement terminates on the later of: (i) the expiration of the last of the Patents or (ii) the expiry of a continuous period of 20 years during which there shall not have been a First commercial sale of any product in any country. Yeda may terminate by written notice, effective immediately, if the Company challenges the validity of any of the Patents. If a challenge is unsuccessful, then in addition to Yeda’s right to termination, the Company shall pay to Yeda liquidated damages in the amount of US$8,000,000. Either the Company or Yeda may terminate the Yeda License Agreement and the License by serving a written notice upon (i) occurrence of a material breach or (ii) the granting of a winding-up order. Additionally, Yeda may terminate for failure to reimburse Yeda for patent application and/or prosecution expenses. |
Our technology portfolio includes a patented platform termed “Veto-Cell” (more formally described as “Anti 3rd party central memory T cell”), which is an immune tolerance biotechnology that enables the selective blocking of immune responses. Specifically, Veto-Cells are specially prepared human cells that selectively protect specific targets from undesirable immune system attack.
We have also secured an exclusive option to license the “Organsource” platform developed by Dr. Reisner and his team under a similar Research & License Agreement. This is a longer-horizon technology that shows significant promise for enabling the sourcing of embryonic cellular material from both animals and humans that can be used to both grow functional major organs in the body of a foreign “host” and regenerate existing diseased or damaged organs. This technology was used to grow pancreases in both rodents and primates, thereby curing them of juvenile diabetes, and has been used to regenerate human lung tissue.
| 53 |
The following table lists the patents and pending patents that Yeda holds and which we have a license to use (or in the case of the organs platform, the option to license to use) in each of the below-referenced countries:
Cell Source currently licenses the following patents held by Yeda:
| Name: VETO CELLS EFFECTIVE IN PREVENTING GRAFT REJECTION AND DEVOID OF GRAFT VERSUS HOST POTENTIAL | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA (Basic) | 6,544,506 | 05-Jan-2000 | 05-Jan-2020 | Granted | Yeda Research and Development Co. Ltd. |
| USA (National Phase) | 7,270,810 | 28-Dec-2000 | 1-Dec-2021 | Granted | Yeda Research and Development Co. Ltd. |
| Europe | 1244803 | 28-Dec-2000 | 28-Dec-2020 | Granted | Yeda Research and Development Co. Ltd. |
| Israel | 150440 | 28-Dec-2000 | 28-Dec-2020 | Granted | Yeda Research and Development Co. Ltd. |
| Name: USE OF ANTI THIRD PARTY CENTRAL MEMORY T CELLS FOR ANTI-LEUKEMIA/LYMPHOMA TREATMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 13/821,255 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 2013-527738 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 2,810,632 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| China | 201180053858.9 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 2013-7008892 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 225102 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | BR 11 2013 005756 4 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2013/002668 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Singapore | 201301743-9 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 11773325.3 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| Hong Kong | 14100513.2 | 08-Sep-2011 | 08-Sep-2031 | Pending | Yeda Research and Development Co. Ltd. |
| 54 |
| Name: ANTI THIRD PARTY CENTRAL MEMORY T CELLS, METHODS OF PRODUCING SAME AND USE OF SAME IN TRANSPLANTATION AND DISEASE TREATMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 14/343,053 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 12769743.1 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 2014-529143 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 2,848,121 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| China | 201280054739.X | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Australia | 2012305931 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 10-2014-7009267 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| New Zealand | 622749 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| South Africa | 2014/01993 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| India | 577/MUMNP/2014 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 231397 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 2014110897 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | BR 11 2014 005355 3 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2014/002771 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Singapore | 11201400513P | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Name: ANTI THIRD PARTY CENTRAL MEMORY T CELLS, METHODS OF PRODUCING SAME AND USE OF SAME IN TRANSPLANTATION AND DISEASE TREATMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| USA | 13/126,472 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 09764302.7 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 212587 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| India | 905/MUMNP/2011 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| China | 200980153053.4 | 29-Oct-2009 | 29-Oct-2029 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 2011121630 | 29-Oct-2009 | 29-Oct-2029 | Granted | Yeda Research and Development Co. Ltd. |
| Name: UNIVERSAL DONOR-DERIVED TOLEROGENIC CELLS FOR INDUCING NON-SYNGENEIC TRANSPLANTATION TOLERANCE | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| US | 11/990,628 | 21-Aug-2006 | 21-Aug-2026 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 06796063.3 | 21-Aug-2006 | 21-Aug-2026 | Pending | Yeda Research and Development Co. Ltd. |
| Europe (Divisional) | 12161171.9 | 21-Aug-2006 | 21-Aug-2026 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 189688 | 21-Aug-2006 | 21-Aug-2026 | Granted | Yeda Research and Development Co. Ltd. |
| 55 |
| Name: A COMBINATION THERAPY FOR A STABLE AND LONG TERM ENGRAFTMENT | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| Singapore | 11201403459X | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2014/007647 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | BR 11 2014 015960 2 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 2014128479 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 233303 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| India | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| South Africa | 2014/05071 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| New Zealand | 627272 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Australia | 2012355990 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| China | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 12859036.1 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| USA | 14/367,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Name: A COMBINATION THERAPY FOR A STABLE AND LONG TERM ENGRAFTMENT USING SPECIFIC PROTOCOLS FOR T/B CELL DEPLETION | |||||
| Country | Patent Number | Filed | Expires | Status | Assignee |
| Singapore | 11201403456U | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Mexico | MX/a/2014/007648 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Brazil | BR 11 2014 015959 9 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Russian Federation | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Israel | 233302 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| India | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| South Africa | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| New Zealand | 627549 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Republic of Korea | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Australia | 2012355989 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| China | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Canada | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Japan | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| Europe | 61/578,917 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| USA | 14/367,923 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. |
| 56 |
Cell Source currently has an option to license, and in the interim is fully funding the following patents:
| Name: METHODS OF TREATING DISEASE BY TRANSPLANTATION OF DEVELOPING ALLOGENEIC OR XENOGENEIC ORGANS OR TISSUES | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| Mexico | 319957 | 04-Mar-2004 | 04-Mar-2024 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Mexico (Divisional) | MX/a/2014/001950 | 04-Mar-2004 | 04-Mar-2024 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Europe | 2216033 | 04-Mar-2004 | 04-Mar-2024 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Name: METHODS OF TREATING DISEASE BY TRANSPLANTATION OF ALLOGENEIC OR XENOGENEIC ORGANS OR TISSUES | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| Europe | 11179593.6 | 04-Mar-2004 | 04-Mar-2024 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Name: THE USE OF DEVELOPING ALLOGENEIC OR XENOGENEIC ORGANS OR TISSUES FOR DISEASE TREATMENT | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| Israel | 170622 | 04-Mar-2004 | 04-Mar-2024 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Name: METHODS OF KIDNEY TRANSPLANTATION UTILIZING DEVELOPING NEPHRIC TISSUE | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| Israel | 218961 | 01-Sep-2002 | 01-Sep-2022 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Name: THERAPEUTIC TRANSPLANTATION USING DEVELOPING, HUMAN OR PORCINE, RENAL OR HEPATIC, GRAFTS | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 7,780,993 | 19-Jan-2005 | 23-Apr-2023 | Granted | Yeda Research and Development Co. Ltd. | |||||
| USA (Divisional) | 12/777,292 | 19-Jan-2005 | 19-Jan-2025 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Name: DISEASE TREATMENT VIA DEVELOPING NON-SYNGENEIC GRAFT TRANSPLANTATION | ||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |
| USA | 11/664,530 | 02-Oct-2005 | 02-Oct-2023 | Pending | Yeda Research and Development Co. Ltd. | |
| Europe | 1809734 | 02-Oct-2005 | 02-Oct-2025 | Granted | Yeda Research and Development Co. Ltd. | |
| Europe (Divisional) | 11193959.1 | 02-Oct-2005 | 02-Oct-2025 | Pending | Yeda Research and Development Co. Ltd. | |
| Israel | 182363 | 02-Oct-2005 | 02-Oct-2025 | Pending | Yeda Research and Development Co. Ltd. | |
| Name: MAMMALIAN FETAL PULMONARY CELLS AND THERAPEUTIC USE OF SAME | ||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |
| USA | 14/363,814 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Europe | 12813990.4 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Japan | 61/568,240 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Canada | 61/568,240 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| China | 61/568,240 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Australia | 2012348574 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Republic of Korea | 10-2014-7018702 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| New Zealand | 627071 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| South Africa | 02014/04958 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| India | 1366/MUMNP/2014 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Israel | 233022 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Russian Federation | 2014127338 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Brazil | 61/568,240 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Mexico | MX/a/2014/006756 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Singapore | 11201402902V | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| Philippines | 1-2014-501309 | 06-Dec-2012 | 06-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |
| 57 |
Patents & Proprietary Rights
Our success will depend in part on our ability to protect our existing product candidates and the products we acquire or license by obtaining and maintaining a strong proprietary position. To develop and maintain our position, we intend to continue relying upon patent protection, orphan drug status, Hatch-Waxman exclusivity, trade secrets, know-how, continuing technological innovations and licensing opportunities. We intend to seek patent protection whenever available for any products or product candidates and related technology we acquire in the future.
We may also seek orphan drug status whenever it is available. If a product which has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and Canada, and 10 years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for a different clinical indication.
It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information made known to the individual during the course of the individual’s relationship with us is to be kept confidential and may not be disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual shall be our exclusive property.
Government Regulation and Product Approval
We have not submitted any drug applications to the FDA and do not have anything pending for approval with the FDA. Cell Source itself has not had any contact with any regulator anywhere regarding treatment approvals or clinical trials associated with regulatory approvals. We are aware that a hospital in Italy has independently requested and received approval to conduct a trial with the same protocol that we plan to use, but we are not mentioned in the application nor in the approval. However, we may indirectly benefit from the outcome of the trial, if successful, although we are not the sponsor of this trial. There are no written or verbal agreements between the hospital and Cell Source regarding the use of the technology. That said, Cell Source is aware and in favor of the hospital plans to use the technology and would find a positive initial outcome encouraging. Since the treatment is being done on compassionate grounds as a non-commercial clinical trial, there is no legal requirement for the hospital to obtain approval to use the treatment protocol.
Cell Source plans to apply for approval for human clinical trials in Italy for its Megadose Drug Combination product in 2015, and in Germany in 2016. It plans to apply for approval for human trials its Anti-Rejection Veto Cell in both Italy and Germany in 2015, and for the Anti-Lymphoma Veto Cell by 2016. Once Cell Source has interim data from European trials (most likely by 2017-2018 in the case of the Megadose Drug Combination) it plans to apply for approval to conduct human trials in the United States, per product, for each of the products as they each, in turn, show initial safety, and possibly efficacy, results in Europe. As of the date of this filing, the company has had no contact with any regulator regarding such approvals.
Regulation by governmental authorities in the U.S. and other countries is a significant factor, affecting the cost and time of our research and product development activities, and will be a significant factor in the manufacture and marketing of any approved products. All of our products require regulatory approval by governmental agencies prior to commercialization. In particular, our products are subject to rigorous pre-clinical and clinical testing and other approval requirements by the FDA and similar regulatory authorities in other countries. Various statutes and regulations also govern or influence the manufacturing, safety, reporting, labeling, transport and storage, record keeping and marketing of our products. The lengthy process of seeking these approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, the necessary regulatory approvals could harm our business.
The regulatory requirements relating to the testing, manufacturing and marketing of our products may change from time to time and this may impact our ability to conduct clinical trials and the ability of independent investigators to conduct their own research with support from us.
The clinical development, manufacturing and marketing of our products are subject to regulation by various authorities in the U.S., the E.U. and other countries, including, in the U.S., the FDA, in Canada, Health Canada, and, in the E.U., the EMEA. The Federal Food, Drug, and Cosmetic Act, the Public Health Service Act in the U.S. and numerous directives, regulations, local laws and guidelines in Canada and the E.U. and elsewhere govern the testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of our products. Product development and approval within these regulatory frameworks takes a number of years and involves the expenditure of substantial resources.
Regulatory approval will be required in all the major markets in which we seek to develop our products. At a minimum, approval requires the generation and evaluation of data relating to the quality, safety, and efficacy of an investigational product for its proposed use. The specific types of data required and the regulations relating to this data will differ depending on the territory, the treatment candidate involved, the proposed indication and the stage of development.
In general, new cell compositions are tested in animals until adequate evidence of safety is established to support the proposed clinical study protocol designs. Clinical trials for new products are typically conducted in three sequential phases that may overlap. In Phase I, the initial introduction of the pharmaceutical into either healthy human volunteers or patients with the disease (typically 20 to 50 subjects), the emphasis is on testing for safety (adverse effects), dosage tolerance, metabolism, distribution, excretion and clinical pharmacology. Phase II involves studies in a limited patient population (typically 50 to 200 patients) to determine the initial efficacy of the pharmaceutical for specific targeted indications, to determine dosage tolerance and optimal dosage and to identify possible adverse side effects and safety risks. Once a treatment protocol shows preliminary evidence of some efficacy and is found to have an acceptable safety profile in Phase II evaluations, Phase III trials are undertaken to more fully evaluate clinical outcomes in a larger patient population in adequate and well-controlled studies designed to yield statistically sufficient clinical data to demonstrate efficacy and safety.
| 58 |
In the U.S., specific pre-clinical data, manufacturing and chemical data, as described above, need to be submitted to the FDA as part of an IND application, which, unless the FDA objects, will become effective thirty (30) days following receipt by the FDA. Phase I studies in human volunteers may commence only after the application becomes effective. Prior regulatory approval for human healthy volunteer studies is also required in member states of the E.U. Currently, in each member state of the E.U., following successful completion of Phase I studies, data are submitted in summarized format to the applicable regulatory authority in the member state in respect of applications for the conduct of later Phase II studies. In many places in Europe, a two tiered approval system mandates approval at the regional level prior to applying for national approval. Regional approval cycle times, including multiple iterations where questions are answered and the specific details of the protocol may be fine-tuned, can last several months prior to applying to the national (federal government level) regulator. The national regulatory authorities in the E.U. typically have between one and three months in which to raise any objections to the proposed study, and they often have the right to extend this review period at their discretion. In the U.S., following completion of Phase I studies, further submissions to regulatory authorities are necessary in relation to Phase II and III studies to update the existing IND. Authorities may require additional data before allowing the studies to commence and could demand that the studies be discontinued at any time if there are significant safety issues. In addition to the regulatory review, a study involving human subjects has to be approved by an independent body. The exact composition and responsibilities of this body will differ from country to country. In the U.S., for example, each study will be conducted under the auspices of an independent institutional review board at each institution at which the study is conducted. This board considers among other things, the design of the study, ethical factors, the privacy of protected health information as defined under the Health Insurance Portability and Accountability Act, the safety of the human subjects and the possible liability risk for the institution. Equivalent rules to protect subjects’ rights and welfare apply in each member state of the E.U. where one or more independent ethics committees, which typically operate similarly to an institutional review board, will review the ethics of conducting the proposed research. These ethical review committees typically exist at the regional level, where approval is required prior to applying for national approval. Other regulatory authorities around the rest of the world have slightly differing requirements involving both the execution of clinical trials and the import/export of pharmaceutical products. It is our responsibility to ensure we conduct our business in accordance with the regulations of each relevant territory.
By leveraging existing pre-clinical and clinical data, we are seeking build upon an existing pre-clinical safety and efficacy database to accelerate our research. In addition, our focus on an end-stage population which has no current treatment options, commercialization, may result in relatively shorter approval cycle times. Approval by the FDA in this category generally has been based on objective response rates and duration of responses rather than demonstration of survival benefit. As a result, trials of drugs to treat end-stage refractory cancer indications have historically involved fewer patients and generally have been faster to complete than trials of drugs for other indications. We are aware that the FDA and other similar agencies are regularly reviewing the use of objective endpoints for commercial approval and that policy changes may impact the size of trials required for approval, timelines and expenditures significantly. The trend over the past few years has been to shorten approval cycles for terminal patients in the U.S. by employing a “fast track” approach.
In order to gain marketing approval, we must submit a dossier to the relevant authority for review, which is known in the U.S. as an NDA and in the E.U. as a marketing authorization application, or MAA. The format is usually specific and laid out by each authority, although in general it will include information on the quality of the chemistry, manufacturing and pharmaceutical aspects of the product as well as the non-clinical and clinical data. Once the submitted NDA is accepted for filing by the FDA, it undertakes the review process that takes ten (10) months, unless an expedited priority review is granted which takes six (6) months to complete. Approval can take several months to several years, if multiple ten (10) month review cycles are needed before final approval is obtained, if at all.
The approval process can be affected by a number of factors. The NDA may be approvable requiring additional pre-clinical, manufacturing data or clinical trials which may be requested at the end of the ten (10) month NDA review cycle, thereby delaying marketing approval until the additional data are submitted and may involve substantial unbudgeted costs. The regulatory authorities usually will conduct an inspection of relevant manufacturing facilities, and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each product, in many cases each drug manufacturing facility must be approved. Further inspections may occur over the life of the product. An inspection of the clinical investigation sites by a competent authority may be required as part of the regulatory approval procedure. As a condition of marketing approval, the regulatory agency may require post-marketing surveillance to monitor for adverse effects or other additional studies as deemed appropriate. After approval for the initial indication, further clinical studies are usually necessary to gain approval for any additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect the marketability of a product.
The FDA offers a number of regulatory mechanisms that provide expedited or accelerated approval procedures for selected drugs in the indications on which we are focusing our efforts. These include accelerated approval under Subpart H of the agency’s NDA approval regulations, fast track drug development procedures and priority review. At this time, we have not determined whether any of these approval procedures will apply to any of our current treatment candidates.
The U.S., E.U. and other jurisdictions may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which, in the U.S., is generally a disease or condition that affects no more than 200,000 individuals. In the E.U., orphan drug designation can be granted if: the disease is life threatening or chronically debilitating and affects no more than fifty (50) in 100,000 persons in the E.U.; without incentive it is unlikely that the drug would generate sufficient return to justify the necessary investment; and no satisfactory method of treatment for the condition exists or, if it does, the new drug will provide a significant benefit to those affected by the condition. If a product that has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and ten (10) years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for different indications. Orphan drug designation must be requested before submitting an NDA or MAA. After orphan drug designation is granted, the identity of the therapeutic agent and its potential orphan use are publicly disclosed. Orphan drug designation does not convey an advantage in, or shorten the duration of, the review and approval process; however, this designation provides an exemption from marketing authorization (NDA) fees.
| 59 |
We are also subject to numerous environmental and safety laws and regulations, including those governing the use and disposal of hazardous materials. The cost of compliance with and any violation of these regulations could have a material adverse effect on our business and results of operations. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, accidental contamination or injury from these materials may occur. Compliance with laws and regulations relating to the protection of the environment has not had a material effect on our capital expenditures or our competitive position. However, we are not able to predict the extent of government regulation, and the cost and effect thereof on our competitive position, which might result from any legislative or administrative action pertaining to environmental or safety matters.
In various countries, animal rights activism has led to either formal or informal boycotting of certain types of animal trials. As we rely on animal experiments as precursors to human trials.
Employees
We currently do not have full-time employees, but retain the services of part-time staff on an independent contractor/consultant and contract-employment basis. However, our Board of Directors intends to negotiate an employment package for our Chief Executive Officer, Itamar Shimrat in the near future. Our ability to manage growth effectively will require us to continue to implement and improve our management systems and to recruit and train new employees. Although we have done so in the past and expect to do so in the future, there can be no assurance that we will be able to successfully attract and retain skilled and experienced personnel. We anticipate that in the near future, other key personnel will enter into employment agreements with the Company on customary terms.
We are not involved in any pending legal proceeding or litigations and, to the best of our knowledge, no governmental authority is contemplating any proceeding to which we are a party or to which any of our properties is subject, which would reasonably be likely to have a material adverse effect on us.
| Name | Age | Title(s) | ||
| Yoram Drucker | 49 | Director (Executive Chairman) | ||
| Itamar Shimrat | 55 | Chief Executive Officer, Chief Financial Officer and Director | ||
| David Zolty | 65 | Director | ||
| Ben Friedman | 56 | Director | ||
| Dennis Brown | 65 | Director |
Yoram Drucker , a Director and Chairman of our Board of Directors, is an Israeli entrepreneur who has previously been involved in the development of two successful cell therapy technology firms. From March 2009 through September 2011, Mr. Drucker served as Chief Executive Officer and Chairman of Rainbow Energy Ltd. From September 2011 through October 2013, Mr. Drucker served as both Chief Executive Officer and Chairman of Cell Source Ltd. From October 2013 to present, Mr. Drucker served as Chairman of Cell Source Ltd. He was a founding member of the cell stem therapy company Brainstorm (NASDAQ: BCLI). He served as COO in 2004 and CEO from 2005 to 2007. He was also among the founders of Pluristem (listed on the NASDAQ), also a cell therapy company, and was a Director in 2004 and 2005. In 2007 he was a seed investor and VP Business Development in a renewable energy technology firm called Millennium Electric TOU Ltd. Since March 2008 he was a Director of a private renewable energy company called Rainbow Energy, where he actively served as CEO from then until November of 2011. From 1996 to 2003 he served as business and marketing consulting and campaign execution in varied industries ranging from real estate development to insurance. He is an honors graduate of the Abudi College of Advertising and Marketing. Yoram brings significant experience in capital markets in the US and in developing Israeli based cell therapy companies from inception through financing over-the-counter and commencing clinical trials. His understanding of both the financial and the technical side of early stage corporate development has and will continue to be of great value to Cell Source.
Itamar Shimrat , CEO, CFO and Director, is a Canadian businessman and a founding member of Cell Source Israel. Since Cell Source Israel’s inception, Mr. Shimrat served as a Director, Chief Financial Officer and, in October 2013, he was appointed Chief Executive Officer. From March 2009 through September 2011, Mr. Shimrat served as Chief Financial Officer and Director of Rainbow Energy Ltd. From September 2011 through October 2013, Mr. Shimrat served as Chief Financial Officer and Director of Cell Source Ltd. From August 2012 to present, Mr. Shimrat served as Director of Step Up – Olim Madrega Inc. From October 2013 to present, Mr. Shimrat served as Chief Executive Officer and Director of Cell Source Ltd.. Previously, Mr. Shimrat served as an Executive Vice President at First International Bank of Israel from March, 2005 until April, 2008. Prior to 2008, he served as a senior manager at McKinsey & Company’s Tel Aviv office after having being elected Partner at Mitchell Madison Group and consulting for Bain & Co. Mr. Shimrat led major profit improvement programs for leading corporations ranging from American Express and Barclays to El Al Airlines. He has been a Director of two private companies: Rainbow Energy Ltd., a company in the renewable energy industry, and Step Up - Olim Madrega Ltd., a company in the wheelchair industry, and also was on the Allocations Committee of Matan, a leading Israeli philanthropic organization. He holds an MBA with Distinction from the Ivey Business School of the University of Western Ontario in Canada. Itamar brings to Cell Source significant knowledge and experience in the area of corporate finance. He also has extensive experience working in foreign environments and cultures and possesses distinctive oral and written presentation skills. This positions him to be effective in both financing and corporate development both domestically and internationally.
| 60 |
David Zolty has been a Director of Cell Source Israel since November, 2011 and of our Board of Directors since June 30, 2014, and is a Canadian businessman who has owned and managed various Canadian enterprises since 1968. From more than five years prior through the present, Mr. Zolty served as Director of Management and Administration for Hightower Investments. In the mid 1970’s David was one of the founders of TNT Appliances, a coin laundry and appliance sales and service company, primarily serving the Canadian burgeoning multi-family apartment industry. The company grew to be the second largest coin laundry in Canada and was sold in and about 2002. While owning and managing TNT, David was also involved in many real estate acquisitions both through TNT and the Zolty family real estate portfolio. Upon David’s father Morris Zoltys’ retirement, David took a larger role in the Zolty family business where David currently holds a 12% ownership interest and has served in various roles therein for more than 5 years. David has received an honors BA and has done his post graduate work at the University of Toronto in the field of Religious Studies. He is also involved in a number of local charities and is a long standing board member of Camp Agudah Toronto, a children’s summer camp which have facilities at Port Carling, Ontario. His extensive business experience and community involvement are an asset to Cell Source.
Ben Friedman , BBA, BGS, LLB, has been a Director of Cell Source Israel since November, 2011 and of our Board of Directors since June 30, 2014, and is a Canadian business executive with over 25 years’ experience in real estate and commerce. From more than five years prior through the present, Mr. Friedman served as Director and Vice President of Ranee Management. Since 1985, he has served as Owner and CEO of Saucham Holdings Ltd., a private real estate holding and development company active throughout Canada. He is, and has been for more than five years, a managing partner and Director of The Zolty Group, a private company specializing in the development and ownership of high rise multi-unit residential buildings in Canada and the United States. He continues to act as Director of numerous private business related enterprises in the high tech, medical, and laser technology fields, and is a Director of an array of non-profit educational and vocational institutions. Mr. Friedman’s experience as both an executive, along with his degrees in both business and law, position him well to help guide Cell Source through its development.
Dr. Dennis M. Brown, PhD, was elected Director of the Company on June 30, 2014. Dr. Brown is a founder and Chief Scientific Officer and director of Del Mar Pharmaceuticals (BC) Ltd. a subsidiary of DelMar Pharmaceuticals, Inc. (OTCQB: DMPI) to which he serves as a director and Chief Scientific Officer. Dr. Brown has more than thirty years of drug discovery and development experience. Since 2000 to the present, Dr. Brown has served as Chairman of Mountain View Pharmaceutical's Board of Directors and is the President of Valent. Dr. Brown has focused over the past 5 years on the development of DelMar Pharmaceuticals, serving as its Chief Scientific Officer since January 25, 2013 and Director since February 11, 2013. His extensive technical expertise, successful track record as an inventor, executive and director in the field of medical technology position him as an authoritative voice on the scientific, intellectual property, finance and commercialization and well as general management issues for Cell Source both now and in the future. In 1999 he founded ChemGenex Therapeutics, which merged with a publicly traded Australian company in 2004 to become ChemGenex Pharmaceuticals (ASX: CXS/NASDAQ: CXSP), of which he served as President and a Director until 2009. He was previously a co-founder of Matrix Pharmaceutical, Inc., where he served as Vice President (VP) of Scientific Affairs from 1985-1995 and as VP, Discovery Research, from 1995-1999. He also previously served as an Assistant Professor of Radiology at Harvard University Medical School and as a Research Associate in Radiology at Stanford University Medical School. He received his B.A. in Biology and Chemistry (1971), M.S. in Cell Biology (1975) and Ph.D. in Radiation and Cancer Biology (1979), all from New York University. Dr. Brown is an inventor of about 34 issued U.S. patents and applications, many with foreign counterparts. Dr. Brown’s scientific knowledge and experience qualifies him to serve on our Board of Directors.
The Company’s directors are elected at the annual meeting of shareholders to hold office until the annual meeting of shareholders for the ensuing year or until their successors have been duly elected and qualified. Officers are elected annually by the Board of Directors and serve at the discretion of the Board.
Board Leadership Structure and Role in Risk Oversight
Due to the small size and early stage of the Company, we have not adopted a formal policy on whether the Chairman and Chief Executive Officer positions should be separate or combined. Following Mr. Buckley’s resignation, Mr. Drucker will serve as the Chairman whereas Mr. Shimrat will serve as the Chief Executive Officer.
Our Board of Directors (“Board”) is primarily responsible for overseeing our risk management processes on behalf of the Company. The Board receives and reviews periodic reports from management, auditors, legal counsel, and others, as considered appropriate regarding our company’s assessment of risks. In addition, the Board focuses on the most significant risks facing our company and our company’s general risk management strategy, and also ensures that risks undertaken by our Company are consistent with the board’s appetite for risk. While the Board oversees our company’s risk management, management is responsible for day-to-day risk management processes. We believe this division of responsibilities is the most effective approach for addressing the risks facing our company and that our board leadership structure supports this approach.
Involvement in Certain Legal Proceedings
Our directors and executive officers have not been involved in any of the following events during the past ten years:
| 1. | any bankruptcy petition filed by or against such person or any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time; |
| 2. | any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); |
| 3. | being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining him from or otherwise limiting his involvement in any type of business, securities or banking activities or to be associated with any person practicing in banking or securities activities; |
| 61 |
| 4. | being found by a court of competent jurisdiction in a civil action, the SEC or the Commodity Futures Trading Commission to have violated a Federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; |
| 5. | being subject of, or a party to, any Federal or state judicial or administrative order, judgment decree, or finding, not subsequently reversed, suspended or vacated, relating to an alleged violation of any Federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| 6. | being subject of or party to any sanction or order, not subsequently reversed, suspended, or vacated, of any self-regulatory organization, any registered entity or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
Code of Ethics
We have not adopted a Code of Business Conduct and Ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions because of the small number of persons involved in the management of the Company.
Nominating Committee
We have not adopted any procedures by which security holders may recommend nominees to our Board of Directors.
Audit Committee
The Board of Directors acts as the Audit Committee and the Board has no separate committees. The Company has no qualified financial expert at this time because it has not been able to hire a qualified candidate. Further, the Company believes that it has inadequate financial resources at this time to hire such an expert.
Executive Compensation
The following table sets forth all compensation paid in respect of the Company’s principal executive officer (“PEO”) and the two (2) most highly compensated executive officers other than the PEO who received compensation in excess of $100,000 per year for 2013 and 2012.
Summary Compensation Table
| Name and Principal Position |
Year | Salary ($) |
Bonus ($) |
Stock Awards ($) |
Option Awards ($) |
Non-Equity Incentive Plan Compensation Earnings ($) |
Non-Qualified Deferred Compensation Earnings ($) |
All Other Compensation ($) |
Total ($) |
|||||||||||||||||||||||||||
| Itamar | 2013 | 107,158 | - | - | - | - | - | - | 107,158 | |||||||||||||||||||||||||||
| Shimrat, CEO | 2012 | 66,328 | - | - | - | 66,328 | ||||||||||||||||||||||||||||||
| Yoram | 2013 | 107,158 | 107,158 | |||||||||||||||||||||||||||||||||
| Drucker | 2012 | 66,583 | 66,583 | |||||||||||||||||||||||||||||||||
Outstanding Equity Awards at Fiscal Year-End
The Company had no outstanding equity awards or equity compensation plan as of March 31, 2014.
Director Compensation
No director of TTSE received any compensation for services as director for TTSE during fiscal year 2013. Additionally, no director of Cell Source Israel has received compensation for services as a director.
| 62 |
Risk Management
The Company does not believe risks arising from its compensation policies and practices for its employees are reasonably likely to have a material adverse effect on the Company.
Compensation Committee Interlocks and Insider Participation
Currently, the Board of Directors does not have any standing audit, nominating or compensation committees, or committees performing similar functions. The directors collectively perform the duties of an audit committee and nominating committee, which prior to the Share Exchange were performed by the Company’s sole Director.
Certain Relationships and Related Transactions
The Company maintains an exclusive worldwide license to certain intellectual property of Yeda, the commercial arm of the Weizmann Institute, which currently owns 1,159,972 shares of Company common stock and warrants to purchase 1,995,376 shares of Company common stock at $0.001 per share. Dr. Reisner, who leads a team at the Weizmann Institute, holds 1,159,972 shares of Company common stock and warrants to purchase 48,459 shares of Company common stock at $0.001 per share. See the section entitled “Intellectual Property” in this prospectus.
In September 2011 and in connection with securing the Yeda License Agreement, Cell Source Israel issued to Yeda and Dr. Reisner Ordinary Shares representing 26% of the then issued and outstanding CSL Ordinary Shares. Cell Source Israel also granted Yeda and Dr. Reisner anti-dilution protections against dilution under 26% of the issued and outstanding CSL Ordinary Shares that would result from issuances pursuant to any capital raises by Cell Source of up to $3,500,000. In connection with the aggregate $3,500,000 subsequently raised by Cell Source Israel pursuant to the Loan Agreements (as defined below), the Note Exchange, the Bridge Exchange (as defined below) and the Private Placement, Yeda and Dr. Reisner exercised their anti-dilution rights. Pursuant to this anti-dilution protection Yeda and Dr. Riesner were entitled to issuances, in the form of any combination of CSL Ordinary Shares and warrants to purchase CSL Ordinary Shares at par value, at their election. Accordingly, Cell Source Israel issued 239,142 CSL Ordinary Shares and warrants to purchase 1,995,376 CSL Ordinary Shares at par value to Yeda and 807,314 CSL Ordinary Shares and warrants to purchase 48,459 CSL Ordinary Shares at par value to Dr. Reisner.
In December 2012 and March 2013, a group of five accredited investors (“Note Investors”), including David Zolty, a director of Cell Source Israel, Solomon Zolty, a director of Cell Source Israel and Phyllis Friedman, the wife of Cell Source Israel’s director Ben Friedman, entered into Convertible Loan Agreements (“Loan Agreements”) pursuant to which the Note Investors loaned Cell Source Israel an aggregate of $510,000 (“Loan Amount”). In accordance with the Loan Agreements, the Note Investors were entitled to receive interest equal to 6% of the Loan Amount per annum and the Loan Amount was payable by Cell Source Israel six (6) months after the receipt of the Loan Amount. In November 2013, the Note Investors elected to convert the Loan Amount into CSL Ordinary Shares equal to 18% of Cell Source Israel’s fully-diluted issued and outstanding capital (“Note Exchange”), which issuance did not dilute the Note Investors’ prior holdings. Accordingly, the Note Investors were issued 2,699,880 CSL Ordinary Shares.
In October 2013, Cell Source Israel and the Note Investors entered into a Bridge Funding Agreement pursuant to which the Note Investors paid $50,000 to Cell Source Israel in exchange for Cell Source Israel’s agreement to issue to the Note Investors an aggregate of 66,667 Ordinary Shares and a warrant to purchase 100,000 Ordinary Shares at an exercise price of $0.75 per share on or prior to the closing of the Private Placement (the “Bridge Exchange”).
Director Independence
None of our directors is independent as that term is defined under the Nasdaq Marketplace Rules.
Security Ownership of Certain Beneficial Owner and Management.
The following table sets forth certain information, as of the date of filing of this prospectus, with respect to the beneficial ownership of the outstanding Common Stock by (i) any holder of more than five (5%) percent; (ii) each of the Company’s executive officers and directors; and (iii) the Company’s directors and executive officers as a group. Except as otherwise indicated, each of the stockholders listed below has sole voting and investment power over the shares beneficially owned.
The address for each Beneficial Owner named is the address of the Company’s principal executive office.
| Name of Beneficial Owner | Number of Common Stock Beneficially Owned | Percentage of Common Stock Beneficially Owned (1) | ||||||
| Directors and Officers: | ||||||||
| Yoram Drucker, Director (Chairman) | 575,004 | 2.44 | % | |||||
| Itamar Shimrat, Chief Executive Officer, Chief Financial Officer and Director | 575,004 | 2.44 | % | |||||
| David Zolty, Director | 1,095,818 | 4.65 | % | |||||
| Ben Friedman, Director (2) | 4,383,344 | 18.59 | % | |||||
| Dennis Brown, Director | 100,000 | * | ||||||
| All directors and executive officers as a group (5 persons) | 6,729,170 | 28.54 | % | |||||
*less than 1%
| (1) | Based on 23,579,256 shares issued and outstanding. | |
| (2) | Mr. Friedman’s beneficial ownership includes shares beneficially owned by his wife, Phyllis Friedman. |
| 63 |
Indemnification of Directors and Officers
Neither our Articles of Incorporation nor Bylaws prevent us from indemnifying our officers, directors and agents to the extent permitted under the Nevada Revised Statute (“NRS”). NRS Section 78.7502 provides that a corporation shall indemnify any director, officer, employee or agent of a corporation against expenses, including attorneys’ fees, actually and reasonably incurred by him in connection with any the defense to the extent that a director, officer, employee or agent of a corporation has been successful on the merits or otherwise in defense of any action, suit or proceeding referred to NRS Section 78.7502(1) or 78.7502(2), or in defense of any claim, issue or matter therein.
NRS Section 78.7502(1) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative, except an action by or in the right of the corporation, by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses, including attorneys’ fees, judgments, fines and amounts paid in settlement actually and reasonably incurred by him in connection with the action, suit or proceeding if he: (a) is not liable pursuant to NRS Section 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful.
NRS Section 78.7502(2) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses, including amounts paid in settlement and attorneys’ fees actually and reasonably incurred by him in connection with the defense or settlement of the action or suit if he: (a) is not liable pursuant to NRS Section 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation. Indemnification may not be made for any claim, issue or matter as to which such a person has been adjudged by a court of competent jurisdiction, after exhaustion of all appeals there from, to be liable to the corporation or for amounts paid in settlement to the corporation, unless and only to the extent that the court in which the action or suit was brought or other court of competent jurisdiction determines upon application that in view of all the circumstances of the case, the person is fairly and reasonably entitled to indemnity for such expenses as the court deems proper.
NRS Section 78.747 provides that except as otherwise provided by specific statute, no director or officer of a corporation is individually liable for a debt or liability of the corporation, unless the director or officer acts as the alter ego of the corporation. The court as a matter of law must determine the question of whether a director or officer acts as the alter ego of a corporation.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by us is against public policy as expressed hereby in the Securities Act and we will be governed by the final adjudication of such issue.
The Company's corporate headquarter is located at 65 Yigal Alon Street, 23rd Floor, Tel Aviv 67433, Israel, and the telephone number at such address is (972) 3 562-1755. Currently our corporate headquarter is located at the offices of our general counsel. We do not own or lease this office space and are provided access to these offices as needed. Because we are a small company with few employees this is adequate for our current needs. The Company’s U.S. contact information is: 57 W. 57th St., Suite 400 New York, NY 10019 and (646) 416-7896. The Company sponsors research at the Weizmann Institute which does not and will not include human clinical trials. In the future, the Company plans to sponsor clinical trials at the University of Würzburg in Germany, as well as at University of Parma, Italy, and at that time the Company will enter into written agreements for the use of their facilities.
| 64 |
The following table details the name of each Selling Stockholder, the number of shares owned by such Selling Stockholders, and the number of shares that may be offered by each Selling Stockholder for resale under this prospectus. The Selling Stockholders may sell up to 9,618,648 shares of our Common Stock from time to time in one or more offerings under this prospectus, including 4,759,324 shares which are issuable upon the exercise of warrants held by certain Selling Stockholders. Because each Selling Stockholder may offer all, some or none of the shares it holds, and because, based upon information provided to us, there are currently no agreements, arrangements, or understandings with respect to the sale of any of the shares, no definitive estimate as to the number of shares that will be held by each Selling Stockholder after the offering can be provided. The following table has been prepared on the assumption that all shares offered under this prospectus will be sold to parties unaffiliated with the Selling Stockholders.
Each of the transactions by which the Selling Stockholders acquired their securities from us was exempt under the registration provisions of the Securities Act.
The 9,618,648 shares of Common Stock referred to above are being registered to permit public sales of the shares, and the Selling Stockholders may offer the shares for resale from time to time pursuant to this prospectus. The Company shareholders may also sell, transfer or otherwise dispose of all or a portion of their shares in transactions exempt from the registration requirements of the Securities Act or pursuant to another effective registration statement covering those shares. We may from time to time include additional Selling Stockholders in supplements or amendments to this prospectus.
None of the Selling Stockholders is a broker dealer or an affiliate of a broker dealer other than as described in the footnotes to the table below.
Beneficial ownership is determined in accordance with the rules of the Securities and Exchange Commission. The Selling Stockholders’ percentage of ownership of our outstanding shares in the table below is based upon 23,579,256 shares of Common Stock outstanding as of October 23, 2014.
| 65 |
| Ownership Before Offering | Ownership After Offering (1) | |||||||||||||||
| Selling Stockholder | Number of Shares of Common Stock Beneficially Owned | Number of Shares of Common Stock Offered | Number of Shares of Common Stock Beneficially Owned | Percentage of Common Stock Beneficially Owned | ||||||||||||
| Adam K. Stern (69) | 133,332 | (2) | 133,332 | (2) | 0 | 0.00 | % | |||||||||
| Akita Capital, LLC (3) | 240,666 | (4) | 240,666 | (4) | 0 | 0.00 | % | |||||||||
| Aton Select Funds Ltd (5) | 720,000 | (6) | 720,000 | (6) | 0 | 0.00 | % | |||||||||
| B&C Scotti Family Trust (7) | 133,334 | (8) | 133,334 | (8) | 0 | 0.00 | % | |||||||||
| Bull Hunter, Inc. (9) | 346,666 | (10) | 346,666 | (10) | 0 | 0.00 | % | |||||||||
| Chris Mahne | 66,666 | (11) | 66,666 | (11) | 0 | 0.00 | % | |||||||||
| CR Financial Holdings, Inc. (12)(69) | 53,334 | (13) | 53,334 | (13) | 0 | 0.00 | % | |||||||||
| Craig Fielder | 100,000 | (14) | 100,000 | (14) | 0 | 0.00 | % | |||||||||
| Daniel Garofalo | 66,666 | (15) | 66,666 | (15) | 0 | 0.00 | % | |||||||||
| Daniel J. Witanek | 26,666 | (16) | 26,666 | (16) | 0 | 0.00 | % | |||||||||
| David Barry | 26,666 | (17) | 26,666 | (17) | 0 | 0.00 | % | |||||||||
| Deepak H. Aggarwal | 140,000 | (18) | 140,000 | (18) | 0 | 0.00 | % | |||||||||
| Dennis M. Brown | 200,000 | (19) | 200,000 | (19) | 0 | 0.00 | % | |||||||||
| Donald G. Bahouth | 200,000 | (20) | 200,000 | (20) | 0 | 0.00 | % | |||||||||
| Emanuel Nisan | 14,000 | (21) | 14,000 | (21) | 0 | 0.00 | % | |||||||||
| Empire Stock Transfer, Inc. (22) | 66,666 | (23) | 66,666 | (23) | 0 | 0.00 | % | |||||||||
| Firerock Global Opportunities Fund, L.P. (24) | 791,666 | (25) | 666,666 | (25) | 125,000 | 0.53 | % | |||||||||
| Fred Weiss Nancy Weiss | 165,000 | (26) | 120,000 | (26) | 45,000 | 0.19 | % | |||||||||
| GRQ Consultants, Inc. 401 K FBO Barry Honig (27) | 908,768 | (28) | 381,068 | (28) | 527,700 | 2.24 | % | |||||||||
| Howard K. Fuguet | 533,332 | (29) | 533,332 | (29) | 0 | 0.00 | % | |||||||||
| Ikona Global Partners (30) | 266,666 | (31) | 266,666 | (31) | 0 | 0.00 | % | |||||||||
| Jeffrey K. Greene | 28,000 | (32) | 28,000 | (32) | 0 | 0.00 | % | |||||||||
| Jennifer Lorenzo | 40,000 | (33) | 40,000 | (33) | 0 | 0.00 | % | |||||||||
| Jerry S. Browner | 26,666 | (34) | 26,666 | (34) | 0 | 0.00 | % | |||||||||
| Jill Strauss | 66,000 | (35) | 48,000 | (35) | 18,000 | 0.08 | % | |||||||||
| John C. Leo (69) | 140,000 | (36) | 140,000 | (36) | 0 | 0.00 | % | |||||||||
| John Chambers (69) | 26,666 | (37) | 26,666 | (37) | 0 | 0.00 | % | |||||||||
| Josephine Viglione | 20,000 | (38) | 20,000 | (38) | 0 | 0.00 | % | |||||||||
| Keith A. Goodman | 66,666 | (39) | 66,666 | (39) | 0 | 0.00 | % | |||||||||
| Lawrence Neil Grossbard | 333,334 | (40) | 333,334 | (40) | 0 | 0.00 | % | |||||||||
| Mark Edward Gaetz | 53,334 | (41) | 53,334 | (41) | 0 | 0.00 | % | |||||||||
| Michael Bonevento | 66,666 | (42) | 66,666 | (42) | 0 | 0.00 | % | |||||||||
| Michael Chill (69) | 26,666 | (43) | 26,666 | (43) | 0 | 0.00 | % | |||||||||
| Michael Leiter | 150,000 | (44) | 150,000 | (44) | 0 | 0.00 | % | |||||||||
| Michael Margolis (69) | 26,666 | (45) | 26,666 | (45) | 0 | 0.00 | % | |||||||||
| Michael Ravallo | 136,000 | (46) | 136,000 | (46) | 0 | 0.00 | % | |||||||||
| Omnibus SA (47) | 266,660 | (48) | 266,660 | (48) | 0 | 0.00 | % | |||||||||
| Phillip J. Lifschitz | 133,332 | (49) | 133,332 | (49) | 0 | 0.00 | % | |||||||||
| Raymond Coppede | 66,000 | (50) | 66,000 | (50) | 0 | 0.00 | % | |||||||||
| Renald J. Anelle | 66,666 | (51) | 66,666 | (51) | 0 | 0.00 | % | |||||||||
| Richard S. Grossbard | 333,334 | (52) | 333,334 | (52) | 0 | 0.00 | % | |||||||||
| Robert D. Crocitto | 54,000 | (53) | 54,000 | (53) | 0 | 0.00 | % | |||||||||
| Robert Freedman | 266,666 | (54) | 266,666 | (54) | 0 | 0.00 | % | |||||||||
| Robert M. Newsome | 140,000 | (55) | 140,000 | (55) | 0 | 0.00 | % | |||||||||
| Samuel Nussbaum | 400,000 | (56) | 400,000 | (56) | 0 | 0.00 | % | |||||||||
| Sandor Capital Master Fund (57) | 510,000 | (58) | 340,000 | (58) | 170,000 | 0.72 | % | |||||||||
| Sichenzia Ross Friedman Ference LLP (59) | 100,000 | (60) | 100,000 | (60) | 0 | 0.00 | % | |||||||||
| Stephen M. Payne | 800,000 | (61) | 800,000 | (61) | 0 | 0.00 | % | |||||||||
| Suntime Enterprises Ltd. (62) | 666,666 | (63) | 666,666 | (63) | 0 | 0.00 | % | |||||||||
| Suzanne Adams | 134,200 | (64) | 97,600 | (64) | 36,600 | 0.16 | % | |||||||||
| Vantage IRA FBO Jerrold Karnell (65) | 66,666 | (66) | 66,666 | (66) | 0 | 0.00 | % | |||||||||
| Viswanathan Ramamurthy Reddy | 30,000 | (67) | 30,000 | (67) | 0 | 0.00 | % | |||||||||
| William R. Lefever | 100,000 | (68) | 100,000 | (68) | 0 | 0.00 | % | |||||||||
| TOTAL: | 10,540,948 | 9,618,648 | 922,300 | |||||||||||||
| 66 |
| (1) | Represents the amount of shares that will be held by the selling stockholders after completion of this offering based on the assumptions that (a) all shares registered for sale by the registration statement of which this prospectus is part will be sold and (b) that no other shares of our Common Stock beneficially owned by the selling stockholders are acquired or are sold prior to completion of this offering by the selling stockholders. |
| (2) | Includes 66,666 shares of common stock and 66,666 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (3) | Gary Gotto holds voting and dispositive power over shares held by Akita Capital, LLC. |
| (4) | Includes 120,333 shares of common stock and 120,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (5) | David Dane holds voting and dispositive power over shares held by Aton Select Funds Ltd. |
| (6) | Includes 360,000 shares of common stock and 360,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (7) | Benjamin J. Scotti holds voting and dispositive power over shares held by B&C Scotti 1998 Family Trust. |
| (8) | Includes 66,667 shares of common stock and 66,667 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (9) | Mark Groussman holds voting and dispositive power over shares held by Bull Hunter, Inc. Mr. Groussman is the President of Flying High Financial Corporation, which entity provided consulting services to Ticket to See, Inc. prior to the Share Exchange. |
| (10) | Includes 173,333 shares of common stock and 173,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (11) | Includes 33,333 shares of common stock and 33,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (12) | Donald Skrdlant holds voting and dispositive power over shares held by CR Financial Holdings, Inc. |
| (13) | Includes 26,667 shares of common stock and 26,667 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (14) | Includes 50,000 shares of common stock and 50,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (15) | Includes 33,333 shares of common stock and 33,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (16) | Includes 13,333 shares of common stock and 13,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (17) | Includes 13,333 shares of common stock and 13,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (18) | Includes 70,000 shares of common stock and 70,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (19) | Since June 30, 2014, Mr. Brown has been a member of our Board of Directors. Mr. Brown’s ownership of 200,000 shares of common stock consists of 100,000 shares of common stock and 100,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (20) | Includes 100,000 shares of common stock and 100,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (21) | Includes 7,000 shares of common stock and 7,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (22) | Patrick Mokros holds voting and dispositive power over shares held by Empire Stock Transfer Inc. |
| (23) | Includes 33,333 shares of common stock and 33,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (24) | Seth Fireman holds voting and dispositive power over shares held by Firerock Global Opportunities Fund, L.P. |
| (25) | Includes 458,333 shares of common stock and 333,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (26) | Includes 105,000 shares of common stock and 60,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (27) | Barry Honig holds voting and dispositive power over shares held by GRQ Consultants, Inc. 401K FBO Barry Honig. |
| (28) | Includes 718,234 shares of common stock and 190,534 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (29) | Includes 266,666 shares of common stock and 266,666 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (30) | Richard Calta holds voting and dispositive power over shares held by Ikona Global Partners. |
| (31) | Includes 133,333 shares of common stock and 133,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (32) | Includes 14,000 shares of common stock and 14,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| 67 |
| (33) | Includes 20,000 shares of common stock and 20,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (34) | Includes 13,333 shares of common stock and 13,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (35) | Includes 42,000 shares of common stock and 24,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (36) | Includes 70,000 shares of common stock and 70,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (37) | Includes 13,333 shares of common stock and 13,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (38) | Includes 10,000 shares of common stock and 10,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (39) | Includes 33,333 shares of common stock and 33,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (40) | Includes 166,667 shares of common stock and 166,667 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (41) | Includes 26,667 shares of common stock and 26,667 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (42) | Includes 33,333 shares of common stock and 33,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (43) | Includes 13,333 shares of common stock and 13,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (44) | Includes 75,000 shares of common stock and 75,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (45) | Includes 13,333 shares of common stock and 13,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (46) | Includes 68,000 shares of common stock and 68,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (47) | Kenneth Ciapala holds voting and dispositive power over shares held by Omnibus S.A. |
| (48) | Includes 133,330 shares of common stock and 133,330 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (49) | Includes 66,666 shares of common stock and 66,666 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (50) | Includes 33,000 shares of common stock and 33,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (51) | Includes 33,333 shares of common stock and 33,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (52) | Includes 166,667 shares of common stock and 166,667 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (53) | Includes 27,000 shares of common stock and 27,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (54) | Includes 133,333 shares of common stock and 133,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (55) | Includes 70,000 shares of common stock and 70,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (56) | Includes 200,000 shares of common stock and 200,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (57) | Richard Lemak holds voting and dispositive power over shares held by Sandor Capital Master Fund. |
| (58) | Includes 340,000 shares of common stock and 170,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (59) | Gregory Sichenzia, Marc J. Ross, Richard A. Friedman, Michael Ference, Thomas A. Rose, Jeffrey Fessler, Harvey Kesner, Benjamin Tan, Andrea Cataneo and Darrin M. Ocasio have shared voting and dispositive power over the shares of Common Stock held by Sichenzia Ross Friedman Ference LLP. |
| (60) | Includes 100,000 shares of common stock issued to Sichenzia Ross Friedman Ference LLP for legal services provided to the Company. |
| (61) | Includes 400,000 shares of common stock and 400,000 shares of common stock issuable upon the exercise of an outstanding warrant. | |
| (62) | Lian Wang holds voting and dispositive power over shares held by Suntime Enterprises Ltd. |
| 68 |
| (63) | Includes 333,333 shares of common stock and 333,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (64) | Includes 85,400 shares of common stock and 48,800 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (65) | Jerrold Karnell holds voting and dispositive power over shares held by Vantage IRA FBO Jerrold Karnell. |
| (66) | Includes 33,333 shares of common stock and 33,333 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (67) | Includes 15,000 shares of common stock and 15,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (68) | Includes 50,000 shares of common stock and 50,000 shares of common stock issuable upon the exercise of an outstanding warrant. |
| (69) | The selling stockholder or the individual deemed to hold voting and dispositive power over the selling stockholder is an affiliate of a broker-dealer. The selling stockholder acquired the securities in the ordinary course of business and at the time of such acquisition, did not have any agreements, plans or understandings, directly or indirectly, with any person to distribute the securities. |
Description of Registrant’s Securities to be Registered.
The Company’s authorized capital stock of 210,000,000 consists of (i) 200,000,000 shares of common stock, par value $0.001 and (ii) 10,000,000 shares of preferred stock, par value $0.001. As of the date of the filing of this prospectus, there are 23,579,256 shares of the Company’s common stock issued and outstanding.
The holders of common stock are entitled to one non-cumulative vote for each share held on all matters submitted to a vote of shareholders. Holders of common stock are entitled to receive ratably such dividends, if any, as may be declared by the Board of Directors out of funds legally available. Upon a liquidation, dissolution or winding up the Company, the holders of common stock are entitled to receive ratably the net assets available after the payment of all debts and other liabilities, and subject further only to the prior rights of any outstanding preferred stock.
The holders of common stock have no preemptive, subscription, redemption or sinking fund conversion rights. Holders of shares of our common stock do not have cumulative voting rights, which means that the holders of more than 50% of the outstanding shares, voting for the election of directors, can elect all of the directors to be elected, if they so choose, and, in that event, the holders of the remaining shares will not be able to elect any of our directors.
Plan of Distribution
The Selling Stockholders, which, as used herein, includes donees, pledgees, transferees or other successors-in-interest selling shares of Common Stock received after the date of this prospectus from a Selling Stockholder as a gift, pledge, partnership distribution or other transfer, may, from time to time, sell, transfer or otherwise dispose of any or all of their shares of Common Stock on any stock exchange, market or trading facility on which the shares are traded or in private transactions. These dispositions may be at fixed prices, at prevailing market prices at the time of sale, at prices related to the prevailing market price, at varying prices determined at the time of sale, or at negotiated prices. We have not been advised of any arrangements by the Selling Stockholders for the sale of any of the Common Stock owned by them.
The Selling Stockholders may use any one or more of the following methods when disposing of shares or interests therein:
| • | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| • | block trades in which the broker-dealer will attempt to sell the shares as agent, but may position and resell a portion of the block as principal to facilitate the transaction; |
| • | crosses, where the same broker acts as an agent on both sides of the trade; |
| • | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| • | an exchange distribution in accordance with the rules of the applicable exchange; |
| • | privately negotiated transactions; |
| • | short sales; |
| • | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
| 69 |
| • | broker-dealers may agree with the Selling Stockholders to sell a specified number of such shares at a stipulated price per share; |
| • | a combination of any such methods of sale; and |
| • | any other method permitted pursuant to applicable law. |
The Selling Stockholders may, from time to time, pledge or grant a security interest in some or all of the shares of Common Stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of Common Stock, from time to time, under this prospectus, or under an amendment to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act amending the list of Selling Stockholders to include the pledgee, transferee or other successors in interest as Selling Stockholders under this prospectus. The Selling Stockholders also may transfer the shares of Common Stock in other circumstances, in which case the transferees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus; provided, however, that prior to any such transfer the following information (or such other information as may be required by the federal securities laws from time to time) with respect to each such selling beneficial owner must be added to the prospectus by way of a prospectus supplement or post-effective amendment, as appropriate: (1) the name of the selling beneficial owner; (2) any material relationship the selling beneficial owner has had within the past three years with us or any of our predecessors or affiliates; (3) the amount of securities of the class owned by such security beneficial owner before the transfer; (4) the amount to be offered for the security beneficial owner’s account; and (5) the amount and (if one percent or more) the percentage of the class to be owned by such security beneficial owner after the transfer is complete.
Any Selling Stockholder and any other person participating in a distribution will be subject to applicable provisions of the Exchange Act and the rules and regulations under that statute, including, without limitation, possibly Regulation M. This may limit the timing of purchases and sales of any of the securities by a Selling Stockholder and any other participating person. Regulation M may also restrict the ability of any person engaged in the distribution of the securities to engage in market-making activities with respect to the securities. All of the foregoing may affect the marketability of the securities and the ability of any person or entity to engage in market-making activities with respect to the securities.
We are required to pay all fees and expenses incident to the registration of the shares, including fees and disbursements of counsel to the Selling Stockholder, but excluding brokerage commissions or underwriter discounts.
The Selling Stockholders, alternatively, may sell all or any part of the shares offered in this prospectus through an underwriter. No Selling Stockholder has entered into any agreement with a prospective underwriter and there is no assurance that any such agreement will be entered into.
A Selling Stockholder may pledge its shares to their brokers under the margin provisions of customer agreements. If a Selling Stockholder defaults on a margin loan, the broker may, from time to time, offer and sell the pledged shares.
If the Selling Stockholder notifies us that it has a material arrangement with a broker-dealer for the resale of the Common Stock, then we would be required to amend the registration statement of which this prospectus is a part, and file a prospectus supplement to describe the agreements between the Selling Stockholder and the broker-dealer.
To the extent required by the Securities Act and the rules and regulations thereunder, the Selling Stockholders and any broker-dealer participating in the distribution of the shares of common stock may be deemed to be “underwriters” within the meaning of the Securities Act, and any commission paid, or any discounts or concessions allowed to, any such broker-dealer may be deemed to be underwriting commissions or discounts under the Securities Act. At the time a particular offering of the shares of common stock is made, a prospectus supplement, if required, will be distributed, which will set forth the aggregate amount of shares of common stock being offered and the terms of the offering, including the name or names of any broker-dealers or agents, any discounts, commissions and other terms constituting compensation from the selling stockholders and any discounts, commissions or concessions allowed or re-allowed or paid to broker-dealers.
LEGAL MATTERS
Sichenzia Ross Friedman Ference LLP, New York, New York, will pass upon the validity of the shares of Common Stock offered by the Selling Stockholders under this prospectus. Sichenzia Ross Friedman Ference LLP is the beneficial holder of 100,000 shares of Common Stock, which shares are being registered pursuant to this prospectus.
The balance sheets of Cell Source Ltd. as of December 31, 2013 and December 31, 2012 and the related statements of operations, changes in stockholders’ deficiency, and cash flows included in this registration statement on Form S-1 have been so included in reliance on the report of Marcum LLP, an independent registered public accounting firm, given upon their authority as experts in accounting and auditing. In July 2014, the Company issued 100,000 shares of common stock to Sichenzia Ross Friedman Ference LLP, our counsel, in lieu of cash payment for legal services.
| 70 |
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the Securities and Exchange Commission a registration statement on Form S-1, together with any amendments and related exhibits, under the Securities Act, with respect to our shares of Common Stock offered by this prospectus. The registration statement contains additional information about us and our shares of Common Stock that the Selling Stockholders are offering in this prospectus.
We file annual, quarterly and current reports and other information with the Securities and Exchange Commission under the Securities Exchange Act. Our Securities and Exchange Commission filings are available to the public over the Internet at the Securities and Exchange Commission’s website at http://www.sec.gov. Access to these electronic filings is available as soon as practicable after filing with the Securities and Exchange Commission. You may also read and copy any document we file at the Securities and Exchange Commission’s public reference room located at 100 F Street, N.E., Washington, D.C. 20549. Please call the Securities and Exchange Commission at 1-800-SEC-0330 for further information on the public reference rooms and their copy charges.
| 71 |
FINANCIAL STATEMENTS
CONTENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
| PAGE: | ||
| Cell Source Limited | 73 |
FOR THE THREE AND SIX MONTHS ENDED JUNE 30, 2014 AND 2013
| Cell Source, Inc. | 106 |
| 72 |
CELL SOURCE LIMITED
FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
| 73 |
| CELL SOURCE LIMITED |
| CONTENTS |
| Report of Independent Registered Public Accounting Firm | 75 |
| Financial Statements | |
| Balance Sheets | 76-77 |
| Statements of Operations | 78 |
| Statements of Stockholders’ Deficit | 79-80 |
| Statements of Cash Flows | 81-82 |
| Notes to Financial Statements | 83-105 |
| 74 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors
of Cell Source Limited
We have audited the accompanying balance sheets of Cell Source Limited (the “Company”) as of December 31, 2013 and 2012 and the statements of operations, stockholders’ deficit and cash flows for the years ended December 31, 2013 and 2012. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Cell Source Limited, as of December 31, 2013 and 2012, and the results of its operations and its cash flows for the years ended December 31, 2013 and 2012 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has had recurring losses, and has a working capital and stockholders' deficit as of December 31, 2013 and 2012. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Marcum LLP
Marcum LLP
| New York, NY | |
| July 1, 2014 |
| 75 |
| CELL SOURCE LIMITED |
| BALANCE SHEETS |
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Assets | ||||||||
| Current Assets | ||||||||
| Cash | $ | 28,878 | $ | 161,323 | ||||
| Other Assets | 63,337 | 35,832 | ||||||
| Total Assets | $ | 92,215 | $ | 197,155 | ||||
The accompanying notes are an integral part of these financial statements.
| 76 |
| CELL SOURCE LIMITED |
| BALANCE SHEETS |
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Liabilities and Stockholders' Deficit | ||||||||
| Current Liabilities | ||||||||
| Accounts payable and accrued expenses | $ | 558,349 | $ | 48,177 | ||||
| Derivative liabilities | 231,200 | 38,300 | ||||||
| Covertible notes net of debt discount of $— and $43,167 at December 31, 2013 and 2012, respectively | — | 258,039 | ||||||
| Total Current Liabilities | 789,549 | 344,516 | ||||||
| Commitments and Contingencies | — | — | ||||||
| Stockholders’ Deficit | ||||||||
| Common shares of $0.01 par value: | ||||||||
| 50,000,000 shares authorized; 14,155,190 and 12,763,818 shares issued and outstanding at December 31, 2013 and 2012, respectively | 141,552 | 127,637 | ||||||
| Additional paid-in capital | 3,102,125 | 1,882,363 | ||||||
| Accumulated deficit | (3,941,011 | ) | (2,157,361 | ) | ||||
| Total Stockholders’ Deficit | (697,334 | ) | (147,361 | ) | ||||
| Total Liabilities and Stockholders’ Deficit | $ | 92,215 | $ | 197,155 | ||||
The accompanying notes are an integral part of these financial statements.
| 77 |
| CELL SOURCE LIMITED |
| STATEMENTS OF OPERATIONS |
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Revenues | $ | — | $ | — | ||||
| Operating Expenses | ||||||||
| Research and development | 1,135,513 | 1,219,515 | ||||||
| General and administrative | 319,857 | 447,264 | ||||||
| Total Operating Expenses | 1,455,370 | 1,666,779 | ||||||
| Operating Loss | (1,455,370 | ) | (1,666,779 | ) | ||||
| Interest Expense | (681,780 | ) | (8,633 | ) | ||||
| Change in Fair Value of Derivative Liability | 353,500 | 13,500 | ||||||
| Net Loss | $ | (1,783,650 | ) | $ | (1,661,912 | ) | ||
| Loss Per Share - Basic and Diluted | $ | (0.14 | ) | $ | (0.14 | ) | ||
| Weighted Average Number of Shares Outstanding | 13,168,636 | 12,067,243 | ||||||
The accompanying notes are an integral part of these financial statements.
| 78 |
| CELL SOURCE LIMITED |
| STATEMENTS OF STOCKHOLDERS' DEFICIT |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
| Additional | Total | |||||||||||||||||||
| Common Stock | Paid-in | Accumulated | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||
| Balance - January 1, 2012 | 10,513,818 | 105,137 | 1,094,863 | (495,449 | ) | 704,551 | ||||||||||||||
| Issuance of common stock for cash | 2,250,000 | 22,500 | 787,500 | — | 810,000 | |||||||||||||||
| Net loss | — | — | — | (1,661,912 | ) | (1,661,912 | ) | |||||||||||||
| December 31, 2012 | 12,763,818 | 127,637 | 1,882,363 | (2,157,361 | ) | (147,361 | ) | |||||||||||||
| Issuance of common stock and warrants for cash | 735,327 | 7,354 | 544,143 | — | 551,497 | |||||||||||||||
| Reclassification of detachable warrants to derivative liability | — | — | (231,200 | ) | — | (231,200 | ) | |||||||||||||
| Issuance of common stock for settlement of convertible notes | 2,699,880 | $ | 26,999 | $ | 794,091 | $ | — | $ | 821,090 | |||||||||||
The accompanying notes are an integral part of these financial statements.
| 79 |
| CELL SOURCE LIMITED |
| STATEMENTS OF STOCKHOLDERS' DEFICIT (CONTINUED) |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
| Deficit | ||||||||||||||||||||
| Accumulated | ||||||||||||||||||||
| Additional | during the | Total | ||||||||||||||||||
| Common Stock | Paid-in | Development | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Stage | Deficit | ||||||||||||||||
| Balance - December 31, 2012 (continued) | ||||||||||||||||||||
| Contribution of services by officers for no consideration | — | — | 76,990 | — | 76,990 | |||||||||||||||
| Exchange of common stock for warrants to founders | (2,043,835 | ) | (20,438 | ) | 20,438 | — | — | |||||||||||||
| Forgiveness of accrued interest | — | — | 15,300 | — | 15,300 | |||||||||||||||
| Net loss | — | — | — | (1,783,650 | ) | (1,783,650 | ) | |||||||||||||
| Balance - December 31, 2013 | 14,155,190 | $ | 141,552 | $ | 3,102,125 | $ | (3,941,011 | ) | $ | (697,334 | ) | |||||||||
The accompanying notes are an integral part of these financial statements.
| 80 |
| CELL SOURCE LIMITED |
| STATEMENTS OF CASH FLOWS |
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Cash Flows from Operating Activities | ||||||||
| Net loss | $ | (1,783,650 | ) | $ | (1,661,912 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Contribution of services by officers | 76,990 | — | ||||||
| Forgiveness of accrued interest | 15,300 | — | ||||||
| Accretion of debt discount | 358,367 | 8,633 | ||||||
| Change in fair value of derivative liability | (353,500 | ) | (13,500 | ) | ||||
| Interest expense | 309,885 | — | ||||||
| Changes in assets and liabilities: | ||||||||
| (Increase) decrease in other assets | (27,505 | ) | 82,188 | |||||
| Increase in accrued expenses and other liabilities | 510,171 | 49,383 | ||||||
| Total Adjustments | 889,708 | 126,704 | ||||||
| Net Cash Used in Operating Activities | (893,942 | ) | (1,535,208 | ) | ||||
| Cash Flows from Financing Activities | ||||||||
| Proceeds from issuance of convertible loan | 210,000 | 300,000 | ||||||
| Proceeds from issuance of common stock and warrants, net | 551,497 | 810,000 | ||||||
| Net Cash Provided by Financing Activities | 761,497 | 1,110,000 | ||||||
| (Decrease) Increase in Cash | (132,445 | ) | (425,208 | ) | ||||
| Cash - Beginning of year | 161,323 | 586,531 | ||||||
| Cash - End of year | $ | 28,878 | $ | 161,323 | ||||
The accompanying notes are an integral part of these financial statements.
| 81 |
| CELL SOURCE LIMITED |
| STATEMENTS OF CASH FLOWS (CONTINUED) |
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Supplemental Disclosures of Cash Flow Information: | ||||||||
| Cash paid for income taxes | $ | — | $ | — | ||||
| Cash paid for interest expense | $ | — | $ | — | ||||
| Non-cash investing and financing transactions: | ||||||||
| Issuance of common stock for settlement of debt | $ | 511,206 | $ | — | ||||
| Reclassification of warrants to derivative liability | $ | 231,200 | $ | — | ||||
The accompanying notes are an integral part of these financial statements.
| 82 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 1 - Organization and Nature of Business
Cell Source Limited ("the Company") was incorporated on September 15, 2011 under the General Corporation Law of Israel to engage in the research and development of cell therapy treatments and a new source of human organs based on research performed at the Weizman Institute of Science in Israel. The Company’s operations and corporate headquarters are located in Israel.
Note 2 - Going Concern
The Company has not generated any revenues since its inception, has recurring net losses, and a working capital deficit as of December 31, 2013 and 2012 of approximately $761,000 and $183,200, respectively, and used cash in operations of approximately $894,000 and $1,535,000 for the years ended December 31, 2013 and 2012, respectively. In addition, the Company has an accumulated deficit from inception of approximately $3,941,000. These conditions raise substantial doubt about the Company’s ability to continue as a going concern.
The financial statements have been prepared on the going concern basis, which assumes the realization of assets and liquidation of liabilities in the normal course of operations. The ability of the Company to continue its operations is dependent on management’s plans, which include the raising of capital through debt and/or equity markets with some additional funding from other traditional financing sources, including term notes, until such time that funds provided by operations are sufficient to fund working capital requirements. The Company may need to incur additional liabilities with certain related parties to sustain the Company’s existence. If the Company were not to continue as a going concern, it would likely not be able to realize on its assets at values comparable to the carrying value or the fair value estimates reflected in the balances set out in the preparation of the financial statements.
| 83 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 2 - Going Concern (continued)
There can be no assurances that the Company will be successful in generating additional cash from equity or other sources to be used for operations. The financial statements do not include any adjustments relating to the recoverability of assets and classification of assets and liabilities that might be necessary. Should the Company not be successful in obtaining the necessary financing to fund its operations, the Company would need to curtail certain or all operational activities and/or contemplate the sale of its assets if necessary.
Note 3 – Share Exchange Transaction
On June 30, 2014, Ticket to See, Inc. (“TTSE”) entered into an Acquisition and Share Exchange Agreement with the Company. Upon the terms and subject to the conditions of the agreement, at the effective date of the share exchange, the Company was merged with and into TSTE, with TSTE continuing as the surviving corporation.
At the closing date, TSTE acquired 100% of the issued and outstanding shares of the Company. At the effective date of the share exchange, each share of the Company's common stock was cancelled and converted automatically into the right to receive common shares of TSTE for an aggregate of 18,245,923 shares common shares of Cell Source Limited, which constituted 78.5% of the post-acquisition outstanding shares of TSTE’s stock at the end of the share exchange. TSTE’s existing shareholders retained a total of 5,000,000 shares of TSTE’s stock, which constituted 21.5% of the post-acquisition outstanding shares of TSTE. Post-acquisition and after share exchange, there was a total of 23,245,923 issued and outstanding shares of TSTE stock, which was recorded as a recapitalization of TSTE.
For accounting purposes, the transaction described above will be treated as a recapitalization of the Company, the accounting acquirer, because the Company shareholders own the majority of TSTE’s outstanding common stock following the transaction and exercise significant influence over the operating and financial policies of the consolidated entity. TSTE was a non-operating company prior to the share exchange. Pursuant to Securities and Exchange Commission rules, the merger or acquisition of a private operating company into a non-operating public company with nominal net assets is considered a capital transaction in substance, rather than a business combination.
| 84 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 4 - Significant Accounting Policies
Use of Estimates
The preparation of the financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period.
Management bases its estimates on historical experience and on various assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. The most significant estimates, among other things, are used in accounting for valuation allowances for deferred income taxes, contingencies, as well as the recording and presentation of convertible notes and the embedded conversion feature and common stock and related warrants issuances. Estimates and assumptions are periodically reviewed and the effects of any material revisions are reflected in the financial statements in the period that they are determined to be necessary. Actual results could differ from those estimates and assumptions.
| 85 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 4 - Significant Accounting Policies (continued)
Cash and Cash Equivalents
The Company considers all highly-liquid instruments with an original maturity of three months or less when purchased to be cash equivalents. As of December 31, 2013 and 2012, the Company did not have any cash equivalents.
Income Taxes
The Company accounts for income taxes using the asset and liability method. Accordingly, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in the tax rate is recognized in income or expense in the period that the change is effective. Income tax benefits are recognized when it is probable that the deduction will be sustained. A valuation allowance is established when it is more likely than not that all or a portion of a deferred tax asset will either expire before the Company is able to realize the benefit, or that future deductibility is uncertain.
Research and Development Costs
Research and development costs are expensed as they are incurred and consist of salaries, stock-based compensation benefits and other personnel related costs, fees paid to consultants, clinical trials and related clinical manufacturing costs, license and milestone fees, and facilities and overhead costs. For the years ended December 31, 2013 and 2012 the Company has recorded a charge of research and development of approximately $1,135,500 and $1,219,500, respectively.
| 86 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 4 - Significant Accounting Policies (continued)
Loss Per Share
Basic loss per share was computed using the weighted average number of outstanding common shares. Diluted loss per share includes the effect of dilutive common stock equivalents from the assumed exercise of warrants and convertible debt. Common stock equivalents would be excluded from the computation of diluted loss per share since their inclusion would be anti-dilutive. Total shares issuable upon the exercise of warrants and convertible debt as of December 31, 2013 and 2012, were comprised as follows:
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Warrants | 2,812,499 | — | ||||||
| Convertible notes | — | 643,199 | ||||||
| Total | 2,812,499 | 643,199 | ||||||
For the year ended December 31, 2013, 2,043,835 warrants were included in loss per share because their exercise price was determined to be nominal.
Fair Value of Financial Instruments
Financial instruments consist of cash, accounts payable, accrued expenses, convertible debt and derivative liabilities. The Company determines the estimated fair value of such financial instruments presented in these financial statements using available market information and appropriate methodologies. These financial instruments are stated at their respective historical carrying amounts, which approximate fair value due to their short term nature, except derivative instruments which are marked to market at the end of each reporting period.
Derivative Financial Instruments
In connection with the issuance of convertible notes, the terms of the convertible notes included an embedded conversion feature; which provided for a conversion of the convertible notes into shares of common stock at a rate which was determined to be variable. In addition, the Company granted warrants to investors whereby the exercise prices contained price protection reset provisions. The Company determined that the conversion feature and exercise prices were derivative instruments pursuant to ASC 815 “Derivatives and Hedging”.
| 87 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 4 - Significant Accounting Policies (continued)
Derivative Financial Instruments (continued)
The accounting treatment of derivative financial instruments requires that the Company record the embedded conversion option and warrants at their fair values as of the inception date of the agreement and at fair value as of each subsequent balance sheet date. Any change in fair value is recorded as non-operating, non-cash income or expense for each reporting period at each balance sheet date. The Company reassesses the classification of its derivative instruments at each balance sheet date. If the classification changes as a result of events during the period, the contract is reclassified as of the date of the event that caused the reclassification.
The fair value of an embedded conversion option that is convertible into at variable amount of shares and warrants that include price protection reset provision features are deemed to be a “down-round protection” and, therefore, do not meet the scope exception for treatment as a derivative under ASC 815, since “down-round protection” is not an input into the calculation of the fair value of the conversion option and warrants and cannot be considered “indexed to the Company’s own stock” which is a requirement for the scope exception as outlined under ASC 815.
The Company determined the fair value of the Binomial Lattice Model and the Black-Scholes Method to be materially the same. The Black-Scholes option valuation model is used to estimate the fair value of the warrants granted. The model includes subjective input assumptions that can materially affect the fair value estimates. The model was developed for use in estimating the fair value of traded options or warrants. The expected volatility is estimated based on the most recent historical period of time equal to the weighted average life of the warrants or options granted.
Foreign Currency Translation
The New Israeli Shekel is the functional currency of the Company. Assets and liabilities are translated based on the exchange rates at the balance sheet date, while revenue and expense accounts are translated at the average exchange rates prevailing during the year. Equity accounts are translated at historical exchange rates. The resulting translation gain and loss adjustments are accumulated as a component of other comprehensive income.
Foreign currency gains and losses resulting from transactions denominated in foreign currencies, including intercompany transactions, are included in results of operations.
| 88 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 4 - Significant Accounting Policies (continued)
Foreign Currency Translation (continued)
Translation gains and losses were immaterial for the years ended December 31, 2013 and 2012. The Company recorded approximately $21,000 and $64,000 of transaction losses for the years ended December 31, 2013 and 2012 which have been included in general and administrative expenses, respectively.
Comprehensive Income (Loss)
The Company reports comprehensive income (loss) and its components in its financial statements. Comprehensive income (loss) consists of net loss and foreign currency translation adjustments affecting stockholders’ deficit that, under U.S. GAAP, are excluded from net loss. The differences between net loss as reported and comprehensive income (loss) have historically been immaterial.
Stock Split
On November 11, 2013, the Company’s Board approved a 32-for-1 forward stock split of its common stock, which was approved at a meeting of stockholders held on November 11, 2013. Each share of common stock outstanding immediately prior to the approval date was combined, reclassified and changed into thirty two of fully paid and non-assessable shares of common stock. All common share and common per share information in these financial statements and accompanying notes have been retroactively adjusted to reflect the forward stock split for all periods presented. Simultaneous with the forward stock split the Company increased its authorized shares from 3,800,000 shares to 50,000,000 shares.
Recent Accounting Standards
In June 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-10, “Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation.” This ASU removes the definition of a development stage entity from the ASC, thereby removing the financial reporting distinction between development stage entities and other reporting entities from GAAP. In addition, the ASU eliminates the requirements for development stage entities to (1) present inception-to-date information in the statements of operations, cash flows, and shareholders’ deficit, (2) label the financial statements as those of a development stage entity, (3) disclose a description of the development stage activities in which the entity is engaged, and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. Early adoption is permitted. The Company has elected to adopt this ASU effective with these Annual Financial Statements and its adoption resulted in the removal of all development stage disclosures.
| 89 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
The Company has implemented all new accounting standards that are in effect and may impact its financial statements and does not believe that there are any other new accounting standards that have been issued that might have a material impact on its financial position or results of operations.
Subsequent Events
The Company evaluates events that have occurred after the balance sheet date but before the financial statements are issued. Based upon the evaluation, the Company did not identify any recognized or non-recognized subsequent events that would require adjustment or disclosure in the financial statements other than as disclosed in Note 14.
| 90 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 5 - Fair Value
The Company determines the estimated fair value of amounts presented in these financial statements using available market information and appropriate methodologies. However, considerable judgment is required in interpreting market data to develop the estimates of fair value. The estimates presented in the financial statements are not necessarily indicative of the amounts that could be realized in a current exchange between buyer and seller. The use of different market assumptions and/or estimation methodologies may have a material effect on the estimated fair value amounts. These fair value estimates were based upon pertinent information available as of December 31, 2013 and 2012 and, as of those dates, the carrying value of all amounts approximates fair value.
The Company has categorized its assets and liabilities at fair value based upon the following fair value hierarchy:
Level 1 - Inputs use quoted prices in active markets for identical assets or liabilities that the Company has the ability to access.
Level 2 - Inputs use other inputs that are observable, either directly or indirectly. These inputs include quoted prices for similar assets and liabilities in active markets as well as other inputs such as interest rates and yield curves that are observable at commonly quoted intervals.
Level 3 - Inputs are unobservable inputs, including inputs that are available in situations where there is little, if any, market activity for the related asset or liability.
In instances where inputs used to measure fair value fall into different levels in the above fair value hierarchy, fair value measurements in their entirety are categorized based on the lowest level input that is significant to the valuation. The Company’s assessment of the significance of particular inputs to these fair measurements requires judgment and considers factors specific to each asset or liability.
Both observable and unobservable inputs may be used to determine the fair value of positions that are classified within the Level 3 category. As a result, the unrealized gains and losses for assets within the Level 3 category presented in the tables below may include changes in fair value that were attributable to both observable (e.g., changes in market interest rates) and unobservable (e.g., changes in historical company data) inputs.
| 91 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 5 - Fair Value (continued)
The following table summarizes the valuation of the Company’s investment by the above fair value hierarchy levels as of December 31, 2013 and 2012 using quoted prices in active markets for identical assets (Level 1), significant other observable inputs (Level 2), and significant unobservable inputs (Level 3):
| Quoted Prices | ||||||||||||||||
| In Active | Significant | |||||||||||||||
| Markets for | Other | Significant | ||||||||||||||
| Identical | Observable | Unobservable | ||||||||||||||
| Liabilities | Inputs | Inputs | ||||||||||||||
| Total | (Level 1) | (Level 2) | (Level 3) | |||||||||||||
| Warrant liability | $ | 231,200 | $ | 231,200 | ||||||||||||
| Balance - December 31, 2013 | $ | 231,200 | $ | 231,200 | ||||||||||||
| Embedded conversion option | $ | 38,300 | $ | 38,300 | ||||||||||||
| Balance - December 31, 2012 | $ | 38,300 | $ | 38,300 | ||||||||||||
Financial assets are considered Level 3 when their fair values are determined using pricing models, discounted cash flow methodologies or similar techniques and at least one significant model assumption or input is unobservable. The Company’s Level 3 liabilities consist of derivative liabilities associated with the convertible debt that contains an indeterminable conversion share price and the tainted warrants as the Company cannot determine if it will have sufficient authorized common stock to settle such arrangements.
Assumptions utilized in the development of Level 3 liabilities as of and during the year ended December 31, 2013 are described as follows.
| December 31, | ||||
| 2012 | 2013 | |||
| Risk-free interest rate | .05% | 1.75% | ||
| Expected life of grants | 0.5 Years | 0.5 - 5 years | ||
| Expected volatility of underlying stock | 98% - 112% | 98% - 117% | ||
| Dividends | $0 | $0 | ||
The expected stock price volatility for the Company’s stock options was determined by the historical volatilities for industry peers and used an average of those volatilities. Risk free interest rates were obtained from U.S. Treasury rates for the applicable periods. The expected life was the contractual life.
| 92 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 5 - Fair Value (continued)
The following table provides a summary of the changes in fair value, including net transfers in and/or out, of all financial assets measured at fair value on a recurring basis using Level III unobservable inputs during the years ended December 31, 2012 and 2013.
| Embedded | ||||||||||||
| Warrant | Conversion | |||||||||||
| Liability | Feature | Total | ||||||||||
| Balance - January 1, 2012 | $ | — | $ | — | $ | — | ||||||
| Included in debt discount | — | 51,800 | 51,800 | |||||||||
| Change in fair value of derivative liability | — | (13,500 | ) | (13,500 | ) | |||||||
| Balance - December 31, 2012 | — | 38,300 | 38,300 | |||||||||
| Included in debt discount | — | $ | 315,200 | 315,200 | ||||||||
| Change in fair value of derivative liability | — | (353,500 | ) | (353,500 | ) | |||||||
| Reclassification of warrants to derivative liability | 231,200 | — | 231,200 | |||||||||
| Balance - December 31, 2013 | $ | 231,200 | $ | — | $ | 231,200 | ||||||
The Company’s significant financial instruments such as cash, accounts payable and accrued expenses and convertible notes payable were deemed to be Level 1 as they approximate fair value due to their short term nature.
Note 6 - Convertible Notes
During the years ended December 31, 2013 and 2012, the Company has issued convertible notes in the amount of $210,000 and $300,000, respectively. The convertible notes accrue interest at annual interest rates of 6% and mature within six months of original note issuance dates of December 5, 2012, March 5, 2013 and March 24, 2013 respectively and may be prepaid without penalty at any time.
The Convertible Notes are also convertible at any time at the option of the holder into shares of the Company’s common stock at a conversion price ranging from 4% to 8% of the total shares of common stock on a fully diluted basis. Therefore, since this embedded conversion feature provides for the settlement of this convertible note with shares of common stock at a rate which is variable in nature, this embedded conversion feature must be classified and accounted for as a derivative financial instrument.
| 93 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 6 - Convertible Notes (continued)
Generally accepted accounting principles require that:
| a) | Derivative financial instruments be recorded at their fair value on the date of issuance and then adjusted to fair value at each subsequent balance sheet date with any change in fair value reported in the statement of operations; and |
| b) | The classification of derivative financial instruments be reassessed as of each balance sheet date and, if appropriate, be reclassified as a result of events during the reporting period then ended. |
The fair value of the embedded conversion feature aggregated to approximately $315,200 and $51,800 for the years ended December 31, 2013 and 2012, respectively, which has been recorded as a debt discount. The debt discount will be amortized over the earlier of (i) the term of the debt or (ii) conversion of the debt, using the straight-line method which approximates the interest method. The amortization of debt discount included as a component of interest expense in the statements of operations for the years ended December 31, 2013 and 2012 was approximately $358,370 and $8,600, respectively.
On November 11, 2013, the Company issued 2,699,880 common shares for settlement of approximately $511,200 of convertible notes which was 770,283 shares in excess of the contractual amount in the conversion provision. The fair value of the shares issued by the Company in excess was $309,890. Accordingly, the Company recorded a charge of this amount to interest expense. (see Note 10). For the year ended December 31, 2013, the note holders forgave approximately $15,300 of interest expense for no consideration and the Company recorded the charge to interest expense as a contribution to equity.
| 94 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 7 – Accounts Payable and Accrued Expenses
As of December 31, 2013 and 2012 accrued expenses consisted of the following:
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Accrued consulting expenses and related expenses | $ | 436,348 | $ | — | ||||
| Professional fees | 109,057 | 30,477 | ||||||
| Accrued payroll | 12,944 | 17,700 | ||||||
| $ | 558,349 | $ | 48,177 | |||||
Note 8 - Consulting Agreements - Related Party
On October 3, 2011, the Company entered into a definitive license agreement and an exclusive option agreement to negotiate a commercial license with Yeda Research and Development Company Ltd. (“Yeda”), a founder and shareholder of the Company. Yeda is the technology transfer and commercial arm of the Weizmann Institute of Science, for research conducted at the Weitzman Institute of Science for an invention comprising methods of bone marrow transplantation and cell therapy utilizing Veto cells. The option to negotiate originally expired on June 20, 2014 and was extended to September 1, 2014.
Under the terms of the agreement, Yeda granted the Company an exclusive worldwide license under the licensed information and the patents for the development, manufacture and sale of the products. In consideration for the grant of the license, the Company has paid and will pay Yeda: (1) on the date of signature of the agreement $210,000; (2) an annual license fee for 3 years in the amount of $800,000 for the period until October 3, 2014; (3) A non-refundable and non-creditable license fee of $50,000 per year during the terms of the agreement, commencing on the first day after the date of termination or expiry of the research period; (4) a royalty of 4% of net future sales by or on behalf of the Company or any sub licensees.
If the Company fails to achieve any of the milestones by the dates set forth in the agreement, Yeda is entitled to terminate the license upon written notice to the Company. Either Yeda or the Company may terminate the agreement and the license after the commitment of a material breach by the other party and in certain other instances as detailed in the agreement.
| 95 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 8 - Consulting Agreements - Related Party (continued)
For the years ended December 31, 2013 and 2012, the Company has recorded a charge to operations of approximately $784,000 and $796,000, respectively, for this consulting arrangement as a component of research and development expense. As of December 31, 2013, approximately $441,700 has been accrued and is payable.
Note 9 - Related Parties
During the year ended December 31, 2013, the officers and founders of the Company contributed approximately $77,000 of services for no consideration.
Note 10 - Stockholders’ Deficit
Common Stock
As of December 31, 2013 the Company is authorized to issue up to 50,000,000 shares of common stock with $0.001 par value. The ordinary shares confer upon their holders the right to participate and vote in general stockholders’ meetings of the Company and to share in the distribution of dividends, if any, declared by the Company.
During the year ended December 31, 2012, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the Company issued 2,250,000 shares in exchange for $810,000 of cash proceeds.
During the year ended December 31, 2013, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company the amount of $551,497 in exchange for 735,327 of common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share.
| 96 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 10 - Stockholders’ Deficit (continued)
Common Stock (continued)
The warrants carried provisions that were deemed to be “down round” price protection features. The Company reclassified approximately $231,200 for the fair value of the warrants to derivative liabilities which will be marked to market at each reporting period.
On November 11, 2013, the Company issued 2,699,880 common shares for settlement of approximately $511,200 of convertible notes which was 770,283 shares in excess of the contractual amount in the conversion provision. The fair value of the shares issued by the Company in excess was $309,890. Accordingly, the Company recorded a charge of this amount to interest expense. (see Note 6).
| 97 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 11 - Warrants
Pursuant to an agreement, on November 11, 2013, certain founders returned to the Company 2,043,835 shares of common stock in exchange for 2,043,835 of warrants. The warrants have an exercise price of $0.001 per share and have a life of 7 years. In addition, the warrants have a cashless exercise provision and were fully vested on the date of the grant. The Company determined that the exchange was a modification of a previously granted equity instrument whereby a gain or loss is calculated as the incremental difference between the fair value of the warrants and the fair value of the shares of common stock returned. The fair value of the warrants was determined using the Black-Scholes fair value model. Since the fair value of the warrants was less than the common stock returned, no incremental charge was required to be recorded for the year ended December 31, 2013. Subsequent to the returning of the shares to the Company retired such shares.
Note 12 - Income Taxes
The income tax provision (benefit) for the years ended December 31, 2013 and 2012 consists of the following:
| 2013 | 2012 | |||||||
| Current | $ | — | $ | — | ||||
| Deferred | ||||||||
| Federal (Israel) | (445,913 | ) | (233,418 | ) | ||||
| State | — | — | ||||||
| Foreign | — | — | ||||||
| Change in Valuation Allowance | 445,913 | 233,418 | ||||||
| Income tax provision | $ | — | $ | — | ||||
| 98 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 12 - Income Taxes (continued)
The reconciliation between the Israeli income tax rate and the Company’s effective rate for the years ended December 31, 2013 and 2012 is as follows:
| 2013 | 2012 | |||||||
| Statutory tax rate | (25.0 | )% | (25.0 | )% | ||||
| Change in valuation allowance | 25.0 | 25.0 | % | |||||
| — | % | — | % | |||||
As of December 31, 2013 and 2012, the Company’s deferred tax assets consisted of the effects of temporary differences attributable to the following:
| 2013 | 2012 | |||||||
| Net loss carry forwards | 679,330 | 233,418 | ||||||
| Total Deferred Tax Asset | 679,330 | 233,418 | ||||||
| Less: valuation allowance | (679,330 | ) | (233,418 | ) | ||||
| Total | $ | — | $ | — | ||||
As of December 31, 2013 and 2012, the Company had approximately $2,717,000 and $934,000, respectively, of net operating loss (“NOL”) carryovers available to offset future taxable income. Utilized losses may be carried over indefinitely.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. In assessing the realization of deferred tax assets, management considers, whether it is “more likely than not”, that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which temporary differences representing net future deductible amounts become deductible.
| 99 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 12 - Income Taxes (continued)
ASC 740, “Income Taxes” requires that a valuation allowance be established when it is “more likely than not” that all, or a portion of, deferred tax assets will not be realized. A review of all available positive and negative evidence needs to be considered, including the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies. After consideration of all the information available, management believes that uncertainty exists with respect to future realization of its deferred tax assets and has, therefore, established a full valuation allowance as of December 31, 2013 and 2012. As of December 31, 2013 and December 31, 2012, the change in valuation allowance was $445,913 and $233,418 respectively.
ASC 740 also clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements and prescribes a recognition threshold and measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more likely than not to be sustained upon examination by taxing authorities. ASC 740 also provides guidance on de-recognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. The Company is required to file income tax returns in Israel. Based on the Company’s evaluation, it has been concluded that there are no material uncertain tax positions requiring recognition in the Company’s financial statements for the years ended December 31, 2013 and 2012.
The Company’s policy for recording interest and penalties associated with unrecognized tax benefits is to record such interest and penalties as interest expense and as a component of selling, general and administrative expense, respectively. There were no amounts accrued for interest or penalties for the years ended December 31, 2013 and 2012. Management does not expect any material changes in its unrecognized tax benefits in the next year. The Company also files tax returns in Israel and is subject to examination by tax authorities beginning with the year ended December 31, 2011.
Note 13 - Commitments and Contingencies
Litigation
Certain conditions may exist as of the date the financial statements are issued, which may result in a loss to the Company, but which will only be resolved when one or more future events occur or fail to occur. The Company assesses such contingent liabilities, and such assessment inherently involves an exercise of judgment.
| 100 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 13 - Commitments and Contingencies (continued)
Litigation (continued)
In assessing loss contingencies related to legal proceedings that are pending against the Company, or unasserted claims that may result in such proceedings, the Company evaluates the perceived merits of any legal proceedings or unasserted claims, as well as the perceived merits of the amount of relief sought or expected to be sought therein.
If the assessment of a contingency indicates that it is probable that a material loss has been incurred and the amount of the liability can be estimated, then the estimated liability would be accrued in the Company’s financial statements. If the assessment indicates that a potentially material loss contingency is not probable, but is reasonably possible, or is probable but cannot be estimated, then the nature of the contingent liability and an estimate of the range of possible losses, if determinable and material, would be disclosed.
Loss contingencies considered remote are generally not disclosed, unless they involve guarantees, in which case the guarantees would be disclosed. There can be no assurance that such matters will not materially and adversely affect the Company’s business, financial position, and results of operations or cash flows. As of December 31, 2013 and 2012, the Company has not accrued any amounts for contingencies.
Credit Risk
Financial instruments that subject the Company to credit risk consist principally of cash deposits at financial institutions in Israel. These amounts are uninsured, however, Company believes that credit risk is limited because the Company routinely assesses the financial strength these institutions. As of December 31, 2013 and 2012, the Company has not experienced any losses and believes it is not exposed to any significant credit risk from cash.
Severance Pay Fund
Assets held for employees’ severance payments represent contributions to insurance policies that are recorded at their current redemption value. The deposited funds include profits and losses accumulated up to the balance sheet date. The deposited funds may be withdrawn only upon the fulfillment of the obligation pursuant to Israel’s Severance Pay Law or labor agreements.
| 101 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
Note 13 - Commitments and Contingencies (continued)
Accrued Severance Pay
Under Israeli law and labor agreements the Company is required to pay severance payments to each employee who was employed by the Company for over one year and has been terminated by the Company or resigned under certain specified circumstances. The Company’s liability for the severance payments is partially covered by deposits with insurance companies in the name of the employee. As of December 31, 2013 and 2012, the Company has accrued $0 and $8,225 for severance pay which has been included in accounts payable and accrued expenses
Note 14 - Subsequent Events
On January 29, 2014, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company the amount of $245,000 in exchange for 326,667 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. The warrants were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features. The Company reclassified $119,100 for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
On January 31, 2014, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company the amount of $56,250 in exchange for 75,000 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. The warrants were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features. The Company reclassified $27,300 for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
On February 3, 2014, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company the amount of $62,500 in exchange for 83,333 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. The warrants were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features. The Company reclassified $30,400 for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
| 102 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
On February 4, 2014, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company the amount of $63,000 in exchange for 84,000 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. The warrants were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features. The Company reclassified $30,600 for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
On February 6, 2014, the Company entered into an investment agreement with an investor. Pursuant to the agreement, the investor contributed to the Company the amount of $75,000 in exchange for 100,000 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. The warrants were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features. The Company reclassified $36,500 for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
On February 7, 2014, the Company entered into an investment agreement with an investor. Pursuant to the agreement, the investor contributed to the Company the amount of $52,500 in exchange for 70,000 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. The warrants were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features. The Company reclassified $19,100 for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
On February 27, 2014, the Company entered into an investment agreement with
an investor. Pursuant to the agreement, the investor contributed to the Company the amount of $50,000 in exchange for 66,667 common
shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder
to purchase 1 share of common stock at an exercise price of $0.75 per share. The warrants were fully vested on the date of the
grant. The warrants carried provisions that were deemed to be down round price protection features. The Company reclassified $30,800
for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
| 103 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
On March 6, 2014, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company the amount of $1,643,496 in exchange for 2,191,328 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share and were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features. The Company reclassified $804,000 for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
On April 2, 2014, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company the amount of $182,100 in exchange for 242,800 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share and were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features.
On April 3, 2014, the Company entered into an investment agreement with two investors. Pursuant to the agreement, the investors contributed to the Company the amount of $55,000 in exchange for 73,333 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share. The warrants carried provisions that were deemed to be down round price protection features.
On April 4, 2014, the Company entered into an investment agreement with two investors. Pursuant to the agreement, the investors contributed to the Company the amount of $317,900 in exchange for 423,867 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share and were fully vested on the date of the grant.. The warrants carried provisions that were deemed to be down round price protection features.
| 104 |
| CELL SOURCE LIMITED |
| NOTES TO FINANCIAL STATEMENTS |
| FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012 |
On April 7, 2014, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company the amount of $265,250 in exchange for 353,666 common shares. Each unit was sold for $0.75 and consisted of 1 share of common stock and 1 five-year warrant, which entitles the holder to purchase 1 share of common stock at an exercise price of $0.75 per share and were fully vested on the date of the grant. The warrants carried provisions that were deemed to be down round price protection features.
| 105 |
CELL SOURCE, INC.
CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE THREE AND SIX MONTHS ENDED JUNE 30, 2014 AND 2013
| 106 |
CONTENTS
| Condensed Consolidated Financial Statements | |
| Condensed Consolidated Balance Sheets | 108 |
| Condensed Consolidated Statements of Operations | 109 |
| Condensed Consolidated Statement of Stockholders’ Deficit | 110 |
| Condensed Consolidated Statements of Cash Flows | 111 |
| Notes to Condensed Consolidated Financial Statements | 112-120 |
| 107 |
CONDENSED CONSOLIDATED BALANCE SHEETS
| June 30, | December 31, | |||||||
| 2014 | 2013 | |||||||
| (Unaudited) | ||||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash | $ | 889,332 | $ | 28,878 | ||||
| Prepaid expenses | 135,000 | - | ||||||
| Other current assets | 59,564 | 63,337 | ||||||
| Total Current Assets | 1,083,896 | 92,215 | ||||||
| Property and equipment, net | 2,557 | - | ||||||
| Total Assets | $ | 1,086,453 | $ | 92,215 | ||||
| Liabilities and Stockholders’ Deficiency | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | 70,626 | $ | 116,649 | ||||
| Accounts payable and accrued expenses - related party | 216,035 | 441,700 | ||||||
| Derivative liabilities | 2,476,800 | 231,200 | ||||||
| Total Current Liabilities | 2,763,461 | 789,549 | ||||||
| Commitments and contingencies (Note 8) | - | - | ||||||
| Stockholders’ Deficiency: | ||||||||
| Preferred stock, $0.001 par value; 10,000,000 shares authorized; no shares issued and outstanding at June 30, 2014 and December 31, 2013 | - | - | ||||||
| Common stock, $0.001 par value; 200,000,000 shares authorized; 23,245,923 and 14,155,262 shares issued and outstanding at June 30, 2014 and December 31, 2013, respectively | 23,246 | 14,155 | ||||||
| Additional paid-in capital | 4,047,077 | 3,229,522 | ||||||
| Accumulated deficit | (5,747,331 | ) | (3,941,011 | ) | ||||
| Total Stockholders’ Deficiency | (1,677,008 | ) | (697,334 | ) | ||||
| Total Liabilities and Stockholders’ Deficiency | $ | 1,086,453 | $ | 92,215 | ||||
See Notes to these Condensed Consolidated Financial Statements
| 108 |
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(Unaudited)
| For The | For The | |||||||||||||||
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2014 | 2013 | 2014 | 2013 | |||||||||||||
| Revenues | $ | - | $ | - | $ | - | $ | - | ||||||||
| Operating Expenses | ||||||||||||||||
| Research and development | 220,995 | 87,436 | 545,412 | 193,806 | ||||||||||||
| Research and development - related party | 398,845 | 329,753 | 611,203 | 402,629 | ||||||||||||
| Selling, general and administrative | 397,338 | 79,100 | 633,305 | 127,853 | ||||||||||||
| Total Operating Expenses | 1,017,178 | 496,289 | 1,789,920 | 724,288 | ||||||||||||
| Loss From Operations | (1,017,178 | ) | (496,289 | ) | (1,789,920 | ) | (724,288 | ) | ||||||||
| Other (Expense) Income | ||||||||||||||||
| Interest expense | - | (362,001 | ) | - | (396,534 | ) | ||||||||||
| Change in fair value of derivative liabilities | 31,100 | 54,300 | (16,400 | ) | 64,500 | |||||||||||
| Total Other Income (Expense) | 31,100 | (307,701 | ) | (16,400 | ) | (332,034 | ) | |||||||||
| Net Loss | $ | (986,078 | ) | $ | (803,990 | ) | $ | (1,806,320 | ) | $ | (1,056,322 | ) | ||||
| Net Loss Per Share | ||||||||||||||||
| - Basic and Diluted | $ | (0.05 | ) | $ | (0.06 | ) | $ | (0.10 | ) | $ | (0.08 | ) | ||||
| Weighted Average Number of | ||||||||||||||||
| Common Shares Outstanding | ||||||||||||||||
| - Basic and Diluted | 20,228,764 | 12,763,818 | 18,774,759 | 12,763,818 | ||||||||||||
See Notes to these Condensed Consolidated Financial Statements
| 109 |
CONDENSED CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDERS’ DEFICIENCY
FOR THE SIX MONTHS ENDED JUNE 30, 2014
(Unaudited)
| Additional | ||||||||||||||||||||
| Common Stock | Paid-In | Accumulated | ||||||||||||||||||
| Shares | Amount | Capital | Deficit | Total | ||||||||||||||||
| Balance - December 31, 2013 | 14,155,262 | $ | 14,155 | $ | 3,229,522 | $ | (3,941,011 | ) | $ | (697,334 | ) | |||||||||
| Issuance of common stock and warrants for cash, net [1] | 4,090,661 | 4,091 | 3,008,755 | - | 3,012,846 | |||||||||||||||
| Reclassification of detachable warrants to derivative liabilities | - | - | (1,499,000 | ) | - | (1,499,000 | ) | |||||||||||||
| Shares retained by public company stockholders in Share Exchange | 5,000,000 | 5,000 | (735,200 | ) | - | (730,200 | ) | |||||||||||||
| Stock-based compensation | - | - | 43,000 | - | 43,000 | |||||||||||||||
| Net loss | - | - | - | (1,806,320 | ) | (1,806,320 | ) | |||||||||||||
| Balance - June 30, 2014 | 23,245,923 | $ | 23,246 | $ | 4,047,077 | $ | (5,747,331 | ) | $ | (1,677,008 | ) | |||||||||
[1] Net of $55,150 of issuance costs.
See Notes to these Condensed Consolidated Financial Statements
| 110 |
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Unaudited)
| For The Six Months Ended | ||||||||
| June 30, | ||||||||
| 2014 | 2013 | |||||||
| Cash Flows From Operating Activities | ||||||||
| Net loss | $ | (1,806,320 | ) | $ | (1,056,322 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Accretion of debt discount | - | 384,034 | ||||||
| Interest expense | - | 12,500 | ||||||
| Change in fair value of derivative liabilities | 16,400 | (64,500 | ) | |||||
| Depreciation | 25 | - | ||||||
| Stock-based compensation | 43,000 | - | ||||||
| Contribution of services by officers | - | 76,990 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses | (135,000 | ) | - | |||||
| Other assets | 3,773 | 26,418 | ||||||
| Accounts payable and accrued expenses | (271,688 | ) | 280,790 | |||||
| Net Cash Used in Operating Activities | (2,149,810 | ) | (340,090 | ) | ||||
| Cash Flows From Investing Activities | ||||||||
| Purchase of propery and equipment | (2,582 | ) | - | |||||
| Net Cash Used in Investing Activities | (2,582 | ) | - | |||||
| Cash Flows From Financing Activities | ||||||||
| Proceeds from issuance of convertible note | - | 210,000 | ||||||
| Proceeds from issuance of common stock and warrants, net [1] | 3,012,846 | - | ||||||
| Net Cash Provided by Financing Activities | 3,012,846 | 210,000 | ||||||
| Net Increase (Decrease) In Cash | 860,454 | (130,090 | ) | |||||
| Cash - Beginning | 28,878 | 161,323 | ||||||
| Cash - Ending | $ | 889,332 | $ | 31,233 | ||||
| Supplemental Disclosures of Cash Flow Information: | ||||||||
| Non-cash investing and financing transactions: | ||||||||
| Reclassification of warrants to derivative liabilities | $ | 1,499,000 | $ | - | ||||
| Reclassification of embedded conversion options to derivative liabilities | $ | - | $ | 315,200 | ||||
[1] Net of $55,150 of issuance costs.
See Notes to these Condensed Consolidated Financial Statements
| 111 |
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 1 – Organization, Operations and Basis of Presentation
Basis of Presentation
The accompanying unaudited condensed consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for interim financial information. Accordingly, they do not include all of the information and footnotes required by U.S. GAAP for complete financial statements. In the opinion of management, such statements include all adjustments (consisting only of normal recurring items) which are considered necessary for a fair presentation of the condensed consolidated financial position of Cell Source, Inc. (“CSI” or the “Company”), formerly known as Ticket To See, Inc. (“TTSI”), as of June 30, 2014 and the condensed consolidated results of its operations and cash flows for the three and six months ended June 30, 2014 and 2013. The results of operations for the three and six months ended June 30, 2014 are not necessarily indicative of the operating results for the full year. It is recommended that these condensed consolidated financial statements be read in conjunction with the financial statements and related disclosures of Cell Source Limited for the year ended December 31, 2013 which were included in the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission (“SEC”) on July 1, 2014.
Organization and Operations
The Company is a Nevada corporation formed on June 6, 2012 under the name TTSI. Prior to the Share Exchange (as defined below), the Company did not have any significant assets or operations.
The Company is the parent company of Cell Source Limited, which was founded in Israel in 2011 in order to commercialize a suite of inventions relating to certain cancer treatments. Cell Source Limited’s target indications include treatment of lymphoma, multiple myeloma and BCLL (which is a common form of leukemia), facilitating transplantation acceptance (initially bone marrow transplantation and subsequently organ transplantation) and ultimately treating a variety of non-malignant diseases. Cell Source Limited’s lead prospective product is its patented Veto-Cell immune system management technology, which is an immune tolerance biotechnology that enables the selective blocking of immune responses. Cell Source Limited’s Veto-Cell immune system management technology is based on technologies patented, owned, and licensed to Cell Source Limited by Yeda Research and Development Company Limited, an Israeli corporation (“Yeda”).
Share Exchange and Reorganization
On May 7, 2014, the Board of Directors and the majority stockholder of TTSI adopted resolutions approving an amendment (the “Amendment”) of the Company’s Articles of Incorporation to increase the number of authorized shares. Prior to the Amendment, the authorized shares of the Company consisted of 75,000,000 shares of common stock, $0.001 par value. The Amendment was filed with the Secretary of State of the State of Nevada on May 20, 2014, which increased the number of shares of common stock that the Company is authorized to issue from 75,000,000 shares to 200,000,000 shares. The Company also authorized 10,000,000 shares of preferred stock, par value $0.001, for designation in one or more series, with such designations, preferences and relative, participating, optional, or other special rights and qualifications, limitations, or restrictions thereof, as may, from time to time, be adopted by the Company’s Board of Directors.
On June 23, 2014, the majority stockholder of TTSI adopted resolutions approving an amendment of the Company’s Articles of Incorporation to change the name of the corporation from Ticket to See, Inc. to Cell Source, Inc. The Amendment was filed with the Secretary of State of the State of Nevada on June 23, 2014, which changed the name of the corporation from Ticket to See, Inc. to Cell Source, Inc., effective June 26, 2014. In connection with the name change, the trading symbol of the Company’s common stock was changed from TTSE to CLCS.
On June 27, 2014, CSI issued five-year warrants to purchase an aggregate of 2,000,000 shares of common stock at a price of $0.75 per share to consultants in exchange for consulting services previously provided to the Company.
| 112 |
CELL SOURCE, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 1 – Organization, Operations and Basis of Presentation – Continued
Share Exchange and Reorganization – Continued
On June 30, 2014 (the “Closing Date”), CSI entered into and closed a Share Exchange Agreement (the “Share Exchange Agreement”) with Cell Source Limited and 100% of the shareholders of Cell Source Limited (the “CSL Shareholders”) whereby Cell Source Limited became the wholly-owned subsidiary of CSI (the “Share Exchange”), and whereby the CSL Shareholders, transferred to the Company all 18,245,923 outstanding shares of Cell Source Limited’s ordinary shares (“CSL Ordinary Shares”) in exchange for an aggregate of 18,245,923 newly issued shares of the Company’s Common Stock, par value $0.001 per share (the “Company Common Stock” or the “Common Stock”). The aggregate of 18,245,923 shares of newly issued Company Common Stock represents 78.5% of the 23,245,923 outstanding shares of Company Common Stock following the Closing Date. In addition, outstanding five (5) year warrants to acquire 4,859,324 CSL Ordinary Shares at an exercise price of $0.75 per share (the “CSL Warrants”) were exchanged for newly issued warrants to purchase shares of Company Common Stock at $0.75 per share (the “Company Warrants”), which Company Warrants contain substantially similar terms as the CSL Warrants. In addition, outstanding seven-year warrants to acquire 2,043,835 CSL Ordinary Shares at $0.001 per share were exchanged for warrants to purchase shares of Company Common Stock at $0.001 per share (the “Researcher Company Warrants”), which Researcher Company Warrants contain substantially similar terms as their warrants to acquire CSL Ordinary Shares. The aggregate of 6,903,159 Company Warrants and Researcher Company Warrants represents 77.5% of the outstanding warrants to purchase Common Stock of the Company following the Closing Date.
For accounting purposes, the Share Exchange will be treated as a recapitalization of the Company, the accounting acquirer, because the Company shareholders own the majority of CSI’s outstanding common stock following the transaction and exercise significant influence over the operating and financial policies of the consolidated entity. CSI was a non-operating company prior to the share exchange. Pursuant to Securities and Exchange Commission rules, the merger or acquisition of a private operating company into a non-operating public company with nominal net assets is considered a capital transaction in substance, rather than a business combination.
Note 2 – Going Concern
The Company has not generated any revenues, has recurring net losses, a working capital deficiency as of June 30, 2014 and December 31, 2013 of approximately $1,680,000 and $697,000, respectively, and used cash in operations of approximately $2,150,000 and $340,000 for the six months ended June 30, 2014 and 2013, respectively. In addition, as of June 30, 2014, the Company had an accumulated deficit of approximately $5,747,000. These conditions raise substantial doubt about the Company’s ability to continue as a going concern.
These unaudited condensed consolidated financial statements have been prepared on the going concern basis, which assumes the realization of assets and liquidation of liabilities in the normal course of operations. The ability of the Company to continue its operations is dependent on management’s plans, which include the raising of capital through debt and/or equity markets with some additional funding from other traditional financing sources, including term notes, until such time that funds provided by operations are sufficient to fund working capital requirements. The Company may need to incur additional liabilities with certain related parties to sustain the Company’s existence. If the Company were not to continue as a going concern, it would likely not be able to realize its assets at values comparable to the carrying value or the fair value estimates reflected in the balances set out in the preparation of the unaudited condensed consolidated financial statements.
There can be no assurances that the Company will be successful in generating additional cash from equity or other sources to be used for operations. The unaudited condensed consolidated financial statements do not include any adjustments relating to the recoverability of assets and classification of assets and liabilities that might be necessary. Based on the Company’s current funding levels, the Company expects to be able to fund its operations through November 2014. Should the Company not be successful in obtaining the necessary financing to fund its operations, the Company would need to curtail certain or all operational activities and/or contemplate the sale of its assets if necessary.
| 113 |
CELL SOURCE, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 3 – Significant Accounting Policies
Principles of Consolidation
For June 30, 2014 and forward, the Company’s financial statements are consolidated and include the accounts of CSI and Cell Source Limited. All significant intercompany transactions have been eliminated in the consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period.
Management bases its estimates on historical experience and on various assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. The most significant estimates, among other things, are used in accounting for allowances for deferred income taxes, contingencies, as well as the recording and presentation of its common stock and related warrant issuances. Estimates and assumptions are periodically reviewed and the effects of any material revisions are reflected in the financial statements in the period that they are determined to be necessary. Actual results could differ from those estimates and assumptions.
Research and Development Costs
Research and development costs are expensed as they are incurred and consist of salaries, benefits and other personnel related costs, fees paid to consultants, clinical trials and related clinical manufacturing costs, license and milestone fees, and facilities and overhead costs.
Loss Per Share
The Company computes basic net loss per share by dividing net loss per share available to common stockholders by the weighted average number of common shares outstanding for the period and excludes the effects of any potentially dilutive securities. Diluted earnings per share, if presented, would include the dilution that would occur upon the exercise or conversion of all potentially dilutive securities into common stock using the “treasury stock” and/or “if converted” methods as applicable. For the three and six months ended June 30, 2014, warrants to purchase 2,043,835 shares of common stock were included in the loss per share denominator because their exercise price was determined to be nominal.
The following securities are excluded from the calculation of weighted average dilutive common shares because their inclusion would have been anti-dilutive:
| June 30, | ||||||||
| 2014 | 2013 | |||||||
| Warrants | 6,859,324 | - | ||||||
| Convertible notes | - | 2,297,487 | ||||||
| Total | 6,859,324 | 2,297,487 | ||||||
Stock-Based Compensation
The Company measures the cost of services received in exchange for an award of equity instruments based on the fair value of the award. For non-employees, the fair value of the award is generally re-measured on financial reporting dates and vesting dates until the service period is complete. The fair value amount is then recognized over the period the services are required to be provided in exchange for the award, usually the vesting period. Because the Company’s common stock historically was not actively traded on a public market, the fair value of the Company’s restricted equity instruments are estimated based on the historical observations of cash prices paid for the Company’s common stock.
| 114 |
CELL SOURCE, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 3 – Significant Accounting Policies - Continued
Derivative Financial Instruments
The fair value of an embedded conversion option that is convertible into a variable amount of shares and warrants that include price protection reset provision features are deemed to be “down-round protection” and, therefore, do not meet the scope exception for treatment as a derivative under ASC 815 “Derivatives and Hedging”, since “down-round protection” is not an input into the calculation of the fair value of the conversion option and warrants and cannot be considered “indexed to the Company’s own stock” which is a requirement for the scope exception as outlined under ASC 815.
The accounting treatment of derivative financial instruments requires that the Company record the embedded conversion option and warrants at their fair values as of the inception date of the agreement and at fair value as of each subsequent balance sheet date. Any change in fair value is recorded as non-operating, non-cash income or expense for each reporting period at each balance sheet date. The Company reassesses the classification of its derivative instruments at each balance sheet date. If the classification changes as a result of events during the period, the contract is reclassified as of the date of the event that caused the reclassification.
The Black-Scholes option valuation model was used to estimate the fair value of the warrants and conversion options. The model includes subjective input assumptions that can materially affect the fair value estimates. The expected volatility is estimated based on the most recent historical period of time equal to the weighted average life of the warrants or conversion options.
Conversion options are recorded as debt discount and are amortized as interest expense over the life of the underlying debt instrument.
Reclassifications
Certain prior period amounts have been reclassified for comparative purposes to conform to the fiscal 2014 presentation. These reclassifications have no impact on the previously reported net loss.
Recent Accounting Standards
In June 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-10, “Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation.” This ASU removes the definition of a development stage entity from the ASC, thereby removing the financial reporting distinction between development stage entities and other reporting entities from GAAP. In addition, the ASU eliminates the requirements for development stage entities to (1) present inception-to-date information in the statements of operations, cash flows, and stockholders’ equity, (2) label the financial statements as those of a development stage entity, (3) disclose a description of the development stage activities in which the entity is engaged, and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. This ASU is effective for annual reporting periods beginning after December 15, 2014, and interim periods therein. Early adoption is permitted. The Company elected to adopt this ASU effective with its Current Report on Form 8-K filed with SEC on July 1, 2014 and the adoption resulted in the removal of previously required development stage disclosures.
In June 2014, the FASB issued ASU No. 2014-12, “Compensation - Stock Compensation (Topic 718): Accounting for Share-Based Payments When the Terms of an Award Provide that a Performance Target Could be Achieved after the Requisite Service Period,” (“ASU 2014-12”). The amendments in ASU 2014-12 require that a performance target that affects vesting and that could be achieved after the requisite service period be treated as a performance condition. A reporting entity should apply existing guidance in Accounting Standards Codification Topic No. 718, “Compensation - Stock Compensation” as it relates to awards with performance conditions that affect vesting to account for such awards. The amendments in ASU 2014-12 are effective for annual periods and interim periods within those annual periods beginning after December 15, 2015. Early adoption is permitted. Entities may apply the amendments in ASU 2014-12 either: (a) prospectively to all awards granted or modified after the effective date; or (b) retrospectively to all awards with performance targets that are outstanding as of the beginning of the earliest annual period presented in the financial statements and to all new or modified awards thereafter. The Company does not anticipate that the adoption of this standard will have a material impact on its condensed consolidated financial statements.
In August 2014, the FASB issued a new accounting standard which requires management to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern for each annual and interim reporting period. If substantial doubt exists, additional disclosure is required. This new standard will be effective for the Company for annual and interim periods beginning after December 15, 2016. Early adoption is permitted. The Company expects to adopt this new standard for the fiscal year ending December 31, 2014 and the Company will continue to assess the impact on its consolidated financial statements.
The Company has implemented all new accounting standards that are in effect and may impact its financial statements and does not believe that there are any other new accounting standards that have been issued that might have a material impact on its financial position or results of operations.
| 115 |
CELL SOURCE, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 4 - Fair Value
The Company determines the estimated fair value of amounts presented in these condensed consolidated financial statements using available market information and appropriate methodologies. However, considerable judgment is required in interpreting market data to develop the estimates of fair value. The estimates presented in the financial statements are not necessarily indicative of the amounts that could be realized in a current exchange between buyer and seller. The use of different market assumptions and/or estimation methodologies may have a material effect on the estimated fair value amounts. These fair value estimates were based upon pertinent information available as of June 30, 2014 and 2013, plus December 31, 2013 and 2012, and, as of those dates, the carrying value of all amounts approximates fair value.
The Company has categorized its assets and liabilities at fair value based upon the following fair value hierarchy:
Level 1 - Inputs use quoted prices in active markets for identical assets or liabilities that the Company has the ability to access.
Level 2 - Inputs use directly or indirectly observable inputs. These inputs include quoted prices for similar assets and liabilities in active markets as well as other inputs such as interest rates and yield curves that are observable at commonly quoted intervals.
Level 3 - Inputs are unobservable inputs, including inputs that are available in situations where there is little, if any, market activity for the related asset or liability.
In instances where inputs used to measure fair value fall into different levels in the above fair value hierarchy, fair value measurements in their entirety are categorized based on the lowest level input that is significant to the valuation. The Company’s assessment of the significance of particular inputs to these fair measurements requires judgment and considers factors specific to each asset or liability.
Both observable and unobservable inputs may be used to determine the fair value of positions that are classified within the Level 3 category. As a result, the unrealized gains and losses for assets within the Level 3 category presented in the tables below may include changes in fair value that were attributable to both observable (e.g., changes in market interest rates) and unobservable (e.g., changes in historical company data) inputs.
The following table summarizes the valuation of the Company’s derivatives by the above fair value hierarchy levels as of June 30, 2014 and December 31, 2013 using quoted prices in active markets for identical assets (Level 1), significant other observable inputs (Level 2), and significant unobservable inputs (Level 3):
| Quoted Prices | ||||||||||||||||
| In Active | Significant | |||||||||||||||
| Markets for | Other | Significant | ||||||||||||||
| Identical | Observable | Unobservable | ||||||||||||||
| Liabilities | Inputs | Inputs | ||||||||||||||
| Total | (Level 1) | (Level 2) | (Level 3) | |||||||||||||
| Warrant liability | $ | 2,476,800 | $ | - | $ | - | $ | 2,476,800 | ||||||||
| Balance - June 30, 2014 | $ | 2,476,800 | $ | - | $ | - | $ | 2,476,800 | ||||||||
| Warrant liability | $ | 231,200 | $ | - | $ | - | $ | 231,200 | ||||||||
| Balance - December 31, 2013 | $ | 231,200 | $ | - | $ | - | $ | 231,200 | ||||||||
Financial assets are considered Level 3 when their fair values are determined using pricing models, discounted cash flow methodologies or similar techniques and at least one significant model assumption or input is unobservable. The Company’s Level 3 liabilities shown in the above table consist of warrants with “down-round protection”, as the Company is unable to determine if it will have sufficient authorized common stock to settle such arrangements. Earlier in 2013, the Company’s Level 3 liabilities consisted of conversion options with “down-round protection”.
| 116 |
CELL SOURCE, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 4 - Fair Value – Continued
Assumptions utilized in the valuation of Level 3 liabilities are described as follows:
| For the Three Months Ended | For the Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2014 | 2013 | 2014 | 2013 | |||||||||||||
| Risk-free interest rate | 1.62% - 1.65% | 0.05% - 0.08% | 1.46% - 1.73 | % | 0.02% - 0.08% | |||||||||||
| Expected term (years) | 4.33 - 5.00 | 0.00 - 0.24 | 4.33 - 5.00 | 0.00 - 0.50 | ||||||||||||
| Expected volatility | 164% - 166% | 65% | 164% - 168 | % | 65% - 99% | |||||||||||
| Expected dividends | 0.00% | 0.00% | 0.00 | % | 0.00% | |||||||||||
The expected term used is the contractual life of the instrument being valued. Since the Company’s stock has not been publicly traded for a sufficiently long period of time, the Company is utilizing an expected volatility based on a review of the historical volatilities, over a period of time, equivalent to the expected life of the instrument being valued, of similarly positioned public companies within its industry. The risk-free interest rate was determined from the implied yields from U.S. Treasury zero-coupon bonds with a remaining term consistent with the expected term of the instrument being valued.
The following table provides a summary of the changes in fair value, including net transfers in and/or out, of all Level 3 liabilities measured at fair value on a recurring basis using unobservable inputs during the six months ended June 30, 2014 and 2013:
| 2014 | 2013 | |||||||
| Balance - January 1, | $ | 231,200 | $ | 38,300 | ||||
| Change in fair value of derivative liability | 16,400 | (64,500 | ) | |||||
| Issuance of derivative liability | 2,229,200 | 434,200 | ||||||
| Balance - June 30, | $ | 2,476,800 | $ | 408,000 | ||||
The Company’s significant financial instruments such as cash, accounts payable and accrued expenses were deemed to approximate fair value due to their short term nature.
Note 5 – Accounts Payable and Accrued Expenses
Accounts payable and accrued expenses consisted of the following:
| June 30, | December 31, | |||||||
| 2014 | 2013 | |||||||
| (unaudited) | ||||||||
| Accrued fees and expenses | $ | 29,332 | $ | 103,705 | ||||
| Accrued payroll | 41,294 | 12,944 | ||||||
| Total | $ | 70,626 | $ | 116,649 | ||||
| 117 |
CELL SOURCE, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 6 – Consulting Agreements – Related Party
On October 3, 2011, the Company entered into a definitive license agreement for Veto Cell technology and also an exclusive option agreement to negotiate an additional license for organ regeneration technology with Yeda Research and Development Company Limited (“Yeda”), a founder and shareholder of the Company. Yeda is the technology transfer and commercial arm of the Weizmann Institute of Science, for research conducted at the Weizmann Institute of Science for an invention comprising methods of bone marrow transplantation and cell therapy utilizing Veto-Cells. The evaluation period with respect to the option to license the organ regeneration technology originally expired on October 3, 2012 and has since been extended to September 1, 2014. See Note 9 – Subsequent Events for information regarding an additional extension of the expiration date.
Under the terms of the agreement, Yeda granted the Company an exclusive worldwide license for the licensed information and the patents for the development, manufacture and sale of the products derived therefrom. In consideration for the grant of the license, the Company has paid and will pay Yeda: (1) $210,000 on October 3, 2011; (2) an annual Research budget commitment for 3 years in the amount of $800,000 for the period until October 3, 2014; (3) a non-refundable and non-creditable license fee of $50,000 per year during the terms of the agreement, commencing on the first day after the date of termination or expiry of the research period (which period has not expired and will be extended), (4) a royalty of 4% of net future sales by or on behalf of the Company or any sub licensees.
If the Company fails to achieve any of the milestones by the dates set forth in the agreement, Yeda is entitled to terminate the license upon written notice to the Company. To date, the Company has met all of the milestones and the next milestone in the agreement is October 3, 2016. Either Yeda or the Company may terminate the agreement and the license after the commitment of a material breach by the other party and in certain other instances as detailed in the agreement.
For the three and six months ended June 30, 2014, the Company recorded a charge to operations of approximately $399,000 and $611,000, respectively, for this consulting arrangement. For the three and six months ended June 30, 2013, the Company recorded a charge to operations of approximately $330,000 and $403,000, respectively, for this consulting arrangement. As of June 30, 2014 and December 31, 2013, approximately $216,000 and $442,000 has been accrued and is payable, respectively.
Note 7 – Stockholders’ Deficiency
Common Stock and Warrant Offerings
During the six months ended June 30, 2014, the Company entered into an investment agreement with a group of investors. Pursuant to the agreement, the investors contributed to the Company aggregate net proceeds of $3,012,846 (gross proceeds of $3,067,996 less issuance costs of $55,150) in exchange for 4,090,661 units. Each unit was sold for $0.75 per unit and consisted of one share of common stock and an immediately vested five-year warrant to purchase one share of common stock at an exercise price of $0.75 per share. The warrants carried provisions that were deemed to be “down round” price protection features. As a result, during the six months ended June 30, 2014, the Company reclassified $1,499,000 for the fair value of the warrants to derivative liabilities and will be marked to market for each reporting period.
Stock-Based Compensation
On July 7, 2014, the Board of Directors resolved to issue 100,000 shares of common stock to a service provider in connection with the June 30, 2014 completion of the Share Exchange. The Company recognized the $43,000 value of the award as stock-based compensation expense during the three months ended June 30, 2014, which was the service period. See Note 9 – Subsequent Events for additional information regarding the common stock award.
| 118 |
CELL SOURCE, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 7 – Stockholders’ Deficiency – Continued
Stock Warrants
A summary of the warrant activity during the six months ended June 30, 2014 is presented below:
| Weighted | ||||||||||||||||
| Weighted | Average | |||||||||||||||
| Average | Remaining | |||||||||||||||
| Number of | Exercise | Life | Intrinsic | |||||||||||||
| Warrants | Price | In Years | Value | |||||||||||||
| Outstanding, December 31, 2013 | 2,812,498 | $ | 0.21 | |||||||||||||
| Granted | 6,090,661 | 0.75 | ||||||||||||||
| Exercised | - | - | ||||||||||||||
| Forfeited | - | - | ||||||||||||||
| Outstanding, June 30, 2014 | 8,903,159 | $ | 0.58 | 5.1 | $ | 815,490 | ||||||||||
| Exercisable, June 30, 2014 | 6,859,324 | $ | 0.75 | 4.7 | $ | - | ||||||||||
The following table presents information related to stock warrants at June 30, 2014:
| Warrants Outstanding | Warrants Exercisable | |||||||||||||
| Weighted | ||||||||||||||
| Outstanding | Average | Exercisable | ||||||||||||
| Exercise | Number of | Remaining Life | Number of | |||||||||||
| Price | Warrants | In Years | Warrants | |||||||||||
| $ | 0.001 | 2,043,835 | - | - | ||||||||||
| $ | 0.750 | 6,859,324 | 4.7 | 6,859,324 | ||||||||||
| 8,903,159 | 4.7 | 6,859,324 | ||||||||||||
Note 8 – Commitments and Contingencies
Litigation
Certain conditions may exist as of the date the condensed consolidated financial statements are issued, which may result in a loss to the Company, but which will only be resolved when one or more future events occur or fail to occur. The Company assesses such contingent liabilities, and such assessment inherently involves an exercise of judgment. In assessing loss contingencies related to legal proceedings that are pending against the Company, or unasserted claims that may result in such proceedings, the Company evaluates the perceived merits of any legal proceedings or unasserted claims, as well as the perceived merits of the amount of relief sought or expected to be sought therein.
If the assessment of a contingency indicates that it is probable that a material loss has been incurred and the amount of the liability can be estimated, then the estimated liability would be accrued in the Company’s condensed consolidated financial statements. If the assessment indicates that a potentially material loss contingency is not probable, but is reasonably possible, or is probable but cannot be estimated, then the nature of the contingent liability and an estimate of the range of possible losses, if determinable and material, would be disclosed.
Loss contingencies considered remote are generally not disclosed, unless they involve guarantees, in which case the guarantees would be disclosed. There can be no assurance that such matters will not materially and adversely affect the Company’s business, financial position, and results of operations or cash flows. As of June 30, 2014 and December 31, 2013, the Company has not accrued any amounts for contingencies.
| 119 |
CELL SOURCE, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Note 9 – Subsequent Events
The Company evaluates events that have occurred after the balance sheet date but before the condensed consolidated financial statements are issued. Based upon the evaluation, the Company did not identify any recognized or non-recognized subsequent events that would require adjustment or disclosure in the condensed consolidated financial statements, other than as disclosed below.
Issuance of Common Stock
On July 29, 2014, the Company issued 100,000 shares of common stock to a service provider in order to satisfy a stock-based compensation award. See Note 7 - Stockholders’ Deficiency – Stock-Based Compensation for additional details.
Extension of Evaluation Period of Yeda’s Organ Regeneration Technology
On August 15, 2014, the Company and Yeda executed an amendment to the exclusive option agreement to negotiate a license for organ regeneration technology which extends the evaluation period through December 31, 2015. The Company has been informed that Yeda is currently investigating the scope of its rights and title to the patents and inventions that comprise the organ regeneration technology (see Note 6 – Consulting Agreements – Related Party).
Warrant Cashless Exercise
In September 2014, the Company issued 233,333 shares of common stock in connection with a cashless exercise of a warrant to purchase 400,000 shares of common stock at $0.75 per share.
| 120 |
PROSPECTUS
CELL SOURCE, INC.
9,618,648 SHARES OF COMMON STOCK
[[♦] [♦], 2014]
Until [[♦], 2014] (the 90th day after the date of this Prospectus), all dealers that buy, sell or trade our common stock, whether or not participating in this offering, may be required to deliver a Prospectus. This delivery requirement is in addition to the dealers’ obligation to deliver a Prospectus when acting as underwriters and with respect to unsold allotments or subscriptions.
No dealer, salesperson or other individual has been authorized to give any information or to make any representations not contained in this Prospectus in connection with the offering covered by this Prospectus. If given or made, such information or representations must not be relied upon as having been authorized by us. This Prospectus does not constitute an offer to sell, or a solicitation of an offer to buy the offered securities in any jurisdiction where, or to any person to whom, it is unlawful to make any such offer or solicitation. Neither the delivery of this Prospectus nor any offer or sale made hereunder shall, under any circumstances, create an implication that there has not been any change in the facts set forth in this Prospectus or in our affairs since the date hereof.
| 121 |
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
We are paying all of the selling stockholders’ expenses related to this offering, except that the selling stockholders will pay any applicable underwriting discounts and commissions. The fees and expenses payable by us in connection with this Registration Statement are estimated as follows:
| Securities and Exchange Commission Registration Fee | $ | 782.38 | ||
| Accounting Fees and Expenses | $ | 200,000* | ||
| Legal Fees and Expenses | $ | 50,000* | ||
| Miscellaneous Fees and Expenses | $ | 5,000* | ||
| Total | $ | 255,782.38* |
* Estimate
Item 14. Indemnification of Directors and Officers
Neither our Articles of Incorporation nor Bylaws prevent us from indemnifying our officers, directors and agents to the extent permitted under the Nevada Revised Statute (“NRS”). NRS Section 78.7502 provides that a corporation shall indemnify any director, officer, employee or agent of a corporation against expenses, including attorneys’ fees, actually and reasonably incurred by him in connection with any the defense to the extent that a director, officer, employee or agent of a corporation has been successful on the merits or otherwise in defense of any action, suit or proceeding referred to NRS Section 78.7502(1) or 78.7502(2), or in defense of any claim, issue or matter therein.
NRS Section 78.7502(1) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative, except an action by or in the right of the corporation, by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses, including attorneys’ fees, judgments, fines and amounts paid in settlement actually and reasonably incurred by him in connection with the action, suit or proceeding if he: (a) is not liable pursuant to NRS Section 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful.
NRS Section 78.7502(2) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses, including amounts paid in settlement and attorneys’ fees actually and reasonably incurred by him in connection with the defense or settlement of the action or suit if he: (a) is not liable pursuant to NRS Section 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation. Indemnification may not be made for any claim, issue or matter as to which such a person has been adjudged by a court of competent jurisdiction, after exhaustion of all appeals there from, to be liable to the corporation or for amounts paid in settlement to the corporation, unless and only to the extent that the court in which the action or suit was brought or other court of competent jurisdiction determines upon application that in view of all the circumstances of the case, the person is fairly and reasonably entitled to indemnity for such expenses as the court deems proper.
NRS Section 78.747 provides that except as otherwise provided by specific statute, no director or officer of a corporation is individually liable for a debt or liability of the corporation, unless the director or officer acts as the alter ego of the corporation. The court as a matter of law must determine the question of whether a director or officer acts as the alter ego of a corporation.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by us is against public policy as expressed hereby in the Securities Act and we will be governed by the final adjudication of such issue.
| 122 |
Item 15. Recent Sales of Unregistered Securities
In September 2011, Cell Source Israel issued an aggregate of 3,673,527 shares of CSL Ordinary Shares to certain founders of Cell Source Israel, including 575,004 CSL Ordinary Shares to Itamar Shimrat, 100,023 CSL Ordinary Shares to Yoram Drucker, 920,830 CSL Ordinary Shares to Yeda and 352,658 CSL Ordinary Shares to Dr. Reisner. In connection with the issuances to the founders, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act.
In November 2011, Cell Source Israel issued an aggregate of 3,750,000 shares of CSL Ordinary Shares to certain accredited investors, including 1,875,000 CSL Ordinary Shares to Ben Friedman’s wife, Phyllis Freidman. Cell Source Israel received gross proceeds of $1,200,000 for the sale of the 3,750,000 CSL Ordinary Shares (the “First Private Placement”). In connection with the issuances to these accredited investors, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act and Rule 506 as promulgated under the Securities Act for transactions not involving a public offering.
In April 2012, Cell Source Israel issued an aggregate of 2,250,000 shares of CSL Ordinary Shares to certain accredited investors, including 1,125,000 CSL Ordinary Shares to Ben Friedman’s wife, Phyllis Freidman. Cell Source Israel received gross proceeds of $800,000 for the sale of the 2,250,000 CSL Ordinary Shares (the “Second Private Placement”). In connection with the issuances to these accredited investors, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act and Rule 506 as promulgated under the Securities Act for transactions not involving a public offering.
In December 2012 and March 2013, a group of five accredited investors (the “Note Investors”), including David Zolty, a director of the Company, and Phyllis Friedman, the wife of the Company's director Ben Friedman, entered into Convertible Loan Agreements (the “Loan Agreements”) pursuant to which the Note Investors loaned Cell Source Israel an aggregate of $510,000 (the “Loan Amount”). In accordance with the Loan Agreements the Note Investors were entitled to receive interest equal to 6% of the Loan Amount per annum and the Loan Amount was payable by Cell Source 6 months after the receipt of the Loan Amount. In November 2013, the Note Investors elected to convert the Loan Amount into CSL Ordinary Shares equal to 18% of Cell Source’s fully-diluted issued and outstanding capital, which issuance did not dilute the Note Investors’ prior holdings (the “Note Exchange”). Accordingly, the Note Investors were issued 2,699,880 CSL Ordinary Shares. In connection with the issuances to these accredited investors, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act and Rule 506 as promulgated under the Securities Act for transactions not involving a public offering.
In October 2013, Cell Source Israel and the Note Investors entered into a Bridge Funding Agreement pursuant to which the Note Investors paid $50,000 to Cell Source Israel in exchange for Cell Source Israel’s agreement to issue to the Note Investors an aggregate of 66,667 CSL Ordinary Shares and a warrant to purchase 100,000 CSL Ordinary Shares at an exercise price of $0.75 per share. In or around November 2013, the Note Investors elected to convert the $50,000 owed to them under the Bridge Funding Agreement into 66,667 CSL Ordinary Shares and warrants to purchase 100,000 CSL Ordinary Shares at an exercise price of $0.75 per share. In connection with the issuances to these accredited investors, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act and Rule 506 as promulgated under the Securities Act for transactions not involving a public offering.
Beginning in November 2013, Cell Source Israel collected and entered into a series of Subscription Agreement with certain accredited investors in a private placement offering. Cell Source Israel held closings of the Private Placement between December 9, 2013 through April 7, 2014, pursuant to which Cell Source Israel sold an aggregate of 4,759,324 Units, at a purchase price of $0.75 per Unit, for gross proceeds of $3,569,475. Each Unit consists of one (1) share of CSL Ordinary Shares and one (1) CSL Warrant. Each CSL Warrant entitled the holder to purchase one (1) share of CSL Ordinary Shares for a five (5) year period at an exercise price of $0.75 per share. In connection with the Private Placement, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act and Rule 506 as promulgated under the Securities Act for transactions not involving a public offering.
In connection with the First Private Placement, the Second Private Placement, the Note Exchange, the Bridge Exchange and the Private Offering (pursuant to which Cell Source Israel sold an aggregate of 4,759,324 CSL Ordinary Shares), Yeda and Dr. Reisner exercised their anti-dilution rights granted to them pursuant to the Yeda License Agreement and the consulting agreement between Dr. Reisner and Cell Source Israel, which anti-dilution rights protected their combined 26% interest in Cell Source Israel against all issuances of CSL Ordinary Shares in connection with financings up to an aggregate of $3,500,000. Pursuant to the anti-dilution protection Yeda and Dr. Riesner were entitled to issuances, in the form of any combination of CSL Ordinary Shares and warrants to purchase CSL Ordinary Shares at par value, at their election. Accordingly, Cell Source Israel issued 239,142 CSL Ordinary Shares and warrants to purchase 1,995,376 CSL Ordinary Shares at par value to Yeda and 807,314 CSL Ordinary Shares and warrants to purchase 48,459 CSL Ordinary Shares at par value to Dr. Reisner. The warrants issued to Yeda and Dr. Reisner have an exercise price of $0.001 per share and have a life of 7 years. In addition, the warrants have a cashless exercise provision and were fully vested on the date of the grant. In connection with the issuances to Yeda and Dr. Reisner, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act.
In July 2014, the Company issued 100,000 shares of common stock to Sichenzia Ross Friedman Ference LLP for legal services rendered, which shares of common stock are being registered pursuant to this Registration Statement. In connection with the issuance for legal services, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act.
In September 2014, the Company issued 233,333 shares of common stock in connection with a cashless exercise of a warrant to purchase 400,000 shares of common stock at $0.75 per share. In connection with the issuance of warrant exercise shares, the Company relied upon the exemption from securities registration provided by Section 4(a)(2) of the Securities Act.
The transactions described above were exempt from securities registration provided by Section 4(a)(2) of the Securities Act and Rule 506 as promulgated under the Securities Act for transactions not involving a public offering and under Regulation S promulgated by the SEC.
| 123 |
Item 16. Exhibits and Financial Statement Schedules.
| Exhibit Number | Description | |
| 2.1 (1) | Share Exchange Agreement, dated June 30, 2014, by and between Cell Source, Ltd., and Ticket to See, Inc. | |
| 3.1 (1) | Articles of Association of Cell Source Limited, dated August 14, 2011, as amended on November 11, 2013 | |
| 3.2 (2) | Articles of Incorporation of Ticket to See, Inc., dated June 6, 2012 | |
| 3.3 (3) | Certificate of Amendment to Articles of Incorporation of Ticket to See, Inc., dated June 23, 2014 | |
| 3.3 (4) | Certificate of Amendment to Articles of Incorporation of Ticket to See, Inc., dated May 20, 2014 | |
| 3.4 (2) | Bylaws of Cell Source, Inc., dated June 6, 2012 | |
| 5.1* | Opinion of Sichenzia Ross Friedman Ference LLP | |
| 10.1 (1) | Form of Subscription Agreement | |
| 10.2 (1) | Form of Registration Rights Agreement | |
| 10.3 (1) | Form of Investor Warrant | |
| 10.4 (1) | Form of Researcher Company Warrant | |
| 10.5 (1) | Form of Company Warrant | |
| 10.6 (1) | Form of Lockup Agreement (included in Exhibit 2.1) | |
| 10.7 (1) | Research and License Agreement by and between Yeda Research and Development Company Limited and Cell Source Limited, dated October 3, 2011 | |
| 10.8 (1) | Amendment to Research and License Agreement | |
| 10.9 (1) | Evaluation and Exclusive Option Agreement by and between Yeda Research and Development Company Limited and Cell Source Limited, dated Oct. 3, 2011 (included in Exhibit 10.7) | |
| 10.10 (1) | Amendment dated April 1, 2014 to Evaluation and Exclusive Option Agreement by and between Yeda Research and Development Company Limited and Cell Source Limited | |
| 10.11 (1) | Second Amendment dated June 22, 2014 to Evaluation and Exclusive Option Agreement by and between Yeda Research and Development Company Limited and Cell Source Limited | |
| 10.12 (1) | Consulting Agreement by and between Cell Source Limited and Professor Yair Reisner | |
| 10.13 (6) | Form of Amendment No. 1 to Registration Rights Agreement | |
| 10.14(7) | Bridge Funding Agreement | |
| 10.15(5) | Third Amendment dated June 22, 2014 to Evaluation and Exclusive Option Agreement by and between Yeda Research and Development Company Limited and Cell Source Limited | |
| 23.1** | Consent of Marcum LLP | |
| 23.2* | Consent of Sichenzia Ross Friedman Ference LLP (included in Exhibit 5.1) | |
| 24.1** | Power of Attorney (included in Registration Statement under "Signatures") | |
| 16.1 (1) | Letter from Paritz & Company, P.A. | |
| 99.1 (1) | Pro forma financial information | |
| 101.INS*** | XBRL Instance Document | |
| 101.SCH*** | XBRL Taxonomy Extension Schema Document | |
| 101.CAL*** | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF*** | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB*** | XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE*** | XBRL Taxonomy Extension Presentation Linkbase Document |
| (1) | Incorporated by reference to the Company's current report on Form 8-K filed with the Securities and Exchange Commission on July 1, 2014 |
| (2) | Incorporated by reference to the Company's Registration Statement on Form S-1 filed with the Securities and Exchange Commission on June 6, 2012 |
| (3) | Incorporated by reference to the Company's Form 8-K filed with the Securities and Exchange Commission on June 26, 2014. |
| (4) | Incorporated by reference to the Company's Form 8-K filed with the Securities and Exchange Commission on June 6, 2014. |
| (5) | Incorporated by reference to the Company's Form 10-Q filed with the Securities and Exchange Commission on August 19, 2014. |
| (6) | Incorporated by reference to the Company's Registration Statement on Form S-1 filed with the Securities and Exchange Commission on August 8, 2014. |
| (7) | Incorporated by reference to the Company’s Registration Statement Form S-1/A filed with the Securities and Exchange Commission on September 23, 2014. |
| *** | Furnished herewith |
| ** | Filed herewith |
| * | To be filed by amendment |
Item 17. Undertakings.
(a) The undersigned registrant hereby undertakes to:
(1) File, during any period in which it offers or sells securities, a post-effective amendment to this Registration Statement to:
(i) Include any prospectus required by Section 10(a)(3) of the Securities Act;
(ii) Reflect in the prospectus any facts or events which, individually or together, represent a fundamental change in the information in the Registration Statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective Registration Statement.
| 124 |
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) The undersigned registrant hereby undertakes that, for the purpose of determining any liability under the Securities Act of 1933, as amended, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) The undersigned registrant hereby undertakes to remove from registration by means of a post-effective amendment any of the securities being registered that remain unsold at the termination of the offering.
(4) The undersigned registrant hereby undertakes that, for the purposes of determining liability to any purchaser:
If the registrant is subject to Rule 430C, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(5) Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended, may be permitted to directors, officers and controlling persons of the undersigned registrant according the foregoing provisions, or otherwise, the undersigned registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933, as amended, and will be governed by the final adjudication of such issue.
| 125 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in Tel Aviv, Israel, on October 24, 2014.
| CELL SOURCE, INC. | ||
| Dated: October 24, 2014 | By: | /s/ Itamar Shimrat |
| Itamar Shimrat | ||
| Chief Executive Officer, Chief Financial Officer and Director | ||
IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney. In accordance with the requirements of the Securities Act of 1933, as amended, this registration statement was signed by the following persons in the capacities and on the dates stated:
| SIGNATURE | TITLE | DATE | |||
| By: | */s/ Yoram Drucker | Chairman | October 24, 2014 | ||
| Yoram Drucker | |||||
| By: | /s/ Itamar Shimrat | Chief Executive Officer, Chief Financial Officer and Director | October 24, 2014 | ||
| Itamar Shimrat | (Principal Executive, Financial and Accounting Officer) | ||||
| By: | */s/ Ben Friedman | Director | October 24, 2014 | ||
| Ben Friedman | |||||
| By: | */s/ Dennis Brown | Director | October 24, 2014 | ||
| Dennis Brown | |||||
| By: | */s/ David Zolty | Director | October 24, 2014 | ||
| David Zolty | |||||
| By: | */s/ Itamar Shimrat | Director | October 24, 2014 | ||
| Itamar Shimrat | |||||
| Attorney-in Fact | |||||
| October 24, 2014 | |||||
| 126 |